Результат интеллектуальной деятельности: КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ БЕССОННИЦЫ
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на композиции и способы для лечения бессонницы. Настоящая заявка испрашивает приоритет на основании заявки на патент США № 62/067443, поданной 23 октября 2014 года в Соединенных Штатах Америки, содержание которой включено в данный документ посредством ссылки.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Рецепторы орексина представляют собой сопряженные с G-белком рецепторы, выявленные главным образом в головном мозге. Их эндогенные лиганды, орексин-A и орексин-В, экспрессируются в нейронах, локализованных в гипоталамусе. Орексин-A представляет собой пептид из 33 аминокислот; орексин-В состоит из 28 аминокислот (Sakurai T. et al., Cell, 1998, 92 573-585). Существуют два подтипа рецепторов орексина, рецептор орексина 1 (далее по тексту называемый как OX1) и рецептор орексина 2 (далее по тексту называемый как OX2); OX1 преимущественно связывается с орексином-A, в то время как OX2 связывается как с орексином-A, так и с -B. Орексины стимулируют потребление пищи у крыс, и было сделано предположение, что передача сигнала орексином может играть роль в центральном механизме обратной связи для регуляции пищевого поведения (Sakurai et al., supra). Также было отмечено, что орексины контролируют состояния сна-бодрствования (Chemelli R.M. et al., Cell, 1999, 98, 437-451). Орексины также могут играть роли в изменениях в головном мозге, связанными с опиоидной и никотиновой зависимостью (S.L. Borgland et al., Neuron, 2006, 49, 598-601; C.J. Winrow et al., Neuropharmacology, 2010, 58, 185-194) и алкогольной зависимостью (J.R. Shoblock et al., Psychopharmacology, 2011, 215, 191-203). Дополнительно было сделано предположение, что орексины играют роль в некоторых стрессовых реакциях (T. Ida et al., Biochem. Biophys. Res. Commun., 2000, 270, 318-323). Было обнаружено, что соединение, такое как
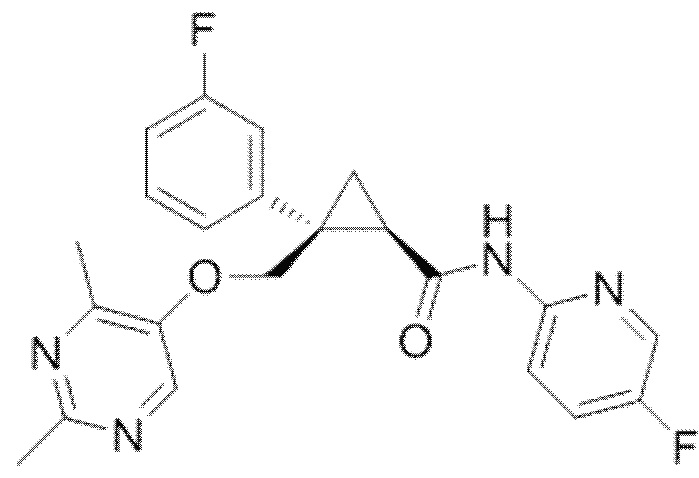
(1R,2S)-2-(((2,4-диметилпиримидин-5-ил)окси)метил)-2-(3-фторфенил)-N-(5-фторпиридин-2-ил)
циклопропанкарбоксамид (далее по тексту называемое соединением A) является сильным антагонистом рецепторов орексина, и его можно применять в лечении нарушений сна, таких как бессонница, а также для других применений с целью терапии.
Формула соединения A
С точки зрения снотворного средства, если дозировка активного фармацевтического ингредиента (далее по тексту называемого как API) в фармацевтическом составе, подлежащем однократному приему на ночь, представляет собой слишком высокую дозу, она потенциально может вызывать остаточную сонливость на следующий день, в то время как однократная доза, являющаяся недостаточной, может вызвать у пациента пробуждение в течение периода нормального сна, даже если пациенты способны засыпать со снотворным. Следовательно, сложно установить правильную дозу с учетом неустойчивого равновесия между легким наступлением сна и предупреждением остаточной сонливости по сравнению с учетом только равновесия между побочными эффектами и эффективностью. Кроме того, даже если были известны доза определенного лекарственного средства от бессонницы, физико-химические свойства API и фармакокинетический (далее по тексту называемый как PK) профиль после введения лекарственного средства, такая информация не будет применима по отношению к другим API от бессонницы, поскольку на нее будет оказывать воздействие ряд факторов, включая механизм действия, путь введения, скорость абсорбции, физико-химическое свойство, такое как растворимость и стабильность в плазме крови, или другие факторы для каждого API. В действительности, зависимость между остаточной сонливостью и характеристиками снотворных средств является не всегда устойчивой (CNS Drugs 2004; 18 (5): 297-328). Взаимосвязь между PK профилем и эффектом сонливости, таким как наступление сна или остаточная сонливость, до сих пор была неизвестна для соединения A.
В уровне техники существует потребность в более эффективных способах лечения бессонницы для достижения быстрого наступления сна, а также поддержания сна на протяжении всего периода сна, с предупреждением при этом остаточной сонливости и/или ухудшения состояния на следующий день, включающих пероральное введение твердой лекарственной формы снотворного средства. Кроме того, в уровне техники существует потребность в фармацевтической композиции, содержащей снотворное средство и по меньшей мере один фармацевтически приемлемый наполнитель для лечения бессонницы с достижением быстрого наступления сна, а также поддержания сна на протяжении всего периода сна, с предупреждением при этом остаточной сонливости и/или ухудшения состояния на следующий день.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является обеспечение способов лечения бессонницы, включающих пероральное введение твердой лекарственной формы лекарственного средства, представляющего собой соединение A.
Дополнительной целью настоящего изобретения является обеспечение фармацевтической композиции, содержащей терапевтически эффективное количество соединения A.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 1 мг до приблизительно 15 мг.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 2 мг до приблизительно 15 мг.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 2 мг до приблизительно 10 мг.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, выбранную из приблизительно 2, 2,5, 4, 5, 8, 10 или 15 мг.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение максимальной концентрации (Cmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 7,2 нг/мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 1 мг до приблизительно 15 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 7,2 нг/мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 5,3 нг/мл после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 16 нг/мл после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 23 нг/мл после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 36 нг/мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение AUC(0-24) от приблизительно 15,9 нг*ч./мл до приблизительно 23,8 нг*ч./мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 17 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 57 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 95 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 159 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 51,1 нг*ч./мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 19 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 128 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 284 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую достижение среднего значения AUC(0-inf) от приблизительно 19,8 нг*ч./мл до приблизительно 53,1 нг*ч./мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в 20 нг*ч./мл в диапазоне от приблизительно 80% до приблизительно 125% после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 149 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 311 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 1 мг до приблизительно 15 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения концентрации в плазме крови соединения А, составляющее приблизительно 20 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной суточной дозы.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 1 мг до приблизительно 15 мг.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую достижение среднего значения максимальной концентрации (Cmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 7,2 нг/мл на каждый 1 мг лекарственного средства после введения субъектам-людям однократной дозы.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 5,3 нг/мл после введения субъектам-людям однократной дозы.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения (Cmax) максимальной концентрации в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 16 нг/мл после введения субъектам-людям однократной дозы.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне приблизительно 80% до приблизительно 125% от 23 нг/мл после введения субъектам-людям однократной дозы.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 36 нг/мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение AUC(0-24) от приблизительно 15,9 нг*ч./мл до приблизительно 23,8 нг*ч./мл на каждый 1 мг лекарственного средства после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне приблизительно 80% до приблизительно 125% от 17 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 57 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную 5 мг суточную дозу, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 95 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 159 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 51,1 нг*ч./мл на каждый 1 мг лекарственного средства после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 19 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 128 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 284 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение AUC(0-inf) от приблизительно 19,8 нг*ч./мл до приблизительно 53,1 нг*ч./мл на каждый 1 мг лекарственного средства после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 20 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 149 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 311 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 1 мг до приблизительно 15 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения концентрации соединения A в плазме крови, составляющее приблизительно 20 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую форму для перорального введения, содержащую фармацевтически приемлемый наполнитель и эффективное количество соединения A, для лечения бессонницы, лекарственную форму, обеспечивающую показатель растворения, составляющий 85% или более, в среде растворения (хлористоводородная кислота, 0,1 моль/л, содержащая 0,5% полисорбата 80, 900 мл, при 37 ± 0,5°C) в течение 30 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой, при скорости вращения лопастей 75 об./мин.) в соответствии с методикой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую форму для перорального введения, содержащую фармацевтически приемлемый наполнитель и эффективное количество соединения A, для лечения бессонницы, лекарственную форму, обеспечивающую показатель растворения, составляющий 85% или более, в среде растворения (хлористоводородная кислота, 0,1 моль/л, 900 мл, при 37 ± 0,5°C) в течение 15 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой, при скорости вращения лопастей 50 об./мин.) в соответствии с методикой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую лактозу в качестве фармацевтически приемлемого наполнителя.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую гидроксипропилцеллюлозу с низкой степенью замещения в качестве фармацевтически приемлемого наполнителя.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую лактозу и гидроксипропилцеллюлозу с низкой степенью замещения в качестве фармацевтически приемлемого наполнителя.
Способ по настоящему изобретению обладает потенциалом в применении для лечения бессонницы с легким наступлением сонливости, но с предупреждением остаточной сонливости и/или ухудшения состояния на следующий день.
Фармацевтическая композиция по настоящему изобретению обладает потенциалом в применения в качестве твердой лекарственной формы для перорального введения для лечения бессонницы.
ПОДРОБНОЕ ОПИСАНИЕ
I. Определения
Для того, чтобы описанное в данном документе настоящее изобретение стало более понятным, следующие значения представлены для целей данного раскрытия.
Выражение "эффективное количество" означает количество лекарственного средства, представляющего собой соединение A, которое способно обеспечить терапевтический эффект у нуждающегося в этом субъекта-человека.
Под термином "лекарственное средство, представляющее собой соединение A" необходимо понимать (1R, 2S)-2-(((2,4-диметилпиримидин-5-ил)окси)
метил)-2-(3-фторфенил)-N-(5-фторпиридин-2-ил)циклопропанкарбоксамид или его фармацевтически приемлемую соль, гидрат, сольват, полиморф, свободное основание или любую их комбинацию.
Под термином "субъект-человек" понимают нормальных здоровых мужчин- или женщин-добровольцев и/или любого индивидуума, у которого обнаружены клинические признаки и симптомы бессонницы, или любые заболевание или расстройство, которое вызывают бессонницу.
Под термином "бессонница", который используют в данном документе, понимают все формулировки, определенные в Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (2013) (здесь и далее именуемое как DSM-V), опубликованное Американской ассоциацией психиатров. В DSM-V перечислены диагностические критерии бессонницы, представляющие собой следующие:
A. Основной жалобой является неудовлетворенность количеством или качеством сна, связанных с одним (или несколькими) из следующих симптомов:
1. Затруднение в наступлении сна. (У детей, это может проявляться в виде затруднения с наступлением сна без вмешательства осуществляющего уход лица.)
2. Затруднение в поддержании сна, характеризующееся частыми пробуждениями или проблемами с возвращением ко сну после пробуждений. (У детей, это может проявляться в виде затруднения с возвращением ко сну без вмешательства осуществляющего уход лица.)
3. Раннее утреннее пробуждение с неспособностью возвращения ко сну.
B. Нарушение сна вызывает клинически значимое расстройство или ухудшение в социальной, профессиональной, образовательной, научной, поведенческой или других важных областях деятельности.
C. Затруднение со сном возникает по меньшей мере в течение 3 ночей в неделю.
D. Затруднение со сном присутствует по меньшей мере в течение 3 месяцев.
E. Затруднение со сном возникает несмотря на достаточную возможность для сна.
F. Бессонница преимущественно не обусловлена другим нарушением сна-бодрствования (например, нарколепсией, нарушением сна, связанным с расстройствами дыхания, нарушением циркадного ритма сна-бодрствования, парасомнией) и она не возникает исключительно при таком нарушении.
G. Бессонница, которая не обусловлена физиологическими эффектами вещества (например, при злоупотреблении лекарственным средством,
терапевтическим препаратом).
H. Сопутствующие психические нарушения и заболевания, не позволяющие в достаточной степени объяснить основную жалобу на бессонницу.
Под бессонницей необходимо понимать нарушение сна, которое характеризуется симптомами, включающими без ограничения затруднение с засыпанием, затруднение с пребыванием во сне, прерывистое бодрствование, и/или слишком раннее пробуждение. Термин также охватывает дневные симптомы, такие как сонливость, беспокойство, ухудшенную концентрацию, ослабленную память и раздражительность. Типы бессонницы, подходящие для лечения с помощью композиций по настоящему изобретению, включают без ограничения кратковременную и хроническую бессонницу. Термин "кратковременна бессонница" относится к бессоннице с продолжительностью от приблизительно двух до приблизительно четырех недель. Термин "хроническая бессонница" относится к бессоннице с продолжительностью по меньшей мере от одного месяца или дольше.
Выражение "биоэквивалент" или "биоэквивалентность" является специальным термином и определяется в соответствии с Approved Drug Products with Therapeutic Equivalence Evaluations, 34th Edition, которое опубликовано Министерством здравоохранения и социальных служб США и известно под названием "Orange Book". Биоэквивалентность разных составов с одним и тем же лекарственным веществом включает эквивалентность по отношению к скорости и степени абсорбции лекарственного средства. Степень и скорость абсорбции тестируемого состава сравнивали с эталонным составом чтобы определить, являются ли эти два состава биоэквивалентными. Стандартное исследование биоэквивалентности проводят перекрестным способом путем всестороннего тестирования, которое включает введение однократных доз исследуемого и эталонного лекарственных средств определенному количеству волонтеров, обычно 12-24 нормальным здоровым взрослым, а затем измеряют уровни лекарственного средства в крови и плазме крови в течение длительного времени. Подробное руководство для установления биоэквивалентности состава с эталонным составом было опубликовано Департаментом по генерическим препаратам FDA, сектором биоэквивалентности.
Два состава, PK параметры которых, такие как Cmax, AUC или Tmax, отличаются на -20%/+25% или менее, как правило, считают "биоэквивалентными". Другой подход для вычисления средней биоэквивалентности включает расчет 90% доверительного интервала для соотношения средних значений (средних геометрических значений популяции) для показателей исследуемых и эталонных продуктов. Для установления BE, рассчитанный доверительный интервал должен находиться в пределах обычно 80-125% для соотношения средних значений показателей продукта. В дополнение к данному общему подходу, другие подходы, включая (1) логарифмическое преобразование фармакокинетических данных, (2) способы оценки последовательных эффектов и (3) способы оценки данных с исключением выбросов, можно использовать для установления биоэквивалентности. Например, в вышеупомянутом пункте (1), доверительный интервал должен находиться в пределах обычно 80-125% для различия в среднем значении логарифмически обращенного PK параметра.
Термин "время сна" относится ко времени, которое субъект проводит спящим. Время сна может быть непрерывным или прерывистым.
"Эффективность сна" относится к общему времени сна, которое субъект получает на протяжении проведенного в постели времени. Эффективность сна измеряют с помощью следующего уравнения: 100*(общее время сна (TST)/общее время, проведенное в постели).
Словосочетание "остаточная сонливость" относится к субъективным ощущениям пациентом сонливости или заторможенности при пробуждении, как правило, на следующее утро после введения снотворного накануне вечером. "Ухудшение состояния на следующий день" относится к ухудшению активности в поведении пациента, которая требует концентрации внимания, включая вождение автомобиля, которое возникает при пробуждении пациента на следующее утро, но при этом уровни лекарственных препаратов от бессонницы в их крови остаются достаточно высокими. Каролинская шкала сонливости (KSS) является одним из нескольких средств, используемых для оценки субъективной сонливости. KSS была исходно разработана как шкала, представляющая собой одномерную шкалу сонливости, и ее валидировали по электроэнцефалографической активности альфа- и тета-ритмов (EEG), а также замедленному движению глаза по электроокулографической (EOG) активности (Åkerstedt and Gillberg, 1990). Другие субъективные тесты для оценки остаточной сонливости или эффекта ухудшения состояния на следующий день включают шкалу сонливости Эпворта (ESS), и шкалу сонливости Стэнфорда (SSS), и оценку активности сна-пробуждения (SWAI). Данные эффекты также могут быть оценены специалистами в данной области с использованием одного или нескольких тестов субъектов-людей для измерения их памяти, их внимательности, обработки информации и психомоторной работоспособности, включая, например, тест замены цифровых символов (DSST), тест психомоторной бдительности (PVT), тест на время принятия решения (CRT), тест латентности ко сну (SLT), визуально-аналоговый тест (VAT), тест на копирование символов (SCT), тест на критическую частоту слияния мельканий (CFF), тест на время простой реакции (визуальный или слуховой; SRT), тест на запоминание слов (WLT), тест на критическое слежение (CTT), тест на распределение внимания (DAT), тест зачеркивания цифр или букв, разделение сна на несколько стадий с помощью полисомнографических (PSG) измерений, тест на непрерывную деятельность (CPT), множественный тест латентности ко сну (MSLT), тест быстрой обработки зрительной информации (RVIP) и другие.
Под термином "лекарственная(лекарственные) форма(формы)" или "фармацевтическая(фармацевтические) лекарственная(лекарственные) форма(формы)" необходимо понимать средства для введения лекарственного вещества (активного фармацевтического ингредиента (API)), или для облегчения дозирования, введения и доставки лекарственного препарата пациенту и другим млекопитающим. Лекарственные формы классифицируют с точки зрения путей введения и мест применения, включая, например, пероральное введение, местное нанесение, ректальное, вагинальное, внутривенное, подкожное, внутримышечное, офтальмическое, назальное, ушное и ингаляционное введение. В качестве альтернативы, лекарственные формы классифицируют с точки зрения физической формы, такой как твердая, полутвердая или жидкая. Кроме того, лекарственные формы подразделяют на основе их формы, функций и характеристик, включая без ограничений таблетку, капсулу или инъекционную форму, как описано в монографии Японская фармакопея 16 издание (JP16) или в Общей главе <1151> Фармацевтические лекарственные формы Фармакопеи США-NF (37)(USP37).
Под термином "наполнитель", как правило, понимают неактивный ингредиент, который используют в качестве среды-носители (например, воду, оболочку капсулы и т.д.), разбавителя или компонента для составления лекарственной формы или фармацевтической композиции, содержащей лекарственное средство, такое как терапевтическое средство. Как правило, данный термин также охватывает неактивный ингредиент, который придает композиции свойство связывания (т.е. связывающее средство), свойство разрыхления (т.е. разрыхлитель), свойство смазывания (смазывающее средство) и/или другое свойство (т.е. растворитель, поверхностно-активное вещество и т.д.).
Термин "среднее значение" относится к среднему геометрическому значению. Фармакокинетические параметры, такие как "среднее значение Cmax" или "среднее значение AUC" относятся к среднему геометрическому значению Cmax или AUC.
Список сокращений и определений терминов, используемых в данной заявке, представлен далее.
AUC: площадь под кривой концентрация в плазме крови-время.
AUC(0-x): площадь под кривой концентрация в плазме крови-время от нуля до x часов после введения дозы.
AUC(0-t): площадь под кривой концентрация в плазме крови-время от нуля до времени достижения последней концентрации, поддающейся количественному определению.
AUC(0-inf): площадь под кривой концентрация в плазме крови-время от нуля до бесконечности.
ANCOVA: ковариационный анализ.
CI: доверительный интервал.
Cmax: максимальная концентрация лекарственного средства.
Cx: концентрация в плазме в x часов после введения дозы.
CV: коэффициент вариаций.
DSST: тест замены цифровых символов.
ECG: электрокардиограмма.
EEG: электроэнцефалограмма.
EMG: электромиограмма.
EOG: электроокулограмма.
KSS: Каролинская шкала сонливости.
LC-MS/MS: жидкостная хроматография-масс-спектрометрия/масс-спектрометрия.
LPS: латентность к постоянному сну, продолжительность времени, измеренная от выключения света до первых 30 секунд записи PSG (период) 20 последовательных периодов без пробуждения.
LS: методика наименьших квадратов.
MAD: многократная нарастающая доза.
MTD: максимально переносимая доза.
PD: фармакодинамика.
PK: фармакокинетика(показатели фармакокинетики).
PSG: полисомнограмма, полисомнография.
PVT: тест психомоторной бдительности.
REM: быстрый сон.
RT: время реакции.
SE: эффективность сна, TST, разделенное на проведенное в постели время (мин.), умноженное на 100.
SAD: однократная нарастающая доза.
SD: среднеквадратичное отклонение.
t1/2: период полувыведения в конечной фазе.
tmax: время достижения максимальной (пиковой) концентрации после введения лекарственного средства.
TST: общее время сна, продолжительность быстрого сна (REM)+не-REM (NREM) сон в течение времени, проведенного в постели (TIB).
WASO: пробуждение после наступления сна, продолжительность бодрствования от наступления устойчивого сна (LPS) до подъема.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг. 1 показаны профили растворения для 1 мг соединения A и капсул по 50 мг.
На фиг. 2 показано профили растворения для 1 мг, 2,5 мг, 5 мг, 10 мг соединения A и таблеток по 25 мг.
На фиг. 3 показано сравнительные профили растворения для капсул с соединением A и таблеток, полученными в условии Ι.
II. Описание вариантов осуществления
В определенных вариантах осуществления настоящее изобретение направлено на способ лечения бессонницы, включающий пероральное введение однократной суточной дозы соединения A в количестве от приблизительно 1 мг до приблизительно 15 мг, и где указанная однократная доза обеспечивает легкое наступление сна, но предупреждает возникновение остаточной сонливости и/или ухудшения состояния на следующий день.
В определенных вариантах осуществления настоящее изобретение направлено на способ лечения бессонницы, включающий пероральное введение однократной суточной дозы соединения A в количестве от приблизительно 1 мг до приблизительно 15 мг для достижения среднего значения максимальной концентрации соединения (Cmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 108 нг/мл. Введение однократной суточной дозы обеспечивает достижение среднего значения AUC(0-24) от приблизительно 15,9 нг*ч./мл до приблизительно 356,4 нг*ч./мл; среднего значения AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 766,5 нг*ч./мл; среднего значения AUC(0-inf) от приблизительно 19.8 нг*ч./мл до приблизительно 796.5 нг*ч./мл; среднего t1/2 от приблизительно 12,7 до приблизительно 60 часов; и обеспечивает среднее значение времени достижения максимальной концентрации в плазме крови (tmax) от приблизительно 1 до приблизительно 3,25 часа.
В другом варианте осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу для обеспечения достижения среднего значения максимальной концентрации (Cmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 7,2 нг/мл на каждый 1 мг соединения A. Введение однократной суточной дозы обеспечивает достижение среднего значения AUC(0-24) от приблизительно 15,9 нг*ч./мл до приблизительно 23,8 нг*ч./мл на каждый 1 мг соединения A; среднего значения AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 51,1 нг*ч./мл на каждый 1 мг соединения A; среднего значения AUC(0-inf) от приблизительно 19,8 нг*ч./мл до приблизительно 53,1 нг*ч./мл на каждый 1 мг соединения A.
При введении субъекту-человеку однократной суточной дозы 1 мг соединения A обеспечивается достижение среднего значения максимальной концентрации (Cmax) в плазме крови приблизительно 5,3 нг/мл; среднего значения AUC(0-24) приблизительно 17,2 нг*ч./мл; среднего значения AUC(0-t) приблизительно 19,1 нг*ч./мл; среднего значения AUC(0-inf) приблизительно 19,8 нг*ч./мл; среднего t1/2 приблизительно 12,7 часов; и обеспечивается среднее значение времени достижения максимальной концентрации соединения в плазме крови (tmax) приблизительно 1 час.
При введении субъекту-человеку однократной суточной дозы 2,5 мг соединения A обеспечивается достижение среднего значения максимальной концентрации (Cmax) в плазме крови от приблизительно 10 нг/мл до приблизительно 18 нг/мл; среднего значения AUC(0-24) от приблизительно 57 нг*ч./мл до приблизительно 60 нг*ч./мл; среднего значения AUC(0-t) от приблизительно 80 нг*ч./мл до приблизительно 95 нг*ч./мл; среднего значения AUC(0-inf) от приблизительно 80 нг*ч./мл до приблизительно 103 нг*ч./мл; среднего t1/2 от приблизительно 30 до приблизительно 37 часов; и обеспечивается среднее значение времени достижения максимальной концентрации соединения в плазме крови (tmax) от приблизительно 1 до 2 часов.
При введении субъекту-человеку однократной суточной дозы 5 мг соединения A обеспечивается достижение среднего значения максимальной концентрации (Cmax) в плазме крови от приблизительно 19 нг/мл до приблизительно 23 нг/мл; среднего значения AUC(0-24) от приблизительно 95 нг*ч./мл до приблизительно 110 нг*ч./мл; среднего значения AUC(0-t) приблизительно 128 нг*ч./мл; среднего значения AUC(0-inf) приблизительно 150 нг*ч./мл; среднего t1/2 приблизительно 31 час; и обеспечивается среднее значение времени достижения максимальной концентрации соединения в плазме крови (tmax) от приблизительно 1 до приблизительно 2 часов.
При введении субъекту-человеку однократной суточной дозы 10 мг соединения A обеспечивается достижение среднего значения максимальной концентрации (Cmax) в плазме крови от приблизительно 30 нг/мл до приблизительно 58 нг/мл; среднего значения AUC(0-24) от приблизительно 160 нг*ч./мл до приблизительно 190 нг*ч./мл; среднего значения AUC(0-t) от приблизительно 280 нг*ч./мл до приблизительно 510 нг*ч./мл; среднего значения AUC(0-inf) от приблизительно 310 нг*ч./мл до приблизительно 530 нг*ч./мл; среднего t1/2 от приблизительно 56 до приблизительно 60 часов; и обеспечивается среднее значение времени достижения максимальной концентрации соединения в плазме крови (tmax) от приблизительно 1 до приблизительно 3,25 часа.
В определенном варианте осуществления при введении субъекту-человеку однократной суточной дозы 1 мг соединения A обеспечивается достижение (1) среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 5,3 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 17 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 19 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 19 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,0 часа.
В другом варианте осуществления при введении субъекту-человеку однократной суточной дозы 2,5 мг соединения A обеспечивается достижение (1) среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 16 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 57 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,0 часа.
В другом варианте осуществления при введении субъекту-человеку однократной суточной дозы 5 мг соединения A обеспечивается достижение (1) среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 23 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 95 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 128 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 149 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,6 часа.
В другом варианте осуществления при введении субъекту-человеку однократной суточной дозы 10 мг соединения A обеспечивается достижение (1) среднего значения максимальной концентрации (Cmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 36 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 159 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 284 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 311 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,0 часа.
В определенных вариантах осуществления при введении однократной суточной дозы 2,5 мг за период приблизительно 14 дней обеспечивается достижение среднего значения максимальной концентрации (Сmax) в плазме крови приблизительно 15 нг/мл; среднего значения AUC(0-24) приблизительно 120 нг*ч./мл; среднего t1/2 приблизительно 44 часа; и среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) приблизительно 2 часа.
В определенных вариантах осуществления при введении однократной суточной дозы 5 мг за период приблизительно 14 дней обеспечивается достижение среднего значения максимальной концентрации (Сmax) в плазме крови приблизительно 24 нг/мл; среднего значения AUC(0-24) приблизительно 190 нг*ч./мл; среднего t1/2 приблизительно 46 часов; и среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) приблизительно 1 час.
В определенных вариантах осуществления при введении однократной суточной дозы 10 мг за период приблизительно 14 дней обеспечивается достижение среднего значения (Сmax) максимальной концентрации в плазме крови приблизительно 47 нг/мл; среднего значения AUC(0-24) приблизительно 360 нг*ч./мл; среднего t1/2 приблизительно 55 часов; и среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) приблизительно 2 часа.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 20 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 18 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 15 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 9,0 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, обеспечивающую среднее значение концентрации соединения A в плазме крови, составляющее от приблизительно 0,4 нг/мл до приблизительно 9,0 нг/мл через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает способы лечения бессонницы, включающие пероральное введение лекарственной формы с терапевтически эффективным количеством соединения A, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 2,5 мг до приблизительно 10 мг, и где указанная однократная суточная доза обеспечивает достижение среднего значения концентрации соединения A в плазме крови от приблизительно 1,8 нг/мл до приблизительно 9,0 нг/мл через 8 часов, или от приблизительно 1,5 нг/мл до приблизительно 5,0 нг/мл через 9 часов, или от приблизительно 2,0 нг/мл до приблизительно 8,0 нг/мл через 10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную дозу, находящуюся в диапазоне от приблизительно 1 мг до приблизительно 15 мг, и где указанная однократная доза обеспечивает легкое наступление сна, но предупреждает возникновение остаточной сонливости и/или ухудшение состояния на следующий день.
Лекарственная форма по настоящему изобретению обеспечивает достижение: 1) среднего значения максимальной концентрации (Сmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 108 нг/мл; 2) среднего значения AUC(0-24) от приблизительно 15,9 нг*ч./мл до приблизительно 356,4 нг*ч./мл; 3) среднего t1/2 от приблизительно 12,7 до приблизительно 60 часов; и 4) среднего значения времени достижения максимальной концентрации соединения в плазме крови (tmax) от приблизительно 1 до приблизительно 3,25 часа после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение максимальной концентрации (Сmax) в плазме крови от приблизительно 3,0 нг/мл до приблизительно 7,2 нг/мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 1 мг соединения A и обеспечивает среднее значение максимальной концентрации (Сmax) в плазме крови в приблизительно 5,3 нг/мл после введения субъекту-человеку однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 2,5 мг соединения A и обеспечивает среднее значение максимальной концентрации (Сmax) в плазме крови от приблизительно 10 нг/мл до приблизительно 18 нг/мл после введения субъекту-человеку однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 5 мг соединения A и обеспечивает среднее значение максимальной концентрации (Сmax) в плазме крови от приблизительно 19 нг/мл до приблизительно 23 нг/мл после введения субъекту-человеку однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 10 мг соединения A и обеспечивает среднее значение максимальной концентрации (Сmax) в плазме крови от приблизительно 30 нг/мл до приблизительно 58 нг/мл после введения субъекту-человеку однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение AUC(0-24) от приблизительно 15,9 нг*ч./мл до приблизительно 23,8 нг*ч./мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 1 мг соединения A и обеспечивает среднее значение AUC(0-24) приблизительно 17 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 2,5 мг соединения A и обеспечивает среднее значение AUC(0-24) от приблизительно 57 нг*ч./мл до приблизительно 60 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 5 мг соединения A и обеспечивает среднее значение AUC(0-24) от приблизительно 95 нг*ч./мл до приблизительно 110 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 10 мг соединения A и обеспечивает среднее значение AUC(0-24) от приблизительно 160 нг*ч./мл до приблизительно 190 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления лекарственная форма обеспечивает среднее значение AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 766,5 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления лекарственная форма содержит от 1 мг до 15 мг соединения A и обеспечивает среднее значение AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 766,5 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение AUC(0-t) от приблизительно 19,1 нг*ч./мл до приблизительно 51,1 нг*ч./мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 1 мг соединения A и обеспечивает среднее значение AUC(0-t) приблизительно 19 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 2,5 мг соединения A и обеспечивает среднее значение AUC(0-t) от приблизительно 80 нг*ч./мл до приблизительно 95 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 5 мг соединения A и обеспечивает среднее значение AUC(0-t) приблизительно 128 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 10 мг соединения A и обеспечивает среднее значение AUC(0-t) от приблизительно 280 нг*ч./мл до приблизительно 510 нг*ч./мл после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления лекарственная форма обеспечивает среднее значение AUC(0-inf) от приблизительно 19,8 нг*ч./мл до приблизительно 796,5 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит от приблизительно 1 мг до приблизительно 15 мг соединения A и обеспечивает среднее значение AUC(0-inf) от приблизительно 19,8 нг*ч./мл до приблизительно 796,5 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение AUC(0-inf) от приблизительно 19,8 нг*ч./мл до приблизительно 53,1 нг*ч./мл на каждый 1 мг соединения A после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 1 мг соединения A и обеспечивает среднее значение AUC(0-inf) приблизительно 19,8 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 2,5 мг соединения A и обеспечивает среднее значение AUC(0-inf) от приблизительно 80 нг*ч./мл до приблизительно 103 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 5 мг соединения A и обеспечивает среднее значение AUC(0-inf) приблизительно 150 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 10 мг соединения A и обеспечивает среднее значение AUC(0-inf) от приблизительно 310 нг*ч./мл до приблизительно 530 нг*ч./мл после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 20 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 18 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 15 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение концентрации соединения A в плазме крови, составляющее приблизительно 9,0 нг/мл или менее через 8-10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма обеспечивает среднее значение концентрации соединения A в плазме крови, составляющее от приблизительно 0,4 нг/мл до приблизительно 9,0 нг/мг через 8-10 часов после введения субъектам-людям однократной дозы.
В дополнительных вариантах осуществления лекарственная форма содержит терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от приблизительно 2,5 мг до приблизительно 10 мг, и где указанная однократная доза обеспечивает достижение среднего значения концентрации соединения A в плазме крови от приблизительно 1,8 нг/мл до приблизительно 9,0 нг/мл через 8 часов, или от приблизительно 1,5 нг/мл до приблизительно 5,0 нг/мл через 9 часов, или от приблизительно 2,0 нг/мл до приблизительно 8,0 нг/мл через 10 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 1 мг соединения A и обеспечивает период полувыведения (t1/2) приблизительно 12,7 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 2,5 мг соединения A и обеспечивает период полувыведения (t1/2) от приблизительно 30 до 37 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 5 мг соединения A и обеспечивает период полувыведения (t1/2) приблизительно 31 час после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 10 мг соединения A и обеспечивает период полувыведения (t1/2) от приблизительно 56 до 60 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 1 мг соединения A и обеспечивает среднее значение времени достижения максимальной концентрации в плазме крови (tmax) приблизительно через 1 час после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 2,5 мг соединения A и обеспечивает среднее значение времени достижения максимальной концентрации в плазме крови (tmax) через от приблизительно 1 часа до приблизительно 2 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 5 мг соединения A и обеспечивает среднее значение времени достижения максимальной концентрации в плазме крови (tmax) от приблизительно 1 до приблизительно 2 часов после введения субъектам-людям однократной дозы.
В определенных вариантах осуществления лекарственная форма содержит 10 мг соединения A и обеспечивает среднее значение времени достижения максимальной концентрации в плазме крови (tmax) через от приблизительно 1 до приблизительно 3,25 часа после введения субъектам-людям однократной дозы.
В определенном варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 1 мг, и где указанная однократная дневная доза обеспечивает достижение (1) среднего значения максимальной концентрации (Сmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 5,3 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 17 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 19 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 19 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,0 часа.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 2,5 мг, и где указанная однократная дневная доза обеспечивает достижение (1) среднего значения максимальной концентрации (Сmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 16 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 57 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 80 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,0 часа.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 5 мг, и где указанная однократная дневная доза обеспечивает достижение (1) среднего значения максимальной концентрации (Сmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 23 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 95 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 128 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 149 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,6 часа.
В другом варианте осуществления настоящее изобретение предлагает лекарственную форму для перорального введения для лечения бессонницы, содержащую терапевтически эффективное количество соединения A и по меньшей мере один фармацевтически приемлемый наполнитель, где указанное терапевтически эффективное количество представляет собой однократную суточную дозу, равную 10 мг, и где указанная однократная дневная доза обеспечивает достижение (1) среднего значения максимальной концентрации (Сmax) в плазме крови в диапазоне от приблизительно 80% до приблизительно 125% от 36 нг/мл; (2) среднего значения AUC(0-24) в диапазоне от приблизительно 80% до приблизительно 125% от 159 нг*ч./мл; (3) среднего значения AUC(0-t) в диапазоне от приблизительно 80% до приблизительно 125% от 284 нг*ч./мл; (4) среднего значения AUC(0-inf) в диапазоне от приблизительно 80% до приблизительно 125% от 311 нг*ч./мл; и/или (5) среднего значения времени достижения максимальной концентрации в плазме крови (tmax) в диапазоне от приблизительно 80% до приблизительно 125% от 1,0 часа.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую фармацевтически приемлемый наполнитель и эффективное количество соединения A, для лечения бессонницы, где указанная лекарственная форма обеспечивает показатель растворения, составляющий 85% или более, в течение 45 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой) в соответствии с методикой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37. Среду растворения (900 мл, 37 ± 0,5°C) выбирали из 0,1 моль/л хлористоводородной кислоты или 0,1 моль/л хлористоводородной кислоты, содержащей 0,5% полисорбата 80. Скорость вращения лопастей выбирали из 50 об./мин. или 75 об./мин.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую фармацевтически приемлемый наполнитель и эффективное количество соединения A, для лечения бессонницы, где указанная лекарственная форма обеспечивает показатель растворения, составляющий 85% или более, в течение 30 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой) в соответствии с методикой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37. Среду растворения (900 мл, 37 ± 0,5°C) выбирали из 0,1 моль/л хлористоводородной кислоты или 0,1 моль/л хлористоводородной кислоты, содержащей 0,5% полисорбата 80. Скорость вращения лопастей выбирали из 50 об./мин. или 75 об./мин.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую фармацевтически приемлемый наполнитель и эффективное количество соединения A, для лечения бессонницы, где указанная лекарственная форма обеспечивает показатель растворения. составляющий 85% или более, в течение 15 минут с начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой) в соответствии с процедурой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37. Среду растворения (900 мл, 37 ± 0,5°C) выбирали из 0,1 моль/л хлористоводородной кислоты или 0,1 моль/л хлористоводородной кислоты, содержащей 0,5% полисорбата 80. Скорость вращения лопастей выбирали из 50 об./мин. или 75 об./мин.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую фармацевтически приемлемый наполнитель и эффективное количество соединения A для лечения бессонницы, где лекарственная форма обеспечивает показатель растворения, составляющий 85% или более, в среде растворения (рН 1,2, 900 мл, 37 ± 0,5°C) в течение 15 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой, при скорости вращения лопастей 50 об./мин.) в соответствии с методикой для немедленного высвобождения лекарственной формы согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую лактозу в качестве фармацевтически приемлемого наполнителя.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую гидроксипропилцеллюлозу с низкой степенью замещения в качестве фармацевтически приемлемого наполнителя.
В определенных вариантах осуществления настоящее изобретение направлено на фармацевтическую лекарственную форму для перорального введения, содержащую лактозу и гидроксипропилцеллюлозу с низкой степенью замещения в качестве фармацевтически приемлемого наполнителя.
Согласно настоящему изобретению, соединение A может находиться в форме свободного основания, фармацевтически приемлемой соли, гидрата, сольвата, полиморфа или любой комбинации вышеупомянутых.
Фармацевтически приемлемые соли могут включать без ограничения
соли неорганических кислот (например, сульфат, нитрат, перхлорат, фосфат, карбонат, бикарбонат, гидрофторид, гидрохлорид, гидробромид, гидроиодид); органические карбоксилаты (например, ацетат, оксалат, малеат, тартрат, фумарат, цитрат); органические сульфонаты (например, метансульфонат, трифторметансульфонат, этансульфонат, бензолсульфонат,
толуолсульфонат, камфорсульфонат); соли аминокислот (например, аспартат, глутамат); соли четвертичного аммония; соли щелочных металлов (например, соль натрия, соль калия); и соли щелочноземельных металлов (например, соль магния, соль кальция).
Способы лечения бессонницы по настоящему изобретению включают соединение A в терапевтически эффективном количестве для лечения бессонницы при введении в соответствии с идеями настоящего изобретения. Эффективное количество представляет собой однократную суточную дозу, находящуюся в диапазоне от 0,5 мг до 100 мг, от 1 мг до 15 мг, от 2 мг до 15 мг или от 2 мг до 10 мг.
Составы
Лекарственные формы по настоящему изобретению содержат соединение A в терапевтически эффективном количестве для лечения бессонницы при введении в соответствии с идеями настоящего изобретения. Стандартная доза эффективного количества в лекарственной форме составляет от 0,5 мг до 100 мг, от 1 мг до 15 мг, от 2 мг до 15 мг, или она выбрана из 2 мг, 2,5 мг, 4 мг, 5 мг, 8 мг, 10 мг или 15 мг. Стандартная доза не ограничена типом лекарственной формы или числом лекарственных форм для однократной дозы.
Лекарственная форма в настоящем изобретении может составлять одну или несколько фармацевтических композиций, содержащих соединение A вместе с фармацевтически приемлемыми наполнителями.
Используемый в данном документе термин "композиция" включает продукт, содержащий конкретный ингредиент в конкретном количестве и какой-либо продукт, полученный непосредственно или опосредованно путем комбинирования определенных ингредиентов в конкретных количествах. Такой термин по отношению к фармацевтической композиции предусматривает включение продукта, содержащего активный ингредиент и инертный ингредиент в виде носителя, и включает каждый продукт, полученный непосредственно или опосредованно путем комбинирования, комплексообразования или агрегации любых двух или более ингредиентов, или путем диссоциации, посредством других типов реакций или взаимодействия одного или нескольких ингредиентов. Таким образом, фармацевтическая композиция в соответствии с настоящим изобретением включает каждую композицию, полученную путем смешивания соединения по настоящему изобретению с фармацевтически приемлемым носителем. Термин "фармацевтически приемлемый" используют для обозначения того, что носитель, разбавитель или среда-носитель должны быть совместимыми с другими ингредиентами препарата и должны быть нетоксичными для потребителя.
Лекарственная форма, не ограниченная указанными выше, предпочтительно представляет собой твердую лекарственную форму; более предпочтительно твердую лекарственную форму для перорального введения; кроме того, предпочтительно лекарственную форму для перорального введения с немедленным высвобождением.
Показатель растворения соединения A в ходе высвобождения из лекарственной формы составляет более 85% в течение 45 минут, предпочтительно в течение 30 минут, более предпочтительно в течение 15 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой, при скорости вращения лопастей) в соответствии с методикой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37. Среду растворения (900 мл, 37 ± 0,5°C) выбирали из 0,1 моль/л хлористоводородной кислоты или 0,1 моль/л хлористоводородной кислоты, содержащей 0,5% полисорбата 80. Скорость вращения лопастей выбирали из 50 об./мин. или 75 об./мин.
Под термином "немедленное высвобождение" в настоящем изобретении необходимо понимать профиль растворения, при котором показатель высвобождения соединения A из лекарственной формы составляет более 80%, предпочтительно более 85% в среде растворения (pH 1,2, 900 мл, 37 ± 0,5°C) в течение 15 минут от начала исследования растворения с использованием аппарата 2 (аппарата с лопастной мешалкой, при скорости вращения лопастей 50 об./мин.) в соответствии с методикой для лекарственной формы с немедленным высвобождением согласно 6.10 Испытание на растворение в JP16 или <711> Растворение в USP37.
Твердые лекарственные формы включают капсулы, гранулы, пастилки для рассасывания, пеллеты, пилюли, порошки, суспензии, таблетки, предпочтительно капсулы, гранулы, пеллеты, пилюли, таблетки.
Фармацевтическую композицию по настоящему изобретению можно получить с использованием стандартных методик и способов производства, общеизвестных в данной области. См., например, монографию Японская фармакопея 16 издание или Общую главу <1151> Фармацевтические лекарственные формы в Фармакопее США-NF (37).
Можно получить фармацевтическую композицию для твердой лекарственной формы по настоящему изобретению, например, порошки получают путем сухого смешивания компонентов. Например, соединение A, один или несколько разбавителей, один или несколько необязательных наполнителей (например, связывающие вещества и/или разрыхлители, а также другие дополнительные необязательные наполнители) смешивают вместе. Компоненты смеси до смешивания или саму смесь можно пропускать через сито с размером ячеек, например, 400-700 мкм. Смазывающее средство, которое можно также подвергнуть просеиванию, затем добавляют к смеси и продолжают смешивать до тех пор пока не получат гомогенную смесь в виде гранул. Затем смесь прессуют в таблетки. В качестве альтернативы, можно использовать влажное гранулирование. Например, активное средство и наполнитель(наполнители) смешивают вместе, например, с использованием гранулятора, при этом порошкообразную смесь гранулируют с малым объемом очищенной воды. Полученные влажные гранулы высушивали и пропускали через измельчитель с получением гранул. Кроме того, разрыхлитель и смазывающее средство добавляют к измельченным гранулам, а после смешивания полученную гомогенную смесь прессуют в таблетки. В качестве альтернативы, среду-носитель, такую как оболочки капсул, заполняли порошками или гранулами для получения капсул. Следует иметь в виду, что модификации методик сухого смешивания и влажного гранулирования, включая порядок добавления компонентов и их просеивание и смешивание до прессования в таблетки, можно осуществлять согласно хорошо известным принципам в данной области.
В случае получения таблеток или гранул, их можно покрывать пленкой на водной основе, например, с помощью нанесения покрытия распылением, если это необходимо.
Примеры используемых в данном документе разбавителей включают лактозу, кукурузный крахмал и кристаллическую целлюлозу, и т.п. Примеры используемых в данном документе связывающих веществ включают гидроксипропилцеллюлозу, гипромеллозу и т.п. Примеры используемых в данном документе разрыхлителей включают гидроксипропилцеллюлозу с низкой степенью замещения, кальциевую соль карбоксиметилцеллюлозы, натриевую соль кроскармеллозы и т.п. Примеры используемых в данном документе смазывающих средств включают стеарат магния, стеарат кальция и т.п. Примеры используемых в данном документе красителей включают оксид титана и т.п. Примеры используемых в данном документе средств для нанесения покрытия включают гидроксипропилцеллюлозу, гипромеллозу, метилцеллюлозу и т.п. Однако не вызывает сомнения, что примеры вышеупомянутых средств не ограничены вышеперечисленным.
Подробное описание предпочтительных вариантов осуществления
ПРИМЕРЫ
Следующие примеры иллюстрируют различные аспекты настоящего изобретения. Они не должны быть истолкованы как ограничивающие каким-либо образом формулу настоящего изобретения.
Пример 1
Исследование с введением однократной дозы (исследование 001)
Данное исследование представляло собой рандомизированное, двойное слепое, плацебо-контролируемое и контролируемое по действующему веществу, непрерывное, с введением однократной дозы исследование. Исследование включало две части, части A (здоровые субъекты) и части B (в целом здоровые субъекты с первичной бессонницей).
Основной целью в данном исследовании была оценка безопасности и переносимости однократных пероральных доз соединения A, которое утром вводили здоровым субъектам, и оценка выбранных фармакодинамических (PD) параметров (например, показателей сна, определенных посредством полисомнографии) в отношении дозозависимого эффекта у субъектов с первичной бессонницей с последующим однократным пероральным введением дозы соединения A вечером примерно за 30 минут до отхода ко сну по сравнению с 10 мг золпидема и плацебо.
Дополнительной целью была оценка безопасности и переносимости однократных доз соединения A для перорального введения у здоровых в целом субъектов с первичной бессонницей, и для оценки фармакокинетики (PK) соединения A с дальнейшим пероральным введением однократных доз у здоровых субъектов и субъектов с первичной бессонницей.
Обе части исследования имели две фазы, фазу предварительной рандомизации и фазу рандомизации. Продолжительность фазы предварительной рандомизации составляла не более 21 дня и состояла из скринингового периода (от дня -21 до дня -3) и периода исходного уровня (от дня -2 до дня -1), в течение которых определяли, соответствует ли каждый субъект критериям для включения его в исследование, при этом исходное состояние оценивали в день -2. В фазе рандомизации субъектов рандомизировали на получающих однократную пероральную дозу каждого соединения A или соответствующего соединению A плацебо (часть A), и/или золпидема, или соответствующего золпидему плацебо (часть B).
Планировали обследовать примерно 160 здоровых субъектов и 250 здоровых в целом субъектов с первичной бессонницей с целью включения в исследование 64 и 60 субъектов, в частности, в часть A и часть B соответственно. В действительности, 160 здоровых субъектов и 281 в здоровых в целом субъектов с первичной бессонницей обследовали с включением в исследование 64 и 58 субъектов в часть A и часть B соответственно.
Для части A 64 здоровых субъекта включали в когорты последовательно, с режимом постепенного повышения дозы, для получения либо соединения A, либо плацебо, при этом субъектов стратифицировали по половому признаку. Каждая когорта включала шесть субъектов, получающих соединение A, и двух субъектов, получающих плацебо. Все исследуемые лекарственные средства вводили в виде однократных доз в одной или нескольких капсулах с соединением A или капсулах с соответствующим соединению A плацебо из расчета по тестируемой дозе. После отбора субъектов подвергали процедурам исходного уровня и рандомизации в день -2. Субъектам вводили лекарственное средство в день 1 утром натощак, через 1 час после пробуждения. Образцы крови для PK анализа отбирали в предварительно определенные временные точки и проводили оценку данных PD. Проводили оценку субъектов в день 1 до введения лекарственного средства и каждые 2 часа от 2 до 12 часов после введения лекарственного средства, а также каждое утро в дни 2-6.
Данные оценки включали Каролинскую шкалу сонливости (KSS), тест замены цифровых символов (DSST) и тест психомоторной бдительности (PVT) для оценки дневной сонливости, уровня концентрации внимания и способности сосредоточиваться. Опросники после пробуждения заполняли после получения данных PD каждое утро в дни 1-6. Время и продолжительность короткого сна записывали в дни 1 и 2. Безопасность контролировали на протяжении всего исследования.
Дозы для части A составляли 1 мг, 2,5 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг и 200 мг соединения A. Повышение на следующий более высокий уровень дозы не осуществляли до тех пор, пока: 1) пока при заслеплении не рассматривали доступные данные по безопасности, переносимости (включая лабораторные показатели и электрокардиограмму [ECG]) и PK от последней прошедшей курс лечения когорты и 2) доступные данные не обосновывали возможность повышения следующей дозы.
Для части B 58 в целом здоровых субъектов с первичной бессонницей рандомизировали в три когорты и их разделяли по половому признаку. Часть B также включала активный контроль (золпидем) и соответствующее плацебо. В каждой когорте находились примерно 12 субъектов в группе соединение A/соответствующее золпидему плацебо, примерно четыре субъекта в группе золпидем/соответствующее соединению A плацебо и примерно четыре субъекта в группе соответствующее соединению A плацебо/соответствующее золпидему плацебо. Введение дозы в части B проводили вечером, за 30 минут до отхода ко сну. Начальная доза для части B представляли собой 3 уровня доз, которые определили как безопасные и хорошо переносимые в части A. Последующие уровни доз в части B определяли на основе результатов PD первой когорты в части B и результатов PD и безопасности из по меньшей мере 3 оформленных когорт с более высокой дозой в части A. Каждую когорту в части B разделяли по меньшей мере на две группы с введением дозы в каждой группе, разнесенным по времени на минимум 2 дня.
После предварительного визита скринингового обследования подходящих для исследования субъектов включали в график для возвращения в клинику на 2 дня в ходе скринингового периода для проведения PSG скрининга /на исходном уровне. Эти два дня следовали по меньшей мере через 3 дня после предварительного визита скринингового обследования и в течение окна от дня -7 до дня -6 (± 2 дня). Первую PSG использовали для скрининга в отношении апноэ во время сна и периодических движений конечностей во сне (PLMS), при этом она служила в качестве PSG первого исходного уровня. Вторую PSG использовали в качестве PSG второго исходного уровня. Определенные показатели PSG использовали для определения того, какой из двух субъектов удовлетворял критериям включения согласно PSG, при этом средние значения измеряемых показателей PSG из этих двух PSG использовали в качестве исходного уровня для данного измерения PD. Субъекты, которые соответствовали критериям включения согласно PSG, возвращались в клинику в день -1 для дополнительных процедур исходного уровня и рандомизации. Субъектам не разрешали спать днем в день 1. Вечером в день 1, после минимум 3-часового голодания, исследуемое лекарственное средство вводили за 30 минут до привычного для субъекта времени отхода ко сну (с выключением освещения), которое рассчитывали согласно дневнику регистрации сна при первой PSG в ходе скринингового периода. Образцы крови для PK анализа отбирали в предварительно определенные временные точки и проводили оценку данных PD. PSG регистрировали в день 1, после введения лекарственного средства. По отношению к субъектам использовали дополнительные анализы PD каждое утро в дни 1-6. Данные оценки включали KSS, DSST и PVT с целью анализа дневной сонливости, уровня концентрации внимания и способности сосредоточиваться. Также проводили KSS и DSST за 5 минут до введения лекарственного средства и через 25 минут после введения лекарственного средства в день 1, непосредственно перед выключением освещения. Заполнение опросников после пробуждения осуществляли после анализа данных PD в течение 15 минут после подъема, на протяжении дней 1-6. Время и продолжительность короткого дневного сна записывали в день 2.
Фармакокинетика
У субъектов в части A для определения концентрации соединения A в плазме крови образцы крови отбирали в день 1 до введения лекарственного средства и через 0,25 (15 минут), 0,5 (30 минут), 1, 2, 3, 4, 5, 6, 9, 12, 24, 48, 72, 96, 120, 168 и 240 часов после перорального введения соединения A, но при этом в первых трех когортах образцы отбирали (группы с 1, 2,5 и 5 мг соединения A) не позже 72 часов. Образцы для PK отбирали преимущественно с помощью постоянно находящегося в вене катетера в первые 12 часов и посредством последующей прямой венепункции.
У субъектов в части B образцы крови для определения концентраций соединения A в плазме крови получали путем прямой венепункции в день 1 до введения лекарственного средства и через 0,5, 9, 12, 24, 36, 60, 84, 108, 156 и 228 часов после введения лекарственного средства.
Некомпартментные PK параметры плазмы крови, которые рассчитывали для соединения A (в виде доступных данных) включали без ограничения: Сmax (максимальная концентрация лекарственного средства); tmax (время достижения максимальной (пиковой) концентрации после введения лекарственного средства); AUC(0-24 ч) (площадь под кривой концентрация в плазме крови-время от нуля до 24 часов); AUC(0-t) (площадь под кривой концентрация x в плазме крови-время от нуля до времени достижения последней концентрации, поддающейся количественному определению); AUC(0-inf) (площадь под кривой концентрация х в плазме крови-время от нуля до бесконечности); t1/2 (период полувыведения в конечной фазе); CL/F (кажущийся общий клиренс лекарственного средства после внесосудистого введения); и V/F (кажущийся объем распределения).
Концентрации соединения A в плазме крови измеряли с использованием валидированной методики жидкостной хроматографии-масс-спектрометрии/масс-спектрометрии (LC-MS/MS).
PK параметры соединения A для субъектов в части A изложены в таблице 1. В части B профили концентрация-время были примерно аналогичны таким профилям их соответствующем дозовым группам в части A.
Таблица 1. PK параметры соединения A у субъектов из части A

Фармакодинамика
У субъектов из части A проводили KSS, DSST и PVT, начиная за 30 минут до введения лекарственного средства в день 1, затем каждые 2 часа в течение 12 часов после введения лекарственного средства, и в дни 2-6, начиная через 30 минут после подъема. Опросники после пробуждения заполняли по утрам в дни 1-6 с последующим проведением оценок PD.
В части A показатели сонливости (KSS, DSST, PVT) указывали на общую зависимость доза-ответ. Фармакодинамический ответ в данных измерениях был, как правило, максимальным на 2 час после введения лекарственного средства, совпадая с Сmax. Продолжительность эффекта коррелировала с дозой, т.е. эффекты были более длительными при использовании более высоких доз.
У субъектов из части B проводили KSS, DSST и PVT в течение 15 минут после пробуждения, в частности, в таком порядке: каждое утро с 1 дня по день 6, с последующим заполнением опросника после пробуждения. KSS и DSST также проводили за 5 минут до введения лекарственного средства и через 25 минут после введения лекарственного средства в день 1 (непосредственно перед выключением освещения).
Кроме того, PSG проводили в ходе скринингового периода в дни -7 и -6 (±2 дня) и после введения лекарственного средства в день 1. 8-часовую диагностическую PSG, состоящую из электроэнцефалограммы (EEG), электроокулограммы (EOG), электромиограммы (EMG), ECG, снятия данных с помощью электродов на нижних конечностях и измерений респираторной функции (поток воздуха, дыхательное усилие и насыщение кислородом), проводили, начиная с привычного для субъекта времени отхода ко сну, которое определено согласно дневнику регистрации сна для 3 ночей непосредственно перед первой PSG в день -7 (±2 дня). Показатели PSG этой ночи использовали для скрининга в отношении апноэ во время сна и PLMS. В день -6 (±2) и после введения лекарственного средства в день 1 проводили стандартные PSG (т.е., без электродов на нижних конечностях и измерений респираторной функции, за исключением насыщения кислородом). Указанные показатели PSG из этих двух PSG, проведенных в ходе скринингового периода, использовали для того, чтобы определить, какой из субъектов соответствовал критериям включения согласно PSG. Средние значения показателей PSG из этих двух проведенных PSG на протяжении периода отбора использовали в качестве значений PSG исходного уровня. Основные PD параметры, такие как PSG LPS, TST, SE и WASO, получали из всех записей PSG.
Результаты полисомнографии указывали на предварительную эффективность соединения A. При дозах в 2,5 и 10 мг LPS уменьшалась почти на 30 минут, а при 25 мг LPS уменьшалась на примерно 45 минут по сравнению с исходным уровнем. При дозах в 2,5 и 10 мг WASO уменьшалось на примерно 30 минут, а при 25 мг WASO уменьшалось на более чем 45 минут по сравнению с исходным уровнем. По сравнению с золпидемом дозы в 2,5 и 10 мг соединения A проявляли аналогичную величину эффекта в отношении LPS и WASO. По сравнению с исходным уровнем SE улучшилась на 11% при дозе соединения A в 2,5 мг, на 13% при дозе 10 мг и на 18% при дозе 25 мг. Это сравнимо с изменением SE на 3% от исходного уровня при лечении с использованием плацебо и на 13% для золпидема. После введения однократной дозы соединения A в 25 мг, SE увеличилась на примерно 90%. Однако анализ данных PD показал, что у некоторых индивидуумов при использовании таких доз проявлялось увеличение показателей остаточной сонливости на следующий день от исходного уровня.
При использовании какой-либо дозы соединения A, золпидема или плацебо, клинически значимые эффекты последействия в отношении KSS, DSST или PVT отсутствовали.
Пример 2
Исследование с использованием многократной нарастающей дозы (исследование 002)
Данное исследование представляло собой одноцентровое, рандомизированное, двойное слепое, плацебо-контролируемое, непрерывное исследование и с использованием многократных доз.
Основной целью данного исследования являлась оценка безопасности, переносимости и фармакокинетики (PK) соединения A после перорального введения многократных доз один раз в сутки вечером в течение 14 дней здоровым взрослым субъектам. Кроме того, целью данного исследования являлось выявление максимально переносимой дозы (MTD) или достаточно высокой переносимой дозы соединения A, обеспечивающей широту терапевтического действия по отношению к предполагаемой терапевтической дозе.
Всего 48 здоровых взрослых субъектов (18-55 лет) включали в 1 из 6 когорт последовательно в порядке постепенного повышения дозы, и их рандомизировали как получавших в течении 14 дней либо соединение A, либо соответствующее соединению A плацебо вечером за 30 минут до привычного времени отхода ко сну и после 3-часового голодания. Каждая когорта включала 6 субъектов, получавших соединение A, и 2 субъектов, получающих плацебо. Образцы крови для PK анализа отбирали в предварительно определенные временные точки и проводили оценки данных PD.
Исследование имело две фазы, фазу предварительной рандомизации и фазу рандомизации. Фаза предварительной рандомизации составляла не более 21 дня и состояла из скринингового периода (от дня -21 до дня -3) и периода исходного уровня (от дня -2 до дня -1), в течение которых определяли, соответствует ли каждый субъект критериям для включения в исследование, и при этом исходное состояние оценивали в день -2. Фаза рандомизации (дни 1-28) состояла из 3 периодов: лечения (дни 1-14), в ходе которого субъектов рандомизировали и они получали перорально суточные дозы либо соединения A, либо соответствующего соединению A плацебо, наблюдения в стационарных условиях (дни 15-19) и наблюдения в амбулаторных условиях (дни 20-28), в течение которых проводили исследования PK и безопасности.
У всех субъектов проводили исследования PD, включая Каролинскую шкалу сонливости (KSS), тест замены цифровых символов (DSST) и тест психомоторной бдительности (PVT), с целью оценки сильной сонливости в промежутке между введением дозы и отходом ко сну, а также остаточной сонливости на следующий день и уровня концентрации внимания, и способности сосредоточиваться. Кроме того, заполнение опросника после полного пробуждения осуществляли ежедневно с целью оценки качества сна в прошедшую ночь.
Начальная доза для данного исследования основывалась на результатах исследования из примера 1. Повышение уровня до следующего более высокого уровня не осуществляли до тех пор, пока данные о безопасности, переносимости (включая лабораторные данные и электрокардиограмму [ECG]) и доступные PK данные, полученные для последней прошедшей исследование когорты, не обосновывали повышение до следующей дозы.
Образцы крови для определения концентраций соединения A в плазме крови собирали в день 1 до введения лекарственного средства и через 0,5 (30 минут), 1, 1,5, 2, 3, 4, 5, 6, 8, 10 и 12 часов после введения лекарственного средства; дни 2-13: до введения лекарственного средства; день 14: до введения лекарственного средства и через 0,5 (30 минут), 1, 1,5, 2, 3, 4, 5, 6, 8, 10 и 12 часов после введения лекарственного средства; день 15: через 24 часа после введения лекарственного средства в день 14; дни 16-19: через 36, 60, 84 и 108 часов после введения лекарственного средства в день 14; день 21, 24, 26, 28: как можно точнее в 156, 228, 276 и 324 часа после введения лекарственного средства в день 14. Концентрации соединения A в плазме крови измеряли таким же способом, как описано в примере 1, и рассчитывали вышеприведенные некомпартментные PK параметры для соединения A в плазме крови.
PD эффекты исследовали путем оценки функций в виде KSS, DSST и PVT после введения лекарственного средства и на следующий день, и с помощью самостоятельного отчета о качестве сна в опроснике после пробуждения. KSS, DSST и PVT проводили начиная с дня -1 за 15 минут до привычного времени отхода ко сну; в дни 1-15 в течение 15 минут после обычного времени пробуждения, и на 1, 2, 4, 8 и 12 часы после привычного времени пробуждения, и в дни 1-14 за 15 минут до введения лекарственного средства, и через 15 минут после введения лекарственного средства. Холтеровское мониторирование продолжительностью 24 часа начинали проводить за 30 минут до отхода ко сну в день -2 и непосредственно перед введением дозы в день 14. Выборки из этих записанных материалов использовали для проведения анализов ECG, в том числе анализа HPQT. Опросники после пробуждения заполняли в период от дня 1 до дня 19.
KSS, PVT и DSST проводили в каждый день лечения, вечером до введения лекарственного средства и после введения лекарственного средства для исследования немедленного эффекта соединения A в отношении сонливости. Для данных временных точек значения KSS, DSST и PVT, получаемые ежедневно за 15 минут до введения лекарственного средства, служили в качестве исходного уровня для значений, получаемых через 15 минут после введения лекарственного средства (ежедневный исходный уровень), для тех же самых дней. KSS, PVT и DSST также проводили в течение последующих дневных часов после каждого вечернего введения лекарственного средства для оценки эффекта соединения A в отношении остаточной сонливости на следующий день. Для этих временных точек проводили исследования в день 1, через 15 минут и 1, 2, 4, 8 и 12 часов после обычного времени пробуждения, служащего в качестве исходного уровня для исследований, которые проводили в соответствующие моменты времени после обычного времени пробуждения за период от дня 2 до дня 15 (согласованный во времени исходный уровень). Опросники после пробуждения заполняли в утренние часы в дни 1-19. Значение перед введением лекарственного средства в день 1 использовали в качестве исходного уровня. Разницу между плацебо и каждой дозой соединения A при изменении относительно исходного уровня в каждой временной точке рассчитывали вместе с 95% доверительными интервалами (CI). Насколько позволяли данные, изучали потенциальный дозозависимый эффект и характер зависимости от времени.
Капсулы с соединением A и капсулы с соответствующим соединению A плацебо были доступны в дозировках 2,5 мг, 10 мг и 50 мг. Все исследуемые лекарственные средства вводили в виде суточных доз с использованием одной или нескольких капсул с соединением A или капсул с соответствующим соединению A плацебо из расчета на тестовую дозу.
Фармакокинетика
PK параметры в день 1 и день 14 изложены в таблице 2 и таблице 3 соответственно.
Таблица 2. PK параметры в день 1
|
Таблица 3. PK параметры в день 14
|
Исходя из графической оценки нормализованных по дозам данных, Сmax увеличивалась несколько меньше, чем соразмерно дозе для обеих исследований в день 1 и день 14. Исходя из нормализованных данных по дозам, AUC(0-24 ч.) увеличилась несколько меньше, чем соразмерно дозе в день 1, но при этом увеличилась приблизительно пропорционально дозе, исходя из оценок в день 14. Периоды полувыведения в конечной фазе для доз 2,5 и 5 мг были аналогичным, составляя в среднем примерно 45 часов. При дозах в 10 мг и выше среднее значение периода полувыведения в конечно фазе составляло примерно 55 часов после последнего дня 14-дневного введения лекарственного средства. Накопление было ниже чем спрогнозированное по периоду полувыведения в конечной фазе. Исходя из накопления, эффективный период полувыведения находился в диапазоне от 16,9 до 24,7 часов для доз, которые находились в диапазоне от 2,5 до 25 мг, и 28,0 и 39,3 часа для доз в 50 и 75 мг соответственно.
Фармакодинамика
Эффект остаточной сонливости на следующий день: при измерении с помощью KSS, PVT и DSST отмечали дозозависимые увеличения как в величине, так и в продолжительности остаточной сонливости на следующий день. В группах с дозами 2,5 мг и 5 мг значимое отличие от группы плацебо отсутствовало, что свидетельствовало об увеличении в группах остаточной сонливости на следующий день в любом исследовании в любое время по сравнению со временем пробуждения в любой день лечения. Отмечали незначительные отличия от группы плацебо у группы с дозой 10 мг во временных точках в пределах 2 часов после времени пробуждения в KSS в дни 2-4. У группы с дозой 25 мг увеличение остаточной сонливости на следующий день было более стойким и несколько большим, чем у группы с дозой в 10 мг. Эффект, опять же, был ограничен временными точками в пределах 2 часов после времени пробуждения, но его наблюдали в KSS и до определенной степени в отклонениях PVT и PVT среднего значения RRT. Отличия от плацебо были наиболее достоверными и большими в день 2 и день 15 по сравнению со всеми другими днями лечения. В группах с дозами 50 мг и 75 мг были достоверные и относительно большие отличия от плацебо по всем исследованиям сонливости. Данные отличия имели большую величину во временных точках в пределах 2 часов после времени пробуждения, но по-прежнему наблюдались через 4 часа и 8 часов после времени пробуждения, в частности, в PVT среднего RRT. Через 12 часов после времени пробуждения отличия от плацебо по любым показателям сонливости в группах с любой дозой в любой день отсутствовали. Для групп с дозой, при которой наблюдали остаточную сонливость на следующий день (т.е., 10 мг и выше), сонливость была относительно больше выраженной в дни 2-4 по сравнению с днями 5-15. Как правило, данную динамику уменьшения сонливости наблюдали на протяжении дней лечения, несмотря на накопление соединения A в плазме крови.
Ни один из пунктов опросника после полного пробуждения не показал наличие систематических изменений в динамике ночного сна в любой группе с введением лекарственного средства или в группе с плацебо, за исключением того, что согласно шкале качества сна проявилась тенденция к тому, что большее число субъектов в обеих группах с введением плацебо и введением соединения A сообщали о "беспокойном" или "очень беспокойном" сне в день 2 и особенно в день 15 по сравнению с другими днями.
Фармакокинетика-фармакодинамика
Для PK-PD разведочного анализа остаточной сонливости на следующий день совокупность PK концентраций полученного модельного соединения A в плазме крови через 8, 9 и 10 часов после вечернего введения в день 1 сопоставляли с изменением от исходного уровня в KSS, отклонениями PVT и DSST через 15 минут, 1 час и 2 часа после полного пробуждения утром в день 2 соответственно. Во всех временных точках, как в KSS, так и в отклонениях PVT, наблюдали большее увеличение от исходного уровня при увеличении концентраций соединения A. Концентрации ниже 30 нг/мл (которые регистрировали при дозах ниже 25 мг) через 9-10 часов после введения лекарственного средства, что соответствует 1-2 часам после утреннего пробуждения, были связаны с минимальным изменением или с отсутствием изменений от исходного уровня в KSS, отклонениях PVT или DSST.
Пример 3
Перекрестное исследование относительной биологической доступности таблетированного состава по сравнению с капсулированным составом (исследование 005)
Проводили одноцентровое, открытое, рандомизированное перекрестное исследование для оценки у здоровых взрослых субъектов биологической доступности однократных пероральных доз соединения A в таблетированном составе по сравнению с однократными пероральными дозами соединения A в капсулированном составе в дозе 2,5, 10 и 25 мг. Другой целью исследования была оценка безопасности и переносимости таблетированных составов с соединением A в дозе 2,5, 10 и 25 мг у здоровых взрослых субъектов. Примерно 36 субъектов рандомизированно разделяли в одну из трех когорт (примерно по 12 субъектов в когорте) и они получали как однократную дозу соединения A в виде капсулированного состава, так и однократную дозу соединения A в виде таблетированного состава в произвольной последовательности при соотношении 1:1. Дозы составляли 2,5 мг, 10 мг и 25 мг.
Исследование включало две фазы: предварительной рандомизации и рандомизации. Фаза предварительной рандомизации длилась не более 21 дня и включала скрининговый период и исходный период A, в течение которых определяли соответствие критериям для включения в исследование и проводили исследования исходного уровня перед введением дозы первого состава. Фаза рандомизации включала два периода лечения (A и B), разделенные исходным периодом B. В первый день периода лечения A субъекты получали однократную пероральную дозу первого состава. После введения дозы первого состава, на протяжении периода лечения А проводили исследования фармакокинетики (PK) и безопасности, затем данный период субъекты завершали 20-дневным "вымыванием". Перед введением дозы второго состава завершали исследование субъектов исходного периода B. Затем субъекты переходили к периоду лечения B и получали однократную пероральную дозу второго состава. На протяжении периода лечения В проводили исследования фармакокинетики и безопасности.
Образцы крови для определения концентраций соединения A в плазме крови отбирали в следующее временные точки: период лечения A (дни 1-15): до и после введения дозы в 0,5 (30 минут), 1, 1,5, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 120, 168, 240 и 336 часов; период лечения B (дни 22-36): до и после введения дозы в 0,5 (30 минут), 1, 1,5, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 120, 168, 240 и 336 часов.
Концентрации соединения A в плазме крови измеряли с использованием валидированной методики жидкостной хроматографии-тандемной масс-спектрометрии (LC-MS/MS).
Некомпартментные способы использовали для расчета следующих PK параметров плазмы крови для соединения A: площадь под кривой концентрация в плазме крови-время от нуля до 8 часов после введения дозы (AUC(0-8)), площадь под кривой концентрация в плазме крови-время от нуля до 72 часов после введения дозы (AUC(0-72)), площадь под кривой концентрация в плазме крови-время от нуля до времени достижения последней концентрации, поддающейся количественному определению (AUC(0-t)), площадь под кривой концентрация в плазме крови-время от нуля, экстраполированное на бесконечность (AUC(0-inf)), максимальная концентрация лекарственного средства в плазме крови (Сmax), период полувыведения в конечно фазе (t1/2), время задержки абсорбции (tlag) и время достижения максимальной (пиковой) концентрации в плазме крови после введения лекарственного средства (tmax). Основными PK параметрами являлись AUC(0-inf) и Сmax. Отдельные PK параметры для соединения A были изложены в виде данных по составу (таблетка или капсула) и по дозе (2,5, 10 или 25 мг). PK параметры, кроме tmax и tlag, были изложены по составу и дозе с использованием описательной статистики: числа субъектов, среднего значения, SD, коэффициента вариации, среднего геометрического, медианы, минимума и максимума. Параметры tmax и tlag изложены для состава и дозы с применением следующей описательной статистики: медианы, минимума, максимума и 90% доверительного интервала (CI) оценки медианной точки. Преобразованные натуральным логарифмом PK параметры для соединения A (AUC(0-inf), Сmax, AUC(0-8), AUC(0-72) и AUC(0-t)) сравнивали отдельно по дозе с моделью смешанных эффектов с последовательностью, периодом лечения и составом как фиксированные эффекты и субъекты сгруппированные внутри последовательности в виде случайного эффекта. Соотношение средних геометрических значений (LS), рассчитанных по методу наименьших квадратов (таблетированный в качестве тестового/капсулированный состав в качестве контрольного) и соответствующего 90% CI рассчитывали путем возведения в степень различия средних значений, полученных методом наименьших квадратов LS, и соответствующих 90% CI.
PK параметры соединения A изложены в таблице 4.
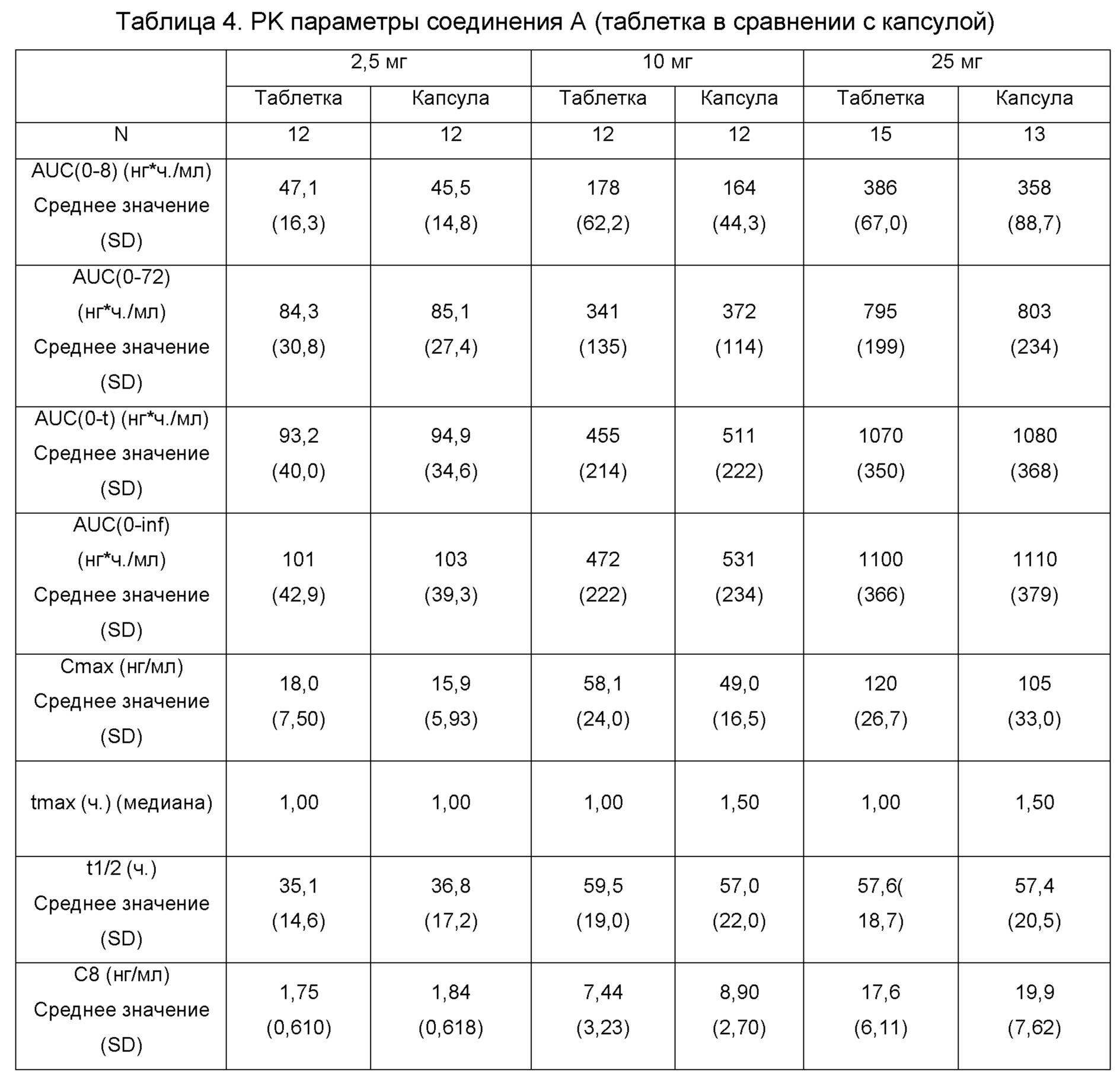
Отличия между составами (лечение A [таблетка] по сравнению с лечением B [капсула]) в AUC(0-8), AUC(0-72), AUC(0-t) и AUC(0-inf) по всем уровням дозы составляли менее 13% в каждом. Отличия между таблетированными и капсульными составами в Сmax по всем уровням дозы составляли менее 16% в каждом. Медиану tmax наблюдали через 1-1,5 часа после введения как таблетированного, так и капсульного составов по всем уровням дозы. Тенденцию к 30-минутной задержке медианы tmax для капсульного состава (лечение B) по сравнению с таблетированным составом (лечение A) наблюдали при повышенных дозах. Задержка абсорбции в случае каждого состава при любом уровне дозы отсутствовала.
В целом, результаты показали, что как скорость, так и степень абсорбции соединения A после введения таблетки сравнимы с контрольной капсулой для всех исследуемых дозировок. Вариабельность полученных PK параметров была также аналогична лечению таблеткой по сравнению с лечением капсулами. Данные результаты подтверждают заключение о том, что относительная биологическая доступность таблетки в при дозировках в 2,5, 10 и 25 мг аналогична соответствующим показателям биологической доступности в случае использования капсулы с соответствующими концентрациями. Таким образом, переход в клинических условиях к таблетированному составу можно совершить без коррекции дозы по отношению к капсуле.
Пример 4 (исследование 201)
Данное исследование являлось многоцентровым, рандомизированным, двойным слепым, с адаптивным планом исследования, а также исследованием зависимости доза-эффект у субъектов с бессонницей. Субъектов рандомизировали в 1 из 6 доз соединения A (1 мг, 2,5 мг, 5 мг, 10 мг, 15 мг и 25 мг) или плацебо.
Основными целями исследования были:
1. Выявление дозы или доз соединения A, которые максимально повышают эффективность и сводят к минимуму остаточную сонливость на следующий день у субъектов с хронической бессонницей в начале лечения путем сравнения эффекта 6 доз соединения A с плацебо с использованием комплексной функции полезности, включающей изменение от исходного уровня в эффективности сна (SE) и изменение от исходного уровня в Каролинской шкале сонливости (KSS) через 1 час после утреннего пробуждения после введения дозы в день 2 и день 3.
2. Сравнение эффекта 6 доз соединения A с плацебо в отношении KSS через 1 час после утреннего пробуждения в день 15 и день 16 у субъектов с хронической бессонницей для того, чтобы подтвердить, что доза или дозы, которые максимально повышают эффективность и сводят к минимуму остаточную сонливость на следующий день в начале лечения, не связаны с неприемлемыми уровнями остаточной сонливости на следующий день при завершении лечения.
Дополнительными целями исследования было проведение оценки:
1. Эффективности в начале лечения:
В общем: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения SE на исходном уровне до среднего значения SE после введения дозы в день 1 и день 2.
Индукция сна: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения латентности к постоянному сну (LPS) на исходном уровне до среднего значения LPS после введения дозы в день 1 и день 2.
Поддержание сна: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения бодрствования после наступления сна (WASO) на исходном уровне до среднего значения WASO после введения дозы в день 1 и день 2.
2. Эффективность по завершению лечения: В общем: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения SE на исходном уровне до среднего значения SE после введения дозы в день 14 и день 15.
Индукция сна: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения LPS на исходном уровне до среднего значения LPS после введения дозы в день 14 и день 15.
Поддержание сна: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения WASO на исходном уровне до среднего значения WASO после введения дозы в день 14 и день 15.
3. Вероятность возникновения эффекта привыкания с начала до завершения лечения:
В общем: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения SE на исходном уровне до среднего значения SE после введения дозы в день 1 и день 2 против изменения от среднего значения SE на исходном уровне до среднего значения SE после введения дозы в день 14 и день 15.
Индукция сна: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения LPS на исходном уровне до среднего значения LPS после введения дозы в день 1 и день 2 против изменения от среднего значения LPS на исходном уровне до среднего значения LPS после введения дозы в день 14 и день 15.
Поддержание сна: сравнение каждого уровня дозы соединения A с плацебо по изменению от среднего значения WASO на исходном уровне до среднего значения WASO после введения дозы в день 1 и день 2 против изменения от среднего значения WASO на исходном уровне до среднего значения WASO после введения дозы в день 14 и день 15.
Общее число подвергнутых скринингу субъектов составило 616, и 291 из этих субъектов рандомизировали в исследовании по группам: 56 в группу с плацебо, 32 в группу с 1 мг, 27 в группу с 2,5 мг, 38 в группу с 5 мг, 32 в группу с 10 мг, 56 в группу с 15 мг и 50 в группу с 25 мг. 291 субъект был включен в множество для полного анализа, множество для анализа безопасности и множество для анализа PD. В группы с дозами активного соединения входило 222 субъекта (приблизительно поровну среди всех доз, 90-100%) и 51 (91,1%) в группу плацебо, которые прошли запланированный курс лечения.
Исследование включало 2 фазы: предварительной рандомизации и рандомизации. Продолжительность фазы предварительной рандомизации составляла не более 21 дня и состояла из скринингового периода (от дня -21 до -2) и исходного периода (день -1). После
исходного периода всех субъектов, соответствующих критериям для включение в исследование, рандомизировали в двойном слепом режиме с тем, чтобы они получали соединение A или плацебо в течение 15 ночей на протяжении периода лечения (дни 1-15). Затем все субъекты получали плацебо в одностороннем слепом режиме на протяжении 2 ночей (дни 16-17) в ходе периода исследования повторного возникновения бессонницы (дни 16-18). Субъекты не получали исследуемое лекарственное средство на протяжении периода последующего наблюдения (дни 19-30). Все субъекты приходили в клинику для прохождения процедур скрининга. На протяжении скринингового периода субъекты ежедневно заполняли дневник регистрации сна. Полисомнографию во время сна проводили в ходе скринингового периода на протяжении 2 ночей подряд между днем -9 и днем -3. Полисомнографические исследования (PSG) продолжительностью 8 часов начинали в середине привычного времени отхода ко сну, рассчитанного по ответам в дневнике регистрации сна, который заполняли за 7 дней перед первым ночным PSG. Данные записи служили как в качестве скрининговых PSG для определения соответствия субъектов критериям включения в исследование, так и PSG исходного уровня. Субъекты могли покидать клинику между ночами проведения скрининговой/PSG исходного уровня.
Все субъекты возвращались в клинику в день -1 для прохождения процедур исходного периода. Они оставались в клинике до дня 3. Утреннее обследование в день 1 предусматривало получение значений исходного уровня для KSS, теста замены цифровых символов (DSST) и индекса времени реакции (RTI). Обследование через 6 часов после пробуждения предусматривали получение значений исходного уровня для теста функции бодрствования (WFB) и профиля эмоционального состояния в краткой форме (POMS-B). Затем субъектов рандомизировали с тем, чтобы они получали 1 из 6 доз соединения A или плацебо на протяжении следующих 15 дней. Исследуемые лекарственные средства представляли собой таблетки, содержащие соответствующее соединению A плацебо или соединение A в количестве 1 мг, 2,5 мг, 5 мг или 10 мг, и которые принимали во внутрь за 30 минут до среднего привычного времени отхода ко сну, которое рассчитывали по ответам в дневнике регистрации сна на протяжении скринингового периода. PSG продолжительностью 8 часов, начиная в то же самое время отхода ко сну, как и при проведении ночью скрининговой PSG и PSG исходного уровня, записывали в первые 2 ночи периода лечения (дни 1 и 2). Дневник регистрации сна продолжали ежедневно заполнять в клинике, при этом проводили исследования тяжести бессонницы (ISI), остаточных эффектов на следующий день (KSS, DSST и RTI), причем субъекты находились в клинике. В указанные дни исследования оценивали концентрации соединения A в плазме крови, тогда как субъекты находились в клинике утром после пробуждения и непосредственно перед введением дозы.
Субъекты продолжали принимать соединение A или плацебо за 30 минут перед их предполагаемым, самостоятельно определенным временем отхода ко сну, и продолжали ежедневно заполнять журнал регистрации сна во время пребывания дома в течение периода лечения. В день 14 периода лечения субъекты возвращались в клинику. Они оставались в клинике в течение 4 ночей и в течение последующих дней до дня 18. Каждую ночь в клинике записывали восьмичасовые PSG, начиная со среднего значения привычного времени отхода ко сну, которое рассчитывали по ответам в дневнике регистрации сна, заполняемого в дни 3-13. Дневник регистрации сна продолжали ежедневно заполнять в клинике, и при этом ISI, KSS, DSST, RTI, проводили в предварительно определенные временные точки на протяжении дневных часов.
После завершения периода лечения все субъекты получали плацебо в одностороннем слепом режиме в последние 2 ночи, проведенные в клинике (дни 16-17). В эти 2 ночи регистрацию 8-часовых PSG начинали в то же самое время отходу ко сну, как и при записи в дни 14 и 15 для исследования возникновения повторного возникновения бессонницы (периода оценки повторного возникновения бессонницы).
В течение периода лечения образцы крови для определения концентраций соединения A в плазме крови отбирали за 30 минут до введения лекарственного средства каждую ночь (за исключением дня 1) в клинике и на протяжении 1 часа после утреннего пробуждения после каждой ночи, проведенной в клинике. Концентрации соединения A в плазме крови измеряли с использованием валидированной методики жидкостной хроматографии-тандемной масс-спектрометрии (LC-MS/MS).
KSS использовали для измерения остаточных эффектов на следующий день в предварительно определенных временных точках. В данном тесте субъекты проводили оценку своей сонливости с использованием KSS 9-балльной вербальной шкалы. Категории и показатели в баллах варьировали от "крайне высокой концентрации внимания" (балл=l), "высокой концентрации внимания" (3), "состояния между высокой концентрацией внимания и сонливостью" (5), "сонного-но без затруднения пребывающего в состоянии бодрствования" (7) до "чрезвычайно сонного-борющегося со сном" (9). Ключевым параметром эффективности в KSS являлся балл от 1 до 9.
Все статические тесты основывались на 5% уровне значимости за исключением байесовских методик, которые использовали для основной конечной точки. Подробности статистических методик и анализов определены в плане статистического анализа (SAP) и в основной части отчета о клиническом исследовании.
Множество для анализа безопасности представляло собой группу субъектов, которые получали по меньшей мере 1 дозу исследуемого лекарственного средства и которые проходили по меньшей мере 1 исследование безопасности после введения лекарственного средства. Множество полного анализа (FAS) представляло собой группу рандомизированных субъектов, которые получали по меньшей мере 1 дозу исследуемого лекарственного средства и характеризовались по меньшей мере 1 измерением показателя основной эффективности после введения лекарственного средства. Множество для анализа PK представляло собой группу рандомизированных субъектов, которые получали по меньшей мере 1 дозу соединения A и характеризовались по меньшей мере 1 поддающейся количественному определению концентрации соединения A. Множество для анализа PD представляло собой группу субъектов, которые характеризовались данными по PD, достаточными для получения по меньшей мере 1 PD параметра. Множество для анализа PK/PD представляло собой группу рандомизированных субъектов, которые получали по меньшей мере 1 дозу соединения A или плацебо и характеризовались по меньшей мере 1 поддающейся количественному определению концентрацией соединения A (активные субъекты) и по меньшей мере 1 оценкой PD после введения препарата.
Отличие от плацебо по меньшей мере на 6% в изменении от исходного уровня среднего значения SE в день 1 и день 2 считали минимальным клинически значимым различием (CSD).
Каждую дозу оценивали по остаточной сонливости на следующий день с использованием KSS. Разницу среднего значения по сравнению с изменением от исходного уровня в KSS через 1 час после времени пробуждения в день 2 и день 3 менее 4 единиц включали в функцию полезности. Полагали, что доза соединения A имеет приемлемый балл KSS в день 15 и день 16, если разница среднего значения изменения от исходного уровня в KSS через 1 час после пробуждения в день 15 и день 16 при этой дозе по отношению к плацебо составляла менее 4 единиц. Необязательно предпочтительный балл KSS для дня 15 и дня 16 определяли как нижнюю границу 90% доверительного интервала (CI), составляющую менее 4 единиц (разница среднего значения изменения от исходного уровня в KSS через 1 час после пробуждения при этой дозе по отношению к плацебо).
Функция полезности: полезность при дозе представляла собой функцию зависимости как для SE, так и для KSS, полученную путем определения 1-размерного компонента для каждой конечной точки с последующим их мультипликативным объединением. Надлежащую полезность определяли как Pr(полезность >1).
Доза с максимальной полезностью (dUmax): доза, при которой получали максимальный показатель полезности, т.е. наилучшая
комбинация эффективности и остаточной сонливости, исходя из указанной выше полезности.
Множество для анализа PK/PD использовали для оценки взаимосвязей между концентрациями соединения A и выбранными PD параметрами. Зависимости между воздействием соединения A и определенными PD конечными точками (например, KSS, DSST, RTI) рассматривали графически, и их в дальнейшем можно было моделировать в виде соповкупности PK/PD. Взаимосвязь между концентрациями соединения A в плазме крови перед введением лекарственного средства (непосредственно) и в течение 1 часа после утреннего пробуждения и определенными PD параметрами анализировали с использованием Nonmem версии 7.2 или более поздней.
Результаты
Сводные статистические данные по изменению от исходного уровня в SE представлены в таблице 5. Все соединения, все дозы являлись статистически значимыми против плацебо в виде изменения среднего значения от исходного уровня в дни 1/2. Дозы соединения A в 2,5 мг и выше являлись статистически значимыми против плацебо в виде изменения среднего значения от исходного уровня в дни 14/15. Статистические данные об увеличении или уменьшении в SE в виде изменения среднего значения от исходного уровня в дни 1/2 по сравнению с днями 14/15 отсутствовали, свидетельствуя о том, что потери лечебного эффекта не было.
В таблице 5 "исходный уровень" определяли как среднее значение скрининга по PSG 1 и 2 на протяжении рандомизации от дня -9 до дня -3. "Различие средних значений, определенное по методике наименьших квадратов LS" относится к различию между средними значениями LS для плацебо и для каждой дозы соединения A, "95% CI" означает 95% CI разл. средних значений LS. "p-величину" анализировали с использованием ковариационного анализа (ANCOVA), при этом в качестве ковариаты выступал исходный уровень.
Таблица 5. Сводные статистические данные по изменению от исходного уровня в SE (%)
|
Сводные статистические данные по изменению от исходного уровня в KSS через 1 час после пробуждения представлены в таблице 6. Различия средних значений LS между плацебо и соединением А в дозах от 1 мг до 15 мг не были статистически значимыми по отношению к среднему значению для дней 2/3. Только различие средних значений LS между плацебо и 25 мг соединения A было статистически значимым по отношению к среднему значению для дней 2/3 (различие средних значений LS 0,47; P=0,0393), указывая на то, что субъекты оценивали себя хуже, чем субъекты, принимавшие плацебо. Данный результат был аналогичным среднему значению для дней 15/16 через 1 час после пробуждения. Исследования через 2 часа после пробуждения для обоих средних значений для дней 2/3 показали статистическую значимость для доз соединения А в 15 мг и 25 мг, тогда как среднее значение для дней 15/16 было статистически значимым для 25 мг соединения A. Не наблюдали статистически значимых различий во временных точках через 15 мин после пробуждения.
В таблице 6 "исходный уровень" определяли как согласованную во времени величину в день 1. "Различие средних значений, определенное по методике наименьших квадратов LS" относится к различию между средними значениями LS для плацебо и для каждой дозы соединения A, "95% CI" означает 95% CI разл. средних значений LS. "p-величину" анализировали с использованием ковариационного анализа (ANCOVA), при этом в качестве ковариаты выступал исходный уровень.
Таблица 6. Сводные статистические данные по изменению от исходного уровня в KSS через 1 час после пробуждения
|
Сводные статистические данные по изменению от исходного уровня в LPS представлены в таблице 7. Вследствие отличного от нормального распределения данные подвергали логарифмическому преобразованию и анализировали с использованием, как уже было указано, ANCOVA.
Соотношение среднего геометрического значения для плацебо и 1 мг соединения A не было статистически значимым для изменения среднего значения от исходного уровня в дни 1/2. Соотношения средних геометрических значений для плацебо и всех других активных доз соединения A продемонстрировали доказательство статистической значимости для изменения среднего значения от исходного уровня в дни 1/2. Аналогичные результаты показаны для изменения среднего значения от исходного уровня в дни 14/15.
10 мг соединения A продемонстрировали статистическое различие в изменении среднего значения от исходного уровня в дни 1/2 по сравнению с днями 14/15 днями, показывая дальнейшее улучшение в LPS с течением времени. При использовании всех других доз, за исключением 10 мг соединения A, другие статистических данных в отношении увеличения или уменьшения в LPS для изменения среднего значения от исходного уровня в дни 1/2 по сравнению с днями 14/15 отсутствовали.
В таблице 7 "исходный уровень" определяли как среднее значение, полученное в ходе скринингового PSG 1 и 2 в течение от -9 до -3 дней рандомизации. "p-величину" анализировали с использованием ковариационного анализа (ANCOVA), при этом в качестве ковариаты выступал исходный уровень.
Таблица 7. Сводные статистические данные по изменению от исходного уровня в LPS (мин.)
|
Сводные статистические данные по изменению от исходного уровня в WASO представлены в таблице 8. Все дозы соединения A в 10 мг и выше являлись статистически значимыми в отношении плацебо для изменения среднего значения от исходного уровня в дни 1/2. Дозы соединения A в 15 мг и выше являлись статистически значимыми в отношении плацебо для изменения среднего значения от исходного уровня в дни 14/15. Статистические данные об увеличении или уменьшении в WASO между изменением среднего значения от исходного уровня в дни 1/2 по сравнению с днями 14/15 отсутствовали.
В таблице 8 "исходный уровень" определяли как среднее значение, полученное в ходе скринингового PSG 1 и 2 на протяжении рандомизации от дня -9 до дня -3. "Различие средних значений, определенное по методике наименьших квадратов LS" относится к различию между средними значениями LS для плацебо и для каждой дозы соединения A, "95% CI" означает 95% CI разл. средних значений LS. "p-величину" анализировали с использованием ковариационного анализа (ANCOVA), при этом в качестве ковариаты выступал исходный уровень.
Таблица 8. Сводные статистические данные по изменению от исходного уровня в WASO (мин.)
|
PK соединения A наилучшим образом описана с помощью 2-компартментной модели с элиминацией из центральной камеры. Кажущийся клиренс соединения A не зависел от дозы и времени, свидетельствуя о линейности в PK. Измерения PD эффектов соединения A включали KSS, RTI, DSST, WFB (RTI, быструю обработку зрительной информации [RVP] и объем зрительно-пространственной памяти [SSP]), POMS, уровни мелатонина и DLMO. В связи с высокой вариабельностью и отличным от нормального распределением в изменении от исходного уровня LPS, было невозможно надежно смоделировать зависимость концентрация-ответ между PK параметрами и LPS для соединения A. Тем не менее, более высокие концентрации соединения A в плазме крови связаны с более значительным уменьшением в LPS до примерно 10 нг/мл. Данный результат согласовывался с результатами эффективности, где LPS уменьшалась при дозах в 2,5 мг и выше. Выше данной концентрации зависимость, по видимому, достигала предельного распределения, указывая на то, что явного дополнительного преимущества от более высоких концентраций соединения A в отношении возникновения сна не было. При моделировании данные WASO наилучшим образом были описаны с помощью линейно-логарифмических взаимосвязей с максимальной наблюдаемой концентрацией (Сmax). Зависимость воздействие-ответ для WASO показала линейно-логарифмическую взаимосвязь с Сmax таким образом, что более высокие концентрации соединения A при Сmax ассоциировались с уменьшения в WASO в большей степени. PK/PD анализы для исследований остаточной сонливости на следующий день (KSS, DSST и RTI) не показали какой-либо явной взаимосвязи с концентрациями соединения A в плазме крови, согласованными во времени. Однако субъекты, у которых концентрации соединения A в плазме крови были выше, чем 20 нг/мл через 1 час после пробуждения, имели незначительно большее увеличение по KSS и повышенный уровень возникновения AE (побочных явлений) в виде сонливости. Данной концентрации, как предполагается, достигнет большинство субъектов, получающих дозы более 10 мг.
Пример 5 (состав)
Капсулы, которые использовали в примерах 1, 2 и 3, представляют собой капсулы размера 2 на основе гипромеллозы, каждая из которых содержала 1 мг, 2,5 мг, 10 мг или 50 мг лекарственного вещества, представляющего собой соединение A. Капсулы с 25 мг соединения A, которые представляют собой капсул размера 2 на основе гипромеллозы, содержащие 25 мг лекарственного вещества, представляющего собой соединение A, также получали только для оценки растворения. Плацебо представляло собой капсулу размера 2 на основе гипромеллозы, содержащую 10 мг микрокристаллической целлюлозы.
Компоненты и композиции таблеток, используемых для примеров 3 и 4, показаны в таблице 9.
Таблица 9. Компоненты и композиции таблеток с соединением A
|
Для получения соединения А в таблетках, покрытых оболочкой, использовали стандартный способ влажного гранулирования. Таблетки соединения A, покрытые оболочкой, получали путем смешивания, влажного гранулирования, высушивания, распределения по размеру, смазывания, таблетирования и проведения процесса нанесения пленки. Соединение A, моногидрат лактозы и гидроксипропилцеллюлозу с низкой степенью замещения смешивали с использованием смесителя. Смесь подвергали влажному гранулированию с использованием смесителя, с постепенным добавлением соответствующего количества водного раствора гидроксипропилцеллюлозы. Влажные гранулы сушили с использованием сушилки. Высушенные гранулы пропускали через 1,0 мм сито с использованием ситовой мельницы. Гидроксипропилцеллюлозу с низкой степенью замещения и стеарат магния отвешивали в зависимости от выхода гранул. Гранулы, гидроксипропилцеллюлозу с низкой степенью замещения и стеарат магния вместе пластифицировали в мешалке. Пластифицированные гранулы, эквивалентные одной таблетке, подвергали прессованию в двояковыпуклые таблетки с использованием таблеточного пресса. Сердцевины таблеток покрывали путем нанесения распылением водной суспензии Opadry RED с использованием машины для нанесения покрытий.
Пример с испытанием
Испытание растворения капсул и таблеток соединения A, полученных в примере 5, осуществляли с использованием аппарата 2 (аппарата с лопастной мешалкой) согласно JP 6.10, USP <711> и Европейской фармакопее. 2.9.3. Капсулы и таблетки подвергали испытанию в 900 мл 0,1 моль/л хлористоводородной кислоты, содержащей 0,5% полисорбата 80, при скорости вращения лопастей 75 об./мин. (Условие Ι). Кроме того, таблетки подвергали испытанию в 900 мл 0,1 моль/л хлористоводородной кислоты при скорости вращения лопастей 50 об./мин. (Условие ΙΙ). Грузило сферической формы из проволоки использовали в испытаниях капсул. Аликвоты сред пропускали через фильтр (размер пор: 0,45 мкм) в заданные временные точки для получения образцов раствора. Стандартные растворы готовили с тем, чтобы концентрации соединения A соответствовали таким концентрациям в образцах растворов при номинальном уровне концентрации. Количество высвобожденного соединения A определяли хроматографически при сравнении со стандартным раствором. Условия растворения и условия HPLC представлены в таблице 10. Испытание проводили с 6 капсулами/таблетками, и их среднее значение определяли в каждом случае. Профили растворения капсул с 1 мг и 50 мг соединения A, полученные в условии Ι, представлены на фиг. 1 и в таблице 11. Профили растворения таблеток с 1 мг, 2,5 мг, 5 мг, 10 мг и 25 мг соединения A, полученные в условии ΙΙ, представлены на фиг. 2 и в таблице 12. Сравнительные профили растворения для капсул и таблеток с соединением A, полученные в условии Ι, представлены на фиг. 3 и в таблице 13. Различия между капсулами и таблетками наблюдали в профилях растворения, которые были вызваны временем задержки, затрачиваемой на разрушение капсул.
Таблица 10. Условия растворения и условия HPLC
|
Таблица 11. Показатели растворения капсул с 1 мг и 50 мг соединения A, полученные в условии I
|
Таблица 12. Показатели растворения таблеток с 1 мг, 2,5 мг, 5 мг, 10 мг и 25 мг соединения A, полученные в условии II
|
Таблица 13. Показатели растворения капсул и таблеток с соединением A, полученные в условии I
|
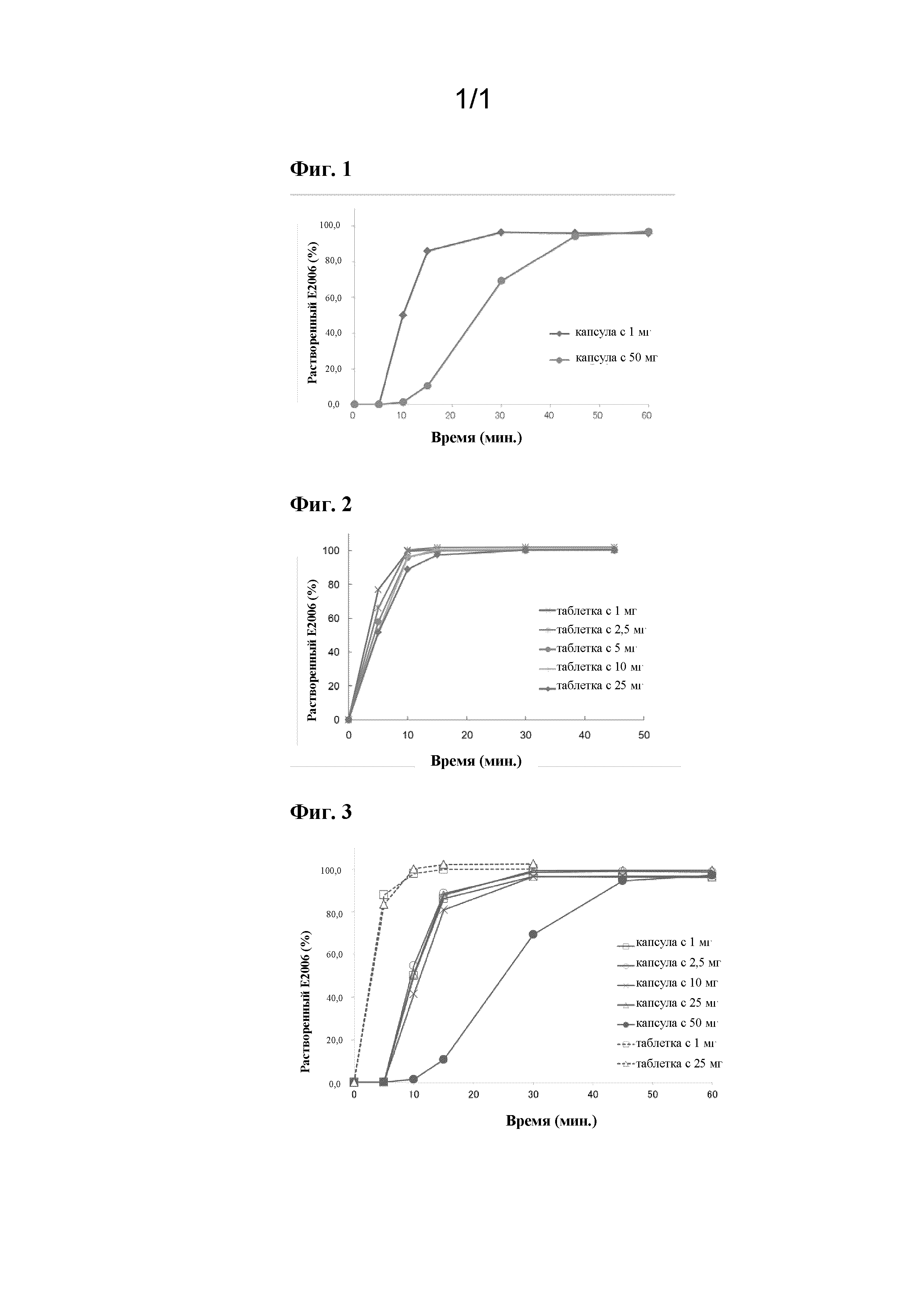
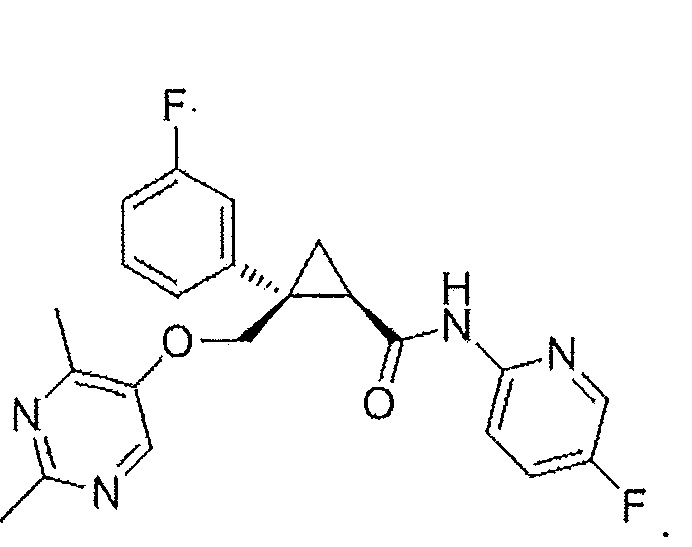
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства
Фармацевтическая композиция на основе конденсированного производного аминодигидротиазина
Способы лечения расстройств циркадного ритма сна
Способы лечения расстройств циркадного ритма сна

