Результат интеллектуальной деятельности: МЕДИКАМЕНТЫ, ПРИМЕНЕНИЯ И СПОСОБЫ
Вид РИД
Изобретение
Настоящее изобретение относится к композиции, содержащей молекулу антитела и агент для применения в лечении рефрактерного рака и/или рецидивного рака. Также изобретение относится к способу лечения рефрактерного рака и/или рецидивного рака, включающему введение молекулы антитела и агента. Также описаны наборы, содержащие молекулу антитела и агенты.
Терапевтические антитела изменили терапию рака, раскрыв новые механизмы действия, задействующие иммунную систему. Например, механизм, при помощи которого антитела могут оказывать терапевтическое действие, состоит в стимуляции уничтожения раковых и других нежелательных клеток путем подключения естественных эффекторных систем, таких как цитотоксические клетки (например, макрофаги) и ферменты (например, комплемент), которые потом нацеливаются на клетку, с которой связана молекула антитела.
Специфическое к CD20 моноклональное антитело (mAb) ритуксимаб было первым антителом, разрешенным к применению в иммунотерапии рака, и, как следствие, широко применялось в случае пациентов с CD20-экспрессирующим В-клеточным раком, включая фолликулярную лимфому (ФЛ), диффузную крупноклеточную В-клеточную лимфому (ДКВКЛ), хронический лимфоцитарный лейкоз (ХЛЛ) и мантийноклеточную лимфому (МКЛ) (обзор приведен в Lim, S.H. et al., Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica 95, 135-143 (2010).
Хотя терапия антителами является эффективной, в некоторых случаях у пациента развивается устойчивость к терапии антителами, что приводит к неэффективности лечения и ухудшению ракового состояния. В других случаях рак может вообще быть невосприимчивым к терапии рака.
К сожалению, до сих пор отсутствует эффективное лечение терапевтическими антителами для рецидивных видов рака и/или рефрактерных видов рака. Учитывая увеличивающееся число разрабатываемых видов терапии антителами для лечения нескольких типов рака, появляется необходимость в понимании устойчивости раковых клеток к этим видам терапии и разработке лекарственных препаратов для преодоления такой устойчивости.
Например, в случае восприимчивых к ритуксимабу лимфом некоторые индивиды демонстрируют устойчивость на первом этапе лечения или становятся устойчивыми к комбинированной терапии, включающей применение ритуксимаба. Кроме ритуксимаба, лимфомы также могут проявлять устойчивость к терапии другими антителами; например, молекулами антитела офатумумаба (который связывается с CD20) или алемтузумаба (который связывается с CD52).
Учитывая сложившуюся ситуацию, авторы настоящего изобретения неожиданно обнаружили, что для лечения рецидивного рака и/или рефрактерного рака можно использовать лечение, включающее применение антитела и агента, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела.
Ранее было показано, что ингибирование взаимодействий между FcγRIIb и терапевтическим антителом ритуксимабом может снижать рециклинг антитела и улучшать эффективность ритуксимаба при лечении хронического лимфоцитарного лейкоза и мантийноклеточной лимфомы (Lim, S.H., et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood (2011) и WO 2012/022985).
Дополнительно известно, что активностью терапевтических антител частично управляет их взаимодействие с Fc-гамма-рецепторами (FcγR). В частности, относительный уровень экспрессии, аффинность и активность FcγR объясняют терапевтическую активность IgG (обзор приведен в Nimmerjahn, F. & Ravetch, J.V. FcgammaRs in health and disease. Curr Top Microbiol Immunol 350, 105-125 (2011), и Nimmerjahn, F. & Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nature reviews 8, 34-47 (2008)).
Однако авторы настоящего изобретения неожиданно продемонстрировали, что для лечения рецидивного рака и/или рефрактерного рака можно использовать комбинированную терапию, включающую применение антитела и агента, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела. Таким образом, в настоящем изобретении предложены способы и применения для лечения подгруппы пациентов с рецидивным раком и/или рефрактерным раком.
Другими словами, авторы настоящего изобретения продемонстрировали, что комбинированную терапию, включающую применение антитела, к которому невосприимчив (т.е. устойчив) рак у субъекта, в комбинации с агентом, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела, можно использовать для лечения рецидивного рака и/или рефрактерного рака путем снижения и/или преодоления устойчивости рака к антителу.
В первом аспекте в изобретении предложена композиция, содержащая:
(i) молекулу антитела, которая специфически связывает антиген клеточной поверхности клетки-мишени, при этом молекула антитела содержит Fc-домен, способный связывать FcγRIIb; в комбинации с
(ii) агентом, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела;
отличающаяся тем, что композиция предназначена для применения в лечении рецидивного рака и/или рефрактерного рака у субъекта, а субъект имеет клетки-мишени, экспрессирующие FcγRIIb.
Во втором аспекте в изобретении предложено применение композиции, содержащей:
(i) молекулу антитела, которая специфически связывает антиген клеточной поверхности клетки-мишени, при этом молекула антитела содержит Fc-домен, способный связывать FcγRIIb; в комбинации с
(ii) агентом, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела;
отличающееся тем, что применение имеет место при производстве медикамента для применения в лечении рецидивного рака и/или рефрактерного рака у субъекта, а субъект имеет клетки-мишени, экспрессирующие FcγRIIb.
В третьем аспекте в изобретении предложен способ лечения рецидивного рака и/или рефрактерного рака у субъекта, включающий введение:
(i) молекулы антитела, которая специфически связывает антиген клеточной поверхности клетки-мишени, при этом молекула антитела содержит Fc-домен, способный связывать FcγRIIb; в комбинации с
(ii) агентом, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела;
отличающийся тем, что субъект выбран на основании наличия у него рецидивного рака и/или рефрактерного рака и наличия у субъекта клеток-мишеней, экспрессирующие FcγRIIb.
В четвертом аспекте в изобретении предложен набор для применения в лечении рецидивного рака и/или рефрактерного рака у субъекта, содержащий:
(i) молекулу антитела, которая специфически связывает антиген клеточной поверхности клетки-мишени, при этом молекула антитела содержит Fc-домен, способный связывать FcγRIIb;
(ii) агент, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела;
(iii) одно или более вещество, выбранное из группы, состоящей из: ритуксимаба; биоаналога ритуксимаба; офатумумаба; обинутузумаба; алемтузумаба; галиксимаба; тозитумомаба; тозитумомаба, конъюгированного с радиоактивным веществом; ибритутомаба; ибритутомаба, конъюгированного с радиоактивным веществом; анти-CD40 антитела; анти-CD19 антитела; анти-CD37 антитела; терапевтического антитела, применяемого для лечения В-клеточного рака;
отличающийся тем, что применение предназначено для лечения рецидивного рака и/или рефрактерного рака у субъекта, а субъект имеет клетки-мишени, экспрессирующие FcγRIIb.
В предпочтительном варианте реализации четвертого аспекта изобретения одно или более вещество содержит или состоит из: ритуксимаба (или биоаналога ритуксимаба) и офатумумаба; или ритуксимаба (или биоаналога ритуксимаба) и обинутузумаба.
В альтернативном предпочтительном варианте реализации четвертого аспекта изобретения одно или более вещество содержит или состоит из: алемтузумаба и анти-CD40 антитела; алемтузумаба и анти-CD19 антитела; алемтузумаба и анти-CD370 антитела; или алемтузумаба и анти-CD40 антитела, и анти-CD19 антитела, и анти-CD37 антитела.
В альтернативном предпочтительном варианте реализации четвертого аспекта изобретения одно или более вещество содержит или состоит из: терапевтического антитела, применяемого для лечения В-клеточного рака, такого как галиксимаб.
Молекулы антител хорошо известны специалистам в области техники иммунологии и молекулярной биологии. Молекула антитела является компонентом гуморальной иммунной системы и представляет собой белок, вырабатываемый эффекторными В-клетками, для выявления и нейтрализации объектов, чужеродных для организма, таких как бактерии и вирусы.
Как правило, молекула антитела содержит две тяжелых цепи и две легких цепи. Тяжелая цепь молекулы антитела содержит один вариабельный домен и три константных домена, а легкая цепь молекулы антитела содержит один вариабельный домен и один константный домен. Вариабельные домены (иногда называемые вместе FV-областью) связываются с мишенью антитела, при этом каждый вариабельный домен содержит три петли, называемые определяющими комплементарность областями (CDR), которые отвечают за связывание мишени.
Соответственно, в термин «молекула антитела» включены все типы и классы молекул антител и их функциональные фрагменты, включая: моноклональные антитела или поликлональные антитела, или синтетические антитела, или рекомбинантно полученные антитела, или мультиспецифические антитела, или биспецифические антитела, или человеческие антитела, или гуманизированные антитела, или химерные антитела, или верблюдизированные антитела, или одноцепочечные Fv (scFv), или одноцепочечные антитела, или фрагменты Fab, или фрагменты F(ab'), или дисульфидно-связанные Fv (sdFv), или интратела, или тяжелые цепи антител, или легкие цепи антитела, или гомодимеры или гетеродимеры тяжелых и/или легких цепей антител, или их антигенсвязывающие функциональные фрагменты или производные, или IgG, или IgG1, или IgG2, или IgG3, или IgG4, или IgA, или IgM, или IgD, или IgE.
Известно, что молекула антитела специфически связывает определенную молекулу-мишень. Иными словами, молекула антитела предпочтительно и избирательно связывает свою мишень, а не молекулу, которая не является мишенью.
Способы оценки связывания белка известны специалисту в области биохимии и иммунологии. Специалисту должно быть понятно, что эти способы можно применять для оценки связывания молекулы антитела с мишенью и/или связывания Fc-домена антитела с Fc-рецептором; а также относительной силы или специфичности, или ингибирования, или предотвращения, или снижения таких взаимодействий. Примерами способов, которые можно применять для оценки связывания белка, являются, например, иммуноанализ, BIAcore, вестерн-блоттинг, радиоиммуноанализ (РИА) и ферментный иммуносорбентный анализ (ELISA) (Смотрите Fundamental Immunology Second Edition, Raven Press, New York, страницы 332-336 (1989), в отношении обсуждения специфичности антител).
Соответственно, под «молекулой антитела, которая специфически связывает», подразумевается, что молекула антитела специфически связывает мишень, но не связывается с не-мишенью или слабее (например, с более низкой аффинностью) связывается с не-мишенью, чем с мишенью.
Также подразумевается, что молекула антитела специфически связывается с мишенью по меньшей мере в два раза сильнее или по меньшей мере в пять раз сильнее, или по меньшей мере в 10 раз сильнее, или по меньшей мере 20 раз сильнее, или по меньшей мере 50 раз сильнее, или по меньшей мере в 100 раз сильнее, или по меньшей мере в 200 раз сильнее, или по меньшей мере в 500 раз сильнее, или по меньшей мере в около 1000 раз сильнее, чем с не-мишенью.
Кроме того, подразумевается, что молекула антитела специфически связывается с мишенью, если она связывается с мишенью с Kд, составляющей по меньшей мере около 10-1 Kд, или по меньшей мере около 10-2 Kд, или по меньшей мере около 10-3 Kд, или по меньшей мере около 10-4 Kд, или по меньшей мере около 10-5 Kд, или по меньшей мере около 10-6 Kд, или по меньшей мере около 10-7 Kд, или по меньшей мере около 10-8 Kд, или по меньшей мере около 10-9 Kд, или по меньшей мере около 10-10 Kд, или по меньшей мере около 10-11 Kд, или по меньшей мере около 10-12 Kд, или по меньшей мере около 10-13 Kд, или по меньшей мере около 10-14 Kд, или по меньшей мере около 10-15 Kд.
Другой важной частью молекулы антитела является Fc-домен (также известный как домен кристаллизующегося фрагмента), который содержит два константных домена каждой из тяжелых цепей молекулы антитела. Fc-домен отвечает за взаимодействия между молекулой антитела и Fc-рецептором.
Fc-рецепторы представляют собой мембранные белки, которые часто можно обнаружить на клеточной поверхности клеток иммунной системы (т.е. Fc-рецепторы находятся на мембране клетки-мишени, также известной как плазматическая мембрана или цитоплазматическая мембрана). Роль Fc-рецепторов состоит в обеспечении связывания антител посредством Fc-домена и интернализации антитела клеткой. В иммунной системе это может приводить к антитело-опосредованному фагоцитозу и антитело-зависимой клеточно-опосредованной цитотоксичности.
Примером Fc-рецептора, включенного в настоящее изобретение, является FcγRIIb (также известный как CD32, CD32B, CD32B1, CD32B2, FcRII, FcγRII или FcRIIB), который представляет собой ингибиторный Fc-рецептор, отвечающий за связывание с антителами и за рециклинг антител.
Соответственно, под «Fc-доменом, способным связывать FcγRIIb» подразумевается, что Fc-домен молекулы антитела согласно изобретению способен связывать FcγRIIb, а связывание предпочтительно по меньшей мере в два раза сильнее, или по меньшей мере в пять раз сильнее, или по меньшей мере в 10 раз сильнее, или по меньшей мере в около 20 раз сильнее, или по меньшей мере в 50 раз сильнее, или по меньшей мере в 100 раз сильнее, или по меньшей мере в 200 раз сильнее, или по меньшей мере в 500 раз сильнее, или по меньшей мере в 1000 раз сильнее, чем связывание молекулы антитела с другим белком или пептидом, или полипептидом, или Fc-рецептором.
Агент согласно изобретению предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела, что предотвращает или снижает интернализацию молекулы антитела клеткой.
Для специалистов в области молекулярной биологии должно быть очевидно, что может представлять собой агент согласно изобретению, и как можно определить агент согласно изобретению. Например, агенты согласно изобретению можно определить, проводя скрининг в отношении агентов, которые блокируют стимуляцию или сигналинг FcγRIIb, о чем свидетельствует фосфорилирование тирозина-293 во внутриклеточном иммунорецепторном тирозиновом ингибирующем мотиве (ITIM) согласно данным вестерн-блоттинга. Например, можно культивировать клетки Raji с молекулой антитела к антигену клеточной поверхности, например, анти-CD20 ритуксимабом, в присутствии или отсутствие испытуемого анти-FcγRIIb агента перед проведением иммуноблоттинга в отношении фосфорилированного FcγRIIb (WO 2012/022985).
В Neubig et al. (2003) Pharmacol. Rev. 55, 597-606, включенной в данный документ посредством ссылки, описаны различные классы молекул, среди которых можно поводить скрининг для определения агентов, которые предотвращают или снижают связывание FcγRIIb с Fc-доменом молекулы антитела.
Агент согласно изобретению может быть небольшим органическим соединением или небольшим неорганическим соединением, или пептидом, или полипептидом, или пептидомиметиком, или нуклеиновой кислотой, или пептидной нуклеиновой кислотой (ПНК), или аптамером, или липидом, или углеводом, или молекулой антитела.
Под «агентом, который предотвращает или снижает связывание FcγRIIb» подразумевается, что агент полностью блокирует связывание FcγRIIb с Fc-доменом молекулы антитела или частично блокирует связывание FcγRIIb с Fc-доменом молекулы антитела.
Под «полностью блокирует» подразумевается отсутствие выявляемого связывания между FcγRIIb и Fc-доменом молекулы антитела. Под «частично блокирует» подразумевается, что выявляемое связывание между FcγRIIb и Fc-доменом молекулы антитела ниже в присутствии агента, чем выявляемое связывание между FcγRIIb и Fc-доменом молекулы антитела в отсутствие агента.
Агент может предотвращать или снижать связывание FcγRIIb посредством стерического несоответствия и/или посредством связывания с молекулой антитела, и/или посредством связывания с Fc-доменом, и/или связывания с FcγRIIb, и/или посредством связывания с молекулой антитела и блокирования контакта с FcγRIIb, и/или посредством связывания с Fc-доменом и блокирования контакта с FcγRIIb, и/или посредством связывания FcγRIIb и блокирования контакта с Fc-доменом.
Также подразумевается, что агент снижает связывание FcγRIIb с Fc-доменом молекулы антитела, если в присутствии агента связывание составляет менее чем около 90% или менее чем около 80%, или менее чем около 70%, или менее чем около 60%, или менее чем около 50%, или менее чем около 40%, или менее чем около 30%, или менее чем около 20%, или менее чем около 10%, или менее чем около 5%, или менее чем около 1% от связывания FcγRIIb с Fc-доменом молекулы антитела в отсутствие агента.
Дополнительно подразумевается, что агент снижает связывание FcγRIIb с Fc-доменом молекулы антитела, если в присутствии агента связывание по меньшей мере в два раза слабее, или по меньшей мере в пять раз слабее, или по меньшей мере в 10 раз слабее, или по меньшей мере в 20 раз слабее, или по меньшей мере в 50 раз слабее, или по меньшей мере в 100 раз слабее, или по меньшей мере в 200 раз слабее, или по меньшей мере в 500 раз слабее, или по меньшей мере в 1000 раз слабее, чем связывание FcγRIIb с Fc-доменом молекулы антитела в отсутствие агента.
Подразумевается, что агент предотвращает связывание FcγRIIb с Fc-доменом молекулы антитела, если в присутствии агента связывание не выявляется или, если связывание все же выявляется, то выявляемое связывание является незначительным.
Примером мишени для молекулы антитела, которая включена в данное изобретение, является антиген клеточной поверхности, который может быть эпитопом (также известным в данном контексте как эпитоп клеточной поверхности) для молекулы антитела. Термины «антиген» и «эпитоп клеточной поверхности» должны быть понятны специалисту в области иммунологии или клеточной биологии.
Под «антигеном клеточной поверхности» подразумевается, что антиген клеточной поверхности расположен на внеклеточной стороне клеточной мембраны, хотя он может быть только временно расположен на внеклеточной стороне клеточной мембраны. Под «временно расположен» подразумевается, что антиген клеточной поверхности может интернализоваться клеткой или высвобождаться из внеклеточной части клеточной мембраны во внеклеточное пространство. Антиген клеточной поверхности может высвобождаться из внеклеточной части клеточной мембраны посредством расщепления, которое может опосредоваться протеазами.
Также подразумевается, что антиген клеточной поверхности может быть связан с клеточной мембраной, хотя он может быть только временно ассоциирован с клеточной мембраной. Под «временно ассоциирован» подразумевается, что антиген клеточной поверхности может высвобождаться из внеклеточной части клеточной мембраны во внеклеточное пространство. Антиген клеточной поверхности может высвобождаться из внеклеточной части клеточной мембраны посредством расщепления, которое может опосредоваться протеазами.
Дополнительно подразумевается, что антиген клеточной поверхности может представлять собой пептид или полипептид, или углевод, или олигосахаридную цепь, или липид; и/или эпитоп, который находится на белке или гликопротеине, или липопротеине.
Заболеванием, лечение которого проводят в соответствии с настоящим изобретением, является рецидивный рак и/или рефрактерный рак.
Рецидивный рак представляет собой рак, лечение которого проводили ранее, и в результате этого лечения субъект полностью или частично выздоровел (т.е. говорят, что субъект находится на стадии ремиссии), но после прекращения лечения рак появился снова или ухудшился. Иными словами, рецидивный рак - это рак, который стал устойчив к лечению после периода времени, в течение которого оно было эффективным, а субъект полностью или частично выздоровел.
Следует понимать, что рак может представлять собой рецидивный рак или рецидивный рак и рефрактерный рак вследствие приобретенной устойчивости. Под «приобретенной устойчивостью» подразумевается, что рак и/или субъект, и/или клетка-мишень не были устойчивы к конкретному виду лечения до его первого применения, но стали устойчивыми после или во время по меньшей мере его первого применения, например: после второго применения; после третьего применения; после четвертого применения; после пятого применения; после шестого применения; после седьмого применения; после восьмого применения; после девятого применения; после десятого применения; после одиннадцатого применения; после двенадцатого применения лечения.
В контексте настоящего изобретения предпочтительно, чтобы рецидивный рак ранее лечили при помощи антитела, и он приобрел устойчивость к этому антителу. Как обсуждается в данном документе, в настоящем изобретении предложены средства для лечения подгруппы пациентов, которые имеют такой рецидивный рак, что означает, что в изобретении предложены средства для лечения субъекта, имеющего рецидивный рак, который устойчив к лечению антителом.
В контексте настоящего изобретения также предпочтительно, чтобы рецидивный рак ранее лечили при помощи определенной в данном документе молекулы антитела, и он приобрел устойчивость к этой молекуле антитела. Как обсуждается в данном документе, в настоящем изобретении предложены средства для лечения подгруппы пациентов, которые имеют такой рецидивный рак, что означает, что в изобретении предложены средства для лечения субъекта, имеющего рецидивный рак, который устойчив к определенной в данном документе молекуле антитела.
Следует понимать, что таким образом в настоящем изобретении предложены средства для лечения рака у субъекта при помощи той же молекулы антитела, к которой рак стал устойчив.
Рефрактерный рак представляет собой рак, лечение которого проводили, но он был невосприимчив к этому лечению, и/или рак, лечение которого проводили, но он прогрессировал во время лечения. Иными словами, рефрактерный рак - это рак, устойчивый к лечению.
Следует понимать, что рак может являться рефрактерным раком вследствие первичной устойчивости. Под «первичной устойчивостью» подразумевается, что рак и/или субъект, и/или клетка устойчивы к конкретному виду лечения, начиная с первого применения.
В контексте настоящего изобретения предпочтительно, чтобы рефрактерный рак ранее лечили при помощи антитела, но он был устойчив к этому антителу. Как обсуждается в данном документе, в настоящем изобретении предложены средства для лечения подгруппы пациентов, которые имеют такой рефрактерный рак, что означает, что в изобретении предложены средства для лечения субъекта, имеющего рефрактерный рак, который устойчив к лечению антителом.
В контексте настоящего изобретения также предпочтительно, чтобы рефрактерный рак ранее лечили при помощи определенной в данном документе молекулы антитела, но он был устойчив к этой молекуле антитела. Как обсуждается в данном документе, в настоящем изобретении предложены средства для лечения подгруппы пациентов, которые имеют такой рефрактерный рак, что означает, что в изобретении предложены средства для лечения субъекта, имеющего рефрактерный рак, который устойчив к определенной в данном документе молекуле антитела.
Следует понимать, что таким образом в настоящем изобретении предложены средства для лечения рака у субъекта при помощи той же молекулы антитела, к которой рак устойчив.
Рецидивный рак и/или рефрактерный рак легко может быть диагностирован специалистом в области медицины и дополнительно обсуждается ниже.
Авторы изобретения неожиданно показали, что лечение молекулой антитела и агентом, который блокирует связывание FcγRIIB с молекулой антитела, можно применять для лечения рецидивного рака и/или рефрактерного рака. Как продемонстрировано в прилагаемых Примерах, молекула антитела и агент согласно изобретению в особенности эффективны для лечения рефрактерного хронического лимфоцитарного лейкоза и/или рецидивного хронического лимфоцитарного лейкоза.
Авторы изобретения полагают, что лечение молекулой антитела и агентом согласно изобретению оказывает действие, так как FcγRIIB снижает эффективность молекул терапевтических антител при лечении рецидивного рака и/или рефрактерного рака вследствие интернализации этих молекул антител. В свете своих открытий авторы изобретения полагают, что такие молекулы антител и агенты согласно изобретению будут эффективны в лечении любого рецидивного рака и/или рефрактерного рака, который можно лечить молекулой терапевтического антитела и в случае которого клетки-мишени субъекта экспрессируют FcγRIIB.
Следует принимать во внимание, что многие виды В-клеточного рака экспрессируют FcγRIIB, хотя и на разных уровнях. Экспрессия FcγRIIB наиболее выражена при хроническом лимфоцитарном лейкозе и мантийноклеточных лимфомах, умеренно - при диффузной крупноклеточной В-клеточной лимфоме и наименее выражена при фолликулярных лимфомах. Однако в некоторых случаях субъекты с раками, которые обычно экспрессируют низкие уровни FcγRIIB (например, фолликулярными лимфомами), могут иметь очень высокие уровни экспрессии FcγRIIB. Уровень экспрессии FcγRIIB при разных типах В-клеточного рака (и, в частности, упомянутых выше) коррелирует с уровнем интернализации молекулы антитела ритуксимаба. Следовательно, считается, что экспрессия FcγRIIB и связанная с ней интернализация молекул антител является общим механизмом, свойственным В-клеточным ракам (Lim et al., 2011). FcγRIIB-зависимую интернализацию молекулы антитела можно блокировать при помощи раскрытых в данном документе антител к FcγRIIB (Примеры, в частности, Фигура 3).
Соответственно, молекулу антитела и агент согласно изобретению можно применять в лечении В-клеточных раков и, в частности, рецидивной мантийноклеточной лимфомы и/или рефрактерной мантийноклеточной лимфомы, и/или рецидивной фолликулярной лимфомы и/или рефрактерной фолликулярной лимфомы, и/или рецидивной диффузной крупноклеточной В-клеточной лимфомы и/или рефрактерной диффузной крупноклеточной В-клеточной лимфомы.
В контексте изобретения для того, чтобы проводить лечение субъекта с рецидивным раком и/или рефрактерным раком, у субъекта должны быть клетки-мишени, экспрессирующие FcγRIIb. Следует понимать, что FcγRIIb является рецептором клеточной поверхности и, следовательно, будет присутствовать на поверхности клеток-мишеней. Специалисту в данной области техники должно быть понятно, что для оценки наличия FcγRIIb можно использовать разные биологические маркеры; например, белок FcγRIIb и/или мРНК FcγRIIb. Для специалиста должно быть очевидно существование известных в данной области техники способов для определения белка FcγRIIb; например, иммуногистохимия, вестерн-блоттинг, метод количественного определения белка по Бредфорду, проточная цитометрия и выявление при помощи антитела AT-10. Для специалиста должно быть очевидно существование известных в данной области техники способов для определения мРНК FcγRIIb; например, нозерн-блоттинг, секвенирование РНК, количественная ПЦР и микроматричная гибридизация.
Для специалиста должно быть очевидно, что, чтобы оценить наличие FcγRIIb, может быть необходимо сравнение уровня биологического маркера FcγRIIb в клетке-мишени с уровнем биологического маркера FcγRIIb в контрольной клетке, которая не экспрессирует FcγRIIb. Такая контрольная клетка может представлять собой контрольную клетку от индивида, который не экспрессирует FcγRIIb, или контрольную клетку, которая не экспрессирует FcγRIIb, от индивида, или контрольную клетку, которая не экспрессирует FcγRIIb, от субъекта, или клетку, которая была генетически сконструирована так, чтобы не экспрессировать FcγRIIb.
Под «клетками-мишенями, экспрессирующие FcγRIIb» подразумевается, что клетки-мишени экспрессируют белок FcγRIIb и/или мРНК FcγRIIb. Также подразумевается, что клетка-мишень может быть определена как экспрессирующая FcγRIIb, если содержание белка FcγRIIb и/или мРНК FcγRIIb более чем в два раза выше, или более чем в пять раз выше, или более чем в 10 раз выше, или более чем в 20 раз выше, или более чем в 50 раз выше, или более чем в 100 раз выше, или более чем в 200 раз выше, или более чем в 500 раз выше, или более чем в 1000 выше в клетке-мишени, чем в контрольной клетке.
Специалисту в области медицины должно быть известно, что лекарственные средства можно модифицировать разными добавками, например, чтобы изменить скорость абсорбции лекарственного средства в организме; и можно модифицировать в разные формы, например, чтобы сделать возможным конкретный способ введения в организм.
Соответственно, подразумевается, что композицию и/или антитело, и/или агент, и/или медикамент согласно изобретению можно комбинировать со вспомогательным веществом и/или фармацевтически приемлемым носителем, и/или фармацевтически приемлемым разбавителем, и/или адъювантом.
Также подразумевается, что композиция и/или антитело, и/или агент, и/или медикамент согласно изобретению могут быть предназначены для парентерального введения, включая водные и/или неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты и/или буферы, и/или бактериостатические агенты, и/или растворенные вещества, которые обеспечивают изотоничность препарата с кровью предполагаемого реципиента; и/или водные и/или неводные стерильные суспензии, которые могут содержать суспендирующие агенты и/или загустители. Композиция и/или антитело, и/или агент, и/или медикамент согласно изобретению могут находиться в однодозовых или многодозовых емкостях, например, запечатанных ампулах и флаконах, и могут храниться в вымороженном (т.е. лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением.
Растворы и суспензии для немедленного применения можно готовить из стерильных порошков и/или гранул, и/или таблеток, таких как описанные ранее.
В случае парентерального введения пациентам дневной дозировочный уровень композиции и/или антитела, и/или агента, и/или медикамента согласно изобретению обычно составляет от 1 пг до 10 мг на взрослого человека в день, которые вводят в виде одной дозы или разделенных доз. В любом случае фактическую дозировку, которая будет наиболее подходящей для каждого отдельного пациента и которая варьируется в зависимости от возраста, массы и восприимчивости конкретного пациента, определяет лечащий врач. Приведенные выше дозировки являются иллюстративными для усредненного случая. Конечно, могут быть отдельные случаи, когда требуются более высокие или низкие диапазоны дозировок, и такие случаи входят в объем данного изобретения. Как правило, композиция и/или медикамент согласно изобретению содержит антитело и/или агент согласно изобретению в концентрации, составляющей приблизительно от 2 мг/мл до 150 мг/мл или приблизительно от 2 мг/мл до 200 мг/мл. В предпочтительном варианте реализации изобретения медикаменты и/или композиции согласно изобретению содержат антитело и/или агент согласно изобретению в концентрации, составляющей 10 мг/мл.
В общем случае для людей предпочтительным способом является пероральное или парентеральное введение композиции и/или антитела, и/или агента, и/или медикамента согласно изобретению, так как оно наиболее удобно. В случае ветеринарного применения композицию и/или антитело, и/или агент, и/или медикамент согласно изобретению вводят в виде подходящего приемлемого препарата в соответствии с традиционной ветеринарной практикой, а наиболее подходящий для конкретного животного режим дозирования и способ введения определяет ветеринарный врач. Таким образом, в настоящем изобретении предложен фармацевтический препарат, содержащий количество антитела и/или агента согласно изобретению, эффективное для лечения различных патологических состояний (как описано выше и дополнительно описано ниже). Предпочтительно композиция и/или антитело, и/или агент, и/или медикамент согласно изобретению приспособлены для доставки способом, выбранным из группы, включающей: внутривенный; внутримышечный; подкожный.
В настоящее изобретение также включены композиция и/или антитело, и/или агент, и/или медикамент, содержащие фармацевтические приемлемые соли присоединения кислот или оснований полипептид-связывающих компонентов согласно настоящему изобретению. Кислоты, которые используются для приготовления солей присоединения кислот вышеупомянутых основных соединений, применяемых в данном изобретении, представляют собой кислоты, которые образуют нетоксичные соли присоединения кислот, т.е. соли, содержащие фармакологически приемлемые анионы, такие как, помимо прочего, гидрохлориды, гидробромиды, гидройодиды, нитраты, сульфаты, бисульфаты, фосфаты, кислые фосфаты, ацетаты, лактаты, цитраты, кислые цитраты, тартраты, битартраты, сукцинаты, малеаты, фумараты, глюконаты, сахараты, бензоаты, метансульфонаты, этансульфонаты, бензенсульфонаты, п-толуолсульфонаты и памоаты [т.е. 1,1'-метилен-бис-(2-гидрокси-3 нафтоаты)]. Для получения фармацевтически приемлемых форм солей агентов в соответствии с настоящим изобретением также можно использовать фармацевтически приемлемые соли присоединения оснований. Химические основания, которые можно использовать в качестве реагентов для приготовления фармацевтически приемлемых основных солей настоящих агентов, которые по природе являются кислыми, представляют собой основания, которые образуют с такими соединениями нетоксичные основные соли. Такие нетоксичные основные соли включают, но не ограничиваются этим, помимо прочего, соли, полученные из таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия) и катионы щелочноземельных металлов (например, кальция и магния), аммониевые или водорастворимые соли присоединения аминов, такие как N-метилглюкамин-(меглюмин), и низшие алканоаммониевые и другие основные соли фармацевтически приемлемых органических аминов. Агенты и/или полипептид-связывающие компоненты согласно изобретению можно лиофилизировать для хранения и восстанавливать в подходящем носителе перед применением. Можно применять любой подходящий способ лиофилизации (например, сушку распылением, сушку осаждением) и/или методы восстановления. Специалистам в данной области техники должно быть понятно, что лиофилизация и восстановление могут приводить к разной степени потери активности антителами (например, среди традиционных иммуноглобулинов антитела IgM имеют тенденцию к большей потере активности, чем антитела IgG) и что может существовать необходимость в корректировке применяемых уровней с целью компенсации. В одном варианте реализации изобретения лиофилизированный (вымороженный) полипептид-связывающий компонент теряет не более чем около 20% или не более чем около 25%, или не более чем около 30%, или не более чем около 35%, или не более чем около 40%, или не более чем около 45%, или не более чем около 50% своей активности (до лиофилизации) после регидратации.
Подразумевается, что субъект может быть млекопитающим или не млекопитающим. Предпочтительно млекопитающий субъект является человеком или является отличным от человека млекопитающим, таким как лошадь или корова, или овца, или свинья, или верблюд, или собака, или кошка. Наиболее предпочтительно млекопитающий субъект является человеком.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом рецидивный рак и/или рефрактерный рак устойчив к лечению антителом.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом рецидивный рак и/или рефрактерный рак устойчив к молекуле антитела, определение которой приведено в (i).
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент предотвращает или снижает связывание FcγRIIb, присутствующего на клетке-мишени, с Fc-доменом молекулы антитела.
Как отмечалось выше, Fc-рецепторы (включая FcγRIIb) представляют собой мембранные белки, которые присутствуют на клетках. Специалисту понятно, что существуют известные в данной области техники способы определения наличия белка на клетке; например, иммуногистохимия и визуализация белков, меченных выявляемой меткой.
Под «FcγRIIb, присутствующим на клетке-мишени» подразумевается, что FcγRIIb представляет собой белок FcγRIIb и/или FcγRIIb присутствует на мембране клетки-мишени, и/или FcγRIIb присутствует в мембране клетки-мишени.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом молекула антитела, которая специфически связывает антиген клеточной поверхности клетки-мишени, причем молекула антитела содержит Fc-домен, способный связывать FcγRIIb, может интернализироваться клеткой-мишенью FcγRIIb-зависимым способом.
Как отмечалось выше, Fc-рецепторы (включая FcγRIIb) опосредуют интернализацию молекулы антитела клеткой, что может приводить к разрушению молекулы антитела. Способ, которым FcγRIIb опосредует интернализацию молекулы антитела клеткой, должен быть известным специалисту в клеточной биологии.
Под «интернализирована клеткой-мишенью» подразумевается, что молекула антитела удалена с клеточной мембраны в клетку-мишень и/или что молекула антитела возвращена в клетку-мишень, и/или молекула антитела интернализирована клеткой-мишенью и разрушена, и/или молекула антитела инкапсулирована в эндосоме клетки-мишени, и/или молекула антитела интернализирована клеткой-мишенью и, таким образом, более не является терапевтически эффективной, и/или молекула антитела поглощена клеткой-мишенью посредством эндоцитоза.
Следует понимать, что молекула антитела может быть интернализирована клеткой посредством большого количества разных процессов. Как обсуждалось выше, идея изобретения состоит в том, что молекула антитела может быть интернализирована посредством связывания Fc-домена молекулы антитела с FcγRIIb. При этом следует понимать, что молекула антитела может быть интернализирована посредством другого процесса, такого как пиноцитоз - процесс, в котором внеклеточные молекулы могут пассивно поглощаться клеткой. Другие процессы, посредством которых молекула антитела может быть поглощена клеткой, должны быть известны специалисту в клеточной биологии.
Под «FcγRIIb-зависимым способом» подразумевается, что молекула антитела интернализирована способом, для которого необходима активность FcγRIIb.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент, который предотвращает или снижает связывание FcγRIIb с Fc-доменом молекулы антитела, дополнительно предотвращает или снижает интернализацию молекулы антитела клеткой-мишенью.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом рефрактерный рак можно охарактеризовать как рефрактерный рак, если субъект проходил лечение против рака и был невосприимчив к нему и/или субъект проходил лечение против рака, но рак прогрессировал в организме субъекта во время указанного лечения.
Под «субъект проходил лечение против рака» подразумевается, что субъект ранее проходил лечение терапевтическим агентом или на данный момент проходит лечение терапевтическим агентом.
Также подразумевается, что если субъект ранее проходил лечение терапевтическим агентом, это лечение происходило около одного дня, или около двух дней, или около трех дней, или около четырех дней, или около пяти дней, или около шести дней, или более чем около недели, или более чем около двух недель, или более чем около трех недель, или более чем около одного месяца, или более чем около двух месяцев, или более чем около трех месяцев, или более чем около четырех месяцев, или более чем около пяти месяцев, или более чем около шести месяцев, или более чем около семи месяцев, или более чем около восьми месяцев, или более чем около девяти месяцев, или более чем около десяти месяцев, или более чем около 11 месяцев, или более чем около одного года, или более чем около двух лет, или более чем около трех лет, или более чем около 4 лет, или более чем около 5 лет, или более чем около 10 лет назад.
Под «терапевтическим агентом» подразумевается один или более терапевтический агент, выбранный из списка, включающего: ритуксимаб, биоаналог ритуксимаба, офатумумаб, обинутузумаб, алемтузумаб, окрелизумаб, галиксимаб, тозитумомаб, ибритутомаб, антитело к CD20, антитело к CD40, антитело к CD19, антитело к CD37, антитело к CD52, хлорамбуцил, циклофосфамид, гиперфракционированный циклофосфамид, флударабин, оксалиплатин, ибрутиниб, нуклеозидный аналог, алкилятор, бендамустин, бортезомиб, леналидомид, винкристин, доксорубицин (также известный как адриамицин), преднизон, идарубицин, мелфалан, кладрибин, цитарабин, дексаметазон, метотрексат, месна, гемцитабин, темсиролимус, митоксантрон, цисплатин, талидомид, этопозид, прокарбазин, флавопиридол, энзастаурин, блеомицин, винбластин, антрациклин, ифосфамид, карбоплатин, метилпреднизолон и дакарбазин; и/или одна или более комбинация терапевтических агентов, выбранная из списка, включающего: флударабин и циклофосфамид (FC); циклофосфамид, винкристин, доксорубицин, липосомальный доксорубицин и преднизон (CHOP); циклофосфамид, винкристин и преднизон (COP, также известный как CVP); ритуксимаб, циклофосфамид, винкристин и преднизон (R-COP, также известный как R-CVP); идарубицин и флударабин; мелфалан, хлорамбуцил и преднизон (MCP); ритуксимаб и хлорамбуцил; ритуксимаб, циклофосфамид, винкристин, доксорубицин и преднизон (R-CHOP); ритуксимаб, флударабин и циклофосфамид (R-FC); месна, ифосфамид, митоксантрон и этопозид; месна, ифосфамид, митоксантрон, этопозид и ритуксимаб; леналидомид и ритуксимаб; ритуксимаб и кладрибин; ритуксимаб, мелфалан, хлорамбуцил и преднизон (R-MCP); цитарабин, циклофосфамид, винкристин, доксорубицин и преднизон; гиперфракционированный циклофосфамид, винкристин, доксорубицин и дексаметазон (гипер-CVAD); ритуксимаб, циклофосфамид, винкристин, доксорубицин и дексаметазон (R-гипер-CVAD); ифосфамид, карбоплатин и этопозид (ICE); ифосфамид, карбоплатин, этопозид и ритуксимаб; гемцитабин, дексаметазон и карбоплатин; гемцитабин, дексаметазон, карбоплатин и ритуксимаб; гемцитабин и оксалиплатин; гемцитабин, оксалиплатин и ритуксимаб; метотрексат и цитарабин; бортезомиб и гемцитабин; ритуксимаб и бортезомиб; флударабин, циклофосфамид и митоксантрон (FCM); ритуксимаб, флударабин, циклофосфамид и митоксантрон (R-FCM, также известный как FCMR); ритуксимаб и флударабин; дексаметазон и гемцитабин; дексаметазон, гемцитабин и цисплатин; дексаметазон, гемцитабин, цисплатин и ритуксимаб; дексаметазон, флударабин, митоксантрон и дексаметазон (RFND); талидомид и ритуксимаб; преднизон, этопозид, прокарбазин и циклофосфамид (PEP-C); ритуксимаб, талидомид, преднизон, этопозид, прокарбазин и циклофосфамид; бендамустин и ритуксимаб (BR); ритуксимаб, циклофосфамид, доксорубицин, винкристин, этопозид и преднизон (R-CHOEP, также известный как EPOCH-R); адриамицин, блеомицин, винбластин и дакарбазин (ABVD); блеомицин, этопозид, адриамицин, циклофосфамид, винкристин, прокарбазин и преднизон (BEACOPP); циклофосфамид, винкристин, прокарбазин и преднизон (COPP); циклофосфамид, этопозид, преднизон и прокарбазин (CEPP); ритуксимаб, циклофосфамид, этопозид, преднизон и прокарбазин (RCEPP); ритуксимаб, циклофосфамид, липосомальный доксорубицин, винкристин и преднизон (RCDOP); ритуксимаб, циклофосфамид, митоксантрон, винкристин и преднизон (RCNOP); ритуксимаб, циклофосфамид, этопозид, винкристин и преднизон; дексаметазон, цисплатин, цитарабин и ритуксимаб (DHAP); этопозид, метилпреднизолон, цитарабин, цисплатин и ритуксимаб (ESHAP); и хлорамбуцил, винбластин, прокарбазин и преднизон (ChlVPP).
Кроме приведенных выше терапевтических агентов специалисту в области медицины известны другие типы и комбинации терапевтических агентов, которые можно использовать для лечения рака и/или рецидивного рака, и/или рефрактерного рака; примеры таких агентов можно найти в Agency for Healthcare Research and Quality Guideline Summary NGC-9392 September 2012, Agency for Healthcare Research and Quality Guideline Summary NGC-9278 2012, McKay et al., 2012, British Journal of Haematology: 12046, Hallek et al., 2008, Blood, 111: 5446-5456, Wang et al., 2013, N. Engl. J. Med., 369(6): 507-516, Byrd et al., N. Engl. J. Med., 369(1): 32-42, и NCCN Guidelines on Non-Hodgkin’s Lymphomas Version 1.2014.
Под «проходил лечение против рака и был невосприимчив к нему» подразумевается, что после прохождения лечения против рака субъект не демонстрирует снижение тяжести симптомов рака и/или субъект не демонстрирует снижение количества симптомов рака, и/или у субъекта отсутствует улучшение в прогнозе течения рака, и/или субъект не демонстрирует снижение в диагностических маркерах рака.
Под «раком, прогрессирующим у субъекта» подразумевается, что во время лечения рака субъект демонстрирует повышение тяжести симптомов рака и/или субъект демонстрирует повышение количества симптомов рака, и/или у субъекта ухудшается прогноз течения рака, и/или субъект демонстрирует повышение в диагностических маркерах рака.
Под «демонстрирует» подразумевается, что субъект демонстрирует симптом рака и/или диагностический маркер рака, и/или симптом рака и/или диагностический маркер рака могут быть определены и/или оценены, и/или количественно оценены.
Для специалиста в области медицины должно быть очевидно, какими должны быть симптомы рака и диагностические маркеры рака, и как можно определить и/или оценить, и/или количественно оценить, существует ли снижение или повышение тяжести симптомов рака или снижение или повышение в диагностических маркерах рака; а также как можно использовать эти симптомы рака и/или диагностические маркеры рака для составления прогноза для течения рака.
Лечение против рака часто применяют в виде курса лечения, что означает, что терапевтический агент вводят в течение некоторого периода времени. Время курса лечения зависит от большого количества факторов, которые могут включать, помимо других причин, тип вводимого терапевтического агента, тип рака, лечение которого проводят, тяжесть рака, лечение которого проводят, и возраст и состояние здоровья субъекта.
Под «во время лечения» подразумевается, что субъект на данный момент проходит курс лечения и/или получает терапевтический агент, и/или проходит курс лечения терапевтическим агентом.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом субъект характеризуют как невосприимчивый, если субъект ранее проходил лечение против рака, но достиг менее чем частичной ремиссии.
Ремиссия является термином, хорошо известным в области техники раковой медицины, который относится к оценке снижения степени рака у субъекта. Частичная ремиссия также является термином, хорошо известным в области техники раковой медицины, который относится к стадии ремиссии, на которой степень рака у субъекта согласно определению по конкретному симптому рака и/или диагностическому маркеру рака снижена на 50% по сравнению с уровнем этого конкретного симптома рака и/или диагностического маркера рака перед лечением. В некоторых ситуациях для того, чтобы субъект был классифицирован, как характеризующийся частичной ремиссией, такое снижение в симптоме рака и/или диагностическом маркере рака должно сохраняться в течение конкретного периода времени. Специалисту в области медицины должно быть понятно, находится ли субъект на стадии ремиссии в отношении рака, и на какой стадии ремиссии находится субъект.
Под «достигает менее чем частичной ремиссии» подразумевается, что симптом рака или диагностический маркер рака был оценен и/или определен, и/или количественно оценен на основании снижения тяжести симптомов рака и/или снижения количества симптомов рака, и/или улучшения прогноза течения рака, и/или снижения в диагностических маркерах рака; а его снижение составляет менее 50% по сравнению с симптомом рака или диагностическим маркером рака до лечения.
Также подразумевается, что оценка и/или определение, и/или количественное определение, достигает ли субъект менее чем частичной ремиссии, основаны на сравнении между симптомом рака и/или диагностическим маркером рака после прекращения лечения и симптомом рака и/или диагностическим маркером рака до начала лечения, и/или симптомом рака и/или диагностическим маркером рака во время лечения. Также подразумевается, что оценку и/или определение, и/или количественное определение можно проводить по меньшей мере один раз или по меньшей мере два раза, или по меньшей мере три раза, или по меньшей мере четыре раза, или по меньшей мере пять раз, или по меньшей мере шесть раз, или по меньшей мере семь раз, или по меньшей мере восемь раз, или по меньшей мере девять раз, или по меньшей мере десять раз, или по меньшей мере 15 раз, или по меньшей мере 20 раз перед тем, как считать, что субъект достигает менее чем частичной ремиссии.
Дополнительно подразумевается, что субъект достигает менее чем частичной ремиссии, если снижение в симптоме рака и/или диагностическом маркере рака после прекращения лечения составляет менее чем около 1 % или менее чем около 5%, или менее чем около 10%, или менее чем около 15%, или менее чем около 20%, или менее чем около 25%, или менее чем около 30%, или менее чем около 35%, или менее чем около 40%, или менее чем около 45%, или менее чем около 50% по сравнению с симптомом рака и/или диагностическим маркером рака до начала лечения и/или симптомом рака и/или диагностическим маркером рака во время лечения.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом рецидивный рак может быть охарактеризован как рецидивный рак, если субъект ранее проходил лечение от рака и ранее был восприимчив к лечению, а впоследствии случился рецидив.
Под «ранее проходил лечение от рака» подразумевается, что субъект ранее проходил лечение терапевтическим агентом, но это лечение было прекращено. Следовательно, подразумевается, что субъект ранее проходил лечение от рака, если прекращение лечения терапевтическим агентом произошло по меньшей мере на около один день, или по меньшей мере на около два дня, или по меньшей мере на около три дня, или по меньшей мере на около четыре дня, или по меньшей мере на около пять дней, или по меньшей мере на около шесть дней, или более чем на около одну неделю, или более чем на около две недели, или более чем на около три недели, или более чем на около один месяц, или более чем на около два месяца, или более чем на около три месяца, или более чем на около четыре месяца, или более чем на около пять месяцев, или более чем на около шесть месяцев, или более чем на около семь месяцев, или более чем на около восемь месяцев, или более чем на около девять месяцев, или более чем на около десять месяцев, или более чем на около 11 месяцев, или более чем на около один год, или более чем на около два года, или более чем на около три года, или более чем на около 4 года, или более чем на около 5 лет, или более чем на около 10 лет ранее.
Для специалиста в области медицины должно быть понятно, восприимчив ли пациент к лечению.
Под «ранее был восприимчив к лечению» подразумевается, что после прекращения лечения существует оцененное и/или определенное, и/или количественно определенное снижение тяжести симптомов рака и/или снижение количества симптомов рака, и/или улучшение прогноза течения рака, и/или снижение в диагностических маркерах рака.
Рецидив является хорошо известным термином в области медицины рака, который относится к ухудшению рака у субъекта после того, как этот рак у субъекта ранее был восприимчив к лечению. Для специалиста в области медицины понятно, когда происходит рецидив рака.
Под «впоследствии рецидивировавший» подразумевается, что субъект демонстрирует повышение тяжести симптомов рака и/или субъект демонстрирует повышение количества симптомов рака, и/или субъект имеет плохой прогноз течения рака, и/или субъект демонстрирует повышение в диагностических маркерах рака после того, как субъект ранее был восприимчив к лечению.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом субъект характеризуют как такой, у которого впоследствии случился рецидив, если субъект (i) достигает по меньшей мере частичной ремиссии после лечения и/или если субъект достигает по меньшей мере полной ремиссии после лечения, но (ii) после прекращения лечения у субъекта прогрессирует рак.
Под «полной ремиссией после лечения» подразумевается, что после прекращения лечения симптомы рака и/или диагностические маркеры рака не выявляются.
Также подразумевается, что в некоторых случаях полная ремиссия после лечения наступает, если симптомы рака и/или диагностические маркеры рака не выявляются в течение по меньшей мере около одного дня, или по меньшей мере около двух дней, или по меньшей мере около трех дней, или по меньшей мере около четырех дней, или по меньшей мере около пяти дней, или по меньшей мере около шести дней, или более чем около одной недели, или более чем около двух недель, или более чем около трех недель, или более чем около одного месяца, или более чем около двух месяцев, или более чем около трех месяцев, или более чем около четырех месяцев, или более чем около пяти месяцев, или более чем около шести месяцев, или более чем около семи месяцев, или более чем около восьми месяцев, или более чем около девяти месяцев, или более чем около десяти, или более чем около 11 месяцев, или более чем около одного года, или более чем около двух лет, или более чем около трех лет, или более чем около 4 лет, или более чем около 5 лет, или более чем около 10 лет, или около 20 лет, или около 30 лет, или около 40 лет, или около 50 лет после прекращения лечения. В частности, подразумевается, что в некоторых случаях полная ремиссия после лечения наступает, если симптомы рака и/или диагностические маркеры рака не выявляются в течение более чем около шести месяцев после прекращения лечения.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом у субъекта впоследствии произошел рецидив через более чем около 1 месяц после прекращения лечения.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом у субъекта впоследствии произошел рецидив через более чем около 1 месяц, или более чем около 2 месяца, или более чем около 3 месяца, или более чем около 4 месяца, или более чем около 5 месяцев, или более чем около 6 месяцев, или более чем около 7 месяцев, или более чем около 8 месяцев, или более чем около 9 месяцев, или более чем около 10 месяцев, или более чем около 11 месяцев, или более чем около 12 месяцев, или более чем около 2 года, или более чем около 3 года, или более чем около 4 года, или более чем около 5 лет, или более чем около 6 лет, или более чем около 7 лет, или более чем около 8 лет, или более чем около 9 лет, или более чем около 10 лет после прекращения лечения. Наиболее предпочтительно у субъекта впоследствии произошел рецидив через более чем около 6 месяцев после прекращения лечения.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом субъект демонстрирует характеристики рецидивного рака по меньшей мере один раз, или по меньшей мере два раза, или по меньшей мере три раза, или по меньшей мере четыре раза, или по меньшей мере пять раз, или по меньшей мере шесть раз, или по меньшей мере семь раз, или по меньшей мере восемь раз, или по меньшей мере девять раз, или по меньшей мере десять раз.
Под «демонстрирует характеристики рецидивного рака» подразумевается, что субъект ранее проходил лечение от рака и ранее был восприимчив к лечению, а впоследствии случился рецидив.
Считается, что рак может рецидивировать потому, что у него развивается устойчивость к лечению, которое ранее применялось для лечения рака у субъекта. Следовательно, субъекта с рецидивным раком часто лечат при помощи терапевтического агента, отличного от терапевтического агента, который ранее использовался для лечения рака у субъекта. Соответственно, в одном варианте реализации изобретения, приведенном непосредственно выше, подразумевается, что каждый следующий раз, когда субъект демонстрирует характеристики рецидивного рака, субъекта лечат при помощи терапевтического агента, отличного от терапевтического агента, которым ранее лечили субъекта, и/или субъекта лечат при помощи терапевтического агента, аналогичного терапевтическому агенту, которым ранее лечили субъекта.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом лечение представляет собой лечение антителом, например, молекулой антитела, определение которой приведено в (i) более ранних вариантов реализации изобретения, которую вводили в отсутствие агента, определение которого приведено в (ii) более ранних вариантов реализации изобретения.
Под «вводили в отсутствие» подразумевается, что молекулу антитела, определение которой приведено в (i), вводили в течение периода времени после и/или до агента, определение которого приведено в (ii). Соответственно, под «в течение периода времени» подразумевается, что молекулу антитела, определение которой приведено в (i), вводили в течение более чем одного часа, или более чем двух часов, или более чем трех часов, или более чем шести часов, или более чем 12 часов, или более чем 18 часов, или более чем 24 часов, или более чем двух дней, или более чем трех дней, или более чем четырех дней, или более чем пяти дней, или более чем шести дней, или более чем семи дней, или более чем двух недель, или более чем трех недель, или более чем четырех недель, или более чем пяти недель, или более чем шести недель, или более чем семи недель, или более чем восьми недель, или более чем трех месяцев, или более чем четырех месяцев, или более чем пяти месяцев, или более чем шести месяцев, или более чем семи месяцев, или более чем восьми месяцев, или более чем девяти месяцев, или более чем десяти месяцев, или более чем 11 месяцев, или более чем 12 месяцев, или более чем 1 года, или более чем двух лет, или более чем трех лет, или более чем четырех лет, или более чем пяти лет, или более чем десяти лет, или более чем 20 лет до и/или после агента, определение которого приведено в (ii).
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом лечение включает применение одного или более терапевтического агента.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом клетка-мишень имеет повышенный уровень экспрессии FcγRIIb.
Специалисту в области молекулярной биологии или клеточной биологии должно быть известно, что означает повышенный уровень экспрессии FcγRIIb, и как можно измерить экспрессию. Например, кроме вышеуказанных способов измерения экспрессии FcγRIIb, уровни экспрессии FcγRIIb можно измерить методом иммуногистохимии образцов биопсии опухоли. Специалисту в данной области техники должно быть понятно, что существует множество способов и методологий для определения уровней экспрессии FcγRIIb.
Под выражением «клетка-мишень имеет повышенный уровень экспрессии FcγRIIb» подразумевается, что клетка-мишень содержит повышенный уровень белка FcγRIIb и/или клетка-мишень содержит повышенный уровень мРНК FcγRIIb по сравнению с контролем, как описано ниже.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом повышенную экспрессию FcγRIIb на клетке-мишени определяют относительно контроля.
Подходящим контролем может быть образец, который имеет нормальный уровень экспрессии FcγRIIb. Выбор правильного контроля и определение нормального уровня экспрессии FcγRIIb может зависеть от большого числа переменных, таких как субъект, тип клетки-мишени в организме субъекта и тип рака. Для специалиста в области молекулярной биологии или клеточной биологии должно быть очевидно, каким должен быть соответствующий контроль и нормальный уровень экспрессии FcγRIIb.
Под «контролем» подразумевается, что контроль представляет собой контрольную клетку и/или контроль представляет собой информацию из базы данных уровней экспрессии FcγRIIb. Также подразумевается, что контрольная клетка принадлежит отличному от клетки-мишени типу клеток и/или тому же типу клеток, что и клетка-мишень. Дополнительно подразумевается, что контрольная клетка получена от контрольного индивида и что этот контрольный индивид может быть самим субъектом и/или индивидом, отличным от субъекта.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом контроль включает контрольные клетки контрольного индивида с нерефрактерным раком и/или нерецидивным раком.
Под «контрольным индивидом с нерефрактерным раком и/или нерецидивным раком» подразумевается, что этот контрольный индивид имеет рак, который не является рецидивным раком и/или рефрактерным раком. Также подразумевается, что ранее контрольный индивид по меньшей мере один раз проходил лечение терапевтическим агентом.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом повышенная экспрессия FcγRIIb по меньшей мере в 2 раза выше в клетке-мишени субъекта по сравнению с контролем.
Также подразумевается, что повышенная экспрессия FcγRIIb в около 10 раз выше или в около 20 раз выше, или в около 30 раз выше, или в около 40 раз выше, или в около 50 раз выше, или в около 60 раз выше, или в около 70 раз выше, или в около 80 раз выше, или в около 90 раз выше, или в около 100 раз выше, или в около 200 раз выше, или в около 300 раз выше, или в около 400 раз выше, или в около 500 раз выше, или в около 600 раз выше, или в около 700 раз выше, или в около 800 раз выше, или в около 900 раз выше, или в около 1000 раз выше в клетке-мишени по сравнению с контролем.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом клетка-мишень представляет собой раковую клетку.
Раковая клетка представляет собой клетку, которая демонстрирует характеристики рака, что означает, что она демонстрирует один или более диагностических маркеров рака. Специалисту в области клеточной биологии и онкологии должно быть понятно, является ли клетка-мишень раковой клеткой.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом клетка-мишень представляет собой В-клетку.
B-клетки (известные также как В-лимфоциты) представляют тип клеток адаптивного иммунного ответа, которые отличаются от других клеток иммунной системы наличием рецепторов В-клеток на клеточной поверхности.
Под «В-клеткой» подразумеваются плазматические В-клетки (также известные как эффекторные В-клетки) и/или В-клетки памяти, и/или клетки B-1, и/или клетки B-2, и/или В-клетки маргинальной зоны, и/или фолликулярные В-клетки, и/или регуляторные В-клетки, и/или наивные В-клетки.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом рецидивный рак и/или рефрактерный рак, и/или раковая клетка, и/или тот же тип рака, и/или рак выбраны из группы, состоящей из: неходжкинской лимфомы; фолликулярной лимфомы; диффузной крупноклеточной В-клеточной лимфомы; мантийноклеточной лимфомы; хронического лимфоцитарного лейкоза; мелкоклеточной лимфоцитарной лимфомы.
Неходжкинская лимфома (НХЛ) представляет общее название для группы разных лимфом, которая включает, помимо прочего, фолликулярную клеточную лимфому, мантийноклеточную лимфому, лимфому маргинальной зоны селезенки, MALT-лимфому, лимфоплазмоцитарную НХЛ (также известную как макроглобулинемия Вальденстрема), мелкоклеточную лимфоцитарную лимфому, хронический лимфоцитарный лейкоз, диффузную крупноклеточную В-клеточную лимфому, диффузную смешанно-клеточную лимфому, лимфому Беркитта, анапластическую крупноклеточную лимфому и диффузную смешанно-клеточную лимфому.
Каждый из вышеописанных раков является хорошо известным, а симптомы и диагностические маркеры рака хорошо описаны, как и терапевтические агенты, применяемые для лечения этих раков. Соответственно, симптомы, диагностические маркеры рака и терапевтические агенты, применяемые для лечения неходжкинской лимфомы, фолликулярной лимфомы, диффузной крупноклеточной В-клеточной лимфомы, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза и мелкоклеточной лимфоцитарной лимфомы должны быть известны специалистам в области медицины.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом невосприимчивость субъекта и/или частичную ремиссию у субъекта, и/или полную ремиссию у субъекта, и/или прогрессирование рака у субъекта определяют, измеряя один или более показателей из группы, включающей:
(i) число лимфоцитов; и/или
(ii) число нейтрофилов; и/или
(iii) число тромбоцитов; и/или
(iv) количество гемоглобина; и/или
(v) процентное содержание опухолевых клеток; и/или
(vi) процентное содержание лимфоцитов костного мозга; и/или
(vii) процентное содержание циркулирующих лимфоцитов; и/или
(viii) наличие и/или отсутствие биомаркеров на лимфоцитах; и/или
(ix) стадирование рака; и/или
(x) гистологическое исследование; и/или
(xi) исследование костного мозга; и/или
(xii) цитогенетическое исследование; и/или
(xiii) оценку лимфатических узлов; и/или
(xiv) физические симптомы; и/или
(xv) снижение раковых клеток в селезенке.
Следует понимать, что (i), (ii), (iii), (iv), (v), (vi), (vii), (viii), (ix), (x), (xi), (xii), (xiii) и (xv) относятся к «диагностическим маркерам рака»; а (xiv) относится к «симптомам рака».
В течение десятилетий исследований и инноваций в области онкологии были получены исчерпывающие характеристики «диагностических маркеров рака» и «симптомов рака» для большого числа видов рака, а также способы определения и оценки диагностических маркеров рака и симптомов рака. Соответственно, специалисты в области онкологии должны понимать, каким образом каждый из вышеприведенных пунктов относится к конкретному виду рака, а на основании определения или оценки конкретных диагностических маркеров рака или симптомов рака для специалистов должно быть очевидно, что субъект не восприимчив к лечению, или что субъект находится на стадии частичной ремиссии, или что субъект находится на стадии полной ремиссии, или что рак у субъекта прогрессирует; примеры чего можно найти в McKay et al., 2012, British Journal of Haematology: 12046, Hallek et al., 2008, Blood, 111: 5446-5456 и NCCN Guidelines on Non-Hodgkin’s Lymphomas Version 1.2014.
Оценка количества диагностических маркеров рака (таких как «число лимфоцитов», «число нейтрофилов», «число тромбоцитов», «количество гемоглобина», «процентное содержание атипичных клеток», «процентное содержание лимфоцитов костного мозга» и «процентное содержание циркулирующих лимфоцитов») основана на подсчете клеток и молекул в организме субъекта. Методы анализа для подсчета таких клеток и молекул хорошо известны в данной области техники.
Под «опухолевыми клетками» подразумеваются неопластические клетки и/или неопластические клетки, которые представляют собой CD19+, CD5+ и CD23+.
Количество биомаркеров может указывать на наличие определенных типов рака. Например, клетки хронического лимфоцитарного лейкоза коэкспрессируют CD5, CD19, CD20 и CD23. При этом уровни CD20 и CD79b ниже по сравнению с нормальными В-клетками.
Под «наличием и/или отсутствием биомаркеров на лимфоцитах» подразумевается, что термин «наличие» включает повышенное количество биомаркера по сравнению с контрольной В-клеткой или выявляемый биомаркер, а отсутствие включает сниженное количество биомаркера по сравнению с контрольной В-клеткой или отсутствие выявляемого биомаркера. Также подразумевается, что контрольная В-клетка может быть получена от «контрольного индивида». Дополнительно подразумевается, что наличие и/или отсутствие биомаркеров на лимфоцитах включает наличие CD5 и/или CD19, и/или CD20, и/или CD23, и/или циклина D1, и/или BCL2, и/или FMC6, и/или CD3, и/или CD10 и/или BCL6, и/или CD21, и/или CD45, и/или Ki-67, и/или IRF4/MUM1, и/или MYC, и/или CD30, и/или CD138, и/или EBER-ISH, и/или ALK, и/или HHV8, и/или каппа/лямбда, и/или CD79b, и/или отсутствие CD20, и/или CD79b, и/или CD10, и/или CD23, и/или BCL6.
Клиническое определение диагноза, прогноза и прогрессирования большого числа видов рака основано на определенной классификации, известной как стадирование. Функция таких систем стадирования состоит в сопоставлении большого числа разных диагностических маркеров рака и симптомов рака для того, чтобы сделать заключение о диагнозе и/или прогнозе, и/или прогрессировании рака. Специалисту в области онкологии должно быть известно, как оценить диагноз и/или прогноз, и/или прогрессирование рака, используя систему стадирования, и какие диагностические маркеры рака и симптомы рака следует для этого использовать.
Под «стадированием рака» подразумевается стадирование по Rai, которое включает стадию 0, стадию I, стадию II, стадию III и стадию IV, и/или стадирование по Binet, которое включает стадию A, стадию B и стадию C, и/или стадирование по Ann Arbour, которое включает стадию I, стадию II, стадию III и стадию IV.
Известно, что рак может приводить к аномалиям в морфологии клеток. Такие аномалии часто повторяемо возникают при определенных видах рака, что означает, что исследование таких изменений в морфологии (известное также как гистологическое исследование) можно использовать для диагностирования или прогнозирования рака. Методы визуализации образцов для исследования морфологии клеток и приготовления образцов для визуализации хорошо известны в данной области техники; например, оптическая микроскопия или конфокальная микроскопия.
Под «гистологическим исследованием» подразумевается наличие мелких, зрелых лимфоцитов и/или наличие мелких, зрелых лимфоцитов с узкой границей цитоплазмы, наличие мелких, зрелых лимфоцитов с плотным ядром, в котором отсутствуют видимые ядрышки, и/или наличие мелких, зрелых лимфоцитов с узкой границей цитоплазмы и с плотным ядром, в котором отсутствуют видимые ядрышки, и/или наличие атипичных клеток, и/или расщепленных клеток, и/или пролимфоцитов.
Хорошо известно, что рак является результатом мутаций ДНК клетки, которые могут приводить к появлению клеток, не подлежащих клеточной гибели или неконтролируемо пролиферирующих. Следовательно, исследование таких мутаций (также известное, как цитогенетическое исследование) может быть полезным инструментом для оценки диагноза и/или прогноза рака. Примером является делеция хромосомной локации 13q14.1, характерная для хронического лимфоцитарного лейкоза. Способы исследования мутаций в клетках хорошо известны в данной области техники; например, флуоресцентная in situ гибридизация (FISH).
Под «цитогенетическим исследованием» подразумевается исследование ДНК в клетке и, в частности, в хромосомах. Цитогенетическое исследование можно использовать для выявления изменений в ДНК, которые могут быть связаны с наличием рефрактерного рака и/или рецидивного рака. Такие изменения могут включать: делеции в длинном плече хромосомы 13, и/или делецию хромосомной локации 13q14.1, и/или трисомию хромосомы 12, и/или делеции в длинном плече хромосомы 12, и/или делеции в длинном плече хромосомы 11, и/или делецию 11q, и/или делеции в длинном плече хромосомы 6, и/или делецию 6q, и/или делеции в коротком плече хромосомы 17, и/или делецию 17p, и/или транслокацию t(11:14), и/или транслокацию (q13:q32), и/или генные перестройки рецепторов антигенов, и/или перестройки BCL2, и/или перестройки BCL6, и/или транслокации t(14:18), и/или транслокации t(11:14), и/или транслокации (q13:q32), и/или транслокации (3:v), и/или транслокации (8:14), и/или транслокации (8:v), и/или транслокации t(11:14) и (q13:q32).
Известно, что у субъектов с раком проявляются определенные физические симптомы, которые часто являются результатом раковой нагрузки на организм. Такие симптомы часто повторно появляются при одном и том же виде рака и, таким образом, могут быть характерными для диагноза и/или прогноза, и/или прогрессирования болезни. Специалист в области медицины должен понимать, какие физические симптомы связаны с конкретными видами рака, и как оценка таких физических симптомов может коррелировать с диагнозом и/или прогнозом, и/или прогрессированием заболевания.
Под «физическими симптомами» подразумевается увеличение печени и/или увеличение селезенки.
Под «невосприимчивостью субъекта» подразумевается, что определение и/или оценка показателей (i), и/или (ii) и/или (iii), и/или (iv), и/или (v), и/или (vi), и/или (vii), и/или (viii), и/или (ix), и/или (x), и/или (xi), и/или (xii), и/или (xiii), и/или (xiv) и/или (xv) из приведенного непосредственно выше варианта реализации изобретения остается без изменений, или появившееся изменение является пренебрежимо малым у субъекта при их сравнении до лечения и после прекращения лечения и/или при сравнении во время лечения и после прекращения лечения, и/или во время лечения.
Под «частичной ремиссией у субъекта» подразумевается, что для показателя «количества лимфоцитов» наблюдается по меньшей мере 50% снижение количества лимфоцитов после прекращения лечения по сравнению с количеством до лечения и/или во время лечения, и/или для показателя «оценка лимфатических узлов» наблюдается по меньшей мере 50% снижение размера одного или более лимфатических узлов после прекращения лечения по сравнению с размером до лечения и/или во время лечения, и/или отсутствие дополнительных увеличенных лимфатических узлов, и/или для показателя «физические симптомы» наблюдается по меньшей мере 50% снижение размера селезенки (в случае увеличения селезенки) после прекращения лечения по сравнению с размером до лечения и/или во время лечения, и/или по меньшей мере 50% снижение размера печени (в случае увеличения печени) после прекращения лечения по сравнению с размером до лечения и/или во время лечения, и/или показатель «количество нейтрофилов» составляет не более 1500 клеток/мкл, и/или показатель «количество тромбоцитов» составляет не более 100000 тромбоцитов/мкл, и/или наблюдается по меньшей мере 50% снижение количества нейтрофилов после прекращения лечения по сравнению с количеством до лечения и/или во время лечения, и/или «количество гемоглобина» составляет не более 11 г/дл, и/или наблюдается по меньшей мере 50% снижение количества гемоглобина после прекращения лечения по сравнению с количеством до лечения и/или во время лечения.
Под «полной ремиссией у субъекта» подразумевается, что «количество лимфоцитов» соответствует количеству клеток, составляющему не более 4000 клеток/мкл, и/или в случае показателя «оценка лимфатических узлов» диаметр лимфатических узлов составляет не более 1,5 см, и/или в случае показателя «физические симптомы» не наблюдается выявляемого увеличения печени и/или увеличения селезенки, и/или количество нейтрофилов составляет не более 1500 клеток/мкл, и/или «количество тромбоцитов составляет не более 100000 тромбоцитов/мкл, и/или «количество гемоглобина» составляет не более 11 г/дл.
Под «прогрессированием рака у субъекта» подразумевается, что для показателя «количества лимфоцитов» наблюдается по меньшей мере 50% увеличение количества лимфоцитов после прекращения лечения по сравнению с количеством до лечения и/или во время лечения и/или это количество составляет по меньшей мере более 5000 клеток/мкл, и/или для показателя «оценка лимфатических узлов» наблюдается увеличение лимфатических узлов до диаметра, составляющего по меньшей мере 1,5 см, и/или по меньшей мере 50% увеличение размера одного или более лимфатических узлов после прекращения лечения по сравнению с размером до лечения и/или во время лечения, и/или для показателя «физические симптомы» наблюдается увеличение печени и/или увеличение селезенки, и/или по меньшей мере 50% увеличение размера селезенки (в случае увеличения селезенки) после прекращения лечения по сравнению с размером до лечения и/или во время лечения, и/или по меньшей мере 50% увеличение размера печени (в случае увеличения печени) после прекращения лечения по сравнению с размером до лечения и/или во время лечения, и/или в случае показателя «количество гемоглобина» уровни гемоглобина снижаются более чем на 20 г/л и/или уровни гемоглобина снижаются до менее чем 100 г/л, и/или для показателя «количество тромбоцитов» наблюдается по меньшей мере 50% снижение количества нейтрофилов после прекращения лечения по сравнению с количеством до лечения и/или во время лечения, и/или оно составляет не более 100000 тромбоцитов/мкл.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент включает: полипептид; или антикалин; или пептид; или антитело; или химерное антитело; или одноцепочечное антитело; или аптамер; или дарпин; или фрагмент антитела Fab или F(ab′)2, или Fv, или ScFv или dAb; или антитело IgG2; или антитело IgG4; или химерную молекулу из IgG2 и IgG4; или вариант антитела, содержащий мутацию N297Q; или вариантное антитело DANA; или небольшую молекулу; или естественный продукт; или аффитело; или пептидомиметик; или нуклеиновую кислоту; или молекулу пептидной нуклеиновой кислоты; или липид; или углевод; или белок на основе модулярного каркаса, включая белки с анкириновыми повторами, или белки с повторами armadillo, или богатые лейцином белки, или белки с тетрапептидными повторами, или сконструированные белки с анкириновыми повторами (DARPin).
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент, определение которого приведено в (ii), представляет собой одну или более молекулу антитела, которая специфически связывает FcγRIIb.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом при этом агент, определение которого приведено в (ii), представляет собой одну или более молекулу антитела, которая не содержит домен, способный рекрутировать эффекторную клетку.
Иммунная система содержит большое количество разных типов клеток, каждый из которых имеет отличную роль, вызывая, способствуя или поддерживая иммунный ответ. Для выполнения своей роли в иммунитете клетка иммунной системы часто реагирует на стимулы, что часто приводит к мобилизации такой клетки в конкретном участке организма или мишени (например, как в случае клетки, которая обладает сигнализацией). Одним из классов клеток иммунной системы являются эффекторные клетки, определение и роль которых должна быть известна специалисту в области иммунологии.
Под «эффекторной клеткой» подразумевается эффекторная Т-клетка и/или эффекторная В-клетка (также известная как плазматическая клетка), и/или эффекторная Т-клетка памяти, и/или эффекторная Т-клетка памяти CD4+, и/или эффекторная Т-клетка памяти CD8+.
Под «доменом, способным рекрутировать эффекторную клетку» подразумевается эпитоп и/или антиген на молекуле антитела, который мобилизует эффекторную клетку к месту нахождения молекулы антитела. Также подразумевается, что домен, способный рекрутировать эффекторную клетку, может представлять собой Fc-домен молекулы антитела и/или антиген, и/или эпитоп на Fc-домене молекулы антитела.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент, определение которого приведено в (ii), представляет собой одну или более молекул антител, которые являются молекулами моноклональных антител и/или молекулами поликлональных антител, и/или молекулами биспецифических антител.
Как отмечено выше, в изобретение включены разные типы и формы антител, которые должны быть известны специалисту в области иммунологии. Хорошо известно, что антитела, применяемые в терапевтических целях, часто модифицируют дополнительными компонентами, которые модифицируют свойства молекулы антитела.
Соответственно, подразумевается, что молекула антитела согласно изобретению (например, молекула моноклонального антитела и/или молекула поликлонального антитела, и/или молекула биспецифического антитела) содержит выявляемый компонент и/или цитотоксический компонент.
Под «выявляемым компонентом» подразумевается один или более компонент из группы, состоящей из: фермента; радиоактивного атома; флуоресцентного компонента; хемилюминесцентного компонента; биолюминесцентного компонента. Выявляемый компонент позволяет визуализировать молекулу антитела in vitro и/или in vivo, и/или ex vivo.
Под цитотоксическим компонентом подразумевается радиоактивный компонент и/или фермент, причем фермент является каспазой, и/или токсин, причем токсин является бактериальным токсином или ядом; при этом цитотоксический компонент способен индуцировать клеточный лизис.
Дополнительно подразумевается, что молекула антитела может находиться в выделенной форме и/или очищенной форме, и/или может быть ПЭГилированной.
В следующих вариантах реализации изобретения SEQ ID NO относятся к последовательностям, указанным ниже в клонах.
Как обсуждалось выше, CDR антитела связываются с мишенью антитела. Отнесение аминокислот к каждой описанной в данном документе CDR сделано в соответствии с определениями по Kabat EA et al. 1991, в "Sequences of Proteins of Immulogical Interest" Fifth Edition, NIH Publication No. 91-3242, pp xv- xvii.
Как должно быть известно специалисту в данной области техники, также существуют другие способы отнесения аминокислот к каждой CDR. Например, это Международная иммуногенетическая информационная система (IMGT(R)) (http://www.imgt.org/ и Lefranc and Lefranc "The Immunoglobulin FactsBook", опубликованная Academic Press, 2001).
Следует принимать во внимание, что молекулы, содержащие три или менее областей CDR (в некоторых случаях даже одну единственную CDR или ее часть), способны сохранять антигенсвязывающую активность антитела, из которого получена(ы) CDR. Например, в Gao et al., 1994, J. Biol. Chem., 269: 32389-93 описано, что целая цепь VL (включая все три CDR) обладает аффинностью в отношении своего субстрата.
Молекулы, содержащие две области CDR, описаны, например, в Vaughan & Sollazzo 2001, Combinatorial Chemistry & High Throughput Screening, 4: 417-430. На странице 418 (правая колонка - 3 Наша стратегия конструирования) описано минитело, содержащее только гипервариабельные области CDR H1 и H2, вперемежку с каркасными областями. Описано, что минитело способно связываться с мишенью. Pessi et al., 1993, Nature, 362: 367-9 и Bianchi et al., 1994, J. Mol. Biol., 236: 649-59 приведены в Vaughan & Sollazzo в виде ссылок и более подробно описывают минитело Н1 и Н2 и его свойства. В Qiu et al., 2007, Nature Biotechnology, 25:921-9 продемонстрировано, что молекула, состоящая из двух связанных CDR, способна связывать антиген. В Quiocho 1993, Nature, 362: 293-4 приведено краткое изложение технологии «минител». В Ladner 2007, Nature Biotechnology, 25:875-7 сообщается, что молекулы, содержащие две CDR, способны сохранять антигенсвязывающую активность.
Молекулы, содержащие одну область CDR, описаны, например, в Laune et al., 1997, JBC, 272: 30937-44, в которой продемонстрировано, что ряд гексапептидов, полученных из CDR, проявляют антигенсвязывающую активность, и отмечено, что синтетические пептиды полной, одиночной CDR проявляют сильную связывающую активность. В Monnet et al., 1999, JBC, 274: 3789-96 показано, что ряд 12-мерных пептидов и ассоциированных с ними каркасных областей обладают антигенсвязывающей активностью, и отмечено, что один CDR3-подобный пептид способен связывать антиген. В Heap et al., 2005, J. Gen. Virol., 86: 1791-1800 сообщается, что «микроантитело» (молекула, содержащая одну CDR) способно связывать антиген, и показано, что кольцевой пептид из анти-ВИЧ антитела обладает антигенсвязывающей активностью и функцией. В Nicaise et al., 2004, Protein Science, 13:1882-91 показано, что одна CDR может обеспечивать антигенсвязывающую активность и аффинность в отношении антигена лизоцима.
Таким образом, молекулы, содержащие три или менее CDR, способны сохранять антигенсвязывающие свойства антител, из которых они получены.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент содержит вариабельную тяжелую цепь (VH), содержащую следующие CDR:
(i) SEQ ID NO: 51 и SEQ ID NO: 52 и SEQ ID NO: 53; или
(ii) SEQ ID NO: 57 и SEQ ID NO: 58 и SEQ ID NO: 59; или
(iii) SEQ ID NO: 63 и SEQ ID NO: 64 и SEQ ID NO: 65; или
(iv) SEQ ID NO: 69 и SEQ ID NO: 70 и SEQ ID NO: 71; или
(v) SEQ ID NO: 75 и SEQ ID NO: 76 и SEQ ID NO: 77; или
(vi) SEQ ID NO: 81 и SEQ ID NO: 82 и SEQ ID NO: 83; или
(vii) SEQ ID NO: 87 и SEQ ID NO: 88 и SEQ ID NO: 89; или
(viii) SEQ ID NO: 93 и SEQ ID NO: 94 и SEQ ID NO: 95; или
(ix) SEQ ID NO: 99 и SEQ ID NO: 100 и SEQ ID NO: 101; или
(x) SEQ ID NO: 105 и SEQ ID NO: 106 и SEQ ID NO: 107; или
(xi) SEQ ID NO: 111 и SEQ ID NO: 112 и SEQ ID NO: 113; или
(xii) SEQ ID NO: 117 и SEQ ID NO: 118 и SEQ ID NO: 119; или
(xiii) SEQ ID NO: 123 и SEQ ID NO: 124 и SEQ ID NO: 125; или
(xiv) SEQ ID NO: 129 и SEQ ID NO: 130 и SEQ ID NO: 131; или
(xv) SEQ ID NO: 135 и SEQ ID NO: 136 и SEQ ID NO: 137; или
(xvi) SEQ ID NO: 141 и SEQ ID NO: 142 и SEQ ID NO: 143; или
(xvii) SEQ ID NO: 147 и SEQ ID NO: 148 и SEQ ID NO: 149; или
(xviii) SEQ ID NO: 153 и SEQ ID NO: 154 и SEQ ID NO: 155; или
(xix) SEQ ID NO: 159 и SEQ ID NO: 160 и SEQ ID NO: 161; или
(xx) SEQ ID NO: 165 и SEQ ID NO: 166 и SEQ ID NO: 167; или
(xxi) SEQ ID NO: 171 и SEQ ID NO: 172 и SEQ ID NO: 173; или
(xxii) SEQ ID NO: 177 и SEQ ID NO: 178 и SEQ ID NO: 179; или
(xxiii) SEQ ID NO: 183 и SEQ ID NO: 184 и SEQ ID NO: 185; или
(xxiv) SEQ ID NO: 189 и SEQ ID NO: 190 и SEQ ID NO: 191.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент содержит вариабельную легкую цепь (VL), содержащую следующие CDR:
(i) SEQ ID NO: 54 и SEQ ID NO: 55 и SEQ ID NO: 56; или
(ii) SEQ ID NO: 60 и SEQ ID NO: 61 и SEQ ID NO: 62; или
(iii) SEQ ID NO: 66 и SEQ ID NO: 67 и SEQ ID NO: 68; или
(iv) SEQ ID NO: 72 и SEQ ID NO: 73 и SEQ ID NO: 74; или
(v) SEQ ID NO: 78 и SEQ ID NO: 79 и SEQ ID NO: 80; или
(vi) SEQ ID NO: 84 и SEQ ID NO: 85 и SEQ ID NO: 86; или
(vii) SEQ ID NO: 90 и SEQ ID NO: 91 и SEQ ID NO: 92; или
(viii) SEQ ID NO: 96 и SEQ ID NO: 97 и SEQ ID NO: 98; или
(ix) SEQ ID NO: 102 и SEQ ID NO: 103 и SEQ ID NO: 104; или
(x) SEQ ID NO: 108 и SEQ ID NO: 109 и SEQ ID NO: 110; или
(xi) SEQ ID NO: 114 и SEQ ID NO: 115 и SEQ ID NO: 116; или
(xii) SEQ ID NO: 120 и SEQ ID NO: 121 и SEQ ID NO: 122; или
(xiii) SEQ ID NO: 126 и SEQ ID NO: 127 и SEQ ID NO: 128; или
(xiv) SEQ ID NO: 132 и SEQ ID NO: 133 и SEQ ID NO: 134; или
(xv) SEQ ID NO: 138 и SEQ ID NO: 139 и SEQ ID NO: 140; или
(xvi) SEQ ID NO: 144 и SEQ ID NO: 145 и SEQ ID NO: 146; или
(xvii) SEQ ID NO: 150 и SEQ ID NO: 151 и SEQ ID NO: 152; или
(xviii) SEQ ID NO: 156 и SEQ ID NO: 157 и SEQ ID NO: 158; или
(xix) SEQ ID NO: 162 и SEQ ID NO: 163 и SEQ ID NO: 164; или
(xx) SEQ ID NO: 168 и SEQ ID NO: 169 и SEQ ID NO: 170; или
(xxi) SEQ ID NO: 174 и SEQ ID NO: 175 и SEQ ID NO: 176; или
(xxii) SEQ ID NO: 180 и SEQ ID NO: 181 и SEQ ID NO: 182; или
(xxiii) SEQ ID NO: 186 и SEQ ID NO: 187 и SEQ ID NO: 188; или
(xxiv) SEQ ID NO: 192 и SEQ ID NO: 193 и SEQ ID NO: 194.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент содержит аминокислотную последовательность вариабельной тяжелой цепи (VH), выбранную из группы, состоящей из: SEQ ID NO: 3; SEQ ID NO: 4; SEQ ID NO: 5; SEQ ID NO: 6; SEQ ID NO: 7; SEQ ID NO: 8; SEQ ID NO: 9; SEQ ID NO: 10; SEQ ID NO: 11; SEQ ID NO: 12; SEQ ID NO: 13; SEQ ID NO: 14; SEQ ID NO: 15; SEQ ID NO: 16; SEQ ID NO: 17; SEQ ID NO: 18; SEQ ID NO: 19; SEQ ID NO: 20; SEQ ID NO: 21; SEQ ID NO: 22; SEQ ID NO: 23; SEQ ID NO: 24; SEQ ID NO: 25; и SEQ ID NO: 26.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент содержит вариабельную аминокислотную последовательность легкой цепи (VL), выбранную из группы, состоящей из: SEQ ID NO: 27; SEQ ID NO: 28; SEQ ID NO: 29; SEQ ID NO: 30; SEQ ID NO: 31; SEQ ID NO: 32; SEQ ID NO: 33; SEQ ID NO: 34; SEQ ID NO: 35; SEQ ID NO: 36; SEQ ID NO: 37; SEQ ID NO: 38; SEQ ID NO: 39; SEQ ID NO: 40; SEQ ID NO: 41; SEQ ID NO: 42; SEQ ID NO: 43; SEQ ID NO: 44; SEQ ID NO: 45; SEQ ID NO: 46; SEQ ID NO: 47; SEQ ID NO: 48; SEQ ID NO: 49; и SEQ ID NO: 50.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент содержит следующие аминокислотные последовательности CDR:
(i) SEQ ID NO: 51 и SEQ ID NO: 52, и SEQ ID NO: 53, и SEQ ID NO: 54, и SEQ ID NO: 55, и SEQ ID NO: 56; или
(ii) SEQ ID NO: 57 и SEQ ID NO: 58, и SEQ ID NO: 59, и SEQ ID NO: 60, и SEQ ID NO: 61, и SEQ ID NO: 62; или
(iii) SEQ ID NO: 63 и SEQ ID NO: 64, и SEQ ID NO: 65, и SEQ ID NO: 66, и SEQ ID NO: 67, и SEQ ID NO: 68; или
(iv) SEQ ID NO: 69 и SEQ ID NO: 70, и SEQ ID NO: 71, и SEQ ID NO: 72, и SEQ ID NO: 73, и SEQ ID NO: 74; или
(v) SEQ ID NO: 75 и SEQ ID NO: 76, и SEQ ID NO: 77, и SEQ ID NO: 78, и SEQ ID NO: 79, и SEQ ID NO: 80; или
(vi) SEQ ID NO: 81 и SEQ ID NO: 82, и SEQ ID NO: 83, и SEQ ID NO: 84, и SEQ ID NO: 85, и SEQ ID NO: 86; или
(vii) SEQ ID NO: 87 и SEQ ID NO: 88, и SEQ ID NO: 89, и SEQ ID NO: 90, и SEQ ID NO: 91, и SEQ ID NO: 92; или
(viii) SEQ ID NO: 93 и SEQ ID NO: 94, и SEQ ID NO: 95, и SEQ ID NO: 96, и SEQ ID NO: 97, и SEQ ID NO: 98; или
(ix) SEQ ID NO: 99 и SEQ ID NO: 100, и SEQ ID NO: 101, и SEQ ID NO: 102, и SEQ ID NO: 103, и SEQ ID NO: 104; или
(x) SEQ ID NO: 105 и SEQ ID NO: 106, и SEQ ID NO: 107, и SEQ ID NO: 108, и SEQ ID NO: 109, и SEQ ID NO: 110; или
(xi) SEQ ID NO: 111 и SEQ ID NO: 112, и SEQ ID NO: 113, и SEQ ID NO: 114, и SEQ ID NO: 115, и SEQ ID NO: 116; или
(xii) SEQ ID NO: 117 и SEQ ID NO: 118, и SEQ ID NO: 119, и SEQ ID NO: 120, и SEQ ID NO: 121, и SEQ ID NO: 122; или
(xiii) SEQ ID NO: 123 и SEQ ID NO: 124, и SEQ ID NO: 125, и SEQ ID NO: 126, и SEQ ID NO: 127, и SEQ ID NO: 128; или
(xiv) SEQ ID NO: 129 и SEQ ID NO: 130, и SEQ ID NO: 131, и SEQ ID NO: 132, и SEQ ID NO: 133, и SEQ ID NO: 134; или
(xv) SEQ ID NO: 135 и SEQ ID NO: 136, и SEQ ID NO: 137, и SEQ ID NO: 138, и SEQ ID NO: 139, и SEQ ID NO: 140; или
(xvi) SEQ ID NO: 141 и SEQ ID NO: 142, и SEQ ID NO: 143, и SEQ ID NO: 144, и SEQ ID NO: 145, и SEQ ID NO: 146; или
(xvii) SEQ ID NO: 147 и SEQ ID NO: 148, и SEQ ID NO: 149, и SEQ ID NO: 150, и SEQ ID NO: 151, и SEQ ID NO: 152; или
(xviii) SEQ ID NO: 153 и SEQ ID NO: 154, и SEQ ID NO: 155, и SEQ ID NO: 156, и SEQ ID NO: 157, и SEQ ID NO: 158; или
(xix) SEQ ID NO: 159 и SEQ ID NO: 160, и SEQ ID NO: 161, и SEQ ID NO: 162, и SEQ ID NO: 163, и SEQ ID NO: 164; или
(xx) SEQ ID NO: 165 и SEQ ID NO: 166, и SEQ ID NO: 167, и SEQ ID NO: 168, и SEQ ID NO: 169, и SEQ ID NO: 170; или
(xxi) SEQ ID NO: 171 и SEQ ID NO: 172, и SEQ ID NO: 173, и SEQ ID NO: 174, и SEQ ID NO: 175, и SEQ ID NO: 176; или
(xxii) SEQ ID NO: 177 и SEQ ID NO: 178, и SEQ ID NO: 179, и SEQ ID NO: 180, и SEQ ID NO: 181, и SEQ ID NO: 182; или
(xxiii) SEQ ID NO: 183 и SEQ ID NO: 184, и SEQ ID NO: 185, и SEQ ID NO: 186, и SEQ ID NO: 187, и SEQ ID NO: 188; или
(xxiv) SEQ ID NO: 189 и SEQ ID NO: 190, и SEQ ID NO: 191, и SEQ ID NO: 192, и SEQ ID NO: 193, и SEQ ID NO: 194.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент содержит следующие аминокислотные последовательности:
(i) SEQ ID NO: 3 и SEQ ID NO: 27; или
(ii) SEQ ID NO: 4 и SEQ ID NO: 28; или
(iii) SEQ ID NO: 5 и SEQ ID NO: 29; или
(iv) SEQ ID NO: 6 и SEQ ID NO: 30; или
(v) SEQ ID NO: 7 и SEQ ID NO: 31; или
(vi) SEQ ID NO: 8 и SEQ ID NO: 32; или
(vii) SEQ ID NO: 9 и SEQ ID NO: 33; или
(viii) SEQ ID NO: 10 и SEQ ID NO: 34; или
(ix) SEQ ID NO: 11 и SEQ ID NO: 35; или
(x) SEQ ID NO: 12 и SEQ ID NO: 36; или
(xi) SEQ ID NO: 13 и SEQ ID NO: 37; или
(xii) SEQ ID NO: 14 и SEQ ID NO: 38; или
(xiii) SEQ ID NO: 15 и SEQ ID NO: 39; или
(xiv) SEQ ID NO: 16 и SEQ ID NO: 40; или
(xv) SEQ ID NO: 17 и SEQ ID NO: 41; или
(xvi) SEQ ID NO: 18 и SEQ ID NO: 42; или
(xvii) SEQ ID NO: 19 и SEQ ID NO: 43; или
(xviii) SEQ ID NO: 20 и SEQ ID NO: 44; или
(xix) SEQ ID NO: 21 и SEQ ID NO: 45; или
(xx) SEQ ID NO: 22 и SEQ ID NO: 46; или
(xxi) SEQ ID NO: 23 и SEQ ID NO: 47; или
(xxii) SEQ ID NO: 24 и SEQ ID NO: 48; или
(xxiii) SEQ ID NO: 25 и SEQ ID NO: 49; или
(xxiv) SEQ ID NO: 26 и SEQ ID NO: 50.
Агенты согласно изобретению также могут содержать константные области (CH) и (CL) SEQ ID NO 1 и SEQ ID NO 2.
В дополнительном варианте реализации изобретения агент способен конкурировать с описанными в данном документе агентами согласно изобретению, например, агентами, содержащими аминокислотные последовательности, приведенные в вышеуказанных вариантах реализации (например, SEQ ID NO: 1-194), за предотвращение или снижение связывания FcγRIIb с Fc-доменом молекулы антитела.
Под «способный конкурировать» за предотвращение или снижение связывания FcγRIIb с Fc-доменом молекулы антитела с агентом (таким как молекула антигена) по определению данного документа подразумевается, что исследуемый агент способен ингибировать или каким-либо другим образом препятствовать, по меньшей мере частично, связыванию агента по определению данного документа с FcγRIIb и предотвращать или снижать связывание FcγRIIb с Fc-доменом молекулы антитела.
Например, агент может быть способен ингибировать связывание описанного в данном документе агента по меньшей мере на около 10%; например, по меньшей мере на около 20% или по меньшей мере на около 30%, по меньшей мере на около 40%, по меньшей мере на около 50%, по меньшей мере на около 60%, по меньшей мере на около 70%, по меньшей мере на около 80%, по меньшей мере на около 90%, по меньшей мере на около 95%, по меньшей мере на около 100% и/или ингибировать способность агента предотвращать или снижать связывание FcγRIIb с Fc-доменом молекулы антитела по меньшей мере на около 10%; например, по меньшей мере на около 20%, по меньшей мере на около 30%, по меньшей мере на около 40%, по меньшей мере на около 50%, по меньшей мере на около 60%, по меньшей мере на около 70%, по меньшей мере на около 80%, по меньшей мере на около 90%, по меньшей мере на около 95%, или по меньшей мере на около 100%.
Конкурентное связывание можно определить способами, хорошо известными специалистам в данной области техники, такими как ферментный иммуносорбентный анализ (ELISA).
Анализ ELISA можно использовать для оценки эпитоп-модифицирующих или блокирующих антител. Дополнительные способы, подходящие для выявления конкурирующих антител, раскрыты в Antibodies: A Laboratory Manual, Harlow & Lane, которая включена в данный документ посредством ссылки (например, смотрите страницы от 567 до 569, от 574 до 576, 583 и от 590 до 612, 1988, CSHL, NY, ISBN 0-87969-314-2).
Агенты согласно изобретению могут содержать следующие константные области (CH и CL):
IgG1-CH [SEQ ID NO: 1]
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
l-CL [SEQ ID NO: 2]
QPKAAPSVTLFPPSSEELQANKATLVCLISDFYPGAVTVAWKADSSPVKAGVETTTPSKQSNNKYAASSYLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
Агенты согласно изобретению могут содержать одну или более последовательностей следующих клонов:
Клон антитела: 1A01
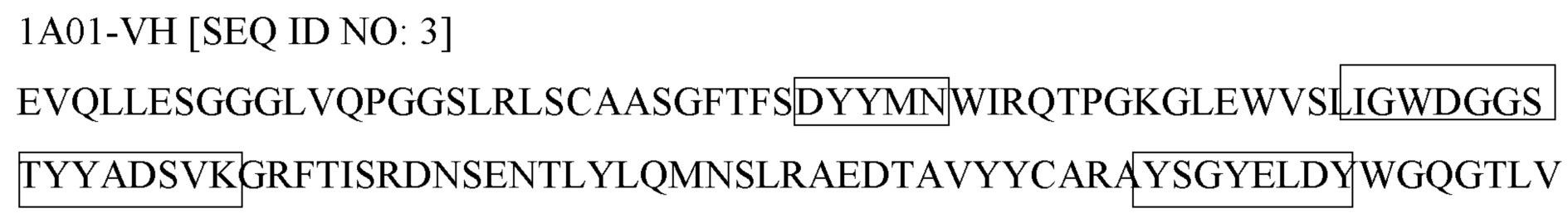
TVSS
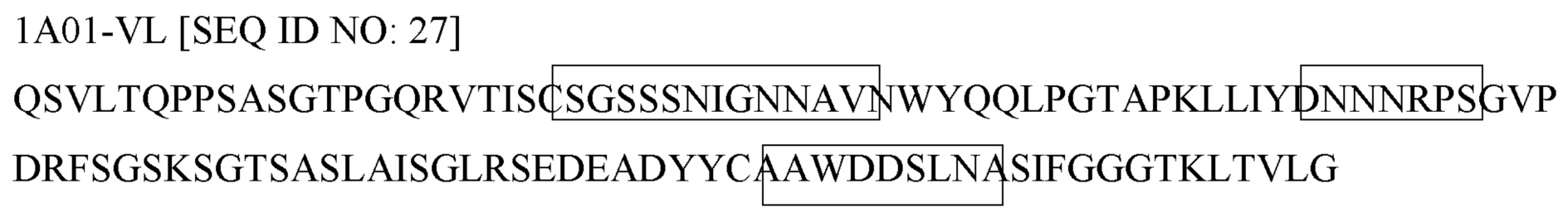
Области CDR
CDRH1: DYYMN [SEQ ID NO: 51]
CDRH2: LIGWDGGSTYYADSVKG [SEQ ID NO: 52]
CDRH3: AYSGYELDY [SEQ ID NO: 53]
CDRL1: SGSSSNIGNNAVN [SEQ ID NO: 54]
CDRL2: DNNNRPS [SEQ ID NO: 55]
CDRL3: AAWDDSLNASI [SEQ ID NO: 56]
Клон антитела: 1B07
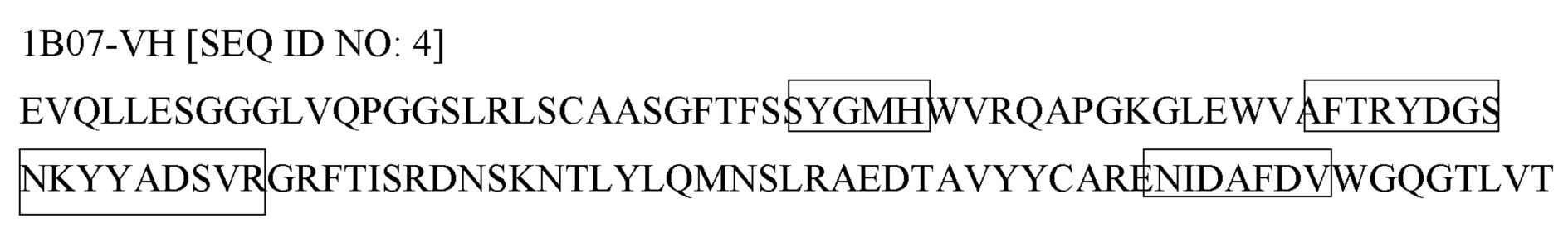
VSS
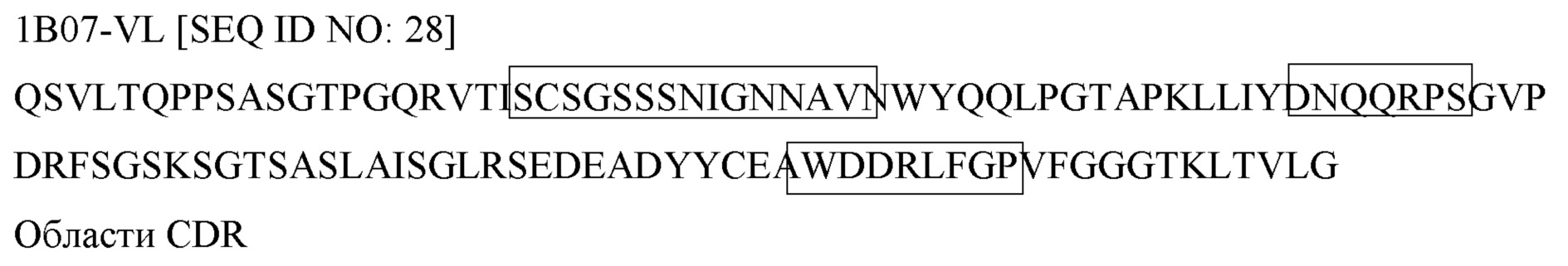
CDRH1: SYGMH [SEQ ID NO: 57]
CDRH2: FTRYDGSNKYYADSVRG [SEQ ID NO: 58]
CDRH3: ENIDAFDV [SEQ ID NO: 59]
CDRL1: SGSSSNIGNNAVN [SEQ ID NO: 60]
CDRL2: DNQQRPS [SEQ ID NO: 61]
CDRL3: WDDRLFGPV [SEQ ID NO: 62]
Клон антитела: 1C04

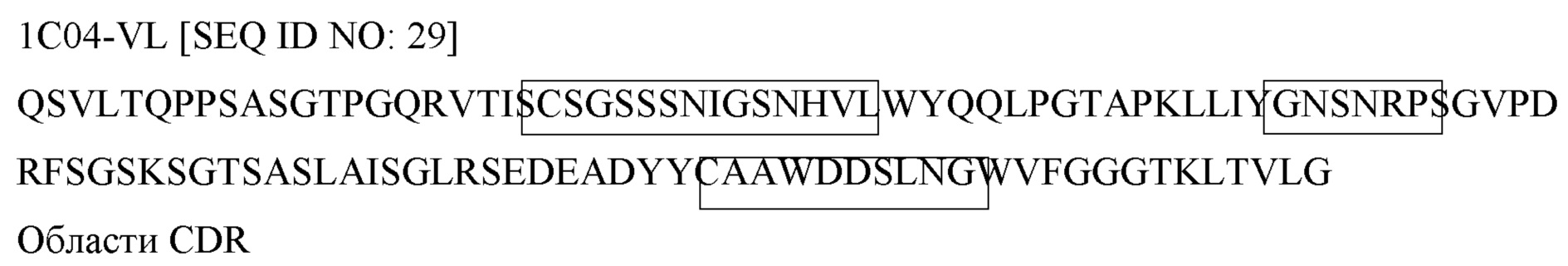
CDRH1: SYAMS [SEQ ID NO: 63]
CDRH2: SISDSGAGRYYADSVEG [SEQ ID NO: 64]
CDRH3: THDSGELLDAFDI [SEQ ID NO: 65]
CDRL1: SGSSSNIGSNHVL [SEQ ID NO: 66]
CDRL2: GNSNRPS [SEQ ID NO: 67]
CDRL3: AAWDDSLNGWV [SEQ ID NO: 68]
Клон антитела: 1E05


CDRH1: TYAMN [SEQ ID NO: 69]
CDRH2: VISYDGSNKNYVDSVKG [SEQ ID NO: 70]
CDRH3: NFDNSGYAIPDAFDI [SEQ ID NO: 71]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 72]
CDRL2: DNNSRPS [SEQ ID NO: 73]
CDRL3: AAWDDSLGGPV [SEQ ID NO: 74]
Клон антитела: 2A09

CDRH1: NAWMS [SEQ ID NO: 75]
CDRH2: YISRDADITHYPASVKG [SEQ ID NO: 76]
CDRH3: GFDYAGDDAFDI [SEQ ID NO: 77]
CDRL1: SGSSSNIGSNAVN [SEQ ID NO: 78]
CDRL2: GNSDRPS [SEQ ID NO: 79]
CDRL3: AAWDDSLNGRWV [SEQ ID NO: 80]
Клон антитела: 2B08

Области CDR
CDRH1: DYYMS [SEQ ID NO: 81]
CDRH2: LIGHDGNNKYYLDSLEG [SEQ ID NO: 82]
CDRH3: ATDSGYDLLY [SEQ ID NO: 83]
CDRL1: SGSSSNIGNNAVN [SEQ ID NO: 84]
CDRL2: YDDLLPS [SEQ ID NO: 85]
CDRL3: TTWDDSLSGVV [SEQ ID NO: 86]
Клон антитела: 2E08

CDRH1: DYYMS [SEQ ID NO: 87]
CDRH2: AIGFSDDNTYYADSVKG [SEQ ID NO: 88]
CDRH3: GDGSGWSF [SEQ ID NO: 89]
CDRL1: SGSSSNIGNNAVN [SEQ ID NO: 90]
CDRL2: DNNKRPS [SEQ ID NO: 91]
CDRL3: ATWDDSLRGWV [SEQ ID NO: 92]
Клон антитела: 5C04

CDRH1: NYGMH [SEQ ID NO: 93]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 94]
CDRH3: WRDAFDI [SEQ ID NO: 95]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 96]
CDRL2: SDNQRPS [SEQ ID NO: 97]
CDRL3: AAWDDSLSGSWV [SEQ ID NO: 98]
Клон антитела: 5C05

CDRH1: TYGMH [SEQ ID NO: 99]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 100]
CDRH3: ENFDAFDV [SEQ ID NO: 101]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 102]
CDRL2: SNSQRPS [SEQ ID NO: 103]
CDRL3: AAWDDSLNGQVV [SEQ ID NO: 104]
Клон антитела: 5D07
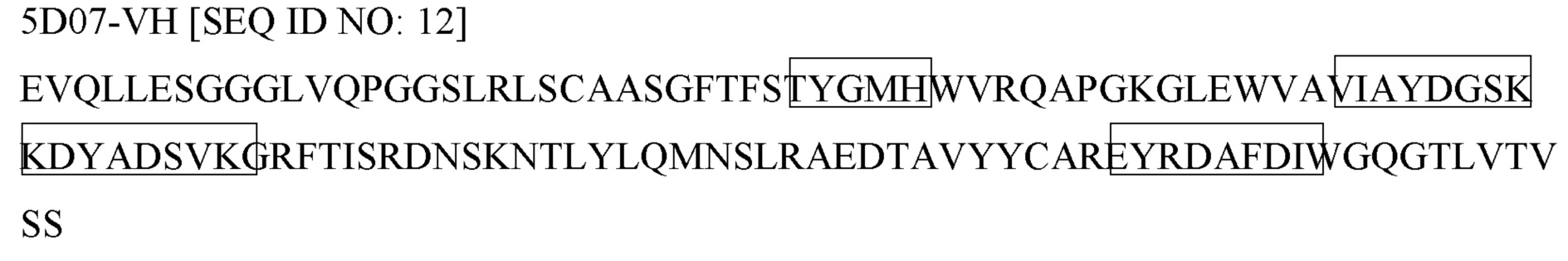
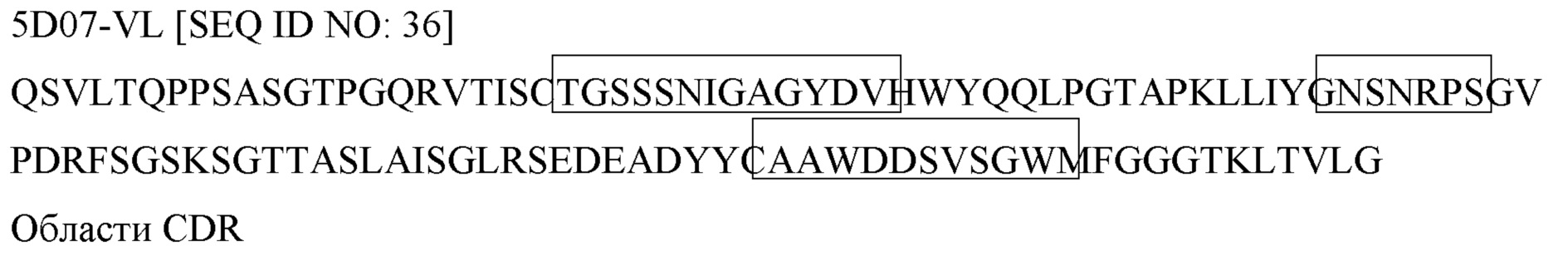
CDRH1: TYGMH [SEQ ID NO: 105]
CDRH2: VIAYDGSKKDYADSVKG [SEQ ID NO: 106]
CDRH3: EYRDAFDI [SEQ ID NO: 107]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 108]
CDRL2: GNSNRPS [SEQ ID NO: 109]
CDRL3: AAWDDSVSGWM [SEQ ID NO: 110]
Клон антитела: 5E12


CDRH1: SYGMH [SEQ ID NO: 111]
CDRH2: VISYDGINKDYADSMKG [SEQ ID NO: 112]
CDRH3: ERKDAFDI [SEQ ID NO: 113]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 114]
CDRL2: SNNQRPS [SEQ ID NO: 115]
CDRL3: ATWDDSLNGLV [SEQ ID NO: 116]
Клон антитела: 5G08

CDRH1: NYGMH [SEQ ID NO: 117]
CDRH2: VISYDGSNRYYADSVKG [SEQ ID NO: 118]
CDRH3: DRWNGMDV [SEQ ID NO: 119]
CDRL1: SGSSSNIGAGYDVH [SEQ ID NO: 120]
CDRL2: ANNQRPS [SEQ ID NO: 121]
CDRL3: AAWDDSLNGPWV [SEQ ID NO: 122]
Клон антитела: 5H06
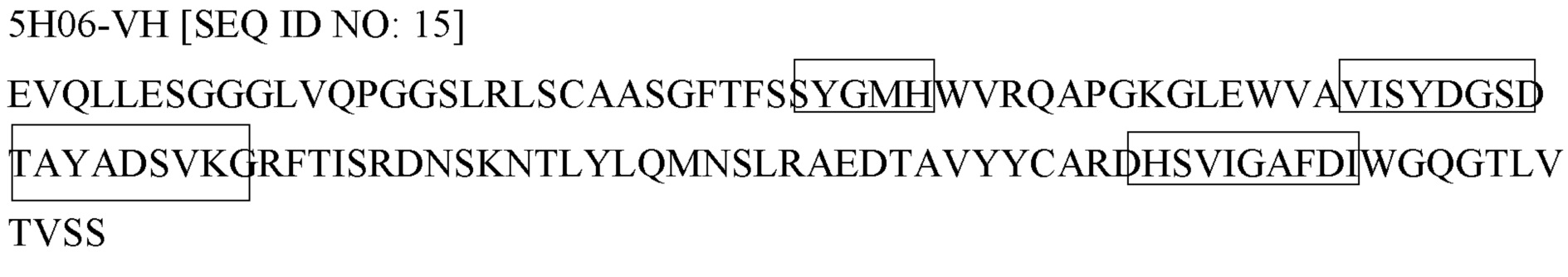
5H06-VL [SEQ ID NO: 39]
CDRH1: SYGMH [SEQ ID NO: 123]
CDRH2: VISYDGSDTAYADSVKG [SEQ ID NO: 124]
CDRH3: DHSVIGAFDI [SEQ ID NO: 125]
CDRL1: SGSSSNIGSNTVN [SEQ ID NO: 126]
CDRL2: DNNKRPS [SEQ ID NO: 127]
CDRL3: SSYAGSNNVV [SEQ ID NO: 128]
Клон антитела: 6A09

CDRH1: SYGMH [SEQ ID NO: 129]
CDRH2: VTSYDGNTKYYANSVKG [SEQ ID NO: 130]
CDRH3: EDCGGDCFDY [SEQ ID NO: 131]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 132]
CDRL2: GNSNRPS [SEQ ID NO: 133]
CDRL3: AAWDDSLNEGV [SEQ ID NO: 134]
Клон антитела: 6B01

Области CDR
CDRH1: NYGMH [SEQ ID NO: 135]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 136]
CDRH3: DQLGEAFDI [SEQ ID NO: 137]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 138]
CDRL2: DNNKRPS [SEQ ID NO: 139]
CDRL3: ATWDDSLSGPV [SEQ ID NO: 140]
Клон антитела: 6C11

Области CDR
CDRH1: DYGMS [SEQ ID NO: 141]
CDRH2: AISGSGSSTYYADSVKG [SEQ ID NO: 142]
CDRH3: GDIDYFDY [SEQ ID NO: 143]
CDRL1: TGSSSNFGAGYDVH [SEQ ID NO: 144]
CDRL2: ENNKRPS [SEQ ID NO: 145]
CDRL3: AAWDDSLNGPV [SEQ ID NO: 146]
Клон антитела: 6C12

Области CDR
CDRH1: SYGMH [SEQ ID NO: 147]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 148]
CDRH3: ERRDAFDI [SEQ ID NO: 149]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 150]
CDRL2: SDNQRPS [SEQ ID NO: 151]
CDRL3: ATWDSDTPV [SEQ ID NO: 152]
Клон антитела: 6D01

Области CDR
CDRH1: SYGMH [SEQ ID NO: 153]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 154]
CDRH3: DHSAAGYFDY [SEQ ID NO: 155]
CDRL1: SGSSSNIGSNTVN [SEQ ID NO: 156]
CDRL2: GNSIRPS [SEQ ID NO: 157]
CDRL3: ASWDDSLSSPV [SEQ ID NO: 158]
Клон антитела: 6G03

Области CDR
CDRH1: SYGMH [SEQ ID NO: 159]
CDRH2: GISWDSAIIDYAGSVKG [SEQ ID NO: 160]
CDRH3: DEAAAGAFDI [SEQ ID NO: 161]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 162]
CDRL2: GNTDRPS [SEQ ID NO: 163]
CDRL3: AAWDDSLSGPVV [SEQ ID NO: 164]
Клон антитела: 6G08
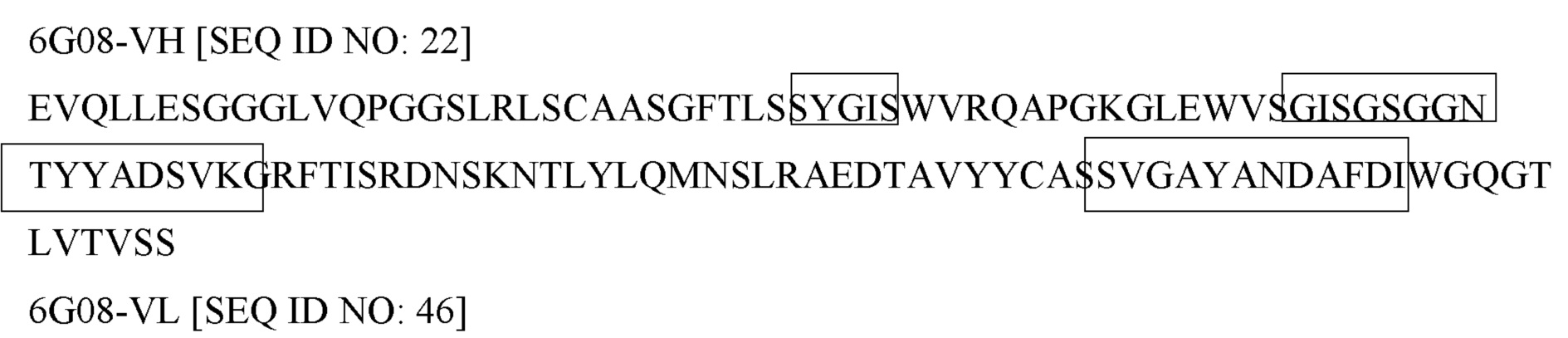
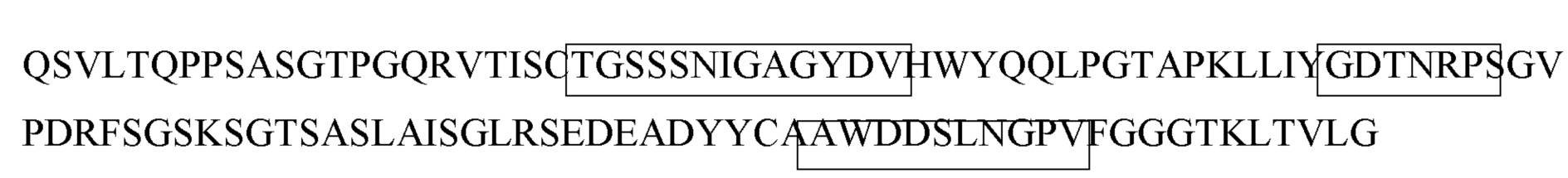
Области CDR
CDRH1: SYGIS [SEQ ID NO: 165]
CDRH2: GISGSGGNTYYADSVKG [SEQ ID NO: 166]
CDRH3: SVGAYANDAFDI [SEQ ID NO: 167]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 168]
CDRL2: GDTNRPS [SEQ ID NO: 169]
CDRL3: AAWDDSLNGPV [SEQ ID NO: 170]
Клон антитела: 6G11

Области CDR
CDRH1: SYGMH [SEQ ID NO: 171]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 172]
CDRH3: ELYDAFDI [SEQ ID NO: 173]
CDRL1: TGSSSNIGAGYDVH [SEQ ID NO: 174]
CDRL2: ADDHRPS [SEQ ID NO: 175]
CDRL3: ASWDDSQRAVI [SEQ ID NO: 176]
Клон антитела: 6H08

Области CDR
CDRH1: NYGMH [SEQ ID NO: 177]
CDRH2: VISYDGSNKYYAD SVKG [SEQ ID NO: 178]
CDRH3: EYKDAFDI [SEQ ID NO: 179]
CDRL1: TGSSSNIGSNTVN [SEQ ID NO: 180]
CDRL2: DNNKRPS [SEQ ID NO: 181]
CDRL3: QAWGTGIRV [SEQ ID NO: 182]
Клон антитела: 7C07

Области CDR
CDRH1: SYGMH [SEQ ID NO: 183]
CDRH2: VISYDGSNKYYADSVKG [SEQ ID NO: 184]
CDRH3: EFGYIILDY [SEQ ID NO: 185]
CDRL1: SGSSSNIGSNTVN [SEQ ID NO: 186]
CDRL2: RDYERPS [SEQ ID NO: 187]
CDRL3: MAWDDSLSGVV [SEQ ID NO: 188]
Клон антитела: 4B02

Области CDR
CDRH1: NHGMH [SEQ ID NO: 189]
CDRH2: VISYDGTNKYYADSVRG [SEQ ID NO: 190]
CDRH3: ETWDAFDV [SEQ ID NO: 191]
CDRL1: SGSSSNIGSNNAN [SEQ ID NO: 192]
CDRL2: DNNKRPS [SEQ ID NO: 193]
CDRL3: QAWDSSTVV [SEQ ID NO: 194]
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент способен конкурировать с агентами, определенными в более ранних вариантах реализации, за предотвращение или снижение связывания FcγRIIb с Fc-доменом молекулы антитела.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент предотвращает или снижает сигналинг FcγRIIb.
Fc-рецепторы могут модулировать поведение клеток посредством клеточной сигнализации. Специалисту в области клеточной биологии должно быть известно, какие нижерасположенные модуляторы клеточной сигнализация активируются и/или деактивируются вследствие сигнализации FcγRIIb, и какое воздействие оказывает на клетку активация и/или деактивация таких модуляторов клеточной сигнализации.
Под выражением «агент предотвращает или снижает сигналинг FcγRIIb» подразумевается, что происходит предотвращение или снижение сигнализации FcγRIIb, когда FcγRIIb связан с Fc-доменом, и/или происходит предотвращение или снижение сигнализации FcγRIIb, когда FcγRIIb не связан с Fc-доменом.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом агент предотвращает или снижает интернализацию молекулы антитела клеткой-мишенью.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом антиген клеточной поверхности выбран из группы, состоящей из: CD19: или его части; CD20: или его части; CD40: или его части; CD52: или его части; Thy-1 (т.е. CD90, кластера дифференцировки 90 (Biofactors. 2009 May-Jun;35(3):258-65)): или его части; Ly-6 (т.е. лимфоцитарного антигена 6 (Mol Biol Rep. 2009 Apr;36(4):697-703)): или его части; CD59 (т.е. регуляторного белка комплемента (Mol Immunol. 2007 Jan;44(l-3):73-81)): или его части; Fas (т.е. FS7-ассоциированного антигена клеточной поверхности, CD95, APO-1 или TNFRSF6 (Adv Exp Med Biol. 2009;647:64-93)): или его части; EGFR (т.е. рецептора эпидермального фактора роста (FEBS J. 2010 Jan;277(2):301-8)): или его части; Her2 (т.е. человеческого рецептора эпидермального фактора роста 2 (Clin Breast Cancer. 2008 Oct; 8(5): 392- 401)): или его части; CXCR4 (т.е. рецептора хемокинов 4 (Biochim Biophys Acta. 2007 Apr;1768(4):952- 63)): или его части; молекул HLA (т.е. молекул человеческого лейкоцитарного антигена (Korean J Lab Med. 2010 Jun;30(3):203)): или его части; GM1 (т.е. ганглиозида, моносиалотетрагексозилганглиозида (J Lipid Res. 2010 Sep;51(9):2731-8)): или его части; CD22 (т.е. Cheson (2008) NEJM 359(6): 613-26): или его части; CD23 (Cheson, 2008): или его части; CD80 (Cheson, 2008): или его части; CD74 (Cheson, 2008): или его части; DRD (Cheson, 2008): или его части.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом молекула антитела, определение которой приведено в (i), специфически связывается с CD20.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом молекула антитела, определение которой приведено в (i), представляет собой антитело к CD20 типа I.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом молекула антитела, определение которой приведено в (i), представляет собой антитело к CD20 типа II.
Существует два типа молекул антител к CD20, которые изначально в 2003 г. были определены изобретателями как принадлежащие двум разным группам (Cragg et al., 2003. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101:1045-1052 и Chan, et al., 2003. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res 63:5480-5489), а после этого, в 2004 г., определены как молекулы антител I и II типа (Cragg and Glennie 2004. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 103:2738-2743). Изначально основанием для этого было то, что анти-CD20 mAb разделялись на два разных типа реагентов на основании своей способности уничтожать ксенографтную лимфому: тип I (например, ритуксимаб и 1F5) задействует систему комплемента; а тип II (например, Bl) - нет. Оба типа молекул антител обеспечивают хорошее продление времени жизни, но снижение активности системы комплемента при введении CVF в значительной степени снижает эффективность ритуксимаба и 1F5, но не оказывает воздействие на Bl. Эти результаты четко показали, что разные молекулы антител к CD20 управляют разными эффекторными механизмами in vivo. Кроме того, они полностью согласуются с более ранней работой, показывающей, что ритуксимаб и 1F5 способны эффективно активировать систему комплемента в результате транслокации CD20 в липидные рафты мембраны клетки-мишени, что не может делать молекула антитела типа Bl (Cragg et al., 2003. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101:1045-1052). Существует хорошая корреляция со способностью молекулы антитела задействовать комплемент и индуцировать перемещение CD20 в липидные рафты (Cragg et al., 2003. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101:1045-1052 и Cragg, and Glennie 2004. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 103:2738-2743). Следовательно, природу типа I и II можно определить по способности перемещать CD20 в липидные рафты. Это можно определить так, как указано ниже. Также существует корреляция со способностью молекулы антитела типа II вызывать более сильную гомотипическую адгезию и непосредственную гибель клеток, но одни эти факторы нельзя применять для определения молекул антител типа I или II (в отличие от анализа гомотипической адгезии Tx-100; смотрите ниже).
Следовательно, различные анти-CD20 молекулы антител можно классифицировать как принадлежащие типу I (например, ритуксимаб и офатумумаб) или типу II (например, тозитумомаб (Bl), GA101 и 11B8) в соответствии с их способностью перераспределять CD20 в плазматической мембране и их активностью в различных эффекторных анализах (Weng and Levy 2009. Genetic polymorphism of the inhibitory IgG Fc receptor FcgammaRIIb is not associated with clinical outcome in patients with follicular lymphoma treated with rituximab. Leuk Lymphoma 50:723-727., Chan et al., 2003. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res 63:5480-5489 и Cragg and Glennie 2004. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 103:2738-2743). Анти-CD20 молекулы антител типа I (например, ритуксимаб, офатумумаб) индуцируют перераспределение CD20 в крупные устойчивые к детергентам микродомены (рафты), в то время как анти-CD20 моноклональные антитела типа II (тозитумумаб-подобные) этого не делают (Beer et al., 2010. Seminars in Haematology 47(2):ppl07-l 14).
Как обсуждается выше, анти-CD20 молекулы антител можно определить как принадлежащие типу I или типу II на основании их способности перераспределять CD20 в липидные рафты. Это делают при помощи анализа нерастворимости Tx-100 или сепарации в градиенте плотности сахарозы и вестерн-блоттинга. Оба способа описаны в Cragg et al Blood 2003 следующим образом:
1. Оценка рафт-ассоциированного антигена по нерастворимости Тритон X-100
В качестве быстрой оценки наличия антигена в микродоменах рафтов мы использовали метод проточной цитометрии на основании нерастворимости Тритон X-100 при низких температурах. Вкратце, клетки промывали в RPMI/1% БСА и пересуспендировали при 2,5 x 106/мл. Затем клетки инкубировали с 10 мкг/мл конъюгированного с FITC mAb в течение 15 минут при 37°C, промывали в холодном PBS/1% БСА/20 мМ азида натрия, а затем делили образец пополам. Одну половину выдерживали на льду для расчета 100% уровней поверхностного антигена, в то время как другую обрабатывали 0,5% Тритон X-100 в течение 15 минут, чтобы определить относительное содержание антигенов в нерастворимой рафтовой фракции. Затем клетки выдерживали при 4°C в течение оставшегося времени анализа, промывали один раз в PBS/БСА/азиде, пересуспендировали и оценивали методом проточной цитометрии, как указано выше. Аналогичные результаты получали, используя непрямые методы детекции. Чтобы определить конститутивный уровень ассоциации антигенов-мишеней с рафтами, клетки сначала обрабатывали 0,5 % Тритон X-100 в течение 15 минут на льду и промывали в PBS/БСА/азиде перед связыванием с FITC-меченым mAb. Чтобы определить, возможно ли перемещение большего количества антигена в нерастворимую фракцию Тритон X-100 посредством дополнительного перекрестного связывания, клетки инкубировали с FITC-mAb как и раньше, промывали, а затем делили на четыре части. Два из этих образцов инкубировали с F(ab')2-фрагментами козьего анти-мышиного Ig в течение 15 минут на льду. После промывания один из перекрестно-связанных и один из не-перекрестно-связанных образцов лизировали при помощи Тритон X-100 и промывали, как указано выше, перед проведением проточной цитометрии.
2. Сепарация в градиенте плотности сахарозы и вестерн-блоттинг - получение фракций липидных рафтов и вестерн-блоттинг
Моноклональное Ab (1 мкг/106 клеток) добавляли к клеткам при 37°C. После 20 минут инкубации клетки пеллетировали и лизировали в охлажденном на льду 1,0 % Тритон X-100 в забуференном МЭС солевом растворе (25 мM МЭС, pH 6,5, 150 мM NaCl, 1 мM фенилметилсульфонилфторида, 5 мкг/мл апротинина, 5 мкг/мл лейпептина, 10 мМ ЭДТК). Затем получали фракции липидных рафтов путем центрифугирования в градиенте плотности сахарозы. Вкратце, лизаты смешивали с эквивалентным объемом 80% сахарозы в лизисном буфере, покрывали прерывистым градиентом плотности 5-30% сахарозы, а затем центрифугировали при 200000 x g в течение 16 ч. Фракции (0,5 мл) собирали и анализировали методом вестерн-блоттинга. 15 мл аликвоты каждой фракции разводили 1:1 в 2x загрузочного буфера, нагревали до 95°C в течение 5 мин и проводили сепарацию в 15% ДСН-ПААГ геле перед переносом на ПВДФ-мембраны, и инкубировали с первичным антителом (например, мышиным анти-CD20 клоном 7D1 для выявления CD20 или анти-Lyn кроличьей полисывороткой; Serotec, UK, для выявления фракций рафтов), за которым следовало ПХ-конъюгированное вторичное антитело (Amersham Biosciences UK Ltd). Блоты визуализировали при помощи ЭХЛ+плюс (Amersham Biosciences UK Ltd).
Для молекул анти-CD20 антитела может требоваться мотив AxP в большой петле CD20 (которые не требуется для офатумумаба и других антител Генмаб). При этом в (Niederfellner, G et al. 2011. Blood 118, 358-367) указано, что молекулы антител типа II связываются с немного другой областью петли CD20 по сравнению с типом I.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом антитело к CD20 типа I представляет собой ритуксимаб или биоаналог ритуксимаба, или офатумумаб.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом антитело к CD20 типа II представляет собой обинутузумаб или тозитумомаб.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом молекула антитела, определение которой приведено в (i), представляет собой антитело к CD52.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом антитело к CD52 представляет собой алемтузумаб.
Предпочтительно в изобретении предложена композиция или набор, при этом композиция или набор содержит один или более терапевтический агент.
Предпочтительно в изобретении предложено применение, при этом композиция дополнительно содержит один или более терапевтический агент.
Предпочтительно в изобретении предложен способ, при этом субъекту дополнительно вводят один или более терапевтический агент.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом субъект имеет рефрактерный рак или рецидивный рак или субъект имеет рефрактерный рак и рецидивный рак.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, при этом субъект имеет рефрактерный хронический лимфоцитарный лейкоз или рецидивный хронический лимфоцитарный лейкоз или субъект имеет рефрактерный хронический лимфоцитарный лейкоз и рецидивный хронический лимфоцитарный лейкоз.
Предпочтительно в изобретении предложена композиция или применение, или способ, или набор, в значительной степени сходный с описанным и/или заявленным в данном документе на основании описания и/или примеров, и/или прилагающихся графических материалов.
Перечисление или обсуждение в тексте данной заявки очевидно ранее опубликованных документов не обязательно следует воспринимать как признание того, что данные документы составляют существующий уровень техники или представляют общедоступные сведения.
Далее описаны предпочтительные, неограничивающие примеры, которые являются вариантами реализации определенных аспектов изобретения, со ссылкой на следующие фигуры:
Фигура 1. Генерация и получение характеристик mAb, способных различать hFcγRIIB и hFcγRIIA. (A) Проводили скрининг клонов scFv в отношении специфического связывания с hFcγRIIB и/или hFcγRIIA. Желтые точки представляют клоны, специфические в отношении hFcγRIIB, которые были выбраны для преобразования в полноразмерный IgG. (B) Анализ связывания hFcγRIIB mAb. hFcγRIIB mAb оценивали в отношении связывания с hFcγRIIB-трансфицированными клетками (красная линия) или hFcγRIIA-трансфицированными клетками (голубая линия). То же mAb оценивали в отношении связывания с hFcγRIIB-трансфицированными клетками в присутствии ИК (3 нM, зеленая линия), что свидетельствует о том, что данное mAb способно конкурировать с ИК за связывание с hFcγRIIB-экспрессирующими клетками. На фигуре приведены данные для одного типового клона hFcγRIIB. (C) Профиль связывания полученного mAb на популяциях МКПК, определенный методом проточной цитометрии. Специфическое к hFcγRIIA mAb проявляло сильное связывание с моноцитами и нейтрофилами, в то время как специфические к hFcγRIIB mAb связывались главным образом с В-клетками, за исключением 6A09, которое также связывалось с моноцитами и нейтрофилами и, вероятно, обладает двойной специфичностью к hFcγRIIA и B. (D) Дозозависимое связывание mAb с В-клетками. mAb добавляли при 0,1, 1 или 10 мкг/мл, а интенсивность окрашивания определяли методом проточной цитометрии. (E) Аффинность hFcγRIIB mAb. Выборку специфических к hFcγRIIB mAb (7C07, 5C04, 5C05) оценивали в отношении их связывания с различными концентрациями слитого белка hFcγRIIB (0 (красный), 0,16 (зеленый), 0,8 (голубой), 4 (розовый), 20 (бирюзовый) и 100 (коричневый) нM) методом поверхностного плазмонного резонанса. Сенсограммы демонстрируют типичные ответы связывания для каждого mAb. Величины KД рассчитывали по 1:1 модели связывания (смотрите Таблицу 2).
Фигура 1(2/2). Терапевтическое действие hFcγRII mAb (AT10) и генерация специфических mAb, способных различать hFcγRIIB и hFcγRIIA. (Вверху) Мышей SCID (5/группу) с ксенографтными клетками Daudi (п.к.) обрабатывали (в.б.), как показано стрелками. На график наносили среднюю массу опухолей ± СОС и анализировали, используя парный t-критерий; p-величины сравнивали для групп, обработанных одним ритуксимабом (Rit) и Rit + AT10 (**p≤0,005). Репрезентативные данные (n = 2). (Внизу) Связывание hFcγRIIB mAb с hFcγRIIB- (красная линия) или hFcγRIIA-трансфицированными клетками (голубая линия) и mAb-зависимое ингибирование связывания ИК с hFcγRIIB-трансфицированными клетками (зеленая линия). Также смотрите Фигуры 7 и 8 и Таблицу 6.
Фигура 2. hFcγRIIB mAb способны блокировать взаимодействие ритуксимаба (Rit) с FcγRIIB на поверхности клеток-мишеней. (A) Способность специфических к hFcγRIIB mAb вызывать фосфорилирование hFcγRIIB ITIM. Клетки Raji обрабатывали специфическим к hFcγRIIB N297Q mAb (10 мкг/мл) при 37°C в течение 30 минут перед лизисом и оценивали методом иммуноблоттинга статус фосфорилирования hFcγRIIB (pFcγRIIB). Изотипический контроль hIgG1 (изо ктрл) и Rit использовали в качестве отрицательного и положительного контролей, соответственно. α-тубулин пробовали в качестве контроля нагрузки. (B) Способность специфических к hFcγRIIB mAb блокировать фосфорилирование hFcγRIIB ITIM, индуцированное Rit. Клетки Raji предварительно обрабатывали N297Q hFcγRIIB mAb (10 мкг/мл) в течение 10 минут перед добавлением 10 мкг/мл Rit при 37°C в течение 30 минут. Обработанные клетки лизировали и оценивали методом иммуноблоттинга в отношении pFcγγRIIB и α-тубулина, как описано выше. Представлены типовые блоты по меньшей мере по 3 экспериментам. (C) Способность специфических к hFcγRIIB mAb блокировать интернализацию, индуцированную Rit. Трансфицированные контрольным вектором или hFcγRIIB клетки Ramos обрабатывали ДТ или N297Q вариантами hFcγRIIB mAb (10 мкг/мл) и AF488-меченым Rit (5 мкг/мл) в указанные временные точки при 37°C. Интернализацию Rit определяли при помощи анализа блокирования и выражали в виде % поверхностной экспрессии CD20. AF488-меченый тозитумомаб использовали в качестве отрицательного контроля, так как он не демонстрирует существенной интернализации после задействования CD20.22 Представлено среднее ± СО по 3 независимым экспериментам. (D) Способность панели mAb блокировать интернализацию Rit (показана на C) коррелировала с их ранжированными аффинностями (R2 = 0,78). (E) Способность панели mAb блокировать интернализацию Rit (показана на C) коррелировала с их способностью блокировать Rit-индуцированное фосфорилирование hFcγRIIB (B) согласно данным, полученным при помощи программного обеспечения Image J densitometry (R2 = 0,79).
Фигура 3. hFcγRIIB mAb 6G11 обладает эффективной цитотоксической активностью in vitro и способно блокировать взаимодействие Rit с hFcγRIIB на поверхности клеток-мишеней ХЛЛ. (A) ИГХ-анализ hFcγRIIB mAb 7C07 и 6G11, проведенный для ряда замороженных тканей. 6G11 демонстрировало очень специфическое связывание с клетками в человеческой селезенке с практически отсутствующим фоновым окрашиванием нелимфоцитарных клеток и слабым или отсутствующим окрашиванием на ткани селезенки от яванского макака или мыши, в то время как 7C07 демонстрировало, как и ожидалось, связывание с лимфоцитами, но также с эндотелиальной выстилкой тканей как человека, так и яванского макака. (B-E) Цитотоксическая способность 6G11 в анализе АЗКЦ (B), ЗГК (C) и АЗКФ (D) на полученных от пациентов клетках первичного ХЛЛ. Клетки ХЛЛ опсонизировали 6G11 (10 мкг/мл); Rit (10 мкг/мл) добавляли в качестве положительного контроля в каждом анализе. Каждая точка представляет среднее по трем образцам от одного пациента с ХЛЛ. (E) Анализ АЗКЦ с применением эффекторных клеток, несущих высоко- или низкоаффинные аллельные варианты hFcγRIIIA (158F и V, соответственно). В обоих случаях 6G11 более эффективно индуцировало АЗКЦ по сравнению с Rit. На фигуре представлено среднее + СО по трем независимым экспериментам с тремя разными донорами ХЛЛ. (F) Способность 6G11 уменьшать интернализацию Rit с поверхности клеток. Шесть образцов ХЛЛ обрабатывали ДТ (левая панель) или N297Q (правая панель) mAb, изотипическим контролем или 6G11 (10-20 мкг/мл) и AF488-меченым Rit (5 мкг/мл) при 37°C до 6 часов. Интернализацию Rit оценивали, как описано выше, представляя % поверхностной экспрессии CD20. (G) Способность 6G11 оставаться на поверхности клеток ХЛЛ. Шесть образцов ХЛЛ обрабатывали ДТ или N297Q 6G11 (10 мкг/мл) до 6 часов и непрямым образом проводили количественную оценку поверхностной экспрессии hFcγRIIB путем окрашивания анти-hF(ab’)2-PE. Величины нормировали относительно окрашивания, проводимого на льду в момент времени 0, и выражали в виде % экспрессии поверхностного hFcγRIIB. Данные представлены в виде блочного графика с усами; p-величины представляют сравнение указанных групп (*p ≤ 0,05). (H и I) Усиление АЗКФ 6G11. CFSE-меченые клетки ХЛЛ опсонизировали Rit в комбинации с изотипическим контролем N297Q или 6G11 в течение 3 часов в культуре, промывали и добавляли к MDM в соотношении 5:1. После совместного культивирования для выявления MDM применяли окрашивание CD206-APC, а результаты анализировали методом проточной цитометрии. Типовые точечные графики показаны на (H), а % вдвойне положительных событий, рассчитанный по относительному количеству MDM, для которых наблюдали фагоцитоз CFSE+ve клеток ХЛЛ, показан на (I). (J) Усиление АЗКЦ 6G11. Клетки ХЛЛ опсонизировали Rit в комбинации с изотипическим контролем N297Q или 6G11 в течение 3 часов, а затем совместно культивировали с NK-клетками. Каждая точка представляет среднее по трем репликам одного образца первичного ХЛЛ. Данные анализировали, используя парный t-критерий (B, C, D, E, I и J); p-величины представляют сравнение указанных групп (*p ≤ 0,05, **p ≤ 0,005, ****p ≤ 0,0001).
Фигура 4. hFcγRIIB mAb 6G11 активно in vivo и стимулирует истощение В-клеток со стороны CD20 mAb. (A и B) Способность 6G11 истощать клетки-мишени hFcγRIIB+ve in vivo. Целевые hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6, меченые высокими или низкими уровнями CFSE, соответственно, были адоптивно перенесены (в.в.) в организм мышей C57BL/6 ДТ и γKO. Через 24 часа мыши получали указанные ДТ или N297Q 6G11 mAb (в.в.); а через 16 часов анализировали находящиеся в циркуляции (A) или в селезенке (B) клетки, чтобы определить соотношение оставшихся клеток-мишеней и не-мишеней. Данные нормировали, чтобы получить контрольное соотношение (NT) мишень:не-мишень 1,0. Каждая точка отображает результат для отдельной мыши, а средние соотношения обозначены горизонтальной линией (± СОС). Данные получены по меньшей мере по 2 независимым экспериментам. (C-E) Способность 6G11 усиливать способность Rit уничтожать клетки-мишени in vivo. (C) Целевые hCD20+/- x hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6, меченые высокими или низкими уровнями CFSE, соответственно, инъецировали (в.в.) мышам-реципиентам C57BL/6 ДТ, как указано выше. Через 24 часа мыши получали указанные mAb, одни или в комбинации (5-10 мкг; в.в.); а через 16 часов проводили анализ селезенок, чтобы определить соотношение мишеней и не-мишеней, и нормировали, как указано выше. Данные получены по меньшей мере по 3 независимым экспериментам. (D) Целевые CFSE+ve hCD20+/- x hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6 инъецировали (в.в.) мышам-реципиентам C57BL/6 hFcγRIIB+/- x mFcγRII-/-, как указано выше. На 1 и 2 день мыши получали ДТ 6G11 (500 мкг; в.в./в.б.), а после этого - Rit (5-50 мкг; в.в.) на 2 день; и через 16 часов проводили анализ селезенок, чтобы определить соотношение мишеней и не-мишеней, и нормировали, как указано выше. Данные получены по меньшей мере по 3 независимым экспериментам. (E) Мыши C57BL/6 hCD20+/- x hFcγRIIB+/- x mFcγRII-/- получали Rit или 6G11 (500 мкг) или 250 мкг каждого mAb в комбинации в.в. на день 0, а число циркулирующих В-клеток оценивали в течение времени, как указано. Столбики указывают средний ± СОС % циркулирующих В-клеток у 12 мышей/группу по меньшей мере по 2 независимым экспериментам, нормированный относительно уровней до обработки. Проводили статистический двухфакторный дисперсионный анализ ANOVA, чтобы сравнить группы обработки; p-величины представляют сравнение указанных групп (**p ≤ 0,01, ***p ≤ 0,001) (F-G) Способность 6G11 усиливать способность GA101gly уничтожать клетки-мишени in vivo. Целевые CFSE+ve hCD20+/- x hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6 инъецировали (в.в.) мышам-реципиентам ДТ и после этого обрабатывали на 1 день GA101gly или 6G11, одним или в комбинации (0,2 мкг; в.в.), и через 16 часов проводили анализ селезенок, чтобы определить соотношение мишеней и не-мишеней, и выражали, как подробно описано выше. Данные получены по 3 независимым экспериментам. (G) Целевые CFSE+ve hCD20+/- x hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6 инъецировали (в.в.) мышам-реципиентам hFcγRIIB+/- x mFcγRII-/- и после этого обрабатывали на 1 и 2 день изотипическим контролем дикого типа или 6G11 (500 мкг; в.б.), затем GA101gly (1 мкг; в.в.) на 2 день, и через 16 часов проводили анализ селезенок, чтобы определить соотношение мишеней и не-мишеней, и выражали, как подробно описано выше. Проводили статистический однофакторный дисперсионный анализ ANOVA, чтобы сравнить группы обработки с NT / изотипическим контролем или обработанные только CD20 mAb/6G11 группы (A-D и F-G); p-величины представляют сравнение указанных групп (*p ≤ 0,05, **p ≤ 0,01, ***p ≤ 0,001).
Фигура 4(2/2). hFcγRIIB mAb 6G11 активно in vivo и стимулирует истощение В-клеток со стороны CD20 mAb. Мыши hCD20+/- x hFcγRIIB+/- x mFcγRII-/- получали Rit ± ДТ или N297Q 6G11 (20 мг/кг) или 10 мг/кг каждого mAb в комбинации на день 0, а число циркулирующих В-клеток оценивали в течение времени. (Вверху) Типовые точечные графики, анализирующие циркулирующие В-клетки до обработки и на 2 день после инъекции mAb. Числа в верхних правых квадрантах указывают % В-клеток по сравнению с уровнями до обработки. (Внизу) Левый график отображает истощение циркулирующих В-клеток со стороны Rit ± ДТ 6G11; верхний правый график отображает истощение циркулирующих В-клеток со стороны Rit ± ДТ или N297Q 6G11; нижний правый график отображает истощение циркулирующих В-клеток со стороны Rit (m2a; 4 мг/кг) ± 6G11 (mIgG1; 20 мг/кг). Представлены средние величины + СО % циркулирующих В-клеток после обработки, нормированные относительно уровней до обработки; до 12 мышей/группу скомбинированы по меньшей мере по 2 независимым экспериментам. Проводили двухфакторный дисперсионный анализ ANOVA. Также смотрите Фигуру 15.
Фигура 5. hFcγRIIB mAb 6G11 стимулирует истощение клеток ХЛЛ пациентов in vivo со стороны Rit. (A-B) Полученные от пациентов клетки первичного ХЛЛ имплантируют мышам NOD/SCID, где они формируют отчетливые пролиферативные кластеры, подобные тем, которые наблюдаются у людей. Мышей облучали перед в.в. инокуляцией 6-10x107 клетками первичного ХЛЛ от пациентов. Через 4-5 дней после инъекции получали селезенки, готовили срезы и окрашивали в отношении наличия (A) hCD19 (красный) или Ki67 (зеленый) и оценивали методом иммунофлуоресценции или (B) hCD20 и Ki67 и оценивали методом ИГХ. (C) Противоопухолевая активность Rit, 6G11 или комбинации у мышей, которым инъецировали человеческие клетки ХЛЛ. Мышей NOD/SCID инокулировали человеческими клетками первичного ХЛЛ, как описано выше, через 4-5 дней после инокуляции мышей обрабатывали 1-10 мг/кг любого из hCD20 mAb, hFcγRII mAb или обоими. Мышам проводили вторую инъекцию через 2-3 и умерщвляли через 2-3 дня после последней обработки, подсчитывали % человеческих клеток ХЛЛ, оставшихся в селезенке, и нормировали относительно количества после обработки изотипическим контрольным mAb. Таким образом оценивали образцы ХЛЛ от 10 разных пациентов. Каждая точка представляет одну мышь. (D и E) Затем рассчитывали относительное количество особей, демонстрирующих полный ответ (D), т.е. мышей с отсутствием выявляемых клеток ХЛЛ в селезенке, и особей, демонстрирующих объективный ответ, т.е. мышей с ≥75% снижением числа клеток ХЛЛ (E), по данным, представленным выше на (C). (F-G) Ответ рефрактерных ХЛЛ-клеток пациентов in vivo. Клетки ХЛЛ от пациентов, ранее определенные как рефрактерные (n = 4; смотрите Таблицу 4), инокулировали, обрабатывали и оценивали как в (C-E). (F) Указывает первичные данные с точками, представляющими отдельных мышей, а (G) указывает частоту объективных ответов среди Rit-рефрактерных пациентов. Чтобы сравнить группы обработки, проводили однофакторный дисперсионный анализ ANOVA; p-величины представляют сравнение указанных групп (*p ≤ 0,05, **p ≤ 0,01, ***p ≤ 0,001).
Фигура 5(2/2). hFcγRIIB mAb 6G11 стимулирует истощение нормальных и злокачественных клеток-мишеней in vivo со стороны терапевтического mAb. (A-B) Селезенку мыши NOD/SCID с имплантированными полученными от пациента клетками первичного ХЛЛ оценивали методом иммунофлуоресценции (A) или методом ИГХ (B); контроль, отсутствие первичного mAb. (C) Мышей с ксенографтными человеческими клетками ХЛЛ (n = 11 пациентов) обрабатывали 1-10 мг/кг hCD20 mAb (Rit), hFcγRIIB mAb (6G11) или обоими, подсчитывали % оставшихся в селезенке клеток ХЛЛ и нормировали относительно количества после обработки изотипическим контролем. (D) Мышей с ксенографтными человеческими клетками ХЛЛ от пациентов, ранее определенными как рефрактерные (n = 4), обрабатывали и оценивали как в (C). (E) CFSE+ hCD20+/- x hFcγRIIB+/- x mFcγRII-/- (мишени) и mFcγRII-/- (не-мишени) спленоциты инъецировали (в.в.) мышам ДТ и обрабатывали GA101gly или 6G11, одним или в комбинации (0,008 мг/кг), и оценивали в отношении уничтожения в селезенке как ранее. Данные получены по 2-3 независимым экспериментам. (F) CFSE+ hCD20+/- x hFcγRIIB+/- x mFcγRII-/- (мишени) и mFcγRII-/- (не-мишени) спленоциты инъецировали hFcγRIIB+/- x mFcγRII-/- мышам-реципиентам и обрабатывали изотипическим контролем ДТ или 6G11 (20 мг/кг), затем GA101gly (0,04 мг/кг) и анализировали как в (E). (G) Мышей с привитыми клетками ХЛЛ (n = 4) обрабатывали GA101 (0, мг/кг), 6G11 (1 мг/кг) или обоими и оценивали как в (C). (H) Мышей с привитыми клетками ХЛЛ (n = 3) обрабатывали алемтузумабом (Alem; 1 мг/кг), 6G11 (1 мг/кг) или обоими и оценивали как в (C). (C-D и G-H) Секторные диаграммы представляют число мышей, несущих полученные от пациентов клетки первичного ХЛЛ NR (черный), OR (голубой) и CR (зеленый) после терапии mAb, как указано в Таблице 1. (C-H) Каждая точка представляет отдельную мышь, а средние соотношения указаны горизонтальной линией. (E и F) Данные анализировали при помощи однофакторного дисперсионного анализа ANOVA (C-D и G-H) и пермутационного статистического критерия. Также смотрите Фигуры 16 и 17 и Таблицы 8 и 4.
Фигура 6. hFcγRIIB mAb 6G11 хорошо переносится и не приводит к токсичности в доклинических in vivo и in vitro системах. (A-D) In vivo эффективность и время полужизни 6G11 после одного введения (Фиг. 17A). Мышей hFcγRIIB+/- x mFcγRII-/- одного возраста и пола (6-7 мышей/группу) инъецировали указанными концентрациями 6G11 mAb (в.в. или в.б.). (A) % циркулирующих В-клеток оценивали в течение времени вплоть до 7 дня методом проточной цитометрии. (B) На 7 день мышей умерщвляли и количественно оценивали число В-клеток в селезенке (выраженное как % лимфоцитов селезенки) методом проточной цитометрии. (C) Сывороточные концентрации 6G11 mAb и (D) титры MAHA после 7-дневного периода оценивали методом MSD (среднее + СОС), как описано в разделе Материалы и методы. (E-F) In vivo эффективность и время полужизни 6G11 после нескольких введений. Мышей hFcγRIIB Tg x mFcγRII-/- (6 мышей/группу) или мышей mFcγRII-/- (3 мышей/группу) одного возраста и пола инъецировали 10 мг/кг ДТ 6G11 mAb в.в. на день 0 с последующими в.б. инъекциями той же дозы на 3, 7 и 10 дни. Через 10 дней (24 день) мышей умерщвляли, а органы исследовали в отношении токсичности (Фиг. 17C). (E) В течение времени брали образцы крови и оценивали количество циркулирующих В-клеток методом проточной цитометрии (среднее + СО). (F) Титры MAHA против 6G11 mAb оценивали как в (D) (среднее + СОС). (G) In vitro ответ цитокинов с применением предварительно культивируемых человеческих МКПК высокой плотности. Человеческие МКПК предварительно культивировали при 1x107/мл в течение 48 часов перед добавлением PBS (NT) или 10 мкг/мл ДТ или N297Q вариантов изотипического контроля, или 6G11 в течение 48 часов. Собирали супернатанты и оценивали концентрации ИФН-γ, ИЛ-6, ИЛ-10 и ФНО-α методом MSD. CD3 mAb (OKT3) использовали в качестве положительного контроля при оптимальной и субоптимальной концентрациях (1 и 0,02 мкг/мл, соответственно). Данные представляют 3 независимых эксперимента с использованием МКПК от 3 независимых здоровых доноров (среднее ± СОС).
Фигура 6(2/2). hFcγRIIB mAb 6G11 хорошо переносится и не приводит к токсичности. In vitro анализ цельной крови для оценки эффективности Rit ± ДТ или N297Q 6G11 в истощении В-клеток крови hFcγRIIB+ (левый график), моноцитов (средний график) или нейтрофилов (правый график). Представлены средние величины (горизонтальные линии), при этом каждая точка представляет отдельного донора. Также смотрите Фигуры 14, 18 и 19 и Таблицу 7.
Фигура 7. hFcγRII mAb (AT10) стимулирует клиренс злокачественных В-клеток со стороны Rit in vivo. (A) 5x106 клеток Daudi или (B) Raji, смешанных в матригеле в соотношении 1:1, инъецировали (п.к.) в бок мышей SCID (до 5 мышей/группу). После этого несущих опухоль мышей инъецировали (в.б.) указанным mAb в еженедельном режиме, начиная с 7 дня, до 4 раз, как указано по оси Х (стрелкой). В течение времени отслеживали рост опухолей и оценивали, используя следующее уравнение: [масса = (длина × ширина2)/2]. На график наносили средний объем опухолей ± СО измерения. Представлены типовые данные по 2 независимым измерениям. (C) 2,5x106 клеток Raji инъецировали (в.в.) мышам SCID (6 мышей/группу). Как и выше, несущих опухоль мышей инъецировали (в.б.) указанным mAb в еженедельном режиме, начиная с 7 дня, до 4 раз. За мышами наблюдали в течение времени и умерщвляли после развития признаков терминальной стадии развития опухоли. Данные анализировали при помощи парного t-критерия; p-величины представляют сравнение указанных групп (*p ≤ 0,05, **p ≤ 0,005).
Фигура 8. Созданные клоны являются высокоспецифичными в отношении hFcγRIIB. (A) Выравнивание аминокислотной (аа) последовательности hFcγRIIA (вверху) по сравнению с высокогомологичным белком hFcγRIIB (внизу). Различия отмечены красным с указанием участка связывания IgG. (B-D) hFcγRIIB-специфические mAbs инкубировали с МКПК, а затем оценивали в отношении связывания с моноцитами CD14+ve (B), В-клетками CD19+ve (C) или Т-клетками CD3+ve (D) методом проточной цитометрии. Сильное и дозозависимое связывание с В-клетками CD19+ve в крови (C) наблюдали для всех клонов за исключением 2B08 и 2E08; при этом в случае связывания с моноцитами CD14+ve наблюдали обратную ситуацию (B). Клон 6A09 проявлял перекрестную реактивность в отношении как моноцитов, так и В-клеток. (D) Ни для одного из mAb не наблюдали окрашивание Т-клеток CD3+ve.
Фигура 9. Оба варианта hFcγRIIB mAb - ДТ и N297Q - одинаково способны блокировать интернализацию Rit с поверхности клеток-мишеней. (A-B) Способность ДТ и/или N297Q (NQ) hFcγRIIB-специфических mAb (клоны 6G11 и 6G08) вызывать фосфорилирование ITIM hFcγRIIB (pFcγRIIB) на выделенных человеческих В-клетках миндалевидной железы (A) и первичных моноцитах периферической крови (B), соответственно. α-тубулин, GAPDH и hFcγRIIB использовали в качестве контролей нагрузки, как указано; представлены типовые блоты. (C) hFcγRIIB-трансфицированные клетки Ramos обрабатывали ДТ или N297Q вариантами (10 мкг/мл) выбранных hFcγRIIB mAb (представляющих выборку антагонистов и агонистов) или изотипическими контролями (изо ктрл) и AF488-меченым Rit (Rit; 5 мкг/мл) в указанные временные точки при 37°C. Интернализацию Rit определяли при помощи анализа блокирования и выражали в виде процента поверхностной экспрессии CD20. AF488-меченый тозитумомаб использовали в качестве отрицательного контроля, так как он не демонстрирует существенной интернализации после задействования CD20.22 Представлено среднее ± СО по 3 независимым экспериментам.
Фигура 10. АЗКЦ-активность hFcγRIIB mAb in vitro. (A) Клетки Raji, предварительно опсонизированные hFcγRIIB mAb, совместно культивировали с NK-клетками, выделенными из периферической крови здоровых доноров, и оценивали АЗКЦ-активность, как описано в разделе Материалы и методы. На фигуре представлено среднее по 4 независимым экспериментам + СО. (B) На основании результатов A, семь hFcγRIIB-специфических mAb дополнительно исследовали в отношении их АЗКЦ-активности для ряда концентраций. Rit (Rit) был включен в качестве положительного контроля. Показатели всех hFcγRIIB mAb превосходили Rit, при этом наилучшие показатели среди mAb были у 7C07 и 6G11, даже при более низких концентрациях. Представлен один типовой эксперимент 2; точки на графике представляют среднее ± СО для образцов в трех повторах.
Фигура 11. ИГХ человеческих тканей, окрашенных ДТ или N297Q hFcγRIIB mAb. ДТ или N297Q варианты 7C07 или 6G11 добавляли к свежим замороженным срезам, полученным из различных тканей. Тканевую реактивность определяли при помощи амплификации Tyramide Signal Amplification (TSA; PerkinElmer) без блокирования перекисью водорода. (A) Окрашивание 7C07 человеческой селезенки. Было показано, что 7C07 неспецифически окрашивает синусоидные капилляры и кровеносные сосуды. (B) ДТ или N297Q варианты 6G11 добавляли к срезам человеческой селезенки и оценивали в отношении связывания, как подробно описано выше. Было показано, что в случае обоих форматов имеет место одинаковое окрашивание клеток лимфоцитов в этой ткани. (C) Окрашивание 6G11 (1 мкг/мл) после TSA-детекции на криоконсервированных поперечных срезах различных человеческих тканей, указанных на срезах. Не наблюдали никакой реактивности.
Фигура 12. Агликозилированный вариант N297Q 6G11 не обладает характерной Fc-зависимой эффекторной активностью и не может индуцировать АЗКЦ in vitro. Человеческие клетки первичного ХЛЛ опсонизировали ДТ или N297Q версиями 6G11 или изотипическим контрольным ДТ hIgG1 (10 мкг/мл), а затем совместно культивировали с клетками NK92 и оценивали АЗКЦ-активность, как описано в разделе Материалы и методы. В отличие от 6G11, N297Q 6G11 mAb не обладал характерной Fc-опосредованной эффекторной функцией, что иллюстрирует АЗКЦ-активность, сопоставимая с опсонизированными изотипическим контролем клетками-мишенями. Каждая точка представляет одного пациента с ХЛЛ; **** p ≤ 0,0001 по парному t-критерию Стьюдента.
Фигура 13. hFcγRIIB более устойчив к mAb-индуцированной интернализации, чем CD20 на человеческих клетках первичного ХЛЛ. Шесть образцов ХЛЛ обрабатывали 5 мкг/мл AF488-меченого Rit (серые столбики) или AF488-меченого ДТ 6G11 (белые столбики) при 37°C в течение 2 или 6 часов перед оценкой интернализации, как это делали ранее; представлен % экспрессии поверхностного CD20 или hFcγRIIB. (B) Шесть образцов ХЛЛ обрабатывали ДТ или N297Q AT10 (10 мкг/мл) в течение 1-6 часов, а количественную оценку поверхностной экспрессии hFcγRIIB проводили непрямым способом путем окрашивания анти-hF(ab’)2-PE. Значения нормировали относительно окрашивания, проводимого на льду во временную точку 0, и выражали в виде % экспрессии поверхностного hFcγRIIB. Данные представлены в виде блочного графика с усами. Для сравнения групп обработки использовали критерий Уилкоксона; p-величины представляют сравнение указанных групп (*p ≤ 0,05).
Фигура 14. Получение и характеристики мышей hFcγRIIB. (A) Полноразмерный hFcγRIIB2 амплифицировали из клеток Raji и лигировали с нативным промотором hFcγRIIB, выделенным их тех же клеток, при помощи ПЦР с перекрывающимися праймерами, чтобы получить указанную конструкцию. (B) Экспрессию hFcγRIIB Tg оценивали методом ПЦР-амплификации в положительных и отрицательных мышиных линиях. (C) Получали мышей, проводили возвратное скрещивание и фенотипирование циркулирующей крови путем окрашивания CD19-PE и hFcγRII (AT10)-FITC mAb и оценивали методом проточной цитометрии. (D) Активирующие и ингибирующие экспрессию рецепторы mFcγRII и/или hFcγRIIB исследовали на BMDM, полученных от указанных мышиных штаммов, используя специфические лабораторные mAb (Tutt et al, работа на стадии подготовки). Данные свидетельствуют об отсутствии компенсирующих изменений в профиле активирующего или ингибирующего mFcγR в присутствии человеческого трансгена. (E) Экспрессию mFcγRII или hFcγRIIB оценивали на нейтрофилах CD19-ve CD11b+ve NK1.1-ve Ly6G+ve из селезенок указанных мышей ДТ или hFcγRIIB+/- x mFcγRII-/-, соответственно. Приведены типовые точечные графики по 3 независимым экспериментам. Как и ожидалось, mFcγRII, но не hFcγRIIB, экспрессируется на нейтрофилах. (F) Замороженные срезы из селезенок мышей ДТ или hFcγRIIB+/- x mFcγRII-/- анализировали при помощи иммунофлуоресценции, а экспрессию hFcγRIIB Tg оценивали и сравнивали с эндогенным рецептором mFcγRII, используя указанные маркеры (AT130-2 и AT10 mAb, соответственно; красный); В-клетки (B220; верхняя панель (зеленый)) и макрофаги (F4/80; нижняя панель (зеленый)).
Фигура 14(4/5). Получение и характеристики мышей hFcγRIIB Tg и оценка ФК, ФД, MABEL и CRS-индуцирующих свойств 6G11 mAb. Связана с Фигурой 14. (D) Стратегия гейтинга для иммунофенотипирования подгрупп мышиных лейкоцитов (Rose et al, 2012). (E) Экспрессию mFcγRII или hFcγRIIB оценивали для циркулирующих (кровяных) В-клеток и В-клеток селезенки, моноцитов/макрофагов и нейтрофилов указанных мышей ДТ или hFcγRIIB+/- x mFcγRII-/- и человеческой крови, соответственно. Приведены типовые точечные графики по 3 независимым экспериментам.
Фигура 14(5/5). Получение и характеристики мышей hFcγRIIB Tg и оценка ФК, ФД, MABEL и CRS-индуцирующих свойств 6G11 mAb. Связана с Фигурой 14. (F) Приведена оценка экспрессии hFcγRIIB на находящихся в циркуляции (крови) и селезенке подгруппах лейкоцитов мышей hFcγRIIB+/- x mFcγRII-/- и на здоровых человеческих лейкоцитах, а также клетках пациентов с ХЛЛ и линиях В-клеток. Приведено среднее ± СО; каждая точка представляет одного донора/один образец. (Средняя панель). Замороженные срезы селезенок мышей ДТ или hFcγRIIB+/- x mFcγRII-/- анализировали при помощи иммунофлуоресценции, а экспрессию hFcγRIIB Tg оценивали и сравнивали с эндогенным mFcγRII, используя указанные маркеры (AT130-2 и AT10 mAb, соответственно; красный); В-клетки (B220; верхняя панель (зеленый)) и макрофаги (F4/80; нижняя панель (зеленый)). (Нижняя панель) Оценка экспрессии hFcγRIIB на эндотелиальных клетках CD31+ в печени мышей hFcγRIIB+/- x mFcγRII-/-.
Фигура 15. hFcγRII mAb AT10 активно in vivo и стимулирует истощение В-клеток со стороны Rit. (A) Целевые hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6, меченые высокими или низкими уровнями CFSE, соответственно, адоптивно переносили (в.в.) в организм мышей-реципиентов C57BL/6 ДТ или γKO. Через 24 часа мыши получали указанные ДТ или F(ab)2-фрагменты hFcγRII mAb AT10 (в.в.; mIgG1); а через 16 часов клетки селезенки анализировали, чтобы определить оставшееся соотношение мишеней и не-мишеней. Данные нормировали, чтобы получить контрольное соотношение (NT) мишень:не-мишень 1,0. Каждая точка отображает результат для отдельной мыши, а средние соотношения указаны горизонтальной линией. Данные получены для 4-6 мышей/группу по меньшей мере по 2 независимым экспериментам. (B) Целевые hCD20+/- x hFcγRIIB+/- x mFcγRII-/- и нецелевые mFcγRII-/- спленоциты C57BL/6, меченые высокими или низкими уровнями CFSE, соответственно, инъецировали (в.в.) мышам-реципиентам C57BL/6 ДТ. Через 24 часа мыши получали указанные mAb, одни или в комбинации (10 мкг; в.в.); а через 16 часов клетки селезенки анализировали, чтобы определить соотношение мишеней и не-мишеней, как указано выше. Данные получены по меньшей мере по 3 независимым экспериментам. (C и D) Системное истощение В-клеток посредством Rit и AT10, одних или в комбинации. Мыши hCD20+/- x hFcγRIIB+/- x mFcγRII-/- C57BL/6 получали 500 мкг Rit (Rit, hIgG1), AT10 (mIgG1) или 250 мкг каждого mAb в комбинации (в.в.) на день 0, а число циркулирующих В-клеток оценивали в течение времени методом проточной цитометрии. (C) Типовые точечные графики по данным проточной цитометрии с анализом циркулирующих В-клеток перед обработкой и на 2 день после инъекции mAb. (D) Столбики указывают средние величины ± СОС циркулирующих В-клеток в указанные дни после инъекции mAb; ≥ 6 мышей/группу в двух независимых экспериментах, нормирование относительно числа циркулирующих В-клеток до обработки (выраженное в виде % циркулирующих В-клеток). Проводили статистический однофакторный (B) и двухфакторный дисперсионный анализ ANOVA (D), чтобы сравнить группы обработки; p-величины представляют сравнение указанных групп (*p ≤ 0,05, **p ≤ 0,01 и ***p ≤ 0,001).
Фигура 16. Клетки ХЛЛ защищены от Rit-зависимого уничтожения взаимодействием со стромальными клетками.
(A) 6-10x107 полученных от пациентов клеток первичного ХЛЛ одновременно в.в. и в.б. переносили в организм мышей NOD/SCID. Через 4-5 дней после инъекции мышей обрабатывали 1 или 2 дозами Rit (Rit) или изотипическим контролем при 10 мг/кг. Через 2 дня мышей умерщвляли и оценивали число оставшихся в селезенке и брюшной полости клеток ХЛЛ методом проточной цитометрии перед нормированием относительно образцов, обработанных изотипическим контролем. Данные показывают, что происходило эффективное уничтожение клеток ХЛЛ в брюшной полости (заштрихованные столбики) после одной дозы mAb, в то время как Rit-индуцированное уничтожение клеток в селезенке (залитые цветом столбики) было неполным даже после двух доз Rit. На фигуре представлена средняя величина для 3-5 мышей/группу + СОС. Анализ данных проводили при помощи t-критерия; p-величины представляют сравнение указанных групп (**p ≤ 0,01, ****p ≤ 0,001).
(B) Противоопухолевая активность Rit, 6G11 или комбинации у мышей с ксенографтами человеческих клеток ХЛЛ. Мышей обрабатывали 1-10 мг/кг hCD20 mAb (Rit), hFcγRIIB mAb (6G11) или обоими, подсчитывали % клеток ХЛЛ, оставшихся в селезенке, и нормировали относительно числа клеток после обработки изотипическим контролем. Приведены средние величины для каждого независимого эксперимента, при этом каждый пациент обозначен отдельным цветом (n = 11 пациентов).
(C) Клетки ХЛЛ от пациентов, ранее определенные как рефрактерные (n = 4; смотрите Таблицу S4), трансплантировали, обрабатывали и оценивали как в (B). Данные анализировали, используя парный однофакторный дисперсионный анализ ANOVA.
Фигура 17. hFcγRIIB mAb 6G11 стимулирует истощение клеток мантийноклеточной лимфомы (МКЛ) со стороны Rit in vivo. (A) Мышей NOD/SCID инокулировали 10x106 клеток МКЛ Jeko-1 MCL и обрабатывали, когда опухоли достигали 4x4 мм, комбинацией 6G11, Rit (Rit) или комбинацией обоих. За мышами наблюдали в течение времени и умерщвляли после развития признаков терминальной стадии развития опухоли. Данные анализировали, используя непарный t-критерий (*p ≤ 0,05 и *** p ≤ 0,001). (B) Мышей NOD/SCID облучали, а затем инокулировали 6-7x106 человеческих клеток первичной МКЛ. Через 4-5 дней после инокуляции мышей обрабатывали 1мг/кг Rit (Rit), 6G11 или обоих. Через 2-3 дня мыши получали вторую инъекцию, а через 2-3 дня их умерщвляли и подсчитывали % оставшихся в селезенке человеческих клеток МКЛ. Данные четко демонстрируют способность обоих mAb и, в частности, комбинации уничтожать человеческие клетки первичной МКЛ. Для сравнения групп обработки проводили однофакторный дисперсионный анализ ANOVA; p-величины представляют сравнение указанных групп (*p ≤ 0,05, *** p ≤ 0,001).
(C и D) Способность 6G11 усиливать способность hCD20 mAb GA101gly типа II уничтожать клетки-мишени in vivo. (C) Спленоциты CFSE+ hCD20+/- x hFcγRIIB+/- x mFcγRII-/- (мишени) и mFcγRII-/- (не-мишени) адоптивно переносили (в.в.) в организм мышей ДТ и обрабатывали GA101gly или 6G11, одним или в комбинации (0,008 мг/кг), и проводили анализ крови, как ранее. Данные получены по 2-3 независимым экспериментам. (D) Спленоциты CFSE+ hCD20+/- x hFcγRIIB+/- x mFcγRII-/- (мишени) и mFcγRII-/- (не-мишени) адоптивно переносили (в.в.) в организм мышей-реципиентов hFcγRIIB+/- x mFcγRII-/- и обрабатывали изотипическим контролем или 6G11 (20 мг/кг), затем GA101gly (0,04 мг/кг) и проводили анализ крови, как ранее. Каждая точка отображает результат для отдельной мыши, а средние соотношения обозначены горизонтальными линиями. (B, C, F и G) Данные анализировали, используя однофакторный дисперсионный анализ ANOVA (*p ≤ 0,05, **p ≤ 0,01, ***p ≤ 0,001 и ****p ≤ 0,0001).
Фигура 18. Оценка ФК, ФД и MABEL-свойств 6G11 in vivo. (A и B) Мышей hFcγRIIB+/- x mFcγRII-/- (6-7 мышей/группу) одного возраста и пола инъецировали 1-100 мг/кг ДТ 6G11 mAb (в.в. или в.б.) и получали образцы, как указано на схематической диаграмме (A). На 7 день эксперимента мышей умерщвляли, а органы оценивали в отношении признаков токсичности. (B) Массу мышей оценивали в течение времени вплоть до 7 дня (168 часов) после инъекции mAb (нормировали относительно 100% массы на день 0, а затем выражали как среднее + СОС для каждой группы). (C и D) Мышей hFcγRIIB Tg x mFcγRII-/- (6 мышей/группу) или мышей mFcγRII-/- (3 мыши/группу) одного возраста и пола инъецировали 10 мг/кг ДТ 6G11 (в.в.) на день 0, затем проводили в.б. инъекцию таких же доз mAb на 3, 7 и 10 день, как указано на схематической диаграмме (C). На 24 день мышей умерщвляли, а органы оценивали в отношении признаков токсичности. (D) Массу мышей измеряли в течение эксперимента и оценивали, как детально описано в (B).
(Нижняя панель) Оценка уничтожения циркулирующих моноцитов и нейтрофилов со стороны ДТ 6G11 (20 мг/кг) у мышей hFcγRIIB+/- x mFcγRII-/- (4 мышей/группу; среднее + СО).
Фигура 19. In vitro анализ высвобождения цитокинов для цельной крови и разведенной крови (20% об/об). (A) Свежую или (B) 5x разведенную (в бессывороточной среде CTL) гепаринизированную кровь от 3 здоровых волонтеров обрабатывали 10 мкг/мл указанного mAb в течение 24 часов при 37°C перед сбором супернатантов для последующего анализа. Количественную оценку цитокинов (ИЛ-1β, ИЛ-2, ИЛ-6, ИЛ-8, ФНО-α и ИФН-γ) проводили методом MSD. Каждая точка обозначает отдельного донора (n = 3).
ПРИМЕРЫ
Пример 1: Экспериментальные данные
Краткое изложение
Терапевтические антитела трансформировали противораковую терапию, открыв новые механизмы действия с привлечением иммунной системы. К сожалению, полное излечение случается редко, а пациенты демонстрируют врожденную или приобретенную устойчивость. В данном документе мы продемонстрировали терапевтический потенциал таргетирования и блокирования человеческого (h) FcγRIIB - рецептора, связанного со снижением чувствительности иммунных клеток и устойчивостью опухолевых клеток к препаратам антител. FcγRIIB-блокирующие антитела предотвращали интернализацию CD20-специфического антитела ритуксимаба, тем самым максимизируя доступность клеточной поверхности и опосредованную иммунными эффекторными клетками противоопухолевую активность in vitro и in vivo. В полностью сингенных мышиных моделях hFcγRIIB Tg hFcγRIIB mAb стимулирует уничтожение В-клеток ритуксимабом. В мышиной модели, оценивающей уничтожение человеческих опухолевых клеток хронического лимфоцитарного лейкоза (ХЛЛ) из предрасположенных к устойчивости стромальных компартментов, совместное введение с ритуксимабом улучшало объективные и полные ответы, включая эксперименты с клетками ХЛЛ от рецидивных/рефрактерных пациентов. Было показано, что основной кандидат - hFcγRIIB-специфическое антитело 6G11 - обладает хорошей иммунореактивностью в отношении мишени, благоприятной фармакокинетикой и не имеет нежелательных эффектов in vivo в случае кумулятивной/синергетической активности в комбинации с ритуксимабом. Эти данные являются основой для дальнейшей клинической разработки hFcγRIIB-specific mAb для иммунотерапии В-клеточных лимфопролиферативных нарушений.
Введение
Биологическая терапия, в общем, и моноклональные антитела (mAb), в частности, представляют постоянно расширяющийся класс терапевтических средств.1 Они совершили революцию в противораковой терапии и стали стандартом лечения наравне с традиционной химиотерапией для нескольких видов злокачественных опухолей. На протяжении более чем десятилетия мы знали, что активность терапевтических mAb регулируется их взаимодействием с Fc-гамма-рецепторами (FcγR). В частности, с относительным уровнем экспрессии, аффинностью и активностью FcγR в значительной степени связана терапевтическая активность IgG (обзор приведен в 2,3). Намного меньше известно о механизмах, лежащих в основе первичной или приобретенной устойчивости к препаратам антител. Хотя было показано, что сигналы типа «не-ешь-меня», такие как CD47, ограничивают опосредованную антителом противораковую активность эффекторных клеток4, подтверждение их клинической значимости все еще находится на ранней стадии. Учитывая возрастающее число видов терапии на основе антител, разрабатываемых для лечения нескольких типов рака, возникает необходимость понимания устойчивости раковых клеток к этим видам терапии и разработке лекарственных препаратов для ее преодоления.
Так как несколько противораковых mAb зависят от включения антитело-зависимых опосредованных иммунными клетками противоопухолевых механизмов в случае доклинической5-8 и клинической эффективности9-11, существует конкретная необходимость в понимании и предотвращении механизмов устойчивости к этим общим связанным с антителами эффекторным механизмам.
Специфический к человеческому (h) CD20 mAb ритуксимаб был первым средством, утвержденным для иммунотерапии рака, вследствие чего его широко применяли в случае пациентов с CD20-экспрессирующими В-клеточными раками, включая фолликулярную лимфому (ФЛ), диффузную крупноклеточную В-клеточную лимфому (ДКВКЛ), хронический лимфоцитарный лейкоз (ХЛЛ) и мантийноклеточную лимфому (МКЛ) (обзор приведен в 12). Интересно, что хотя ритуксимаб эффективен при ФЛ и ДКВКЛ, где он улучшает общую выживаемость, в случае ХЛЛ и МКЛ наблюдали только умеренную восприимчивость. Кроме того, даже в случае восприимчивых к ритуксимабу лимфом, некоторые индивиды демонстрируют устойчивость при первом лечении или приобретают устойчивость к включающей ритуксимаб комбинированной терапии, делая его идеальной системой для изучения механизмов устойчивости к mAb.
Недавние исследования продемонстрировали, что ингибиторный Fc-гамма-рецептор IIB (FcγRIIB/CD32B) способствует интернализации ритуксимаба с В-клеток-мишеней 32, 33. Как основной IgG-связывающий иммунный рецептор, экспрессируемый на В-клетках, FcγRIIB «всасывает» ритуксимаб с поверхности В-клетки, эффективно подавляя все mAb-зависимые противораковые механизмы иммунных клеток (13 и находящаяся на стадии подготовки работа). В противоположность этому, так называемые анти-CD20 mAb типа II, такие как недавно утвержденный обинутузумаб (GA101), не настолько чувствительны к этому процессу, возможно, вследствие своей неспособности перераспределять CD20 в липидных рафтах (Cragg et al., 2003; Lim et al., 2011). FcγRIIB-опосредованная интернализация ритуксимаба коррелирует с клинической восприимчивостью в следующем порядке ХЛЛ > МКЛ > ФЛ и ДКВКЛ 32, 33. В контексте роли FcγRIIB в устойчивости к Ab ретроспективный анализ пациентов с МКЛ, которых лечили при помощи иммунохимиотерапии ритуксимабом, продемонстрировал большую выживаемость среди пациентов с FcγRIIB-отрицательными по сравнению с FcγRIIB-положительными результатами опухолевой биопсии.13 Сходную слабую восприимчивость наблюдали у пациентов с ФЛ, экспрессирующих высокие уровни FcγRIIB, которые получали монотерапию ритуксимабом (14 и поданная работа).
В следующем Примере описана разработка антител к hFcγRIIB, способных блокировать интернализацию ритуксимаба, и исследован их терапевтический потенциал in vitro и in vivo. Получали трансгенных (Tg) мышей, коэкспрессирующих hCD20 и hFcγRIIB специфическим в отношении типа человеческих клеток и тканей образом, которых использовали в исследованиях для экспериментальной проверки концепции и для оценки ФК/ФД и токсикологических параметров. Затем действие hFcγRIIB mAb, используемых одних или в комбинации с ритуксимабом или другими CD20 mAb, в предотвращении или преодолении устойчивости оценивали на этих и других уникальных мышиных моделях, в которых человеческие клетки ХЛЛ (сохраняющие фенотип пациентов, т.е. ритуксимаб-восприимчивые или рецидивные/рефрактерные) можно исследовать in vivo в соответствующих предрасположенных к устойчивости тканевых компартментах, содержащих человеческие стромальные клетки.
Материалы и методы
Животные и клетки
Мыши с человеческим (h) CD20 Tg, γ-цепью-/- и мышиным (m) FcγRIIB-/- были описаны ранее (Beers et al., 2008), а их генотип подтвержден методом ПЦР и/или проточной цитометрии. Мышей выращивали и содержали в местных вивариях в соответствии с министерским руководством. Эксперименты с животными были согласованы с местным этическим комитетом и проводились по министерским лицензиям PPL30/1269 и M90-11. Мыши с человеческим (h) CD20 Tg, γ-цепью-/- и FcγRIIB-/- были описаны ранее15, а их генотип подтвержден методом ПЦР и/или проточной цитометрии. Самок мышей BALB/c и C57BL/6 возрастом от десяти до двенадцати недель держали на корме Harlan UK Limited (Blackthorn, Oxon, UK) и содержали в местных вивариях. Мышей CB-17 SCID приобретали у Charles River, а затем выращивали и содержали в местных вивариях. В случае ксенографтных исследований с первичными опухолевыми клетками (смотрите ниже) 6-8-недельных самок мышей NOD SCID держали на корме Taconic (Bomholt, Denmark) и содержали в местных вивариях.
Клинические образцы
Одобрение этического комитета для применения клинических образцов было получено Трастом НСЗ университетских госпиталей Саутгемптона от этического комитета по научным исследованиям Саутгемптона и юго-западного Гэмпшира или этическим комитетом университетского госпиталя Сконе. Информированное согласие было предоставлено в соответствии с Хельсинской Декларацией. Образцы получали в Университете Саутгемптона по лицензии Ассоциации по вопросам человеческих тканей, раковом отделении банка тканей или приобретали через отдел гематологии и отдел онкологии университетского госпиталя Сконе, Лунд. Оценку образцов ХЛЛ и МКЛ проводили в виде одноклеточных суспензий, которые выделяли, очищали в градиенте фиколла и проводили криоконсервацию для последующего анализа или использовали свежими в ксенографтных исследованиях.
Культивирование клеток
Культивирование клеток проводили в дополненной среде RPMI (RPMI, содержащей 2 мM глутамина, 1 мМ пирувата, 100 МЕ/мл пенициллина и стрептомицина и 10 % ФТС [Myoclone]) (GIBCO BRL, Paisley, Scotland). В-клетки из мышиной селезенки очищали методом негативной селекции, используя наборы для выделения В-клеток MACS (Miltenyi Biotec, UK), и культивировали в той же самой среде. Клеточные линии получали от ECACC и поддерживали в не содержащей антибиотики дополненной среде RPMI. Нормальные человеческие периферические В-клетки очищали методом негативной селекции, используя наборы для выделения В-клеток MACS (Miltenyi Biotec, UK).
Получение моноцитарных макрофагов (MDM) и макрофагов костного мозга (BMDM)
Человеческие MDM были дифференцированы из человеческой крови, полученной от Национальной службы хранения крови, общий госпиталь Саутгемптона (Саутгемптон, ВБ), или от центров крови в госпитале Хальмстада или университетского госпиталя Сконе (Швеция). Вкратце, адгезивные моноциты CD14+ культивировали в RPMI, содержащей пенициллин (100 Е/мл), стрептомицин (100 мкг/мл), 10% ФТС и 25-100 нг/мл рекомбинантного человеческого макрофагального колониестимулирующего фактора с низким содержанием эндотоксинов (M-CSF; R&D Systems, US или полученного в лаборатории), как было описано ранее.16 Половину среды заменяли свежей M-CSF каждые 2 дня до сбора. На 7-10 день культивирования собирали MDM после короткой инкубации с холодным PBS.
BMDM получали из клеток, выделенных из костного мозга бедренной и большеберцовой кости мышей, как сообщалось ранее.17 Вкратце, клетки костного мозга культивировали в RPMI 1640 (Life Technologies Invitrogen, Paisley, U.K.), обогащенной 10% ФТС, 2 мМ глутамина и 1 мМ пирувата, пенициллином и стрептомицином (каждый в концентрации 100 мкг/мл), и 20% кондиционированной клетками L929 среде (содержащей M-CSF). Клетки культивировали при 37°C, 5% CO2 в течение 10-14 дней перед применением. Дифференцировку макрофагов подтверждали стандартным морфологическим исследованием и/или проточной цитометрией в отношении экспрессии CD11b и F4/80.
Антитела и реагенты
mAb обычно получали из культурального супернатанта гибридомы или стабильно трансфицированных клеток CHO-k1. IgG очищали на протеине А, степень очистки оценивали при помощи электрофореза (Beckman EP system; Beckman), а отсутствие агрегации подтверждали ВЭЖХ. Фрагменты F(ab’)2 получали, как было описано ранее.18 hFcγRII mAb AT10 было описано ранее.19 Ритуксимаб был подарен онкологической фармацией общего госпиталя Саутгемптона или приобретен в фармации университетского госпиталя в Лунде, Швеция. Антитела против фосфорилированного hFcγRIIB (клон EP926Y) (Origene, US) и α-тубулина (клеточная сигнализация, US) использовали для иммуноблоттинга. AF647-меченый IgG1, анти-CD3-PE, анти-CD19-perCp-Cy5.5 и анти-CD56-AF488 (BD Biosciences) использовали для мечения МКПК. Для иммунофенотипирования МКПК использовали FcγRIIB mAb, меченый PE при помощи набора для мечения zenon (Molecular Probes), в сочетании с анти-CD3-FITC, анти-CD19-PerCP-Cy5.5 и анти-CD56-APC.
Проточная цитометрия
Флуоресцентно конъюгированные mAb приобретали у BD Biosciences, eBiosciences, AbD Serotec или получали в лаборатории. Проточную цитометрию проводили так, как описано ранее20, с оценкой образцов при помощи FACScan, FACSCalibur или FACSCanto II и анализом данных при помощи CellQuest Pro или FACSDiva (все от BD Biosciences).
Получение hFcγRIIB mAb
hFcγRIIB mAb выявляли, проводя скрининг библиотеки фагового дисплея n-CoDeR®scFv. Внеклеточный домен hFcγRIIB и hFcγRIIA сливали с mIgG3-Fc (hFcγRIIA/B-Fc), чтобы использовать в качестве мишеней и не-мишеней, соответственно, и получали во временно трансфицированных клетках HEK293 с последующей очисткой на протеине A. Проводили три последовательных этапа отбора. Перед 1-м отбором проводили предварительный отбор против покрытого мышиного IgG3κ и биотинилированного hFcγRIIA-Fc, нагруженных на стрептавидиновые частицы Dynabeads. Связывающие фаги элюировали путем расщепления трипсином и амплифицировали на планшетах, используя стандартные процедуры.21 Амплифицированные фаги с этапа отбора 1 использовали для отбора против hFcγRIIB, покрывающего протравленные полистироловые сферы (Polysciences, US). Предварительный отбор для этапа отбора 2 проводили против избыточного покрытого стрептавидина. Этап отбора 3 проводили методом предельного разведения, например, используя биотинилированный hFcγRIIB в разных концентрациях. hFcγRIIA использовали в качестве конкурирующего элемента на всех этапах отбора. Фагмиды с этапа отбора 3 конвертировали в формат для получения scFv и использовали при проведении последующего скрининга, когда оценивали специфическое связывание с растворимыми или связанными с клетками антигенами, а также ингибирование связывания иммунного комплекса. Для оценки рекомбинантного и связанного с клеточной поверхностью FcγRIIB использовали три разных коммерческих антитела против hFcγRII (MCA1075XZ, AbD Serotec; MAB1330, R&D Systems; AF1330, R&D Systems). Для оценки связанного с клеточной поверхностью FcγRIIB при помощи проточной цитометрии и флуоресцентной микроматричной технологии (FMAT) также использовали мышиное анти-hFcγRII-APC (BD Pharmingen). Во все эксперименты в качестве отрицательных контролей были включены соответствующие изотипические контроли. Для оценки связанных с клетками антигенов клетки CHO-k1 котрансфицировали FcγRIIA-pIRO или FcγRIIB-pIRO вместе с pIRESpuro, используя FuGENE (Roche). Для отбора трансфицированных клеток использовали пиромицин (InvivoGen) в концентрации 10 мкг/мл. Затем отдельные клоны, полученные методом предельного разведения, окрашивали антителом hFcγRII (BD Biosciences) и соответствующим изотипическим контролем, после чего проводили анализ методом проточной цитометрии и FMAT, чтобы отобрать клоны с наибольшей экспрессией, которые затем использовались в дальнейших экспериментах. Для первичного скрининга scFv-, FcγRIIA- и FcγRIIB-трансфицированные клетки CHO-k1 высевали в планшеты FMAT. Добавляли экспрессируемые E. coli scFv, после чего добавляли дегликозилированное мышиное анти-HIS антитело (R&D Systems) и дегликозилированный анти-мышиный-Cy5 (GE Healthcare). Выявление окрашенных клеток проводили при помощи системы для выявления 8200 (Applied Biosystems). Положительные клоны первичного скрининга повторно экспрессировали и еще раз исследовали в отношении связывания с клетками, трансфицированными FcγRIIA и FcγRIIB. Клоны ScFv, которые специфически связывали FcγRIIA или FcγRIIB и ингибировали связывание ИК, секвенировали в отношении CDR H1, H2 и H3, используя стандартные процедуры для выявления уникальных клонов. Уникальные клоны очищали на центрифужных колонках Ni-NTA (GE Healthcare) после периплазматического получения с лизозимом (Sigma) из 10 мл экспрессионного препарата в E.coli. В ДТ и N297Q hIgG1 варианты было преобразовано всего 17 клонов. VH и VL амплифицировали при помощи ПЦР и вставляли в экспрессионные векторы, содержащие константные области тяжелой и легкой цепи антитела, соответственно. После этого клетки HEK 293EBNA (Life Technologies), адаптированные для роста суспензии, трансфицировали, используя PEI, а затем проводили инкубацию клеточной суспензии с перемешиванием при 37°C перед разведением питательным раствором (UltraPepSoy, Sheffield Bio-Science) и сбором через 6 дней после трансфекции. Собранную культуральную среду стерильно фильтровали и применяли к колонке, упакованной MabSelect (GE Healthcare), соединенной с системой ÄKTA Purifier. Колонку промывали загрузочным буфером и элюировали буфером с низким рН. Элюированное антитело стерильно фильтровали и диализовали до получения приемлемого препаратного буфера, используя мембрану для диализа Spectra/Por 4 (Spectrum Laboratories Inc). После диализа материал стерильно фильтровали и хранили при 4°C. Затем очищенный IgG оценивали в отношении связывания с трансфицированными клетками CHO-k1, а также клеточными линиями и МКПК, экспрессирующими FcγRIIB естественным образом.
Блокирование связывания иммунного комплекса (ИК)
ИК формировались при смешивании hIgG1, специфического в отношении FITC, с FITC-БСА-биотином или FITC-БСА в молярном соотношении 10:1. Перед применением смесь предварительно инкубировали в течение 1 часа при комнатной температуре. Супернатанты (10 мкл) экспрессируемых E. coli клонов ScFv, которые специфически связывали FcγRIIA и FcγRIIB, добавляли к экспрессирующим FcγRIIA и FcγRIIB клеткам CHO-k1 и оставляли для связывания на 1 час. ИК добавляли при 3 нМ. Связанные ИК выявляли при помощи Strep Alexa Fluor (AF)-647 (Life Technologies, UK) с последующей проточной цитометрией, а блокирование количественно оценивали по исчезновению сигнала AF674.
Антитело-зависимые клеточный фагоцитоз (АЗКФ) и клеточная цитотоксичность (АЗКЦ)
Анализ фагоцитоза (АЗКФ) проводили в основном, как подробно было описано ранее.23 После созревания в макрофаги клетки собирали, используя охлажденный PBS или аккутазу (Sigma) или трипсин/ЭДТК (Life Technologies, UK), и повторно высевали при 5x104 клеток/лунку в 96-луночные планшеты и инкубировали в течение 2-4 часов или в течение ночи при 37°C. После этого клетки ХЛЛ метили 5 мкМ сукцинимидилового сложного эфира карбоксифлуоресцеина (CFSE, Molecular Probes) в течение 15 минут при 37 °C. После промывания клетки ХЛЛ инкубировали с опсонизированными mAb при 10 мкг/мл в течение 30 минут, а после этого добавляли к культурам макрофагов в соотношении 5:1. Через 1 час клетки собирали путем соскребания и окрашивали APC-меченым CD206 (BD Biosciences) или hFcγRIII mAb (3G8, лабораторное) в течение 15 минут на льду, чтобы распознать MDM. Клетки собирали и анализировали методом проточной цитометрии с минимальным числом собранных макрофагов, соответствующим 5000. Процентную долю клеток с положительными результатами двойного окрашивания определяли как фагоцитарный потенциал. Подтверждение фагоцитоза обычно проводили при помощи конфокальной микроскопии.23
Анализ АЗКЦ проводили двумя способами: используя первичные NK-клетки CD56+ve, MACS-выделенные (Miltenyi Biotec, Germany) из периферической крови здоровых волонтеров; или клеточную линию NK-92, стабильно трансфицированную для экспрессии аллеля CD16-158V вместе с ЗФБ (приобретенную у Conkwest, San Diego, CA).24 В случае анализа с применением первичных NK-клеточных эффекторов, клетки-мишени предварительно инкубировали с mAb при 1-10 мкг/мл в течение 30 минут перед смешиванием с NK-клетками. Клетки инкубировали в течение 3-4 часов в среде RPMI 1640 + GlutaAMX (Life Technologies), содержащей 10 мМ буфера ГЭПЭС, 1 мМ пирувата натрия и 10% ФБС (все от Gibco) в соотношении эффектор:клетка-мишень 20:1. Лизис определяли, используя набор CellaTOX (Cell Technology Inc) в соответствии с инструкцией производителя, а результирующую биолюминесценцию считывали на люминометре Victor2V. В случае применения линии NK-клеток NK-клетки инкубировали с мишенями в соотношениях от 1:1 до 5:1 избытка в течение 4 часов, а лизис определяли методом проточной цитометрии. Вкратце, в конце инкубации клеточную суспензию инкубировали с 9 нМ красного красителя фиксированных клеток SYTOX (Life Technologies) в течение 20 минут в темноте, а затем клетки анализировали методом проточной цитометрии.
Для исследования того, как ингибирование интернализации влияет на АЗКФ и АЗКЦ, клетки ХЛЛ инкубировали с ритуксимабом, одним или в комбинации с изотипическим контролем или мутированным вариантом 6G11 N297Q, в течение 3-4 часов перед совместным культивированием с эффекторами, чтобы обеспечить время для интернализации антител.
Анализ интернализации и мечение AF488
mAb метили AF488 в соответствии с инструкциями производителя (Life Technologies, UK). Для определения интернализации проводили анализ гашения, как подробно было описано ранее.23 Вкратце, образцы клеток (2-4 x 105 клеток/лунку) инкубировали с меченым AF488 mAb (5 мкг/мл) в течение заданного времени, промывали, пересуспендировали и инкубировали при 4oC в течение 30 минут в присутствии или отсутствии анти-AF488 гасящего антитела (Life Technologies, UK). Затем образцы оценивали методом проточной цитометрии. Результаты представлены в виде % поверхностно-доступных mAb, который обратно пропорционален количеству интернализированных mAb.
Вестерн-блоттинг
Вестерн-блоттинг проводили, как было описано ранее.26 Вкратце, 2,5-5 x 106 клеток промывали, обрабатывали и лизировали в буфере onyx, содержащем коктейль из ингибиторов протеаз и фосфатаз. Затем образцы сепарировали методом ДСН-ПААГ, а белки незамедлительно переносили на ПВДФ-мембрану. Мембраны блокировали 5% нежирным сухим молоком, инкубировали с разведенными соответствующим образом первичными антителами, промывали, а затем инкубировали с конъюгированным с пероксидазой хрена антикроличьим или антимышиным IgG (Sigma Aldrich, UK). Полосы визуализировали посредством инкубации с усиленной хемилюминесценцией (ECL; GE Healthcare, UK) и применения светочувствительной пленки (Hyperfilm ECL; GE Healthcare, UK). Исследование оптической плотности проводили, используя программное обеспечение Image J в соответствии с инструкциями производителя.
Иммунотерапия in vivo
Анализ методом адоптивного переноса: Как было подробно описано ранее (Beers et al., 2010b), ~2x107 спленоцитов/мл окрашивали как мишени или не-мишени с 5 мкМ и 0,5 мкМ CFSE, соответственно, промывали и смешивали в соотношении 1:1, и инъецировали в.в. мышам-реципиентам (~5x106 клеток/мышь). Затем на 1 и/или 2 день мышей инъецировали в.в. и/или в.б. и через 1 день отбирали для исследования лейкоцитов крови и селезенки методом проточной цитометрии.
Адоптивный перенос: Анализ методом адоптивного переноса проводили в основном так, как было описано ранее.27 Вкратце, ~2 x 107 спленоцитов/мл от релевантных мышей C57BL/6 окрашивали как мишени (Т) или не-мишени (NT) 5 мкМ и 0,5 мкМ CFSE, соответственно, в течение 10 минут при комнатной температуре, промывали, смешивали в соотношении NT:T 1:1 и внутривенно (в.в.) инъецировали на -1 день мышам-реципиентам (~5 x 106 спленоцитов/мышь). Затем мышам инъецировали в.в. mAb на день 0 и через 1 день отбирали для исследования их крови и спленоцитов в отношении клеток T и NT. В некоторых адоптивных экспериментах мышам-реципиентам инъецировали mAb на день 0 и день 1 (в.в. и/или в.б. путями) и через 1 день отбирали для исследования их крови и спленоцитов в отношении клеток T и NT. Популяцию В-клеток определяли по параметрам FSC-H и SSC-H и CD19-положительности методом проточной цитометрии.
Уничтожение В-клеток: Для анализа системного истощения В-клеток мышам с разным генотипом давали одну дозу hCD20, hFcγRII или обоих mAb в.в. (250-500 мкг), а затем в течение времени оценивали долю В-клеток, оставшихся в крови или органах, методом проточной цитометрии или иммуногистохимии (МГХ), как это делали ранее.27
Первичные человеческие ксенографтные модели: От пациентов с ХЛЛ или МКЛ на стадии лейкоза получали образцы крови и использовали для ксенографтных исследований в течение 24 часов после сбора. Вкратце, при помощи Ficoll Paque PLUS выделяли МКПК и после тщательной промывки клетки пересуспендировали в стерильном PBS при 3-5x108 клеток/мл. Мышей облучали 1 Гр в течение 1-5 часов перед проведением в.в. инъекции 200 мкл клеточной суспензии, что соответствует 6-10x107 клеток/мышь. На 4-5 день после инъекции клеток мышей обрабатывали 1-10 мг/кг любого из hCD20 mAb, hFcγRII mAb или обоими. Через 2-3 дня мыши получали вторую инъекцию, а еще через 2-3 дня мышей умерщвляли. Выделяли селезенки, делили на две части, половину замораживали в OCT, а вторую перерабатывали в одноклеточную суспензию. После этого лизировали красные кровяные тельца, используя буфер для лизиса (Gibco), перед инкубацией с окрашивающими клеточную поверхность mAb в течение 30 минут на льду. Человеческие клетки идентифицировали и количественно оценивали как hCD45-положительные и лейкозные клетки посредством окрашивания hCD5 и hCD19 (BD Biosciences).
In vivo истощение лейкоцитов
Системное истощение лейкоцитов периферической крови мышей оценивали в течение времени методом проточной цитометрии после инъецирования мышей hFcγRIIB+/- x mFcγRII-/- C57BL/6 20 мг/кг изотипического контроля или ДТ 6G11 (в.в.). Уровни подгрупп лейкоцитов нормировали относительно уровней до дозирования и выражали в виде %.
In vivo ФД, ФК и иммуногенность
Сывороточные концентрации 6G11 (ФК) и MAHA определяли при помощи ЭХЛ-иммуноанализа. 6G11 mAb выявляли в сыворотке мышей, используя биотинилированную анти-6G11 поликлональную козью сыворотку и метку стрептавидин-сульфо (Meso Scale Diagnostics, US). Количественную оценку уровней mAb в сыворотке проводили путем сравнения с известной стандартной кривой, сгенерированной для 6G11. ФК-параметры 6G11 (Cмакс, Tмакс, ППК, CL(F), Vz(F), Vss и t1/2) оценивали, используя некомпартментную модель при помощи программного приложения Phoenix™ WinNonlin v.6.2 (Pharsight Corp, CA, US). Cмакс и Tмакс получали непосредственно из профиля сывороточная концентрация-время.
Для выявления в мышиной сыворотке наличия MAHA, направленного на 6G11, использовали качественный и полуколичественный иммуноанализ ЭХЛ. Образцы разводили в аналитическом буфере с биотином и меченым сульфо-меткой 6G11. После инкубации образцы переносили на предварительно заблокированный стрептавидином планшет. Сигнал оценивали, используя технологию на основе MSD, при этом люминесцентный сигнал пропорционален уровню анти-6G11, присутствующего в образце. Аффинно очищенную козью анти-6G11 сыворотку использовали для получения положительных контрольных образцов.
In vivo исследования фармакодинамики (ФД), фармакокинетики (ФК), минимального ожидаемого биологического действия (MABEL) и иммуногенности
ФД, ФК и MABEL при введении одной дозы: Мышей hFcγRIIB+/- x mFcγRII-/- одного пола и возраста инъецировали одной дозой, составляющей 1, 10 или 100 мг/кг ДТ 6G11, проверяя как в.в., так и в.б. пути введения при дозе 10 мг/кг (Фиг. 17A). Серийные образцы крови брали непосредственно после инъекции и вплоть до 168 часов до окончания эксперимента. В течение всего времени животных проверяли в отношении любых признаков нарушений, потери массы, токсичности или патологии. Уничтожение В-клеток hFcγRIIB+ve (ФД) и другие параметры (например, ФК 6G11 и наличие мышиных античеловеческих антител [MAHA]) исследовали следующим образом.
Иммуногенность при повторных дозах: Мышей hFcγRIIB+/- x mFcγRII-/- и мышей mFcγRII-/- одного пола и возраста многократно инъецировали 10 мг/кг ДТ 6G11, как указано на Фиг. 17C, в течение 10-дневного периода (за первым в.в. введением следовали три дозы, доставляемые в.б.). Образцы крови, полученные перед дозированием, анализировали в отношении циркулирующих В-клеток (CD19+ve и B220+ve) методом проточной цитометрии. Животных проверяли в течение всего времени и на 14 дней позже, как указано выше. Сывороточную концентрацию 6G11 (ФК) и MAHA определяли следующим образом.
ФК-анализ 6G11: Для определения концентрации 6G11 в мышиной сыворотке применяли иммуноанализ ЭХЛ. Овечьим анти-hIgG (сайт связывания) или козьим анти-hλ (AbD Serotec) (в присутствии ритуксимаба) покрывали 96-луночный планшет MSD, а затем блокировали, используя 0,45% рыбий желатин. 6G11 mAb выявляли путем последовательного добавления биотинилированной анти-6G11 поликлональной козьей сыворотки и метки стрептавидин-сульфо (Meso Scale Diagnostics, MSD), обеспечивая смывание любого несвязанного материала. Добавляли буфер считывания T (MSD), содержащий триполиамин, а метка sTag, связанная с 6G11, давала хемилюминесцентный сигнал при приложении напряжения. Количественную оценку уровней mAb проводили путем сравнения с известной стандартной кривой, сгенерированной для 6G11.
ФК 6G11 оценивали, используя некомпартментную модель при помощи программного приложения Phoenix™ WinNonlin v.6.2 (Pharsight Corp, Mountain View, CA, US). Определяли ФК-параметры, включая Cмакс, Tмакс, ППК, CL(F), Vz(F), Vss и t1/2. Cмакс и Tмакс получали непосредственно из профиля сывороточная концентрация-время (Таблица 3).
In vitro анализ истощения для цельной крови
Свежую гепаринизированную человеческую кровь разводили 5x в среде CTL (Cell Technology Limited, Germany) и высевали в 96-луночные планшеты с 20 мкг/мл Rit и/или ДТ или N297Q 6G11; или оставляли NT на 24 часа перед тем, как клетки собирали и окрашивали античеловеческими CD3-PerCP-Cy5.5/FITC, CD19-PE, CD14-APC и CD66b-FITC mAb, чтобы выявить Т-клетки, В-клетки, моноциты и нейтрофилы для проточной цитометрии, соответственно. Рассчитывали соотношение между подгруппами лейкоцитов и клетками CD3+.
Таблица 3
|
Анализ иммуногенности (MAHA): Для выявления в мышиной сыворотке наличия MAHA, направленного на hIgG1 6G11, использовали качественный и полуколичественный иммуноанализ ЭХЛ. Образцы разводили в аналитическом буфере с биотином и меченым сульфо-меткой 6G11. После инкубации образцы переносили на предварительно заблокированный стрептавидином планшет. Добавляли буфер считывания T (MSD), содержащий триполиамин, а сигнал оценивали, используя технологию на основе MSD, при этом люминесцентный сигнал пропорционален уровню анти-6G11, присутствующего в образце. Аффинно очищенную козью анти-6G11 сыворотку использовали для получения положительных контрольных образцов.
In vitro анализ высвобождения цитокинов
МКПК очищали путем центрифугирования в градиенте плотности Lymphoprep (Axis-Shield, Oslo, Norway). Клетки предварительно инкубировали в культуре с высокой плотностью28 (1x107/мл) в течение 48 часов в бессывороточной среде CTL (Cell Technology Limited, Germany) в 24-луночном планшете при высокой плотности (1,5 мл/лунку). После этого клетки промывали, пересуспендировали в бессывороточной среде CTL (1x106 /мл) и высевали в 96-луночный планшет (100 мкл/лунку), который предварительно был покрыт 0,02 или 1 мкг/мл OKT3 или оставлен необработанным (NT). ДТ и N297Q варианты hFcγRIIB mAb (клон 6G11) или контрольные mAb соответствующего изотипа (hIgG1) добавляли в концентрации 10 мкг/мл, а клетки инкубировали еще в течение 48 часов. Цитокины (ИЛ-6, -10, ИФН-γ и ФНО-α) количественно оценивали в обработанных супернатантах МКПК методом анализа MSD V-Plex (Meso Scale Discovery, Rockville, USA) в соответствии с инструкциями производителя.
Иммуногистохимия (ИГХ)
ИГХ-окрашивание тканей человека и других животных проводили следующим образом. Из различных источников получали органы, замораживали, а затем готовили срезы перед иммуноокрашиванием клонами 6G11 и 7C07. Тканевую реактивность определяли при помощи амплификации Tyramide Signal Amplification (TSA; PerkinElmer) без блокирования перекисью водорода. Включали антиген-отрицательные ткани, чтобы выявить какое-либо неспецифическое в отношении hFcγRIIB связывание.
Статистический анализ
Для сравнения различий между экспериментальными группами in vitro применяли двухсторонний t-критерий. Для оценки разницы в выживаемости между экспериментальными группами in vivo строили кривые Каплана-Мейера и анализировали, используя лог-ранговый критерий. В случае in vivo экспериментов, включающих более двух групп, использовали двухфакторный или однофакторный дисперсионный анализ ANOVA. В случае разницы в объективном или полном ответе in vivo, использовали критерий хи-квадрат. Статистический анализ проводили, используя программное обеспечение GraphPadPrism (версия 5 для Windows).
Получение трансгенных мышей, экспрессирующих hFcγRIIB и hCD20
Трансген hFcγRIIB2 конструировали из геномной ДНК и кДНК Raji посредством ПЦР-реакций с перекрытиями, используя праймеры, нацеленные на разные экзонные и интронные соединения. Экспрессионная кассета содержит промотор hFcγRIIB 22, экзон 1/2, интрон 2-3 и экзоны 3-7 (2,4 т.п.о.) и была изначально клонирована в pcDNA3 (Invitrogen) при помощи сайтов NotI и XbaI и лигирована в BGH полиА последовательность вектора. Конструкцию, длина которой (включая последовательность полиА) составляла 3517 п.о., дополнительно субклонировали в вектор pBC-SK (Stratagene) при помощи сайтов NotI и SmaI. Функциональная экспрессия конструкции была подтверждена во временно трансфицированных клетках IIA1.6 методом проточной цитометрии с применением AT10-FITC. Очищенную экспрессионную кассету, в которой отсутствовали векторные последовательности, микроинъецировали в мужские пронуклеусы зигот FVB/N. Скрининг трансгенных мышей проводили методом ПЦР (амплифицируя экзоны 3-7 фрагмента кДНК из геномной ДНК, выделенной из краев ушей) или проточной цитометрии периферической крови с применением AT10-FITC. Полученных в результате Tg-положительных основателей возвратно скрещивали с мышами C57BL/6 или BALB/c на протяжении >10 поколений. Трансгенных мышей hFcγRIIB с отсутствием соответствующего мышиного рецептора получали путем скрещивания с мышами, несущими мышиный FcγRIIB-/-. Затем полученных в результате мышей скрещивали с трансгенными в отношении hCD20 мышами, чтобы получить потомство hCD20 x hFcγRIIB+/- x mFcγRIIB-/-.
Поверхностный плазмонный резонанс
Для определения аффинности связывания hFcγRIIB mAb использовали анализатор BIAcore T100 (GE Healthcare, UK), как было описано в другой работе.25 mAb иммобилизовали на сенсорном чипе CM5 (GE Healthcare, UK), используя стандартное аминное сопряжение в соответствии с инструкциями производителя. Растворимый hFcγRIIB (0,16-100 нM; R & D Systems, UK) инъецировали в течение 5 минут, а диссоциацию отслеживали в течение 10 минут. Фоновое связывание с контрольными проточными клетками отслеживали и вычитали автоматически. Величины KД рассчитывали по 1:1 модели связывания, используя программное обеспечение BIAcore T100 Evaluation.
Иммунотерапия in vivo
Опухолевая модель с ксенографтной подкожно вводимой линией клеток: Мышам SCID (3-6 мыши/группу) подкожно инъецировали 5x106 клеток Daudi или Raji в матрице матригель со сниженным содержанием факторов роста (BD Biosciences, UK) на день 0, а затем еженедельно подкожно обрабатывали терапевтическим mAb на 7, 14, 21 и 28 день. Мышам NOD.SCID подкожно инъецировали 10x106 клеток Jeko-1 на день 0. Отслеживали рост опухолей, а когда опухоли достигали 4x4 мм, мышей рандомизировали и обрабатывали терапевтическим mAb при 10 мг/кг дважды в неделю. Рост опухолей отслеживали в течение времени, используя калиперы, а размер опухолей оценивали, используя следующее уравнение:
[Масса = (длина × ширина2)/2]
Опухолевая модель с ксенографтной внутривенно вводимой линией клеток: Мышам SCID (6/группу) инъецировали в.в. 2,5x106 клеток Raji (BD Biosciences, UK) на день 0, а затем еженедельно обрабатывали вплоть до 4 раз терапевтическим mAb на 7, 14, 21 и 28 день. Рост опухолей отслеживали регулярно, исследуя мышей в отношении каких-либо признаков паралича.
In vitro анализ высвобождения цитокинов
Свежую или 5x разведенную (в бессывороточной среде CTL) гепаринизированную кровь культивировали в 96-луночных планшетах (с U-образным дном) и обрабатывали 10 мкг/мл mAb в течение 24 часов при 37 °C перед сбором супернатантов для последующего анализа. Цитокины (ИЛ-1β, -2, -4, -6, -10, ИФН-γ и ФНО-α) ) количественно оценивали в супернатантах цельной или разведенной крови методом анализа MSD V-Plex (Meso Scale Discovery, Rockville, USA) в соответствии с инструкциями производителя.
Флуоресцентная микроскопия
Ткани для приготовления срезов замораживали в среде OCT (RA Lamb, Thermo Shandon) и помещали в изопентан на сухой лед. 10 мкм замороженные срезы фиксировали в ацетоне, блокировали 5% нормальной козьей сывороткой и инкубировали с mAb к hFcγRII/CD32 (клон AT10, полученный в лаборатории), mFcγRII/CD32 (клон AT130-2, полученный в лаборатории29) В-клетками (крысиный антимышиный CD45R/B220; BD Pharmingen, UK), фолликулярными дендритными клетками (крысиный антимышиный FDC; BD Pharmingen, UK) и макрофагами (крысиный антимышиный F4/80; AbD Serotec, UK) с последующим добавлением DyLight594-конъюгированного козьего анти-hIgG (Abcam, Cambridge, UK) и AF488-конъюгированного козьего анти-крысиного IgG (Life Technologies, UK). Срезы помещали на Vectashield (Vector Laboratories, UK) и получали изображения при помощи инвертированного микроскопа с системой для флуоресценции при полном внутреннем отражении CKX41, оборудованного камерой для цветной съемки CC12, управляемой программным обеспечением Cell B, с использованием линз для объектива Plan Achromat 10x 0,25 (все от Olympus, UK). Файлы изображений RGB (TIFF) переносили в Adobe Photoshop (CS6) и преобразовывали красные/зеленые слои для получения изображения в градациях серого.
Протокол окрашивания нейтрофилов
Образцы крови или селезенки получали от мышей C57BL/6, mFcγRII -/- и mFcγRII -/- X hFcγRIIB+/- одного пола и возраста. Селезенки гомогенизировали путем пропускания через 100 мкм клеточное сито (BD), затем промывали в полной среде RPMI и пересуспендировали в 5 мл PBS, 200 мкл клеточной суспензии на пробирку использовали для проточной цитометрии. Образцы окрашивали 10 мкг/мл антимышиного FcγRII (AT130-2 F(ab’)2-FITC), античеловеческого FcγRII (AT10 F(ab’)2-FITC) или нерелевантного контроля (3G8 F(ab’)2-FITC) (все получены и помечены в лаборатории) плюс следующее: антимышиный CD19 (1D3-PE, лабораторный), антимышиный NK1.1 (PK136-AlexaFluor 647), антимышиный CD11b (M1/70-Pacific Blue), антимышиный CD11c (N418-PE-Cy7), антимышиный Ly-6-G (1A8-APC-Cy7), антимышиный Ly-6C (HK1.4-PerCP-Cy5.5; все от BioLegend, если не указано иное). Клетки окрашивали в течение 30 минут при 4°C, затем добавляли 1 мл лизисного буфера для эритроцитов (AbD Serotec, UK), клетки центрифугировали, затем один раз промывали и анализировали на FACs Canto. Дебрис и клетки CD11chigh исключали, нейтрофилы представляли собой CD19- CD11b+ NK1.1- Ly-6G+.
Результаты
Получение и характеристики mAb, специфических в отношении Fc-связывающего домена hFcγRIIB
Недавно сообщалось, что устойчивость к ритуксимабу, CD20 mAb типа I, у некоторых пациентов с лимфомой можно частично объяснить его интернализацией с опухоли и тем, что экспрессия ингибиторного FcγRIIB/CD32B на поверхности В-клетки-мишени стимулирует этот процесс.13,23 В соответствии с этой гипотезой, in vivo совместное введение AT10 с ритуксимабом приводило к аддитивным/синергетическим противоопухолевым ответам в двух разных ксенографтных моделях лимфомы, в которых hCD20 и hFcγRIIB коэкспрессируются на опухолях (Фиг. 7).
Внеклеточный домен hFcγRIIB на ~98% гомологичен внеклеточному домену hFcγRIIA, ключевому активаторному FcγR (Фиг. 8A). Так как эти два рецептора опосредуют противоположные функции, для терапевтическиго антитела крайне важно быть высокоспецифичным в отношении hFcγRIIB. AT10 не удовлетворяет этому критерию, так как он связывается с hFcγRIIA и hFcγRIIB с одинаковой аффинностью.19 Кроме того, он имеет мышиное происхождение (IgG1), что ограничивает его трансляционный потенциал. Чтобы создать специфические к hFcγRIIB антитела с терапевтическим потенциалом в отношении людей, мы использовали собственную запатентованную библиотеку фагового дисплея человеческих антител n-CoDeR®,30 проводя отбор по связыванию с hFcγRIIB и против связывания с hFcγRIIA (или наоборот), чтобы получить моноспецифические реагенты (Фиг. 1). Полученные в результате mAb оценивали в отношении их способности избирательно связывать FcγRIIB (Фиг. 1A) и блокировать связывание иммунного комплекса (ИК) с hFcγRIIB, но не hFcγRIIA (Фиг. 1B). Скрининг в отношении связывания с отдельными подгруппами человеческих МКПК (нейтрофилы, моноциты, В-клетки, Т-клетки и NK-клетки), выделенными В-клетками (Фиг. 1C и D; Фиг. 8B-D), злокачественными человеческими В-клеточными линиями или спленоцитами трансгенных мышей (hFcγRIIB+/- x mFcγRII-/-; данные не показаны) продемонстрировал высокую специфичность антител в отношении hFcγRIIB или hFcγRIIA. Относительную аффинность этих mAb в отношении hFcγRIIB определяли методом ELISA (Таблица 1), а подгруппа, оценивая методом поверхностного плазмонного резонанса демонстрировала KД в отношении связывания с hFcγRIIB в диапазоне 2 x 10-6 - 2 x 10-8 M (Фиг. 1E и Таблица 2). На основании вышеприведенных данных было выявлено и утверждено 14 высокоспецифичных hFcγRIIB mAb. Хотя точная специфичность для каждого mAb определена не была, они все блокируют связывание ИК и не демонстрируют перекрестную реактивность с FcγRIIA, что четко указывает на то, что связывание происходит в области связывающего углубления IgG с высокой концентрацией остатков, разных для FcγRIIA и B (Фигуры 7 и 8).
Таблица 1. Данные по EC50 для hFcγRIIA и hFcγRIIB mAb
|
Таблица 2. Данные по аффинности и авидности Biacore для отобранных клонов hFcγRIIB mAb
|
Антагонистическое hFcγRIIB mAb блокирует интернализацию ритуксимаба
Инкубация В-клеток hFcγRIIB+ve с ритуксимабом индуцирует ингибиторную сигналинг через hFcγRIIB, которая связана с фосфорилированием цитоплазматического ITIM-мотива hFcγRIIB.31-33 На основании иммунной ингибиторной функции этого рецептора мы предполагаем, что mAb, способное как предотвращать интернализацию CD20, так и блокировать активацию/фосфорилирование hFcγRIIB, может представлять терапевтический интерес. Поэтому мы провели исследование 14 специфических к hFcγRIIB mAb в отношении их способности регулировать фосфорилирование FcγRIIB в В-клетках Raji неходжкинской лимфомы. Чтобы оценить антитело-опосредованное действие в зависимости от вариабельного домена, мы сконструировали варианты антител, несущие мутацию N297Q в константной области, которые не могут связываться с FcγR посредством константного Fc-домена (смотрите ниже).34 Кратковременная обработка (30 минут) клеток Raji hFcγRIIB mAb привела к двум разным ответам; mAb, которые индуцировали высокие уровни фосфорилирования hFcγRIIB ITIM (например, клоны 5C04 и 6G08), и те, которые оказывали слабое действие или не оказывали действие, такие как клоны 6G11 и 7C07 (Фиг. 2A). Аналогичные наблюдения имели место для клеток первичного ХЛЛ, миндалин и моноцитов (Фигура 9B и 9C, данные не показаны). Эти данные демонстрируют, что было успешно получено hFcγRIIB mAb, способное блокировать связывание иммунного комплекса с hFcγRIIB, не активируя этот рецептор.
Далее мы исследовали, может ли hFcγRIIB mAb блокировать взаимодействие между hFcγRIIB и ритуксимабом и предотвращать связанную с ним интернализацию ритуксимаба. Некоторые mAb, такие как 6G08, оставались агонистичными, стимулируя фосфорилирование рецептора. В противоположность этому, два mAb (6G11 и 7C07), называемые в данном документе антагонистичными, были способны практически полностью предотвращать фосфорилирование FcγRIIB, индуцируемое после связывания ритуксимаба на клетках Raji и клетках первичного ХЛЛ (Фиг. 2B, данные не показаны), подобно AT10.32 При помощи анализа гашения методом проточной цитометрии23 мы определили, что те же mAb были способны эффективно блокировать интернализацию ритуксимаба с поверхности hFcγRIIB-трансфицированных клеток Ramos (Фиг. 2C). Следует отметить, что отсутствие интернализации в присутствии этих mAb было сходно с наблюдаемым для mAb типа II, тозитумомабом, и клеток Ramos, в которых отсутствовал hFcγRIIB (Фиг. 2C). Оба ДТ и N297Q варианты обладали одинаковой активностью в этом анализе, что свидетельствует о зависимом от вариабельного домена (Fv) антитела действии (Фиг. 9) и демонстрирует, что антагонистические эффекты сохранялись в формате ДТ hIgG1. Последующий анализ выявил, что способность панели mAb блокировать интернализацию ритуксимаба была напрямую связана с их относительной аффинностью в отношении hFcγRIIB; R2 = 0,78 (Фиг. 2D), что, в свою очередь, коррелировало с их способностью блокировать фосфорилирование hFcγRIIB после стимуляции ритуксимабом; R2 = 0,79 (Фиг. 2E). Таким образом, высокоаффинное антагонистическое hFcγRIIB mAb предотвращало hFcγRIIB-опосредованное удаление ритуксимаба с поверхности клетки-мишени.
Антагонистическое hFcγRIIB mAb индуцирует Fc:FcγR-зависимую цитотоксичность и блокирует интернализацию ритуксимаба, тем самым проявляя высокую противоопухолевую активность in vitro
Открытие того, что антагонистическое действие некоторых mAb сохранялось в формате ДТ hIgG1, который продуктивно задействует активаторные FcγR-экспрессирующие иммунные клетки, дает основание предположить, что этим mAb может быть свойственна Fc:FcγR-зависимая противоопухолевая активность. Поэтому мы исследовали наши hFcγRIIB mAb в отношении подобных проявлений. Инкубация опсонизированных клеток-мишеней Raji с первичными NK-клетками продемонстрировала, что антагонистические mAb 7C07 и 6G11 также обладают наивысшей цитотоксической активностью, превышающей проявляемую самим ритуксимабом (Фиг. 10A и B). Установив, что эти два клона обладают наиболее высокой аффинностью, наиболее сильной активностью в отношении блокирования интернализации ритуксимаба и проявления АЗКЦ, далее мы оценивали их связывание с панелью человеческих и животных тканей, экспрессирующих FcγR. Оба клона, 7C07 и 6G11, специфически окрашивали лимфоциты в селезенке и миндалинах человека (Фиг. 3A, данные не показаны). Ни 7C07, ни 6G11 не реагировали с лимфоцитами яванских макаков, крыс, кроликов или мышей, что свидетельствует о том, что эти mAb не являются перекрестно-реактивными с FcγRIIB других видов (Фиг. 3A, данные не показаны). Однако клон 7C07, но не 6G11, также окрашивал синусоидные капилляры тканей селезенки и лимфатических узлов от людей и других видов, что свидетельствует о нежеланной перекрестной реактивности (Фиг. 3A и Фиг. 11A). Было показано, что ДТ и N297Q mAb окрашивают одинаково (Фиг. 11B), а в других человеческих тканях не наблюдалось никакой непредвиденной перекрестной реактивности (Фиг. 11C). На сновании этого профиля реактивности клон 6G11 был отобран как основной клинический кандидат.
Свойственную 6G11 цитотоксическую активность дополнительно исследовали в анализе АЗКЦ, ЗГК и АЗКФ, используя широкую панель полученных от пациентов образцов первичного ХЛЛ. В каждом анализе была продемонстрирована значительная активность на уровне, существенно превышающем наблюдаемый для ритуксимаба (Фиг. 3B-D). Кроме того, 6G11 было более эффективно в индукции гибели клеток по сравнению с ритуксимабом при проведении анализа с эффекторными NK-клетками, экспрессирующими высоко- или низкоаффинные варианты hFcγRIIIA (158 V или F, соответственно) (Фиг. 3E).
После этого мы исследовали способность 6G11 предотвращать интернализацию ритуксимаба с поверхности клеток ХЛЛ. Оба варианта 6G11, ДТ и N297Q (которым самим не свойственна Fc-зависимая эффекторная активность; Фиг. 12), были способны в значительной степени предотвращать интернализацию ритуксимаба (Фиг. 3F). Интересно, что в отличие от CD20 и, как сообщалось ранее17, задействование hFcγRIIB на поверхности клеток ХЛЛ со стороны ДТ или N297Q hIgG1 mAb (6G11 или AT10) не приводило к высоким уровням интернализации hFcγRIIB при оценивании прямым или непрямым способом (Фиг. 3G и Фиг. 13A и B).
Чтобы узнать, может ли 6G11 также усиливать цитотоксическую активность ритуксимаба путем предотвращения его интернализации, мы совместно инкубировали клетки первичного ХЛЛ с ритуксимабом и N297Q-вариантом 6G11 и оценивали АЗКФ MDM и АЗКЦ NK-клеток. Неожиданно было показано, что N297Q 6G11 в значительной степени стимулирует способность MDM поглощать ритуксимаб-опсонизированные клетки ХЛЛ по сравнению с применением одного ритуксимаба (Фиг. 3H и I). Аналогичное повышение активности наблюдали в анализе АЗКЦ с NK-клетками (Фиг. 3J). NK-клетки не экспрессируют FcγRIIB, подтверждая, что любое повышение, вызываемое hFcγRIIB mAb, связано с действием на клетки-мишени в результате ингибирования интернализации ритуксимаба. Эти данные подтверждают, что блокирование hFcγRIIB на поверхности В-клеток-мишеней ингибирует интернализацию комплекса mAb:Ag:hFcγRIIB и повышает их возможность стать мишенями для уничтожения эффекторными клетками.
Вместе эти наблюдения позволяют предположить, что 6G11 может вызывать противоопухолевую активность посредством двойного механизма - свойственной ему цитотоксической активности и усиления активности ритуксимаба путем предотвращения его удаления с клеточной поверхности.
6G11 обладает характерной активностью, связанной с истощением В-клеток, и усиливает истощение со стороны ритуксимаба у иммунно-компетентных трансгенных мышей hCD20+ve hFcγRIIB+ve
Как обсуждалось выше, hFcγRIIB экспрессируется как на В-клетках-мишенях, где он опосредует нежелательное удаление ритуксимаба с клеточной поверхности, так и на ключевых иммунных эффекторных клетках, таких как макрофаги, где он ослабляет противораковые ответы антител.5-7,35 Чтобы понять влияние системного нацеливания hFcγRIIB на соответствующие hFcγRIIB-экспрессирующие типы клеток, мы получали мышей, экспрессирующих ген hFcγRIIB под управлением промотора hFcγRIIB (Фиг. 14A и B). Экспрессия и перераспределение hFcγRIIB в организме трансгенных мышей очень напоминает экспрессию в человеческих тканях, при этом сильная экспрессия происходит на В-лимфоцитах, BMDM и моноцитах, но не на нейтрофилах, в отличие от mFcγRII (Фиг. 14C-F, данные не показаны). Кроме того, эквивалентная экспрессия сохранялась для фона mFcγRII-/-, что позволило нам исследовать влияние hFcγRIIB в отсутствие осложнений, связанных с эндогенным мышиным ингибиторным FcγR. Важно, что у этих мышей сохранялась антагонистическая активность 6G11 (Фигура 14(5/5). Кроме того, экспрессия hFcγRIIB на эндотелиальных клетках была ниже, чем у мышей ДТ и более походила на экспрессию у людей (Фигура 14(5/5).
Чтобы убедиться в безопасности применения 6G11 in vivo, мы провели исследование с увеличением дозы, обрабатывая мышей hFcγRIIB+/- x mFcγRII-/- 1, 10 или 100 мг/кг 6G11 (Фигура 18). Ни одна из обработанных мышей не страдала от нежелательных явлений, таких как острые эффекты, нарушения или потеря массы (Фигура 18). Исследование тканей на 7 день не выявило признаков какой-либо сильной токсичности в органах (почки, головной мозг, селезенка, печень, легкие). Значительное истощение В-клеток hFcγRIIB+ наблюдали как в крови (Фигура 6A), так и в селезенке (Фигура 6B) при дозах выше 1 мг/кг с эквивалентной активностью в группах, обрабатываемых 10 и 100 мг/кг. 6G11 представляет собой полностью человеческое mAb, легко выявляемое в сыворотке мышей, но иммуногенное по своей природе, поэтому мы одновременно оценивали его время полужизни и наличие признаков ответов со стороны мышиного античеловеческого антитела (MAHA). При дозах >1 мг/кг преодолевалось мишень-опосредованное выведение, которое влияет на ФК-профиль, а в группах, обрабатываемых 10 и 100 мг/кг, наблюдали слабое или отсутствующее влияние связывания мишеней (Фигура 6C). Однако в течение 7 дней наблюдали значительное количество MAHA, что приводило к быстрому выведению mAb (Фигура 6D). На основании временных точек до появления значительного количества MAHA мы оценили, что время полужизни mAb должно соответствовать диапазону в 2-4 дня для доз 10 и 100 мг/кг (Фигура 6C, D и Таблица 7).
Также мы провели исследование с повторным дозированием для лучшей имитации того, как 6G11 можно доставлять в клинических условиях, вводя его 4 раза в течение 24-дневного периода (Фигура 18), мышей при этом исследовали, как и ранее. Истощение циркулирующих В-клеток наблюдали в группе hFcγRIIB+/- x mFcγRII-/-, но не в контрольной группе mFcγRII-/-, инъекции проводили многократными дозами (10 мг/кг) 6G11 (Фигура 6E). Аналогично, мыши не страдали от потери массы (Фигура 18) или нежелательных явлений, а также не наблюдались признаки сильной токсичности (данные не показаны). Как и раньше, у мышей hFcγRIIB+/- x mFcγRII-/- наблюдали значительные ответы со стороны MAHA в течение 1 недели, которые приводили к быстрому выведению 6G11 из сыворотки (Фигура 6F). В противоположность этому в контрольной группе mFcγRII-/- не было выявлено MAHA, что указывает на совместную зависимость от ксеногенного mAb и поверхностного антигена, необходимых для индукции MAHA (Фигура 6F).
Чтобы исследовать, может ли происходить уничтожение клеток hFcγRIIB+, отличных от В-клеток, после обработки 6G11, проводили анализ уничтожения с цельной кровью человека (Фигура 6(2/2)) и in vivo эксперименты с трансгенными мышами hFcγRIIB (Фигура S4N). В-клетки, но не моноциты или нейтрофилы, уничтожались 6G11 IgG1, но не N297Q 6G11. Такое же отсутствие истощения моноцитов и нейтрофилов наблюдали в комбинации с ритуксимабом (Фигура 6(2/2). Далее мы использовали недавно разработанный in vitro анализ синдрома высвобождения цитокинов (СВЦ) (АВЦ) (Hussain et al., 2015), чтобы оценить потенциальное влияние 6G11 на мононуклеарные клетки периферической крови (МКПК) человека, которые сделали чувствительными к стимуляции посредством культивирования при высокой плотности (Romer et al., 2011), который дает возможность выявлять значительные уровни ИФН-γ, ФНО-α и/или ИЛ-8 после добавления нескольких специфичностей mAb (CD3, CD28 или CD52), ранее демонстрировавших, что могут вызывать СВЦ. Применение ДТ или N297Q 6G11 в течение 48 ч не привело к значительному высвобождению цитокинов, в отличие от в 500 раз меньших доз CD3 mAb (Фигура 6). Аналогичные результаты были получены при проведении АВЦ для цельной или разведенной крови (Фигура 19).
Вместе эти данные демонстрируют отсутствие нежелательных явлений и свидетельствуют о терапевтически релевантном ФК-профиле 6G11 mAb, подтверждая исследования эффективности.
6G11 обладает характерной активностью и стимулирует истощение вследствие действия ритуксимаба у иммунокомпетентных мышей
Потенциал истощения при нацеливании на hFcγRIIB, одного антитела или в комбинации с ритуксимабом, оценивали, используя hIgG1 6G11 и mIgG1 AT10. Сначала, в кратковременном анализе адоптивного переноса23, в котором спленоциты CFSE+ve hFcγRIIB+/- x mFcγRII-/- инъецировали реципиентам ДТ, после чего обрабатывали 6G11 (Фиг. 4A и B) или AT10 (Фиг. 15A), наблюдали эффективное истощение перенесенных клеток в циркуляции и селезенке. Интересно, что введения такой низкой дозы, как 1 мкг mAb, было достаточно, чтобы вызвать ~50% истощений В-клеток. Истощение полностью зависело от взаимодействия Fc:активаторный FcγR, так как с фрагментами F(ab’)2 и вариантами N297Q такой активности не наблюдалось; мишени hFcγRIIB+ve также не были уничтожены у мышей γ цепь-/- с отсутствием активаторных FcγR (Фиг. 4 A и B и Фиг. 15A). Эти данные подтверждают, что антагонистические hFcγRII mAb способны уничтожать клетки-мишени in vivo в зависимости от взаимодействия с активаторными FcγR на иммунных эффекторных клетках, как было отмечено ранее.36
Чтобы расширить наш анализ на комбинированную терапию с ритуксимабом, посредством скрещивания получали мышей hCD20+/-15,37 x hFcγRIIB+/- x mFcγRII-/-. Комбинация ритуксимаба и 6G11 (или AT10) приводила к более сильному истощению В-клеток hCD20+/- x hFcγRIIB+/- x mFcγRII-/- по сравнению с одним mAb в кратковременном анализе с переносом мишеней в организм реципиентов ДТ (Фиг. 4C и Фиг. 15B, соответственно). Аналогично, происходило более значительное истощение адоптивно перенесенных В-клеток hCD20+/- x hFcγRIIB+/- x mFcγRII-/- при комбинировании ритуксимаба с 6G11 в случае мышей-реципиентов hFcγRIIB+/- x mFcγRII-/- (экспрессирующих hFcγRIIB также на эффекторных клетках) (Фиг. 4D).
Далее мы исследовали влияние комбинации на циркулирующие В-клетки на мышиной модели hCD20+/- x hFcγRIIB+/- x mFcγRII-/-. Как и раньше, комбинация приводила к значительно более сильному истощению циркулирующих В-клеток по сравнению с монотерапией (Фиг. 4E и Фиг. 15C и D), продемонстрировав это свойство в полностью сингенной системе, в которой hFcγRIIB экспрессируется как на клетках-мишенях, так и на эффекторных клетках. Влияние комбинирования ритуксимаба и ДТ hIgG1 6G11 (или AT10) было большим, чем ожидалось для аддитивной активности (Фигура 4(2/2) и 15, соответственно), судя по ответам, наблюдаемым с отдельными mAb, применяемыми поодиночке в два раза больших дозах.
Чтобы оценить, какие из эффектов 6G11, внутренние (истощение В-клеток) или внешние (усиление ритуксимаба), были более важны для этой активности, мы использовали N297Q 6G11 в случае мышей hCD20+/- x hFcγRIIB+/- x mFcγRII-/-, и показали, что хотя один N297Q 6G11 неактивен в отношении уничтожения, он значительно усиливает уничтожение В-клеток ритуксимабом (Фигура 4(2/2). Аналогичные результаты наблюдали для mIgG1 6G11. Как ожидалось и наблюдалось для AT10 (Фигура 15), mIgG1 6G11 демонстрировало слабую одиночную активность уничтожения в случае мышей hCD20+/- x hFcγRIIB+/- x mFcγRII-/- (Hamaguchi et al., 2006), но, как и N297Q hIgG1 6G11, было способно значительно усиливать мышиную IgG2a-версию ритуксимаба в отношении уничтожения В-клеток (Фигура 15). Более того, вследствие наличия мышиного Fc, оно активно не выводится MAHA, облегчая более долговременную оценку комбинированной терапии. Эти данные демонстрируют долговременный благоприятный эффект антагонизирования hFcγRIIB-функции для усиления уничтожающей активности ритуксимаба in vivo.
Вместе эти исследования подтвердили двойной механизм действия in vivo для 6G11, включающий внутреннюю противоопухолевую функцию, сопряженную со стимуляцией противоопухолевой активности ритуксимаба посредством предотвращения интернализации.
6G11 усиливает опосредованное hCD20 mAb типа II истощение В-клеток in vivo
Чтобы исследовать, является ли 6G11 эффективным также в комбинации с hCD20 mAb типа II, которые не интернализируются в той же степени, что и CD20 mAb типа I 13,23,33, и которые недавно были утверждены для применения при ХЛЛ, мы проводили эксперименты на модели адоптивного переноса. Как и в случае ритуксимаба, монотерапия GA101gly (гликозилированный обинутузумаб) и 6G11 приводила к умеренному истощению В-клеток-мишеней CFSE+ve hCD20+/- x hFcγRIIB+/- x mFcγRII-/- в циркуляции и селезенке, в то время как комбинированная терапия существенно повышала истощение клеток-мишеней как в селезенке, так и в циркуляции у мышей-реципиентов ДИ т hFcγRIIB+/- x mFcγRII-/- (Фиг. 4F и G; данные не показаны). Эти данные позволяют предположить, что 6G11 также должен быть эффективным в комбинации с другими mAb, нацеленными непосредственно на В-клетки.
6G11 усиливает ритуксимаб-опосредованное истощение клеток первичного ХЛЛ и улучшает объективные и полные противоопухолевые ответы in vivo
Хотя ритуксимаб успешно применяли для улучшения лечения нескольких В-клеточных злокачественных опухолей, он обладает ограниченной активностью в отношении контролирования ХЛЛ. Несмотря на начальное периферическое истощение в крови, эффективная опухолевая циторедукция во вторичных лимфоидных тканях и так называемых центрах пролиферации является менее успешной. Следовательно, чтобы лучше оценить противоопухолевую активность 6G11 против клеток ХЛЛ, мы разработали животную модель, в которой человеческие клетки первичного ХЛЛ вводят во вторичные лимфоидные органы (селезенку и костный мозг), где они пролиферируют в кластерах вместе с поддерживающими человеческими Т-клетками, тем самым близко имитируя ситуацию при человеческом заболевании (Фиг. 5 A и B). В этой модели ритуксимаб эффективен в отношении истощения клеток ХЛЛ, находящихся в брюшной полости, но не полностью эффективен в отношении истощения клеток ХЛЛ в центрах пролиферации, находящихся в селезенке (Фиг. 16).
Когда мышей с привитыми клетками первичного ХЛЛ обрабатывали только ритуксимабом или 6G11, наблюдали существенное снижение опухолевой массы в селезенке по сравнению с животными, обработанными изотипическим контрольным mAb (Фиг. 5C). Обработка комбинацией ритуксимаба и 6G11 приводила к еще более высоким уровням истощения (Фиг. 5C). Более того, когда эти данные оценивали в соответствии с критерием уровней ответов, число частичных и полных ответов после комбинированной терапии было существенно выше, чем в случае обработанных изотипическим контролем мышей или монотерапии (Фиг. 5D и 5E, соответственно).
Кроме того, число объективных (ОО; определяемый как >75% снижение числа клеток ХЛЛ в селезенке) и полных ответов (ПО; определяемый как <0,1 % клеток ХЛЛ в селезенке) после комбинированной терапии было существенно выше, чем в случае обработанных изотипическим контролем мышей или монотерапии (Фигуры 5 и 5(2/2), и Таблица 5). Эти данные демонстрируют повышенную эффективность комбинированной терапии ритуксимабом/6G11 против клеток первичного ХЛЛ in vivo.
Затем мы исследовали способность 6G11 лечить мышей с привитыми клетками ХЛЛ, выделенными из рефрактерных в отношении ритуксимаба, офатумумаба (hCD20 mAb) и/или алемтузумаба (hCD52 mAb) пациентов (Таблица 4). Ксенографтных мышей обрабатывали 6G11 или ритуксимабом в виде монотерапии, или двумя mAb в комбинации. Как и ожидалось, обработка одним ритуксимабом была неэффективной (Фигуры 5 и 5(2/2) и Таблица 8), и у >95% мышей не вырабатывался ОО. В противоположность этому, одно 6G11 демонстрировало существенное истощение клеток ХЛЛ, но не улучшало ОО (Фигуры 5 и 5(2/2) и Таблица 5). При этом примечательно, что совместное введение ритуксимаба с 6G11 приводило к робастному истощению (Фигуры 5 и 5(2/2) и Таблица 8) с >25% ОО (Таблица 5). Эти данные позволяют предположить, что, кроме усиления активности ритуксимаба у пациентов с ответом, 6G11 может быть активно против рефрактерных в отношении лечения клеток ХЛЛ.
Комбинированная терапия ритуксимабом и 6G11 преодолевает клетки рефрактерного ХЛЛ in vivo
После этого мы исследовали способность лечить мышей с привитыми клетками ХЛЛ, выделенными из рефрактерных в отношении ритуксимаба и/или алемтузумаба (hCD52 mAb) пациентов в присутствии или отсутствии ритуксимаба (Таблица 4). Как и ожидалось, истощение клеток одним ритуксимабом было неэффективным, но существенно улучшалось в присутствии 6G11 (Фиг. 5F). Более того, у большего числа мышей с привитыми клетками рефрактерного ХЛЛ, получавших комбинированную терапию, наблюдали частичные ответы (Фиг. 5G). Эти данные позволяют надеяться, что 6G11 может усиливать эффективность ритуксимаба как в чувствительных к ритуксимабу, так и в рефрактерных группах пациентов.
Таблица 4. Информация по пациентам, предыдущему лечению и ответам пациентов, клинически определенных как рефрактерные в отношении ритуксимаба. Она сравнивается с относительными ответами в in vivo модели, в которой число клеток ХЛЛ в селезенке обрабатываемых изотипическим контролем мышей принято за 100% +СО
|
Комбинированная терапия ритуксимабом и 6G11 обладает in vivo активностью против клеток первичной МКЛ
Чтобы оценить способность 6G11 нацеливаться на другие типы злокачественных В-клеток, мы использовали модели с иммунодефицитными мышами с привитыми клетками Jeko МКЛ или свежевыделенными клетками первичной МКЛ (Фиг. 17A и B, соответственно). Когда мышам прививали клетки Jeko МКЛ, монотерапия 6G11 или ритуксимабом не приводила к долгосрочной выживаемости, в то время как комбинация была эффективна и привела к излечению ~30% мышей на протяжении до 100 дней. Аналогично клеткам первичного ХЛЛ, клетки первичной МКЛ благоприятным образом отвечали на комбинированную терапию (Фиг. 17B).
6G11 хорошо переносится, имеет терапевтически релевантную фармакокинетику in vivo и не приводит к цитокиновой буре in vitro
Далее, чтобы убедиться в безопасности лечения 6G11 mAb, мы проводили исследование с увеличением дозы, обрабатывая группы мышей hFcγRIIB+/- x mFcγRII-/- однократным введением 1, 10 или 100 мг/кг 6G11, исследуя как в.в., так и в.б. пути введения при дозе 10 мг/кг (Фиг. 18A). Ни одна из обработанных мышей не страдала от нежелательных явлений, таких как острые эффекты, нарушения или потеря массы (Фиг. 17B). Исследование тканей на 7 день не выявило признаков какой-либо сильной токсичности в органах (почки, головной мозг, селезенка, печень, легкие) (данные не показаны). Истощение В-клеток hFcγRIIB+ve наблюдали как в крови (Фиг. 6A), так и в селезенке (Фиг. 6B) при дозах выше 1 мг/кг с эквивалентной активностью в группах, обрабатываемых 10 и 100 мг/кг, что указывает на эффективность. 6G11 представляет собой полностью человеческое mAb, легко выявляемое в сыворотке мышей, но иммуногенное по своей природе, поэтому мы одновременно оценивали его время полужизни и наличие признаков ответов со стороны мышиного античеловеческого антитела (MAHA) (Фиг. 6C и D). Эти данные свидетельствуют о том, что при дозах выше 1 мг/кг преодолевалось мишень-опосредованное выведение, которое влияет на ФК-профиль, а в группах, обрабатываемых 10 и 100 мг/кг, наблюдали слабое или отсутствующее влияние связывания мишеней. Однако в течение 7 дней наблюдали значительное количество MAHA (увеличившееся на 4 порядка между 4 и 7 днями в группе, обрабатываемой 10 мг/кг в.в.; Фигура 6D), что приводило к быстрому выведению mAb. На основании первых 4 дней после введения до появления значительного количества MAHA мы оценили, что время ½ для антитела должно соответствовать диапазону в 2-4 дня для доз 10 и 100 мг/кг (Фиг. 6C и D и Таблица 3).
Также мы провели исследование с повторным дозированием для лучшей имитации того, как 6G11 можно доставлять в клинических условиях, вводя его в течение 10-дневного периода (первое в.в. введение, за которым следуют 3 дозы, доставляемые в.б.; Фиг. 17C), при этом животных проверяли в течение всего времени и на 14 дней позже (всего 24 дня получения mAb) в отношении каких-либо признаков нарушений, потери массы, токсичности или патологий. Как и раньше, истощение циркулирующих В-клеток наблюдали в группе hFcγRIIB+/- x mFcγRII-/-, но не в контрольной группе mFcγRII-/-, инъекции проводили многократными дозами (10 мг/кг) 6G11 (Фиг. 6E). Аналогично, мыши не страдали от потери массы (Фиг. 17B), не страдали от нежелательных явлений, а также не наблюдались признаки сильной токсичности (Фиг. 17D; данные не показаны). Как и раньше, у мышей hFcγRIIB+/- x mFcγRII-/- наблюдали значительные ответы со стороны MAHA в течение 1 недели, которые приводили к быстрому выведению 6G11 из сыворотки (Фиг. 6F). Интересно, что в контрольной группе mFcγRII-/- не было выявлено MAHA, что указывает на то, что наличие поверхностного антигена необходимо для индукции MAHA (Фиг. 6F).
Наконец, мы использовали недавно разработанный in vitro анализ синдрома высвобождения цитокинов (СВЦ), чтобы оценить потенциальное влияние 6G11 на человеческие МКПК, которые сделали чувствительными к стимуляции посредством культивирования при высокой плотности.28 Эта аналитическая система способна выявлять значительные уровни ИФН-γ, ФНО-α и/или ИЛ-8 после добавления нескольких специфичностей mAb (CD3, CD28 или CD52), ранее демонстрировавших, что могут вызывать СВЦ (Фигура 6F и работа на стадии подготовки). Применение в этом анализе ДТ или N297Q 6G11 в течение 48 ч не привело к значительному высвобождению цитокинов, в отличие от в 500 раз меньших доз CD3 mAb (Фиг. 6G). Аналогичные результаты были получены при проведении анализа цитокинов в цельной крови (Фиг. 18), что вместе свидетельствует о хорошем профиле безопасности для 6G11 перед проведением испытаний на людях.
6G11 усиливает терапевтическую активность других клинически релевантных антител in vivo
Недавние наблюдения свидетельствуют о том, что FcγRIIB-зависимая интернализация может лежать в основе устойчивости к нескольким клинически релевантным антителам помимо ритуксимаба (Vaughan et al., 2014); (Pallasch et al., 2014). Поэтому мы исследовали комбинацию 6G11 с недавно утвержденным hCD20 mAb обинутузумабом (GA101) и клинически сертифицированным, специфическим к hCD52 mAb алемтузумабом. Специфичность обинутузумаба в отношении hCD20 позволила нам исследовать его действие в сингенной мышиной модели. Монотерапия GA101 и 6G11 приводила к умеренному истощению В-клеток в селезенке и циркуляции, в то время как комбинация значительно усиливала истощение у мышей ДТ (Фигура 5(2/2) и hFcγRIIB+/- x mFcγRII-/- (Фигура 6F и S6G). Комбинирование 6G11 и GA101 значительно усиливало истощение опухолевых клеток селезенки в мышиной модели с ксенотрансплантатами, полученными от пациентов с ХЛЛ (Фигура 5(2/2) и Таблицы 5 и 8). Хотя специфичность алемтузумаба в отношении hCD52 исключала исследование на сингенной модели hCD20, наблюдалось значительное улучшение терапевтической активности, когда 6G11 и алемтузумаб комбинировали в ХЛЛ-мышиной модели, когда у >90% обрабатываемых комбинацией мышей развивался ОО (Фигура 5(2/2) и Таблицы 5 и 8).
Эти данные свидетельствуют о том, что 6G11 может преодолевать устойчивость к лекарственным препаратам mAb для многих мишеней.
Таблица 5: Ответы на лечение mAb в ксенотрансплантатах от пациентов с первичным ХЛЛ
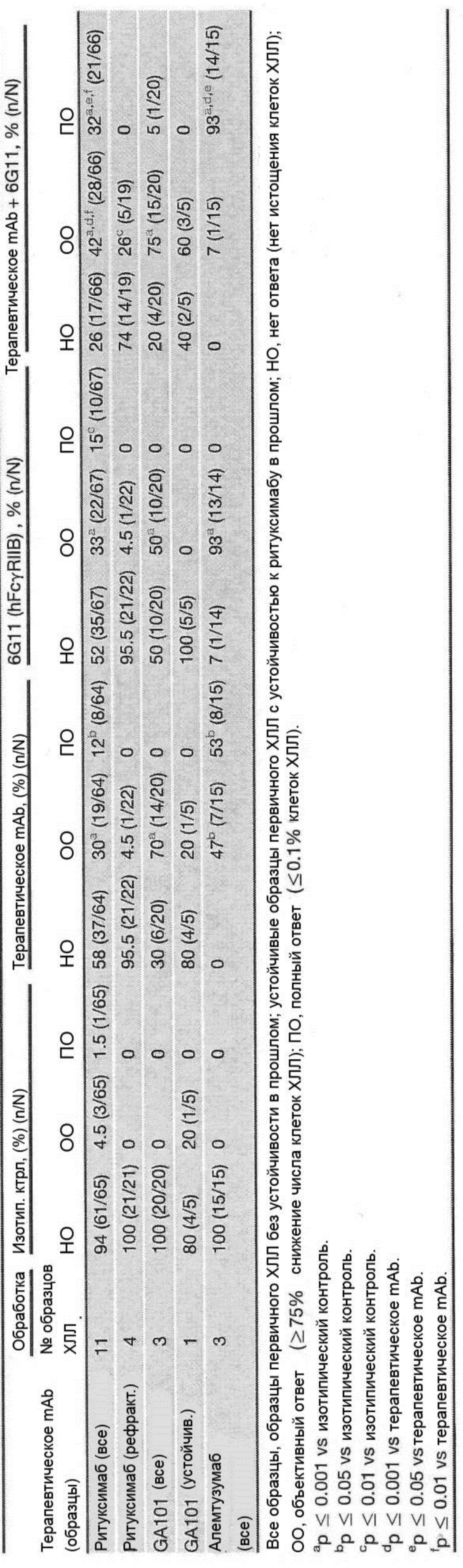
Таблица 6: связана с Фигурой 1. Определение аффинности hFcγRIIA и hFcγRIIB mAb методом ELISA и/или ППР
|
a нс: нет связывания,
b но: не определено.
Таблица 7, связана с Фигурой 14. Оценка фармакокинетических (ФК) параметров ДТ 6G11 mAb у мышей hFcγRIIB+/- x mFcγRII-/-
|
a время полужизни mAb,
b Объем распределения, связанный с терминальной фазой.
c Объем распределения в стационарном состоянии (стационарное состояние, состояние равновесия, получаемое в конце определенного количества введений),
d Выведение; объем очищенной плазмы (т.е. более не содержащей каких-либо рассматриваемых лекарственных препаратов) в единицу времени,
e Площадь под кривой; соответствует интегралу от плазменной концентрации лекарственного препарата за конечный интервал времени,
f Пиковая плазменная концентрация лекарственного препарата после введения,
g Время до достижения Cмакс.
Таблица 8, связанная с Фигурами 5 и 5(2/2). Детализированные р-величины, анализируемые при помощи пермутационного статистического критерия, для сравнения обработки разными mAb в in vivo моделях с полученными от пациентом ксенотрансплантатами
Ксенотрансплантаты от любых пациентов с ХЛЛ
|
Ксенотрансплантаты от пациентов с рефрактерным в отношении ритуксимаба ХЛЛ
|
Ксенотрансплантаты от пациентов с МКЛ
|
НД: нет данных.
Обсуждение
Раковые клетки являются сильно пролиферирующими, геномно нестабильными по своей природе и имеют высокую предрасположенность к мутациям. Эти аспекты обеспечивают идеальную обстановку для клеточной эволюции под влиянием набора традиционных противораковых лекарственных препаратов, когда генетически отличные, устойчивые к лечению клоны становятся основной причиной неэффективности лечения. Эта способность четко прослеживается в механизмах устойчивости, которые развиваются против направленной на повреждение ДНК химиотерапии38 и низкомолекулярных ингибиторов, нацеленных на разные сигнальные пути, таких как иматиниб, эрлотиниб и ибрутиниб.39-43 Такие механизмы включают мутации в рецепторах-мишенях или компенсаторные изменения в нижеследующих сигнальных белках.44 Другие включают множественную лекарственную устойчивость, достигаемую путем повышающей регуляции эффлюксных насосов (обзор приведен в 45). Неэффективность традиционных и нацеленных низкомолекулярных ингибиторов привела к поиску средств для преодоления этих типов устойчивости, а также альтернативных видов терапии, включая те, которые задействуют иммунную систему.
На сегодняшний день моноклональные антитела (mAb) являются наиболее успешными образцами этого подхода, при этом лидирующими в данной области являются Ab, применяемые при гематологических нарушениях. В-клеточные раки представляют наиболее клинически изученный тип рака в отношении терапии антителами. После утверждения ритуксимаба в 1997 г. и недавнего утверждения второго (офатумумаб) и третьего (обинутузумаб) поколения hCD20 mAb эти агенты стали основой медицинского обеспечения для лечения В-клеточных раков. Однако понятно, что иммунотерапия mAb также подвержена как первичной, так и приобретенной устойчивости. На данный момент установлено, что опухоли влияют на свое микроокружение путем разрушения стромальных и миелоидных клеток для поддержания своего выживания и роста.46 По меньшей мере два механизма устойчивости, связанные с терапией mAb, вызваны ингибиторным FcγRIIB (обзор приведен в 47). Кроме ингибиторного действия на эффекторные клетки, описанного Clynes5, недавно было показано, что ритуксимаб и другие hCD20 mAb типа I задействуют hFcγRIIB посредством сшивки биполярным антителом на поверхности В-клетки, что приводит к интернализации комплекса mAb:CD20:FcγRIIB 13,23,48, тем самым ограничивая его способность задействовать важные Fc-зависимые эффекторные функции (23 и Tipton et al., не опубликовано). В данном примере описано получение hFcγRIIB mAb, которые способны блокировать оба этих механизма устойчивости и, таким образом, способны раскрывать полный потенциал других терапевтических mAb и помогать преодолевать устойчивость к терапии антителами in vivo.
Неожиданно, совместное введение hFcγRIIB mAb не только улучшает объективные и полные ответы мышей с привитыми клетками ХЛЛ от восприимчивых к ритуксимабу пациентов, но также преодолевает устойчивый к лечению mAb фенотип клеток ХЛЛ от рецидивных/рефрактерных пациентов, устойчивых к ритуксимабу или нацеленному на hCD52 mAb алемтузумабу.
Недавно было сделано предположение, что роль FcγRIIB опухолевых клеток состоит в устойчивости к терапии hCD52 mAb в избранном микроокружении (Pallasch et al., 2014). Повышенная регуляция hFcγRIIB была обнаружена в устойчивых к алемтузумабу лейкозных В-клетках костного мозга по сравнению с более восприимчивыми компартментами селезенки, а опосредованный кшРНК нокдаун hFcγRIIB улучшал терапию hCD52 mAb в устойчивой в ином случае ткани. Эти открытия согласуются с наблюдениями в нашей модели ХЛЛ. Хотя клетки ХЛЛ в восприимчивых компартментах брюшной полости легко уничтожались hCD20 mAb, уничтожение в устойчивом микроокружении требует блокирования опухолевого FcγRIIB. В соответствии с высокой экспрессией FcγRIIB, лежащей в основе снижения активности mAb в устойчивых тканевых компартментах, блокирование FcγRIIB усиливало действие алемтузумаба, а также hCD20 mAb типа II в нашей модели ХЛЛ. Ранее мы показали, что интернализация hCD20 mAb типа II эффективно происходит только в присутствии высоких уровней FcγRIIB (Vaughan et al., 2014). Вместе эти наблюдения демонстрируют, что FcγRIIB-опосредованная устойчивость касается нескольких разных клинически утвержденных антител, что указывает на широкий терапевтический потенциал комбинирования с hFcγRIIB mAb.
Ранее было показано, что CD20 mAb типа I проявляют намного большую тенденцию к интернализации опухолевыми клетками-мишенями, чем тип II 13,23,33, причем интернализация последних проходит эффективно только в присутствии высоких уровней FcγRIIB.33 Следовательно, приведенные здесь данные о том, что связанная с истощением В-клеток активность антител CD20 типа II так же само улучшается путем совместной обработки 6G11 in vivo, являются многообещающими. Hemann и его сотрудники недавно выяснили роль FcγRIIB опухолевых клеток в устойчивости к терапии hCD52 mAb в избранном микроокружении.49 Повышенная регуляция FcγRIIB была обнаружена в устойчивом к алемтузумабу костном мозге по сравнению с более восприимчивыми компартментами селезенки, а опосредованный миРНК нокдаун FcγRIIB в лейкозных клетках улучшал терапевтическое действие hCD52 mAb в устойчивой в ином случае ткани. Эти открытия согласуются с наблюдениями в нашей модели ХЛЛ. Хотя одиночные клетки ХЛЛ в восприимчивых компартментах селезенки легко уничтожались hCD20 mAb, уничтожение кластеризованных пролиферирующих клеток ХЛЛ в устойчивом микроокружении требует блокирования опухолевого FcγRIIB. Важно отметить, что эти наблюдения (данный документ и 33) демонстрируют, что FcγRIIB-опосредованная устойчивость касается нескольких разных клинически утвержденных антител и подтвержденных мишеней, что указывает на широкий терапевтический потенциал комбинирования с hFcγRIIB mAb.
Устойчивость к терапии антителами наблюдают при нескольких типах рака. Среди В-клеточных раков ХЛЛ и МКЛ хуже лечатся схемами, включающими применение hCD20 mAb, по сравнению с ФЛ и ДКВКЛ, что свидетельствует о присущих первым механизмах устойчивости. Кроме того, отдельные пациенты с ФЛ и ДКВКЛ демонстрируют первичную устойчивость к терапии hCD20 mAb, а у части пациентов с разными видами В-клеточного рака, изначально восприимчивых к включающей применение антител иммунной терапии, развивается устойчивость, а лечение более не оказывает благоприятное воздействие. Предыдущие наблюдения (32,14 и неопубликованные данные) указывают на то, что FcγRIIB-опосредованная интернализация антител может быть общим механизмом, лежащим в основе устойчивости этих разных В-клеточных раков и индивидов. В данном документе мы представляем данные о том, что совместное введение с hFcγRIIB mAb усиливает противоопухолевую активность ритуксимаба против опухолевых клеток ХЛЛ и МКЛ in vivo, что согласуется с наблюдением, что FcγRIIB-опосредованная интернализация антител является терапевтически релевантным механизмом устойчивости, общим для разных В-клеточных раков.
Этот Пример дополнительно демонстрирует, что hFcγRIIB mAb обладает свойственной ему противоопухолевой активностью. Ранее Veri et al. разработали hFcγRIIB-специфические mAb, но не исследовали их активность против раковых мишеней in vivo.50 Впоследствии Rankin et al. показали, что нацеливание на hFcγRIIB на злокачественных человеческих В-клетках может быть эффективным в качестве монотерапии. Однако в этом исследовании36 использовали иммунодефицитные ксенографтные системы, в которых вследствие отсутствия перекрестной реактивности Ab с мышиным рецептором антиген-мишень экспрессировался только на опухоли, но не на важных иммунных эффекторных клетках, исключая возможность оценить суммарное действие hFcγRIIB mAb. В настоящей работе, используя два разных клона mAb, один полностью человеческий, полученный при помощи фагового дисплея, а один мышиный, полученный при помощи традиционной технологии гибридомы, мы продемонстрировали в иммунокомпетентных мышиных моделях, в которых hFcγRIIB экспрессируется как на В-клетках-мишенях, так и на эффекторах, что антагонистические антитела hFcγRIIB обладают свойственной им противоопухолевой активностью. Важно отметить, что используя вдвойне трансгенных мышей, экспрессирующих hCD20 и hFcγRIIB, мы дополнительно продемонстрировали, что блокирование mAb опосредованной hFcγRIIB интернализации усиливает терапевтическую активность ритуксимаба, когда обе мишени экспрессируются в иммунокомпетентных хозяевах клеточно- или тканеспецифическим образом аналогично тому, как это происходит в организме человека. Интересно, что в данном эксперименте наблюдался явно выраженный и очевидный синергетический эффект совместного введения антагонистических hFcγRIIB mAb и CD20 mAb (ритуксимаба или обинутузумаба). Можно предположить, что причиной повышения активности является блокирование mAb иммунной подавляющей функции hFcγRIIB в эффекторных клетках, что во многом сходно с тем, что наблюдалось при генетическом удалении hFcγRIIB в сходных иммунокомпетентных моделях.6,7 В любом случае и в соответствии с осуществлением FcγRIIB-опосредованного ингибирования и предотвращением интернализации ритуксимаба (вместо прямого нацеливания), которые являются основным механизмом повышения терапевтической активности ритуксимаба со стороны hFcγRIIB mAb, мы обнаружили, что уничтожение В-клеток было менее эффективным в случае AT10 по сравнению с одним 6G11 (возможно, вследствие разницы в изотипе), но комбинация AT10 и ритуксимаба была так же эффективна в повышении активности ритуксимаба. Следует отметить, что долгое время считалось, что ХЛЛ субоптимально лечится ритуксимабом51 и другими анти-CD20 mAb типа I 52,53, для чего необходимы более высокие дозы. Вместе с нашими предыдущими данными13 наши новые результаты свидетельствуют о том, что комбинированная терапия hFcγRIIB mAb может не только быть способом предотвращения устойчивости, но, возможно, также и способом снижения дозы hCD20 mAb, необходимой в этих случаях.
Таким образом, в общую in vivo терапевтическую активность 6G11 могут делать вклад по меньшей мере три разных механизма: первичная цитотоксичность; предотвращение интернализации терапевтического mAb; и нейтрализация FcγRIIB-ингибиторной сигнализации в иммунных клетках. Несколько наблюдений дают основание предположить, что блокирование интернализации и ингибиторной сигнализации рецепторов является наиболее важным для преодоления лекарственной устойчивости. Во-первых, наблюдался явно выраженный и очевидный синергетический эффект при совместном введении мышам антагонистического hFcγRIIB и hCD20 mAb в случае, когда hFcγRIIB экспрессировался как на клетках-мишенях, так и на эффекторных клетках. Во-вторых, мы обнаружили, что хотя уничтожение В-клеток со стороны одного AT10 было неэффективным, комбинация AT10 и ритуксимаба эффективно повышала активность ритуксимаба. И наконец, наши данные по N297Q hIgG1 6G11, у которого отсутствует способность задействовать активаторный FcγR и которое не обладает непосредственной цитотоксической активностью, подтверждают, что блокирование активности FcγRIIB является ключевым механизмом, обеспечивающим эффективность такого подхода. Это свидетельствует о том, что блокирующие функции mAb к FcγRIIB могут воспроизводить усиленные противораковые ответы mAb, наблюдаемые после генетического удаления FcγRIIB (Clynes et al., 2000). Однако мы не исследовали прямым образом относительную важность для активности антагонизирования функции FcγRIIB на клетках-мишенях в сравнении с эффеторными клетками; такие исследования образуют базис нашей текущей работы.
Следует отметить, что долгое время считалось, что ХЛЛ субоптимально лечится ритуксимабом (O'Brien et al., 2001) и офатумумабом (Coiffier et al., 2008; Coiffier et al., 2006), для чего необходимы более высокие дозы. Вместе с нашими предыдущими данными наши настоящие результаты свидетельствуют о том, что комбинированная терапия hFcγRIIB mAb может не только быть способом предотвращения устойчивости, но, возможно, также и способом снижения дозы или уменьшения длительности терапии hCD20 mAb.
Кроме обладания значительной активностью терапевтическое mAb также должно хорошо переноситься и иметь терапевтически релевантную фармакокинетику (ФК). Очень высокая специфичность 6G11 в отношении hFcγRIIB с отсутствием связывания с 98 % гомологичного человеческого hFcγRIIA и пренебрежимо малой перекрестной реактивностью с животными видами, обычно применяемыми в токсикологических исследованиях, побудили нас исследовать параметры безопасности и ФК/ФД для мышей, экспрессирующих человеческий hFcγRIIB на уровнях и в клеточных типах и тканях, аналогичных человеческим. Эти исследования показали, что 6G11 хорошо переносился, а животные не демонстрировали признаков нарушений, потери массы, токсичности или патологий. Эксперименты с титрованием доз продемонстрировали, что дозы, составляющие 10 мг/кг или более, являются достаточными для насыщения hFcγRIIB in vivo, что приводит к эквивалентному истощению В-клеток, и способны поддерживать долговременное блокирование или устранение рецептора. В противоположность этому, доза, составляющая 1 мг/кг, была субоптимальной, быстро выводилась и существенно не уничтожала В-клетки селезенки. При дозах, превышающих насыщение рецептора, терминальное время полужизни 6G11 у мышей было оценено в 2-4 дня. Хотя отсутствие перекрестной реактивности с отличными от человека видами приматов исключало межвидовой пересчет, эти фигуры свидетельствуют о терапевтически релевантном ФК-профиле, типичном для hIgG1 mAb со временем полужизни порядка недель у человека.
Интересно, что как в человеческой крови, так и in vivo в организме трансгенных мышей hFcγRIIB обработка 6G11 приводила к специфическому уничтожению В-клеток. Хотя моноциты в обеих системах экспрессируют hFcγRIIB, они не были существенно уничтожены. Имеющиеся на данный момент сведения позволяют предположить, что макрофаги и/или моноциты являются ключевыми эффекторными клетками, ответственными за терапию mAb (Beers et al., 2010); (Biburger et al., 2011; Gul et al., 2014), и, таким образом, наши данные свидетельствуют о том, что ключевые эффекторы не уничтожаются hFcγRIIB mAb.
Ранее мы исследовали эквивалентную панель антимышиных FcγRII-специфических mAb25 и наблюдали, что они имеют ограниченное терапевтическое действие вследствие их быстрого поглощения in vivo, главным образом, неопухолевыми клетками организма-хозяина.17 Важно, что сходные уровни интернализации не наблюдали с человеческими клетками-мишенями, по меньшей мере in vitro, что согласуется с более ранними исследованиями.36 В данной работе мы расширили эти наблюдения и продемонстрировали, что то же самое наблюдалось для образцов первичного человеческого ХЛЛ, и что у мышей, экспрессирующих трансген hFcγRIIB, не наблюдалось быстрого и интенсивного поглощения mAb. Эти данные подтверждают наше более раннее предположение, что мышиный и человеческий ингибиторные FcγRII(B) имеют разные свойства в отношении их способности к интернализации и функционирования в качестве антигенного накопителя, и позволяют предположить, что hFcγRIIB mAb, такие как 6G11, будут эффективны в организме человека.
В целом, этот Пример демонстрирует in vivo подтверждение концепции того, что hFcγRIIB mAb преодолевают первичную и приобретенную устойчивость опухолевых клеток к препаратам mAb; преодолевая клетки рецидивного/рефрактерного ХЛЛ. Наши данные способствуют клинической разработке hFcγRIIB mAb для терапии FcγRIIB-экспрессирующих В-клеточных раков.
Эти данные способствуют клинической разработке hFcγRIIB mAb для терапии FcγRIIB-экспрессирующих В-клеточных раков. Кроме того, аналогично распространению CD20 mAb на другие заболевания, есть основания предполагать их применимость в других терапевтических схемах, например, в случае аутоиммунности, когда FcγRIIB-экспрессирующие мишени могут быть подвержены манипуляциям, например, при системном амилоидозе легкой цепи, когда PC-мишени экспрессируют высокие уровни FcγRIIB (Zhou et al., 2008), и ревматоидном артрите, когда активацию В-клеток можно снизить посредством агонизма FcγRIIB (Baerenwaldt et al., 2011; Mauri and Jury, 2010).
Ссылки
1. Reichert, J.M. & Dhimolea, E. The future of antibodies as cancer drugs. Drug Discov Today 17, 954-963 (2012).
2. Nimmerjahn, F. & Ravetch, J.V. FcgammaRs in health and disease. Curr Top Microbiol Immunol 350, 105-125 (2011).
3. Nimmerjahn, F. & Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nature reviews 8, 34-47 (2008).
4. Chao, M.P., et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699-713 (2010).
5. Clynes, R.A., Towers, T.L., Presta, L.G. & Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nature medicine 6, 443-446 (2000).
6. Minard-Colin, V., et al. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood 112, 1205-1213 (2008).
7. Hamaguchi, Y., Xiu, Y., Komura, K., Nimmerjahn, F. & Tedder, T.F. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. The Journal of experimental medicine 203, 743-753 (2006).
8. Beers, S.A., et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191-5201 (2010).
9. Dyer, M.J., Hale, G., Hayhoe, F.G. & Waldmann, H. Effects of CAMPATH-1 antibodies in vivo in patients with lymphoid malignancies: influence of antibody isotype. Blood 73, 1431-1439 (1989).
10. Weng, W.K. & Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 21, 3940-3947 (2003).
11. Cartron, G., et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99, 754-758 (2002).
12. Lim, S.H., et al. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica 95, 135-143 (2010).
13. Lim, S.H., et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood (2011).
14. Lee, C.A.-K., Margaret; Cogliatti, Sergio; Crowe, Susanne; Cragg, Mark S; Schmitz, Shu-Fang H; Ghielmini, Michele and Peter W Johnson. Expression of Inhibitory Fc Receptor (FcgRIIB) Is a Marker of Poor Response to Rituximab Monotherapy in Follicular Lymphoma (FL). ASH abstract 50396 (2012).
15. Beers, S.A., et al. Type II (tositumomab) anti-CD20 monoclonal antibody out performs type I (rituximab-like) reagents in B-cell depletion regardless of complement activation. Blood 112, 4170-4177 (2008).
16. Roghanian, A., et al. Filament-associated TSGA10 protein is expressed in professional antigen presenting cells and interacts with vimentin. Cellular immunology 265, 120-126 (2010).
17. Williams, E.L., et al. Immunotherapy Targeting Inhibitory Fcgamma Receptor IIB (CD32b) in the Mouse Is Limited by Monoclonal Antibody Consumption and Receptor Internalization. J Immunol 191, 4130-4140 (2013).
18. Glennie, M.J., McBride, H.M., Worth, A.T. & Stevenson, G.T. Preparation and performance of bispecific F(ab' gamma)2 antibody containing thioether-linked Fab' gamma fragments. J Immunol 139, 2367-2375 (1987).
19. Greenman, J., et al. Characterization of a new monoclonal anti-Fc gamma RII antibody, AT10, and its incorporation into a bispecific F(ab')2 derivative for recruitment of cytotoxic effectors. Mol Immunol 28, 1243-1254 (1991).
20. Tutt, A.L., et al. Monoclonal antibody therapy of B cell lymphoma: signaling activity on tumor cells appears more important than recruitment of effectors. J Immunol 161, 3176-3185 (1998).
21. Olsson, N., et al. Proteomic analysis and discovery using affinity proteomics and mass spectrometry. Mol Cell Proteomics 10, M110 003962 (2011).
22. Nishimura, T., et al. Characterization of the human Fc gamma RIIB gene promoter: human zinc-finger proteins (ZNF140 and ZNF91) that bind to different regions function as transcription repressors. Int Immunol 13, 1075-1084 (2001).
23. Beers, S.A., et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191-5201 (2010).
24. Binyamin, L., et al. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol 180, 6392-6401 (2008).
25. Williams, E.L., et al. Development and characterisation of monoclonal antibodies specific for the murine inhibitory FcgammaRIIB (CD32B). European journal of immunology (2012).
26. Walshe, C.A., et al. Induction of cytosolic calcium flux by CD20 is dependent upon B Cell antigen receptor signaling. The Journal of biological chemistry 283, 16971-16984 (2008).
27. Beers, S.A., Chan, C.H., French, R.R., Cragg, M.S. & Glennie, M.J. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin Hematol 47, 107-114 (2010).
28. Romer, P.S., et al. Preculture of PBMCs at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood 118, 6772-6782 (2011).
29. Williams, E.L., et al. Development and characterisation of monoclonal antibodies specific for the murine inhibitory FcgammaRIIB (CD32B). European journal of immunology 42, 2109-2120 (2012).
30. Hallborn, J. & Carlsson, R. Automated screening procedure for high-throughput generation of antibody fragments. Biotechniques Suppl, 30-37 (2002).
31. Cragg MS, A.A., O'Brien L, Tutt A, Chan HTC, Anderson VA, Glennie MJ. Opposing properties of CD20 mAb. in Leukocyte Typing VII 95-97 (Oxford University Press, Oxford, 2002).
32. Lim, S.H., et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood 118, 2530-2540 (2011).
33. Vaughan, A.T., et al. Inhibitory FcgammaRIIb (CD32b) becomes activated by therapeutic mAb in both cis and trans and drives internalization according to antibody specificity. Blood 123, 669-677 (2014).
34. Tao, M.H. & Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol 143, 2595-2601 (1989).
35. Montalvao, F., et al. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. The Journal of clinical investigation (2013).
36. Rankin, C.T., et al. CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood 108, 2384-2391 (2006).
37. Hu, C.Y., et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. The Journal of clinical investigation 117, 3857-3867 (2007).
38. Rossi, D., et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood (2014).
39. Zhang, J., Yang, P.L. & Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9, 28-39 (2009).
40. Pao, W., et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2, e73 (2005).
41. Yun, C.H., et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences of the United States of America 105, 2070-2075 (2008).
42. Shah, N.P., et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117-125 (2002).
43. Corbin, A.S., Buchdunger, E., Pascal, F. & Druker, B.J. Analysis of the structural basis of specificity of inhibition of the Abl kinase by STI571. The Journal of biological chemistry 277, 32214-32219 (2002).
44. Engelman, J.A., et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (New York, N.Y 316, 1039-1043 (2007).
45. Cragg, M.S., Harris, C., Strasser, A. & Scott, C.L. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 9, 321-326 (2009).
46. Hanahan, D. & Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 144, 646-674 (2011).
47. Williams, E.L., et al. Overcoming resistance to therapeutic antibodies by targeting Fc Receptors. Resistance to Immunotherapeutic Antibodies in Cancer: Strategies to Overcome Resistance. Pubs. Springer In Press(2013).
48. Vaughan, A.T., et al. Inhibitory FcγRIIb (CD32b) becomes activated by therapeutic mAb in both cis and trans and drives internalization according to antibody specificity. Blood (2013).
49. Pallasch, C.P., et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 156, 590-602 (2014).
50. Veri, M.C., et al. Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology 121, 392-404 (2007).
51. O'Brien, S.M., et al. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol 19, 2165-2170 (2001).
52. Coiffier, B., et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1-2 study. Blood 111, 1094-1100 (2008).
53. Coiffier, B., et al. Significant Correlation between Survival Endpoints and Exposure to Ofatumumab (HuMax-CD20) in Chronic Lymphocytic Leukemia. ASH Annual Meeting Abstracts 108, 2842- (2006).
54. Shawn Rose, Alexander Misharin, Harris Perlman, 2012, A novel Ly6c/Ly6G-based strategy to analyse the mouse splenic myeloid compartment, Cytometry Part A 81(4): 343-50
55. Beers, S. A., French, R. R., Chan, H. T., Lim, S. H., Jarrett, T. C., Vidal, R. M., Wijayaweera, S. S., Dixon, S. V., Kim, H., Cox, K. L., et al. (2010). Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191-5201.
56. Biburger, M., Aschermann, S., Schwab, I., Lux, A., Albert, H., Danzer, H., Woigk, M., Dudziak, D., and Nimmerjahn, F. (2011). Monocyte subsets responsible for immunoglobulin G-dependent effector functions in vivo. Immunity 35, 932-944.
57. Coiffier, B., Lepretre, S., Pedersen, L. M., Gadeberg, O., Fredriksen, H., van Oers, M. H., Wooldridge, J., Kloczko, J., Holowiecki, J., Hellmann, A., et al. (2008). Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1-2 study. Blood 111, 1094-1100.
58. Coiffier, B., Tilly, H., Pedersen, L. M., Plesner, T., Frederiksen, H., van Oers, M. H. J., Wooldridge, J., Kloczko, J. S., Holowiecki, J., Hellmann, A., et al. (2006). Significant Correlation between Survival Endpoints and Exposure to Ofatumumab (HuMax-CD20) in Chronic Lymphocytic Leukemia. ASH Annual Meeting Abstracts 108, 2842-.
59. Cragg, M. S., Morgan, S. M., Chan, H. T., Morgan, B. P., Filatov, A. V., Johnson, P. W., French, R. R., and Glennie, M. J. (2003). Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101, 1045-1052.
60. Gul, N., Babes, L., Siegmund, K., Korthouwer, R., Bogels, M., Braster, R., Vidarsson, G., Ten Hagen, T. L., Kubes, P., and van Egmond, M. (2014). Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. The Journal of clinical investigation 124, 812-823.
61. Hamaguchi, Y., Xiu, Y., Komura, K., Nimmerjahn, F., and Tedder, T. F. (2006). Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. The Journal of experimental medicine 203, 743-753.
62. Hussain, K., Hargreaves, C. E., Roghanian, A., Oldham, R. J., Chan, H. T., Mockridge, C. I., Chowdhury, F., Frendeus, B., Harper, K. S., Strefford, J. C., et al. (2015). Upregulation of FcgammaRIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood 125, 102-110.
63. Lim, S. H., Vaughan, A. T., Ashton-Key, M., Williams, E. L., Dixon, S. V., Chan, H. T., Beers, S. A., French, R. R., Cox, K. L., Davies, A. J., et al. (2011). Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood 118, 2530-2540.
64. O'Brien, S. M., Kantarjian, H., Thomas, D. A., Giles, F. J., Freireich, E. J., Cortes, J., Lerner, S., and Keating, M. J. (2001). Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol 19, 2165-2170.
65. Pallasch, C. P., Leskov, I., Braun, C. J., Vorholt, D., Drake, A., Soto-Feliciano, Y. M., Bent, E. H., Schwamb, J., Iliopoulou, B., Kutsch, N., et al. (2014). Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 156, 590-602.
66. Romer, P. S., Berr, S., Avota, E., Na, S. Y., Battaglia, M., ten Berge, I., Einsele, H., and Hunig, T. (2011). Preculture of PBMCs at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood 118, 6772-6782.
67. Vaughan, A. T., Iriyama, C., Beers, S. A., Chan, C. H., Lim, S. H., Williams, E. L., Shah, V., Roghanian, A., Frendeus, B., Glennie, M. J., and Cragg, M. S. (2014). Inhibitory FcgammaRIIb (CD32b) becomes activated by therapeutic mAb in both cis and trans and drives internalization according to antibody specificity. Blood 123, 669-677.
Пример 2 - Типовые фармацевтические лекарственные формы
Хотя композицию и/или антитело, и/или агент, и/или медикамент согласно изобретению можно вводить сами по себе, предпочтительно, чтобы они были представлены в виде фармацевтической лекарственной формы вместе с одним или более приемлемыми носителями. Носитель(и) должен(ны) быть «приемлемым(и)» в смысле совместимости с композицией и/или антителом, и/или агентом, и/или медикаментом согласно изобретению и не вредными для реципиентов. Как правило, носителем является вода или солевой раствор, которые должны быть стерильными и апирогенными.
Следующие примеры иллюстрируют медикаменты и фармацевтические композиции в соответствии с изобретением, в которых активным ингредиентом является молекула антитела и/или агент согласно изобретению.
{PRIVATE}
Пример A: Таблетка
|
Таблетки готовят из вышеуказанных ингредиентов путем влажной грануляции с последующей компрессией.
Пример B: Офтальмологический раствор
|
Пример C: Таблеточные формы
Следующие лекарственные формы, А и В, готовят путем влажной грануляции ингредиентов с раствором повидона с последующим добавлением стеарата магния и компрессией.
Форма A
|
Форма B
|
Форма C
|
Следующие лекарственные формы, D и E, готовят путем прямой компрессии смешиваемых ингредиентов. Лактоза, применяемая в форме Е, принадлежит направляющему компрессионному типу.
Форма D
|
Форма E
|
Форма F (Форма с контролируемым высвобождением)
Данную лекарственную форму готовят путем влажной грануляции ингредиентов (ниже) с раствором повидона с последующим добавлением стеарата магния и компрессией.
|
|
|
Высвобождение лекарства происходит в течение периода, составляющего около 6-8 часов, и завершается через 12 часов.
Пример D: Капсульные формы
Форма A
Капсульную лекарственную форму готовят, смешивая ингредиенты Формы D в Примере C, выше, и помещая их в двухсоставную жесткую желатиновую капсулу. Форму B (infra) готовят сходным способом.
Форма B
|
Форма C
|
Капсулы готовят, расплавляя макрогол 4000 BP, диспергируя активный ингредиент в расплавленном веществе и наполняя расплавом двухсоставную жесткую желатиновую капсулу.
Форма D
|
Капсулы готовят, диспергируя активный ингредиент в лецитине и арахисовом масле и наполняя дисперсией мягкие, эластичные желатиновые капсулы.
Форма E (Капсула с контролируемым высвобождением)
Следующую капсульную форму с контролируемым высвобождением готовят, экструдируя ингредиенты a, b и c при помощи экструдера с последующим окатыванием экструдата и сушкой. Затем сухие пеллеты покрывают контролирующей высвобождение мембраной (d) и наполняют двухсоставную жесткую желатиновую капсулу.
|
Пример E: Инъецируемая форма
|
Активный ингредиент растворяют в большей части фосфатного буфера (35-40°C), затем доводят до необходимого объема и фильтруют через стерильный микропористый фильтр в стерильный 10 мл флакон из желтого стекла (тип 1) и запечатывают стерильной крышкой и колпачком.
Пример F: Внутримышечная инъекция
|
Активный ингредиент растворяют в гликофуроле. Затем добавляют и растворяют бензиловый спирт, а воду добавляют до 3 мл. Затем смесь фильтруют через стерильный микропористый фильтр и запечатывают в стерильных 3 мл стеклянных флаконах (тип 1).
Пример G: Сироп-суспензия
|
Бензоат натрия растворяют в части очищенной воды и добавляют раствор сорбита. Добавляют и диспергируют активный ингредиент. В глицероле диспергируют загуститель (диспергируемую целлюлозу). Смешивают две дисперсии и доводят до необходимого объема очищенной водой. При необходимости дополнительное загустевание достигают путем добавочного разделения суспензии.
Пример H: Суппозиторий
|
*Активный ингредиент используют в виде порошка, когда по меньшей мере 90% частиц имеют диаметр 63 мкм или менее.
Одну пятую Witepsol H15 расплавляют в котле с паровой рубашкой максимум при 45°C. Активный ингредиент просеивают через 200 мкм сито и добавляют к расплавленной основе, перемешивая при помощи устройства silverson, оснащенного режущей головкой, до достижения гомогенной дисперсии. Поддерживая смесь при 45°C, в суспензию добавляют остаток Witepsol H15 и перемешивают, чтобы гарантировать гомогенность смеси. Всю суспензию пропускают через 250 мкм сито из нержавеющей стали и охлаждают до 40°C с постоянным перемешиванием. При температуре от 38°C до 40°C 2,02 г смеси помещают в подходящие пластиковые формы. Суппозиториям дают остыть при комнатной температуре.
Пример I: Пессарии
|
Вышеуказанные ингредиенты непосредственно смешивают, а пессарии готовят путем прямой компрессии полученной в результате смеси.















































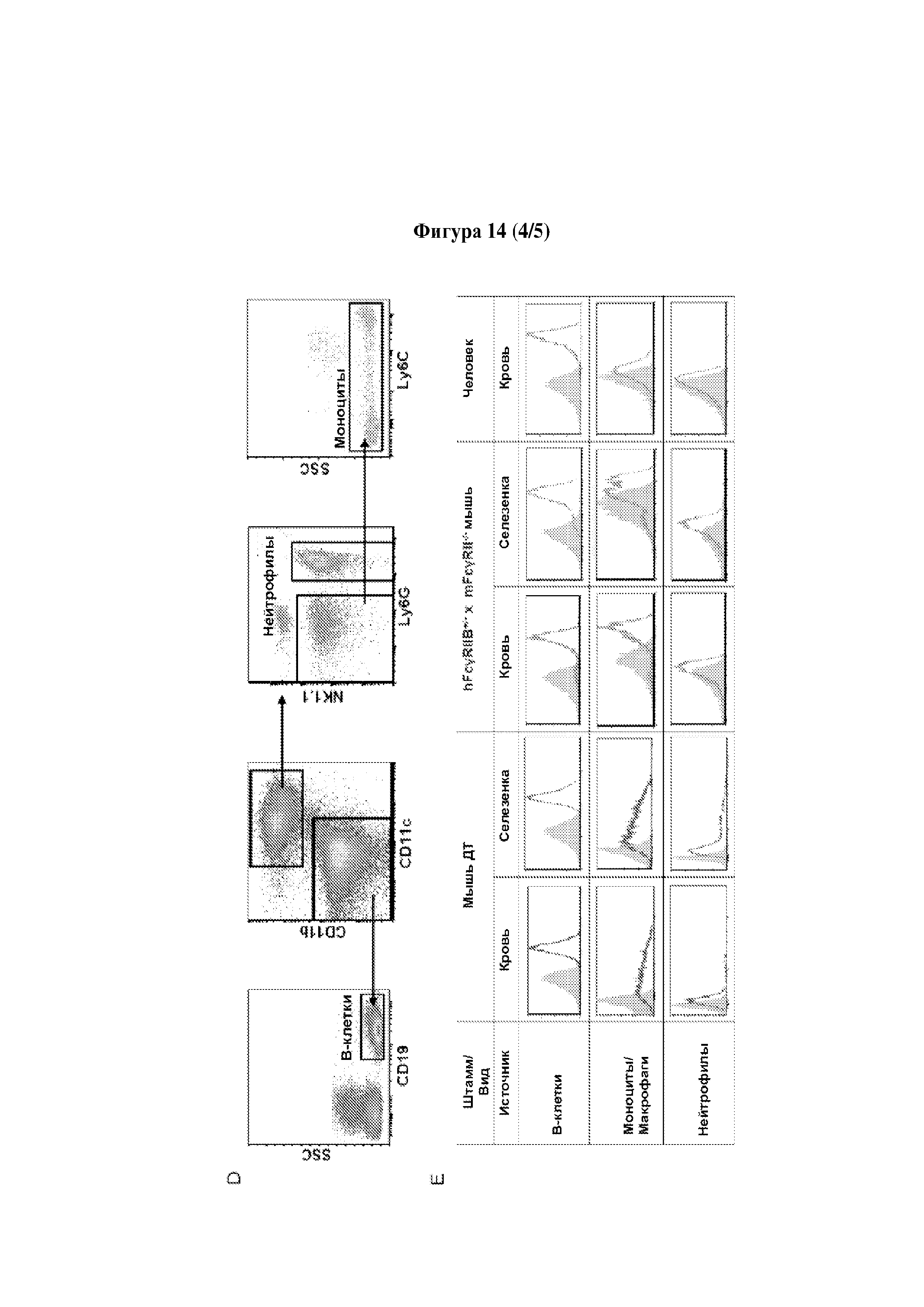

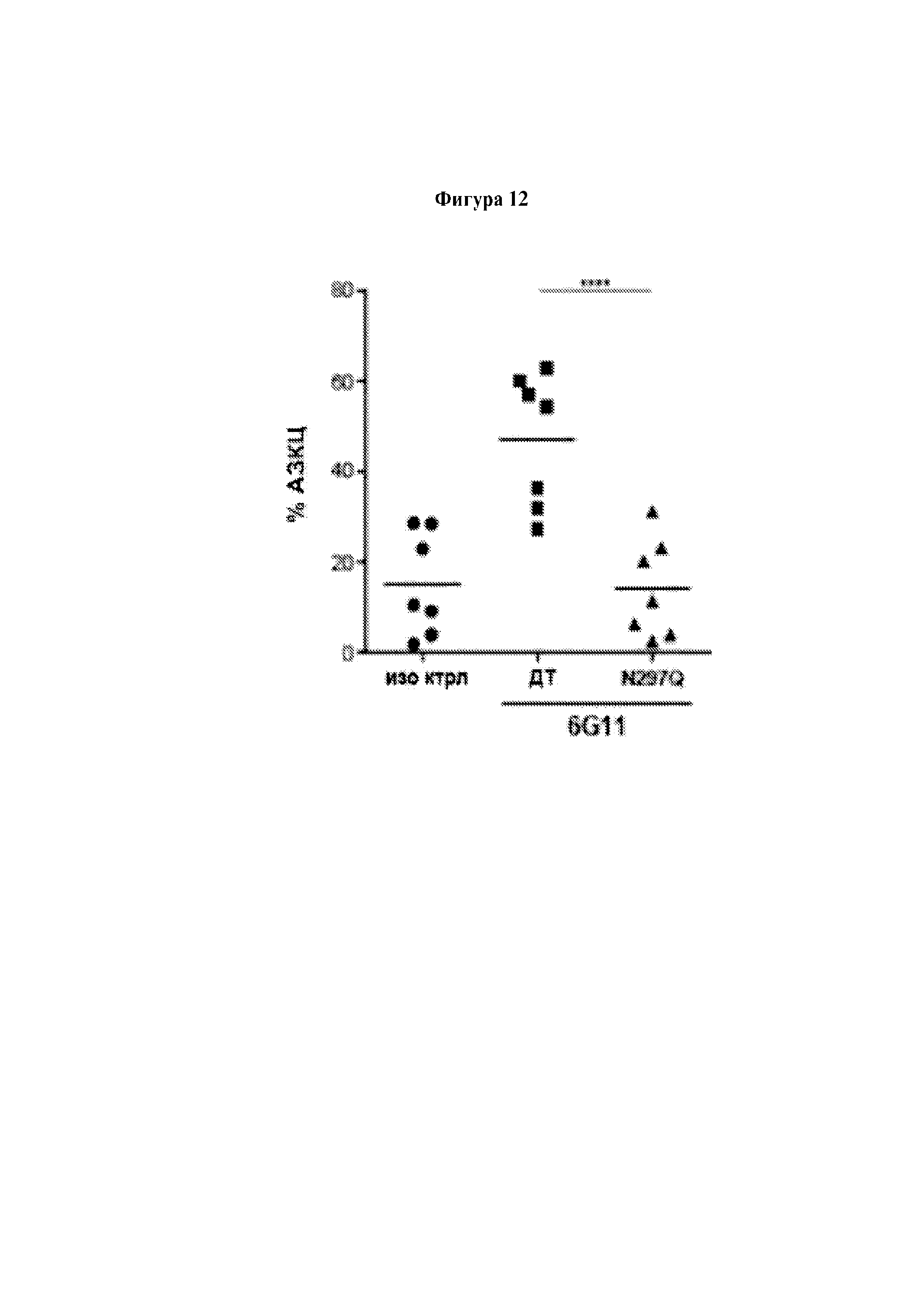
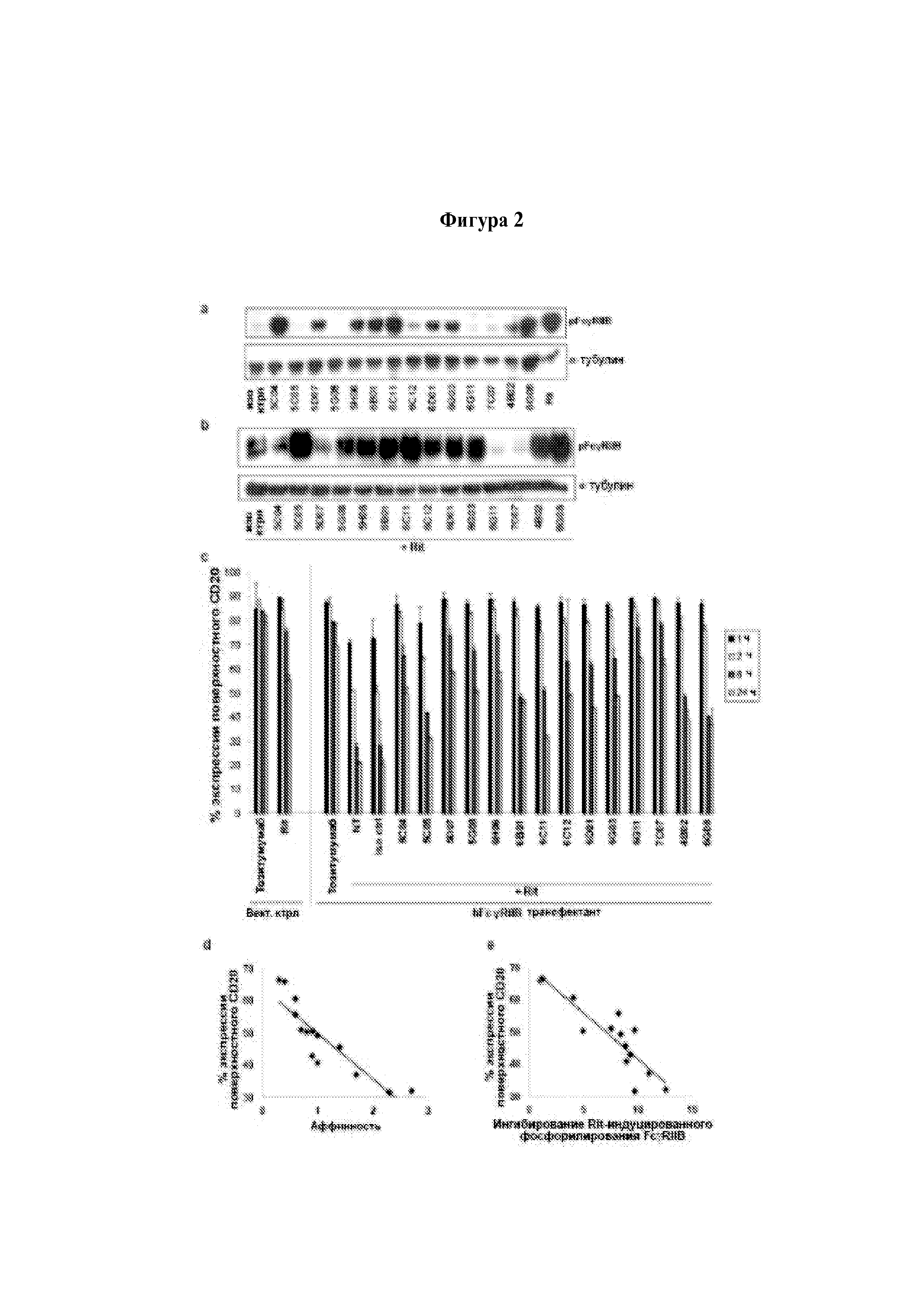
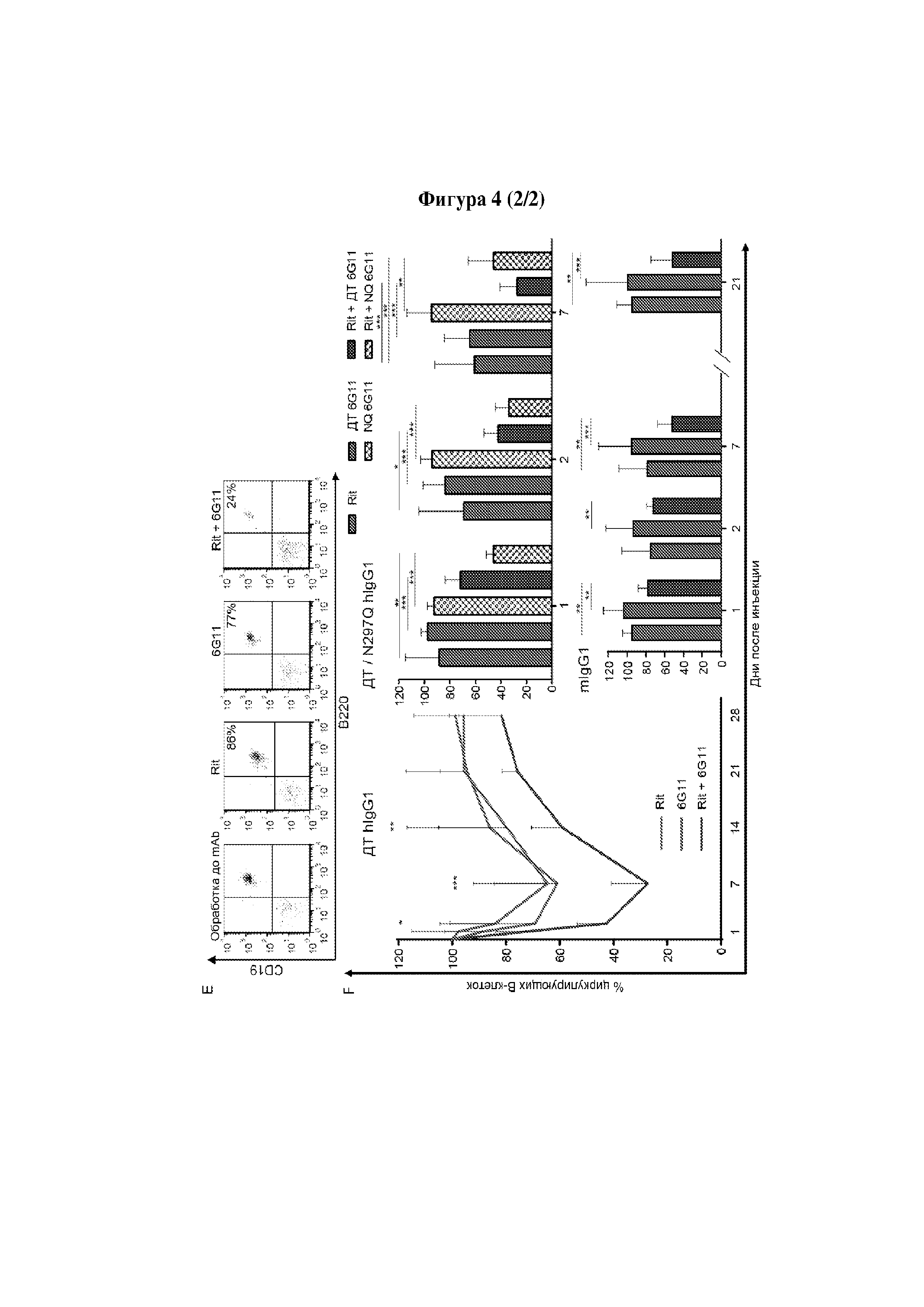

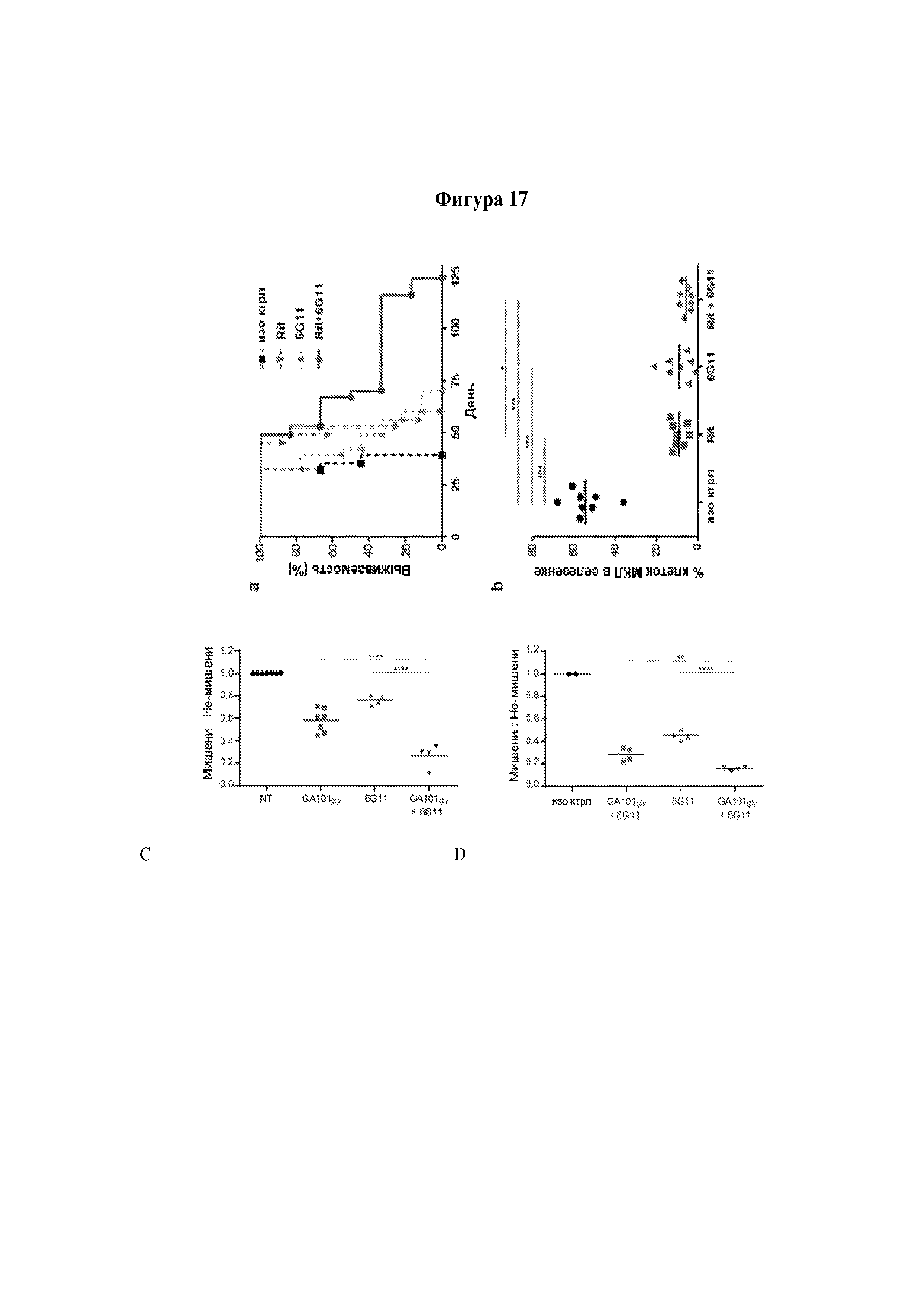
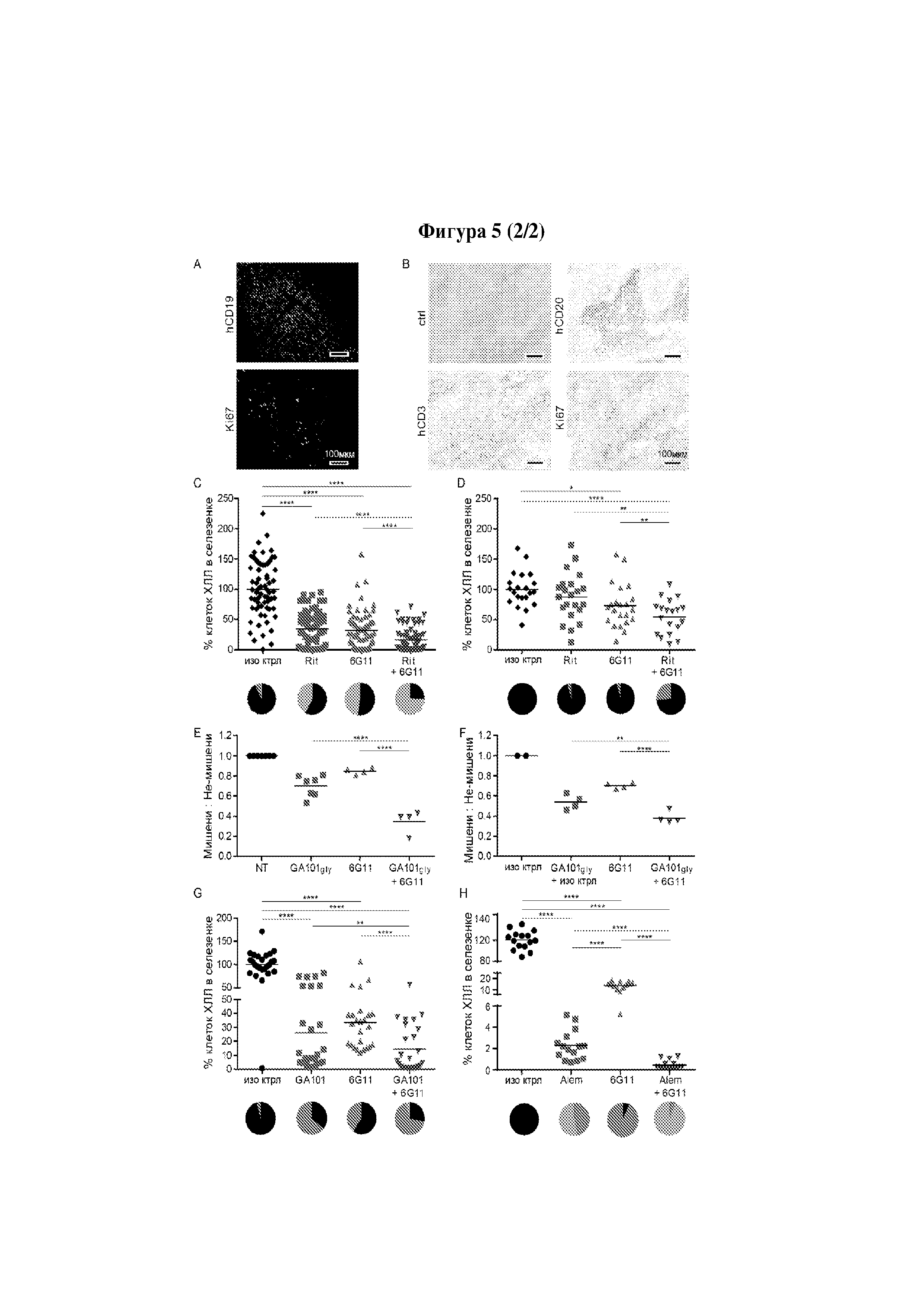

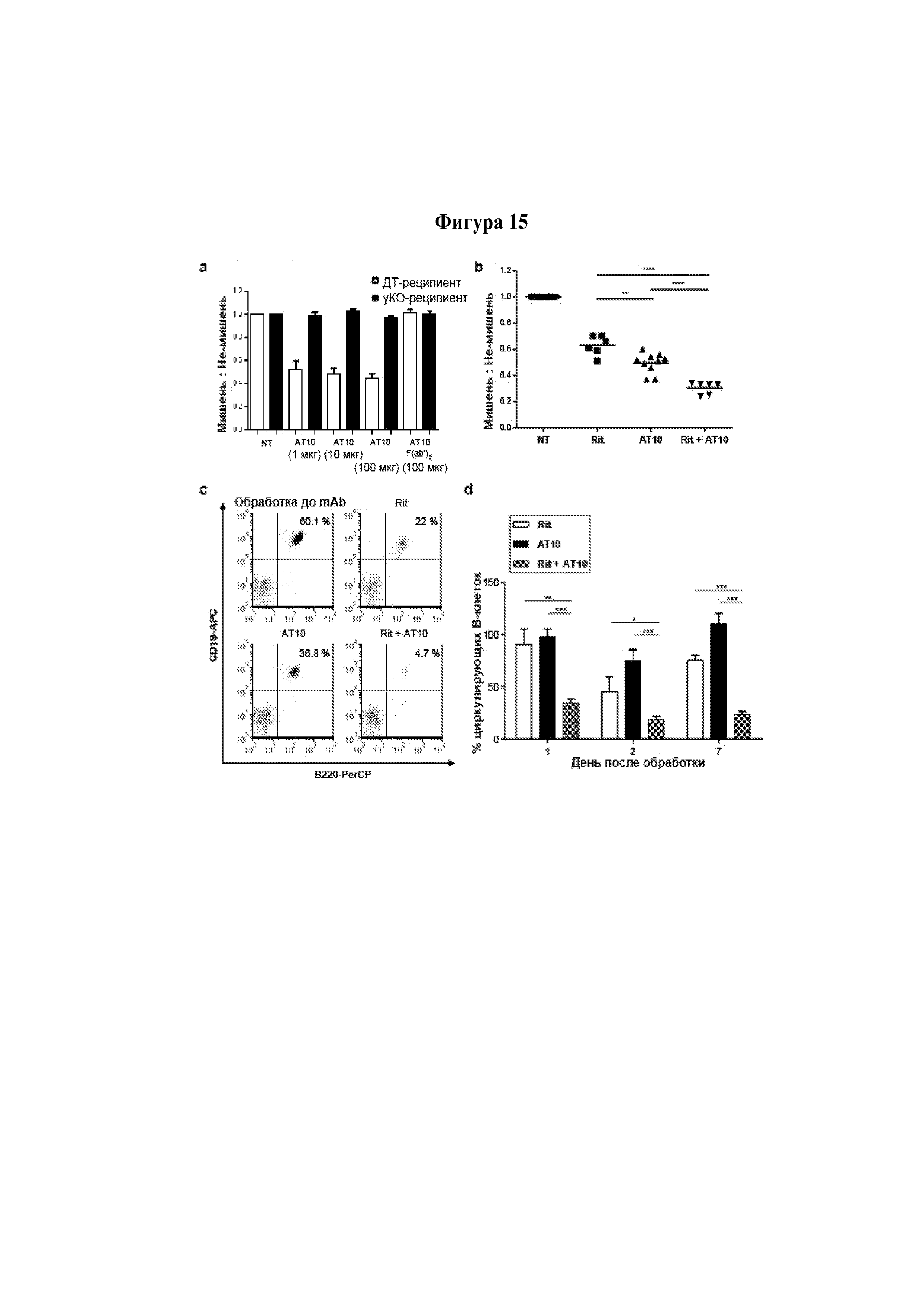
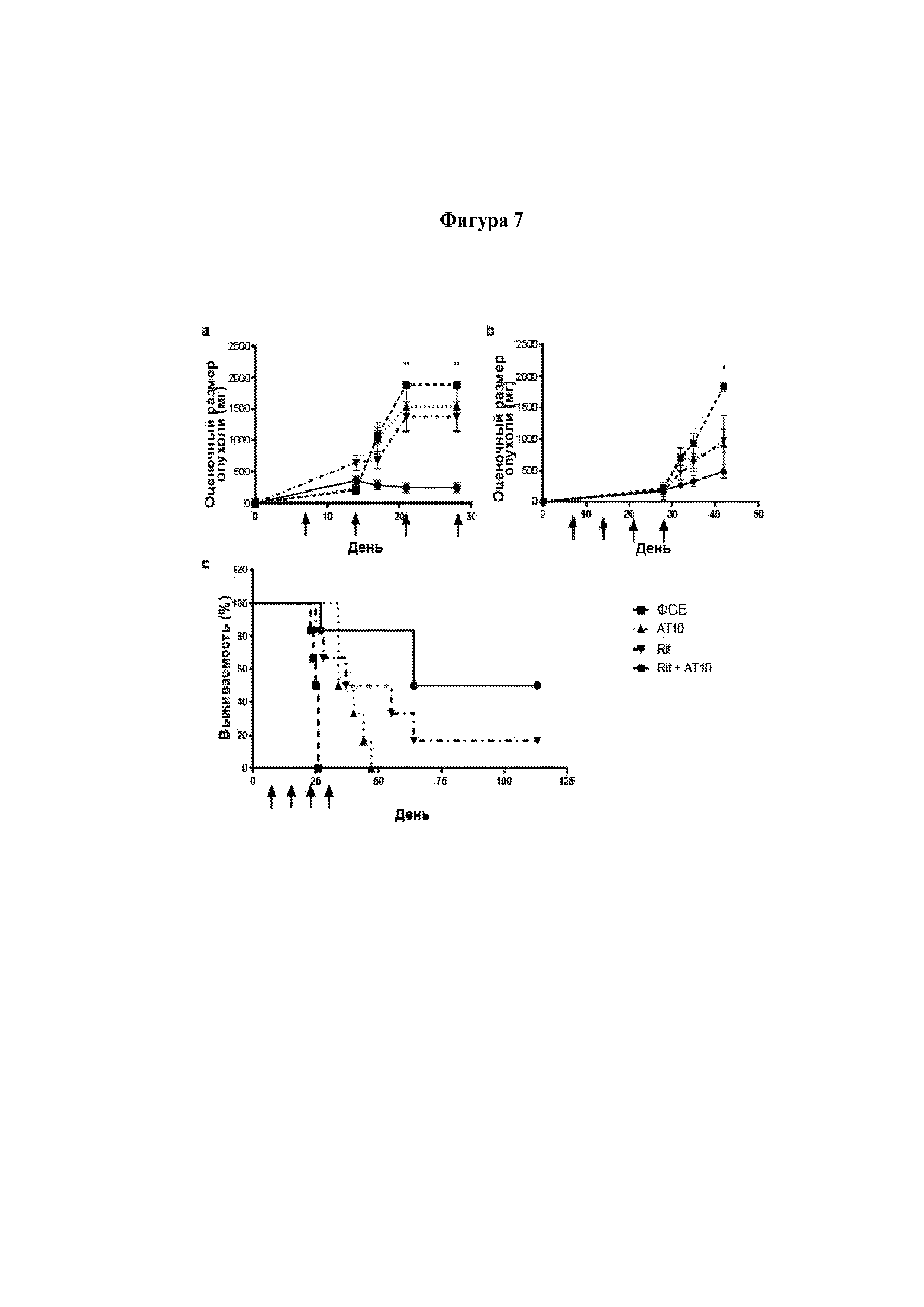
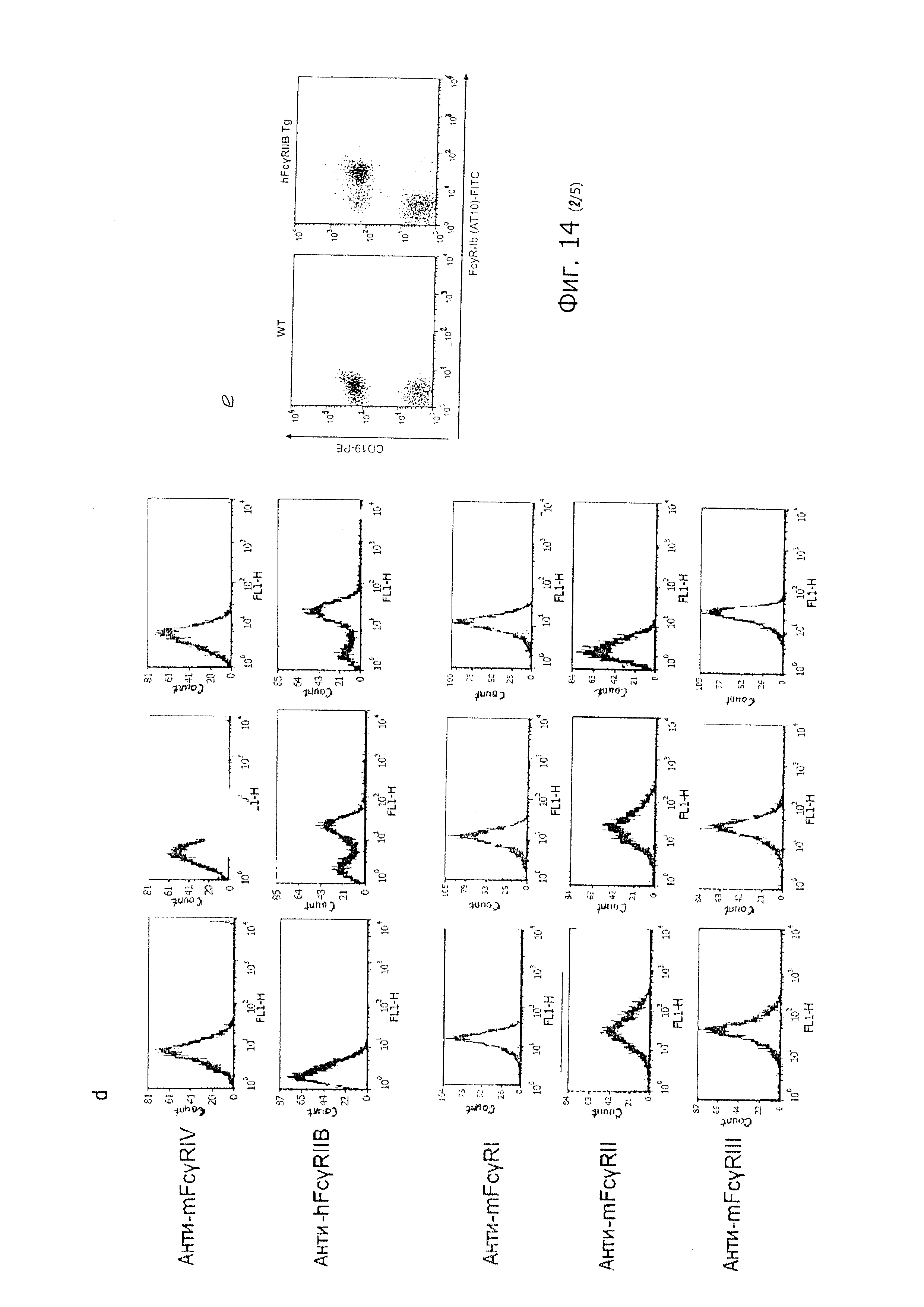
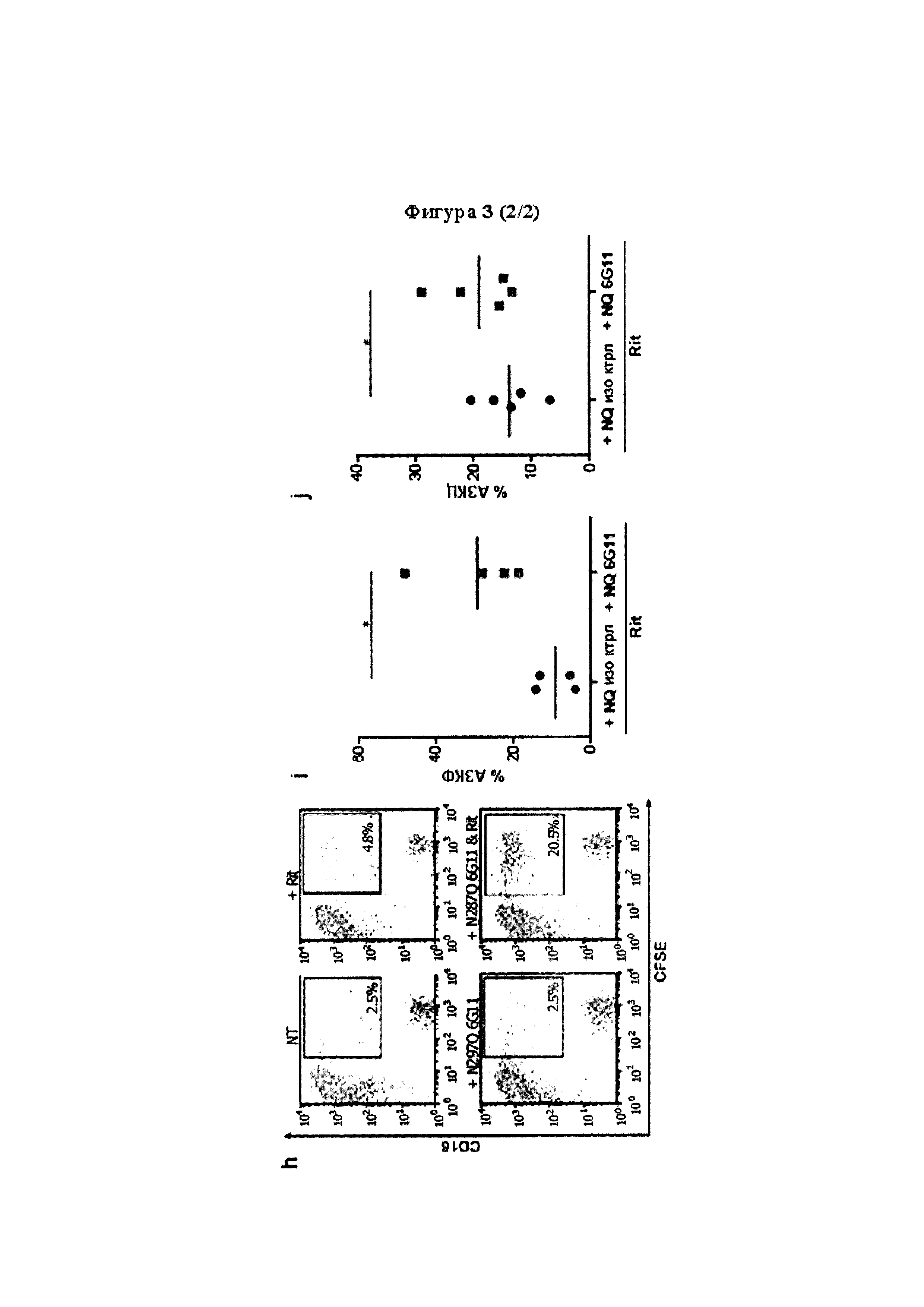

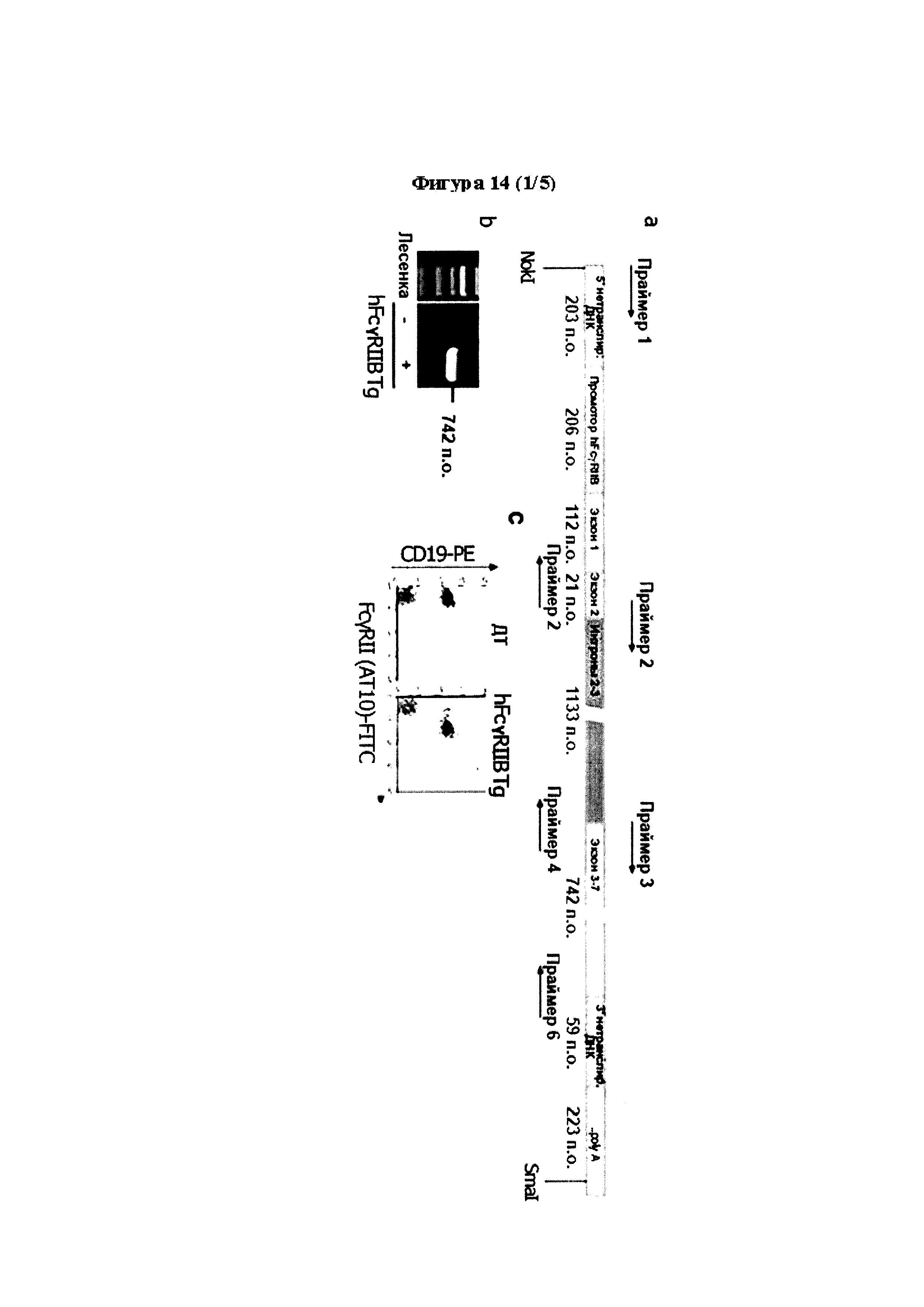
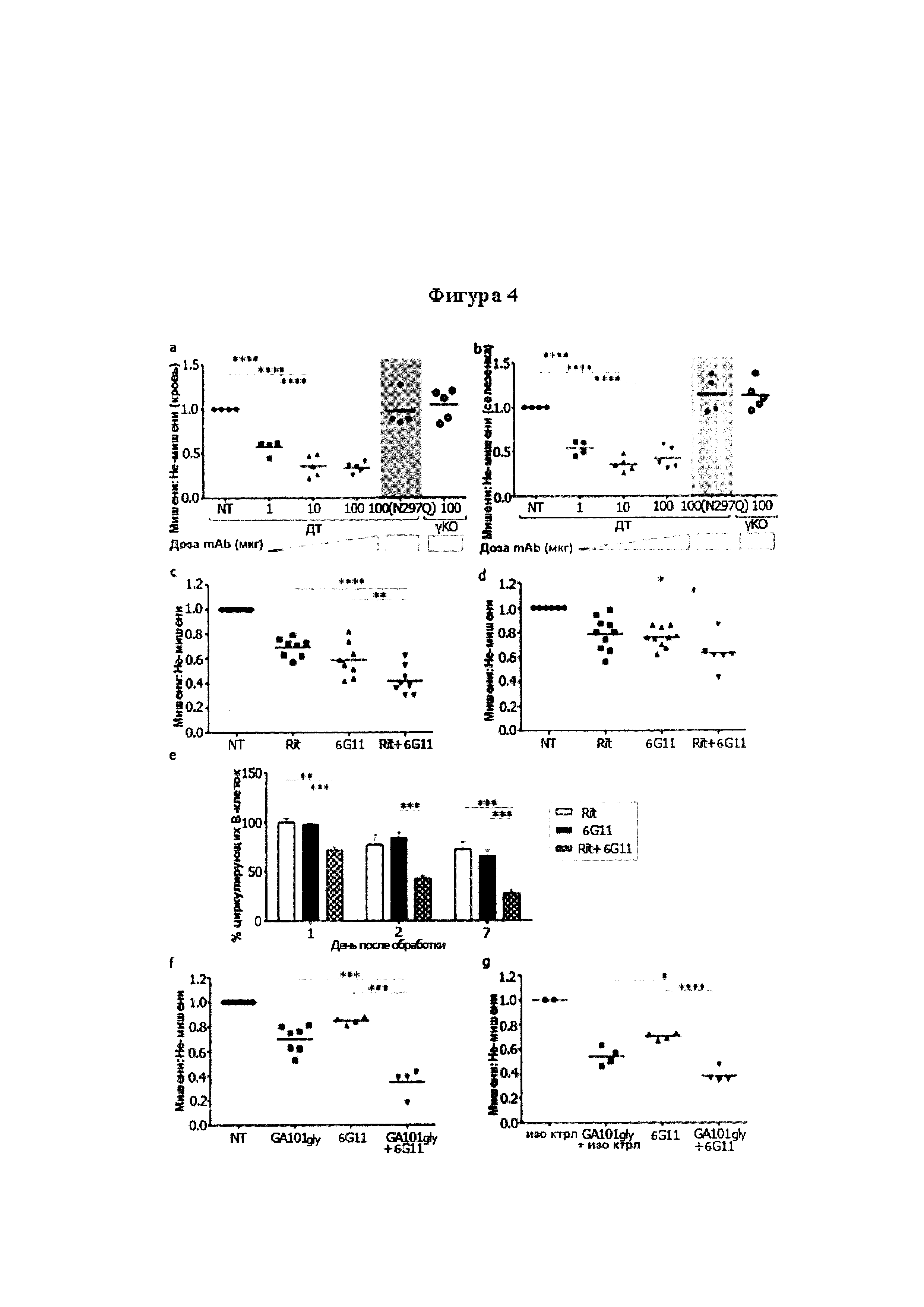
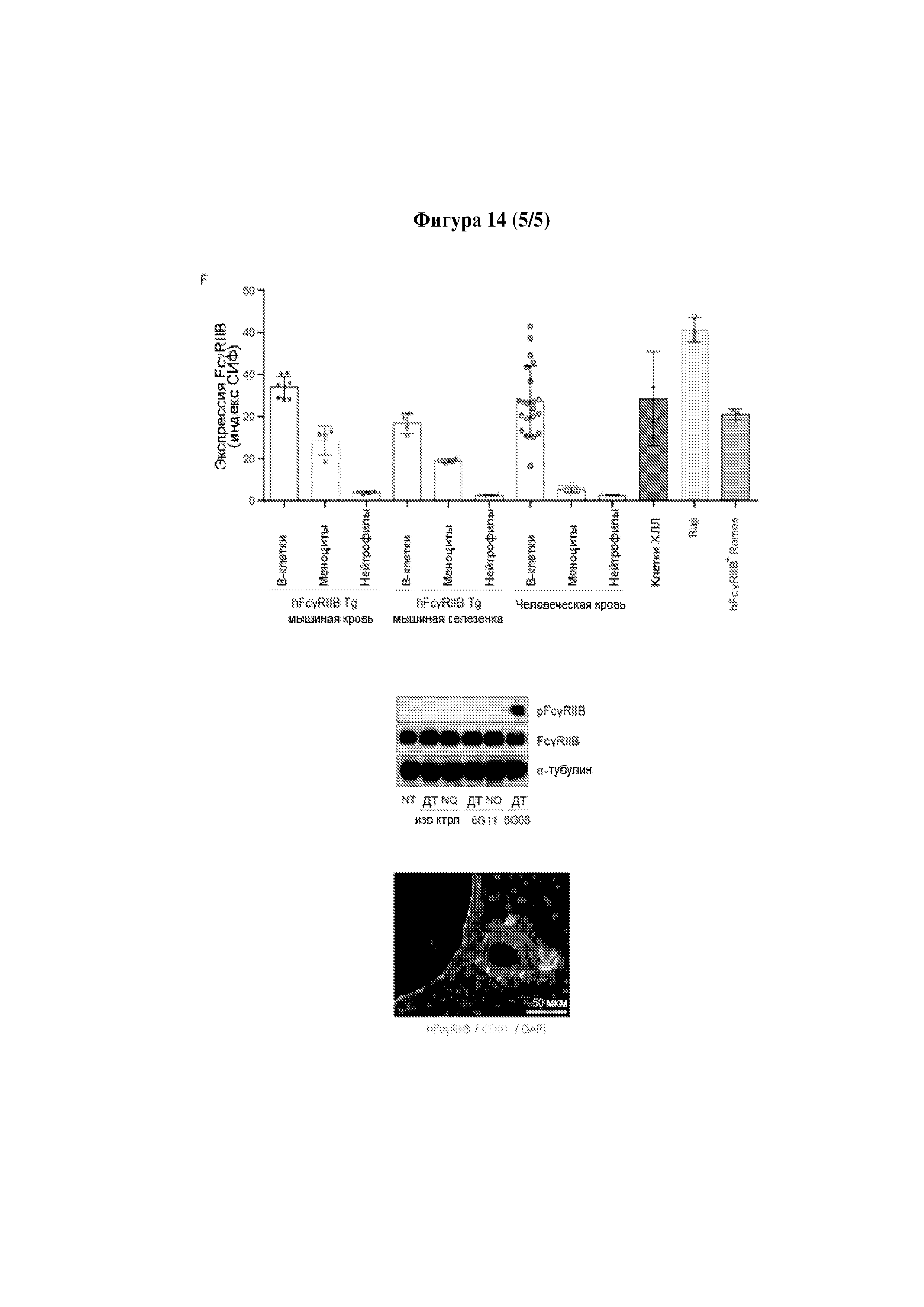
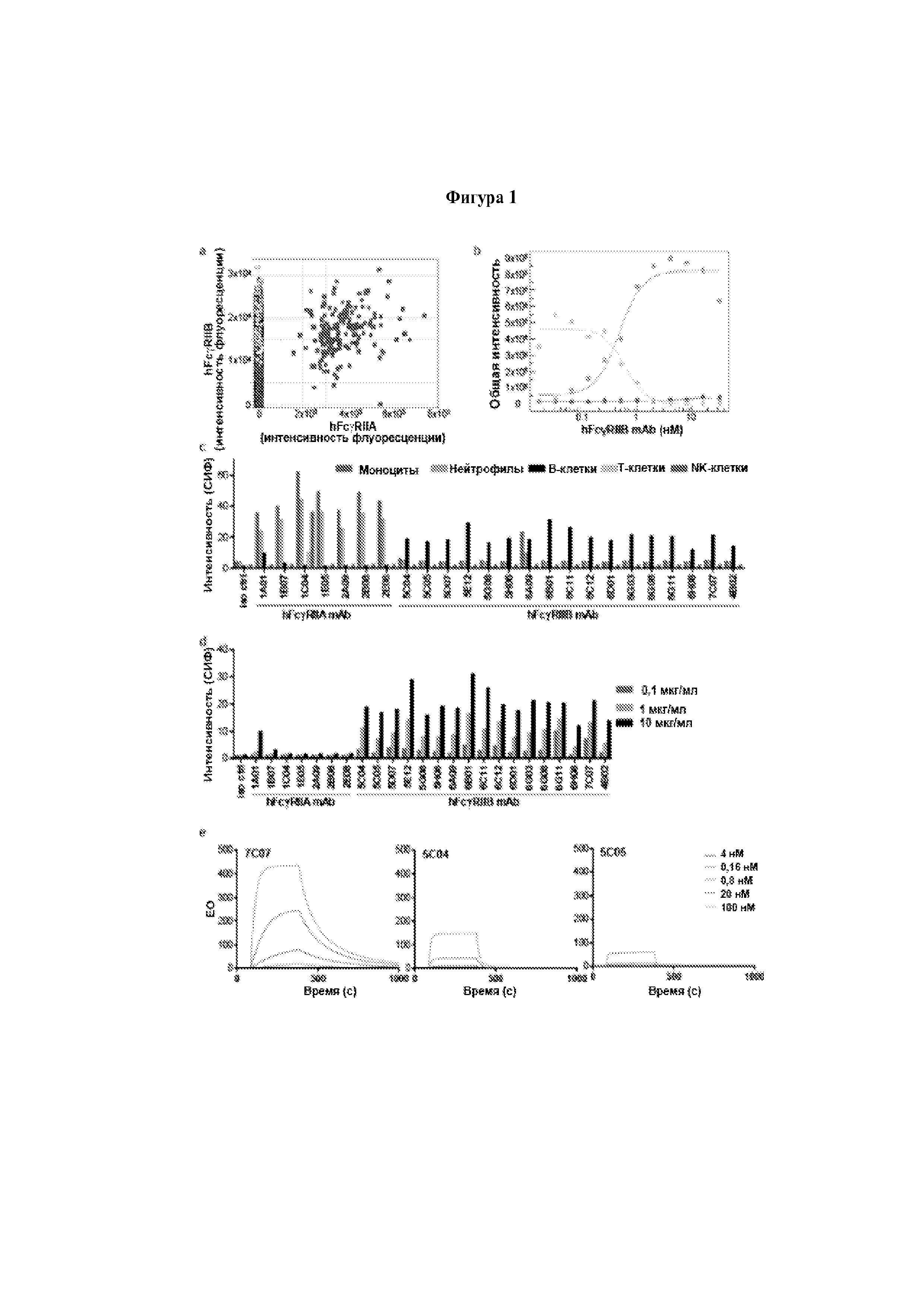
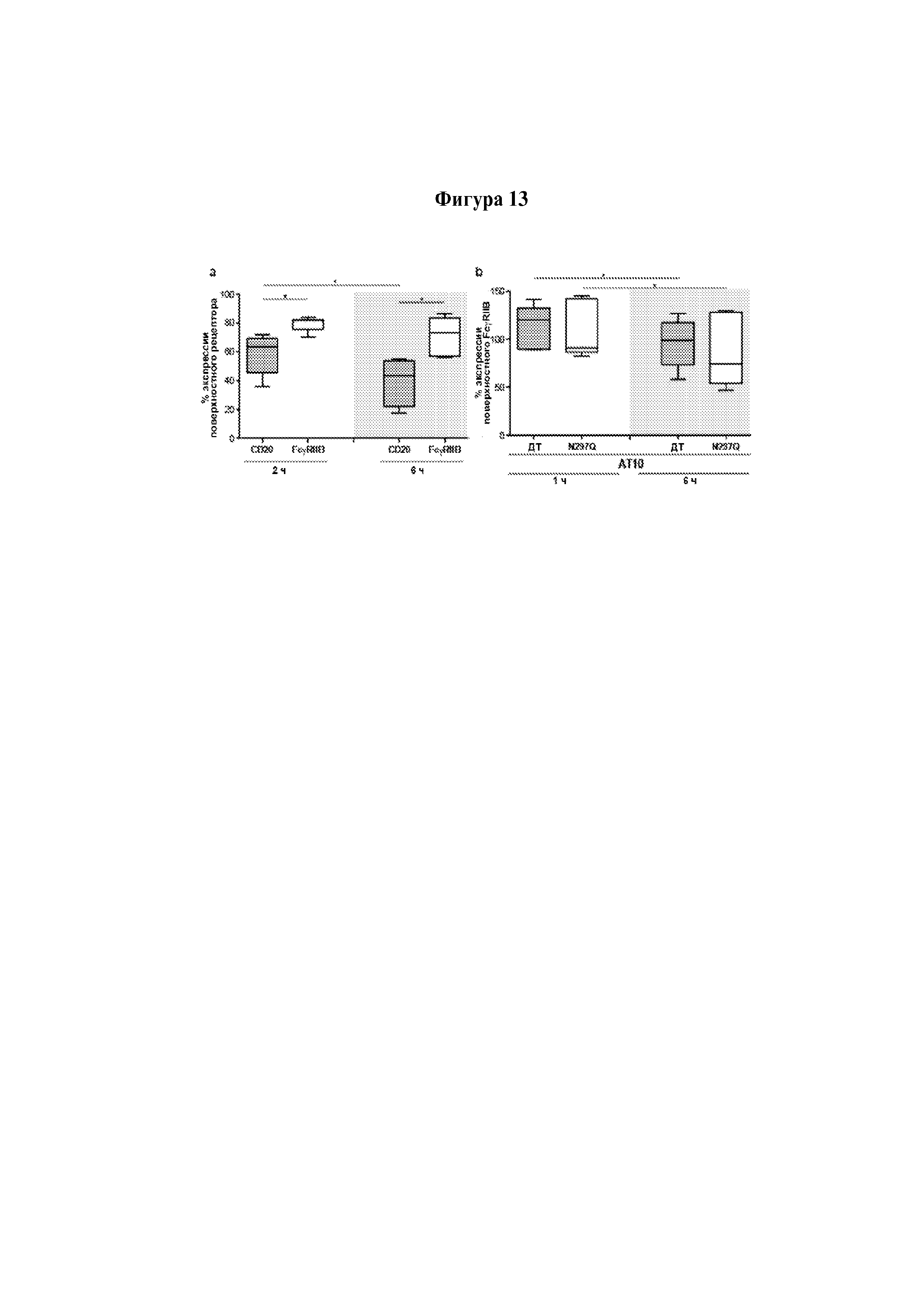
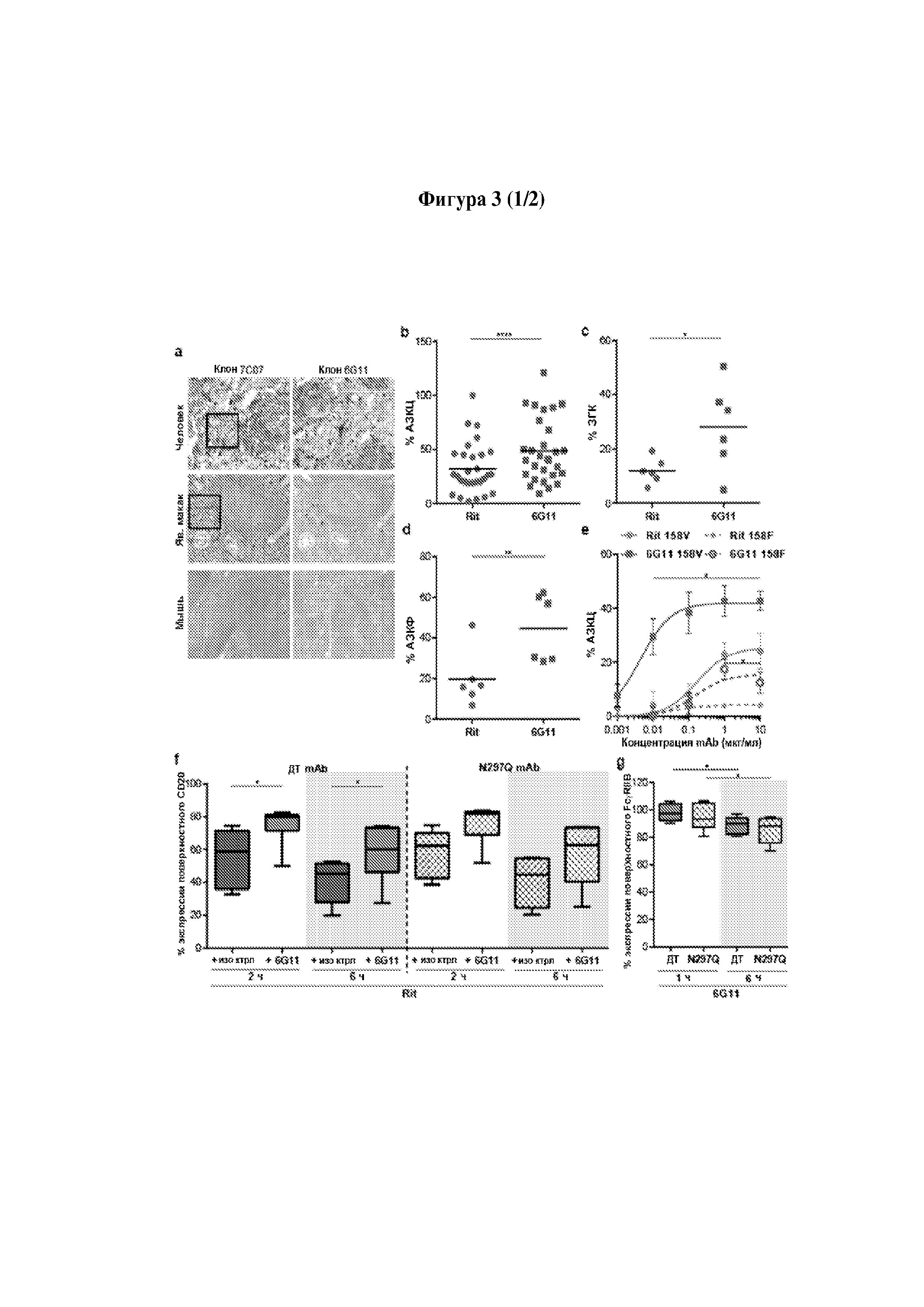
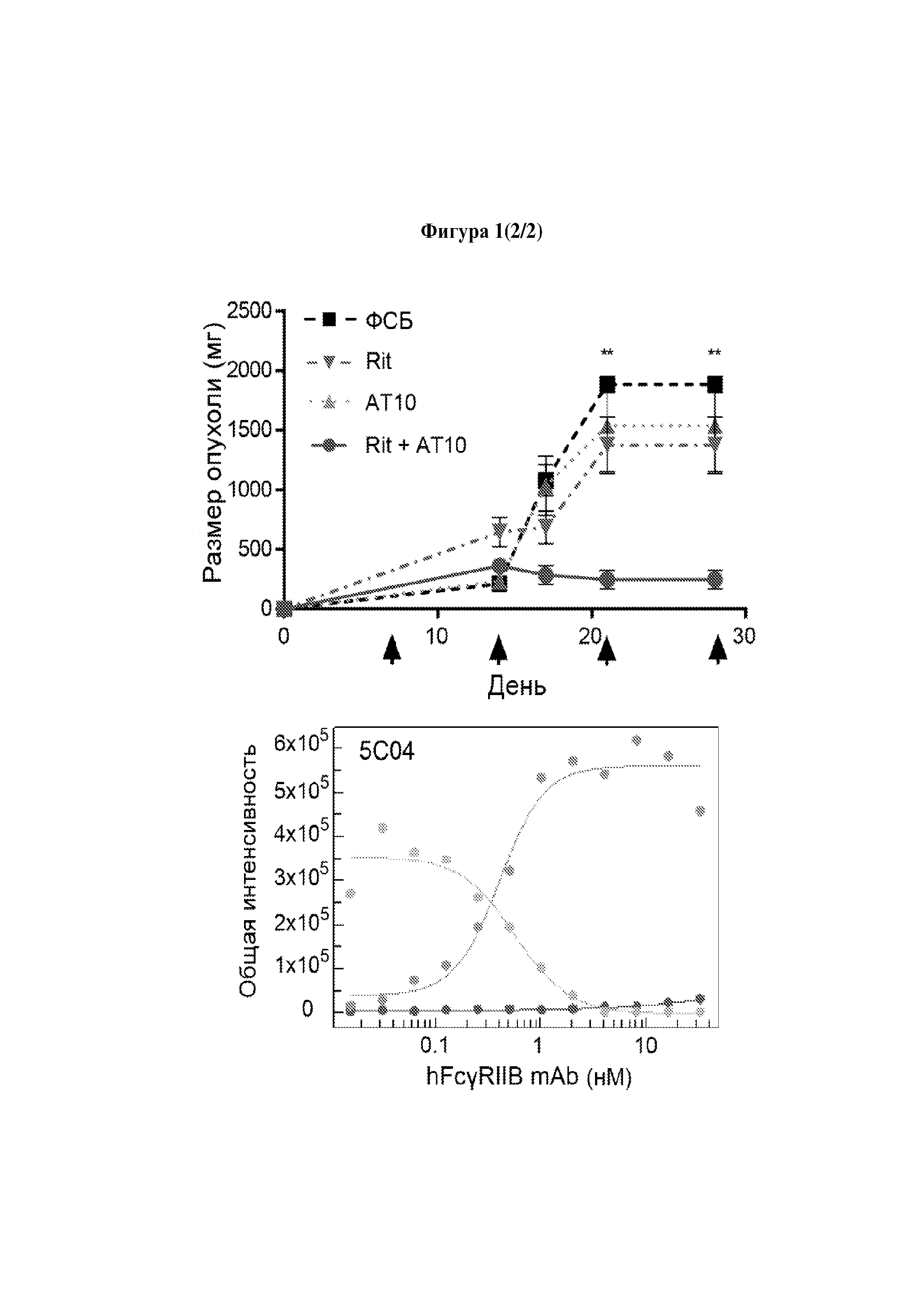



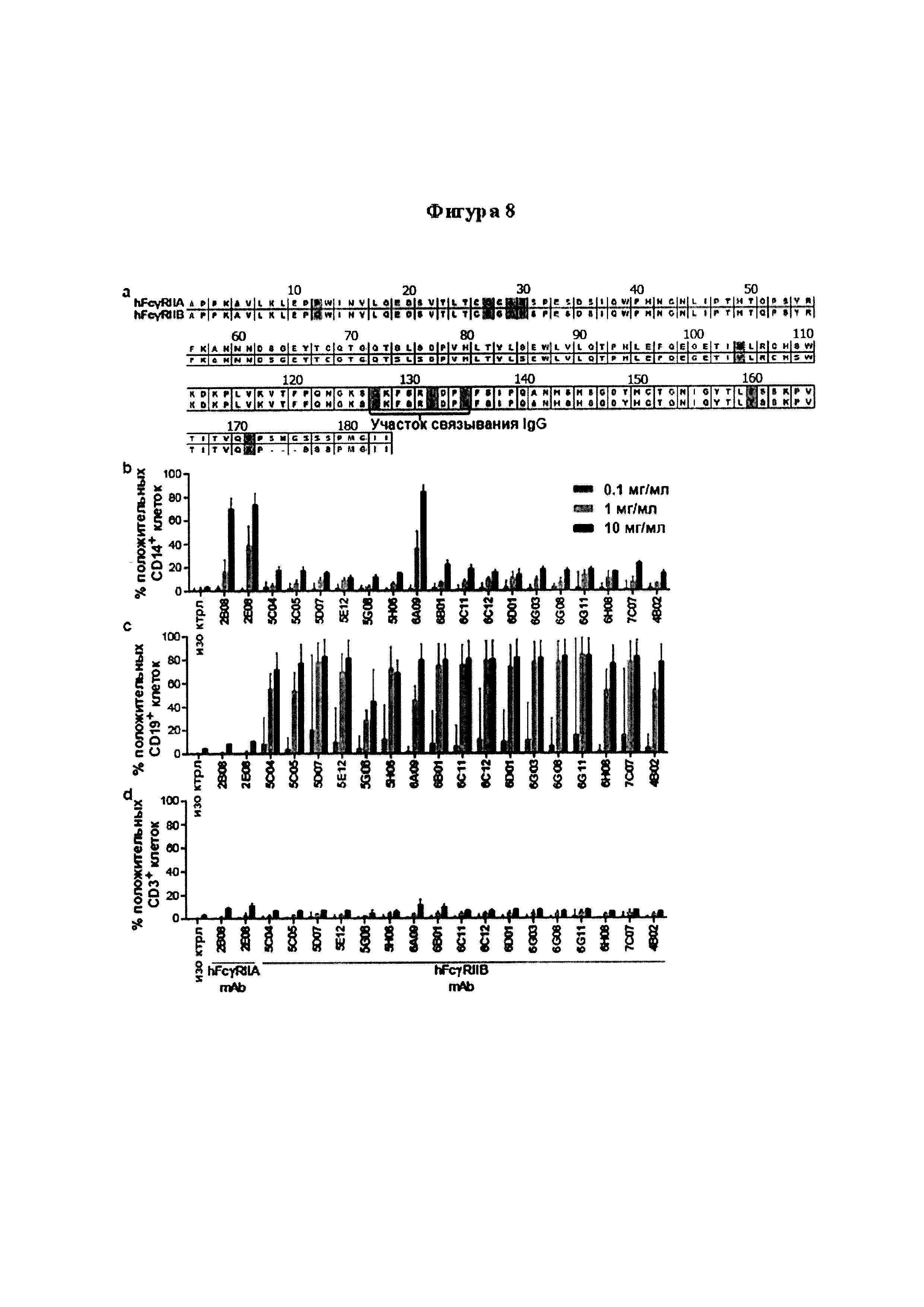
Биологические материалы и их применение
Способы скрининга и их применения
Биологические материалы и их применение
Биологические материалы и их применение
Способы скрининга и их применения
Биологические материалы и их применение

