Результат интеллектуальной деятельности: ПРИМЕНЕНИЕ ТИАОКСОСОЕДИНЕНИЙ ДЛЯ УМЕНЬШЕНИЯ СОДЕРЖАНИЯ АРО С3
Вид РИД
Изобретение
Настоящее изобретение относится к способу уменьшения содержания аполипопротеина C-III (apoC-III) мРНК или белка у нуждающегося в нем субъекта, включающему введение субъекту фармацевтически эффективного количества соединения формулы (I):
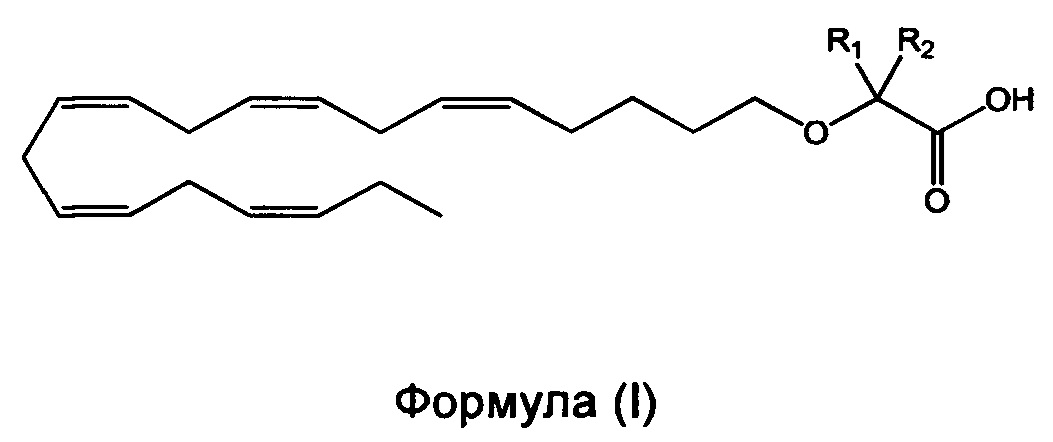
или его фармацевтически приемлемой соли или сложного эфира,
в которой R1 и R2 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6-алкильных групп при условии, что R1 и R2 оба не означают водород. Такие способы, соединения и композиции применимы для лечения патологических состояний, вызванных, связанных с или осложненных повышенным содержанием apoC-III в печени и/или плазме, таких как гипертриглицеридемия (ГТГ), гиперхиломикронемия, дислипидемия, панкреатит, и проводят предупреждение или лечение сердечно-сосудистого заболевания или метаболического нарушения, или его симптома.
Пищевые полиненасыщенные жирные кислоты (ППНЖК), включая омега-3-жирные кислоты, влияют на различные физиологические процессы, обеспечивающие нормальное состояние здоровья, и хронические заболевания, такие как регулирование содержания липидов в плазме, сердечнососудистые и иммунные функции, воздействие инсулина, развитие нейронов и зрительная функция.
Омега-3-жирные кислоты, например, (5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаеновая кислота (ЭПК) и (4Z,7Z,10Z,13Z,16Z,19Z)-докоза-4,7,10,13,16,19-гексаеновая кислота (ДГК), регулируют содержание липидов в плазме, сердечно-сосудистые и иммунные функции, воздействие инсулина, развитие нейронов и зрительную функцию. Показано, что омега-3-жирные кислоты благоприятно влияют на факторы риска сердечно-сосудистых заболеваний, например, гипертензии и гипертриглицеридемии (ГТГ), и на фактор свертывания крови VII комплекса активности фосфолипида.
В WO 2010/128401 раскрыто, что 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановая кислота благоприятно влияет на липидные профили и подавляет внутриартериальное развитие атеросклероза, снижает общий холестерин и увеличивает содержание ЛВП (липо-протеины высокой плотности) холестерина по сравнению с контролем. Эти результаты показывают, что 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановую кислоту и ее производные можно использовать для предупреждения или лечения различных патологических состояний, таких как воспаление, гиперлипидемические патологические состояния, ожирение, жировая инфильтрация печени, атеросклероз, периферическая резистентность к инсулину и/или диабетические патологические состояния. Кроме того, применение 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановой кислоты и ее производных для лечения различных заболеваний или патологических состояний раскрыто в WO 2012/059818.
Точнее, в WO2012/059818 описан способ лечения или предупреждения по меньшей мере одного заболевания или патологического состояния, выбранного из группы, включающей повышенное содержание Аро В, первичная гиперхолестеринемия (гетерозиготная семейная и несемейная) и первичная дисбеталипопротеинемия (Fredrickson Type III) у нуждающегося в нем субъекта, включающий введение субъекту фармацевтически эффективного количества соединения формулы (I). Однако, хотя уже установлено, что на связанные с Аро В и Аро Е (дисбеталипопротеинемия) пути благоприятно влияют соединения формулы (I), данные 2 клинических исследований разных групп пациентов неожиданно показали, что содержание дополнительного аполипопротеина, apoC-III, также значительно снижается соединениями формулы (I).
ApoC-III является гликопротеином, продуцируемым преимущественно печенью, функция которого предположительно включает промотирование сборки и секреции обогащенных триглицеридами частиц ЛОНП клеток печени в обогащенных липидом состояниях (Sundaram М et al., J Lipid Research, vol. 51, 2010). В плазме он в значительной степени ассоциирован с липопротеинами очень низкой плотности (ЛОНП), липопротеинами высокой плотности (ЛВП) и хиломикронами. Увеличение содержания ароС-III индуцирует развитие гипертриглицеридемии. Механизмы, по которым экспрессия apoC-III увеличивает содержание триглицеридов в плазме, частично опосредуются путем ингибирования липопротеинлипазы и печеночной липазы; таким образом, задерживается катаболизм обогащенных триглицеридами частиц. Также считают, что ApoC-III ингибирует поступление в печень обогащенных триглицеридами частиц. Клиническая важность apoC-III была установлена в исследованиях, показавших, что носители редких мутаций, которые нарушают функцию apoC-III, одновременно обладают более низкими содержаниями ТГ (триглицериды) и сниженным риском коронарного/ишемического заболевания сердца (N Engl J Med. 2014 Jul 3; 371(1): 22-31, Loss-of-function mutations in APOC3, triglycerides, и coronary disease).
Длинноцепочечные омега-3-жирные кислоты, ЭПК и ДГК, широко применяются для лечения ГТГ. С учетом недавно установленной роли apoC-III, как главного регулятора содержания триглицеридов и как генетически установленной мишени для предупреждения ишемической болезни сердца, было исследовано влияние омега-3-жирных кислот разных форм и составов на содержание apoC-III в плазме. Например, в US 2014/0221486 заявлен способ уменьшения содержания apoC-III у субъекта, которого лечат статином, и обладающего исходным содержанием триглицеридов натощак, равным примерно 200 мг/дл до примерно 499 мг/дл, или у субъекта, обладающего исходным содержанием триглицеридов натощак, равным по меньшей мере примерно 500 мг/дл, путем введения субъекту фармацевтической композиции, содержащей от примерно 1 г до примерно 4 г этилэйкозапентаеноата в сутки. В US 2013/0177643 заявлен способ уменьшения содержания apoC-III в сыворотке или плазме, включающий введение фармацевтической композиции, содержащей: ЭПК, в основном в форме свободной кислоты, в количестве, равном по меньшей мере примерно 50% (а/а); ДГК, в основном в форме свободной кислоты, в количестве, равном по меньшей мере примерно 15% (а/а); ДПК, в основном в форме свободной кислоты, в количестве, равном по меньшей мере примерно 1% (а/а); в количестве и в течение времени, достаточного для уменьшения содержания apoC-III в сыворотке или плазме по сравнению с содержанием до лечения. Другой пример приведен в US 2014/0094520, в котором заявлен способ уменьшения значения липидного параметра у субъекта по сравнению с исходным значением липидного параметра, где липидный параметр выбран из группы, включающей, в частности, apoC-III, включающий введение субъекту композиции, содержащей жирные кислоты, где по меньшей мере 50 мас. % жирных кислот составляют омега-3-жирные кислоты, соли, эфиры или их производные, где омега-3-жирные кислоты включают эйкозапентаеновую кислоту (ЭПК), докозапентаеновую кислоту (ДПК) и где отношение содержания докозагексаеновой кислоты к содержанию ДГК к содержанию ЭПК (ДГК : ЭПК) составляет менее 1:10, и где отношение содержания ДГК к содержанию ДПК (ДГК : ДПК) составляет менее 2:1.
Эффективное уменьшение содержания apoC-III в печени/плазме при пероральной доставке омега-3/омега-3-производного образует привлекательную возможность лечения выбранной группы пациентов, если возможно обеспечение клинически значимого результата. Хотя пока не определено, какая степень уменьшения содержания apoC-III является "клинически значимой", исследования субъектов с мутациями с потерей функции apoC-III показали, что содержания apoC-III, на 46% меньшие, чем у субъектов, не являющихся носителями мутаций, соответствуют на 40% меньшему риску ишемической болезни сердца (ИБС) (N Engl J Med. 2014 Jul 3; 371(1): 22-31, Loss-of-function mutations in APOC3, triglycerides, and coronary disease). В дополнение к уменьшению концентраций apoC-III, носители мутаций также обладали на 39% меньшими концентрациями ТГ, чем неносители. С учетом того, что утрата функции означает пожизненное воздействие, возможно, что лечение, направленное на уменьшение содержания apoC-III на более короткие период времени, должно быть направлено на уменьшение содержания apoC-III, до как можно более близкого (или более высокого), чем связанное с потерей функции мутаций, если необходимо оказать благоприятное воздействие на ИБС. Поскольку влияние на содержание apoC-III, обеспечиваемое природными омега-3-липидами, является относительно умеренным (см. пример 26), соединения, которые более активно уменьшают содержание apoC-III, могут обеспечить не только превосходное уменьшение содержания триглицеридов, но и превосходные кардиозащитные воздействия.
Настоящее изобретение относится к способу уменьшения содержания аполипопротеина C-III (apoC-III) мРНК или белка у нуждающегося в нем субъекта, включающему введение субъекту фармацевтически эффективного количества соединения формулы (I):
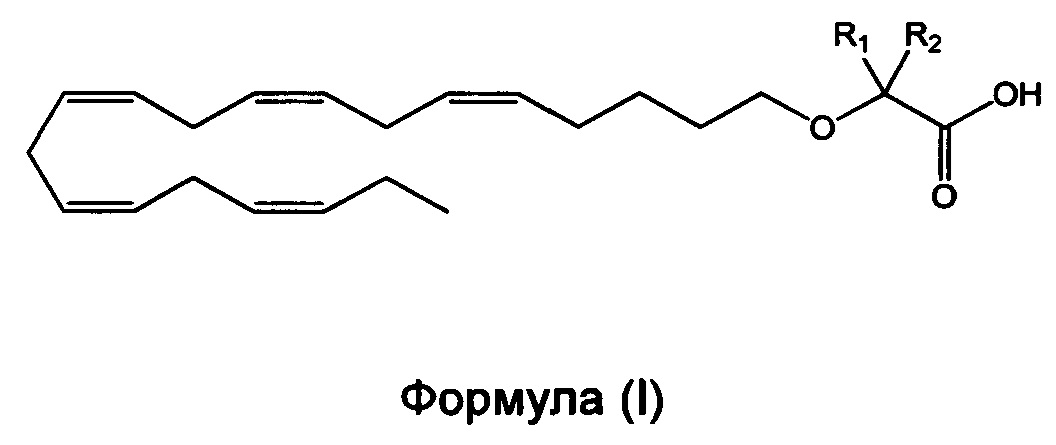
или его фармацевтически приемлемой соли или сложного эфира,
в которой R1 и R2 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C2-алкильных групп при условии, что R1 и R2 оба не означают водород.
Целый ряд метаболических заболеваний или патологических состояний тесно связан с повышенным риском сердечно-сосудистых проявлений. Такие заболевания или патологические состояния включают, но не ограничиваются только ими, сахарный диабет типа I и типа II, метаболический синдром, дислипидемические патологические состояния, такие как гиперхолестеринемия, гиперлипидемия, смешанная дислипидемия, гипертриглицеридемия, гиперхиломикронемия и различные семейные дислипидемии.
По меньшей мере в одном варианте осуществления заболевание или патологическое состояние выбрано из группы, включающей любое из следующих: гипертриглицеридемия (ГТГ), гиперхиломикронемия, дислипидемия и панкреатит, и проводят предупреждение или лечение одного или большего количества следующих: сердечно-сосудистое заболевание или метаболическое нарушение, или его симптом.
Настоящее изобретение также включает способ уменьшения содержания apoC-III у нуждающегося в нем субъекта, способ включает введение субъекту фармацевтически эффективного количества 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутановой кислоты:
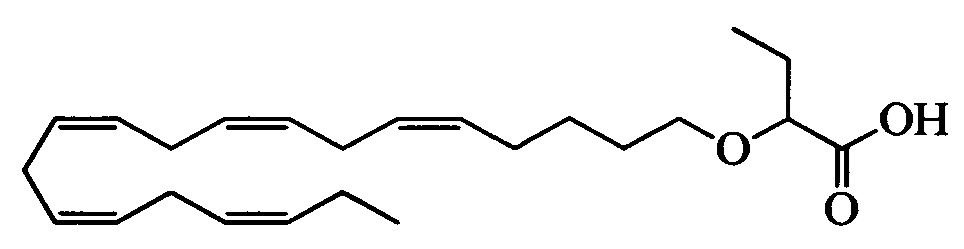
или ее фармацевтически приемлемой соли или сложного эфира.
Краткое описание чертежей
На фиг. 1 представлена относительная степень экпрессирования гена печени apoC-III для соединения формулы (I), контроля и эталонного соединения.
Конкретные объекты настоящего изобретения более подробно описаны ниже. Термины и определения, использующиеся в настоящем изобретении и описанные в настоящем изобретении, обладают значениями, использующимися в настоящем изобретении.
Термины в единственном числе включат термины во множественном числе, если из контекста не следует иное.
Термины "приблизительно" и "примерно" означают почти такое же значение, как указанное число или значение. При использовании в настоящем изобретении термины "приблизительно" и "примерно" обычно следует понимать, как отличающиеся на ±5% от указанного количества, частоты или значения.
Термины "лечить", "лечение" включают любое терапевтическое применение, которое благоприятно для человека или млекопитающего, не являющегося человеком. Медицинское и ветеринарное лечение входят в объем настоящего изобретения. Лечение может быть направлено на имеющееся патологическое состояние или может быть профилактическим, т.е. предупредительным.
Термины "введение", "вводить" при использовании в настоящем изобретении означают (1) предоставление, введение, дозирование и/или назначение практикующим врачом или его уполномоченным представителем, или по его указанию соединения или композиции, соответствующих настоящему изобретению, и (2) введение, прием или потребление самим человеком или пациентом, или млекопитающим, не являющимся человеком, соединения или композиции, соответствующих настоящему изобретению.
Термин "фармацевтически эффективное количество" означает количество, достаточное для обеспечения желательных фармакологических и/или терапевтических эффектов, т.е. количество раскрытого соединения, которое эффективно для назначенной цели. Хотя потребности отдельного субъекта/пациента могут меняться, определение оптимальных диапазонов для эффективных количеств раскрытого соединения находится в компетенции специалиста в данной области техники. Обычно режим дозирования для лечения заболевания или/или патологического состояния соединениями, раскрытыми в настоящем изобретении, можно определить в соответствии с различными факторами, такими как тип, возраст, масса, пол, диета и/или состояние здоровья субъекта/пациента.
Термин "фармацевтическая композиция" означает соединение, соответствующее настоящему изобретению, в любой форме, пригодной для использования в медицине.
Соединения формулы (I) могут существовать в различных стереоизомерных формах, включая энантиомеры, диастереоизомеры или их смеси. Следует понимать, что в объем настоящего изобретения входят все оптические изомеры соединений формулы (I) и их смеси. Следовательно, соединения формулы (I), которые существуют в виде диастереоизомеров, рацематов и/или энантиомеров, входят в объем настоящего изобретения.
Настоящее изобретение относится к способу уменьшения содержания аполипопротеина C-III (apoC-III) мРНК или белка у нуждающегося в нем субъекта, включающему введение субъекту фармацевтически эффективного количества соединения формулы (I):
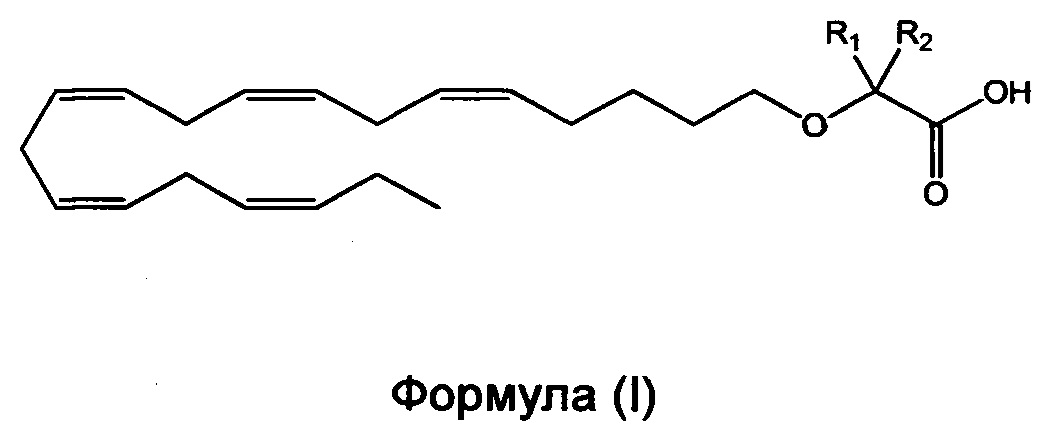
или его фармацевтически приемлемой соли или сложного эфира,
в которой R1 и R2 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6-алкильных групп при условии, что R1 и R2 оба не означают водород.
По меньшей мере в одном варианте осуществления, настоящее изобретение относится к применению фармацевтически эффективного количества соединения формулы (I):
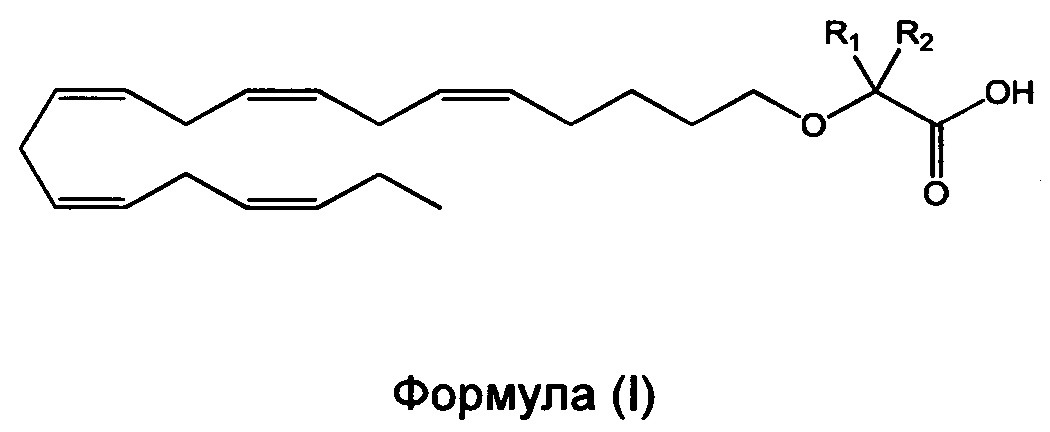
или его фармацевтически приемлемой соли или сложного эфира, для уменьшения содержания аполипопротеина C-III (apoC-III) мРНК или белка у нуждающегося в нем субъекта,
в которой R1 и R2 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических С1-С6-алкильных групп при условии, что R1 и R2 оба не означают водород.
По меньшей мере в одном варианте осуществления R1 и R2 выбраны из атома водорода, метильной группы, этильной группы, н-пропильной группы и изопропильной группы.
По меньшей мере в одном варианте осуществления R1 и R2 выбраны из атома водорода, метильной группы и этильной группы.
По меньшей мере в одном варианте осуществления один из R1 и R2 означает атом водорода и второй из R1 и R2 выбран из C1-С3-алкильной группы. В одном варианте осуществления один из R1 и R2 означает атом водорода и второй из R1 и R2 выбран из метильной группы или этильной группы.
По меньшей мере в одном варианте осуществления соединение находится в своих различных стереоизомерных формах, таких как энантиомер (R или S), диастереоизомер или их смеси.
По меньшей мере в одном варианте осуществления соединение находится в рацемической форме.
В случаях, когда соединение формулы (I) является солью противоиона и обладает по меньшей мере одним стереогенным центром или является сложным эфиром спирта, обладающим по меньшей мере одним стереогенным центром, соединение может обладать множеством стереогенных центров. В этих случаях соединения, соответствующие настоящему изобретению, могут существовать в виде диастереоизомеров. Таким образом, по меньшей мере в одном варианте осуществления соединения, соответствующие настоящему изобретению, содержатся в виде по меньшей мере одного диастереоизомера.
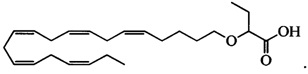
По меньшей мере в одном варианте осуществления соединением, соответствующим настоящему изобретению, является 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутановая кислота:
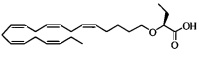
По меньшей мере в одном варианте осуществления соединение, соответствующее настоящему изобретению, находится в его S- и/или R-форме, представленных формулами:
 и
и 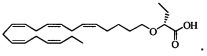
По меньшей мере в одном варианте осуществления заболевание или патологическое состояние выбрано из группы, включающей любое из следующих: гипертриглицеридемия (ГТГ), гиперхиломикронемия, дислипидемия и панкреатит и проводят предупреждение или лечение одного или большего количества следующих: сердечно-сосудистое заболевание или метаболическое нарушение, или его симптом. В одном варианте осуществления заболевание или патологическое состояние выбрано из группы, включающей любое из следующих: гиперхиломикронемия, панкреатит и проводят предупреждение или лечение одного или большего количества следующих: сердечно-сосудистое заболевание или метаболическое нарушение, или его симптом. В одном варианте осуществления заболевание или патологическое состояние выбрано из группы, включающей любое из следующих: гиперхиломикронемия и панкреатит.
Соединения формулы (I) можно получить, как описано, например, в заявке РСТ WO 2010/128401, поданной 7 мая 2010 г., и в соответствии с приведенными ниже примерами 1-23.
Примеры 1-23 являются типичными и специалист в данной области техники должен понимать, как применять общие методики для получения других соединений, входящих в объем формулы (I). Соединения, соответствующие настоящему изобретению, могут находиться в форме фармацевтически приемлемой соли или сложного эфира. Например, соединения формулы (I) могут находиться в форме сложных эфиров, таких как фосфолипид, глицерид или C1-C6-алкиловый сложный эфир. По меньшей мере в одном варианте осуществления, сложный эфир выбран из глицерида или C1-C6-алкилового сложного эфира. По меньшей мере в одном варианте осуществления, сложный эфир выбран из триглицерида, 1,2-диглицерида, 1,3-диглицерида, 1-моноглицерида, 2-моноглицерида, метилового эфира, этилового эфира, пропилового эфира, изопропилового эфира, н-бутилового эфира и трет-бутилового эфира. По меньшей мере в одном варианте осуществления, соединение формулы (I) находится в виде метилового эфира, этилового эфира, изопропилового эфира, н-бутилового эфира или трет-бутилового эфира, например, в виде метилового эфира или этилового эфира. По данным исследований гидролиза in vitro в подходящих биологических средах показано, что сложные эфиры, описывающиеся формулой (I) (т.е. этиловый эфир и бутиловый эфир), будут быстро гидролизоваться в желудочно-кишечном тракте.
Соли, подходящие для настоящего изобретения, включают, но не ограничиваются только ими, соли NH4+; ионов металлов, таких как Li+, Na+, K+, Mg2+ или Са2+; протонированных первичных аминов, таких как трет-бутиламмоний, (3S,5S,7S)-адамантан-1-аммоний, 1,3-дигидрокси-2-(гидроксиметил)пропан-2-аммоний, протонированного аминопиридина (например, пиридин-2-аммоний); протонированного вторичного амина, такого как диэтиламмоний, 2,3,4,5,6-пентагидрокси-N-метилгексан-1-аммоний, N-этилнафталин-1-аммоний, протонированного третичного амина, такого как 4-метилморфолин-4-ий, протонированного четвертичного амина, такого как 2-гидрокси-N,N,N-триметилэтан-1-аминий, и протонированного гуанидина, такого как амино((4-амино-4-карбоксибутил)амино)метаниминий или протонированного гетероцикла, такого как 1Н-имидазол-3-ий. Дополнительные примеры подходящих солей включают соли дипротонированного диамина, такого как этан-1,2-диамминий или пиперазин-1,4-диий. Другие соли, соответствующие настоящему изобретению, могут содержать протонированный хитозан:
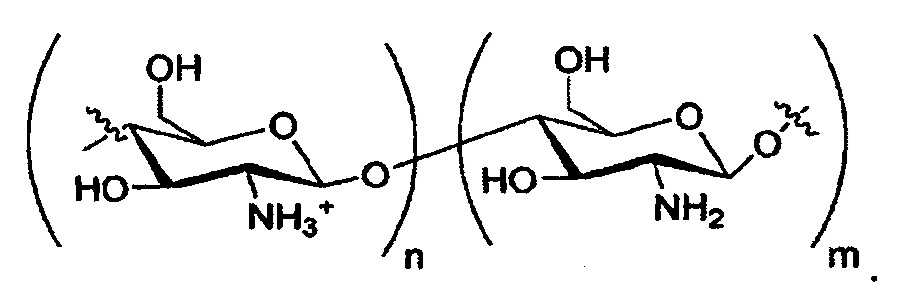
По меньшей мере в одном варианте осуществления соли выбраны из натриевой соли, кальциевой соли и соли с холином.
Настоящее изобретение относится к способу уменьшения содержания apoC-III у нуждающегося в нем субъекта, включающему введение субъекту фармацевтически эффективного количества соединения формулы (I). Субъектом может быть человек или млекопитающее, не являющееся человеком. Соединения, раскрытые в настоящем изобретении, можно вводить в качестве лекарственного средства, например, в фармацевтической композиции.
По меньшей мере в одном варианте осуществления, настоящее изобретение относится к способу уменьшения содержания apoC-III у субъекта, которого лечат статином, и обладающего исходным содержанием триглицеридов натощак, равным от примерно 200 мг/дл до примерно 499 мг/дл, путем введения субъекту фармацевтически эффективного количества соединения формулы (I). В другом варианте осуществления настоящее изобретение относится к применению фармацевтически эффективного количества соединения формулы (I) для приготовления лекарственного средства для уменьшения содержания apoC-III у субъекта, которого лечат статином, и обладающего исходным содержанием триглицеридов натощак, равным примерно 200 мг/дл до примерно 499 мг/дл. Содержание apoC-III можно уменьшить по меньшей мере примерно на 20%, по меньшей мере примерно на 25%, по меньшей мере примерно на 30% или по меньшей мере примерно на 35%.
По меньшей мере в одном варианте осуществления, настоящее изобретение относится к способу уменьшения содержания apoC-III у субъекта, обладающего исходным содержанием триглицеридов натощак, равным от примерно 200 мг/дл до примерно 499 мг/дл, путем введения субъекту фармацевтически эффективного количества соединения формулы (I). В другом варианте осуществления настоящее изобретение относится к применению фармацевтически эффективного количества соединения формулы (I) для приготовления лекарственного средства для уменьшения содержания apoC-III у субъекта, обладающего исходным содержанием триглицеридов натощак, равным примерно 200 мг/дл до примерно 499 мг/дл. Содержание apoC-III можно уменьшить по меньшей мере примерно на 20%, по меньшей мере примерно на 25%, по меньшей мере примерно на 30% или по меньшей мере примерно на 35%.
По меньшей мере в одном варианте осуществления, настоящее изобретение относится к способу уменьшения содержания apoC-III у субъекта, которого лечат статином, и обладающего исходным содержанием триглицеридов натощак, равным более 500 мг/дл, путем введения субъекту фармацевтически эффективного количества соединения формулы (I). В другом варианте осуществления настоящее изобретение относится к применению фармацевтически эффективного количества соединения формулы (I) для приготовления лекарственного средства для уменьшения содержания apoC-III у субъекта, которого лечат статином, и обладающего исходным содержанием триглицеридов натощак, равным более 500 мг/дл. Содержание apoC-III можно уменьшить по меньшей мере примерно на 25%, по меньшей мере примерно на 30%, по меньшей мере примерно на 35% или по меньшей мере примерно на 40%.
По меньшей мере в одном варианте осуществления, настоящее изобретение относится к способу уменьшения содержания apoC-III у субъекта, обладающего исходным содержанием триглицеридов натощак, равным более 500 мг/дл, путем введения субъекту фармацевтически эффективного количества соединения формулы (I). В другом варианте осуществления настоящее изобретение относится к применению фармацевтически эффективного количества соединения формулы (I) для приготовления лекарственного средства для уменьшения содержания apoC-III у субъекта, обладающего исходным содержанием триглицеридов натощак, равным более 500 мг/дл. Содержание apoC-III можно уменьшить по меньшей мере примерно на 25%, по меньшей мере примерно на 30%, по меньшей мере примерно на 35% или по меньшей мере примерно на 40%.
Настоящее изобретение также относится к способу уменьшения содержания apoC-III у субъекта, обладающего исходным содержанием натощак ЛНП (липопротеины низкой плотности)-холестерина, равным по меньшей мере 2,5 ммоль/л (~97 мг/дл), путем введения субъекту фармацевтически эффективного количества соединения формулы (I). В другом варианте осуществления настоящее изобретение относится к применению фармацевтически эффективного количества соединения формулы (I) для приготовления лекарственного средства для уменьшения содержания apoC-III у субъекта, обладающего исходным содержанием натощак ЛНП-холестерина, равным по меньшей мере 2,5 ммоль/л (~97 мг/дл). Содержание apoC-III можно уменьшить по меньшей мере примерно на 25%, по меньшей мере примерно на 30%, по меньшей мере примерно на 35% или по меньшей мере примерно на 40%.
По меньшей мере в одном варианте осуществления настоящее изобретение относится к способу уменьшения содержания apoC-III у нуждающегося в нем субъекта, включающему введение субъекту фармацевтически эффективного количества антидислипидемического средства, такого как, например, статин и соединение формулы (I).
Композиция, раскрытая в настоящем изобретении, может содержать по меньшей мере одно соединение формулы (I) и необязательно по меньшей мере один неактивный фармацевтический ингредиент, т.е. наполнитель. Неактивные ингредиенты можно солюбилизировать, суспендировать, загущать, разбавлять, эмульгировать, стабилизировать, консервировать, защищать, окрашивать, ароматизировать, и/или превращать активные ингредиенты в применимый и эффективный препарат, так чтобы он мог быть безопасным, удобным и/или в другом отношении приемлемым для применения. Примеры инертных наполнителей включают, но не ограничиваются только ими, растворители, носители, разбавители, связующие, наполнители, подсластители, ароматизаторы, модификаторы pH, модификаторы вязкости, антиоксиданты, средства, увеличивающие объем, влагоудерживающие средства, разрыхляющие агенты, замедляющие растворение агенты, ускорители всасывания, смачивающие агенты, абсорбенты, смазывающие вещества, окрашивающие агенты, диспергирующие агенты и консерванты. Инертные наполнители могут исполнять более чем одну роль или функцию, или их можно разделить более чем на одну группу; разделение на группы является только описательным и не предназначено для наложения ограничений. В некоторых вариантах осуществления, например, по меньшей мере один наполнитель можно выбрать из группы, включающей кукурузный крахмал, лактозу, глюкозу, микрокристаллическую целлюлозу, стеарат магния, поливинилпирролидон, лимонную кислоту, винную кислоту, воду, этанол, глицерин, сорбит, полиэтиленгликоль, пропиленгликоль, цетилстеариловый спирт, карбоксиметилцеллюлозу и жирные вещества, такие как твердый жир, или подходящие их смеси. В некоторых вариантах осуществления композиции, раскрытые в настоящем изобретении, содержат по меньшей мере одно соединение формулы (I) и по меньшей мере один фармацевтически приемлемый антиоксидант, например, токоферол, такой как альфа-токоферол, бета-токоферол, гамма-токоферол и дельта-токоферол, или их смеси, БГА, такой как 2-трет-бутил-4-гидроксианизол и 3-трет-бутил-4-гидроксианизол или их смеси, и БГТ (3,5-ди-трет-бутил-4-гидрокситолуол) или их смеси.
Композиции, раскрытые в настоящем изобретении, можно приготовить в формах для перорального введения, таких как, например, таблетки или капсулы из мягкого или твердого желатина. Дозированная форма может обладать любой формой, подходящей для перорального введения, такой как сферическая, овальная, эллипсоидальная, кубическая, правильной и/или неправильной формой. Обычные технологии, известные в данной области техники, можно использовать для приготовления соединений, соответствующих настоящему изобретению. В некоторых вариантах осуществления композиция может находиться в форме желатиновой капсулы или таблетки.
Подходящая суточная доза соединения формулы (I) может находиться в диапазоне от примерно 5 мг до примерно 2 г. Например, в некоторых вариантах осуществления суточная доза составляет от примерно 50 мг до примерно 1 г, от примерно 100 мг до примерно 1 г, от примерно 50 мг до примерно 800 мг, от примерно 100 мг до примерно 800 мг, от примерно 100 мг до примерно 600 мг. По меньшей мере в одном варианте осуществления суточная доза составляет от примерно 200 мг до примерно 600 мг. Соединения можно вводить, например, один, два или три раза в сутки. По меньшей мере в одном варианте осуществления соединение формулы (I) вводят в количестве, находящемся в диапазоне от примерно 200 мг до примерно 800 мг на дозу. По меньшей мере в одном варианте осуществления, соединение формулы (I) вводят один раз в сутки.
Авторы настоящего изобретения установили, что соединения формулы (I), такие как 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутановая кислота, обладают примечательно хорошей фармацевтической активностью. Неожиданно установлено, что соединения формулы (I), раскрытые в настоящем изобретении, обладают улучшенной биологической активностью по сравнению с природными омега-3-жирными кислотами, такими как ЭПК и ДГК, при уменьшении содержания apoC-III.
Например, в некоторых вариантах осуществления соединения формулы (I) могут уменьшить медианное содержание apoC-III в плазме или в печени по меньшей мере на 25-30% по сравнению с исходным содержанием, т.е. обеспечить более значительное уменьшение, чем обеспечиваемое при использовании имеющихся комбинаций ЭПК/ДГК/ДПК. Поскольку показано, что соединения формулы (I) уменьшают содержание apoC-III мРНК в доклинических моделях (и таким образом предположительно также влияют на продуцирование/секрецию в печени), можно полагать, что добавление лекарственных средств, уменьшающих содержание липидов, которые уменьшают содержание apoC-III путем увеличенного потребления в печени частиц apo-B, например, статинов или ингибиторов PCSK-9, окажут дополнительные воздействия, уменьшающие содержание apoC-III в плазме.
Примеры
Настоящее изобретение можно дополнительно описать с помощью приведенных ниже неограничивающих примеров, в которых можно использовать стандартные методики, известные подготовленному химику, и методики, аналогичные описанным в этих примерах, если это является подходящим. Следует понимать, что специалист в данной области техники должен представлять себе дополнительные варианты осуществления, согласующиеся с приведенным настоящим изобретением.
Если не указано иное, реакции проводили при комнатной температуре, обычно в диапазоне 18-25°C с использованием растворителей квалификации "для ВЭЖХ (высокоэффективная жидкостная хроматография)" в безводных условиях. Выпаривание проводили путем выпаривания в роторном испарителе в вакууме. Колоночную хроматографию проводили во флэш-режиме на силикагеле. Химические сдвиги в ядерном магнитном резонансе (ЯМР) регистрировали на приборе Bruker Avance DPX 200 или 300, или на приборе AVII 400 и мультиплетности пиков обозначены следующим образом: s, синглет; d, дублет; dd, дублет дублетов; t, триплет; q, квадруплет; p, пентаплет; m, мультиплет; br, уширенный. Масс-спектры регистрировали на масс-спектрометре GI956A (электрораспыление, 3000 В) с переключением между режимами ионизации с образованием положительных и отрицательных ионов. Приведенные выходы являются иллюстративными и необязательно указывают максимальный достигаемый выход.
Пример 1: Получение трет-бутил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутаноата:
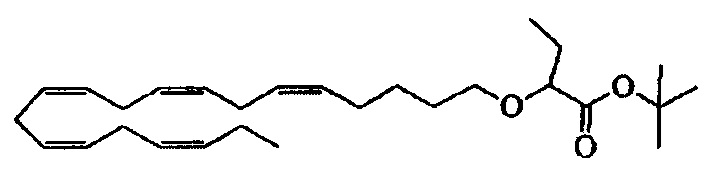
Тетрабутиламмонийхлорид (0,55 г, 1,98 ммоля) добавляли к раствору (5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ола (3,50 г, 12,1 ммоля) в толуоле (35 мл) при комнатной температуре в атмосфере азота. Водный раствор гидроксида натрия (50% (мас./мас.), 11,7 мл) добавляли при энергичном перемешивании при комнатной температуре, затем трет-бутил-2-бромбутират (5,41 г, 24,3 ммоля). Полученную смесь нагревали при 50°C и дополнительное количество трет-бутил-2-бромбутирата добавляли через 1,5 ч (2,70 г, 12,1 ммоля), 3,5 ч (2,70 г, 12,1 ммоля) и 4,5 ч (2,70 г, 12,1 ммоля) и перемешивали в течение всего 12 ч. После охлаждения до комнатной температуры добавляли воду со льдом (25 мл) и образовавшиеся две фазы разделяли. Органическую фазу промывали смесью NaOH (5%) и рассолом, сушили (MgSO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием смесей гептана и этилацетата увеличивающейся полярности (100:0 -> 95:5) в качестве элюента. Концентрирование соответствующих фракций давало 1,87 г (выход 36%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3): δ 0,85-1,10 (m, 6Н), 1,35-1,54 (m, 11Н), 1,53-1,87 (m, 4Н), 1,96-2,26 (m, 4Н), 2,70-3,02 (m, 8Н), 3,31 (dt, 1Н), 3,51-3,67 (m, 2Н), 5,10-5,58 (m, 10H).
Пример 2: Получение 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановой кислоты (соединение А):
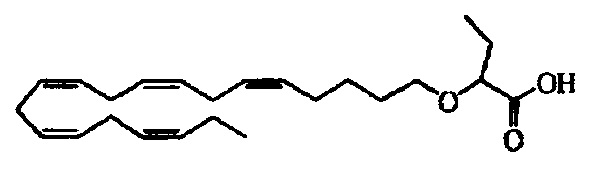
трет-Бутил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)-бутаноат (19,6 г, 45,5 ммоля) растворяли в дихлорметане (200 мл) и помещали в атмосферу азота. Добавляли трифторуксусную кислоту (50 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли воду и водную фазу дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана, этилацетата и муравьиной кислоты (90:10:1 -> 80:20:1) в качестве элюента. Концентрирование соответствующих фракций давало 12,1 г (выход 71%) искомого соединения в виде масла. 1Н-ЯМР (300 МГц, CDCl3): δ 0,90-1,00 (m, 6Н), 1,50 (m, 2Н), 1,70 (m, 2Н), 1,80 (m, 2Н), 2,10 (m, 4Н), 2,80-2,90 (m, 8Н), 3,50 (m, 1Н), 3,60 (m, 1Н), 3,75 (t, 1Н), 5,30-5,50 (m, 10H); МС (масс-спектр) (электрораспыление): 373,2 [М-Н]-.
Пример 3: Получение (4S,5R)-3-((S)-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутаноил)-4-метил-5-фенилоксазолидин-2-она и (4S,5R)-3-((R)-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенил-окси)бутаноил)-4-метил-5-фенилоксазолидин-2-она:
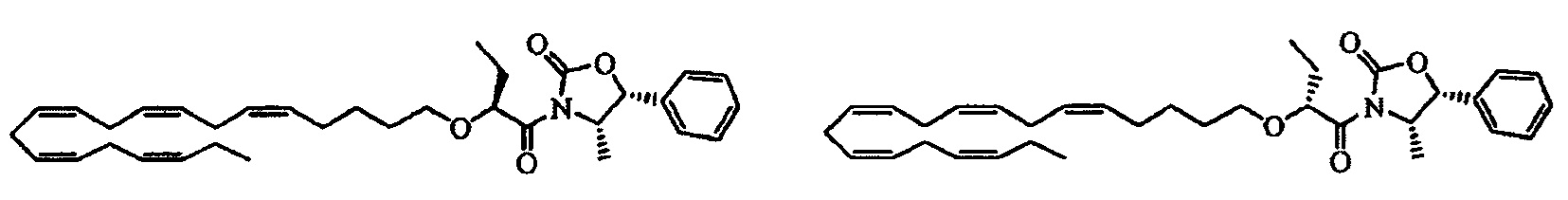
ДМАП (диметиламинопиридин) (1,10 г, 8,90 ммоля) и ДЦК (1,90 г, 9,30 ммоля) добавляли к смеси 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановой кислоты (3,20 г, 8,50 ммоля) в сухом дихлорметане (100 мл), выдерживаемой при 0°C в атмосфере азота. Полученную смесь перемешивали при 0°C в течение 20 мин. (4S,5R)-4-метил-5-фенилоксазолидин-2-он (1,50 г, 8,50 ммоля) добавляли и полученную мутную смесь перемешивали при температуре окружающей среды в течение 5 дней. Смесь фильтровали и концентрировали при пониженном давлении и получали неочищенный продукт, содержащий искомый продукт в виде смеси двух диастереоизомеров. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 15% этилацетата в гептане в качестве элюента. Эти два диастереоизомера разделяли и соответствующие фракции концентрировали. (4S,5R)-3-((S)-2-((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаенилокси)бутаноил)-4-метил-5-фенилоксазолидин-2-он элюировался первым и получали 1,1 г (выход 40%) в виде масла. (4S,5R)-3-((R)-2-((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаенилокси)бутаноил)-4-метил-5-фенилоксазолидин-2-он получали в количестве, равном 0,95 г (выход 34%), в виде масла.
(4S,5R)-3-((S)-2-((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаенилокси)-бутаноил)-4-метил-5-фенилоксазолидин-2-он (Е1): 1Н-ЯМР (300 МГц, CDCl3): δ 0,90 (d, 3H), 1,00 (t, 3H), 1,07 (t, 3H), 1,45-1,57 (m, 2Н), 1,62-1,76 (m, 3H), 1,85-1,95 (m, 1Н), 2,05-2,15 (m, 4Н), 2,87 (m, 8Н), 3,39 (m, 1Н), 3,57 (m, 1Н), 4,85-4,92 (m, 2Н), 5,30-5,45 (m, 10H), 5,75 (d, 1Н), 7,32 (m, 2Н), 7,43 (m, 3H).
(4S,5R)-3-((R)-2-((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаенилокси)-бутаноил)-4-метил-5-фенилоксазолидин-2-он (Е2): 1Н-ЯМР (300 МГц, CDCl3): δ 0,98 (d, 3H), 0,99 (t, 3H), 1,08 (t, 3H), 1,40-1,52 (m, 2Н), 1,55-1,75 (m, 3H), 1,80-1,90 (m, 1Н), 2,05-2,15 (m, 4Н), 2,84 (m, 8Н), 3,39 (m, 1Н), 3,56 (m, 1Н), 4,79 (пентет, 1Н), 4,97 (dd, 1Н), 5,30-5,45 (m, 10H), 5,71 (d, 1Н), 7,33 (m, 2Н), 7,43 (m, 3H).
Пример 4: Получение (S)-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановой кислоты:
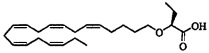
Пероксид водорода (35% в воде, 0,75 мл, 8,54 ммоля) и моногидрат гидроксида лития (0,18 г, 4,27 ммоля) добавляли к раствору (4S,5R)-3-((S)-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутаноил)-4-метил-5-фенилоксазолидин-2-она (1,10 г, 2,13 ммоля) в тетрагидрофуране (12 мл) и воде (4 мл), выдерживаемому при 0°C в атмосфере азота. Реакционную смесь перемешивали при 0°C в течение 30 мин. Добавляли 10% Na2SO3 (водный раствор) (30 мл), значение pH устанавливали равным ~2 с помощью 2М HCl и смесь дважды экстрагировали гептаном (30 мл). Объединенный органический экстракт сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (98:8 -> 1:1) в качестве элюента. Концентрирование соответствующих фракций давало 0,48 г (выход 60%) искомого соединения в виде масла. 1Н-ЯМР (300 МГц, CDCl3): δ 0,90-1,00 (m, 6Н), 1,48 (m, 2Н), 1,65 (m, 2Н), 1,85 (m, 2Н), 2,10(m, 4Н), 2,80-2,90 (m, 8Н), 3,55 (m, 1Н), 3,60 (m, 1Н), 3,88 (t, 1Н), 5,35-5,45 (m, 10H); МС (электрораспыление): 373,3 [М-Н]-; [α]D -37° (с=0,104, этанол).
Пример 5: Получение (R)-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутановой кислоты:
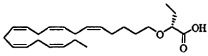
Пероксид водорода (35% в воде, 0,65 мл, 7,37 ммоля) и моногидрат гидроксида лития (0,15 г, 3,69 ммоля) добавляли к раствору (4S,5R)-3-((R)-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)бутаноил)-4-метил-5-фенилоксазолидин-2-она (0,95 г, 1,84 ммоля) в тетрагидрофура не (12 мл) и воде (4 мл), выдерживаемому при 0°C в атмосфере азота. Реакционную смесь перемешивали при 0°C в течение 30 мин. Добавляли 10% Na2SO3 (водный раствор) (30 мл), значение pH устанавливали равным 2 с помощью 2М HCl и смесь дважды экстрагировали гептаном (30 мл). Объединенный органический экстракт сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (98:8 -> 50:50) в качестве элюента. Концентрирование соответствующих фракций давало 0,19 г (выход 29%) искомого соединения в виде масла. 1Н-ЯМР (300 МГц, CDCl3): δ 0,90-1,00 (m, 6Н), 1,48 (m, 2Н), 1,65 (m, 2Н), 1,85 (m, 2Н), 2,10 (m, 4Н), 2,80-2,90 (m, 8Н), 3,55 (m, 1Н), 3,60 (m, 1Н), 3,88 (t, 1Н), 5,35-5,45 (m, 10H); МС (электрораспыление): 373,3 [М-Н]-; [α]D -31° (с=0,088, этанол).
Пример 6: Получение трет-бутил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)пропаноата:
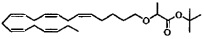
Смесь (5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ола, (1,00 г, 3,47 ммоля), тетрабутиламмонийхлорида (0,24 г, 0,87 ммоля) и трет-бутил-2-бромпропионата (3,62 г, 17,3 ммоля) растворяли в толуоле (36 мл) и помещали в атмосферу азота. Водный раствор гидроксида натрия (50%, 8 мл) медленно добавляли при энергичном перемешивании и полученную смесь перемешивали при температуре окружающей среды в течение 20 ч. Добавляли воду и смесь трижды экстрагировали эфиром. Объединенный органический экстракт промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 2% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 1,40 г (выход 90%) искомого соединения в виде масла. 1Н-ЯМР (300 МГц, CDCl3): δ 0,95 (t, 3H), 1,41 (d, 3H), 1,48 (s, 9Н), 1,48-1,66 (m, 4Н), 2,05 (m, 4Н), 2,83 (m, 8Н), 3,35 (m, 1Н), 3,55 (m, 1Н), 3,79 (q, 1Н), 5,32-5,44 (m, 10H).
Пример 7: Получение 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)пропановой кислоты:
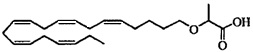
Трифторуксусную кислоту (2 мл) добавляли к раствору 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)пропаноата (1,40 г, 3,36 ммоля) в дихлорметане (10 мл), выдерживаемому в атмосфере азота, и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли диэтиловый эфир (50 мл) и органическую фазу промывали водой (30 мл), сушили (Na2SO4) и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана, этилацетата и муравьиной кислоты (95:5:0,25 -> 80:20:1) в качестве элюента. Концентрирование соответствующих фракций давало 0,67 г немного загрязненного продукта. Это вещество растворяли в гептане (15 мл), трижды промывали водой (5 мл), сушили (Na2SO4), фильтровали и концентрировали и получали 0,50 г (выход 41%) искомого соединения в виде масла. 1Н-ЯМР (300 МГц, CDCl3): δ 0,99 (t, 3H), 1,40-1,48 (m, 5Н), 1,67 (m, 2Н), 2,09 (m, 4Н), 2,80-2,60 (m, 8Н), 3,53 (m, 2Н), 4,01 (q, 1Н), 5,31-5,47 (m, 10H); МС (электрораспыление): 359,2 [М-Н]-.
Пример 8: Получение трет-бутил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)-2-метилпропаноата:
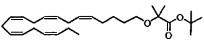
Смесь (5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ола (0,83 г, 3,14 ммоля), тетрабутиламмонийхлорида (0,24 г, 0,85 ммоля) и трет-бутил-2-бромизобутирата (3,50 г, 15,7 ммоля) растворяли в толуоле (15 мл) и помещали в атмосферу азота. Водный раствор гидроксида натрия (50%, 5 мл) медленно добавляли при энергичном перемешивании при комнатной температуре. Полученную смесь нагревали при 60°C и перемешивали в течение 6 ч. Смесь охлаждали, добавляли воду и трижды экстрагировали эфиром. Объединенный органический экстракт промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 5-10% этилацетата в гептане в градиентном режиме в качестве элюента. Концентрирование соответствующих фракций давало 0,60 г (выход 44%) искомого соединения в виде масла. МС (электрораспыление): 453,3 [M+Na]+.
Пример 9: Получение 2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)-2-метилпропановой кислоты:
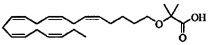
Трифторуксусную кислоту (5 мл) добавляли к раствору трет-бутил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилокси)-2-метил-пропаноат (600 мг, 1,39 ммоля) в дихлорметане (20 мл) в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли воду и водную фазу дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием смеси гептана, этилацетата и муравьиной кислоты (80:20:1) в качестве элюента. Соответствующие фракции концентрировали и остаток (135 мг) дополнительно очищали с помощью флэш-хроматографии на силикагеле с использованием в градиентном режиме 5-10% смеси этилацетата и муравьиной кислоты (95:5) в гептане в качестве элюента. Концентрирование соответствующих фракций давало 80 мг немного загрязненного продукта. Это вещество растворяли в гептан (5 мл), дважды промывали водой (5 мл), сушили (Na2SO4), фильтровали и концентрировали и получали 40 мг (выход 8%) искомого соединения в виде масла. 1Н-ЯМР (300 МГц, CDCl3): δ 0,99 (t, 3H), 1,47 (s, 6Н), 1,64 (m, 2Н), 2,07 (m, 4Н), 2,81-2,88 (m, 8Н), 3,46 (t, 2Н), 5,29-5,44 (m, 10H); МС (электрораспыление): 373,3 [М-Н]-.
Пример 10: Получение трет-бутил-2-этил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутаноата:
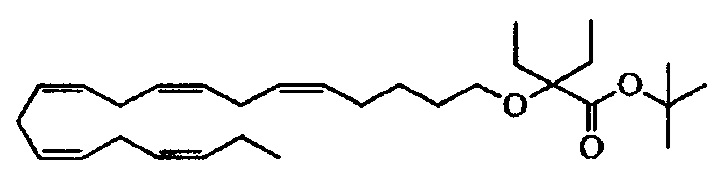
трет-Бутил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)-бутаноат (480 мг, 1,11 ммоля) по каплям добавляли в течение 30 мин к раствору диизопропиламина лития (ДАЛ) (2,0 М, 750 мкл, 1,50 ммоля) в сухом тетрагидрофуране (10 мл), выдерживаемому при -70°C в атмосфере азота. Реакционную смесь перемешивали в течение 30 мин. Одной порцией добавляли этилйодид (312 мг, 2,00 ммоля) и полученную смесь нагревали до температуры окружающей среды в течение 1 ч. Реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Смесь выливали в насыщенный NH4Cl (водный раствор) (50 мл) и экстрагировали гептаном (2×50 мл). Объединенные органические фазы последовательно промывали рассолом (50 мл), 0,25 М HCl (50 мл) и рассолом (50 мл), сушили (MgSO4), фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (100:0 -> 95:5) в качестве элюента. Концентрирование соответствующих фракций давало 343 мг (выход 67%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3): δ 0,84 (t, 6Н), 0,99 (td, 3H), 1,35-1,55 (m, 11Н), 1,54-1,69 (m, 2Н), 1,68-1,87 (m, 4Н), 1,99-2,24 (m, 4Н), 2,74-2,99 (m, 8Н), 3,31 (t, 2Н), 5,23-5,52 (m, 10H); МС (электрораспыление): 401,3 [М-1]-.
Пример 11: Получение 2-этил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутановой кислоты:
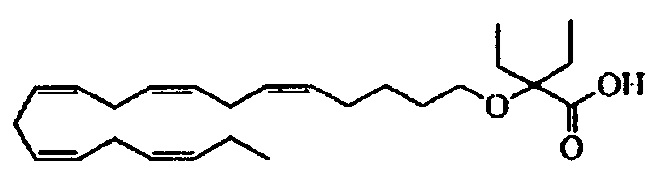
Смесь муравьиной кислоты (5 мл) и трет-бутил-2-этил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-илокси)бутаноат (250 мг, 0,55 ммоля) энергично перемешивали в атмосфере азота при комнатной температуре в течение 4,5 ч. Муравьиную кислоту удаляли в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (100:0 -> 80:20) в качестве элюента. Концентрирование соответствующих фракций давало 163 мг (выход 74%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3): δ 0,86 (t, 6Н), 0,99 (t, 3H), 1,36-1,57 (m, 2Н), 1,68 (dd, 2Н), 1,73-1,98 (m, 4Н), 2,11 (tt, 4Н), 2,70-3,01 (m, 8Н), 3,39 (t, 2Н), 5,20-5,56 (m, 10H). МС (электрораспыление): 481,4 [M+Na]+.
Пример 12: Получение этил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата:
Дициклогексилметандиимин (ДЦК) (304 мг, 1,47 ммоля) и N,N-диметилпиридин-4-амин (ДМАП) (10 мг, 0,067 ммоля) при перемешивании добавляли к раствору 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты (501,3 мг, 1,335 ммоля) в дихлорметане (ДХМ) (4 мл) при 0°C в атмосфере N2. Смесь перемешивали в течение 5 мин, затем добавляли этанол (EtOH) (0,16 мл, 2,67 ммоля). Полученную смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (100:0 → 99:1) в качестве элюента. Концентрирование соответствующих фракций давало 473 мг (выход 88%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, хлороформ-d) δ 0,95 (2×t, 6Н), 1,37-1,48 (m, 2Н), 1,54-1,79 (m, 4Н), 2,01-2,10 (m, 4Н), 2,77-2,84 (m, 8Н), 3,27-3,34 (m, 1Н), 3,53-3,60 (m, 1Н), 3,69-3,73 (dd, 1Н), 4,13-4,24 (m, 2Н), 5,25-5,33 (m, 10H), МС (электрораспыление); 425,3 [M+Na]+; МСВР (масс-спектроскопия высокого разрешения) (электрораспыление): Найдено 425,3021 [M+Na]+, рассчитано для [C26H42O3+Na]+ 425,3031.
Пример 13: Получение изопропил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
ДЦК (310 мг, 1,47 ммоля) и ДМАП (9 мг, 0,067 ммоля) при перемешивании добавляли к раствору 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты (501 мг, 1,335 ммоля) в ДХМ (дихлорметан) (4 мл) при 0°C в атмосфере N2. Смесь перемешивали в течение 5 мин, затем добавляли изопропанол (iPrOH) (0,16 мл, 2,67 ммоля). Полученную смесь перемешивали при комнатной температуре в течение 20 ч. Смесь фильтровали и концентрировали в вакууме. К остатку добавляли гептан (50 мл), фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 1% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 496 мг (выход 89%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3): δ 0,97 (2×t, 6Н), 1,25 (m, 6Н), 1,42-1,50 (m, 2Н), 1,61-1,70 (m, 2Н), 1,70-1,77 (m, 2Н), 2,05-2,12 (m, 4Н), 2,79-2,86 (m, 8Н), 3,29-3,34 (m, 1Н), 3,54-3,59 (m, 1Н), 3,67-3,71 (m, 1Н), 5,06-5,10 (m, 1Н), 5,31-5,42 (m, 10Н); МС (электрораспыление): 439,3 [M+Na]+.
Пример 14: Получение метил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата:
Серную кислоту (0,049 мл, 0,918 ммоля) добавляли к раствору 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты ((385 мг, 1,028 ммоля) в метаноле (20 мл) при комнатной температуре в атмосфере N2 и полученную смесь перемешивали при комнатной температуре в течение ночи. МС (электрораспыление): 389,3 [М+1]+.
Пример 15: Получение бутил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата:
Серную кислоту (0,049 мл, 0,918 ммоля) добавляли к раствору 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты ((33 г, 88 ммоля) в бутан-1-оле (400 мл, 4,37 моля) при комнатной температуре в атмосфере N2 и реакционную смесь перемешивали в течение 120 ч. Добавляли гептан (400 мл) и этилацетат (400 мл) и раствор промывали насыщенным водным раствором. NaHCO3 (3×300 мл) и водой (2×300 мл). Объединенную водную фазу экстрагировали смесью гептан/эфир (1:1) (2×300 мл). Объединенную органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (99:1 → 96:4) в качестве элюента. Концентрирование соответствующих фракций давало 26,3 г (выход 67%) искомого соединения в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ 0,93-1,02 (m, 9Н), 1,36-1,51 (m, 4Н), 1,60-1,70 (m, 4Н), 1,72-1,84 (m, 2Н), 2,05-2,16 (m, 4Н), 2,78-2,92 (m, 8Н), 3,28-3,39 (m, 1Н), 3,54-3,65 (m, 1Н), 3,70-3,82 (m, 1Н), 4,08-4,24 (m, 2Н), 5,27-5,48 (m, 10H), МС (электрораспыление): 453,2 [M+Na]+.
Пример 16: Получение 2,3-дигидроксипропил 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата:
Стадия а) Получение (2,2-диметил-1,3-диоксолан-4-ил)метил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
2-(((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (25 г, 66,7 ммоля) и ДМАП (8,15 г, 66,7 ммоля) добавляли к раствору 2,2-диметил-1,3-диоксолан-4-метанола (7,54 мл, 60,7 ммоля) в хлороформе (150 мл) в атмосфере азота. Затем при температуре окружающей среды по каплям добавляли раствор ДЦК (13,77 г, 66,7 ммоля) в хлороформе (65 мл). Смесь перемешивали в течение ночи и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 10% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 19,6 г (выход 66%) искомого продукта в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,99 (t, 6Н), 1,37-1,40 (m, 3H), 1,41-1,53 (m, 5Н), 1,59-1,71 (m, 2Н), 1,72-1,85 (m, 2Н), 2,05-2,14 (m, 4Н), 2,74-2,95 (m, 8Н), 3,31-3,38 (m, 1Н), 3,57-3,65 (m, 1Н), 3,72-3,86 (m, 2Н), 4,07-4,12 (m, 1Н), 4,15-4,27 (m, 2Н), 4,29-4,40 (m, 1Н), 5,23-5,50 (m, 10H). МС (электрораспыление): 511,3 [M+Na]+.
Стадия b) Получение 2,3-дигидроксипропил 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
К раствору (2,2-диметил-1,3-диоксолан-4-ил)метил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата (27,5 г, 56,3 ммоля) в диоксане (280 мл) при комнатной температуре в атмосфере азота добавляли водный раствор HCl (37% (мас./мас.), 28 мл, 341 ммоля) и смесь перемешивали в течение 60 мин. Затем смесь осторожно выливали в насыщенный водный раствор NaHCO3 (500 мл) и экстрагировали с помощью EtOAc (2×300 мл). Органическую фазу промывали с помощью 1М HCl (200 мл), рассолом (200 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием смеси гептана и этилацетата (50:50) в качестве элюента. Концентрирование соответствующих фракций давало 19 г искомого продукта в виде масла, содержащего ~10% изомера 1,3-дигидроксипропан-2-ил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата. Вещество смешивали с 1,35 г из другой партии, затем дополнительно очищали с помощью препаративной ВЭЖХ. В изократическом режиме в качестве элюента использовали смесь 17:83 от вода/ацетонитрил (9:1) до ацетонитрила (100%). Концентрирование соответствующих фракций давало 11,3 г (выход 38%) искомого продукта в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,97-1,03 (m, 6Н), 1,41-1,51 (m, 2Н), 1,59-1,69 (m, 2Н), 1,72-1,87 (m, 2Н), 2,05-2,14 (m, 5Н), 2,56 (s, 1Н), 2,73-2,94 (m, 8Н), 3,33-3,40 (m, 1Н), 3,55-3,68 (m, 2Н), 3,69-3,77 (m, 1Н), 3,79-3,85 (m, 1Н), 3,93-4,03 (m, 1Н), 4,15-4,37 (m, 2Н), 5,25-5,51 (m, 10H). МС (электрораспыление): 471,3 [M+Na]+.
Пример 17: Получение 1,3-дигидроксипропан-2-ил 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
Стадия а) Получение оксиран-2-илметил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
Смесь 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты (800 мг, 2,14 ммоля), глицидола (0,17 мл, 2,56 ммоля), 1-(3-диметиламинопропил)-3-этил карбодиимидгидрохлорида (EDC*HCl) (491 мг, 2,56 ммоля) и 4-диметиламинопиридин (ДМАП) (313 мг, 2,56 ммоля) в сухом ДХМ (7 мл) перемешивали при комнатной температуре в атмосфере N2. Реакционную смесь концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (99:1 → 95:5) в качестве элюента. Концентрирование соответствующих фракций давало 527 мг (выход 57%) искомого продукта в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ 0,94-0,98 (m, 6Н), 1,40-1,44 (m, 2Н), 1,57-1,64 (m, 2Н), 1,70-1,82 (m, 2Н), 2,02-2,12 (m, 4Н), 2,63 (bs, 1Н), 2,78-2,84 (m, 9Н), 3,20 (bs, 1Н), 3,30-3,35 (m, 1Н), 3,55-3,61 (m, 1Н), 3,77-3,80 (m, 1Н), 3,94-4,01 (m, 1Н), 4,42-4,48 (m, 1Н), 5,36-5,26 (m, 10H). МС (электрораспыление): 453,3 [M+Na]+.
Стадия b) Получение 2-((2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноил)окси)пропан-1,3-диилбис(2,2,2-трифторацетата)
Трифторуксусный ангидрид (ТФУА) (0,55 мл, 3,96 ммоля) в сухом ДХМ (3 мл) порциями добавляли к предварительно охлажденному раствору оксиран-2-ил метил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата (286 мг, 0,66 ммоля) в сухом ДХМ (3 мл) при -20°C в атмосфере N2. Охлаждающую баню удаляли и смесь перемешивали в течение 19 ч при температуре окружающей среды, затем реакционную смесь концентрировали в вакууме. Остаток растворяли в толуоле (6 мл) и пропускали через слой диоксида кремния (6,5 г) при элюировании толуолом (150 мл). Концентрирование в вакууме приводило к 357 мг (выход 84%) искомого соединения в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ 0,95 (2×t, 6Н), 1,38-1,45 (m, 2Н), 1,57-1,63 (m, 2Н), 1,66-1,78 (m, 2Н), 2,09-2,02 (m, 4Н), 2,78-2,84 (m, 8Н), 3,27-3,33 (m, 1Н), 3,51-3,56 (m, 1Н), 3,77 (dd, 1Н), 4,30-4,53 (m, 2Н), 4,60-4,69 (m, 2Н), 5,17-5,43 (m, 10H), 5,43-5,55 (m, 1Н). МС (электрораспыление): 661,1 [M+Na]+.
Стадия с) Получение 1,3-дигидроксипропан-2-ил-2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
Раствор пиридина (0,4 мл, 4,95 ммоля) и метанола (0,3 мл, 7,41 ммоля) в смеси пентан/ДХМ (2:1) (4,5 мл) по каплям добавляли к раствору 2-((2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноил)окси)пропан-1,3-диилбис(2,2,2-трифторацетата) (354 мг, 0,552 ммоля) в смеси пентан/ДХМ (2:1) (5 мл), охлажденному до -20°C, в атмосфере N2. Охлаждающую баню удаляли и смесь перемешивали в течение 3 ч при комнатной температуре, затем концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (95:5 → 90:10 → 80:20 → 50:50) в качестве элюента. Концентрирование соответствующих фракций давало 223 мг искомого продукта в виде неочищенного масла. Очистка с помощью препаративной ВЭЖХ давало 58 мг (выход 22%) искомого соединения в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ 0,95 (t, 3H), 0,96 (t, 3H), 1,38-1,45 (m, 2Н), 1,54-1,64 (m, 2Н), 1,67-1,84 (m, 2Н), 2,01-2,09 (m, 4Н), 2,45 (bs, 2Н), 2,83-2,77 (m, 8Н), 3,36-3,30 (m, 1Н), 3,60-3,55 (m, 1Н), 3,84-3,78 (m, 5Н), 4,98-4,93 (m, 1Н), 5,65-5,09 (m, 10H). МС (электрораспыление): 471,1 [M+Na]+.
Пример 18: Получение 3-гидроксипропан-1,2-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата)
Стадия а) Получение трет-бутил((2,2-диметил-1,3-диоксолан-4-ил)метокси)диметилсилана
трет-Бутил-хлордиметилсилан (14,41 г, 91 ммоля) добавляли к раствору (2,2-диметил-1,3-диоксолан-4-ил)метанола (10 г, 76 ммоля) и имидазола (7,73 г, 114 ммоля) в ТГФ (тетрагидрофуран) (100 мл) при температуре окружающей среды в атмосфере азота. Смесь перемешивали в течение 1,5 ч, выливали в воду (200 мл) и экстрагировали трет-бутилметиловым эфиром (2×150 мл). Фазы разделяли и органический слой промывали водой (100 мл), рассолом (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 3% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 18 г (выход 97%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,02-0,05 (m, 6H), 0,85-0,89 (m, 9H), 1,31-1,35 (m, 3H), 1,35-1,40 (m, 3H), 3,50-3,60 (m, 1H), 3,63-3,72 (m, 1H), 3,75-3,85 (m, 1H), 3,96-4,05 (m, 1H), 4,07-4,18 (m, 1H). МС (электрораспыление): 229,2 [M+Na]+.
Стадия b) Получение 3-((трет-бутилдиметилсилил)окси)пропан-1,2-диола
К раствору трет-бутил((2,2-диметил-1,3-диоксолан-4-ил)метокси)диметилсилана в хлороформе (60 мл) добавляли FeCl3×6H2O, адсорбированный на силикагеле (30 г, 11,9 ммоля), и смесь перемешивали в течение ночи. Смесь фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (50:50 → 25:75) в качестве элюента. Концентрирование соответствующих фракций давало 760 мг (9%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,09-0,12 (m, 6Н). 0,91-0,95 (m, 9Н), 2,11-2,17 (m, 1Н), 2,60 (d, 1Н), 3,57-3,85 (m, 5Н). МС (электрораспыление): 229,2 [M+Na]+.
Стадия с) Получение 3-((трет-бутилдиметилсилил)окси)пропан-1,2-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)-бутаноата)
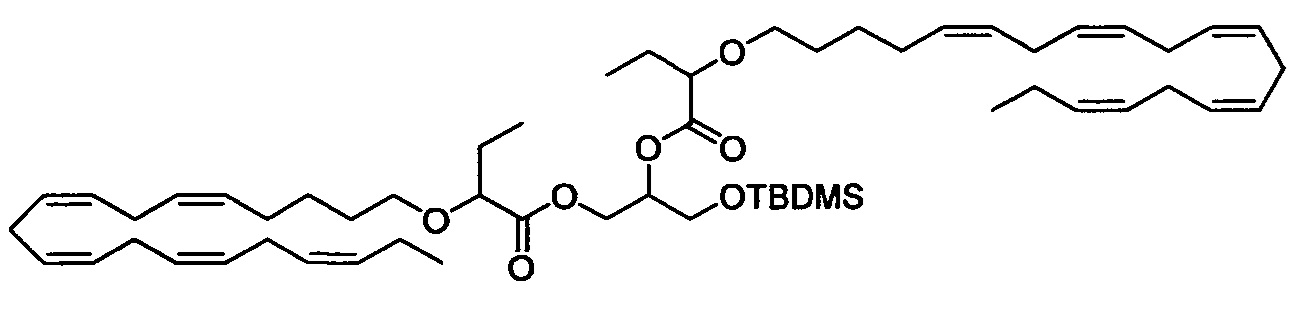
К раствору 3-((трет-бутилдиметилсилил)окси)пропан-1,2-диола (0,91 г, 4,41 ммоля) в ДМФ (диметилформамид) (20 мл) в атмосфере N2 при температуре окружающей среды добавляли 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (3,47 г, 9,3 ммоля), ДМАП (1,13 г, 9,3 ммоля), 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (ДКИ) (1,776 г, 9,26 ммоля) и сухой ДХМ (60 мл). Смесь перемешивали в течение ночи, затем реакционную смесь разбавляли диэтиловым эфиром (200 мл). Смесь промывали с помощью 1М HCl (100 мл) и рассолом (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 3% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 2,26 г (выход 56%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,08 (s, 6Н), 0,90 (d, 9Н), 0,95-1,03 (m, 12Н), 1,40-1,52 (m, 4Н), 1,58-1,69 (m, 4Н), 1,70-1,83 (m, 4Н), 2,04-2,15 (m, 8Н), 2,77-2,92 (m, 16Н), 3,27-3,37 (m, 2Н), 3,57-3,67 (m, 2Н), 3,72-3,80 (m, 4Н), 4,14-4,32 (m, 1Н), 4,41-4,56 (m, 1Н), 5,09-5,22 (m, 1Н), 5,23-5,49 (m, 20Н). МС (электрораспыление): 941,6 [M+Na]+.
Стадия d) Получение 3-гидроксипропан-1,2-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата)
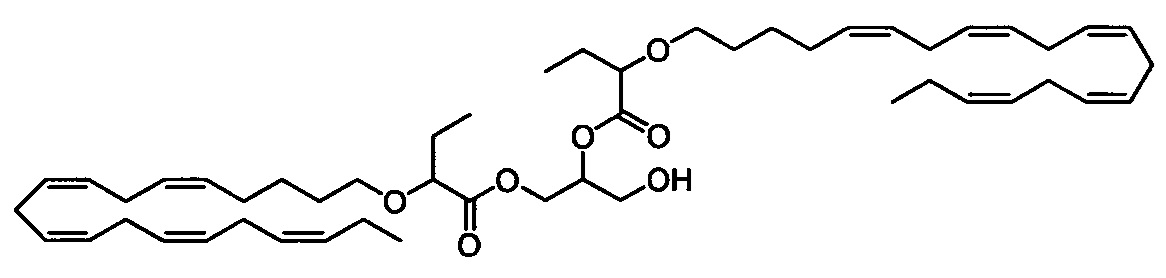
К раствору 3-((трет-бутилдиметилсилил)окси)пропан-1,2-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата) (2,26 г, 2,46 ммоля) в диоксане (100 мл) добавляли водный раствор HCl (37% (мас./мас., 2 мл) и смесь перемешивали в течение 3 ч в атмосфере азота при температуре окружающей среды, затем концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 15% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 0,83 г (выход 42%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,96-1,03 (m, 12Н), 1,40-1,53 (m, 4Н), 1,58-1,68 (m, 4Н), 1,70-1,85 (m, 4Н), 1,87-2,01 (m, 1Н), 2,05-2,15 (m, 8Н), 2,75-2,95 (m, 16Н), 3,28-3,41 (m, 2Н), 3,56-3,65 (m, 2Н), 3,73-3,85 (m, 4Н), 4,24-4,37 (m, 1Н), 4,42-4,53 (m, 1Н), 5,14-5,23 (m, 1Н), 5,26-5,51 (m, 20Н). МС (электрораспыление): 827,5 [M+Na]+.
Пример 19: Получение 2-гидроксипропан-1,3-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата):
Стадия а) 2-оксопропан-1,3-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноат)
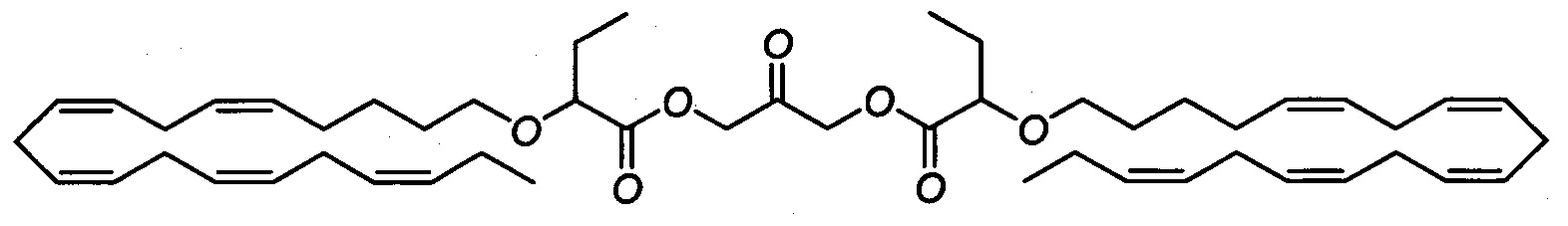
2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (5,0 г, 13,4 ммоля) и ДМАП (1,63 г, 13,4 ммоля) добавляли к раствору димера 1,3-дигидроксиацетона (1,145 г, 6,36 ммоля) в хлороформе (25 мл) в атмосфере азота. Затем при температуре окружающей среды по каплям добавляли раствор ДЦК (2,75 г, 13,35 ммоля) в хлороформе (10 мл). Смесь перемешивали в течение ночи при комнатной температуре, затем концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием обладающих увеличивающейся полярностью смесей гептана и этилацетата (90:10 → 88:12) в качестве элюента. Концентрирование соответствующих фракций давало 2,4 г (выход 47%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,97-1,06 (m, 12Н). 1,38-1,53 (m, 4Н), 1,57-1,73 (m, 4Н), 1,73-1,96 (m, 4Н), 2,03-2,17 (m, 8Н), 2,76-2,92 (m, 16Н), 3,35-3,42 (m, 2Н), 3,63-3,70 (m, 2Н), 3,89 (dd, 2Н), 4,75-4,93 (m, 4Н), 5,27-5,49 (m, 20Н). МС (электрораспыление): 827,5 [M+Na]+.
Стадия b) 2-гидроксипропан-1,3-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноат)
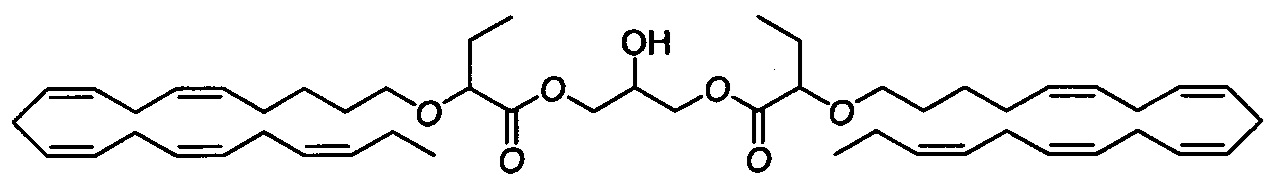
NaBH4 (0,336 г, 8,87 ммоля) осторожно добавляли к раствору 2-оксопропан-1,3-диилбис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата) (3,24 г, 4,03 ммоля) в ТГФ (55 мл) и воде (4 мл) при 0°C. Смесь перемешивали в течение 15 мин при 0°C. Затем осторожно добавляли уксусную кислоту (1 мл) и затем этилацетат (100 мл). Смесь промывали водой (100 мл), насыщенным водным раствором NaHCO3 (100 мл) и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток объединяли с другой партией вещества и затем очищали с помощью флэш-хроматографии на силикагеле с использованием 15% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 1,62 г (выход 50%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,97-1,03 (m, 12Н), 1,41-1,52 (m, 4Н), 1,58-1,69 (m, 6Н), 1,71-1,87 (m, 4Н), 2,05-2,14 (m, 8Н), 2,38-2,42 (m, 1Н), 2,78-2,92 (m, 16Н), 3,32-3,39 (m, 2Н), 3,57-3,64 (m, 2Н), 3,80-3,84 (m, 2Н), 4,05-4,34 (m, 5Н), 5,26-5,49 (m, 20Н). МС (электрораспыление): 827,5 [M+Na]+.
Пример 20: Получение пропан-1,2,3-триилтрис(2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата)
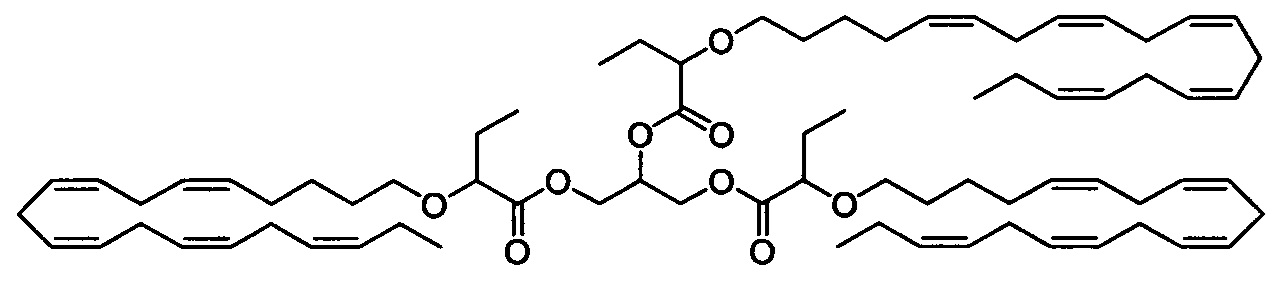
2-(((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (4,0 г, 10,7 ммоля), 4-диметиламинопиридин (1,305 г, 10,7 ммоля), 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (2,047 г, 10,7 ммоля) и сухой ДХМ (30 мл) добавляли к раствору глицерина (0,173 мл, 2,373 ммоля) в ДМФ (10 мл) в атмосфере N2 при комнатной температуре. Смесь перемешивали в течение ночи, затем реакционную смесь разбавляли диэтиловым эфиром (250 мл). Смесь промывали 1М водным раствором HCl (100 мл) и рассолом (100 мл), затем сушили (Na2SO4), фильтровали и выпаривали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием 5% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций давало 2,1 г (73%) искомого соединения в виде масла. 1Н ЯМР (300 МГц, CDCl3) δ 0,91-1,05 (m, 18Н), 1,40-1,52 (m, 6Н), 1,57-1,69 (m, 6Н), 1,69-1,86 (m, 6Н), 2,01-2,17 (m, 12Н), 2,69-2,96 (m, 24Н), 3,27-3,38 (m, 3H), 3,53-3,67 (m, 3H), 3,73-3,81 (m, 3H), 4,17-4,27 (m, 2Н), 4,37-4,54 (m, 2Н), 5,28-5,47 (m, 30Н). МС (электрораспыление): 1183,8 [M+Na]+.
Пример 21: Получение 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата кальция
2-(((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бута новую кислоту (1,87 г, 4,99 ммоля, 93%) смешивали с СаСО3 (0,25 г, 2,50 ммоля). Добавляли воду (1 мл) и смесь перемешивали механической мешалкой при КТ в течение 1 ч. Выделялся CO2. Образовывалась плотная и однородная паста. При перемешивании добавляли ацетон (7 мл). Отделялось твердое вещество. Твердое вещество отфильтровали и сушили азотом, герметизировали и хранили в холодильнике при 4°C. Выход: 1,86 г (95%).
Твердое вещество дополнительно не исследовали с помощью аналитических или спектроскопических методик, но немногочисленные эксперименты показывали, что образовывалась кальциевая соль:
- Твердое вещество плавилось на плитке при температуре ниже 100°C. Не определялась резкая температура плавления
- Вещество не выделяло CO2 при добавлении кислоты, а "растворялось" и осаждалось в виде масла.
Пример 22: Получение 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата натрия
2-(((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (1,87 г, 4,99 ммоля, 93%) смешивали с NaHCO3 (0,420 г, 5,00 ммоля). Добавляли воду (1 мл) и смесь перемешивали механической мешалкой при КТ в течение 1 ч. Выделялся CO2 и образовывалась плотная однородная паста. При перемешивании в реакционную колбу добавляли этанол (7 мл). Натриевая соль, образовавшаяся из 2-(((5Z,8Z,11Z,14Z,17Z)-Эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты не переходила в раствор при добавлении этанола (7 мл). Небольшие количества непрореагировавшего NaHCO3 отфильтровывали и раствор выпаривали досуха. Неочищенное немного вязкое масло дважды выпаривали с 96% этанолом для удаления следов воды.
Пример 23: Получение 2-гидрокси-N,N,N-триметилэтан-1-аминий 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутаноата
Гидроксид холина (327,7 мкл) в воде пипеткой помещали в сцинтилляционный сосуд вместе с примерно 2,5 мл МТБЭ (метил-трет-бутиловый эфир) и 7,5 мл н-гептана. В камере с атмосферой азота 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (500 мг, 95,8%) переносили в сосуд. В камере с атмосферой азота в сосуд медленно и при перемешивании добавляли примерно 1,0 мл воды. Затем сосуд герметизировали. Реакционную смесь перемешивали в течение примерно 30 мин. Образовавшаяся соль 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой кислоты с холином представляла собой твердое гелеподобное вещество, которое отфильтровывали на воронке Бюхнера. Влажное вещество 3 раза промывали на фильтре с использованием 1 мл МТБЭ. Промытое вещество выглядело, как твердое гелеподобное вещество.
Пример 24: Доклиническое исследование
Исследование регуляции apoC-III в дислипидемической модели на мышах (трансгенные мыши APOE*3Leiden)
Трансгенные мыши APOE*3Leiden экспрессируют вариант аполипопротеина Е3 человека (АРОЕ3), APOE*3Leiden, в дополнение к аполипопротеину С1 человека (АРОС1). Трансгенные мыши APOE*3Leiden обладают повышенными содержаниями в плазме холестерина и триглицеридов, в основном ограниченными фракцией липопротеина размера ЛОНП/ЛНП (Van den Maagdenberg AMJM et al., Transgenic mice carrying apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia, J Biol Chem 1993; 268: 10540-10545). В отличие от обычных мышей дикого типа трансгенные мыши APOE*3Leiden сильно реагируют на корм и противогиполипидемические лекарственные средства, влияющие на содержания в плазме ЛОНП и хиломикронов (Van Vlijmen В et al., Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice, J Clin Invest 1994; 93: 1403-1410; Groot PHE, et al., Quantitative assessment of aortic atherosclerosis in apoE3Leiden transgenic mice and its relationship to serum cholesterol exposure, Arterioscler Thromb Vasc Biol 1996; 16: 926-933). Следовательно, эта модель является подходящей для оценки влияния лекарственных средств, уменьшающих содержание липидов.
В этом исследовании самкам трансгенных мышей APOE*3Leiden давали полусинтетический кормовой рацион типа Вестерна (WTD; 15% масла какао, 40% сахарозы и 0,25% холестерина; все содержания в мас./мас.). После предоставления этого рациона в течение 4 недель содержание холестерина в плазме достигало немного повышенных значений, равных примерно 12-15 ммоль/л. Затем мышей разделяли на группы по 10 мышей в каждой, подобранные по содержанию в плазме холестерина, триглицеридов и массе тела (t=0). Исследуемые вещества изучали при дозе, равной 0,3 ммоль/кг массы тела/сутки и вводили перорально в виде смеси в WTD. Через 4 недели всех животных умерщвляли и собирали сыворотку и ткани. Затем ткани печени хранили в реагенте RNAlater (Qiagen) при -80°C. Ткани гомогенизировали в RLT-буфере с дитиотреитолом (Qiagen) и РНК выделяли с использованием набора RNeasy (Qiagen) в соответствии с инструкциями изготовителя. Качество выделенной РНК исследовали с помощью Bioanalyser (Agilent) и установлено, что значения показателя RIN (показатель целостности РНК) равно от 8,1 до 9,5, что указывает на хорошее качество. кДНК синтезировали с использованием набора "РНК в кДНК" (Applied Biosystems). Экспрессию гена определяли с использованием Low Density Arrays (LDA, специфических для РНК мышей (Applied Biosystems)). Каждый образец исследовали трижды, и результаты приведены в виде среднего значения по сравнению с контролем (WTD без добавок). Кратность изменения экспрессии гена рассчитывали по методике ΔΔCt с использованием Rplp0 в качестве гена "домашнего хозяйства" и среднего значения для контрольных образцов в качестве калибратора.
Результаты, представленные на фиг. 1, показывают, что мыши, которым давали соединение А (пример 2), характеризовались значительно меньшей экспрессией apoC-III, чем мыши, которым давали стандартный WTD (Р<0,05, Т-критерий Стьюдента). Влияние соединения А более значительно, чем влияние эталонного соединения 12, производное ЭПК, полученного в соответствии с примером 20 в WO 2010/008299, обладающего следующей структурой:
Эталонное соединение 12
Кроме того, определяли способность обоих соединений уменьшать содержание ТГ в плазме. Оба соединения уменьшали содержание ТГ на 69% по сравнению с контролем. Это подтверждает отсутствие прямой корреляции между наблюдаемым уменьшением содержания apoC-III и уменьшением содержания ТГ.
Пример 25: Клинические исследования
Способность соединения А уменьшать содержание apoC-III продемонстрирована в двух 12-недельных исследованиях и одном 4-недельном исследовании пациентов, страдающих дислипидемией. Все три исследования продемонстрировали клинические и статистически значимые уменьшения содержания apoC-III при лечении соединением А.
Пример 25А: Группа пациентов, страдающих от тяжелой гипертриглицеридемии
В этом исследовании изучали пациентов, у которых содержания триглицеридов в плазме натощак превышали 500 мг/дл. Главной задачей настоящего исследования являлось изучение эффективности соединения А (пример 2), вводимого по 600 мг один раз в сутки (ОРС) перорально путем оценки выраженного в процентах изменения содержания триглицеридов (ТГ) по сравнению с исходным содержанием после 12 недель лечения.
Одной из вторичных задач являлось изучение влияния соединения А на содержание apoC-III в плазме.
Это многоцентровое подтверждающее исследование фазы II включало 6--8-недельный период скрининга (который включал 4- или 6-недельный период питания и стабилизации образа жизни/выведения средства и 2-недельный период оценки ТГ) и 12-недельный период двойного слепого рандомизированного проводимого в параллельных группах при контроле с помощью плацебо лечения.
После подтверждения содержания ТГ натощак включенные в исследование субъекты проходили 12-недельный период рандомизированного двойного слепого лечения. После визита 4 (неделя 0) субъекты случайным образом в соотношении 1:1 распределяли в одну из следующих групп лечения: соединение А 600 мг ОРС или плацебо ОРС.
В этом исследовании рандомизировали приблизительно 43 субъектов на группу лечения (всего приблизительно 86 субъектов). Стратификацию проводили по исходному содержанию ТГ (≤700 мг/дл или >700 мг/дл), применению статина при рандомизации и полу.
В группы для этого исследования входили мужчины и женщины (женщины, способные к деторождению должны были использовать эффективные методы предупреждения беременности) в возрасте от 18 до 79 лет включительно. Субъекты, получающие стабильно снижающее содержание липидов лечение статином, и субъекты, не получающие стабильно снижающее содержание липидов лечение статином, считались подходящими для включения в исследование. Требовалось, чтобы субъекты обладали средним содержанием ТГ натощак ≥500 мг/дл и ≤1500 мг/дл при переходе от визита 2 к визиту 3 или от визита 3 к визиту 3.1 до рандомизации.
Включенная в исследование группа пациентов (ITT) включала всех рандомизированных субъектов, которые получали по меньшей мере 1 дозу исследуемого препарата, для которых проводилось определение исходной эффективности и для которых проводилось по меньшей мере 1 определение эффективности после рандомизации. Группа ITT пациентов являлась группой для первичного анализа. Все определения эффективности проводили для группы пациентов ITT.
Сводная статистика (n, среднее, стандартное отклонение [СО], медианное, минимальное и максимальное) для измерений в исходном состоянии и после исходного состояния, изменения в процентах или изменения по сравнению с исходным значением приведены для группы лечения и при визите для всех анализируемых параметров эффективности.
Первичный анализ эффективности проводили с использованием модели ковариационного анализа (ANCOVA) с применением факта проведения лечения, пола и применения лечения статином при рандомизации в качестве факторов и исходного содержания ТГ в качестве коварианта. Приведены среднеквадратичные средние, стандартные погрешности и двусторонние 95% доверительные интервалы (ДИ) для каждой группы лечения и для сопоставления соединения А и плацебо.
Модель ANCOVA использовали для анализа вторичных параметров эффективности с применением факта проведения лечения, пола и применения лечения статином при рандомизации в качестве факторов и исходного значения соответствующего параметра эффективности в качестве коварианта.
Группа пациентов, включенная в настоящее исследование, включала мужчин (69,0%) и женщин (31,0%) со средним возрастом 52,5 лет. Приблизительно 21% субъектов в обеих группах лечения во время исследования получали лечение статином. Все остальные не являющиеся статинами лекарственные средства, изменяющие содержание липидов, исключались при скрининге. Среднее соблюдение режима приема исследуемых лекарственных средств во время исследования составляло 96,5% для группы плацебо и 99,9% для группы, получавшей соединение А, 600 мг.
В группе пациентов ITT среднеквадратичное (СК) изменение содержания apoC-III в процентах составляло -38,0% (-47,5, -28,5) по сравнению с исходным значением и -34,7% (-46,5, -22,8) по сравнению с плацебо.
Пример 25В: Группа пациентов, страдающих от смешанной дислипидемии
В этом исследовании изучали пациентов, у которых содержания ТГ в плазме натощак составляли от 200 до 499 мг/дл и содержания холестерина липопротеинов, не обладающих высокой плотностью (не-ЛВП-С), составляли более 130 мг/дл, уже получавших лечение статинами. Главной задачей настоящего исследования являлось изучение эффективности соединения А (пример 2) 600 мг ОРС перорально путем оценки выраженного в процентах изменения содержания триглицеридов не-ЛВП-С по сравнению с исходным содержанием после 12 недель лечения. Одной из вторичных задач являлось изучение влияния соединения А на содержание ароС-III в плазме.
Это многоцентровое подтверждающее исследование фазы II включало 6--8-недельный период скрининга (который включал 4- или 6-недельный период питания и стабилизации образа жизни/выведения средства и 2-недельный период оценки ТГ и не-ЛВП-С) и 12-недельный период двойного слепого рандомизированного проводимого в параллельных группах при контроле с помощью плацебо лечения.
После подтверждения содержания ТГ и не-ЛВП-С натощак включенные в исследование субъекты проходили 12-недельный период рандомизированного двойного слепого лечения. После визита 4 (неделя 0) субъекты случайным образом в соотношении 1:1 распределяли в одну из следующих групп лечения: соединение А 600 мг ОРС или плацебо ОРС.
В группы для этого исследования входили мужчины и женщины (женщины, способные к деторождению должны были использовать эффективные методы предупреждения беременности) в возрасте от 18 до 79 лет включительно. Субъекты, получающие стабильно снижающее содержание липидов лечение статином, и субъекты, не получающие стабильно снижающее содержание липидов лечение статином, считались подходящими для включения в исследование. Требовалось, чтобы субъекты обладали средним содержанием ТГ натощак, равным от 200 до 499 мг/дл, и содержанием не-ЛВП-С, равным более 130 мг/дл, при переходе от визита 2 к визиту 3 или от визита 3 к визиту 3.1 до рандомизации.
Включенная в исследование группа пациентов (ITT) включала всех рандомизированных субъектов, которые получали по меньшей мере 1 дозу исследуемого препарата, для которых проводилось определение исходной эффективности и для которых проводилось по меньшей мере 1 определение эффективности после рандомизации. Группа ITT пациентов являлась группой для первичного анализа. Все определения эффективности проводили для группы пациентов ITT.
Сводная статистика (n, среднее, стандартное отклонение [СО], медианное, минимальное и максимальное) для измерений в исходном состоянии и после исходного состояния, изменения в процентах или изменения по сравнению с исходным значением приведены для группы лечения и при визите для всех анализируемых параметров эффективности.
Первичный анализ эффективности проводили с использованием модели ANCOVA с применением рандомизации в качестве фактора и исходного содержания не-ЛВП-С в качестве коварианта. Приведены среднеквадратичные, стандартные погрешности и двусторонние 95% ДИ для каждой группы лечения и для сопоставления соединения А и плацебо.
Первичный анализ эффективности был основан на 12-недельной полной группе пациентов.
Группа пациентов, включенная в настоящее исследование, включала мужчин (58,4%) и женщин (46,1%) со средним возрастом 58,3 лет. Во время исследования требовалось, чтобы все субъекты получали лечение статином (с использованием или без использования эзетимиба). Все остальные не являющиеся статинами лекарственные средства, изменяющие содержание липидов, исключались при скрининге. Среднее соблюдение режима приема исследуемых лекарственных средств во время исследования составляло 97,2% для группы плацебо и 95,3% для группы, получавшей соединение А.
Исходное среднее содержание не-ЛВП-С для группы пациентов, участвующих в исследовании, составляло 165,9 мг/дл; исходное медианное содержание ТГ составляло 262,0 мг/дл.
В 12-недельной полной группе пациентов СК среднее изменение содержания apoC-III в процентах составляло - 32,5% (-38,4, -26,6) по сравнению с исходным значением и - 20,8% (-28,8, -12,7) по сравнению с плацебо.
Пример 25А относится к исследованиям пациентов, обладающих очень большим содержанием триглицеридов (ТГ 500-2000 мг/дл). Пример 25В относится к исследованиям пациентов, постоянно получающих статин, страдающих от смешанной дислипидемии и стойкой гипертриглицеридемии (ТГ 200-499 мг/дл). Исследования, включенные в этот раздел, обладают сходным дизайном и сравнимыми группами пациентов.
Пример 25С: Группа пациентов, страдающих от гиперхолестеринемии
В этом исследовании изучали субъекты, у которых содержание ЛНП-С натощак составляло по меньшей мере 2,5 ммоля (~97 мг/дл). Задачей настоящего исследования являлось изучение фармакодинамики и способности уменьшать содержание липидов для соединения А (пример 2) после 4 недель лечения у субъектов мужского пола, страдающих гиперхолестеринемии, для которых отменили постоянное лечение статином.
Группа пациентов для этого исследования состояла из мужчин в возрасте от 18 до 65 лет любого этнического происхождения, обладающих ИМТ (индекс массы тела), равным от 18,0 до 35,0 кг/м2.
Эта фаза Ib исследования включала 4-5-недельный период скрининга и 4-недельный период двойного слепого рандомизированного при контроле с помощью плацебо лечения.
Все субъекты должны были получать снижающее содержание липидов лечение статином в течение по меньшей мере 3 месяцев до первого визита при скрининге и постоянную дозу статина в течение по меньшей мере 4 недель до первого визита при скрининге.
Лечение статином прекращали при первом визите при скрининге и не проводили в течение всего периода скрининга. После прекращения приема статина в течение по меньшей мере 21 дня у субъекта должно было наблюдаться содержание ЛНП-С, равное по меньшей мере 2,5 ммоль/л (~97 ммоль/л) при втором визите при скрининге, и увеличение содержания ЛНП-С по меньшей мере на 20% между первым визитом при скрининге и вторым визитом при скрининге до рандомизации.
После подтверждения квалификационных содержаний ЛНП-С натощак включенные в исследование субъекты проходили 4-недельный период двойного слепого рандомизированного при контроле с помощью плацебо лечения. Субъектов случайным образом в соотношении 3:1 распределяли в одну из следующих групп лечения: соединение А 600 мг ОРС (N=18) или плацебо ОРС (N=6).
Содержание липидов в крови определяли в конце периода скрининга и после 4 недель лечения. Диагностические фармакодинамические исследования включали определение содержания ЛНП-С, ЛОНП-С, ТС, ТГ, ЛВП-С, не-ЛВП-С и Аро-В. Также определяли влияние соединения А на содержание Apo-C-III.
Сводная статистика для исходного состояния приведена в виде средних значений с коэффициентом вариаций. Средние изменения по сравнению с исходным значением с 95% доверительными интервалами приведены для группы лечения для анализа параметров эффективности.
Анализы проводили с использованием модели ковариационного анализа (ANCOVA) для изменения по сравнению с исходным значением и исходное значение включали в качестве коварианта.
Группа пациентов, включенная в настоящее исследование, включала мужчин (100%) со средним возрастом 55 лет, средней массой, равной 85 кг, и средним ИМТ, равным 27,9 кг/м2.
Среднее изменение содержания apoC-III в процентах после лечения соединением А составляло -42% по сравнению с исходным значением. Это значение являлось статистически значимым.
Пример 26: Сопоставление уменьшения содержания apoC-III, обеспечиваемое при использовании ЭПК/ДГК, с обеспечиваемым соединением А
(а) Влияние препаратов ЭПК/ДГК по сравнению с соединением А на содержание apoC-III в плазме и другие параметры липидов у субъектов, у которых имеется тяжелая ГТГ
Исследование MARINE:
В двойном слепом рандомизированном при контроле с помощью плацебо исследовании изучали влияние этилового эфира эйкозапентаеновой кислоты (концентрат с содержанием >96 мас. %) (Vascepa) на содержание apoC-III у 229 пациентов, обладающих содержанием ТГ в плазме натощак, равным 500-2000 мг/дл. Vascepa 4 г/сутки в течение 12 недель уменьшал медианные содержания apoC-III от 25,6 мг/дл до 19,7 мг/дл, что соответствовало медианному изменению по сравнению с исходным значением, составляющему -10,1% [Journal of Clinical Lipidology 2014; 8(3): 313-314, Icosapent Ethyl (eicosapentaenoic acid ethyl ester): Effects on Apolipoprotein C-III in patients from the MARINE и ANCHOR studies.] (таблица 1).
Исследование EVOLVE:
В двойном слепом рандомизированном при контроле с помощью плацебо исследовании изучали влияние комбинации ЭПК и ДГК в виде свободных жирных кислот (55 мас. % ЭПК и 20 мас. % ДГК) (Epanova) на содержание apoC-III у 399 пациентов, обладающих содержанием ТГ в плазме натощак, равным 500-2000 мг/дл. Epanova 4 г/сутки в течение 12 недель приводил к медианному изменению содержания apoC-III по сравнению с исходным значением, составляющему -15% [Circulation 2012; 126: А19030, Abstract 19030: Apolipoprotein C-III is Significantly Reduced by Prescription Omega-3 Free Fatty Acids (Epanova) in Patients with Severe Hypertriglyceridemia и Changes Correlate with Increases in LDL-C: A Sub-analysis of the EVOLVE trial] (таблица 1).
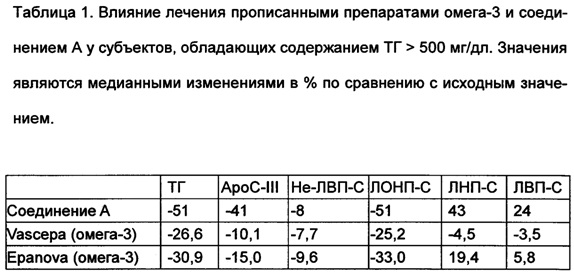
(b) Влияние препаратов ЭПК/ДГК по сравнению с соединением А на содержание apoC-III в плазме и другие параметры липидов на постоянно принимающих статин субъектов, у которых имеется смешанная дислипидемия и стойкая гипертриглицеридемия
Исследование ANCHOR:
В двойном слепом рандомизированном при контроле с помощью плацебо исследовании изучали влияние этилового эфира эйкозапентаеновой кислоты (Vascepa) на содержание apoC-III у 702 постоянно принимающих статин пациентов, у которых имеется смешанная дислипидемия и стойкая гипертриглицеридемия, обладающих содержанием ТГ в плазме натощак, равным 200-499 мг/дл. Vascepa 4 г/сутки в течение 12 недель уменьшал медианные содержания apoC-III от 15,2 мг/дл до 13,7 мг/дл, что соответствовало медианному изменению по сравнению с исходным значением, составляющему -9,4% [Journal of Clinical Lipidology 2015, in press, http://dx.doi.Org/10.1016/j.jacl.2014.11.009, Effects of icosapent ethyl on lipoprotein particle concentration and size in statin-treated patients with persistent high triglycerides (the ANCHOR study)] (таблица 2).
Исследование ESPRIT:
В двойном слепом рандомизированном при контроле с помощью плацебо исследовании изучали влияние комбинации ЭПК и ДГК в виде свободных жирных кислот (Epanova) на содержание apoC-III у 647 постоянно принимающих статин пациентов, у которых имеется смешанная дислипидемия и стойкая гипертриглицеридемия, обладающих содержанием ТГ в плазме натощак, равным 200-499 мг/дл. Epanova 4 г/сутки в течение 12 недель приводил к медианному изменению содержания apoC-III по сравнению с исходным значением, составляющему -13,1% [JACC 2013; 61: Е1468, А highly bioavailable omega-3 fatty acid reduces non-high density lipoprotein cholesterol in high-risk patients treated with a statin and residual hypertriglyceridemia (ESPRIT trial)] (таблица 2).
Исследование COMBOS:
В двойном слепом рандомизированном исследовании изучали влияние комбинации этиловых эфиров (ЭЭ) ЭПК и ДГК (46,5 мас. % ЭЭ ЭПК и 37,5 мас. % ЭЭ ДГК) (Lovaza) на содержание ароС-III у 256 постоянно принимающих статин пациентов, у которых имеется смешанная дислипидемия и стойкая гипертриглицеридемия, обладающих содержанием ТГ в плазме натощак, равным 200-400 мг/дл. Lovaza 4 г/сутки в течение недель приводил к медианному изменению содержания apoC-III по сравнению с исходным значением, составляющему -7,8% [Clinical Therapeutics 2007; 29(7): 1354-1367, Efficacy and tolerability of adding prescription Omega-3 fatty acids 4 g/d to simvastatin 40 mg/d in hypertriglyceridemic patients: An 8-week, randomized, double-blind, placebo-controlled study] (таблица 2).

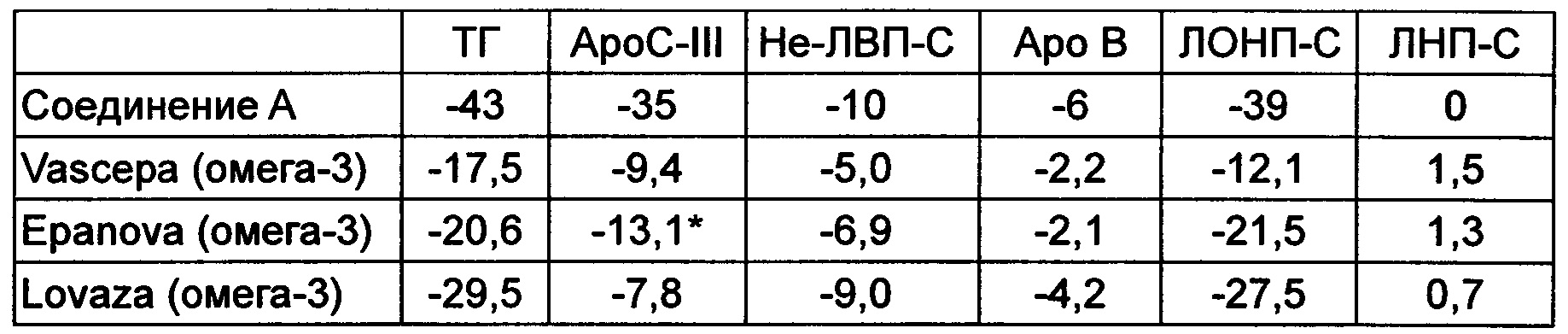
* Значение содержания ApoC-III для Epanova является средним изменением в % по сравнению с исходным значением
Сводка данных по сравнительным уменьшениям содержания apoC-III в плазме при использовании ЭПК/ДГК по сравнению с соединением А
Хотя прямые сравнительные исследования не были завершены, сравнимые группы пациентов и дизайны исследовании образуют исходные положения, по которым можно сопоставить эффективность соединения А по сравнению с ЭПК/ДГК для уменьшения содержания apoC-III в плазме. Имеются два примечательных фактора, отличающие природные омега-3-жирные кислоты от соединения А.
Первым является превосходная активность соединения А, которая обеспечивает медианные уменьшения содержания apoC-III, составляющие 35 и 41% в группах пациентов, у которые имеется смешанная дислипидемия и тяжелая ГТГ соответственно. Это следует сопоставить с уменьшениями содержания apoC-III, составляющих лишь 7,8-15% в исследованиях ЭПК/ДГК.
Вторым отличающим фактором является низкая необходимая доза соединения А (600 мг ОРС) в отличие от равной 4 г дозы при исследованиях ЭПК/ДГК. В пересчете на грамм это различие даже больше для соединения А и ясно демонстрирует активность этой молекулы при уменьшении содержания apoC-III в плазме по сравнению с ЭПК/ДГК. Как отмечено выше, доклинические модели показывают, что уменьшение содержания apoC-III не зависит от уменьшения содержания ТГ (фиг. 1).
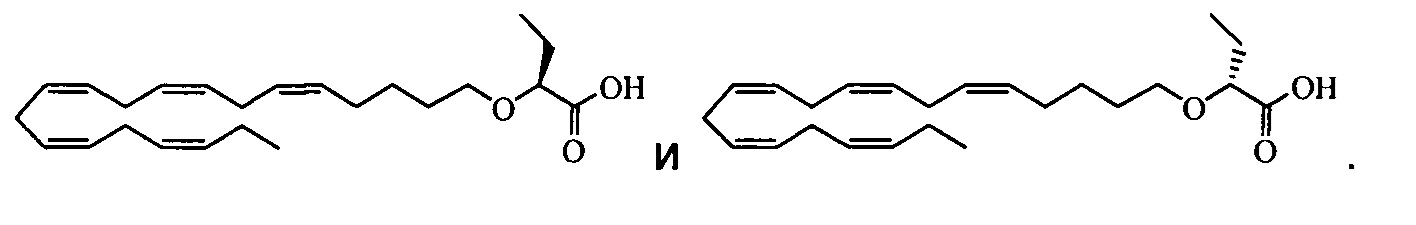
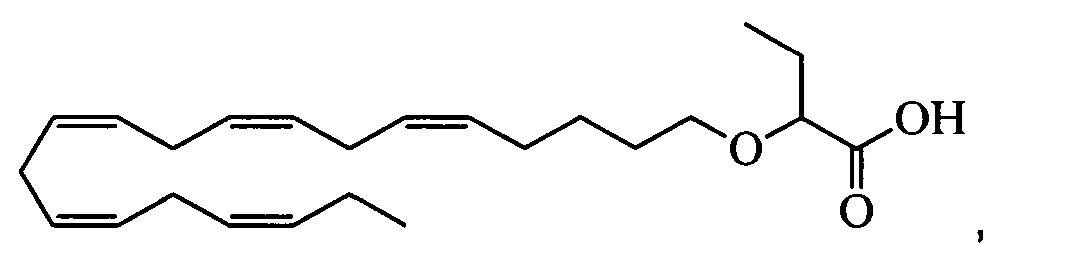
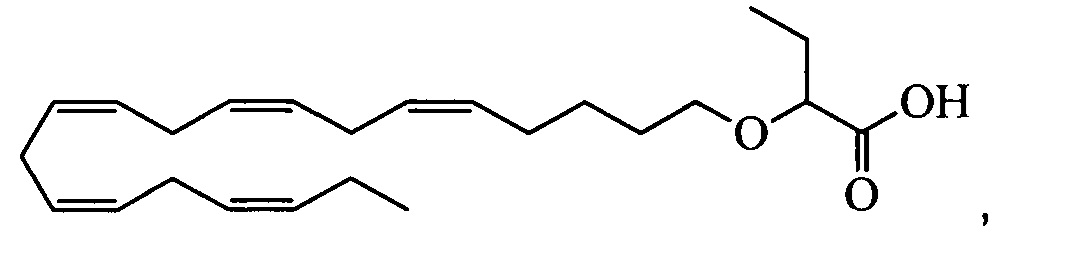
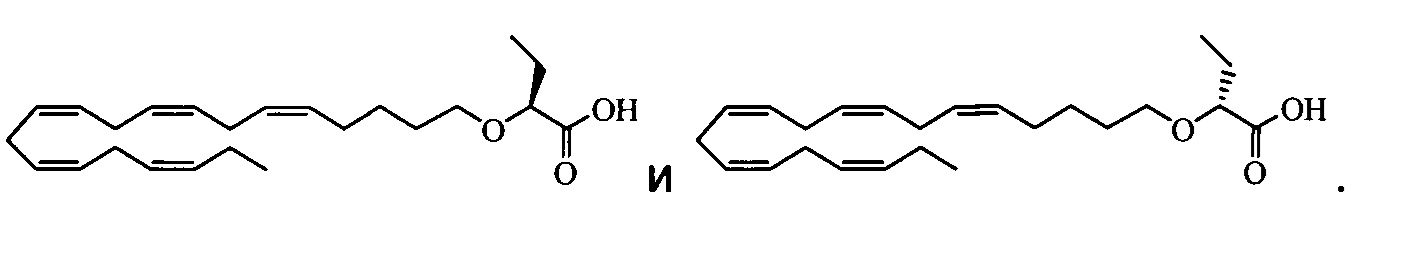
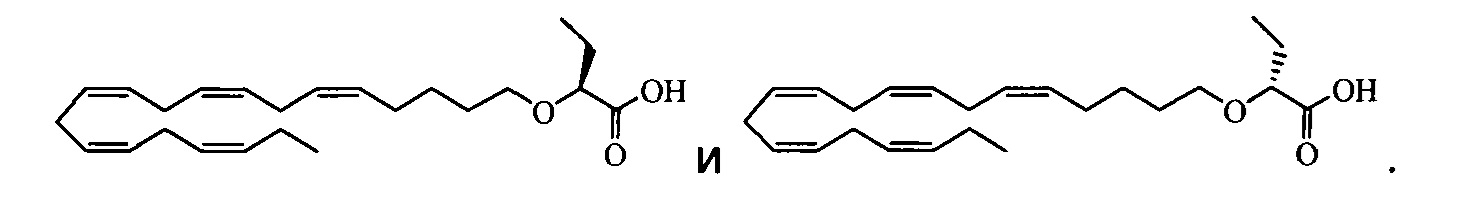
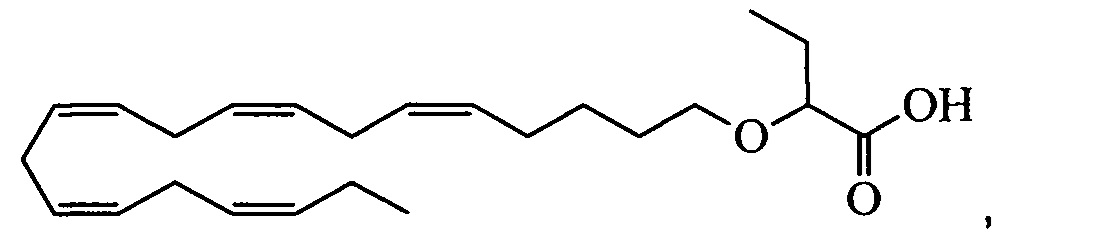
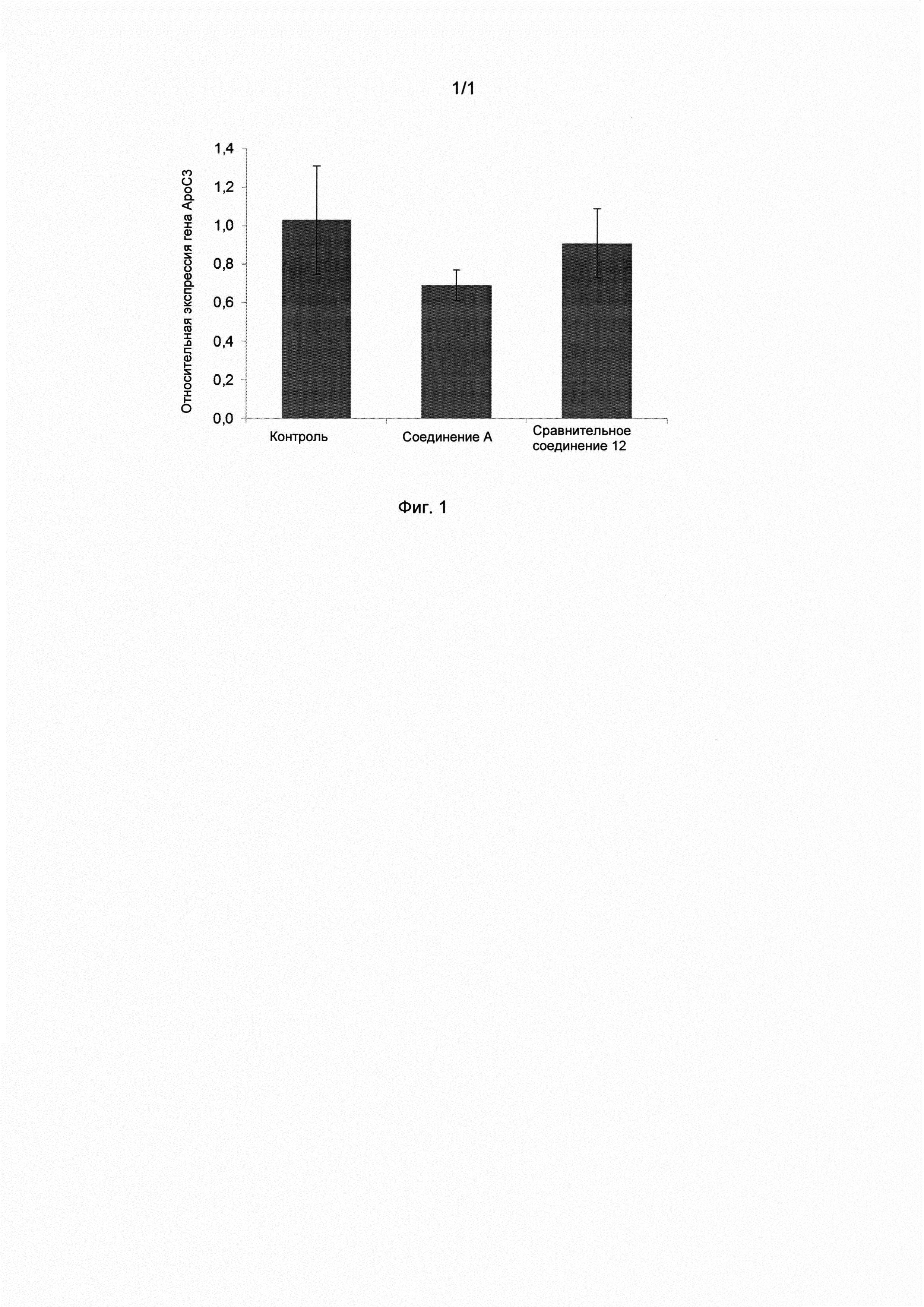
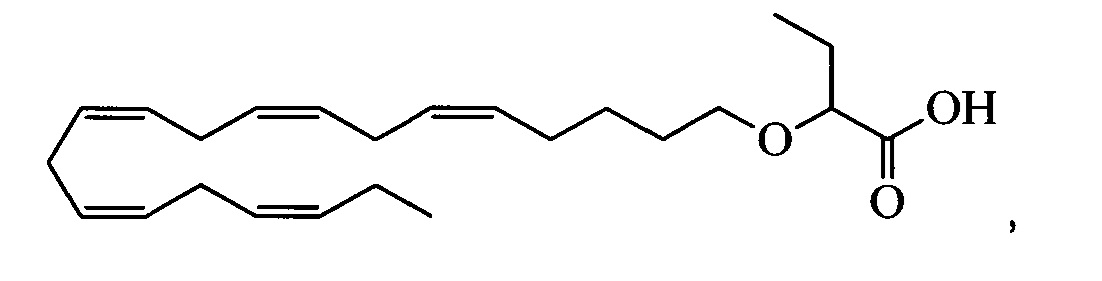
Новые липидные соединения

