Результат интеллектуальной деятельности: ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики, в частности, настоящее изобретение относится к индазольным соединениям, представляющим собой ингибиторы киназы FGFR, к их получению и применению.
УРОВЕНЬ ТЕХНИКИ
Рецепторная тирозинкиназа играет ключевую роль в различных аспектах жизнедеятельности опухоли, таких как возникновение, развитие, прорастание, метастазирование и лекарственная устойчивость опухоли вследствие активации аномальной экспрессии рецепторной тирозинкиназы или генной мутации. Она стала важной мишенью в научных исследованиях и разработках противоопухолевых лекарственных средств. Рецепторы фактора роста фибробластов (англ. fibroblast growth factor receptors, FGFR) являются важными представителями семейства рецепторов тирозинкиназы и в целом включают четыре подтипа: FGFR1, FGFR2, FGFR3 и FGFR4. Их лиганд представляет собой фактор роста фибробластов (FGF). Из-за амплификации гена, мутации, слияния или индукции лиганда и по другим причинам различные члены FGFR непрерывно активируются, вызывая пролиферацию опухолевых клеток, прорастание, миграцию, способствуя ангиогенезу и способствуя возникновению и развитию опухоли. FGFR сильно экспрессированы и чрезмерно активированы в различных опухолях, а также тесно связаны с неблагоприятным прогнозом у субъектов с немелкоклеточным раком легкого, раком молочной железы, раком желудка, раком мочевого пузыря, раком эндометрия, раком предстательной железы, раком шейки матки, раком толстой кишки, раком пищевода, глиобластомой, миеломой, рабдомиосаркомой и подобными. Исследования показали, что амплификация FGFR1 является причиной 20% случаев немелкоклеточного рака легкого, в частности плоскоклеточной карциномы. Проведенные in vitro исследования пролиферации и сигнальных путей штаммов клеток рака легкого с пролиферацией FGFR1 показали, что селективные ингибиторы FGFR могут эффективно ингибировать активацию сигнального пути FGFR1 и пролиферацию клеток. При раке молочной железы амплификация хромосомной области (8р11-12), где локализуется FGFR1, встречается у примерно 10% ER-положительных пациентов и связана с высоким уровнем экспрессии мРНК FGFR1 и неблагоприятным прогнозом для пациентов.
Амплификация или мутация гена FGFR2 приводят к аномальной активации сигнального пути FGFR2, который главным образом связан с раком желудка, трижды негативным раком молочной железы, раком эндометрия и др. Доля амплификации FGFR2 в ткани рака желудка составляет 5%-10%. Анализ 313 случаев рака желудка показал, что амплификация FGFR2 была в значительной степени связана с размером опухоли, местной инфильтрацией, метастазами в лимфатические узлы и отдаленными метастазами, а кроме того, рак желудка с амплификацией FGFR2, как правило, представляет собой прогрессирующую опухоль с неблагоприятным прогнозом, при этом общая выживаемость пациентов относительно низка. Амплификация FGFR2 является причиной 4% случаев рефрактерного трижды негативного рака молочной железы. Рак эндометрия представляет собой часто встречающуюся опухоль гинекологического репродуктивного тракта, при этом мутации FGFR2 являются причиной примерно 12% случаев рака эндометрия. При неинвазивном раке мочевого пузыря мутации FGFR3 являются причиной 50%-60% случаев, а при инвазивном раке мочевого пузыря мутации FGFR3 являются причиной 10%-15% случаев заболевания. Генная перегруппировка FGFR3t (4; 14) (р16.3; q32) является причиной 15-20% случаев множественной миеломы. В то же время различные подтипы FGFR и их лиганды (FGF), такие как FGFR2, FGFR3, FGFR4, FGF19, FGF2, FGF5, FGF8 и FGF9, показали аномальную экспрессию и активацию при раке печени. Многочисленные доклинические и клинические исследования показали, что аномальная активация оси FGF/FGFR играет важную роль при раке печени. Невозможно игнорировать тот факт, что аномальная активация оси FGF/FGFR тесно связана с устойчивостью к лекарственным средствам, представляющим собой ингибиторы EGFR, ингибиторы реваскуляризации и эндокринную терапию. Таким образом, разработка ингибиторов FGFR становится очень важной задачей в исследованиях и разработке противораковых лекарственных средств.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является получение нового ингибитора FGFR.
В первом аспекте настоящего изобретения предложено соединение формулы I или его фармацевтически приемлемая соль
где:
L выбран из группы, состоящей из: Н, тетрагидропиранила (ТГП);
каждый X независимо выбран из группы, состоящей из: Cl, F, Н и CN;
каждый из W, Y и Z независимо выбран из: N или СН;
кольцо А отсутствует, представляет собой незамещенную или замещенную 5-8-членную ариленовую группу или незамещенную или замещенную 5-8-членную гетероариленовую группу, при этом указанная гетероариленовая группа содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода или серы незамещенное или замещенное 3-12-членное насыщенное гетероциклическое кольцо или карбоциклическое кольцо, при этом указанное гетероциклическое кольцо содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода или серы;
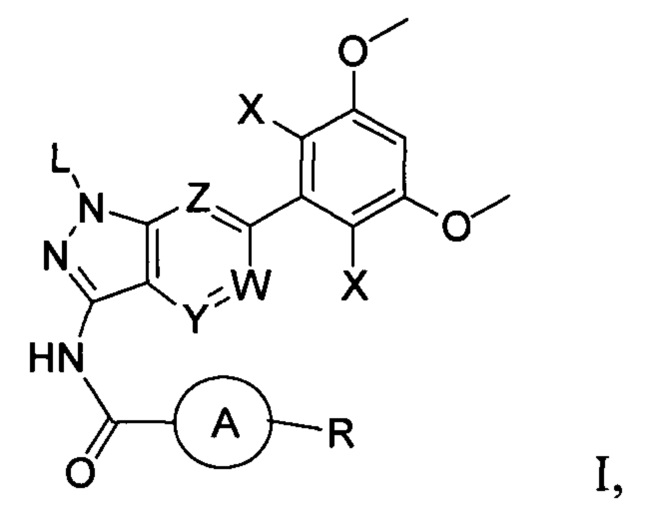
R представляет собой Н или замещенную или незамещенную группу, выбранную из группы, состоящей из:
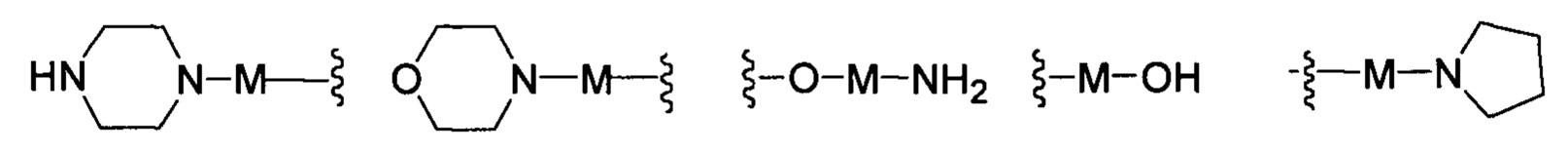
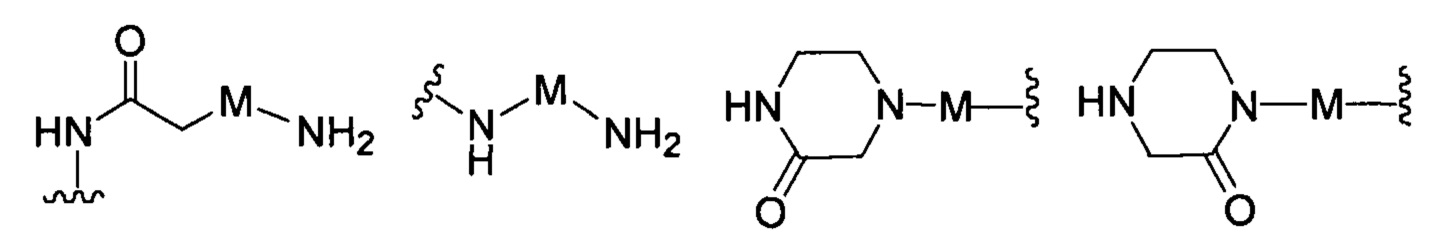
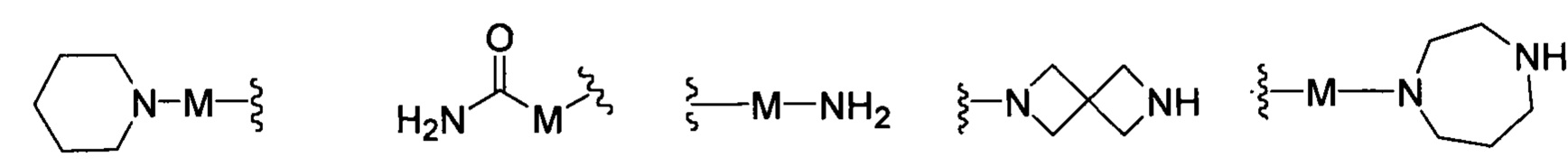 ;
;
где М выбран из группы, состоящей из: замещенного или незамещенного С1-С6-алкилена, замещенного или незамещенного С6-С10-арилена, замещенного или незамещенного С1-С10-гетероарилена, или М отсутствует;
при этом термин "замещенный" в каждом случае означает, что один или более атомов водорода в указанной группе замещены заместителем, выбранным из группы, состоящей из: галогена, незамещенного или галогенированного С1-С6-алкила, незамещенной или галогенированной С1-С6-алкоксигруппы, незамещенной или галогенированной С1-С6-алкоксиалкильной группы, незамещенной или галогенированной С3-С8-циклоалкильной группы, незамещенной или галогенированной С2-С6-алкилкарбонильной группы, незамещенного или галогенированного С1-С6-алкилен-гидрокси, незамещенной или С1-С6-алкил-замещенной аминогруппы.
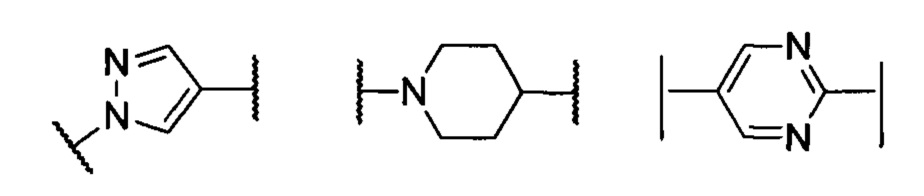
В другом предпочтительном варианте реализации настоящего изобретения указанное кольцо А представляет собой гетероарил или насыщенное гетероциклическое кольцо, выбранное из группы, состоящей из следующих структур, или кольцо А отсутствует:
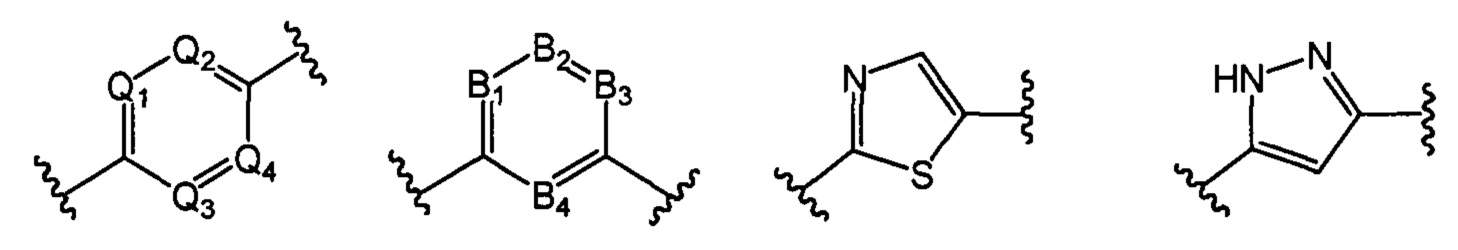
 ;
;
где каждый из Q1, Q2, Q3 и Q4 независимо выбран из: N или СН;
каждый из B1, В2, В3 и В4 независимо выбран из: N или СН.
В другом предпочтительном варианте реализации настоящего изобретения указанное кольцо А представляет собой гетероарил или насыщенное гетероциклическое кольцо, выбранное из группы, состоящей из следующих структур, или кольцо А отсутствует:
 .
.
В другом варианте реализации настоящего изобретения R представляет собой Н, замещенную или незамещенную группу, выбранную из группы, состоящей из:


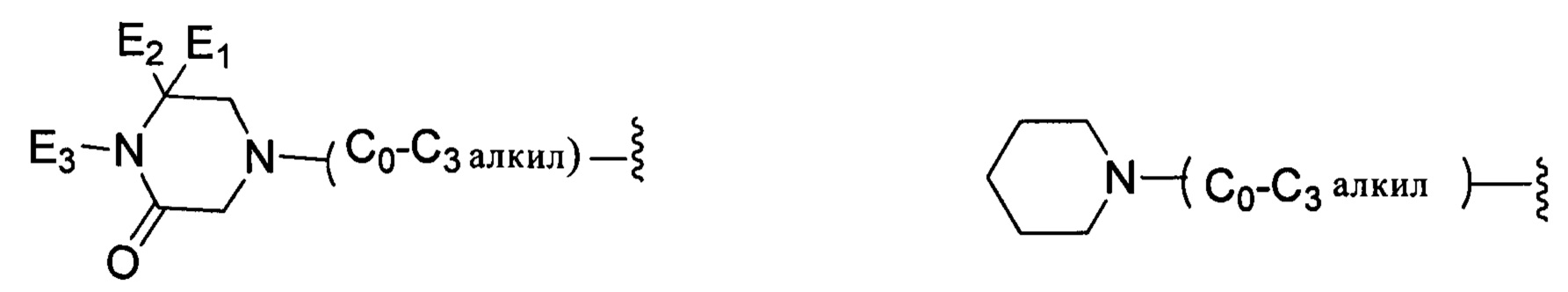

 ;
;
где:
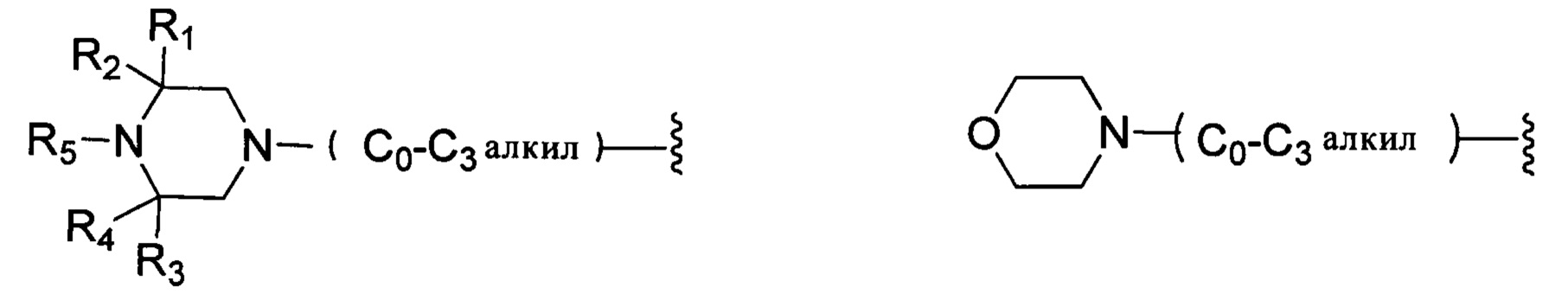
каждый из R1, R2, R3 и R4 независимо выбран из группы, состоящей из: Н, галогена, С1-С6 неразветвленного или разветвленного алкила, галогенированного С1-С6 неразветвленного или разветвленного алкила;
R5 выбран из группы, состоящей из: Н, неразветвленного или разветвленного С1-С6-алкила, неразветвленного или разветвленного С1-С6-алкилкарбонила, неразветвленного или разветвленного С1-С6-алкилен-гидрокси, С1-С6 алкоксиалкила, незамещенной или алкил-замещенной аминогруппы, С1-С8 циклоалкильной группы.
каждый из R6, R7, R8, R9, R10, R11 и R12 независимо выбран из группы, состоящей из: Н, С1-С6 неразветвленного или разветвленного алкила, С1-С6 неразветвленного или разветвленного алкилкарбонила, С1-С6 неразветвленной или разветвленной спиртовой группы (алкилен-гидрокси);
каждый из G1, G2, G3 и G4 независимо выбран из группы, состоящей из: Н, галогена, С1-С6 неразветвленного или разветвленного алкила, галогенированного С1-С6 неразветвленного или разветвленного алкила;
G5 выбран из группы, состоящей из: Н, неразветвленного или разветвленного С1-С6-алкила, неразветвленного или разветвленного С1-С6-алкилкарбонила, неразветвленного или разветвленного С1-С6-алкил-гидрокси, С1-С6-алкоксиалкила, незамещенной или алкил-замещенной аминогруппы, С1-С8-циклоалкильной группы;
каждый из E1, Е2 независимо выбран из группы, состоящей из: Н, галогена, неразветвленного или разветвленного алкила, галогенированного неразветвленного или разветвленного С1-С6алкила;
Е3 выбран из группы, состоящей из: Н, неразветвленного или разветвленного С1-С6-алкила, неразветвленного или разветвленного С1-С6-алкилкарбонила, неразветвленного или разветвленного С1-С6-алкилен-гидрокси, С1-С6-алкоксиалкила, незамещенной или алкил-замещенной аминогруппы, С1-С8-циклоалкильной группы;
каждый из R13, R14, R15 и R16 независимо выбран из группы, состоящей из: Н, неразветвленного или разветвленного С1-С6-алкила, неразветвленного или разветвленного С1-С6-алкилкарбонила, неразветвленной или разветвленной С1-С6 спиртовой группы (алкилен-гидрокси) или R13 и R14, или R15 и R16 присоединены к атому углерода так, что образуется 5- или 7-членное кольцо;
С0-С3 алкил обозначает отсутствие заместителя или алкилен с 1-3 атомами углерода;
С1-С6 алкил представляет собой алкилен с 1-6 атомами углерода.
В другом предпочтительном варианте реализации настоящего изобретения
L выбран из группы, состоящей из: Н, тетрагидропиранила (ТГП);
каждый X независимо выбран из группы, состоящей из: Н, Cl, F и CN;
каждый из W, Y и Z независимо выбран из: N или СН;
кольцо А представляет собой незамещенную или замещенную 6-членную арильную группу или незамещенную или замещенную 5- или 6-членную гетероарильную группу, при этом указанная гетероарильная группа содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода и серы;
М выбран из группы, состоящей из: незамещенного или замещенного С1-С4 алкилена или М отсутствует; при этом термин "замещенный" означает, что один или более атомов водорода в указанной группе замещены заместителем, выбранным из группы, состоящей из: галогена, незамещенного или галогенированного С1-С4-алкила, незамещенной или галогенированной С1-С6-алкоксигруппы, незамещенного или галогенированного С2-С6-алкоксиалкила, незамещенной или галогенированной С3-С8-циклоалкильной группы, незамещенной или галогенированной С2-С4-алкилкарбонильной группы, незамещенного или галогенированного С1-С4-алкил-гидрокси, незамещенной или С1-С6-алкил-замещенной аминогруппы.
В другом предпочтительном варианте реализации настоящего изобретения
L представляет собой Н;
каждый X независимо выбран из группы, состоящей из: Н, Cl и F;
каждый из W, Y и Z независимо выбран из: N или СН;
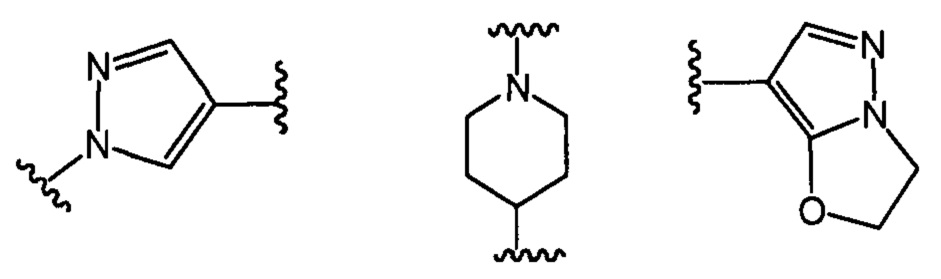
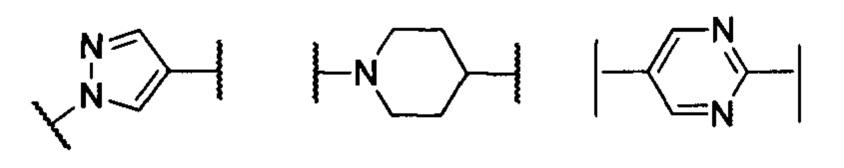
кольцо А представляет собой группу, выбранную из: фенила, пиразолила, пиридила, тиазолила, пиримидинила, пиразинила, пиперидинила или А отсутствует;
М выбран из группы, состоящей из незамещенной или замещенной С1-С3-алкиленовой группы, или М отсутствует;
при этом термин «замещенный» в каждом случае означает, что один или более атомов водорода на указанной группе замещены заместителем, выбранным из группы, состоящей из: галогена, незамещенного или галогенированного С1-С4-алкила, незамещенной или галогенированной С2-С6-алкоксигруппы, незамещенной или галогенированной С2-С6-алкоксиалкильной группы, незамещенной или галогенированной С3-С8-циклоалкильной группы, незамещенной или галогенированной С2-С6-алкилкарбонильной группы, незамещенного или галогенированного С1-С4- алкил-гидрокси, незамещенной или С1-С4-алкил-замещенной аминогруппы.
В другом предпочтительном варианте реализации настоящего изобретения соединение формулы I выбрано из приведенной ниже таблицы А:
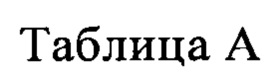
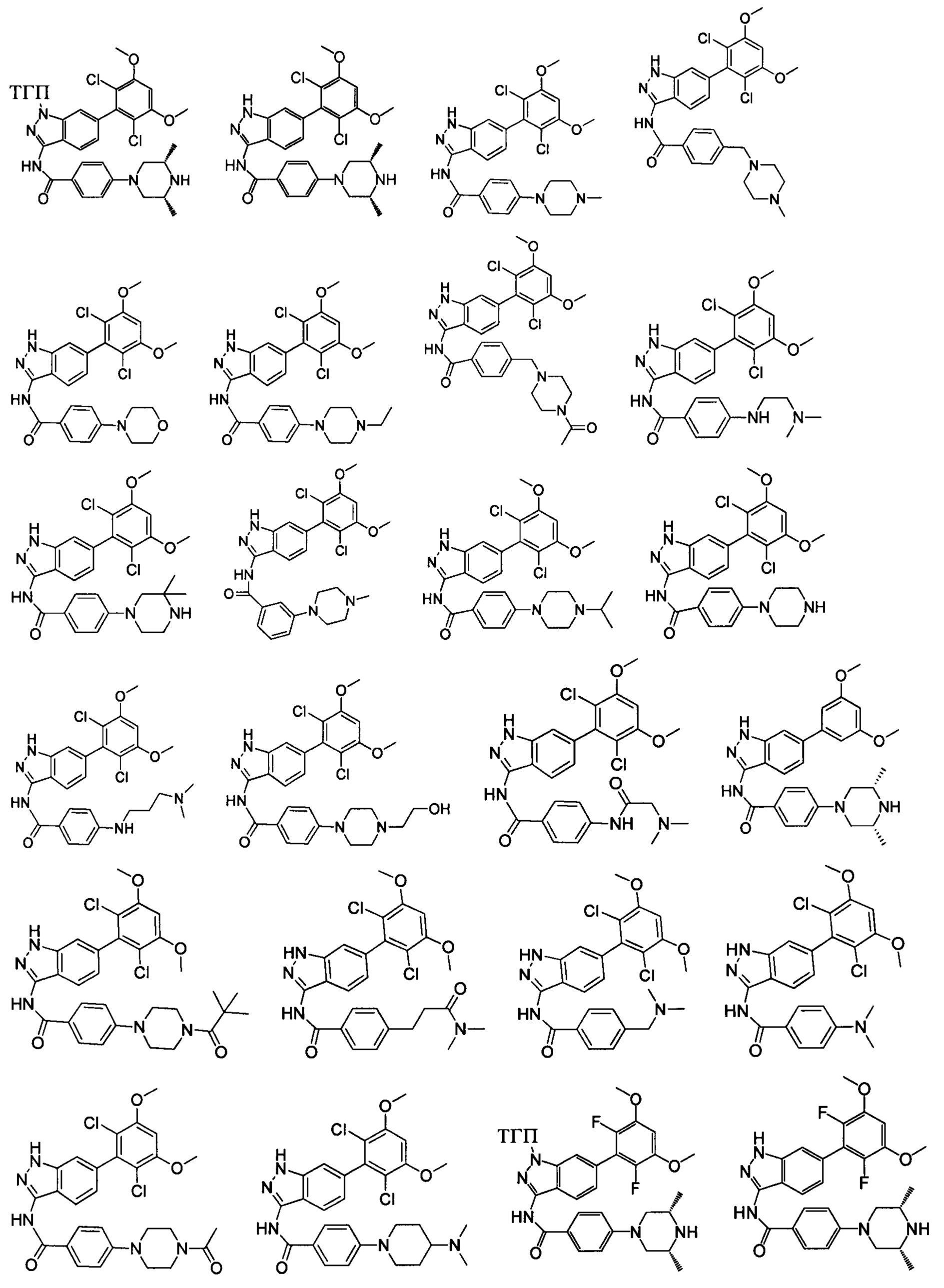
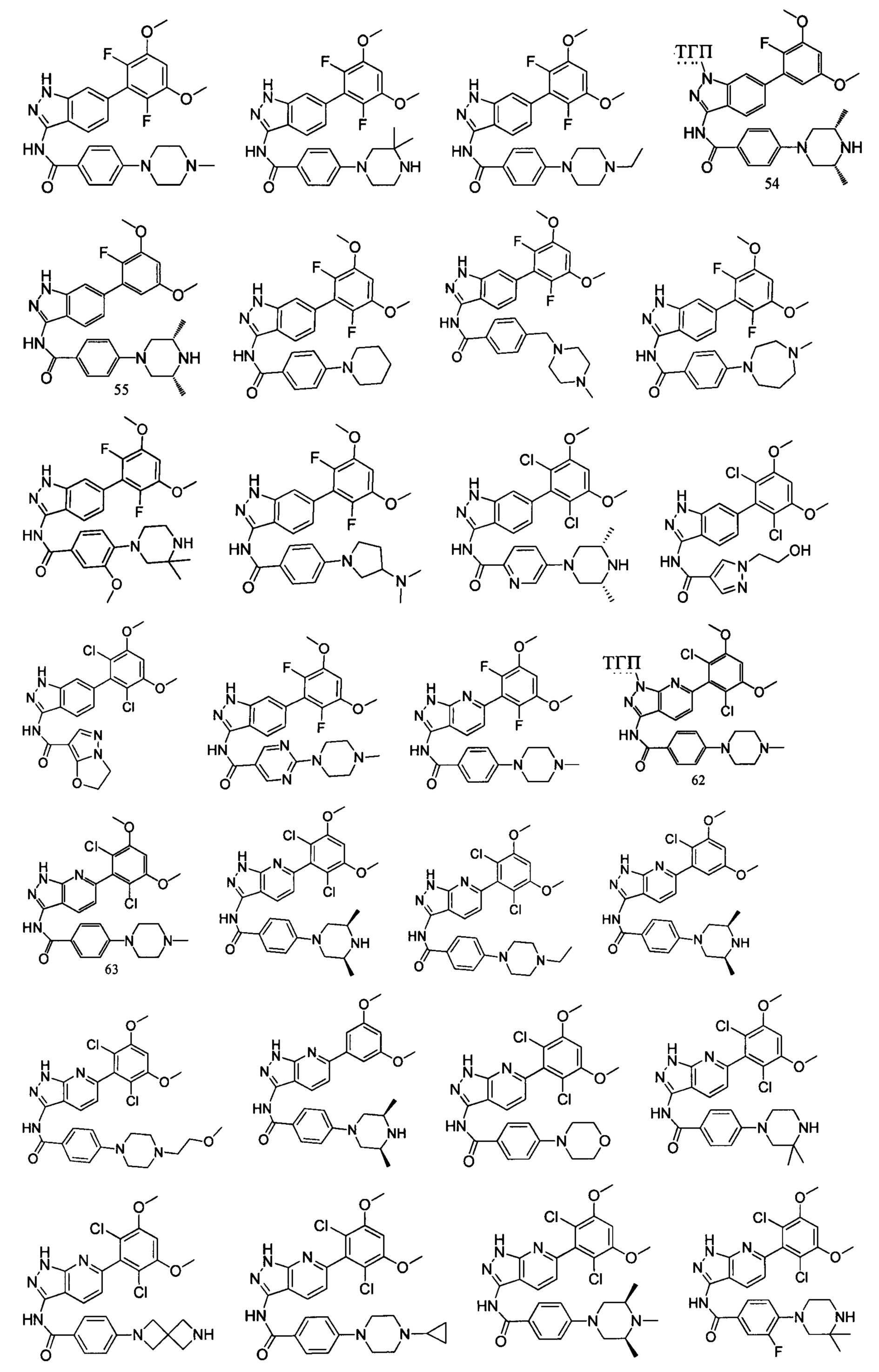
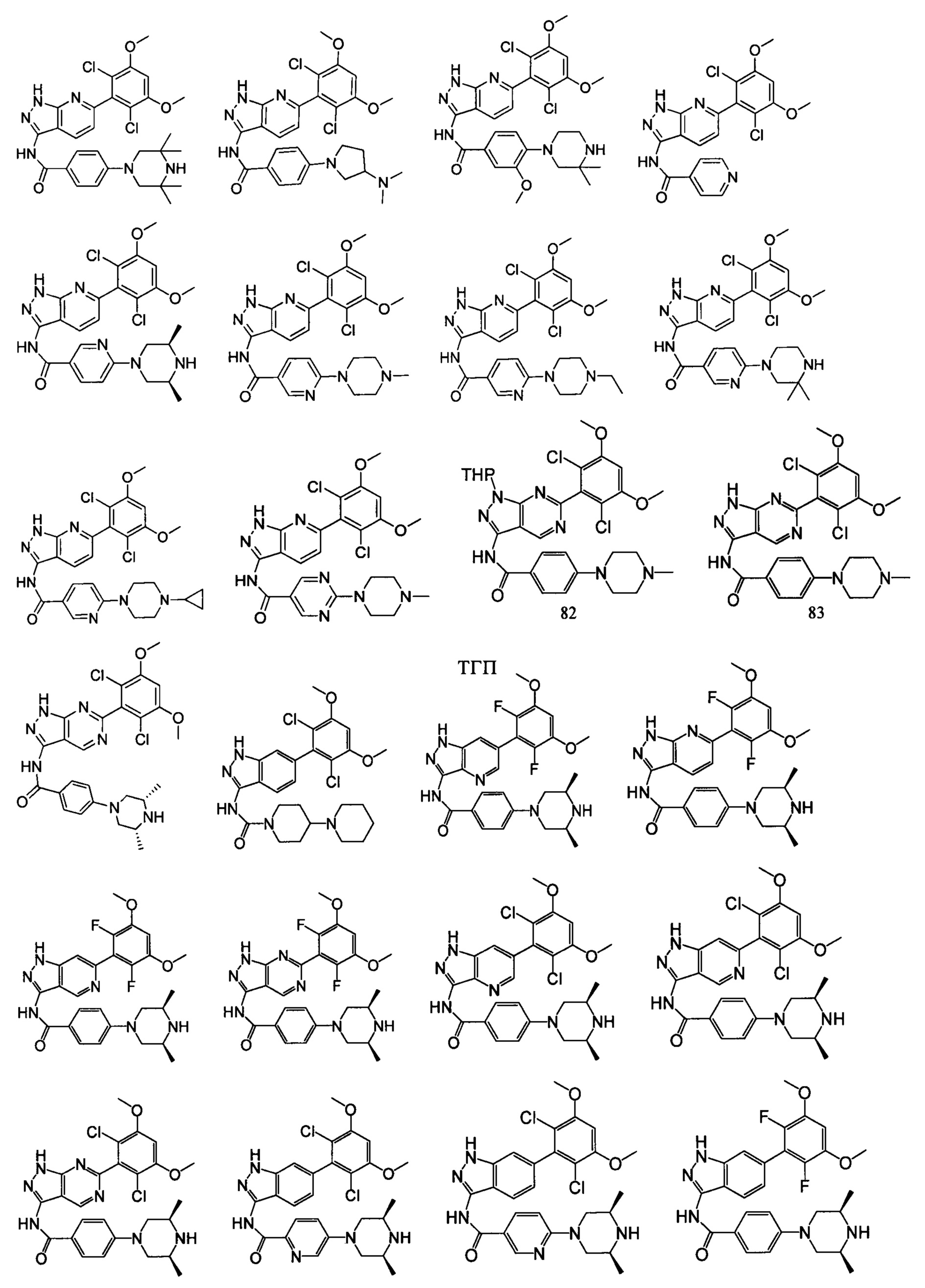
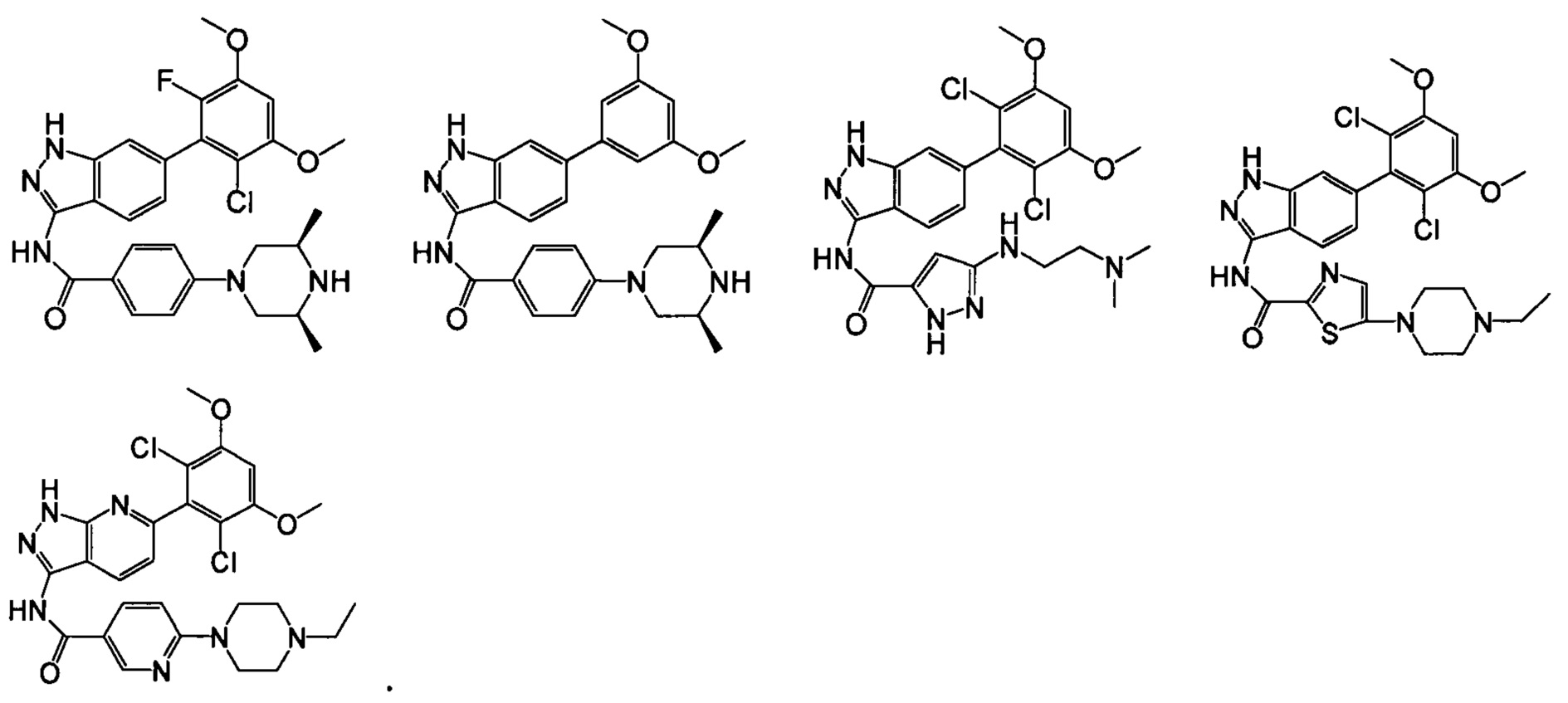
В другом предпочтительном варианте реализации настоящего изобретения L, X, W, Y, Z, кольцо А или R представляют собой соответствующие группы в конкретных соединениях, описанных в примерах.
Во втором аспекте настоящего изобретения предложен способ получения соединения по первому аспекту настоящего изобретения, включающий следующие стадии:
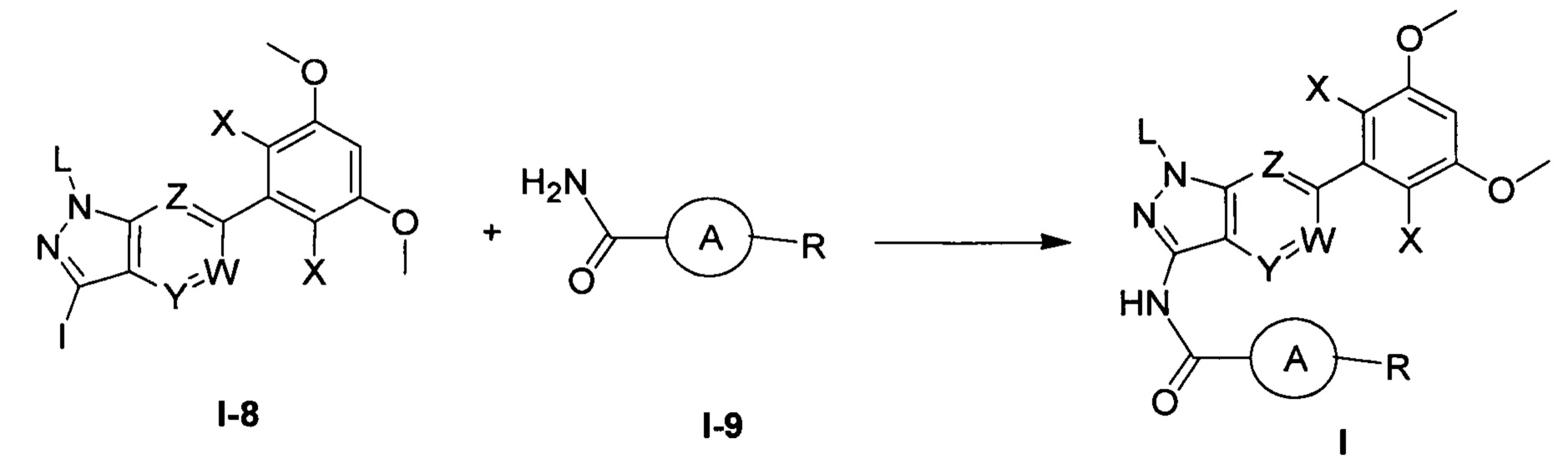
(а) взаимодействия соединения формулы I-8 с соединением формулы I-9 в инертном растворителе с получением соединения формулы I;
при этом группы в указанных выше формулах являются такими, как определено в п. 1 формулы изобретения.
В другом предпочтительном варианте реализации настоящего изобретения на стадии (а) указанное взаимодействие проводят в присутствии соли меди, предпочтительно указанная соль меди выбрана из группы, состоящей из: CuI, Cu, CuCl, Cu2O, CuO, Cu(ОАс)2, CuSO4⋅5H2O, Cu(асас)2 (Cu(II) ацетилацетонат), CuCl2, CuSCN или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения на стадии (а) указанное взаимодействие проводят в присутствии лиганда, предпочтительно указанный лиганд представляет собой бидентатный аминный лиганд, более предпочтительно указанный лиганд выбран из группы, состоящей из: N1,N2-диметил-этилендиамина, (1R,2R)-(-)-N,N'-диметил-1,2-циклогександиамина или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения на стадии (а) указанное взаимодействие проводят в присутствии основания, предпочтительно указанное основание представляет собой неорганическое основание, более предпочтительно указанное основание выбрано из группы, состоящей из: K2CO3, K3PO4, Cs2CO3 или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанный инертный растворитель выбран из группы, состоящей из: толуола, диоксана, ТГФ, ДМФА или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанный способ дополнительно включает следующие стадии:
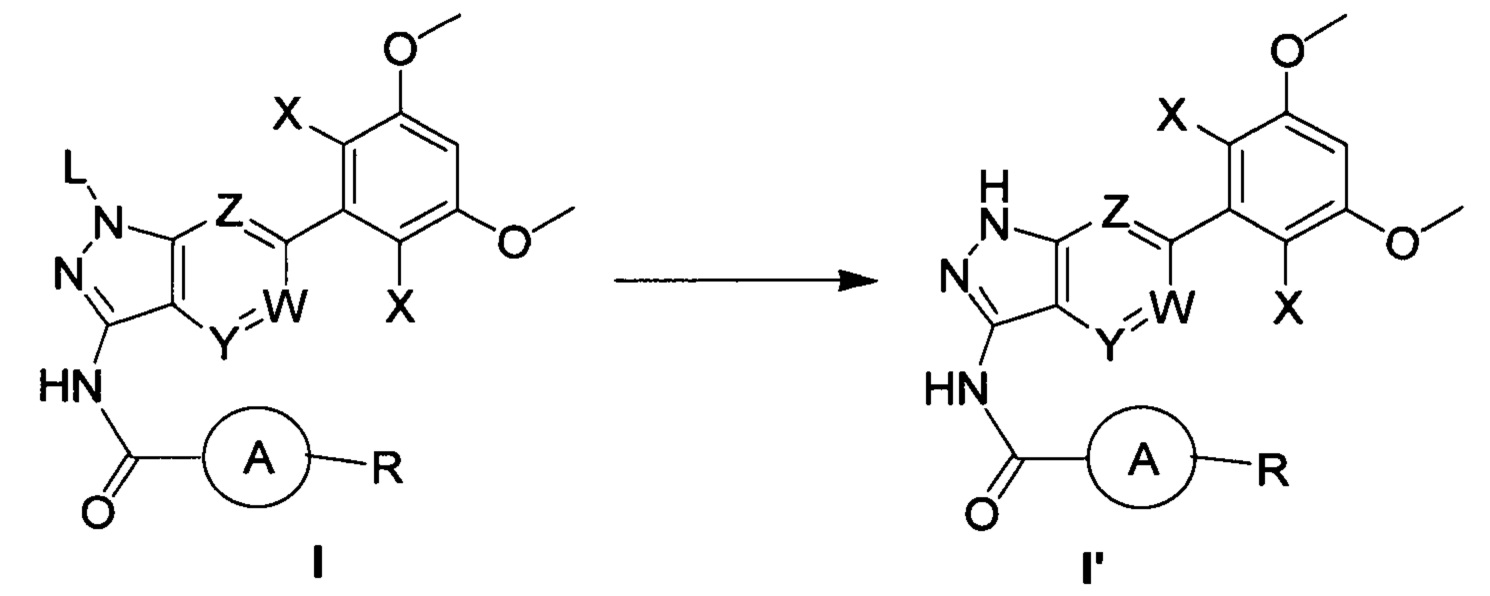
(b) удаление защитной группы из соединения формулы I в инертном растворителе с получением соединения формулы I';
где L выбран из группы, состоящей из тетрагидропиранила (ТГП);
при этом остальные группы яляются такими, как определено выше.
В другом предпочтительном варианте реализации настоящего изобретения на стадии (b) указанное взаимодействие осуществляют в присутствии кислоты, предпочтительно указанная кислота выбрана из группы, состоящей из: соляной кислоты, п-толуолсульфоновой кислоты, TFA или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения на стадии (b) указанный инертный растворитель выбран из группы, состоящей из: дихлорметана, метанола, этанола, изопропанола, н-бутанола, t-бутанола, изобутанола или их комбинации.
В третьем аспекте настоящего изобретения предложено применение соединения по первому аспекту настоящего изобретения для:
(a) получения лекарственного средства для лечения заболеваний, связанных с активностью или величиной экспрессии киназы FGFR;
(b) получения ингибиторов киназы FGFR;
(c) нетерапевтического ингибирования активности киназы FGFR in vitro;
(d) нетерапевтического ингибирования пролиферации опухолевых клеток in vitro; и/или
(e) лечения заболеваний, связанных с активностью или величиной экспрессии киназы FGFR.
В другом предпочтительном варианте реализации настоящего изобретения указанное заболевание, связанное с активностью или величиной экспрессии FGFR, представляет собой опухоль, предпочтительно опухоль, выбранную из группы, состоящей из: рака эндометрия, рака молочной железы, рака желудка, рака мочевого пузыря, миеломы, рака печени.
В другом варианте реализации настоящего изобретения указанная киназа FGFR выбрана из группы, состоящей из: FGFR1, FGFR2, FGFR3 или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанные опухолевые клетки представляют собой клеточную линию лейкоза, предпочтительно клеточную линию миелобластного лейкоза, более предпочтительно клеточную линию острого миелобластного лейкоза KG1.
В четвертом аспекте настоящего изобретения предложена фармацевтическая композиция, при этом указанная фармацевтическая композиция содержит: (i) эффективное количество соединения формулы I или его фармацевтически приемлемой соли и (ii) фармацевтически приемлемый носитель.
В другом предпочтительном варианте реализации настоящего изобретения указанное эффективное количество означает терапевтически эффективное или эффективное ингибирующее количество, предпочтительно от 0,01 до 99,99%.
В другом предпочтительном варианте реализации настоящего изобретения указанную фармацевтическую композицию применяют для ингибирования активности киназы FGFR.
В другом предпочтительном варианте реализации настоящего изобретения указанную фармацевтическую композицию применяют для лечения заболевания, связанного с активностью или величиной экспрессии киназы FGFR.
В пятом аспекте настоящего изобретения предложен способ ингибирования активности киназы FGFR, включающий следующий этап: введение эффективного ингибирующего количества соединения формулы I согласно первому аспекту или его фармацевтически приемлемой соли субъекту, нуждающемуся в ингибировании, или введение эффективного ингибирующего количества фармацевтической композиции согласно четвертому аспекту настоящего изобретения субъекту, нуждающемуся в ингибировании.
В другом предпочтительном варианте реализации настоящего изобретения указанное ингибирование представляет собой нетерапевтическое ингибирование in vitro.
В другом предпочтительном варианте реализации настоящего изобретения указанное эффективное ингибирующее количество составляет от 0,001 до 500 нмоль/л, предпочтительно от 0,01 до 200 нмоль/л, когда эффективное ингибирующее количество соединения формулы I по п. 1 или его фармацевтически приемлемую соль вводят субъекту, нуждающемуся в ингибировании.
В шестом аспекте настоящего изобретения предложен способ лечения заболевания, связанного с активностью или величиной экспрессии киназы FGFR, указанный способ включает: введение субъекту, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I, как описано в первом аспекте настоящего изобретения, или фармацевтической композиции, как описано в четвертом аспекте настоящего изобретения.
В другом предпочтительном варианте реализации настоящего изобретения указанное заболевание, связанное с активностью или величиной экспрессии FGFR, представляет собой опухоль, предпочтительно опухоль, выбранную из группы, состоящей из: рака эндометрия, рака молочной железы, рака желудка, рака мочевого пузыря, миеломы, рака печени.
В седьмом аспекте настоящего изобретения предложен способ ингибирования опухолевых клеток in vitro, включающий: введение субъекту, нуждающемуся в ингибировании, эффективного ингибирующего количества соединения формулы I согласно первому аспекту настоящего изобретения или фармацевтической композиции согласно четвертому аспекту настоящего изобретения.
Следует понимать, что в объеме настоящего изобретения каждый из технических признаков, подробно описанный выше и ниже в настоящем документе (например, признаки, описанные в примерах) можно комбинировать друг с другом, тем самым составляя новые или предпочтительные технические решения, которые для краткости не обязательно указаны в настоящем документе.
ВАРИАНТЫ РЕАЛИЗАЦИИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
В результате долгих и тщательных исследований авторами настоящего изобретения был получен класс соединений, имеющих структуру формулы I, и было обнаружено, что они способны ингибировать киназу FGFR. Указанные соединения обладают ингибирующей активностью при очень низких концентрациях (даже при настолько низких, как ≤ 100 нмоль/л) в отношении ряда киназ FGFR, и, следовательно, оказывают превосходную ингибирующую активность и могут применяться для лечения заболеваний, связанных с активностью или уровнем экспрессии киназы FGFR, таких как опухоли. Таким образом, настоящим изобретением выполнены поставленные задачи.
ТЕРМИНЫ
При использовании в настоящем документе термин «С1-С6-алкил» относится к неразветвленному или разветвленному алкилу, содержащему от 1 до 6 атомов углерода, такому как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил или к подобным.
Термин «С1-С6-алкилен» относится к группам, образованным С1-С6-алкилом, как описано выше, в которых отсутствует один атом водорода, таким как -СН2-, -СН2-СН2- или к подобным.
Термин «С6-С10-арилен» относится к группам, образованным при удалении одного атома водорода из арилов, содержащих 6-10 атомов углерода, таким как моноциклический или бициклический арилен, таким как фенилен, нафтилен или к подобным.
Термин «шестичленная арильная группа» обозначает фенил.
Термин «5-8-членный арил» относится к заместителю, представляющему собой 5-8-членное ненасыщенное карбоциклическое кольцо, такому как фенил или к подобным.
Термин «5-8-членный гетероарил» относится к заместителю, представляющему собой 5-8-членную ненасыщенную кольцевую систему, содержащую один или более гетероатомов, выбранных из О, S, N или Р, такому как пиридил, тиенил или к подобным.
Термин «насыщенное 3-12-членное карбоциклическое кольцо» обозначает насыщенные карбоциклические кольца, содержащие от 3 до 12 атомов углерода, например, циклогексил или подобные.
Термин «3-12-членное гетероциклическое кольцо» относится к заместителю, представляющему собой 3-12-членную насыщенную кольцевую систему, содержащую один или более гетероатомов, выбранных из О, S, N или Р, такому как пиперидил, пирролил или к подобным.
Термин «галоген» относится к F, Cl, Br и I.
Согласно настоящему изобретению термины «содержит» или «включает» означают, что различные компоненты можно применять вместе в смеси или в композиции согласно настоящему изобретению. Таким образом, термин «содержит» охватывает фразы «в основном состоит из» и «состоит из».
В настоящем изобретении термин «фармацевтически приемлемый» компонент относится к веществам, которые подходят для применения для человека и/или животных без нежелательных вредных побочных реакций (таких как токсичность, стимуляция или аллергия), то есть к веществам с разумным соотношением польза/риск.
Согласно настоящему изобретению термин «эффективное количество» относится к такому количеству терапевтического агента, в котором указанный агент может лечить, облегчать или предотвращать целевое заболевание или состояние или обладать лечебным или профилактическим эффектом, поддающимся обнаружению. Точное эффективное количество соединения для определенного субъекта будет зависеть от роста и веса субъекта и состояния его здоровья, от природы заболевания и степени его развития, а также от терапевтического агента и/или комбинации терапевтических агентов, выбранных для введения. Таким образом, нецелесообразно указывать точное эффективное количество заранее. Тем не менее, для конкретной ситуации указанное эффективное количество можно определить в ходе обычных манипуляций, выполняемых на основании обоснованных решений лечащего врача.
Согласно настоящему изобретению, если не указано иное, термин «замещенный» означает, что один или более атомов водорода в группе замещены заместителем, выбранным из группы, состоящей из: галогена, незамещенного или галогенированного С1-С6 алкила, незамещенной или галогенированной С2-С6 ацильной группы, незамещеного или галогенированного С1-С6 алкил-гидрокси.
Если не указано иное, все соединения согласно настоящему изобретению включают все возможные оптические изомеры, такие как отдельные хиральные соединения или смесь различных хиральных соединений (т.е. рацемат). В соединениях согласно настоящему изобретению каждый хиральный атом углерода необязательно может иметь R конфигурацию или S конфигурацию или смесь R конфигурации и S конфигурации.
При употреблении в настоящем документе термин «соединение согласно настоящему изобретению» относится к соединению формулы I. Термин также охватывает различные кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы I.
Употребляемый в настоящем документе термин «фармацевтически приемлемая соль» относится к соли, подходящей для применения в качестве лекарственного средства, которая образуется соединением согласно настоящему изобретению с кислотой или основанием. Указанные фармацевтически приемлемые соли включают неорганические и органические соли. Предпочтительным типом солей являются соли, образованные соединениями согласно настоящему изобретению с кислотами. Подходящие солеобразующие кислоты включают, но не ограничиваются следующими: неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, фтористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота; органические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислота, метансульфоновая кислота, толуолсульфоновая кислота, бензолсульфоновая кислота и подобные; и кислые аминокислоты, такие как аспарагиновая кислота и глутаминовая кислота.
Соединение формулы I
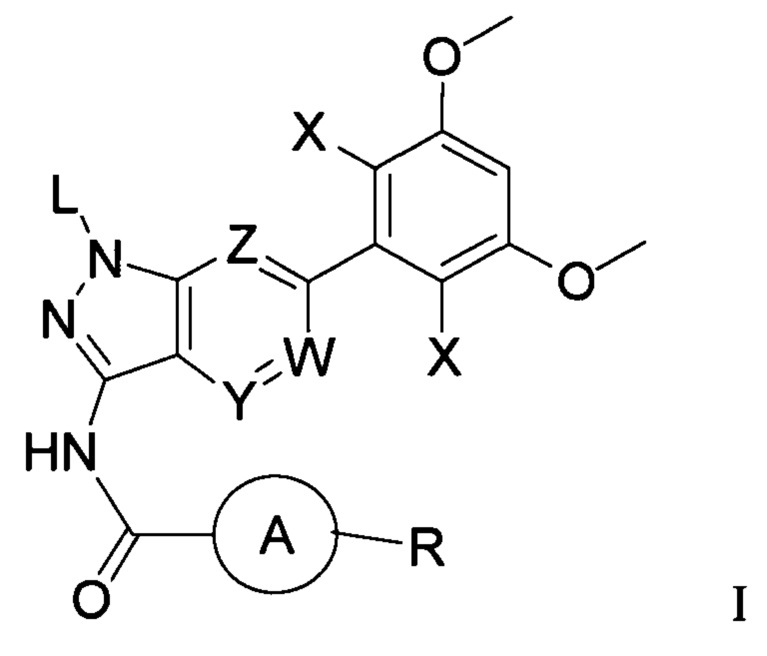
где:
L выбран из группы, состоящей из: Н, тетрагидропиранила (ТГП);
каждый X независимо выбран из группы, состоящей из: Cl, F, Н и CN;
каждый из W, Y и Z независимо выбран из: N или СН;
кольцо А отсутствует, представляет собой незамещенную или замещенную 5-8-членную арильную группу или незамещенную или замещенную 5-8-членную гетероарильную группу, при этом указанная гетероарильная группа содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода или серы; незамещенное или замещенное 3-12-членное насыщенное гетероциклическое кольцо или карбоциклическое кольцо, при этом указанное гетероциклическое кольцо содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода или серы; или
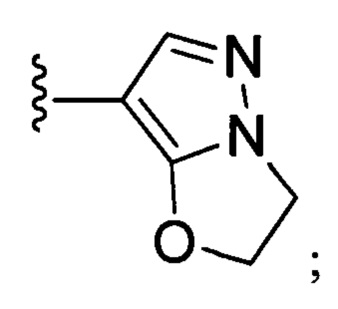
R представляет собой Н или замещенную или незамещенную группу, выбранную из группы, состоящей из:
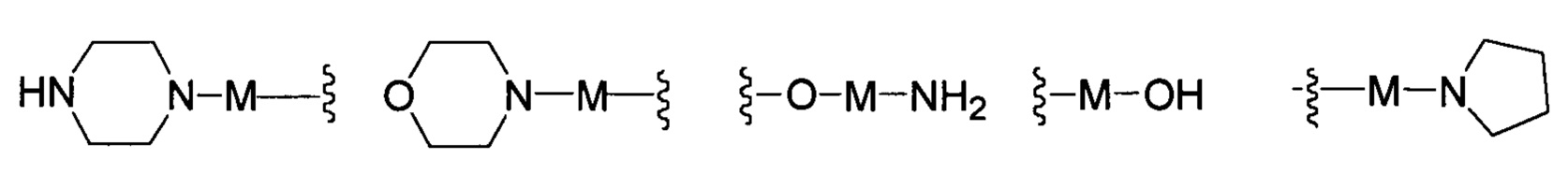
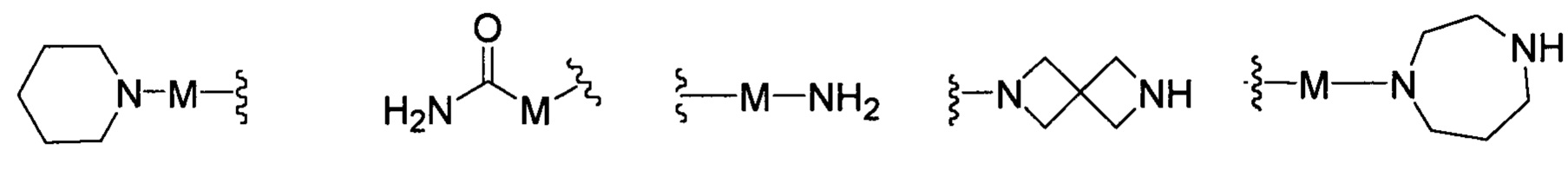 ;
;
где М выбран из группы, состоящей из: замещенного или незамещенного С1-С6-алкилена, замещенного или незамещенного С6-С10-арилена, замещенного или незамещенного С1-С10-гетероарилена или М отсутствует;
при этом термин «замещенный» означает, что один или более атомов водорода на указанной группе замещены заместителями, выбранными из группы, состоящей из: галогена, незамещенного или галогенированного С1-С6 алкила, незамещенной или галогенированной С2-С6 ацильной группы, незамещеного или галогенированного С1-С6 алкил-гидрокси.
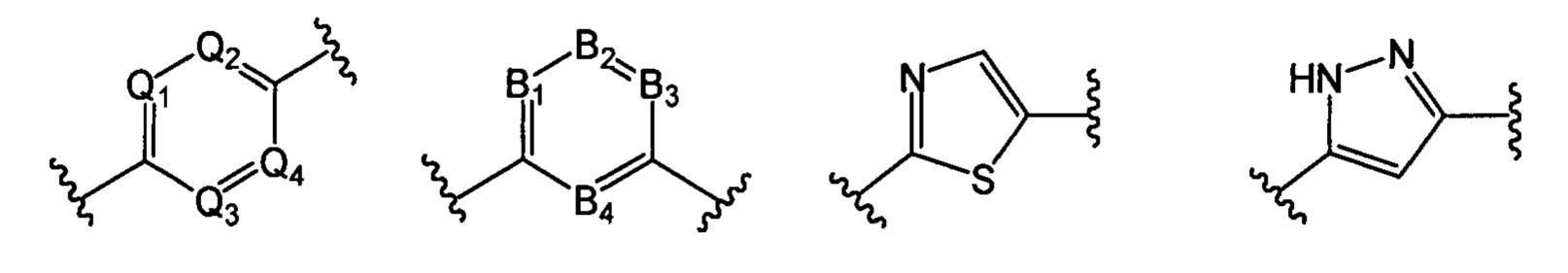
В другом предпочтительном варианте реализации настоящего изобретения кольцо А представляет собой гетероарильную группу, выбранную из группы, состоящей из:
 ;
;
где каждый из Q1, Q2, Q3 и Q4 независимо выбран из: N или СН;
каждый из B1, В2, В3 и В4 независимо выбран из: N или СН.
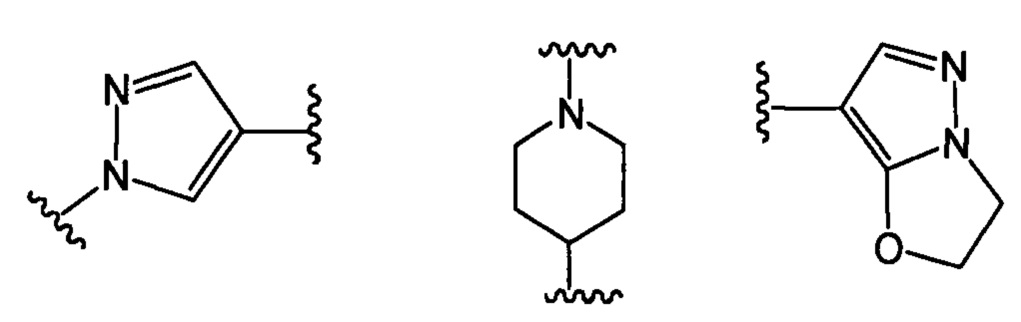
В другом предпочтительном варианте реализации настоящего изобретения кольцо А представляет собой гетероарильную группу, выбранную из группы, состоящей из:
 .
.
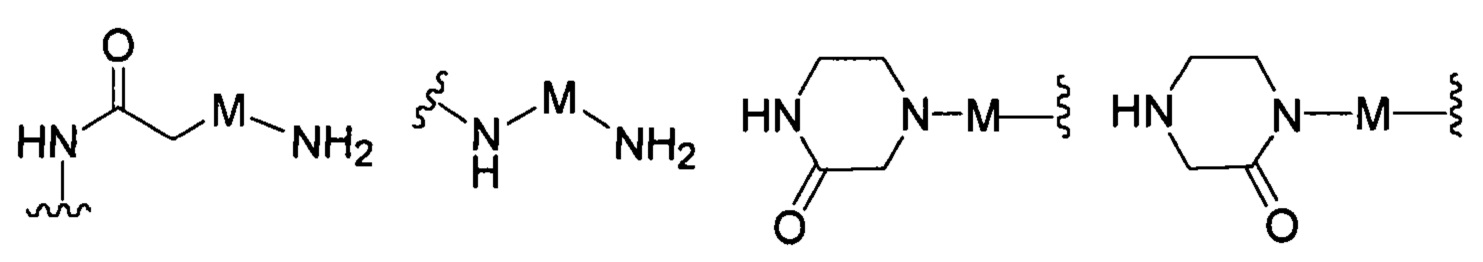
В другом предпочтительном варианте реализации настоящего изобретения R представляет собой замещенную или незамещенную группу, выбранную из группы, состоящей из:
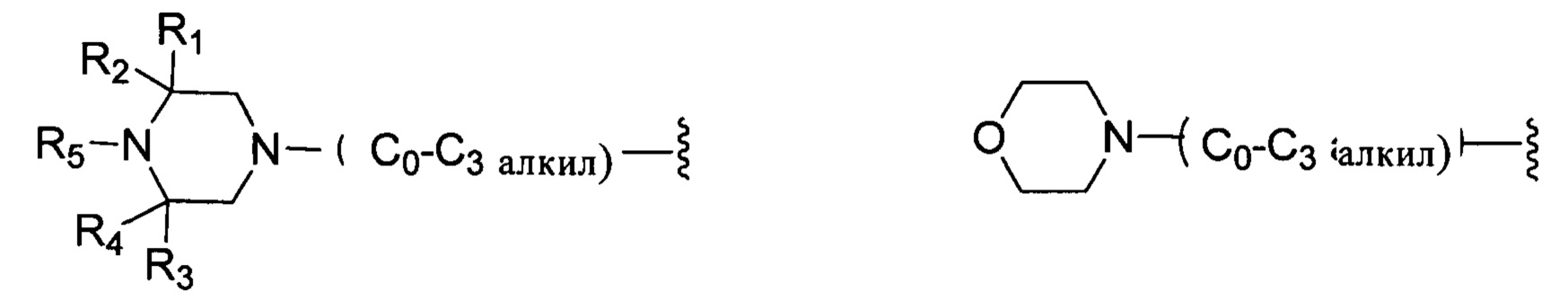


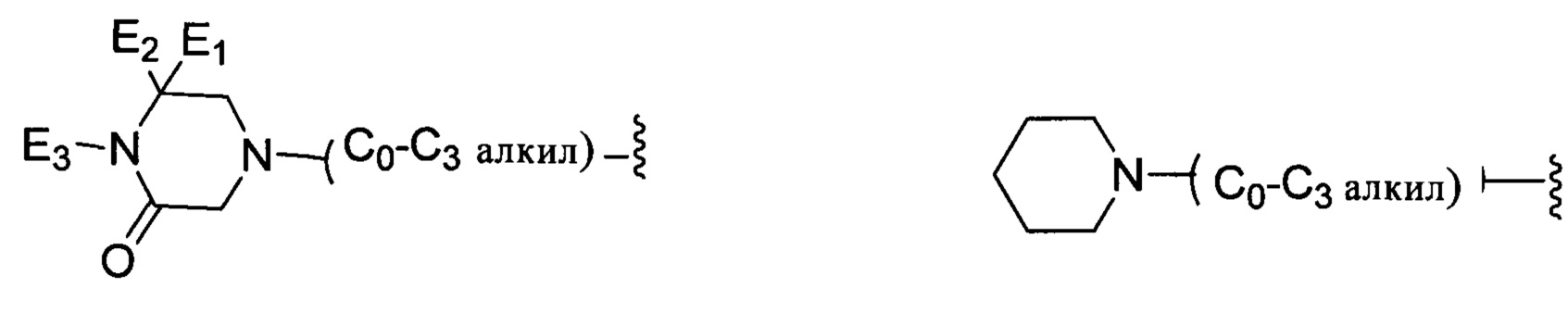 ;
;
где:
каждый из R1, R2, R3 и R4 независимо выбран из группы, состоящей из: Н, галогена, С1-С6 неразветвленного или разветвленного алкила, галогенированного С1-С6 неразветвленного или разветвленного алкила;
R5 выбран из группы, состоящей из: Н, С1-С6 неразветвленного или разветвленного алкила, С1-С6 неразветвленного или разветвленного ацила, С1-С6 неразветвленного или разветвленного алкилен-гидрокси;
каждый из R6, R7, R8, R9, R10, R11 и R12 независимо выбран из группы, состоящей из: Н, С1-С6 неразветвленного или разветвленного алкила, С1-С6 неразветвленной или разветвленной ацильной группы, С1-С6 неразветвленной или разветвленной спиртовой группы (алкилен-гидрокси);
каждый из G1, G2, G3 и G4 независимо выбран из группы, состоящей из: Н, галогена, С1-С6 неразветвленного или разветвленного алкила, галогенированного С1-С6 неразветвленного или разветвленного алкила, или
G5 выбран из группы, состоящей из: Н, С1-С6 неразветвленного или разветвленного алкила, С1-С6 неразветвленного или разветвленного ацила, С1-С6 неразветвленного или разветвленного алкил-гидрокси;
каждый из E1 и Е2 независимо выбран из группы, состоящей из: Н, галогена, неразветвленного или разветвленного алкила, галогенированного С1-С6 неразветвленного или разветвленного алкила;
Е3 выбран из группы, состоящей из: Н, С1-С6 неразветвленного или разветвленного алкила, С1-С6 неразветвленного или разветвленного ацила, С1-С6 неразветвленного или разветвленного алкилен-гидрокси;
каждый из R13, R14, R15 и R16 независимо выбран из группы, состоящей из: Н, С1-С6 неразветвленного или разветвленного алкила, С1-С6 неразветвленной или разветвленной ацильной группы, С1-С6 неразветвленной или разветвленной спиртовой группы (алкилен-гидрокси);
С0-С3 алкил обозначает отсутствие заместителя или алкилен, содержащий 1-3 атомов углерода;
С1-С6 алкил представляет собой алкилен, содержащий 1-6 атомов углерода.
В другом варианте реализации настоящего изобретения L выбран из группы, состоящей из: Н, тетрагидропиранила (ТГП);
каждый X независимо выбран из группы, состоящей из: Н, Cl, F и CN;
каждый из W, Y и Z независимо выбран из: N или СН;
кольцо А представляет собой незамещенную или замещенную 6-членную арильную группу или незамещенную или замещенную 5- или 6-членную гетероарильную группу, где указанная гетероарильная группа содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода или серы;
М выбран из группы, состоящей из незамещенной или замещенной С1-С4-алкиленовой группы, или М отсутствует;
при этом термин «замещенный» означает, что один или более атомов водорода на указанной группе замещены заместителями, выбранными из группы, состоящей из: галогена, незамещенного или галогенированного С1-С4 алкила, незамещенной или галогенированной С2-С4 ацильной группы, незамещеного или галогенированного С1-С4 алкил-гидрокси.
В другом предпочтительном варианте реализации настоящего изобретения L представляет собой Н;
каждый X независимо выбран из группы, состоящей из: Н, Cl и F;
каждый из W, Y и Z независимо выбран из: N или СН;
кольцо А представляет собой группу, выбранную из группы, состоящей из: фенила, пиразолила, пиридила, тиазолила или пиперидинила; или А отсутствует;
М выбран из группы, состоящей из незамещенной или замещенной С1-С3-алкиленовой группы, или М отсутствует;
при этом термин «замещенный» означает, что один или более атомов водорода на указанной группе замещены заместителями, выбранными из группы, состоящей из: галогена, незамещенного или галогенированного С1-С4 алкила, незамещенной или галогенированной С2-С4 ацильной группы, незамещеного или галогенированного С1-С4 алкил-гидрокси.
В другом предпочтительном варианте реализации настоящего изобретения соединение формулы I выбрано из группы, состоящей из:
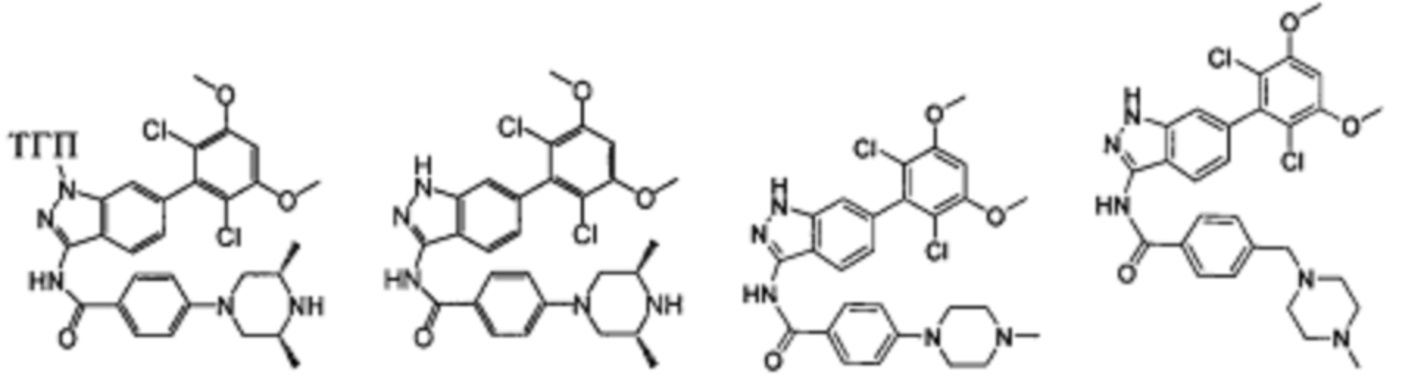
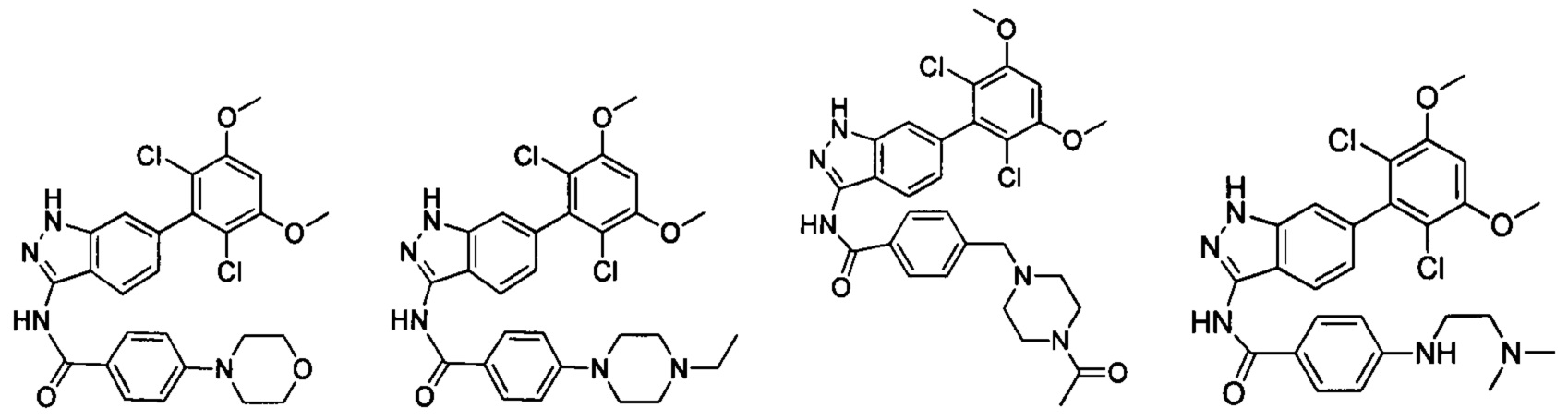
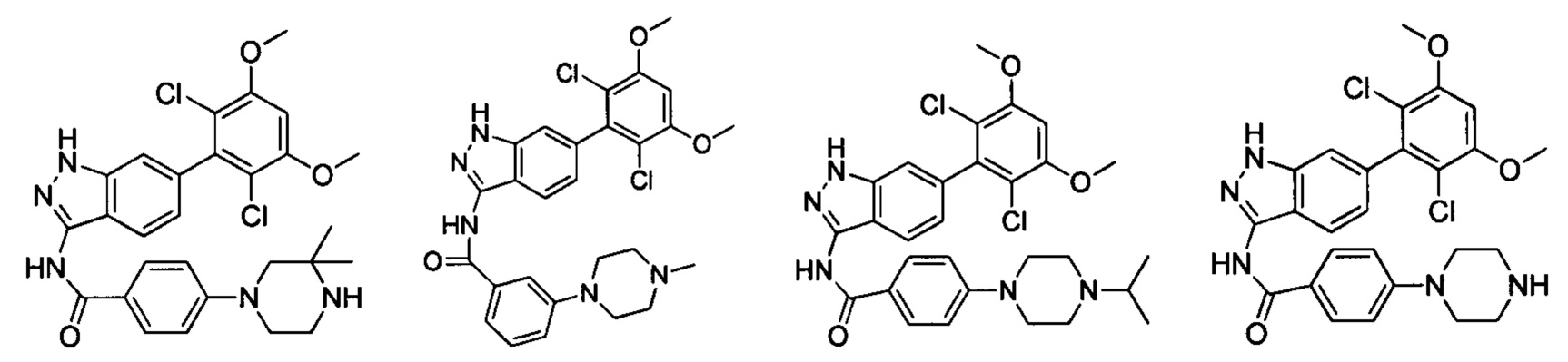
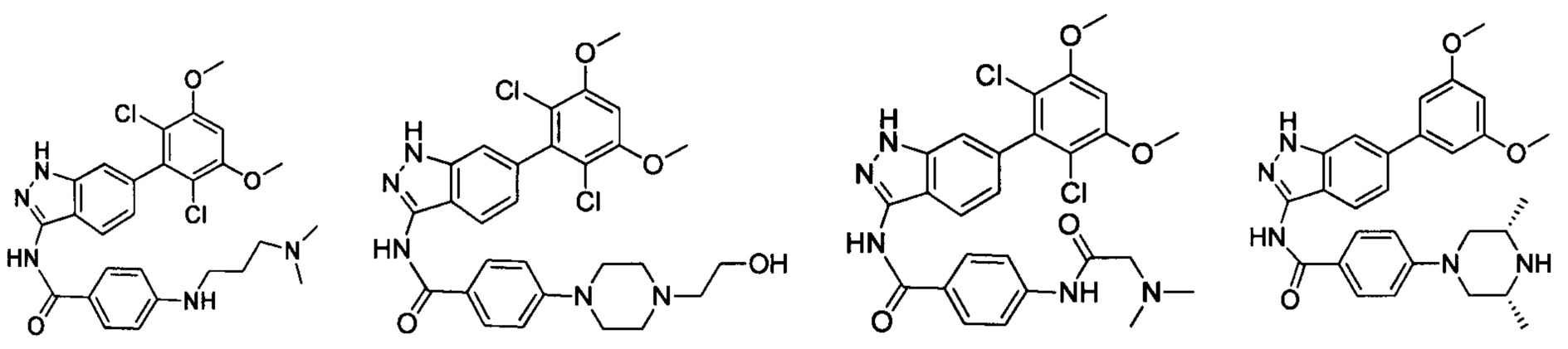
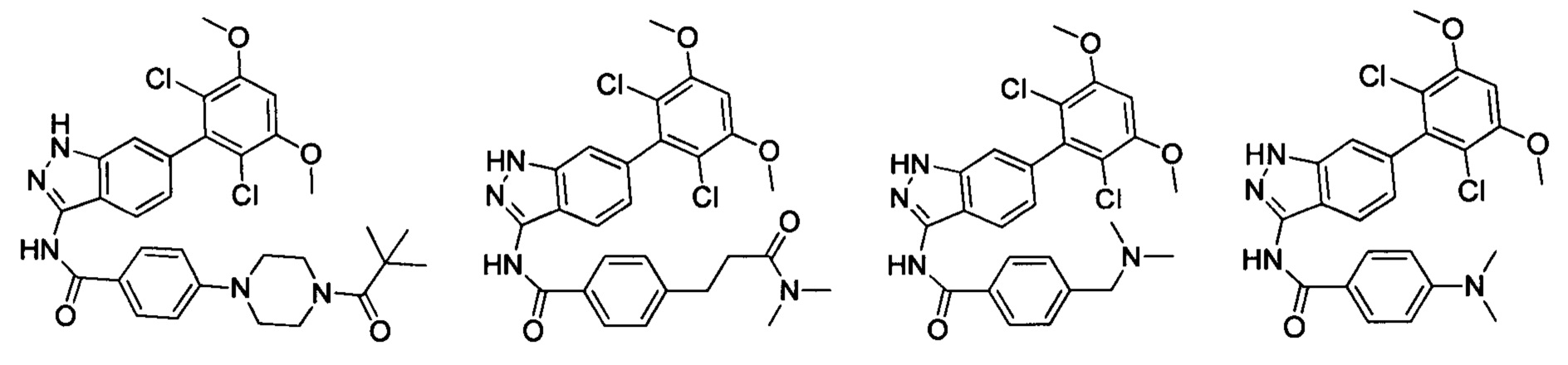
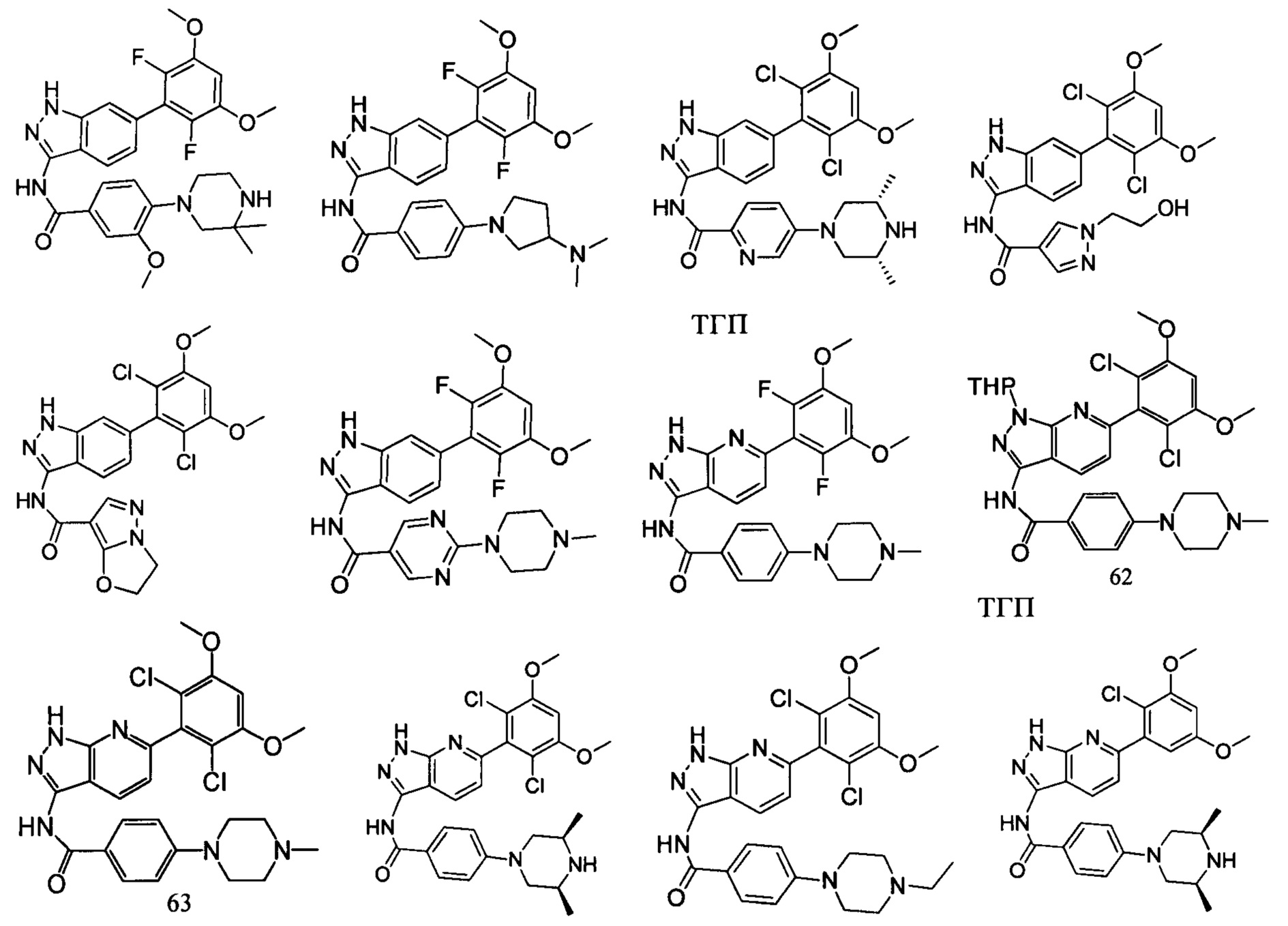
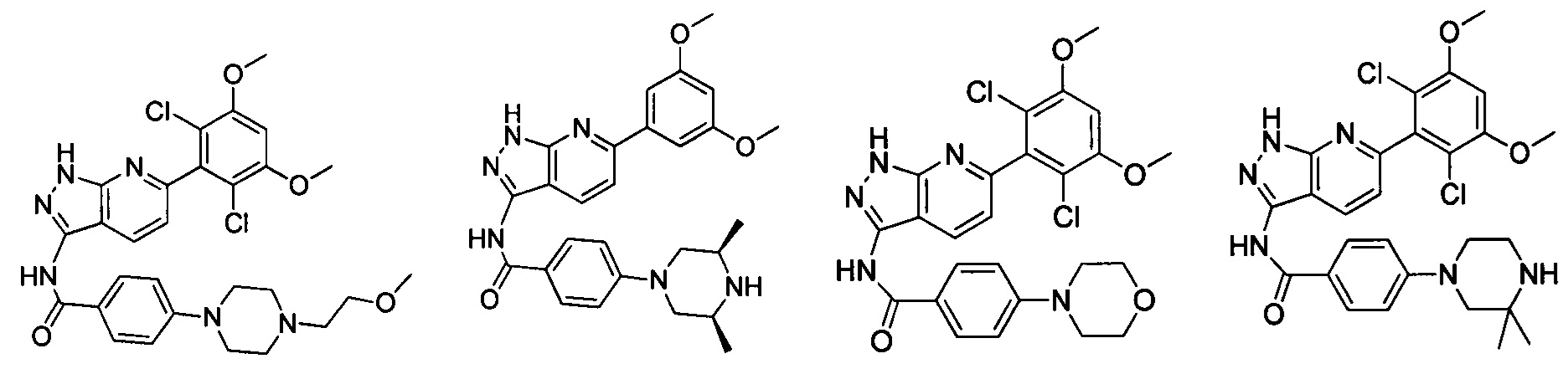
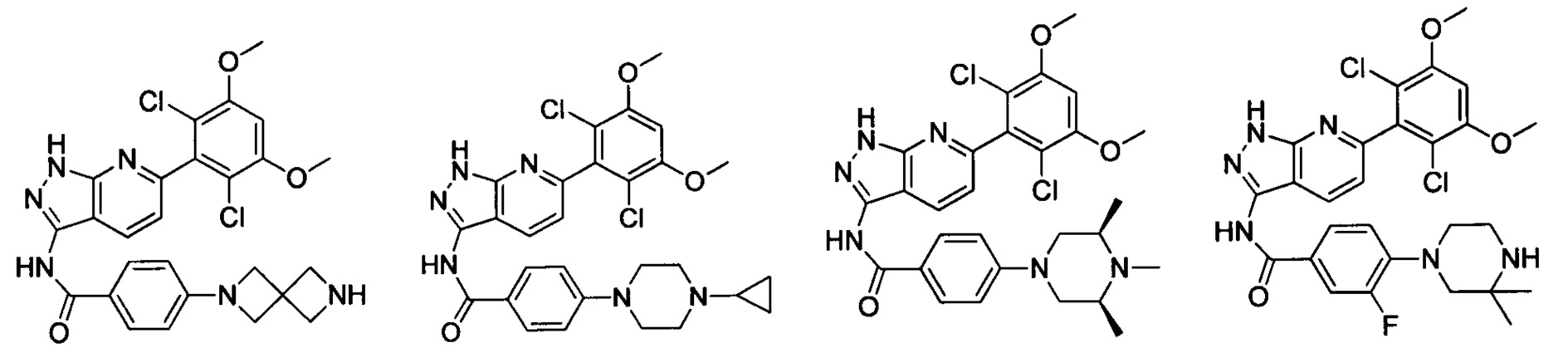
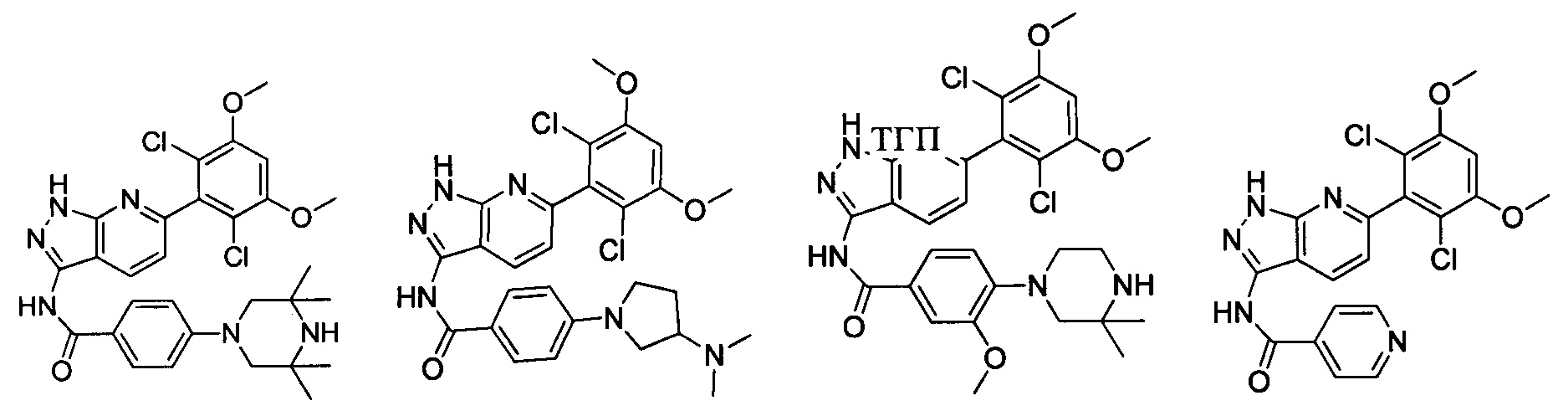
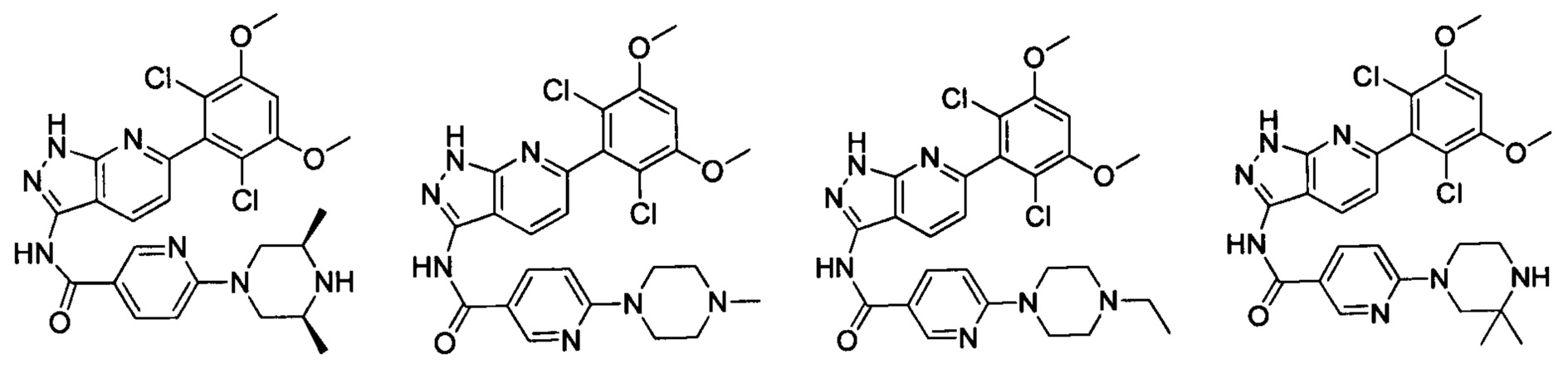
 .
.
Получение соединений формулы I
Способ получения
Далее более подробно описаны способы получения соединений формулы (I), но такие конкретные способы не ограничивают никоим образом настоящее изобретение. Кроме того, соединения согласно настоящему изобретению можно легко получать путем произвольного сочетания различных способов синтеза, описанных в настоящем документе, или способов, известных в данной области техники, такие комбинации может легко выполнить средний специалист в области техники, к которой относится настоящее изобретение.
Способы получения соединений формулы I-8 и соединений формулы I-9, используемых в настоящем изобретении, представляют собой способы, которые уже известны в данной области техники. Обычно в процессе получения каждое взаимодействие проводят в инертном растворителе при температуре от комнатной температуры до температуры образования жидкости при нагревании с обратным холодильником. Время реакции обычно составляет от 0,1 до 60 часов, предпочтительно от 0,5 до 48 часов.
Один предпочтительный способ получения соединения формулы I включает следующие стадии:
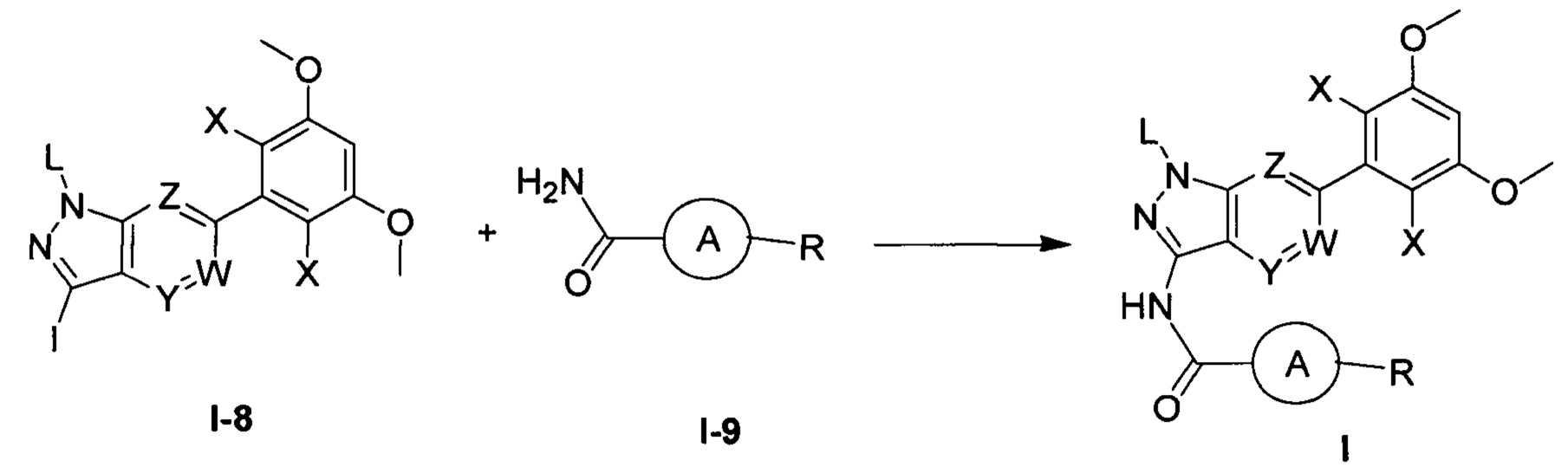
(а) взаимодействие соединения формулы I-8 с соединением формулы I-9 в инертном растворителе с получением соединения формулы I.
В приведенных выше формулах группы такие, как определено выше.
В другом предпочтительном варианте реализации настоящего изобретения указанное взаимодействие проводят в присутствии соли меди; предпочтительно, указанная соль меди выбрана из группы (но не ограничивается ею), состоящей из: CuI, Cu, CuCl, Cu2O, CuO, Cu(ОАс)2, CuSO4⋅5H2O, Cu(асас)2 (Cu(II) ацетилацетонат), CuCl2, CuSCN или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанное взаимодействие проводят в присутствии лиганда, предпочтительно указанный лиганд представляет собой бидентатный аминный лиганд, более предпочтительно указанный лиганд выбран из группы (но не ограничивается ею), состоящей из: N1,N2-диметил-этилендиамина, (1R,2R)-(-)-N,N'-диметил-1,2-циклогександиамина или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанное взаимодействие проводят в присутствии основания; предпочтительно указанное основание представляет собой неорганическое основание; более предпочтительно основание выбрано из группы (но не ограничивается ею), состоящей из: K2CO3, K3PO4, Cs2CO3 или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения инертный растворитель выбран из группы (но не ограничивается ею), состоящей из: толуола, диоксана, ТГФ, ДМФА или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанный способ дополнительно включает следующие стадии:
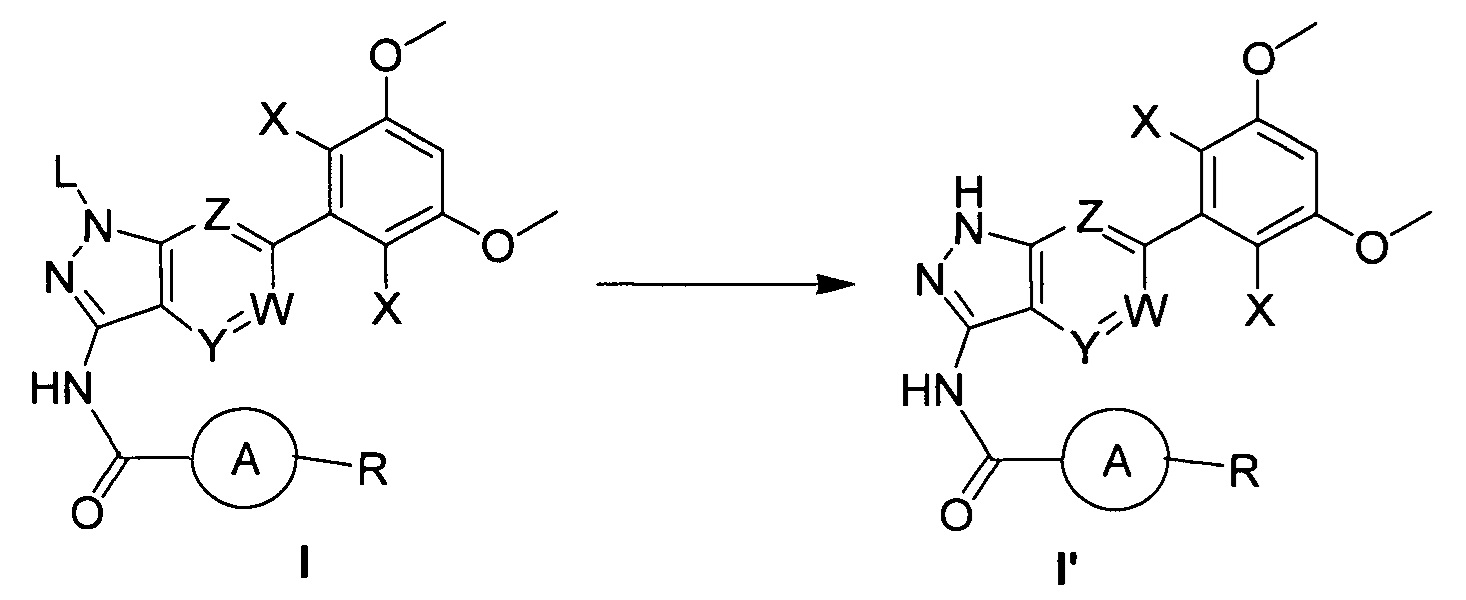
(b) удаление защитной группы из соединения формулы I в инертном растворителе с получением соединения формулы I'; при этом другие группы являются такими, как определено выше.
В другом предпочтительном варианте реализации настоящего изобретения указанное взаимодействие осуществляют в присутствии кислоты, предпочтительно указанная кислота выбрана из группы (но не ограничивается ею), состоящей из: соляной кислоты, п-толуолсульфоновой кислоты, TFA или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанный инертный растворитель выбран из группы (но не ограничивается ею), состоящей из: дихлорметана, метанола, этанола, изопропанола, н-бутанола, т-бутанола, изобутанола или их комбинации.
Предпочтительный способ получения включает следующие стадии:
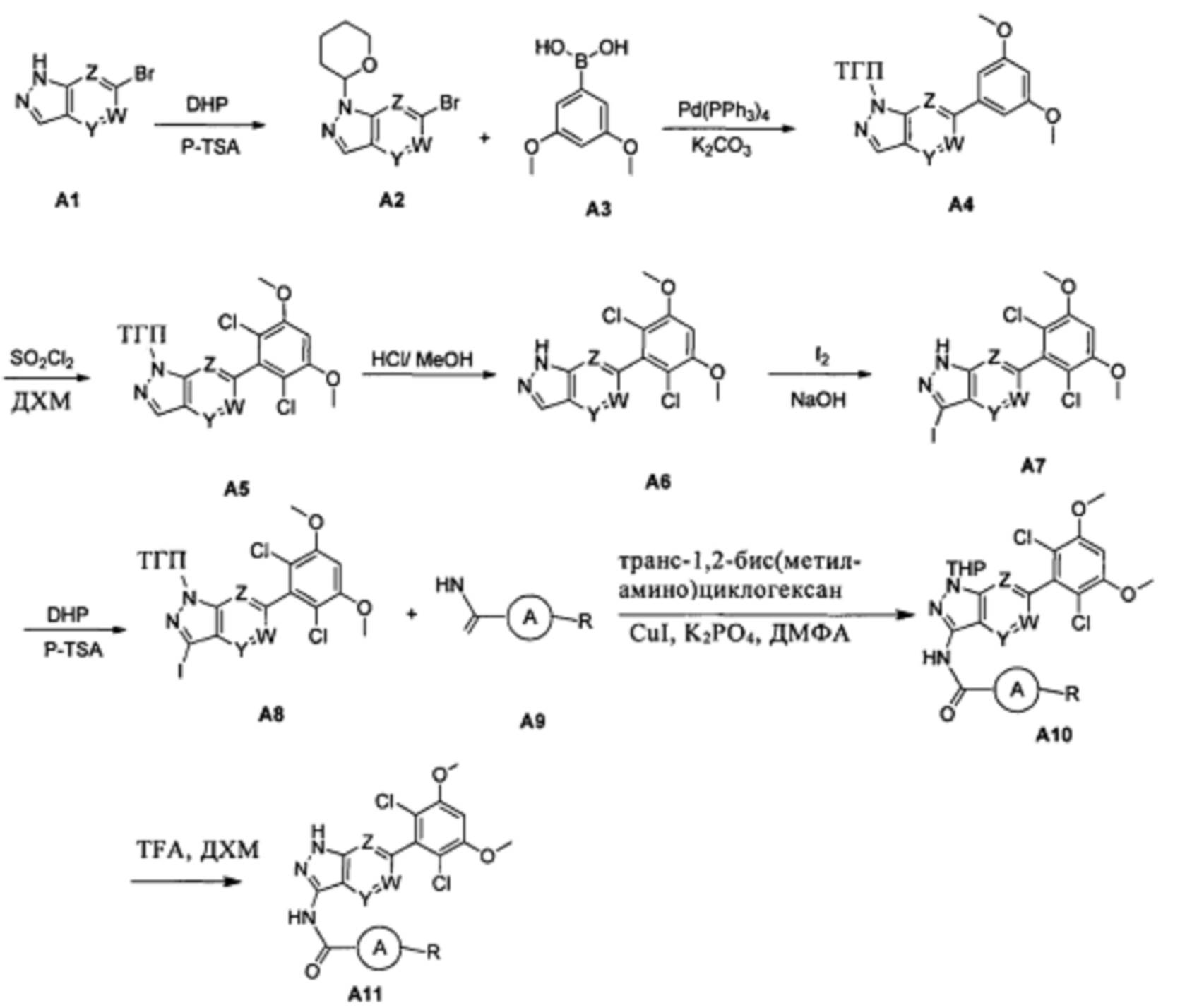
(1) Соединение А2 можно получить при взаимодействии соединения А1 и DHP в инертном растворителе (ДХМ, ТГФ и т.д.) с катализом при помощи кислоты (например, но не ограничиваясь ими: п-толуолсульфоновой кислоты, трифторуксусной кислоты).
(2) Соединение А4 можно получить путем проведения реакции сочетания Сузуки из соответствующей бороновой кислоты или сложного эфира в инертном растворителе (таком как диоксан и вода, толуол и вода, ДМСО, ТГФ, ДМФА) и в присутствии катализатора (например, тетракис(трифенилфосфин)палладия, трис(дибензилиденацетон)дипалладия (Pd2(dba)3), бис(дибензилиденацетон)палладия, дихлорбис(трифенилфосфин)палладия, трифенилфосфин палладия ацетата, бис(три-о-бензилфосфин)палладия дихлорида, 1,2-бис(дифенилфосфино)этан палладия дихлорида, и т.д.) и основания (такого как карбонат калия, фторид калия, фторид цезия, фторид натрия, фосфат калия, фосфата калия гидрат, карбонат натрия, бикарбонат натрия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламин, диизопропилэтиламин, пиридин или их комбинации и т.д.) в течение периода времени (например, от 1 до 4 часов);
(3) Соединение А5 можно получить из соединения А4 в инертном растворителе (дихлорметан, ТГФ, ацетонитрил), по каплям медленно добавляя SO2Cl2 при перемешивании при комнатной температуре.
(4) Соединение А6 можно получить путем удаления защитной группы из соединения А5 при добавлении кислоты (такой как соляная кислота, п-толуолсульфоновая кислота, TFA) в инертном растворителе (таком как дихлорметан, метанол, этанол, изопропанол, н-бутанол, трет-бутанол, изобутанол).
(5) Соединение А7 можно получить путем добавления соединения А6, йода и NaOH в инертном растворителе (1,4-диоксан, ДМФА и т.д.) при перемешивании при комнатной температуре.
(6) Соединение А8 можно получить при взаимодействии соединения А7 и DHP в инертном растворителе (ДХМ, ТГФ и т.д.), катализируемого при помощи кислоты (например, но не ограничиваясь ими: п-толуолсульфоновой кислоты, трифторуксусной кислоты).
(7) Соединение А10 можно получить путем проведения реакции амидирования соединения А8 и соединения А9. Предпочтительно указанное взаимодействие проводят в присутствии одного или более из следующих реагентов: соли меди, которая может представлять собой, но не ограничивается ими: CuI, Cu, CuCl, Cu2O, CuO, Cu(ОАс)2, CuSO4⋅5H2O, Cu(асас)2 (Cu(II) ацетилацетонат), CuCl2, CuSCN или их комбинации; лиганда, который может представлять собой бидентатные аминные лиганды, включая, но не ограничиваясь: N1,N2-диметил-этилендиамин, (1R,2R)-(-)-N,N'-диметил-1,2-циклогександиамин; основания, которое может представлять собой, без ограничения, неорганические основания, такие как K2CO3, K3PO4, Cs2CO3, и реакционный растворитель может представлять собой, но не ограничивается ими: толуол, диоксан, ТГФ и ДМФА.
(8) Соединение А11 можно получить путем удаления защитной группы из соединения А10 с применением соответствующей кислоты (такой как, но не ограничиваясь ими: соляная кислота, п-толуолсульфоновая кислота, TFA) в инертном растворителе (таком как дихлорметан, метанол, этанол, изопропанол, н-бутанол, трет-бутанол, изобутанол и т.д.).
Применение соединений формулы I
Соединения формулы I можно применять для одного или более из следующего:
(а) получения лекарственного средства для лечения заболеваний, связанных с активностью или величиной экспрессии киназы FGFR;
(b) получения ингибиторов киназы FGFR;
(c) нетерапевтического ингибирования активности киназы FGFR in vitro;
(d) нетерапевтического ингибирования пролиферации опухолевых клеток in vitro;
(e) лечения заболеваний, связанных с активностью или величиной экспрессии киназы FGFR.
В другом предпочтительном варианте реализации настоящего изобретения указанное заболевание, связанное с активностью или величиной экспрессии FGFR, представляет собой опухоль, предпочтительно опухоль, выбранную из группы, состоящей из: рака эндометрия, рака молочной железы, рака желудка, рака мочевого пузыря, миеломы, рака печени.
В другом варианте реализации настоящего изобретения киназа FGFR выбрана из группы, состоящей из: FGFR1, FGFR2, FGFR3 или их комбинации.
В другом предпочтительном варианте реализации настоящего изобретения указанные опухолевые клетки представляют собой клеточную линию лейкоза, предпочтительно клеточную линию миелобластного лейкоза, более предпочтительно клеточную линию острого миелобластного лейкоза KG1.
Соединение формулы I можно применять для получения фармацевтической композиции, содержащей: (i) эффективное количество соединения формулы I или его фармацевтически приемлемой соли и (ii) фармацевтически приемлемый носитель.
В другом предпочтительном варианте реализации настоящего изобретения эффективное количество означает терапевтически эффективное или эффективное ингибирующее количество.
В другом предпочтительном варианте реализации настоящего изобретения указанную фармацевтическую композицию применяют для ингибирования активности киназы FGFR.
В другом предпочтительном варианте реализации настоящего изобретения указанную фармацевтическую композицию применяют для лечения заболеваний, связанных с активностью или величиной экспрессии киназы FGFR.
Кроме того, соединение формулы I согласно настоящему изобретению также можно применять в способе ингибирования активности киназы FGFR, при этом указанный способ включает следующий этап: введение эффективного ингибирующего количества соединения формулы I по п. 1 формулы изобретения или его фармацевтически приемлемой соли субъекту, нуждающемуся в ингибировании, или введение эффективного ингибирующего количества фармацевтической композиции по п. 7 формулы изобретения субъекту, нуждающемуся в ингибировании.
В другом предпочтительном варианте реализации настоящего изобретения указанное ингибирование представляет собой нетерапевтическое ингибирование in vitro.
В другом предпочтительном варианте реализации настоящего изобретения указанное эффективное ингибирующее количество составляет от 0,001 до 500 нмоль/л, предпочтительно от 0,01 до 200 нмоль/л, когда эффективное ингибирующее количество соединения формулы I по п. 1 или его фармацевтически приемлемой соли вводят субъекту, нуждающемуся в ингибировании.
В частности, настоящее изобретение также относится к способу лечения заболеваний, связанных с активностью или экспрессией киназы FGFR, при этом указанный способ включает: введение субъекту, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I или фармацевтической композиции, содержащей соединение формулы I в качестве активного ингредиента.
В другом предпочтительном варианте реализации настоящего изобретения указанное заболевание, связанное с активностью или величиной экспрессии FGFR, представляет собой опухоль, предпочтительно опухоль, выбранную из группы, состоящей из: рака эндометрия, рака молочной железы, рака желудка, рака мочевого пузыря, миеломы, рака печени.
Фармацевтическая композиция и ее введение
Соединения согласно настоящему изобретению обладают превосходной активностью в отношении ингибирования киназ FGFR, таких как киназы FGFR1, FGFR2 и FGFR3. Таким образом, соединения согласно настоящему изобретению и кристаллические формы, их фармацевтически приемлемые неорганические или органические соли, гидраты или сольваты, а также фармацевтические композиции, содержащие соединение согласно настоящему изобретению в качестве основного активного ингредиента, можно применять для лечения, предотвращения и облегчения заболеваний, связанных с активностью или уровнем экспрессии FGFR. Согласно предшествующему уровню техники, соединения согласно настоящему изобретению можно применять для лечения следующих заболеваний: рака эндометрия, рака молочной железы, рака желудка, рака мочевого пузыря, миеломы, рака печени и подобного.
Фармацевтическая композиция согласно настоящему изобретению содержит соединение согласно настоящему изобретению или его фармакологически приемлемую соль в безопасном и эффективном диапазоне доз и фармакологически приемлемое вспомогательное вещество или носитель. При этом «безопасная и эффективная доза» означает, что количество соединения является достаточным для заметного улучшения состояния, не вызывая при этом серьезных побочных эффектов. Обычно указанная фармацевтическая композиция содержит 1-2000 мг соединения согласно настоящему изобретению на дозу, предпочтительно 5-200 мг соединения согласно настоящему изобретению на одну дозу. Предпочтительно «доза» представляет собой капсулу или таблетку.
Термин «фармацевтически приемлемый носитель» обозначает один или более совместимых твердых или жидких наполнителей или желеобразных материалов, которые подходят для применения человеком и должны иметь достаточную чистоту и достаточно низкую токсичность. Термин «совместимый» означает, что каждый компонент в композиции можно смешивать с соединением согласно настоящему изобретению и друг с другом без значительного снижения эффективности указанного соединения. Некоторые примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (такие как натрий карбоксиметилцеллюлоза, натрий этилцеллюлоза, ацетат целлюлозы и т.д.), желатин, тальк, твердые смазки (такие как стеариновая кислота, стеарат магния), сульфат кальция, растительные масла (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (например, Tween®), смачивающий агент (например, додецилсульфат натрия), красители, вкусоароматические агенты, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и т.д.
Нет каких-либо специальных ограничений, касающихся режима введения соединений или фармацевтических композиций согласно настоящему изобретению, и типичный режим введения включает (но не ограничивается ими): пероральный, внутриопухолевый, ректальный, парентеральный (внутривенный, внутримышечный или подкожный) и местное введение.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В указанных твердых лекарственных формах указанные активные соединения смешаны с по меньшей мере одним обычным инертным вспомогательным веществом (или носителем), таким как цитрат натрия или Ca2HPO4 или смешаны с любым из следующих компонентов: (а) наполнителями или компатибилизаторами, например, крахмалом, лактозой, сахарозой, глюкозой, маннитом и кремниевой кислотой; (b) связующими веществами, например, гидроксиметил целлюлозой, альгинатами, желатином, поливинилпирролидоном, сахарозой и аравийской камедью; (с) увлажнителями, такими как глицерин; (d) дезинтегрирующими агентами, такими как агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (е) замедляющими растворение агентами, такими как парафин; (f) ускорителями абсорбции, например, соединениями четвертичного аммония; (g) смачивающими агентами, такими как цетиловый спирт и глицеринмоностеарат; (h) адсорбентами, например, каолином; и (i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия или их смесью. В капсулах, таблетках и пилюлях лекарственные формы могут также содержать буферные агенты.
Твердые лекарственные формы, такие как таблетки, сахарные драже, капсулы, пилюли и гранулы можно получать с применением материалов покрытий и оболочек, таких как энтеросолюбильные покрытия и другие материалы, известные в данной области техники. Они могут содержать непрозрачный агент, и, кроме того, активное соединение (соединения) или соединение (соединения) в композиции может высвобождаться в отсроченном режиме в конкретной части желудочно-кишечного тракта. Примеры инкапсулирующих компонентов включают полимеры и воски. При необходимости активные соединения и одно или более из указанных выше вспомогательных веществ могут образовывать микрокапсулы.
Жидкие лекарственные формы для перорального введения содержат фармацевтически приемлемые эмульсии, растворы, суспензии или сиропы. В дополнение к активным соединениям указанные жидкие лекарственные формы могут содержать обычные инертные разбавители, известные в данной области техники, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид, а также масла, в частности, хлопковое масло, арахисовое масло, кукурузное масло, оливковое масло, касторовое масло и кунжутное масло или их комбинация.
Кроме этих инертных разбавителей, указанные композиции могут также содержать вспомогательные вещества, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластитель, вкусоароматические агенты и специи.
В дополнение к активным соединениям суспензия может содержать суспендирующие агенты, например, этоксилированный изооктадеканол, сложные эфиры полиоксиэтилена сорбитола и сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия и агар или их комбинации.
Композиции для парентерального введения при помощи инъекций могут включать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, которые можно повторно растворять в стерильных растворах для инъекций или дисперсиях. Подходящие водные и неводные носители, разбавители, растворители или вспомогательные вещества включают воду, этанол, полиолы и любые подходящие смеси указанных веществ.
Лекарственные формы для местного введения соединения согласно настоящему описанию включают мази, порошки, пластыри, спреи и ингаляторы. Активные ингредиенты смешивают с физиологически приемлемыми носителями и любыми консервантами, буферами или пропеллентом, если необходимо, в стерильных условиях.
Соединения согласно настоящему изобретению можно вводить отдельно или в комбинации с любым другим фармацевтически приемлемым соединением (соединениями).
При применении фармацевтических композиций безопасное и эффективное количество соединения согласно настоящему изобретению вводят млекопитающему (например, человеку), нуждающемуся в лечении, при этом доза для введения представляет собой фармацевтически эффективную дозу. Для человека весом 60 кг суточная доза обычно составляет от 1 до 2000 мг, предпочтительно от 5 до 500 мг. Естественно, конкретная доза также зависит от различных факторов, таких как способ введения, состояние здоровья субъекта, что находится в компетенции лечащего врача.
Основные преимущества настоящего изобретения включают:
1. Получение соединения формулы I.
2. Получение структурно нового ингибитора FGFR, состава и его применения, причем указанный ингибитор может ингибировать активность различных киназ FGFR даже тогда, когда ингибитор присутствует в очень низких концентрациях.
3. Получение фармацевтической композиции для лечения заболеваний, связанных с активностью киназы FGFR.
Настоящее изобретение далее будет проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что эти примеры приведены лишь в иллюстративных целях и не предназначены для ограничения объема настоящего изобретения. Экспериментальные способы, для которых в нижеследующих примерах не указаны конкретные условия, обычно осуществляют в обычных условиях или в соответствии с инструкциями изготовителя. Если не указано иное, приведенные части и проценты рассчитаны по массе.
Во всех примерах:
Прибор ЖХ-МС: Pump Agilent 1100. УФ детектор: Agilent 1100 DAD
Масс-спектрометр API 3000
Хроматографическая колонка: Waters sunfire CI8, 4,6×50 мм, 5 мкм
Мобильная фаза: А - ацетонитрил, В - H2O (0,1% FA)
Пример 1
Путь синтеза I
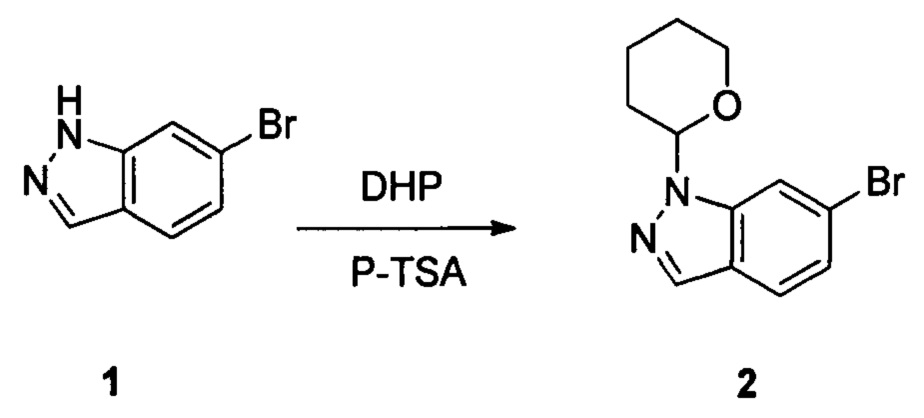
Соединение 1 (10,00 г, 51,0 ммоль), p-TSA (1,75 г, 10,2 ммоль) и дихлорметан (100,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл, по каплям медленно добавляли DHP (8,56 г, 102,0 ммоль), перемешивали при комнатной температуре в течение 4,0 ч. После завершения взаимодействия реакционный раствор разбавляли 100,0 мл воды и дважды экстрагировали дихлорметаном 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия и растворитель выпаривали в роторном испарителе с получением соединения 2 (8,90 г, 62%). ЖХ-МС: 281 (М+Н)+, время удержания = 1,626 минут.
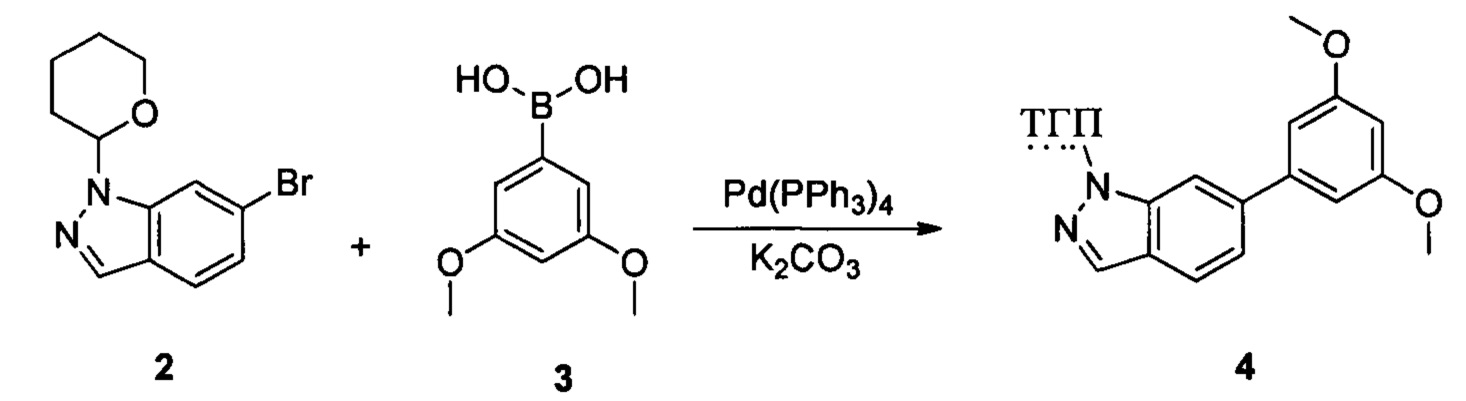
Соединение 2 (8,90 г, 31,7 ммоль), соединение 3 (5,77 г, 31,7 ммоль), Pd(PPh3)4 (3,66 г, 3,17 ммоль), K2CO3 (8,75 г, 63,4 ммоль), 1,4-диоксан (60,0 мл) и воду (15,0 мл) последовательно добавляли в сухую круглодонную колбу объемом 250 мл при комнатной температуре, перемешивали до достижения равномерного диспергирования в системе. Указанную реакционную смесь нагревали с обратным холодильником в течение 4,0 часов в защитной атмосфере азота. Реакционный раствор охлаждали до комнатной температуры и растворитель выпаривали в роторном испарителе с получением неочищенного продукта. Посредством колоночной хроматографии (этилацетат : петролейный эфир = 1:10) получали соединение 4 (8,10 г, 76%). ЖХ-МС: 339 (М+Н)+, время удержания = 1,626 минут.
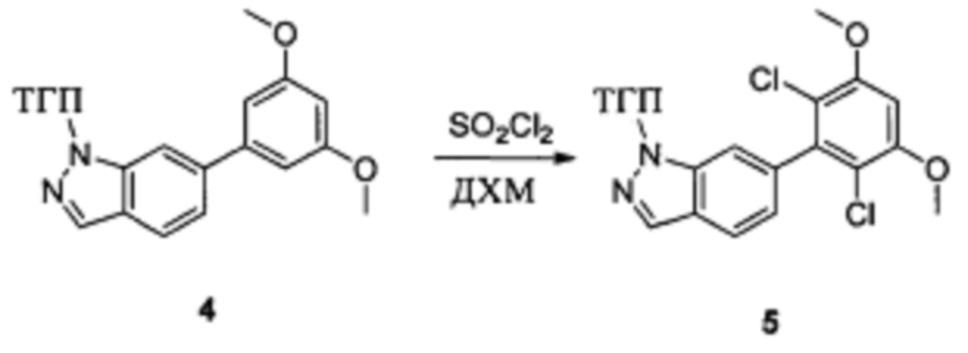
Соединение 4 (7,90 г, 23,4 ммоль) и дихлорметан (50,0 мл) добавляли в сухую 250 мл круглодонную колбу, по каплям медленно добавляли SO2Cl2 (7,15 г, 46,7 ммоль) и перемешивали при комнатной температуре в течение 4,0 часов. Реакционный раствор разбавляли 50,0 мл воды, дважды экстрагировали дихлорметаном 200 мл и промывали насыщенным раствором NaHCO3. Органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением соединения 5 (8,50 г, 89%). ЖХ-МС: 407 (М+Н)+, время удержания = 1,798 минут.
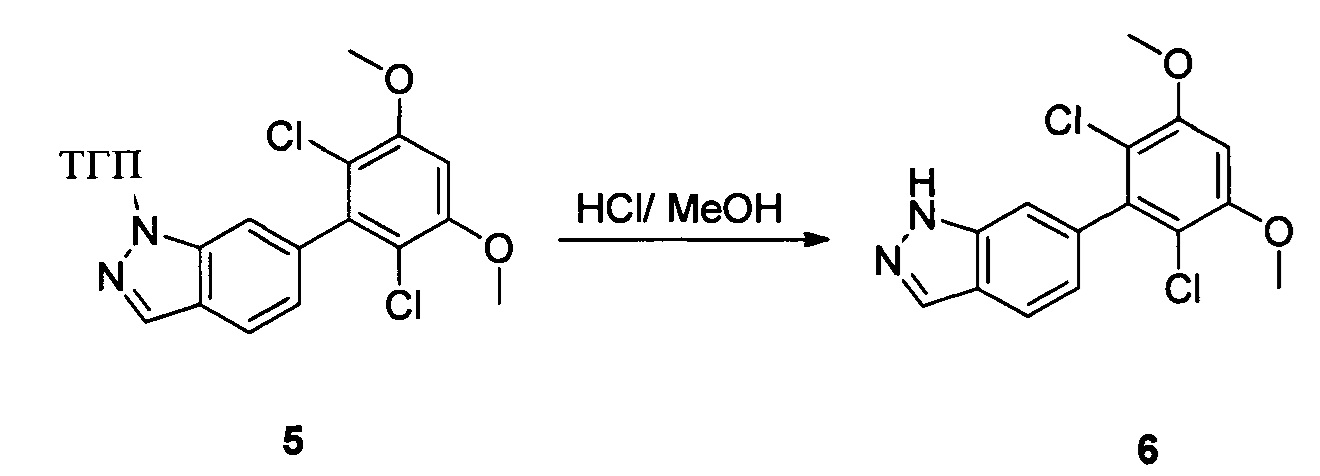
Соединение 5 (8,50 г, 20,9 ммоль) и раствор хлористоводородной кислоты в метаноле (1 М) (80,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл, нагревали с обратным холодильником в течение 16,0 часов. Растворитель выпаривали в роторном испарителе с получением 7,50 г соединения 6, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: 323 (М+Н)+, время удержания = 1,592 минут.
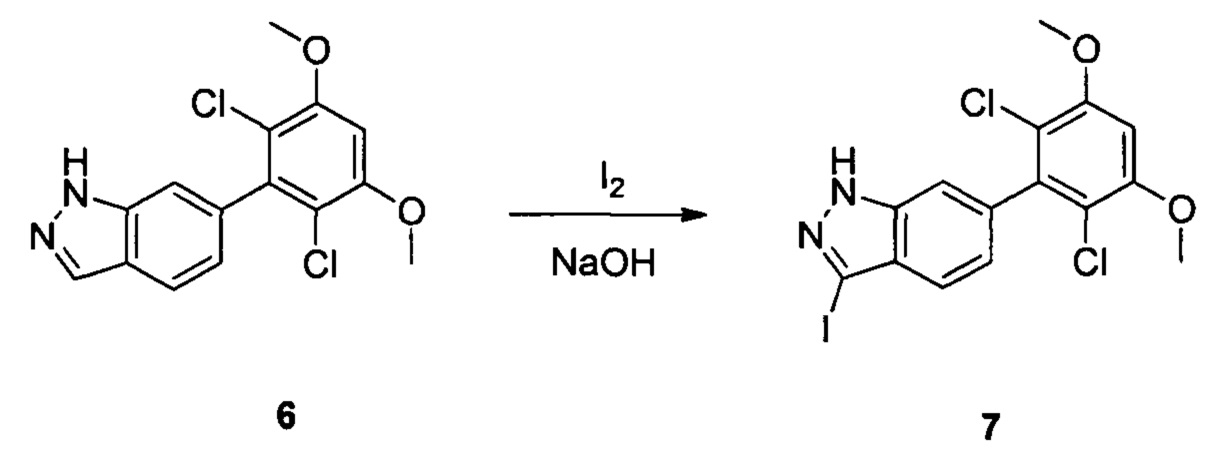
Соединение 6 (7,40 г, 23,0 ммоль), йод (11,68 г, 46,0 ммоль), NaOH (1,84 г, 46,0 ммоль) и 1,4-диоксан (60,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл. Смесь перемешивали при комнатной температуре в течение 2,0 часов. После завершения взаимодействия в реакционный раствор добавляли 200 мл воды и дважды экстрагировали дихлорметаном 200 мл. Органические фазы промывали насыщенным раствором тиосульфата натрия, объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением соединения 7 (9,50 г, 92%). ЖХ-МС: 449 (М+Н)+, время удержания = 1,644 минут.
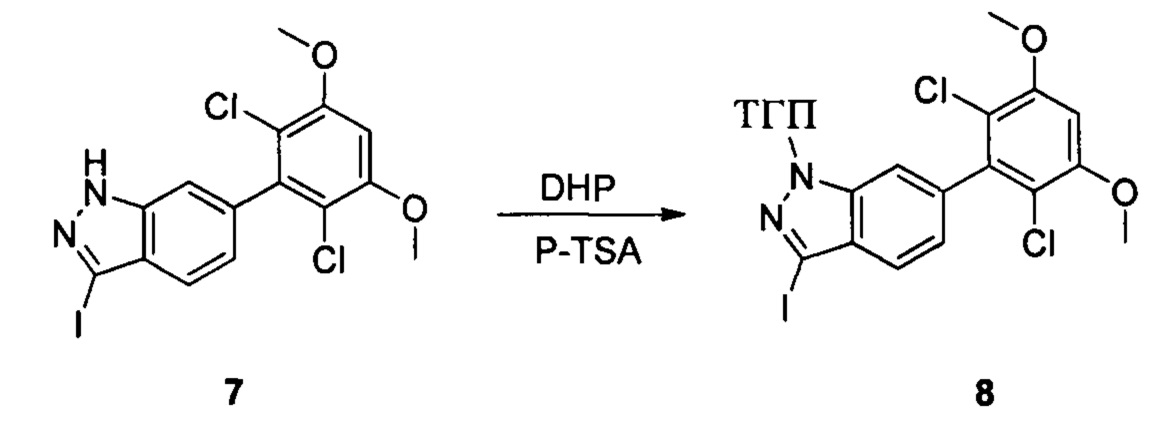
Соединение 7 (9,30 г, 20,76 ммоль), p-TSA (0,71 г, 4, 152 ммоль) и дихлорметан (50,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл, по каплям медленно добавляли DHP (3,48 г, 41,52 ммоль), перемешивали при комнатной температуре в течение 4,0 часов. Реакционный раствор разбавляли 50,0 мл воды, дважды экстрагировали дихлорметаном 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением соединения 8 (7,70 г, 70%). ЖХ-МС: 281 (М+Н)+, время удержания = 2,165 минут.
1Н ЯМР (CDCl3, 400 МГц) δ (ppm) 7,56 (d, 1Н, J=8,0 Гц), 7,45 (s, 1Н), 7,09 (d, 1H, J=8,0 Гц), 6,65 (s, 1Н), 5,70 (t, 1H, J=4,0 Гц), 4,03 (s, 1H), 3,98 (s, 6Н), 3,69 (t, 1H, J=8,0 Гц), 2,53 (t, 1Н, J=10 Гц), 2,12 (m, 2Н), 1,68-1,74 (m, 2Н), 1,56-1,63 (m, 1Н).
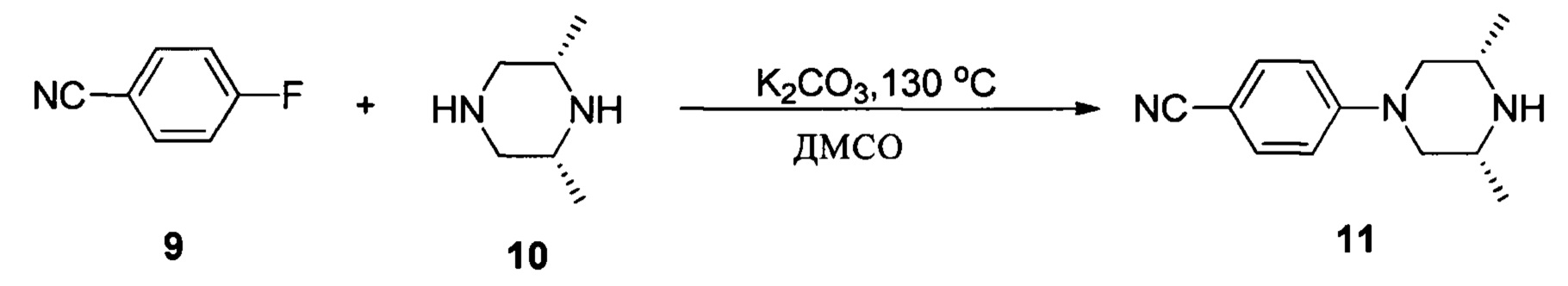
Соединение 9 (10 г, 82,6 ммоль) добавляли в одногорлую колбу объемом 500 мл, затем добавляли 200 мл ДМСО. При комнатной температуре добавляли (2R,6S)-2,6-диметилпиперазин (14 г, 124 ммоль) и K2CO3 (28,5 г, 206,5 ммоль) и равномерно перемешивали. Реакционную смесь нагревали до температуры 130°С и проводили взаимодействие в течение 8 часов. После завершения указанного взаимодействия смесь выливали в 1 л воды и трижды экстрагировали этилацетатом (150 мл * 3). Органические фазы промывали насыщенным солевым раствором 100 мл, сушили над безводным сульфатом натрия и растворитель выпаривали в роторном испарителе с получением 15 г желтоватого твердого вещества, выход составил 85%.
1Н ЯМР (400 МГц, CDCl3) δ (ppm) 7,48 (2Н, d, J=8,8 Гц), 6,85 (2Н, d, J=8,0 Гц), 3,65 (2Н, dd, J1=12,0 Гц, J2=2,0 Гц), 2,95-3,01 (2Н, m), 2,42 (2Н, t, J=11,4 Гц), 1,15 (6Н, d, J=6,4 Гц).
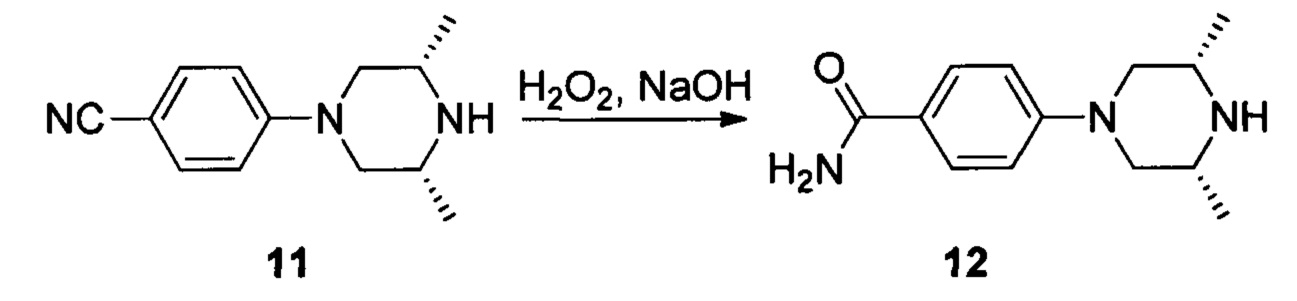
Соединение 11 (9,03 г, 42 ммоль) растворяли в 175 мл этанола, затем к раствору при комнатной температуре последовательно добавляли NaOH (6,0 N, 105 мл) и Н2О2 (16,1 мл). Смесь нагревали до температуры 50°С и перемешивали в течение 5 часов. После завершения указанного взаимодействия раствор охлаждали до 0°С и доводили до рН 7 с помощью 3N серной кислоты. Органическую фазу выпаривали в роторном испарителе, перемешивали при 0°С в течение 30 минут и фильтровали с получением 6,5 г белого твердого вещества, выход составил 66%.
1Н ЯМР (400 МГц, ДМСО-d6): δ (ppm) 7,76 (2Н, d, J=8 8 Гц), 7,73 (1Н, br), 7,04 (1H, br), 6,97 (2H, d, J=8,8 Гц), 3,85 (2H, d, J=11,2 Гц), 3,08 (2H, br), 2,45 (2H, t, J=11,6 Гц), 1,15 (6H, d, J=6,4 Гц).
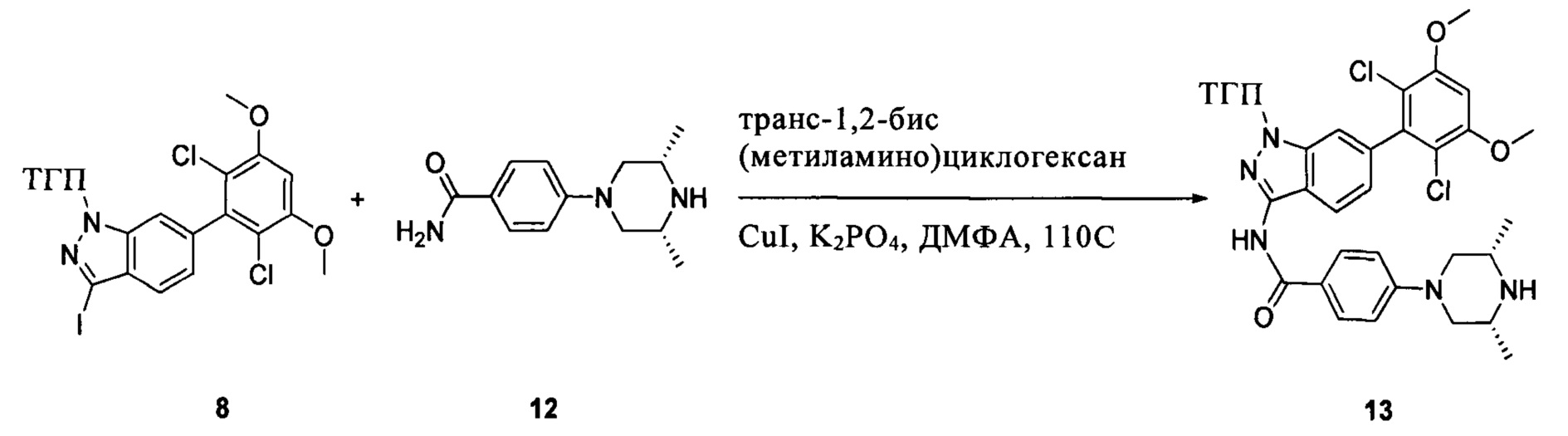
Соединение 8 (2,0 г, 3,75 ммоль) растворяли в 20 мл безводного ДМФА, затем к раствору при комнатной температуре последовательно добавляли транс-N,N'-диметил-1,2-циклогександиамин (107 мг, 0,75 ммоль), CuI (36 мг, 0,19 ммоль), K3PO4 (1,6 г, 7,5 ммоль) и соединение 12 (1,05 г, 4,5 ммоль). Указанный раствор три раза продували азотом, нагревали до 110°С и перемешивали в течение 16 часов. После завершения указанного взаимодействия растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (дихлорметан: метанол = 40:1) с получением белого твердого вещества (0,98 г), выход составил 41%.
1Н ЯМР (CDCl3, 400 МГц) δ (ppm) 8,46 (s, 1Н), 8,22 (d, 1Н, J=8,0 Гц), 7,86 (d, 2Н, J=8,0 Гц), 7,35 (s, 1Н), 7,01 (d, 1Н, J=8,0 Гц), 6,94 (d, 2Н, J=8,0 Гц), 6,62 (s, 1Н), 5,61 (m, 1Н), 3,96 (s, 6Н), 3,71 (m, 3Н), 3,06 (m, 2Н), 2,48 (m, 3Н), 2,06 (m, 3Н), 1,70 (m, 3Н), 1,20 (s, 3Н), 1,18 (s, 3Н).
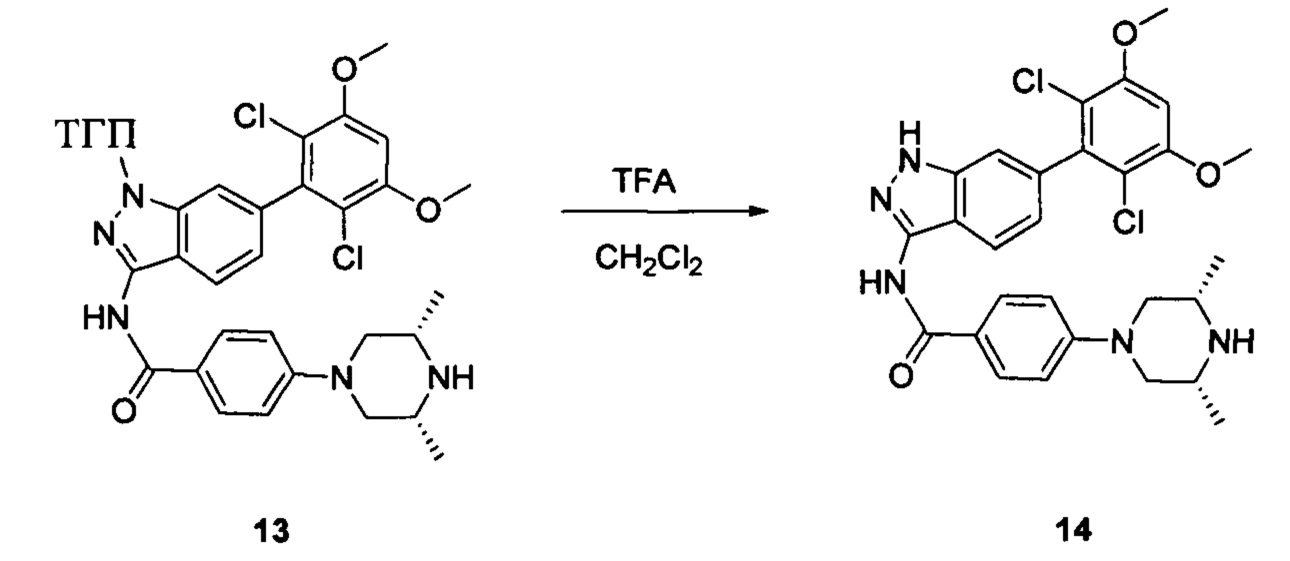
Соединение 13 (0,86 г, 1,35 ммоль) растворяли в 10 мл дихлорметана и при комнатной температуре к раствору добавляли трифторуксусную кислоту (5 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. После завершения указанного взаимодействия растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (дихлорметан : метанол = 20:1) с получением белого твердого вещества (0,61 г), выход составил 82%.
1Н ЯМR (CDCl3, 400 МГц) δ (ppm) 8,79 (s, 1Н), 8,15 (d, 1H, J=8,0 Гц), 7,86 (d, 2H, J=8,0 Гц), 7,26 (s, 1H), 6,99 (d, 1H, J=8,0 Гц), 6,89 (d, 2H, J=8,0 Гц), 6,62 (s, 1H), 3,96 (s, 6H), 3,65 (m, 2H), 3,06 (m, 2H), 1,17 (s, 3H), 1,15 (s, 3H).
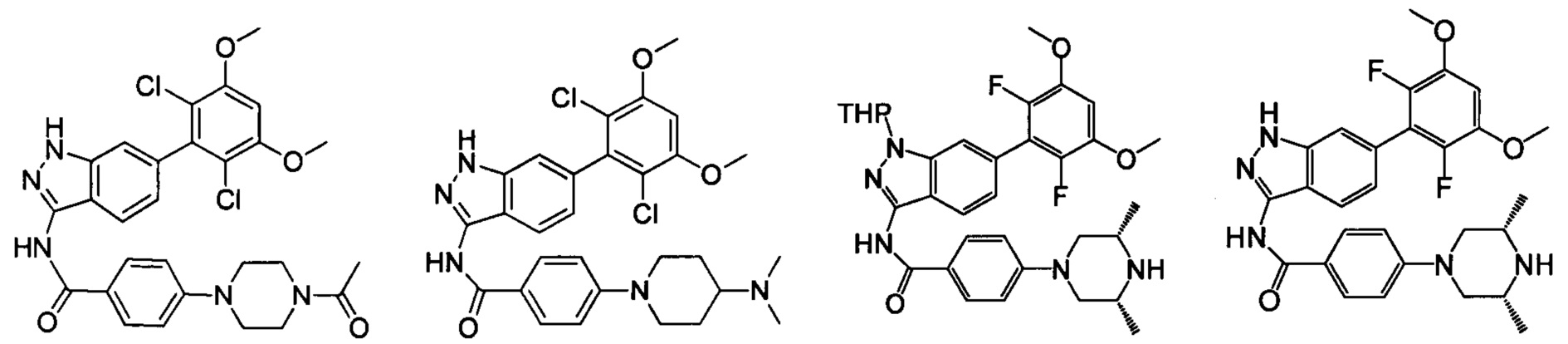
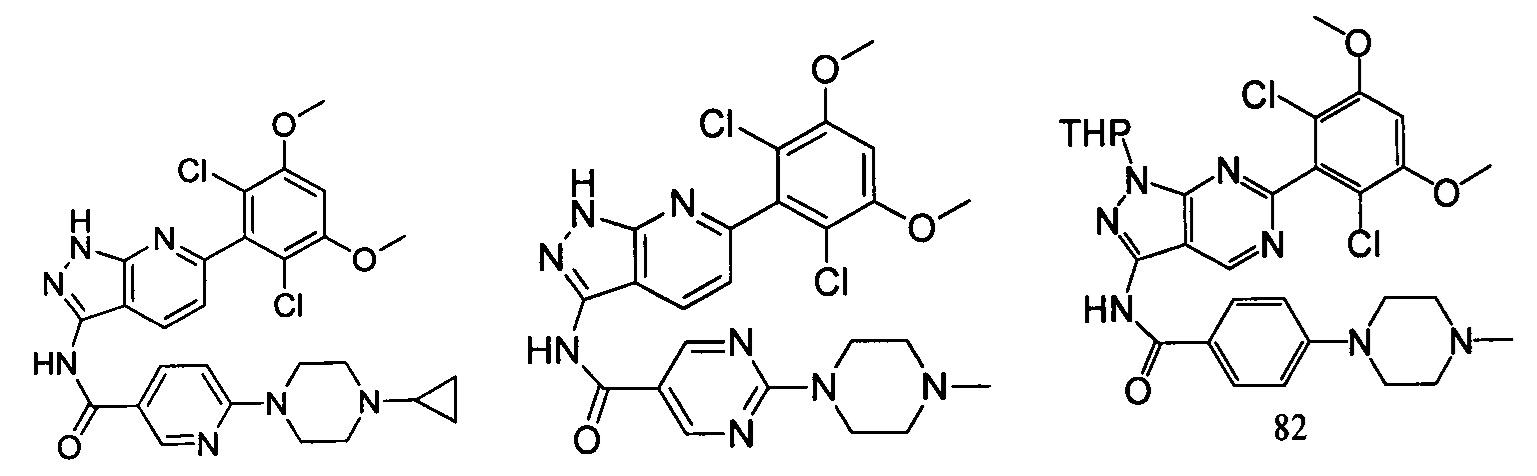
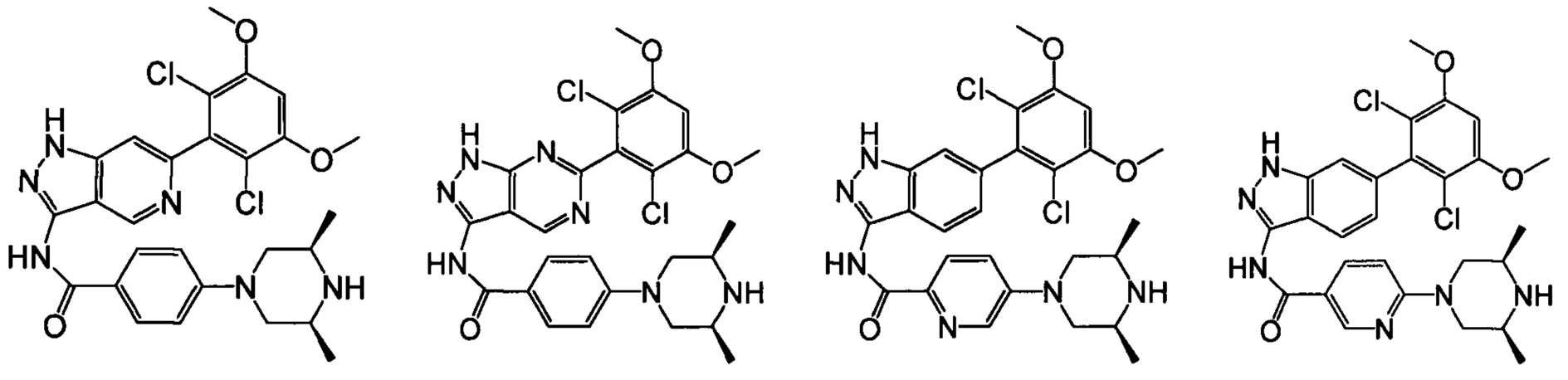
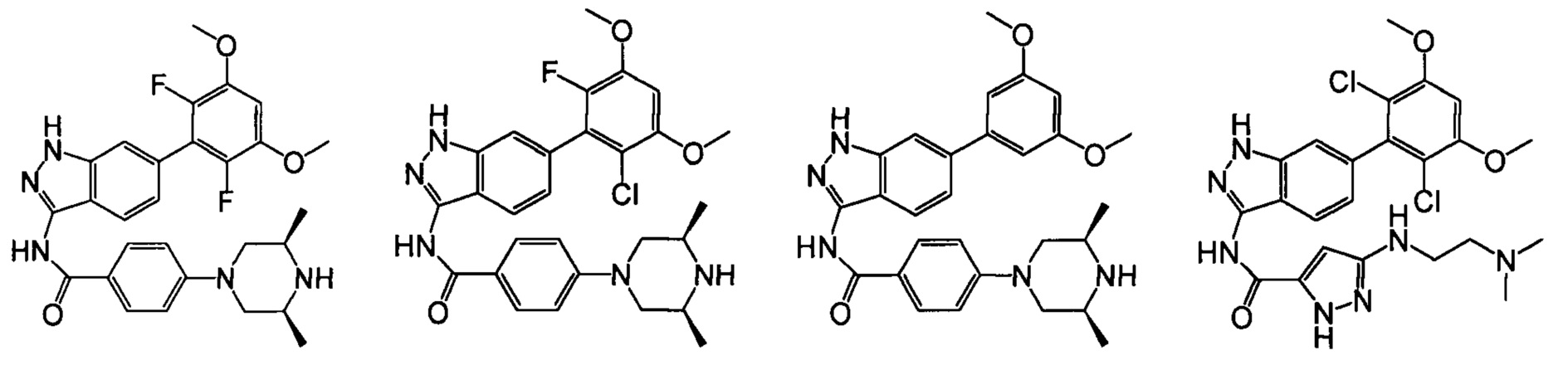
Аналогичными способами были получены следующие соединения:
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-метилпиперазин-1-ил)бензамид
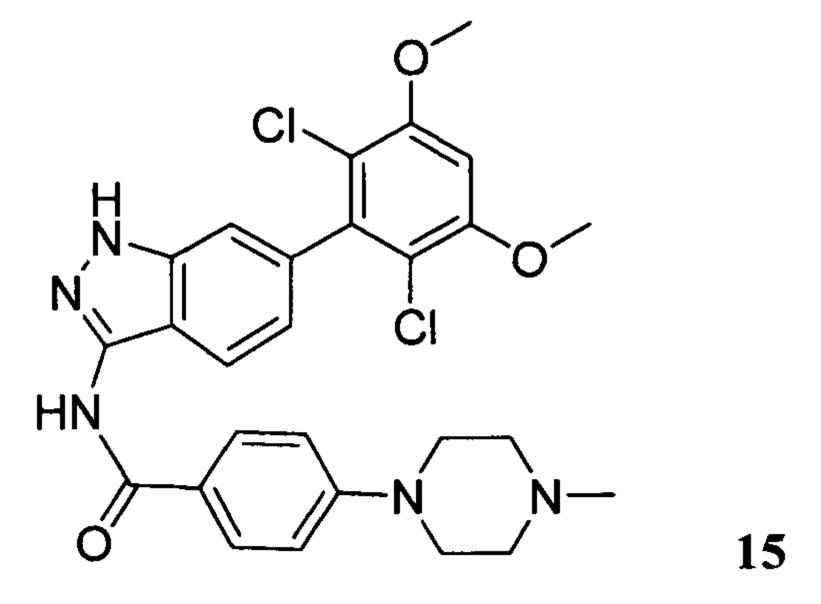
1Н ЯМР (CDCl3, 400 МГц) δ (ppm) 8,83 (s, 1Н), 8,21 (d, 1H, J=8,0 Гц), 7,89 (d, 2H, J=8,0 Гц), 7,23 (s, 1H), 6,99 (d, 1H, J=8,0 Гц), 6,91 (d, 2H, J=8,0 Гц), 6,61 (s, 1H), 3,95 (s, 6H), 3,33 (m, 4H), 2,56 (m, 4H), 2,35 (s, 3H).
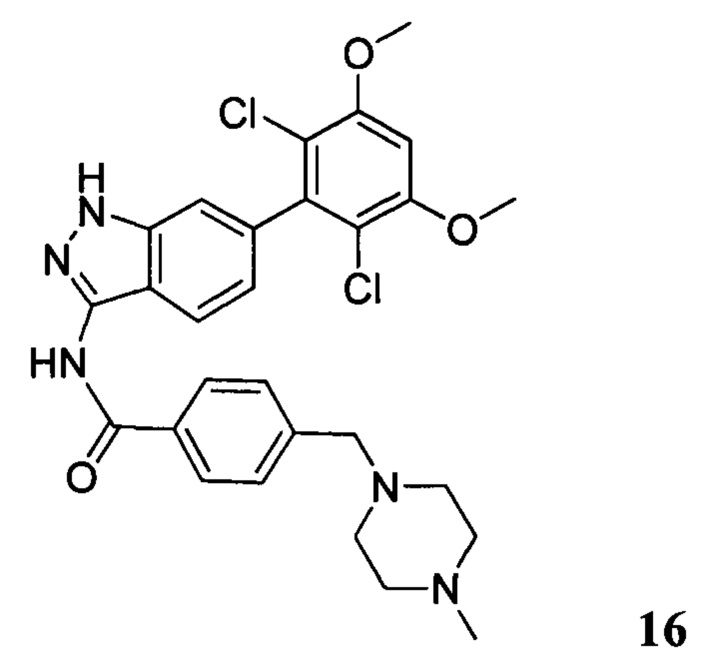
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1H-индазол-3-ил)-4-((4-метилпиперазин-l-ил)метил)бензамид
1H ЯМР (MeOD, 400 МГц) δ (ppm) 8,03 (d, 2H, J=8,0 Гц), 7,84 (d, 1H, J=8,0 Гц), 7,53 (d, 2H, J=8,0 Гц), 7,29 (s, 1H), 6,93 (d, 1H, J=8,0 Гц), 6,87 (s, 1H), 3,97 (s, 6H), 3,68 (s, 2H), 2,98 (br, 4H), 2,56 (br, 4H), 2,54 (s, 3H).
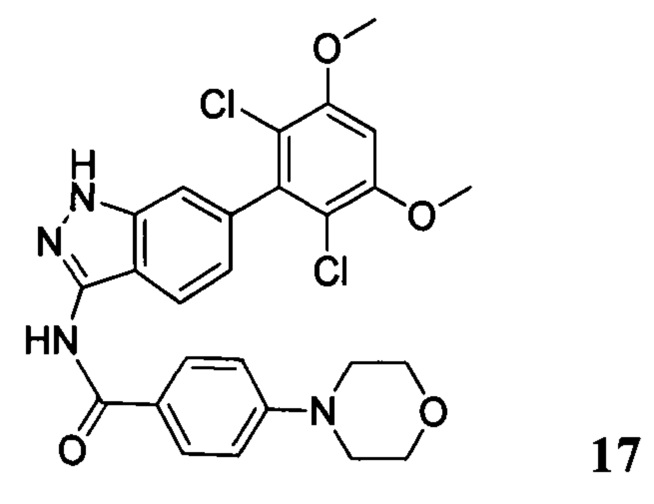
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-морфолинобензамид
1H ЯМР (MeOD, 400 МГц) δ (ppm) 8,02 (d, 2Н, J - 8,0 Гц), 7,97 (d, 1Н, J=8,0 Гц), 7,32 (s, 1H), 7,07 (d, 2H, J=8,0 Гц), 7,99 (d, 1H, J=8,0 Гц), 6,88 (s, 1H), 3,98 (s, 6H), 3,84 (t, 4H, J=5,2 Гц), 3,33 (t, 4H, J - 5,2 Гц).
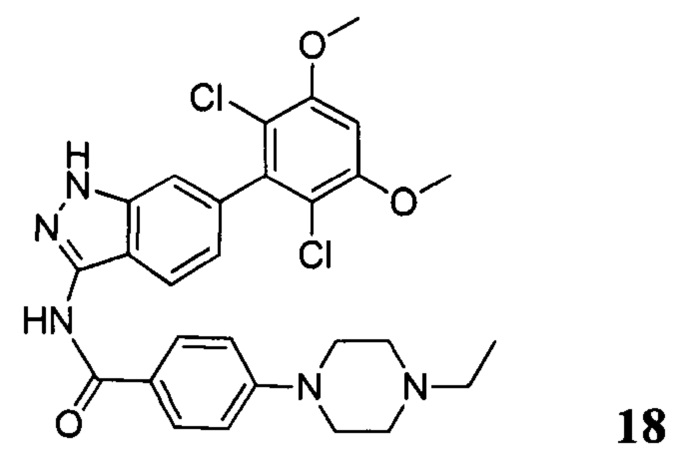
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-этилпиперазин-1-ил)бензамид
1H ЯМР (MeOD, 400 МГц) δ (ppm) 8,09 (m, 3Н), 7,36 (s, 1H), 7,20 (m, 2H), 7,99 (m, 1H), 6,89 (s, 1H), 3,97 (s, 6H), 3,27 (m, 2H), 1,4 (t, 3H, J=6,4 Гц).
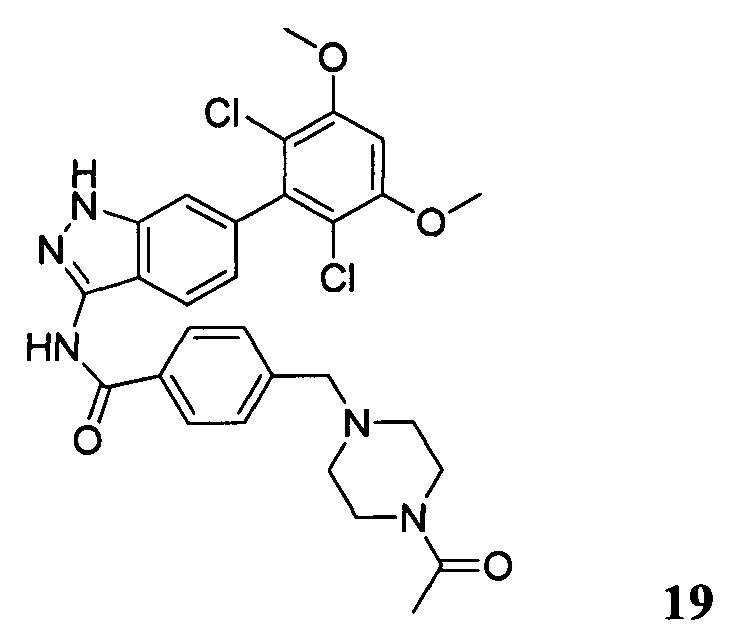
4-((4-ацетил-1-ил)метил)-N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)бензамид
1Н ЯМР (ДМСО-d6, 400 МГц) δ (ppm) 12,88 (s, 1Н), 10,81 (s, 1H), 8,06 (d, 2H, J=8,0 Гц), 7,77 (d, 1H, J=8,0 Гц), 7,78 (d, 2H, J=8,0 Гц), 7,29 (s, 1H), 7,00 (s, 1H), 6,85 (d, 1H, J=8,0 Гц), 3,97 (s, 6H), 3,59 (s, 2H), 2,40 (t, 2H, J=4,8 Гц), 2,33 (t, 2H, J=4,8 Гц), 1,98 (s, 3H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-((2-(диметиламино)этил)амино)бензамид
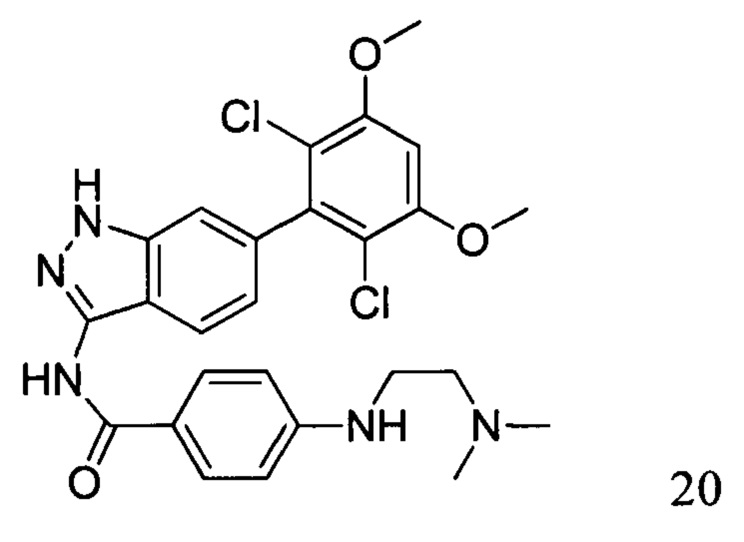
1H ЯМР (400 МГц, ДМСО-d6/D2O) δ (ppm) 7,87 (2Н, s), 7,71 (1H, s), 7,29 (1Н, s), 7,10-6,60 (4Н, m), 3,91 (6Н, s), 3,70 (2Н, s), 3,48 (2Н, s), 2,80 (6Н, s).
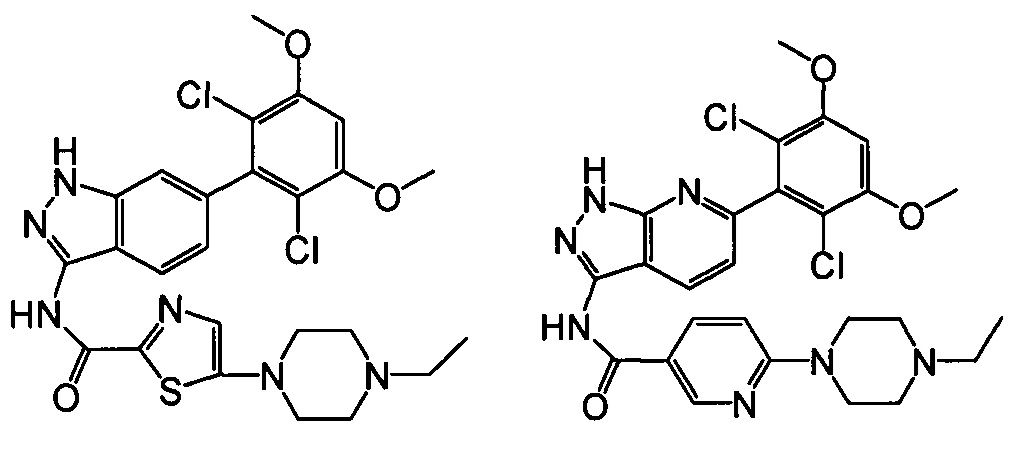
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-5-((3R,5S)-3,5-диметил-1-ил)пиридинамид
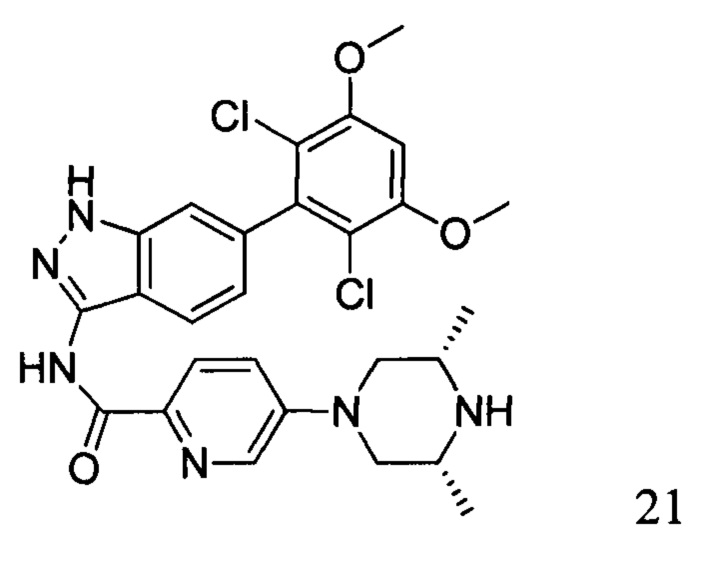
1Н ЯМР (ДМСО-d6, 400 МГц) δ (ppm) 12,89 (1H, br), 10,48 (1Н, s), 9,06 (1Н, br), 8,51 (2Н, br), 8,05 (1Н, d, J=8,8 Гц), 7,94 (1H, d, J=8,4 Гц), 7,60-7,63 (1H, m), 7,29 (1Н, s), 7,01 (1Н, s), 4,22 (2Н, d, J=14,4 Гц), 3,97 (6Н, s), 2,84 (2Н, t, J=12,6 Гц), 2,54-2,58 (2Н, m), 1,30 (6Н, d, J=6,4 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(3,3-диметил-1-ил)бензамид
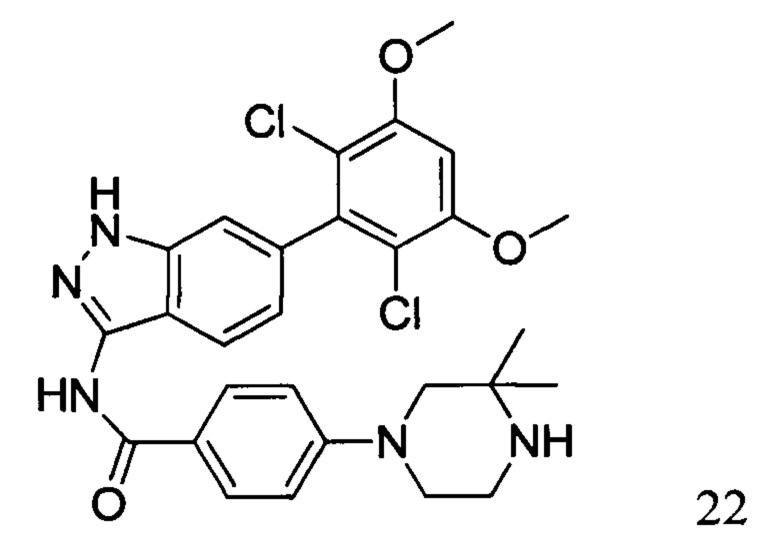
1Н ЯМР (400 МГц, ДМСО-d6) δ (ppm) 12,85 (s, 1Н), 10,57 (s, 1H), 8,01 (2H, d, J=8,8 Гц), 7,76 (1H, d, J=8,4 Гц), 7,29 (s, 1H), 7,07 (2H, d, J=8,8 Гц), 7,01 (1H, s), 6,85 (d, 1H, J=8,4 Гц), 3,98 (s, 6H), 3,18-3,50 (m, 6H), 1,31 (s, 6H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-3-(4-метил-1-ил)бензамид
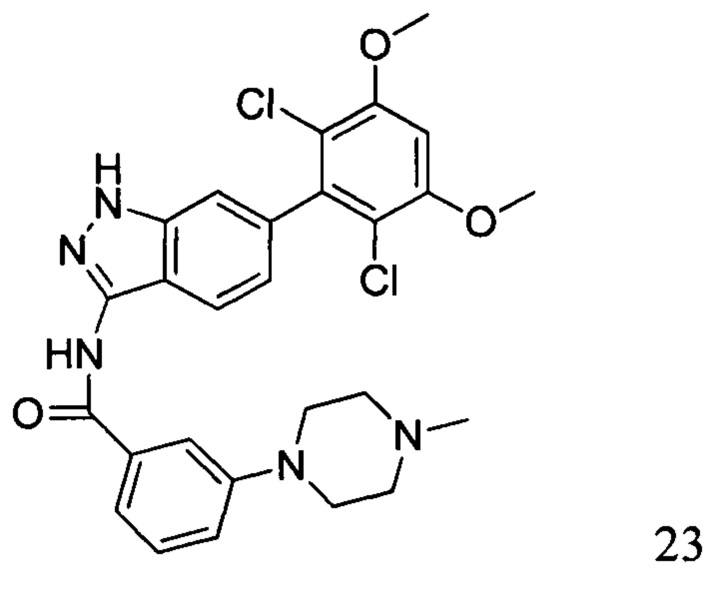
1H ЯМР (400 МГц, ДМСО-d6) δ (ppm) 12,92 (1H, s), 10,83 (1H, s), 9,60-9,20 (1H, m), 7,78 (1H, d, J=8,4 Гц), 7,68 (1H, s), 7,58 (1H, d, J=7,6 Гц), 7,44 (1H, t, J=8,0 Гц), 7,30 (1H, s), 7,26 (1H, dd, J1=1,6 Гц, J2=8,0 Гц), 7,00 (1H, s), 6,87 (1H, d, J=8,4 Гц), 3,97 (6H, s), 3,30-3,10 (4H, m), 3,10-2,80 (4H, m), 2,88 (3H, s).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-изопропил-1-ил)бензамид
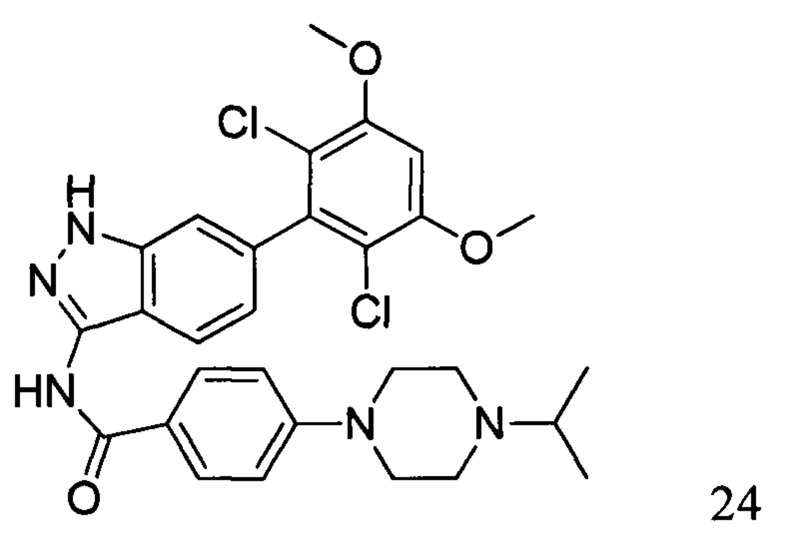
1H ЯМР (MeOD, 400 МГц) δ (ppm) 8,08 (br, 2H), 7,99 (br, 1H), 7,33 (br, 1H), 7,18 (br, 2H), 7,02 (br, 1H), 6,89 (s, 1H), 4,11-4,17 (m, 2H), 3,57-3,64 (m, 3H), 3,19-3,37 (m, 4H), 1,42 (6H, d, J=6,4 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(пиперазин-1-ил)бензамид
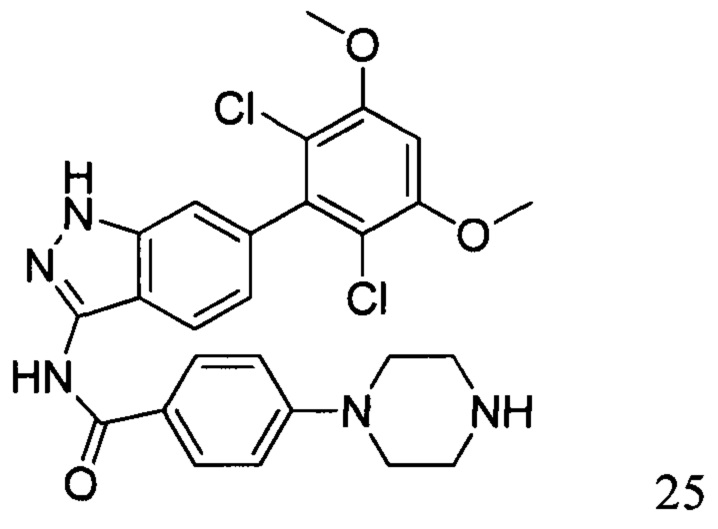
1Н ЯМР (MeOD, 400 МГц) δ (ppm) 8,00 (2H, d, J=9,2 Гц), 7,83 (1H, d, J=8,4 Гц), 7,28 (s, 1H), 7,10 (2H, d, J=9,2 Гц), 6,90 (1H, d, J=8,4 Гц), 6,88 (s, 1H), 3,97 (s, 6H), 3,48-3,51 (m, 4H), 3,18-3,25 (m, 4H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-((3-(диметиламино)пропил)амино)бензамид
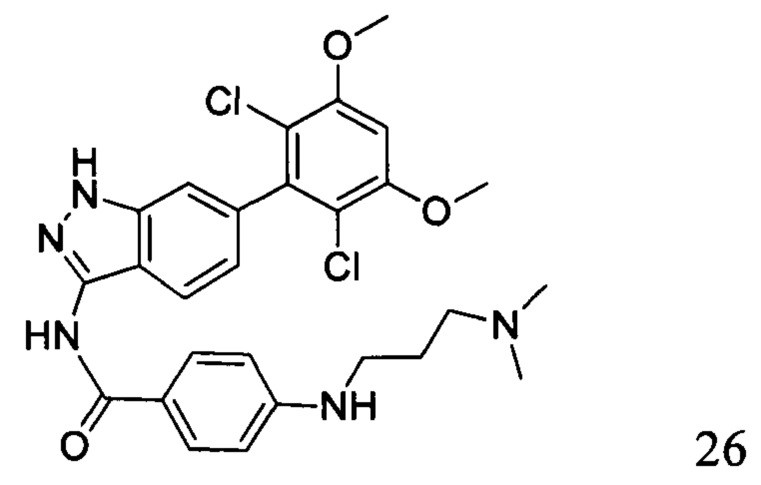
1H ЯМР (400 МГц, ДМСО-d6) δ (ppm) 12,87 (1H, br), 10,39 (s, 1H), 9,57 (1H, br), 7,80 (2H, d, J=8,8 Гц), 7,75 (1H, d, J=8,4 Гц), 7,27 (s, 1H), 6,84 (1H, d, J=8,4 Гц), 6,66 (2H, d, J=8,8 Гц), 3,98 (6H, s), 3,20 (4H, t, J=6,8 Гц), 2,80 (s, 6H), 1,89-1,96 (m, 2H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-(2-гидроксиэтил)пиперазинил)бензамид
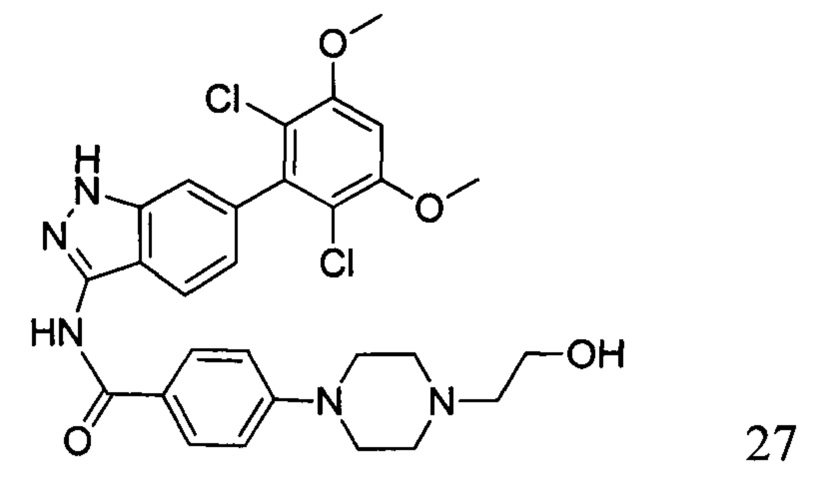
1H ЯМР (400 МГц, ДМСО-d6) δ (ppm) 12,85 (1H, br), 10,62 (1H, s), 9,73 (1H, br), 8,05 (2H, d, J=8,8 Гц), 7,76 (1H, d, J=8,4 Гц), 7,29 (1H, s), 7,13 (2H, d, J=8,8 Гц), 6,87 (1H, s), 6,86 (1H, d, J=8,4 Гц), 4,06-4,08 (m, 2H), 3,98 (6H, s), 3,80 (2H, t, J=4,8 Гц), 3,56-3,60 (m, 2H), 3,14-3,28 (m, 6H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(2-(диметиламино)ацетиламино)бензамид
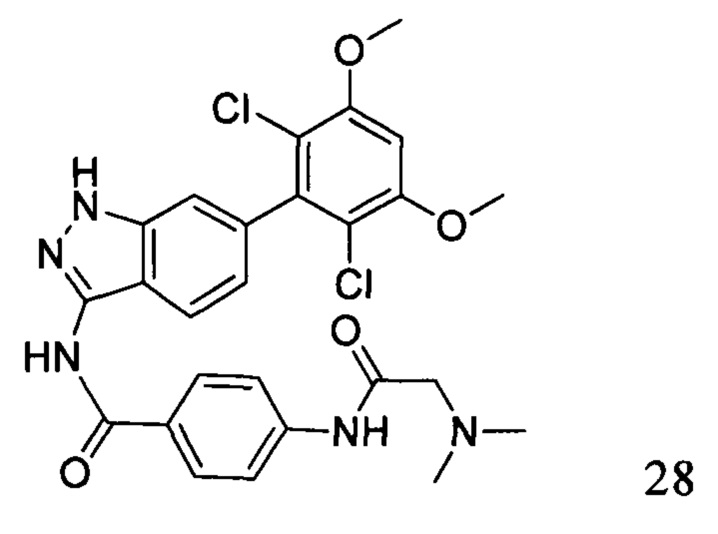
1H ЯМР (400 МГц, ДМСО-d6) δ (ppm) 12,92 (1H, s), 10,78 (1Н, s), 10,08 (1H, s), 8,12 (2Н, d, J=8,8 Гц), 7,89 (2Н, d, J=8,8 Гц), 7,83 (1Н, d, J=8,4 Гц), 7,34 (1Н, s), 7,06 (1Н, s), 6,91 (1Н, d, J=8,4 Гц), 4,03 (6Н, s), 3,18 (s, 2Н), 2,35 (s, 6Н).
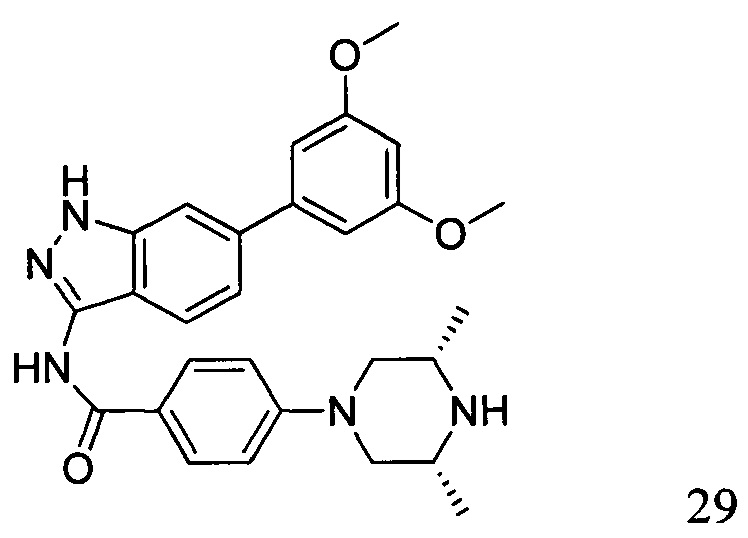
N-(6-(3,5-диметоксифенил)-1Н-индазол-3-ил)-4-((3R,5S)-3,5-диметил пиперазин-1-ил)бензамид
1Н ЯМР (d6- ДМСО, 400 МГц) δ ppm 12,78 (s, 1Н), 10,50 (s, 1H), 7,97 (d, 2H, J=9,2 Гц), 7,76 (d, 1H, J=8,4 Гц), 7,66 (s, 1H), 7,36 (d, 1H, J=8,4 Гц), 7,01 (d, 2H, J=8,8 Гц), 6,84 (d, 2H, J=2,0 Гц), 6,53 (s, 1H), 3,83 (s, 6H), 3,76 (d, 2H, J=6,8 Гц), 2,83 (s, 2H), 2,24 (t, 3H, J=6,8 Гц), 1,23 (s, 4H), 1,04 (d, 6H, J=6,4 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-пивалоилпиперазин-1-ил)бензамид
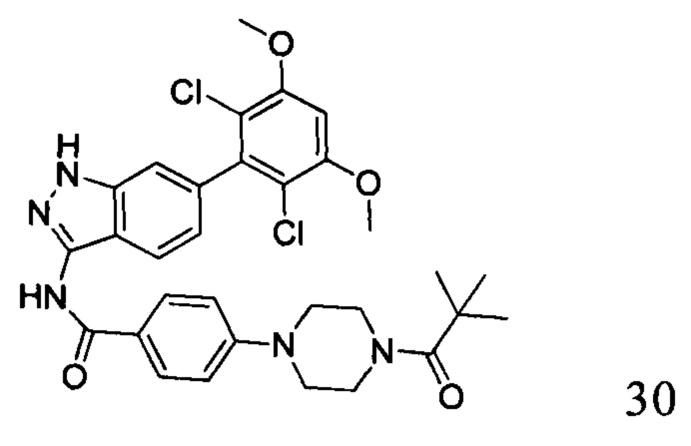
1H ЯМР (d6- ДМСО, 400 МГц) δ ppm 12,81 (s, 1H), 10,6 (s, 1H), 8,01 (d, 2H, J=8,8 Гц), 7,75 (d, 1H, J=4,4 Гц), 7,27 (s, 1H), 7,03 (d, 2H, J=8,8 Гц), 7,00 (s, 1H), 6,84 (d, 1H, J=8,8 Гц), 4,00 (s, 6H), 3,70-3,72 (m, 4H), 3,30-3,32 (m, 4H), 1,23 (s, 9H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-1-(2-гидроксиэтил)-1Н-пиразол-4-формамид
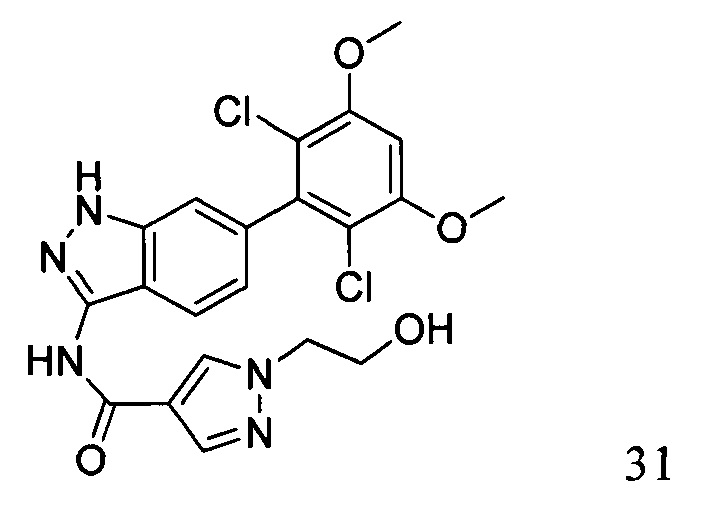
1H ЯМР (d6- ДМСО, 400 МГц) δ ppm 12,84 (brs, 1Н), 10,53 (s, 1H), 8,41 (s, 1H), 8,11 (s, 1H), 7,80 (d, 1H, J=8,4 Гц), 7,26 (s, 1H), 7,01 (d, 1H, J=3,2 Гц), 6,84 (d, 1H, J=8,4 Гц), 4,21 (t, 2H, J=5,2 Гц), 3,94 (s, 6H), 3,77 (t, 2H, J=5,2 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-2,3-дигидроимидазо[5,1-В]оксазол-7-формамид
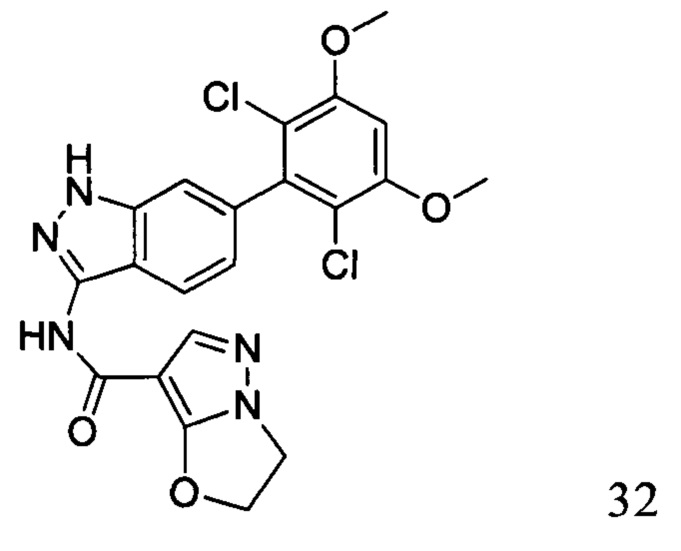
1H ЯМР (d6- ДМСО, 400 МГц) δ ppm 12,80 (brs, 1Н), 10,18 (s, 1Н), 8,07 (s, 1H), 7,77 (d, 1H, J=8,0 Гц), 7,25 (s, 1H), 7,00 (s, 1H), 6,83 (dd, 1H, J1=0,8 Гц, J2=8,4 Гц), 5,23 (t, 2H, J=7,6 Гц), 4,33 (t, 2Н, J=8,0 Гц), 3,97 (s, 6Н).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(3-(диметиламино)-3-оксопропил)бензамид
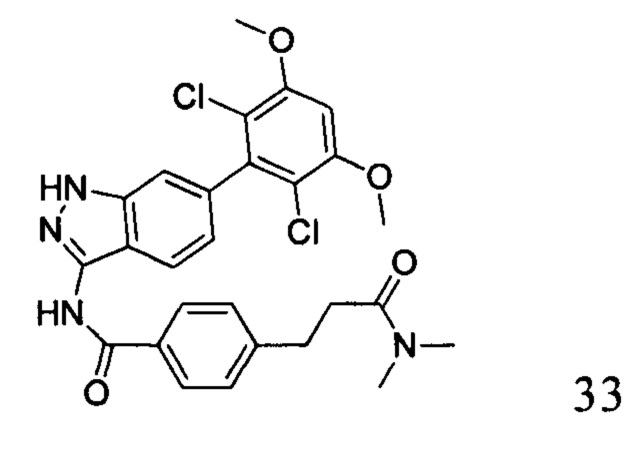
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,90 (s, 1Н), 10,78 (s, 1H), 8,00 (d, 2H, J=8,4 Гц), 7,77 (d, 1H, J=8,4 Гц), 7,42 (d, 2H, J=8,0 Гц), 7,03 (s, 1H), 7,01 (d, 1H), 6,86 (d, 1H, J=8,4 Гц), 3,98 (s, 6H), 2,91 (t, 2H, J=14,4 Гц), 2,84 (s, 3H), 2,67 (t, 2H, J=14,4 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(3-(диметиламино)метил)бензамид
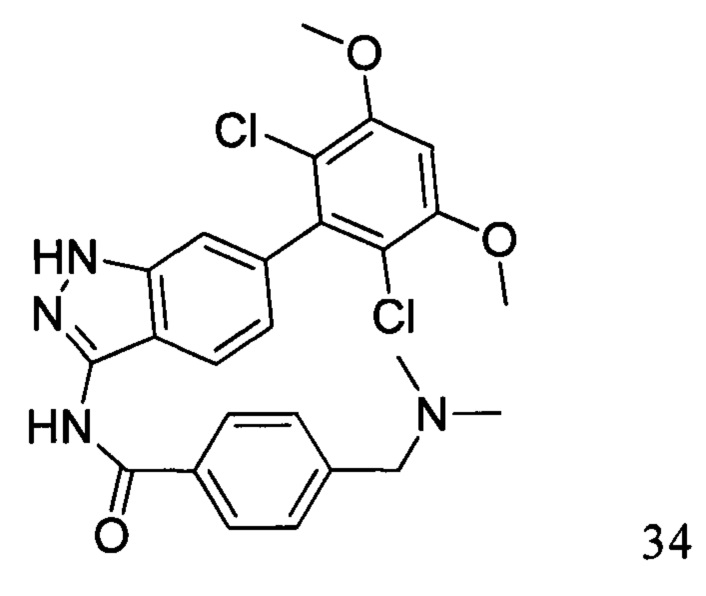
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,95 (s, 1H), 10,97 (s, 1H), 9,85 (d, 1H, J=2,8 Гц), 8,17 (d, 2H, J=8,0 Гц), 7,78 (d, 1H, J=8,0 Гц), 7,66 (d, 2H, J=8,4 Гц), 7,31 (s, 1H), 7,00 (s, 1H), 6,87 (d, 1H, J=8,4 Гц), 4,39 (d, 2H, J=4,4 Гц), 3,97 (s, 6H), 2,79 (t, 6H, J=3,6 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(диметиламино)бензамид
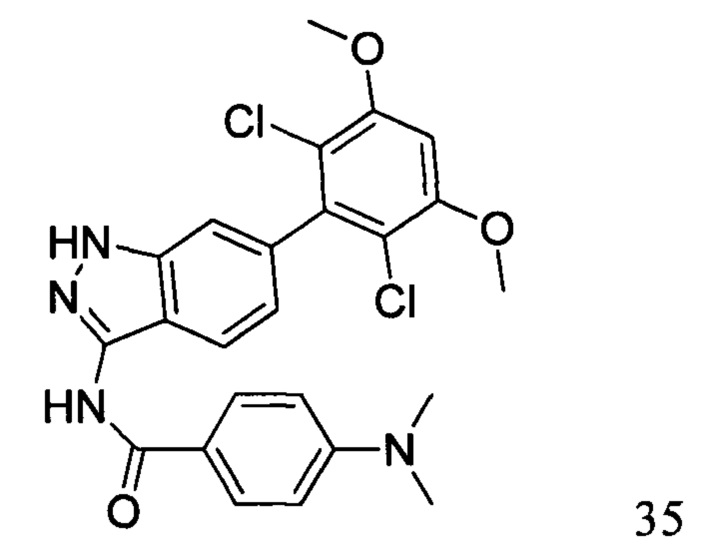
1Н ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,82 (s, 1H), 10,47 (s, 1H), 7,98 (d, 2H, J=6,4 Гц), 7,76 (d, 1H, J=8,0 Гц), 7,28 (s, 1H), 7,03 (s, 1H), 7,01 (s, 1H), 6,84 (d, 1H, J=8,4 Гц), 6,78 (d, 2H, J=6,8 Гц), 3,98 (s, 6H), 3,18 (s, 6H).
4-(4-ацетилпиперазин-1-ил)-N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)бензамид
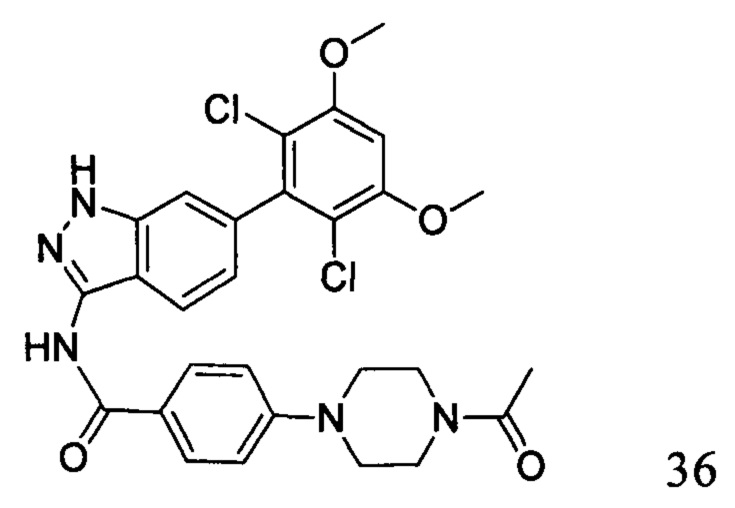
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,89 (s, 1H), 10,62 (s, 1H), 8,06 (d, 2H, J=8,8 Гц), 7,82 (d, 1H, J=8,4 Гц), 7,34 (s, 1H), 7,10 (d, 2H, J=8,8 Гц), 7,06 (s, 1H), 6,84 (d, 1H, J=8,4 Гц), 4,03 (s, 6H), 3,65-3,66 (m, 4H), 3,38-3,43 (m, 4H), 2,10 (s, 3H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-(диметиламино)пиперидин-1-ил)бензамид
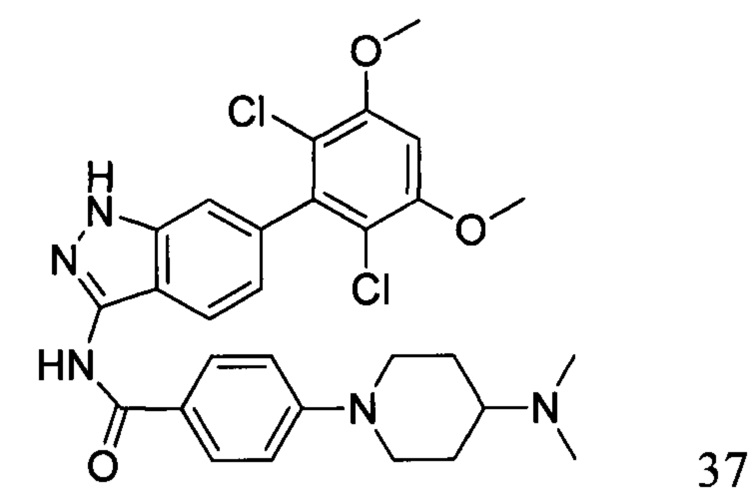
1H ЯМР (CDCl3, 400 МГц) δ ppm 8,48 (s, 1Н), 8,23 (d, 1H, J=8 Гц), 7,88 (d, 2H, J=8,8 Гц), 7,02 (d, 1H, J=8,8 Гц), 6,79 (d, 2H, J=8,4 Гц), 6,63 (s, 1H), 3,97 (s, 6H), 3,93 (d, 2H, J=14 Гц), 2,84-2,87 (m, 2H), 2,33 (m, 7H), 1,95 (d, 4H, J=12 Гц). ЖХ-МС: 568 (M+H)+, время удержания = 1,25 минут
Путь синтеза II
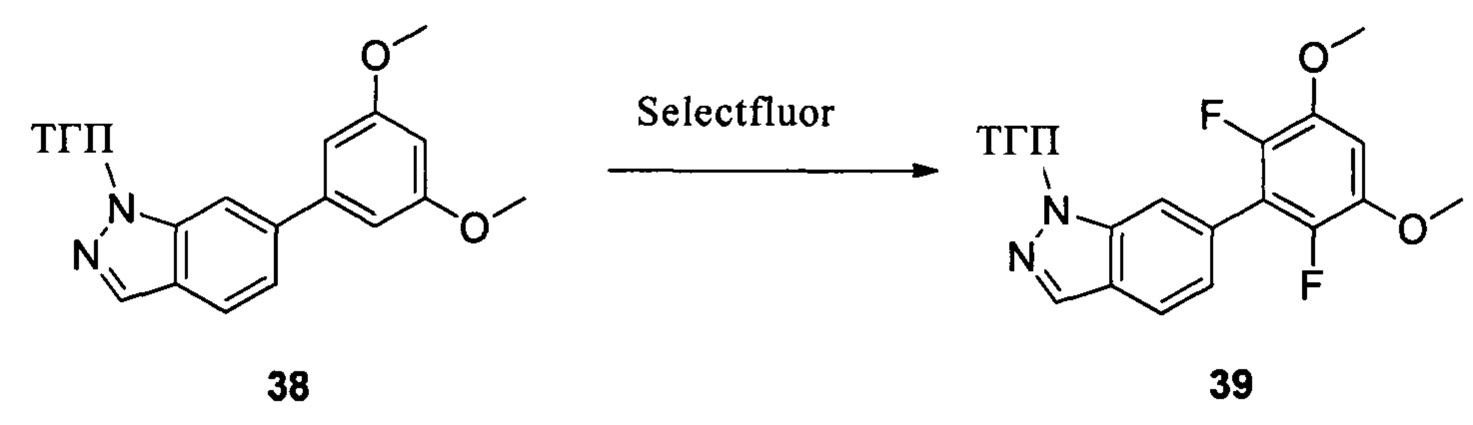
Соединение 38 (1,20 г, 3,55 ммоль) и ацетонитрил (20,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл. Порциями добавляли реагент Selectfluor (2,51 г, 7,1 ммоль) в защитной атмосфере азота при 0°С и перемешивали при комнатной температуре в течение 18,0 часов. Реакционный раствор разбавляли водой и экстрагировали этилацетатом. Органическую фазу последовательно промывали водой, насыщенным раствором NaHCO и насыщенным солевым раствором. Сушили над безводным сульфатом натрия и растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который подвергали колоночной хроматографии (этилацетат : петролейный эфир = 1:8) с получением неочищенного соединения 39 (629 мг, 47%). ЖХ-МС: 374,9(М+Н)+, время удержания = 1,243 мин.
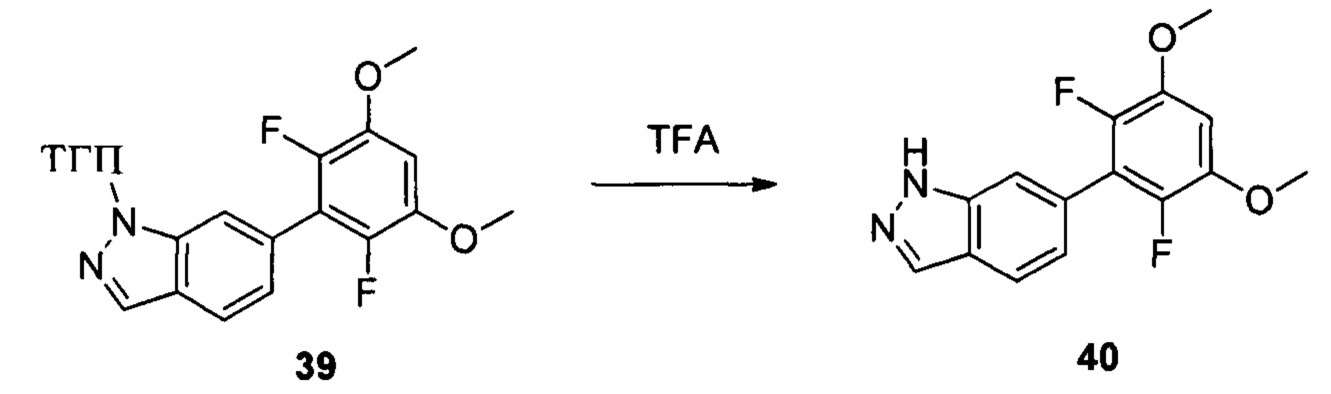
Соединение 39 (629 мг, 1,68 ммоль) и дихлорметан (10,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл, и на ледяной бане по каплям медленно добавляли TFA (2 мл), перемешивали при комнатной температуре в течение 3,0 часов. Растворитель выпаривали в роторном испарителе, остаток разбавляли водой со льдом и рН доводили до 8 с помощью насыщенного раствора NaHCO3, затем экстрагировали этилацетатом и органическую фазу промывали водой. Сушили над безводным сульфатом натрия и растворитель выпаривали в роторном испарителе с получением неочищенного продукта, представляющего собой соединение 40 (460 мг, 94%). ЖХ-МС: 291,0(М+Н)+, время удержания = 1,233 минут.
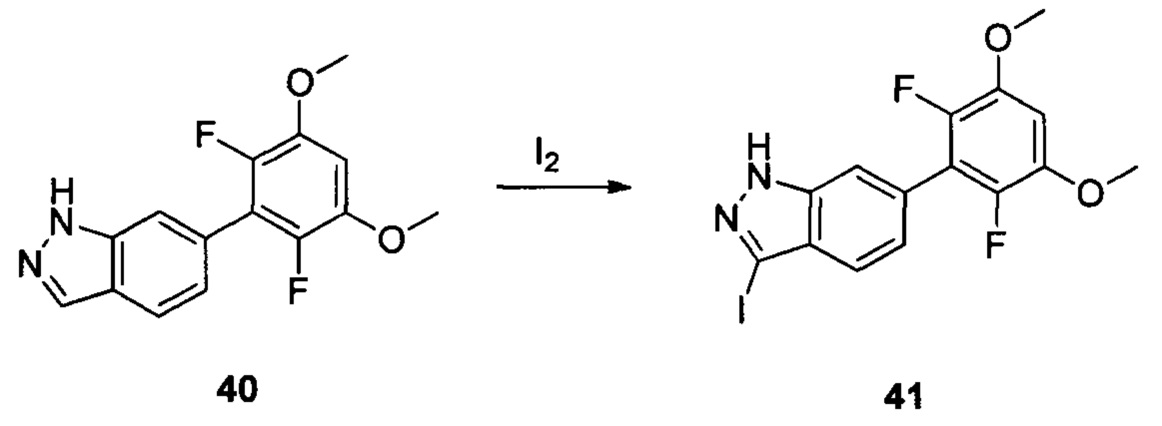
Соединение 40 (460 г, 1,59 ммоль), водный раствор NaOH (5,3 г, 3N) и 1,4-диоксан (6,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл. По каплям добавляли йод (484,0 мг, 1,90 ммоль) в 1,4-диоксане при температуре 0°С. Перемешивали при комнатной температуре в течение 18 часов. Промывали насыщенным раствором тиосульфата натрия и экстрагировали этилацетатом, затем органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и сушили в роторном испарителе с получением неочищенного продукта 41 (633,0 мг, 95,6%), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС: 416,9(М+Н)+, время удержания = 1,540 минут.
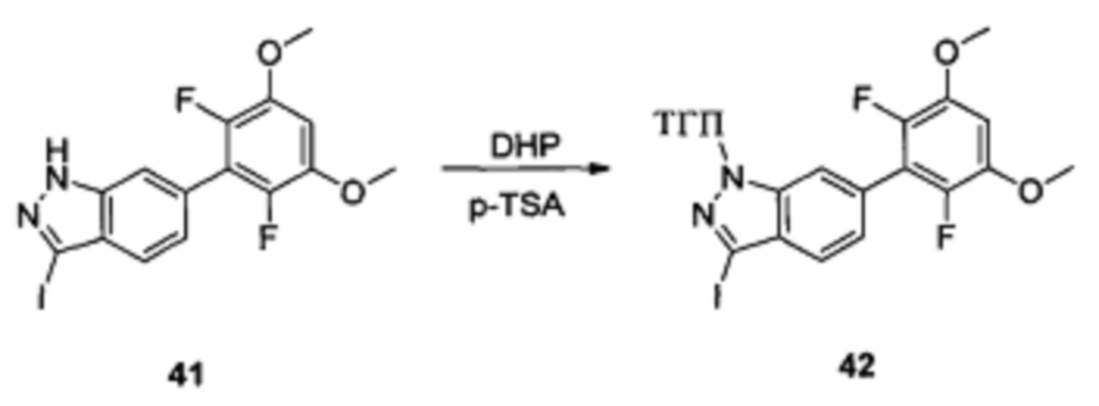
Соединение 41 (633 г, 1,52 ммоль), p-TSA (58 г, 0,30 ммоль) и дихлорметан (6,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл, по каплям медленно добавляли DHP (256 г, 3,04 ммоль), перемешивали при комнатной температуре в течение 18,0 часов. Реакционный раствор разбавляли 20,0 мл воды, дважды экстрагировали дихлорметаном 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (этилацетат : петролейный эфир = 1:12) с получением соединения 42 (260,0 мг, 34%).
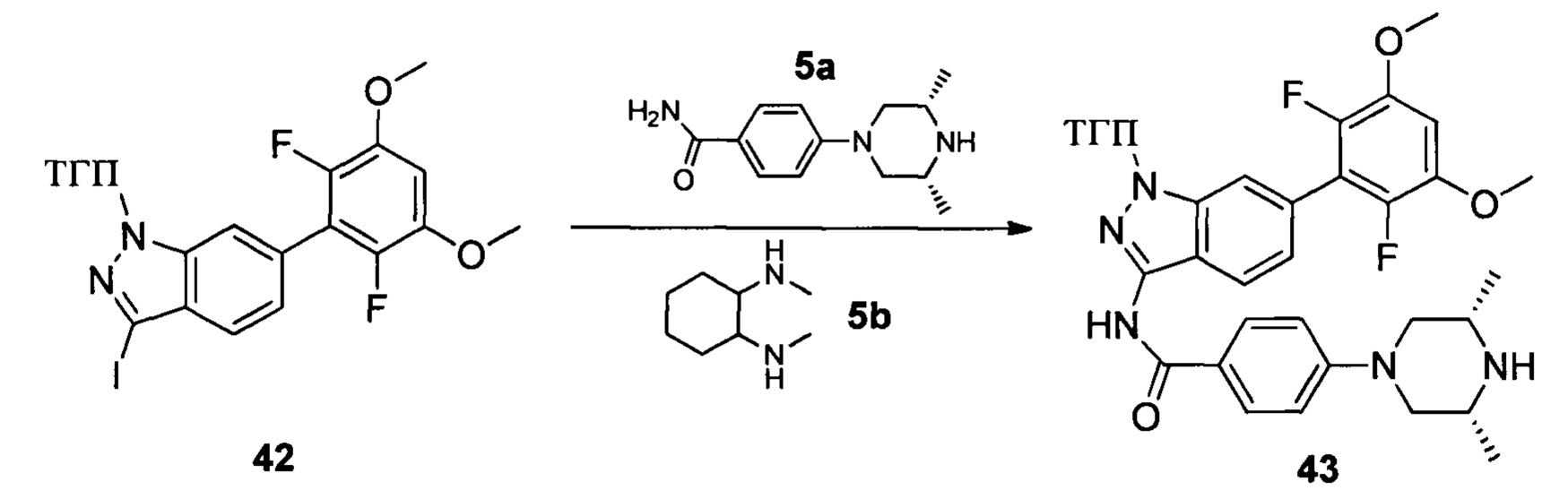
Соединение 42 (260 мг, 0,52 ммоль), соединение 5а (145 мг, 0,62 ммоль), соединение 5b (148,0 мг, 1,04 ммоль), K3PO4 (331,0 мг, 1,56 ммоль), CuI (99 мг, 0,52 ммоль) и высушенный ДМФА (3,0 мл) добавляли в сухую 25 мл трехгорлую колбу и перемешивали при 120°С в течение 6,0 часов. Реакционную смесь разбавляли 20,0 мл воды и экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и сушили в роторном испарителе с получением неочищенного продукта 43 (290 мг, 92%), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС: 606,1 (М+Н)+, время удержания = 1,063 минут.
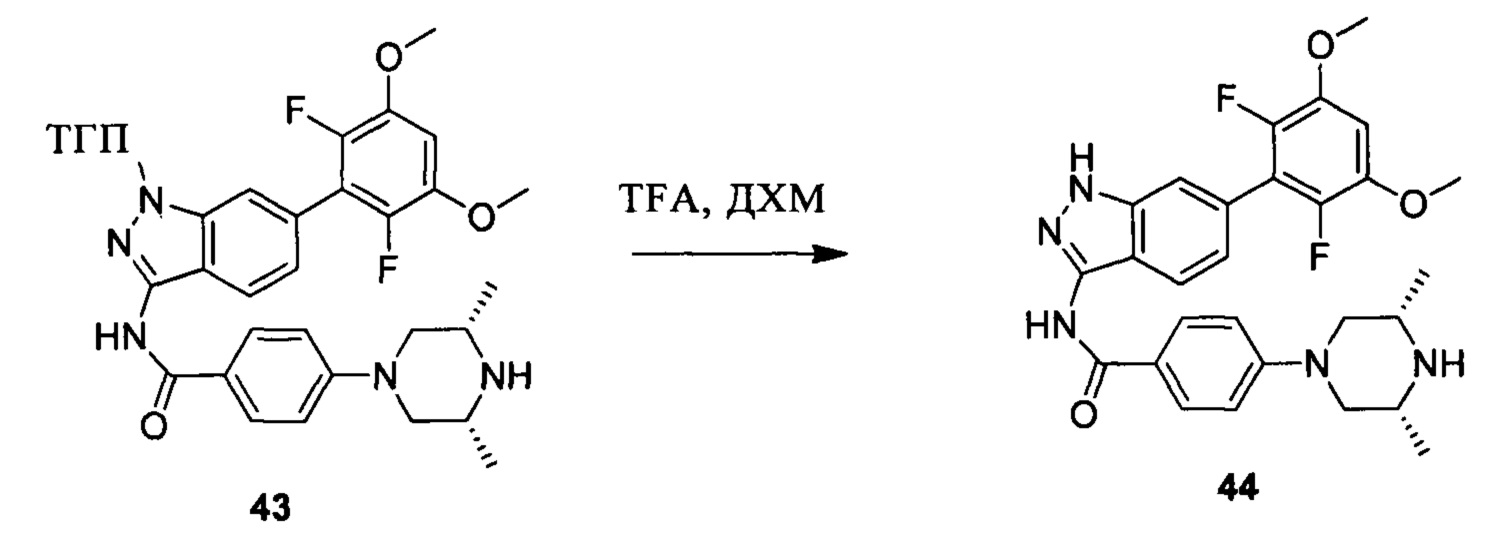
Соединение 43 (290,0 мг, 0,48 ммоль) и дихлорметан (10,0 мл) добавляли в сухую круглодонную колбу объемом 25 мл, по каплям медленно добавляли TFA (2,0 мл) на ледяной бане и перемешивали при комнатной температуре в течение 4,0 часов. Растворитель сушили в роторном испарителе с получением неочищенного продукта, который подвергали кислотной препаративной ВЭЖХ с получением соединения 44 (50,2 мг, 21%, соль TFA). ЖХ-МС: 522,1 (М+Н)+, время удержания = 1,227 минут.
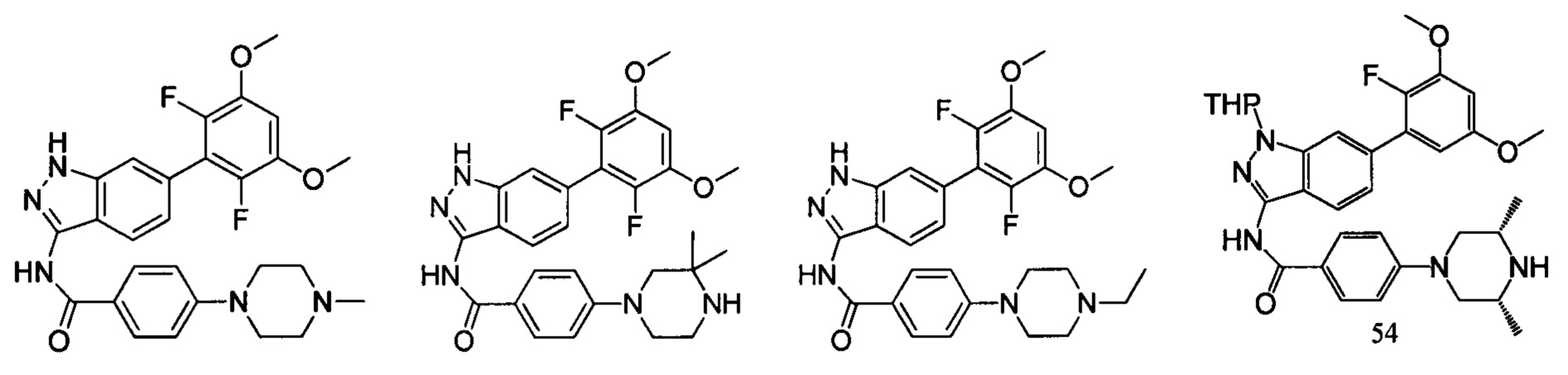
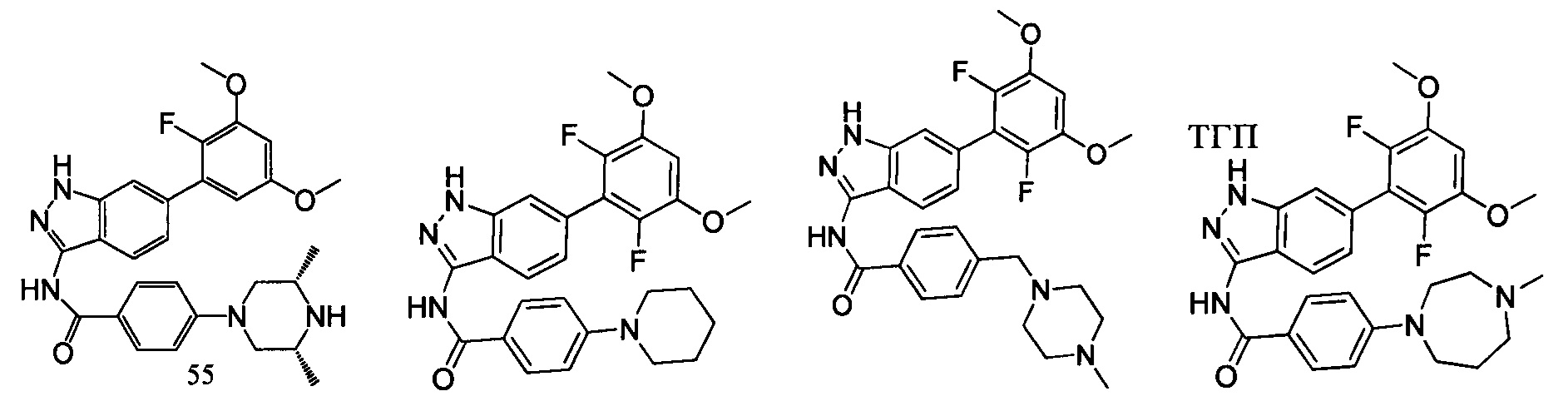
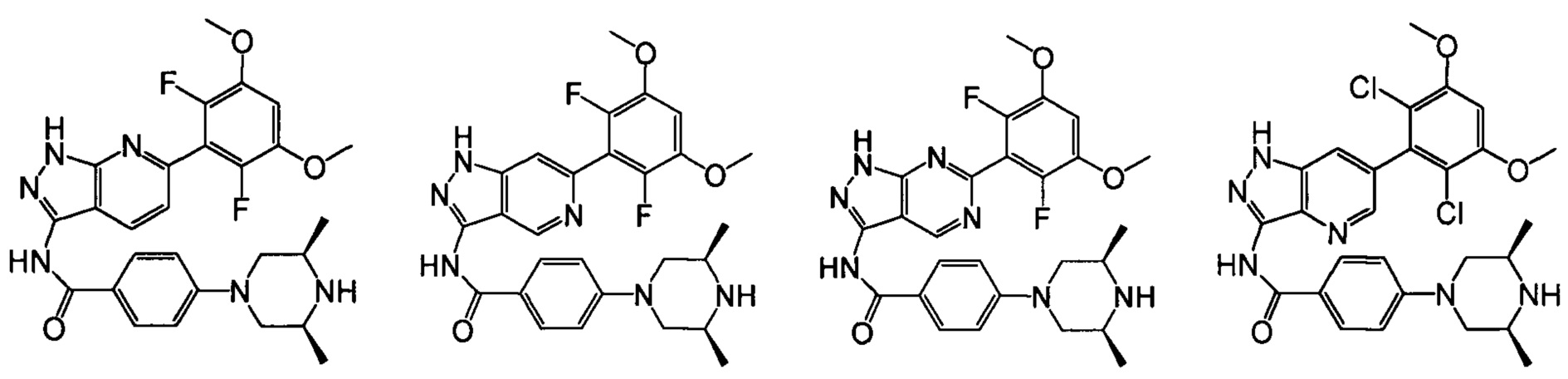
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,91 (brs, 1Н), 10,64 (d, 1H, J=3,2 Гц), 9,01 (m, 1H), 8,45 (m, 1H), 8,04 (d, 2H, J=8,8 Гц), 7,78 (d, 1H, J=8,4 Гц), 7,52 (s, 1H), 7,09 (m, 4H), 4,12 (d, 2H, J=13,8 Гц), 3,92 (s, 6H), 2,77 (m, 3H), 1,29 (d, 6H, J=6,4 Гц).
Аналогичными способами были получены следующие соединения:
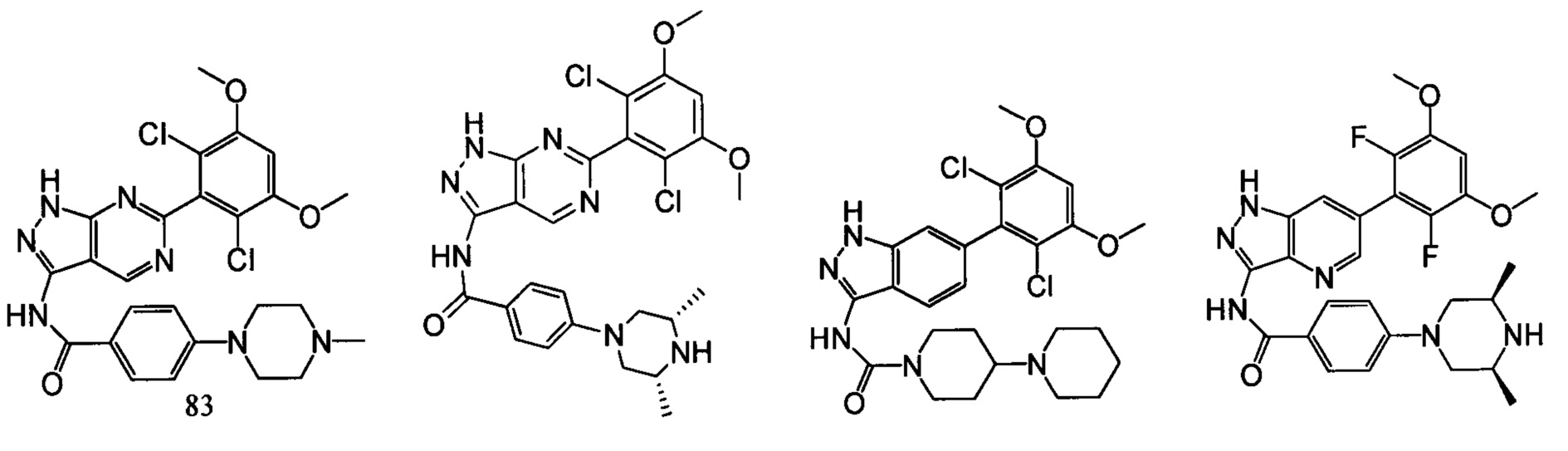
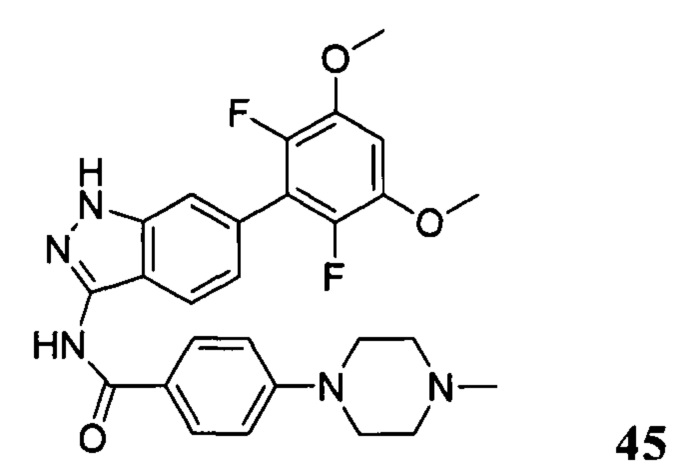
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-метилпиперазин-1-ил)бензамид
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,95 (s, 1Н), 10,62 (s, 1Н), 9,78 (s, 1Н), 8,04 (d, 2H, J=8,8 Гц), 7,79 (d, 1H, J=8,4 Гц), 7,52 (s, 1H), 7,02-7,18 (m, 4H), 4,08 (d, 2H, J=12,0 Гц), 3,92 (s, 6H), 3,47-3,63 (m, 2H), 3,01-3,27 (m, 4H), 2,88 (s, 3H).
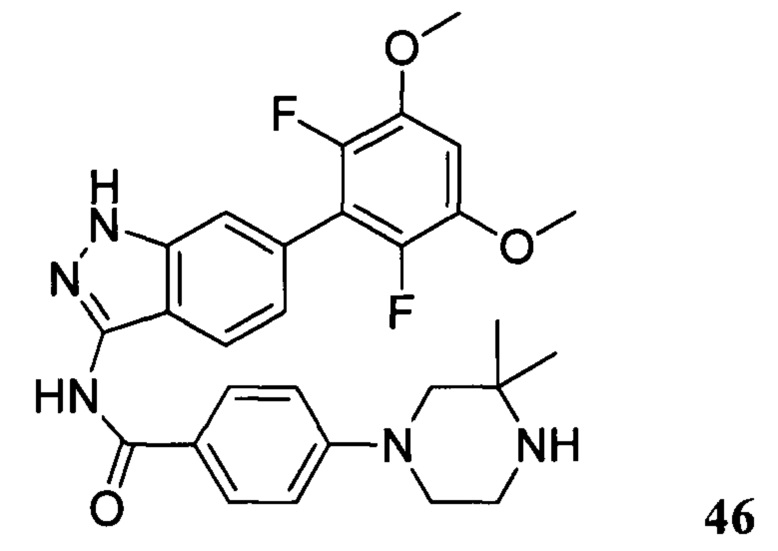
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(3,3-диметилпиперазин-1-ил)бензамид
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,91 (s, 1H), 10,63 (s, 1H), 8,91 (s, 2H), 8,03 (d, 2H, J=9,2 Гц), 7,78 (d, 1H, J=8,0 Гц), 7,52 (s, 1H), 7,02-7,16 (m, 4H), 3,86 (s, 6H), 3,51 (m, 2H), 3,43 (m, 2H), 3,30 (s, 2H), 1,26 (t, 3H, J=7,2 Гц).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(4-этилпиперазин-1-ил)бензамид
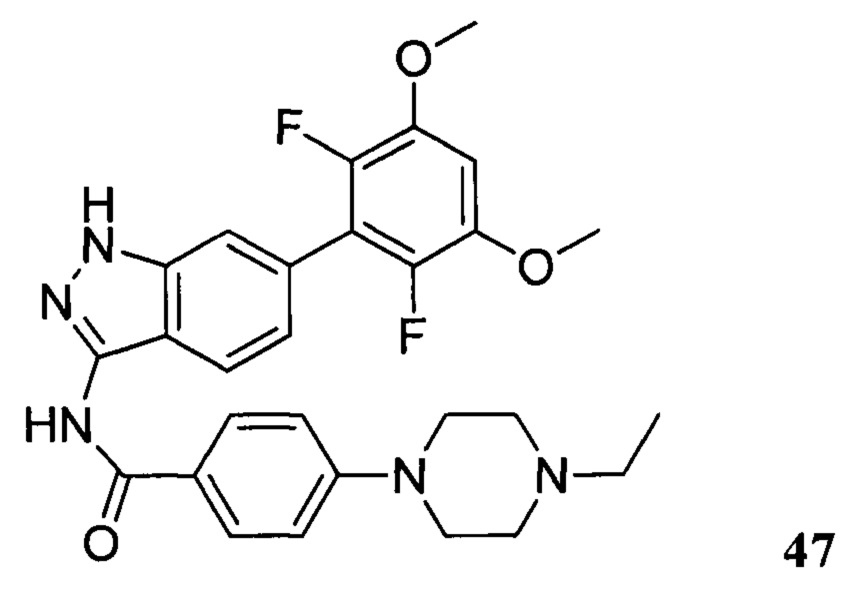
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,95 (s, 1H), 10,62 (s, 1H), 9,78 (s, 1H), 8,04 (d, 2H, J=8,8 Гц), 7,79 (d, 1H, J=8,4 Гц), 7,52 (s, 1H), 7,02-7,18 (m, 4H), 4,08 (d, 2H, J=12,0 Гц), 3,92 (s, 6H), 3,47-3,63 (m, 2H), 3,21-3,22 (m, 2H), 3,01-3,27 (m, 4H), 1,27 (t, 3Н, J=7,2 Гц).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)бензамид
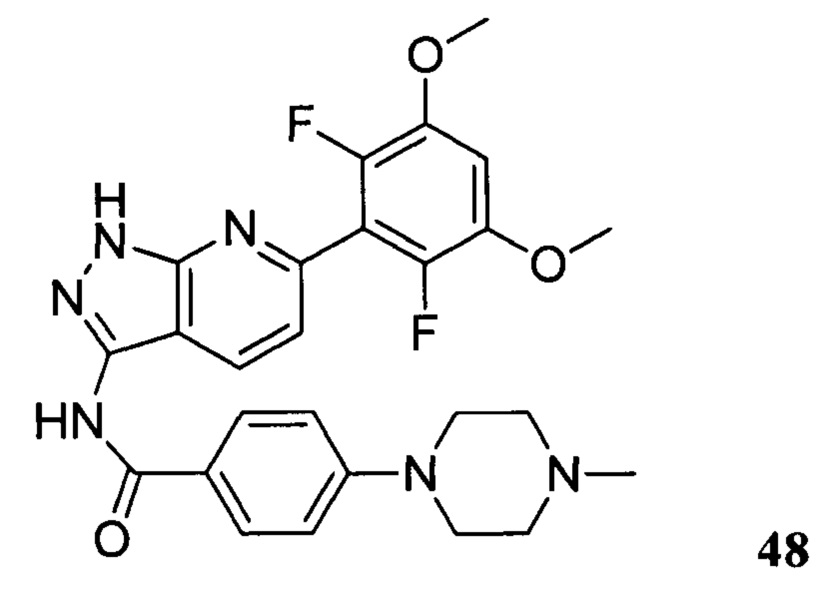
1Н ЯМР (d-MeOD, 400 МГц) δ ppm 8,50 (d, 1H, J=8,4 Гц)), 7,99 (d, 2Н, J=8,8 Гц), 7,30 (d, 1Н, J=8,4 Гц), 7,08 (d, 2H, J=8,8 Гц), 7,01 (s, 1H), 3,93 (s, 6H), 3,45 (s, 4H), 2,80 (s, 4H), 2,49 (s, 3Н). ЖХ-МС: 509 (M+H)+, время удержания = 1,18 минут.
Путь синтеза III
Соединение 49 (1,20 г, 3,55 ммоль) и ацетонитрил (20,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл. Порциями добавляли реагент Selectfluor (2,51 г, 7,1 ммоль) при 0°С в защитной атмосфере N2 и перемешивали при комнатной температуре в течение 18,0 часов. Реакционный раствор разбавляли водой и экстрагировали этилацетатом. Органическую фазу последовательно промывали водой, насыщенным раствором NaHCO и насыщенным солевым раствором. Сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали при помощи колоночной хроматографии (этилацетат : петролейный эфир = 1:8) с получением неочищенного соединения 50 (629 мг, 47%). ЖХ-МС: 374,9 (М+Н)+, время удержания = 1,243 минут.
Соединение 50 (629 мг, 1,68 ммоль) и дихлорметан (10,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл, по каплям медленно добавляли TFA (2 мл) на ледяной бане и перемешивали при комнатной температуре в течение 3,0 часов. Растворитель испаряли в роторном испарителе и остаток разбавляли водой со льдом. Значение рН доводили до 8 при помощи насыщенного раствора NaHCO3 и указанную смесь экстрагировали этилацетатом. Органическую фазу промывали водой. Сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением неочищенного продукта, представляющего собой соединение 51 (460 мг, 95%). ЖХ-МС: 291,0 (М+Н)+, время удержания = 1,233 минут.
Соединение 51 (460 г, 1,59 ммоль), водный раствор NaOH (5,3 г, 3N) и 1,4-диоксан (6,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл. По каплям добавляли йод (484,0 мг, 1,90 ммоль) в 1,4-диоксане при температуре 0°С. Смесь перемешивали при комнатной температуре в течение 18 часов, промывали насыщенным раствором тиосульфата натрия и экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и сушили в роторном испарителе с получением неочищенного продукта 52 (633 мг, 96%), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС: 416,9 (М+Н)+, время удержания = 1,540 минут.
Соединение 52 (633 г, 1,52 ммоль), p-TSA (58 г, 0,30 ммоль) и дихлорметан (6,0 мл) добавляли в сухую круглодонную колбу объемом 50 мл, по каплям медленно добавляли DHP (256 г, 3,04 ммоль) и перемешивали при комнатной температуре в течение 18,0 часов. Реакционный раствор разбавляли 20,0 мл воды, дважды экстрагировали дихлорметаном 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (этилацетат : петролейный эфир = 1:12) с получением соединения 53 (265,0 мг, 26%).
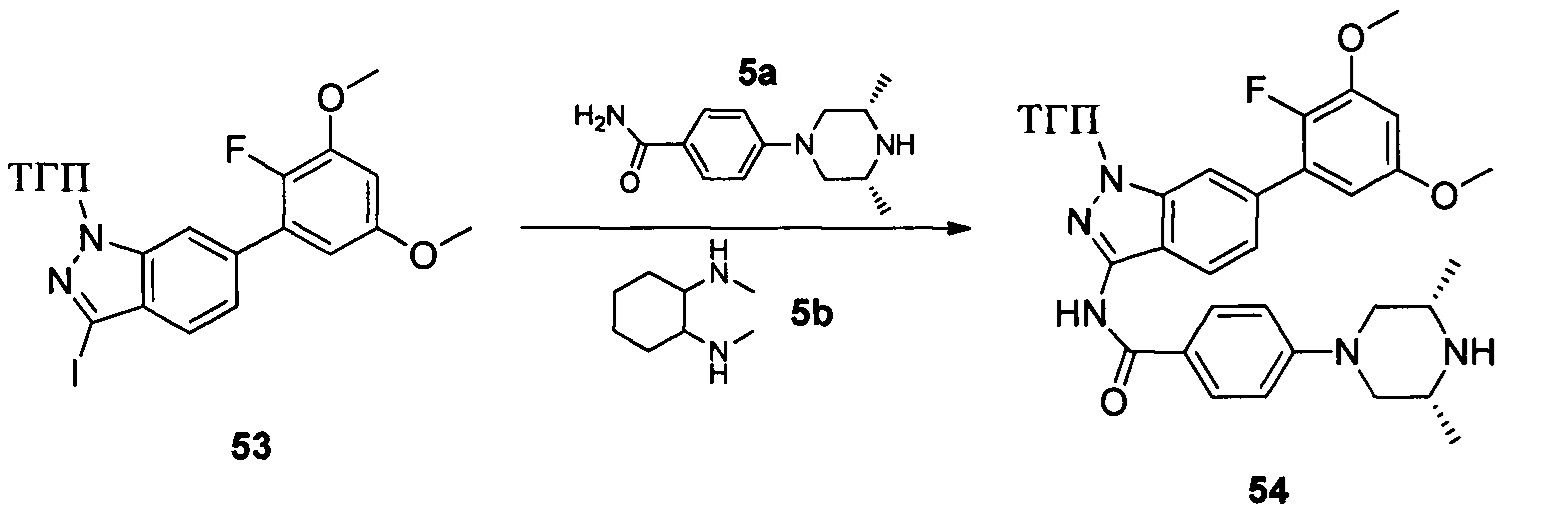
Соединение 53 (194 мг, 0,40 ммоль), соединение 5а (112 мг, 0,48 ммоль), соединение 5b (114,0 мг, 0,81 ммоль), K3PO4 (256,0 мг, 1,21 ммоль), CuI (76 мг, 0,40 ммоль) и высушенный ДМФА (3,0 мл) добавляли в сухую трехгорлую колбу объемом 25 мл и перемешивали при 120°С в течение 6,0 часов. Реакционную смесь разбавляли 30,0 мл воды и экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и сушили в роторном испарителе с получением неочищенного продукта 54 (240 мг, 99%), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС: 588,1 (М+Н)+, время удержания = 1,320 минут.
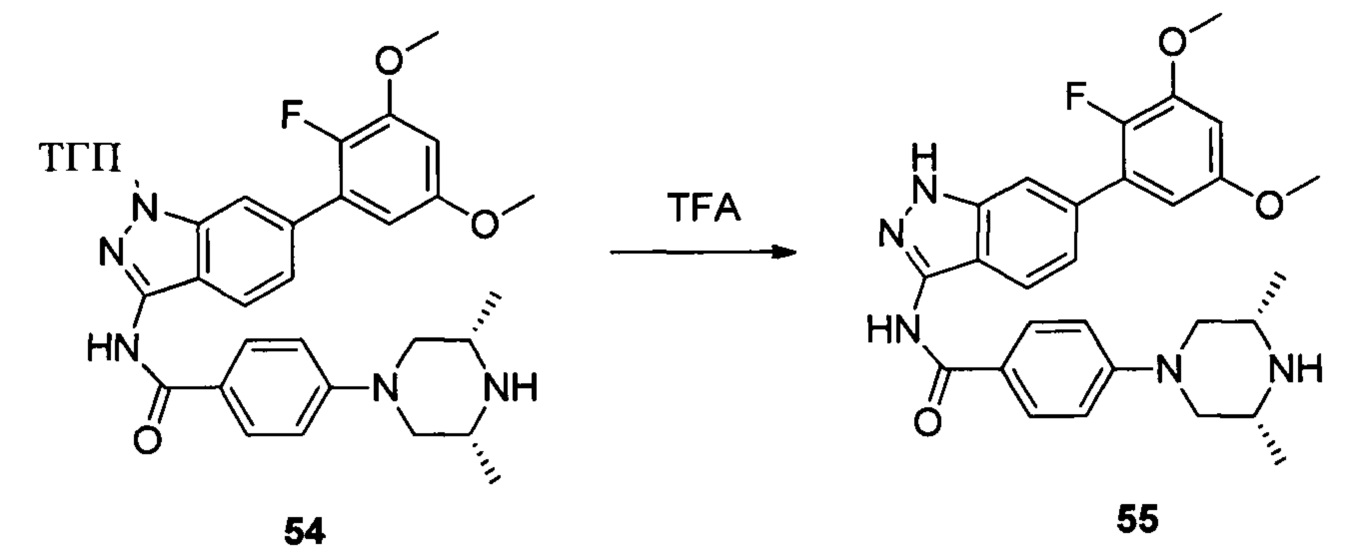
Соединение 54 (290,0 мг, 0,41 ммоль) и дихлорметан (7,5 мл) добавляли в сухую круглодонную колбу объемом 25 мл, по каплям медленно добавляли TFA (1,5 мл) на ледяной бане и перемешивали при комнатной температуре в течение 4,0 часов. Растворитель сушили в роторном испарителе с получением неочищенного продукта, который подвергали кислотной препаративной ВЭЖХ с получением соединения 55 (70,8 мг, 34%, соль TFA). ЖХ-МС: 522,1 (М+Н)+, время удержания = 1,227 минут.
1Н ЯМР (d6-ДМСО, 400 МГц) δ ppm 12,95 (brs, 1Н), 10,61 (s, 1H), 9,17 (m, 1H), 8,57 (m, 1H), 8,03 (d, 2H, J=9,2 Гц), 7,76 (m, 1H), 7,59 (s, 1H), 7,22 (d, 1H, J=8,8 Гц), 7,13 (d, 2H, J=9,2 Гц), 6,76 (dd, 1H, J1=2,8 Гц, J2=6,8 Гц), 6,61 (m, 1H), 4,01 (d, 2H, J=12,4 Гц), 3,88 (s, 1H), 3,84 (s, 3H), 3,81 (s, 3H), 3,30-3,50 (m, 2H), 2,75 (t, 2H, J=6,4 Гц), 1,29 (d, 6H, J=6,4 Гц).
Путь синтеза IV
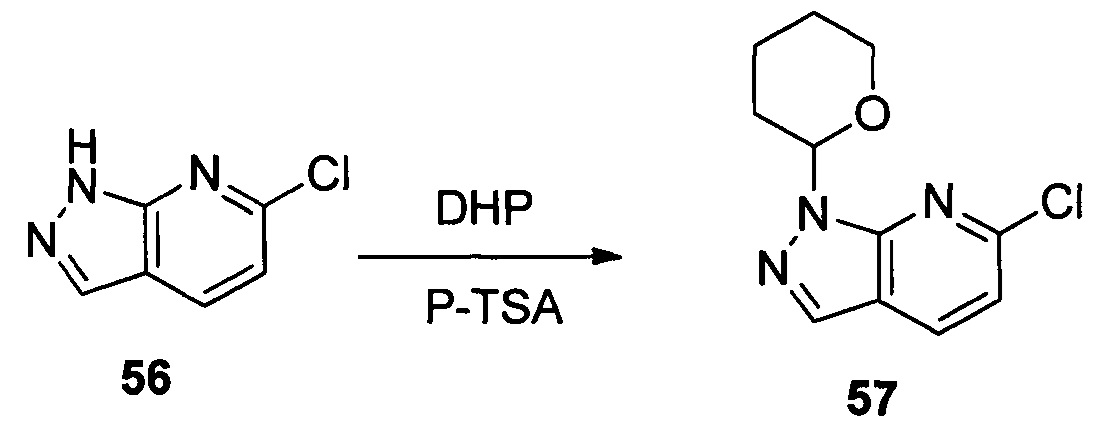
Соединение 56 (10,00 г, 51,0 ммоль) p-TSA (1,75 г, 10,2 ммоль) и дихлорметан (100,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл, по каплям медленно добавляли DHP (8,56 г, 102,0 ммоль), перемешивали при комнатной температуре в течение 4,0 часов. После завершения взаимодействия указанный раствор разбавляли 100,0 мл воды, дважды экстрагировали дихлорметаном 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия и растворитель выпаривали в роторном испарителе с получением соединения 57 (8,90 г, 62%). ЖХ-МС: 281 (М+Н)+, время удержания = 1,626 минут.
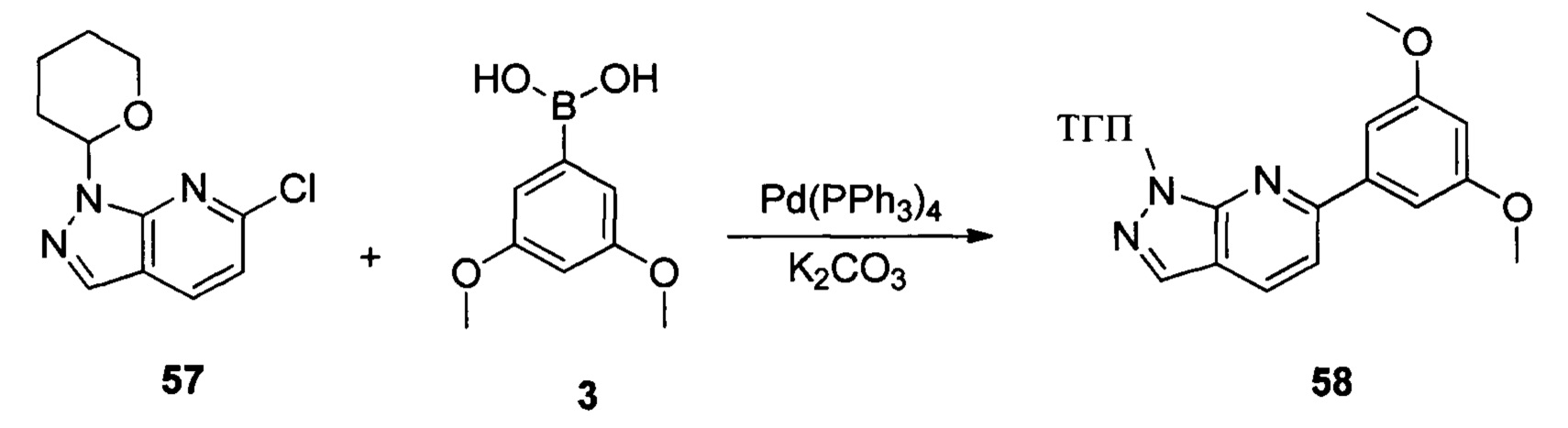
Соединение 57 (8,90 г, 31,7 ммоль), соединение 3 (5,77 г, 31,7 ммоль), Pd(PPh3)4 (3,66 г, 3,17 ммоль), K2CO3 (8,75 г, 63,4 ммоль), 1,4-диоксан (60,0 мл) и воду (15,0 мл) последовательно добавляли в сухую круглодонную колбу объемом 250 мл при комнатной температуре и равномерно перемешивали. Реакционную смесь нагревали с обратным холодильником в течение 4,0 часов в защитной атмосфере азота. Реакционный раствор охлаждали до комнатной температуры и растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали при помощи колоночной хроматографии (этилацетат : петролейный эфир = 1:10) с получением неочищенного соединения 58 (8,10 мг, 76%). ЖХ-МС:339 (М+Н)+, время удержания = 1,626 минут.
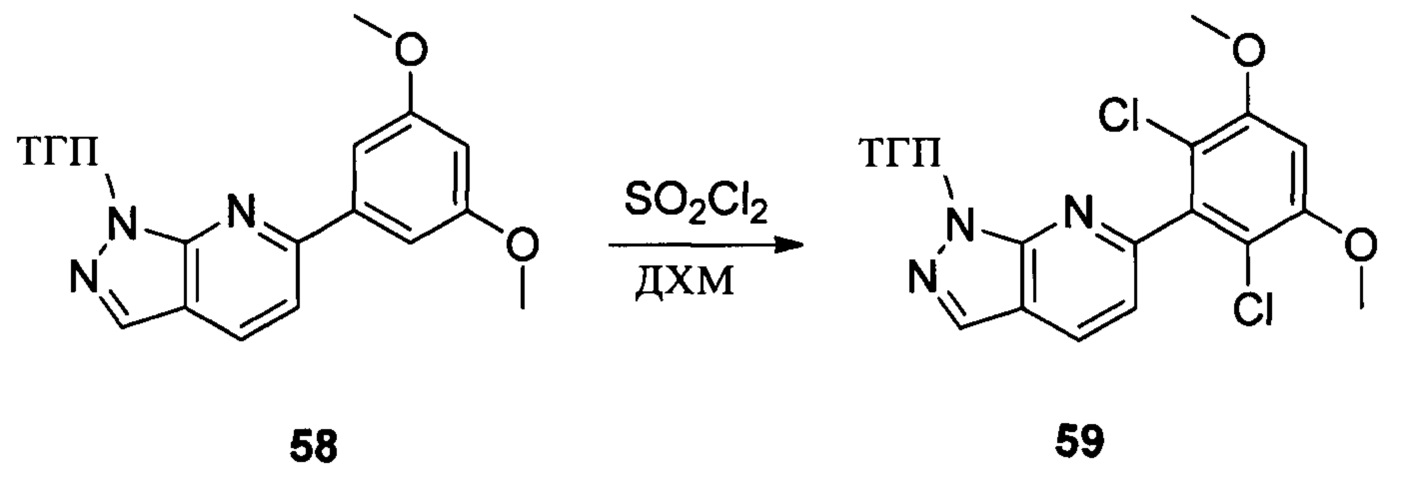
Соединение 58 (7,90 г, 23,4 ммоль) и дихлорметан (50,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл, по каплям медленно добавляли SO2Cl2 (7,15 г, 46,7 ммоль), перемешивали при комнатной температуре в течение 4,0 часов. Реакционный раствор разбавляли 50,0 мл воды, дважды экстрагировали дихлорметаном 200 мл и промывали насыщенным раствором NaHCO3. Органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением соединения 59 (8,50 г, 89%). ЖХ-МС: 407 (М+Н)+, время удержания = 1,798 минут.
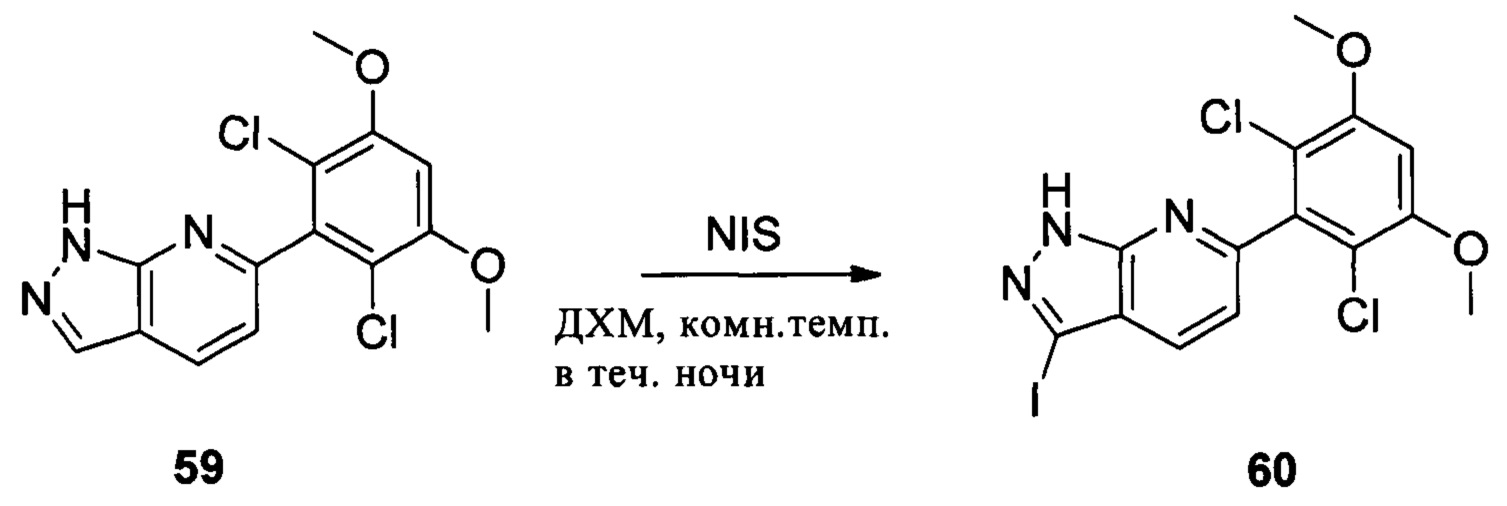
Соединение 59 (0,972 г, 3 ммоль), NIS (1,6 г, 7 ммоль) и ДХМ (10,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл. Смесь перемешивали при комнатной температуре в течение 2,0 часов. После завершения взаимодействия в реакционный раствор добавляли 200 мл воды и дважды экстрагировали дихлорметаном 200 мл. Органические фазы промывали насыщенным тиосульфатом натрия, объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением соединения 60 (1,1 г, 82%). ЖХ-МС: 450 (М+Н)+, время удержания = 1,644 минут.
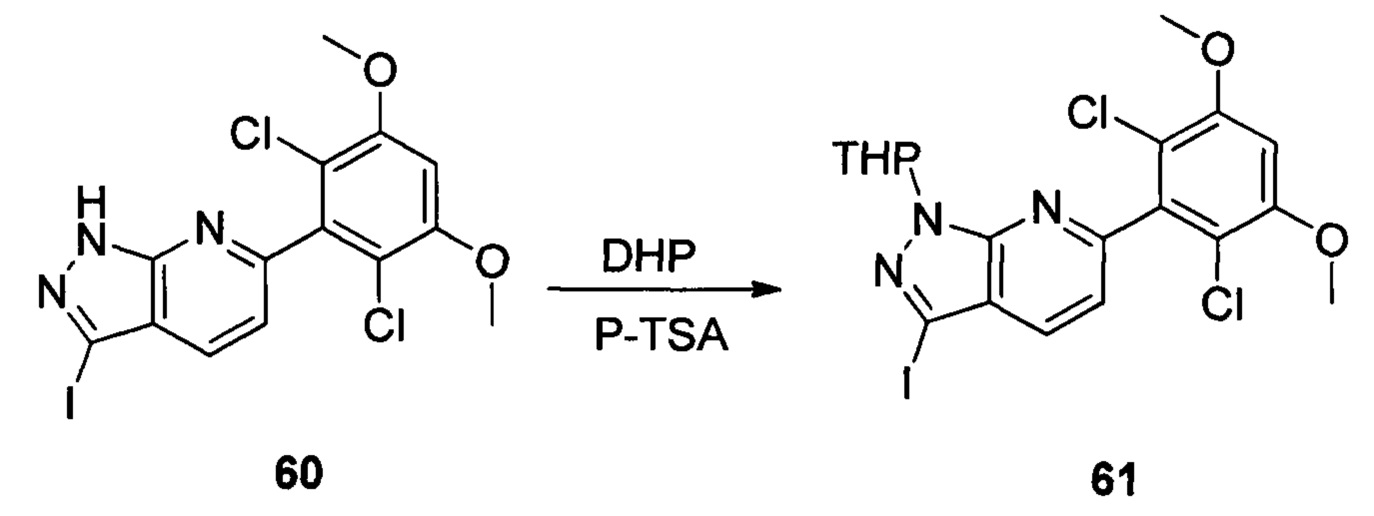
Соединение 60 (9,30 г, 20,76 ммоль), p-TSA (0,71 г, 4, 152 ммоль) и дихлорметан (50,0 мл) добавляли в сухую круглодонную колбу объемом 250 мл, по каплям медленно добавляли DHP (3,48 г, 41,52 ммоль), перемешивали при комнатной температуре в течение 4,0 часов. Реакционный раствор разбавляли 50,0 мл воды, дважды экстрагировали дихлорметаном 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель выпаривали в роторном испарителе с получением соединения 61 (7,70 г, 70%). ЖХ-МС: 281 (М+Н)+, время удержания = 2,165 минут.
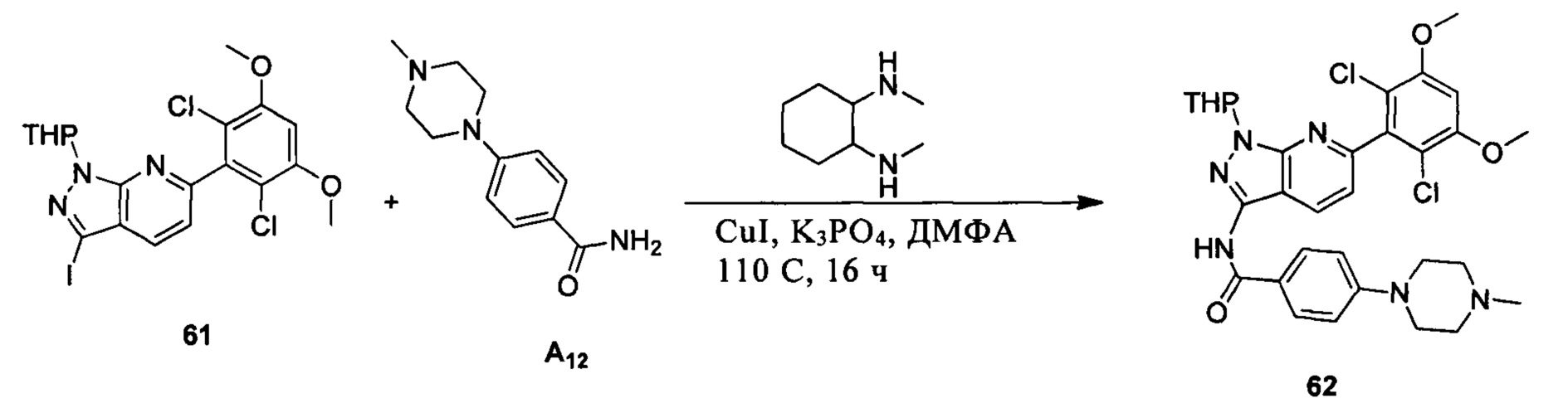
Соединение 61 (2,0 г, 3,75 ммоль) растворяли в 20 мл безводного ДМФА, затем в раствор при комнатной температуре последовательно добавляли транс-N,N'-диметил-1,2-циклогександиамин (107 мг, 0,75 ммоль), CuI (36 мг, 0,19 ммоль), K3PO4 (1,6 г, 7,5 ммоль) и А12 (1,05 г, 4,5 ммоль). Указанный раствор три раза продували азотом, нагревали до 110°С и перемешивали в течение 16 часов. После завершения указанного взаимодействия растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (дихлорметан : метанол = 40:1) с получением белого твердого вещества (0,98 г), выход составил 41%.
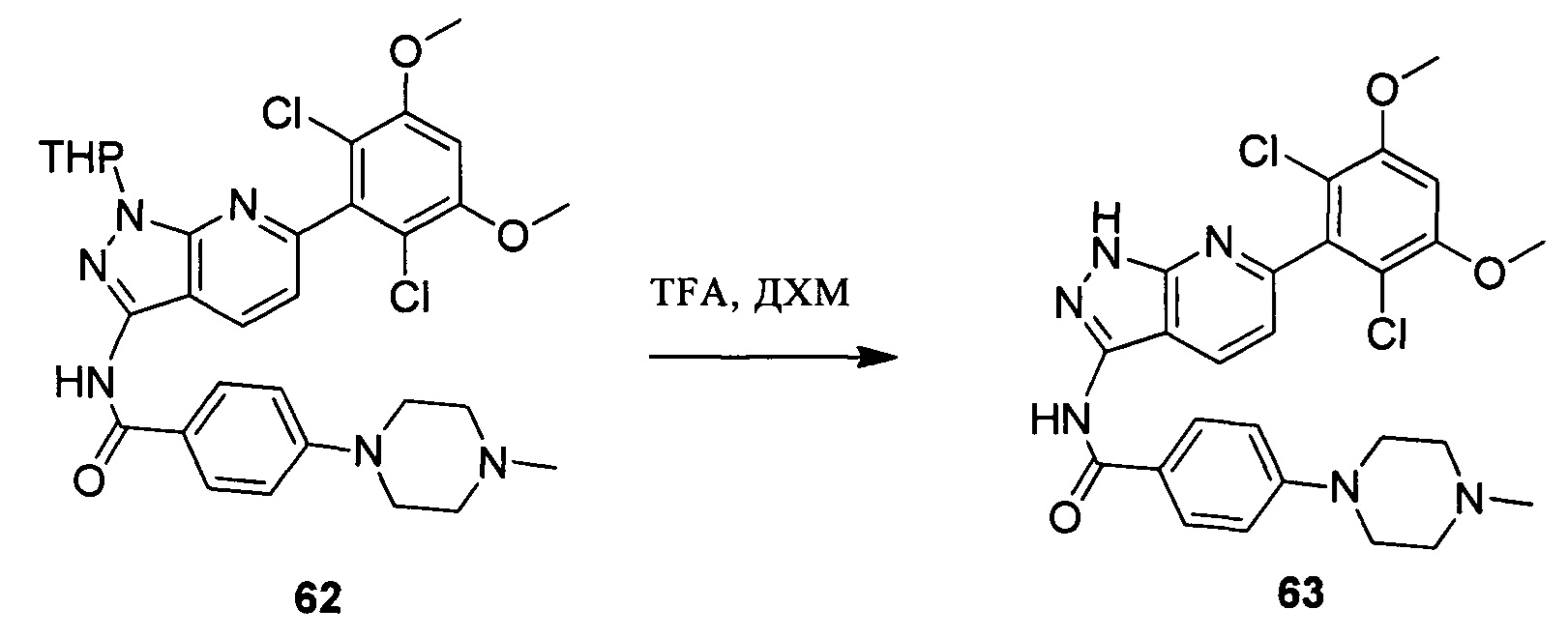
Соединение 62 (0,86 г, 1,35 ммоль) растворяли в 10 мл дихлорметана и при комнатной температуре к раствору добавляли трифторуксусную кислоту (5 мл). Смесь перемешивании при комнатной температуре в течение 4 часов. После завершения указанного взаимодействия растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (дихлорметан : метанол = 20:1) с получением белого твердого вещества 63 (0,61 г), выход составил 82%.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)бензамид
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 13,37 (s, 1Н), 10,81 (s, 1H), 8,39 (d, 1H, J=8,4 Гц), 8,01 (d, 2H, J=8,8 Гц), 7,08-7,02 (m, 4H), 3 98 (s, 6H), 3,32 (m, 4H), 2,45 (m, 4H), 2,23 (s, 3H).
Аналогичными способами были получены следующие соединения:
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-((3S,5R)-3,5-диметилпиперазин-1-ил)бензамид
1Н ЯМР (CDCl3, 400 МГц) δ ppm 10,76 (br s, 1H), 8,97 (s, 1H), 8,87 (d, 1H, J=8,4 Гц), 7,90 (d, 2H, J=8,8 Гц), 7,16 (d, 1H, J=8,4 Гц), 6,95 (d, 2H, J=8,8 Гц), 6,66 (s, 1H), 3,97 (s, 6H), 3,70 (d, 2H, J=12,0 Гц), 3,02 (m, 2H), 2,46 (t, 2H, J=11,2 Гц), 1,16 (d, 6H, J=6,4 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-этилпиперазин-1-ил)бензамид
1H ЯМР (CDCl3, 400 МГц) δ ppm 10,13 (s, 1H), 8,86 (d, 1H, J=8,4 Гц), 8,66 (s, 1H), 7,90 (d, 2H, J=8,8 Гц), 7,16 (d, 1H, J=8,4 Гц), 6,95 (d, 2H, J=8,8 Гц), 6,67 (s, 1H), 3,97 (s, 6H), 3,41-3,79 (m, 4H), 2,64-2,60 (m, 4H), 2,49 (q, 2H, J=7,2 Гц), 1,15 (t, 3H, J=7,2 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-((3R,5S)-3,5-диметилпиперазин-1-ил)бензамид
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 13,36 (s, 1Н), 10,77 (s, 1Н), 8,34 (d, 1H, J=8,4 Гц), 7,98 (d, 2H, J=8 Гц), 7,30 (d, 1H, J=8 Гц), 7,00 (d, 2H, J=8,8 Гц), 6,82 (d, 1H, J=2,8 Гц), 6,73 (d, 1H, J=2,8 Гц), 3,91 (s, 3H), 3,84 (s, 3H), 3,75 (d, 2H, J=10,4 Гц), 2,81 (s, 2H), 2,26 (s, 1H), (d, 2H, J=11,2 Гц), 1,04 (s, 3H), 1,03 (s, 3Н). ЖХ-МС: 521 (M+H)+, время удержания = 1,233 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-(диметиламино)пиперидин-1-ил)бензамид
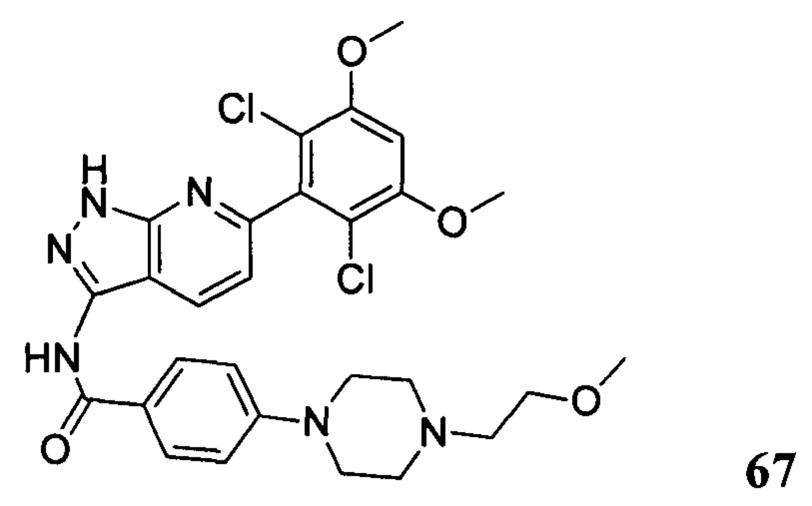
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 10,81 (s, 1Н), 8,381 (d, 1H, J=8,4 Гц), 8,00 (d, 2H, J=8,8 Гц), 7,01-7,07 (m, 4H), 3,99 (s, 6H), 3,49-3,51 (m, 3H), 3,26 (s, 4H), 3,17 (s, 3H), 2,55-258 (m, 5H). ЖХ-МС: 585 (M+H)+, время удержания = 1,225 минуты.
N-(6-(3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(((3R,5S)-3,5-диметилпиперазин-1-ил))бензамид
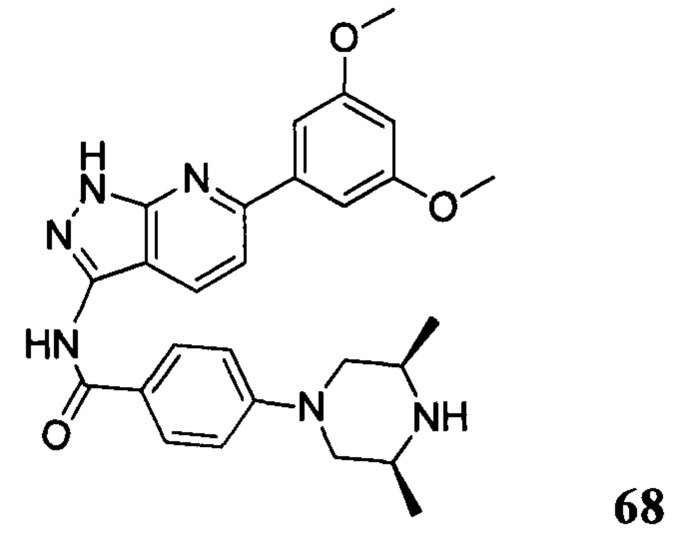
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 13,34 (s, 1Н), 10,77 (s, 1H), 8,36 (d, 1H, J=8,8 Гц), 7,98 (d, 2H, J=8,8 Гц), 7,74 (s, 1H), 7,31 (d, 2H, J=1,6 Гц), 7,00 (d, 2H, J=8,8 Гц), 6,62 (s, 1H), 3,84 (s, 6H), 3,75 (d, 2H, J=11,2 Гц), 2,81 (s, 2H), 2,26 (s, 1H), 2,22 (d, 2H, J=10,8 Гц), 1,04 (d, 6H, J=6 Гц). ЖХ-МС: 487 (M+H)+, время удержания = 1,113 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)пиридин формамид
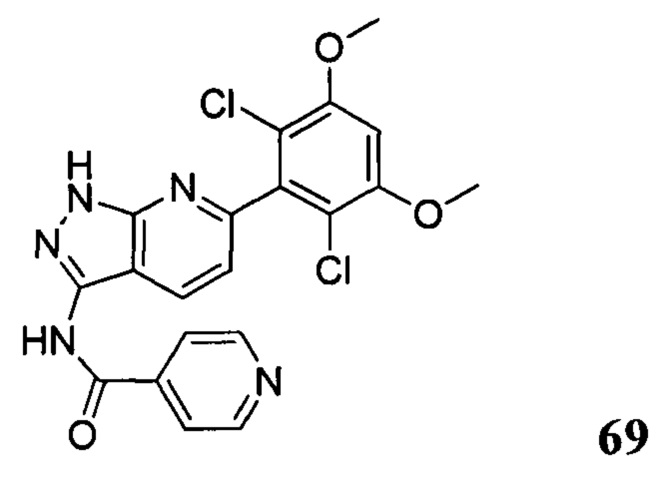
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 8,74 (s, 2Н), 8,41 (d, 1Н, J=8,0 Гц), 8,02 (d, 2H, J=5,6 Гц), 7,04 (s, 2Н), 3,98 (s, 6Н). ЖХ-МС: 445 (М+Н)+, время удержания = 1,409 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-морфолинобензамид
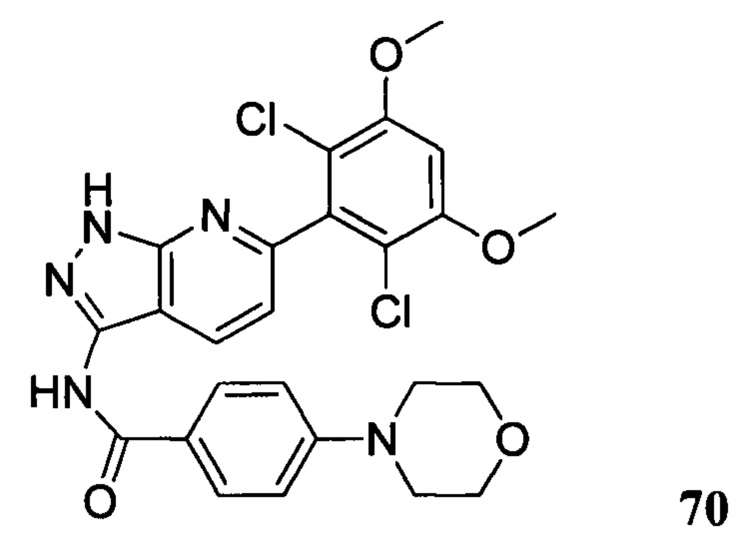
1Н ЯМР (d6-ДМСО, 400 МГц) δ ppm 13,42 (s, 1Н), 10,86 (s, 1Н), 8,41 (d, 1H, J=7,2 Гц), 8,05 (s, 3Н), 7,07 (s, 4H), 4,00 (s, 6H), 3,77 (s, 4H), 3,30 (s, 4H). ЖХ-МС: 528 (M+H)+ время удержания = 1,643 минуты.
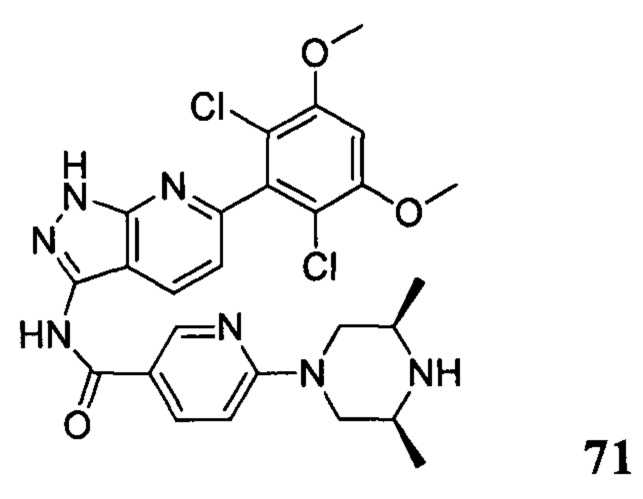
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 13,47 (brs, 1Н), 11,03 (s, 1H), 9,28 (s, 1H), 8,89 (s, 1H), 8,71 (s, 1H), 8,41 (d, J=8,0 Гц, 1H), 8,28-8,30 (m, 1H), 7,05-7,12 (m, 3H), 4,66 (d, J=9,2 Гц, 2H), 3,98 (s, 6H), 3,46 (brs, 2H), 2,83-2,92 (m, 2H), 1,30 (d, J=6,4 Гц, 6H).
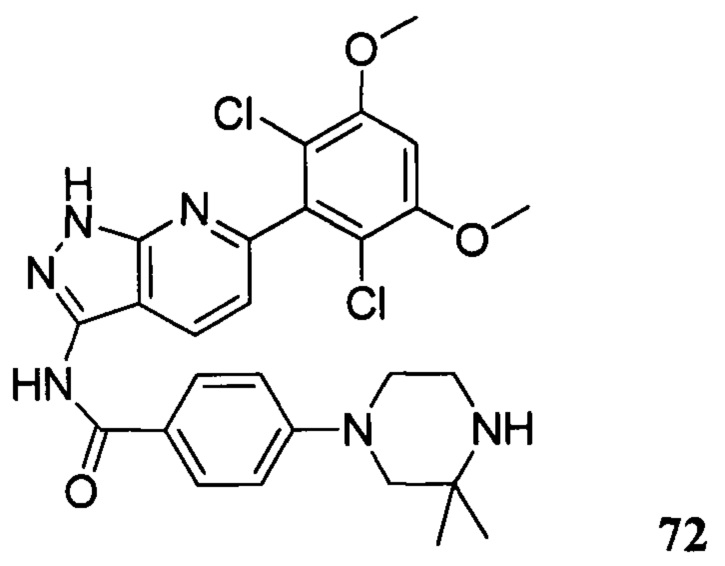
1Н ЯМР (d-MeOD, 400 МГц) δ ppm 8,52 (d, J=8,0 Гц, 1H), 8,03 (d, J=8,0 Гц, 2H), 7,15-7,13 (m, 3H), 6,94 (s, 1H), 3,99 (s, 6H), 3,58-3,57 (m, 2H), 3,44-3,40 (m, 4H), 1,50 (s, 6H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(2,6-диазаспиро[3.3]гептан-2-ил)бензамид
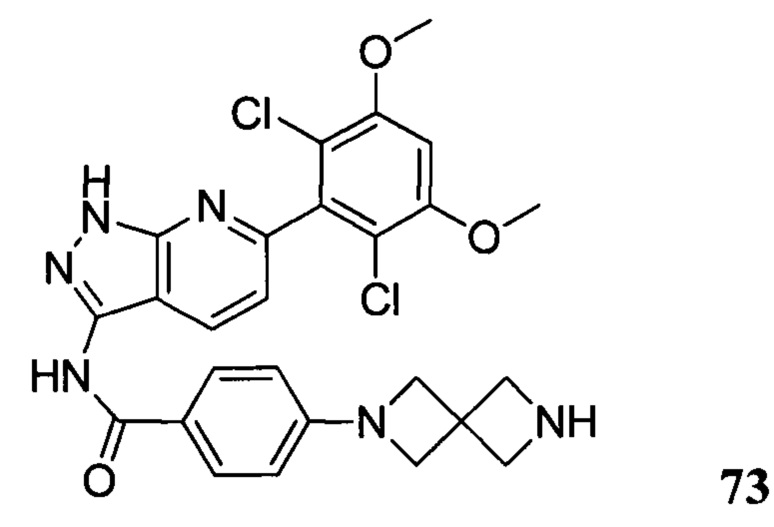
1H ЯМР (d-MeOD, 400 МГц) δ ppm 8,49 (d, 1H, J=8,4 Гц), 7,94 (d, 2H, J=8,8 Гц), 7,12 (d, 1H, J=8,4 Гц), 6,94 (s, 1H), 6,55 (d, 2H, J=8,4 Гц), 4,08 (s, 4H), 3,98 (s, 6H), 3,83 (s, 4H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-6-(4-метилпиперазин-1-ил)никотинамид
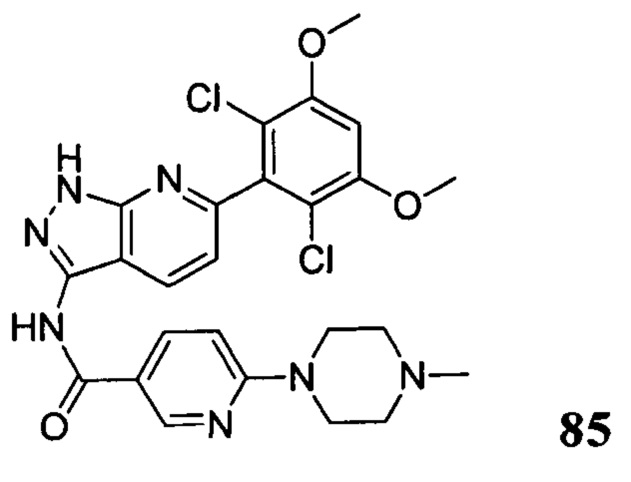
1H ЯМР (ДМСО-d6, 400 МГц) δ ppm 8,84 (s, 1H), 8,37 (d, 1H, J=8,4 Гц), 8,20 (d, 1H, J=7,6 Гц), 7,03 (s, 2H), 6,92 (d, 1H, J=8,8 Гц), 3,98 (s, 6H), 3,63 (s, 4H), 2,41 (t, 4H, J=4,0 Гц), 2,22 (s, 3Н). ЖХ-МС: 542 2 [M+H]+, время удержания = 1,18 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-6-(4-этилпиперазин-1-ил)никотинамид
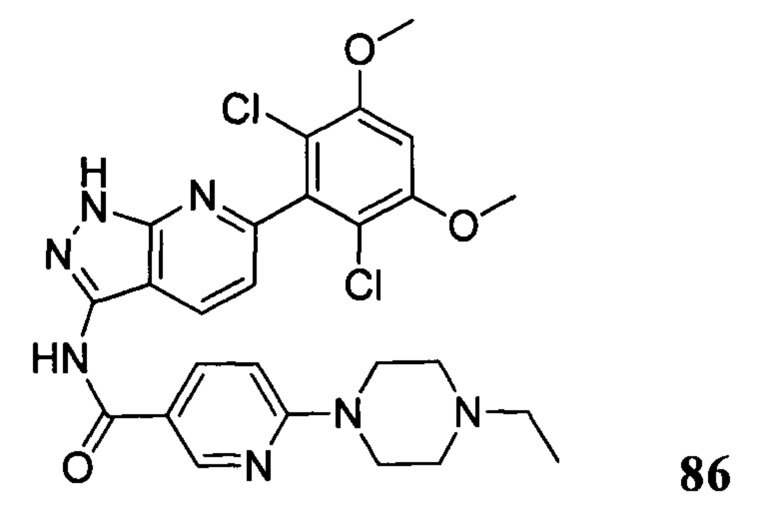
1H ЯМР (ДМСО-d6, 400 МГц) δ ppm 13,40 (s, 1H), 10,91 (s, 1H), 8,84 (d, 1H, J=2,4 Гц), 8,41 (d, 1H, J=8,4 Гц), 8,19 (dd, 1H, J1=J2=2,4 Гц), 7,09 (t, 2H, J=12,4 Гц), 6,93 (d, 1H, J=9,2 Гц), 3 98 (s, 6H), 3,65 (t, 4H, J=3,6 Гц), 2,45 (t, 4H, J=4,8 Гц), 2,39 (q, 2H, J=7,2 Гц), 1,05 (t, 3H, J=6,8 Гц). ЖХ-МС: 556,2 [M+H]+, время удержания = 1,19 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-6-(3,3-диметилпиперазин-1-ил)никотинамид
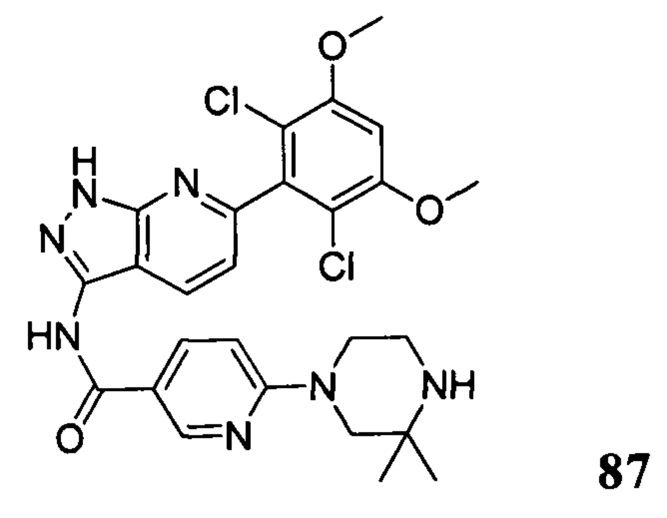
1Н ЯМР (ДМСО-d6, 400 МГц) δ ppm 13,39 (s, 1Н), 10,86 (s, 1H), 8,81 (d, 1H, J=2,0 Гц), 8,40 (d, 1H, J=8,0 Гц), 8,16 (dd, 1H, J1=2,4 Гц, J2=2,4 Гц), 7,08 (t, 2H, J=8,4 Гц), 6,90 (d, 1H, J=9,2 Гц), 3,99 (s, 6H), 3,60 (t, 2H, J=4,0 Гц), 3,43 (s, 2H), 2,82 (t, 2H, J=4,4 Гц), 1,04 (s, 6H). ЖХ-МС: 556,2 [M+H]+ время удержания = 1,21 минуты.
6-(4-циклопропилпиперазин-1-ил)-N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)никотинамид
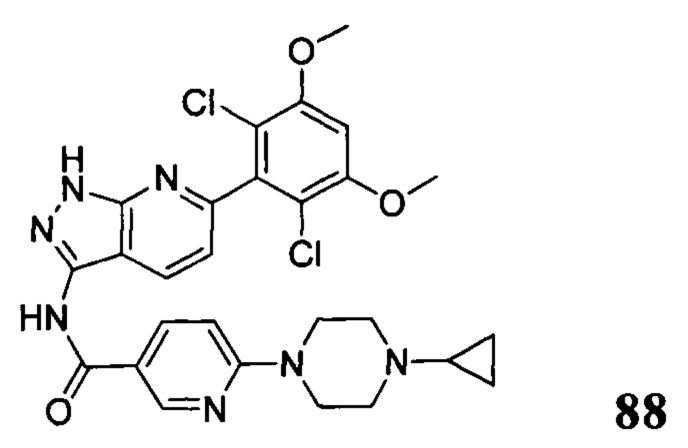
1Н ЯМР (ДМСО-d6, 400 МГц) δ ppm 13,40 (s, 1Н), 10,92 (s, 1H), 8,85 (d, 1H, J=1,6 Гц), 8,41 (d, 1H, J=8,4 Гц), 8,20 (dd, 1H, J1=2,0 Гц, J2=1,6 Гц), 7,08 (t, 2H, J=8,0 Гц), 6,94 (d, 1H, J=9,2 Гц), 3,98 (s, 6H), 3,62 (s, 4H), 2,62 (s, 4H), 1,66 (d, 1H, J=3,6 Гц), 0,46 (d, 2H, J=4,4 Гц), 0,38 (d, 2H, J=2,4 Гц). ЖХ-МС: 568,2 [M+H]+, время удержания = 1,21 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-циклопропилпиперидин-1-ил)бензамид
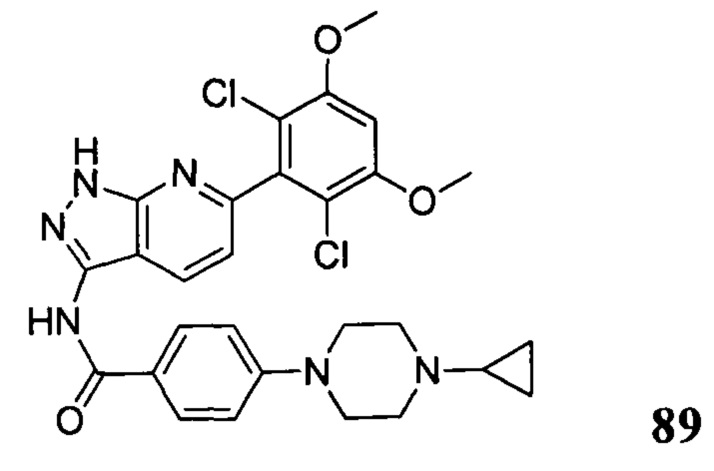
1H ЯМР (d6-ДМСО, 400 МГц) δ ppm 8,34 (d, 1H, J=8 Гц), 8,00 (d, 2H, J=8,8 Гц), 7,02 (t, 4H, J=7,8 Гц), 3,98 (s, 6H), 3,26 (s, 4H), 2,67 (s, 4H), 1,66 (s, 1Н), 0,45 (d, 2H, J=4,8 Гц), 0,363 (s, 2H). ЖХ-МС: 567 (М+Н)+, время удержания = 1,22 минуты.
Путь синтеза V
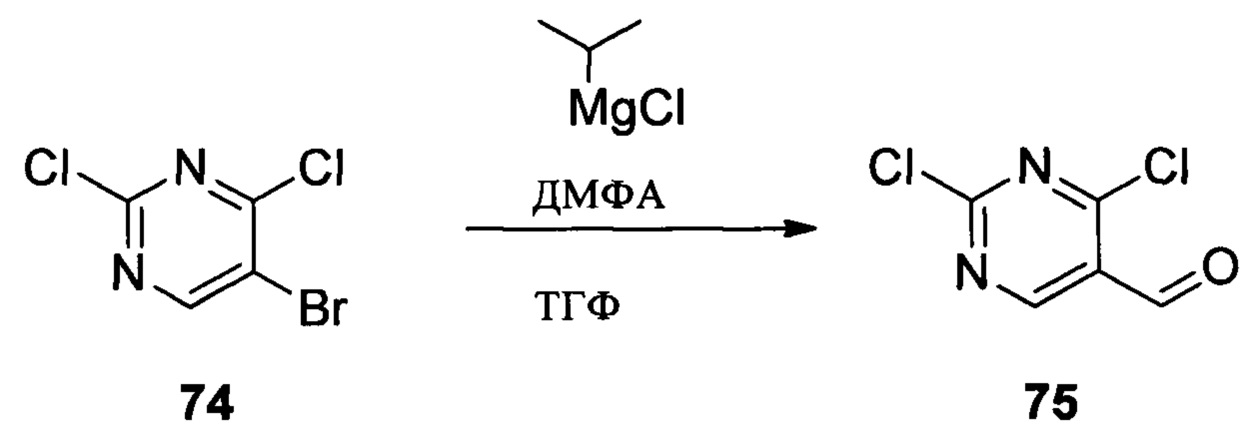
В защитной атмосфере азота соединение 74 (10,00 г, 43,9 ммоль) и тетрагидрофуран (100,0 мл) добавляли в сухую трехгорлую колбу объемом 250 мл и раствор комплекса изопропилмагний хлорид - лития хлорид (2М) (24,2 мл, 48,3 ммоль) по каплям медленно добавляли при температуре -78°С, перемешивали при температуре -78°С - 35°С в течение 0,5 часа и затем по каплям медленно добавляли ДМФА (9,6 г, 131,6 ммоль) при -78°С, перемешивали при температуре -35°С в течение 4 часов. Реакцию гасили насыщенным раствором хлорида аммония при температуре -78°С, указанный реакционный раствор разбавляли 100,0 мл воды и дважды экстрагировали этилацетатом 200 мл. Органические фазы объединяли и сушили над безводным сульфатом натрия и растворитель выпаривали роторном испарителе с получением соединения 75 (2,1 г, 31%) в виде белого твердого вещества.
1H ЯМР (ДМСО-d6, 400 МГц) δ ppm 10,19 (s, 1H), 9,12 (s, 1H).
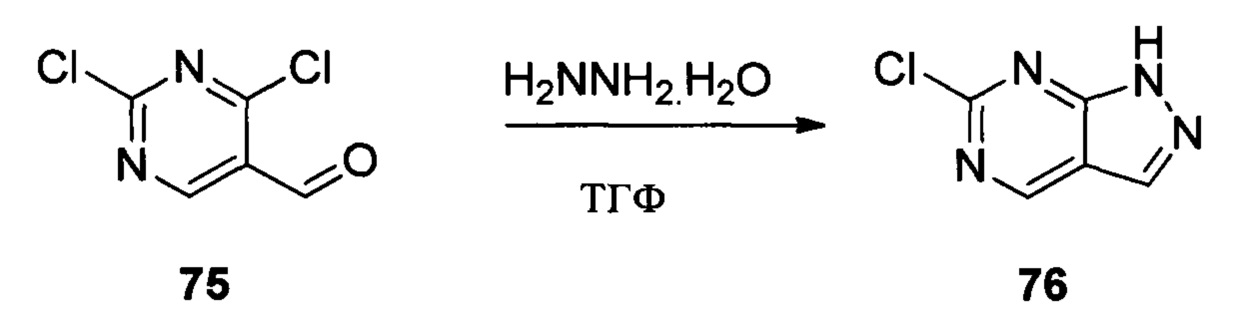
Соединение 75 (2,0 г, 11,3 ммоль) растворяли в ТГФ (10 мл) и добавляли гидрат гидразина (1,33 г, 22,6 ммоль) в ТГФ (20 мл) при температуре 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часа и очищали на колонке с силикагелем с получением соединения 76 в виде желтого твердого вещества (1,0 г, 57%). ЖХ-МС: 155 (М+Н)+, время удержания = 0,929 минут.
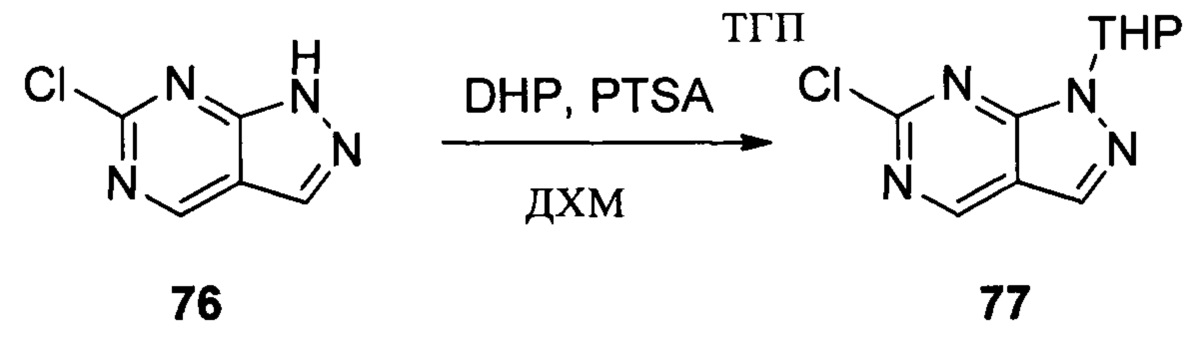
Соединение 76 (1,0 г, 6,5 ммоль), DHP (1,1 г, 13 ммоль) и PTSA (224 мг, 1,3 ммоль) растворяли в 30 мл метиленхлорида в течение ночи при комнатной температуре и полученную смесь очищали при помощи колоночной хроматографии на силикагеле с получением твердого вещества желтого цвета, представляющего собой соединение 77 (1,0 г, 65%). ЖХ-МС: 239 (М+Н)+, время удержания = 1,32 минут.
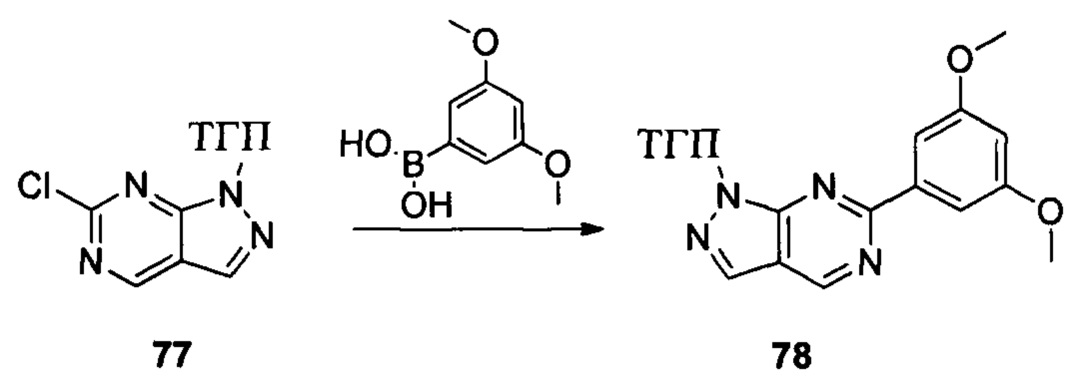
Соединение 77 (900 мг, 3,8 ммоль), 3,5-диметоксифенилбороновую кислоту (826 мг, 4,5 ммоль), Pd(dppf)Cl2 (415 мг, 0,57 ммоль) и фосфат калия (960 мг, 4,5 ммоль) растворяли в 1,4-диоксане (12 мл), проводили взаимодействие в микроволновой печи при 110°С в течение 90 минут и очищали на колонке с силикагелем с получением твердого вещества желтого цвета, представляющего собой соединение 78 (950 мг, 74%). ЖХ-МС: 341 (М+Н)+, время удержания = 1,803 минут.
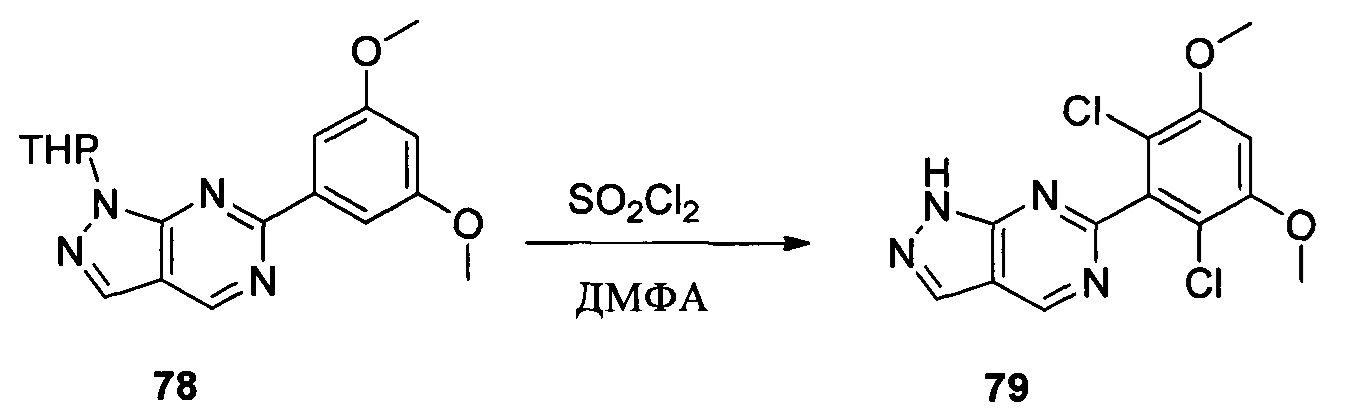
Соединение 78 (500 мг, 1,5 ммоль) растворяли в 10 мл метиленхлорида. Добавляли сульфонилхлорид (446 мг, 3,3 ммоль) при 0°С и перемешивали при комнатной температуре в течение 4 часов. ЖХ-МС показала отсутствие остаточного материала. Реакционный раствор гасили небольшим количеством воды и очищали на колонке с силикагелем с получением белого твердого соединения 79 (460 мг, 84%). ЖХ-МС: 325 (М+Н)+, время удержания = 1,24 минут.
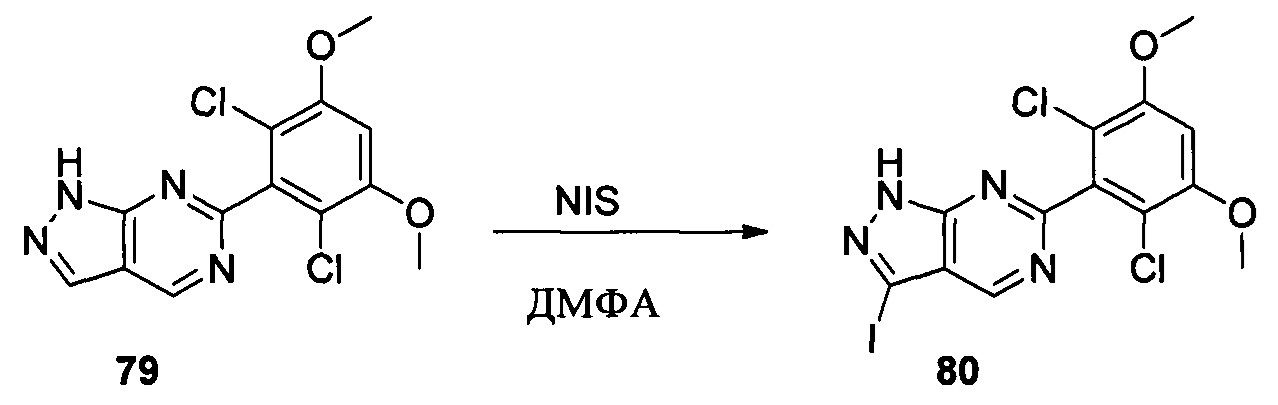
Соединение 79 (400 мг, 1,23 ммоль) и NIS (554 мг, 2,46 ммоль) растворяли в 5 мл ДМФА при 80°С в течение ночи. Реакционную смесь сушили в роторном испарителе и очищали над силикагелем с получением соединения 80 (370 мг, 67%). ЖХ-МС: 450 (М+Н)+, время удержания = 1,577 минут.
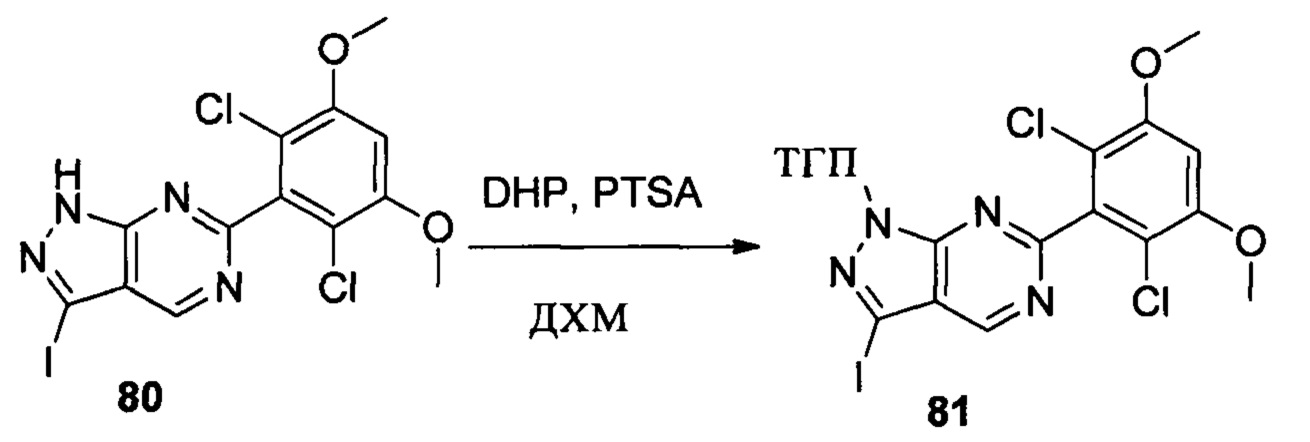
Соединение 80 (370 мг, 0,82 ммоль), DHP (138 мг, 1,64 ммоль) и PTSA (28 мг, 0,16 ммоль) растворяли в 10 мл дихлорметана и перемешивали в течение 4 часов при комнатной температуре. Смесь очищали на колонке с получением твердого вещества желтого цвета, представляющего собой соединение 81 (450 мг, 100%). ЖХ-МС: 534 (М+Н)+, время удержания = 1,964 минут.
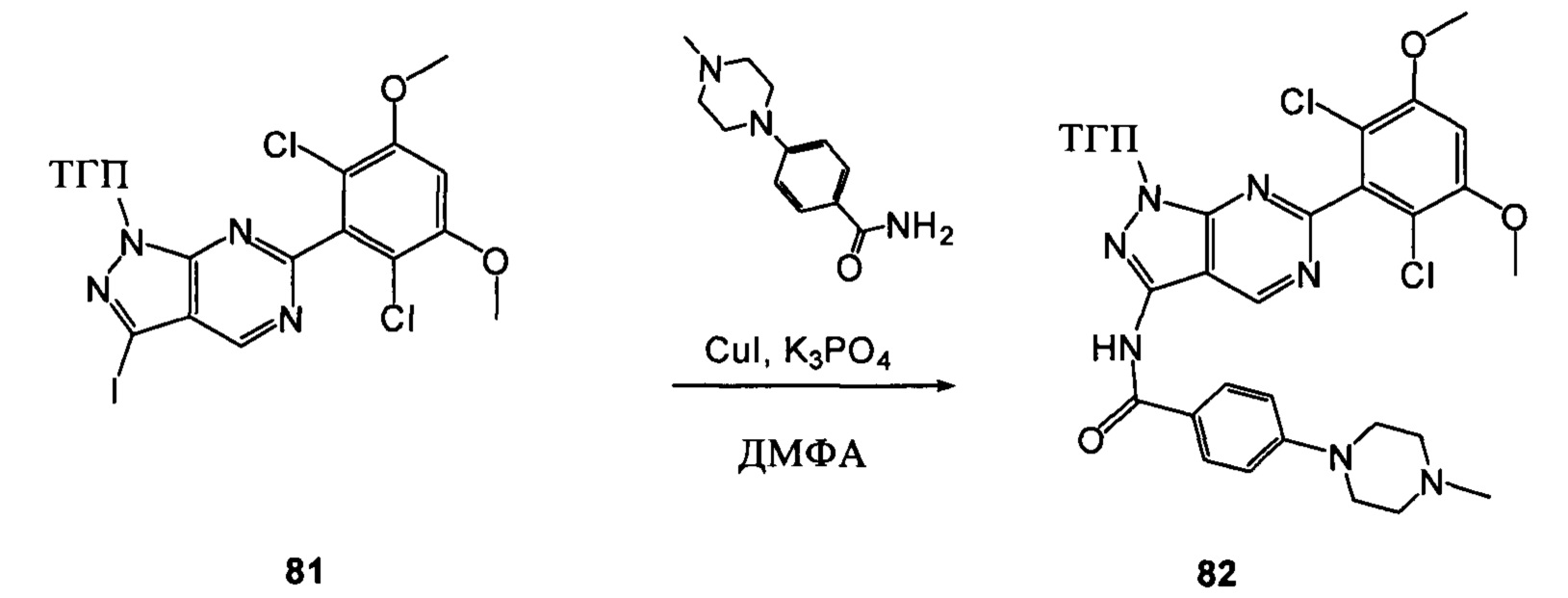
Соединение 81 (100 мг, 0,19 ммоль), 4-(4-метилпиперазин-1-ил)бензамид (82 мг, 0,37 ммоль), CuI (7 мг, 0,037 ммоль), K3PO4 (119 мг, 0,56 ммоль) и N,N-диметил-1,2-циклогександиамин (5 мг, 0,037 ммоль) растворяли в 3 мл ДМФА и перемешивали в течение ночи при температуре 110°С, сушили в роторном испарителе и очищали на колонке с получением коричневого масла, представлявшего собой соединение 82 (55 мг, выход 47%). ЖХ-МС: 626 (М+Н)+, время удержания = 1,37 минут.
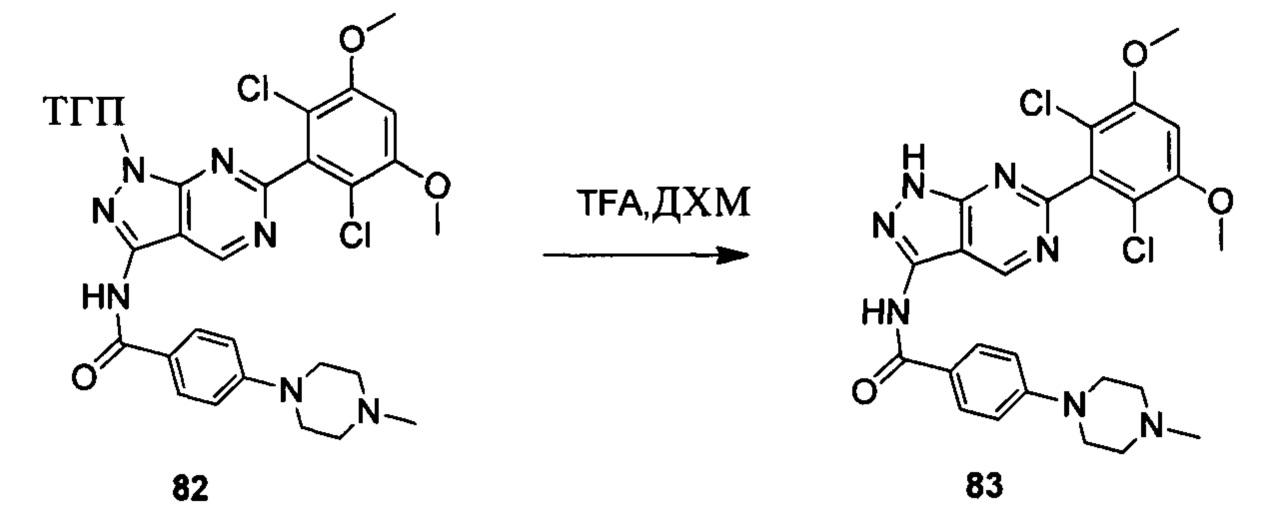
Соединение 82 (55 мг, 0,088 ммоль) и 2 мл трифторуксусной кислоты растворяли в 2 мл дихлорметана в течение ночи при комнатной температуре. После завершения взаимодействия растворитель выпаривали в роторном испарителе с получением неочищенного продукта, который очищали с помощью предварительной ТСХ и затем предварительной ВЭЖХ с получением 4 мг желтого твердого продукта, представляющего собой соединение 83, выход составил 8%. ЖХ-МС: 542(М+Н)+, время удержания = 1,27 минут.
1Н ЯМР (d-MeOD, 400 МГц) δ ppm 9,76 (s, 1Н), 8,06 (d, 2Н, J=8,8 Гц), 7,16 (d, 2Н, J=8,8 Гц), 6,96 (s, 1Н), 4,13 (d, 2Н, J=10 Гц), 3,99 (s, 6Н), 3,63 (d, 2Н, J=2,8 Гц), 3 21-3,17 (m, 4Н), 2,99 (s, 3Н).
Аналогичными способами были получены следующие соединения:
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1H-пиразоло[3,4-d]пиримидин-3-ил)-4-((3R,5S)-3,5-диметилпиперазин-1-ил)бензамид
1Н ЯМР (d-MeOD, 400 МГц) δ ppm 9,76 (s, 1Н), 8,05 (d, 2H, J=8,8 Гц), 7,17 (d, 2H, J=8,8 Гц), 6,96 (s, 1H), 3,99 (s, 6H), 3,53-3,49 (m, 2H), 3,42-3,38 (m, 2H), 2,86-2,80 (m, 2H), 1,48 (d, 6H, J=5,2 Гц). ЖХ-МС: 556 (M+H)+, время удержания = 133 минуты.
Следующие соединения можно получить тем же способом синтеза, что и соединение 63:
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-6-(4-метилпиперазин-1-ил)никотинамид
1H ЯМР (ДМСО-d6, 400 МГц) δ ppm 8,84 (s, 1Н), 8,37 (d, 1H, J=8,4 Гц), 8,20 (d, 1Н, J=7,6 Гц), 7,03 (s, 2H), 6,92 (d, 1H, J=8,8 Гц), 3,98 (s, 6H), 3,63 (s, 4H), 2,41 (t, 4H, J=4,0 Гц), 2,22 (s, 3Н). ЖХ-МС: 542,2 [M+H]+, время удержания = 1,18 минут.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1H-пиразоло[3,4-b]пиридин-3-ил)-6-(4-этилпиперазин-1-ил)никотинамид
1Н ЯМР (ДМСО-d6, 400 МГц) δ ppm 13,40 (s, 1H), 10,91 (s, 1H), 8,84 (d, 1H, J=2,4 Гц), 8,41 (d, 1H, J=8,4 Гц), 8,19 (dd, 1H, J1=J2=2,4 Гц), 7,09 (t, 2H, J=12,4 Гц), 6,93 (d, 1H, J=9,2 Гц), 3,98 (s, 6H), 3,65 (t, 4H, J=3,6 Гц), 2,45 (t, 4H, J=4,8 Гц), 2,39 (q, 2H, J=7,2 Гц), 1,05 (t, 3H, J=6,8 Гц). ЖХ-МС: 556,2 [M+H]+, время удержания = 1,19 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-6-(3,3-диметилпиперазин-1-ил)никотинамид
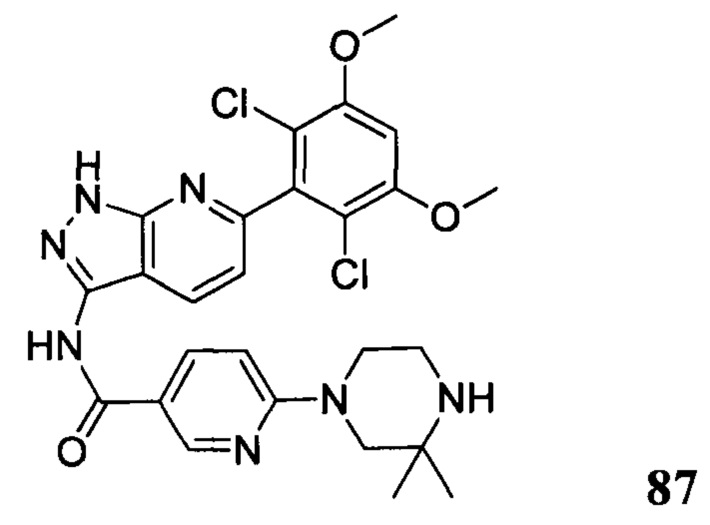
1Н ЯМР (ДМСО-d6, 400 МГц) δ ppm 13,39 (s, 1Н), 10,86 (s, 1H), 8,81 (d, 1H, J=2,0 Гц), 8,40 (d, 1H, J=8,0 Гц), 8,16 (dd, 1H, J1=2,4 Гц, J2=2,4 Гц), 7,08 (t, 2H, J=8,4 Гц), 6,90 (d, 1H, J=9,2 Гц), 3,99 (s, 6H), 3,60 (t, 2H, J=4,0 Гц), 3,43 (s, 2H), 2,82 (t, 2H, J=4,4 Гц), 1,04 (s, 6H). ЖХ-МС: 556,2 [M+H]+, время удержания = 1,21 минут.
6-(4-циклопропилпиперазин-1-ил)-N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)никотинамид
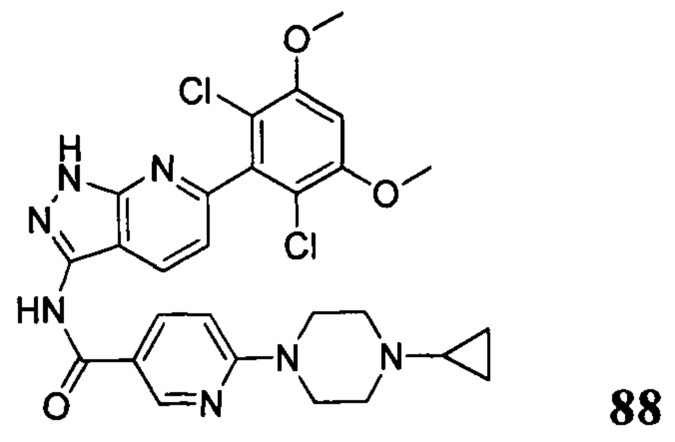
1Н ЯМР (ДМСО-d6, 400 МГц) δ ppm 13,40 (s, 1Н), 10,92 (s, 1Н), 8,85 (d, 1H, J=1,6 Гц), 8,41 (d, 1H, J=8,4 Гц), 8,20 (dd, 1H, J1=2,0 Гц, J2=1,6 Гц), 7,08 (t, 2H, J=8,0 Гц), 6,94 (d, 1H, J=9,2 Гц), 3,98 (s, 6H), 3,62 (s, 4H), 2,62 (s, 4H), 1,66 (d, 1H, J=3,6 Гц), 0,46 (d, 2H, J=4,4 Гц), 0,38 (d, 2H, J=2,4 Гц). ЖХ-МС: 568,2 (M+H)+, время удержания = 1,21 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-циклопропилпиперидин-1-ил)бензамид
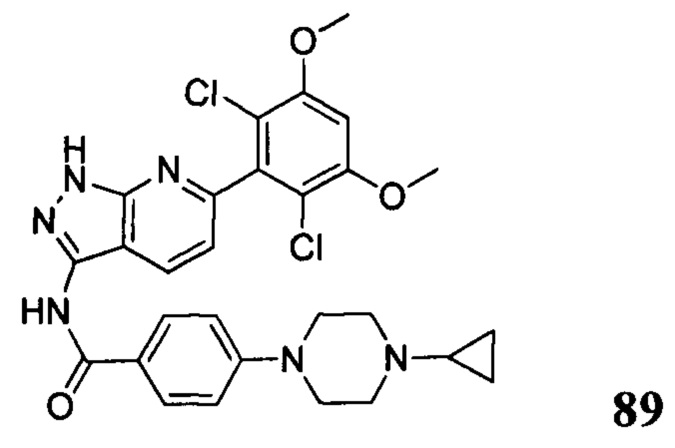
1Н ЯМР (d6-ДМСО, 400 МГц) δ ppm 8,34 (d, 1Н, J=8 Гц), 8,00 (d, 2H, J=8,8 Гц), 7,02 (t, 4H, J=7,8 Гц), 3,98 (s, 6H), 3,26 (s, 4H), 2,67 (s, 4H), 1,66 (s, 1H), 0,45 (d, 2H, J=4,8 Гц), 0,363 (s, 2H). ЖХ-МС: 567 (M+H)+, время удержания = 1,22 минуты.
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-((3S,5R)-3,4,5-триметилпиперидин-1-ил)бензамид
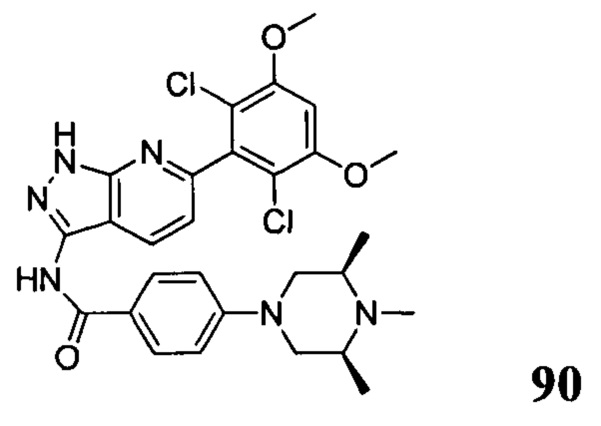
1Н ЯМР (400 МГц, ДМСО-d6): 13,41 (s, 1Н), 10,90 (s, 1Н), 9,38 (s, 1Н), 8,39 (d, 1Н, J=8,0 Гц), 8,06 (d, 2Н, J=8,8 Гц), 7,16 (d, 2Н, J=9,2 Гц), 7,09 (d, 1Н, J=12,4 Гц), 7,05 (s, 1Н), 4,19 (d, 2Н, J=13,6 Гц), 3,99 (s, 6Н), 3,35-3,44 (m, 2Н), 2,90-2,95 (m, 2Н), 2,88 (d, 3Н, J=4,4 Гц), 1,39 (d, 6Н, J=6,4 Гц).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(3,3-диметилпиперазин-1-ил)-3-фторбензамид
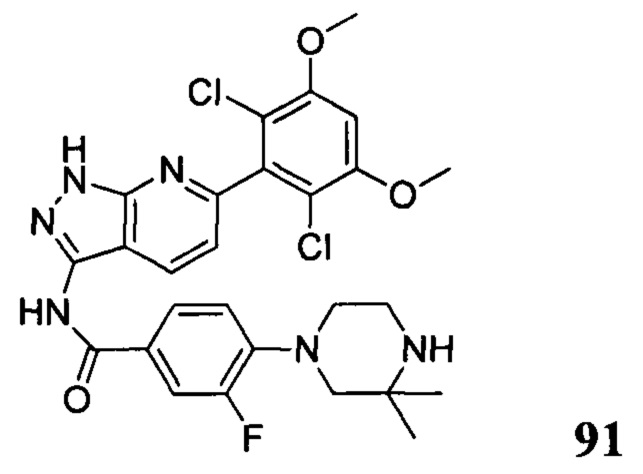
1H ЯМР (400 МГц, ДМСО-d6): 13,48 (s, 1Н), 11,10 (s, 1H), 8,86 (brs, 1H), 8,39 (d, 1H, J=8,0 Гц), 7,96-7,92 (m, 2H), 7,25 (t, 1H, J=8,8 Гц), 7,10 (d, 1H, J=2,8 Гц), 7,05 (s, 1H), 3,99 (s, 6H), 3,35 (s, 4H), 3,18 (s, 2H), 1,41 (s, 6H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-2-(4-метилпиперазин-1-ил)пиримидин-5-формамид
1Н ЯМР (400 МГц, ДМСО-d6): 13,46 (s, 1Н), 11,08 (s, 1Н), 9,00 (s, 2H), 8,42 (d, 1H, J=8,4 Гц), 8,33 (s, 1H), 7,09 (d, 1H, J=8,4 Гц), 7,05 (s, 1H), 3,99 (s, 6H), 3,85-3,88 (m, 4H), 2,38-2,40 (m, 4H), 2,23 (s, 3H),
Следующие соединения были получены теми же способами, что и соединение 44:
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(пиперидин-1-ил)бензамид
1Н ЯМР (400 МГц, ДМСО-d6): 12,85 (s, 1Н), 10,38 (s, 1Н), 7,87 (d, 2Н, J=8,8 Гц), 7,77 (d, 1Н, J=8,4 Гц), 7,50 (s, 1Н), 7,08 (t, 2Н, J=8,4 Гц), 6,62 (d, 2Н, J=8,8 Гц), 6,34 (d, 1Н, J=6,4 Гц), 3,92 (s, 6Н), 3,77-3,82 (m, 1Н), 1,91-2,00 (m, 2Н), 1,65-1,73 (m, 2Н), 1,55-1,62 (m, 2Н), 1,45-1,51 (m, 2Н).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-((4-метилпиперазин-1-ил)метил)бензамид
1H ЯМР (400 МГц, ДМСО-d6): 13,01 (s, 1Н), 10,91 (s, 1H), 8,10 (s, 2H), 7,79 (s, 1H), 7,54 (s, 3H), 7,08 (s, 2H), 3,91 (s, 6H), 3,83 (s, 2H), 3,36-3,48 (m, 2H), 2,95-3,14 (m, 4H), 2,79 (s, 3H), 2,50-2,58 (m, 2H).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1H-индазол-3-ил)-4-(4-метил-1,4-диазепан-1-ил)бензамид
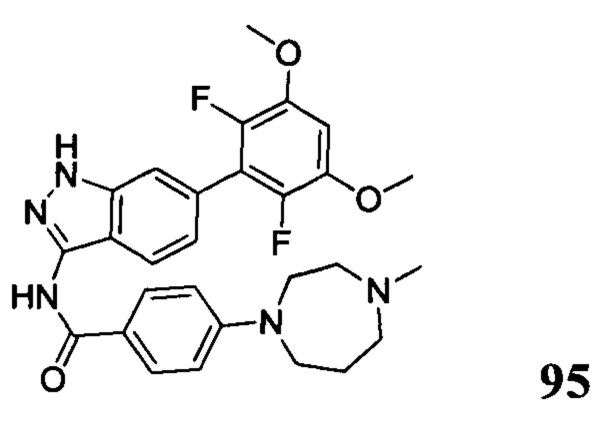
1Н ЯМР (400 МГц, ДМСО-d6): 13,91 (s, 1Н), 10,56 (s, 1H), 9,81 (s, 1H), 8,02 (d, 2H, J=8,4 Гц), 7,78 (d, 1H, J=8,4 Гц), 7,52 (s, 1H), 7,06-7,09 (m, 2H), 6,89 (d, 2H, J=8,8 Гц), 3,92 (s, 6H), 3,85-3,96 (m, 2H), 3,67-3,74 (m, 1H), 3,45-3,58 (m, 3H), 3,12-3,26 (m, 1H), 2,86 (d, 3H, J=3,6 Гц), 2,15-2,25 (m, 2H).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-2-(4-метилпиперазин-1-ил)пиримидин-5-формамид
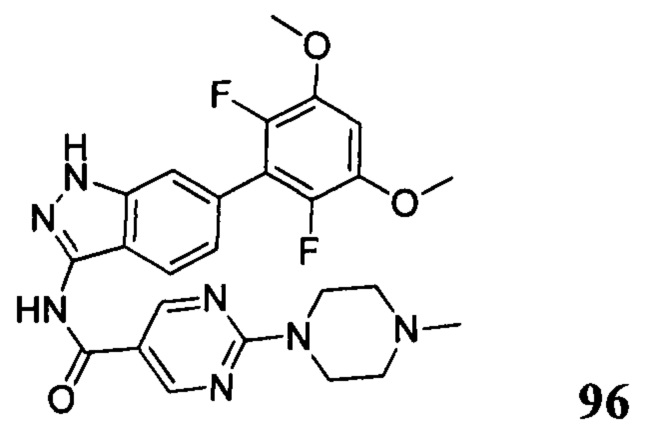
1H ЯМР (400 МГц, ДМСО-d6): 12,99 (s, 1H), 10,95 (s, 1H), 9,08 (s, 2H), 7,84 (d, 1H, J=8,4 Гц), 7,54 (s, 1H), 7,06-7,11 (m, 2H), 4,75-4,95 (m, 2H), 3,92 (s, 6H), 3,35-3,55 (m, 2H), 3,05-3,35 (m, 4H), 2,84 (s, 3H).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(3,3-диметилпиперазин-1-ил)-3-метоксибензамид
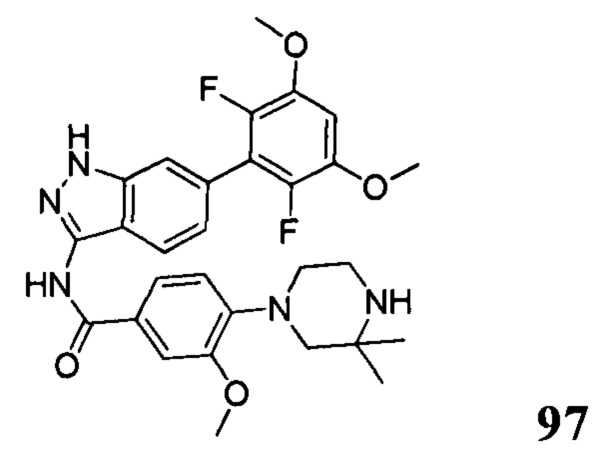
1Н ЯМР (400 МГц, MeOD): 7,86 (d, 1Н, J=8,4 Гц), 7,69-7,72 (m, 2H), 7,54 (s, 1H), 7,18 (d, 1H, J=8,4 Гц), 7,09 (d, 1H, J=8,0 Гц), 6,94 (t, 1H, J=8,0 Гц), 3,98 (s, 3H), 3,93 (s, 6H), 3,42-3,45 (m, 2H), 3,35-3,39 (m, 2H), 3,19 (s, 2H), 1,54 (s, 6H).
N-(6-(2,6-дифтор-3,5-диметоксифенил)-1Н-индазол-3-ил)-4-(3-(диметиламино)пирролидин-1-ил)бензамид
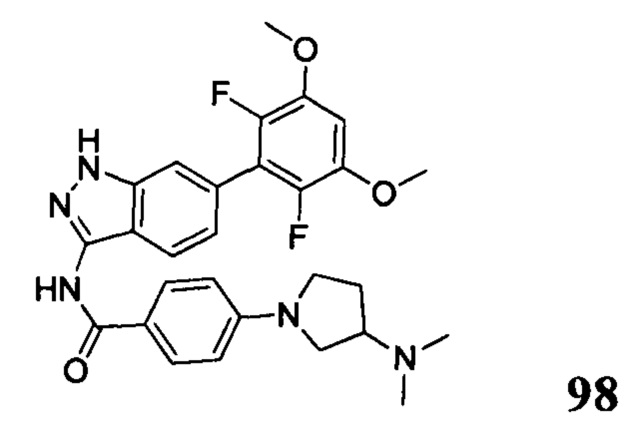
1Н ЯМР (400 МГц, ДМСО-d6): 12,88 (s, 1Н), 10,51 (s, 1Н), 8,00-8,01 (m, 2H), 7,78-7,80 (m, 1H), 7,51 (s, 1H), 7,08 (s, 2H), 6,65-6,67 (m, 2H), 3,92 (s, 6H), 3,62-3,71 (m, 1H), 3,51-3,60 (m, 1H), 2,50 (s, 6H), 2,26-2,35 (m, 2H), 1,97-2,15 (m, 1H).
Следующие соединения можно получить теми же способами синтеза, что и соединение 63:
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(3,3,5,5-тетраметилпиперридин-1-ил)бензамид
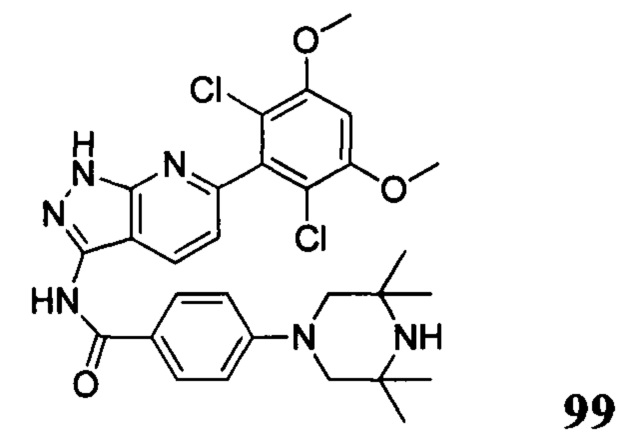
1H ЯМР (400 МГц, MeOD): 8,51 (d, 1Н, J=8,4 Гц), 8,03 (d, 1H, J=7,6 Гц), 7,12-7,22 (m, 3H), 6,95 (s, 1H), 3,99 (s, 6H), 3,47 (s, 4H), 1,52 (s, 12H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(3-(диметиламино)пирролидин-1-ил)бензамид
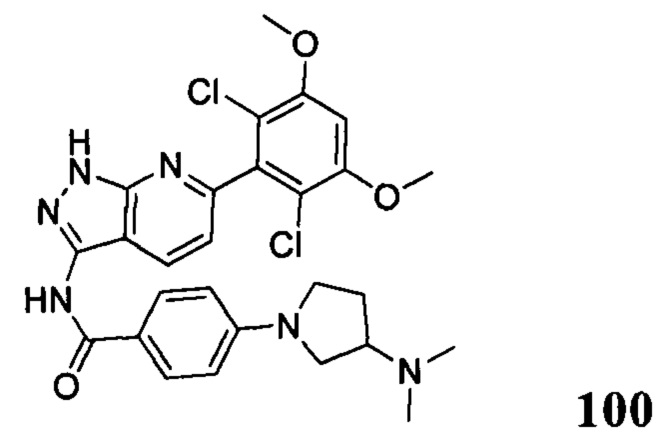
1H ЯМР (400 МГц, MeOD): 8,27 (d, 1H, J=8,4 Гц), 8,01 (d, 2H, J=8,4 Гц), 7,12 (d, 1H, J=8,4 Гц), 6,94 (s, 1H), 6,79 (d, 2H, J=8,4 Гц), 4,06-4,13 (m, 1H), 3,99 (s, 6H), 3,85-3,93 (m, 1H), 3,70-3,75 (m, 1H), 3,61-3,65 (m, 1H), 3,46-3,52 (m, 1H), 3,01 (s, 6H), 2,60-2,67 (m, 1H), 2,26-2,36 (m, 1H).
N-(6-(2,6-дихлор-3,5-диметоксифенил)-1H-пиразоло[3,4-b]пиридин-3-ил)-4-(3,3-диметилпиперазин-1-ил)-3-метоксибензамид
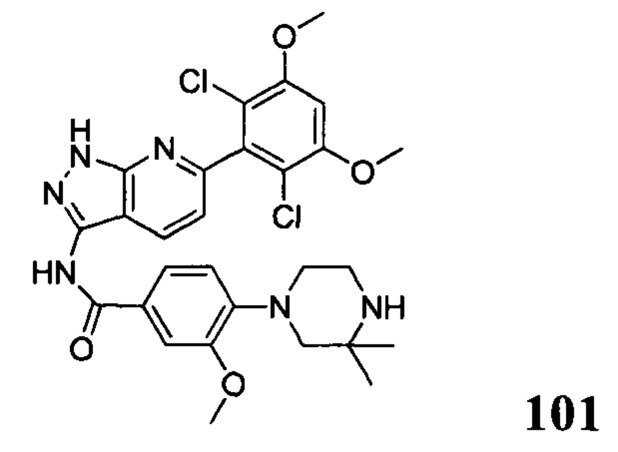
1Н ЯМР (400 МГц, ДМСО-d6): 13,46 (s, 1Н), 11,07 (s, 1Н), 8,41 (d, 1H, J=8,4 Гц), 7,23-7,50 (m, 2H), 7,06-7,11 (m, 3H), 3,99 (s, 6H), 3,92 (s, 3H), 3,29 (s, 2H), 3,25 (s, 2H), 3,07 (s, 2H), 2,58 (s, 1H), 1,41 (s, 6H).
Пример 2: Влияние соединений на активность киназ FGFR1, FGFR2, FGFR3, KDR на молекулярном уровне
1. Экспериментальный метод
Субстрат для ферментативной реакции Poly(Glu,Tyr)4:l разбавляли фосфатно-солевым буферным раствором (PBS) без ионов калия (10 мМ натрий фосфатного буфера, 150 мМ NaCl, рН 7,2-7,4) до 20 мкг/мл, 125 мкл/лунка для покрытия ферментного планшета, взаимодействие проводили при температуре 37°С в течение 12-16 часов. После удаления жидкости из лунок планшет промывали три раза по 5 минут 200 мкл/лунку T-PBS (PBS, содержащий 0,1% Tween-20) в течение 5 минут. Ферментный планшет сушили в печи при температуре 37°С в течение 1-2 часов.
50 мкл раствора АТФ, разбавленного реакционным буфером (50 мМ HEPES, рН 7,4, 50 мМ MgCl2, 0,5 мМ MnCl2, 0,2 мМ Na3VO4, 1 мМ DTT) добавляли в каждую лунку в конечной концентрации 5 мкМ. Соединения разбавляли до соответствующей концентрации в ДМСО, 1 мкл/лунку или в лунку помещали ДМСО в соответствующей концентрации (лунки с отрицательным контролем). Реакцию инициировали добавлением каждого рекомбинантного белка с киназным доменом, разбавленного в 49 мкл реакционного буфера. В каждом эксперименте присутствовали две контрольные лунки без АТФ. Реакционную смесь помещали в аппарат для встряхивания (100 об/мин) для проведения взаимодействия при температуре 37°С в течение 1 часа. Планшеты три раза промывали T-PBS. Добавляли 100 мкл/лунку основного разбавленного раствора антитела PY99 и проводили взаимодействие в аппарате для встряхивания (100 об/мин) при температуре 37°С в течение 0,5 часа. Планшеты три раза промывали T-PBS. Добавляли 100 мкл/лунку вторичного разбавленного раствора, помеченного пероксидазой хрена козьего антимышиного IgG, и проводили взаимодействие в аппарате для встряхивания (100 об/мин) при температуре 37°С в течение 0,5 часа. Планшеты три раза промывали T-PBS. Добавляли 100 мкл/лунку 2 мг/мл OPD проявляющего раствора (разбавленного 0,1М буфером лимонная кислота-цитрат натрия, содержащим 0,03% Н2О2 (рН=5,4)) и проводили взаимодействие в темноте в течение 1-10 минут при температуре 25°С. (Для растворения OPD требовалось воздействие ультразвука, а проявляющий раствор должен быть приготовлен на месте). Реакцию гасили с помощью 50 мкл/лунку 2М H2SO4 и считывали данные при 490 нм с помощью настраиваемого считывателя микропланшетов SPECTRA МАХ 190.
Показатель ингибирования образцов определяли по следующей формуле:

Значения IC50 получали при помощи регрессионного анализа с четырьмя параметрами с использованием программного обеспечения, идущего в комплекте с указанным считывателем микропланшетов.
2. Результаты
Некоторые из значений IC50 приведены в следующей таблице. Символом + обозначены значения IC50, составляющие менее 100 нм, символом ++ обозначены значения IC50 от 100 нм до 500 нм и «нет данных» обозначает, что данных нет.


Данные результаты показали, что соединения согласно настоящему изобретению могут эффективно ингибировать активность различных видов киназы FGFR при чрезвычайно низкой концентрации (≤100 нм).
Пример 3. Влияние указанных соединений на пролиферацию опухолевых клеток, опосредованную FGFR1
1. Экспериментальный метод
Ингибирование роста клеточной линии острого миелобластного лейкоза KG1 (штамм клеток FGFR1-зависимой опухоли; слитный белок FGFR1 в клетке экспрессировался в цитоплазме, приобретен у банка клеток АТСС) указанными соединениями проводилось при помощи комплекта для подсчета клеток ССК-8 (Dojindo). Конкретные стадии заключались в следующем: Клетки KG1 в фазе логарифмического роста высевали в 96-луночный культуральный планшет в соответствующей плотности, 90 мкл на лунку. После культивирования в течение ночи добавляли соединения в различных концентрациях и обработку продолжали в течение 72 часов, устанавливали контрольную группу с растворителем (отрицательный контроль). После обработки в течение 72 часов наблюдали действие соединений на пролиферацию клеток при помощи набора для подсчета клеток ССК-8 (Dojindo). В каждую лунку добавляли 10 мкл реагента ССК-8. После инкубирования в течение 2-4 часов в инкубаторе при температуре 37°С считывали данные при помощи считывателя микропланшетов Spectramax 190 при длине волны 450 нм. Долю ингибирования (%) роста опухолевых клеток указанными соединениями рассчитывали по следующей формуле: Доля ингибирования (%) = (OD лунки отрицательного контроля - OD лунки с соединением) / OD лунки отрицательного контроля × 100%. Значения IC50 получали при помощи регрессионного анализа с четырьмя параметрами с использованием программного обеспечения, идущего в комплекте с указанным считывателем микропланшетов.
2. Результаты
Значения IC50 для некоторых соединений приведены в таблице ниже. Символом + обозначены значения IC50, составляющие менее 200 нм, а символом ++ обозначены значения IC50, составляющие от 200 нм до 1000 нм.
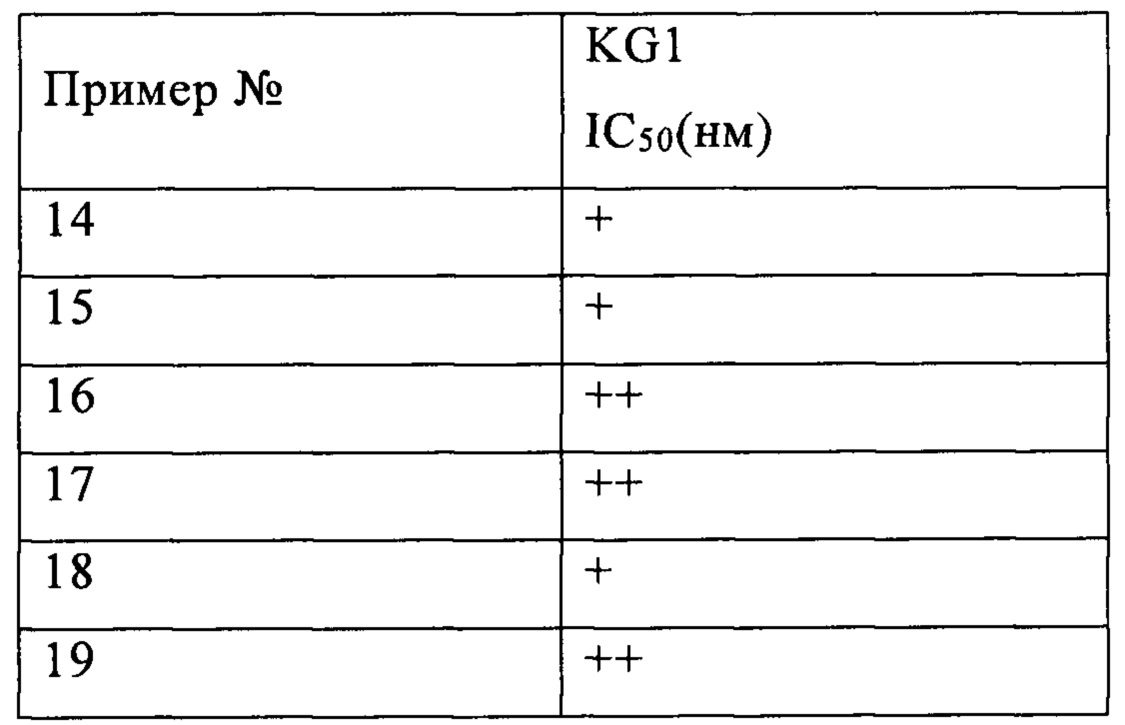
Результаты показывают, что соединения согласно настоящему изобретению могут эффективно ингибировать пролиферацию опухолевых клеток при чрезвычайно низких концентрациях (≤1000 нм, предпочтительно ≤200 нм).
Все публикации, упомянутые в настоящем документе, включены в настоящий документ посредством ссылки, как если бы ссылка на каждую из них была упомянута по отдельности. Кроме того, следует понимать, что после прочтения вышеизложенного специалисты в данной области техники могут произвести различные изменения и модификации настоящего изобретения. Эти эквиваленты также попадают в объем изобретения, определенный прилагаемой формулой изобретения.
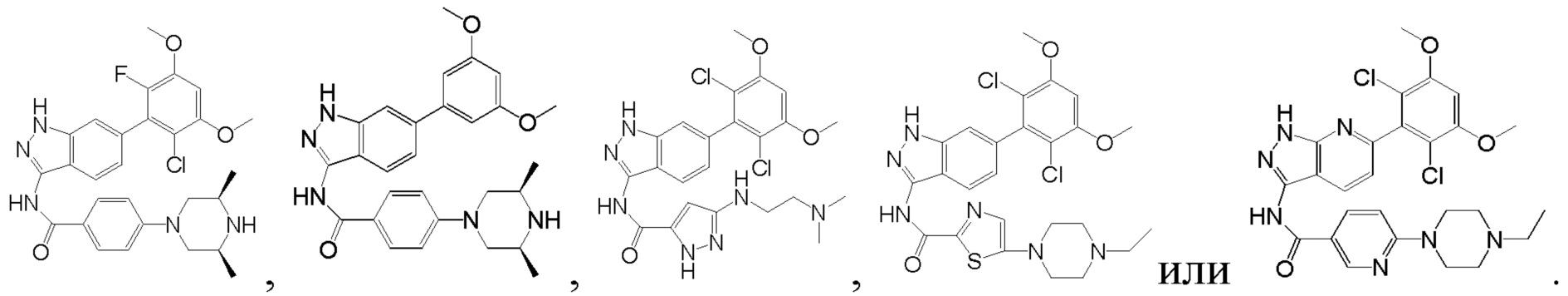
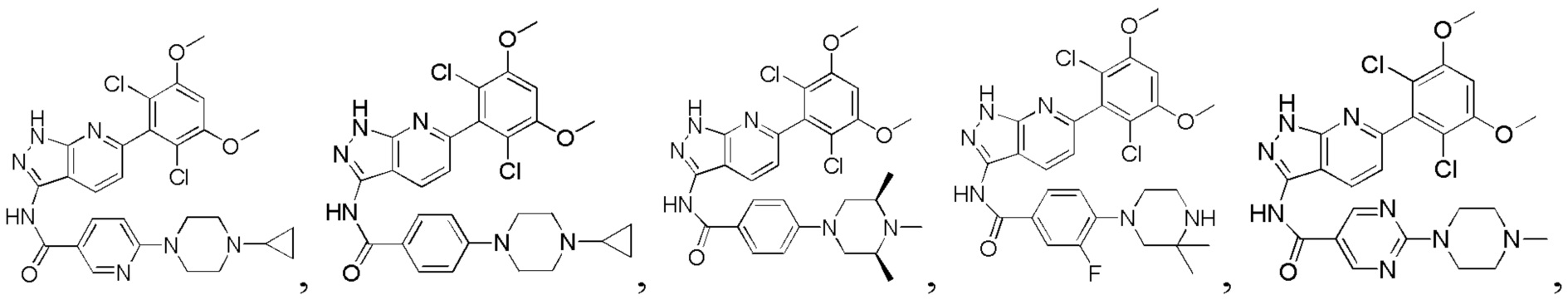
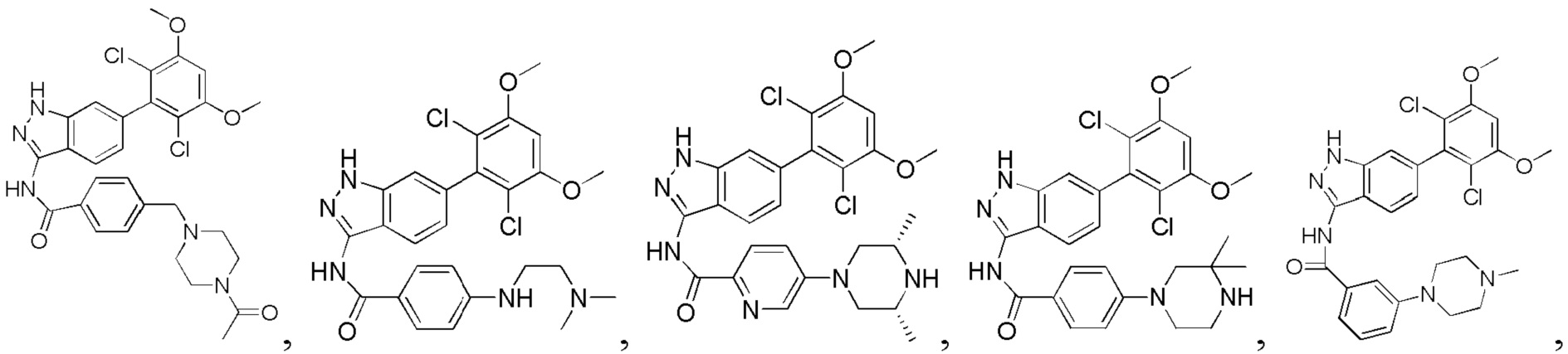
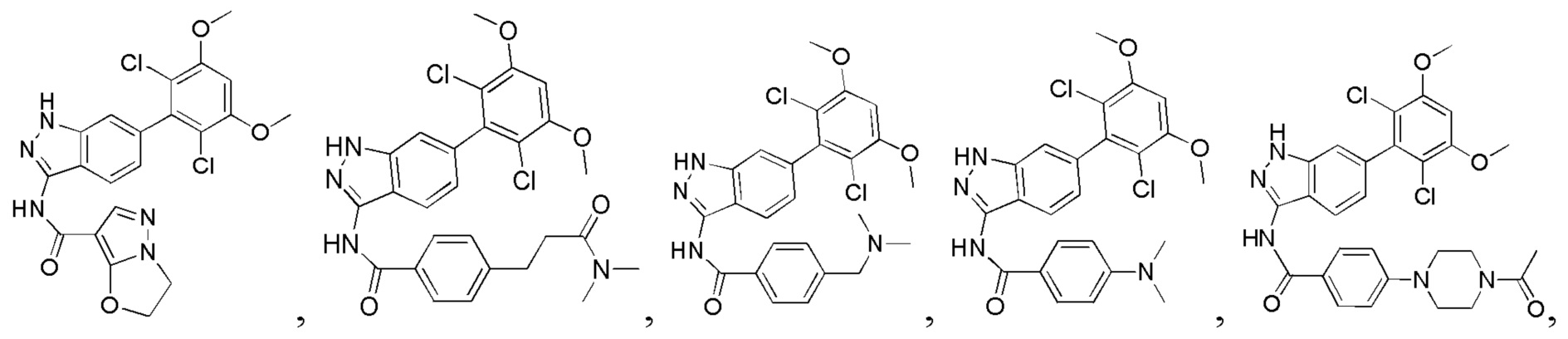
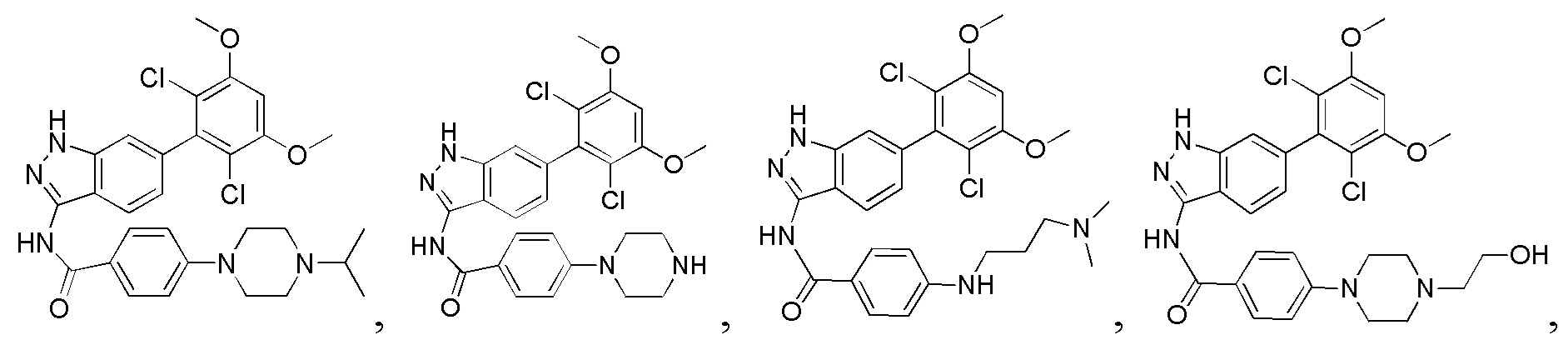
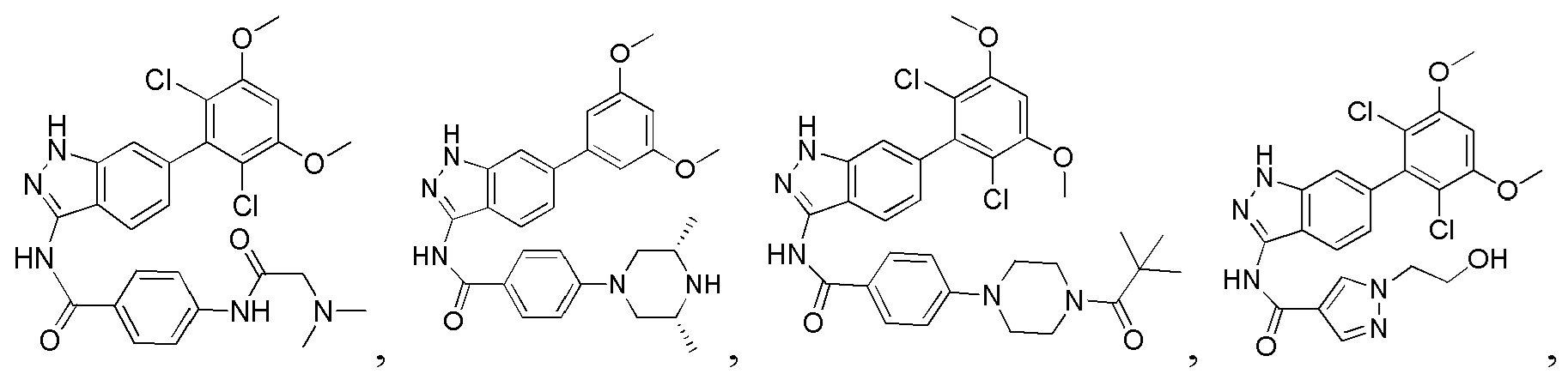
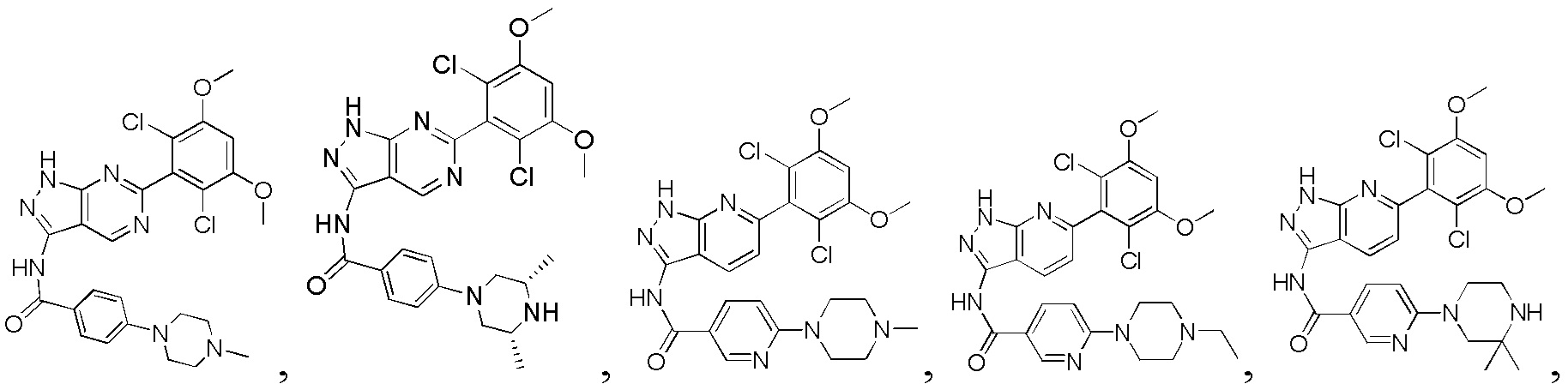
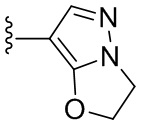
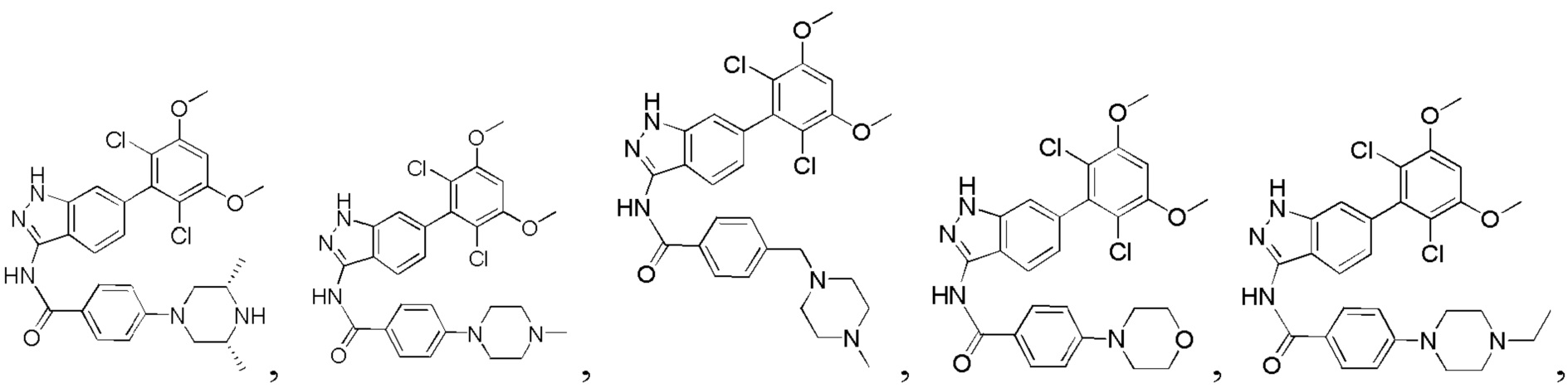
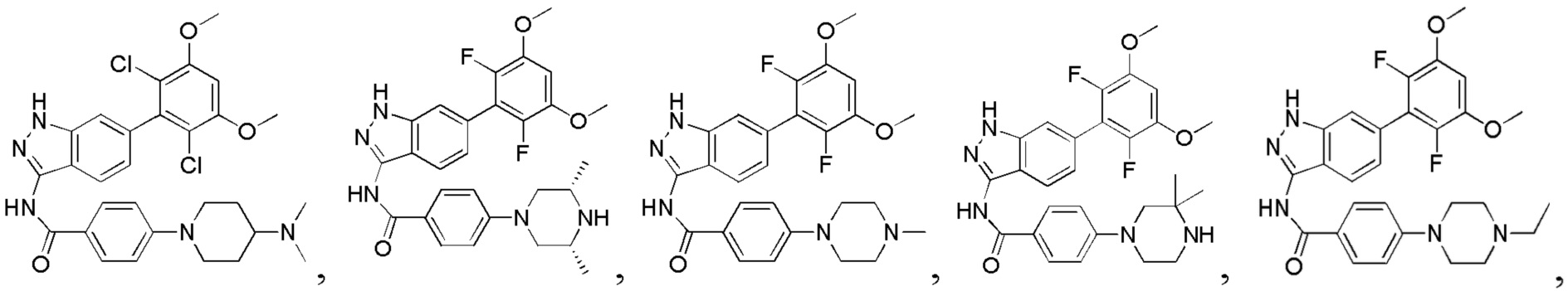
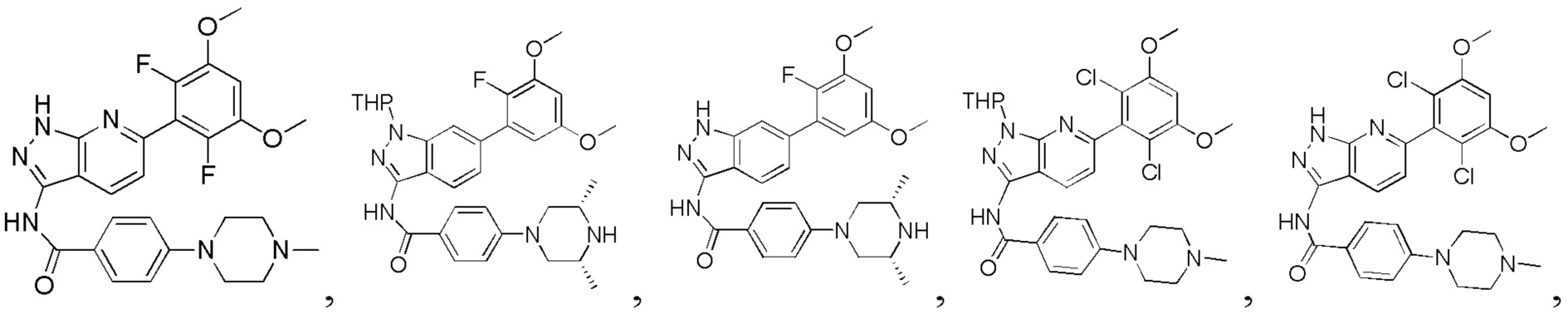
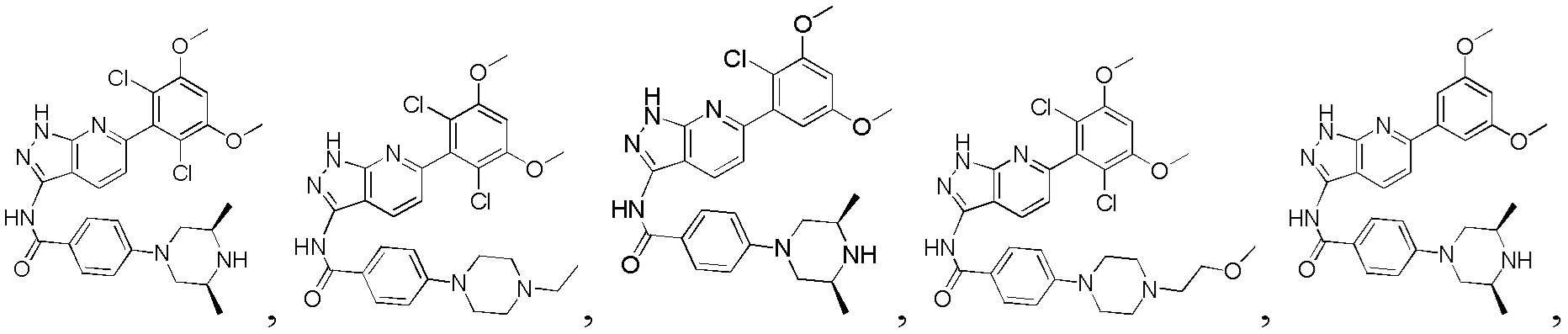
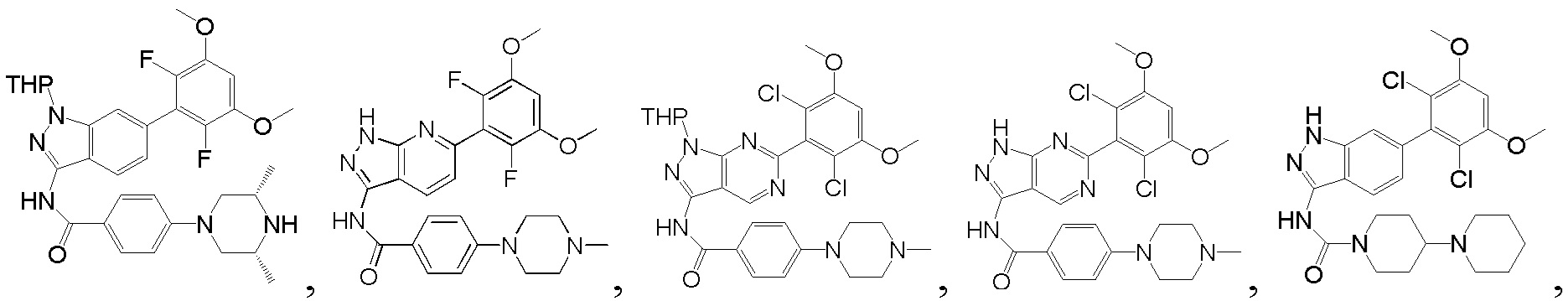
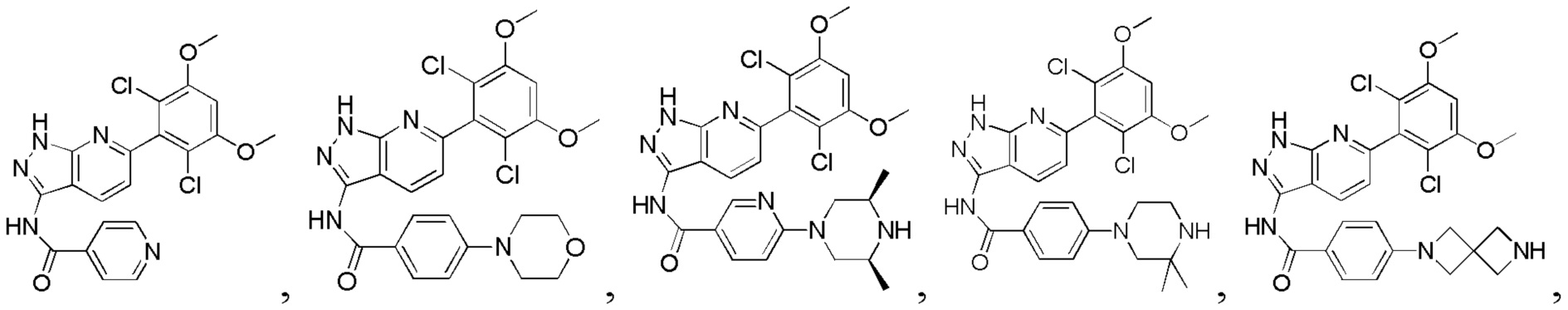
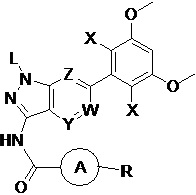
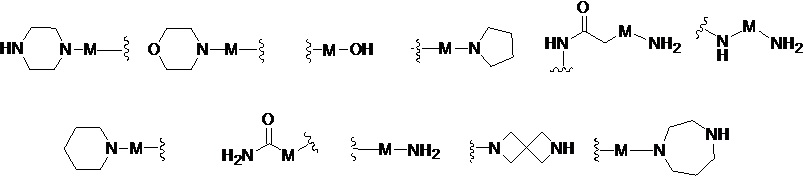
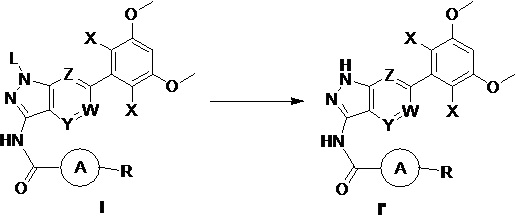
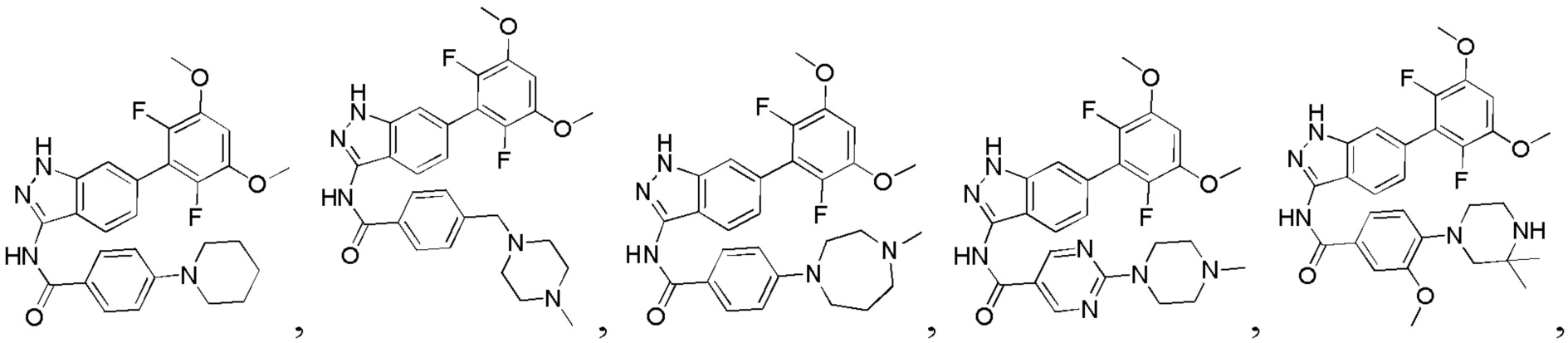
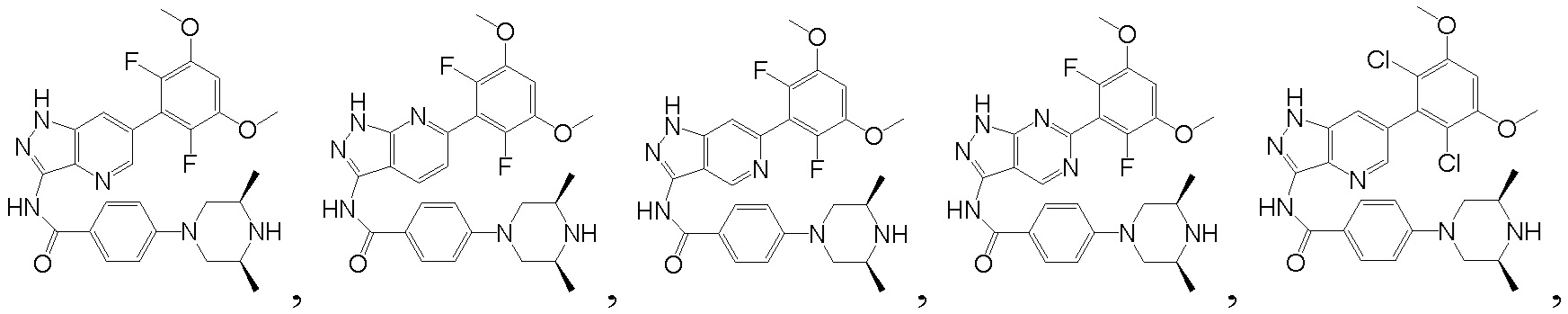
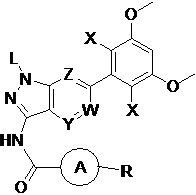
Способ синтеза производных 9-аллилкамптотецина
Способ и устройство в сети радиодоступа
Способ, устройство и система управления доступом
Способ и система для отображения веб-страницы
Пирроло[2,1-f][1,2,4]триазиновое соединение, способ его получения и применения
Способ и устройство гидрообработки углеводородного масла
Способ и устройство для гидрообработки риформата
Способ и устройство оптимизации ресурса памяти
Способ многоантенной передачи данных, базовая станция, пользовательское оборудование и система
Мобильный терминал и способ запуска съемки на мобильном терминале
Соединение нафтиламида, способ его получения и применение

