Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ
Вид РИД
Изобретение
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] По данной заявке испрашивается приоритет на основании заявки PCT/US2015/018085, поданной 27 февраля 2015 года, которая включена во всей своей полноте в настоящий документ посредством ссылки.
СПРАВКА ОТНОСИТЕЛЬНО ФИНАНСИРУЕМЫХ ИЗ ФЕДЕРАЛЬНОГО БЮДЖЕТА НАУЧНО-ИССЛЕДОВАТЕЛЬСКИХ И КОНСТРУКТОРСКИХ РАБОТ
[0002] Настоящее изобретение было осуществлено без государственной поддержки.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ МАТЕРИАЛОВ, ПОДАННЫХ В ЭЛЕКТРОННОМ ВИДЕ
[0003] Отсутствует
СПРАВКА ОТНОСИТЕЛЬНО ПРЕДВАРИТЕЛЬНЫХ РАСКРЫТИЙ, СДЕЛАННЫХ АВТОРОМ ИЗОБРЕТЕНИЯ ИЛИ СОАВТОРОМ ИЗОБРЕТЕНИЯ
[0004] Предварительные раскрытия настоящего изобретения отсутствуют.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0005] В целом, настоящее изобретение относится к использованию соединений для лечения целого ряда нарушений, заболеваний или патологических состояний и, более конкретно, к использованию замещенных производных пиримидина для модулирования протеинкиназ и для лечения заболеваний, опосредованных протеинкиназами.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0006] Протеинкиназы составляют большие семейства структурно родственных белковоподобных ферментов, которые отвечают за регуляцию многочисленных путей передачи сигналов в эукариотической клетке. Протеинкиназы, содержащие сходный каталитический домен из 250-300 аминокислот, катализируют фосфорилирование целевых белковых субстратов. Как таковые, протеинкиназы входят в число самых многообещающих низкомолекулярных лекарственных мишеней.
[0007] Киназы можно классифицировать на семейства по субстратам фосфорилирования (например, белок-тирозин, белок-серин/треонин, липиды, и т.д.). Фосфорилирование тирозина представляет собой центральное событие в регуляции целого ряда биологических процессов, таких как пролиферация, миграция, дифференцировка и выживание клеток. Некоторые семейства рецепторных и нерецепторных тирозинкиназ регулируют указанные события путем катализа переноса фосфата от АТФ к остатку тирозина белковых мишеней в конкретной клетке. Было определено, что фрагменты последовательности, как правило, соответствуют каждому из указанных семейств киназ [Hanks et al., FASEB J., (1995), 9, 576-596; Knighton et al., Science, (1991), 253, 407-414; Garcia-Bustos et al., EMBO J., (1994),13:2352-2361]. Примеры киназ среди протеинкиназ включают без ограничения: abl, Akt, bcr-abl, Blk, Brk, Btk, c-kit, c-Met, c-src, c-fms, CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, cRaf1, CSF1R, CSK, EGFR, ErbB2, ErbB3, ErbB4, Erk, Fak, fes, FGFR1, FGFR2, FGFR3, FGFR4, FGFR5, Fgr, flt-1, Fps, Frk, Fyn, Hck, IGF-1R, INS-R, Jak, KDR, Lck, Lyn, MEK, p38, PDGFR, PIK, PKC, PYK2, ros, Tie, Tie-2, TRK, Yes и Zap70.
[0008] Исследования указывают на то, что протеинкиназы играют центральную роль в регуляции и поддержании широкого ряда процессов в клетке. Например, активность киназ действует в качестве молекулярных переключателей, регулирующих пролиферацию, активацию и/или дифференцировку клеток. Неконтролируемая или избыточная активность киназ, как у мутантных киназ, так и у киназ дикого типа, наблюдалась при многих болезненных состояниях, включая доброкачественные и злокачественные нарушения пролиферации, а также заболевания, возникающие вследствие неправильной активации иммунной системы (аутоиммунные нарушения), отторжение аллотрансплантата или заболевания «трансплантат против хозяина».
[0009] Сообщалось, что многие заболевания ассоциированы с аномальными ответами клеток, вызванными событиями, опосредованными протеинкиназами. Такие заболевания включают аутоиммунные заболевания, воспалительные заболевания, заболевания костей, метаболические заболевания, неврологические и нейродегенеративные заболевания, злокачественные опухоли, сердечно-сосудистые заболевания, аллергии и бронхиальную астму, болезнь Альцгеймера и заболевания, связанные с гормонами. Кроме того, специфические рецепторные протеинтирозинкиназы эндотелиальных клеток, такие как VEGF-2 и Tie-2, опосредуют процесс ангиогенеза и участвуют в поддержании прогрессирования злокачественных опухолей и других заболеваний с вовлечением неконтролируемой васкуляризации. Соответственно, в медицинской химии прилагались значительные усилия в попытке обнаружить ингибиторы протеинкиназ, которые эффективны в качестве терапевтических средств.
[0010] Многие злокачественные опухоли характеризуются нарушениями путей передачи сигналов в клетках, которые приводят к неконтролируемому росту и пролиферации злокачественных клеток. Рецепторные тирозинкиназы (RTK) играют ключевую роль в указанных путях передачи сигналов, передавая внеклеточные молекулярные сигналы в цитоплазму и/или ядро клетки. RTK представляют собой трансмембранные белки, которые, как правило, включают внеклеточный лиганд-связывающий домен, трансмембранный домен и каталитический цитоплазматический тирозинкиназный домен. Считается, что связывание лиганда с внеклеточной частью усиливает димеризацию, приводя к трансфосфорилированию и активации внутриклеточного тирозинкиназного домена (Schlessinger et al. Neuron 1992; 9:383-391).
[0011] Принимая во внимание нехватку существующих вариантов лечения большинства ассоциированных с протеинкиназами состояний, по-прежнему существует большая потребность в новых терапевтических средствах, которые ингибируют протеинкиназы. В частности, существует потребность в высокоактивных ингибиторах киназ, которые также нетоксичны и специфичны к определенным протеинкиназам.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
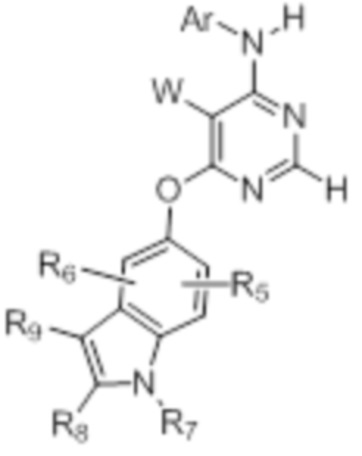
[0012] Соединение формулы
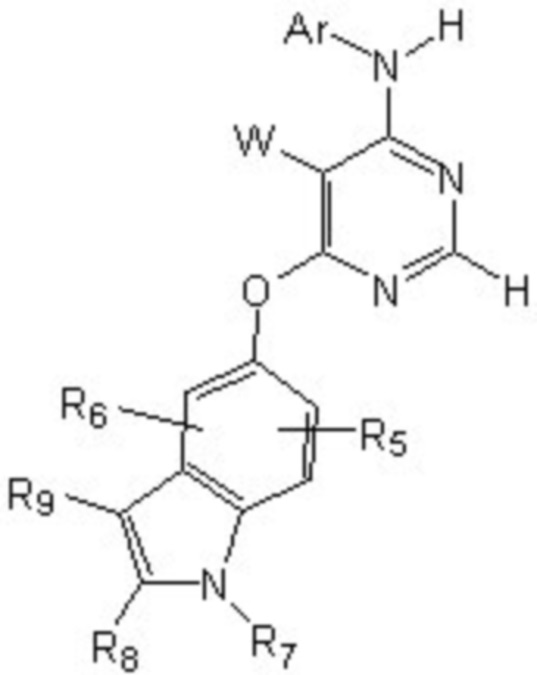
или его фармацевтически приемлемая соль, где:
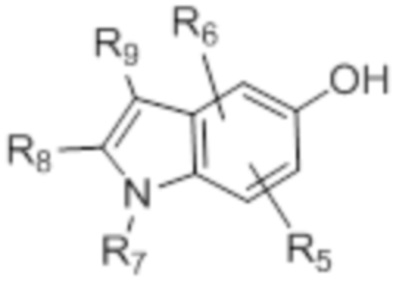
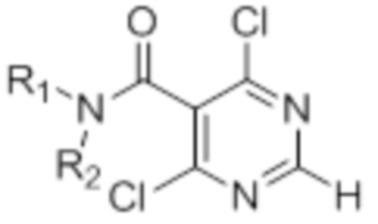
[0013] W выбирают из: F, Cl, Br, I, CN, C1-C4алкила, C1-C6алкокси, C2-C6алкенила, CF3, CF2H, CFH2, C2-C6алкинила, CON(R1)R2.
[0014] R1 и R2 представляют собой водород, алкил, циклоалкил, алкенил, алкинил, алкилтио, арил, арилалкил.
[0015] Ar представляет собой гетероарил или арил, каждый из которых замещен 0-4 заместителями, независимо выбранными из:
[0016] (1) галогена, гидрокси, амино, амида, циано, -COOH, -SO2NH2, оксо, нитро и алкоксикарбонила; и
[0017] (2) NR1
[0018] (3) групп формулы (Ia):
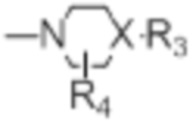 (Ia)
(Ia)
[0019]
[0020] где:
[0021] R4 представляет собой водород, C1-C4алкил, оксо;
[0022] X представляет собой CH, когда R3 представляет собой водород; или X-R3 представляет собой O; или X представляет собой N, R3 представляет собой группы, выбранные из водорода, C1-C6алкила, C2-C6алкенила, C2-C6алкинила, C3-C10арила или гетероарила, (C3-C7циклоалкил)C1-C4алкила, C1-C6галогеналкила, C1-C6алкокси, C1-C6алкилтио, C2-C6алканоила, C1-C6алкоксикарбонила, C2-C6алканоилокси, моно- и ди-(C3-C8циклоалкил)аминоC0-C4алкила, (4-7-членный гетероцикл)C0-C4алкила, C1-C6алкилсульфонила, моно- и ди-(C1-C6алкил)сульфонамидо, и моно- и ди-(C1-C6алкил)-аминокарбонила, каждый из которых замещен 0-4 заместителями, независимо выбранными из галогена, гидрокси, циано, амино, -COOH и оксо.
[0023] Заместители на индоле являются следующими: R5 и R6 независимо выбирают из: водорода, F, Cl, Br, CN, C1-C4алкила, C1-C6алкокси.
[0024] R7, R8 и R9 независимо выбирают из водорода, C1-C4алкила, C2-C6алкенила, C2-C6алкинила, C3-C10арила или гетероарила, C1-C6алкокси, C1-C6алкилтио, C2-C6алканоила, C1-C6алкоксикарбонила, C2-C6алканоилокси.
[0025] Фармацевтическая композиция, содержащая, по меньшей мере, одно соединение по п. I или его фармацевтически приемлемые соли, их гидраты, сольваты, кристаллические соли и отдельные диастереоизомеры, и фармацевтически приемлемый носитель.
[0026] Соответственно, целью настоящего изобретения является предоставление противоопухолевого средства, содержащего замещенные производные пиримидина, описанные формулой (I), его фармацевтически приемлемых составов, способов получения новых соединений и композиций для использования соединений. Соединения и композиции, содержащие соединения формулы (I), находят свое применение для лечения целого ряда заболеваний.
[0027] Комбинированная терапия, описанная в настоящем документе, может обеспечиваться путем приготовления замещенных производных пиримидина формулы (I) и другого терапевтического средства в виде отдельных фармацевтических составов с последующим введением их пациенту одновременно, полуодновременно, по отдельности или через определенные промежутки времени.
[0028] Настоящее изобретение относится к способам использования определенных химических соединений, таких как ингибиторы киназ, для лечения различных заболеваний, нарушений и патологий, например, злокачественной опухоли, сердечно-сосудистых заболеваний, таких как инфаркт миокарда (MI), инсульт или ишемия. Соединения, описанные в настоящем изобретении, могут блокировать ферментативную активность некоторых или многих представителей семейства FGFR киназ, в дополнение к блокированию
[0029] активности других рецепторных или нерецепторных киназ.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0030] На фигуре 1 отражена кривая дозовой зависимости для соединения 19.
[0031] На фигуре 2 отражена противоопухолевая активность соединения 19.
[0032] На фигуре 3 отражена токсичность соединения 19 на основании снижения веса крыс.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0033] Настоящее изобретение относится к соединениям, характеризующимся общей формулой (I)
(I),
или их фармацевтически приемлемой соли, где:
[0034] W выбирают из: F, Cl, Br, I, CN, C1-C4алкила, C1-C6алкокси, C2-C6алкенила, CF3, CF2H, CFH2, C2-C6алкинила, CON(R1)R2.
[0035] R1 и R2 представляют собой водород, алкил, циклоалкил, алкенил, алкинил, алкилтио, арил, арилалкил.
[0036] Ar представляет собой гетероарил или арил, каждый из которых замещен 0-4 заместителями, независимо выбранными из:
(1) галогена, гидрокси, амино, амида, циано, -COOH, -SO2NH2, оксо, нитро и алкоксикарбонила; и
(2) NR1
[0037] (3) групп формулы (Ia):
(Ia)
[0038] Соединение формулы
[0039] или его фармацевтически приемлемая соль, где:
W выбирают из: F, Cl, Br, I, CN, C1-C4алкила, C1-C6алкокси, C2-C6алкенила, CF3, CF2H, CFH2, C2-C6алкинила, CON(R1)R2.
1. R1 и R2 представляют собой представляют собой водород, алкил, циклоалкил, алкенил, алкинил, алкилтио, арил, арилалкил.
2. Ar представляет собой гетероарил или арил, каждый из которых замещен 0-4 заместителями, независимо выбранными из:
(1) галогена, гидрокси, амино, амида, циано, -COOH, -SO2NH2, оксо, нитро и алкоксикарбонила; и
(2) NR1
(3) групп формулы (Ia):
(Ia)
где:
R4 представляет собой водород, C1-C4алкил, оксо;
X представляет собой CH, когда R3 представляет собой водород; или X-R3 представляет собой O; или X представляет собой N, R3 представляет собой группы, выбранные из водорода, C1-C6алкила, C2-C6алкенила, C2-C6алкинила, C3-C10арила или гетероарила, (C3-C7циклоалкил)C1-C4алкила, C1-C6галогеналкила, C1-C6алкокси, C1-C6алкилтио, C2-C6алканоила, C1-C6алкоксикарбонила, C2-C6алканоилокси, моно- и ди-(C3-C8циклоалкил)аминоC0-C4алкила, (4-7-членный гетероцикл)C0-C4алкила, C1-C6алкилсульфонила, моно- и ди-(C1-C6алкил)сульфонамидо, и моно- и ди-(C1-C6алкил)-аминокарбонила, каждый из которых замещен 0-4 заместителями, независимо выбранными из галогена, гидрокси, циано, амино, -COOH и оксо.
3. Заместители на индоле являются следующими:
R5 и R6 независимо выбирают из: водорода, F, Cl, Br, CN, C1-C4алкила, C1-C6алкокси.
4. R7, R8 и R9 независимо выбирают из водорода, C1-C4алкила, C2-C6алкенила, C2-C6алкинила, C3-C10арила или гетероарила, C1-C6алкокси, C1-C6алкилтио, C2-C6алканоила, C1-C6алкоксикарбонила, C2-C6алканоилокси.
5. Фармацевтическая композиция, содержащая, по меньшей мере, одно соединение по п. I или его фармацевтически приемлемые соли, их гидраты, сольваты, кристаллические соли и отдельные диастереоизомеры, и фармацевтически приемлемый носитель.
где:
R4 представляет собой водород, C1-C4алкил, оксо;
X представляет собой CH, когда R3 представляет собой водород; или X-R3 представляет собой O; или X представляет собой N, R3 представляет собой группы, выбранные из водорода, C1-C6алкила, C2-C6алкенила, C2-C6алкинила, C3-C10арила или гетероарила, (C3-C7циклоалкил)C1-C4алкила, C1-C6галогеналкила, C1-C6алкокси, C1-C6алкилтио, C2-C6алканоила, C1-C6алкоксикарбонила, C2-C6алканоилокси, моно- и ди-(C3-C8циклоалкил)аминоC0-C4алкила, (4-7-членный гетероцикл)C0-C4алкила, C1-C6алкилсульфонила, моно- и ди-(C1-C6алкил)сульфонамидо, и моно- и ди-(C1-C6алкил)-аминокарбонила, каждый из которых замещен 0-4 заместителями, независимо выбранными из галогена, гидрокси, циано, амино, -COOH и оксо.
R5 и R6 независимо выбирают из: водорода, F, Cl, Br, CN, C1-C4алкила, C1-C6алкокси.
R7, R8 и R9 независимо выбирают из водорода, C1-C4алкила, C2-C6алкенила, C2-C6алкинила, C3-C10арила или гетероарила, C1-C6алкокси, C1-C6алкилтио, C2-C6алканоила, C1-C6алкоксикарбонила, C2-C6алканоилокси.
[0040] Термин «галоген-» или «галоген» относится к фтору, хлору, брому или йоду.
[0041] Термин «алкил», используемый в настоящем документе по отдельности или как часть другой группы, относится к одновалентному полученному из алкана (углеводорода) радикалу, содержащему от 1 до 12 атомов углерода, если не указано иное. Алкильные группы могут быть замещены по любому доступному для замещения положению. Алкильная группа, замещенная другой алкильной группой, также называется «разветвленной алкильной группой». Примеры алкильных групп включают метил, этил, пропил, изопропил, н-бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, и т.д. Примеры заместителей включают без ограничения одну или несколько из следующих групп: алкил, арил, галоген (такой как F, Cl, Br, I), галогеналкил (такой как CCl3 или CF3), алкокси, алкилтио, гидрокси, карбокси (-COOH), алкилоксикарбонил (-C(O)R), алкилкарбонилокси (-OCOR), амино (-NH2), карбамоил (-NHCOOR- или -OCONHR-), мочевина (-NHCONHR-) или тиол (-SH). Согласно некоторым предпочтительным вариантам осуществления настоящего изобретения, алкильные группы замещены, например, амино, гетероциклоалкилом, таким как морфолин, пиперазин, пиперидин, азетидин, гидроксилом, метокси или гетероарильными группами, такими как пирролидин.
[0042] Термин «циклоалкил», используемый в настоящем документе по отдельности или как часть другой группы, относится к полностью насыщенным или частично ненасыщенным углеводородным кольцам, содержащим от 3 до 9, предпочтительно от 3 до 7, атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил и циклогексил, и т.д. Кроме того, циклоалкил может быть замещенным. Замещенный циклоалкил относится к кольцам, содержащим 1, 2 или 3 заместителя, выбранных из группы, состоящей из галогена, алкила, замещенного алкила, алкенил, алкинила, нитро, циано, оксо (=O), гидрокси, алкокси, тиоалкила, -CO2H, -C(=O)H, CO2-алкила, -C(=O)алкила, кето, =N-OH, =N-O-алкила, арила, гетероарила, гетероцикло, -NR'R'', -C(=O)NR'R'', -CO2NR'R'', -C(=O)NR'R'', -NR'CO2R'', -NR'C(=O)R'', -SO2NR'R'' и -NR'SO2R'', где каждый из R' и R'' независимо выбирают из галогена, алкила, замещенного алкила и циклоалкила, или R' и R'' формируют вместе гетероцикл или гетероарильное кольцо.
[0043] Термин «алкенил», используемый в настоящем документе по отдельности или как часть другой группы, относится к неразветвленному, разветвленному или циклическому углеводородному радикалу, содержащему от 2 до 12 атомов углерода и, по меньшей мере, одну углерод-углеродную двойную связь. Примеры таких групп включают винил, аллил, 1-пропенил, изопропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 1-пентенил, 2-пентенил, 3-пентенил, 4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 1-гептенил, и т.д. Алкенильные группы также могут быть замещены по любому доступному для замещения положению. Примеры заместителей алкенильных групп включают таковые, перечисленные выше для алкильных групп, и в особенности включают C3-C7 циклоалкильные группы, такие как циклопропил, циклопентил и циклогексил, которые могут быть замещены, например, амино, оксо, гидроксилом, и т.д.
[0044] Термин «алкинил» относится к неразветвленным или разветвленным алкиновым группам, которые содержат одну или несколько ненасыщенных углерод-углеродных связей, по меньшей мере, одна из которых представляет собой тройную связь. Алкинильные группы включают C2-C8алкинильные, C2-C6алкинильные и C2-C4алкинильные группы, которые содержат от 2 до 8, от 2 до 6 или от 2 до 4 атомов углерода, соответственно. Примеры алкинильной группы включают этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил и гексенил. Алкинильные группы также могут быть замещены по любому доступному для замещения положению. Примеры заместителей алкинильных групп включают таковые, перечисленные выше для алкильных групп, такие как амино, алкиламино, и т.д. Числа, приводимые в нижнем индексе после символа «C», определяют число атомов углерода, которое может содержать конкретная группа.
[0045] Термин «алкокси», по отдельности или как часть другой группы, означает описанную выше алкильную группу, присоединенную посредством кислородной связи (-O-). Предпочтительные алкоксигруппы содержат от 1 до 8 атомов углерода. Примеры таких групп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, н-гексилокси, циклогексилокси, н-гептилокси, н-октилокси и 2-этилгексилокси.
[0046] Термин «алкилтио» относится к описанной выше алкильной группе, присоединенной посредством серного мостика. Предпочтительные алкокси и алкилтиогруппы представляют собой группы, в которых алкильная группа присоединена посредством гетероатомного мостика. Предпочтительные алкилтиогруппы содержат от 1 до 8 атомов углерода. Примеры таких групп включают метилтио, этилтио, н-пропилтиол, н-бутилтиол, и т.д.
[0047] Используемый в настоящем документе термин «оксо» относится к кетогруппе (C=O). Оксогруппа, которая представляет собой заместитель неароматического атома углерода, приводит к преобразованию -CH2- до -C(=O)-.
[0048] Термин «алкоксикарбонил», используемый в настоящем документе по отдельности или как часть другой группы, означает алкоксигруппу, присоединенную посредством карбонильной группы. Алкоксикарбонильный радикал представлен формулой: -C(O)OR, где группа R представляет собой неразветвленную C1-C6алкильную группу, циклоалкил, арил или гетероарил.
[0049] Термин «арилалкил», используемый в настоящем документе по отдельности или как часть другой группы, означает ароматическое кольцо, присоединенной посредством описанной выше алкильной группы (такое как бензил).
[0050] Термин «арил», используемый в настоящем документе по отдельности или как часть другой группы, относится к моноциклическим или бициклическим ароматическим кольцам, например, к фенилу, замещенному фенилу и т.д., а также к конденсированным группам, например, к нафтилу, фенантренилу и т.д. Таким образом, арильная группа содержит, по меньшей мере, одно кольцо, содержащее, по меньшей мере, 6 атомов, и до пяти подобных колец, содержащих в своем составе до 20 атомов с чередующимися (резонирующими) двойными связями между смежными атомами углерода или подходящими гетероатомами. Арильные группы могут быть необязательно замещены одной или несколькими группами, включая без ограничения галоген, такой как I, Br, F, или Cl; алкил, такой как метил, этил, пропил, алкокси, такой как метокси или этокси, гидрокси, карбокси, карбамоил, алкилоксикарбонил, нитро, алкенилокси, трифторметил, амино, циклоалкил, арил, гетероарил, циано, алкилS(O)m (m=0, 1, 2), или тиол.
[0051] Термин «ароматический» относится к циклически конъюгированному молекулярному фрагменту со стабильностью, значительно превышающей (по причине делокализации) таковую для гипотетической делокализованной структуры, такой как структура Кекуле.
[0052] Термин «амино», используемый в настоящем документе по отдельности или как часть другой группы, относится к -NH2. «Амино» может быть необязательно замещен одним или двумя заместителями, которые могут быть одинаковыми или разными, такими как алкил, арил, арилалкил, алкенил, алкинил, гетероарил, гетероарилалкил, циклогетероалкил, циклогетероалкилалкил, циклоалкил, циклоалкилалкил, галогеналкил, гидроксиалкил, алкоксиалкис, тиотио, карбонил или карбоксил. Указанные заместители могут быть дополнительно замещены карбоновой кислотой, причем любым из представленных в этом документе алкила и арила. Согласно некоторым вариантам осуществления, аминогруппы замещены карбоксилом или карбонилом с формированием производных N-ацила и N-карбамоила.
[0053] Термин «алкилсульфонил» относится к группам формулы (SO2)-алкил, в которых атом серы является точкой присоединения. Предпочтительно, алкилсульфонильные группы включают C1-C6алкилсульфонильные группы, которые содержат от 1 до 6 атомов углерода. Одной типовой алкилсульфонильной группой является метилсульфонил.
[0054] Термин «гетероатом» относится к любому атому, отличному от атома углерода, например, N, O или S.
[0055] Термин «гетероарил», используемый в настоящем документе по отдельности или как часть другой группы, относится к замещенным или незамещенным ароматическим 5-6-членным моноциклическим группам, 9- или 10-членным бициклическим группам и 11-14-членным трициклическим группам, которые содержат, по меньшей мере, один гетероатом (O, S или N), по меньшей мере, в одном из колец. Каждое кольцо гетероарильной группы, содержащее гетероатом, может содержать 1 или 2 атома кислорода или серы и/или от 1 до 4 атомов азота при условии, что общее число гетероатомов в каждом кольце составляет 4 или менее, и сто каждое кольцо содержит, по меньшей мере, 1 атом углерода.
[0056] Термин «гетероцикл» или «гетероциклоалкил», используемый в настоящем документе по отдельности или как часть другой группы, относится к циклоалкильной группе (неароматической), в которой один из атомов углерода в кольце заменен на гетероатом, выбранный из O, S или N. «Гетероцикл» содержит от 1 до 3 конденсированных, связанных мостиковой связью или спироколец, по меньшей мере, одно из которых представляет собой гетероциклическое кольцо (т.е., один или несколько кольцевых атомов представляют собой гетероатом, а оставшиеся кольцевые атомы представляют собой атом углерода). Гетероциклическое кольцо может быть необязательно замещено, что означает, что гетероциклическое кольцо может быть замещено по одному или нескольким доступным для замещения положениям кольца одной или несколькими группами, независимо выбранными из алкила (предпочтительно, низшего алкила), гетероциклоалкила, гетероарила, алкокси (предпочтительно, низшего алкокси), нитро, моноалкиламино (предпочтительно, низшего алкиламино), диалкиламино (предпочтительно, алкиламино), циано, галогена, галогеналкила (предпочтительно, трифторметила), алканоила, аминокарбонила, моноалкиламинокарбонила, диалкиламинокарбонила, алкиламидо (предпочтительно, низшего алкиламидо), алкоксиалкила (предпочтительно, низшего алкокси и низшего алкила), алкоксикарбонила (предпочтительно, низшего алкоксикарбонила), алкилкарбонилокси (предпочтительно, низшего алкилкарбонилокси) и арила (предпочтительно, фенила), причем упомянутый арил необязательно замещен галогеном, низшей алкильной и низшей алкоксигруппами. Гетероциклическая группа, как правило, может быть присоединена посредством любого кольца или атома заместителя при условии формирования стабильного соединения. N-связанные гетероциклические группы присоединены посредством составного атома азота.
[0057] Обычно, гетероциклическое кольцо содержит 1-4 гетероатомов; согласно определенным вариантам осуществления, каждое гетероциклическое кольцо содержит 1 или 2 гетероатома в кольце. Каждое гетероциклическое кольцо, как правило, содержит от 3 до 8 кольцевых атомов (кольца, содержащие от до 7 кольцевых атомов, изложены в определенных вариантах осуществления), а гетероциклы, содержащие конденсированные, связанные мостиковой связью или спирокольца, обычно содержат от 9 до 14 кольцевых атомов, которые состоят из атомов углерода и содержат 1, 2 или 3 гетероатома, выбранных из азота, кислорода и/или серы.
[0058] Примеры «гетероцикла» или «гетероциклоалкильных» групп включают пиперазин, пиперидин, морфолин, тиоморфолин, пирролидин, имидазолидин и тиазолид.
[0059] Используемый в настоящем документе термин «заместитель» относится к молекулярному фрагменту, который ковалентно связан с атомом представляющей интерес молекулы. Например, «заместитель кольца» может представлять собой фрагмент, такой как галоген, алкильная группа, галогеналкильная группа или другая группа, рассматриваемая в настоящем документе, которая ковалентно связана с атомом (предпочтительно, с атомом углерода или азота), который представляет собой кольцевой атом.
[0060] Термин «необязательно замещенный» означает, что арил или гетероциклил, или другая группа, могут быть замещены по одному или нескольким доступным для замещения положениям одной или несколькими группами, независимо выбранными из алкила (предпочтительно, низшего алкила), алкокси (предпочтительно, низшего алкокси), нитро, моноалкиламино (предпочтительно, с 1-6 атомами углерода), диалкиламино (предпочтительно, с 1-6 атомами углерода), циано, галогена, галогеналкила (предпочтительно, трифторметила), алканоила, аминокарбонила, моноалкиламинокарбонила, диалкиламинокарбонила, алкиламидо (предпочтительно, низшего алкиламидо), алкоксиалкила (предпочтительно, низшего алкокси и низшего алкила), алкоксикарбонила (предпочтительно, низшего алкоксикарбонила), алкилкарбонилокси (предпочтительно, низшего алкилкарбонилокси) и арила (предпочтительно, фенила), причем упомянутый арил необязательно замещен галогеном, низшей алкильной и низшей алкоксигруппами. Необязательное замещение также обозначают выражением «замещенный от 0 до X заместителями», где X представляет собой максимальное число возможных заместителей. Определенные необязательно замещенные группы замещены от 0 до 2, 3 или 4 независимо выбранных заместителей.
[0061] Штрих («-»), который не располагается между двумя буквами или символами, используют для указания точки присоединения заместителя. Например, -CONH2 присоединен посредством атома углерода.
[0062] Обозначенный пунктиром цикл, который расположен внутри гетероциклического кольца, используют для обозначения конъюгированной системы. Связи между двумя атомами могут представлять собой одинарную связь или двойную связь. Термин «киназа» относится к любому ферменту, который катализирует добавление фосфатных групп к остатку белка; например, серин/треонин-киназы катализируют добавление фосфатных групп к остаткам серина и треонина.
[0063] Термин «терапевтически эффективное количество» относится к количеству соединения или фармацевтической композиции, которое вызывает биологический или медицинский ответ со стороны ткани, системы, животного или человека, которые наблюдаются исследователем, ветеринаром, врачом или другим клиницистом, например, восстановление и поддержание васкулостаза или профилактика нарушения или утраты васкулостаза; снижение опухолевой массы; снижение заболеваемости и/или смертности.
[0064] Термин «фармацевтически приемлемый» относится к тому факту, что носитель, разбавитель или наполнитель является совместимым с другими ингредиентами состава и не является вредным для его реципиента.
[0065] Термины «введение соединения» или «осуществление введения соединения» относятся к действию по предоставлению соединения согласно настоящему изобретению или фармацевтической композиции нуждающемуся в лечении субъекту.
[0066] Термин «защищенная» означает, что группа находится в модифицированной форме для предотвращения нежелательных побочных реакций в защищаемом положении. Подходящие защитные группы для соединений согласно настоящему изобретению станут понятны из настоящего описания с учетом уровня подготовки в данной области техники, и со ссылкой на стандартные учебники, такие как Greene, T. W. et al., Protective Groups in Organic Synthesis, John Wiley & Sons, New York (1999).
[0067] Термин «фармацевтически приемлемая соль» соединения, используемый в настоящем документе, представляет собой соль кислоты или основания, которая применима для использования в контакте с тканями людей или животных, не обладая чрезмерной токсичностью или канцерогенностью и предпочтительно не вызывая раздражение, аллергическую реакцию или другую проблему или осложнение. Такие соли включают соли неорганических и органических кислот с остатками оснований, таких как амины, а также соли щелочей или органических веществ с остатками кислот, таких как карбоновые кислоты. Конкретные фармацевтически приемлемые соли включают без ограничения соли кислот, таких как соляная, фосфорная, бромистоводородная, яблочная, гликолевая, фумаровая, серная, сульфамовая, сульфаниловая, муравьиная, толуолсульфоновая, метансульфоновая, бензолсульфоновая, этандисульфоновая, 2-гидроксиэтилсульфоновая, азотная, бензойная, 2-ацетоксибензойная, лимонная, винная, молочная, стеариновая, салициловая, глутаминовая, аскорбиновая, памовая, янтарная, фумаровая, малеиновая, пропионовая, гидроксималеиновая, йодистоводородная, фенилуксусная, алкановая, такая как уксусная, HOOC-(CH2)n-COOH, где n равно 0-4, и т.д. По аналогии, фармацевтически приемлемые катионы включают без ограничения натрий, калий, кальций, алюминий, литий и аммоний. Средним специалистам в данной области техники будут очевидны дополнительные фармацевтически приемлемые соли соединений, представленных в настоящем документе. В общем, фармацевтически приемлемая соль кислоты или основания может быть синтезирована из исходного соединения, которое содержит щелочной или кислотный фрагмент, любым общепринятым химическим способом. Вкратце, такие соли могут быть получены путем осуществления взаимодействия указанных соединений в форме свободной кислоты или свободного основания со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе, или в их смеси; как правило, предпочтительно использование неводной среды, такой как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Очевидно, что каждое соединение формулы I может быть необязательно включено в состав в виде гидрата, сольвата или нековалентного комплекса. Кроме того, под объем настоящего изобретения подпадают различные кристаллические формы и полиморфы. Настоящим документом также предусмотрены пролекарства соединений формулы I.
[0068] Предпочтительные группы W в формуле (I) представляют собой: F, Cl, Br, CN, CF3, CF2H, CFH2, CH3, OCH3, NH2 и представленный ниже перечень:
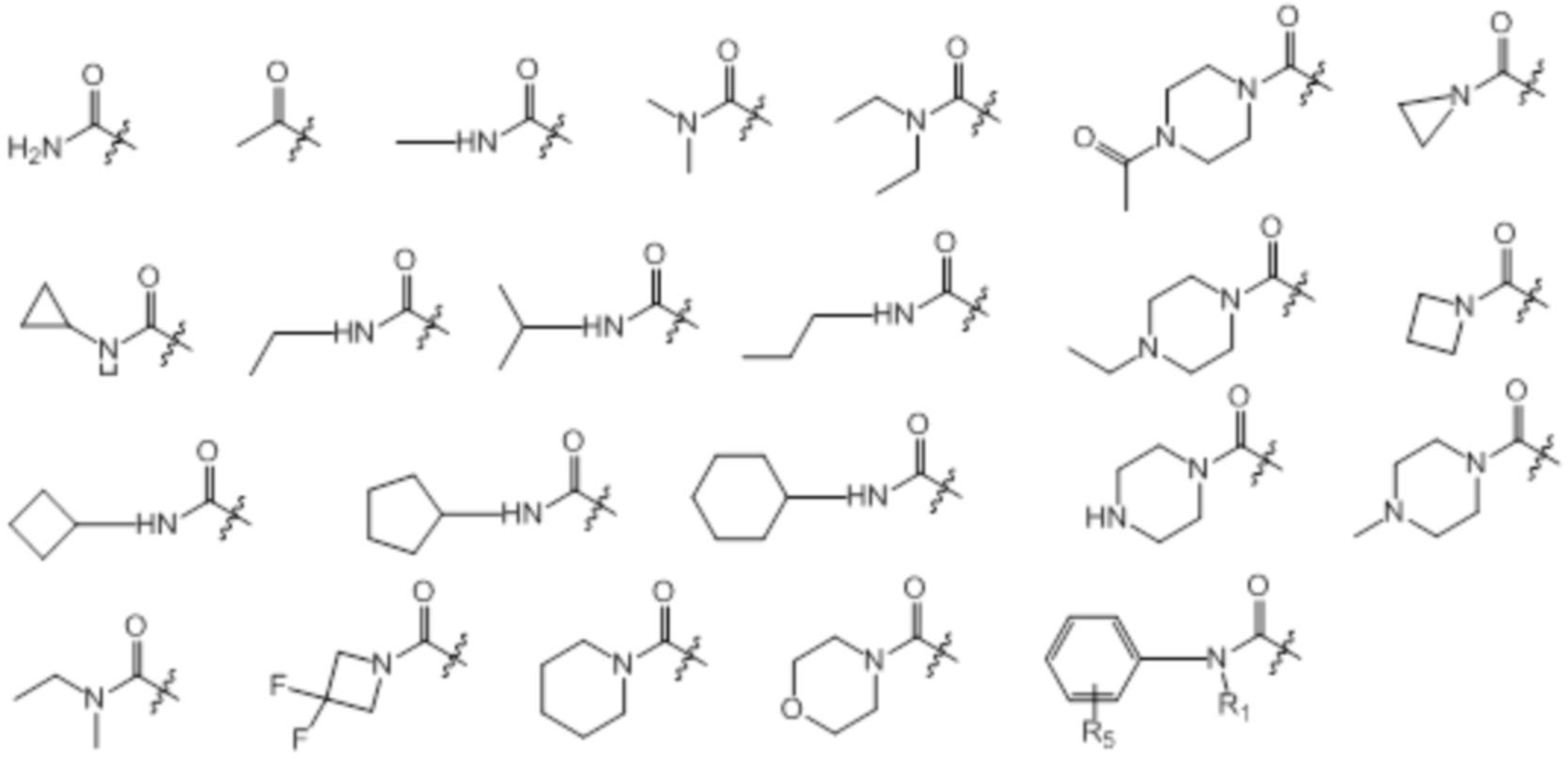
[0069] Предпочтительные замещенные индольные группы формулы (I) перечислены ниже:


[0070] Предпочтительные группы Ar в формуле (I) являются следующими:
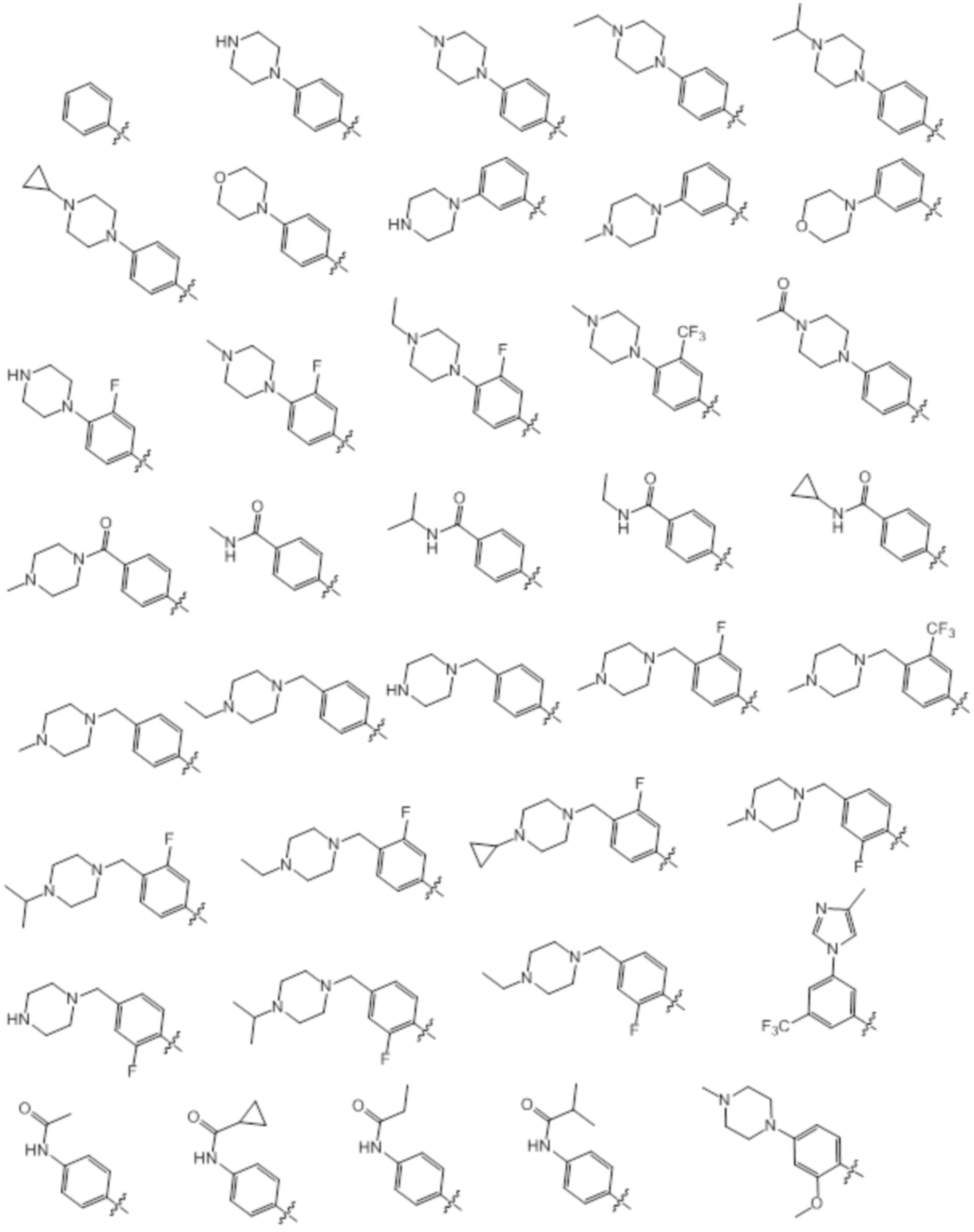
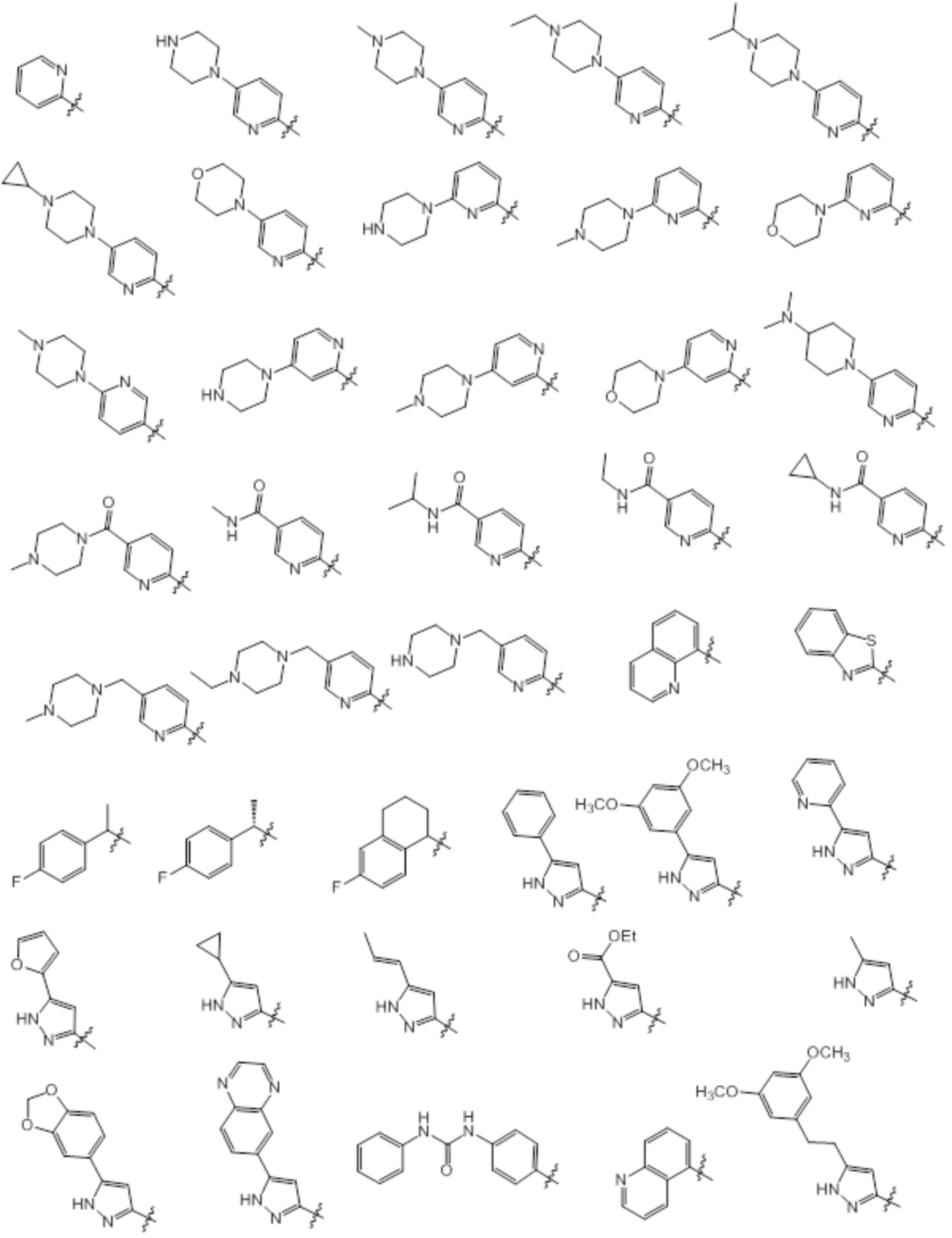
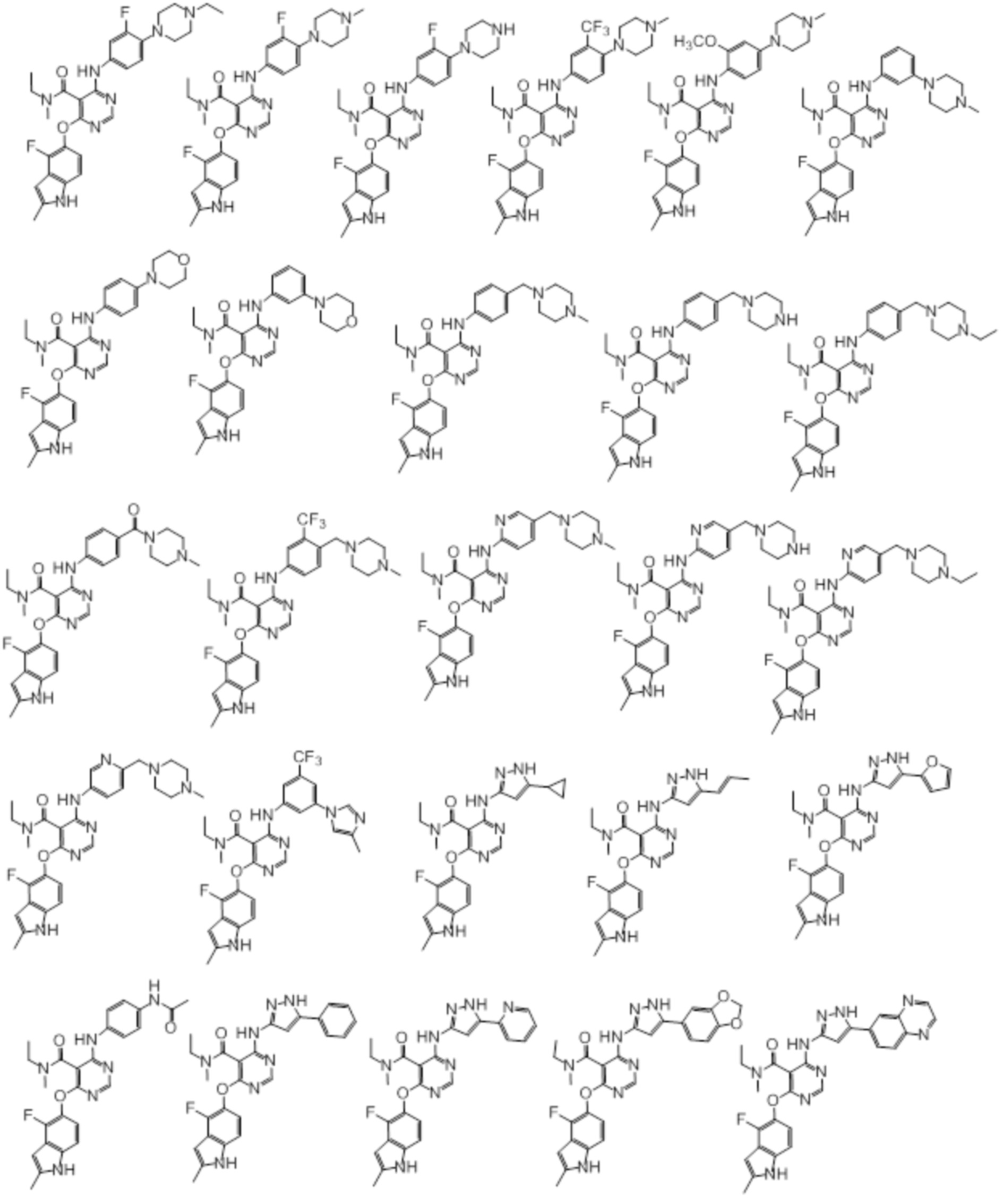
[0071] Примеры конкретных соединений согласно настоящему изобретению представляют собой соединения, определенные ниже:
[0072]
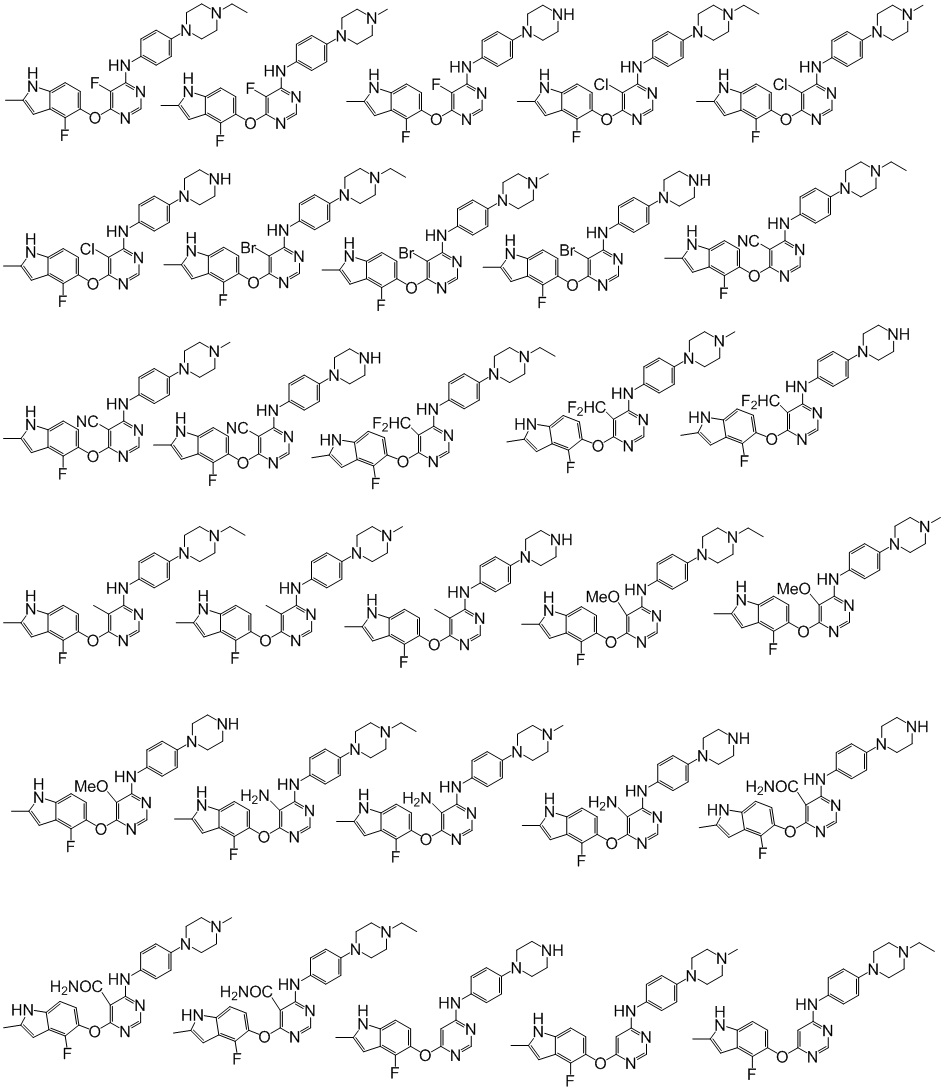
[0073]

[0074]

[0075]

[0076]

[0077]
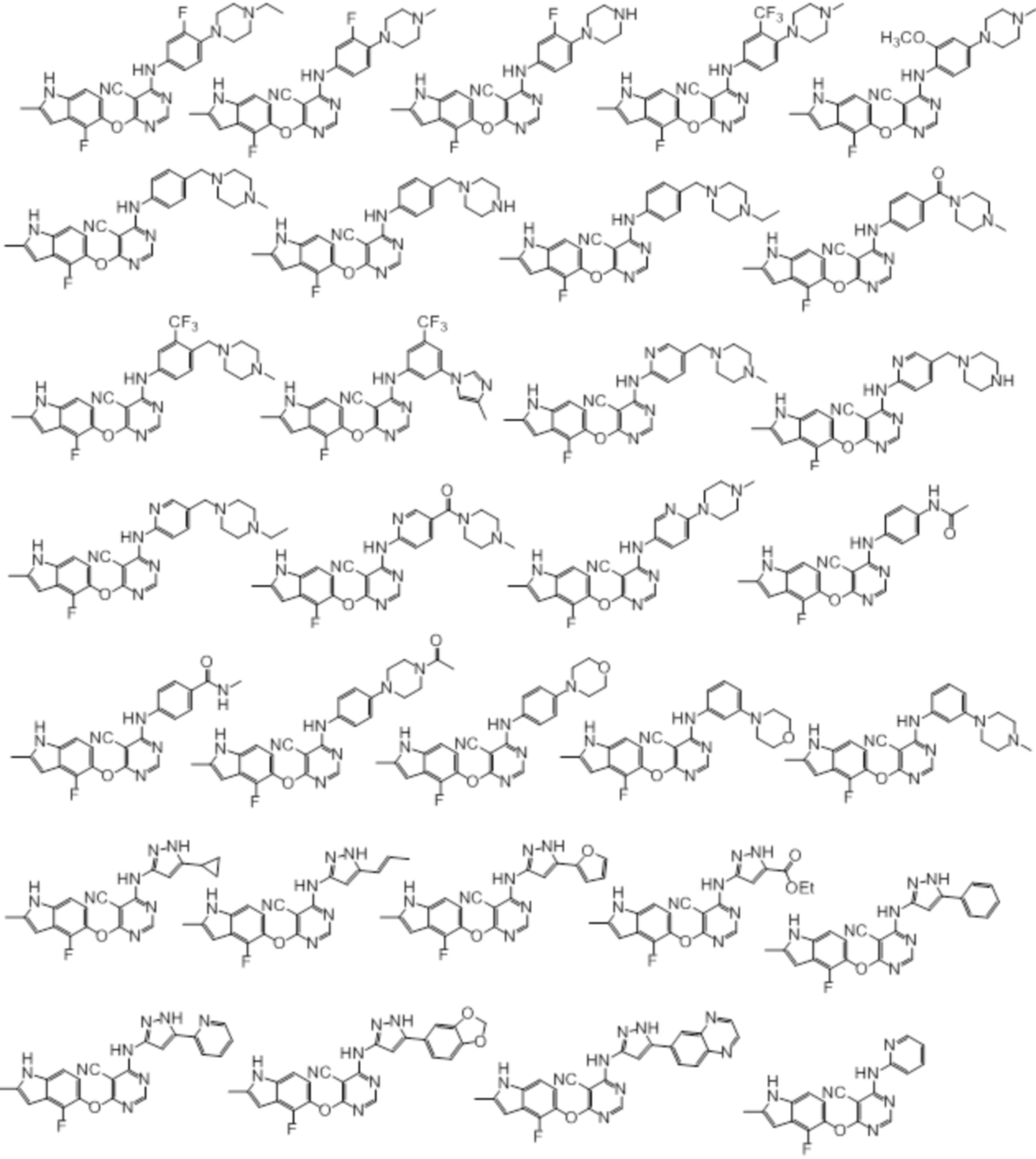
[0078]


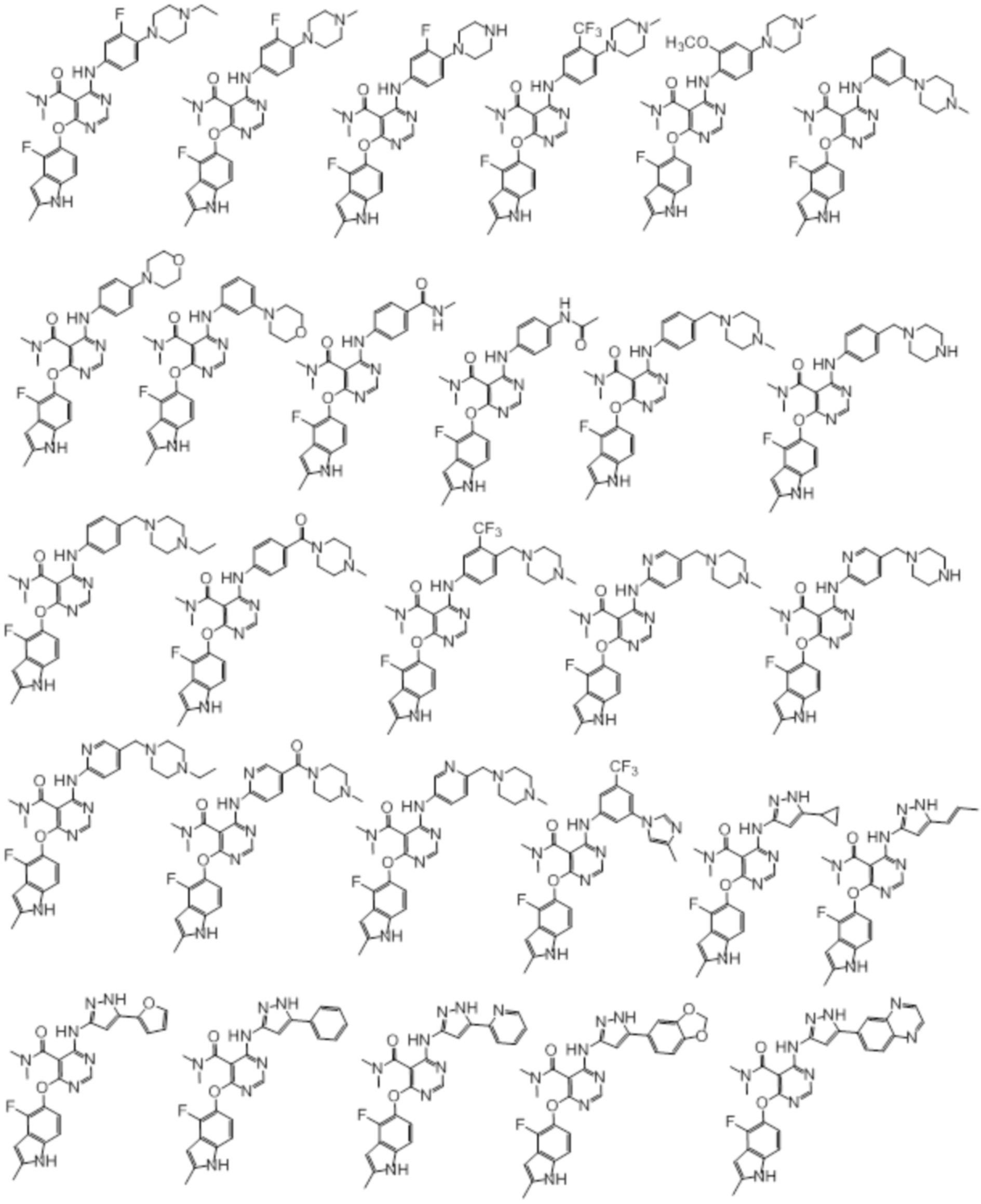
[0079]
[0080]
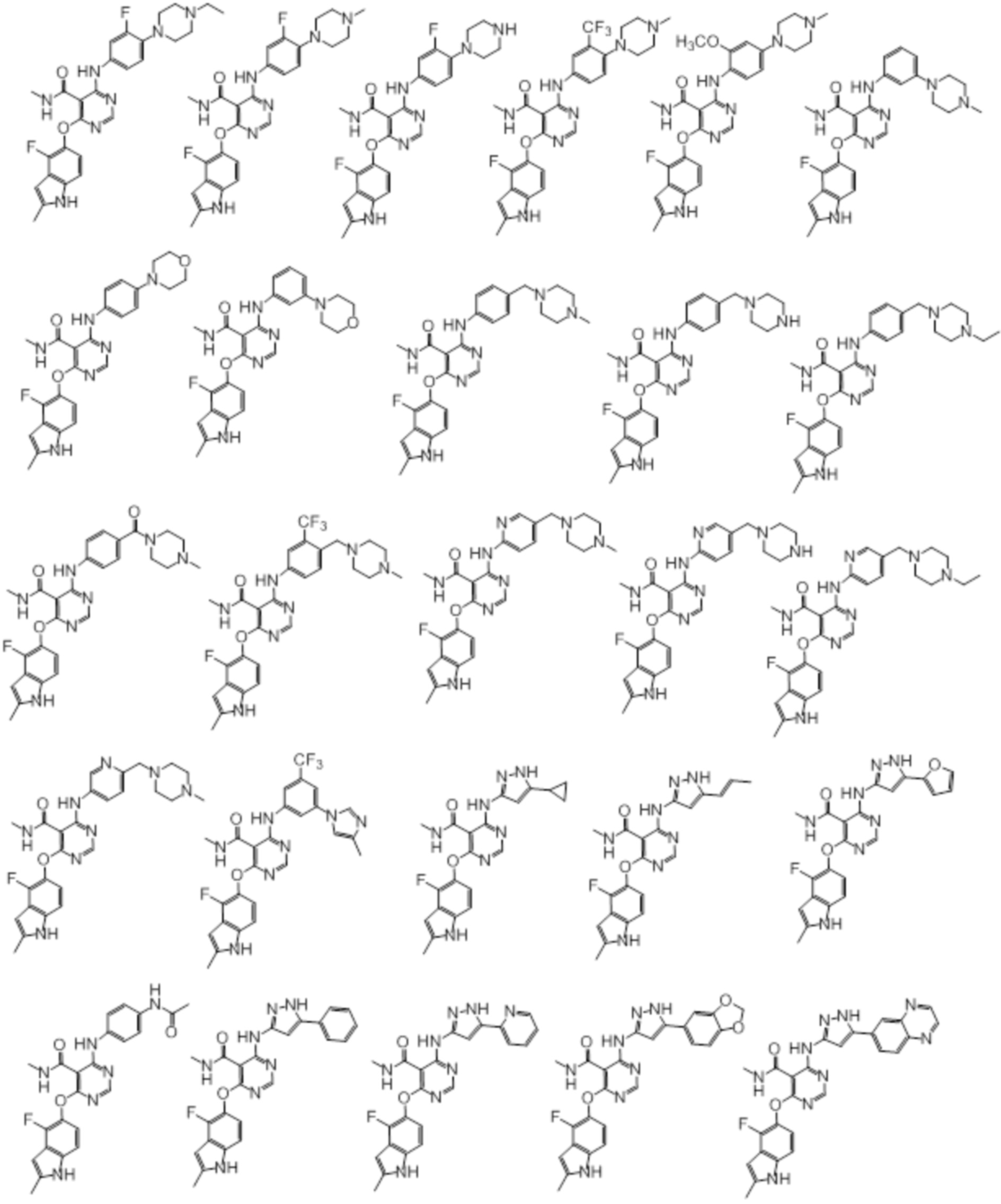
[0081]
[0082]

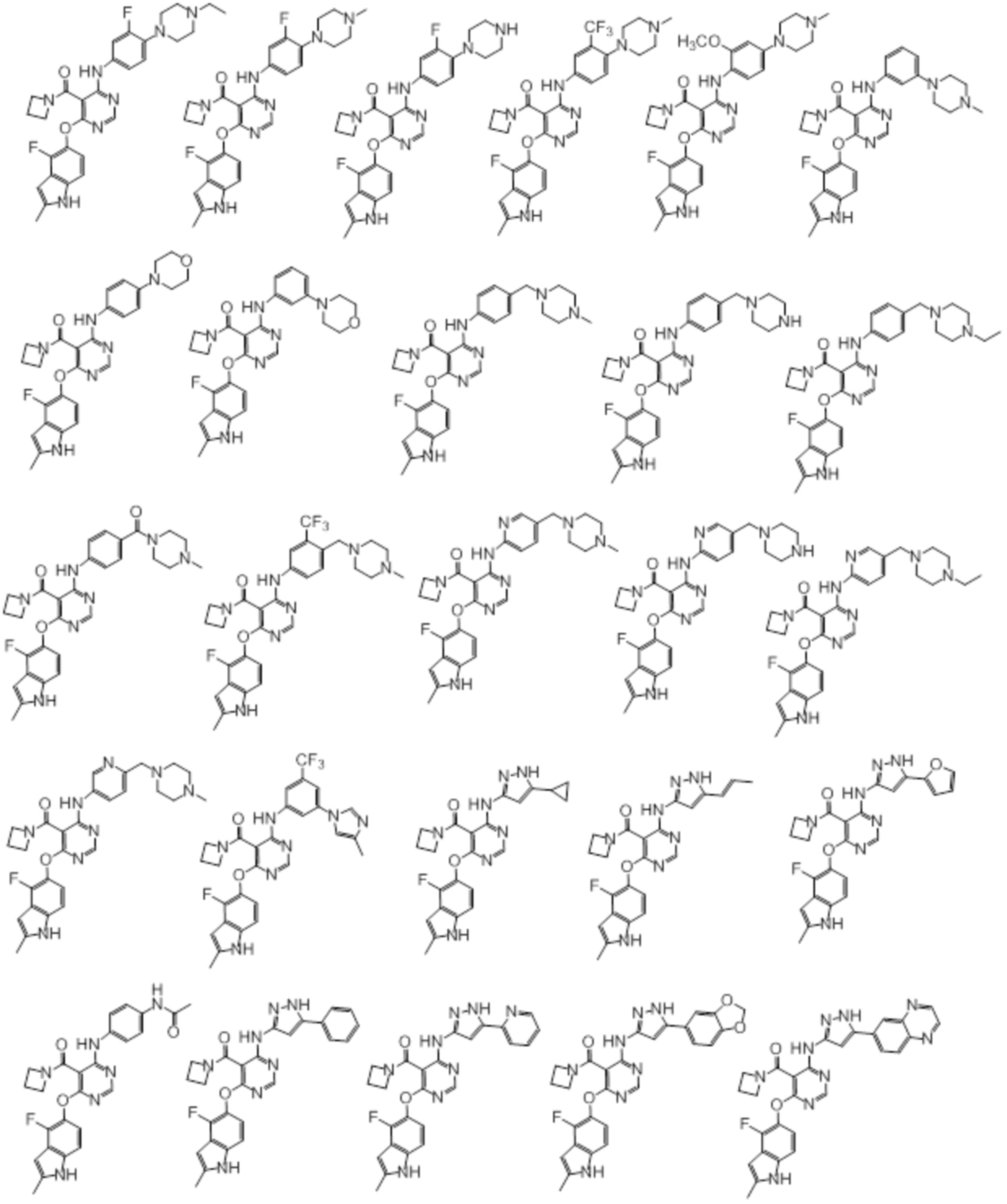
[0083]

[0084]

[0085]

[0086]

[0087]
[0088]


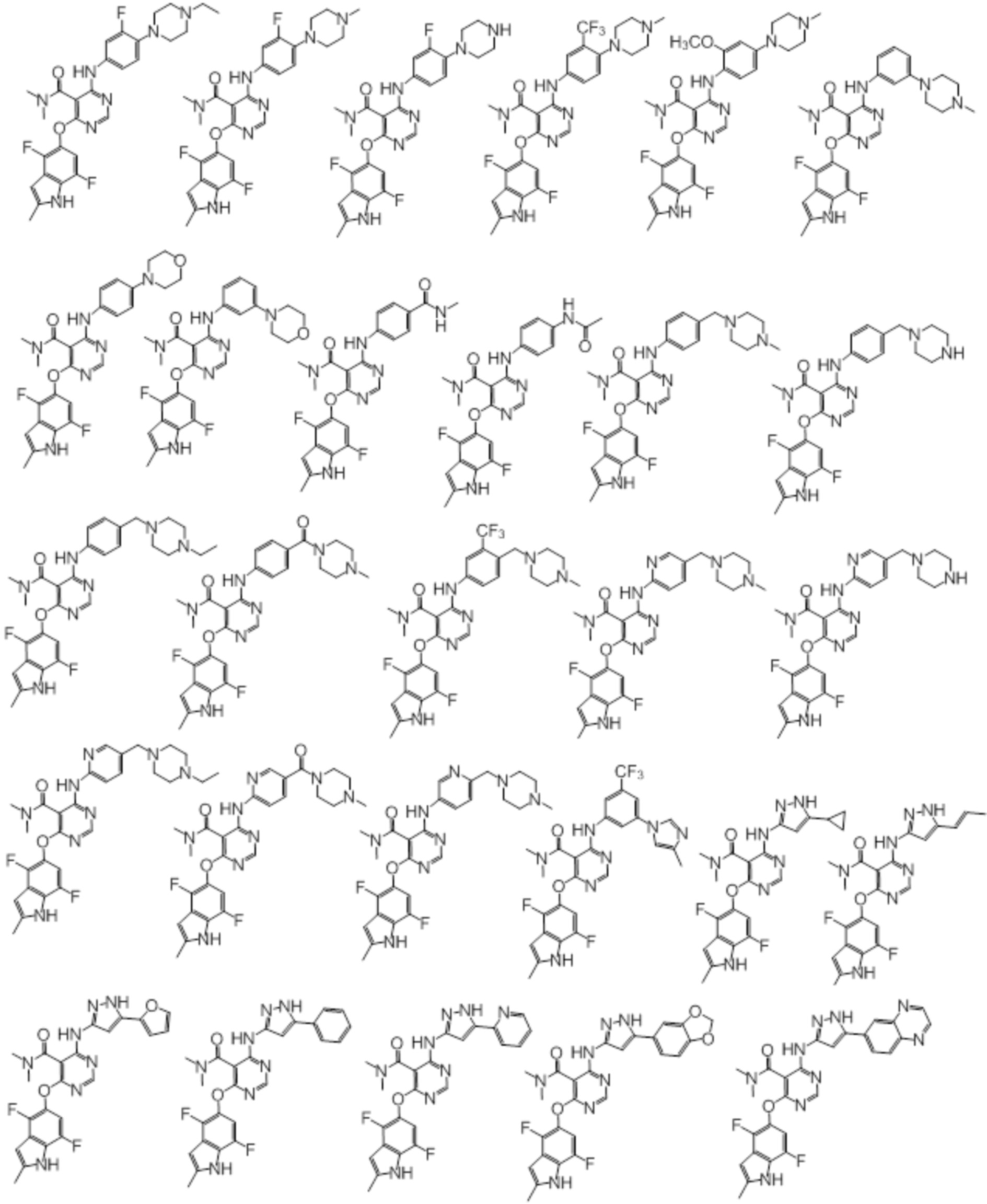
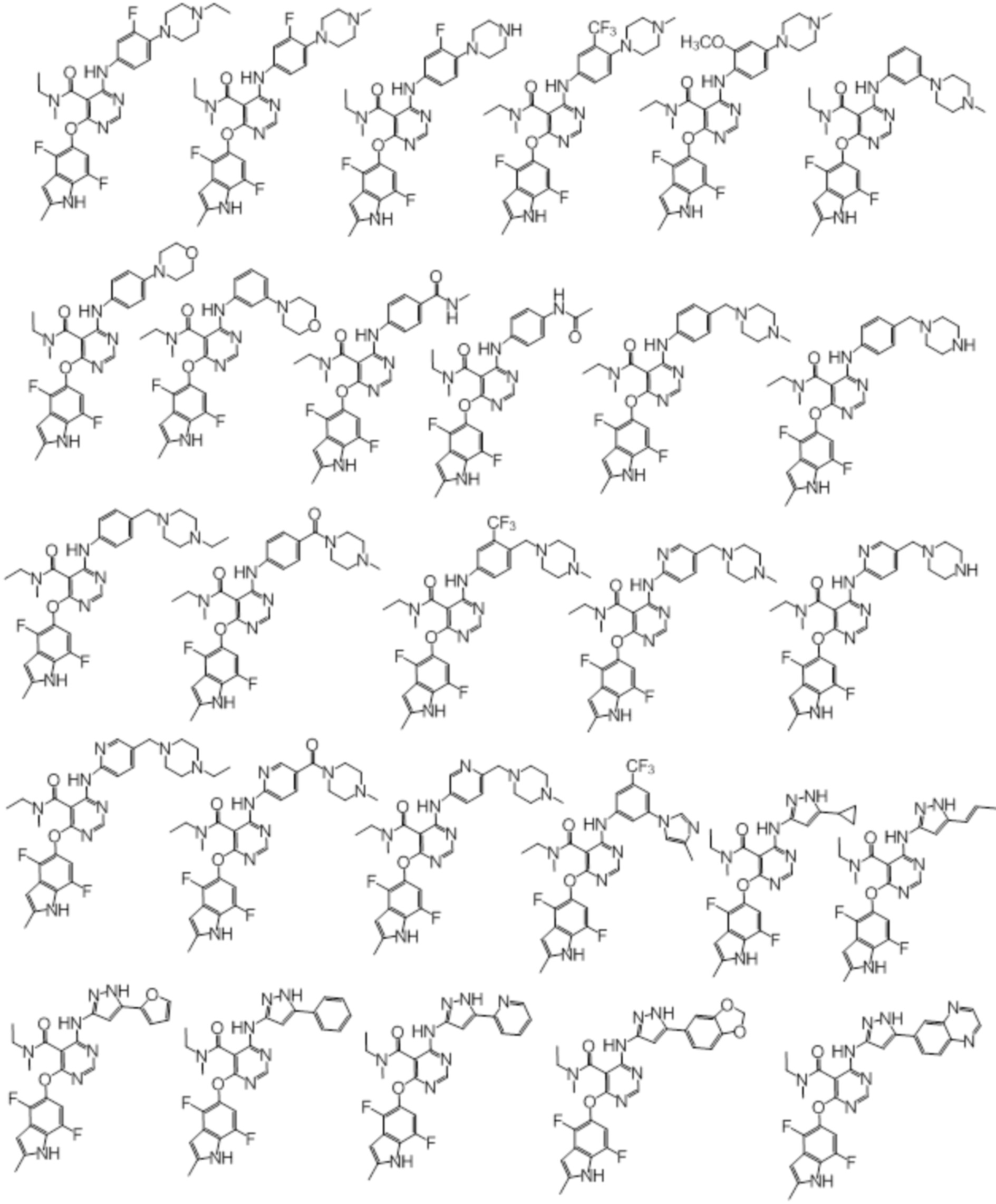
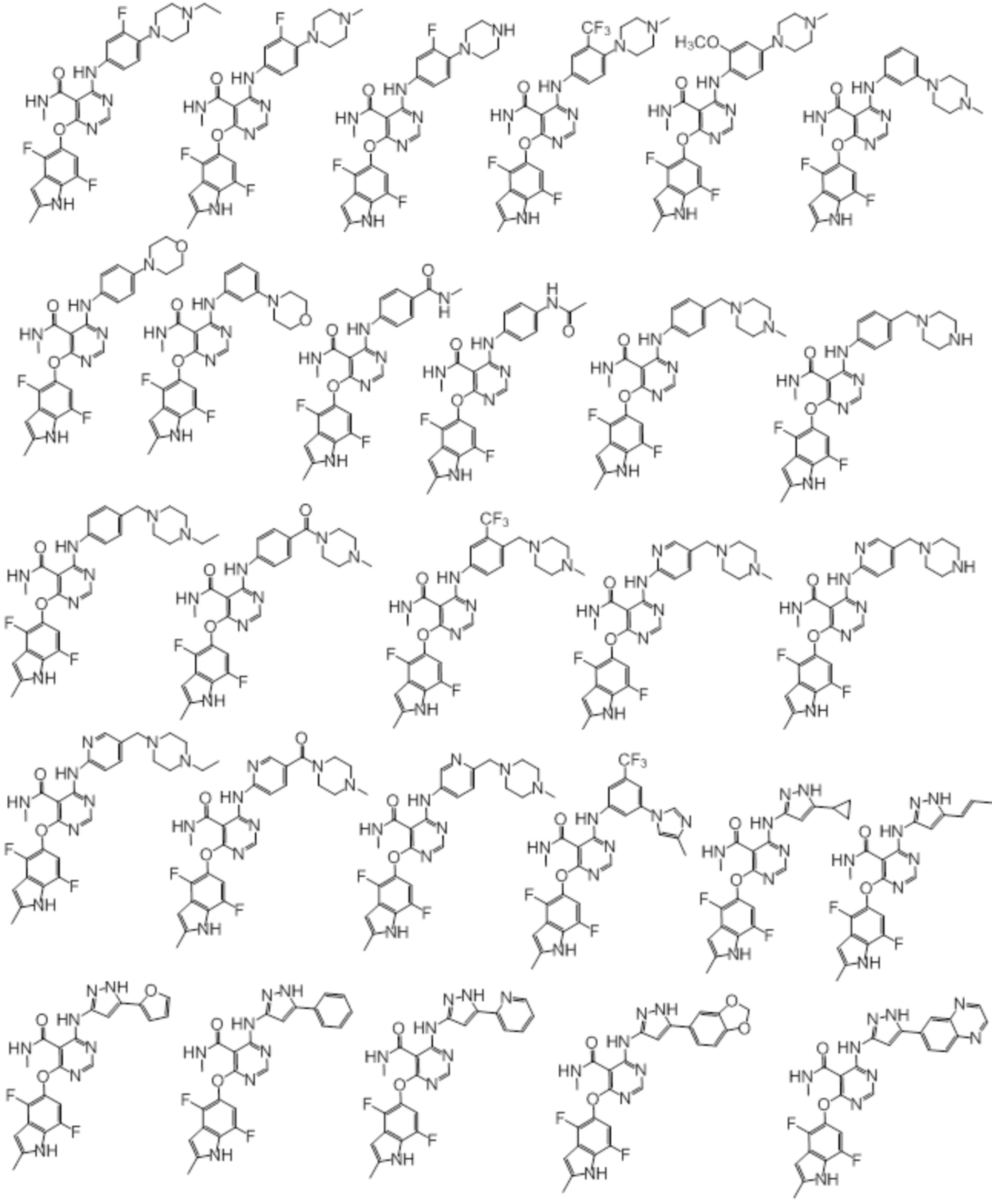
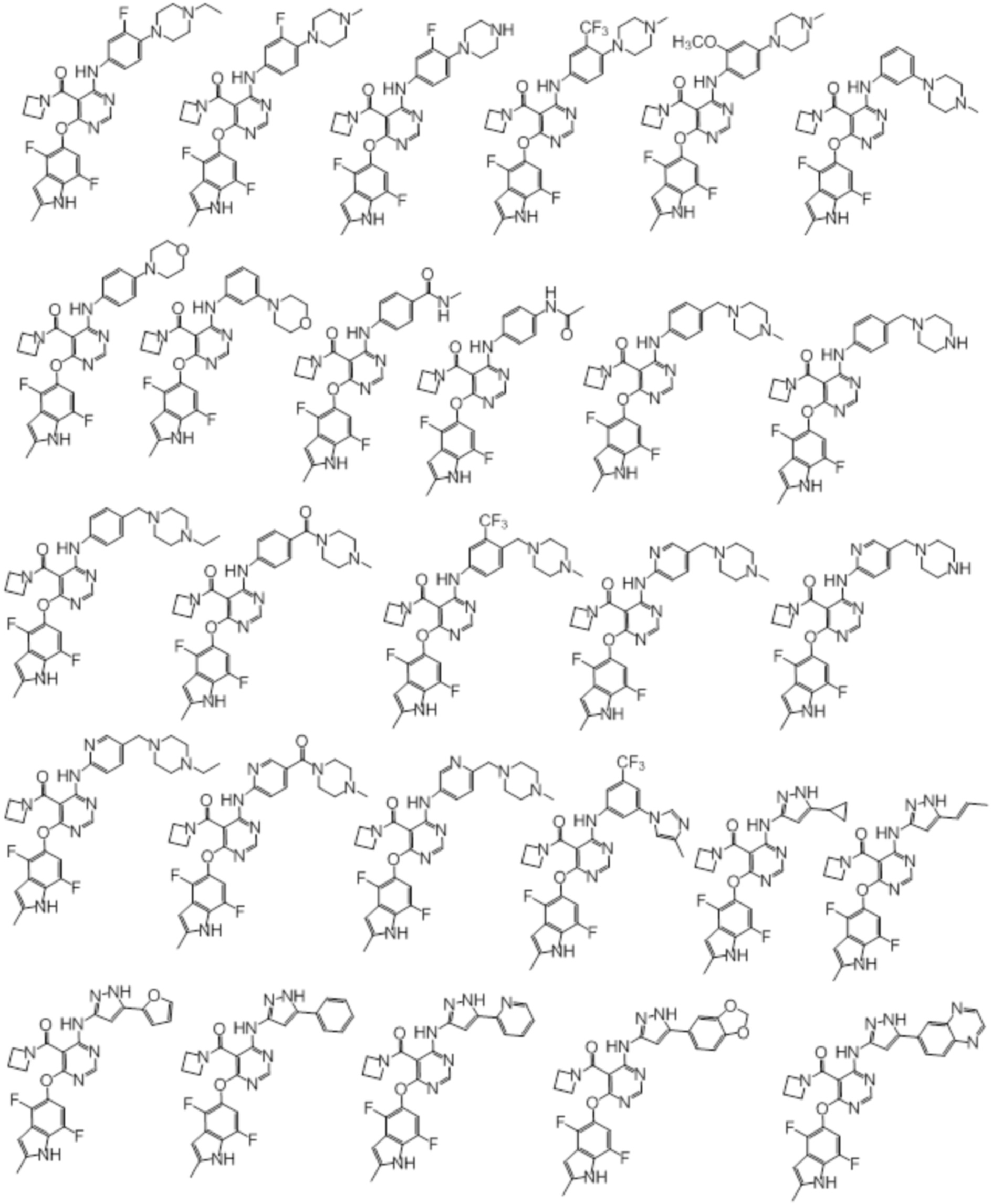
[0089] Согласно другому варианту осуществления предусмотрен способ получения соединений согласно настоящему изобретению. Соединения согласно настоящему изобретению, в общем, могут быть получены с использованием 4,6-дихлорпиримидина с различными заместителями в 5-положении. Соединение (I) может содержать различные стереоизомеры, геометрические изомеры, таутомеры, и т.д. Все возможные изомеры и их смеси включены в настоящее изобретение, и соотношение компонентов в смеси конкретно не ограничено.
[0090] Соединения-производные пиримидина формулы (I) в настоящем изобретении могут быть синтезированы из коммерчески доступных предшественников с использованием общепринятых методик. Например, может быть использован путь синтеза, сходный с представленными на любой из схем, вместе со способами синтеза, известными в области органического синтеза, или их вариациями, известным специалистам в данной области техники. Каждая переменная характеристика в последующих схемах относится к любой группе, соответствующей описанию соединений, представленных в настоящем документе.
[0091] В последующих схемах термин «восстановление» относится к процессу восстановления нитрогруппы до аминогруппы или к процессу преобразования сложноэфирной функциональной группы до спирта. Восстановление нитрогруппы может проводиться целым рядом способов, хорошо известных специалистам в области органического синтеза, включая без ограничения каталитического гидрирование, восстановление SnCl2 и восстановление хлоридом титана. В последующих схемах термин «гидролиз» относится к взаимодействию субстрата или реагента с водой. Более конкретно, термин «гидролиз» относится к процессу преобразования сложноэфирной или нитритной функциональной группы до карбоновой кислоты. Этот процесс может катализироваться целым рядом кислот или оснований, хорошо известных специалистам в области органического синтеза.
[0092] Соединения формулы (I) могут быть получены с использованием известных химических реакций и методик. Последующие общие способы получения представлены с целью помочь специалисту в данной области техники синтезировать ингибиторы, а более подробные примеры, представленные в экспериментальной части, описывают рабочие примеры.
[0093] Пропенилпиразоламин, определенный формулой (III), не является коммерчески доступным. Он может быть получен несколькими способами, описанными ранее (см., например, предварительную патентную заявку США № 61/555,738).
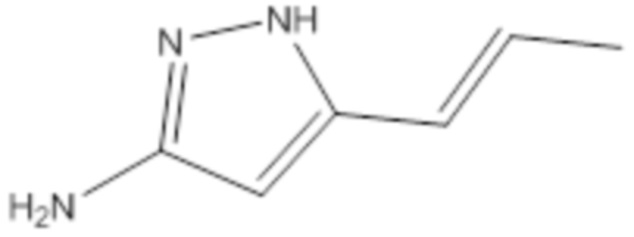 (II)
(II)
[0094] Предшественники замещенного индол-5-ола, определенные формулой (III), могут быть приобретены у поставщиков или синтезированы из коммерчески доступных предшественников с использованием общепринятых методик. (WO2004/009542, P33-38; Journal of Medicinal Chemistry, 2006, Vol 49, No. 7, P2143-2146; Org. Lett. Vol 10, No 12, 2008, P 2369-2372; WO00/47212, P245-250; WO2009/036055 A1, P57).
[0095] В частности, о предшественнике 4,7-дифториндол-5-оле, определенном формулой (IIIa), ранее не сообщалось, и он может быть получен несколькими способами, описанными ранее (WO2014/145403 A1).
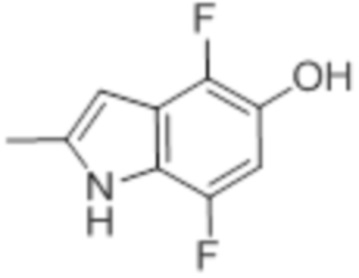
(III) (IIIa)
[0096] Предшественники 5-замещенных 4,6-дихлорпиримидинов, определенные формулой (IV), могут быть приобретены у поставщиков. В частности, предшественник, определенный формулой (IVa), может быть синтезирован из коммерчески доступных предшественников с использованием общепринятых методик (международная заявке согласно PCT 2010141406, 09 декабря 2010 года, соединение 310F).
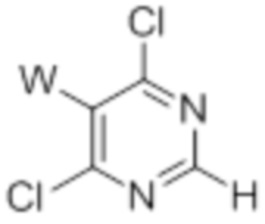
(IV) (IVa)
[0097] В общем, предшественники ArNH2 могут быть приобретены у поставщиков. Предшественники ArNH2, определенные формулой (V) могут быть приобретены у поставщиков синтезированы из коммерчески доступных предшественников с использованием общепринятых методик. (J. Med. Chem. 2010, 53, 7938-7957, конкретно, P7949).
 (V)
(V)
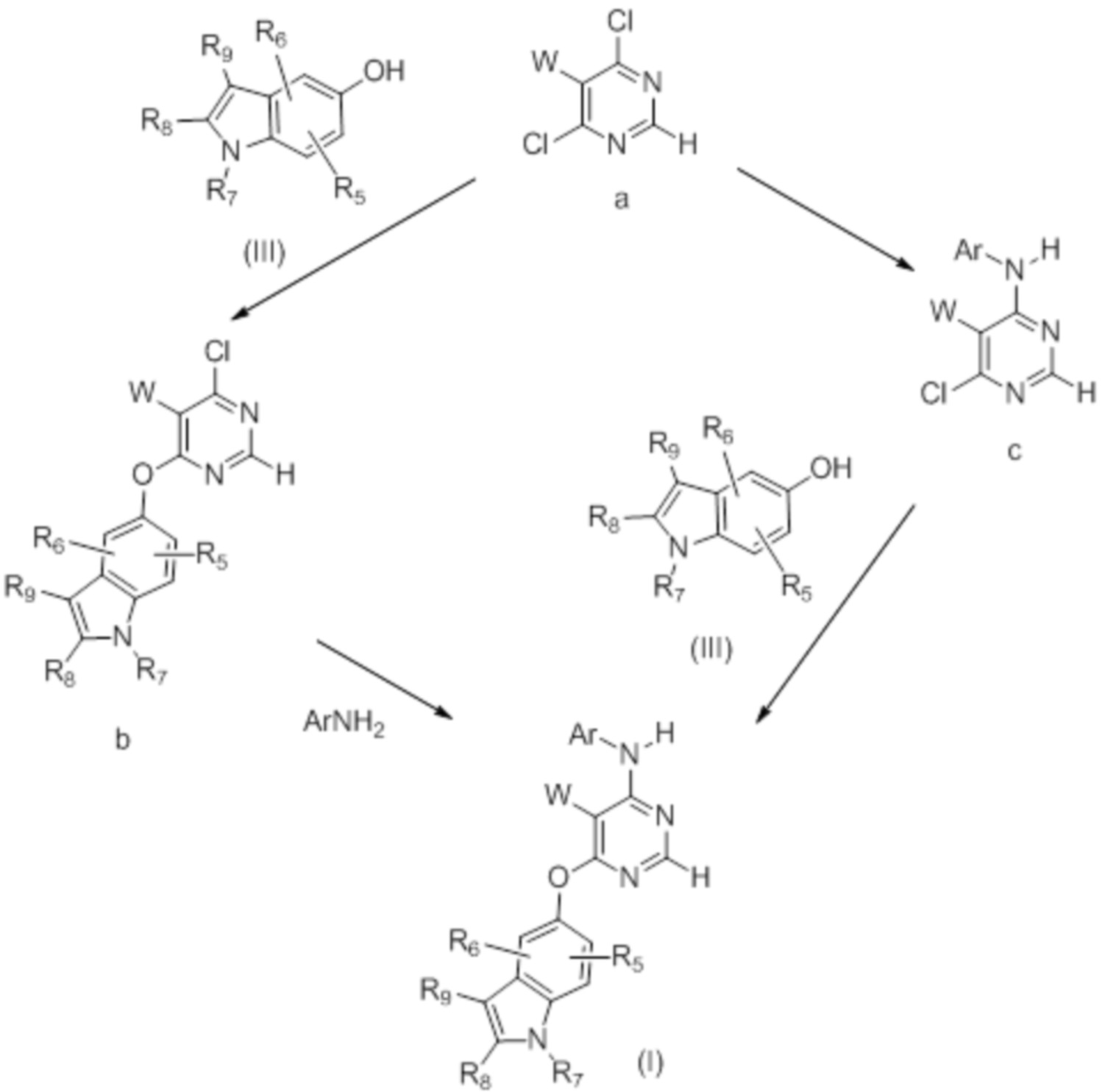
[0098] Получение соединений формулы (I) в настоящем изобретении может осуществляться способами, перечисленными на схеме 1.
 (I)
(I)
[0099] Как представлено на схеме 1, производное пиримидина (I) может быть синтезировано путем осуществления взаимодействия 5-замещенного 4,6-дихлорпиримидина с замещенным индол-5-олом с получением монохлорпиримидинового промежуточного продукта b, который может вступать во взаимодействие с ArNH2 с получением конечного соединения (I). Реакция может быть многостадийной или однореакторной. Для получения производных пиримидина также может быть использована альтернативная последовательность.
Схема 1
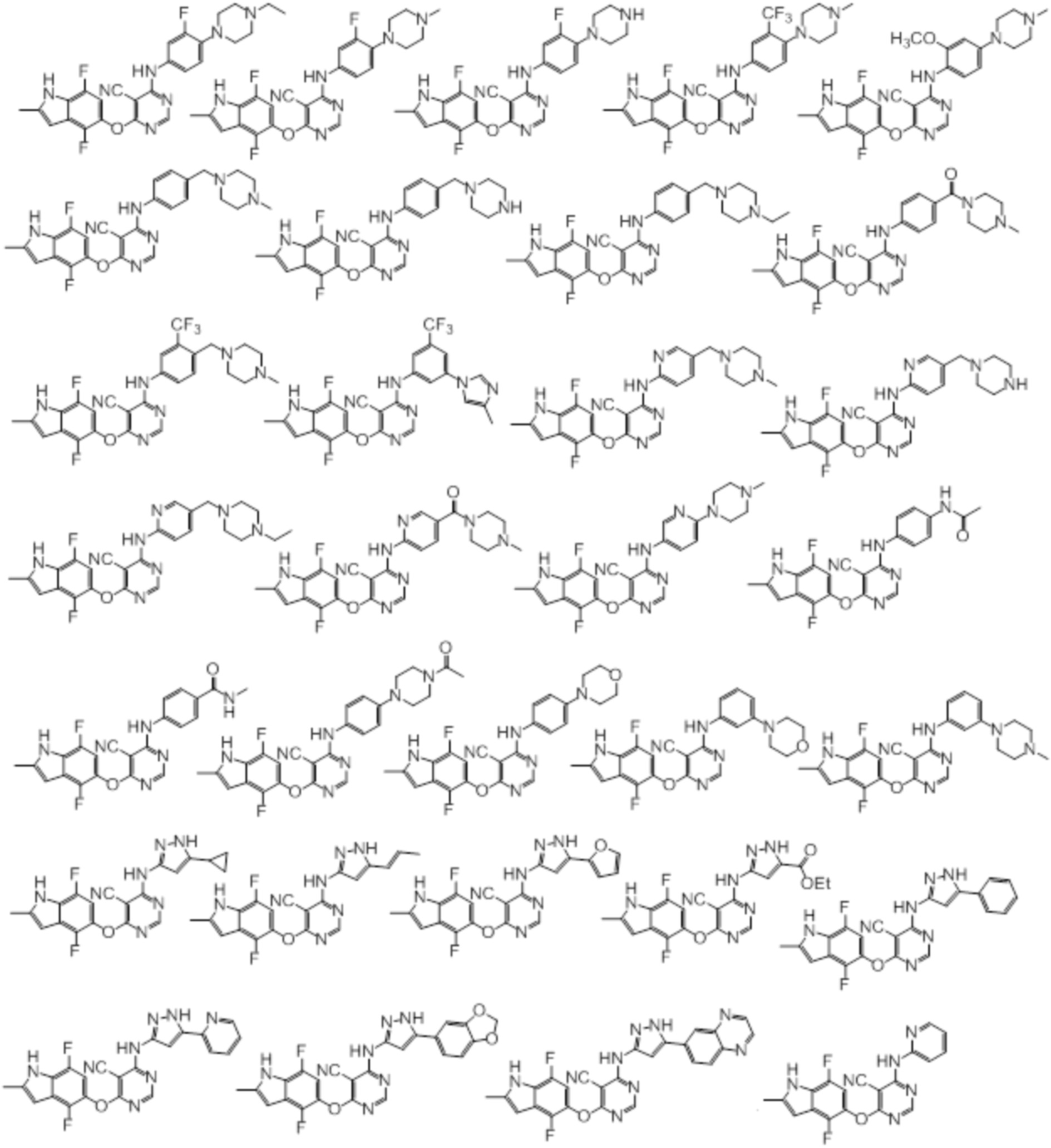
Как представлено на схеме 2, конечные соединения, определенные формулой (I-b), могут быть синтезированы из соответствующих предшественников, в которых W представляет собой CN.
Схема 2
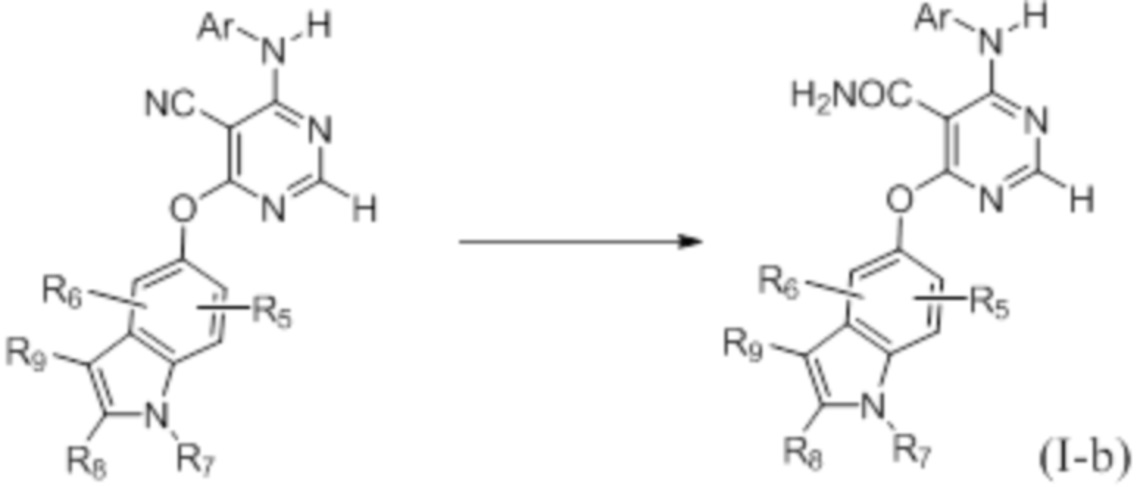
[00100] Реакцию предпочтительно проводят в присутствии инертного растворителя. Особые ограничения на природу растворителя не налагаются при условии, что он не оказывает неблагоприятного эффекта на реакцию или на участвующие в реакции реагенты, и что он способен растворять реагенты, по меньшей мере, до некоторой степени. Примеры подходящих растворителей включают: алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, в особенности ароматические и алифатические углеводороды, такие как хлористый метилен, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол и дихлорбензолы; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; нитросоединения, которые могут представлять собой нитроалканы или нитроараны, такие как нитроэтан и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, которые могут представлять собой амиды жирных кислот, такие как формамид, диметилформамид, диметилацетамид и гексаметилфосфорный триамид; и сульфоксиды, такие как диметилсульфоксид и сульфолан.
[00101] Реакция может протекать в широком диапазоне температур, и точное значение температуры реакции не является критичным для настоящего изобретения. В общем, авторы изобретения находят удобным проведение реакции при температуре от -50°C до 100°C.
[00102] Настоящее изобретение относится к композициям, которые представляют собой составы из одного или нескольких активных лекарств и фармацевтически приемлемого носителя. В этой связи, настоящее изобретение относится к композиции для введения субъекту-млекопитающему, которая может включать соединение формулы I или его фармацевтически приемлемые соли.
[00103] Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислотно-аддитивных солей включают ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формат, фумарат, глюкогептаноат, глицерофосфат, гликолят, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая кислота, хотя и не являются фармацевтически приемлемыми сами по себе, могут быть использованы для получения солей, применимых в качестве промежуточных продуктов, при получении соединений согласно настоящему изобретению и их фармацевтически приемлемых кислотно-аддитивных солей.
[00104] Соли, полученные из подходящих оснований, включают соли щелочных металлов (например, натрия и калия), щелочноземельных металлов (например, магния), аммония и соли N+(C1-4алкил)4. Настоящее изобретение также предусматривает кватернизацию любых основных азотсодержащих групп соединений, раскрытых в настоящем документе. Путем такой кватернизации могут быть получены водо- или жирорастворимые или диспергируемые продукты.
[00105] Композиции согласно настоящему изобретению можно вводить перорально, парентерально, посредством ингаляционного спрея, местно, ректально, назально, буккально, вагинально или посредством имплантируемого резервуара. Используемый в настоящем документе термин «парентерально» включает подкожную, внутривенную, внутримышечную, внутрисуставную, интрасиновиальную, интрастернальную, интратекальную, внутрипеченочную, внутриочаговую и интракраниальную инъекцию или инфузионные методики. Предпочтительно, композиции вводят перорально, интраперитонеально или внутривенно.
[00106] Фармацевтически приемлемые композиции согласно настоящему изобретению можно вводить перорально в любой перорально приемлемой лекарственной форме, включая без ограничения капсулы, таблетки, пастилки, эликсиры, суспензии, сиропы, облатки, жевательные резинки, водные суспензии или растворы.
[00107] Пероральные композиции могут содержать дополнительные ингредиенты, такие как: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; наполнитель, такой как крахмал или лактоза; разрыхлитель, такой как альгиновая кислота, кукурузный крахмал и т.п.; смазка, такая как стеарат магния; глидант, такой как коллоидный диоксид кремния; и подсластитель, такой как сахароза или сахарин, или вкусоароматизатор, такой как перечная мята, метилсалицилат или апельсиновый вкусоароматизатор. Если лекарственная форма представляет собой капсулу, то она может дополнительно содержать жидкий носитель, такой как жирное масло. Другие лекарственные формы могут содержать другие различные вещества, которые модифицируют физическую форму лекарственной формы, такие как, например, покровные средства. Таким образом, таблетки или пилюли могут быть покрыты сахаром, шеллаком или другими кишечнорастворимыми покрытиями. В дополнение к активным ингредиентам, сироп может содержать сахарозу в качестве подсластителя и определенные консерванты, красители и пигменты и вкусоароматизаторы. Вещества, используемые для приготовления указанных различных композиций, должны быть фармацевтически или ветеринарно чистыми и нетоксичными в используемых количествах.
[00108] С целью парентерального терапевтического введения, активный ингредиент можно включать в состав раствора или суспензии. Растворы или суспензии могут также включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфат натрия; хелатирующие средства, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и средства для регулирования тоничности, такие как хлорид натрия или декстроза. Парентеральный препарат может быть заключен в ампулы, одноразовые шприцы или многодозовые флаконы, изготовленные из стекла или пластика.
[00109] Фармацевтические формы, подходящие для инъекционного применения, включают стерильные растворы, дисперсии, эмульсии и стерильные порошки. Конечная форма должна быть стабильной в условиях производства и хранения. Кроме того, конечная фармацевтическая форма должна быть защищена от контаминации, а потому должна быть способна ингибировать рост микроорганизмов, таких как бактерии или грибы. Может вводиться однократная внутривенная или интраперитонеальная доза. В качестве альтернативы, может применяться медленная длительная инфузия или множество коротких ежесуточных инфузий, как правило продолжающихся от 1 до 8 суток. Также может быть использовано дозирование через сутки или дозирование раз в несколько суток.
[00110] Стерильные инъекционные растворы могут быть приготовлены путем включения соединения в необходимом количестве в состав одного или нескольких подходящих растворителей, в которые при необходимости могут быть добавлены другие ингредиенты, перечисленные выше или известные специалистам в данной области техники. Стерильные инъекционные растворы могут быть приготовлены путем включения соединения в необходимом количестве в состав подходящего растворителя с различными другими ингредиентами, при необходимости. Затем могут следовать стерилизационные процедуры, такие как фильтрование. Как правило, дисперсии приготавливают путем включения соединения в состав стерильной основы, которая также содержит дисперсионную среду и другие указанные выше необходимые соединения. В случае стерильного порошка, предпочтительные способы включают вакуумную сушку или лиофилизацию, при которых добавляют любые необходимые ингредиенты.
[00111] Подходящие фармацевтические носители включают стерильную воду; солевой раствор, декстрозу; декстрозу в воде или солевом растворе; продукты конденсации касторового масла и этиленоксида, содержащие приблизительно от 30 приблизительно до 35 моль этиленоксида на моль касторового масла; жидкую кислоту; низшие алканолы; масла, такие как кукурузное масло, арахисовое масло, кунжутное масло и т.п., вместе с эмульгаторами, такими как моно- или диглицериды жирной кислоты, или фосфатид, например, лецитин и т.п.; гликоли; полиалкиленгликоли; водную среду в присутствии способствующего суспендированию средства, например карбоксиметилцеллюлозы; альгинат натрия; поли(винилпирролидон); и т.п., по отдельности или вместе с подходящими способствующими диспергированию средствами, такими как лецитин; полиоксиэтиленстеарат; и т.п. Носитель также может содержать адъюванты, такие как консерванты, стабилизаторы, увлажнители, эмульгаторы и т.п. вместе с усилителем проницаемости. Во всех случаях, указанная конечная форма должна быть стерильна, а также должна быть способна легко проходить через инъекционное устройство, такое как полая игла. Подходящая вязкость может быть достигнута и сохранена посредством правильного выбора растворителей или наполнителей. Кроме того, могут применяться молекулярные или дисперсные покрытия, такие как лецитин, может проводиться правильный выбор размера частиц в дисперсиях или использоваться вещества со свойствами сурфактантов.
[00112] В соответствии с настоящим изобретением предусмотрены композиции, содержащие производные пиримидина, и способы, применимые для доставки in vivo производных пиримидина в форме наночастиц, которые подходят для любого из вышеупомянутых путей введения.
[00113] Патенты США №№ 5,916,596, 6,506,405 и 6,537,579 рассматривают получение наночастиц из биосовместимых полимеров, таких как альбумин. Поэтому, в соответствии с настоящим изобретением, предусмотрены способы получения наночастиц согласно настоящему изобретению посредством методики выпаривания растворителя из эмульсии типа «масло-в-воде», приготовленной в условиях высоких сил сдвига (например, обработка ультразвуком, гомогенизация высокого давления или т.п.)
[00114] В качестве альтернативы, фармацевтически приемлемые композиции согласно настоящему изобретению можно вводить в форме суппозиториев для ректального введения. Такие композиции могут быть приготовлены путем смешивания средства с подходящим нераздражающим наполнителем, который является твердым при комнатной температуре, но жидким при ректальной температуре, а потому будет разжижаться в прямой кишке, высвобождая лекарство. Такие вещества включают какао-масло, пчелиный воск и полиэтиленгликоли.
[00115] Фармацевтически приемлемые композиции согласно настоящему изобретению также можно вводить местно, особенно если мишень для обработки включает области, легкодоступные для местного применения, включая заболевания глаз, кожи или нижние отделы кишечника. Подходящие местные составы легко можно приготовить для каждой из этих областей или органов.
[00116] Местное применение для нижних отделов кишечника может быть осуществлено посредством состава ректального суппозитория (см. выше) или в виде подходящего состава клизмы. Также можно использовать чрескожные пластыри для местного применения.
[00117] Для местного применения фармацевтически приемлемые композиции могут быть составлены в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителях. Носители для местного введения соединений согласно настоящему изобретению включают без ограничения минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, соединение полиоксипропилена, эмульгирующий воск или воду. В качестве альтернативы, фармацевтически приемлемые композиции могут быть составлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители включают без ограничения минеральное масло, сорбитмоностеарат, полисорбат 60, воск сложных эфиров цетила, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[00118] Для офтальмологического применения фармацевтически приемлемые композиции могут быть составлены в виде микронизированных суспензий в изотоническом стерильном солевом растворе с регулируемым значением рН или, предпочтительно, в виде растворов в изотоническом стерильном солевом растворе с регулируемым значением рН, как в присутствии, так и в отсутствие консерванта, такого как бензалкония хлорид. В качестве альтернативы, для офтальмологического применения фармацевтически приемлемые композиции могут быть составлены в виде мази, такой как вазелин.
[00119] Фармацевтически приемлемые композиции, согласно настоящему изобретению, также можно вводить посредством назального аэрозоля или ингаляции. Такие композиции приготавливают в соответствии с методиками, хорошо известными в области техники фармацевтических составов, и они могут быть приготовлены в виде растворов в солевом растворе, с использованием бензилового спирта или других подходящих консервантов, усилителей всасывания для усиления биодоступности, фторуглеродов и/или других общепринятых солюбилизаторов или средств, способствующих диспергированию.
[00120] Наиболее предпочтительно, фармацевтически приемлемые композиции согласно настоящему изобретению составляют для перорального введения.
[00121] В соответствии с настоящим изобретением соединения согласно настоящему изобретению можно использовать для лечения заболеваний, ассоциированных с пролиферацией или гиперпролиферацией клеток, таких как злокачественные опухоли, которые включают без ограничения опухоли назальной полости, параназальных синусов, носоглотки, ротовой полости, ротоглотки, гортани, гортаноглотки, слюнных желез и параганглиом. Соединения согласно настоящему изобретению также можно использовать для лечения злокачественных опухолей печени и желчных протоков (в частности, гепатоцеллюлярной карциномы), злокачественных опухолей кишечника, в частности, колоректального рака, рака яичников, мелкоклеточного и немелкоклеточного рака легких, рака молочной железы, сарком (включая фибросаркому, злокачественную фиброзную гистиоцитому, эмбриональную рабдомиосаркому, лейомиосаркому, нейрофибросаркому, остеосаркому, синовиальную саркому, липосаркому и альвеолярную саркому мягких тканей), новообразований центральной нервной системы (в частности, злокачественной опухоли головного мозга) и лимфом (включая лимфому Ходжкина, лимфоплазмацитоидную лимфому, фолликулярную лимфому, лимфому лимфоидной ткани слизистых оболочек, лимфому из клеток мантии, крупноклеточную лимфому В-клеточной линии, лимфому Беркитта и Т-клеточную анапластическую крупноклеточную лимфому).
[00122] Соединения и способы согласно настоящему изобретению, как при введении по отдельности, так и в сочетании с другими средствами (например, химиотерапевтическими средствами или белковыми терапевтическими средствами, описанными ниже), также применимы для лечения целого ряда нарушений, включая без ограничения, например: инсульт, сердечно-сосудистое заболевание, инфаркт миокарда, застойную сердечную недостаточность, кардиомиопатию, миокардит, ишемическую болезнь сердца, болезнь коронарных артерий, кардиогенный шок, сосудистый шок, легочную гипертензию, отек легких (включая кардиогенный отек легких), экссудативный плеврит, ревматоидный артрит, диабетическую ретинопатию, пигментный ретинит и ретинопатии, включая диабетическую ретинопатию и ретинопатию недоношенных, воспалительные заболевания, рестеноз, бронхиальную астму, острый или респираторный дистресс-синдром взрослых (ARDS), волчанку, транссудацию, защиту от ишемического или реперфузионного повреждения, такого как ишемическое или реперфузионное повреждение, возникающее в процессе трансплантации органов, индукцию транспланционной толерантности; ишемическое или реперфузионное повреждение после ангиопластики; артрит (такой как ревматоидный артрит, псориатический артрит и остеоартрит); рассеянный склероз; воспалительное заболевание кишечника, включая язвенный колит и болезнь Крона; волчанку (системная красная волчанка); заболевания «трансплантат против хозяина»; заболевания Т-клеточно-опосредованной гиперчувствительности, включая контактную гиперчувствительность, гиперчувствительность замедленного типа и глютен-чувствительная энтеропатия (целиакия); сахарный диабет I типа; псориаз; контактный дерматит (включая дерматит, вызванный ядовитым сумахом); тиреоидит Хашимото; синдром Шегрена; аутоиммунный гипертиреоидизм, такой как болезнь Грейвса; болезнь Аддисона (аутоиммунное заболевание надпочечников); аутоиммунное полигландулярное заболевание (также известное как аутоиммунный полигландулярный синдром); аутоиммунную алопецию; пернициозную анемию; витилиго; аутоиммунный гипопитуатаризм; синдром Гийена-Барре; другие аутоиммунные заболевания; злокачественные опухоли, включая таковые, при которых киназы, такие как киназы Src-семейства, активированы или оверэкспрессируются, такие как карцинома толстого кишечника и тимома, или злокачественные опухоли, при которых киназная активность способствует росту или выживанию опухоли; гломерулонефрит, сывороточную болезнь; крапивницу; аллергические заболевания, такие как респираторные аллергии (бронхиальная астма, поллиноз, аллергический ринит) или кожные аллергии; фунгоидный микоз; острые воспалительные ответы (такие как острый или респираторный дистресс-синдром взрослых и ишемическое реперфузионное повреждение); дерматомиозит; очаговую алопецию; хроническую солнечную эритему; экзему; болезнь Бехчета; ладонно-подошвенный пустулез; гангренозную пиодермию; синдром Сезари; атопический дерматит; системный склероз; очаговую склеродермию; периферическую ишемию конечностей и ишемическое заболевание конечностей; заболевание кости, такое как остеопороз, остеомаляция, гиперпаратиреоидизм, болезнь Паджета и почечная остеодистрофия; синдромы транссудации, включая синдромы транссудации, индуцированные химиопрепаратами или иммуномодуляторами, такими как IL-2; повреждение или травму спинного и головного мозга; глаукому; заболевания сетчатки, включая макулярную дегенерацию; витреоретинальное заболевание; панкреатит; васкулиты, включая васкулит, болезнь Кавасаки, облитерирующий тромбангиит, грануломатоз Вегенера и болезнь Бехчета; склеродермию; преэкслампсию; талассемию; саркому Капоши; болезнь фон Гиппеля Линдау; и т.п.
[00123] В соответствии с настоящим изобретением, соединения согласно настоящему изобретению можно использовать для лечения заболеваний, ассоциированных с нежелательной пролиферацией или гиперпролиферацией клеток, включающего выявление животного, страдающего от указанного заболевания или состояния, и введение указанному животному композиции, содержащей соединение формулы (I), где заболевание или состояние ассоциировано с киназой.
[00124] Настоящее изобретение также относится к способам лечения животного, пораженного вышеупомянутыми заболеваниями и состояниями. Количество соединений согласно настоящему изобретению, которые могут быть скомбинированы с веществами-носителями для получения композиции в стандартной лекарственной форме, будет варьироваться в зависимости от подвергаемого лечению хозяина и конкретного способа введения. Предпочтительно, композиции должны быть составлены так, чтобы принимающему указанные композиции пациенту могла быть введена дозировка в пределах 0,01-100 мг/кг массы тела в сутки ингибитора.
[00125] Согласно одному аспекту, соединения согласно настоящему изобретению вводят субъекту, нуждающемуся в таком лечении, в сочетании с химиотерапевтическим средством, противовоспалительным средством, антигистаминами, химиотерапевтическим средством, иммуномодулятором, терапевтическим антителом или ингибитором протеинкиназ, например, ингибитором тирозинкиназы.
[00126] Способ включает введение одного или нескольких соединений согласно настоящему изобретению пораженному млекопитающему. Способ может дополнительно включать введение второго активного средства, такого как цитотоксическое средство, включая алкилирующие средства, факторы некроза опухоли, интеркаляторы, ингибиторы микротубулина и ингибиторы топоизомеразы. Второе активное средство может быть введено совместно в одной и той же композиции или в виде второй композиции. Примеры подходящих вторых активных средств включают без ограничения цитотоксическое лекарство, такое как активицин; акларубицин; акодазола гидрохлорид; акронин; адозелезин; алдеслейкин; алтретамин; амбомицин; аметантрона ацетат; аминоглютетимид; амсакрин; анастрозол; антрамицин; аспарагиназа; асперлин; азацитидин; азетепа; азотомицин; батимастат; бензодепа; бикалутамид; бизантрена гидрохлорид; биснафида димезилат; бизелезин; блеомицина сульфат; бреквинар натрия; бропиримин; бусульфан; кактиномицин; калустерон; карацемид; карбетимер; карбоплатин; кармустин; карубицина гидрохлорид; карзелезин; цедефингол; хлорамбуцил; циролемицин; цисплатин; кладрибин; криснатола мезилат; циклофосфамид; цитарабин; дакарбазин; дактиномицин; даунорубицина гидрохлорид; децитабин; дексормаплатин; дезагуанин; дезагуанин мезилат; диазиквон; доцетаксел; доксорубицин; доксорубицина гидрохлорид; дролоксифен; дролоксифена цитрат; дромостанолона пропионат; дуазомицин; эдатрексат; эфломитина гидрохлорид; эльсамитруцин; энлоплатин; энпромат; эпипропидин; эпирубицина гидрохлорид; эрбулозол; эзорубицина гидрохлорид; эстрамустин; эстрамустина фосфат натрия; этанидазол; эфирированное масло 131; этопозид; этопозида фосфат; этоприн; фадрозола гидрохлорид; фазарабин; фенретинид; флоксуридин; флударабина фосват; фторурацил; фторцитабин; фосквидон; фостриецин натрия; гемцитабин; гемцитабина гидрохлорид; золото Au198; гидроксимочевина; идарубицина гидрохлорид; ифосфамид; илмофозин; интерферон альфа-2a; интерферон альфа-2b; интерферон альфа-n1; интерферон альфа-n3; интерферон бета-Ia; интерферон гамма-Ib; ипроплатин; иринотекана гидрохлорид; ланреотида ацетат; летрозол; лейпролида ацетат; лиарозола гидрохлорид; лометрексол натрия; ломустин; лозоксантрона гидрохлорид; мазопрокол; майтансин; мехлорэтамин гидрохлорид; мегестрола ацетат; мелегестрола ацетат; мелфалан; меногарил; меркаптопурин; метотрексат; метотрексат натрия; метоприн; метуредепа; митиндомид; митокарцин; митокромин; митогиллин; митомалцин; митомицин; митоспер; митотан; митоксантрона гидрохлорид; микофеноловая кислота; нокодазол; ногаламицин; ормаплатин; оксизуран; паклитаксел; пегаспаргаза; пелиомицин; пентамустин; пепломицина сульфат; перфосфамид; пипоброман; пипосульфан; пироксантрона гидрохлорид; пликамицин; пломестан; порфимер натрия; порфиромицин; преднимустин; прокарбазина гидрохлорид; пуромицин; пуромицина гидрохлорид; пиразофурин; рибоприн; роглетимид; сафмгол; сафингола гидрохлорид; семустин; симтразен; спарфосат натрия; спарсомицин; спирогермания гидрохлорид; спиромустин; спироплатин; стрептонигрин; стрептозоцин; стронция хлорид Sr 89; сулофенур; талисомицин; таксан; таксоид; текогалан натрия; тегафур; телоксантрона гидрохлорид; темопорфин; тенипозид; тероксирон; тестолактон; тиамиприн; тиогуанин; тиотепа; тиазофурин; тирапазамин; топотекана гирохлорид; торемифена цитрат; трестолона ацетат; трицирибина фосфат; триметрексат; триметрексат глюкуронат; трипторелин; тубулозола гидрохлорид; урациловый иприт; уредепа; вапреотид; вертепорфин; винбластина сульфат; винкрситина сульфат; виндезин; виндезина сульфат; винепидина сульфат; винглицината сульфат; винлейрозина сульфат; винорелбина тартрат; винрозидина сульфат; винзолидина сульфат; ворозол; зениплаин; зиностатин; и зорубицина гидрохлорид.
[00127] В соответствии с настоящим изобретением, соединения и композиции могут быть использованы в субцитотоксических количествах в сочетании с другими средствами с целью достижения высокоселективной активности при лечении не неопластических нарушений, таких как болезнь сердца, инсульт и нейродегенеративные заболевания (Whitesell et al., Curr Cancer Drug Targets (2003), 3(5), 349-58).
[00128] Типовые терапевтические средства, которые можно вводить в сочетании с соединениями согласно настоящему изобретению, включают ингибиторы EGFR, такие как гефитиниб, эрлотиниб и цетуксимаб. Ингибиторы Her2 включают канертиниб, EKB-569 и GW-572016. Также включены ингибиторы Src, дасатиниб, а также касодекс (бикалутамид), тамоксифен, ингибиторы киназы MEK-1, ингибиторы киназы MARK, ингибиторы PI3 и ингибиторы PDGF, такие как иматиниб, ингибиторы Hsp90, такие как 17-AAG и 17-DMAG. Также включены антиангиогенные и антиваскулярные средства, которые прерывая кровоток к солидным опухолям, делают клетки злокачественных опухолей неактивными, лишая их питания. Также может использоваться эмаскуляция, которая также делает андроген-зависимые карциномы непролиферативными. Также включены ингибиторы IGF1R, ингибиторы нерецепторных и рецепторных тирозинкиназ и ингибиторы интегрина.
[00129] Фармацевтическая композиция и способ согласно настоящему изобретению могут дополнительно включать другие белковые терапевтические средства, такие как цитокины, иммуномодулирующие средства и антитела. Используемый в настоящем документе термин «цитокин» охватывает хемокины, интерлейкины, лимфокины, монокины, колоний-стимулирующие факторы и рецептор-ассоциированные белки и их функциональные фрагменты. Используемый в настоящем документе термин «функциональный фрагмент» относится к полипептиду или пептиду, который обладает биологической функцией или активностью, которую определяют посредством определенного функционального метода исследования. Цитокины включают эндотелиальный активирующий моноциты полипептид II (EMAP-II), CSF гранулоцитов и макрофагов (GM-CSF), CSF гранулоцитов (G-CSF), CSF макрофагов (M-CSF), IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-12 и IL-13, интерфероны и т.п., которые ассоциированы с конкретной биологической, морфологической или фенотипической альтерацией в клетке или клеточном механизме.
[00130] Другие терапевтические средства для комбинированной терапии включают циклоспорины (например, циклоспорин А), CTLA4-Ig, антитела, такие как ICAM-3, анти-IL-2 рецептор (Anti-Tac), анти-CD45RB, анти-CD2, анти-CD3 (OKT-3), анти-CD4, анти-CD80, анти-CD86, средства, блокирующие взаимодействие между CD40 и gp39, такие как антитела, специфичные для CD40 и для gpn39 (т.е., CD154), рекомбинантные белки, состоящие из CD40 и gp39 (CD40Ig и CD8gp39), ингибиторы, такие как ингибиторы ядерной транслокации, функции NF-κB, такие как деоксиспергуалин (DSG), ингибиторы биосинтеза холестерина, такие как ингибиторы HM:G CoA редуктазы (ловастатин и симвастатин), нестероидные противовоспалительные средства (NSAID), такие как ибупрофен, и ингибиторы циклооксигеназы, такие как рофекоксиб, стероиды, такие как преднизон или дексаметазон, соединения золота, антипролиферативные средства, такие как метотрексат, FK506 (такролимус, програф), микофенолята мофетил, цитотоксические лекарства, такие как азатиоприн и циклофосфамид, ингибиторы TNF-α, такие как тенидап, анти-TNF-антитела или растворимый рецептор TNF, и рапамицин (сиролимус или рапамун) или их производные.
[00131] Если другие терапевтические средства используются в сочетании с соединениями согласно настоящему изобретению, то их можно использовать, например, в количествах, указанных в Настольном справочнике врача (PDR), или определенных иным образом специалистом в данной области техники.
[00132] Далее представлены химическая структура и синтез 2 «промежуточных продуктов», применимых для синтеза 61 «соединения». Указанные 61 соединение будут протестированы на их способность ингибировать ряд киназ.
[00133] Все синтезы проводили в безводных условиях (т.е. в безводных растворителях) в атмосфере аргона, если не указано иное, с использованием высушенного в печи оборудования и с использованием стандартных методик работы с чувствительными к воздушной среде веществами. Водные растворы бикарбоната натрия(NaHCO3) и хлорида натрия (солевой раствор) были насыщенными.
Аналитическую тонкослойную хроматографию (TLC) проводили на пластинах Merck Kiesel gel 60 F254 с визуализацией ультрафиолетом и/или проявлением анисовым альдегидом, перманганатом калия или фосфорномолибденовой кислотой.
ЯМР-спектры: Спектры протонного (1H) ядерного магнитного резонанса регистрировали при 400 МГц. Данные представляли следующим образом: химический сдвиг, мультиплетность (с=синглет, д=дублет, т=триплет, кв=квартет, квинт=квинтет, дд=дублет дублетов, м=мультиплет, уш.с=уширенный синглет), константа взаимодействия (Дж/Гц) и результат интегрирования. Константы взаимодействия получали и рассчитывали непосредственно из спектра и приводили нескорректироваными.
Масс-спектры низкого разрешения: Использовали ионизацию электрораспылением (ES+). Регистрировали протонированный исходный ион (M+H) или исходный ион добавления натрия (M+Na) или фрагмент с наибольшей массой. Если не указано иное, то аналитический градиент состоял из 10% ACN в воде с повышением до 100% ACN в течение 5 минут.
Высокоэффективную жидкостную хроматографию (HPLC) использовали для анализа чистоты производных. HPLC проводили на колонке Phenomenex Synergi Polar-RP, 4u, 80A, 150×4.6 мм, с использованием системы Shimadzu, оснащенной фотодиодным матричным детектором SPD-M10A. Подвижная фаза A представляла собой воду, и подвижная фаза B представляла собой ацетонитрил с градиентом от 20% до 80% B в течение 60 минут с повторным уравновешиванием при A/B (80/20) в течение 10 минут. УФ-обнаружение проводили при 220 и 254 нм.
Последующее демонстрирует химическую структуру и синтез двух промежуточных соединений («промежуточный продукт 1» и «промежуточный продукт 2»), известных средним специалистам в данной области техники как пригодные для синтеза производных пиримидина, которые могут обладать ингибирующей активностью в отношении киназ.
Промежуточный продукт 1
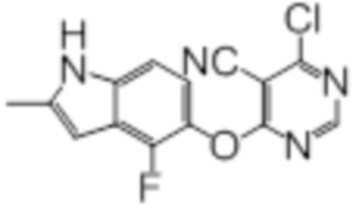
К раствору 4-фтор-2-метил-1H-индол-5-ола (200 мг, 1,21 ммоль) в смеси ацетонитрила (4 мл) и N,N-диметилформамида (1 мл) добавляли карбонат калия (200 мг, 1,45 ммоль). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, после чего добавляли суспензию 4,6-дихлорпиримидин-5-карбонитрила (221 мг, 1,27 ммоль) в 3 мл ацетонитрила. Эту смесь перемешивали при комнатной температуре в течение 1 ч. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь разбавляли водой и этилацетатом. Слои разделяли, и дважды экстрагировали водную фазу этилацетатом. Объединенные органические фазы промывали однократно водой, затем солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали фильтрат в условиях вакуума с получением целевого продукта в виде коричневых твердых веществ (365 мг, выход 99%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,46 (уш.с, 1H), 8,83 (с, 1H), 7,17 (д, J=8,8 Гц, 1H), 7,00 (т, J=7,6 Гц, 1H), 6,27 (с, 1H), 2,41 (с, 3H); ESI-MS: рассчит. для (C14H8ClFN4O) 302, обнаружено 303 (MH+).
Промежуточный продукт 2
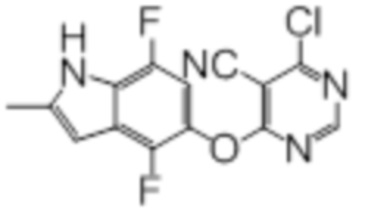
К раствору 4,7-дифтор-2-метил-1H-индол-5-ола (500 мг, 2,73 ммоль) в смеси ацетонитрила (9 мл) и N,N-диметилформамида (1 мл) добавляли карбонат калия (453 мг, 3,28 ммоль). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре, после чего добавляли суспензию 2,4-дихлор-5-цианопиримидина (499 мг, 2,87 ммоль) в смеси ацетонитрил/DMF (2,5 мл/2,5 мл). Эту смесь перемешивали при 0°C в течение 2 ч. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь разбавляли водой/солевым раствором и этилацетатом. Слои разделяли, и дважды экстрагировали водную фазу этилацетатом. Объединенные органические фазы промывали однократно водой и трижды солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали фильтрат в условиях вакуума с получением целевого соединения в виде пурпурных липких твердых веществ (890 мг, выход 100%, содержит некоторое количество DMF). Этот продукт использовали без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 11,91 (уш.с, 1H), 8,84 (с, 1H), 7,03 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,35 (уш.с, 1H), 2,39 (с, 3H); ESI-MS: рассчит. для (C14H7ClF2N4O) 320, обнаружено 321 (MH+).
Далее представлена химическая структура и способ синтеза 61 производного пиримидина (соединения 1-61), которые являются видами родового соединения, раскрытого в настоящем документе.
Соединение 1
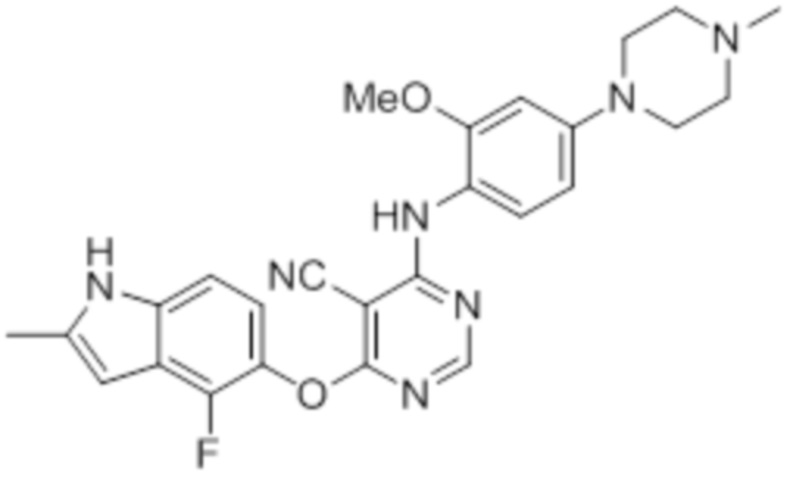
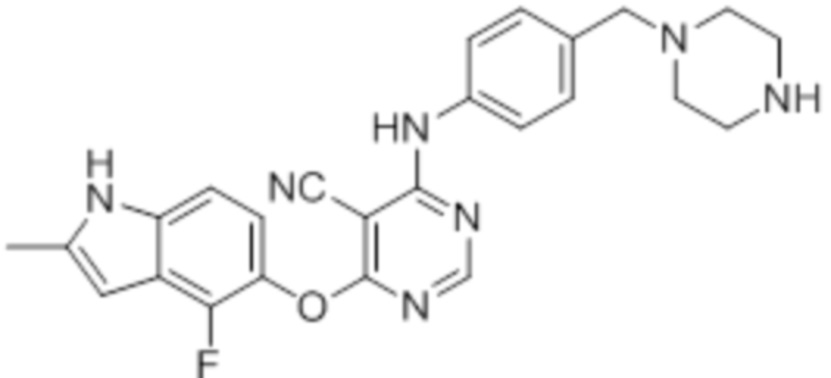
Смесь промежуточного продукта 1 (100 мг, 0,33 ммоль), 2-метокси-4-(4-метилпиперазин-1-ил)анилина (73 мг, 0,33 ммоль) и DIPEA (0,08 мл, 0,49 ммоль) в DMSO (5 мл) перемешивали при комнатной температуре в течение 30 мин. После проверки методом TLC, смесь добавляли к воде (100 мл). После охлаждения на бане со льдом, твердые вещества собирали путем фильтрования, промывали водой. Неочищенный продукт очищали методом колоночной хроматографии (силикагель, 0-15% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (64 мг, выход 40%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,34 (уш.с, 1H), 9,45 (уш.с с, 1H), 8,15 (с, 1H), 7,13 (м, 2H), 6,93 (м, 1H), 6,65 (м, 1H), 6,50 (м, 1H), 6,23 (с, 1H), 3,77 (с, 3H), 3,18 (м, 4H), 2,46 (м, 4H), 2,39 (с, 3H), 2,23 (с, 3H); ESI-MS: рассчит. для C26H26FN7O2 487, обнаружено 488 (MH+). Время удерживания согласно HPLC: 18,63 мин. Чистота: 96%.
Соединение 2
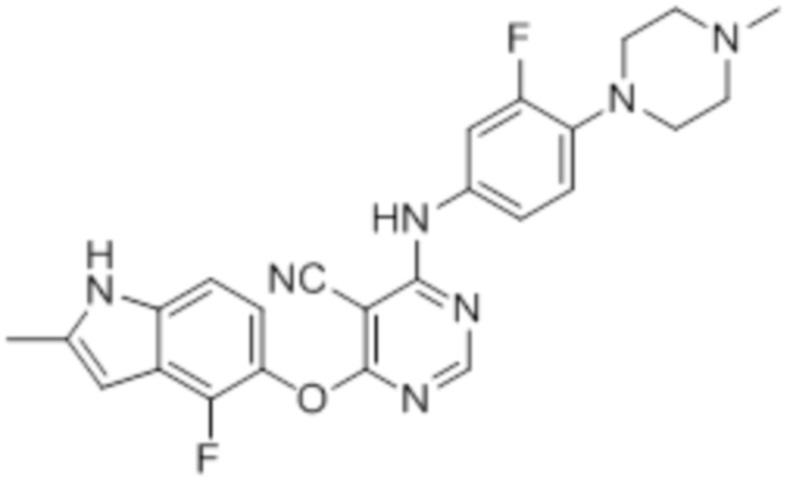
Смесь промежуточного продукта 1 (100 мг, 0,33 ммоль), 3-фтор-4-(4-метилпиперазин-1-ил)анилина (69 мг, 0,33 ммоль) и DIPEA (0,15 мл, 0,82 ммоль) в DMSO (5 мл) перемешивали при комнатной температуре в течение 30 мин. После проверки методом TLC, смесь добавляли к воде (100 мл). После охлаждения на бане со льдом, твердые вещества собирали путем фильтрования, промывали водой. Неочищенный продукт очищали методом колоночной хроматографии (силикагель, 0-15% MeOH в DCM) с получением целевого продукта в виде светло-коричневых твердых веществ (90 мг, выход 57%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 10,0 (с, 1H), 8,30 (с, 1H), 7,41 (м, 1H), 7,26 (м, 1H), 7,13 (м, 1H), 6,98 (м, 2H), 6,24 (уш.с с, 1H), 3,00 (м, 4H), 2,50 (м, 4H), 2,40 (с, 3H), 2,23 (с, 3H); ESI-MS: рассчит. для C25H23F2N7O 475, обнаружено 476 (MH+). Время удерживания согласно HPLC: 19,15 мин. Чистота: 95%.
Соединение 3
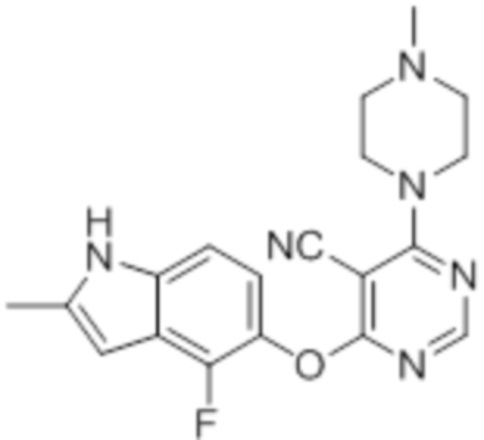
Смесь промежуточного продукта 1 (100 мг, 0,33 ммоль), 4-((4-метилпиперазин-1-ил)метил)анилина (68 мг, 0,33 ммоль) и DIPEA (0,15 мл, 0,82 ммоль) в DMSO (5 мл) перемешивали при комнатной температуре в течение 30 мин. После проверки методом TLC, смесь добавляли к воде (100 мл). После охлаждения на бане со льдом, твердые вещества собирали путем фильтрования, промывали водой. Неочищенный продукт очищали методом колоночной хроматографии (силикагель, 0-15% MeOH в DCM) с получением целевого продукта в виде не совсем белых твердых веществ (41 мг, выход 26%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,34 (уш.с с, 1H), 8,24 (м, 1H), 7,12 (м, 1H), 6,92 (м, 1H), 6,22 (с, 1H), 3,95 (м, 4H), 2,41s (м, 4H), 2,39 (с, 3H), 2,24 (с, 3H); ESI-MS: рассчит. для C19H19FN6O 366, обнаружено 367 (MH+). Время удерживания согласно HPLC: 12,36 мин. Чистота: 98%.
Соединение 4
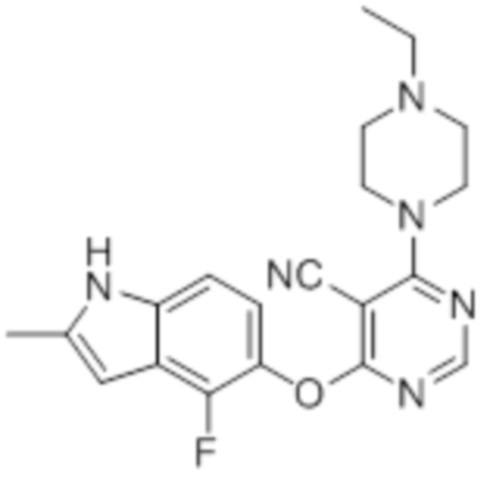
Смесь промежуточного продукта 1 (100 мг, 0,33 ммоль), 4-((4-этилпиперазин-1-ил)метил)анилина (72 мг, 0,33 ммоль) и DIPEA (0,15 мл, 0,82 ммоль) в DMSO (5 мл) перемешивали при комнатной температуре в течение 30 мин. После проверки методом TLC, смесь добавляли к воде (100 мл). После охлаждения на бане со льдом, твердые вещества собирали путем фильтрования, промывали водой. Неочищенный продукт очищали методом колоночной хроматографии (силикагель, 0-15% MeOH в DCM) с получением целевого продукта в виде не совсем белых твердых веществ (18 мг, выход 11%) 1H-ЯМР (400 МГц, DMSO-d6) δ 11,34 (уш.с с, 1H), 8,24 (с, 1H), 7,12 (м, 1H), 6,92 (м, 1H), 6,23 (с, 1H), 3,96 (м, 4H), 2,52 (м, 3H), 2,45 (м, 4H), 1,23 (м, 2H), 1,050 (м, 3H); ESI-MS: рассчит. для C20H21FN6O 380, обнаружено 381 (MH+). Время удерживания согласно HPLC: 13,77 мин. Чистота: 93%.
Соединение 5
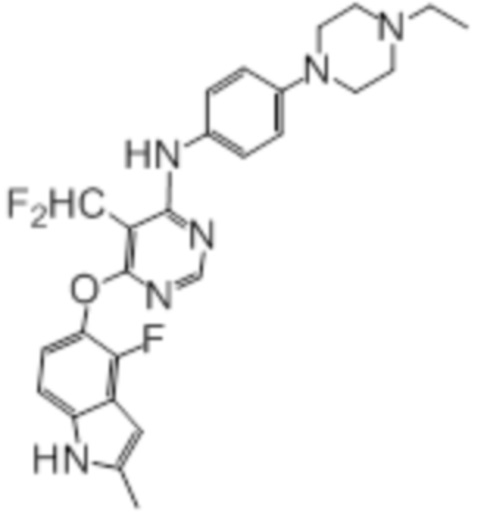
Стадия 1: К раствору фториндола (415 мг, 2,51 ммоль) и 4,6-дихлор-5-(дифторметил)пиридина (500 мг, 2,51 ммоль) в DMSO (3 мл) при комнатной температуре добавляли карбонат калия (695 мг, 5,03 ммоль), и нагревали смесь при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали расходование исходного вещества. Реакционную смесь добавляли в колбу с водой/солевым раствором (50 мл/50 мл), смесь перемешивали при комнатной температуре в течение 1 часа, а затем охлаждали на бане со льдом. Твердые вещества собирали путем фильтрования и промывали водой с получением целевого продукта в виде желтых твердых веществ (803 мг, выход 98%). Дополнительную очистку не проводили, и использовали продукт на следующей реакционной стадии. 1H-ЯМР (400 МГц, DMSO-d6) δ 11,39 (уш.с, 1H), 8,71 (с, 1H), 7,51 (т, J=52,4 Гц, 1H), 7,15 (д, J=8,8 Гц, 1H), 6,96 (т, J=7,6 Гц, 1H), 6,25 (с, 1H), 2,40 (с, 3H); ESI-MS: рассчит. для (C14H9ClF3N3O) 327, обнаружено 328 (MH+).
Стадия 2: Смесь полученного выше промежуточного продукта (125 мг, 0,61 ммоль), 4-(4-этилпиперазин-1-ил))анилина (200 мг, 0,61 ммоль) и DIPEA (0,27 мл, 1,52 ммоль) в DMSO (3,0 мл) перемешивали при 100°C в течение 2 ч, а затем при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь добавляли к смеси вода/нас. NH4Cl (50 мл/50 мл), и перемешивали при комнатной температуре в течение 30 мин. Значение pH смеси корректировали до ~6 с использованием 2н HCl. Охлаждали при 4°C, твердые вещества собирали путем фильтрования, промывали водой с получением липкого неочищенного продукта. Неочищенный продукт растворяли введением в DCM/MeOH (2 мл/2 мл), сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали на колонке (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (103 мг, выход 34%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,32 (уш.с, 1H), 10,49 (уш.с, 1H), 8,93 (с, 1H), 8,12 (с, 1H), 7,36 (д, J=8,8 Гц, 2H), 7,10 (д, J=8,8 Гц,1H), 7,00 (д, J=9,2 Гц, 2H), 6,86 (т, J=7,6 Гц, 1H), 6,21 (с, 1H), 3,80 (уш.с, 2H), 3,55 (уш.с, 2H), 3,10 (м, 6H), 2,39 (с, 3H), 1,28 (т, J=7,2 Гц, 3H); ESI-MS: рассчит. для (C26H27F3N6O) 496, обнаружено 497 (MH+).
Соединение 6
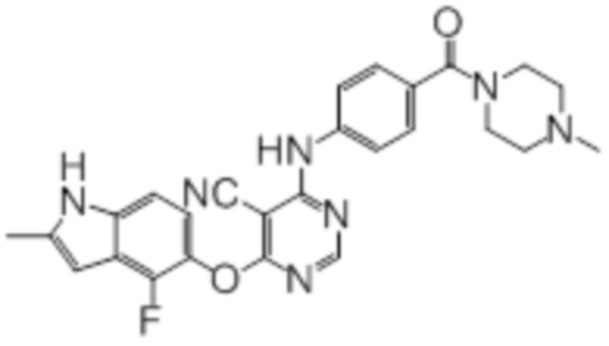
В колбу загружали промежуточный продукт 1 (150 мг, 0,5 ммоль), 1-(4-аминобензоил)-4-метилпиперазин (109 мг, 0,5 ммоль), TFA (50 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (160 мг, выход 66%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 10,17 (уш.с, 1H), 8,34 (с, 1H), 7,63 (д, J=8,8 Гц, 2H), 7,39 (д, J=8,8 Гц, 2H), 7,13 (д, J=8,4 Гц,1H), 6,96 (т, J=7,6 Гц, 1H), 6,24 (с, 1H), 3,80-3,40 (уш.с, 4H), 2,40 (с, 3H), 2,36-2,24 (уш.с, 4H), 2,19 (с, 3H); ESI-MS: рассчит. для (C26H24FN7O2) 485, обнаружено 486 (MH+).
Соединение 7
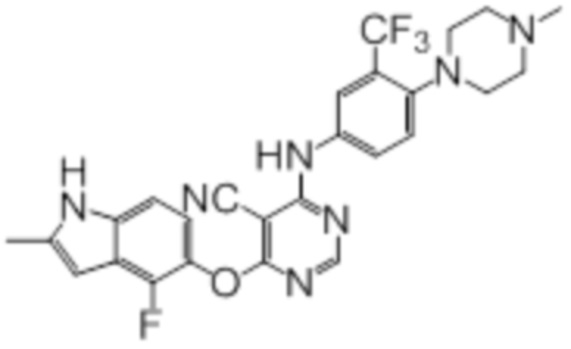
В колбу загружали промежуточный продукт 1 (100 мг, 0,33 ммоль), 4-(4-метилпиперазин-1-ил)-3-(трифторметил)анилин (86 мг, 0,33 ммоль), TFA (50 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (103 мг, выход 60%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 10,18 (уш.с, 1H), 8,33 (с, 1H), 7,90-7,80 (м, 2H), 7,55 (д, J=8,8 Гц, 1H), 7,13 (д, J=8,8 Гц,1H), 6,95 (т, J=7,2 Гц, 1H), 6,24 (с, 1H), 2,85 (м, 4H), 2,49-2,32 (м, 7H), 2,23 (с, 3H); ESI-MS: рассчит. для (C26H23F4N7O) 525, обнаружено 526 (MH+).
Соединение 8
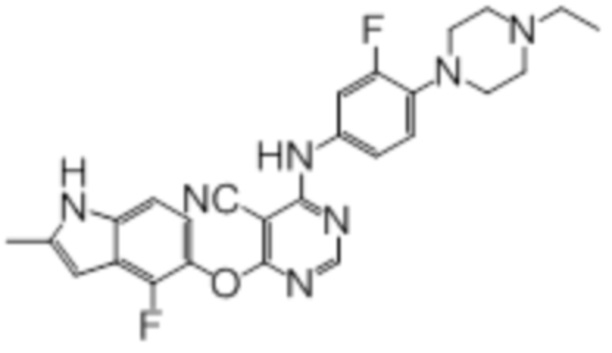
В колбу загружали промежуточный продукт 1 (100 мг, 0,33 ммоль), 4-(4-этилпиперазин-1-ил)-3-фторанилин (74 мг, 0,33 ммоль), TFA (50 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (120 мг, выход 74%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 9,99 (уш.с, 1H), 8,29 (с, 1H), 7,39 (дд, J=2,0 Гц, J=14,4 Гц, 1H), 7,26 (д, J=8,8 Гц, 1H), 7,13 (д, J=8,4 Гц,1H), 7,01 (т, J=9,2 Гц, 1H), 6,95 (т, J=8,0 Гц, 1H), 6,24 (с, 1H), 3,00 (м, 4H), 2,60-2,45 (м, 4H), 2,40 (м, 5H), 1,02 (т, J=7,2 Гц, 3H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
Соединение 9
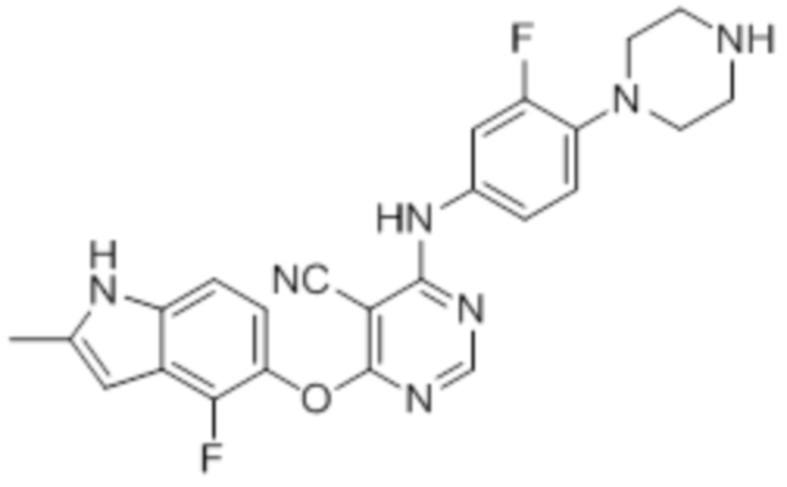
Смесь промежуточного продукта 1 (100 мг, 0,33 ммоль), трет-бутил-4-(4-амино-2-фторфенил)пиперазин-1-карбоксилата (97 мг, 0,33 ммоль) и DIPEA (0,15 мл, 0,82 ммоль) в DMSO (5 мл) перемешивали при комнатной температуре в течение 30 мин. После проверки методом TLC, смесь добавляли к воде 100 мл). После охлаждения на бане со льдом, твердые вещества собирали путем фильтрования, промывали водой. После сушки на воздухе при комнатной температуре в течение ночи, твердые вещества суспендировали введением в DCM/MeOH (10/1, 5 мл), и добавляли 1 мл TFA. Смесь перемешивали при комнатной температуре в течение ночи. После концентрирования, остаток растворяли введением в DCM/MeOH (8/2, 15 мл), и добавляли нас. раствор бикарбоната натрия приблизительно до pH~7. Органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом колоночной хроматографии (силикагель, 0-15% MeOH в DCM) с получением целевого продукта в виде не совсем белых твердых веществ (60 мг, выход 39%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 8,30 (с, 1H), 7,43 (м, 1H), 7,28 (м, 1H), 7,14 (м, 1H), 7,02 (м, 1H), 6,94 (м, 1H), 6,24 (с, 1H), 2,97 (м, 8H), 2,40 (м, 3H); ESI-MS: рассчит. для C24H21F2N7O 461, обнаружено 462 (MH+). Время удерживания согласно HPLC: неизвестно
Соединение 10
Смесь промежуточного продукта 1 (100 мг, 0,33 ммоль), трет-бутил-4-(4-аминобензил)пиперазин-1-карбоксилата (96 мг, 0,33 ммоль) и DIPEA (0,15 мл, 0,82 ммоль) в DMSO (5 мл) перемешивали при комнатной температуре в течение 30 мин. После проверки методом TLC, смесь добавляли к воде (100 мл). После охлаждения на бане со льдом, твердые вещества собирали путем фильтрования, промывали водой. После сушки на воздухе при комнатной температуре в течение ночи, твердые вещества суспендировали введением в DCM/MeOH (10/1, 5 мл), и добавляли 1 мл TFA. Смесь перемешивали при комнатной температуре в течение ночи. После концентрирования, остаток растворяли введением в DCM/MeOH (8/2, 15 мл) и добавляли нас. раствор бикарбоната натрия приблизительно до pH~7. Органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом колоночной хроматографии (силикагель, 0-15% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (15 мг, выход 10%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,48 (уш.с, 1H), 10,06 (с, 1H), 8,90 (м, 2H), 8,28 (м, 1H), 7,48 (м, 2H), 7,29 (м, 2H), 7,13 (м, 1H), 6,94 (м, 1H), 6,23 (с, 1H), 3,53 (м, 2H), 3,09 (м, 4H), 2,54 (м, 4H), 2,40 (м, 3H); ESI-MS: рассчит. для C25H24FN7O 457, обнаружено 458 (MH+). Время удерживания согласно HPLC: 13,75 мин. Чистота: 91%.
Соединение 11
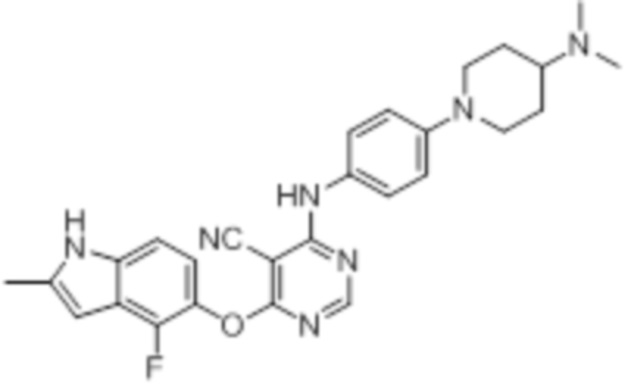
Стадия 1: Смесь 1-фтор-4-нитробензола (3,00 г, 21,3 ммоль), N,N-диметилпиперидин-4-амина дигидрохлорида (4,70 г, 23,4 ммоль) и DIPEA (15 мл, 86,1 ммоль) перемешивали при 95°C в течение 18 ч. Смесь охлаждали до комнатной температуры, разбавляли смесью EtOAc/гексаны (1/1 100 мл), промывали дважды водн. глюконатом кальция (по 100 мл каждый раз, 50% насыщенность), органический слой разделяли и сушили над безводным Na2SO4 и концентрировали с получением целевого продукта в виде красного масла (4,1 г, выход 77%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 8,03 (д, J=9,6 Гц, 1H), 7,01 (д, J=9,6 Гц, 1H), 4,04-4,01 (м, 2H), 3,02-2,95 (м, 2H), 2,39-2,32 (м, 1H), 2,17 (с, 6H), 1,85-1,81 (м, 2H), 1,44-1,34 (м, 2H); MS (ESI): рассчит. для C13H19N3O2: 249, обнаружено: 250 (MH+).
Стадия 2: Смесь N,N-диметил-1-(4-нитрофенил)пиперидин-4-амина (1,5 г, 6,0 ммоль), дигидрата хлорида олова(II) (6,8 г, 30 ммоль) и метанола (100 мл) перемешивали при 70°C в течение 19 ч. Смесь охлаждали до комнатной температуры, разбавляли смесью EtOAc/гексаны (4/1, 200 мл), промывали 5M водн. NaOH (200 мл), органический слой разделяли и сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-20% MeOH в DCM (об./об.) в качестве элюента с получением целевого 1-(4-аминофенил)-N,N-диметилпиперидин-4-амина в виде светло-желтого твердого вещества (340 мг, выход 26%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 6,67 (д, J=8,8 Гц, 2H), 6,47 (д, J=8,8 Гц, 2H), 4,53 (уш.с с, 2H), 3,37-3,34 (м, 2H), 2,48-2,42 (м, 2H), 2,18 (с, 6H), 2,11-2,05 (м, 1H), 1,80-1,77 (м, 2H), 1,51-1,41 (м, 2H); MS (ESI): рассчит. для C13H21N3: 219, обнаружено: 220 (MH+).
Стадия 3: К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (200 мг, 0,661 ммоль) и 1-(4-аминофенил)-N,N-диметилпиперидин-4-амина (142 мг, 0,647 ммоль) в безводном DMSO (2,0 мл) добавляли TEA (0,28 мл; 1,98 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 19 ч. Полученную смесь разбавляли смесью EtOAc/гексаны (4/1, 100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл; 50% насыщенность). Органический слой разделяли и сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации из EtOAc с получением целевого продукта в виде желтого твердого вещества (254 мг, выход 81%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,35 (уш.с с, 1H), 9,83 (уш.с с, 1H), 8,21 (с, 1H), 7,30-7,28 (м, 2H), 7,13-7,11 (м, 1H), 6,95-6,91 (м, 2H), 6,23 (с, 1H), 3,71-3,68 (м, 2H), 2,69-2,62 (м, 2H), 2,40 (с, 3H), 2,24-2,21 (м, 1H), 2,19 (с, 6H), 1,84-1,81 (м, 2H), 1,52-1,42 (м, 2H); MS (ESI): рассчит. для C27H28FN7O: 485, обнаружено: 486 (MH+).
Соединение 12
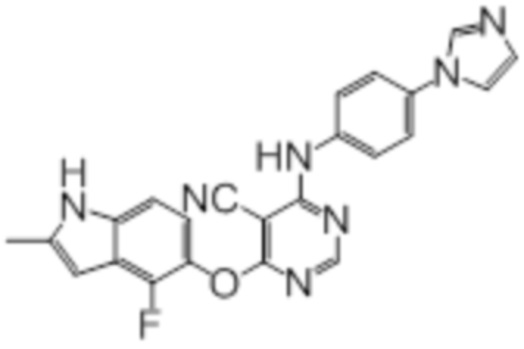
Смесь промежуточного продукта 1 (150 мг, 0,50 ммоль), 4-имидазол-1-илфениламина (91 мг, 0,57 ммоль) и DIPEA (0,22 мл, 1,245 ммоль) в DMSO (3,0 мл) перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь добавляли к смеси нас. NH4Cl/вода (25 мл/50 мл), и перемешивали при комнатной температуре в течение 30 мин. Значение pH смеси корректировали до ~6 с использованием 2н HCl. После охлаждения на льду в течение 1 ч, твердые вещества собирали путем фильтрования, промывали водой с получением неочищенного продукта. Неочищенный продукт очищали на колонке с силикагелем (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (140 мг, выход 66%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 10,16 (с, 1H), 8,31 (с, 1H), 8,24 (с, 1H), 7,66 (м, 5H), 7,14 (м, 2H), 6,95 (т, J=7,6 Гц, 1H), 6,24 (с, 1H), 2,40 (с, 3H); ESI-MS: рассчит. для (C23H16FN7O) 425, обнаружено 426 (MH+).
Соединение 13
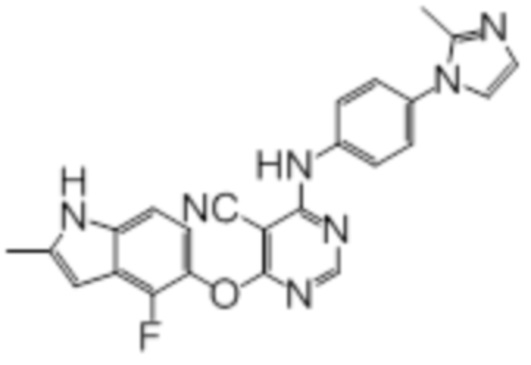
Смесь промежуточного продукта 1 (150 мг, 0,50 ммоль), 4-(2-метилимидазол-1-илфениламина (99 мг, 0,57 ммоль) и DIPEA (0,22 мл, 1,245 ммоль) в DMSO (3,0 мл) перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь добавляли к смеси нас. NH4Cl/вода (25 мл/50 мл), и перемешивали при комнатной температуре в течение 30 мин. Значение pH смеси корректировали до ~6 с использованием 2н HCl. После охлаждения на льду в течение 1 ч, твердые вещества собирали путем фильтрования, промывали водой с получением неочищенного продукта. Неочищенный продукт очищали на колонке с силикагелем (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (108 мг, выход 50%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 10,16 (уш.с, 1H), 8,33 (с, 1H), 7,70 (д, J=8,8 Гц, 2H), 7,42 (д, J=8,8 Гц, 2H), 7,28 (уш.с, 1H), 7,14 (д, J=8,4 Гц, 1H), 6,96 (т, J=7,6 Гц, 1H), 6,25 (с, 1H), 2,41 (с, 3H), 2,30 (с, 3H); ESI-MS: рассчит. для (C24H18FN7O) 439, обнаружено 440 (MH+).
Соединение 14
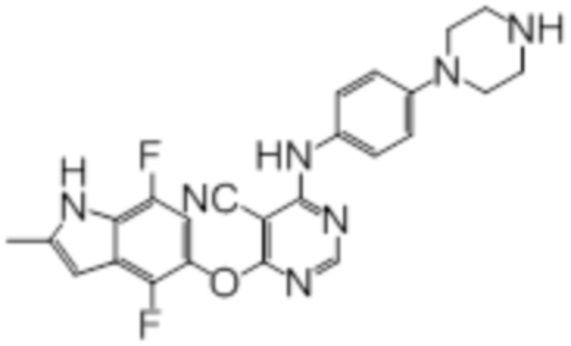
Смесь промежуточного продукта 2 (150 мг, 0,47 ммоль), трет-бутил-4-(4-аминофенил)пиперазин-1-карбоксилата (149 мг, 0,54 ммоль) и DIPEA (0,21 мл, 1,17 ммоль) в DMSO (3,0 мл) перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь добавляли к смеси нас. NH4Cl/вода (25 мл/50 мл), и перемешивали при комнатной температуре в течение 30 мин. Значение pH смеси корректировали до ~6 с использованием 2н HCl. После охлаждения на льду в течение 1 ч, твердые вещества собирали путем фильтрования, промывали водой с получением неочищенного продукта. Неочищенный продукт суспендировали в DCM (10 мл), и добавляли 1 мл TFA (смесь становилась прозрачным раствором). Смесь перемешивали при комнатной температуре в течение ночи. К смеси добавляли фосфат калия в воде (pH~8), и экстрагировали DCM/MeOH. Объединенную органическую фазу промывали солевым раствором, концентрировали и очищали на колонке с силикагелем (5-15% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (97 мг, выход 45%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,90 (уш.с, 1H), 8,24 (с, 1H), 7,32 (д, J=8,8 Гц, 2H), 7,00 (дд, J =5,6 Гц, J=10,4 Гц,1H), 6,94 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,14 (м, 4H), 2,99 (м, 4H), 2,41 (с, 3H) (пик NH может располагаться под пиком растворителя); ESI-MS: рассчит. для (C24H21F2N7O) 461, обнаружено 462 (MH+).
Соединение 15
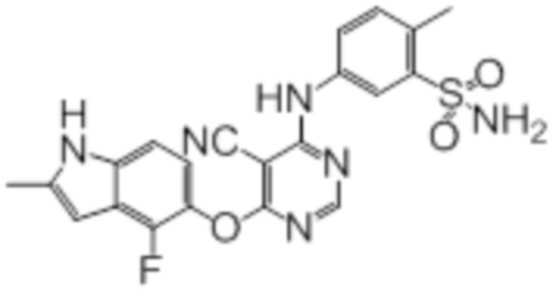
В колбу загружали промежуточный продукт 1 (100 мг, 0,33 ммоль), 5-амино-2-метилбензолсульфонамид (77 мг, 0,41 ммоль), TFA (50 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (79 мг, выход 53%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 10,25 (уш.с, 1H), 8,30 (с, 1H), 8,07 (уш.с, 1H), 7,69 (дд, J=2,4 Гц, J=8,4 Гц, 1H), 7,39 (уш.с, 2H), 7,37 (д, J=8,4 Гц, 1H), 7,13 (д, J=8,8 Гц,1H), 6,95 (т, J=7,6 Гц, 1H), 6,24 (с, 1H), 2,57 (с, 3H), 2,40 (с, 3H); ESI-MS: рассчит. для (C21H17FN6O3S) 452, обнаружено 453 (MH+).
Соединение 16
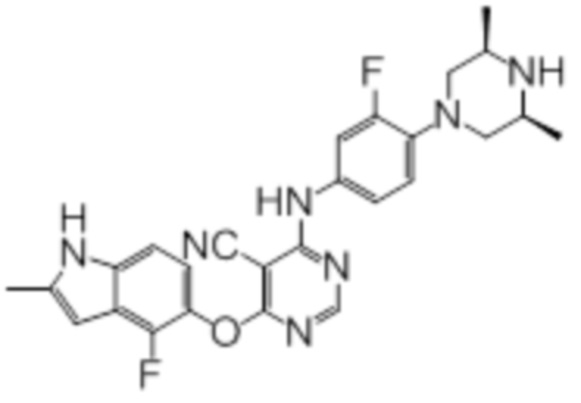
[00134] В колбу загружали промежуточный продукт 1 (100 мг, 0,33 ммоль), 4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-фторанилин (92 мг, 0,41 ммоль), TFA (50 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (118 мг, выход 73%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,35 (уш.с, 1H), 9,98 (уш.с, 1H), 8,29 (с, 1H), 7,40 (дд, J=2,4 Гц, J=14,4 Гц, 1H), 7,26 (дд, J=2,0 Гц, J=8,4 Гц, 1H), 7,12 (д, J=8,8 Гц, 1H), 6,95 (м, 2H), 6,24 (с, 1H), 3,17 (д, J=9,6 Гц, 2H), 2,91 (м, 2H), 2,40 (с, 3H), 2,19 (м, 3H), 1,00 (с, 3H), 0,98 (с, 3H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00135]
[00136] Соединение 17
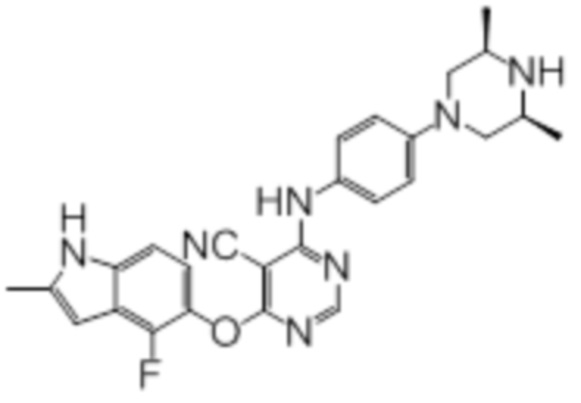
[00137] В колбу загружали промежуточный продукт 1 (100 мг, 0,33 ммоль), 4-((3S,5R)-3,5-диметилпиперазин-1-ил)анилин (85 мг, 0,41 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (122 мг, выход 78%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 9,84 (уш.с, 1H), 8,21 (с, 1H), 7,28 (д, J=8,8 Гц, 2H), 7,12 (д, J=8,8 Гц, 1H), 6,92 (м, 3H), 6,24 (с, 1H), 3,51 (м, 2H), 2,84 (м, 2H), 2,40 (с, 3H), 2,11 (м, 3H), 1,03 (с, 3H), 1,01 (с, 3H); ESI-MS: рассчит. для (C26H26FN7O) 471, обнаружено 472 (MH+).
[00138]
[00139] Соединение 18
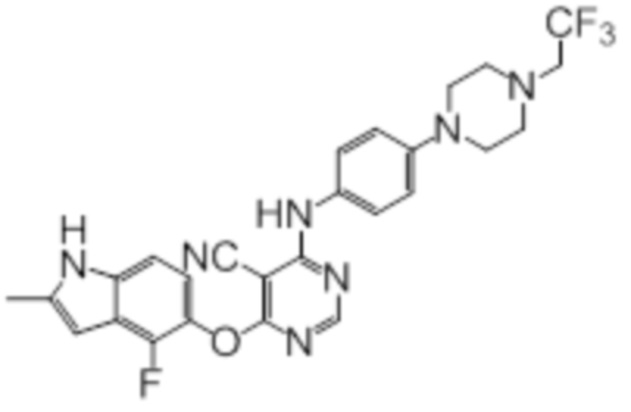
[00140] В колбу загружали промежуточный продукт 1 (100 мг, 0,33 ммоль), 4-(4-(2,2,2-трифторэтил)пиперазин-1-ил)анилин (86 мг, 0,33 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% EtOAc в DCM) с получением целевого продукта в виде желтых твердых веществ (73 мг, выход 42%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,36 (уш.с, 1H), 9,86 (уш.с, 1H), 8,21 (с, 1H), 7,31 (д, J=8,8 Гц, 2H), 7,12 (д, J=8,4 Гц, 1H), 6,94 (м, 3H), 6,24 (с, 1H), 3,23 (кв, J=10 Гц, 2H), 3,13 (м, 4H), 2,76 (м, 4H), 2,34 (с, 3H); ESI-MS: рассчит. для (C26H23F4N7O) 525, обнаружено 526 (MH+).
[00141]
[00142] Соединение 19
[00143] В колбу загружали промежуточный продукт 2 (100 мг, 0,31 ммоль), 4-((3S,5R)-3,5-диметилпиперазин-1-ил)анилин (80 мг, 0,39 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (146 мг, выход 95%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,86 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,90 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,51 (м, 2H), 2,84 (м, 2H), 2,41 (с, 3H), 2,11 (м, 2H), 1,02 (д, J=6,4 Гц, 6H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00144]
[00145] Соединение 20
[00146] В колбу загружали промежуточный продукт 2 (100 мг, 0,31 ммоль), 4-(4-(2,2,2-трифторэтил)пиперазин-1-ил)анилин (101 мг, 0,39 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 100°C в течение 4 ч. Реакционную смесь подщелачивали добавлением насыщенного водного раствора бикарбоната натрия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% EtOAc в DCM) с получением целевого продукта в виде желтых твердых веществ (101 мг, выход 60%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,88 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,8 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,8 Гц, 1H), 6,94 (д, J=9,2 Гц, 2H), 6,34 (с, 1H), 3,25 (кв, J=10,0 Гц, 2H), 3,14 (м, 4H), 2,76 (м, 4H), 2,41 (с, 3H); ESI-MS: рассчит. для (C26H22F5N7O) 543, обнаружено 544 (MH+).
[00147]
[00148] Соединение 21
[00149]
[00150] 
[00151]
[00152] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (200 мг, 0,661 ммоль) и 3-морфолиноанилина (124 мг, 0,694 ммоль) в безводном DMSO (2,0 мл) добавляли TEA (0,28 мл; 1,98 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 18 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл; 50% насыщенность). Органический слой разделяли и сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации из EtOAc с получением целевого продукта в виде коричневого твердого вещества (123 мг, выход 42%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,36 (уш.с с, 1H), 9,89 (уш.с с, 1H), 8,28 (с, 1H), 7,23-7,19 (м, 1H), 7,14-7,10 (м, 2H), 7,05-7,03 (м, 1H), 6,96-6,92 (м, 1H), 6,79-6,77 (м, 1H), 3,75-3,73 (м, 4H), 3,11-3,09 (м, 4H), 2,40 (с, 3H); MS (ESI): рассчит. для C24H21FN6O2: 444, обнаружено: 445 (MH+).
[00153]
[00154] Соединение 22
[00155]
[00156] 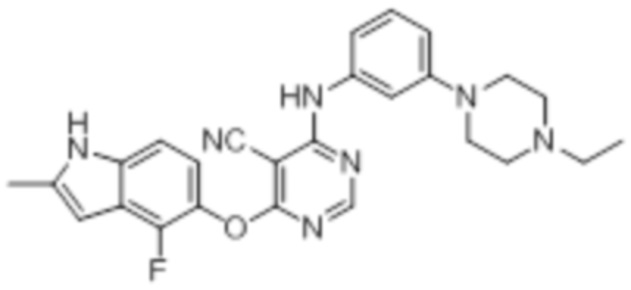
[00157]
[00158] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (200 мг, 0,661 ммоль) и 3-(4-этилпиперазин-1-ил)анилина (193 мг, 0,694 ммоль) в безводном DMSO (2,0 мл) добавляли TEA (0,28 мл; 1,98 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 18 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл; 50% насыщенность). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации из EtOAc с получением целевого продукта в виде коричневого твердого вещества (221 мг, выход 69%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,36 (уш.с с, 1H), 9,86 (уш.с с, 1H), 8,28 (с, 1H), 7,20-7,08 (м, 3H), 7,02-6,92 (м, 2H), 6,78-6,75 (м, 1H), 3,14-3,12 (4H), 2,48 (м, 4H); заслонен сигналом DMSO), 2,40 (с, 3H), 2,37 (кв, J=7,2 Гц, 2H), 1,03 (т, J=7,2 Гц, 3H); MS (ESI): рассчит. для C26H26FN7O: 471, обнаружено: 472 (MH+).
[00159]
[00160] Соединение 23
[00161]
[00162] 
[00163]
[00164] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (200 мг, 0,330 ммоль) и анилина (33 мкл, 0,363 ммоль) в безводном DMSO (2,0 мл) добавляли TEA (0,14 мл; 0,99 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 18 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл; 50% насыщенность). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-100% EtOAc в гексанах (об./об.) с получением целевого продукта в виде не совсем белого твердого вещества (93 мг, выход 78%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,36 (уш.с с, 1H), 10,04 (уш.с с, 1H), 8,29 (с, 1H), 7,53 (д, J=7,6 Гц, 2H), 7,39-7,35 (м, 2H), 7,20-7,12 (м, 2H), 6,97-6,93 (м, 1H), 6,24 (с, 1H), 2,40 (с, 3H); MS (ESI): рассчит. для C20H14FN5O: 359, обнаружено: 360 (MH+).
[00165]
[00166] Соединение 24
[00167]
[00168] 
[00169]
[00170] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и анилина (33 мкл, 0,363 ммоль) в безводном DMSO (2,0 мл) добавляли TEA (0,14 мл; 0,99 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 18 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл; 50% насыщенность). Органический слой разделяли и сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-100% EtOAc в гексанах (об./об.) с получением целевого продукта в виде не совсем белого твердого вещества (93 мг, выход 78%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,35 (уш.с с, 1H), 9,82 (уш.с с, 1H), 8,21 (д, J=0,8 Гц, 1H), 7,29 (д, J=8,0 Гц, 2H), 7,12 (д, J=8,8 Гц, 1H), 6,95-6,90 (м, 3H), 6,23 (д, J=0,8 Гц, 1H), 3,14-3,11 (м, 4H), 2,40 (с, 3H), 1,65-1,51 (м, 6H); MS (ESI): рассчит. для C25H23FN6O: 442, обнаружено: 443 (MH+).
[00171]
[00172] Соединение 25
[00173]
[00174] 
[00175]
[00176] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и N-(4-аминофенил)ацетамида (54 мг, 0,36 ммоль) в безводном DMSO (2,0 мл) добавляли TEA (0,14 мл; 0,99 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 18 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл; 50% насыщенность). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации из EtOAc с получением целевого N-(4-((5-циано-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-4-ил)амино)фенил)ацетамида в виде не совсем белого твердого вещества (88 мг, выход 64%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,37 (уш.с с, 1H), 9,99 (уш.с с, 1H), 9,98 (уш.с с, 1H), 8,25 (с, 1H), 7,57-7,55 (м, 2H), 7,42-7,40 (м, 2H), 7,14-7,11 (м, 1H), 6,96-6,92 (м, 1H), 6,24 (с, 1H), 2,40 (с, 3H), 2,04 (с, 3H); MS (ESI): рассчит. для C22H17FN6O2: 416, обнаружено: 417 (MH+).
[00177]
[00178] Соединение 26
[00179]
[00180] 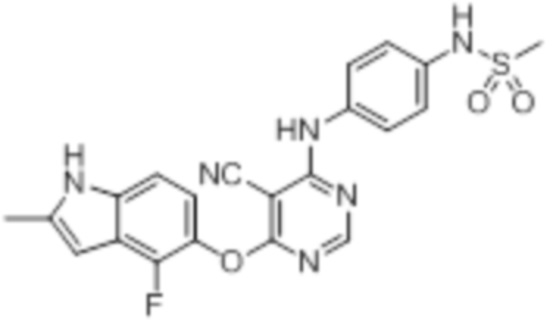
[00181]
[00182] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и N-(4-аминофенил)метансульфонамида (68 мг, 0,36 ммоль) в безводном изопропаноле (5,0 мл) добавляли TFA (0,05 мл; 0,65 ммоль), и энергично перемешивали полученную смесь при 80°C в атмосфере аргона в течение 22 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации из EtOAc с получением целевого продукта в виде не совсем белого твердого вещества (97 мг, выход 65%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,35 (уш.с с, 1H), 10,02 (уш.с с, 1H), 9,71 (уш.с с, 1H), 8,27 (с, 1H), 7,49-7,46 (м, 2H), 7,21-7,11 (м, 3H), 6,96-6,92 (м, 1H), 6,24 (с, 1H), 2,98 (с, 3H), 2,40 (с, 3H); MS (ESI): рассчит. для C21H17FN6O3S: 452, обнаружено: 453 (MH+).
[00183]
[00184] Соединение 27
[00185]
[00186] 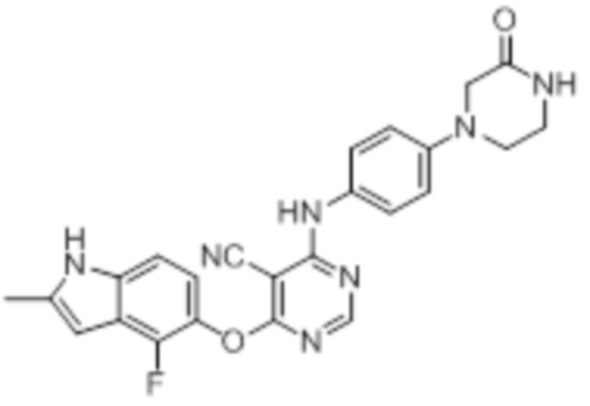
[00187]
[00188] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и 4-(4-аминофенил)пиперазин-2-она (69 мг, 0,36 ммоль) в безводном DMSO (3,0 мл) добавляли TEA (0,14 мл; 0,99 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 27 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде желтого твердого вещества (88 мг, выход 64%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,34 (уш.с с, 1H), 9,86 (уш.с с, 1H), 8,22 (с, 1H), 8,04 (уш.с с, 1H), 7,35-7,33 (м, 2H), 7,12 (д, J=8,8 Гц, 1H), 6,95-6,91 (м, 2H), 6,24-6,23 (м, 1H), 3,71 (с, 2H), 3,41-3,38 (м, 2H), 3,32-3,29 (м, 2H; заслонен сигналом воды), 2,40 (с, 3H); MS (ESI): рассчит. для C24H20FN7O2: 457, обнаружено: 458 (MH+).
[00189]
[00190] Соединение 28
[00191]
[00192] 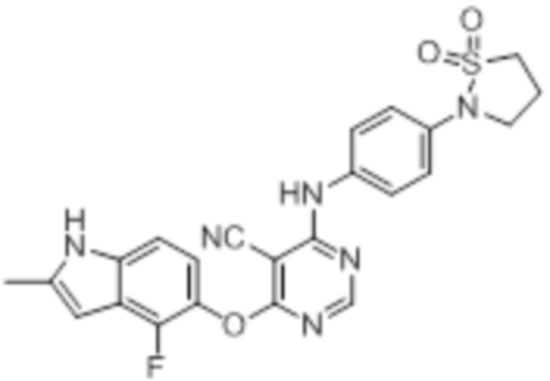
[00193]
[00194] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и 2-(4-аминофенил)изотиазолидин-1,1-диоксида (77 мг, 0,36 ммоль) в безводном изопропанол (5,0 мл) добавляли TFA (0,05 мл; 0,65 ммоль), и перемешивали полученную смесь при 80°C в атмосфере аргона в течение 22 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде не совсем белого твердого вещества (97 мг, выход 65%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,37 (уш.с с, 1H), 10,06 (уш.с с, 1H), 8,26 (с, 1H), 7,52-7,49 (м, 2H), 7,22-7,19 (м, 2H), 7,12 (д, J=8,4 Гц, 1H), 6,97-6,93 (м, 1H), 3,74 (т, J=6,4 Гц, 2H), 3,51 (т, J=7,2 Гц, 2H), 2,44-2,37 (м, 2H), 2,40 (с, 3H); MS (ESI): рассчит. для C23H19FN6O3S: 478, обнаружено: 479 (MH+).
[00195]
[00196] Соединение 29
[00197]
[00198] 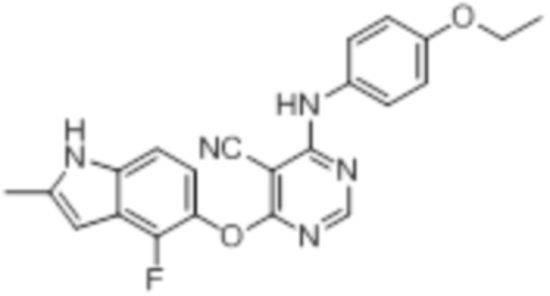
[00199]
[00200] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и 4-этоксианилина (47 мкл, 0,36 ммоль) в безводном DMSO (3,0 мл) добавляли TEA (0,14 мл; 0,99 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 23 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-100% EtOAc в гексанах (об./об.) с получением целевого продукта в виде желтого твердого вещества (111 мг, выход 83%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,35 (уш.с с, 1H), 9,90 (уш.с с, 1H), 8,22 (с, 1H), 7,37 (д, J=9,2 Гц, 1H), 7,12 (д, J=8,8 Гц, 1H), 6,95-6,90 (м, 3H), 6,23 (с, 1H), 4,02 (кв, J=6,8 Гц, 2H), 2,40 (с, 3H), 1,33 (т, J=6,8 Гц, 3H); MS (ESI): рассчит. для C22H18FN5O2: 403, обнаружено: 404 (MH+).
[00201]
[00202] Соединение 30
[00203] 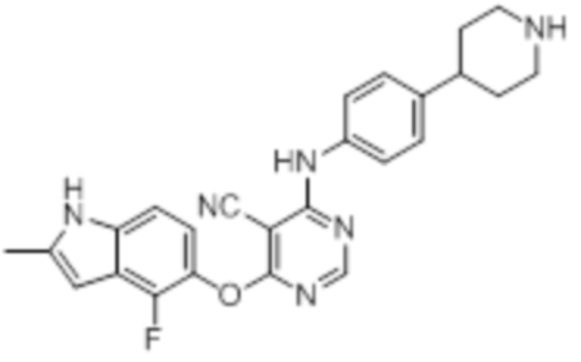
[00204]
[00205] Стадия 1: К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (200 мг, 0,661 ммоль) и трет-бутил-4-(4-аминофенил)пиперидин-1-карбоксилата (201 мг, 0,727 ммоль) в безводном DMSO (3,0 мл) добавляли TEA (0,28 мл; 1,98 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение 20 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли и сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-100% EtOAc в гексанах (об./об.) с получением целевого продукта в виде желтого твердого вещества (322 мг, выход 90%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,36 (уш.с с, 1H), 9,98 (уш.с с, 1H), 8,25 (с, 1H), 7,42 (д, J=8,4 Гц, 2H), 7,23 (д, J=8,8 Гц, 2H), 7,12 (д, J=8,4 Гц, 1H), 6,96-6,92 (м, 1H), 6,24-6,23 (м, 1H), 4,11-4,06 (м, 1H), 2,84-2,78 (м, 2H), 2,70-2,64 (м, 2H), 2,40 (с, 3H), 1,77-1,74 (м, 2H), 1,54-1,41 (м, 2H), 1,42 (с, 9H); MS (ESI): рассчит. для C30H31FN6O3: 542, обнаружено: 442 (M-BOC+H+).
[00206]
[00207] Стадия 2: Смесь трет-бутил-4-(4-((5-циано-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-4-ил)амино)фенил)пиперидин-1-карбоксилата (200 мг, 0,369 ммоль) перемешивали в 8% TFA в DCM (10 мл), и перемешивали полученную смесь при комнатной температуре в течение 17 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3), в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc. Маточный раствор концентрировали с получением целевого продукта в виде стекловидного белого твердого вещества (60 мг, выход 40%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,37 (уш.с с, 1H), 9,99 (уш.с с, 1H), 8,26 (с, 1H), 7,44-7,42 (м, 2H), 7,23-7,20 (м, 2H), 7,13 (д, J=8,8 Гц, 1H), 6,96-6,92 (м, 1H), 6,24-6,23 (м, 1H), 3,13-3,10 (м, 2H), 2,72-2,60 (м, 3H), 2,40 (с, 3H), 1,77-1,72 (м, 2H), 1,62-1,52 (м, 2H); MS (ESI): рассчит. для C25H23FN6O: 442, обнаружено: 443 (MH+).
[00208]
[00209] Соединение 31
[00210] 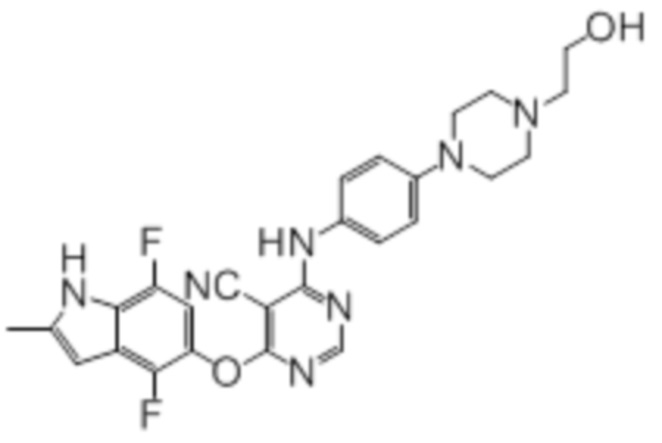
[00211]
[00212] Стадия 1: К раствору 4-фторнитробензола (2,0 г, 14,16 ммоль) в AcN (15 мл) добавляли 2-(пиперазин-1-ил)этанол (1,85 г, 14,17 ммоль) и DIEA (2,97 мл, 17,01 ммоль). Смесь нагревали с обратным холодильником в течение 15 ч (в герметизированной пробирке). После охлаждения, полученный раствор вливали в воду (300 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Твердые вещества собирали путем фильтрования и промывали водой с получением целевого продукта в виде не совсем белых твердых веществ (2,74 г, выход 72%). ESI-MS рассчит. для (C12H17N3O3) 251, обнаружено 252 [M+H]+.
[00213]
[00214] Стадия 2: Раствор полученного выше продукта (2,74 г) в метаноле (50 мл) гидрировали в присутствии 10% Pd/C (270 мг) с использованием баллона с H2. спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали до получения целевого продукта (2,70 г) в виде светло-желтых твердых веществ. Этот продукт использовали непосредственно на следующей стадии без дополнительной очистки. ESI-MS: рассчит. для (C13H21N3) 219, обнаружено 220 (MH+).
[00215] Стадия 3: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), 2-(4-(4-аминофенил)пиперазин-1-ил)этанол (30,4 мг, 0,16 ммоль), DIPEA (60 мкл), DMSO (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (42 мг, выход 54%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,86 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,90 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,51 (м, 2H), 2,84 (м, 2H), 2,41 (с, 3H), 2,11 (м, 2H), 1,03 (с, 3H), 1,01 (с, 3H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00216]
[00217]
[00218]
[00219] Соединение 32
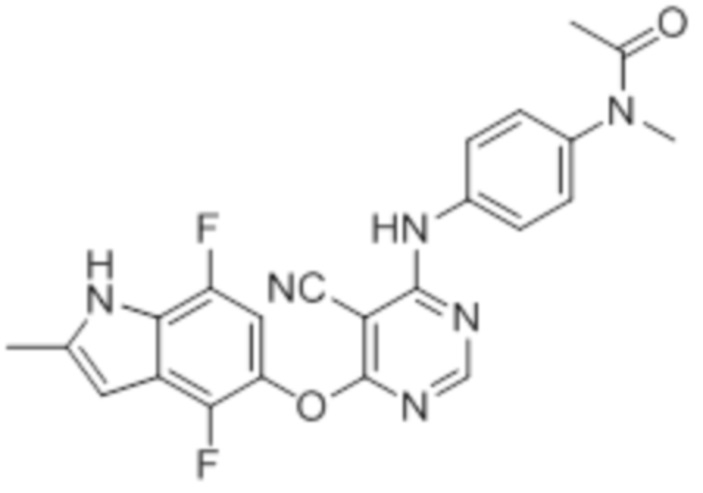
[00220] В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), 2-(4-(4-аминофенил)пиперазин-1-ил)этанол (30,4 мг, 0,16 ммоль), DIPEA (60 мкл), DMSO (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (42 мг, выход 54%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,86 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,90 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,51 (м, 2H), 2,84 (м, 2H), 2,41 (с, 3H), 2,11 (м, 2H), 1,03 (с, 3H), 1,01 (с, 3H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00221]
[00222] Соединение 33
[00223] 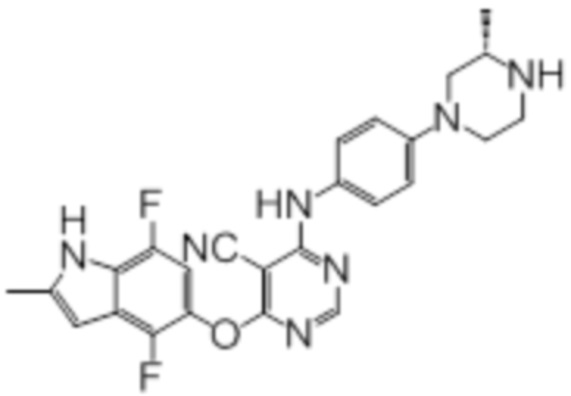
[00224]
[00225] Стадия 1: К раствору 4-фторнитробензола (0,5 г, 3,54 ммоль) в AcN (30 мл) добавляли (S)-трет-бутил-2-метилпиперазин-1-карбоксилат (0,71 г, 3,54 ммоль) и DIEA (0,74 мл, 4,25 ммоль). Смесь нагревали с обратным холодильником в течение 15 ч (в герметизированной пробирке). После охлаждения, полученный раствор вливали в воду (300 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь экстрагировали EtOAc (3×30 мл) и безводным Na2SO4, и концентрировали в условиях вакуума. Полученный неочищенный продукт очищали с использованием флэш-системы Teledyne-Isco и EtOAc/Hex (0-30% этилацетат в гексане) с получением трет-бутил-(S)-4-(4-нитрофенил)-2-метилпиперазин-1-карбоксилата в виде светло-желтых твердых веществ (710 мг, 62%) в виде белых твердых веществ с желтоватым или сероватым оттенком. 1H-ЯМР (400 МГц, DMSO-d6) δ 8,01 (д, J=9,6 Гц, 2H), 6,97 (д, J =9,2 Гц, 2H), 3,84 (м, 2H), 2,95-2,50 (м, 4H), 2,40 (м, 1H), 2,30 (уш.с, 1H), 1,00 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C11H15N3O2) 221, обнаружено 222 (MH+).
[00226]
[00227] Стадия 2: Раствор трет-бутил-(S)-4-(4-нитрофенил)-2-метилпиперазин-1-карбоксилата в метаноле (30 мл) гидрировали в присутствии 10% Pd/C (70 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением указанного в заголовке трет-бутил-(S)-4-(4-аминофенил)-2-метилпиперазин-1-карбоксилата (0,62 г, 97%) в виде светло-желтых твердых веществ. Этот продукт использовали непосредственно на следующей стадии без дополнительной очистки. ESI-MS: рассчит. для (C13H21N3) 219, обнаружено 220 (MH+).
[00228]
[00229] Стадия 3: Смесь промежуточного продукта 2 (150 мг, 0,47 ммоль), (S)-трет-бутил-2-метилпиперазин-1-карбоксилата (LN927, 0,05 г, 0,016 ммоль) и DIPEA (60 мкл, 0,037 ммоль) в DMSO (3,0 мл) перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь добавляли к смеси нас. NH4Cl/вода (25 мл/50 мл), и перемешивали при комнатной температуре в течение 30 мин. Значение pH смеси корректировали до ~6 с использованием 2н HCl. После охлаждения на льду в течение 1 ч, твердые вещества собирали путем фильтрования и промывали водой с получением неочищенного продукта. Неочищенный продукт суспендировали в DCM (10 мл), и добавляли 1 мл TFA (смесь становилась прозрачным раствором). Смесь перемешивали при комнатной температуре в течение ночи. К смеси добавляли фосфат калия в воде (pH~8), и экстрагировали DCM/MeOH. Объединенную органическую фазу промывали солевым раствором, концентрировали и очищали на колонке с силикагелем (5-15% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (97 мг, выход 45%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,90 (уш.с, 1H), 8,24 (с, 1H), 7,32 (д, J=8,8 Гц, 2H), 7,00 (дд, J =5,6 Гц, J=10,4 Гц,1H), 6,94 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,14 (м, 4H), 2,99 (м, 4H), 2,41 (с, 3H) (пик NH может располагаться под пиком растворителя); ESI-MS: рассчит. для (C24H21F2N7O) 461, обнаружено 462 (MH+).
[00230]
[00231] Соединение 34
[00232] 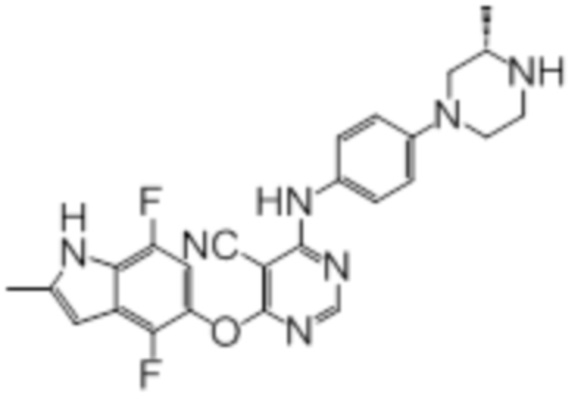
[00233]
[00234] Стадия 1: К раствору 4-фторнитробензола (0,5 г, 3,54 ммоль) в AcN (30 мл) добавляли (R)-трет-бутил-2-метилпиперазин-1-карбоксилат (0,71 г, 3,54 ммоль) и DIEA (0,74 мл, 4,25 ммоль). Смесь нагревали с обратным холодильником в течение 15 ч (в герметизированной пробирке). После охлаждения, полученный раствор вливали в воду (300 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь экстрагировали EtOAc (3×30 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Полученный неочищенный продукт очищали с использованием флэш-системы Teledyne-Isco и EtOAc/Hex (0-30% этилацетат в гексане) с получением целевого трет-бутил-(S)-2-метил-4-(4-нитрофенил)пиперазин-1-карбоксилата в виде светло-желтых твердых веществ (720 мг, 63%) в виде не совсем белых твердых веществ. 1H-ЯМР (400 МГц, DMSO-d6) δ 8,01 (д, J=9,6 Гц, 2H), 6,97 (д, J =9,2 Гц, 2H), 3,84 (м, 2H), 2,95-2,50 (м, 4H), 2,40 (м, 1H), 2,30 (уш.с, 1H), 1,00 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C11H15N3O2) 221, обнаружено 222 (MH+).
[00235]
[00236] Стадия 2: Раствор трет-бутил-(S)-2-метил-4-(4-нитрофенил)пиперазин-1-карбоксилата (0,7 г) в метаноле (30 мл) гидрировали в присутствии 10% Pd/C (70 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением указанного в заголовке трет-бутил-(S)-4-(4-аминофенил)-2-метилпиперазин-1-карбоксилата (0,61 г, 95%) в виде светло-желтых твердых веществ. Этот продукт использовали непосредственно на следующей стадии без дополнительной очистки. ESI-MS: рассчит. для (C13H21N3) 219, обнаружено 220 (MH+).
[00237]
[00238] Стадия 3: Смесь промежуточного продукта 3 (150 мг, 0,47 ммоль), трет-бутил-(S)-4-(4-аминофенил)-2-метилпиперазин-1-карбоксилата (0,05 г, 0,016 ммоль) и DIPEA (60 мкл, 0,037 ммоль) в DMSO (3,0 мл) перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали завершение реакции. Смесь добавляли к смеси нас. NH4Cl/вода (25 мл/50 мл), и перемешивали при комнатной температуре в течение 30 мин. Значение pH смеси корректировали до ~6 с использованием 2н HCl. После охлаждения на льду в течение 1 ч, твердые вещества собирали путем фильтрования и промывали водой с получением неочищенного продукта. Неочищенный продукт суспендировали в DCM (10 мл), и добавляли 1 мл TFA (смесь становилась прозрачным раствором). Смесь перемешивали при комнатной температуре в течение ночи. К смеси добавляли фосфат калия в воде (pH~8), и экстрагировали DCM/MeOH. Объединенную органическую фазу промывали солевым раствором, концентрировали и очищали на колонке с силикагелем (5-15% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (97 мг, выход 45%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,90 (уш.с, 1H), 8,24 (с, 1H), 7,32 (д, J=8,8 Гц, 2H), 7,00 (дд, J =5,6 Гц, J=10,4 Гц,1H), 6,94 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,14 (м, 4H), 2,99 (м, 4H), 2,41 (с, 3H) (пик NH может располагаться под пиком растворителя); ESI-MS: рассчит. для (C24H21F2N7O) 461, обнаружено 462 (MH+).
[00239]
[00240] Соединение 35
[00241] 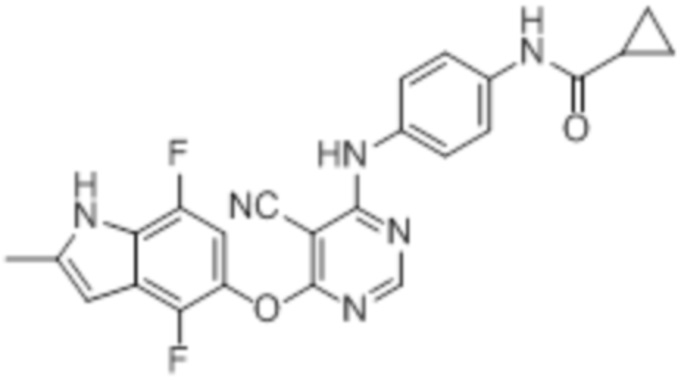
[00242]
[00243] Стадия 1: Смесь 4-нитроанилина (1,0 г, 7,24 ммоль) и циклопропилкарбонилхлорида (0,83 г, 7,96 ммоль) растворяли в безводном THF (30 мл). К полученной выше смеси добавляли DIPEA (3,16 мл, 18,10 ммоль), и перемешивали при комнатной температуре в течение ночи. Смесь перемешивали при комнатной температуре в течение 30 мин. Полученную неочищенную смесь экстрагировали EtOAc (3×30 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Полученный неочищенный продукт очищали с использованием флэш-системы Teledyne-Isco и EtOAc/Hex (0-30% этилацетат в гексане) с получением целевого (4-нитрофенил)циклопропан-карбоксамида (880 мг, 62%) в виде не совсем белых твердых веществ. ESI-MS: рассчит. для (C10H10N2O3) 206, обнаружено 207 (MH+).
[00244] Стадия 2: Раствор (4-нитрофенил)циклопропан-карбоксамида (0,7 г) в метаноле (30 мл) гидрировали в присутствии 10% Pd/C (70 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением указанного в заголовке (4-аминофенил)циклопропанкарбоксамида
[00245] в виде светло-желтых твердых веществ. Этот продукт использовали непосредственно на следующей стадии без дополнительной очистки.
[00246] Стадия 3: В колбу загружали FA425_1 (100 мг, 0,312 ммоль), N-(4-аминофенил)циклопропанкарбоксамид (LN933_1, 0,055 г, 0,312 ммоль), DIPEA (60 мкл), DMSO (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде светло-желтых твердых веществ (85 мг, выход 54%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,86 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,90 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,51 (м, 2H), 2,84 (м, 2H), 2,41 (с, 3H), 2,11 (м, 2H), 1,03 (с, 3H), 1,01 (с, 3H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00247]
[00248] Соединение 36
[00249] 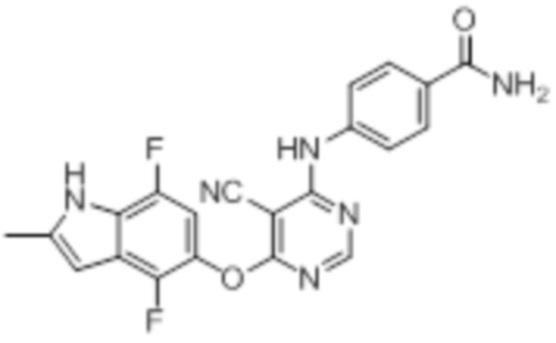
[00250]
[00251] К смеси 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,330 ммоль) и 4-аминобензамида (47 мг, 0,34 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,13 мл; 0,93 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в атмосфере аргона в течение ~17 ч. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0,5-15% MeOH в DCM (об./об.) с получением целевого продукта в виде белого твердого вещества (42 мг, выход 32%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,85 (уш.с с, 1H), 10,22 (уш.с с, 1H), 8,35 (с, 1H), 7,91 (уш.с с, 1H), 7,86 (д, J=8,8 Гц, 2H), 7,62 (д, J=8,4 Гц, 2H), 7,29 (уш.с с, 1H), 7,04-7,00 (м, 1H), 6,35 (с, 1H), 2,42 (с, 3H); MS (ESI): рассчит. для C21H14F2N6O2: 420, обнаружено: 421 (MH+).
[00252] Соединение 37
[00253]
[00254] 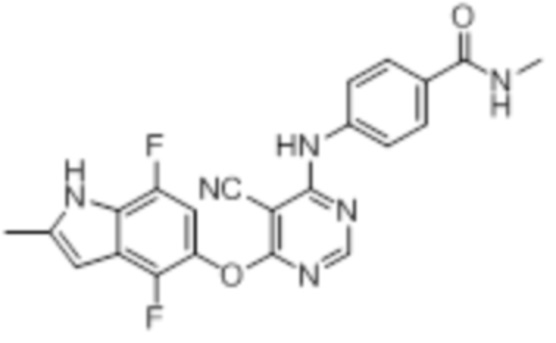
[00255]
[00256] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 4-амино-N-метилбензамида (45 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0,5-15% MeOH в DCM (об./об.) с получением целевого продукта в виде белого твердого вещества (18 мг, выход 17%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,85 (уш.с с, 1H), 10,23 (уш.с с, 1H), 8,37-8,34 (м, 2H), 7,81 (д, J=8,8 Гц, 2H), 7,64-7,62 (м, 2H), 7,04-7,00 (м, 1H), 6,35 (с, 1H), 2,78 (д, J=4,0 Гц, 3H), 2,42 (с, 3H); MS (ESI): рассчит. для C22H16F2N6O2: 434, обнаружено: 435 (MH+).
[00257]
[00258] Соединение 38
[00259]
[00260] 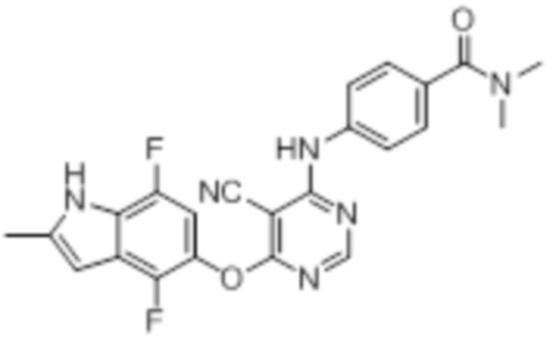
[00261]
[00262] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 4-амино-N,N-диметилбензамида (49 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации (DCM) с получением целевого продукта в виде белого твердого вещества (49 мг, выход 44%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,85 (уш.с с, 1H), 10,20 (уш.с с, 1H), 8,33 (с, 1H), 7,60 (д, J=8,4 Гц, 2H), 7,41 (д, J=8,4 Гц, 2H), 7,04-7,00 (м, 1H), 6,35 (с, 1H), 2,97 (с, 6H), 2,42 (с, 3H); MS (ESI): рассчит. для C23H18F2N6O2: 448, обнаружено: 449 (MH+).
[00263]
[00264] Соединение 39
[00265]
[00266] 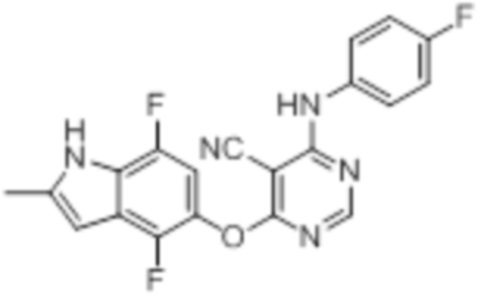
[00267]
[00268] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 4-фторанилина (33 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации (DCM) с получением целевого продукта в виде белого твердого вещества (65 мг, выход 66%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,84 (уш.с с, 1H), 10,10 (уш.с с, 1H), 8,29 (с, 1H), 7,54-7,51 (м, 2H), 7,23-7,29 (м, 2H), 7,03-6,99 (м, 1H), 6,34 (с, 1H), 2,42 (с, 3H); MS (ESI): рассчит. для C20H12F3N5O: 395, обнаружено: 396 (MH+).
[00269]
[00270] Соединение 40
[00271]
[00272] 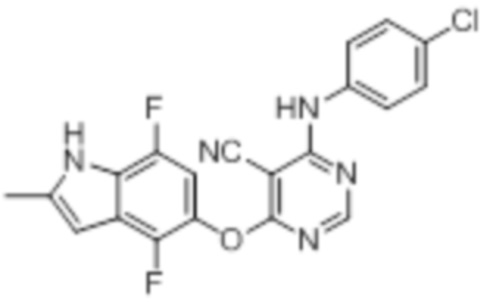
[00273]
[00274] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 4-хлоранилина (33 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации (DCM) с получением целевого продукта в виде белого твердого вещества (65 мг, выход 44%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,85 (уш.с с, 1H), 10,15 (уш.с с, 1H), 8,34 (д, J=1,2 Гц, 1H), 7,59-7,57 (м, 2H), 7,43 (д, J=8,0 Гц, 2H), 7,04-7,00 (м, 1H), 6,35 (с, 1H), 2,42 (с, 3H); MS (ESI): рассчит. для C20H12ClF2N5O: 411, обнаружено: 412 (MH+).
[00275]
[00276] Соединение 41
[00277]
[00278] 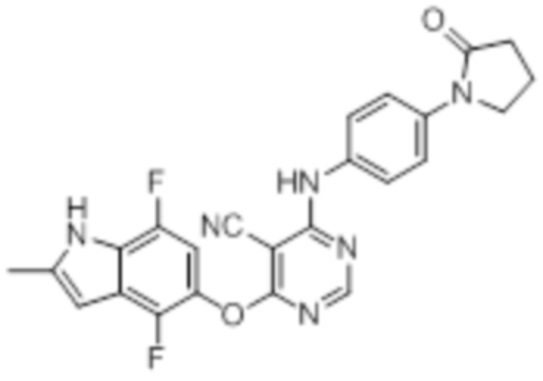
[00279]
[00280] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 1-(4-аминофенил)пирролидин-2-она (53 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали путем кристаллизации (DCM) с получением целевого продукта в виде белого твердого вещества (56 мг, выход 49%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,34 (уш.с с, 1H), 10,07 (уш.с с, 1H), 8,26 (с, 1H), 7,64 (д, J=8,8 Гц, 2H), 7,48 (д, J=8,8 Гц, 2H), 7,03-6,99 (м, 1H), 6,34 (с, 1H), 3,84 (т, J=6,8 Гц, 2H), 2,50-2,48 (м, 2H; заслонен сигналом DMSO), 2,42 (с, 3H), 2,11-2,03 (м, 2H); MS (ESI): рассчит. для C24H18F2N6O2: 460, обнаружено: 461 (MH+).
[00281]
[00282] Соединение 42
[00283]
[00284] 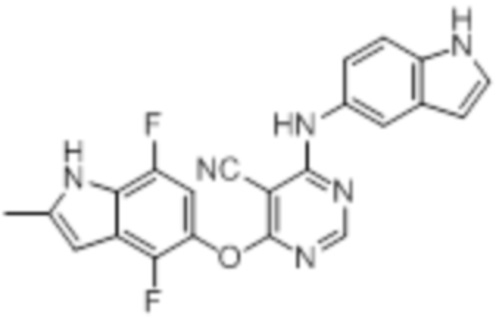
[00285]
[00286] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 1H-индол-5-амина (40 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0,5-15% MeOH в DCM (об./об.) с получением целевого продукта в виде белого твердого вещества (32 мг, выход 33%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,83 (уш.с с, 1H), 11,14 (уш.с с, 1H), 10,00 (уш.с с, 1H), 8,21 (с, 1H), 7,58 (с, 1H), 7,39-7,36 (м, 2H), 7,14-7,11 (м, 1H), 7,03-6,99 (м, 1H), 6,44-6,42 (м, 1H), 6,34 (с, 1H), 5,76 (д, J=0,8 Гц, 1H), 2,41 (с, 3H); MS (ESI): рассчит. для C22H14F2N6O: 416, обнаружено: 417 (MH+).
[00287]
[00288] Соединение 43
[00289] 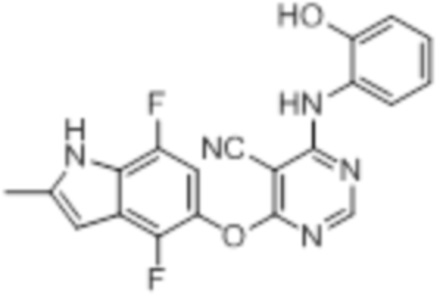
[00290]
[00291] К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и 1H-индол-5-амина (40 мг, 0,30 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,122 мл; 0,875 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение ~3 суток. Полученную смесь разбавляли EtOAc (100 мл) и промывали нас. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0,5-15% MeOH в DCM (об./об.) с получением целевого продукта в виде белого твердого вещества (32 мг, выход 33%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,84 (уш.с с, 1H), 9,76 (уш.с с, 1H), 9,57 (уш.с с, 1H), 8,24 (с, 1H), 7,40 (д, J=8,0 Гц, 1H), 7,13-7,09 (м, 1H), 7,02-6,98 (м, 1H), 6,91 (д, J=8,0 Гц, 1H), 6,84-6,80 (м, 1H), 6,34 (д, J=3,2 Гц, 1H), 2,41 (с, 3H); MS (ESI): рассчит. для C20H13F2N5O2: 393, обнаружено: 394 (MH+).
[00292]
[00293] Соединение 44
[00294]
[00295] 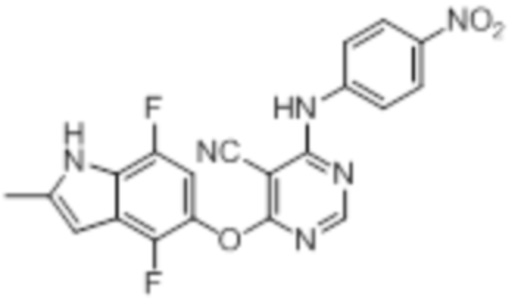
[00296]
[00297] Смесь 4-хлор-6-((4-фтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,31 ммоль), 4-нитроанилина (47 мг, 0,34 ммоль), Pd(OAc)2 (10 мг, 0,045 ммоль), Xantphos (45 мг, 0,078 ммоль), K2CO3 (150 мг, 1,09 ммоль) и безводного диоксана (12 мл) герметично закрывали в пробирке для микроволновой обработки и дегазировали аргоном в течение 10 мин. Смесь затем нагревали до 120°C в течение 20 мин в условиях обработки микроволновым излучением. Смесь охлаждали до комнатной температуры и распределяли между EtOAc и водн. NaHCO3 (по ~100 мл каждого; 50% насыщенность NaHCO3), органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток поглощали EtOAc, фильтровали и промывали дополнительным количеством EtOAc с получением целевого продукта в виде светло-желтого твердого вещества (54 мг, выход 41%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,86 (уш.с с, 1H), 10,59 (уш.с с, 1H), 8,47 (с, 1H), 8,26-8,23 (м, 2H), 7,92-7,88 (м, 2H), 7,06-7,02 (м, 1H), 6,36 (с, 1H), 2,42 (с, 3H); MS (ESI): рассчит. для C20H12F2N6O3: 422, обнаружено: 423 (MH+).
[00298]
[00299]
[00300] Соединение 45
[00301]
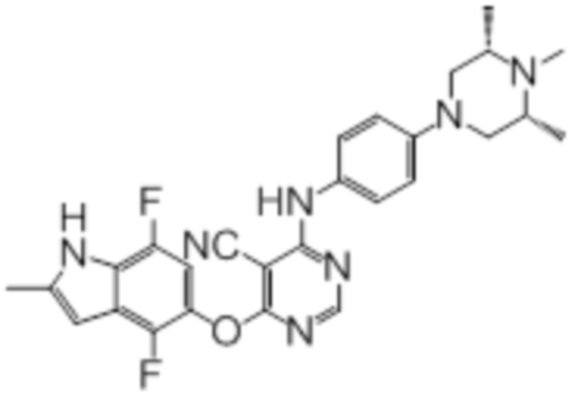
[00302]
[00303] Стадия 1: К раствору 4-фторнитробензола (3 г, 21,26 ммоль) в AcN (60 мл) добавляли цис-2,6-диметилпиперазин (2,55 г, 22,32 ммоль) и DIEA (3,90 мл, 22,32 ммоль). Смесь перемешивали и нагревали с обратным холодильником в течение 18 ч. Полученную смесь охлаждали до комнатной температуры и упаривали с получением. Добавляли EtOAc и воду. Смесь трижды экстрагировали EtOAc. Органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток кристаллизовали из EtOAc с получением желтых твердых веществ побочного продукта обратного присоединения (300 мг). Маточный раствор концентрировали до минимального объема растворителей, а затем добавляли гексаны с формированием желтого осадка. Твердые вещества собирали путем фильтрования и промывали гексанами с получением целевого (3R,5S)-3,5-диметил-1-(4-нитрофенил)пиперазина в виде светло-желтых твердых веществ (2,60 г, выход 52%). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,02 (д, J=9,6 Гц, 2H), 7,02 (д, J =9,2 Гц, 2H), 3,90 (м, 2H), 2,78 (м, 2H), 2,40 (м, 2H), 1,03 (д, J=5,6 Гц, 6H); ESI-MS: рассчит. для (C12H17N3O2) 235, обнаружено 236 (MH+).
[00304] Стадия 2: К раствору (3R,5S)-3,5-диметил-1-(4-нитрофенил)пиперазина (600 мг, 2,55 ммоль) в DMF (6 мл) порциями добавляли гидрид натрия (60%, 122 мг, 3,06 ммоль), и перемешивали смесь при комнатной температуре в течение 30 мин. Затем, добавляли йодметан (0,19 мл г, 3,06 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали расходование исходного вещества. Смесь порциями вливали в холодную воду, и экстрагировали смесь EtOAc (3×50 мл). Объединенную органическую фазу промывали водой, сушили над сульфатом натрия и концентрировали с получением 4-((3R,5S)-3,4,5-триметилпиперазин-1-ил)нитробензола в виде желтого твердого вещества (100 мг, выход 15%). Дополнительную очистку не проводили. ESI-MS: рассчит. для (C13H19N3O2) 249, обнаружено 250 (MH+).
[00305]
[00306] Стадия 3: Раствор (3R,5S)-3,5-диметил-1-(4-нитрофенил)пиперазина (~100 мг) в метаноле (20 мл) гидрировали в присутствии 10% Pd/C (10 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением целевого 4-((3R,5S)-3,4,5-триметилпиперазин-1-ил)анилина (51 мг, выход 9% в 2 этапа) в виде красных твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. ESI-MS: рассчит. для (C13H21N3) 219, обнаружено 220 (MH+).
[00307]
[00308] Стадия 4: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), 4-((3R,5S)-3,4,5-триметилпиперазин-1-ил)анилин (38 мг, 0,17 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (51 мг, выход 65%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,84 (уш.с, 1H), 9,87 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,8 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,91 (д, J=9,2 Гц, 2H), 6,34 (с, 1H), 3,54 (д, J=11,2 Гц, 2H), 2,41 (с, 3H), 2,34 (д, J=11,6 Гц, 2H), 2,24 (м, 2H), 2,19 (с, 3H),1,07 (д, J=6,0 Гц, 6H); ESI-MS: рассчит. для (C27H27F2N7O) 503, обнаружено 504 (MH+).
[00309]
[00310] Соединение 46
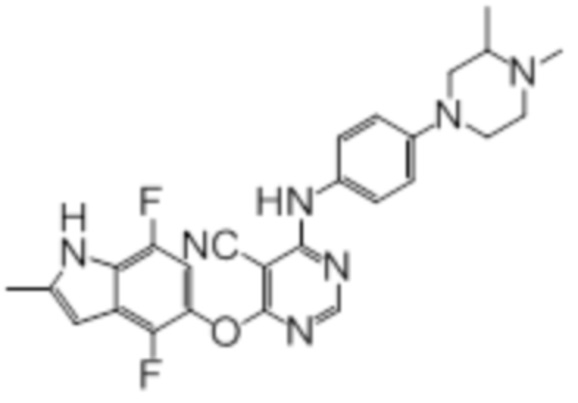
[00311] Стадия 1: К раствору 4-фторнитробензола (1,80 г, 12,76 ммоль) в AcN (18 мл) добавляли 2-метилпиперазин (3,19 г, 31,89 ммоль) и DIEA (3,34 мл, 19,14 ммоль). Смесь перемешивали при 75°C в течение 3 ч (в герметизированной пробирке). Полученную смесь охлаждали до комнатной температуры, а затем переносили в воду (300 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, а затем охлаждали на льду. Твердые вещества собирали путем фильтрования и промывали водой с получением неочищенного продукта, который очищали методом колоночной хроматографии (0-10% MeOH в DCM) с получением целевого 3-метил-1-(4-нитрофенил)пиперазина в виде желтых твердых веществ (2,03 г, выход 72%). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,01 (д, J=9,6 Гц, 2H), 6,97 (д, J =9,2 Гц, 2H), 3,84 (м, 2H), 2,95-2,50 (м, 4H), 2,40 (м, 1H), 2,30 (уш.с, 1H), 1,00 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C11H15N3O2) 221, обнаружено 222 (MH+).
[00312]
[00313] Стадия 2: К раствору 3-метил-1-(4-нитрофенил)пиперазина (600 мг, 2,71 ммоль) в DMF (6 мл) порциями добавляли гидрид натрия (60%, 130 мг, 3,25 ммоль), и перемешивали смесь при комнатной температуре в течение 30 мин. Затем, добавляли йодметан (0,20 мл г, 3,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали расходование исходного вещества. Смесь порциями вливали в холодную воду, и экстрагировали смесь EtOAc (3×50 мл). Объединенную органическую фазу промывали водой, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (0-10% MeOH в DCM) с получением желтого масла. ESI-MS: рассчит. для (C12H17N3O2) 235, обнаружено 236 (MH+).
[00314]
[00315] Стадия 3: Раствор 4-(3,4-диметилпиперазин-1-ил)нитробензола в метаноле (~20 мл) гидрировали в присутствии Pd/C (25 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали до целевого 4-(3,4-диметилпиперазин-1-ил)анилина (398 мг, выход 72% в 2 этапа) в виде коричневых твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 6,65 (д, J=8,4 Гц, 2H), 6,45 (д, J =8,0 Гц, 2H), 4,51 (уш.с, 2H), 3,17 (м, 2H), 2,74 (м, 1H), 2,57 (м,1H), 2,30-2,00 (м, 5H), 0,98 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C12H19N3) 205, обнаружено 206 (MH+).
[00316]
[00317]
[00318] Стадия 4: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), 4-(3,4-диметилпиперазин-1-ил)анилин (35 мг, 0,17 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (60 мг, выход 79%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,88 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,8 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,90 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,51 (м, 2H), 2,83-2,70 (м, 2H), 2,41 (с, 3H), 2,36 (т, J=10,4 Гц, 1H), 2,30-2,05 (м, 5H), 1,03 (д, J=6,4 Гц, 3H); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00319]
[00320] Соединение 47
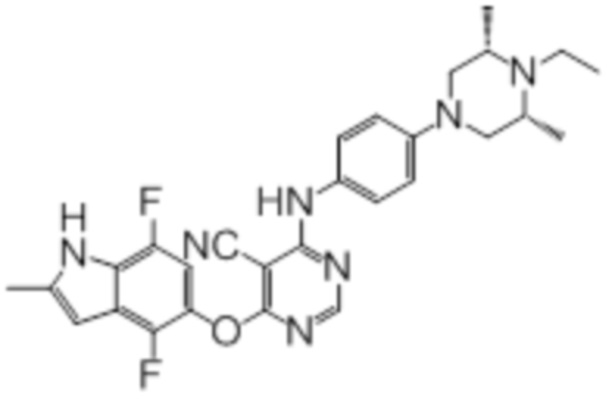
[00321]
[00322] Стадия 1: К раствору 4-фторнитробензола (3 г, 21,26 ммоль) в AcN (60 мл) добавляли цис-2,6-диметилпиперазин (2,55 г, 22,32 ммоль) и DIEA (3,90 мл, 22,32 ммоль). Смесь перемешивали и нагревали с обратным холодильником в течение 18 ч. Полученную смесь охлаждали до комнатной температуры и упаривали с получением. Добавляли EtOAc и воду. Смесь трижды экстрагировали EtOAc. Органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток кристаллизовали из EtOAc с получением желтых твердых веществ побочного продукта обратного присоединения (300 мг). Маточный раствор концентрировали до минимального объема растворителей, а затем добавляли гексаны с формированием желтого осадка. Твердые вещества собирали путем фильтрования и промывали гексанами с получением целевого (3R,5S)-3,5-диметил-1-(4-нитрофенил)пиперазина в виде светло-желтых твердых веществ (2,60 г, выход 52%). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,02 (д, J=9,6 Гц, 2H), 7,02 (д, J =9,2 Гц, 2H), 3,90 (м, 2H), 2,78 (м, 2H), 2,40 (м, 2H), 1,03 (д, J=5,6 Гц, 6H); ESI-MS: рассчит. для (C12H17N3O2) 235, обнаружено 236 (MH+).
[00323]
[00324]
[00325] Стадия 2: К раствору (3R,5S)-3,5-диметил-1-(4-нитрофенил)пиперазина (650 мг, 2,76 ммоль) и йодэтана (453 мг, 2,90 ммоль) в DMF (6 мл) добавляли карбонат калия (573 мг, 4,15 ммоль), и перемешивали смесь при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали расходование исходного вещества. Смесь вливали в холодную воду и экстрагировали DCM (3×15 мл). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (0-10% MeOH в DCM) с получением желтого масла. ESI-MS: рассчит. для (C14H21N3O2) 263, обнаружено 264 (MH+).
[00326]
[00327] Стадия 3: Раствор полученного выше нитробензола в метаноле (~30 мл) гидрировали в присутствии Pd/C (50 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением целевого 4-((3R,5S)-4-этил-3,5-диметилпиперазин-1-ил)анилина (520 мг, выход 81% в 2 этапа) в виде коричневых твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 6,65 (д, J=7,2 Гц, 2H), 6,47 (д, J =7,6 Гц, 2H), 4,52 (уш.с, 2H), 3,20 (д, J=10,8 Гц, 2H), 2,81 (кв, J=7,2 Гц, 2H), 2,66 (м, 2H), 2,18 (т, J=10,4 Гц, 2H), 1,00 (д, J=6,4 Гц, 6H), 0,85 (т, J=7,2 Гц, 3H); ESI-MS: рассчит. для (C14H23N3) 233, обнаружено 234 (MH+).
[00328]
[00329]
[00330] Стадия 4: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), 4-((3R,5S)-4-этил-3,5-диметилпиперазин-1-ил)анилин (40 мг, 0,17 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (65 мг, выход 80%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,84 (уш.с, 1H), 9,87 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,8 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,90 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,54 (д, J=11,6 Гц, 2H), 2,85 (кв, J=7,2 Гц, 2H), 2,67 (м, 2H), 2,41 (с, 3H), 2,33 (т, J=10,8 Гц, 2H), 1,05 (д, J=6,0 Гц, 6H), 0,86 (т, J=7,2 Гц, 3H); ESI-MS: рассчит. для (C28H29F2N7O) 517, обнаружено 518 (MH+).
[00331]
[00332]
[00333] Соединение 48
[00334]
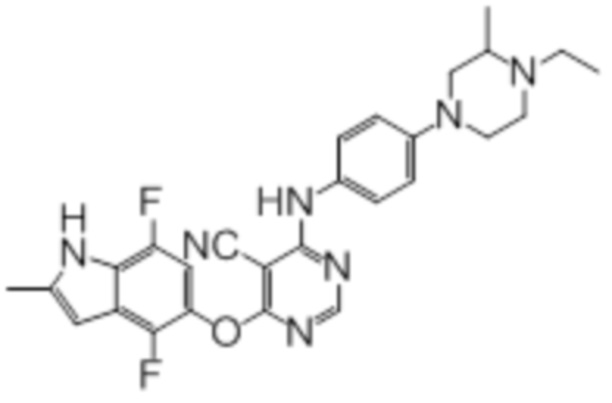
[00335] Стадия 1: К раствору 4-фторнитробензола (1,80 г, 12,76 ммоль) в AcN (18 мл) добавляли 2-метилпиперазин (3,19 г, 31,89 ммоль) и DIEA (3,34 мл, 19,14 ммоль). Смесь перемешивали при 75°C в течение 3 ч (в герметизированной пробирке). Полученную смесь охлаждали до комнатной температуры, а затем переносили в воду (300 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, а затем охлаждали на льду. Твердые вещества собирали путем фильтрования и промывали водой с получением неочищенного продукта, который очищали методом колоночной хроматографии (0-10% MeOH в DCM) с получением целевого 3-метил-1-(4-нитрофенил)пиперазина в виде желтых твердых веществ (2,03 г, выход 72%). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,01 (д, J=9,6 Гц, 2H), 6,97 (д, J =9,2 Гц, 2H), 3,84 (м, 2H), 2,95-2,50 (м, 4H), 2,40 (м, 1H), 2,30 (уш.с, 1H), 1,00 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C11H15N3O2) 221, обнаружено 222 (MH+).
[00336]
[00337] Стадия 2: К раствору 3-метил-1-(4-нитрофенил)пиперазина (610 мг, 2,76 ммоль) и йодэтана (452 мг, 2,89 ммоль) в DMF (6 мл) добавляли карбонат калия (572 мг, 4,14 ммоль), и перемешивали смесь при комнатной температуре в течение ночи. Проводили проверку методом TLC, и обнаруживали расходование исходного вещества. Смесь вливали в холодную воду и экстрагировали DCM (3×15 мл). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (0-10% MeOH в DCM) с получением 1-этил-2-метил-4-(4-нитрофенил)пиперазина в виде желтого масла. ESI-MS: рассчит. для (C13H19N3O2) 249, обнаружено 250 (MH+).
[00338] Стадия 3: Раствор 1-этил-2-метил-4-(4-нитрофенил)-пиперазина в метаноле (~30 мл) гидрировали в присутствии Pd/C (50 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением целевого продукта (560 мг, выход 92% в 2 этапа) в виде коричневого масла. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 6,66 (д, J=8,4 Гц, 2H), 6,48 (д, J =8,0 Гц, 2H), 4,53 (уш.с, 2H), 3,15 (м, 2H), 2,85-2,70 (м, 2H), 2,60 (м, 1H), 2,48-2,20 (м, 4H), 1,00 (д, J=6,0 Гц, 3H), 0,97 (т, J=7,2 Гц, 3H); ESI-MS: рассчит. для (C13H21N3) 219, обнаружено 220 (MH+).
[00339]
[00340] Стадия 4: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), амин (38 мг, 0,17 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (63 мг, выход 80%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,88 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,8 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,91 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,44 (м, 2H), 3,00-2,70 (м, 3H), 2,50-2,20 (м, 7H), 1,05 (д, J=5,2 Гц, 3H), 0,98 (т, J=7,2 Гц, 3H); ESI-MS: рассчит. для (C27H27F2N7O) 503, обнаружено 504 (MH+).
[00341]
[00342] Соединение 49
[00343] 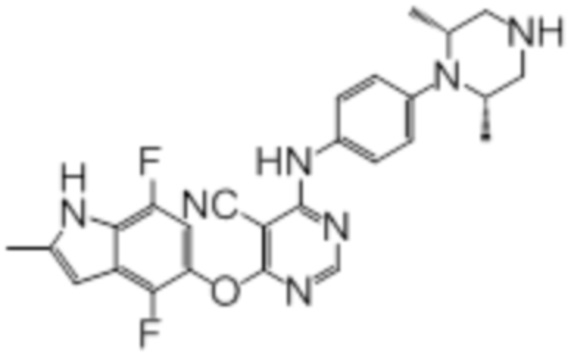
[00344] Стадия 1: Раствор (2S,6R)-2,6-диметил-1-(4-нитрофенил)пиперазина (290 мг, 1,23 ммоль) в метаноле (~20 мл) гидрировали в присутствии Pd/C (28 мг) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением целевого продукта (265 мг, выход 100%) в виде пурпурных твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 6,72 (д, J=7,2 Гц, 2H), 6,50 (д, J =7,2 Гц, 2H), 4,60 (уш.с, 2H), 3,50-3,20 (м, 6H), 2,55 (м,1H), 1,26 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C12H19N3) 205, обнаружено 206 (MH+).
[00345]
[00346] Стадия 2: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), 4-((2S,6R)-2,6-диметилпиперазин-1-ил)анилин (35 мг, 0,17 ммоль), TFA (50 мкл), изопропанол (5 мл). Реакционную смесь нагревали до 100°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (50 мг, выход 65%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,87 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,8 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,91 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,51 (д, J=9,6 Гц, 2H), 2,84 (м, 2H), 2,41 (с, 3H), 2,11 (т, J=10,8 Гц, 2H), 1,02 (д, J=6,0 Гц, 6H), (NH, исчезающий); ESI-MS: рассчит. для (C26H25F2N7O) 489, обнаружено 490 (MH+).
[00347]
[00348]
[00349] Соединение 50
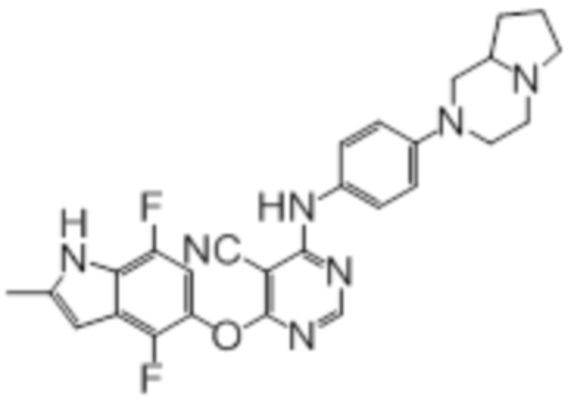
[00350] Стадия 1: К раствору 4-фторнитробензола (0,61 мл, 5,71 ммоль) в AcN (6 мл) добавляли октагидропирроло[1,2-a]пиразин (600 мг, 4,75 ммоль) и DIEA (1,2 мл, 7,13 ммоль). Смесь перемешивали при 75°C в течение ночи (в герметизированной пробирке). Полученную смесь охлаждали до комнатной температуры, а затем концентрировали. Неочищенный продукт очищали методом колоночной хроматографии (0-5% MeOH в DCM) с получением целевого соединения в виде желтого масла (1,09 г, выход 92%). ESI-MS: рассчит. для (C13H17N3O2) 247, обнаружено 248 (MH+).
[00351]
[00352] Стадия 2: Раствор полученного выше нитробензола (1,09 г, 4,41 ммоль) в метаноле (30 мл) гидрировали в присутствии 10% Pd/C (0,08 г) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением целевого продукта (0,93 г, 4,28 ммоль, 92%) в виде красных твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 6,69 (д, J=6,8 Гц, 2H), 6,47 (д, J =6,8 Гц, 2H), 4,53 (уш.с, 2H), 3,41 (д, J=10,8 Гц, 1H), 3,27 (д, J=10,8 Гц, 1H), 2,98 (м, 2H), 2,58 (т, J=10,8 Гц, 1H), 2,30-2,00 (м, 4H), 1,85-1,65 (м, 3H), 1,32 (м, 1H); ESI-MS: рассчит. для (C13H19N3) 217, обнаружено 218 (MH+).
[00353]
[00354] Стадия 3: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), анилин (39 мг, 0,18 ммоль), TFA (25 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (56 мг, выход 72%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,88 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,93 (д, J=8,8 Гц, 2H), 6,34 (с, 1H), 3,76 (д, J=10,8 Гц, 1H), 3,62 (д, J=11,6 Гц, 1H), 3,03 (м, 2H), 2,73 (т, J=11,6 Гц, 1H), 2,41 (с, 3H), 2,40-2,30 (м, 1H), 2,22 (т, J=8,0 Гц, 1H), 2,12-2,08 (м, 2H), 1,90-1,60 (м, 3H), 1,42-1,32 (м, 1H); ESI-MS: рассчит. для (C27H25F2N7O) 501, обнаружено 502 (MH+).
[00355]
[00356] Соединение 51
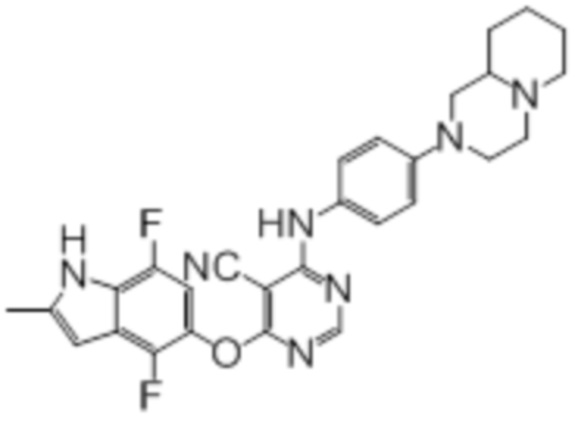
[00357] Стадия 1: К раствору 4-фторнитробензола (0,55 мл, 5,13 ммоль) в AcN (6 мл) добавляли октагидро-1H-пиридо[1,2-a]пиразин (600 мг, 4,28 ммоль) и DIEA (1,1 мл, 6,42 ммоль). Смесь перемешивали при 75°C в течение ночи (в герметизированной пробирке). Полученную смесь охлаждали до комнатной температуры, а затем концентрировали. Неочищенный продукт очищали методом колоночной хроматографии (0-5% MeOH в DCM) с получением целевого продукта в виде желтого масла (1,03 г, выход 92%). ESI-MS: рассчит. для (C14H19N3O2) 261, обнаружено 262 (MH+).
[00358] Стадия 2: Раствор полученного выше нитробензола (1,03 г, 3,94 ммоль) в метаноле (30 мл) гидрировали в присутствии 10% Pd/C (0,08 г) с использованием баллона с H2. Спустя 16 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (3×15 мл). Фильтрат концентрировали с получением целевого продукта (0,89 г, 7,85 ммоль, 92%) в виде красных твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 6,65 (д, J=6,8 Гц, 2H), 6,47 (д, J =6,8 Гц, 2H), 4,52 (уш.с, 2H), 3,23 (д, J=10,8 Гц, 1H), 3,16 (д, J=10,8 Гц, 1H), 2,72 (м, 2H), 2,56 (т, J=11,2 Гц, 1H), 2,19 (м, 2H), 1,93 (т, J=10,8 Гц, 2H), 1,75-1,35 (м, 4H), 1,30-1,00 (м, 2H); ESI-MS: рассчит. для (C14H21N3) 231, обнаружено 232 (MH+).
[00359] Стадия 3: В колбу загружали промежуточный продукт 2 (50 мг, 0,16 ммоль), полученный выше анилин (42 мг, 0,18 ммоль), TFA (25 мкл), изопропанол (3 мл). Реакционную смесь нагревали до 100°C в течение ночи. Реакционную смесь подщелачивали добавлением насыщенного водного раствора ортофосфата калия, а затем экстрагировали DCM (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали методом флэш-хроматографии (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (62 мг, выход 77%). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,83 (уш.с, 1H), 9,87 (уш.с, 1H), 8,23 (с, 1H), 7,30 (д, J=8,0 Гц, 2H), 7,00 (дд, J=5,6 Гц, J=10,4 Гц, 1H), 6,91 (д, J=7,6 Гц, 2H), 6,33 (с, 1H), 3,70-3,40 (м, 2H), 2,90-2,60 (м, 3H), 2,41 (с, 3H), 2,40-2,10 (м, 2H), 2,00-1,90 (м, 2H), 1,80-1,10 (м, 5H); ESI-MS: рассчит. для (C28H27F2N7O) 515, обнаружено 516 (MH+).
[00360]
[00361]
[00362] Соединение 52
[00363] 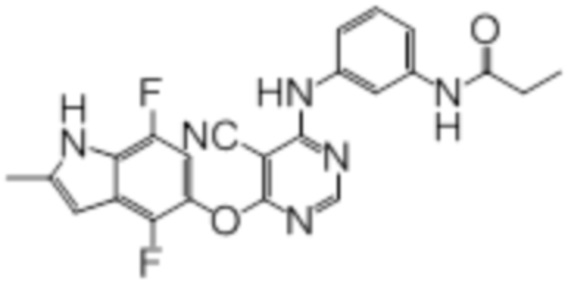
[00364] Смесь QW823 (неочищ., 70 мг, 0,22 ммоль), N-(3-аминофенил)пропанамида (43 мг, 0,26 ммоль) и DIPEA (0,08 мл, 0,44 ммоль) в DMSO (2 мл) перемешивали при комнатной температуре в течение 2 часов. Проводили проверку методом TLC, и обнаруживали завершение реакции. Добавляли этилацетат (15 мл), а затем NH4Cl (20 мл). После разделения, водную фазу экстрагировали EtOAc (15 мл × 1). Объединенную органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали на колонке с силикагелем (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (57 мг выход, 58%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,84 (уш.с, 1H), 10,10 (уш.с, 1H), 9,91 (с, 1H), 8,31 (с, 1H), 7,84 (с, 1H), 7,37 (д, J=8,4 Гц, 1H), 7,27 (т, J=8,0 Гц, 1H), 7,18 (д, J=8,0 Гц, 1H), 7,00 (дд, J =5,2 Гц, J=10,4 Гц,1H), 6,34 (с, 1H), 2,42 (с, 3H), 2,32 (кв, J=7,6 Гц, 2H), 1,08 (т, J=7,6 Гц, 3H); ESI-MS: рассчит. для (C23H18F2N6O2) 448, обнаружено 449 (MH+).
[00365]
[00366] Соединение 53
[00367] 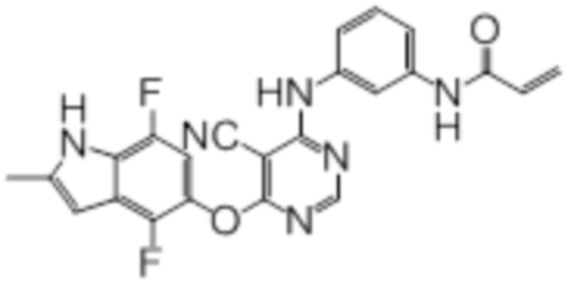
[00368] Стадия 1: К раствору 3-нитроанилина (5,00 г, 36,2 ммоль) в безводном THF (50 мл) добавляли TEA (7,50 мл, 54,3 ммоль). Смесь перемешивали при к.т. в течение 10 мин, а затем при 0°C по каплям добавляли акрилоилхлорид (7,38 мл, 90,50 ммоль). Смесь затем перемешивали при к.т. в течение 4 ч. Полученную смесь гасили добавлением бикарбоната натрия и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали в условиях вакуума. Неочищенный продукт кристаллизовали из смеси EtOAc/гексаны с получением целевого продукта в виде желтых твердых веществ (3,37 г, выход 48%). 1H-ЯМР (400 МГц, DMSO-d6) δ 10,65 (уш.с, 1H), 8,71 (с, 1H), 8,00-7,90 (м, 2H), 7,63 (т, J=8,0 Гц, 1H), 6,50-6,30 (м, 2H), 5,84 (д, J=10,0 Гц, 1H); ESI-MS: рассчит. для (C9H8N2O3) 192, обнаружено 193 (MH+).
[00369]
[00370] Стадия 2: К раствору N-(3-нитрофенил)акриламида (2,06 г, 10,72 ммоль) в смеси MeOH (40 мл) и THF (40 мл) добавляли дигидрат хлорида олова(II) (12,09 г, 53,60 ммоль). Смесь перемешивали при к.т. в течение ночи, а затем концентрировали. Остаток обрабатывали насыщенным водн. раствором Na2CO3 до pH=10-11. Смесь экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток использовали непосредственно на следующей реакционной стадии без дополнительной очистки (желтое масло, 1,49 г, выход 85%). 1H-ЯМР (400 МГц, DMSO-d6) δ 9,81 (уш.с, 1H), 6,99 (с, 1H), 6,95 (т, J=9,2 Гц, 1H), 6,76 (д, J =7,6 Гц, 1H), 6,41 (м, 1H), 6,30-6,10 (м, 2H), 5,6 (д, J=10,4 Гц, 1H), 5,07 (уш.с, 2H); ESI-MS: рассчит. для (C9H10N2O) 162, обнаружено 163 (MH+).
[00371]
[00372] Стадия 3: Смесь промежуточного продукта 2 (неочищ., 70 мг, 0,22 ммоль), анилина (43 мг, 0,26 ммоль) и DIPEA (0,08 мл, 0,44 ммоль) в DMSO (2 мл) перемешивали при комнатной температуре в течение 2 часов. Проводили проверку методом TLC, и обнаруживали завершение реакции. Добавляли этилацетат (15 мл), а затем NH4Cl (20 мл). После разделения, водную фазу экстрагировали EtOAc (15 мл × 1). Объединенную органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали на колонке с силикагелем (0-10% MeOH в DCM) с получением целевого продукта в виде желтых твердых веществ (50 мг, выход 51%),1H-ЯМР (400 МГц, DMSO-d6) δ 11,84 (уш.с, 1H), 10,20 (уш.с, 1H), 10,14 (с, 1H), 8,32 (с, 1H), 7,93 (с, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,31 (т, J=8,0 Гц, 1H), 7,23 (д, J=8,0 Гц, 1H), 7,00 (дд, J =5,2 Гц, J=10,4 Гц,1H), 6,46 (дд, J=10,0 Гц, J=17,2 Гц, 1H), 6,34 (с, 1H), 6,26 (д, J=16,8 Гц, 1H), 5,76 (д, J=10,0 Гц, 1H), 2,42 (с, 3H); ESI-MS: рассчит. для (C23H16F2N6O2) 446, обнаружено 447 (MH+).
[00373]
[00374]
[00375] Соединение 54
[00376]
[00377] 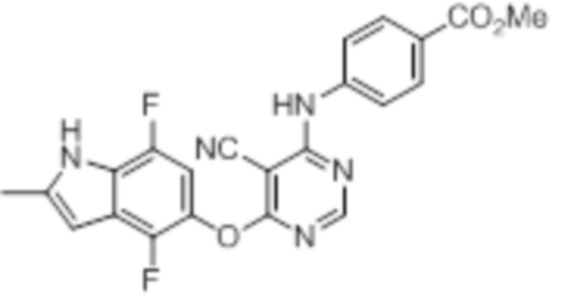
[00378]
[00379] Смесь 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,31 ммоль), метил-4-аминобензоата (47 мг, 0,34 ммоль), Pd(OAc)2 (10 мг, 0,045 ммоль), Xantphos (45 мг, 0,078 ммоль), K2CO3 (150 мг, 1,09 ммоль) и безводного диоксана (12 мл) герметично закрывали в пробирке для микроволновой обработки и дегазировали аргоном в течение 10 мин. Смесь затем нагревали до 120°C в течение 20 мин в условиях обработки микроволновым излучением. Смесь охлаждали до комнатной температуры и распределяли между EtOAc и водн. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде белого твердого вещества (68 мг, выход 50%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,85 (уш.с с, 1H), 10,39 (уш.с с, 1H), 8,38-8,37 (м, 1H), 7,95-7,93 (м, 2H), 7,75-7,73 (м, 2H), 7,04-7,00 (м, 1H), 6,35 (с, 1H), 3,84 (с, 3H), 2,42 (с, 3H); MS (ESI): рассчит. для C22H15F2N5O3: 435, обнаружено: 436 (MH+).
[00380]
[00381] Соединение 55
[00382] 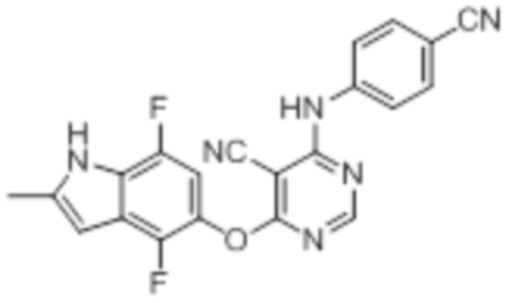
[00383]
[00384] Смесь 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (100 мг, 0,31 ммоль), 4-аминобензонитрила (40 мг, 0,34 ммоль), Pd(OAc)2 (10 мг, 0,045 ммоль), Xantphos (45 мг, 0,078 ммоль), K2CO3 (150 мг, 1,09 ммоль) и безводного диоксана (12 мл) герметично закрывали в пробирке для микроволновой обработки и дегазировали аргоном в течение 10 мин. Смесь затем нагревали до 120°C в течение 20 мин в условиях обработки микроволновым излучением. Смесь охлаждали до комнатной температуры и распределяли между EtOAc и водн. NaHCO3 (~100 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде белого твердого вещества (58 мг, выход 45%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,85 (уш.с с, 1H), 10,48 (уш.с с, 1H), 8,38 (с, 1H), 7,81-7,77 (м, 4H), 7,04-7,00 (м, 1H), 6,35 (д, J=0,8 Гц, 1H), 2,42 (с, 3H); MS (ESI): рассчит. для C21H12F2N6O: 402, обнаружено: 403 (MH+).
[00385] Соединение 56
[00386]
[00387] 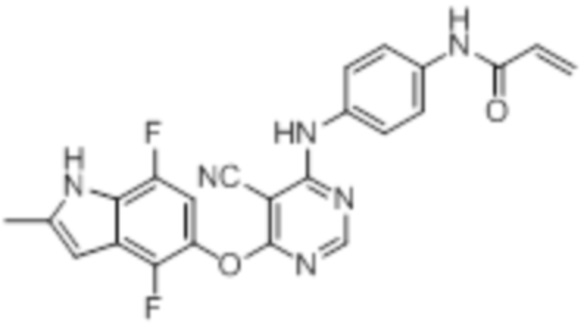
[00388] Стадия 1: К смеси 4-нитроанилина (3,0 г, 22 ммоль) и TEA (9,2 мл, 66 ммоль) в THF (100 мл) добавляли акрилоилхлорид (1,9 мл; 24 ммоль), смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 17 ч. Затем, дополнительно добавляли TEA (9,2 мл, 66 ммоль) и акрилоилхлорид (1,9 мл; 24 ммоль), и перемешивали смесь дополнительно в течение 2 ч. Смесь распределяли между EtOAc и водн. NaHCO3 (~100 мл; 50% насыщенность NaHCO3), органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде желтого твердого вещества (1,9 г, выход 45%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 10,76 (уш.с с, 1H), 8,25-8,23 (м, 2H), 7,93-7,91 (м, 2H), 6,51-6,44 (м, 1H), 6,37-6,32 (м, 1H), 5,88-5,85 (м, 1H); MS (ESI): рассчит. для C9H8N2O3: 192, обнаружено: 193 (MH+).
[00389] Стадия 2: К суспензии N-(4-нитрофенил)акриламида (800 мг, 4,16 ммоль) в смеси EtOH/вода (5/1, 21 мл) добавляли железный порошок (469 мг, 8,40 ммоль) и насыщенны водн. NH4Cl (2,1 мл), и перемешивали смесь при 80°C в течение 3 ч. Затем, дополнительно добавляли железный порошок (500 мг, 8,95 ммоль) и NH4Cl (700 мг, 13,1 ммоль), и перемешивали смесь дополнительно в течение 17 ч. Смесь распределяли между EtOAc и водн. NaHCO3 (по 100 мл каждого), органический слой разделяли, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0,5-15% MeOH в DCM (об./об.) с получением целевого продукта в виде желтого твердого вещества (216 мг, выход 32%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 9,71 (уш.с с, 1H), 7,30 (д, J=8,4 Гц, 2H), 6,51 (д, J=8,0 Гц, 2H), 6,40-6,33 (м, 1H), 6,18-6,13 (м, 1H), 5,66-5,63 (м, 1H); MS (ESI): рассчит. для C9H10N2O: 162, обнаружено: 163 (MH+).
[00390] Стадия 3: К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (230 мг, 0,71 ммоль) и N-(4-аминофенил)акриламида (120 мг, 1,0 ммоль) в безводном DMSO (5,0 мл) добавляли DIPEA (0,43 мл; 2,5 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение 20 ч. Полученную смесь разбавляли MeOH/EtOAc (1/9, 100 мл) и промывали нас. NH4Cl (~100 мл; 50% насыщенность NH4Cl), а затем солевым раствором (100 мл). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде не совсем белого твердого вещества (219 мг, выход 69%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,84 (уш.с с, 1H), 10,18 (уш.с с, 1H), 10,04 (уш.с с, 1H), 8,29 (д, J=0,4 Гц, 1H), 7,66 (д, J=8,8 Гц, 2H), 7,46 (д, J=8,8 Гц, 2H), 7,03-6,99 (м, 1H), 6,47-6,41 (м, 1H), 6,34-6,24 (м, 2H), 5,77-5,74 (м, 1H), 2,42 (с, 3H); MS (ESI): рассчит. для C23H16F2N6O2: 446, обнаружено: 447 (MH+).
[00391]
[00392] Соединение 57
[00393]
[00394] 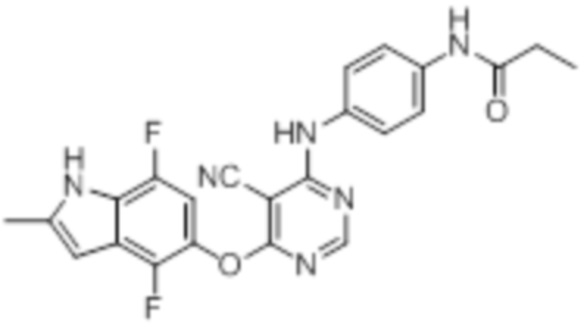
[00395] Стадия 1: К раствору N-(4-нитрофенил)пропионамида (1,00 г, 5,15 ммоль) в MeOH (100 мл) добавляли 10% Pd/C (100 мг), и перемешивали полученную смесь в атмосфере H2 (1 атм) в течение 21 ч. Смесь затем фильтровали через Celite® и концентрировали с получением целевого продукта в виде оранжевого масла (858 мг, выход ~100%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 9,39 (уш.с с, 1H), 7,20 (д, J=8,8 Гц, 2H), 6,48 (д, J=8,4 Гц, 2H), 4,79 (уш.с с, 2H), 2,22 (кв, J=7,6 Гц, 2H), 1,05 (т, J=7,6 Гц, 3H).
[00396] Стадия 2: К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и N-(4-аминофенил)пропионамида (69 мг, 0,42 ммоль) в безводном DMSO (1,5 мл) добавляли TEA (0,10 мл; 0,75 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение 20 ч. Полученную смесь разбавляли MeOH/EtOAc (1/9, 100 мл) и промывали нас. NH4Cl (~10 мл; 50% насыщенность NH4Cl), а затем солевым раствором (10 мл). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого продукта в виде не совсем белого твердого вещества (69 мг, выход 62%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,83 (уш.с с, 1H), 10,00 (уш.с с, 1H), 9,89 (уш.с с, 1H), 8,27 (д, J=0,4 Гц, 1H), 7,58 (д, J=8,8 Гц, 2H), 7,41 (д, J=8,8 Гц, 2H), 7,03-6,99 (м, 1H), 6,34 (с, 1H), 2,41 (с, 3H), 2,32 (кв, J=7,6 Гц, 2H), 1,09 (т, J=7,6 Гц, 3H); MS (ESI): рассчит. для C23H18F2N6O2: 448, обнаружено: 449 (MH+).
Соединение 58
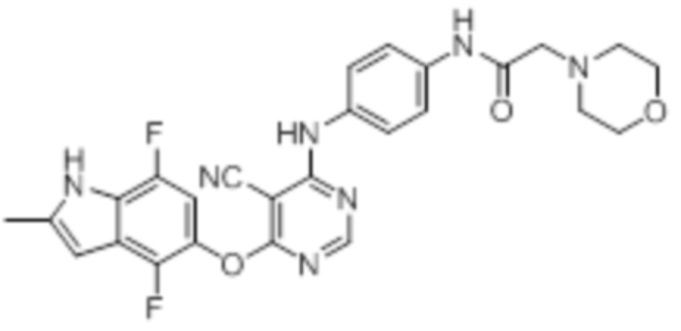
[00397] Стадия 1: К смеси 4-нитроанилина (5,0 г, 36 ммоль) и TEA (5,0 мл, 36 ммоль) в THF (100 мл), перемешанной при 0°C, добавляли 2-хлорацетилхлорид (1,9 мл; 24 ммоль), и оставляли смесь нагреваться до комнатной температуры и перемешиваться в течение 17 ч. Смесь распределяли между MeOH/EtOAc (1/9) и водн. NaHCO3 (~100 мл; 50% насыщенность NaHCO3), органический слой промывали нас. солевым раствором (~100 мл; 50% насыщенность), разделяли, сушили над безводным Na2SO4 и концентрировали, в результате чего формировался осадок. Осадок фильтровали и промывали EtOAc с получением целевого 2-хлор-N-(4-нитрофенил)ацетамида в виде желтого твердого вещества (6,17 г, выход 80%),1H-ЯМР (DMSO-d6, 400 МГц) δ 10,90 (уш.с с, 1H), 8,25 (д, J=8,4 Гц, 2H), 7,84 (д, J=9,2 Гц, 2H), 4,34 (с, 2H); MS (ESI): рассчит. для C8H7ClN2O3: 214, обнаружено: слабый сигнал.
[00398] Стадия 2: Смесь 2-хлор-N-(4-нитрофенил)ацетамида (500 мг, 2,33 ммоль) и морфолина (2,0 мл, 23 ммоль) в изопропаноле (~5 мл) перемешивали при 80°C в течение ~17 ч. Смесь распределяли между EtOAc и водн. NaHCO3 (по ~10 мл каждого; 50% насыщенность NaHCO3), органический слой разделяли и сушили над безводным Na2SO4 с получением целевого продукта в виде желтого твердого вещества (323 мг, выход 52%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 10,36 (уш.с с, 1H), 8,24-8,21 (м, 2H), 7,92-7,89 (м, 2H), 3,63 (д, J=3,2 Гц, 4H), 3,31 (уш.с с, 4H), 3,21 (с, 2H); MS (ESI): рассчит. для C12H15N3O4: 265, обнаружено: 266 (MH+).
[00399] Стадия 3: К раствору 2-морфолино-N-(4-нитрофенил)ацетамида (320 мг, 1,21 ммоль) в MeOH (70 мл) добавляли 10% Pd/C (80 мг), и перемешивали полученную смесь в атмосфере H2 (1 атм) в течение 23 ч. Смесь затем фильтровали через Celite® и концентрировали с получением целевого продукта в виде оранжевого масла (314 мг, выход ~100%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 9,28 (уш.с с, 1H), 7,22 (д, J=8,4 Гц, 2H), 6,50 (д, J=8,8 Гц, 2H), 4,86 (уш.с с, 2H), 3,64-3,62 (м, 4H), 3,04 (с, 2H), 2,51-2,47 (м, 4H); MS (ESI): рассчит. для C12H17N3O2: 235, обнаружено: 236 (MH+).
[00400] Стадия 4: К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и N-(4-аминофенил)-2-морфолиноацетамида (68 мг, 0,29 ммоль) в безводном DMSO (1,5 мл) добавляли DIPEA (0,15 мл; 0,87 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение 1,5 суток. Полученную смесь разбавляли MeOH/EtOAc (1/9, 10 мл) и промывали нас. NH4Cl (~10 мл; 50% насыщенность NH4Cl), а затем солевым раствором (10 мл). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали с получением целевого продукта в виде не совсем белого твердого вещества (142 мг, выход ~100%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,84 (с, 1H), 10,03 (с, 1H), 9,77 (с, 1H), 8,28 (с, 1H), 7,63-7,61 (м, 2H), 7,44 (д, J=8,8 Гц, 2H), 7,03-6,99 (м, 1H), 6,34 (уш.с с, 1H), 3,65-3,63 (м, 4H), 2,54-2,52 (м, 4H; заслонен сигналом DMSO), 2,41 (с, 3H); MS (ESI): рассчит. для C26H23F2N7O3: 519, обнаружено: 520 (MH+).
Соединение 59
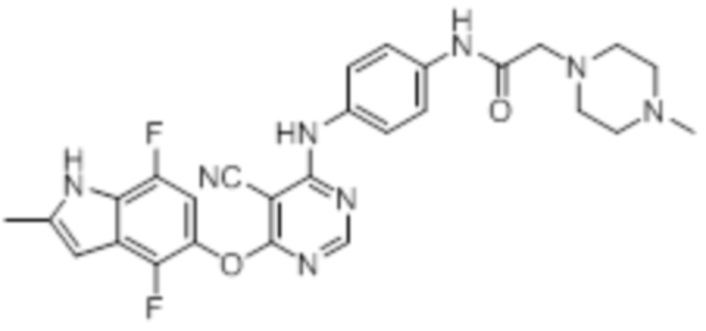
[00401] Стадия 1: Смесь 2-хлор-N-(4-нитрофенил)ацетамида (500 мг, 2,33 ммоль) и N-метилпиперазина (2,6 мл, 23 ммоль) в изопропаноле (~5 мл) перемешивали при 80°C в течение ~17 ч. Смесь распределяли между EtOAc и водн. NaHCO3 (по ~10 мл каждого; 50% насыщенность NaHCO3), органический слой разделяли и сушили над безводным Na2SO4 с получением целевого продукта в виде желтого твердого вещества (513 мг, выход 79%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 10,32 (уш.с с, 1H), 8,23-8,21 (м, 2H), 7,91-7,89 (м, 2H), 3,31 (с, 2H), 2,51 (уш.с с, 4H), 2,33 (уш.с с, 4H), 2,17 (с, 3H); MS (ESI): рассчит. для C13H18N4O3: 278, обнаружено: 279 (MH+).
[00402] Стадия 2: К раствору 2-(4-метилпиперазин-1-ил)-N-(4-нитрофенил)ацетамида (500 мг, 1,80 ммоль) в MeOH (70 мл) добавляли 10% Pd/C (75 мг), и перемешивали полученную смесь в атмосфере H2 (1 атм) в течение 19 ч. Смесь затем фильтровали через Celite® и концентрировали с получением целевого продукта в виде не совсем белого твердого вещества (456 мг, выход ~100%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 9,21 (уш.с с, 1H), 7,22 (д, J=8,8 Гц, 2H), 6,49 (д, J=8,4 Гц, 2H), 4,86 (уш.с с, 2H), 3,01 (с, 2H), 2,50 (уш.с с, 4H), 2,36 (уш.с с, 4H), 2,16 (с, 3H); MS (ESI): рассчит. для C13H20N4O: 248, обнаружено: 249 (MH+).
[00403] Стадия 3: К смеси 4-хлор-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (80 мг, 0,25 ммоль) и N-(4-аминофенил)-2-(4-метилпиперазин-1-ил)ацетамида (71 мг, 0,29 ммоль) в безводном DMSO (1,5 мл) добавляли DIPEA (0,15 мл; 0,87 ммоль), и энергично перемешивали полученную двухфазную смесь при комнатной температуре в течение 18 ч. Полученную смесь разбавляли MeOH/EtOAc (1/9, 10 мл) и промывали нас. NH4Cl (~10 мл; 50% насыщенность NH4Cl), а затем солевым раствором (10 мл). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали, и очищали полученный остаток методом колоночной флэш-хроматографии на силикагеле с использованием 0-20% MeOH в DCM (об./об.) с получением целевого продукта в виде не совсем белого твердого вещества (54 мг, выход 41%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,84 (с, 1H), 10,03 (с, 1H), 9,71 (с, 1H), 8,28 (с, 1H), 7,63-7,60 (м, 2H), 7,45-7,42 (м, 2H), 7,03-6,99 (м, 1H), 6,35 (уш.с с, 1H), 2,55-2,52 (м, 4H; заслонен сигналом DMSO), 2,41 (с, 3H), 2,40-2,37 (м, 4H), 2,17 (с, 3H); MS (ESI): рассчит. для C27H26F2N8O2: 532, обнаружено: 533 (MH+).
[00404] Соединение 60
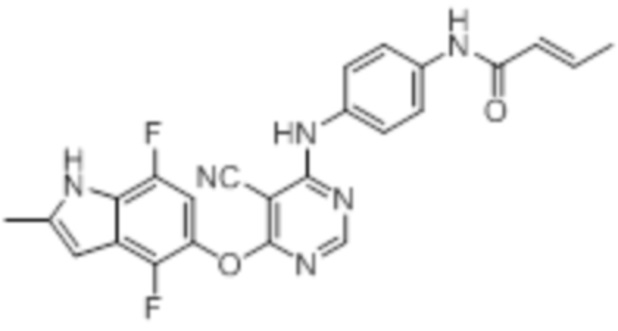
[00405] К смеси 4-((4-аминофенил)амино)-6-((4,7-дифтор-2-метил-1H-индол-5-ил)окси)пиримидин-5-карбонитрила (64 мг, 0,16 ммоль) и пиридина (39 мг, 0,49 ммоль) в DCM (3,0 мл) добавляли кротоноилхлорид (51 мг, 0,49 ммоль). Полученную смесь энергично перемешивали при комнатной температуре в течение 1 ч, после чего она становилась гомогенной. Полученную смесь разбавляли EtOAc (10 мл) и промывали водн. NaHCO3 (~10 мл; 50% насыщенность NaHCO3). Органический слой разделяли, сушили над безводным Na2SO4 и концентрировали с получением целевого продукта в виде не совсем белого твердого вещества (51 мг, выход 68%). 1H-ЯМР (DMSO-d6, 400 МГц) δ 11,84 (уш.с с, 1H), 10,03 (с, 1H), 9,82 (с, 1H), 8,29 (с, 1H), 7,68-7,65 (м, 2H), 7,46-7,43 (м, 2H), 7,03-6,99 (м, 1H), 6,34 (д, J=0,8 Гц, 1H), 5,80 (с, 1H), 5,52 (с, 1H), 2,42 (с, 3H), 1,96 (с, 3H); MS (ESI): рассчит. для C27H26F2N8O2: 532, обнаружено: 533 (MH+).
Соединение 61
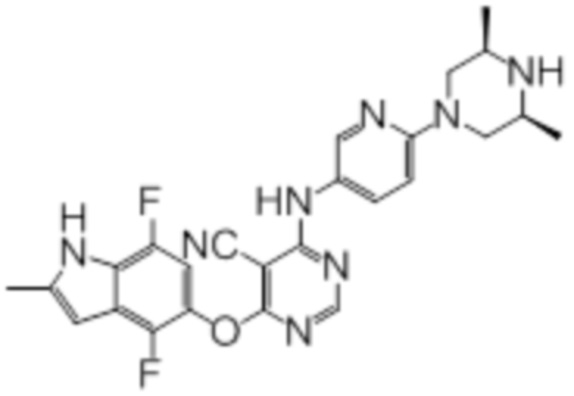
[00406] Стадия 1: К раствору 2-хлор-5-нитропиридина (10,00 г, 63,07 ммоль) и цис-2,6-диметилпиперазина (9,00 г, 78,84 ммоль) в DMSO (50 мл) добавляли карбонат калия (10,90 г, 78,84 ммоль). Смесь перемешивали при 50°C в течение 18 ч. Полученную смесь охлаждали до комнатной температуры и добавляли в колбу с водой/солевым раствором (600 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. а затем охлаждали до 0°C. Твердые вещества собирали путем фильтрования, промывали водой (100 мл × 3). Твердые вещества растирали с гексаном, собирали путем фильтрования и дополнительно сушили в условиях вакуума с получением продукта в виде желтых твердых веществ (13,82 г, выход 92%). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,93 (д, J=2,8 Гц, 1H), 8,17 (дд, J =2,8 Гц, J=9,6 Гц, 1H), 6,94 (д, J=9,6 Гц, 1H), 4,40 (уш.с, 2H), 2,69 (м, 2H), 2,49-2,30 (м, 3H), 1,02 (д, J=6,4 Гц, 3H); ESI-MS: рассчит. для (C11H16N4O2) 236, обнаружено 237 (MH+).
[00407] Стадия 2: Раствор QW910 (13,72 г, 58,07 ммоль) в метаноле (350 мл) гидрировали в присутствии 10% Pd/C (0,60 г) с использованием баллона с H2 (3×). спустя 48 ч, реакционную смесь фильтровали через слой Celite® и промывали метанолом (250 мл). Фильтрат концентрировали с получением целевого продукта (12,00 г, выход 100%) в виде пурпурных твердых веществ. Этот продукт использовали непосредственно на следующей реакционной стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 7,58 (д, J=2,8 Гц, 1H), 6,88 (дд, J =8,8 Гц, J=2,8 Гц, 1H), 6,58 (д, J=8,8 Гц, 1H), 4,50 (уш.с, 2H), 3,80 (м, 2H), 2,75 (м, 2H), 2.,02 (м, 3H), 0,98 (д, J=6,0 Гц, 3H); ESI-MS: рассчит. для (C11H18N4) 206, обнаружено 207 (MH+).
[00408] Стадия 3: К раствору анилина, полученного на стадии 2 (141 мг, 0,68 ммоль), в изопропаноле (3 мл) добавляли TFA (50 мкл) и энергично встряхивали. Добавляли раствор промежуточного продукта 2 (200 мг, 0,62 ммоль) в изопропаноле (5 мл). Реакционную смесь нагревали до 85°C в течение 16 ч. После охлаждения до комнатной температуры, добавляли гексаны (~8 мл), охлаждали на льду и фильтровали с получением пурпурных твердых веществ. Твердые вещества суспендировали в смеси вода (50 мл)/MeOH (5 мл), и добавляли нас. NaHCO3 (~20 мл) (pH >8). Смесь перемешивали при комнатной температуре в течение 45 мин, а затем охлаждали на льду. Твердые вещества собирали путем фильтрования, промывали водой (~5 мл) и гексанами (10 мл). Продукт получали в виде пурпурных твердых веществ (263 мг, 86%). Чистота согласно HPLC составляла 95%, и дополнительная очистка не проводилась. 1H-ЯМР (400 МГц, DMSO-d6) δ 11,85 (уш.с, 1H), 9,93 (уш.с, 1H), 8,24 (с, 1H), 8,17 (д, J=2,4 Гц, 1H), 7,62 (дд, J=2,4 Гц, J =9,2 Гц, 1H), 7,00 (дд, J=5,2 Гц, J =10,4 Гц, 1H), 6,87 (д, J=9,2 Гц, 1H), 6,34 (с, 1H), 4,22 (д, J=12 Гц, 2H), 2,90 (уш.с, 2H), 2,46-2,00 (м, 6H), 1,11 (д, J=6,4 Гц, 6H); ESI-MS: рассчит. для (C25H24F2N8O) 490, обнаружено 491 (MH+).
ПРИМЕРЫ
[00409] Последующие примеры представлены для дополнительной иллюстрации настоящего изобретения и, безусловно, не должны быть истолкованы, как ограничивающие его объем каким бы то ни было образом.
Пример 1
[00410] В данном примере типовые соединения из числа раскрытых выше соединений 1-61 протестированы на ингибирующую активность в отношении киназ. Значительное число указанных соединений обобщенно обладает ингибирующей активностью в отношении киназ, в отношении широкого спектра киназ.
[00411] Протоколы методов анализа киназ хорошо известны специалистам в данной области техники. Более конкретно, буферная композиция являлась следующей: 20 мМ MOPS, 1 мМ EDTA, 0,01% Brij-35, 5% глицерин, 0,1% β-меркаптоэтанол, 1 мг/мл BSA. Тестируемые соединения сначала растворяли в DMSO до желаемой концентрации, затем последовательно разбавляли для получения буфера для анализа киназ. В конечной реакции объем из 25 мкл FGFR1(h) (5-10 мЕд) и KDR(h) (5-10 мЕд) инкубировали с 8 мМ MOPS pH 7,0, 0,2 мМ EDTA, 200 мкМ LRRASLG (Кемптид), 10 мМ ацетата магния и [γ33P-ATP]. Реакцию инициировали добавлением смеси MgATP. После инкубирования в течение 40 минут при комнатной температуре, реакцию останавливали добавлением 5 мкл 3% раствора фосфорной кислоты. Затем, 10 мкл реакционной смеси наносили на P30 filtermat, промывали трижды в течение 5 минут 50 мМ фосфорной кислотой и однократно метанолом, после чего высушивали и проводили сцинтилляционные измерения. Лунки, содержащие субстрат без киназ, и лунки, содержащие фосфопептидный контроль, использовали для определения значений, соответствующих 0% и 100% фосфорилирования, соответственно.
[00412] В таблице 1 представлены репрезентативные данные по ингибированию киназ соединениями согласно настоящему изобретению. Киназы FGFR1 и KDR известны специалистам в данной области техники как ассоциированные со злокачественной опухолью.
Таблица 1. Ингибирование киназной активности двух киназ, ассоциированных со злокачественной опухолью.
|
Пример 2
[00413] Был проведен целый ряд исследований для анализа последствий ингибирования тирозинкиназ в линиях клеток. Для этого 1000 клеток высевали в концентрации 27 мкл на лунку в 384-луночные микропланшеты, которые затем помещали на ночь в инкубатор с увлажненным CO2 при температуре 37°C. На следующий день добавили 3 мкл на лунку 10-кратно концентрированного лекарства, и возвращали планшеты в инкубатор на 72 часа. После 72-часового инкубирования, планшеты извлекли и добавили 6 мкл на лунку реагента для анализа жизнеспособности CellTiterblue (Promega). Планшеты возвращали в инкубатор на 3 часа, после чего измерения считывали флуоресценцию на планшетном ридере Victor X3 (Perkin Elmer). Данные анализировали с использованием Excel (Microsoft), и определяли значения GI50 с использованием Prism (Graphpad).
[00414] Для фосфо-FGFR использовали следующий протокол метода анализа. Высевали 25000 клеток в объеме 90 мкл на лунку в 96-луночные микропланшеты, которые затем помещали на ночь в инкубатор с увлажненным CO2 при температуре 37°C. 96-луночные ELISA планшеты (Mesoscale Discovery) покрывали иммобилизованным антителом (R&D Systems Duo-Set) в количестве 4 мкг/мл, 30 мкл на лунку. На следующий день добавляли 10 мкл на лунку 10-кратно концентрированного лекарства, и возвращали планшеты в инкубатор на 20 минут. Планшеты для ELISA промывали с использованием автоматизированной машины для мойки планшетов (BioTek Instruments). Через 30 минут клетки переворачивали и мягко постукивали для удаления излишка среды, и немедленно помещали на лед. В лунку добавляли 30 мкл/лунку лизирующего реагента mPer (Thermo Scientific) с ингибиторами протеаз и фосфатаз. После 15 минут на льду лизаты смешивали, и переносили 30 мкл в планшет для ELISA. Планшеты инкубировали в течение 2 часов, промывали и добавляли детекторное антитело в количестве 30 мкл на лунку. Через 1 час планшеты промывали и добавляли 30 мкл детекторного реагента SulfoTag (MesoScale Discovery). Через 1 час планшеты промывали и добавляли считывающий раствор в количестве 150 мкл на лунку. Электрохемилюминесценцию определяли на Mesoscale Discovery Sector Imager 2000. Данные анализировали с использованием Excel (Microsoft), и определяли значения EC50 с использованием Prism (Graphpad).
[00415] В таблице 2 представлены репрезентативные данные GI50 по ингибированию выбранных линий клеток злокачественных опухолей. Специалистам в данной области техники следует понимать, что каждая линия клеток представляет собой суррогат для конкретного типа злокачественной опухоли. Этот пример подтверждает, что ингибиторы протеинкиназ могут оказывать эффекты на пролиферацию клеток. Специалисты в данной области техники могут быть удивлены специфичностью ингибирования киназ. Все ингибиторы киназ, протестированные в настоящем документе, были ассоциированы со злокачественной опухолью.
Таблица 2. Ингибирование пролиферации
|
Пример 3
[00416] В этом примере противоопухолевую активность соединений 14, 17, 19, 45 и 48 согласно настоящему изобретению тестировали с использованием известной в данной области техники модели ксенотрансплантата AML. (Величина «Т/С» относится к соотношению размера опухоли у животных, подвергающихся лечению, к размеру опухоли у контрольных животных, не подвергавшихся лечению. Величина «BWC» относится к «изменению массы тела»).
Таблица 3. Противоопухолевая эффективность ведущих соединений в отношении TG1a ксенотрансплантата AML человека
|
Qdx10 - один раз в сутки в течение 10 суток
Пример 4
[00417] В этом примере продемонстрирована биологическая активность и фармацевтическая пригодность варианта осуществления настоящего изобретения, соединения 19. Этот пример служит в качестве примера характеристик исследуемых соединений, раскрытых в настоящей заявке. Тем не менее, этот пример никоим образом не подразумевается как ограничивающий объем настоящего изобретения.
[00418] Среди всех этапов разработки лекарства самым трудным является обнаружение перспективных соединений (см., например, Malo et al., "Statistical practice in high-throughput screening data analysis," Nature Biotechnology, 2006, 24, 167-75.) Ингибиторы киназ также часто характеризуются антипролиферативным эффектом. Привлекательной характеристикой ингибиторов киназ, например, соединений примера 2, является их способность ингибировать мутантные формы киназ, которые, как было показано, важны для трансформации нормальных клеток в клетки злокачественных опухолей. Ингибирующая активность соединения 19 в отношении киназ тестировали с использованием метода анализа киназ, описанного в примере 3. В таблицах 4 и 5 представлена ингибирующая активность соединения 19 в отношении киназ. Соединение 19 неожиданно является высоко активным в отношении рецепторных киназ фактора роста фибробластов дикого типа и их мутантных форм. Предполагается, что мутации в тестируемых киназах играют важную роль в трансформации некоторых злокачественных опухолей. Кроме того, в таблице 4 также представлено, что соединение 19 умеренно селективно в отношении рецепторных киназ фактора роста фибробластов.
[00419] Среди всех этапов разработки лекарства, самым трудным является обнаружение перспективных соединений.
Таблица 4. PHD
|
[00420] На фигуре 1 представлена кривая дозовой зависимости ингибирования киназ для соединения 19 в отношении специфических киназ в модельной системе с использованием клеток BaF3, созданных методом генной инженерии для экспрессии специфических киназ. Указанные на фигуре 1 киназы входят в состав класса рецепторных киназ фактора роста фибробластов (FGFR1-FGFR4). Ингибирующая активность соединения 19 в отношении киназ FGFR1-FGFR4 также представлена в таблице 5. Таким образом, соединение 19 демонстрирует значительную ингибирующую активность в отношении киназ.
Таблица 5. Активность соединения 19 в отношении рецепторных киназ фактора роста фибробластов
|
[00421] Соединение 19 также обладает значительными антипролиферативными свойствами. В таблице 6 обобщены результаты для соединения 19, протестированного согласно методу анализа пролиферации, описанному в примере 2.
[00422] Как представлено в таблице 6, соединение 19 обладает значительной антипролиферативной активностью в отношении некоторых, но не всех протестированных линий клеток. Примечательно, что общей чертой чувствительных клеток является экспрессия ими одного из рецепторов фактора роста фибробластов. В данном методе анализа соединение 19 особенно эффективно в отношении линии KG1a, которая представляет собой линию клеток, полученную из формы острого миелоидного лейкоза (AML).
Таблица 6. Активность соединения 19 в отношении клеток
|
[00423] Фармакокинетический профиль соединения 19 у крыс представлен в таблице 7. Специалистам в данной области техники следует понимать, что хотя это и не является абсолютно предсказуемым, указанные результаты могут быть сходными у других животных, включая, например, пациентов-людей. Фармакокинетический профиль соединения 19 соответствует соединению 19, используемому в качестве терапевтического средства.
Таблица 7. Фармакокинетический профиль соединения 19 у крыс
|
[00424] Время полужизни в процессе метаболизма для соединения 19 было определено в модельных системах людей, крыс и мышей и представлено в таблице 7. Специалистам в данной области техники следует понимать, что приведенные результаты указывают на то, что соединение 19 может быть довольно стабильным in vivo.
Таблица 8. Метаболическая стабильность соединения 19
|
[00425] В таблице 9 раскрыты данные по токсичности соединения 19 в различных тестах на токсичность, хорошо известных средним специалистам в данной области техники. Представленные результаты не отражают ничего, что могло бы свидетельствовать против проведения фазы I клинических испытаний соединения 19.
Таблица 9. Оценка токсичности соединения 19
|
[00426] К удивлению, было обнаружено, что соединение 19 обладает значительной противоопухолевой активностью в модели AML на «голых» мышах Nude. На фигуре 2 показано, что обе тестируемые дозировки соединения 19 привели практически к полной супрессии роста опухолевых клеток. На фигуре 3 показано, что масса животного, используемая в качестве суррогантного маркера токсичности, обнаруживала отсутствие различий между контрольными животными и животными, подвергавшимися обработке соединением 19. Указанные результаты были подтверждены в исследовании дозовой зависимости в такой же модели животных. (Следует отметить, что самая высокая доза соединения 19 (20 мг/кг) приводит к значительной потере массы, что скорее предполагает наличие общей токсичности, чем специфического эффекта на клетки злокачественной опухоли.)
[00427] Таким образом, все данные о характере соединения 19 полностью соответствуют соединению, разрабатываемому в качестве антипролиферативного/противоопухолевого средства.
[00428] Все ссылки, включая публикации, патентные заявки и патенты, процитированные в настоящем документе, включены в него посредством ссылки в том же объеме, как если бы каждая ссылка была по отдельности и прямо указана как подлежащая включению посредством ссылки и была представлена в настоящем документе во всей ее полноте.
[00429] Если в настоящем документе не указано иное, или это прямо не противоречит контексту, то использование определенных и неопределенных артиклей и термина «по меньшей мере» или подобных ссылок в контексте описания изобретения (в особенности, в контексте последующей формулы изобретения) должно истолковываться как охватывающее как единственное, так и множественное числа. Если в настоящем документе не указано иное, или это прямо не противоречит контексту, то использование термина «по меньшей мере» с последующим перечнем одного или нескольких понятий (например, «по меньшей мере, один из A и B») должно истолковываться как означающее одно понятие, выбранное из перечисленных понятий (A или B), или сочетание двух или более из перечисленных понятий (A и B). Если в настоящем документе не указано иное, то термины «заключающий в своем составе», «обладающий», «включающий» и «содержащий» должны истолковываться как неограничивающие термины (т.е., означающие «включающий без ограничения»). Если в настоящем документе не указано иное, то описание в настоящем документе диапазонов значений служит исключительно в качестве краткого способа индивидуальной ссылки на каждое отдельное значение, находящее внутри диапазона, и каждое отдельное значение включено в настоящее описание, как если бы оно было по отдельности процитировано в настоящем документе. Если в настоящем документе не указано иное, или это иным образом прямо не противоречит контексту, то все способы, описанные в настоящем документе, могут выполняться в любом удобном порядке. Если не указано иное, то использование абсолютно всех примеров или типовых формулировок (например, «такой как»), представленных в настоящем документе, предназначено исключительно с целью лучшего освещения настоящего изобретения, и не должно служить ограничением объема настоящего изобретения. Ни одна из формулировок в описании не должна истолковываться как указывающая на то, что какой-либо не указанный в формуле изобретения элемент является необходимым для практического использования изобретения.
[00430] В настоящем документе описаны предпочтительные варианты осуществления настоящего изобретения, включая наилучший известный авторам изобретения способ осуществления настоящего изобретения. Вариации указанных предпочтительных вариантов осуществления станут очевидны средним специалистам в данной области техники после прочтения представленного выше описания. Авторы изобретения ожидают, что специалисты будут использовать такие вариации в соответствующих случаях, и авторы изобретения предполагают применение настоящего изобретения на практике в дополнительных областях, прямо не указанных в настоящем документе. Соответственно, настоящее изобретение включает в себя все модификации и эквиваленты объекта изобретения, перечисленные в прилагаемой формуле изобретения, как разрешенные действующим законодательством. Более того, если в настоящем документе не указано иное, или это иным образом прямо не противоречит контексту, то настоящим изобретением охватывается любое сочетание описанных выше элементов во всех их возможных вариациях.
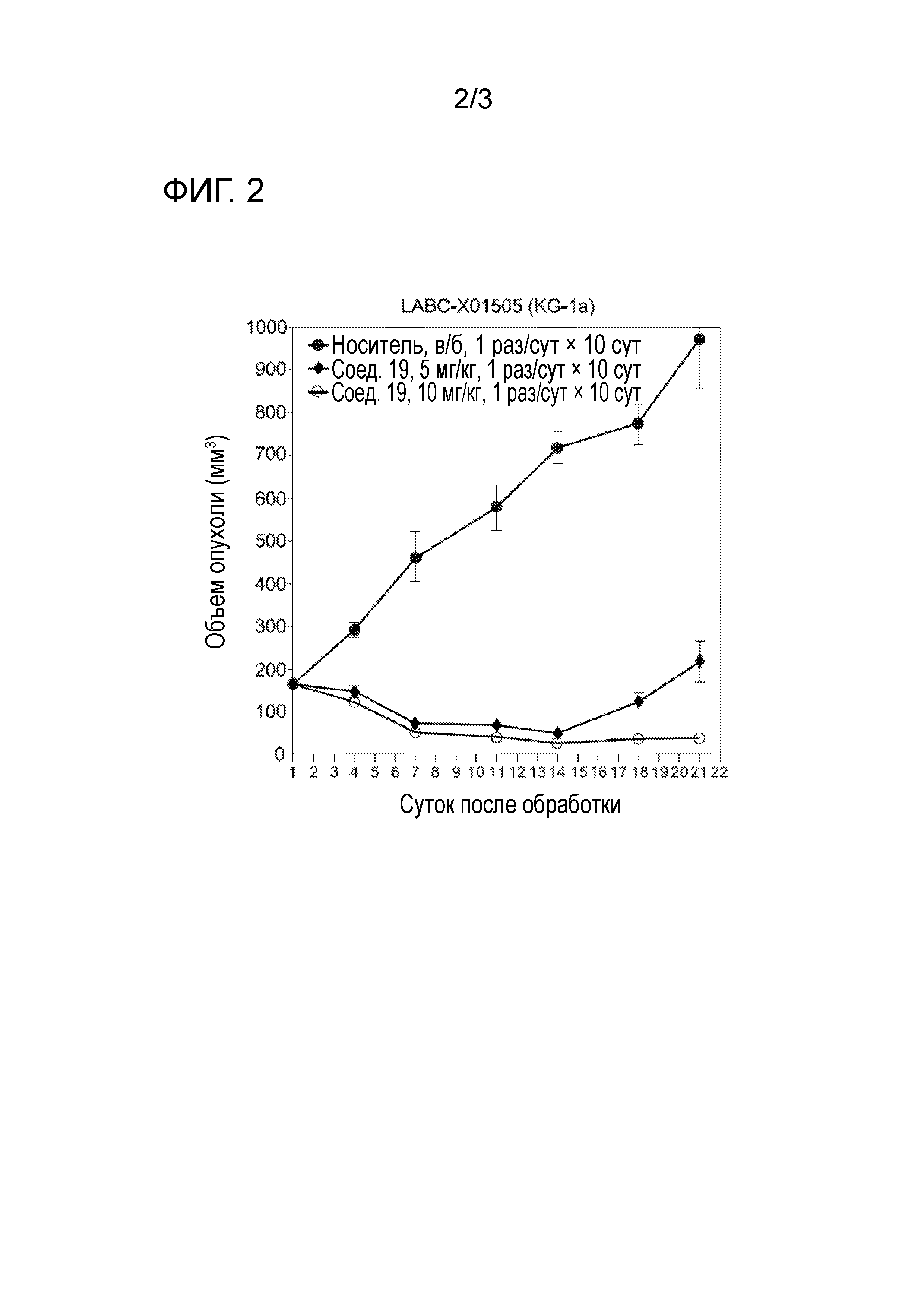
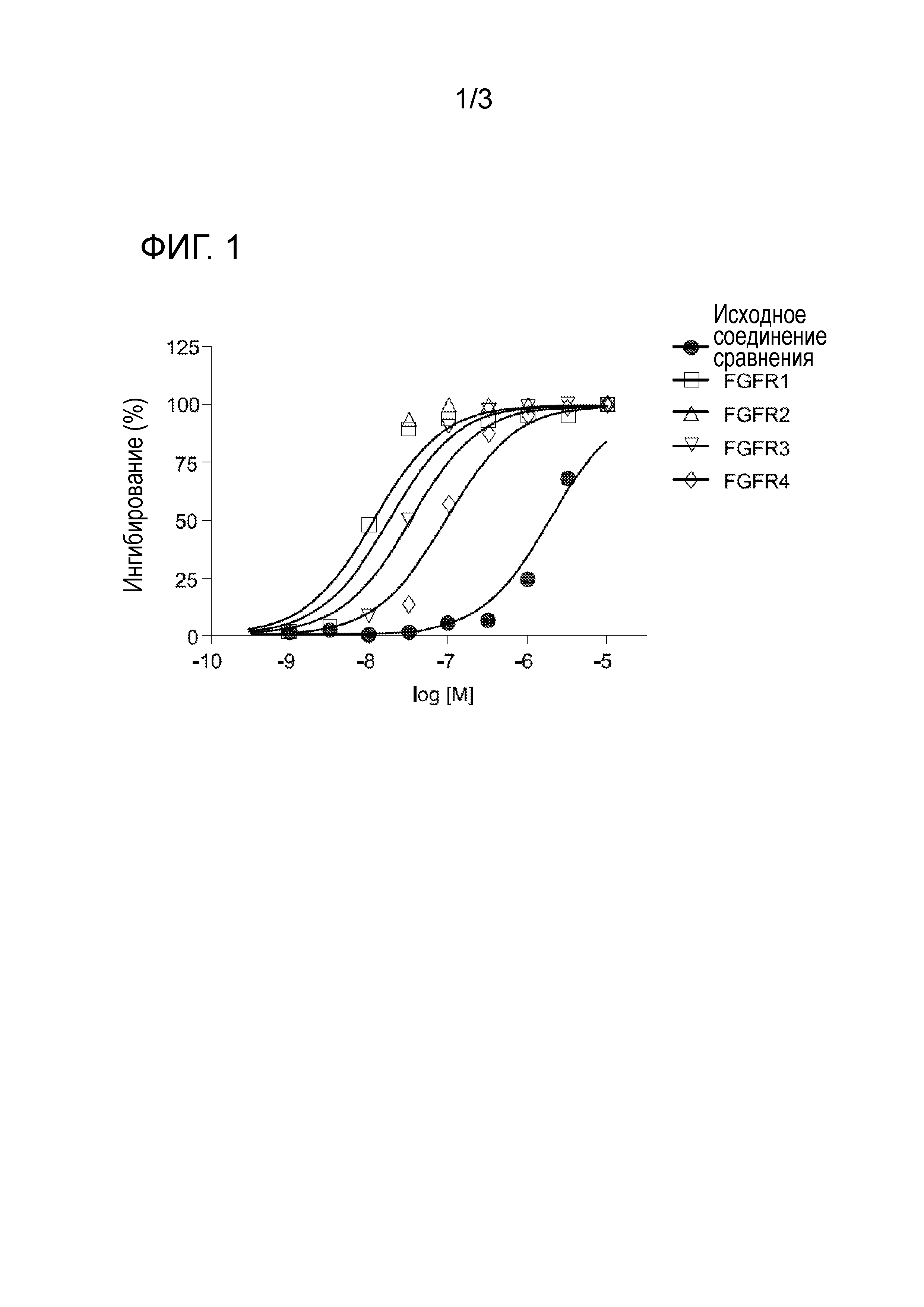
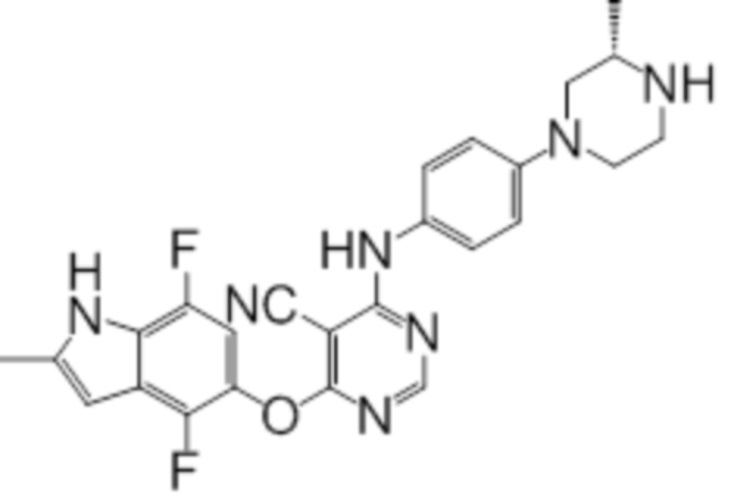
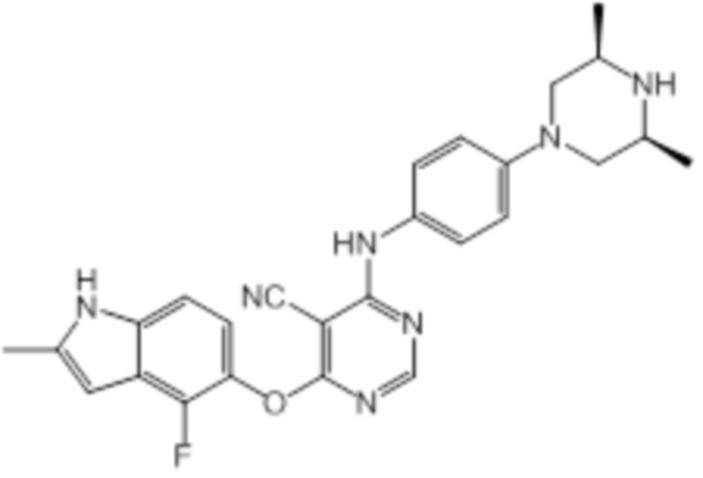
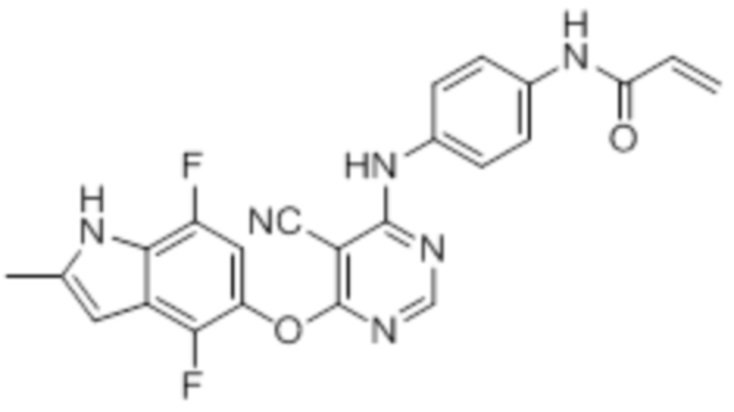
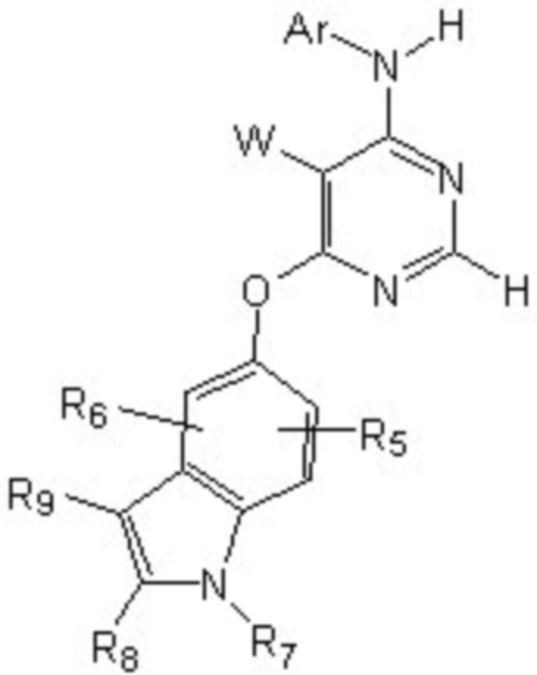
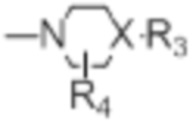
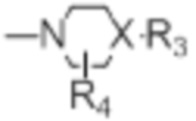
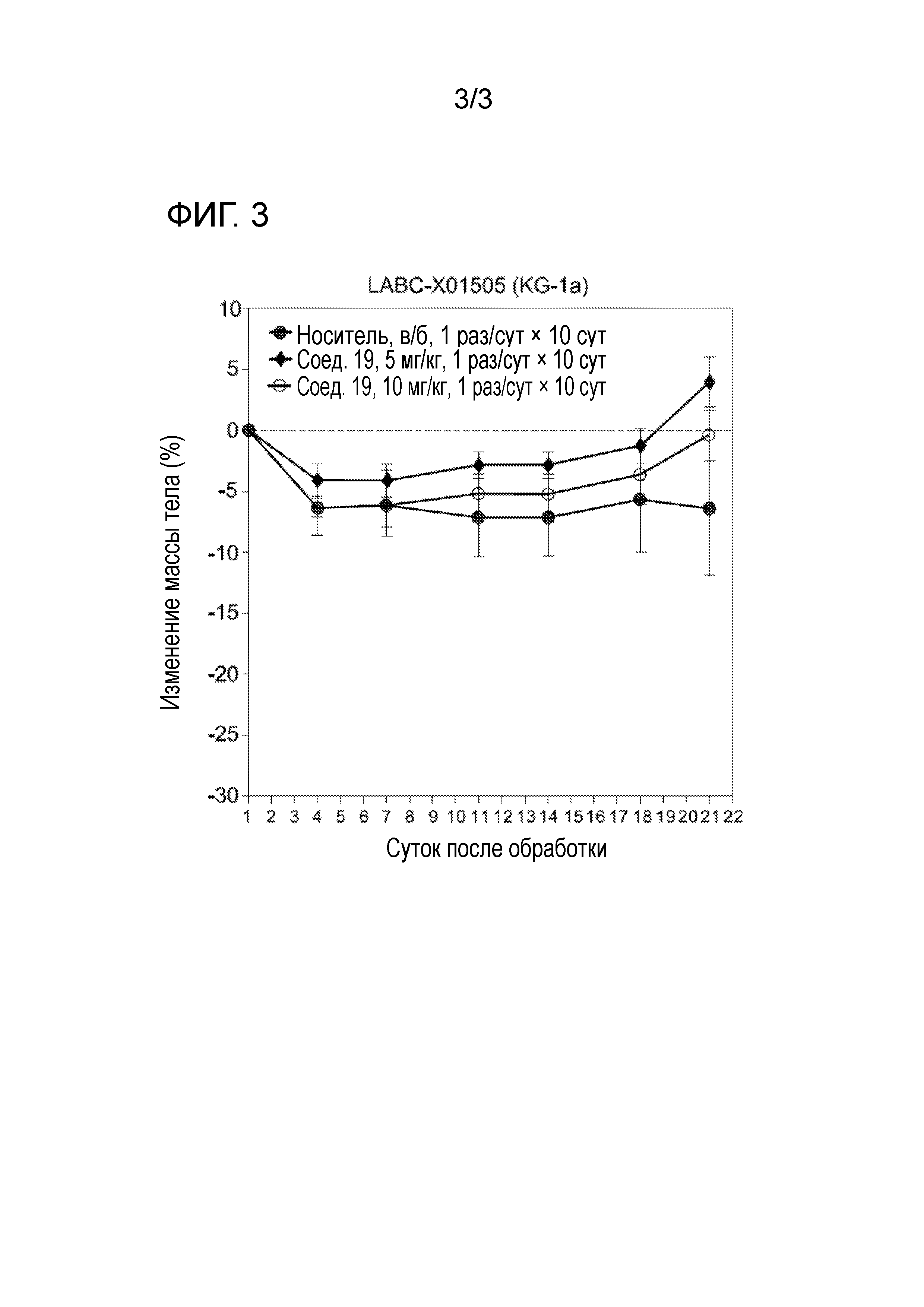
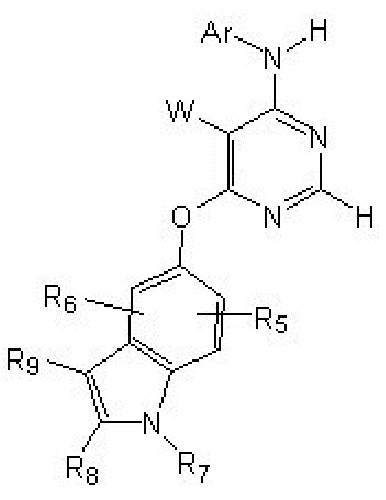
Производные пиримидина в качестве ингибиторов киназы и их терапевтические применения
Способ и система обеспечения обратной связи пространственной информации состояния канала на основании произведения кронекера
Комбинированная терапия композициями наночастиц таксана и ингибиторами хэджхог
Мини-антитела j591 и цис-диатела для направленной доставки простата-специфичного мембранного антигена (psma) человека и способы их применения
Замещенные производные индол-5-ола и их терапевтическое применение

