Результат интеллектуальной деятельности: ПОЛИАРИЛЕНОВОЕ ВОЛОКНО, ХАРАКТЕРИЗУЮЩЕЕСЯ УЛУЧШЕННОЙ ГИДРОЛИТИЧЕСКОЙ СТАБИЛЬНОСТЬЮ
Вид РИД
Изобретение
Высокопрочные синтетические полимерные материалы широко используются в областях применения, в которых требуется долговечная высокая прочность. Во время использования изделия, содержащие такие синтетические полимеры, могут быть подвергнуты воздействию повышенных температур, высокой влажности и/или воды. Такие условия возникают вследствие воздействия окружающей среды, например, в тропических условиях, непосредственного вхождения в контакт с водой, машинным оборудованием, телом человека или вследствие воздействия тепла, вырабатываемого во время динамического нагружения (гистерезиса, внутреннего/внешнего трения). Такие высокотехнологические синтетические материалы включают, например, арамиды и жесткие стержневидные ароматические гетероциклические полимеры, такие как полибензазол и другие.
Условия окружающей среды и условия использования могут оказывать пагубное воздействие на прочность синтетических волокон. Это в особенности имеет место при воздействии на волокна условий, которые благоприятствуют гидролизу, таких как, например, предельное значение рН, присутствие влаги и повышенная температура. Такие условия могут иметь место, например, в армированных гибких выкидных линиях, стояках и трубах шлангокабелей, которые используются при операциях по разработке подводных нефтяных и газовых месторождений. Как это представляется, при воздействии повышенной температуры и высокой относительной влажности восприимчивыми к ухудшению качества являются, в частности, синтетические полимеры, содержащие карбоксилатные концевые группы.
Например, остаточная прочность нити из ароматического полиамида при 70°С и 50%-ной относительной влажности по истечении трех лет может уменьшиться до 70% от первоначального значения. Таким образом, для синтетических материалов, которые должны использоваться в течение нескольких лет, улучшение долговечности имеет важное значение.
Улучшение гидролитической стабильности синтетических волокон было описано. Публикация US 5,003,036 относится к арамидному сополимеру, где, по меньшей мере, 10% терефталоильных мономеров заменяют на моно- и/или дихлортерефталоильные группы. В публикации US 5,091,456 было описано арамидное волокно, содержащее от 0,5 до 3% конкретной фторспиртовой добавки. Для такого полимера было сообщено об улучшенном сохранении прочности. Публикация US 5,543,492 направлена на арамид, содержащий от 2 до 8% (моль.) алкил- и/или алкокси-замещенных диаминовых или дикислотных мономерных элементарных звеньев. Также в публикации US 4,011,203 описывается арамидный сополимер, включающий фенилендиамин, терефталоил и пиперазин, который обладает улучшенными теплостойкостью, ударной вязкостью и химической стойкостью. Во множестве данных ссылок сополимер получают в результате полимеризации п-фенилендиамина, терефталоилдихлорида и третьего мономера.
Недостатки введения сомономеров связаны с их непосредственным воздействием на свойства материала, которые зачастую оказывают воздействие на характеристики при растяжении сополимеров в сопоставлении с гомополимерами. Кроме того, для синтезирования сополимеров должны быть приобретены или получены высокочистые сомономеры, иногда обладающие сложными молекулярными структурами. Пользователь ограничен в выборе оптимального полимера.
Цель настоящего изобретения заключается в получении полиариленового волокна, характеризующегося улучшенной гидролитической стабильностью, и в преодолении ограничений предшествующего уровня техники. Также цель настоящего изобретения заключается в предложении волокна, которое объединяет высокий предел прочности при растяжении и высокий модуль упругости с хорошей гидролитической стабильностью. Кроме того, в данном изобретении предлагается прядильный раствор и способ получения полиариленового волокна, характеризующегося улучшенной гидролитической стабильностью.
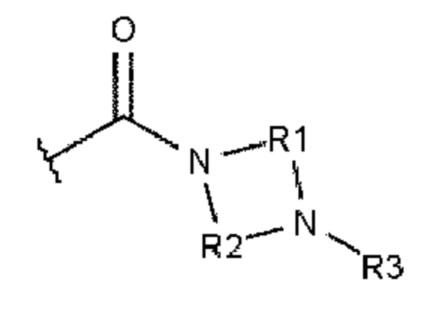
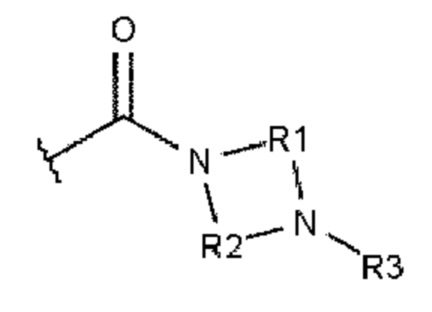
Как это к удивлению было установлено, гидролитическая стабильность полиариленовых волокон может быть намного улучшена. Этого добиваются при использовании полиариленового волокна, содержащего 0,1-15%, при расчете на массу волокна, ароматического соединения или комбинации из ароматических соединений и характеризующегося тем, что каждое ароматическое соединение содержит ароматическое ядро и, по меньшей мере, одного из заместителей А или В, где А описывается формулой 1:
А=
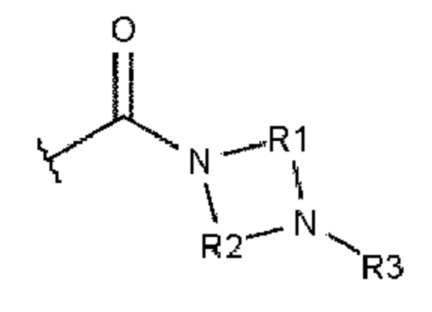 ,
,
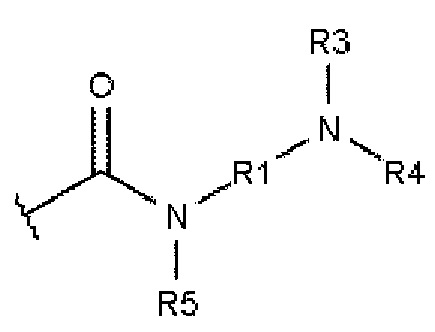
а В описывается формулой 2:
В=
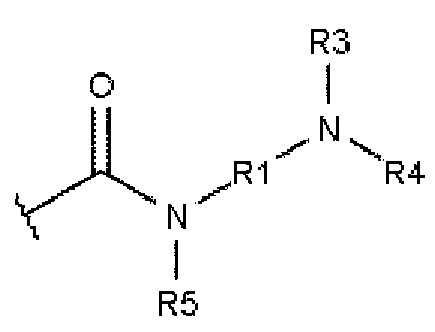 ,
,
где R1 и R2 независимо друг от друга выбирают из алкандиила, содержащего от 2 до 10 атомов углерода;
где R3 и R4 независимо друг от друга выбирают из -Н, алкила, содержащего от 1 до 10 атомов углерода, гомоциклоалкила, содержащего от 3 до 10 атомов углерода, или гетероциклоалкила, содержащего от 1 до 10 атомов углерода;
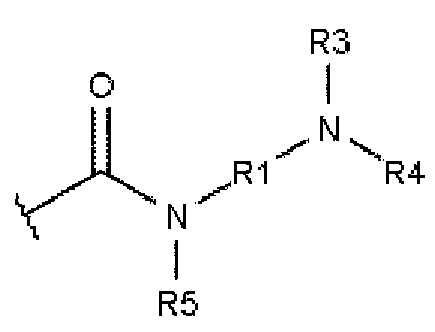
где R5 выбирают из: -Н, алкила, содержащего от 1 до 10 атомов углерода, гомоциклоалкила, содержащего от 3 до 10 атомов углерода, гетероциклоалкила, содержащего от 1 до 10 атомов углерода, и карбонила (-С(=О)-), который образует структуру фталимидо-кольца, сконденсированную с ароматическим ядром.
В основном заместители А и В ароматического соединения представляют собой карбоксамиды.
Настоящее изобретение направлено на синтетические полиариленовые полимерные материалы, говоря более конкретно, лиотропные жидкокристаллические полимеры, предпочтительно полимеры, содержащие фениленовые группы, более предпочтительно полимеры, выбираемые из арамидов (ароматических полиамидов, полиарамидов) и жестких стержневидных ароматических полимеров, более предпочтительно жестких стержневидных ароматических гетероциклических полимеров. Жесткие стержневидные ароматические полимеры содержат то, что на современном уровне техники известно под наименованием жестких спейсерных сегментов. Жесткие спейсеры зачастую содержат другое циклическое элементарное звено или функциональные концевые группы, такие как -NH-, -CO-, -O-, -COO-, -N=N- и/или -CH=CH-. В общем случае жесткие стержневидные полимеры содержат высоко-пара-ориентированные ароматические группы, и волокна, полученные из данных полимеров, характеризуются высоким модулем упругости при растяжении.
Множество данных полимеров подвергают переработке при использовании кислотных растворителей, и поэтому они склонны проявлять гидролитическую нестабильность.
Полиарилены, использующиеся для полиариленовых профилированных волокон изобретения, которые могут обеспечивать достижение повышенной гидролитической стабильности, включают арамиды (ароматические полиамиды) и жесткие стержневидные ароматические гетероциклические полимеры, такие как полибензазол, полигидрохинон-диимидазопиридин, и сополимеры данных полимеров.
В контексте настоящего описания изобретения термин «арамид» относится к ароматическому полиамиду, состоящему из ароматических фрагментов, непосредственно связанных друг с другом при использовании амидных фрагментов. Способы синтезирования арамидов для специалистов в соответствующей области техники известны и обычно включают поликонденсацию между ароматическими диаминами и ароматическими диацилгалогенидами. Арамиды могут существовать в мета- и пара-форме, обе из которых могут быть использованы в настоящем изобретении. Предпочтительным считается использование арамида, где, по меньшей мере, 85% связей между ароматическими фрагментами представляют собой пара-арамидные связи. В качестве типичных представителей данной группы могут быть упомянуты поли(пара-фенилентерефталамид), поли(4,4'-бензанилидтерефталамид), поли(пара-фенилен-4,4'-бифенилендикарбоксамид) и поли(пара-фенилен-2,6-нафталиндикарбоксамид), 5,4'-диамино-2-фенилбензимидазол или сополи(пара-фенилен/3,4'-оксидифенилентерефталамид) или их сополимеры. Предпочтительным считается использование арамида, где, по меньшей мере, 90%, говоря более конкретно, по меньшей мере, 95%, связей между ароматическими фрагментами представляют собой пара-арамидные связи. В особенности предпочтительным является использование поли(пара-фенилентерефталамида), также обозначаемого как РРТА.
Жесткие стержневидные ароматические гетероциклические полимеры включают полиазолы, такие как полибензазолы и полипиридазолы, и тому подобное, они могут представлять собой гомополимеры или сополимеры. Подходящие для использования полиазолы представляют собой полибензазолы, такие как полибензоксазол (РВО), полибензотиазол (РВТ), полибензимидазол (PBI) и РВО-подобные полимеры, такие как, например, поли(п-фенилен-2,6-бензобисоксазол) и полигидрохинон-диимидазопиридин. Полибензоксазол представляет собой полимер, содержащий оксазольное кольцо, связанное с ароматической группой, которая необязательно представляет собой бензольное кольцо. РВО-подобные полимеры включают широкий спектр полимеров, каждый из которых содержит элементарное звено в виде множества оксазольных колец, связанных с поли(фениленбензобисоксазолом) и ароматическими группами. Подобными аналогичными структурами могут обладать полимеры PBI и РВТ.
В случае полибензазола, представляющего собой полибензимидазол, предпочтительно им будет являться поли[5,5'-би-1Н-бензимидазол]-2,2'-диил-1,3-фенилен. В случае полибензазола, представляющего собой полибензотиазол, предпочтительно им будет являться полибензобистиазол, а более предпочтительно им будет являться поли(бензо[1,2-d:4,5-d']бистиазол-2,6-диил-1,4-фенилен. В случае полибензазола, представляющего собой полибензоксазол, предпочтительно им будет являться полибензобисоксазол, а более предпочтительно им будет являться поли(бензо[1,2-d:4,5-d']бисоксазол-2,6-диил-1,4-фенилен. В некоторых вариантах осуществления предпочтительные полипиридазолы представляют собой жесткие стержневидные полипиридобисазолы, в том числе поли(пиридобисимидазол), поли(пиридобистиазол) и поли(пиридобисозазол). Предпочтительный поли(пиридобисозазол) представляет собой поли(1,4-(2,5-дигидрокси)фенилен-2,6-пиридо[2,3-d:5,6-d']бисимидазол.
Жесткие стержневидные ароматические гетероциклические полимеры также включают смеси, сополимеры или блок-полимеры для двух и более вышеупомянутых представителей.
Полиариленовое волокно, соответствующее изобретению, содержит, по меньшей мере, один из описанных выше полимеров, оно также может содержать и комбинацию из таких полимеров. Обычно полиариленовое волокно содержит, по меньшей мере, 60% (масс.) любого одного или любой комбинации из полиариленов, использующихся в изобретении, (при расчете на массу волокна).
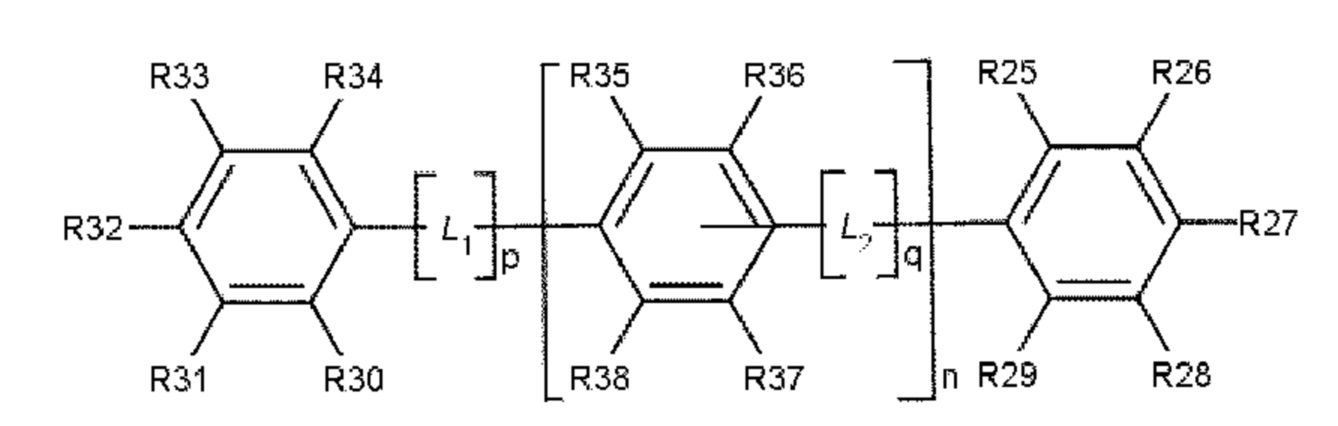
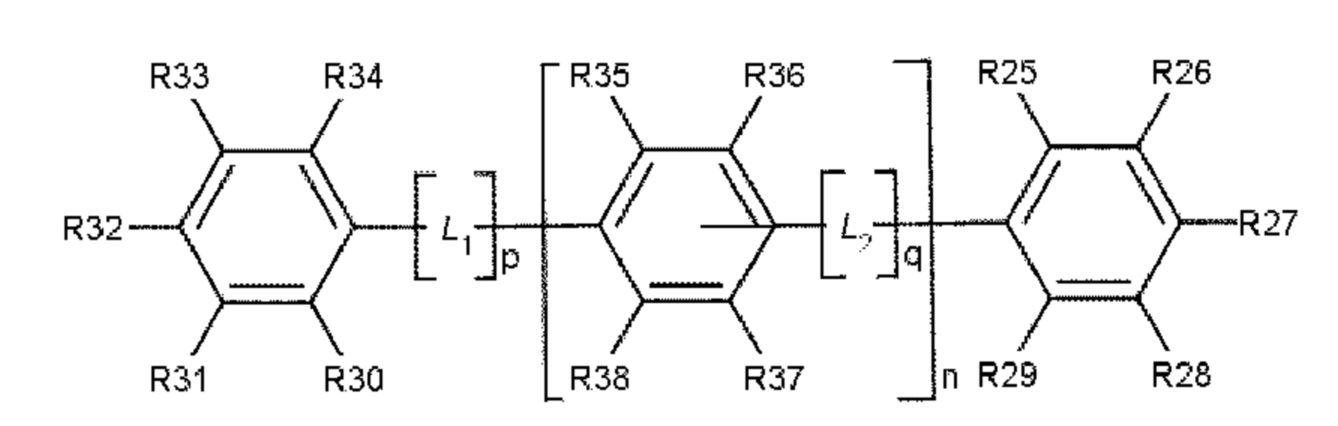
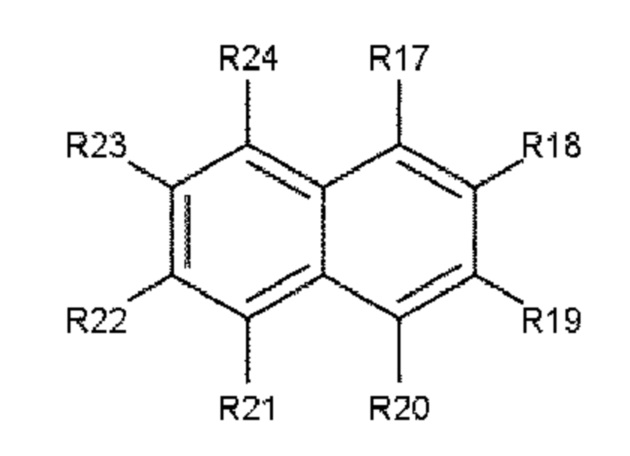
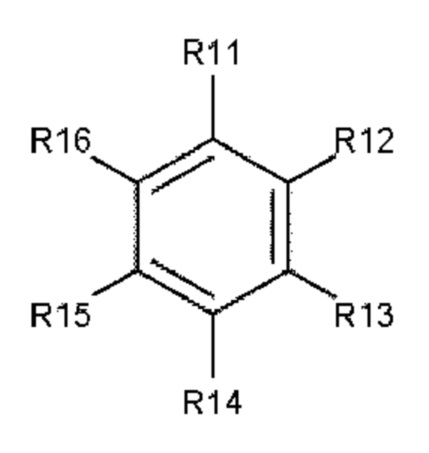
В одном варианте осуществления полиариленовое волокно содержит ароматическое соединение, содержащее ароматическое ядро, выбираемое из: ядра 1 (формула 3), ядра 2 (формула 4) и ядра 3 (формула 5).
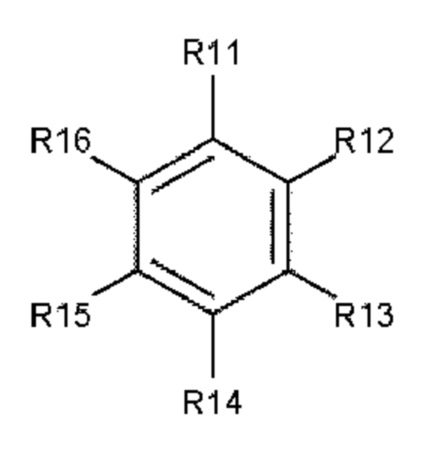
Формула 3: ядро 1
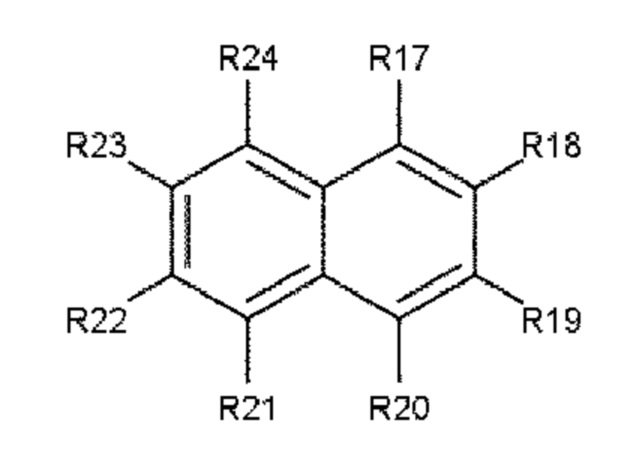
Формула 4: ядро 2
Формула 5: ядро 3
где R11, R12, R13, R14, R15, R16, R17, R18, R19, R20, R21, R22, R23, R24, R25, R26, R27, R28, R29, R30, R31, R32, R33, R34, R35, R36, R37 и R38 независимо друг от друга выбирают из следующих далее одновалентных радикалов: дополнительный заместитель А или В, -Н, атом галогена, -NO2, -CN, -OR3, -NR3R4, -SR3, алкил, содержащий от 1 до 10 атомов углерода, гомоциклоалкил, содержащий от 3 до 10 атомов углерода, гетероциклоалкил, содержащий от 1 до 10 атомов углерода, перфторалкил, содержащий от 1 до 10 атомов углерода, и (замещенный) карбонил;
где L1 и L2 в каждом случае независимо друг от друга выбирают из следующих далее двух- и трехвалентных радикалов: карбонил (-С(=О)-), -О-, -S-, -SO2-, алкандиил, содержащий от 1 до 6 атомов углерода, перфторалкандиил, содержащий от 1 до 6 атомов углерода, циклоалкандиил, содержащий от 3 до 10 атомов углерода, перфторциклоалкандиил, содержащий от 3 до 10 атомов углерода, иминокарбонил (-C(=O)NR5-, -R5NC(=O)-), ацилокси (-С(=О)О- или -ОС(=О)-); и где р и q составляют 0 или 1, а n составляет 0, 1, 2 или 3.
В контексте данного изобретения следующие далее термины определяют следующим образом:
«Алкил» обозначает одновалентный насыщенный линейный или разветвленный гидрокарбильный радикал, содержащий от 1 до 10 атомов углерода.
«Алкандиил» обозначает двухвалентный насыщенный линейный или разветвленный гидрокарбильный радикал, содержащий от 2 до 10 атомов углерода.
«Гетероциклоалкил» обозначает неароматический одновалентный моноциклический или полициклический радикал, содержащий от 1 до 10 атомов углерода и, по меньшей мере, один гетероатом, выбираемый из N, O, S. Гетероциклоалкильная группа может содержать в кольце одну или несколько двойных связей углерод-углерод или двойных связей углерод-гетероатомы до тех пор, пока кольцо не будет становиться ароматическим вследствие их присутствия.
«Гомоциклоалкил» обозначает одновалентный насыщенный циклический гидрокарбильный радикал, содержащий от 3 до 10 атомов углерода.
«Перфтор-» обозначает замещение всех атомов водорода в соответствующем фрагменте атомами фтора.
Карбонильная группа является группой -(С=О)-. Сюда включаются замещенные карбонильные группы. Примерами замещенных карбонильных групп являются: сложные эфиры (-С(=О)О-), амиды (-С(=О)N-), кетоны (-С(=О)С-), альдегиды (-С(=О)Н) и карбоновая кислота (-С(=О)ОН). В одном варианте осуществления соседние карбонильные заместители могут образовывать фталимидный фрагмент. Алкокси-фрагмент описывается формулой -OR3. Тиоалкильный заместитель описывается формулой -SR3. Замещенные амины описываются как -NR3R4, а структура фталимидо-кольца представляет собой фрагмент, описывающийся формулой 6, где ароматическое кольцо представляет собой ароматическое ядро ароматического соединения. Фталимидо-фрагмент может быть образован из заместителя В в результате присоединения второй карбонильной группы в положении R5, при этом N является дополнительно замещенным в соответствии с описанием изобретения для заместителя В. В альтернативном варианте, фталимидо-фрагмент может представлять собой часть фрагмента L1 или L2 ядра 3.
Формула 6: фталимидо-фрагмент
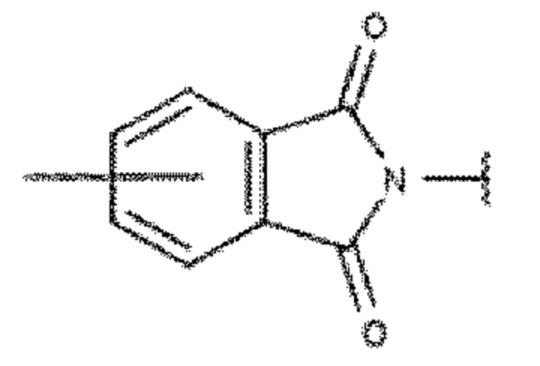
Для специалистов в соответствующей области техники известны различные способы получения фталимидо-фрагмента. Фталимиды обычно получают в результате дегидратации соседних фрагментов карбоксамида (-C(=O)NHR-) и карбоновой кислоты (-С(=О)ОН).
Один или несколько несоседних атомов углерода алкильных и алкандиильных заместителей соединения, соответствующего изобретению, могут быть замещены гетероатомами (N, O, S) с образованием, соответственно, фрагментов вторичного амина, простого эфира и простого тиоэфира. В таком варианте осуществления совокупное количество атомов основной цепи не увеличивается, а вместо этого один из атомов углерода замещается гетероатомом.
Один или несколько атомов водорода в алкильном, алкандиильном или циклоалкильном заместителях могут быть замещены атомами галогена или гетероатомами (-NR3R4, -OR3, -SR3, где R3 представляет собой Н) с образованием, соответственно, (замещенных) аминов, спиртов и тиолов (где R3 представляет собой алкил).
В случае включения в L1 или L2 алифатического фрагмента упомянутый фрагмент может быть линейным или разветвленным.
В общем случае заместители R (R1-R38) каждого соединения, соответствующего данному изобретению, выбирают для увеличения гидрофобности соединения.
В одном предпочтительном варианте осуществления, по меньшей мере, одно из соединений, использующихся в полиариленовых профилированных конструкциях настоящего изобретения, является замещенным при использовании, по меньшей мере, одного заместителя А, где R1 и R2 упомянутого заместителя А представляют собой -С2Н4-.
Независимо или в комбинации с данным вариантом осуществления R3 упомянутого заместителя А представляет собой метил.
В одном предпочтительном варианте осуществления полиариленовое волокно, соответствующее данному изобретению, содержит ароматическое соединение, содержащее заместитель В, где R1 упомянутого заместителя В представляет собой -С2Н4-.
Независимо или в комбинации с этим R3 и R4 упомянутого заместителя В представляют собой метил.
Независимо или в комбинации с таким R1 такой R3 и такие R4, R5 упомянутого заместителя В предпочтительно представляют собой атом водорода.
В одном предпочтительном варианте осуществления полиариленовое волокно содержит, по меньшей мере, одно ароматическое соединение, содержащее ядро 3, где L1 и L2 в каждом случае представляют собой иминокарбонильную группу, где n, p и q составляют 1. Данный вариант осуществления также может иметь место и в комбинации с вариантами осуществления, представленными в настоящем документе прежде.
Таким образом, предпочтительным является полиариленовое волокно, где заместитель А ароматического соединения представляет собой карбоксамид, произведенный из 1-алкилпиперазина, предпочтительно из 1-метилпиперазина.
В еще одном предпочтительном варианте осуществления заместитель В ароматического соединения представляет собой карбоксамид, произведенный из N,N-диалкилэтилендиамина.
В еще одном предпочтительном варианте осуществления используют ароматическое соединение, состоящее из ядра 1, ядра 2 или ядра 3, где заместители R11, R12, R13, R14, R15, R16, R17, R18, R19, R20, R21, R22, R23, R24, R25, R26, R27, R28, R29, R30, R31, R32, R33, R34, R35, R36, R37 и R38 независимо друг от друга выбирают из атома водорода или атома галогена.
Соединение, использующееся в данном изобретении, например, может представлять собой:
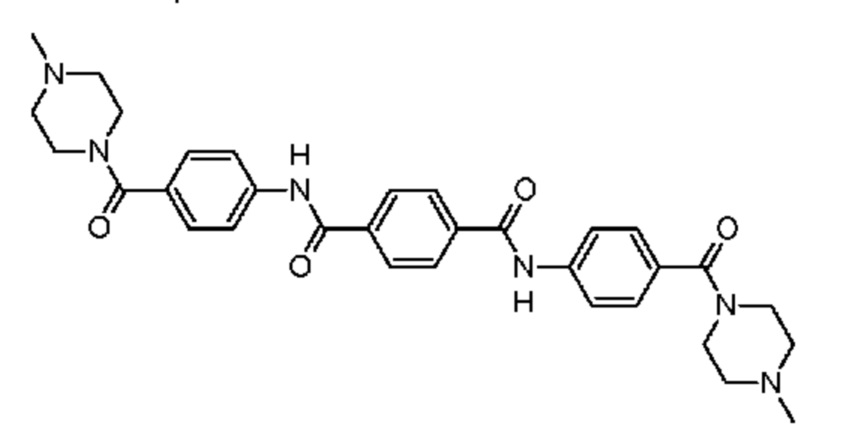
N1,N4-бис[4-(4-метилпиперазин-1-карбонил)фенил]бензол-1,4-дикарбоксамид,

1-метил-4-[4-(4-метилпиперазин-1-карбонил)бензоил]пиперазин или
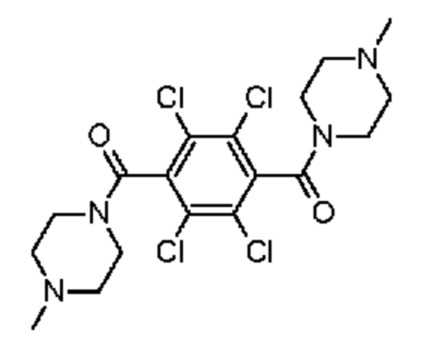
1-метил-4-[2,3,5,6-тетрахлор-4-(4-метилпиперазин-1-карбонил)бензоил]пиперазин.
Другие неограничивающие примеры включают:
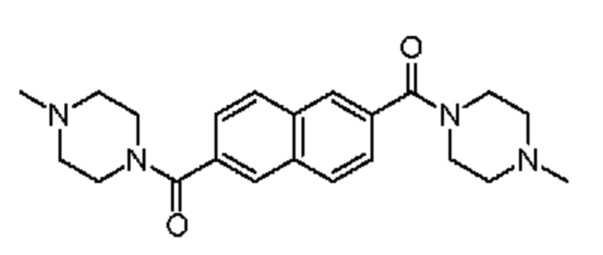
1-метил-4-[6-(4-метилпиперазин-1-карбонил)нафталин-2-карбонил]пиперазин,
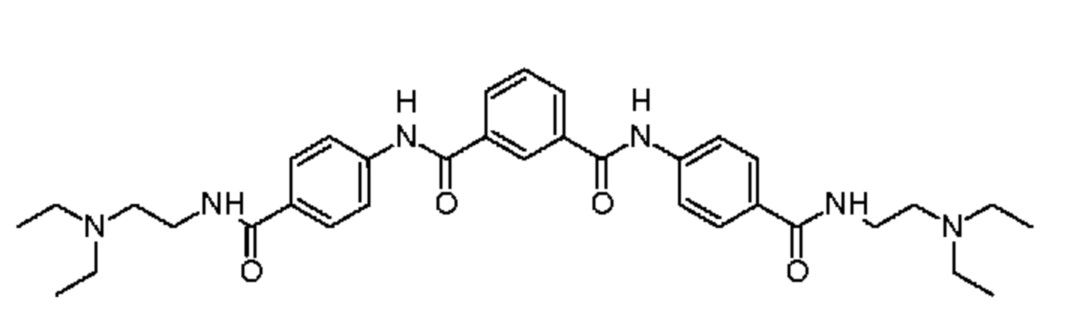
1-N,3-N-бис(4-{[2-(диэтиламино)этил]карбамоил}фенил)бензол-1,3-дикарбоксамид,
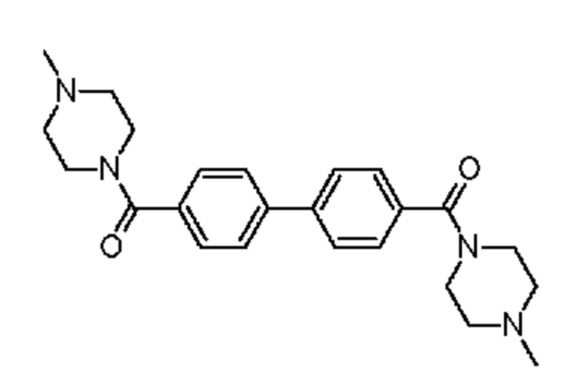
1-метил-4-{4-[4-(4-метилпиперазин-1-карбонил)фенил]бензоил}пиперазин,
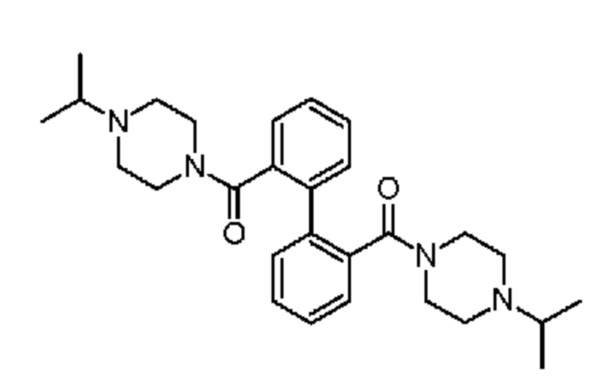
1-(пропан-2-ил)-4-(2-{2-[4-(пропан-2-ил)пиперазин-1-карбонил]фенил}бензоил)пиперазин и
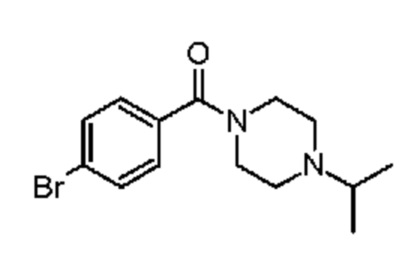
1-(4-бромбензоил)-4-(пропан-2-ил)пиперазин.
Ароматические соединения, содержащие фрагмент А или фрагмент В, использующиеся в изобретении, являются химически инертными в том смысле, что полиариленовый полимер и добавленное ароматическое соединение действительно образуют смесь.
Это значит то, что в случае присутствия ароматического соединения, содержащего фрагмент А или фрагмент В, во время реакции полимеризации соединение не будет исполнять функцию мономера, который встраивается в полимер. Поэтому в настоящем изобретении образуется не сополимер, а композиция, содержащая полиариленовый полимер и соединение или комбинацию из соединений.
В целях предотвращения изменения первоначальной окраски волокна в результате введения нейтрализующего ароматического соединения ароматическое соединение предпочтительно не должно представлять собой пигмент или краситель. Пигменты и красители представляют собой известные добавки, но обычно не оказывают выгодного воздействия на гидролитическую стабильность, как это делают ароматические соединения, использующиеся в настоящем изобретении.
Количество соединения или комбинации из соединений в полиариленовом волокне выбирают на основании молекулярной массы соединения.
Совокупная концентрация соединения или комбинации из соединений в полиариленовом волокне варьируется в диапазоне от 0,1 до 15% (масс.) при расчете на массу полиариленового волокна, предпочтительно 1-10% (масс.), более предпочтительно 2-6% (масс.).
Предпочтительно любое соединение, использующееся в изобретении, имеет молекулярную массу в диапазоне от 200 до 1000 г/моль, предпочтительно от 300 до 800 г/моль, более предпочтительно от 400 до 600 г/моль.
Для целей настоящего документа термин «волокно» определяют как гибкую макроскопически гомогенную конструкцию. Волокна характеризуются высоким соотношением между длиной и шириной поверхности поперечного сечения, перпендикулярной данной длине. Поперечное сечение волокна может иметь любой профиль, например, круглый или прямоугольный. Термин «волокно», соответствующий настоящему изобретению, включает нижеследующее, но не ограничивается только этим: филаменты (в том числе монофиламенты и мультифиламентный жгут), непрерывная нить, ленты, волокнистая масса, фибриллы, фибриды, штапельное волокно и короткое резаное волокно. В настоящем изобретении ленту определяют как предмет, длина которого, то есть, наибольший размер предмета, является большей, чем ширина - предпоследний по величине размер предмета - и толщина, то есть, наименьший размер предмета, в то время, как ширина, в свою очередь, является большей, чем толщина. Говоря более конкретно, соотношение между длиной и шириной в общем случае составляет, по меньшей мере, 2. В зависимости от ширины ленты данное соотношение может быть более значительным, например, составляющим, по меньшей мере, 4 или, по меньшей мере, 6. Максимальное соотношение не является критическим параметром для настоящего изобретения и будет зависеть от параметров переработки.
Полиариленовое волокно, соответствующее изобретению, также охватывает и варианты осуществления, где волокно содержит различные полиариленовые полимеры в соответствии с представленным выше определением изобретения. Например, волокно, соответствующее изобретению, может включать арамидные филаменты и филаменты, полученные из жесткого стержневидного ароматического гетероциклического полимера, где либо один, либо оба из двух филаментов, выбираемых из арамидных филаментов и жестких стержневидных ароматических гетероциклических полимерных филаментов, содержат ароматическое соединение, соответствующее настоящему изобретению.
Полиариленовое волокно, соответствующее изобретению, может быть использовано в широком спектре продуктов.
Примеры включают армированные трубы для транспортирования нефти/газа или муниципального отопления, шлангокабели, тросы, кабели, стропы, шланги системы охлаждения в автомобилях, покрышки, транспортерные ленты и стойкие к проникновению изделия, такие как, например, пуленепробиваемые или непрорезаемые изделия.
Одна цель данного изобретения заключается в предложении полиариленового волокна, характеризующегося улучшенной гидролитической стабильностью. Гидролитическая стабильность относится к пределу прочности на разрыв, который сохраняется после воздействия на волокно условий, которые в результате приводят к гидролизу. Гидролитическую стабильность определяют в результате измерения предела прочности на разрыв и определения момента времени и периода времени, когда в предписанных условиях по температуре и низкому значению рН сохраняются значения 90% (t0,9) или 80% (t0,9) от первоначального предела прочности на разрыв. Таким образом, периода времени, когда волокно после воздействия предписанных условий по температуре (90°С) и низкому значению рН (рН 4) в течение указанного времени характеризуется значениями 90% (t0,9) или 80% (t0,8) от предела прочности на разрыв в сопоставлении с тем, что имеет место для волокна, которое было подвергнуто воздействию стандартных условий окружающей среды (воздух, 20°С, 65%-ная относительная влажность). Для лент может быть использован тот же самый метод. Пленка может быть разрезана на ленты, для которых впоследствии может быть проведено измерение тем же самым образом.
Полиариленовое волокно, соответствующее данному изобретению, предпочтительно характеризуется гидролитической стабильностью t0,9, составляющей, по меньшей мере, 3 недели, предпочтительно, по меньшей мере, 6 недель, более предпочтительно, по меньшей мере, 9 недель, а еще более предпочтительно, по меньшей мере, 12 недель, и гидролитической стабильностью t0,8, составляющей, по меньшей мере, 10 недель, предпочтительно, по меньшей мере, 15 недель, более предпочтительно, по меньшей мере, 20 недель, а еще более предпочтительно, по меньшей мере, 25 недель, где гидролитическую стабильность t0,9 определяют как период времени вплоть до достижения равенства пределом прочности на разрыв волокна при воздействии раствора при рН 4 и 90°С 90% в сопоставлении с пределом прочности на разрыв волокна при воздействии воздуха, характеризующегося температурой 20°С и 65%-ной относительной влажностью. Гидролитическую стабильность t0,8 определяют как период времени вплоть до достижения равенства пределом прочности на разрыв волокна при воздействии раствора при рН 4 и 90°С 80% в сопоставлении с пределом прочности на разрыв волокна при воздействии воздуха, характеризующегося температурой 20°С и 65%-ной относительной влажностью.
Подробное описание метода определения гидролитической стабильности представлено в экспериментальном разделе, но оно может быть применено и к изобретению в общем случае.
Волокно, характеризующееся 100%-ным пределом прочности на разрыв, является тем же самым волокном, что и волокно, подвергнутое воздействию гидролитических условий, и, таким образом, содержит те же самые ароматическое соединение или комбинацию из соединений.
Таким образом, например, полиариленовое волокно, соответствующее изобретению, может быть подвергнуто воздействию условий окружающей среды при 90°С и рН 4 в течение 3 недель, и предел прочности на разрыв волокон остается на уровне 90% в сопоставлении с пределом прочности на разрыв полиариленового волокна, подвергнутого воздействию стандартных условий окружающей среды.
Предел прочности на разрыв определяют в соответствии с документом ASTMD7269 после кондиционирования волокна при 20°С и 65%-ной относительной влажности в течение 14 часов в соответствии с документом ASTMD1776.
Гидролитическая стабильность t0,9 композиций в отсутствие соединений, описанных в настоящем изобретении, обычно составляет менее, чем 3 недели, а гидролитическая стабильность t0,8 составляет менее, чем 10 недель и в общем случае не может достигать значений, соответствующих достижениям настоящего изобретения. В одном варианте осуществления гидролитическая стабильность t0,9 полиариленового волокна настоящего изобретения увеличивается на, по меньшей мере, 50%, предпочтительно, по меньшей мере, 100%, более предпочтительно, по меньшей мере, 200%, (в неделях в сопоставлении с гидролитической стабильностью полиариленового волокна, не содержащего никаких описанных соединений).
Изобретение также направлено на способ изготовления полиариленового волокна, характеризующегося улучшенной гидролитической стабильностью. Данный способ включает следующие далее стадии:
i) получение композиции, содержащей полиариленовый полимер, растворитель и ароматическое соединение или комбинацию из ароматических соединений,
ii) переработка композиции для получения полиариленового волокна, характеризующегося тем, что каждое ароматическое соединение содержит, по меньшей мере, одного из заместителей А или В, где А описывается формулой 1
А=
,
а В описывается формулой 2:
В=
,
где R1 и R2 независимо друг от друга выбирают из алкандиила, содержащего от 2 до 10 атомов углерода;
где R3 и R4 независимо друг от друга выбирают из -Н, алкила, содержащего от 1 до 10 атомов углерода, гомоциклоалкила, содержащего от 3 до 10 атомов углерода, или гетероциклоалкила, содержащего от 1 до 10 атомов углерода;
где R5 выбирают из: -Н, алкила, содержащего от 1 до 10 атомов углерода, гомоциклоалкила, содержащего от 3 до 10 атомов углерода, гетероциклоалкила, содержащего от 1 до 10 атомов углерода, и карбонила (-С(=О)-), который образует структуру фталимидо-кольца, сконденсированную с ароматическим ядром.
Композиция может быть подвергнута переработке различным образом.
Она может быть переработана в волокна, которые впоследствии могут переработаны в ленты. Для получения волокон изобретения могут быть использованы обычные способы прядения, хорошо известные для специалистов в соответствующей области техники, такие как, например, мокрое прядение, предпочтительно мокрое прядение с воздушным зазором.
Однако, возможным также является и использование аэромеханического прядения, при котором поток воздуха или коагулянта соударяется потоком прядильного раствора и преобразует его в капли. Еще один способ переработки представляет собой роторное прядение, при котором прядильный раствор вводят в ротор, вращающийся с высокой скоростью, выдавливают через маленькие отверстия и коагулируют на стенке при использовании текущего коагулянта (роторно-статорное коагулирование). При использовании аэромеханического прядения и роторного прядения возможным является непосредственное получение волокнистой массы, фибрилл и фибрид.
Обычное мокрое прядение включает следующие далее стадии: получение прядильного раствора, фильтрование, прядение, коагулирование, промывание, нейтрализация и высушивание. Такой способ хорошо известен и был описан, например, в публикации WO2010094620.
Композиция, содержащая растворитель, полиариленовый полимер и ароматическое соединение или комбинацию из ароматических соединений, может быть получена различным образом. В одном варианте осуществления смешивают полимер, растворитель и ароматическое соединение или комбинацию из ароматических соединений. Однако, возможным также является и добавление ароматического соединения или комбинации из ароматических соединений во время прохождения реакции полимеризации, которая приводит к получению полиарилена.
Растворитель выбирают в зависимости от выбранного полимера. Например, для пара-арамида обычно используют серную кислоту, в то время как для полибензазолов может быть использована фосфорная кислота.
Предпочтительно после переработки полиариленовое волокно нагревают при температуре в диапазоне от 250 до 500°С, предпочтительно при температуре в диапазоне от 300 до 450°С, более предпочтительно от 350 до 400°С. Продолжительность тепловой обработки может варьироваться в диапазоне от субсекунд до минут или даже долее в случае проведения тепловой обработки в автономном режиме. Предпочтительно во время нагревания прикладывают натяжение.
В одном предпочтительном варианте осуществления тепловую обработку проводят при использовании бесконтактной печи, продуваемой азотом.
Таким образом, в способе настоящего изобретения волокно, полученное после стадии ii), нагревают при температуре в диапазоне от 250 до 500°С, предпочтительно при температуре в диапазоне от 300 до 450°С, более предпочтительно при температуре в диапазоне от 350 до 400°С.
Тепловая обработка может индуцировать рост кристаллов, что может оказывать дополнительное положительное воздействие на гидролитическую стабильность. Рост размеров кристаллов может быть определен в результате измерения наблюдаемого размера кристаллов L110 при использовании широкоугольной дифракции рентгеновского излучения.
Например, в особенности выгодными считаются значения L110 для арамидного волокна в диапазоне от 45 до 130 Å, предпочтительно от 60 до 120 Å, более предпочтительно от 80 до 110 Å. Поэтому настоящее изобретение также направлено и на арамидное волокно, соответствующее изобретению и характеризующееся размером кристаллов L110 в диапазоне от 45 до 130 Å, предпочтительно от 60 до 120 Å, более предпочтительно от 80 до 110 Å.
Например, в особенности выгодными считаются значения L110 для полибензазольного волокна в диапазоне от 55 до 130 Å, предпочтительно от 65 до 125 Å, более предпочтительно от 80 до 120 Å. Поэтому настоящее изобретение также направлено и на полибензазольное волокно, соответствующее изобретению и характеризующееся размером кристаллов L110 в диапазоне от 55 до 130 Å, предпочтительно от 65 до 125 Å, более предпочтительно от 80 до 120 Å.
При использовании для изготовления полиариленового волокна способов прядения должен быть получен прядильный раствор.
Поэтому настоящее изобретение также направлено на прядильный раствор, содержащий полиариленовый полимер и от 0,1 до 15%, при расчете на массу полиариленового полимера в прядильном растворе, ароматического соединения или комбинации из ароматических соединений, характеризующийся тем, что каждое соединение содержит ароматическое ядро и, по меньшей мере, одного из заместителей А или В, где А описывается формулой 1:
А=
,
а В описывается формулой 2:
В=
,
где R1 и R2 независимо друг от друга выбирают из алкандиила, содержащего от 2 до 10 атомов углерода;
где R3 и R4 независимо друг от друга выбирают из -Н, алкила, содержащего от 1 до 10 атомов углерода, гомоциклоалкила, содержащего от 3 до 10 атомов углерода, или гетероциклоалкила, содержащего от 1 до 10 атомов углерода;
где R5 выбирают из: -Н, алкила, содержащего от 1 до 10 атомов углерода, гомоциклоалкила, содержащего от 3 до 10 атомов углерода, гетероциклоалкила, содержащего от 1 до 10 атомов углерода, и карбонила (-С(=О)-), который образует структуру фталимидо-кольца, сконденсированную с ароматическим ядром.
Для пара-арамида, в особенности полимера РРТА, растворитель для такого прядильного раствора представляет собой серную кислоту. Для полибензазолов он обычно представляет собой фосфорную кислоту. Предпочтительно соединения, описанные в настоящем изобретении, являются стабильными в кислотных растворителях и нерастворимыми или только слаборастворимыми в водных растворах таким образом, что во время или после коагулирования вымывание соединения не происходит ни на каком уровне или происходит только на низком уровне.
В случае полиарилена, представляющего собой арамид, концентрация арамида при расчете на массу прядильного раствора будет варьироваться в диапазоне от 12 до 25% (масс.), предпочтительно от 17 до 22% (масс.). Наиболее предпочтительной является концентрация арамида в диапазоне от 18 до 20% (масс.).
В прядильном растворе присутствуют либо одно ароматическое соединение, либо комбинация из ароматических соединений в соответствии с представленным выше описанием изобретения. Совокупная концентрация соединения или комбинации из соединений варьируется в диапазоне от 0,1 до 15% (масс.) при расчете на массу арамида в прядильном растворе, предпочтительно 1-10% (масс.), более предпочтительно 2-6% (масс.).
В случае использования нескольких соединений концентрация комбинации (определенная в результате суммирования значений индивидуальных концентраций) должна находиться в диапазоне вышеупомянутых концентраций.
Различные варианты осуществления, описанные для соединений, которые могут содержаться в полиариленовом волокне, также являются возможными вариантами осуществления соединения или комбинации из соединений, использующихся в прядильном растворе и в способе производства полиариленового волокна.
Говоря более конкретно, изобретение также относится и к прядильному раствору, содержащему полиарилены и ароматическое соединение (соединения) в соответствии с определением изобретения в пунктах 2-13 формулы изобретения, и к способу, где используются полиарилены и ароматические соединения в соответствии с определением изобретения в пунктах 2-13 формулы изобретения.
Следующие далее примеры описывают изобретение более подробно, но никоим образом не ограничивают объем изобретения.
Подробности экспериментов и примеры
1. Определение линейной плотности и характеристик при растяжении
Массу при расчете на единицу измерения (линейную плотность) для нитей измеряют в результате взвешивания известной длины нити в соответствии с документом ASTM D1907 «Test Method for Linear Density of Yarn (Yarn Number) by the Skein Method». Единица измерения линейной плотности=г/10000 м или 0,1 мг/м.
Характеристики при растяжении нитей определяют в соответствии с документом ASTM D7269 «Standard Test Methods for Tensile Testing of Aramid Yarns», таким образом, кондиционируя образцы при 20°С и 65%-ной относительной влажности в течение 14 часов в соответствии с документом ASTM D1776 «Practice for Conditioning and Testing Textiles». В отличие от данного стандарта какого-либо предварительного кондиционирования при 45°С в течение 3 часов не использовали. Линейную плотность и характеристики при растяжении лент измеряют по аналогии с этим при том условии, что данные образцы не будут скрученными.
2. Определение гидролитической стабильности
Гидролитическую стабильность нитей определяют в печах с принудительной циркуляцией воздуха. Нити, снабженные защитным кручением, наматывают без натяжения на стеклянный стержень и размещают в стеклянной трубке, заполненной буферным раствором при рН 4 на основе лимонной кислоты, гидроксида натрия и хлористо-водородной кислоты. После вставления стеклянную трубку закрывают и размещают в печи при 90°С. Через регулярные интервалы времени образцы извлекают из печи. После охлаждения нить прополаскивают при использовании водопроводной воды и высушивают в течение 3 часов при 45°С. После высушивания нить кондиционируют в течение, по меньшей мере, 14 часов при 20°С и 65%-ной относительной влажности.
В соответствии с документом ASTM D7269 измеряют предел прочности на разрыв. Предел прочности на разрыв образца, подвергнутого воздействию гидролитических условий, выражают в виде процентной доли по отношению к пределу прочности на разрыв образца той же самой нити, намотанной на стеклянный стержень и подвергнутой воздействию воздуха, характеризующегося температурой 20°С и 65%-ной относительной влажностью. Предел прочности на разрыв нити, подвергнутой воздействию воздуха, задают составляющим 100%.
Предел прочности на разрыв обычно измеряют по истечении 1, 2, 4, 8, 13, 26 и 52 недель после воздействия.
В результате интерполирования определяют время, за которое предел прочности на разрыв нити уменьшается до 90% (t0,9) и 80% (t0,8) от первоначального значения.
3. Определение поперечного размера кристаллов
а) Получение образца и измерение
Измерение для образца, в данном примере являющегося образцом арамидного волокна, проводят при использовании дифрактометра Bruker D8 Advance с геометрией θ/2θ. Дифрактометр снабжают оптикой с параллельными пучками и точечным детектором (сцинтилляционным счетчиком). Оптика состоит из первичного фокусирующего зеркала Гебеля на 60 мм (параболическое многослойное устройство на основе Ni/C), создающего излучение Cu-Kα (дублет Kα1/Kα2, длина волны Kα=1,5418 Å) и щелей Soller на 0,12°. Образец нити наматывают на держатель образца при толщине 0,3 мм, удостоверяясь в параллельности филаментов. Держатель образца устанавливают в дифрактометре при параллельности оси нити оси гониометра. Для определения размера кристаллитов измеряют интенсивность экваториального рассеяния рентгеновского излучения при экваториальных отражениях 110 и 200 в зависимости от угла дифракции 2θ в геометрии отражения. Установки для генератора составляют 40 кВ, 35 мА. Параметры сканирования: диапазон 3-43° (2θ), размер шага: 0,02° (2θ), время/шаг: 8 сек. Угол дифракции 2θ является углом между первичным пучком рентгеновского излучения и дифрагированным пучком рентгеновского излучения.
b) Оценка
Размер кристаллитов в соответствии с количественной оценкой при использовании параметра L110 обратно пропорционален ширине пика β отражения 110 в соответствии с формулой Шеррера:
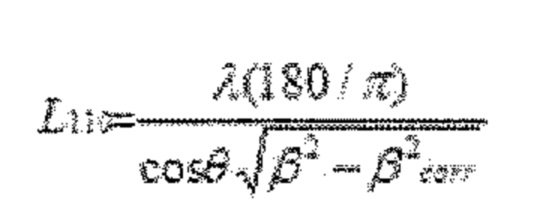 ,
,
где:
λ представляет собой длину волны использующегося рентгеновского излучения (1,5418 Å).
θ представляет собой половину угла дифракции.
βcorr. представляет собой аппаратурное уширение в градусах.
Полную ширину на половине высоты максимума (ПШПВ) β левого основного пика определяют при использовании аппроксимирования профиля. Левый основной пик представляет собой отражение 110. При аппроксимировании профиля пики 110 и 200 аппроксимируют совместно при использовании линейного фона. Пики аппроксимируют при использовании симметричной функции Пирсона VII. Используют аппаратурное уширение, полученное в результате измерения образца порошкообразного кремния. Ширину пика (ПШПВ) у пика при приблизительно 2θ=28° принимают за аппаратурное уширение βcorr.. Параметр размера кристаллитов L110 впоследствии рассчитывают в соответствии с формулой Шеррера. Для полибензазолов при определении размера кристаллитов L используют аналогичную методику в отношении основного экваториального пика в области 2θ=16,2°.
4. Синтезирование соединений, использующихся в изобретении
а) Синтезирование N1,N4-бис[4-(4-метилпиперазин-1-карбонил)фенил]бензол-1,4-дикарбоксамида (1)
Синтез соединения 1 представлен на схеме 1 и состоит из трех последовательных стадий реакции а1-а3, каждая из которых описывается ниже.
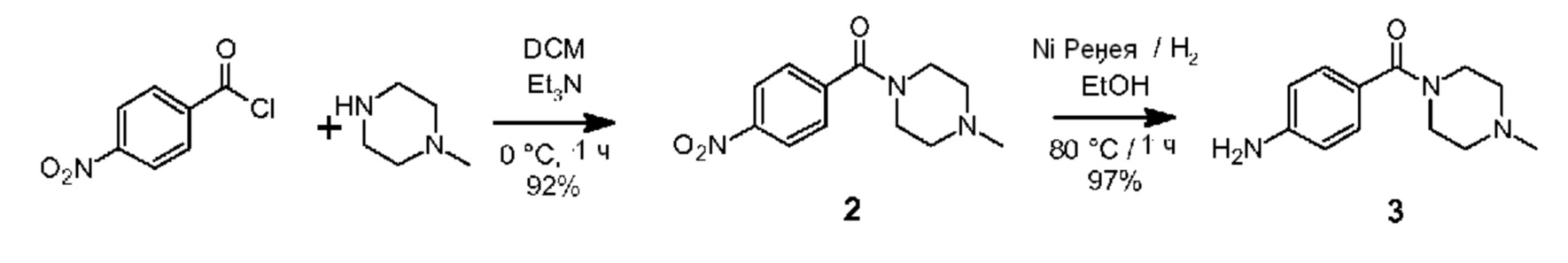
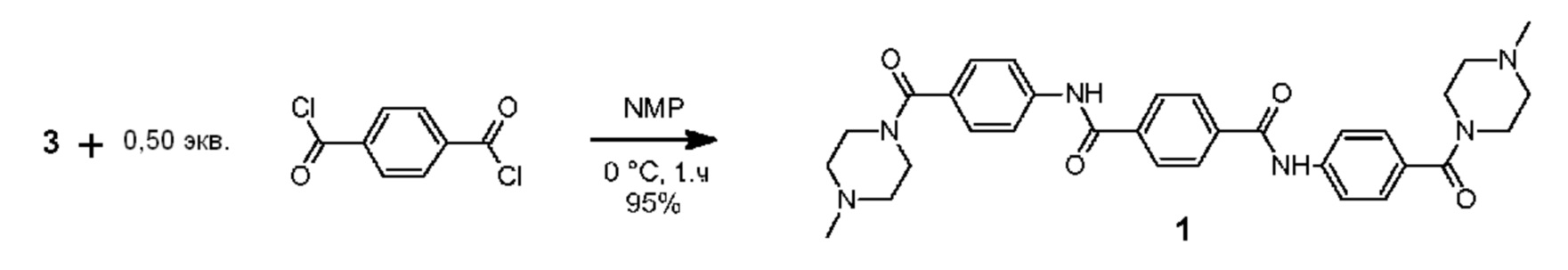
Схема 1
а1) Синтезирование (4-метилпиперазин-1-ил)(4-нитрофенил)метанона (2)
3-горлую колбу на 100 мл снабжали якорем магнитной мешалки, впускным и выпускным отверстием для азота и мембранной перегородкой. Установку тщательно высушивали при использовании теплового фена и обеспечивали ее охлаждение до комнатной температуры при продувании азота. После этого в сухом дихлорметане (50 мл) растворяли 4-нитробензоилхлорид (4,64 г, 25,0 ммоль) и раствор размещали на 10 минут в водяной бане со льдом. Через 10-минутный интервал времени покапельно добавляли N-метилпиперазин (2,58 г, 25,8 ммоль, 1,03 экв.) при одновременном интенсивном перемешивании. Получающуюся в результате суспензию перемешивали в течение 1 часа при 0°С, после чего добавляли триэтиламин (5,01 г, 50,0 ммоль, 2,0 экв.) и перемешивание продолжали при комнатной температуре в течение 1 часа. Реакционную смесь переводили в делительную воронку и промывали при использовании деминерализованной воды (3 × 50 мл). Органический слой высушивали над Na2SO4, офильтровывали через бумажный фильтр и все летучие соединения удаляли при пониженном давлении, используя роторный испаритель, для получения желательного продукта в виде бледно-оранжевого твердого вещества (5,77 г, 23,1 ммоль, 92%).
а2) Синтезирование (4-метилпиперазин-1-ил)(4-аминофенил)метанона (3)
(4-метилпиперазин-1-ил)(4-нитрофенил)метанон (2) каталитически восстанавливали при использовании никеля Ренея в качестве катализатора в автоклаве на 0,5 л от компании Büchi. В реактор загружали раствор нитро-соединения 2 (25,2 г, 101,1 ммоль) в 96%-ном этаноле (249,2 г) и суспензию никеля Ренея (2,05 г, 0,22 экв. (масс.)) в воде (9 мл). Впоследствии реакторную емкость продували при использовании N2 (3 ×) и Н2 (3 ×), после чего содержимое реактора нагревали до 80°С и давление в реакторе увеличивали до 10 бар. Скорость перемешивания увеличивали до 1500 об/мин и реакции давали возможность протекать в течение 60 минут для обеспечения полного восстановления. Содержимое реактора охлаждали до 50°С и реактор опорожняли в емкость через фильтр (размер пор 2 мкм). Непосредственно после этого другое количество нитро-соединения 2 (21,0 г) в 96%-ном этаноле (210 г) восстанавливали при использовании той же самой партии катализатора. После удаления второй партии реактор прополаскивали при использовании этанола (130 г) и все летучие соединения объединенных фракций удаляли при пониженном давлении, используя роторный испаритель, для получения масла с окраской в диапазоне от розовой до бледно-коричневой, которое кристаллизовалось при стоянии. Впоследствии твердое вещество высушивали в течение ночи в вакууме при 50°С для получения сырого амина 3 (39,5 г, 97%) в виде твердого бледно-коричневого остатка.
а3) Синтезирование N1,N4-бис(4-(4-метилпиперазин-1-карбонил)фенил)терефталамида
Высушенную в печи 3-горлую колбу на 1000 мл снабжали механическим перемешивающим устройством, впускным и выпускным отверстием для азота и охлаждали до комнатной температуры при продувании азота. В колбе размещали амин 3 (38,1 г, 173 ммоль), который растворяли в сухом N-метилпирролидин-2-оне (NMP) (503 г). Раствор охлаждали в течение 20 минут на водяной бане со льдом, после чего в колбу одной порцией добавляли твердые чешуйки терефталоилдихлорида (17,53 г, 86,3 ммоль, 0,50 экв.) при одновременном перемешивании при 500 об/мин. Капельную воронку прополаскивали при использовании соединения NMP (40 г) и суспензию перемешивали в течение 1 часа при 0°С с последующим выдерживанием в течение 0,5 часа при комнатной температуре. В реакционную смесь медленно добавляли концентрированный водный раствор гидроксида аммония (94 г, 30% (масс.)) и впоследствии реакционную смесь выливали в деминерализованную воду (3,5 л). Значение рН раствора доводили до 10 в результате добавления дополнительного количества гидроксида аммония (130 г, 30% (масс.)). Мелкому белому осадку давали возможность отстояться в течение приблизительно 0,5 часа, после чего прозрачный верхний слой декантировали. Осадок суспендировали, отфильтровывали через фильтр Millipore (Durapore GV, размер пор 0,22 мкм) при одновременном приложении вакуума и промывали при использовании деминерализованной воды (3 × 250 мл). Сырой продукт дополнительно очищали в результате неоднократного (4 ×) суспендирования продукта в теплой высокочистой воде MilliQ (1,0 л, Т=65°С) в течение приблизительно 1 часа, охлаждения до приблизительно 36-38°С, фильтрования через фильтр Millipore, промывания при использовании воды MilliQ (2 × 250 мл) и высушивания на воздухе при отсасывании в течение 5-15 минут. Влажный продукт высушивали в течение ночи в вакууме при 50°С для получения желательного соединения в виде беловатого мелкого порошка (46,4 г, 95%).
b) Синтезирование 1-метил-4-[4-(4-метилпиперазин-1-карбонил)бензоил]пиперазина
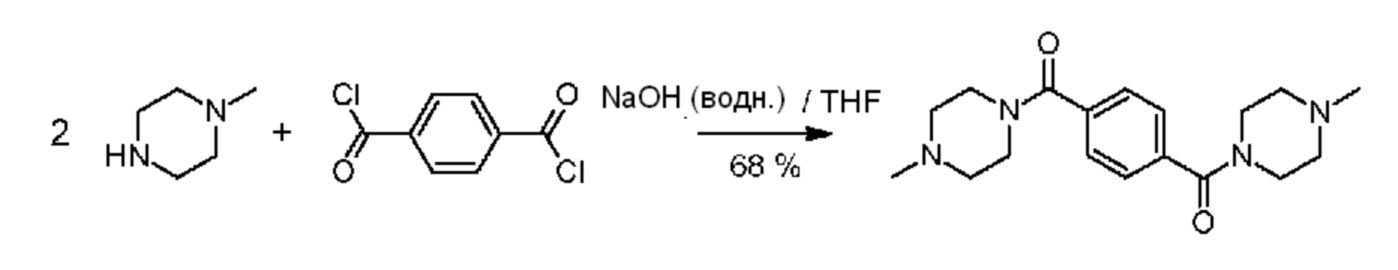
Трехгорлую колбу на 500 мл снабжали механическим перемешивающим устройством и капельной воронкой. В колбу загружали раствор N-метилпиперазина (26,58 г, 0,265 моль) в растворе NaOH при 2,0 моль/л (166,2 г, 0,308 ммоль NaOH). Колбу охлаждали в течение 30 минут в водяной бане со льдом при одновременном перемешивании. После этого в колбу покапельно добавляли раствор терефталоилдихлорида (24,36 г, 0,120 моль) в сухом тетрагидрофуране (55,2 г) при одновременном интенсивном перемешивании. Скорость добавления была такой, что внутренняя температура оставалась ниже 5°С. Реакционную смесь разбавляли в результате добавления деминерализованной воды, а значение рН устанавливали на 8 в результате осторожного добавления раствора HCl при 37% (масс.). Реакционную смесь разделяли на две равные части. Из каждой части реакционной смеси выделяли продукт в результате экстрагирования при использовании дихлорметана (2 × 400 мл+3 × 100 мл). Объединенные органические соли промывали при использовании рассола и высушивали над Na2SO4. После фильтрования все летучие соединения удаляли при пониженном давлении в роторном испарителе, а сырой продукт дополнительно очищали в результате растирания в порошок в диэтиловом простом эфире с последующим высушиванием в вакууме для получения титульного соединения в виде мелкого белого порошка (27,16 г, 68%).
5. Прядение арамидного волокна, соответствующего изобретению
i) Получение песчанистого прядильного раствора
Партии песчанистого прядильного раствора получали в 6-литровом смесителе от компании Drais. Сначала 2008 г раствора серной кислоты при 99,8% (масс.) смешивали с 15 г ароматической добавки в течение 2 часов при - 20°С. После этого добавляли и смешивали в течение 2 часов 477 г поли(п-фенилентерефталамида) (РРТА). Впоследствии охлаждение прекращали и смесь перемешивали в течение еще 10-12 часов. Получающийся в результате песчанистый прядильный раствор содержал 19,1% (масс.) соединения РРТА и 0,6% (масс.) добавки. Справочную партию при 19,7% (масс.) соединения РРТА получали сопоставимым образом, но без добавления добавки к серной кислоте. Совокупный уровень содержания твердого вещества в обоих прядильных растворах составлял 19,7% (масс.).
ii) Прядение из прядильного раствора
Песчанистые прядильные растворы, полученные в позиции i), дозировали в двухчервячный экструдер и экструдировали при 85°С, что приводило к получению расплавленного жидкокристаллического раствора. Данный раствор перекачивали через фильеру со 106 отверстиями с диаметром в 59 микронов. После экструдирования через прядильные отверстия жидкие филаменты вытягивали в воздушном зазоре, составляющем приблизительно 5 нм, и впоследствии они поступали в коагуляционную ванну, состоящую из деминерализованной воды. Коагулированные волокна промывали при использовании дополнительного количества воды для удаления остаточной серной кислоты, нейтрализовали при использовании раствора NaOH, промывали при использовании дополнительного количества воды, высушивали при 250°С или 160°С и наматывали на бобины. Совокупный способ прядения осуществляли в режиме реального времени при скорости наматывания 160 м/мин.
Пример 1 - Арамидные профилированные конструкции, соответствующие изобретению
Образец 1
Прядильный раствор, содержащий 19,1% (масс.) пара-арамида и 0,6% (масс.) N1,N4-бис[4-(4-метилпиперазин-1-карбонил)фенил]бензол-1,4-дикарбоксамида (при расчете на массу прядильного раствора), получали и подвергали переработке в соответствии с представленным выше описанием изобретения, нить высушивали при 250°С.
Сравнительный образец 1
Прядильный раствор, содержащий 19,7% (масс.) пара-арамида, получали и подвергали переработке в соответствии с вышеупомянутым представлением изобретения, никакого ароматического соединения не добавляли, нить высушивали при 250°С.
Впоследствии в соответствии с описанием изобретения испытаниям подвергали гидролитическую стабильность t0,9 и t0,8 волокон, полученных в качестве образца 1 и сравнительного образца 1. Результаты продемонстрированы в таблице 1.
Таблица 1: гидролитическая стабильность для сравнительного примера 1 и примера 1 (соответствующего изобретению)
|
Гидр. стаб.: гидролитическая стабильность, определенная в соответствии с описанием изобретения, уровень массового процентного содержания добавки получают при расчете на массу волокна.
Гидролитическая стабильность t0,9 и t0,8 волокон, содержащих добавку, значительно улучшается, соответственно, от 2 до 8 недель и от 10 до 23 недель. Это означает то, что полиариленовые профилированные конструкции, соответствующие изобретению, сохраняют свою прочность намного дольше, чем обычные полиариленовые волокна, и поэтому могут быть использованы намного дольше в условиях, индуцирующих гидролиз.
Модуль упругости полиариленового волокна может быть улучшен в результате проведения тепловой обработки в автономном режиме. Для определения этого получали следующие далее образцы:
Образец 2
Волокна пряли так же, как и в случае образца 1, но высушивали при 160°С вместо 250°С.
Сравнительный образец 2
Волокна пряли так же, как и в случае сравнительного образца 1, но высушивали при 160°С вместо 250°С.
Образец 3
Волокна образца 2 подвергали тепловой обработке при 400°С при натяжении 1,6 сН/дтекс в зоне 1 и при 120°С при натяжении 0,11 сН/дтекс в зоне 2.
Образец 4
Волокна образца 1 подвергали тепловой обработке при 450°С при натяжении 1,5 сН/дтекс в зоне 1 и при 120°С при натяжении 0,11 сН/дтекс в зоне 2.
Таблица 2: Результаты по тепловой обработке
|
Способ формования и промывки арамидного волокна и регенерации серной кислоты
Арамидные частицы, содержащие пероксидный инициатор радикало-цепной полимеризации
Гибкая сплошная лента из комплексной нити и способ ее изготовления
Внутрисосудистый катетер, содержащий армирующую микроленту
Способ получения полиолефиновых пленок
Многотоннажный процесс полимеризации полиарамида, содержащего 5(6)-амино-2-(п-аминофенил)бензимидазол (dapbi)
Изделия для баллистической защиты, включающие ленты
Сшитый арамидный полимер
Пулестойкие изделия, содержащие удлиненные элементы
Способ получения высокомолекулярных полиэтиленовых волокон
Способ прядения волокон из графеновых лент
Углеродное нанотрубчатое волокно, имеющее низкое удельное сопротивление
Корд, включающий многоволоконную пара-арамидную нить, включающую некруглые волокна

