Результат интеллектуальной деятельности: ДЕЗОКСИНУКЛЕОЗИДНАЯ ТЕРАПИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ НЕСБАЛАНСИРОВАННЫМИ ПУЛАМИ НУКЛЕОТИДОВ, В ТОМ ЧИСЛЕ СИНДРОМОВ ИСТОЩЕНИЯ МИТОХОНДРИАЛЬНОЙ ДНК
Вид РИД
Изобретение
ПРАВИТЕЛЬСТВЕННАЯ ПОДДЕРЖКА
Данное изобретение сделано при поддержке правительства в виде HD080642, присужденного Национальным Институтом Здравоохранения (НИЗ). Правительство обладает определенными правами на данное изобретение.
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
В данной заявке заявляется приоритет по предварительной заявке на патент США №62/180,194, поданной 17 июня 2015 года, полное содержание которой включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Изобретение в целом относится к фармакологической терапии генетического заболевания человека, в частности заболеваний, характеризующихся несбалансированными пулами нуклеотидов, например, синдромов истощения митохондриальной ДНК и, более конкретно, дефицита тимидинкиназы 2 (ТК2). Фармакологическая терапия включает введение, по меньшей мере, одного дезоксинуклеозида или его смесей. С целью лечения дефицита TK2 фармакологическая терапия предполагает введение дезокситимидина (дТ) или дезоксицитидина (дЦ), или их смесей. Такое введение одного или более дезоксинуклеозидов применимо к другим нарушениям несбалансированных пулов нуклеозидов, особенно тех, которые обнаруживаются при синдроме истощения митохондриальной ДНК.
УРОВЕНЬ ТЕХНИКИ
Митохондриальные заболевания являются клинически гетерогенными заболеваниями из-за дефектов митохондриальной дыхательной цепи (ДЦ) и окислительного фосфорилирования, дефектов биохимических путей, которые преобразуют энергию электронов в аденозинтрифосфат (АТФ). Дыхательная цепь состоит из четырех ферментов со многими субъединицами (комплексы I-IV), которые переносят электроны с целью образования градиента протонов через внутреннюю мембрану митохондрий, в результате чего поток протонов через комплекс X обеспечивает синтез АТФ (DiMauro и Schon, 2003 год; DiMauro и Hirano, 2005 год). Кофермент Q10 (КоQ10) является важной молекулой, которая перемещает электроны из комплексов I и II в комплекс III и обратно. Дыхательная цепь является уникальной в эукариотических, например, млекопитающих, клетках в результате контроля двух геномов, митохондриальной ДНК (мтДНК) и ядерной ДНК (яДНК). Как следствие, мутации в любом геноме могут вызывать митохондриальные заболевания. Большинство митохондриальных заболеваний затрагивают множество органов тела и, как правило, являются смертельными в детском возрасте или в раннем взрослом возрасте. На данный момент не существует доказанных эффективных методов лечения митохондриальных заболеваний, только поддерживающие терапии, такие как введение КоQ10 и его аналогов для усиления активности дыхательной цепи и для детоксикации активных форм кислорода (АФК), которые являются токсичными побочными продуктами ферментов дыхательной цепи, с нарушенной функцией.
Синдром истощения митохондриальной ДНК (MDS), который относится к подгруппе митохондриальных заболеваний, является частым случаем тяжелой детской энцефаломиопатии, которая с молекулярной стороны характеризуется уменьшением количества копий митохондриальной ДНК (мтДНК) в тканях и недостаточным синтезом митохондриальных комплексов ДЦ (Hirano и соавт. 2001). Мутации в нескольких ядерных генах были определены как причины детского MDS, в том числе: TK2, DGUOK, POLG, POLG2, SCLA25A4, MPV17, RRM2B, SUCLA2, SUCLG1, TYMP, OPA1, и C10orf2 (PEO1). (Bourdon и соавт. 2007; Copeland 2008; Elpeleg и соавт. 2005; Mandel и соавт. 2001; Naviaux и Nguyen 2004; Ostergaard и соавт. 2007; Saada и соавт. 2003; Sarzi и соавт. 2007; Spinazzola и соавт, 2006). Кроме того, мутации в этих ядерных генах также могут вызывать множественные делеции мтДНК с или без истощения мтДНК (Béhin и соавт. 2012; Garone и соавт. 2012; Longley и соавт. 2006; Nishino и соавт. 1999; Paradas и соавт. 2012; Ronchi и соавт. 2012; Spelbrink и соавт. 2001; Tyynismaa и соавт. 2009; Tyynismaa и соавт. 2012; Van Goethem и соавт. 2001).
Одним из этих генов является ТК2, который кодирует тимидинкиназу (TK2), митохондриальный фермент, необходимый для фосфорилирования пиримидиновых нуклеозидов (тимидина и дезоксицитидина) для получения дезокситимидинмонофосфата (дТМФ) и дезоксицитидинмонофосфата (дЦМФ) (Saada и соавт. 2001). Мутации в ТК2 нарушают митохондриальные пути реутилизации нуклеозида/нуклеотида, необходимые для синтеза дезоксинуклеотидтрифосфата (дНТФ), который является строительными блоками для репликации и репарации мтДНК.
Дефицит ТК2 был впервые описан в 2001 году Saada и его коллегами (Saada и соавт. 2001) у четырех пострадавших детей из четырех разных семей, страдающих от тяжелой, изнуряющей миопатии. После раннего развития без осложнений, в возрасте 6-36 месяцев у пациентов развилась гиперКФКемия (КФК - креатинфосфокиназа), тяжелая мышечная гипотония с последующей потерей спонтанной активности. Болезнь быстро прогрессировала, в результате чего два пациента были подключены к системе механической вентиляции легких в возрасте 3-х лет, а другие два пациента уже были мертвы к моменту отчета.
После первого описания, шестьдесят дополнительных пациентов были зарегистрированы в литературе, и по меньшей мере двадцать шесть пациентов были диагностированы, но не зарегистрированы (Alston и соавт. 2013; Bartesaghi и соавт. 2010; Béhin и соавт. 2012; Blakely и соавт. 2008; Carrozzo и соавт. 2003; Chanprasert и соавт. 2013; Collins и соавт. 2009; Galbiati и соавт. 2006; Gotz и соавт. 2008; Leshinsky-Silver и соавт. 2008; Lesko и соавт. 2010; Mancuso и соавт. 2002; Mancuso и соавт. 2003; Marti и соавт. 2010; Oskoui и соавт. 2006; Paradas и соавт. 2012; Roos и соавт. 2014; Tulinius и соавт. 2005; Tyynismaa и соавт. 2012; Vilà и соавт. 2003; Wang и соавт. 2005), в сумме было девяносто пациентов, 53 мужчины и 37 женщин.
Двадцать шесть недавно диагностированных пациентов были идентифицированы с помощью секвенирования ДНК нового поколения. Такое большое количество вновь выявленных случаев свидетельствует о том, что дефицит ТК2 является диагностированным нарушением.
Дефицит ТК2 имеет широкий клинический и молекулярно-генетический спектр симптомов, при этом у большинства пациентов симптомы проявляются в раннем детстве с последующим губительным течением болезни, в то время как другие пациенты имеют медленно прогрессирующую слабость на протяжении десятилетий.
Лечение дефицита ТК2, как и большинства MDS (синдромов истощения митохондриальной ДНК) и митохондриальных расстройств, было ограничено поддерживающей терапией. В то время как введение дезокситимидинмонофосфата (дТМФ) и дезоксицитидинмонофосфата (дЦМФ) улучшало состояние как нокин мутантных мышей ТК2, так и пациентов с дефицитом TK2 (Заявка США серийный № 15/082,207, которая включена в данный документ во всей своей полноте), по-прежнему существует потребность в терапевтическом воздействии по отношению к дефициту ТК2.
Кроме того, существует потребность в лечении других форм MDS и других заболеваний, характеризующихся несбалансированными пулами нуклеотидов. Например, несколько менделевских расстройств с истощением мтДНК или множественными деланиями, или и с тем и с другим, характеризуются несбалансированными пулами дезоксинуклеотидтрифосфатов, которые приводят к дефектам репликации мтДНК. Одно такое нарушение, мутации DGUOK, нарушают функционирование внутримитохондриального фермента дезоксигуанозинкиназы, которая в нормальных условиях фосфорилирует дезоксипуриновые нуклеозиды дезоксгуанозин и дезоксицитидин для получения дезоксигуанозинмонофосфата (дГМФ) и дезоксицитидинмонофосфата (дЦМФ). Другие ядерные гены, которые нарушают митохондриальные пулы дНТФ, включают в себя TYMP, RRM2B, SUCLA2, SUCLG1 и MPV17. Терапии, которые восстанавливают баланс пула дНТФ, были бы полезны для лечения этих расстройств.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В некоторых вариантах реализации данное изобретение относится к способу лечения заболевания или нарушения, характеризующегося несбалансированными пулами нуклеотидов, который включает введение субъекту, имеющему для этого показания, терапевтически эффективного количества композиции, содержащей один или более дезоксинуклеозидов.
Заболевания или нарушения, характеризующиеся несбалансированными пулами нуклеотидов, которые могут лечиться по способу данного изобретения, включают, но без ограничений, те, которые характеризуются мутациями в следующих генах: TK2; DGUOK; TYMP; RRM2B; SUCLA2; SUCLG1; и MPV17.
В предпочтительном варианте реализации изобретения нарушение представляет собой синдром истощения митохондриальной ДНК (MDS). В более предпочтительном варианте реализации изобретения MDS включает в себя нарушения миопатической формы, характеризующейся мутациями в ТК2, энцефаломиопатической формы, характеризующейся мутациями в SUCLA2, нейрогастроинтестинальной энцефалопатической формы, характеризующейся мутациями в TYMP и гепатопатической формы, характеризующейся мутациями в DGUOK, POLG, а также MPV17. В более предпочтительном варианте реализации изобретения нарушение представляет собой дефицит тимидинкиназы 2, характеризующийся мутацией(ами) в гене ТК2.
Все синдромы истощения митохондриальной ДНК можно лечить с помощью способа данного изобретения, который включает в себя введение дезоксинуклеозидов. Примеры MDS, которые можно лечить с помощью способа данного изобретения, включают, но без ограничений, дефекты в: гене DGUOK, кодирующем дезоксигуанозинкиназу, дГК; гене RRM2B, кодирующем p53R2, индуцируемую p53 малую субъединицу рибонуклеотидредуктазы, РНР; а также гене TYMP, кодирующем тимидинфосфорилазу, ТФ.
В предпочтительном варианте реализации изобретения дезоксинуклеозид представляет собой или дезокситимидин (дТ) либо дезоксицитидин (дЦ), или их смеси. Дезоксиаденозин (дА) и дезоксигуанозин (дГ), по отдельности или вместе, также могут быть использованы в способе данного изобретения. Один дезоксинуклеозид (то есть, дТ, дЦ, дА или дГ) и смеси двух или более любого из четырех дезоксинуклеозидов могут быть использованы в способе данного изобретения.
Предпочтительные дозы дезоксинуклеозида(ов) составляют от около 100 до около 1000 мг/кг/день, более предпочтительно от около 300 до около 800 мг/кг/день и наиболее предпочтительно от около 250 до около 600 мг/кг/день. Если композиция содержит один дезоксинуклеозид, то дозы являются дозой одного дезоксинуклеозида. Если композиция содержит более одного дезоксинуклеозида, дозы могут быть дозой каждого дезоксинуклеозида или всех дезоксинуклеозидов в композиции.
Введение дезоксинуклеозида(ов) может выполнятся один раз в день, два раза в день, три раза в день, четыре раза в день, пять раз в день, до шести раз в день, предпочтительно с равными промежутками времени.
Предпочтительными способами введения являются пероральный, интратекальный, внутривенный и энтеральный.
Введение дезоксинуклеозида(ов) следует начинать, как только возникает подозрение нарушения, характеризующегося несбалансированными пулами нуклеотидов, например, MDS, и продолжаться в течение всей жизни пациента. Тест на установление диагноза таких нарушений, включая дефицит ТК2, является известным в данной области техники.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
В целях иллюстрации изобретения на чертежах изображены некоторые варианты реализации изобретения. Тем не менее, изобретение не ограничивается точной последовательностью и способами вариантов реализации, изображенными на иллюстрациях
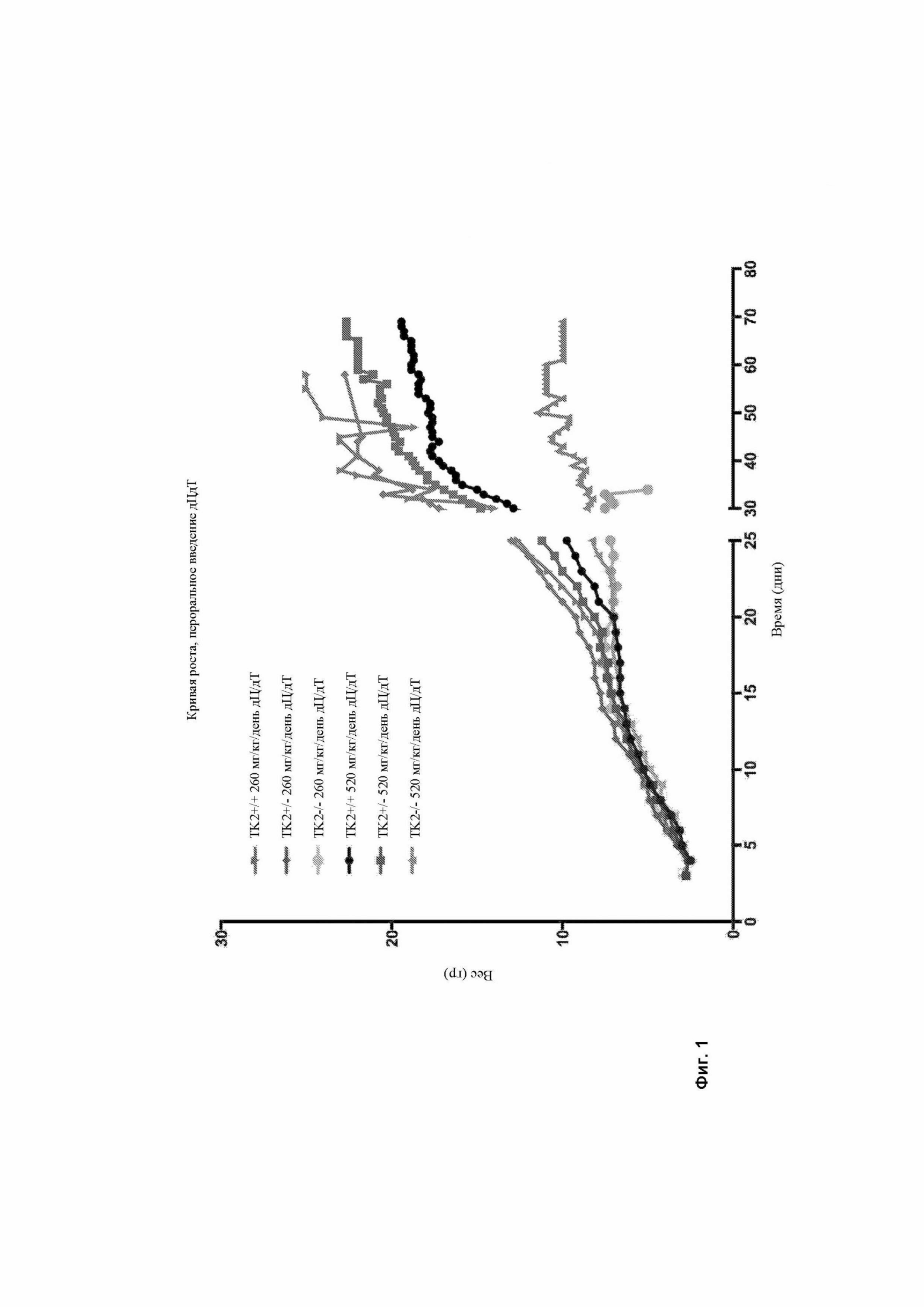
Фигура 1 изображает кривую роста мышей дикого типа (Tk2+/+ и Tk2+/-), а также Tk2-/-, получавших лечение в виде 260 мг/кг/день или 520 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ) через 4 дня после рождения. Каждый символ представляет среднее значение веса в каждый момент времени. N каждой группы указывается на рисунке.
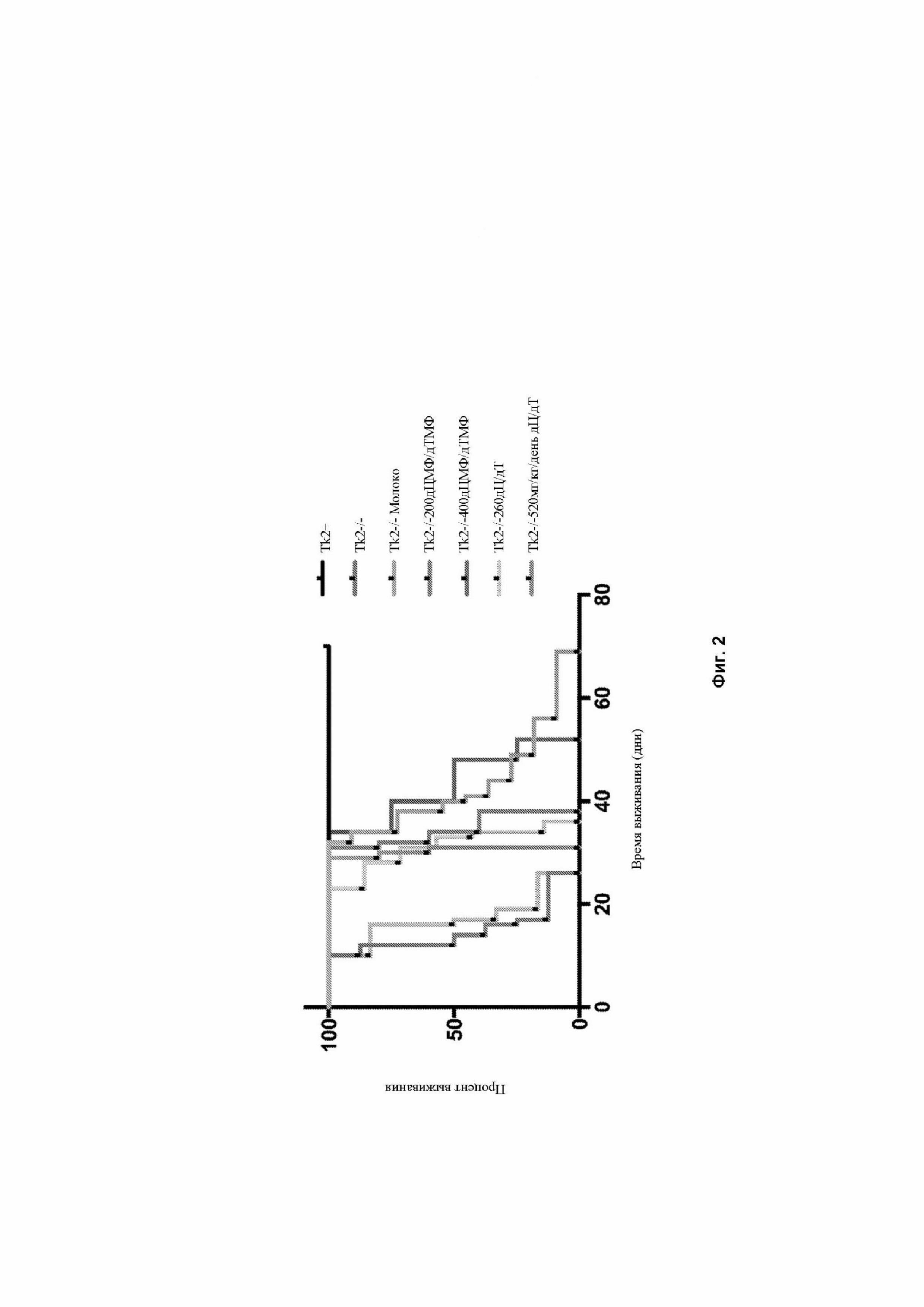
Фигура 2 изображает кривую выживаемости мышей дикого типа (Tk2+/+) и Tk2-/- подвергающихся лечению следующим образом: Tk2-/-молоко против Tk2-/-200 мг/кг/день дЦМФ+дТМФ, p=0,0013; Tk2-/-молоко против Tk2-/- 260 мг/кг/день дЦ+дТ, p=0,0006; Tk2-/-молоко против Tk2-/- 520 мг/кг/день дЦ+дТ, p<0,0001; Tk2-/- 260 мг/кг/день дЦ=дТ против Tk2-/-520 мг/кг/день дЦдТ, p=0,0009, на 4-й день после рождения. N каждой группы указывается на рисунке. p – значения, определенные критериями Кокса-Мантеля
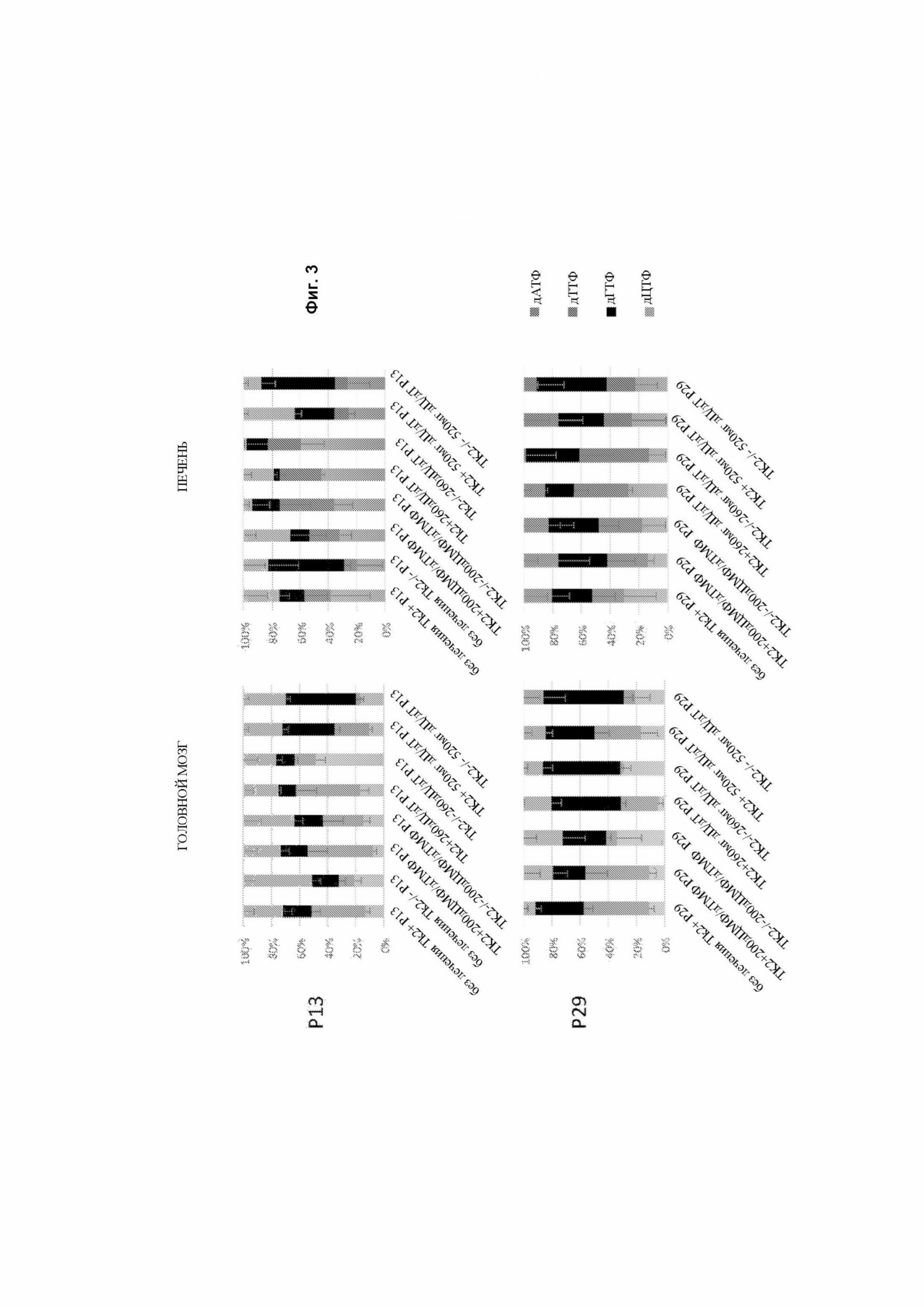
На фигуре 3 представлены графики относительных пропорций дНТФ в митохондриях, выделенных из ткани мозга и печени дикого типа (Tk2+/+), а также Tk2-/-, не повергающихся лечению или получавших лечение в виде 200 мг/кг/день дЦМФ и дАМФ, или 260 мг/кг/день, или 520 мг/кг/день дезоксицитидином (дЦ) и дезокситимидином (дТ) в возрасте 13 дней после рождения (верхние панели) и 29 дней после рождения (нижние панели).
На фигуре 4 представлены графики, показывающие соотношение мтДНК/яДНК в головном мозге, печени, кишечнике и мышцах у мышей Tk2 дикого типа (Tk2+/+) (левая панель) по сравнению с мышами Tk2-/-, не повергающихся лечению или получавших лечение в виде 260 мг/кг/день или 520 мг/кг/день дезоксицитидином (дЦ) и дезокситимидином (дТ), в возрасте 13 и 29 дней после рождения. Данные представлены как среднее ± стандартное отклонение (СО) процента копий мтДНК относительно Tk2+, p-значения были оценены с помощью тестов Манна-Уитни. (*p<0,05, **p<0,01, ***p<0,001, ****p<0,0001).
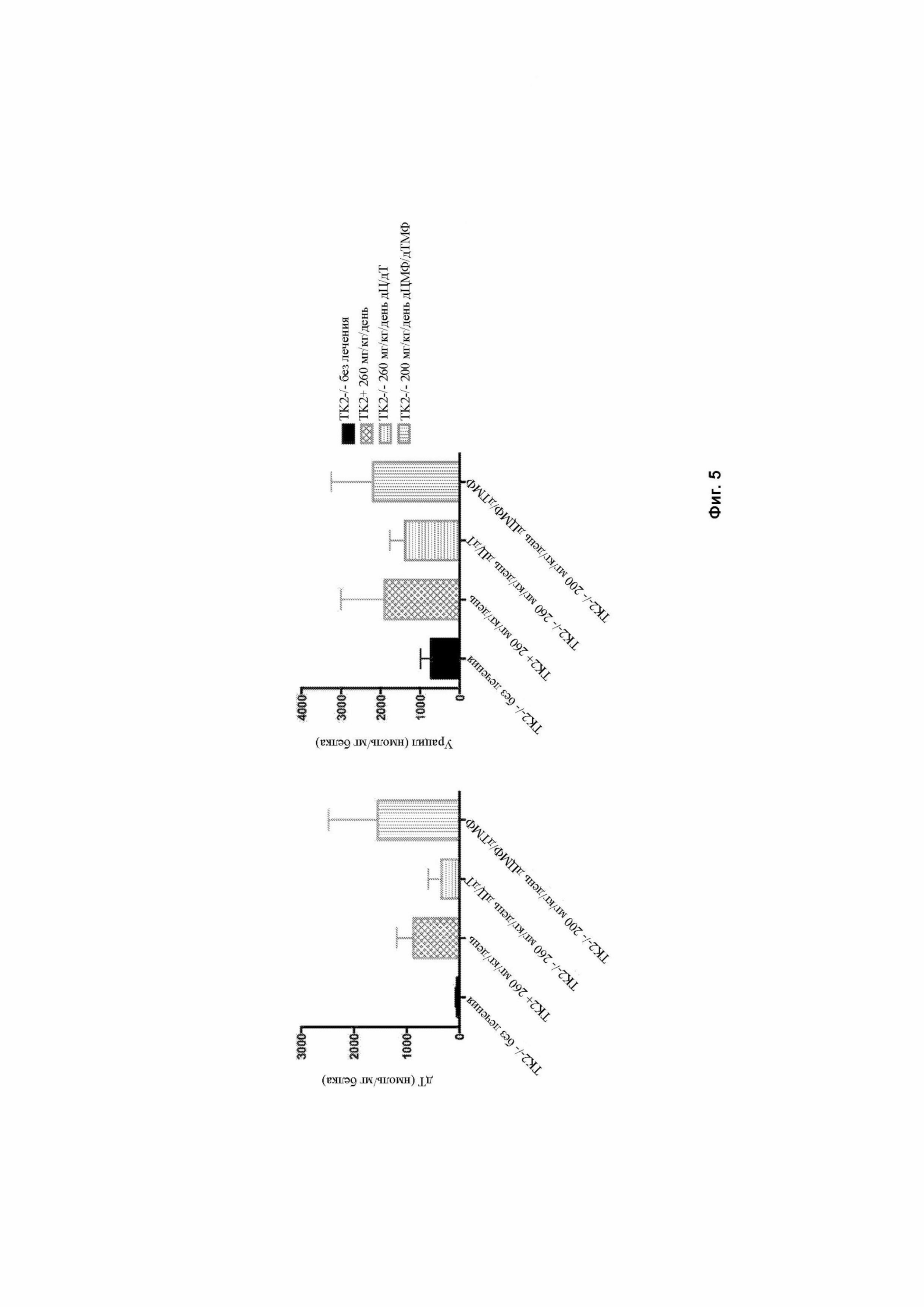
На фигуре 5 представлены графики отображающие результаты измерения с помощью ВЭЖХ (высокоэффективной жидкостной хроматографии) дТ и урацила в плазме мышей дикого типа, которых не подвергали лечению (Tk2+/+), мышей дикого типа (Tk2+/+), получавших лечение в виде 260 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ), мышей Tk2-/-, получавших лечение в виде 260 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ), и мышей Tk2-/-, получавших лечение в виде 200 мг/кг/день дЦМФ и дТМФ, через 30 минут после лечения. Данные выражаются как среднее ± СО.
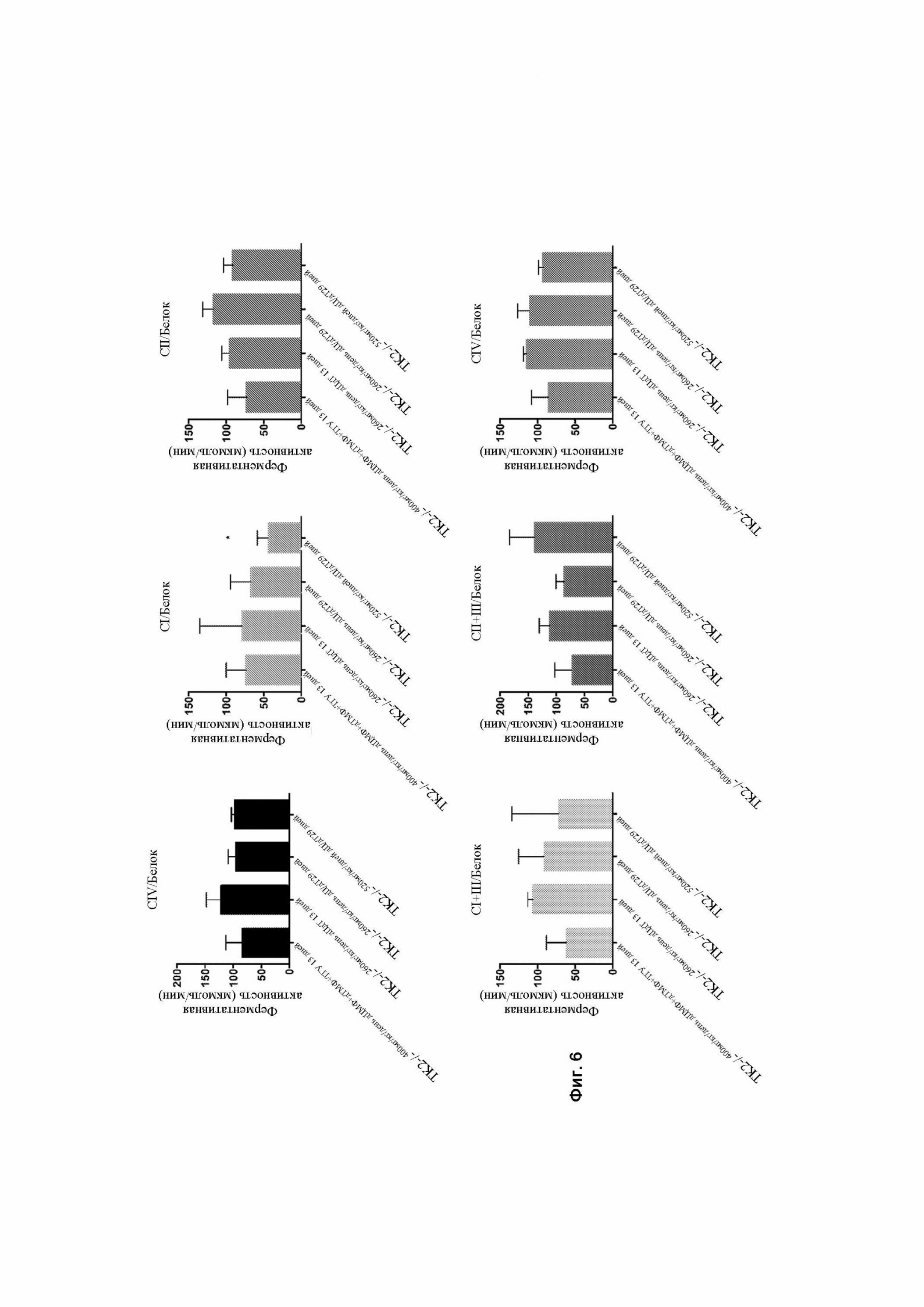
На фигуре 6 представлены графики уровней активности ферментов дыхательной цепи у мышей Tk2-/-, получавших лечение в виде 400 мг/кг/день дЦМФ и дТМФ и тетрагидроуридина (ТГУ) через 13 дней после рождения, 260 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ) через 13 и 29 дней после рождения или 520 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ) через 29 дней после рождения. Данные представлены как процент активности ферментов дыхательной цепи (RCE) в тканях мышей Tk2-/-, нормализованные к уровням белка и относительно Tk2+ для каждого лечения. p-значения определенны с помощью тестов Манна-Уитни. *p<0,05.
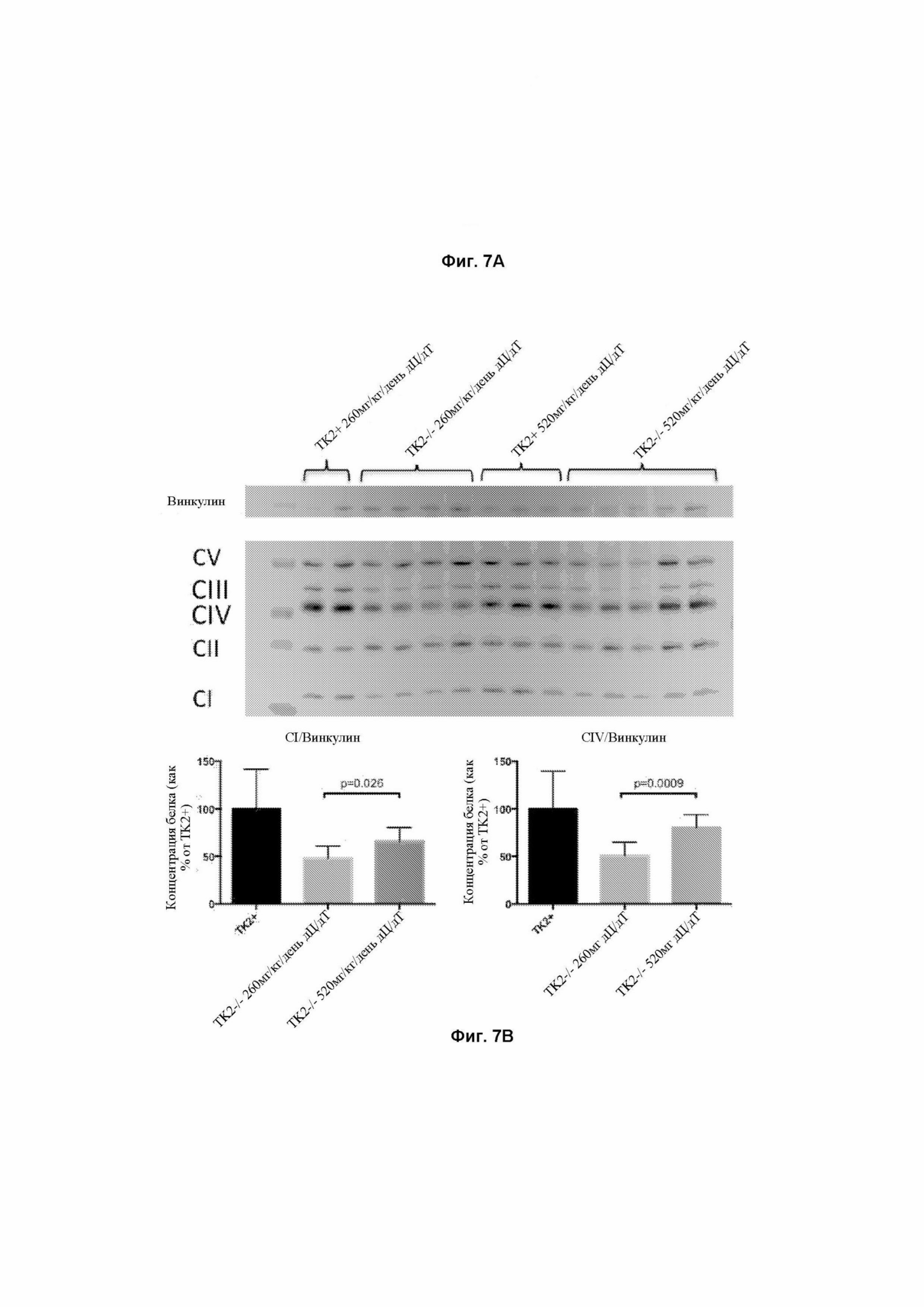
На фигуре 7А показан иммуноблот белков дыхательной цепи у мышей дикого типа, получавших лечение в виде 260 мг/кг/день или 520 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ), и мышей Tk2-/-, получавших лечение в виде 260 мг/кг/день или 520 мг/кг/день дезоксицитидина (дЦ) и дезокситимидина (дТ) через 29 дней после рождения. На фигуре 7В представлены графики, показывающие уровни RCE, нормализованные относительно комплекса II, представленные в процентах от уровней RCE у мышей TK2+/+. p-значения определенны с помощью тестов Манна-Уитни.
Сокращения: ЦС = цитрат-синтаза; CI = НАДФ-дегидрогеназа; CII = сукцинатдегидрогеназа; CIII = цитохром с-редуктаза; CIV = цитохром с-оксидаза (ЦО); CI + III = NADH-цитохром с-редуктаза; CII + III = сукцинатдегидрогеназа-цитохром с -редуктаза.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение основано на неожиданном открытии того, что синдромы истощения митохондриальной ДНК, включая дефицит ТК2, можно лечить дезоксинуклеозидами. Как показано в приведенных в данном документе результатах, введение дезоксинуклеозидов значительно улучшило состояние как мышиной модели дефицита ТК2, так и пациентов с дефицитом ТК2.
Определения
Термины, используемые в этом описании, как правило, имеют свои обычные значения в уровне техники, в контексте данного изобретения и в конкретном контексте, в котором используется каждый термин. Определенные термины обсуждаются ниже в тексте или в другом месте описания для предоставления дополнительных указаний практику в описании способов изобретения и способах их использования. Более того, следует понимать, что одно и то же может быть сказано более чем одним способом. Следовательно, альтернативный язык и синонимы могут быть использованы для любого одного или более терминов, обсуждаемых в данном документе, и не иметь какого-либо особого значения для определения или обсуждения этого термина в данном документе. Для определенных терминов предоставлены синонимы. Перечисление одного или более синонимов не исключает использования других синонимов. Использование примеров в любом месте описания, включая примеры любых терминов, обсуждаемых в данном документе, является только иллюстрацией и никоим образом не ограничивает объем и значение изобретения или любого термина, в качестве примера. Подобным же образом изобретение не ограничивается его предпочтительными вариантами реализации.
Термин "субъект", используемый в этой заявке, означает млекопитающих. Млекопитающие включают псовых, кошачьих, грызунов, быков, лошадей, свиней, овец и приматов. Таким образом, изобретение может быть использовано в ветеринарной медицине, например, для лечения домашних животных, сельскохозяйственных животных, лабораторных животных в зоологических парках и животных в дикой природе. Изобретение является особенно желательным в качестве медицинских применений для людей.
Термин "пациент", используемый в данной заявке, означает человека. В некоторых вариантах реализации данного изобретения у "пациента" известно или подозревается наличие заболевания или нарушения, характеризующегося несбалансированными пулами нуклеотидов, митохондриальным заболеванием, синдромом истощения митохондриальной ДНК или дефицитом ТК2.
Фраза "терапевтически эффективное количество" используется в данном документе для обозначения количества, достаточного для улучшения клинически значимого состояния у субъекта, или задержки, или минимизации, или смягчения одного или более симптомов, связанных с заболеванием или нарушением, или приводящего к желаемому полезному изменению физиологии у субъекта.
Термины "лечить", "лечение" и тому подобные, относятся к способу замедления, облегчения, улучшения или смягчения, по меньшей мере, одного из симптомов заболевания или нарушения, или устранению болезни или нарушения после ее начала.
Термины "предотвращать", "предотвращение" и тому подобные, относятся к действию до явно выраженного начала заболевания или нарушения, с целью предотвращения развития заболевания или нарушения, или минимизации степени заболевания или нарушения, или замедления ее развития.
Термин "имеющий для этого показания" относится к субъекту, у которого известно или подозревается наличие, или который подвергается риску заболевания или нарушения, характеризующегося несбалансированными пулами нуклеотидов, митохондриальным заболеванием, синдромом истощения митохондриальной ДНК или дефицитом ТК2.
Используемый в данном документе термин "агент" означает вещество, которое производит или является способным производить эффект, и будет включать, но без ограничений, химические вещества, лекарственные препараты, биологические препараты, малые органические молекулы, антитела, нуклеиновые кислоты, пептиды и белки.
Используемый в данном документе термин "дезоксинуклеозид" означает дезокситимидин или дТ, дезоксицитидин или дЦ, дезоксиаденозин или дА, и дезоксигуанозин или дГ. Полное имя и общая аббревиатура для каждого будут использоваться взаимозаменяемо. Такие дезоксинуклеозиды также включают в себя физиологически функциональные производные дезоксинуклеозидов.
Используемый в данном документе термин "физиологически функциональное производное" относится к соединению (например, предшественнику лекарственного средства), которое преобразуется in vivo для получения дезоксинуклеозида. Преобразование может происходить с помощью различных механизмов (например, с помощью метаболических или химических процессов), таких как, например, гидролиз в крови. Пролекарства являются такими производными, а обсуждение применения пролекарств приведено в Т. Higuchi и W. Stella, "Pro-drugs as Novel Delivery Systems," том14 A. C. S. Symp osium Series, и в Bioreversible Carriers in Drug Design, ред. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987 год.
Используемый в данном документе термин "нежелательная реакция" представляет собой побочный эффект, вызванный введением лекарственного средства. В большинстве случаев введение дезоксинуклеозидов не вызывало нежелательных реакций. Наиболее ожидаемой нежелательной реакцией будет незначительная желудочно-кишечная непереносимость.
Термин "около" или "приблизительно" означает пределы приемлемого диапазона ошибок для конкретного значения, определяемые обычным специалистом в данной области техники, что будет частично зависеть от того, как измеряется или определяется значение, то есть, ограничения измерительной системы, то есть, степени точности, требуемой для конкретной цели, например, лекарственной формы. Например, термин "около" может означать значения в пределах 1 или более чем 1 стандартного отклонения, на практике в данной области техники. В альтернативном варианте, "около" может означать диапазон до 20 %, предпочтительно до 10 %, более предпочтительно до 5 % и еще более предпочтительно до 1 % от заданного значения. В альтернативном варианте, особенно в отношении биологических систем или процессов, этот термин может означать пределы порядка величины, предпочтительно в 5 раз и более предпочтительно в 2 раза от значения. В случаях, когда конкретные значения описаны в заявке и в формуле изобретения, если не указано иное, следует предполагать, что термин "около" означает пределы приемлемого диапазона ошибок для конкретного значения.
Введение дезоксинуклеозидов для лечения синдрома истощения митохондриальной ДНК
Синдром истощения митохондриальной ДНК (мтДНК) включает в себя несколько тяжелых аутосомных заболеваний, характеризующихся уменьшением числа копий мтДНК в пораженных тканях. Большинство ядерных генов, которые приводят к MDS, кодируют белки, вовлеченные в механизм репликации мтДНК, или участвующие в метаболизме дезоксирибонуклеозидтрифосфатов (дНТФ).
Одной из форм MDS является дефицит тимидинкиназы или ТК2. ТК2, которая кодируется ядерным геном, ТК2, является белком матрикса митохондрий, который фосфорилирует тимидиновые и дезоксицитидиновые нуклеозиды для получения дезокситимидинмонофосфата (дТМФ) и дезоксицитидинмонофосфата (дЦМФ), которые, в свою очередь, преобразуются в дезоксинуклеотидтрифосфаты (дНТФ), необходимые для синтеза митохондриальной ДНК. Как обсуждалось в разделе уровня техники, аутосомно-рецессивные мутации TK2 вызывают губительную нервно-мышечную слабость с тяжелым истощением митохондриальной ДНК (мтДНК) у младенцев и детей, а также прогрессирующую внешнюю офтальмоплегию со множественными делециями мтДНК у взрослых. Многие пациенты не могут передвигаться и испытывают необходимость в механической вентиляции и питательной трубке определенного типа. В таких нарушениях определенным образом участвует центральная нервная система с симптомами, которые включают в себя судороги, энцефалопатию, нарушение когнитивных функций и потерю слуха. Менее 7% пациентов живут более 42 лет.
На основании клинических и молекулярных генетических данных пациентов, диагностированных таким образом, были идентифицированы три проявления болезни: i) инфантильная миопатия (возраст ≤ 1-го года) с началом слабости в первый год жизни, с тяжелым истощением мтДНК и ранней смертностью; ii) миопатия в детском возрасте (возраст > 1-11 лет) с тяжелым истощением мтДНК; и iii) поздняя миопатия (возраст ≥ 12 лет) с легкой слабостью в начале и медленной прогрессией к потере движения, респираторной недостаточности или и тому, и другому, часто с хроническим прогрессирующим внешним офтальмопарезом в подростковом или в зрелом возрасте в сочетании с множественными делециями мтДНК, сокращенным количеством копий мтДНК или и тем, и другим. В общем, см. Garone и соавт. (2016 год) в процессе подготовки.
Попытки изучить патогенез и пробные методы лечения дефицита ТК2 с использованием культивируемых фибробластов пациентов оказались безуспешными, поскольку реплицирующиеся клетки не смогли продемонстрировать истощение мтДНК. Напротив, мышиная модель гомозиготного нокин мутанта Tk2 H126N (Tk2)-/-), демонстрирует фенотип, который поразительно похож на человеческую инфантильную энцефаломиопатию, вызванную мутациями TK2, которая характеризуется началом в возрасте 10 дней с уменьшенной способностью к передвижению, нестабильной походкой, крупноамплитудным тремором, замедлением роста и истощением митохондриальной ДНК (мтДНК), быстро развивающейся до ранней смерти в возрасте от 14 до 16 дней, что является периодом времени, аналогичным инфантильной форме болезни у человека (Akman и соавт. 2008 год; Dorado и соавт. 2011 год).
Исследования, изложенные в данном документе, на нокин Tk2 мышах, показали, что пероральное введение дЦ/дТ пролонгировало задержку начала клинических симптомов дефицита ТК2 и продлевало жизнь мышей в два-три раза (пример 2).
Дополнительные эксперименты показали тканеспецифические реакции. Измерение уровней пула дНТФ в экстрактах митохондрий показало, что дЦТФ был восстановлен в головном мозге, а дТТФ был восстановлен в печени (пример 3). Измерение истощения мтДНК показало, что и дЦМФ + дТФМ и дЦ + дТ терапии восстановили количество копий мтДНК в печени, мышцах и тканях (пример 4). Ранее предполагалось, что образование гематоэнцефалического барьера может поставить под угрозу биодоступность лечения мозга. Тем не менее, измерения с помощью ВЭЖХ показали, что каталитические продукты этих соединений были обнаружены в более высоких концентрациях после лечения нуклеотидами монофосфатов и дезоксинуклеозидов, что свидетельствует о том, что они способны пересекать гематоэнцефалический барьер. Измерения истощения мтДНК также показали полное сохранение количества копий мтДНК в кишечнике.
Таким образом, изложенные в данном документе эксперименты с использованием мышиной модели дефицита Tk2, демонстрируют, что введение дезоксинуклеозидов является эффективным и безопасным для лечения данного заболевания. Кроме того, как показано в примере 5, введение дТ и дЦ значительно улучшало симптомы дефицита ТК2 у пациентов.
Таким образом, данное изобретение включает введение, по меньшей мере, одного дезоксинуклеозида пациенту, имеющему для этого показания. В одном варианте реализации данное изобретение включает в себя введение, по меньшей мере, одного дезокспиримидина. В дополнительном варианте реализации изобретения дезоксипиримидин является выбранным из дЦ, дТ и их смесей. В еще одном варианте реализации данное изобретение включает в себя введение, по меньшей мере, одного дезоксипурина. В дополнительном варианте реализации изобретения дезоксипурин является выбранным из дА, дГ и их смесей.
У пациентов, которые получат пользу от введения дезоксинуклеозидов, будет диагностироваться дефицит ТК2. Этим пациентам можно вводить, по меньшей мере, один дезоксипиримидин, дЦ или дТ или их смеси.
Параллельный дефект дезоксигуанозинкиназы (дГК), обусловленный аутосомно-рецессивными мутациями в DGUOK с дефицитами дГМФ и дАМФ, вызывает истощение мтДНК, обычно проявляющееся в виде раннего детского гепатоцепербального заболевания (Mandel и соавт. 2001). Этим пациентам пошло бы на пользу введение, по меньшей мере, одного дезоксипурина, дГ или дА, или их смесей.
Другие формы MDS, а также другие нарушения, связанные с несбалансированными пулами нуклеотидов, можно лечить путем введения конкретных дезоксинуклеозидов, то есть, дА, дГ, дЦ или дТ, или их смесей. Эти нарушения включают в себя, но без ограничений, дефициты, связанные с RRM2B (кодирующем p53R2, индуцибельную p53 малую субъединицу рибонуклеотидредуктазы, РНР) и мутациями в TYMP (кодирующем тимидинфосфорилазу, ТФ), которые приводят к митохондриальной нейрогастроинтестинальной энцефаломиопатии (МНГИЭ). Дополнительные ядерные гены, которые нарушают митохондриальные пулы дНТФ, включают в себя, но без ограничений, SUCLA2, SUCLG1, а также MPV17. Нарушения, связанные с этими генами, также можно лечить путем введения одного или более дезоксинуклеозидов.
Кроме того, по мере выяснения механизмов других форм MDS и других расстройств, квалифицированным практиком может быть определен подходящий для лечения дезоксинуклеозид(ы).
Пациенты, которые демонстрируют фенотип, рассмотренный выше при дефиците ТК2, включая наиболее типичное проявление прогрессирующей мышечной болезни, характеризующейся генерализованной гипотонией, слабостью проксимальной мышцы, потерей ранее приобретенных моторных навыков, плохим питанием и проблемами дыхания, могут быть протестированы для окончательного определения заболевания.
Если клиническое проявление очень напоминает синдром истощения мтДНК, должно быть выполнено молекулярно-генетическое тестирование с использованием группы известных генов, которые вызывают синдром истощения мтДНК (Chanprasert и соавт. 2012 год). Ген ТК2 является единственным геном, в котором, как известно, мутации вызывают связанный с ТК2 синдром истощения митохондриальной ДНК. Такое тестирование может включать в себя анализ последовательности всех кодирующих областей и участков перехода экзон/интрон гена ТК2 на наличие вариантов последовательности и делеций/дупликаций. Если при анализе последовательностей идентифицируются сложные гетерозиготные или гомозиготные вредные мутации, то подтверждается диагноз дефицита ТК2, и, таким образом, субъект получит пользу от применения дезоксинуклеозидной терапии. Если анализ последовательности не идентифицирует две смешанные гетерозиготные или гомозиготные вредные мутации, то для определения и/или подтверждения диагноза дефицита ТК2 следует рассматривать анализ делеций/дупликаций.
Последующие тесты для определения и/или подтверждения диагноза дефицита ТК2 могут включать в себя тестирование концентрации креатинкиназы (КК) в сыворотке крови, электромиографию, гистопатологию скелетной мышцы, содержание (количество копий) митохондриальной ДНК (мтДНК) и активность цепи транспорта электронов (ЦТЭ) в скелетной мышце. Дефицит ТК2 определяется и/или подтверждается если в этих тестах обнаружен один или более из следующих признаков. Повышенная концентрация КК по сравнению с здоровым контролем может указывать на дефицит ТК2. Может быть проведена биопсия скелетной мышцы с последующим анализом содержания мтДНК в скелетной мышце. Если биопсия скелетных мышц свидетельствует о заметном отклонение в размерах волокон, переменных саркоплазматических вакуолях, переменном увеличении соединительной ткани и разорванных красных волокнах, а также повышенной активности сукцинатдегидрогеназы (СДГ) и от низкой до отсутствующей активности цитохром с-оксидазы (ЦО), а количество копий мтДНК является значительно сниженным (как правило, менее 20 % от здоровых контролей, соответствующего возраста и ткани), таким образом может быть установлен и/или подтвержден диагноз дефицита ТК2 (Chanprasert и соавт. 2012 год).
Кроме того, дефицит TК2 наследуется аутосомно-рецессивным способом. Таким образом, с целью диагностирования болезни, потомок пострадавшего пациента может быть проверен как можно раньше после рождения.
Во всех этих примерах терапию дезоксинуклеозидами следует начинать как можно скорее после диагноза дефицита ТК2.
Фармацевтические композиции, способы введения и дозировка
Данное изобретение охватывает введение дезоксинуклеозидов, более конкретно, одного или более дезоксинуклеозидов.
Наиболее предпочтительными способами введения являются пероральный, интратекальный и парентеральный, в том числе внутривенный. Дезоксинуклеозиды должны быть в соответствующей форме для выбранного введения.
Дезоксинуклеозиды легко растворяются в жидкости (такой как вода, смесь или молоко), тогда как форма свободной кислоты не растворяется в жидкости.
Такие фармацевтические композиции, содержащие один из более дезоксинуклеозидов для введения, могут содержать терапевтически эффективное количество дезоксинуклеозидов и фармацевтически приемлемый носитель. Фраза "фармацевтически приемлемый" относится к химическим соединениям и композициям, которые являются физиологически переносимыми и обычно не вызывают аллергическую или подобную нежелательную реакцию, такую как нарушение желудка, головокружение и тому подобное, при введении человеку и одобрены регуляторным ведомством Федерального правительства или правительством штата, или приведены в Фармакопее США или другой общепризнанной фармакопее с целью использования на животных и, более конкретно, на людях. "Носитель" относится к разбавителю, адъюванту, вспомогательному веществу или носителю, с которым вводится терапевтическое средство. Такие фармацевтические носители могут представлять собой стерильные жидкости, такие как физиологические растворы в воде и масле, в том числе нефтяные масла, масла животного, растительного или синтетического происхождения, такие как ореховое масло, соевое масло, минеральное масло, кунжутное масло и тому подобное. В случае если фармацевтическая композиция вводится внутривенно физиологический раствор является предпочтительным носителем. Солевые растворы и водные растворы декстрозы и глицерина также могут быть использованы в качестве жидких носителей, особенно для инъекционных препаратов. Подходящие фармацевтические вспомогательные вещества включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, моностеарат глицерина, тальк, хлорид натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду, этанол и тому подобные. При желании композиция может также содержать незначительные количества увлажнителей или эмульгаторов, или буферизующих средств для поддержания рН.
Пероральное введение является предпочтительным методом введения. Дезоксинуклеозиды могут быть добавлены в любую форму жидкости, которую пациент будет потреблять, включая, но без ограничений, молоко, как из груди человека, так и коровье, молочную смесь и воду.
Кроме того, фармацевтические композиции, предназначенные для перорального введения, могут представлять собой капсулы, таблетки, порошки, гранулы, растворы, сиропы, суспензии (на неводной или водной основе) или эмульсии. Таблетки или твердые желатиновые капсулы могут содержать лактозу, крахмал или их производные, стеарат магния, сахарин натрия, целлюлозу, карбонат магния, стеариновую кислоту или их соли. Мягкие желатиновые капсулы могут содержать растительные масла, воски, жиры, полутвердые или жидкие полиолы. Растворы и сиропы могут содержать воду, полиолы и сахара. Действующее вещество, предназначенное для перорального введения, может быть покрыто или смешано с материалом, который замедляет время растворения и/или абсорбцию действующего вещества в желудочно-кишечном тракте. Таким образом, пролонгированное высвобождение может длиться в течение многих часов, и при необходимости действующее вещество может быть защищено от деградации в желудке. Фармацевтические композиции для перорального введения могут быть приготовлены для облегчения высвобождения действующего вещества в конкретном участке желудочно-кишечного тракта ввиду специфических ферментативных состояний или рН.
Интратекальное введение является еще одной предпочтительной формой введения для устранения любых проблем касательно преодолевания гематоэнцефалического барьера дезоксинуклеозидами (Galbiati и соавт. 2006 год; Gotz и соавт. 2008 год). Интратекальное введение включает в себя инъекцию лекарственного средства в спинномозговой канал, более конкретно в субарахноидальное пространство, таким образом, что лекарственное средство достигает спинномозговой жидкости. Этот способ традиционно применяется для спинальной анестезии, химиотерапии и обезболивания. Интратекальное введение может быть выполнено с помощью люмбальной пункции (болюсная инъекция) или с помощью системы порта-катетера (болюс или инфузия). Катетер чаще всего вставляют между пластинками поясничных позвонков, а кончик катетера проводят в дуральное пространство до желаемого уровня (как правило, L3-L4). Интратекальные лекарственные формы чаще всего используют воду и физиологический раствор в качестве вспомогательного вещества, но также используются ЭДТК и липиды.
Другой предпочтительной формой введения является парентеральное введение, в том числе внутривенное введение. Фармацевтические композиции, предназначенные для парентерального введения, в том числе внутривенного введения, включают в себя водные и неводные стерильные инъекционные растворы или суспензии, которые могут содержать антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, которые делают композицию по сути изотоничной по отношению к крови субъекта. Другие компоненты, которые могут присутствовать в таких композициях, включают в себя воду, спирты, полиолы, глицерин и растительные масла. Композиции, предназначенные для парентерального введения, могут быть представлены в виде однодозовых или многодозовых контейнеров, таких как герметичные ампулы и флаконы, и могут храниться в сублимированном (лиофилизированном) состоянии, требующем только добавления стерильного носителя непосредственно перед применением. Экстемпоральные растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток. Подходящие носители, которые могут быть применены для обеспечения парентеральных лекарственных форм согласно данному изобретению, хорошо известны специалистам в данной области техники. Это может быть: Вода для инъекций USP; водные носители, такие как хлорид натрия для инъекций, раствор Рингера для инъекций, декстроза для инъекций, декстроза и хлорид натрия для инъекций и лактатный раствор Рингера для инъекций; смешиваемые с водой носители, такие как этиловый спирт, полиэтиленгликоль и полипропиленгликоль; и неводные носители, такие как кукурузное масло, хлопковое масло, арахисовое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат.
Кроме того, поскольку некоторые пациенты к моменту начала лечения дезоксинуклеозидами могут получать энтеральное питание, дезоксинуклеозиды могут быть введены через гастрономическую питательную трубку или другие средства энтерального питания.
Другие способы введения включают слизистое, такое как назальное, сублингвальное, вагинальное, буккальное или ректальное введение; или трансдермальное введение субъекту.
Фармацевтические композиции, предназначенные для назального и ингаляционного введения, могут содержать твердые носители, такие как порошки, которые можно вводить путем быстрой ингаляции через нос. Композиции для назального введения могут содержать жидкие носители, такие как спреи или капли. В альтернативном варианте, ингаляция непосредственно через легкие может быть выполнена путем глубокого вдоха или путем установки ротового мундштука. Такие композиции могут содержать водные или масляные растворы активного компонента. Композиции для ингаляции могут поставляться в специально приспособленных устройствах, включая, но без ограничений, аэрозоли под давлением, распылители или инсуффляторы, которые могут быть сконструированы таким образом, чтобы обеспечить заданные дозировки активного компонента.
Фармацевтические композиции, предназначенные для ректального введения, могут быть представлены в виде суппозиториев или клизм. Фармацевтические композиции, предназначенные для вагинального введения, могут быть представлены в форме пессариев, тампонов, кремов, гелей, паст, пен или спреев.
Фармацевтические композиции, предназначенные для трансдермального введения, могут быть предоставлены в виде отдельных пластырей, предназначенных для поддержания непосредственного контакта с эпидермисом реципиента в течение длительного периода времени.
Дезоксинуклеозидная терапия включает в себя введение одного или более дезоксинуклеозидов, выбранных из группы, состоящей из дезокситимидина (дТ), дезоксицитидина (дЦ), дезоксиаденозина (дА) и дезоксигуанозина (дГ).
Квалифицированный практик может определить, какие дезоксинуклеозиды являются полезными исходя из дефицита. Практикующий специалист в данной области техники также может определить, следует ли вводить смеси дезоксинуклеозидов и в каком отношении. При необходимости введения двух дезоксинуклеозидов, они могут находиться в соотношении 50/50 каждый, например, дЦ и дТ, или в соотношениях около 5/95, 10/90, 15/85, 20/80, 25/75, 30/70, 35/65, 40/60, 45/55, 55/45, 60/40, 65/35, 70/30, 75/25, 80/20, 85/15, 90/10 и 95/5.
В качестве примера, дТ и дЦ вводятся в смеси в равных количествах при дефиците ТК2.
Выбор терапевтически эффективной дозы будет определяться специалистом в данной области с учетом нескольких факторов, которые будут известны среднему специалисту в данной области техники. К таким факторам относится конкретная форма дезоксинуклеозида и его фармакокинетические параметры, такие как биодоступность, биотрансформация и период полувыведения, которые будут определены в ходе обычных процедур разработки, обычно используемых для получения разрешения контролирующего органа для фармацевтического соединения. Другие факторы, которые берутся во внимание при рассмотрении дозы, включают в себя состояние или заболевание, подлежащее лечению, или пользу, которая должна быть достигнута для нормального индивидуума, массу тела пациента, способ введения, является ли введение однократным или постоянным, препараты сопутствующей терапии и другие факторы, которые, как известно, влияют на эффективность вводимых фармацевтических веществ. Таким образом, точная доза должна определяться на основе оценки специалиста в данной области техники и определенной ситуации каждого пациента, а также в соответствии со стандартными клиническими методами.
Предпочтительная доза составляет от около 100 мг/кг/день до около 1000 мг/кг/день. Дополнительная предпочтительная доза составляет от около 200 мг/кг/день до около 800 мг/кг/день. Дополнительная предпочтительная доза составляет от около 250 мг/кг/день до около 400 мг/кг/день. Такие размеры дозы представляют собой отдельные дезоксинуклеозиды или композиции со смесью одного или более дезоксинуклеозидов, например, дТ и дЦ. Например, доза может содержать 400 мг/кг/день только дТ. В дополнительном примере доза может содержать смесь из 200 мг/кг/день дТ и 200 мг/кг/день дЦ. В дополнительном примере доза может содержать 400 мг/кг/день смеси дТ и дЦ.
Введение дезоксинуклеозидов может происходить один раз в день, два раза в день, три раза в день, четыре раза в день, пять раз в день, до шести раз в день, предпочтительно с равными промежутками времени. Например, когда дезоксинуклеозиды вводятся четыре раза в день, дозы должны вводиться в 8:00, 12:00, 16:00 и 20:00.
В случае внутривенного или интратекального введения дозы также могут быть уменьшены. Для введений такого типа предпочтительная доза составляет от около 50 мг/кг/день до около 500 мг/кг/день.
Как показано в примере 5, дозы могут регулироваться с целью оптимизации реакций у субъекта. Например, дезоксинуклеозиды сначала можно вводить со 100 мг/кг/день, а затем увеличивать с течением времени до 200 мг/кг/день, до 400 мг/кг/день, до 800 мг/кг/день и до 1000 мг/кг/день, в зависимости от реакции и переносимости субъекта.
С целью улучшения состояния, перед увеличением дозировки над субъектом может проводиться наблюдение. Реакцию субъекта на терапевтическое введение дезоксинуклеозидов можно контролировать путем наблюдения за силой и контролем над мышцами субъекта, его двигательной активностью, а также изменениями в росте и весе. Если один или более из этих параметров увеличивается после введения, лечение может быть продолжено. Если один или более из этих параметров остается таким же или уменьшается, доза дезоксинуклеозидов может быть увеличена.
Как показано в примерах, дезоксинуклеозиды хорошо переносятся. Любые наблюдаемые нежелательные реакции были незначительными и представляли собой в основном диарею, вздутие живота и другие желудочно-кишечные проявления. Над субъектом также может проводиться наблюдение с целью выявления любых нежелательных реакций, таких как желудочно-кишечная непереносимость, например, диарея. Если после введения наблюдается одна или более нежелательная реакция, то дозировка может быть уменьшена. Если таких нежелательных реакций не наблюдается, то дозировка может быть увеличена. Кроме того, как только дозировка уменьшена из-за нежелательной реакции, и нежелательная реакция больше не наблюдается, дозировка может быть увеличена.
Дезоксинуклеозиды также могут быть введены совместно с другими препаратами. Такие препараты будут включать в себя лекарственные средства для лечения симптомов конкретной формы MDS. В частности, при дефиците ТК2, дТ и дЦ могут быть введены совместно с ингибитором широко распространенных нуклеозидных катаболических ферментов, в том числе, но без ограничений, ингибиторами ферментов, такими как тетрагидроуридин (ингибитор цитидиндезаминазы) и иммуциллин H (ингибитор пуриновой нуклеозидфосфорилазы), и типирацил (ингибитор тимидинфосфорилазы). Такие ингибиторы известны и используются для лечения некоторых видов рака.
ПРИМЕРЫ
Данное изобретение является более понятным со ссылкой на следующие, не имеющие ограничительного характера примеры, которые представлены для более полной иллюстрации предпочтительных вариантов реализации изобретения. Они никоим образом не должны толковаться как ограничивающие широкий объем данного изобретения.
Пример 1 – Материалы и методы
Мышиная модель дефицита ТК2
Ранее сообщалось о гомозиготной мыши с нокин мутацией Tk2 H126N (Tk2-/-), которая проявляет фенотип, поразительно похожий на человеческую инфантильную энцефаломиопатию (Akman и соавт. 2008 год). Между днем 10 и 13 после рождения, мыши Tk2-/- стремительно развивают фатальную энцефаломиопатию, характеризующуюся уменьшенной способностью к передвижению, нестабильной походкой, крупноамплитудным тремором, замедлением роста и стремительными прогрессированием до ранней смерти в возрасте от 14 до 16 дней. Молекулярные и биохимические анализы данной мышиной модели продемонстрировали, что патогенез заболевания является связанным с потерей активности фермента и последующими дисбалансами пула дНТФ с пониженными уровнями дТТФ в головном мозге и уровнями дТТФ и дЦТФ в печени, что, в свою очередь, приводит к истощению мтДНК и дефектам ферментов дыхательной цепи, содержащим субъединицы, которые кодируются мтДНК, что является наиболее заметным в головном и спинном мозге.
Все эксперименты проводились в соответствии с протоколом, одобренным Комитетом по содержанию и использованию лабораторных животных медицинского центра Колумбийского университета, и были согласованы с руководством Национального института здравоохранения по содержанию и использованию лабораторных животных. Мышей содержали и разводили в соответствии с международными стандартными условиями, с циклом из 12-часов света и 12-часов темноты, и забивали в возрасте 4, 13 и 29 дней.
Органы (головной мозг, спинной мозг, печень, сердце, почку, мышцы квадрицепса, легкие и желудочно-кишечный тракт) удаляли и либо замораживали в жидкой фазе изопентана, предварительно охлаждая его около точки замерзания (-160°С) сухим льдом, либо фиксировали в 10 % забуференном нейтральном формалине и заливали в парафин с использованием стандартных процедур. Залитые в парафин ткани затем окрашивали гематоксилином и эозином (Г-Э) с целью морфологического исследования или обрабатывали для исследований иммуноокрашивания с помощью глиального фибриллярного кислого белка (ГФКБ), ЦО I или субъединицы комплекса I, как подробно описано в дополнительных процедурах. Как гетерозиготные так и гомозиготные мыши дикого типа считались контрольной группой (Tk2+), поскольку ранее между ними не было обнаружено клинических и биохимических различий (Akman и соавт. 2008; Dorado и соавт. 2011).
Введение препаратов и экспериментальный план
Дезоксицитидин (дЦ) и дезокситимидин (дТ) вводили в 50 мкл смеси молока Esbilac для маленьких домашних животных (Pet-Ag) с помощью ежедневного перорального введения через желудочный зонд нокин мышам Tk2 H126N (Tk2-/-) и контролю дикого типа (Tk2+), совпадающего по возрасту, используя 2 дозы, 260 мг/кг/день и 520 мг/кг/день, от дня 4 до дня 29 после рождения. В возрасте 21-го дня мышей отделяли от матери, и лечение продолжали введением дЦ и дТ в питьевой воде с использованием эквимолярных доз 1,6 мМ и 3,2 мМ, соответственно. Группу мутантов Tk2 отрицательного контроля, не получавших лечение, и контрольных мышей дикого типа взвешивали и внимательно осматривали для сравнения.
Оценка фенотипа
Вес тела оценивали ежедневно, поскольку ранее отмечалось, что неспособность набирать вес является первым признаком заболевания (Akman и соавт. 2008).
Для определения степени безопасности и эффективности дТ/дЦ, сопоставляли время выживания, возраст на момент начала болезни, тип и тяжесть симптомов, возникновение побочных действий и соразмерность окончания лечения из-за нежелательных явлений у мышей Tk2, которые получали лечение и не получали его. Общее поведение, время выживания и вес тела мышей оценивали ежедневно, начиная с 4-го дня после рождения.
Анализ пула дНТФ с помощью полимеразы
Ткани гомогенизировали на льду в 10 объемах (масса/объем) холодного буфера, стабилизирующего микротрубочки (MTSE) (210 мМ маннита, 70 мМ сахарозы, 10 мМ Трис-HCl, pH 7,5, 0,2 мМ этиленгликоль-тетрауксусной кислоты (ЭГТК), 0,5% БСА) и центрифугировали при 1000 g в течение 5-ти минут при 4°С, затем трижды центрифугирования при 13000 g в течение 2-х минут при 4°С. Супернатант осаждали 60% метанолом, выдерживали 2 часа при -80°С, кипятили 3 минуты, хранили при -80°C (от 1 часа до ночи) и центрифугировали при 20800 g в течение 10 минут при 4°C. Супернатанты выпаривали досуха, а осадок ресуспендировали в 65 мкл воды и хранили при -80°С до анализа. Для сведения к минимуму интерференции рибонуклеотидов были определены совокупные пулы дНТФ, как сообщалось (Ferraro и соавт. 2010 год; Marti и соавт. 2012a). Вкратце, 20 мкл объемных реакций получали путем смешивания 5 мкл образца или стандартного дНТФ с 15 мкл рабочего буферного раствора [0,025 U/мл ДНК-полимеразы ThermoSequenase (GE Healthcare, Пискатауэй, штат Нью-Джерси, США) или Taq-полимеразы (Life Technologies, Нью-Йорк, США), 0,75 мкМ 3H-дТТФ или 3H-дАТФ (Moravek Biochemicals), 0,25 мкМ определенного олигонуклеотида, 40 мМ Трис-HCl, pH 7,5, 10 мМ MgCl2, 5 мМ DTT]. Через 60 минут при 48°С 18 мл реакции наносили на фильтры Ватмана DE81, сушили на воздухе и три раза промывали в течение 10 минут с 5% Na2HPO4, раз в дистиллированной воде и раз в абсолютном этаноле. Сохраненная радиоактивность определялась сцинтилляционным методом.
Измерения нуклеозидов методом ВЭЖХ
Уровни дезокситимидина (дТ), дезоксиуридина (дУ), урацила (У) и тимина (Т) оценивали с помощью метода ВЭЖХ с градиентным элюированием, следуя описанной ранее методике (Lopez и соавт. 2009 год; Marti и соавт. 2012b) с незначительными изменениями. Вкратце, депротеинизированные образцы инъецировали в систему ВЭЖХ Alliance (Waters Corporation) с колонкой для обращенно-фазовой хроматографии Alltima C18NUC (Alltech) при постоянной скорости потока 1,5 мл/мин (за исключением тех случаев, когда это указано) с использованием четырех буферов: элюент А (20 мМ фосфата калия, рН 5,6), элюент В (вода) и элюент С (метанол). Образцы элюировали в течение 60 минут со следующим градиентом:0-5 мин, 100% элюента А; 5-25 мин, 100-71% элюента А, 29% элюента В; 25-26 мин, 0-100% элюента C; 26-30 мин, 100% элюента C; 30-31 мин, 0-100% элюента B; 31-35 мин, 100% элюента В (1,5-2 мл/мин); 35 - 45 мин, 100 % элюента В (2 мл/мин); 45-46 мин, 100% элюента В (2-1,5 мл/мин); 46-47 мин, 0-100 % элюента C; 47-50 мин, 100 % элюента C; 50-51 мин, 0-100% элюента А; и 51-60 мин, 100% элюента А.
Абсорбцию элюатов наблюдали при 267 нм, а пики дТмд и дУрд определяли количественно путем сравнения их площадей пиков с калибровочной кривой, полученной с использованием водных стандартов. Для однозначной идентификации пиков дезокситимидина, дезоксиуридина, урацила и тимина в каждом образце, вторую аликвоту обрабатывали избытком очищенной E. coli TP (Sigma) для конкретного исключения дТ и дУ. Предел чувствительности этого метода составляет 0,05 ммоль/л для всех нуклеозидов. Результаты были выражены в виде нмоль/мг белка.
ОТ-кПЦР: количественная оценка митохондриальной ДНК
ПЦР в реальном времени проводили с использованием праймеров и зондов к гену ЦО I (мтДНК) мыши и глицеральдегид-3-фосфатдегидрогеназы мыши (ГАФДГ, яДНК) (Applied Biosystems, Invitrogen, Фостер-Сити, штат Калифорния, США), как описано с использованием метода ddCt в ПЦР-системе Step One Plus Real Time (Applied Biosystems) (Dorado и соавт. 2011). Значения мтДНК нормализовали к значениям яДНК и выражали в процентах относительно дикого типа (100%).
Уровни белка митохондриальной дыхательной цепи
Тридцать микрограммов полных экстрактов головного мозга или мозжечка подвергали электрофорезу в ПААГ-геле ДСН-12%, переносили на Immun-BlotTM ПВДФ-мембраны (ПВДФ - поливинилиденфторид) (Biorad, Геркулес, штат Калифорния, США) и зондировали с MitoProfile® Total OXPHOS Rodent WB Antibody Cocktail (MitoSciences, Юджин, штат Орегон, США). Взаимодействие протеин-антитело детектировали с помощью мышиного антитела против IgG мыши, конъюгированного с пероксидазой (Sigma-Aldrich, Сент-Луис, штат Миссури, США), используя систему детектирования вестерн-блоттинга AmershamTM ECL Plus (GE Healthcare Life Sciences, Великобритания). Количественную оценку белков проводили с использованием программного обеспечения NIH ImageJ 1,37V. Среднее значение уровня серого цвета вычислялось в выбранных областях как сумма значений серого для всех пикселей в выбранной области, деленная на количество пикселей.
Измерение активности митохондриальных ферментов дыхательной цепи с помощью спектрофотометрического анализа
Анализ митохондриальных ферментов ДЦ проводили в ткани головного мозга, следуя описанной ранее методике (DiMauro и соавт. 1987 год).
Статистические методы
Данные выражаются как среднее ± СО, по меньшей мере, трех экспериментов на группу. Тест Гехана-Бреслоу-Вилкоксона использовался для сравнения доли выживаемости каждой группы мышей. Значение p<0,05 считалось статистически значимым.
Пример 2 - Введение дЦ/дТ мышам Tk2-/- задерживало клиническое начало дефицита ТК2 и увеличивало выживаемость
Дозу 260 и 520 мг/кг/день каждого дезоксинуклеозида (дЦ/дТ) вводили мышам Tk2-/-. Такие дозы дезоксинуклеозидов являлись молярным эквивалентом 400 и 800 мг/кг/день дЦМФ + дТМФ, соответственно.
Мыши, которые перорально получали лечение в виде дЦ + дТ (260 или 520 мг/кг/день с 4-го дня после рождения), производили впечатление нормальных до 21-го дня после рождения (фигура 1). После 21-го дня мутантные мыши, получавшие дозу 260 мг/кг/день (Tk2-/- 260 мг/кг/день дС/дТ), перестали набирать вес и демонстрировали умеренный тремор головы, а также слабость, которые приводили к смерти на 31-й ± 4,3 день после рождения (фигура 2).
Мутантные мыши, получавшие 520 мг/кг/день дЦ + дТ (Tk2-/- 520 мг/кг/день дЦ/дТ) продолжали набирать вес еще одну неделю, но впоследствии демонстрировали ухудшение состояния, подобное Tk2-/- 260 мг/кг/день дЦ/дТи погибали на 43-й ± 10 день после рождения. Эти результаты являются сопоставимыми с результатами, показанными мышами Tk2-/-, получавшими пероральное лечение в виде 200 или 400 мг/кг/день дЦМФ/дТМФ. За Tk2+ 260 мг/кг/день дЦ/дТ и Tk2+ 520 мг/кг/день дЦ/дТ наблюдали до 60-го дня после рождения. Никаких побочных действий не наблюдалось.
Как показано, продолжительность жизни Tk2-/- была значительно увеличена. Мыши Tk2-/-, не получавшие лечение имели среднюю продолжительность жизни 13 дней, тогда как мыши, которые получали лечение выживали в среднем 31 и 40 дней с дозой 260 и 520 мг/кг/день, соответственно (фигура 2). Интересно, что одна из мышей прожила до 56-го дня после рождения, что, на сегодняшний день, было самой длительной продолжительностью жизни для модели мыши нокин Tk2.
Пример 3. Пероральное введение дЦ/дТ улучшает молекулярные аномалии в мозге и печени
Измерение дНТФ в митохондриальном экстракте показало, что оба Tk2-/- 260 мг/кг/день дЦ/дТ и Tk2-/- 520 мг/кг/день дЦ/дТ не полностью устраняли дисбалансы пула митохондриальных дНТФ на 13-й день после рождения и проявляли непостоянные эффекты в тканях с полным восстановлением дефицита дЦТФ в головном мозге, тогда как дТТФ корригировался в печени. И наоборот, дефициты дТТФ в головном мозге и дЦТФ в печени оставались существенными, несмотря на добавление дезоксинуклеозида (фигура 3).
У мышей Tk2-/- 260 мг/кг/день дЦ/дТ и Tk2-/- 520 мг/кг/день дЦ/дТ лечение предотвратило истощение мтДНК в сердце, печени, почках, кишечнике и мышцах на 13-й день после рождения (фигура 4). И наоборот, количество копий мтДНК, на 13-й день после рождения, было только частично восстановлено в головном мозге дозозависимым образом с отношением мтДНК/яДНК относительно контрольного головного мозга, достигая 39 % с 260 мг/кг/день дЦ + дТ и 52 % с 520 мг/кг/день. Измерения оснований дТ и урацила в головном мозге с помощью ВЭЖХ показали более высокие уровни у животных, получавших лечение в виде дЦ + дТ или с дЦМФ + дТМФ (фигура 5), дополнительно указывая на то, что как дезоксинуклеозиды, так и дезоксинуклеозидные монофосфаты преодолевают гематоэнцефалический барьер. На 29-й день после рождения, истощение мтДНК было частично восстановлено при терапии 260 и 520 мг/кг/день дЦ + дТ в сердце (40 и 35 %), печени (46 и 45 %), почках (38 и 42 %) и мышцах (24 и 35%), но, что удивительно, было полностью восстановлено в кишечнике (82 и 84 %) (фигура 4).
Пример 4. Пероральное введение дЦ/дТ улучшает биохимические аномалии в головном мозге
Активность ферментов дыхательной цепи (RCE) и уровни белка были полностью восстановлены в головном мозге с TK2-/-260 мг/кг/день дЦ/дТ на 13-й день после рождения (фигура 6). Активности RCE также были восстановлены на 29-й день после рождения, и лишь незначительное снижение активности комплекса I наблюдалось с ТК2-/- 520 мг/кг/день дЦ/дТ (фигура 6). Уровни белка RCE в головном мозге частично восстанавливались на 29-й день после рождения с более высокими уровнями в ТК2-/- 520 мг/кг/день дЦ/дТ чем в TK2-/- 260 мг/кг/день дЦ/дТ (фигура 7). Эти различия в уровнях белка согласуются с различиями в истощении мтДНК в головном мозге, получавших лечение мутантных мышей на 29-й день после рождения и, вероятно, учитываются в случае пролонгированного выживания, наблюдаемого с более высокой дозой.
Пример 5. Введение дЦ/дТ пациентам с дефицитом ТК2 было эффективным
Симптомы, дозирование и результаты пациентов с дефицитом ТК2, которые получили терапию дезоксинуклеозидом под наблюдением и контролем изобретателей, приведены ниже.
Пациент 1
Этот пациент родился в США в феврале 2011 года. Его симптомы проявились через 12 месяцев в виде пониженного тонуса мышц и амиотонии головы. Он никогда не ходил. У него также есть слабость дыхательной мускулатуры, и он был переведен на механическую вентиляцию через 19 месяцев после рождения, которой он до сих пор пользуется 24 часа в сутки. Он также пользуется питательной трубкой с 19 месяцев.
Ранее он принимал дЦМФ и дТМФ в количестве 100 мг/кг/день, а затем 200 мг/кг/день. Благодаря такой терапии он мог хватать небольшие предметы, а его вес увеличился с 10,4 кг до 19,5 кг.
В октябре 2015 года он начал с 260 мг/кг/день дЦ и дТ, дозу которых увеличил до 340 мг/кг/день дЦ и дТ. Через два месяца он двигал руками и головой лучше, мог стоять 5 минут при поддержке человеком, начиная кашлять, а его сердечный ритм был медленнее (от 140-170 ударов в минуту в день, до 100-120 ударов в минуту в день).
23 марта 2016 года доза была увеличена до 400 мг/кг/день дЦ и дТ. Через 6 недель после этой терапии он показал дальнейшие улучшения: он мог сидеть в кресле около 5 часов в день; стоял в вертикализаторе в течение 1,5 часов; пытался схватить и держать маленькие плюшевые игрушки; нажимал кнопки компьютера; развязывал свои подгузники и нацеливал свой пенис с целью обмочить человека, меняющего его подгузники; и держал свои колени согнутыми в течение нескольких секунд.
Единственной нежелательной реакцией, наблюдающейся во время лечения, был диарея.
Пациент 2
Этот пациент родился в Испании в 1987 году. Он начал проявлять симптомы в возрасте 3-х лет, включая слабость проксимальных мышц. Он потерял способность передвигаться в возрасте 13-ти лет и использовал механическую вентиляцию 24 часа в сутки. Ранее он принимал дАМФ и дЦМФ в дозе 200 мг/кг/день и демонстрировал увеличение массы тела и уменьшение времени механический вентиляции легких с 24-х до 22-х часов в день.
Он был на дезоксинуклеозидовой терапии с июня 2015 года, принимая 400 мг/кг/день дЦ и дТ, и продемонстрировал улучшение мышечной силы, его вес и дыхание стабилизировались, и он наслаждается лучшим качеством жизни.
Единственными нежелательными реакциями, которые наблюдались во время лечения, была диарея и выпадение волос.
Пациент 3
Этот пациент родился в Испании в 1985 году. Его симптомы начались с 6 лет с лицевой, проксимальной и аксиальной мышечной слабости. Он начал с 200 мг/кг/день дТ и дЦ в июне 2015 года, и на сегодняшний день его состояние изменилось в лучшую сторону с улучшениями в тесте шестиминутной ходьбы, во времени, необходимом для того чтобы встать и уйти, и в поднимании вверх и вниз на 4 шага.
Единственной нежелательной реакцией, наблюдающейся во время лечения, был диарея.
Пациент 4
Этот пациент родился в Испании в феврале 2009 года. Его симптомы проявились в возрасте шести месяцев в виде потери веса и отставании в физическом развитии. Он начал с 230 мг/кг/день дЦ и дТ в июле 2015 года. К январю 2016 года он показал улучшение своего состояния и аппетита.
Нежелательные реакции не наблюдались.
Пациент 5
Этот пациент родился в Испании в 1957 году и начал проявлять симптомы ортопноэ и слабость диафрагмы в возрасте 50 лет. Ночью он использовал систему двухфазной вентиляции с положительным давлением в дыхательных путях (BiPAP). Он начал с 200 мг/кг/день дЦ и дТ в ноябре 2015 года.
Нежелательные реакции не наблюдались.
Пациент 6
Этот пациент родился в Испании в октябре 2011 года и начал проявлять симптомы через 15 месяцев после рождения, включая гипотонию и слабость. Он потерял способность к передвижению в возрасте 22-х месяцев и имел слабость дыхательной мускулатуры. К нему начали применять механическую вентиляцию через 16 месяцев после рождения, и он в данное время пользуется BiPAP двенадцать часов в день. Ранее он принимал дЦМФ и дАМФ в количестве 100 мг/кг/день, которое было увеличено до 400 мг/кг/день. Его сила за шкалой Egen Klassification улучшилась (от 28/30 до 13/30), а его вес увеличился с 9,8 кг до 12,3 кг.
Он начал дезоксинуклеозидную терапию в апреле 2015 года по 400 мг/кг/день дЦ и дТ. В октябре 2015 года его сила по шкале Egen Klassification изменилась с 13/30 до 11/30, а его вес увеличился до 16,5 кг с 12,3 кг.
Нежелательные реакции не наблюдались.
Пациент 7
Этот пациент родился в Испании в ноябре 2012 года. Он начал проявлять симптомы через 17 месяцев после рождения, в том числе слабость и гипотонию. Он потерял способность к передвижению в возрасте 22-х месяцев и к нему начали применять механическую вентиляцию через 29 месяцев после рождения. Ранее он принимал дЦМФ и дАМФ в количестве 100 мг/кг/день, которое было увеличено до 400 мг/кг/день. Его сила за шкалой Egen Klassification улучшилась (от 30/30 до 24/30), а его вес увеличился с 11 кг до 15,7 кг.
Он начал дезоксинуклеозидную терапию в апреле 2015 года с дозы 400 мг/кг/день дТ и дЦ. В ноябре 2015 года его сила по шкале Egen Klassification изменилась с 24/30 до 19/30, а его вес увеличился до 17 кг с 15,7 кг.
Нежелательные реакции не наблюдались.
Пациент 8
Этот пациент родилась в Чили в сентябре 1989 года и начала проявлять симптомы через 11 месяцев в виде частых падений и прогрессирующего нарушения походки. Она потеряла способность ходить без посторонней помощи в возрасте около 4 лет. Ранее она пользовалась нуклеотидной терапией и демонстрировала улучшение в своей способности к передвижению, в том числе ходьбе без посторонней помощи, длительном стоянии, подъеме по лестнице, посещении тренажерного зала и посещении по личным потребностям.
Она перешла на дезоксинуклеозидную терапию в феврале 2016 года с дозы 260 мг/кг/день дЦ и дТ, а затем увеличила дозу до 400 мг/кг/день дЦ и дТ в мае 2016 года и продолжала демонстрировать улучшение состояния.
Нежелательные реакции не наблюдались.
Пациент 9
Этот пациент родился в Гватемале в сентябре 1989 года. Он начал с 130 мг/кг/день дЦ и дТ в августе 2015 года и увеличил дозу до 260 мг/кг/день дЦ и дТ в феврале 2016 года. Он показал улучшения в своей энергичности.
Нежелательные реакции не наблюдались.
ССЫЛКИ НА ПУБЛИКАЦИИ
Akman и соавт. (2008) Thymidine kinase 2 (H126N) knock in mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum. Mol. Genet. 17:2433-2440
Alston и соавт. (2013) Late-onset respiratory failure due to TK2 mutations causing multiple mtDNA deletions. Neurology 81:2051-3
Bartesaghi и соавт. (2010) Loss of thymidine kinase 2 alters neuronal bioenergetics and leads to neurodegeneration. Hum. Mol. Genet. 19:1669-77
Béhin и соавт. (2012) Adult cases of mitochondrial DNA depletion due to TK2 defect An expanding spectrum. Neurology 78:644-648
Blakely и соавт. (2008) Novel mutations in the TK2 gene associated with fatal mitochondrial DNA depletion myopathy. Neuromuscular Disorders 18:557-560
Bourdon и соавт. (2007) Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nature Genetics 39:776-780
Carrozzo и соавт. (2003) Mutation analysis in 16 patients with mtDNA depletion. Hum. Mutat. 21:453-454
Chanprasert и соавт. (2013) Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol. Genet. Metab. 110:153-61
Chanprasert и соавт. (2012) TK2-Related Mitochondrial DNA Depletion Syndrome, Myopathic Form. GeneReviews® Internet, 6 декабря 2012 года.
Collins и соавт. (2009) Progressive myofiber loss with extensive fibro-fatty replacement in a child with mitochondrial DNA depletion syndrome and novel thymidine kinase 2 gene mutations. Neuromuscular Disorders 19:784-787
Copeland (2008) Inherited mitochondrial diseases of DNA replication. Ann. Rev. Med. 59:131-146
DiMauro и соавт. (1987) Cytochrome c oxidase deficiency in Leigh syndrome. Ann. Neurol. 22:498-506
DiMauro, Schon. (2003) Mitochondrial respiratory-chain diseases. New England Journal of Medicine 348:2656-2668
DiMauro, Hirano. (2005) Mitochondrial encephalomyopathies:an update. Neuromuscul. Disord. 15:276-286
Dorado и соавт. (2011) Onset and organ specificity of Tk2 deficiency depends on Tk1 down-regulation and transcriptional compensation. Hum. Mol. Genet. 20:155-64
Elpeleg и соавт. (2005) Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am. J. Hum. Genet. 76:1081-1086
Ferraro и соавт. (2010) Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Research 38:e85
Galbiati и соавт. (2006) New mutations in TK2 gene associated with mitochondrial DNA depletion. Pediatr. Neurol. 34:177-185
Garone и соавт. (2012). MPV17 Mutations Causing Adult-Onset Multisystemic Disorder With Multiple Mitochondrial DNA Deletions. Arch Neurol 69:1648-1651
Garone и соавт. (2014). Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol Med 6:1016-1027
Garone и соавт. (2016) в процессе подготовки, "Phenotypic Spectrum and Retrospective Natural History of Thymidine Kinase 2 Deficiency"
Gotz и соавт. (2008) Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain 131:2841-2850
Hirano и соавт. (2001) Defects of intergenomic communication:autosomal disorders that cause multiple deletions and depletion of mitochondrial DNA. Semin. Cell. Develop. Biol. 12:417-427
Hirano и соавт. (2004) MtDNA maintenance and stability genes:MNGIE and mtDNA depletion syndromes. В:редакции Köhler, Bauer. Mitochondrial Function and Biogenetics. Berlin:Springer-Verlag. ст. 177-200
Leshinsky-Silver и соавт. (2008) A defect in the thymidine kinase 2 gene causing isolated mitochondrial myopathy without mtDNA depletion. Eur. J. Paediatr. Neurol. 12:309-13
Lesko и соавт. (2010) Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscul. Disord. 20:198-203
Longley и соавт. (2006). Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 78:1026-1034
Lopez и соавт. (2009) Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase deficient mice. Human Molecular Genetics 18:714-22
Mancuso и соавт. (2003) Mitochondrial myopathy of childhood associated with mitochondrial DNA depletion and a homozygous mutation (T77M) in the TK2 gene. Arch. Neurol. 60:1007-9
Mancuso и соавт. (2002) Mitochondrial DNA depletion:mutations in thymidine kinase gene with myopathy and SMA. Neurology. 59:1197-202
Mandel и соавт. (2001) The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nature Genet. 29:337-341
Martí и соавт. (2010) Hearing loss in a patient with the myopathic form of mitochondrial DNA depletion syndrome and a novel mutation in the TK2 gene. Pediatr. Res. 68:151-4
Martí и соавт. (2012a) Measurement of mitochondrial dNTP pools. Methods Mol. Biol. 837:135-148
Martí и соавт. (2012b) Assessment of thymidine phosphorylase function:measurement of plasma thymidine (and deoxyuridine) and thymidine phosphorylase activity. Methods Mol. Biol. 837:121-133
Naviaux, Nguyen. (2004) POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann. Neurol. 55:706-712
Nishino и соавт. (1999). Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283:689-692.
Oskoui и соавт. (2006) Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch. Neurol. 63:1122-1126.
Ostergaard и соавт. (2007) Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am. J. Hum. Genet. 81:383-387
Paradas и соавт. (2012) TK2 mutation presenting as indolent myopathy. Neurology 29:504-506
Ronchi и соавт. (2012). Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 135:3404-3415.
Roos и соавт. (2014) Mitochondrial DNA depletion in single fibers in a patient with novel TK2 mutations. Neuromuscul. Disord. 24:713-20
Saada и соавт. (2001) Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nature Genet. 29:342-344
Saada и соавт. (2003) Mitochondrial deoxyribonucleoside triphosphate pools in thymidine kinase 2 deficiency. Biochem. Biophys. Res. Commun. 310:963-966
Sarzi и соавт. (2007) Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann. Neurol. 62:579-587
Spelbrink и соавт. (2001). Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nature Genet. 28:223-231
Spinazzola и соавт. (2006) MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nature Genet. 38:570-575
Tulinius и соавт. (2005) Novel mutations in the thymidine kinase 2 gene (TK2) associated with fatal mitochondrial myopathy and mitochondrial DNA depletion. Neuromuscul. Disord. 15:412-415
Tyynismaa и соавт. (2012) Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum. Mol. Genet. 21:66-75
Tyynismaa и соавт. (2009). A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am. J. Hum. Genet. 85:290-295
Van Goethem и соавт. (2001) Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature Genet. 28:211-212.
Vilà и соавт. (2003) Reversion of mtDNA depletion in a patient with TK2 deficiency. Neurology 60:1203-1205
Wang и соавт. (2005) Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol. Genet. Metab. 84:75-82.

Способ лечения синдрома прадера-вилли

