Результат интеллектуальной деятельности: СПОСОБ ОБРАБОТКИ ВОДНОГО АЗОТНОКИСЛОГО РАСТВОРА, ПОЛУЧЕННОГО ПРИ РАСТВОРЕНИИ ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА, ВЫПОЛНЯЕМЫЙ В ОДНОМ ЦИКЛЕ И НЕ ТРЕБУЮЩИЙ КАКОЙ-ЛИБО ОПЕРАЦИИ, ВКЛЮЧАЮЩЕЙ ВОССТАНОВИТЕЛЬНУЮ РЕЭКСТРАКЦИЮ ПЛУТОНИЯ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Изобретение относится к способу обработки водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте, позволяющему экстрагировать, разделять и очищать от загрязнителей уран и плутоний, содержащиеся в этом растворе, в одном цикле, и не прибегая к операции восстановительной реэкстракции плутония.
Данный способ найдет применение в переработке отработавшего ядерного топлива, содержащего уран, особенно оксиды урана - UOX, или оксиды урана и плутония, в частности, смешанные оксиды урана и плутония - МОХ.
Предшествующий уровень техники
Пурекс-процесс (PUREX), применяющийся на всех заводах по переработке отработавшего ядерного топлива во всем мире (La Hague во Франции, Rokkasho в Японии, Sellafield в Соединенном Королевстве, и т.д.) использует три-н-бутил фосфат (или ТБФ) в качестве экстрагента для извлечения урана и плутония путем жидкостно-жидкостной экстракции из водных растворов, полученных при растворении этих видов топлива в азотной кислоте.
В этом способе ТБФ применяют в 30 об.% (об./об.) растворе в органическом разбавителе (гидрированном тетрапропилене (или ТПГ) или н-додекане). Этот органический раствор обычно называют «растворителем» в области, к которой относится изобретение.
Извлечение урана и плутония в PUREX процессе проводят в несколько циклов:
- первый цикл очистки урана и плутония (называемый «1CUPu», предназначенный для очистки урана и плутония от америция, кюрия и продуктов деления, с разделением урана и плутония на два водных потока в данном первом цикле посредством восстановительной реэкстракции плутония;
- второй цикл очистки урана (называемый «2CU»), предназначенный для полной очистки от загрязнителей урана для достижения требований, изложенных в стандартах ASTM (Американского общества по испытанию материалов) для конечного продукта урана; и
- второй, и на некоторых заводах третий цикл очистки плутония (соответственно, называемый «2CPu» или «3CPu»), предназначенный для полной очистки от загрязнителей плутония для достижения требований, изложенных в стандартах ASTM для конечного продукта урана для его концентрирования перед преобразованием в оксид.
Уровни эффективности PUREX процесса являются удовлетворительными, и отзывы об опыте, приобретенном с момента запуска установок, использующих этот способ, являются положительными.
Однако применение ТБФ имеет пределы, ограничивающие возможность достижения с этим экстрагентом целей простоты, компактности и повышенной безопасности, установленных для будущих заводов по переработке отработавшего ядерного топлива, в частности, направленных на устранение циклов 2CU, 2CPu и 3CPu из PUREX процесса.
Эти пределы являются следующими:
- коэффициенты очистки урана и плутония от некоторых продуктов деления (технеция и рутения) и трансурановых элементов (Np) являются недостаточными в конце первого цикла очистки, следовательно, невозможно достичь с ТБФ схемы, приводящей к конечным продуктам, удовлетворяющим вышеупомянутым требованиям, в одном цикле;
- разделение урана и плутония на два водных потока требует восстановления плутония (IV) в плутоний (III) (поскольку с ТБФ коэффициент разделения урана (VI) и плутония (IV) является недостаточным, независимо от кислотности водного раствора, используемого для достижения такого разделения), и в результате требует использования высоких количеств восстанавливающих и анти-азотнокислых агентов, которые генерируют нестабильные и активные формы при деградации, и по этой причине ограничивают применение с точки зрения безопасности;
- продукты деградации ТБФ оказывают влияние на уровни производительности процесса; в частности, ди-н-бутил-фосфат (или ДБФ) приводит к образованию комплексов металлов, некоторые из которых являются нерастворимыми и могут вызывать удерживание плутония в растворителе; следовательно, необходимо проводить операцию, известную как «барьер Pu», которая осуществляется после восстановительной реэкстракции плутония, и которая предназначена для завершения этой реэкстракции;
- риск образования 3-й фазы, индуцированной присутствием плутония, ограничивает осуществление схемы концентрирования плутония (риск критичности) или схемы, обеспечивающей переработку отработавшего ядерного топлива с высоким содержанием плутония, такого как МОХ топливо, полученное из легководных реакторов или реакторов на быстрых нейтронах;
- реэкстракция урана из растворителя, в который он был предварительно экстрагирован, является неполной, если её проводят при комнатной температуре, следовательно, необходимо выполнить эту реэкстракцию при температуре 50°С (что соответствует максимальной температуре, допускаемой точкой воспламенения растворителя); однако, даже при этой температуре реэкстракцию урана проводят при разбавлении (отношение органического/водного потоков (О/В) составляет меньше 1);
- растворимость ТБФ, которая является довольно большой в водной фазе (до 300 мг/л, в зависимости от кислотности водной фазы), требует промывания органическим разбавителем водной фазы из-за разницы циклов экстракции для восстановления ТБФ, солюбилизированного в этих водных фазах; и
- сжигание отработанного ТБФ и его продуктов деградации приводит к образованию вторичных отходов, включающих твердые фосфатные остатки.
Таким образом, в перспективе будущих заводов по переработке отработавшего ядерного топлива, которые являются более простыми и более компактными, чем современные заводы, и кроме того, обладают повышенной безопасностью, авторы изобретения разработали способ, который при выполнении также PUREX процесса для извлечения и очистки от загрязнителей урана и плутония, содержащихся в водных азотнокислых растворах, полученных при растворении отработавшего ядерного топлива, позволяет преодолеть все ограничения, связанные с применением ТБФ в качестве экстрагента, и который, в частности, включает только один производственный цикл и не содержит какой-либо операции для восстановительной реэкстракции плутония.
Изложение сущности изобретения
Эта задача решается изобретением, которое предлагает способ переработки в одном цикле водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте, где водный раствор содержит уран, плутоний, америций, кюрий и продукты деления, включая технеций, где цикл включает:
(а) совместную экстракцию урана и плутония из водного раствора, включающую по меньшей мере один контакт в экстракторе указанного водного раствора с органическим растворителем, включающим от 1 моль/л до 2 моль/л N,N-ди(2-этилгексил)-3,3-диметилбутанамида или смеси N,N-ди(2-этилгексил)-изобутанамида и N,N-ди(2-этилгексил)-н-бутанамида в качестве экстрагента в растворе в органическом разбавителе, с последующим разделением водного и органического растворов;
(b) очистку органического раствора, полученного на этапе (а), от америция, кюрия и продуктов деления, включающую по меньшей мере одно контактирование в экстракторе указанного органического раствора с водным раствором, включающим от 0,5 моль/л до 6 моль/л азотной кислоты, с последующим разделением органического и водного растворов;
(с) разделение урана и плутония, содержащихся в органическом растворе, полученном на этапе (b), на водный раствор и органический раствор, где водный раствор содержит либо плутоний без урана, либо смесь плутония и урана, а органический раствор содержит уран без плутония, где это разделение включает:
(с1) реэкстракцию плутония в степени окисления +IV и фракции урана из органического раствора, полученного на этапе (b), включающую по меньшей мере одно контактирование в экстракторе органического раствора с водным раствором, содержащим от 0,1 моль/л до 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов;
(с2) экстракцию всей или части урановой фракции, содержащейся в водном растворе, полученном на этапе (с1), включающую по меньшей мере одно контактирование в экстракторе водного раствора, полученного на этапе (с1), с органическим раствором, идентичным органическому раствору, используемому на этапе (а), с последующим разделением водного и органического растворов;
(d) очистку органического раствора, полученного на этапе (с1), от технеция, включающую:
(d1) реэкстракцию технеция в степени окисления +IV из органического раствора, полученного на этапе (с1), включающую по меньшей мере одно контактирование в экстракторе этого органического раствора с водным раствором, содержащим от 0,1 моль/л до 3 моль/л азотной кислоты и по меньшей мере один восстанавливающий агент, способный восстанавливать технеций из степени окисления +VII до степени окисления +IV, или комплексообразующий агент, способный к стабилизации технеция в водной фазе, с последующим разделением органического и водного растворов;
(d2) экстракцию урановой фракции, содержащейся в водном растворе, полученном на этапе (d1), включающую по меньшей мере одно контактирование в экстракторе указанного водного раствора с органическим раствором, идентичным органическому раствору, используемому на этапе (а), с последующим разделением водного и органического растворов;
(е) реэкстракцию урана из органического раствора, полученного на этапе (d1), включающую по меньшей мере одно контактирование в экстракторе органического раствора, полученного на этапе (d1), с водным раствором, содержащим не более 0,05 моль/л азотной кислоты, с последующим разделением органического и водного растворов; и
(f) регенерацию органической фазы, полученной на этапе (е);
где получают первый и второй водные растворы, очищенные от америция, кюрия и продуктов деления, включая технеций, где первый водный раствор включает плутоний без урана или смесь плутония и урана, а второй водный раствор включает уран без плутония.
Таким образом, способ настоящего изобретения основан на применении в качестве экстрагента определенного N,N-диалкиламида или смеси двух определенных N,N-диалкиламидов, где эти определенные N,N-диалкиламиды выбраны из:
- N,N-ди(2-этилгексил)-3,3-диметилбутанамида (или DEHDMBA) формулы: (CH3)3-C-CH2-C(O)-N-(CH2-CH(C2H5)C4H9)2;
- N,N-ди(2-этилгексил)-изобутанамида (или DEHiBA) формулы: (CH3)2-CH-C(O)-N-(CH2-CH(C2H5)C4H9)2; и
- N,N-ди(2-этилгексил)-н-бутанамида (или DEHBA) формулы: C3H7-C(O)-N-(CH2-CH(C2H5)C4H9)2.
Необходимо отметить, что N,N-диалкиламиды (также называемые «моноамидами») представляют семейство экстрагентов, которые в значительной степени изучались в качестве возможной альтернативы ТБФ для переработки отработавшего ядерного топлива. Впервые разработанное в США в 1950-х, это семейство экстрагентов позже стало предметом исследований для различных ученых в Европе, Индии, Японии и Китае в 1980-х. Три французских заявки (FR-A-2591213, FR-A-2642561 и FR-A-2642562, далее упомянутые как ссылки [1], [2] и [3]), относящиеся к применению N,N-диалкиламидов в качестве экстрагентов для переработки отработавшего ядерного топлива, были поданы в 1980-х, и одна из них, а именно ссылка [1], предусматривает возможное разделение урана и плутония без выполнения восстановительной реэкстракции плутония с применением N,N-диалкиламидов, разветвленных на их карбонильной функциональной стороне.
Однако, как известно авторам изобретения, в литературе нет данных о том, что использование в качестве экстрагентов подходящим образом выбранных N,N-диалкиламидов может позволить разработать способ переработки отработавшего ядерного топлива, который, включая только один цикл и не включая какой-либо операции восстановительной реэкстракции плутония, действует так же, как PUREX процесс с точки зрения извлечения и очистки от загрязнений (дезактивации) урана и плутония, содержащихся в водных азотнокислых растворах, полученных при растворении этого топлива. Тем более, указанный способ никогда не предлагался в литературе.
В вышеизложенном тексте и далее термины «водный раствор» и «водная фаза» являются эквивалентными и применяются взаимозаменяемо, и подобным образом, термины «органический раствор» и «органическая фаза» являются эквивалентными и применяются взаимозаменяемо.
Далее, выражения «от … до …», «в диапазоне от … до …» и «между … и …» являются эквивалентными и используются для того, чтобы показать, что указанные пределы включены.
В соответствии с изобретением, органический раствор, используемый на этапе (а) и следовательно, те, которые используются на этапах (с2) и (d2), поскольку органические растворители, используемые на этапах (а), (с2) и (d2), имеют один и тот же состав, предпочтительно включают от 1,3 моль/л до 1,4 моль/л, и более предпочтительно 1,35 моль/л DEHDMBA, или также от 1,35 моль/л до 1,45 моль/л, и еще более предпочтительно 1,4 моль/л смеси DEHiBA и DEHBA, в этом случае молярное отношение DEHiBA/DEHBA предпочтительно составляет от 1,75 до 1,85, и более предпочтительно 1,80.
Особо предпочтительно, органический раствор, используемый на этапе (а), и следовательно, на этапах (с2) и (d2), включает 0,9 моль/л DEHiBA и 0,5 моль/л DEHBA.
Как указывалось ранее, водный раствор, используемый на этапе (b), может включать от 0,5 моль/л до 6 моль/л азотной кислоты.
Однако предпочтительно, чтобы этот водный раствор содержал от 4 моль/л до 6 моль/л азотной кислоты для облегчения реэкстракции рутения и технеция из органического раствора, полученного на этапе (а). В этом случае этап (b) предпочтительно также включает нейтрализацию (уменьшение кислотности) органического раствора, включающую по меньшей мере одно контактирование органического раствора с водным раствором, содержащим от 0,1 моль/л до 1 моль/л, и предпочтительно 0,5 моль/л азотной кислоты, с последующим разделением органического и водного растворов.
В соответствии с изобретением, контактирование органического и водного растворов в экстракторе, в котором осуществляют этап (с1), включает циркуляцию этих растворов при отношении потоков О/В, предпочтительно составляющем больше 1, предпочтительно 3 или больше, и более предпочтительно 5 или больше, для достижения концентрирующей реэкстракции плутония, т.е. реэкстракции плутония, приводящей к водному раствору, в котором концентрация плутония превышает концентрацию этого элемента в органическом растворе, из которого его реэкстрагируют.
Восстанавливающий агент (агенты) в водном растворе, используемом на этапе (d1), предпочтительно выбирают из нитрата урана (также называемого «U (IV)», гидразиния нитрата (также называемого «гидразина нитрат»), гидроксиламмония нитрата (также называемого «гидроксиламина нитрат»), ацетальдоксима и их смесей, таких как смесь урана нитрата и гидразиния нитрата, смесь урана нитрата и гидроксиламмония нитрата, или смесь урана нитрата и ацетальдоксима, предпочтительно смесь урана нитрата и гидразиния нитрата, или смесь урана нитрата и гидроксиламмония нитрата, которые предпочтительно применяются в концентрации в диапазоне от 0,1 моль/л до 0,3 моль/л, и обычно 0,2 моль/л.
Кроме того, этап (d1), который можно проводить при комнатной температуре, предпочтительно проводят, однако, при температуре в диапазоне от 30 до 40°С, и предпочтительно при 32°С для стимуляции кинетики реэкстракции технеция, при этом способствуя ограничению феномена повторного окисления данного элемента в водной фазе. Таким образом, экстрактор, в котором осуществляют этап (d1), предпочтительно нагревают до температуры от 30°С до 40°С.
В соответствии с изобретением, предпочтительно этап (d2) дополнительно включает подкисление водного раствора, полученного на этапе (d1), где это подкисление включает добавление азотной кислоты в экстрактор, в котором проводят этап (d2), для доведения концентрации азотной кислоты до значения по меньшей мере 2,5 моль/л.
Этап (е) можно проводить при комнатной температуре. Однако его предпочтительно проводить при температуре в диапазоне от 40°С до 50°С, также для стимуляции реэкстракции урана. Таким образом, экстрактор, в котором осуществляют этап (е), предпочтительно нагревают до температуры от 40°С до 50°С.
Независимо от температуры, при которой проводят этап (е), контактирование органического и водного растворов в экстракторе, в котором осуществляют этап (е), включает циркуляцию этих растворов с отношением потоков О/В больше 1, так чтобы достичь концентрирующей реэкстракции урана, т.е. реэкстракции урана, приводящей к водному раствору, в котором концентрация урана превышает концентрацию этого элемента в органическом растворе, из которого его реэкстрагируют.
Как указано ранее, способ настоящего изобретения дополнительно включает этап (f) для регенерации органического раствора, полученного на этапе (е), где эта регенерация предпочтительно включает по меньшей мере одно промывание органического раствора основным водным раствором, с последующим по меньшей мере одним промыванием органического раствора водным раствором азотной кислоты.
Предпочтительно, органический раствор, полученный на этапе (f), разделяют на первую и вторую фракцию, где первая фракция образована органическим раствором с этапа (а), а вторая фракция образована органическим раствором с этапа (с2).
Способ из настоящего изобретения, в дополнение к тому, что уже упомянуто, имеет следующие преимущества:
- реэкстракцию урана проще проводить, чем с помощью PUREX процесса, поскольку её можно выполнять как при комнатной температуре, так и при нагревании, с применением отношения потоков О/В больше 1, таким образом, обеспечивая концентрирующую реэкстракцию урана, которая невозможна для PUREX процесса;
- благодаря тому, что не проводится какой-либо реакции восстановления плутония, и таким образом, устраняется какой-либо риск повторного окисления плутония, реэкстракцию плутония также легче осуществлять, чем при PUREX процессе, и можно проводить с большим концентрированием, чем для этого последнего способа; важность этих преимуществ еще более велика, так как в будущем заводам по переработке отработавшего ядерного топлива придется перерабатывать топливо с более высоким содержанием плутония (такое, как МОХ топливо из легководных реакторов или реакторов на быстрых нейтронах), чем топливо, перерабатываемое в настоящее время;
- продукты деградации (посредством гидролиза и радиолиза) N,N-диалкиламидов являются менее проблематичными, чем продукты деградации ТБФ, поскольку они являются водорастворимыми и не образуют комплексов, которые могут удерживать плутоний;
- N,N-диалкиламиды, как правило, имеют растворимость в водной фазе в 100-200 раз ниже растворимости ТБФ, и таким образом, можно предусмотреть отмену или по меньшей мере снижение числа промываний в органическом разбавителе водных растворов, полученных в результате способа по настоящему изобретения, по сравнению с таким числом, представленным в PUREX процессе;
- поскольку N,N-диалкиламиды и продукты их деградации включают только атомы углерода, водорода, кислорода и азота, они полностью поддаются сжиганию, и таким образом, не производят обременительных вторичных отходов, в отличие от ТБФ и его продуктов деградации.
Другие характеристики и преимущества изобретения станут понятными из следующего дополнительного описания, со ссылкой на прилагаемые фигуры.
Однако это дополнительное описание приведено только для иллюстрации предмета обсуждения изобретения, и ни при каких обстоятельствах не должно рассматриваться как ограничивающее этот предмет обсуждения.
Краткое описание фигур
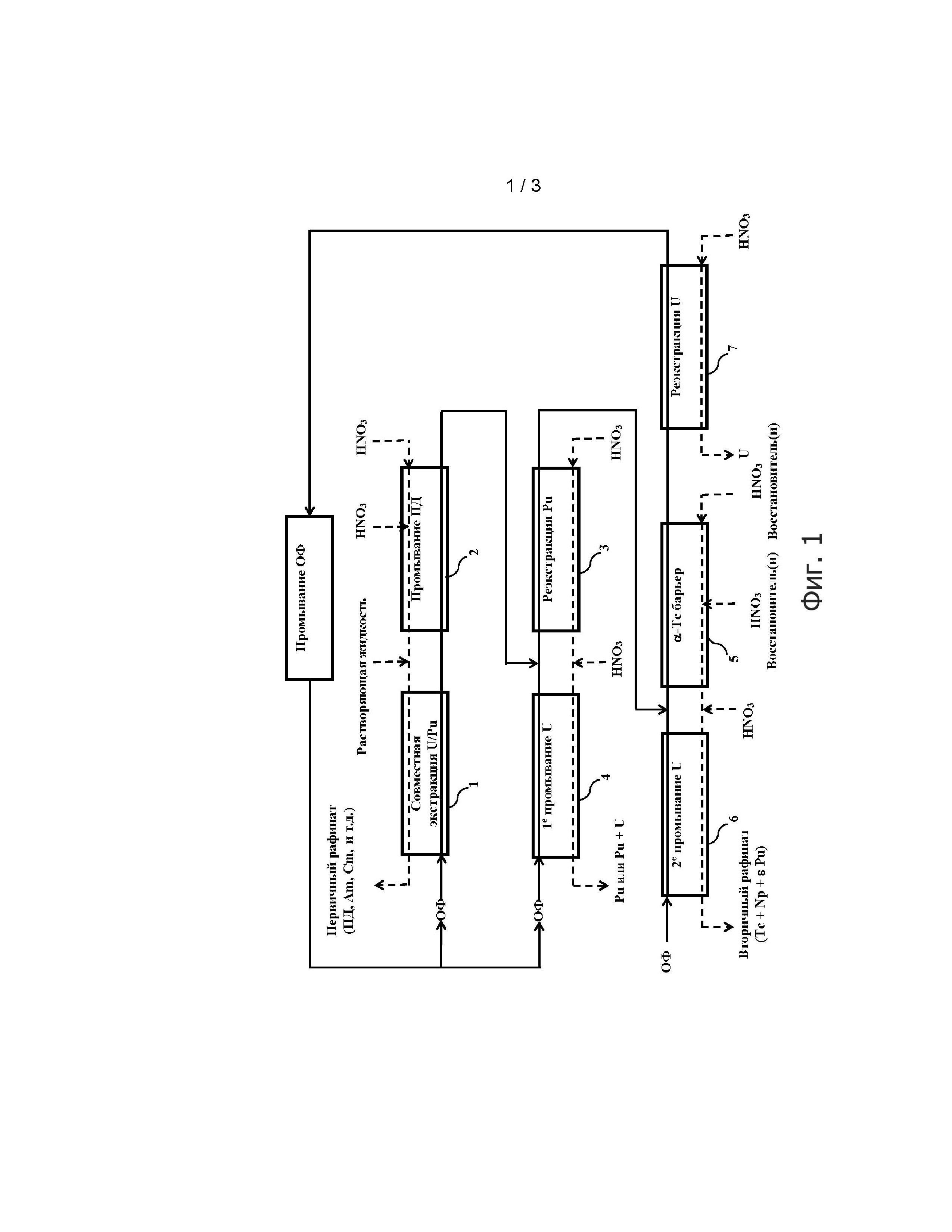
Фигура 1 демонстрирует поточную диаграмму способа настоящего изобретения; на этой фигуре прямоугольники 1-7 представляют многоступенчатые экстракторы, такие как те, которые обычно применяются для переработки отработавшего ядерного топлива (смесители-отстойники, пульсационные колонны или отжимные центрифуги).
Фигура 2 схематически иллюстрирует условия установки и эксплуатации, используемые для теста, предназначенного для валидации этапа «α-Tc барьер» способа настоящего изобретения в экстракторе.
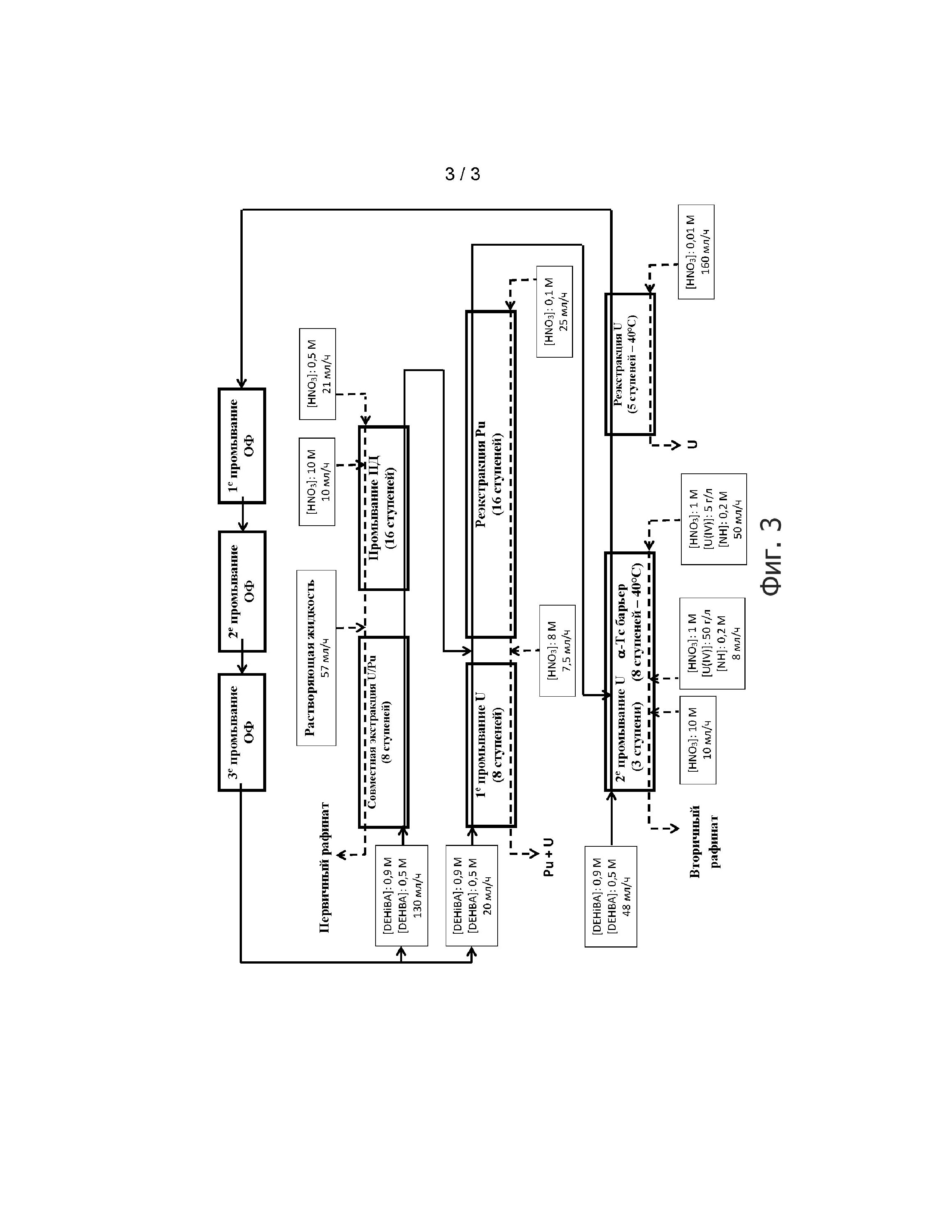
Фигура 3 схематически иллюстрирует условия установки и эксплуатации, используемые для двух тестов, предназначенных для валидации способа настоящего изобретения в целом в экстракторах.
На Фигурах 1-3 органические фазы представлены сплошными линиями, а водные фазы представлены пунктирными линиями.
Подробное описание частных вариантов осуществления
I - Поточная диаграмма способа настоящего изобретения
Вначале приводится ссылка на Фигуру 1, где изображена поточная диаграмма способа настоящего изобретения.
Как показано на этой Фигуре, способ включает 8 этапов.
Первый из этих этапов, обозначенный как «совместная экстракция U/Pu» на Фигуре 1, предназначен для достижения объединенной экстракции урана и плутония, где первый из них находится в степени окисления +VI, а второй в степени окисления +IV, из водного раствора, полученного при растворении отработавшего ядерного топлива в азотной кислоте.
Такой раствор, как правило, включает от 3 до 6 моль/л HNO3, уран, плутоний, минорные актиноиды (америций, кюрий и нептуний), продукты деления (La, Ce, Pr, Nd, Sm, Eu, Gd, Mo, Zr, Ru, Tc, Rh, Pd, Y, Cs, Ba, и т.д.) и несколько продуктов коррозии, таких как железо.
«Этап совместной экстракции U/Pu» проводят путем циркуляции растворяющей жидкости в экстракторе 1, в противоточном потоке к органической фазе (обозначенной как «ОФ» на Фигуре 1), включающей:
- либо DEHDMBA в растворе в органическом разбавителе, в этом случае концентрация этого моноамида в органической фазе составляет от 1 моль/л до 2 моль/л, предпочтительно от 1,3 моль/л до 1,4 моль/л, и предпочтительно 1,35 моль/л;
- либо смесь DEHiBA и DEHBA в растворе в органическом разбавителе, в этом случае концентрация этой смеси (которая, таким образом, соответствует общей концентрации моноамидов) в органической фазе составляет от 1 моль/л до 2 моль/л, предпочтительно от 1,3 моль/л до 1,5 моль/л, и предпочтительно 1,4 моль/л, с молярным отношением DEHiBA/DEHBA, предпочтительно составляющим от 1,7 до 1,9, и предпочтительно 1,8; это дает, например, 0,9 моль/л DEHiBA на 0,5 моль/л DEHBA при концентрации смеси 1,4 моль/л.
Органический разбавитель является алифатическим, прямоцепочечным или разветвленным углеводородом, таким как н-додекан, ТПГ; изопарафиновый разбавитель, выпускаемый TOTAL под торговой маркой Isane IP185T; предпочтение отдается ТПГ.
Второй этап способа, обозначенный как «промывание ПД» на Фигуре 1, предназначен для извлечения из органической фазы, полученной от «совместной экстракции U/Pu», фракции продуктов деления, которые были экстрагированы из растворяющей жидкости совместно с ураном и плутонием.
Для этой цели этап «промывания ПД» включает одну или несколько операций промывания органической фазы, полученной при «совместной экстракции U/Pu», где каждую операцию промывания выполняют путем циркуляции этой органической фазы в экстракторе 2, в противоточном потоке к водному азотнокислому раствору, имеющему концентрацию, по возможности, в диапазоне от 0,5 моль/л до 6 моль/л HNO3, но предпочтительно от 4 моль/л до 6 моль/л HNO3, и более предпочтительно от 4 до 5 моль HNO3, для облегчения реэкстракции рутения и технеция.
Если этап «промывания ПД» проводят с одним или несколькими водными растворами с сильной кислотностью, т.е. как правило, 3 моль/л HNO3 или больше, то этот этап дополнительно включает нейтрализацию (уменьшение кислотности) органической фазы, которую проводят путем циркуляции этой органической фазы в противоточном потоке к слабокислому водному азотнокислому раствору, т.е. включающему от 0,1 до 1 моль/л HNO3, например, водному раствору, включающему 0,5 моль/л HNO3, для предотвращения отведения слишком большого количества кислоты к экстрактору, предназначенного для третьего этапа, обозначенного как «реэкстракция Pu» на Фигуре 2, что может нарушать производительность этого третьего этапа.
Этап «реэкстракции Pu», который представляет первый этап разделения U/Pu, предназначен для извлечения плутония в степени окисления +IV, и таким образом, без восстановления этого плутония, из органической фазы, полученной при «промывании ПД».
Этот этап проводят путем циркуляции этой органической фазы в экстракторе 3 в противоточном потоке к водному раствору, содержащему от 0,1 моль/л до 0,5 моль/л HNO3, и предпочтительно с применением отношения потоков О/В больше 1, предпочтительно 3 или больше, и более предпочтительно 5 или больше, так чтобы плутоний (IV) реэкстрагировался концентрирующим образом.
Реэкстракция плутония (IV), выполняемая на этапе «реэкстракции Pu», сопровождается реэкстракцией фракции урана (VI), который также содержится в органической фазе, полученной при «промывании ПД».
Четвертый этап способа, обозначенный как «1-е промывание U» на Фигуре 1, и представляющий второй этап разделения U/Pu, таким образом, предназначен для экстракции из водной фазы, полученной при «реэкстракции Pu»:
- либо всего урана, содержащегося в водной фазе, если необходимо, чтобы разделение U/Pu приводило к водному раствору, содержащему плутоний без урана, и к органическому раствору, содержащему уран без плутония;
- либо количества урана, которое после «1-го промывания U» позволяет получать водный раствор, содержащий уран и плутоний в предварительно выбранном отношении, если необходимо, чтобы разделение U/Pu приводило к водному раствору, содержащему смесь плутония и урана в этом отношении, и к органическому раствору, содержащему уран без плутония.
В обоих случаях «1-е промывание U» проводят путем циркуляции водной фазы, полученной при «реэкстракции Pu» в экстракторе 4, в противоточном потоке к органической фазе, имеющей идентичный состав с органической фазой, используемой при «совместной экстракции U/Pu». Количество экстрагируемого урана вначале регулируют путем действия в первую очередь на отношение потоков О/В, во-вторых, на кислотность водной фазы; экстракция урана увеличивается при повышении отношения потока органической фазы/водной фазы и усилении кислотности водной фазы. Таким образом, добавление HNO3 большей или меньшей концентрации к водной фазе, циркулирующей в экстракторе 4, можно проводить в зависимости от кислотности, которую необходимо обеспечить в этой водной фазе.
Пятый этап, обозначенный как «α-барьер Тс» на Фигуре 1, предназначен для извлечения из органической фазы, полученной при «реэкстракции Pu», фракции технеция, который был экстрагирован при «совместной экстракции U/Pu», но не был реэкстрагирован при «промывании ПД», с целью очистки этой органической фазы от технецию.
Это также позволяет проводить реэкстракцию из органической фазы, полученной при «реэкстракции Pu», фракции нептуния, которая была экстрагирована при «совместной экстракции U/Pu», и которая следует за технецием до «α-Тс барьера», а также следов плутония, которые все еще может содержать эта органическая фаза.
Этот этап проводят путем циркуляции органической фазы, полученной при «реэкстракции Pu» в экстракторе 5, в противоточном потоке к водному азотнокислому раствору с низкой кислотностью, т.е. содержащему от 0,1 моль/л до 3 моль/л HNO3, и предпочтительно 1 моль/л HNO3, и содержащему один или несколько восстанавливающих агентов, обеспечивающих восстановление технеция, который присутствует в органической фазе в степени окисления +VII, до технеция (IV), не экстрагируемого N,N-диалкиламидами; нептуния (VI) до нептуния (IV) или нептуния (V), не экстрагируемого N,N-диалкиламидами при низкой кислотности; и плутония (IV) до плутония (III), который меньше экстрагируется N,N-диалкиламидами при низкой кислотности, чем плутоний (IV), без восстановления урана (VI).
В качестве восстанавливающих агентов можно применять урана нитрат (или U(IV)), гидразиния нитрат (или HN), гидроксиламмония нитрат (или HAN), ацетальдоксим, или их смесь, такую как смесь U(IV)/HN, U(IV)/HAN или U(IV)/ацетальдоксима, предпочтительной является смесь U(IV)/HN или U(VI)/HAN. Можно добавлять глюконовую кислоту к водному раствору для уменьшения явления повторного окисления технеция в водной фазе, и таким образом, ограничения потребления восстанавливающего агента (агентов).
Этот этап можно проводить при комнатной температуре (т.е. 20-25°С), но предпочтительно его проводят при температуре в диапазоне от 30°С до 40°С, и более предпочтительно при 32°С для стимуляции кинетики реэкстракции технеция при ограничении явления повторного окисления технеция в водной фазе, и таким образом, для ограничения риска обратной экстракции реэкстрагированного технеция в органическую фазу.
Шестой этап, обозначенный как «2-е промывание U» на Фигуре 1, предназначен для экстракции из водной фазы, полученной от «α-Тс барьера», урана, который был реэкстрагирован вместе с технецием на предыдущем этапе, так чтобы избежать получения на этапе «α-Тс барьера» слишком большой потери урана в водной фазе.
Его проводят путем циркуляции водной фазы, полученной от «α-Тс барьера» в экстракторе 6, в противоточном потоке к органической фазе, имеющей идентичный состав с органическими фазами, используемыми для «совместной экстракции U/Pu» и «1-го промывания U», после подкисления этой водной фазы путем добавления концентрированной азотной кислоты, например, 10 М, для того чтобы стимулировать экстракцию урана.
Седьмой этап, обозначенный как «реэкстракция U» на Фигуре 1, предназначен для извлечения урана (VI) из органической фазы, полученной на этапе «α-Тс барьера».
Его проводят путем циркуляции органической фазы, полученной от «α-Тс барьера» в экстракторе 7, в противоточном потоке к водному азотнокислому раствору низкой кислотности, т.е. содержащему не более 0,05 моль/л HNO3, например, водному раствору, содержащему 0,01 моль/л HNO3. Этот этап можно проводить при комнатной температуре (т.е. 20-25°С), но предпочтительно его проводят при нагревании (т.е., как правило, при температуре 40-50°С) с применением отношения потоков О/В больше 1, так чтобы уран (VI) реэкстрагировался с концентрированием.
После этих 7 этапов мы получаем:
- два рафината, соответствующих водным фазам, соответственно выходящим из экстракторов 1 и 6, где первый включает продукты деления и америций и кюрий (Первичный рафинат» на Фигуре 1), а второй включает технеций, нептуний, и возможно, следы плутония («Вторичный рафинат» на Фигуре 1);
- водную фазу, выходящую из экстрактора 4, включающую либо очищенный от загрязнителей (дезактивированный) плутоний, либо смесь очищенного от загрязнителей (дезактивированного) плутония и очищенного от загрязнителей (дезактивированного) урана, называемую «поток Pu» или «поток Pu+U», соответственно;
- водную фазу, выходящую из экстрактора 7, включающую очищенный от загрязнителей (дезактивированный уран), называемую «поток U»; и
- органическую фазу, выходящую из экстрактора 7, которая больше не содержит какого-либо плутония или урана, но может содержать определенный ряд примесей и продуктов распада (образованных при гидролизе и радиолизе) экстрагента, которые могут накапливаться в предыдущих этапах.
Таким образом, восьмой этап, обозначенный как «промывание ОФ» на Фигуре 1, предназначен для регенерации этой органической фазы путем её обработки посредством одного или нескольких промываний основным водным раствором, например, первого промывания водным раствором 0,3 моль/л натрия карбоната, с последующим вторым промыванием водным раствором 0,1 моль/л натрия гидроксида, затем с одним или несколькими промываниями водным раствором азотной кислоты, обеспечивающим повторное подкисление органической фазы, например, водным раствором, содержащим 2 моль/л HNO3, где каждое промывание выполняют путем циркуляции указанной органической фазы в экстракторе в противоточном потоке к водному промывающему раствору.
Как можно видеть на Фигуре 1, органическая фаза, регенерированная таким способом, может быть возвращена обратно к экстракторам 1 и 4 для повторного ввода в цикл переработки.
II - ЭКСПЕРИМЕНТАЛЬНОЕ ПОДТВЕРЖДЕНИЕ
II.1 - Определение в тестовых пробирках коэффициентов распределения урана, плутония и продуктов деления, в водных растворах, полученных при растворении гранул отработавшего ядерного топлива в HNO3.
Первая серия тестов: сравнения моноамидов и ТБФ
Экстракцию проводили в экспериментальных пробирках, с применением:
- органических фаз: фаз, включающих либо смесь DEHiBA/DEHBA, содержащую 1,2 моль/л DEHiBA и 0,3 моль/л DEHBA в ТПГ, либо 1,1 моль/л DEHDMBA в ТПГ, либо 30 об.% ТБФ в ТПГ; и
- в качестве водных фаз: аликвоты водного раствора, ранее полученного путем растворения гранул облученного МОХ топлива в азотной кислоте.
Этот водный раствор содержал 3,15 моль/л HNO3, а его составляющие элементы приведены в Таблице 1 ниже.
Таблица 1
|
Каждую органическую фазу приводили в контакт при перемешивании с аликвотой водного раствора в течение 30 минут при 25°С. Использовали объемное отношение О/В 4. Эти фазы затем отделяли друг от друга после центрифугирования.
Концентрации урана и плутония, и активность америция и продуктов деления измеряли в органической и водной фазах, разделенных таким способом, путем колориметрии для урана, α-спектрометрии для плутония и γ-спектрометрии для америция и продуктов деления.
В Таблице II ниже приведены коэффициенты распределения, определенные по измеренным концентрациям и активностям.
Таблица II
|
Эти результаты показали, что при используемых концентрациях уран (VI) и плутоний (IV) хуже экстрагируются органическими фазами, содержащими смесь DEHiBA/DEHBA или DEHDMBA в ТПГ, чем органической фазой, состоящей из ТБФ и ТПГ. Однако они также показали, что продукты деления и трехвалентные актиноиды, такие как америций, также хуже экстрагируются органическими фазами на основе моноамидов, что делает возможным достижение очень эффективной дезактивации урана и плутония в отношении этих продуктов деления и трехвалентных актиноидов посредством этапа «промывания ПД» из способа в соответствии с настоящим изобретением.
Вторая серия тестов со смесью DEHiBA/DEHBA в качестве экстрагента
Тесты, предназначенные для имитации в пробирках осуществлений в экстракторах этапов «совместной экстракции U/Pu», «1-го промывания ПД», «реэкстракции Pu» (две стадии), «α-Тс барьера», и «реэкстракции U» способа в соответствии с настоящим изобретением проводили с использованием водного раствора, ранее полученного путем растворения в азотной кислоте гранул различного облученного топлива UOX-BWR типа (из ядерного реактора на кипящей воде) и UOX-REP типа (из водо-водяного энергетического реактора).
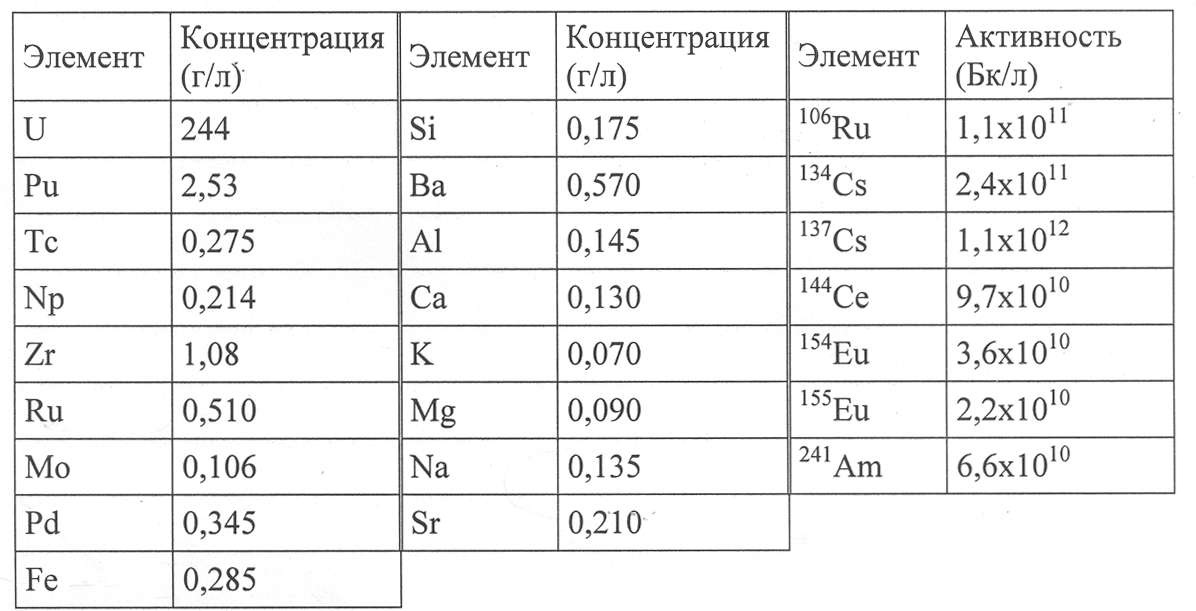
Этот водный раствор содержал 4,3 моль/л HNO3, а его составляющие элементы приведены в Таблице III ниже.
Таблица III
Тесты проводили следующим образом.
Этап «совместной экстракции U/Pu»: водный раствор приводили в контакт при перемешивании с органической фазой, содержащей 0,9 моль/л DEHiBA и 0,5 моль/л DEHBA в ТПГ, предварительно уравновешенной с 6 моль/л HNO3, в течение 15 минут при 25°C, с объемным отношением О/В 2,5. Затем водную и органическую фазу разделяли после центрифугирования.
Концентрации урана и плутония и активности америция и β-γ изотопов измеряли в каждой из органических и водных фаз, разделенных таким способом, посредством рентгеновской флуоресценции для урана и плутония, и посредством γ-спектрометрии для β-γ изотопов.
Концентрации Tc, Np, Zr, Mo и Fe можно было измерить только в водной фазе посредством ICP-AES (масс-спектрометрии с индуктивно-связанной плазмой), а концентрации этих элементов в органической фазе устанавливали по разнице между исходными концентрациями указанных элементов в водной фазе и концентрациями, измеренными при равновесии после экстракции.
Этап «промывания ПД»: органическую фазу, полученную после «этапа совместной экстракции U/Pu», приводили в контакт при перемешивании с водным раствором, содержащим 2 моль/л HNO3 в течение 15 минут при 25°C, с объемным отношением О/В 2. Водную и органическую фазы затем разделяли после центрифугирования и анализировали, как описано ранее.
Этап «реэкстракции Pu»: органическую фазу, полученную после этапа «промывания ПД», приводили в контакт при перемешивании, последовательно 2 раза (с обновлением водной фазы) с водным раствором, содержащим 0,1 моль/л HNO3 и 140 г/л урана (это позволяет урану оставаться в органической фазе и предотвращает его перенос в водную фазу), в течение 15 минут при 25°С, с объемным отношением О/В 2. Затем разделяли водную и органическую фазу после центрифугирования, и анализировали, как описано ранее.
Этап «α-Тс барьера»: органическую фазу, полученную после этапа «реэкстракции Pu», приводили в контакт при перемешивании с водным раствором, содержащим 1,5 моль/л HNO3, 5 г/л урана (IV) и 0,2 моль/л гидроксиламмония нитрата (HAN), в течение 30 минут при 25°C, с объемным отношением О/В 1,5. Затем разделяли водную и органическую фазу после центрифугирования, и анализировали, как описано ранее.
Этап «реэкстракции U»: органическую фазу, полученную после этапа «α-Тс барьера», приводили в контакт при перемешивании с водным раствором, содержащим 0,01 моль/л HNO3 в течение 15 минут при 45°C, с объемным отношением О/В 0,5. Затем разделяли водную и органическую фазу после центрифугирования, и анализировали, как описано ранее.
Все рабочие условия, используемые для каждого этапа, суммированы в Таблице IV ниже, в то время как результаты, полученные после каждого контактирования, с точки зрения кислотности водной фазы, обозначенной как [H+]вод., концентрации урана в органической фазе, обозначенной как [U]орг., и коэффициентов распределения, обозначенных как D, приведены в Таблице V ниже.
Таблица IV
|
Таблица V
Эти результаты подтверждают, что органическая фаза, содержащая 0,9 моль/л DEHiBA и 0,5 моль/л DEHBA в ТПГ, обеспечивает экстракцию урана (VI) и плутония (IV), которая является количественной и избирательной в отношении главных продуктов деления. Высокие коэффициенты распределения (>1) были получены для урана (VI) и плутония (IV) с 5,8 моль/л HNO3, несмотря на сильное насыщение урана (86 г/л) в органической фазе, с коэффициентами разделения U/ПД и Pu/ПД больше 3000, в частности, в отношении рутения.
Они также подтверждают, что плутоний (IV) можно избирательно реэкстрагировать из органической фазы (DPu=0,07) на этапе «реэкстракции Pu» с использованием водного азотнокислого раствора слабой кислотности ([HNO3] =0,16M), в то время как уран предпочтительно остается в органической фазе. Уран можно затем количественно реэкстрагировать из органической фазы (DU=0,06) на этапе «реэкстракции U» с применением водного раствора очень слабой кислотности ([HNO3] =0,01M), нагретого до 45°C.
II.2 - Осуществление этапа «α-Тс барьера» способа в соответствии с настоящим изобретением посредством теста, проведенного в экстракторах.
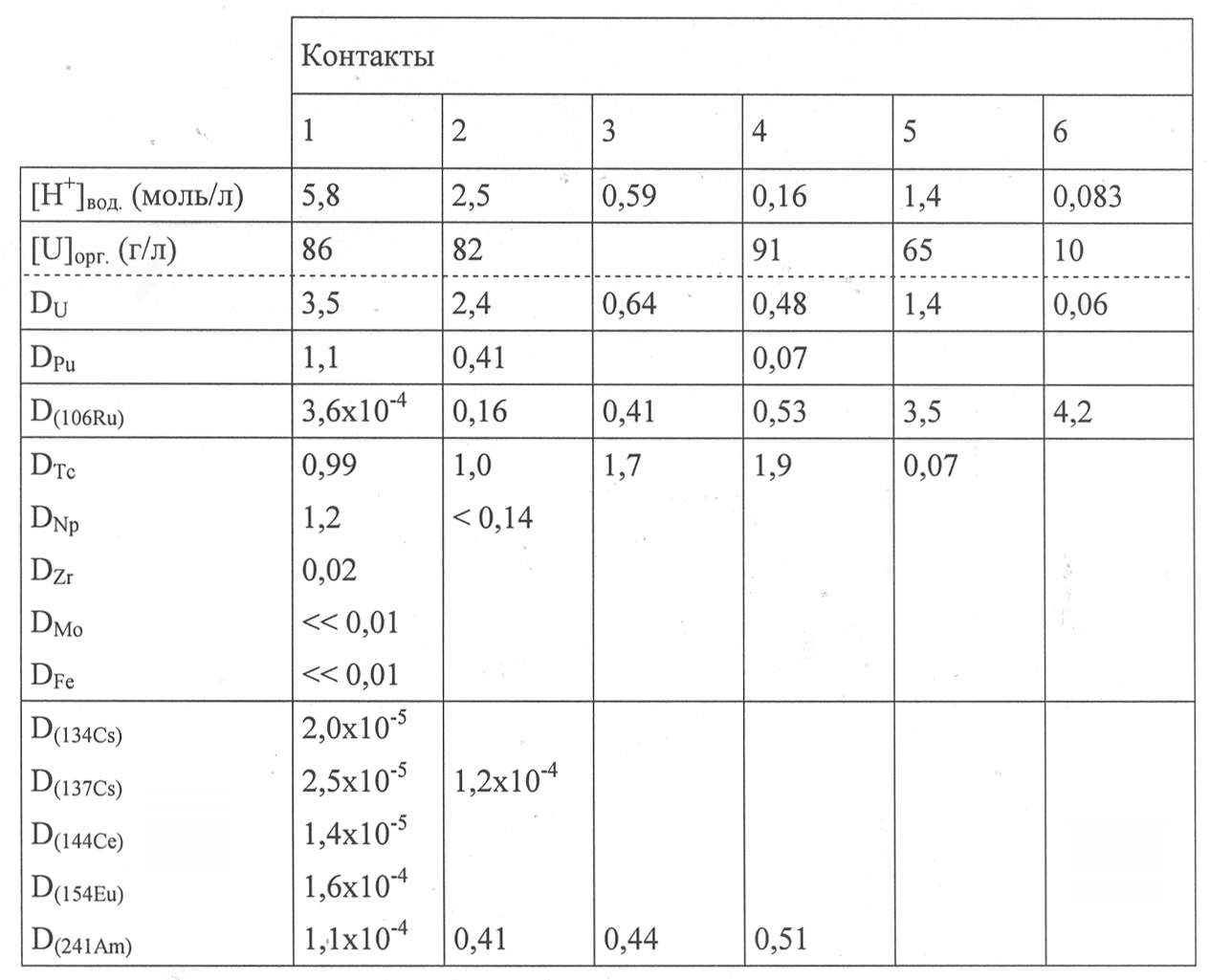
Ссылка на Фигуру 2 иллюстрирует условия установки и работы для выполнения теста, предназначенного для подтверждения этапа «α-Тс барьера» способа в соответствии с настоящим изобретением в экстракторах.
Как можно видеть на этой Фигуре, этот тест включает:
- этап, обозначенный как «Экстракция» на Фигуре 2, проводили в первом 8-ступенчатом смесителе-отстойнике, предназначенном для экстракции урана и технеция из водного раствора, обозначенного как «Загрузка» на Фигуре 2, включающего 320 г/л урана, 279 мг/л технеция 99m и 0,57 моль/л HNO3, с применением органической фазы, включающей 0,9 моль/л DEHiBA и 0,5 моль/л DEHBA в ТРН; состав загрузки и рабочие условия, при которых проводили «Экстракцию», были выбраны так, чтобы получить после этого этапа органическую фазу, имеющую состав, подобный составу, который, вероятно, будет иметь органическая фаза, полученная от этапа «реэкстракции Pu» в способе по изобретению;
- этап, обозначенный как «α-Тс барьер» на Фигуре 2, проводимый в последних 8 ступенях второго смесителя-отстойника, имеющего 11 ступеней, и предназначенный для извлечения технеция из органической фазы, полученной с этапа «Экстракция» с применением водного раствора, содержащего 1 моль/л HNO3, 5 г/л U(IV) и 0,2 моль/л гидразиния нитрата (HN);
- этап, обозначенный как «промывание U» на Фигуре 2, проводимый в первых 3 ступенях второго смесителя-отстойника, и предназначенный для обратной экстракции в органическую фазу урана, который был совместно реэкстрагирован с технецием на этапе «α-Тс барьера», чтобы ограничить утечку урана в поток технеция; этот этап проводили с применением органической фазы, имеющий тот же самый состав, как у фазы, используемой на этапе «Экстракции»;
- этап, обозначенный как «реэкстракция U» на Фигуре 2, проводимый в третьем смесителе-отстойнике, имеющем 5 ступеней, и предназначенный для реэкстракции урана из органической фазы, полученной от «α-Тс барьера» с применением водного раствора, содержащего 0,01 моль/л HNO3; и
- три промывания органической фазы, полученной от «реэкстракции U», соответственно обозначенные как «1-е промывание ОФ», «2-е промывание ОФ» и «3-е промывание ОФ» на Фигуре 2, которые выполнялись в 3 отжимных центрифугах и состояли из промывания этой органической фазы последовательно водным раствором, содержащим 0,3 моль/л натрия карбоната, водным раствором, содержащим 0,1 моль/л натрия гидроксида, и водным раствором, содержащим 2 моль/л HNO3 для повторного подкисления органической фазы с целью её повторного применения на этапах «Экстракции» и «Промывания U».
Водный раствор, содержащий 1 моль/л HNO3, 50 г/л U(IV) и 0,2 моль/л HN, добавляли на 5-й ступени второй установки (которая, таким образом, соответствует 2-й ступени этапа «α-Тс барьера») для поддержания минимальной концентрации U(IV) на первых двух ступенях «α-Тс барьера», поскольку U(IV) частично потребляется со временем в циклах повторного окисления/восстановления технеция азотной (и азотистой) кислотой и U(IV).
Водный раствор, содержащий 10 моль/л HNO3, также добавляли на 3-й ступени второй установки для повышения кислотности водной фазы, циркулирующей на 3 ступенях, предназначенных для «Промывания U», от 1 моль/л до 2,5 моль/л, и таким образом, стимуляции обратной экстракции урана в органическую фазу.
Отношение потоков О/В 1 применяли на 3 ступенях «Промывания U», в то время как отношение потоков О/В 4 применяли на 8 ступенях «α-Тс барьера» для достижения концентрирующей реэкстракции технеция. Температуру 8 ступеней «α-Тс барьера» и 5 ступеней «Промывания U» устанавливали на 40°С для стимуляции кинетики реэкстракции технеция U(IV), при этом ограничивая явление повторного окисления этого элемента, катализируемое под действием высокой температуры.
Анализ проводили в течение 8,5 часов (из которых 3 при равновесии), начиная от времени загрузки в установку, предназначенную для этапа «Экстракции».
Образцы отбирали каждые два часа для проверки достижения термодинамического равновесия, после чего органическую и водную фазы удаляли и исследовали в конце анализа.
Результаты представлены в Таблице VI ниже.
Таблица VI
|
Эти результаты показывают, что технеций, количественно экстрагируемый в органической фазе посредством смеси DEHiBA/DEHBA при «Экстракции», затем количественно реэкстрагируется из этой фазы, избирательно по отношению к урану, на стадиях «α-Тс барьера» посредством восстановления из степени окисления +VII до степени окисления +IV раствором U(IV)/HN.
Стабильность концентрации U(IV) на ступенях «α-Тс барьера», наблюдаемая посредством оперативного спектрофотометрического мониторинга при анализе, подтвердила, что можно было предотвратить явление повторного окисления технеция и избыточное потребление U(IV), таким образом, обеспечивая эффективную реэкстракцию технеция из органической фазы по всей продолжительности теста.
99,8% технеция, исходно содержащегося в загрузке, было извлечено в водную фазу, полученную при «Промывании U»; 0,17% было найдено в водной фазе, полученной при «Реэкстракции U», и 0,02% в органической фазе, полученной при этой реэкстракции.
Коэффициент дезактивации (очистки) урана по отношению к технецию (FDU/Tc), рассчитанный путем деления отношения концентрации урана к концентрации технеция в водной фазе, полученной при «Реэкстракции U», на отношение концентрации урана к концентрации технеция в загрузке, составил 538 в конце теста. Таким образом, было достигнуто целевое значение FDU/Tc 153 для водной фазы, полученной на этапе «Реэкстракции U» способа настоящего изобретения, что соответствует максимальной концентрации технеция 5 мкг/г урана в этой водной фазе.
II.3 - Осуществление способа настоящего изобретения в целом посредством двух тестов в экстракторах.
Ссылка на Фигуру 3 иллюстрирует условия установки и работы, используемые для двух тестов, предназначенных для подтверждения способа настоящего изобретения в целом в экстракторах.
Для этих тестов способ настоящего изобретения применяли для обработки водного азотнокислого раствора, полученного при растворении отработавшего ядерного топлива для получения первого водного потока, включающего смесь очищенного плутония и очищенного урана, и второго водного потока, включающего очищенный уран.
Эти тесты проводили с водным раствором, ранее полученным при растворении в азотной кислоте гранул от трех различных видов отработавшего ядерного топлива: 75% растворенного топлива было UOX3 (выгорание топлива = 65 МВт·сутки/т, с охлаждением 4 года), остальные 25% состояли из UOX с выгоранием топлива 37 МВт·сутки/т, с охлаждением 7 лет, и МОХ с выгоранием топлива 25 МВт·сутки/т, с охлаждением 18 лет. Растворяющая жидкость содержала 4,9 моль/л HNO3. Элементарный состав приведен в Таблице VII ниже.
Таблица VII
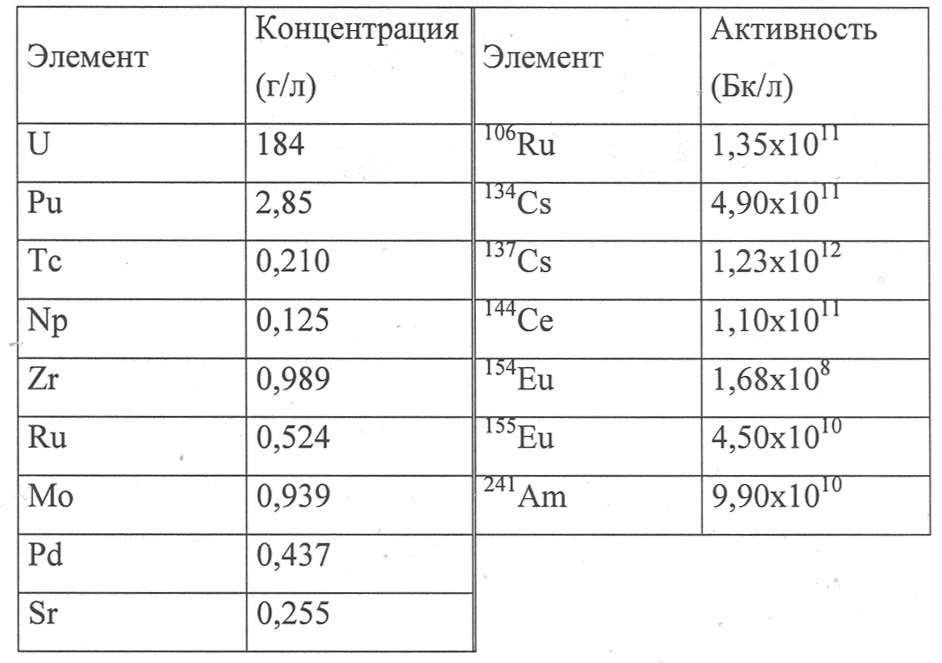
Использованные органические фазы включали смесь DEHiBA/DEHBA в пропорции 0,9 моль/л DEHiBA и 0,5 моль/л DEHBA в ТПГ.
Для этого использовали экранированную линию, включающую:
- первый смеситель-отстойник из 8 ступеней для «этапа совместной экстракции U/Pu»;
- второй смеситель-отстойник из 16 ступеней для этапа «промывания ПД», где последние четыре ступени этой установки предназначены для нейтрализации органической фазы;
- третий смеситель-отстойник из 16 ступеней для этапа «реэкстракции Pu»;
- четвертый смеситель-отстойник из 8 ступеней для «1-го этапа промывания U»;
- пятый смеситель-отстойник из 11 ступеней, в котором первые три ступени были предназначены для «2-го промывания U», а последние 8 ступеней для этапа «α-Тс барьера»;
- шестой смеситель-отстойник из 5 ступеней для этапа «реэкстракции U»; и
- три отжимных центрифуги, предназначенных для трех промываний органической фазы, соответственно обозначенных как «1-е промывание ОФ», «2-е промывание ОФ» и «3-е промывание ОФ» на Фигуре 3, и состоящих из промывания этой органической фазы последовательно водным раствором, содержащим 0,3 моль/л натрия карбоната, водным раствором, содержащим 0,1 моль/л натрия гидроксида, и водным раствором, содержащим 2 моль/л HNO3 для повторного подкисления органической фазы с целью её повторного использования на этапах «совместной экстракции U/Pu» и «1-го промывания U».
В соответствии с пунктом II.2 выше:
- водный раствор, содержащий 1 моль/л HNO3, 50 г/л U(IV) и 0,2 моль/л HN, добавляли на 5-ю ступень второй установки (которая, таким образом, соответствует 2-й стадии этапа «α-Тс барьера») для поддержки минимальной концентрации U(IV) на двух первых стадиях «α-Тс барьера»; в то время как
- водный раствор, содержащий 10 моль/л HNO3, также добавляли на 3-ю ступень пятой установки с 11 ступенями, для повышения концентрации азотной кислоты в водной фазе, циркулирующей на 3 ступенях, предназначенных для «2-го промывания U», от 1 моль/л до 2,5 моль/л, и таким образом, облегчения обратной экстракции урана в органическую фазу.
Кроме того, водный раствор, содержащий 8 моль/л HNO3, добавляли к водному раствору, полученному при «реэкстракции Pu», когда он поступал в пятую установку, предназначенную для «1-го промывания U» для повышения концентрации азотной кислоты и для облегчения обратной экстракции урана в органическую фазу.
Температуру 8 ступеней «α-Тс барьера» и 5 ступеней «реэкстракции U» устанавливали на 40°С.
Отношения потоков О/В, используемые на этапах «реэкстракции Pu» и реэкстракции U» составили, соответственно, 6 и 1,24.
Вначале первый тест проводили в течение 80 часов.
После этого теста собирали различные водные и органические фазы, и анализировали для оценки производительности способа.
Результаты этих тестов приведены в Таблице VIII ниже.
Таблица VIII
|
*радиологический контроль
Эти результаты показывают, что уран и плутоний количественно экстрагируются (более 99,99% и 99,96%, соответственно) из растворяющей жидкости, а затем извлекаются в водные фазы, полученные с этапа «1-го промывания U» (или потока Pu+U) и с этапа «реэкстракции U» (или потока U).
Высокая избирательность смеси DEHiBA/DEHBA была подтверждена очень хорошей очисткой этих фаз от наиболее проблематичных продуктов деления (Ru, Cs, Tc, и т.д.).
На выходе от «промывания ПД» очистка органической фазы от рутения-106, вносящему основной вклад в остаточную β-активность конечных продуктов, является эффективной, поскольку коэффициенты дезактивации достигают 8x105, в соответствии с измерениями, проведенными для образцов органической фазы, взятых на последней стадии «промывания ПД».
Несмотря на превышение β-γ активности в потоке U при расчетевследствие проблем контаминации на экранированной линии (в частности, с подтверждением измерениями радиационных контролей), активность рутения-106, измеренная в этом водном растворе, практически достигает требований ASTM C788-03 для очистки урана, полученного в форме уранила нитрата, от технеция (8x105 Мэкв.Бк/кг U для целевого значения 3x105 Мэкв.Бк/кг U).
Что касается общей β-γ активности, требование ASTM не выполняется с учетом сильной контаминации цезием на экранированной линии, где радиационный контроль достигает такого же уровня, как при измерении активности цезия-137 в потоке U.
Однако можно скорее обосновать органические активности, измеренные в органической фазе, полученной от «промывания ПД». Тот факт, что смесители-отстойники, расположенные после экстрактора, предназначенного для этапа «промывания ПД», проявляют значительно большие γ-активности, чем те, которые измерены в органической фазе, полученной на этом этапе, может быть связан только с вкладом γ-активности за счет контаминации из-за многочисленных операций, проводимых при тестировании установок, предназначенных для «1-го промывания U», «реэкстракции Pu», «α-Т барьера» и «реэкстракции U».
Таблица IX внизу показывает, что если учитывать активности, измеренные в Мэкв.Бк в органической фазе, полученной на этапе «промывания ПД», на грамм U или на грамм Pu, содержащегося в этой фазе (менее нарушенной при контаминациях, чем поток Pu+U и поток U), можно достичь требований ASTM C757-90 для потока Pu+U, и очень приблизиться к требованиям для водного раствора, полученного из потока U (ASTM C788-03).
Таблица IX
|
Поток U очень хорошо очищен по отношению к:
- плутонию, поскольку концентрация плутония в потоке U составляет 67 мкг/л, т.е. FD U/Pu 12400, а остаточная активность Pu составляет 1,5x105 Бк/г U при требовании ASTM стандарта 125 Бк/г U;
- нептунию, поскольку концентрация нептуния в потоке U составляет 34 мкг/л, т.е. FD U/Np 1070, а остаточная активность Np 17 Бк/г U при требовании ASTM 125 Бк/г U; и
- технецию, поскольку концентрация технеция в потоке U составляет примерно 270 мкг/л, т.е. FDU/Tc 230, а остаточное количество Тс 5 мкг Tc/г U при требовании ASTM 5 мкг Tc/г U, особенно при получении посредством этапа «α-Тс барьера», применяемого после разделения U/Pu.
Таким образом, выполняются требования ASTM для потока U, в частности, с учетом того, что:
- активности источников β-γ и концентрация Pu, измеренные в потоке U, по существу обусловлены контаминацией образцов, взятых на экранированной линии;
- превращение уранила нитрата в оксид U3O8 обеспечивает дополнительную дезактивацию по отношению к продуктам деления (чистота урана эффективно анализируется на конечном оксиде); и
- того факта, что тест, для ограничений, связанных с чувствительностью анализа рутения -106, в частности, проводили на разбавленном растворе отработавшего ядерного топлива, слабо охлажденного, и следовательно, имеющего очень высокую активность источников β-γ, т.е. в наихудших и ограничивающих условиях, по сравнению с обычными операциями на заводах по переработке отработавшего ядерного топлива.
Поток Pu+U также хорошо очищен от продуктов деления. Как и в случае потока U, с учетом того, что общую активность β-γ измеряли в потоке Pu+U в конце теста, требование ASTM не выполняется по причинам, связанным с проблемами контаминации смесителей-отстойников, предназначенных для «реэкстракции Pu» и «1-го промывания U». Однако, если в качестве основы использовать активность β-γ, измеренную в органической фазе, полученной на этапе «промывания ПД», требование ASTM по общей γ-активности выполняется (4x104 экв.Бк/кгPu для целевого значения 105 Мэкв.Бк/гPu).
Поток Pu+U очень хорошо очищен от технеция, поскольку концентрация технеция в потоке Pu+U составляет 4,2 мг/л, т.е. FDPu/Tc 121 и остаточное количество Тс 609 мкг/гPu, значительно ниже предела 6000 мкг/г Pu по требованиям ASTM для плутония оксида.
Наконец, концентрация урана в потоке Pu+U, измеренная в конце теста, была выше целевого отношения Pu/U, но это обусловлено дисфункцией потока водного раствора HNO3, добавленного в водный раствор, полученный при «реэкстракции Pu», при поступлении в четвертую установку, предназначенную для «1-го промывания U».
Таким образом, второй тест проводили в той же самой установке и при тех же самых рабочих условиях, но с коррекцией этой дисфункции. Этот тест проводили в течение 74 часов. В конце этого теста достигалось термодинамическое равновесие, и собирали и анализировали различные водные и органические фазы.
Этот второй тест не только подтвердил хорошую очистку потоков U и Pu+U от продуктов деления, но также возможность разделения U/Pu без восстановления плутония. Плутоний количественно реэкстрагировали и извлекали в конце теста, и поток Pu+U содержал 5,45 г/л Pu и 2,07 г/л U, т.е. отношение U/Pu составило 0,38, что соответствовало целевому отношению Pu/U.
Тесты, описанные выше, показали возможность извлечения, разделения и дезактивации (очистки от загрязнений) урана и плутония, содержащихся в водном азотнокислом растворе, полученном при растворении отработавшего ядерного топлива, в одном цикле переработки без необходимости восстановительной реэкстракции плутония, и с коэффициентами очистки урана и плутония, в частности, от источников β-γ, которые являются такими, что нет необходимости предусматривать дополнительные циклы очистки урана и плутония.
Цитированные ссылки
[1] FR-A-2 591 213
[2] FR-A-2 642 561
[3] FR-A-2 642 562
Устройство и способ измерения скорости счета
Способ дезактивации жидких радиоактивных отходов от одного или нескольких радиоактивных химических элементов путем отделения твердой фазы от жидкой с использованием контура рециркуляции
Способы приготовления оксалата актиноидов и приготовления соединений актиноидов
Способ определения спектрального и пространственного распределения фотонов тормозного излучения и соответствующее устройство
Устройство и способ осаждения пророшков смеси и порошков для формирования объекта с градиентным составом
Способ неинтрузивного обнаружения химического элемента
Способ электрокинетической дезактивации твердой пористой среды
Анемометрический зонд с одной или несколькими проволочками и способ его осуществления
Способ определения изотопного отношения делящегося вещества, содержащегося в камере деления
Способ изготовления набора пластин для теплообменника

