Результат интеллектуальной деятельности: СПОСОБ ДЕКАРБОКСИЛИРУЮЩЕЙ КЕТОНИЗАЦИИ ЖИРНЫХ КИСЛОТ ИЛИ ПРОИЗВОДНЫХ ЖИРНОЙ КИСЛОТЫ
Вид РИД
Изобретение
Данное изобретение относится к способу производства длинноцепных внутренних кетонов через декарбоксилирующую кетонизацию жирных кислот или производных жирных кислот.
Превращение кислот в соответствующие кетоны декарбоксилирующей кетонизацией является хорошо известным способом, который применяется коммерчески.
Способ может проводиться в газовой фазе при температурах обычно превышающих 350°C, и обычно выше 400°C для жирных кислот в присутствии каталитических количества соединений оксидов металлов (например, MgO, ZrO2, Al2O3, CeO2, MnO2, TiO2).
Проведение реакции в газовой фазе с жирными кислотами с высокой температурой кипения является трудным, так как испарение реагентов требует очень высоких температур, что негативно влияет на селективность процесса и приводит к образованию нежелательных побочных продуктов.
Проведение процесса в жидкой фазе имеет определенные преимущества над реакцией в газовой фазе, например, обычно большую производительность, снижение производственных затрат и лучшую селективность, которая важна для последующей обработки реакционной смеси.
Патент Германии DE 295 657 относится к способу производства кетонов, в котором монокарбоновые кислоты, имеющие температуру кипения более 300°C, нагревают в жидкой фазе с небольшими количествами каталитически активных соединений металла, силикагелями или силикатами до температур, незначительно превышающих 300°C. Органическую кислоту смешивают с каталитически активными видами и затем нагревают до желаемой температуры реакции. Описано, что способ дает желаемые кетоны с хорошим выходом и чистотой.
Способ, описанный в DE 295 657, не дает желаемые кетоны с хорошим выходом, однако, если исходные жирные кислоты содержат жирные кислоты или производные жирных кислот, имеющие температуру кипения менее 300°C (что относится к линейным жирным кислотам, содержащим 12 атомов углерода или менее, таким как лауриновая кислота, каприновая кислота, каприловая кислота и т.д.), в более чем незначительных количествах.
Патент Германии DE 259 191 относится к способу производства кетонов нагреванием высших жирных кислот с тонкоизмельченными металлами и снижением температуры из-за того, что кетон начинает расщепляться. В примере стеариновую кислоту нагревают с порошком чугуна до температуры 360°C и выдерживают при 360°C в течение около 4 ч, и затем продукт охлаждают и полученный кетон выделяют. Количество порошка чугуна составляет 10% масс. по отношению к массе стеариновой кислоты, что соответствует стехиометрическим количествам. Снова, способ, описанный в этой ссылке, дает только низкие количества кетонов, если жирные кислоты, содержащие 12 атомов углерода или менее, применяются в качестве исходного материала или они присутствуют в исходном материале в более чем незначительных количествах.
EP2468708 относится к декарбоксилирующей перекрестной кетонизации смесей арил- и алкилкарбоновых кислот с применением железных катализаторов, таких как магнетитные нанопорошки, с получением алкиларилкетонов. Согласно заявленному способу, смесь ароматических монокарбоновых кислот, вторую монокарбоновую кислоту, выбранную из бензильных или алифатических монокарбоновых кислот, и содержащий железо катализатор нагревают в не водном растворителе до температуры, по меньшей мере, 220°C, в течение, по меньшей мере, 10 ч, при непрерывном удалении воды и двуокиси углерода. После окончания реакции полученную смесь отгоняют при пониженном давлении, и продукт реакции получают в дистилляте. Применение не водного растворителя считается существенным. Время реакции более 10 часов, однако, не подходит для синтеза в промышленном масштабе.
В докторской диссертации Christoph Oppel ("New methods of ketone synthesis, University of Kaiserslautern 2012), одного из изобретателей указанного выше EP 2468708, описаны эксперименты с кетонизацией лауриновой кислоты с металлическими медиаторами. Реакцию проводят при 340°C с различными соединениями металла, включая Fe и MgO, и получают кетон 12-трикозанон с хорошим выходом. Реакцию проводят в закрытых сосудах, насыщенных азотом. Образовавшиеся вода и двуокись углерода приводят к возрастанию давления внутри закрытой системы. и температура реакции 340°C также вносит вклад в возрастание давления, так как лауриновая кислота при этой температуре является газообразной. Применение такого способа в промышленном масштабе потребует применения автоклавов, которые являются дорогими. Количество металлического медиатора в примерах, данное в таблице на странице 88 докторской диссертации, составляет 50% моль. по отношению к общему количеству кислоты, что соответствует стехиометрическим соотношениям, и общее количество реагентов объединяют изначально и нагревают вместе.
Хотя способы, описанные в известном уровне техники и указанные в ссылках здесь, дают кетоны с хорошим выходом, некоторые из них не эффективны при применением исходных жирных кислот, содержащих 12 атомов углерода или менее, или смеси жирных кислот, содержащей значительные количества жирных кислот, содержащих 12 атомов углерода или менее. Более того, для некоторых из указанных выше способов, их применение в промышленных масштабах ограничиваются проблемами и необходимостью применения дорогостоящих аппаратов. Таким образом, до сих пор существует необходимость в коммерчески приемлемом способе производства кетонов из жирных кислот или их производных.
Таким образом, задачей данного изобретения является разработка практичного и простого в применении способа синтеза кетонов декарбоксилирующей кетонизацией жирных кислот или производных жирных кислот в жидкой фазе в открытой реакционной системе, особенно с исходным применением жирных кислот с 12 атомами углерода или менее или смесей жирных кислот, содержащих, по меньшей мере, 10% моль. жирных кислот с 12 атомами углерода или менее или их производных по отношению к общему количеству карбоновых кислот.
Эта задача достигается способом по пункту 1, который включает способ декарбоксилирующей кетонизации жирных кислот, производных жирных кислот или их смесей в жидкой фазе с соединениями металла в качестве катализатора, отличающийся тем, что:
a) на первой стадии элементарный металл или соединение металла и жирную кислоту, производное жирной кислоты или их смесь, содержащую, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных жирных кислот, содержащих 12 атомов углерода или менее, по отношению к общему количеству жирных кислот или производных жирных кислот, смешивают в молярном отношении от 1:0,8 до 1:3,5 (молярное отношение металл:эквивалент карбоксильной группы) и подвергают взаимодействию в течение времени P1 от 5 мин до 24 ч при температуре T1 от 100°C до 270°C по существу при отсутствии добавляемых растворителей, и
b) затем температуру повышают до температуры T2, которая строго выше 270°C и вплоть до 400°C, и дополнительные жирные кислоты, производные жирных кислот или их смесь, содержащую, и, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных таких жирных кислот по отношению к общему количеству жирных кислот или производных жирных кислот, добавляют в течение времени P2 от 5 мин до 24 ч по существу при отсутствии добавляемых растворителей, до тех пор, пока отношение жирной кислоты, производных жирных кислот или их смеси к металлу не достигнет интервала от 6:1 до 99:1.
Определенные предпочтительные варианты способа в соответствии с данным изобретением представлены в зависимых пунктах формулы изобретения.
Например, вариант, представленный в пункте 4 (далее вариант E*), относится к способу декарбоксилирующей кетонизации жирных кислот, производных жирных кислот или их смесей в жидкой фазе с соединениями металла в качестве катализатора, отличающемуся тем, что:
a) на первой стадии элементарный металл или соединение металла и жирную кислоту, производное жирной кислоты или их смесь, содержащую, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных жирных кислот, содержащих 12 атомов углерода или менее, по отношению к общему количеству жирных кислот или производных жирных кислот, смешивают в молярном отношении от 1:0,8 до 1:3,5 (молярное отношение металл:эквивалент карбоксильной группы) и подвергают взаимодействию в течение от 5 мин до 24о мин при температуре от 180°C до 270°C по существу при отсутствии добавляемых растворителей, и
b) затем температуру повышают до температуры от 280°C до 320°C, и дополнительные жирные кислоты, производные жирных кислот или их смесь, содержащую, и, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных таких жирных кислот по отношению к общему количеству жирных кислот или производных жирных кислот, добавляют в течение времени 1 ч до 24 ч по существу при отсутствии добавляемых растворителей, до тех пор, пока отношение жирной кислоты, производных жирных кислот или их смеси к металлу не достигнет интервала от 6:1 до 99:1.
Другие варианты способа в соответствии с данным изобретением представлены в подробном описании ниже.
Температура T1
Температура T1 составляет от 100°C до 270°C.
Температура T1 предпочтительно составляет, по меньшей мере, 180°C, более предпочтительно, по меньшей мере, 210°C и еще более предпочтительно, по меньшей мере, 230°C.
Кроме того, температура T1 может быть не более 260°C.
Температура T1 может быть от 180°C до 270°C или от 210°C до 260°C. Хорошие результаты получают при T1 от 230°C до 270°C, в частности, от 240°C до 260°C.
Температура T2
Температура T2 строго выше 270°C и вплоть до 400°C.
Температура T2 может быть строго ниже 280°C. Однако предпочтительно она составляет, по меньшей мере, 280°C, более предпочтительно, по меньшей мере, 290°C и еще более предпочтительно, по меньшей мере, 300°C. Она может быть строго выше 320°C.
Температура T2 может быть строго выше 360°C. Однако она обычно составляет не более 360°C и часто не более 340°C. Она может быть не более 320°C.
Температура T2 может быть от 280°C до 320°C. Температура T2 также может быть строго выше 320°C и вплоть до 360°C.
Хорошие результаты получают, когда T2 варьируется от 280°C до 360°C, в частности, от 300°C до 340°C.
Разница температур T2 минус T1 (T2-T1)
Разница температур T2 минус T1 предпочтительно составляет, по меньшей мере, 3°C. Предпочтительно она составляет, по меньшей мере, 10°C, более предпочтительно, по меньшей мере, 30°C и еще более предпочтительно, по меньшей мере, 45°C.
Кроме того, T2-T1
предпочтительно составляет, не более 100°C. Она может быть не более 85°C, не более 70°C или не более 55°C.
Хорошие результаты получают, когда T2-T1 варьируется от 30°C до 100°C, в частности, от 45°C до 85°C.
Определенные сочетания температуры T1 и температуры T2
В первом варианте, T1 составляет от 230°C до 270°C, а T2 составляет от 280°C до 400°C, предпочтительно, от 290°C до 360°C, и более предпочтительно, от 300°C до 340°C.
Во втором варианте, T2 строго ниже 280°C, а T1 составляет от 180°C до 270°C, предпочтительно, от 230°C до 270°C, и более предпочтительно, от 240°C до 260°C.
В третьем варианте, T2 составляет от 280°C до 320°C, а T1 составляет от 180°C до 270°C, предпочтительно, от 230°C до 270°C, и более предпочтительно, от 240°C до 260°C.
В четвертом варианте, T2 точно выше 320°C и вплоть до 360°C, а T1 составляет от 180°C до 270°C, предпочтительно, от 230°C до 270°C, и более предпочтительно, от 240°C до 260°C.
В пятом варианте, T2 точно выше 360°C, а T1 составляет от 180°C до 270°C, предпочтительно, от 230°C до 270°C, и более предпочтительно, от 240°C до 260°C.
Период времени P1
Период времени P1 может варьироваться до значительной степени в зависимости исключительно от природы простого металла или соединения металла. В любом случае, период времени P1 составляет от 5 мин до 24 ч.
Период времени P1 составляет, предпочтительно, по меньшей мере, 10 мин и более предпочтительно, по меньшей мере, 20 мин.
Кроме того, период времени P1 составляет, предпочтительно, не более 12 ч, более предпочтительно, не более 8 ч и еще более предпочтительно, не более 5 ч.
Хорошие результаты получают при периоде времени P1 от 10 мин до 8 ч, в частности, от 20 мин до 5 ч.
Каждый указанный нижний предел, верхний предел или интервал для периода времени P1 должен рассматриваться как недвусмысленно описанный в сочетании с каждым указанным нижним пределом, верхним пределом или интервалом, ранее указанным для температуры T1.
Период времени P2
Период времени P2 также может варьироваться до значительной степени в зависимости исключительно от общего применяемого количества кислоты или производного кислоты. В любом случае, период времени P2 составляет от 5 мин до 24 ч.
Период времени P2 предпочтительно составляет, по меньшей мере, 30 мин, более предпочтительно, по меньшей мере, 1 ч, и еще более предпочтительно, по меньшей мере, 2 ч.
Кроме того, период времени P2 составляет предпочтительно не более 16 ч, и более предпочтительно, не более 8 ч.
Хорошие результаты получают, когда период времени P2 составляют от 1 ч до 16 ч, в частности, от 2 ч до 8 ч.
Каждый определенный нижний предел, верхний предел или интервал периода времени P2 должен рассматриваться как недвусмысленно описанный в сочетании с каждым указанным нижним пределом, верхним пределом или интервалом, ранее указанным для температуры T2.
Первая стадия
На первой стадии способа в соответствии с данным изобретением элементарный металл (или смесь элементарных металлов) или соединение металла (или смесь соединений металла) и жирную кислоту, производное жирных кислот или их смесь, содержащую, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных таких жирных кислот по отношению к общему количеству жирных кислот или производных жирных кислот, смешивают в молярном отношении от 1:1,0 до 1:3,0 (молярное отношение металл:эквивалент карбоксилатной группы) и подвергают взаимодействию в течение периода времени P1 при температуре T1 по существу при отсутствии добавляемых растворителей, предпочтительно, в отсутствие растворителей.
Например, в варианте E*, на первой стадии способа, элементарный металл (или смесь элементарных металлов) или соединение металла (или смесь соединений металла) и жирную кислоту, производное жирных кислот или их смесь, содержащую, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных таких жирных кислот по отношению к общему количеству жирных кислот или производных жирных кислот, смешивают в молярном отношении от 1:1,0 до 1:3,0 (молярное отношение металл:эквивалент карбоксилатной группы) и подвергают взаимодействию в течение от 5 до 240 мин, предпочтительно от 10 до 180 мин, и даже более предпочтительно, от 15 до 120 мин, при температуре от 180 до 270°C, предпочтительно от 190 до 260°C, и даже более предпочтительно, от 210 до 260°C по существу при отсутствии добавляемых растворителей, предпочтительно, в отсутствие растворителей. В данном варианте E* показано, что время реакции от 15 до 60 минут при температуре реакции от 220 до 260°C иногда является предпочтительным.
Количество атомов углерода всегда относится к соответствующему количеству в свободной кислоте; если применяют производные, число атомов углерода может быть выше.
Подходящие металлы в способе в соответствии с данным изобретением выбирают из группы, включающей Mg, Ca, Al, Ga, In, Ge, Sn, Pb, As, Sb, Bi, Cd и переходные металлы, имеющие атомное число от 21 до 30. Подходящие соединения металлов включают оксиды указанных выше металлов, нафтенаты указанных выше металлов или ацетаты указанных выше металлов. Предпочтительны магний, железо и их оксиды, и, в частности, порошок железа.
Термин жирные кислоты относится к карбоновым кислотам, содержащим, по меньшей мере, 4 атома углерода. Термин производные жирных кислот относится к ангидридам, полученным конденсацией 2 жирных кислот или к сложным эфирам, полученным конденсацией жирных кислот со спиртами.
Подходящими производными жирных кислот являются сложные эфиры и ангидриды жирных кислот, но применение свободных жирных кислот как таковых обычно предпочтительно. Сложные эфиры или ангидриды в ходе реакции превращаются в кислоты, которые затем взаимодействуют с металлом или соединением металла. Особенно в случае сложных эфиров, однако, в качестве побочных продуктов образуются спирты, которые позднее необходимо удалять, что требует дополнительных работ и затрат. Однако если сложные эфиры получены из низших спиртов, таких как метанол, этанол, пропанол или бутанол, спирты удаляют по мере образования в ходе реакции, благодаря реакционной дистилляции.
Жирные кислоты или производные жирных кислот могут применяться в форме так называемых фракций жирных кислот или производных жирных кислот, которые могут быть получены гидролизом или алкоголизом различных природных жиров и масел. Следовательно, эти фракции могут содержать различные количества разных линейных жирных кислот или производных линейных жирных кислот с различной длиной цепи. Только в качестве примеров, могут быть упомянуты фракции жирной кислоты, полученные из кокосового масла и содержащие в основном C12-C18 жирные кислоты. Специалисту в данной области техники хорошо известны другие фракции жирных кислот, получаемые из различных источников, и он выберет наиболее подходящие исходные материалы в зависимости от желаемых кетонов.
Жирные кислоты, содержащие 12 атомов углерода или менее, предпочтительно от 8 до 12 атомов углерода, или производные таких кислот (сложные эфиры или ангидриды) составляют, по меньшей мере, 10% моль. и, предпочтительно, по меньшей мере, 15% моль. от общего молярного количества смеси жирной кислоты или смеси производных жирных кислот, применяемых в качестве исходного материала. Эти кислоты дают кетоны, имеющие общее количество атомов углерода 23 или менее, которые оказались полезными во множестве областей применения. Не существует определенного верхнего предела количества таких жирных кислот или производных жирных кислот из кислот, содержащих 12 атомов углерода или менее, т.е. исходный материал может также полностью состоять из таких жирных кислот или производных жирных кислот.
С учетом вышеизложенного, предпочтительными жирными кислотами для применения в способе в соответствии с данным изобретением являются гексановая кислота, изостеариновая кислота, каприловая кислота, каприновая кислота, лауриновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, арахидиновая кислота, бегеновая кислота, лигноцериновая кислота, церотиновая кислота или их смеси, и предпочтительными производными жирных кислот являются сложные эфиры и ангидриды этих кислот.
Понятно, что когда одну и только одну жирную кислоту или производное жирной кислоты применяют в качестве исходного материала, она должна иметь 12 атомов углерода или менее.
Жирные кислоты могут содержать одну или более двойных связей в цепи, например, как в олеиновой кислоте, линолевой кислоте, линоленовой кислоте, эруковой кислоте, пальмитолеиновой кислоте или их смесях.
Если исходным материалом является одна жирная кислота, в качестве продукта реакции получают симметричный кетон; если исходным материалом являются фракции жирных кислот, описанная выше, получают все кетоны, образованные сочетанием различных алкильных групп исходных кислот, и распределение различных смешанных кетонов обычно отвечает статистическому биноминальному закону. Уравнение реакции может быть суммировано следующим образом:

где Rn и Rm представляет алькильные группы жирных кислот, присутствующих во фракции. Очевидно, что, например, если присутствуют три различные кислоты, может быть получено всего шесть различных кетонов; три симметричных кетона, где Rn и Rm являются идентичными, и три смешанных кетона с разными группами Rn и Rm.
В соответствии с предпочтительным вариантом, металлом является порошок железа или соединением металла является оксид железа(II) или смешанный оксид железа(II) и железа(III), такой как, например, магнетит. Порошок железа обладает экономическими преимуществами, так как он дешев и легкодоступен.
Во время первой стадии способа в соответствии с данным изобретением образуется карбоксилат металла в качестве промежуточного соединения, который на следующей стадии расщепляется на желаемый кетон и оксид металла, который является активным катализатором для последующего превращения кислоты или производного кислоты, добавляемых последовательно или непрерывно на второй стадии к смеси, содержащей желаемый кетон.
Если на первой стадии применяют металл, указанный металл взаимодействует с жирной кислотой до карбоксилата металла с одновременным образованием газообразного водорода. Если применяют оксид металла на первой стадии, образование карбоксилата сопровождается одновременным образованием воды. Общее уравнение для образования карбоксилата на первой стадии (для металла, имеющего валентность 2 в качестве примера) может быть представлено следующим образом:
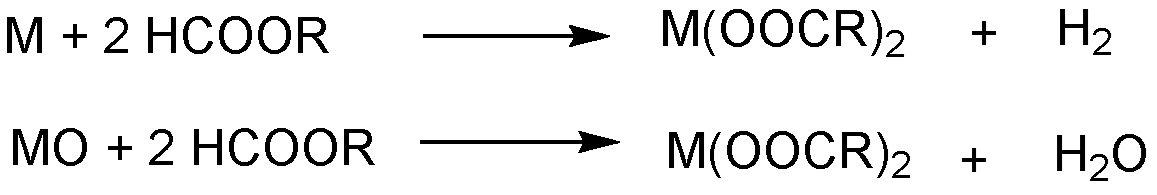
Молярное отношение металла или соединения металла к общему количеству карбоксильных групп в исходном материале на первой стадии составляет от 1:0,8 до 1:3,5, и обычно предпочтительно применять молярное отношение, достаточное для образования соответствующего карбоксилата металла и для превращения всей присутствующей кислоты или производного кислоты в карбоксилат металла, т.е. по существу, не оставляющее свободных карбоксильных групп после образования карбоксилата после первой стадии. Таким образом, для двухвалентного металла молярное отношение металла к карбоксильным группам составляет, предпочтительно около 1:2, так как два эквивалента необходимы для образования дикарбоксилата металла двухвалентного металла. Если вместо простого металла применяют оксид металла, указанное выше молярное отношение рассчитывают с количеством простого металла в оксиде. Молярное количество карбоксильных групп рассчитывают, принимая во внимание количество таких групп в жирной кислоте или производном жирной кислоты, которое применяют в качестве исходного материала. Таким образом, например, ангидрид кислоты содержит две карбоксилатных функциональных группы и может давать две карбоксильные группы для образования карбоксилата металла.
Образование карбоксилата металла на первой стадии может удобным образом отслеживаться in situ ИК анализом. Полоса абсорбции карбонила регулируется длинноволновым сдвигом в карбоксилате металла, что позволяет отслеживать развитие реакции.
Согласно особенно предпочтительному варианту способа в соответствии с данным изобретением, в качестве металла применяют порошок железа, так как он является дешевым и легкодоступным.
Вторая стадия
На второй стадии способа в соответствии с данным изобретением температуру повышают до температуры T2, при которой карбоксилат металла расщепляется преимущественно до желаемого кетона, оксида металла и двуокиси углерода.
Например, в варианте E*, на второй стадии способа температуру повышают от 280 до 320°C, при которой карбоксилат металла расщепляется преимущественно до желаемого кетона, оксида металла и двуокиси углерода.
Дополнительные жирные кислоты, производные жирных кислот или их смесь, содержащая, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных таких жирных кислот по отношению к общему количеству жирных кислот или производных жирных кислот, добавляют на второй стадии, по существу при отсутствии добавляемого растворителя, предпочтительно, в отсутствие растворителя. Они могут быть добавлены последовательно или непрерывно, и преимущественно их добавляют со скоростью, позволяющей избежать образования значительных количеств свободной кислоты в реакционной системе. Снова, развитие реакции и превращение исходных материалов в карбоксилаты в качестве промежуточных соединений, и кетоны в качестве конечных продуктов, может удобным образом отслеживаться подходящими способами, такими как ИК анализ.
Во время второй стадии, дополнительные жирные кислоты, производные жирных кислот или их смеси добавляют в течение периода времени P2, который зависит, в частности, от применяемого общего количества кислоты или производного кислоты.
Например, в варианте E*, период времени P2 составляет от 1 ч до 24 ч, предпочтительно, от 2 ч до 12 ч, и особенно предпочтительно, от 2 до 8 часов.
Общее количество жирной кислоты (жирной кислоты или производного жирной кислот), добавляемое на второй стадии реакции таково, что общее молярное отношение металла к количеству карбоксильных групп, достигаемое в конце второй стадии, составляет от 1:6 до 1:99, т.е. количество соединения металла составляет от около 1% моль. до около 14% моль., и предпочтительно, от 2 до около 10% моль. от общего количества жирных кислот или производных жирных кислот, т.е. металл или соединение металла на самом деле функционирует как катализатор и не применяется в курсе реакции. Для большинства способов, описанных в известном уровне техники в жидкой фазе, металл или соединение металла применяют в количестве, превышающем 50% моль., и во многих случаях даже превышающем эквимолярные количества. Такие высокие количества металла не являются необходимыми в данном изобретении, что является как техническим, так и экономическим преимуществом способа в соответствии с данным изобретением над известным уровнем техники.
В соответствии с данным изобретением, температура T2 точно выше 270°C и вплоть до 400°C. Согласно варианту E* в соответствии с данным изобретением, температура на второй стадии реакции составляет от 280 до 320°C и, предпочтительно, от 285 до 310°C.
То, что было сказано в отношении композиции исходной жирной кислоты на первой стадии способа в соответствии с данным изобретением, также применимо ко второй стадии.
Способ в соответствии с данным изобретением проводят в негерметичной системе, т.е. без применения избыточного давления. Побочные продукты вода и двуокись углерода могут непрерывно удаляться в ходе реакции. Подходящее оборудование известно специалистам в данной области техники, и они применяют наиболее подходящие уставки оборудования для конкретной ситуации. Только в качестве примера, так называемая ловушка Дина-Старка может применяться для удаления воды во время реакции, и такое удаление является предпочтительным вариантом данного изобретения.
Способ в соответствии с данным изобретением проводят практически при отсутствии добавляемого растворителя. Желаемый кетон, образующийся во время реакции, в основном действует как растворитель для реакции. Так как получаемый кетон в основном имеет более высокую температуру кипения, чем жирные кислоты или производные жирных кислот, применяемые в качестве исходного материала, это позволяет поводить реакцию в жидкой фазе, как желательно, без добавления внешнего растворителя, который необходимо удалять в конце реакции, что является дорогим и трудоемким и, поэтому, нежелательно.
Период времени P12
Дополнительные жирные кислоты, производные жирных кислот или их смеси могут быть добавлены в период времени P2 в указанных выше условиях, сразу же после повышения температуры до T2 (конкретный вариант, соответствующий P12, определенному ниже, равному 0).
Альтернативно, после повышения температуры до T2 и до добавления дополнительных жирных кислот, производных жирных кислот или их смесей в течение периода времени P2, указанная температура может поддерживаться на уровне температуры T2 в период времени P12 (>0).
Период времени P12 составляет, предпочтительно, по меньшей мере, 30 мин, и более предпочтительно, по меньшей мере, 1 ч.
Кроме того, период времени P12 составляет, предпочтительно, не более 5 ч, и более предпочтительно, не более 3 ч.
Хорошие результаты, в частности, получают при P12 от 30 мин до 300 мин, особенно от 1 ч до 3 ч.
Период времени P23
Сразу после добавления дополнительных жирных кислот, производных жирных кислот или их смесей в течение периода времени P2, температура может быть снижена, возможно, до температуры T3, которая, предпочтительно, составляет от около 5°C до около 50°C (конкретный вариант соответствует P23, определенному ниже, равному 0). Температурой T3 может быть комнатная температура или температура слегка выше комнатной температуры.
Альтернативно, после добавления дополнительных жирных кислот, производных жирных кислот или их смеси в течение периода времени P2, температура может сохраняться на уровне температуры T2 в течение периода времени P23 (>0).
Период времени P23 предпочтительно составляет, по меньшей мере, 30 мин, и более предпочтительно, по меньшей мере, 1 ч.
Кроме того, период времени P23 предпочтительно составляет не более 5 ч, и более предпочтительно, не более 3 ч.
Хорошие результаты получают, в частности, если P23 варьируется от 30 мин до 300 мин, особенно от 1 ч до 3 ч.
Восстановление кетона жирной кислоты и рециклирование металлических соединений
Как только производное жирной кислоты или жирная кислота, добавляемая на второй стадии способа в соответствии с данным изобретением, преобразуется, желаемый кетон может быть легко получен, например, дистилляцией при пониженном давлении. Также можно использовать преимущества ферромагнитных свойств металлических соединений, образованных во время реакции (таких как оксиды железа) для отделения металлических соединений от кетона с помощью магнитного поля. Другой метод отделения кетоновых продуктов от металлических соединений включает простую фильтрацию, так как металлические соединения не растворяются в кетонах, полученных в качестве продукта реакции. Специалист в данной области техники знает типовые методики, поэтому более детальное описание не требуется.
Весь процесс может преимущественно проводиться в атмосфере инертного газа, и подходящими инертными газами являются, например, азот или аргон, в качестве двух примеров.
Согласно другому предпочтительному варианту данного изобретения, после отделения желаемого кетона, оставшийся остаток, состоящий в основном из металлических соединений (например, продукт на дне после дистилляции), может повторно применяться напрямую на втором цикле добавления жирных кислот или производных жирных кислот, превращаемых в желаемые кетоны жирных кислот. В целом, количества не более одного мольного процента металла или металлического соединения, по отношению к количеству эквивалентов карбоновой кислоты, достаточно для получения желаемых кетонов с хорошим выходом. Было обнаружено, что вплоть до четырех циклов возможно проводить без значительной потери каталитической активности металла или металлического соединения (см. пример 1).
Следовательно, в другом предпочтительном варианте способа в соответствии с данным изобретением, в конце стадии b) металлические соединения отделяют от продуктов с применением обычных методов, и затем рециклируют для преобразования другой партии/порции/части жирных кислот или производных жирных кислот или их смеси, содержащей, по меньшей мере, 10% моль. жирных кислот, содержащих 12 атомов углерода или менее, или производных таких жирных кислот по отношению к общему количеству жирных кислот или производных жирных кислот.
Выход желаемых кетонов после второй стадии обычно превышает 60 процентов, более предпочтительно, 70%, и может достигать более 90%.
Применение кетона жирной кислоты для синтеза вторичных жирных спиртов
Кетоны жирной кислоты, получаемые способом в соответствии с данным изобретением, преимущественно используют для производства соответствующих вторичных жирных спиртов. Для получения таких спиртов кетоны жирной кислоты, полученные в способе в соответствии с данным изобретением, подвергают реакции гидрирования. Реакцию обычно проводят с применением катализаторов на основе гетерогенных переходных металлов на подложке в автоклаве с водородом в качестве гидрирующего агента.
Только в качестве примера, катализаторами могут быть палладиевые катализаторы на подложке из углеродных материалов. Реакцию гидрирования обычно проводят под давлением водорода от 500 до 5000 кПа при температуре в интервале от 120 до 200°C без применения добавляемого растворителя.
Применение вторичных жирных спиртов для синтеза внутренних олефинов
Вторичные спирты, полученные как описано выше, могут быть далее превращены во внутренние олефины реакцией дегидрирования.
Предпочтительно, дегидрирование проводят по существу при отсутствии добавляемого растворителя, предпочтительно, в отсутствие добавляемого растворителя, с применением оксида алюминия, предпочтительно, η-Al2O3, в качестве катализатора, при температуре в интервале от 250 до 350°C и в течение времени от 30 мин до 6 ч.
Внутренние олефины, получаемые после дегидрирования, как описано выше, демонстрируют очень низкую степень изомеризации двойной связи. Двойная связь образуется рядом со спиртовой группой, которую удаляют, и поэтому олефины являются внутренними олефинами, имеющими двойную связь в основном в середине цепи. Очевидно, что структура получаемого олефина в основном определяется структурой исходных спиртов. Реакцию дегидрирования обычно проводят в инертной атмосфере.
Сульфонирование внутренних олефинов
Внутренние олефины, полученные после дегидрирования, описанного выше, могут быть сульфонированы далее щелочных гидролизом с получением сульфонатов внутренних олефинов, которые применяют в качестве поверхностно-активных веществ.
Согласно первой альтернативе, сульфонирование может быть проведено с применением реактора с падающей пленкой, возможно, в лабораторном реакторе пленочного типа. Этот реактор может быть оборудован охлаждающим кожухом, в который подают холодную воду для предотвращения повышения температуры в реактора из-за высокой экзотермичности реакции. Для этой реакции температуру охлаждающего кожуха обычно устанавливают от около 0° до 8°C.
Поток газа, содержащий смесь сульфонирующего агента (например, безводного SO3), разбавленную тщательно высушенным инертным газом (например, азотом или воздухом) в концентрации обычно от 0,5 до 10, предпочтительно, от 1 до 5% об./об. (особенно предпочтительно, около 2,5% об./об.), подвергают взаимодействию с падающей пленкой из жидких олефинов. Поток газовой и жидкой фаз устанавливают так, чтобы время пребывания составляло от 10 секунд до 10 мин, предпочтительно, от 1 мин до 6 мин (например, 3 минуты) в реакторе, и мольное отношение SO3:внутренний олефин составляло от 0,7:1 до 1,5:1, предпочтительно, от 0,8:1 до 1,2:1, и наиболее предпочтительно, от 0,9:1 до 1,1:1 (например, наиболее предпочтительно, 1,05:1).
При применении смеси внутренних олефинов с различными длинами цепей (и, следовательно, различной молекулярной массой), общий молярный расход внутренних олефинов может быть рассчитан с применением средней молекулярной массы смеси олефинов.
После реакции сульфонирования смесь, выходящую из реактора (состоящую в основном из β-сультонов), может выстаиваться для проведения транс-сульфонирования и для увеличения превращения исходных олефинов.
Затем полученная смесь может быть нейтрализована с применением водного раствора основания (например, NaOH) в реакторе, который, предпочтительно, оборудован механической мешалкой. Гидролиз проводят нагреванием смеси при механическом перемешивании. Во время этой стадии способа, β-сульфон превращают в желаемые сульфонаты внутренних олефинов через реакцию раскрытия кольца.
Реакции сульфонирования, расщепления и гидролиза могут проводиться с применением ЯМР анализа. В конце процесса количество воды в среде может быть скорректировано для получения водного раствора сульфонатов внутренних олефинов с желаемой концентрацией активного вещества.
Согласно 2-му варианту, сульфонирование может проводиться в (периодическом) реакторе, оборудованном механической мешалкой в жидкой фазе с применением полученного in situ сульфонирующего реагента, например, ʺSO3-диоксанаʺ. Этот вариант описан в качестве примера.
В круглодонной колбе смешивают безводный диоксан и безводный трихлорметан (пропорция смеси от 1:2 до 1:5 об./об.) и охлаждают до температуры в интервале от -5 до 10°C, предпочтительно, до около 0°C. Затем медленно добавляют жидкий SO3 (2 молярных эквивалента) при перемешивании в течение 10 минут с получением комплекса SO3-диоксан, который осаждается из смеси в виде белых кристаллов.
Внутренние олефины (1 эквивалент) затем медленно добавляют при перемешивании при температуре от -5 до 10°C, предпочтительно, около 0°C, в реакционную среду в течение времени от 0,3 до 3 ч, предпочтительно, в течение приблизительно 1 часа, и смесь нагревают вплоть до комнатной температуры. В это время цвет смеси меняется от светло-желтого до темно-коричневого, и ЯМР анализ показывает практически полное завершение внутренних олефинов (около 94% олефинов превращаются в сультоны). Все летучие соединения (CHCl3 и диоксан) затем удаляют в вакууме.
Затем к остатку добавляют 2,4 эквивалента водного раствора NaOH (10% масс.), и полученную смесь перемешивают при комнатной температуре в течение приблизительно 1 часа для полной нейтрализации.
Затем проводят гидролиз перемешиванием полученной реакционной смеси при 95°C в течение ночи. ЯМР анализ показывает полное превращение сультонов в сульфонаты внутренних олефинов.
В конце процесса количество воды корректируют для получения водного раствора сульфонатов внутренних олефинов с подходящей концентрацией активного вещества, например, 30% масс.
Таким образом, способ в соответствии с данным изобретением дает легкий доступ к внутренним кетонам, которые являются универсальными исходными материалами для множества продуктов, как описано выше.
Способ дает желаемые кетоны с высоким выходом и только незначительными количествами (если они есть) нежелательных побочных продуктов, которые могут быть легко отделены от реакционной смеси.
Кетоны могут быть отделены от реакционной смеси удобными и экономичными способами, и каталитический материал может применяться для нескольких каталитических циклов без значительного ухудшения каталитической активности.
Представленные ниже примеры показывают эффективность способа и дополнительно объясняют способ в соответствии с данным изобретением.
Пример 1 - Синтез 12-трикозанона (дикетон лауриновой кислоты)
Реакцию проводят под аргоном в круглодонной колбе, оборудованной механической мешалкой, аппаратом Дина-Старка и капельной воронкой. В реактор помещают 700 мг железного порошка и 20г лауриновой кислоты вводят в капельную воронку.
Первое частичное количество 5 г кислоты добавляют в реактор, и температуру доводят до 250°C. Смесь перемешивают при этой температуре в течение 30 минут, в это время цвет среды меняется на черный и выделяется газообразный H2.
Затем температуру повышают до 300°C, смесь перемешивают в течение 1 ч 30 мин и оставшееся количество лауриновой кислоты (15 граммов) медленно добавляют в реактор в течение 4 ч 30 мин со скоростью потока, которая позволяет сохранять концентрацию лауриновой кислоты в реакционной среде очень низкой (без аккумулирования свободной кислоты в растворе).
В конце реакции капельную воронку меняют на дистилляционный аппарат, и продукты отгоняют при 290°C-340°C под давлением 5 кПа.
Затем дистилляционный аппарат меняют на капельную воронку, содержащую новую порцию 20 г жирных кислот, и повторяют описанные выше операции в другом цикле. Дополнительное железо не требуется. Остаток в колбе после дистилляции эффективно превращает следующую порцию кислот.
Всего 4 цикла проводят без потери эффективности, что позволяет снизить концентрацию железа до менее 1% масс. по отношению к превращенным жирным кислотам.
Превращение, селективность и выход (измеренный газовой хроматографией (ГХ) и фактический/изолированный) даны в таблице 1 ниже.
Таблица 1 (все значения в % от теоретического)
|
Данные показывают превосходную селективность и выход желаемого кетона.
Пример 2 - Фракция кокосовых жирных кислот в качестве исходного материала
Превращение 400 г кокосовых жирных кислот, имеющих следующее распределение массы: C12: 55%, C14: 21%, C16: 13%, C18: 12%.
Превращение проводят с применением 6,4 г железного порошка (1,6% масс.) в 2 цикла с использованием всего 200 г жирных кислот для каждого цикла.
Реакцию проводят под аргоном в 1-л круглодонной колбе, оборудованной механической мешалкой, аппаратом Дина-Старка и капельной воронкой.
В 250 мл капельную воронку вводят 200 г кокосовых жирных кислот, которые держат в расплавленном виде с помощью внешнего нагревателя.
6,4 г железного порошка распределяют в реакторе и первую порцию жирных кислот (около 58 мл) добавляют в реактор. Смесь перемешивают (500 об./мин) при 250°C в течение 30 минут для превращения металлического железа в соли железа. В это время цвет смеси меняется на черный и выделяется водород. Затем температуру повышают до 300°C-320°C для превращения в жирные кетоны. Смесь перемешивают при этой температуре в течение 1 ч 30 мин, и оставшуюся часть жирных кислот медленно добавляют в реактор в течение 5 часов со скоростью потока, которая позволяет сохранять низкую концентрацию жирных кислот в растворе (без аккумулирования свободной кислоты в растворе). В конце реакции капельную воронку меняют на дистилляционный аппарат, и жирные кетоны выделяют дистилляцией (290°C-340°C, 5 кПа).
Первую партию из 141 г жирного кетона выделяют в виде белого воска.
Остаток в колбе реактора, состоящий в основном из солей железа, применяют для превращения оставшихся 200 г жирных кислот во втором цикле. Для этого дистилляционный аппарат меняют на капельную воронку, содержащую 200 г расплавленных жирных кислот, и повторяют стадии процесса, описанные выше.
Общий выход реакции после этих 2 циклов составляет: 79%, выделенный в виде белого воска.
Пример 3 - Превращение внутренних кетонов во вторичные спирты
В этом примере описано гидрирование кетонов, полученных в соответствии с данным изобретением для получения соответствующих вторичных спиртов жирных кислот. Реакцию проводят без растворителя с применением гетерогенного Pd/C (3%) в качестве катализатора в автоклаве, оборудованном турбинной мешалкой Раштона.
Гидрирование проводят с фракцией внутренних жирных кетонов, полученной реакцией конденсации, проводимой с фракцией C12-C18 кокосовых жирных кислот по методике, описанной в примере 2.
Реакцию проводят в 750 мл автоклаве, оборудованном турбинной мешалкой Раштона. 28 г Pd/C (3%) и 280 г жирных кетонов вводят в реактор, который герметично закрывают. Затем температуру доводят до 80°C, и смесь перемешивают при 1000 об./мин. Атмосферу реактора продувают 3 раза 4 МПа азота, затем 3 раза 3 МПа водорода. Затем температуру повышают до 150°C, и смесь перемешивают при этой температуре, поддерживая 3 МПа водорода до завершения реакции (отслеживают ГХ анализом). В конце реакции смесь охлаждают до 80°C, и реактор продувают азотом. 1-ю партию продукта (180 г) получают фильтрацией, и оставшуюся часть экстрагируют с применением 400 мл горячего толуола. После выпаривания растворителя получают общее количество 247 г белого твердого вещества, соответствующее фактическому выходу 88%.
Пример 4 - Дегидрирование вторичных спиртов до внутренних олефинов
В этом примере вторичные жирные спирты, полученные в примере 3, дегидрируют при ограниченной изомеризации C=C связи. Реакцию проводят без растворителя и с применением Al2O3-η в качестве катализатора. Воду, образующуюся во время реакции, улавливают аппаратом Дина-Старка.
Полученные олефины являются внутренними олефинами с длинной прямой цепью с C=C двойной связью, расположенной вокруг середины цепи. Структура олефина в основном определяется структурой исходных спиртов, полученных на 2-й стадии. Если исходная фракция жирных кислот не очень широкая (например, фракция C12-C18), полученный кетон и спирты, получаемые гидрированием, являются практически симметричными с -OH, расположенной почти в середине цепи. Поэтому после гидрирования C=C двойная связь расположена близко к середине цепи.
Реакцию проводят под аргоном.
47 г фракции внутренних спиртов, полученной в примере 3, затем 4,7 г Al2O3-η добавляют в круглодонную колбу, оборудованную аппаратом Дина-Старка и магнитной мешалкой. Затем смесь перемешивают при 300°C в течение 2 часов. После завершения реакции продукт экстрагируют 150 мл горячего толуола. После выпаривания растворителя продукт получают в виде бледно-желтой жидкости (39 г), соответствующей фактическому выходу 87%.
Продукт включает фракцию внутренних жирных олефинов с C=C связью, расположенной в середине цепи. Если начинают с фракции кокосовых жирных кислот C12-C18 с четным количеством атомов углерода, полученные олефины имеют нечетное количество атомов углерода. Массовое распределение олефинов в зависимости от массового распределения исходных жирных кислот соответствует приблизительно биномиальному распределению.
Пример 5 - Сравнительный пример
Лауриновую кислоту смешивают с 12,5% моль. железного порошка и нагревают до 298°C (температура кипения лауриновой кислоты) и хранят при этой температуре в течение 5 часов. Затем определяют композицию реакционного продукта. Выход 12-трикозанона составляет только 18% и образуется значительное количество ундекана (8%). Более того, все еще присутствуют значительные количества непрореагировавшей лауриновой кислоты (общее превращение лауриновой кислоты составляет 46%).
Этот сравнительный пример показывает, что добавлением полного количества кислоты в одну стадию и не постепенно не дает желаемые кетоны с удовлетворительным выходом, кроме того образуется значительное количество нежелательных побочных продуктов.
Пример 6 - Синтез нонадекан-10-она (дикетон C10 каприновой кислоты)
Реакцию проводят под аргоном в 250-мл круглодонной колбе, оборудованной механической мешалкой, аппаратом Дина-Старка и капельной воронкой. В реакторе распределяют 2,0 г (35,8 ммоль) железного порошка и 50 г (290,4 ммоль) каприновой кислоты вводят в капельную воронку.
Первую часть количества 12,5 г каприновой кислоты добавляют в реактор и температуру доводят до 250°C. Смесь перемешивают при этой температуре в течение 1 ч 45 мин. В это время цвет среды меняется на черный и выделяется газообразный H2. Анализ ИСОПФ неочищенной смеси показал завершение образования промежуточного карбоксилата железа.
Затем температуру повышают до 315°C, и смесь перемешивают в течение 1 ч 30 мин для превращения комплекса карбоксилата железа в кетон, CO2 и оксид железа.
Оставшееся количество каприновой кислоты (37,5 г) затем медленно добавляют в реактор в течение 5 ч со скоростью потока, которая позволяет сохранять концентрацию каприновой кислоты в реакционной среде очень низкой (без аккумулирования свободной кислоты в растворе). На практике это может осуществляться последовательным медленным добавлением частей по 12,5 г каприновой кислоты каждые 1,5 ч.
После завершения добавления каприновой кислоты смесь перемешивают при 315°C до тех пор, пока промежуточный комплекс железа не перестанет определяться ИСОПФ.
После завершения реакции смесь охлаждают при комнатной температуре и 200 мл CHCl3 добавляют в неочищенную среду. Смесь перемешивают при 40°C для солюбилизации продукта (нонадекан-10-он). Полученную суспензию фильтруют на слое двуокиси кремния и элюируют 1,5 л хлороформа. Выпаривание растворителя дает 39,7 г (140,5 ммоль) продукта нонадекан-10-она в виде аналитически чистого желтого порошка (97% фактический выход).
Пример 7 - Синтез фракции C15-C35 кетонов, начиная с фракции C8-C18 кокосовых насыщенных жирных кислот
Реакцию проводят под аргоном в 750-мл реакторе, оборудованном механической мешалкой, аппаратом Дина-Старка и капельной воронкой. В реакторе распределяют 6,8 г (0,12 моль) железного порошка и 200 г (0,97 моль) фракции кокосовых насыщенных жирных кислот (со следующим распределением: C8: 7% масс., C10: 8% масс., C12: 48% масс., C14: 17% масс., C16: 10% масс., C18: 10% масс.) вводят в капельную воронку.
Первую часть количества 50 г жирных кислот добавляют в реактор и температуру доводят до 250°C. Смесь перемешивают при этой температуре в течение 4 ч. В это время цвет среды меняется на черный и выделяется газообразный H2. Анализ ИСОПФ неочищенной смеси показал завершение образования промежуточных комплексов карбоксилата железа.
Затем температуру повышают до 330°C, и смесь перемешивают при этой температуре в течение 2 ч. В это время промежуточные комплексы карбоксилата железа расщепляются на жирные кетоны, оксид железа и CO2.
Оставшиеся жирные кислоты (150 г) медленно вводят в реактор в течение 5 ч с такой скоростью потока, что температура реакционной среды не снижается ниже 320°C и которая позволяет сохранять концентрацию жирных кислот в реакционной среде очень низкой. Средняя скорость потока добавления около 25 г жирных кислот/час оказалась удовлетворительной. На практике, это достигается постепенным медленным добавлением (1 час на добавление) 3 частей по 50 г расплавленных жирных кислот, затем перемешивают в течение 1 часа при 330°C между добавлениями.
В конце третьего и последнего добавления, неочищенную среду перемешивают при 330°C в течение 2 ч, и развитие реакции отслеживают ИСОПФ.
После завершения реакции (комплекс железа не определяется ИСРПФ) смесь охлаждают при комнатной температуре и 400 мл CHCl3 добавляют в неочищенную среду. Смесь перемешивают при 40°C для солюбилизации продукта (C15-C35 кетонов). Полученную суспензию фильтруют на слое двуокиси кремния (400 г) и элюируют 3 л хлороформа. Выпаривание растворителя дает 161 г (0,46 моль) продукта C15-C35 кетонов в виде аналитически чистого белого воска (95% фактический выход).
Если описание любых патентов, заявок на патенты и публикаций, которые включены сюда в качестве ссылок, вступает в противоречие с описание данной заявки до такой степени, что может сделать определение неясным, данное описание будет предпочтительным.
Полиамидная огнестойкая композиция
Способ получения соединений, содержащих нитрильные группы
Композиция на основе оксида циркония, оксида титана или смешанного оксида циркония и титана, нанесенная на носитель из оксида алюминия или оксигидроксида алюминия, способы ее получения и ее применение в качестве катализатора
Способ получения соединений, содержащих функциональные нитрильные группы
Вязкоупругая композиция с улучшенной вязкостью
Композиция, содержащая лантансодержащий перовскит на подложке из алюминия или из оксигидроксида алюминия, способ получения и применение в катализе
Способ получения соединений, содержащих нитрильные группы
Способ получения алкилгидропероксидных соединений
Новый способ получения осажденных кремнеземов, осажденные кремнеземы с особой морфологией, гранулометрическим составом и пористостью и их применение, в частности, для усиления полимеров
Способ получения дифторуксусной кислоты
Способ получения соединений, содержащих функциональные нитрильные группы
Способ получения миндальных ароматических соединений и ароматических альдегидных соединений

