Результат интеллектуальной деятельности: КЛАСС БИФУНКЦИОНАЛЬНЫХ СОЕДИНЕНИЙ СО СТРУКТУРОЙ СОЛИ ЧЕТВЕРТИЧНОГО АММОНИЯ
Вид РИД
Изобретение
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к классу соединений с бифункциональной структурой соли четвертичного аммония с активностью агониста β2-адренорецептора и антагониста мускаринового рецептора (М-рецептора). Настоящее изобретение также относится к фармацевтическим композициям, включающим в себя такие соединения-соли четвертичного аммония, способам получения таких соединений-солей четвертичного аммония и их промежуточных соединений, а также к применениям в лечении легочных нарушений.
Предшествующий уровень техники настоящего изобретения
Астма и хроническое обструктивное заболевание легких (COPD) являются наиболее распространенными заболеваниями из легочных нарушений, при этом COPD является четвертым по распространенности заболеванием со смертельным исходом в мире, и предполагается, что оно станет третьим к 2020 году. COPD обычно ассоциировано с курением сигарет, и пациенты также легко могут страдать от других загрязнений окружающей среды, таких как связанное с профессиональной деятельностью вредное воздействие пыли, газа, дыма и смога.
Бронхолитические средства являются наиболее предпочтительным вариантом при лечении астмы и COPD. Обычные бронхолитические средства включают в себя агонист β2-адренорецептора (например, альбутерол, формотерол, сальметерол и индакатерол) и антагонист М-рецептора (например, бромид гликопиррония, бромид ипратропия, бромид тиотропия и т.д.). Дополнительно к единому препарату препарат с соединениями, состоящими из агониста β2-адренорецептора и антагониста М-рецептора, также применяют для лечения заболеваний легких, таких как астма и COPD, и он характеризуется лучшим терапевтическим эффектом. Например, в патенте США US 6433027 раскрыта терапевтическая композиция, включающая в себя антагонист М-рецептора в виде бромида тиотропия и агонист β2-адренорецептора в виде фумарата формотерола.
Кроме того, о применении соединений, характеризующихся как агонистической активностью в отношении β2-адренорецептора, так и антагонистической активностью в отношении М-рецептора, для лечения астмы и COPD, сообщается во многих литературных источниках, таких как международные заявки WO 2004074246, WO 2009098448, WO 2010004517, WO 2008096127, WO 2008149110, WO 2008017827, WO 2010126025, WO 2010015792, WO 2005111004, WO 2010015792, WO 2008041095, WO 2005051946, WO 2011012896, WO 2010123766 и т.д. Химическая структура этих соединений, характеризующихся как агонистической активностью в отношении β2-адренорецептора, так и антагонистической активностью в отношении М-рецептора (сокращенно обозначаемые МАВА), состоит из трех фрагментов, которые представляют собой фрагмент с антагонистической активностью в отношении М-рецептора, фрагмент с агонистической активностью в отношении β2-адренорецептора и линкерный фрагмент. Типичное МАВА-соединение имеет следующую структурную формулу:
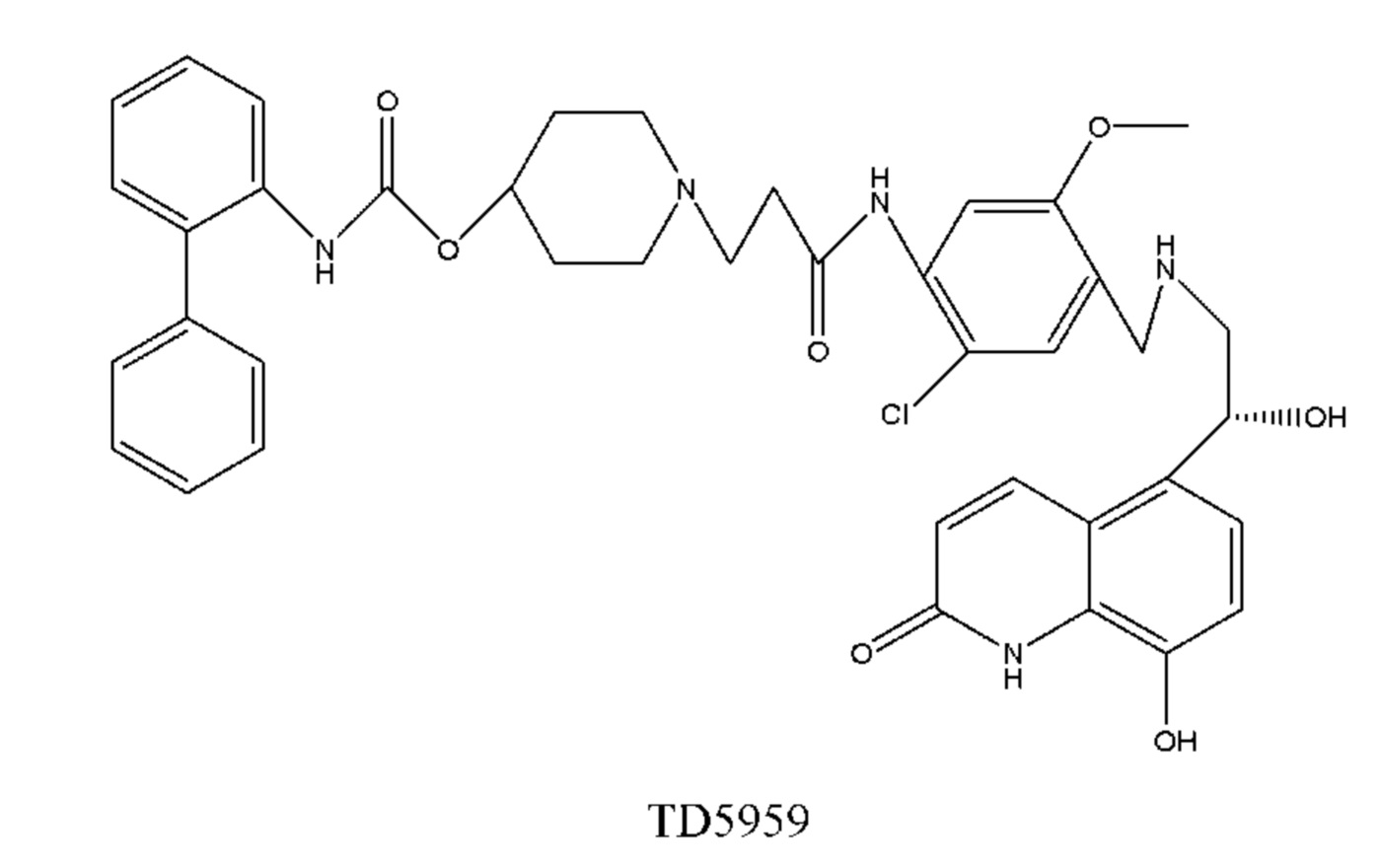
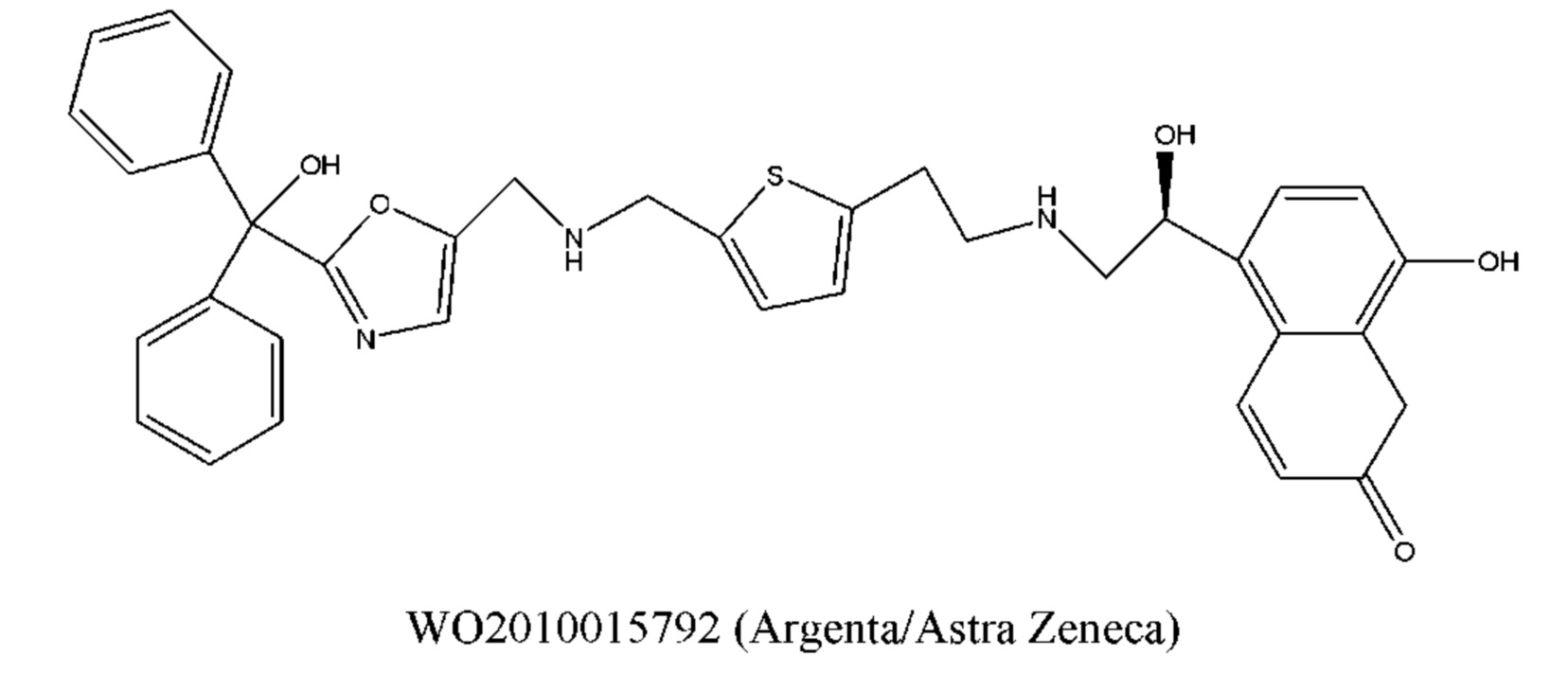
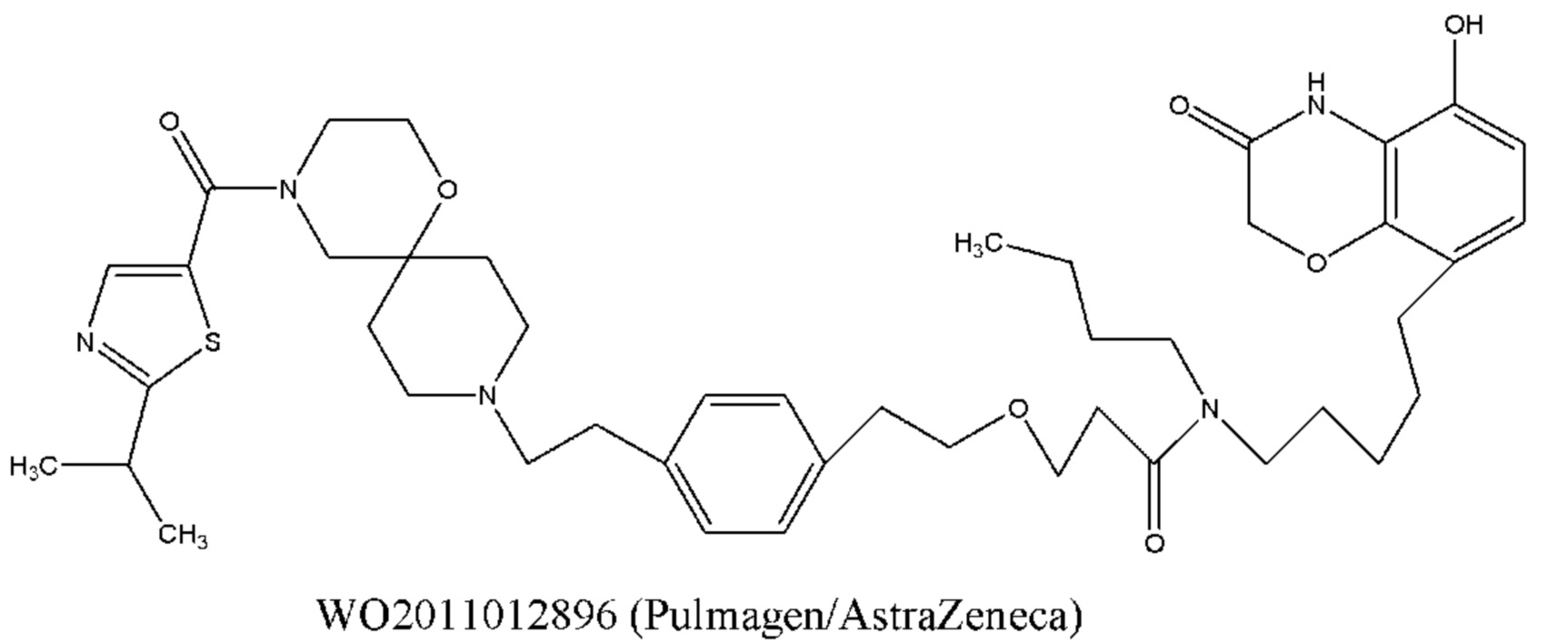
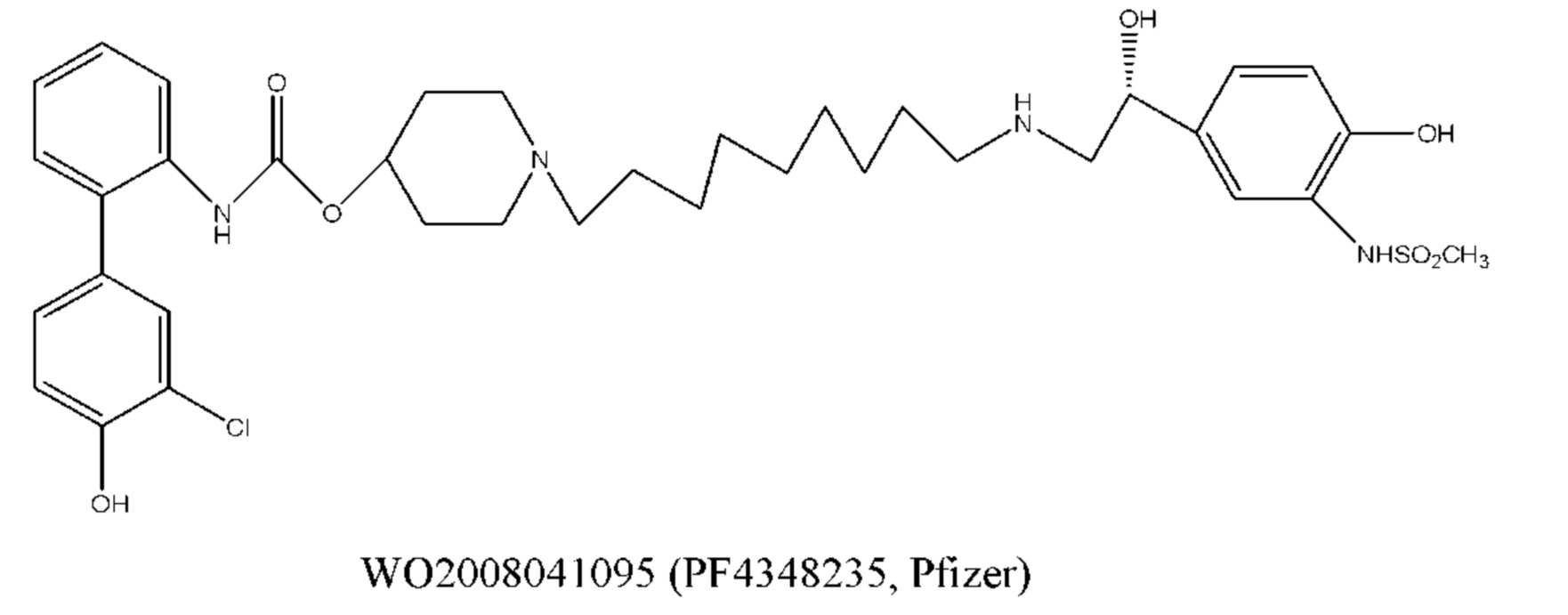
Такие бифункциональные соединения способны оказывать бронходилатирующее воздействие посредством двух отдельных механизмов действия и имеют фармакокинетические свойства единой молекулы. Клиническое исследование фазы II иллюстративного соединения TD5959 показало, что соединение оказывает хорошее воздействие на умеренное и тяжелое COPD. Тем не менее, такие бифункциональные соединения, описываемые в литературе, в настоящее время являются менее селективными в отношении подтипа М-рецептора. Недостаток предшествующего уровня техники заключается в том, что подавляющее большинство таких бифункциональных соединений в литературе представляют собой соединения со структурой типа третичного амина, которые могут проходить через гематоэнцефалический барьер, оказывают другие центральные побочные эффекты и не характеризуются явной селективностью в отношении подтипа М3-рецептора, блокируют М2-рецептор, создавая много побочных реакций, таких как повышенная частота сердечных сокращений, повышенное кровяное давление и обострение астмы. Соли четвертичного аммония трудно проходят через гематоэнцефалический барьер, и проглатываемая часть ингаляционных препаратов характеризуются низкой биологической доступностью. Таким образом, идеальным выбором является создание и синтез МАВА-соединения со структурой соли четвертичного аммония. Тем не менее, в международных заявках WO 2004074246 и WO 2008017827 сообщалось о ряде соединений, полученных посредством связывания групп, связывающих атомы N у двух активных единиц известного активного антагониста М-рецептора и агониста β2-адренорецептора, но результаты скрининга биологической активности не являются идеальными. В международной заявке WO 2004074246 показано, что активность антагонистического фрагмента М-рецептора существенно снижалась в 100 раз. В международной заявке WO 2008017827 описывается активность агонистического фрагмента β2-адренорецептора, которая существенно снижается более чем в 100 раз, и такое МАВА не достигает желаемых результатов, что привело к отказу от подобного способа для поиска идеального МАВА в виде соли четвертичного аммония, с того времени не сообщалось о подобных исследованиях.
Краткое раскрытие настоящего изобретения
Цель настоящего изобретения заключается в обеспечении соединения-соли четвертичного аммония, характеризующегося агонистической активностью в отношении β2-адренорецептора и антагонистической активностью в отношении М-рецептора, и его фармацевтически приемлемой соли, сольвата, оптического изомера и предшественника. С этой целью с помощью непрерывных изменений в активных единицах в виде антагонистов М-рецепторов и агониста β2-адренорецептора и связывающих группах в результате обширных исследований и скрининга согласно настоящему изобретению был неожиданно открыт новый класс идеального МАВА-соединения со структурой соли четвертичного аммония. Предварительные эксперименты на животных доказали, что активность таких соединений удовлетворяет требованиям для обеспечения более заметных преимуществ, чем МАВА из уровня техники: (1) в сравнении с предшествующим уровнем техники доза, в которой внутривенная инъекция соединения согласно настоящему изобретению вызывает повышение частоты сердечных сокращений, более чем в 10 раз превышает таковую для соединения из предшествующего уровня техники; (2) соединение согласно настоящему изобретению характеризуется сильным пресистемным метаболизмом в печени, но является стабильным в легких и не может накапливаться в достаточной дозе для токсичного воздействия, даже если оно попадает в кровеносную систему; и (3) степень соответствия между антагонистической активностью в отношении М-рецептора и агонистической активностью в отношении β2-рецептора у соединения согласно настоящему изобретению является практически идеальной. В сравнении с предшествующим уровнем техники соединение согласно настоящему изобретению характеризуется высокой селективностью в отношении подтипа М-рецептора, имеет характеристики быстрого действия, продолжительного времени действия, более низкие токсические и побочные эффекты, и вследствие структуры соли четвертичного аммония оно трудно проходит через гематоэнцефалический барьер, легко метаболизируется и менее склонно к побочным эффектам, связанным с сердечно-сосудистой системой. Структура этого класса соединений представлена в виде формулы I:
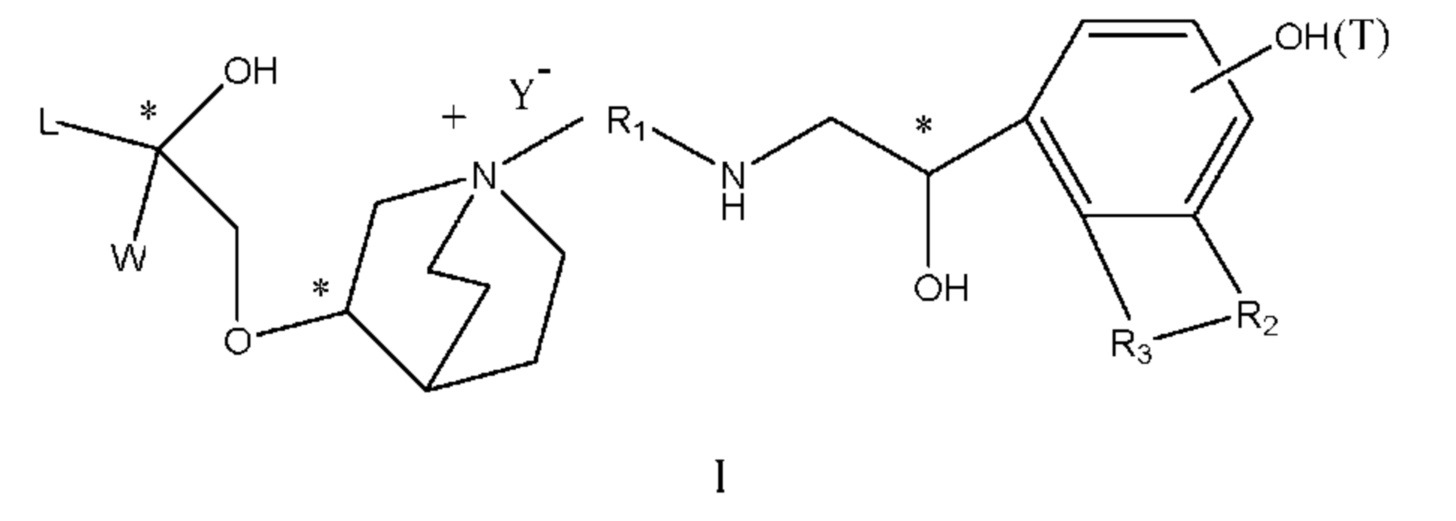
Углероды, помеченные *, в формуле I все являются (R) конфигурации.
L представляет собой (4-10С) ар ил или гетероарил, где гетероатом гетероарила выбран из N, О и S, и вышеуказанные группы могут быть незамещенными или необязательно замещенными одним или несколькими заместителями, выбранными из галогена, -OR1, -SR1, -NR1R2, -NHCOR1, -CONR1R2, -CN, -NO2, -COOR1, -CF3 и C1-C4 неразветвленного или разветвленного гидрокарбила.
R1, R2 могут быть атомом водорода, С1-С4 неразветвленным или разветвленным гидрокарбилом.
L предпочтительно представляет собой незамещенную фенильную группу, пиридильную группу, фурильную группу или тиенильную группу.
W независимо выбран из замещенного или незамещенного (3-7С)циклоалкила, заместитель выбран из галогена, (1-4С)алкила, (1-4С)алкокси, алкоксигидрокарбила и гетероцикла. Предпочтительно, W представляет собой незамещенный (3-7С)циклоалкил и наиболее предпочтительно W представляет собой циклобутил, циклопентил и циклогексил.
R1 представляет собой дивалентную группу -(R1a)d-(A1)e-(R1b)f-, где d, е и f каждый независимо выбран из 0, 1,2 или 3 и количество примыкающих атомов в самой короткой цепи между двумя атомами азота, к которому R1 присоединен, находится в диапазоне от 3 до 14.
R1a и R1b каждый независимо выбран из (1-10С)алкилена, (2-10С)алкенилена, (1-4С)алкиленокси, алкиленоксиалкила, алкиленамидо, алкиленацилокси, алкиленамино и т.п., где каждый из алкилена, алкенилена, алкиленокси, алкиленоксиалкила, алкиленамино, алкиленацилокси, алкиленамидо является незамещенным или замещен заместителями, независимо выбранными из (1-4С)алкила, хлора, фтора, гидрокси, фенила и замещенного фенила, R1a и R1b могут быть одинаковыми или разными.
A1 независимо выбран из (3-7С)циклоалкилена, (2-7С)алкилена, (6-10С)арилена, (4-9С)гетероарилена и (3-8С)гетероциклоалкилена и т.п., где циклоалкилен может быть незамещенным или замещен 1-4 заместителями, независимо выбранными из (1-6С)алкила. Каждый из арилена, гетероарилена и гетероциклоалкилена может быть незамещенным или замещен 1-3 заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси, -S-(1-4С)алкила, -S(O)-(1-4С)алкила, -S(O)2-(1-4С)алкила, -С(O)-O-(1-4С)алкила, -NH-(1-4С)алкила, -N=[(1-4С)алкил]2, карбокси, нитро, циано, амидо, сложноэфирной группы, трифторметила и трифторметокси.
В частности, R1a и R1b в дивалентной группе R1 каждый независимо выбран из (1-10С)алкилена, (1-4С)алкиленокси, алкиленамидо и т.п. A1 независимо выбран из (6-10С)арилена и т.п., где арилен может незамещенными или замещен 1-2 заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси, карбокси, нитро, циано, амидо или сложноэфирной группы.
R1 дополнительно выбран из: -(СН2)3-, -(СН2)4-, -(СН2)8-, -(СН2)9-, -(СН2)10-, -(СН2)2O(СН2)2-, -(СН2)2O(СН2)4-, -(СН2)3O(СН2)4-, -(СН2)4O(СН2)4-, -(СН2)5O(СН2)4-, -(СН2)2O(СН2)2O(СН2)2-, -(СН2)2O(СН2)3O(СН2)2-, -CH2O(СН2)5ОСН2-, -(СН2)2O(СН2)2O(СН2)2O(СН2)2-, -(СН2)2O(фен-1,4-илен)(СН2)2-, -(СН2)3O(фен-1,4-илен)СН2-, -(СН2)3О(3,5-дихлорфен-1,4-илен)СН2-, -(СН2)2CONH(2-метокси-5-хлорфен-1,4-илен)СН2-, -(СН2)2CONH(3-метилфен-1,4-илен)СН2-, -(СН2)2CONH(2-метоксифен-1,4-илен)СН2-, -(СН2)2CONH(фен-1,4-илен)СН2-, -(СН2)3CONH(фен-1,4-илен)СН(СН3)-, -(СН2)3ОСН2(3-метоксифен-1,4-илен)СН(СН3)-, -(СН2)3O(фен-1,4-илен)С(СН3)2-, -(СН2)3О(фен-1,4-илен)СН(СН2СН3)-, -(СН2)3O(фен-1,4-илен)(СН2)2-, -(СН2)3O(фен-1,4-илен)(СН2)3-, -(СН2)3O(фен-1,4-илен)СН2СН(СН3)-, -(СН2)2ОСН2(фен-1,4-илен)CH2O(СН2)2-, -(СН2)2ОСН2(фен-1,4-илен)СН(СН3)-, -(СН2)2O(фен-1,4-илен)O(СН2)2-, -(СН2)3O(2-метоксифен-1,4-илен)O(СН2)3-, -(СН2)3O(фен-1,4-илен)O(СН2)3-, -(СН2)3O(фен-1,4-илен)СН2С(СН3)2-, -(СН2)2O(фен-1,4-илен)СН2С(СН3)2-, -(СН2)2O(фен-1,4-илен)СН2СН(СН3)-, -(СН2)2O(фен-1,4-илен)(СН2)2- и -(СН2)2O(СН2)3O(СН2)2-.
R2 выбран из -N(R2a)C(R2b)(O), -C(R2c)(R2d)OR2e, -N(R2f)-, -О- и т.п. R3 выбран из водорода, -C(R3a)=C(R3b)-C(O)-, -OC(R3c)(R3d)C(O)-, -N(R3e)CH(R3f)C(O)-, -C(R3g)(R3h)S(O)2-, -SCO- и т.п., при условии, что если R3 представляет собой водород, R2 выбран из -N(R2a)C(R2b)(O) и -C(R2c)(R2d)OR2e и если R3 выбран из -C(R3a)=C(R3b)-C(O)-, -OC(R3c)(R3d)C(O)-, -N(R3e)CH(R3f)C(O)-, -C(R3g)(R3h)S(O)2- и -SCO-, R2 выбран из -N(R2f) - и -О-.
R2a-2f и R3a-3h каждый независимо выбран из водорода или (1-4С)алкила.
R2 предпочтительно выбран из -NHCHO, -СН2ОН, -NH- и -О-.
R3 предпочтительно выбран из: водорода, -CH=CH-C(O)-, -ОСН2С(O)-, -NHCH2C(O)-, -CH2S(O)2- и -SCO-.
Т представляет собой положение гидрокси на бензольном кольце и выбран из орто-или мета-положения R2 на бензольном кольце.
Y- выбран из фармацевтически приемлемых кислотных радикалов, включая неорганические кислотные радикалы, такие как Br-, Cl-, I-, бикарбонат, карбонат, бисульфат, сульфат, нитрат, фосфат, гидрофосфат, дигидрофосфат и фосфит; и органические кислотные радикалы, такие как формиат, ацетат, пропионат, изобутират, метансульфонат, пара-толуолсульфонат, бензоат, оксалат, тартрат, фумарат, малонат, сукцинат, суберат, манделат, фталат, бензолсульфонат, цитрат, глюкуронат, галактонат и аминокислотный радикал. Предпочтительно Y- представляет собой Br- или Cl-.
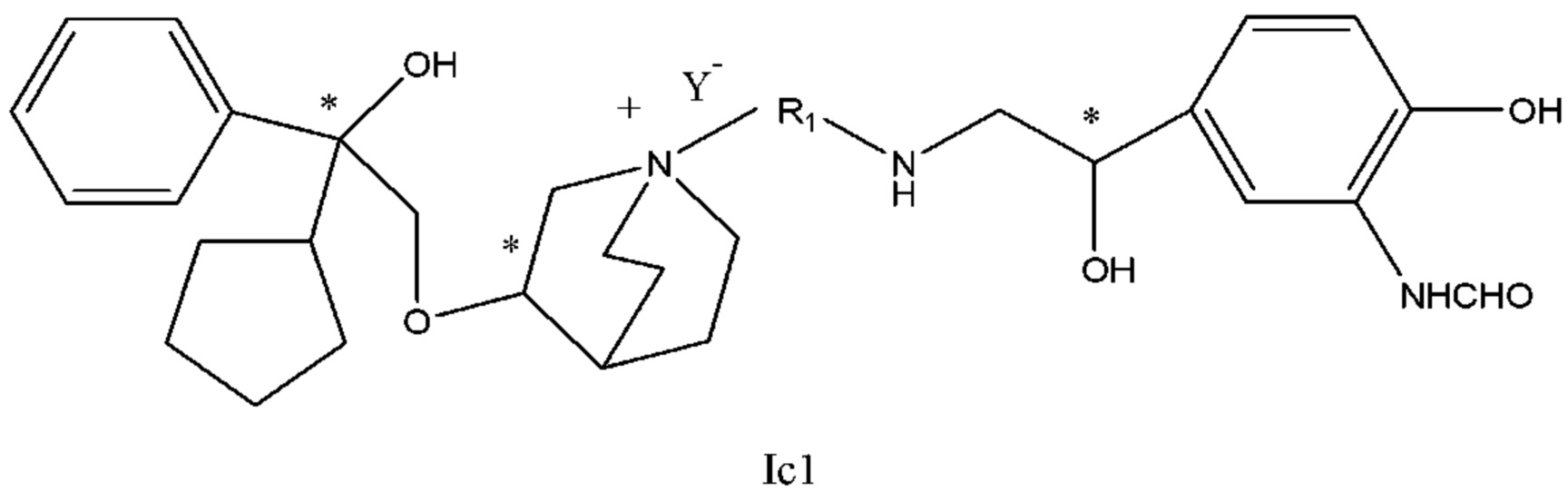
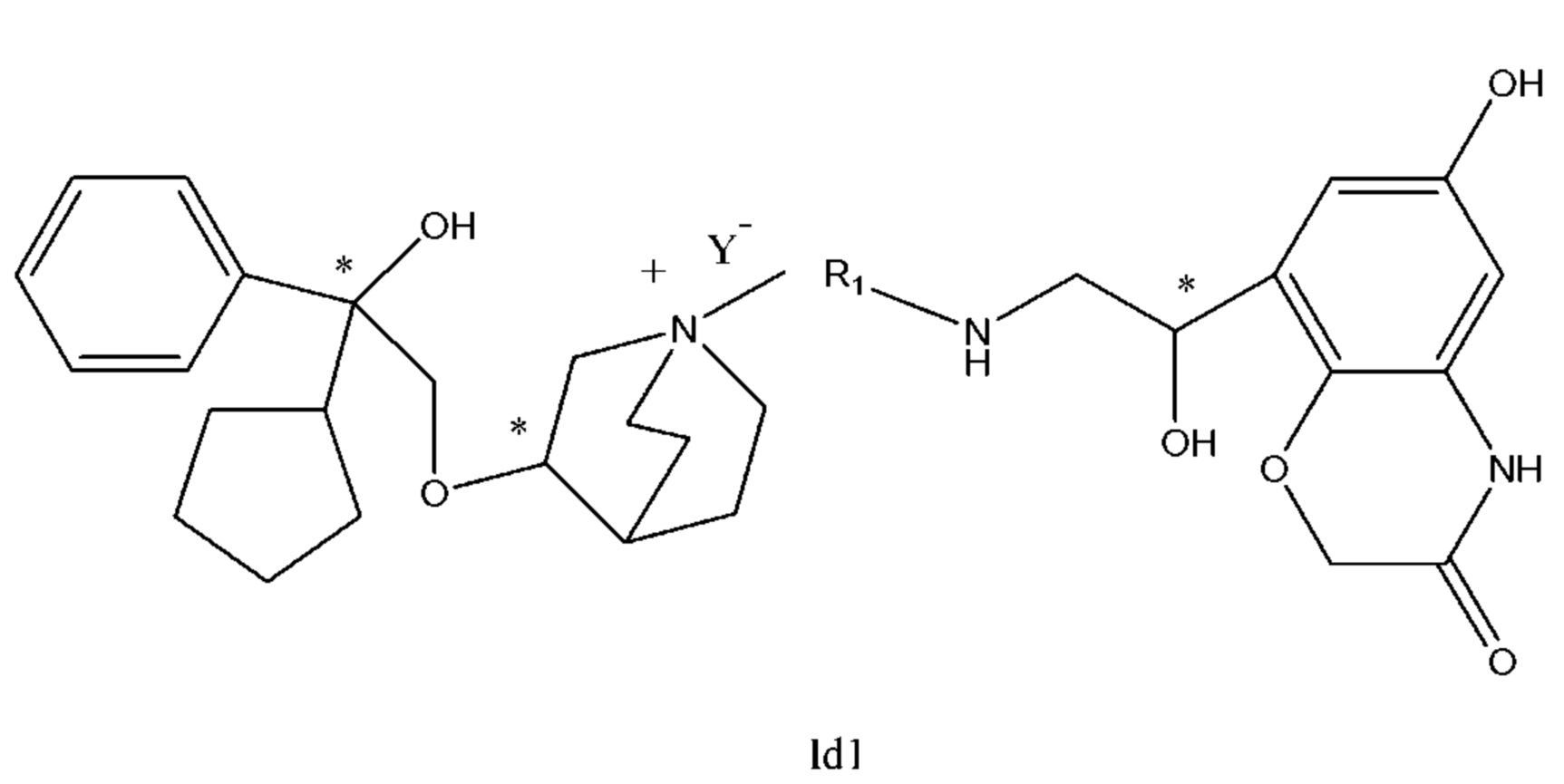
Настоящее изобретение также включает в себя соединения, представленные следующими структурными формулами Ia-Id:
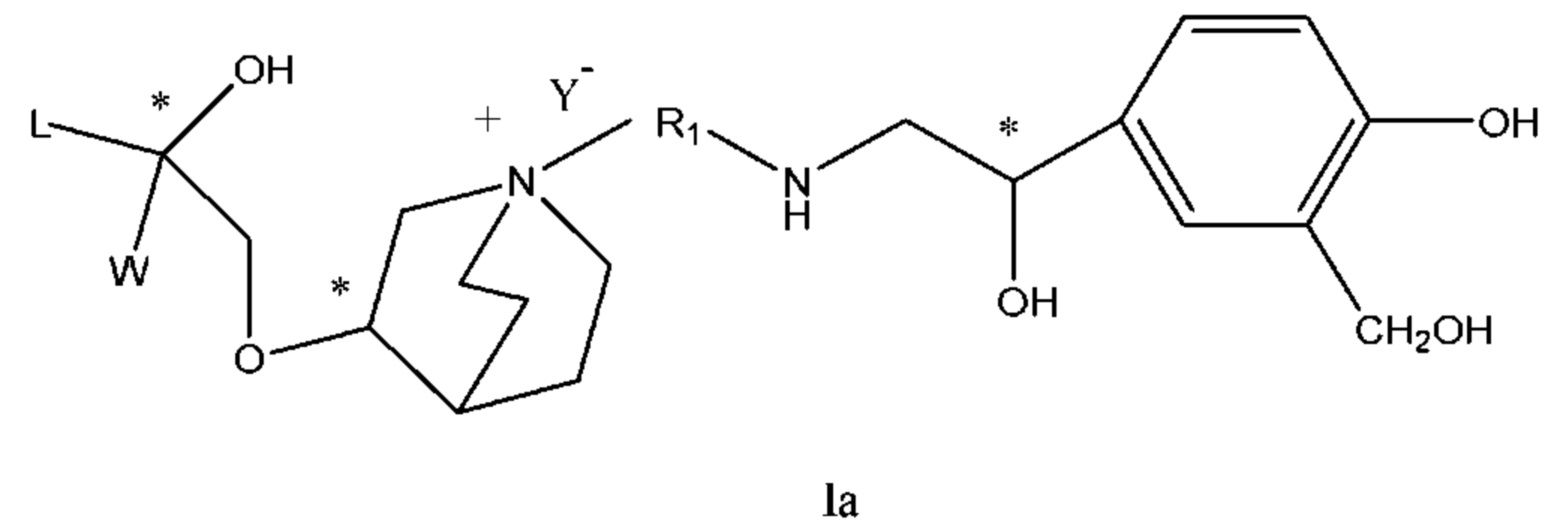
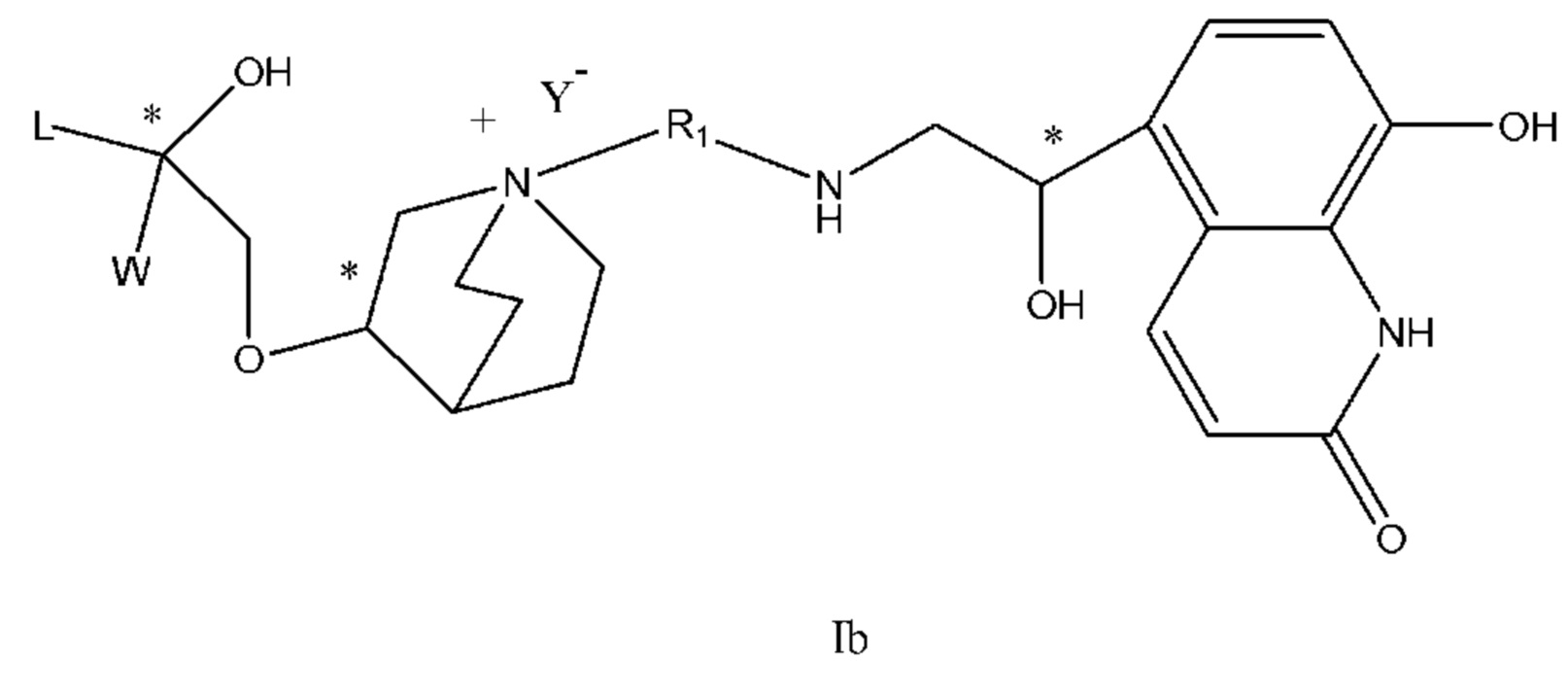
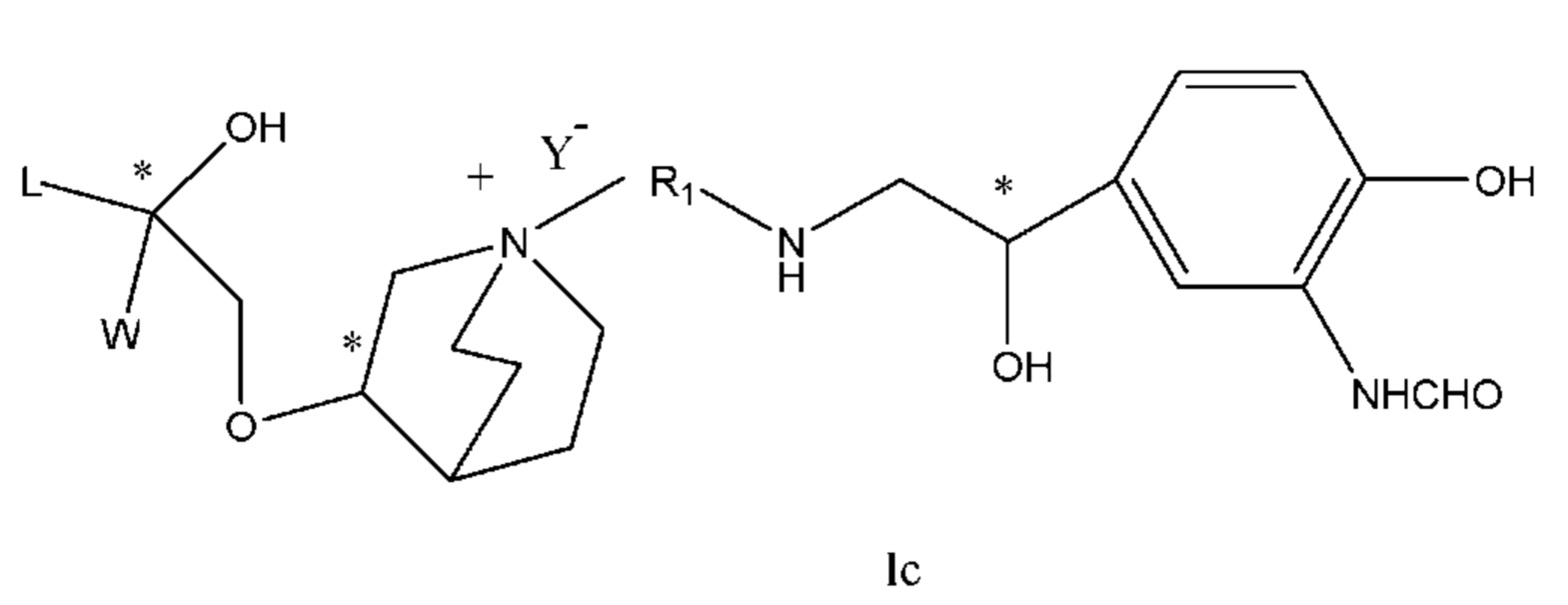
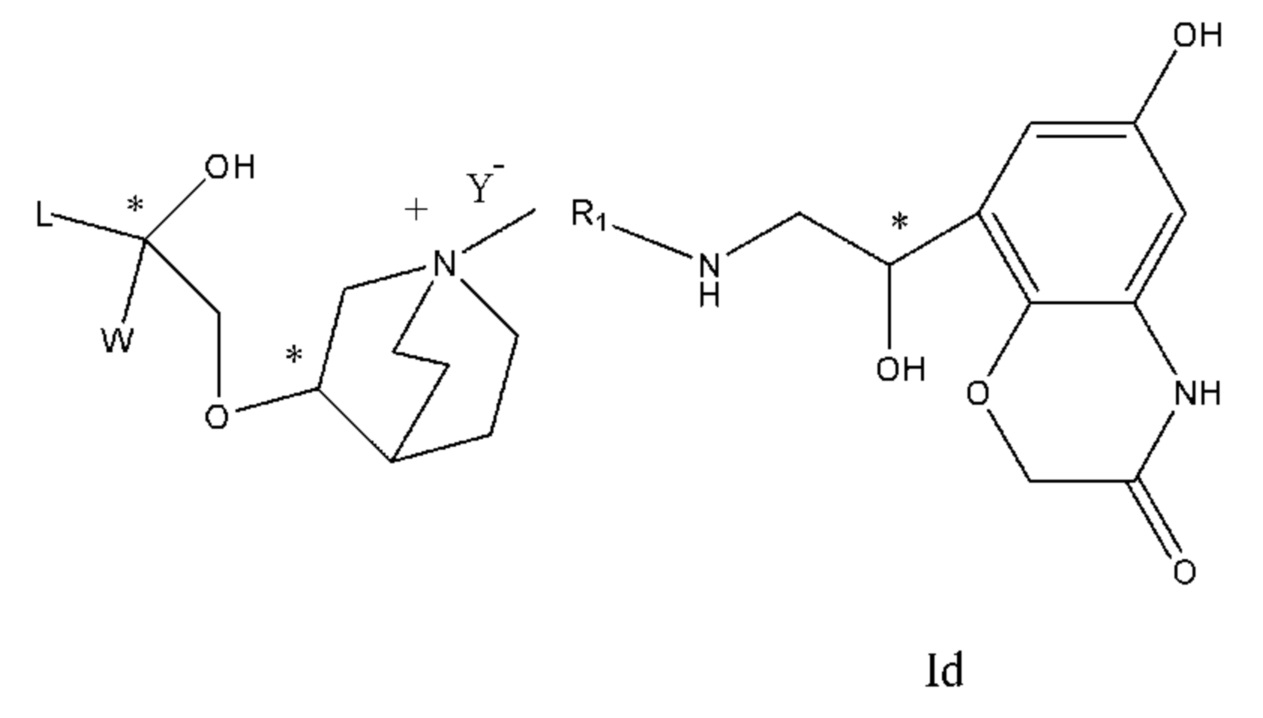
В формулах Ia-Id: углероды, помеченные *, являются (R) конфигурации и L, W, R1, Y соответствуют группам, определенным выше.
Соединения четвертичной аммониевой соли по настоящему изобретению обладают антагонизмом М рецептора и агонизмом β2 адреноцрецептора. Их легко получали из исходных веществ рынка и получали описанными ниже общими способами и также могут быть получены с применением другой информации, легко доступной специалистам настоящей области техники. Конкретные варианты осуществления и связанные способы описаны в настоящем изобретении и соответствующие соединения могут не только быть получены с применением способов настоящего изобретения специалистами настоящей области техники, а также были получены с применением других веществ, способов и исходных веществ. Если не отмечено иное, в дополнение к условиям общего или предпочтительных способов (т.е., температура реакции, давление, время, используемый растворитель, молярное отношение реагентов, и т.п.), представленных в настоящем изобретении, могут быть использованы другие способы и условия. В то время, как оптимальные реакционные условия меняются в зависимости от конкретных реагентов или растворителей, специалисты настоящей области техники могут легко определить такие реакционные условия традиционными процедурами оптимизации.
Более того, специалистам настоящей области техники будет очевидно, что традиционные защитные группы являются необходимыми для предотвращения нежелательных химических реакций конкретных функциональных групп от взаимодействия с достижением заданной реакции. Подходящие защитные группы для конкретных функциональных групп, а также подходящие условия для введения и снятия защитных групп с таких функциональных групп, хорошо известны из области техники. При необходимости в настоящем изобретении также могут быть использованы защитные группы, отличные от этих. Условия для введения и снятия защитных групп с различных функциональных групп описаны подробно в различных литературных источниках.
Настоящее изобретение относится к способу получения соединения формулы I и его фармацевтически приемлемой соли, сольвата или их смесям и к применению новых промежуточных соединений при получении таких соединений. Способ получения соединения формулы I или его фармацевтически приемлемой соли, сольвата или оптического изомера включает в себя:
(a) осуществление взаимодействия промежуточного соединения 1 или его соли с X1-R1-X2 с образованием промежуточного соединения 2;
(b) осуществление взаимодействия промежуточного соединения 2 с промежуточным соединением 3 с образованием промежуточного соединения 4 с защитными группами;
(c) снятие защитных групп с промежуточного соединения 4 или другого соединения формулы I с защитными группами с получением соединения формулы I; и
(d) обмен соединения формулы I с основной анионообменной смолой с получением гидроксида формулы I и затем осуществление взаимодействия с различными кислотами с образованием четвертичных аммонийных солей с различными кислотными радикалами; или обмен соединения формулы I с конкретной анионообменной смолой с получением четвертичной аммонийной соли с конкретным кислотным радикалом; или осуществление взаимодействия галогенида формулы I с оксидом серебра с образованием гидроксида формулы I и затем осуществление взаимодействия с другими кислотами с образованием четвертичных аммонийных солей с различными кислотными радикалами; или осуществление взаимодействия галогенида формулы I с солью серебра с получением четвертичной аммонийной соли с соответствующим кислотным радикалом.
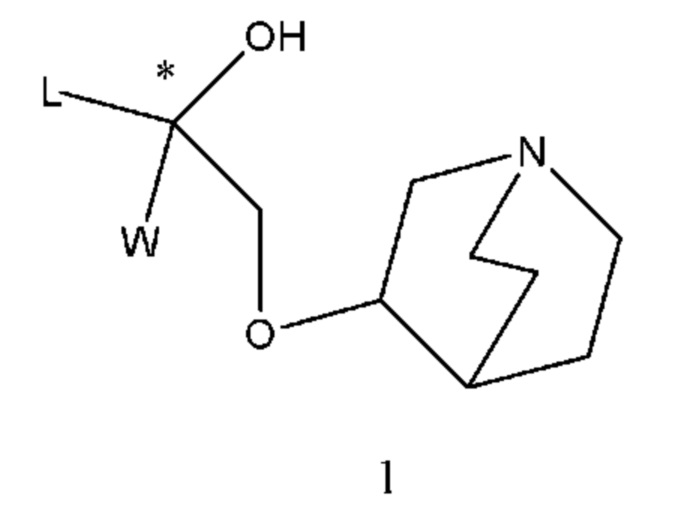
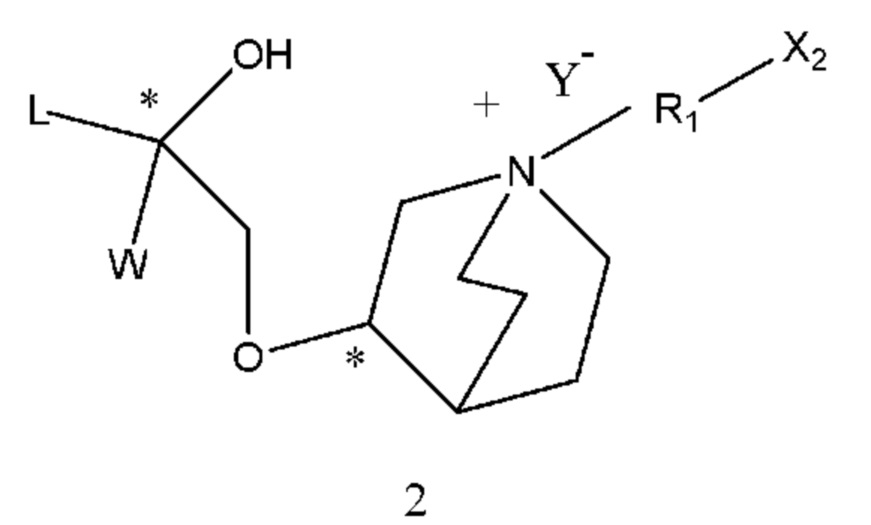
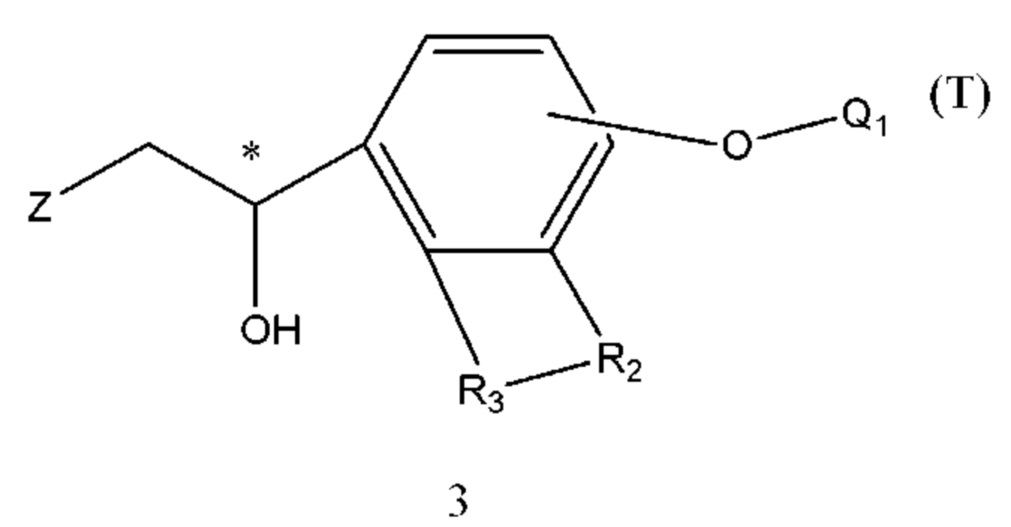
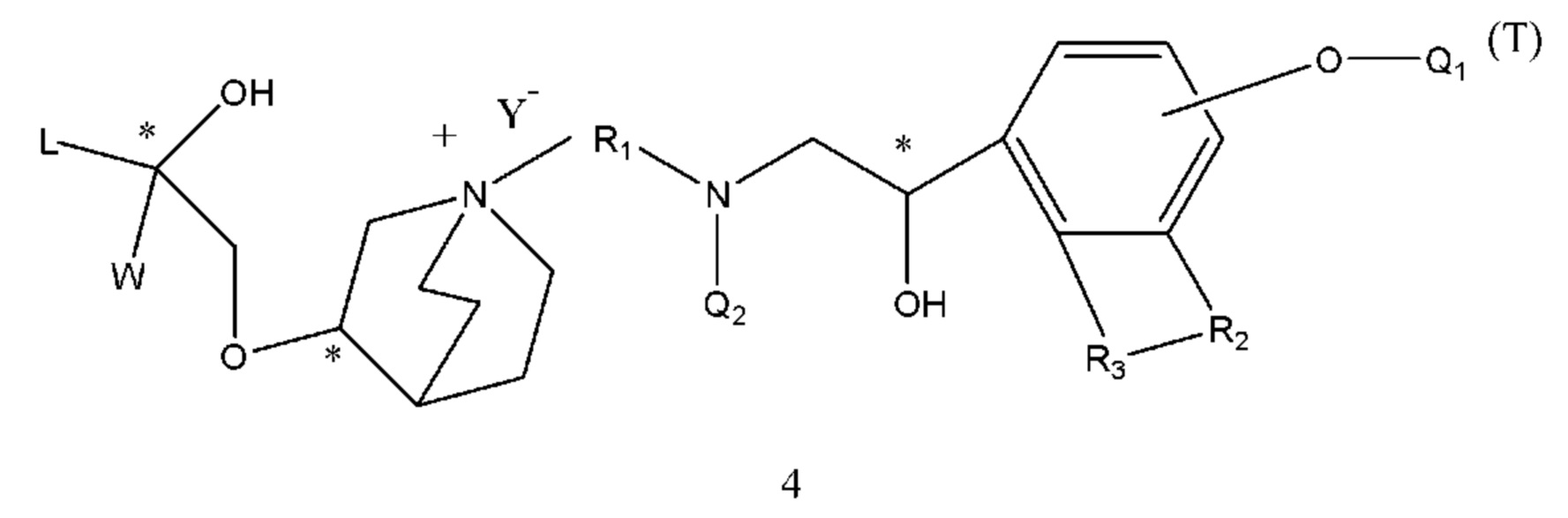
В промежуточных соединениях 1, 2, 3, 4 углероды, помеченные *, обозначенные как R конфигурации, далее такие же, как и определено выше.
Т представляет собой положение группы на фенильном кольце и выбран из орто- и мета-положения R2 на бензольном кольце.
Q1 представляет собой водород или гидроксизащитную группу, которая выбрана из силильных эфиров, таких как триметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил и т.п., сложных эфиров (ацильных групп), таких как фор мил, ацетил и т.п., и арилметильных групп, таких как бензил, пара-метоксибензил, 9-флуоренилметил, бензгидрил и т.п. Q2 представляет собой водород или аминозащитную группу, которая выбрана из бензила (Bn), трет-бутоксикарбонила (Boc), бензилоксикарбонила (Cbz), 9-флуоренилметоксикарбонила (Fmoc), формила, ацетила и т.п. X1 и Х2 в соединении X1-R1-Х2 независимо выбраны из NHQ2, галогена, такого как хлор, бром и йод, и сульфонатов, таких как метансульфонат, пара-толуолсульфонат и карбонил. Z выбран из NHQ2, галогена, такого как хлор, бром и йод, и сульфонатов, таких как метансульфонат и пара-толуолсульфонат, при условии, что если Х2 представляет собой галоген или сульфонат, Z представляет собой -NHQ2, если Х2 представляет собой -NHQ2, Z представляет собой галоген или сульфонат, и если Х2 представляет собой карбонил, Z представляет собой -NQ2 и Q2 представляет собой водород. R представляет собой (1-6С)алкил, фенил или замещенный фенил, предпочтительно метил, этил, пара-толил или фенил. R1, R2, R3, Y такие же, как определено выше.
В вышеуказанном способе, если одно из исходных веществ является солью, соль обычно нейтрализовали до или в течение реакции и такую нейтрализацию обычно проводили с основанием, молярный эквивалент которого равен молярному эквиваленту соли.
Если Q1 и Q2 представляют собой защитные группы, они могут быть удалены способами, хорошо известными специалистам настоящей области техники.
Реакция между соединением 1 и X1-R1-X2 на стадии (а) представляет собой реакцию замещения, в которой нуклеофильный атом азота в соединении 1 замещен X1 в X1-R1-X2 с образованием четвертичной аммонийной соли.
Стадию (а) обычно проводили в протонном растворителе, биполярном растворителе или инертном растворителе, таком как метанол, этанол, ацетонитрил, ацетон, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.п. Реакцию обычно проводили в диапазоне 10-100°С до существенного завершения реакции. Разделение продукта проводили обычным способом очистки, таким как экстракция, перекристаллизация, колоночная хроматография и т.п.
Получение промежуточного соединения 1 проводили способом, сообщенным в литературе (WO 2015007073 A1), иными словами получали из промежуточного соединения 5 и промежуточного соединения 6 в присутствии сильного основания.
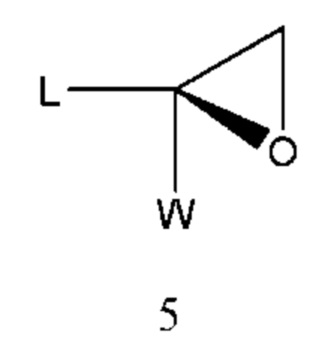
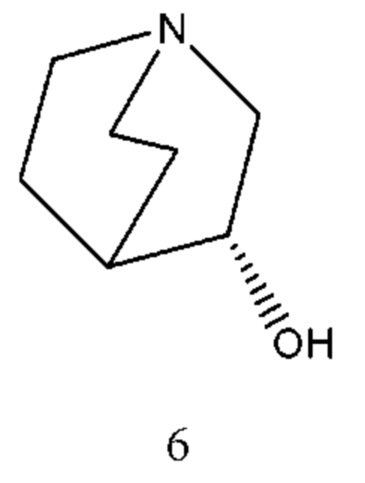
Молярное соотношение двух промежуточных соединений составляет промежуточное соединение 5 : промежуточное соединение 6=1:1-2, предпочтительно 1:1,2. Реакцию проводили в биполярном растворителе, апротонном растворителе или инертном растворителе и обычным растворителем обычно является ацетонитрил, ацетон, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.п. Температура реакции находилась в диапазоне 10-100°С, предпочтительно 70-100°С. Сильное основание выбрано из гидрида натрия, амида натрия и т.п. Молярное соотношение сильного основания к промежуточному соединению 6 составляло 1-2:1, предпочтительно 1,2:1. Время реакции составляло 2-10 часов, предпочтительно 5-8 часов.
Промежуточное соединение 6 может быть коммерчески доступным.
Промежуточное соединение 5 может быть получено из промежуточного соединения 7:
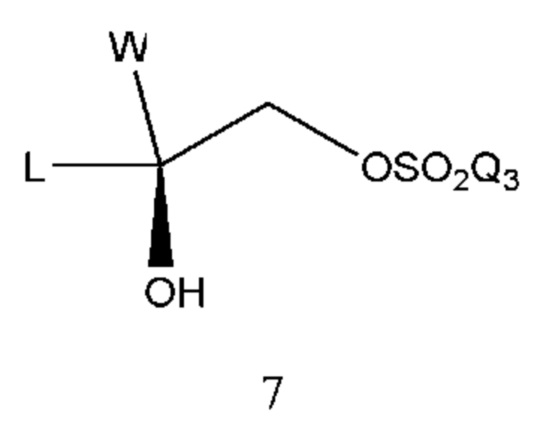
В промежуточном соединении 7 Q3 выбран из метила, фенила, пара-толила и т.п. При помощи промежуточного соединения 7 удаляли одну молекулу сульфоновой кислоты в молекуле в щелочной среде с образованием промежуточного соединения эпоксисоединения 5 и реакционный растворитель выбран из метанола, этанола, ацетона, N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида и т.п. Основание выбрано из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия и т.п. Время реакции составляло 2-10 часов, предпочтительно 3-6 часов. Температура реакции составляла 10-100°С, предпочтительно 20-50°С.
Получение промежуточного соединения 7 проводили из промежуточного соединения 8:
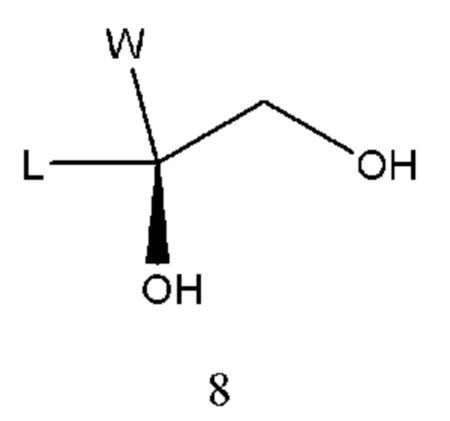
Промежуточное соединение 8 взаимодействовало с сульфонилхлоридом в щелочной среде с образованием промежуточного соединения 7. Используемым в такой реакции растворителем является инертный растворитель, такой как дихлорметан, хлороформ, ацетонитрил, ацетон, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.п. Основание выбрано из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия, пиридина, триэтиламина, N-метилморфолина и т.п. Время реакции составляет 2-10 часов, предпочтительно 3-6 часов. Температура реакции составляла 10-100°С, предпочтительно 20-50°С.
Промежуточное соединение 8 получали из промежуточного соединения 9 в R конфигурации:
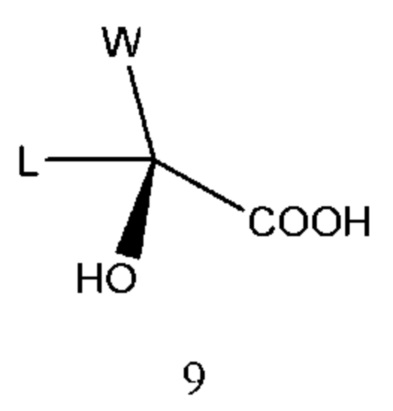
Промежуточное соединение 9 восстанавливали восстановителем, таким как боргидрид натрия, в присутствии кислоты Льюиса с прямым получением промежуточного соединения 8. Реакционным растворителем являлся инертный растворитель, такой как дихлорметан, хлороформ, ацетонитрил, ацетон, N,N-диметилформамид, N,N-диметилацетамид или их смесь. Кислота Льюиса представляет собой трихлорид алюминия, тетрахлорид олова, тетрахлорид титана или т.п. Время реакции составляло 2-10 часов, предпочтительно 3-6 часов. Температура реакции составляла 10-100°С, предпочтительно 20-50°С.
Промежуточное соединение 9 получали хиральным расщеплением его рацемата, используемый способ расщепления ссылался на патент WO 9942460 и CN 100408549 C.
Способы получения различных типов соединений, представленных формулой X1-R1-Х2, описаны подробно в разделах получения и примеров.
На стадии (b) промежуточное соединение 2 и промежуточное соединение 3 подвергали реакции нуклеофильного замещения с образованием промежуточного соединения 4, т.е., азотный атом амина подвергали действию уходящей группы. Реакцию обычно проводили в протонном растворителе, биполярном растворителе или инертном растворителе, таком как метанол, этанол, ацетонитрил, ацетон, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.п. Реакцию обычно проводили при температуре в диапазоне 10-100°С, предпочтительно 60-100°С до существенного завершения реакции.
Если Х2 в промежуточном соединении 2 представляет собой -OSO2R, R определен выше и структурная формула промежуточного соединения 2 представляет собой формулу 10:
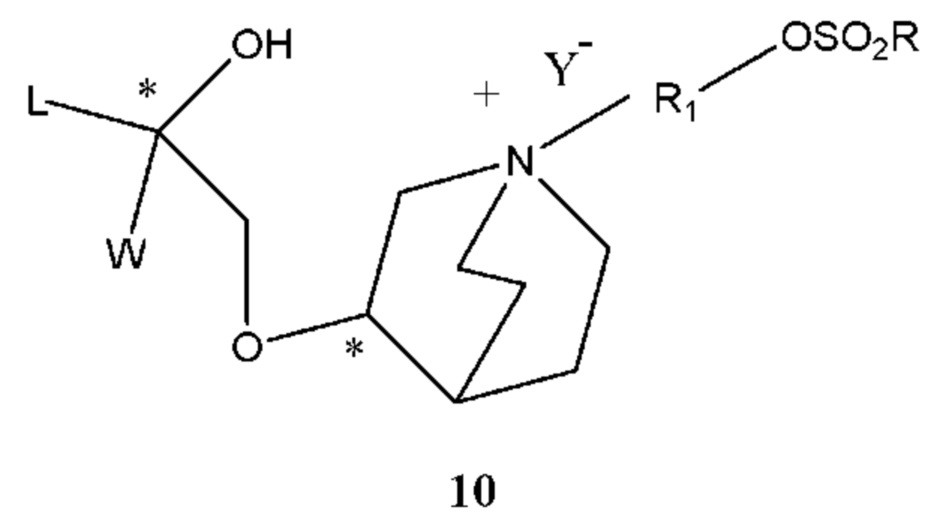
Получение соединения формулы 10 проводили путем осуществления взаимодействия соединения формулы 11 с сульфонилхлоридом в щелочной среде. Растворителем реакции является дихлорметан, тетрагидрофуран, этиловый эфир, изопропиловый эфир, ацетонитрил, ацетон или их смесь. Температурой реакции являлась комнатная температура. Основание выбрано из карбоната калия, карбоната натрия, гидроксида натрия, бикарбоната натрия, бикарбоната калия, триэтиламина, N-метилморфолина, диизопропиламина и т.п. Сульфонилхлорид выбран из метансульфонилхлорида и пара-толуолсульфонилхлорида. Молярное соотношение формулы 11 к сульфонилхлориду составляет 1:1-2, предпочтительно 1:1,05. Молярное соотношение промежуточного соединения 2 к основанию составляет 1:1-2, предпочтительно 1:1,2.
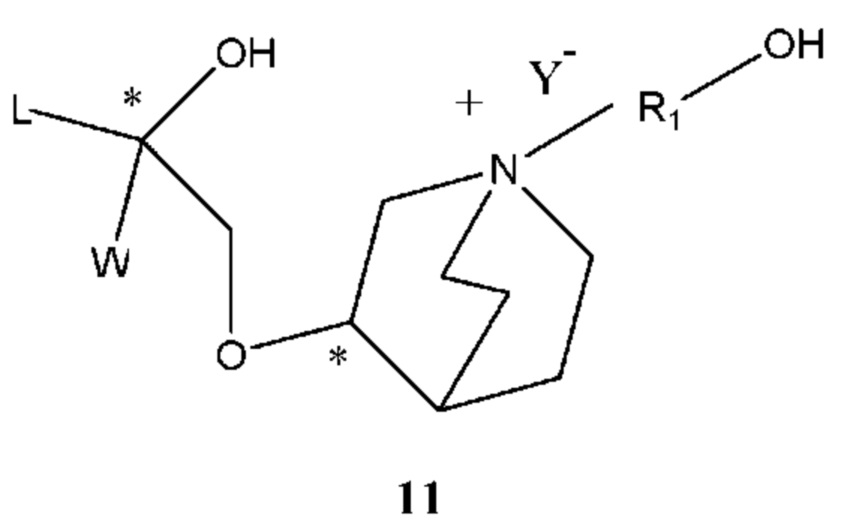
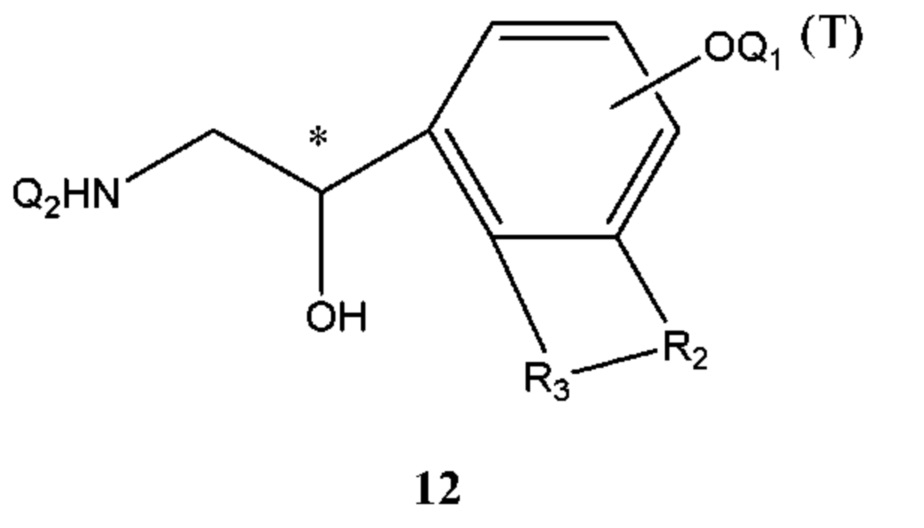
В промежуточном соединении 3 Z представляет собой -NHQ2, Q1 и Q2 определены выше, структурной формулой промежуточного соединения 3 является формула 12:
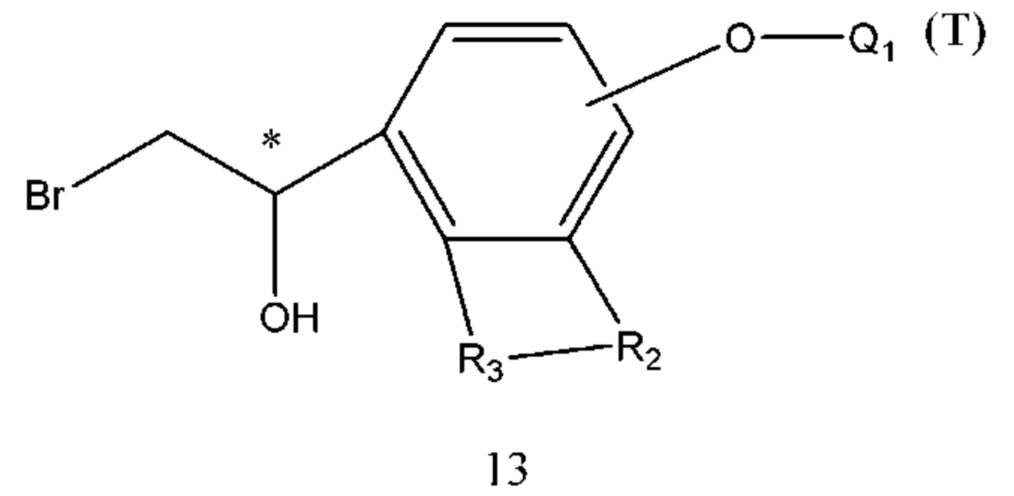
В промежуточном соединении 3, если Z представляет собой уходящую группу, такую как Br-, и Q1 определен выше, структурная формула промежуточного соединения 3 представляет собой формулу 13:
В соединении формулы 12, если R3 представляет собой водород, R2 представляет собой -CH2OQ4 и Т находится в орто-положении, Q1 и Q2 таковы, как соответственно описано выше. Q4 представляет собой водород или гидроксизащитную группу, которая выбрана из силильных эфиров, таких как триметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил и т.п., сложных эфиров (ацильных групп), таких как фор мил, ацетил и т.п., и арилметильных групп, таких как бензил, пара-метоксибензил, 9-флуоренилметил, бензгидрил и т.п. Структурная формула соединения формулы 12 представляет собой формулу 14:
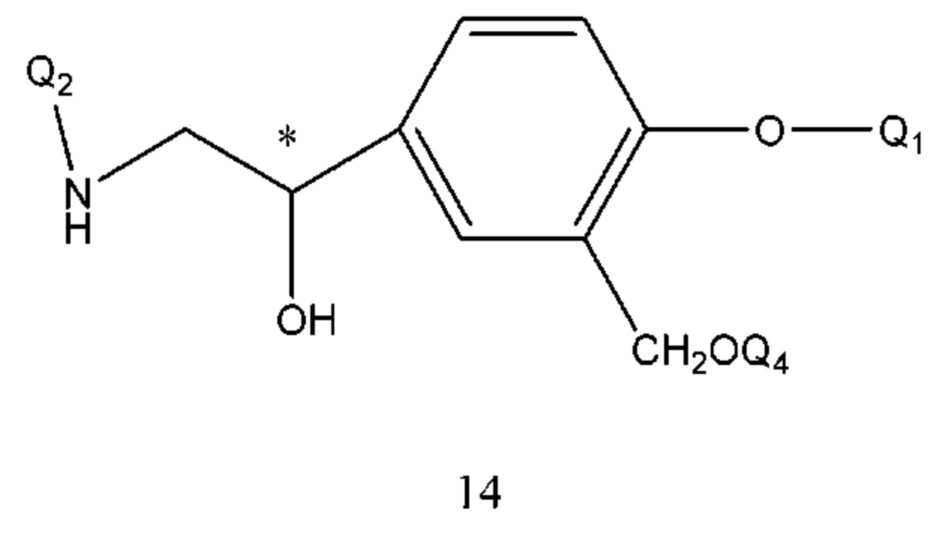
Получение соединения формулы 14 проводили путем осуществления взаимодействия соединения формулы 15 с Q2NH2 в щелочной среде под действием давления или при нагревании. Растворителем реакции является спирт, вода или Q2NH2. Температура реакции составляла 50-120°С, предпочтительно 80-110°С. Время реакции составляло 1-10 часов, предпочтительно 4-6 часов.
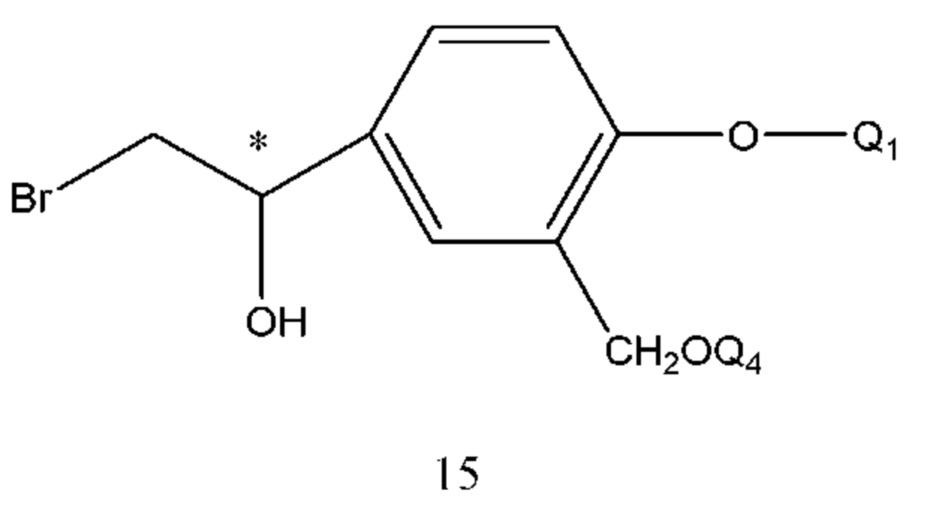
В соединении формулы 15 Q4 описан выше. Соединение формула 15 представляет собой продукт в R конфигурации, полученный хиральным восстановлением соединения формулы 16 с применением диметилсульфида борана или тетрагидрофуранового раствора в присутствии хирального катализатора. Растворитель реакции выбран из тетрагидрофурана, диоксана, этилового эфира, дихлорметана и т.п. Время реакции составляло 1-6 часов. Температура реакции составляла -5-50°С, предпочтительно 0-40°С.
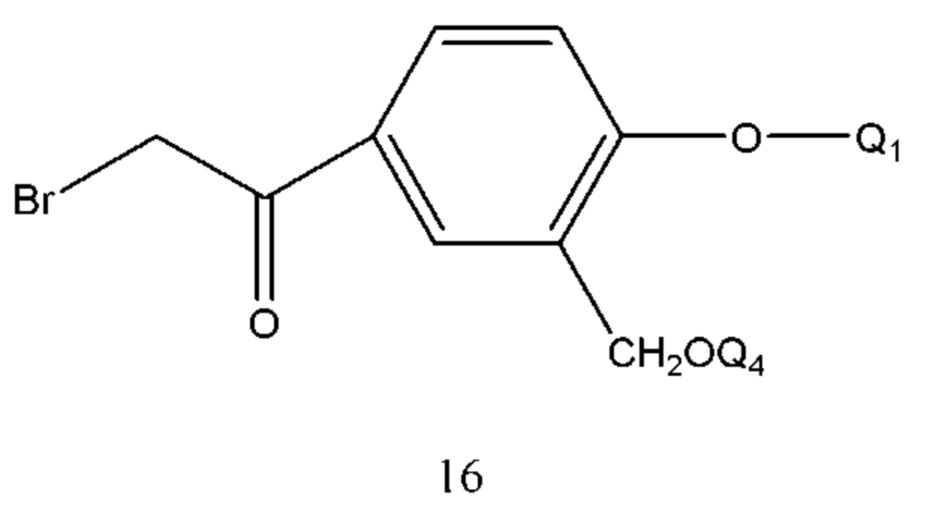
В соединении формулы 16 Q1 и Q4 такие же, как определено выше, и способ его получения проводили путем осуществления взаимодействия соединения формулы 17 с бромом в растворе. Растворитель реакции выбран из тетрагидрофурана, диоксана, этилового эфира, изопропилового эфира, дихлорметана, хлороформа и их смеси. Время реакции составляло 1-6 часов. Температура реакции составляла -5-50°С, предпочтительно -5-30°С.
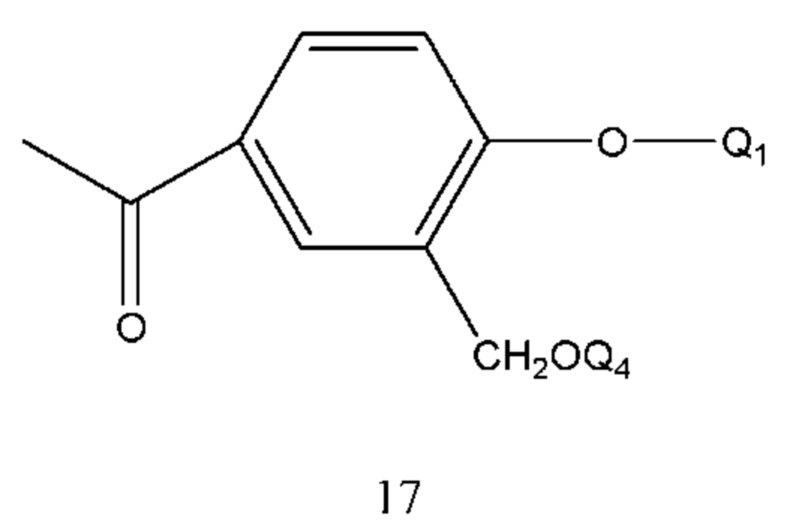
В соединении формулы 17 Q1 и Q4 такие же, как определено выше, и способ его получения проводили путем осуществления взаимодействия 4-гидрокси-3-гидроксиметилацетофенона с соединением, содержащим гидроксизащитную группу, в щелочной среде, например, с бензилбромидом.
В соединении формулы 12, если R3 представляет собой водород, R2 представляет собой -NHCHO и Т находится в орто-положении, Q1 и Q2 таковы, как соответственно описано выше и их структурная формула представляет собой формулу 18:
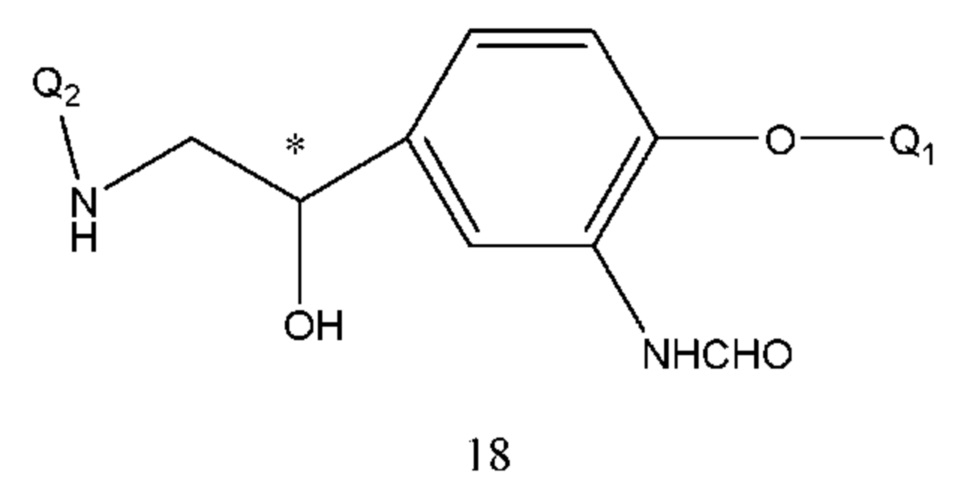
Соединение формулы 18 получали путем осуществления взаимодействия соединения формулы 19 с Q2NH2 под действием давления или при нагревании. Растворитель реакции выбран из спиртов, воды, тетрагидрофурана, диоксана или Q2NH2. Температура реакции составляла 50-120°С, предпочтительно 80-110°С. Время реакции составляло 1-10 часов.
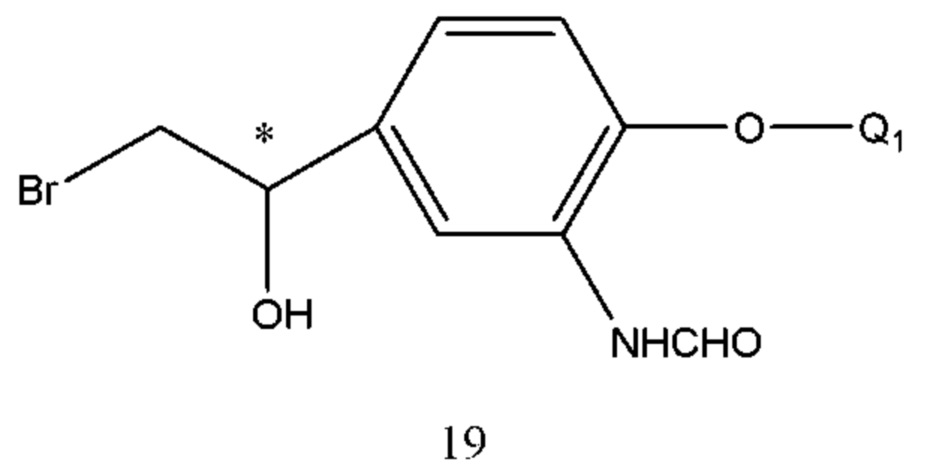
В соединении формулы 19 Q1 такой же, как определено выше, и его получение проводили хиральным восстановлением формулы 20 с применением диметил сульфид а борана или тетрагидрофуранового раствора в присутствии хирального катализатора с получением R конфигурации. Условия реакции такие же, что и для получения соединения 15.
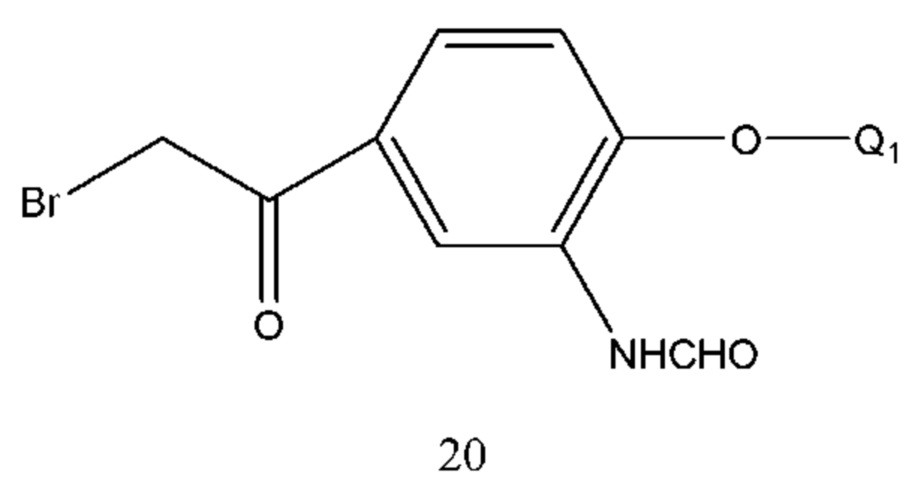
В соединении формулы 20 Q1 такой же, как определено выше, соединение формулы 20 получали из соединения формулы 21 и безводной муравьиной кислоты. Конденсирующее средство реакции выбрано из 1,3-дициклогексилкарбодиимида (DCC), диизопропилкарбодиимида (DIC), 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI) и т.п. Соединение формулы 20 также может быть получено путем осуществления взаимодействия смешанного ангидрида, полученного из уксусного ангидрида и муравьиной кислоты, с соединением формулы 21. Растворитель реакции выбран из дихлорметана, тетрагидрофурана, безводной муравьиной кислоты или их смеси. Время реакции составляло 2-8 часов. Температура реакции составляла 5-50°С, предпочтительно температура составляла 5-30°С.
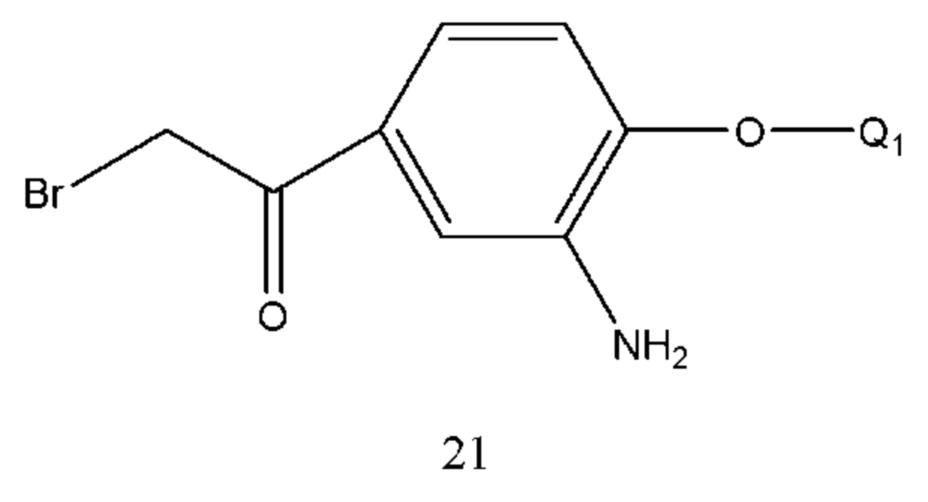
В соединении формулы 21 Q1 такой же, как определено выше, и соединение формулы 21 получали восстановлением соединения формулы 22 металлом и хлоридом аммония. Растворитель реакции выбран из воды, спиртов, таких как метанол, этанол и т.п. Металл выбран из восстановительного железного порошка, цинковой пыли и т.п.
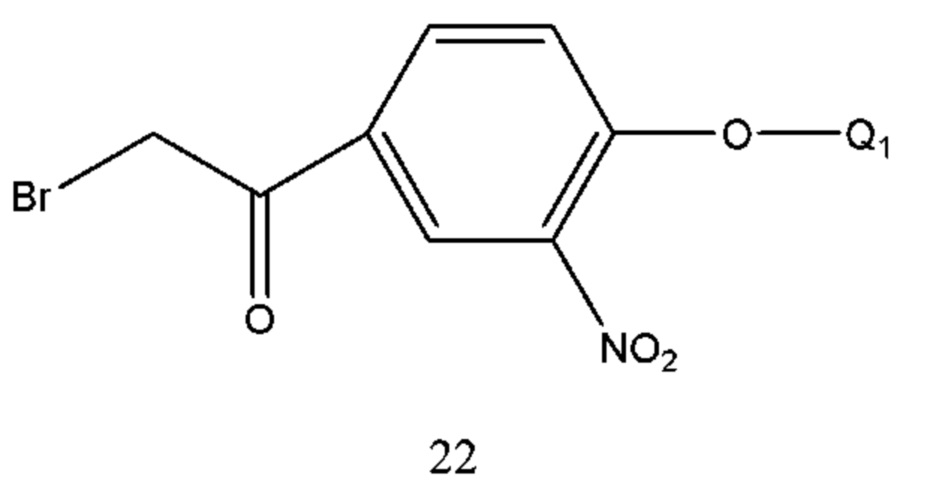
В соединении формулы 22 Q1 такой же, как определено выше, и соединение формулы 22 получали путем осуществления взаимодействия соединения формулы 23 с бромом в растворе. Растворителем реакции являлся тетрагидрофуран, диоксан, этиловый эфир, изопропиловый эфир, дихлорметан, хлороформ или т.п. Время реакции составляло 1-6 часов. Температура реакции составляла -5-50°С, предпочтительно температура составляла 5-30°С.
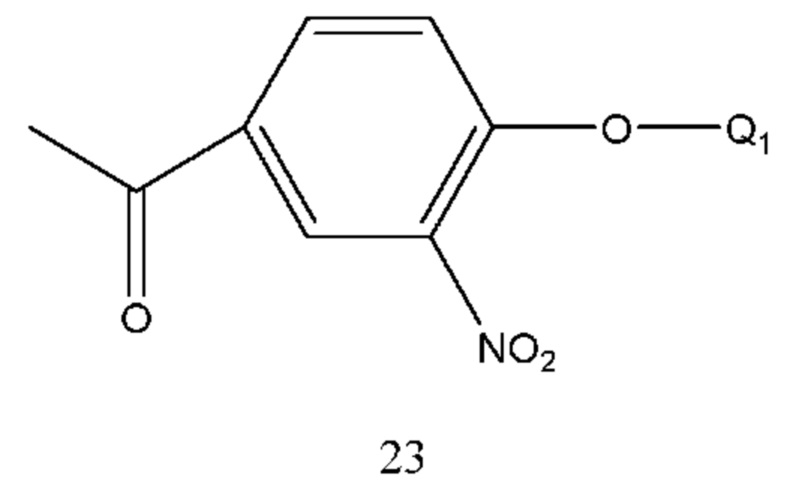
В соединении формулы 23 Q1 такой же, как определено выше и соединение формулы 23 получали путем осуществления взаимодействия 4-гидрокси-3-нитроацетофенона с соединением, содержащим гидроксизащитную группу, в щелочной среде, например, путем осуществления взаимодействия с бензилбромидом.
В соединении формулы 12 Q1 и Q2 такие же, как определено выше. Если R2 представляет собой -NH-, R3 представляет собой -СН=СН-СО- и Т находится в орто-положении, соединение, представленное формулой 12, представляет собой соединение формулы 24:
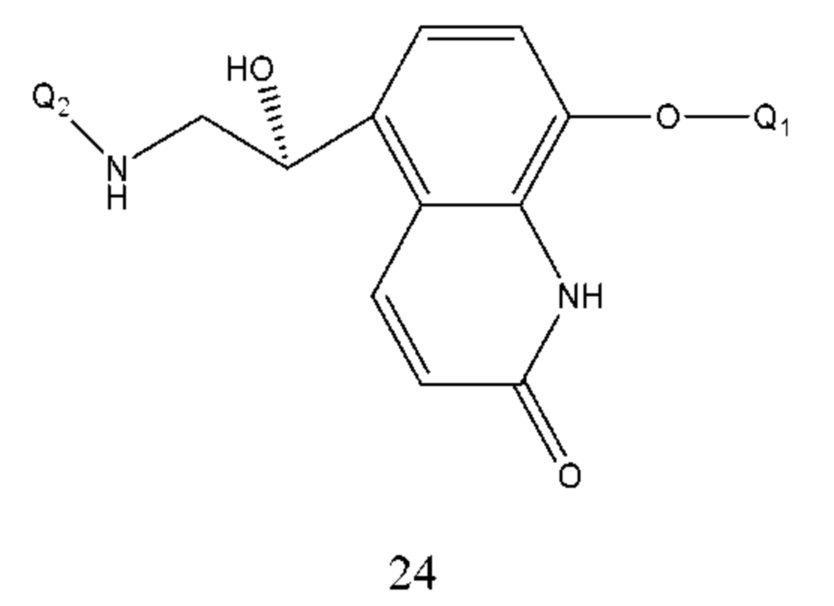
Способ получения соединения формулы 24 проводили путем реакции нуклеофильного присоединения соединения формулы 25 и Q2NH2 и растворитель реакции выбран из метанола, этанола, ацетонитрила, тетрагидрофурана, диоксана, этилового эфира, изопропилового эфира и их смеси, предпочтительно метанола, этанола, ацетонитрила, тетрагидрофурана, диоксана. Температура реакции составляла 10-100°С. Время реакции составляло 2-8 часов. Q1 и Q2 соответственно описаны выше.
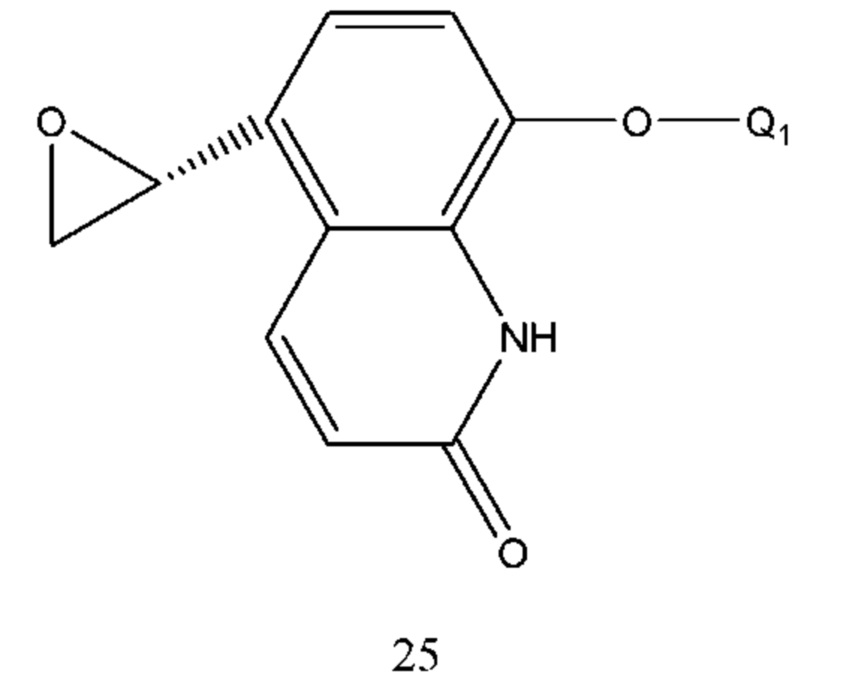
В соединении формулы 25 Q1 такой же, как определено выше и соединение формулы 25 циклизировали из соединения формулы 26 в щелочной среде. Основание выбрано из карбоната калия, карбоната натрия, гидроксида натрия, гидроксида калия и т.п. Растворителем реакции является вода, метанол, этанол, ацетонитрил, ацетон, бутанон или т.п., предпочтительно метанол, этанол, ацетон или воду. Температура реакции составляла 10-100°С, предпочтительно температура составляла 10-30°С. Время реакции составляло 1-5 часов.
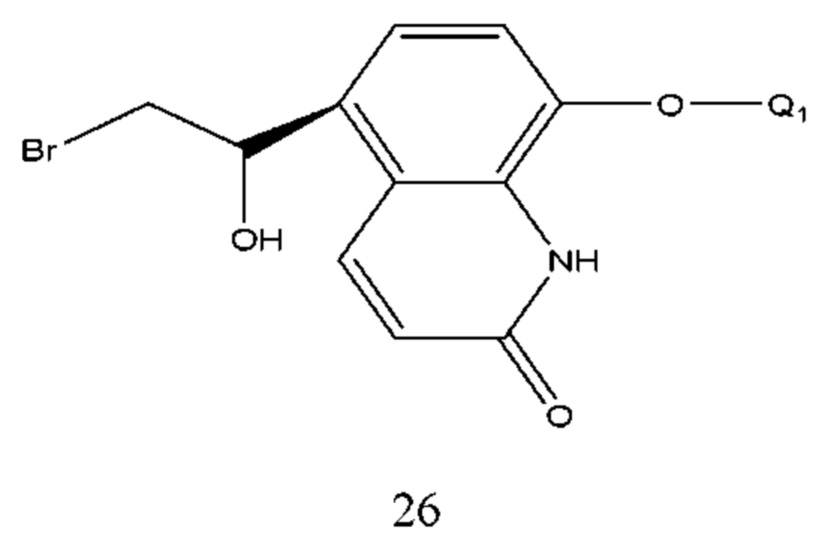
В структурной формуле соединения формулы 26 Q1 такой же, как определено выше. Его получение проводили селективным восстановлением формулы 27 при помощи борана в присутствии хирального катализатора с образованием спирта в R конфигурации. Используемым в реакции катализатором является (1R, 2S)-(+)-инданол. Растворитель реакции выбран из N,N-диметилформамида, N,N-диэтилацетамида, диметилсульфоксида, дихлорметана, хлороформа, тетрагидрофурана, этилового эфира или изопропилового эфира и т.п., предпочтительно тетрагидрофурана или этилового эфира. Молярное соотношение соединения формулы 27 к (1R, 2S)-(+)-инданолу составляло 1:0,01-0,2. Молярное соотношение соединения формулы 27 к борану составляло 1:1,1-2,5. Температура реакции составляла 0-50°С, предпочтительно температура составляла 0-30°С. Время реакции составляло 4-10 часов.
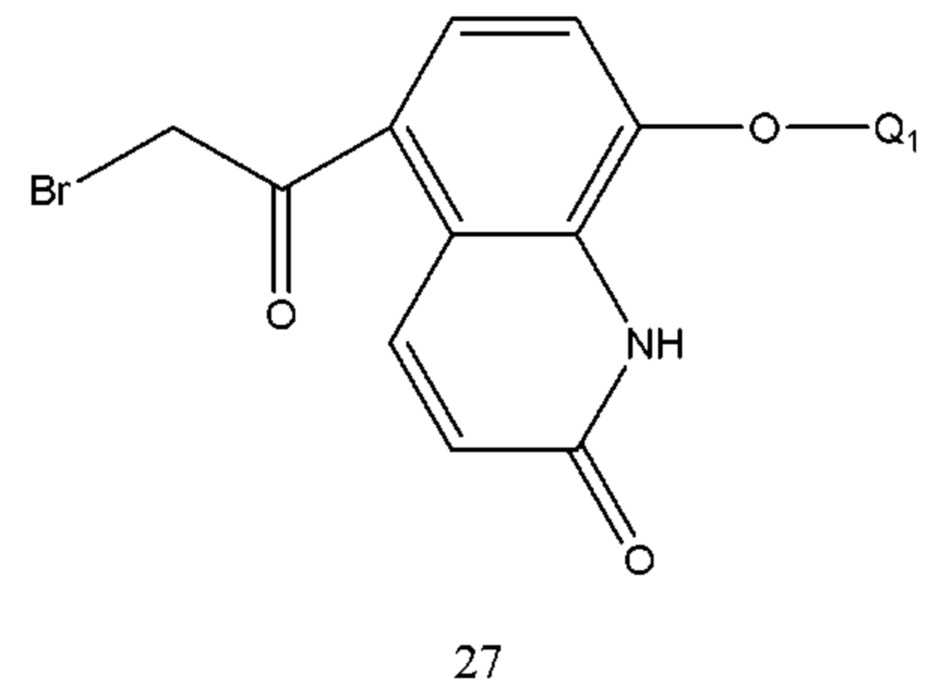
В соединении формулы 27 Q1 такой же, как определено выше. Способ его получения проводили путем осуществления взаимодействия соединения формулы 28 с бромом в растворе. Растворитель реакции выбран из тетрагидрофурана, диоксана, этилового эфира, изопропилового эфира, дихлорметана, хлороформа и т.п. Время реакции составляло 1-6 часов. Температура реакции составляла -5-50°С, предпочтительно температура составляла от 0 до 30°С.
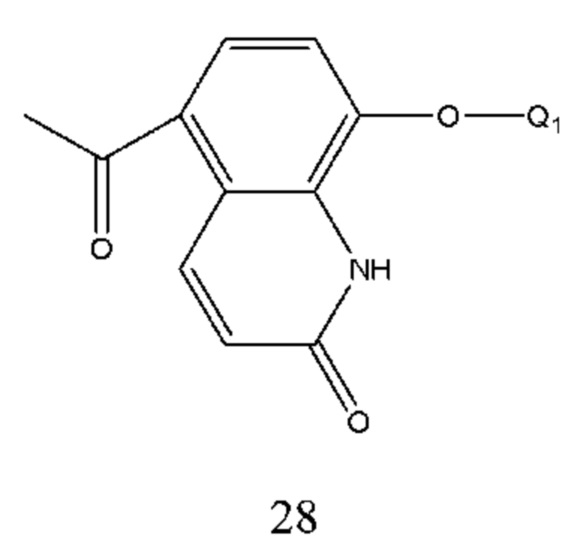
При получении соединения формулы 28 брали 5-ацетил-8-гидроксихинолин в качестве исходного вещества, которое реагировало с Q1X в щелочной среде с образованием соединения формулы 28. X представляет собой уходящую группу, выбранную из галогена, такого как хлор, бром и йод, сульфоната, такого как метансульфонат и пара-толуолсульфонат. Основание выбрано из карбоната калия, карбоната натрия и т.п. Растворитель реакции выбран из метанола, этанола, ацетона, бутанона, тетрагидрофурана, диоксана, дихлорметана, хлороформа, этилового эфира, изопропилового эфира, ацетонитрила и т.п., предпочтительно ацетона, тетрагидрофурана или ацетонитрила.
В соединении формулы 12 Q1 и Q2 такие же, как определено выше. Если R2 представляет собой -NH-, R3 представляет собой -О-СН2-СО- и Т находится в мета-положении, соединение, представленной формулой 12, представляет собой соединение формулы 29:
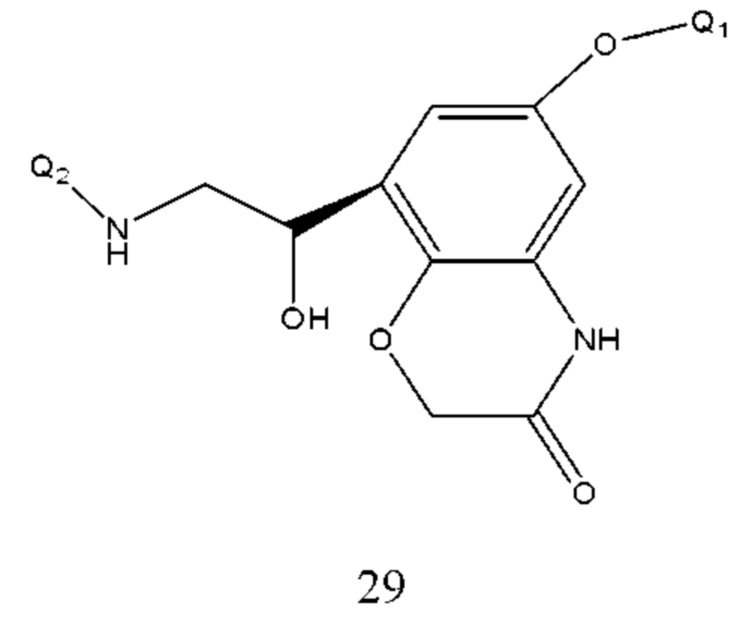
Способ получения проводили реакцией нуклеофильного присоединения такого соединения формулы 30 и Q2NH2 и условия реакции подобны условиям для соединения формулы 24. Q1 и Q2 соответственно описаны выше.
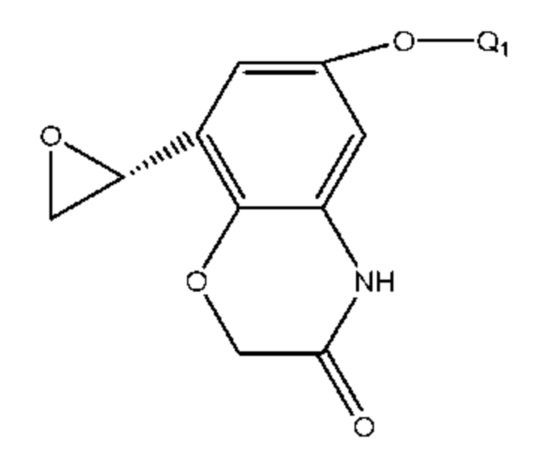
Соединение формулы 30 получали из соединения формулы 31 и способ ее получения подобный способу получения соединения формулы 25, где Q1 такой же, как определено выше.
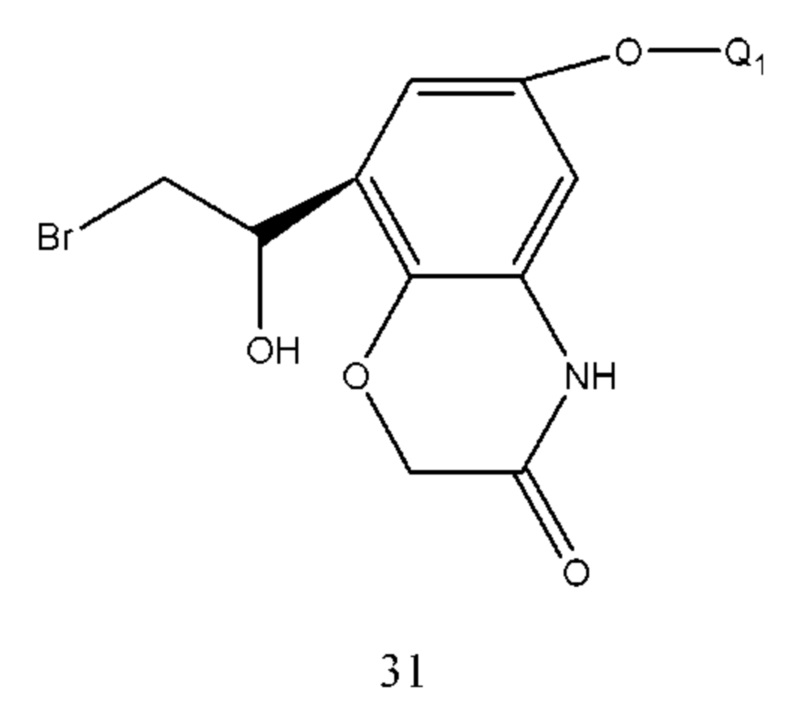
Соединение формулы 31 получали из соединения формулы 32 и способ ее получения подобный способу получения соединения формулы 26, где Q1 такой же, как определено выше.
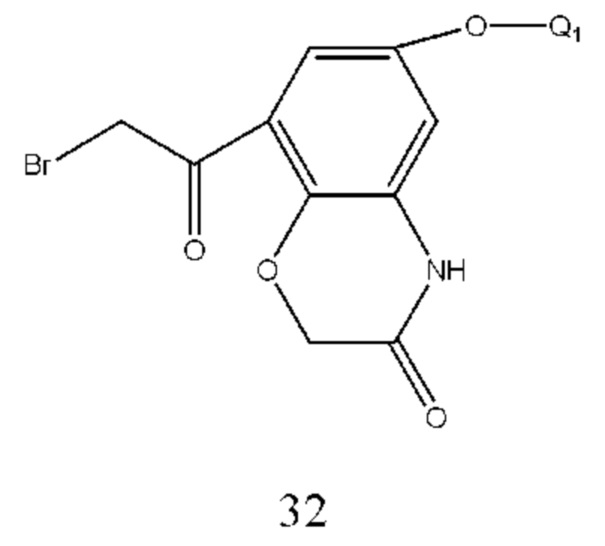
Соединение формулы 32 получали из соединения формулы 33 и способ ее получения относится к способу получения соединения формулы 27, где Q1 такой же, как определено выше.
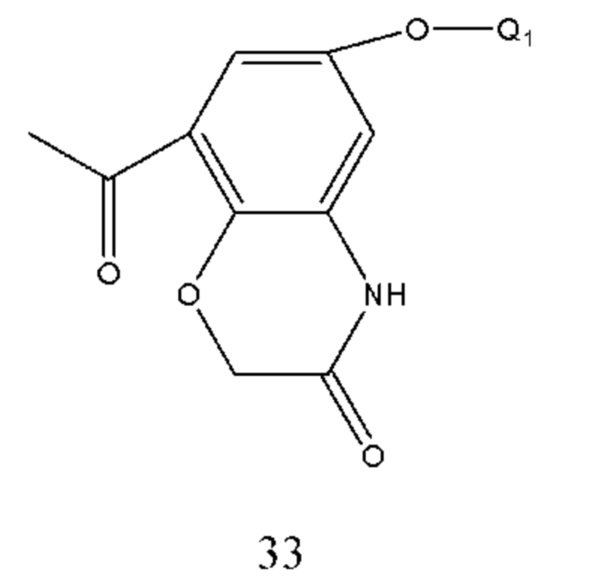
Соединение формулы 33 получали путем осуществления взаимодействия соединения формулы 34 с хлорацетилхлоридом в щелочной среде. Условия получения: молярное соотношение соединения формулы 34 к хлорацетилхлориду составляло 1:1-2, предпочтительно 1:1,05. Основание выбрано из карбоната калия, карбоната натрия, бикарбоната калия, бикарбоната натрия или низкоконцентрированного раствора гидроксида натрия и т.п. и молярное соотношение соединения формулы 34 к основанию составляло 1:1-3, предпочтительно 1:1,5. Температура реакции составляла 10-100°С, предпочтительно 20-60°С.
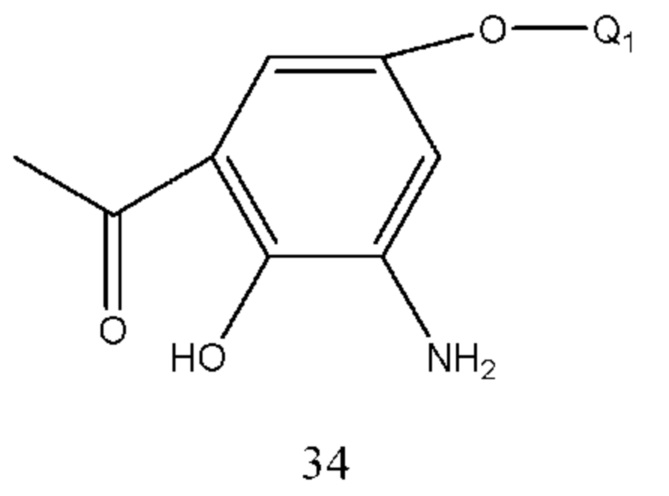
Соединение формулы 34 получали из соединения формулы 35, в котором нитрогруппа восстановлена до аминогруппы. Способ получения может применять способ гидрогенизации и оксид платины использовали в качестве катализатора для селективной гидрогенизации. Растворитель реакции выбран из метанола, этанола, тетрагидрофурана и их смеси. Температура составляла 10-50°С, предпочтительно 20-30°С. Давление составляло 0,5-4 МПа. Необязательно, восстановительный железный порошок использовали для селективного восстановления при помощи водного раствора хлорида аммония, таким образом нитрогруппу восстанавливали до аминогруппы. Растворитель выбран из метанола, этанола, тетрагидрофурана и их смеси. Молярное соотношение соединения формулы 34 к хлориду аммония составляло 1:1-4, предпочтительно 1:2. Молярное соотношение соединения формулы 35 к восстановительному железному порошку составляло 1:1-5, предпочтительно 1:2.
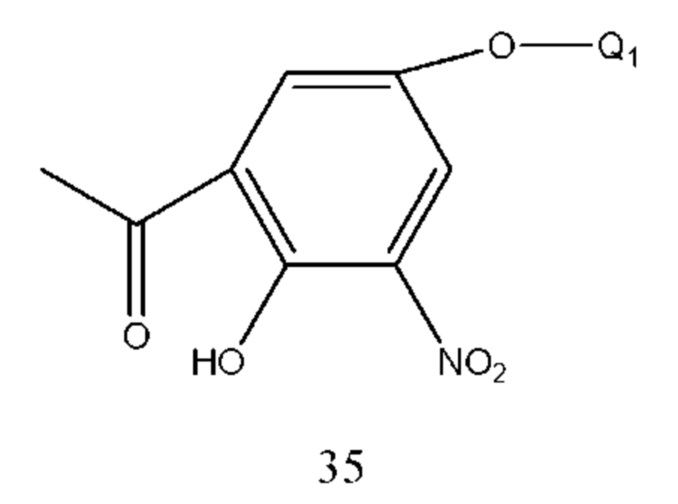
Соединение формулы 35 получали нитрированием соединения формулы 36. Растворителем реакции являлась ледяная уксусная кислота и температура реакции составляла-5-50°С, предпочтительно 0-30°С.
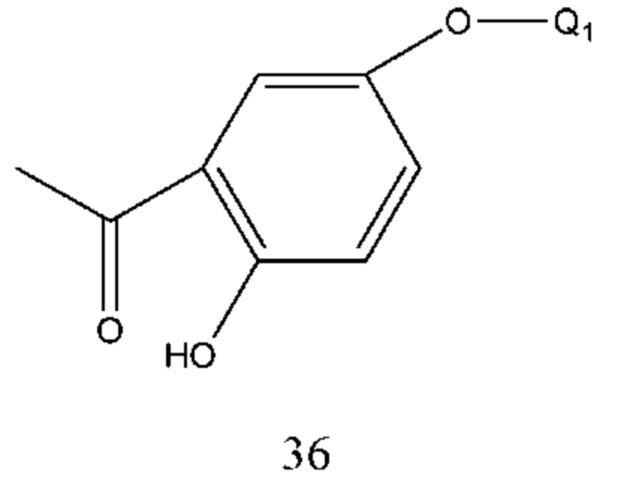
Соединение формулы 36 получали путем осуществления взаимодействия 2,4-дигидроксиацетофенона с Q1X в слабой щелочной среде и Q1X такой же, как определено выше. Основание выбрано из бикарбоната натрия, бикарбоната калия, их смеси и т.п. Молярное соотношение 2,4-дигидроксиацетофенона к основанию составляло 1:1. Молярное соотношение 2,4-дигидроксиацетофенона к Q1X составляло 1:1. Температура реакции составляла 10-50°С и время реакции составляло 2-5 часов.
На стадии (с) промежуточное соединение 4 или другое заданное соединение с защитной группой восстанавливали с образованием заданного соединения. В реакции может быть использован любой подходящий восстановитель, например, в каталитическом гидрировании. Катализатор выбран из Pd/C, никеля Ренея, оксида платины и их смеси и реагента гидрида металла, такого как триацетилборгидрид натрия и т.п. Растворитель реакции выбран из метанола, этанола и их смеси.
На стадии (d) соединение формулы I обменивали с основной анионообменной смолой с получением гидроксида формулы I, такой как ОН- смола, а затем осуществляли взаимодействие с различными кислотами с получением четвертичных аммонийных солей с различными кислотными радикалами, включая различные соли кислотных радикалов, как упомянуто выше. Необязательно, соединение формулы I обменивали с конкретной анионообменной смолой с получением четвертичной аммонийной соли с конкретным кислотным радикалом. Необязательно, осуществляли взаимодействие галогенида формулы I с оксидом серебра с образованием гидроксида формулы I, а затем осуществляли взаимодействие с другими кислотами с образованием четвертичных аммонийных солей с различными кислотными радикалами. Необязательно, осуществляли взаимодействие галогенида формулы I с солью серебра, такой как сульфат серебра или нитрат серебра, с получением четвертичной аммонийной соли с соответствующим кислотным радикалом.
Согласно конкретным вариантам осуществления определенные конкретные соединения формулы I получали способом (a1)-(d1). Этот способ включает в себя соединения, обладающие следующей структурной формулой, или их необязательные фармацевтически приемлемые соли или сольваты или их оптические изомеры и их смеси.
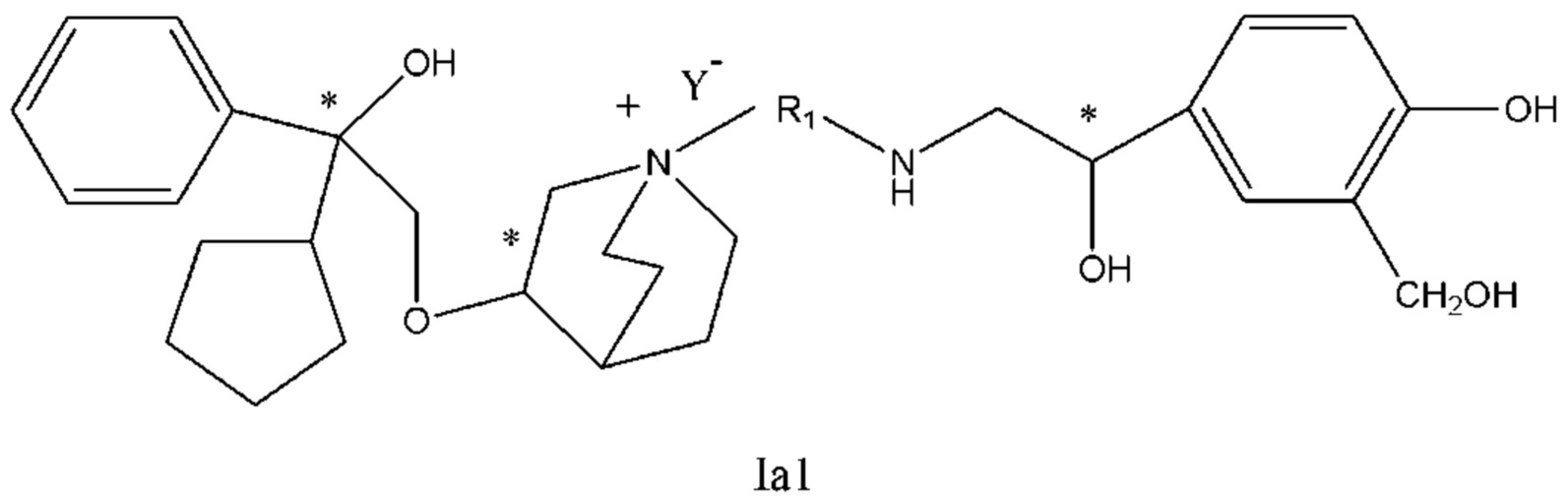
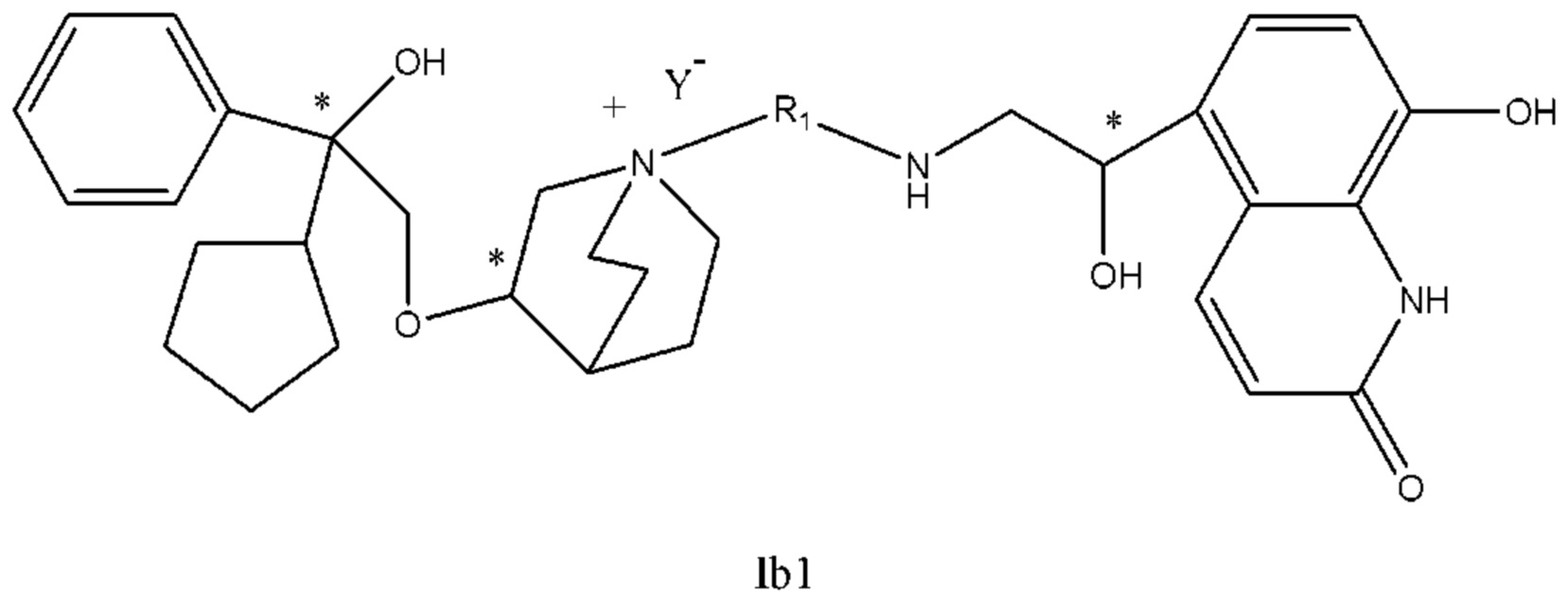
С другой стороны, настоящее изобретение относится к фармацевтической композиции соединения формулы I, которая включает в себя приемлемый фармацевтический носитель, и фармацевтическая композиция может избирательно содержать другие терапевтические ингредиенты, такие как стероидное противовоспалительное лекарственное средство, ингибитор фосфодиэстеразы (PDE-4) и их фармацевтически приемлемые соли, сольваты и терапевтически эффективное количество оптических изомеров.
Соединение формулы I согласно настоящему изобретению обычно применяют в виде композиций или препаратов для пациентов. Эти композиции можно применять у пациентов с помощью любого приемлемого пути введения, в том числе, без ограничения, ингаляционного, перорального, назального, местного (в том числе трансдермального) и парентерального введения. То есть любую форму соединения согласно настоящему изобретению, подходящую для любого конкретного способа введения (в том числе свободное основание, его фармацевтически приемлемые соли или сольваты и т.д.), можно применять в фармацевтической композиции согласно настоящему изобретению.
Фармацевтическая композиция согласно настоящему изобретению обычно содержит терапевтически эффективное количество соединения согласно настоящему изобретению или его фармацевтически приемлемой соли или сольвата. Обычно такая фармацевтическая композиция содержит от приблизительно 0,001% до приблизительно 100% по массе активного ингредиента.
В настоящем изобретении можно применять любые традиционные носители или вспомогательные средства, и выбор конкретного носителя или вспомогательного средства или комбинации носителя и вспомогательного средства зависит от способа введения, или медицинского состояния, или типа заболевания при лечении конкретного пациента. Технология получения фармацевтической композиции для конкретного способа находится в рамках квалификации специалистов в данной области техники. Кроме того, носитель или вспомогательное средство или комбинация носителя и вспомогательного средства могут быть приобретены у коммерческого поставщика.
Типичные примеры фармацевтически приемлемых носителей включают в себя, без ограничения: (1) сахариды, такие как глюкоза, лактоза, сахароза и т.д.; (2) крахмалы, такие как кукурузный крахмал; (3) целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, ацетат целлюлозы и т.д.; (4) тальк; (5) вспомогательные средства, такие как какао-масло и воски; (6) масла, такие как оливковое масло, соевое масло и т.д.; (7) спирты, такие как этанол, пропиленгликоль, глицерин, сорбит, полиэтиленгликоль, маннит и т.д.; (8) сложные эфиры, такие как этилолеат и этиллаурат; (9) апирогенную воду; (10) изотоничный солевой раствор; (11) фосфатный буферный раствор; (12) газы-вытеснители под давлением, такие как хлорфторуглероды, гидрофторуглероды и т.д.; и (13) другие нетоксичные смешиваемые вещества, применяемые в фармацевтических композициях.
Композицию согласно настоящему изобретению обычно получают посредством тщательного смешивания соединения согласно настоящему изобретению с необязательным одним или несколькими носителями. Если необходимо, однородную смесь, полученную согласно настоящему изобретению, можно пластифицировать или загружать в таблетки, капсулы, пилюли, банки или картриджи с применением традиционного оборудования и способов.
Фармацевтическая композиция согласно настоящему изобретению является подходящей для ингаляционного введения. Композиции для ингаляционного введения обычно имеют форму аэрозолей или порошка для ингаляции. Такие композиции обычно вводят с применением хорошо известных устройств для введения, таких как небулайзер, ингалятор отмеренных доз (MDI) или ингалятор сухого порошка (DPI) или другие подобные устройства для ингаляции.
Композицию, содержащую активный ингредиент согласно настоящему изобретению, распыляют и вводят с помощью небулайзера. Распыляющие устройства обычно могут создавать высокоскоростной поток воздуха для распыления фармацевтической композиции, содержащей активный ингредиент, подлежащий ингаляции в дыхательные пути пациентов. Таким образом, активный ингредиент обычно растворяют в подходящем растворителе с получением раствора и помещают в небулайзер. Необязательно, активный ингредиент является тонко измельченным и присутствует в комбинации с подходящим носителем для образования суспензии тонко измельченных частиц, подходящих для ингаляции. Тонкое измельчение обычно определяется как условия, когда диаметр менее 10 мкм имеют 90% или большее количество частиц. Подходящие распыляющие устройства являются коммерчески доступными.
Типичные фармацевтические композиции, применяемые с небулайзером, включают в себя изотоничный водный раствор или этанольный раствор, содержащий от 0,05 мкг/мл до 10 мг/мл соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или оптических изомеров.
Фармацевтическую композицию, предполагаемую согласно настоящему изобретению, вводят посредством ингаляции с применением ингалятора сухого порошка. Введение с помощью ингаляторов сухого порошка обычно осуществляют таким образом, чтобы активный ингредиент образовывал свободнотекучий порошок во вдыхаемом пациентом воздушном потоке во время ингаляции. Таким образом, активный ингредиент обычно составляют вместе с подходящим вспомогательным средством для получения свободнотекучего порошка, например, с лактозой в качестве вспомогательного средства.
Типичные фармацевтические композиции для ингаляторов сухого порошка включают в себя сухую лактозу, имеющую размер частиц приблизительно от 1 мкм до 100 мкм, и вышеупомянутые тонко измельченные частицы соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или оптического изомера.
Состав в виде сухого порошка можно получить посредством сухого перемешивания активного ингредиента со вспомогательным средством или без вспомогательного средства, а затем фармацевтическую композицию загружают в дозатор сухого порошка, или в картридж для ингаляции, или в капсулу для применения с устройством для введения сухого порошка.
Устройства для введения сухого порошка являются коммерчески доступными.
Фармацевтическую композицию, содержащую активный ингредиент согласно настоящему изобретению, вводят посредством ингаляции с применением ингалятора отмеренных доз. В таком устройстве для ингаляции отмеренных доз применяется находящийся под давлением газ-вытеснитель, чтобы выпустить отмеренное количество активных ингредиентов или их фармацевтически приемлемых солей. Таким образом, фармацевтическая композиция, вводимая с помощью ингалятора отмеренных доз, содержится в растворе или суспензии, вытесняемой при разжижении.
Типичные фармацевтические композиции для ингаляторов отмеренных доз включают в себя от 0,001% до приблизительно 3% по массе соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или оптического изомера, от приблизительно 0% до приблизительно 40% сорастворителя в виде этанола или диолов, предпочтительно, от 5% до приблизительно 30%, и приблизительно от 0% до 3% по массе поверхностно-активного вещества. Остальная часть представляет собой гидрофторалкановый (HFA) газ-вытеснитель.
Такую композицию обычно получают посредством добавления охлажденных или находящихся под давлением гидрофторалканов в подходящий контейнер, содержащий активный ингредиент, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для получения суспензии активный ингредиент тонко измельчают, а затем смешивают с газом-вытеснителем. Препарат затем помещают в аэрозольный баллон, образующий часть ингалятора отмеренных доз. Суспензионный препарат также можно получать с помощью метода распылительной сушки с образованием покрытия из поверхностно-активного вещества на поверхности микрочастиц активного ингредиента.
Способы и препараты для получения частиц, которые можно вводить посредством ингаляции, и другие примеры, подходящие для ингаляционного введения, обсуждаются в литературных источниках.
Композиция согласно настоящему изобретению является подходящей для перорального введения. Фармацевтическая композиция для перорального введения может представлять собой капсулу, таблетку, пилюлю, порошок, гранулу, плоскую капсулу и пилюлю с сахарным покрытием, или ее можно превратить в водный или неводный раствор или суспензию, или превратить в эмульсию типа «вода в масле» или «масло в воде», или превратить в сироп. Все они содержат заранее определенное количество активного ингредиента в виде соединения согласно настоящему изобретению.
Если вводят твердую лекарственную форму, композиция согласно настоящему изобретению включает в себя соединение согласно настоящему изобретению в качестве активного ингредиента и один или несколько фармацевтически приемлемых носителей для лекарственных средств, если необходимо, например, (1) наполнители или сухие разбавители, такие как крахмал, сахароза, кремниевая кислота и т.д.; (2) адгезивы, такие как карбоксиметилцеллюлоза, поливинилпирролидон и т.д.; (3) увлажняющее средство, такое как глицерин; (4) разрыхлители, такие как карбонат кальция, крахмал и т.д.; (5) смазывающие средства, такие как стеарат магния, тальк, твердый полиэтиленгликоль или их смеси; и (6) абсорбент, такой как каолин и т.д.
Разрыхляющие средства, увлажнители, покровные средства, подсластители, антиоксиданты, отдушки, ароматизирующие средства и консерванты также могут присутствовать в композиции согласно настоящему изобретению. Фармацевтически приемлемые антиоксиданты включают в себя, без ограничения, следующие вещества: водорастворимые антиоксиданты, такие как сульфит натрия, аскорбиновая кислота, гидрохлорид цистеина и т.д.; жирорастворимые антиоксиданты, такие как пропилгаллат, альфа-токоферол и т.д.; и средство, хелатирующее металлы, такое как лимонная кислота, сорбит и этилендиаминтетрауксусная кислота (EDTA) и т.д.
Покровные средства для таблеток, капсул и пилюль включают в себя, без ограничения, ацетатфталат целлюлозы (САР), карбоксиметилэтилцеллюлозу (СМЕС) и т.п.
Композицию согласно настоящему изобретению также можно составлять в средство для медленного высвобождения, чтобы контролировать медленное высвобождение активных ингредиентов, например, карбоксиметилцеллюлозу или другие полимерные матрицы, липосомы или микросферы в разных соотношениях применяют для получения средства с медленным контролируемым высвобождением.
Подходящие жидкие лекарственные формы для перорального введения включают в себя суспензии, сиропы, эмульсии, микроэмульсии и растворы и т.д. Жидкие лекарственные формы содержат активные ингредиенты и инертные разбавители, такие как вода и другие растворители, солюбилизаторы и эмульгаторы и т.д. Типичные представители являются следующими: масла (оливковое масло и т.д.), глицерин, полиэтиленгликоль, сорбитановые сложные эфиры жирных кислот или их смеси.
Фармацевтическая композиция согласно настоящему изобретению также может представлять собой смесь, образованную соединением формулы I или его фармацевтически приемлемой солью, сольватом или оптическим изомером и их смесями с другими лекарственными средствами для терапии с совместным введением. Например, фармацевтическая композиция согласно настоящему изобретению включает в себя: одно или несколько других бронхолитических средств, таких как ингибитор PDE3, агонист β2-адренорецептора и т.д.; противовоспалительные средства, такие как стероидные противовоспалительные средства, нестероидные противовоспалительные средства, ингибиторы PDE4 и т.д.; антагонисты М-рецептора; противоинфекционные средства, такие как антибиотики, действующие на грамотрицательные и грамположительные бактерии, противовирусные лекарственные средства и т.д.; антигистаминное средство; ингибитор протеазы и блокаторы афферентных путей, такие как агонисты D2. Другие терапевтические средства можно применять в форме фармацевтически приемлемых солей или сольватов. Кроме того, другие терапевтические средства также можно применять в форме оптических изомеров.
Типичный агонист β2-адренорецептора, который можно применять в комбинации с соединением согласно настоящему изобретению (дополнительно к соединениям согласно настоящему изобретению), включает в себя, без ограничения, сальметерол, сальбутамол, левалбутерол, формотерол, индакатерол, виландт, арформотерол, сальмефамол, фенотерол, изоэтарин, метапротеренол, битолтерол, пирбутерол и т.д. или их фармацевтически приемлемые соли.
Типичные стероидные противовоспалительные средства (отличные от соединения согласно настоящему изобретению), которые можно применять в комбинации с соединением согласно настоящему изобретению, включают в себя, без ограничения, метилпреднизолон, преднизолон, дексаметазон, пропионат флутиказона, сложный эфир беклометазона, будесонид, флунизолид, сложный эфир мометазона, триамцинолон, рофлепонид, циклезонид и т.д. или их фармацевтически приемлемую соль. При применении стероидные противовоспалительные средства будут присутствовать в композиции в терапевтически эффективном количестве, обычно количество стероида составляет от 0,05 мкг до 500 мкг.
Другие подходящие композиции включают в себя композиции, образуемые соединением, представленным формулой I согласно настоящему изобретению, и другими противовоспалительными лекарственными средствами. Например, типичные иллюстративные лекарственные средства из числа нестероидных противовоспалительных лекарственных средств являются следующими: NSAID, такие как недокромил натрия, кромогликат натрия и т.д., ингибиторы фосфодиэстеразы (PDE), такие как теофиллин, ингибиторы PDE4, смешанные ингибиторы PDE3/PDE4 и т.д., антагонисты лейкотриенов, такие как монтелукаст, ингибиторы протеазы, антагонисты цитокинов и ингибиторы синтеза цитокинов.
Типичные антагонисты М-рецептора, которые можно применять в комбинации с соединениями согласно настоящему изобретению (дополнительно к соединениям согласно настоящему изобретению), включают в себя, без ограничения, гликопирролат, бромид ипратропия, бромид тиотропия, атропин, сульфат атропина, оксид атропина, нитрат метилатропина, гидробромид гоматропина, гидробромид скополамина, бромид окситропия, бромид метантелина, бромид пропантелина, метилбромид анизотропина, бромид клидиния, йодид изопропамида, бромид мепензолата, пирензепин, телензепин, метоктрамин и т.д. или их фармацевтически приемлемые соли.
Антигистаминные лекарственные средства, которые можно применять в комбинации с соединениями согласно настоящему изобретению, включают в себя, без ограничения, этаноламины, такие как фумарат клемастина, малеат карбиноксамина, гидрохлорид дифенгидрамина, дименгидринат и т.д., этилендиамины, такие как малеат пириламина, гидрохлорид трипеленнамина и цитрат трипеленнамина и т.д., алкиламины, такие как хлорфенирамин, акривастин и т.д., пиперазины, такие как гидрохлорид гидроксизина, устойчивый к кислотам гидроксизин, гидрохлорид циклизина, лактат циклизина, гидрохлорид меклизина, гидрохлорид цетиризина и т.д., пиперидины, такие как астемизол, гидрохлорид левокабастина, лоратадин и его аналоги, терфенадин, гидрохлорид фексофенадина, гидрохлорид азеластина и т.д., а также их фармацевтически приемлемые соли.
Эффективные терапевтические дозы других лекарственных средств, вводимых в комбинации с соединениями согласно настоящему изобретению, находятся в диапазоне от приблизительно 0,005 мг до приблизительно 10 мг каждый раз.
Настоящее изобретение также относится к применению соединения формулы I или его фармацевтически приемлемой соли, сольвата, оптического изомера или их смеси для получения лекарственного препарата для лечения респираторных заболеваний, в том числе COPD, астмы, ринита и т.п.
Соединение согласно настоящему изобретению характеризуется как агонистической активностью в отношении β2-адренорецептора, так и антагонистической активностью в отношении М-рецептора, поэтому оно является подходящим для лечения заболеваний, опосредуемых β2-адренорецептором и М-рецептором. То есть заболеваний, которые могут быть ослаблены посредством применения агонистов β2-адренорецепторов и антагонистов М-рецептора. Такое заболевание включает в себя легочные нарушения или заболевания, связанные с обратимой обструкцией дыхательных путей, такие как COPD, астма, легочный фиброз и т.д.
Настоящее изобретение относится к способу лечения заболеваний легких, который включает в себя введение эффективной дозы соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или оптического изомера пациенту, нуждающемуся в лечении. При лечении заболеваний соединение согласно настоящему изобретению обычно вводят посредством ингаляции в виде ежесуточных многократных доз, ежесуточных разовых доз или еженедельных разовых доз. Дозировка составляет от приблизительно 1,0 мкг до приблизительно 200 мкг каждый раз.
При введении посредством ингаляции соединение согласно настоящему изобретению обеспечивает бронхоэктаз, таким образом, настоящее изобретение относится к способу обеспечения бронхоэктаза у пациента, включающему в себя введение эффективной дозы соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или оптического изомера пациенту, нуждающемуся в лечении. Дозировка составляет от приблизительно 1,0 мкг до приблизительно 200 мкг каждый раз.
Настоящее изобретение относится к способу лечения хронического обструктивного заболевания легких или астмы, который включает в себя введение эффективной дозы соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или оптического изомера пациенту, нуждающемуся в лечении. При лечении COPD или астмы его вводят ежесуточно в виде многократных доз и разовых доз, причем дозировка находится в диапазоне от приблизительно 1,0 мкг до приблизительно 200 мкг каждый раз.
Когда соединение согласно настоящему изобретению применяют в лечении заболеваний легких, соединение согласно настоящему изобретению можно необязательно вводить в комбинации с другими терапевтическими средствами. В особенности, в случае, когда соединение согласно настоящему изобретению комбинируют со стероидными противовоспалительными лекарственными средствами, композиции согласно настоящему изобретению с двумя активными ингредиентами могут обеспечить тройную терапию, а именно агонистический эффект в отношении β2-адренорецептора, антагонистический эффект в отношении М-рецептора и противовоспалительный эффект. Композицию, содержащую два активных ингредиента согласно настоящему изобретению, обычно проще приготовить по сравнению с композицией, содержащей три активных ингредиента. Поэтому двухкомпонентная композиция превосходит трехкомпонентную композицию. Фармацевтическая композиция согласно настоящему изобретению может содержать терапевтически эффективное количество стероидного противовоспалительного средства.
Соединение согласно настоящему изобретению проявляет агонистическую активность в отношении β2-адренорецептора и антагонистическую активность в отношении М-рецептора. Что касается прочих свойств, соединениями, которые представляют особый интерес, являются соединения со значением Ki для константы ингибирования рецептора подтипа М3 и ЕС50 для агонистической активности в отношении β2-адренорецептора менее 100 нМ, в особенности, соединения, у которых оба значения составляют менее 10 нМ. Кроме того, в экспериментах in vitro или подобных экспериментах также нужно обратить внимание на такие соединения с подобным значением Ki для константы ингибирования рецептора подтипа М3 и значением ЕС50 для агонистической активности в отношении β2-адренорецептора. Например, можно обратить внимание на соединения, у которых отношение значения Ki для константы ингибирования рецептора подтипа М3 к значению ЕС50 для агонистической активности в отношении β2-адренорецептора составляет от приблизительно 1:30 до приблизительно 30:1, конкретно, от 1:20 до 20:1, более конкретно, от приблизительно 1:10 до приблизительно 10:1 и, еще более конкретно, от 1:5 до приблизительно 5:1.
Настоящим изобретением также предполагается способ лечения COPD, включающий в себя введение эффективной дозы соединения, характеризующегося антагонистической активностью в отношении М3-рецептора и агонистической активностью в отношении β2-адренорецептора, пациенту, нуждающемуся в лечении.
В определенных конкретных примерах соединение согласно настоящему изобретению может характеризоваться слабой антагонистической активностью в отношении М-рецептора или агонистической активностью в отношении связывания с β2-адренорецептором, но его при этом все еще можно применять отдельно как антагонист М-рецептора или агонисты β2-адренорецепторов.
При описании соединения, композиции, способа и процесса по настоящему изобретению, если не отмечено иное, следующие термины обладали следующими значениями.
Термин «алкил» относится к неразветвленным или разветвленным незамещенным насыщенным углеводородам. Если не отмечено иное, такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Приводимые в качестве примера алкильные группы включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и т.п.
Термин «алкокси» относится к моновалентной группе (алкил)-О-, где алкил определен в настоящем описании. Приводимые в качестве примера алкоксигруппы включают в себя метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси и т.п.
Термин «арилен» относится к дивалентной группе ароматического кольца, включая замещенные и незамещенные ароматические кольца. Приводимые в качестве примера ариленовые группы включают в себя 1,4-фенилен, 2-метокси-1,4-фенилен, 2,5-фурилиден и т.п.
Термин «гетероциклилен» относится к дивалентной группе гетероатома циклических углеводородов, включая замещенные и незамещенные гетероциклоалканы. Типичные примеры включают в себя 2,3-тетрагидрофурилиден, 2,4-тетрагидропирролилен и т.п.
Термин «алкиленамидная группа» относится к дивалентной группе, содержащей как алкильные, так и амидные группы, включая замещенные и незамещенные алкиленамидные группы. Приводимые в качестве примера алкиленамидные группы включают в себя 2-оксопропиламин-1,4-илиден, 2-оксоэтиламин-1,3-илиден и т.п.
Термин «галоген» относится к фтору, хлору, брому и йоду.
Термин «фармацевтически приемлемая соль» относится к соли, которая может быть использована для введения пациенту. Эта соль может быть получена из фармацевтически приемлемых неорганических и органических оснований и также может быть получена из фармацевтически приемлемых неорганических и органических кислот, включая соли активных соединений, которые получены с относительно нетоксичными кислотами или основаниями в соответствии с конкретными заместителями, которые находятся на описанном в настоящем изобретении соединении. Примеры солей, полученных из фармацевтически приемлемых неорганических оснований, включают в себя без ограничения соли аммония, кальция, калия, натрия и т.п. Соли, полученные из фармацевтически приемлемых органических оснований, включают в себя соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.п., например, без ограничения бетаин, кофеин, холин и т.п. Если соединение по настоящему изобретению содержит относительно основные функциональные группы, соли могут быть получены приведением в контакт такого соединения в свободной форме с достаточным количеством требуемой кислоты отдельно или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают в себя соли, полученные из неорганических кислот, включая без ограничения нитраты, карбонаты, бикарбонаты, фосфаты, сульфаты, бисульфаты, гидрохлориды, гидроброматы и т.п.; и соли, полученные из относительно нетоксичных органических кислот, таких как без ограничения уксусная кислота, янтарная кислота, фумаровая кислота, миндальная кислота, бензолсульфоновая кислота, пара толуолсульфоновая кислота, лимонная кислота, виннокаменная кислота, метансульфоновая кислота и т.п.
Формы соединения, включенные в настоящее изобретение, и его соль могут быть превращены друг в друга традиционными способами из области техники. Например, аммонийные соли могут быть разделены на свободные формы приведением в контакт солей с основаниями или кислотами традиционным способом. Соединение в свободной форме добавляли к кислоте или основанию с получением других солевых форм. Некоторые физические свойства свободной формы соединения, такие как растворимость в полярных растворителях, отличаются от свойств различных солевых форм, но в целях изобретения соль обладает тем же терапевтическим эффектом, что и исходная форма соединения.
В дополнение к солевой форме настоящее изобретение относится к соединениям в форме пролекарственных сложных эфиров. «Пролекарства» описанных в настоящем изобретении соединений представляют собой такие соединения, которые подвержены химическим изменениям в физиологической среде с получением соединений по настоящему изобретению.
«Группа прекурсора» относится к типу защитной группы, которая может превращать лекарственное средство в пролекарство, если функциональная группа, используемая для маскировки активного лекарственного средства, превращена в «фрагмент прекурсора». Группы прекурсора обычно соединены с функциональными группами лекарственных средств при помощи связей, которые могут быть расщеплены при конкретных условиях использования. Таким образом, группа прекурсора являются частью элемента прекурсора, который отщеплялся при условиях конкретного применения для высвобождения функциональных групп.
Конкретные примеры подходящих групп прекурсоров и их соответствующих фрагментов прекурсоров будут очевидны специалистам настоящей области техники.
Определенные соединения по настоящему изобретению содержат асимметрический атом углерода (центр оптического вращения) или двойную связь. Их рацемат, диастереомер, геометрический изомер и оптический изомер включены в объем настоящего изобретения. Такие изомеры могут быть разделены или асимметрически синтезированы традиционными способами с получением «оптически чистых» изомеров, т.е., которые в основном не содержат его других изомеров. Например, если необходим конкретный энантиомер соединения по настоящему изобретению, он может быть получен асимметрическим синтезом или дериватизацией хиральным вспомогательным элементом, где полученную смесь диастереомеров разделяли и вспомогательную группу отщепляли с получением чистого требуемого соединения. Необязательно, различные диастереоизомеры, полученные согласно их множественному хиральному центру, разделяли при помощи подготовительной колонки, например, соединения 61, 70, 82, 98 и другие соединения содержат хиральный атом углерода в своих R1 структурах, т.е., разделение проводили таким способом. Альтернативно, если молекула содержит основную функциональную группу, такую как аминогруппа, или кислотную функциональную группу, такую как карбоксильная группа, соль асимметрического изомера была образована с соответствующим образом вращательно активной кислотой или основанием, а затем образованный таким образом диастереомер отделяли фракционной кристаллизацией или способами хроматографии, хорошо известными из области техники, а затем чистый энантиомер восстанавливали.
Термин «сольват» относится к композиту или полимеру, образованному одной или несколькими молекулами соединения в формуле I или его фармацевтически приемлемой соли и одной или несколькими молекулами растворителя. Такой сольват обычно представляет собой кристалл раствора и растворителя с фиксированным молярным соотношением. Приводимые в качестве примера растворители включают в себя, например, этанол, уксусную кислоту, изопропанол, N, N-диметилформамид, тетрагидрофуран, диметилсульфоксид и воду. В общем, сольватированные формы являются эквивалентными не сольватированным формам и включены в объем настоящего изобретения.
Термин «терапевтически эффективное количество» относится к количеству, достаточному для проведения лечения путем введения пациенту, нуждающемуся в таком лечении.
Термин «уходящая группа» относится к функциональной группе или атому, замещенному другой группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. Приводимая в качестве примера уходящая группа включает в себя, например, хлор, бром, йод и т.п.; сульфонатные группы, такие как метансульфонат, пара-толуолсульфонат, пара-бромбензолсульфонат, пара-нитробензолсульфонат и т.п.; и ацилоксигруппы, такие как ацетилокси, трифторацетилокси и т.п.
Термин «аминозащитная группа» относится к подходящей аминогруппе для предотвращения нежелательной необратимой реакции аминогруппы в процессе реакции и защитная группа может быть впоследствии удалена без затрагивания других частей молекулярной структуры. Приводимые в качестве примера аминозащитные группы включают в себя без ограничения бензил (Bn), трет-бутилоксикарбонил(Вос), бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), формил, ацетил и т.п.
Термин «гидроксизащитная группа» относится к защитной группе, подходящей для предотвращения гидроксигрупп от прохождения нежелательных реакций. Приводимая в качестве примера гидроксизащитная группа включает в себя без ограничения силильные эфиры, такие как триметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил и т.п.; сложные эфиры (ацильные группы), такие как формил, ацетил и т.п.; и арилметильные группы, такие как бензил, пара-метоксибензил, 9-флуоренилметил, бензгидрил и т.п. Кроме того, две гидроксигруппы могут быть защищены защитными группами, такими как пропиленгликолевый эфир, образованный осуществлением взаимодействия ацетона и диола.
Подробное описание предпочтительного варианта осуществления
Следующие Подготовительные примеры и Примеры иллюстрируют конкретные варианты осуществления настоящего изобретения, которые никоим образом не предназначены для ограничения объема настоящего изобретения, если иное не указано особо.
Подготовительный пример 1
(R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанол
(a) 4-гидрокси-3-хлорметилацетофенон
В трехгорлую колбу емкостью 5000 мл помещали 360 г (2,637 моль) 4-гидроксиацетофенона, добавляли 775,5 г (10,34 моль) водного раствора формальдегида, и при перемешивании добавляли 3216 г концентрированной соляной кислоты. Твердое вещество полностью растворялось, температуру реакционной смеси фиксировали при 20°С путем охлаждения на бане с холодной водой, и подавали HCl. Реакцию продолжали при перемешивании, реакционный раствор становился красным, и в осадок выпадало твердое вещество. Реакцию продолжали при перемешивании в течение 5 часов. Реакционную смесь вливали в воду со льдом, перемешивали в течение 30 минут, фильтровали для сбора твердого вещества, 5 раз промывали водой, каждый раз по 1000 мл, а затем 2 раза промывали петролейным эфиром. Твердое вещество сушили в печи при 50°C с получением 400 г 4-гидрокси-3-хлорметилацетофенона в виде красного твердого вещества с выходом 82,1%.
(b) 4-гидрокси-3-ацетилоксиметилацетофенон
В трехгорлую колбу емкостью 2000 мл помещали 397,5 г (2,153 моль) 4-гидрокси-3-хлорметилацетофенона, добавляли 1000 мл ледяной уксусной кислоты, и добавляли при перемешивании 215 г (2,62 моль) ацетата натрия. Реакционную смесь нагревали до 100°С, реакционный раствор становился коричневым, реакцию продолжали при перемешивании при этой температуре в течение 3 часов, и реакция останавливалась. Реакционную смесь охлаждали до комнатной температуры. Реакционную смесь вливали в воду со льдом, и 3 раза экстрагировали дихлорметаном, каждый раз по 600 мл. Органическую фазу объединяли и 3 раза промывали водой. Органическую фазу сушили над безводным сульфатом магния. Осушитель фильтровали и удаляли. Дихлорметан удаляли в условиях пониженного давления. Остаточное твердое вещество растворяли в 200 мл этилацетата путем нагревания, охлаждали и кристаллизовали. Твердое вещество фильтровали и собирали с получением 211 г 4-гидрокси-3-ацетилоксиметилацетофенона в виде белого твердого вещества с выходом 47%.
(c) 4-бензилокси-3-ацетилоксиметилацетофенон
В трехгорлую колбу емкостью 3000 мл помещали 315 г (1,51 моль) 4-гидрокси-3-ацетилоксиметилацетофенона, и добавляли 1800 мл DMF (N,N-диметилформамид) для растворения. Реакционную смесь охлаждали до внутренней температуры 10°С, добавляли 215 г безводного карбоната калия, при этой температуре по каплям добавляли 291 г (1,65 моль) бензилбромида, и завершали добавление по каплям в течение 2 часов. Температуру реакционной смеси повышали до 30°С, и продолжали реакцию в течение 10 часов. Карбонат калия удаляли путем фильтрования, и удаляли из раствора N,N-диметилформамид в условиях пониженного давления. К остатку добавляли 1500 мл воды, и 3 раза экстрагировали этиловым эфиром, каждый раз по 1000 мл. Этилэфирные экстракты объединяли. Этилэфирный слой 3 раза промывали водой, каждый раз по 1000 мл, и сушили этилэфирный слой над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли этиловый эфир. Остаток растворяли путем нагревания с 150 мл этанола, и оставляли охлаждаться для кристаллизации. Твердое вещество фильтровали и собирали с получением 334 г 4-бензилокси-3-ацетилоксиметилацетофенона в виде белого твердого вещества с выходом 74%.
(d) 4-бензилокси-3-гидроксиметилацетофенон
В трехгорлую колбу емкостью 3000 мл помещали 238 г (0,798 моль) 4-бензилокси-3-ацетилоксиметилацетофенона, добавляли 1900 мл метанола, и растворяли исходные вещества путем нагревания и перемешивания. Затем, добавляли 83,11 г (1,995 моль) гидроксида натрия. Реакционную смесь нагревали с обратным холодильником в течение 1 часа. Реакцию останавливали, удаляли растворитель в условиях пониженного давления, добавляли 1000 мл воды, и 3 раза экстрагировали смесь дихлорметаном, каждый раз по 800 мл. Дихлорметановые слои объединяли и сушили над безводным сульфатом магния. Сульфат магния удаляли путем фильтрования. Раствор концентрировали в условиях пониженного давления. Часть дихлорметана удаляли, затем добавляли равный объем этилацетата, и помещали раствор в холодильник для кристаллизации. Твердое вещество фильтровали и собирали с получением 172,56 г 4-бензилокси-3-гидроксиметилацетофенона в виде белого твердого вещества с выходом 84,4%.
(e) 4-бензилокси-3-бензилоксиметилацетофенон
В трехгорлую колбу емкостью 2 л помещали 172,56 г (0,673 моль) 4-бензилокси-3-гидроксиметилацетофенона, растворенного в 1000 мл тетрагидрофурана, и нагревали до внутренней температуры 30°С. Отдельными порциями добавляли гидрид натрия, всего 34,65 г (1,0 моль). После добавления, реакцию продолжали при перемешивании в течение 20 минут, и при этой температуре по каплям добавляли 176,28 г (1,0 моль) бензилбромида в течение 1 часа. После добавления, реакцию продолжали при перемешивании в течение 10 часов. Растворитель удаляли в условиях пониженного давления. К остатку добавляли 800 мл воды, и 3 раза экстрагировали этилацетатом, каждый раз по 600 мл. Экстрагированный раствор объединяли, сушили над безводным сульфатом магния, а затем концентрировали досуха в условиях пониженного давления после удаления осушителя путем фильтрования. Остаток растворяли в этаноле для кристаллизации, твердое вещество фильтровали и собирали с получением 164,15 г белого 4-бензилокси-3-бензилоксиметилацетофенона с выходом 70,4%.
(f) 4-бензилокси-3-бензилоксиметилбромацетофенон
В трехгорлую колбу емкостью 3 л помещали 178,5 г (500,1 ммоль) 4-бензилокси-3-бензилоксиметилацетофенона. Добавляли 2200 мл дихлорметана и перемешивали в течение 30 мин. По каплям добавляли 88 г (510 ммоль) брома при внутренней температуре 20-25°С. Добавление по каплям завершали в течение 2 часов, и проводили реакцию в течение 30 мин, так что получали большое количество твердых продуктов. Методом TLC анализа обнаруживали, что исходные вещества в незначительной степени не прореагировали. Твердое вещество удаляли путем фильтрования. Твердое вещество растворяли в 2000 мл дихлорметана и промывали насыщенным водным раствором бикарбоната натрия (1000 мл × 3 раза). Органический слой сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель вакуумированием с водоструйным насосом в условиях пониженного давления (20°С) с получением твердого вещества. Твердое вещество перекристаллизовывали с абсолютным спиртом и охлаждали досуха с получением 180,5 г продукта с выходом 83,1%.
(g) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бромэтанол
В трехгорлую колбу емкостью 2 л помещали 0,360 г (2,37 ммоль) (1R,2S)-(+)-1-амино-2-инденола. Добавляли 145 мл THF, и перемешивали при 20-25°С. Добавляли 3,3 мл (34,8 ммоль) диметилсульфид-борана и перемешивали в течение 20 мин, и в то же время при 20-25°С по каплям добавляли раствор 130,72 г (300,0 ммоль) 4-бензилокси-3 бензилоксиметилбромацетофенона в 1396 мл THF и раствор 24,54 мл (258,8 ммоль) диметилсульфид-борана в 364 мл TFIF. Добавление по каплям завершали в течение 3 часов. Реакционную смесь поддерживали при температуре 20-25°С, и проводили реакцию в течение 30 мин под защитой атмосферы азота. Методом TLC анализа обнаруживали, что реакция завершена. С внешним охлаждением на бане со льдом (<10°С) по каплям добавляли 145,4 мл метанола, и после добавления по каплям реакционную смесь поддерживали на бане со льдом (<10°С) и перемешивали в течение 10 минут. Растворитель удаляли при 40°С вакуумированием с водоструйным насосом в условиях пониженного давления. Добавляли 500 мл воды и 500 мл этилацетата, и перемешивали при комнатной температуре до тех пор, пока образуются пузырьки газа. Раствор перемешивали при комнатной температуре в течение 5 минут и переносили в делительную воронку. Органический слой отделяли, водный слой дополнительно экстрагировали этилацетатом (150 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и растворитель удаляли при 40°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением неочищенного продукта. Методом колоночной хроматографии неочищенного продукта получали 113,1 г маслянистого вязкого продукта с выходом 86,0%. 1H-ЯМР (м.д.): (CD3Cl), 7,52-7,38 (5Н), 7,36-7,29 (5Н), 7,27-7,19 (2Н), 6,88 (д, 1H), 5,19 (с, 2Н), 5,10 (с, 2Н), 4,92 (т, 1H), 3,86-3,79 (м, 1H), 3,66-3,52 (м, 1H), 2,18 (ушир, 1H). Анализ методом HPLC (хиральная колонка, колонка Daicel AD-H, 4,6 мм × 250 мм), 96,831%) (R-конфигурация), 2,54% (S-конфигурация)
(h) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанол
В трехгорлую колбу емкостью 500 мл помещали 87,4 г (200,0 ммоль) промежуточного (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бромэтанола. Добавляли 162,3 г (1,517 моль) бензиламина и 100 мл диоксана. Реакцию проводили в течение 3 часов при 100-110°С на масляной бане. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли при 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. Добавляли 500 мл этилацетата и 500 мл воды и перемешивали, водный слой корректировали до рН 8-9 добавлением бикарбоната натрия, водный слой переносили в делительную воронку для отделения органического слоя, и дополнительно экстрагировали водный слой этилацетатом (300 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель при 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением маслянистого продукта. К упомянутому выше маслянистому продукту добавляли 160 мл этилацетата, перемешивали для растворения, охлаждали и кристаллизовали на бане со льдом. Белое твердое вещество фильтровали и промывали небольшим количеством этилацетата. Продукт сушили на воздухе при 60°С в течение 2 часов с получением 64,8 г кристаллизованного продукта с выходом реакции 75,5%. 1H-ЯМР (м.д., DMSO (d6)): 7,52-7,38 (5Н), 7,36-7,09 (10Н), 7,27-7,19 (2Н), 6,88 (д, 1H), 5,19 (с, 2Н), 5,10 (с, 2Н), 4,92 (т, 1H), 3,86-3,79 (м, 1H), 3,66-3,52 (м, 1H), 2,18 (ушир, 1H)
Подготовительный пример 2
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октан
(a) R-циклопентилминдальная кислота
В трехгорлую колбу емкостью 5 л добавляли 320 г (1,453 моль) рацемата циклопентилминдальной кислоты, 2670 мл ацетонитрила и 211 мл воды, перемешивали и нагревали для растворения. Температура реакционной смеси составляла 40-45°С, и в этот момент твердое вещество полностью растворялось. При этой температуре добавляли 141,53 г (0,725 моль) сложного метилового эфира D-тирозина для осаждения твердого вещества. Нагревание продолжали до тех пор, пока реакционная смесь нагревалась с обратным холодильником, твердое вещество полностью растворялось, поэтому формировался чистый и прозрачный раствор. Нагревание останавливали, реакционную смесь охлаждали до 0°С на бане со льдом, и продолжали реакцию при перемешивании в течение 4 часов для осаждения большого количества кристаллических веществ. Твердое вещество собирали путем фильтрования, 3 раза промывали ацетонитрилом (3×150 мл), и сушили с получением 230,69 г соли R-циклопентилминдальной кислоты и сложного метилового эфира D-тирозина с выходом 76,6%.
В трехгорлую колбу емкостью 5 л добавляли 230,69 г полученной выше соли R-циклопентилминдальной кислоты и сложного метилового эфира D-тирозина и 2000 мл толуола, и добавляли 1000 мл воды. При перемешивании добавляли 66 мл концентрированной соляной кислоты, нагревали до 40°С на водяной бане до полного растворения твердого вещества и охлаждали. Водную фазу и органическую фазу разделяли на делительной воронке. К водной фазе добавляли 20 мл концентрированной соляной кислоты, и дважды экстрагировали (2×250 мл) толуолом. Органические фазы объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления, и к остатку добавляли 500 мл н-гексана, и перемешивали с получением большого количества твердого вещества. Твердое вещество фильтровали и сушили при 45°С в течение 3 часов с получением 134,4 г R-циклопентилминдальной кислоты в виде белого твердого вещества с выходом 83,4%.
(b) (R)-2-гидрокси-2-фенил-2-циклопентилэтанол
В реакционный котел емкостью 10 л добавляли 134 г (0,608 моль) R-циклопентилминдальной кислоты и 2 л метилгликолевого эфира и перемешивали для растворения. Реакционную смесь охлаждали до -5°С, медленно добавляли 243,21 г (1,824 моль) трихлорида алюминия, температура повышалась, отдельными порциями добавляли 92 г (2,43 моль) боргидрида натрия, и завершали добавление в течение получаса. Температуру реакционной смеси повышали до 50°С, и постоянно перемешивали реакционную смесь в течение 4 часов. После завершения реакции, смесь охлаждали приблизительно до 10°С, и по каплям добавляли 1,1 л 2 М соляной кислоты, контролируя неизменность температуры реакционной смеси. После завершения добавления, для экстрагирования 3 раза использовали изопропиловый эфир (3×500 мл). Органические фазы объединяли, 3 раза промывали насыщенным раствором бикарбоната натрия (3×200 мл) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления, добавляли петролейный эфир для измельчения остатка, твердое вещество фильтровали и собирали с получением 99,88 г (R)-2-гидрокси-2-фенил-2-циклопентилэтанола с выходом 79,6%.
(c) пара-толуолсульфонат (R)-2-гидрокси-2-фенил-2-циклопентилэтанола
В трехгорлую колбу емкостью 2 л помещали 350 мл дихлорметана. Добавляли 98,88 г (0,479 моль) (R)-2-гидрокси-2-фенил-2-циклопентилэтанола, и перемешивали для растворения. Реакционную смесь охлаждали до -5°С, добавляли 121,17 г (1,198 моль) N-метилморфолин, и по каплям добавляли 91,32 г (0,479 моль) раствора пара-толуолсульфонилхлорида в 400 мл дихлорметана. Добавление по каплям завершали в течение 1 часа, и продолжали реакцию при этой температуре в течение 4 часов. Реакционную смесь 3 раза промывали водой (3×500 мл) для разделения органической фазы, и сушили органическую фазу над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления, добавляли изопропиловый эфир для измельчения остатка, твердое вещество фильтровали и собирали с получением 140 г пара-толуолсульфоната (R)-2-гидрокси-2-фенил-2-циклопентилэтанола с выходом 81,1%.
(d) (R)-1-фенил-1 -циклопентилэтиленоксид
В трехгорлую колбу емкостью 2 л помещали 700 мл диметилсульфоксида, добавляли 139 г (0,386 моль) пара-толуолсульфоната (R)-2-гидрокси-2-фенил-2-циклопентилэтанола, температуру реакционной смеси повышали до 30°С, и твердое вещество полностью растворялось при перемешивании. Добавляли 20,07 г (0,502 моль) твердого гидрида натрия (60%). После добавления, температуру реакционной смеси повышали до 50°С, продолжали реакцию в течение 3 часов, и исходные вещества полностью исчезали. Реакционный раствор охлаждали до 10°С, по каплям добавляли 300 мл воды, и 3 раза добавляли изопропиловый эфир для экстрагирования (3×500 мл). Органические фазы объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 70 г (R)-1-фенил-1-циклопентилэтиленоксида в виде твердого вещества с выходом 96,3%.
(е) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октан
В реакционный котел емкостью 2 л помещали 500 мл диметилсульфоксида. Добавляли 47,5 г (0,373 моль) R-(-)-3-хининовый спирт и перемешивали, но твердое вещество оставалось нерастворимым. Добавляли 7,5 г (0,313 моль) гидрида натрия (60%) до тех пор, пока образуются пузырьки газа. Реакционную смесь нагревали до 80°С, и перемешивали реакционную смесь при этой температуре в течение 1 часа. В течение приблизительно 1 часа по каплям добавляли раствор 70 г (0,372 моль) (R)-1-фенил-1-циклопентилэтиленоксида в 100 мл диметилсульфоксида. После завершения добавления, реакцию продолжали при этой температуре в течение 2 часов. Реакционную смесь охлаждали до 20°С, и по каплям добавляли 785 мл воды. Добавление завершали, смесь 3 раза экстрагировали этилацетатом (3×500 мл), органические фазы объединяли, и 3 раза промывали органические фазы водой (3×200 мл). Органические фазы отделяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 88,5 г твердого (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана с выходом 75,2%.
Подготовительный пример 3
8-бензилокси-5-[(R)-2-бензиламино-1-гидроксиэтил]-1Н-хинолин-2-он
(a) 8-ацетилокси-(1Н)-хинолин-2-он
В трехгорлую колбу емкостью 500 мл добавляли 80 г (0,5 моль) 8-гидроксихинолин-N-оксида. Добавляли 300 мл (3,15 моль) уксусного ангидрида, перемешивали и нагревали до 90-100°С. Реакцию продолжали при перемешивании в течение 4 часов, поддерживая указанную температуру, и охлаждали реакционную смесь на бане со льдом для осаждения твердого вещества. Твердое вещество собирали путем фильтрования, дважды промывали ледяным уксусным ангидридом (2×50 мл) и сушили в условиях вакуума с получением 82,2 г 8-ацетилокси-(1Н)-хинолин-2-она в виде твердого вещества с выходом 81,0%.
(b) 5-ацетил-8-гидрокси-(1Н)-хинолин-2-он
В трехгорлую колбу емкостью 500 мл добавляли 200 мл 1,2-дихлорэтана, а затем добавляли 61 г (0,30 моль) 8-ацетилокси-(1Н)-хинолин-2-она для суспендирования в растворителе, реакционную смесь нагревали до 80°С, отдельными порциями добавляли 120 г (0,90 моль) трихлорида алюминия, и продолжали реакцию при этой температуре в течение 1,5 часов для полного преобразования до 5-ацетил-8-гидрокси-(1Н)-хинолин-2-она. Затем, реакционную смесь вливали в 1 л горячей воды при 80°С, и выдерживали при этой температуре в течение 30 минут. Затем, реакционную смесь фильтровали горячей. Твердое вещество измельчали, промывали водой и сушили в условиях вакуума при 80°С. Получали 50,3 г светло-желтого 5-ацетил-8-гидрокси-(1Н)-хинолин-2-она с выходом 82,6%.
(c) 5-ацетил-8-бензилокси-(1Н)-хинолин-2-он
В реакционный сосуд емкостью 500 мл добавляли 32,52 г (0,16 моль) 5-ацетил-8-гидрокси-(1Н)-хинолин-2-она, добавляли 200 мл N,N-диметилформамида для растворения, добавляли 27,2 г (0,20 моль) безводного карбоната калия, при перемешивании по каплям добавляли 27,4 г (0,16 моль) бензилбромида, и перемешивали реакционную смесь при комнатной температуре в течение 4 часов. Карбонат калия удаляли путем фильтрования. Остаток растворяли в 1500 мл дихлорметана, 3 раза промывали водой (3×300 мл) и сушили над безводным сульфатом магния. Дихлорметан удаляли путем фильтрования и концентрирования, остаток измельчали с ацетоном, твердое вещество фильтровали и промывали смесью ацетон/вода (1/1, 2×35 мл) с получением 42,8 г 5-ацетил-8-бензилокси-(1Н)-хинолин-2-она с выходом 91,5%.
(d) 8-бензилокси-5-бромацетил-(1Н)-хинолин-2-он
В трехгорлую колбу емкостью 1 л добавляли 40,0 г (0,137 моль) 5-ацетил-8-бензилокси-(1Н)-хинолин-2-она, добавляли 500 мл дихлорметана и перемешивали для растворения. Реакционный раствор охлаждали приблизительно до 0°С на бане со льдом, и добавляли 0,267 г (0,002 моль) безводного хлорида алюминия. Затем, по каплям добавляли 20 г (0,15 моль) брома, добавление завершали приблизительно в течение 30 минут, и повышали температуру реакционной смеси до комнатной температуры. При этой температуре, реакцию продолжали при перемешивании в течение 4 часов, и методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакционную смесь промывали 3 раза насыщенным раствором бикарбоната натрия (3×100 мл), и сушили органическую фазу над безводным сульфатом магния в течение 3 часов. Сульфат магния удаляли путем фильтрования, и удаляли растворитель путем концентрирования в условиях пониженного давления с получением 42,2 г 8-бензилокси-5-(2-бромацетил)-(1Н)-хинолин-2-она в виде твердого вещества с выходом 82,9%.
(e) 8-бензилокси-5-[(R)-2-бром-1-гидроксиэтил]-1Н-хинолин-2-он
В трехгорлую колбу емкостью 500 мл помещали 0,12 г (0,79 ммоль) (1R,2S)-(+)-1-амино-2-инденола. Добавляли 50 мл THF, и перемешивали при 20-25°С. Добавляли 1,1 мл (11,6 ммоль) диметилсульфид-борана, и перемешивали в течение 20 мин. Одновременно, при 20-25°С в течение 3 часов по каплям добавляли раствор 37,2 г (100,0 ммоль) 8 бензилокси-5-(2-бромацетил)-(1Н)-хинолин-2-она в 500 мл THF и 8,1 мл (86,3 ммоль) диметилсульфид-борана и 120 мл THF. Реакционную смесь выдерживали при температуре 20-25°С, и проводили реакцию в течение 30 мин под защитой атмосферы азота. Методом TLC анализа обнаруживали, что реакция завершена. С внешним охлаждением на бане со льдом (<10°С), по каплям добавляли 50 мл метанола, реакционную смесь выдерживали на бане со льдом (<10°С) и перемешивали в течение 10 минут после добавления по каплям. Растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. Добавляли 200 мл воды и 200 мл этилацетата и перемешивали при комнатной температуре до тех пор, пока выделяются пузырьки газа, перемешивали при комнатной температуре в течение 5 минут, и переносили в делительную воронку. Органический слой отделяли. Водный слой дополнительно экстрагировали этилацетатом (50 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт растворяли в 400 мл ацетонитрила. Добавляли 3 г активированного угла. Нагревание с обратным холодильником продолжали в течение 5 минут, фильтрование проводили, пока раствор был горячим, и при охлаждении в осадок выпадали кристаллы. Твердое вещество фильтровали и собирали с получением 31,8 г 8-бензилокси-5-[(R)-2-бром-1-гидроксиэтил]-1H-хинолин-2-она с выходом 85,5%.
(f) 8-бензилокси-5-[(R)-2-бензиламино-1-гидроксиэтил]-(1Н)-хинолин-2-он
В трехгорлую колбу емкостью 500 мл помещали 29,9 г (80,0 ммоль) промежуточного 8-бензилокси-5-[(R)-2-бром-1-гидроксиэтил]-(1Н)-хинолин-2-она. Добавляли 40,3 г (0,51 моль) бензиламина и 30 мл диоксана. Реакцию проводили в течение 3 часов при 100-110°С на масляной бане. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. Добавляли 150 мл этилацетата и 200 мл воды и перемешивали, добавляли бикарбонат натрия для корректировки водного слоя до рН 8-9, водный слой переносили в делительную воронку для отделения органического слоя, и дополнительно экстрагировали водный слой этилацетатом (100 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением маслянистого продукта. К полученному выше маслянистому продукту добавляли 160 мл этилацетата и перемешивали для растворения, охлаждали и кристаллизовали на бане со льдом. Белое твердое вещество фильтровали и промывали небольшим количеством этилацетата. Продукт сушили на воздухе при 60°С в течение 2 часов с получением 25 г кристаллического продукта с выходом реакции 81,2%.
Подготовительный пример 4
(R)-2-бензиламино-1-[(4-бензилокси-3-формамидо)фенил]этанол
(a) (R)-2-бром-1-[(4-бензилокси-3-нитро)фенил]этанол
В трехгорлую колбу емкостью 2 л помещали 0,132 г (0,87 ммоль) (1R,2S)-(+)-1-амино-2-инденола. Добавляли 180 мл THF, и перемешивали приблизительно при 10°С. Добавляли 9,2 мл (97,0 ммоль) диметилсульфид-борана, и перемешивали в течение 25 мин. Температуру реакционной смеси поддерживали при 5-10°С, и в этот момент по каплям добавляли раствор 63,6 г (180,16 ммоль) 4-бензилокси-3-нитро-бромацетофенона в 600 мл THF и раствор 8,1 мл (86,3 ммоль) диметилсульфид-борана в 120 мл THF. Добавление по каплям завершали в течение 3 часов. Реакционную смесь поддерживали при 5-10°С, и проводили реакцию в течение 30 мин под защитой атмосферы азота. Методом TLC анализа обнаруживали, что реакция завершена. С внешним охлаждением на бане со льдом (<10°С) по каплям добавляли 50 мл метанола, и после добавления по каплям реакционную смесь выдерживали на бане со льдом (<10°С) и перемешивали в течение 10 минут. Растворитель удаляли при 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. К остатку добавляли 200 мл воды и 200 мл этилацетата, перемешивали при комнатной температуре до тех пор, пока выделяются пузырьки газа, перемешивали при комнатной температуре в течение 5 минут, и переносили в делительную воронку. Органический слой отделяли. Водный слой дополнительно экстрагировали этилацетатом (100 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением маслянистого продукта. Маслянистый продукт измельчали с н-гексаном и охлаждали до отделения твердого вещества. Твердое вещество фильтровали и собирали с получением 57,9 г (R)-2-бром-1-[(4-бензилокси-3-нитро)фенил]этанола с выходом 90,5%.
(b) (R)-2-бром-1-[(4-бензилокси-3-амино)фенил]этанол
В реакционный котел для гидрирования емкостью 1 л помещали 55 г (156,15 ммоль) (R)-2-бром-1-[(4-бензилокси-3-нитро)фенил]этанола. Добавляли 600 мл метанола, перемешивали и растворяли. После этого, добавляли 5 г 10% палладированного угля, подавали водород, поддерживали давление при 0,4 МПа, и поддерживали реакционную температуру не выше 5°С. В указанных условиях, реакцию проводили в течение 12 часов. Реакционную смесь фильтровали. Палладированный уголь 3 раза промывали метанолом (3×100 мл), и удаляли растворитель в условиях пониженного давления. Остаток растворяли в этилацетате/петролейном эфире (1/1), охлаждали и кристаллизовали при 0°С, фильтровали для сбора твердого вещества, и сушили с получением 41,9 г (R)-2-бром-1-[(4-бензилокси-3-амино)фенил]этанола с выходом 83,3%.
(c) (R)-2-бром-1-[(4-бензилокси-3-формиламино)фенил]этанол
В трехгорлую колбу емкостью 500 мл добавляли 88 г безводной муравьиной кислоты, охлажденной до 0-5°С.При перемешивании по каплям добавляли 19,6 г (18,7 ммоль) уксусного ангидрида, реакционную температуру поддерживали при 0-5°С, реакционный раствор перемешивали в течение 15 минут, к реакционному раствору добавляли 40,0 г (12,4 ммоль) (R)-2-бром-1-[(4-бензилокси-3-амино)фенил]этанола, и продолжали реакцию при этой температуре в течение 10 часов, так что исходный (R)-2-бром-1-[(4-бензилокси-3-амино)фенил]этанол полностью прореагировали. Избыток муравьиной кислоты удаляли в условиях пониженного давления. Остаток растворяли в 400 мл дихлорметана. Дихлорметановый слой 3 раза промывали водой (3×100 мл). Органическую фазу сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в 300 мл метанола путем нагревания. Для кристаллизации проводили охлаждение, твердое вещество фильтровали и собирали с получением 30,2 г (R)-2-бром-1-[(4-бензилокси-3-формиламино)фенил]этанола с выходом 72,8%.
(d) (R)-2-бензиламино-1-[(4-бензилокси-3-формиламино)фенил]этанол
В трехгорлую колбу емкостью 500 мл с 40,3 г (0,51 моль) бензиламина и 30 мл диоксана добавляли 30,0 г (85,7 ммоль) промежуточного (R)-2-бром-1-[(4-бензилокси-3-формиламино)фенил]этанола. Реакцию проводили в течение 3 часов при 100-110°С на масляной бане. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли вакуумированием с водоструйным насосом в условиях пониженного давления (45°С, -0,095 МПа). Добавляли 250 мл этилацетата и 200 мл воды и перемешивали, добавляли бикарбонат натрия для корректировки водного слоя до рН 8-9, и переносили водный слой в делительную воронку для отделения органического слоя. Водный слой дополнительно экстрагировали этилацетатом (100 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением маслянистого продукта. К упомянутому выше маслянистому продукту добавляли 260 мл этилацетата, и перемешивали для растворения, охлаждали и кристаллизовали на бане со льдом. Белое твердое вещество фильтровали и промывали небольшим количеством этилацетата. Продукт сушили на воздухе при 60°С в течение 2 часов с получением 19,65 г кристаллического продукта с выходом реакции 80,1%.
Подготовительный пример 5
8-[(1R)-1-гидрокси-2-бензиламин]этил-5-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он (формула 28)
(a) 2-гидрокси-5-бензилоксиацетофенон
В трехгорлую колбу емкостью 1 л добавляли 152 г (1,0 моль) 2,5-дигидроксиацетофенона. Добавляли 400 мл абсолютного этанола, перемешивали для растворения. При комнатной температуре добавляли 92,4 г (1,1 моль) бикарбоната натрия и перемешивали в течение 5 минут. Температуру реакционной смеси поддерживали при 0-10°С. По каплям добавляли 180 г (1,05 моль) бензилбромида, и завершали добавление в течение 20 минут. При этой температуре, реакцию продолжали при перемешивании в течение 3 часов, и методом тонкослойной хроматографии обнаруживали, что исходный 2,5-дигидроксиацетофенон полностью прореагировал. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в 400 мл дихлорметана, и 3 раза промывали насыщенным водным раствором лимонной кислоты (3×150 мл). Раствор в дихлорметане сушили над безводным сульфатом магния. Безводный сульфат магния удаляли путем фильтрования. Дихлорметан удаляли из раствора в условиях пониженного давления. Остаток растворяли в 300 мл абсолютного этанола, который охлаждали и кристаллизовали с получением 213,7 г 2-гидрокси-5-бензилоксиацетофенона в виде белого твердого вещества с выходом 88,3%. Химическая чистота составляла 99,0% (HPLC).
(b) 2-гидрокси-3-нитро-5-бензилоксиацетофенон
В трехгорлую колбу емкостью 2 л добавляли 210 г (0,868 моль) 2-гидрокси-5-бензилоксиацетофенона и 900 мл ледяной уксусной кислоты для растворения, реакционную смесь охлаждали до 10-20°С, по каплям добавляли 64,3 мл (0,90 моль) 63% концентрированной азотной кислоты, и завершали добавление в течение 20 минут. Реакцию продолжали при перемешивании при этой температуре в течение 1 часа, и реакция завершалась. Реакционную смесь вливали в 900 мл воды со льдом, и непрерывно перемешивали в течение 1 часа для осаждения кристаллов. Твердое вещество фильтровали, 3 раза промывали водой со льдом, и сушили в условиях вакуума при 50°С с получением 224 г 2-гидрокси-3-нитро-5-бензилоксиацетофенона в виде твердого вещества с выходом 89,9%. Химическая чистота составляла 99,2% (HPLC).
(c) 2-гидрокси-3-амино-5-бензилоксиацетофенон
В реакционный котел для гидрирования емкостью 2 л помещали 220 г (0,767 моль) твердого 2-гидрокси-3-нитро-5-бензилоксиацетофенона, добавляли 800 мл диоксана, и добавляли 11 г оксида платины. При комнатной температуре подавали водород, и поддерживали давление при 3 МПа до прекращения поглощения водорода. Катализатор удаляли путем фильтрования и использовали непосредственно в последующей реакции без обработки.
(d) 8-ацетил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он
В трехгорлую колбу емкостью 3 л добавляли 272 г (2,0 моль) безводного карбоната калия, добавляли фильтрат с предшествующей стадии (с) и перемешивали. При комнатной температуре по каплям добавляли 112,5 г (0,90 моль) хлорацетилхлорида, растворенного в 800 мл диоксана, и завершали добавление в течение 30 минут. Реакционную смесь нагревали до 70°С, и продолжали реакцию при перемешивании в течение 2 часов. Растворитель удаляли в условиях пониженного давления, остаток добавляли в 1 л воды, и растворяли с 1 л дихлорметана. Водную фазу экстрагировали для разделения органической фазы. Водную фазу дважды экстрагировали дихлорметаном (2×500 мл). Органические фазы объединяли и 3 раза промывали водой (3×500 мл). Органическую фазу сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования. Часть дихлорметана удаляли в условиях пониженного давления до тех пор, пока объем органических фаз не составил приблизительно 500 мл. Путем добавления 1500 мл петролейного эфира и перемешивания, в органических фазах в осадок выпадали кристаллы. Кристаллы фильтровали и сушили при 50°С с получением 156,7 г 8-ацетил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она в виде твердого вещества с общим выходом 68,8% в двух партиях. Химическая чистота составляла 99,1% (HPLC).
(e) 8-(бромацетил)-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он
В трехгорлую колбу емкостью 2 л добавляли 148,5 г (0,50 моль) твердого 8-ацетил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она, добавляли 1000 мл диоксана и перемешивали для растворения. Реакционный раствор охлаждали приблизительно до 0°С на бане со льдом, и добавляли 1,0 г (0,01 моль) безводного трихлорида алюминия. Затем, по каплям добавляли 176 г (0,55 моль) брома, и завершали добавление приблизительно в течение 30 минут. Температуру реакционной смеси повышали до комнатной температуры. При этой температуре, реакцию продолжали при перемешивании в течение 4 часов, и методом тонкослойной хроматографии обнаруживали, что реакция завершена. Растворитель удаляли из реакционной смеси в условиях пониженного давления. Остаток растворяли в 1000 мл дихлорметана и 3 раза промывали насыщенным раствором бикарбоната натрия (3×100 мл). Органическую фазу сушили над безводным сульфатом магния в течение 3 часов. Сульфат магния удаляли путем фильтрования, фильтрат концентрировали в условиях пониженного давления для удаления растворителя, и измельчали полученный маслянистый продукт с петролейным эфиром с получением 161,2 г 8-(бромацетил)-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она в виде твердого вещества с выходом 85,7%. Химическая чистота составляла 98,3% (HPLC).
(f) 8-[(1R)-1-гидрокси-2-бром]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он
В трехгорлую колбу емкостью 3 л помещали 0,308 г (2,61 ммоль) (1R,2S)-(+)-1-амино-2-инденола. Добавляли 380 мл THF и перемешивали приблизительно при 10°С. Добавляли 21,4 мл (0,226 моль) диметилсульфид-борана и перемешивали в течение 25 мин. Температуру реакционной смеси поддерживали при 5-10°С, и в этот момент по каплям добавляли раствора 158,2 г (0,42 моль) твердого 8-(бромацетил)-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она в 1200 мл THF и раствор 18,9 мл (0,20 моль) диметилсульфид-борана в 120 мл THF, и завершали добавление по каплям в течение 3 часов. Реакционную смесь поддерживали при 5-10°С, и проводили реакцию в течение 30 мин под защитой атмосферы азота. Методом TLC анализа обнаруживали, что реакция завершена. С внешним охлаждением на бане со льдом (<10°С) по каплям добавляли 120 мл метанола, и после добавления по каплям реакционную смесь выдерживали на бане со льдом (<10°С) и перемешивали в течение 10 минут. Растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. К остатку добавляли 500 мл воды и 500 мл этилацетата, и перемешивали при комнатной температуре до тех пор, пока выделялись пузырьки газа. Смесь перемешивали при комнатной температуре в течение 5 мин и переносили в делительную воронку для отделения органического слоя. Водный слой дополнительно экстрагировали этилацетатом (100 мл × 3 раза), органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением маслянистого продукта. Маслянистый продукт измельчали с н-гексаном и охлаждали для осаждения твердого вещества. Твердое вещество фильтровали и собирали с получением 144,5 г 8-[(1R)-1-гидрокси-2-бром]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она с выходом 91,0%. Химическая чистота составляла 98,5% (HPLC), и значение э.и. составляло 95%.
(g) 8-[(1R)-1-гидрокси-2-бензиламин]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он
В трехгорлую колбу емкостью 1000 мл помещали 143,6 г (0,38 моль) промежуточного 8-[(1R)-1-гидрокси-2-бром]этил-5-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она. Добавляли 321,0 г (3,0 моль) бензиламина и 120 мл диоксана. Реакцию проводили в течение 3,5 ч при 100-110°С на масляной бане. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли вакуумированием с водоструйным насосом в условиях пониженного давления (45°С, -0,095 МПа). Добавляли 1050 мл дихлорметана и 400 мл воды и перемешивали, добавляли бикарбонат натрия для коррекции водного слоя до рН 8-9, и переносили водный слой в делительную воронку для отделения органического слоя. Водный слой дополнительно экстрагировали дихлорметаном (200 мл × 3 раза). Органические слои объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления с получением маслянистого продукта. К упомянутому выше маслянистому продукту добавляли 460 мл этилацетата, и перемешивали для растворения. Реакционную смесь охлаждали и кристаллизовали на бане со льдом. Белое твердое вещество фильтровали и промывали небольшим количеством этилацетата. Продукт сушили на воздухе при 60°С в течение 2 часов с получением 123,6 г кристаллического 8-[(1R)-1-гидрокси-2-бензиламин]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она с выходом реакции 83,6%. Чистота изомера составила 98,5%.
Пример 1
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(R)-2-(3-гидроксиметил-4-гидрокси)фенил-2-гидроксиэтиламино]пропил}-1-азабицикло[2,2,2]октилония бромид
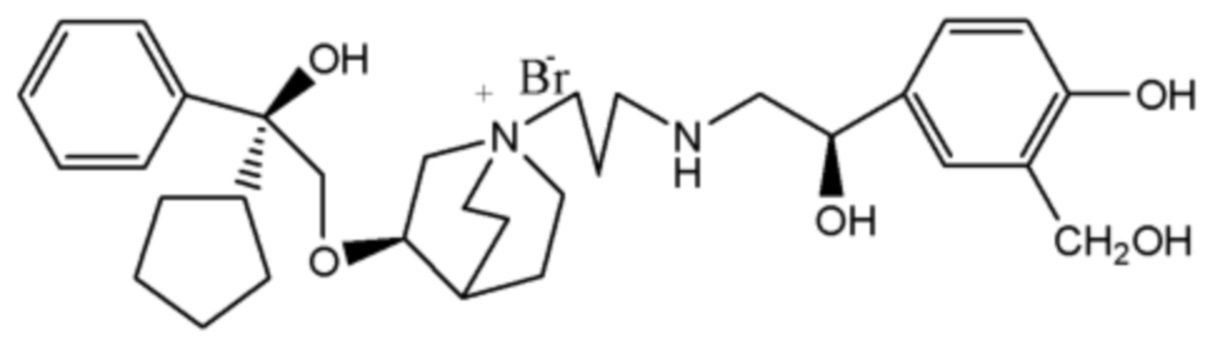
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(3-бромпропил)-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 250 мл добавляли 30 мл абсолютного этанола и 6,0 г (19,02 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 2), перемешивали и растворяли, а затем добавляли 153,6 г (760,8 ммоль) 1,3-дибромпропана, перемешивали и проводили реакцию при комнатной температуре в течение 12 часов. Этанол и избыток 1,3-дибромпропана удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и н-гексаном (1/2) с получением 9,5 г (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(3-бромпропил)-1-азабицикло[2,2,2]октилония бромида в виде твердого вещества с выходом 96,1%. MS (m/z) 436,3, 438,3.
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(R)-2-(3-бензилоксиметил-4-бензилокси)фенил-2-гидроксиэтилбензиламино]пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,396 г (5,283 ммоль) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанола (Подготовительный пример 1) и 2,733 г (5,283 ммоль) продукта (а). Добавляли 20 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,475 г (10,566 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 80°С, и проводили реакцию при этой температуре в течение 5 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 3,0 г целевого продукта. Выход составил 63,8%. MS (m/z) 809,9.
(с) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(R)-2-(3-гидроксиметил-4-гидрокси)фенил-2-гидроксиэтиламино]пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,5 г (4,83 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,5 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. Спустя 10 часов, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с этанолом. Получали 1,6 г целевого соединения в виде твердого вещества с выходом 53,5%. 1H-ЯМР (δ м.д.) (DMSO-d6) 7,19-7,24 (м, 5Н), 6,95-7,01 (м, 3Н), 5,0 (с, 1H), 4,80 (с, 2Н), 4,74 (м, 1H), 3,93 (с, 2Н), 3,68 (д, 2Н), 3,15 (м, 1H), 2,90 (м, 1H), 2,84 (м, 1H), 2,55 (м, 2Н), 2,43 (с, 1H), 2,36 (м, 2Н), 2,21 (с, 1H), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C32H47BrN2O5: 619,63, обнаружено: 539,27 (M-Br).
Пример 2
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(3-{(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}пропил)-1-азабицикло[2,2,2]октилония бромид
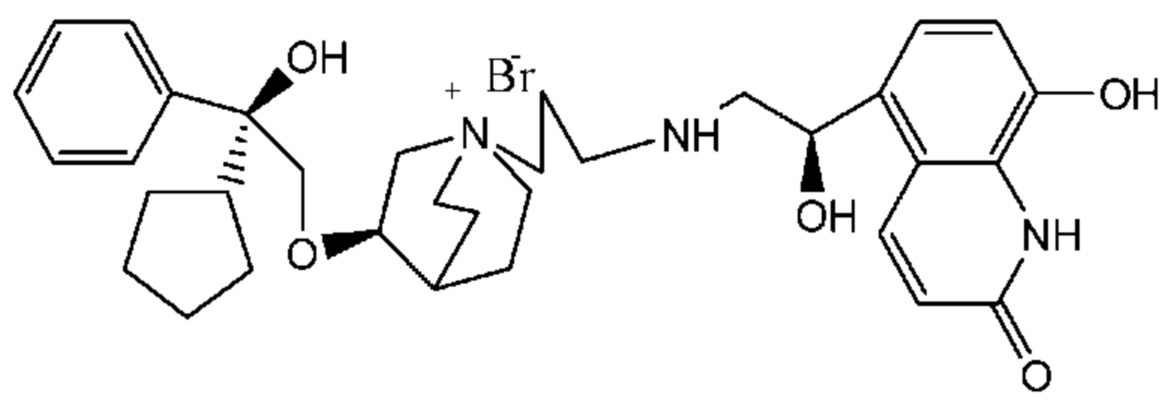
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(3-бромпропил)-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(3-{(R)-[2-гидрокси-2-(8-бензилокси-2-оксо-1,2-дигидрохинолин-5-ил)]этилбензиламино}пропил)-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (b).
В реакционную колбу емкостью 50 мл добавляли 2,0 г (5,0 ммоль) 8-бензилокси-5-[(R)-2-бензиламино-1-гидроксиэтил]-(1Н)-хинолин-2-она (Подготовительный пример 2) и 2,587 г (5,0 ммоль) продукта (а). Добавляли 20 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,475 г (10,566 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 80°С. Реакцию проводили при этой температуре в течение 5 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,3 г целевого продукта. Выход составил 54,9%. MS (m/z) 758,65.
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(3-{(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}пропил)-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (с).
В реакционный котел для гидрирования помещали 2,2 г (2,62 ммоль) продукта (b), добавляли 20 мл метанола для растворения, добавляли 0,4 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. Спустя 10 часов, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,3 г целевого соединения в виде твердого вещества с выходом 90,0%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,0 (с, 1H), 7,36 (д, 1H), 7,19-7,24 (м, 5Н), 6,71 (д, 1H), 6,57 (д, 1H), 6,52 (д, 1H), 5,01 (с, 1H), 4,79 (с, 2Н), 4,73 (м, 1H), 3,93 (с, 2Н), 3,68 (д, 2Н), 3,15 (м, 1H), 2,90 (м, 1H), 2,84 (м, 1H), 2,54 (м, 2Н), 2,43 (с, 1H), 2,36 (м, 2Н), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,82 (м, 3Н), 1,75 (м, 1H), 1,68 (м, 2Н), 1,55-1,61 (м, 6Н), 1,35-1,46 (м, 6Н). Рассчитано для MS (m/z) C34H46BrN3O5: 656,65, обнаружено: 576,26 (M-Br).
Пример 3
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{4-[(R)-2-(3-гидроксиметил-4-гидрокси)фенил-2-гидроксиэтиламино]бутил}-1-азабицикло[2,2,2]октилония бромид
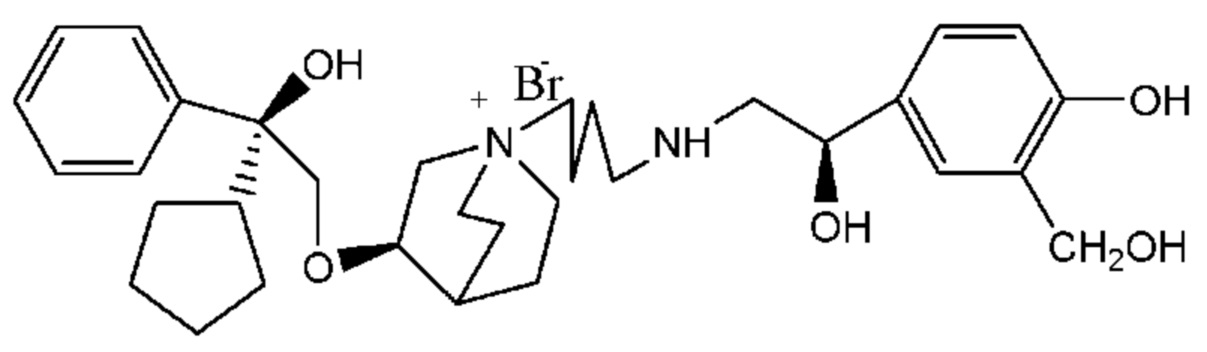
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(4-бромбутил)-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (а).
В трехгорлую колбу емкостью 250 мл добавляли 25 мл абсолютного этанола и 0,5 г (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (15,85 ммоль) (Подготовительный пример 2), перемешивали и растворяли. Затем, добавляли 78,4 мл (634,0 ммоль) 1,4-дибромбутана, перемешивали и проводили реакцию при комнатной температуре в течение 18 часов. Этанол и избыток 1,4-дибромбутана удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и н-гексаном (1/2) с получением 8,1 г (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(4-бромбутил)-1-азабицикло[2,2,2]октилония бромида в виде твердого вещества с выходом 96,1%. MS (m/z) 450,3, 452,3.
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{4-[(R)-2-(3-бензилоксиметил-4-бензилокси)фенил-2-гидроксиэтилбензиламино]бутил}-1-азабицикло[2,2,2]октилония бромид
В реакционную бутыль емкостью 50 мл добавляли 4,096 г (7,253 ммоль) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанола (Подготовительный пример 1) и 3,854 г (7,253 ммоль) продукта (а). Добавляли 30 мл N,N-диметилформамида. После перемешивания реакционной смеси в течение 10 минут, добавляли 3,038 г (21,759 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 55-60°С, и проводили реакцию при этой температуре в течение 12 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 3,24 г целевого продукта. Выход составил 48,0%. MS (m/z) 823,811.
(с) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{4-[(R)-2-(3-гидроксиметил-4-гидрокси)фенил-2-гидроксиэтиламино]бутил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,8 г (3,11 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,7 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. Спустя 12 часов, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с этанолом. Получали 1,5 г целевого соединения в виде твердого вещества с выходом 76,5%. 1H-ЯМР (δ м.д.) (DMSO-d6) 7,18-7,23 (м, 5Н), 6,96-7,01 (м, 3Н), 5,0 (с, 1H), 4,80 (с, 2Н), 4,74 (м, 1H), 3,93 (с, 2Н), 3,68 (д, 2Н), 3,15 (м, 1H), 2,90 (м, 1H), 2,83 (м, 1H), 2,55 (м, 2Н), 2,43 (с, 1H), 2,35 (м, 2Н), 2,21 (с, 1H), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 8Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C33H49BrN2O5: 633,66, обнаружено: 553,38 (M-Br).
Пример 4
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(4-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}бутил-1-азабицикло[2,2,2]октилония бромид
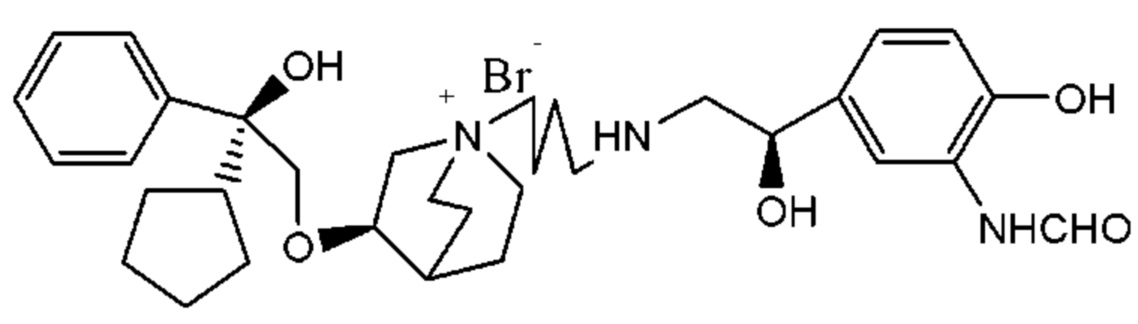
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(4-бромбутил)-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 3 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(4-{(R)-[2-гидрокси-2-(3-формамидо-4-бензилокси)фенил]этилбензиламино}бутил-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 3 (b).
В реакционную колбу емкостью 50 мл добавляли 1,882 г (5,0 ммоль) (R)-2-бензиламино-1-[(4-бензилокси-3-формамидо)фенил]этанола (Подготовительный пример 4) и 2,66 г (5,0 ммоль) продукта (а), добавляли 30 мл ацетонитрила. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,5 г (11,0 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 55-60°С, и проводили реакцию при этой температуре в течение 12 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл этанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 3,04 г целевого продукта. Выход составил 73,6%. MS (m/z) 746,911.
(с) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(4-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}бутил-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 3 (с).
В реакционный котел для гидрирования помещали 2,9 г (3,42 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,6 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа и температура представляла собой комнатную температуру. Спустя 15 часов, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с этанолом. Получали 1,5 г целевого соединения в виде твердого вещества с выходом 67,8%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,21 (с, 1H), 7,40 (м, 1H), 7,18-7,23 (м, 5Н), 6,66-6,71 (м, 2Н), 5,0 (с, 1H), 4,74 (м, 1H), 4,0 (с, 1H), 3,92 (с, 2Н), 3,68 (д, 2Н), 3,15 (м, 1H), 2,91 (м, 1H), 2,83 (м, 1H), 2,54 (м, 2Н), 2,42 (с, 1H), 2,35 (м, 2Н), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 8Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C33H48BrN3O5: 646,65, обнаружено: 566,28.
Пример 5
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(9-{-[(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}нонил)-1-азабицикло[2,2,2]октилония бромид
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(9-{-[(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}нонил)-1-азабицикло[2,2,2]октилония метансульфонат
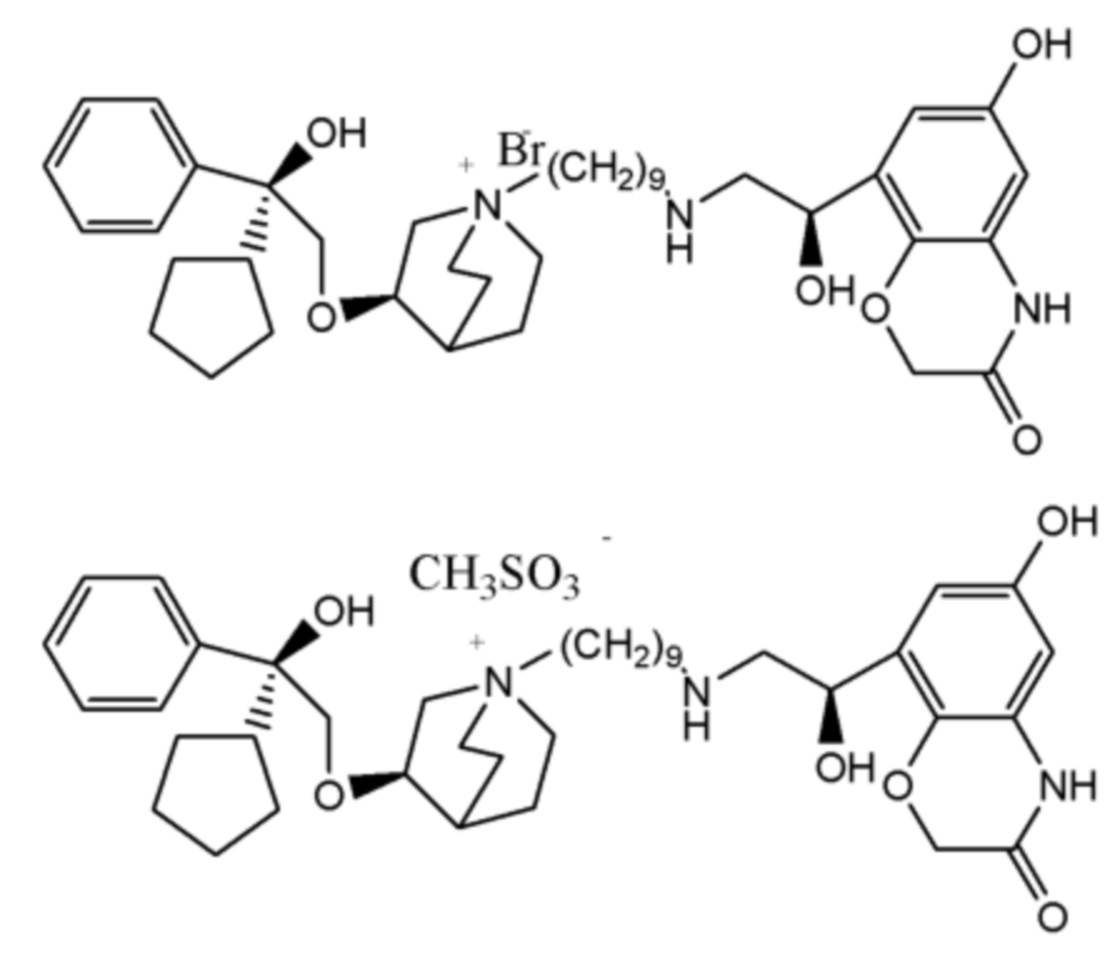
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(9-бромнонил)-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (а).
В трехгорлую колбу емкостью 250 мл добавляли 35 мл абсолютного этанола и 5,0 г (15,85 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 2), перемешивали и растворяли. Затем, добавляли 60,69 г (212,2 ммоль) 1,9-дибромнонана, перемешивали и проводили реакцию при комнатной температуре в течение 18 часов. Этанол и избыток 1,9-дибромнонана удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и петролейным эфиром (1/3) с получением 9,1 г целевого продукта с выходом 95,4%. MS (m/z) 520,18, 522,18.
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-9-{(R)-[2-гидрокси-2-(5-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этилбензиламино}нонил)-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,02 г (5,0 ммоль) 8-[(1R)-1-гидрокси-2-бензиламин]этил-6-бензилокси-2Н-1,4-бензоксазин-34Н)-она (Подготовительный пример 5) и 3,0 г (5,0 ммоль) продукта (а), добавляли 30 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,0 г (7,35 ммоль) безводного карбоната калия. Температуру реакционной смеси повышали до 65-70°С. Реакцию проводили при этой температуре в течение 12 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 3,56 г целевого продукта. Выход составил 77,0%. MS (m/z) 844,35 (M-Br).
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(9-{-[(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}нонил)-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 3,4 г (3,67 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,6 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 15 часов реакции, поглощение водорода останавливалось, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,5 г целевого соединения в виде твердого вещества с выходом 54,8%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,0 (с, 1H), 7,15-7,20 (м, 5Н), 6,60 (д, 1H), 6,20 (д, 1H), 5,0 (с, 1H), 4,88 (с, 2Н), 4,73 (м, 1H), 3,93 (с, 1H), 3,68 (д, 1H), 3,14 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,55 (м, 2Н), 2,36 (м, 2Н), 2,21 (с, 1H), 2,10 (с, 1H), 2,0 (м, 2Н), 1,96 (м, 1H), 1,79-1,80 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 6Н), 1,34-1,46 (м, 10Н), 1,27-1,29 (м, 10Н); Рассчитано для MS (m/z) C39H58BrN3O6: 744,80, обнаружено: 664,35 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(9-{-[(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}нонил)-1-азабицикло[2,2,2]октилония метансульфонат
В 10 мл смеси метанол/вода (об./об., 1:1) растворяли 0,56 г (0,75 ммоль) продукта (с), добавляли 0,10 г оксида серебра, перемешивали при комнатной температуре в течение 30 минут, полученное твердое вещество удаляли путем фильтрования, твердое вещество промывали 5 мл воды, фильтраты объединяли, концентрировали в условиях пониженного давления для удаления части растворителя, добавляли 82 мг метансульфоновой кислоты, перемешивали в течение 10 минут, к раствору добавляли 0,2 г активированного угля, непрерывно перемешивали в течение 5 минут, фильтровали, фильтрат концентрировали досуха, и кристаллизовали остаток с 3 мл изопропанола с получением 0,45 г целевого продукта с выходом 59,2%. C40H61N3O9S элементный анализ (%) (расчетное значение) С 63,10 (63,21), Н 7,97 (8,09), N 5,36 (5,53).
Следующие соединения могут быть синтезированы с использованием способов и исходных веществ, сходных с описанными выше.
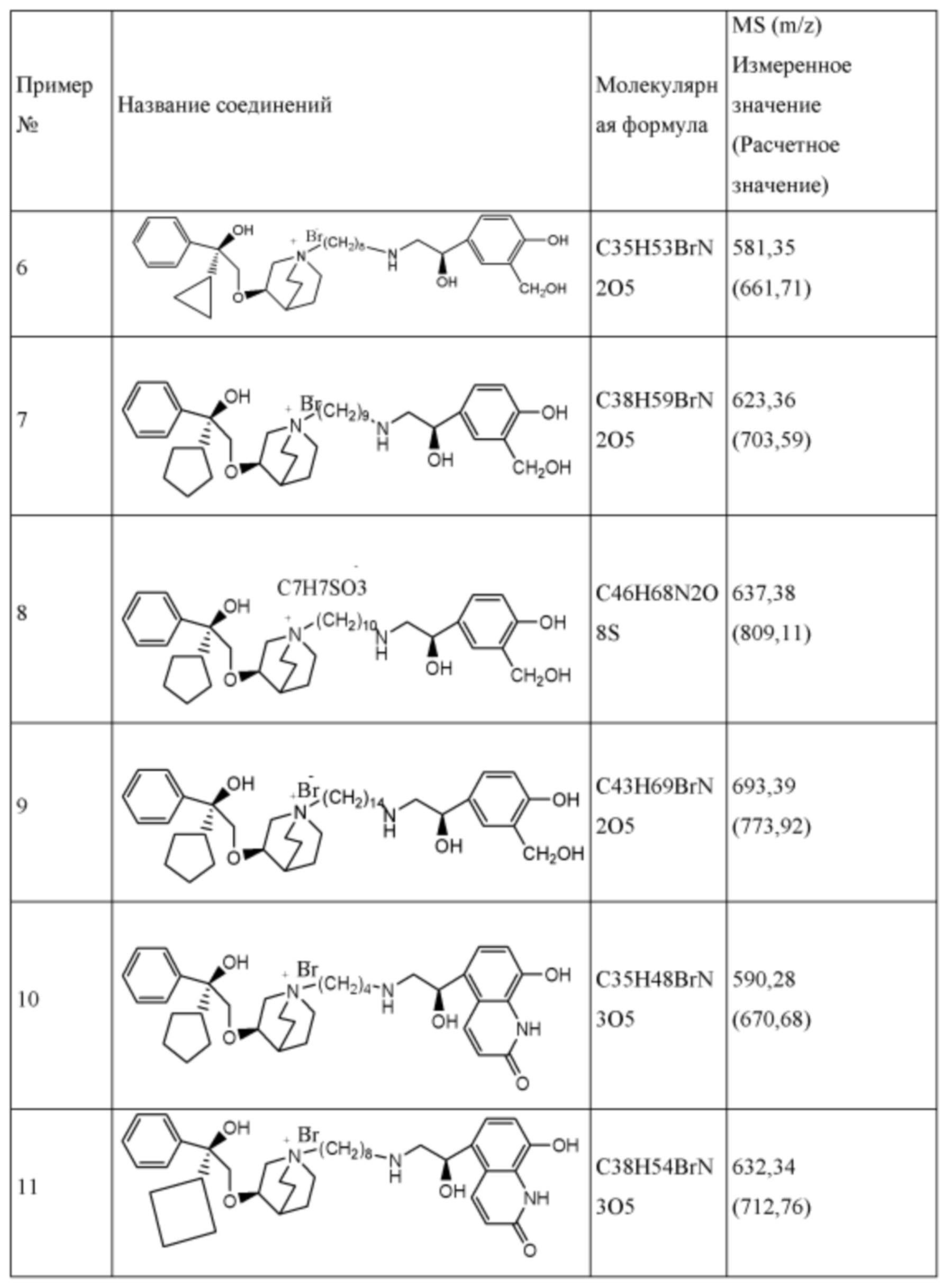
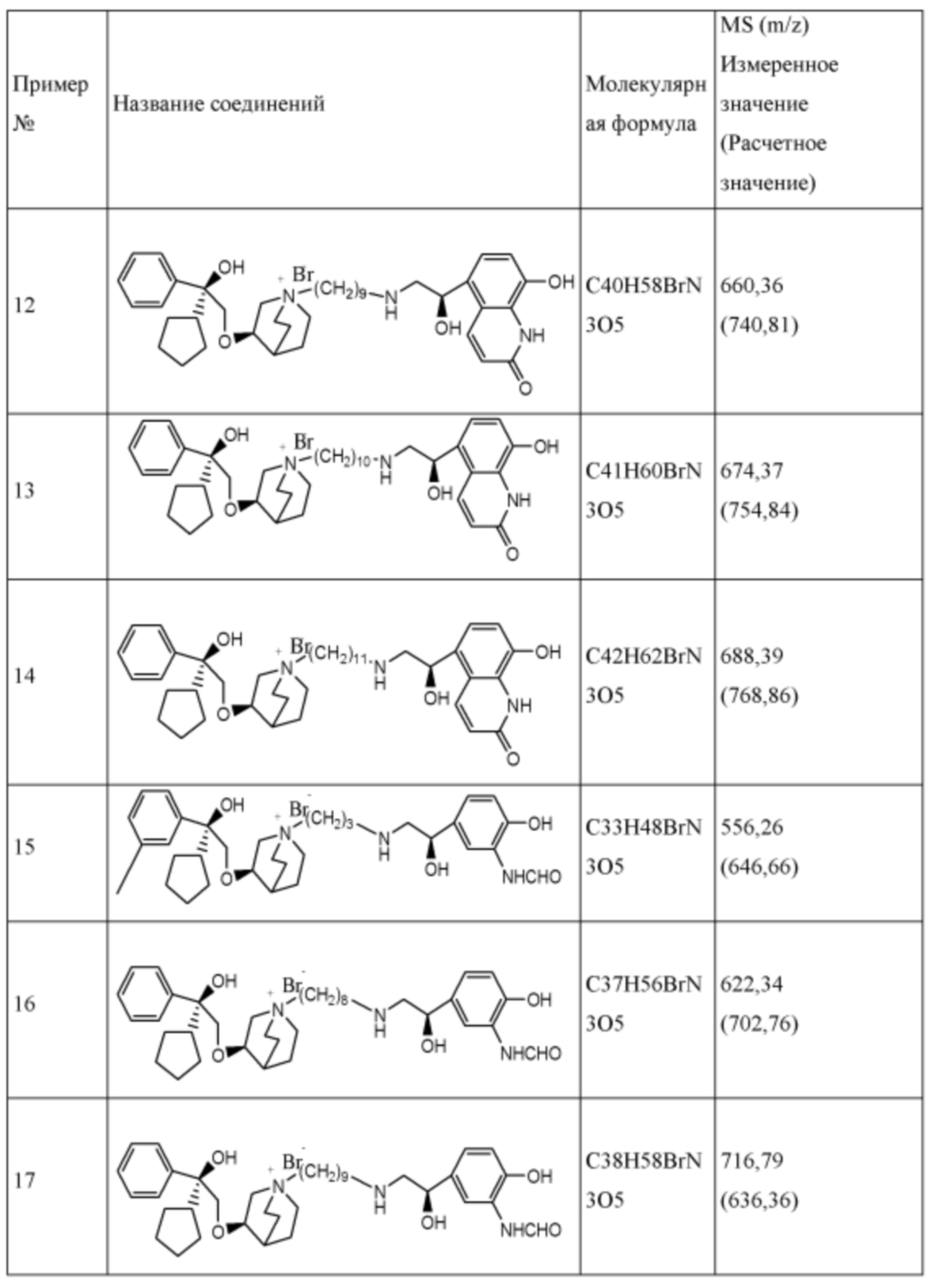
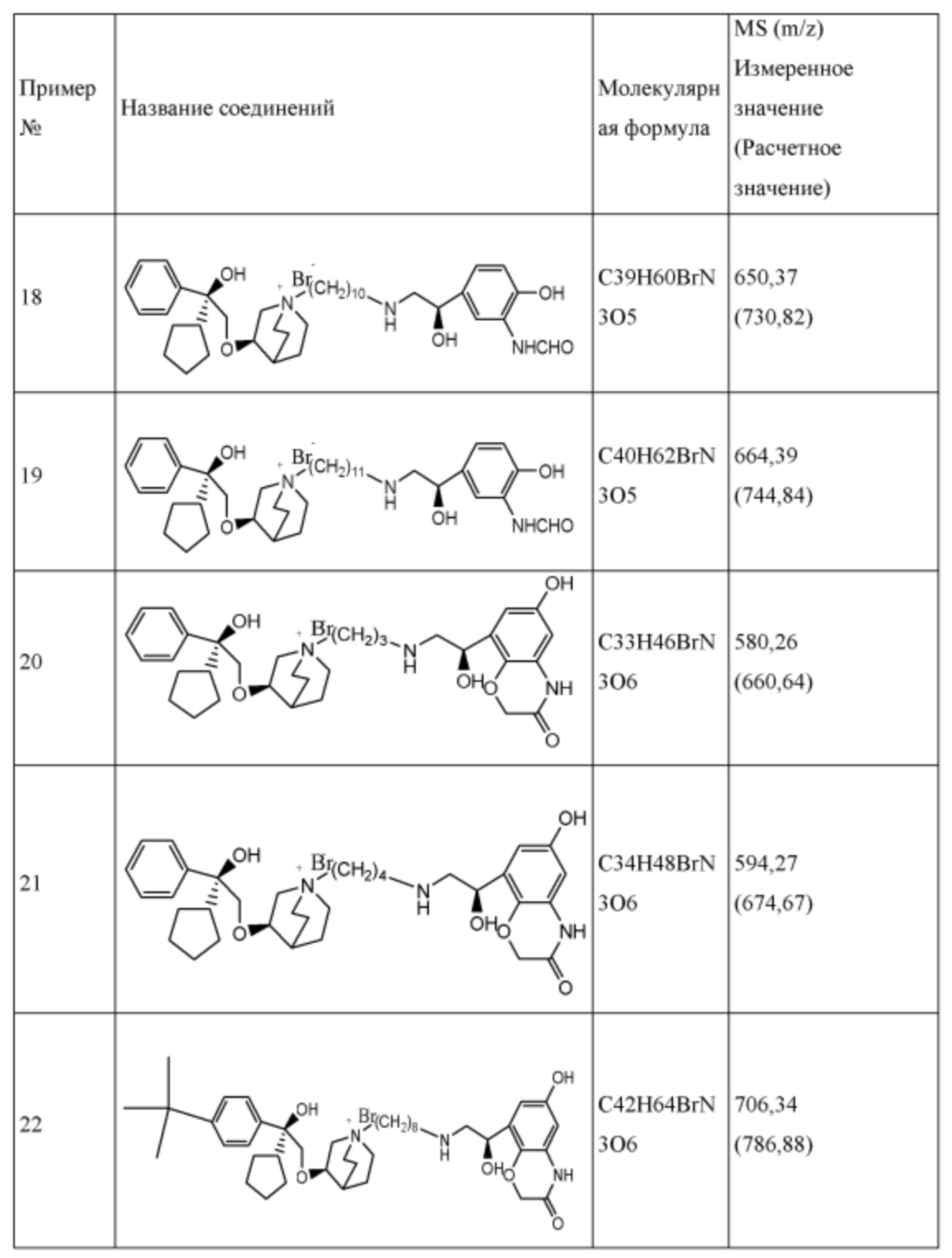
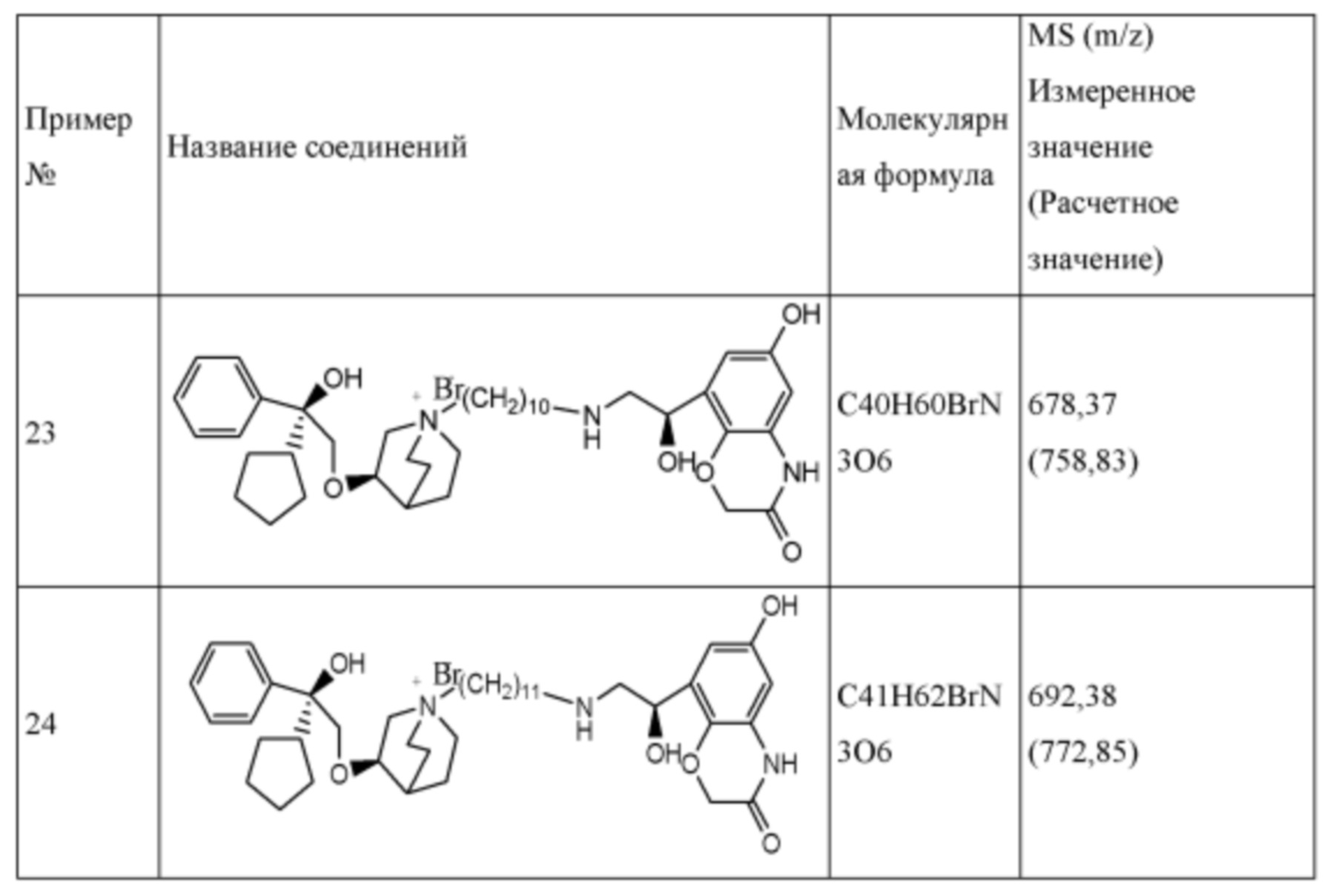
Подготовительный пример 6
4-трет-бутоксиформиламино-1-бутанол
В трехгорлую колбу емкостью 3 л помещали 318 г (3,0 моль) карбоната натрия. Добавляли 500 мл диоксана и 500 мл воды и растворяли. В реакционную колбу единоразово добавляли 178 г (2,0 моль) 4-амино-1-бутанола, и равномерно перемешивали. Температуру реакционной смеси поддерживали приблизительно при 10°С, и по каплям добавляли раствор 545 г (2,50 моль) ди-трет-бутилдикарбоната в 500 мл диоксана. Реакционную температуру поддерживали, и продолжали перемешивание в течение 4 часов. Диоксан удаляли в условиях пониженного давления. Водную фазу 3 раза экстрагировали дихлорметаном (3×1000 мл), сушили над безводным сульфатом магния, и удаляли растворитель в условиях пониженного давления с получением 343 г целевого продукта с выходом 90,7%.
Подготовительный пример 7
4-трет-бутоксиформиламинобутил-2-бромэтиловый эфир
В трехгорлую колбу емкостью 500 мл добавляли 37,8 г (0,20 моль) 4-трет-бутоксиформиламино-1-бутанола и растворяли в 200 мл тетрагидрофурана. Добавляли 8,0 г (0,20 моль) гидрида натрия (60%), перемешивали в течение 15 минут. По каплям добавляли 75,2 г (0,40 моль) 1,2-дибромэтана. После добавления, температуру реакционной смеси повышали до 50°С, и продолжали реакцию в течение 4 часов. Растворитель и избыток 1,2-дибромэтана удаляли в условиях пониженного давления. Остаток растворяли в 300 мл этилацетата, 3 раза промывали водой (3×100 мл) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 48,5 г целевого продукта с выходом 77,2%.
Пример 25
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}бутокси)этил]-1-азабицикло[2,2,2]октилония бромид
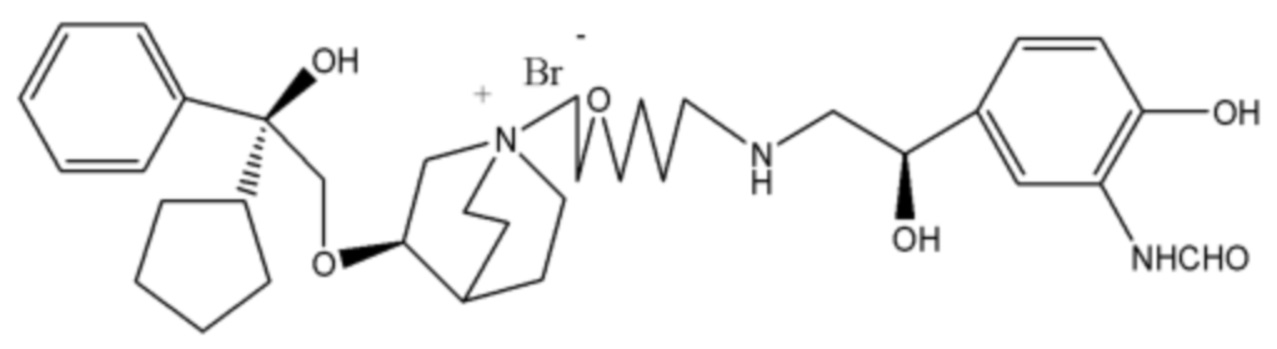
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-трет-бутоксиформамидобутокси)этил]-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 250 мл добавляли 25 мл абсолютного этанола и 5,0 г (15,85 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 2), перемешивали и растворяли. Затем, добавляли 16,2 г (50,0 ммоль) 4-трет-бутоксиформиламинобутил-2-бромэтилового эфира (Подготовительный пример 7), перемешивали, и проводили реакцию при комнатной температуре в течение 24 часов. Этанол удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и н-гексаном (2/1) с получением 6,1 г целевого продукта в виде твердого вещества с выходом 63,1%. MS (m/z) 531,3 (M-Br).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-аминобутокси)этил]-1-азабицикло[2,2,2]октилония бромид
В одногорлую колбу емкостью 50 мл помещали 5,9 г (8,92 ммоль) продукта (а). Добавляли 30 мл 2 М раствора HBr в диоксане, перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли в условиях пониженного давления, оставшееся твердое вещество нейтрализовали добавлением N-метилморфолина и подвергали колоночной хроматографии с получением 3,3 г целевого продукта с выходом 72,3%. MS (m/z) 431,3 (M-Br).
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-{(R)-[2-гидрокси-2-(3-формамидо-4-бензилокси)фенил]этиламино}бутокси)этил]-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 50 мл помещали 3,20 г (9,39 ммоль) промежуточного (R)-2-бром-1-[(4-бензилокси-3-формиламино)фенил]этанола (Подготовительный пример 4). Добавляли 3,20 г (6,26 ммоль) продукта (b), 2,76 г (20 ммоль) безводного карбоната калия и 5 мл диоксана. Реакцию проводили на масляной бане при 100-110°С в течение 3 часов. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли вакуумированием с водоструйным насосом в условиях пониженного давления (45°С, -0,095 МПа). Остаток измельчали со смесью петролейного эфира и дихлорметана (3/1) с получением твердого вещества. Твердое вещество кристаллизовали с изопропанолом с получением 3,8 г целевого продукта с выходом 77,7%. MS (m/z) 700,3 (M-Br)
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}бутокси)этил]-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 3,50 г (4,48 ммоль) продукта (с), добавляли 30 мл метанола для растворения, добавляли 0,5 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 10 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с этанолом. Получали 1,8 г целевого соединения в виде твердого вещества с выходом 58,2%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,20 (с, 1H), 7,40 (м, 1H), 7,18-7,23 (м, 5Н), 6,64-6,71 (м, 2Н), 5,0 (с, 1H), 4,74 (м, 1H), 4,0 (с, 1H), 3,93 (с, 1H), 3,67 (д, 1H), 3,37-3,47 (м, 4Н), 3,15 (м, 1H), 2,91 (м, 1H), 2,83 (м, 1H), 2,53-2,55 (м, 4Н), 2,25 (м, 2Н), 2,10 (с, 1H), 1,96-2,0 (м, 3Н), 1,75-1,81 (м, 3Н), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C35H52BrN3O6: 690,71, обнаружено: MS (m/z) 610,30 (M-Br).
Подготовительный пример 8
4-трет-бутоксиформамидбутил-5-бромпентиловый эфир
Указанное выше соединение получали в соответствии со способом Подготовительного примера 7.
Подготовительный пример 9
4-трет-бутоксиформамидбутил-4-бромбутиловый эфир
Указанное выше соединение получали в соответствии со способом Подготовительного примера 7.
Подготовительный пример 10
4-трет-бутоксиформамидбутил-3-бромпропиловый эфир
Указанное выше соединение получали в соответствии со способом Подготовительного примера 7.
Подготовительный пример 11
2-трет-бутоксиформамидэтил-2-бромэтиловый эфир
Указанное выше соединение получали в соответствии со способом Подготовительного примера 7. Пример 26
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[5-(4-{(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-З (4Н)-он-8-ил)]этиламино}бутокси)пентил]-1-азабицикло[2,2,2]октилония бромид
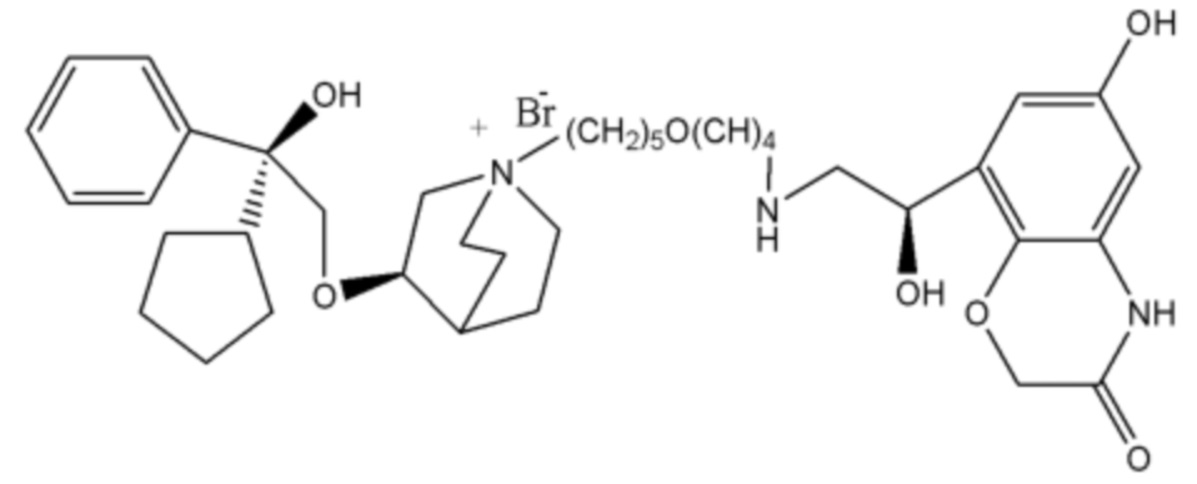
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[5-(4-трет-бутоксиформамидобутокси)пентил]-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 250 мл добавляли 25 мл абсолютного этанола и 5,0 г (15,85 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 2), перемешивали для растворения. Затем, добавляли 17,58 г (52,0 ммоль) 4-трет-бутоксиформамидобутил-5-бромпентилового эфира (Подготовительный пример 8). Реакционную смесь перемешивали для проведения реакции при 40°С в течение 4 часов. Этанол удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и н-гексаном (2/1) с получением 6,0 г целевого продукта в виде твердого вещества с выходом 60,1%. MS (m/z) 573,3 (M-Br).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-аминобутокси)пентил]-1-азабицикло[2,2,2]октилония бромид
В одногорлую колбу емкостью 50 мл помещали 5,9 г (9,02 ммоль) продукта (а), добавляли 30 мл 2 М раствора HBr в диоксане, и перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли в условиях пониженного давления, оставшееся твердое вещество нейтрализовали добавлением N-метилморфолина и подвергали колоночной хроматографии с получением 3,2 г целевого продукта с выходом 64,2%. MS (m/z) 473,3 (M-Br).
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[5-(4-{(R)-[2-гидрокси-2-(6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}бутокси)пентил]-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 50 мл помещали 4,20 г (11,2 ммоль) промежуточного 8-[(1R)-1-гидрокси-2-бром]этил-5-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она (Подготовительный пример 5 (f)). Добавляли 3,10 г (5,60 ммоль) продукта (b), 2,76 г (20 ммоль) карбоната калия и 5 мл диоксана. Реакцию проводили на масляной бане при 100-110°С в течение 3 ч. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. Остаток измельчали со смесью петролейного эфира и дихлорметана (3/1) с получением твердого вещества. Твердое вещество кристаллизовали с изопропанолом с получением 3,9 г целевого продукта с выходом 81,8%. MS (m/z) 770,4 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[5-(4-{(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}бутокси)пентил]-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 3,80 г (4,47 ммоль) продукта (с), добавляли 30 мл метанола для растворения, добавляли 0,5 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 10 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,9 г целевого соединения в виде твердого вещества с выходом 55,8%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,0 (с, 1H), 7,18-7,20 (м, 5Н), 6,65 (д, 1H), 6,20 (д, 1H), 5,0 (с, 1H), 4,88 (с, 2Н), 4,74 (м, 1H), 3,93 (д, 1H), 3,68 (д, 1H), 3,37-3,40 (м, 4Н), 3,15 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,55 (м, 2Н), 2,36 (т, 2Н), 2,10 (с, 1H), 1,98-2,0 (м, 3Н), 1,75-1,81 (м, 4Н), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 12Н), 1,29 (м, 2Н); Рассчитано для MS (m/z) C39H58BrN3O7: 760,80, обнаружено: MS (m/z) 680,35 (M-Br).
Пример 27
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[4-(4-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}бутокси)бутил-1-азабицикло[2,2,2]октилония бромид
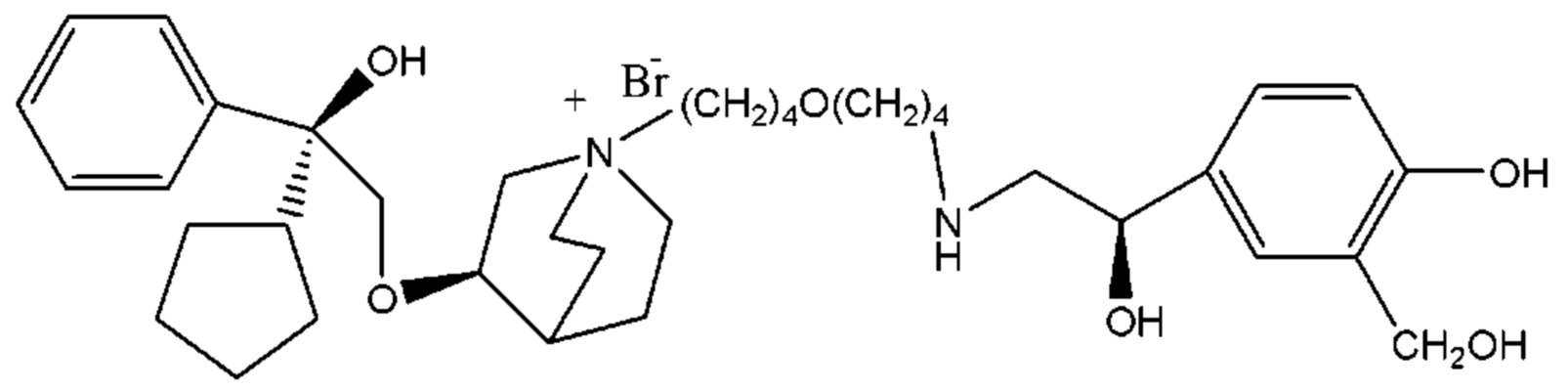
(а) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[4-(4-трет-бутоксиформамидобутокси)бутил-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 25 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[4-(4-аминобутокси)бутил-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 25 (b).
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[4-(4-{(R)-[2-(3-бензилоксигидроксиметил-4-бензилокси)фенил-2-гидрокси]этиламино}бутокси)бутил-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 50 мл помещали 3,50 г (8,19 ммоль) промежуточного (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бромэтанола (Подготовительный пример 1 (g)). Добавляли 3,20 г (5,09 ммоль) продукта (b), 2,76 г (20 ммоль) карбоната калия и 5 мл диоксана. Реакцию проводили в течение 3 часов при 100-110°С на масляной бане. Методом TLC анализа обнаруживали, что промежуточный продукт прореагировал полностью. Растворитель удаляли при температуре ниже 45°С вакуумированием с водоструйным насосом в условиях пониженного давления. Остаток измельчали со смесью петролейного эфира и дихлорметана (3/1) с получением твердого вещества. Твердое вещество кристаллизовали с изопропанолом с получением 4,1 г целевого продукта с выходом 93,8%. MS (m/z) 777,4 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[4-(4-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}бутокси)бутил-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 4,0 г (4,10 ммоль) продукта (с), добавляли 30 мл метанола для растворения, добавляли 0,8 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 20 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с этанолом. Получали 1,6 г целевого соединения в виде твердого вещества с выходом 55,4%. 1H-ЯМР (δ м.д.) (DMSO-d6) 7,17-7,20 (м, 5Н), 6,95 (м, 2Н), 6,64 (д, 1H), 5,0 (с, 1H), 4,79 (с, 2Н), 4,74 (м, 1H), 3,93 (д, 1H), 3,67 (д, 1H), 3,37-3,47 (т, 4Н), 3,14 (м, 1H), 2,90 (м, 1H), 2,84 (м, 1H), 2,53-2,55 (м, 2Н), 2,36 (м, 2Н), 1,96-2,0 (м, 5Н), 1,74-1,81 (м, 4Н), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 14Н); Рассчитано для MS (m/z) C37H57BrN2O6: 705,76, обнаружено: 625,34 (M-Br).
Следующие соединения могут быть получены из описанных выше исходных веществ в соответствии со следующими способами.
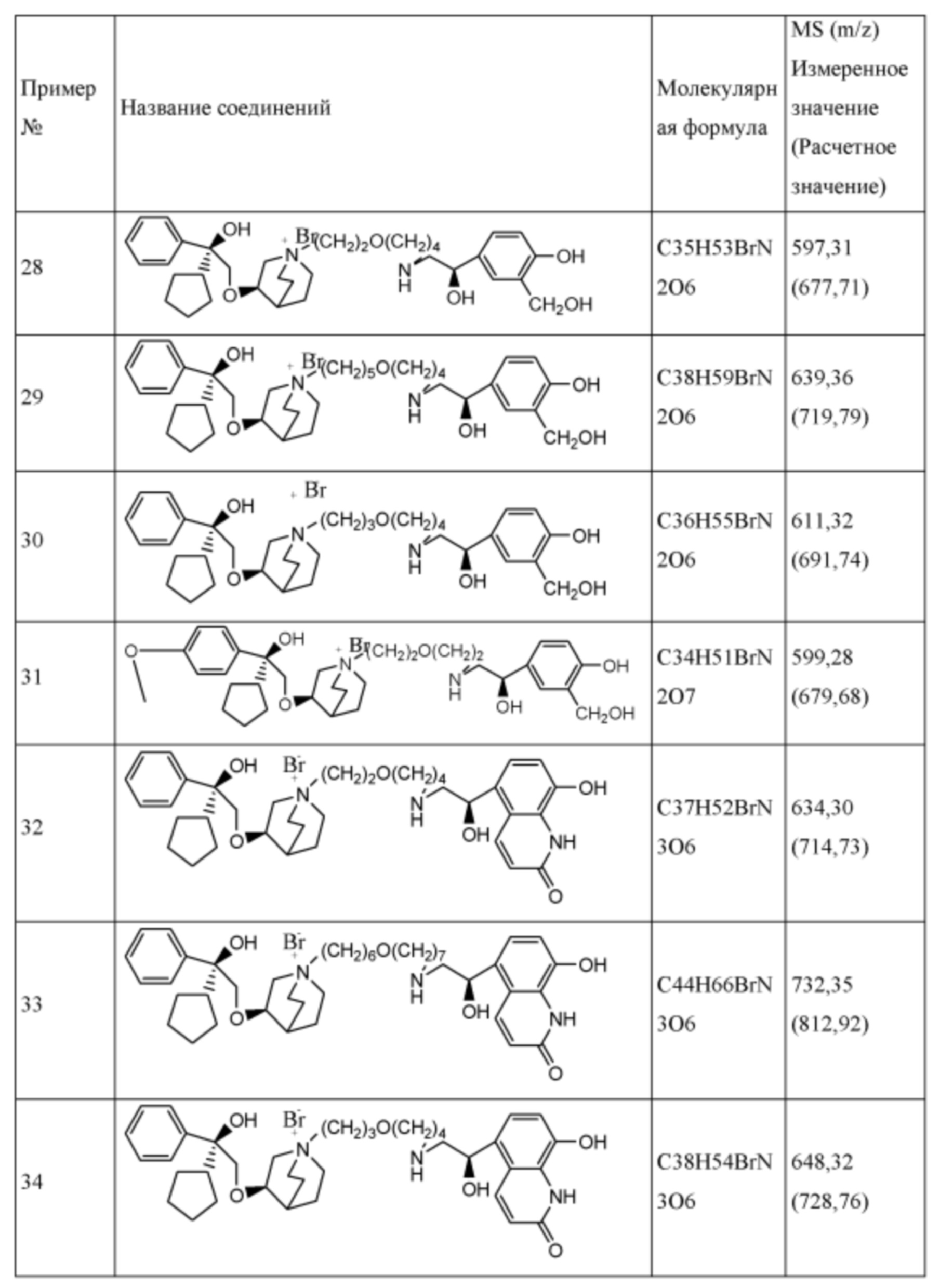
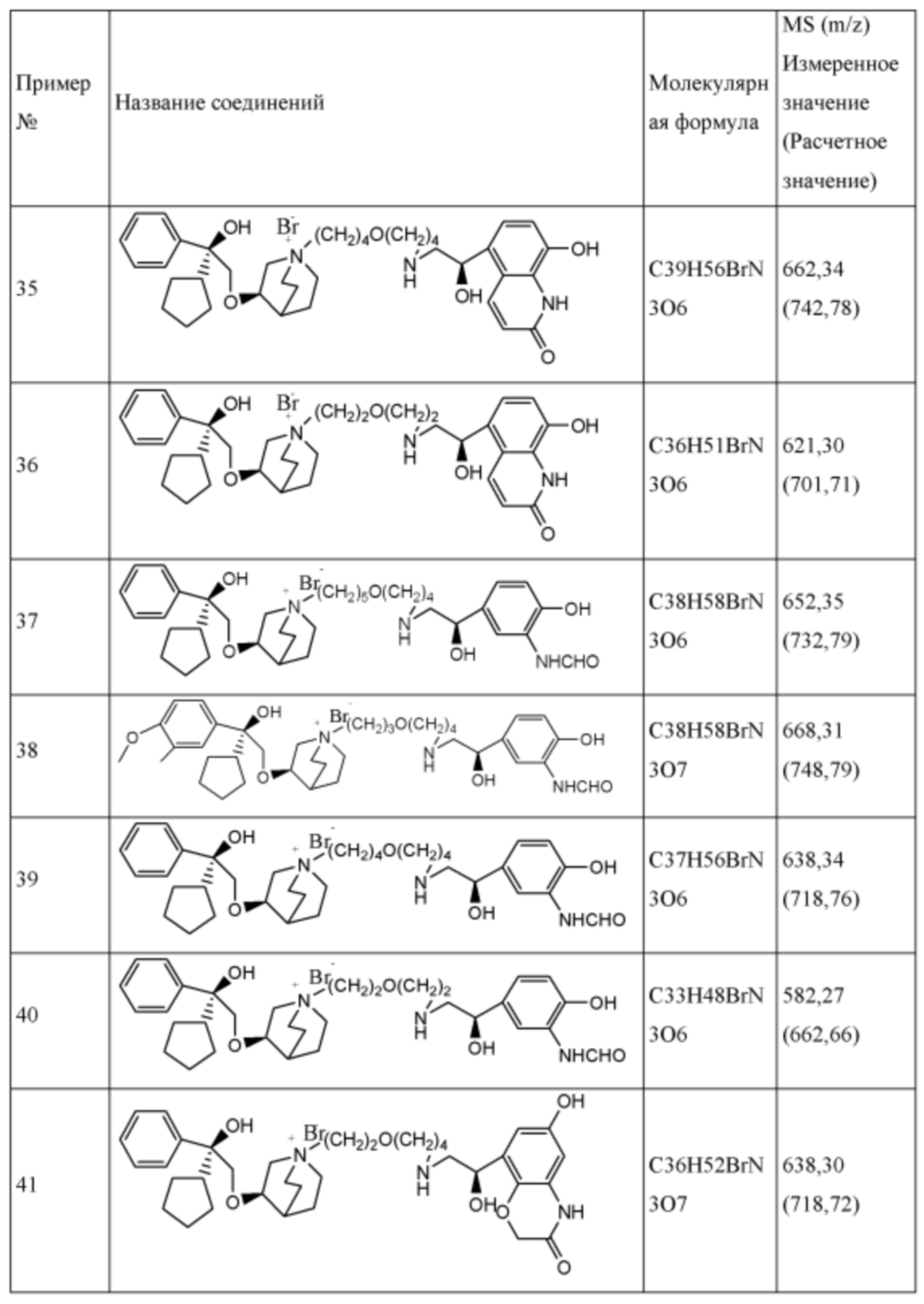
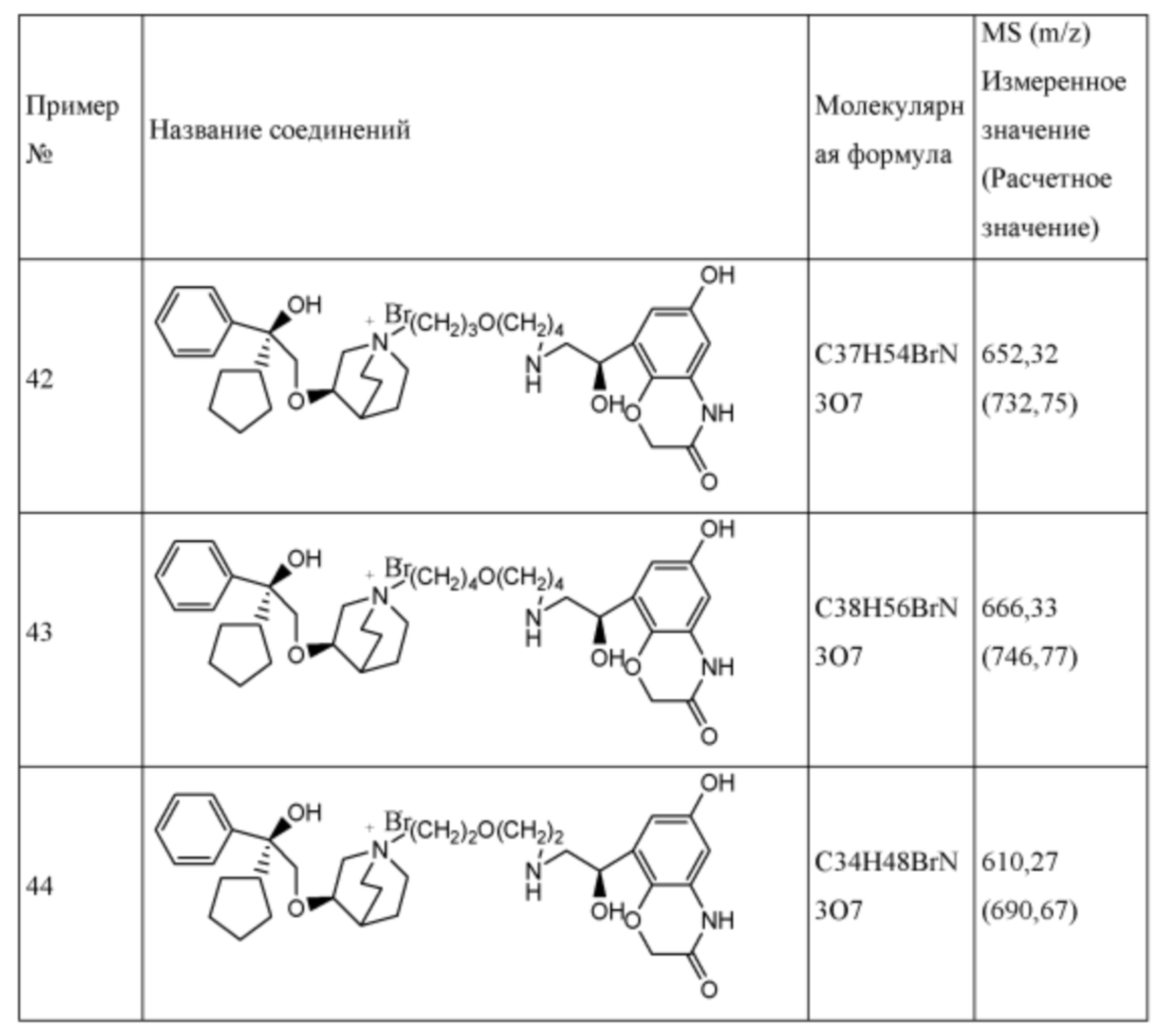
Подготовительный пример 12
1,3-бис(2-бромэтокси)пропан
В 500 мл тетрагидрофурана растворяли 38 г (0,50 моль) 1,3-пропандиола. Добавляли 40 г (1,0 моль) гидрида натрия (60%). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут. По каплям добавляли 752 г (4,0 моль) 1,2-дибромэтана. После добавления, проводили нагрев реакционной смеси с обратным холодильником в течение 3 часов, растворитель и избыток 1,2-дибромэтана удаляли в условиях пониженного давления. Остаток растворяли в 500 мл дихлорметана, 3 раза промывали водой (3×200 мл) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 80 г целевого продукта с выходом 61%.
Подготовительный пример 13
бис(2-бромэтоксиэтил)эфир
В 400 мл тетрагидрофурана растворяли 42,4 г (0,40 моль) бис(2-гидроксиэтил)эфира. Добавляли 33,6 г (1,05 моль) гидрид натрия (60%). Реакционную смесь перемешивали при комнатной температуре в течение 25 минут. По каплям добавляли 752 г (4,0 моль) 1,2-дибромэтан. После добавления, проводили нагрев реакционной смеси с обратным холодильником в течение 5 часов, растворитель и избыток 1,2-дибромэтан удаляли в условиях пониженного давления. Остаток растворяли в 400 мл дихлорметана, 3 раза промывали водой (3×200 мл) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 70 г целевого продукта с выходом 54,7%.
Пример 45
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(2-{2-[2-(2-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этокси)этокси]этокси}этил)-1-азабицикло[2,2,2]октилония бромид
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(2-{2-[2-(2-этокси)этокси]этокси}бромэтил)-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 250 мл добавляли 30 мл абсолютного этанола, 6,0 г (19,02 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 2), перемешивали и растворяли. Затем, добавляли 243,6 г (760,8 ммоль) бис(2-бромэтоксиэтил)эфир (Подготовительный пример 13). Реакционную смесь перемешивали для проведения реакции при комнатной температуре в течение 10 часов. Этанол удаляли в условиях пониженного давления, избыток бис(2-бромэтоксиэтил)эфира в остатке удаляли методом колоночной хроматографии, и кристаллизовали неочищенный продукт с дихлорметаном и н-гексаном (1/1) с получением 9,3 г целевого продукта с выходом 76,9%. MS (m/z) 654,2, 656,2.
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(2-{2-[2-(2-{(R)-[2-(3-бензилоксиметил-4-бензилокси)фенил-2-гидрокси]этиламино}этокси)этокси]этокси}-этил)-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,396 г (5,283 ммоль) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанола (Подготовительный пример 1) и 3,46 г (5,283 ммоль) продукта (а). Добавляли 20 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,475 г (10,566 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 80°С, и проводили реакцию при этой температуре в течение 3 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,8 г целевого продукта. Выход составил 57,7%. Рассчитано для MS (m/z) C51H69BrN2O8: 918,0, обнаружено: 838,4 (M-Br).
(с) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-(2-{2-[2-(2-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этокси)этокси]этокси}этил)-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,4 г (2,61 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,5 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 20 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,3 г целевого соединения в виде твердого вещества с выходом 67,5%. 1H-ЯМР (δ м.д.) (DMSO-d6) 7,18-7,21 (м, 5Н), 6,59-6,95 (м, 3Н), 5,0 (с, 1H), 4,79 (с, 2Н), 4,74 (м, 1H), 3,93 (д, 1H), 3,68 (д, 1H), 3,47-3,54 (м,12Н), 3,15 (м, 1H), 2,90 (м, 1H), 2,83 (м, 1H), 2,72 (т, 2Н), 2,53 (м, 2Н), 2,36 (м, 1H), 2,21 (с, 1H), 2,10 (с, 1H), 1,96-2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,74 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,33-1,45 (м, 6Н); Рассчитано для MS (m/z) C37H57BrN2O8: 737,76, обнаружено: 657,3 (M-Br).
Пример 46
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[2-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}этокси)этокси]этил}-1-азабицикло[2,2,2]октилония хлорид
(а) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[2-(2-хлорэтокси)этокси]этил}-1-азабицикло[2,2,2]октилония хлорид
В трехгорлую колбу емкостью 250 мл добавляли 30 мл абсолютного этанола, 6,0 г (19,02 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 2), перемешивали и растворяли. Затем, добавляли 142,3 г (760,8 ммоль) 1,2-бис(2-хлорэтокси)этан (Shanghai Hanhong Chemical). Реакционную смесь нагревали до 45°С, перемешивали и проводили реакцию в течение 6 часов. Этанол удаляли в условиях пониженного давления, избыток 1,2-бис(2-хлорэтокси)этана в остатке удаляли методом колоночной хроматографии, и кристаллизовали неочищенный продукт с дихлорметаном и н-гексаном (3/1) с получением 8,3 г целевого продукта с выходом 86,8%. Рассчитано для MS (m/z) C26H41ClNO4: 502,51, обнаружено: 466,24 (М-Cl), 468,23 (М-Cl).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[2-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-бензилокси)фенил]этиламино}этокси)этокси]этил}-1-азабицикло[2,2,2]октилония хлорид
В реакционную колбу емкостью 50 мл добавляли 1,882 г (5,0 ммоль) (R)-2-бензиламино-1-[(4-бензилокси-3-формамидо)фенил]этанола (Подготовительный пример 4) и 2,568 г (5,0 ммоль) продукта (а). Добавляли 30 мл ацетонитрила. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,5 г (11,0 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 75-80°С, и проводили реакцию при этой температуре в течение 8 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,4 г целевого продукта. Выход составил 63,8%. Рассчитано для MS (m/z) C42H58ClN3O7: 752,38, обнаружено: 716,84 (М-Cl).
(c) (R)-(-)-3 -[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[2-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}этокси)этокси]этил}-1-азабицикло[2,2,2]октилония хлорид
В реакционный котел для гидрирования помещали 2,1 г (2,79 ммоль) продукта (b), добавляли 25 мл метанола для растворения, добавляли 0,4 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 10 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,1 г целевого соединения в виде твердого вещества с выходом 59,5%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,20 (с, 1H), 7,40 (дд, 1H), 7,19-7,21 (м, 5Н), 6,64-6,76 (м, 2Н), 5,01 (с, 1H), 4,73 (м, 1H), 4,0 (с, 1H), 3,93 (д, 2Н), 3,68 (д, 2Н), 3,47-3,54 (м, 8Н), 3,15 (м, 1H), 2,90 (м, 1H), 2,84 (м, 1H), 2,72 (т, 2Н), 2,53 (д, 2Н), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,68 (м, 2Н), 1,55-1,61 (м, 6Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C35H52ClN3O7: 662,26, обнаружено: 626,74 (М-Cl).
Следующие соединения могут быть получены из представленных выше исходных веществ в соответствии со способами Примеров 45 и 46:
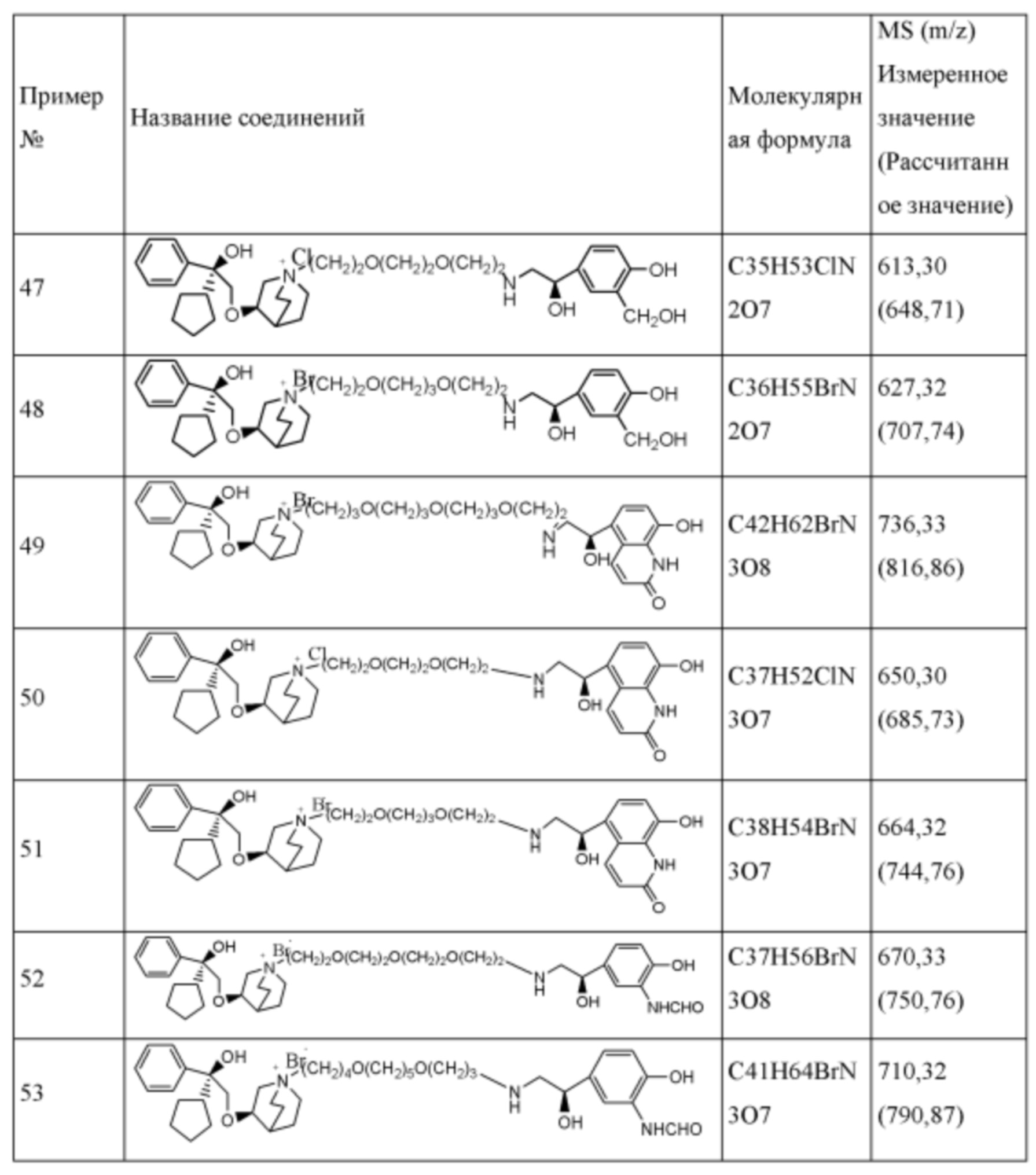
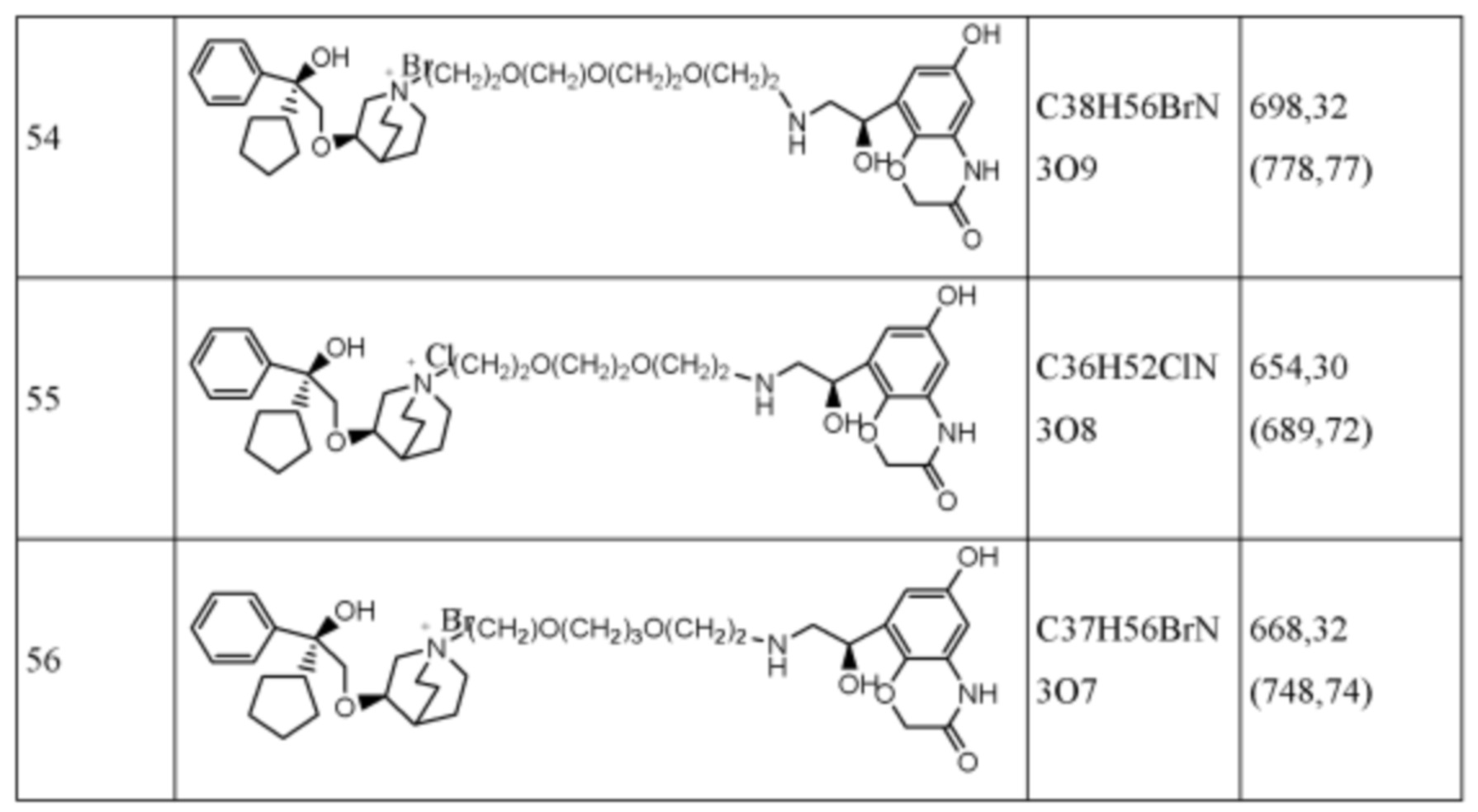
Подготовительный пример 14
2-[(4-бромэтокси)фенил]этиловый спирт
(a) 4-[2-гидроксиэтил]фенол
В 500 мл метанола, охлажденного на бане со льдом, растворяли 166 г (1,0 моль) метил-пара-гидроксифенилацетата. Отдельными порциями добавляли 56,75 г (1,50 моль) боргидрид натрия, и завершали добавление приблизительно в течение 2 часов. Реакцию продолжали при перемешивании в течение 2 часов. Методом тонкослойной хроматографии обнаруживали, что исходные вещества полностью прореагировали. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в воде, которую корректировали до рН 2 добавлением 2 М соляной кислоты. Раствор 3 раза экстрагировали дихлорметаном (300 мл × 3). Экстракты объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления, и кристаллизовали остаток с этилацетатом и петролейным эфиром (1/5). Получали 120,0 г целевого соединения с выходом 86,9%.
(b) 2-[(4-бромэтокси)фенил]этиловый спирт
В 300 мл этанола растворяли 30 г (0,217 моль) продукта (а). Добавляли 22,4 (0,26 моль) бикарбоната натрия. Одновременно добавляли 188 г (1,0 моль) 1,2-дибромэтана. Реакционную смесь нагревали и перемешивали. Реакцию проводили при 50°С в течение 5 часов. Растворитель и избыток 1,2-дибромэтана удаляли в условиях пониженного давления. Остаток растворяли в 300 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования. Растворитель удаляли в условиях пониженного давления с получением 35,0 г целевого соединения с выходом 65,8%. m/z 245,01, 247,05 (М+Н).
Подготовительный пример 15
2-[(4-бромпропокси)фенил]этиловый спирт
В 300 мл этанола растворяли 30 г (0,217 моль) продукта Подготовительного примера 14 (а). Добавляли 22,4 г (0,26 моль) бикарбоната натрия. Одновременно добавляли 202 г (1,0 моль) 1,3-дибромпропана. Реакционную смесь нагревали и перемешивали. Реакцию проводили при 50°С в течение 6 часов. Растворитель и избыток 1,2-дибромпропана удаляли в условиях пониженного давления. Остаток растворяли в 300 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования. Растворитель удаляли в условиях пониженного давления с получением 40 г целевого соединения с выходом 71,1%. m/z 259,03, 261,04 (М+Н).
Подготовительный пример 16
3-(4-бромпропоксифенил)пропанол
(a) 4-(3-гидроксипропил)фенол
В 200 мл метанола, охлажденного на бане со льдом, растворяли 45,0 г (0,25 моль) метил-пара-гидроксифенилпропионата. Отдельными порциями добавляли 14,2 г (0,375 моль) боргидрида натрия, и завершали добавление приблизительно в течение 1 часа. Реакцию продолжали при перемешивании при комнатной температуре в течение 2 часов. Методом тонкослойной хроматографии обнаруживали, что исходные вещества полностью прореагировали. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в воде, которую корректировали до рН 2 добавлением 2 М соляной кислоты. Раствор 3 раза экстрагировали дихлорметаном (200 мл × 3). Экстракты объединяли и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и петролейным эфиром (1/5). Получали 32,0 г целевого соединения с выходом 84,2%.
(b) 3-(4-бромпропоксифенил)пропанол
В 300 мл этанола растворяли 30 г (0,197 моль) продукта (а). Добавляли 22,4 (0,26 моль) бикарбоната натрия. Одновременно добавляли 161,6 г (0,80 моль) 1,3-дибромпропана. Реакционную смесь нагревали и перемешивали. Реакцию проводили при 50°С в течение 5 часов. Растворитель и избыток 1,3-дибромпропана удаляли в условиях пониженного давления. Остаток растворяли в 250 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 25 г целевого соединения с выходом 46,5%. m/z 273,03, 275,04 (М+Н).
Подготовительный пример 17
4-бромпропоксибензиловый спирт
Указанное выше соединение получали в соответствии со способом Подготовительного примера 16 (b), выход составил 75,1%, m/z 245,01, 247,0 (М+Н).
Подготовительный пример 18
1-(4-бромпропоксифенил)ацетон
В 300 мл этанола растворяли 45 г (0,30 моль) 1-(4-гидроксифенил)ацетона. Добавляли 62,1 г (0,45 моль) карбоната калия. Одновременно добавляли 161,6 г (0,80 моль) 1,3-дибромпропана. Реакционную смесь перемешивали. Реакцию проводили при комнатной температуре в течение 5 часов, и удаляли растворитель и избыток 1,3-дибромпропана в условиях пониженного давления. Остаток растворяли в 250 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 44,8 г целевого соединения с выходом 52,5%. m/z 285,01, 287,02 (М+Н).
Подготовительный пример 19
1-(4-бромэтоксифенил)ацетон
Указанное выше соединение получали в соответствии со способом Подготовительного примера 18. Выход составил 62,3%, m/z 271,03, 273,02 (М+Н).
Подготовительный пример 20
4-бромпропокси-3,5-дихлорбензиловый спирт
(a) Метил-4-гидрокси-3,5-дихлорбензоат
В 300 мл метанола растворяли 62,1 г (0,30 моль) 4-гидрокси-3,5-дихлорбензойной кислоты. Добавляли 3 г пара-толуолсульфоновой кислоты. Реакционную смесь нагревали с обратным холодильником в течение 6 часов. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в 500 мл дихлорметана, 3 раза промывали водой (200 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель с получением 56,3 г целевого продукта с выходом 84,9%. m/z 219,01 (М-Н).
(b) 4-гидрокси-3,5-дихлорбензиловый спирт
В 300 мл метанола, охлажденного на бане со льдом, растворяли 56 г (0,253 моль) продукта (а). Отдельными порциями добавляли 15,13 г (0,40 моль) боргидрида натрия, и завершали добавление приблизительно в течение 1,5 часов. Реакцию продолжали при перемешивании в течение 2 часов; методом тонкослойной хроматографии обнаруживали, что исходные вещества полностью прореагировали. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в воде, которую корректировали до рН 2 добавлением 2 М соляной кислоты. Раствор 3 раза экстрагировали дихлорметаном (200 мл × 3). Экстракты объединяли, и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления, и кристаллизовали остаток с дихлорметаном и петролейным эфиром (1/5). Получали 30,3 г целевого соединения с выходом 62,0%. m/z 192,02 (М-Н).
(с) 4-бромпропокси-3,5-дихлорбензиловый спирт
Указанное выше соединение получали в соответствии со способом Подготовительного примера 16 (b), выход составил 68,5%, m/z 311,03 (М-Н).
Подготовительный пример 21
4-(3-бромпропокси)ацетофенон
В 400 мл метанола растворяли 40,8 г (0,30 моль) 4-гидроксиацетофенона. Добавляли 62,1 г (0,45 моль) карбоната калия. Одновременно добавляли 161,6 г (0,80 моль) 1,3-дибромпропана. Реакционную смесь перемешивали. Реакцию проводили при комнатной температуре в течение 10 часов. Растворитель и избыток 1,3-дибромпропана удаляли в условиях пониженного давления. Остаток растворяли в 250 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 44,8 г целевого соединения с выходом 58,1%. m/z 257,01, 259,01 (М+Н).
Подготовительный пример 22
2-(4-бромпропоксифенил)-2-пропанол
(a) 2-(4-бензилоксифенил)-2-пропанол
В 300 мл тетрагидрофурана растворяли 62,3 г (0,25 моль) 4-бензилоксибромбензола. Под защитной атмосферой аргона добавляли 6,3 г (0,263 моль) металлического магния. Добавляли каталитическое количество йода. Реакционную смесь нагревали с обратным холодильником в течение 30 минут. По каплям добавляли 58 г (1 моль) ацетона. После добавления, продолжали нагревание реакционной смеси с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры. При охлаждении на бане со льдом, по каплям добавляли 132 мл 2 М соляной кислоты, и удаляли тетрагидрофуран и избыток ацетона в условиях пониженного давления. Оставшуюся водную фазу 3 раза экстрагировали этиловым эфиром (200 мл × 3) и сушили над безводным сульфатом магния в течение ночи. Осушитель удаляли путем фильтрования, и концентрировали фильтрат досуха с получением 38,8 г целевого продукта с выходом 68,0%. m/z 228,11.
(b) 2-(4-гидроксифенил)-2-пропанол
В 150 мл метанола растворяли 35 г (0,153 моль) продукта (а), добавляли 2,4 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 4 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, и концентрировали фильтрат досуха в условиях пониженного давления с получением 18,8 г целевого продукта с выходом 80,7%. m/z 152,08.
(с) 2-(4-бромпропоксифенил)-2-пропанол
В 200 мл метанола растворяли 15,8 г (0,104 моль) продукта (b). Добавляли 31,1 г (0,225 моль) карбоната калия. Одновременно добавляли 80,8 г (0,40 моль) 1,3-дибромпропана. Реакционную смесь перемешивали. Реакцию проводили при комнатной температуре в течение 20 часов, и удаляли растворитель и избыток 1,3-дибромпропана в условиях пониженного давления. Остаток растворяли в 250 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 24,8 г целевого соединения с выходом 87,3%. m/z 272,04, 274,0.
Подготовительный пример 23
4-(3-бромпропокси)пропионилбензол
Указанное выше соединение получали в соответствии со способом Подготовительного примера 22 (с), выход составил 65,8%, m/z 270,02, 272,02.
Подготовительный пример 24
1-[4-(2-бромэтокси)фенил]-2-метил-2-пропанол
(a) 1-[(4-бензилокси)фенил]-2-метил-2-пропанол
В 600 мл тетрагидрофурана растворяли 131,50 г (0,50 моль) 4-бензилоксибензилбромид. Под защитной атмосферой аргона добавляли 12,6 г (0,526 моль) металлического магния. Добавляли каталитическое количество йода. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. По каплям добавляли 116 г (2,0 моль) ацетона. После добавления, реакцию продолжали при перемешивании при комнатной температуре в течение 4 часов. Реакционную смесь охлаждали на бане со льдом, по каплям добавляли 266 мл 2 М соляной кислоты, и удаляли тетрагидрофуран и избыток ацетона в условиях пониженного давления. Оставшуюся водную фазу экстрагировали дихлорметаном 3 раза (400 мл × 3) и сушили над безводным сульфатом магния в течение ночи. Осушитель удаляли путем фильтрования, и концентрировали фильтрат досуха с получением 91,2 г целевого продукта с выходом 75. 28%. m/z 242,31.
(b) 1-[(4-гидрокси)фенил]-2-метил-2-пропанол
В 400 мл метанола растворяли 88,0 г (0,363 моль) продукта (а), добавляли 4,4 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 5 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, и концентрировали фильтрат досуха в условиях пониженного давления. Остаток кристаллизовали с дихлорметаном/петролейным эфиром (1:1) с получением 53,4 г целевого продукта с выходом 88,6%. m/z 166,1.
(с) 1-[4-(2-бромэтокси)фенил]-2-метил-2-пропанол
В 200 мл метанола растворяли 20,8 г (0,125 моль) продукта (b). Добавляли 31,1 г (0,225 моль) карбоната калия. Одновременно добавляли 75,2 г (0,40 моль) 1,2-дибромэтана. Реакционную смесь перемешивали, нагревали до 50°С для проведения реакции в течение 20 часов и фильтровали. Растворитель и избыток 1,2-дибромэтана удаляли в условиях пониженного давления. Остаток растворяли в 250 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 21,9 г целевого соединения с выходом 80,3%. m/z 272,04, 274,0.
Подготовительный пример 25
1-[4-(2-бромпропокси)фенил]-2-метил-2-пропанол
Указанное выше соединение получали в соответствии со способом Подготовительного примера 24 (b), выход составил 78,9%, m/z 286,05, 288,0.
Подготовительный пример 26
3-метил-4-(3-бромпропокси)ацетофенон
В 300 мл метанола растворяли 30,38 г (0,20 моль) 4-гидрокси-3-метилацетофенона. Добавляли 31,1 г (0,225 моль) карбоната калия. Одновременно добавляли 121,2 г (0,60 моль) 1,3-дибромпропана. Реакционную смесь перемешивали, и проводили реакцию при комнатной температуре в течение 20 часов. Растворитель и избыток 1,3-дибромпропана удаляли в условиях пониженного давления. Остаток растворяли в 250 мл дихлорметана, 3 раза промывали водой (100 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 28,3 г целевого соединения с выходом 52,2%. m/z 270,02, 272,0.
Подготовительный пример 27
3-метокси-4-(3-бромпропокси)ацетофенон
Указанное выше соединение получали в соответствии со способом Подготовительного примера 26, выход составил 65,8%, m/z 286,02, 288,0.
Подготовительный пример 28
(R)-(-)-3-[(R)-2-бензилоксикарбоксил-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октан
В 300 мл дихлорметана, охлажденного на бане со льдом, растворяли 31,45 г (0,10 моль) продукта (е) Подготовительного примера 2. При перемешивании в течение 5 минут добавляли 20,0 г триэтиламина. Контролировали, чтобы реакционная температура была ниже 10°С. По каплям добавляли 20,40 г (0,12 моль) бензилоксиформилхлорида. Реакцию проводили при этой температуре в течение 4 часов. Реакционную смесь фильтровали. Фильтрат разбавляли 200 мл дихлорметана, 3 раза промывали насыщенным раствором бикарбоната натрия (200 мл × 3) и сушили над безводным сульфатом магния. Часть (приблизительно 350 мл) растворителя удаляли в условиях пониженного давления. Путем добавления 200 мл петролейного эфира фильтрат охлаждали и кристаллизовали с получением 40,50 г целевого соединения с выходом 90,1%. MS 449,26.
Пример 57
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-{[(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этил)фенокси]этил}-1-азабицикло[2,2,2]октилония бензоат
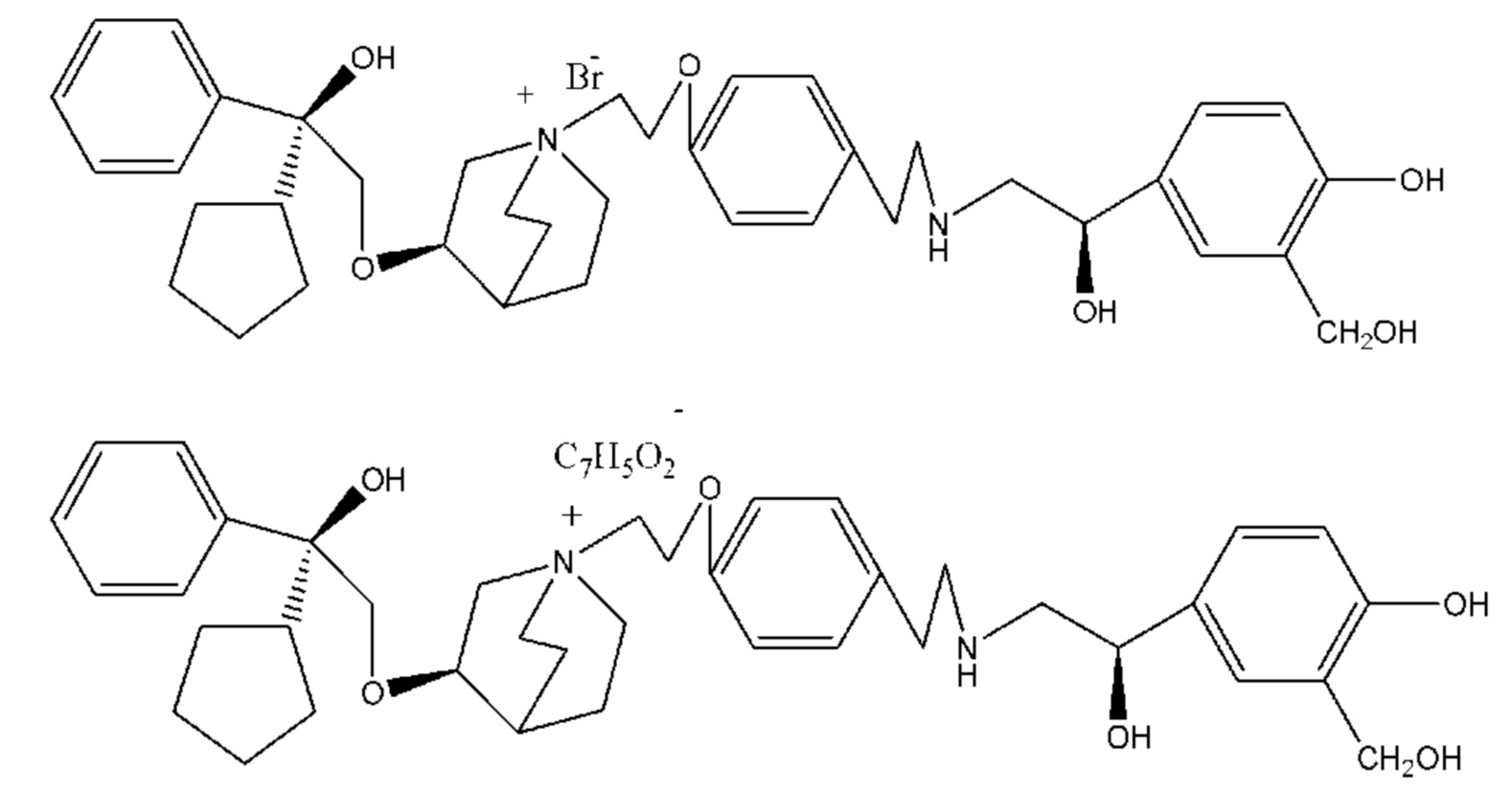
(а) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-[2-(4-гидроксиэтилфенокси)этил]-1-азабицикло[2,2,2]октилония бромид
В трехгорлую колбу емкостью 250 мл добавляли 30 мл абсолютного этанола и (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2]октана (Подготовительный пример 28), перемешивали и растворяли. Затем, добавляли 69,9 г (285,3 ммоль) (4-бромэтокси)фенилэтилового спирта (Подготовительный пример 14). Реакционную смесь перемешивали, и проводили реакцию при комнатной температуре в течение 12 часов. Этанол удаляли в условиях пониженного давления, избыток (4-бромэтокси)фенилэтилового спирта в остатке удаляли методом колоночной хроматографии, и кристаллизовали неочищенный продукт с дихлорметаном и н-гексаном (2/1) с получением 20,61 г целевого продукта с выходом 75,4%. MS (m/z) 614,23.
(b) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-[2-(4-метилсульфонилоксиэтилфенокси)этил]-1-азабицикло[2,2,2]октилония бромид
В 150 мл дихлорметана растворяли 19,3 г (31,4 ммоль) продукта (а). Добавляли 5,0 г (50,0 ммоль) N-метилморфолина. Реакционную смесь охлаждали на бане со льдом. По каплям добавляли 5,50 г (48,0 ммоль) метилсульфонилхлорид. Реакцию продолжали при перемешивании при этой температуре в течение 4 часов. Растворитель удаляли в условиях пониженного давления, и получали 18,8 г целевого продукта методом колоночной хроматографии с выходом 87,8%, MS (m/z) 682,21.
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-{[(R)-[2-(3-бензилоксиметил-4-бензилокси)фенил-2-гидрокси]этиламино}этил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,396 г (5,283 ммоль) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанола (Подготовительный пример 1) и 3,57 г (5,283 ммоль) продукта (b). Добавляли 20 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, Добавляли 1,475 г (10,566 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 80°С, и проводили реакцию при этой температуре в течение 5 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления. К остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,85 г целевого продукта. Выход составил 51,87%. Рассчитано для MS (m/z) C61H61BrN2O8: 1040,0, обнаружено: 959,4 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-{[(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,60 г (2,50 ммоль) продукта (с), добавляли 18 мл метанола для растворения, добавляли 0,5 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 24 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,2 г целевого соединения в виде твердого вещества с выходом 66,0%. 1H-ЯМР (δ м.д.) (DMSO-d6) 7,19-7,21 (м, 5Н), 7,01 (д, 2Н), 6,59-6,95 (м, 5Н), 5,0 (с, 1H), 4,79 (с, 2Н), 4,74 (м, 1H), 4,04 (т, 2Н), 3,93 (д, 1H), 3,68 (д, 1H), 3,15 (м, 1H), 2,67-2,90 (м, 8Н), 1,96-2,0 (м, 5Н), 1,79-1,81 (м, 3Н), 1,74 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,45 (м, 6Н); Рассчитано для MS (m/z) C39H53BrN2O6: 725,75, обнаружено: 645,32 (M-Br).
(е) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этил)фенокси]этил}-1-азабицикло[2,2,2]октилония бензоат
В 50 мл воды растворяли 0,545 г (0,75 ммоль) продукта (d), адсорбировали и элюировали с сильнощелочной анионообменной колонки. Элюент концентрировали до 50 мл в условиях пониженного давления. К реакционной смеси добавляли раствор 100 мг бензойной кислоты в 2 мл этанола, перемешивали в течение 5 минут, и концентрировали досуха в условиях пониженного давления. Остаток кристаллизовали с изопропанолом. Получали 0,42 г целевого продукта с выходом 72,9%. Элементный анализ (%) (расчетное значение) С 71,98 (72,04), Н 7,52 (7,62), N 3,61 (3,65).
Пример 58
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(1-{(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}метил)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
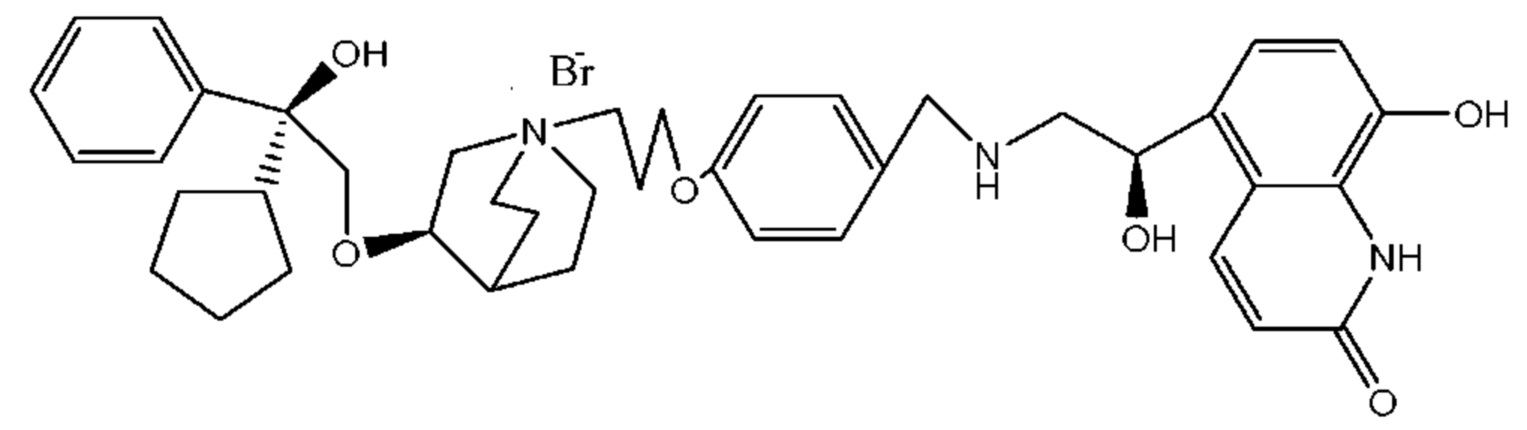
(a) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-[3-(4-гидроксиметилфенокси)пропил]-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-[3-(4-метилсульфонилоксиметилфенокси)пропил]-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (b).
(c) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(1-{(R)-[2-гидрокси-2-(8-бензилокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}метил)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,0 г (5,0 ммоль) 8-бензилокси-5-[(R)-2-бензиламино-1-гидроксиэтил]-(1Н)-хинолин-2-она (Подготовительный пример 2) и 3,20 г (5,0 ммоль) продукта (b). Добавляли 20 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,475 г (10,566 ммоль) безводного карбоната калия. Температуру реакционной смеси повышали до 80°С. Реакцию проводили при этой температуре в течение 5 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,6 г целевого продукта. Выход составил 52,6%. Рассчитано для MS (m/z) C56H64BrN3O8: 986,89, обнаружено: 906,35 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(1-{(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}метил)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,40 г (2,43 ммоль) продукта (с), Добавляли 20 мл метанола для растворения, добавляли 0,5 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 15 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,08 г целевого соединения в виде твердого вещества с выходом 58,2%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,01 (с, 1H), 7,35 (д, 1H), 7,19-7,21 (м, 5Н), 6,95 (д, 2Н), 6,71 (д,1Н), 6,57-6,65 (м, 3Н), 6,52 (д, 1H), 5,01 (с, 1H), 4,74 (м, 1H), 3,94 (м, 3Н), 3,81 (т, 2Н), 3,68 (д, 1H), 3,15 (м, 1H), 2,90 (м, 1H), 2,84 (м, 1H), 2,36 (м, 2Н), 2,10 (с, 1H), 2,0 (м, 4Н), 1,79-1,82 (м, 5Н), 1,75 (м, 1H), 1,68 (м, 2Н), 1,56-1,60 (м, 6Н), 1,35-1,46 (м, 6Н). Рассчитано для MS (m/z) C41H52BrN3O6: 762,772, обнаружено: 682,30 (M-Br).
Пример 59
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(3,5-дихлор-4-(1-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}метил)фенокси)пропил}-1-азабицикло[2,2,2]октилония бромид
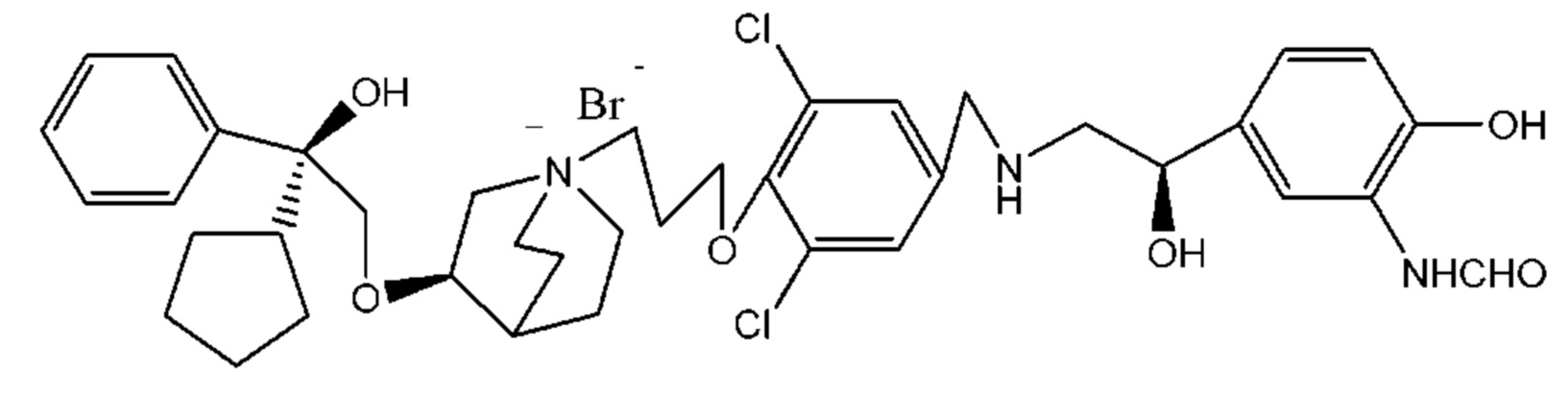
(a) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-{3-[3,5-дихлор-4-(гидроксиметил)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-{3-[3,5-дихлор-4-(метансульфонилоксиметил)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (b).
(c) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-{3-[(3,5-дихлор-4-(1-{(R)-[2-гидрокси-2-(3-формамидо-4-бензилокси)фенил]этиламино}метил)-фенокси)пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 1,882 г (5,0 ммоль) (R)-2-бензиламино-1-[(4-бензилокси-3-формамидо)фенил]этанола (Подготовительный пример 4) и 3,69 г (5,0 ммоль) продукта (b), добавляли 30 мл ацетонитрила. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,5 г (11,0 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 65-70°С. Реакцию проводили при этой температуре в течение 10 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,92 г целевого продукта. Выход составил 56,6%. Рассчитано для MS (m/z) C54H62BrCl2N3O8: 1031,76, обнаружено: 950,27 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(3,5-дихлор-4-(1-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}метил)фенокси)пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,80 г (2,71 ммоль) продукта (с), добавляли 30 мл метанола для растворения, добавляли 0,8 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 15 часов реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,38 г целевого соединения в виде твердого вещества с выходом 63,1%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,20 (с, 1H), 7,40 (м, 1H), 7,18-7,21 (м, 5Н), 6,84 (д, 2Н), 6,64-6,76 (м, 2Н), 5,01 (с, 1H), 4,73 (м, 1H), 4,01 (с, 1H), 3,93 (м, 3Н), 3,81 (д, 2Н), 3,68 (д, 1H), 3,15 (м, 1H), 2,90 (м, 1H), 2,83 (м, 1H), 2,36 (м, 2Н), 2,10 (с, 2Н), 1,98-2,0 (м, 3Н), 1,79-1,81 (м, 5Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 6Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C39H50BrCl2N3O6: 807,64, обнаружено: 726,22 (M-Br).
Пример 60
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{R)[-2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}изобутил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
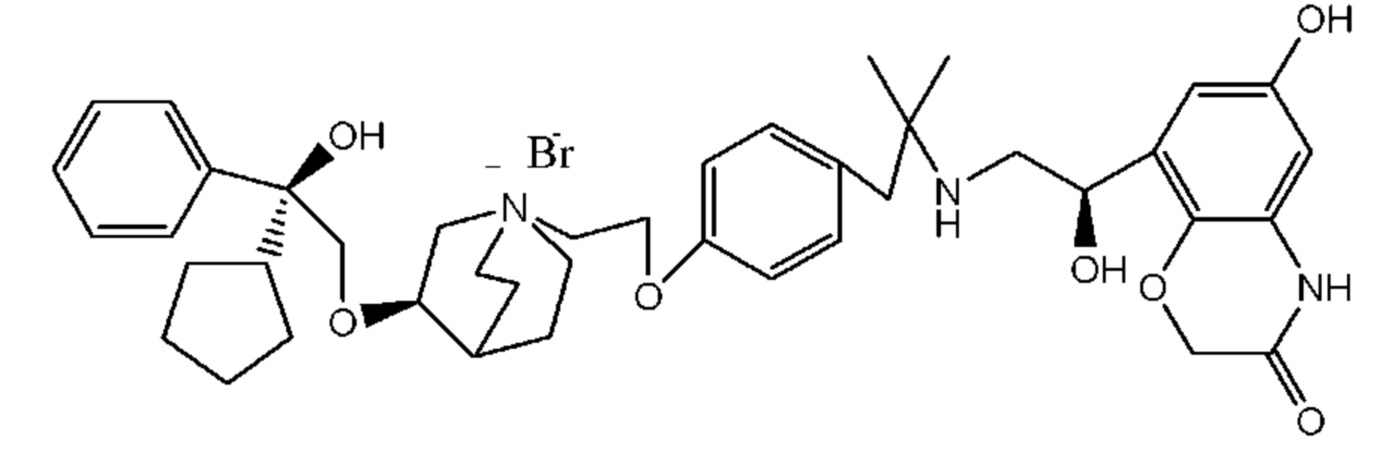
(a) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-гидрокси-2-метил)пропилфенокси]этил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-метилсульфонилокси-2-метил)пропилфенокси]этил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (b).
(c) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(2-{(R)-[2-гидрокси-2-[(6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}изобутил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,02 г (5,0 ммоль) 8-[(1R)-1-гидрокси-2-бензиламин]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он
(Подготовительный пример 5) и 3,57 г (5,0 ммоль) продукта (b). 30 мл диоксана добавляли. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,0 г (7,35 ммоль) безводного карбоната калия. Температуру реакционной смеси повышали до 55-60°С. Реакцию проводили при этой температуре в течение 10 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 10 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,98 г целевого продукта. Выход составил 58,5%.
Рассчитано для MS (m/z) C57H68BrN3O9: 1018,93, обнаружено: 938,38 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенилэтокси]-1-{2-[4-(2-{(R)-2-гидрокси-2-[(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}изобутил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,80 г (2,75 ммоль) продукта (с), добавляли 20 мл метанола для растворения, добавляли 0,8 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 12 часов реакции, поглощение водорода останавливалось, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,46 г целевого соединения в виде твердого вещества с выходом 66,8%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,01 (с, 1H), 7,18-7,20 (м, 5Н), 7,01 (д, 2Н), 6,72 (д, 2Н), 6,65 (д, 1H), 6,20 (д, 1H), 5,01 (с, 1H), 4,87 (с, 2Н), 4,74 (м, 1H), 4,04 (т, 2Н), 3,93 (с, 1H), 3,68 (д, 1H), 3,14 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,78 (т, 2Н), 2,59 (с, 2Н), 2,10 (м, 2Н), 2,0 (м, 2Н), 1,96 (м, 1H), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 6Н), 1,15 (с, 6Н). Рассчитано для MS (m/z) C42H56BrN3O7: 794,93, обнаружено: 714,33 (M-Br).
Подготовительный пример 29
(R)-1-[(4-гидрокси-3-гидроксиметил)фенил]-2-аминоэтанол
В реакционном котле для гидрирования в 300 мл метанола растворяли 44 г (0,10 моль) продукта (g) Подготовительного примера 1, добавляли 6,6 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, поглощение водорода останавливалось после проведения реакции гидрирования при комнатной температуре в течение 20 часов, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 15,6 г целевого соединения в виде твердого вещества с выходом 85,1%. MS (m/z) 182,09 (М-Н).
Подготовительный пример 30
8-гидрокси-5-[(R)-2-амино-1-гидроксиэтил]-1Н-хинолин-2-он
Указанное выше соединение получали в соответствии со способом Подготовительного примера 29 с использованием продукта (f) Подготовительного примера 3.
Подготовительный пример 31
(R)-2-амино-1-[(4-гидрокси-3-формамидо)фенил]этанол
Указанное выше соединение получали в соответствии со способом Подготовительного примера 29 с использованием продукта (d) Подготовительного примера 4.
Подготовительный пример 32
8-[(1R)-1-гидрокси-2-амино]этил-6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он
Указанное выше соединение получали в соответствии со способом
Подготовительного примера 29 с использованием продукта (g) Подготовительного примера 5.
Пример 61
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси]-1-{2-[4-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}пропил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
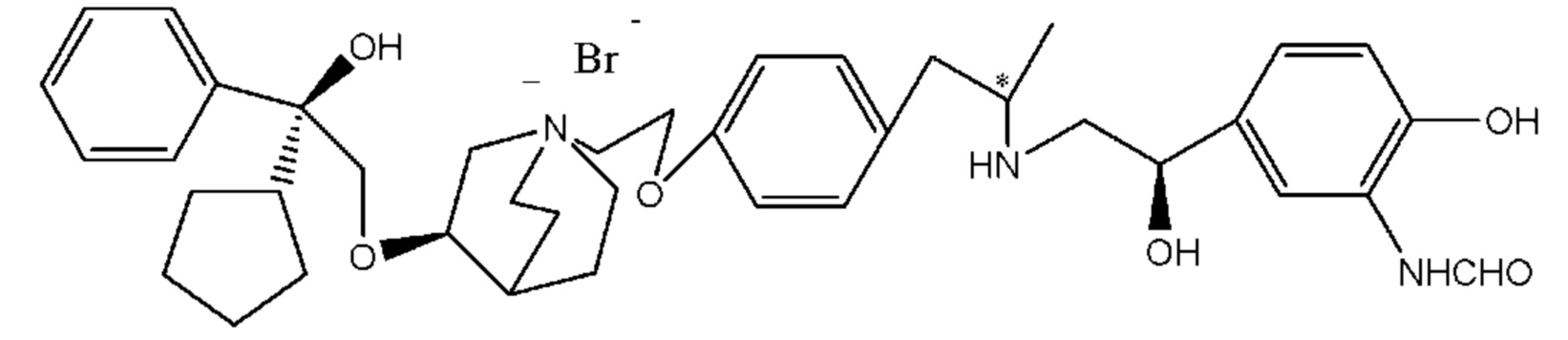
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[2-(4-ацетонилфенокси)этил]-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси]-1-{2-[4-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}пропил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 3 (b).
В реакционную колбу емкостью 50 мл добавляли 0,981 г (5,0 ммоль) (R)-2-амино-1-[(4-гидрокси-3-формамидо)фенил]этанола (Подготовительный пример 31) и 2,86 г (5,0 ммоль) продукта (а), добавляли 50 мл дихлорметана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,27 г (6,0 ммоль) триацетилборгидрида натрия, и добавляли 1 каплю уксусной кислоты для катализа. В реакционной смеси проводили реакцию в течение 24 часов при комнатной температуре, и методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл этанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 1,56 г целевого продукта. Выход составил 73,6%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,20 (с, 1H), 7,40 (м, 1H), 7,18-7,20 (м, 5Н), 7,01 (д, 2Н), 6,72 (д, 2Н), 6,66-6,71 (м, 2Н), 5,0 (с, 1H), 4,74 (м, 1H), 4,04 (т, 2Н), 4,0 (с, 1H), 3,92 (д, 1H), 3,67 (д, 1H), 3,15-3,18 (м, 2Н), 2,91 (м, 1H), 2,78 (т, 2Н), 2,54 (м, 2Н), 2,76-2,51 (дд, 2Н), 2,10 (с, 1H), 1,96-2,0 (м, 4Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 6Н), 1,34-1,46 (м, 6Н), 1,10 (д, 3Н); Рассчитано для MS (m/z) C40H54BrN3O6: 752,78, обнаружено: 671,32 (M-Br).
(с) Получение соединения 61-S и соединения 61-R
В 5 мл метанола растворяли 0,80 г продукта (b), разделяли и очищали на препаративной колонке, проводили градиентное элюирование смесью ацетонитрила и воды, со сбором в указанном порядке компонентов 61-S и 61-R, растворитель удаляли в условиях пониженного давления, и кристаллизовали остаток с этанолом с получением 0,28 г соединения (61-S) и 0,31 г соединения (63-R).
Пример 62
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(1-{(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}пропилиден)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
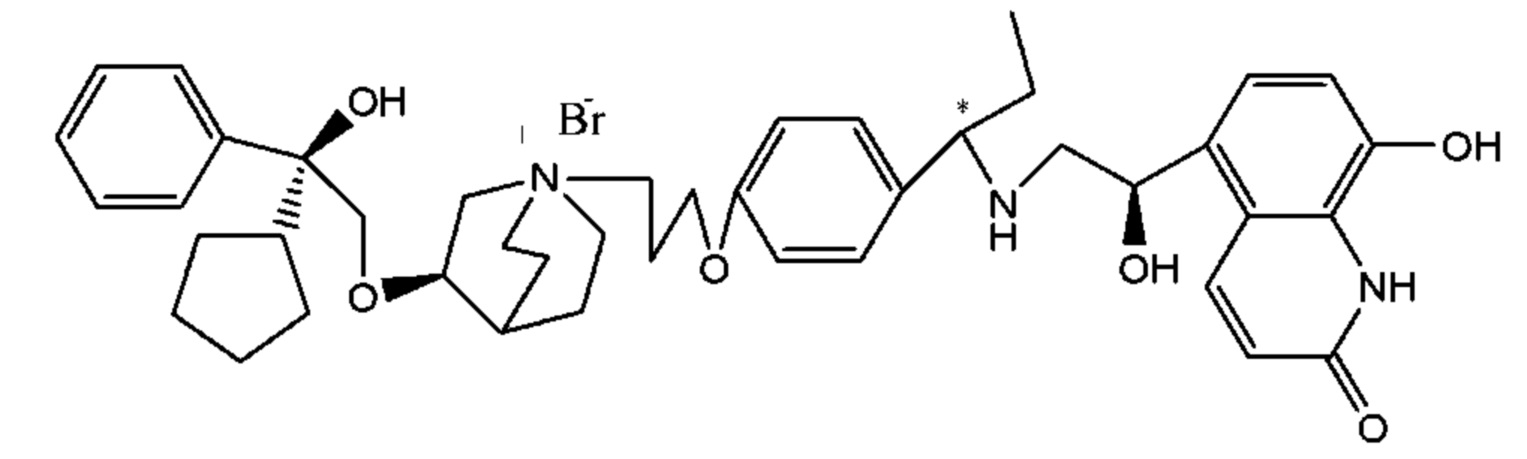
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-[3-(4-пропионилфенокси)пропил]-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(1-{(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}пропилиден)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
В 50 мл дихлорметана суспендировали 1,27 г (6,0 ммоль) триацетилборгидрида натрия, добавляли 1,10 г (5,0 ммоль) 8-гидрокси-5-[(R)-2-амино-1-гидроксиэтил]-1Н-хинолин-2-она (Подготовительный пример 30) и 3,0 г (5,0 ммоль) продукта (а), и добавляли 1 каплю уксусной кислоты для катализа. Реакцию продолжали при перемешивании в течение 20 часов при комнатной температуре. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 8 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,3 г целевого продукта. Выход составил 58,2%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,01 (с, 1H), 7,36 (д, 1H), 7,19-7,21 (м, 5Н), 7,02 (д, 2Н), 6,71 (м, 3Н), 6,56 (д, 1H), 6,52 (д, 1H), 5,01 (с, 1H), 4,74 (м, 1H), 3,94-3,90 (м, 4Н), 3,67 (д, 1H), 3,16 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,36 (м, 2H), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 5Н), 1,75-1,74 (м, 3Н), 1,69 (м, 2Н), 1,55-1,61 (м, 6Н), 1,35-1,46 (м, 6Н), 0,96 (т, 3Н). Рассчитано для MS (m/z) C43H56BrN3O6: 790,82, обнаружено: 719,32 (M-Br).
(с) Получение соединения 62-S и соединения 62-R
В 10 мл метанола растворяли 1,10 г продукта (b), разделяли и очищали на препаративной колонке. Проводили градиентное элюирование смесью ацетонитрила и воды со сбором в указанном порядке элюента с компонентами 62-S и 62-R. Растворитель удаляли в условиях пониженного давления, и кристаллизовали остаток с изопропанолом с получением 0,35 г соединения (62-S) и 0,30 г соединения (62-R), соответственно.
Пример 63
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(4-(3-метил-1-{(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}этилиден)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
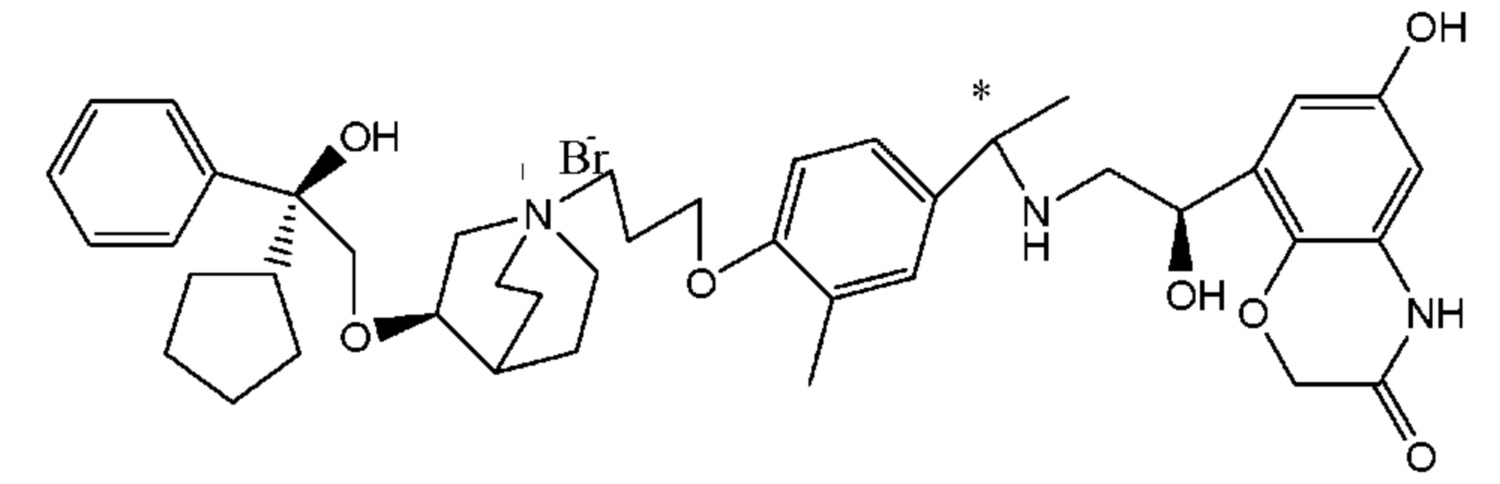
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(4-ацетил-3-метил)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[(4-(3-метил-1-{(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}этилиден)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
В 50 мл дихлорметана суспендировали 1,27 г (6,0 ммоль) триацетилборгидрида натрия, добавляли 1,12 г (5,0 ммоль) 8-гидрокси-5-[(R)-2-амино-1-гидроксиэтил]-(1Н)-хинолин-2-он (Подготовительный пример 32) и 2,93 г (5,0 ммоль) продукта (а), и добавляли 1 каплю уксусной кислоты для катализа. Реакцию продолжали при перемешивании в течение 18 часов при комнатной температуре. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 6 мл этанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 1,86 г целевого продукта. Выход составил 46,8%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,02 (с, 1H), 7,19-7,21 (м, 5Н), 6,82 (м, 2Н), 6,65-6,60 (м, 2Н), 6,20 (д, 1H), 5,0 (с, 1H), 4,88 (с, 2Н), 4,74 (м, 1H), 4,08 (кв, 1H), 3,94 (м, 3Н), 3,68 (д, 1H), 3,15 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,35 (м, 4Н), 2,09 (м, 1H), 2,0 (м, 2Н), 1,96 (м, 1H), 1,79-1,81 (м, 5Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 9Н). Рассчитано для MS (m/z) C42H56BrN3O7: 794,81, обнаружено: 714,30 (M-Br).
(с) Получение соединения 63-S и соединения 63-R
В 3 мл метанола растворяли 0,60 г продукта (b), разделяли и очищали на препаративной колонке. Проводили градиентное элюирование смесью ацетонитрила и воды со сбором компонентов 63-S и 63-R. Растворитель удаляли в условиях пониженного давления, и кристаллизовали остаток с изопропанолом с получением 0,146 г соединения 63-S и 0,187 г соединения 63-R, соответственно.
Следующие соединения могут быть синтезированы с использованием исходных веществ, описанных выше, и сходных способов.
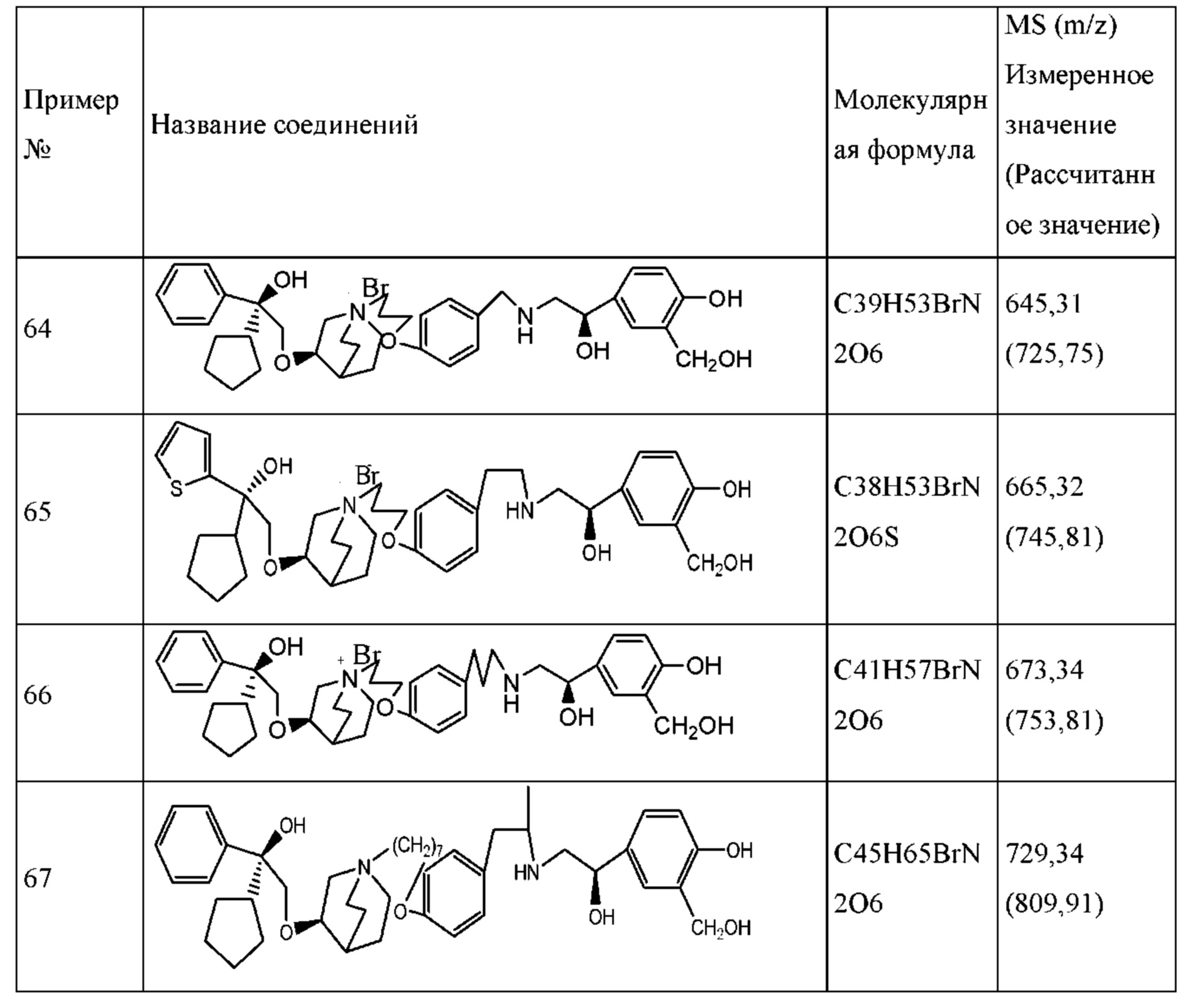
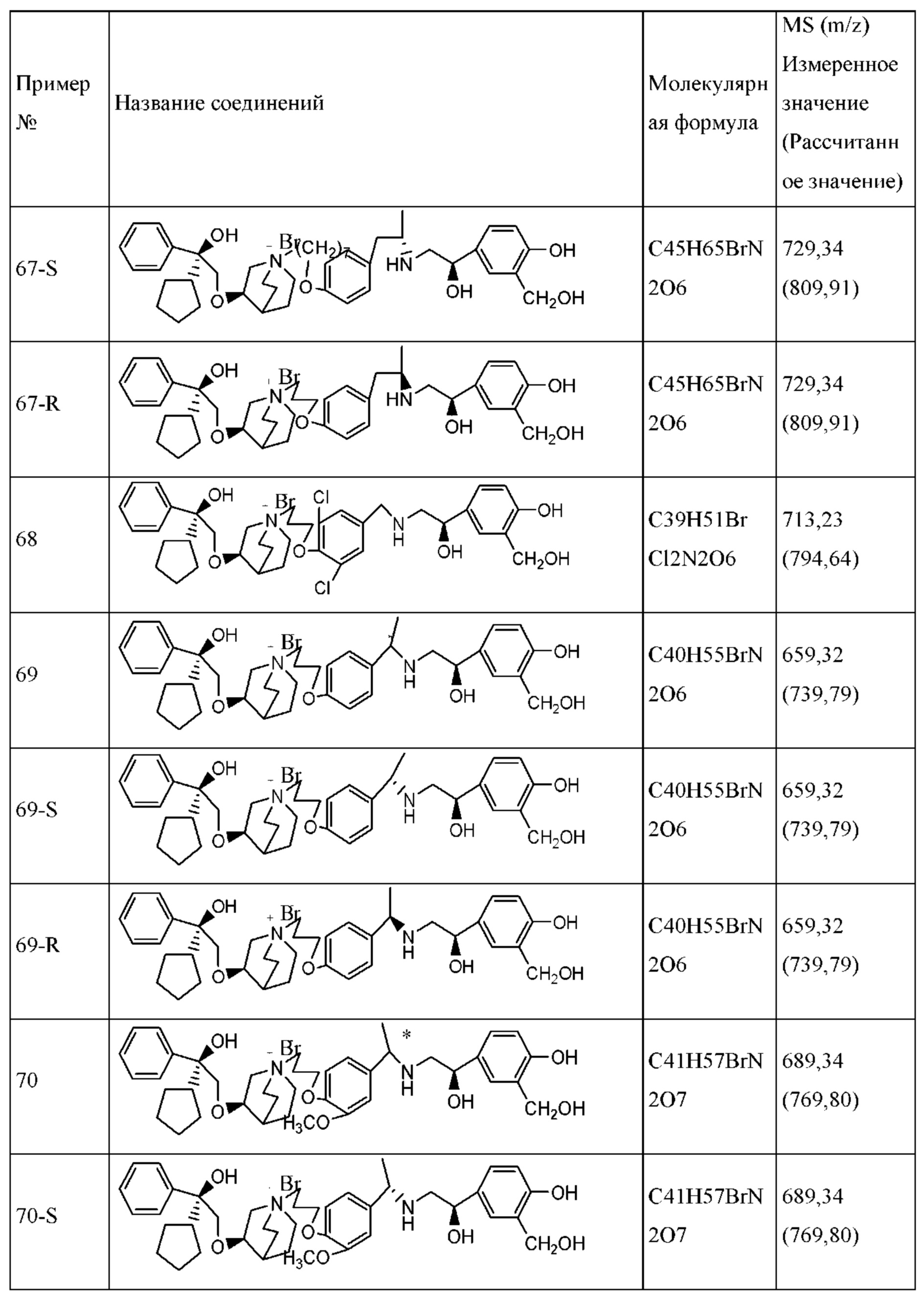
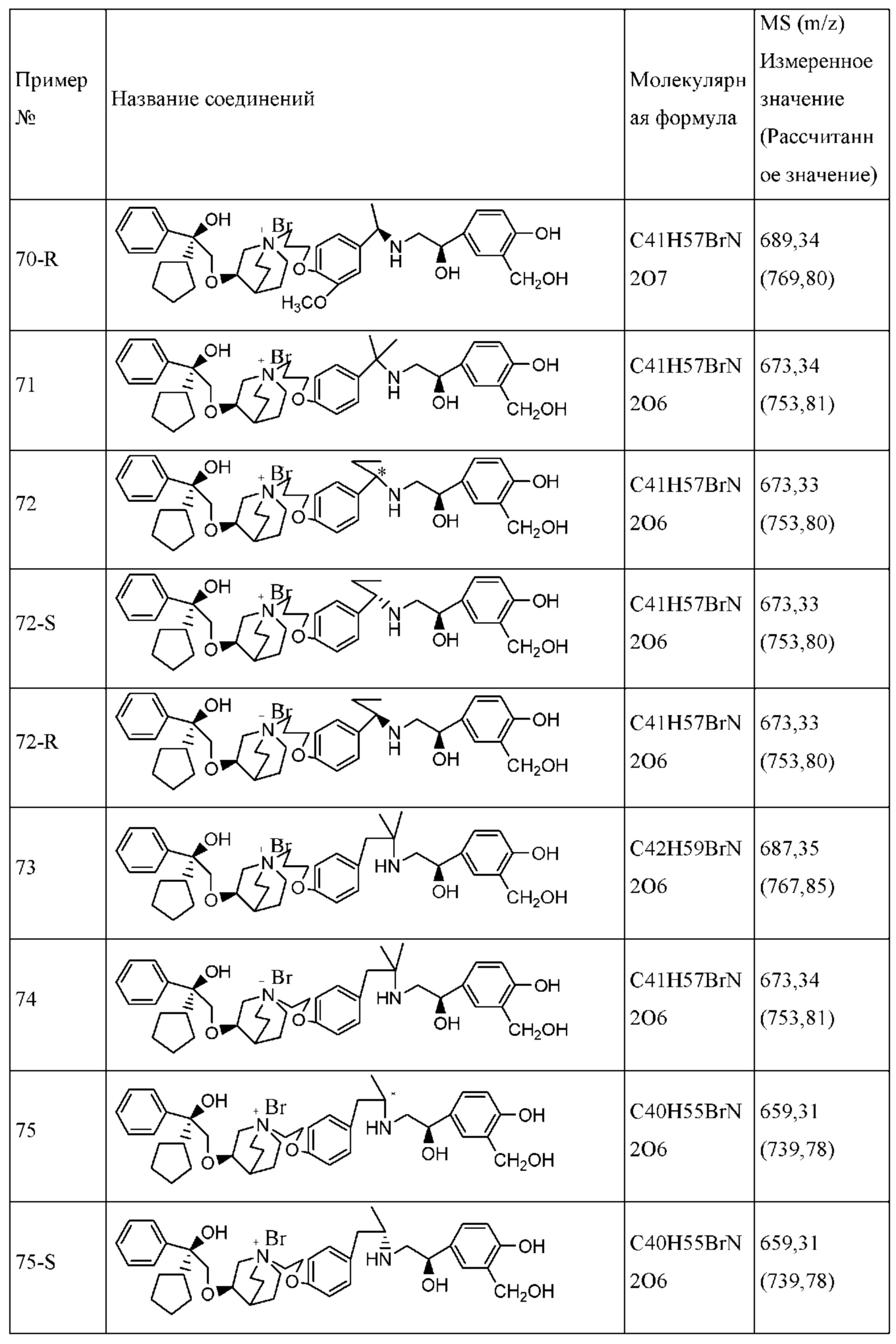
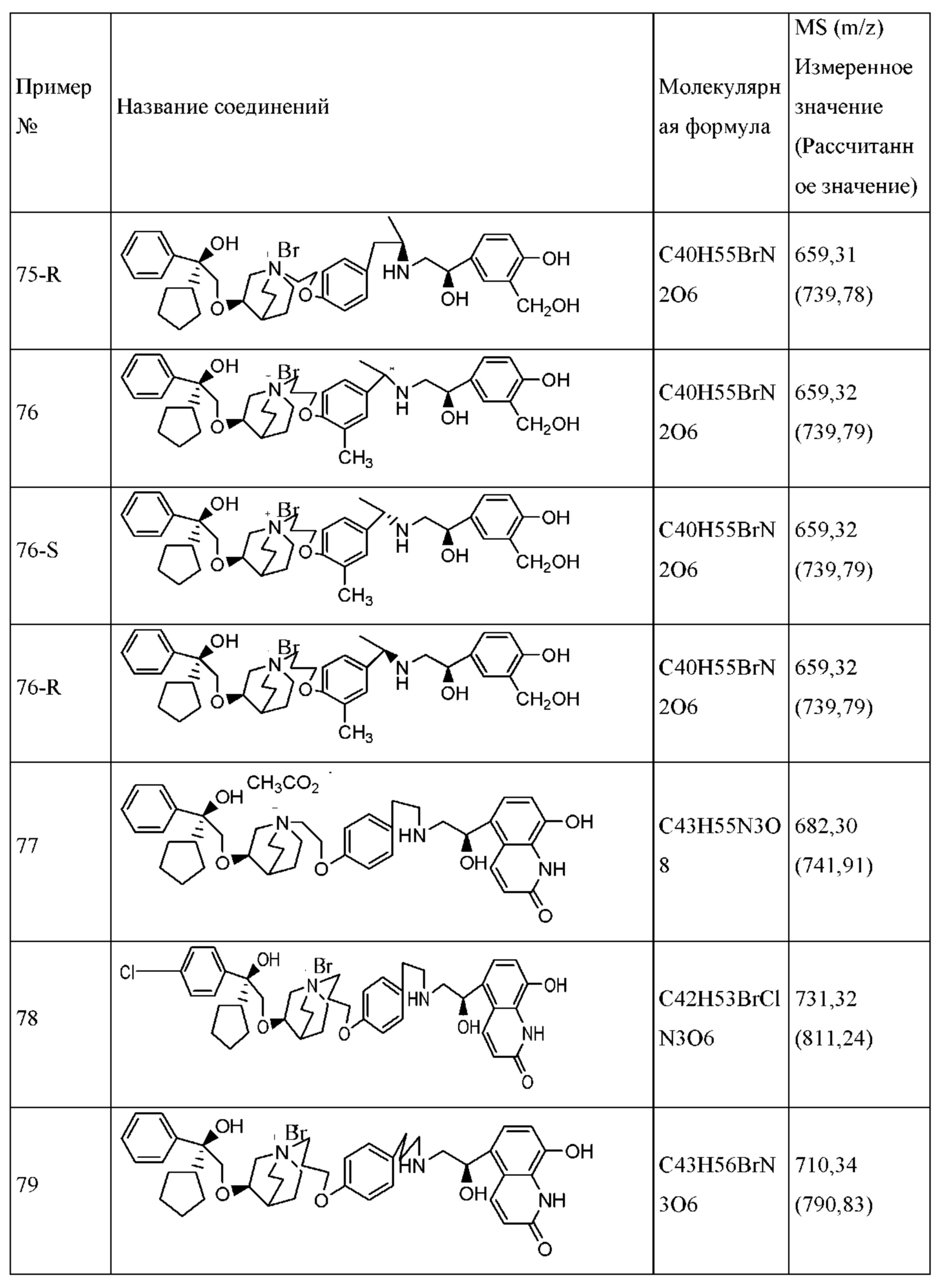
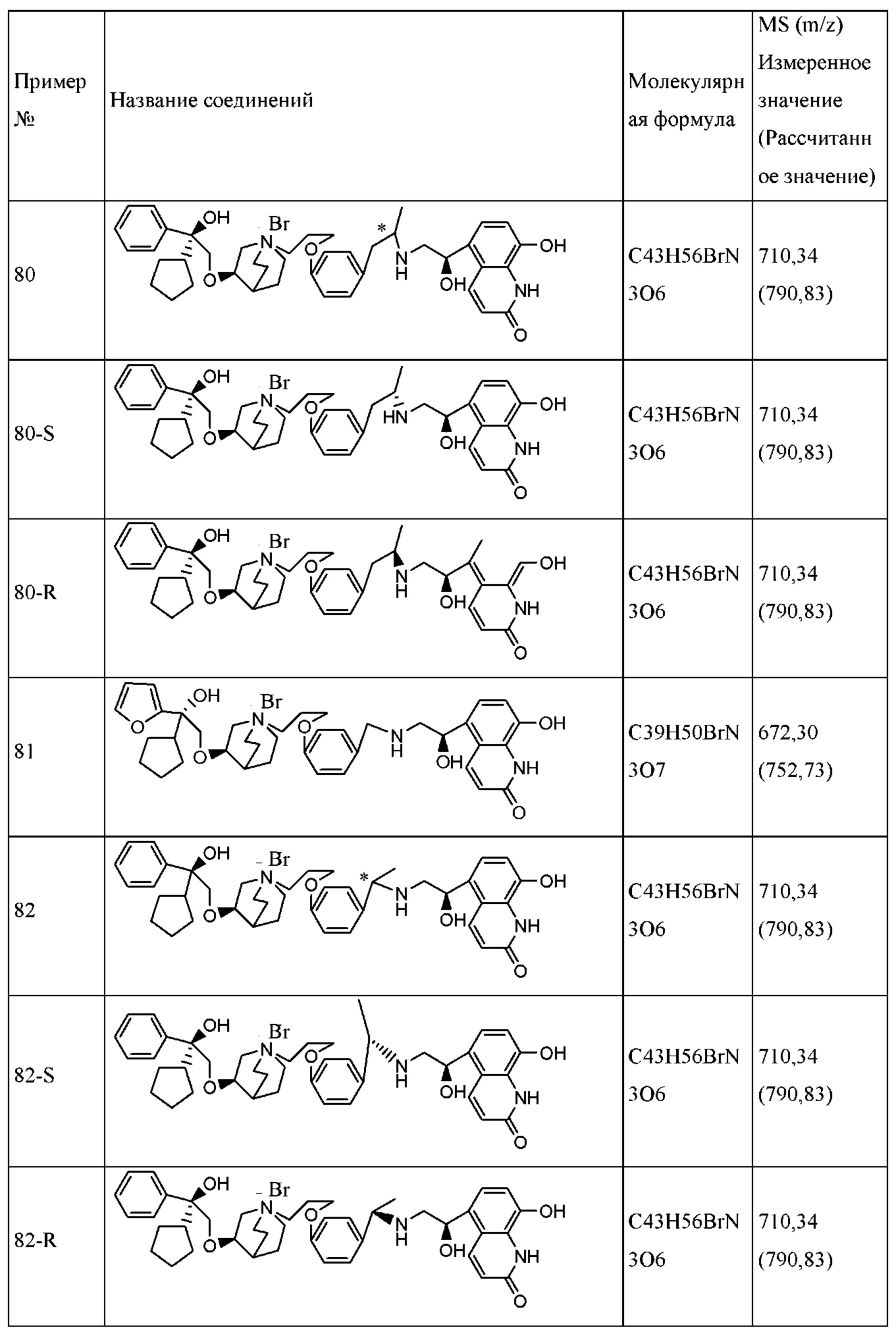
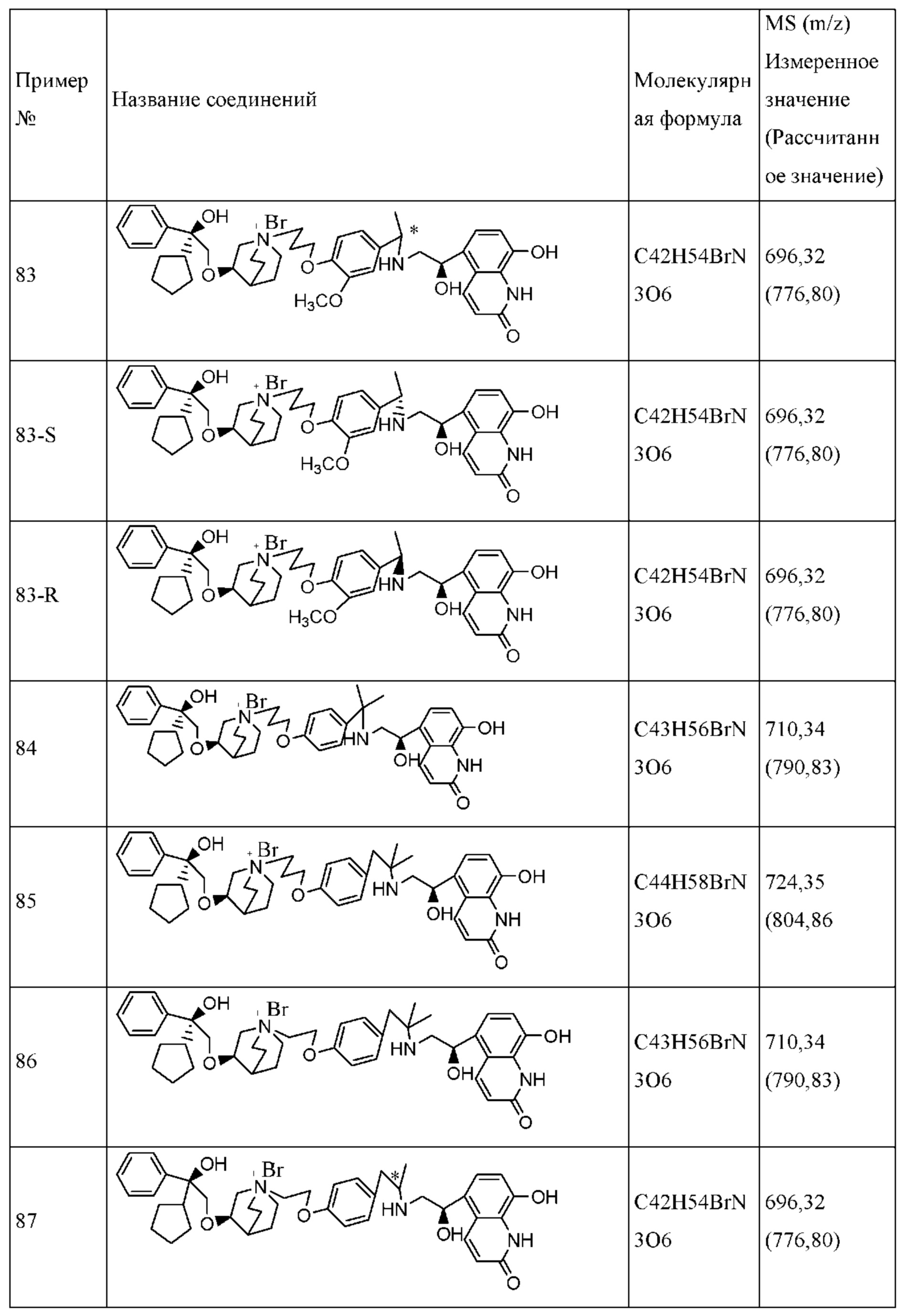
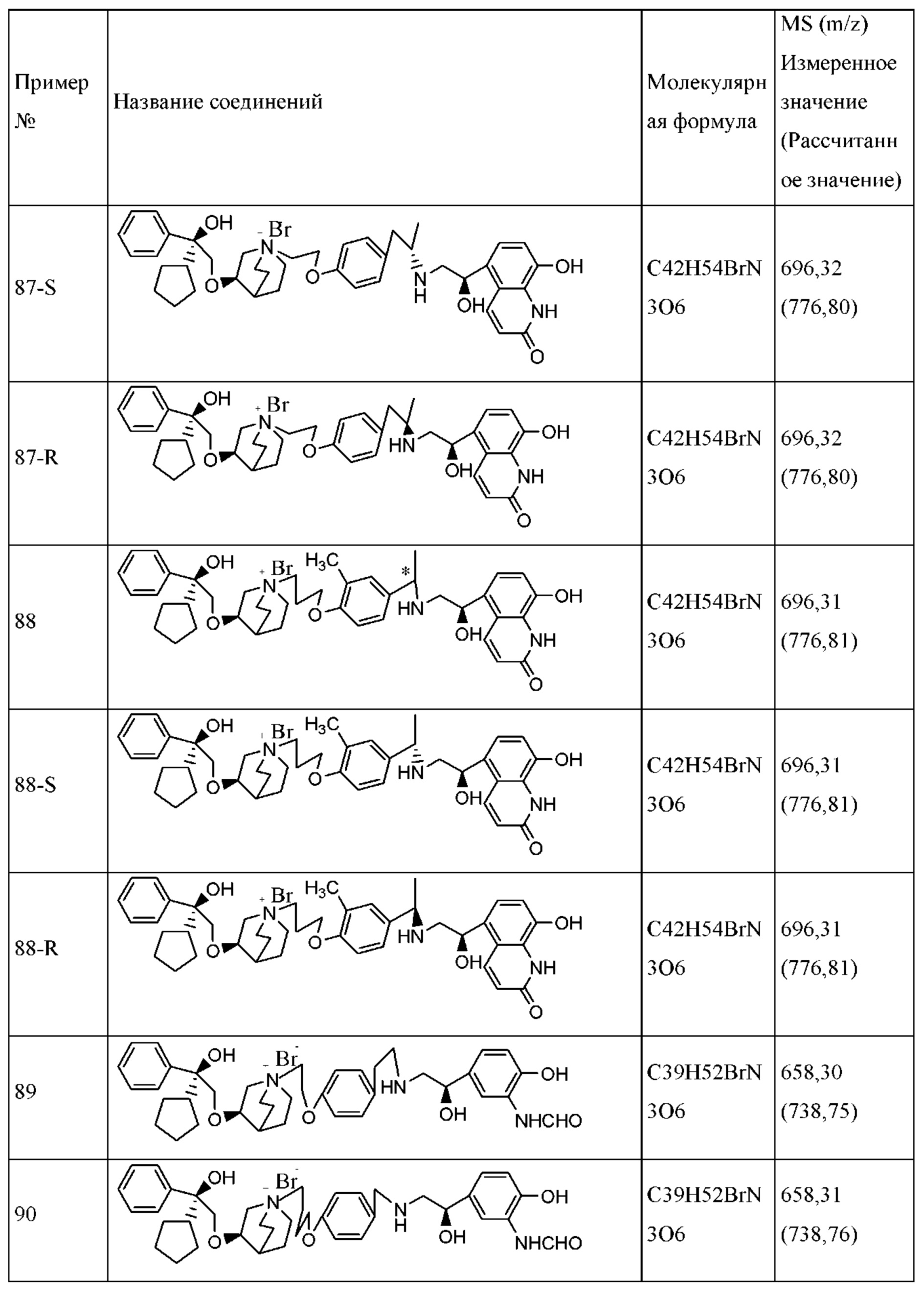
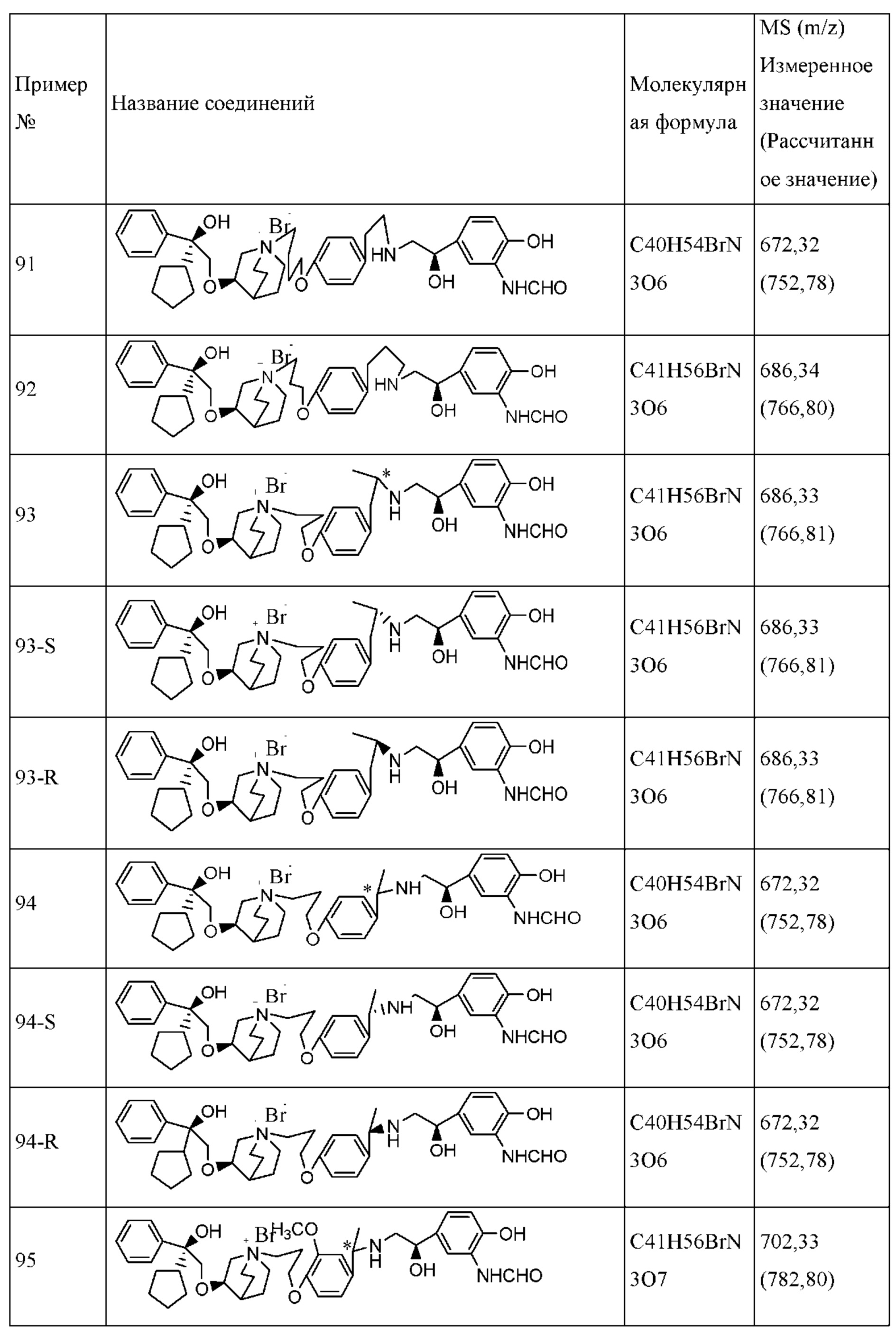
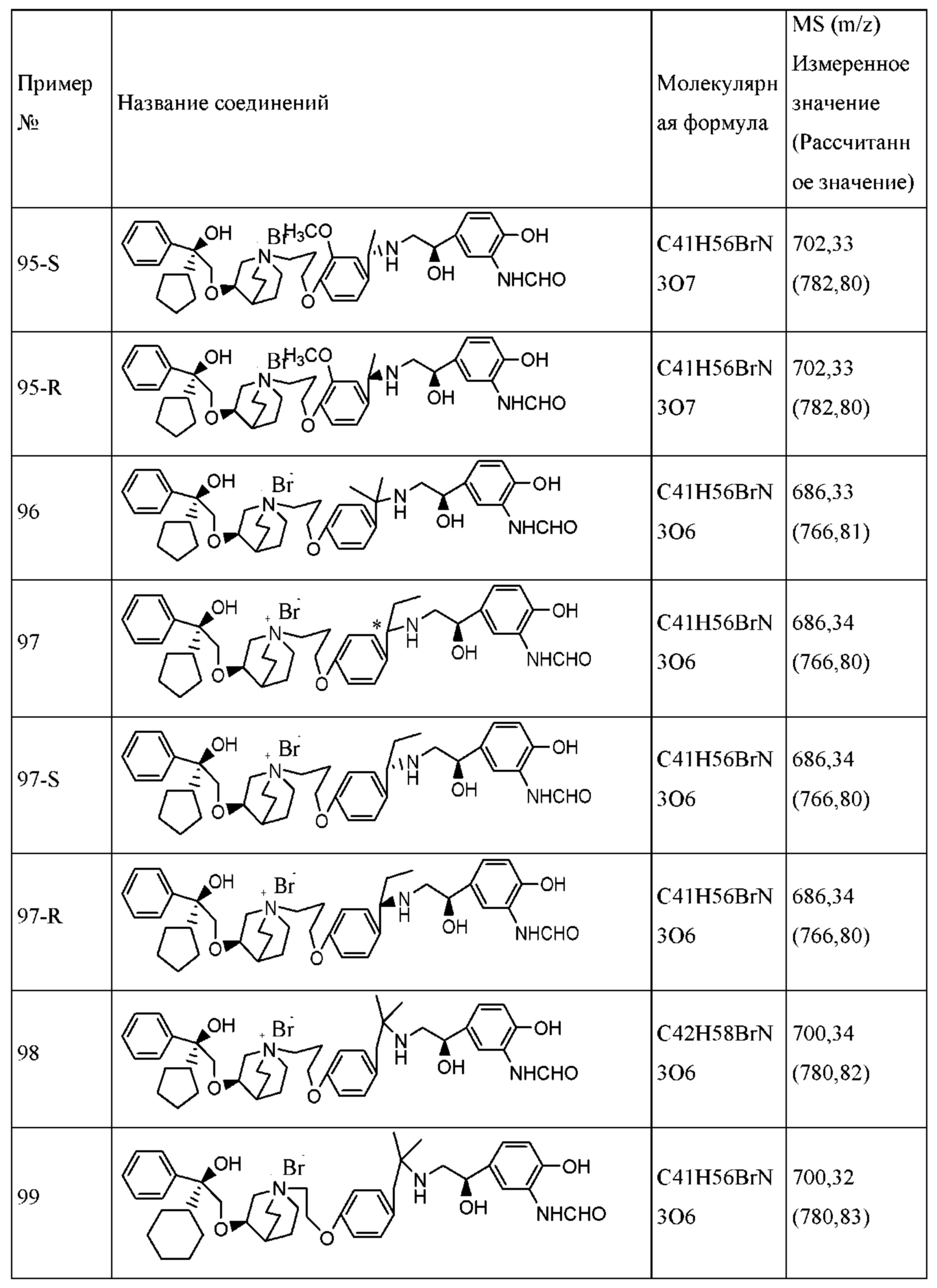
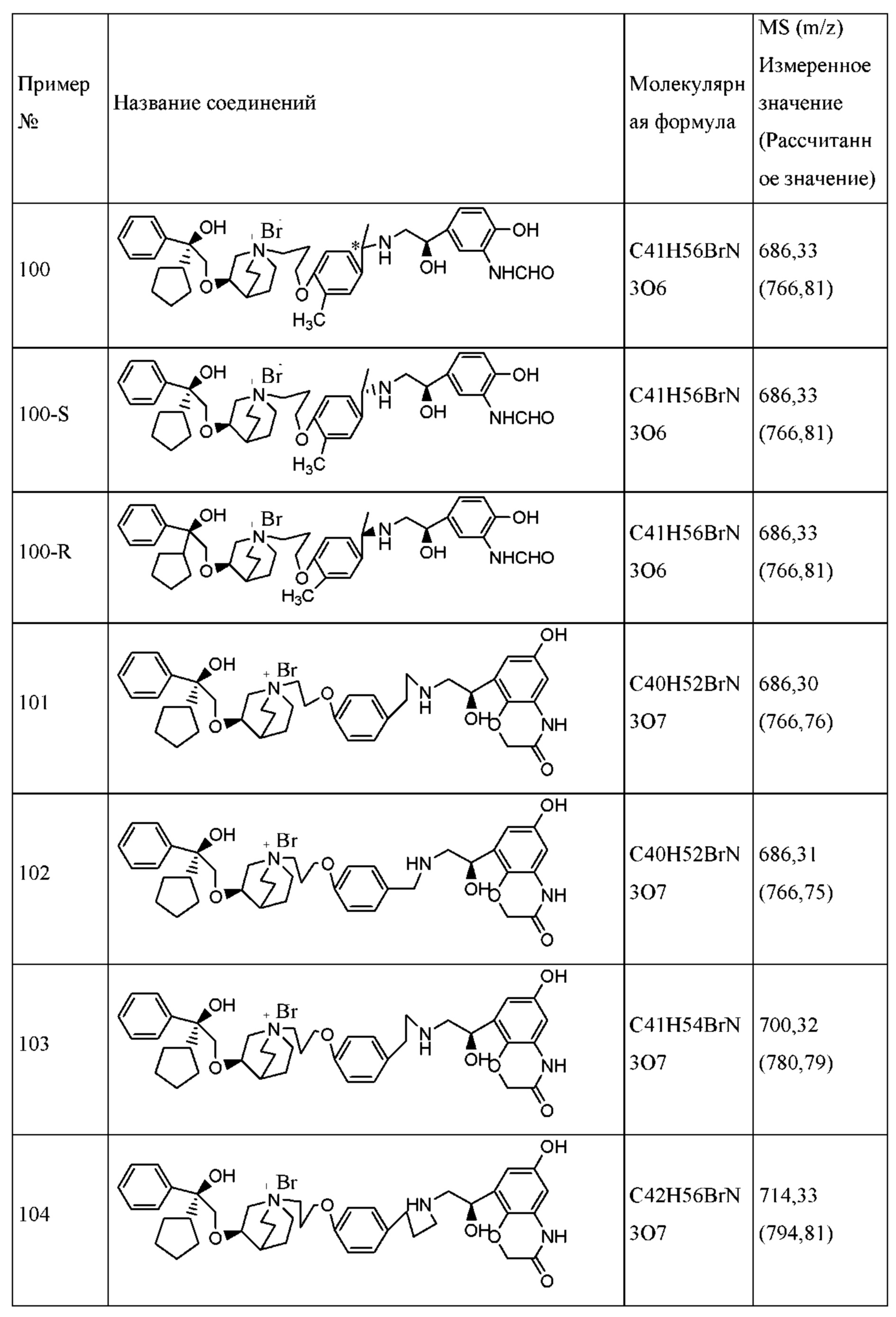
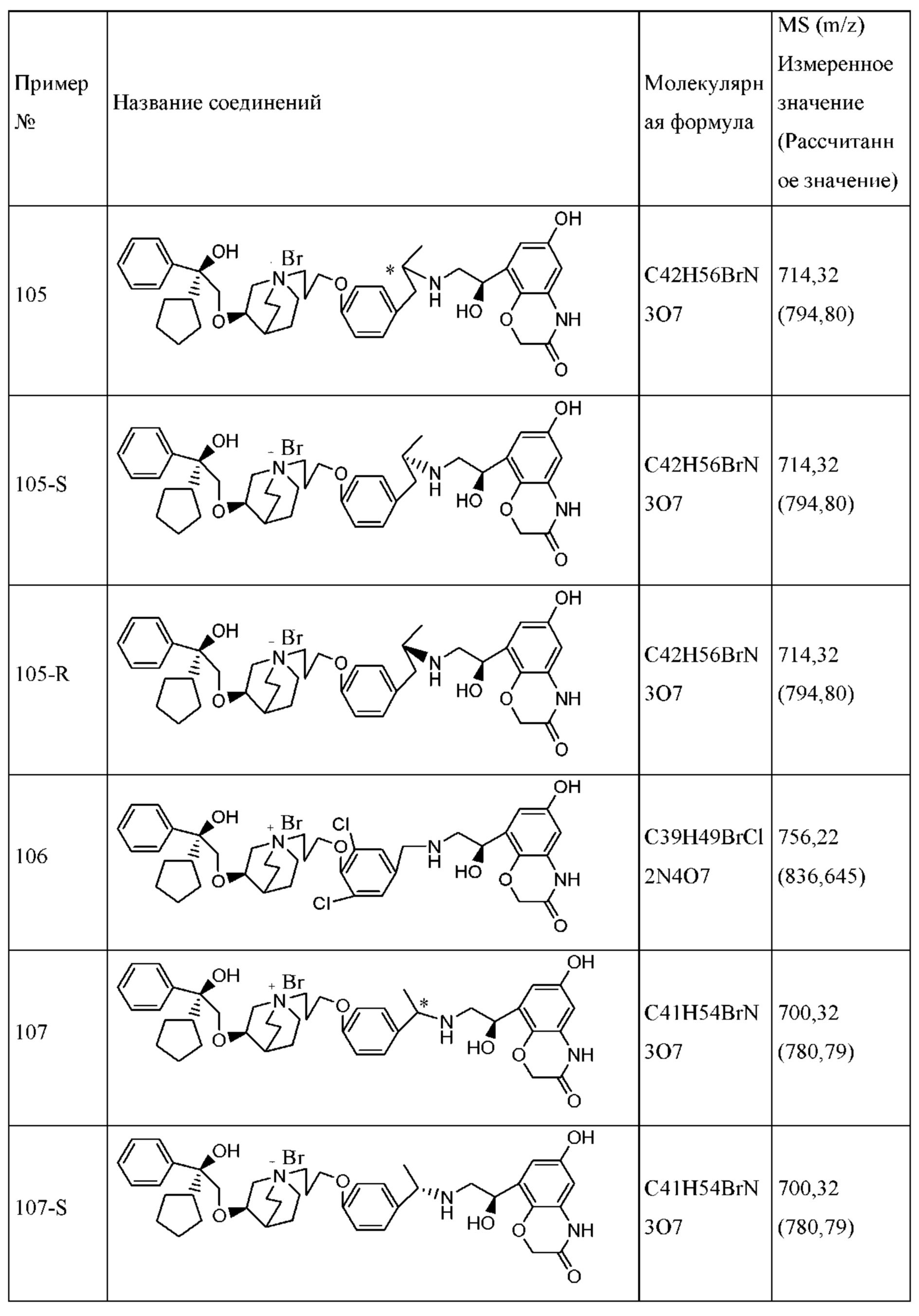
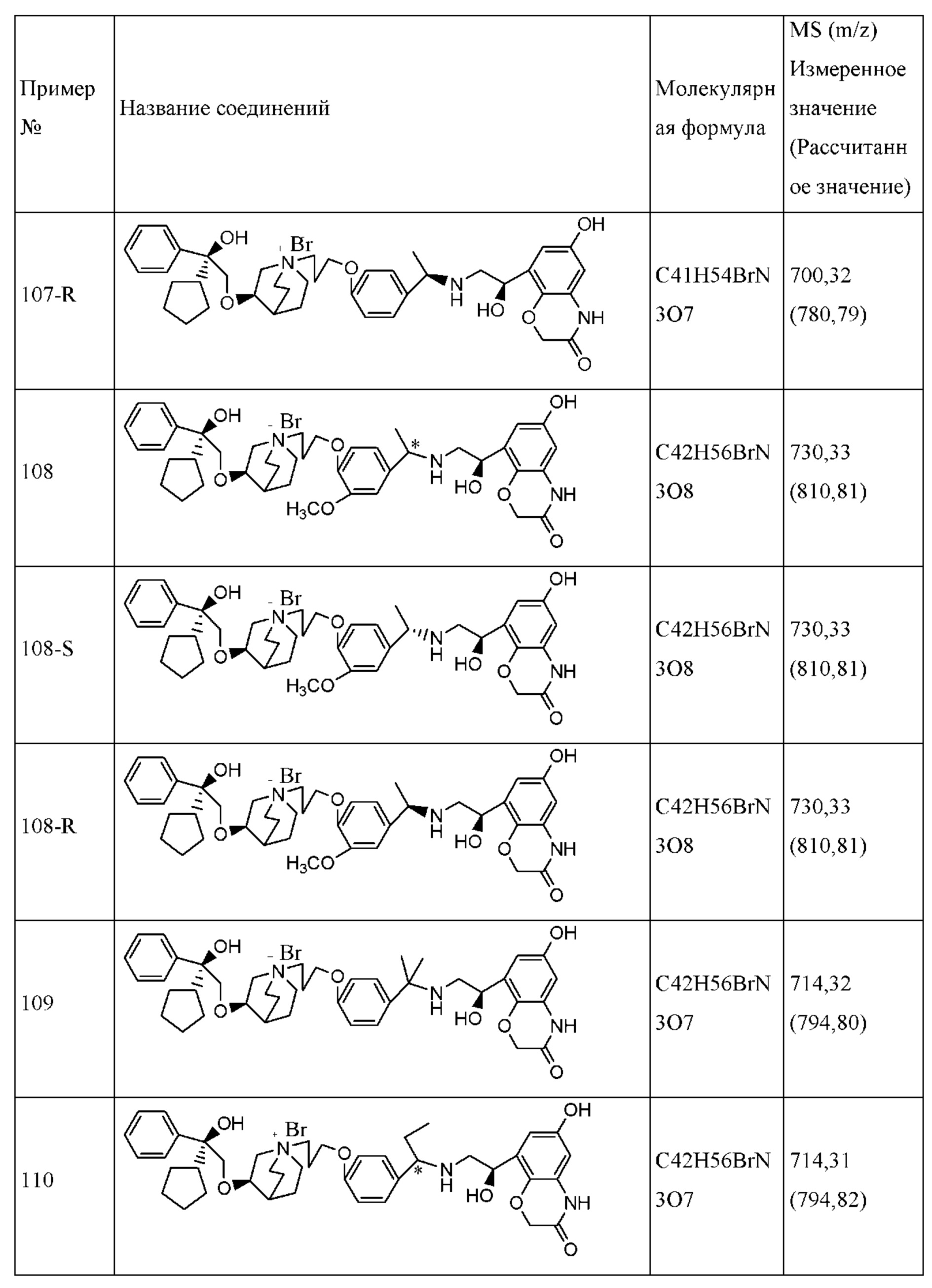
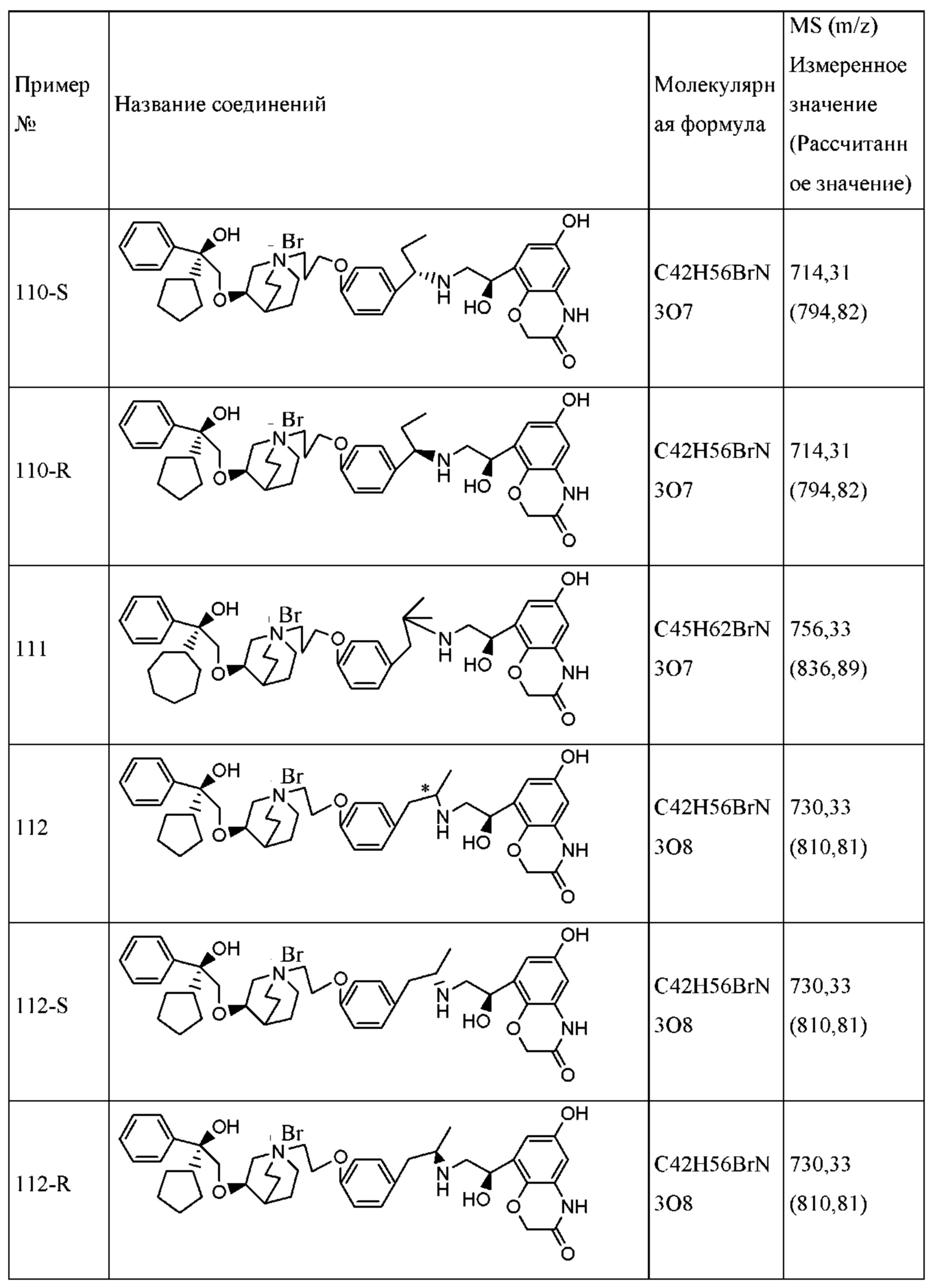
Подготовительный пример 33
1,4-бис(3-бромпропокси)бензол
В 800 мл метанола растворяли 110,1 г (1,0 моль) 1,4-гидрохинона, добавляли 606 г (3,0 моль) 1,3-дибромпропана и 276 г (2,0 моль) безводного карбоната калия. Реакционную смесь нагревали с обратным холодильником в течение 6 часов, и останавливали реакцию. Твердое вещество удаляли путем фильтрования, и удаляли из раствора растворитель досуха в условиях пониженного давления. Добавляли 500 мл этилацетата для растворения, и 3 раза промывали водой (200 мл × 3). Органическую фазу сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и концентрировали фильтрат досуха. Получали 209 г целевого продукта с выходом 59,4%, m/z 351,95.
Подготовительный пример 34
2-метокси-1,4-бис(3-бромпропокси)бензол
В 600 мл метанола растворяли 98,1 г (0,70 моль) 2-метокси-1,4-гидрохинона. Добавляли 404 г (2,0 моль) 1,3-дибромпропана и 207 г (1,50 моль) безводного карбоната калия. Реакционную смесь нагревали с обратным холодильником в течение 8 часов, и останавливали реакцию. Твердое вещество удаляли путем фильтрования. Метанол и избыток 1,3-дибромпропана удаляли из раствора досуха в условиях пониженного давления. Добавляли 400 мл этилацетата для растворения, 3 раза промывали водой (200 мл × 3). Органическую фазу сушили над безводным сульфатом магния в течение ночи. Осушитель удаляли путем фильтрования, и концентрировали фильтрат досуха. Получали 189 г целевого продукта с выходом 49,5%, m/z 381,96.
Подготовительный пример 35
1,4-бис(2-бромэтокси)бензол
Указанное выше соединение получали в соответствии со способом Подготовительного примера 32.
Подготовительный пример 36
2-метокси-5-хлор-4-(3-бромпропионамидо)бензиловый спирт
(а) Метил-2-метокси-5-хлор-4-аминобензоата гидрохлорид
В трехгорлую колбу емкостью 1000 мл помещали 500 мл абсолютного метанола, охлажденного до -5°С на бане с солью на льду. По каплям добавляли 130 мл тионилхлорида. После завершения добавления по каплям, при перемешивании добавляли 100,5 г (0,50 моль) 2-метокси-5-хлор-4-аминобензойной кислоты, твердое вещество полностью не растворялось, реакционную смесь естественным образом нагревали до комнатной температуры, реакцию продолжали при перемешивании в течение 24 часов, твердое вещество фильтровали и собирали. Твердое вещество растворяли путем нагревания с 250 мл метанола, замораживали для кристаллизации, фильтровали для сбора твердого вещества и сушили с получением 94,9 г целевого продукта с выходом 75,7%.
(b) Метил-2-метокси-5-хлор-4-(3-бромпропионамидо)бензоат
В 300 мл DMF растворяли 94,0 г (0,373 моль) продукта (а). Добавляли 37,4 г (0,373 моль) N-метилморфолина. Реакционную смесь охлаждали и перемешивали в течение 10 минут на бане со льдом. Добавляли 57,1 г (0,373 моль) 3-бромпропионовой кислоты и 103 г (0,50 моль) дициклогексилкарбодиимида. После завершения добавления, реакционную смесь нагревали до комнатной температуры. Реакцию продолжали при перемешивании в течение 24 часов, и удаляли растворитель в условиях пониженного давления. Остаток растворяли в 300 мл этилацетата, твердое вещество удаляли путем фильтрования, и 3 раза промывали твердое вещество 200 мл этилацетата. Этилацетатные растворы объединяли, 3 раза промывали насыщенным раствором бикарбоната натрия (150 мл × 3), 3 раза промывали водой (150 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и концентрировали фильтрат досуха с получением 108,3 г целевого продукта с выходом 82,8%, m/z 348,97, 350,97.
(c) 2-метокси-5-хлор-4-(3-бромпропионамидо)бензиловый спирт
В 400 мл метанола, охлажденного на бане со льдом, растворяли 107 г (0,306 моль) продукта (b). Отдельными порциями добавляли 18,9 г (0,50 моль) боргидрида натрия, и завершали добавление приблизительно в течение 1 часа. После завершения добавления, реакцию продолжали при перемешивании при этой температуре в течение 2 часов. Реакционную смесь постепенно нагревали до комнатной температуры, и сохраняли в реакционных условиях в течение 4 часов. Растворитель удаляли в условиях пониженного давления. Остаток растворяли в 500 мл дихлорметана, 3 раза промывали водой (150 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, и концентрировали фильтрат досуха с получением 77,9 г целевого продукта с выходом 78,9%. m/z 320,98, 322,97.
Подготовительный пример 37
2-метокси-4-(3-бромпропионамидо)бензиловый спирт
Указанное выше соединение получали в соответствии со способом Подготовительного примера 36.
Подготовительный пример 38
3-метил-4-(3-бромпропионамидо)бензиловый спирт
Указанное выше соединение получали в соответствии со способом Подготовительного примера 36.
Подготовительный пример 39
4-(3-бромпропионамидо)бензиловый спирт
Указанное выше соединение получали в соответствии со способом
Подготовительного примера 36.
Подготовительный пример 40
1,1-диметокси-1-[4-(2-бромэтоксиметил)фенил]этан
(a) 1,1-диметокси-1-(4-метоксиформил)фенилэтан
В 400 мл метанола растворяли 89 г (0,50 моль) метил-4-ацетилбензоата. Подавали 20 г хлороводорода. Реакционную смесь нагревали до 50°С, проводили реакцию в течение 4 часов, и реакция останавливалась. Избыток метанола и газообразного хлороводорода удаляли в условиях пониженного давления. Остаток растворяли в 400 мл дихлорметана, 3 раза промывали насыщенным раствором бикарбоната натрия (150 мл × 3), 3 раза промывали водой (150 мл × 3) и сушили над безводным сульфатом магния. Осушитель удаляли путем фильтрования, растворитель удаляли в условиях пониженного давления с получением 100,80 г целевого продукта с выходом 90,0%, m/z 224,1.
(b) 1,1-диметокси-1-(4-гидроксиметил)фенилэтан
В 300 мл метанола, охлажденного на бане со льдом, растворяли 98,0 г (0,437 моль) продукта (а). Отдельными порциями добавляли 37,6 г (1,0 моль) боргидрида натрия, и завершали добавление в течение 2 часов. После завершения добавления, реакционную смесь нагревали до комнатной температуры, и продолжали реакцию при перемешивании в течение 2 часов. Метанол удаляли в условиях пониженного давления, остаток растворяли в 400 мл дихлорметана, дихлорметановый слой 3 раза промывали водой (150 мл × 3), органическую фазу сушили над безводным сульфатом магния, осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 68,80 г целевого продукта с выходом 80,2%, m/z 196,1.
(c) 1,1-диметокси-1-[4-(2-бромэтоксиметил)фенил]этан
В 300 мл тетрагидрофурана растворяли 67,0 г (0,341 моль) продукта (b). Добавляли 15,0 г (0,375 моль) гидрида натрия (60%). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут. По каплям добавляли 263,2 г (1,40 моль) 1,2-дибромэтана. После завершения добавления, реакционную смесь нагревали с обратным холодильником в течение 4 часов. Растворитель удаляли в условиях пониженного давления, остаток растворяли в 400 мл дихлорметана, и 3 раза промывали дихлорметановый слой водой (150 мл × 3). Органическую фазу сушили над безводным сульфатом магния, осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 88,90 г целевого продукта с выходом 86,0%, m/z 302,0 (М-Н).
Подготовительный пример 41
1,4-бис(2-бромэтоксиметил)бензол
(а) 1,4-бис(гидроксиметил)бензол
В 400 мл метанола, охлажденного на бане со льдом, растворяли 138,0 г (0,75 моль) диметилтерефталат. Отдельными порциями добавляли 75,20 г (2,0 моль) боргидрида натрия, и завершали добавление в течение 2 часов. После добавления, реакционную смесь нагревали с обратным холодильником в течение 4 часов. Метанол удаляли в условиях пониженного давления, остаток растворяли в 500 мл дихлорметана, дихлорметановый слой 3 раза промывали водой (150 мл × 3), органическую фазу сушили над безводным сульфатом магния, осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 80,80 г целевого продукта с выходом 77,9%, m/z 138,07.
(b) 1,4-бис(2-бромэтоксиметил)бензол
В 500 мл тетрагидрофурана растворяли 78,0 г (0,565 моль) продукта (а). Добавляли 24,86 г (0,622 моль) гидрида натрия, перемешивали при комнатной температуре в течение 15 минут. По каплям добавляли 526,4 г (2,80 моль) 1,2-дибромэтана. После завершения добавления, реакционную смесь нагревали с обратным холодильником в течение 8 часов. Растворитель и избыток 1,2-дибромэтана удаляли в условиях пониженного давления, и растворяли остаток в 800 мл дихлорметана. Дихлорметановый слой 3 раза промывали водой (250 мл × 3), органическую фазу сушили над безводным сульфатом магния, осушитель удаляли путем фильтрования, и удаляли растворитель в условиях пониженного давления с получением 138,80 г целевого продукта с выходом 69,8%, m/z 352,0.
Пример 113
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(3-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}пропокси)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
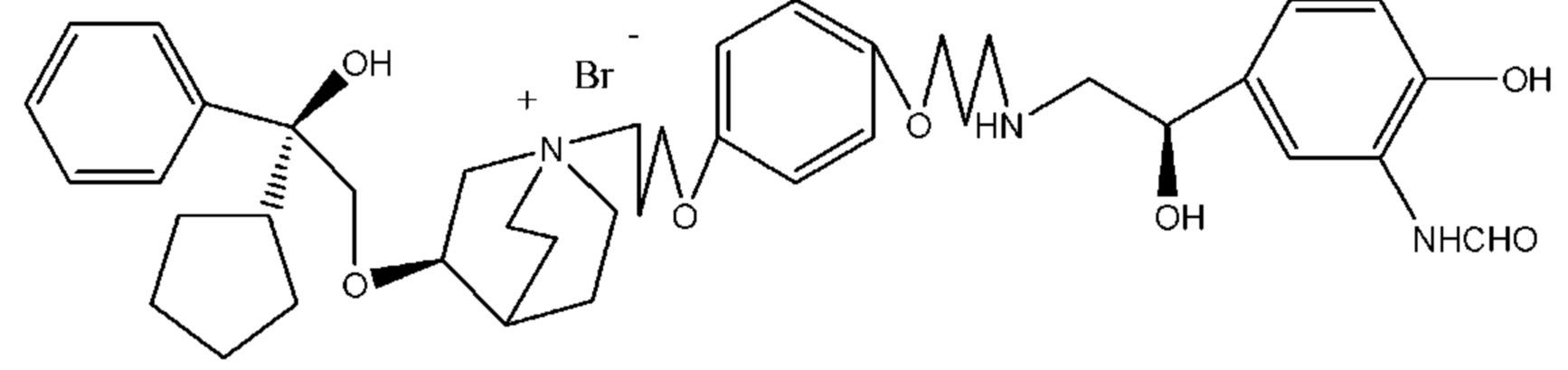
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(3-бромпропокси)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии с Примером 3 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(3-{(R)-[2-гидрокси-2-(3-формамидо-4-бензилокси)фенил]этилбензиламино}пропокси)фенокси]-пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 1,882 г (5,0 ммоль) (R)-2-бензиламино-1-[(4-бензилокси-3-формамидо)фенил]этанола (Подготовительный пример 4) и 3,38 г (5,0 ммоль) продукта (а), добавляли 30 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,5 г (11,0 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 55-60°С, и проводили реакцию при этой температуре в течение 12 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 10 мл изопропанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 3,26 г целевого продукта. Выход составил 67,7%. Рассчитано для MS (m/z) C55H68BrN3O7: 963,0, обнаружено: 883,4 (M-Br).
(с) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-(3-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}пропокси)фенокси]пропил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 3,10 г (3,22 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,6 г 10% Pd-C, подавали водород, давление поддерживали при 0,4-0,43 МПа, и температура представляла собой комнатную температуру. После 20 реакции, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с изопропанолом. Получали 1,38 г целевого соединения в виде твердого вещества с выходом 54,7%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,21 (с, 1H), 7,40 (м, 1H), 7,18-7,23 (м, 5Н), 6,66-6,71 (м, 2Н), 5,0 (с, 1H), 4,74 (м, 1H), 4,0 (с, 1H), 3,92 (с, 2Н), 3,68 (д, 2Н), 3,15 (м, 1H), 2,91 (м, 1H), 2,83 (м, 1H), 2,54 (м, 2Н), 2,42 (с, 1H), 2,35 (м, 2Н), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 8Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C41H56BrN3O7: 782,8, обнаружено: 702,3 (M-Br).
Пример 114
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этиоксил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
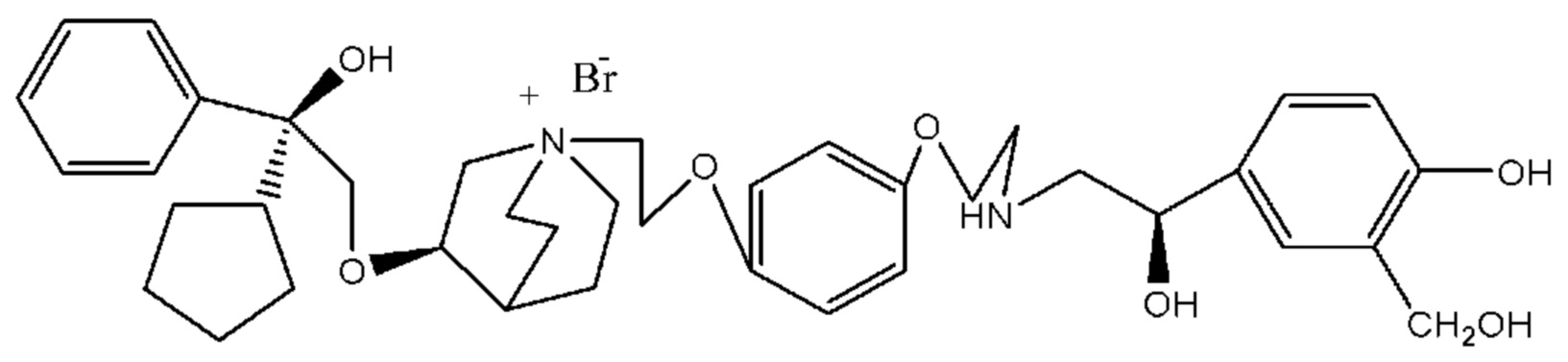
(а) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-бромэтиоксил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (а)
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-(3-бензилоксиметил-4-бензилокси)фенил-2-гидрокси]этилбензиламино}этиоксил)фенокси]-этил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,40 г (5,28 ммоль) (R)-1-[(4-бензилокси-3-бензилоксиметил)фенил]-2-бензиламиноэтанола (Подготовительный пример 1) и 3,38 г (5,28 ммоль) продукта (а), добавляли 20 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,475 г (10,60 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 65-70°С, и проводили реакцию при этой температуре в течение 8 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, остаток растворяли в изопропаноле и замораживали для кристаллизации, твердое вещество фильтровали и собирали с получением 3,42 г целевого продукта. Выход составил 64,0%. Рассчитано для MS (m/z) C60H71BrN2O7: 1012,1, обнаружено: 932,4 (M-Br).
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-(3-гидроксиметил-4-гидрокси)фенил-2-гидрокси]этиламино}этиоксил)фенокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 3,30 г (3,26 ммоль) продукта (b), добавляли 20 мл метанола для растворения, добавляли 0,8 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 10 часов реакции, реакцию останавливали. Катализатор удаляли путем фильтрования. Фильтрат концентрировали досуха в условиях пониженного давления. Остаток растворяли в изопропаноле и замораживали для кристаллизации. Получали 1,38 г целевого соединения в виде твердого вещества с выходом 57,1%. 1H-ЯМР (δ м.д.) (DMSO-d6) 7,19-7,22 (м, 5Н), 6,95-7,01 (м, 2Н), 6,66 (д, 2Н), 6,63-6,59 (м, 3Н), 5,0 (с, 1H), 4,79 (с, 2Н), 4,74 (м, 1H), 4,04 (т, 2Н), 4,06 (т, 2Н), 3,93 (д, 1H), 3,68 (д, 1H), 3,15 (м, 1H), 2,97 (т, 2Н), 2,90 (м, 1H), 2,74 (м, 1H), 2,78 (т, 2Н), 2,21 (с, 1H), 2,10 (с, 1H), 2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 6Н); Рассчитано для MS (m/z) C39H53BrN2O7: 741,8, обнаружено: 661,3 (M-Br).
Пример 115
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[5-хлор-2-метокси-4-({(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}метил)анилино]оксопропил}-1-азабицикло[2,2,2]октилония бромид
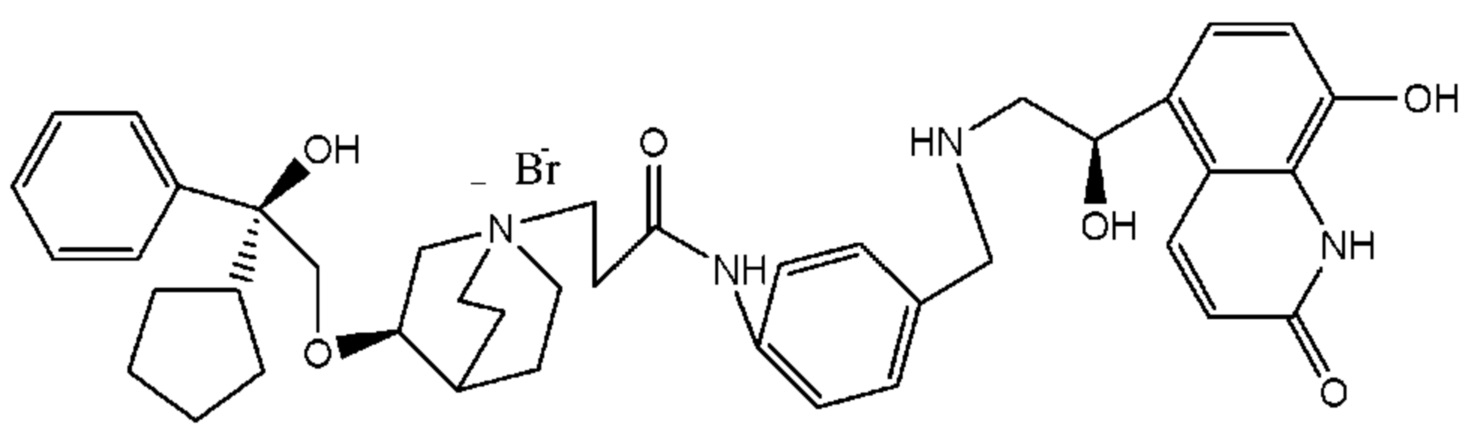
(a) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил)]этокси-1-{3-[(5-хлор-2-метокси-4-гидроксиметил)анилино]оксопропил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-{2-[5-хлор-2-метокси-4-метансульфонилоксиметил)анилино]оксопропил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (b).
(c) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-{2-[5-хлор-2-метокси-4-({(R)-[2-гидрокси-2-(8-бензилокси-2-оксо-1,2-дигидрохинолин-5-ил)]этилбензиламино}метил)анилино]оксопропил)-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,0 г (5,0 ммоль) 8-бензилокси-5-[(R)-2-бензиламино-1-гидроксиэтил]-(1Н)-хинолин-2-он (Подготовительный пример 2) и 4,28 г (5,0 ммоль) продукта (b), добавляли 30 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,475 г (10,57 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 65-70°С. Реакцию проводили при этой температуре в течение 5 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления. Остаток растворяли в 10 мл этанола, замораживали и кристаллизовали. Твердое вещество фильтровали и сушили с получением 2,96 г целевого продукта. Выход составил 51,2%. Рассчитано для MS (m/z) C64H70BrClN4O9: 1154,49, обнаружено: 1073,86 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[5-хлор-2-метокси-4-({(R)-[2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)]этиламино}метил)анилино]оксопропил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,88 г (2,49 ммоль) продукта (с), добавляли 20 мл метанола для растворения, добавляли 0,7 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 18 часов реакции, поглощение водорода останавливалось, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, остаток растворяли и кристаллизовали с этанолом. Получали 1,48 г целевого соединения в виде твердого вещества с выходом 70,7%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,10 (с, 1H), 8,01 (с, 1H), 7,36 (д, 1H), 7,18-7,21 (м, 5Н), 6,97 (д, 1H), 6,94 (д, 1H), 6,71 (д, 1H), 6,52 (д, 1H), 5,0 (с, 1H), 4,74 (м, 1H), 3,93 (м, 1H), 3,81 (д, 2Н), 3,73 (с, 3Н), 3,68 (д, 1H), 3,15 (м, 1H), 2,90 (м, 1H), 2,84 (м, 1H), 2,74 (т, 2Н), 2,33 (т, 2Н), 2,05 (с, 1H), 2,0 (м, 4Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,68 (м, 2Н), 1,56-1,60 (м, 6Н), 1,35-1,46 (м, 6Н). Рассчитано для MS (m/z) C42H52BrClN4O7: 840,24, обнаружено: 759,77 (M-Br).
Пример 116
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-({(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}метил)анилино]оксопропил}-1-азабицикло[2,2,2]октилония бромид
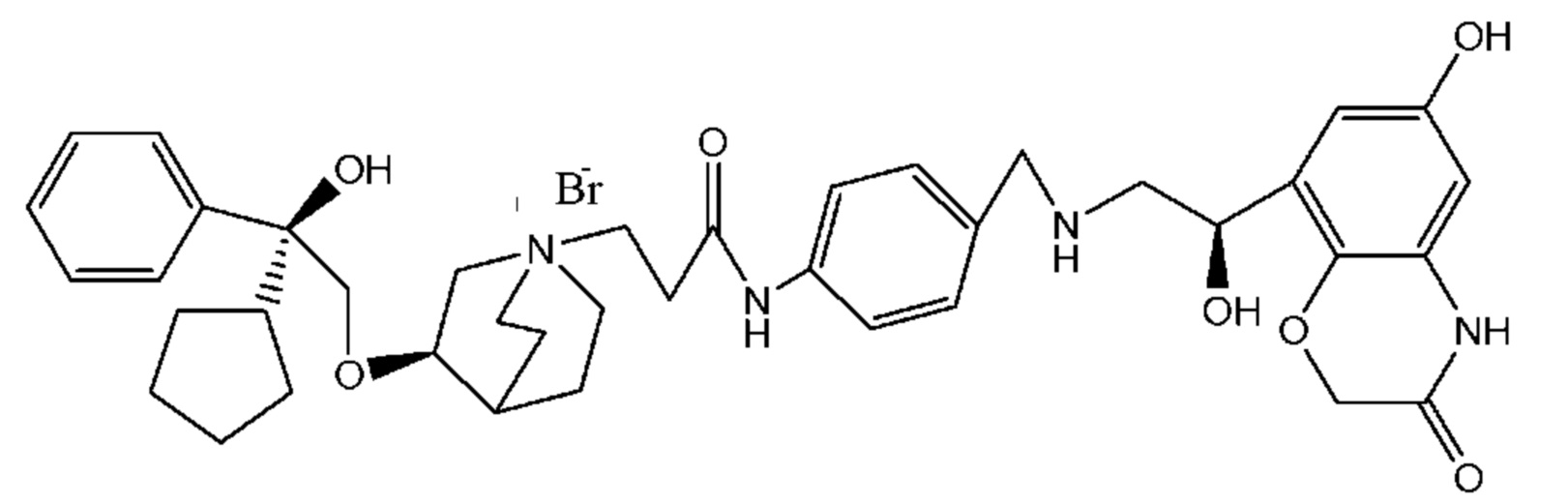
(a) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-[3-(4-гидроксиметиланилино)оксопропил]-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (а).
(b) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-[3-(4-метилсульфонилоксиметиланилино)оксопропил]-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 57 (b).
(c) (R)-(-)-3-[(R)-2-бензилоксиформокси-2-циклопентил-2-фенил]этокси-1-{2-[4-({(R)-[2-гидрокси-2-(6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этилбензиламино}метил)анилиноформил]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,02 г (5,0 ммоль) 8-[(R)-1-гидрокси-2-бензиламин]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она (Подготовительный пример 5) и 3,96 г (5,0 ммоль), добавляли 30 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,0 г (7,35 ммоль) безводного карбоната калия, температуру реакционной смеси повышали до 65-70°С. Реакцию проводили при этой температуре в течение 10 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 10 мл изопропилового спирта для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 2,88 г целевого продукта. Выход составил 60,0%.
Рассчитано для MS (m/z) C62H69BrN4O9: 1094,0, обнаружено: 1014,38 (M-Br).
(d) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{3-[4-({(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}метил)анилино]оксопропил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 2,70 г (2,47 ммоль) продукта (с), добавляли 20 мл метанола для растворения, добавляли 0,6 г 10% Pd-C, подавали водород, и поддерживали давление при 0,43 МПа. После проведения реакции при комнатной температуре в течение 16 часов, поглощение водорода останавливалось, и останавливали реакцию. Катализатор удаляли путем фильтрования, фильтрат концентрировали досуха, и кристаллизовали остаток с изопропанолом. Получали 1,26 г целевого соединения в виде твердого вещества с выходом 65,4%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,05 (с, 1H), 8,0 (с, 1H), 7,52 (д, 2Н), 7,19-7,21 (м, 5Н), 7,04 (д, 2Н), 6,65 (д, 1H), 6,20 (д, 1H), 5,0 (с, 1H), 4,88 (с, 2Н), 4,74 (м, 1H), 3,93 (д, 1H), 3,81 (д, 2Н), 3,68 (д, 1H), 3,15 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,74 (т, 2Н), 2,33 (т, 2Н), 2,10 (м, 2Н), 2,0 (м, 2Н), 1,96 (м, 1H), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,56-1,60 (м, 6Н), 1,34-1,46 (м, 6Н). Рассчитано для MS (m/z) C40H51BrN4O7: 779,76, обнаружено: 700,33 (M-Br).
Пример 117
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}этилиден)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
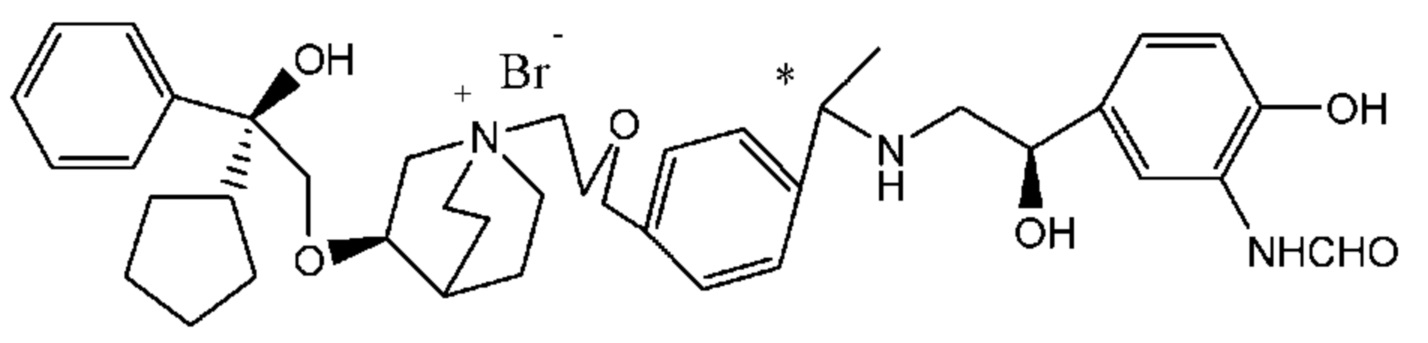
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил)]этокси-1-{2-[4-(1,1-диметоксиэтил)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (а).
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-гидрокси-2-(3-формамидо-4-гидрокси)фенил]этиламино}этилиден)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 250 мл добавляли 0,981 г (5,0 ммоль) (R)-2-амино-1-[(4-гидрокси-3-формамидо)фенил]этанола (Подготовительный пример 30) и 3,09 г (5,0 ммоль) продукта (а), добавляли 50 мл дихлорметана. После перемешивания реакционной смеси в течение 10 минут, добавляли 2,54 г (12,0 ммоль) триацетилборгидрида натрия, и добавляли 1 каплю уксусной кислоты для катализа. В реакционной смеси проводили реакцию в течение 24 часов при комнатной температуре, и методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, и реакционную смесь фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 5 мл этанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 1,62 г целевого продукта. Выход составил 43,0%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,21 (с, 1H), 7,40 (м, 1H), 7,18-7,20 (м, 5Н), 7,14 (д, 2Н), 7,05 (д, 2Н), 6,76 (дд, 1H), 6,64 (дд, 1H), 5,0 (с, 1H), 4,74 (м, 1H), 4,63 (с, 2Н), 4,08 (м, 1H), 4,0 (с, 1H), 3,93 (д, 1H), 3,68 (д, 1H), 3,47 (т, 2Н), 3,15 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,53 (т, 2Н), 2,10 (с, 1H), 1,96-2,0 (м, 3Н), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 6Н), 1,34-1,46 (м, 6Н), 1,38 (д, 3Н); Рассчитано для MS (m/z) C40H54BrN3O6: 752,78, обнаружено: 672,32 (M-Br).
(с) Расщепление изомера 117-S и изомера 117-R
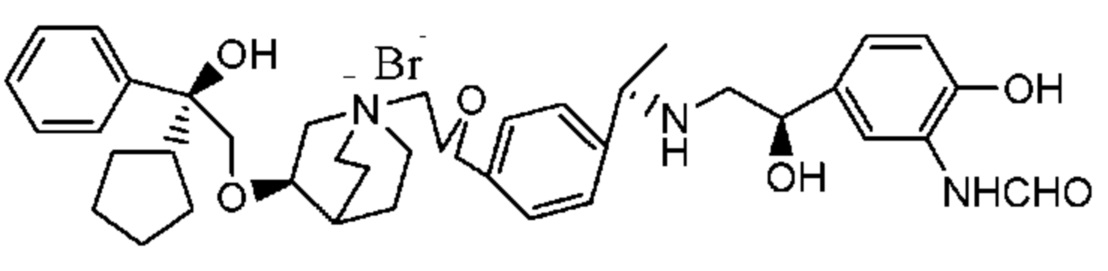
изомер 117-S
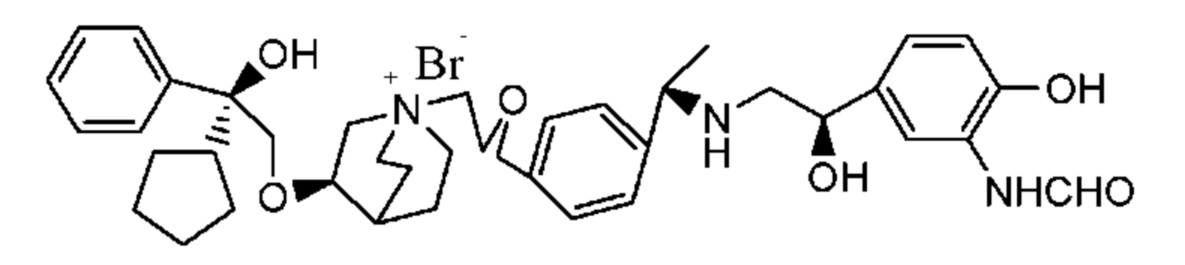
изомер 117-R
В метаноле растворяли 0,80 г продукта (b), разделяли на препаративной колонке, проводили градиентное элюирование метанолом и водой, содержащие S-изомер и R-изомер пики собирали, соответственно, сушили в условиях пониженного давления, остаток перекристаллизовывали с этанолом с получением 0,24 г S-изомера и 0,28 г R-изомера.
Пример 118
(R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}этоксиметил)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
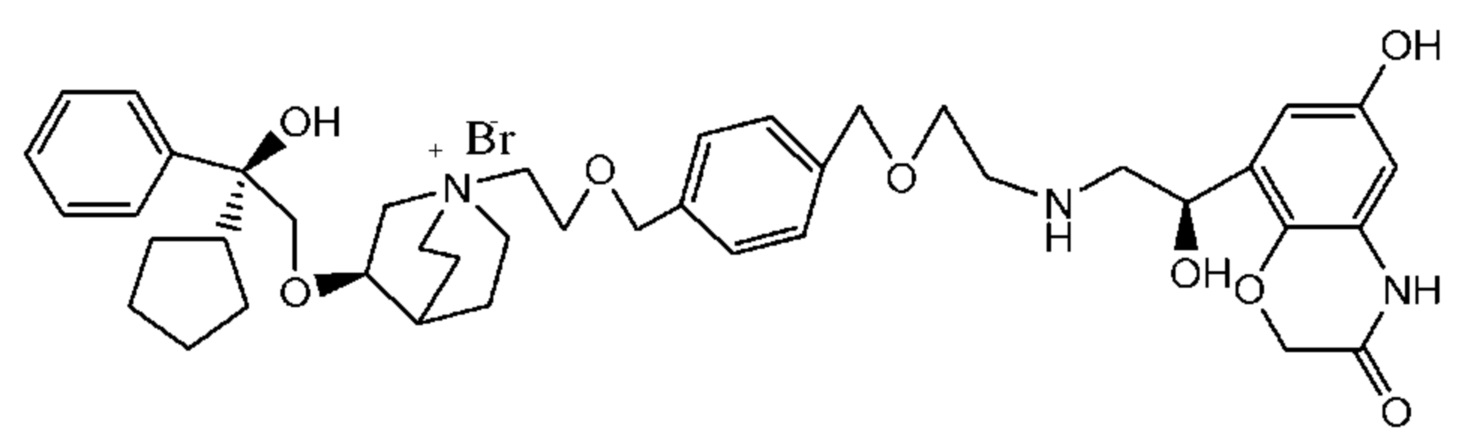
(a) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-бромэтоксиметил)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
Указанное выше соединение получали в соответствии со способом Примера 1 (а).
В трехгорлую колбу емкостью 250 мл добавляли 35 мл абсолютного этанола, добавляли 5,0 г (15,85 ммоль) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-азабицикло[2,2,2] октана (Подготовительный пример 2). После перемешивания реакционной смеси для растворения, добавляли 44,66 г (126,3 ммоль) 1,4-бис(2-бромэтоксиметил)бензола (Подготовительный пример 40). Реакцию проводили при перемешивании при комнатной температуре в течение 18 часов. Этанол удаляли в условиях пониженного давления, и подвергали остаток колоночной хроматографии с получением 8,50 г целевого продукта с выходом 80,4%. MS (m/z) 686,18, 688,17.
(b) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-2-гидрокси-2-(5-бензилокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этилбензиламино}-этоксиметил)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционную колбу емкостью 50 мл добавляли 2,02 г (5,0 ммоль) 8-[(1R)-1-гидрокси-2-бензиламин]этил-6-бензилокси-2Н-1,4-бензоксазин-3(4Н)-она (Подготовительный пример 5) и 3,38 г (5,0 ммоль) продукта (а), добавляли 30 мл диоксана. После перемешивания реакционной смеси в течение 10 минут, добавляли 1,0 г (7,35 ммоль) безводного карбоната калия, и повышали температуру реакционной смеси до 75-80°С. Реакцию проводили при этой температуре в течение 12 часов. Методом тонкослойной хроматографии обнаруживали, что реакция завершена. Реакцию останавливали, реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Растворитель удаляли из фильтрата в условиях пониженного давления, к остатку добавляли 15 мл этанола для кристаллизации вымораживанием, твердое вещество фильтровали и собирали с получением 3,36 г целевого продукта. Выход составил 67,8%. MS (m/z) 901,45.
(c) (R)-(-)-3-[(R)-2-гидрокси-2-циклопентил-2-фенил]этокси-1-{2-[4-(2-{(R)-[2-гидрокси-2-(6-гидрокси-2Н-1,4-бензоксазин-3(4Н)-он-8-ил)]этиламино}этоксиметил)бензилокси]этил}-1-азабицикло[2,2,2]октилония бромид
В реакционный котел для гидрирования помещали 3,20 г (3,23 ммоль) продукта (b), добавляли 30 мл метанола для растворения, добавляли 0,6 г 10% Pd-C, подавали водород, давление поддерживали при 0,43 МПа, и температура представляла собой комнатную температуру. После 18 часов реакции, поглощение водорода останавливалось, реакцию останавливали, катализатор удаляли путем фильтрования, фильтрат концентрировали досуха в условиях пониженного давления, и кристаллизовали остаток с этанолом. Получали 1,48 г целевого соединения в виде твердого вещества с выходом 56,5%. 1H-ЯМР (δ м.д.) (DMSO-d6) 8,0 (с, 1H), 7,15-7,20 (м, 5Н), 7,12 (д, 4Н), 6,65 (д, 1H), 6,20 (д, 1H), 5,0 (с, 1H), 4,88 (с, 2Н), 4,73 (м, 1H), 4,63 (с, 4Н), 3,93 (д, 1H), 3,68 (д, 1H), 3,49 (т, 2Н), 3,47 (т, 2Н), 3,15 (м, 1H), 2,91 (м, 1H), 2,84 (м, 1H), 2,72 (т, 2Н), 2,53 (т, 2Н), 2,10 (с, 1H), 2,0 (м, 2Н), 1,96 (м, 1H), 1,79-1,81 (м, 3Н), 1,75 (м, 1H), 1,69 (м, 2Н), 1,55-1,61 (м, 6Н), 1,34-1,46 (м, 6Н). Рассчитано для MS (m/z) C42H56BrN3O8: 810,8, обнаружено: 730,3 (M-Br).
Следующие соединения могут быть получены с использованием исходных веществ, синтезированных выше, и способов синтеза:
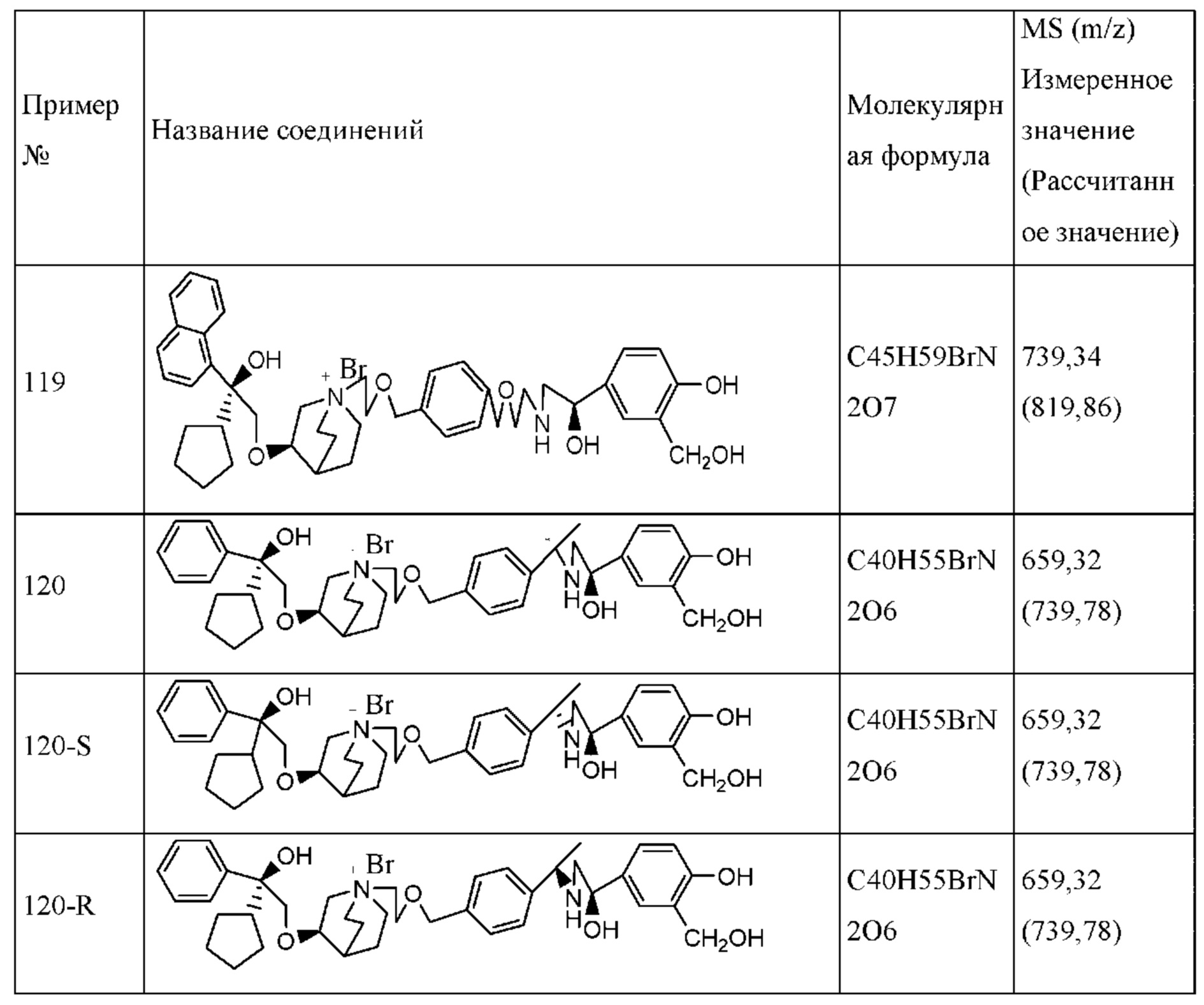
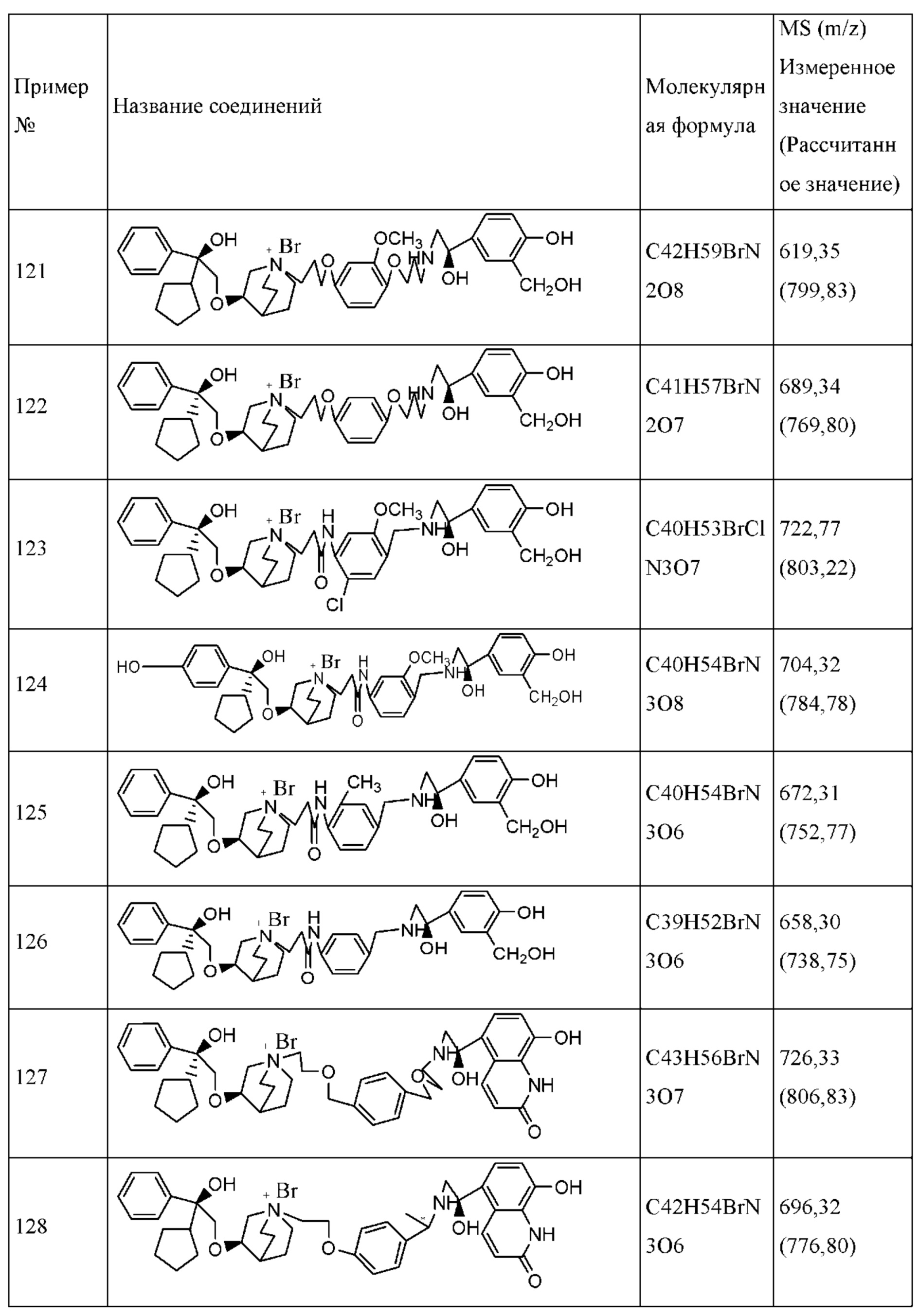
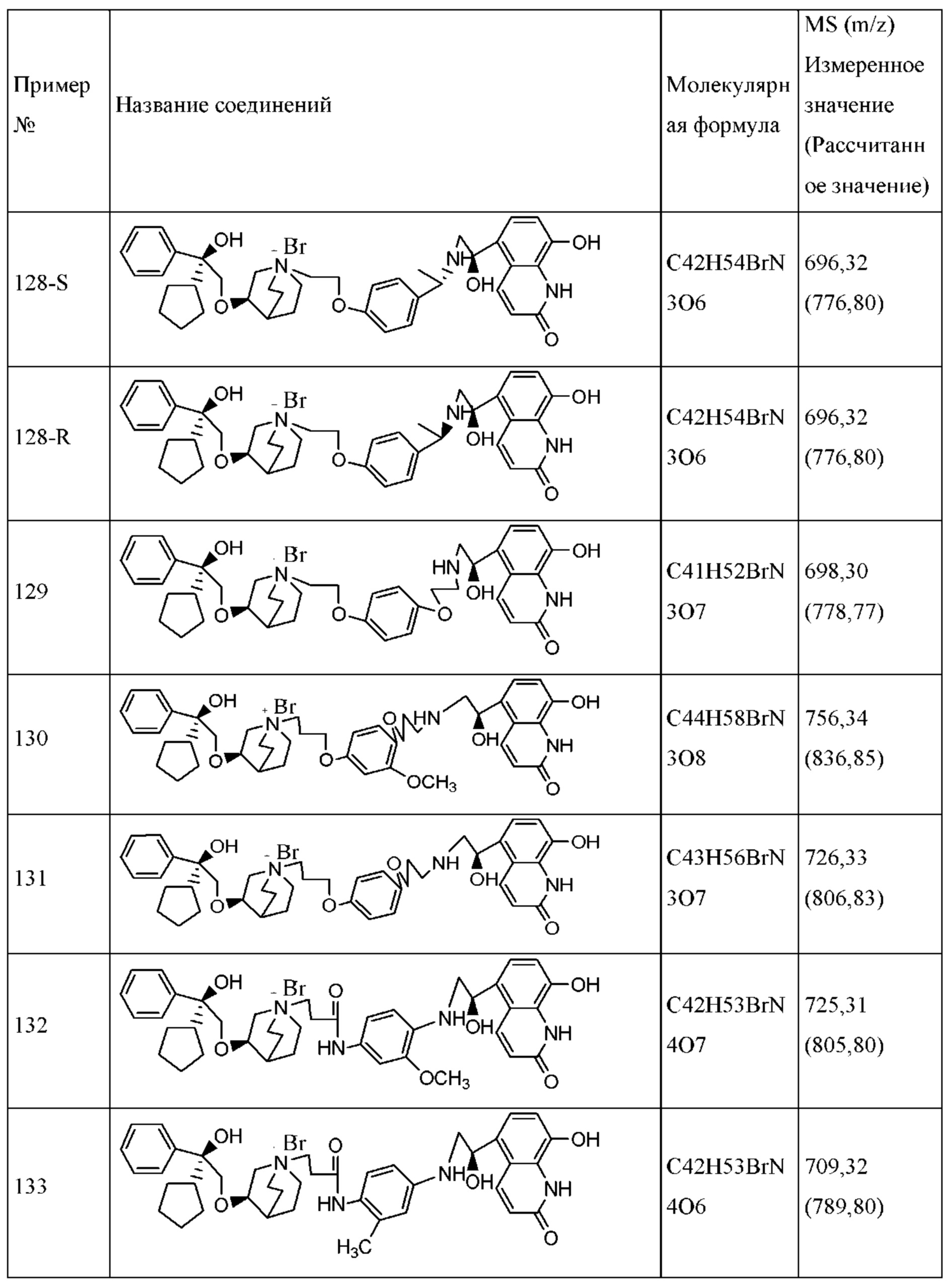
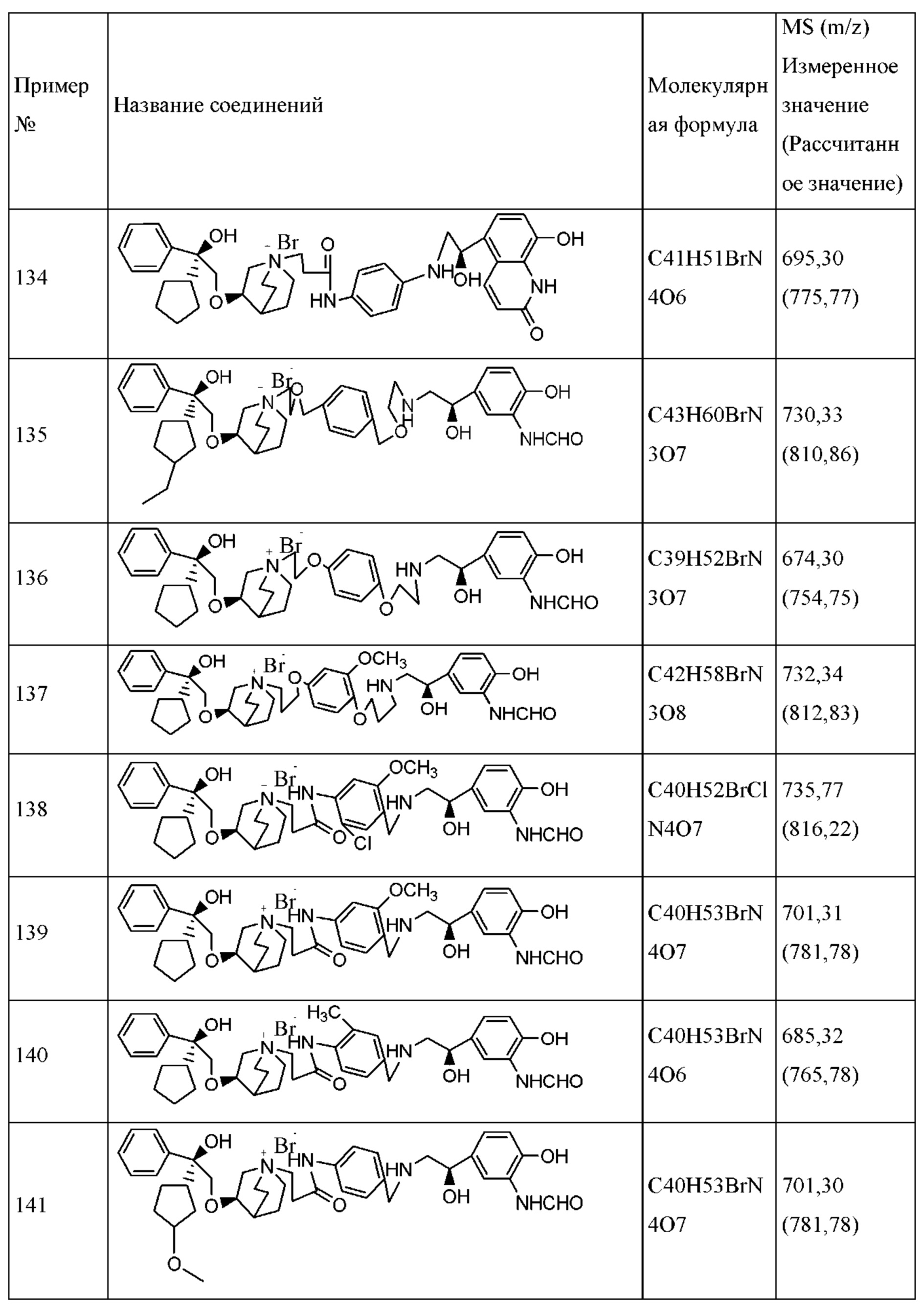
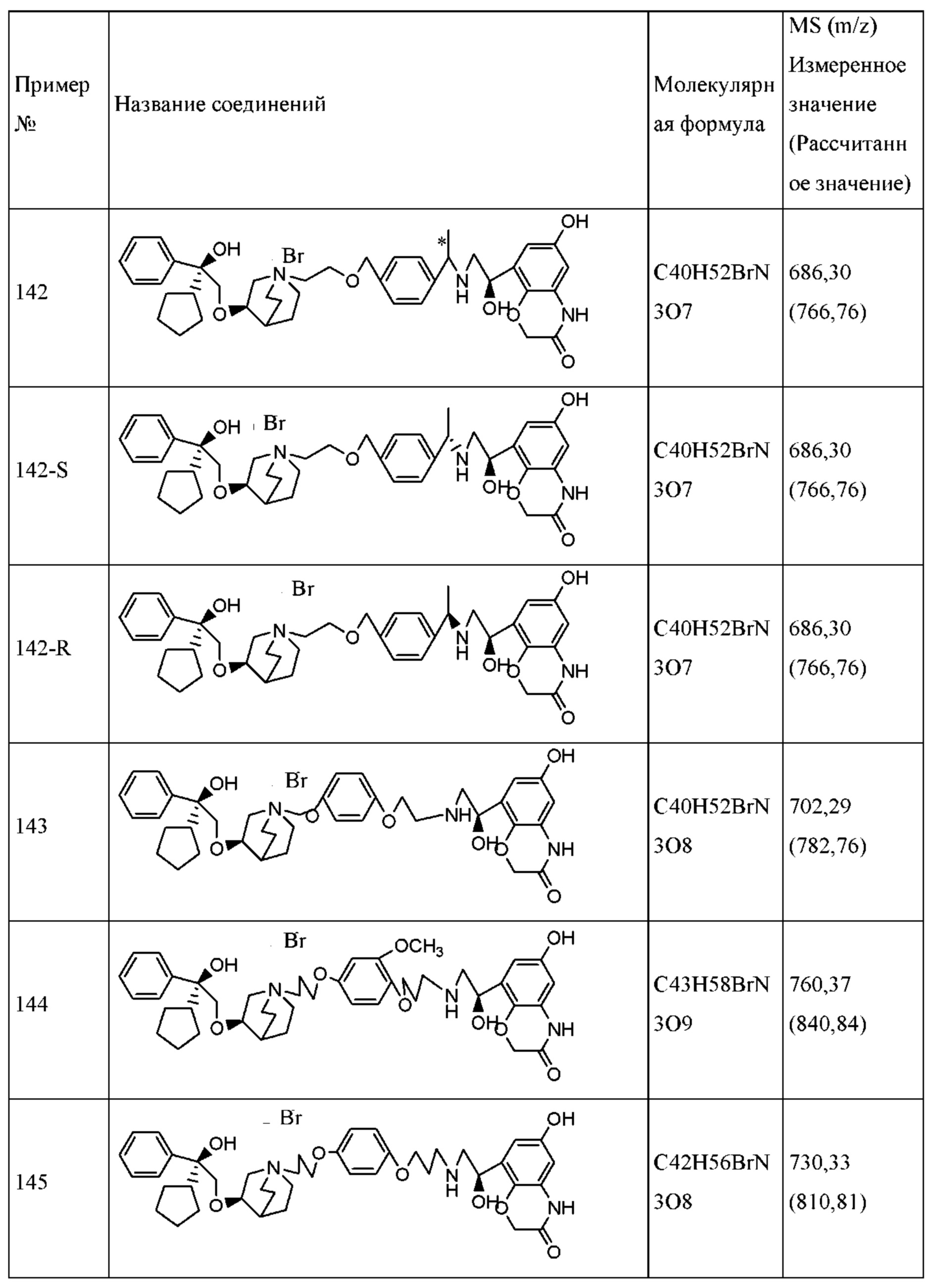
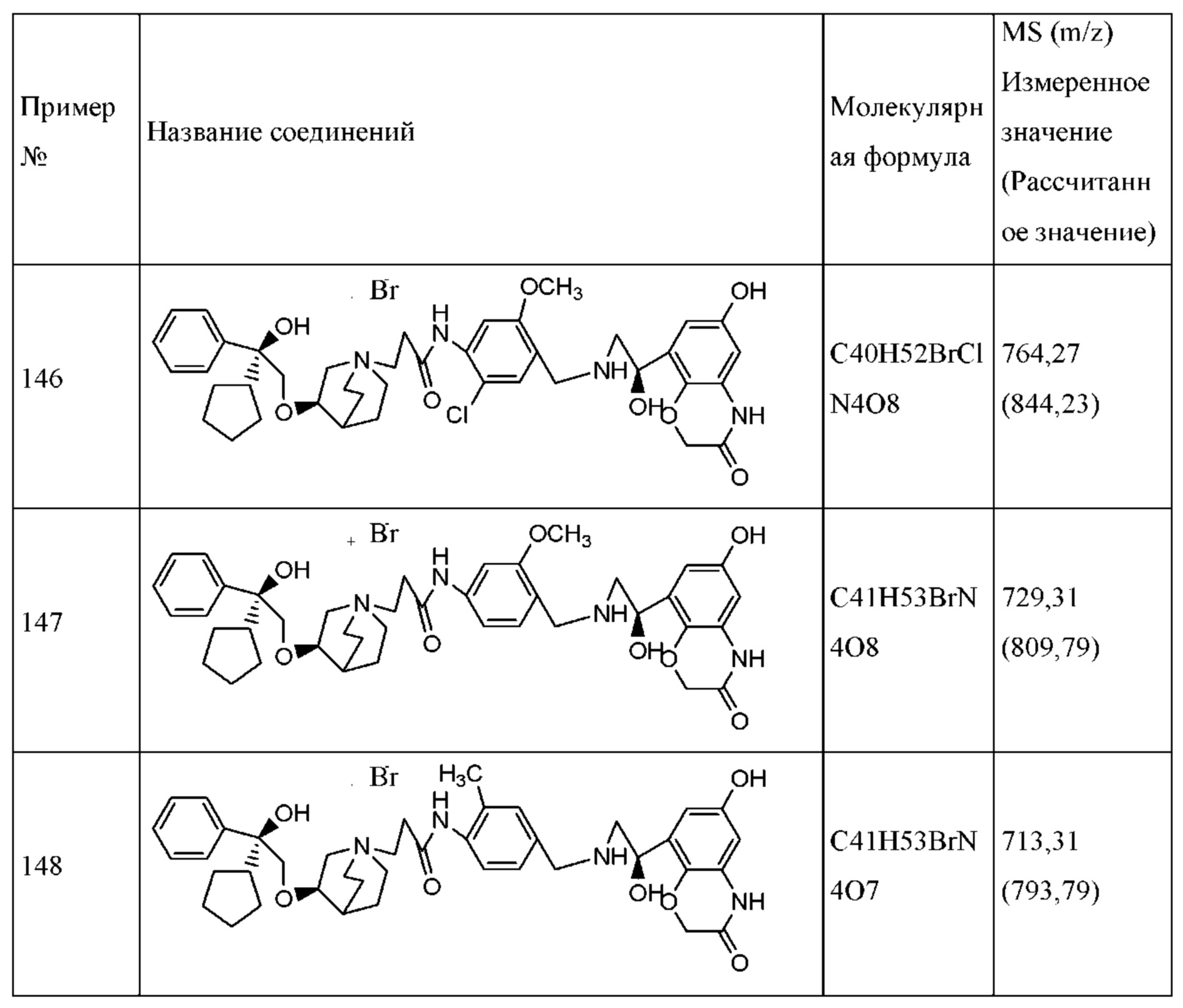
Экспериментальный пример 1. Интенсивность ответа в виде сокращения гладких мышц трахеи у морской свинки, индуцированного антагонизмом в отношении карбахола (CCh), на соединения согласно настоящему изобретению
Получение выделенного образца гладких мышц трахеи морской свинки. После того как морская свинка получала анестезию уретаном, трахею между гортанью и килем быстро вынимали и помещали в раствор Кребса-Гензеляйта (K-Н) (состав (г/л): NaCl 6,92, KCl 0,35, CaCl2 0,28, KH2PO4 0,16, MgSO4 7H2O 0,4568, NaHCO3 2,1, глюкоза 2,0) с добавлением смеси газов 5% СО2 и 95% О2. После того как рыхлую соединительную ткань и жир вокруг трахеи отделяли, трахею выделяли таким образом, чтобы получить пластинку трахеи шириной приблизительно 3 мм и длиной 20 мм и с двумя связанными концами, и ее помещали в ванну с постоянной температурой 37°С (рН 7,4), содержащую 5 мл K-Н раствора, с непрерывным добавлением смеси газов, содержащей 5% СО2 и 95% О2. Верхний конец соединяли с датчиком мышечного напряжения и к образцу прикладывали напряжение в состоянии покоя 1,0 г. Изменения в мышечном напряжении фиксировали, питательный раствор меняли каждые 20 мин. и эксперимент начинали после 60 мин. уравновешивания.
Способ введения. После того как пластинку трахеи стабилизировали, в ванну добавляли 3×10-6 моль/л CCh. Когда напряжение сокращения пластинки трахеи достигало пикового уровня, иллюстративные соединения, бромид ипратропия и бромид тиотропия добавляли в ванну Magnus согласно совокупной схеме дозирования в дозировке 10-9~10-5 моль, соответственно. Наблюдали диастолическое состояние пластинки трахеи. Если ответ не наблюдался (ниже пороговой концентрации), следующую дозу продолжали добавлять по порядку. Если какой-либо ответ наблюдался, добавляли следующую дозу до тех пор, пока не достигалось диастолическое плато. Вышеуказанную операцию повторяли до тех пор, пока кривая сокращения не достигала минимума. Наконец, 10-6 моль/л изопротеренола добавляли для получения максимального расслабления и кривую записывали.
Статистические методы. Статистический анализ осуществляли с применением пакета статистического программного обеспечения Sigma Stat, данные анализировали с применением дисперсионного анализа. Критерий Стьюдента-Ньюмана-Кейлса (SNK) применяли при сравнении ряда средних значений из нескольких выборок. Уровень значимости отличий в статистическом анализе составлял α=0,05 (двусторонний). EC50 (95% доверительный интервал) рассчитывали с помощью программного обеспечения POMS версии 2.0 от Shanghai Science and Technology Press.
3×10-6 моль/л CCh могут заставлять гладкие мышцы трахеи морской свинки давать устойчивый и стабильный ответ в виде сокращения. При совокупной схеме дозирования соединения, подлежащие исследованию, и контрольные лекарственные средства в виде бромида ипратропия и бромида тиотропия добавляли в ванну Magnus, и каждое из иллюстративных соединений и каждое из контрольных лекарственных средств было способно расслаблять сокращение гладких мышц трахеи, индуцированное CCh. Результаты показаны в таблице 1.
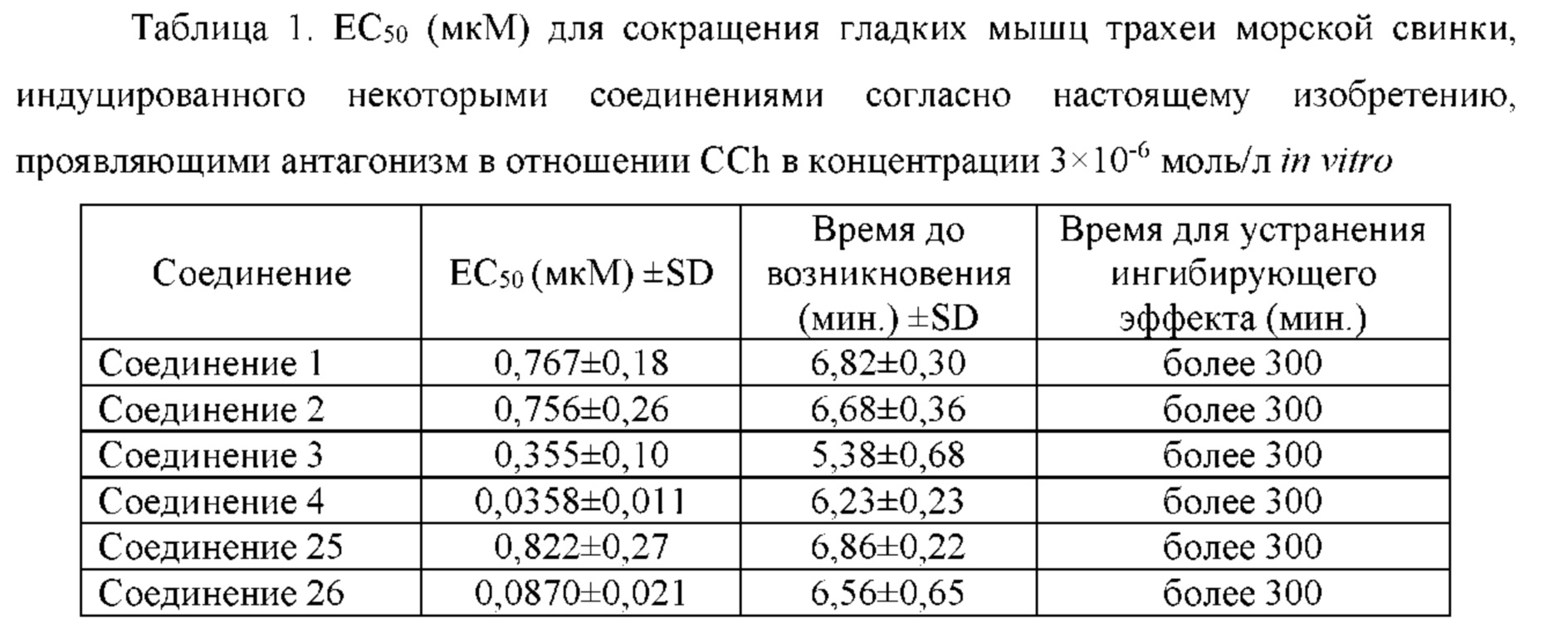
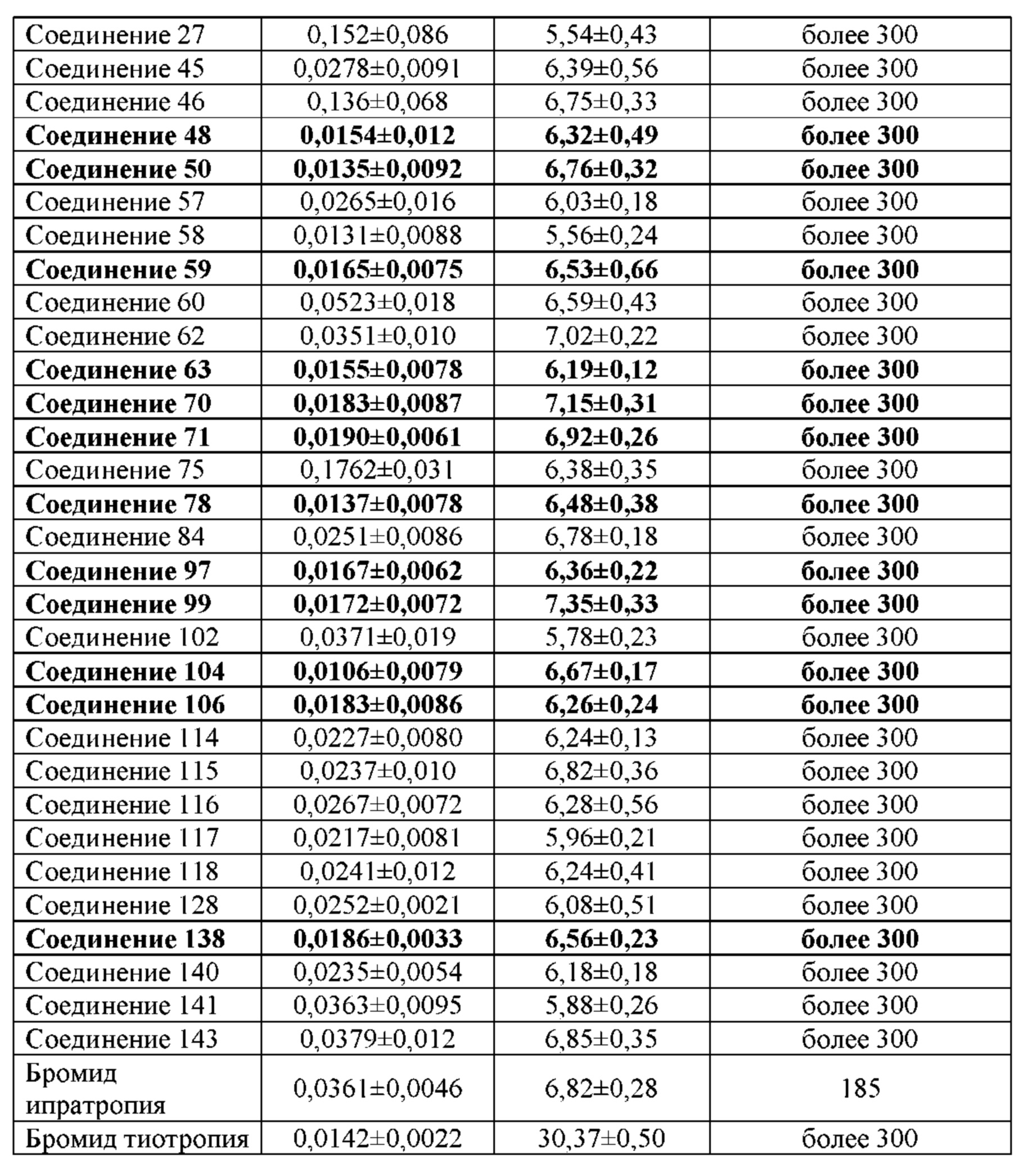
Результаты в таблице 1 показывают, что все соединения, предполагаемые настоящим изобретением, характеризуются эффектом блокирования индуцированного карбахолом сокращения гладких мышц трахеи у морской свинки, значительное число соединений характеризуются более сильным блокирующим эффектом, чем средство-положительный контроль в виде бромида ипратропия, и некоторые соединения имеют силу действия, сравнимую со средством-положительным контролем в виде бромида тиотропия. Время до возникновения ответа на соединение согласно настоящему изобретению и бромид ипратропия было меньшим, чем для бромида тиотропия, напротив, время до возникновения ответа на бромид тиотропия является большим. Соединения согласно настоящему изобретению и бромид тиотропия характеризуется большой продолжительностью действия, а бромид ипратропия характеризуется малой продолжительностью действия.
Экспериментальный пример 2. Определение силы антагонизма и продолжительности действия ответа в виде бронхоконстрикции в трахее морской свинки, индуцированного соединением согласно настоящему изобретению, проявляющего антагонизм в отношении метахолина (Mch)
Определение дыхательного объема, скорости потока в дыхательных путях и транспульмонарного давления. 1,5 г/кг уретана вводили в виде интраперитонеальной инъекции морской свинке для анестезии. Морскую свинку фиксировали на спине, подвергали трахеальной интубации и вставляли постоянную иглу, в то время как яремную наружную вену отделяли. Морскую свинку помещали в плетизмограф для регистрации изменения объемов всего тела, тупоконечную иглу для интубации в полость грудной клетки вводили между 4~5 ребрами передней части груди морской свинки, и можно измерить внутригрудное давление (с использованием отрицательного значения водяного столба на водяном манометре и колебания при дыхании морской свинки в качестве отметок). После стабилизации значения дыхательного объема, скорости потока в дыхательных путях и транспульмонарного давления у морской свинки перед введением Mch записывали с помощью системы для сбора и обработки биологических сигналов MedLab в качестве базовых значений. 10 мкг/кг массы тела Mch вводили в виде внутривенной инъекции. Наблюдали изменения скорости потока в дыхательных путях, дыхательного объема и транспульмонарного давления у морской свинки в течение 5 минут. Расчет Raw и Cdyn: рассчитывали изменения в виде повышенного процента значения Raw и пониженного процентного значения Cdyn после ингаляции Mch.
Формулы для расчета Raw и Cdyn представляли собой соответственно:
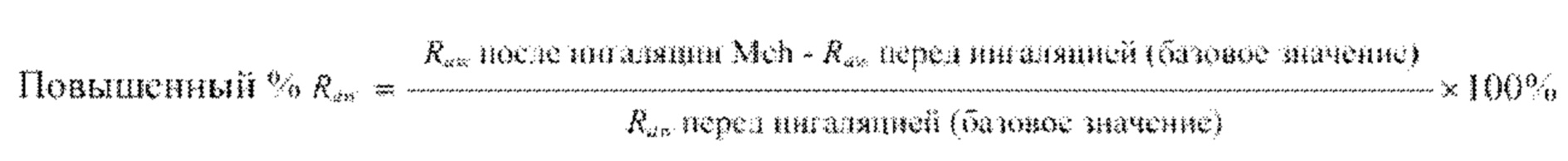

Зависимость доза-эффект. В случае каждого из иллюстративных соединений 27 морских свинок произвольно делили на три группы: группа с 0,1 мкг/кг соединений [48, 70, 99, 138] согласно настоящему изобретению, группа с 0,3 мкг/кг соединений [48, 70, 99, 138] согласно настоящему изобретению и группа с 1 мкг/кг соединений [48, 70, 99, 138] согласно настоящему изобретению, и 15 морских свинок присутствовало в контрольной группе со средой. Сопротивление дыхательных путей (Raw) и динамическую податливость легких (Cdyn) измеряли в течение 5 минут после закапывания в дыхательные пути лекарственных средств в вышеуказанной концентрации в течение 30 мин. и внутривенной инъекции 10 мкг/кг массы тела Mch для возбуждения.
Зависимость время-эффект. После того как морские свинки получали анестезию, 0,5 мкг/кг и 1 мкг/кг соединений [48, 70, 99, 138] согласно настоящему изобретению закапывали в дыхательные пути. Отдельно через 0,25 часа, 0,5 часа, 1 час, 1,5 часа, 2 часа, 4 часа, 6 часов, 12 часов и 24 часа после введения Mch в дозе 10 мкг/кг массы тела вводили в виде внутривенной инъекции для возбуждения и измеряли сопротивление дыхательных путей (Raw) и динамическую податливость легких (Cdyn) в течение 5 мин.
Зависимость доза-эффект. Вводимые дозировки соединений [48, 70, 99 и 138] согласно настоящему изобретению составляли 0,1 мкг/кг, 0,3 мкг/кг и 1 мкг/кг, соответственно. Через 30 мин. после закапывания в дыхательные пути Mch в дозе 10 мкг/кг массы тела вводили в виде внутривенной инъекции для возбуждения и измеряли сопротивление дыхательных путей (Raw) и динамическую податливость легких (Cdyn) в течение 5 мин. Результаты представлены в таблице 2 ниже.
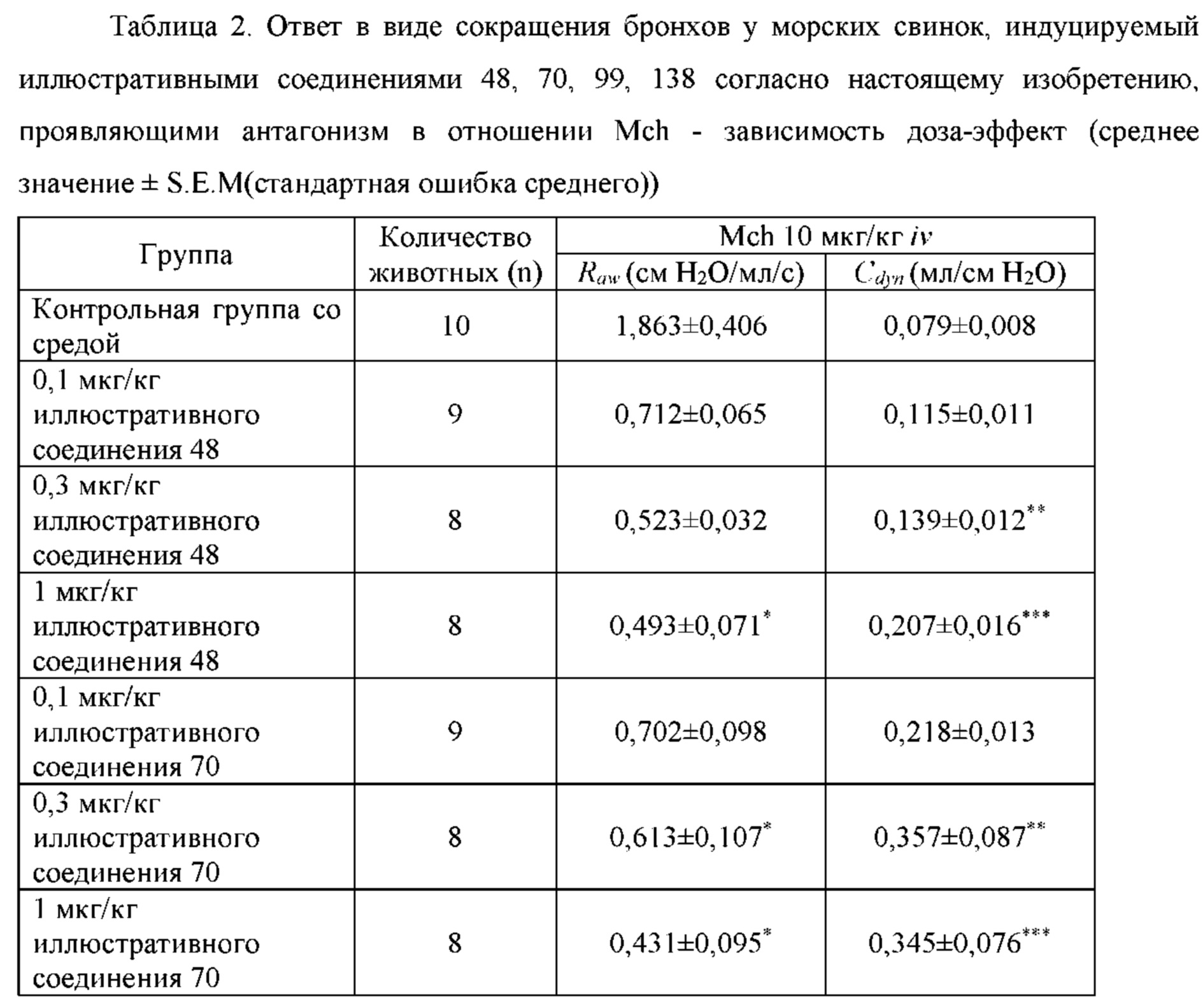
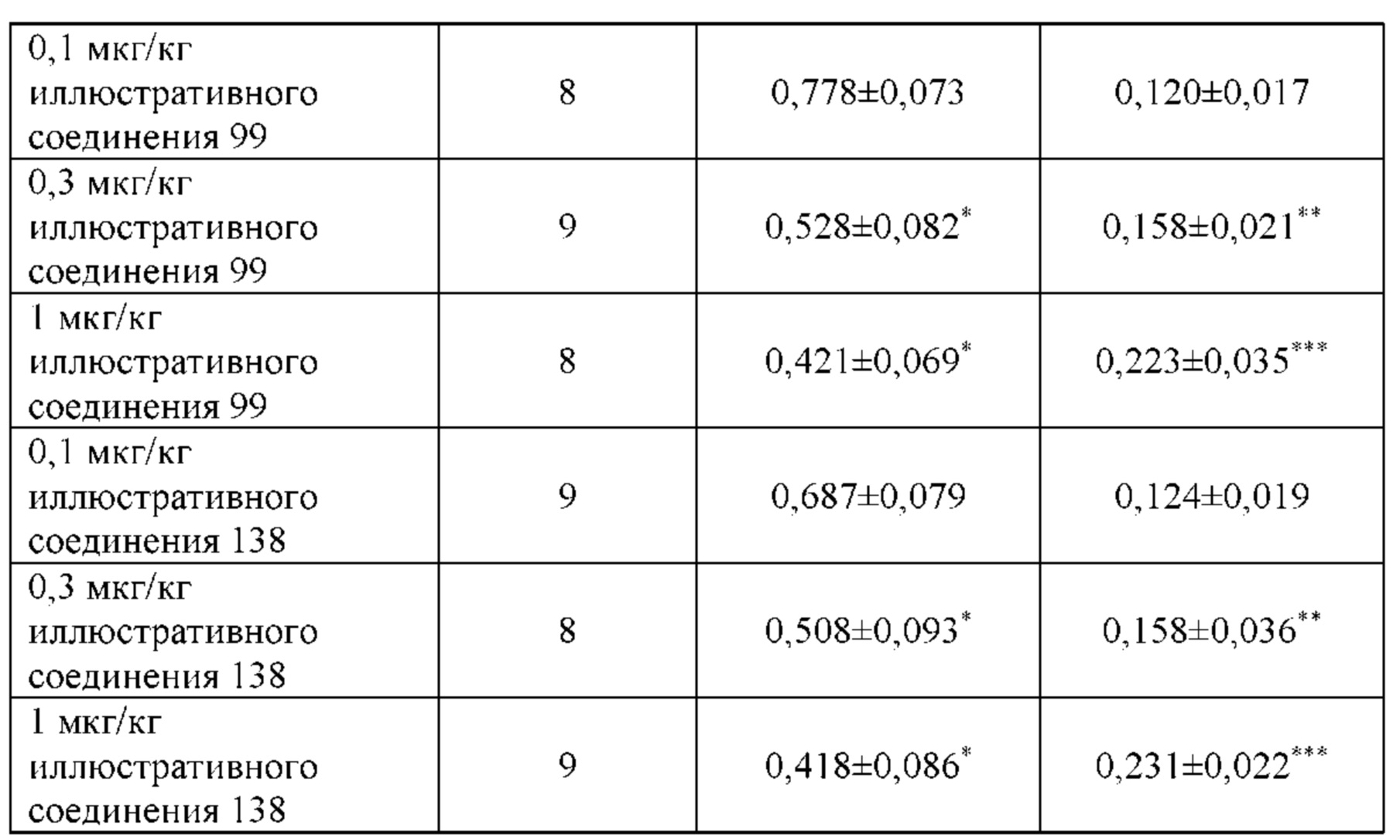
Статистические методы: однофакторный дисперсионный анализ. Сравнение между группами осуществляли с помощью метода Бонферрони. Для сравнения с контрольной группой со средой использовали * Р<0,05, ** Р<0,01, *** Р<0,001.
После внутривенной инъекции Mch в дозе 10 мкг/кг морской свинке сопротивление дыхательных путей повышалось на 320%, динамическая податливость легких снижалась на 71%. В дыхательные пути морским свинкам закапывали 0,1 мкг/кг, 0,3 мкг/кг и 1 мкг/кг соединений [48, 70, 99, 138], соответственно, которые ингибировали повышение сопротивления дыхательных путей и снижение динамической податливости легких дозозависимым образом. Показатели ингибирования соединением 48 повышения сопротивления дыхательных путей в трех группах дозировок составляли 66,2% (р<0,01), 88,3% (р<0,001) и 91,6% (р<0,001), соответственно. Показатели ингибирования снижения динамической податливости легких составляли 32,2% (р>0,05), 48,9% (р<0,001) и 50,1% (р<0,001), соответственно. Показатели ингибирования соединением 70 повышения сопротивления дыхательных путей в трех группах дозировок составляли 69,6% (р<0,01), 86,5% (р<0,001) и 92,1% (р<0,001), соответственно. Показатели ингибирования снижения динамической податливости легких составляли 34,2% (р>0,05), 47,8% (р<0,001) и 49,3% (р<0,001), соответственно. Показатели ингибирования соединением 99 повышения сопротивления дыхательных путей в трех группах дозировок составляли 73,5% (р<0,01), 86,8% (р<0,001) и 91,9% (р<0,001), соответственно. Показатели ингибирования для динамической податливости легких составляли 35,0% (р>0,05), 48,8% (р<0,001) и 49,7% (р<0,001), соответственно. Показатели ингибирования соединением 138 повышения сопротивления дыхательных путей в трех группах дозировок составляли 74,8% (р<0,01), 88,9% (р<0,001) и 92,6% (р<0,001), соответственно. Показатели ингибирования снижения динамической податливости легких составляли 31,0% (р>0,05), 48,2% (р<0,001) и 49,9% (р<0,001), соответственно.
Зависимость время-эффект. После внутривенной инъекции Mch в дозе 10 мкг/кг сопротивление дыхательных путей у морских свинок повышалось на 328%. После того как соединения [48, 70, 99, 138] в дозе 1 мкг/кг закапывали в дыхательные пути морским свинкам в течение 0,25 часа, показатель ингибирования повышения сопротивления дыхательных путей составлял 80% или более. Максимальный показатель ингибирования может достигать 90% или больше спустя 1 час. При продлении срока наблюдения показатель ингибирования повышения сопротивления дыхательных путей спустя 24 часа все еще составлял 85% или более, и присутствовали статистически значимые различия по сравнению с контрольной группой со средой (р<0,01~0,001). Для того чтобы подтвердить правильность результатов, дозировку вводимых соединений [48, 70, 99, 138] снижали до 0,5 мкг/кг. Результаты показали, что после того как соединения [48, 70, 99, 138] в дозе 0,5 мкг/кг закапывали в дыхательные пути морским свинкам в течение 12 часов, показатель ингибирования в отношении сопротивления дыхательных путей составлял 85% или более (р<0,001), спустя 24 часа показатель ингибирования в отношении сопротивления дыхательных путей составлял 75% или более (р<0,01). Не присутствовало значимого отличия в показателе ингибирования в отношении сопротивления дыхательных путей между дозой 1 мкг/кг соединений [48, 70, 99, 138] и их дозой 0,5 мкг/кг спустя 12 часов. Тем не менее, спустя 24 часа не присутствовало значимого отличия в показателе ингибирования в отношении сопротивления дыхательных путей между дозой 1 мкг/кг соединений [48, 70, 99, 138] и их дозой 0,5 мкг/кг (р<0,05). Вышеуказанные результаты говорят о том, что соединения [48, 70, 99, 138] в дозе 1 мкг/кг и в дозе 0,5 мкг/кг являлись длительно действующими трахеальными дилататорами, у которых период действия после введения посредством закапывания в дыхательные пути составлял более чем 24 часа.
Экспериментальный пример 3. Исследование активности и селективности соединений согласно настоящему изобретению в отношении человеческих рекомбинантных β1- и β2-адренорецепторов, экспрессируемых в HEK и СНО клетках
Для того чтобы измерить активность и селективность агонистов в отношении человеческих β1- и β2-адренорецепторов, измеряли накопление цАМФ экспрессируемыми рецепторами в каждой рекомбинантной клеточной линии. цАМФ определяли с помощью радиоиммунологического анализа гомологии, то есть клетки промывали PSB буферным раствором и суспендировали в буферном растворе при комнатной температуре, чтобы выращивать 30000-40000 клеток на лунку. Исследуемое соединение разводили PSB буферным раствором, содержащим 0,1% BSA и исследовали в 11 последовательно уменьшающихся концентрациях в диапазоне от 100 мкМ до 0,1 пМ. Для анализа β1-дренорецептора 10 нМ ICI118551 добавляли для блокирования экспрессии β2-дренорецептора в HEK293 клетках. После того как соединение добавляли к суспендированным клеткам, планшет для сцинтилляционного анализа инкубировали при 37°С в течение 10 минут, реакцию завершали с использованием ледяного раствора буфера для выявления и радиоактивность в нем измеряли после хранения при 4°С в течение ночи.
Агонистическую активность (значение pEC50) анализировали с помощью регрессии с применением пакета программного обеспечения с моделью S-типа для изучения зависимости доза-эффект (GraphPad Software, Сан-Диего, Калифорния). Селективность рассчитывали на основании соотношения измеренного цАМФ β1- и β2-адренорецепторов, которое выражено как селективность β1/β2=(ЕС50(β1)/EC50(β2)).
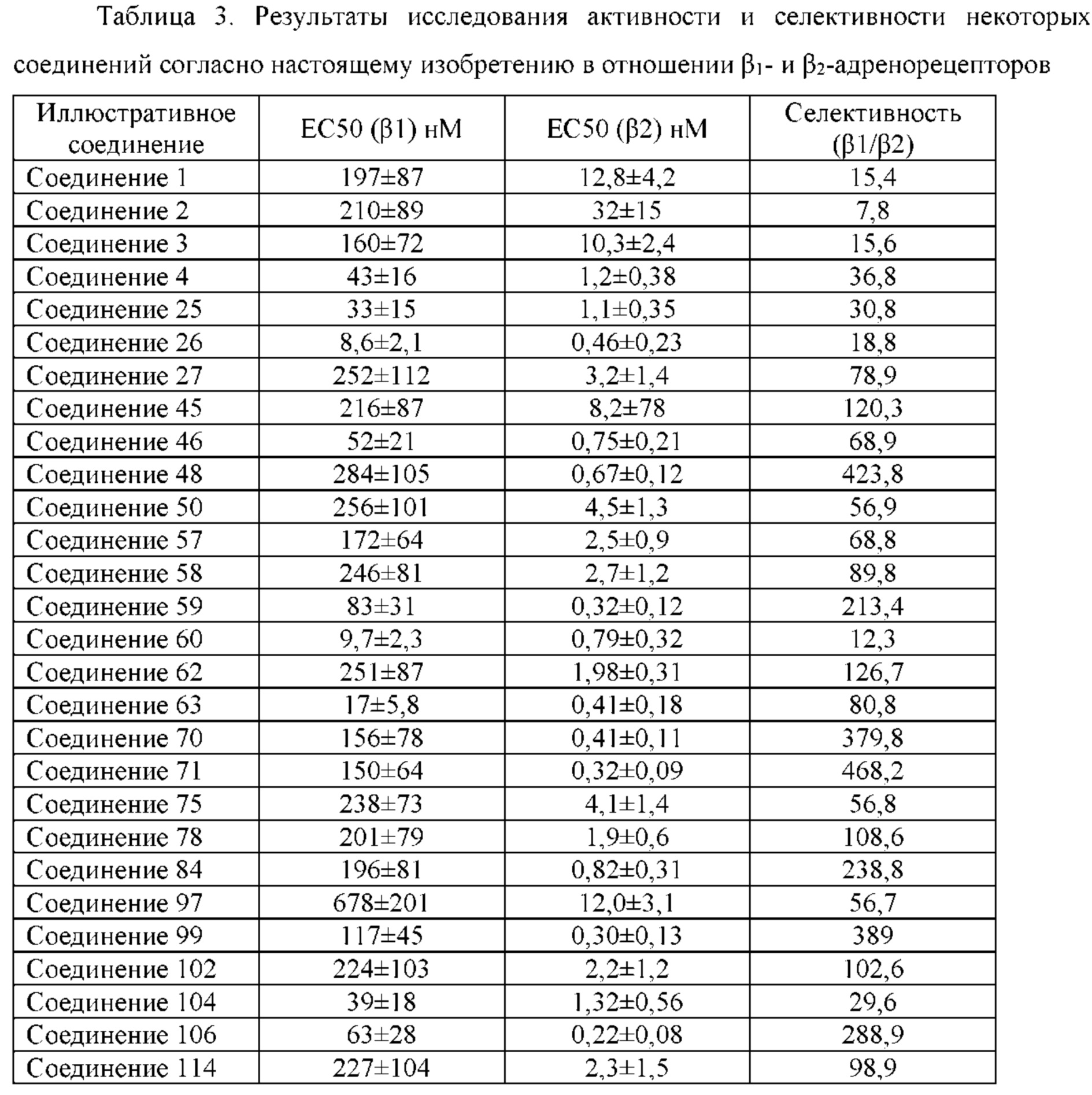
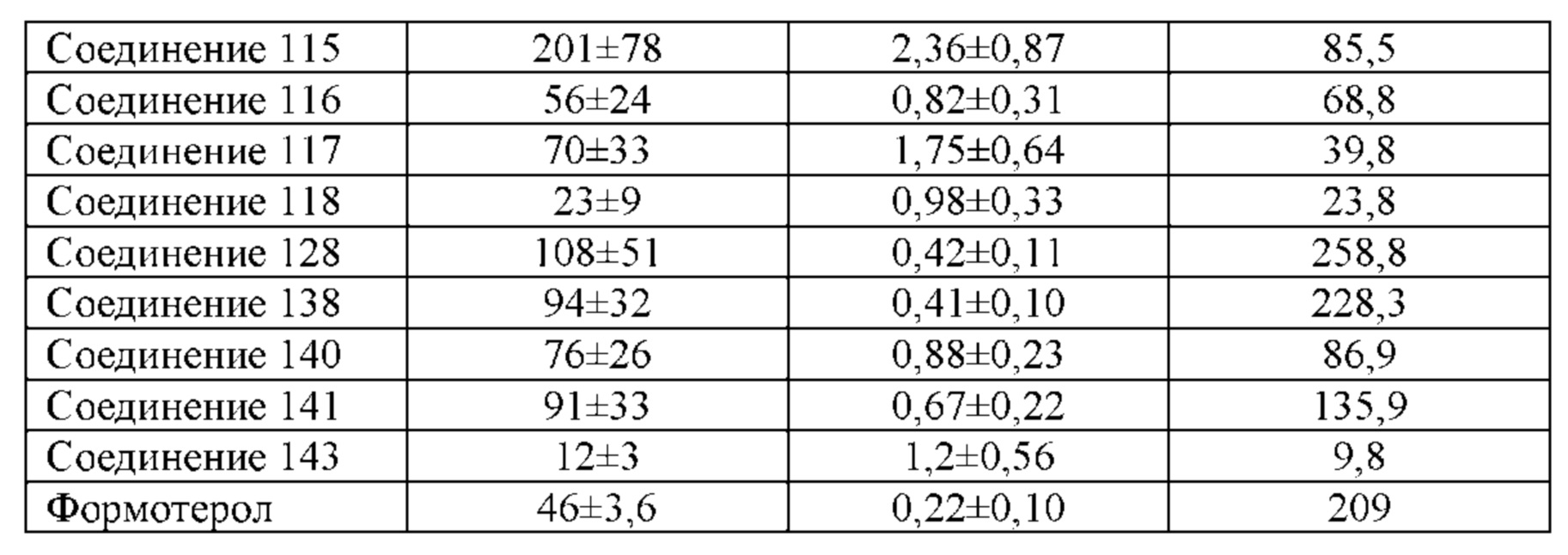
Результат анализа. Соединения согласно настоящему изобретению характеризуются хорошей агонистической активностью и функциональной селективностью в отношении β2-адренорецептора.
Экспериментальный пример 4. Исследование по определению активности и селективности in vitro у соединений согласно настоящему изобретению в отношении подтипов M1, М2, М3 человеческих рекомбинантных рецепторов, экспрессируемых в СНО клетках
Трансфицированные CHOm1, CHOm2 и CHOm3 клетки культивировали в среде DMEM (содержащей 15% фетальной бычьей сыворотки, 4 мМ L-глутамин, 1% заменимых аминокислот и 1% антибиотика/противогрибкового средства) в инкубаторе при 37°С и 5% СО2, соответственно.
Как упоминается в литературном источнике [Нu Ya 'er, Shi Ju, Xia Zongqin; Some pharmacological and biological properties of CHOm2 cells transfected with M2 receptor cDNA [J]. Nuclear Science и Technology, 1999, 11(22): 642-646], когда клетки в колбе с культурой росли и пролиферировали с образованием монослоя клеток, и дно колбы было покрыто приблизительно на 90% (приблизительно через 48 часов после инокуляции), среду сливали и дважды промывали PBS буферным раствором (рН 7,4). Затем клетки соскабливали с ледяным фосфатно-буферным раствором (рН 7,7, содержащим 5 ммоль/л MgCl2). Собранные клетки гомогенизировали с использованием стеклянного гомогенизатора с тефлоновым пестиком, гомогенат центрифугировали при 20000r × 20 мин. при низкой температуре и осадок гомогенизировали с реакционным буферным раствором с образованием суспензии мембранных белков. Количество белка, добавляемого в каждую из реакционных пробирок, составляло: m1 (приблизительно 0,05 мг), m2(0,05 мг), m3 (0,1 мг), соответственно, концентрация [3H]-QNB составляла 0,1-2,16 нмоль/л. 1 мкмоль/л атропина добавляли в пробирку для неспецифического связывания. Общий объем реакционной смеси составлял 300 мкл. Затем обеспечивали реакцию полученного в результате раствора при 25°С в течение 5 часов, и гасили ее ледяным реакционным буферным раствором, и собирали на мембрану из стекловолокна с применением мультиголовочного устройства для сбора из ячеек. После высушивания при 80°С лист фильтрующей мембраны помещали в пузырек для жидкостной сцинтилляции с последующим добавлением 5 мл жидкого сцинтилляционного средства, а затем поддерживали в темном месте в течение ночи, причем ерш (количество импульсов счета в минуту) измеряли с помощью жидкостного сцинтилляционного спектрометра. Значения Bmax и Kd рассчитывали с помощью программного обеспечения Graphpad prism.
3H-QNB, который является неселективным в отношении М-рецептора, добавляли в каждую экспериментальную пробирку в качестве метящего лиганда, и конечная концентрация в каждой пробирке составляла m1, m3 (1,042 нмоль/л), m2 (1,81 нмоль/л). Одновременно добавляли различные концентрации бромида тиотропия, бромида ипратропия и каждого исследуемого соединения, конечная концентрация составляла 10-10~10-4 моль/л, в общей сложности 11 дозировок. Добавляли одинаковое количество мембранных белков (такое же как в эксперименте с насыщением, упомянутом выше). Общий объем реакционной смеси составлял 300 мкл, и осуществляли реакцию конкурентного связывания. Еще одну пробирку для неспецифического связывания использовали для измерения неспецифического связывания с большим количеством атропина (конечная концентрация 1 мкмоль/л). После того как реакцию осуществляли при 25°С в течение 5 часов и завершали добавлением ледяного реакционного буферного раствора, продукт реакции собирали на фильтрующей мембране из стекловолокна с применением мультиголовочного устройства для сбора из ячеек. После высушивания при 80°С фильтрующую мембрану помещали в пузырек для жидкостной сцинтилляции, добавляли 5 мл жидкого сцинтилляционного средства и поддерживали в темном месте в течение ночи. Cpm (количество импульсов счета в минуту) измеряли с использованием жидкостного сцинтилляционного счетчика. Значения Ki у каждого соединения в отношении трех подтипов М-рецептора рассчитывали с помощью программного обеспечения Graphpad prism. Селективность в отношении подтипа М-рецептора сравнивали у бромида тиотропия, бромида ипратропия и каждого исследуемого соединения.

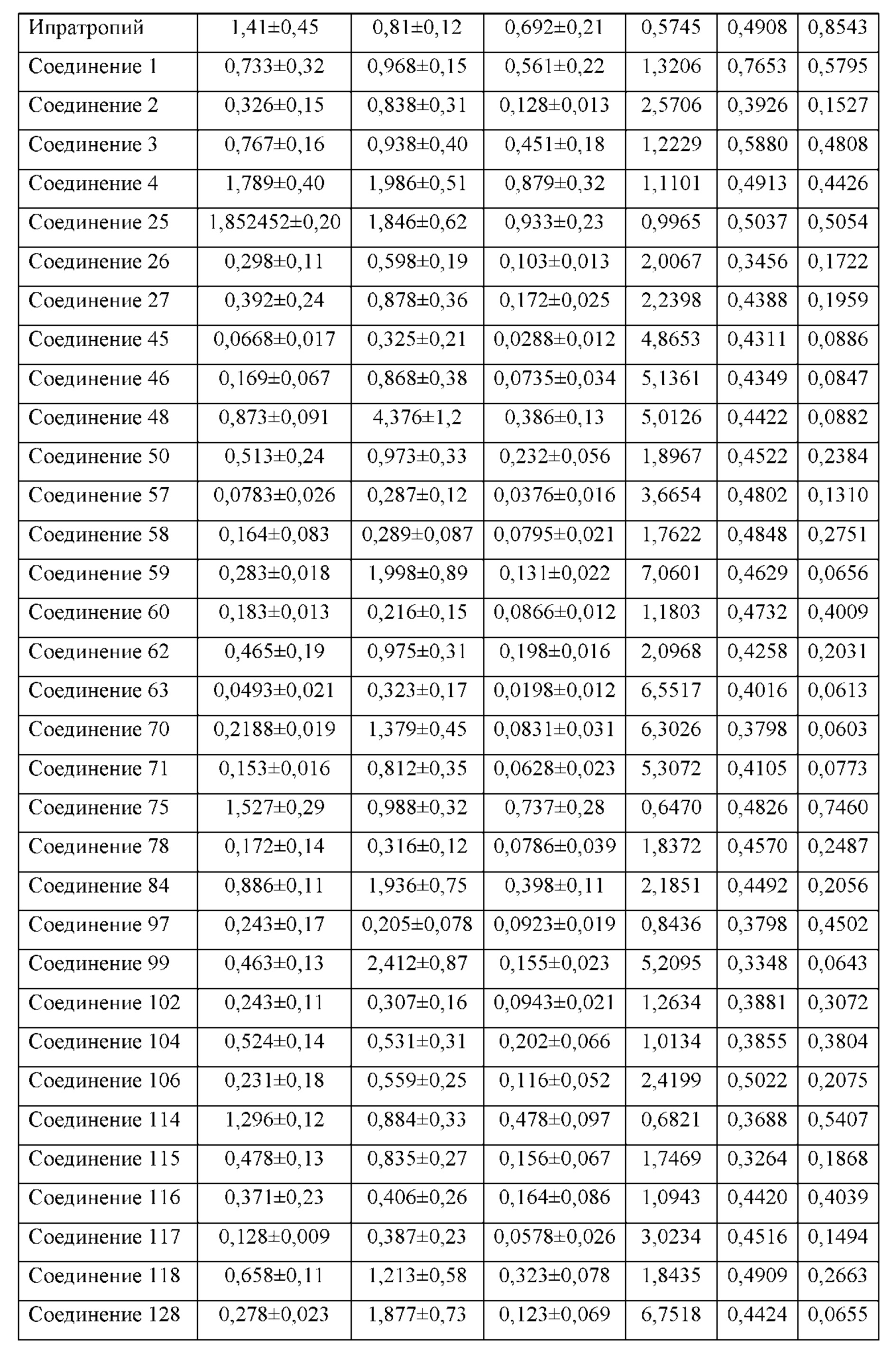
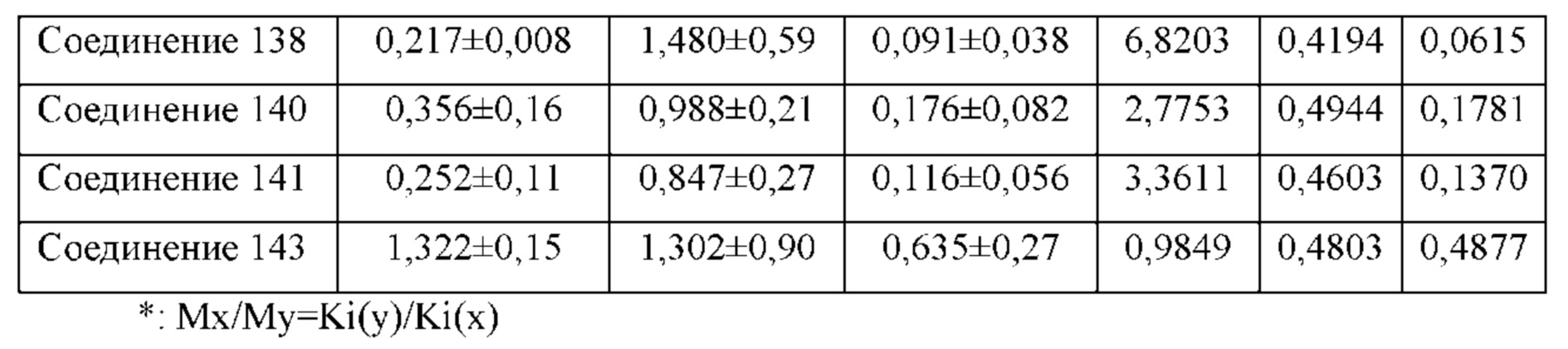
Результаты эксперимента показывают, что соединения согласно настоящему изобретению характеризуются наиболее высокой селективностью в отношении М3-рецептора, затем следует селективность в отношении M1-рецептора, и наиболее низкая селективность в отношении М2-рецептора, и соединения оказывают сильное воздействие на М3- и M1-рецепторы. По сравнению с предшествующим уровнем техники настоящее изобретение обладает очевидными преимуществами в лечении ринита, гиперчувствительности дыхательных путей, хронического бронхита, COPD и других заболеваний.
Экспериментальный пример 5. Активность в отношении МАВА, М-рецептора и β2-адренорецептора определяли в выделенной трахее морской свинки
(1) Эффект сокращения гладких мышц трахеи морской свинки, индуцированной иллюстративными соединениями, проявляющими антагонизм в отношении карбахола (CCh)
Получение выделенного образца гладких мышц трахеи морской свинки: также как и в экспериментальном примере 1.
Способ введения. После того как пластинку трахеи стабилизировали, добавляли карбахол с конечной концентрацией 3×10-6 ммоль/л и введение начинали, когда величина давления в пластинке трахеи повышалась до стабильного плато. Экспериментальных животных делили на холостую группу и группу введения каждого исследуемого образца. Раствор в DMSO, приготовленный из K-Н раствора и 50 мкл раствора каждого исследуемого образца, добавляли в каждой группе, соответственно. В группе введения исследуемого образца добавляли 10-9, 0,3×10-8, 10-8, 0,3×10-7, 10-7, 0,3×10-6 и 10-6 моль/л исследуемого соединения и контрольного вещества в совокупной концентрации. В холостой группе добавляли соответствующий разведенный раствор в DMSO. Medlab использовали для записи степени расслабления гладких мышц в пластинке трахеи. Наконец, изопротеренол в конечной концентрации 10-5 моль/л добавляли в качестве 100% расслабления. Результаты показаны в таблице 5.
(2) Блокирующий эффект иллюстративных соединений в отношении М-рецептора в гладких мышцах трахеи морской свинки
Получение выделенного образца гладких мышц трахеи морской свинки: также как и в экспериментальном примере 1.
Способ введения. После того как пластинку трахеи стабилизировали, раствор гидрохлорида пропанолола в конечной концентрации 10-5 моль/л добавляли для блокирования β-рецептора в течение 10 мин., добавляли карбахол в конечной концентрации 3×10-6 моль/л (наблюдение антагонизма соединения в отношении М-рецептора после того, как пропанолол блокирует β-рецептор) и введение начинали, когда величина давления в пластинке трахеи повышалась до стабильного плато. Эксперименты делили на холостую группу, холостую группу + пропанолол, группу введения каждого исследуемого образца и группу введения каждого исследуемого образца + пропанолол. Раствор в DMSO, приготовленный из K-Н раствора и 50 мкл раствора исследуемого образца, добавляли в каждой из холостой группы и группы введения каждого исследуемого образца, соответственно. В холостой группе + пропанолол и группе введения каждого исследуемого образца + пропанолол добавляли 50 мкл раствора пропанолола в течение 10 мин., соответственно, а затем добавляли раствор в DMSO, приготовленный из K-Н раствора и 50 мкл раствора исследуемого образца. В группе введения исследуемого образца добавляли соединения, подлежащие исследованию, в конечных концентрациях 10-9, 0,3×10-8, 10-8, 0,3×10-7, 10-7, 0,3×10-6 и 10-6 моль/л в совокупной концентрации и в холостой группе добавляли соответствующий разведенный раствор в DMSO. В группе введения каждого исследуемого образца + пропанолол добавляли пропанолол в течение 10 мин., а затем добавляли соединения, подлежащие исследованию, в конечных концентрациях 10-9, 0,3×10-8, 10-8, 0,3×10-7, 10-7, 0,3×10-6 и 10-6 моль/л в совокупной концентрации. После того как в холостой группе + пропанолол добавляли пропанолол в течение 10 мин., добавляли соответствующий разведенный раствор в DMSO. Medlab использовали для записи степени расслабления гладких мышц трахеи, наконец, добавляли нитропруссид натрия в конечной концентрации 2×10-4 моль/л в качестве 100% расслабления. Результаты показаны в таблице 5.
(3) Антагонизм иллюстративных соединений в отношении β2-рецептора в гладких мышцах трахеи морской свинки
Получение выделенного образца гладких мышц трахеи морской свинки: также как и в экспериментальном примере 1.
Способ введения. После того как пластинки трахеи стабилизировали, раствор гистамина в конечной концентрации 3×10-5 моль/л добавляли в течение 10 мин., добавляли карбахол в конечной концентрации 3×10-6 моль/л (наблюдение антагонизма в отношении β-рецептора) и введение начинали, когда величина давления в пластинке трахеи повышалась до стабильного плато. Эксперименты делили на холостую группу, холостую группу + гистамин, группу введения каждого исследуемого образца и группу введения каждого исследуемого образца + гистамин. Раствор в DMSO, приготовленный из K-Н раствора и 50 мкл раствора исследуемого образца, добавляли в каждой из холостой группы и группы введения каждого исследуемого образца, соответственно. В холостой группе + гистамин и группе введения каждого исследуемого образца + гистамин добавляли 50 мкл раствора гистамина в течение 10 мин., соответственно, затем добавляли раствор в DMSO, приготовленный из K-Н раствора и 50 мкл раствора исследуемого образца. В группе введения исследуемого образца добавляли соединения, подлежащие исследованию, в конечных концентрациях 10-9, 0,3×10-8, 10-8, 0,3×10-7, 10-7, 0,3×10-6 и 10-6 моль/л в совокупной концентрации и в холостой группе добавляли соответствующий разведенный раствор в DMSO. В группе введения каждого исследуемого образца + гистамин добавляли гистамин в течение 10 мин., а затем добавляли исследуемые соединения в конечных концентрациях 10-9, 0,3×10-8, 10-8, 0,3×10-7, 10-7, 0,3×10-6 и 10-6 моль/л в совокупной концентрации. После того как в холостой группе + гистамин добавляли гистамин в течение 10 мин., добавляли соответствующий разведенный раствор в DMSO. Medlab использовали для записи степени расслабления гладких мышц в пластинке трахеи. Наконец, теофиллин в конечной концентрации 2×10-4 моль/л добавляли в качестве 100% расслабления. Результаты показаны в таблице 5.
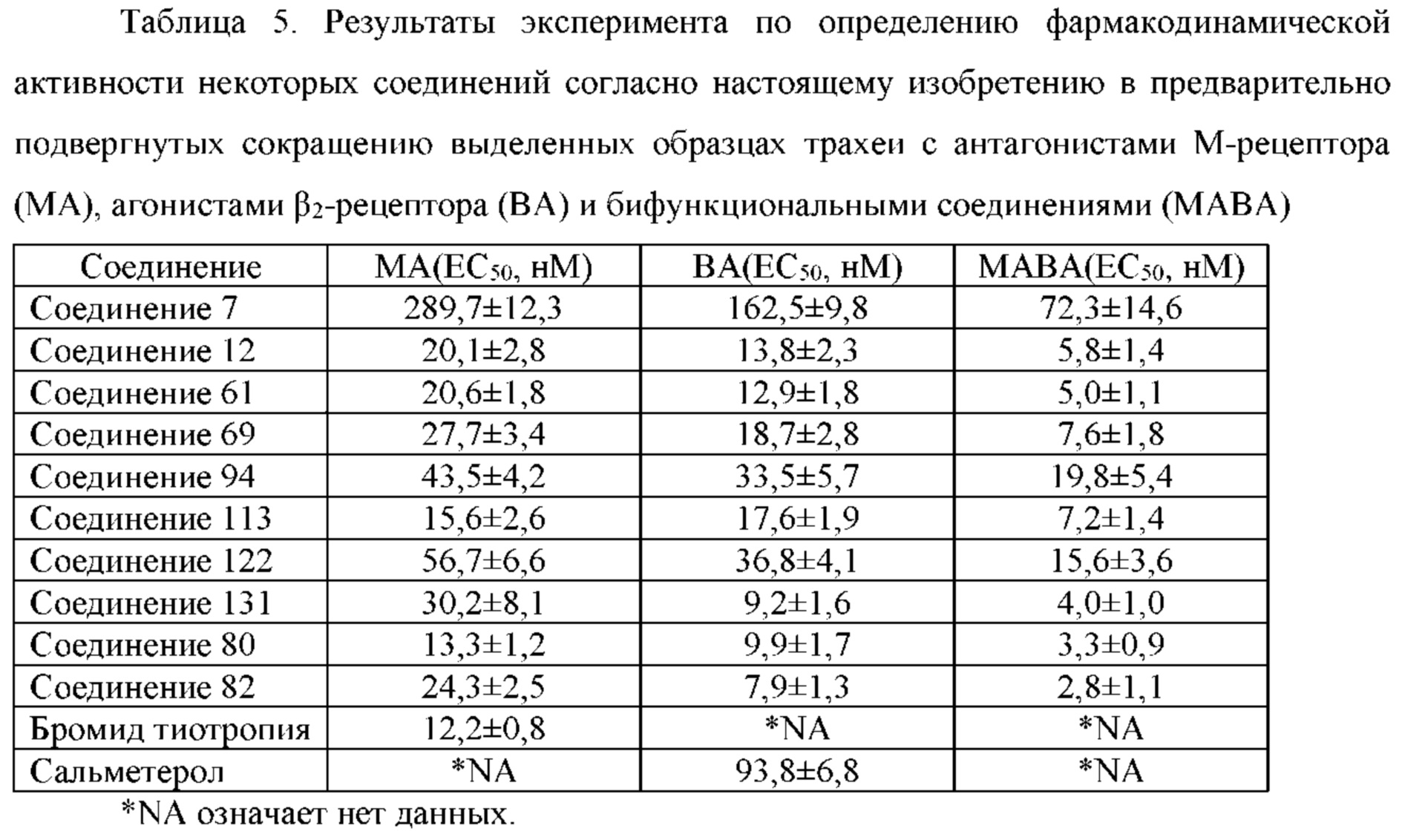
Результаты показывают, что соединения согласно настоящему изобретению одновременно обладают функцией сильного блокирования М-рецептора и функцией, агостической в отношении β-рецептора, и эти две функции у большого количества соединений имеют степень соответствия МА:ВА, которая, по существу, достигает 1-1,5:1. Степень соответствия у иллюстративного соединения 113 практически достигает 1:1.
Экспериментальный пример 6. Исследование острой токсичности соединений согласно настоящему изобретению у собак породы бигль при однократном скармливании
Исследуемые соединения:
Иллюстративные соединения 7, 12, 61, 69, 80, 82, 94, 113, 122 и 131 согласно настоящему изобретению
Контрольные соединения:
(1) Соединение из примера 1 в WO 2010126025: трифторацетат 4-({[5-(2-{[4-({[(2R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил]амино}метил)фенил]карбамоил}этил)-2-фенилфенил]карбамоил}окси)-1,1-диметилпиперидин-1-ония (полученное согласно способу из литературы).
(2) Соединение из примера 1 в WO 2010004517: 5-[(1R)-2-({9-[4-({3-[(R)-циклогексил(гидрокси)бензил)-1Н-1,2,4-триазол-1-ил}метил)пиперидин-1-ил]нонил}амино)-1-гидроксиэтил]-8-гидроксихинолин-2(1Н)-он (полученное согласно способу из литературы).
Способ исследования.
Собак породы бигль произвольно делили на две группы, половина самцов и половина самок, и им вводили в виде внутривенной инъекции (iv) 7,5% этанольный раствор соединений согласно настоящему изобретению и контрольного соединения. Каждое соединение использовали в двух группах дозировки 0,01 мг/кг и 0,10 мг/кг. Изменения частоты сердечных сокращений наблюдали после введения (30 мин. - 4 часа после введения), и результаты показаны в таблице 6.
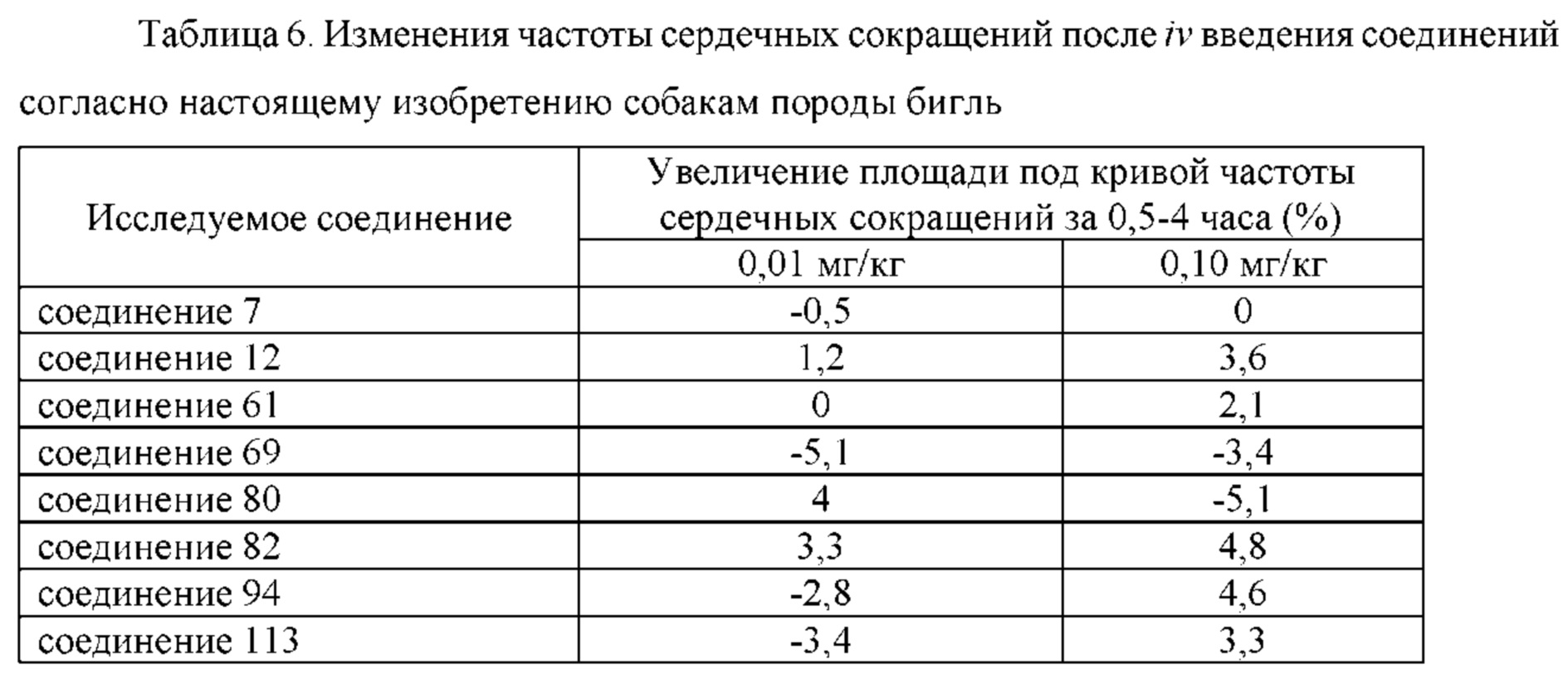
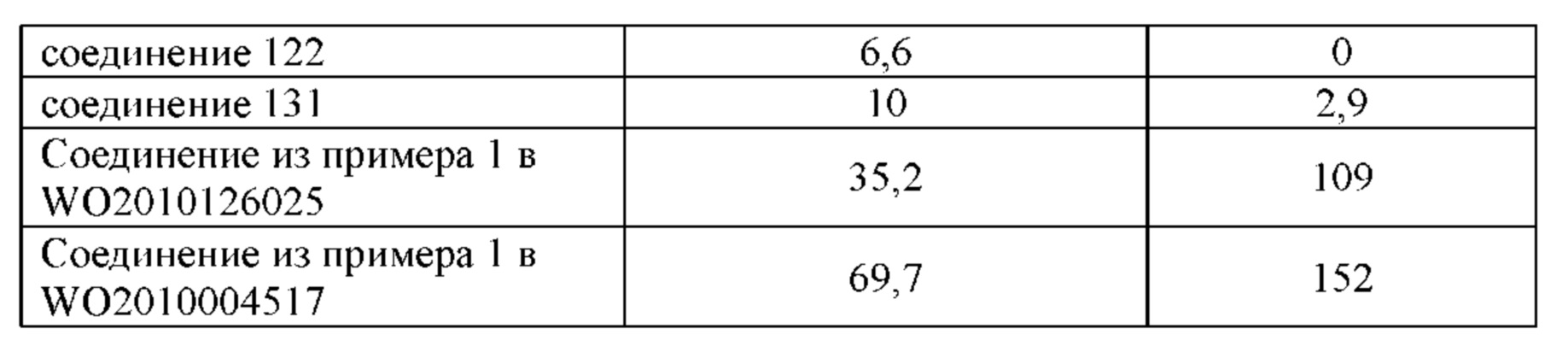
Как можно увидеть из таблицы 6, частота сердечных сокращений у собак породы бигль не изменялась существенно после iv введения соединений согласно настоящему изобретению в дозировке 0,1 мг/кг, в то время как контрольное соединение существенно повышает частоту сердечных сокращений у собак породы бигль в дозе 0,01 мг/кг. Таким образом, соединения согласно настоящему изобретению характеризуются более низкой токсичностью и побочными эффектами.
Экспериментальный пример 7. Исследования стабильности соединений согласно настоящему изобретению в печеночных микросомах и гомогенате легких in vitro
1. Исследуемое соединение в тесте с печеночными микросомами
Иллюстративные соединения 7, 12, 61, 69, 80, 82, 94, 113, 122 и 131 согласно настоящему изобретению
Контрольное соединение:
(1) соединение из примера 1 в WO 2010126025, (2) соединение из примера 1 в WO 2010004517.
Способ проведения экспериментов: иллюстративные соединения согласно настоящему изобретению и контрольное соединение соответственно инкубировали с печеночными микросомами SD крыс и собак породы бигль при 37°С в течение 0, 2, 5, 10, 15, 20, 30 мин. и образцы анализировали соответственно с помощью LC-MS/MS, когда реакцию завершали. Соотношение площади у образца в момент времени 0 мин. принимали за 100%, и оставшийся процент соединения после инкубирования в различные моменты времени получали в каждый момент времени посредством сравнения с образцом. С натуральным логарифмом этого процента на оси ординат и временем на оси абсцисс строили график разброса данных, моделируя прямую линию с отрицательным наклоном k. Параметры рассчитывали согласно формуле.
t1/2 (мин.)=0,693/k
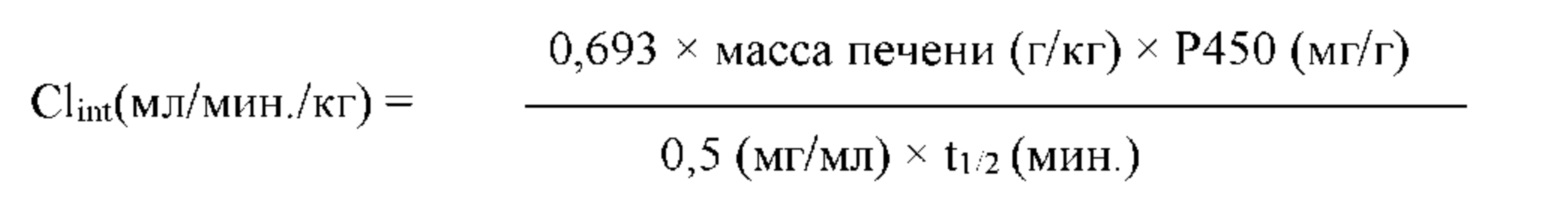
Масса печени означает массу печени (г) на килограмм массы тела, и Р450 означает количество (мг) фермента Р450 на грамм печени.
Значения для различных образцов показаны в таблице ниже.

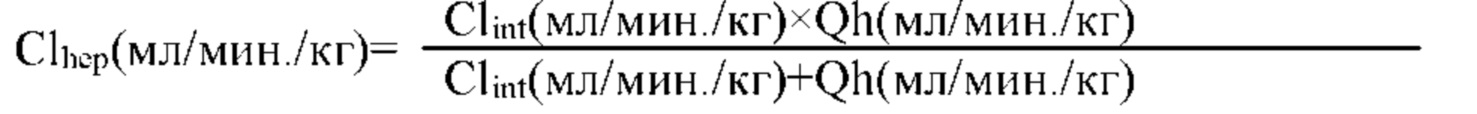
Qh представляет собой печеночный кровоток, и значения для различных видов представлены в таблице выше.
Показатель выведения из печени (ERh) рассчитывали согласно ERh=Clhep/Qh, и результаты показаны в таблице 7.
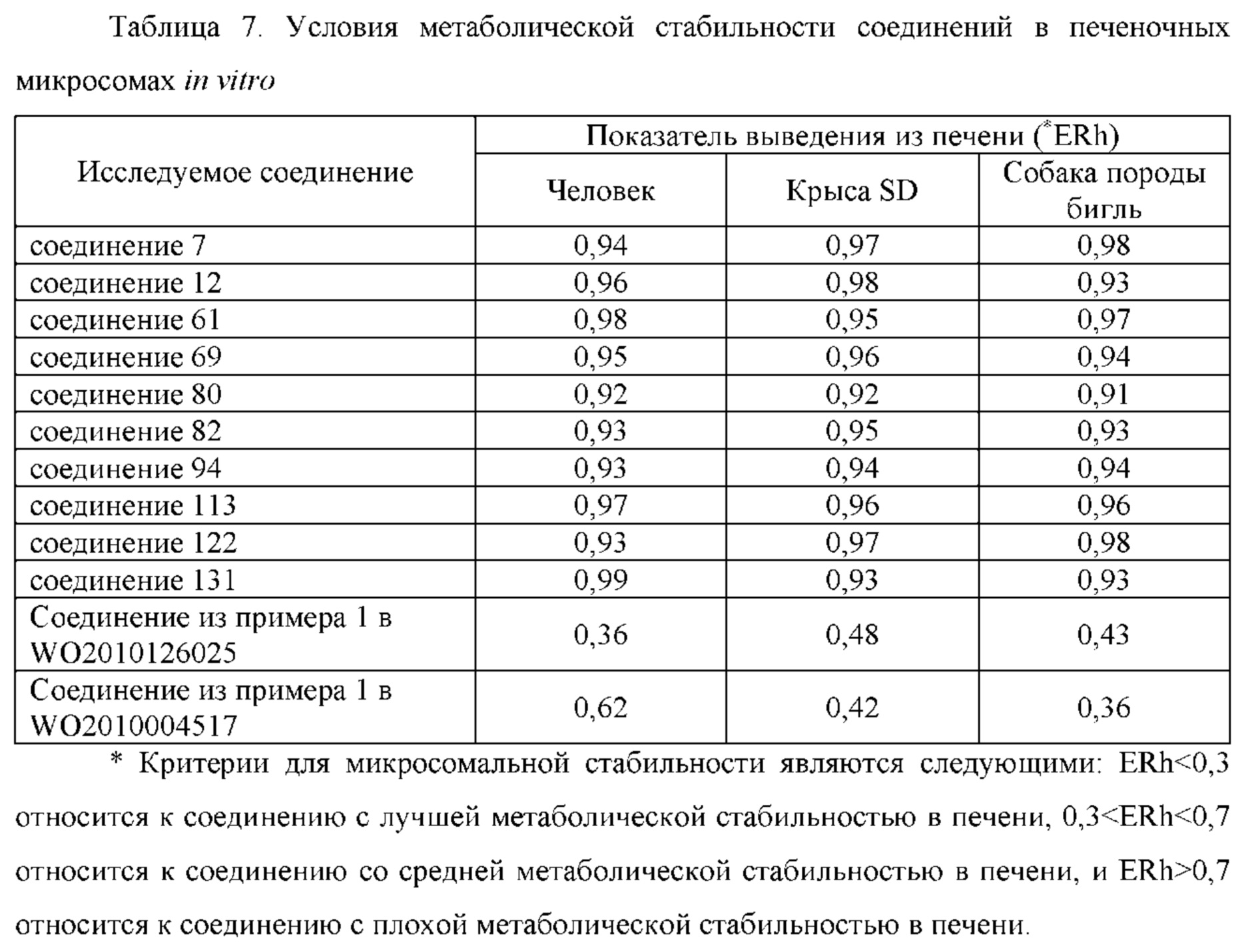
Как показано в таблице 7, соединения согласно настоящему изобретению характеризуются плохой метаболической стабильностью в печеночных микросомах SD крыс и собак породы бигль, метаболизируются быстрее и трудно накапливаются in vivo, в то время как контрольное соединение характеризуется лучшей метаболической стабильностью и легче накапливается in vivo.
2. Исследуемые соединения в гомогенате легких
Иллюстративные соединения 7, 12, 61, 69, 80, 82, 94, 113, 122 и 131 согласно настоящему изобретению
Соединения согласно настоящему изобретению соответственно инкубировали с гомогенатом легких собак породы бигль в течение 8 часов на водяной бане при 37°С, и образец анализировали с помощью LC-MS/MS, когда реакцию завершали. Соотношение площади у образца в момент времени 0 мин. принимали за 100%, и оставшийся процент соединений после инкубирования в течение 8 часов получали посредством сравнения с образцом после 8 часов. Результаты показаны в таблице 8.
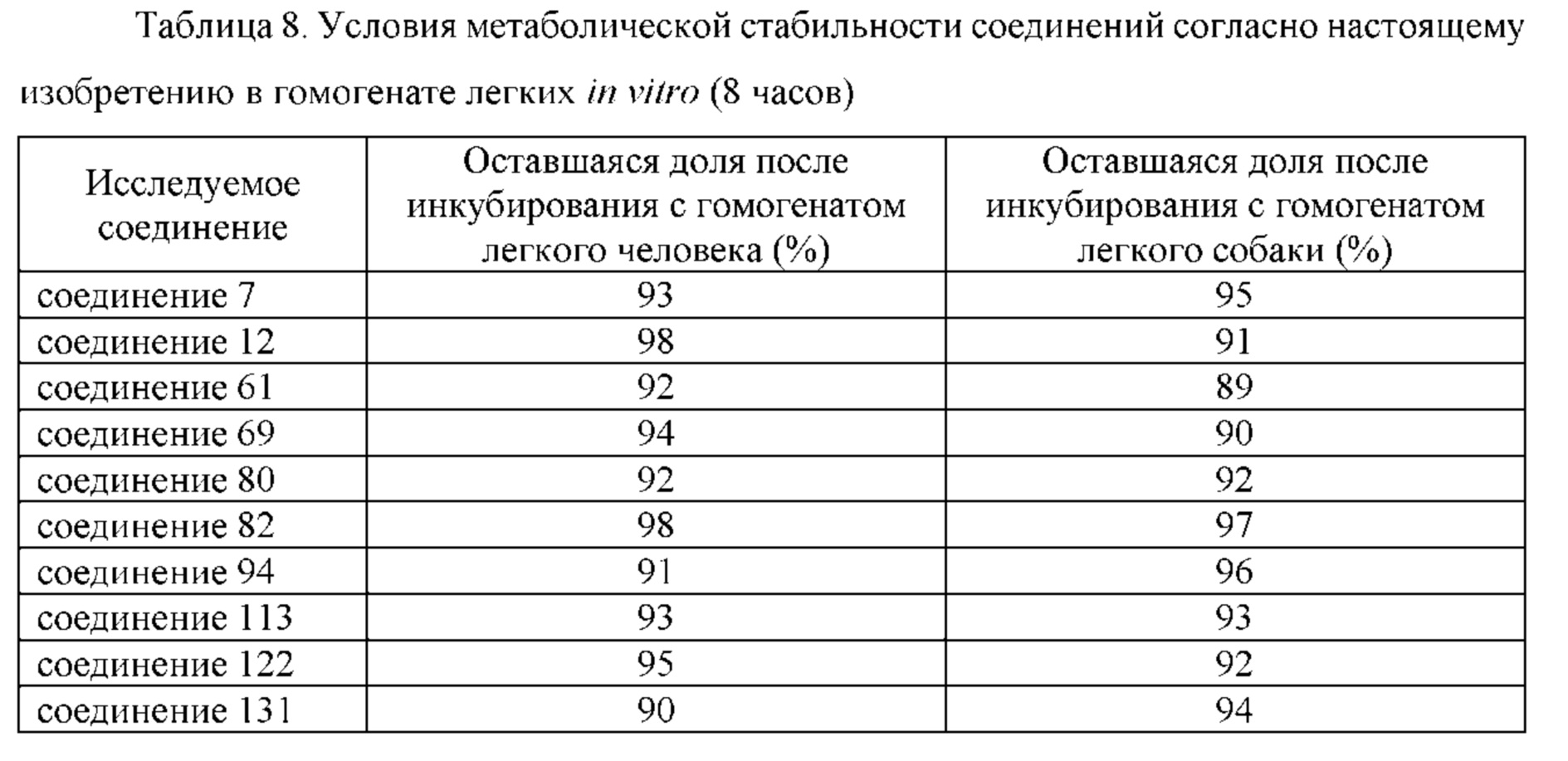
Вывод. Вышеуказанные исследования демонстрируют, что соединения согласно настоящему изобретению являются стабильными в легких и, следовательно, характеризуются продолжительным эффектом в легком. Как можно увидеть из таблицы 7, соединения согласно настоящему изобретению характеризуются плохой метаболической стабильностью в печеночных микросомах SD крыс и собак породы бигль, метаболизируются быстрее и трудно накапливаются in vivo, в то время как контрольное соединение характеризуется лучшей метаболической стабильностью и легче накапливается. Это показывает, что соединения согласно настоящему изобретению не могут накапливаться в эффективной дозе для токсичного действия.
Пример получения 1
Препарат в виде сухого порошка для ингаляционного введения получали следующим способом:
Каждый компонент и его количество в композиции составляли:
Соединение 59 по настоящему изобретению 0,20 мг
Лактоза 30 мг
Соединение по настоящему изобретению тонко измельчали, достаточным образом смешивали с тонкоизмельченной лактозой и смесь загружали в желатиновый всасывающий картридж и вводили при помощи порошкового ингалятора.
Пример получения 2
Препарат в виде сухого порошка для применения в устройстве для ингаляции для сухого порошка получали следующим способом:
Соединение 70 по настоящему изобретению тонко измельчали и равномерно смешивали с тонкоизмельченной лактозой с образованием препарата. Комбинированное соотношение препарата составляло 1:200 и композицию препарата загружали в устройство для ингаляции для сухого порошка, способное переносить от 10 мкг до 100 мкг соединения по настоящему изобретению.
Пример получения 3
Препарат в виде сухого порошка для применения для ингаляционного введения в ингаляторе отмеренных доз получали следующим способом:
10 г тонкоизмельченного соединения 99 по настоящему изобретению с частицами среднего размера менее чем 10 мкм диспергировали в 200 мл смягченном водном растворе, содержащем 0,2 г лецитина, с образованием суспензии, содержащей 5 масс. % соединения по настоящему изобретению и 0,1 масс. % лецитина, и суспензию сушили распылением с образованием частиц среднего размера менее чем 1,5 мкм. Частицы загружали в 1,1,1,2-тетрафторэтановые картриджи под давлением.
Пример получения 4
Фармацевтическую композицию для применения в ингаляторе отмеренных доз получали следующим способом:
5 г тонкоизмельченного соединения 138 по настоящему изобретению с частицами среднего размера менее чем 10 мкм диспергировали в 100 мл смягченном водном коллоидном растворе, содержащем 0,5 г трегалозы и 0,5 г лецитина, с образованием суспензии, содержащей 5 масс. % соединения по настоящему изобретению, 0,5% трегалозы и 0,5 масс. % лецитина. Суспензию сушили распылением с образованием частиц среднего размера менее чем 1,5 мкм. Частицы загружали в 1,1,1,2-тетрафторэтановые картриджи под давлением.
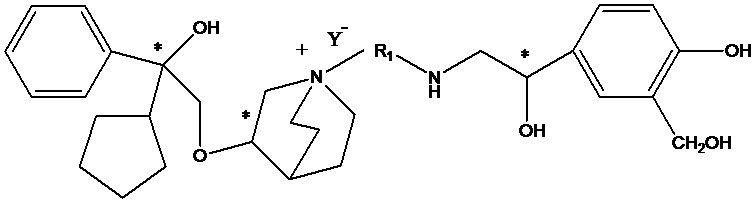
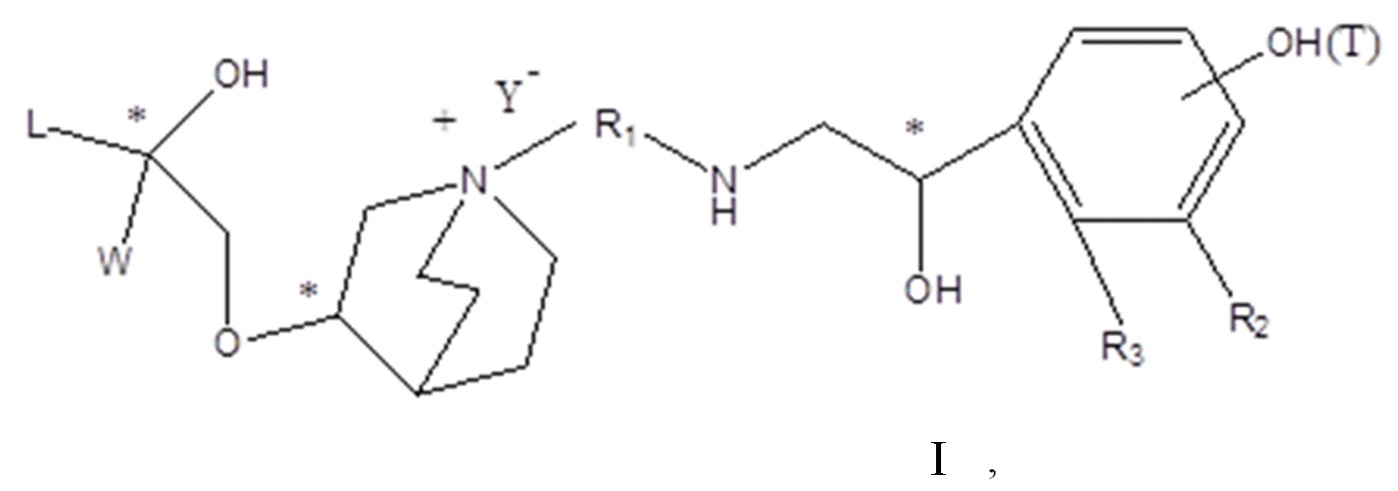
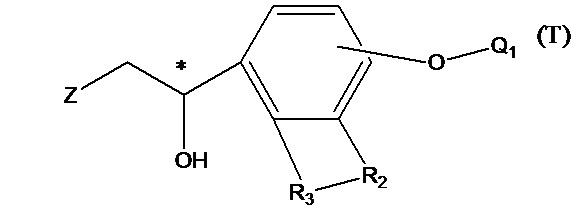
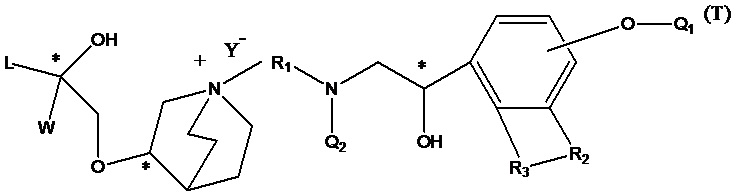
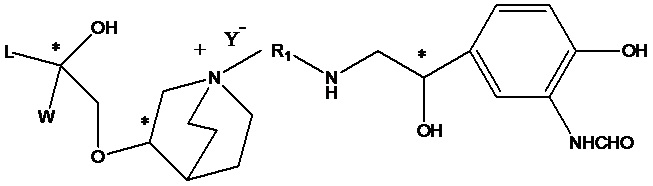
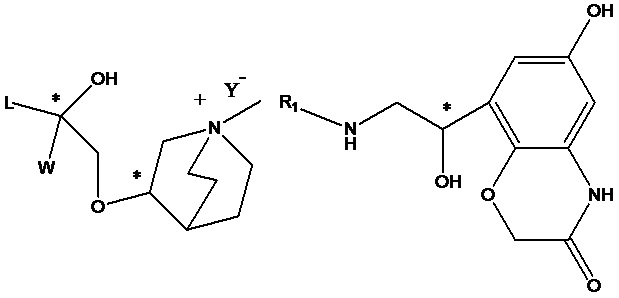
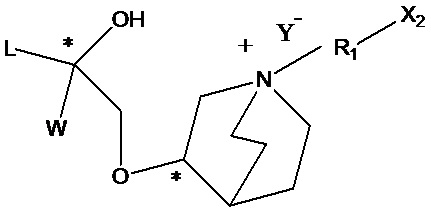
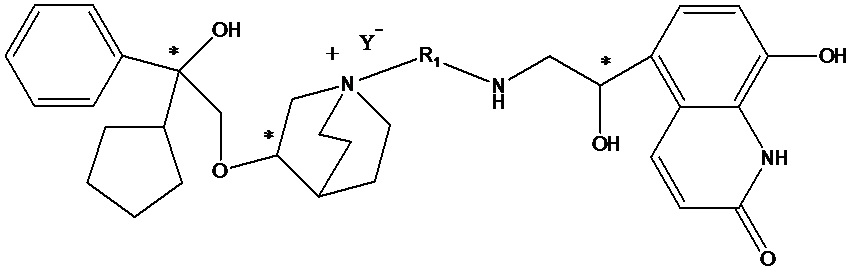
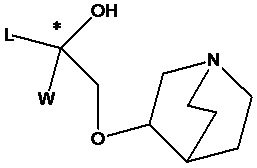
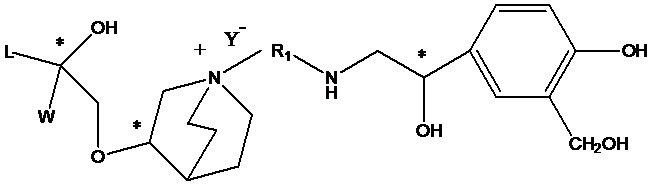
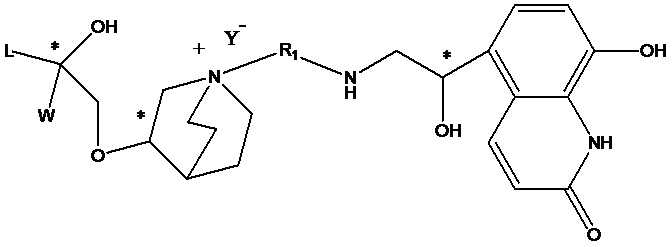
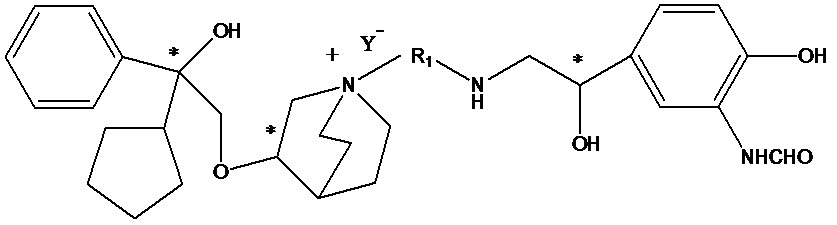
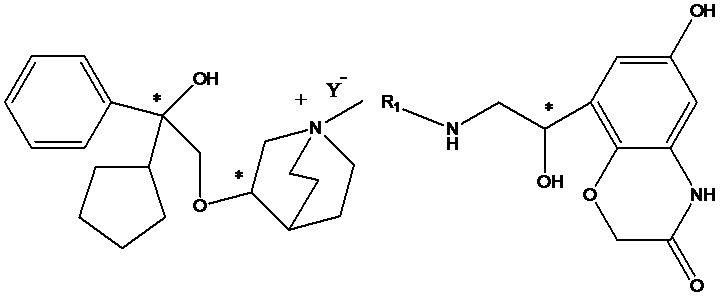
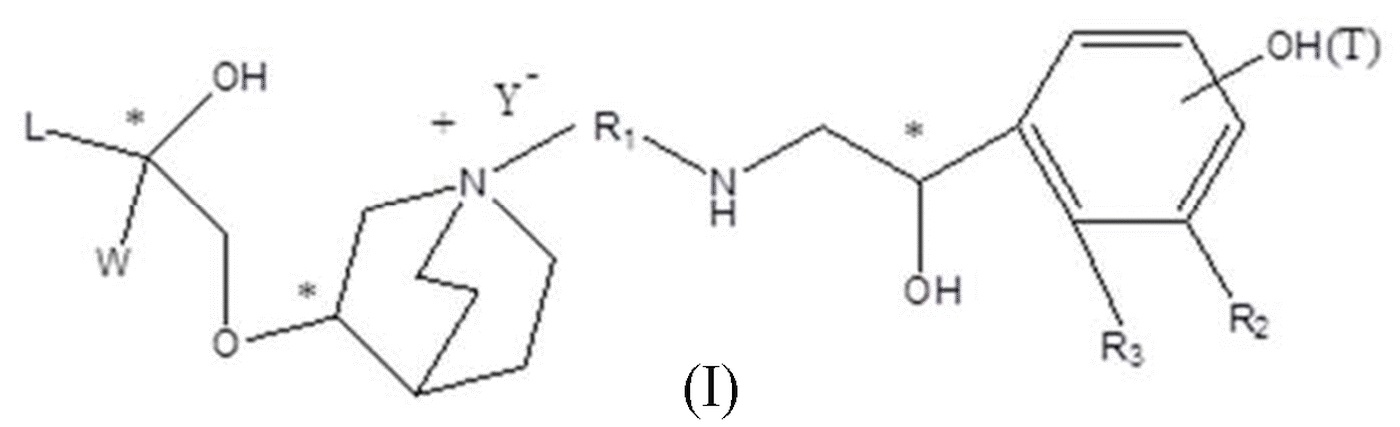
Способ получения ибрутиниба
Способ и оборудование для обнаружения услуг на основе сетевых функций

