Результат интеллектуальной деятельности: Производные хромена в качестве ингибиторов фосфоинозитид-3-киназ
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, ингибирующим фосфоинозитид-3-киназы (в дальнейшем PI3K); в частности, изобретение относится к соединениям, представляющим собой производные хромена, способам получения таких соединений, содержащим их фармацевтическим композициям и их терапевтическому применению.
Более конкретно, соединения по изобретению представляют собой ингибиторы активности или функции класса I PI3K; точнее, они представляют собой ингибиторы активности или функции изоформ PI3Kα, PI3Kβ, PI3Kδ и/или PI3Kγ класса I PI3K.
Следовательно, соединения по изобретению могут быть полезными в лечении многих расстройств, ассоциированных с механизмами ферментов PI3K, таких как респираторные заболевания, включая астму, хроническую обструктивную болезнь легких (COPD) и кашель; аллергические заболевания, включая аллергический ринит и атопический дерматит; аутоиммунные заболевания, включая ревматоидный артрит и рассеянный склероз; воспалительные расстройства, включая воспалительное заболевание кишечника; сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гематологические злокачественные заболевания; муковисцидоз; нейродегенеративные заболевания; панкреатит; мультиорганная недостаточность; заболевания почек; агрегация тромбоцитов; рак; подвижность сперматозоидов; трансплантация органов и, в частности, отторжение при трансплантации; отторжение трансплантата; повреждения легких и боль, включая боль, ассоциированную с ревматоидным артритом или остеоартритом, боль в спине, общую воспалительную боль, постгепатическую невралгию, диабетическую невропатию, воспалительную невропатическую боль, тригеминальную невралгию и центральную боль.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
В биохимии киназа представляет собой тип фермента, который переносит фосфатные группы от высокоэнергетических донорных молекул, таких как АТР (аденозинтрифосфат), к специфическим субстратам, при этом процесс называется фосфорилированием. Точнее, ферменты PI3K являются липидными ферментами киназами, которые могут фосфорилировать фосфоинозитиды (PIs) по 3'-гидроксильной группе инозитольного кольца (Panayotou et al., Trends Cell Biol, 2: 358-60 (1992)). Хорошо известно, что PIs, локализованные в плазматических мембранах, могут действовать в качестве вторичных мессенджеров в сигнальных каскадах посредством стыкующих белков, содержащих домен гомологии с плекстрином (РН), FYVE, РХ и другие фосфолипид-связывающие домены (Vanhaesebroeck В et al., Annu. Rev. Biochem, 70, 535-602, 2001; Katso R et al. Annu. Rev. Cell Dev. Biol., 17, 615-675, 2001).
Следовательно, PIs могут действовать в качестве вторичных мессенджеров во многих клеточных процессах, включая сигнальную трансдукцию, регуляцию мембранного переноса и транспорта, формирование цитоскелета, выживание и гибель клеток и многие другие функции.
PIs могут быть связаны с липидным бислоем клеточной мембраны посредством двух жирных кислот, которые присоединяются к цитозольному инозитольному кольцу через глицерофосфатный линкер. Инозитольное кольцо PIs может фосфорилироваться ферментами PI3K, что приводит к регуляции клеточного роста, выживания и пролиферации. По этой причине фосфорилирование PIs посредством ферментов PI3K является одним из наиболее значимых событий сигнальной трансдукции, ассоциированных с активацией поверхностных рецепторов клеток млекопитающих (Cantley LC, Science, 296, 1655-7, 2002; Vanhaesebroeck В et al. Annu. Rev. Biochem, 70, 535-602, 2001).
Ферменты PI3K были разделены на три класса: PI3K класса I, PI3K класса II и PI3K класса III, исходя из гомологии последовательностей, структуры, партнеров по связыванию, способа активации и субстратного предпочтения (Vanhaesebroeck В et al., Exp. Cell Res., 253(1), 239-54, 1999; и Leslie NR et al., Chem. Rev., 101(8), 2365-80, 2001).
PI3K класса I превращают фосфоинозитид-(4,5)-дифосфат (PI(4,5)P2) в фосфоинозитид-(3,4,5)-трифосфат (PI(3,4,5)P3), который действует в качестве вторичного мессенджера. Сигнальный каскад, активированный вследствие повышения внутриклеточных уровней PI(3,4,5)P3, отрицательно регулируется посредством действия 5'-специфических и 3'-специфических фосфатаз (Vanhaesebroeck В et al., Trends Biochem. Sci., 22(7), 267-72, 1997; Katso R et al., Annu. Rev. Cell Dev. Biol., 17, 615-75, 2001; и Toker A, Cell. Mol. Life Sci., 59(5), 761-79, 2002).
Ферменты PI3K класса II представляют собой совсем недавно идентифицированный класс PI3K, при этом их точная функция до сих пор является неясной.
Класс III ферментов PI3K состоит из единственного члена семейства, который структурно относится к ферментам PI3K класса I и, по-видимому, важен в эндоцитозе и везикулярном переносе. Однако существуют некоторые данные, показывающие, что PI3K класса III могут быть значимыми в иммунных процессах клеток, таких как фагоцитоз и передача сигнала с Toll-подобных рецепторов (TLR).
Ферменты PI3K класса I, исходя из механизмов их активации, можно дополнительно разделить на класс IA и класс IB.
Более подробно, класс IA ферментов PI3K содержит три близкородственные изоформы: PI3Kα, PI3Kβ и PI3Kδ, тогда как класс IB содержит только изоформу PI3Kγ. Эти ферменты представляют собой гетеродимеры, состоящие из каталитической субъединицы, известной как р110, с четырьмя типами изоформ: альфа (α), бета (β), дельта (δ) и гамма (γ), конститутивно связанной с регуляторной субъединицей. Первые две изоформы р110 (α и β) повсеместно экспрессируются и вовлечены в дифференцировку и пролиферацию клеток. Вследствие этого, было проведено глубокое исследование ферментов PI3Kα и PI3Kβ в качестве мишеней для разработки новых химиотерапевтических агентов.
В свою очередь, изоформы р110δ и р110γ главным образом экспрессируются в лейкоцитах и важны в активации иммунного ответа, например миграции лейкоцитов, активации В- и Т-клеток и дегрануляции тучных клеток. Следовательно, изоформы PI3Kδ и PI3Kγ являются очень значимыми в воспалительных респираторных заболеваниях.
На сегодняшний день производные ингибиторов ферментов PI3K, известных в данной области техники, могли в общем случае ингибировать указанные изоформы (изоформы альфа α, бета β, дельта δ и гамма γ), и они могли действовать на отдельные функции, выполняемые указанными конкретными изоформами в различных заболеваниях.
По этой причине была проведена тщательная разработка анализов специфической активности ингибиторов класса IA в отношении одной конкретной изоформы PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ по сравнению с другой для того, чтобы распознать подходящий профиль лечения расстройств, ассоциированных с механизмами ферментов PI3K. Такие расстройства могли, например, включать респираторные заболевания, выбранные из идиопатического хронического кашля, кашлевого варианта астмы, кашля, ассоциированного с опухолью грудной клетки или раком легких, вирусного или поствирусного кашля, синдрома кашля верхних дыхательных путей (UACS) или вызванного постназальным затеканием кашля, или кашля, ассоциированного с гастроэзофагеальной рефлюксной болезнью как кислотной, так и некислотной, астмы, хронического бронхита, хронической обструктивной болезни легких (COPD), интерстициального заболевания легких, идиопатического фиброза легких (IPF), застойной болезни сердца, саркоидоза, инфекций (таких как коклюш), вирусных инфекций, включая вирусные инфекции дыхательных путей и вирусное обострение респираторных заболеваний; невирусные респираторные инфекции, включая аспергиллез и лейшманиоз; аллергические заболевания, включая аллергический ринит и атопический дерматит; аутоиммунные заболевания, включая ревматоидный артрит и рассеянный склероз; воспалительные расстройства, включая воспалительное заболевание кишечника; сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гематологические злокачественные заболевания; нейродегенеративные заболевания; панкреатит; мультиорганную недостаточность; заболевания почек; агрегацию тромбоцитов; рак; подвижность сперматозоидов; отторжение при трансплантации; отторжение трансплантата; повреждения легких и боль, включая боль, ассоциированную с ревматоидным артритом или остеоартритом, боль в спине, общую воспалительную боль, постгепатическую невралгию, диабетическую невропатию, воспалительную невропатическую боль (травму), тригеминальную невралгию и центральную боль.
Принимая во внимание множество патологических ответов, опосредуемых ферментами PI3K, существует постоянная потребность в ингибиторах ферментов PI3K, которые могут быть полезными в лечении многих расстройств. Таким образом, настоящее изобретение относится к новым соединениям, представляющим собой ингибиторы изоформ PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ ферментов PI3K класса I, которые, по указанным выше причинам, часто могут иметь терапевтически желательные характеристики.
В частности, соединения по изобретению могут обладать гораздо большей селективностью в отношении изоформы δ или обеих изоформ γ и δ фермента PI3K, чем в отношении других изоформ того же фермента.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на соединения формулы (I)
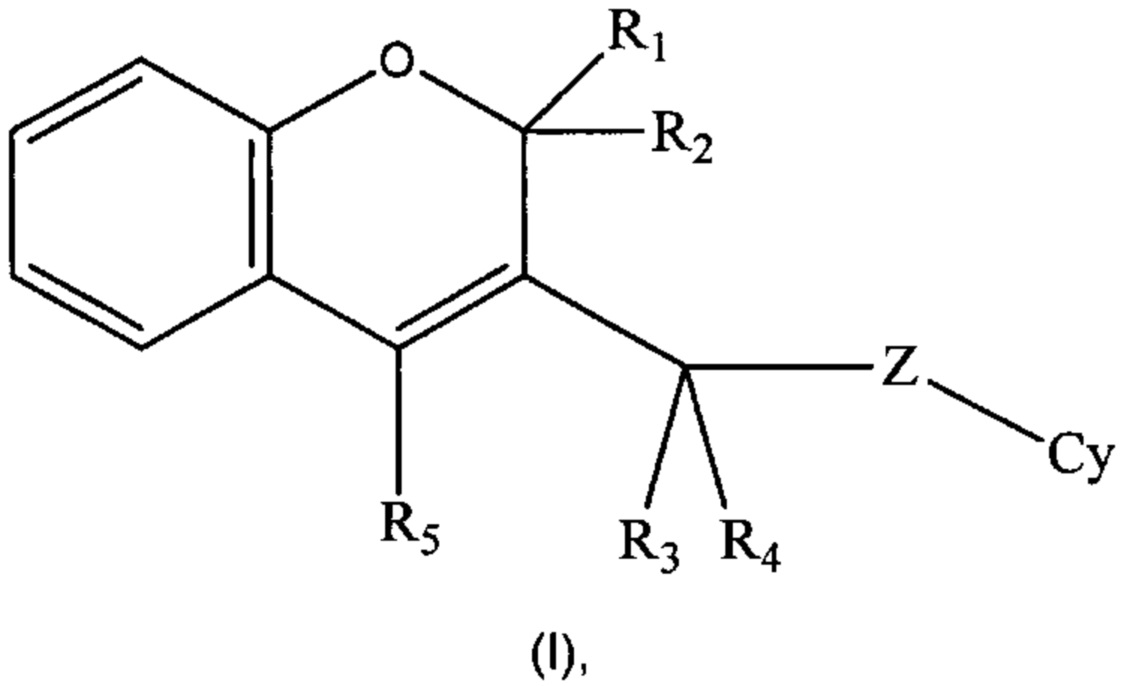
где R1, R2, R3, R4, R5, Cy, Z такие, как раскрыто ниже в подробном описании изобретения, действующие в качестве ингибиторов фосфоинозитид-3-киназ, на способы их получения, фармацевтические композиции, содержащие их или как таковые или в комбинации с одним или более активными ингредиентами в смеси с одним или более фармацевтически приемлемыми носителями.
В изобретении дополнительно предложено подходящее устройство для доставки фармацевтической композиции соединения по изобретению.
В одном аспекте настоящего изобретения предложено применение соединения по изобретению для изготовления лекарственного средства.
В еще одном аспекте настоящего изобретения предложено применение соединения по изобретению для изготовления лекарственного средства для предупреждения и/или лечения любого заболевания, характеризующегося повышенной активностью фермента фосфоинозитид-3-киназы (PI3K) и/или когда желательно ингибирование активности PI3K, в частности, посредством селективного ингибирования изоформы фермента дельта или обеих изоформ дельта и гамма по сравнению с изоформами фермента альфа и бета.
Кроме того в настоящем изобретении предложен способ предупреждения и/или лечения любого заболевания, когда желательно ингибирование фермента PI3K, где указанный способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению.
В частности, соединения по изобретению как таковые или в комбинации с другими активными ингредиентами можно вводить для предупреждения и/или лечения заболевания дыхательных путей, характеризующегося обструкцией дыхательных путей с воспалением, такого как, например, кашель, астма, COPD и IPF.
Понятно, что соединения по изобретению подпадают под более общую формулу:
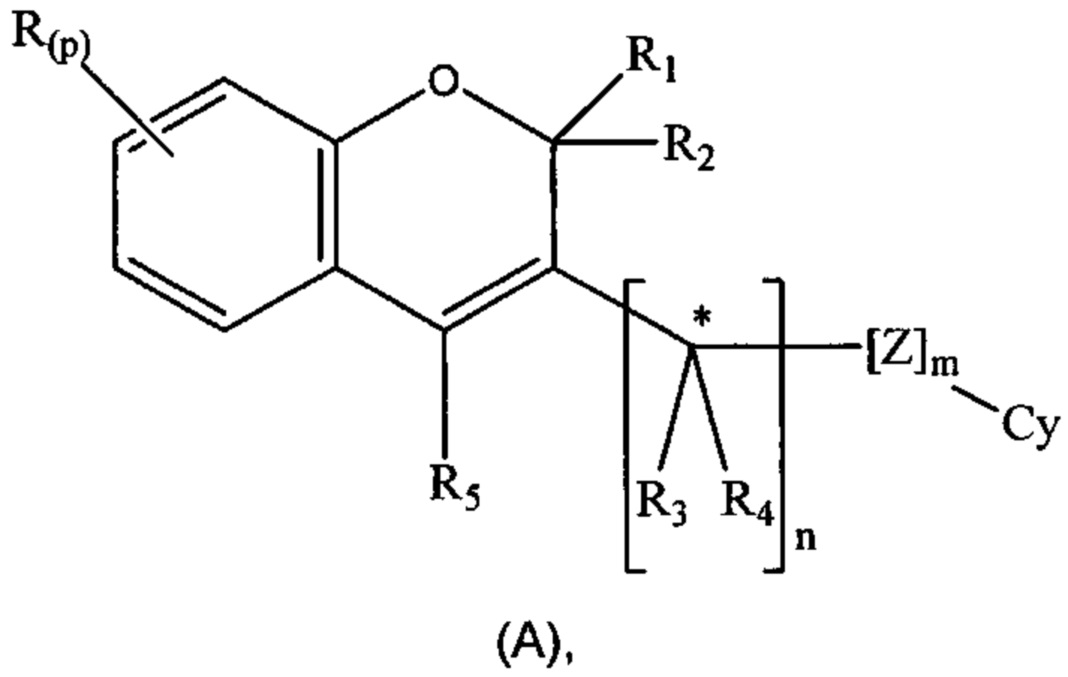
где m равен нулю или 1; n равен 1; р равен нулю.
Соединения, подпадающие под такую более широкую формулу, известны в данной области техники, например из международной заявки на патент WO 2014/164942 и из заявки на патент США 2014/0005247.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение направлено на класс соединений, действующих в качестве ингибиторов фосфоинозитид-3-киназ (PI3K).
Указанный класс соединений ингибирует активность или функцию класса I PI3K; более конкретно, они представляют собой производные ингибиторов активности или функции изоформ PI3Kα, PI3Kβ, PI3Kγ и/или PI3Kδ класса I PI3K.
Настоящее изобретение относится к соединениям формулы (I):
где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 и R4, одинаковые или разные, в каждом случае независимо выбраны из группы, состоящей из Н, (С1-C6)алкила и (С1-C6)галогеналкила;
R5 выбран из группы, состоящей из замещенного или незамещенного арила и замещенного или незамещенного гетероарила;
Z отсутствует или представляет собой NH;
Cy выбран из группы, состоящей из замещенного или незамещенного гетероарила;
или их фармацевтически приемлемым солям.
Термин "фармацевтически приемлемые соли" при использовании в данном описании изобретения относится к производным соединений формулы (I), когда исходное соединение модифицируют соответствующим образом путем превращения любой из свободной кислотной или основной группы, если она присутствует, в соответствующую соль присоединения любого основания или кислоты, обычно предназначенных в качестве фармацевтически приемлемых.
Таким образом, подходящие примеры указанных солей могут включать соли присоединения неорганической или органической кислоты к основным остаткам, таким как аминогруппы, а также соли присоединения неорганического или органического основания к кислотным остаткам, таким как карбоксильные группы.
Катионы неорганических оснований, которые можно соответствующим образом использовать для получения охватываемых изобретением солей, включают ионы щелочных или щелочноземельных металлов, таких как калий, натрий, кальций или магний.
Соли, полученные путем взаимодействия основного соединения, действующего в качестве основания, с неорганической или органической кислотой с образованием соли, включают, например, соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, камфорсульфоновой кислоты, уксусной кислоты, щавелевой кислоты, малеиновой кислоты, фумаровой кислоты, янтарной кислоты и лимонной кислоты.
ОПРЕДЕЛЕНИЯ
Термин "атомы галогена" при использовании в данном описании изобретения включает фтор, хлор, бром и йод, предпочтительно хлор или фтор.
Термин "(С1-Сх)алкил", где х представляет собой целое число больше 1, относится к алкильным группам с линейной цепью или разветвленной цепью, где число входящих в состав атомов углерода находится в диапазоне от 1 до х. Особенно предпочтительными алкильными группами являются метил, этил, н-пропил, изопропил и трет-бутил.
Выражение "(C1-Сх)галогеналкил" относится к определенным выше группам "(С1-Сх)алкил", где один или более атомов водорода заменены одним или более атомами галогенов, которые могут быть одинаковыми или отличаться друг от друга.
Примеры указанных (С1-Сх)галогеналкильных групп могут, таким образом, включать галогенированные, полигалогенированные и полностью галогенированные алкильные группы, например трифторметильные или дифторметильные группы.
Аналогично, термины "(С1-Сх)гидроксиалкил" или "(С1-Сх)аминоалкил" относятся к определенным выше группам "(C1-Сх)алкил", где один или более атомов водорода заменены одной или более чем одной гидрокси- (ОН) или аминогруппой, соответственно.
В настоящем описании, если не предусмотрено иное, определение аминоалкила охватывает алкильные группы, замещенные одной или более аминогруппами -NR10R11.
Что касается заместителя R10 и R11, то, согласно приведенному ниже дополнительному пояснению, когда R10 и R11 взяты вместе с атомом азота, то они связаны с образованием 5-6-членного гетероциклического радикала, при этом по меньшей мере один дополнительный кольцевой атом углерода в указанном гетероциклическом радикале может быть заменен по меньшей мере одним гетероатомом или гетерогруппой (например, N, NH, S или О) или может иметь группу-заместитель -оксо (=O). Указанный гетероциклический радикал может быть дополнительно возможно замещен в подходящих точках кольца, а именно: по атому углерода или по гетероатому либо гетерогруппе, доступным для замещения. Таким образом, примерами указанных гетероциклических радикалов являются 1-пирролидинил, 1-пиперидинил, 1-пиперазинил, 4-морфолинил, пиперазин-4-ил-2-он, 4-метилпиперазин-1-ил и 3-(гидроксиметил)азетидин-1-ил.
Термин "(C3-Cy)циклоалкил", где у представляет собой целое число больше 3, относится к насыщенным циклическим углеводородным группам, содержащим от 3 до у кольцевых атомов углерода. Неограничивающие примеры включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "арил(С1-Сх)алкил" относится к арильному кольцу, связанному с алкильными группами с линейной цепью или с разветвленными алкильными группами, где число входящих в состав атомов углерода находится в диапазоне от 1 до х, например фенилметилу (то есть бензилу), фенилэтилу или фенилпропилу.
Производное выражение "(С3-Cz)гетероциклоалкил" относится к насыщенным или частично ненасыщенным моно-, би- или трициклическим (С3-Cz)циклоалкильным группам, где z представляет собой целое число больше 3, предпочтительно от 5 до 11 кольцевых атомов, в которых по меньшей мере один кольцевой атом углерода заменен по меньшей мере одним гетероатомом или гетерогруппой (например N, NH, S или О); в определение включены мостиковые моно-, би- или три-циклические кольцевые системы. Неограничивающие примеры (C3-Cz)гетероциклоалкила представлены пирролидинильным, имидазолидинильным, тиазолидинильным, пиперазинильным, пиперидинильным, морфолинильным, тиоморфолинильным, дигидро- или тетрагидро-пиридинильным, тетрагидропиранильным, пиранильным, 2Н- или 4Н-пиранильным, дигидро- или тетрагидро-фуранильным, 1,3-диоксолан-2-ильным, 8-азабицикло[3.2.1]окт-2-ен-3-ильным радикалами и тому подобными. (С3-Cz)гетероциклоалкильные группы, как определено выше, могут быть возможно дополнительно замещены в подходящих точках кольца, а именно: по атому углерода или по гетероатому либо гетерогруппе, доступным для замещения. Например, тетрагидро-пиридинильные группы, когда дополнительно замещены, могут быть замещены по группе -NH, например в следующих случаях: 1-бензил-1,2,3,6-тетрагидропиридин-4-ил, 1-(циклопропилметил)-1,2,3,6-тетрагидропиридин-4-ил, 1-ацетил-1,2,3,6-тетрагидропиридин-4-ил, 1-(пиридин-4-илметил)-1,2,3,6-тетрагидропиридин-4-ил.
Термин "(С2-Сх)алкенил" относится к линейным или разветвленным, конъюгированным или неконъюгированным углеродным цепям с одной или более двойными связями в цис или транс конфигурации, где число атомов находится в диапазоне от 2 до х.
Аналогично, термин "(С5-Cy)циклоалкенил", где y представляет собой целое число больше 5, относится к циклическим углеводородным группам, содержащим от 5 до y кольцевых атомов углерода и одну или две двойные связи, где циклоалкенил может быть дополнительно возможно замещен одной или более группами, например аминогруппами.
Термин "(С2-Сх)алкинил" относится к линейным или разветвленным углеродным цепям с одной или более тройными связями, где число атомов находится в диапазоне от 2 до х.
Аналогично, термин "(С2-Сх)аминоалкинил" относится к определенным выше группам "(С2-Сх)алкинил", где один или более атомов водорода заменены одной или более чем одной аминогруппой, и где аминогруппа может быть дополнительно возможно замещена одной или более (С1-C6)алкильными группами.
Выражение "арил" относится к моно, би- или трициклическим кольцевым системам, которые имеют от 6 до 20, предпочтительно от 6 до 15 кольцевых атомов, где по меньшей мере одно кольцо является ароматическим.
Выражение "гетероарил" относится к моно-, би- или трициклическим кольцевым системам с 5-20, предпочтительно 5-15 кольцевыми атомами, в которых по меньшей мере одно кольцо является ароматическим и в которых по меньшей мере один кольцевой атом представляет собой гетероатом или гетероароматическую группу (например, N, NH, S или О).
Примеры подходящих арильных или гетероарильных моноциклических кольцевых систем включают, например, фенильный, тиенильный (в этом документе также называемый тиофен-ильным или тиофен-илом), пирролильный, пиразолильный, имидазолильный, изоксазолильный, оксазолильный, изотиазолильный, тиазолильный, пиридинильный, пиримидинильный, пиразинильный, фуранильный радикалы и тому подобные.
Примеры подходящих арильных или гетероарильных бициклических кольцевых систем включают нафталинильный, бифениленильный, пуринильный, птеридинильный, пиразолопиримидинильный, бензотриазолильный, хинолинильный, изохинолинильный, индолильный, изоиндолильный, бензотиофенильный, бензодиоксинильный, дигидробензодиоксинильный, инденильный, дигидроинденильный, дигидробензодиоксепинильный, бензооксазинильный радикалы и тому подобные.
Примеры подходящих арильных или гетероарильных трициклических кольцевых систем включают флуоренильные радикалы, а также бензоконденсированные производные вышеупомянутых гетероарильных бициклических кольцевых систем.
Термин "(С1-Сх)алканоил" относится к алкилкарбонильным группам (например, (С1-Сх)алкил(СО), где х представляет собой целое число больше 1), где группа "алкил" имеет значение, определенное выше. Неограничивающие примеры включают ацетил, пропаноил, бутаноил.
Выражение "арилкарбонил" относится к группам арил-(СО)-, где группа "арил" имеет значение, определенное выше. Неограничивающий пример представлен бензоилом.
Термин "арил(С2-Сх)алканоил" относится к группе арил(С2-Сх)алкил-карбонил, где х представляет собой целое число больше 2, где арил и алкил имеют значения, определенные выше. Неограничивающие примеры представлены фенилацетильным, фенилпропаноильным или фенилбутаноильным радикалами.
Аналогично, выражения "арил(С1-Сх)алкил", "гетероарил(С1-Сх)алкил" и "(С3-Cy)циклоалкил(C1-Сх)алкил" относятся к "(С1-Сх)алкилу", замещенному, соответственно, одной или более арильными, гетероарильными или (С3-Cy)циклоалкильными группами, как определено выше.
Примеры, например, арил(С1-С6)алкила включают фенилметил, в этом документе также называемый бензилом. Примеры, например, гетероарил(С1-С6)алкила включают пиридин-4-илметил. Примеры, например, (С3-C7)циклоалкил(С1-С6)алкила включают циклопропилметил.
При использовании в данном описании изобретения выражение "кольцевая система" относится к моно- или бициклическим кольцевым системам, которые могут быть насыщенными, частично ненасыщенными или ненасыщенными, таким как арил, (С3-С7)циклоалкил, (С3-С6)гетероциклоалкил или гетероарил.
При использовании в данном описании изобретения группировка оксо представлена в виде (О) как альтернатива другому общепринятому обозначению, например (=O). Таким образом, исходя из общей формулы, карбонильная группа в этом документе предпочтительно представлена в виде С(O) в качестве альтернативы другим общепринятым обозначениям, таким как СО, (СО) или С(=O). В общем случае, заключенная в скобки группа представляет собой боковую группу, не включенную в цепь, и скобки используют, когда это считается полезным, чтобы помочь устранению неоднозначности линейных химических формул; например, сульфонильная группа -SO2- также может быть представлена в виде -S(O)2-, чтобы устранить неоднозначность, например, по отношению к сульфиновой группе -S(O)O-.
Специалисту в данной области техники будет очевидно, что соединения формулы (I) могут содержать по меньшей мере один стереогенный центр, когда R3 и R4 разные, а именно: изображенный в формуле (IA) в виде атома углерода (*) со звездочкой, и, следовательно, могут существовать в виде оптических стереоизомеров.
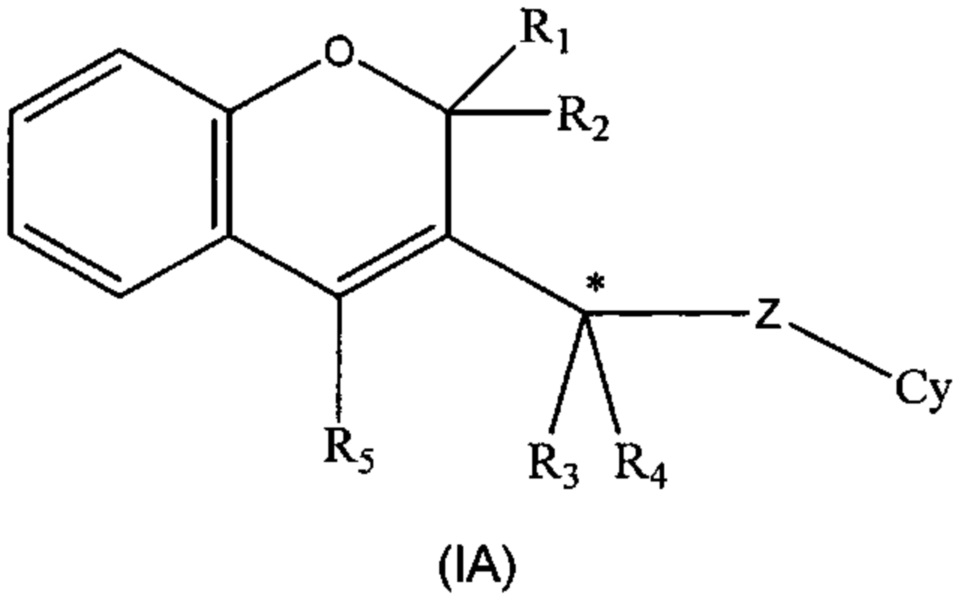
В тех случаях, когда соединения по изобретению имеют по меньшей мере один такой стереогенный центр, они могут, соответственно, существовать в виде энантиомеров. В тех случаях, когда соединения по изобретению обладают двумя или более стереогенными центрами, они дополнительно могут существовать в виде диастереоизомеров. Следует понимать, что все такие отдельные энантиомеры, диастереоизомеры и их смеси в любой пропорции включены в объем настоящего изобретения. Абсолютную конфигурацию (R) или (S) атома углерода (*), когда он является стереогенным центром, определяют на основе правил для номенклатуры Кана-Ингольда-Прелога, основанных на приоритетах групп.
Атропоизомеры представляют собой стереоизомеры, полученные в результате затрудненного вращения вокруг одинарных связей, когда барьер стерического напряжения вращению является достаточно высоким, чтобы позволить выделить конформеры (Bringmann G et al, Angew. Chemie Int. Ed., 44 (34), 5384-5427, 2005).
Oki определил атропоизомеры как конформеры, которые превращаются друг в друга с полупериодом более 1000 секунд при заданной температуре (Oki М, Topics in Stereochemistry, 14, 1-82, 1983).
Атропоизомеры отличаются от других хиральных соединений тем, что во многих случаях они могут быть термически уравновешенными, тогда как для других форм хиральности обычно возможна только химическая изомеризация.
Разделение атропоизомеров возможно посредством способов хирального разделения, таких как селективная кристаллизация. В атропоэнантиоселективном или атропоселективном синтезе один атропоизомер образуется за счет другого. Атропоселективный синтез можно проводить, используя хиральные вспомогательные вещества, подобные катализатору Кори-Бакши-Шибаты (CBS), асимметричному катализатору, полученному из пролина, или методами, основанными на термодинамическом равновесии, когда реакция изомеризации благоприятствует одному атропоизомеру по сравнению с другим.
Рацемические формы соединений формулы (I), также как и индивидуальные атропоизомеры (по существу не содержащие соответствующего им энантиомера) и обогащенные стереоизомерами смеси атропоизомеров включены в объем настоящего изобретения.
В предпочтительном воплощении настоящее изобретение направлено на соединения формулы (IA), как они определены выше, где R3 имеет такое же значение, как указано выше, за исключением Н, R4 представляет собой Н, и абсолютной конфигурацией хирального углерода (*) является (R)-конфигурация.
В еще одном воплощении предпочтительной конфигурацией углерода (*) является (S)-конфигурация.
В предпочтительном воплощении соединения формулы (I), описанные в настоящем изобретении, присутствуют в виде смесей энантиомеров и/или диастереоизомеров в любой пропорции.
Первая предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н и (C1-С6)алкила;
R4 представляет собой Н;
R5 выбран из замещенного или незамещенного арила и замещенного или незамещенного гетероарила;
Z и Cy являются такими, как определено выше.
Более предпочтительная группа соединений представляет собой соединения формула (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н, метила, этила и пропила;
R4 представляет собой Н;
R5 выбран из фенила, 2-, 3- или 4-пиридинила, 5-тиазолила, 2-, 3-, 4- или 5-тиенила, 1Н-пиразол-4-ила, 2-, 4-, 5- или 6-пиримидинила, все из которых возможно замещены одной или более группами, выбранными из (C1-C6)алкила, (С1-C6)гидроксиалкила, замещенного или незамещенного (C1-C6)аминоалкила;
Z и Cy являются такими, как определено выше.
Еще более предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н, метила, этила и пропила;
R4 представляет собой Н;
R5 выбран из фенила, 2-, 3-, 4- или 5-тиенила, все из которых возможно замещены одной или более группами, выбранными из пиперазин-4-илметила, (4-метилпиперазин-1-ил)метила, пиперидин-1-илметила, гидроксиметила, диметиламинометила и (3-(гидроксиметил)азетидин-1-ил)метила;
Z и Cy являются такими, как определено выше.
Вторая предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н, метила, этила и пропила;
R4 представляет собой Н;
R5 выбран из замещенного или незамещенного арила и замещенного или незамещенного гетероарила;
Z отсутствует (значением Z является связь) или представляет собой группу NH;
Cy представляет собой гетероарил, выбранный из группы 9Н-пурин-6-ила, 1Н-пиразоло[3,4-d]пиримидин-1-ила, 2-, 4-, 5- или 6-пиримидинила, все из которых возможно замещены одной или более группами, выбранными из галогена, CN, NR10R11, возможно замещенного арила и возможно замещенного гетероарила, выбранных из фенила, 2-, 3-, 4-, 5-, 6-пиридинила;
R10, R11, одинаковые или разные, в каждом случае независимо выбраны из группы, состоящей из Н, (C1-C6)аминоалкила, (C1-C6)гидроксиалкила и (C1-C6)алкила, или R10 и R11, взятые вместе с атомом азота, с которым они связаны, могут образовывать 5-6-членный гетероциклический радикал.
Вторая более предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н, метила или этила;
R4 представляет собой Н;
R5 выбран из арила, представляющего собой фенил, гетероарила, выбранного из 2-, 3-, 4- или 5-тиенила, все из которых возможно замещены одной или более группами, выбранными из пиперазинометила, (4-метилпиперазин-1-ил)метила, пиперидин-1-илметила, гидроксиметила, диметиламинометила и (3-(гидроксиметил)азетидин-1-ил)метила;
Z отсутствует или представляет собой NH;
Cy представляет собой гетероарил, выбранный из группы 9Н-пурин-6-ила, 1Н-пиразоло[3,4-d]пиримидин-1-ила, 2-, 4-, 5- или 6-пиримидинила; все из которых возможно замещены одной или более группами, выбранными из Cl, Br, F, I, CN; NH2, арила, выбранного из 3-фтор-5-гидроксифенила, 3-хлор-5-гидроксифенила и 3-циано-5-гидроксифенила, гетероарила, выбранного из 6-, 5-, 4-гидроксипиридин-3-ила, (2,2,2-трифтор-1-(пиридин-3-ил)этанол)-5-ила.
Третья более предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н;
R3 выбран из Н, метила или этила;
R4 представляет собой Н;
R5 выбран из замещенного или незамещенного (C3-C6)гетероциклоалкила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила, как определено выше;
Z отсутствует;
Cy представляет собой 1Н-пиразоло[3,4-d]пиримидин-1-ил, возможно и независимо замещенный одной или более группами, выбранными из галогена, NR10R11, (C1-C6)алкила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила, как определено выше;
или их фармацевтически приемлемые соли или сольваты.
Еще более предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н;
R3 выбран из Н, метила или этила;
R4 представляет собой Н;
R5 выбран из группы фенила, 2-, 3-, 4- или 5-тиенила, все из которых возможно замещены одной или более группами, выбранными из замещенного или незамещенного (С1-C6)аминоалкила;
Z отсутствует;
Cy представляет собой 1Н-пиразоло[3,4-d]пиримидин-1-ил, возможно замещенный одной или более группами, независимо выбранными из галогена, NR10R11, фенила и гетероарила, представляющего собой пиридинил; где указанные фенил и гетероарил в свою очередь дополнительно возможно и независимо замещены одной или более группами, выбранными из галогена, -ОН, -CN; NR10R11 (C1-C6)галогеналкила, (C1-C6)гидроксиалкила;
R10, R11 являются такими, как определено выше;
или их фармацевтически приемлемые соли или сольваты.
Четвертая предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н и (C1-C6)алкила;
R4 представляет собой Н;
R5 выбран из замещенного или незамещенного арила или замещенного или незамещенного гетероарила;
Z отсутствует или представляет собой NH;
Cy представляет собой замещенный или незамещенный гетероарил.
Более предпочтительная группа соединений представляет собой соединения формулы (I), где:
R1 и R2 оба представляют собой Н или объединены с образованием оксогруппы (=O);
R3 выбран из Н и метила;
R4 представляет собой Н;
R5 представляет собой фенил или тиенил; где указанные фенил или тиенил возможно замещены группой, выбранной из замещенного или незамещенного (C1-C6)аминоалкила или (C1-C6)гидроксиалкила;
Z отсутствует или представляет собой NH;
Cy представляет собой гетероарил, выбранный из группы, состоящей из 9Н-пурин-6-ила, 1Н-пиразоло[3,4-d]пиримидин-1-ила и 2-, 4-, 5- или 6-пиримидинила, все из которых возможно замещены одной, двумя или тремя группами, выбранными из CN, NH2, возможно замещенного арила и возможно замещенного гетероарила, выбранных из 3-фтор-5-гидроксифенила, 3-хлор-5-гидроксифенила, 3-циано-5-гидроксифенила, 3-гидрокси-5-пиридила и (2,2,2-трифтор-1-(пиридин-3-ил)этанол)-5-ила.
Согласно конкретным воплощениям в настоящем изобретении предложены соединения, перечисленные ниже:
3-((4-амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-4-фенил-2Н-хромен-2-он,
3-(1-(4-амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-4-фенил-2Н-хромен-2-он,
3-(4-амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол,
5-(4-амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ол,
3-((9Н-пурин-6-иламино)метил)-4-фенил-2Н-хромен-2-он,
3-(1-(9Н-пурин-6-иламино)этил)-4-фенил-2Н-хромен-2-он,
N-((4-фенил-2Н-хромен-3-ил)метил)-9Н-пурин-6-амин,
4-амино-6-((4-фенил-2Н-хромен-3-ил)метиламино)пиримидин-5-карбонитрил,
4-амино-6-(1-(4-фенил-2Н-хромен-3-ил)этиламино)пиримидин-5-карбонитрил,
1-(5-(4-амино-1-(1-(4-фенил-2Н-хромен-2-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ил)-2,2,2-трифторэтан-1-ол,
3-(4-амино-1-(1-(4-фенил-2Н-хромен-2-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-гидроксибензонитрил,
3-(4-амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-хлорфенол,
3-(4-амино-1-(1-(4-(5-((4-метилпиперазин-1-ил)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол,
3-(4-амино-1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол,
3-(4-амино-1-(1-(4-(5-(гидроксиметил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол,
5-(4-амино-1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ол,
и их фармацевтические приемлемые соли.
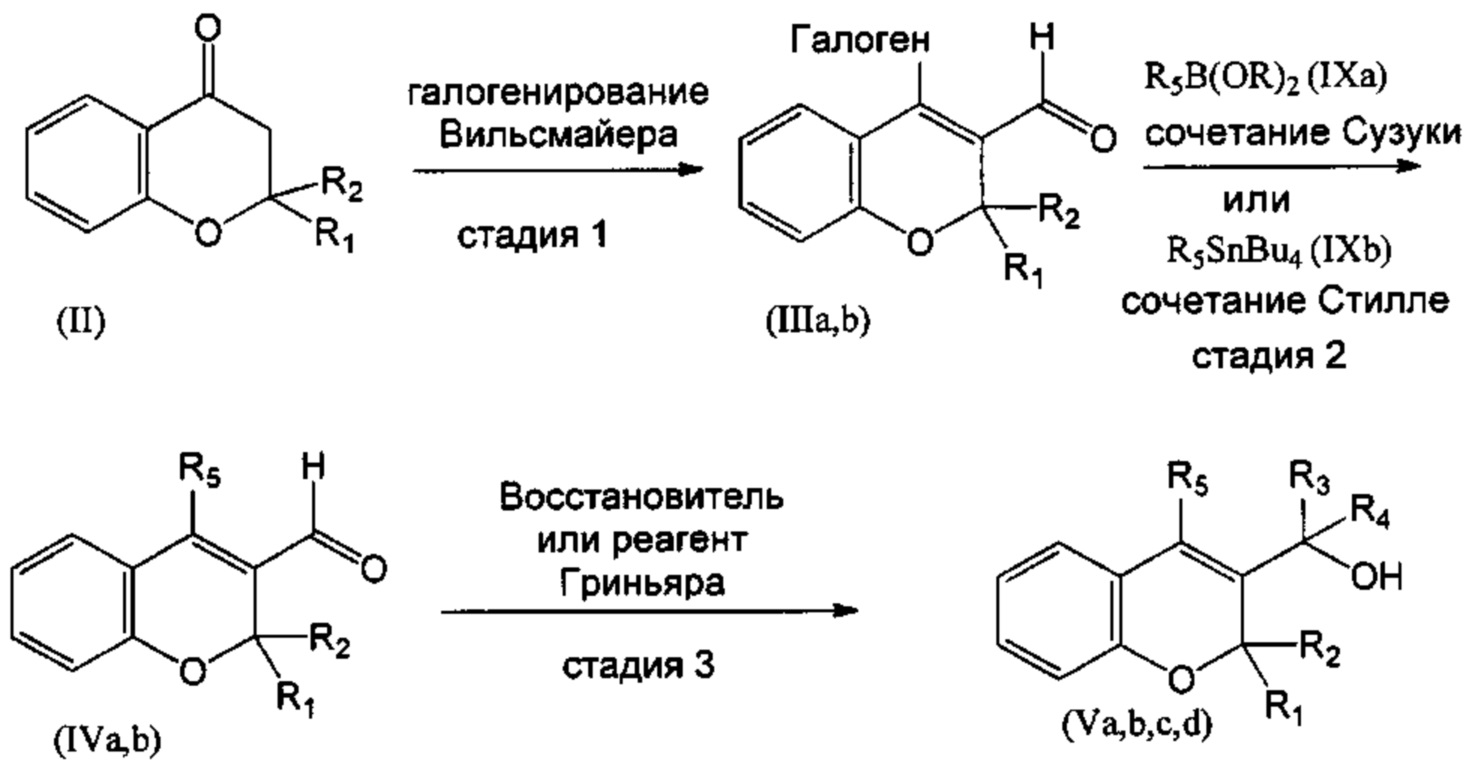
Соединения формулы (I), в том числе все соединения, перечисленные в данном описании выше, обычно могут быть получены по методикам, приведенным в изображенных ниже Схемах, с использованием общеизвестных способов.
В одном воплощении настоящего изобретения соединение (Va), где R1=R2=R3=R4=Н, в соответствии со СХЕМОЙ 1 может быть получено из соединения (II), такого как, например, имеющийся в продаже хроман-4-он. В действительности, соединение (II) можно превратить в галогеновое производное (IIIa),I, где R1=R2=Н (СХЕМА 1, стадия 1), путем реакции галогенирования Вильсмайера с галогенирующими агентами, такими как POCl3 (оксихлорид фосфора) в присутствии подходящего формамида, такого как DMF (диметилформамид). Затем соединение (IIIa) можно превратить в (IVa), где R1=R2=Н, посредством реакций перекрестного сочетания в присутствии палладиевого катализатора, подобных реакции сочетания Сузуки, с подходящим борорганическим реагентом (IXa) (СХЕМА 1, стадия 2) и, окончательно, в соединение (Va) путем восстановления гидридным реагентом, таким как борогидрид натрия (СХЕМА 1, стадия 3).
Аналогично, соединения (Vb), где R1 и R2 объединены с образованием оксогруппы (=O), R3=R4=Н, могут быть получены из соединения (IIIb), где R1 и R2 объединены с образованием оксогруппы (=O), такого как, например, имеющийся в продаже 4-хлор-2-оксо-2Н-хромен-3-карбальдегид. Затем соединение (IIIb) можно превратить в (IVb), где R1 и R2 объединены с образованием оксогруппы (=O), посредством реакций перекрестного сочетания в присутствии палладиевого катализатора, подобных реакции сочетания Стилле, с подходящим органостаннаном (IXb) (СХЕМА 1, стадия 2) и, окончательно, в соединение (Vb) путем восстановления гидридным реагентом, таким как борогидрид натрия (СХЕМА 1, стадия 3).
Соединения (Vc), где R1=R2=R3=Н, R4=Me, могут быть получены из соединения (IVa), где R1=R2=Н, путем добавления реагента Гриньяра, подобного метилмагнийбромиду (СХЕМА 1, стадия 3).
Аналогично, соединения (Vd), где R1 и R2 объединены с образованием оксогруппы (=O), R3=Н, R4=Me, могут быть получены из соединения (IVb), где R1 и R2 объединены с образованием оксогруппы (=O), путем добавления реагента Гриньяра, такого как метилмагнийбромид (СХЕМА 1, стадия 3).
СХЕМА 1
Соединения (Va), где R1=R2=R3=R4=Н, и соединения (Vc), где R1=R2=R3=Н, R4=Me, потом можно превратить в (VIa), где R1=R2=R3=R4=Н, Z=NH, и (VIc), где R1=R2=R3=Н, R4=Me, Z=NH, посредством реакции азидирования с DPPA (дифенилфосфорилазид) с последующим восстановлением подходящим восстановителем, таким как LiAlH4 (СХЕМА 2). Соединения (Ib), где R1=R2=R3=R4=H, и (Ic), где R1=R2=R3=H, R4=Me, затем получали из соединений (VIa) и (VIc) путем взаимодействия с подходящим галогенсодержащим гетероциклом (X), таким как 4-амино-6-хлорпиримидин-5-карбонитрил и 6-хлор-9Н-пурин, в присутствии подходящего основания, подобного DIEA (СХЕМА 2). Следуя этому пути синтеза, получали соединения 4-амино-6-((4-фенил-2Н-хромен-3-ил)метиламино)пиримидин-5-карбонитрил (пример 1), N-((4-фенил-2Н-хромен-3-ил)метил)-9Н-пурин-6-амин (пример 2), 4-амино-6-(1-(4-фенил-2Н-хромен-3-ил)этиламино)пиримидин-5-карбонитрил (пример 3).
СХЕМА 2
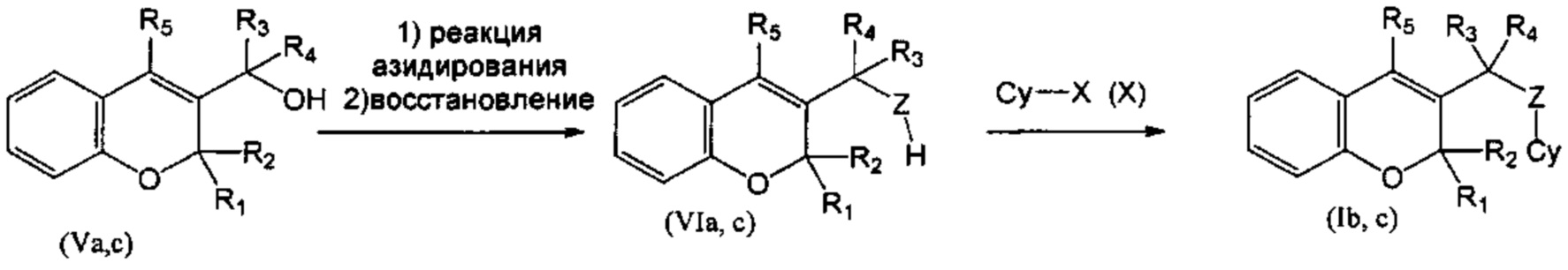
Соединение (Id), где R1 и R2 объединены с образованием оксогруппы (=O), R3=R4=Н, Z=NH, и (Ie), где R1 и R2 объединены с образованием оксогруппы (=O), R3=Н, R4=Me, Z=NH, могут быть синтезированы, как показано на Схеме 3, из соединений (Vb) и (Vd), которые превращали в (VIIb) и (VIId), где Y представляет собой уходящую группу (Lg), такую как галогенидный атом, путем взаимодействия с подходящим галогенирующим агентом, таким как PBr3, и, окончательно, подвергали взаимодействию с нуклеофилом (XI) на основе азота в присутствии основания, подобного NaH (гидрид натрия) (СХЕМА 3). Следуя этому пути синтеза, получали соединения 3-((9Н-пурин-6-иламино)метил)-4-фенил-2Н-хромен-2-он (пример 8) и 3-(1-(9Н-пурин-6-иламино)этил)-4-фенил-2Н-хромен-2-он (пример 9).
СХЕМА 3
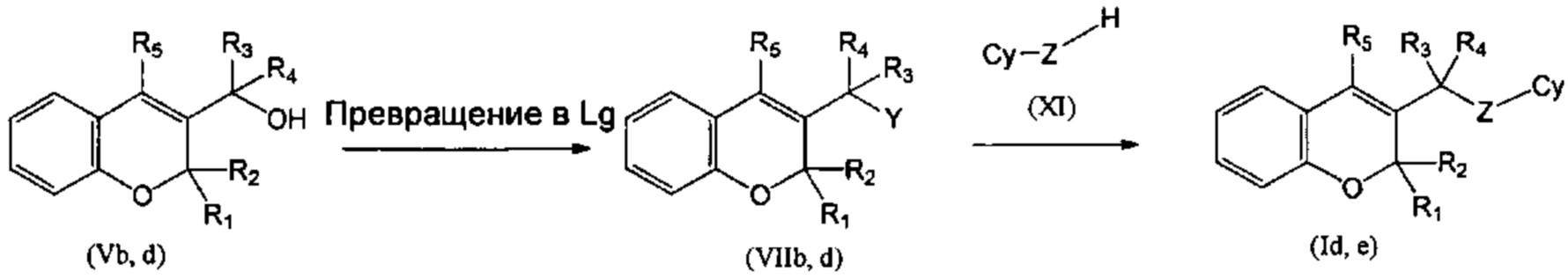
Соединение VIIIb, где R1 и R2 объединены с образованием оксогруппы (=O), и R3=R4=Н, Z отсутствует, и соединение VIIId, где R1 и R2 объединены с образованием оксогруппы (=O) и R3=Н, R4=Me, Z отсутствует, могут быть синтезированы, как показано в Схеме 4, из соединений (Vb) и (Vd), которые превращали в (VIIb) и VIId), где группа Y представляет собой подходящую уходящую группу, такую как галогенидный атом, путем взаимодействия с подходящим галогенирующим агентом, таким как PBr3, а затем подвергали взаимодействию с имеющимся в продаже 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амином в присутствии подходящего основания, подобного K2CO3 (Схема 4, стадия 1 и 2). Соединение (VIIIc), где R1=R2=R3=Н, R4=Me, Z отсутствует, может быть синтезировано из соединения (Vc), где R1=R2=R3=Н, R4=Me, путем реакции Мицунобу с диалкилазадикарбоксилатом, подобным DIAD (диизопропилазадикарбоксилат), в присутствии фосфина, такого как PPh3 (трифенилфосфин), и имеющегося в продаже 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амина (СХЕМА 4, стадия 3).
СХЕМА 4
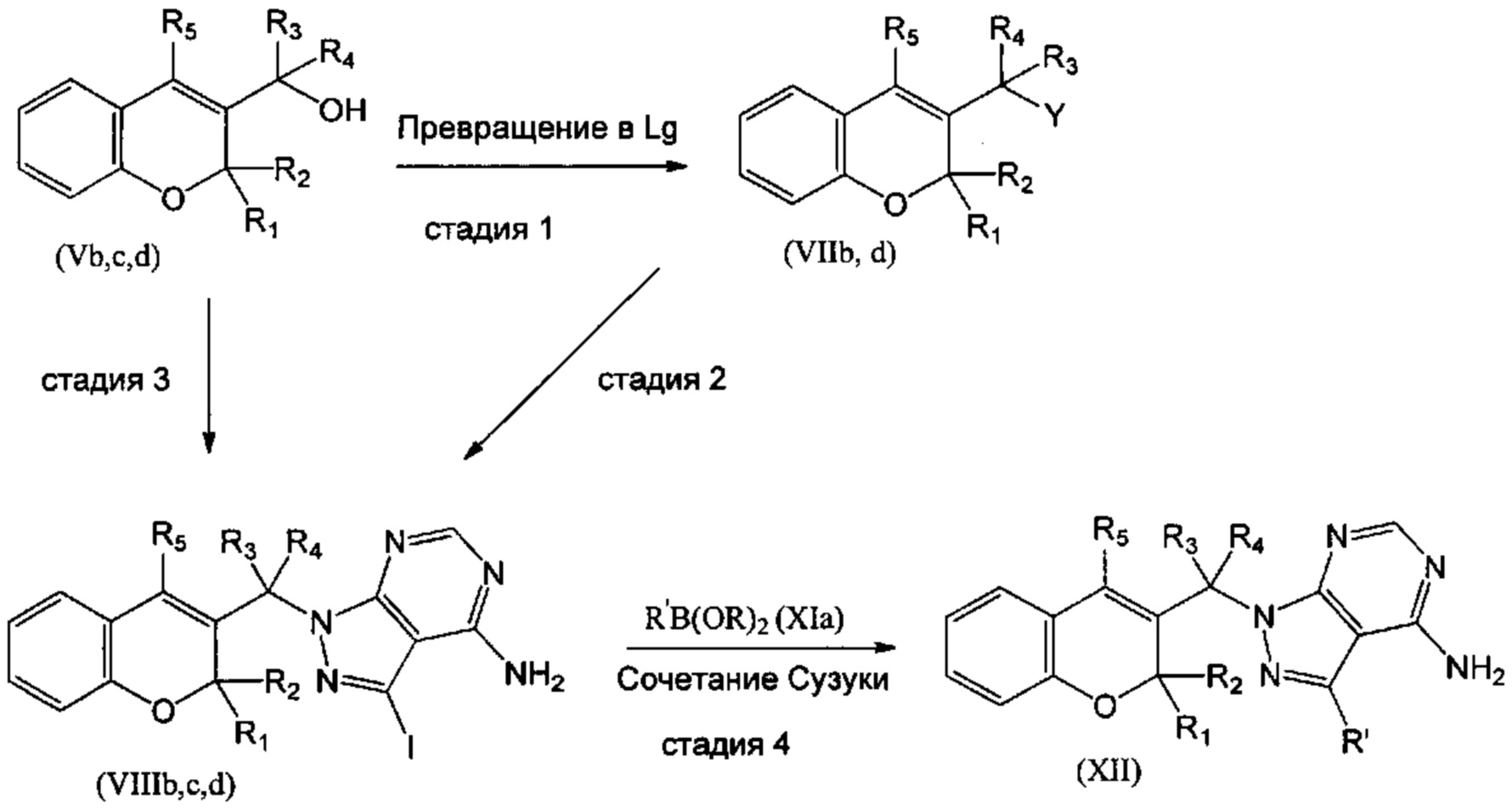
Затем соединения (VIIIb,c,d) можно превратить в соединения (XII) посредством сочетания Сузуки с подходящим борорганическим реагентом (XIa) (СХЕМА 4, стадия 4).
Следуя этому пути синтеза, получали соединения 3-(4-амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-хлорфенол (пример 7с), 3-(4-амино-1-(1-(4-фенил-2Н-хромен-2-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-гидроксибензонитрил (пример 7b), 1-(5-(4-амино-1-(1-(4-фенил-2Н-хромен-2-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ил)-2,2,2-трифторэтан-1-ол (пример 7а), 3-((4-амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-4-фенил-2Н-хромен-2-он (пример 7), 3-(1-(4-амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-4-фенил-2Н-хромен-2-он (пример 6), 3-(4-амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол (пример 4), 5-(4-амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ол (пример 5).
В конкретном случае, когда R5 представляет собой гетероароматическое кольцо, замещенное различными группировками, подобное, например, 4,4,5,5-тетраметил-2-(тиофен-2-ил)-1,3-диоксоланильной группировке, соединения XIIb, где R1=R2=R3=Н, R4=Me, Z отсутствует, могут быть получены из соединения XIII путем последовательности из трех стадий (СХЕМА 5). Соединения XIV могут быть получены из соединения XII путем гидролиза ацетальной группировки посредством взаимодействия с водным раствором неорганической кислоты, подобной HCl (СХЕМА 5, стадия 1). Соединения XV, где R1=R2=R3=Н, R4=Me, Z отсутствует, и W=NMe2 или N-метилпиперазин, или ОН, могут быть получены из соединения XIV путем восстановительного аминирования в присутствии амина, подобного 1-метилпиперазину или диметиламину, и подходящего гидридного донора, подобного триацетоксиборогидриду натрия (СХЕМА 5, стадия 2). Соединения (XV), где R1=R2=R3=Н, R4=Me, Z отсутствует и W=NMe2 или N-метилпиперазин, или ОН, затем можно превратить в соединения (XIIb) посредством сочетания Сузуки с подходящим борорганическим реагентом (XIa) (СХЕМА 5, стадия 3).
СХЕМА 5
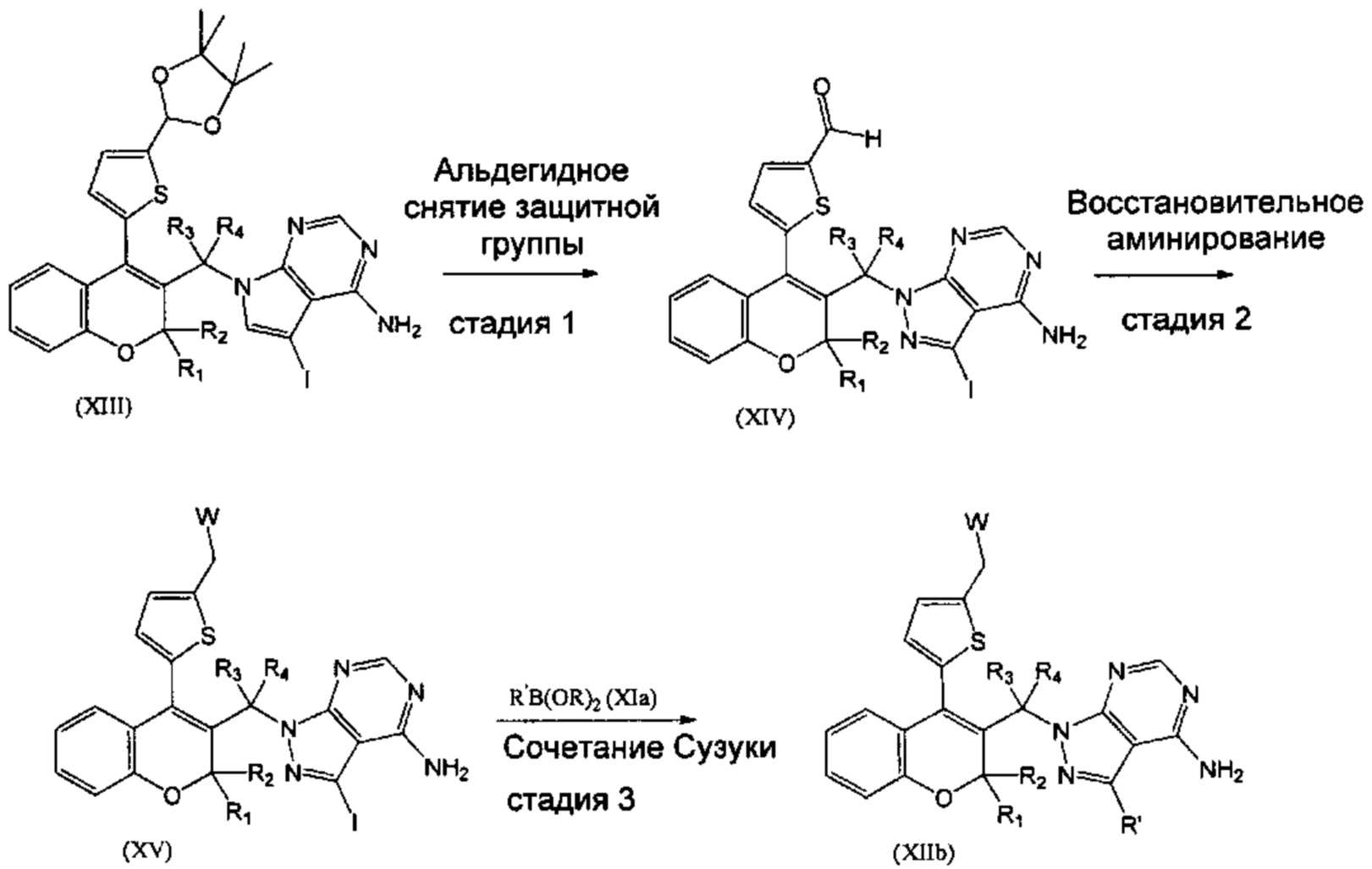
Следуя этому пути синтеза, получали соединения 3-(4-амино-1-(1-(4-(5-((4-метилпиперазин-1-ил)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол (Пример 10), 3-(4-амино-1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол (Пример 11), 3-(4-амино-1-(1-(4-(5-(гидроксиметил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол (Пример 12), 5-(4-амино-1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ол (Пример 13).
Некоторые соединения могут содержать защищенную гидроксильную или азотную группу, такую как, например, силиловый или бензиловый простой эфир, которую затем удаляли по хорошо известной методике.
Соединения по изобретению являются ингибиторами киназной активности, в частности PI3-киназной активности. В общем, соединения, представляющие собой ингибиторы PI3K, могут быть полезны в лечении многих расстройств, ассоциированных с механизмами ферментов PI3K. Таким образом, их можно использовать в изготовлении лекарственного средства для лечения указанных расстройств.
В одном воплощении расстройства, которые можно лечить соединениями по настоящему изобретению, включают респираторные заболевания, выбранные из идиопатического хронического кашля, кашлевого варианта астмы, кашля, ассоциированного с опухолью грудной клетки или раком легких, вирусного или поствирусного кашля, синдрома кашля верхних дыхательных путей (UACS) или вызванного постназальным затеканием кашля, или кашля, ассоциированного с гастроэзофагеальной рефлюксной болезнью (как кислотным, так и некислотным рефлюксом), астмы, хронического бронхита, хронической обструктивной болезни легких (COPD), интерстициального заболевания легких (такого как идиопатический фиброз легких (IPF)), застойной болезни сердца, саркоидоза, инфекций (таких как коклюш), астмы, хронической обструктивной болезни легких (COPD) и идиопатического фиброза легких (IPF); вирусные инфекции (включая вирусные инфекции дыхательных путей и вирусное обострение респираторных заболеваний); невирусные респираторные инфекции, включая аспергиллез и лейшманиоз; аллергические заболевания, включая аллергический ринит и атопический дерматит; аутоиммунные заболевания, включая ревматоидный артрит и рассеянный склероз; воспалительные расстройства, включая воспалительное заболевание кишечника; сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гематологические злокачественные заболевания; нейродегенеративные заболевания; панкреатит; мультиорганную недостаточность; заболевания почек; агрегацию тромбоцитов; рак; подвижность сперматозоидов; отторжение при трансплантации; отторжение трансплантата; повреждения легких и боль, включая боль, ассоциированную с ревматоидным артритом или остеоартритом, боль в спине, общую воспалительную боль, постгепатическую невралгию, диабетическую невропатию, воспалительную невропатическую боль (травму), тригеминальную невралгию и центральную боль.
В еще одном воплощении расстройство, которое можно лечить посредством соединения по настоящему изобретению, выбрано из группы, состоящей из идиопатического хронического кашля, кашлевого варианта астмы, кашля, ассоциированного с опухолью грудной клетки или раком легких, вирусного или поствирусного кашля, синдрома кашля верхних дыхательных путей (UACS), вызванного постназальным затеканием кашля, кашля, ассоциированного с гастроэзофагеальной рефлюксной болезнью (как кислотным, так и некислотным рефлюксом), астмы, хронического бронхита, хронической обструктивной болезни легких (COPD) и интерстициального заболевания легких (такого как идиопатический фиброз легких (IPF).
В еще одном воплощении расстройство выбрано из группы астмы, хронической обструктивной болезни легких (COPD), идиопатического фиброза легких (IPF), кашля и хронического кашля.
Способы лечения по изобретению включают введение безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом. При использовании в данном описании изобретения, "безопасное и эффективное количество" в отношении соединения формулы (I) или его фармацевтически приемлемой соли или другого фармацевтически активного агента означает количество соединения, достаточное для лечения состояния пациента, однако довольно низкое, чтобы избежать серьезных побочных эффектов, и при этом оно может быть в рабочем порядке определено специалистом в данной области. Соединения формулы (I) или их фармацевтически приемлемые соли можно вводить один раз или согласно режиму дозирования, когда число доз вводят с различными интервалами времени в течение заданного периода времени. Обычные суточные дозировки могут варьироваться в зависимости от конкретного выбранного пути введения.
В изобретении также предложены фармацевтические композиции соединений формулы (I) в смеси с одним или более фармацевтически приемлемыми носителями или эксципиентами, например, такими, которые описаны в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений по настоящему изобретению и их фармацевтических композиций можно осуществлять согласно потребностям пациента, например, перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и посредством инфузии), посредством ингаляции, ректально, вагинально, местно, локально, чрескожно и путем окулярного введения.
Различные твердые пероральные лекарственные формы можно использовать для введения соединений по изобретению, включая такие твердые формы, как таблетки, желатиновые капсулы, капсулы, каплеты, гранулы, таблетки для рассасывания и нерасфасованные порошки. Соединения по настоящему изобретению можно вводить взятыми отдельно или комбинированными с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и известными эксципиентами, включая суспендирующие агенты, солюбилизаторы, буферные агенты, связывающие вещества, разрыхлители, консерванты, красители, корригенты, смазывающие вещества и тому подобные. Капсулы, таблетки и гели с пролонгированным действием также являются выгодными для введения соединений по настоящему изобретению.
Различные жидкие пероральные лекарственные формы также можно использовать для введения соединений по изобретению, включая водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать подходящие известные инертные разбавители, такие как вода, и подходящие известные эксципиенты, такие как консерванты, увлажняющие агенты, подсластители, корригенты, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по настоящему изобретению можно вводить посредством инъекции, например внутривенно, в форме изотонического стерильного раствора. Также возможны другие препараты.
Суппозитории для ректального введения соединений по изобретению могут быть получены путем смешивания соединения с подходящим эксципиентом, таким как кокосовое масло, салицилаты и полиэтиленгликоли.
Композиции для вагинального введения могут быть в форме крема, геля, пасты, пены или в форме спрея, содержащих в дополнение к активному ингредиенту подходящие носители, которые также известны.
Для местного введения фармацевтическая композиция может быть в форме кремов, мазей, линиментов, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, подходящих для введения на/в кожу, глаз, ухо или нос. Местное введение также может включать чрескожное введение при помощи таких средств, как трансдермальные пластыри.
Для лечения заболеваний дыхательных путей соединения по изобретению предпочтительно вводят посредством ингаляции.
Ингалируемые препараты включают ингалируемые порошки, содержащие пропеллент дозированные аэрозоли или не содержащие пропеллент ингалируемые композиции.
Для введения в виде сухого порошка можно использовать одно- или многодозовые ингаляторы, известные из предшествующего уровня техники. В этом случае порошок можно засыпать в желатиновые, пластиковые или другие капсулы, картриджи или блистерные упаковки или в резервуар.
К порошкообразным соединениям по изобретению можно добавлять разбавитель или носитель, обычно нетоксичный и химически инертный к соединениям по изобретению, например лактозу или любые другие добавки, подходящие для улучшения вдыхаемой фракции.
Ингаляционные аэрозоли, содержащие газ-пропеллент, такой как гидрофторалканы, могут содержать соединения по изобретению либо в растворе, либо в диспергированной форме. Работающие на пропелленте композиции также могут содержать другие ингредиенты, такие как сорастворители, стабилизаторы и, возможно, другие эксципиенты.
Не содержащие пропеллент ингалируемые композиции, содержащие соединения по изобретению, могут быть в форме растворов или суспензий в водной, спиртовой или водно-спиртовой среде, и их можно доставлять посредством струйного или ультразвукового распылителей, известных из предшествующего уровня техники, или посредством распылителей, создающих "мягкий" туман, таких как Respimat®.
Соединения по изобретению можно вводить в виде единственного активного агента или в комбинации с другими фармацевтически активными ингредиентами, включая те, которые используют в настоящее время в лечении респираторных расстройств, например бета2-агонисты, антимускариновые агенты, кортикостероиды, ингибиторы митоген-активируемых киназ (Р38 МАР-киназ), ингибиторы бета-субъединицы киназы ядерного фактора каппа-В (IKK2), ингибиторы эластазы нейтрофилов человека (HNE), ингибиторы фосфодиэстеразы 4 (PDE4), модуляторы лейкотриенов, нестероидные противовоспалительные агенты (NSAID) и регуляторы слизи.
Таким образом, в изобретении предложены фармацевтические композиции, содержащие соединения по изобретению с комбинации с такими фармацевтически активными ингредиентами, в смеси с одним или более фармацевтически приемлемыми носителями или эксципиентами.
Дозировки соединений по изобретению зависят от целого ряда факторов, включающих конкретное заболевание, подлежащее лечению, серьезность симптомов, путь введения, периодичность интервалов между приемами лекарственного средства, конкретное применяемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения.
Предпочтительно, соединения формулы (I) можно вводить, например, в дозировке, содержащей от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки.
Когда соединения формулы (I) вводят посредством ингаляции, их предпочтительно дают в дозировке, содержащей от 0,001 до 500 мг/сутки, предпочтительно от 0,1 до 200 мг/сутки.
Следующие примеры иллюстрируют изобретение без ограничения его объема.
Основные экспериментальные подробности
Флэш-хроматографию выполняют, используя систему Isolera MPLC (изготовленную Biotage) с применением предварительно упакованного силикагеля или картриджей с обращенной фазой (поставляемых Biotage).
Большая часть соединений, описанных в следующих ниже Примерах, были получены из стереохимически чистых исходных веществ, например с э.и. (энантиомерный избыток) 95%.
Стереохимия соединений в Примерах, когда она указана, была присвоена исходя из допущения, что абсолютная конфигурация в разрешенных стереогенных центрах исходных веществ сохраняется в условиях любой последующей реакции.
В следующих ниже методиках после каждого исходного вещества иногда предлагается ссылка на номер соединения. Она предлагается только для помощи специалисту-химику. Исходное вещество не обязательно могло быть получено из партии, на которую делается ссылка.
Когда делается ссылка на использование "подобной" или "аналогичной" методики, специалисту в данной области техники будет ясно, что такая методика может включать незначительные изменения, например, температуры реакции, количества реагента/растворителя, времени реакции, условий обработки или условий хроматографической очистки.
Химические названия соединений генерировали с помощью программного обеспечения Structure То Name Enterprise 10.0 Cambridge.
Сокращения:
Et2O означает диэтиловый эфир; Et3N означает триэтиламин; DMF означает диметилформамид; EtOAc означает этилацетат; КТ означает комнатную температуру; THF означает тетрагидрофуран; DCM означает дихлорметан; МеОН означает метиловый спирт; EtOH означает этиловый спирт; TFA означает трифторуксусную кислоту; LC-MS означает жидкостную хроматографию/масс-спектрометрию; MPLC означает жидкостную хроматографию среднего давления; dppf означает 1,1'-бис(дифенилфосфино)-ферроцен; S-Phos-Pd-G2 означает хлор(2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий (II); DIEA означает N,N-диизопропилэтиламин; ACN означает ацетонитрил; DMSO означает диметилсульфоксид; UPLC означает сверхэффективную жидкостную хроматографию.
Описание характеристик ЯМР:
1Н-ЯМР спектры получали на спектрометре Varian MR-400, работающем при 400 МГц (частота протона), оснащенном: самоэкранируемой, с z-градиентной катушкой, 5 мм, 1Н/nX широкополосной измерительной головкой для обратного детектирования, канальным блоком с дейтериевой цифровой стабилизацией, блоком квадратурного цифрового детектора с передатчиком с частотным сдвигом. Химические сдвиги выражены в виде значений δ в м.д. относительно триметилсилана (TMS) в качестве внутреннего стандарта. Константы взаимодействия (значения J) приведены в герцах (Гц), а мультиплетности описаны с использованием следующих сокращений: s - синглет, d - дублет, t - триплет, q - квартет, m - мультиплет, br - уширенный, nd - не определяли.
LC/UV/MS аналитические способы
LC/UV/MS - Способ 1
Прибор для LC: Acquity Waters UPLC (или эквивалентный), соединенный с 2996 PDA детектором (детектором с фотодиодной матрицей)
Колонка: Acquity UPLC ВЕН С18 1,7 мкм, 50×2,1 мм
Температура колонки (°C) 30,0
Подвижные фазы: 95:5 H2O:ACN+(0,1% НСООН) (А); 5:95 H2O:ACN+(0,1% НСООН) (В)
Поток (мл/мин) 0,6 (разделение в MS 1:6)
Время остановки (мин) 3,5
Градиент:
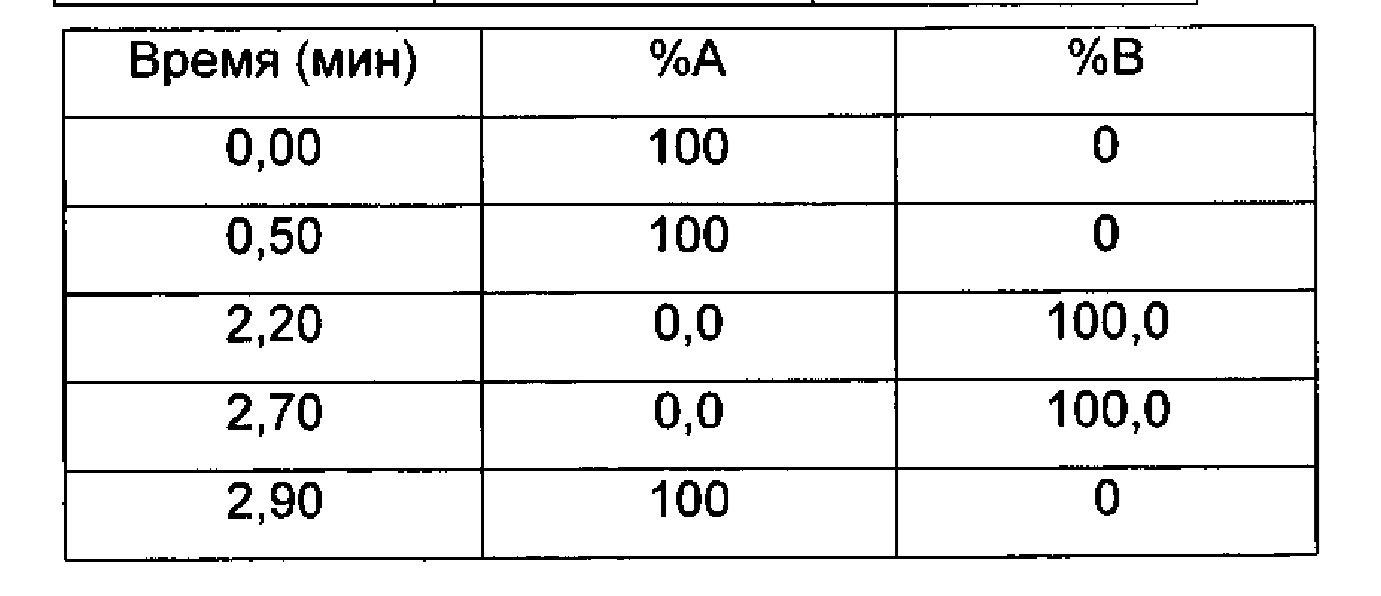
UV (ультрафиолетовое) детектирование: BPI детектирование (начальная длина волны 210 нм, конечная длина волны 400 нм, частота отбора проб спектры/сек = 20)
Объем вводимой пробы (мкл) 1,00
Растворители образца: соотношение DMSO : МеОН : ACN 1:3:3
LC/UV/MS - Способ 2
Прибор для LC: Acquity Waters UPLC (или эквивалентный), соединенный с 2996 PDA детектором
Колонка: Acquity UPLC ВЕН С18 1,7 мкм, 50×2,1 мм
Температура колонки (°C) 30,0
Подвижные фазы: 95:5 H2O:ACN+(0,1% НСООН) (А); 5:95 H2O:ACN+(0,1% НСООН) (В)
Поток (мл/мин) 0,6 (разделение в MS 1:6)
Время остановки (мин) 12,0
Градиент:
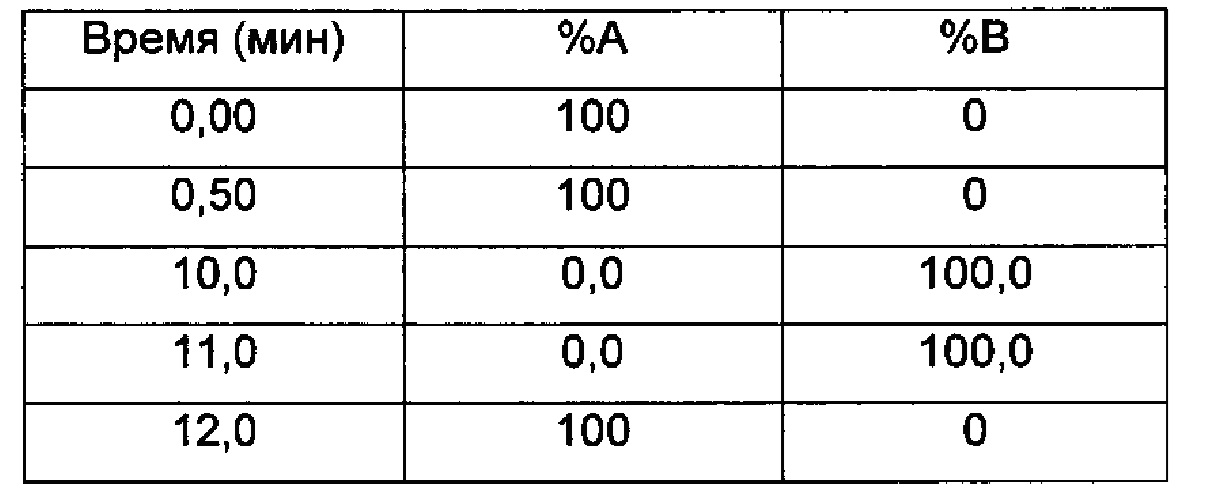
UV детектирование: BPI детектирование (начальная длина волны 210 нм, конечная длина волны 400 нм, частота отбора проб спектры/сек = 20)
Объем вводимой пробы (мкл) 1,00
Растворители образца: соотношение DMSO : МеОН : ACN 1:3:3
LC/UV/MS - Способ 3 и 3а
Прибор для LC: Acquity Waters UPLC (или эквивалентный), соединенный с 2996 PDA детектором
Колонка: Acquity UPLC CSH С18 1,7 мкм, 130А, 50×2,1 мм
Температура колонки (°C) 50,0
Подвижные фазы: 0.025М HCOONH4, рН 3 (A); ACN+0,1% НСООН (В)
Поток (мл/мин) 0,35 (разделение в MS 1:3 в способе 3) (разделение 1:10 в способе 3а)
Время остановки (мин) 10
Градиент:
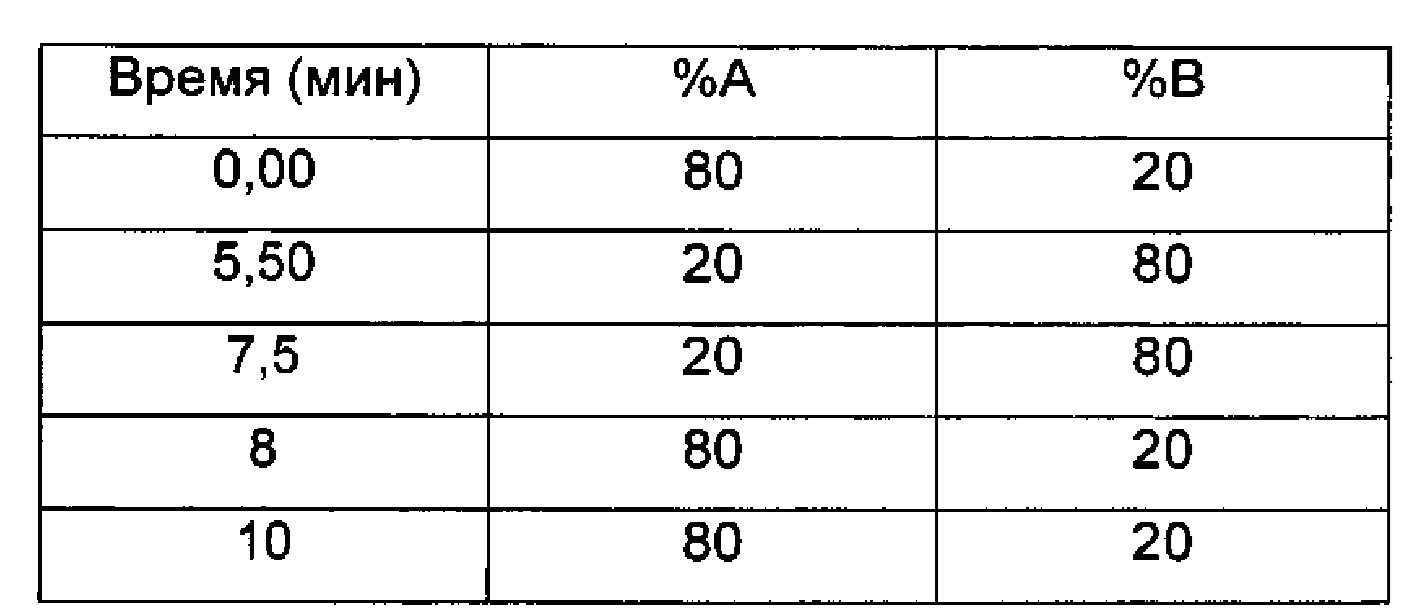
UV детектирование: BPI детектирование (начальная длина волны 210 нм, конечная длина волны 400 нм, частота отбора проб спектры/сек = 20)
Объем вводимой пробы (мкл) 2,00
Растворители образца: H2O/ACN 80/20
LC/UV/MS - Способ 4
Прибор для LC: Acquity Waters UPLC (или эквивалентный), соединенный с 2996 PDA детектором
Колонка: Acquity UPLC ВЕН С18 1,7 мкм, 50×2,1 мм
Температура колонки (°C) 40,0
Подвижные фазы: 95:5 H2O:ACN+(0,1% НСООН) (А); 5:95 H2O:ACN+(0,1% НСООН) (В)
Поток (мл/мин) 1 мл/мин
Время остановки (мин) 2
Градиент:
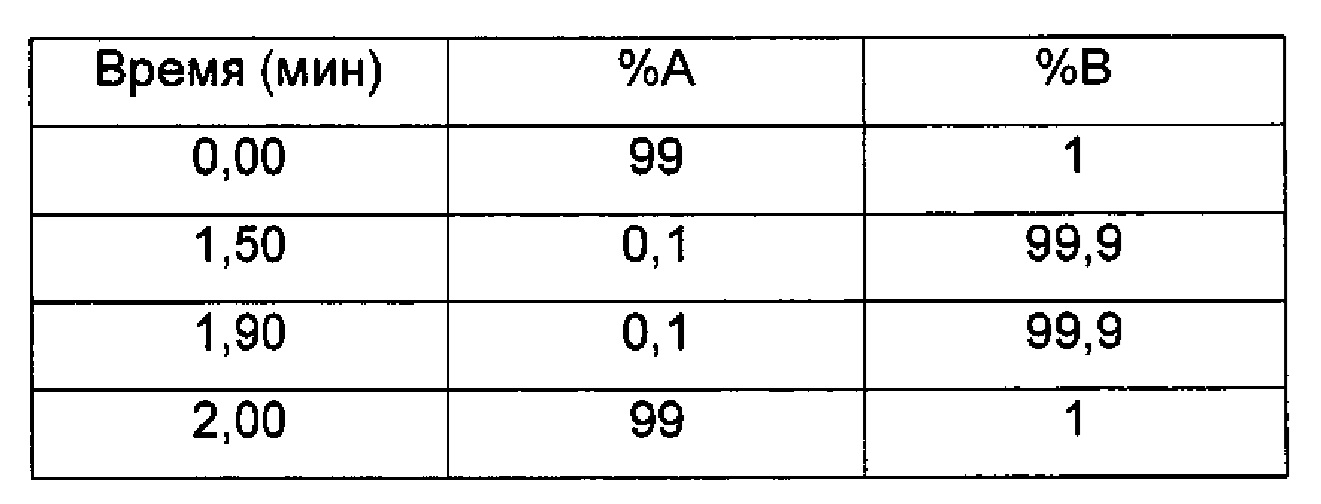
UV детектирование: BPI детектирование (начальная длина волны 210 нм, конечная длина волны 400 нм, частота отбора проб спектры/сек = 20)
Объем вводимой пробы (мкл) 1,00
LC/UV/MS - Способ 5
Прибор для UPLC: Waters Acquity
Колонка: Kinetex 1,7u PFP 100°, 100×2,1 мм (Phenomenex)
Температура колонки (°C): 55
Подвижные фазы: 0,025М HCOONH4, рН 3 (A); ACN (В)
Поток (мл/мин): 0,45 (разделение в MS 1:3)
Время остановки (мин): 10
Градиент:
Время (мин) %А %В
Градиент:
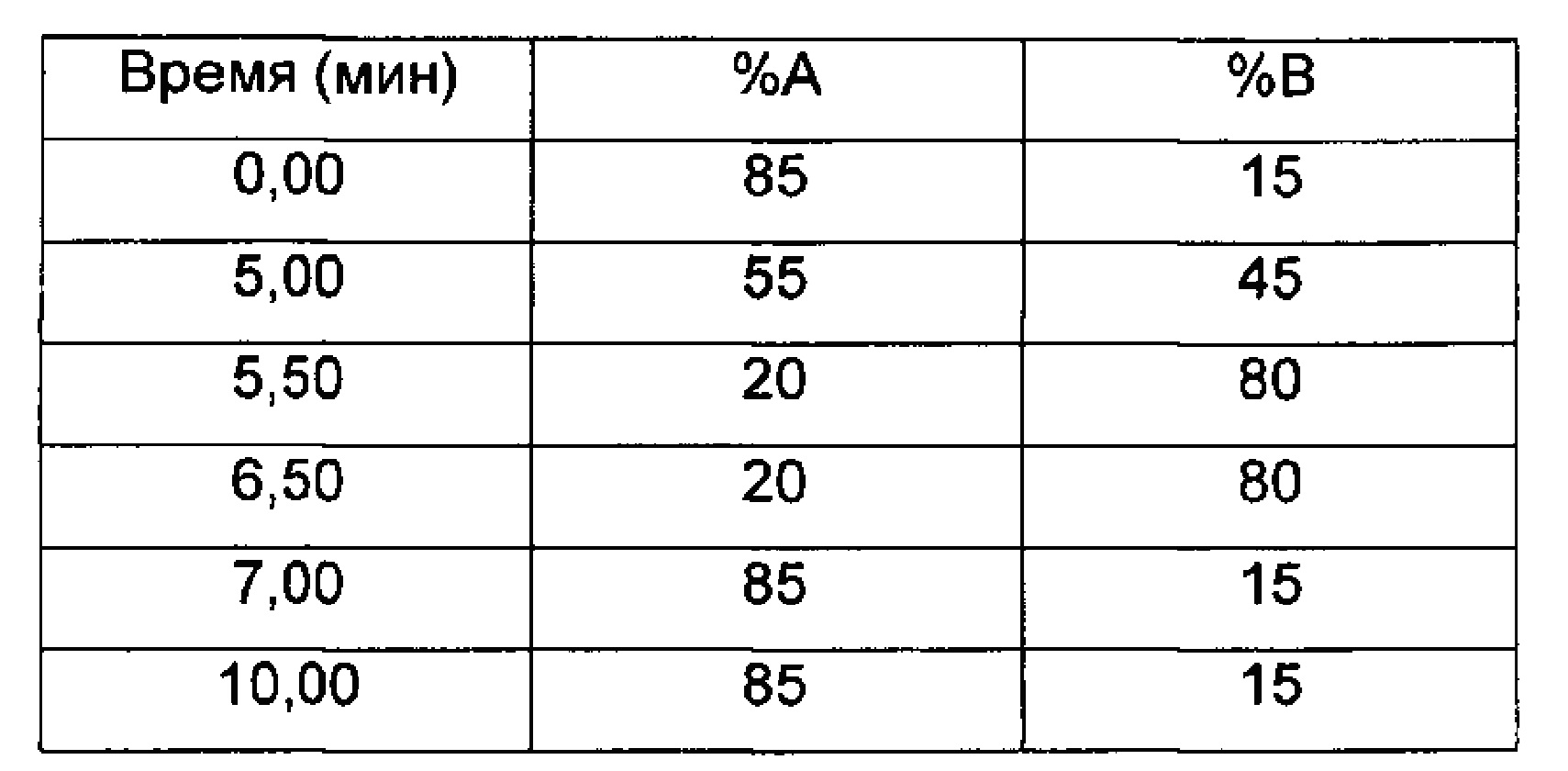
УФ-детектирование: DAD (детектор с диодной матрицей)
Диапазон обнаружения УФ (нм): 210-400
Объем вводимой пробы (мкл): 2
Растворитель образца: Ацетонитрил
LC/UV/MS - Способ 6
Прибор для LC: Acquity Waters UPLC (или эквивалентный), соединенный с 2996 PDA детектором
Колонка: Acquity UPLC CSH С18 1,7 мкм, 130А, 50×2,1 мм
Температура колонки (°C) 40,0
Подвижные фазы: 95:5 H2O:ACN+(0,1% НСООН) (А); 5:95 H2O:ACN+(0,1% НСООН) (В)
Поток (мл/мин) 1 мл/мин
Время остановки (мин) 2
Градиент:
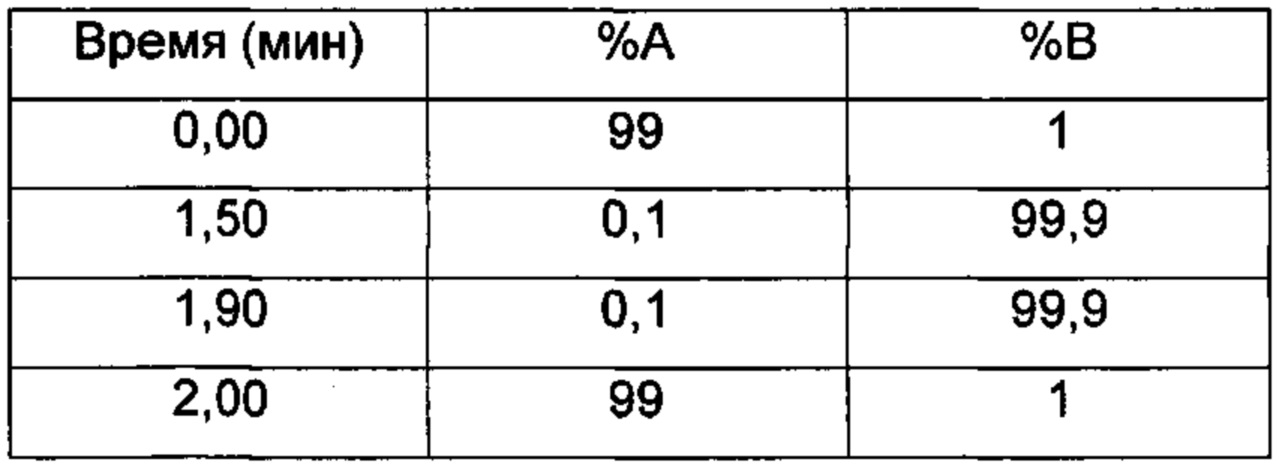
UV детектирование: BPI детектирование (начальная длина волны 210 нм, конечная длина волны 400 нм, частота отбора проб спектры/сек = 20)
Объем вводимой пробы (мкл) 1,00
ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И ПРИМЕРЫ
Промежуточное соединение А1: 4-хлор-2Н-хромен-3-карбальдегид
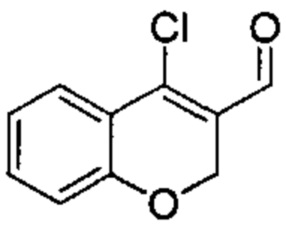
Хроман-4-он (2 г, 13,50 ммоль) растворяли в DCM (45 мл) и DMF (1,6 мл, 20,25 ммоль), затем по каплям под азотом добавляли POCl3 (3,77 мл, 40,5 ммоль) и смесь нагревали с обратным холодильником в течение 6 ч и при КТ в течение ночи. Реакционную смесь разбавляли DCM, промывали водой, насыщенным NaClводн., сушили над Na2SO4 и упаривали досуха. Неочищенное вещество подвергали хроматографии на силикагеле со смесями гексан\EtOAc с получением указанного в заголовке соединения (2,1 г, выход 80%) в виде желтого твердого вещества.
UPLC-MS: 1,99 мин, 194,8 [М+Н]+, способ 1.
Промежуточное соединение В1: 4-фенил-2Н-хромен-3-карбальдегид
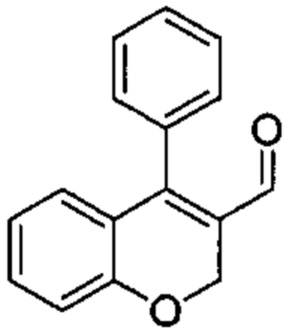
4-Хлор-2Н-хромен-3-карбальдегид (Промежуточное соединение А1, 1 г, 5,14 ммоль), фенилбороновую кислоту (0,75 г, 6,17 ммоль) и Cs2CO3 (2,0 г, 6,17 ммоль) растворяли в DMF (19 мл) и дезоксигенировали под аргоном в течение 5 мин, после чего добавляли Pd(PPh3)2Cl2 (0,18 г, 0,257 ммоль) и смесь нагревали под азотом при 45°C в течение 1 ч и при 65°C в течение 4 ч. Реакционную смесь оставляли охлаждаться до КТ, распределяли между AcOEt и 1М HClводн., промывали дважды водой, один раз насыщенным NaClводн., затем органический слой сушили над Na2SO4 и упаривали досуха. Неочищенное вещество подвергали хроматографии на силикагеле со смесями гексан\EtOAc с получением указанного в заголовке соединения (0,9 г, выход 74%) в виде желтого твердого вещества.
UPLC-MS: 2,14 мин, 237,0 [M+H]+, способ 1.
Промежуточное соединение В2: 2-оксо-4-фенил-2Н-хромен-3-карбальдегид
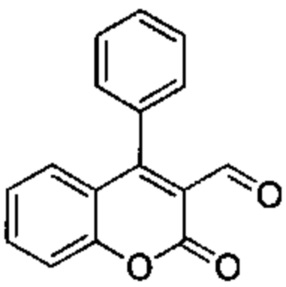
4-Хлор-2-оксо-2Н-хромен-3-карбальдегид (600 мг, 2,88 ммоль), Pd-бис(дифенилфосфин)хлорид (202 мг, 0,288 ммоль), трибутил(фенил)станнан (1,47 г, 4,03 ммоль), фторид цезия (1,31 г, 8,63 ммоль) подвергали взаимодействию в диоксане (5,9 мл) при 90°C в течение 2 ч. Реакционную смесь распределяли между NH4Clводн. (100 мл) и AcOEt (30 мл), органический слой промывали рассолом, сушили над Na2SO4 и сушили при пониженном давлении, неочищенное вещество подвергали хроматографии на силикагеле со смесями гексан\EtOAc с получением указанного в заголовке соединения (407 мг, выход 57%).
UPLC-MS: 4,55 мин, 251 [М+Н]+, способ 2.
Промежуточное соединение В3: 4-(5-(4,4,5,5-тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-2Н-хромен-3-карбальдегид
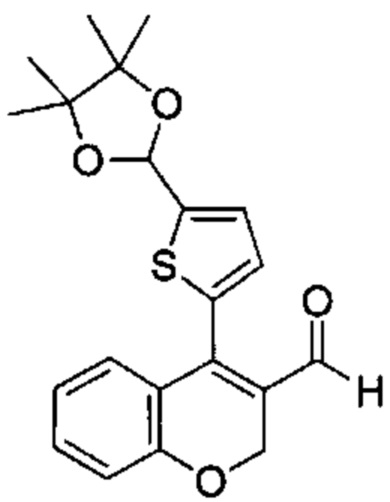
4-Хлор-2Н-хромен-3-карбальдегид (Промежуточное соединение А1, 3,5 г, 17,98 ммоль), 4,4,5,5-тетраметил-2-(5-(4,4,5,5-тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-1,3,2-диоксаборолан (5,07 г, 14,99 ммоль; получали, как описано в WO 2015/091685 на стр. 138) и K3PO4 H2O (10,35 г, 45 ммоль) растворяли в THF (100 мл) и воде (100 мл). В течение 15 мин барботировали Ar, после чего добавляли XPhos Pd G2 (0,825 г, 1,049 ммоль). Барботирование продолжали в течение следующих 10 мин, мутную коричневую смесь перемешивали в атмосфере Ar при КТ в течение ночи. Смесь распределяли между этилацетатом и водой, органическую фазу обезвоживали над сульфатом натрия и растворитель выпаривали с получением указанного в заголовке соединения (5,4 г, 97%) в виде красного масла. Неочищенное вещество использовали на следующей стадии синтеза без дополнительной очистки.
UPLC-MS: 2,64 мин, 371,2 [(М+Н)]+, 272,16 [(М-100)]+, способ 6 @
Промежуточное соединение С1: (4-фенил-2Н-хромен-3-ил)метанол
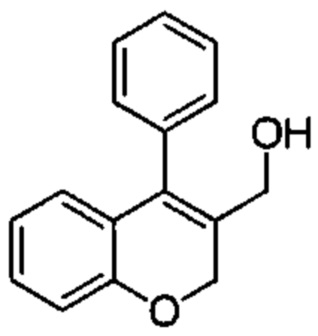
4-Фенил-2Н-хромен-3-карбальдегид (Промежуточное соединение В1, 400 мг, 1,69 ммоль) и NaBH4 (256 мг, 6,77 ммоль) подвергали взаимодействию в EtOH (8 мл) под азотом в течение 30 мин при КТ. Реакционную смесь гасили путем добавления 1М HClводн. (10 мл) и смесь распределяли между AcOEt и 1М HClводн.. Органический слой промывали насыщенным NaClводн., сушили над Na2SO4 и упаривали досуха с получением указанного в заголовке соединения (382 мг, выход 95%) в виде бесцветного масла.
UPLC-MS: 1,94 мин, 220,9 [(М+Н)-H2O]+, способ 1.
Промежуточное соединение С2: 3-(гидроксиметил)-4-фенил-2Н-хромен-2-он
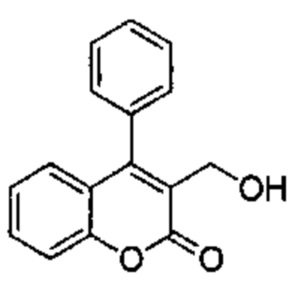
2-Оксо-4-фенил-2Н-хромен-3-карбальдегид (Промежуточное соединение В2, 191 мг, 0,763 ммоль) и тетрагидроборат натрия (29 мг, 0,763 ммоль) подвергали взаимодействию в метаноле (7,5 мл) при КТ. Реакционную смесь распределяли между AcOEt/NH4Clводн. 5% 1/1 (10 мл). Органический слой сушили над Na2SO4 и сушили при пониженном давлении с получением указанного в заголовке соединения (180 мг, 93%), которое использовали на следующей стадии без какой-либо дополнительной очистки.
UPLC-MS: 1,76 мин, 235 [(М+Н)-H2O]+, способ 1.
Промежуточное соединение С3: 1-(4-фенил-2Н-хромен-3-ил)этанол
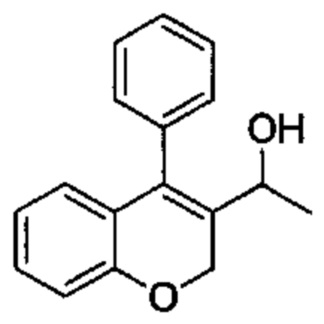
4-Фенил-2Н-хромен-3-карбальдегид (Промежуточное соединение В1, 520 мг, 2,20 ммоль) растворяли в безводном THF (8 мл) при 0°C, затем по каплям в течение 5 мин добавляли 3М метилмагнийбромид в Et2O (1,47 мл, 4,40 ммоль). Смесь перемешивали при 0°C в течение 15 мин, затем гасили путем добавления насыщенного NH4Clводн. и экстрагировали с помощью AcOEt. Органический слой промывали насыщенным NaClводн., сушили над Na2SO4 и упаривали досуха с получением указанного в заголовке соединения (503 мг, выход 91%) в виде желтого масла.
UPLC-MS: 2,00 мин, 234,8 [(М+Н)-H2O]+, способ 1.
Промежуточное соединение С4: 3-(1-гидроксиэтил)-4-фенил-2Н-хромен-2-он
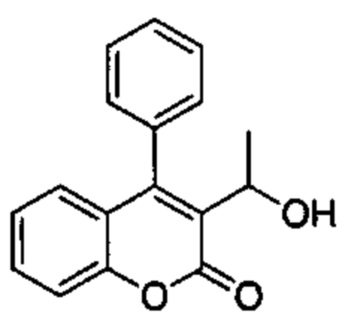
2-Оксо-4-фенил-2Н-хромен-3-карбальдегид (Промежуточное соединение В2, 585 мг, 2,34 ммоль) и 3М метилмагнийбромид в THF (0,86 мл, 2,57 ммоль) подвергали взаимодействию в сухом THF (7,5 мл) при -15°C в течение 15 мин. Реакционную смесь распределяли между NH4Clводн. (1 мл) и AcOEt (2 мл). Фазы разделяли, органический слой сушили над сульфатом натрия и упаривали при пониженном давлении с получением неочищенного вещества, которое подвергали хроматографии на силикагеле со смесями гексан\EtOAc с получением указанного в заголовке соединения (250 мг, 40%).
UPLC-MS: 1,06 мин, 249 [М+Н]+, способ 4.
Промежуточное соединение С5: 1-(4-(5-(4,4,5,5-тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-2Н-хромен-3-ил)этан-1-ол
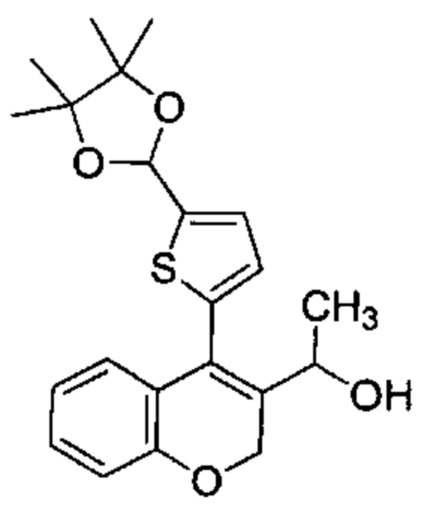
4-(5-(4,4,5,5-Тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-2Н-хромен-3-карбальдегид (Промежуточное соединение В3, 5,55 г, 14,98 ммоль) и 3М метилмагнийбромид в THF (9,99 мл, 30,0 ммоль) подвергали взаимодействию в сухом THF (200 мл) при 0°C в течение 2 ч. Реакционную смесь распределяли между NН4Clводн. (1 мл) и AcOEt (2 мл). Фазы разделяли, органический слой сушили над сульфатом натрия и упаривали при пониженном давлении с получением неочищенного вещества, которое подвергали хроматографии на силикагеле со смесями гексан\EtOAc с получением указанного в заголовке соединения (5,55 г, 96%).
UPLC-MS: 1,31 мин, 369,2 [(М+Н)-H2O]+, способ 6.
Промежуточное соединение D1: 3-(бромметил)-4-фенил-2Н-хромен-2-он
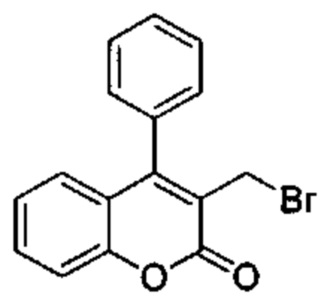
3-(Гидроксиметил)-4-фенил-2Н-хромен-2-он (Промежуточное соединение С2, 160 мг, 0,634 ммоль), 1М трибромфосфин в DCM (1,08 мл, 1,08 ммоль), подвергали взаимодействию в DCM (1,6 мл) при КТ. Растворители выпаривали с получением указанного в заголовке соединения (239 мг), которое использовали на следующей стадии без какой-либо дополнительной очистки.
UPLC-MS: 2,17 мин, 234 [М-Br]+, способ 1.
Промежуточное соединение D2: 3-(1-бромэтил)-4-фенил-2Н-хромен-2-он
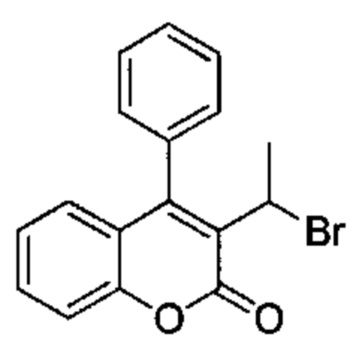
3-(1-Гидроксиэтил)-4-фенил-2Н-хромен-2-он (Промежуточное соединение С4, 250 мг, 0,94 ммоль) и 1М PBr3 в DCM (1,57 мг, 1,57 ммоль) подвергали взаимодействию в DCM (2,5 мл) при КТ в течение 3 ч. Растворитель выпаривали при пониженном давлении и неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 30 g SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (203 мг, 66%).
UPLC-MS: 2,24 мин, 330,66, 328,66 [(М+Н)]+, способ 1.
Промежуточное соединение Е1: гидрохлорид (4-фенил-2Н-хромен-3-ил)метанамина
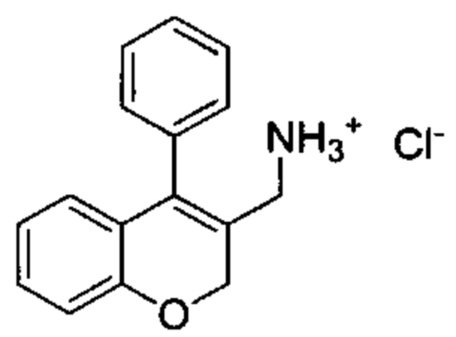
(4-Фенил-2Н-хромен-3-ил)метанол (Промежуточное соединение С1) DBU (0,48 мл, 3,19 ммоль) и дифенилфосфорилазид (0,69 мл, 3,19 ммоль) подвергали взаимодействию в THF (8 мл) при КТ в течение 3 ч. Реакционную смесь распределяли между водой и AcOEt, органическую фазу промывали 1М HClводн., насыщенным NaClводн., сушили над Na2SO4 и упаривали досуха. Неочищенное вещество растворяли в безводном THF (10 мл), затем по каплям под азотом добавляли 1М раствор LiAlH4 в THF (3,19 мл, 3,19 ммоль). Реакционную смесь перемешивали в течение 1 ч при КТ и гасили путем добавления AcOEt и 1М HClводн.. Водный слой нейтрализовывали 1М NaОНводн. до рН приблизительно 9-10 и экстрагировали дважды с помощью AcOEt. Органический слой сушили над Na2SO4 и упаривали досуха. Неочищенное масло растворяли в Et2O с 2 мл 4М HCl в диоксане и смесь упаривали досуха с получением указанного в заголовке соединения (290 мг, выход 66%) в виде желтого твердого вещества.
UPLC-MS: 1,45 мин, 220,9 [(M+H)-NH3]+, способ 1.
Промежуточное соединение Е2: гидрохлорид 1-(4-фенил-2Н-хромен-3-ил)этанамина
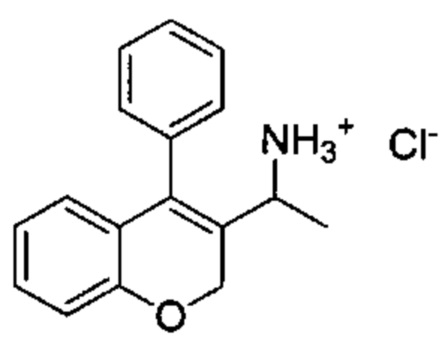
Указанное в заголовке соединение получали аналогично промежуточному соединению Е1 из 1-(4-фенил-2Н-хромен-3-ил)этанола (Промежуточное соединение С3, 350 мг, 1,39 ммоль) с получением указанного в заголовке соединения (22 мг, выход 6%).
UPLC-MS: 1,55 мин, 235,0 [(M+H)-NH3]+, способ 1.
Промежуточное соединение F1: 3-йод-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин
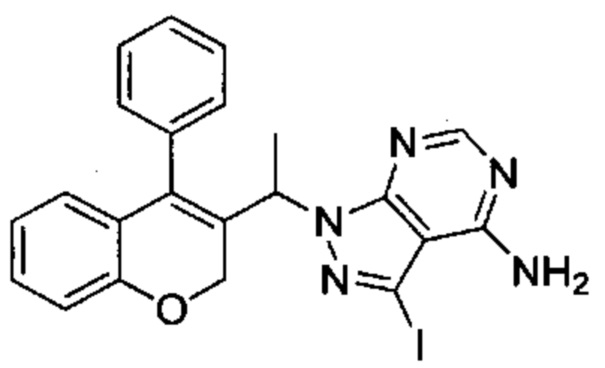
(4-Фенил-2Н-хромен-3-ил)этанол (Промежуточное соединение С3, 1,60 г, 6,34 ммоль), 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин (1,99 г, 7,61 ммоль) и PPh3 (2,0 г, 7,61 ммоль) перемешивали в THF (42 мл) в течение 5 мин при КТ, после чего по каплям при 0°C добавляли DIAD (1,22 мл, 6,28 ммоль). Реакционную смесь перемешивали при 0°C в течение 5 мин и при КТ в течение 1 ч, затем распределяли между AcOEt и 1М HClводн.. Органический слой промывали три раза водой, один раз насыщенным NaClводн., сушили над Na2SO4 и упаривали досуха. Неочищенное вещество подвергали хроматографии на силикагеле со смесями гексан\EtOAc с получением указанного в заголовке соединения (667 мг, выход 21%) в виде желтоватого твердого вещества.
UPLC-MS: 1,32 мин, 496,0 [М+Н]+, способ 4.
Промежуточное соединение F2: 3-(1-(4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-4-фенил-2Н-хромен-2-он
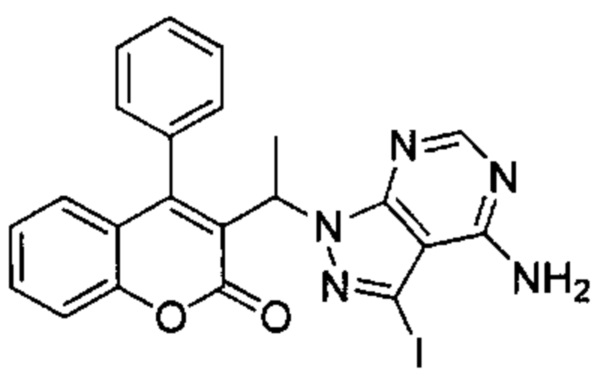
3-(1-Бромэтил)-4-фенил-2Н-хромен-2-он (Промежуточное соединение D2, 101 мг, 0,307 ммоль), 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин (104 мг, 0,40 ммоль) и K2CO3 (55 м, 0,399 ммоль) подвергали взаимодействию в DMF (1 мл) при 60°C в течение 18 ч. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке С18 (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (75 мг, 48%).
UPLC-MS: 1,15 мин, 510 [М+Н]+, способ 4.
Промежуточное соединение F3: 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-4-фенил-2Н-хромен-2-он
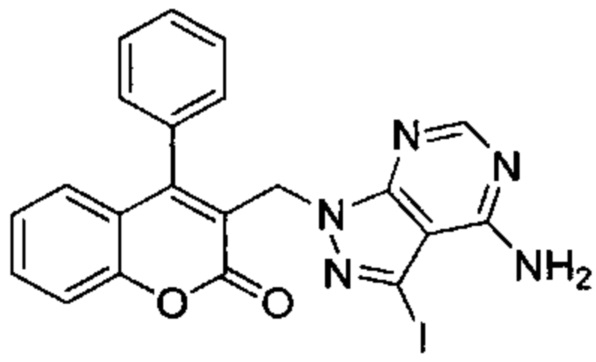
3-(Бромметил)-4-фенил-2Н-хромен-2-он (Промежуточное соединение D1, 102 мг, 0,324 ммоль), K2CO3 (54 мг, 0,40 ммоль) и 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин (101 мг, 0,39 ммоль) подвергали взаимодействию в DMF (1,2 мл, 15,50 ммоль) при 80°C в течение 3 ч. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке С18 (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (20 мг, 12,5%).
UPLC-MS: 1,84 мин, 496 [М+Н]+, способ 4.
Промежуточное соединение F4: 3-йод-1-(1-(4-(5-(4,4,5,5-тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин
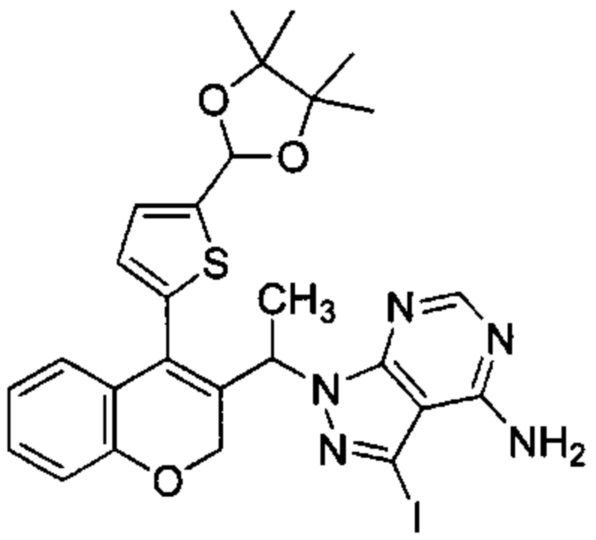
1-(4-(5-(4,4,5,5-Тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-2Н-хромен-3-ил)этанол (Промежуточное соединение С5, 2 г, 5,17 ммоль), 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин (1,621 г, 6,21 ммоль) перемешивали в THF (42 мл) в течение 5 мин при КТ, после чего добавляли PPh3 (1,629 г, 6,21 ммоль), а затем DIAD (1,257 мл, 5,17 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 5 мин и при КТ в течение ночи, затем распределяли между AcOEt и насыщенным NH4Clводн.. Органический слой промывали насыщенным NaClводн., сушили над Na2SO4 и упаривали досуха. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке С18 (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (193 мг, 5%).
UPLC-MS: 1,46 мин, 629,33 [М+Н]+, 529,4 [(М+Н)-100]+, способ 6.
Промежуточное соединение G1: 5-(3-(1-(4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-2Н-хромен-4-ил)тиофен-2-карбальдегид
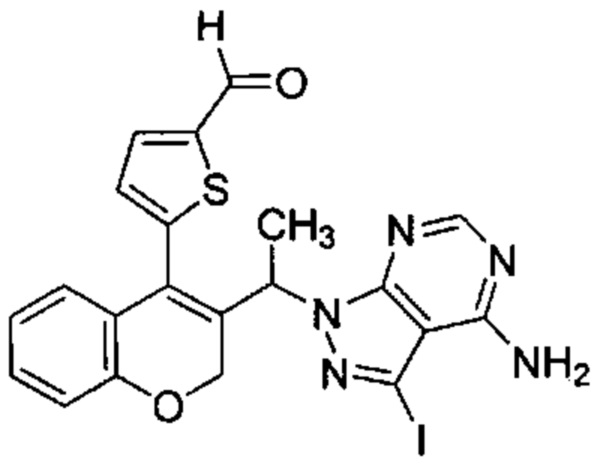
3-Йод-1-(1-(4-(5-(4,4,5,5-тетраметил-1,3-диоксолан-2-ил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин (Промежуточное соединение F4, 193 мг, 0,307 ммоль) растворяли в ACN (12 мл), затем добавляли 1М HClводн. до рН менее 2 и смесь перемешивали при КТ в течение ночи. ACN выпаривали с получением указанного в заголовке продукта (150 мг, 92%), который непосредственно использовали на следующей стадии без дополнительной очистки.
UPLC-MS: 1,17 мин, 529,73 [М+Н]+, способ 6.
Промежуточное соединение Н1: 3-йод-1-(1-(4-(5-((4-метилпиперазин-1-ил)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин
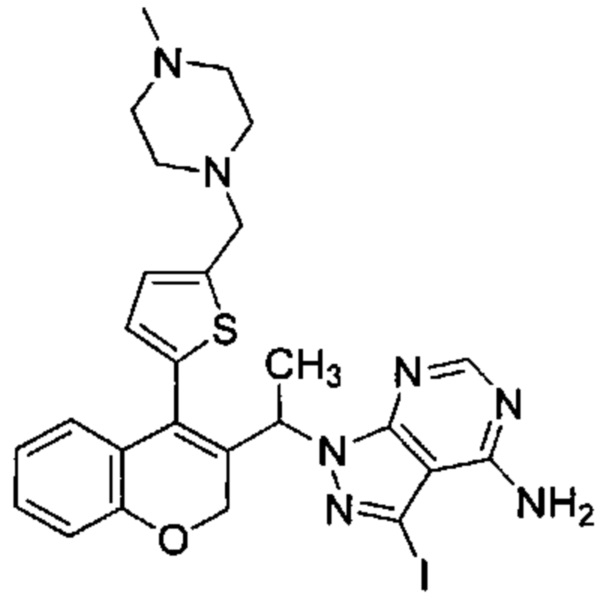
В статической атмосфере Ar 5-(3-(1-(4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-2Н-хромен-4-ил)тиофен-2-карбальдегид (Промежуточное соединение G1, 162 мг, 0,306 ммоль) суспендировали в смеси DCM\DMF (15 мл\5 мл), затем добавляли 1-метилпиперазин (0,175 мл, 1,530 ммоль), уксусную кислоту (0,088 мл, 1,530 ммоль) и смесь перемешивали при КТ в течение 10 минут. Затем добавляли триацетоксигидроборат натрия (0,370 мл, 1,530 ммоль) и смесь перемешивали при КТ в течение 4 ч. После дополнительного добавления 1-метилпиперазина (0,175 мл, 1,530 ммоль) и триацетоксигидробората натрия (0,370 мл, 1,530 ммоль) реакционную смесь перемешивали при КТ в течение ночи. Растворитель выпаривали, смесь распределяли между изопропилацетатом и 1М NaOHводн., затем органическую фазу промывали водой и насыщенным раствором NaCl. Растворитель удаляли и неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке С18 (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (42 мг, 22,4%).
UPLC-MS: 0,71 мин, 613,70 [М+Н]+, способ 6.
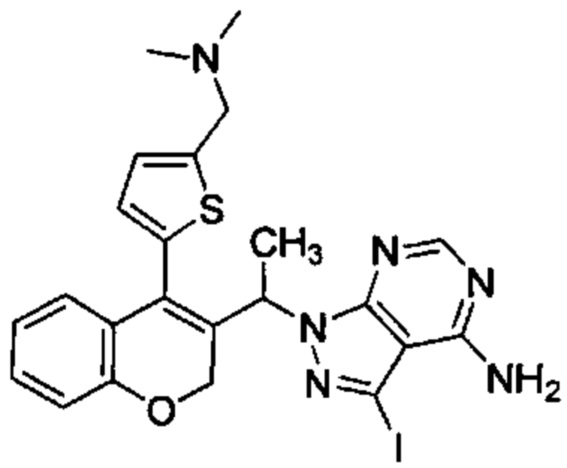
Промежуточное соединение Н2: 1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин
и
Промежуточное соединение Н3: (5-(3-(1-(4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-2Н-хромен-4-ил)тиофен-2-ил)метанол
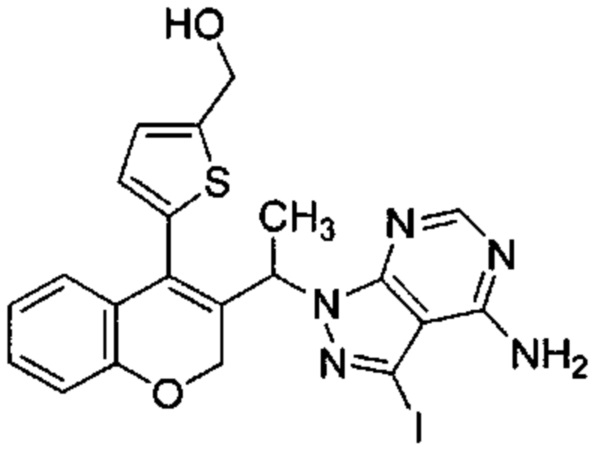
В статической атмосфере Ar 5-(3-(1-(4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-2Н-хромен-4-ил)тиофене-2-карбальдегид (Промежуточное соединение G1, 177 мг, 0,334 ммоль) суспендировали в смеси DCM\диоксан\ацетонитрил (15 мл/2,5 мл/2,5 мл), затем добавляли 2,0 М диметиламин в THF (0,018 мл, 0,334 ммоль), уксусную кислоту (0,096 мл, 1,672 ммоль) и смесь перемешивали при КТ в течение 10 минут. Затем добавляли триацетоксигидроборат натрия (0,404 мл, 1,672 ммоль) и смесь перемешивали при КТ в течение 4 ч. После дополнительного добавления 2,0 М диметиламина в THF (0,018 мл, 0,334 ммоль) и триацетоксигидробората натрия (0,404 мл, 1,672 ммоль) реакционную смесь перемешивали при КТ в течение ночи. Добавляли 1М HClводн. и смесь перемешивали в течение 10 мин. Органические растворители удаляли и неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке С18 (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением
Промежуточного соединения Н2: (58,5 мг, 31%) UPLC-MS: 0,68 мин, 558,32 [М+Н]+, способ 6 и
Промежуточного соединения Н3: (53,5 мг, 30%), UPLC-MS: 1,06 мин, 531,47 [М+Н]+, способ 6.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ:
Пример 1
4-Амино-6-((4-фенил-2Н-хромен-3-ил)метиламино)пиримидин-5-карбонитрил
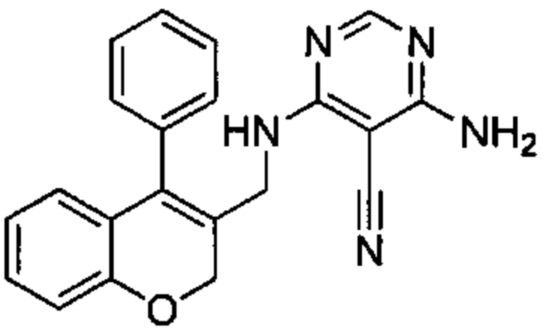
Гидрохлорид 1-(4-фенил-2Н-хромен-3-ил)метанамина (Промежуточное соединение Е1, 80 мг, 0,292 ммоль), 4-амино-6-хлорпиримидин-5-карбонитрил (54 мг, 0,351 ммоль) и DIEA (0,10 мл, 0,584 ммоль) подвергали взаимодействию в диоксане (15 мл) при 80°C в течение 3,5 ч, затем гасили путем добавления 1М HClводн. (1 мл). Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 60g SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (38 мг, выход 36%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 7.85 (s, 1 Н), 7.36-7.57 (m, 4 Н), 7.05-7.33 (m, 5 Н), 6.71-6.90 (m, 2 Н), 6.41-6.58 (m, 1 Н), 4.68-4.85 (m, 2 Н), 3.72-4.04 (m, 2 Н).
UPLC-MS: 5,26 мин, 356,1 [М+Н]+, способ 5.
Пример 2
N-((4-Фенил-2Н-хромен-3-ил)метил)-9Н-пурин-6-амин
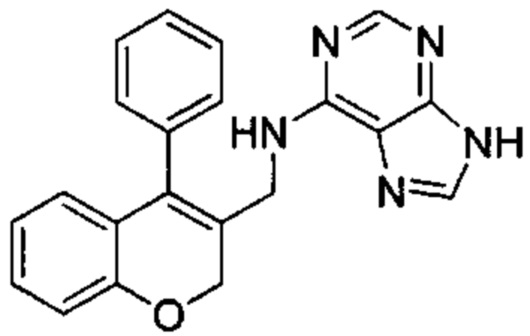
Указанное в заголовке соединение получали аналогично соединению примера 1 из гидрохлорида 1-(4-фенил-2Н-хромен-3-ил)метанамина (Промежуточное соединение Е1, 80 мг, 0,292 ммоль) и 6-хлор-9Н-пурина (54 мг, 0,351 ммоль) с получением указанного в заголовке соединения (18 мг, 0,051 ммоль, выход 18%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 12.59-13.04 (bs, 1 Н), 7.98-8.26 (m, 2 Н), 7.73-7.91 (m, 1 Н), 7.28-7.59 (m, 6 Н), 7.11 (m, 1 Н), 6.66-6.95 (m, 2 Н), 6.53 (d, J=7,06 Гц, 1 Н), 4.82 (s, 2 Н), 3.99-4.28 (m, 2 Н).
UPLC-MS: 4,82 мин, 356,1 [М+Н]+, способ 5.
Пример 3
4-Амино-6-(1-(4-фенил-2Н-хромен-3-ил)этиламино)пиримидин-5-карбонитрил
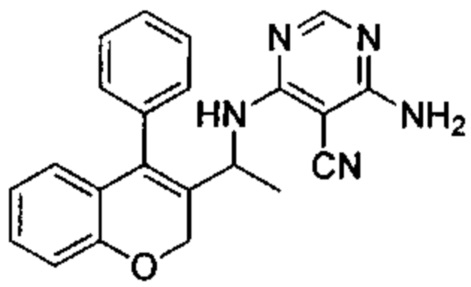
Указанное в заголовке соединение получали аналогично соединению примера 1 из гидрохлорида 1-(4-фенил-2Н-хромен-3-ил)этанамина (Промежуточное соединение Е2, 20 мг, 0,080 ммоль) и 4-амино-6-хлорпиримидин-5-карбонитрила (32 мг, 0,207 ммоль) с получением указанного в заголовке соединения (7 мг, выход 25%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 7.95 (s, 1 Н), 7.34-7.57 (m, 4 Н), 7.01-7.31 (m, 5 Н), 6.71-6.89 (m, 2 Н), 6.34-6.48 (m, 1 Н), 4.96-5.05 (m, 1 Н), 4.70-4.82 (m, 1 Н), 4.54-4.67 (m, 1 Н), 1.27 (d, J=7,06 Гц, 3 Н).
UPLC-MS: 5,64 мин, 370,1 [М+Н]+, способ 3а.
Пример 4
3-(4-Амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол
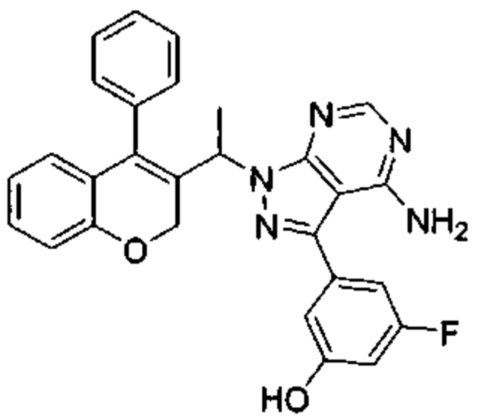
3-Йод-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин (Промежуточное соединение F1, 60 мг, 0,121 ммоль), (3-фтор-5-гидроксифенил)бороновую кислоту (38 мг, 0,242 ммоль), S-Phos Pd G2 (8,73 мг, 0,012 ммоль) и K3PO4 H2O (118 мг, 0,363 ммоль) диспергировали в THF (2 мл) и дезоксигенировали под аргоном в течение 5 мин, после чего добавляли воду (0,5 мл) и смесь нагревали с помощью микроволнового излучения в течение 40 мин при 85°C. Реакционную смесь разбавляли AcOEt и промывали дважды водой, один раз насыщенным NaClводн., органический слой сушили над Na2SO4 и сушили при пониженном давлении. Неочищенное вещество подвергали хроматографии на силикагеле со смесями DCM/AcOEt с получением указанного в заголовке соединения (37 мг, выход 64%) в виде желтоватого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.19 (s, 1 Н), 8.16 (s, 1 Н), 7.43-7.57 (m, 3 Н), 7.27 (d, J=7,06 Гц, 2 Н), 7.06-7.20 (m, 1 Н), 6.75-6.97 (m, 4 Н), 6.67 (dt, J=11,03, 2,21 Гц, 1 Н), 6.47 (dd, J=7,72, 1,54 Гц, 1 Н), 5.55-5.73 (m, 1 Н), 4.67-5.23 (m, 2 Н), 1.66 (d, J=7,06 Гц, 3 Н).
UPLC-MS: 5,39 мин, 480,0 [М+Н]+, способ 3а.
Пример 5
5-(4-Амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ол
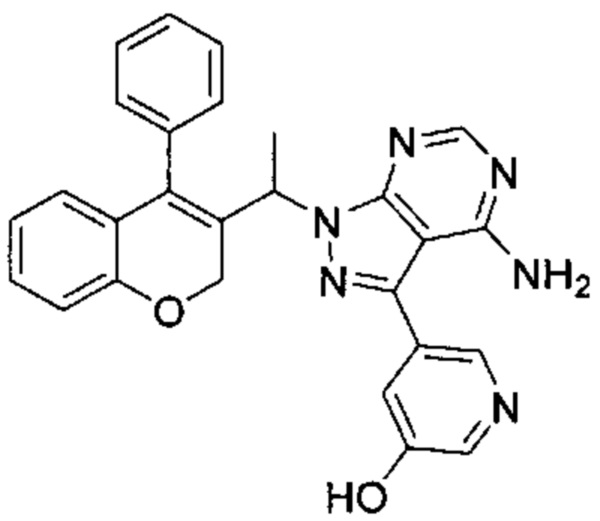
3-Йод-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин (Промежуточное соединение F1 (60 мг, 0,121 ммоль)), 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ол (53,6 мг, 0,242 ммоль), S-Phos Pd G2 (8,73 мг, 0,012 ммоль) и K3PO4 H2O (118 мг, 0,363 ммоль) диспергировали в THF (2 мл) и дезоксигенировали под Ar в течение 5 мин, после чего добавляли воду (0,5 мл), затем реакционную смесь нагревали с помощью микроволнового излучения в течение 80 мин при 85°C. Реакцию гасили путем добавления 2М HClводн. (5 мл) и неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (48 мг, выход 86%) в виде желтоватого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.98-10.46 (bs, 1 Н), 8.33 (d, J=1,32 Гц, 1 Н), 8.22 (d, J=2,65 Гц, 1 Н), 8.17 (s, 1 Н), 7.40-7.55 (m, 4 Н), 7.27 (d, J=6,62 Гц, 2 Н), 7.14 (m, 1 Н), 6.74-6.88 (m, 2 Н), 6.48 (dd, J=7,94, 1,32 Гц, 1 Н), 5.63 (d, J=7,06 Гц, 1 Н), 4.57-5.28 (m, 2 Н), 1.67 (d, J=7,06 Гц, 3 Н).
UPLC-MS: 4,27 мин, 462,9 [М+Н]+, способ 3а.
Пример 6
3-(1-(4-Амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-4-фенил-2Н-хромен-2-он
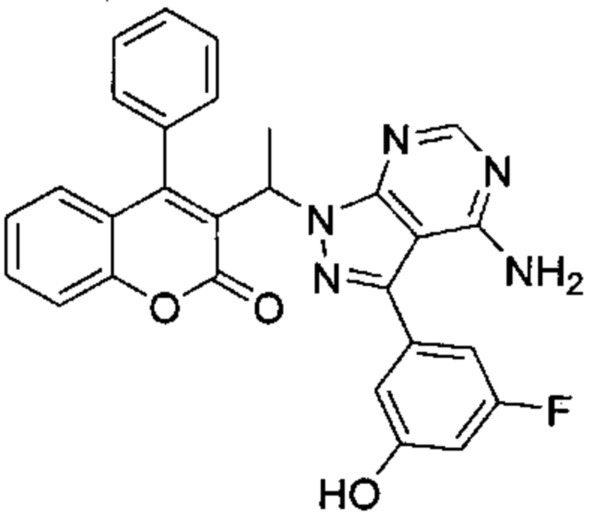
3-(1-(4-Амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-4-фенил-2Н-хромен-2-он (Промежуточное соединение F2 (75 мг, 0,147 ммоль)), 3-фтор-5-гидроксифенилбороновую кислоту (46 мг, 0,295 ммоль), PdCl2(dppf) (12,9 мг, 0,018 ммоль) и карбонат калия (41 мг, 0,295 ммоль) подвергали взаимодействию в диоксане (730 мкл) при 80°C в течение 1 ч. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (62 мг, 85%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.24 (s, 1 Н), 8.05 (s, 1 Н), 7.58-7.66 (m, 1 Н), 7.33-7.53 (m, 5 Н), 7.17-7.28 (m, 1 Н), 6.91-6.96 (m, 1 Н), 6.79-6.83 (m, 1 Н), 6.69-6.79 (m, 2 Н), 6.59-6.67 (m, 1 Н), 5.52-5.73 (m, 1 Н), 1.79-2.00 (m, 3 Н).
UPLC-MS: 4,45 мин, 494 [М+Н]+, способ 3.
Пример 7
3-((4-Амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-4-фенил-2Н-хромен-2-он
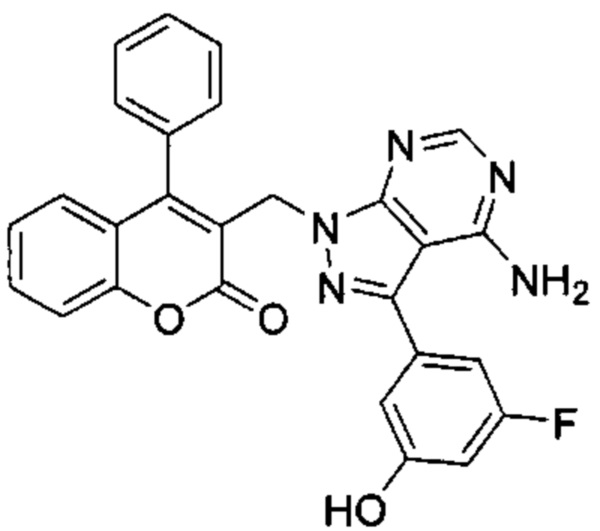
3-((4-Амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-4-фенил-2Н-хромен-2-он (Промежуточное соединение F3 (20 мг, 0,04 ммоль)), 3-фтор-5-гидроксифенилбороновую кислоту (12,6 мг, 0,08 ммоль), карбонат цезия (26,3 мг, 0,08 ммоль) и Pd(PPh3)4 (3,7 мг, 3,23 мкмоль) подвергали взаимодействию в DMF (200 мкл) при микроволновом излучении при 120°C в течение 1 ч. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (9 мг, 47%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.14 (s, 1 Н), 7.18-7.75 (m, 15 Н), 5.36 (s, 2 Н).
UPLC-MS: 4,00 мин, 480 [М+Н]+, способ 3.
Пример 7а
1-(5-(4-Амино-1-(1-(4-фенил-2Н-хромен-2-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ил)-2,2,2-трифторэтан-1-ол
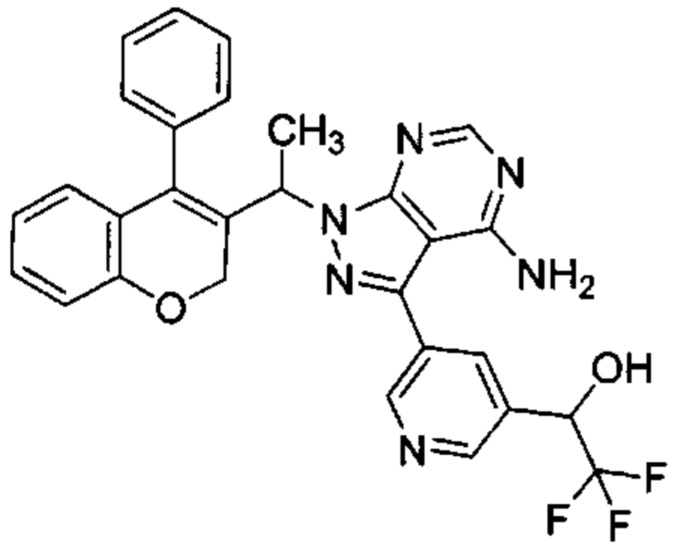
3-Йод-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин (Промежуточное соединение F1 (50 мг, 0,101 ммоль)), 2,2,2-трифтор-1-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)этанол (92 мг, 0,303 ммоль) и K3PO4 Н2О (70 мг, 0,303 ммоль) диспергировали в THF (3,75 мл) и дезоксигенировали под Ar в течение 5 мин, после чего добавляли воду (1,25 мл). Реакционную смесь нагревали при 70°C и затем добавляли S-Phos Pd G2 (7,27 мг, 10,09 мкмоль). Реакционную смесь перемешивали в течение 30 мин.
Затем смесь гасили с помощью 1М HClводн., растворитель выпаривали и остаток подвергали обращенно-фазовой хроматографии. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (0,042 г, 0,072 ммоль, выход 71,6%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.83-8.94 (m, 1 Н), 8.67-8.81 (m, 1 Н), 8.14-8.27 (m, 2 Н), 7.42-7.62 (m, 3 Н), 7.23-7.35 (bs, 2 Н), 7.09-7.21 (m, 2 Н), 6.69-6.92 (m, 2 Н), 6.42-6.58 (m, 1 Н), 5.56-5.74 (m, 1 Н), 5.33-5.54 (m, 1 Н), 5.00-5.23 (m, 1 Н), 4.67-4.87 (m, 1 Н), 1.58-1.86 (m, 3 Н).
UPLC-MS: 1,19 мин, 545 [М+Н]+, способ 6.
Пример 7b
3-(4-Амино-1-(1-(4-фенил-2Н-хромен-2-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-гидроксибензонитрил
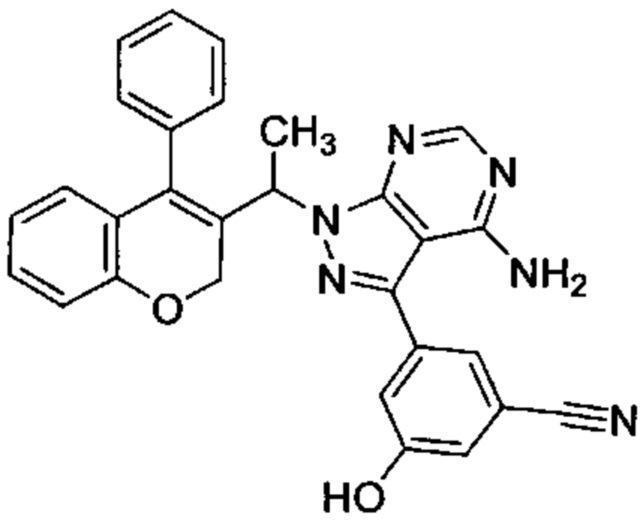
Указанное в заголовке соединение получали аналогично соединению Примера 7 из 3-йод-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амина (Промежуточное соединение F1 (30 мг, 0,061 ммоль)), 3-гидрокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензонитрила (45 мг, 0,182 ммоль), K3PO4 H2O (42 мг, 0,182 ммоль) и SPhos Pd G2 (4,36 мг, 6,06 мкмоль) с получением указанного в заголовке соединения (7,8 мг, 0,016 ммоль, выход 26,5%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.30-10.61 (bs, 1 Н), 8.05-8.23 (m, 1 Н), 7.42-7.59 (m, 4 Н), 7.33-7.42 (m, 1 Н), 7.19-7.33 (m, 3 Н), 7.07-7.19 (m, 1 Н), 6.71-6.87 (m, 2 Н), 6.38-6.53 (m, 1 Н), 5.57-5.71 (m, 1 Н), 4.67-5.22 (m, 2 Н), 1.61-1.72 (m, 3 Н).
UPLC-MS: 1,26 мин, 487,2 [М+Н]+, способ 6.
Пример 7с
3-(4-Амино-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-хлорфенол
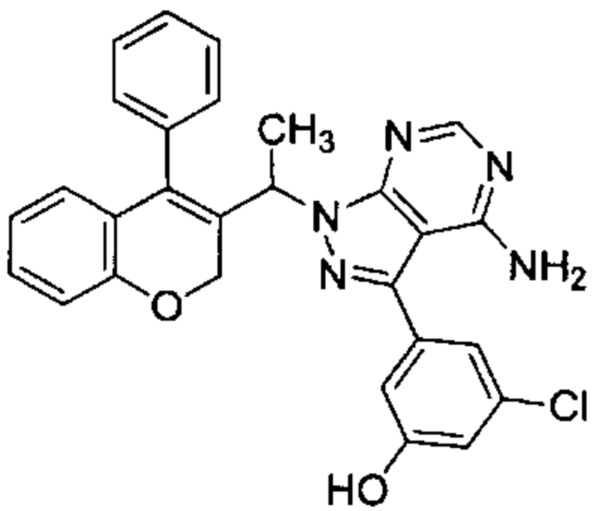
Указанное в заголовке соединение получали аналогично соединению Примера 7 из 3-йод-1-(1-(4-фенил-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амина (Промежуточное соединение F1 (30 мг, 0,061 ммоль)), 3-хлор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола (15 мг, 0,061 ммоль), K3PO4 H2O (42 мг, 0,182 ммоль) и SPhos Pd G2 (4,36 мг, 6,06 мкмоль) с получением указанного в заголовке соединения (10,7 мг, 0,022 ммоль, выход 35,6%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.09 (s, 1 Н), 8.15 (s, 1 Н), 7.36-7.60 (m, 3 Н), 7.22-7.34 (m, 2 Н), 7.08-7.17 (m, 2 Н), 6.99-7.08 (m, 1 Н), 6.72-6.94 (m, 3 Н), 6.40-6.53 (m, 1 Н), 5.44-5.71 (m, 2 Н), 1.56-1.76 (m, 3 Н).
UPLC-MS: 1,37 мин, 496,1 [М+Н]+, способ 6.
Пример 8
3-((9Н-Пурин-6-иламино)метил)-4-фенил-2Н-хромен-2-он
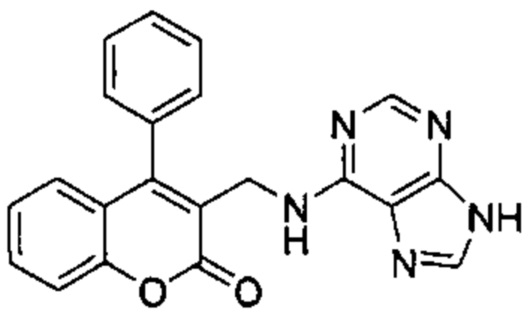
Трет-бутил-9-тритил-9Н-пурин-6-илкарбамат (70 мг, 0,222 ммоль) и 50%-ную дисперсию NaH в минеральном масле (9,7 мг, 0,244 ммоль) растворяли в DMF (0,5 мл) при 0°C. Затем добавляли раствор 3-(бромметил)-4-фенил-2Н-хромен-2-она (Промежуточное соединение D1 (73 мг, 0,143 ммоль)) в DMF (0,5 мл). Реакционную смесь перемешивали при 0°C в течение 5 мин и при 80°C в течение 1 ч. Затем реакционную смесь разбавляли EtOAc (20 мл) и промывали 0,2М HCl, насыщ. NaCl, сушили над Na2SO4 и концентрировали при пониженном давлении. Добавляли TFA (1,5 мл) в DCM (2 мл) и смесь перемешивали в течение 1 ч, затем сушили при пониженном давлении с получением неочищенного вещества, которое очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 30g SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (4 мг, 5%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 12.73-12.98 (bs, 1 Н), 7.98-8.16 (m, 2 Н), 7.36-7.70 (m, 8 Н), 7.18-7.35 (m, 1 Н), 6.89-7.05 (m, 1 Н), 4.00-4.59 (m, 2 Н).
UPLC-MS: 1,69 мин, 370 [М+Н]+, способ 1.
Пример 9
3-(1-(9Н-Пурин-6-иламино)этил)-4-фенил-2Н-хромен-2-он
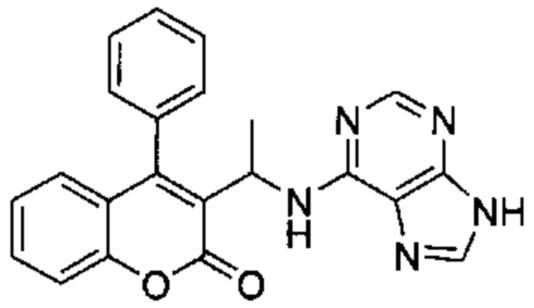
Указанное в заголовке соединение получали аналогично соединению Примера 8 из 3-(1-бромэтил)-4-фенил-2Н-хромен-2-она (Промежуточное соединение D2 (63 мг, 0,191 ммоль) и трет-бутил-9-тритил-9Н-пурин-6-илкарбамата (101 мг, 0,21 ммоль) с получением указанного в заголовке соединения (18 мг, 25%).
1Н ЯМР (400 МГц, DMSO) δ млн-1 13.64-13.85 (bs, 1 Н), 8.74-9.07 (m, 2 Н), 8.33-8.56 (m, 4 Н), 8.11-8.32 (m, 3 Н), 8.01-8.11 (m, 1 Н), 7.77-7.90 (m, 1 Н), 7.65-7.73 (m, 1 Н), 5.75-6.17 (m, 1 Н), 2.20-2.52 (m, 3 Н).
UPLC-MS: 3,47 мин, 384 [М+Н]+, способ 3.
Пример 10
3-(4-Амино-1-(1-(4-(5-((4-метилпиперазин-1-ил)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол
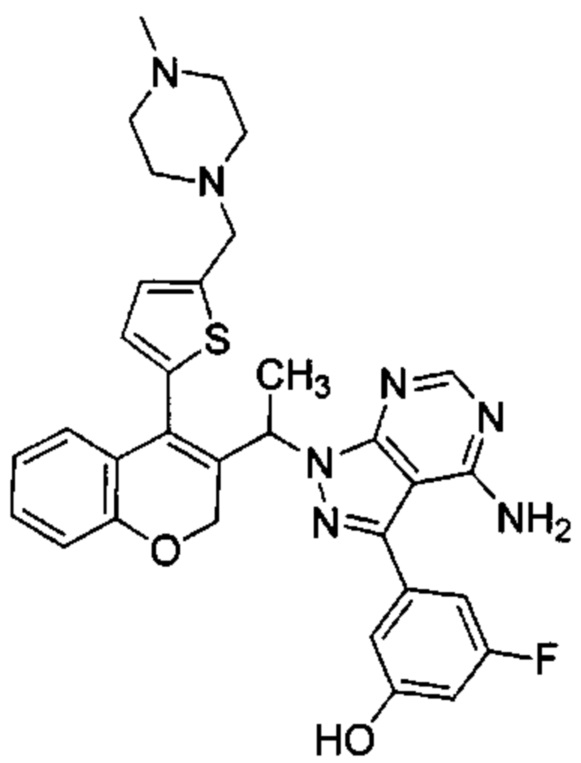
3-Фтор-5-гидроксифенилбороновую кислоту (10,67 мг, 0,068 ммоль), 3-йод-1-(1-(4-(5-((4-метилпиперазин-1-ил)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-4-амин (Промежуточное соединение Н1, 42 мг, 0,068 ммоль) и гидрат фосфата калия (47,3 мг, 0,205 ммоль) диспергировали в THF (3,75 мл) и дезоксигенировали под Ar в течение 5 мин, после чего добавляли воду (1,25 мл). Реакционную смесь нагревали при 70°C и затем добавляли S-Phos Pd G2 (7,27 мг, 10,09 мкмоль). Реакционную смесь перемешивали в течение 3 ч, после чего добавляли дополнительные эквиваленты катализатора и основания. Реакционную смесь перемешивали в течение следующих 3 ч при 70°C.
Затем смесь гасили с помощью 1М HClводн., растворитель выпаривали и остаток подвергали обращенно-фазовой хроматографии. Неочищенное вещество очищали посредством обращенно-фазовой хроматографии на колонке Biotage С18 SNAP (фаза А, вода 95%, ACN 5%, муравьиная кислота 0,1%; фаза В, ACN 95%, вода 5%, муравьиная кислота 0,1%) с получением указанного в заголовке соединения (12,8 мг, выход 31,3%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.04-10.33 (m, 1 Н), 8.15 (d, J=7,9 Гц, 2 Н), 6.55-7.26 (m, 10 Н), 5.84-6.00 (m, 1 Н), 4.63-5.15 (m, 2 Н), 3.55-3.85 (m, 2 Н), 2.33 (m, 8 Н), 2.16 (s, 3 Н), 1.68 (d, J=7,0 Гц, 3 Н).
UPLC-MS: 0,75 мин, 598,16 [М+Н]+, способ 6.
Пример 11
3-(4-Амино-1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол
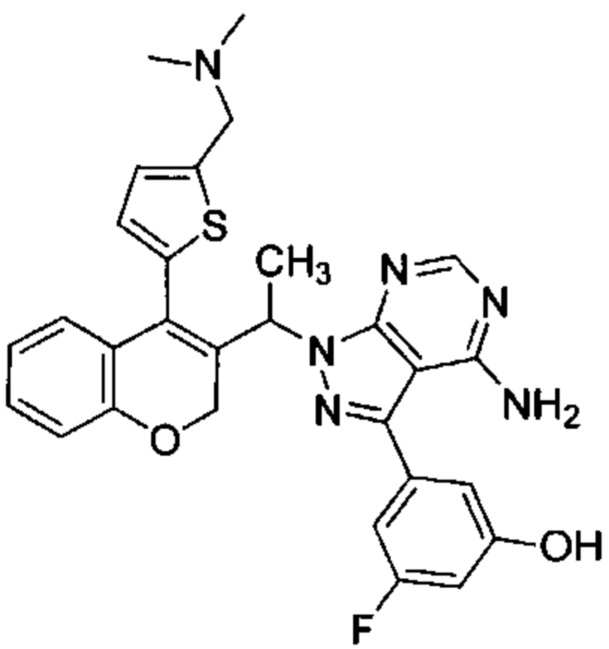
Указанное в заголовке соединение получали аналогично соединению Примера 7 из 1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амина (Промежуточное соединение Н2, 60 мг, 0,107 ммоль), 3-фтор-5-гидроксифенилбороновой кислоты (16,75 мг, 0,107 ммоль), гидрата фосфата калия (74,2 мг, 0,322 ммоль) и SPhos Pd G2 (7,74 мг, 10,74 мкмоль) с получением указанного в заголовке соединения (3,7 мг, выход 6,3%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.17 (s, 1 Н), 7.12-7.19 (m, 1 Н), 6.92 (s, 1 Н), 6.80-6.90 (m, 4 Н), 6.77 (br d, J=7,89 Гц, 2 Н), 6.65 (br d, J=10,09 Гц, 1 Н), 6.48 (s, 1 Н), 5.88 (br d, J=7,45 Гц, 1 Н), 5.10 (d, J=14,47 Гц, 1 Н), 4.81 (br d, J=14,47 Гц, 1 H), 3.63 (s, 2 Н), 1.67 (d, J=7,45 Гц, 3 H).
UPLC-MS: 0,70 мин, 542,78 [М+Н]+, способ 6.
Пример 12
3-(4-Амино-1-(1-(4-(5-(гидроксиметил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенол
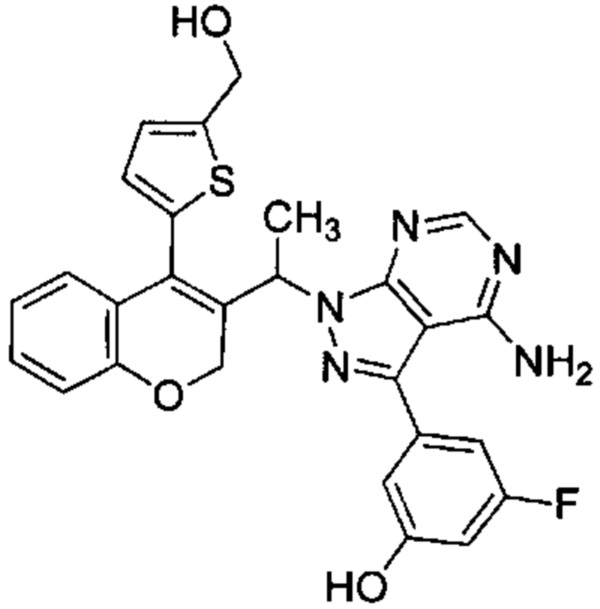
Указанное в заголовке соединение получали аналогично соединению Примера 7 из (5-(3-(1-(4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)этил)-2Н-хромен-4-ил)тиофен-2-ил)метанола (Промежуточное соединение Н3, 53,5 мг, 0,101 ммоль), 3-фтор-5-гидроксифенилбороновой кислоты (47,1 мг, 0,302 ммоль), гидрата фосфата калия (69,6 мг, 0,302 ммоль) и SPhos Pd G2 (7,26 мг, 10,07 мкмоль) с получением указанного в заголовке соединения (10,0 мг, выход 19,6%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.05-10.36 (bs, 1 Н), 8.20 (s, 1 Н), 6.47-7.25 (m, 11 Н), 5.89 (d, J=7,0 Гц, 1 Н), 5.55 (br. s., 1 Н), 5.13 (d, J=14,9 Гц, 1 Н), 4.19-4.82 (m, 3 Н), 1.71 (d, J=7,5 Гц, 3 Н).
UPLC-MS: 1,03 мин, 515,93 [М+Н]+, способ 6.
Пример 13
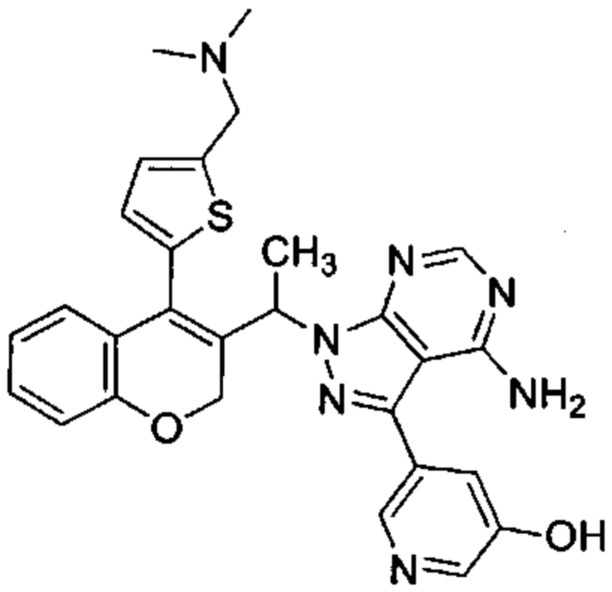
5-(4-Амино-1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)пиридин-3-ол
Указанное в заголовке соединение получали аналогично соединению Примера 7 из 1-(1-(4-(5-((диметиламино)метил)тиофен-2-ил)-2Н-хромен-3-ил)этил)-3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амина (Промежуточное соединение Н2, 30 мг, 0,054 ммоль), 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ола (35,6 мг, 0,161 ммоль), гидрата фосфата калия (37,1 мг, 0,161 ммоль) и SPhos Pd G2 (3,87 мг, 5,37 мкмоль) с получением указанного в заголовке соединения (28,2 мг, выход 11,7%) в виде твердого вещества.
ФАРМАКОЛОГИЧЕСКАЯ АКТИВНОСТЬ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ
Определение ингибирующей активности в отношении фермента PI3K in vitro в бесклеточном анализе
Рекомбинантные человеческие белки PI3Kα, PI3Kβ, PI3Kγ и PI3Kδ приобретали у Millipore Ltd (Billerica, MA). Соединения растворяли в концентрации 0,5 мМ в DMSO и тестировали при разных концентрациях на их активность против PI3Ks, используя ADP-Glo™ Kinase Assay (Promega, Madison WI) согласно инструкциям изготовителя.
Вкратце, киназные реакции выполняли в 384-луночных белых планшетах (Greiner Bio-One GmbH, Frickenhausen). В каждую лунку вводили по 0,1 мкл тестируемых соединений и 2,5 мкл 2х реакционного буферного раствора (40 мМ Tris (трис(гидроксиметиламинометан)) рН 7,5, 0,5 мМ EGTA (этиленгликольтетрауксусная кислота), 0,5 мМ Na3VO4, 5 мМ β-глицерофосфата, 0,1 мг/мл BSA (бычий сывороточный альбумин), 1 мМ DTT (дитиотреитол)), содержащего 50 мкМ субстратов PI и PS (натриевая соль L-α-фосфатидилинозитола и L-α-фосфатидил-L-серин, Sigma-Aldrich, St. Louis МО) и рекомбинантные белки PI3K (PI3Kγ 0,25 нг/мкл, PI3Kδ 1 нг/мкл, PI3Kα 0,125 нг/мкл, PI3Kβ 1 нг/мкл).
Реакции начинали путем добавления 2,5 мкл 2х раствора АТР в каждую лунку (конечные концентрации: PI3Kγ АТР 30 мкМ; PI3Kδ АТР 80 мкМ; PI3Kα АТР 50 мкМ; PI3Kβ АТР 100 мкМ) и инкубировали в течение 60 мин при комнатной температуре. Потом инкубировали каждую реакционную смесь киназы в течение 40 мин с 5 мкл реагента ADP-Glo™, делающего возможным инактивацию неиспользованного АТР. Затем в каждую лунку добавляли реагент детектирования киназы (10 мкл) для превращения ADP (аденозинтрифосфат) в АТР и делали возможным измерение вновь синтезированного АТР, используя люциферин-люциферазную реакцию. После 60 мин инкубирования измеряли сигнал люминесценции, используя Wallac EnVision® многоканальный ридер (PerkinElmer, Waltham MA).
Обработку кривых и расчет IC50 выполняли, используя четырехпараметрическую логистическую модель в XLfit (IDBS, Guilford, UK) для Microsoft Excel (Microsoft, Redmont, WA).
Соединения по изобретению показали IC50 ниже 1 мкМ, по отношению к дельта-субъединице PI3K, предпочтительно ниже 100 нМ.
Определение ингибирующей активности в отношении фермента PI3K in vitro в анализе РВМС
Мононуклеарные клетки периферической крови человека (РВМС) приобретали у Lonza (Basel, СН), промывали и ресуспендировали в среде RPMI 1640 (вода-в-масле феноловый красный), дополненной 10% FBS (фетальная бычья сыворотка), 2 мМ глутамина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина (Life Technologies, Carlsbad СА). РВМС высевали с плотностью 105 клеток/лунка в 96-луночные планшеты, покрытые 6 мкг/мл антитела к человеческим CD3 (Biolegend, San Diego СА).
Клетки обрабатывали в RPMI (вода-в-масле феноловый красный), дополненной 10% FBS, разными концентрациями ингибиторов PI3K (от 10-12М до 10-5М, конечная концентрация DMSO 0,2%), совместно стимулировали с 3 мкг/мл антитела к человеческим CD28 (BD Biosciences, San Jose СА) и инкубировали в течение 72 ч в атмосфере из 95% воздуха и 5% CO2 при 37°C. Человеческие IL-6 и IL-17 измеряли в супернатантах, используя наборы для количественного ELISA (твердофазного иммуноферментного анализа) парных антител (от Life Technologies, Carlsbad СА и R&D Systems, Minneapolis MN, соответственно) согласно инструкциям изготовителя.
Значения IC50 определяли по кривым концентрация-ответ посредством нелинейного регрессионного анализа с использованием программного обеспечения Graph Pad Prism v.6 (GraphPad Software, La Jolla CA).
Соединения по изобретению показали IC50 ниже 1 мкМ по отношению к дельта-субъединице PI3K.
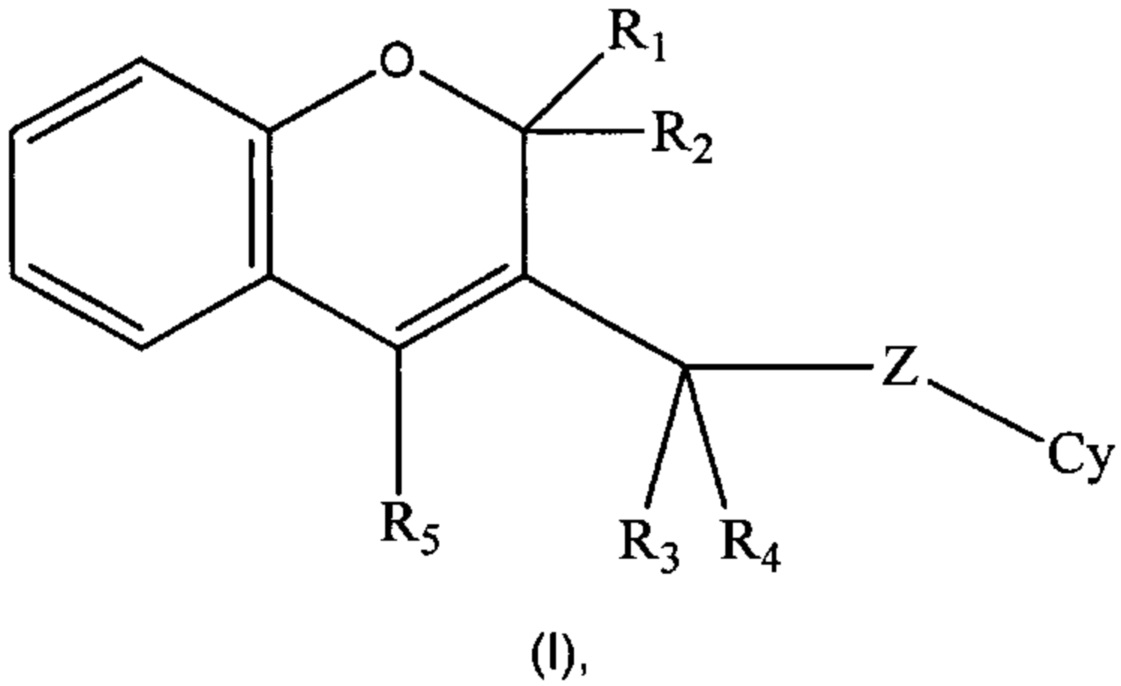
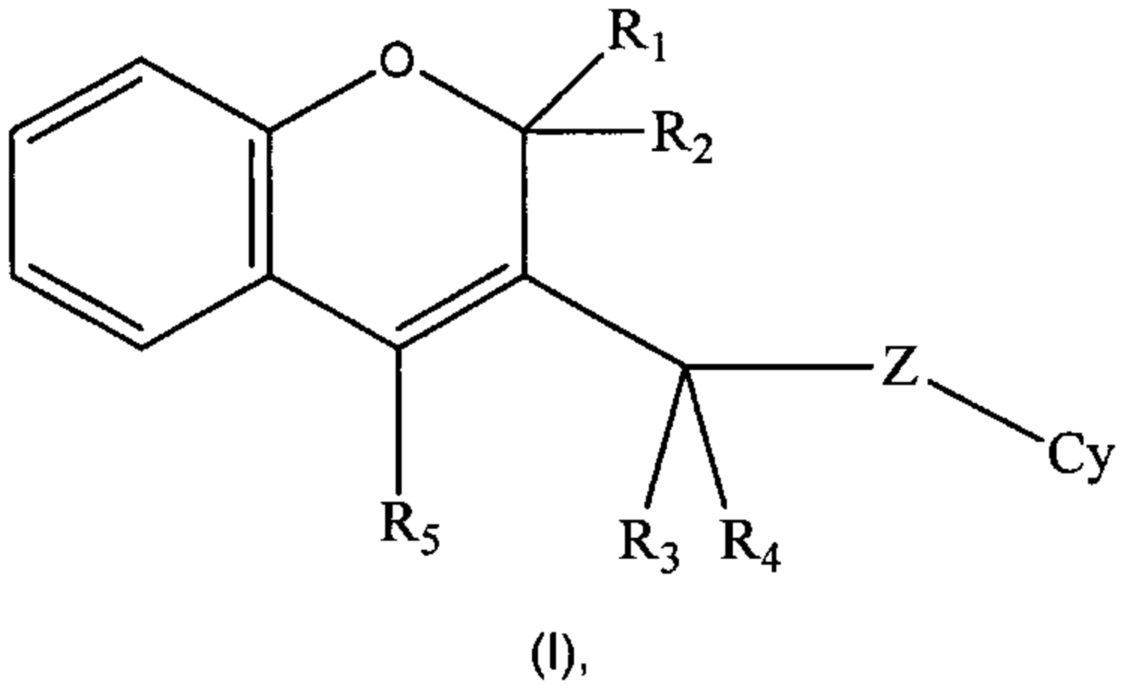
Пиримидопиридазиновые производные, пригодные в качестве р38 марк
Терапевтическая комбинация, включающая легочный сурфактант и антиоксидантные ферменты
Комбинированное лечение хронического обструктивного заболевания легких
Производные 1-(2-фторбифенил-4-ил)-алкилкарбоновой кислоты для лечения транстиретинового амилоидоза
Аэрозольная препаративная форма для лечения по поводу хронического обструктивного легочного заболевания
Производные сложного аминоэфира алкалоида и их лекарственные композиции
Новые полиморфы и соли
Применение стеарата магния в составах сухого порошка для ингаляции
Алкалоидные производные на основе сложных аминоэфиров и композиции лекарственных средств, содержащие их
Производные глицина и их применение в качестве антагонистов мускариновых рецепторов
Пиридазиноновые производные в качестве ингибиторов фосфоинозитид-3-киназ

