Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА
Вид РИД
Изобретение
Настоящее изобретение относится к области медицины и предусматривает новый класс производных пиразоло[3,4-с]пиридина, фармацевтические композиции, их содержащие, способы их получения и их применение в медицине.
Уровень техники
Тромботическое заболевание вызывается тромбозом и эмболией. В некоторых патологических условиях в кровеносном сосуде из компонентов крови могут образоваться сгустки крови. Сгустки крови отрываются от мест, где они образуются, и частично или полностью блокируют вены или подающие кровь артерии в процессе их перемещения вместе с потоком крови, вызывая ряд патологических процессов, таких как сосудистая или системная ишемия, аноксия и некроз. Распространенное тромботическое заболевание тромбоз, включая инфаркт миокарда, церебральный тромбоз, глубокий венозный тромбоз, легочную эмболию и тромбоэмболию периферических артерий, серьезно ухудшают жизнь людей и качество их жизни. Коронарная болезнь сердца представляет собой серьезную разновидность тромботических заболеваний и включает инфаркт миокарда и стенокардию. Каждый год около 0.8-1.5 миллионов людей заболевают коронарной болезнью сердца в Китае. Коронарная болезнь сердца является четвертой по счету ведущей причиной смертности, в то время как цереброваскулярная болезнь занимает второе место. Кроме того, хотя нет конкретных статистических данных о случаях
глубокого венозного тромбоза, по предварительным подсчетам количество пациентов с глубоким венозным тромбозом может достигать в Китае одного миллиона. Более того, с улучшением качества жизни людей, ожиданием значительного увеличения национальной средней продолжительности жизни и возрастающей доли пожилых людей количество случаев возникновения глубокого венозного тромбоза будет постепенно увеличиваться, и эта болезнь станет распространенной.
Тромбоз вызывается активацией двух систем, а именно факторов коагуляции и тромбоцитов. Факторы коагуляции представляют собой ряд белковых компонентов, участвующих в процессе свертывания крови. В процессе ангиофагии или в некоторых патологических условиях эти белки активируются и слипаются с тромбоцитами с образованием сгустков крови. В организме существуют две системы коагуляции, а именно, эндогенная и экзогенная. Первая относится к системе, когда кровь контактирует с аномальной поверхностью для активации фактора коагуляции XII. Последняя относится к повреждению тканей, когда фактор коагуляции III высвобождается и, следовательно, активируется фактор коагуляции VII. Обе системы могут запускать ряд цепных реакций и сходиться у фактора коагуляции X, что в конце концов приведет к активации протромбина и образованию фибрина.
В последние годы в клинике широко используется антитромботическая терапия с применением гепарина, аспирина и варфарина. Среди этих лекарств варфарин ингибирует посттрансляционное созревание факторов коагуляции VII, IX, X и протромбина и он оказался эффективным в случае венозного и артериального тромбоза. Однако его использование ограничено из-за его узкого терапевтического индекса, что замедляет терапевтический эффект, многочисленных взаимодействий с пищевыми продуктами и лекарствами и необходимости мониторинга и регулировки величин доз. Гепарин также является ценным лекарством при терапии тромболитических заболеваний, но обычный гепарин не может орально абсорбироваться, а его инъекция является неудобной. Следовательно, в Китае более эффективные оральные антитромботические лекарства найдут большой сбыт.
Фактор коагуляции X представляет собой хорошую мишень для антитромботического лечения. Во-первых, фактор коагуляции расположен выше (апстрим) тромбина в амплификации (усилении) каскада коагуляции. Одна молекула фактора коагуляции X может активировать сотни молекул тромбина. Следовательно, говоря теоретически, было бы более эффективно ингибировать фактор коагуляции X, чем ингибировать тромбин. Во-вторых, ингибирование фактора коагуляции X не влияет на тромбин, который уже был активирован. Обратимо действующие ингибиторы фактора коагуляции X могут ингибировать образование тромбина неполностью, в то время как небольшое количество тромбина может активировать тромбоциты для поддержки процесса гемостаза. Таким образом, ингибирование фактора коагуляции X может приводить к появлению относительно умеренных вредных явлений при кровотечении, чем ингибирование тромбина. Это было подтверждено на животных моделях. В-третьих, непрямой ингибитор фактора коагуляции X фондапаринукс оказался успешным при проведении клинических испытаний, демонстрируя, что ингибирование факторов коагуляции действительно является эффективным средством лечения тромбоза.
В процессе превращения протромбина в тромбин фактор Ха является самой важной мишенью для лекарства в каскаде коагуляции. Ингибиторы фактора Ха могут прочно прикрепляться к активному сайту фактора Ха, приводя к инактивации фактора Ха, который является свободным или соединен с фибрином таким образом, что имеет место действие антикоагулянта. По сравнению с низкомолекулярным гепарином ингибиторы фактора Ха могут значительно снижать развитие венозного тромбоза и не увеличивают вероятность возникновения кровотечения. По сравнению с варфарином ингибиторы фактора Ха являются удобными, они не требуют регулировки дозы и повседневного наблюдения, и они мало взаимодействуют с пищевыми продуктами и лекарствами, поэтому могут вводиться совместно с другими лекарствами.
В настоящее время был опубликован ряд заявок, относящихся к ингибиторам фактора Ха, включая WO 2001047919, WO 2008006479, WO 2007137801, WO 2006047528 и т.д. Кроме того, на зарубежных рынках появились некоторые ингибиторы фактора коагуляции X, включая ривароксабан фирмы Bayer, апиксабан фирмы Bristol-Myers Squibb (BMS) и т.п. Апиксабан разрабатывался совместно фирмами BMS и Pfizer. Он является другим прямым оральным ингибитором фактора Ха после ривароксабана и пригоден для профилактики венозного тромбоза элективного тазобедренного сочленения у взрослых или тотальной артропластики коленного сустава и был внесен в список лекарств в Европейском союзе в июле 2011 г.
Хотя тенденция к кровотечению, вызываемая ингибиторами фактора Ха, проявляется меньше, чем в случае традиционных антикоагулянтов, основной клинической неблагоприятной реакцией все еще остается кровотечение. Следовательно, в области исследований в фокусе остается уменьшение риска кровотечения и расширение терапевтического окна.
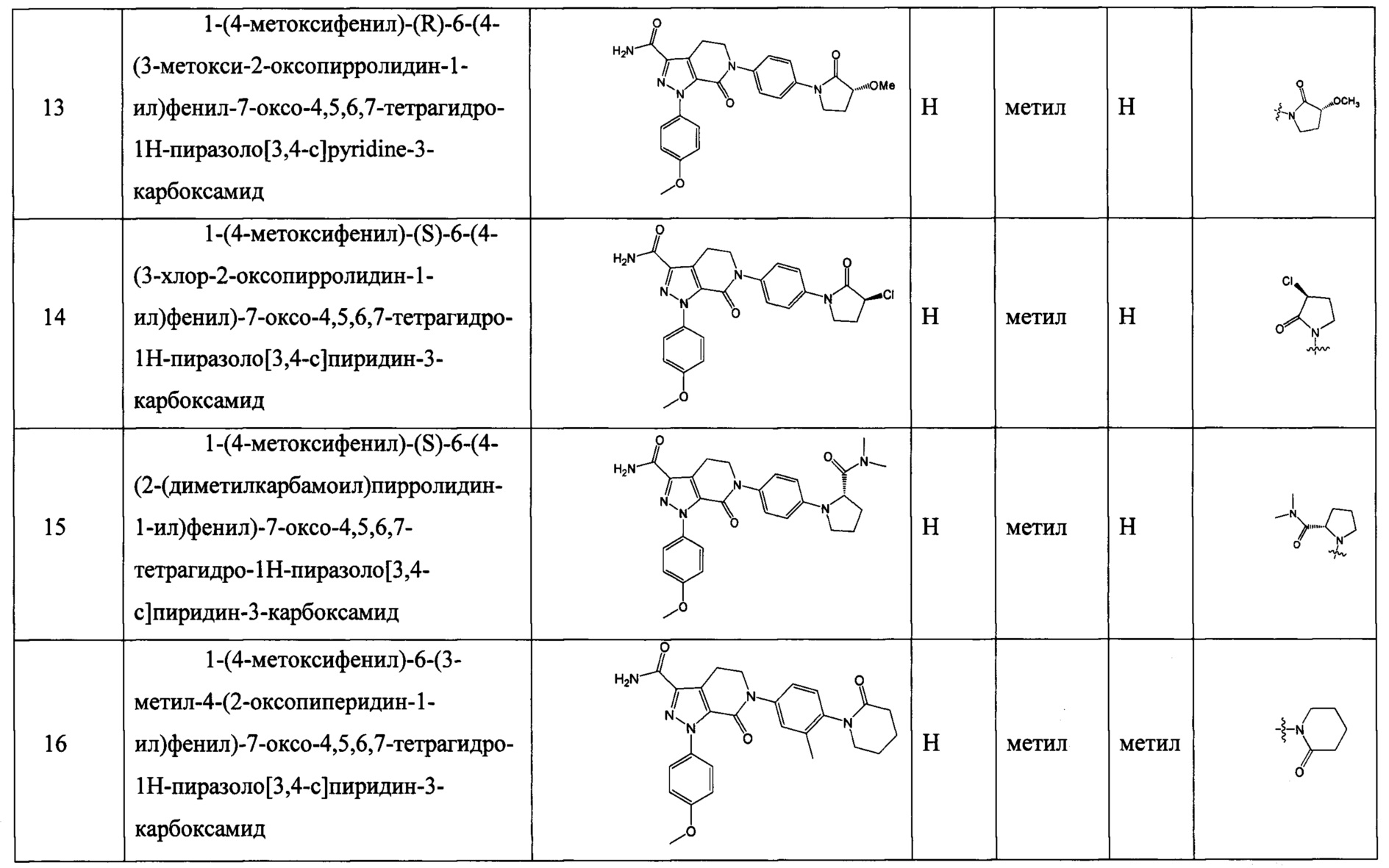
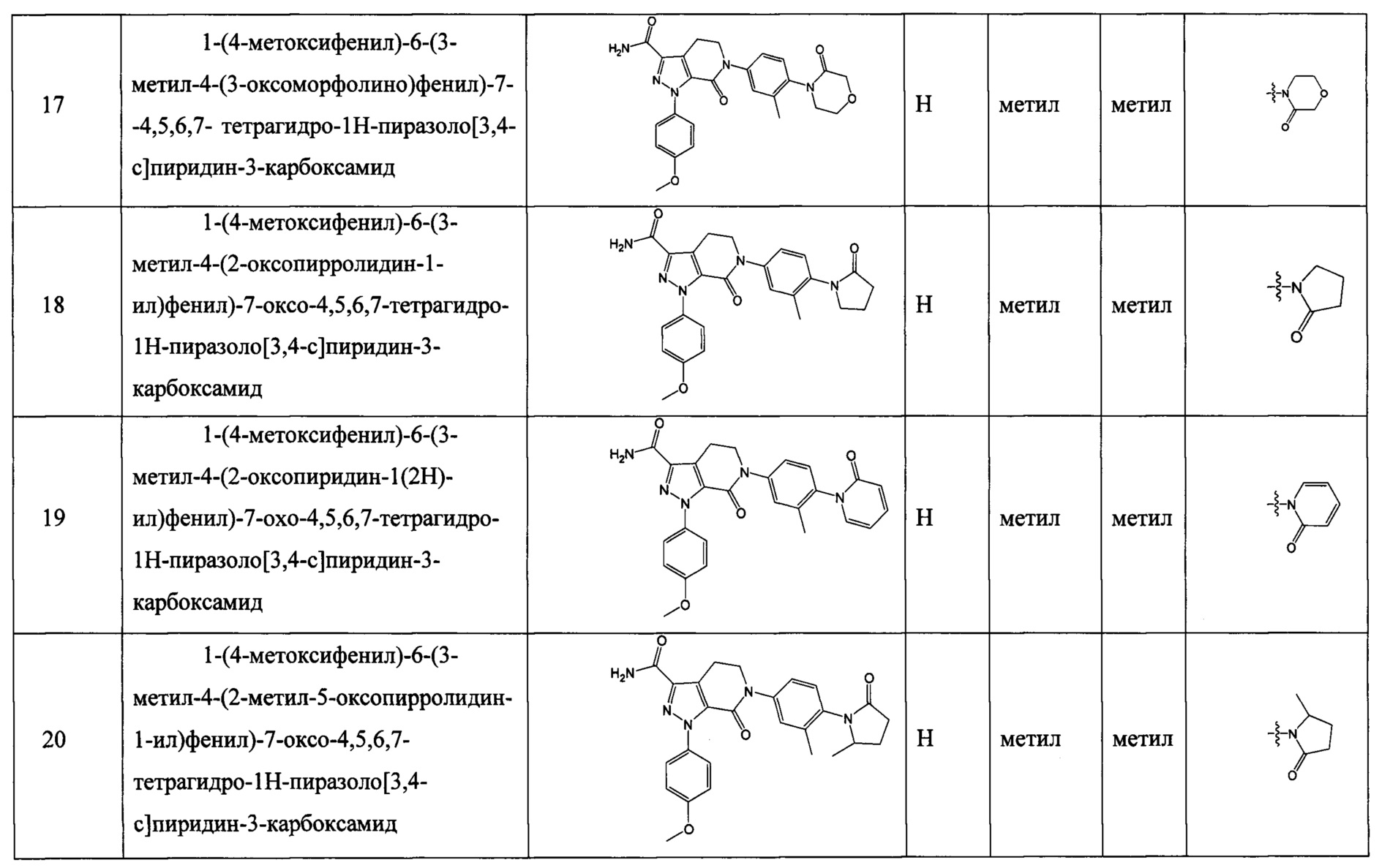
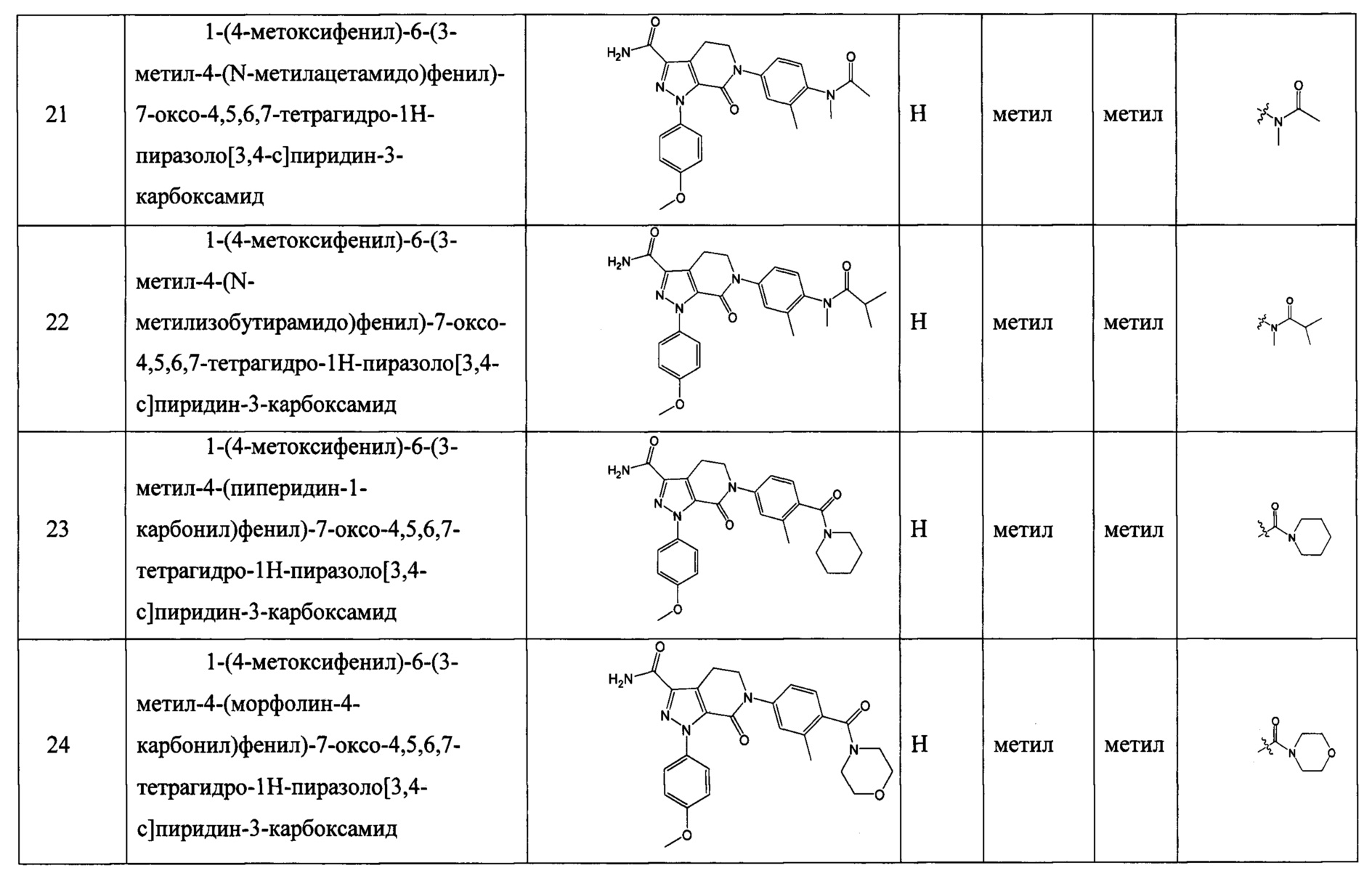
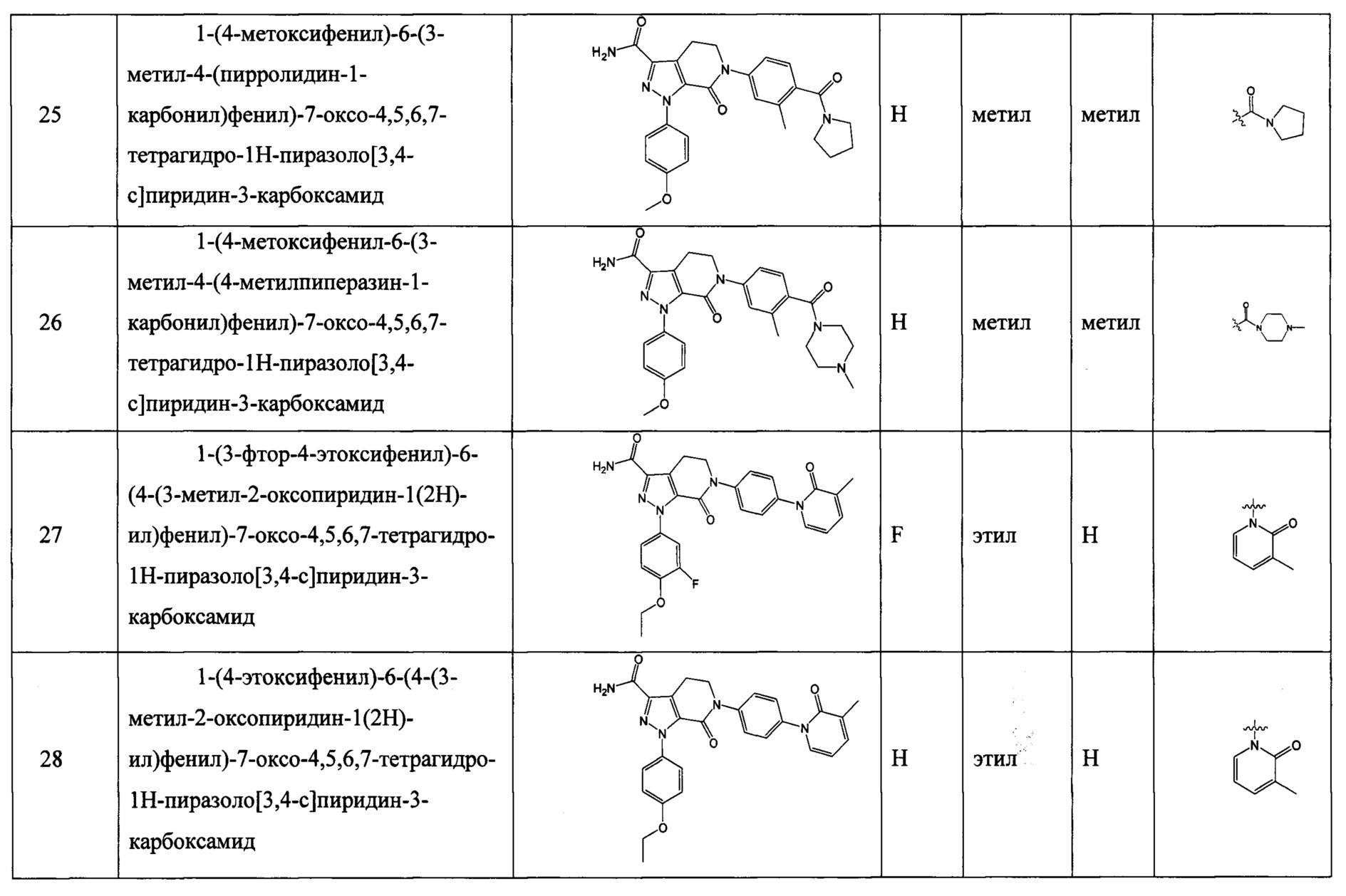
Хотя ряд ингибиторов фактора Ха с антитромботическим действием был описан, все еще остается необходимость создания новых лекарств с большей эффективностью и меньшим риском возникновения кровотечения.
Раскрытие изобретения
Настоящее изобретение предусматривает новые соединения с хорошим антитромботическим действием и меньшим риском возникновения кровотечения.
Конкретно, эти соединения являются соединениями, предусмотренными техническими решениями 1-13.
Техническое решение 1.
Соединение формулы (I), его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
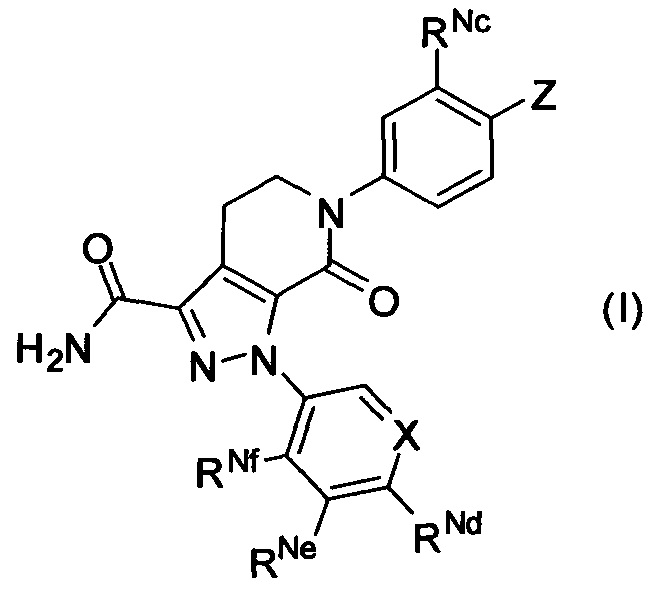
где
X выбран из СН и N;
Z выбран из  ;
;
RNa и RNb каждый независимо выбран из водорода, C1-6 алкила, С2-6 алкенила, С2-6 алкинила, C1-6 алкокси-C0-6 алкила, (C0-6 алкил)(C0-6 алкил)N-C1-6 алкила, (С2-6 алкилен)N-C1-6 алкила или карбамоил-C1-6 алкила; или
RNa и RNb вместе с атомами, к которым они присоединены, образуют 5, 6 или 7-членный циклический фрагмент,
причем
5, 6 или 7-членный циклический фрагмент замещен одним радикалом RNg, где RNg выбран из водорода, С1-4 алкила, гидроксила, С1-4 алкокси, галогена, оксо и амино,
5, 6 или 7-членный циклический фрагмент, кроме атома N, присоединенного к RNb, содержит 0, 1, 2, 3 или 4 гетероатома, выбранные из N, О и S,
5, 6 или 7-членный циклический фрагмент содержит 0, 1, 2 или 3 двойные связи;
RNc выбран из водорода, C1-6 алкила и C1-6 алкокси;
RNd выбран из водорода, C1-6 алкокси, галогензамещенной C1-6 алкокси, карбамоил-C1-6 алкила и C1-6 алкокси-C1-6 алкила;
RNe выбран из водорода, галогена, C1-6 алкокси, галогензамещенной C1-6 алкокси, карбамоил-C1-6 алкила и C1-6 алкокси-C1-6 алкила;
RNf выбран из водорода, галогена, C1-6 алкокси, галогензамещенной C1-6 алкокси, карбамоил-C1-6 алкила и C1-6 алкокси-C1-6 алкила;
при условии, что соединение формулы (I) не включает соединения, которые исключены из п. 1 формулы изобретения.
Техническое решение 2.
Соединение формулы (I) согласно предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
X обозначает СН.
Техническое решение 3.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
Z выбран из  ;
;
RNa и RNb вместе с атомами, к которым они присоединены, образуют 5, 6 или 7-членный циклический фрагмент,
причем
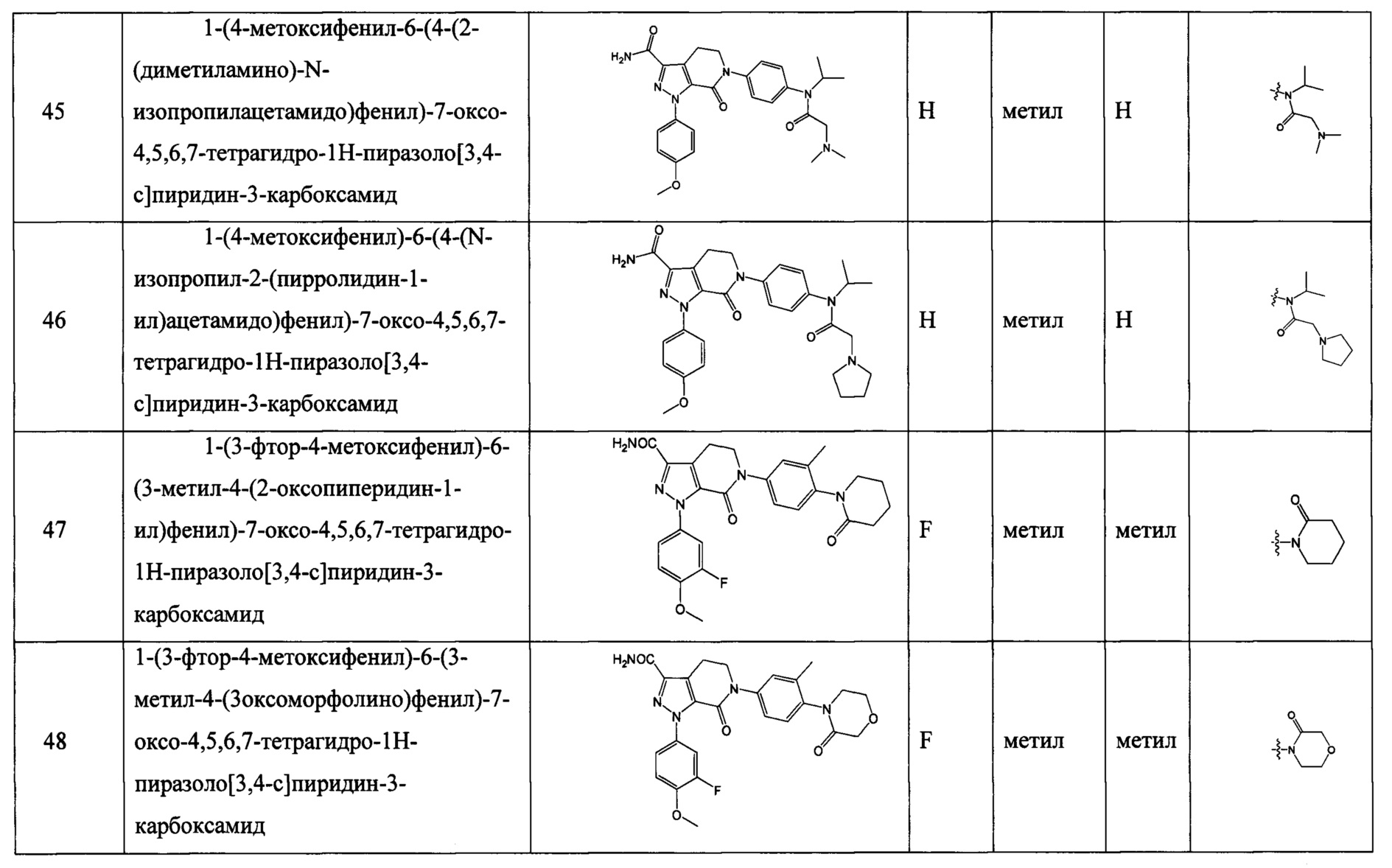
5, 6 или 7-членный циклический фрагмент замещен одним радикалом RNg, где RNg выбран из водорода, С1-4 алкила, гидроксила, С1-4 алкокси, галогена, оксо и амино,
5, 6 или 7-членный циклический фрагмент, кроме атома N, присоединенного к RNb, содержит 0, 1, 2, 3 или 4 гетероатома, выбранные из N, О и S,
5, 6 или 7-членный циклический фрагмент содержит 0, 1, 2 или 3 двойные связи.
Техническое решение 4.
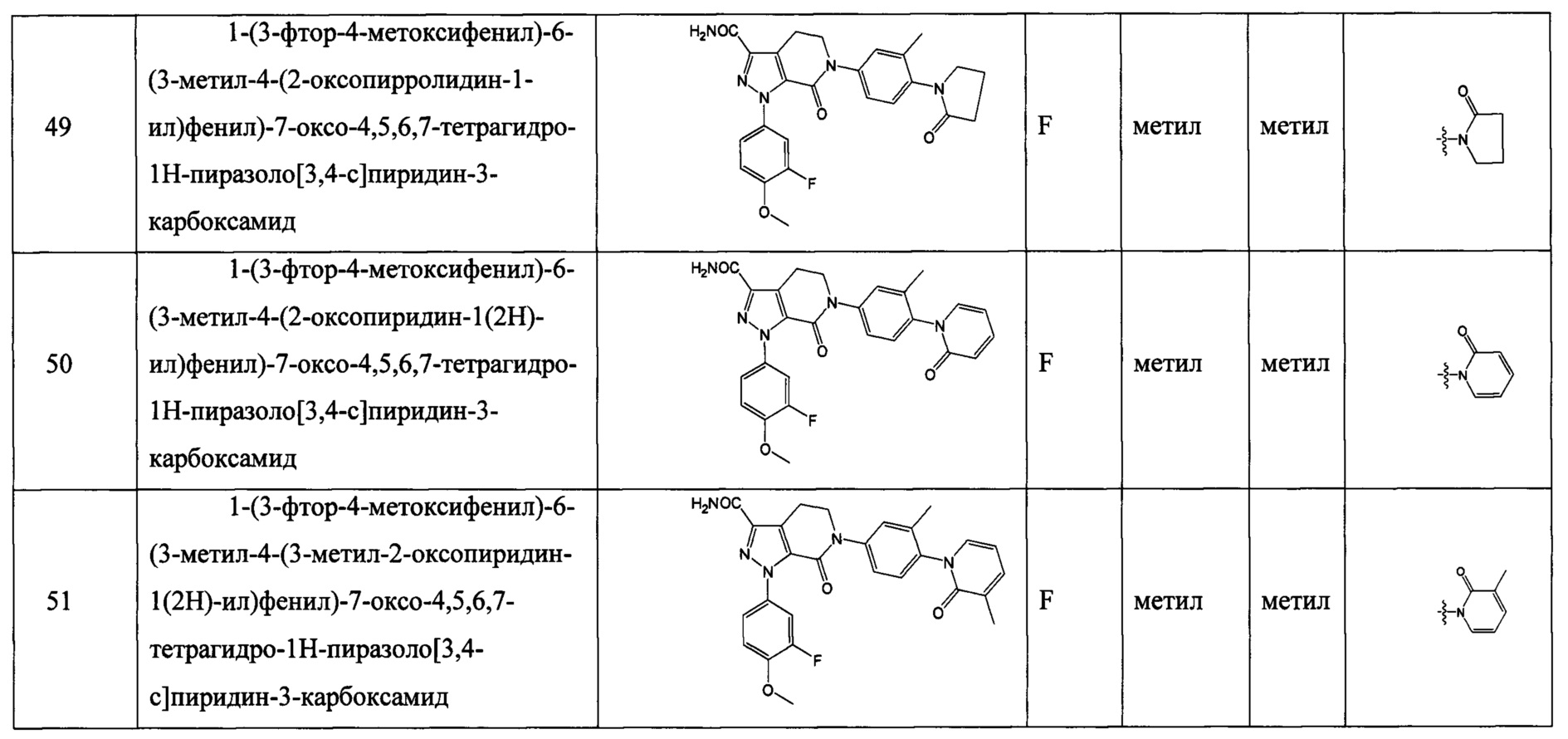
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
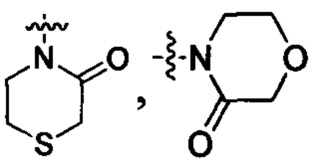
Z выбран из:
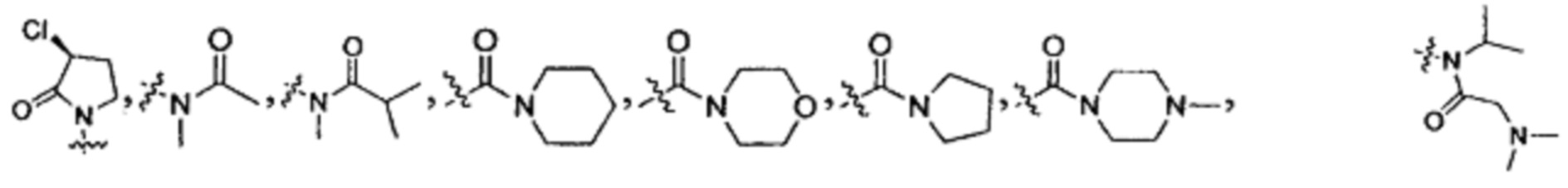 , и
, и
 .
.
Техническое решение 5.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль: где
RNc выбран из водорода и метила.
Техническое решение 6.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
RNd выбран из С1-3 алкокси.
Техническое решение 7.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
RNc выбран из водорода, хлора и фтора.
Техническое решение 8.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
RNf выбран из водорода, хлора и фтора.
Техническое решение 9.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
Z выбран из 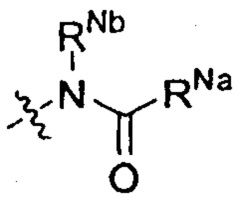 ;
;
RNa и RNb вместе с атомами, к которым они присоединены, образуют 5, 6 или 7-членный циклический фрагмент,
причем
5, 6 или 7-членный циклический фрагмент замещен одним радикалом RNg, где RNg выбран из водорода, С1-4 алкила, гидроксила, С1-4 алкокси, галогена, оксо и амино,
5, 6 или 7-членный циклический фрагмент, кроме атома N, присоединенного к RNb, содержит 0, 1, 2, 3 или 4 гетероатома, выбранные из N, О и S,
5, 6 или 7-членный циклический фрагмент содержит 0, 1, 2 или 3 двойных связи,
RNd выбран из C1-6 алкокси,
по меньшей мере один из RNc, RNg, RNe и RNf не является водородом.
Техническое решение 10.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
X обозначает СН;
Z выбран из 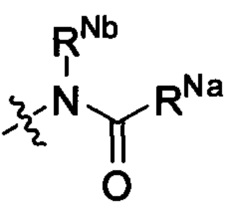 ;
;
RNa и RNb вместе с атомами, к которым они присоединены, образуют 5, 6 или 7-членный циклический фрагмент, причем
5, 6 или 7-членный циклический фрагмент замещен одним радикалом RNg, где RNg выбран из водорода и метила,
5, 6 или 7-членный циклический фрагмент, кроме атома N, присоединенного к RNb, содержит 0, 1, 2, 3 или 4 гетероатома, выбранные из N, О и S,
5, 6 или 7-членный циклический фрагмент содержит 0, 1, 2 или 3 двойных связи,
RNd выбран из этокси;
по меньшей мере один из RNc, RNg, RNe и RNf не является водородом.
Техническое решение 11.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
X обозначает СН;
Z выбран из 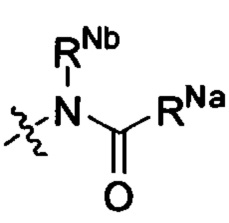 ;
;
RNa и RNb вместе с атомами, к которым они присоединены, образуют 5, 6 или 7-членный циклический фрагмент,
причем
5, 6 или 7-членный циклический фрагмент замещен одним радикалом RNg, где RNg выбран из водорода и метила,
5, 6 или 7-членный циклический фрагмент, кроме атома N, присоединенного к RNb, содержит 0, 1, 2, 3 или 4 гетероатома, выбранные из N, О и S,
5, 6 или 7-членный циклический фрагмент содержит 0, 1, 2 или 3 двойные связи,
RNd выбран из этокси;
RNc обозначает метил.
Техническое решение 12.
Соединение формулы (I) согласно любому предыдущему решению, его таутомер или его оптический изомер, или его фармацевтически приемлемая соль:
где
Z выбран из: 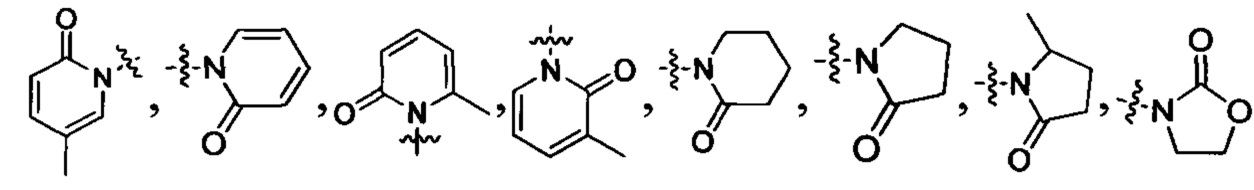 ,
,
 .
.
Техническое решение 13.
Следующие соединения, их таутомеры или их оптические изомеры, или их фармацевтически приемлемые соли:
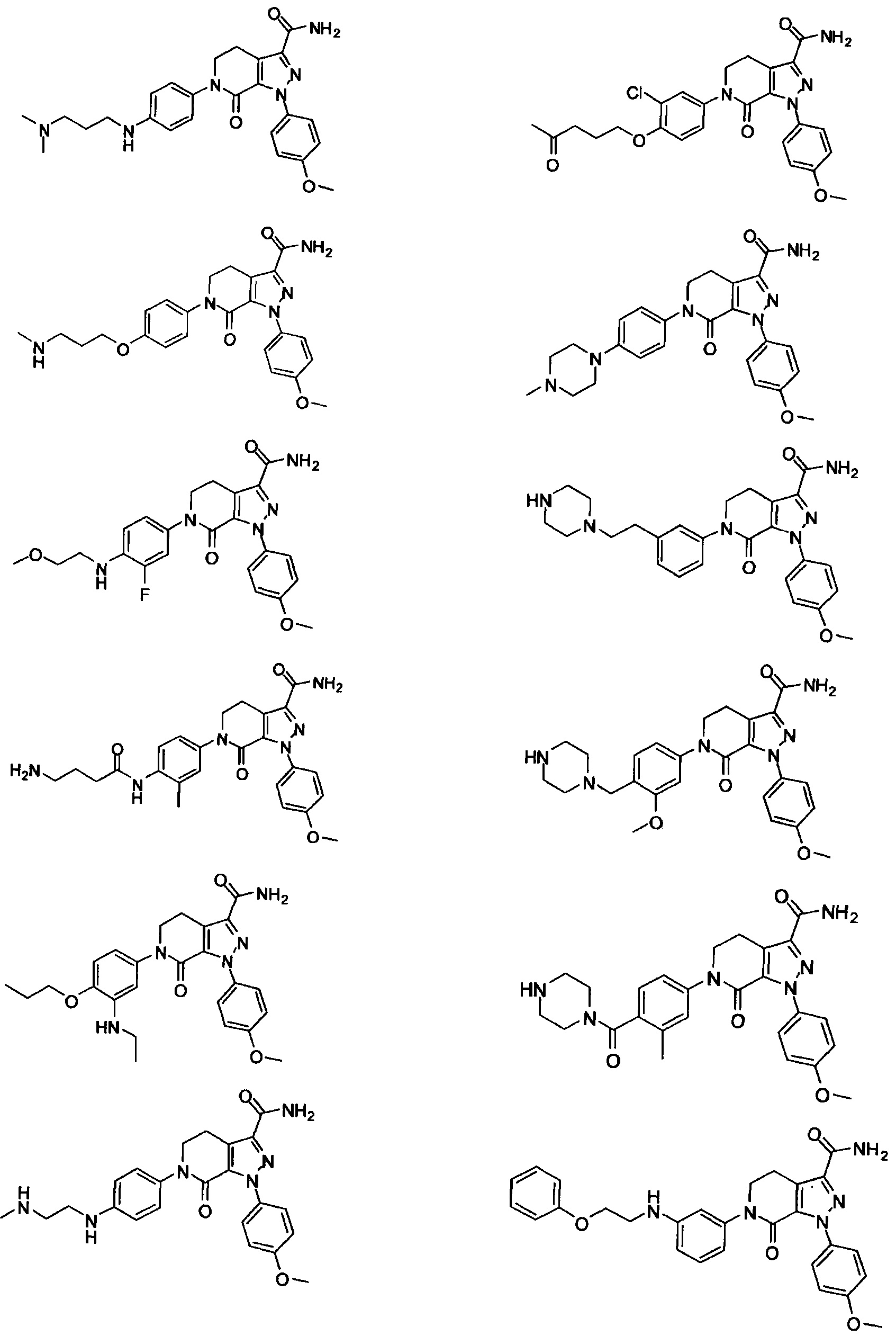
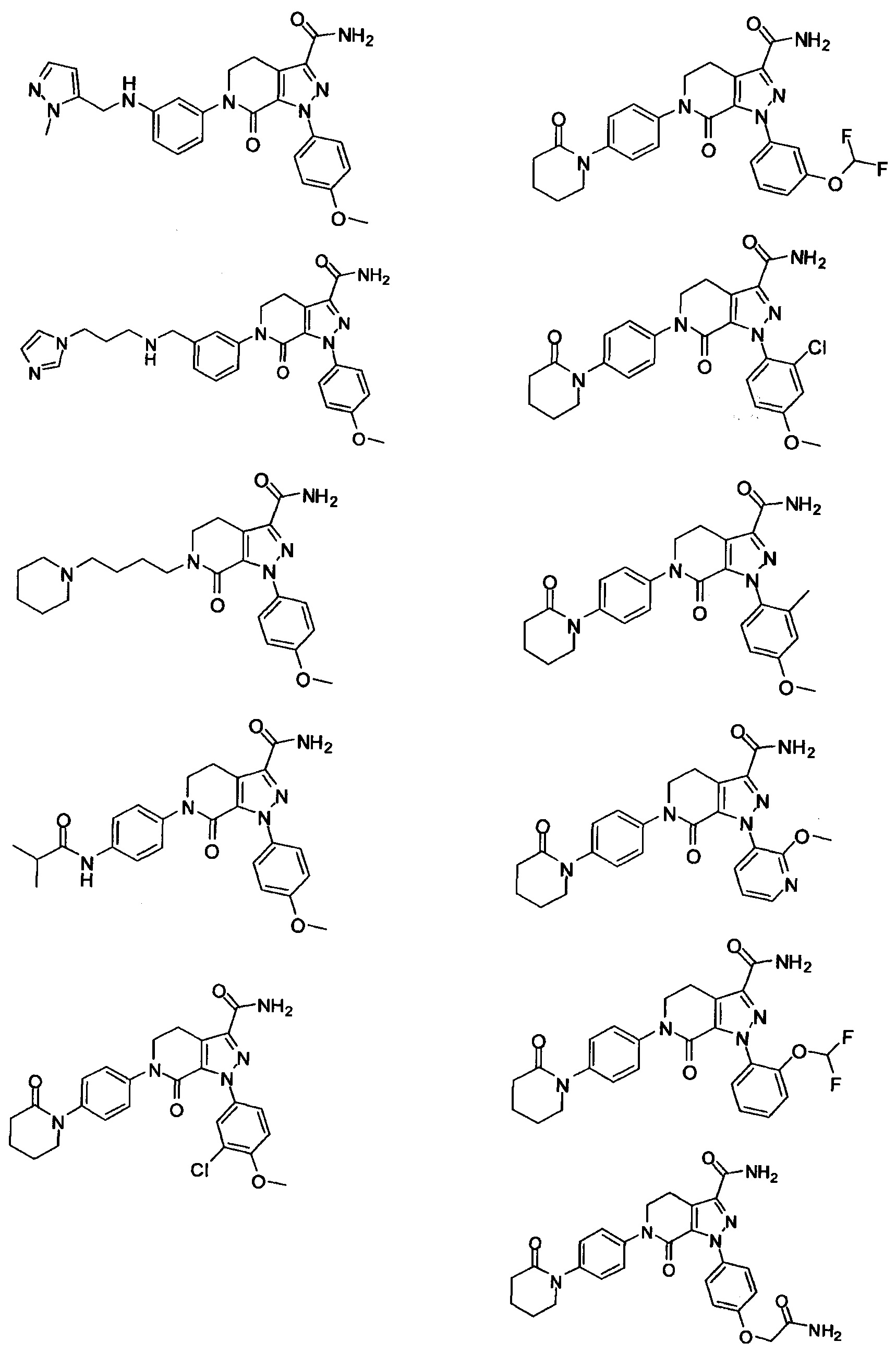
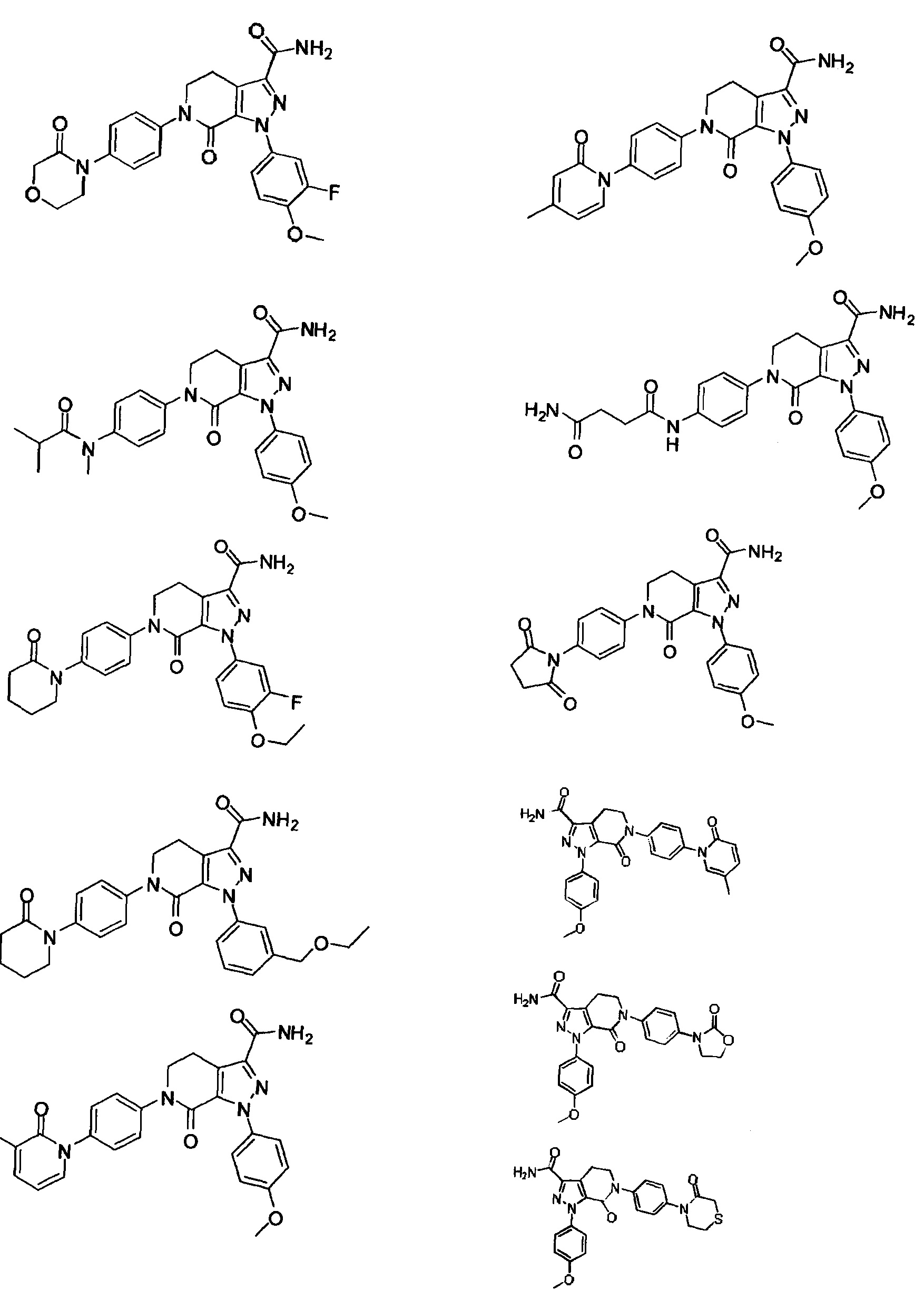
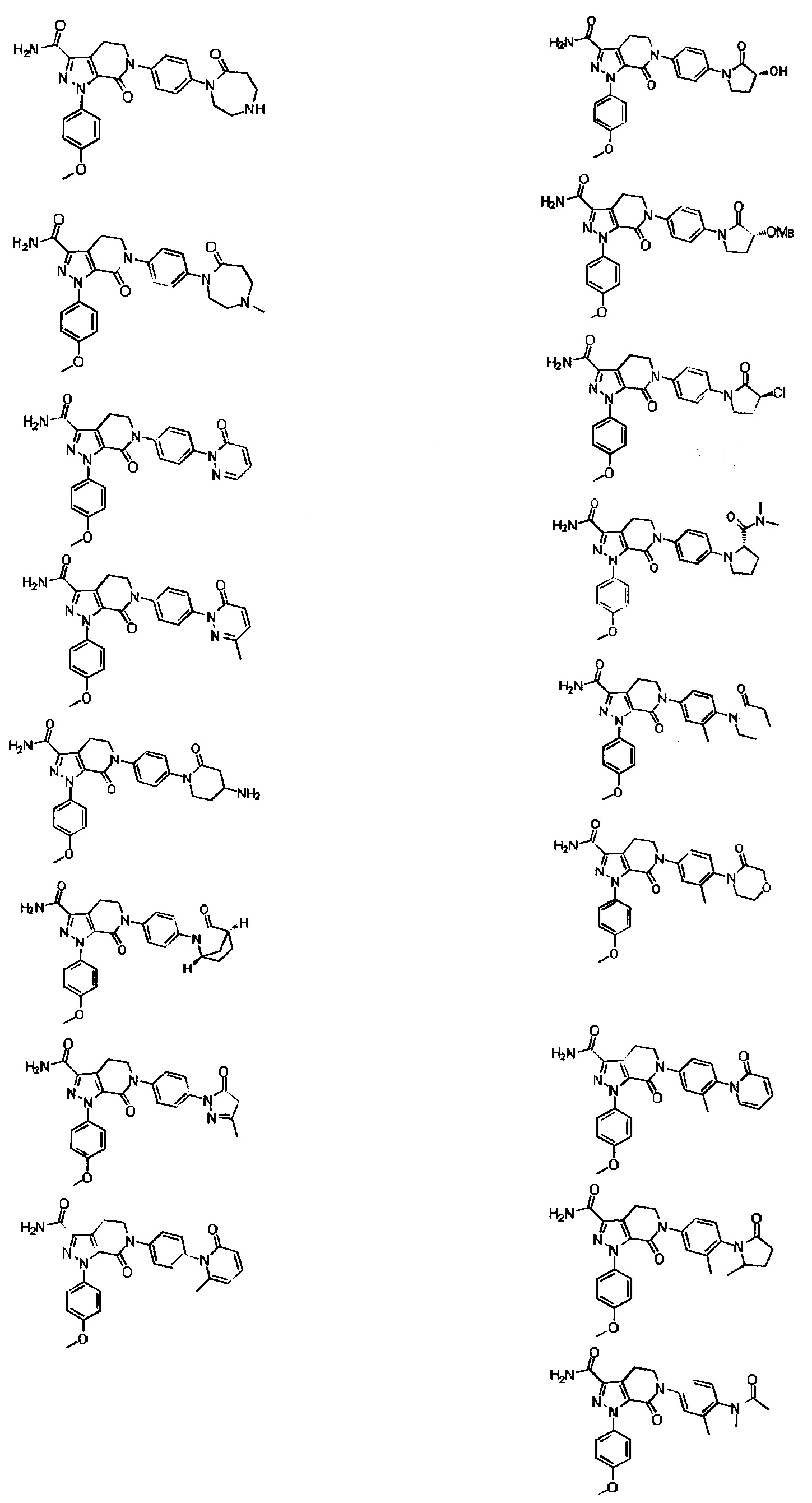
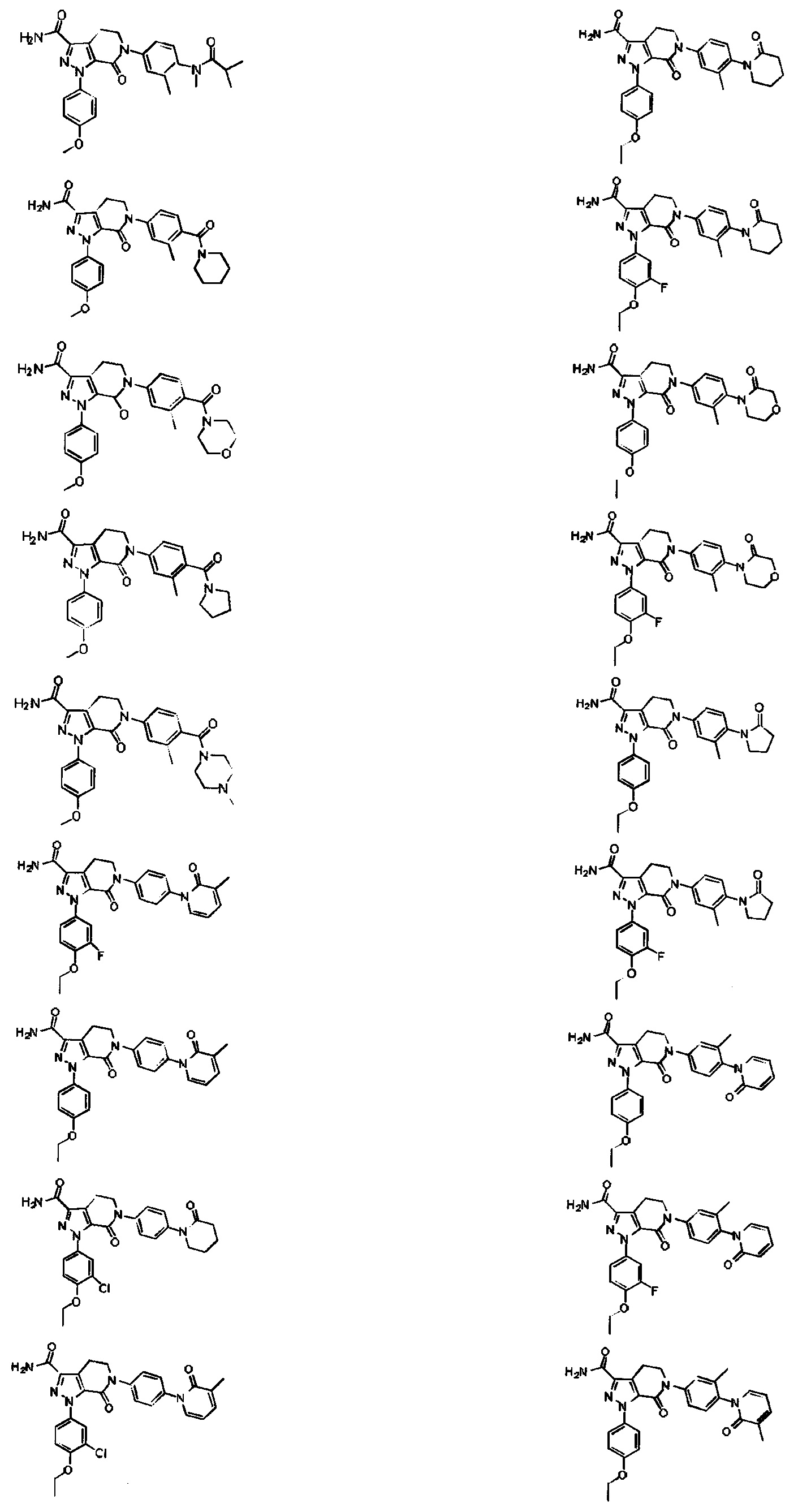
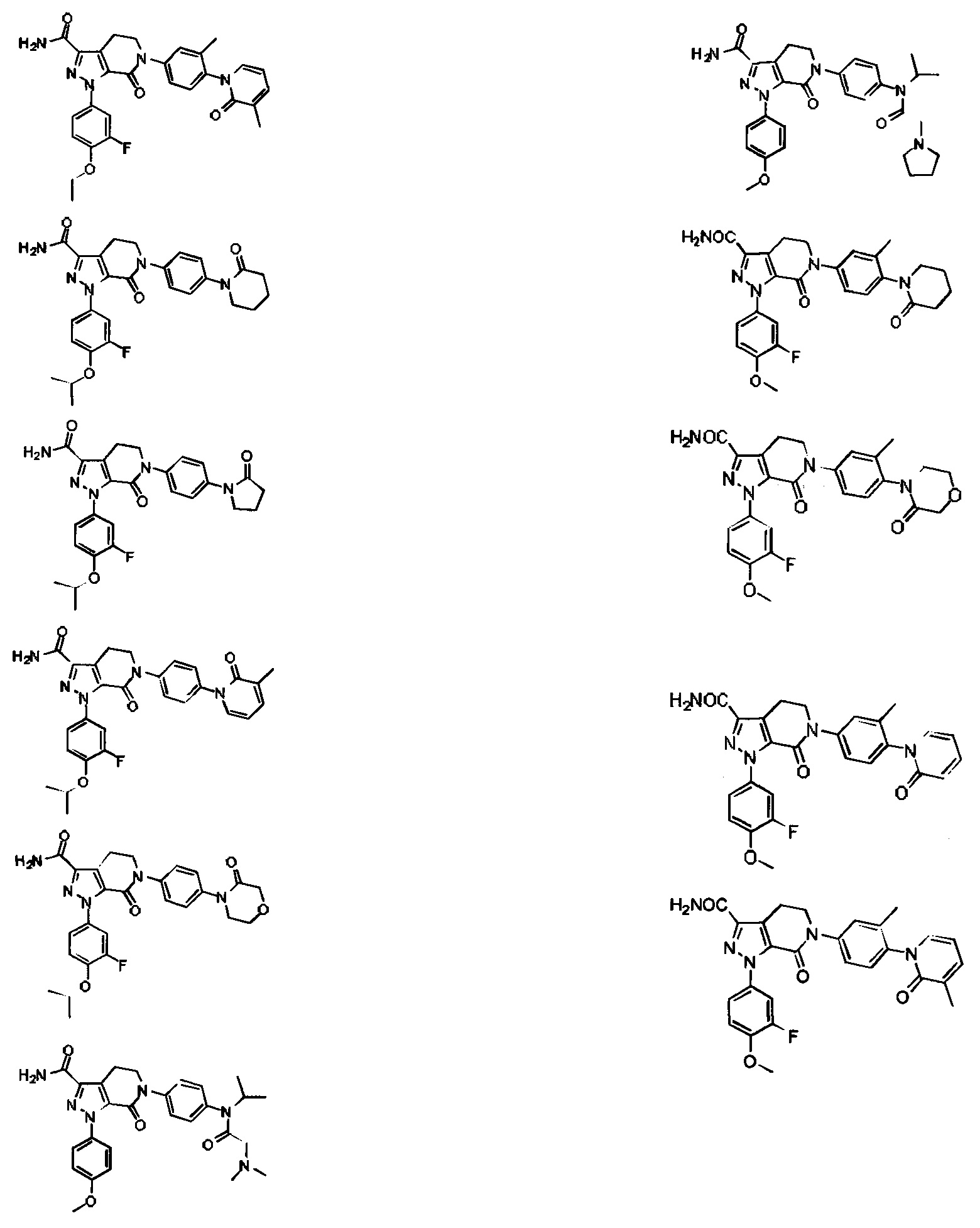
Настоящее изобретение предусматривает также новую фармацевтическую композицию с хорошим антитромботическим эффектом и пониженным риском возникновения кровотечения, содержащую соединение формулы (I) согласно любому техническому решению 1-13, его таутомер или его оптический изомер, или его фармацевтически приемлемую соль.
Настоящее изобретение предусматривает также применение соединения формулы (I) согласно любому техническому решению 1-13, его таутомера или его оптического изомера, или его фармацевтически приемлемой соли, или фармацевтической композиции согласно вышеуказанному техническому решению для приготовления лекарственного средства для профилактики и/или лечения заболевания, которые ингибируют положительное действие фактора Ха; при этом указанное заболевание выбрано, например, из тромбоэмболии и диссеминированного внутрисосудистого свертывания крови; например, указанное заболевание выбрано из инфаркта миокарда, стенокардии, реокклюзии и рестеноза после ангиопластики или аортокоронарного шунтирования, инсульта, преходящих парциальных припадков, окклюзионного заболевания периферических артерий, легочной эмболии или глубокого венозного тромбоза.
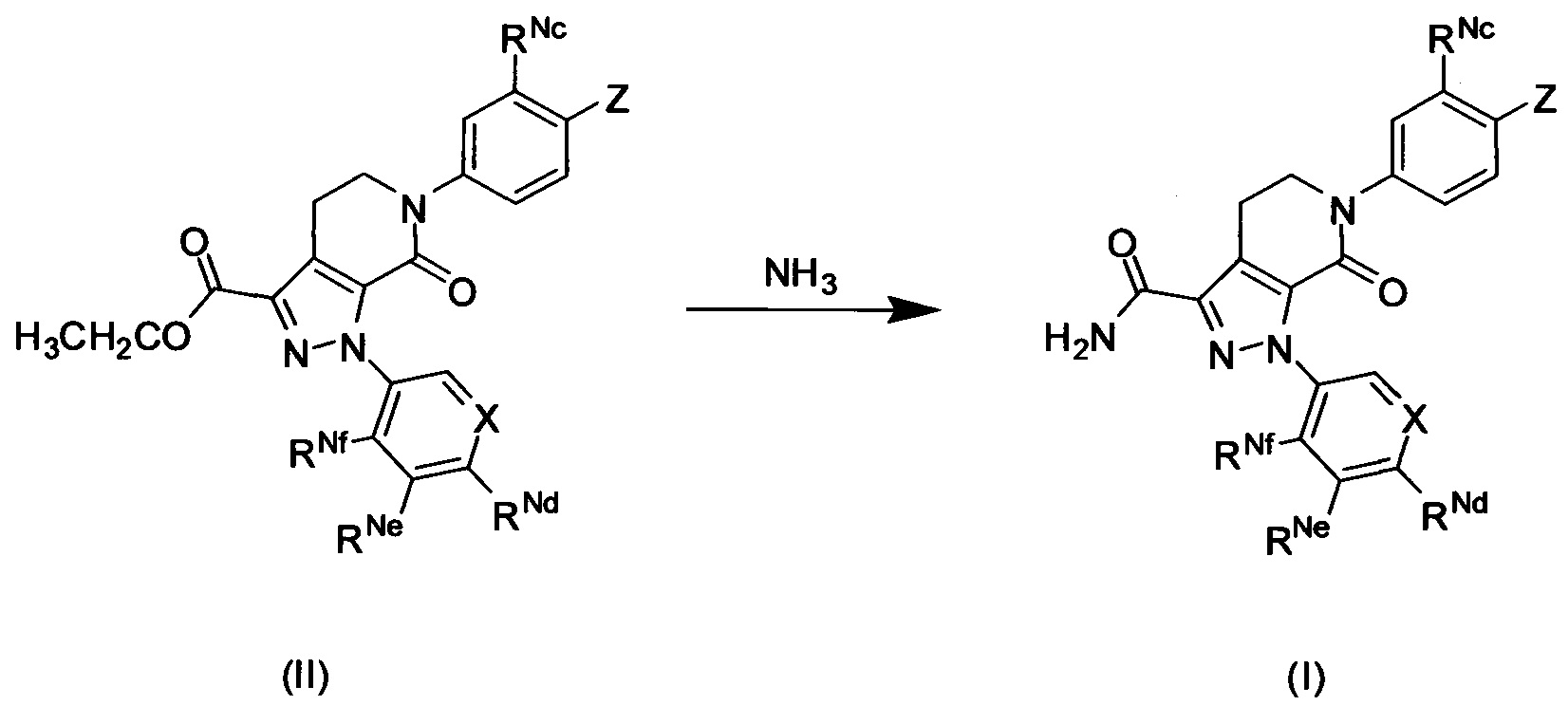
Данное изобретение предусматривает также способ получения соединения формулы (I) в соответствии с техническим решением 1, его таутомера или его оптического изомера, или его фармацевтически приемлемой соли, при этом такой способ включает аммонификацию соединения формулы (II) с получением соединения формулы (I):
Настоящее изобретение предусматривает также применение соединения формулы (I) согласно любому техническому решению 1-13, его таутомера или его оптического изомера, или его фармацевтически приемлемой соли, или фармацевтической композиции согласно вышеуказанному техническому решению для приготовления лекарственного средства для профилактики и/или лечения заболевания, которые ингибируют положительное действие фактора Ха в случае низкого риска возникновения кровотечения (например, риска кровотечения, меньшего по сравнению с применением апиксабана); при этом указанное заболевание выбрано, например, из тромбоэмболии и диссеминированного внутрисосудистого свертывания крови; например, указанное заболевание выбрано из инфаркта миокарда, стенокардии, реокклюзии и рестеноза после ангиопластики или аортокоронарного шунтирования, инсульта, преходящих парциальных припадков, окклюзионного заболевания периферических артерий, легочной эмболии или глубокого венозного тромбоза.
Осуществление изобретения
Соединения по изобретению могут существовать в виде геометрических изомеров. Соединения по изобретению могут содержать углерод-углеродные двойные связи или углерод-азотные двойные связи в Е- или Z-конфигурации, причем обозначение "Е" представляет старшие заместители на противоположных сторонах углерод-углеродных или углерод-азотных двойных связей, и термин "Z" представляет старшие заместители на той же самой стороне углерод-углеродных или углерод-азотных двойных связей по определению Правил приоритета (старшинства) Кана-Ингольда-Прелога. Соединения согласно данному изобретению могут также существовать в виде смеси "Е" и "Z" изомеров. Заместители вокруг циклоалкила или гетероциклоалкила обозначаются как цис- или транс-конфигурация.
Соединения согласно данному изобретению могут содержать асимметрично замещенные атомы углерода в R- или S-конфигурации, причем обозначения "R" и "S" указываются в соответствии с рекомендациями IUPAC 1974 Recommendations for Section Е, Fundamental Stereochemistry, Pure Appl. Chem. (1976) 45, 13-10. Соединения, содержащие асимметрично замещенные атомы углерода с равными количествами R- и S-конфигураций являются рацемическими при этих атомах. Атомы с избытком одной конфигурации в сравнении с другой конфигурацией считаются атомами с конфигурацией, находящейся в большем количестве, предпочтительно, в избытке около 85%-90%, более предпочтительно, в избытке около 95%- 99%, и еще более предпочтительно в избытке более примерно 99%. Соответственно, данное изобретение охватывает рацемические смеси, относительные и абсолютные стереоизомеры и смеси стереоизомеров.
Соединения согласно настоящему изобретению, содержащие группы NH, С(O)ОН, ОН или SH, могут включать фрагменты, образующие пролекарства. Фрагменты, образующие пролекарства, удаляются при протекании метаболических процессов, и при этом высвобождаются соединения, содержащие свободные гидроксильные группы, аминогруппы или карбоксильные группы in vivo. Пролекарства полезны для регулирования таких фармакокинетических свойств соединений, как растворимость и/или гидрофобность, абсорбция в желудочно-кишечном тракте, биодоступность, тканевая проницаемость и скорость клиренса.
Пролекарства являются производными активного лекарства, предназначенными для улучшения некоторых идентифицированных нежелательных физических или биологических свойств. Физическими свойствами обычно являются растворимость (слишком большая или недостаточная растворимость в липидах или воде) или связанная с этим стабильность, в то время как проблемными биологическими свойствами являются слишком быстрый метаболизм или плохая биодоступность, которая может быть отнесена к физико-химическим свойствам.
Пролекарства обычно получают путем: а) получения сложного эфира, гемиэфиров, эфиров угольной кислоты, нитратов, амидов, гидроксамовых кислот, карбаматов, иминов, оснований Манниха, фосфатов, фосфатных эфиров и енаминов активного лекарства, b) введения в молекулу лекарства функциональных групп: азогрупп, гликозидных групп, пептидных и групп простых эфиров, с) применения аминалей, гемиаминалей, полимеров, солей, комплексов, фосфорамидов, ацеталей, гемиацеталей и кеталей лекарства. Например, см. публикацию Andrejus Korolkovas's, "Essentials of Medicinal Chemistry", John Wiley-Interscience Publications, John Wiley and Sons, New York (1988), pp. 97-118, которая полностью включена в данную заявку посредством отсылки. Сложные эфиры могут быть получены из субстратов, содержащих или гидроксильную группу, или карбоксильную группу, способами, известными специалистам в данной области. Типичными реакциями этих соединений являются реакции замещения, приводящие к замене одного из гетероатомов другим атомом. Амиды могут быть получены подобным образом из субстратов, содержащих или аминогруппу, или карбоксильную группу. Сложные эфиры могут также взаимодействовать с аминами или аммиаком с образованием амидов. Другим способом получения амидов является совместный нагрев карбоновых кислот и аминов.
Соединения по изобретению могут существовать в формах, являющихся меченными или обогащенными изотопами, содержащих один или более атомов, имеющих атомную массу или массовое число, отличающиеся от атомной массы или массового числа атомов, наиболее часто встречающихся в природе. Изотопы могут быть радиоактивными или нерадиоактивными. Изотопы таких атомов как водород, углерод, фосфор, сера, фтор, хлор и йод, включают, но без ограничения, 2Н, 3Н, 13С, 14С, 15N, 18O, 32Р, 35S, 18F, 36Cl и 125I. Соединения, которые содержат другие изотопы этих и/или других атомов, также входят в объем данного изобретения.
В данной заявке термин "таутомер" обозначает соединение в состоянии химического равновесия между одной формой и другой формой, быстро превращающимися одна в другую при перемещении протона и электронном сдвиге. Один вид формы является приоритетным, например, в случае кето-енольной таутомерии.
В данной заявке термин "оптический изомер" обозначает вещество с идентичной молекулярной структурой, идентичными физическими и химическими свойствами, но с другой оптической активностью.
В данной заявке термин "соль" обозначает соль, выбранную из гидрохлорида, гидробромида, сульфата, сульфита, фосфата, мезилата, п-толуолсульфоната, малеата, тартрата, малата, фумарата, цитрата и т.п.
В данной заявке термин "C1-6 алкил" обозначает линейный или разветвленный насыщенный углеводородный радикал, содержащий 1-6 атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, втор.бутил, изобутил, трет-бутил и т.д.
В данной заявке термин "С2-6 алкенил" обозначает линейный или разветвленный ненасыщенный углеводородный радикал, содержащий 2-6 атомов углерода и содержащий по меньшей мере одну двойную связь, включая, но без ограничения, этенил, 1-пропенил, 2-пропенил (аллил), изопропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил и т.д.
В данной заявке термин "С2-6 алкинил" обозначает линейный или разветвленный ненасыщенный углеводородный радикал, содержащий 2-6 атомов углерода и содержащий по меньшей мере одну тройную связь, включая, но без ограничения, этинил, пропинил, бутинил и т.д.
В данной заявке термин "галоген" обычно обозначает фтор, хлор, бром или йод. В данной заявке термин " С1-6 алкокси" обозначает " С1-6 алкил-О-", где С1-6 алкил определен выше.
В данной заявке термин "карбамоил" обозначает "NH2-CO-".
В данной заявке термин "С2-6 алкилен" обозначает двухвалентный радикал, полученный из С2-6 алкана при удалении двух атомов водорода. С2-6 алкилен включает, но без ограничения, 1,2-этилен, 1,3-пропилен, 1,4-бутилен и т.д.
В данной заявке термин "С0-6 алкил" обозначает сочетание химической связи и C1-6 алкила.
В данной заявке термин "С1-6 алкокси- С0-6 алкил" обозначает С0-6 алкил, замещенный С1-6 алкокси. С1-6 алкокси-С0-6 алкил обозначает С1-6 алкокси.
В данной заявке термин "(С0-6 алкил)( С0-6 алкил)N- С1-6 алкил" обозначает С1-6 алкил, замещенный аминогруппой, при этом указанная аминогруппа далее замещена двумя группами С0-6 алкил, независимо друг от друга, причем С0-6 алкильное замещение обозначает отсутствие замещения.
В данной заявке термин "(С2-6 алкилен)N- С1-6 алкил" обозначает С1-6 алкил, замещенный аминогруппой, при этом атом N в указанной аминогруппе соединен с указанным С2-6 алкиленом с образованием насыщенного кольца.
В данной заявке термин "карбамоил-С1-6 алкил" обозначает С1-6 алкил, замещенный карбамоилом.
В данной заявке термин "5, 6 или 7-членный циклический фрагмент" обозначает кольцо, содержащее 5-7 членов, причем указанное кольцо содержит по меньшей мере один атом азота в качестве члена кольца, и кроме указанного атома азота указанный 5, 6 или 7-членный циклический фрагмент может также содержать 0, 1, 2, 3 или 4 гетероатома, выбранные из N, О и S; указанный 5, 6 или 7-членный циклический фрагмент содержит 0, 1, 2 или 3 двойные связи; указанный 5, 6 или 7-членный циклический фрагмент может быть замещен оксогруппой; указанный 5, 6 или 7-членный циклический фрагмент может быть в виде моноциклического кольца или мостикового кольца. Указанное 5, 6 или 7-членное циклическое кольцо включает, но без ограничения, пиррол, дигидропиррол, пирролидин, пиридин, дигидропиридин, тетрагидропиридин, пиперидин, морфолин, пиперазин, азациклогептан, 2-аза-бицикло[2,2,1]гептан и т.п.
Согласно другому аспекту настоящего изобретения предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) согласно данному изобретению, его таутомер или его оптический изомер, или его фармацевтически приемлемую соль.
Фармацевтическая композиция может вводиться перорально, например, в виде гранул, таблеток или капсул, или путем парентеральной инъекции, например, внутривенной инъекции, подкожной инъекции, внутримышечной инъекции или интратекальной инъекции, или путем трансфузии, например, в виде стерильных растворов, суспензий или эмульсий, или путем местного нанесения, например, в виде мази или крема, или путем ректального введения, например, в виде суппозитория. Обычно вышеупомянутые композиции могут быть приготовлены обычными способами с традиционными эксципиентами.
Согласно другому аспекту настоящего изобретения оно предусматривает применение соединения формулы (I) согласно данному изобретению, его таутомера или его оптического изомера, или его фармацевтически приемлемой соли согласно данному изобретению для приготовления лекарственного средства для профилактики и/или лечения заболевания, которые ингибируют положительную активность фактора Ха. Например, указанное заболевание выбрано, например, из тромбоэмболии и диссеминированного внутрисосудистого свертывания крови; например, указанное заболевание выбрано из инфаркта миокарда, стенокардии, реокклюзии и рестеноза после ангиопластики или аортокоронарного шунтирования, инсульта, преходящих парциальных припадков, окклюзионного заболевания периферических артерий, легочной эмболии или глубокого венозного тромбоза.
Согласно другому аспекту настоящего изобретения оно предусматривает способ получения соединения формулы (I) согласно данному изобретению, его таутомера или его оптического изомера, или его фармацевтически приемлемой соли, включающий аммонификацию соединения формулы (II) с получением соединения формулы (I):
Соединения согласно данному изобретению характеризуются хорошими антитромботическими свойствами и пониженным риском возникновения кровотечения. Эти характеристики могут быть определены и подтверждены при проведении следующего анализа биологической активности.
I. Определение анти-Ха активности
Материалы:
Фермент: фактор Ха (MERK)
Субстрат: CS-1122 (Hyphen)
Буфер: 50 мМ TrisHCl, 150 мМ NaCl, РН8.3
Методы:
100 мкл буфера, 50 мкл соединений с разными концентрациями (разведенные буфером) и 50 мкл (0.1 мк/мл) фермента, фактора Ха (разведенного буфером) добавляли в 96-луночный планшет. Через 15 мин добавляли 50 мкл хромогенного субстрата (2.5 мг/мл) CS-11 (22). Проводили мониторинг гидролиза субстрата путем измерения абсорбции при длине волны 405 нм при температуре 37°С в течение периода времени до 30 мин (с интервалом в 30 с), используя микропланшетный спектрофотометр. Величины IC50 определяли по методу Блисса. Эти величины соединений определяли, как описано ниже.

II Модель кровотечения из хвоста у крыс
Самцов крыс SD распределяли рандомизированно и вводили им лекарство или носитель. После введения крыс анестезировали путем внутрибрюшинной инъекции 10% хлоралгидрата. В моменты времени от 0.5 до 4 ч (Тмакс PK) после введения хвосты у анестезированных крыс рассекали на расстоянии 3 мм от кончика и погружали вертикально в физиологический раствор при температуре 37°С. Регистрировали момент времени, когда прекращался непрерывный поток крови в течение 30 с. Пролонгирование времени кровотечения выражали как отношение величины у обработанных крыс к средней величине у крыс, которым вводили носитель. Действие соединений определяли, как описано ниже.

III. Модель артериовенозного шунтирования у крыс
Две полиэтиленовые трубки длиной 10 см (внутренний и внешний диаметр - 1.50 и 2.88 мм, соответственно) соединяли с центральной частью (длина 8 см, внутренний и внешний диаметр - 2.11 и 3.77 мм, соответственно), содержащей шелковую нить длиной 6 см, заполняли гепарином (3.125 мк/мл) и помещали между правой сонной артерией и левой яремной веной у крыс.
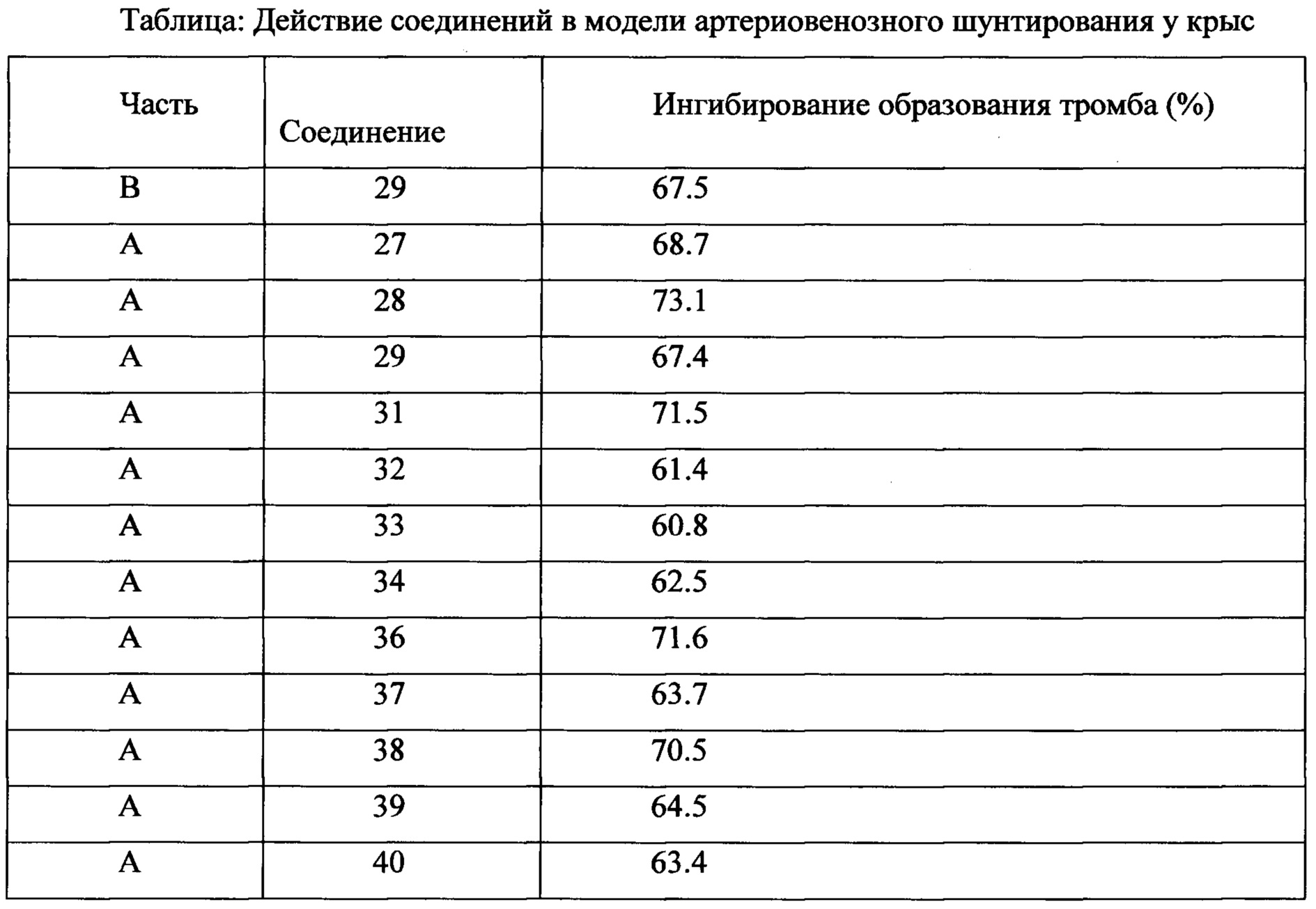
Самцов крыс SD анестезировали путем внутрибрюшинной инъекции 10% хлоралгидрата после оральной дозы, меняли фиксацию, затем изолировали правую сонную артерию и левую внешнюю яремную вену, проксимальная и дистальная нити сонной артерии были завязаны. Шприц с иглой канюлировали в сонную артерию и прикрепляли, другую трубку вставляли в правую внешнюю яремную вену. Крысы получали оральную дозу за 2-4 ч перед открытием шунта. Кровь текла из правой сонной артерии в полиэтиленовую трубку и затем возвращалась в левую внешнюю яремную вену. Затем шунт отсоединяли через 15 мин, и шелковую нить вместе с прилипшим тромбом немедленно вынимали и взвешивали. Определяли вес тромба во влажном состоянии. Затем шелковую нить высушивали при 60°С в течение 4 ч, чтобы определить сухой вес тромба. Антитромботическое действие агентов выражали как отношение степени ингибирования образования тромба у крыс, получивших лекарство, к степени ингибирования образования тромба у крыс, получивших носитель. Результаты показали, что соединения по изобретению обладали сильной антитромботической активностью (% ингибирования > 60%, даже > 70%).
Примеры
В следующем разделе способ получения соединений согласно настоящему изобретению иллюстрируется примерами. Соединения синтезировали способом, описанным в данной заявке, или же эти соединения были коммерчески доступными у следующих производителей: J&K Chemicals, Beijing InnoChem Science & Technology Co., Ltd., Aladdin, Alfa Aesar, Accela ChemBio Co., Ltd.
Сокращения названий соединений в примерах имеют следующие значения:
Вос трет-бутоксикарбонил
DCM метиленхлорид
DEAD диэтилазодикарбоксилат
DIPEA N,N-диизопропилэтиламин
DMF диметилформамид
DMSO диметилсульфоксид
ЕА этилацетат
EDCI 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид
HOBt гидроксибензотриазол
MeOH метанол
РЕ петролейный эфир
THF тетрагидрофуран
NIS N-йодсукцинимид
Prep-HPLC препаративная высокоэффективная жидкостная хроматография
(Вос)2О ди-трет-бутилдикарбонат
NBS N-бромсукцинимид
HATU O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
МТВЕ метил-трет-бутиловый эфир
NIS N-йодсукцинимид
KTB трет-бутоксид калия
TEA триэтиламин
MsCl метансульфонилхлорид
EG этандиол (этиленгликоль)
DMAP4-диметиламинопиридин
Часть А
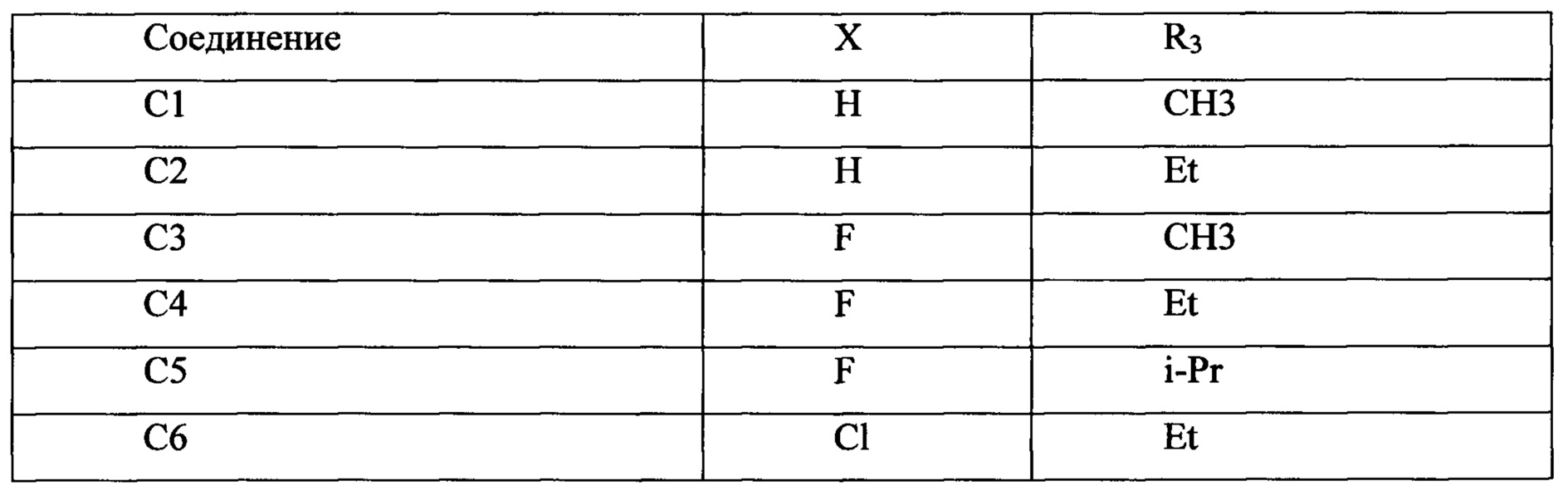
(1) Способ получения соединений C1-С6.
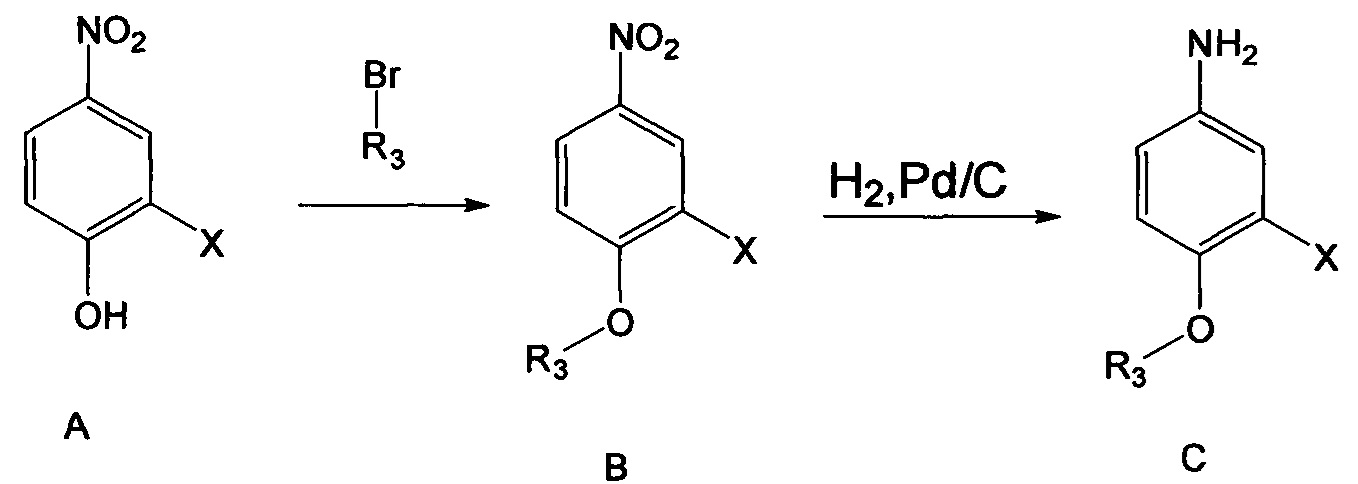
Соединение А было исходным коммерчески доступным веществом.
Получение соединения В: В реакционный сосуд добавляли соединение А (например, 61.8 ммоля), бромалкан (например, 154.6 ммоля), триэтиламин (например, 154.6 ммоля) и ацетонитрил (например, 80 мл). Полученную смесь нагревали до 50°С и
перемешивали для взаимодействия в течение 6 ч. После завершения реакции полученную смесь концентрировали и добавляли очищенную воду и этилацетат. Эту смесь перемешивали и экстрагировали, органическую фазу отделяли и концентрировали, получая масло с выходом > 95%.
Соединения С1 и С2 были коммерчески доступны.
Получение соединений С3-С6: в реакционный сосуд добавляли исходное вещество В (например, 59.5 ммол), Pd/C (например, 3.0 г) и метанол (например, 200 мл). Смесь гидрировали в течение 2 ч при нормальном давлении при комнатной температуре. После завершения реакции смесь отфильтровывали и концентрировали в вакууме с получением масла с выходом около 95%.
(2) Получение соединения F: соединение F, то есть соединения F1-F51, было/были получены согласно следующим методам 1-4.
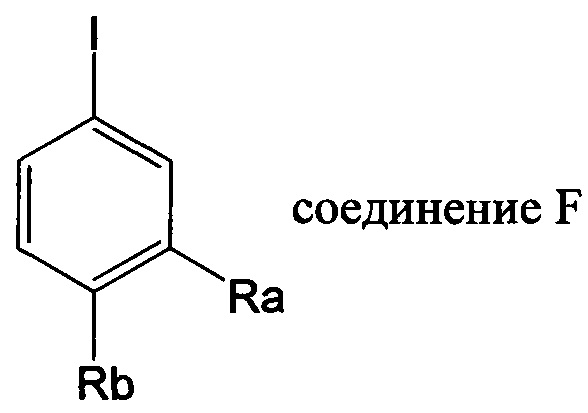
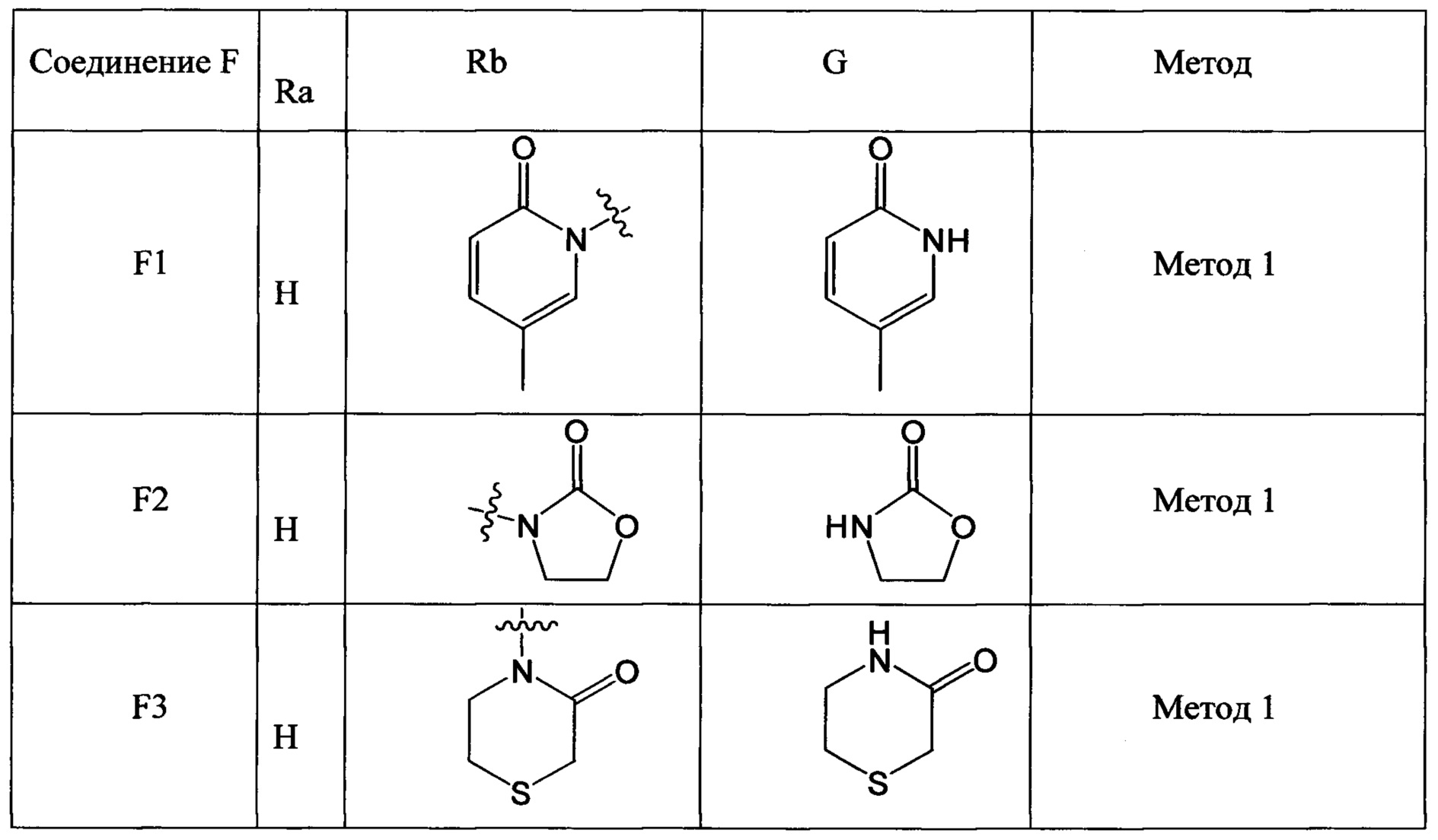
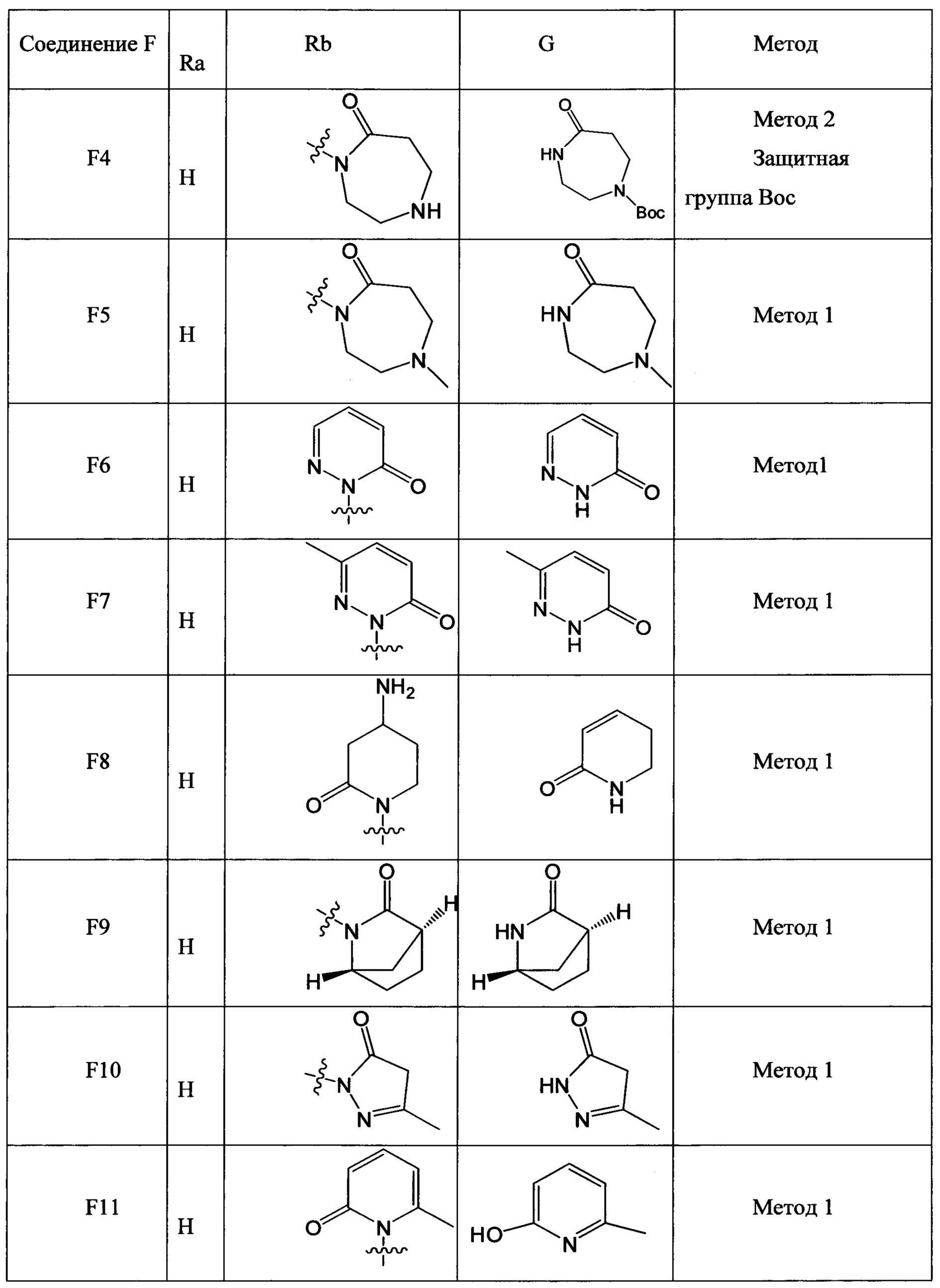
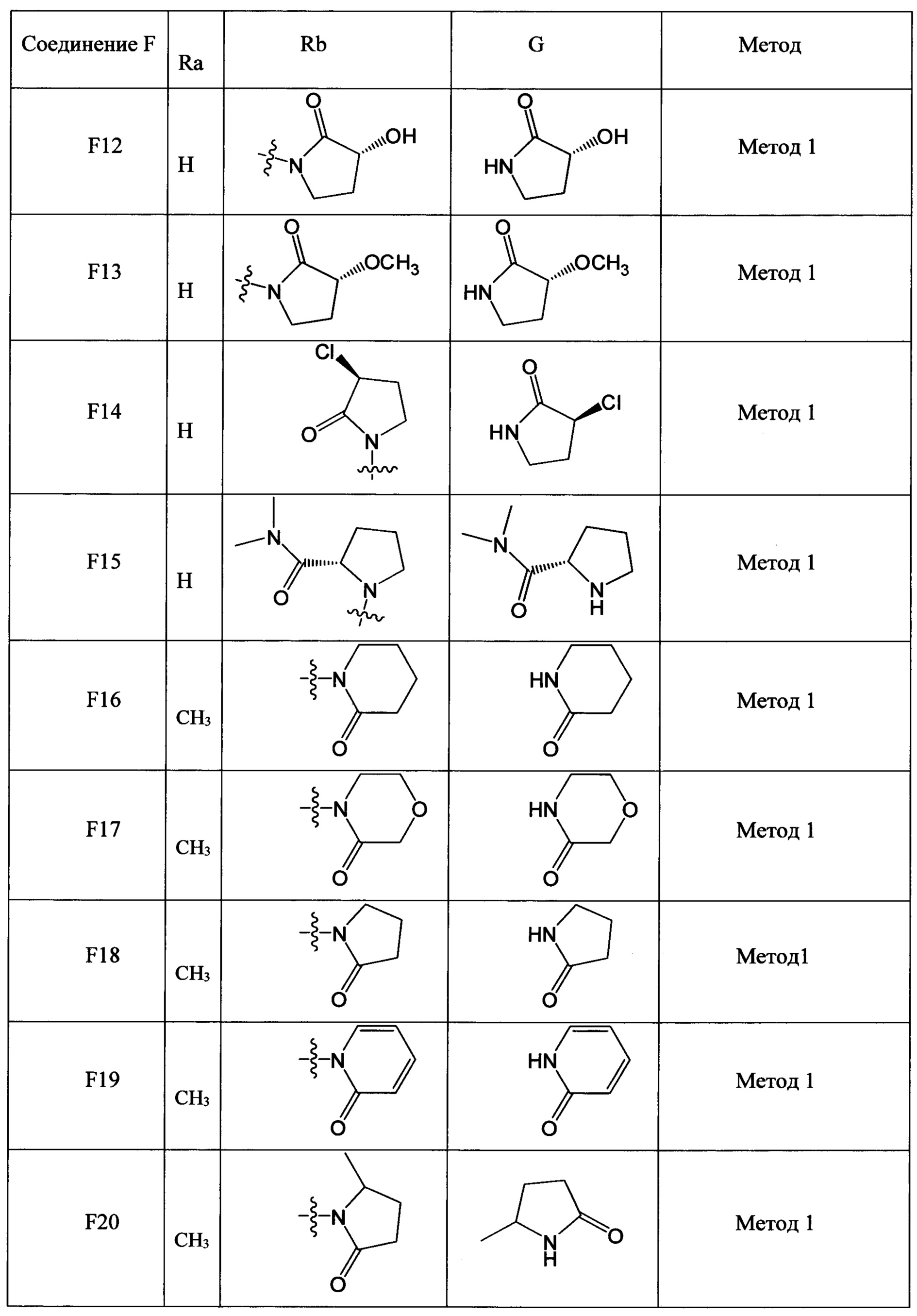
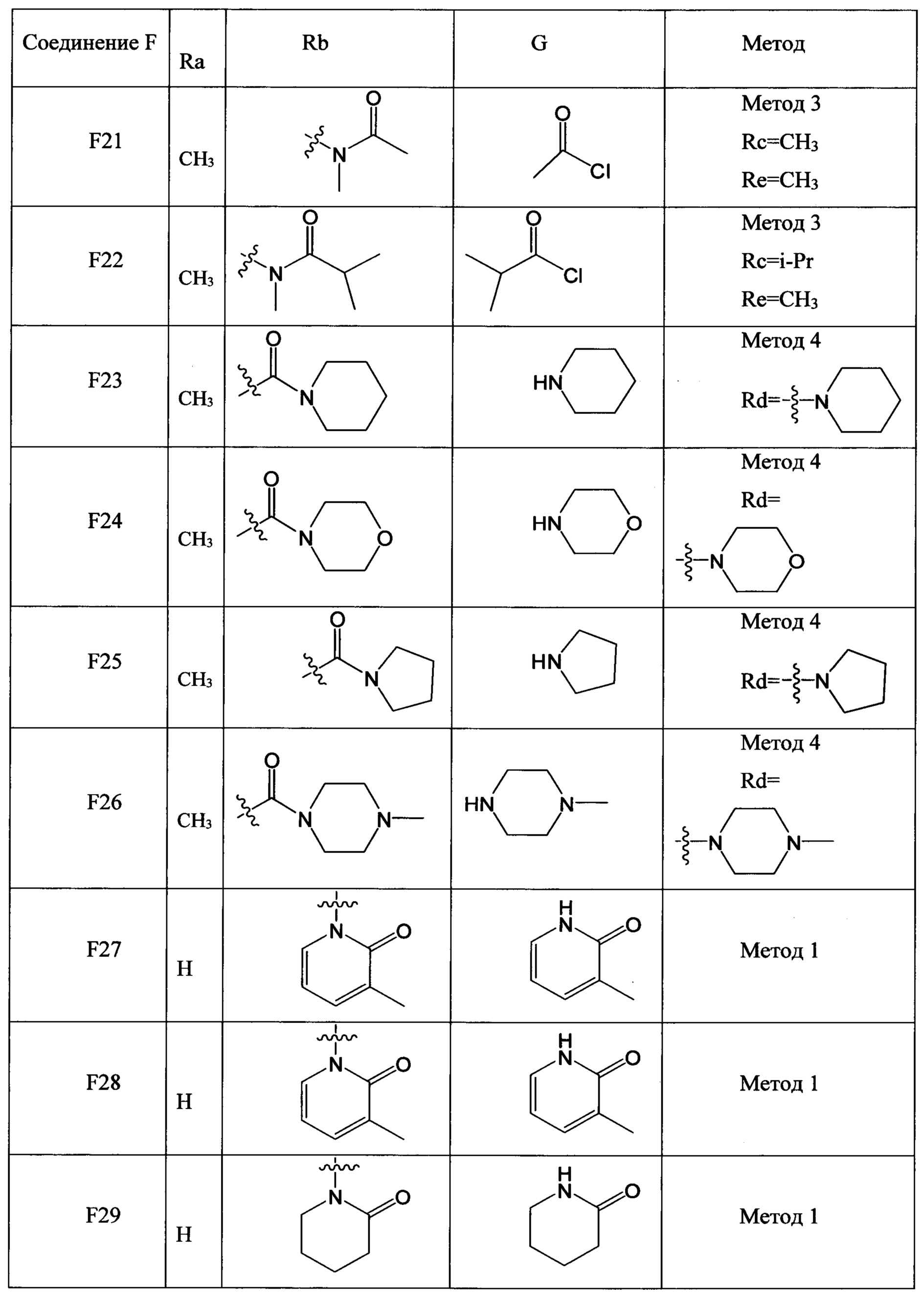
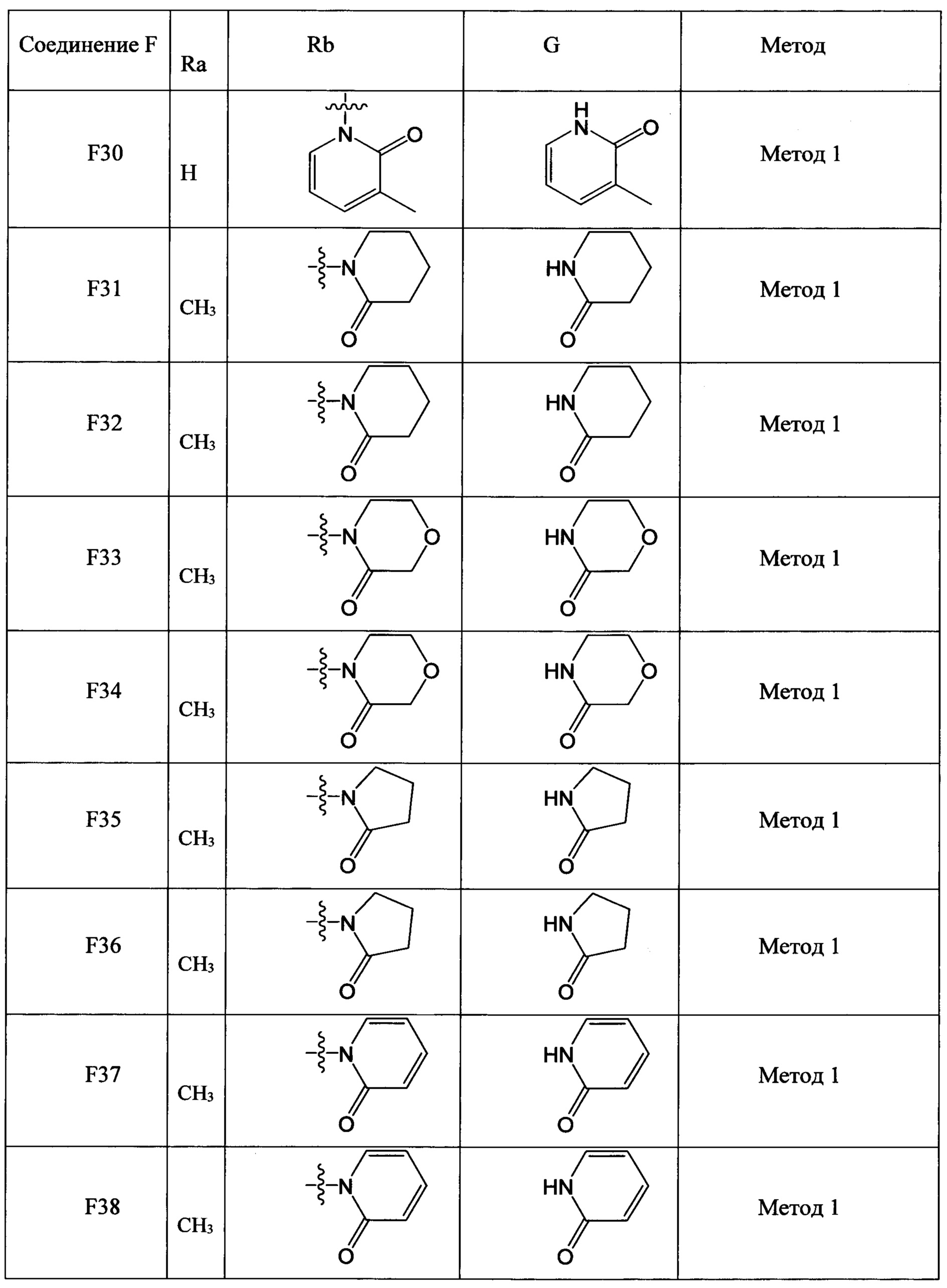
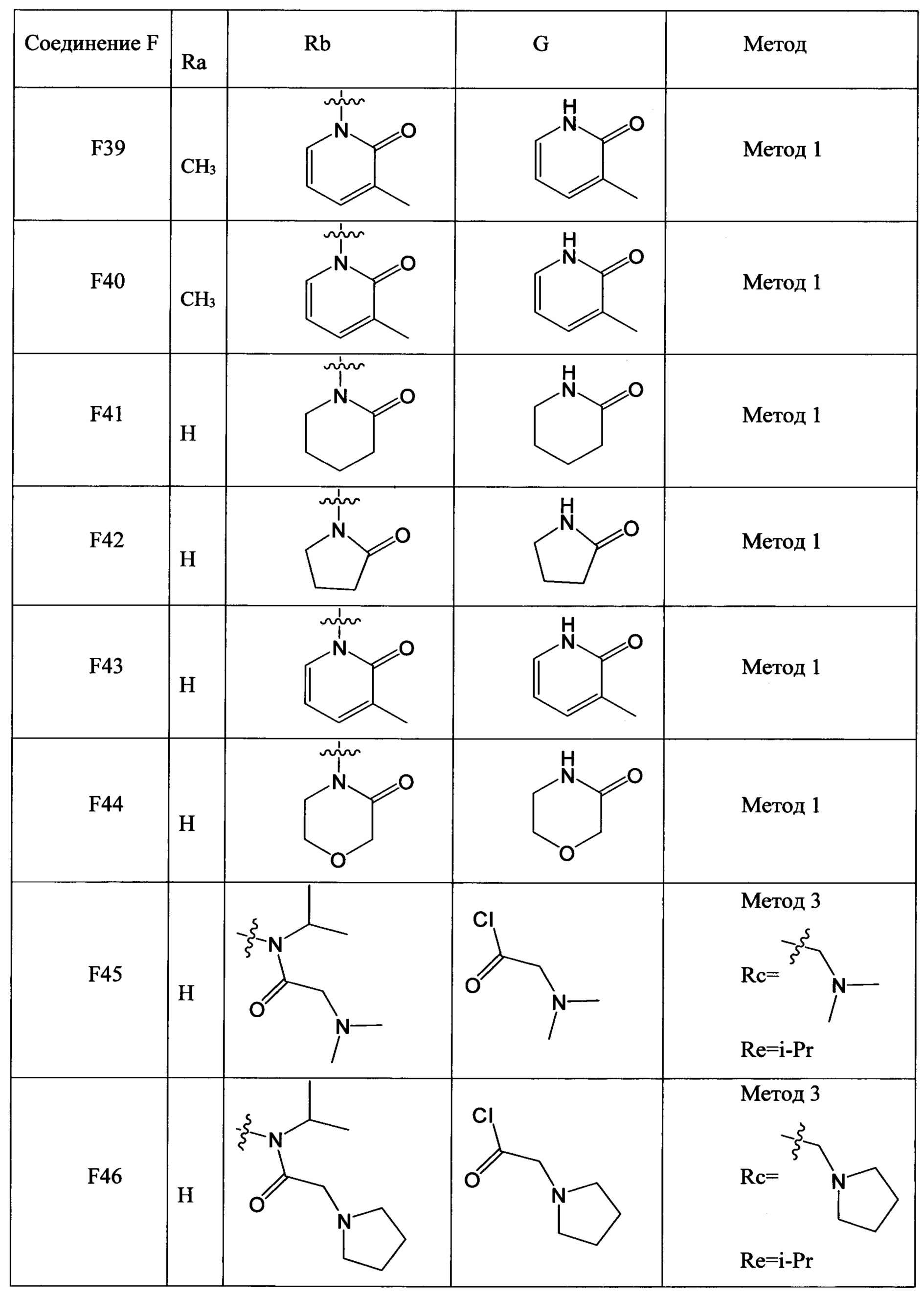
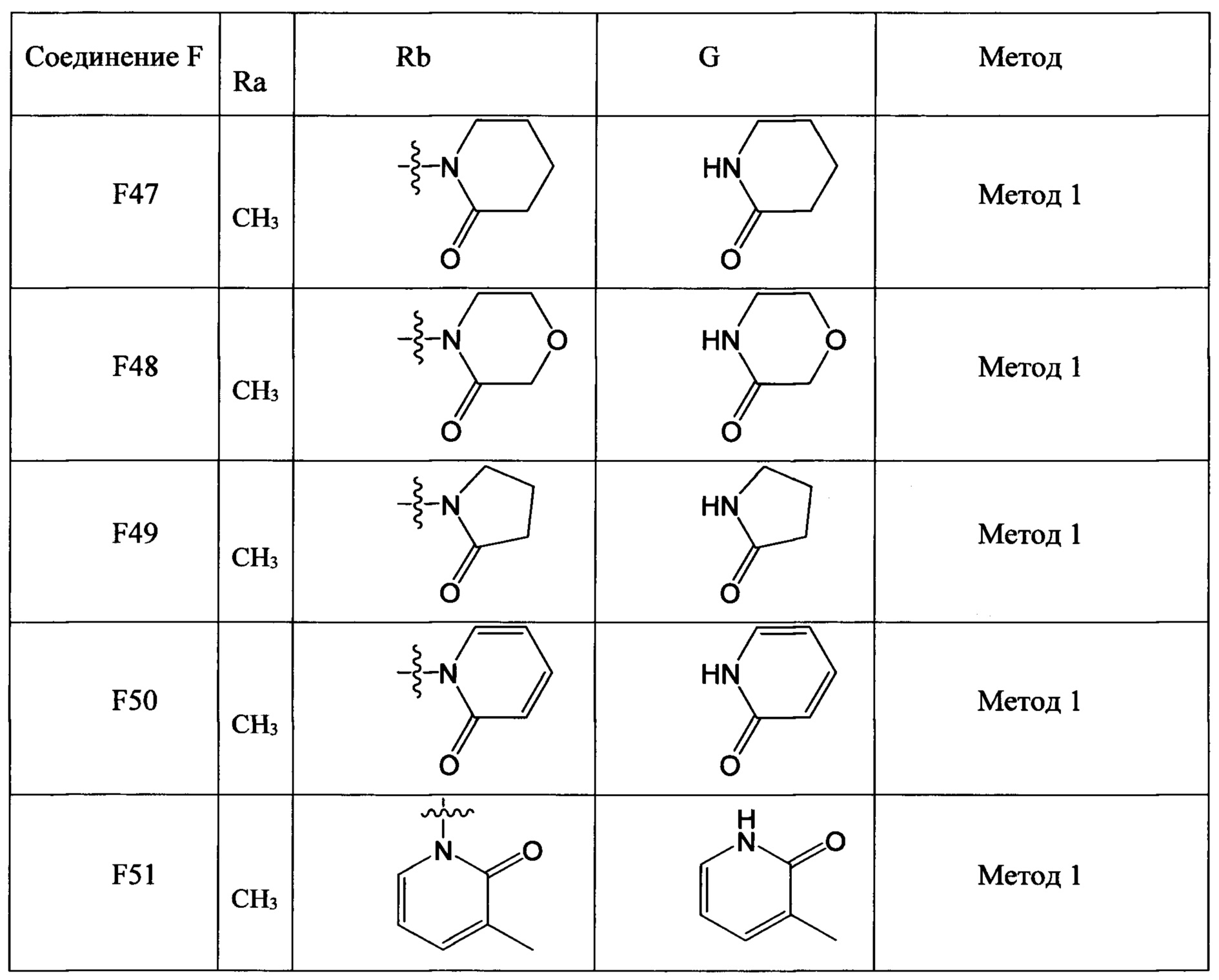
Метод 1
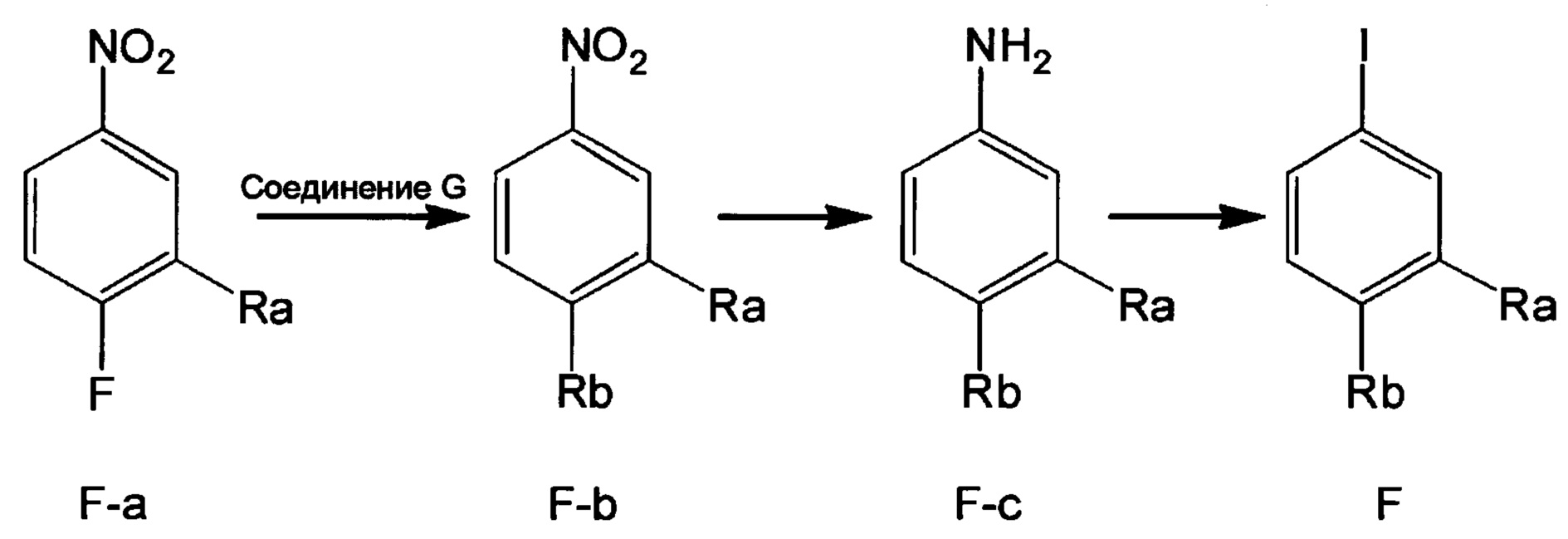
Стадия 1: Получение соединения F-b
В колбу объемом 50 мл добавляли соединение G (например, 40.0 ммолей) и трет-бутоксид калия (например, 40.0 ммолей), затем добавляли DMF (например, 30 мл). Смесь перемешивали в течение 1 ч при 0°С и добавляли соединение F-a (например, 20.0 ммолей). Полученную смесь нагревали до 90°С и проводили реакцию в течение 6 ч. После завершения реакции охлаждали реакционную смесь до комнатной температуры, добавляли очищенную воду и этилацетат. Органическую фазу отделяли и концентрировали с получением продукта с выходом около 70%.
Стадия 2: Получение соединения F-c
В колбу объемом 50 мл добавляли соединение F-b (например, 10.0 ммолей), Pd/C (например, 0.5 г) и метанол (например, 20 мл). Полученную смесь гидрировали при комнатной температуре и нормальном давлении в течение 4 ч. После завершения реакции реакционную смесь отфильтровывали и концентрировали фильтрат в вакууме с получением масла с выходом > 98%.
Стадия 3: Получение соединения F
В колбу объемом 50 мл добавляли соединение F-c (например, 8.0 ммолей), очищенную воду (например, 10 мл) и концентрированную соляную кислоту (например, 1.7 мл, 20.0 ммолей). Полученную смесь охлаждали при перемешивании до температуры 0-5°С. По каплям добавляли к смеси водный раствор нитрита натрия (например, 10 мл), пока поддерживалась температура 0-5°С. После окончания добавления по каплям смесь перемешивали в течение 20 мин при той же температуре. Затем к реакционной смеси добавляли йодид натрия (2.99 г, 20.0 ммолей) и полученную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции к реакционной смеси добавляли этилацетат. Затем разделяли водную фазу и органическую фазу. Водную фазу экстрагировали этилацетатом. Органические фазы соединяли и концентрировали с получением продукта с выходом 80%.
Метод 2
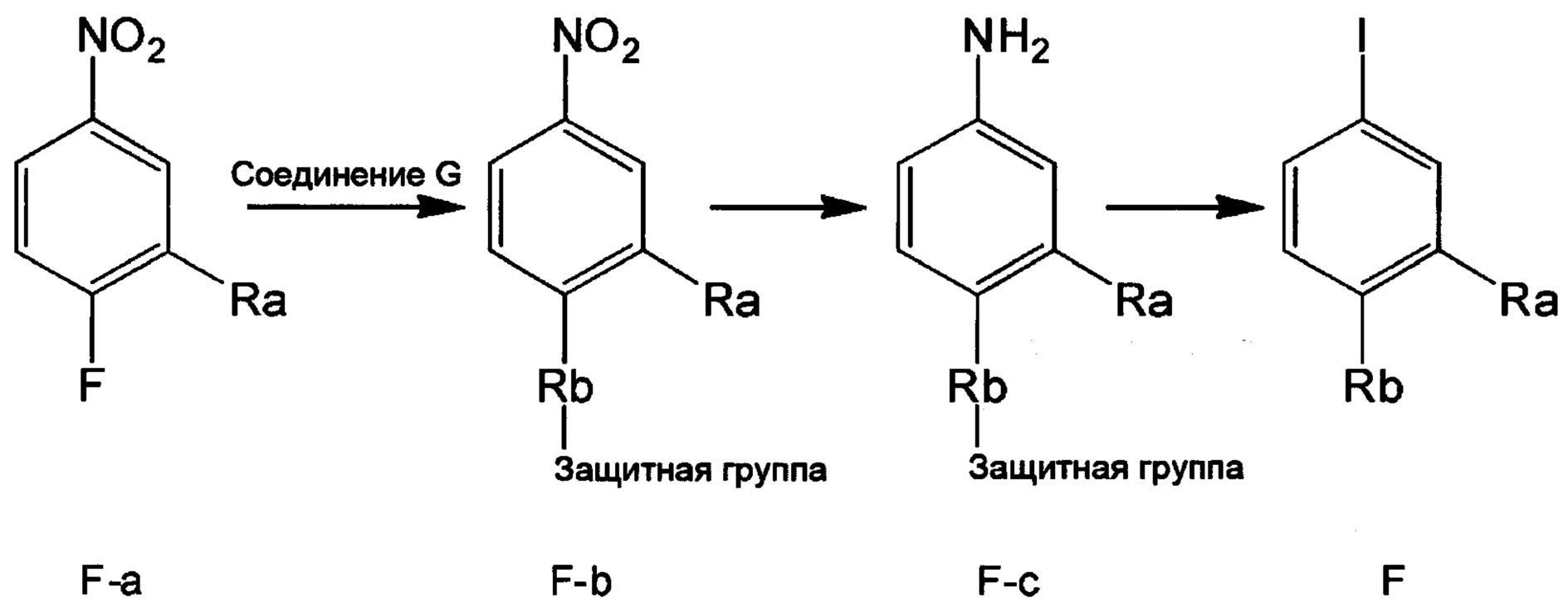
Стадия 1: получение соединения F-b, как описано в Методе 1.
Стадия 2: получение соединения F-c, как описано в Методе 1.
Стадия 3: получение соединения F.
В колбу объемом 50 мл добавляли соединение F-c (например, 8.0 ммолей), очищенную воду (например, 10 мл) и концентрированную соляную кислоту (например, 1.7 мл, 20.0 ммолей). Полученную смесь охлаждали при перемешивании до температуры 0-5°С. По каплям добавляли к смеси водный раствор нитрита натрия (например, 10 мл), пока поддерживалась температура 0-5°С. После окончания добавления по каплям смесь перемешивали в течение 20 мин при той же температуре. Затем к реакционной смеси добавляли йодид натрия (2.99 г, 20.0 ммолей) и полученную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции к реакционной смеси добавляли этилацетат. Затем разделяли водную фазу и органическую фазу. Водную фазу экстрагировали этилацетатом. Органические фазы соединяли и отфильтровывали. К фильтрату добавляли трифторуксусную кислоту (например, 10.0 ммолей). Полученную смесь перемешивали при комнатной температуре в течение 4 ч. К смеси добавляли воду и экстрагировали полученный продукт. Органическую фазу концентрировали в вакууме с получением продукта с выходом около 80%.
Метод 3
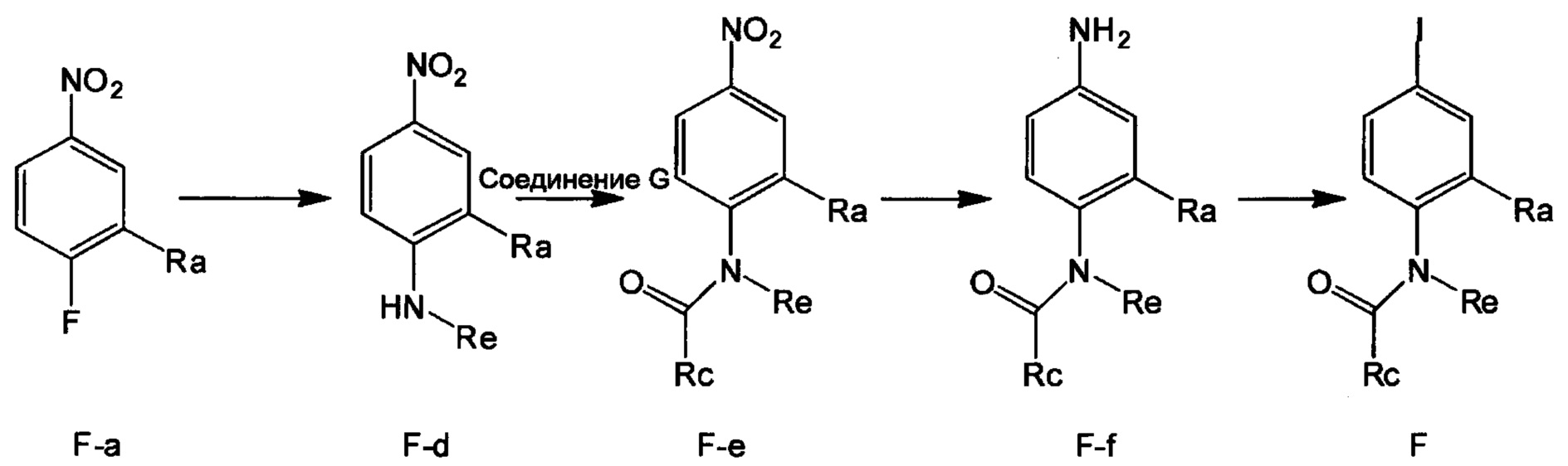
Стадия 1: Получение соединения F-d
В колбу объемом 50 мл добавляли соединение F-a (например, 20.0 ммол), водный раствор алкиламина (такого, как метиламин и изопропиламин) (например, 60.0 ммолей) и карбоната калия (например, 5.5 г, 39.8 ммоля), затем добавляли DMF (например, 30 мл). Полученную смесь нагревали до 50°С и проводили реакцию в течение 4 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры и выливали в очищенную воду. Полученную смесь отфильтровывали, получали продукт с выходом около 80%.
Стадия 2: Получение соединения F-e
В колбу объемом 50 мл добавляли соединение F-d (например, 5.0 ммолей), триэтиламин (например, 1.0 ммоль) и дихлорметан (например, 20 мл). Полученную смесь охлаждали до 0°С и по каплям добавляли соединение G (например, 6.0 ммолей). После окончания добавления по каплям смесь перемешивали в течение 1 ч при комнатной температуре. К полученной смеси добавляли 5% водный раствор карбоната натрия (например, 40 мл). Органическую фазу отделяли и концентрировали с получением продукта с выходом около 90%.
Стадия 3: Получение соединения F-f
В колбу объемом 50 мл добавляли соединение F-e (например, 3.8 ммоля), Pd/C (например, 0.2 г) и метанол (например, 20 мл). Смесь гидрировали при комнатной температуре и нормальном давлении в течение 4 ч. После завершения реакции реакционную смесь отфильтровывали и концентрировали фильтрат с получением продукта с выходом > 95%.
Стадия 4: Получение соединения F
В колбу объемом 50 мл добавляли соединение F-f (например, 3.9 ммоля), очищенную воду (например, 10 мл) и концентрированную соляную кислоту (например, 0.8 мл, 9.6 ммоля). Полученную смесь при перемешивании охлаждали до 0-5°С. К смеси добавляли по каплям водный раствор нитрита натрия (например, 10 мл) при поддержании температуры равной 0-5°С. После окончания добавления по каплям смесь перемешивали в течение 20 мин при поддержании указанной температуры. Затем к реакционной смеси добавляли йодид натрия (например, 1.17 г, 7.8 ммоля) и полученную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции к реакционной смеси добавляли этилацетат. Затем разделяли водную фазу и органическую фазу. Водную фазу экстрагировали этилацетатом. Органические фазы соединяли и концентрировали с получением продукта с выходом около 80%.
Метод 4
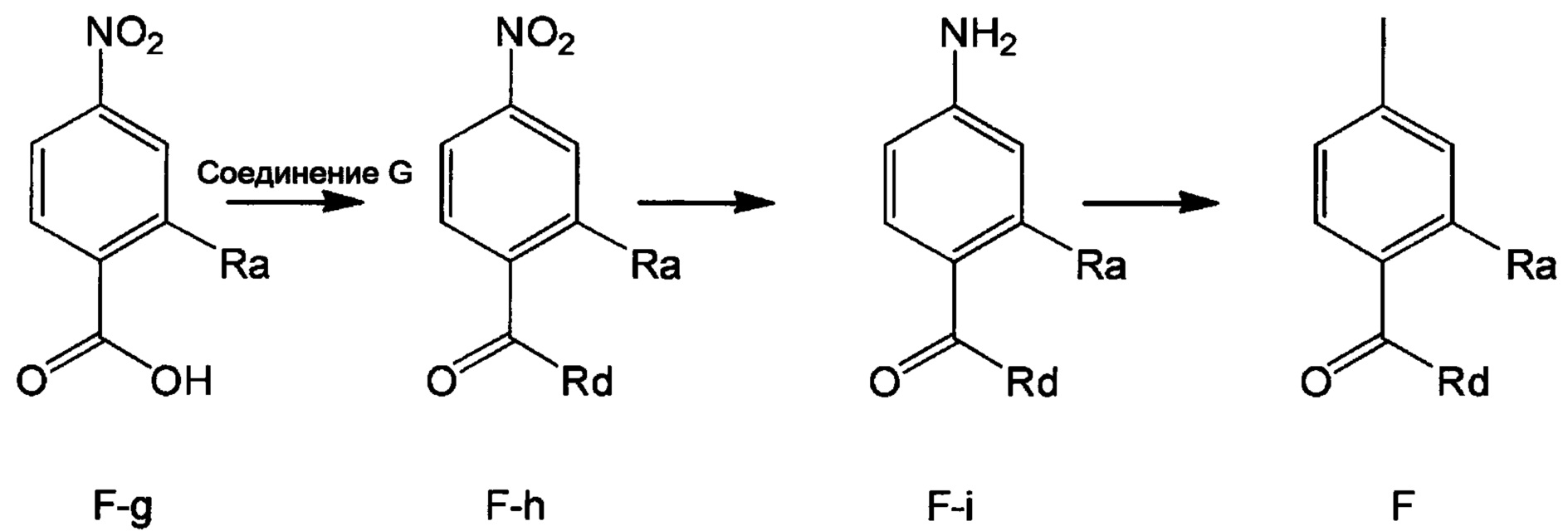
Стадия 1: Получение соединения F-h
В колбу объемом 50 мл добавляли соединение F-g (например, 5.52 ммоля) и тионилхлорид (например, 16.8 г, 141.2 ммоля). Смесь нагревали до 50°С и перемешивали для реакции в течение 2 ч. После завершения реакции реакционную смесь концентрировали в вакууме. После окончания концентрирования к смеси добавляли DCM (например, 30 мл), и при 0°С добавляли соединение G (например, 6.07 ммоля). После окончания его добавления смесь перемешивали в течение 1 ч при комнатной температуре и добавляли очищенную воду (например, 30 мл). Органическую фазу отделяли от полученной смеси и концентрировали с получением продукта с выходом около 95%.
Стадия 2: Получение соединения F-i
В колбу объемом 100 мл добавляли соединение F-h (например, 5.24 ммоля), Pd/C (например, 0.5 г) и метанол (например, 50 мл). Смесь гидрировали при комнатной температуре и нормальном давлении в течение 4 ч. После завершения реакции реакционную смесь отфильтровывали и фильтрат концентрировали в вакууме с получением продукта с выходом > 95%.
Стадия 3: Получение соединения F
В колбу объемом 50 мл добавляли соединение F-i (например, 3.91 ммоля), очищенную воду (например, 10 мл) и концентрированную соляную кислоту (например, 0.8 мл, 9.6 ммоля.
Смесь охлаждали до 0-5°С при перемешивании. К полученной смеси добавляли по каплям водный раствор нитрита натрия (например, 10 мл), поддерживая температуру равной 0-5°С. После окончания его добавления по каплям смесь перемешивали в течение 20 мин при поддержании той же температуры. Затем к реакционной смеси добавляли йодид натрия (например, 1.17 г, 7.8 ммоля) и полученную смесь перемешивали в течение 2 ч. После завершения реакции к реакционной смеси добавляли этилацетат. Разделяли водную фазу и органическую фазу. Водную фазу экстрагировали этилацетатом. Органические фазы соединяли и концентрировали с получением продукта с выходом около 50%.
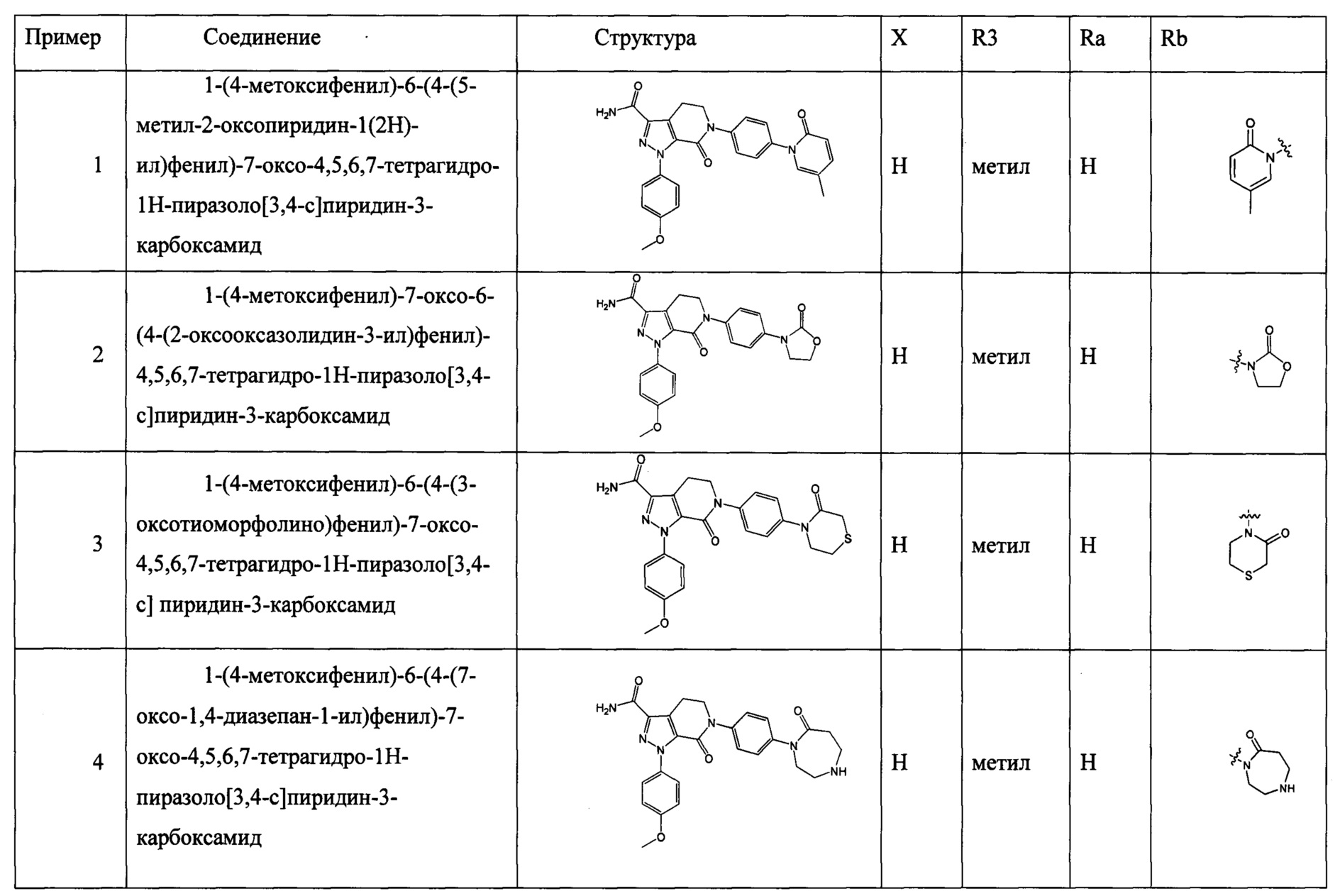
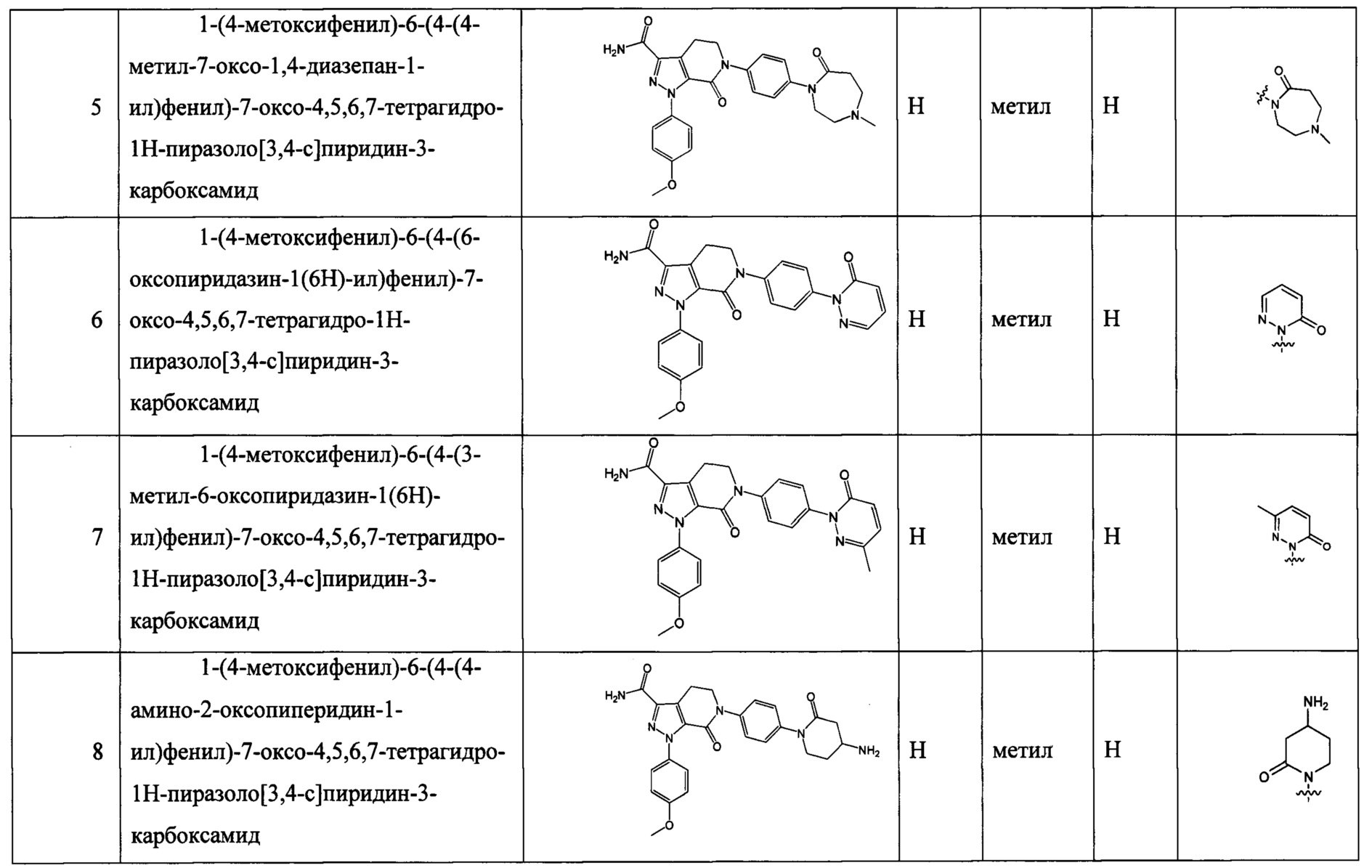
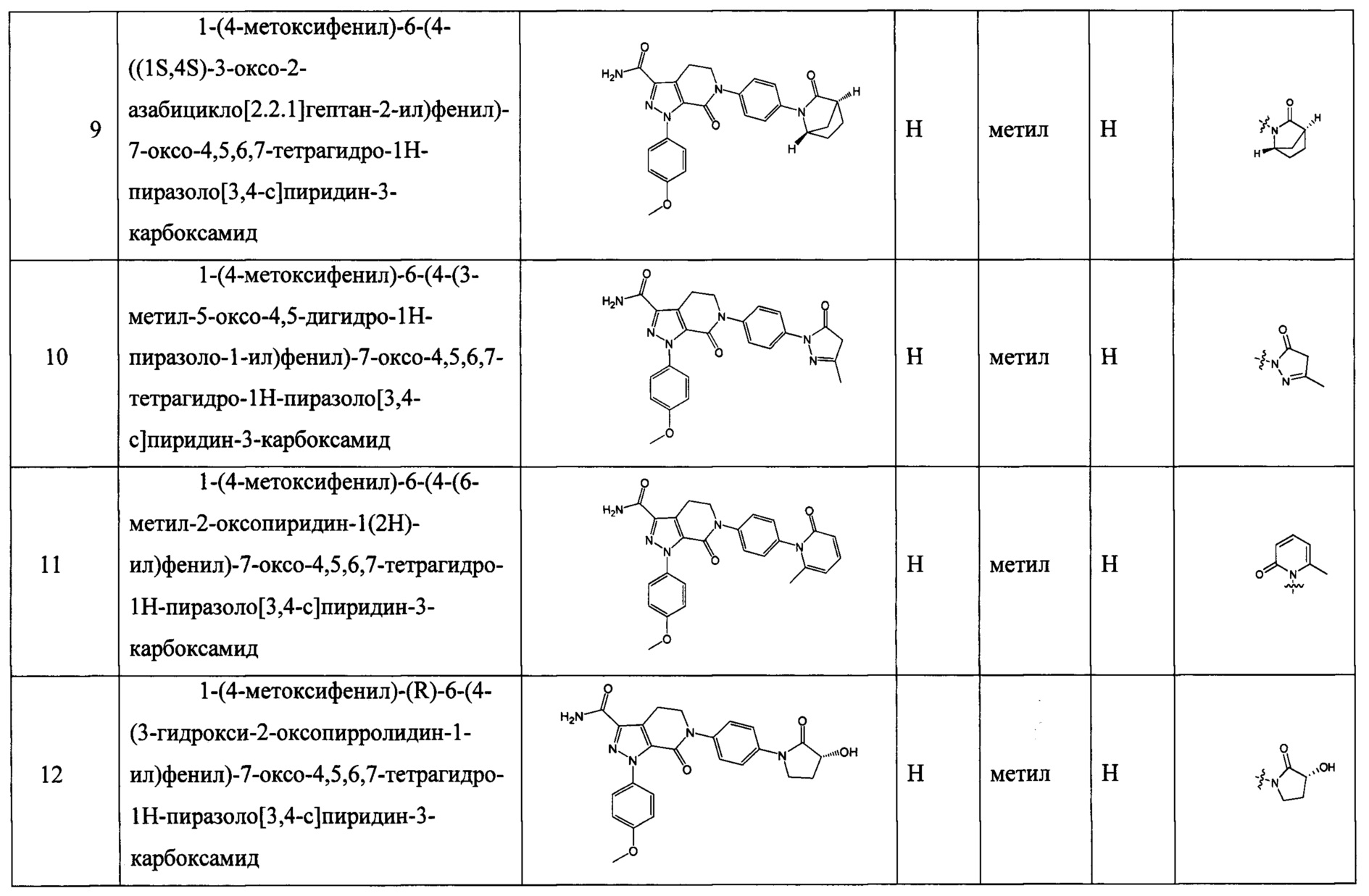
(3) Получение соединения формулы I по данному изобретению, где R3, X, Ra и Rb имеют значения, указанные в следующей Таблице.
Способ синтеза:
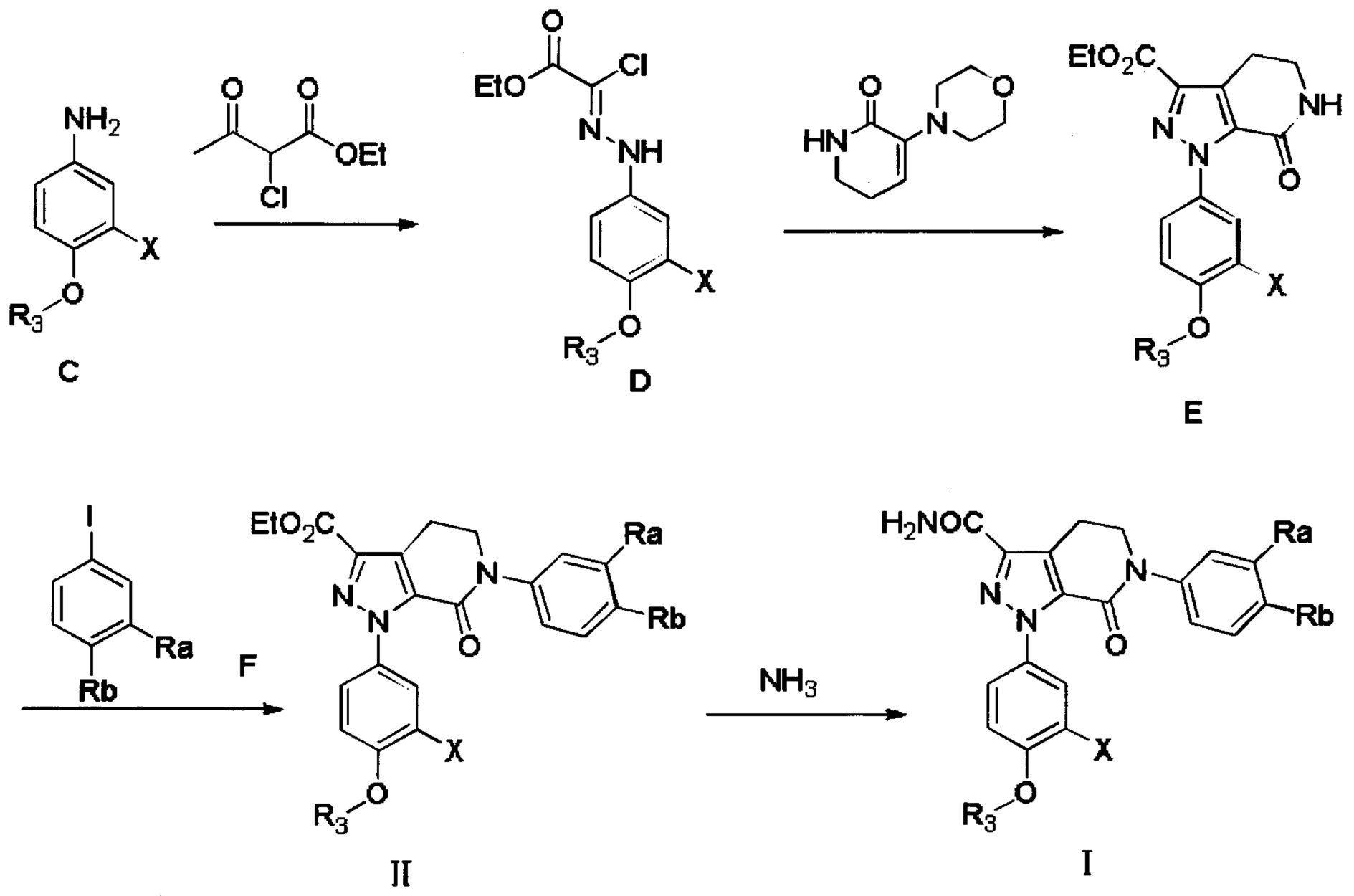
Стадия 1: Получение соединения D
В колбу добавляли соединение С (например, 63.2 ммоля) и затем добавляли очищенную воду (например, 60 мл). Эту смесь при перемешивании охлаждали до температуры -5-0°С и добавляли концентрированную соляную кислоту (например, 26 мл). К полученной смеси по каплям добавляли водный раствор нитрита натрия (например, 30 мл), поддерживая температуру равной -5-0°С. После окончания его добавления смесь перемешивали в течение 20 мин при указанной температуре. К полученной смеси добавляли раствор этил-2-хлор-3-оксобутаноата (например, 10.7 г, 65.1 ммоля) в этаноле (например, 100 мл) и раствор ацетата натрия (например, 15.5 г, 189.0 ммолей) в воде (60 мл). После окончания добавления перемешивали смесь в течение 0.5 ч. Затем смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. Во время реакции выделялся твердый продукт. После завершения реакции реакционную смесь отфильтровывали и сушили остаток на фильтре с получением твердого продукта желтого цвета с выходом около 70%.
Стадия 2: Получение соединения Е
При комнатной температуре в колбу (например, объемом 100 мл) добавляли соединение D (например, 4.7 ммоля), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (например, 0.94 г, 5.2 ммоля) и затем добавляли толуол (например, 20 мл)и триэтиламин. После окончания добавления смесь нагревали до 100°С и нагревали с обратным холодильником в течение 12 ч. Полученный продукт охлаждали до комнатной температуры и концентрировали. К остатку добавляли дихлорметан (например, 20 мл) и по каплям при комнатной температуре добавляли трифторуксусную кислоту (например, 5.0 мл). Полученную смесь для осуществления реакции перемешивали в течение 2 ч. После завершения реакции реакционную смесь концентрировали в вакууме. После окончания концентрирования к смеси добавляли этилацетат и очищенную воду (q.s.). Смесь перемешивали, отделялся твердый продукт. Полученную смесь отфильтровывали. Остаток на фильтре сушили в вакууме, получали продукт с выходом около 30%.
Стадия 3: получение соединения II
В колбу (50 мл) добавляли соединение Е (например, 1.3 ммоля), соединение F (например, 1.4 ммоля) и карбонат калия (например, 376 мг, 2.7 ммоля), затем добавляли DMSO (например, 10 мл). В атмосфере азота добавляли йодид меди (например, 114 мг, 0.6 ммоля) и 1,10-фенантролин (например, 110 мг, 0.6 ммоля). Эту смесь нагревали до 120°С и перемешивали в атмосфере азота в течение 12 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры и добавляли очищенную воду. Полученный продукт экстрагировали этилацетатом. Органическую фазу концентрировали с получением продукта с выходом около 75%.
Стадия 4: Получение соединения I
В герметичную трубку добавляли соединение II (например, 0.45 ммоля) и затем добавляли этиленгликоль (например, 10 мл). Полученную смесь перемешивали. Затем в указанную трубку вводили газообразный аммиак в течение 0.5 ч. Трубку снова герметизировали. Смесь нагревали до 120°С и проводили реакцию в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и затем выливали в холодную воду. Выделялся твердый продукт. Смесь отфильтровывали. Остаток на фильтре сушили, получали целевой продукт.
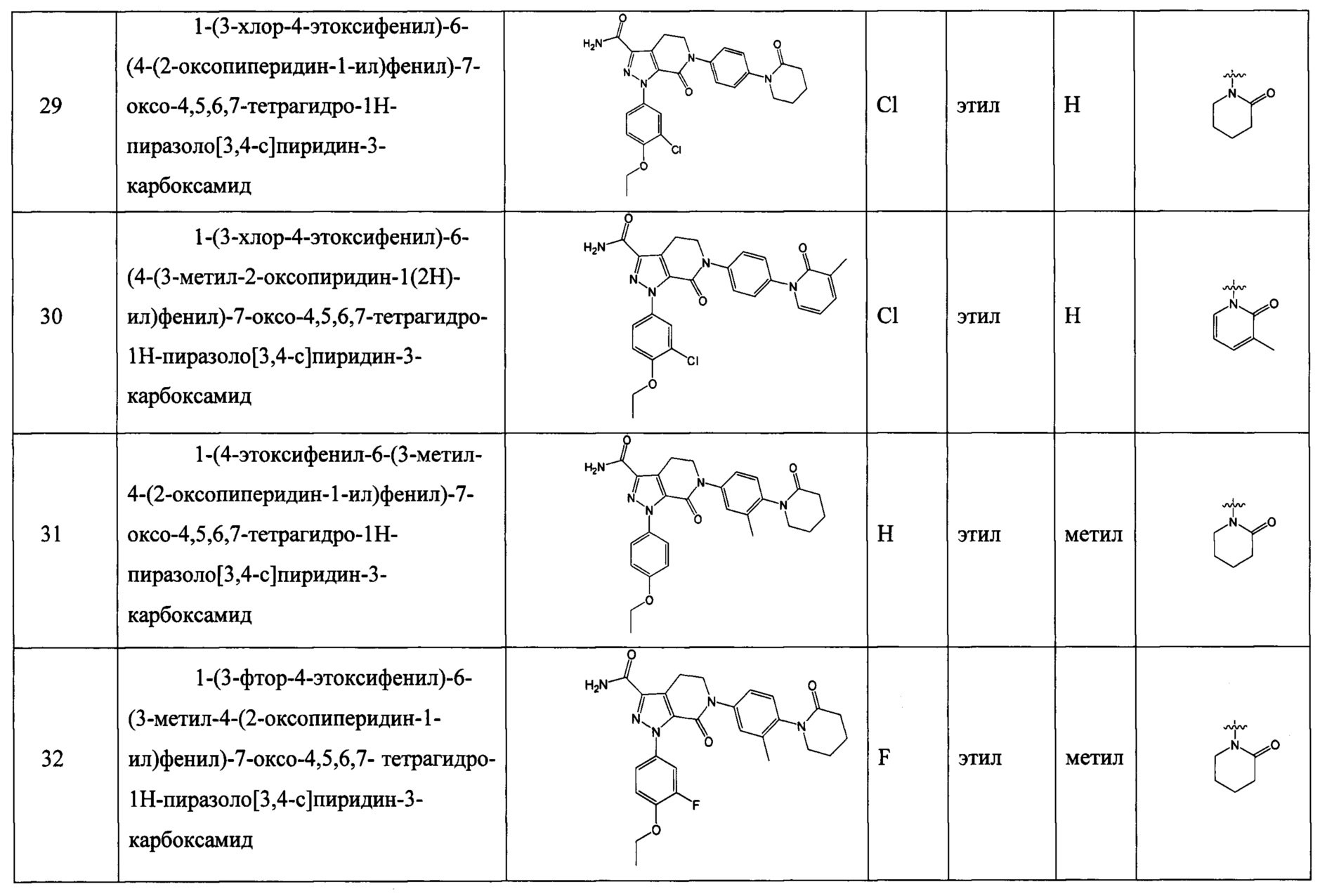
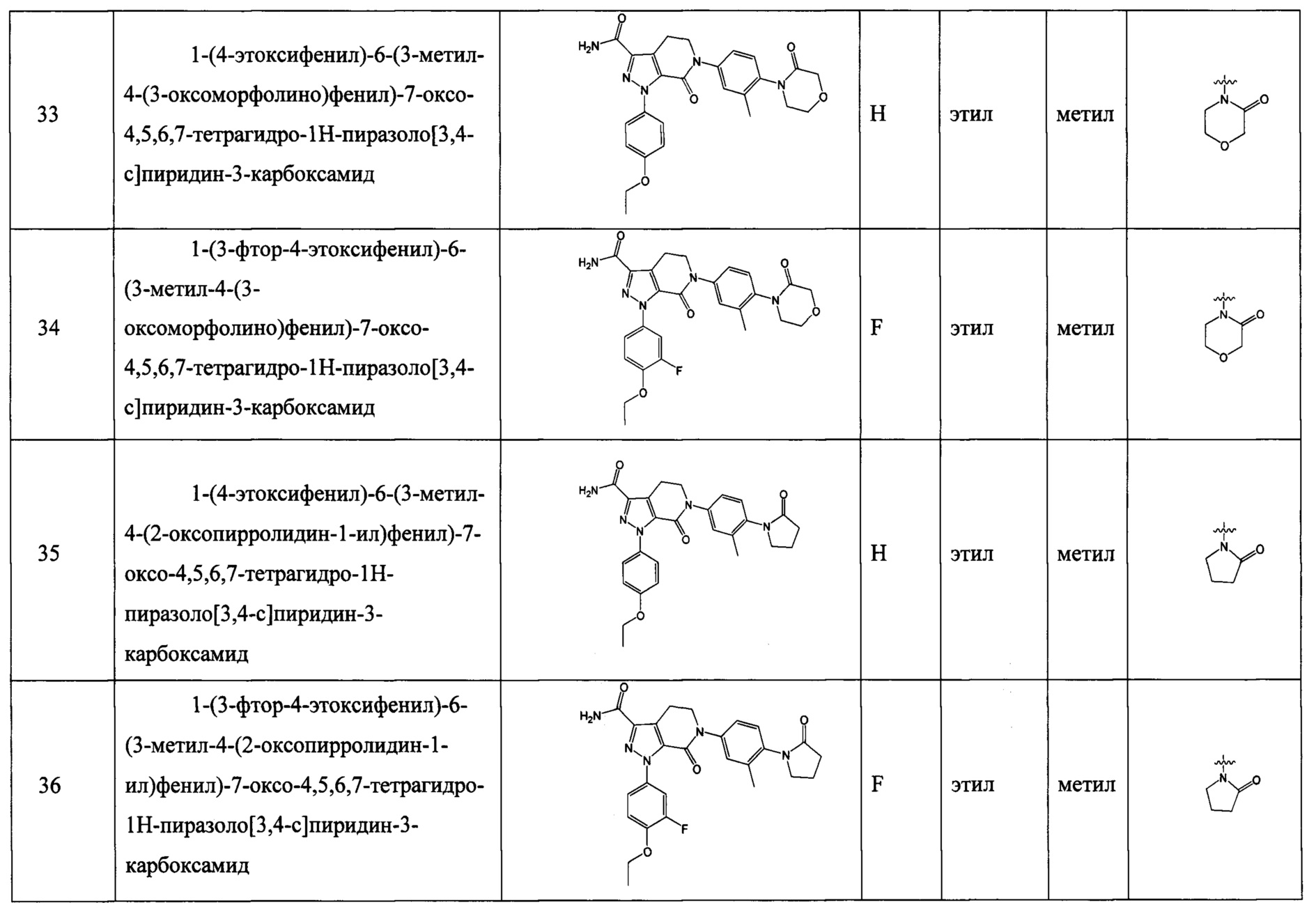
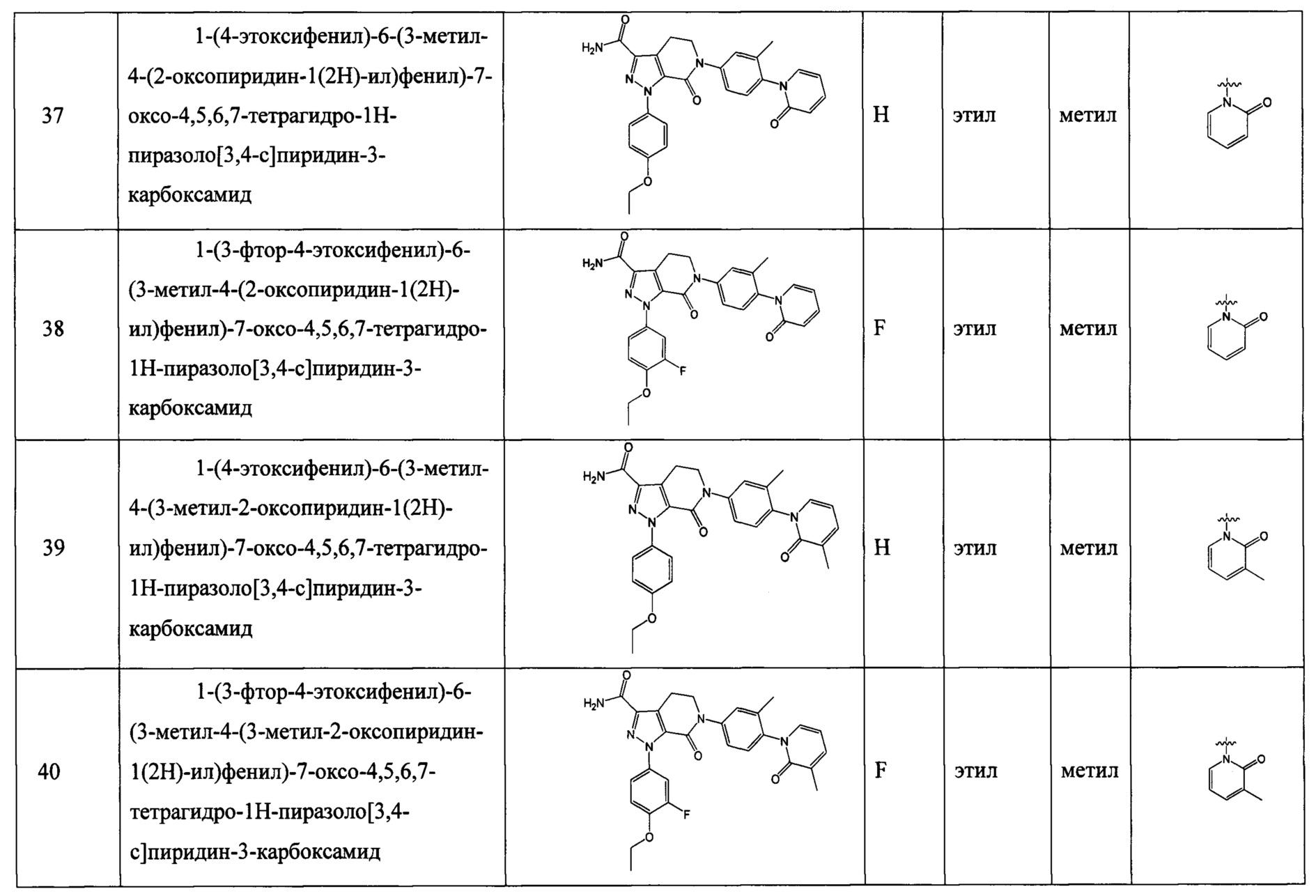
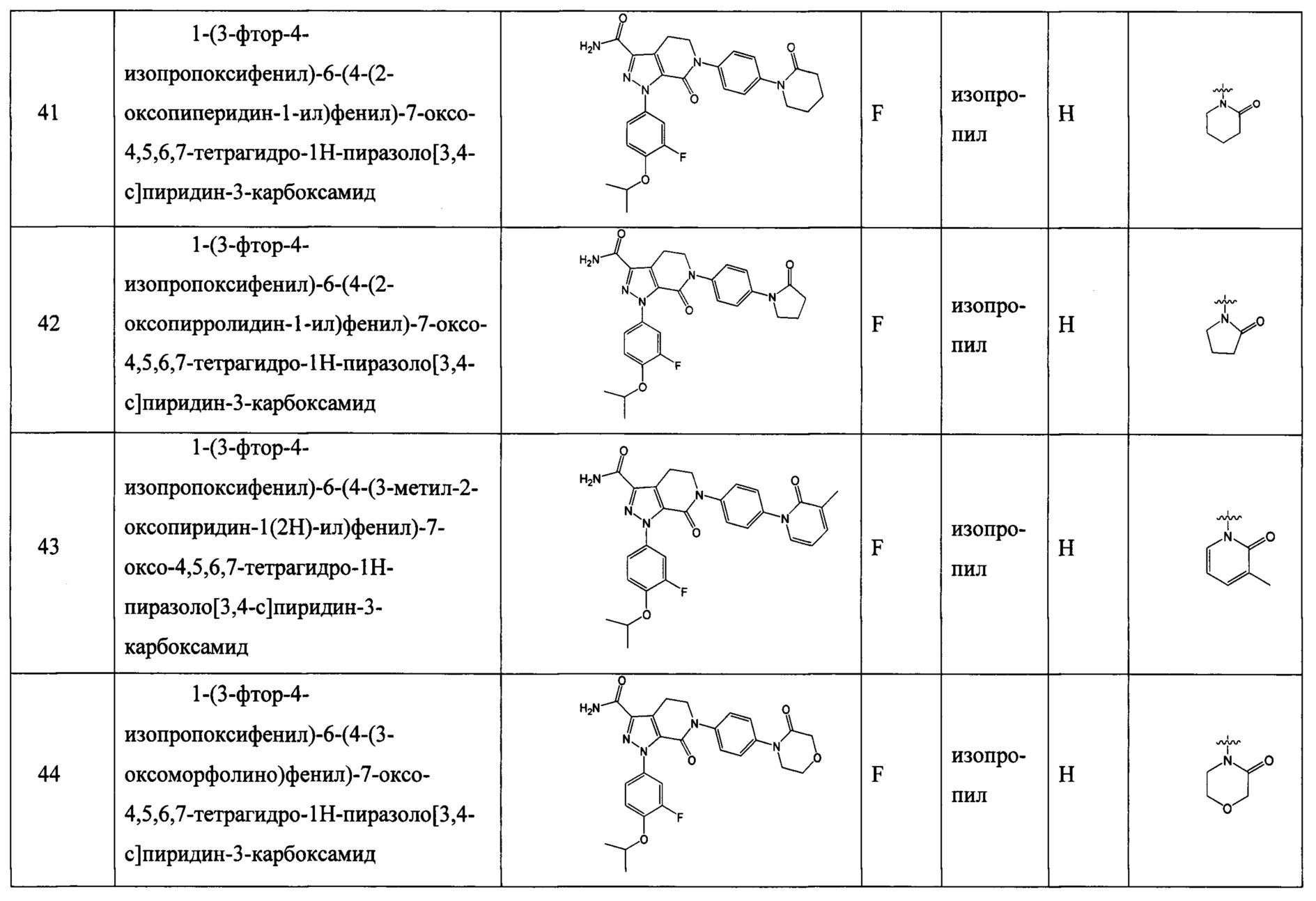
Конкретные физические характеристики:
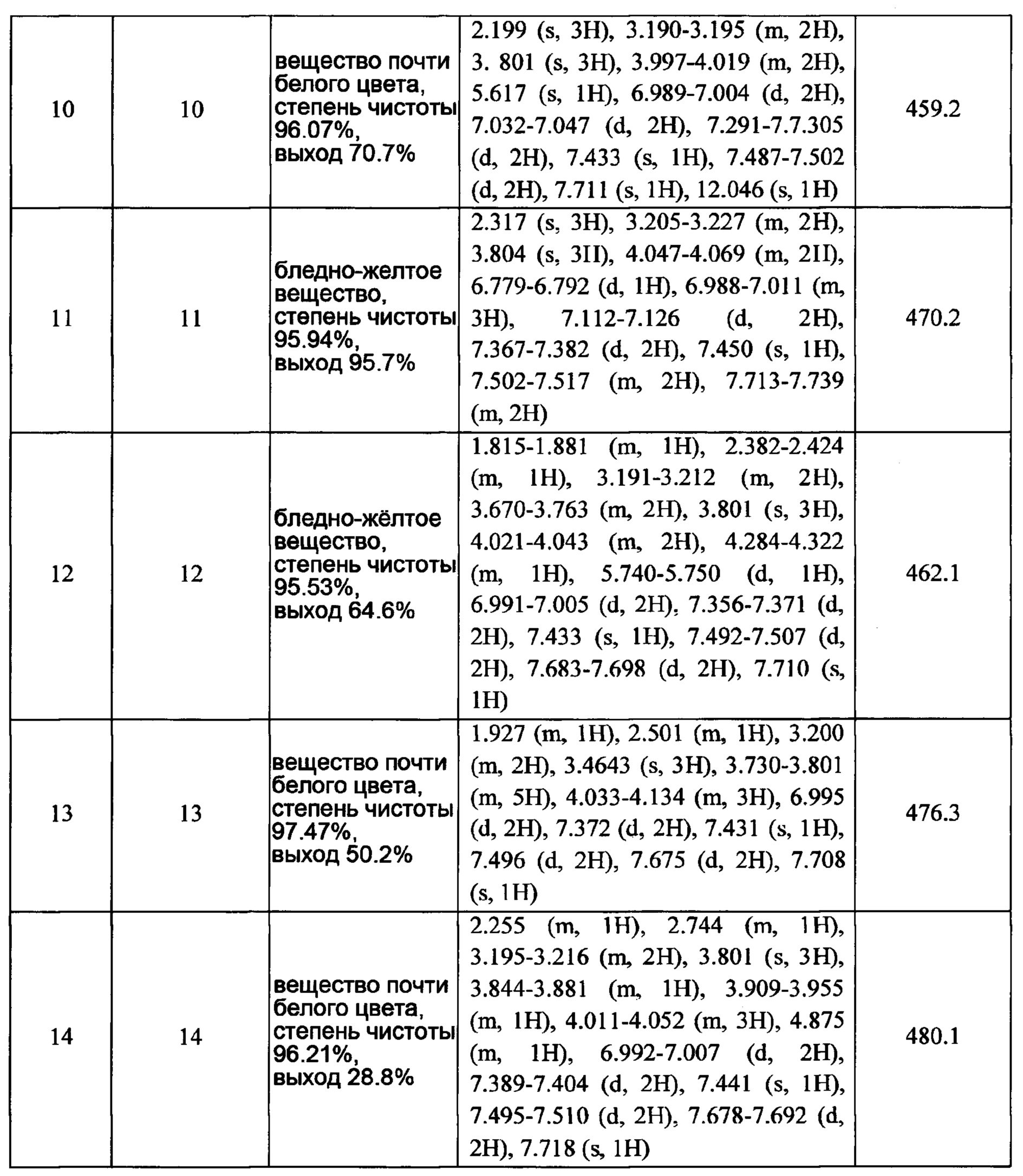
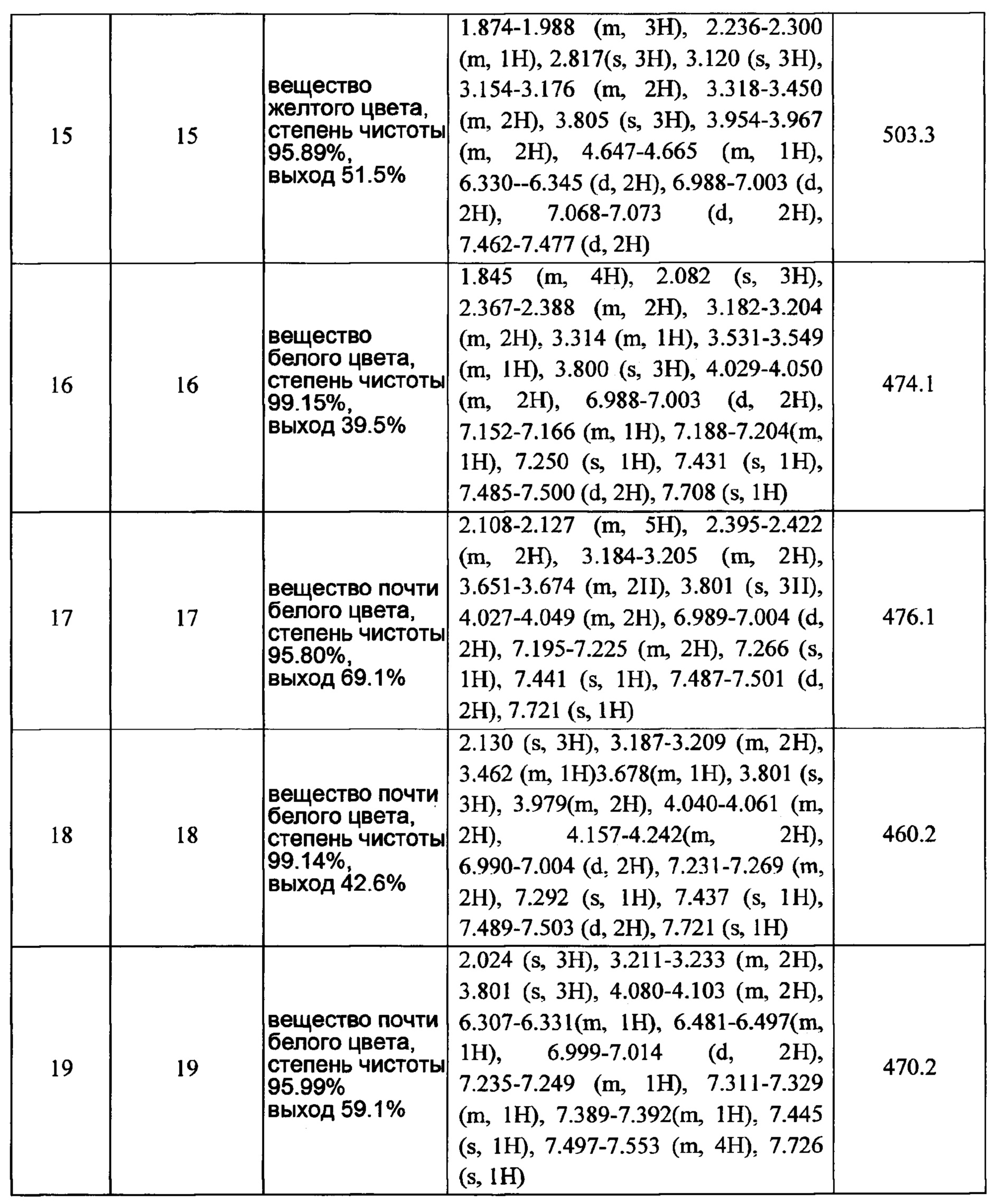
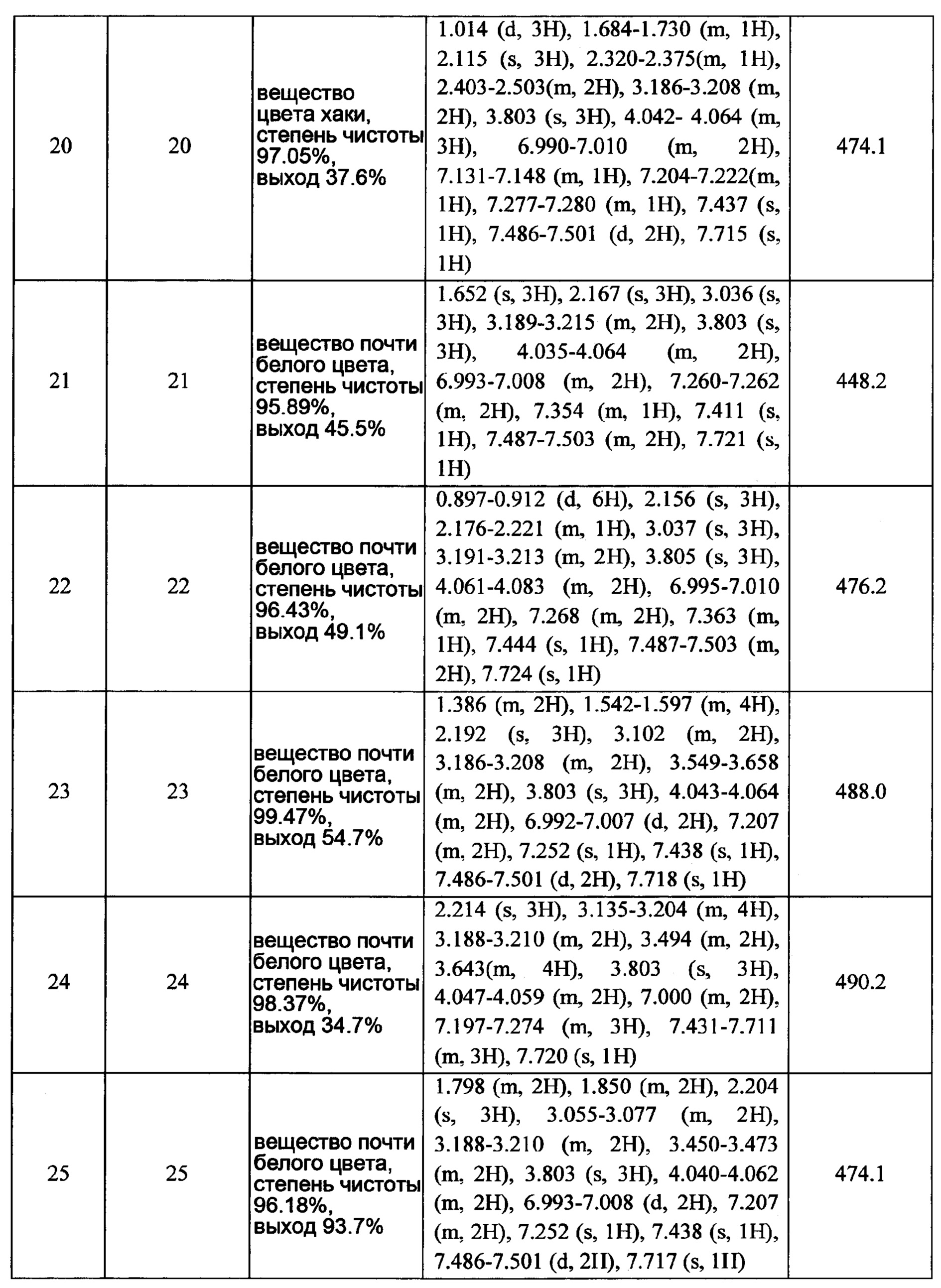
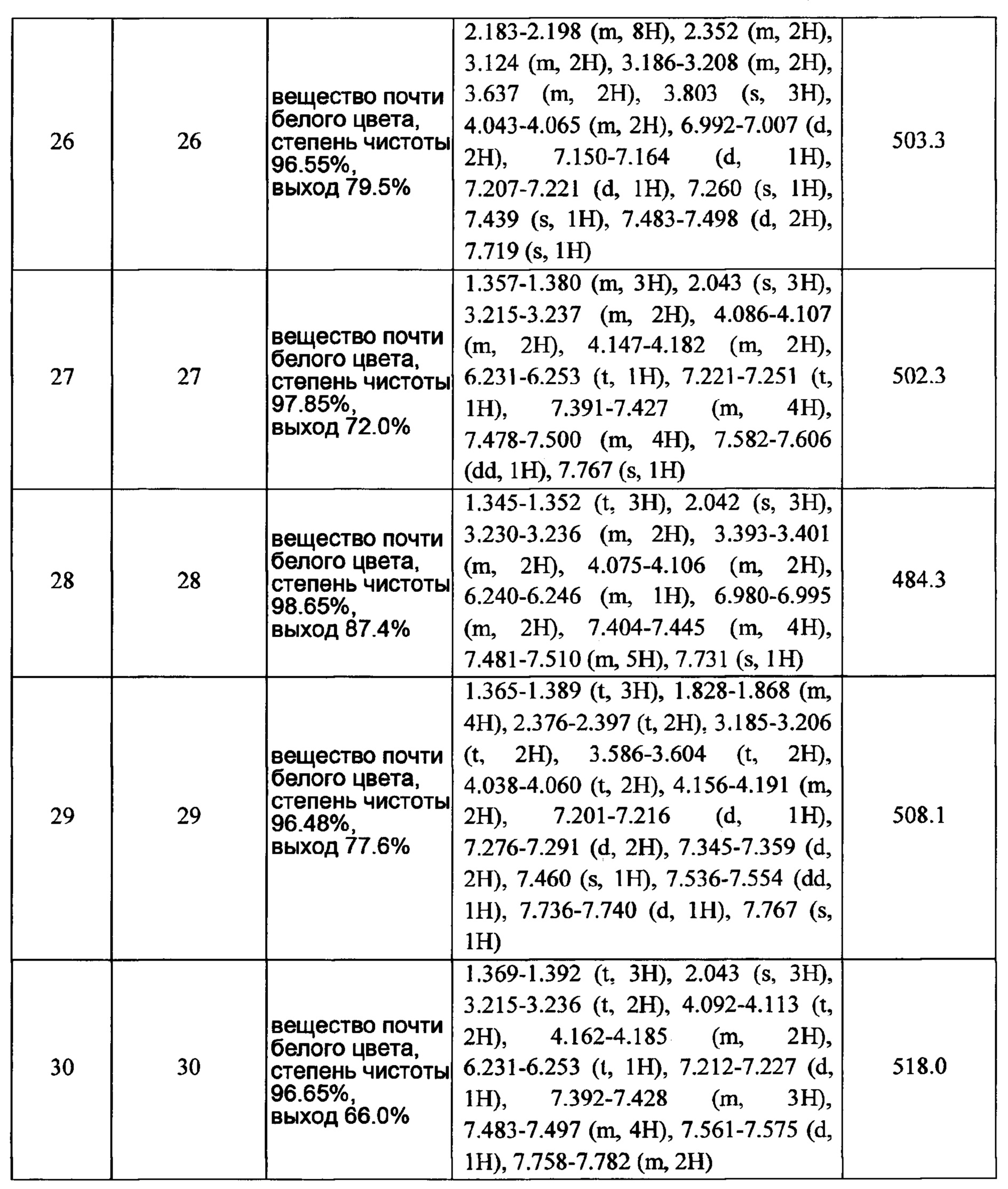
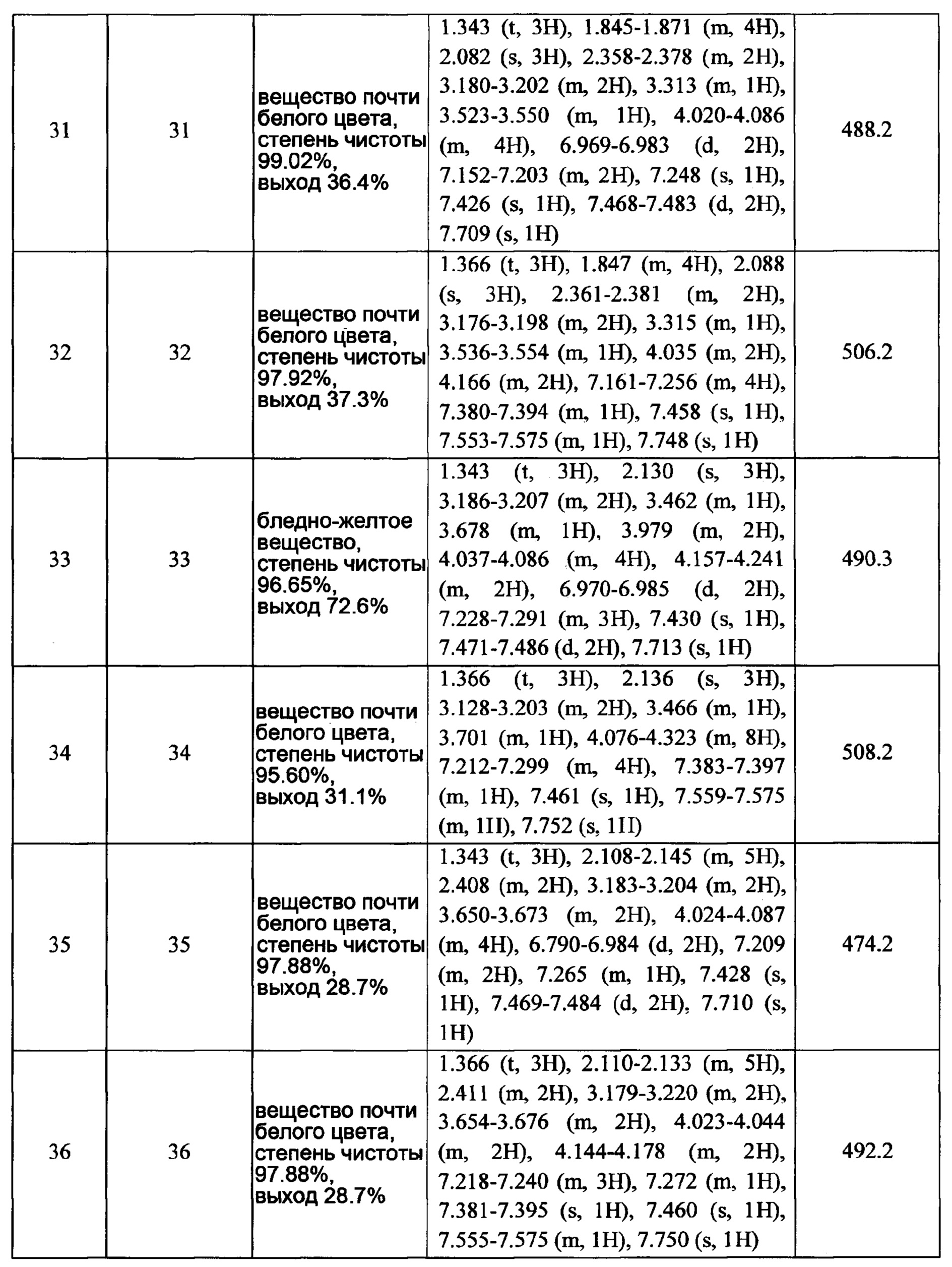
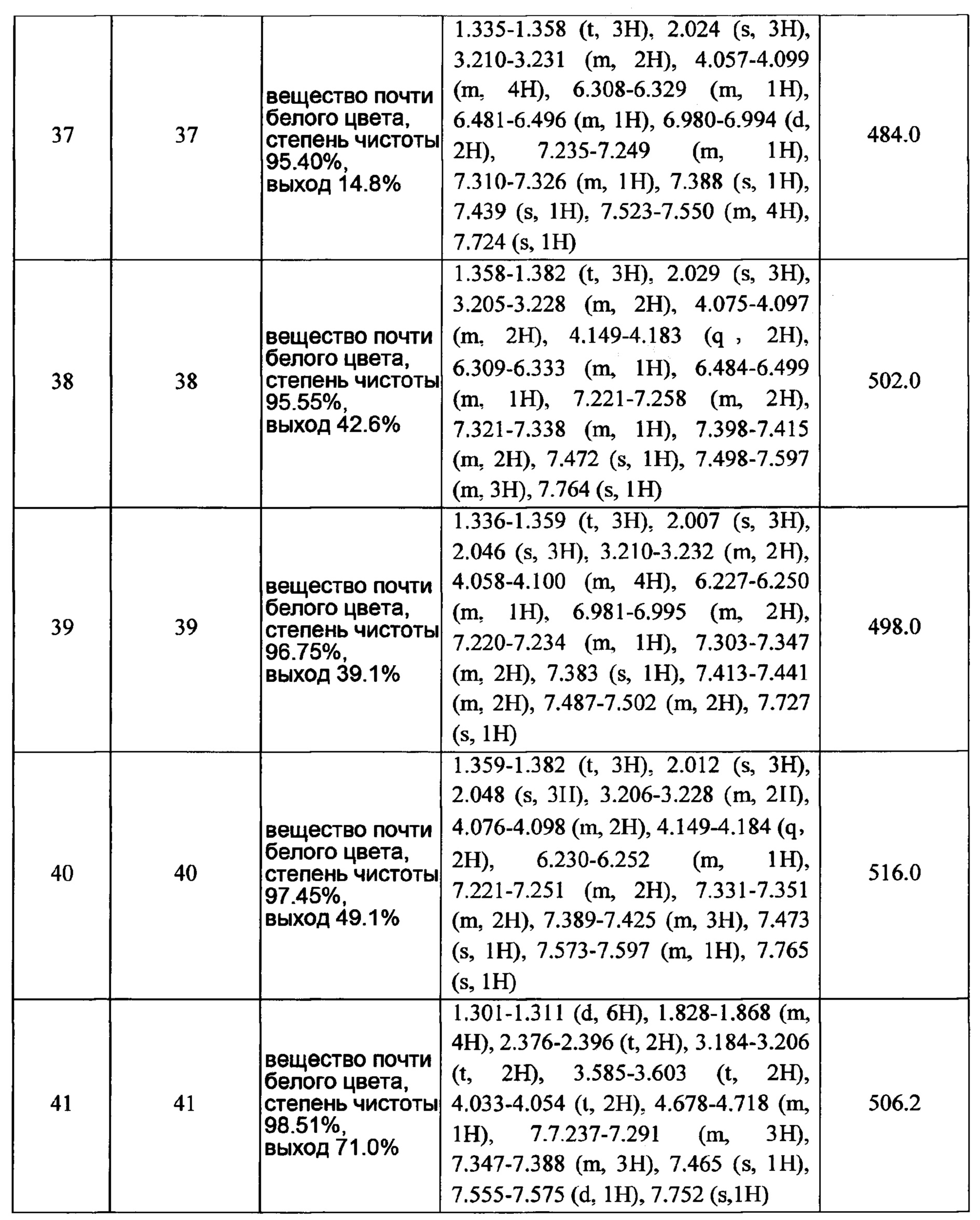
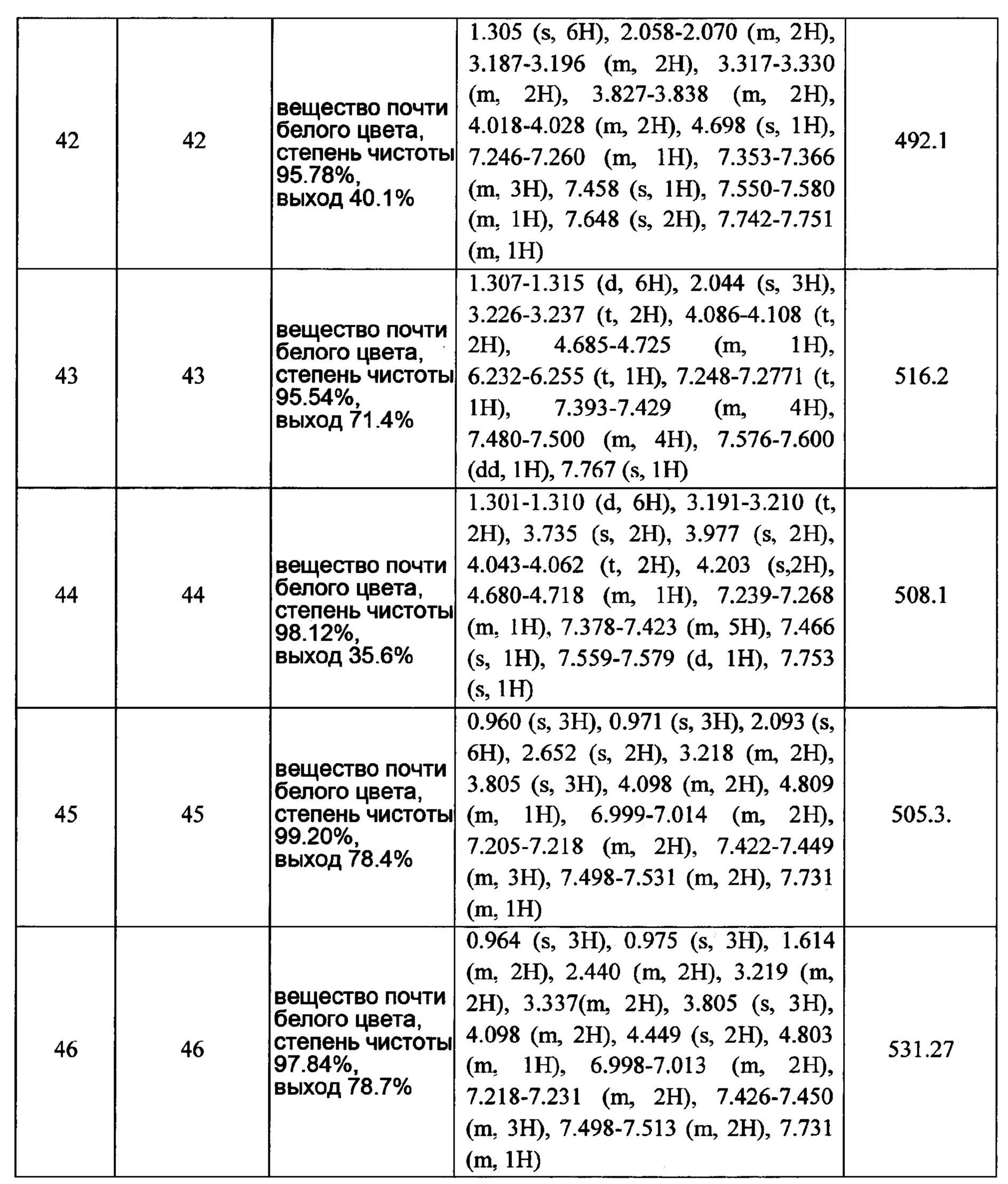
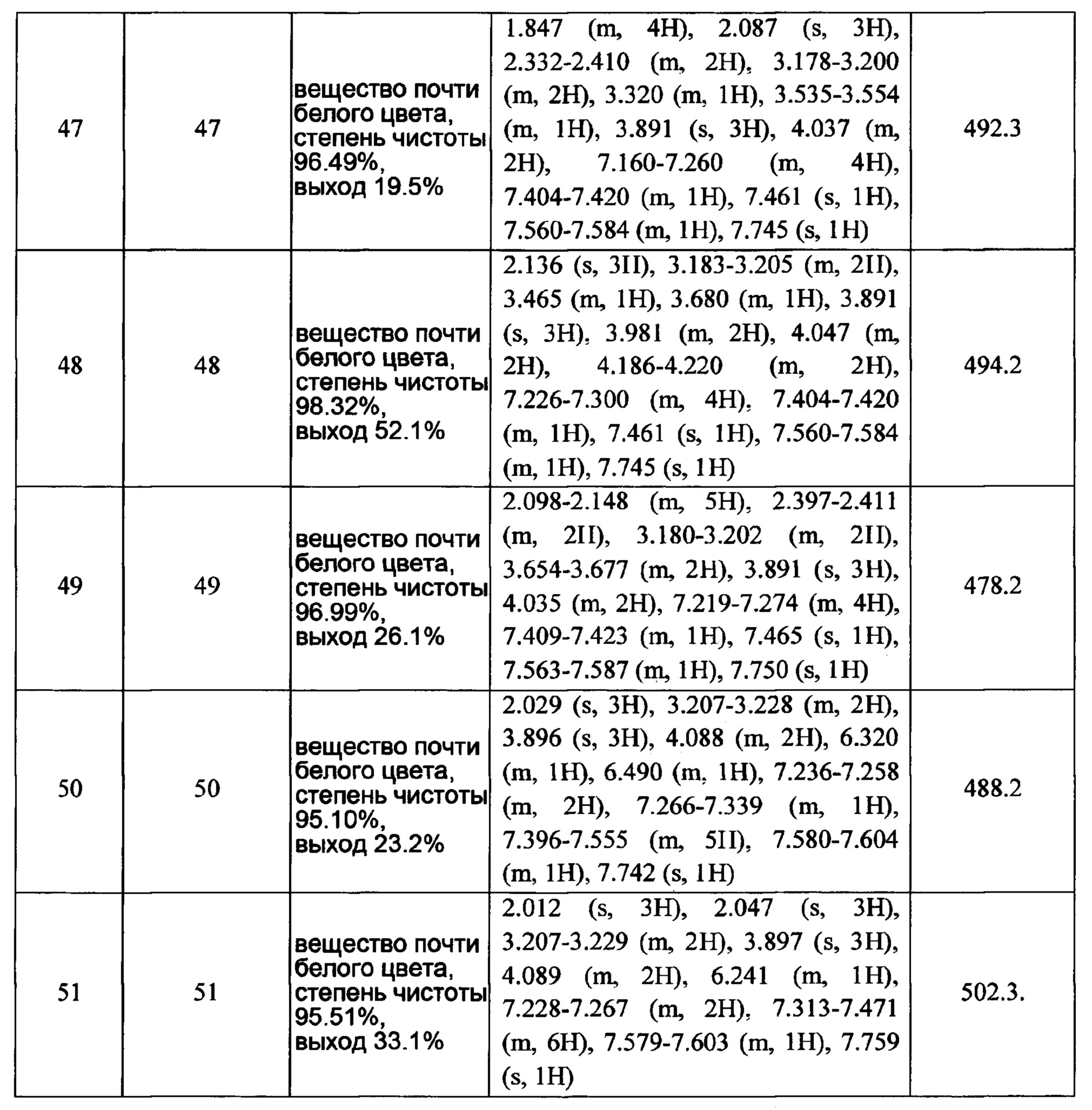
Часть В
Данное изобретение предусматривает также следующие соединения 1-35, соответствующие соединениям, описанным в примерах 1-35.
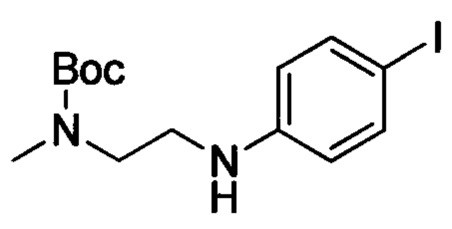
Промежуточное соединение 1: трет-бутил (3-(диметиламино)пропил)(4-йодфенил)карбамат
Стадия 1: Получение трет-бутил (4-йодфенил)карбамата
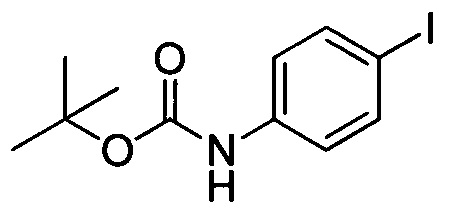
4-йоданилин (5.0 г, 22.83 ммоля) растворяли в DMF (50 мл). В реакционную смесь добавляли DIPEA (2.5 мл). Затем на ледяной бане добавляли по каплям (Вос)2O (11.0 г, 50.04 ммоля). После перемешивания при комнатной температуре в течение 14 ч реакционную смесь выливали в ледяную воду (250 мл). Полученный продукт экстрагировали DCM. Органические фазы концентрировали в вакууме, получали сырой продукт. Этот сырой продукт смешивали с гексаном при нагревании с образованием суспензии, затем суспензию охлаждали и отфильтровывали с получением 3.5 г продукта белого цвета с выходом 48.0%.
Стадия 2: Получение трет-бутил (3-(диметиламино)пропил)(4-йодфенил)карбамата
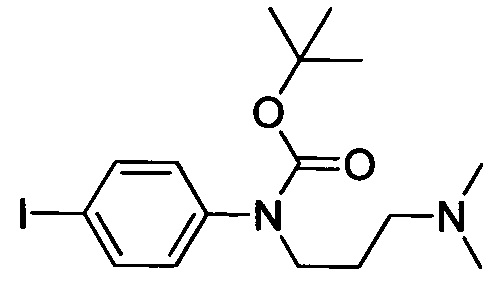
трет-Бутил (4-йодфенил)карбамат (2.0 г, 6.27 ммоля), 3-хлоро-1-(N,N-диметил)пропиламин (0.9 г, 7.40 ммоля), карбонат цезия (3.0 г, 9.21 ммоля) и йодид калия (0.1 г, 0.63 ммоля) диспергировали в DMF (20 мл) при комнатной температуре. Полученную смесь нагревали до 80°С и осуществляли реакцию в течение 48 ч. Реакционную смесь выливали в ледяную воду (100 мл). Полученный продукт экстрагировали ЕА. Органические фазы концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=20:1), получали 1.0 г титульного соединения в виде твердого продукта желтого цвета с выходом 40%.
Промежуточное соединение 2: 3-хлор-5,6-дигидропиридин-2(1Н)-он
Стадия 1: Получение 3,3-дихлорпиперидин-2-она
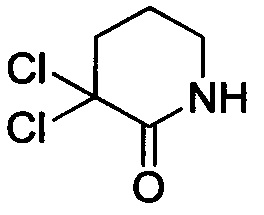
В колбу добавляли пиперидин-2-он (20 г, 20.2 ммоля) и хлороформ (500 мл). Затем к полученной смеси отдельными порциями добавляли пентахлорид фосфора (168 г, 80.7 ммоля) при температуре 0-5°С. После окончания добавления нагревали реакционную смесь до 66°С и проводили реакцию с обратным холодильником в течение 12 ч. После окончания реакции реакционную смесь охлаждали до комнатной температуры и медленно выливали в смесь воды со льдом (1.5 л). Полученный продукт экстрагировали DCM и концентрировали органические вытяжки в вакууме с получением 31.0 г твердого продукта белого цвета, то есть, сырого 3,3-дихлорпиперидин-2-она с выходом 91.3%, который использовали на следующей стадии без очистки.
Стадия 2: 3-хлор-5,6-дигидропиридин-2(1Н)-он
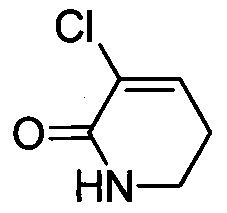
3,3-дихлорпиперидин-2-он (31.0 г, 0.18 ммоля) растворяли при комнатной температуре в DMF (200 мл). К полученной смеси добавляли карбонат лития (33.3 г, 0.45 ммоля). Смесь нагревали до 120°С и перемешивали в течение 12 ч для проведения реакции. После окончания реакции растворитель удаляли при выпаривании. Остаток разбавляли DCM (300 мл) и фильтровали с отсасыванием. Фильтрат концентрировали в вакууме с получением 45.0 г сырого титульного соединения в виде коричневого масла.
Промежуточное соединение 3: получение трет-бутил-(3-(4-йодфенокси)пропил)(метил)карбамата
Стадия 1: 3-(метиламино)пропан-1-ол
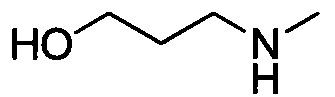
3-бромпропан-1-ол (10.0 г, 71.94 ммоля) медленно по каплям добавляли в водный раствор метиламина (50 мл) на ледяной бане. После окончания добавления по каплям реакционную смесь выдерживали при комнатной температуре для осуществления реакции в течение 14 ч. Полученную реакционную смесь концентрировали в вакууме с получением 6 г титульного соединения в виде желтого масла, который использовали непосредственно на следующей стадии.
Стадия 2: трет-бутил (3-гидроксипропил)(метил)карбамат
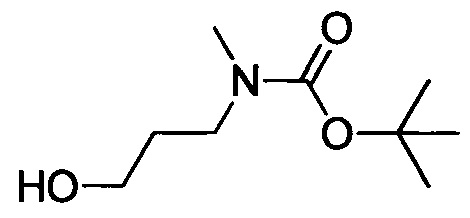
3-(метиламино)пропан-1-ол (6.0 г, 67.3 ммол) растворяли в DCM (100 мл) при комнатной температуре. К полученной смеси добавляли триэтиламин (29.0 г, 0.287 моля). Затем к полученной смеси по каплям медленно добавляли Boc2O (22.0 г, 0.101 моля) на ледяной бане. После окончания добавления по каплям реакционную смесь выдерживали при комнатной температуре для осуществления реакции в течение 14 ч. Полученную реакционную смесь концентрировали в вакууме с получением сырого продукта, этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=50:1), получали 6.7 г титульного соединения в виде безводного масла.
Стадия 3: трет-бутил (3-(4-йодфенокси)пропил)(метил)карбамат
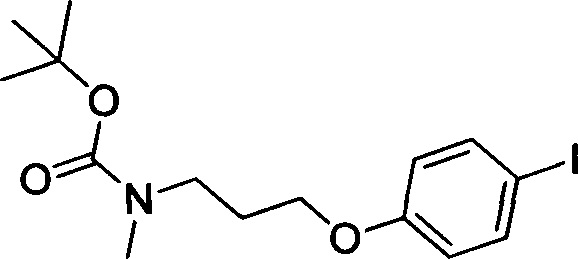
4-Йодфенол (2.0 г, 9.09 ммоля), трет-бутил-(3-гидроксипропил)(метил)карбамат (2.06 г, 10.89 ммоля) и трифенилфосфин (3.6 г, 13.64 ммоля) последовательно добавляли при комнатной температуре к безводному THF (20 мл). К полученной смеси по каплям на ледяной бане медленно добавляли DEAD (2.3 г, 13.21 ммоля). После окончания добавления по каплям реакционную смесь выдерживали для осуществления реакции в течение 14 ч. Затем полученную реакционную смесь концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (DCM:MeOH=10:1) с получением 2.2 г титульного соединения в виде бесцветного масла с выходом 61.9%.
Промежуточный продукт 4: Получение трет-бутил (4-бром-2-фторфенил)(2-метоксиэтил)карбамата
Стадия 1:4-бром-2-фторанилин
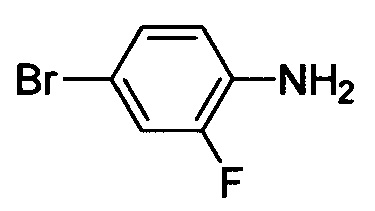
2-фторанилин (3.0 г, 27.0 ммол) растворяли при комнатной температуре в DMF (15 мл). К полученной смеси по каплям на ледяной бане в атмосфере N2 медленно добавляли раствор NBS (5.3 г, 29.7 ммоля) в DMF (15 мл). После окончания добавления по каплям реакционную смесь выдерживали при постоянной температуре в течение 1 ч. полученную смесь выливали в ледяную воду (100 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки концентрировали под вакуумом с получением 5.0 г титульного соединения в виде коричневого масла, который использовали на следующей стадии без очистки.
Стадия 2: трет-бутил (4-бром-2-фторфенил)карбамат
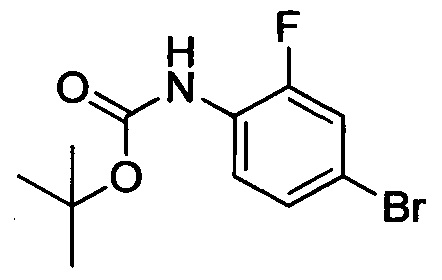
4-бром-2-фторанилин (2 г, 10.47 ммоля) растворяли при комнатной температуре в DMF (20 мл). К смеси добавляли DIEPA (1 мл). На ледяной бане медленно по каплям добавляли Вос2О (4.6 г, 20.93 моля). После окончания этого добавления реакционную смесь выдерживали для осуществления реакции в течение 14 ч. Затем реакционную смесь выливали в ледяную воду (100 мл). Полученный продукт экстрагировали ЕА. Органические фазы концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=50:1) с получением 0.8 г титульного соединения в виде твердого продукта желтого цвета с выходом 26.3%.
Стадия 3: трет-бутил (4-бром-2-фторфенил)(2-метоксиэтил)карбамат
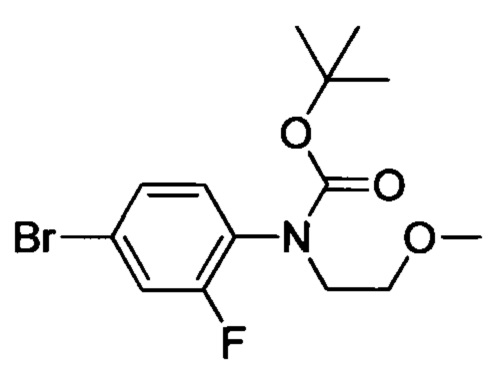
трет-Бутил (4-бром-2-фторфенил)карбамат (1.6 г, 5.51 ммоля), 1-бром-2-метоксиэтан (1.1 г, 7.91 ммоля), карбонат цезия (3.6 г, 139.4 ммоля) и йодид калия (300 мг, 1.81 ммоля) последовательно растворяли при комнатной температуре в DMSO (20 мл). Полученную смесь нагревали до 120°С и выдерживали в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, выливали в ледяную воду (100 мл). Полученный продукт экстрагировали ЕА. Органические фазы концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=20:1) с получением 1.6 г титульного соединения в виде бесцветного масла с выходом 83.5%.
Промежуточный продукт 5: трет-бутил (4-((4-йод-2-метилфенил)амино)-4-оксобутил)карбамат
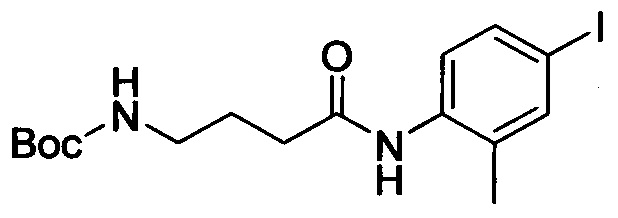
4-йод-2-метиланилин (4.46 г, 19.1 ммоля), карбонат калия (5.3 г, 38.4 ммоля), 4-((третбутоксикарбонил)амино)бутановую кислоту (7 г, 34.4 ммоля) и HATU (8.7 г, 22.9 ммоля) добавляли последовательно при комнатной температуре к ацетонитрилу (50 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Полученный продукт концентрировали в вакууме и затем добавляли воду. Полученную смесь экстрагировали ЕА. Органические фазы концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (DCM:MeOH=50:1) с получением 6.72 г титульного соединения в виде твердого продукта белого цвета с выходом 84.2%.
Промежуточное соединение 6: N-этил-5-йод-2-пропоксианилин
Стадия 1: 4-йод-2-нитрофенол
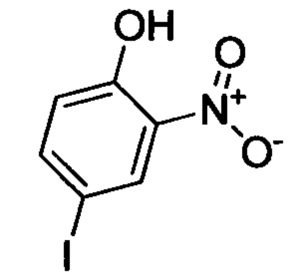
4-йодфенол (7.0 г, 31.8 ммоля) при комнатной температуре растворяли в уксусной кислоте (50 мл). Полученную смесь охлаждали до 0°С. К охлажденной смеси по каплям добавляли азотную кислоту (2.4 мл). Полученную смесь нагревали до комнатной температуры и перемешивали для проведения реакции в течение 3 ч. Реакционную смесь выливали в ледяную воду (200 мл), перемешивали и отфильтровывали с отсасыванием. Остаток на фильтре высушивали в вакууме с получением 8.0 г титульного соединения в виде твердого продукта желтого цвета, который использовали на следующей стадии без очистки.
Стадия 2: 4-йод-2-нитро-1-пропоксибензол
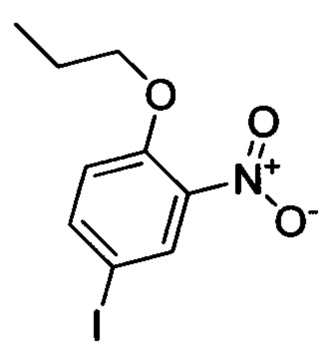
4-йод-2-нитрофенол (8.0 г, 30.2 ммоля), карбонат калия (8.3 г, 60.1 ммоля) и 1-йодпропан (10.3 г, 60.4 ммоля) при комнатной температуре последовательно добавляли к ацетонитрилу (100 мл). Полученную смесь нагревали до 80°С и перемешивали для проведения реакции в течение 14 ч. Полученный продукт выливали в ледяную воду (100 мл). Затем проводили экстракцию ЕА. Органические вытяжки концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=50:1) с получением 5.1 г титульного соединения в виде желтого масла с выходом 50.0%.
Стадия 3: 5-йод-2-пропоксианилин
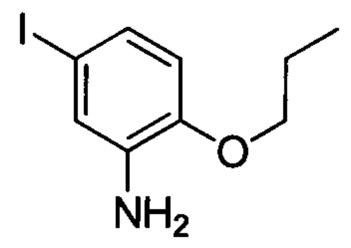
4- йод-2-нитро-1-пропоксибензол (5.1 г, 16.6 ммоля), Fe (2.79 г, 49.8 ммоля) и хлорид аммония (4.44 г, 83.0 ммоля) последовательно добавляли при комнатной температуре к раствору этанола (50 мл) в воде. Полученную смесь нагревали до 60°С и перемешивали для проведения реакции в течение 3 ч. Затем смесь фильтровали с отсасыванием. Фильтрат экстрагировали этилацетатом. Органические фазы концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=50:1) с получением 2.7 г титульного соединения в виде коричневого масла с выходом 58.7%.
Стадия 4: N-этил-5-йод-2-пропоксианилин
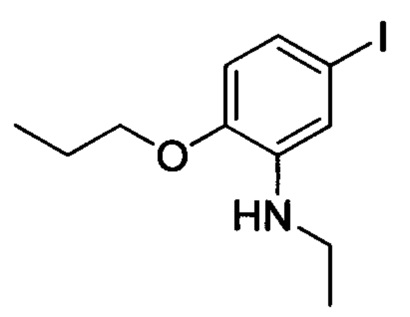
5-йод-2-пропоксианилин (2.0 г, 7.2 ммоля), ацетальдегид (0.95 г, 21.6 ммол) и сульфат магния (3.0 г, 25.0 ммоля) добавляли в МеОН (50 мл). Полученную смесь перемешивали для проведения реакции в течение 2 ч. Затем реакционную смесь охлаждали до 0°С. К охлажденной смеси отдельными порциями добавляли цианборгидрид натрия (3.8 г, 60.4 ммоля). Полученную смесь перемешивали для проведения реакции в течение 14 ч. полученный продукт реакции концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=3:1) с получением 0.94 г титульного соединения в виде коричневого масла с выходом 42.8%.
Промежуточное соединение 7: Получение трет-бутил 2-((4-йодфенил)амино)этил)(метил)карбамата
Стадия 1: 2-((трет-бутоксикарбонил)(метил)амино)уксусной кислоты
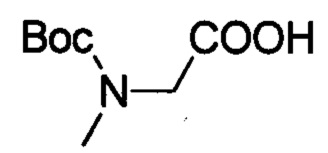
2-(метиламино)уксусную кислоту (10.0 г, 112.2 ммоля), Вос2О (29.4 г, 134.8 ммоля) и гидроксид натрия (20.0 г, 0.5 мол) при комнатной температуре добавляли в смесь воды (100 мл) и 1,4-диоксана (200 мл). Полученную смесь перемешивали для проведения реакции в течение 14 ч. Затем смесь выпаривали для удаления 1,4-диоксана. В оставшейся водной фаза разбавленной соляной кислотой регулировали рН до величины 2. Смесь экстрагировали ЕА. Органические вытяжки концентрировали в вакууме с получением 12.0 г титульного соединения в виде коричневого масла с выходом 56.6%.
Стадия 2: трет-бутил(2-((4-йодфенил)амино)-2-оксоэтил)(метил)карбамат
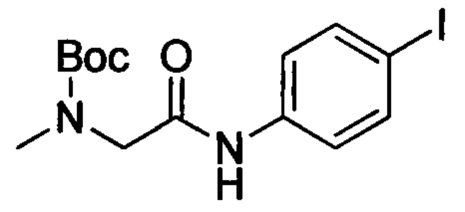
2-((трет-бутоксикарбонил)(метил)амино)уксусную кислоту (12.0 г, 63.8 ммоля), 4-йоданилин (11.6 г, 53.2 ммоля), EDCl (20.3 г, 105 ммоля), HOBt (1.43g, 10.6 ммоля) и триэтиламин (16 г, 159 ммоля) при комнатной температуре последовательно добавляли к THF (150 мл). Полученную смесь перемешивали для проведения реакции в течение 14 ч. Полученный продукт концентрировали. Остаток разбавляли водой (100 мл). Полученный продукт экстрагировали ЕА. Органические фазы концентрировали в вакууме с получением сырого продукта. Этот сырой продукт смешивали с МТВЕ (200 мл) с образованием суспензии. Суспензию отфильтровывали с отсасыванием. Остаток на фильтре высушивали в вакууме с получением 8.0 г соединения в виде белого твердого продукта с выходом 32.2%.
Стадия 3: трет-бутил-(2-((4-йодфенил)амино)этил)(метил)карбамат
трет-Бутил (2-((4-йодфенил)амино)-2-оксоэтил)(метил)карбамат (2.0 г, 5.12 ммоля) растворяли в THF (20 мл). Смесь охлаждали до 0°С и к охлажденной смеси по каплям добавляли раствор борана в тетрагидрофуране (16 мл). После окончания добавления полученную смесь нагревали до 50°С и перемешивали для проведения реакции в течение 14 ч. Затем полученную смесь охлаждали до 0°С и к охлажденной смеси добавляли МеОН (0.82 г, 25.6 ммоля). Полученную смесь нагревали до 80°С и перемешивали в течение 2 ч. Смесь выпаривали в вакууме и остаток растворяли в растворе метилбензола (10 мл) в н-BuOH (10 мл). Эту смесь перемешивали для проведения реакции в течение 14 ч при температуре 100°С. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=10:1) с получением 1.6 г титульного соединения в виде желтого масла с выходом 83.1%.
Промежуточное соединение 8: 5-(4-бром-2-хлорфенокси)пентан-2-он
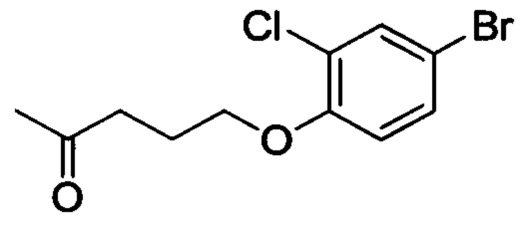
4-бром-2-хлорфенол (5.0 г, 24.1 ммоля), карбонат калия (9.97 г, 72.3 ммоля) и 5-хлорпентан-2-он (4.36 г, 36.1 ммоля) при комнатной температуре последовательно добавляли к DMF (50 мл). Полученную смесь нагревали до 80°С и перемешивали для прохождения реакции в течение 14 ч. Полученную смесь охлаждали до комнатной температуры и затем выливали в воду (300 мл). Полученный продукт экстрагировали ЕА. Органические вытяжки концентрировали в вакууме с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=20:1) с получением 2.0 г титульного соединения в виде масла бледно-желтого цвета с выходом 28.6%.
Промежуточный продукт 9: 1-(4-йодфенил)-4-метилпиперазин
Стадия 1: 1-(4-йодфенил)пиперазин
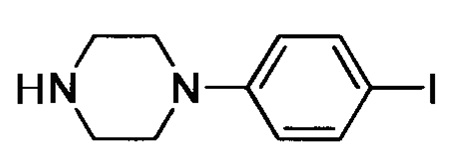
4-йоданилин (5.0 г, 22.8 ммоля) и карбонат калия (10 г, 72.4 ммоля) добавляли к н-ВиОН (50 мл) при комнатной температуре. Смесь охлаждали до 0°С. К охлажденной смеси отдельными порциями добавляли бис-(2-хлорэтил)амина гидрохлорид (5.0 г, 28.0 ммоля). После окончания добавления реакционную смесь нагревали до 100°С и перемешивали для проведения реакции в течение 14 ч. Полученную смесь охлаждали до комнатной температуры и отфильтровывали с отсасыванием. Остаток на фильтре сушили в вакууме с получением 2.5 г титульного соединения в виде темно-желтого твердого продукта с выходом 38.0%.
Стадия 2: 1-(4-йодфенил)-4 метилпиперазин
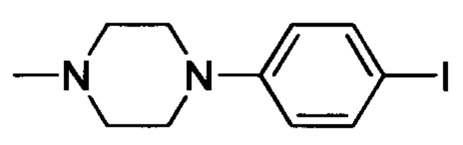
1-(4-йодфенил)пиперазин (1.0 г, 3.5 ммоля) и гидрид натрия (0.13 г, 5.4 ммоля) добавляли при комнатной температуре к THF (10 мл). Смесь перемешивали и проводили реакцию в атмосфере азота в течение 0.5 ч. Смесь охлаждали до 0°С. К охлажденной смеси по каплям добавляли CH3I (0.54 г, 3.8 ммоля). После окончания добавления смесь перемешивали и проводили реакцию при комнатной температуре в течение 2 ч. Реакцию прекращали добавлением воды. Полученную смесь экстрагировали ЕА. Органические фазы концентрировали с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (DCM:MeOH=20:1) с получением 150 г титульного соединения в виде твердого продукта желтого цвета с выходом 14.2%.
Промежуточное соединение 10: трет-бутил-4-(3-бромфенетил)пиперазин-1-карбоксилат
Стадия 1: 2-(3-бромфенил)уксусная кислота
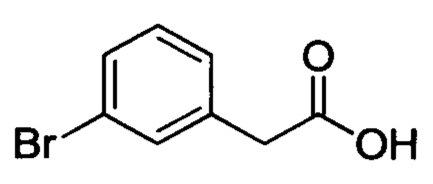
Этил-2-(3-бромфенил)ацетат (5.0 г, 20.6 ммоля) и гидроксид лития (1.0 г, 41.8 ммоля) при комнатной температуре добавляли к смеси THF (30 мл) и воды (20 мл). Эту смесь перемешивали и проводили реакцию при комнатной температуре в течение 1 ч. Затем доводили рН смеси до значения 2 добавлением 2N водного раствора HCl. Полученную смесь экстрагировали ЕА. Органические фазы концентрировали с получением 4.1 г титульного соединения в виде твердого продукта белого цвета.
Стадия 2: 2-(3-бромфенил)ацетилхлорид
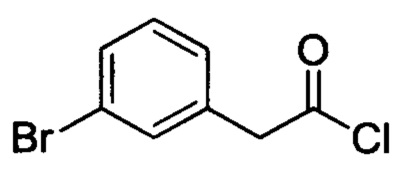
2-(3-бромфенил)уксусную кислоту (4.1 г, 19.1 ммоля) и DMF (1 мл) добавляли к DCM (40 мл). Полученную смесь охлаждали до 0°С. К охлажденной смеси по каплям добавляли оксалилхлорид (5.6 г, 43.7 ммол). После окончания добавления смесь перемешивали и проводили реакцию при комнатной температуре в течение 2яч. Полученную реакционную смесь концентрировали, получали 4.3 г титульного соединения в виде жидкости желтого цвета, которую использовали непосредственно на следующей стадии.
Стадия 3: трет-бутил-4-(2-(3-бромфенил)ацетил)пиперазин-1-карбоксилат
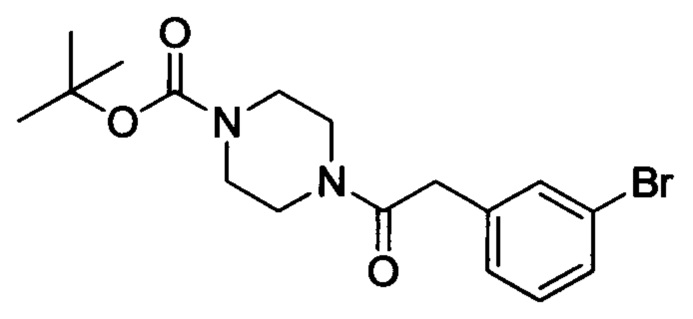
2-(3-бромфенил)ацетилхлорид (4.3 г, 18.4 ммоля) и триэтиламин (4.8 г, 47.1 ммоля) добавляли к THF (50 мл). Полученную смесь охлаждали до 0°С. К охлажденной смеси по каплям добавляли 1-Вос-пиперазин (5.7 г, 30.6 ммол). После окончания этого добавления смесь перемешивали и проводили реакцию при комнатной температуре в течение 14 ч, затем выливали в воду (50 мл). Полученную смесь экстрагировали ЕА. Органические фазы концентрировали, получали 7.0 г титульного соединения в виде жидкости желтого цвета, которую использовали непосредственно на следующей стадии.
Стадия 4: трет-бутил 4-(3-бромфенетил)пиперазин-1-карбоксилат
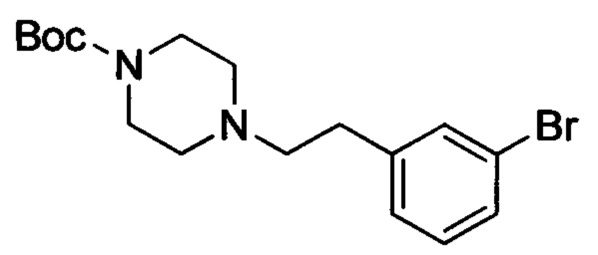
трет-Бутил 4-(2-(3-бромфенил)ацетил)пиперазин-1-карбоксилат (7.0 г, 18.3 ммоля) растворяли в THF (70 мл) при комнатной температуре. К этой реакционной смеси по каплям в атмосфере азота добавляли раствор боран-тетрагидрофуран (37 мл, 33.6 ммоля). Затем полученную смесь нагревали до 50°С и проводили реакцию при перемешивании в течение 14 ч. Полученную смесь охлаждали до комнатной температуры. К охлажденной смеси по каплям добавляли МеОН (20 мл). Затем полученную смесь нагревали до 80°С и перемешивали в течение 2 ч. Затем смесь концентрировали. К остатку добавляли н-BuOH (10 мл) и метилбензол (40 мл). Эту смесь перемешивали в течение 14 ч при 80°С. Затем смесь концентрировали с получением сырого продукта. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (DCM:MeOH=50:1) с получением 3.9 г титульного соединения в виде желтого масла с выходом 57.7%.
Промежуточное соединение 11: трет-бутил-4-(4-бром-2-метоксибензил)пиперазин-1-карбоксилат
Стадия 1: трет-бутил-4-(4-бром-2-гидроксибензоил)пиперазин-1-карбоксилат
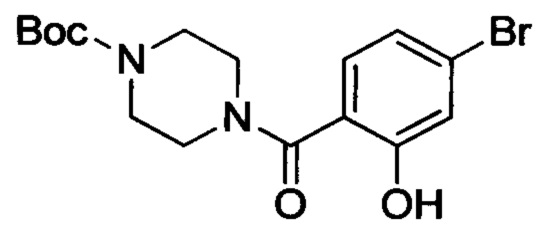
4-бром-2-гидроксибензойную кислоту (5.0 г, 23 ммол), EDCI (8.8 г, 46 ммоля), HOBt (257 мг, 1.9 ммоля) и N-Boc-пиперазин (4.7 г, 25 ммоля) растворяли при комнатной температуре в DMF (50 мл). Смесь перемешивали в течение 3 ч для проведения реакции. Затем реакционную смесь выливали в ледяную воду (70 мл). Выделялся твердый продукт белого цвета. Смесь отфильтровывали с отсасыванием. Остаток на фильтре высушивали, получая 5.3 г титульного соединения в виде твердого продукта белого цвета с выходом 60.0%.
Стадия 2: трет-бутил-4-(4-бром-2-метоксибензоил)пиперазин-1-карбоксилат
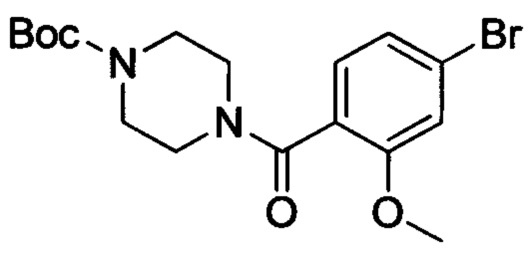
NaH (1.0 г, 41.7 ммоля) растворяли при комнатной температуре в DMF (30 мл). После продувки азота смесь охлаждали до 0°С. К охлажденной смеси добавляли трет-бутил-4-(4-бром-2-гидроксибензоил)пиперазин-1-карбоксилат (5 г, 13 ммоля). Затем полученную смесь оставляли нагреваться до комнатной температуры и проводили реакцию при перемешивании в течение 2 ч. Затем к полученной смеси медленно по каплям добавляли CH3I (2.3 г, 16 ммоля). После окончания этого добавления проводили реакцию при комнатной температуре в течение 2 ч и затем охлаждали до 0°С. Реакцию обрывали добавлением ледяной воды (100 мл). Выделялся твердый продукт, смесь отфильтровывали с отсасыванием. Остаток на фильтре высушивали, получая 4.5 г титульного соединения в виде твердого продукта желтого цвета с выходом 87%.
Стадия 3: трет-бутил-4-(4-бром-2-метоксибензил)пиперазин-1-карбоксилат
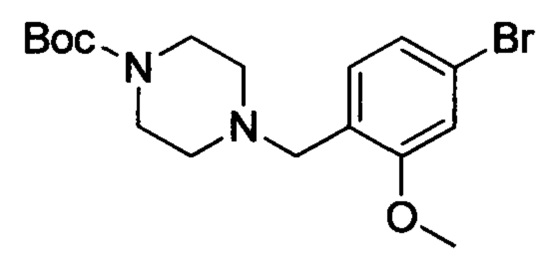
трет-Бутил-4-(4-бром-2-метоксибензоил)пиперазин-1-карбоксилат (4.5 г, 11 ммоля) растворяли в THF (30 мл). После продувки азотом смесь охлаждали до 0°С. К охлажденной смеси по каплям добавляли раствор боран-тетрагидрофуран (33 мл). После окончания этого добавления смесь нагревали до 60°С и проводили реакцию с обратным холодильником в течение 14 ч. Затем полученную смесь концентрировали. К концентрированной смеси добавляли смесь толуола (30 мл) и изопропанола (30 мл). Полученную смесь нагревали до 80°С и проводили реакцию при перемешивании в течение 8 ч. После завершения реакции реакционную смесь концентрировали, получали сырой продукт. Этот сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=5:1), получали титульное соединение в виде твердого продукта белого цвета с выходом 54.3%.
Промежуточное соединение 12: трет-бутил-4-(4-бром-2-метилбензоил)пиперазин-1-карбоксилат
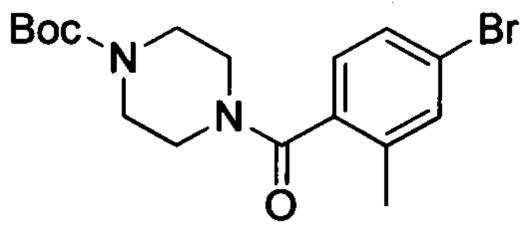
4-бром-2-метилбензойную кислоту (2.15 г, 10 ммолей), EDCI (3.80 г, 20 ммолей), HOBt (112 мг, 1 ммоль) и N-Boc-пиперазин (2.05 г, 11 ммолей) добавляли к DMF (30 мл) при комнатной температуре. Проводили реакцию при перемешивании при комнатной температуре в течение 3 ч. В полученную смесь выливали ледяную воду (30 мл). Выделялся продукт белого цвета, смесь отфильтровывали с отсасыванием. Полученный на фильтре остаток сушили с получением 2.0 г титульного соединения в виде продукта белого цвета с выходом 52.4%.
Промежуточное соединение 13: 3-бром-N-(2-феноксиэтил)анилин
Стадия 1: 2-феноксиацетальдегид
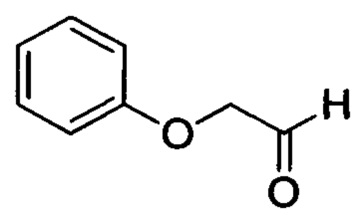
3-феноксипропан-1,2-диол (5.04 г, 30 ммолей) растворяли в DCM (90 мл). К полученной смеси в атмосфере азота на ледяной бане отдельными порциями медленно добавляли перйодат натрия (8.40 г, 39.3 ммоля). Затем проводили реакцию при перемешивании в течение 2 ч. К реакционной смеси добавляли воду (120 мл). Полученный продукт экстрагировали DCM. Органические фазы концентрировали в вакууме с получением 3.63 г титульного соединения в виде бледно-желтого масла с выходом 89.0%.
Стадия 2: 3-бром-N-(2-феноксиэтил)анилин
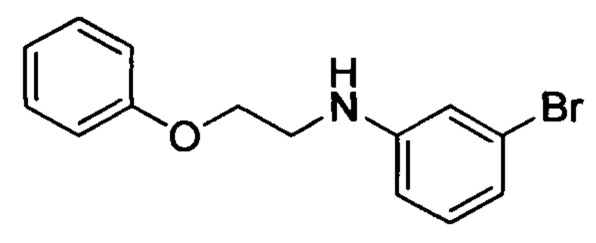
2-феноксиацетальдегид (3.2 г, 23.5 ммоля), 3-броманилин (4.25 г, 24.7 ммоля) и уксусную кислоту (5 мл) растворяли в THF (50 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. К реакционной смеси добавляли триацетоксиборгидрид натрия (12.45 г, 58.75 ммоля). Реакционную смесь перемешивали в течение ночи. После завершения реакции добавляли воду. Затем смесь экстрагировали DCM. Органические фазы концентрировали в вакууме с получением 3 г титульного соединения, которое использовали непосредственно на следующей стадии без очистки.
Промежуточное соединение 14: 3-бром-N-((1-метил-1Н-пиразол-5-ил)метил)анилин
Стадия 1: 1-метил-1Н-пиразоло-5-карбоновая кислота
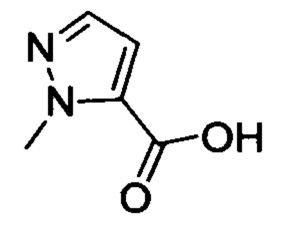
1-метил-1Н-пиразол (2.0 г, 24.4 ммоля) растворяли в THF (30 мл) при комнатной температуре. Эту смесь охлаждали до температуры -78°С в атмосфере азота. К полученной смеси медленно добавляли н-BuLi (10.72 мл, 26.8 ммоля). Полученную смесь перемешивали в течение 2 ч при -78°С и затем нагревали до комнатной температуры. Затем смесь перемешивали еще один час. Затем, поддерживая указанную температуру, в реакционную смесь вводили сухой газ СО2 в течение 5 мин. К полученной смеси добавляли воду (30 мл) для обрыва реакции и разбавления смеси. Полученную смесь экстрагировали DCM. Регулировали величину рН водной фазы, осаждалось большое количество твердого продукта. Полученную смесь отфильтровывали с отсасыванием. Остаток на фильтре сушили, получали 1.5 г титульного соединения в виде твердого продукта белого цвета с выходом 48.8%.
Стадия 2: N-(3-бромфенил)-1-метил-1Н-пиразол-5-карбоксамид
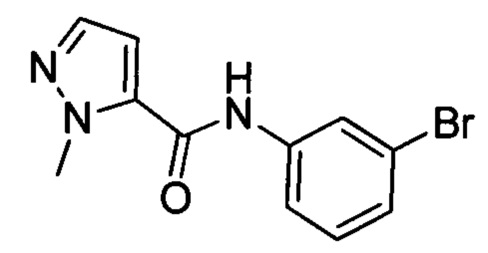
1-метил-1Н-пиразол-5-карбоновую кислоту (4 г, 31.7 ммоля) и DMF (0.2 мл) растворяли при комнатной температуре в ЕА (70 мл). Полученную смесь нагревали до 35°С. К нагретой смеси добавляли по каплям SOCl2 (4 мл) и раствор 3-броманилина (5.5 г, 31.7 ммоля) в ЕА (20 мл). После окончания добавления проводили реакцию при перемешивании в течение 14 ч при 35°С. Затем полученную смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли воду (60 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки концентрировали в вакууме с получением 6 г титульного соединения в виде продукта желтого цвета, которое использовали на следующей стадии без очистки.
Стадия 3: 3-бром-N-((1-метил-1Н-пиразоло-5-ил)метил)анилин
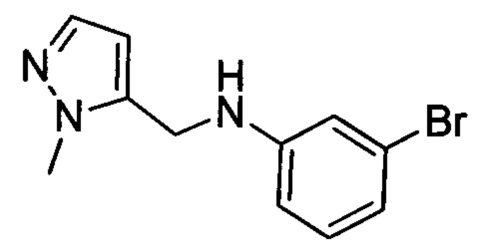
N-(3-бромфенил)-1-метил-1Н-пиразол-5-карбоксамид (1.0 г, 3.6 ммоля) растворяли при комнатной температуре в THF (20 мл). К этой смеси отдельными порциями на ледяной бане добавляли LiAlH4 (0.35 г, 9 ммолей). После окончания добавления проводили реакцию при перемешивании при 40°С в течение 14 ч. После завершения реакции добавляли воду, и экстрагировали реакционную смесь ЕА. Органические фазы концентрировали в вакууме с получением 0.8 г титульного соединения в виде твердого продукта желтого цвета с выходом 83.5%.
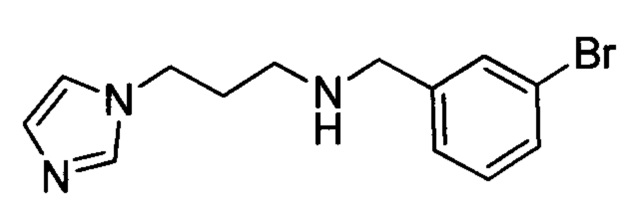
Промежуточное соединение 15: N-(3-бромбензил)-3-(1Н-имидазол-1-ил)пропан-1-амин
3-бромбензальдегид (2.0 г, 10.8 ммоля) и 3-(1Н-имидазол-1-ил)пропан-1-амин (1.48 г, 11.8 ммоля) растворяли при комнатной температуре в МеОН (20 мл). Смесь охлаждали до 0°С на ледяной бане. К охлажденной смеси медленно отдельными порциями добавляли цианборгидрид натрия (1.0 г, 16.2 ммол). После окончания его добавления проводили реакцию смеси при комнатной температуре в течение 3 ч. Полученную смесь концентрировали и добавляли к ней воду (100 мл). Полученную смесь экстрагировали DCM. Полученные органические фазы концентрировали с получением сырого продукта. Сырой продукт очищали методом колоночной хроматографии на силикагеле (DCM/MeOH=10/1), получали 1.57 г титульного соединения в виде желтого масла с выходом 49.4%.
Промежуточное соединение 16: N-(4-йодфенил)изобутирамид
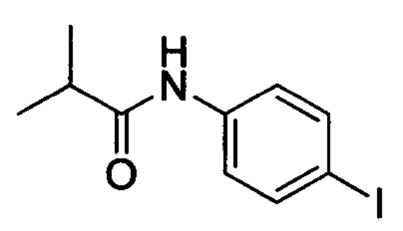
4-йоданилин (5.0 г, 22.8 ммоля) растворяли в ацетонитриле (25 мл). Полученную смесь охлаждали до температуры ниже 0°С. К охлажденной смеси добавляли пиридин (1.85 мл). Затем к полученной смеси по каплям добавляли изобутилхлорид (2.3 мл, 22.8 ммоля). После окончания его добавления осуществляли реакцию смеси в течение 3 ч. После завершения реакции реакционную смесь выливали в ледяную воду (70 мл). Выделялся твердый продукт. Полученную смесь фильтровали с отсасыванием. Остаток на фильтре сушили с получением 6.4 г титульного соединения в виде продукта белого цвета с выходом 97.1%.
Промежуточное соединение 17: 1-(4-йодфенил)пирролидин-2-он
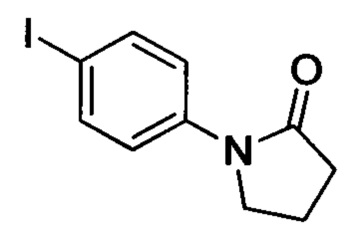
В колбу при комнатной температуре последовательно добавляли N-фенилпирролидин-2-он (5.0 г, 31 ммоля), NIS (10.4 г, 46 ммолей), карбонат цезия (340 мг) и уксусную кислоту (100 мл). Полученную смесь нагревали до 100°С и проводили реакцию в течение 4 ч. Затем реакционную смесь выливали в воду (650 мл). Затем экстрагировали реакционную смесь ЕА. Органические фазы промывали насыщенным водным раствором бикарбоната натрия и водой, сушили над безводным сульфатом натрия и отфильтровывали с отсасыванием. Фильтрат концентрировали, получая 6.15 г титульного соединения в виде коричневого твердого продукта с выходом 69.1%.
Промежуточное соединение 18: этил 2-(4-аминофенокси)ацетат
Стадия 1: этил 2-(4-нитрофенокси)ацетат
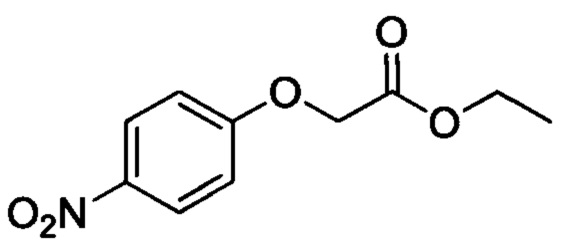
4-нитрофенол (5.0 г, 36.0 мл), этилхлорацетат (8.8 г, 72.0 ммоля) и триэтиламин (9.1 г, 90.0 ммолей) последовательно добавляли при комнатной температуре к ацетонитрилу (30 мл). После окончания добавления полученную смесь нагревали до 80°С и проводили реакцию при перемешивании в течение 6 ч. После завершения реакции полученную смесь концентрировали в вакууме. К остатку добавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки концентрировали в вакууме досуха, получали 6.0 г титульного соединения в виде твердого продукта светло-желтого цвета с выходом 74.1%.
Стадия 2: этил 2-(4-аминофенокси)ацетат
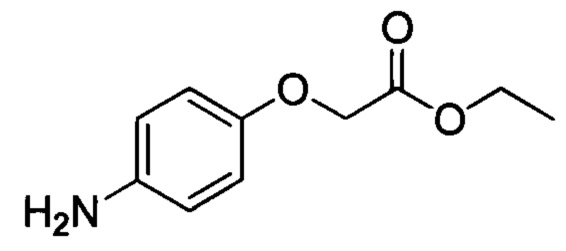
Этил 2-(4-нитрофенокси)ацетат (6.0 г, 26.6 ммоля) и Pd/C (1.0 г) добавляли при комнатной температуре к метанолу (30 мл). Смесь гидрировали в течение 2 ч при комнатной температуре. После завершения реакции полученную смесь отфильтровывали. Фильтрат концентрировали в вакууме, получали 5.0 г титульного соединения с выходом 95.9%.
Промежуточное соединение 19: N-(4-йодфенил)-N-метилизобутирамид
Стадия 1: N-(4-йодфенил)изобутирамид
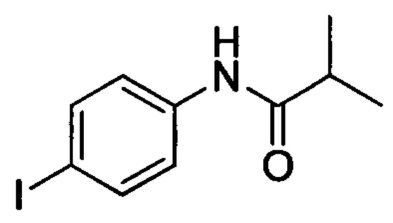
4-йоданилин (5.0 г, 22.8 ммоля) и триэтиламин (2.42 г, 24.0 ммоля) последовательно при комнатной температуре добавляли к ацетонитрилу (45 мл). Полученную смесь при перемешивании охлаждали до температуры от 0 до 5°С. К охлажденной смеси по каплям добавляли изобутирхлорид (2.55 г, 24.0 ммоля). После окончания добавления полученную смесь перемешивали в течение 0.5 ч, поддерживая указанную температуру. После завершения реакции к полученной смеси добавляли воду. Выделялся твердый продукт. Смесь отфильтровывали и высушивали с получением 6.2 г титульного соединения с выходом 94.1%.
Стадия 2: N-(4-йодфенил)N-метилизобутирамид
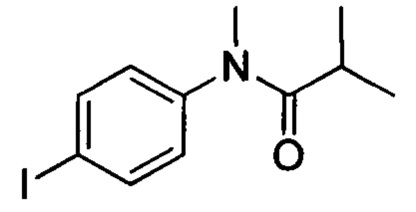
N-(4-йодфенил)изобутирамид (5.78 г, 20 ммолей) и трет-бутоксид калия (6.73 г, 60 ммолей) при комнатной температуре добавляли к THF (50 мл). Затем к этой смеси по каплям при перемешивании добавляли йодметан (5.67 г, 40 ммолей). Затем смесь перемешивали в течение 2 ч при комнатной температуре. После завершения реакции реакционную смесь выливали в водный 5% раствор 5% соляной кислоты (100 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки концентрировали с получением 5.3 г титульного соединения с выходом 87.4%.
Промежуточное соединение 20: 4-этокси-3-фторанилин
Стадия 1: 1-этокси-2-фтор-4-нитробензол
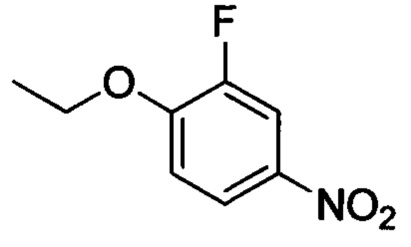
2-фтор-4-нитрофенол (5.0 г, 31.8 ммоля), бромэтан (8.67 г, 79.6 ммоля) и триэтиламин (8.05 г, 79.6 ммоля) добавляли последовательно при комнатной температуре в ацетонитрил (40 мл). После окончания добавления полученную смесь нагревали до 50°С и проводили реакцию при перемешивании в течение 6 ч. После завершения реакции реакционную смесь концентрировали в вакууме. К остатку добавляли воду. Полученную смесь экстрагировали ЕА. Органические фазы концентрировали в вакууме с получением 5.3 г титульного соединения в виде светло-желтого твердого продукта с выходом 89.9%.
Стадия 2: 4-этокси-3-фторанилин
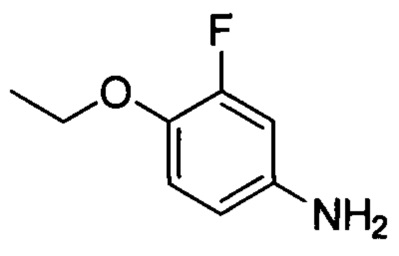
1-этокси-2-фтор-4-нитробензол (5.3 г, 28.6 ммоля) и Pd/C (0.75 г) при комнатной температуре последовательно добавляли к метанолу (100 мл). Смесь гидрировали при комнатной температуре в течение 2 ч. После завершения реакции полученную смесь отфильтровывали. Фильтрат концентрировали в вакууме, получали 3.9 г титульного соединения в виде масла с выходом 88.6%.
Промежуточное соединение 21: 3-(этоксиметил)анилин
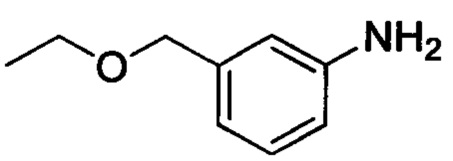
(3-нитрофенил)метанол (3.3 г, 21.5 ммоля), бромэтан (3.5 г, 32.3 ммоля) и гидроксид калия (2.4 г, 43.0 ммолей) при комнатной температуре последовательно добавляли к DMSO (20 мл). После окончания добавления полученную смесь перемешивали и проводили реакцию при комнатной температуре в течение 6 ч. После завершения реакции к реакционной смеси добавляли воду и ЕА. Полученную смесь перемешивали и оставляли стоять для образования водной фазы и органической фазы. Водную фазу экстрагировали ЕА. Соединенные органические вытяжки концентрировали в вакууме. К остатку последовательно добавляли воду (30 мл), THF (30 мл), порошок Fe (3.6 г, 64.5 ммоля) и хлорид аммония (0.5 г). После окончания добавления смесь нагревали до 75°С и проводили реакцию в течение 1 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры. Полученную смесь экстрагировали ЕА. Органические вытяжки концентрировали в вакууме с получением 3.3 г титульного соединения в виде масла.
Промежуточное соединение 22: этил 6-(4-йодфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
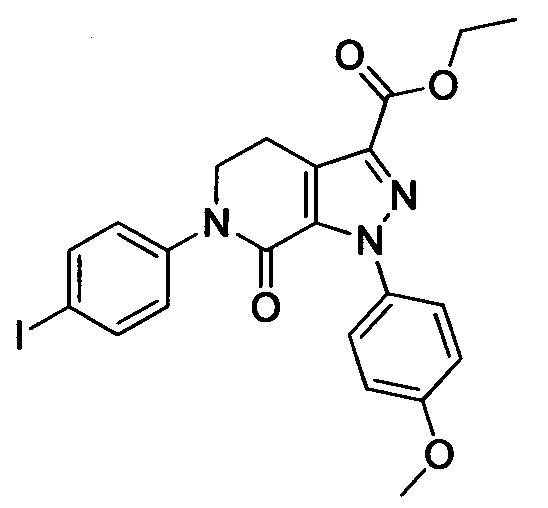
Этил 6-(4-аминофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (8.0 г, 19.7 ммоля) и концентрированную соляную кислоту (3.3 мл, 39.4 ммоля) при комнатной температуре добавляли в воду (100 мл). Эту смесь перемешивали и охлаждали до температуры от 0 до 5°С. К охлажденной смеси добавляли отдельными порциями нитрит натрия (1.63 г, 23.6 ммоля). После окончания добавления смесь перемешивали при 5-10°С в течение 1 ч. Затем к этой смеси добавляли йодид натрия (4.43 г, 29.6 ммоля), полученную смесь перемешивали при комнатной температуре в течение 4 ч. После завершения реакции реакционную смесь экстрагировали этил ацетатом. Органические фазы концентрировали в вакууме с получением сырого продукта. Сырой продукт очищали методом колоночной хроматографии на силикагеле (РЕ:ЕА=1:1) с получением 5.2 г титульного соединения с выходом 51.0%.
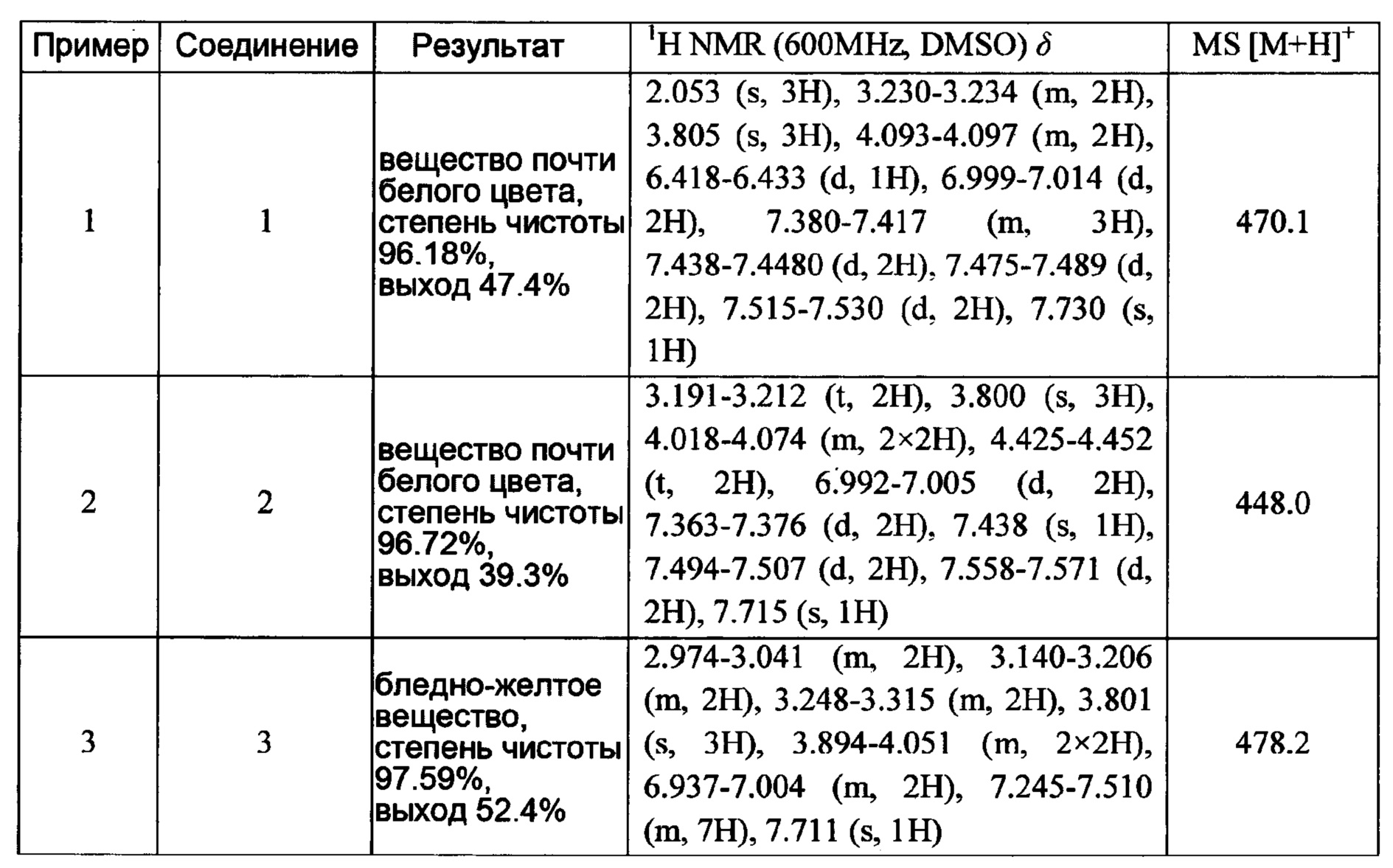
Пример 1:
6-(4-((3-(Диметиламино)пропил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(4-метоксифенил)гидразоно)ацетат
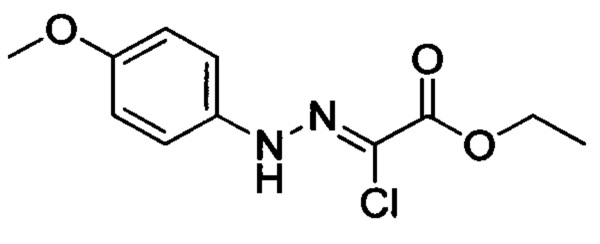
В колбу при комнатной температуре последовательно загружали 4-метоксианилин (30.0 г, 244 ммоля) и воду (100 мл). Смесь перемешивали и охлаждали до температуры от -5 до 0°С. К полученной смеси последовательно прибавляли концентрированную соляную кислоту (35 мл) и водный раствор нитрита натрия (50 мл). По окончании прибавления смесь перемешивали в течение 0.5 ч, поддерживая указанную температуру. Затем к этой смеси по каплям прибавляли раствор этил 2-хлор-3-оксобутаноата (40.2 г, 244 ммоля) в этаноле (200 мл) и раствор ацетата натрия (60.0 г, 732 ммоля) в воде (500 мл). По завершении прибавления смесь перемешивали в течение 0.5 ч при температуре от -5 до 0°С. Затем смесь нагревали до комнатной температуры и проводили реакцию при перемешивании в течение 6 ч. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=10:1), получали 31.6 г титульного соединения, выход 50.4%.
Стадия 2: Этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
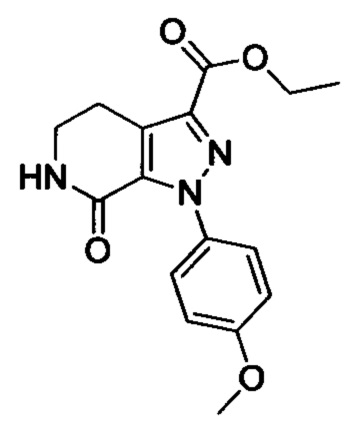
При комнатной температуре в колбу последовательно загружали толуол (500 мл), 3-хлор-5,6-дигидропиридин-2(1Н)-он (35g, сырой продукт), этил-2-хлор-2-(2-(4-метоксифенил)гидразоно)ацетат (30.8 г, 0.120 ммоля) и триэтиламин (24.2 г, 0.240 ммоля). Полученную смесь при перемешивании нагревали до кипения и проводили реакцию при кипячении в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в раствор ЕА (500 мл) в воде (500 мл). Смесь фильтровали с отсасыванием.
Осадок на фильтре сушили в вакууме, получали 25.0 г титульного соединения в виде твердого вещества желтого цвета с выходом 66.1%.
Стадия 3: Этил 6-(4-((трет-бутоксикарбонил)(3-(диметиламино)пропил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
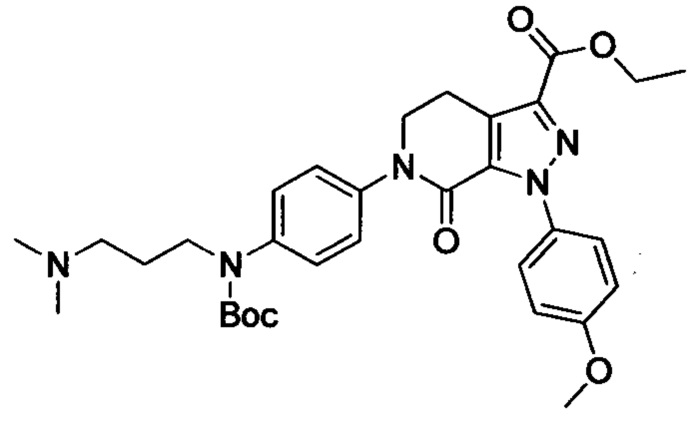
трет-Бутил (3-(диметиламино)пропил)(4-иодфенил)карбамат (930 мг, 2.30 ммоля), этил-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (604 мг, 1.92 ммоля), карбонат калия (530 мг, 3.83 ммоля), 1,10-фенантролин (138 мг, 0.77 ммоля) и иодид меди (73 мг, 0.38 ммоля) последовательно диспергировали в DMSO (10 мл) при комнатной температуре. Смесь нагревали при 120°С в атмосфере азота при перемешивании в течение 14 ч. По завершении реакции реакционную смесь охлаждали и выливали в воду со льдом (50 мл). Полученную смесь экстрагировали DCM. Объединенные органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=20:1), получали 1.0 г титульного соединения в виде коричневого масла с выходом 88.0%.
Стадия 4: трет-Бутил (4-(3-карбамоил-1-(4-метокси)-7-оксо-4,5-дигидро-1Н-пиразоло [3,4-с] пиридин-6(7Н)-ил)фенил)(3-(диметиламино)пропил)карбамат
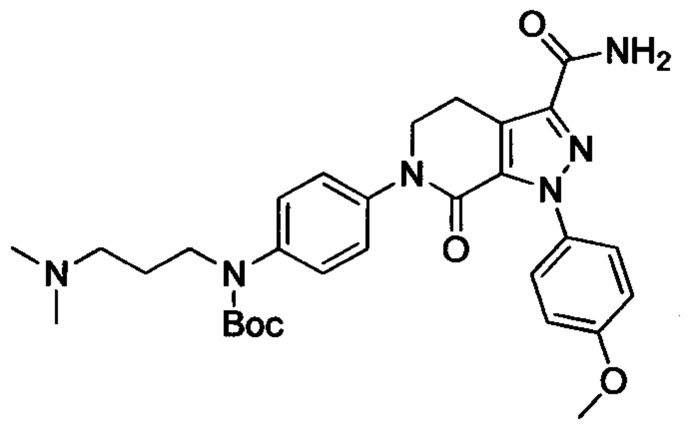
Этил 6-(4-((трет-бутоксикарбонил)(3-(диметиламино)пропил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (600 мг, 1.01 ммоля) при комнатной температуре растворяли в растворе газообразного аммиака в этиленгликоле (15 мл). Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. По завершении реакции реакционную смесь выливали в воду со льдом (100 мл). Полученную смесь экстрагировали DCM. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=50:1), получали 250 мг титульного соединения в виде желтого масла. Выход 44.0%.
Стадия 5: 6-(4-((3 -(Диметиламино)пропил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
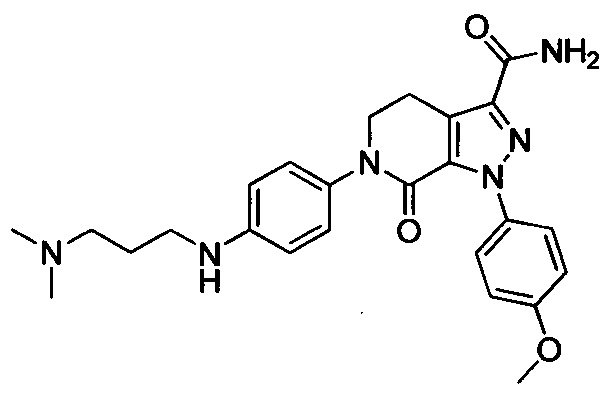
трет-Бутил (4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)(3-(диметиламино)пропил)карбамат (250 мг, 0.44 ммоля) растворяли в растворе HCl/ЕА (10 мл). Реакцию проводили при комнатной температуре в течение 14 ч, а затем с помощью раствора гидроксида натрия доводили значение рН до нейтрального. Полученную смесь упаривали в вакууме, получали твердое вещество серого цвета. Этот твердый продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=20:1), получали 180 мг (выход 88.5%) титульного соединения в виде твердого вещества белого цвета.
1H-NMR (400 MHz, DMSO_d6&D20) δ: 7.52-7.46 (m, 2H), 7.23-7.17 (m, 2H), 7.03-6.97 (m, 2H), 6.91-6.84 (m, 2H), 3.97 (t, 2H), 3.80 (s, 3Н), 3.23-3.11 (m, 6H), 2.77 (s, 6H), 2.02-1.90 (m, 2H);
MS 463 [M+H]+.
Пример 2: 1-(4-Метоксифенил)-6-(4-(3-(метиламино)пропокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-(3-((трет-бутоксикарбонил)(метил)амино)пропокси)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
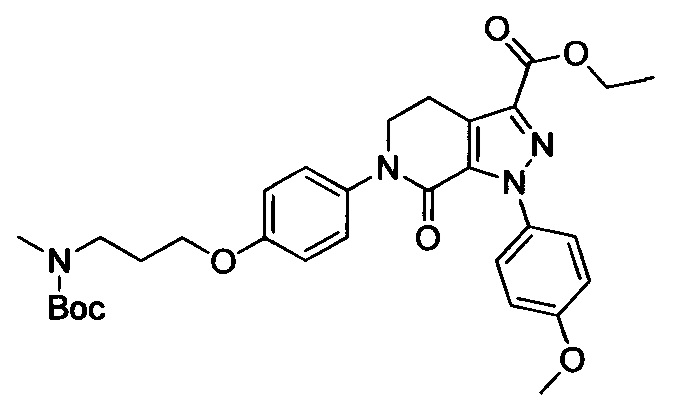
трет-Бутил (3-(4-иодфенокси)пропил)(метил)карбамат (932 мг, 2.38 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (500 мг, 1.59 ммоля), карбонат калия (438 мг, 3.18 ммоля), 1,10-фенантролин (114 мг, 0.64 ммоля) и иодид меди (60 мг, 0.32 ммоля) последовательно диспергировали в DMSO (10 мл) при комнатной температуре. Смесь нагревали при 120°С в атмосфере азота при перемешивании в течение 14 ч. Реакционную смесь охлаждали и выливали в воду со льдом (50 мл). Полученную смесь экстрагировали DCM. Объединенные органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=50:1), получали 600 мг титульного соединения в виде желтого масла. Выход 65.2%.
Стадия 2: трет-Бутил (3-(4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло [3,4-с]пиридин-6(7Н)-ил)фенокси)пропил)(метил)карбамат
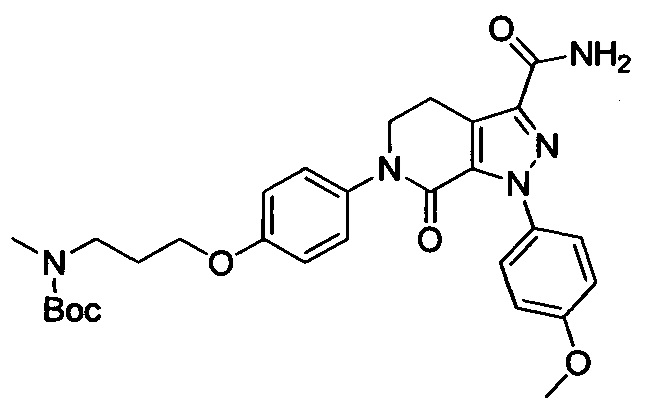
Этил 6-(4-(3-((трет-бутоксикарбонил)(метил)амино)пропокси)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (400 мг, 0.69 ммоля) при комнатной температуре растворяли в растворе аммиак газ/EG (15 мл). Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. По завершении реакции реакционную смесь выливали в воду со льдом (100 мл). Полученную смесь экстрагировали DCM. Органические вытяжки упаривали в вакууме, получали 330 мг титульного соединения в виде твердого вещества желтого цвета, которое использовали на следующей стадии без очистки.
Стадия 3: 1-(4-Метоксифенил)-6-(4-(3-(метиламино)пропокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
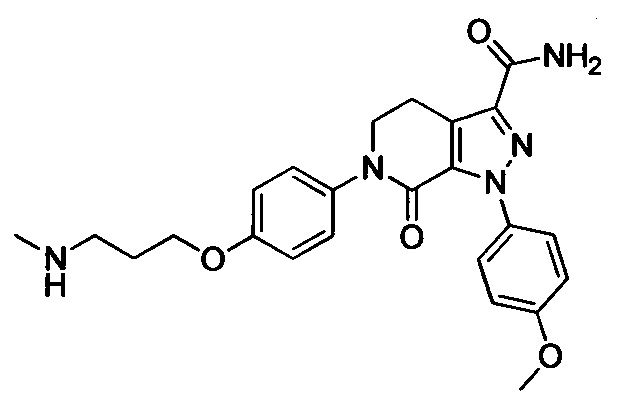
трет-Бутил (3-(4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенокси)пропил)(метил)карбамат (300 мг, 0.55 ммоля) растворяли в растворе HCl/ЕА (10 мл). Реакцию смеси проводили при комнатной температуре в течение 14 ч, а затем доводили рН до нейтрального значения с помощью водного раствора гидроксида натрия. Полученную смесь упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH=20:1), получали 220 мг титульного соединения в виде твердого вещества белого цвета 89.1%.
1H-NMR (400 MHz, DMSO_d6) δ: 8.61 (brs, 2H), 7.71 (s, 1H), 7.52-7.41 (m, 2H), 7.43 (s, 1H), 7.30-7.24 (m, 2H), 7.03-6.92 (m, 4H), 4.06 (t, 2H), 3.98 (t, 2H), 3.80 (s, 3H), 3.19 (t, 2H), 3.09-3.00 (m, 2H), 2.58 (t, 3H), 2.10-2.01 (m, 2H);
MS 450 [M+H]+.
Пример 3: 6-(3-Фтор-4-((2-метоксиэтил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-((трет-бутоксикарбонил)(2-метоксиэтил)амино)-3-фторфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
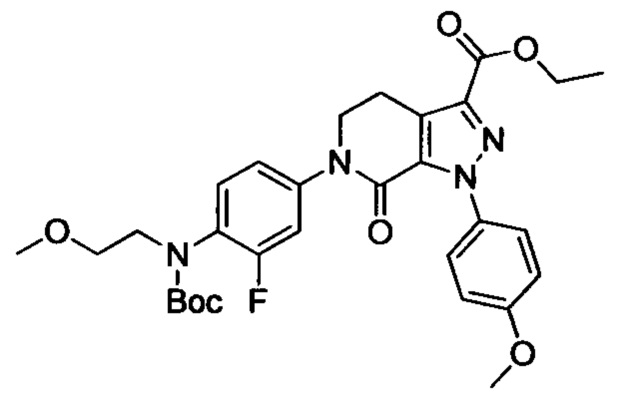
Трет-Бутил (4-бром-2-фторфенил)(2-метоксиэтил)карбамат (862 мг, 2.48 ммоля), этил-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (600 мг, 1.91 ммоля), карбонат калия (1050 мг, 7.62 ммоля), иодид калия (700 мг, 4.22 ммоля), иодид меди (145 мг, 0.76 ммоля) и N,N'-диметил-1,2-этандиамин (84 мг, 0.95 ммоля) последовательно растворяли в DMSO (10 мл) при комнатной температуре. Смесь нагревали при 120°С и перемешивали в течение ночи в атмосфере азота. Затем реакционную смесь охлаждали до комнатной температуры и выливали в воду со льдом (50 мл). Полученную смесь экстрагировали DCM. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=80:1), получали 1.0 г титульного соединения в виде твердого вещества желтого цвета. Выход 89.9%.
Стадия 2: трет-Бутил (4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-фторфенил)(2-метоксиэтил)карбамат
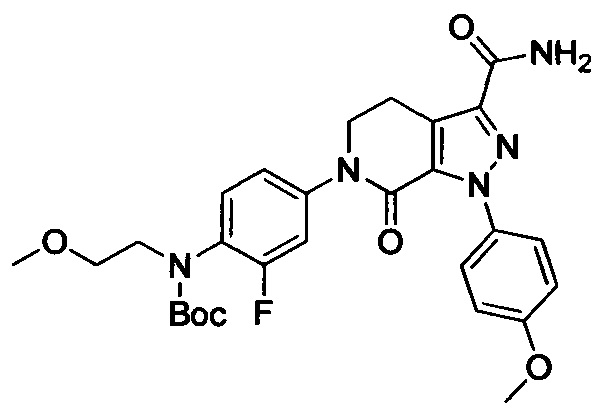
Этил 6-(4-((трет-бутоксикарбонил)(2-метоксиэтил)амино)-3-фторфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.0 г, 1.72 ммоля) при комнатной температуре растворяли в растворе аммиак газ/EG (15 мл). Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. По завершении реакции реакционную смесь выливали в воду со льдом (100 мл). Полученную смесь экстрагировали DCM. Органические вытяжки упаривали в вакууме, получали 800 мг титульного соединения в виде твердого вещества желтого цвета, которое использовали на следующей стадии без очистки.
Стадия 3: 6-(3-Фтор-4-((2-метоксиэтил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
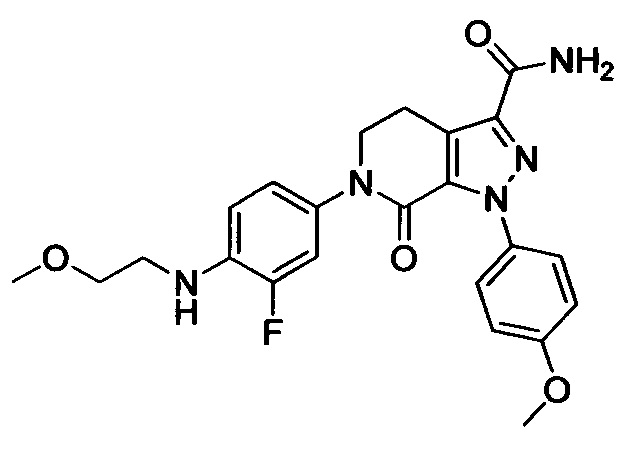
трет-Бутил (4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло-[3,4-с]пиридин-6(7Н)-ил)-2-фторфенил)(2-метоксиэтил)карбамат (300 мг, 0.54 ммоля) растворяли в растворе HCl/ЕА (10 мл). Реакцию проводили при комнатной температуре в течение 14 ч, а затем водным раствором гидроксида натрия доводили рН до нейтрального значения. Полученную смесь упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH=25:1), получали 220 мг титульного соединения в виде твердого вещества белого цвета. Выход 90.0%.
1H-NMR (400 MHz, DMSO_d6) δ: 7.69 (s, 1Н), 7.53-7.46 (m, 2H), 7.42 (s, 1H), 7.09 (dd, 1H), 7.03-6.95 (m, 3H), 6.77 (t, 1H), 5.80 (br s, 4H), 3.95 (t, 2H), 3.80 (s, 3H), 3.49 (t, 2H), 3.27 (t, 5H), 3.17 (t, 2H);
MS 454 [M+H]+.
Пример 4: 6-(4-(4-Аминобутанамидо)-3-метилфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-(4-((трет-бутоксикарбонил)амино)бутанамидо)-3-метилфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
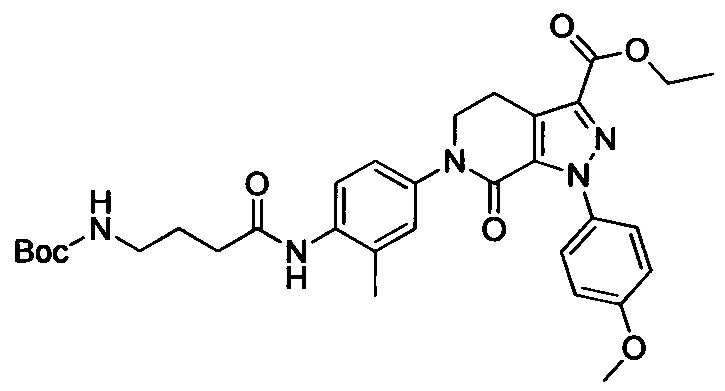
трет-Бутил-(4-((4-иод-2-метилфенил)амино)-4-оксобутил)карбамат (1.27 г, 3.0 ммоля), карбонат калия (0.7 г, 5.0 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.8 г, 2.5 ммоля), иодид меди (0.12 г, 0.63 ммоля) и 1,10-фенантролин (0.18 г, 10 ммолей) последовательно растворяли в DMSO (10 мл) при комнатной температуре. Смесь нагревали до 120°С и проводили реакцию при этой температуре при перемешивании в течение 14 час в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры и выливали в воду со льдом (40 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали хроматографией на колонке с силикагелем (DCM:MeOH=50:1), получали 0.72 г титульного соединения в виде твердого вещества желтого цвета. Выход 47.5%.
Стадия 2: трет-бутил (4-((4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-метилфенил)амино)-4-оксобутил) карбамат
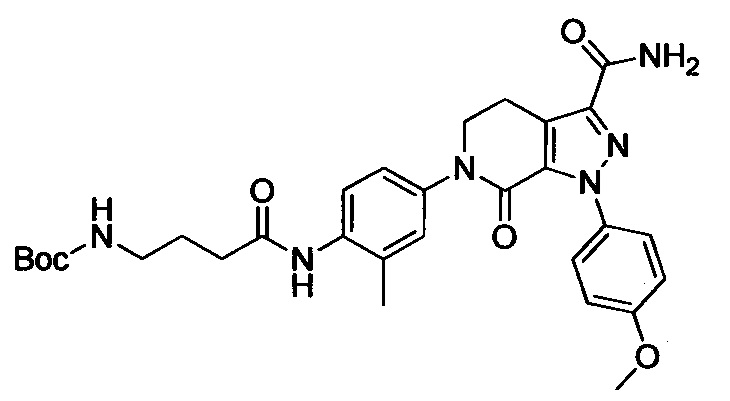
Этил 6-(4-(4-((трет-бутоксикарбонил)амино)бутанамидо)-3-метилфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.72 г, 1.18 ммоля) растворяли при комнатной температуре в растворе аммиак газ/EG (15 мл) Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в воду (30 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали с помощью преп.HPLC, получали 400 мг титульного соединения в виде твердого вещества желтого цвета, выход 58.8%.
Стадия 3: 6-(4-(4-Аминобутанамидо)-3-метилфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
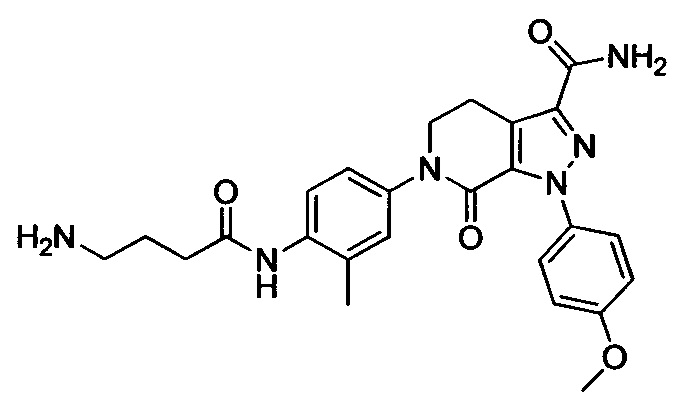
трет-Бутил-(4-((4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-метилфенил)амино)-4-оксобутил)карбамат (400 мг, 0.69 ммоля) растворяли в растворе HCl/ЕА (10 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, а затем упаривали. Остаток растворяли в воде. Полученную смесь очищали с помощью преп.-HPLC, получали 200 мг (выход 60.6%) титульного соединения в виде твердого вещества телесного цвета.
1H-NMR (400 MHz, DMSO-d6) δ: 9.37 (br s, 1H), 7.72 (s, 1H), 7.49 (d, 2H), 7.44 (s, 1H), 7.37 (d, 1H), 7.19 (s, 1H), 7.13 (d, 1H), 7.00 (d, 2H), 4.04-4.00 (m, 2H), 3.81 (s, 3H), 3.21-3.18 (m, 2H), 2.65-2.62 (m, 2H), 2.39-2.36 (m, 2H), 2.18 (s, 3H), 1.75-1.69 (m, 2H).
MS 477 [M+H]+.
Пример 5: 6-(3-(Этиламино)-4-пропоксифенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(3-(этиламино)-4-пропоксифенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
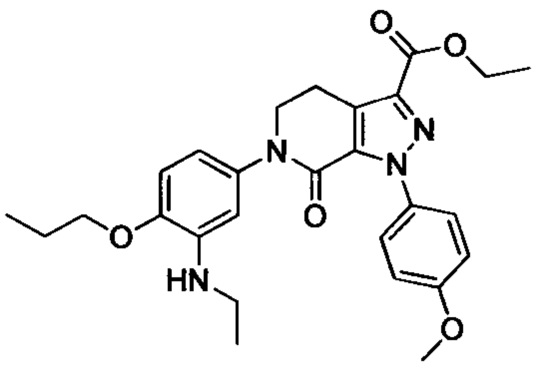
N-Этил-5-иод-2-пропоксианилин (0.94 г, 3.1 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.69 г, 2.2 ммоля), 1,10-фенантролин (0.16 г, 0.88 ммоля) и карбонат калия (0.61 г, 4.4 ммоля) последовательно прибавляли к DMSO (5 мл) при комнатной температуре. Смесь нагревали до 120°С и проводили реакцию при этой температуре при перемешивании в течение 14 час в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры и выливали в воду со льдом (50 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали препаративной (преп.)-HPLC, получали 400 мг (выход 36.9%) титульного соединения в виде твердого вещества желтого цвета.
Стадия 2: 6-(3-(Этиламино)-4-пропоксифенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1 Н-пиразоло [3,4-с]пиридин-3-карбоксамид
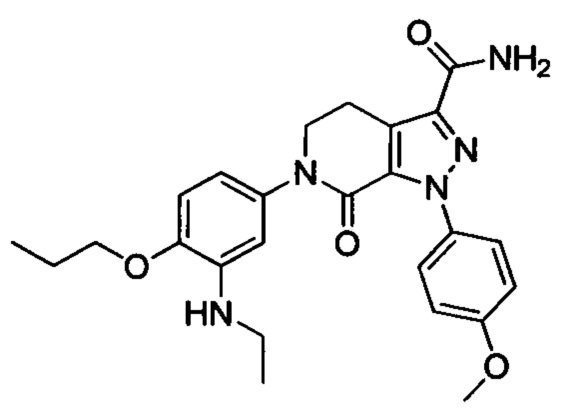
Этил 6-(3-(этиламино)-4-пропоксифенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (250 мг, 0.51 ммоля) растворяли при комнатной температуре в растворе аммиак газ/EG. Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, а затем выливали в воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали 150 мг титульного соединения в виде твердого вещества желтого цвета, выход 63.5%.
1H-NMR (400 MHz, DMSO-d6) δ: 7.69 (s, 1Н), 7.48 (d, 2H), 7.42 (s, 1H), 6.99 (d, 2H), 6.67 (d, 1H), 6.48-6.44 (m, 2H), 4.64 (t, 1H), 3.97-3.89 (m, 4H), 3.80 (s, 3H), 3.18-3.15 (m, 2H), 3.08-3.04 (m, 2H), 1.77-1.72 (m, 2H), 1.14 (t, 3H), 0.99 (t, 3H).
MS 464 [M+H]+.
Пример 6: 1-(4-Метоксифенил)-6-(4-((2-(метиламино)этил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-((2-((трет-бутоксикарбонил)(метил)амино)этил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
трет-Бутил-(2-((4-иодфенил)амино)этил)(метил)карбамат (1.14 г, 3.0 ммоля), карбонат калия (0.7 г, 5.0 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.8 г, 2.5 ммоля), иодид меди (0.1 г, 0.53 ммоля) и 1,10-фенантролин (0.18 г, 1.0 ммоля) последовательно прибавляли к DMSO (10 мл) при комнатной температуре. Смесь нагревали до 120°С и проводили реакцию при этой температуре при перемешивании в течение 14 час в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры и выливали в воду со льдом (40 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=50:1), получали 0.5 г титульного соединения в виде твердого вещества желтого цвета (выход 35.5%).
Стадия 2: трет-Бутил-(2-((4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)амино)этил)(метил)карбамат
Этил 6-(4-((2-((трет-бутоксикарбонил)(метил)амино)этил)амино)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.5 г, 0.89 ммоля) растворяли в растворе аммиак газ/EG (10 мл) при комнатной температуре. Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, а затем выливали в воду (30 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали преп.-HPLC, получали 406 мг титульного соединения в виде твердого вещества желтого цвета. Выход 85.3%.
Стадия 3: 1-(4-Метоксифенил)-6-(4-((2-(метиламино)этил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
трет-Бутил-(2-((4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)амино)этил)(метил)карбамат (406 мг, 0.76 ммоля) растворяли в растворе HCl/ЕА (10 мл). Реакцию проводили при перемешивании при комнатной температуре в течение 2 ч, рН доводили до нейтрального с помощью водного раствора гидроксида натрия. Полученную смесь упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH=20:1), получали 220 мг титульного соединения в виде твердого вещества светло-фиолетового цвета, выход 66.7%.
1H-NMR (400 MHz, DMSO-d6&D2O) δ: 7.48 (d, 2H), 7.12 (d, 2H), 7.01 (d, 2H), 6.69 (d, 2H), 3.96-3.93 (m, 2H), 3.81(s, 3Н), 3.38-3.35 (m, 2H), 3.19-3.16 (m, 2H), 3.10-3.07 (m, 2H), 2.59 (s, 3Н).
MS 435 [M+H]+
Пример 7: 6-(3-Хлор-4-((4-оксопентил)окси)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил-6-(3-хлор-4-((4-оксопентил)окси)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
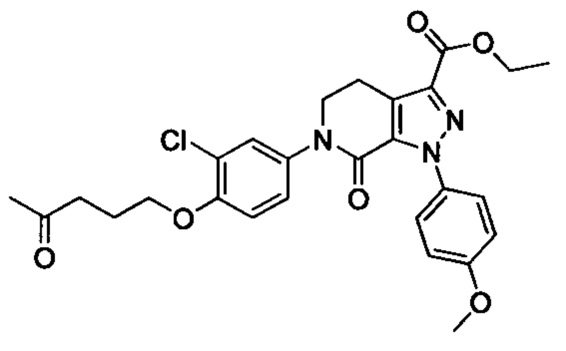
5-(4-Бром-2-хлорфенокси)пентан-2-он (1.75 г, 6.0 ммоля), карбонат калия (2.54 г, 18.4 ммоля), этил-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.45 г, 4.6 ммоля), иодид меди (0.35 г, 1.84 ммоля), N,N'-диметил-1,2-этандиамин (200 мг, 2.3 ммоля) и иодид калия (1.68 г, 10.1 ммоля) последовательно прибавляли к DMSO (10 мл) при комнатной температуре. Смесь нагревали до 120°С и проводили реакцию при этой температуре при перемешивании в течение 14 час в атмосфере азота. Реакционную смесь выливали в воду со льдом (100 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали с помощью преп.-HPLC, получали 280 мг (выход 8.9%) титульного соединения в виде твердого вещества белого цвета.
Стадия 2: 6-(3-Хлор-4-((4-оксопентил)окси)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
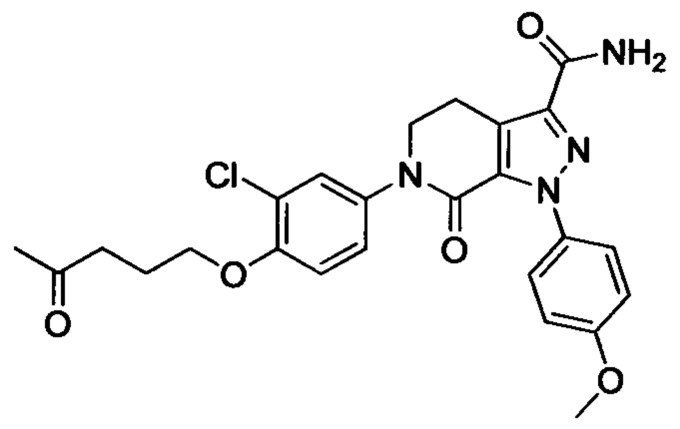
Этил 6-(3-хлор-4-((4-оксопентил)окси)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (270 мг, 0.51 ммоля) растворяли в смеси аммиак газ/EG. Смесь помещали в герметично закрывающуюся пробирку, герметизировали ее и проводили реакцию при 100°С в течение 14 ч. Реакционную смесь выливали в воду (30 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали 150 мг титульного соединения в виде твердого вещества светло-желтого цвета с выходом 59.2%.
1H-NMR (400 MHz, DMSO-d6) δ: 7.71 (s, 1Н), 7.55-7.35 (m, 4H), 7.27 (dd, 1H), 7.14 (d, 1H), 7.00 (d, 2H), 4.05-3.97 (m, 4H), 3.80 (s, 3H), 3.21-3.17 (m, 2H), 2.62 (t, 2H), 2.12 (s, 3H), 1.95-1.91 (m, 2H)
MS: 497.8 [M+H]+
Пример 8: 1-(4-Метоксифенил)-6-(4-(4-метилпиперазин-1-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксамид
Стадия 1: Этил 1-(4-метоксифенил)-6-(4-(4-метилпиперазин-1-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
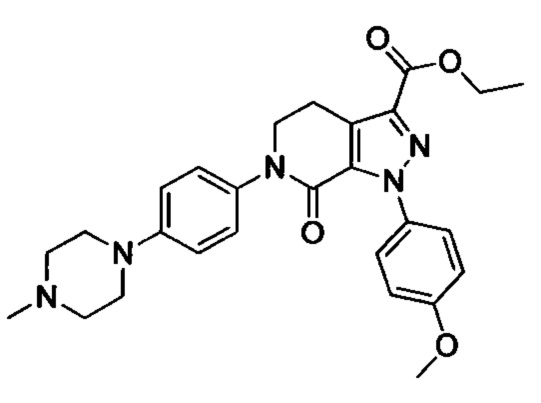
1-(4-Иодфенил)-4-метилпиперазин (500 мг, 1.7 ммоля), карбонат калия (381 мг, 2.8 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (435 мг, 1.4 ммоля), иодид меди (26 мг, 0.14 ммоля) и 1,10-фенантролин (50 мг, 0.28 ммоля) последовательно добавляли к DMSO (5 мл) при комнатной температуре. Смесь нагревали до 120°С и проводили реакцию при этой температуре при перемешивании в течение 14 час в атмосфере азота. Реакционную смесь выливали в воду со льдом (20 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле, получали 400 мг титульного соединения в виде твердого вещества светло-желтого цвета. Выход 48.0%.
Стадия 2: 1-(4-Метоксифенил)-6-(4-(4-метилпиперазин-1-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
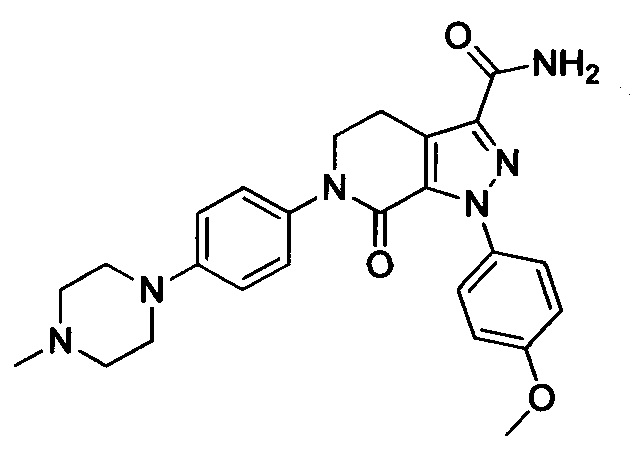
Этил 1-(4-метоксифенил)-6-(4-(4-метилпиперазин-1-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1H-пиразоло[3,4-с]пиридин-3-карбоксилат (350 мг, 0.7 ммоля) растворяли в смеси аммиак газ/EG. Реакцию проводили в герметических условиях при перемешивании при 100°С в течение 14 ч. Реакционную смесь выливали в воду (30 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали 140 мг титульного соединения в виде желтого порошка, выход 43.4%.
1H-NMR (400 MHz, DMSO-d6) δ: 7.69 (s, 1H), 7.49 (d, 2H), 7.42 (s, 1H), 7.17 (d, 2H), 7.00 (d, 2H), 6.92 (d, 2H), 3.96 (t, 2H), 3.80 (s, 3H), 3.20-3.07 (m, 6H), 2.47-2.41 (m, 4H), 2.21 (s, 3H).
MS: 461.1 [M+H]+
Пример 9:
1-(4-Метоксифенил)-7-оксо-6-(3-(2-(пиперазин-1-ил)этил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(3-(2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)этил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
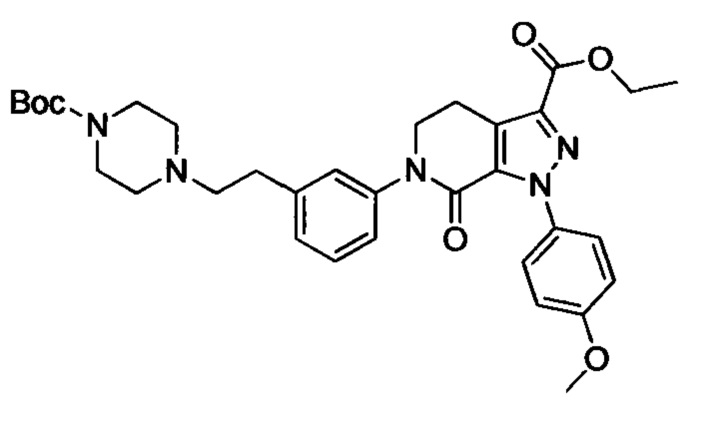
трет-Бутил 4-(3-брофенетил)пиперазин-1-карбоксилат (1.0 г, 2.7 ммоля), карбонат натрия (750 мг, 5.4 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (700 мг, 2.2 ммоля), иодид меди (210 мг, 1.1 ммоля), N,N'-диметил-1,2-этандиамин (120 мг, 1.4 ммоля) и иодид калия (900 мг, 5.4 ммоля) последовательно добавляли к DMSO (10 мл) при комнатной температуре. Реакцию проводили при перемешивании при 120°С в течение 14 ч в атмосфере азота. Реакционную смесь выливали в воду со льдом (30 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали, получали сырой продукт. Сырой продукт очищали с помощью преп.-HPLC, получали 800 мг титульного соединения в виде жидкости желтого цвета, выход 49.1%.
Стадия 2: трет-бутил 4-(3-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенетил)пиперазин-1-карбоксилат
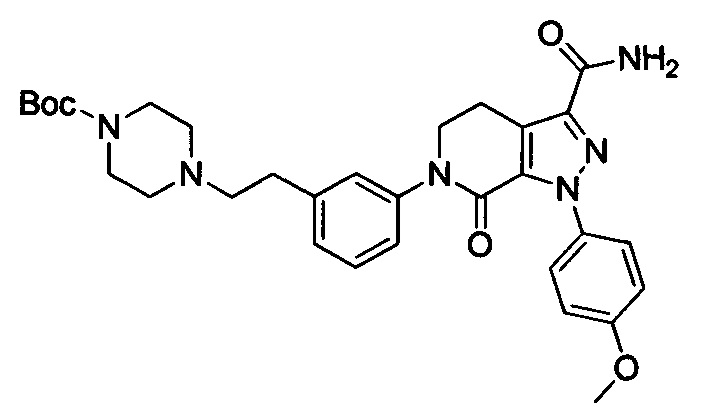
Этил 6-(3-(2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)этил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (800 мг, 1.3 ммоля) растворяли в смеси аммиак газ/EG. Реакцию проводили в герметических условиях при перемешивании при 100°С в течение 14 ч. Реакционную смесь выливали в воду (30 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали 400 мг титульного соединения в виде желтого порошка, выход 53.6%.
Стадия 3: 1-(4-Метоксифенил)-7-оксо-6-(3-(2-(пиперазин-1-ил)этил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
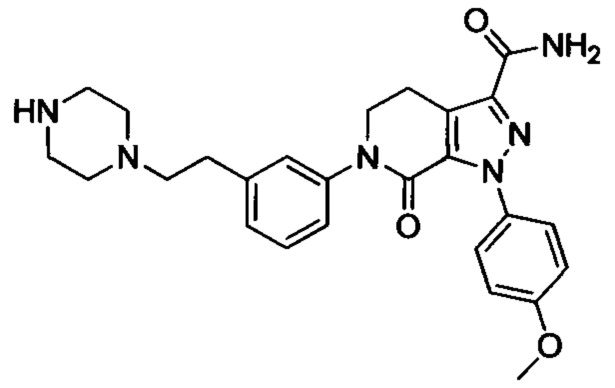
трет-Бутил 4-(3-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенетил)пиперазин-1-карбоксилат (400 мг, 0.7 ммоля) растворяли в ЕА (4 мл). У этой смеси по каплям прибавляли раствор HCl/МТВЕ (5 мл). По завершении прибавления смесь перемешивали при комнатной температуре в течение 2 ч, рН доводили до нейтрального, упаривали в вакууме, получали сырой продукт в виде масла. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=20:1), получали 150 мг титульного соединения в виде твердого вещества светло-коричневого цвета, выход 45.2%.
1H-NMR (400 MHz, DMSO-d6&D2O) δ: 7.49 (d, 2H), 7.38 (t, 1H), 7.30 (s, 1H), 7.26 (d, 1H), 7.20 (d, 1H), 7.02 (d, 2H), 4.06 (t, 2H), 3.81 (s, 3H), 3.50-3.16 (m, 12H), 3.06-2.96 (m, 2H).
MS 475 [M+H]+.
Пример 10: 6-(3-Метокси-4-(пиперазин-1-илметил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3 -карбоксамид
Стадия 1: Этил 6-(4-((4-(трет-бутоксикарбонил)пиперазин-1-ил)метил)-3-метоксифенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
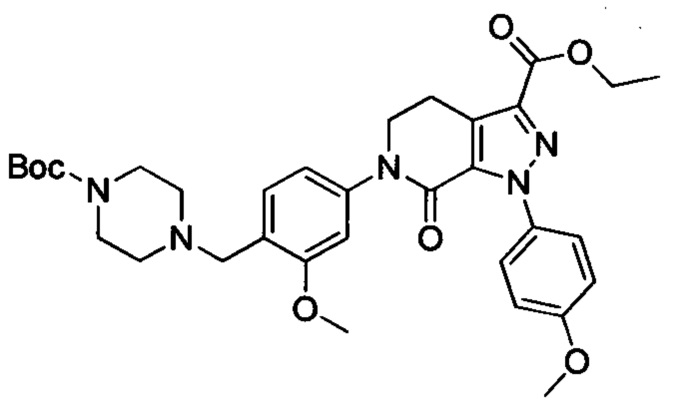
трет-Бутил 4-(4-бром-2-метоксибензил)пиперазин-1-карбоксилат (2.0 г, 5.1 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с] пиридин-3-карбоксилат (1.6 г, 5.1 ммоля), карбонат калия (1.4 г, 10 ммолей), иодид меди (0.2 г, 1.1 ммоля) и N,N'-диметил-1,2-этандиамин (0.1 г, 1.1 ммоля) последовательно добавляли к DMSO (25 мл) при комнатной температуре. После продувания азотом смесь нагревали до 100°С и проводили реакцию при этой температуре в течение 14 ч. Реакционную смесь выливали в воду (100 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получая сырой продукт. Сырой продукт очищали с помощью преп.-HPLC, получали 600 мг титульного соединения в виде твердого вещества белого цвета, выход 19.0%.
Стадия 2: трет-бутил 4-(4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-метоксибензил)пиперазин-1-карбоксилат
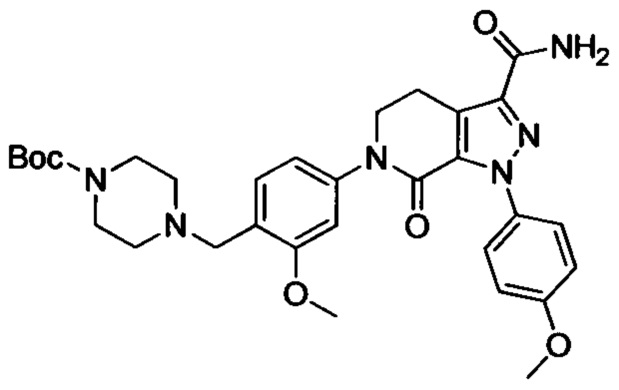
Этил 6-(4-((4-(трет-бутоксикарбонил)пиперазин-1-ил)метил)-3-метоксифенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.6 г, 0.99 ммоля) помещали в герметизируемую пробирку, а затем добавляли NH3/EG (3 мл). После растворения содержимое герметизированной пробирки нагревали до 100°С и при этой температуре проводили реакцию в течение 14 ч. По завершении реакции реакционную смесь выливали в холодную воду (50 мл). Выпадал осадок белого цвета. Смесь фильтровали с отсасыванием. Осадок на фильтре сушили, получали 400 мг титульного соединения в виде твердого вещества белого цвета, выход 68.5%.
Стадия 3: 6-(3-Метокси-4-(пиперазин-1-илметил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
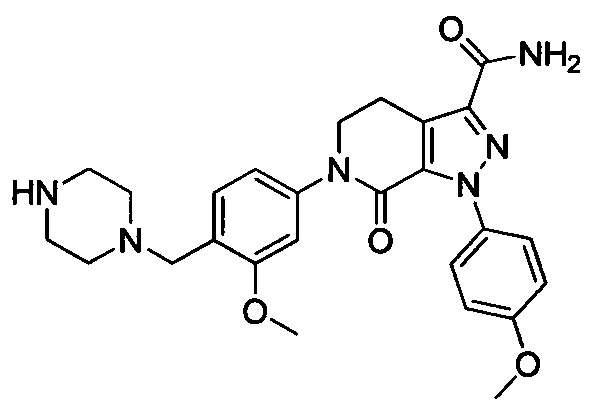
трет-Бутил 4-(4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-метоксибензил)пиперазин-1-карбоксилат (400 мг, 0.7 ммоля) растворяли в МеОН (10 мл). В реакционную смесь вводили HCl газ до завершения реакции. рН реакционной смеси доводили до нейтрального значения. Полученную смесь упаривали в вакууме и очищали колоночной хроматографией на силикагеле (DCM:MeOH=30:1), получали 160 мг титульного соединения в виде светло-желтого порошка, выход 46.6%.
1Н-NMR (400 MHz, DMSO-d6&D2O) δ: 7.54-7.48 (m, 3Н), 7.14 (d, 1H), 7.07-6.99 (m, 3H), 4.33 (s, 2H), 4.10 (t, 2H), 3.84 (s, 3H), 3.81 (s, 3H), 3.49-3.33 (m, 8H), 3.23 (t, 2H).
MS: 491.2 [M+H]+
Пример 11: 1-(4-Метоксифенил)-6-(3-метил-4-(пиперазин-1-карбонил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-(4-(трет-бутоксикарбонил)пиперазин-1-карбонил)-3-метилфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
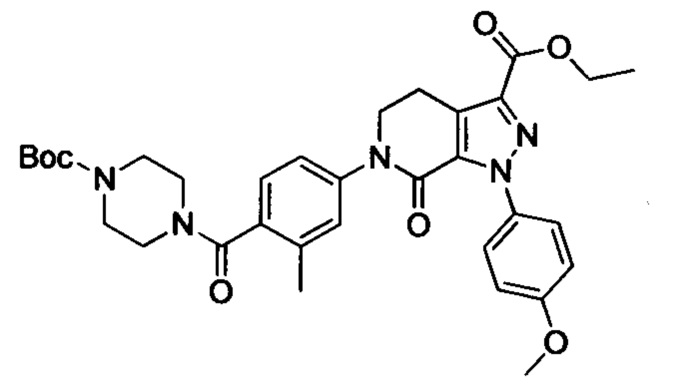
трет-Бутил 4-(4-бром-2-метилбензоил)пиперазин-1-карбоксилат (1.9 г, 5.0 ммолей), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.57 г, 5.0 ммолей), карбонат калия (1.4 г, 10.0 ммолей), иодид меди (0.2 г, 1.0 ммоль) и N,N'-диметил-1,2-этандиамин (0.1 г, 1.1 ммоля) последовательно прибавляли к DMSO (15 мл) при комнатной температуре. Смесь доводили до 120°С и перемешивали при этой температуре в течение 14 ч в атмосфере азота. Реакционную смесь выливали в воду (20 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали с помощью преп.-HPLC, получали 500 мг титульного соединения в виде твердого вещества белого цвета, выход 16.2%.
Стадия 2: трет-Бутил 4-(4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-метилбензоил)пиперазин-1-карбоксилат
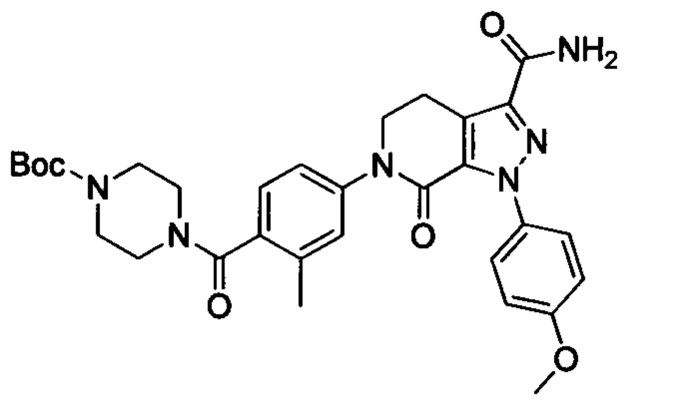
Этил 6-(4-(4-(трет-бутоксикарбонил)пиперазин-1-карбонил)-3-метилфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.5 г, 0.81 ммоля) помещали в герметизируемую пробирку, а затем добавляли NH3/EG (3 мл). После растворения содержимое герметизированной пробирки нагревали до 100°С и проводили реакцию при этой температуре в течение 14 ч. По завершении реакции реакционную смесь выливали в холодную воду (50 мл). Выпадал осадок. Смесь фильтровали с отсасыванием. Осадок на фильтре сушили, получали 300 мг титульного соединения в виде твердого вещества светло-желтого цвета, выход 63.0%.
Стадия 3: 1-(4-Метоксифенил)-6-(3-метил-4-(пиперазин-1-карбонил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
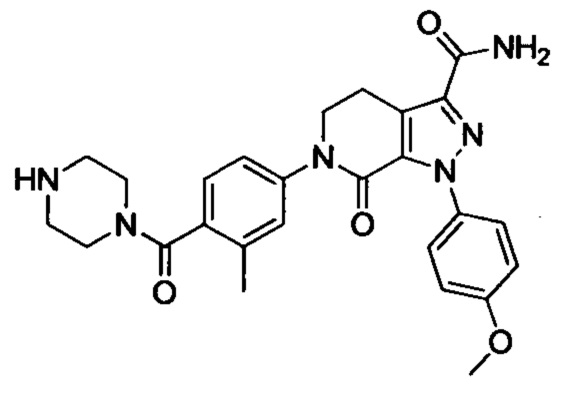
трет-Бутил 4-(4-(3-карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)-2-метилбензоил)пиперазин-1-карбоксилат (300 мг, 0.51 ммоля) растворяли в МеОН (10 мл). В эту реакционную смесь вводили HCl газ до завершения реакции. Смесь упаривали в вакууме. Остаток растворяли в воде и очищали преп.-HPLC, получали 110 мг титульного соединения в виде твердого вещества белого цвета, выход 44.2%.
1H-NMR (400 MHz, DMSO d6) δ: 7.72 (s, 1H), 7.49 (d, 2H), 7.44 (s, 1H), 7.27-7.16 (m, 3H), 7.00 (d, 2H), 4.05 (t, 2H), 3.80 (s, 3H), 3.60-3.52 (m, 2H), 3.20 (t, 2H), 3.07-3.00 (m, 2H), 2.78-2.68 (m, 2H), 2.63-2.53 (m, 2H), 2.20 (s, 3H).
MS: 489.3[M+H]+
Пример 12: 1-(4-Метоксифенил)-7-оксо-6-(3-((2-феноксиэтил)амино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
Стадия 1: 1-(4-Метоксифенил)-7-оксо-6-(3-((2-феноксиэтил)амино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоновая кислота
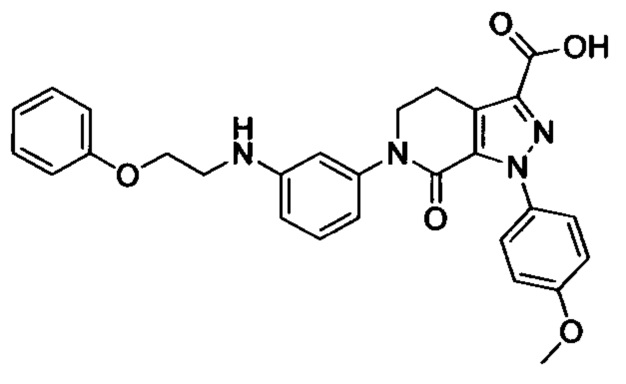
3-Бром-N-(2-феноксиэтил)анилин (2.0 г, 6.8 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (2.1 г, 6.8 ммоля), фосфат калия (5.0 г, 23.5 ммоля), иодид меди (260 мг, 1.36 ммоля) и N,N'-диметил-1,2-этандиамин (250 мг, 2.8 ммоля) растворяли в DMSO (30 мл) при комнатной температуре. После продувания азотом смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (250 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получая 3.1 г титульного соединения в виде коричневого масла, которые использовали сразу же на следующей стадии.
Стадия 2: Этил 1-(4-метоксифенил)-7-оксо-6-(3-((2-феноксиэтил)амино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
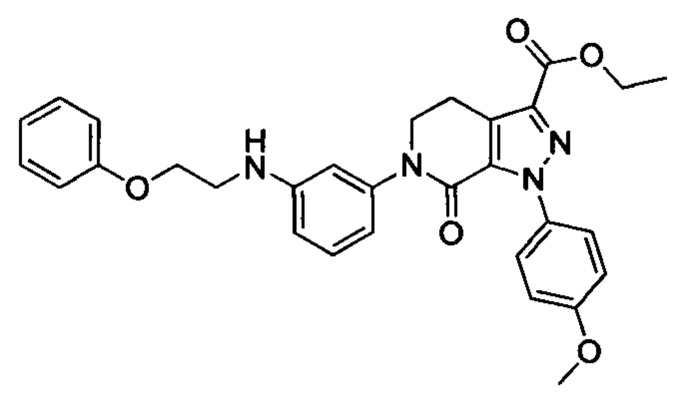
1-(4-Метоксифенил)-7-оксо-6-(3-((2-феноксиэтил)амино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоновую кислоту (2.0 г, 4.0 ммоля) растворяли в EtOH (20 мл) при комнатной температуре. К полученному раствору, помещенному в баню с водой и льдом, по каплям добавляли SOCl2 (1.4 г, 11.8 ммоля). По окончании прибавления смесь нагревали до 40°С и при этой температуре проводили реакцию, перемешивая в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры и упаривали. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH=50:1), получали 300 мг титульного соединения в виде твердого вещества коричневого цвета, выход 14.2%.
Стадия 3: 1-(4-Метоксифенил)-7-оксо-6-(3-((2-феноксиэтил)амино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
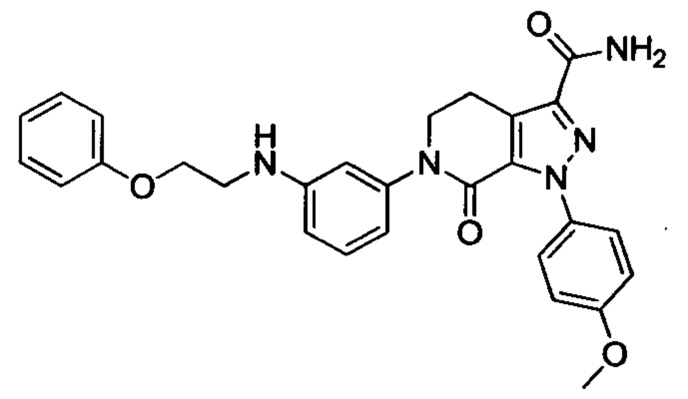
Этил 1-(4-метоксифенил)-7-оксо-6-(3-((2-феноксиэтил)амино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.1 г, 0.19 ммоля) помещали в герметизируемую пробирку, а затем добавляли NH3/EG (4.5 мл). После растворения содержимое пробирки нагревали при 100°С в течение 14 ч. По завершении реакции реакционную смесь выливали в холодную воду (10 мл). Выпадал твердый осадок. Смесь фильтровали с отсасыванием. Осадок на фильтре сушили, получали 40 мг титульного соединения в виде твердого вещества телесного цвета, выход 42.4%.
1H-NMR (400 MHz, DMSO-d6) δ: 7.70 (s, 1H), 7.50 (d, 1H), 7.42 (s, 1H), 7.31-7.23 (m, 2H), 7.08 (t, 1H), 7.02-6.89 (m, 5H), 6.61 (s, 1H), 6.56-6.47 (m, 2H), 5.88 (t, 1H), 4.09 (t, 2H), 3.98 (t, 2H), 3.79 (s, 3H), 3.40 (q, 2H),3.17(t,2H)
MS: 498.2 [M+H]+
Пример 13: 1-(4-Метоксифенил)-6-(3-(((1-метил-1Н-пиразол-5-ил)метил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: 1-(4-метоксифенил)-6-(3-(((1-метил-1Н-пиразол-5-ил)метил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоновая кислота
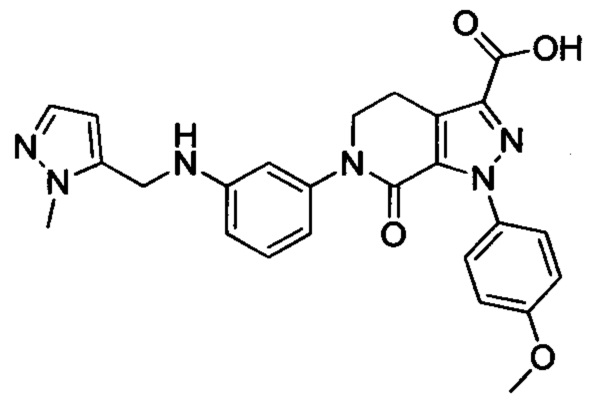
3-Бром-N-((1-метил-1Н-пиразол-5-ил)метил)анилин (2.0 г, 7.5 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (2.4 г, 7.5 ммоля), фосфат калия (4.0 г, 18.8 ммоля), иодид меди (285 мг, 1.5 ммоля) и N,N'-диметил-1,2-этандиамин (265 мг, 3 ммоля) последовательно добавляли к DMSO (30 мл) при комнатной температуре. После продувания азотом смесь нагревали до 120°С и при этой температуре перемешивали в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (250 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получая 2.9 г титульного соединения в виде коричневого масла, которые использовали сразу же на следующей стадии.
Стадия 6: Этил 1-(4-метоксифенил)-6-(3-(((1-метил-1Н-пиразол-5-ил)метил)амино) фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
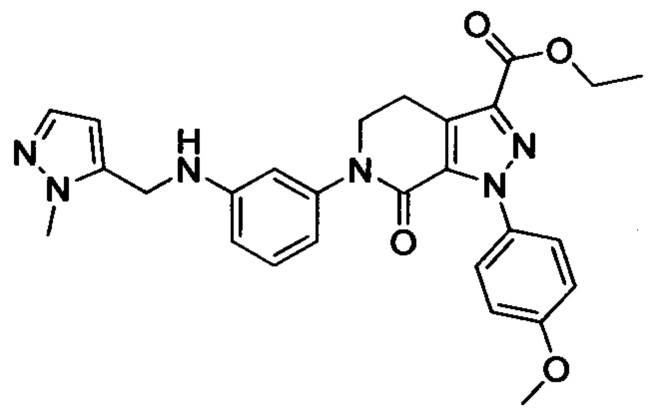
1-(4-Метоксифенил)-6-(3-(((1-метил-1Н-пиразол-5-ил)метил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоновую кислоту (2.9 г, 6.1 ммоля) растворяли в МеОН (40 мл) при комнатной температуре. К полученной смеси, помещенной в баню лед плюс вода, по каплям прибавляли SOCl2 (2.2 г, 18.5 ммоля). По завершении реакции смесь нагревали до 40°С и перемешивали при этой температуре в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры и упаривали растворитель, получали сырой продукт. Этот сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=100:1), получали 500 мг титульного соединения в виде твердого вещества белого цвета, выход 16.4%.
Стадия 3: 1-(4-Метоксифенил)-6-(3-(((1-метил-1Н-пиразол-5-ил)метил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
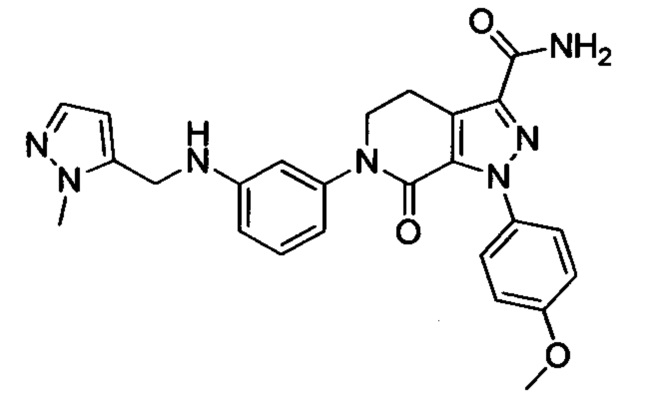
В герметизируемой пробирке этил 1-(4-метоксифенил)-6-(3-(((1-метил-1Н-пиразол-5-ил)метил)амино)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (500 мг, 1.0 ммоль) растворяли в NH3/EG (20 мл) при комнатной температуре. Содержимое герметизированной пробирки нагревали до 100°С и перемешивали при этой температуре в течение 14 ч. К этой смеси прибавляли воду (100 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали, получали сырой продукт. Сырой продукт очищали преп.-HPLC, получали 120 мг титульного соединения в виде твердого вещества светло-коричневого цвета, выход 25.4%.
1H-NMR (400 MHz, DMSO-d6) δ: 7.70 (s, 1Н), 7.48 (dd, 2H,), 7.42 (s, 1H), 7.28 (d, 1H), 7.07 (t, 1H), 6.99 (dd, 2H), 6.64 (t, 1H), 6.54-6.51 (m, 2H), 6.19 (d, 1H), 6.14 (t, 1H), 4.27 (d, 2H), 3.97 (t, 2H), 3.79 (d, 6H), 3.17 (t,2H).
MS 472 [M+H]+.
Пример 14: 6-(3-(((3 (1Н-Имидазол-1-ил)пропил)амино)метил)фенил)-1-(4-метокси)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(3-(((3-(1Н-имидазол-1-ил)пропил)амино)метил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
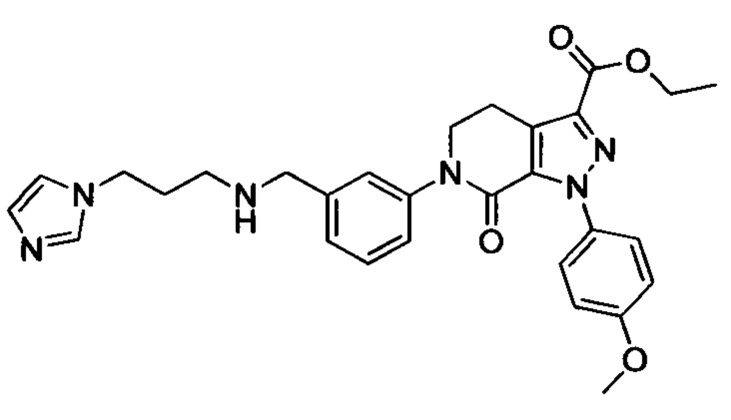
N-(3-Бромбензил)-3-(1Н-имидазол-1-ил)пропан-1-амин (706 мг, 2.4 ммоля) и этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (500 мг, 1.6 ммоля) прибавляли к DMSO (5 мл) при комнатной температуре. Затем к полученной смеси последовательно прибавляли при комнатной температуре иодид калия (797 мг, 4.8 ммоля), карбонат калия (883 мг, 6.4 ммоля), иодид меди (122 мг, 0.64 ммоля) и N,N'-диметил-1,2- этандиамин (70.4 мг, 0.8 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 14 час в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, а затем выливали в воду (50 мл). Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=10:1), получали 480 мг титульного соединения в виде твердого вещества желтого цвета, выход 56.9%.
Стадия 2: 6-(3-(((3-(1Н-Имидазол-1-ил)пропил)амино)метил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
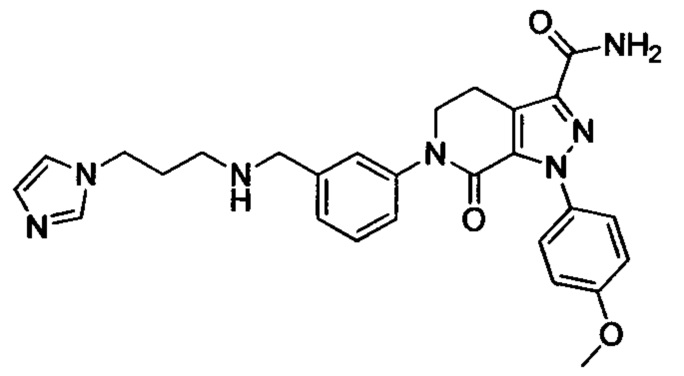
Этил 6-(3-(((3-(1Н-имидазол-1-ил)пропил)амино)метил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (480 мг, 0.9 ммоля) поместили в герметизируемую пробирку, а затем добавляли NH3/EG (4 мл). После герметизации пробирки полученную смесь нагревали при 100°С в течение 14 ч. Полученную смесь выливали в воду (100 мл). Экстрагировали с помощью DCM. Органические вытяжки упаривали, получали сырой продукт. Сырой продукт очищали препаративной HPLC, получали ПО мг титульного соединения в виде желтого вязкого воскообразного вещества, выход 24.2%.
1H-NMR (400 MHz, DMSO d6) δ: 7.71 (s, 1H), 7.56 (s, 1H), 7.52-7.46 (m, 2H), 7.43 (s, 1H), 7.35-7.29 (m, 2H), 7.24-7.17 (m, 2H), 7.11 (s, 1H), 7.01-6.96 (m, 2H), 6.85 (s, 1H), 4.07-3.96 (m, 4H), 3.79 (s, 3H), 3.74-3.67 (m, 2H), 3.20 (t, 2H), 2.48-2.41 (m, 2H), 1.90-1.80 (m, 2H).
MS 500 [M+H]+
Пример 15: 1-(4-Метоксифенил)-7-оксо-6-(4-(пиперидин-1-ил)бутил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-ацетоксибутил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксилат
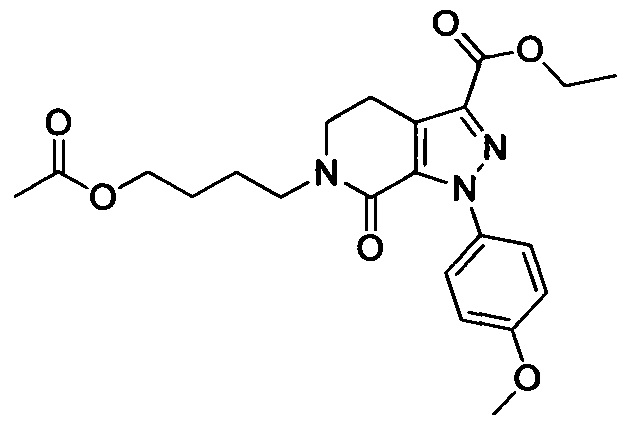
Этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.0 г, 3.2 ммоля) растворяли в DMF (40 мл) при комнатной температуре. К полученной смеси последовательно прибавляли KTB (трет-бутоксид калия) (0.7 г, 6.2 ммоля) и 4-бромбутил ацетат (0.7 г, 3.6 ммоля). Реакцию проводили при комнатной температуре в течение 16 ч. К полученной смеси добавляли воду (100 мл), чтобы остановить реакцию. Полученную смесь экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:MeOH=200:1), получали 470 мг титульного соединения в виде коричнево-желтого масла, выход 34.2%.
Стадия 2: 4-(3-Карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)бутил ацетат
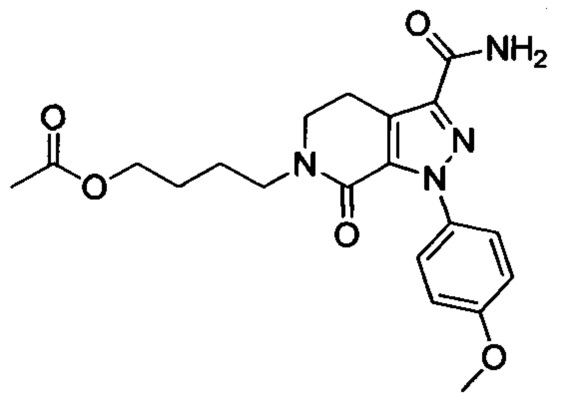
Этил 6-(4-ацетоксибутил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (470 мг, 1.1 ммоля) поместили в герметизируемую пробирку, а затем добавляли NH3/EG (4 мл). После герметизации пробирки полученную смесь нагревали при 100°С в течение 17 ч. Реакционную смесь выливали в воду (100 мл). Полученную смесь экстрагировали с помощью DCM. Органические вытяжки упаривали, получали сырой продукт. Сырой продукт очищали препаративной HPLC, получали 370 мг титульного соединения в виде желтого масла, которое использовали сразу же на следующей стадии.
Стадия 3: 4-(3-Карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)бутил метансульфонат
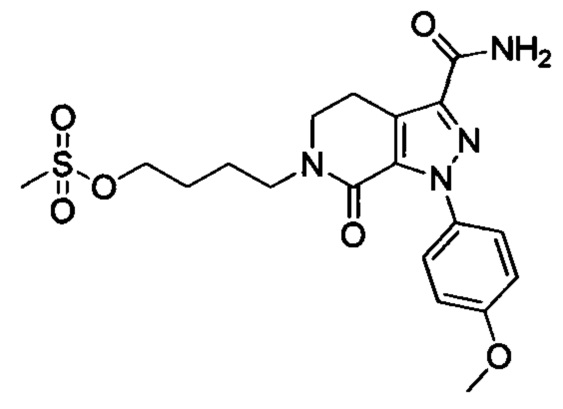
4-(3-Карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)бутил ацетат (370 мг, 0.93 ммоля) растворяли в DCM (5 мл). К полученной смеси прибавляли TEA (208 мг, 2.06 ммоля), а затем по каплям, при охлаждении в бане со льдом, прибавляли MsCl (142 мг, 1.23 ммоля). Реакцию проводили в течение 14 ч при комнатной температуре. Реакционную смесь выливали в воду (100 мл). Полученную смесь экстрагировали DCM. Органические вытяжки упаривали в вакууме, получали 400 мг титульного соединения в виде желтого масла, которое использовали сразу же на следующей стадии.
Стадия 4: 1-(4-Метоксифенил)-7-оксо-6-(4-(пиперидин-1-ил)бутил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
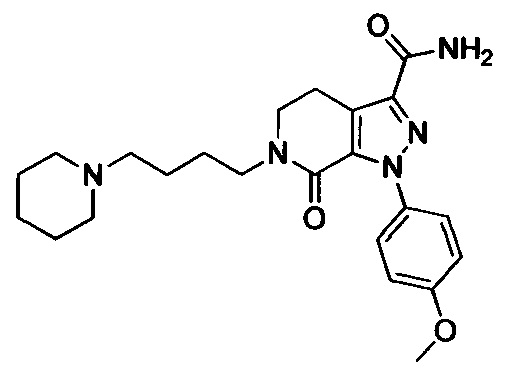
4-(3-Карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)бутил метансульфонат (400 мг, 0.92 ммоля) и пиперидин (140 мг, 1.65 ммоля) растворяли в 1,4-диоксане (5 мл). К этой смеси добавляли карбонат калия (303 мг, 2.2 ммоля). Смесь нагревали до 80°С и проводили реакцию при этой температуре в течение 14 ч. Реакционную смесь выливали в воду (100 мл). Экстрагировали с помощью ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали с помощью преп.-HPLC, получали 170 мг титульного соединения в виде коричневого вязкого воскообразного вещества, выход 43.6%.
1H-NMR (400 MHz, DMSO d6) δ: 7.64 (s, 1H), 7.49-7.41 (m, 2H), 7.38 (s, 1H), 7.04-6.97 (m, 2H), 3.81 (s, 3H), 3.61 (t, 2H), 3.42-3.34 (m, 2H), 3.02 (t, 2H), 2.35-2.12 (m, 6H), 1.56-1.40 (m, 6H), 1.40-1.27 (m, 4H).
MS: 425.9 [M+H]+
Пример 16: 6-(4-Изобутирамидофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-изобутирамидофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
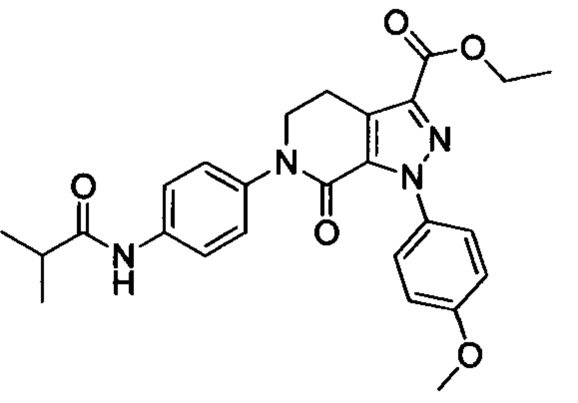
Этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (4.0 г, 12.7 ммоля) и N-(4-иодфенил)изобутирамид (3.8 г, 13.1 ммоля) прибавляли к DMSO (60 мл) при комнатной температуре. К этой смеси в атмосфере азота прибавляли последовательно карбонат калия (3.7 г, 26.7 ммоля), иодид меди (1.1 г, 5.77 ммоля) и 1,10-фенантролин (1.1 г, 6.10 ммоля). Смесь нагревали до 120°С и при этой температуре проводили реакцию в течение 12 ч. По завершении реакции к реакционной смеси прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:3), получали 4.0 г соединения в виде твердого вещества белого цвета, выход 66.2%.
Стадия 2: 6-(4-Изобутирамидофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
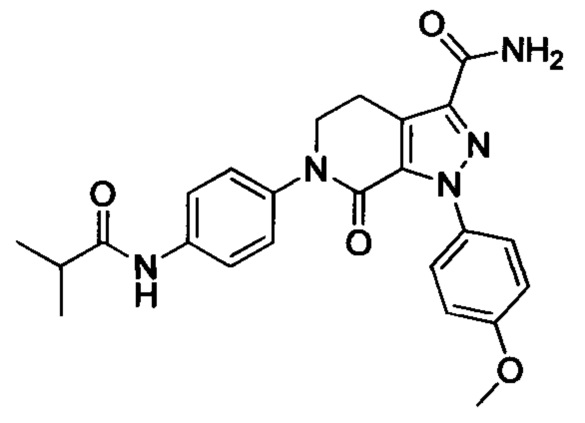
Этил 6-(4-изобутирамидофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (2.0 г, 4.2 ммоля) помещали в герметизируемую пробирку. Добавляли этиленгликоль (16 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 130°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры. Выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 1.2 г титульного соединения в виде твердого вещества белого цвета, выход 63.9%.
1H-NMR (600MHz, DMSO) δ 1.095 (d, 6H), 2.555-2.601 (m, 1H), 3.178-3.200 (m, 2H), 3.802 (s, 3H), 3.991-4.013 (m, 2H), 6.996 (d, 2H), 7.260 (d, 2H), 7.428 (s, 1H), 7.491 (d, 2H), 7.596 (d, 2H), 7.703 (s, 1H), 9.860 (s, 1H)
MS 448.0 [M+H]+
Пример 17: 1-(4-Метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 1-(4-метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
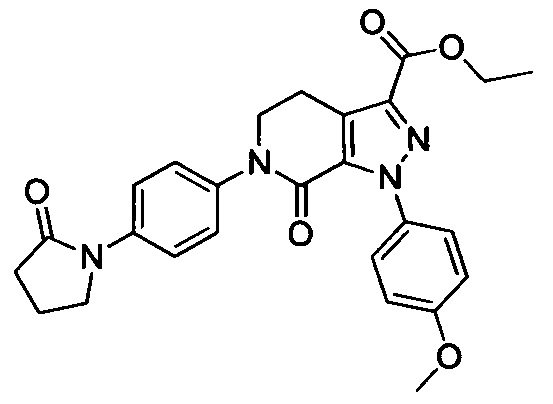
Этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (4.0 г, 12.7 ммоля) и 1-(4-иодфенил)пирролидин-2-он (3.8 г, 13.3 ммоля) добавляли к DMSO (60 мл) при комнатной температуре. К этой смеси последовательно прибавляли карбонат калия (3.7 г, 26.7 ммоля), иодид меди (1.1 г, 5.77 ммоля) и 1,10-фенантролин (1.1 г, 6.10 ммоля) в атмосфере азота. Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции к смеси добавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=от 1:1 до 1:5), получали 4.0 г титульного соединения в виде твердого вещества белого цвета 66.6%.
Стадия 2: 1-(4-Метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
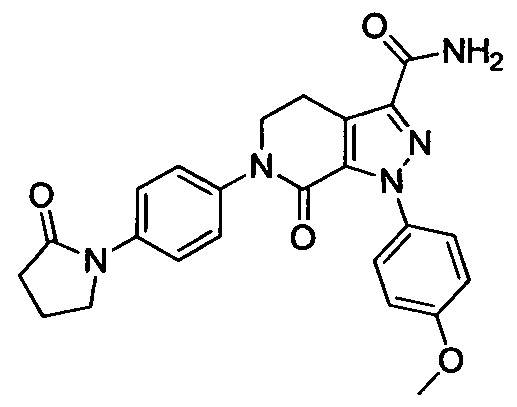
Этил 1-(4-метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.5 г, 3.2 ммоля) помещали в герметизируемую пробирку. Добавляли этиленгликоль (16 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 130°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 1.0 г титульного соединения в виде твердого вещества белого цвета, выход 71.4%.
1H NMR (600MHz, DMSO) δ 2.029-2.080 (m, 2H), 2.477-2.504 (m, 2H), 3.187-3.209 (m, 2H), 3.798 (s, 3H), 3.809-3.833 (m, 2H), 4.011-4.033 (m, 2H), 6.995 (d, 2H), 7.346 (d, 2H), 7.438 (s, 1H), 7.499 (d, 2H), 7.650 (d, 2H), 7.712 (s, 1H)
MS 446.2 [M+H]+
Пример 18: 1-(3-Хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(3-хлор-4-метоксифенил)гидразоно)ацетат
3-Хлор-4-метоксианилин (10 г, 63.5 ммоля) диспергировали в воде (500 мл) при комнатной температуре. Смесь охлаждали до температуры от -5 до 0°С. К этой смеси последовательно прибавляли концентрированную соляную кислоту (20 мл), водный раствор нитрита натрия (30 мл), раствор этил 2-хлор-3-оксобутаноата (10.8 г, 65.6 ммоля) в этаноле (100 мл) и раствор ацетата натрия (15.6 г, 0.19 ммоля) в воде (150 мл). После прибавления смесь нагревали до комнатной температуры и при этой температуре проводили реакцию при перемешивании в течение 6 ч. По завершении реакции смесь фильтровали. Осадок на фильтре сушили в вакууме, получали 15.0 г титульного соединения, выход 81.1%.
Стадия 2: Этил 1-(3-хлор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксилат
Этил 2-хлор-2-(2-(3-хлор-4-метоксифенил)гидразоно)ацетат (10.0 г, 34.3 ммоля), 3-хлор-5,6-дигидропиридин-2(1Н)-он (6.0 г, сырой продукт) и триэтиламин (20 мл) последовательно прибавляли к толуолу (100 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 3 ч. Смесь охлаждали до комнатной температуре и упаривали в вакууме. К полученному остатку добавляли ЕА и воду и оставляли стоять. При стоянии смесь разделялась на две фазы. Водный слой экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Этот сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.5 г титульного соединения, выход 12.5%.
Стадия 3: Этил 1-(3-хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксилат
1-(4-Иодфенил)пиперидин-2-он (1.22 г, 4.0 ммоля), этил 1-(3-хлор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.3 г, 3.72 ммоля) и карбонат калия (1.08 г, 7.81 ммоля)последовательно прибавляли к DMSO (26 мл) при комнатной температуре. К этой смеси прибавляли иодид меди (330 мг, 1.74 ммоля) и 1,10-фенантролин (310 мг, 1.74 ммоля) в атмосфере азота. Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции к смеси прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.5 г титульного соединения в виде твердого вещества светло-желтого цвета, выход 77.2%.
Стадия 4: 1-(3-Хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
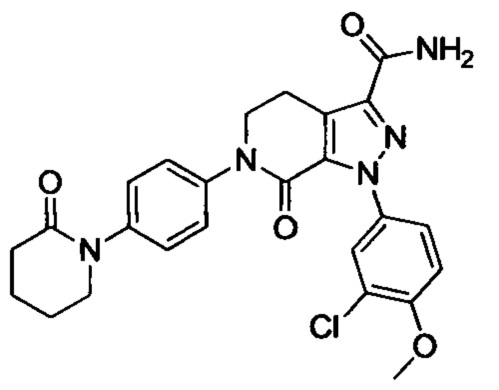
Этил 1-(3-хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.5 г, 2.87 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (16 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 130°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 0.74 г титульного соединения в виде твердого вещества почти белого цвета, выход 52.2%.
1H-NMR(600MHz, DMSO) δ 1.826-1.866 (m, 4Н), 2.376-2.397 (m, 2Н), 3.198-3.220 (m, 2Н), 3.586-3.604 (m, 2Н), 4.053-4.074 (m, 2Н), 7.171 (s, 1H), 7.266-7.294 (m, 4Н), 7.357-7.372 (d, 1Н), 7.505-7.529 (m, 4H) 7.788 (s, 1H)
MS 496.0 [M+H]+
Пример 19: 1-(6-Метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(2-метоксипиридин-5-ил)гидразоно)ацетат
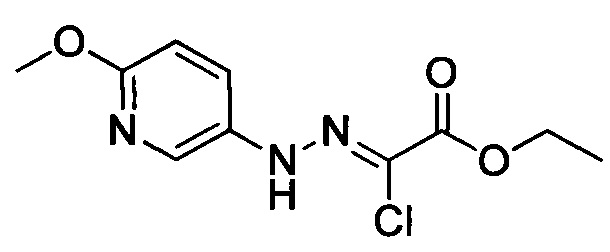
2-Метокси-5-аминопиридин (3.0 г, 24.2 ммоля) диспергировали в воде (15 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К охлажденной смеси последовательно прибавляли концентрированную соляную кислоту (6 мл), водный раствор нитрита натрия (9 мл), раствор этил 2-хлор-3-оксобутаноата (4.08 г, 24.9 ммоля) в этаноле (30 мл), раствор ацетата натрия (5.94 г, 72.3 ммоля) в воде (90 мл). По окончании прибавления смесь нагревали до комнатной температуры и проводили реакцию, перемешивая при этой температуре в течение 6 ч. По завершении реакции полученную смесь фильтровали с отсасыванием. Осадок на фильтре сушили в вакууме и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=10:1), получали 4.3 г титульного соединения, выход 69.0%.
Стадия 2: Этил 1-(6-метоксипиридин-3-ил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
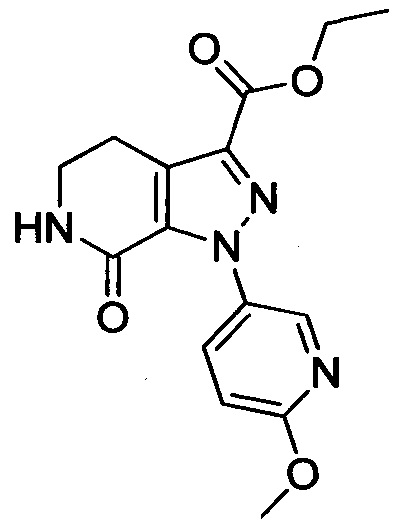
Этил 2-хлор-2-(2-(2-метоксипиридин-5-ил)гидразоно)ацетат (2.58 г, 10 ммолей), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (1.82 г, 10 ммоля) и триэтиламин (3.8 мл) последовательно добавляли к толуолу (26 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 3 ч. Затем смесь охлаждали до комнатной температуры и досуха упаривали в вакууме. К этому остатку прибавляли DCM (30 мл). К смеси при комнатной температуре по каплям прибавляли трифторуксусную кислоту (2.5 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции к полученному раствору прибавляли воду. Полученную смесь экстрагировали с помощью DCM. Органические вытяжки упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.8 г титульного соединения, выход 56.9%.
Стадия 3: Этил 1-(6-метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
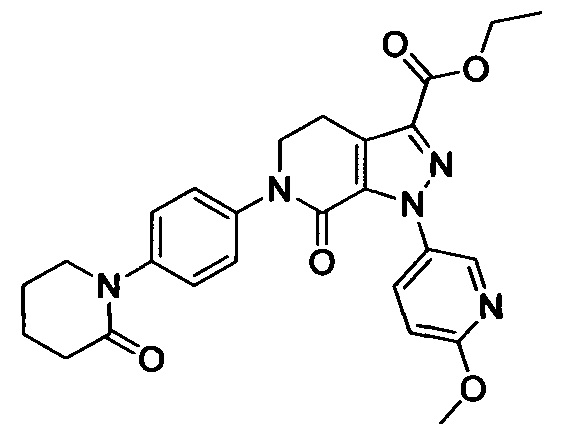
При комнатной температуре 1-(4-иодфенил)пиперидин-2-он (1.9 г, 6.3 ммоля), этил 1-(6-метоксипиридин-3-ил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.8 г, 5.7 ммоля) и карбонат калия (1.7 г, 11.97 ммоля) последовательно добавляли к DMSO (25 мл). В атмосфере азота к этой смеси прибавляли иодид меди (520 мг, 2.7 ммоля) и 1,10-фенантролин (490 мг, 2.7 ммоля). Смесь нагревали до 120°С и при этой температуре проводили реакцию в течение 12 ч. По завершении реакции смесь охлаждали до комнатной температуры. К охлажденной смеси прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.65 г титульного соединения в виде масла, выход 23.3%.
Стадия 4: 1-(6-Метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксипиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
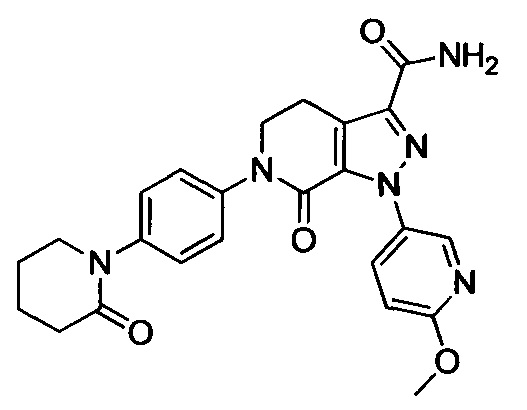
Этил 1-(6-метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.65 г, 1.3 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (16 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 120°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 150 мг титульного соединения в виде порошка почти белого цвета, выход 24.5%.
1H-NMR(600MHz, DMSO)δ 1.829-1.850 (m, 4Н), 2.378-2.386 (m, 2Н), 3.209-3.219 (m, 2Н), 3.585-3.593 (m, 2Н), 3.895-3.904 (m, 2Н), 4.05-4.059 (m, 2Н), 6.917-6.930 (m, 2Н), 7.279-7.351 (m, 4Н), 7.483(s, 1H),7.766(s, 1H), 7.4949-7.958 (s, 1H), 8.405 (s, 1H);
MS 461.2[M+H]+
Пример 20: 1-(3-(Дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(3-(дифторметокси)фенил)гидразоно)ацетат
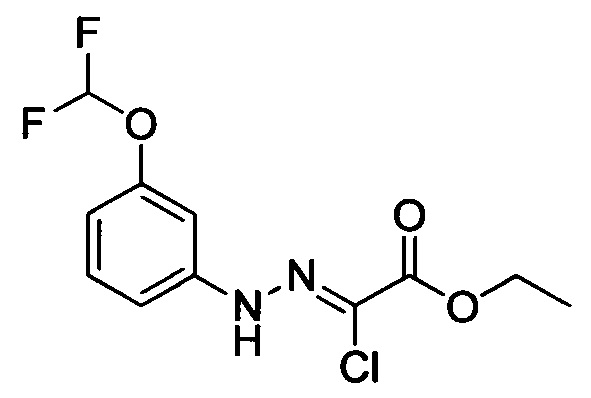
К 3-(дифторметокси)анилину (10.0 г, 62.8 ммоля) добавили воду (50 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К охлажденной смеси добавляли концентрированную соляную кислоту (16.7 мл), водный раствор нитрита натрия (28 мл), раствор этил 2-хлор-3-оксобутаноата (10.6 г, 64.7 ммоля) в этаноле (100 мл) и раствор ацетата натрия (15.5 г, 188.4 ммоля) в воде (120 мл). Полученную смесь нагревали до комнатной температуры и проводили реакцию, перемешивая при этой температуре в течение 6 ч. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме, получали 16.0 г титульного соединения с выходом 87.3%.
Стадия 2: Этил 1-(3-(дифторметокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксилат
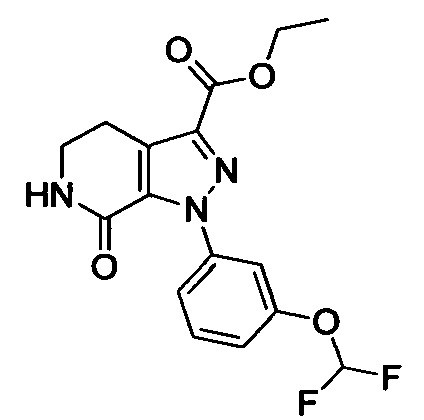
Этил 2-хлор-2-(2-(3-(дифторметокси)фенил)гидразоно)ацетат (2.9 г, 10.0 ммолей), 3- морфолино-5,6-дигидропиридин-2(1Н)-он (1.8 г, 10.0 ммолей) и триэтиламин (2 мл) прибавляли последовательно к толуолу (30 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 3 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме. Остаток добавляли к DCM (25 мл). Полученную смесь охлаждали до 0°С. К охлажденной смеси добавляли трифторуксусную кислоту (3 мл). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (DCM:МеОН=1:1), получали 0.9 г титульного соединения в виде твердого вещества песочного цвета (защитного цвета, цвета хаки), выход 25.7%.
Стадия 3: Этил 1-(3-(дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
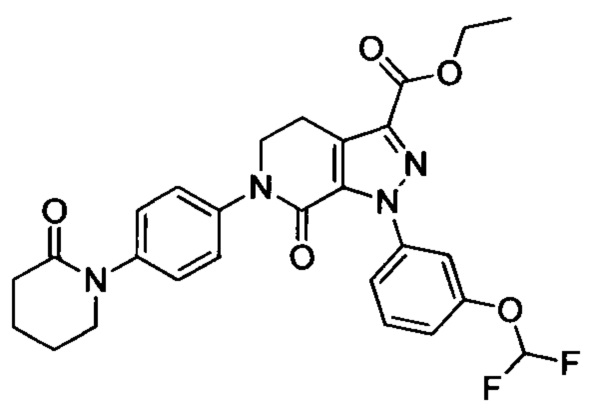
1-(4-Иодфенил)пиперидин-2-он (0.85 г, 2.82 ммоля), этил 1-(3-(дифторметокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.9 г, 2.56 ммоля) и карбонат калия (0.74 г, 5.38 ммоля) последовательно прибавляли к DMSO (20 мл) при комнатной температуре. К полученной смеси прибавляли иодид меди (230 мг, 1.2 ммоля) и 1,10-фенантролин (220 мг, 1.2 ммоля) в атмосфере азота. Эту смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции к смеси добавляли воду. Экстрагировали с помощью ЕА. Органические вытяжки упаривали, получали сырой продукт. Этот сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.74 г титульного соединения в виде твердого вещества белого цвета, выход 55.1%.
Стадия 4: 1-(3-(Дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
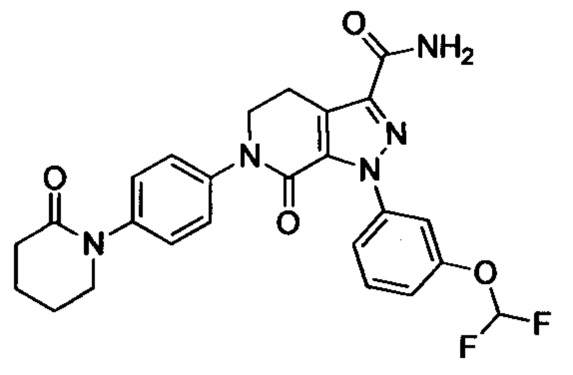
Этил 1-(3-(дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.74 г, 1.41 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (16 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 120°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 450 мг титульного соединения в виде твердого вещества почти белого цвета, выход 64.5%.
1H-NMR (600MHz, DMSO) δ 1.826-1.866 (m, 4H), 2.376-2.397 (m, 2H), 3.198-3.220 (m, 2H), 3.586-3.604 (m, 2H), 4.053-4.074 (m, 2H), 7.171 (s, 1H,), 7.266-7.294 (m, 4H), 7.357-7.372 (d, 1H), 7.505-7.529 (m, 4H) 7.788 (s, 1H)
MS 496.0 [M+H]+
Пример 21: 1-(2-Хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Step 1: Этил 2-хлор-2-(2-(2-хлор-4-метоксифенил)гидразоно)ацетат
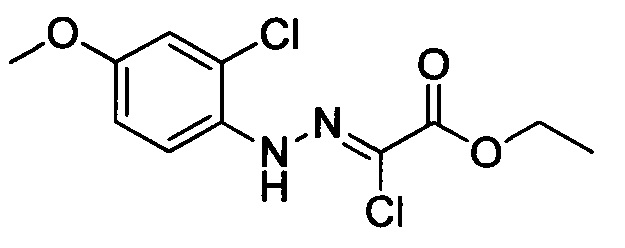
При комнатной температуре 2-хлор-4-метоксианилин (5.0 г, 31.7 ммоля) добавляли в воду (15 мл). Полученную смесь охлаждали до температуры от -5 до 0°С. К смеси последовательно добавляли концентрированную соляную кислоту (6 мл), водный раствор нитрита натрия (9 мл), раствор этил 2-хлор-3-оксобутаноата (5.22 г, 31.7 ммоля) в этаноле (30 мл) и раствор ацетата натрия (7.80 г, 95.1 ммоля) в воде (90 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем смесь нагревали до комнатной температуры и проводили реакцию при перемешивании в течение 6 ч. В процессе реакции наблюдали выпадение осадка. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=10:1), получали 7.9 г титульного соединения, выход 85.5%.
Стадия 2: Этил 1-(2-хлор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
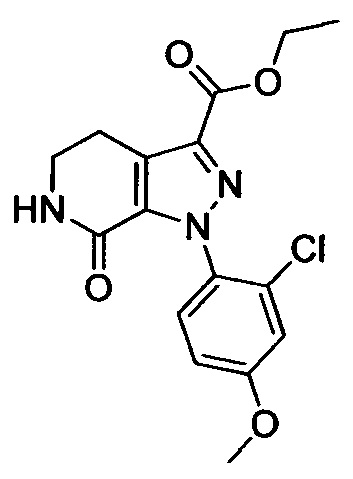
Этил 2-хлор-2-(2-(2-хлор-4-метоксифенил)гидразоно)ацетат (5.82 г, 20 ммолей), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (3.64 г, 20.0 ммолей) и триэтиламин (6.07 г, 60 ммолей) последовательно добавляли к толуолу (26 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (30 мл) и трифторуксусную кислоту (2.5 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции реакционную смесь промывали водой. Водную фазу экстрагировали DCM. Затем органические вытяжки упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.8 г титульного соединения, выход 11.4%.
Стадия 3: Этил 1-(2-хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
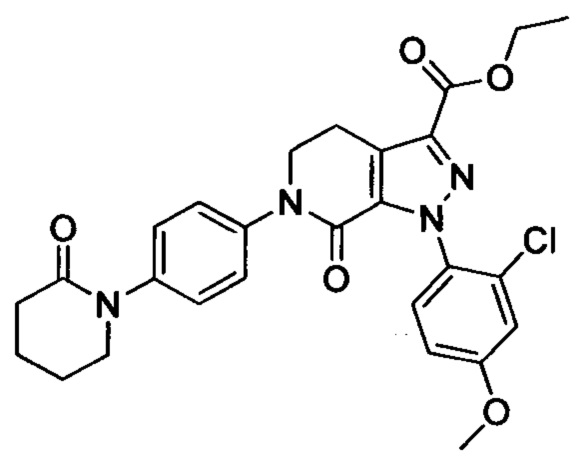
При комнатной температуре к DMSO (25 мл) последовательно прибавляли 1-(4-иодфенил)пиперидин-2-он (0.94 г, 3.1 ммоля), этил 1-(2-хлор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.70 г, 2.0 ммоля) и карбонат калия (0.58 г, 4.2 ммоля). К полученной смеси добавляли иодид меди (180 мг, 0.94 ммоля) и 1,10-фенантролин (170 мг, 0.94 ммоля) в атмосфере азота. Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры. К охлажденной смеси прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.5 г титульного соединения в виде масла, выход 47.8%.
Стадия 4: 1-(2-Хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
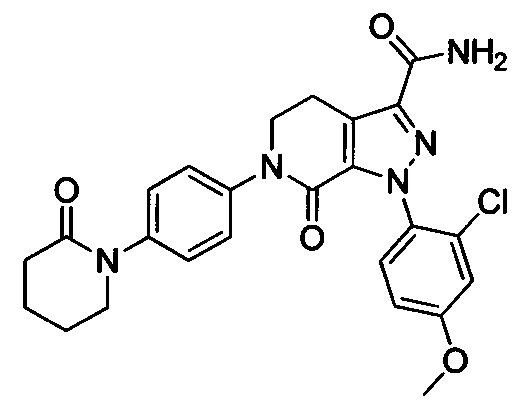
Этил 1-(2-хлор-4-метоксифенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.5 г, 0.95 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (16 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 120°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 420 мг титульного соединения в виде твердого вещества почти белого цвета, выход 89.5%.
1Н NMR (600MHz, DMSO) δ 1.830-1.853 (m, 4Н), 2.378 (m, 2Н), 3.23 (m, 2Н), 3.579 (m, 2Н), 3.834 (s, 3Н), 4.053 (m, 2Н), 7.014-7.018 (m, 1Н), 7.195 (m, 1H), 7.262-7.320 (m, 4H), 7.453(m, 1H), 7.508-7.521(m, 1H), 7.773 (s, 1H)
MS 494.2 [M+H]+
Пример 22: 1-(4-Метокси-2-метилфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(4-метокси-2-метилфенил)гидразоно)ацетат
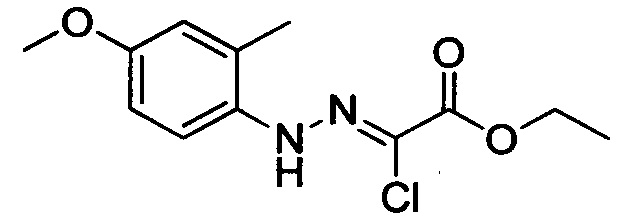
При комнатной температуре 4-метокси-2-метиланилин (5.0 г, 36.4 ммоля) прибавляли в воду (15 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К этой смеси последовательно добавляли концентрированную соляную кислоту (6 мл), водный раствор нитрита натрия (9 мл), раствор этил 2-хлор-3-оксобутаноата (6.0 г, 36.4 ммоля) в этаноле (30 мл) и раствор ацетата натрия (8.96 г, 109.2 ммоля) в воде (90 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. В процессе реакции выпадал осадок. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме. Остаток очищали хроматографией на колонке с силикагелем (РЕ:ЕА=10:1), получали 5.5 г титульного соединения.
Стадия 2: Этил 1-(4-метокси-2-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
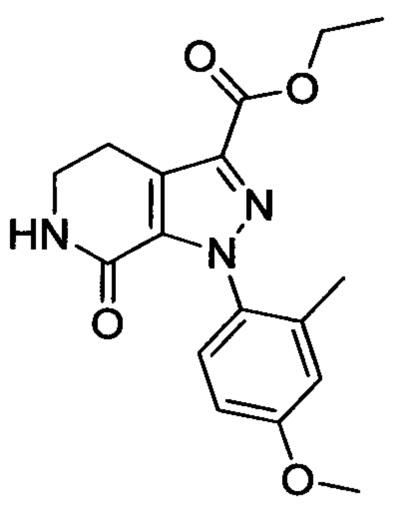
Этил 2-хлор-2-(2-(4-метокси-2-метилфенил)гидразоно)ацетат (5.41 г, 20 ммолей), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (3.64 г, 20.0 ммолей) и триэтиламин (6.07 г, 60 ммолей) последовательно добавляли к толуолу (26 мл) при комнатной температуре, нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (30 мл) и трифторуксусную кислоту (2.5 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции реакционную смесь промывали водой и полученную водную фазу экстрагировали DCM. Затем органические вытяжки упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.3 г титульного соединения, выход 19.7%.
Стадия 3: Этил 1-(4-метокси-2-метилфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
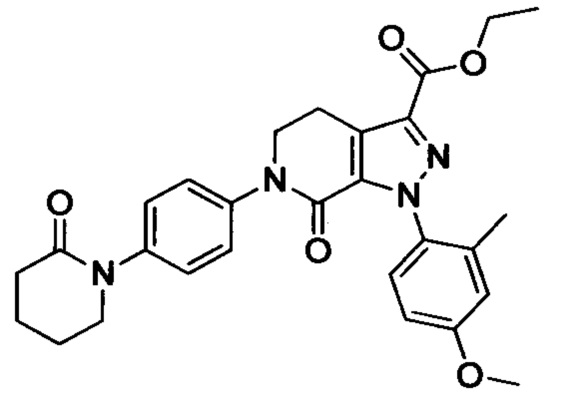
1-(4-Иодфенил)пиперидин-2-он (0.99 г, 3.3 ммоля), этил 1-(4-метокси-2-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.99 г, 3.0 ммоля) и карбонат калия (0.87 г, 6.3 ммоля) последовательно прибавляли к DMSO (25 мл) при комнатной температуре. В атмосфере азота добавляли иодид меди (271 мг, 1.41 ммоля) и 1,10-фенантролин (254 мг, 1.41 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.1 г титульного соединения в виде масла, выход 73.3%.
Стадия 4: 1-(4-Метокси-2-метилфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
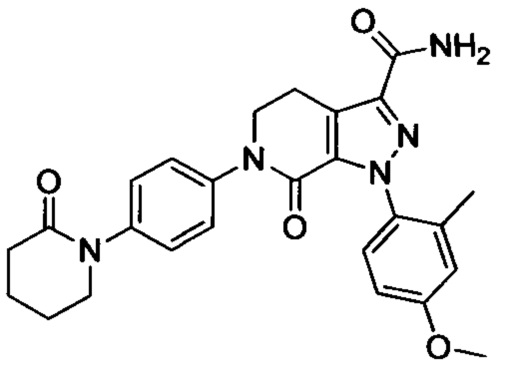
Этил 1-(4-метокси-2-метилфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.0 г, 1.99 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (20 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Реакцию проводили при 120°С в течение 5 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 750 мг титульного соединения в виде твердого вещества почти белого цвета, выход separated 79.4%.
1Н NMR (600MHz, DMSO) δ 1.817-1.855 (m, 4Н), 1.988 (s, 3Н) 2.365-2.385 (m, 2Н), 3.214-3.236 (m, 2Н), 3.564-3.581 (m, 2Н), 3.782 (s, 3Н), 4.037-4.059 (m, 2Н), 6.814-6.828 (m, 1Н), 6.893 (s, 1H), 7.235-7.264 (m, 3H), 7.307-7.321 (m, 2H), 7.408 (s, 1H), 7.729 (s, 1H)
MS 474.2 [M+H]+
Пример 23: 1-(2-Метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(2-метоксипиридин-3-ил)гидразоно)ацетат
2-Метоксипиридин-3-амин (5.0 г, 40.3 ммоля) прибавляли в воду (15 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К смеси последовательно прибавляли концентрированную соляную кислоту (6 мл), водный раствор нитрита натрия (9 мл), раствор этил 2-хлор-3-оксобутаноата (6.63 г, 40.3 ммоля) в этаноле (30 мл) и раствор ацетата натрия (9.91 г, 120.9 ммоля) в воде (90 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. В процессе реакции выпадал осадок. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме. Остаток очищали хроматографией на колонке с силикагелем (РЕ:ЕА=10:1), получали 6.3 г титульного соединения, выход 60.5%.
Стадия 2: Этил 1-(2-метоксипиридин-3-ил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
Этил 2-хлор-2-(2-(2-метоксипиридин-3-ил)гидразоно)ацетат (5.15 г, 20 ммолей), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (3.64 г, 20 ммолей) и триэтиламин (6.07 г, 60 ммолей) последовательно прибавляли к толуолу (26 мл) при комнатной температуре. Эту смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (30 мл) и трифторуксусную кислоту (2.5 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции реакционную смесь промывали водой и полученную водную фазу экстрагировали DCM. Затем органические вытяжки упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.6 г титульного соединения, выход 9.5%.
Стадия 3: Этил 1-(2-метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
К DMSO (25 мл) последовательно прибавляли 1-(4-иодфенил)пиперидин-2-он (0.60 г, 2.0 ммоля), этил 1-(2-метоксипиридин-3-ил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.60 г, 1.9 ммоля) и карбонат калия (0.55 г, 4.0 ммоля) при комнатной температуре. К полученной смеси прибавляли иодид меди (171 мг, 0.89 ммоля) и 1,10-фенантролин (160 мг, 0.89 ммоля) в атмосфере азота. Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.6 г титульного соединения в виде масла, выход 64.2%.
Стадия 4: 1-(2-Метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
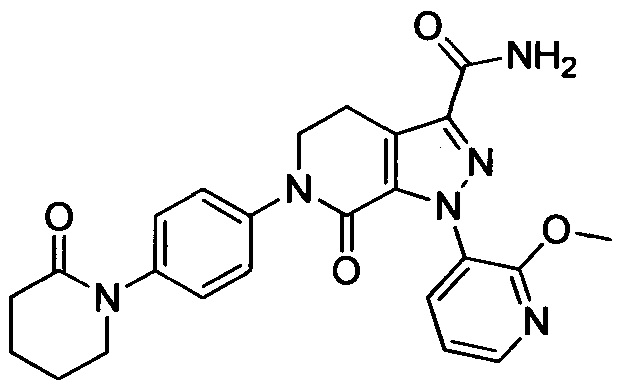
Этил 1-(2-метоксипиридин-3-ил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.6 г, 1.22 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок.
Смесь фильтровали. Осадок на фильтре сушили, получали 340 мг титульного соединения в виде твердого вещества почти белого цвета, выход 60.5%.
1H-NMR (600MHz, DMSO)δ: 1.818-1.856 (m, 4Н), 2.375-2.380 (m, 2Н), 3.219-3.222 (m, 2Н), 3.575-3.581 (m, 2Н), 3.828 (s, 3Н), 4.052 (s, 2Н), 7.128-7.148 (m, 1Н), 7.261-7.276 (d, 2H), 7.314-7.328 (d, 2H), 7.454 (s, 1H), 7.756 (s, 1H), 7.874-7.886 (m, 1H), 8.279-8.285 (d, 1H)
MS 461.2 [M+H]+
Пример 24: 1-(2-(Дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(2-(дифторметокси)фенил)гидразоно)ацетат
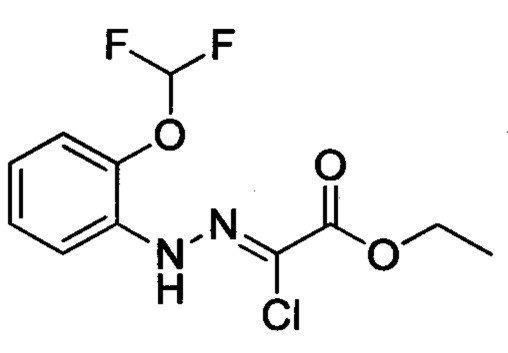
2-(Дифторметокси)анилин (24.0 г, 150 ммоля) прибавляли к воде (120 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К смеси последовательно прибавляли концентрированную соляную кислоту (40 мл), водный раствор нитрита натрия (66 мл), раствор этил 2-хлор-3-оксобутаноата (25.2 г, 150 ммолей) в этаноле (120 мл) и раствор ацетата натрия (36.9 г, 450 ммоля) в воде (360 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. В процессе реакции выпадал осадок. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме. Остаток очищали хроматографией на колонке с силикагелем (РЕ:ЕА=10:1), получали 35.0 г титульного соединения, выход 79.7%.
Стадия 2: Этил 1-(2-(дифторметокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксилат
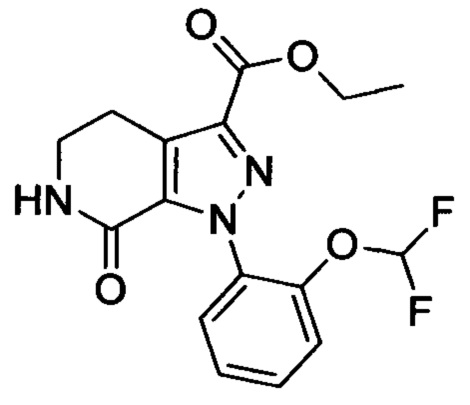
Этил 2-хлор-2-(2-(2-(дифторметокси)фенил)гидразоно)ацетат (5.0 г, 17.1 ммоля), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (3.12 г, 17.1 ммоля) и триэтиламин (5.19 г, 51.3 ммоля) последовательно добавляли к толуолу (60 мл) при комнатной температуре. Эту смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку медленно добавляли DCM (50 мл) и трифторуксусную кислоту (5.0 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции реакционную смесь промывали водой. Водную фазу экстрагировали DCM. Органические вытяжки упаривали в вакууме досуха. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.9 г титульного соединения, выход 15.0%.
Стадия 3: Этил 1-(2-(дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
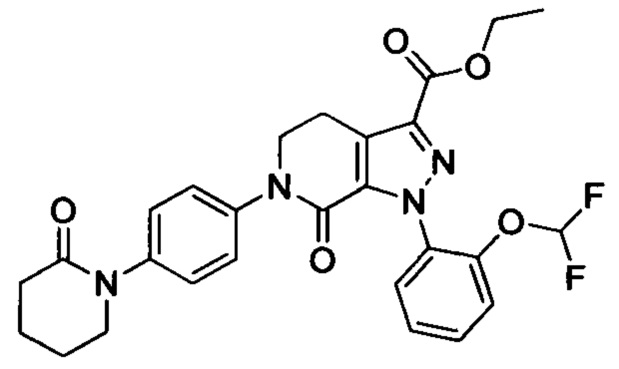
При комнатной температуре к DMSO (20 мл) последовательно добавляли 1-(4-иодфенил)пиперидин-2-он (0.85 г, 2.82 ммоля), этил 1-(2-(дифторметокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.90 г, 2.6 ммоля) и карбонат калия (0.75 г, 5.5 ммоля). В атмосфере азоте к смеси прибавляли иодид меди (235 мг, 1.2 ммоля) и 1,10-фенантролин (216 мг, 1.2 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.9 г титульного соединения в виде масла, выход 66.2%.
Стадия 4: 1-(2-(Дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
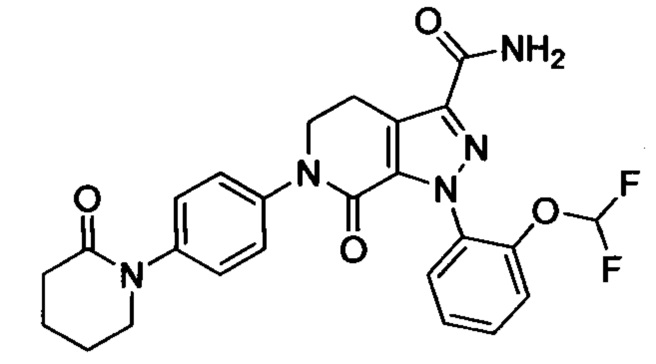
Этил 1-(2-(дифторметокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.9 г, 1.72 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 420 мг титульного соединения в виде твердого вещества песочного цвета, выход 49.3%.
1H-NMR(600MHz, DMSO)δ: 1.819-1.857 (m, 4Н), 2.376-2.383 (m, 2Н), 3.225-3.231 (m, 2Н), 3.576-3.562 (m, 2Н), 4.038-4.044 (m, 2Н), 7.088 (s, 1Н), 7.210-7.317 (m, 5H), 7.371-7.422 (m, 1H), 7.481 (s, 1H), 7.567-7.620 (m, 2H), 7.758 (s, 1H)
MS 496.1 [M+H]+
Пример 25: 1-(4-(2-Амино-2-оксоэтокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(4-(2-этокси-2-оксоэтокси)фенил)гидразоно)ацетат
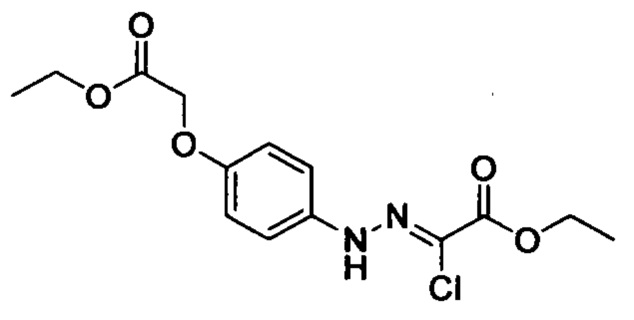
При комнатной температуре этил 2-(4-аминофенокси)ацетат (5.0 г, 25.6 ммоля) прибавляли к воде (28 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К этой смеси последовательно прибавляли концентрированную соляную кислоту (11 мл), водный раствор нитрита натрия (18 мл), раствор этил 2-хлор-3-оксобутаноата (4.5 г, 27.3 ммоля) в этаноле (47 мл) и раствор ацетата натрия (6.3 г, 76.8 ммоля) в воде (28 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. В процессе реакции выпадал осадок. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме и очищали хроматографией на колонке с силикагелем (РЕ:ЕА=10:1), получали 4.2 г титульного соединения, выход 50.0%.
Стадия 2: Этил 1-(4-(2-этокси-2-оксоэтокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
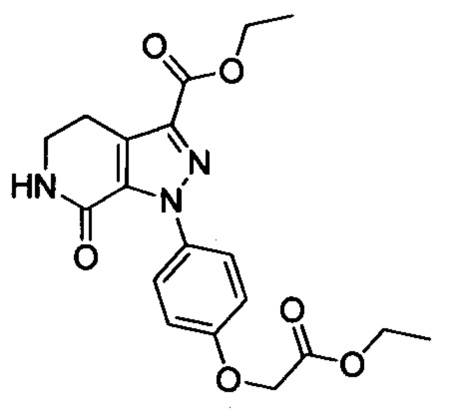
Этил 2-хлор-2-(2-(4-(2-этокси-2-оксоэтокси)фенил)гидразоно)ацетат (4.23 г, 12.9 ммоля), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (2.57 г, 14.1 ммоля) и триэтиламин (3.91 г, 38.7 ммоля) последовательно прибавляли к толуолу (25 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (50 мл) и трифторуксусную кислоту (5.0 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции реакционную смесь промывали водой. Водную фазу экстрагировали DCM. Органические вытяжки упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.32 г титульного соединения, выход 26.4%.
Стадия 3: Этил 1-(4-(2-этокси-2-оксоэтокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
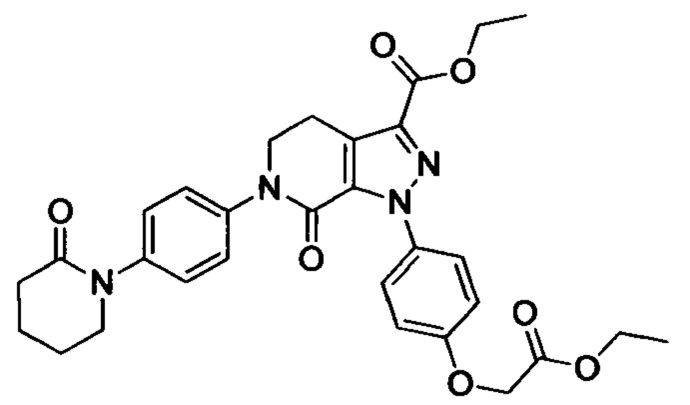
При комнатной температуре к DMSO (25 мл) последовательно прибавляли 1-(4-иодфенил)пиперидин-2-он (1.11 г, 3.7 ммоля), этил 1-(4-(2-этокси-2-оксоэтокси)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.32 г, 3.4 ммоля) и карбонат калия (0.98 г, 7.1 ммоля). К полученной смеси в атмосфере азота прибавляли иодид меди (310 мг, 1.6 ммоля) и 1,10-фенантролин (290 мг, 1.6 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.44 г титульного соединения, выход 23.1%.
Стадия 4: 1-(4-(2-Амино-2-оксоэтокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил) фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
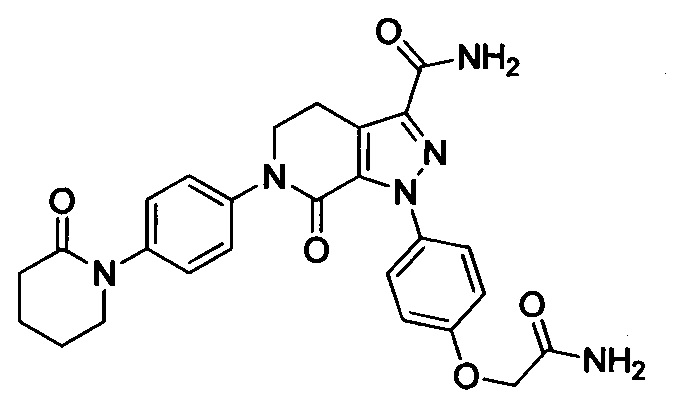
Этил 1-(4-(2-этокси-2-оксоэтокси)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.44 г, 0.78 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 210 мг титульного соединения в виде твердого вещества светло-желтого цвета, выход 53.6%.
1H-NMRC 600MHz, DMSO)δ: 1.825-1.864 (m, 4Н), 2.386-2.389 (m, 2Н), 3. 201 (m, 2Н), 3.582-3.600 (m, 2Н), 4.145 (m, 2Н), 4.952 (s, 2Н), 6.860 (s, 1Н), 7.276-7.288 (d, 2H), 7.358-7.367 (d, 2H), 7.388 (s, 1H), 7.440 (s, 1H), 7.484-7.498 (d, 2H), 7.572-7.583 (d, 2H), 7.750 (s, 1H)
MS 503.1 [M+H]+
Пример 26: 1-(3-Фтор-4-метоксифенил)-7-оксо-6-(4-(3-оксоморфолино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(3-фтор-4-метоксифенил)гидразоно)ацетат
При комнатной температуре 3-фтор-4-метоксианилин (21.0 г, 0.15 моля) добавляли в воду (100 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К полученной смеси последовательно добавляли концентрированную соляную кислоту (45 мл), водный раствор нитрита натрия (70 мл), раствор этил 2-хлор-3-оксобутаноата (24.7 г, 0.15 моля) в этаноле (150 мл) и раствор ацетата натрия (36.9 г, 0.45 моля) в воде (150 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем полученную смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. В процессе реакции выпадал осадок. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме и очищали хроматографией на колонке с силикагелем (РЕ:ЕА=10:1), получали 36.5 г титульного соединения, выход 88.6%.
Стадия 2: Этил 1-(3-фтор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
Этил 2-хлор-2-(2-(3-фтор-4-метоксифенил)гидразоно)ацетат (8.0 г, 29.1 ммоля), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (6.0 г, 31.9 ммоля) и триэтиламин (8.6 г, 85.2 ммоля) последовательно добавляли к толуолу (60 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (100 мл) и трифторуксусную кислоту (6.0 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции реакционную смесь промывали водой. Водную фазу экстрагировали DCM. Органические вытяжки объединяли, сушили Na2SO4 и фильтровали. Фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.32 г титульного соединения, выход 29.6%.
Стадия 3: Этил 1-(3-фтор-4-метоксифенил)-7-оксо-6-(4-(3-оксоморфолино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
При комнатной температуре к DMSO (20 мл) последовательно прибавляли 1-(4-иодфенил)пиперидин-2-он (0.9 г, 3.0 ммоля), этил 1-(3-фтор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.0 г, 3.0 ммоля) и карбонат калия (0.87 г, 6.3 ммоля). В атмосфере аргона к полученной смеси прибавляли иодид меди (270 мг, 1.4 ммоля) и 1,10-фенантролин (255 мг, 1.4 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.0 г титульного соединения, выход 65.5%.
Стадия 4: 1-(3-Фтор-4-метоксифенил)-7-оксо-6-(4-(3-оксоморфолино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
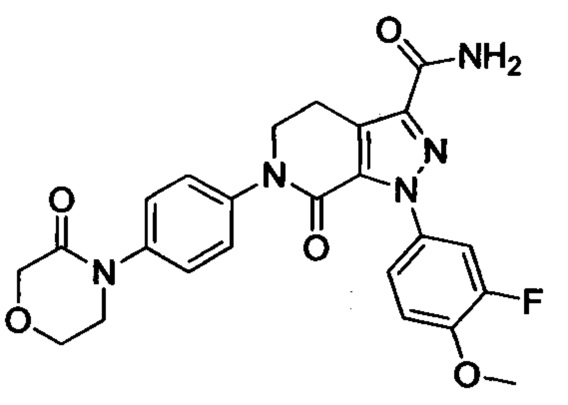
Этил 1-(3-фтор-4-метоксифенил)-7-оксо-6-(4-(3-оксоморфолино)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.0 г, 2.0 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали и сушили, получали 360 мг титульного соединения в виде твердого вещества светло-желтого цвета, выход 37.5%.
1H NMR (600MHz, DMSO) δ: 3.156-3.162 (m, 2H), 3.220-3.230 (m, 3Н), 3.434-3.594 (m, 2H), 3.576-3.594 (m, 2H), 3.829 (s, 2H), 3.892-3.923 (m, 2H), 6.595-6.609 (d, 2H), 7.043-7.057 (d, 2H), 7.219-7.241 (m, 1H), 7.289-7.304 (m, 1H), 7.417 (s, 1H), 7.571-7.599 (dd, 1H), 7.763 (1H,s)
MS 480.1 [M+H]+
Пример 27: 1-(3-Фтор-4-метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 1-(3-фтор-4-метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
При комнатной температуре к DMSO (20 мл) последовательно прибавляли 1-(4-иодфенил)пирролидин-2-он (0.95 г, 3.3 ммоля), этил 1-(3-фтор-4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.05 г, 3.1 ммоля) и карбонат калия (0.87 г, 6.3 ммоля). В атмосфере азота к полученной смеси добавляли иодид меди (270 мг, 1.4 ммоля) и 1,10-фенантролин (250 мг, 1.4 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.8 г титульного соединения в виде масла, выход 52.4%.
Стадия 2: 1-(3-Фтор-4-метоксифенил)-7-оксо-6-(4-(2-оксопирролидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Этил 1-(3-фтор-4-метоксифенил)-7-оксо-6-(4-(2-оксопирро лидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.8 г, 1.6 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали и сушили, получали 500 мг титульного соединения в виде твердого вещества песочного цвета, выход 67.4%.
1H NMR (600MHz, DMSO) δ: 2.053-2.064 (m, 2H), 3.189-3.198 (m, 2H), 3.301-3.395 (m, 1H), 3.821-3.832 (m, 5H), 4.017-4.027 (m, 2H), 4.441 (s, 1H), 7.234-7.247 (m, 1H), 7.352-7.467 (m, 4H), 7.581-7.749 (m, 4H)
MS 464.0 [M+H]+
Пример 28: 1-(4-Метоксифенил)-6-(4-(N-метилизобутирамидо)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 1-(4-метоксифенил)-6-(4-(N-метилизобутирамидо)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксилат
При комнатной температуре к DMSO (5 мл) последовательно прибавляли N-(4-иодфенил)-N-метилизобутирамид (1.00 г, 3.3 ммоля), этил 1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.95 г, 3.0 ммоля), 1,10-фенантролин (0.25 г, 1.41 ммоля) и карбонат калия (0.87 г, 6.3 ммоля). В атмосфере азота смесь нагревали при 120°С в течение 12 ч. Реакционную смесь выливали в воду со льдом (50 мл). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали преп.-HPLC, получали 0.15 г титульного соединения в виде твердого вещества песочного цвета, выход 10.2%.
Стадия 2: 1-(4-Метоксифенил)-6-(4-(N-метилизобутирамидо)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
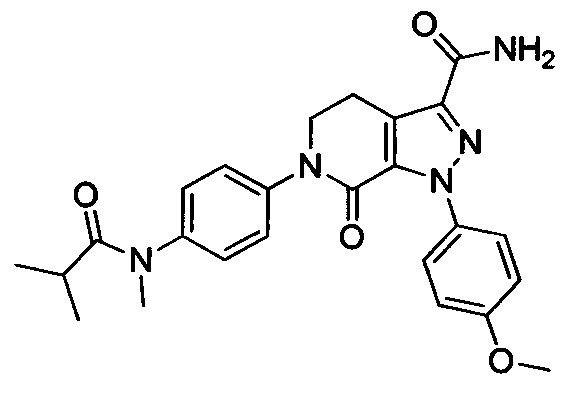
Этил 1-(4-метоксифенил)-6-(4-(N-метилизобутирамидо)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.15 г, 0.3 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (20 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали и сушили, получали 75 мг титульного соединения в виде твердого вещества почти белого цвета, выход 54.2%.
1H NMR (600MHz, DMSO) δ: 0.915-0.924 (m, 6Н), 3.124 (s, 3Н), 3.200-3.221 (m, 2Н), 3.550-3.558(m, 1Н) 3.804 (s, 3Н), 4.070-4.092 (m, 2H), 6.995-7.009 (m, 2H), 7.333-7.346 (m, 2H), 7.426-7.447(m, 3H), 7.496-7.511 (m, 2H), 8.405 (1H,s)
MS 462.2 [M+H]+
Пример 29: 1-(4-Этокси-3-фторфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(4-этокси-3-фторфенил)гидразоно)ацетат
При комнатной температуре 4-этокси-3-фторанилин (3.8 г, 24.5 ммоля) прибавляли в воду (25 мл). Полученную смесь перемешивали и охлаждали до температуры от -5 до 0°С. К образовавшейся смеси последовательно прибавляли концентрированную соляную кислоту (10 мл), водный раствор нитрита натрия (12 мл), раствор этил 2-хлор-3-оксобутаноата (4.2 г, 25.5 ммоля) в этаноле (40 мл) и раствор ацетата натрия (6.1 г, 74.4 ммоля) в воде (25 мл). По окончании прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем полученную смесь нагревали до комнатной температуры и перемешивали в течение 6 ч. В процессе реакции выпадал осадок. По завершении реакции реакционную смесь фильтровали. Осадок на фильтре сушили в вакууме и очищали хроматографией на колонке с силикагелем (РЕ:ЕА=10:1), получали 5.3 г титульного соединения, выход 74.6%.
Стадия 2: Этил 1-(4-этокси-3-фторфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
Этил 2-хлор-2-(2-(4-этокси-3-фторфенил)гидразоно)ацетат (5.3 г, 18.4 ммоля), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (3.4 г, 18.6 ммоля) и триэтиламин (5.6 г, 55.2 ммоля) последовательно прибавляли к толуолу (60 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (40 мл) и трифторуксусную кислоту (5.0 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции полученную смесь промывали водой. Водный слой экстрагировали DCM. Органические вытяжки упаривали в вакууме и остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 4.9 г титульного соединения, выход 76.5%.
Стадия 3: Этил 1-(4-этокси-3-фторфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
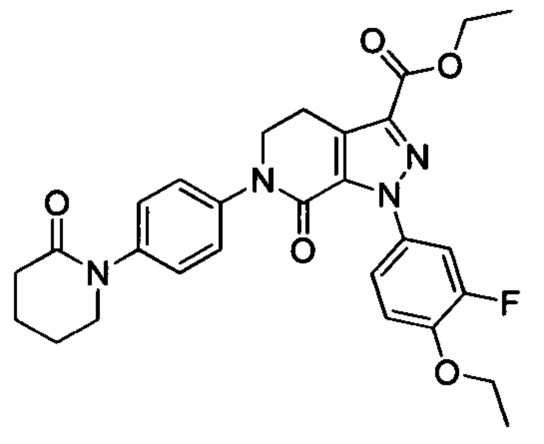
При комнатной температуре 1-(4-иодфенил)пиперидин-2-он (2.5 г, 8.3 ммоля), этил 1-(4-этокси-3-фторфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (2.4 г, 6.9 ммоля) и карбонат калия (2.1 г, 15.1 ммоля) последовательно добавляли к DMSO (35 мл). В атмосфере азота к полученной смеси прибавляли иодид меди (650 мг, 3.4 ммоля) и 1,10-фенантролин (613 мг, 3.4 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции полученную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.0 г титульного соединения, выход 26.7%.
Стадия 4: 1-(4-Этокси-3-фторфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
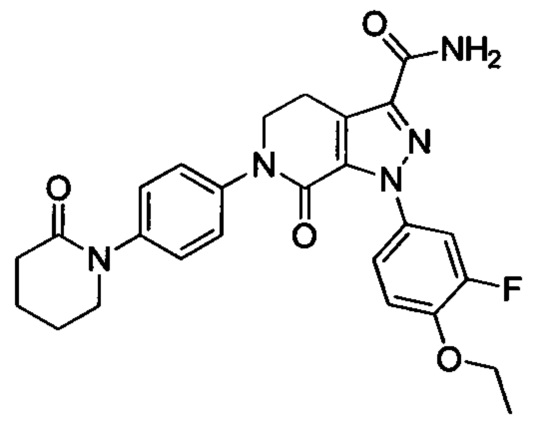
Этил 1-(4-этокси-3-фторфенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (1.0 г, 1.9 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 550 мг титульного соединения в виде твердого вещества песочного цвета, выход 58.3%.
1H NMR (600MHz, DMSO) δ: 1.362-1.368 (m, 3Н), 1.825-1.864 (m, 4Н), 2.386-2.389 (m, 2Н), 3.195-3.201 (m, 2Н), 3.582-3.600 (m, 2Н), 4.031-4.052 (m, 2Н), 4.141-4.175 (m, 2Н), 7.224-7.239 (m, 1Н), 7.275-7.289 (d, 2H), 7.348-7.362 (d, 2H), 7.388-7.402 (d, 1H), 7.470 (s, 1H), 7.564-7.588 (dd, 1H), 7.757 (s, 1H)
MS 492.4 [M+H]+
Пример 30: 1-(4-Метоксифенил)-7-оксо-6-(4-(2-оксазепан-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 6-(4-(6-бромгексанамидо)фенил)-1-(4-метоксифенли)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
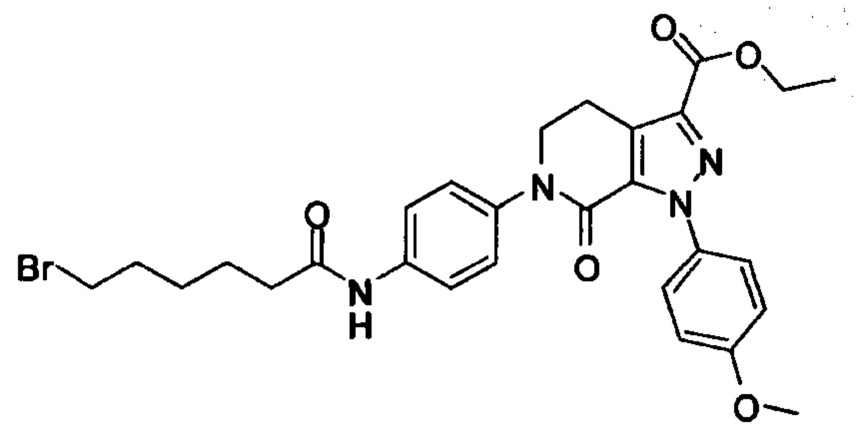
6-Бромгексановую кислоту (2.02 г, 10.4 ммоля), 4-диметиламинопиридин (0.20 г, 1.64 ммоля) и этил 6-(4-аминофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (4.00 г, 9.8 ммоля) прибавляли к DCM (40 мл) при комнатной температуре. Реакцию проводили при перемешивании при комнатной температуре в течение 4 ч. По завершении реакции полученную смесь упаривали в вакууме, получали 2.70 г титульного соединения в виде светло-желтого твердого вещества, выход 47.2%.
Стадия 2: Этил 1-(4-метоксифенил)-7-оксо-6-(4-(2-оксазепан-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
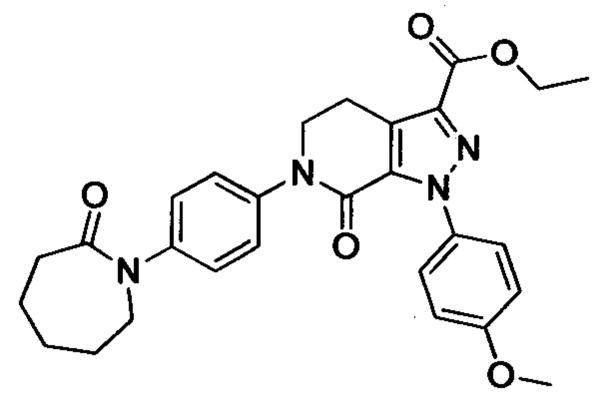
трет-Бутоксид калия (0.62 г, 5.5 ммоля) и этил 6-(4-(6-бромгексанамидо)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (2.60 г, 4.4 ммоля) последовательно прибавляли к THF (30 мл) при температуре от 0 до 5°С. Смесь нагревали до комнатной температуры и проводили реакцию в течение 3 ч. По завершении реакции реакционную смесь прибавляли в ледяной раствор разбавленной соляной кислоты (0.5 моля/л). Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (РЕ:ЕА=1:1), получали 270 мг титульного соединения в виде твердого вещества светло-желтого цвета, выход 11.9%.
Стадия 3: 1-(4-Метоксифенил)-7-оксо-6-(4-(2-оксазепан-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с] пиридин-3-карбоксамид
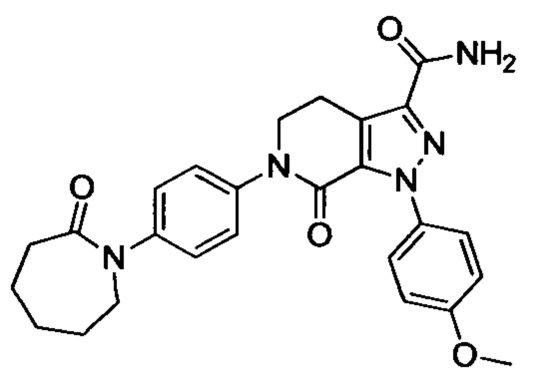
Этил 1-(4-метоксифенил)-7-оксо-6-(4-(2-оксазепан-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.25 г, 0.5 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (20 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 150 мг титульного соединения в виде твердого вещества почти белого цвета, выход 63.4%.
1H NMR (600MHz, DMSO) δ 1.727 (m, 6Н), 2.586-2.602 (m, 2Н), 3.189-3.211 (m, 2Н), 3.714-3.723 (m, 2Н), 3.805 (s, 3Н), 4.029-4.051 (m, 2Н), 6.988-7.002 (d, 2Н), 7.198-7.212 (d, 2Н), 7.322-7.336 (d, 2Н), 7.433 (s, 1Н), 7.492-7.507 (d, 2H), 7.711 (s, 1H)
MS 474.2 [M+H]+
Пример 31: 1-(3-(Этоксиметил)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 2-хлор-2-(2-(3-(этоксиметил)фенил)гидразоно)ацетат
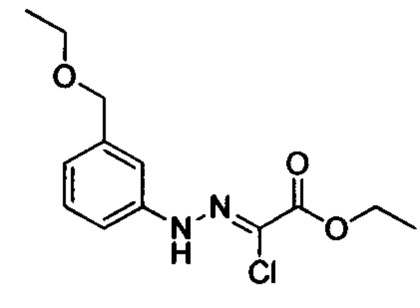
При комнатной температуре 3-(этоксиметил)анилин (3.3 г, 22 ммоля) прибавляли к воде (20 мл). Смесь перемешивали и охлаждали до температуры от -5 до 0°С. К полученной смеси последовательно добавляли концентрированную соляную кислоту (10 мл), водный раствор нитрита натрия (15 мл), раствор этил 2-хлор-3-оксобутаноата (3.7 г, 23 ммоля) в этаноле (20 мл) и раствор ацетата натрия (5.4 г, 66 ммоля) в воде (20 мл). После окончания прибавления смесь перемешивали при пониженной температуре в течение 0.5 ч. Затем полученную смесь нагревали до комнатной температуры и проводили реакцию при перемешивании в течение 4 ч. По завершении реакции реакционную смесь экстрагировали DCM и органические вытяжки упаривали в вакууме, получали 4.5 г титульного соединения в виде масла, выход 71.8%.
Стадия 2: Этил 1-(3-(этоксиметил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
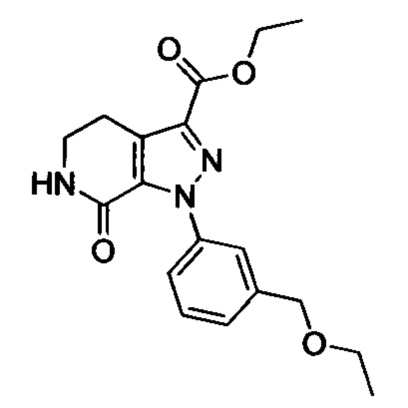
Этил 2-хлор-2-(2-(3-(этоксиметил)фенил)гидразоно)ацетат (4.0 г, 14.1 ммоля), 3-морфолино-5,6-дигидропиридин-2(1Н)-он (3.1 г, 16.9 ммоля) и триэтиламин (4.3 г, 42.3 ммоля) последовательно добавляли к толуолу (40 мл) при комнатной температуре. Смесь нагревали до 110°С и проводили реакцию при кипячении в течение 12 ч. Смесь охлаждали до комнатной температуры и упаривали в вакууме досуха. К остатку добавляли DCM (40 мл) и трифторуксусную кислоту (5.0 мл). Полученную смесь перемешивали в течение 2 ч. По завершении реакции полученную смесь промывали водой. Водный слой экстрагировали DCM. Органические вытяжки упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 1.2 г титульного соединения, выход 24.8%.
Стадия 3: Этил 1-(3-(этоксиметил)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
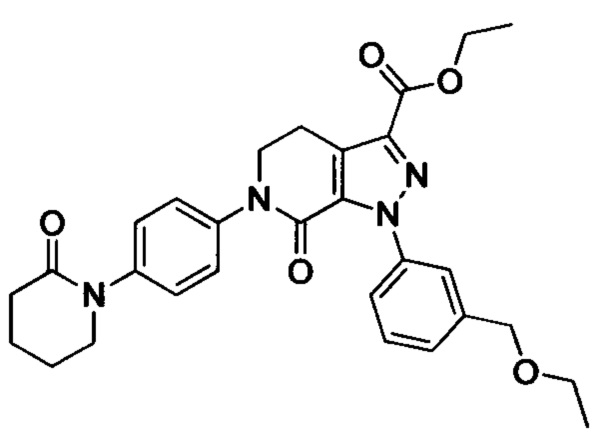
При комнатной температуре 1-(4-иодфенил)пиперидин-2-он (0.80 г, 2.6 ммоля), этил 1-(3-(этоксиметил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.77 г, 2.2 ммоля) и карбонат калия (0.68 г, 4.9 ммоля) последовательно добавляли к DMSO (10 мл). В атмосфере азота к полученной смеси прибавляли иодид меди (210 мг, 1.1 ммоля) и 1,10-фенантролин (197 мг, 1.1 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.2 г титульного соединения, выход 16.8%.
Стадия 4: 1-(3-(Этоксиметил)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
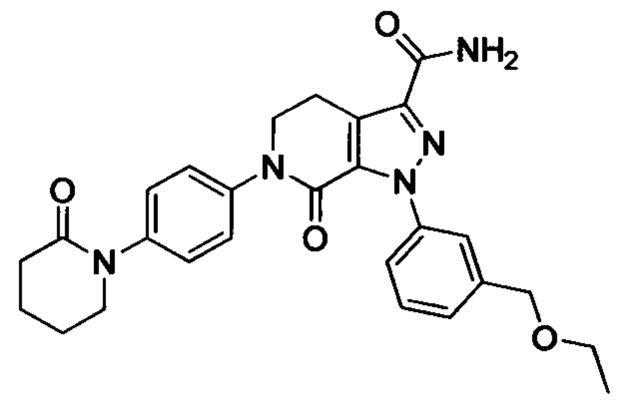
Этил 1-(3-(этоксиметил)фенил)-7-оксо-6-(4-(2-оксопиперидин-1-ил)фенил)-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.2 г, 0.39 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (10 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 110 мг титульного соединения в виде твердого вещества почти белого цвета, выход 56.4%.
1H NMR (600MHz, DMSO) δ 1.134-1.166 (m, 3Н), 1.830-1.859 (m, 4Н), 2.378-2.387 (d, 2Н), 3.205-3.214 (d, 2Н), 3.492-3.512 (m, 2Н), 3.586-3.594 (d, 2Н), 4.063-4.072 (d, 2Н), 4.498-4.506 (d, 2Н), 7.265-7.287 (m, 2Н), 7.341-7.397 (m, 3Н), 7.418-7.452 (m, 2Н), 7.463-7.532 (m, 2Н), 7.753 (s, 1Н)
MS 488.5 [M+H]+
Пример 32: 1-(4-Метоксифенил)-6-(4-(3-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 1-(4-метоксифенил)-6-(4-(3-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
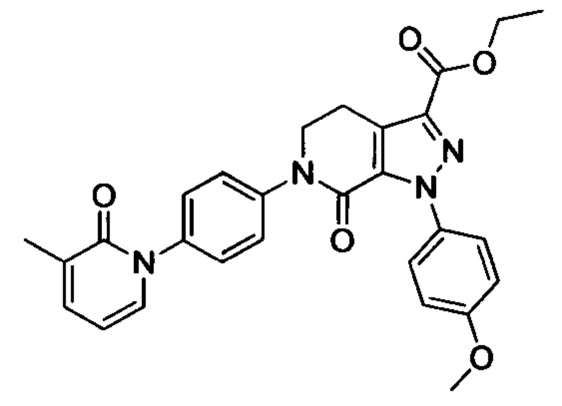
При комнатной температуре этил 6-(4-иодфенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.52 г, 1.0 ммоля), 3-метилпиридин-2(1Н)-он (0.16 г, 1.5 ммоля) и карбонат калия (0.29 г, 2.1 ммоля) последовательно добавляли к DMSO (25 мл). В атмосфере азота к полученной смеси добавляли иодид меди (91 мг, 0.47 ммоля) и 1,10-фенантролин (88 мг, 0.49 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.25 г титульного соединения в виде твердого вещества песочного цвета, выход 50.2%.
Стадия 2: 1-(4-Метоксифенил)-6-(4-(3-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксамид
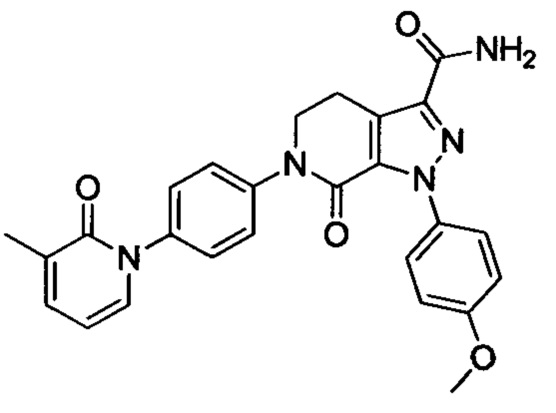
Этил 1-(4-метоксифенил)-6-(4-(3-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.25 г, 0.5 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (20 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 145 мг титульного соединения в виде твердого вещества почти белого цвета, выход 61.8%.
1H NMR (600MHz, DMSO) δ 2.035-2.049 (m, 3Н), 3.228-3.239 (m, 2H), 3.799-3.814 (m, 3Н), 4.100-4.111 (m, 2H), 6.234-6.248 (m, 1H), 6.993-7.022 (m, 2H), 7.398-7.536 (m, 9H), 7.754 (s, 1H)
MS 470.2 [M+H]+
Пример 33: 1-(4-Метоксифенил)-6-(4-(4-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Стадия 1: Этил 1-(4-метоксифенил)-6-(4-(4-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
При комнатной температуре к DMSO прибавляли (25 мл) этил 6-(4-иодфенил)-1-(4-метоксифенил)-7-оксо- 4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.52 г, 1.0 ммоль), 4- метилпиридин-2(1Н)-он (0.16 г, 1.5 ммоля) и карбонат калия (0.29 г, 2.1 ммоля). В атмосфере азота к полученной смеси прибавляли иодид меди (91 мг, 0.47 ммоля) и 1,10-фенантролин (88 мг, 0.49 ммоля). Смесь нагревали до 120°С и проводили реакцию при этой температуре в течение 12 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры и прибавляли воду. Полученную смесь экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:1), получали 0.34 г титульного соединения в виде твердого вещества песочного цвета, выход 68.3%.
Стадия 2: 1-(4-Метоксифенил)-6-(4-(4-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
Этил 1-(4-метоксифенил)-6-(4-(4-метил-2-оксопиридин-1(2Н)-ил)фенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.34 г, 0.68 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (20 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 210 мг титульного соединения в виде твердого вещества почти белого цвета, выход 65.8%.
1Н NMR (600MHz, DMSO) δ 2.181 (s, 3Н), 3.218-3.239 (m, 2Н), 3.804 (s, 3Н), 4.083-4.105 (m, 2Н), 6.177-6.189 (m, 1Н), 6.290 (s, 1H), 6.997-7.012 (m, 2H), 7.387-7.401 (m, 2H), 7.449-7.483 (m, 3H), 7.512-7.527 (m, 3H), 7.729 (s, 1H)
MS 470.2 [M+H]+
Пример 34: N1-(4-(3-Карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1H-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)сукцинамид
Стадия 1: 4-((4-(3-(Этоксикарбонил)-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)амино)-4-оксобутановая кислота
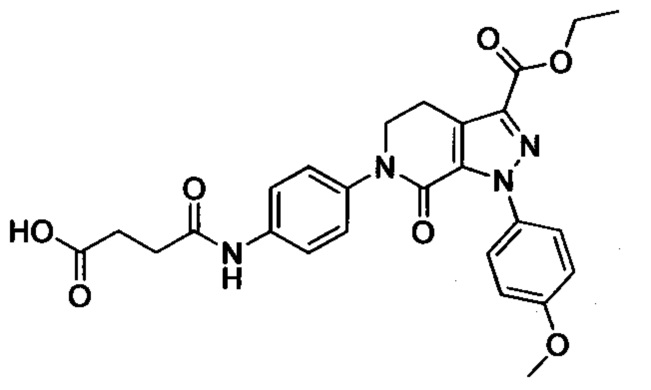
Янтарную кислоту (2.4 г, 20 ммолей), EDCI (11.4 г, 60 ммолей), DMAP (0.4 г, 3.3 ммоля) и этил 6-(4-аминофенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (8.2 г, 20 ммолей) последовательно прибавляли к DCM (60 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 3 ч. По завершении реакции реакционную смесь промывали разбавленной соляной кислотой (0.5 моля/л) и водой, сушили безводным Na2SO4 и фильтровали с отсасыванием. Фильтрат упаривали в вакууме, получали 2.0 г титульного соединения в виде твердого вещества светло-желтого цвета, выход 19.7%.
Стадия 2: Этил 6-(4-(2,5-диоксопирролидин-1-ил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат
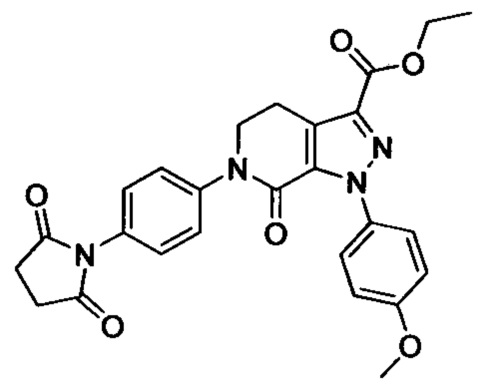
В колбу последовательно добавляли 4-((4-(3-(этоксикарбонил)-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)амино)-4-оксобутановую кислоту (2.0 г, 4.0 ммоля), уксусный ангидрид (20 мл) и ацетат натрия (0.4 г) при комнатной температуре. Реакцию проводили 1 ч при комнатной температуре. По завершении реакции к реакционной смеси добавляли ЕА и воду. Полученная смесь при стоянии разделялась на две фазы. Водную фазу экстрагировали ЕА. Органические вытяжки упаривали в вакууме, получали 1.8 г титульного соединения в виде твердого вещества, выход 92.1%.
Стадия 3: N1-(4-(3-Карбамоил-1-(4-метоксифенил)-7-оксо-4,5-дигидро-1Н-пиразоло[3,4-с]пиридин-6(7Н)-ил)фенил)сукцинамид
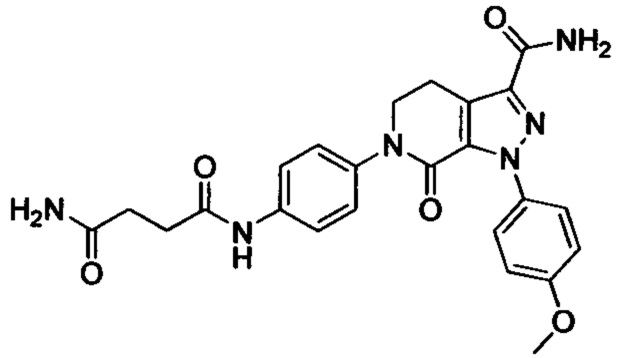
Этил 6-(4-(2,5-диоксопирролидин-1-ил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксилат (0.9 г, 1.8 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (15 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 200 мг титульного соединения в виде твердого вещества песочного цвета, выход 23.3%.
1Н NMR (600MHz, DMSO) δ 2.370-2.394 (m, 2Н), 3.176-3.197 (m, 2Н), 3.801 (s, 3Н), 3.992-4.014 (m, 2Н), 6.760 (s, 1Н), 6.989-7.003 (d, 2H), 7.251-7.265 (d, 2H), 7.324 (s, 1H), 7.430 (s, 1H), 7.483-7.498 (d, 2H), 7.568-7.582 (d, 2H), 7.707 (s, 1H), 9.980 (s, 1H)
MS 477.5 [M+H]+
Пример 35: 6-(4-(2,5-Диоксопирролидин-1-ил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-карбоксамид
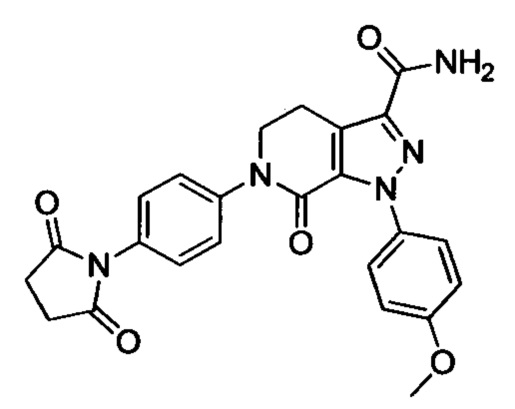
Этил 6-(4-(2,5-диоксопирролидин-1-ил)фенил)-1-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло [3,4-с] пиридин-3-карбоксилат (0.9 г, 1.8 ммоля) помещали в герметизируемую пробирку. В эту пробирку добавляли этиленгликоль (15 мл). В герметизируемую пробирку вводили газообразный аммиак в течение 0.5 ч, а затем эту пробирку герметизировали. Смесь нагревали до 120°С и реакцию проводили при этой температуре в течение 3 ч. По завершении реакции реакционную смесь охлаждали до комнатной температуры, а затем выливали в холодную воду. Выпадал твердый осадок. Смесь фильтровали. Осадок на фильтре сушили, получали 100 мг титульного соединения в виде твердого вещества песочного цвета, выход 12.1%.
1Н NMR (600MHz, DMSO) δ 3.141-3.163 (m, 2Н), 3.799 (s, 3Н), 3.909-3.920 (m, 2Н), 5.097 (m, 2Н), 6.527-6.541 (d, 2Н), 6.949-6.997 (m, 4Н), 7.409 (s, 1Н), 7.469-7.484 (d, 2H), 7.680 (s, 1H);
MS 460.2 [M+H]+.
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/d8f9b3121ca1ff375e414bad7043f1ba.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/cd53bbdb426e80e33cd53a731240fde7.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/11218e695beef3a7adf9a2f665e48fa5.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/9f42695b0a2d24921a899e4304b6c878.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/3c3d2b832be40c207f3da6aac9ce8b67.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/67a6de12c67200a9537fdab89d742c13.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/7fb9bbeddee7a0439ff356188560dce7.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/c4c2ffd92b16bf1109527067e935c491.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/29192880d10f2017ea103e883df3f05e.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/a72290324b917d7e838e6b37e21d00c6.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/244cd93e2191389e7642ba6bfab9adfa.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/b3b3023bd6fd2f028581764dd28a8cf8.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/a20815a2038c97a6a8ad08744d93769e.jpg)
![ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА](https://fips.edrid.ru/images/rid/98/31/ad/2d88d22413ffbbb0078ae75f165ad24d.jpg)
Способ разделения смеси оптических изомеров пинокембрина, в частности рацемата пинокембрина
Липосома, имеющая внутреннюю водную фазу, содержащую соль сульфобутилового эфира циклодекстрина
Кристаллическая форма эртапенема натрия и способ ее получения
Производные хиназолина
Очищенные терапевтические наночастицы и способы их получения
Соединение ингибитора brd4 в твердой форме, способ его получения и применение
Соль ингибитора lsd1 и её полиморфная форма
Лактамное соединение в качестве агониста рецептора fxr
Способ получения промежуточного соединения эртапенема
Кристаллическая форма эртапенема натрия и способ ее получения
Изоляционный состав, изоляционное изделие, способ их изготовления и комплектующее изделие для электрического кабеля на их основе

