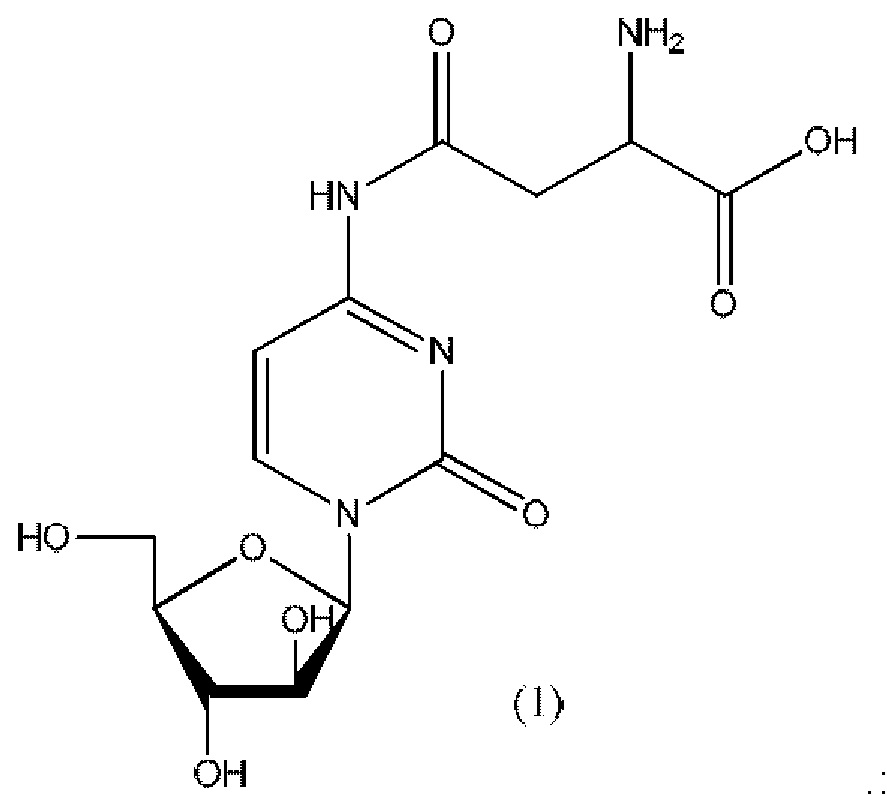
Результат интеллектуальной деятельности: КОНЪЮГАТЫ ЦИТАРАБИНА ДЛЯ ЛЕЧЕНИЯ РАКА
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к конъюгатам, содержащим цитарабин и аминокислоту, для применения для лечения рака. В частности, настоящее изобретение относится к конъюгатам цитарабина и аспарагиновой кислоты для применения для лечения опухолей системы крови у пациентов с ослабленным здоровьем.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Антипролиферативные лекарственные средства
Антипролиферативные лекарственные средства, также известные как антиметаболиты, противоопухолевые агенты или лекарственные средства, ковалентно связывающие ДНК, действуют путем ингибирования основных метаболических путей и обычно применяются для лечения злокачественных заболеваний. Однако их применение в качестве терапевтических агентов ограничено из-за высокой токсичности для нормальных клеток и серьезных нежелательных явлений. Нежелательные побочные эффекты включают анемию, рвоту и облысение из-за цитотоксического воздействия на быстро делящиеся нормальные клетки, такие как стволовые клетки в костном мозге, эпителиальные клетки кишечного тракта, клетки волосяных фолликулов и т.д.
Другой серьезной проблемой, связанной с антипролиферативными лекарственными средствами, является устойчивость опухолей к лекарственным средствам, как первичная, так и приобретенная. Например, хотя начальная доля пациентов, выходящих после лечения L-аспарагиназой в полную клиническую ремиссию, довольно высока у пациентов с острым лимфобластным лейкозом (ОЛЛ), рецидив заболевания и связанная с этим резистентность к лекарственным средствам представляют собой значительную клиническую проблему. Исследования продемонстрировали увеличение экспрессии аспарагинсинтетазы (AS) в резистентных к аспарагиназе клетках, что привело к возникновению гипотезы, что повышенная активность AS обеспечивает выживаемость злокачественных клеток за счет резистентности к лекарственным средствам.
Нуклеотидные/нуклеозидные аналоги
Нуклеозидные аналоги конкурируют со своими физиологическими «эквивалентами» за включения в нуклеиновые кислоты и играют важную роль в лечении острого лейкоза. Наиболее важными из них являются арабинозные нуклеозиды, это уникальный класс антиметаболитов, первоначально выделенных из губки Cryptothethya crypta, которые теперь получают синтетическим путем. Они отличаются от физиологических дезоксирибонуклеозидов присутствием 2'-ОН-группы в цис-конфигурации относительно N-гликозильной связи между цитозином и сахаром арабинозидом. Несколько арабинозных нуклеозидов оказывают полезное противоопухолевое и противовирусное действие. Наиболее активным цитотоксическим агентом этого класса является цитозин-арабинозид (цитарабин или ара-С). Цитарабин (cytarabine) в настоящее время применяется для лечения рака белых кровяных клеток, такого как острый миелоидный лейкоз (ОМЛ), острый лимфобластный лейкоз (ОЛЛ), хронический миелоидный лейкоз (ХМЛ), хронический лимфобластный лейкоз (ХМЛ) и миелодиспластические синдромы (МДС). Однако цитарабин в высокой степени токсичен и имеет серьезные побочные эффекты, такие как токсичность для мозжечка и подавление костного мозга. Поэтому лечение цитарабином пожилых пациентов и пациентов с дисфункцией печени, почек или мозжечка ограничено или, часто, недоступно.
Одной из задач разработки аналогов в области антиметаболитов цитидина был поиск соединений, которые сохраняют ингибирующую активность ара-С, но устойчивы к дезаминированию. Был разработан ряд деаминаз-устойчивых аналогов, в том числе цикло-цитидин (Но DHW, 1974) и N4-бегеноил ара-С (Kodama et al., 1989), которые показали противолейкемическую активность в некоторых клинических испытаниях, но при этом имели нежелательные побочные эффекты (Woodcock et al., 1980). Другими репрезентативными соединениями являются N4-пальмитоил-ара-С, 2'-азидо-2'-дезокси-ара-С, 5'-(кортизон 21-фосфориловый) сложный эфир ара-С, 5'-ациловые эфиры ара-С (например, сложный эфир 5'-пальмитата), N4-бегеноил-ара-С, конъюгат ара-С с поли-Н5(2-гидроксиэтил)-L-глутамином, дигидро-5-азацитидином, 5-аза-арабинозилцитозином, 5-аза-2'-дезоксицитидином и 2'-2'-дифторооксицитидином (Hartel et al., 1990; Heineman et al., 1988).
Гемцитабин (2,2-дифтородеоксицитидин, dFdC) является наиболее важным аналогом цитидина, который вводили в клинические испытания после ара-С. Он включен в стандартную терапию первой линии для пациентов с раком поджелудочной железы, раком легкого и переходно-клеточным раком мочевого пузыря.
Нуклеотидные аналоги также имеют и другие применения, не относящиеся к раку. Например, флуцитозин (Flucytosine), аналог фторированного цитозина, применяется в качестве противогрибкового агента.
Аминокислоты и пролиферативное заболевание
Аспарагин - это заменимая аминокислота, в которой нуждаются быстро пролиферирующие клетки. Клетки млекопитающих могут синтезировать аспарагин из аспартата, используя АТФ-зависимый фермент аспарагинсинтетазу (ЕС 6.3.5.4), которая переносит аминогруппу из амида глутамина в β-карбоксил аспартата в ходе взаимодействия, которое может быть представлено следующим образом: Глутамин+аспартат+АТФ+Н2О=глутамат+аспарагин+AMP+PPi.
Для поддержания метаболизма и пролиферации злокачественных клеток часто требуется большее количество аминокислот, включая аспарагин. Чтобы удовлетворить потребность в большом количестве аминокислот, злокачественные клетки развивают способность активно переносить аминокислоты из окружающей их среды. Кроме того, дефицит аспарагинсинтетазы встречается в некоторых опухолях, что заставляет их полагаться на поступление аспарагина из других источников, таких как сыворотка. Это наблюдение привело к применению фермента L-аспарагиназы (СЕ 3.5.1.1) в качестве химиотерапевтического агента. L-аспарагиназа гидролизует L-аспарагин до аспартата и аммиака, что приводит к обеднению сыворотки в отношении L-аспарагина и ингибированию роста опухоли. L-аспарагиназу применяют главным образом для лечения острого лимфобластного лейкоза (ОЛЛ), также она проявляет некоторую активность против других опухолей системы крови, включая острый нелимфоцитарный лейкоз.
L-аспарагиназа для применения в клинических условиях доступна в двух немодифицированных (нативных) формах, полученных очисткой из бактериальных источников, и в качестве ПЭГилированного соединения. В патенте США №4179377 описана ПЭГилированная L-аспарагиназа, где фермент связан с ПЭГ, имеющим молекулярную массу от 500 до 20000 дальтон.
Пониженная регуляция аспарагиновой синтетазы in vivo может обеспечить эффективный механизм ингибирования роста опухоли. Однако клетки реагируют на нехватку аминокислот путем согласованного увеличения мРНК аспарагинсинтетазы, белковой и ферментативной активности, которая включает в себя транскрипционный контроль гена аспарагинсинтетазы.
Первоначально метаболический подход использовали для ингибирования активности аспарагинсинтетазы путем образования аналогов L-аспарагина и L-аспарагиновой кислоты. Аналоги, включая 5-карбоксамидо-4-амино-3-изоксазолидон (Stammer et al., 1978), N-замещенные сульфонамиды и N'-замещенные сульфонилгидразиды, были получены в виде серных аналогов L-аспарагина (Brynes S et al., 1978а, Brynes S et al., 1978b). В патенте США №4348522 описана соль PALA, N-фосфонацетил-L-аспарагиновой кислоты, которая, как было показано, проявляет противоопухолевую активность и в настоящее время находится в клинических испытаниях в качестве комбинированной химиотерапии колоректального рака и рака поджелудочной железы.
Аспарагиновую кислоту-ара-С применяли в качестве исходного материала для синтеза конъюгатов пептида Т-ара-С для направленного взаимодействия на CD4-позитивные клетки (Manfredini et al., 2000).
Применение пролекарств для получения желаемых характеристик, таких как повышенная биодоступность или повышенная сайт-специфичность, является признанной концепцией в области фармацевтического развития. Например, прямое или косвенное конъюгирование лекарственного средства с антителом создает стабильный конъюгат, который можно доставить в определенное место с минимальной диссоциацией лекарственного средства. Направленное взаимодействие лекарственных средств для максимальной эффективности может сочетаться с механизмом селективного высвобождения указанного лекарственного средства.
В патенте США №4296105 описаны производные доксорубицина, связанные с необязательно замещенной аминокислотой в гидроксигруппе аминокислотного остатка, которые in vitro обладают более высокой противоопухолевой активностью и меньшей токсичностью, чем доксорубицин.
В патенте США №5962216 описаны активируемые опухолями пролекарства, которые не способны проникнуть в клетку до тех пор, пока они не расщепляются фактором или факторами, выделяемыми клеткой-мишенью.
В патенте США №5650386 представлены композиции, содержащие по меньшей мере один активный агент и по меньшей мере одну модифицированную не-альфа аминокислоту или полиаминокислоту, которая действует как носитель активного агента. Модификация аминокислот включает ацилирование или сульфирование по меньшей мере одной свободной аминогруппы.
В патентах США №№6623731; 6428780 и 6344213 описаны нековалентные смеси, содержащие модифицированные аминокислоты в качестве носителей для биологически активных агентов.
В патенте США №5106951 описан конъюгат, содержащий ароматическое лекарственное средство, нековалентно интеркалированное между двумя ароматическими боковыми цепями на олигопептиде, и антитело или фрагмент антитела, ковалентно присоединенный к олигопептиду, для направленного взаимодействия с раковыми клетками.
В патенте США №6617306 описан носитель для доставки in vivo терапевтического агента, при этом указанные носитель и терапевтический агент связаны дисульфидной связью. В патенте США №6617306 описано, что указанный носитель содержит полимер и по меньшей мере одно тиольное соединение, конъюгированное с полимером, так что тиольная группа тиольного соединения и тиольная группа терапевтического агента образуют дисульфидную связь.
В публикации международной патентной заявки № WO 00/33888 описаны расщепляемые противоопухолевые и противовоспалительные соединения, содержащие терапевтический агент, способный проникать в клетку-мишень, олигопептид, стабилизирующую группу и необязательный линкер.
В публикации международной патентной заявки №2005/072061 и в патентах США №№7989188 и 8993278 некоторые из изобретателей настоящего изобретения описывают соединения, содержащие лекарственное средство, ковалентно связанное с функциональной группой боковой цепи аминокислоты, указанные соединения применяют для направленного взаимодействия лекарственных средств с неопластическими клетками.
Остается неудовлетворенной потребность в способах лечения рака, которые включают введение пациентам с раковыми заболеваниями соединений, имеющих противоопухолевую активность и при этом пониженную цитотоксичность к нормальным тканям.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к пролекарствам, содержащим цитарабин, конъюгированный с одной аминокислотой, выбранной из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина, для применения для лечения неопластических заболеваний у пациентов с ослабленным здоровьем.
Настоящее изобретение частично основано на неожиданном обнаружении того, что соединение, обозначенное в настоящем документе как Астарабин®, то есть конъюгат цитарабина и аспарагиновой кислоты, где цитарабин ковалентно присоединен к карбоксильной группе боковой цепи аспарагиновой кислоты, при введении пожилым пациентам с острым лимфоцитарным лейкозом (ОЛЛ) или острым миелоидным лейкозом (ОМЛ) повышает выживаемость таких пациентов.
Известно, что максимальный уровень дозы цитарабина, которую можно вводить пациентам в возрасте 75 или более лет с лейкозом или лимфомой, составляет 20 мг/м2 площади поверхности тела субъекта. Однако большинство пациентов в этом возрасте не могут переносить даже такую низкую дозу цитарабина из-за серьезных побочных эффектов и, следовательно, таким пациентам цитарабин не вводят совсем. В настоящее время впервые выявлено, что пролекарство астарабин (Astarabine®), вводимое пациентам в возрасте 75 или более лет с ОМЛ или ОЛЛ, является безопасным и хорошо переносится в суточной дозе примерно 2 г/м2 или даже в более высоких дозах. Пролекарство астарабин® было эффективным для ослабления или даже устранения недавно диагностированного лейкоза, а также вторичного лейкоза, у пожилых пациентов.
Кроме того, описывается, что астарабин® можно вводить пожилым пациентам, то есть, пациентам 60 лет и старше, в дозах, которые значительно выше (например, в 5-30 раз выше), чем максимальный уровень дозы цитарабина, и астарабин® в таких дозах не вызывает значительных нежелательных явлений, связанных с лекарственным средством, во время или после лечения.
В одном варианте реализации настоящее изобретение относится к способу лечения неопластического заболевания, включающему введение субъекту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество соединения или его фармацевтически приемлемой соли, где указанное соединение имеет общую формулу (I):
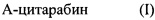
где А обозначает аминокислотный остаток, указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина;
где цитарабин присоединен к А через функциональную группу боковой цепи А; и
при этом указанный субъект представляет собой субъекта с ослабленным здоровьем, который не подлежит лечению цитарабином.
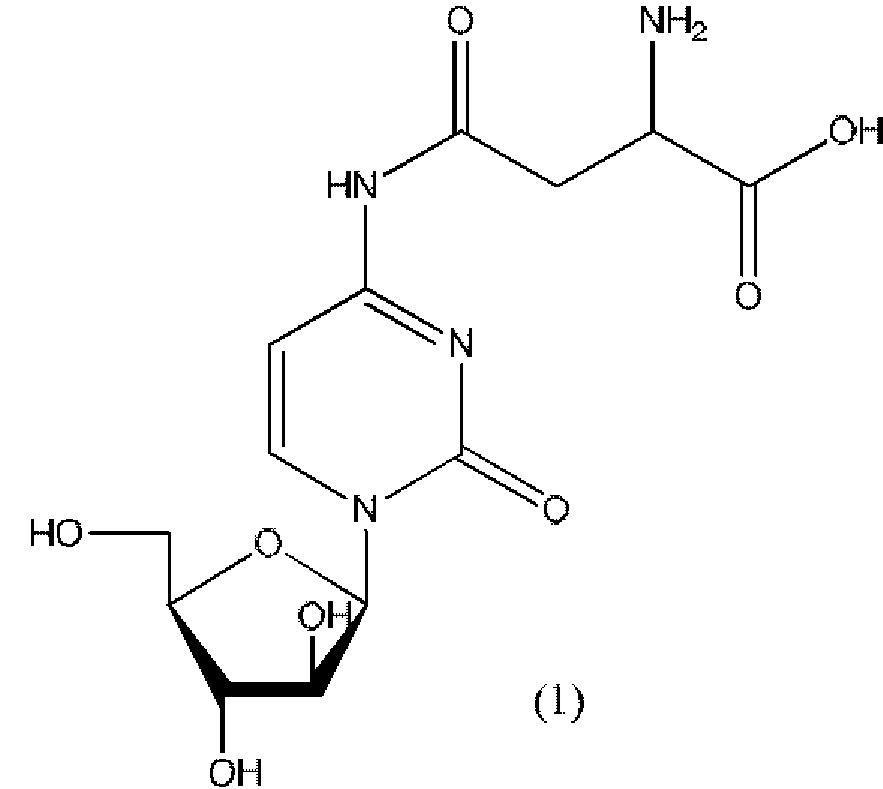
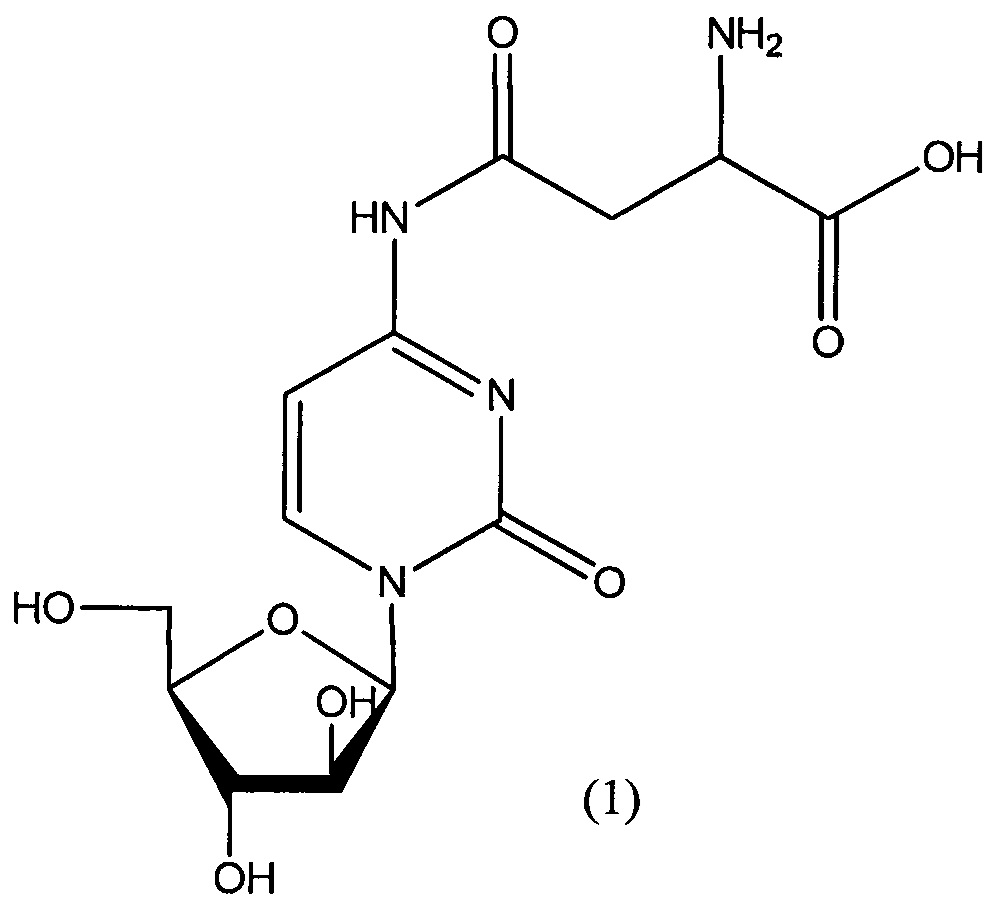
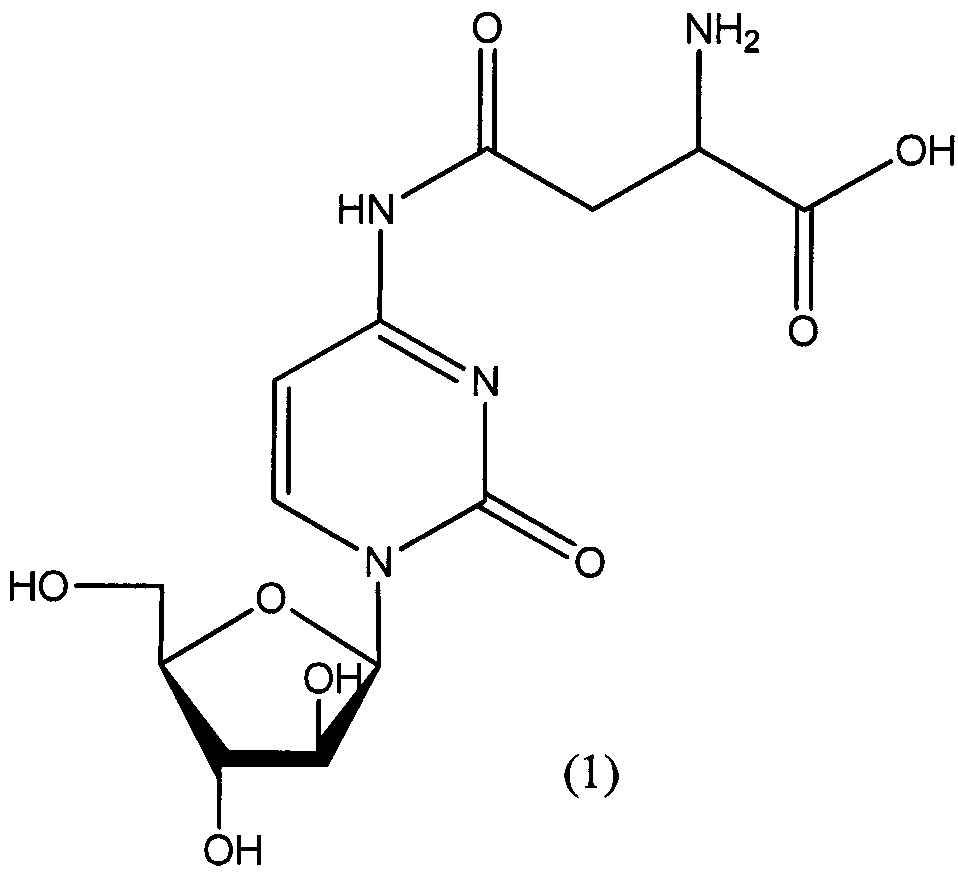
Согласно одному варианту реализации настоящего изобретения А представляет собой остаток аспарагиновой кислоты, и указанное соединение представлено структурой формулы (1), обозначенной как астарабин®:
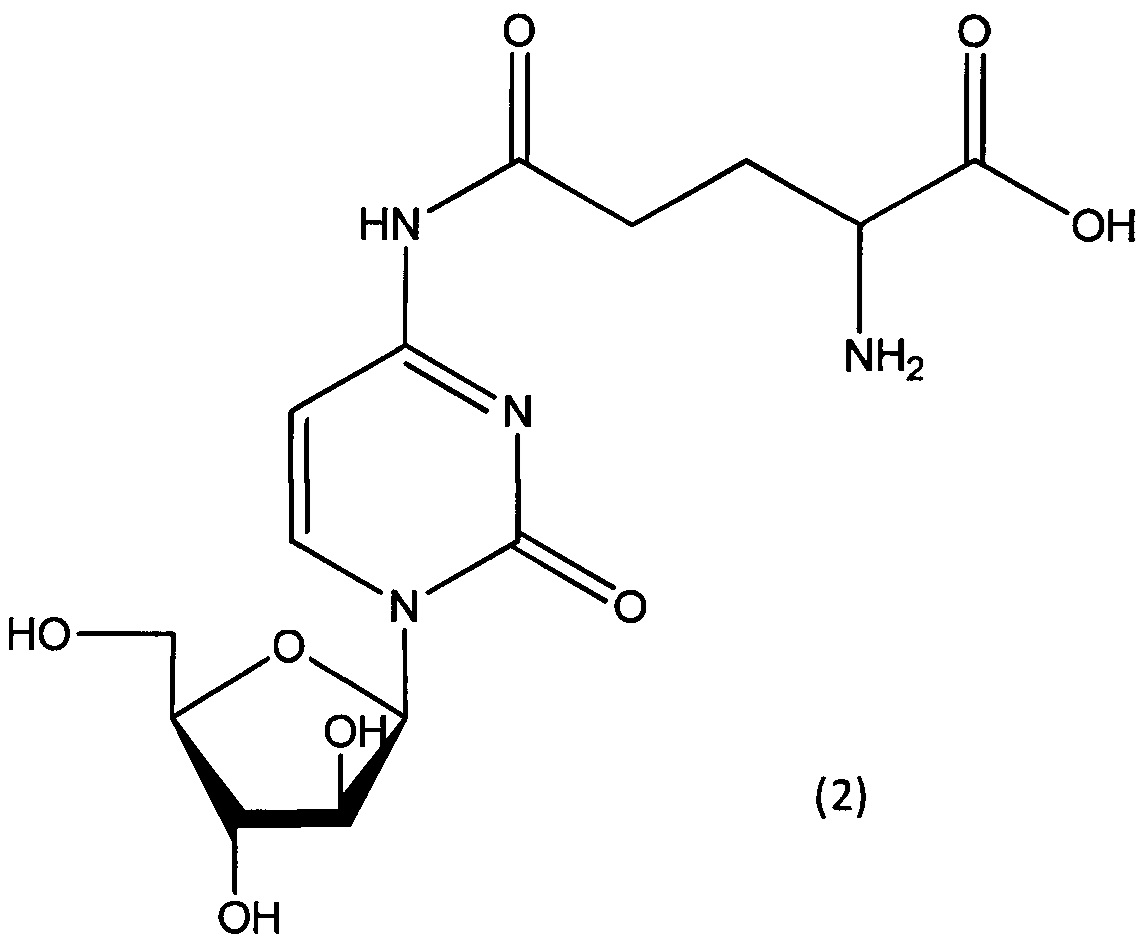
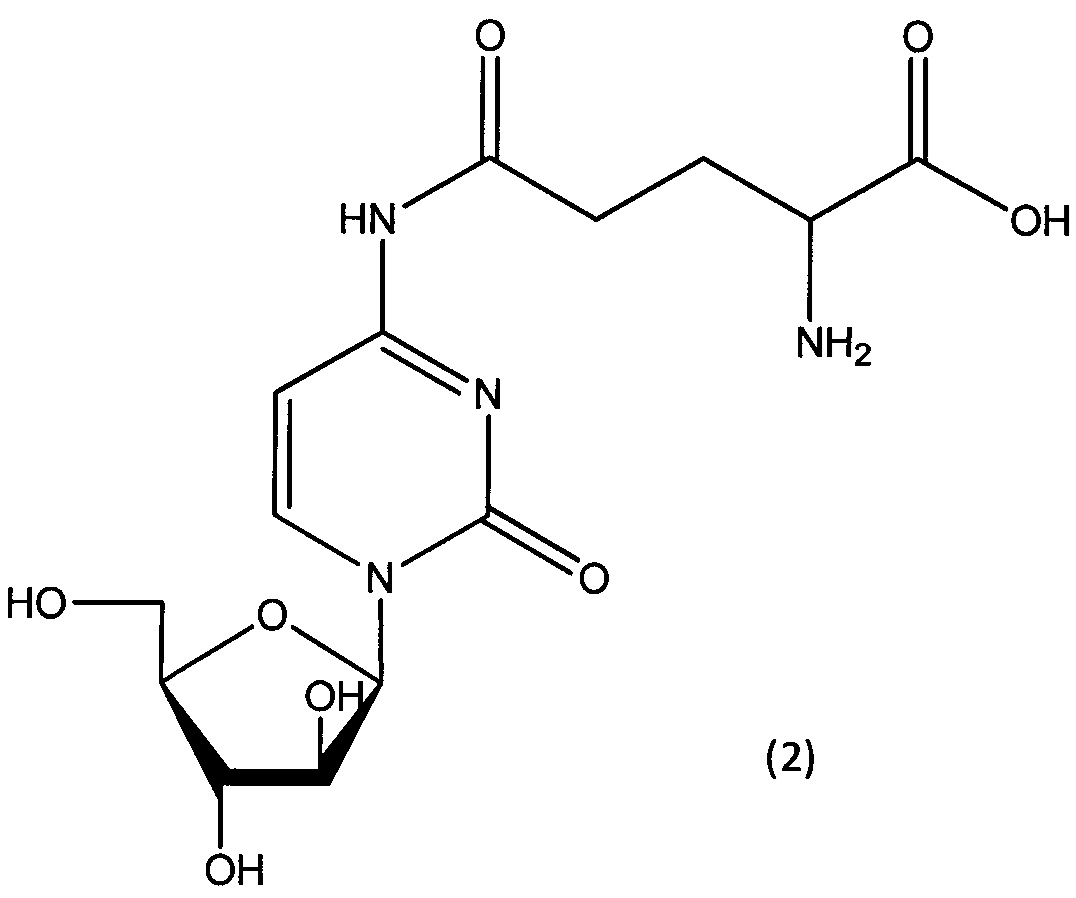
Согласно другому варианту реализации настоящего изобретения А представляет собой остаток глутаминовой кислоты, и указанное соединение представлено структурой формулы (2):
Согласно некоторым вариантам реализации настоящего изобретения указанная фармацевтически приемлемая соль представляет собой соль органической или неорганической кислоты или остатка кислоты. Согласно дополнительным вариантам реализации настоящего изобретения указанная кислота выбрана из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Согласно одному варианту реализации настоящего изобретения указанное соединение представляет собой астарабин®, а его фармацевтически приемлемая соль представляет собой ацетат. Согласно другому варианту реализации настоящего изобретения указанное соединение представляет собой астарабин®, а его фармацевтически приемлемая соль представляет собой гидрохлоридную соль.
Согласно дополнительным вариантам реализации настоящего изобретения указанное неопластическое заболевание выбрано из группы, состоящей из опухолей системы крови (гематологических опухолей) и опухолей, не относящихся к системе крови. Согласно дополнительным вариантам реализации настоящего изобретения указанная опухоль системы крови выбрана из группы, состоящей из лейкозов, лимфом, миелом и миелодиспластических синдромов (МДС). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно другим вариантам реализации настоящего изобретения указанный лейкоз выбран из группы, состоящей из острого миелоидного лейкоза (ОМЛ), острого лимфобластного лейкоза (ОЛЛ), хронического миелоидного лейкоза (ХМЛ) и хронического лимфобластного лейкоза (ХЛЛ). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно другим вариантам реализации настоящего изобретения указанный ОМЛ выбран из группы, состоящей из недавно диагностированного ОМЛ, вторичного ОМЛ и рецидивирующего/резистентного ОМЛ. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения указанная лимфома выбрана из лимфомы Ходжкина и неходжкинской лимфомы (НХЛ).
Согласно некоторым вариантам реализации настоящего изобретения указанный субъект с ослабленным здоровьем выбран из группы, состоящей из пожилых пациентов, субъектов с дисфункцией печени, субъектов с дисфункцией почек, субъектов с дисфункцией поджелудочной железы, субъектов с дисфункцией костного мозга, субъектов с дисфункцией мозжечка, субъектов с иммунологическим расстройством, субъектов с дисфункцией любого другого органа, ограничивающей применение цитарабина, субъектов с рецидивной или рефрактерной опухолью системы крови, и любой их комбинации. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения указанный пожилой субъект представляет собой субъекта в возрасте 50 или более лет, например 60, 70, 75 или более лет. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения астарабин® вводят в суточной дозе по меньшей мере в 2, 3, 5, 10, 15, 20 или по меньшей мере в 30 раз больше, чем максимальный уровень дозы цитарабина. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения астарабин® вводят в суточной дозе в диапазоне от примерно 0,3 г/м2 до примерно 10 г/м2, например, в суточной дозе примерно 0,3 г/м2, 0,5 г/м2, 0,8 г/м2, 1 г/м2, 1,5 г/м2, 2 г/м2, 2,3 г/м2, 2,5 г/м2, 3 г/м2, 3,5 г/м2, 4 г/м2 или 4,5 г/м2 площади поверхности тела субъекта, или любое значение из указанного диапазона. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно другим вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят парентерально. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят внутривенно, внутрибрюшинно, внутримышечно, подкожно, внутрикожно, трансдермально или внутривенно. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Согласно определенному варианту реализации настоящего изобретения указанную фармацевтическую композицию вводят внутривенно. Согласно иллюстративному варианту реализации настоящего изобретения указанную фармацевтическую композицию вводят при помощи внутривенной инфузии в течение периода времени от 15 минут до 3 часов, например, от 30 минут до одного часа.
Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере 3 суток, например, в течение 3 суток, 4, 5,6, 8,10,12 или 14 суток подряд или в течение любого целого количества суток из указанного диапазона. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение 6 последовательных суток по меньшей мере два раза в месяц. Согласно другим дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в двое суток в течение по меньшей мере одной недели, по меньшей мере двух недель, трех недель или по меньшей мере одного месяца.
Согласно иллюстративному варианту реализации настоящего изобретения указанное соединение представляет собой астарабина ацетат, указанный субъект представляет собой субъекта в возрасте 75 лет, имеющего ОЛЛ или ОМЛ, и указанную фармацевтическую композицию вводят в суточной дозе, которая по меньшей мере в два раза превышает максимальный уровень дозы цитарабина.
Согласно другому иллюстративному варианту реализации настоящего изобретения указанное соединение представляет собой астарабина гидрохлорид, указанный субъект представляет собой субъекта в возрасте 75 лет, имеющего ОЛЛ или ОМЛ, и указанную фармацевтическую композицию вводят в суточной дозе, которая по меньшей мере в два раза превышает максимальный стандартный уровень дозы цитарабина.
В другом варианте реализации настоящее изобретение относится к способу лечения неопластического заболевания, включающему введение субъекту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество соединения или его фармацевтически приемлемой соли, где указанное соединение представлено структурой формулы I:
где А обозначает аминокислотный остаток, указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина;
где цитарабин присоединен к А через функциональную группу боковой цепи А; и
при этом нежелательные явления от указанного соединения снижены по сравнению с нежелательными явлениями от неконъюгированного цитарабина, так что указанное соединение можно вводить в дозе, по меньшей мере в два раза превышающей максимальный уровень дозы цитарабина, и при этом субъект не будет испытывать дозолимитирующую токсичность.
Согласно одному варианту реализации настоящего изобретения А представляет собой остаток аспарагиновой кислоты, и указанное соединение представлено структурой формулы (1), обозначенной как астарабин®:
Согласно другому варианту реализации настоящего изобретения А представляет собой остаток глутаминовой кислоты, и указанное соединение представлено структурой формулы (2):
Согласно некоторым вариантам реализации настоящего изобретения указанная фармацевтически приемлемая соль представляет собой соль органической или неорганической кислоты или остатка кислоты. Согласно дополнительным вариантам реализации настоящего изобретения указанная кислота выбрана из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Согласно определенному варианту реализации настоящего изобретения указанное соединение представляет собой астарабин®, а его фармацевтически приемлемая соль представляет собой ацетат. Согласно другому варианту реализации настоящего изобретения указанное соединение представляет собой астарабин®, а его фармацевтически приемлемая соль представляет собой гидрохлоридную соль.
Согласно дополнительным вариантам реализации настоящего изобретения указанное неопластическое заболевание выбрано из группы, состоящей из опухолей системы крови и опухолей, не относящихся к системе крови. Согласно дополнительным вариантам реализации настоящего изобретения указанная опухоль системы крови выбрана из группы, состоящей из лейкозов, лимфом, миелом и миелодиспластических синдромов (МДС). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно другим вариантам реализации настоящего изобретения указанный лейкоз выбран из группы, состоящей из острого миелоидного лейкоза (ОМЛ), острого лимфобластного лейкоза (ОЛЛ), хронического миелоидного лейкоза (ХМЛ) и хронического лимфобластного лейкоза (ХЛЛ). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно другим вариантам реализации настоящего изобретения указанный ОМЛ выбран из группы, состоящей из недавно диагностированного ОМЛ, вторичного ОМЛ и рецидивирующего/резистентного ОМЛ. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения указанная лимфома выбрана из лимфомы Ходжкина и неходжкинской лимфомы (НХЛ).
Согласно некоторым вариантам реализации настоящего изобретения указанный субъект представляет собой субъекта с ослабленным здоровьем, который не подлежит лечению цитарабином. Согласно дополнительным вариантам реализации настоящего изобретения указанный субъект с ослабленным здоровьем выбран из группы, состоящей из пожилых пациентов, субъектов с дисфункцией печени, субъектов с дисфункцией почек, субъектов с дисфункцией поджелудочной железы, субъектов с дисфункцией костного мозга, субъектов с дисфункцией мозжечка, субъектов с иммунологическим расстройством, субъектов с дисфункцией любого другого органа, ограничивающей применение цитарабина, субъектов с рецидивной или рефрактерной опухолью системы крови, и любой их комбинации. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения указанный пожилой субъект представляет собой субъекта в возрасте 50 или более лет, например 60, 70, 75 или более лет. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации настоящего изобретения астарабин® вводят в суточной дозе по меньшей мере в от 2 до 30 раз больше, чем максимальный уровень дозы цитарабина, такой как в 3, 5, 10, 15, 20 или 30 раз больше. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительному варианту реализации настоящего изобретения астарабин® вводят в суточной дозе в диапазоне от примерно 0,3 г/м2 до примерно 10 г/м2, например, в суточной дозе примерно 0,3 г/м2, 0,5 г/м2, 0,8 г/м2, 1 г/м2, 1,5 г/м2, 2 г/м2, 2,3 г/м2, 2,5 г/м2, 3 г/м2, 3,5 г/м2, 4 г/м2 или 4,5 г/м2 площади поверхности тела субъекта, или любое значение из указанного диапазона. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно другим вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят парентерально. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят внутривенно, внутрибрюшинно, внутримышечно, подкожно, внутрикожно, трансдермально или внутривенно. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Согласно определенному варианту реализации настоящего изобретения указанную фармацевтическую композицию вводят внутривенно, предпочтительно при помощи инфузии.
Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере 3 суток, например, в течение 3 суток, 4, 5, 6, 8,10, 12 или 14 суток подряд или в течение любого целого количества суток из указанного диапазона. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение 6 последовательных суток один или два раза в месяц. Согласно другим дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в двое суток в течение по меньшей мере одной недели, по меньшей мере двух недель, трех недель или по меньшей мере одного месяца.
Согласно иллюстративному варианту реализации настоящего изобретения указанное соединение представляет собой астарабина ацетат и указанную фармацевтическую композицию вводят в суточной дозе, которая по меньшей мере в два раза превышает максимальный уровень дозы цитарабина.
Согласно другому иллюстративному варианту реализации настоящего изобретения указанное соединение представляет собой астарабина гидрохлорид и указанную фармацевтическую композицию вводят в суточной дозе, которая по меньшей мере в два раза превышает максимальный уровень дозы цитарабина.
Согласно дополнительному аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль, где указанное соединение представлено структурой формулы I:
где А обозначает аминокислотный остаток, указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина; и
где цитарабин присоединен к А через функциональную группу боковой цепи А;
для применения для лечения неопластического заболевания у субъекта с ослабленным здоровьем, который не подлежит лечению цитарабином в соответствиями с основными идеями настоящего изобретения.
Согласно дополнительному аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль, для применения для лечения неопластического заболевания, где указанное соединение представлено структурой формулы I:
где А обозначает аминокислотный остаток, указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина;
где цитарабин присоединен к А через функциональную группу боковой цепи А; и при этом нежелательные явления от указанного соединения снижены по сравнению с нежелательными явлениями от неконъюгированного цитарабина, так что указанное соединение вводят в дозе, по меньшей мере в два раза превышающей максимальный уровень дозы цитарабина, и при этом субъект не испытывает дозолимитирующую токсичность.
Эти и другие варианты реализации настоящего изобретения станут понятны в сочетании с приведенными ниже фигурами, описанием и формулой изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
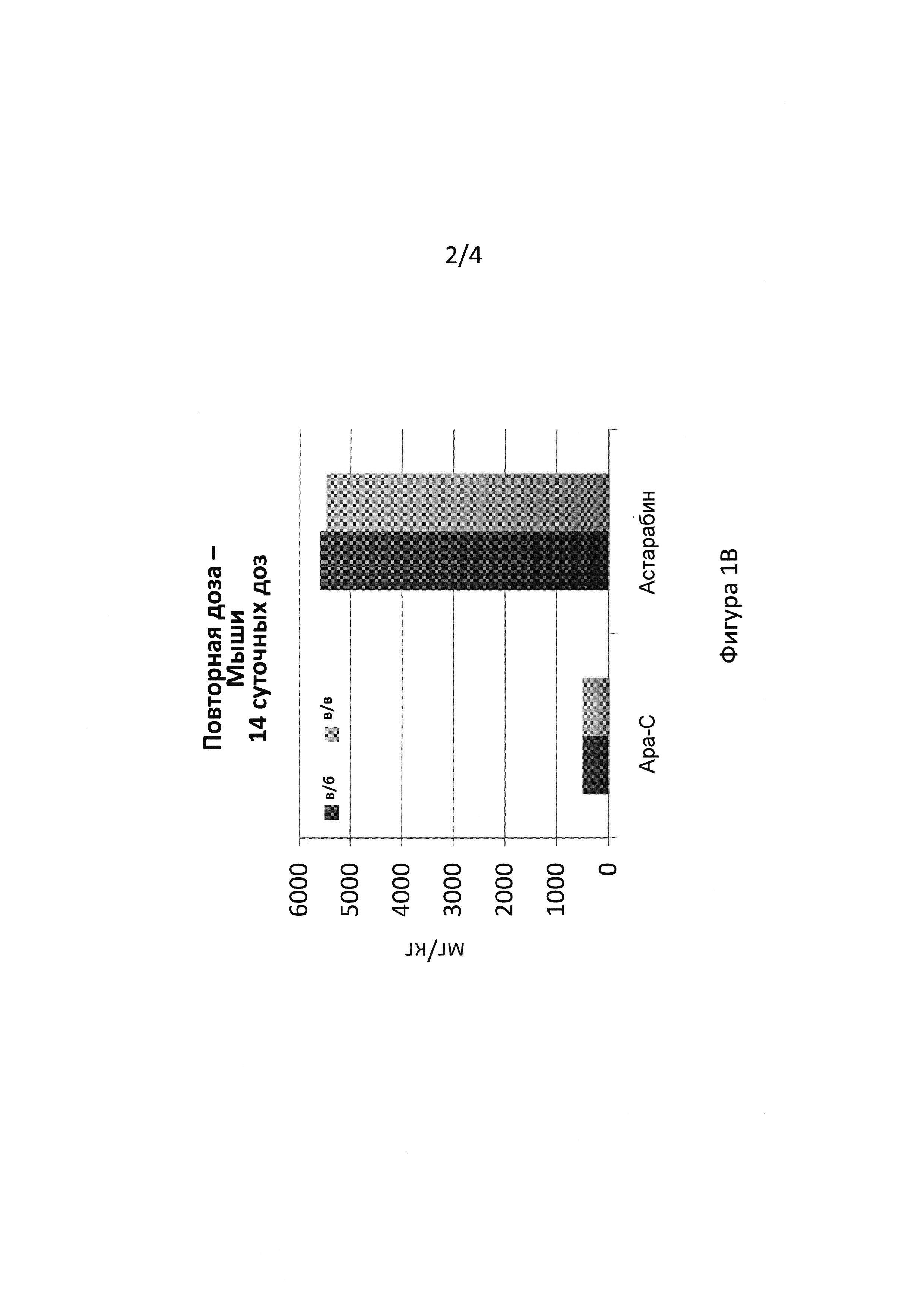
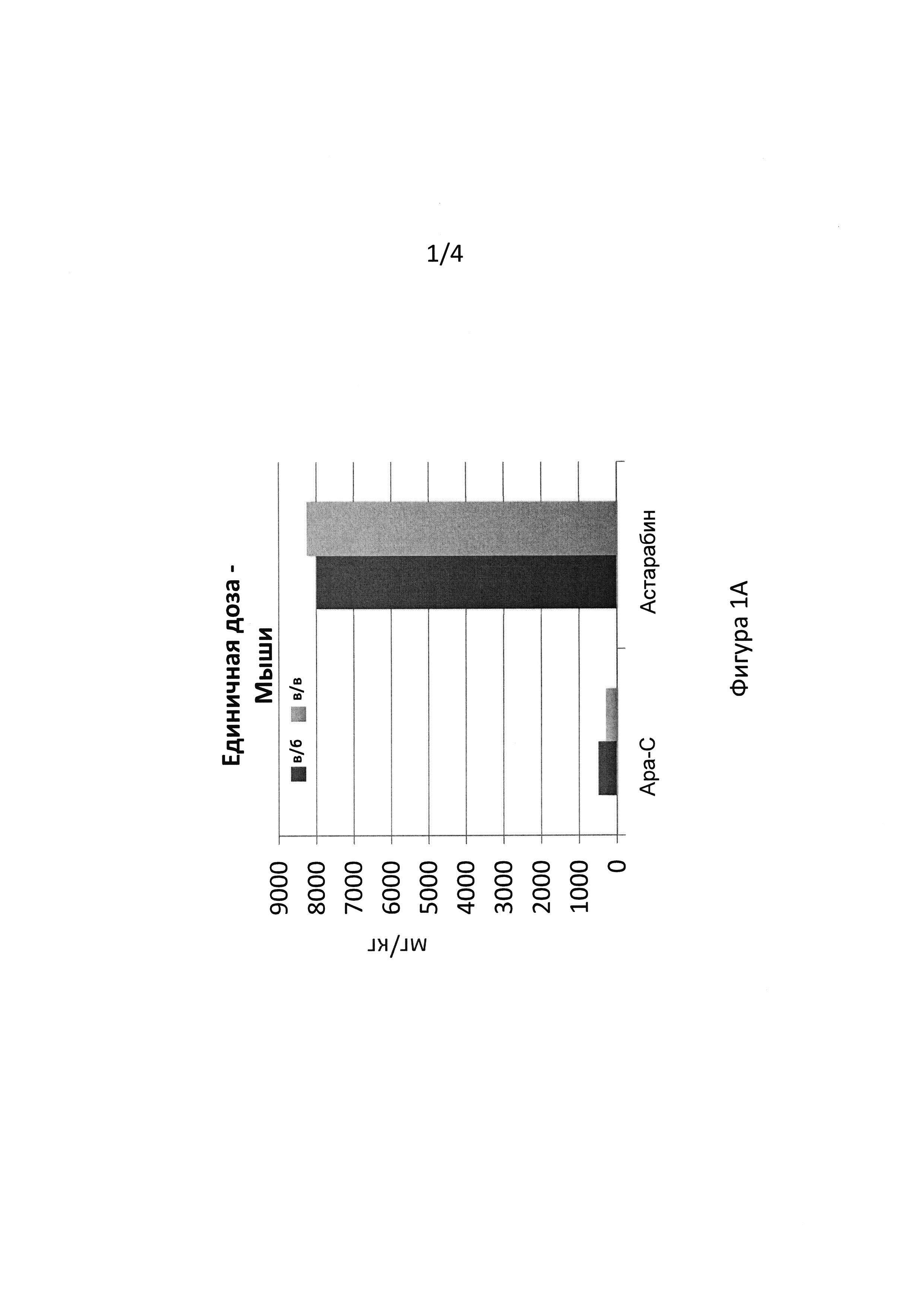
На фиг. 1А-В приведены измерения in vivo максимально переносимой дозы (МПД) астарабина®. Исследования переносимости/токсичности in vivo использовали для определения МПД в случае однократной дозы астарабина® у мышей (фиг. 1А) или в случае 14 суточных повторных доз астарабина® у мышей (фиг. 1В). МПД оценивали для внутривенного (в/в) или внутрибрюшинного (в/б) введения. Результаты сравниваются с МПД цитарабина.
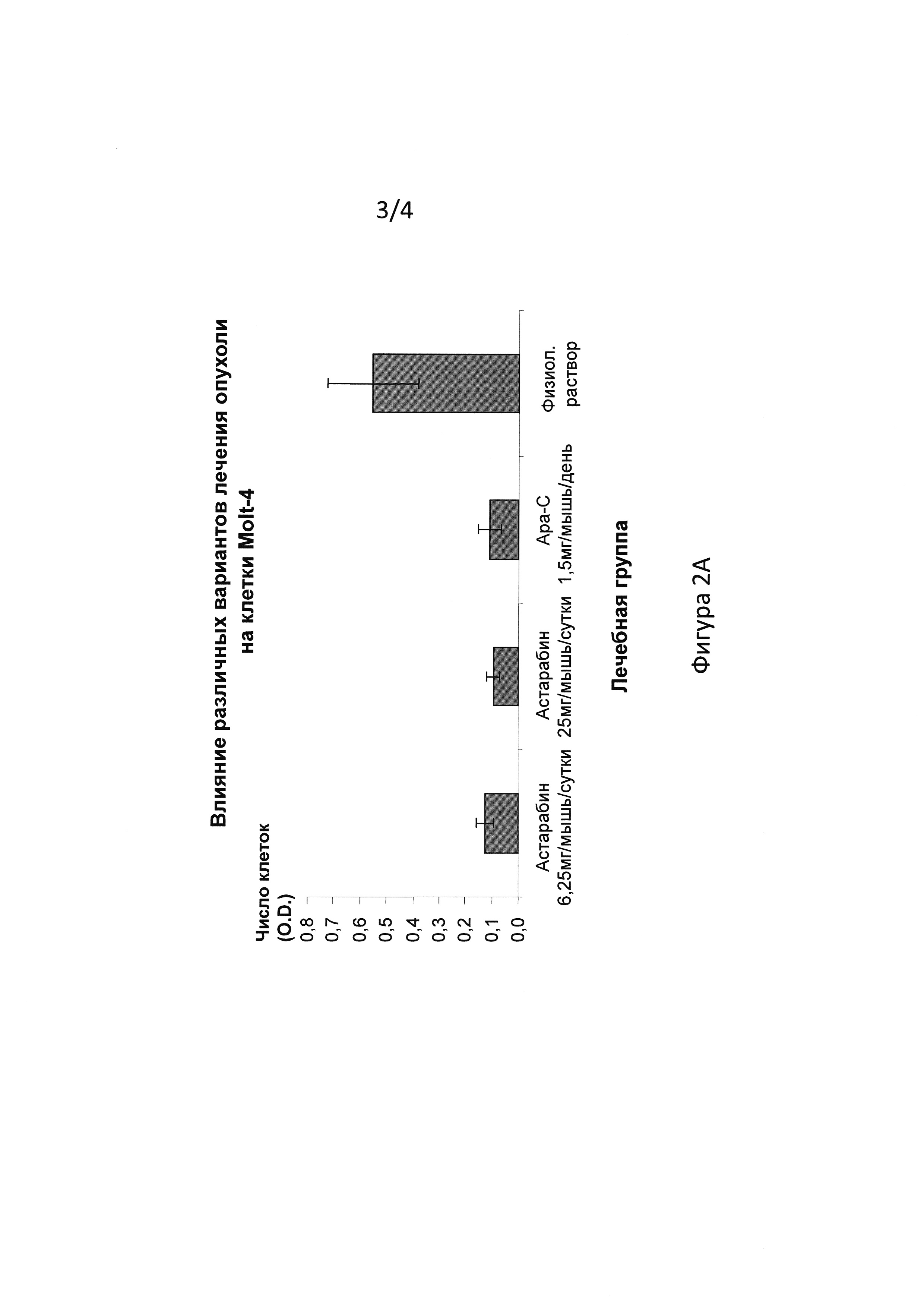
На фиг. 2А-В показано влияние астарабина® на рост лейкоза человека в модели с полыми волокнами, трансплантированными бестимусным мышам in vivo. Моделям лейкоза у мышей вводили 7 последовательных суточных доз одного из следующих вариантов: астарабин® в дозе 6,25 мг на мышь в сутки, астарабин® в дозе 25 мг/мышь/сутки, Ара-С в дозе 1,5 мг/мышь/сутки или физиологический раствор (контроль). Измеряли рост лейкоза in vivo. На фиг. 2А показан рост клеток лейкоза Molt-4. На фиг. 2В показан рост клеток лейкоза CCRF-CEM.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам лечения неопластического заболевания, включающим введение пациентам с ослабленным здоровьем, особенно пожилым пациентам в возрасте от 75 лет и старше, конъюгата цитарабина, ковалентно связанного с одной аминокислотой, при этом таких пациентов нельзя лечить с помощью неконъюгированного цитарабина из-за его тяжелых неблагоприятных явлений, и поэтому им предоставляется только поддерживающая терапия. Настоящее изобретение удовлетворяет давно существующую потребность в лечении пациентов с ослабленным здоровьем, особенно пожилых пациентов, у которых были диагностированы опухоли системы крови, но которые не могут получать лечение цитарабином. Конъюгаты согласно настоящему изобретению позволяют лечить таких больных раком при помощи цитарабина в дозах, которые были бы токсичны при введении цитарабина в неконъюгированной форме.
Определения
Термин субъекты «с ослабленным здоровьем», используемый в настоящем документе, относится к подгруппе субъектов, которые являются ослабленными или здоровье которых нарушено настолько, что они не могут переносить неконъюгированный цитарабин из-за серьезных нежелательных явлений. Субъекты с ослабленным здоровьем включают, но не ограничиваются ими, субъектов, страдающих от или имеющих дисфункцию почек, дисфункцию печени, дисфункцию поджелудочной железы, дисфункцию костного мозга, дисфункцию мозжечка, иммунологическое расстройство, расстройство любого другого органа, ткани или системы, которая ограничивает применение цитарабина, и их комбинацию.
Используемый в настоящем документе термин «неконъюгированный цитарабин» относится к свободному цитарабину или его аналогу или производному, который не присоединен ковалентно к аминокислоте.
Термины «дисфункция почек», «дисфункция печени», «дисфункция поджелудочной железы», «дисфункция костного мозга» и «дисфункция мозжечка» относятся к состоянию, в котором функция органа/ткани, например, почек, печени, поджелудочной железы, костного мозга и мозжечка, уменьшается относительно нормального состояния. В целом, дисфункция органа/ткани - это состояние, характеризующееся тем, что любое одно или более измеренных значений параметров, по которым проводят оценку функции органа, находятся вне диапазона нормальных значений (контрольных значений).
Термины «максимальный уровень дозы» и «рекомендуемая максимальная доза» цитарабина используются в настоящем документе взаимозаменяемо и относятся к дозировке цитарабина, например, к суточной дозе, одобренной Управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) для введения человеку, причем эта доза не вызывает неприемлемых нежелательных явлений и зависит от возраста и физического состояния субъекта, так что субъекту в возрасте 50 или менее лет обычно можно вводить суточную дозу цитарабина до 3 г/м2, субъекту от 50 до 60 лет обычно можно вводить суточную дозу цитарабина до 1,5 г/м2, субъекту в возрасте от 60 до 75 лет обычно можно вводить суточную дозу цитарабина в пределах от 0,1 г/м2 до 0,5 г/м2, а субъекту в возрасте от 75 и более лет можно вводить суточную дозу цитарабина до 20 мг/м2 от площади поверхности тела субъекта. Однако следует отметить, что большинству субъектов в возрасте от 75 и более лет вводить цитарабин нельзя из-за тяжелых нежелательных явлений от его приема.
Термин «дозолимитирующая токсичность» определяется в соответствии с критериями оценки степени тяжести наиболее частых нежелательных явлений, версия 3.0 (СТСАЕ). Дозолимитирующая токсичность возникает при введении соединения субъекту, если в течение цикла лечения лекарственным средством наблюдается любое из следующих событий: нейтропения 4 степени (т.е. абсолютное количество нейтрофилов (АКН)≤500 клеток/мм3) в течение 5 или более дней подряд или фебрильная нейтропения (т.е. температура≤38,5°С с АКН≤1000 клеток/мм3); тромбоцитопения 4 степени (т.е. ≤25000 клеток/мм3 или кровотечение, требующее переливания тромбоцитов); утомление 4 степени или снижение показателя работоспособности по шкале ECOG на две единицы; тошнота, диарея, рвота и/или миалгия 3 степени или выше, несмотря на адекватное/максимальное медицинское вмешательство; не связанные с анализом крови токсические эффекты 3 степени или выше (кроме утомления); задержка повторного лечения более чем на 2 недели из-за задержки восстановления от токсичности, связанной с лечением указанным соединением; клинически значимое токсическое действие на сердце 2 степени или более (например, снижение фракции выброса в покое до 40% - ≤50% или сокращение доли до 15% - ≤24%, кардиотропонин Т≥0,05 нг/мл).
Термин «терапевтически эффективное количество» соединения обозначает количество соединения, которое является достаточным для обеспечения благоприятного эффекта у субъекта, которому вводят указанное соединение. Эффективное количество соединения может варьироваться в зависимости от таких факторов, как состояние заболевания, возраст, пол и вес человека.
Термины «лечение», «лечить» и т.п. подразумевают замедление, остановку или устранение прогрессирования заболевания. Эти термины также включают облегчение, уменьшение, ослабление, устранение или снижение одного или нескольких симптомов заболевания, даже если указанное заболевание фактически не устранено и даже если прогрессирование заболевания само по себе не замедляется или не устраняется. Термин «субъект» относится к млекопитающему, предпочтительно к человеку.
Термин «остаток лекарственного средства» относится к лекарственному средству за исключением функциональной группы, которая используется для присоединения аминокислоты с получением конъюгата аминокислота-лекарственное средство A-D. Аналогично, термин «остаток аминокислоты» относится к аминокислоте за исключением функциональной группы, которая используется для присоединения лекарственного средства с получением конъюгата аминокислота-лекарственное средство A-D.
В настоящем документе термин «примерно» применительно к числовому значению следует понимать, как указанное значение +/- 10%.
В настоящем изобретении применяют аминокислоты, которые доступны коммерчески или могут быть получены обычными методами синтеза. В тех случаях, когда нет прямого указания, можно применять L- или D-изомер.
В одном варианте реализации настоящее изобретение относится к способу лечения неопластического заболевания, включающему введение субъекту с ослабленным здоровьем, который не подлежит лечению цитарабином, фармацевтической композиции, содержащей терапевтически эффективное количество соединения или его фармацевтически приемлемой соли, где указанное соединение имеет общую формулу (I):
где А обозначает аминокислотный остаток, указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина; и
где цитарабин присоединен к А через функциональную группу боковой цепи А.
Термин «фармацевтически приемлемая соль» лекарственного средства относится к соли в соответствии с принципами ИЮПАК. Фармацевтически приемлемая соль представляет собой неактивный ингредиент в форме соли в сочетании с лекарственным средством. Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к солям соединений общей формулы (I), формулы (II), например, к соединениям (1) и (2), или к любой другой соли, охватываемой общими формулами, которая по существу нетоксична для живых организмов. Типичные фармацевтически приемлемые соли включают соли, полученные при взаимодействии соединений согласно настоящему изобретению с фармацевтически приемлемым минералом, основанием, кислотой или солью. Соли кислот также известны как соли присоединения кислоты (см. ниже). Фармацевтически приемлемые соли известны в данной области техники (Stahl and Wermuth, 2011, Handbook of pharmaceutical salt, Second edition).
Согласно некоторым вариантам реализации настоящего изобретения указанная аминокислота может иметь свободные немодифицированные амино- (N-) и карбоксильные (С-) концы, или один или оба из N- и С-концов могут быть модифицированы. Таким образом, соединение согласно настоящему изобретению может быть представлено общей формулой II:

А' обозначает боковую цепь аминокислоты, указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина;
D обозначает остаток цитарабина или его аналога или производного;
каждый из R1, R2 и R2 независимо выбран из группы, состоящей из водорода и низшего алкила; и
X выбран из группы, состоящей из карбоксила, амида и гидразида.
Согласно предпочтительным вариантам реализации настоящего изобретения каждый из R1, R2 и R3 представляет собой водород.
Согласно другому предпочтительному варианту реализации настоящего изобретения X представляет собой карбоксильную группу.
Согласно определенному варианту реализации настоящего изобретения каждый из R1, R2 и R3 представляет собой водород, и X представляет собой карбоксил.
Согласно некоторым вариантам реализации настоящего изобретения X представляет собой амидную группу, присутствующую в виде карбоксиамида.
Аналоги цитарабина включают, но не ограничиваются ими, N4-бегеноил-ара-С, N4-пальмитоил-ара-С, 2'-азидо-2'-дезокси-ара-С, 5'-(кортизон 21-фосфориловый)сложный эфир ара-С, 5'-ациловые эфиры ара-С, N4бегеноил-ара-С, конъюгаты ара-С с поли-Н5(2-гидроксиэтил)-L-глутамином, дигидро-5-азацитидином, 5-аза-арабинозилцитозином и 5-аза-2'-дезоксицитидином.
Согласно некоторым вариантам реализации настоящего изобретения фармацевтически приемлемая соль соединения согласно настоящему изобретению представлена общей формулой (III):

где А представляет собой аминокислотный остаток, и указанная аминокислота выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина и глутамина;
D представляет собой цитарабин или его аналог или производное, и
Y представляет собой фармацевтически приемлемую органическую или неорганическую кислоту или кислотный остаток.
Согласно некоторым вариантам реализации настоящего изобретения Y представляет собой фармацевтически приемлемую кислоту, выбранную из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно иллюстративному варианту реализации настоящего изобретения солевая форма конъюгата астарабина® представляет собой ацетат или гидрохлорид.
Без связи с какой-либо теорией или механизмом действия, конъюгаты аминокислота-цитарабин согласно настоящему изобретению переносятся в раковые клетки при помощи аминокислотных транспортеров, тем самым минуют механизмы множественной лекарственной резистентности (МЛР), и в клетках эти конъюгаты расщепляются с высвобождением цитарабина, который останавливает рост клетки или убивает ее. Поскольку в раковых клетках были обнаружены свободный цитарабин и его метаболиты, конъюгаты согласно настоящему изобретению действуют как пролекарства.
Фармацевтические композиции
Настоящее изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно из соединений согласно настоящему изобретению и фармацевтически приемлемый носитель или разбавитель, а также необязательно дополнительно одно или более вспомогательных веществ.
Термин «фармацевтически приемлемый» означает одобренный регулирующим органом федерального или государственного управления или перечисленный в Фармакопее США или другой общепризнанной фармакопее для использования у животных и, более конкретно, у людей.
Термин «носитель» относится к разбавителю, адъюванту, вспомогательному веществу или носителю, с которым вводится терапевтическое соединение. Такими фармацевтическими носителями могут быть стерильные жидкости, такие как вода и масла, в том числе масла нефтяного, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, минеральное масло, кунжутное масло и тому подобное, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители.
Для внутривенного введения терапевтического соединения предпочтительным носителем является вода. Также можно применять физиологические растворы и водные растворы декстрозы и глицерина.
Согласно определенному варианту реализации настоящего изобретения состав астарабина® для внутривенного введения представляет собой водный изотонический раствор с осмолярностью примерно 300 мОсм и рН 4-8. Таким образом, фармацевтически приемлемый носитель для астарабина® может представлять собой буферизированный физиологический раствор, буферизированный раствор декстрозы или буферизированный раствор глицерина с осмолярностью примерно 300 мОсм и рН 4-8.
Буфер раствора астарабина® может представлять собой фармацевтически приемлемую моноионную буферную систему или полиионную буферную систему, имеющую pK ионизации в диапазоне 4-8.
Для регулирования рН раствора астарабина® можно применять различные буферы с pK 4-8, такие как, например, ACES (N-(ацетамидо)-2-аминоэтансульфоновая кислота); ацетатат; N-(2-ацетамидо)-2-иминодиуксусная кислота; BES (N,N-бис[2-гидроксиэтил]-2-аминоэтансульфоновая кислота); бицин (2-(бис(2-гидроксиэтил)амино)уксусная кислота); бис-трисметан (2-[бис(2-гидроксиэтил)амино]-2-(гидроксиметил)пропан-1,3-диол); бис-триспропан (1,3-бис(трис(гидроксиметил)метиламино)пропан); карбонат; цитрат; 3,3-диметилглутарат; DIPSO (3-N,N-бис(2-гидроксиэтил)амино]-2-гидроксипропансульфоновая кислота); N-этилморфолин; глицерин-2-фосфат; глицин; глицин-амид; HEPBS (N-(2-гидроксиэтил)пиперазин-N'-4-бутансульфоновая кислота); HEPES (N-(2-гидроксиэтил)пиперазин-N'-2-этансульфоновая кислота); HEPPS (N-(2-гидроксиэтил)пиперазин-N'-(3-пропансульфоновая кислота)); HEPPSO (N-(2-гидроксиэтил)пиперазин-N'-(2-гидроксипропансульфоновая кислота); гистидин; гидразин; имидазол; малеат; 2-метилимидазол; MES (2-(N-морфолино)этансульфоновая кислота); MOBS (4-(N-морфолино)-бутансульфоновая кислота); MOPS (3-(N-морфолино)-пропансульфоновая кислота, MOPSO (3-(N-морфолино)-2-гидропропансульфоновая кислота), оксалат, фосфат; пиперазин; PIPES (1,4-пиперазин-диэтансульфоновая кислота); POPSO (пиперазин-N,N'-бис(2-гидроксипропансульфоновая кислота)); сукцинат; сульфит; TAPS (3-[[1,3-дигидрокси-2-(гидроксиметил)пропан-2-ил]амино]пропан-1-сульфоновая кислота); TAPSO (3-[[1,3-дигидрокси-2-(гидроксиметил)пропан-2-ил]амино]-2-гидроксипропан-1-сульфоновая кислота); винная кислота; TES (2-[[1,3-дигидрокси-2-(гидроксиметил)пропан-2-ил]амино]этансульфоновая кислота); ТНАМ (Tris) (2-амино-2-гидроксиметилпропан-1,3-диол); и трицин (N-(2-гидрокси-1,1-бис(гидроксиметил)этил)глицин).
Согласно некоторым вариантам реализации настоящего изобретения указанный буфер представляет собой буфер производного сульфоновой кислоты, включая, но не ограничиваясь ими, ACES, BES, DIPSO, HEPBS, HEPES, HEPPS, HEPPSO, MES, MOBS, MOPS, MOPSO, PIPES, POPSO, сульфит, TAPS, TAPSO и TES.
Согласно дополнительным вариантам реализации настоящего изобретения указанный буфер представляет собой буфер производного карбоновой кислоты, включая, но не ограничиваясь ими, ацетатат, N-(2-ацетамидо)-2-иминодиуксусную кислоту, 2-(бис(2-гидроксиэтил)амино)уксусную кислоту, карбонат, цитрат, 3,3-диметилглутарат, лактат, малеат, оксалат, сукцинат и винную кислоту.
Согласно дополнительным вариантам реализации настоящего изобретения указанный буфер представляет собой буфер производного аминокислоты, включая, но не ограничиваясь ими, бициновый, глициновый, глицин-амидный, гистидиновый и трициновый буфер.
Согласно другим вариантам реализации настоящего изобретения указанный буфер представляет собой буфер производного фосфорной кислоты, включая, но не ограничиваясь ими, глицерол-2-фосфатный и фосфатный буфер.
Альтернативно, буферизированный физиологический раствор для состава астарабина® может представлять собой, например, сбалансированный солевой раствор Хэнкса, сбалансированный солевой раствор Эрла, сбалансированный солевой раствор Гейза, буферизированный HEPES физиологический раствор, буферизированный фосфатом физиологический раствор, Plasma-lyte, раствор Рингера, Рингера ацетат, Рингера лактат, цитратно-солевой буфер или трис-буферизированный физиологический раствор.
Буферизованный раствор декстрозы для состава астарабина® может представлять собой, например, раствор кислый цитратный раствор на основе декстрозы или раствор Эллиотта В.
Согласно иллюстративным вариантам реализации настоящего изобретения растворы для инъекции астарабина® представляют собой Plasma-Lyte А или соединение лактат натрия, приобретенное у Baxter.
Композиции могут принимать форму растворов, суспензий, эмульсии, таблеток, пилюль, капсул, порошков, композиций с замедленным высвобождением и тому подобного. Композицию можно приготовить в виде суппозиториев, с традиционными связующими и носителями, такими как триглицериды, микрокристаллическая целлюлоза, трагакантовая камедь или желатин.
Фармацевтическая композиция может дополнительно содержать фармацевтические вспомогательные вещества, включая, но не ограничиваясь ими, хлорид натрия, хлорид калия, хлорид магния, глюконат натрия, ацетат натрия, хлорид кальция, лактат натрия и тому подобное. Если требуется, указанная композиция может также содержать незначительные количества сахарных спиртов, смачивающих или эмульгирующих агентов и агентов, регулирующих рН. Также предусмотрены антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; и агенты для регулировки тоничности, такие как хлорид натрия или декстроза.
Фармацевтические композиции для парентерального введения также могут быть приготовлены в виде суспензий активных соединений. Такие суспензии можно получать в виде масляных или водных суспензий для инъекций. Для масляных суспензий для инъекций можно применять подходящие липофильные растворители или носители, в том числе жирные масла, такие как кунжутное масло, или эфиры синтетических жирных кислот, такие как этилолеат, триглицериды или липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрийкарбоксиметилцеллюлоза, сорбит или декстран. Указанная суспензия необязательно может также содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость соединений, чтобы обеспечить получение высококонцентрированных растворов.
Для трансмукозального и трансдермального введения в состав можно включать вещества, обеспечивающие проникновение (пенетранты), соответствующие барьеру, через который необходимо проникнуть. В данной области известны такие обеспечивающие проникновение вещества, включая, например, ДМСО или полиэтиленгликоль.
Для перорального введения соединения можно легко приготовить путем объединения активных соединений с фармацевтически приемлемыми носителями и вспомогательными веществами, хорошо известными в данной области. Такие носители позволяют приготовить соединения согласно настоящему изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального приема субъектом. Фармакологические составы для перорального применения можно получать с применением твердого вспомогательного вещества, необязательно измельчения полученной смеси и обработки смеси гранул после добавления подходящих вспомогательных веществ, если оно требуется, с получением таблеток или драже. Подходящими вспомогательными веществами являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозные составы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбометилцеллюлоза натрия; и/или физиологически приемлемые полимеры, такие как поливинилпирролидон (ПВП). При желании можно добавлять дезинтегрирующие агенты, такие как поперечно-сшитый поливинилпирролидон, агар или альгиновую кислоту или ее соль, такую как альгинат натрия.
Кроме того, если желательно предотвратить воздействие соединений согласно настоящему изобретению на желудочную среду, может быть полезным энтеросолюбильное покрытие.
Фармацевтические композиции для перорального применения включают твердые капсулы, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами.
В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, можно добавлять стабилизаторы.
Фармацевтические композиции согласно настоящему изобретению можно получать способами, хорошо известными в данной области, например, при помощи обычных процессов смешивания, растворения, гранулирования, измельчения, пульверизации, дражирования, левигации, эмульгирования, инкапсуляции, захвата или лиофилизации.
Дозировка во вводимой композиции будет зависеть от многих факторов, включая субъекта, подлежащего лечению, стадии рака, пути введения и оценки врача, назначающего лечение.
Терапевтическое применение
Настоящее изобретение относится к способу лечения неопластического заболевания или вирусного заболевания, включающему введение субъекту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество соединения общей формулы (I), (II), например, соединения (1) или (2) или его фармацевтически приемлемой соли, как описано выше, и фармацевтически приемлемый носитель.
Неопластическое заболевание может быть выбрано из опухолей системы крови или опухолей, не относящихся к системе крови.
Опухоли системы крови включают лейкозы, лимфомы, миеломы и миелодиспластические синдромы (МДС), включая, но не ограничиваясь ими, миелоидный лейкоз, лимфоцитарный лейкоз, например, острый миелоидный лейкоз (ОМЛ), хронический миелоидный лейкоз (ХМЛ), острый лимфоцитарный лейкоз (ОЛЛ), хронический лимфоцитарный лейкоз (ХЛЛ), лимфому Ходжкина, неходжкинскую лимфому, множественную миелому и макроглобулинемию Вальденстрема.
Термин «миелодиспластические синдромы» (МДС) относится к гетерогенной группе гемопоэтических злокачественных новообразований, характеризующихся цитопениями крови, неэффективным гематопоэзом и гиперпластическим костным мозгом. МДС - это прелейкемические состояния, которые примерно в 30-40% случаев трансформируются в острый миелоидный лейкоз (ОМЛ). Если не предлагается аллогенная трансплантация стволовых клеток, ДМС обычно считается неизлечимым состоянием.
Опухоли, не относящиеся к системе крови, также известные как солидные опухоли, включают, но не ограничиваются ими, следующие: саркома, карцинома, фибросаркома, миксосаркома, липосаркома, хондросаркома, остеогенная саркома, хордома, ангиосаркома, эндотелиосаркома, мезотелиома, опухоль Юинга лейомиосаркома, рабдомиосаркома, плоскоклеточная карцинома, базальноклеточная карцинома, аденокарцинома, карцинома потовых желез, карцинома сальных желез, папиллярная карцинома, папиллярная аденокарцинома, цистаденокарцинома, медуллярная карцинома, бронхогенная карцинома, почечно-клеточная карцинома, гепатома, карцинома желчных протоков, хориокарцинома, семинома, эмбриональная карцинома, опухоль Вильмса, рак шейки матки, опухоль яичка, карцинома легкого, мелкоклеточная карцинома легкого, карцинома мочевого пузыря, эпителиальная карцинома, астроцитома, саркома Капоши и меланома. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Опухоли, не относящиеся к системе крови включают рак органов, где рак органа включает, но не ограничивается ими, рак молочной железы, рак мочевого пузыря, рак толстой кишки, рак прямой кишки, рак эндометрия, рак почки, рак легких, рак шейки матки, рак поджелудочной железы, рак предстательной железы, рак яичка, рак щитовидной железы, рак яичников, рак головного мозга, включая эпендимому, глиому, глиобластому, медуллобластому, краниофарингиому, пинеалому, неврому слухового нерва, гемангиобластому, олигодендроглиому, менангиому, нейробластому, ретинобластому и их метастазы. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Кроме того, настоящее изобретение относится к способу лечения инфекции, вызванной вирусным агентом, представляющим собой вирус, вызывающий рак. Таким образом, настоящее изобретение относится к способу лечения вирусной инфекции, вызванной вирусным онкогеном. Неограничивающие примеры таких вирусов включают вирус папилломы человека, вирус гепатита В, вирус гепатита С, вирус Эпштейна-Барр, Т-лимфотропный вирус человека, герпесвирус, связанный с саркомой Капоши и полиомавирус клеток Меркель. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Вирусное заболевание может быть вызвано другими вирусами, включая, но не ограничиваясь ими, вирусом иммунодефицита человека (ВИЧ), вирусом простого герпеса (ВПГ), цитомегаловирусом (ЦМВ) и вирусом ветряной оспы (VZV). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Согласно одному варианту реализации настоящего изобретения указанное вирусное заболевание представляет собой вирусный герпетический энцефалит.
Способ согласно настоящему изобретению может быть полезен для лечения неопластического заболевания у субъекта, имеющего иммунологическое заболевание или расстройство. Иммунологические заболевания или расстройства включают, но не ограничиваются ими, ревматоидный артрит (РА), псориатический артрит, системную красную волчанку (СКВ), волчаночный нефрит, воспалительное заболевание кишечника (ВЗК), синдром раздраженной толстой кишки, диабет I типа, иммунную тромбоцитопеническую пурпуру, рассеянный склероз, синдром Шегрена, тиреоидит Хашимото, болезнь Грейвса, первичный билиарный цирроз, гранулематоз Вегенера, туберкулез и макроглобулемию Вальденстрема.
Способ согласно настоящему изобретению может быть полезен для лечения неопластического заболевания у субъектов с дисфункцией органов, таких как дисфункция печени, дисфункция почек, дисфункция поджелудочной железы, дисфункция костного мозга и дисфункция мозжечка.
Термин «дисфункция печени» относится к состоянию, в котором функция печени снижается по сравнению с нормальным состоянием. В целом, дисфункция печени представляет собой состояние, характеризующееся тем, что любое одно или более значений контрольных параметров для оценки функции печени (например, уровни в крови AST, ALT, ALP, ТТТ, ZTT, общий билирубин, общий белок, альбумин, лактатдегидрогеназа, холинэстераза и тому подобное) находятся вне диапазона нормальных значений (контрольных значений). Дисфункция печени характерна для таких заболеваний, как, например, молниеносный гепатит, хронический гепатит, вирусный гепатит, алкогольный гепатит, фиброз печени, цирроз печени, рак печени, аутоиммунный гепатит, аллергическая гепатопатия и первичный билиарный цирроз.
Дисфункция почек характерна для таких заболеваний, как, например, острая почечная недостаточность, гломерулонефрит, хроническая почечная недостаточность, азотемия, уремия, иммунное почечное заболевание, острый нефритический синдром, быстро прогрессирующий нефритический синдром, нефротический синдром, болезнь Бергера, хронический нефритический/протеинурический синдром, тубулоинтерстициальное заболевание, нефротоксические расстройства, некроз почек, атероэмболическая болезнь почек, кортикальный некроз почек, злокачественный нефроангиосклероз, тромбоз почечной вены, почечный тубулярный ацидоз, почечная глюкозурия, нефрогенный несахарный диабет, синдром Барттера, синдром Лиддла, поликистозная болезнь почек, интерстициальный нефрит, острый гемолитический уремический синдром, ювенильный нефронофтиз, спонгиозная почка, наследственный нефрит и наследственная онихоартроостеодисплазия.
Дисфункция поджелудочной железы характерна для таких заболеваний, как, например, диабет, гипергликемия, нарушение толерантности к глюкозе и резистентность к инсулину.
Дисфункция костного мозга характерна для таких заболеваний, как, например, остеомиелит, нарушение кроветворения, ион-дефицитная анемия, пернициозная анемия, мегалобластоз, гемолитическая анемия и апластическая анемия.
Дисфункция мозжечка характерна для моторных и нейро-поведенческих расстройств, таких как, например, гипотония, дизартрия, дизметрия, дисдиадохокинезия, нарушение рефлексов и дрожание при произвольных движениях.
Фармацевтические композиции согласно настоящему изобретению можно вводить любым подходящим способом введения, выбранным из группы, состоящей из парентерального и перорального введения. Согласно некоторым вариантам реализации настоящего изобретения введение осуществляется посредством парентерального введения. В различных вариантах реализации настоящего изобретения способ введения представляет собой внутривенный, внутриартериальный, внутримышечный, подкожный, внутрибрюшинный, интрацеребральный, интрацеребровентрикулярный, интратекальный или внутрикожный путь введения. Например, фармацевтические композиции можно вводить системно, например, путем внутривенной (в/в) или внутрибрюшинной (в/б) инъекции или инфузии. Согласно определенному варианту реализации настоящего изобретения указанную фармацевтическую композицию вводят при помощи внутривенной инфузии в течение периода времени от 30 минут до 2 часов, например, в течение 1 часа. Композиции согласно настоящему изобретению можно вводить локально и они могут дополнительно содержать дополнительный активный агент и/или вспомогательное вещество.
Токсичность и терапевтическая эффективность соединений, описанных в настоящем документе, можно определять при помощи стандартных фармацевтических процедур в культурах клеток или у подопытных животных, например, путем определения IC50 (концентрация, обеспечивающая 50% ингибирование роста клеток) и МПД (максимальная переносимая доза у тестируемых животных) для требуемого соединения. Данные, полученные с помощью анализов клеточной культуры и исследований на животных, можно использовать при разработке диапазона доз для применения у людей. Точный состав, способ введения и дозировку может выбрать лечащий врач с учетом состояния пациента (см., например, Fingl, et al., 1975, "The Pharmacological Basis of Therapeutics", глава 1, стр. 1).
Соединение, например, астарабин®, можно вводить в суточной дозе в пределах от примерно 0,3 г/м2 до примерно 10 г/м2 площади поверхности тела субъекта. Согласно некоторым вариантам реализации настоящего изобретения соединение, например, астарабин®, можно вводить в суточной дозе в пределах от примерно 0,5 г/м2 до примерно 5 г/м2 площади поверхности тела субъекта. Согласно некоторым вариантам реализации настоящего изобретения соединение можно вводить в суточной дозе в пределах от примерно 0,5 г/м2 до примерно 4.5 г/м2 площади поверхности тела субъекта. Согласно некоторым вариантам реализации настоящего изобретения соединение, например, астарабин®, вводят в суточной дозе, составляющей примерно 0,3, 0,5, 0,8, 1, 1,5, 2, 2,3, 2,5, 3, 3,5, 4, 4,5, 5, 6, 7, 8, 9, 10 г/м2 площади поверхности тела субъекта. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно некоторым вариантам реализации настоящего изобретения астарабин® вводят при помощи внутривенной инфузии в суточной дозе в пределах от 0,3 г/м2 до 4,5 г/м2 площади поверхности тела субъекта.
Согласно некоторым вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят по меньшей мере один раз в месяц. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят по меньшей мере два раз в месяц. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят по меньшей мере один раз в неделю. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят по меньшей мере два раза в неделю. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере одной недели. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят по меньшей мере один раз в сутки в течение по меньшей мере одной недели или до тех пор, пока субъект не будет излечен.
Согласно некоторым вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере 2, 3, 4, 5, 6, 8, 10, 12 или по меньшей мере 14 последовательных дней один раз в месяц. Альтернативно указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере 2, 3, 4, 5, 6 или 12 дней два раза в месяц или, альтернативно, указанную фармацевтическую композицию вводят каждый день или два раза в неделю, пока пациент не будет излечен.
В некоторых вариантах реализации настоящего изобретения, в которых фармацевтическую композицию применяют для предотвращения рецидива рака, указанную фармацевтическую композицию можно регулярно вводить в течение продолжительных периодов времени в соответствии с инструкциями врача.
В некоторых случаях может быть целесообразным вводить большую ударную дозу с последующими периодическими (например, вводимыми каждую неделю) поддерживающими дозами в течение периода лечения. Соединения можно также доставлять при помощи систем доставки с медленным высвобождением, помпами и другими известными системами доставки для непрерывной инфузии. Режимы дозирования могут варьироваться для обеспечения желаемых уровней циркуляции конкретного соединения на основе его фармакокинетики. Таким образом, дозы рассчитывают так, чтобы поддерживать желаемый уровень циркуляции терапевтического агента.
Обычно эффективная доза определяется активностью и эффективностью соединения и состоянием субъекта, а также массой тела или площадью поверхности тела подлежащего лечению субъекта. Доза и режим дозирования также определяются наличием, характером и степенью любых неблагоприятных побочных эффектов, которые сопровождают введение соединений у конкретного субъекта.
Следующие примеры следует рассматривать просто как иллюстративные и не ограничивающие настоящее изобретение. Специалисту в данной области техники будет очевидно, что можно произвести множество модификаций, перестановок и вариантов, не выходящих за рамки объема настоящего изобретения.
ПРИМЕР 1
Действие астарабина (Astarabine®) на различные клеточные линии
Оценивали влияние астарабина® на различные клеточные линии. Вкратце, различные клеточные линии были получены от АТСС или ЕСАСС. Гематологические клетки выращивали в среде RPMI, содержащей 10-20% FBS и 1% глутамина. Клетки солидных опухолей выращивали в среде DMEM, содержащей 10-20% FBS и 1% глутамина. Клетки высевали в 96-луночные планшеты, 50000 клеток/мл, 0,2 мл на лунку. Тестируемые соединения разводили в физиологическом растворе или PBS и добавляли в конечных концентрациях от 0,1 нМ до 10 мкМ, объем 20 мкл. Исследование проводили в трех повторах, в качестве контроля применяли PBS. Планшеты инкубировали в течение 72 ч при температуре 37°С, 5% СО2. По окончании периода воздействия проводили анализ МТТ с использованием реагента МТТ [3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид]. МТТ добавляли в каждую лунку в концентрации 5 мг/мл, объем 0,02 мл. Планшеты инкубировали при температуре 37°С в течение 3 часов. Планшеты центрифугировали при 3500 об/мин в течение 5 минут и аспирировали супернатант. Каждую из пеллет, содержащих кристаллы МТТ, растворяли в 0,2 мл ДМСО. Абсорбцию определяли с использованием считывателя ELISA при длине волны 570 нм.
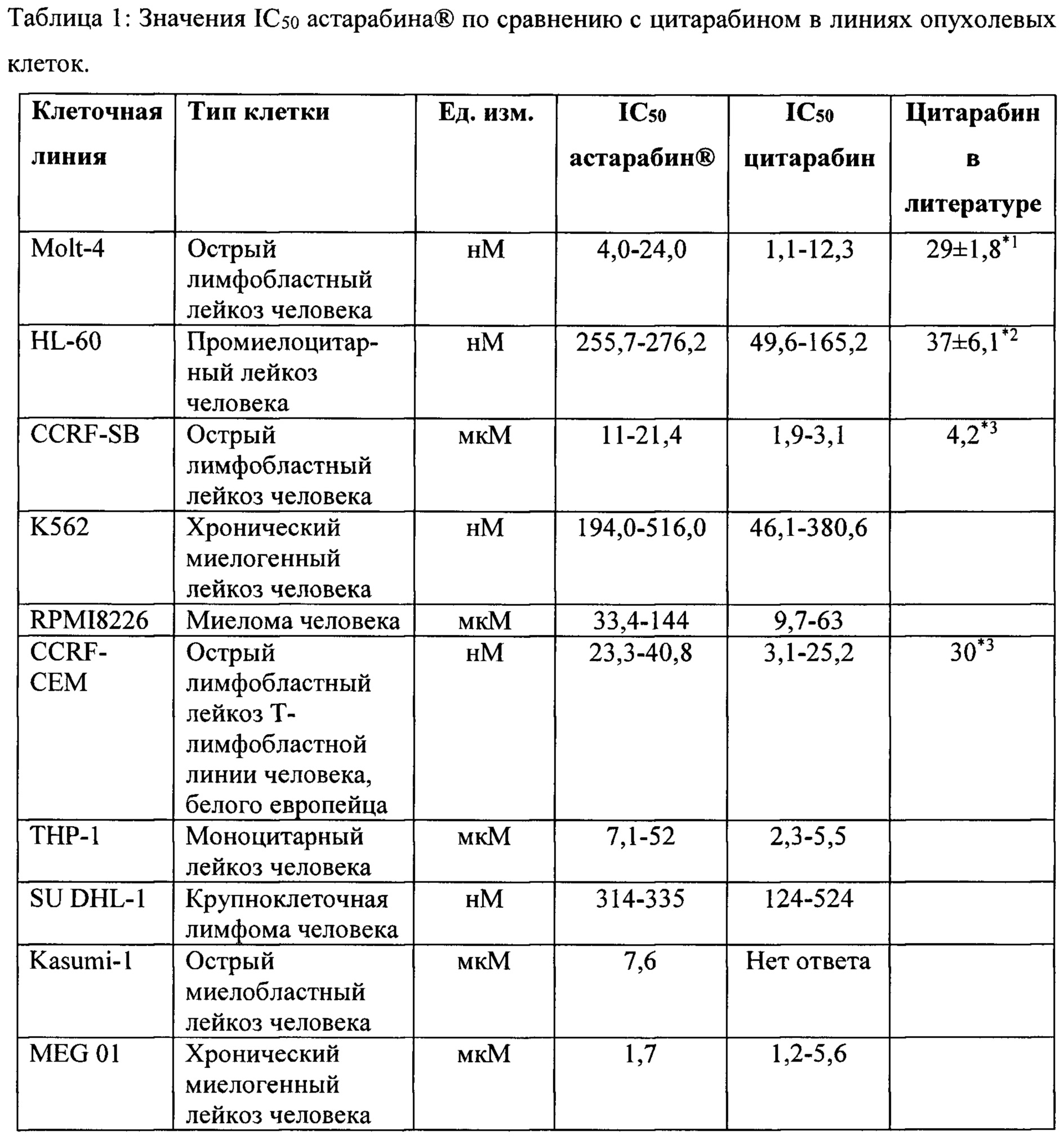
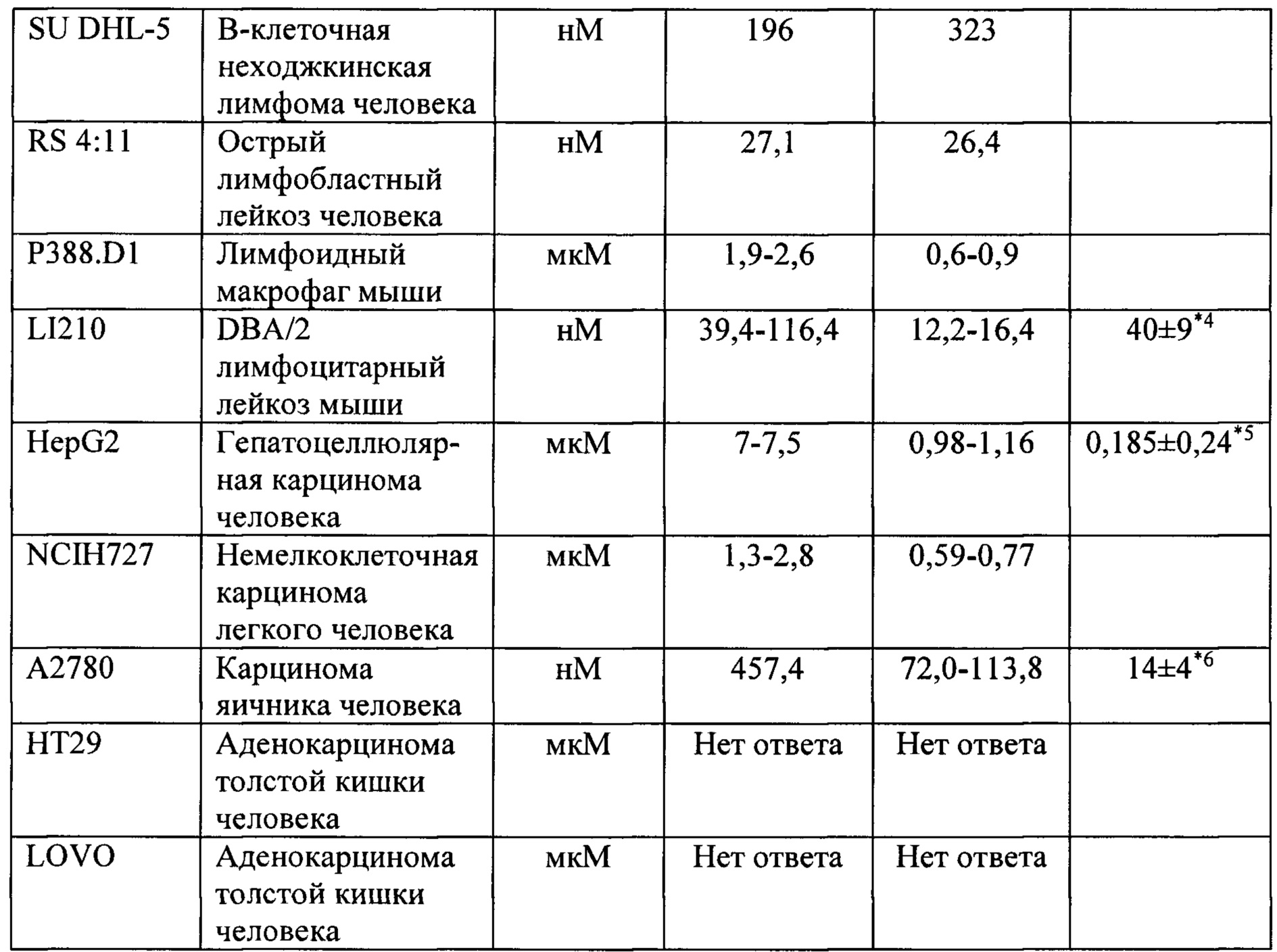
Результаты приведены в Таблице 1.
*1 - Ogbomo et al., Neoplasia 10(12): 1402-1410, 2008;
*2 - Qin et al., Clin, Cancer Res. 13(14): 4225-4232, 2007;
*3 - Manfredini et al., Bioorg. Med. Chem. 8: 539-547, 2000;
*4 - Breistol et al., Cancer Res. 59: 2944-2949, 1999;
*5 - Groveet al., Cancer Res. 55: 3008-3011, 1995;
*6 - Ruiz Van Haperen, Cancer Res. 54: 4138-4143, 1994.
Результаты показали, что большинство анализируемых клеточных линий опухолей системы крови чувствительны к астарабину®, тогда как клеточные линии солидных опухолей менее чувствительны.
При помощи анализа метаболитов астарабина® в клеточных линиях лейкоза человека были обнаружены свободные метаболиты цитарабина, что указывает на то, что астарабин® является пролекарством цитарабина.
ПРИМЕР 2
Максимальная переносимая доза и эффективность Астарабина® у мышей
Максимально переносимую дозу (МПД) астарабина® вводили одной или несколькими инъекциями внутрибрюшинно (в/б) или внутривенно (в/в).
Единичная доза в/б: Мышам IRC в возрасте 10 недель в/б вводили однократную дозу астарабина® в диапазоне от 1428 до 8271 мг/кг (соответственно, от 50 до 300 мг/мышь). Клиническая оценка включала наблюдения за подвижностью и потреблением пищи и воды. Поведение мышей во всех группах было нормальным. Через 7 дней мышей отбирали для общего анализа крови и биохимического анализа, а затем подвергали вскрытию.
Для образцов крови мышей, которым вводили высокую дозу астарабина® (8271 мг/кг), были получены нормальные результаты после восстановительного периода 7 дней. Все ткани у всех мышей были указаны как нормальные, без каких-либо отклонений, обнаруженных макроскопически. Концентрация астарабина® 8271 мг/кг, введенная внутрибрюшинно, считалась как МПД астарабина® острого токсического действия при однократном введении в/б.
Повторная доза в/б: Астарабин® в/б вводили IRC мышам в течение 7 дней в повышенных дозах от 156 до 781 мг/кг/сутки (от 5 до 25 мг/мышь/сутки, соответственно).
Всех мышей проверяли ежедневно во время введения лекарственного средства и в восстановительный период. Клиническая оценка включала наблюдения за подвижностью и потреблением пищи и воды. Поведение мышей во всех группах было нормальным во время периода введения и в восстановительный период. Общий и биохимический анализы крови в конце эксперимента были нормальными. Общие вскрытия не показали аномальных результатов.
Концентрация астарабина® 781 мг/кг/сутки в течение 7 дней, т.е. кумулятивная доза 5467 мг/кг, рассматривалась как МПД (максимальная переносимая доза) у мышей.
Единичная доза в/в: Мышам IRC в/в однократно вводили астарабин® в дозе от 1000 до 8000 мг/кг. За мышами наблюдали в течение 14 дней. Результаты показали, что астарабин® в дозе 8000 мг/кг переносили самцы и самки мышей ICR. При этой дозе, т.е. 8000 мг/кг, следующие параметры были в норме: клинические признаки, масса тела, потребление пищи и анализ крови. При этом уровне дозы не было смертей, относящихся к лекарственному средству. На этом основании сделан вывод, что 8000 мг/кг является МПД астарабина® у мышей при однократном в/в введении.
Повторная доза в/в: Мышам IRC ежедневно в/в вводили астарабин® в течение 14 дней в дозировке от 400 мг/кг/сутки до 1200 мг/кг/сутки. Основываясь на результатах этого 14-дневного исследования повторяющихся доз, вводимых в/в, было сделано заключение, что максимальная переносимая доза для астарабина в/в составляет 400 мг/кг/сутки, вводимая в течение 14 последовательных дней. Общая введенная доза для этой группы составляла 5600 мг/кг. Все измеренные параметры, такие как клинические наблюдения, нейроповеденческие функции, двигательная активность, масса тела, потребление пищи, анализ крови и анализ мочи, находились в нормальном диапазоне по сравнению с группой мышей, не получавших лечение. Таким образом, астарабин® в концентрации 400 мг/кг/сутки был принят за уровень, не вызывающий вредного воздействия (NOAEL) у мышей. Результаты исследований переносимости/токсичности in vivo для определения МПД для однократной дозы у мышей приведены на фиг. 1А и для повторных доз у мышей приведены на фиг. 1В.
Затем изучали эффективность астарабина® in vivo в модели с полыми волокнами (модели Molt-4 и CCRF-CEM), трансплантированными бестимусным мышам. Астарабин® вводили в дозах 6,25 мг/мышь/сутки и 25 мг/мышь/сутки внутрибрюшинно (в/б) ежедневно в течение 7 дней подряд; цитарабин вводили в дозе 1,5 мг/мышь/сутки. Рост клеток лейкоза измеряли с использованием МТТ анализа, как описано в примере 1 выше.
Результаты показали, что астарабин® в дозе 6,25 мг/мышь/сутки и 25 мг/мышь/сутки или цитарабин в дозе 1,5 мг/мышь/сутки ингибировал рост клеток Molt-4 на 76,9%, 82,8% и 81,1%, соответственно, в модели лейкоза Molt-4 с полыми волокнами in vivo. Подобно этому, астарабин® в дозе 6,25 мг/мышь/сутки и 25 мг/мышь/сутки или цитарабин в дозе 1,5 мг/мышь/сутки ингибировал рост клеток CCRF-CEM на 71,9%, 72,0% и 73,3%, соответственно, в модели лейкоза CCRF-CEM in vivo. Эти результаты показывают терапевтическую эффективность астарабина® против клеток лейкоза человека CCRM-CEM и Molt-4. Эффективность астарабина® была аналогична эффективности цитарабина. На фиг. 2А показан ответ в модели лейкоза Molt-4. На фиг. 2В показан ответ в модели лейкоза CCRF-CEM.
Результаты показывают, что астарабин® обладает улучшенной переносимостью лекарственного средства и сниженной в 10-20 раз токсичностью у животных по сравнению с приведенной в литературе токсичностью цитарабина, и сохраняет при этом свою противоопухолевую активность. Следовательно, астарабин® может служить более безопасной, нетоксичной альтернативой цитарабину для лечения различных лейкозов.
ПРИМЕР 3
Состав и введение астарабина
Лекарственное средство астарабин® поставляется в виде стерильного лиофилизированного порошка в стеклянных ампулах по 1 г на ампулу. Указанное лекарственное средство растворяли в 10 мл стерильной воды для инъекций с получением конечной концентрации астарабина® 100 мг/мл. После полного растворения в воде лекарственное средство дополнительно разбавляли инъекционным/инфузионным раствором. Вводимая доза рассчитывалась в г/м2 в зависимости от возраста и состояния пациента.
A. Дозировка лекарственного средства 0.5 г/м2:
Дозу для введения для пациента рассчитывали по следующей формуле: Дозировка лекарственного средства в г/м2 × площадь поверхности тела (ППТ) пациента в м2 = дозировка, вводимая пациенту в граммах (г).
Пример расчета: 0,5 г/м2 × 1,72 м2=0,86 г (860 мг).
Таким образом, 8,6 мл (860 мг) раствора 100 мг/мл астарабина® добавляли к 500 мл стерильного буферизованного физиологического раствора для инъекций, например, лактата Рингера, 273 мОсм/л, рН=6,5. Указанный состав астарабина® вводили пациенту путем инфузии в течение 1 часа.
B. Дозировка лекарственного средства 4,5 г/м2.
Пример расчета: 4,5 г /м2 × 1,65 м2=7,425 г
74,25 мл (7,425 г) раствора 100 мг/мл астарабина® добавляли к 500 мл стерильного буферизованного физиологического раствора для инъекций, например, Plasma-Lyte-A, 294 мОсм/л, рН=7,4, и вводили пациенту путем инфузии в течение 1 часа.
ПРИМЕР 4
Эффективность астарабина® у пожилых пациентов
Было проведено клиническое исследование для оценки эффективности и безопасности применения астарабина® у пациентов с ОМЛ и ОЛЛ.
Дизайн исследования:
Открытое одноцентровое клиническое исследование фазы I/IIa без контрольной группы включало пациентов в возрасте 18 или более лет с рецидивным или рефрактерным острым лейкозом, или пациентов, не подлежащих интенсивной терапии по оценке лечащего врача. Исследование было одобрено Rambam IRB (разрешение №0384-11).
Пациентов разделили на 4 когорты возрастающих доз астарабина®, каждая когорта включала 3 пациента. Лечение проводилось как одночасовая однодневная инфузия в течение 6 последовательных дней.
Дозы астарабина® для каждой инфузии:
- Для возраста≤50 лет: 0,5 г/м2, 1,5 г/м2, 3 г/м2, 4,5 г/м2
- Для возраста≥50 лет: 0,3 г/м2, 0,8 г/м2, 1,5 г/м2, 2,3 г/м2
Результаты
Результаты по 10 пациентам представлены в таблице 2. У девяти пациентов был ОМЛ, из них у пяти пациентов был рецидивный/рефрактерный ОМЛ, а у четырех пациентов недавно был диагностирован вторичный ОМЛ, непригодный для интенсивной терапии. У одного пациента недавно диагностировали ОЛЛ. Средний возраст составлял 80 лет.
В конце 10-месячного периода:
Четыре пациента были живы, двое из которых были в непрерывной полной ремиссии (CR) через 7 и 9 месяцев после лечения.
Четыре пациента умерли от прогрессирования заболевания, один умер внезапно через 7 дней после проведения лечения, считается, что это событие не связано с лечением. Никаких значительных неблагоприятных явлений во время или после терапии не отмечалось, кроме повышения температуры тела у пациентов с нейтропенией.
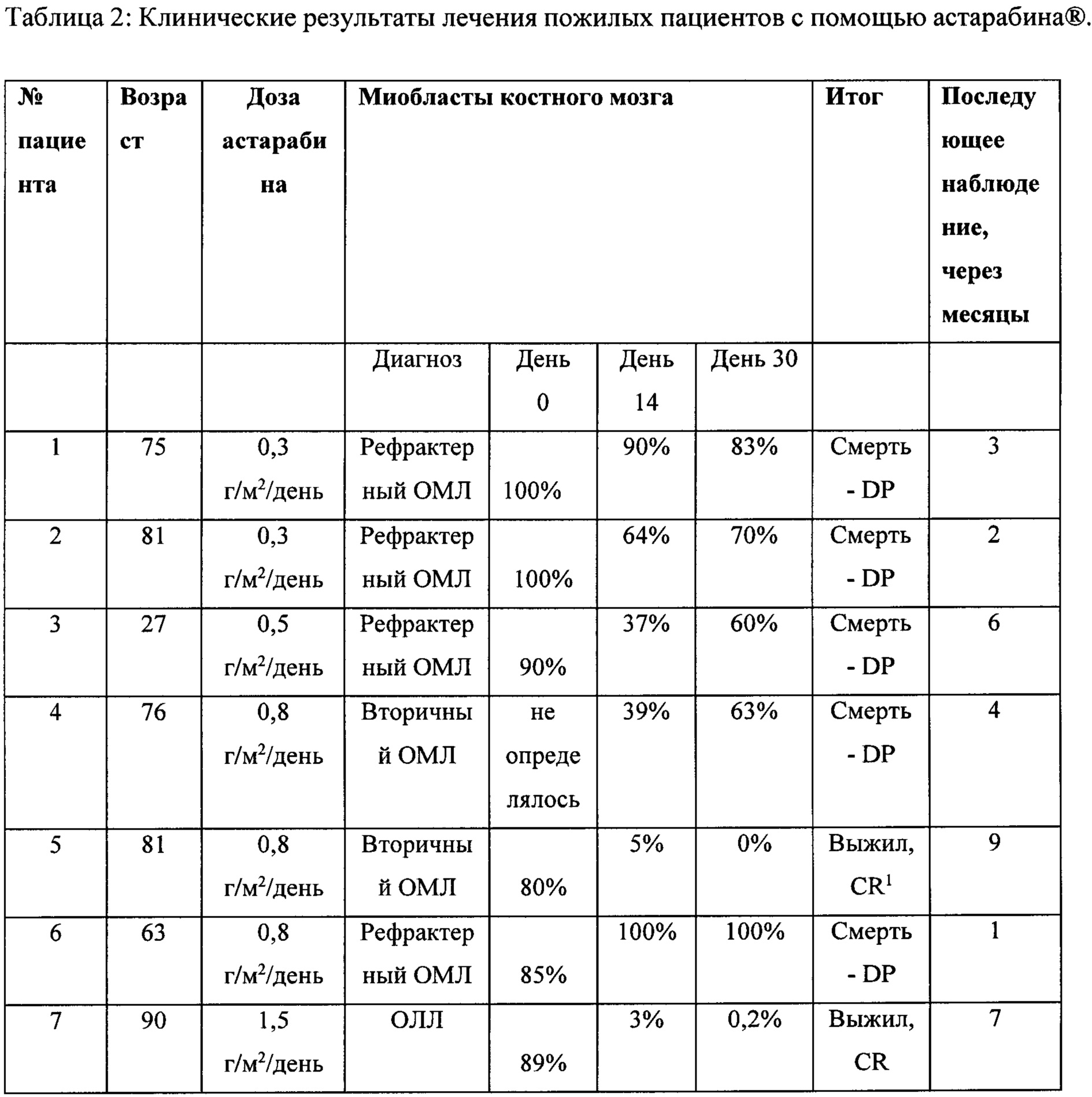
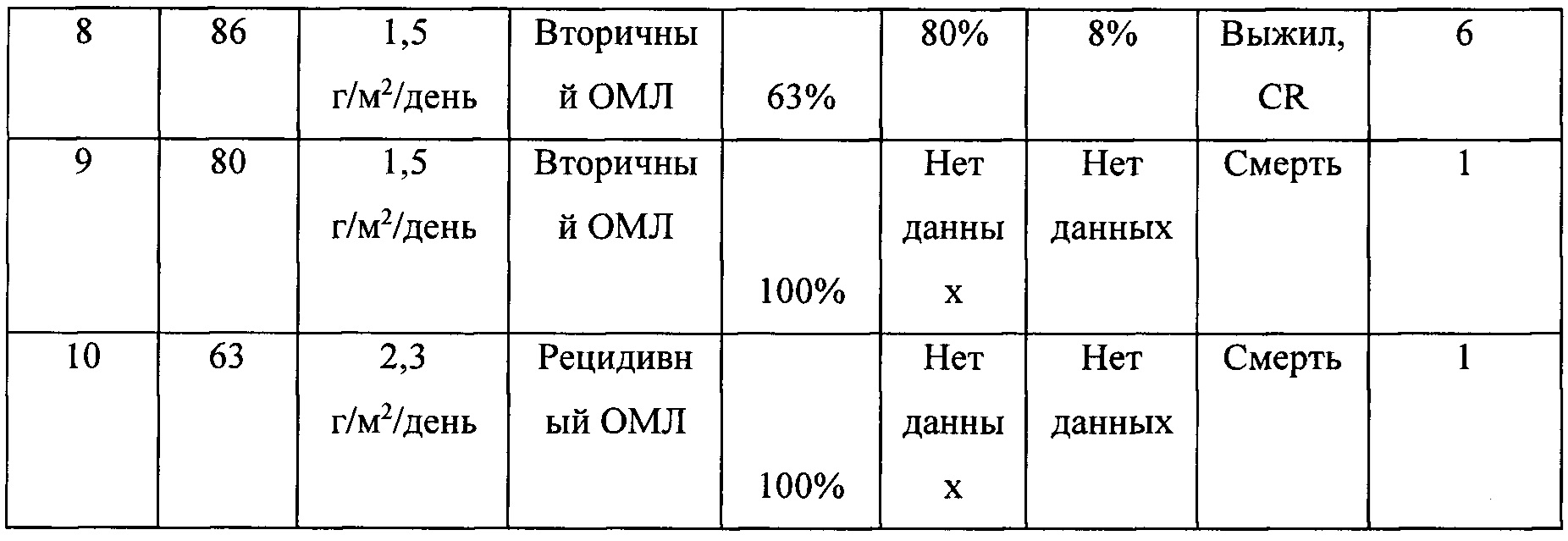
* Каких-либо значительных нежелательных явлений, связанных с лекарственным средством, не наблюдалось.
PR - частичная ремиссия; CR - полная ремиссия; DP - прогрессирование заболевания;
1 рецидив через 9 месяцев CR; планируется следующий цикл астарабина®.
Результаты показывают, что астарабин®, пролекарство цитарабина, безопасен и хорошо переносится даже пациентами в возрасте от 80 и более лет. Примечательно, что введение астарабина® приводило к ремиссии у трех из десяти пациентов с острым лейкозом, и указанное соединение было особенно эффективным у пациентов, у которых впервые диагностировали лейкоз.
Специалистам в данной области техники понятно, что настоящее изобретение не ограничено настоящим описанием. Скорее, объем настоящего изобретения включает в себя как комбинации, так и подкомбинации различных признаков, описанных выше, а также их изменения и модификации. Следовательно, настоящее изобретение не ограничено описанными вариантами реализации настоящего изобретения, и объем и суть изобретения будут более понятны из прилагаемой формулы изобретения.