Результат интеллектуальной деятельности: ПРИМЕНЕНИЕ НОВЫХ СОЕДИНЕНИЙ ДЛЯ СЕЛЕКТИВНОГО ЭКСТРАГИРОВАНИЯ РЕДКОЗЕМЕЛЬНЫХ МЕТАЛЛОВ ИЗ ВОДНЫХ РАСТВОРОВ, СОДЕРЖАЩИХ ФОСФОРНУЮ КИСЛОТУ, И СООТВЕТСТВУЮЩИЙ СПОСОБ ЭКСТРАКЦИИ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение касается области экстракции редкоземельных металлов из водных фаз, содержащих соответствующие редкоземельные металлы.
Более конкретно, оно касается применения по меньшей мере одного специфического соединения в качестве экстрагирующего агента для экстрагирования по меньшей мере одного редкоземельного металла из водной фазы, в которой присутствует указанный по меньшей мере один редкоземельный металл.
Настоящее изобретение касается также способа извлечения по меньшей мере одного редкоземельного металла, присутствующего в кислой водной фазе, в котором применяется указанное специфическое соединение.
Кислая водная фаза, из которой редкоземельный металл (или металлы) можно экстрагировать или извлекать, представляет собой водный раствор, содержащий фосфорную кислоту. Такой водный раствор может, в частности, представлять собой раствор, получаемый при кислотном растворении концентрата руды или отходов, содержащих указанный по меньшей мере один редкоземельный металл.
Настоящее изобретение находит применение, в частности, в обработке природных руд и/или промышленных отходов с целью повторного использования входящих в их состав редкоземельных металлов.
Наконец, настоящее изобретение касается некоторых специфических соединений, применение которых указано выше.
Предшествующий уровень техники
Редкоземельные металлы объединяют группу металлов, отличающихся близкими свойствами, а именно скандий (Sc), иттрий (Y) и все лантаноиды, последние из которых соответствуют химическим элементам в периодической таблице Менделеева, имеющим атомные номера от 57 (лантан La) до 71 (лютеций Lu).
Редкоземельные металлы обычно разделяют на две категории: цериевые редкоземельные металлы или легкие редкоземельные металлы, которые объединяют Sc, Y, а также металлы от лантана (La) до неодима (Nd), и тяжелые редкоземельные металлы или группа иттрия, которые объединяют металлы от прометия (Pm) до лютеция (Lu).
Свойства редкоземельных металлов в частности связаны с их электронной конфигурацией, в особенности со специфичностью их 4f электронного подуровня, который делает возможными многочисленные оптические переходы и обуславливает особые магнитные и каталитические свойства. В свете своих необычных свойств, редкоземельные металлы применяются в многочисленных высокотехнологичных областях, таких как лазеры, постоянные магниты, батареи или экономичные лампы.
Вследствие этого, редкоземельные металлы формируют часть так называемых “технологических” металлов, поставки которых являются стратегическими, но находятся под угрозой из-за эффекта роста мировых потребностей в этих специфических металлах.
Среди источников редкоземельных металлов можно указать месторождения бастнезита в крепкой породе, а также аллювиальные отложения монацита и ксенотима. Помимо этих минералов, содержащих наивысшие концентрации редкоземельных металлов, можно указать другие заслуживающие внимания руды, такие как фосфатные руды или апатиты, которые определенно беднее лантанидами, но их добыча и переработка может быть прибыльной в будущем. В следующем далее тексте настоящей заявки, указанные минералы, содержащие редкоземельные металлы в больших или меньших количествах, обозначают выражением “природные минералы”.
Поскольку редкоземельные металлы также очень широко представлены в технологическом оборудовании, повторное использование после переработки промышленных отходов от такого оборудования, и в частности электрических и электронных отходов, обозначаемых “EEEW” или “3EW”, представляет собой нетрадиционный и альтернативный источник редкоземельных металлов. В следующем далее тексте настоящей заявки, такие промышленные отходы, содержащие редкоземельные металлы, обозначают выражением “промышленные минералы”.
Способы, применяемые в настоящее время для выделения редкоземельных металлов из таких природных или промышленных минералов, состоят в проведении химической обработки таких минералов, после предварительного измельчения, с помощью кислотных или основных реагентов, с получением минерального концентрата.
Затем полученный минеральный концентрат подвергают химическому расщеплению, чтобы добиться солюбилизации содержащихся в них редкоземельных металлов. Это химическое расщепление обычно осуществляют с помощью одного или больше кислотных реагентов, среди которых азотная кислота, серная кислота, фосфорная кислота или хлористоводородная кислота.
Получаемый при этом так называемый “раствор от кислотного гидролиза” затем подвергают гидрометаллургической обработке, основанной на методике жидкость-жидкостной экстракции, которая состоит в контактировании водной фазы, представляющей собой раствор от кислотного гидролиза, с органической фазой, содержащей один или больше экстрагирующих агентов, что обеспечивает экстракцию редкоземельных металлов, при этом такая экстракция должна предпочтительно быть эффективной и селективной одновременно.
Среди экстрагирующих агентов, промышленно применяющихся для экстракции редкоземельных металлов, можно указать бис-(2-этилгексил)фосфорную кислоту (или HDEHP), 2-этилгексил-2-этилгексилфосфоновую кислоту (или HEHEHP), а также три-н-бутил фосфат (или TBP), при этом TBP является наиболее широко применяющимся экстрагирующим агентом на сегодняшний день.
Независимо от того, что HDEHP позволяет экстрагировать редкоземельные металлы из сильно кислых (pH<1) водных фаз, в то время когда HEHEHP позволяет экстрагировать редкоземельные металлы из кислых водных фаз, в которых 2<pH<3, три указанные экстрагирующие агента HDEHP, HEHEHP и TBP характеризуются селективным экстрагированием тяжелых лантаноидов, соответствующих лантаноидам с атомными номерами 61 или больше (до 71).
Таким образом, применение этих экстрагирующих агентов позволяет экстрагировать не все редкоземельные металлы, а только тяжелые редкоземельные металлы. Кроме того, применение экстрагирующих агентов HDEHP и HEHEHP требует определенных диапазонов концентрации кислоты, присутствующей в водной фазе.
Недавняя публикация S. Ansari et al. (“Chemistry of Diglycolamides: Promising Extractants for Actinide Partitioning”, Chemical Reviews, 2012, 112, 1751-1772), обозначенная как [1] в конце настоящего описания, описывает применение дигликольамидов (или DGA), среди которых N,N,N',N'-тетра-n-октилдигликольамид (или TODGA), как соединений, подходящих для экстрагирования лантаноидов и актиноидов, присутствующих в водной фазе, содержащей азотную кислоту.
Однако, в публикации [1] описано, что экстракция с помощью TODGA может приводить к образованию третьей фазы, что совершенно неприемлемо для применения данного способа экстракции в промышленном масштабе. Для устранения образования третьей фазы, в публикации [1] предлагается комбинировать TODGA с фазомодификатором, в данном случае TBP. В таком варианте требуется применение уже не одного, а двух экстрагирующих агентов. В то же время, такая система с двумя экстрагирующими агентами, для которых к тому же требуется определение оптимального соотношения, очевидно менее удобна, чем система с одним экстрагирующим агентом.
В еще более свежей публикации H. Narita et al. ("Separation of Rare Earth Elements from Base Metals in Concentrated HNO3, H2SO4 and HCl Solutions with Diglycolamide", Solvent Extraction Research and Development, Japan, 2013, 20, 115-121), обозначенной как [2], авторы описывают применение N,N'-диметил-N,N'-ди-н-октилдигликольамида (или MODGA) в качестве экстрагирующего агента для неодима Nd(III) и диспрозия Dy(III), содержащихся в водных растворах, содержащих, помимо них, железо Fe(III), никель Ni(II) и различные мольные концентрации, от 0.1 моль/л до 7 моль/л, азотной кислоты, хлористоводородной кислоты или серной кислоты. Согласно экспериментальным результатам, изложенным в публикации [2], наиболее эффективная и наиболее селективная экстракция Nd и Dy, в сравнении с Fe и Ni, достигается для водных растворов, содержащих азотную кислоту.
Хотя экстракция дигликольамидами редкоземельных металлов из кислых водных фаз известна из публикаций [1] и [2], тем не менее отмечено, что была описана только экстракция из водных фаз, содержащих хлористоводородную кислоту, серную кислоту или, более часто, азотную кислоту.
В то же время, и как проиллюстрировано ниже в Примере 8, авторы настоящего изобретения обнаружили, что экстракцию, описанную в публикации [1], с TODGA в качестве экстрагирующего агента, и экстракцию, осуществляемую из водной фазы, содержащей азотную кислоту, нельзя перенести на экстракцию, осуществляемую из водной фазы, содержащей фосфорную кислоту.
На основе этого наблюдения авторы настоящего изобретения поставили цель разработать новое семейство экстрагирующих агентов, которые имеют более высокое сродство ко всем или к части редкоземельных металлов и которые могут вследствие этого применяться для экстракции по меньшей мере одного редкоземельного металла из кислой водной фазы, в которой присутствует этот редкоземельный металл или металлы, при этом водная фаза содержит в качестве кислоты фосфорную кислоту, а не азотную, хлористоводородную или серную кислоту.
Экстракция с помощью указанного нового семейства экстрагирующих агентов должна позволять экстрагировать большинство редкоземельных металлов, присутствующих в водной фазе, или, наоборот, некоторые редкоземельные металлы, например в зависимости от их атомного номера. Экстракция с помощью этих экстрагирующих агентов должна также быть селективной по отношению к элементам, которые также с большой вероятностью присутствуют в кислой водной фазе, но не являются редкоземельными металлами.
Авторы настоящего изобретения также поставили цель, состоящую в том, что экстракция может предпочтительно применяться в максимально широком диапазоне концентраций фосфорной кислоты в водной фазе.
Авторы настоящего изобретения также поставили цель, состоящую в том, что указанную экстракцию можно предпочтительно применять с одним экстрагирующим агентом и без образования третьей фазы, что позволяет предусмотреть успешный перенос соответствующего способа экстрагирования на промышленный уровень.
Раскрытие изобретения
Перечисленные выше и другие цели достигнуты, в первую очередь, посредством применения по меньшей мере одного специфического соединения в качестве экстрагирующего агента для экстракции по меньшей мере одного редкоземельного металла из водной фазы, в которой присутствует указанный по меньшей мере один редкоземельный металл, при этом водная фаза дополнительно содержит фосфорную кислоту.
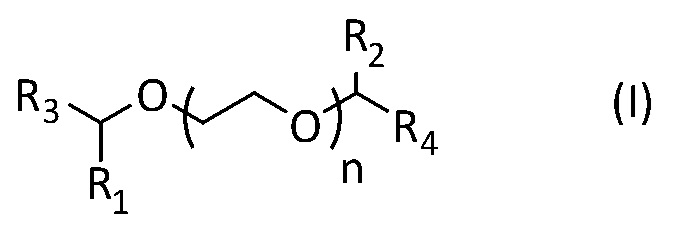
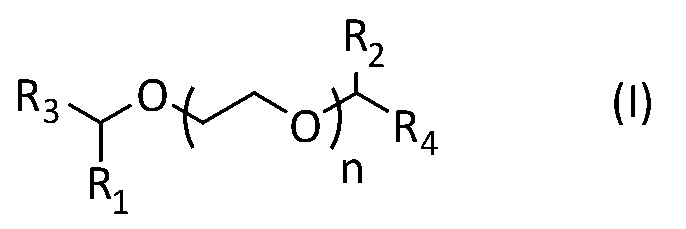
Это специфическое соединение, применение которого является предметом настоящего изобретения, представляет собой соединение, имеющее изображенную ниже формулу (I):
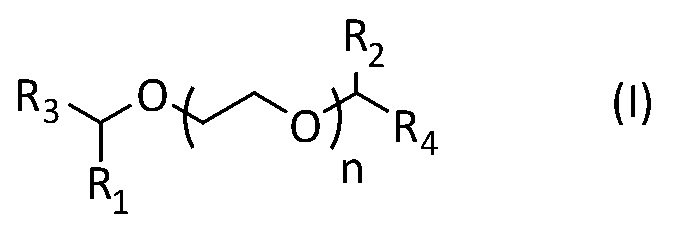
где
- n представляет собой целое число, равное 0, 1 или 2,
- R1 и R2 представляют собой, независимо друг от друга, атом водорода, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную,
- один из R3 и R4 имеет изображенную ниже формулу (II):
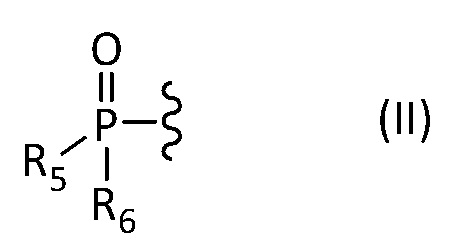
где R5 и R6 представляют собой, независимо друг от друга, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, C3 - C8 циклическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, гидроксильную группу -OH или алкоксильную группу -OR, где R представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, и
- оставшийся из R3 и R4 имеет:
либо изображенную ниже формулу (II'):
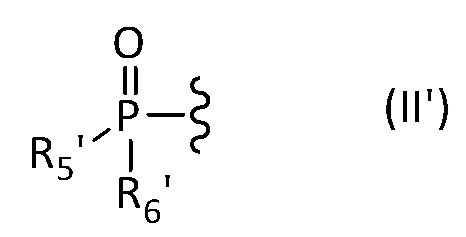
в которой R5' и R6' представляют собой, независимо друг от друга, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, C3 - C8 циклическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, гидроксильную группу -OH или алкоксильную группу -OR', где R’ представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную,
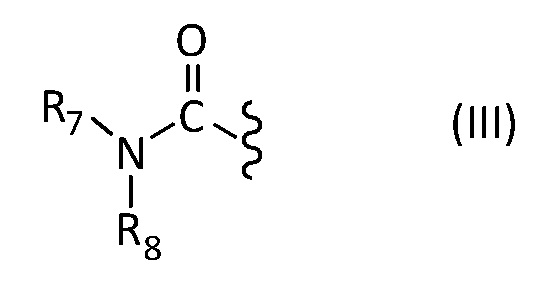
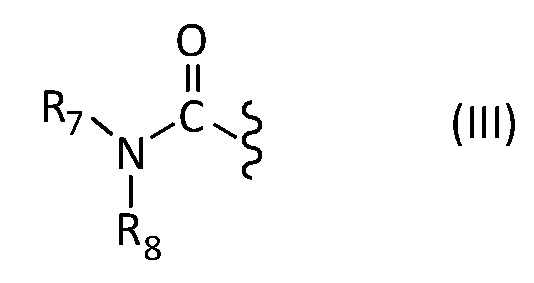
либо изображенную ниже формулу (III):
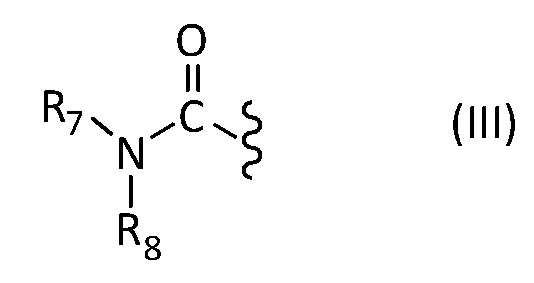
в которой R7 и R8 представляют собой, независимо друг от друга, атом водорода, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную.
Выше и ниже по тексту:
- термин “C1 - C12 алифатическая углеводородная группа, насыщенная или ненасыщенная, линейная или разветвленная”, означает любую алкильную, алкенильную или алкинильную группу, с линейной или разветвленной цепью, которая содержит в сумме от 1 до 12 атомов углерода;
- термин “C3 - C8 циклическая алифатическая углеводородная группа, насыщенная или ненасыщенная, линейная или разветвленная”, означает любую неароматическую углеводородную группу, содержащую по меньшей мере один цикл, где указанный цикл содержит от 3 до 8 атомов углерода, указанный цикл может быть насыщенным или, наоборот, иметь один или больше ненасыщенных фрагментов, и указанные один или больше циклов могут быть разветвленными, где разветвленная цепь (цепи) содержат от 1 до 6 атомов углерода. Так, эта группа может, в частности, представлять собой циклоалкильную группу (циклопропан, циклопентан, циклогексан и т.д.), циклоалкенильную группу (циклопропенил, циклопентенил, циклогексенил и т.д.) или циклоалкинильную группу;
- термин “C3 - C8 циклическая углеводородная группа, насыщенная или ненасыщенная, линейная или разветвленная” означает любую неароматическую углеводородную группу, содержащую по меньшей мере один цикл, как описано у предыдущем абзаце, а также любую ароматическую углеводородную группу, содержащую по меньшей мере один цикл, содержащий от 3 до 8 атомов углерода и удовлетворяющий правилу ароматичности Хюккеля, т.е. число делокализованных π электронов равно 4n + 2. Указанные один или больше циклов могут быть разветвленными, где разветвленная цепь (цепи) содержат от 1 до 6 атомов углерода. Так, эта группа может, в частности, представлять собой циклоалкильную группу, циклоалкенильную группу, циклоалкинильную группу или циклоароматическую группу, такую как фенильная или бензильная группа.
Кроме того, выражение “от… до…”, которое применяется для определения диапазонов и которое используется далее в настоящем тексте, следует понимать как определяющее не только величину интервала, но также значения границ интервала.
Авторы настоящего изобретения неожиданно обнаружили, что применение соединений, имеющих приведенную выше общую формулу (I), делает возможным эффективную и селективную экстракцию по меньшей мере одного присутствующего редкоземельного металла, и предпочтительно по меньшей мере одного присутствующего лантаноида, из водного раствора, содержащего, кроме того, фосфорную кислоту.
Несмотря на то, что некоторые из соединений, имеющих приведенную выше общую формулу (I), уже исследовались в тестах экстракции, как описано в публикации, обозначенной как [3], M. Iqbal et al. ("Synthesis and evaluation of ligands with mixed amide and phosphonate, phosphinoxide, and phosphonothioate sites for An(III)/Ln(III) extraction", New J. Chem., 2012, 36, 2048-2059), эти экстракции проводились в условиях, аналогичных условиям, описанным в публикациях [1] и [2], а именно из водных фаз, содержащих азотную кислоту, а не фосфорную кислоту.
В предпочтительном варианте, соединение, имеющее общую формулу (I), применение которого является предметом настоящего изобретения, представляет собой соединение, в которой n=0, и имеющее изображенную ниже общую формулу (I'):
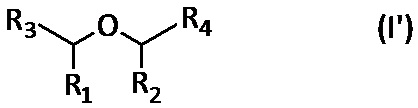
где R1, R2, R3 и R4 имеют указанные выше значения.
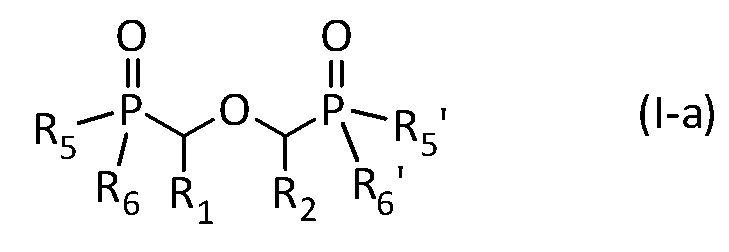
В первом более предпочтительном варианте, настоящее изобретение касается применения соединения, имеющего изображенную ниже общую формулу (I-a):
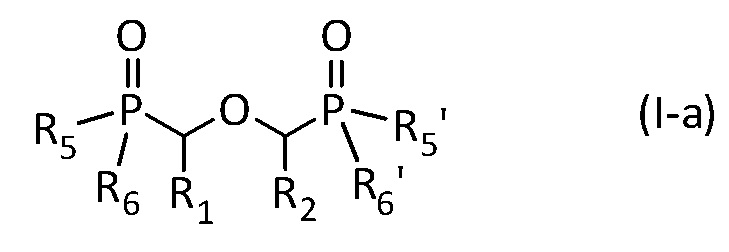
где R1, R2, R5, R6, R5' и R6' имеют указанные выше значения, при этом R5 и/или R6 представляют собой гидроксильную группу -OH.
В предпочтительном варианте, в приведенной выше формуле (I-a):
- один из R5 и R6 представляет собой гидроксильную группу -OH, и оставшийся из R5 и R6 представляет собой алкоксильную группу -OR, где R представляет собой C2 - C8, предпочтительно C4, алкильную группу, линейную или разветвленную, и
- R5' и R6' представляют собой, независимо друг от друга, C4 - C10, предпочтительно C8, алкильную группу, линейную или разветвленную.
Соединение, имеющее формулу (I-a), в которой R5, R6, R5' и R6' имеют указанные выше значения, соответствует дифункциональному соединению, содержащему фосфинатную группу -PO(OH)(OR) и фосфиноксидную группу -POR5'R6'.
Также в предпочтительном варианте, в приведенной выше формуле (I-a), и в первом альтернативном варианте осуществления, R1 и R2 каждый представляют собой атом водорода.
Как показано ниже в Примере 9, применение такого дифункционального соединения, содержащего фосфинатную группу и фосфиноксидную группу, в котором R1=R2=H, позволяет экстрагировать тяжелые редкоземельные металлы (такие как иттербий Yb) эффективным и особенно селективным образом, как в отношении редкоземельных металлов с низкими атомными номерами (таких как La, Nd, Gd и Dy), так и примесей (представленных во всех примерах железом Fe), и независимо от концентрации фосфорной кислоты.
Также в предпочтительном варианте, в приведенной выше формуле (I-a), и во втором альтернативном варианте осуществления, по меньшей мере один из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Так, и в первом аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-a), R1 и R2 каждый представляют собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-a), один из R1 и R2 представляет собой атом водорода, и оставшийся из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
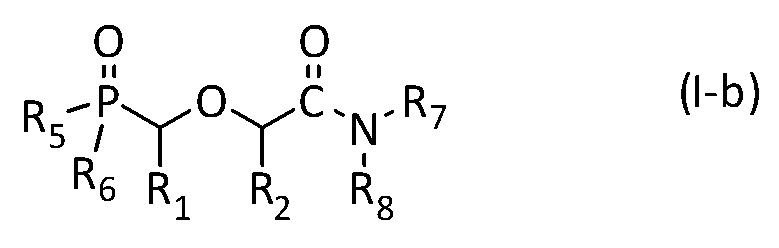
Во втором более предпочтительном варианте, настоящее изобретение касается применения соединения, имеющего изображенную ниже общую формулу (I-b):
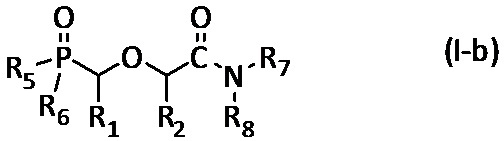
где R1, R2, R5, R6, R7 и R8 имеют указанные выше значения, при этом R5 и/или R6 представляют собой гидроксильную группу -OH.
В предпочтительном варианте, в приведенной выше формуле (I-b):
- один из R5 и R6 представляет собой гидроксильную группу -OH, и оставшийся из R5 и R6 представляет собой C4 - C10, предпочтительно C8, алкильную группу, линейную или разветвленную, или алкоксильную группу -OR, где R представляет собой C2 - C8, предпочтительно C4, алкильную группу, линейную или разветвленную, и
- R7 и R8 предпочтительно представляют собой, независимо друг от друга, C4 - C10, предпочтительно C8, алкильную группу, линейную или разветвленную.
Так, в случае, когда один из R5 и R6 представляет собой -OH, оставшийся из R5 и R6 представляет собой описанную выше алкильную группу, линейную или разветвленную, и R7 и R8 имеют указанные выше значения, соединение, имеющее формулу (I-b) соответствует дифункциональному соединению, содержащему фосфинатную группу -PO(OH)R6 или -PO(OH)R5 и амидную группу -COR7R8.
Так, в случае когда один из R5 и R6 представляет собой -OH, оставшийся из R5 и R6 представляет собой описанную выше алкоксильную группу -OR, и R7 и R8 имеют указанные выше значения, соединение, имеющее формулу (I-b) соответствует дифункциональному соединению, содержащему фосфонатную группу -PO(OH)(OR) и амидную группу -COR7R8.
Также в предпочтительном варианте, в приведенной выше формуле (I-b), и в первом альтернативном варианте осуществления, R1 и R2 каждый представляют собой атом водорода.
Как показано ниже в Примерах 11 и 12, применение такого дифункционального соединения, содержащего фосфонатную группу и амидную группу, в котором R1=R2=H, позволяет экстрагировать вместе все протестированные тяжелые редкоземельные металлы (Gd, Dy и Yb), а также некоторые легкие редкоземельные металлы (Nd, но не La), и независимо от концентрации фосфорной кислоты в водной фазе, кроме того экстракция Yb особенно селективна относительно примесей (Fe).
Также в предпочтительном варианте, в приведенной выше формуле (I-b), и во втором альтернативном варианте осуществления, по меньшей мере один из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Так, в первом аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-b), R1 и R2 каждый представляют собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-b), один из R1 и R2 представляет собой атом водорода, и оставшийся из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором аспекте указанного второго альтернативного варианта осуществления, соединение, имеющее формулу (I-b), применение которого является предметом настоящего изобретения, может содержать разветвление в альфа-положении относительно простоэфирной группы, либо со стороны фосфонатной группы (в этом случае R2=H), либо со стороны амидной группы (в этом случае R1=H).
В этом втором аспекте, соединение, имеющее формулу (I-b), предпочтительно таково, что R2 представляет собой атом водорода.
В типичном случае, и учитывая описанное выше, соединения, имеющие общую формулу (I) и, из их числа, соединения, имеющие общие формулы (I-a) и (I-b), применяют по настоящему изобретению в качестве экстрагирующих агентов для экстрагирования по меньшей мере одного редкоземельного металла из водной фазы, в которой присутствует указанный по меньшей мере один редкоземельный металл, где водная фаза содержит, кроме прочего, фосфорную кислоту.
В предпочтительном альтернативном варианте осуществления настоящего изобретения, водная фаза представляет собой раствор от кислотного гидролиза, осуществляемого обычно с применением одной или больше неорганических кислот, концентрата природной руды или твердых бытовых отходов, содержащий указанный по меньшей мере один редкоземельный металл. Присутствие фосфорной кислоты в данном растворе от кислотного гидролиза объясняется применением фосфорной кислоты в качестве неорганической кислоты и/или ее образованием in situ вследствие присутствия прекурсоров такой кислоты, таких как природные фосфаты.
Следует напомнить, что твердые бытовые отходы могут, в частности, являться продуктом переработки промышленных отходов, таких как отходы от электрического и электронного оборудования (3EW).
В другом предпочтительном альтернативном варианте осуществления настоящего изобретения, водная фаза содержит по меньшей мере 0.1 моль/л, предпочтительно от 0.2 моль/л до 8 моль/л, и более предпочтительно от 0.5 моль/л до 5 моль/л фосфорной кислоты.
В другом предпочтительном альтернативном варианте осуществления настоящего изобретения, экстрагирование проводят по методике экстрагирования жидкости жидкостью, в которой водную фазу, содержащую редкоземельный металл (металлы), предпочтительно лантаноид (лантаноиды), и фосфорную кислоту, вводят в контакт с органической фазой, содержащей одно или больше из указанных выше соединений, предпочтительно одно из указанных выше соединений.
В другом предпочтительном альтернативном варианте осуществления настоящего изобретения, экстрагирование жидкости жидкостью проводят с помощь органической фазы, содержащей по меньшей мере 10-3 моль/л, предпочтительно от 5⋅10-3 моль/л до 1 моль/л, и более предпочтительно от 10-2 моль/л до 10-1 моль/л соединения в растворе органического растворителя.
Указанный органический растворитель предпочтительно имеет алифатическую природу и может, в частности, представлять собой н-додекан, гидрированный тетрапропилен (HTP), керосин или изопарафин, такой как Isane®IP 185, поставляемый компанией Total.
Настоящее изобретение касается, во-вторых, способа извлечения по меньшей мере одного редкоземельного металла, и предпочтительно по меньшей мере одного лантаноида, присутствующего в водной фазе, дополнительно содержащей фосфорную кислоту.
По настоящему изобретению, данный способ извлечения включает следующие стадии:
a) экстрагирование по меньшей мере одного редкоземельного металла, предпочтительно по меньшей мере одного лантаноида, из водной фазы посредством введения указанной водной фазы в контакт с органической фазой, содержащей по меньшей мере одно описанное выше соединение, с последующим отделением водной фазы от органической фазы; и
b) выделение по меньшей мере одного редкоземельного металла, предпочтительно по меньшей мере одного лантаноида, в органической фазе, полученной после стадии a), посредством контакта указанной органической фазы с водной фазой, с последующим отделением органической фазы от водной фазы.
На стадии a) указанного способа по настоящему изобретению, соединение, присутствующее в органической фазе, представляет собой описанное выше соединение, при этом предпочтительные отличительные черты данного соединения могут браться по отдельности или в комбинации.
Аналогично, водные и органические фазы, применяемые на стадии a) способа по настоящему изобретению, могут быть такими как описано выше относительно применения указанного соединения, и могут иметь предпочтительные отличительные черты, описанные выше для данной водной и органической фаз, по отдельности или в комбинации.
В предпочтительном альтернативном варианте осуществления способа по настоящему изобретению, на стадии a) в водную фазу добавляют по меньшей мере одну соль, такую как нитратная или сульфатная соль щелочного или щелочно-земельного металла, которая позволяет повысить ионную силу водного раствора.
Введение такой соли улучшает экстракционные характеристики для всех редкоземельных металлов (см. примеры 11 и 12 далее). Такая соль может, в частности, быть выбрана из нитрата натрия, лития или калия. Предпочтительно, такая соль представляет собой нитрат натрия.
Молярная концентрация соли(солей) в водной фазе составляет от 0.01 моль/л до 4 моль/л, предпочтительно от 0.05 моль/л до 3 моль/л и, более предпочтительно, от 0.1 моль/л до 2 моль/л.
В альтернативном предпочтительном варианте осуществления способа по настоящему изобретению, водная фаза, применяющаяся на стадии a), представляет собой раствор от кислотного гидролиза, осуществляемого с применением фосфорной кислоты, концентрата природной руды или твердых бытовых отходов, содержащий по меньшей мере один редкоземельный металл.
Настоящее изобретение касается, в-третьих, соединений, которые, насколько известно авторам настоящего изобретения, до настоящего момента не были описаны.
В первом варианте, соединение по настоящему изобретению имеет изображенную ниже формулу (I-a):
где
- R1 и R2 представляют собой, независимо друг от друга, атом водорода, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную,
- один из R5 и R6 представляет собой гидроксильную группу -OH, и оставшийся из R5 и R6 представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, C3 - C8 циклическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, гидроксильную группу -OH или алкоксильную группу -OR, где R представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, и
- R5' и R6' представляют собой, независимо друг от друга, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, C3 - C8 циклическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, гидроксильную группу -OH или алкоксильную группу -OR', где R’ представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную.
В предпочтительном варианте, в приведенной выше формуле (I-a):
- один из R5 и R6 представляет собой гидроксильную группу -OH, и оставшийся из R5 и R6 представляет собой алкоксильную группу -OR, где R представляет собой C2 - C8, предпочтительно C4, алкильную группу, линейную или разветвленную, и
- R5' и R6' представляют собой, независимо друг от друга, C4 - C10, предпочтительно C8, алкильную группу, линейную или разветвленную.
Как уже указано выше, соединение, имеющее формулу (I-a), в которой R5, R6, R5' и R6' имеют указанные выше значения, соответствует дифункциональному соединению, содержащему фосфонатную группу -PO(OH)(OR) и фосфиноксидную группу -POR5'R6'.
Также в предпочтительном варианте, в приведенной выше формуле (I-a), и в первом альтернативном варианте осуществления, R1 и R2 каждый представляют собой атом водорода.
Также в предпочтительном варианте, в приведенной выше формуле (I-a), и во втором альтернативном варианте осуществления, по меньшей мере один из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Так, в первом аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-a), R1 и R2 каждый представляют собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-a), один из R1 и R2 представляет собой атом водорода, и оставшийся из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором варианте, соединение по настоящему изобретению имеет изображенную ниже формулу (I-b):
где
- R1 и R2 представляют собой, независимо друг от друга, атом водорода, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную,
- один из R5 и R6 представляет собой гидроксильную группу -OH, и оставшийся из R5 и R6 представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, C3 - C8 циклическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, гидроксильную группу -OH или алкоксильную группу -OR, где R представляет собой C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную, и
- R7 и R8 представляют собой, независимо друг от друга, атом водорода, C1 - C12 алифатическую углеводородную группу, насыщенную или ненасыщенную, линейную или разветвленную, или C3 - C8 циклическую алифатическую углеводородную группу, насыщенную или ненасыщенную, необязательно разветвленную.
В предпочтительном варианте, в приведенной выше формуле (I-b), R5 и R6 такие, что:
- один из R5 и R6 представляет собой гидроксильную группу -OH, и оставшийся из R5 и R6 представляет собой C4 - C10, предпочтительно C8, алкильную группу, линейную или разветвленную, или алкоксильную группу -OR, где R представляет собой C2 - C8, предпочтительно C4, алкильную группу, линейную или разветвленную, и
- R7 и R8 представляют собой, независимо друг от друга, C4 - C10, предпочтительно C8, алкильную группу, линейную или разветвленную.
Так, как уже указано выше, соединение, имеющее формулу (I-b) может соответствовать дифункциональному соединению, которое содержит:
- либо фосфинатную группу -PO(OH)R6 или -PO(OH)R5 и амидную группу -COR7R8, в случае где один из R5 и R6 представляет собой -OH, а оставшийся из R5 и R6 представляет собой описанную выше алкильную группу,
- либо фосфонатную группу -PO(OH)(OR) и амидную группу -COR7R8, в случае где один из R5 и R6 представляет собой -OH, а оставшийся из R5 и R6 представляет собой описанную выше алкоксильную группу -OR.
Также в предпочтительном варианте, в приведенной выше формуле (I-b) и в первом альтернативном варианте осуществления, R1 и R2 каждый представляют собой атом водорода.
Также в предпочтительном варианте, в приведенной выше формуле (I-b) и во втором альтернативном варианте осуществления, по меньшей мере один из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Так, в первом аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-b), R1 и R2 каждый представляют собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором аспекте указанного второго альтернативного варианта осуществления, в приведенной выше формуле (I-b), один из R1 и R2 представляет собой атом водорода, и оставшийся из R1 и R2 представляет собой C1 - C10, предпочтительно C1 - C8, алкильную группу, линейную или разветвленную.
Во втором аспекте указанного второго альтернативного варианта осуществления, соединение, имеющее формулу (I-b), применение которого является предметом настоящего изобретения, может содержать разветвление в альфа-положении относительно простоэфирной группы, либо со стороны фосфонатной группы (в этом случае R2=H), либо со стороны амидной группы (в этом случае R1=H).
В этом втором аспекте, соединение, имеющее формулу (I-b), предпочтительно такое, что R2 представляет собой атом водорода.
Другие отличительные особенности и преимущества настоящего изобретения станут понятны при прочтении приведенных ниже примеров, которые относятся к синтезу соединений по настоящему изобретению, а также к тестам, демонстрирующим способность этих соединений экстрагировать все или часть редкоземельных металлов из водных растворов, содержащих фосфорную кислоту, в которых содержатся редкоземельные металлы.
Следует отметить, что данные примеры приведены только в целях иллюстрации предмета настоящего изобретения и никоим образом не ограничивают объем настоящего изобретения.
Осуществление изобретения
Примеры
Синтез соединений по настоящему изобретению
Пример 1. Синтез бис-фосфиноксида
В данном синтезе реагирует спирт-фосфиноксид с хлорид-фосфиноксидом, по реакции Вильямсона, в присутствии гидрида натрия (NaH) в тетрагидрофуране (ТГФ).
Соответствующая реакция, обозначенная (1), выглядит следующим образом:
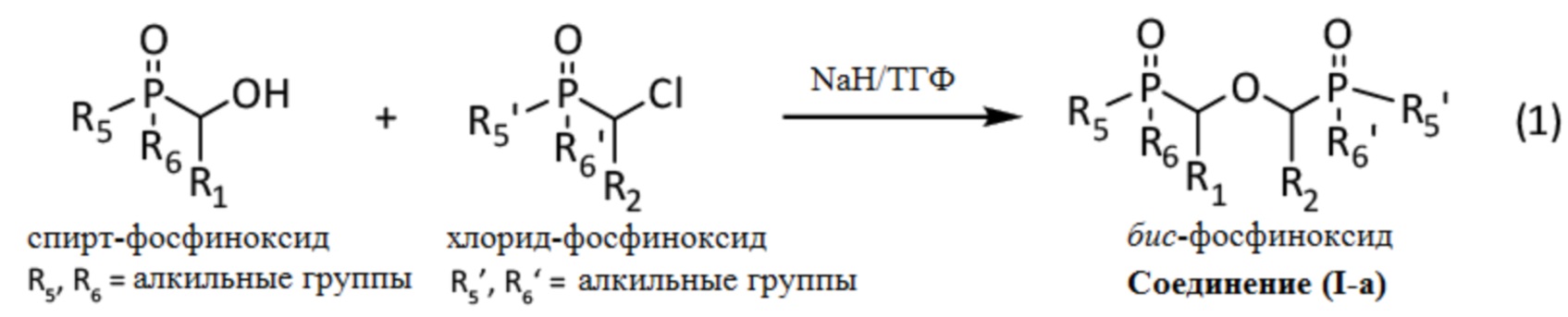
Более конкретно, целевой синтез представляет собой синтез бис-ди-октил фосфиноксида, который соответствует соединению, имеющему формулу (I-a), где R1 = R2 = H и R5 = R6 = R5' = R6' = н-C8H17 = н-октил (далее по тексту сокращенно именуется "Oct").
Бис-ди-октил фосфиноксид получают согласно приведенной ниже реакции (2):
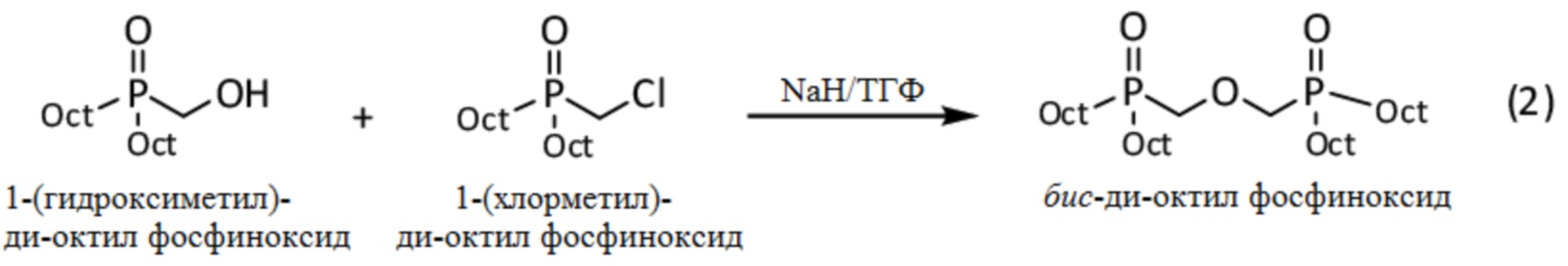
1.1. Синтез 1-(гидроксиметил)-ди-октил фосфиноксида
Синтез 1-(гидроксиметил)-ди-октил фосфиноксида проводят исходя из ди-октил фосфиноксида, а сам указанный ди-октил фосфиноксид синтезируют из фосфита.
1.1.1. Синтез ди-октил фосфиноксида
Ди-октил фосфиноксид синтезируют из диэтилфосфита, согласно изображенной ниже реакции (3):
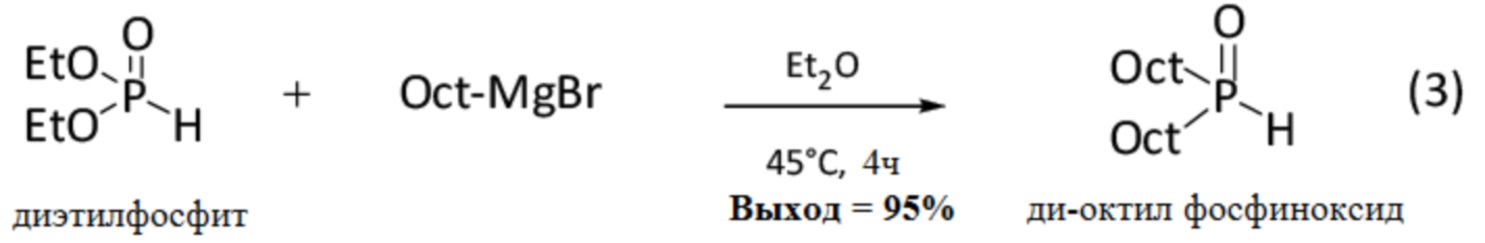
Методика проведения реакции (3) выглядит следующим образом: в 2 моль/л суспензию н-октилмагний бромида (обозначается Oct-MgBr) в эфире (100 мл, т.е. около 3 экв.) добавляют по каплям, при 0°C и при перемешивании, диэтилфосфит (1 экв., т.е. 13.5 г или 70 ммоль). По окончании добавления, смесь медленно доводят до комнатной температуры, затем до 45°C на 4 часа. Смесь затем подкисляют с помощью прикапывания 25%-ного водного раствора серной кислоты H2SO4 при 0°C. После окончания выделения газа, добавляют равное количество воды и эфира в полученную смесь. Органическую фазу затем отделяют и последовательно промывают 10%-ным раствором карбоната калия K2CO3 (2 раза), водой (2 раза), затем насыщенным раствором хлорида натрия (2 раза). Органическую фазу затем сушат над сульфатом натрия Na2SO4, фильтруют и упаривают, получая ди-октил фосфиноксид (95% выход), имеющий вид белого порошка.
Данные анализа для полученного ди-октил фосфиноксида приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 7.38 (д, 1H, P-H); 1.82-1.53 (м, 8H, CH2-CH2-P); 1.41-1.22 (м, 20H, CH2); 0.85 (м, 6H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 35.3.
1.1.2. Синтез 1-(гидроксиметил)-ди-октил фосфиноксида
1-(гидроксиметил)-ди-октил фосфиноксид синтезируют из ди-октил фосфиноксида, получение которого описано выше (см. 1.1.1), согласно изображенной ниже реакции (4):
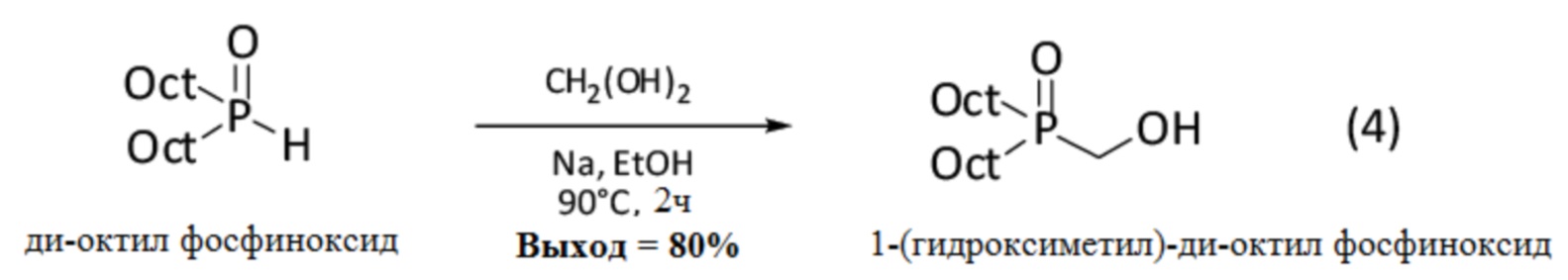
Методика проведения реакции (4) выглядит следующим образом: в раствор ди-октил фосфиноксида (1 экв.) в этаноле (0.5 моль/л) добавляют при перемешивании натрий Na (предварительно активированный в метаноле) затем параформальдегид (1.2 экв.). Смесь кипятят (80°C) в течение 2 часов. Растворитель затем упаривают. Смесь растворяют в дихлорметане (ДХМ), затем промывают водой (2 раза) и насыщенным водным раствором хлорида натрия (2 раза). Органическую фазу сушат над Na2SO4, фильтруют и упаривают, получая 1-(гидроксиметил)-ди-октил фосфиноксид (80% выход) в виде очень вязкого масла.
Данные анализа для полученного 1-(гидроксиметил)-ди-октил фосфиноксида приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 3.89 (с, 2H, CH2-OH); 1.80-1.72 (м, 4H, CH2-P); 1.64-1.55 (м, 4H, CH2-CH2-P); 1.42-1.27 (м, 20H, CH2); 0.90 (м, 6H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 49.1.
1.2. Синтез 1-(хлорметил)-ди-октил фосфиноксида
Синтез 1-(хлорметил)-ди-октил фосфиноксида проводят исходя из 1-(гидроксиметил)-ди-октил фосфиноксида, получение которого описано выше (см. 1.1.2), согласно изображенной ниже реакции (5):
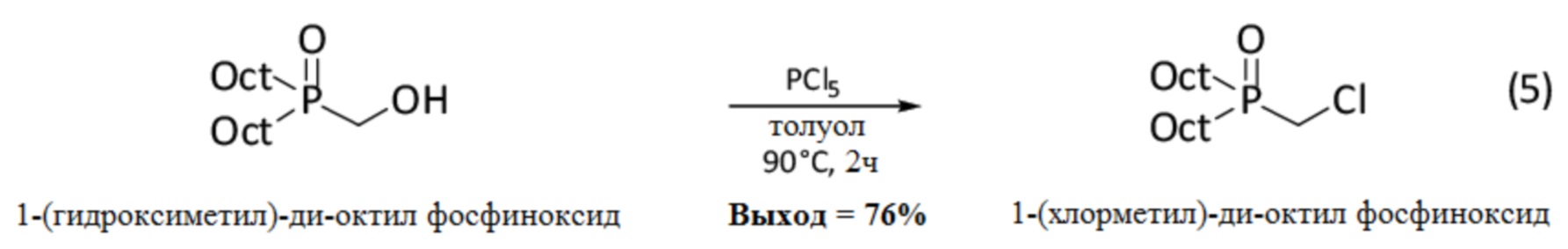
Методика проведения реакции (5) выглядит следующим образом: в раствор 1-(гидроксиметил)-ди-октил фосфиноксида (1 экв.) в безводном толуоле (0.5 моль/л) добавляют, при 0°C и при перемешивании, пентахлорид фосфора PCl5 (2 экв.) небольшими порциями. По окончании добавления, смесь медленно доводят до комнатной температуры и кипятят (110°C) в течение 2 часов. Избыток PCl5 нейтрализовывают прикапыванием воды при 0°C. По окончании выделения газа, добавляют воду в количестве, составляющем половину объема толуола, и смесь перемешивают в течение 10 минут. Органическую фазу затем отделяют и промывают раствором гидроксида натрия NaOH (0.5M) и насыщенным водным раствором хлорида натрия. Затем его сушат над Na2SO4, фильтруют и упаривают, получая 1-(хлорметил)-ди-октил фосфиноксид (76% выход) в виде вязкого масла.
Данные анализа для полученного 1-(хлорметил)-ди-октил фосфиноксида приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 3.58 (д, 2H, J = 8 Гц, CH2-Cl); 1.91-1.82 (м, 4H, CH2-P); 1.74-1.56 (м, 4H, CH2-CH2-P); 1.48-1.26 (м, 20H, CH2); 0.90 (м, 6H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 49.3.
1.3. Синтез бис-ди-октил фосфиноксида
Синтез бис-ди-октил фосфиноксида проводят посредством реакции 1-(гидроксиметил)-ди-октил фосфиноксида и 1-(хлорметил)-ди-октил фосфиноксида, синтез которых описан выше (см. выше соответственно 1.1 и 1.2), согласно изображенной ниже реакции (2'):
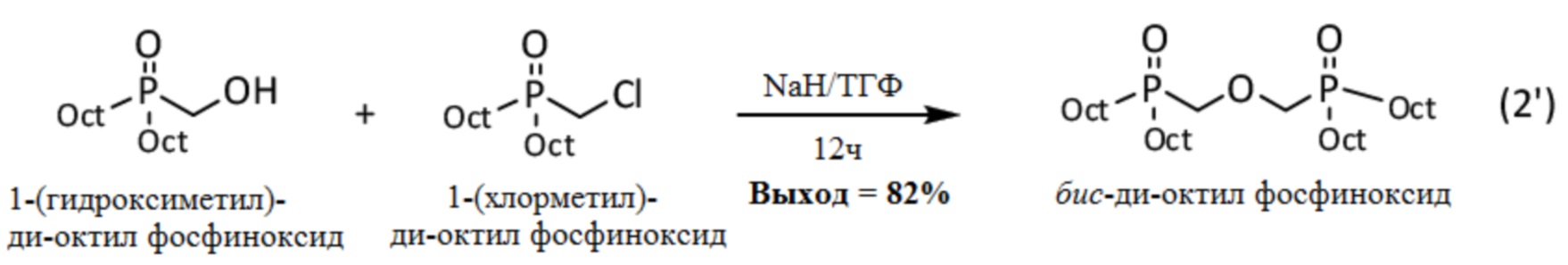
Методика проведения реакции (2') выглядит следующим образом: в суспензию гидрида натрия NaH (4 экв., предварительно промыт 2 раза пентаном) в безводном ТГФ (1 моль/л), добавляют по каплям раствор 1-(гидроксиметил)-ди-октил фосфиноксида в ТГФ (1 экв при 0.5 моль/л). По окончании добавления, смесь перемешивают в течение 1 часа, затем добавляют по каплям раствор 1-(хлорметил)-ди-октил фосфиноксида (1.2 экв.) в ТГФ. Смесь перемешивают в течение 12 часов. Полученный раствор затем подкисляют с помощью раствора хлористоводородной кислоты HCl (3M), который добавляют по каплям при 0°C. Растворитель упаривают. Полученный остаток растворяют в этилацетате, затем промывают раствором HCl (3M) (2 раза) и водой (2 раза). Органическую фазу сушат над Na2SO4, фильтруют и упаривают на роторном испарителе. Полученный сырой продукт очищают перекристаллизацией из этилацетата, получая бис-ди-октил фосфиноксид (82% выход), имеющий вид белого порошка.
Данные анализа для полученного бис-ди-октил фосфиноксида приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 3.90 (д, 4H, J = 6.4 Гц, CH2-O); 1.78-1.57 (м, 16H, CH2-CH2-P); 1.42-1.27 (м, 40H, CH2); 0.90 (м, 12H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 46.1.
Пример 2. Синтез фосфонат-фосфиноксида
В данном синтезе спирт-фосфонат реагирует с хлорид-фосфиноксидом по реакции Вильямсона, в присутствии гидрида натрия (NaH) и иодида калия (KI) в тетрагидрофуране (ТГФ).
Соответствующая реакция, обозначенная (1бис), выглядит следующим образом, при этом одна из алкоксильных групп R5 и R6 во время реакции гидролизуется:

Более конкретно, целью синтеза является бутил (((диоктаноилфосфорил)-2-оксоэтокси)метил)-фосфонат, который соответствует соединению, имеющему формулу (I-a), в которой R1 = R2 = H, один из R5 и R6 представляет собой -OH, а оставшийся из R5 и R6 представляет собой н-C4H9O (далее сокращенно обозначается “BuO”) и R5' = R6'= н-октил (далее сокращенно обозначается "Oct").
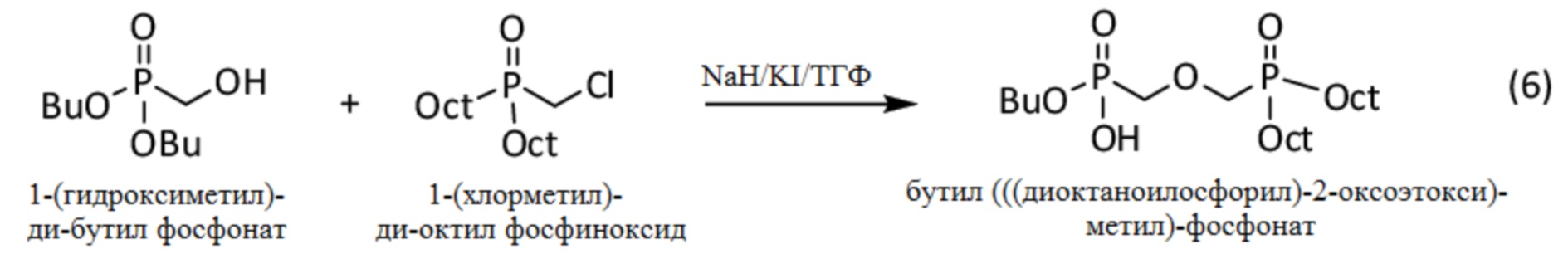
Бутил (((диоктаноилфосфорил)-2-оксоэтокси)метил)-фосфонат получают согласно приведенной ниже реакции (6):
2.1. Синтез 1-(гидроксиметил)-ди-бутил-фосфоната
Синтез 1-(гидроксиметил)-ди-бутил-фосфоната проводят исходя из фосфита, в данном случае дибутил фосфита, согласно изображенной ниже реакции (7):
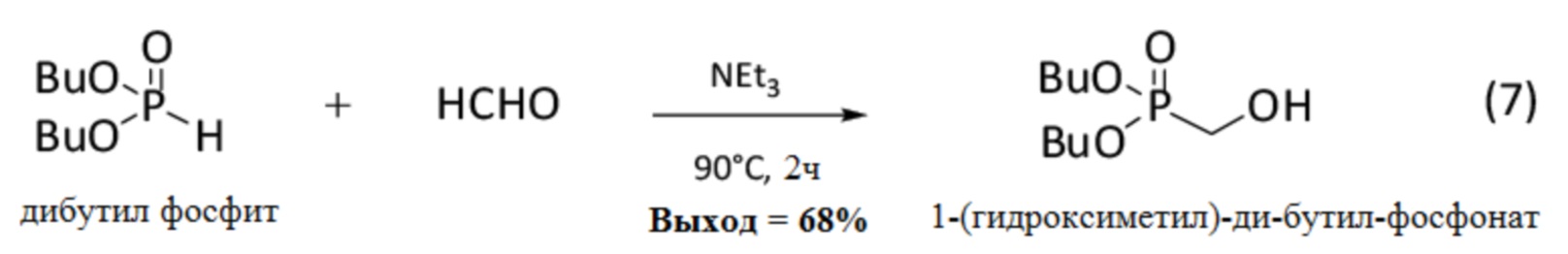
Методика проведения реакции (7) выглядит следующим образом: смесь дибутил фосфита (1 экв.) и формальдегида (1.2 экв.) в триэтиламине NEt3 кипятят (90°C) при перемешивании в течение 2 часов. Затем триэтиламин упаривают. Остаток растворяют в ДХМ, затем промывают насыщенным раствором NaHCO3 (2 раза) и насыщенным водным раствором хлорида натрия (2 раза). Органическую фазу сушат над Na2SO4 и упаривают на роторном испарителе. Избыток формальдегида отгоняют в термошкафу при пониженном давлении. Полученный сырой продукт очищают методом флэш-хроматографии на колонке с силикагелем (элюент: циклогексан/этилацетат от 100/50 до 50/100, об/об), получая 1-(гидроксиметил)-ди-бутил-фосфонат (68% выход) в виде вязкого масла.
Данные анализа для полученного 1-(гидроксиметил)-ди-бутил-фосфоната приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.84 (с, 1H, OH); 4.14-4.08 (м, 2H, O-CH2); 3.92 (д, 4H, J = 6 Гц, CH2-OH); 1.71-1.64 (м, 4H, O-CH2-CH2); 1.47-1.37 (м, 4H, O-CH2-CH2-CH2); 0.95 (т, 6H, J = 7.4 Гц, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 24.5.
2.2. Синтез 1-(хлорметил)-ди-октил фосфиноксида
Синтез 1-(хлорметил)-ди-октил фосфиноксида проводят согласно реакции (5) и описанной выше в пункте 1.2 методике.
2.3. Синтез бутил (((диоктаноилфосфорил)-2-оксоэтокси)метил)-фосфоната
Синтез бутил (((диоктаноилфосфорил)-2-оксоэтокси)метил)-фосфоната проводят посредством реакции 1-(гидроксиметил)-ди-бутил фосфоната и 1-(хлорметил)-ди-октил фосфиноксида, синтез которых описан выше (см. выше соответственно 2.1 и 1.2), согласно изображенной ниже реакции (6'):
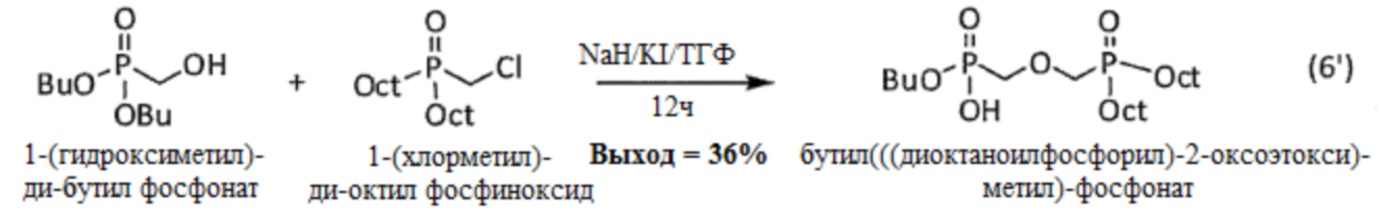
Методика проведения реакции (6') выглядит следующим образом: в суспензию гидрида натрия NaH (4 экв., предварительно промыт 2 раза пентаном) в безводном ТГФ (1 моль/л) в присутствии иодида калия KI (1 экв.) добавляют по каплям раствор 1-(гидроксиметил)-ди-бутил фосфоната в ТГФ (1 экв при 0.5 моль/л). По окончании добавления смесь перемешивают в течение 1 часа, затем добавляют по каплям раствор 1-(хлорметил)-ди-октил фосфиноксида (1.2 экв.) в ТГФ. Смесь перемешивают в течение 12 часов. Реакционную смесь подкисляют с помощью раствора хлористоводородной кислоты HCl (3M), который прикапывают при 0°C. Растворитель затем упаривают. Остаток от упаривания растворяют в этилацетате и промывают раствором HCl (3M) (2 раза) и водой (2 раза). Органическую фазу сушат над Na2SO4, фильтруют и упаривают на роторном испарителе. Полученный сырой продукт затем очищают перекристаллизацией из этилацетата, получая бутил (((диоктаноилфосфорил)-2-оксоэтокси)метил)-фосфонат (36% выход), имеющий вид белого порошка.
Данные анализа для полученного бутил (((диоктаноилфосфорил)-2-оксоэтокси)метил)-фосфоната приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 9.68 (с, 1H, OH); 4.17-4.05 (м, 4H, CH2-O и O-CH2-P-O); 3.95 (д, 2H, J = 7.6 Гц, P-CH2-O); 1.86-1.78 (м, 4H, CH2-P); 1.71-1.55 (м, 6H, CH2-CH2-P и O-CH2-CH2); 1.47-1.25 (м, 22H, CH2); 0.97-0.87 (м, 9H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 51.9; 19.8.
HRMS (EI+): вычислено для C22H48O5P2: 454.3055; найдено: 454.3055.
Пример 3. Синтез фосфината
Этил октаноил-фосфинат или октил этоксифосфинат синтезируют из триэтилфосфита, согласно изображенной ниже реакции (8):
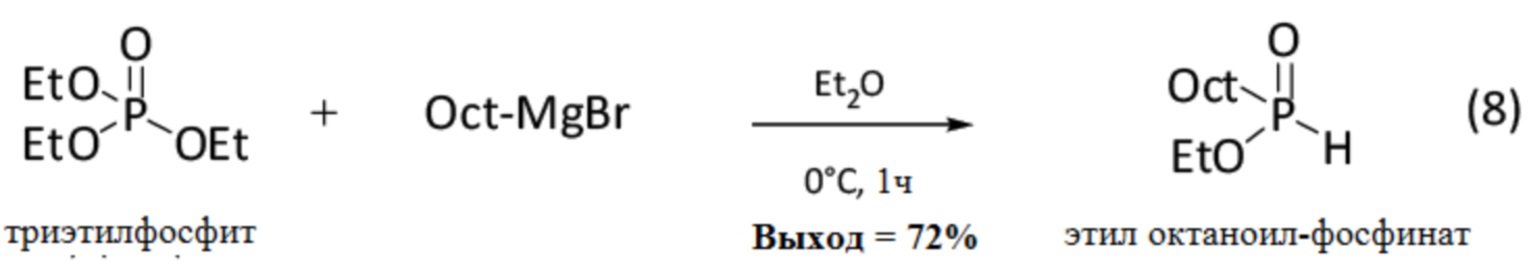
Методика проведения реакции (8) выглядит следующим образом: в 2 моль/л суспензию н-октил магнийбромида (обозначается Oct-MgBr) в эфире (20 мл, т.е. около 1.5 экв.) добавляют по каплям, в течение 1 часа, при 0°C и в атмосфере аргона, триэтилфосфит (10 мл или 57.5 ммоль). По окончании добавления смесь интенсивно перемешивают, затем добавляют 1M раствор HCl (50 мл) до полного растворения солей (необязательно может потребоваться добавление эфира). Смесь медленно доводят до комнатной температуры, затем выдерживают при 45°C в течение 4 часов. Смесь экстрагируют 100 мл эфира (2 раза). Органическую фазу затем отделяют и последовательно промывают с использованием 100 мл 1M раствора HCl, 100 мл воды, затем 100 мл насыщенного водного раствора хлорида натрия. Органическую фазу затем сушат над MgSO4, и растворитель упаривают. После очистки методом флэш-хроматографии на колонке с силикагелем (элюент: циклогексан/этилацетат), получают этил октаноил-фосфинат (72% выход) в виде бесцветного масла.
Данные анализа для полученного этил октаноил-фосфината приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 7.04 (д, 1H, P-H); 4.22-4.05 (м, 2H, O-CH2); 1.81-1.73 (м, 2H, P-CH2); 1.65-1.54 (м, 2H, P-CH2-CH2); 1.42-1.24 (м, 13H, CH2 и O-CH2-CH3); 0.89 (т, 3H, J = 7 Гц, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 39.0.
Пример 4. Синтез спирт-фосфонатов
Спирт-фосфонат можно получить реакцией фосфита с альдегидом согласно изображенной ниже реакции (9):
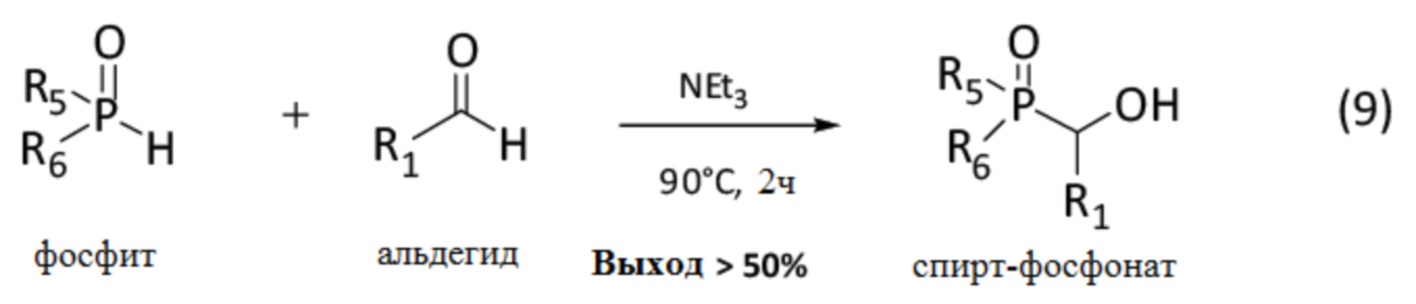
Так, как уже было показано вые в разделе 2.1, 1-(гидроксиметил)-ди-бутил фосфонат синтезировали реакцией дибутил фосфита (R5 = R6 = C4H9O) с формальдегидом (R1 = H).
Другие спирт-фосфонаты синтезировали согласно следующей методике: смесь фосфита (1 экв.) и альдегида (1.2 экв.) в триэтиламине кипятили (90°C) при перемешивании в течение 2 часов. Триэтиламин затем упаривают. Остаток от упаривания растворяют в ДХМ, затем промывают насыщенным раствором NaHCO3 (2 раза) и насыщенным водным раствором хлорида натрия (2 раза). Органическую фазу сушат над Na2SO4, после чего упаривают на роторном испарителе. Избыток формальдегида упаривают в термошкафу при пониженном давлении. Полученный сырой продукт затем очищают методом флэш-хроматографии на колонке с силикагелем (элюент: циклогексан/этилацетат от 100/50 до 50/100, об/об) получая соответствующий спирт-фосфонат (с выходом выше 50%) в виде вязкого масла или белого порошка, в зависимости от конкретного случая.
4.1. Синтез 1-(гидроксинонил)-ди-бутил-фосфоната
1-(гидроксинонил)-ди-бутил-фосфонат представляет собой спирт-фосфонат, в котором R5 = R6 = н-C4H9O (BuO), и R1 = н-C8H17 (Oct). Таким образом, он имеет следующую формулу:
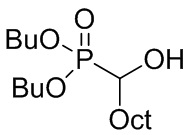
Данные анализа для полученного соединения, синтезированного с 58% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.13-4.04 (м, 4H, O-CH2); 3.85-3.80 (м, 1H, CH); 1.82-1.59 (м, 6H, CH2); 1.42-1.24 (м, 16H, CH2); 0.93-0.84 (м, 9H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 25.4.
4.2. Синтез 1-(гидроксиэтил)-ди-бутил-фосфоната
1-(гидроксиэтил)-ди-бутил-фосфонат представляет собой спирт-фосфонат, в котором R5 = R6 = н-C4H9O (BuO), и R1 = CH3. Таким образом, он имеет следующую формулу:
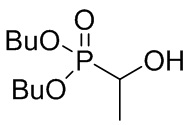
Данные анализа для полученного соединения, синтезированного с 54% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.87 (с, 1H, OH); 4.07-3.95 (м, 5H, CH и O-CH2); 1.62-1.54 (м, 4H, O-CH2-CH2); 1.38-1.28 (м, 7H, O-CH2-CH2-CH2 и CH3); 0.86 (т, 6H, J = 7.4 Гц, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 25.8.
4.3. Синтез 1-(гидроксиметил)-ди-этилгексил-фосфоната
1-(гидроксиметил)-ди-этилгексил-фосфонат представляет собой спирт-фосфонат, в котором R5 = R6 = 2-этилгексил (обозначается EtHex), и R1 = H. Таким образом, он имеет следующую формулу:
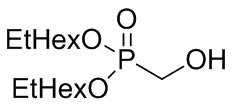
Данные анализа для полученного соединения, синтезированного с 78% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.04-3.98 (м, 4H, O-CH2); 3.92 (д, 2H, J = 6 Гц, CH2-OH); 1.60-1.54 (м, 2H, CH); 1.44-1.28 (м, 16H, CH2); 0.91 (т, 6H, J = 7.2 Гц, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 24.2.
Пример 5. Синтез спирт-фосфината
Спирт-фосфинат можно получить реакцией фосфината с альдегидом согласно изображенной ниже реакции (9'):
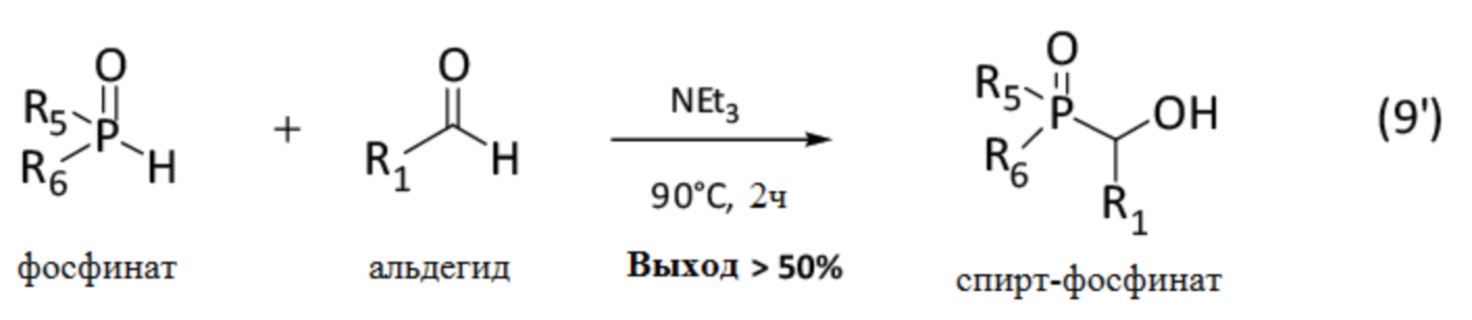
Так, 1-(гидрометил)-этил октаноил-фосфината получают реакцией этилоктаноил-фосфината, синтезированного согласно Примеру 3, с формальдегидом.
1-(гидроксиметил)-этилоктаноил-фосфинат представляет собой спирт-фосфинат, в котором R5 = н-C8H17 (Oct), R6 = C2H5O (EtO), и R1 = H. Таким образом, он имеет следующую формулу:
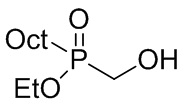
Данные анализа для полученного соединения, синтезированного с 38% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.98 (с, 1H, OH); 4.14-4.08 (м, 2H, O-CH2); 3.88-3.84 (м, 2H, CH2-OH); 1.84-1.77 (м, 2H, P-CH2); 1.64-1.56 (м, 2H, P-CH2-CH2); 1.40-1.27 (м, 13H, CH2 и O-CH2-CH3); 0.88 (т, 3H, J = 7.2 Гц, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 25.8.
Пример 6. Синтез амидо-фосфонатов
Синтез амидо-фосфоната проводят реакцией спирт-фосфоната с гадлогенид-амидом по реакции Вильямсона, в присутствии гидрида натрия (NaH) и иодида калия (KI) в тетрагидрофуране (ТГФ).
Соответствующая реакция, обозначенная (10), выглядит следующим образом, при этом одна из алкоксильных групп R5 и R6 гидролизуется в ходе реакции:
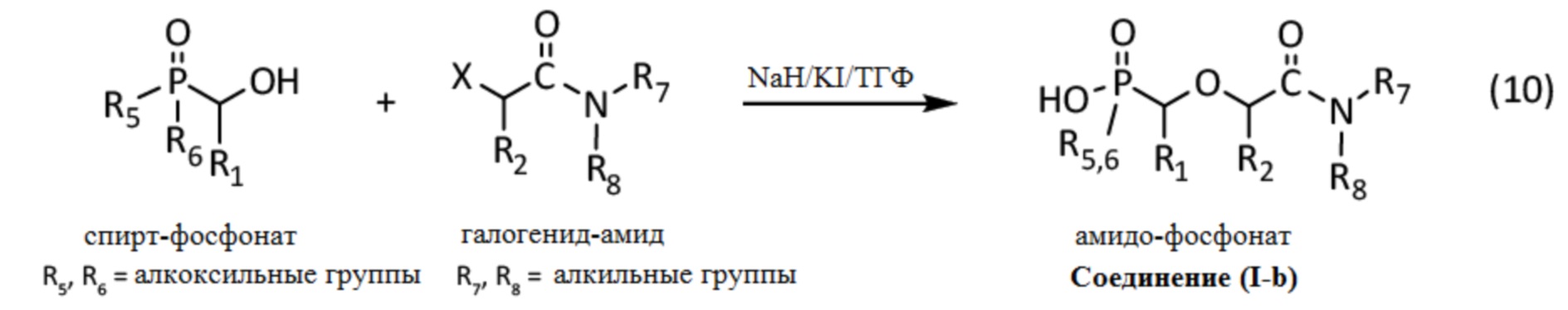
Далее будет описан синтез нескольких амидо-фосфонатов. Все эти синтезы проводили согласно следующей методике: в суспензию NaH (4 экв., предварительно промыт 2 раза пентаном) в безводном ТГФ (1 моль/л) добавляют при перемешивании KI (1 экв.). Суспензию охлаждают до 0°C и добавляют по каплям раствор спирт-фосфоната в ТГФ (1 экв, 0.5 моль/л). По окончании добавления смесь перемешивают в течение 1 часа при 0°C, затем добавляют по каплям раствор галогенид-амида (1.2 экв.) в ТГФ. Смесь перемешивают в течение 6 часов. Затем раствор подкисляют насыщенным раствором хлорида аммония (NH4Cl), который добавляют по каплям при 0°C. По окончании выделения газа, половину объема полученного раствора добавляют в ТГФ, и смесь перемешивают в течение 15 минут. Растворитель затем упаривают. Остаток от упаривания растворяют в этилацетате, промывают насыщенным раствором NH4Cl (2 раза) и водой (2 раза). Органическую фазу сушат над Na2SO4, упаривают на роторном испарителе. Полученный сырой продукт очищают методом флэш-хроматографии на колонке с силикагелем (элюент: этилацетат/метанол от 100/0 до 80/20, об/об) получая соответствующий амидо-фосфонат в виде вязкого масла.
6.1. Синтез бутил (((N,N-диоктилкарбамоил)-2-оксоэтокси)метил)-фосфоната
Синтез бутил (((N,N-диоктилкарбамоил)-2-оксоэтокси)метил)-фосфоната проводят реакцией спирт-фосфоната, в котором R5 = R6 = н-C4H9O (BuO), и R1 = H (см. 2.1 выше), с галогенид-амидом, в котором X = Cl, R7 = R8 = н-C8H17 (Oct), и R2 = H.
Соответствующая реакция, обозначенная (11), выглядит следующим образом:
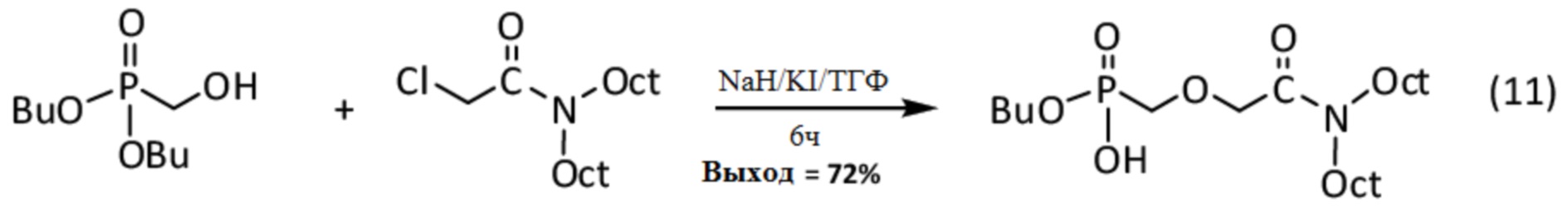
Данные анализа для полученного амидо-фосфоната, синтезированного с 72% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 10.9 (с, 1H, OH); 4.34 (с, 2H, CO-CH2-O); 4.14-4.08 (м, 2H, O-CH2); 3.95 (д, 2H, J = 8 Гц, P-CH2-O); 3.30 (т, 2H, J = 7.8 Гц, CH2-N); 3.13 (т, 2H, J = 7.8 Гц, CH2-N); 1.70-1.63 (м, 2H, O-CH2-CH2); 1.58-1.50 (м, 4H, CH2-CH2-N); 1.44-1.37 (м, 2H, O-CH2-CH2-CH2); 1.32-1.22 (м, 20H, CH2); 0.94-0.86 (м, 9H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 21.5.
HRMS (EI+): вычислено для C23H48NO5P: 449.3348; найдено: 449.3348.
6.2. Синтез бутил (((N,N-диэтилгексилкарбамоил)-2-оксоэтокси)метил)-фосфоната
Синтез бутил (((N,N-диэтилгексилкарбамоил)-2-оксоэтокси)метил)-фосфоната осуществляют реакцией спирт-фосфоната, в котором R5 = R6 = н-C4H9O (BuO), и R1 = H (см. 2.1 выше), с галогенид-амидом, в котором X = Cl, R7 = R8 = 2-этилгексил (обозначается EtHex или HexEt), и R2 = H.
Соответствующая реакция, обозначенная (12), выглядит следующим образом:
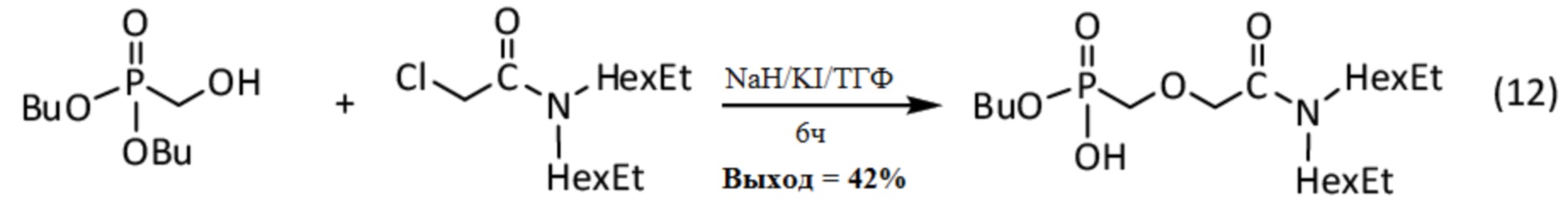
Данные анализа для полученного амидо-фосфоната, синтезированного с 42% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 7.96 (с, 1H, OH); 4.37 (с, 2H, CO-CH2-O); 4.17-4.07 (м, 2H, O-CH2); 3.92 (д, 2H, J = 8 Гц, P-CH2-O); 3.40-3.20 (м, 2H, CH2-N); 3.06-3.0 (м, 2H, CH2-N); 1.72-1.56 (м, 4H, O-CH2-CH2 и CH); 1.47-1.37 (м, 2H, O-CH2-CH2-CH2); 1.36-1.18 (м, 16H, CH2); 0.96-0.86 (м, 15H, CH3)
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 20.6.
HRMS (EI+): вычислено для C23H48NO5P: 449.3348; найдено: 449.3348.
6.3. Синтез бутил (((N,N-диэтилгексилкарбамоил)-2-оксоэтокси)нонил)-фосфоната
Синтез бутил (((N,N-диэтилгексилкарбамоил)-2-оксоэтокси)нонил)-фосфоната осуществляют реакцией спирт-фосфоната, в котором R5 = R6 = н-C4H9O (BuO), и R1 = н-C8H17 (Oct) (см. 4.1 выше), с галогенид-амидом, в котором X = Cl, R7 = R8 = н-C8H17 (Oct), и R2 = H.
Соответствующая реакция, обозначенная (13), выглядит следующим образом:
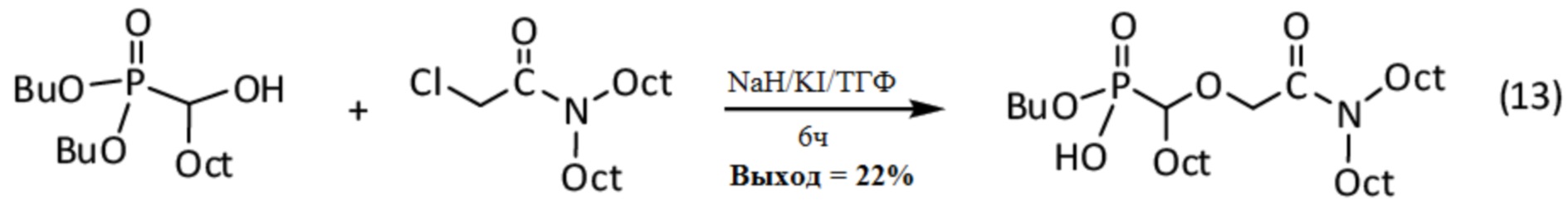
Данные анализа для полученного амидо-фосфоната, синтезированного с 22% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 9.85 (с, 1H, OH); 4.61 (д, 1H, J = 15.4 Гц, CO-CH2-O); 4.19 (д, 1H, J = 15.4 Гц, CO-CH2-O); 4.16-4.07 (м, 2H, O-CH2); 3.62-3.58 (м, 1H, P-CH-O); 3.31 (т, 2H, J = 7.8 Гц, CH2-N); 3.11 (т, 2H, J = 7.8 Гц, CH2-N); 1.96-1.74 (м, 2H, CH2-CH-P); 1.70-1.60 (м, 2H, O-CH2-CH2); 1.59-1.47 (м, 4H, CH2-CH2-N); 1.45-1.35 (м, 2H, O-CH2-CH2-CH2); 1.34-1.20 (м, 32H, CH2); 0.94-0.86 (м, 12H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 22.1.
HRMS (EI+): вычислено для C31H64NO5P: 562.4600; найдено: 562.4604.
6.4. Синтез бутил (((N,N-диэтилгексилкарбамоил)-2-оксоэтокси)этил)-фосфоната
Синтез бутил (((N,N-диэтилгексилкарбамоил)-2-оксоэтокси)этил)-фосфоната осуществляют реакцией спирт-фосфоната, в котором R5 = R6 = н-C4H9O (BuO), и R1 = CH3 (см. 4.2 выше), с галогенид-амидом, в котором X = Cl, R7 = R8 = н-C8H17 (Oct), и R2 = H.
Соответствующая реакция, обозначенная (14), выглядит следующим образом:
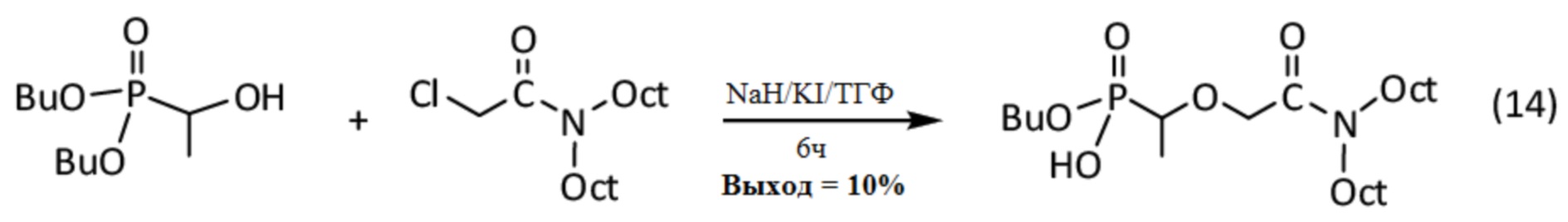
Данные анализа для полученного амидо-фосфоната, синтезированного с 10% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.48 (д, 1H, J = 16 Гц, CO-CH2-O); 4.19 (д, 1H, J = 15.4 Гц, CO-CH2-O); 4.21-4.10 (м, 3H, CO-CH2-O и O-CH2); 3.77-3.71 (м, 1H, P-CH-O); 3.33 (т, 2H, J = 7.6 Гц, CH2-N); 3.10 (т, 2H, J = 7.6 Гц, CH2-N); 1.73-1.65 (м, 2H, O-CH2-CH2); 1.60-1.38 (м, 9H, CH2-CH2-N и O-CH2-CH2-CH2 и CH3-CH); 1.37-1.21 (м, 20H, CH2); 0.97-0.87 (м, 9H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 22.3.
HRMS (EI+): вычислено для C24H50NO5P: 463.3505; найдено: 463.3507.
6.5. Синтез бутил (((N,N-диэтилгексилкарбамоил)-1-оксопропан-2-ил)метил)-фосфоната
Синтез бутил (((N,N-диэтилгексилкарбамоил)-1-оксопропан-2-ил)метил)-фосфоната осуществляют реакцией спирт-фосфоната, в котором R5 = R6 = н-C4H9O (BuO), и R1 = H (см. 2.1 выше), с галогенид-амидом, в котором X = Br, R7 = R8 = н-C8H17 (Oct), и R2 = CH3.
Соответствующая реакция, обозначенная (15), выглядит следующим образом:
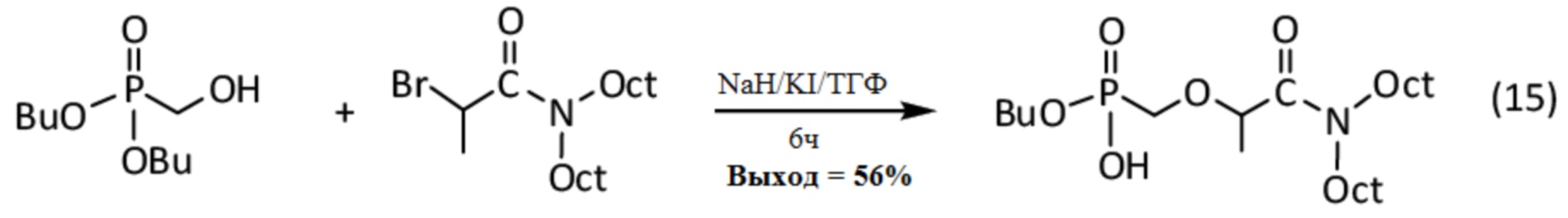
Данные анализа для полученного амидо-фосфоната, синтезированного с 56% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 10.6 (с, 1H, OH); 4.47-4.42 (м, 1H, CH); 4.14-4.08 (м, 2H, O-CH2); 3.76 (дд, 2H, J = 8.8 и 3.8 Гц, P-CH2-O); 3.49-3.41 (м, 1H, CH2-N); 3.30-3.16 (м, 3H, CH2-N); 1.71-1.64 (м, 2H, O-CH2-CH2); 1.61-1.48 (м, 4H, CH2-CH2-N); 1.47-1.37 (м, 5H, O-CH2-CH2-CH2 и CH3-CH); 1.34-1.24 (м, 20H, CH2); 0.96-0.86 (м, 9H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 21.8.
HRMS (EI+): вычислено для C24H50NO5P: 463.3505; найдено: 463.3507.
6.6. Синтез этилгексил (((N,N-диоктилкарбамоил)-2-оксоэтокси)метил)-фосфоната
Синтез этилгексил (((N,N-диоктилкарбамоил)-2-оксоэтокси)метил)-фосфоната осуществляют реакцией спирт-фосфоната, в котором R5 = R6 = 2-этилгексил (обозначается EtHex или HexEt), и R1 = H (см. 4.3 выше), с галогенид-амидом, в котором X = Cl, R7 = R8 = н-C8H17 (Oct), и R2 = H.
Соответствующая реакция, обозначенная (16), выглядит следующим образом:
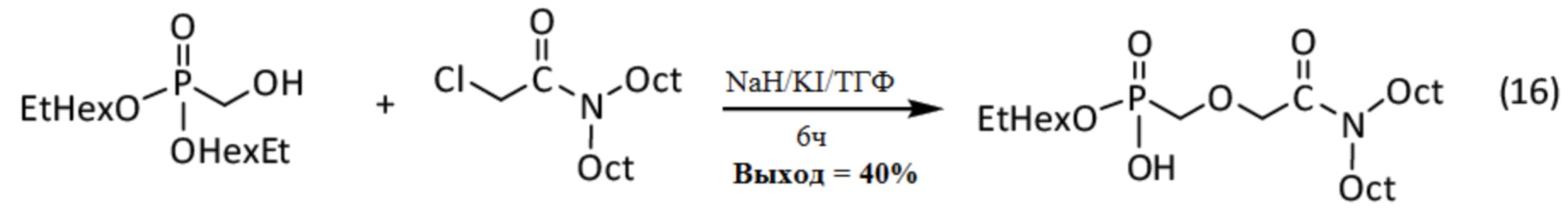
Данные анализа для полученного амидо-фосфоната, синтезированного с 40% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 10.4 (с, 1H, OH); 4.35 (с, 2H, CO-CH2-O); 4.08-3.98 (м, 2H, O-CH2); 3.96 (д, 2H, J = 8 Гц, P-CH2-O); 3.32 (т, 2H, J = 7.6 Гц, CH2-N); 3.12 (т, 2H, J = 7.6 Гц, CH2-N); 1.63-1.49 (м, 5H, CH2-CH2-N и CH); 1.48-1.23 (м, 28H, CH2); 0.92-0.88 (м, 12H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 21.2.
HRMS (EI+): вычислено для C27H56NO4P: 489.4025; найдено: 489.4024.
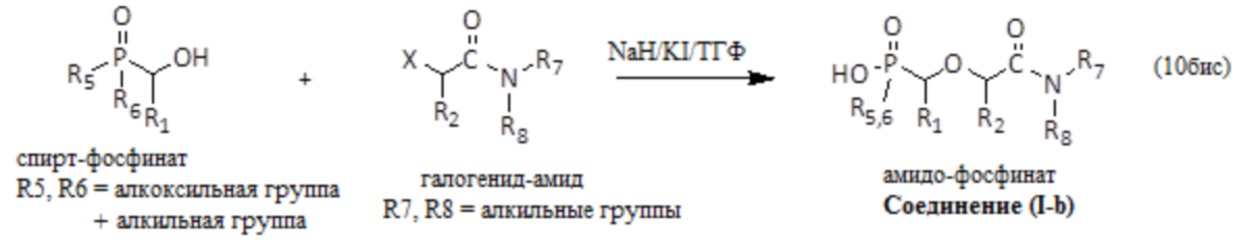
Пример 7. Синтез амидо-фосфината
Синтез амидо-фосфината осуществляют реакцией спирт-фосфината с галогенид-амидом по реакции Вильямсона, в присутствии гидрида натрия (NaH) и иодида калия (KI) в тетрагидрофуране (ТГФ).
Соответствующая реакция, обозначенная (10бис), выглядит следующим образом, при этом алкоксильная группа из числа R5 и R6 гидролизуется в ходе реакции:
Данная реакция проиллюстрирована синтезом октил (((N,N-диоктилкарбамоил)-2-оксоэтокси)метил)-фосфината, который получают реакцией спирт-фосфината, в котором R5(или R6) = н-C8H17 (Oct), R6(или R5) = C2H5O (EtO), и R1 = H (см. Пример 5 выше), с галогенид-амидом, в котором X = Cl, R7 = R8 = н-C8H17 (Oct), и R2 = H.
Данный синтез проводят согласно методике, описанной выше в разделе 6, для синтеза амидо-фосфонатов, при этом в данном случае раствор спирт-фосфоната в ТГФ заменяют на раствор спирт-фосфината.
Соответствующая реакция, обозначенная (17), выглядит следующим образом:
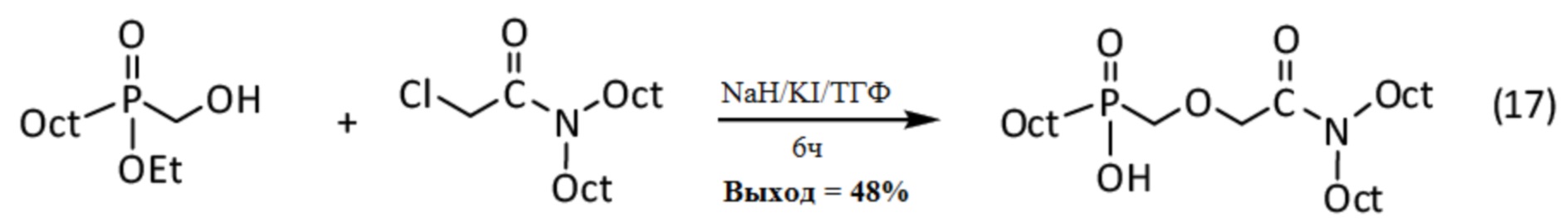
Данные анализа для полученного амидо-фосфоната, синтезированного с 48% выходом, приведены далее:
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 4.34 (с, 2H, CO-CH2-O); 3.88 (д, 2H, J = 6 Гц, P-CH2-O); 3.32 (т, 2H, J = 7.8 Гц, CH2-N); 3.12 (т, 2H, J 7, Гц, CH2-N); 1.88-1.80 (м, 2H, CH2-P); 1.72-1.62 (м, 2H, CH2-CH2-P); 1.61-1.49 (м, 4H, CH2-CH2-N); 1.44-1.20 (м, 28H, CH2); 0.93-0.88 (м, 9H, CH3).
31P ЯМР (160 МГц, CDCl3) δ (м.д.): 48.6.
HRMS (EI+): вычислено для C27H56NO4P: 489.4025; найдено: 489.4024.
Экстрагирующие свойства соединений по настоящему изобретению
Методики определения экстрагирующих свойств
Экстрагирующие свойства описанных соединений оценивали посредством измерения коэффициентов распределения веществ в растворе, с помощью ICP-OES спектрометрии (индуктивно-связанная плазма - оптическая эмиссионная спектрометрия), после разбавления водных растворов до содержания в диапазоне измерений (между 0 и 20 м.д.), до и после контакта с органической фазой.
* Если не указано иное или при экстракции не появляется третьей фазы, коэффициент распределения металлического элемента M между органической фазой и водной фазой, обозначаемый DM, определяют по следующему уравнению:
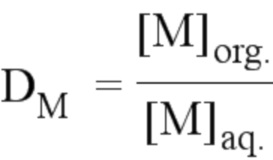
где [M]org. = концентрация металлического элемента в органической фазе при экстракционном равновесии (в мг/л), и
[M]aq. = концентрация металлического элемента в водной фазе при экстракционном равновесии (в мг/л).
* Селективность экстракции систематически оценивают по коэффициенту селективности, обозначаемому FS, и определяют согласно следующему уравнению:
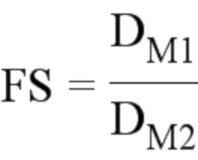
Где DM1 = коэффициент распределения металлического элемента M1, и
DM2 = коэффициент распределения металлического элемента M2 (главным образом железо).
* Эффективность экстрагирования выражают как процент извлечения, обозначаемый E(%), и определяют согласно следующему уравнению:
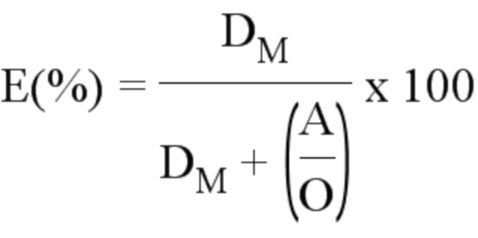
где DM = коэффициент распределения металлического элемента M, и
A/O = соотношение объемов водной и органической фаз.
Методики
Для максимального приближения к условиям, применяемым при экстракции выщелачивающих растворов, тесты экстракции проводили с кислыми водными фазами, содержащими несколько металлических элементов, обозначаемых M, и органическими фазами, содержащими экстрагирующий агент. Более конкретно, металлические элементы M представляют собой переходный металл (Fe) и лантаноиды, обозначаемые Ln, количество которых в сумме равно 5 (La, Nd, Gd, Dy и Yb).
Составы водной и органической фаз до контакта приведены ниже, при этом единица измерения "M", применяемая далее по тексту, соответствует сокращению единицы измерения "моль/л" по системе СИ:
Водные фазы:
1 мM каждого из лантаноидов La, Nd, Gd, Dy и Yb, и 50 мM Fe,
от 0.5 M до 5 M H3PO4, с опциональной модификацией ионной силы посредством добавления от 0.1 M до 2 M NaNO3.
Органические фазы:
от 10 мM до 100 мM экстрагирующего агента в додекане; следует отметить, что молярная концентрация экстрагирующего агента по меньшей мере в два раза выше общей молярной концентрации лантаноидов.
Каждый водный раствор вводили в контакт с органическим раствором, содержащим тестируемый экстрагирующий агент в додекане.
Контакт проводили в соотношении “объем на объем” (то есть, объемы органического раствора Vорг и водного раствора Vводн одинаковы, Vорг = Vводн), посредством перемешивания на магнитной мешалке в течение 20 минут, в пробирке объемом 15мл и при комнатной температуре (21-22°C). После центрифугирования разделяли полученные водные и органические фазы, затем каждую фазу анализировали методом ICP-OES.
Результаты экспериментов
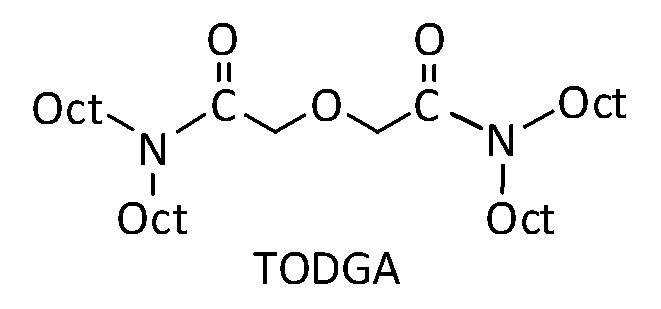
Пример 8. Экстрагирование с помощью TODGA (сравнительный пример)
Для данного примера применяли следующие условия:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM экстрагирующего агента в додекане, где экстрагирующий агент представляет собой TODGA (экстрагирующий агент, описанный в источнике [1]), имеющий формулу:
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 1 ниже:
Таблица 1
|
Как показано выше в таблице 1, все полученные значения процентов экстрагирования E(%) ниже 2%, вне зависимости от концентрации фосфорной кислоты в водной фазе.
Полученные результаты ясно показывают, что TODGA абсолютно не способен экстрагировать лантаноиды из водной фазы, содержащей фосфорную кислоту, в отличие от случая, когда водная фаза содержит азотную кислоту (см. источник [1]).
Пример 9. Экстракция фосфонат-фосфиноксидом
Для данного примера применяли следующие условия:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM экстрагирующего агента в додекане, где экстрагирующий агент представляет собой следующее соединение (см. 2.3 выше):
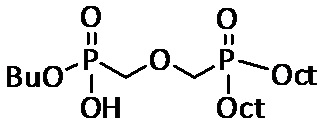
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 2.1 ниже:
Таблица 2.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены в таблице 2.2 ниже:
Таблица 2.2
|
Вычисленные значения коэффициентов селективности FS для элемента Yb по сравнению с каждым элементом M, обозначенные FSYb/M, приведены в таблице 2.3 ниже:
Таблица 2.3
|
Экстрагирующий агент, применявшийся в представленном примере, эффективно экстрагирует Yb из среды, содержащей фосфорную кислоту, особенно при ее концентрации 0.5 M.
Хотя эффективность экстрагирования Yb ниже при 5 M [H3PO4] (39%), такая экстракция, с другой стороны, особенно селективна относительно других лантаноидов (FSYb/Dy > 60 и FSYb/Ln > 200 для Ln = La, Nd, Gd), а также относительно железа (FSYb/Fe = 158) при данном значении молярной концентрации.
Эффективность экстрагирования Yb повышается при низкой кислотности (например, E(%) = 91% при 0.5 M [H3PO4]), но наблюдается снижение коэффициента селективности относительно железа (FSYb/Fe = 16 при 0.5 M [H3PO4]).
Пример 10. Экстракция амидо-фосфинатом
Для данного примера применяли следующие условия:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
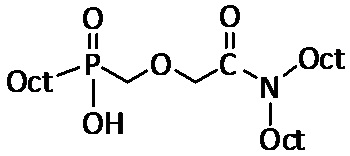
Органическая фаза: 10 мM экстрагирующего агента в додекане, где экстрагирующий агент представляет собой следующее соединение (см. Пример 7 выше):
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 3.1 ниже:
Таблица 3.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены в таблице 3.2 ниже:
Таблица 3.2
|
Экстрагирующий агент, применявшийся в представленном примере, эффективно экстрагирует Yb из среды с 0.5М фосфорной кислоты, при E(%) = 60%; следует отметить, что при этой молярной концентрации [H3PO4] снижается селективность относительно железа (FSYb/Fe = 2.98).
Пример 11. Экстракция первым амидо-фосфонатом
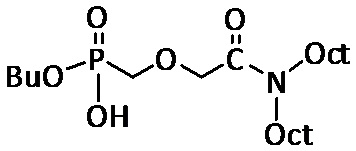
В примере 11, применяющийся экстрагирующий агент представляет собой следующее соединение (см. 6.1 выше):
11.1. Первую серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM описанного выше экстрагирующего агента в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 4.1 ниже:
Таблица 4.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены в таблице 4.2 ниже:
Таблица 4.2
|
11.2. Вторую серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в фосфорной кислоте ([H3PO4] = 5 M).
Органическая фаза: различные концентрации экстрагирующего агента ([экстрагирующий агент] = от 10 мM до 40 мM) в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 4.3 ниже:
Таблица 4.3
|
11.3. Третью серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в фосфорной кислоте ([H3PO4] = 5 M), опционально с добавлением нитрата натрия ([NaNO3] = от 0 до 2 M).
Органическая фаза: 15 мM описанного выше экстрагирующего агента в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 4.4 ниже:
Таблица 4.4
|
Экстрагирующий агент, применявшийся в представленном примере, эффективно экстрагирует все протестированные лантаноиды (за исключением La); более эффективно экстрагируются лантаноиды с наиболее высокими атомными номерами (Dy и Yb).
Данный экстрагирующий агент также позволяет получить хорошую селективность относительно железа (FSNd/Fe = 60, и FSLn/Fe выше (>) или намного выше (>>) 200 для Ln = Gd, Dy, Yb), в среде с 5 M и 3 M фосфорной кислоты.
Повышение молярной концентрации данного экстрагирующего агента, а также добавление NaNO3, позволяет увеличить эффективность экстрагирования всех лантаноидов, в особенности лантаноидов с низкими атомными номерами (La и Nd), при этом сохраняя хорошие коэффициенты разделения относительно железа.
Пример 12. Экстракция вторым амидо-фосфонатом
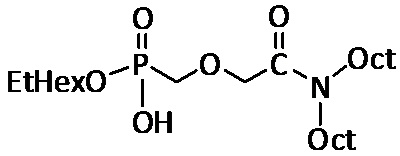
В примере 12, применяющийся экстрагирующий агент представляет собой следующее соединение (см. 6.6 выше):
12.1. Первую серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM описанного выше экстрагирующего агента в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 5.1 ниже:
Таблица 5.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены в таблице 5.2 ниже:
Таблица 5.2
|
12.2. Вторую серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в фосфорной кислоте ([H3PO4] = 5 M).
Органическая фаза: различные концентрации экстрагирующего агента ([экстрагирующий агент] = от 10 мM до 40 мM) в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 5.3 ниже:
Таблица 5.3
|
12.3. Третью серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в фосфорной кислоте ([H3PO4] = 5 M), опционально с добавлением нитрата натрия ([NaNO3] = от 0 до 2 M).
Органическая фаза: 15 мM описанного выше экстрагирующего агента в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 5.4 ниже:
Таблица 5.4
|
Экстрагирующий агент, применяющийся в данном примере, остается эффективным в экстрагировании Yb из среды с фосфорной кислотой, при этом эффективность экстракции обратно пропорционально молярной концентрации [H3PO4].
Как в Примере 11, наблюдается, что увеличение молярной концентрации данного экстрагирующего агента, а также добавление NaNO3, позволяет увеличить эффективность экстракции всех лантаноидов, в особенности лантаноидов с низкими атомными номерами (La и Nd), при этом сохраняя хорошие коэффициенты разделения относительно железа.
Пример 13. Экстракция третьим амидо-фосфонатом
В примере 13, применяющийся экстрагирующий агент представляет собой следующее соединение (см. 6.3 выше):

13.1. Первую серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM описанного выше экстрагирующего агента в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены ниже в таблице 6.1:
Таблица 6.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены ниже в таблице 6.2:
Таблица 6.2
|
Вычисленные значения коэффициентов селективности FS для элемента Yb по сравнению с каждым элементом M, обозначенные FSYb/M, приведены ниже в таблице 6.3:
Таблица 6.3
|
13.2. Вторую серию экстракций проводили в следующих условиях:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в фосфорной кислоте ([H3PO4] = 5 M).
Органическая фаза: различные концентрации экстрагирующего агента ([экстрагирующий агент] = от 10 мM до 40 мM) в додекане.
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены ниже в таблице 6.4:
Таблица 6.4
|
Пример 14. Экстракция четвертым амидо-фосфонатом
Для данного примера применяли следующие условия:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM экстрагирующего агента в додекане, где экстрагирующий агент представляет собой следующее соединение (см. 6.4 выше):
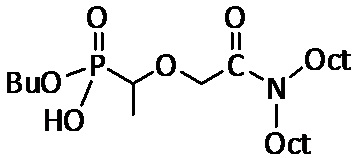
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены в таблице 7.1 ниже:
Таблица 7.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены в таблице 7.2 ниже:
Таблица 7.2
|
Вычисленные значения коэффициентов селективности FS для элемента Yb по сравнению с каждым элементом M, обозначенные FSYb/M, приведены в таблице 7.3 ниже:
Таблица 7.3
|
Пример 15. Экстракция пятым амидо-фосфонатом
Для данного примера применяли следующие условия:
Водная фаза: 1 мM каждого лантаноида (La, Nd, Gd, Dy, Yb) + 50 мM Fe в различных концентрациях фосфорной кислоты ([H3PO4] = от 0.5 M до 5 M).
Органическая фаза: 10 мM экстрагирующего агента в додекане, где экстрагирующий агент представляет собой следующее соединение (см. 6.5 выше):
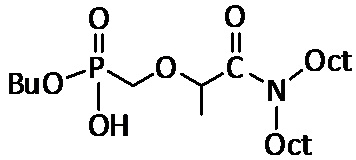
Вычисленные значения коэффициентов распределения D и процентов экстрагирования E(%) приведены ниже в таблице 8.1:
Таблица 8.1
|
Вычисленные значения коэффициентов селективности FS для каждого лантаноида Ln по сравнению с Fe, обозначенные FSLn/Fe, приведены ниже в таблице 8.2:
Таблица 8.2
|
Вычисленные значения коэффициентов селективности FS для элемента Yb по сравнению с каждым элементом M, обозначенные FSYb/M, приведены ниже в таблице 8.3:
Таблица 8.3
|
Результаты в Примерах 13 - 15 показывают, что наличие разветвления в альфа-положении относительно фосфонатной группы или амидной группы позволяет экстрагировать Yb эффективно и селективно при всех молярных концентрациях [H3PO4] (от 0.5 M до 5 M). Фактически обнаружилось, что лантаноиды с атомными номерами меньше, чем у Yb (Ln = La, Nd, Gd, Dy), не экстрагируются или экстрагируются в очень небольшой степени.
Таким образом, экстрагирующие агенты, содержащие разветвление в альфа-положении относительно фосфонатной группы или амидной группы, обладают интересными экстрагирующими характеристиками в плане экстракции Yb, при хорошей селективности относительно Fe. При повышении молярной концентрации экстрагирующего агента до значений между 15 мM и 25 мM, достигается эффективность экстракции Yb от 71% до 80%, при сохранении хороших коэффициентов разделения (FSYb/Fe > 200), при высокой кислотности ([H3PO4] = 5 M).
При сравнении результатов из Примеров 13 и 14 с результатами из Примера 15 можно отметить, что экстрагирующий агент, содержащий разветвление в альфа-положении относительно фосфонатной группы (R1 = Oct в Примере 13 или R1 = CH3 в Примере 14), обладает более высокой эффективностью экстракции Yb по сравнению со значением для экстрагирующего агента, содержащего разветвление в альфа-положении относительно амидной группы (R2= CH3 в Примере 15), особенно при низких молярных концентрациях [H3PO4] (0.5 M и 1 M). Оптимальные значения эффективности/ селективности наблюдаются при 1 M и 3 M молярных концентрациях [H3PO4].
Кроме того, когда экстрагирующий агент содержит разветвление в альфа-положении относительно фосфонатной группы (R1 ≠ H), наблюдается увеличение эффективности экстракции Yb, когда данное разветвление соответствует метилу (R1 = CH3 в Примере 14), по сравнению со случаем, когда данное разветвление соответствует н-октилу (R1 = Oct в Примере 13).
Список литературы
[1] S. Ansari et al., Chem. Rev., 2012, 112, 1751-1772.
[2] H. Narita et al., Solvent Extraction Research and Development, Japan, 2013, 20, 115-121.
[3] M. Iqbal et al., New J. Chem., 2012, 36, 2048-2059.
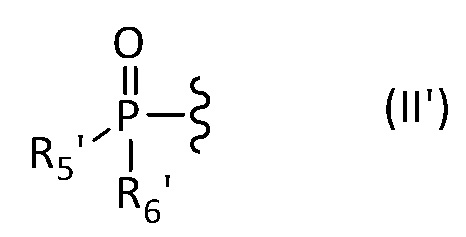
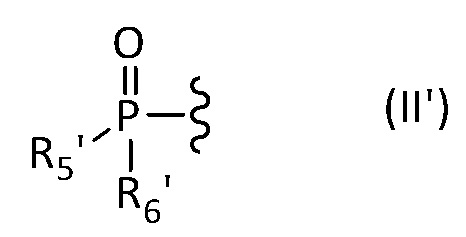
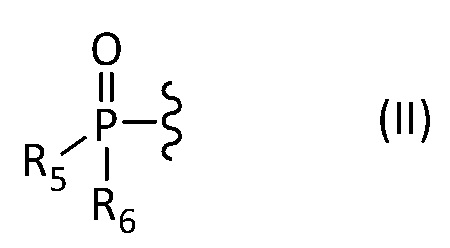
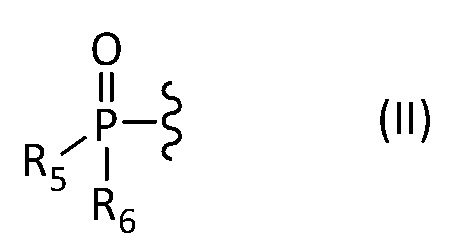
Модифицированные 2'- и 3'-нуклеозиды и их применение для получения лекарственного средства для лечения инфекций flaviviridae
Устройство и способ измерения скорости счета
Способ дезактивации жидких радиоактивных отходов от одного или нескольких радиоактивных химических элементов путем отделения твердой фазы от жидкой с использованием контура рециркуляции
Способы приготовления оксалата актиноидов и приготовления соединений актиноидов
Способ определения спектрального и пространственного распределения фотонов тормозного излучения и соответствующее устройство
Устройство и способ осаждения пророшков смеси и порошков для формирования объекта с градиентным составом
Способ неинтрузивного обнаружения химического элемента
Способ электрокинетической дезактивации твердой пористой среды
Фотогальванический модуль, содержащий прозрачный проводящий электрод переменной толщины и способы изготовления такого модуля
Анемометрический зонд с одной или несколькими проволочками и способ его осуществления

