Результат интеллектуальной деятельности: НОВЫЙ ПЕПТИД-ИНГИБИТОР PI3Kγ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ОРГАНОВ ДЫХАНИЯ
Вид РИД
Изобретение
Область техники
Настоящее описание относится к новому пептидному ингибитору PI3Kγ для лечения патологий дыхательной системы.
Предшествующий уровень техники
Астма представляет собой хроническое респираторное заболевание, характеризующееся воспалением, гиперчувствительностью дыхательных путей (AHR - airway hyperresponsiveness) и отеком слизистых оболочек, что в совокупности приводит к эпизодической бронхоконстрикции и обструкции дыхательных путей. Эффективность современных противоастматических видов лечения является неудовлетворительной, и астма остается нерешенной глобальной проблемой.
Тонус мускулатуры дыхательных путей определяется тонким балансом между активацией просократительных и прорасслабляющих сигнальных путей в гладкомышечных клетках. Сокращение главным образом запускается ацетилхолином, основным нейромедиатором в парасимпатической нервной системе в дыхательных путях, который активирует М3 мускариновые рецепторы, приводя к мобилизации внутриклеточного и внеклеточного кальция (Са2+). Наоборот, расслабление дыхательных путей достигается катехоламин-опосредованной активацией β2-адренорецепторов (β2-AR), которые стимулируют продукцию циклического АМФ (аденозинмонофосфат) (цАМФ) и последующую модуляцию ключевых эффекторов гомеостаза Са2+. В соответствии с прорасслабляющим действием цАМФ, агонисты β2-AR обеспечивают облегчение симптомов бронхоспазмов у пациентов с астмой. Однако их эффективность ограничена во времени, главным образом, вследствие десенсибилизации β2-AR, которая происходит после повторного воздействия агонистов. Аналогично, ингибирование деградации цАМФ ингибиторами фосфодиэстеразы 4 (PDE4), основные ферменты, ответственные за гидролиз цАМФ в дыхательных путях, прошло клиническое испытание, но демонстрирует неприемлемые побочные эффекты, такие как рвота, тошнота, диарея и потеря веса, вследствие неселективного ингибирования PDE4 в центральной нервной системе.
Таким образом, выявление новых ферментов, которые регулируют гомеостаз цАМФ, а также новых стратегий манипулирования путем сигнальной трансдукции β2-AR/цАМФ в гладкомышечных клетках, является желательным для лечения респираторных заболеваний. Кроме того, один и тот же подход мог бы также быть использован в терапевтических целях в других патологических контекстах, таких как кистозный фиброз, где необходимо повышать уровни цАМФ в эпителиальных клетках дыхательных путей.
В респираторном эпителии продукция цАМФ после β2-AR является необходимой для обеспечения открытия цАМФ-зависимого хлоридного канала (регулятор трансмембранной проводимости при кистозном фиброзе (CFTR)). Мутации в гене, который кодирует данный белок, являются основной причиной кистозного фиброза (CF). Среди них, делеция фенилаланина 508 (ΔF508) является наиболее распространенной перестройкой у пациентов с CF и приводит к дефектам как в мембранной экспрессии, так и открытии канала. Был разработан целый ряд лекарственных средств на основе корректоров и потенциаторов CFTR, которые восстанавливают мембранную экспрессию и цАМФ-опосредованное открытие данного канала, соответственно, но их эффективность является неудовлетворительной. В частности, потенциаторы CFTR, для того, чтобы быть эффективными, требуют высоких концентраций внутриклеточного цАМФ. Таким образом, лекарственные средства, которые способны стимулировать уровни цАМФ, могут представлять собой новые стратегии повышения эффективности современных доступных видов лечения или прямой коррекции функциональных дефектов CFTR при CF.
Ранее проведенные исследования показали, что фосфоинозитид 3-киназа γ (PI3Kγ) контролирует компартментализацию цАМФ после β2-AR. В кардиомиоцитах, PI3Kγ действует как якорный белок (АKАР) (1), который связывает протеинкиназу А (РКА) с несколькими изоформами PDE3 и PDE4. PI3Kγ-ассоциированная РКА, в свою очередь, фосфорилирует и стимулирует активацию PDE и последующее снижение уровня цАМФ после β2-AR, ограничивая, в конечном счете, аритмогенное высвобождение Са2+(2). Несмотря на то, что было разработано несколько ингибиторов киназной активности PI3Kγ, в настоящее время отсутствуют способы селективного воспрепятствования активности адаптерного или якорного белка PI3Kγ.
Краткое описание изобретения
С учетом вышеизложенного, таким образом, существует потребность в улучшенных и более эффективных решениях для лечения заболеваний дыхательной системы, по сравнению с известными терапиями.
Согласно изобретению указанная выше цель достигается, благодаря решению, о котором особым образом напоминает прилагаемая формула изобретения, которая составляет неотъемлемую часть настоящего описания.
Одно воплощение настоящего изобретения относится к слитому пептиду, содержащему:
i) аминокислотную последовательность, как определено в SEQ ID No.: 1, или родственный гомолог, обладающий по меньшей мере 90% идентичностью с SEQ ID No.: 1, и обладающий способностью последовательности SEQ ID No.: 1 ингибировать киназонезависимую функцию PI3Kγ, и
ii) пептид, обладающий способностью проникать в клетку, для применения в качестве лекарственного средства, в частности, для лечения респираторных заболеваний.
Другое воплощение настоящего изобретения относится к продукту, содержащему i) слитый пептид, как определено выше, и ii) потенциатор регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) и/или корректор регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) в виде комбинированного препарата для последовательного, одновременного или раздельного применения для лечения респираторных заболеваний, предпочтительно кистозного фиброза.
Согласно настоящему описанию предложено in vitro и in vivo экспериментальное доказательство эффективности лечения патологий дыхательной системы посредством введения слитого пептида, содержащего остатки 126-150 PI3Kγ человека (SEQ ID No.: 1) или ее гомологов. Слитый пептид по настоящему описанию фактически способен ингибировать взаимодействие между РКА и PI3Kγ и, следовательно, уменьшать активность PDE, ассоциированных с PI3Kγ, увеличивая уровни цАМФ и снижая поступление Са2+ через потенциал-зависимые кальциевые каналы (VOCC). Кроме того, слитый пептид, описанный в данном документе, повышает уровни цАМФ in vitro в дыхательных путях и функционирует в качестве бронходилятора при введении внутритрахеальным путем здоровым мышам и мышам с астмой. Наконец, слитый пептид осуществляет функцию потенциатора CFTR, увеличивая уровень цАМФ и, таким образом, усиливая проводимость CFTR в отношении иона хлора (Cl-) в линиях эпителиальных клеток бронхов, экспрессирующих CFTR дикого типа или ΔF508 мутантный CFTR, что является наиболее часто встречающейся мутацией у пациентов с кистозным фиброзом.
Краткое описание графических материалов
Сейчас изобретение будет подробно описано, исключительно в качестве иллюстративного и неограничивающего примера, со ссылкой на прилагаемые графические материалы, где:
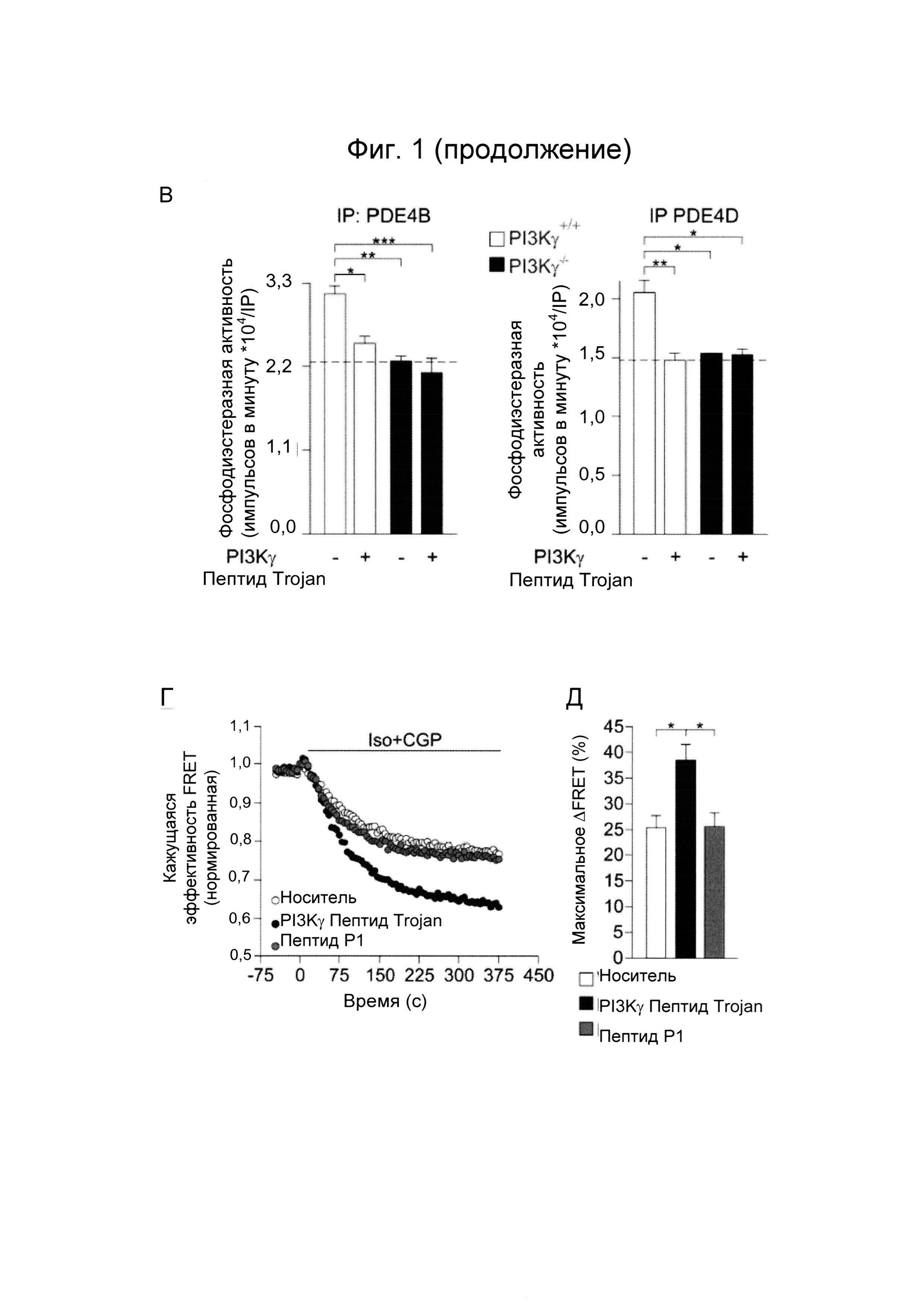
- Фиг. 1. Пептид Trojan, полученный из PI3Kγ, ингибирует активность PDE и усиливает β2-AR/цАМФ сигнализацию в гладкомышечных клетках дыхательных путей. А) Схематичное представление PI3Kγ ингибиторного пептида, проницаемого в клеточные мембраны. Проводили слияние области 126-150 PI3Kγ человека с последовательностью Пенетратина 1 Antennapedia. Б) Внутриклеточная локализация PI3Kγ ингибиторного пептида Trojan. hBSMC инкубировали с пептидом, меченным флуоресцеином (FITC, 50 мкМ) и анализировали внутриклеточную флуоресценцию, спустя 30 минут от начала обработки. Представлено окрашивание филаментного актина (левые панели) и флуоресценция FITC (средние панели) с относительным увеличением (правые панели). В) Фосфодиэстеразная активность с осаждением антителами против PDE4B и против PDE4D в гладкомышечных клетках трахеи, выделенных из PI3Kγ+/+ и PI3Kγ-/- животных и обработанных либо носителем, либо PI3Kγ ингибиторным пептидом Trojan (50 мкМ, 30 минут; n больше или равно 4 независимых экспериментов). Г) hBSMC трансфицировали зондом FRET в отношении цАМФ, ICUE3 и предварительно обрабатывали либо носителем, либо PI3Kγ ингибиторным пептидом, либо контрольным пептидом Пенетратин 1 (50 мкМ, 30 минут) перед активацией β2-AR изопротеренолом (ISO; 100 нМ) и селективным антагонистом β1-AR, CGP-20712A (CGP, 100 нМ). Представлены репрезентативные FRET кривые n больше или равно 3 независимых экспериментов. Д) Максимальное изменение в сигнале FRET (%) кривых, как измерено в Г. *Р менее 0,05, **Р менее 0,01, ***Р менее 0,001 с помощью однофакторного дисперсионного анализа с последующим тестом Бонферрони.
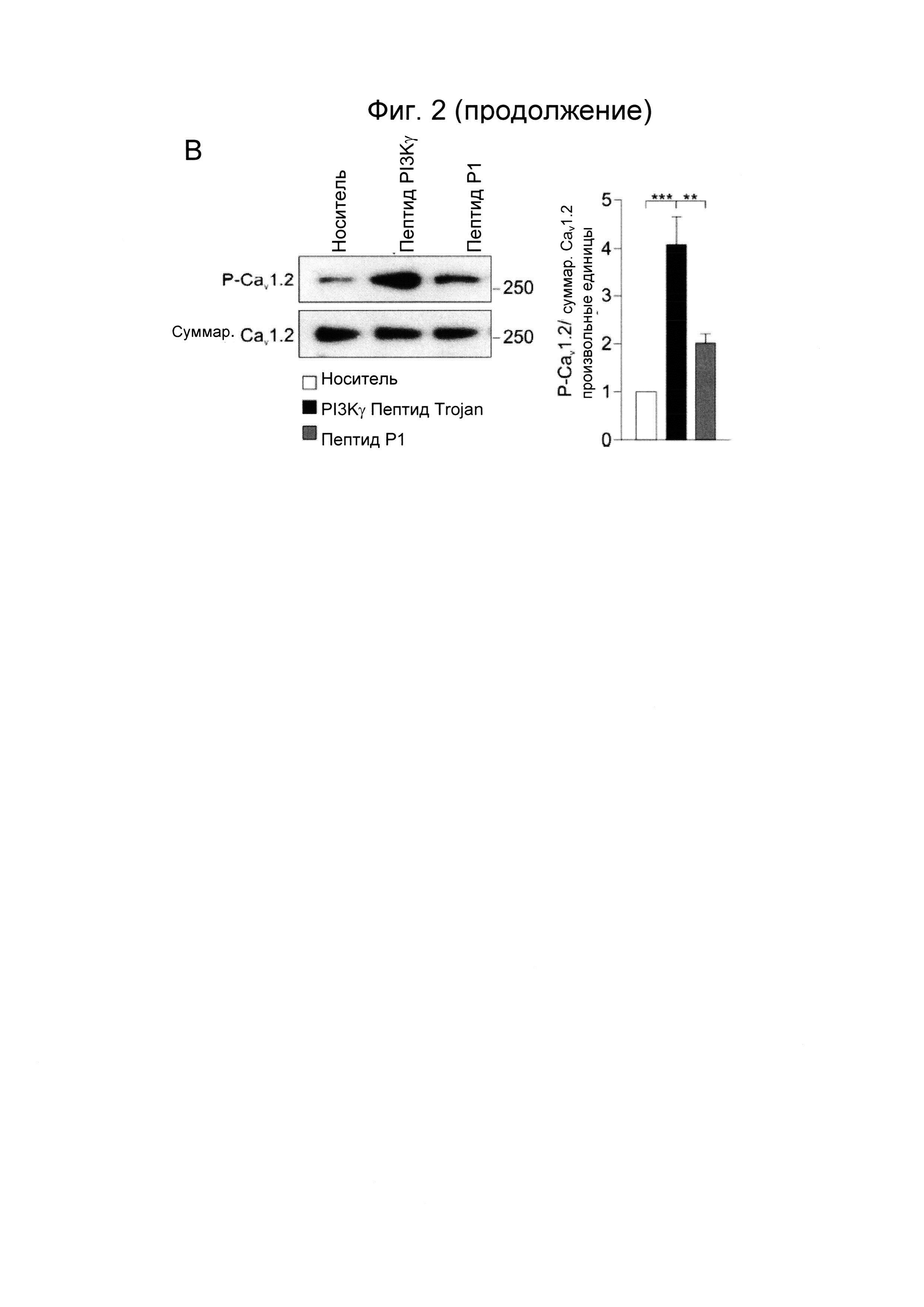
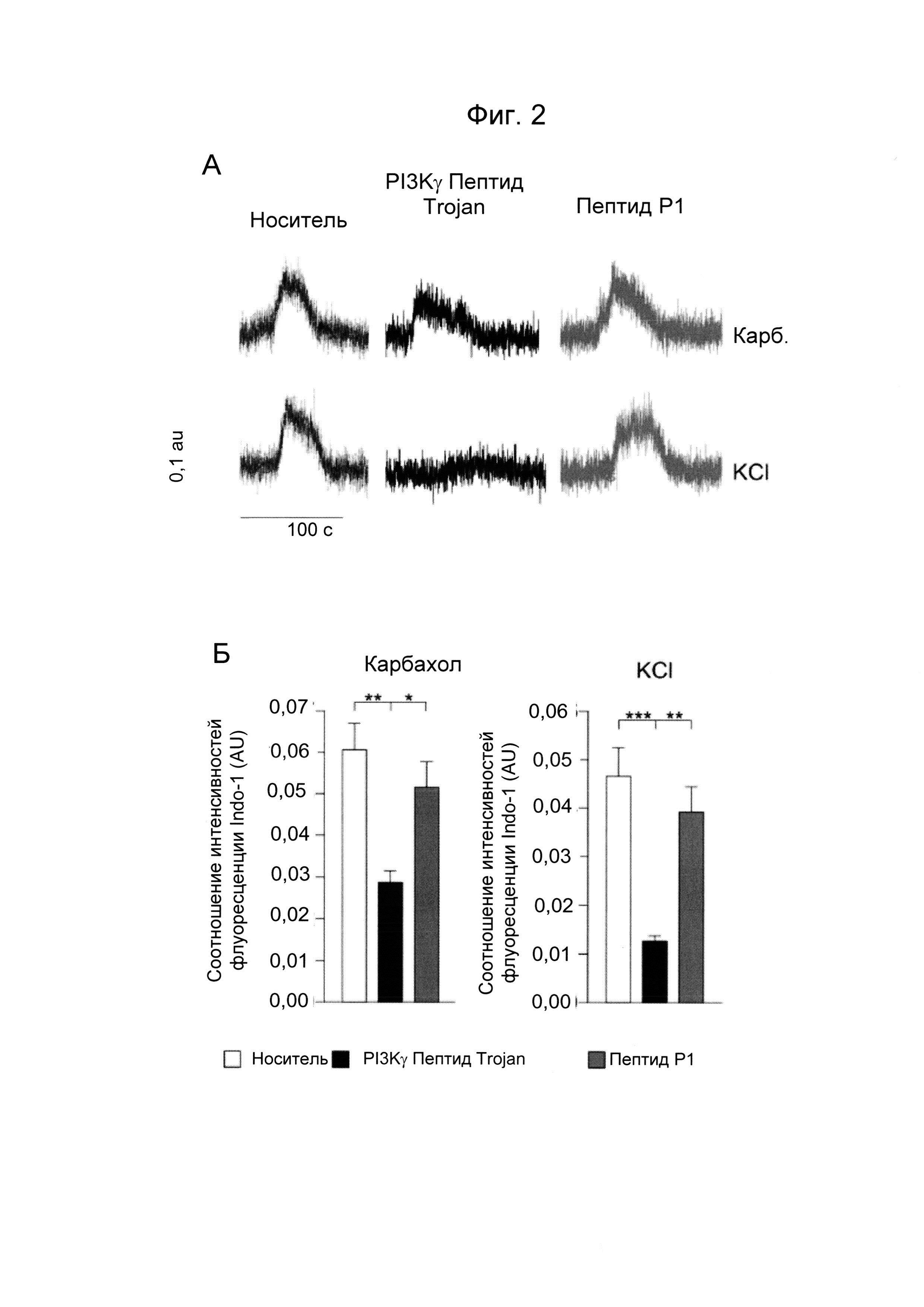
Фиг. 2. PI3Kγ ингибиторный пептид Trojan ингибирует поступление кальция через канал L-типа в гладкомышечных клетках дыхательных путей человека. А) Репрезентативные кривые коротких одиночных импульсов Са2+, записанных в hBSMC, предварительно обработанных носителем, PI3Kγ ингибиторным пептидом Trojan или контрольным пептидом Р1 (50 мкМ, 30 минут) перед стимуляцией мускариновым агонистом карбахолом (Carb, 10 мкМ, верхняя панель) и деполяризирующим раствором (40 мМ KCl, нижняя панель). Б) Максимальное изменение соотношения интенсивностей флуоресценции индикатора INDO-1 (AU (единицы поглощения)) коротких одиночных импульсов Са2+, показанных на А. В) цАМФ-зависимое фосфорилирование субъединицы Cav1.2 (Ser-1928) кальциевого канала L-типа (LTCC) в hBSMC, обработанных PI3Kγ ингибиторным пептидом Trojan или контрольным пептидом Р1 (50 мкМ, 30 минут). Показаны репрезентативные изображения и относительная количественная оценка n более и равно 3 независимых экспериментов Вестерн-блоттинга. **Р менее 0,01 и ***Р менее 0,001 с помощью однофакторного дисперсионного анализа с последующим тестом Бонферрони.
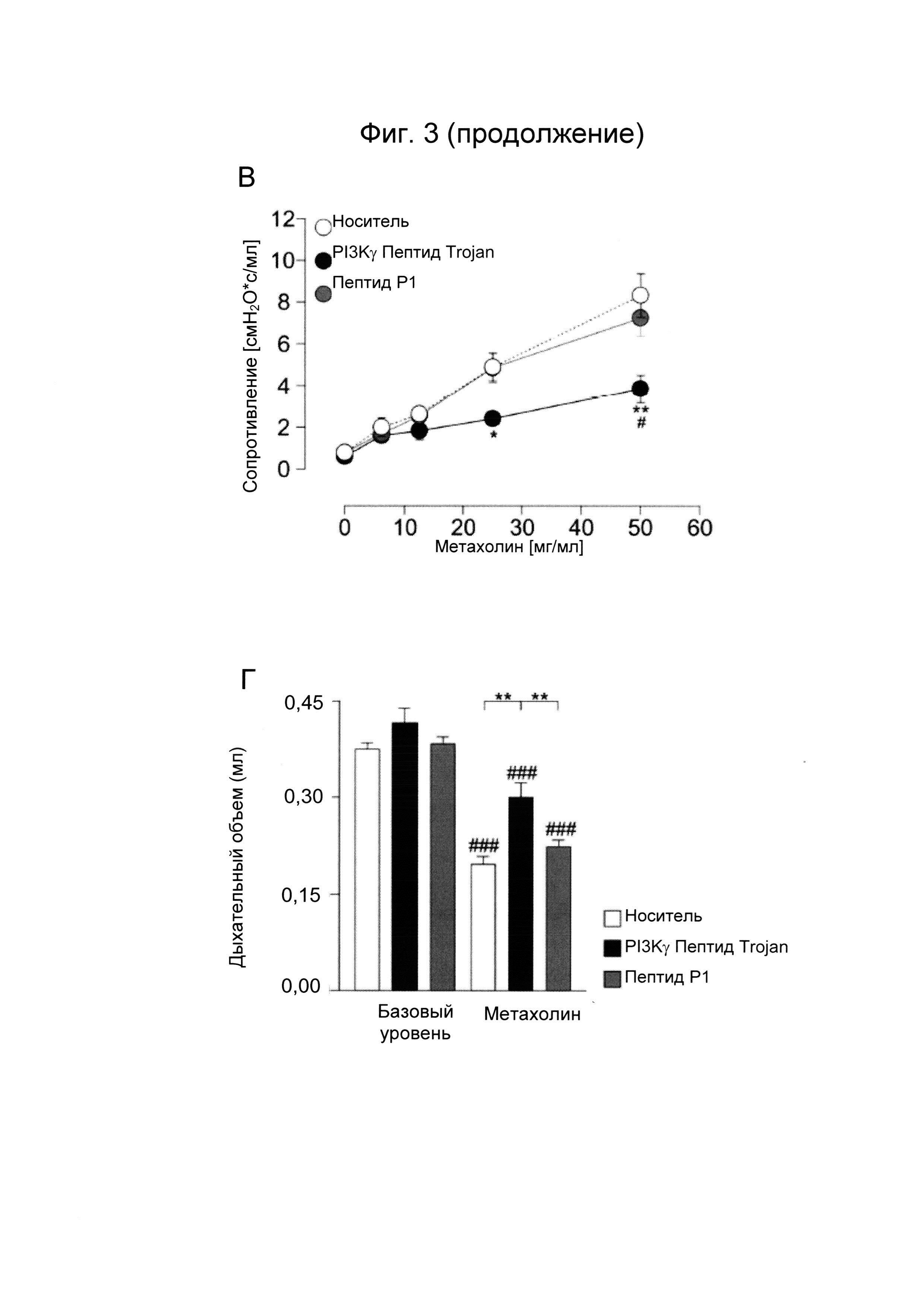
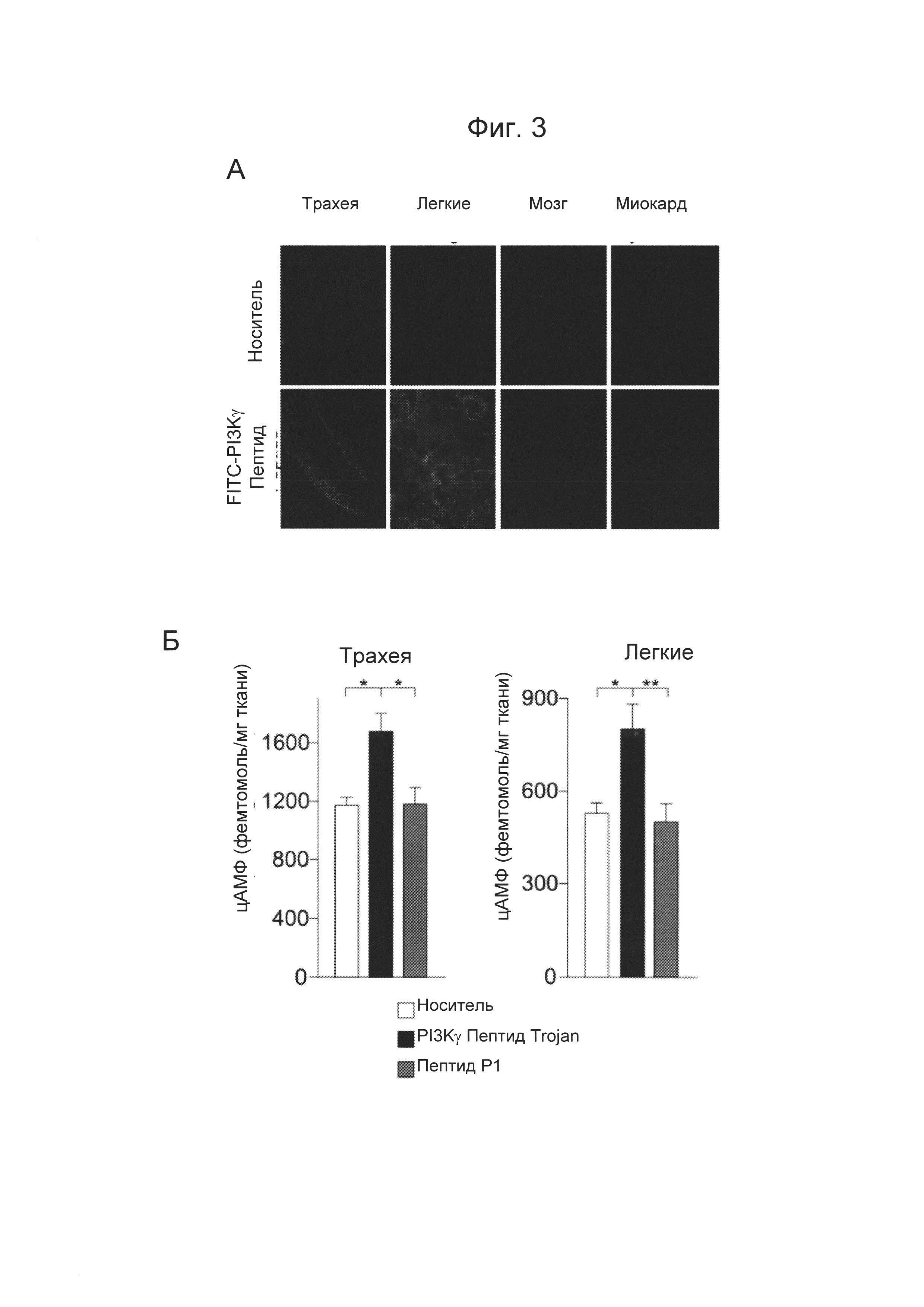
- Фиг. 3. PI3Kγ ингибиторный пептид Trojan повышает уровни цАМФ в дыхательных путях in vivo и снижает гиперчувствительность дыхательных путей у здоровых мышей и мышей с астмой. A) PI3Kγ ингибиторный пептид Trojan метили FITC и вводили внутритрахеальным путем мышам линии BALC/c (1,5 мкг/мышь). Флуоресценцию в легком и трахее анализировали посредством конфокальной микроскопии, спустя 30 минут после обработки. Контрольным мышам вводили по капле равный объем раствора, используемого для FITC мечения. Представлены FITC флуоресцентные изображения срезов трахеи, легких, мозга и миокарда животных, обработанных носителем (верхние панели) или PI3Kγ ингибиторным пептидом Trajan (нижние панели). Б) Уровни цАМФ в целой трахее (левая панель) и легких (правая панель) мышей, обработанных, как описано в А. *Р менее 0,05 и **Р менее 0,01 с помощью однофакторного дисперсионного анализа с последующим тестом Бонферрони. В) Гиперчувствительность дыхательных путей измеряли как среднее сопротивление легкого у здоровых мышей, анестезированных и вентилируемых, обработанных спреем носителя, PI3Kγ ингибиторным пептидом Trojan (1,5 мкг) или контрольным пептидом Р1 (эквимолярное количество) перед воздействием возрастающих доз метахолина. *Р менее 0,05 и **Р менее 0,01, по сравнению с носителем; #Р менее 0,05 по сравнению с Р1 с помощью двухфакторного дисперсионного анализа с последующим тестом Бонферрони. Г) Гиперчувствительность дыхательных путей измеряли как изменение дыхательного объема у мышей с астмой, анестезированных и вентилируемых, в ответ на метахолин (500 мг/кг, внутривенное введение). Животных, сенсибилизированных к овальбумину, обрабатывали носителем, PI3Kγ ингибиторным пептидом Trojan (150 мкг) или контрольным пептидом Р1 (эквимолярное количество) в течение 30 минут перед введением метахолина. **Р менее 0,01, по сравнению с носителем и контрольным пептидом; ###Р менее 0,001, по сравнению с базовым уровнем с использованием однофакторного дисперсионного анализа с последующим тестом Бонферрони.
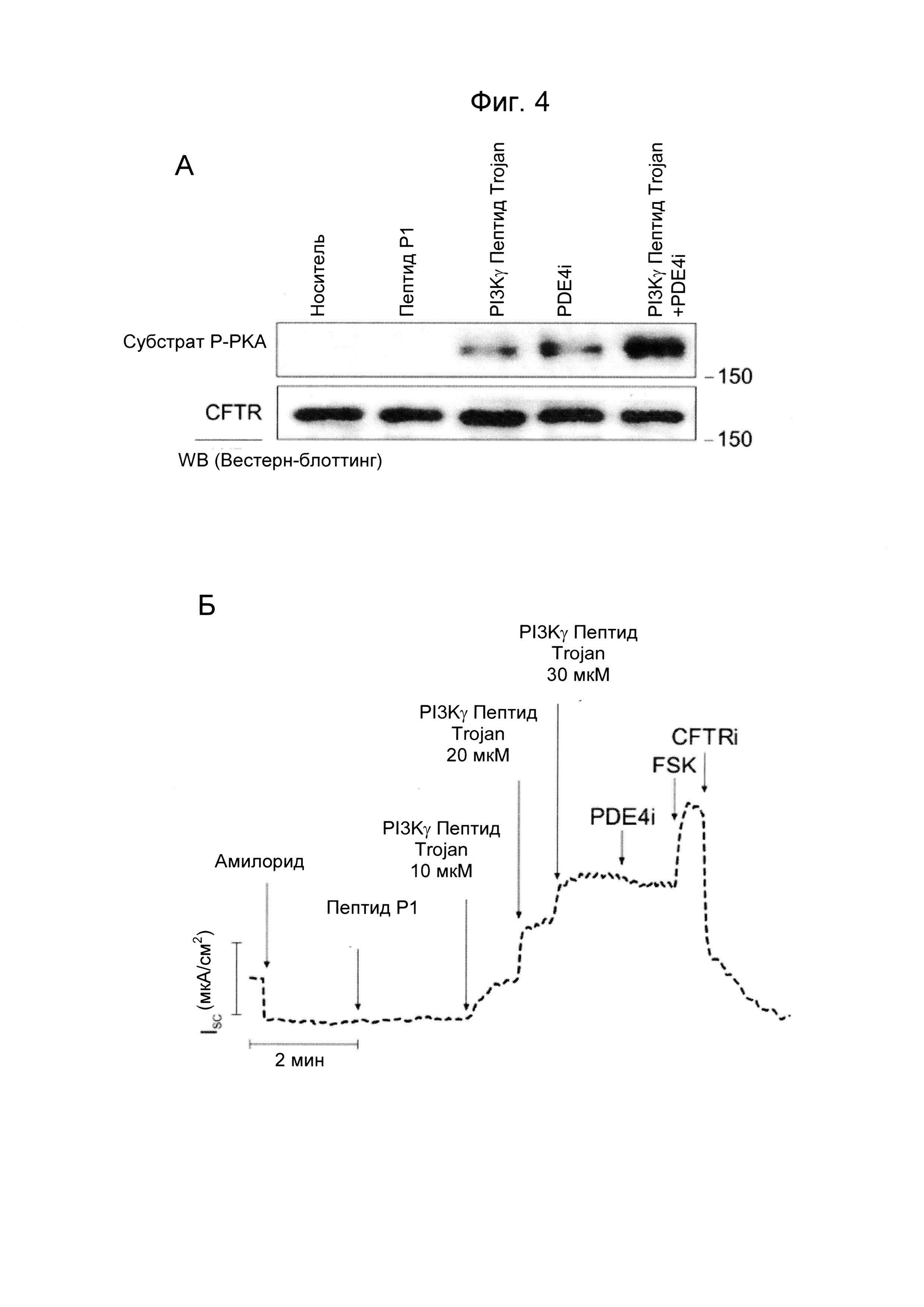
- Фиг. 4. Пептид Trojan, полученный из PI3Kγ, повышает уровень цАМФ-зависимого фосфорилирования и проводимость CFTR дикого типа в отношении ионов хлора. А) цАМФ-опосредованное фосфорилирование CFTR в эпителиальных клетках дыхательных путей человека (NuLi-1), обработанных носителем (дорожка 1), контрольным пептидом Р1 (25 мкМ, дорожка 2), PI3Kγ ингибиторным пептидом Trojan (25 мкМ, дорожка 3), только ингибитором PDE4 Ролипрам (PDE4i; 10 мкМ, дорожка 4) или вместе с пептидом Trojan, полученным из PI3Kγ (дорожка 5), в течение 30 минут. Показаны репрезентативные изображения выявления с помощью Вестерн-блоттинга CFTR иммунопреципитаций и фосфорилирования посредством РКА n≥3 независимых экспериментов. Б) Репрезентативная кривая CFTR токов, измеренных в камерах Уссинга в культурах клеток NuLi-1. Следующие обработки применяли в указанные моменты времени: амилорид (ингибитор канала ENAC, 10 мкМ), контрольный пептид Р1 (30 мкМ), возрастающие концентрации пептида Trojan, полученного из PI3Kγ (10 мкМ, 20 мкМ и 30 мкМ), ингибитор PDE4 Ролипрам (PDE4i; 10 мкМ), форсколин (FSK, 10 мкМ) и ингибитор CFTR 172 (CFTRi; 20 мкМ).
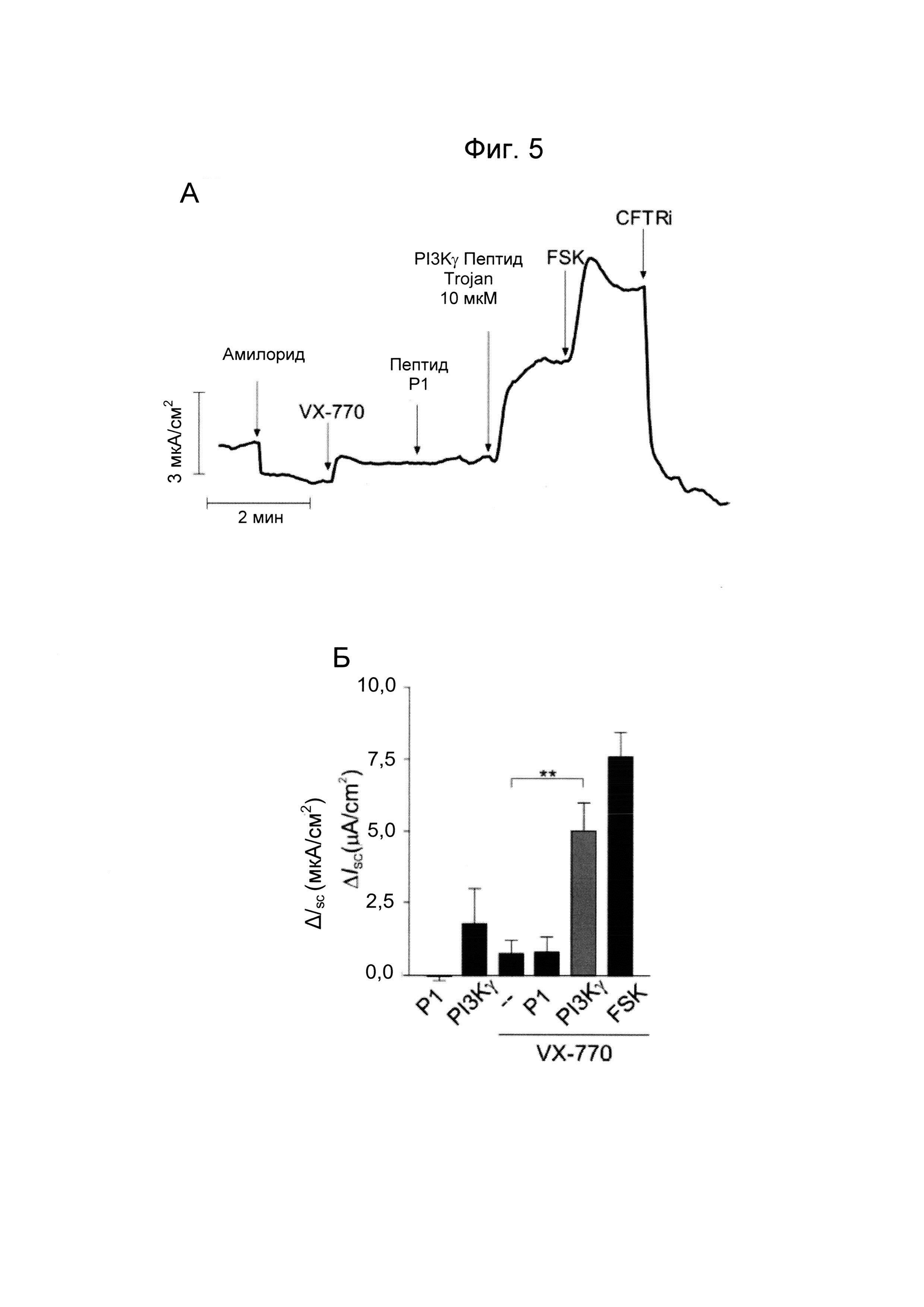
- Фиг. 5. Пептид Trojan, полученный из PI3Kγ, повышает проводимость CFTR в эпителиальных клетках дыхательных путей с мутацией ΔF508. А) Репрезентативная кривая токов CFTR, измеренных в камерах Уссинга в культурах эпителиальных клеток дыхательных путей с мутацией ΔF508 (CuFi-1), обработанных корректором VX-809 (20 мкМ; 24 часа). В указанные моменты времени применяли следующие обработки: амилорид (ингибитор канала ENAC, 10 мкМ), потенциатор CFTR VX-770 (10 мкМ), контрольный пептид Р1 (10 мкМ), PI3Kγ ингибиторный пептид Trojan (10 мкМ), форсколин (FSK, 10 мкМ) и ингибитор CFTR 172 (CFTRi; 20 мкМ). Б) Средние изменения токов в ответ на указанные обработки. **P<0,01 с помощью однофакторного дисперсионного анализа с последующим тестом Бонферрони.
- Фиг. 6. Последовательности проникающих полипептидов, подходящих для получения слитых пептидов по настоящему описанию.
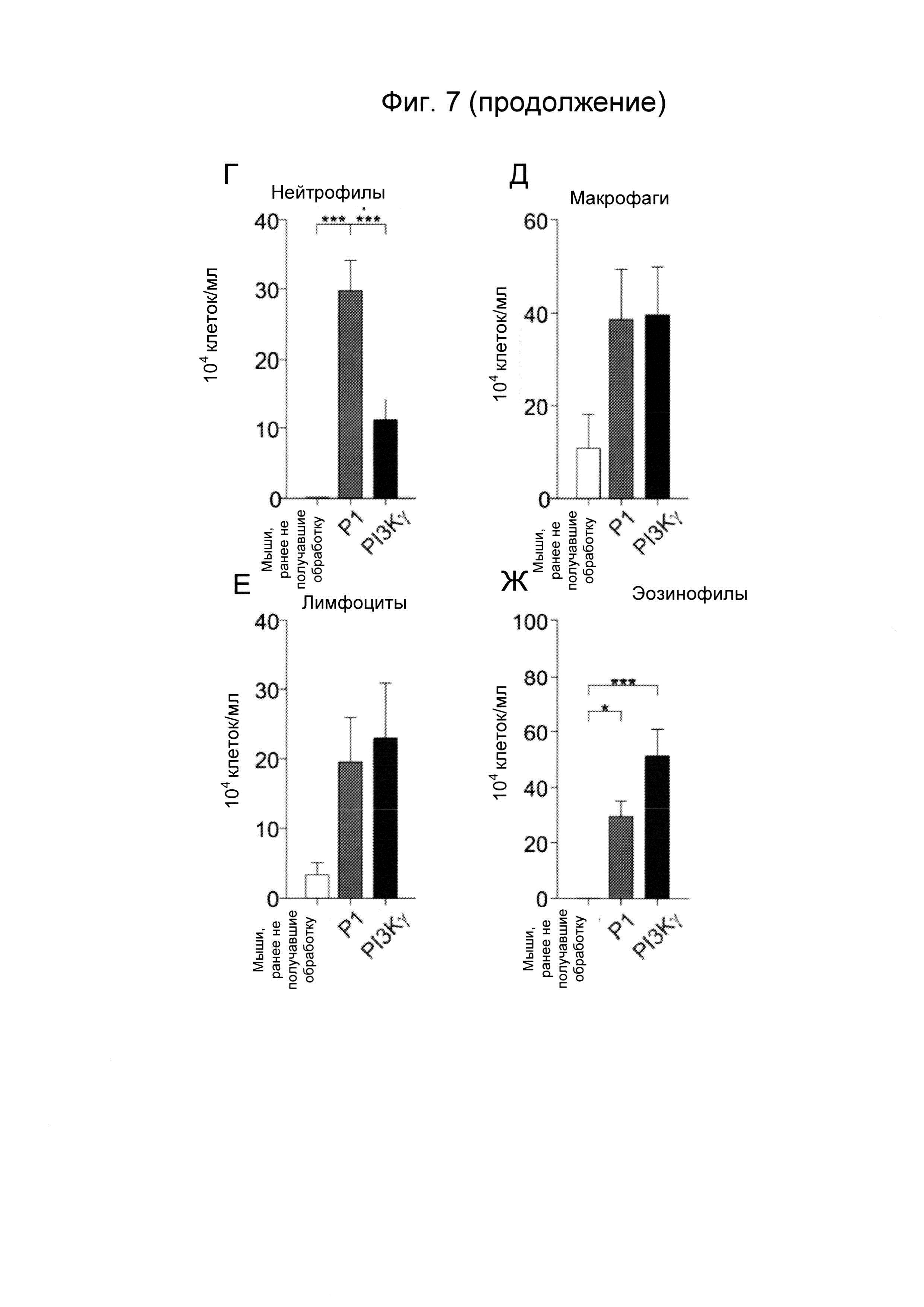
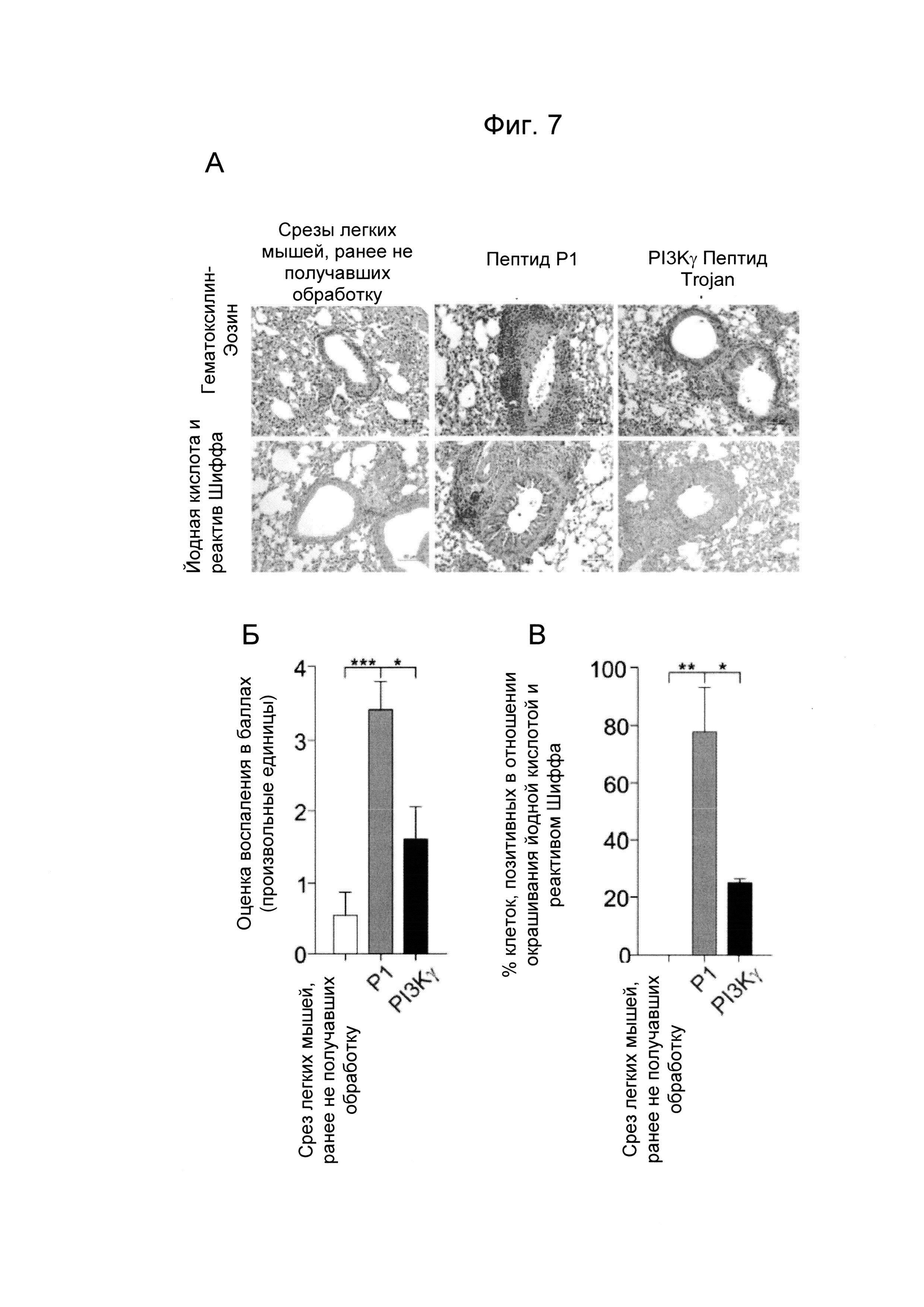
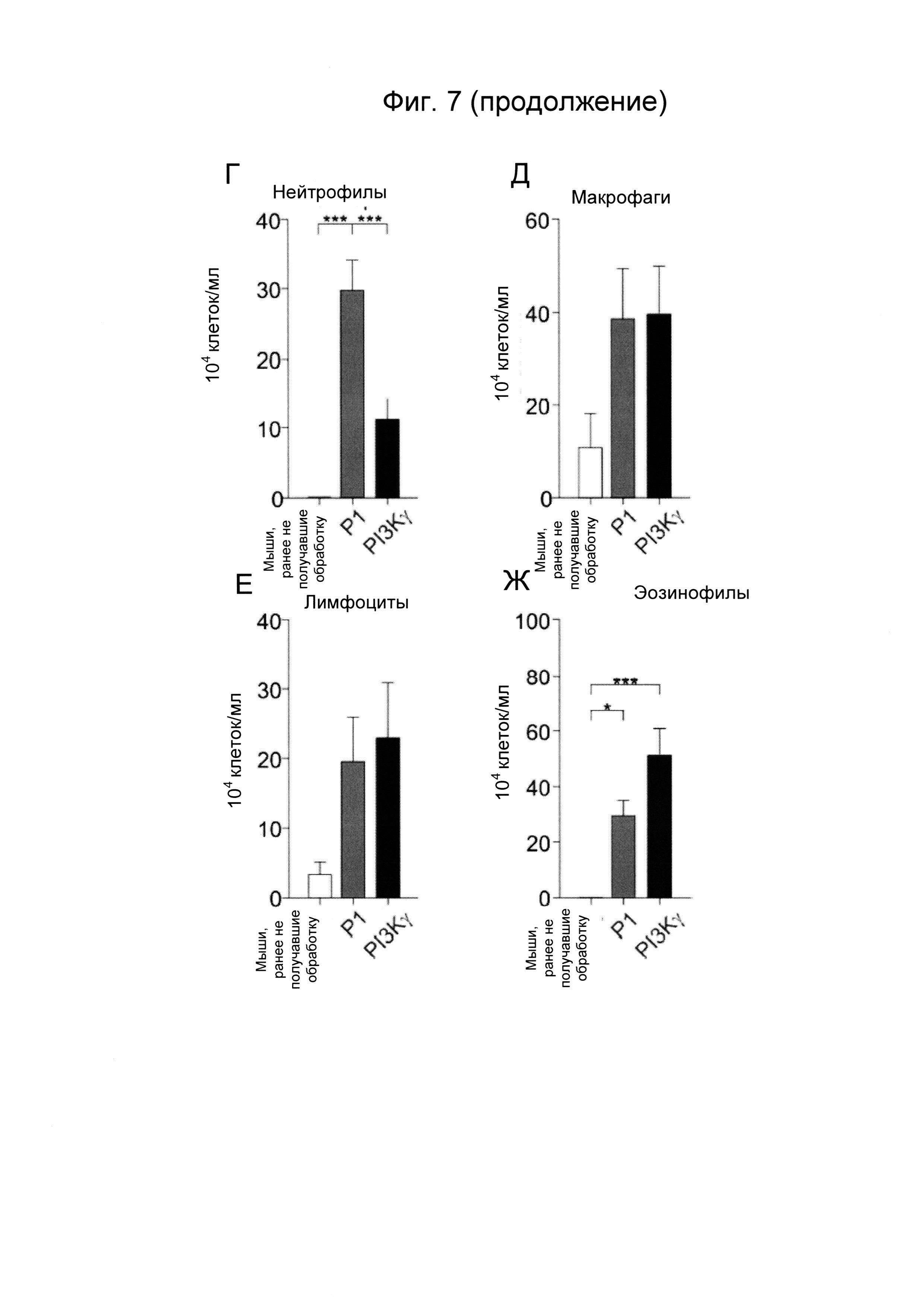
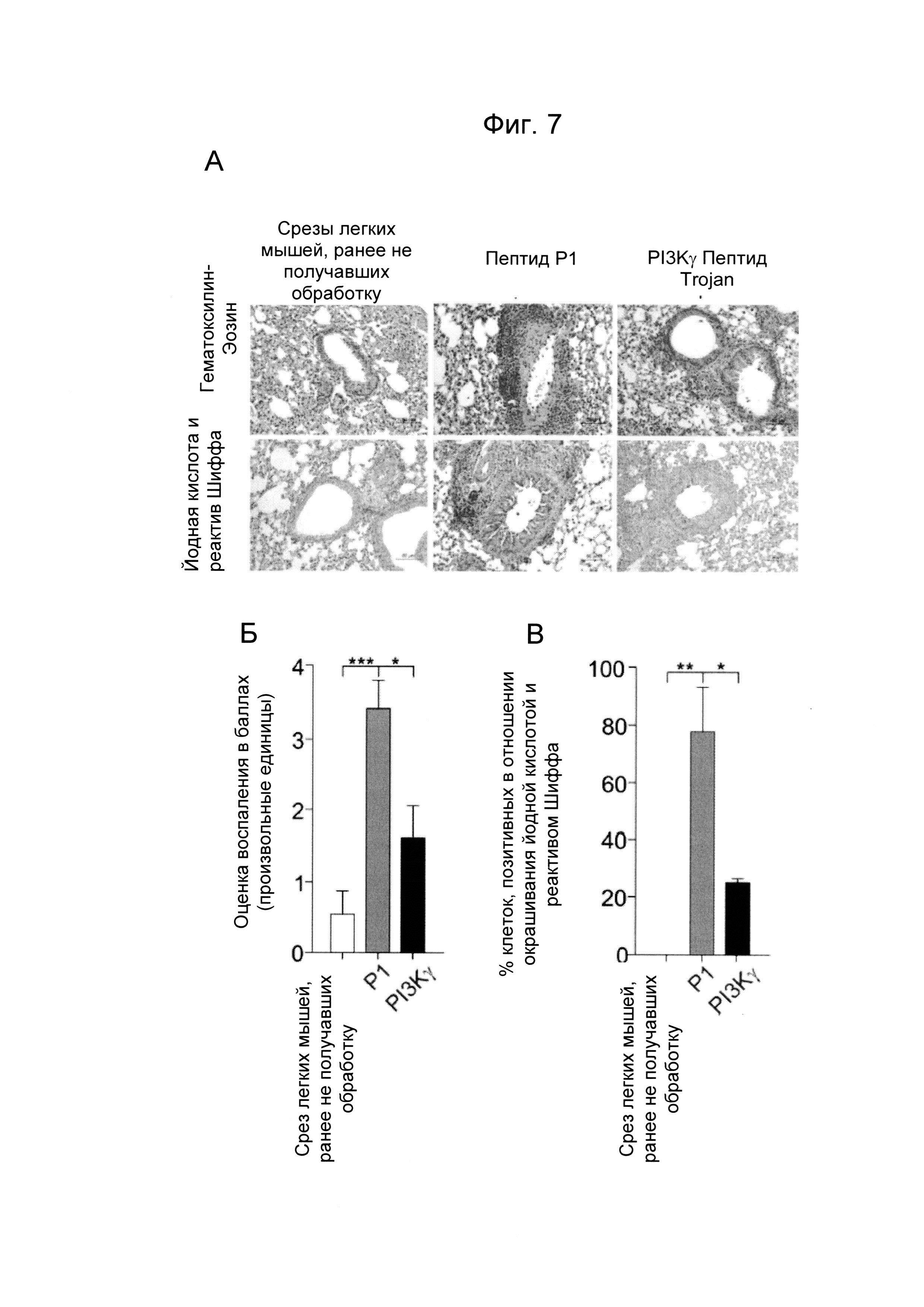
- Фиг. 7. Пептид Trojan, полученный из PI3Kγ, уменьшает легочное воспаление у мышей с астмой. А) Репрезентативные изображения окрашивания гематоксилин-эозином (верхние изображения) и йодной кислотой и реактивом Шиффа (нижние изображения) срезов легкого ранее не получавших обработку мышей и мышей, сенсибилизированных овальбумином и предварительно обработанных пептидом Trojan, полученным из PI3Kγ (25 мкг/мышь/инъекция) или контрольным пептидом Р1 (эквимолярные количества) перед каждым интраназальным введением овальбумина. Срезы окрашивали гематоксилин-эозином для анализа морфологии ткани и уровня воспаления и йодной кислотой и реактивом Шиффа для определения наличия бокаловидных клеток. Б) Полуколичественный анализ степени перибронхиального воспаления в срезах легкого, как показано на А). В) Процент эпителиальных клеток, позитивных в отношении окрашивания йодной кислотой и реактивом Шиффа в эпителии срезов легкого, как показано на А). Г)-Ж) Число нейтрофилов (Г), макрофагов (Д), лимфоцитов (Е) и эозинофилов (Г) в бронхоальвеолярном лаваже мышей, предварительно обработанных пептидом Trojan, полученным из PI3Kγ, или контрольным пептидом Р1, как описано в А). *Р менее 0,05, **Р менее 0,01 и ***Р менее 0,001 с помощью однофакторного дисперсионного анализа с последующим тестом Бонферрони.
Подробное описание изобретения
Сейчас изобретение будет подробно описано, исключительно в качестве иллюстративного и неограничивающего примера, со ссылкой на единственный графический материал, в котором схематично представлена система экстракции под давлением, пригодная для применения в целях осуществления способа, описанного в данном документе.
В следующем описании многочисленные конкретные подробности представлены для обеспечения полного понимания воплощений. Воплощения могут быть реализованы на практике без одной или более чем одной конкретной детали или с другими способами, компонентами, материалами и т.д. В других случаях хорошо известные структуры, материалы или операции не показаны или описаны подробно для того, чтобы избежать затруднения понимания определенных аспектов воплощений.
По всему объему настоящего описания ссылка на «одно воплощение» или «воплощение» означает, что конкретный признак, структура или характеристика, описанная в связи с воплощением, включена в по меньшей мере одно воплощение. Таким образом, появление выражений «в определенном воплощении» или «в воплощении» в разных местах по всему объему настоящего описания не обязательно всегда относится к одному и тому же воплощению. Кроме того, определенные признаки, структуры или характеристики можно объединять любым подходящим образом в одном или более чем одном воплощении.
Названия, используемые в данном документе, служат только для удобства и не разъясняют цель или значение воплощений.
Одно воплощение настоящего описания относится к слитому пептиду, содержащему: i) аминокислотную последовательность, как определено в SEQ ID No.: 1, или родственный гомолог, обладающий по меньшей мере 90% идентичностью с SEQ ID No.: 1 и обладающий способностью последовательности SEQ ID No.: 1 ингибировать киназонезависимую функцию PI3Kγ, и
ii) пептид, обладающий способностью проникать в клетку, для применения в качестве лекарственного средства, в частности, для лечения респираторных заболеваний.
Другое воплощение настоящего изобретения относится к продукту, содержащему i) слитый пептид, как определено выше, и ii) потенциатор регулятора трансмембранной проводимости при кистозном фиброзе и/или корректор регулятора трансмембранной проводимости при кистозном фиброзе в виде комбинированного препарата для последовательного, одновременного или раздельного применения для лечения респираторных заболеваний, предпочтительно кистозного фиброза.
Применение продукта, содержащего слитый пептид, как описано выше, корректор регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) и потенциатор регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) в виде комбинированного препарата для последовательного, одновременного или раздельного применения особенно подходит для лечения пациентов с кистозным фиброзом, которые несут мутацию ΔF508 CFTR.
Настоящее описание относится к продукции и терапевтическим применениям слитого пептида, проницаемого в отношении клеточных мембран, который ингибирует взаимодействие между PI3Kγ и активатором PDE, РКА. Слитый пептид по настоящему описанию снижает активность конкретной PDE, усиливая сигнализацию пути сигнальной трансдукции β2-AR/цАМФ и вызывая цАМФ-опосредованные прорасслабляющее действие, как в гладкомышечных клетках дыхательных путей человека и in vivo, так и в доклинической модели аллергической астмы. Кроме того, слитый пептид с PI3Kγ ингибирующей активностью усиливает тот же сигнальный путь в эпителиальных клетках дыхательного пути и стимулирует цАМФ-зависимое открытие канала CFTR дикого типа (чьи нуклеотидные и аминокислотные последовательности доступны в базе данных последовательностей GenBank в виде номеров доступа NM_000492.3 и NP_000483.3, соответственно) и CFTR с мутацией ΔF508 (последовательность данной мутации доступна в базе данных последовательностей GenBank в виде номера доступа S64640.1), которая является главной причиной кистозного фиброза.
В общем, результаты, показанные в данном документе, демонстрируют возможность использования слитого пептида или его гомологов, обладающих способностью ингибировать PI3Kγ, в качестве локальной терапии посредством ингаляции, для лечения респираторных заболеваний, таких как аллергическая астма и кистозный фиброз.
Несмотря на то, что за последнее десятилетие было разработано много ингибиторов киназной активности PI3Kγ, многие из которых в настоящее время проходят клинические испытания в отношении лечения неопластических заболеваний, не существует способов, которые делают возможным селективное воспрепятствование киназонезависимой активности фермента или скорее функции РКА- или АKАР-якорного белка.
Ранее показано, что пептид, который содержит сайт связывания PI3Kγ с РКА, состоящий из остатков 126-150 PI3Kγ человека, заменяет взаимодействие между данными двумя белками и снижает активность PI3Kγ-связанной PDE, PDE3B в исследованиях взаимодействия in vitro (1).
В настоящем изобретении показано, абсолютно неожиданным образом, что PI3Kγ 126-150 пептид (SEQ ID No.: 1 - KATHRSPGQIHLVQRHPPSEESQAF), конъюгированный с пептидом, проницаемым в отношении клеточных мембран, таким как, например, Пенетратин 1 Antennapedia (SEQ ID No.: 3) (3), может быть использован в качестве ингибитора киназонезависимой функции PI3Kγ in vivo. PI3Kγ ингибиторный слитый пептид проникает в гладкомышечные клетки дыхательных путей и усиливает сигнальный путь β2-AR/цАМФ. В частности, данные показывают, что слитый пептид увеличивает уровни цАМФ и ограничивает гиперчувствительность дыхательных путей в доклинической модели аллергической астмы и что он эффективно достигает нижние дыхательные пути при введении местно внутритрахеальным путем. Эти данные, таким образом, демонстрируют клиническое применение PI3Kγ ингибиторного слитого пептида в аэрозольной терапии для лечения респираторных заболеваний.
Современная ингаляционная терапия бронхообструктивных заболеваний основана на применении агонистов β2-AR, таких как сальбутамол и формотерол, и ингибиторов PDE4, таких как Рофлумиласт, который был недавно одобрен для лечения хронической обструктивной болезни легких (COPD).
Несмотря на то, что интенсивное лечение агонистами β-AR обеспечивает очевидные благоприятные клинические эффекты, длительное или повторное воздействие данных лекарственных средств может приводить к значительному снижению и/или полной потере их эффективности у пациентов с астмой вследствие агонист-зависимой десенсибилизации мембраны β-AR.
Согласно настоящему описанию предложено решение данной проблемы и предложено применение ингибиторного пептида, который воздействует на активность фермента PI3Kγ и, по существу, не действует непосредственно на стимуляцию β2-AR, а усиливает каскад расположенных после него событий сигнализации. Таким образом, PI3Kγ ингибиторный пептид предлагает уникальную возможность модулировать β2-AR-зависимые цАМФ домены, обеспечивая бронхорасслабляющее действие, аналогично действию, опосредованному агонистами β2-AR, без индукции инактивации рецептора.
Данные, предложенные в данном документе, демонстрируют, что ингибирование РKА-заякоривающей функции PI3Kγ снижает активность PDE только в клетках, экспрессирующих PI3Kγ (PI3Kγ+/+), но не в клетках, не содержащих данный фермент (PI3Kγ-/-), указывая, таким образом, на то, что слитый пептид по настоящему описанию ингибирует PDE, исключительно регулируемые PI3Kγ.
PI3Kγ ингибиторный пептид, таким образом, представляет уникальный инструмент, который обеспечивает селективное в отношении изоформы ингибирование PDE и делает возможным ограничение основных побочных эффектов, которые ассоциированы с неселективным ингибиторами PDE4, такими как Рофлумиласт, возникающих главным образом вследствие ингибирования изоформ, которые не экспрессируются в дыхательной системе.
Несмотря на то, что ингибиторы PDE4 характеризуются важным прорасслабляющим действием в выделенных клетках, они не являются оптимальными бронходиляторами in vivo, где они главным образом выполняют противовоспалительные функции.
Результаты, предложенные в данном документе, демонстрируют, что слитый пептид со способностью ингибировать киназонезависимую функцию PI3Kγ, предмет настоящего описания, имеет сильную бронхорасширяющую функцию in vivo у здоровых животных и в доклинической модели аллергической астмы. Данные эффекты, которые можно объяснить способностью пептида препятствовать каталитической активности многочисленных изоформ PDE, не только включая PDE4, но также PDE3.
В соответствии с настоящими данными, было показано, что ингибирование PDE3 и PDE4 может быть аддитивным или синергетическим. В частности, ингибиторы PDE4 и PDE3 являются неэффективными при использовании по отдельности, но действуют синергетически при ингибировании гладкомышечного сокращения. Таким образом, ингибирование киназонезависимой активности PI3Kγ обеспечивает уникальный инструмент для одновременного ингибирования конкретных изоформ PDE3 и PDE4, в частности, изоформ, немаловажным образом участвующих в регуляции сократительной способности гладкой мускулатуры бронхов.
Так как PDE3 и PDE4 не только экспрессируются в дыхательных путях, но также в миокарде и центральной нервной системе, системное ингибирование даже выбранных изоферментов может вызывать значительные побочные эффекты. В частности, ингибирование PDE3 и PDE4 in vivo может оказывать проаритмогенное, рвотное и проанорексигенное действие.
Настоящее описание показывает, что слитый пептид, обладающий способностью ингибировать киназонезависимую активность PI3Kγ, является терапевтически эффективным, и что аэрозольная композиция ингибиторного пептида PI3Kγ эффективно распределяется в нижних дыхательных путях. Кроме того, применение молекулы пептида обеспечивает более широкий профиль терапевтических эффектов/побочных эффектов, по сравнению с небольшими молекулами, такими как ингибиторы PDE, которые могут быстро диффундировать в другие ткани, находящиеся за пределами дыхательной системы.
Данные, предложенные в данном документе, демонстрируют, что флуоресцентный вариант PI3Kγ ингибиторного пептида накапливается в трахее и легких после внутритрахеального введения, не достигая миокарда и мозга.
На основе этих данных, таким образом, возможно сделать вывод о том, что ингаляционная терапия на основе пептида, проницаемого в клеточные мембраны, который селективно ингибирует киназонезависимую активность фермента PI3Kγ, является высокоэффективной. Данный терапевтический подход можно применять для лечения разных респираторных заболеваний, от астмы до кистозного фиброза, где необходимы агенты, способные повышать уровень внутриклеточного цАМФ после β2-AR.
Результаты, представленные в данном документе, демонстрируют, что ингибиторный пептид PI3Kγ не только повышает уровни цАМФ в гладкой мышце, но также в эпителиальном компартменте дыхательных путей, таким образом, открывая возможность использования данного соединения также для стимуляции цАМФ-опосредованного открытия канала CFTR, который является дефектным у пациентов с кистозным фиброзом.
Данные, предложенные в данном документе, также показывают, что ингибирование PI3Kγ действует синергетически с известным, клинически прогрессивным, потенциатором CFTR, VX-770, в результате повышения проводимости одной из наиболее распространенных мутантных форм CFTR при кистозном фиброзе. Эти данные впервые демонстрируют способность PI3Kγ-ингибирующей молекулы увеличивать активность известного потенциатора CFTR, VX-770, который, как известно, стимулирует проводимость разных мутантных форм CFTR, за исключением наиболее распространенного мутанта при кистозном фиброзе - ΔF508.
Слитый пептид по настоящему описанию, таким образом, может быть использован в комбинации с потенциатором регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) и/или корректором регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) в виде комбинированного препарата для последовательного, одновременного или раздельного применения в лечении респираторных заболеваний, характеризующихся дефектным цАМФ-опосредованным открытием CFTR, например, у пациентов с кистозным фиброзом.
Комбинированное применение потенциатора регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) и корректора регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), вместе со слитым пептидом по настоящему описанию, особенно подходит для лечения пациентов с кистозным фиброзом, несущих мутацию CFTR ΔF508, кому необходимо введение корректора CFTR для обеспечения экспрессии мутантного CFTR на мембране.
Потенциаторы регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), которые могут быть преимущественно использованы в комбинации со слитым пептидом по настоящему описанию, например, представляют собой: Ивакафтор или VX-770 (N-(2,4-ди-трет-бутил-5-гидроксифенил)-1,4-дигидро-4-оксохинолин-3-карбоксамид) и VX-532 (4-метил-2-(5-фенил-1Н-пиразол-3-ил)-фенол).
Корректоры регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), которые могут быть преимущественно использованы в комбинации со слитым пептидом по настоящему описанию и с потенциатором регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), например, представляют собой: VX-809 (3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойная кислота) и VX-661 ((R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2 метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид).
В общем, данное исследование демонстрирует терапевтический потенциал слитого пептида, который селективно ингибирует киназонезависимую активность PI3Kγ.
Молекулу можно использовать для лечения респираторных заболеваний, включая аллергическую астму, где лекарственные средства, предназначенные для повышения внутриклеточных уровней цАМФ и для стимуляции расслабления гладкой мускулатуры бронхов, являются крайне желательными.
Кроме того, данное соединение можно применять для пациентов с кистозным фиброзом, когда агенты, которые повышают концентрации цАМФ, являются ключевыми инструментальными средствами для стимуляции цАМФ-опосредованного открытия дефектного CFTR.
Кроме того, пептид-опосредованное ингибирование PI3Kγ можно применять ко всем патологическим состояниям, характеризующимся гипофункциональным CFTR, включая COPD, где воздействие сигаретного дыма, как было показано, изменяет активность CFTR.
Наконец, через функциональный блок PDE4, слитый пептид по настоящему описанию, обладающий способностью ингибировать PI3Kγ, способен оказывать важные противовоспалительные действия. Экспериментальное доказательство, о котором сообщается в данном документе, демонстрирует, что данный пептид действительно ограничивает перибронхиальное воспаление, ассоциированное с аллергической астмой. Таким образом, очевидно, что ингибирование киназонезависимой активности PI3Kγ может обеспечивать множество независимых благоприятных терапевтических эффектов при лечении респираторных заболеваний, одновременно действуя в качестве бронходилятора, потенциатора CFTR и противовоспалительного агента.
Ниже, изобретение будет подробно описано в качестве неограничивающего примера, со ссылкой на слитый пептид, имеющий последовательность, показанную в SEQ ID No.: 2 (ниже в данном документе называемый «пептидом Trojan, полученным из PI3Kγ»), содержащий аминокислотную последовательность SEQ ID No.: 1, и пептид, проникающий в клетку, соответствующий Пенетратину 1 Antennapedia (SEQ ID No.: 3, описанная в (3)).
Ясно, что объем данного описания никоим образом не ограничивается конкретной последовательностью слитого пептида SEQ ID No.: 2, так как слитый пептид по настоящему описанию может содержать: i) последовательности, обладающие гомологией с SEQ ID No.: 1 по меньшей мере 90% и обладающие способностью SEQ ID No.: 1 ингибировать киназонезависимую функцию PI3Kγ, и ii) последовательности пептида, проникающего в клетку, например, выбранного из полипептида HIV-TAT, пептидов Antennapedia, содержащих гомеодомен, также известных как проникающие пептиды или pAntp, пептида R7, пептида KALA, буфорина 2, MAP, транспортана, транспортана 10, pVEC или пептида MPG. Последовательности, соответствующие полипептидам, проникающим в клетки, упомянутым выше, показаны на Фиг. 6 и перечислены в SEQ ID No.:3-12.
Кроме того, при получении пептида Trojan, полученного из PI3Kγ, осуществляли слияние пептида, обладающего способностью проникать в клетку, с N-концом SEQ ID No.:1. Однако возможно получать слитый пептид, который попадает в объем настоящего описания, посредством создания слияния пептида, обладающего способностью проникать в клетку, с С-концом SEQ ID No.:1.
Материалы и методы
Определение способности пептида, гомологичного SEQ ID No.: 1, ингибировать киназонезависимую функцию PI3Kγ
Для определения способности пептида-гомолога, обладающего по меньшей мере 90% идентичностью с SEQ ID No.: 1, ингибировать функцию АКАР PI3Kγ, использовали ранее описанный конкурентный анализ (1). Пептид-гомолог ресуспендировали в фосфатно-солевом буферном растворе (PBS) до конечной концентрации 50 мкМ или 250 мкМ. Клетки HEK293 (номер АТСС (Американская коллекция типовых культур): CRL-1573™) трансфицировали, с использованием метода на основе фосфата кальция, конструкцией, сделанной из экспрессионного вектора pcDNA3.1 (Life Technologies, Карлсбад, штат Калифорния, США; код продукта V790-20), в котором кДНК PI3Kγ человека клонировали (SEQ ID No.: 13) с использованием рестриктаз BamHI и XbaI (New England Biolabs, Ipswich, MA, США). Конструкция pcDNA3.1-PI3Kγ находится в свободном доступе у Dr. Emilio Hirsch в университете Турина, Турин, Италия.
Через 48 часов после трансфекции клетки лизировали в холодном лизирующем буфере, содержащем 120 ммоль/л NaCl, 50 ммоль/л Tris-HCl (рН 8,0), ингибиторы протеазы с полным ингибированием (Roche Applied Science, Индианаполис, штат Индиана) и ингибиторы фосфатазы (50 ммоль/л фторид натрия, 1 ммоль/л ортованадат натрия и 10 ммоль/л пирофосфат натрия). Спустя 30 мин инкубации на льду, лизаты центрифугировали при 13000 об/мин в течение 10 мин при 4°С, и супернатант инкубировали с пептидом в течение 30 минут при комнатной температуре. После инкубации регуляторную субъединицу РKА (РKА RII) иммунопреципитировали посредством инкубирования белкового экстракта с 30 мкл смеси 1:1, белка А и сефарозы (Amersham Biosciences, Бакингемшир, Великобритания) и антителом против РKА RII С20 (Santa Cruz Biotechnology Inc., Даллас, штат Техас, США; код продукта: sc-908) в течение 2 ч при 4°С при встряхивании. Иммунные комплексы тщательно промывали лизирующим буфером, и ассоциацию PI3Kγ с РКА RII анализировали Вестерн-блоттингом с использованием моноклонального антитела против PI3Kγ (в свободном доступе у Dr. Emilio Hirsch в университете Турина, Турин, Италия).
Животные
Мышей, нокаутированных по PI3Kγ (PI3Kγ-/-), и нокин-мышей (knock-in мыши), экспрессирующих каталитически неактивную форму PI3Kγ (PI3KγKD/KD), генерировали, как описано ранее (4, 5). Мутантных мышей скрещивали с животными с генетическим фоном C57BI/6J для 15 поколений, и в качестве контролей использовали мышей C57BI/6J (PI3Kγ+/+). Для исследований гиперчувствительности дыхательных путей у мышей с астмой и здоровых мышей использовали самок мышей линии BALB/C. Для всех экспериментов использовали мышей в возрасте от 8 до 12 недель. Мышей содержали в группах со свободным доступом к пище (стандартная диета) и к воде, в контролируемой системе, согласно которой обеспечивается цикл 12 часов света и 12 часов темноты. Животных использовали в соответствии с основными принципами и ведомственными нормами обращения с животными, одобренными местным комитетом по контролю этических норм обращения с животными.
Клеточная культура и трансфекция
Гладкомышечные клетки бронхов человека (hBSMC) приобретали в Lonza (СС-2576, Lonza Walkersville, Inc. США), выращивали в среде Игла в модификации Дульбекко (DMEM, Gibco, Карлсбад, штат Калифорния) и дополняли 10% фетальной телячьей сывороткой (FBS) и 5 мМ пенициллином/стрептомицином (Gibco, Карлсбад, штат Калифорния). Для экспериментов использовали клетки вплоть до пассажа 15.
Гладкомышечные клетки бронхов человека (hBSMC) трансфицировали плазмидой, кодирующей зонд FRET для цАМФ, ICUE3 (описано в патенте США US 8236523 В2) (2), посредством электропорации с помощью прибора Нуклеофектор (АМАХА, Гейтерсберг, штат Мэриленд) согласно протоколу производителя. Кратко, 1×106 клеток ресуспендировали в 100 мкл раствора для нуклеофекции (VPI-1004, АМАХА, Гейтерсберг, штат Мэриленд), смешивали с 1 мкг pcDNA3-ICUE3 (описано в патенте США US 8236523 В2) и подвергали электропорации в приборе для нуклеофекции Amaxa Biosystems (Program А-033). Эксперименты по визуализации живых клеток проводили через 24 часа после трансфекции.
Линии эпителиальных клеток дыхательных путей человека, экспрессирующие CFTR дикого типа (NuLi-1) или с мутацией ΔF508 (CuFi-1), приобретали у АТСС (код продукта NuLi-1: АТСС® CRL-4011™; код продукта CuFi-1: АТСС® CRL-4013™). Клетки культивировали в среде Игла в модификации Дульбекко (DMEM, Gibco, Карлсбад, штат Калифорния) с добавлением 10% фетальной телячьей сыворотки (FBS), 30 мкг/мл пенициллина, 100 мкг/мл стрептомицина и 300 мкг/мл гигромицина В (Gibco, Карлсбад, штат Калифорния).
Экстракция белков и иммунопреципитация
Для экстракции белков из гладкомышечных клеток трахеи мыши (mTSMC) и гладкомышечных клеток бронхов человека (hBSMC) клетки обрабатывали указанными лекарственными средствами/пептидами и сразу же лизировали в холодном лизирующем буфере, содержащем 120 ммоль/л NaCl, 50 ммоль/л Tris-HCl (рН 8,0), ингибиторы протеазы с полным ингибированием (Roche Applied Science, Индианаполис, штат Индиана) и ингибиторы фосфатаз (50 ммоль/л фторида натрия, 1 ммоль/л ортованадата натрия и 10 ммоль/л пирофосфата натрия).
Спустя 30 мин инкубации на льду, лизаты центрифугировали при 13000 об/мин в течение 10 мин при 4°С и либо использовали для Вестерн-блоттинга, либо подвергали иммунопреципитации и измерениям активности фосфодиэстеразы.
Для анализов иммунопреципитации экстракты белков предварительно инкубировали с 30 мкл 1:1 смеси белка А или G и сефарозы (Amersham Biosciences, Бакингемшир, Великобритания) и затем инкубировали с 20 мкл смеси 1:1 белка А или G и сефарозы и 1 мг первичного антитела на каждый мг белка, в течение 2 часов при 4°С. Иммунные комплексы тщательно промывали лизирующим буфером и использовали для Вестерн-блоттинга или подвергали измерениям активности фосфодиэстеразы.
FRET визуализация и анализ
Измерения уровней цАМФ проводили на гладкомышечных клетках бронхов человека (hBSMC), которые экспрессируют зонд FRET ICUE3, как описано ранее (2). Кратко, клетки поддерживали в растворе K+-Рингера, содержащем (в ммоль/л) 121,6 NaCl, 5,4 KCl, 1,8 MgCl2, 1,8 CaCl2, 4 NaHCO3, 0,8 NaH2PO4, 5 D-глюкозу, 5 пируват натрия, 10 ГЭПЭС при рН 7,4. Снятия показаний FRET осуществляли перед и после добавления 100 нмоль/л изопротеренола (Iso) и 100 нмоль/л CGP-20712А (CGP) с использованием системы SP5 Leica TCS (Leica Microsystems Inc., Буффало Гров, Иллинойс, США) с аргоновым лазером и с иммерсионными объективами 63x. Для возбуждения CFP (голубой флуоресцентный белок) и YFP (желтый флуоресцентный белок) использовали длины волн 458 и 514 нм, соответственно. Изображения получали каждые 4 секунды, без какой - либо линии среды, при скорости сканирования 400 МГц и разрешении 512×512 пикселей. Эффективность FRET рассчитывали с использованием программного обеспечения «Метод 3», предоставленного приложением мастера Leica для FRET визуализации и сенсибилизированного излучения, согласно чему: EA(i)=В/А, где EA(i) представляет собой кажущуюся эффективность FRET; А и В представляют собой интенсивности канала CFP и FRET, соответственно. Для визуализации hBSNC обработанные пептидами клетки, экспрессирующие FRET индикатор ICUE3, предварительно инкубировали с 50 мкМ пептида Trojan, полученного из PI3Kγ (SEQ ID No.: 2 - RQIKIWFQNRRMKWKKGKATHRSPGQIHLVQRHPPSEESQAF), или контрольным пептидом Р1 (SEQ ID No.: 3 - RQIKIWFQNRRMKWKK) в течение 30 минут перед обработкой Iso и CGP.
Анализ активности фосфодиэстеразы
Активность фосфодиэстеразы в иммунопреципитатах измеряли согласно двухстадийному способу Thompson и Appleman, как описано ранее (2), с небольшими модификациями. Кратко, иммунопреципитаты анализировали в общем объеме 200 мкл реакционной смеси, содержащей 40 ммоль/л Tris-HCl (рН 8,0), 1 ммоль/л MgCl2, 1,4 ммоль/л 2-меркаптоэтанола и 0,1 пКи [3Н] цАМФ (Amersham Bioscience, Бакингемшир, Великобритания) в течение 40 мин при 33°С. Для остановки реакции образцы кипятили при 95°С в течение 3 мин. Продукт реакции 5'-АМФ затем гидролизовали посредством инкубации смеси с 50 мкг змеиного яда от Crotalus Atrox в течение 15 мин при 37°С (Sigma-Aldrich, Сент-Луис, штат Миссури). Полученный аденозин отделяли посредством анионообменной хроматографии 400 мкл суспензии смолы Dowex AG1-X8 (Bio-Rad, Сеграте, Милан, Италия), водой и 100% этанолом в равных долях. Количество аденозина с радиоактивной меткой в супернатанте количественно оценивали посредством сцинтилляционных измерений (Ultima Gold liquid scintillation from Perkin Elmer, Уолтем, штат Массачусетс).
Выделение гладкомышечных клеток трахей мыши
Гладкомышечные клетки трахеи мыши культивировали из эксплантов трахеи с использованием ранее описанных способов с модификациями. Целую трахею от гортани до бронхов удаляли и помещали в стерильную чашку Петри, содержащую сбалансированный солевой раствор Хенкса при комнатной температуре и 2х концентрацию антибиотика-противогрибкового средства (Gibco, Карлсбад, штат Калифорния, код продукта: 15240-062). С использованием препаровальной лупы удаляли дополнительную окружающую ткань, сегмент трахеи делили продольно и нарезали на квадраты, размером 2-3 мм. Все сегменты единственной трахеи затем помещали внутренней поверхностью ко дну 60 мм стерильного планшета для культуры клеток. После адгезии эксплантов к планшету для покрытия эксплантов добавляли 2,5 мл среды Игла в модификации Дульбекко (DMEM, Gibco, Карлсбад, штат Калифорния) с добавлением 20% фетальной телячьей сыворотки. Экспланты инкубировали при 37°С в увлажненной атмосфере с 95% воздухом и 5% СО2. Трое суток после выращивания концентрации FBS и антибиотика-противогрибкового средства уменьшались до 10% и 1х, соответственно. Сегменты трахеи удаляли, когда клетки становились локально сливающимися. Как только 60 мм планшет состоял из сливающихся клеток, клетки отделяли посредством трипсинизации и переносили в один 60 мм планшет. Гладкомышечные клетки трахеи далее подразделяли на несколько пассажей в соотношении 1:2. Более чем 90% данных клеток представляли собой гладкомышечные клетки, как определено иммунофлуоресценцией, проводимой с антителом, специфичным в отношении актина гладкой мускулатуры. Все эксперименты проводили на слившихся клетках при пассаже 3.
Измерения кальциевых коротких единичных импульсов
hBSMC загружали индикатором кальция Indo-1-AM (2 мкМ, Invitrogen, Карлсбад, штат Калифорния) при 37°С в течение 40 минут в присутствии контрольного пептида Р1, пептида Trojan, полученного из PI3Kγ, или носителя. Клетки промывали раствором Тироде, содержащим (в ммоль/л): 5 ГЭПЭС, 154 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5,5 D-глюкозу, при рН 7,35 и помещали на инвертированный микроскоп. Клетки держали в растворе Тироде и обрабатывали KCl деполяризующим раствором, содержащим (в ммоль/л): 5 ГЭПЭС, 118 NaCl, 40 KCl, 2 CaCl2, 1 MgCl2, 5,5 D-глюкозу, при рН 7,35. Кальциевые короткие единичные импульсы анализировали как соотношение флуоресцентных сигналов, измеренных при 400 нм и 490 нм после возбуждения клеток, загруженных Indo-1-АМ, при 350 нм. Эксперименты записывали и анализировали с помощью программного обеспечения Igor® с использованием функций, добавленных Jason Rothman (Neuromatic, www.thinkrandom.com).
Измерения токов ионов хлора в камере Уссинга
Для измерения токов ионов хлора в нормальных и ΔF508 первичных эпителиальных клетках бронхов человека клетки культивировали на 1,12 см2 вставках Snapwell. В камерах Уссинга устанавливали фильтры, и градиент ионов хлора создавали посредством инкубирования клеток в базолатеральном буфере с высоким содержанием ионов хлора, содержащем (в ммоль/л): 140 NaCl, 5 KCl, 0,36 K2HPO4, 0,44 КН2РО4, 1,3 CaCl2, 0,5 MgCl2, 4,2 NaHCO3, 10 ГЭПЭС, и 10 глюкозы при рН 7,4, и апикальном буфере с низким содержанием ионов хлора, содержащем (в ммоль/л): 133,3 Na-глюконат, 5 K-глюконат, 2,5 NaCl, 0,36 К2НРО4, 0,44 КН2РО4, 5,7 CaCl2, 0,5 MgCl2, 4,2 NaHCO3, 10 ГЭПЭС и 10 маннит при рН 7,4. Буферы аэрировали смесью 95% O2 и 5% CO2, и во время эксперимента температуру поддерживали при 37°С. Культуры поддерживали при напряжении 0 мВ с использованием зажима EVC4000 MultiChannel V/I (World Precision Instruments, Сарасота, штат Флорида, США). После периода стабилизации 30 минут в конкретные моменты времени добавляли лекарственные средства, в то время как ток непрерывно записывали.
Экстракция цАМФ и количественная оценка
Легкие и трахею эксплантировали из животных после умерщвления, измельчали в жидком азоте и использовали для холодной экстракции цАМФ 6% трихлоруксусной кислотой. На образцы воздействовали ультразвуком в течение 10 секунд и центрифугировали при 13000 об/мин при 4°С в течение 15 минут. Супернатанты промывали четыре раза пятью объемами диэтилового эфира, насыщенного водой, и лиофилизировали. Содержание цАМФ детектировали с помощью системы для иммуноферментного анализа Amersham cAMP BioTrak (GE Healthcare Life Sciences, Питтсбург, США, код продукта: RPN225) в соответствии с протоколом производителя.
Анализ эффективности трансдукции пептида Trojan, полученного из PI3Kγ in vivo
hBSMC инкубировали с пептидом Trojan, полученным из PI3Kγ (SEQ ID No.: 2), конъюгированным с флуоресцеином (50 мкМ), или носителем в течение 30 минут, фиксировали 4% параформальдегидом (PFA) в течение 10 минут и пермеабилизировали фосфатно-солевым буферным раствором (PBS) + 0,5% Тритон (Sigma-Aldrich, Сент-Луис, штат Миссури) в течение 5 минут при комнатной температуре. Затем клетки инкубировали с PBS, содержащим 3% бычий сывороточный альбумин (BSA, Sigma-Aldrich, Сент-Луис, штат Миссури) и фаллоидин-Alexa 488 (1:1000, Ttrermo Fisher Scientific, Уолтем, штат Массачусетс, США), в течение 30 минут и закрепляли на предметных стеклах реактивом ProLong® Antifade (Thermo Fisher Scientific, Уолтем, штат Массачусетс, США). Красные (актин) и FITC (пептид) флуоресцентные изображения получали с помощью Zeiss Observer-Z1, оборудованного модулем Apotome (Carl Zeiss, Оберкохен, Германия).
Мышам дикого типа линии BALB/C инъецировали внутритрахеальным путем 1,5 мкг пептида Trojan, полученного из PI3Kγ, конъюгированного с флуоресцеином или носителем, в конечном объеме 70 мкл PBS. Спустя 30 минут, животных анестезировали, трахею и легкие продували PBS, экстрагировали и замораживали в ОСТ. Криосрезы 10 мкм получали посредством криостата Leica СМ1850 (Leica Microsystems GmbH, Вецлар, Германия), и флуоресценцию получали с помощью Zeiss Observer-Z1, оснащенного модулем Apotome (Carl Zeiss, Оберкохен, Германия).
Иммунизация овальбумином
Овальбумин (100 мкг) (OVA, Sigma-Aldrich, Сент-Луис, штат Миссури), в комплексе с сульфатом алюминия-калия (1 мг, квасцы) вводили интраперитонеально (и/п) в сутки 1 и 14 и интраназально (и/н) (50 мкг OVA в 50 мкл PBS) в сутки 14, 25, 26 и 27. Контрольные мыши получали только и/п инъекции квасцов и и/н инъекции PBS. Гиперчувствительность дыхательных путей, вызванную вдыхаемым метахолином, измеряли через 24 часа после конечной дозы OVA (Сутки 28).
Измерение реактивности дыхательных путей
Способ 1. Мышей, сенсибилизированных к овальбумину, анестезировали (пентобарбитал натрия, 70-90 мг/кг, и/п), проводили трахеотомию и подключали к вентилятору FlexiVent для небольших животных (SCIREQ, Монреаль, Квебек, Канада). Для индукции сокращения дыхательных путей мышей подвергали действию возрастающих концентраций метахолина в виде аэрозоля (10-9 - 10-4 М, конечная концентрация).
Способ 2: Мышей, сенсибилизированных к овальбумину, обрабатывали посредством внутритрахеального введения по капле 70 мкл фосфатно-солевого буферного раствора (PBS), содержащего носитель или 150 мкг пептида Trojan, полученного из PI3Kγ, или эквимолярных количеств контрольного пептида Р1. Гиперчувствительность дыхательных путей оценивали через 30 минут после обработки в соответствии с ранее опубликованным протоколом (6). Кратко, мышей анестезировали (пентобарбитал натрия, 70-90 мг/кг, и/п), проводили трахеотомию и осуществляли вентиляцию при положительном давлении в конце выдоха 10 см H2O, положительном давлении в конце выдоха (PEEP) 3 см H2O, скорости дыхания 90 вдохов/мин в окружающем воздухе. Давление открытия дыхательных путей (Рао) со стороны эндотрахеальной трубки и давление внутри камеры измеряли с помощью датчиков давления (Special Instruments, Digima Clic; Нердлинген, Германия). Газовый поток измеряли посредством пневмотахографа (Special Instruments, Digima Clic; Нердлинген, Германия). Дыхательный объем рассчитывали как интеграл сигнала потока. Переменные механической вентиляции записывали с использованием программного обеспечения ICU-Lab (KleisTEK Advanced Electronic Systems, Бари, Италия). Гиперчувствительность дыхательных путей оценивали как изменение в дыхательном объеме после обработки 500 мг/кг метахолина, введенного внутривенно.
Анализ воспаления дыхательных путей у мышей с астмой
Мышей BALB/C дикого типа обрабатывали, посредством внутритрахеального введения по капле, 25 мкг пептида Trojan, полученного из PI3Kγ, или эквимолярными количествами контрольного пептида Р1 в конечном объеме 70 мкл фосфатно-солевого буферного раствора (PBS) перед каждым интраназальным введением овальбумина (сутки 14, 25, 26 и 27 протокола иммунизации овальбумином). Через 24 часа после конечной инъекции (сутки 28) мышей анестезировали (пентобарбитал натрия, 70-90 мг/кг, и/п), и трахеи надрезали и канюлировали. Дыхательные пути промывали 2,5 мл фосфатно-солевого буферного раствора (PBS). Общее число клеток в бронхоальвеолярном лаваже (BAL) определяли посредством гемоцитометра Нейбауэра. Объем 50 мкл BAL центрифугировали на цитоспиновых покровных стеклах при 400 об/мин при комнатной температуре в течение 5 минут и окрашивали системой Diff-Quick (LabAids, Ронконкома, США). Всего 100 клеток на покровное стекло отсчитывали и классифицировали как нейтрофилы, макрофаги, лимфоциты и эозинофилы на основе морфологических критериев. Эритроциты и эпителиальные клетки не учитывали, и результаты выражали в количестве клеток/мл.
Для оценки перибронхиального воспаления легкие группы животных, которые не подвергались эксплантированию бронхоальвеолярного лаважа, фиксировали в растворе 4% параформальдегида (PFA) в течение 24 часов при 4°С и заключали в парафин. Срезы, которые были толщиной 5 мкм, депарафинировали, окрашивали раствором гематоксилин-эозина (Bio-Optica, Милан, Италия), дегидрировали и закрепляли стеклянными покровными стеклами. Степень перибронхиального воспаления классифицировали следующим образом: 0 - нормально; 1 - мало воспалительных клеток; 3 - толстое кольцо воспалительных клеток.
Для оценки наличия бокаловидных клеток срезы легкого окрашивали йодной кислотой и реактивом Шиффа (PAS) (Bio-Optica, Милан, Италия), и процентное содержание PAS-позитивных клеток рассчитывали посредством подсчета числа PAS-позитивных эпителиальных клеток и общего числа эпителиальных клеток.
Антитела, реактивы и плазмиды
PDE4B и PDE4D иммунопреципитировали, как описано ранее (2), используя поликлональные кроличьи антитела, находящиеся в свободном доступе у Dr. Marco Conti в калифорнийском университете Сан-Франциско (Сан-Франциско, Калифорния, США). Имеющиеся в продаже антитела, специфичные в отношении PDE4B и PDE4D, можно приобрести у Abeam (Abeam, Cambridge, штат
Массачусетс, США): антитело против PDE4B, код продукта: ab14611; антитело против PDE4D, код продукта: ab14614. Кроличьи поликлональные антитела против Cav1.2 и фосфо-Cav1.2 находятся в доступном доступе у Dr. William A. Catterall в Вашингтонском университете, Сиэтл, штат Вашингтон, США.
Антитело, которое распознает субстраты, фосфорилированные РКА (субстрат Р-РКА), приобретали в Cell Signaling Technology (Данверс, штат Массачусетс, США; код продукта: # 9621), и антитело против CFTR клон М3А7 от Millipore (Биллерика, штат Массачусетс, США; код продукта: 05-583).
ICUE3-pcDNA3 была описана ранее (2).
Все из изопротеренола, CGP-20712A, карбахола, ролипрама, амилорида и форсколина приобретали в Sigma (Sigma-Aldrich, Сент-Луис, штат Миссури). Корректор CFTR VX-809, потенциатор CFTR VX-770 и ингибитор CFTR 172 приобретали в Selleckchem (Хьюстон, штат Техас, США).
Контрольный пептид Р1 (SEQ ID No.: 3 - RQIKIWFQNRRMKWKK) и пептид Trojan, полученный из PI3Kγ (SEQ ID No.: 2 RQIKIWFQNRRMKWKKGKATHRSPGQIHLVQRHPPSEESQAF), синтезировали посредством GenScript (GenScript, Пискатауэй, штат Нью-Джерси, США) и Chinapeptides (Chinapeptides Co. Ltd., Шанхай, Китай).
Статистический анализ
Для статистического анализа использовали программное обеспечение Prism (GraphPad Software Inc., Ла-Холья, штат Калифорния, США). Значения р рассчитывали с использованием критерия Стьюдента, однофакторного и двухфакторного дисперсионного анализа с последующим тестом Бонферрони, в соответствующих случаях. На всех фигурах, на графиках представлено среднее значение плюс - минус стандартная ошибка по меньшей мере 3 независимых экспериментов.
Результаты
Пептид Trojan, полученный из PI3Kγ, усиливает β2-AR/цАМФ сигнализацию в гладкомышечных клетках бронхов
Стратегии увеличения уровней цАМФ после β2-AR/цАМФ в гладкомышечной мускулатуре бронхов представляют большой интерес для лечения респираторных заболеваний. Для исследования терапевтического потенциала ингибирования PI3Kγ-зависимой β2-AR-цAMФ сигнализации конструировали пептидный ингибитор киназонезависимой активности PI3Kγ, последовательность которого показана в SEQ ID No.: 2. Предыдущее исследование показывает, что пептид, содержащий 126-150 остатки PI3Kγ человека (SEQ ID No.: 1), ингибирует РКА заякоривание и снижает активность PI3Kγ-связанной фосфодиэстеразы 3В in vitro (1). Для ингибирования РКА заякоривающей активности PI3Kγ in vivo ингибиторный слитый пептид, проницаемый в клеточные мембраны, получали посредством связывания 126-150 домена PI3Kγ человека (SEQ ID No.: 1) с пептидом Trojan Пенетратина 1 Antennapedia (SEQ ID No.: 3 - Фиг. 1A). Вариант PI3Kγ ингибиторного пептида Trojan, меченного флуоресцеином (FITC), накапливался в цитоплазме гладкомышечных клеток бронхов человека (hBSMC) через 30 минут после инкубации, демонстрируя, что ингибитор эффективно трансдуцируется in vivo в выделенных клетках (Фиг. 1Б). Кроме того, ингибиторный пептид PI3Kγ значительно снижал каталитическую активность PDE4B и PDE4D в гладкомышечных клетках трахеи мышей дикого типа (PI3Kγ+/+), но не животных, лишенных фермента (PI3Ky-/-) (Фиг. 1В), демонстрируя способность пептида селективно мешать PI3Kγ-зависимому заякориванию РКА. В соответствии с этими данными, предварительная обработка hBSMC PI3Kγ ингибиторным пептидом Trojan повышала аккумуляцию цАМФ на 35% после стимуляции β2-AR, в то время как уровни цАМФ оставались неизменными в клетках, обработанных контрольным пептидом Р1 (Фиг. 1Г и Д).
Так как цАМФ, как известно, индуцирует снижение внутриклеточных уровней Са2+ и, как следствие, стимулирует расслабление гладкой мускулатуры, анализировали способность PI3Kγ ингибиторного пептида Trojan модифицировать концентрации Са2+ в hBSMC. Максимальный пик коротких одиночных импульсов Са2+, вызванных мускариновым агонистом карбахолом, был значительно меньше в hBSMC, предварительно обработанных PI3Kγ ингибитором, по сравнению с клетками, которые подвергались действию носителя или контрольного пептида Р1 (Фиг. 2А-Б). Первичный механизм, посредством которого циклические нуклеотиды ограничивают внутриклеточный Са2+ в гладкомышечных клетках, заключается в ингибировании потенциал-зависимых Са2+ каналов (VOCC). Кроме того, ранее было продемонстрировано, что PI3Kγ представляет собой ключевой регулятор цАМФ вблизи данных Са2+ каналов (2). Для проверки того, уменьшает ли ингибирование заякоривающей активности PI3Kγ поступление входящего потока Са2+ через VOCC, hBSMC подвергали действию раствора, содержащего KCI, способного деполяризовать мембрану и, вследствие этого, активировать VOCC. Входящий поток Са2+, индуцированный KCI, полностью прекращался под действием PI3Kγ ингибиторного пептида Trojan, в то время как он оставался неизменным в hBSMC, обработанных контрольным пептидом Р1 или носителем (Фиг. 2А-Б). Для дальнейшей поддержки данных результатов цАМФ-опосредованное фосфорилирование порообразующей субъединицы Са2+ канала Cav1.2, как было обнаружено, является значимо сильнее в hBSMC, обработанных ингибитором PI3Kγ, по сравнению с контрольными клетками, обработанными носителем или пептидом Р1 (Фиг. 2В).
В общем, эти данные указывают на то, что пептид Trojan, который ингибирует РКА заякоривающую функцию PI3Kγ, представляет собой новый способ ингибирования выбранных PDE и усиления цАМФ сингализации после β2-AR в гладкомышечных клетках.
Пептид Trojan, полученный из PI3Kγ, ограничивает гиперчувствительность дыхательных путей у здоровых мышей и мышей с астмой
Для определения эффективности трансдукции PI3Kγ ингибиторного пептида in vivo в дыхательных путях меченную флуоресцеином (FITC) форму пептида вводили по капле внутритрахеальным путем мышам BALB/C дикого типа. Флуоресценцию FITC выявляли в трахее и легких, но не в мозге или в миокарде, через 30 минут после введения (Фиг. 3А); это демонстрирует, что пептид эффективно распространяется в дыхательном пути животных. Кроме того, уровни цАМФ были на 30% выше в трахее и в легких мышей, которым был инъецирован PI3Kγ ингибиторный пептид Trojan, по сравнению с контрольными животными, обработанными фосфатно-солевым буферным раствором (PBS) или контрольным пептидом Р1 (Фиг. 3Б). Согласно усиленной аккумуляции цАМФ в дыхательном пути гиперчувствительность дыхательных путей, индуцированная мускариновым агонистом метахолином, была значительно ослаблена PI3Kγ ингибиторным пептидом, но не контрольным пептидом Р1, у здоровых мышей (Фиг. 5В). Для проверки того, подавляет ли внутритрахеальное введение пептида Trojan, полученного из PI3Kγ, гиперчувствительность дыхательных путей, ассоциированную с аллергической астмой, получали доклиническую модель овальбумин (OVA)-индуцированной астмы. Уменьшение дыхательного объема, вызванное метахолином, было значимо ослаблено у животных, обработанных PI3Kγ ингибиторным пептидом, по сравнению с контрольными мышами, которые получали PBS или пептид Р1 (Фиг. 3Г).
В своей совокупности, эти данные демонстрируют способность пептида Trojan, полученного из PI3Kγ, повышать уровни цАМФ in vivo в дыхательных путях и функционировать в качестве бронходилятора.
Пептид Trojan, полученный из PI3Kγ, повышает уровни цАМФ и усиливает проводимость CFTR в эпителиальных клетках бронхов, экспрессирующих CFTR дикого типа или мутантный ΔF508 CFTR
Впоследствии, исследовали способность PI3Kγ ингибиторного пептида Trojan повышать уровни цАМФ не только в гладкомышечных клетках, но также в эпителиальных клетках дыхательных путей. С данной целью авторы изобретения анализировали цАМФ-опосредованное фосфорилирование регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), основного хлоридного канала (Cl-) в эпителии дыхательного пути, активация которого является цАМФ-зависимой. Уровень фосфорилирования CFTR был значимо выше в нормальных эпителиальных клетках бронхов человека (Nuli-1), обработанных пептидом Trojan, полученным из PI3Kγ, по сравнению с контрольными клетками, подверженными действию носителя или контрольного пептида Р1 (Фиг. 4А). В частности, ингибирование заякоривающей функции PI3Kγ дополнительно усиливало цАМФ-зависимое фосфорилирование CFTR, вызванное известным ингибитором PDE4 (Фиг. 4Б), что указывает на то, что PI3Kγ ингибиторный пептид не только ингибирует PDE4, но также другие изоформы PDE, как известно, ассоциированные с PI3Kγ, такие как PDE3 (2).
Для проверки того, коррелирует ли повышенный уровень фосфорилирования CFTR с более высокой проводимостью в отношении Cl-, проводили измерения токов Cl- в камерах Уссинга в клетках NuLi-1, экспрессирующих CFTR дикого типа. CFTR-зависимые токи значимо увеличивались после применения пептида Trojan, полученного из PI3Kγ, в то время как контрольный пептид Р1 не изменял проводимость (Фиг. 4В). Эти данные, таким образом, обнаруживают новую роль PI3Kγ ингибиторного пептида в качестве потенциатора CFTR.
Молекулы с активностью потенциатора CFTR требуются для стимуляции открытия дефектного CFTR при лечении кистозного фиброза (CF). Для определения того, способен ли пептид Trojan, полученный из PI3Kγ, корректировать дефектную Cl- проводимость мутантного CFTR, CFTR-зависимые токи измеряли в эпителиальных клетках бронхов, экспрессирующих мутантный CFTR ΔF508-CFTR (CuFi-1). Ингибирование PI3Kγ синергетически повышало активность известного потенциатора CFTR, VX-770, приводя к увеличению CFTR токов примерно в 5 раз, по сравнению с основной активностью (Фиг. 4А и Б). Наоборот, контрольный пептид Р1 не изменял активность CFTR в присутствии VX-770 (Фиг. 4А и Б).
В общем, эти данные обнаруживают новую функцию PI3Kγ ингибиторного пептида в качестве потенциатора CFTR, который стимулирует цАМФ-зависимое открытие канала дикого типа и мутантного CFTR (ΔF508-CFTR).
Пептид Trojan, полученный из PI3Kγ, ограничивает воспаление легких у мышей с астмой
Хорошо известно, что увеличение цАМФ в лейкоцитах стимулирует противовоспалительный ответ. Таким образом, оценивали способность пептида Trojan, полученного из PI3Kγ, повышать концентрации цАМФ в данном типе клеток и, таким образом, ограничивать воспаление, ассоциированное с хроническими респираторными заболеваниями. Мыши, предварительно обработанные контрольным пептидом Р1, перед каждой интраназальной инъекцией овальбумина демонстрировали увеличение перибронхиального воспаления (Фиг. 7А и Б) и числа бокаловидных клеток, содержащих слизь и позитивных в отношении окрашивания йодной кислотой и реактивом Шиффа (PAS) (Фиг. 7А и В), по сравнению с контрольными животными, не сенсибилизированными к овальбумину (ранее не получавшими обработку). Пептид Trojan, полученный из PI3Kγ, значимо уменьшал как перибронхиальный воспалительный инфильтрат (Фиг. 7А и Б), так и число бокаловидных клеток (Фиг. 7А и В). Согласно данным результатам, число нейтрофилов, находящихся в бронхоальвеолярном лаваже, было существенно меньше у животных, обработанных PI3Kγ ингибиторным пептидом Trojan, по сравнению с контролями, которые получали пептид Р1 (Фиг. 7Г). Напротив, другие популяции лейкоцитов, например, макрофаги, лимфоциты и эозинофилы оставались неизменными (Фиг. 7Д-Е-Ж).
В общем, данное экспериментальное подтверждение демонстрирует способность пептида Trojan, полученного из PI3Kγ, селективно ингибировать нейтрофилию, ассоциированную с хроническими респираторными заболеваниями, и, вследствие этого, функционировать в качестве противовоспалительного лекарственного средства.
Ссылки
1. A. Perino et al., Mol Cell 42, 84 (Apr 8, 2011).
2. A. Ghigo et al., Circulation 126, 2073 (Oct 23, 2012).
3. M. Delia Peruta, C. Giagulli, C. Laudanna, A. Scarpa, C. Sorio, Mol Cancer 9, 61 (2010).
4. E. Hirsch et al., Science 287, 1049 (Feb 11, 2000).
5. E. Patrucco et al., Cell 118, 375 (Aug 6, 2004).
6. V. Fanelli et al., Intensive Care Med 36, 1935 (Nov, 2010).