Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ 4-АМИНО-ИМИДАЗОХИНОЛИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ АГОНИСТАМИ TLR7 И/ИЛИ TLR8
Вид РИД
Изобретение
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым 4-амино-имидазохинолиновым производным, обладающим фармацевтической активностью, их производству, фармацевтическим композициям их содержащим и их потенциальному применению в качестве лекарственных средств.
В частности, настоящее изобретение относится к соединениям формулы
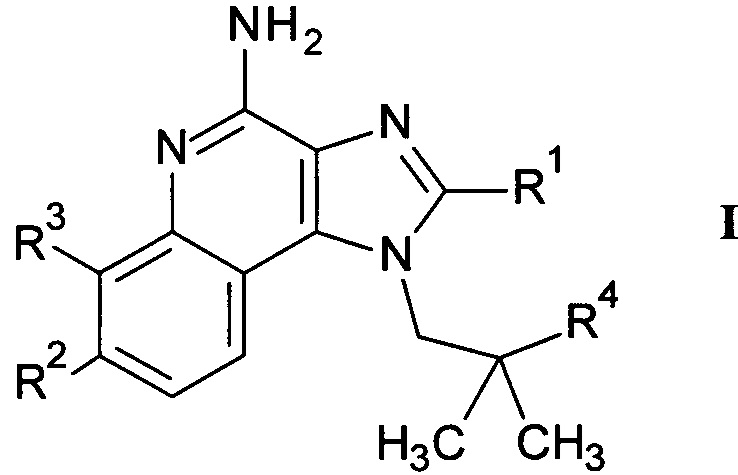
где R1-R4 и X являются такими, как описано далее, или их фармацевтически приемлемым солям.
Указанные соединения являются агонистами TLR. В частности, соединения представляют собой агонисты TLR7 и/или TLR8 и более конкретно агонисты обоих рецепторов TLR7 и TLR8. Таким образом, он могут применяться для лечения и предотвращения рака, аутоиммунных и инфекционных заболеваний. Например, они могут быть полезны при вакцинации против заболеваний, таких как рак, аутоиммунные или инфекционные заболевания.
Toll-подобные рецепторы (TLR) являются семейством трансмембранных белков, которые экспрессируются на клетках иммунной системы, таких как дендритные клетки, макрофаги, моноциты, Т-клетки, В-клетки, NK клетки и тучные клетки, а также на эндотелиальных и эпителиальных клетках (Kawai et al., Immunity, 2011, 34, 637-650, Kawai et al., Nat. Immunol., 2010, 11, 373-384). Рецепторы TLR, которые распознают бактриальные компоненты и компоненты грибов, экспрессируются на клеточной поверхности (т.е. TLR1, 2, 4, 5 и 6), при этом другие рецепторы TLR, которые распознают вирусные или микробные нуклеиновые кислоты, такие как TLR3, 7, 8 и 9 локализуются в эндолизосомальной/фагосомально мембране (Henessy et al. Nat. Rev. Drug Discovery 2010, 9, 293-307). Активация TLR приводит к индукции и высвобождению провоспалительных цитокинов, со специфичной последовательностью активации и ответом, зависящих от конкртеного рецептора TLR и типа клетки. TLR7 и TLR8 оба экспрессируются в моноцитах и макрофагах, при этом TLR7 также сильно экспрессируется в плазмацитоидных дендритных клетках, a TLR8 в миелоидных дендритных клетках и тучных клетках. Оба рецептора активируются с помощью оцРНК и их активация стимулирует продукцию таких цитокинов как ИЛ-6, ИЛ-12, ФНО-α и ИФН-γ, а также дополнительных ко-стимуляторных молекул и хемокиновых рецепторов. В зависимости от типа клеток, интерфероны типа I, ИФНα (из плазмацитоидных дендритных клеток) и ИФНβ, также продуцируются клетками после активации агонистами TLR7/8 (Uematsu et al., J. Biol. Chem., 2007, 282, 15319-15323).
Низкомолекулярные агонисты обоих рецепторов TLR7 и TLR8, а также аналоги, модифицированные для применения в качестве адъювант-вакцин или конъюгатов были разработаны в множестве патентов (например, WO 1992015582, US 2003187016, WO 2005076783, WO 2007024612, WO 2009111337, WO 2010093436, WO 2011017611, WO 2011068233, WO 2011139348, WO 2012066336, WO 2012167081, WO 2013033345, WO 2013067597, WO 2013166110, и US 2013202629). Клинический опыт был получен с использованием только агонистов TLR7. Некоторое количество ранних соединений продемонстрировали противовирусные и противораковые свойства. Например, TLR7 агонист имихимод (ALDARA™) был одобрен Управлением по контролю за продуктами и лекарствами США в качестве местного средства для лечения остроконечных бородавок, поверхностной базальноклдеточной карциномы и солнечного кератоза. Однако системное введение ранних агонистов TLR7, таких как резиквимод было отклонено ввиду недопустимой кардиотоксичности, наблюдаемой после общей стимуляции хемокинов при терапевтических дозах (Holldack, Drug Discovery Today, 2013, 1-4). Знания об агонистах TLR8 менее доступны и главным образом сводятся к данным ранних смешанных агонистов TLR7/8, таких как резиквимод и, по последним данным, соединениям, описанным компанией VentiRX Pharmaceuticals (т.е. WO 2010054215, WO 2012045090). В настоящее время существует необходимость в дополнительных низкомолекулярных агонистах TLR7 и TLR8, особенно в обладающих улучшенной эффективностью.
Настоящее изобретение направлено на 1Н-имидазо[4,5-с]хинолин-4-амин-2-метилпропан-2-илокси производные с улучшенной клеточной эффективностью по сравнению с известными агонистами TLR7 и/или TLR8 этого типа для применения при лечении рака, предпочтительно солидных опухолей и лимфом, и для другого применения, включая лечение некоторых кожных состояний или заболеваний, таких как атопический дерматит, лечения инфекционных заболеваний, предпочтительно вирусных заболеваний, и для применения в качестве адъювантов в вакцинах, приготовленных для применения в противораковой терапии или посредством десенсибилизации рецепторов продолжительной стимуляцией при лечении аутоиммунных заболеваний.
В частности, в настоящем изобретении раскрыты 1Н-имидазо[4,5-с]хинолин-4-амин-2-метилпропан-2-илокси производные, которые дериватизированы непосредственно по третичному спирту аминоэтиловым или глициновым остатком. Вследствие слабой реактивности третичного спирта эти производные не получали ранее в процессе синтеза. Неожиданно, эти новые соединения показали высокую клеточную эффективность в отношении TLR7, которая сравнима или даже выше, чем у резиквимода самого по себе, при том, что ближайшие аналоги, которые были описаны ранее, такие как 1-(2-(2-Аминоэтокси)этил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин (Пример 69 из US 20030187016) не показали необходимой активности. В дополнение и также неожиданно описанные 1Н-имидазо[4,5-с]хинолин-4-амин-2-метилпропан-2-илокси производные являются сильными агонистами рецепторов TLR8, с эффективностью, сравнимой или даже более высокой, чем у агонистов TLR8 из других классов химических веществ и более лучше, чем у резиквимода самого по себе. Так, соединения настоящего изобретения соответствуют необходимости активации обоих рецепторов TLR7 и TLR8 с улучшенной эффективностью.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к 4-амино-имидазохинолиновым производным формулы
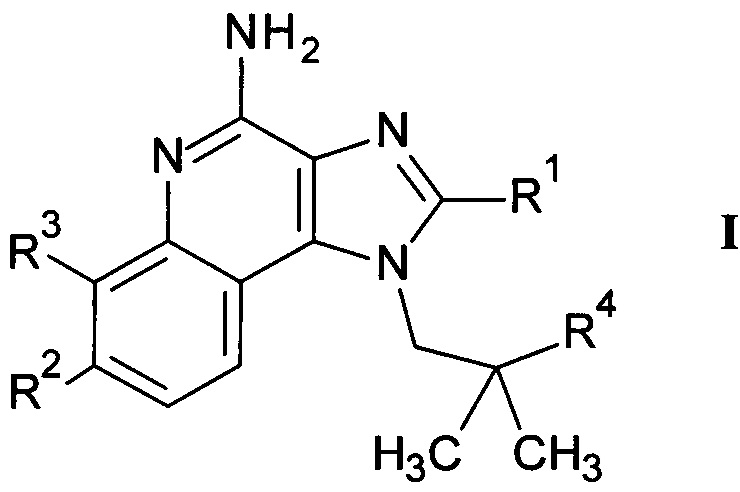
где
R1 представляет собой С1-7-алкил или С1-7-алкокси-С1-7-алкил;
R2 выбран из группы, состоящей из водорода, галогена, гидроксила, гидрокси-С1-7-алкила, алкокси-С1-7-алкила, карбоксила, карбоксил-С1-7-алкила, карбоксил-С2-7-алкенила, аминокарбонил-С1-7-алкила, аминокарбонил-С2-7-алкенила, С1-7-алкиламино-карбонил-С1-7-алкила, С1-7-алкиламино-карбонил-С1-7-алкенила, С1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкоксикарбонил-С2-7-алкенила, С1-7-алкил-сульфонил-С1-7-алкила, сульфамоил-С1-7-алкила, С1-7-алкил-сульфамоил-С1-7-алкила,
фенила, указанный фенил является незамещенным или замещенным одной, двумя или тремя группами, выбранными из группы, состоящей из С1-7-алкила, C1-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, C1-7-алкокси, циано, карбоксила, C1-7-алкоксикарбонила, C1-7-алкоксикарбонил-С1-7-алкила, C1-7-алкилсульфонила, гидрокси-C1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-С1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, C1-7-алкиламино, ди-С1-7-алкиламино и нитро, и
фенокси, указанная феноксигруппа является незамещенной или замещенной одной, двумя или тремя группами, выбранными из группы, состоящей из C1-7-алкила, С1-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, C1-7-алкокси, циано, карбоксила, C1-7-алкоксикарбонила, С1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкил-сульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-C1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, C1-7-алкиламино, ди-С1-7-алкиламино и нитро;
R3 представляет собой водород или галоген;
R4 выбран из группы, состоящей из
-O-(CH2)m-NHR5, и
-O-(CO)-(CH2)n-NHR6,
где
m выбран из 1, 2 или 3,
n выбран из 1 или 2,
R5 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, C1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, карбоксил-С1-7-алкила и C1-7-алкоксикарбонил-амино-С1-7-алкил-карбонила, и
R6 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, C1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, карбоксил-С1-7-алкила и C1-7-алкоксикарбонил-амино-С1-7-алкил-карбонила,
или их фармацевтически приемлемым солям.
Настоящее изобретение также относится к способу производства соединений формулы I.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение формулы I, как описано выше и фармацевтически приемлемый носитель и/или адъювант.
Дополнительным аспектом настоящего изобретения является применение соединений формулы I в качестве терапевтически активных веществ для лечения заболеваний, которые могут быть опосредованы агонистами TLR, в частности агонистами TLR7 и/или TLR8, более конкретно рецепторов TLR7 и TLR8. Настоящее изобретение, таким образом, относится к способу лечения заболеваний, которые могут быть опосредованы агонистами TLR, таких как, например, рак и аутоиммунные или инфекционные заболевания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иначе, все технические и научные термины, используемые в настоящем изобретении, имеют тот же смысл, как обычно понимается квалифицированным специалистом в области техники, к которой относится данное изобретение. Кроме того, следующие далее определения приведены для иллюстрации и определения значения и объема различных терминов, используемых для описания изобретения.
Номенклатура, используемая в настоящем изобретении, основана на систематической номенклатуре ИЮПАК, если не указано иное.
Термин "соединение (соединения) данного изобретения» и «соединение (соединения) по настоящему изобретению" относится к соединениям формулы I и их сольватам или солям (например, фармацевтически приемлемые соли).
Термин "заместитель" обозначает атом или группу атомов заменяющих атом водорода на исходной молекуле.
Термин "галоген" означает фтор, хлор, бром и йод, фтор, при этом хлор и бром представляют особый интерес. Более конкретно, галоген относится к фтору и хлору.
Термин "алкил", отдельно или в комбинации с другими группами, относится к разветвленному или прямоцепочечному одновалентному насыщенному алифатическому углеводородному радикалу из от одного до двадцати атомов углерода, в частности от одного до шестнадцати атомов углерода, более конкретно одного до десяти атомов углерода. Термин "С1-10-алкил" относится к разветвленному или прямоцепочечному одновалентному насыщенному алифатическому углеводородному радикалу из от одного до десяти атомов углерода, такому как, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, 1,1,3,3-тетраметил-бутил и т.п. Более конкретно, термин "алкил" включает в себя также низшие алкильные группы, как описано ниже.
Термин "низший алкил" или "C1-7-алкил", отдельно или в комбинации, означает прямоцепочечную или разветвленную алкильную группу с 1-7 атомами углерода, в частности, прямоцепочечную или разветвленную алкильную группу с 1-6 атомами углерода, и более конкретно прямоцепочечную или разветвленную алкильную группу с 1-4 атомами углерода. Примерами неразветвленной и разветвленной С1-7 алкильной группы являются метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изомерные пентилы, изомерные гексилы и изомерные гептилы, в частности метил и этил.
Термин "низший алкенил" или "С2-7-алкенил" означает прямоцепочечный или разветвленный углеводородный остаток, содержащий олефиновую связь и от 2 до 7, в частности от 3 до 6, предпочтительно от 3 до 4 атомов углерода. Примерами алкенильных групп являются этенил, 1-пропенил, 2-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил и изобутенил, в частности, этенил.
Термин "циклоалкил" или "С3-7-циклоалкил" означает насыщенную моноциклическую углеводородную группу, содержащую от 3 до 7 атомов углерода, такую как циклопропил, циклобутил, циклопентил, циклогексил или циклогептил, более конкретно циклопропил. Кроме того, термин «циклоалкил» включает в себя также бициклические углеводородные группы, содержащие от 3 до 10 атомов углерода. Бициклический означает циклоалкильную группу, состоящую из двух насыщенных карбоциклов, имеющих один или более общих атомов углерода. Примерами бициклических циклоалкилов являются [2,2,1]гептанил или бицикло[2.2.2]октанил.
Термин "низший алкокси" или "С1-7-алкокси" относится к группе R'-O-, где R' представляет собой низший алкил, а термин "низший алкил" имеет значение, данное ранее. Примерами низших алкоксигрупп являются метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси, в частности метокси.
Термин "низший алкоксиалкил" или "С1-7-алкокси-С1-7-алкил" относится к низшим алкильным группам, как определено выше, где по крайней мере один из атомов водорода низшей алкильной группы замещен низшей алкокси группой. Среди низших алкоксиалкильных групп особый интерес представляют метоксиметил, 2-метоксиэтил и 2-этоксиэтил, с наиболее предпочтительной 2-этоксиэтильной группой.
Термин гидрокси или гидроксил означает группу -ОН.
Термин "низший гидроксиалкил" или "гидрокси-С1-7-алкил" относится к низшим алкильным группам, как определено выше, где по крайней мере один из атомов водорода низшей алкильной группы замещен гидроксильной группой. Среди особо интересных низших гидроксиалкильных групп находятся гидроксиметил или гидроксиэтил.
Термин "низший галогеналкил" или "галоген-С1-7-алкил" относится к низшим алкильным группам, как определено выше, где по крайней мере один из атомов водорода низшей алкильной группы замещен атомом галогена, в частности, фтором или хлором, особо фтором. Среди низших галогеналкильных групп особый интерес представляют трифторметил, дифторметил, трифторэтил, 2,2-дифторэтил, фторметил и хлорметил, и предпочтительным является трифторметил.
Термин "низший галогеналкокси" или "галоген-C1-7-алкокси" относится к низшим алкоксигруппам, как определено выше, где по крайней мере один из атомов водорода низшей алкоксигруппы заменен на атом галогена, в частности, фтор или хлор, особо фтор. Среди низших галогеналкоксигрупп особый интерес представляют трифторметокси, дифторметокси, фторметокси и хлорметокси, более конкретно трифторметокси.
Термин "карбоксил" означает группу -СООН.
Термин "низший карбоксилалкил" или "карбоксил-С1-7-алкил" относится к низшим алкильным группам, как определено выше, где по крайней мере один из атомов водорода низшей алкильной группы замещен карбоксильной группой. Среди низших карбоксилалкильных групп особый интерес представляют карбоксилметил (-СН2-СООН) и карбоксилэтил (-СН2-СН2-СООН).
Термин "низший алкоксикарбонил" или "C1-7-алкоксикарбонил" относится к группе-COOR, где R представляет собой низший алкил, а термин "низший алкил" имеет данное ранее значение. Особо интересными низшими алкоксикарбонильными группами являются этоксикарбонил или метоксикарбонил.
Термин "низший алкоксикарбонилалкил" или "С1-7-алкоксикарбонил-С1-7-алкил" означает низшие алкильные группы, как определено выше, где один из атомов водорода низшей алкильной группы замещен С1-7-алкоксикарбонилом. В частности низшей алкоксикарбонилалкильной группой является -СН2-СООСН3.
Термин "низший алкилкарбонил" или "С1-7-алкилкарбонил" означает группу -C(O)-R, где R представляет собой низшую алкильную группу, как определено выше. Низшей алкилкарбонильной группой особого интереса является метилкарбонил или ацетил.
Термин "низший алкоксикарбонилалкил" или "С1-7-алкоксикарбонил-С1-7-алкил" означает низшие алкильные группы, как определено выше, где по меньшей мере один из атомов водорода низшей алкильной группы замещен C1-7-алкоксикарбонилом. В частности низшей алкоксикарбонилалкильной группой является -СН2-СООС2Н5.
Термин "низший алкоксикарбонилалкенил" или "С1-7-алкоксикарбонил-С2-7-алкенил" относится к низшим алкенильным группам, как определено выше, где по меньшей мере один из атомов водорода низшей алкенильной группы заменен на низшую алкоксикарбонильную группу. Среди наиболее предпочтительных низших алкоксикарбонил-алкенильных групп находится -(СН2)2-СООС2Н5.
Термин "низший алкилсульфонил" или "С1-7-алкилсульфонил" означает группу -S(O)2-R, где R представляет собой низшую алкильную группу, как определено выше. Низшей алкилсульфонильной группой особого интереса является метилсульфонил.
Термин "низший алкилсульфонилалкил" или "С1-7-алкилсульфонил-C1-7-аллкил" означает низшие алкильные группы, как определено выше, где один из атомов водорода низшей алкильной группы заменен на С1-7-алкилсульфонил. Конкретная низшая алкилсульфонилалкильная группа представляет собой -СН2-S(O)2-CH3.
Термин "низший гидроксиалкилсульфонил" или "гидрокси-С1-7-алкилсульфонил" относится к низшим алкилсульфонильным группам, как определено выше, где по меньшей мере один из атомов водорода низшей алкилсульфонильной группы заменен на гидрокси группу. Среди наиболее предпочтительных низших гидроксиалкилсульфонилных групп находится гидроксиэтилсульфонил.
Термин "низший алкоксиалкилсульфонил" или "С1-7-алкокси-С1-7-алкилсульфонил" относится к низшим алкилсульфонильным группам, как определено выше, где по меньшей мере один из атомов водорода низшей алкилсульфонильной группы заменен на низшую алкокси группу. Среди наиболее предпочтительных низших алкоксиалкилсульфонилных групп находятся метоксиэтилсульфонил или этоксиэтилсульфонил.
Термин "низший алкоксикарбонилалкилсульфонил" или "C1-7-алкоксикарбонил-С1-7-алкил-сульфонил" относится к низшим алкилсульфонильным группам, как определено выше, где по меньшей мере один из атомов водорода низшей алкилсульфонильной группы заменен на низшую алкоксикарбонильную группу. Среди наиболее предпочтительных низших алкоксикарбонил-алкилсульфонильных групп находится -S(O)2-(CH2)2-COOCH3.
Термин "карбоксиалкилсульфонил" или "карбокси-С1-7-алкилсульфонил" относится к низшим алкилсульфонильным группам, как определено выше, где по меньшей мере один из атомов водорода низшей алкилсульфонильной группы заменен на карбоксильную группу. Среди наиболее предпочтительных низших карбоксил-алкилсульфонильных групп находится -S(O)2-(CH2)3-COOH или -S(O)2-(СН2)4-СООН.
Термин "сульфамоил" или "аминосульфонил" означает группу -S(O)2-NH2.
Термин "низший сульфамоилалкил" или "С1-7-алкил-сульфамоил" означает группу -S(O)2-NH-R, где R представляет собой низший алкил, а термин "низший алкил" имеет данное ранее значение. Примером низшей алкилсульфамоильной группы является метилсульфамоил (метиламиносульфонил).
Термин "низший сульфамоилалкил" или "сульфамоил-С1-7-алкил" означает низшую алкильную группу, как определено выше, где один из атомов водорода низшей алкильной группы заменен на группу -S(O)2-NH2.
Термин "низший алкилсульфамоилалкил" или "C1-7-алкил-сульфамоилалкил" означает низшую алкильную группу, как определено выше, где один из атомов водорода низшей алкильной группы заменен на группу -S(O)2-NH-R, где R представляет собой низший алкил, и термин "низший алкил" обладает данным ранее значением.
"Амино" относится к группе-NH2. Термин "С1-7-алкиламино" означает группу -NHR, где R представляет собой низший алкил, а термин "низший алкил" имеет данное ранее значение. Термин "ди-С1-7-алкиламино" означает группу -NRR', где R и R' являются низшими алкильными группами, как определено выше.
Термин "низший аминоалкил" или "амино-С1-7-алкил" относится к низшим алкильным группам, как определено выше, где по меньшей мере один из атомов водорода низшей алкильной группы заменен на амино группу. Среди наиболее предпочтительных низших аминоалкильных групп находятся аминометил или 2-аминоэтил.
Термин "аминокарбонил" относится к группе -CO-NH2.
Термин "низший аминоалкилкарбонил" или "амино-С1-7-алкил-карбонил" относится к группе -CO-R", где R" представляет собой низшую аминоалкильную группу, как здесь определено ранее.
Термин "низший аминокарбонилалкил" или "аминокарбонил-С1-7-алкил" означает низшие алкильные группы, как определено выше, где один из атомов водорода низшей алкильной группы замещен на аминокарбонил. Низшей аминокарбонильной группой особого интереса является -CH2-CONH2.
Термин "низший алкиламинокарбонилалкил" или "С1-7-алкил-аминокарбонил-C1-7-алкил" относится к низшей алкильной группе, как определено выше, где один из атомов водорода низшей алкильной группы заменен на группу -CONH-R, где R представляет собой низший алкил, как здесь определено ранее.
С1-7-алкоксикарбонил-амино-С1-7-алкил-карбонил
Термин "низший алкоксикарбониламиноалкилкарбонил" или "C1-7-алкоксикарбонил-амино-С1-7-алкил-карбонил" относится к низшим аминоалкилкарбонильным группам, как определено выше, где по меньшей мере один из атомов водорода амино группы заменен на низшую алкоксикарбонильную группу. Среди наиболее предпочтительных низших алкоксикарбонил-алкилсульфонильных групп находится -CO-(CH2)5-NH-COOC(CH3)3.
Термин "циано" относится к группе -CN.
Термин "нитро" относится к группе -NO2.
Термин "фенилкарбонил" означает группу -CO-Phe, где Phe обозначает возможно замещенную фенильную группу.
Термин "гетероарил", в общем, относится к ароматическому 5- или 6-членному кольцу, которое состоит из одного, двух, трех или четырех атомов, выбранных из азота, кислорода и/или серы, таким как пиридил, пиразинил, пиримидинил, 2,4-диоксо-1Н-пиримидинил, пиридазинил, 2-оксо-1,2-дигидропиридинил, пирролил, оксазолил, оксадиазолил, изоксазолил, тиадиазолил, тетразолила, пиразолил, имидазолил, фуранил, тиазолил, изотиазолил, триазолил, тетразолила, тиенил, азепинил, диазепинил. Термин "гетероарил" дополнительно относится к бициклическим ароматическим группам, содержащим от 5 до 12 кольцевых атомов, в которых один или оба кольца могут содержать один, два или три атома, выбранных из азота, кислорода или серы, таким как хинолинил, изохинолинил, циннолинил, хиназолинил, пиразоло[1,5-а]пиридил, имидазо[1,2-а]пиридил, хиноксалинил, бензофуранил, бензотиенил, бензотиазолил, бензотриазолил, индолил и индазолил. Более конкретно, "гетероарил" относится к ароматическому 6-членному кольцу, выбранному из группы, состоящей из пиридила, пиразинила, пиримидинила и пиридазинила, более конкретно пиридилу.
Термин "оксо" означает, что С-атом гетероарильного кольца может быть замещен =O, таким образом означая, что гетероарильное кольцо может содержать одну или более карбонильных (-СО-) групп.
Термин "гетероарилкарбонил" означает группу -CO-Het, где Het представляет собой возможно замещенную гетероарильную группу, как определено выше.
Термин "фармацевтически приемлемый" означает из материала, который используется при получении фармацевтической композиции, который, как правило, безопасен, нетоксичен и ни биологически, ни каким-либо другим образом не является нежелательным и является приемлемым для фармацевтического применения в ветеринарии а также для человека.
Соединения формулы I могут образовывать фармацевтически приемлемые соли. Термин "фармацевтически приемлемые соли" относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются ни биологически, ни каким-либо другим образом нежелательными. Фармацевтически приемлемые соли включают в себя как кислотно-аддитивные соли, так и соли присоединения основания. Солями являются, например, кислотно-аддитивные соли соединений формулы I с физиологически совместимыми минеральными кислотами, такими как соляная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, серная кислота, сернистая кислота или фосфорная кислота, или с органическими кислотами, такими как метансульфоновая, этансульфокислота, п-толуолсульфоновая кислота, муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, молочная кислота, трифторуксусная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, малоновая кислота, винная кислота, бензойной кислота, коричная кислота, миндальная кислота, эмбоновая кислота, янтарная кислота или салициловая кислота. Кроме того, фармацевтически приемлемые соли могут быть получены с добавлением неорганического или органического основания к свободной кислоте. Соли, полученные из неорганических оснований, включают, без ограничения, соли натрия, калия, лития, аммония, кальция, магния, цинка, меди, марганца и алюминия и т.п. Соли, полученные из органических оснований, включают, без ограничения, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как изопропиламиновые, триметиламиновые, диэтиламиновые, триэтиламиновые, трипропиламиновые, этаноламиновые, лизиновые, аргининовые, гистидиновые, кофеиновые, прокаиновые, гидрабаминовые, холиновые, бетаиновые, этилендиаминовые, глюкозаминовые, метилглюкаминовые, теоброминовые, пиперазиновые, N-этилпиперидиновые, пиперидиновые и полиаминовые смолы. Соединение формулы I также может присутствовать в виде цвиттерионов. Фармацевтически приемлемыми солями соединений формулы I особого интереса являются соли натрия или соли третичных аминов.
Соединения формулы I также могут быть сольватированы, например, гидратированы. Сольватация может быть проведена в ходе производственного процесса или может иметь место, например, вследствие гигроскопических свойств изначально безводного соединения формулы I (гидратация). Термин "фармацевтически приемлемые соли" также включает в себя физиологически приемлемые сольваты.
Термин "агонист" означает соединение, которое усиливает активность других соединений или сайтов для рецепторов, как определено, например, в Goodman и Gilman's "The Pharmacological Basis of Therapeutics, 7th ed." на с. 35, Macmillan Publ. Company, Canada, 1985. «Полный агонист" обладает полным ответом, в то время как "частичный агонист" действует меньше, чем полная активация даже тогда, когда взаимодействует со всей популяцией рецепторов. "Обратный агонист" обладает эффектом противоположным агонисту, хотя связывается с тем же рецептор-связывающим сайтом.
Термин "половины максимальной эффективной концентрации" (ЕС50) означает концентрацию конкретного соединения в плазме, необходимую для получения 50% от максимума определенного эффекта in vivo.
Термин "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое при введении субъекту, (i) лечит или предотвращает конкретное заболевание, состояние или расстройство, (ii), смягчает, улучшает или устраняет один или более симптомов конкретного заболевания, состояния или расстройства, или (iii) предотвращает или задерживает начало одного или более симптомов конкретного заболевания, состояния или расстройства, описанных в настоящем изобретении. Терапевтически эффективное количество будет варьироваться в зависимости от соединения, состояния заболевания, которое лечат, тяжести или лечения при заболевании, возраста и относительного здоровья субъекта, пути и формы введения, мнения лечащего или ветеринарного врача, и других факторов.
Более детально, настоящее изобретение относится к соединениям формулы
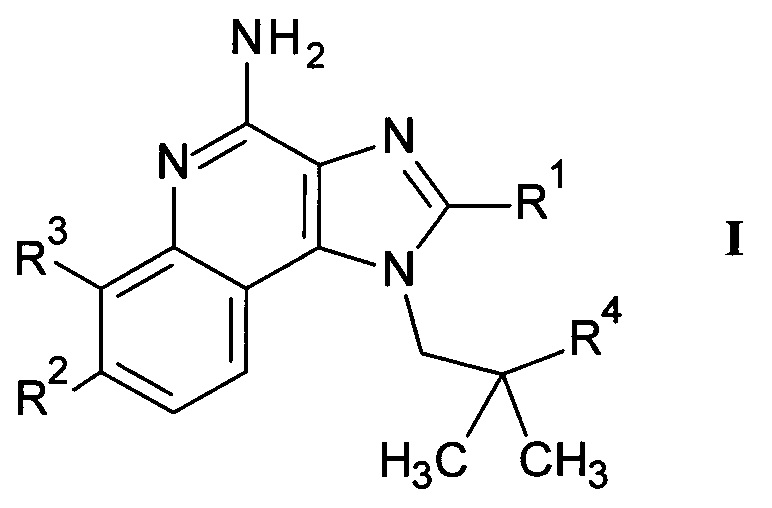
где
R1 представляет собой C1-7-алкил или С1-7-алкокси-С1-7-алкил;
R2 выбран из группы, состоящей из водорода, галогена, гидроксила, гидрокси-С1-7-алкила, алкокси-С1-7-алкила, карбоксила, карбоксил-С1-7-алкила, карбоксил-С2-7-алкенила, аминокарбонил-С1-7-алкила, аминокарбонил-С2-7-алкенила, С1-7-алкиламино-карбонил-С1-7-алкила, С1-7-алкиламино-карбонил-С2-7-алкенила, C1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкоксикарбонил-С2-7-алкенила, C1-7-алкил-сульфонил-С1-7-алкила, сульфамоил-С1-7-алкила, С1-7-алкил-сульфамоил-С1-7-алкила,
фенила, указанный фенил является незамещенным или замещенным одной, двумя или тремя группами, выбранными из группы, состоящей из C1-7-алкила, C1-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, C1-7-алкокси, циано, карбоксила, C1-7-алкоксикарбонила, C1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкилсульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-C1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, C1-7-алкиламино, ди-С1-7-алкиламино и нитро, и
фенокси, указанная феноксигруппа является незамещенной или замещенной одной, двумя или тремя группами, выбранными из группы, состоящей из С1-7-алкила, С1-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, С1-7-алкокси, циано, карбоксила, С1-7-алкоксикарбонила, С1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкил-сульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-С1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, С1-7-алкиламино, ди-С1-7-алкиламино и нитро;
R3 представляет собой водород или галоген;
R4 выбран из группы, состоящей из
-O-(CH2)m-NHR5, и
-O-(CO)-(CH2)n-NHR6,
где
m выбран из 1, 2 или 3,
n выбран из 1 или 2,
R5 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, C1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, карбоксил-С1-7-алкила и C1-7- алкоксикарбонил-амино-С1-7-алкил-карбонила, и
R6 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, С1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, карбоксил-С1-7-алкила и C1-7-алкоксикарбонил-амино-С1-7-алкил-карбонила,
или их фармацевтически приемлемым солям.
В одном аспекте настоящее изобретение относится к соединениям формулы
где
R1 представляет собой C1-7-алкил или С1-7-алкокси-С1-7-алкил;
R2 выбран из группы, состоящей из водорода, гидроксила, гидрокси-С1-7-алкила, С1-7-алкокси-С1-7-алкила, карбоксила, карбоксил-С1-7-алкила, аминокарбонил-С1-7-алкила, С1-7-алкиламино-карбонил-С1-7-алкила, алкоксикарбонил-С1-7-алкила, С1-7-алкил-сульфонил-С1-7-алкила, сульфамоил-C1-7-алкила, С1-7-алкил-сульфамоил-С1-7-алкила,
фенила, указанный фенил является незамещенным или замещенным одной, двумя или тремя группами, выбранными из группы, состоящей из C1-7-алкила, C3-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, C1-7-алкокси, циано, карбоксила, C1-7-алкоксикарбонила, C1-7-алкоксикарбонил-С1-7-алкила, C1-7-алкилсульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-С1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, C1-7-алкиламино, ди-С1-7-алкиламино и нитро, и
фенокси, указанная феноксигруппа является незамещенной или замещенной одной, двумя или тремя группами, выбранными из группы, состоящей из C1-7-алкила, C1-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, С1-7-алкокси, циано, карбоксила, С1-7-алкоксикарбонила, С1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкил-сульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-С1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, С1-7-алкиламино, ди-С1-7-алкиламино и нитро;
R3 представляет собой водород или галоген;
R4 выбран из группы, состоящей из
-O-(CH2)m-NHR5, и
-O-(CO)-(CH2)n-NHR6,
где
m выбран из 1, 2 или 3,
n выбран из 1 или 2,
R5 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, C1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, и карбоксил-С1-7-алкила, и
R6 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, С1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, и карбоксил-С1-7-алкила,
или их фармацевтически приемлемым солям.
В одном аспекте настоящее изобретение относится к соединениям формулы I, где R1 представляет собой С1-7-алкокси-С1-7-алкил. Более конкретно, R1 представляет собой этоксиэтил.
В другом аспекте, настоящее изобретение относится к соединениям формулы I, где R3 представляет собой водород.
В дополнительном аспекте настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -O-(CH2)m-NHR5 и где m выбран из 1, 2 или 3 и где R5 выбран из группы, состоящей из водорода, гидрокси-C1-7-алкила, амино-C1-7-алкила, C1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, карбоксил-С1-7-алкила и C1-7-алкоксикарбонил-амино-С1-7-галкил-карбонила. В частности, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -O-(CH2)m-NHR5 и где m выбран из 1, 2 или 3 и где R5 выбран из группы, состоящей из водорода, гидрокси-C1-7-алкила, амино-С1-7-алкила, C1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, и карбоксил-С1-7-алкила.
В частности, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -O-(CH2)m-NHR5 и где m представляет собой 2 и R5 является таким, как здесь определено ранее.
Более конкретно, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -O-(CH2)m-NHR5 и где m представляет собой 2 и где R5 выбран из группы, состоящей из водород, гидрокси-С1-7-алкила, С1-7-алкилкарбонила, гетероарилкарбонила и C1-7-алкоксикарбонил-амино-С1-7-алкил-карбонила, более конкретно, где R5 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, С1-7-алкилкарбонила и С1-7-алкоксикарбонил-амино-C1-7-алкил-карбонила. Более конкретно, настоящее изобретение относится к соединению формулы I, где R4 представляет собой -О-(CH2)m-NHR5 и где m представляет собой 2 и R5 представляет собой водород.
Настоящее изобретение также относится к соединениям формулы I, где R4 представляет собой -О-(CH2)m-NHR5 и где m выбран из 1, 2 или 3 и где R5 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, С1-7-алкилкарбонила и гетероарилкарбонила. Более конкретно, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -О-(CH2)m-NHR5 и где m выбран из 1, 2 или 3, и где R5 представляет собой водород.
В другом аспекте, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -О-(CO)-(CH2)n-NHR6, и где n выбран из 1 или 2, и где R6 выбран из группы, состоящей из водорода, гидрокси-С1-7-алкила, амино-С1-7-алкила, С1-7-алкилкарбонила, фенилкарбонила, гетероарилкарбонила, карбоксила, карбоксил-С1-7-алкила и C1-7-алкоксикарбонил-амино-С1-7-алкил-карбонила.
В частности, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -O-(CO)-(CH2)n-NHR6 и где n представляет собой 1 или 2 и где R6 является таким, как здесь определено ранее. Более конкретно, настоящее изобретение относится к соединениям формулы I, где R4 представляет собой -О-(CO)-(CH2)n-NHR6 и где n представляет собой 1, и R6 представляет собой водород.
В дополнительном аспекте настоящее изобретение также относится к соединениям формулы I, где R4 представляет собой -O-(CO)-(CH2)n-NHR6 и где n представляет собой 1 или 2, и где R6 представляет собой водород.
Настоящее изобретение также относится к соединениям формулы I, где R2 выбран из группы, состоящей из водорода, галогена, гидроксила, гидрокси-С1-7-алкила, алкокси-С1-7-алкила, карбоксила, карбоксил-С1-7-алкила, карбоксил-С2-7-алкенила, аминокарбонил-С1-7-алкила, аминокарбонил-С2-7-алкенила, C1-7-алкиламино-карбонил-С1-7-алкила, С1-7-алкиламино-карбонил-С2-7-алкенила, C1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкоксикарбонил-С2-7-алкенила, C1-7-алкил-сульфонил-С1-7-алкила, сульфамоил-С1-7-алкила и С1-7-алкил-сульфамоил-С1-7-алкила.
В другом аспекте, настоящее изобретение относится к соединениям формулы I, где R2 выбран из группы, состоящей из водорода, галогена, C1-7-алкоксикарбонил-С1-7-алкила и С1-7-алкоксикарбонил-С2-7-алкенила.
В конкретном аспекте настоящее изобретение относится к соединениям формулы I, где R2 представляет собой водород.
В другом аспекте, настоящее изобретение относится к соединениям формулы I, где R2 представляет собой фенил, указанный фенил является незамещенным или замещенным одной, двумя или тремя группами, выбранными из группы, состоящей из C1-7-алкила, С3-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, С1-7-алкокси, циано, карбоксила, С1-7-алкоксикарбонила, С1-7-алкоксикарбонил-С1-7-алкила, C1-7-алкилсульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-С1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, C1-7-алкиламино, ди-C1-7-алкиламино и нитро, или к соединениям формулы I, где R2 представляет собой фенокси, указанная феноксигруппа является незамещенной или замещенной одной, двумя или тремя группами, выбранными из группы, состоящей из C1-7-алкила, С3-7-циклоалкила, галогена, галоген-С1-7-алкила, галоген-С1-7-алкокси, гидрокси, гидрокси-С1-7-алкила, С1-7-алкокси, циано, карбоксила, С1-7-алкоксикарбонила, С1-7-алкоксикарбонил-С1-7-алкила, С1-7-алкил-сульфонила, гидрокси-С1-7-алкилсульфонила, С1-7-алкокси-С1-7-алкилсульфонила, карбоксил-С1-7-алкилсульфонила, С1-7-алкокси-карбонил-С1-7-алкилсульфонила, амино, С1-7-алкиламино, ди-С1-7-алкиламино и нитро.
Конкретными соединениями формулы I в соответствии с настоящим изобретением являются следующие:
1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-аминоацетат,
N-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этил)никотинамид,
N-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этил)ацетамид,
3-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этиламино)пропан-1-ол,
трет-бутил 6-(2-(1-(4-амино-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этиламино)-6-оксогексилкарбамат,
этил (Е)-3-[4-амино-1-[2-(2-аминоэтокси)-2-метилпропил]-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил]проп-2-еноат,
этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноат,
этил 3-(4-амино-1-(2-(2-аминоацетокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноат,
1-(2-(2-аминоэтокси)-2-метилпропил)-2-пентил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(2-(2-аминоэтокси)-2-метилпропил)-7-бромо-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин;
или их фармацевтически приемлемым солям.
В частности, настоящее изобретение относится к следующим соединениям формулы I:
1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-аминоацетат,
этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноат,
или их фармацевтически приемлемым солям.
Более конкретно, настоящее изобретение относится к соединению формулы I, которое представляет собой
1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин;
и его фармацевтически приемлемые соли.
Более конкретно, настоящее изобретение относится к соединениям формулы I, выбранным из группы, состоящей из:
1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-аминоацетат,
этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноат,
и их фармацевтически приемлемые соли.
В частности, настоящее изобретение относится к следующим солям соединений формулы I:
этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноата гидрохлориду,
1-(2-(2-аминоэтокси)-2-метилпропил)-7-бромо-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амину;
или их фармацевтически приемлемым солям.
Следует понимать, что соединения общей формулы I в настоящем изобретении могут быть дериватизированы по функциональным группам с получением производных, которые способны превращаться обратно в исходное соединение в естественных условиях. Физиологически приемлемые и метаболически лабильные производные, которые способны превращаться в исходные соединения общей формулы I в естественных условиях, также входят в объем данного изобретения.
Дополнительным аспектом настоящего изобретения является способ получения соединений формулы I, как определено выше, который содержит
а) взаимодействие соединения формулы II
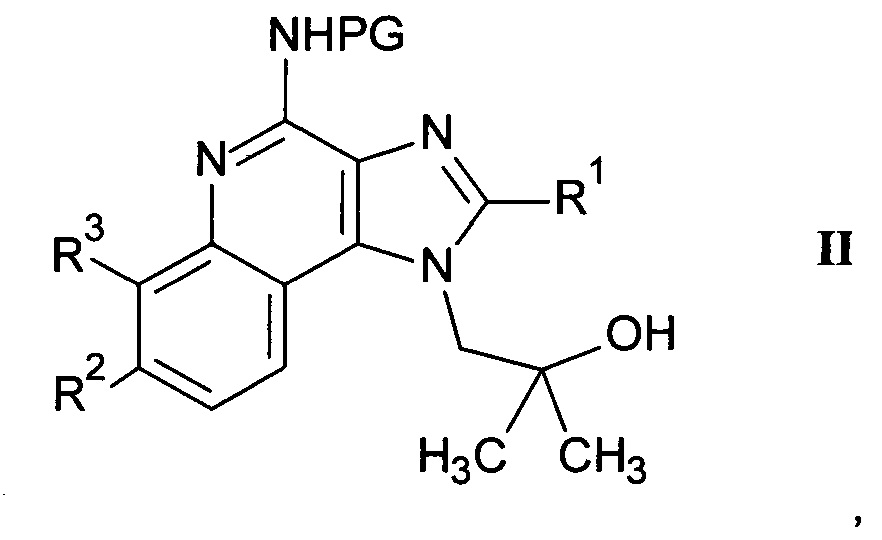
где R1, R2 и R3 являются такими, как определено здесь ранее, a PG представляет собой защитную группу, с соединением формулы III
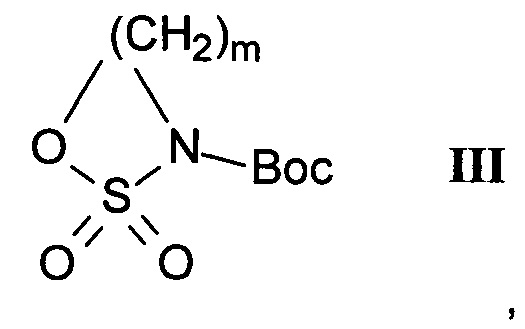
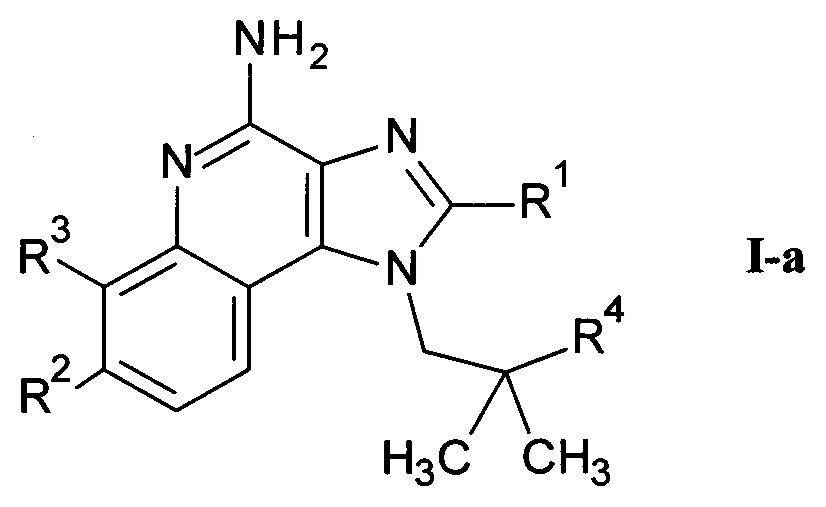
где m является таким, как здесь определено ранее, в щелочной среде и удаление защитных групп PG и Вос в кислой среде с получением соединения формулы I-a
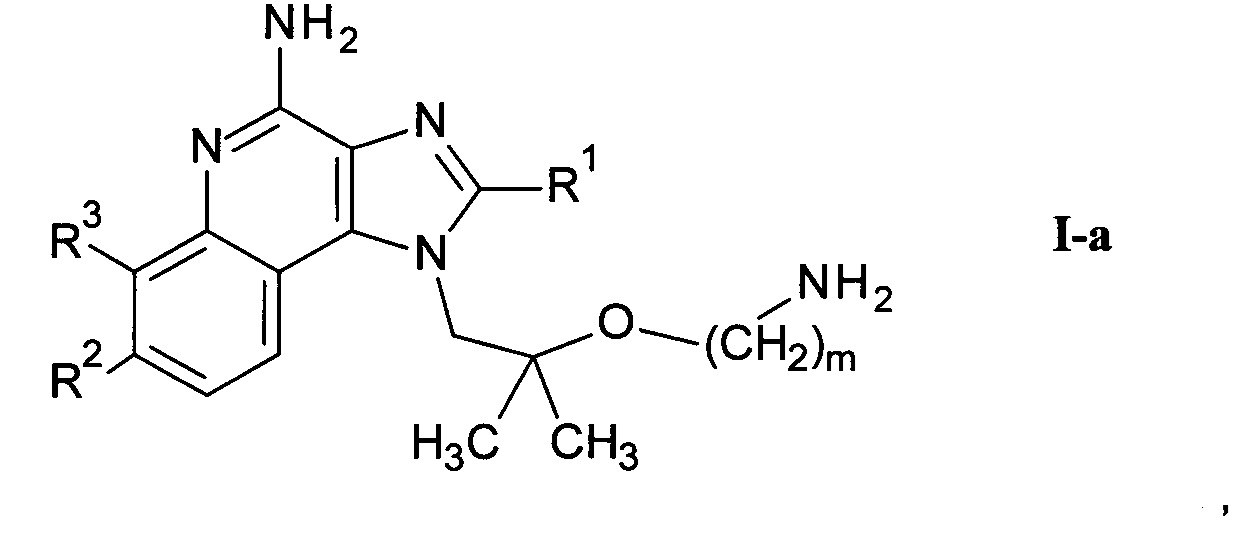
где R1-R3 и m являются такими, как определено здесь ранее, и возможно дальнейшее связывание соединения формулы I-a со спиртом или кислотой формулы R5-OH или и альдегидом формулы R5=O с получением соединения формулы I-c
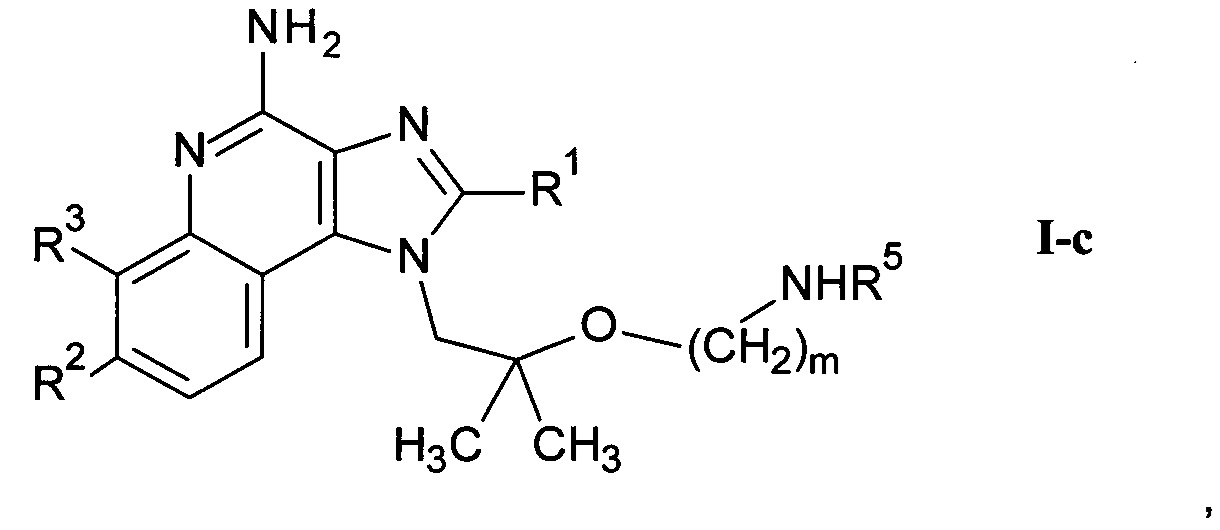
где R1-R3, m и R5 являются такими, как определено здесь ранее, и, если необходимо,
конвертирование полученного соединения в фармацевтически приемлемую соль, или
b) взаимодействие соединения формулы II-a
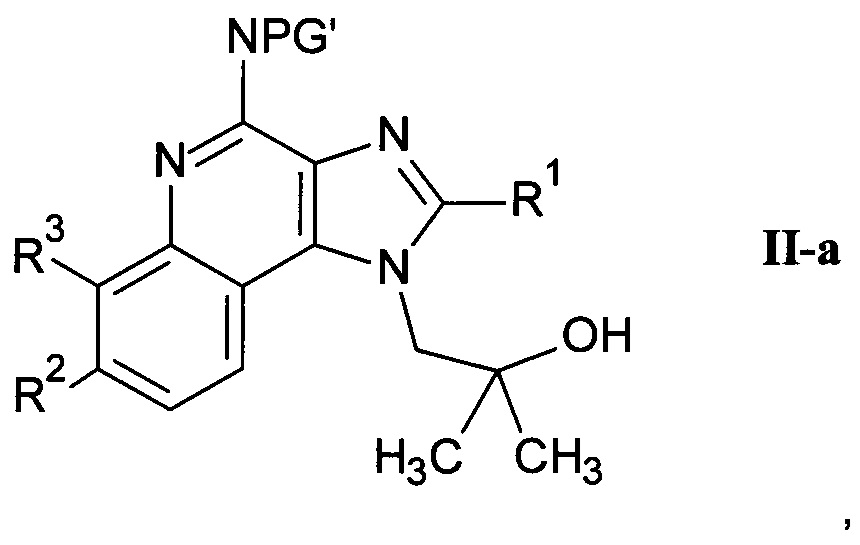
где R1, R2 и R3 являются такими, как определено здесь ранее, a PG' представляет собой защитную группу, с карбоновой кислотой формулы IV
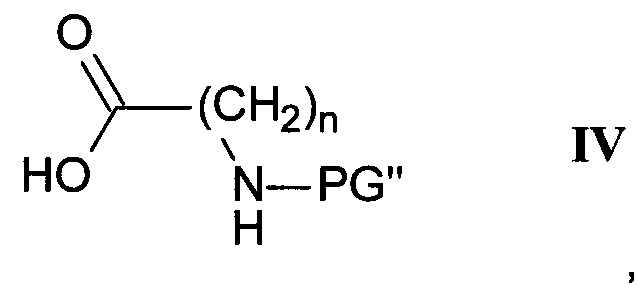
где n является таким, как определено здесь ранее, a PG" представляет собой защитную группу, в присутствии этерификационного агента и удаление защитных групп PG' и PG" с помощью слабого восстановителя с получением соединения формулы I-b
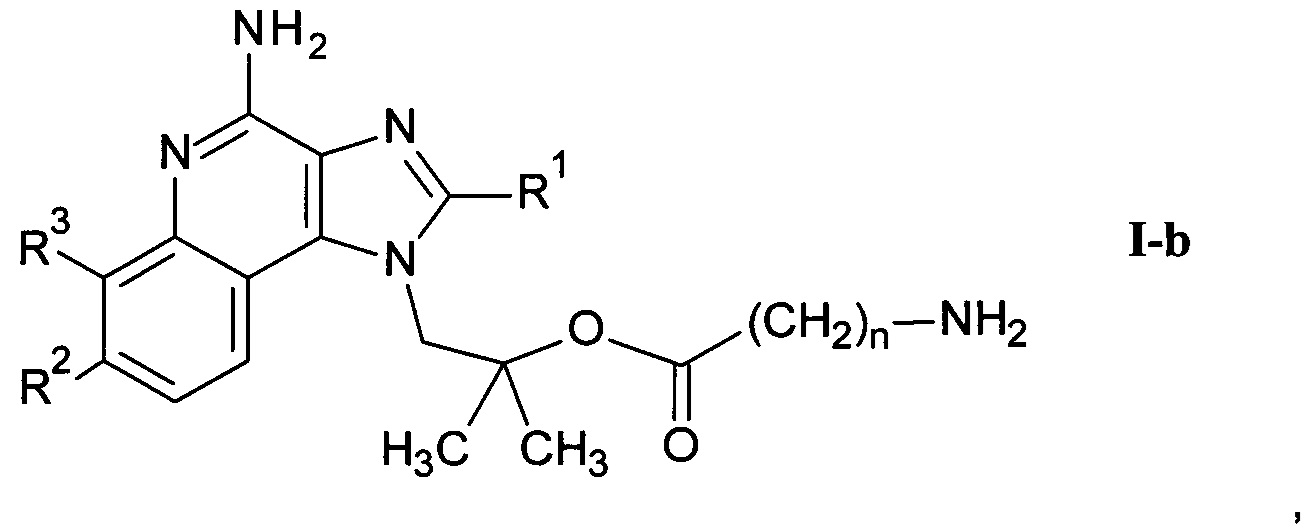
где R1-R3 и n являются такими, как определено здесь ранее, и возможно дальнейшее связывание соединения формулы I-a со спиртом или кислотой формулы R6-OH или и альдегидом формулы R6=O с получением соединения формулы I-d
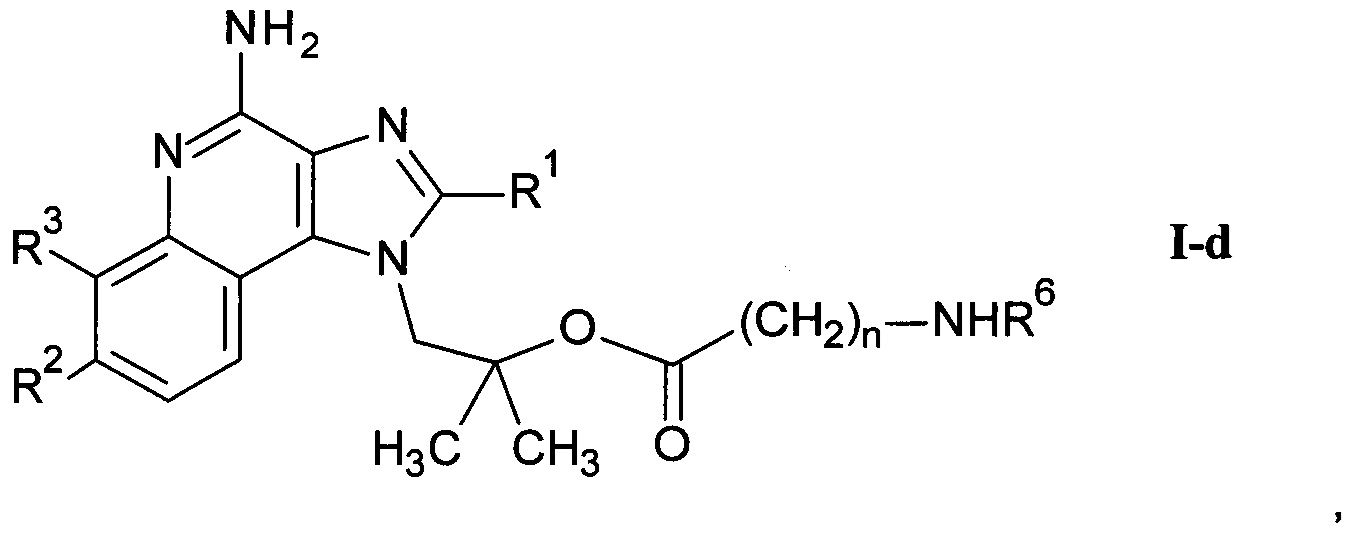
где R1-R3, m и R6 являются такими, как определено здесь ранее, и, если необходимо,
конвертирование полученного соединения в фармацевтически приемлемую соль.
В частности, подходящая защитная группа PG представляет собой амино-защитную группу, выбранную из тритила (TRT), или двойную защиту с использованием изоиндолин-1,3-диона, бис-бензил или бис-карбоксибензил (бис-Z) защитной группы.
"В щелочной среде" означает присутствие основания, такого как гидрид натрия или трет-бутилат калия. Взаимодействие проводят в подходящем растворителе, таком как например N,N-диметилформамид (ДМФ), диметилацетамид (DMA), дихлорметан или диоксан, при температурах между 0°С и комнатной температурой.
"Удаление защитных групп PG и Воc в кислотной среде" означает обработку защищенного соединения кислотами в подходящем растворителе, например, можно использовать трифторуксусную кислоту (ТФУ) в растворителе, таком как дихлорметан (ДХМ).
Подходящие защитные группы PG' и PG" являются защитными группами, которые образуют кольцо вместе с атомом азота амино группы. В частности, PG' или PG" вместе с атомом азота, к которым они присоединены, образуют изоиндолин-1,3-дион или означают бис-бензил или бис-карбоксибензил защитную группу.
Этерификационный агент представляет собой соединение, которое облегчает реакцию этерификации. Конкретным этерификационным агентом является N,N-диизопропил-карбодиимид. Взаимодействие, в частности, проводится в инертном растворителе, таком как ДХМ.
"Удаление защитных групп PG' и PG" с помощью слабого восстановителя" означает, в частности, обработку защищенного соединения гидразином/водой в инертном растворителе, таком как ТГФ.
Настоящее изобретение, кроме того, относится к соединениям формулы I как определено выше, полученным способом, как определено выше.
Синтез соединений общей формулы I может, например, быть проведен в соответствии со следующими схемами. Если не указано иного, R1-R3 и X являются такими, как определено выше.
Способ получения исходных соединений формулы AG дан на Схеме 1, и указанный путь синтеза приведен в WO 2013/033345 (Univ. of Minnesota).
Соединения АВ могут быть получены из подходящим образом замещенных ортоэфиров АА посредством конденсации с 2-амино-пропандинитрилом и 1-амино-2-метил-пропан-2-олом в инертном растворителе, например ТГФ, в присутствии основания, такого как например триэтиламин. Подходящим образом замещенные ортоэфиры АА являются коммерчески доступными, могут быть синтезированы квалифицированным в уровне техники специалистом или показаны в экспериментальной части.
Соединения АС могут быть получены из соединений АВ посредством диазотирования/йодирования как известно в уровне техники; в частности, посредством использования йодометана в качестве источника йода и изоамилнитрита в качестве источника нитрита в инертном растворителе, таком как хлороформ, при температурах от 0°С до точки кипения растворителя, предпочтительно при температуре 80°С.
Соединения формулы АС могут быть связаны с соединениями формулы AD, в которых М обозначает уходящую группу металла и R' обозначает водород и/или подходящую защитную группу, посредством способов, известных в уровне техники, с получением соединений формулы АЕ. Подходящими уходящими группами металлов могут быть бороновые кислоты, боронатные эфиры, и трифторбораты, но также и уходящие группы на основе олова или цинка. IB частности, бороновые кислоты или боронатные эфиры могут быть использованы в реакции сочетания Сузуки-Мияуры с использованием палладиевого катализатора, такого как Pd(OAc)2 в присутствии трифенилфосфина, в инертном растворителе, таком как DME, вместе с подходящим основанием, таки как карбонат натрия. Температура реакции может варьироваться от комнатной температуры до температуры кипения растворителя, при этом комнатная температура является подходящей для множества случаев.
Если соединения АЕ являются защищенными по анилин-амино группе (один R' не равен Н), депротекция с получением соединений вида AF может быть выполнена посредством способов, известных в уровне техники, с кислотным отщеплением защитной группы, с использованием ТФУ в качестве предпочтительного выбора.
Соединения вида AG могут быть получены из соединений AF посредством термической конденсации (замыкание кольца) в присутствии кислотного катализатора. Это может быть просто выполнено посредством нагревания соединений AF в инертном растворителе, таком как диоксан, в присутствии кислоты, такой как HCl, в течение подходящего времени, например в течение 2 часов при 90°С.
Схема 1
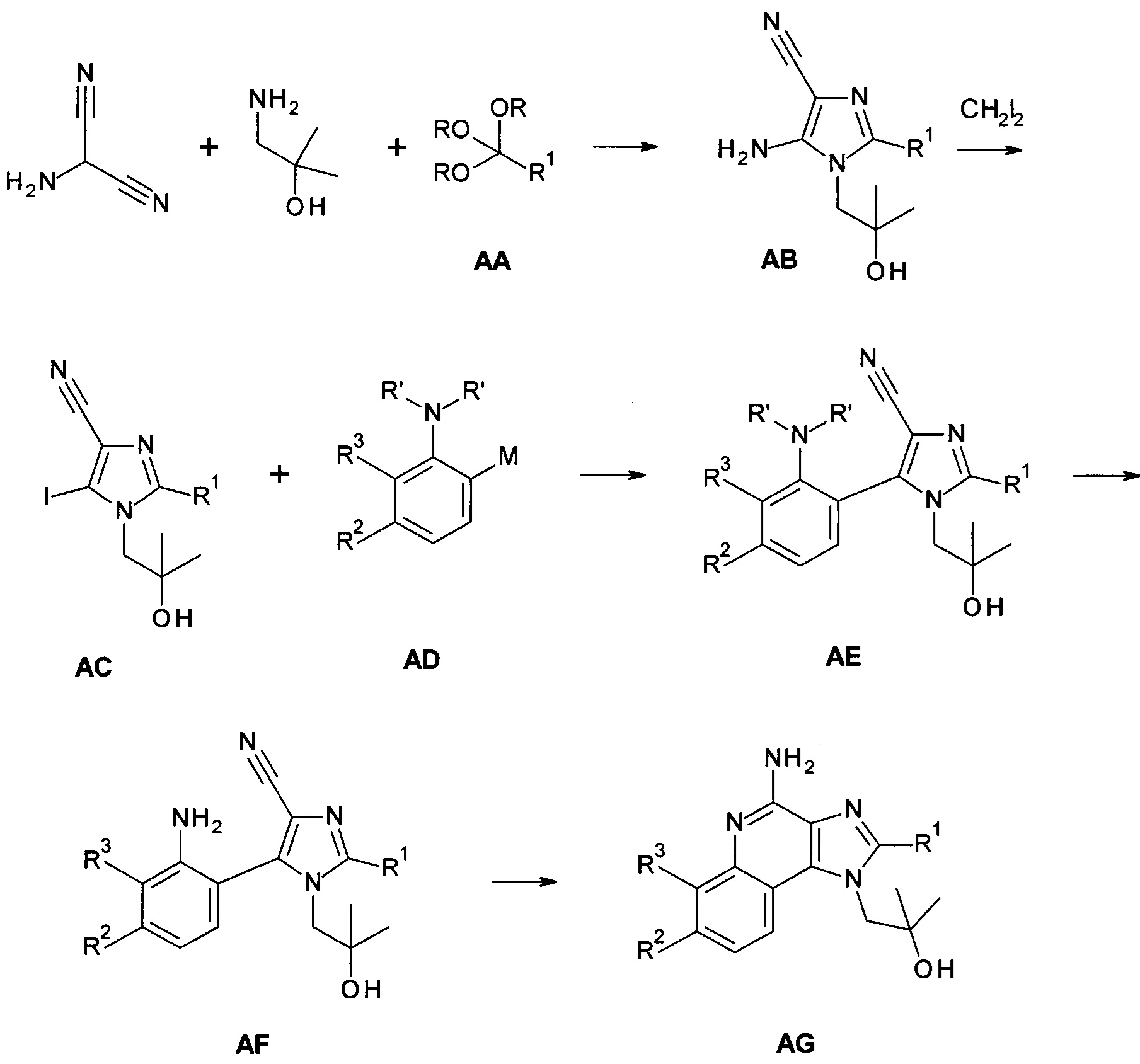
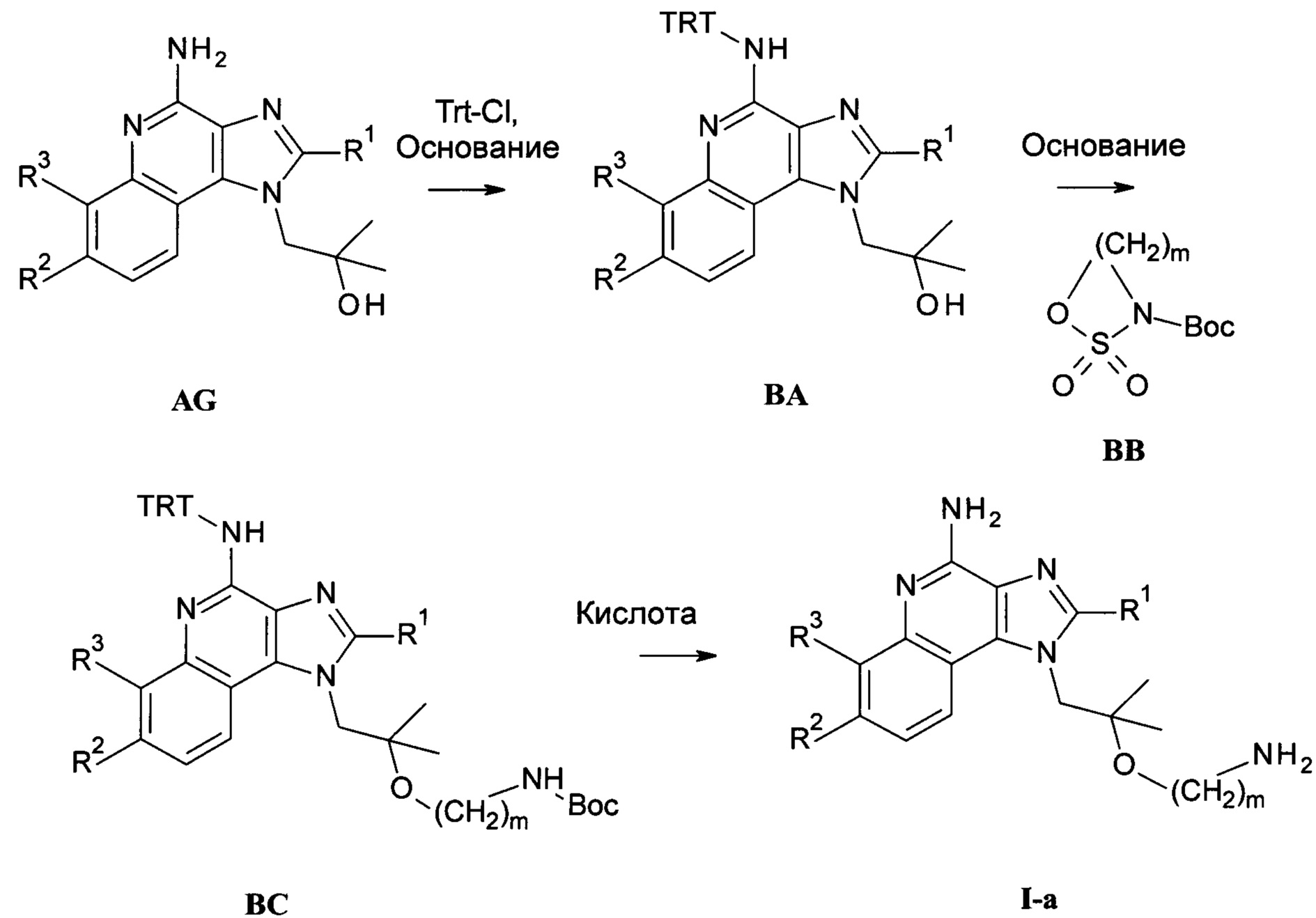
Следуя методике в соответствии со схемой 2, соединения AG могут быть использованы в качестве исходного вещества для синтеза соединений I-a, где R4 представляет собой -O-(СН2)m-NH2.
Соединение ВА может быть получено из AG посредством взаимодействия с тритилхлоридом в присутствии основания в инертном растворителе при повышенной температуре в присутствии или без микроволнового излучения. Подходящей комбинацией основание-растворитель является, например, триэтиламин или DIEA и ацетонитрил, особенно если реакция проводится при повышенной температуре в микроволновой печи.
Соединения общей формулы ВС могут быть получены из соединений общей формулы ВА посредством взаимодействия с Вос-сульфамидатом ВВ в подходящем растворителе, таком как ДМФ, в присутствии подходящего основания, такого как гидрид натрия или трет-бутилат калия. Взаимодействие предпочтительно проводится при от 0°С до комнатной температуры.
Соединения общей формулы I-а могут быть получены из соединений общей формулы ВС посредством удаления защитных групп с помощью обработки кислотами в подходящем растворителе. Одной из таких кислот является ТФУ с или без дополнительного присутствия ДХМ, используемая при комнатной температуре.
Схема 2
Следуя методике в соответствии со схемой 3, соединение AG может быть использовано в качестве исходного вещества для синтеза соединений I-b, где R4 представляет собой -O-(CO)-(CH2)n-NH2.
Схема 3
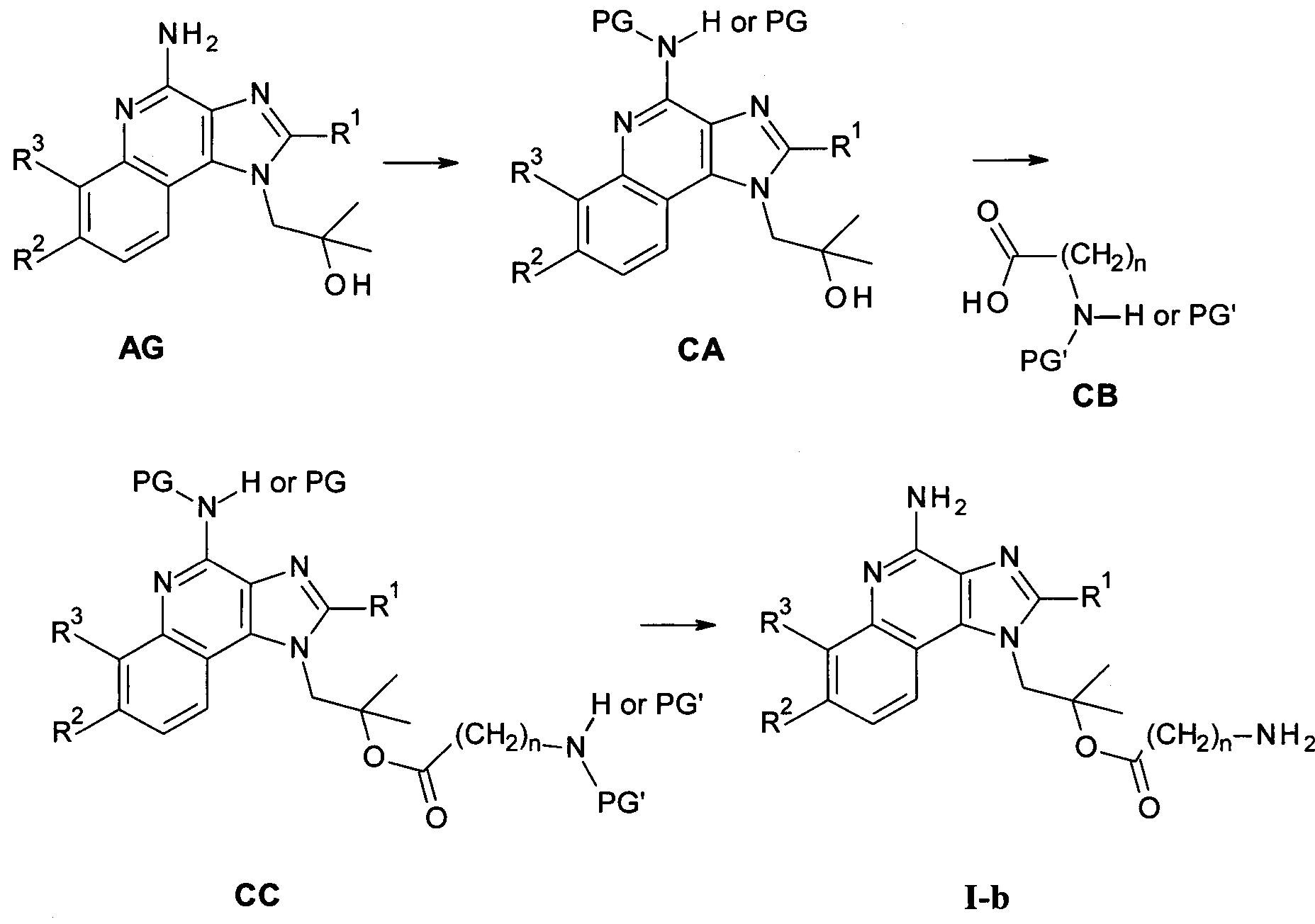
Соединение СА может быть получено из AG посредством введения подходящей защитной группы PG с помощь способов, известных в уровне техники. Такие защитные группы могут представлять собой, например, изоиндолин-1,3-дион (фталил). Особенно, соединение СА может взаимодействовать с фталоилхлоридом в присутствии основания, такого как 1,4-диазабицикло[2,2,2]октан, в инертном растворителе с высокой температурой кипения, таком как толуол, при повышенной температуре с получением соединения вида СА с изоиндолин-1,3-дионом в качестве защитной группы.
Соединение СС может быть получено из соединений СВ и СА посредством этерификации с использованием одного из множества способов, описанных в уровне техники. Преимущественно, такая этерификация может быть выполнена посредством объединения СВ и СА в инертном растворителе, таком как ДХМ, в присутствии N,N-диизопропил-карбодиимида при повышенной температуре. Подходящие карбоновые кислоты СВ являются коммерчески доступными, могут быть получены методиками, известными в уровне техники, или показаны в качестве примера в экспериментальной части.
Соединения общей формулы I-b могут быть получены из соединений общей формулы СС посредством удаления защитных групп посредством способов, известных в уровне техники. В частности, изоиднолин-1,3-дион может быть удален посредством обработки гидразином/водой в инертном растворителе, таком как ТГФ, при комнатной температуре.
Следуя методике в соответствии со схемой 4, соединения I-c и I-d могут быть получены из I-а или I-b посредством способов, известных в уровне техники, как например амидное связывание (R5-OH или R6-OH являются кислотами) или восстановительное аминирование (R5=O или R6=O являются альдегидами) как отражено в экспериментальной части более подробно.
Схема 4
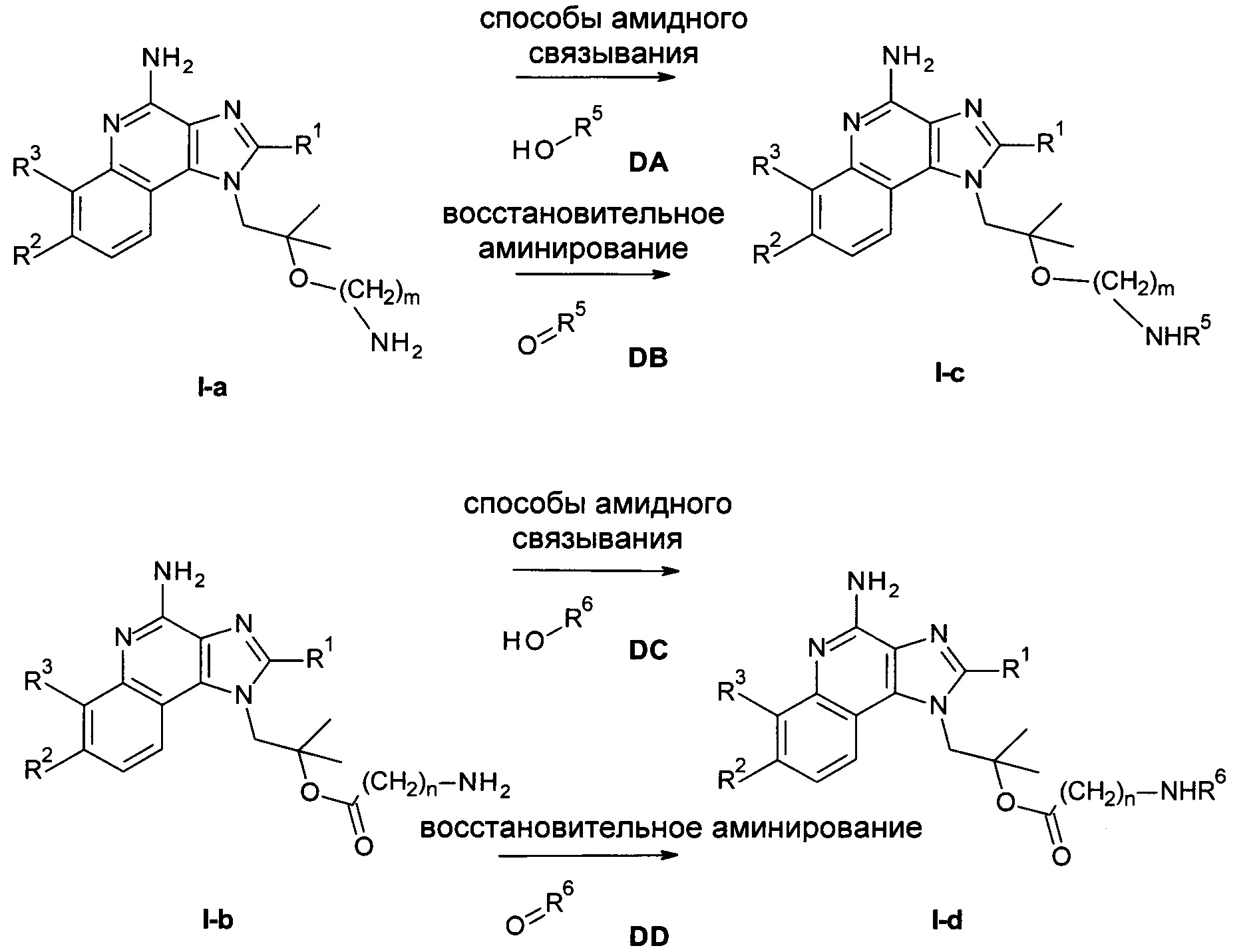
Если один из исходных материалов или соединений формулы АА, AD, AG, DA, DB, DC или DD содержат одну или более функциональных групп, которые не являются стабильными или являются реактивными в условиях реакции одной или нескольких реакционных стадий, соответствующие защитные группы (как описано, например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition) могут быть введены перед критической стадией, используя способы, хорошо известные в данной области. Такие защитные группы могут быть удалены на более поздней стадии синтеза с использованием стандартных способов, описанных в литературе.
Если одно или более соединений формулы АА, AD, AG, DA, DB, DC или DD содержат хиральные центры, соединения формулы I-а могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в области техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацемические соединения могут, например, быть разделены на их антиподы через диастереомерные соли посредством кристаллизации или посредством разделения антиподов с помощью специальных хроматографических способов, с использованием как хирального адсорбента, так и хирального элюента.
Как здесь описано ранее, соединения формулы I настоящего изобретения могут применяться в качестве лекарственного средства для лечения заболеваний, которые опосредованы агонистами TLR, в частности для лечения заболеваний, которые опосредованы агонистами TLR7 и/или TLR8, более конкретно для лечения заболеваний, которые опосредованы агонистами TLR7 и TLR8.
Соединения, определенные в настоящем изобретении являются агонистами рецепторов TLR7 и/или TLR8 в клеточном анализе in vitro. Более конкретно, соединения настоящего изобретения являются агонистами обоих рецепторов TLR7 и TLR8. Соответственно, соединения настоящего изобретения, как ожидается, будут потенциально полезными агентами при лечении заболеваний или медицинских состояний, для которых активация иммунной системы через агонисты TLR7 и/или TLR8 будет благоприятной, более конкретно при лечении заболеваний или медицинских состояний, для которых активация иммунной системы через агонисты обоих TLR7 и/или TLR8 будет благоприятной. Например, следующие заболевания и состояния можно лечить соединениями настоящего изобретения.
Соединения формулы I настоящего изобретения применяются в онкологии, то есть они могут применяться при лечении распространенных видов рака, включая рак мочевого пузыря, рак головы и шеи, рак предстательной железы, колоректальный рак, рак почки, рак молочной железы, рак легкого, рак яичников, рак шейки матки, рак поджелудочной железы, рак кишечника и толстой кишки, рак желудка, рак щитовидной железы, меланомы, рака кожи и опухолей головного мозга и злокачественных опухолей, затрагивающих костный мозг, таких как лейкозы и рак лимфопролиферативных систем, таких как болезнь Ходжкина и неходжкинская лимфома; включая предотвращение и лечение метастатического рака и рецидивов опухоли, и паранеопластических синдромов.
Соединения формулы I настоящего изобретения также применяются при лечении аутоиммунных заболеваний. "Аутоиммунное заболевание" представляет собой заболевание или нарушение, возникающее у и направленное против собственных тканей индивидуума или органов или ее выделение или манифестация или результат его состояние. "Аутоиммунное заболевание" может быть органоспецифическим заболеванием (т.е. иммунный ответ специфически направлен против системы органов, таких, как эндокринной системы, кроветворной системы, кожи, сердечно-легочной системы, желудочно-кишечного тракта и печени, почечной системы, щитовидной железы, ушей, нервно-мышечной системы, центральной нервной системы и т.д.) или системное заболевание, которое может повлиять на несколько систем органов (например, системная красная волчанка (SLE), ревматоидный артрит, полимиозит и т.д.). В конкретном аспекте аутоиммунное заболевание связано с кожей, мышечной тканью, и/или соединительной тканью
Конкретные аутоиммунные заболевания включают аутоиммунные ревматические расстройства (такие как, например, ревматоидный артрит, синдром Шегрена, склеродермия, красная волчанка, такая как SLE и волчаночный нефрит, полимиозит / дерматомиозит, криоглобулинемия, синдром антифосфолипидного антитела и псориатический артрит), аутоиммунные расстройства желудочно-кишечного тракта и печени (такие как, например, воспалительные заболевания кишечника, неспецифический язвенный колит и болезнь Крона), аутоиммунный гастрит и пернициозную анемию, аутоиммунный гепатит, первичный билиарный цирроз печени, первичный склерозирующий холангит и целиакии), васкулит (такой как, например, ANCA-негативный васкулит и ANCA-ассоциированный васкулит, в том числе васкулит Чарга-Стросса, гранулематоз Вегенера и микроскопический полиангиит), аутоиммунные неврологические расстройства (такие как, например, рассеянный склероз, синдром пляшущих глаз, миастению, оптиконевромиелит, болезнь Паркинсона, болезнь Альцгеймера и аутоиммунные полинейропатии), почечные расстройства (такие как, например, гломерулонефрит, синдром Гудпасчера и болезнь Бергера), аутоиммунные дерматологические расстройства (такие как, например, псориаз, крапивница, аллергическая сыпь, пузырчатка обыкновенная, буллезный пемфигоид и кожная красная волчанка), гематологические расстройства (такие как, например, тромбоцитопеническая пурпура, тромбическая тромбоцитопеническая пурпура, посттрансфузионная пурпура и аутоиммунная гемолитическая анемия), атеросклероз, увеит, аутоиммунные заболевания слуха (такие как, например, заболевания внутреннего уха и потеря слуха), болезнь Бехчета, болезнь Рейно, трансплантация органов, и аутоиммунные эндокринные расстройства (такие как, например, диабетические, связанные с аутоиммунными заболеваниями, такие как инсулин-зависимый сахарный диабет (ИЗСД), болезнь Аддисона и аутоиммунные заболевания щитовидной железы (например, болезнь Грейвса и тиреоидит)), аллергические состояния и ответы, пищевые аллергии, лекарственную аллергию, аллергию на насекомых, редкие аллергические расстройства, такие как мастоцитоз, аллергические реакции, экзему, включая аллергический или атопический дерматит, астму, такую как бронхиальная астма и аутоиммунная астма, состояния с участием инфильтрации Т-клетками и хронические воспалительные ответы
Соединения формулы I настоящего изобретения также применяются при лечении инфекционных заболеваний. Таким образом, они могут быть полезны при лечении вирусных заболеваний, в частности заболеваний, вызванных инфекцией вирусами, выбранными из группы, состоящей из вирусов папилломы, таких как вирус папилломы человека (ВПЧ), а также тех, которые вызывают остроконечные бородавки, простые бородавки и подошвенные бородавки, вирус простого герпеса, контагиозный моллюск, вирус гепатита В (HBV), вирус гепатита С (ВГС), вирус денге, вирус оспы, вирус иммунодефицита человека (ВИЧ), цитомегаловирус (ЦМВ), вирус ветряной оспы (VZV), риновирус, энтеровирус, аденовирус, коронавирус (например, атипичная пневмония), гриппа, свинки и парагриппа.
Они также могут применяться при лечении бактериальных заболеваний, в частности заболеваний, вызванных бактериальной инфекцией, выбранной из группы, состоящей из микобактерий, таких как микобактерии туберкулеза, микобактерии микоза и микобактерии лепры. Соединения формулы I настоящего изобретения дополнительно могут быть полезным при лечении других инфекционных заболеваний, таких как хламидиоз, грибковых заболеваний, в частности грибковых заболеваний, выбранных из группы, состоящей из кандидоза, аспергиллеза и криптококкового менингита и паразитарных заболеваний, таких как пневмоцистная пневмония, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция и лейшманиоз.
Таким образом, выражение "заболевания, опосредованные агонистами TLR" означает заболевания, которые можно лечить путем активации иммунной системы с помощью агонистов TLR7 и/или TLR8, такие как рак и инфекционные заболевания. В частности, выражение "заболевания, опосредованные агонистами TLR" означает рак или аутоиммунные заболевания или инфекционные заболевания, выбранные из группы, состоящей из вирусных заболеваний, бактериальных болезней, грибковых заболеваний и паразитарных заболеваний.
В конкретном аспекте, выражение "которые опосредованы агонистами TLR" относится к раку, выбранному из группы, состоящей из рака мочевого пузыря, рак головы и шеи, рак предстательной железы, колоректальный рак, рак почки, рак молочной железы, рак легкого, рак яичников, рак шейки матки, рак поджелудочной железы, рак кишечника и толстой кишки, рак желудка, рак щитовидной железы, меланомы, рака кожи и опухолей головного мозга и злокачественных опухолей, затрагивающих костный мозг, таких как лейкозы и рак лимфопролиферативных систем, таких как болезнь Ходжкина и неходжкинская лимфома; включая предотвращение и лечение метастатического рака и рецидивов опухоли, и паранеопластических синдромов
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение формулы I как определено выше, и фармацевтически приемлемый носитель и/или адъювант. Более конкретно, настоящее изобретение относится к фармацевтическим композициям, полезным для лечения заболеваний, которые опосредованы агонистами TLR.
Дополнительно, настоящее изобретение относится к соединениям формулы I, как определено выше, для применения в качестве терапевтически активных веществ, в частности в качестве терапевтически активных веществ для лечения заболеваний, которые опосредованы агонистами TLR. В частности, настоящее изобретение относится к соединениям формулы I для применения при лечении рака или аутоиммунных заболеваний или инфекционных заболеваний, выбранных из группы, состоящей из вирусных заболеваний, бактериальных заболеваний, грибковых заболеваний и паразитических заболеваний.
В другом аспекте, настоящее изобретение относится к способу лечения заболеваний, которые опосредованы агонистами TLR, который включает введение терапевтически активного количества соединения формулы I человеку или животному. В частности, настоящее изобретение относится к способу лечения опухолей и инфекционных заболеваний, выбранных из группы, состоящей из вирусных заболеваний, бактериальных заболеваний, грибковых заболеваний и паразитических заболеваний.
Настоящее изобретение, кроме того, относится к применению соединений формулы I, как определено выше для лечения заболеваний, которые опосредованы агонистами TLR.
В дополнение, настоящее изобретение относится к применению соединений формулы I, как определено выше, для получения лекарственных средств для лечения заболеваний, которые опосредованы агонистами TLR. В частности, настоящее изобретение относится к применению соединений формулы I как определено выше, для получения лекарственного средства для лечения опухолей или аутоиммунных заболеваний или инфекционных заболеваний, выбранных из группы, состоящей из вирусных заболеваний, бактериальных заболеваний, грибковых заболеваний и паразитических заболеваний.
В дополнительном аспекте соединения формулы I могут быть комбинированы с одним или более способом лечения в схеме лечения опухоли.
Комбинированная терапия включает, в дополнение к введению соединения по изобретению, дополнительное применение одного или нескольких способов, которые эффективны при лечении рака. Такие способы включают, без ограничения, химиотерапевтические агенты, иммунотерапевтические агенты, антиангиогенные агенты, цитокины, гормоны, антитела, полинуклеотиды, радиоактивные и фотодинамические терапевтические агенты. В конкретном аспекте комбинированная терапия может быть использована для предотвращения рецидива рака, ингибирования метастазированию, или ингибирования роста и/или распространения рака или метастазов. Как здесь используется, "в комбинации с" означает, что соединение формулы I вводят в виде части схемы лечения, которая содержит один или несколько дополнительных способов лечения, как упоминалось выше. Таким образом, изобретение также относится к способу лечения опухоли, который включает введение терапевтически активного количества соединения формулы I в комбинации с одним или несколькими другими фармацевтически активными соединениями человеку или животному.
Соединения формулы I могут применяться самостоятельно или в комбинации одним или более дополнительным способом лечения при лечении аутоиммунных заболеваний.
Комбинированная терапия включает, в дополнение к введению соединения по изобретению, дополнительное применение одного или нескольких способов, которые нацелены на предотвращение или лечение аутоиммунных заболеваний. Такие способы включают, без ограничения, химиотерапевтические агенты, иммунотерапевтические агенты, антиангиогенные агенты, цитокины, гормоны, антитела, полинуклеотиды, радиоактивные и фотодинамические терапевтические агенты. Как здесь используется, "в комбинации с" означает, что соединение формулы I вводят в виде части схемы лечения, которая содержит один или несколько дополнительных способов лечения, как упоминалось выше. Таким образом, изобретение также относится к способу лечения аутоиммунного заболевания, который включает введение терапевтически активного количества соединения формулы I в комбинации с одним или несколькими другими фармацевтически активными соединениями человеку или животному.
Соединения формулы I могут применяться самостоятельно или в комбинации одним или более дополнительным способом лечения при лечении инфекционных заболеваний.
Комбинированная терапия включает, в дополнение к введению соединения по изобретению, дополнительное применение одного или нескольких способов, которые нацелены на предотвращение или лечение инфекционных заболеваний. Такие способы включают, без ограничения, противовирусные агенты, антибиотики и противогрибковые агенты. Как здесь используется, "в комбинации с" означает, что соединение формулы I вводят в виде части схемы лечения, которая содержит один или несколько дополнительных способов лечения, как упоминалось выше. Таким образом, изобретение также относится к способу лечения инфекционного заболевания, который включает введение терапевтически активного количества соединения формулы I в комбинации с одним или несколькими другими фармацевтически активными соединениями человеку или животному.
ФАРМАКОЛОГИЧЕСКИЙ ТЕСТ
Следующие тесты проводились с целью определить активность соединений формулы I:
Для тестирования активности относительно TLR8 и TLR7, применяли клетки HEK-Blue TLR8 или TLR7 человека, соответственно, (Invivogen, San Diego, СА, USA) трансфицированные репортерным конструктом SEAP (секретируемая эмбриональная щелочная фосфатаза), в которых экспрессия репортера регулировалась промотором NF-κВ после стимуляции в течение 24 ч. Репортерная активность определялась с применением набора Quanti Blue (Invivogen, San Diego, Ca, USA) при длине волны 640 нм.
Значения ЕС50 определялись с использованием анализа Activity Base (ID Business Solution, Limited).
Соединения в соответствии с формулой I обладали активностью (значение ЕС50) в вышеуказанном анализе для TLR8 человека в диапазоне от 0.01 нМ до 11 мкМ, более конкретно от 0.01 нМ до 3 мкМ и в вышеуказанном анализе для TLR7 человека в диапазоне от 0.01 нМ до 1 мкМ, в частности от 0.01 нМ до 0.3 мкМ и более конкретно от 0.01 нМ до 0.1 мкМ.
Например, следующие соединения показали следующие значение ЕС50 в вышеприведенном анализе:
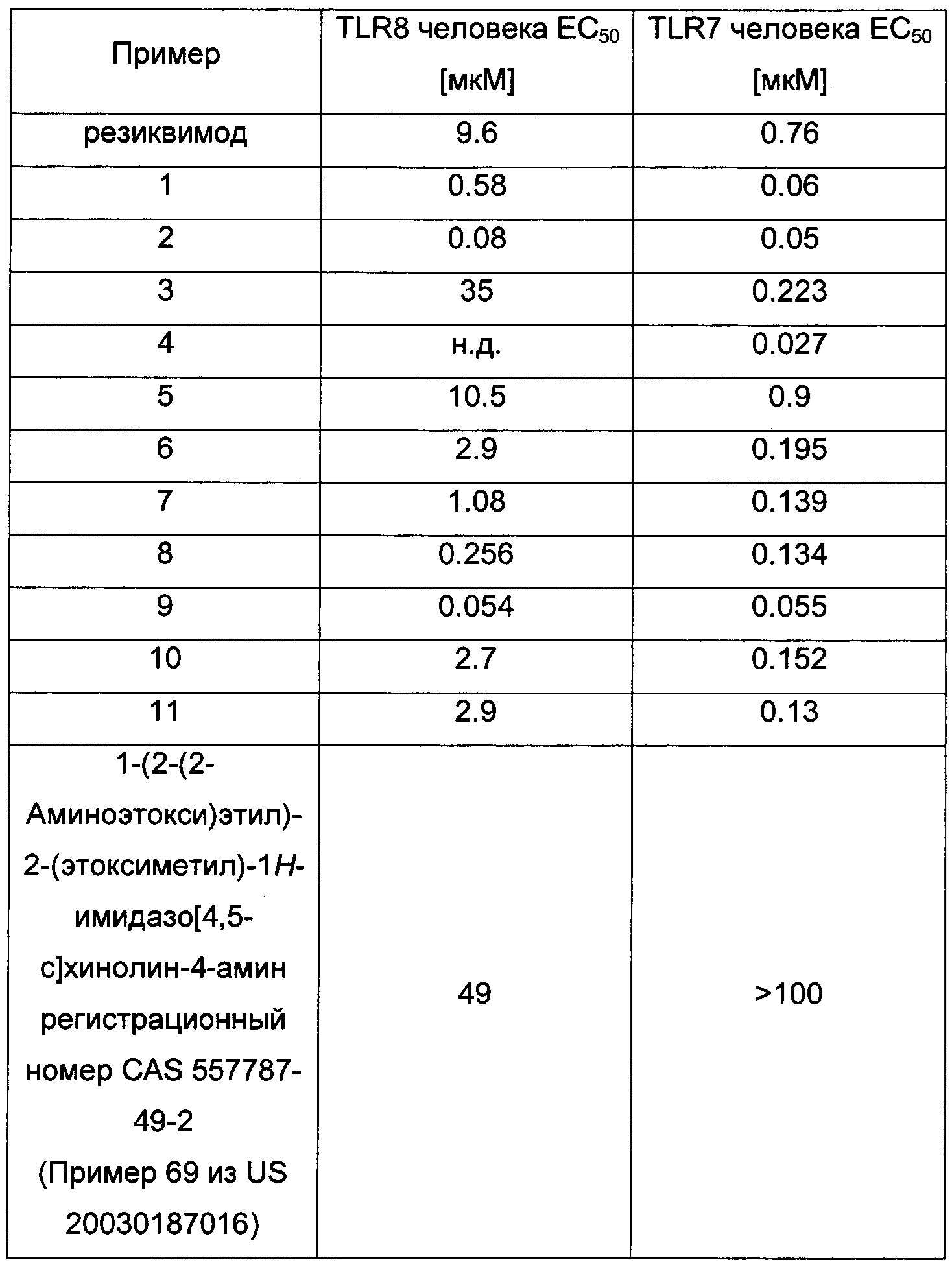
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Соединения формулы I и их фармацевтически приемлемые соли могут применяться в качестве лекарственных средств, например, в виде фармацевтических препаратов для энтерального, парентерального или местного введения. Соединения формулы I и их фармацевтически приемлемые соли могут быть введены системно (например, парентерально) или местно (например, топикальные или интралезиональные инъекции). В некоторых случаях фармацевтическая композиция вводится местно, парентерально, перорально, вагинально, внутриматочно, интраназально или путем ингаляции. Как здесь описано, некоторые ткани могут быть предпочтительными целями для агонистов TLR. Таким образом, введение агониста TLR в лимфатические узлы, селезенку, костный мозг, кровь, а также ткани, зараженные вирусом, являются предпочтительными местами введения.
В одном аспекте, фармацевтическую композицию, содержащую соединения формулы I или его фармацевтически приемлемые соли вводят парентерально. Парентеральные пути введения включают в себя, без ограничения, трансдермальные, трансмукозальные, носоглоточные, легочные и прямые инъекции. Парентеральное введение путем инъекции может осуществляться любым способом парентеральной инъекции, включая, без ограничения, внутривенный (IV), в том числе болюсную и капельную (например, быструю или медленную), внутрибрюшинный (IP), внутримышечный (IM), подкожный (SC) и внутрикожный (ID) путь. Трансдермальное и трансмукозальное введение может быть осуществлено путем, например, включение носителя (например, диметилсульфоксид, ДМСО), путем применения электрических импульсов (например, электрофорез) или их сочетания. Доступны различные устройства, которые могут быть использованы для трансдермального введения. Композиции соединений формулы I, пригодные для парентерального введения обычно готовят в USP воде или в воде для инъекций и могут дополнительно включать в себя рН-буферы, соли, наполнители, консерванты и другие фармацевтически приемлемые эксципиенты.
Трансдермальное введение осуществляется путем использования крема, полоскания, геля и т.д., способных дать проникнуть агонисту TLR в кожу и попасть в кровь. Композиции, пригодные для трансдермального введения включают, без ограничения, фармацевтически приемлемые суспензии, масла, кремы и мази, применяемые непосредственно на кожу или включенные в защитный носитель, такой как трансдермальное устройство (так называемый "пластырь"). Примеры подходящих кремов, мазей и т.д., можно найти, например, в Physician' s Desk Reference. Трансдермальный перенос может быть также осуществлен посредством ионофореза, например с использованием коммерчески доступных пластырей, которые обеспечивают своему продукту непрерывное прохождение через неповрежденную кожу в течение периода нескольких дней или более. Использование этого способа предоставляет контролируемую передачу фармацевтических композиций в относительно больших концентрациях, позволяет инфузию комбинированных препаратов и позволяет одновременное использование промотора абсорбции. Введение с помощью трансдермальных и трансмукозальных путей может быть непрерывным или пульсирующим.
Легочное введение осуществляется путем ингаляции, и включает пути доставки, такие как интраназальный, трансбронхиальный и трансальвеолярный пути. Предложены препараты соединений формулы I, пригодные для введения путем ингаляции, включая, но не ограничиваясь, жидкие суспензии для формирования аэрозолей, а также порошковые формы для систем доставки для ингаляции сухого порошка. Устройства, пригодные для введения путем ингаляции, включают, без ограничения, распылители, испарители, небулайзеры и устройства доставки сухого порошка для ингаляции. Другие способы доставки в слизистую дыхательных путей включают доставку жидких композиций, такую как каплями в нос. Введение путем ингаляции предпочтительно осуществляют в виде дискретных доз (например, через дозирующий ингалятор), хотя доставка, аналогичная инфузии может быть достигнута путем использования ингалятора.
Соединения формулы I и их фармацевтически приемлемые соли также могут быть введены перорально, например в виде таблеток, таблеток покрытых оболочкой, драже, твердых и мягких желатиновых капсул.
Производство фармацевтических препаратов может быть осуществлено способами, знакомыми любому квалифицированному специалисту в данной области посредством использования описанных соединений формулы I и их фармацевтически приемлемых солей, возможно в комбинации с другими терапевтически ценными веществами, в галеновых формах введения вместе с подходящими, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими носителями и, при желании, обычными фармацевтическими адъювантами.
Подходящими материалами для носителей являются не только неорганические вещества-носители, но также и органические вещества-носители. Так, например, лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли могут быть использованы в качестве материалов-носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры и полутвердые и жидкие полиолы (в зависимости от природы активного ингредиента, однако, в случае мягких желатиновых капсул может не требоваться никакого носителя) Подходящими материалами носителей для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар и т.п. Подходящими материалами носителей для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин и растительные масла. Подходящими носителями для суппозиториев являются, например, природные или гидрогенизированные масла, воски, жиры и полужидкие или жидкие полиолы. Подходящими материалами носителей для местных препаратов являются глицериды, полусинтетические и синтетические глицериды, гидрогенизированные масла, жидкие воски, жидкие парафины, жидкие жирные спирты, стиролы, полиэтиленгликоли и производные целлюлозы.
Обычные стабилизаторы, консерванты, увлажняющие агенты и эмульгаторы, улучшающие консистенцию средства, улучшающие аромат средства, соли для изменения осмотического давления, буферные вещества, растворители, красители и маскирующие агенты и антиоксиданты могут использоваться в качестве фармацевтических адъювантов.
Дозировка соединений формулы I может варьироваться в широких пределах в зависимости от заболевания, которые подлежат лечению, возраста и индивидуального состояния пациента и способа введения, и, конечно, должны подбираться под отдельные требования в каждом конкретном случае. Для взрослых пациентов рассматриваются суточные дозы от 1 до 1000 мг, особенно от 1 до 300 мг. В зависимости от тяжести заболевания и точного фармакокинетического профиля соединение может вводиться ежедневно одной или несколькими единичными дозированными формами, например, от 1 до 3 единичных дозированных форм.
Фармацевтические препараты обычно содержат около 1-500 мг, предпочтительно 1-100 мг соединения формулы I.
Следующие примеры С1-С3 иллюстрируют обычные композиции настоящего изобретения, но служат только в качестве его образца.
Пример С1
Покрытые оболочкой таблетки, содержащие следующие ингредиенты, могут быть изготовлены обычными способами:


Активный ингредиент просеивают и смешивают с микрокристаллической целлюлозой и смесь гранулируют с раствором поливинилпирролидона в воде. Гранулят смешивают с карбоксиметилкрахмалом натрия и стеаратом магния и прессуют с получением ядер весом 120 или 350 мг соответственно. Ядра покрывают водным раствором / суспензией вышеупомянутого пленочного покрытия.
Пример С2
Капсулы, содержащие следующие ингредиенты могут быть изготовлены обычными способами:
|
Компоненты просеивают и смешивают и помещают в капсулы 2-го размера.
Пример С3
Раствор для инъекций обладает следующим составом:
|
Активный ингредиент растворили в смеси Полиэтиленгликоля 400 и воды для инъекций (часть). РН довели до 5,0 уксусной кислотой. Объем довели до 1,0 мл добавлением остаточного количества воды. Раствор отфильтровали, разлили в ампулы с использованием необходимого избытка и стерилизовали.
Следующие примеры предназначены для более подробной иллюстрации настоящего изобретения. Они, однако, не должны рассматриваться как каким-либо образом ограничивающие изобретение.
Примеры
Пример 1
1-(2-(2-Аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин
а) 1-(2-(Этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ол
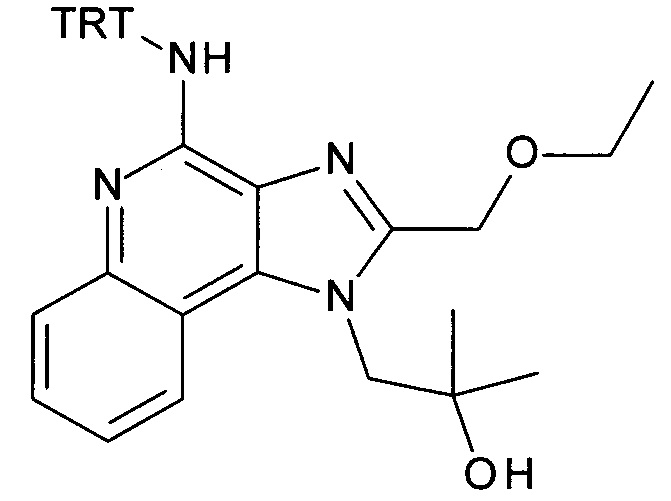
4-амино-2-(этоксиметил)-α,α-диметил-1H-имидазо[4,5-с]хинолин-1-этанол (CAN 144875-48-9, 1.6 г, 5.09 ммоль) объединили с ацетонитрилом (60 мл) с получением белой суспензии. Затем триэтиламин (1.77 мл, 12.7 ммоль) и тритилхлорид (1.7 г, 6.11 ммоль) добавили в атмосфере аргона, при перемешивании. Реакционную смесь облучали в микроволновой печи при 100°С в течение 30 минут. После перемешивания смеси продукт преципитировался и его выделили посредством фильтрации при 0°С, промыли холодным ацетонитрилом и высушили с получением соединения, указанного в заголовке (2.37 г, 83%), в виде белого осадка; MS (ESI): 557.5 (МН+).
b) трет-бутил 2-(1-(2-(этоксиметил)-4-(тритиламино)-1H-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этилкарбамат
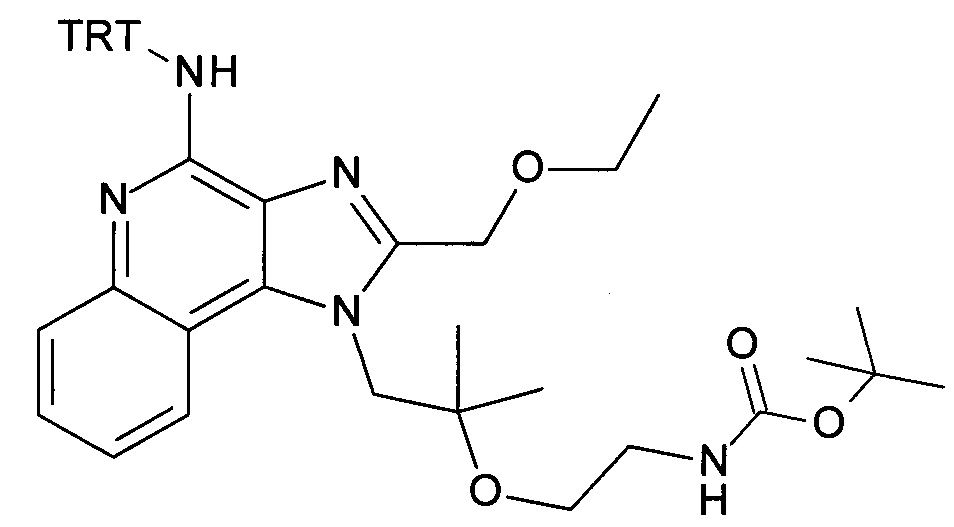
Дисперсию гидрида натрия в масле 60% (173 мг, 4.32 ммоль) объединили с ДМФ (15 мл) с получением бесцветной суспензии. Смесь охладили до 0°С при перемешивании, и при этой температуре раствор 1-(2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ола (1.85 г, 3.32 ммоль) в ДМФ (15 мл) добавили по каплям в течение периода 10 мин. Затем реакционную смесь перемешивали в течение 1 часа при 0°С и в течение 30 минут при комнатной температуре с получением желтого раствора. К этому раствору добавили при 0°С 2,2-диоксид-1,2,3-оксатиазолидин-3-карбоновая кислота-1,1-диметилэтиловый эфир (CAN 459817-82-4, 964 мг, 4.32 ммоль). Температуре дали достигнуть комнатной температуры и смесь перемешивали в течение ночи. Смесь влили в воду со льдом и экстрагировали этилацетатом. Органические слои промыли водой/солевым раствором (2:1), объединили, высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель, 0-100% этилацетата в гептане) с получением соединения, указанного в заголовке (1.47 г, 63%) в виде белой пены; LC-MS (Площадь УФ пика, ESI) 98.7%, 700.3850 (МН+).
с) 1-(2-(2-Аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин
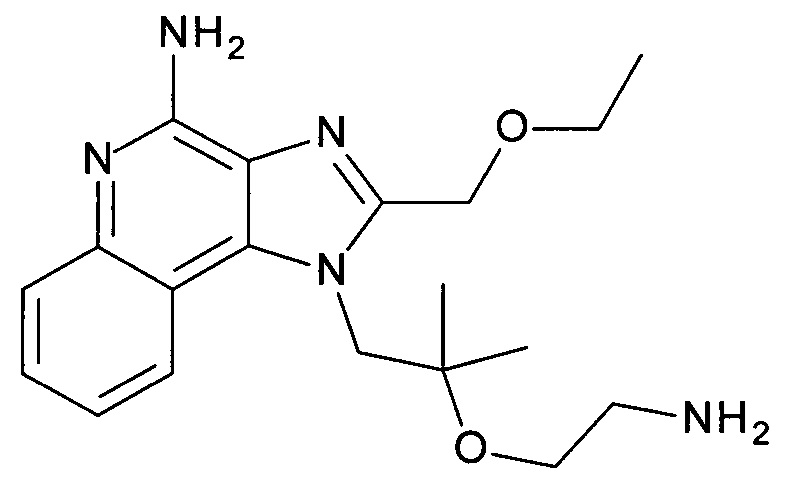
трет-бутил 2-(1-(2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этилкарбамат (1.45 г, 2.07 ммоль) объединили дихлорметаном (ДХМ, 12 мл) с получением бесцветного раствора. ТФУ (6.0 мл, 77.9 ммоль) добавили, и смесь перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь охладили до 0°С, добавили 2н раствор гидроксида натрия (40 мл), и щелочной раствор экстрагировали дихлорметаном. Органические слои объединили, высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель, 0-10% метанол в ДХМ) с получением соединения, указанного в заголовке (0.62 г, 83%), в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 97.5%, 358.2238 (МН+).
Пример 2
1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-аминоацетат
а) 2-(2-(Этоксиметил)-1-(2-гидрокси-2-метилпропил)-1Н-имидазо[4,5-с]хинолин-4-ил)изоиндолин-1,3-дион
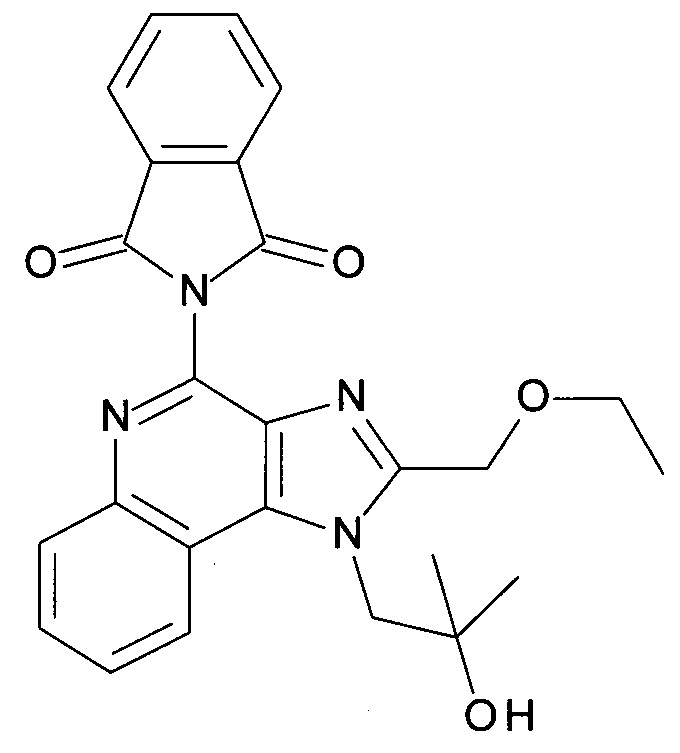
4-амино-2-(этоксиметил)-α,α-диметил-1Н-имидазо[4,5-с]хинолин-1-этанол (CAN 144875-48-9, 3.0 г, 9.54 ммоль) объединили с толуолом (21.0 мл) с получением белой суспензии. 1,4-Диазабицикло[2.2.2]октан (3.21 г, 28.6 ммоль) и фталоилхлорид (1.65 мл, 11.5 ммоль) добавили при перемешивании и реакционную смесь перемешивали при 110°С в течение 4 часов. После охлаждения смесь разбавили этилацетатом (300 мл) и промыли с помощью 1 н соляной кислоты. Фазы разделили и водный слой экстрагировали этилацетатом. Органические слои объединили, высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. После перемешивания остатка с этилацетатом (50 мл) продукт преципитировался, его отфильтровали и высушили под вакуумом (1.9 г). Маточную жидкость сконцентрировали и получили после флеш-хроматографии (силикагель, 50 г, 0% - 100% EtOAc в гептане) другую часть продукта (0.59 г). В сумме 2.49 г (59%) соединения, указанного в заголовке, выделили в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 96%, 445.2 (МН+).
b) 1-(4-(1,3-Диоксоизоиндолин-2-ил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-(1,3-диоксоизоиндолин-2-ил)ацетат
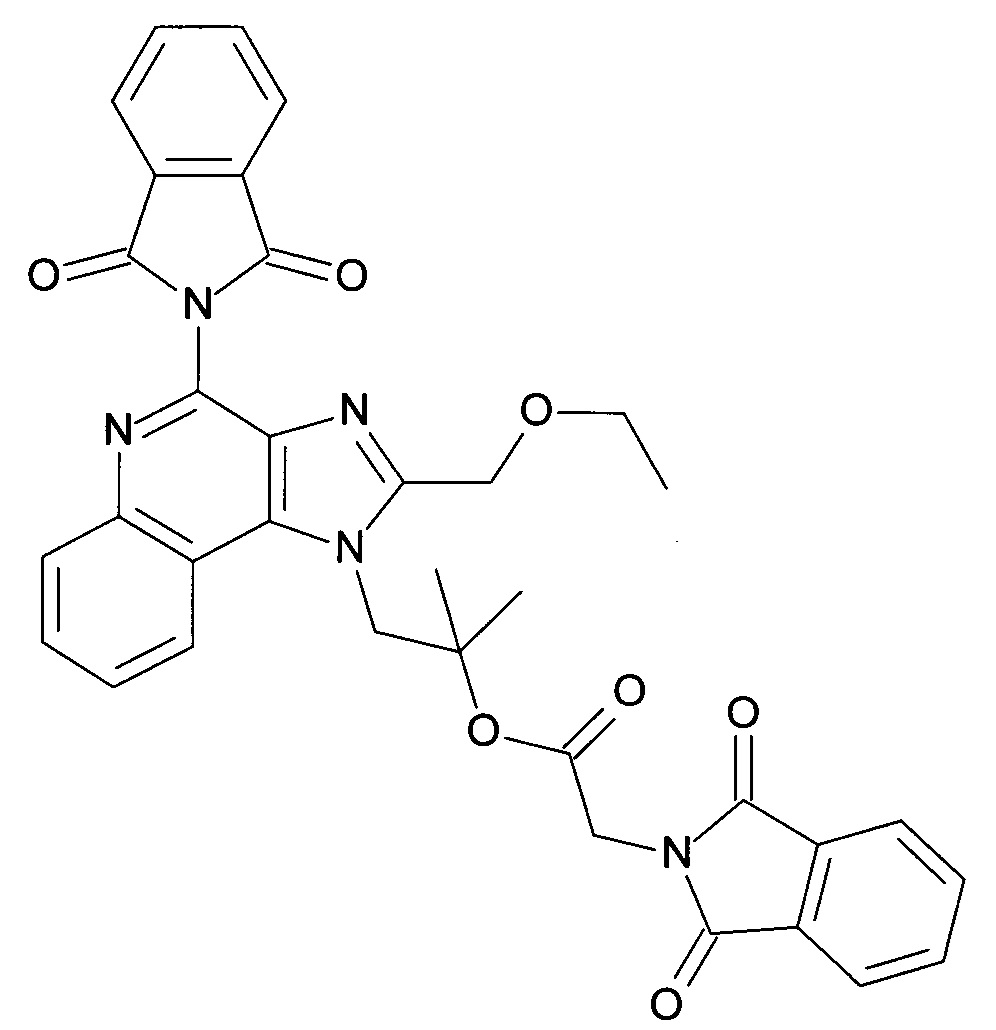
2-(2-(Этоксиметил)-1-(2-гидрокси-2-метилпропил)-1H-имидазо[4,5-с]хинолин-4-ил)изоиндолин-1,3-дион (1600 мг, 3.6 ммоль), 1,3-дигидро-1,3-диоксо-2H-изоиндол-2-уксусную кислоту (CAN 4702-13-0, 2.22 г, 10.8 ммоль) и 4-(1-пирролидинил)-пиридин (800 мг, 5.4 ммоль) объединили с помощью ДХМ (36 мл) с получением белой суспензии. Добавили N,N-диизопропилкарбодиимид (1.68 мл, 10.8 ммоль) и молекулярные сита. Реакционную смесь нагрели до 50°С и перемешивали в течение 2 часов и, после охлаждения, отфильтровали. Фильтрат разбавили с помощью ДХМ (150 мл) и промыли 1 н соляной кислотой и водой. Водные фазы экстрагировали с помощью ДХМ, органические фазы объединили, высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Диизопропилмочевину удалили после тритурирования с помощью ДХМ/метанола посредством фильтрации, фильтрат сконцентрировали и остаток очистили с помощью флеш-хроматографии (силикагель, 0% - 100% этилацетат в гептане) с получением светло-желтого осадка. Кристаллизация из ДМСО дала первую часть продукта (1.95 г), и маточная жидкость после концентрирования и препаративной ВЭЖХ другие 0.13 г продукта. В сумме 2.08 г (91%) соединения, указанного в заголовке, выделили в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 95.9%, 632.2157 (МН+).
с) 1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-аминоацетат
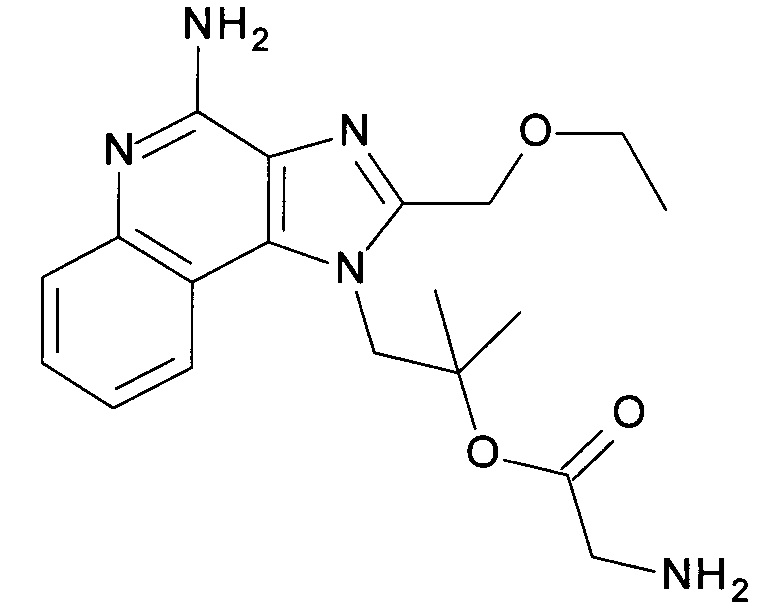
1-(4-(1,3-Диоксоизоиндолин-2-ил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ила 2-(1,3-диоксоизоиндолин-2-ил)ацетат (300 мг, 475 мкмоль) объединили с ТГФ (5 мл) с получением белой суспензии. К этой суспензии добавили гидразин в воде (1000 мкл, 11.2 ммоль) и раствор перемешивали в течение 0.75 часа при комнатной температуре. Смесь охладили до 0°С, добавили 1 н соляную кислоту (30 мл) и ДХМ (50 мл). Белый преципитат (фталилгидразид) удалили посредством фильтрации и органическую фазу отбросили. Водную фазу лиофилизировали и остаток растворили с помощью ацетонитрила (20 мл) и DIEA (рН 10). После эвапорации растворителя, остаток очистили с помощью препаративной ВЭЖХ (Gemini NX 3u 50×4.6 мм; ацетонитрил/вода/Еt3N) с получением соединения, указанного в заголовке (111 мг, 63%) в виде аморфного белого осадка; LC-MS (Площадь УФ пика, ESI) 99%, 372.4 (МН+).
Пример 3
N-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этил)никотинамид
Никотиновую кислоту (11.4 мг, 92.3 мкмоль) объединили с ДМФ (1.0 мл) с получением бесцветного раствора. К этому раствору добавили TBTU (29.6 мг, 92.3 мкмоль), и DIEA (54.2 мг, 71.8 мкл). В заключение добавили 1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин (30 мг, 83.9 мкмоль) и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Летучие компоненты удалили в глубоком вакууме при 40°С. Остаток растворили в смеси этилацетата (5 мл) и метанола (0.5 мл). Добавили 1 н раствор гидроксида натрия (1.5 мл) и через несколько минут перемешивания слои разделили. Водный слой экстрагировали этилацетатом. Органические фазы объединили, высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель-NH2, 0% - 10% метанол в ДХМ) с получением продукта, указанного в заголовке (33 мг, 85%), в виде белой пены; LC-MS (Площадь УФ пика, ESI) 88%, 463.2459 (МН+).
Пример 4
N-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этил)ацетамид
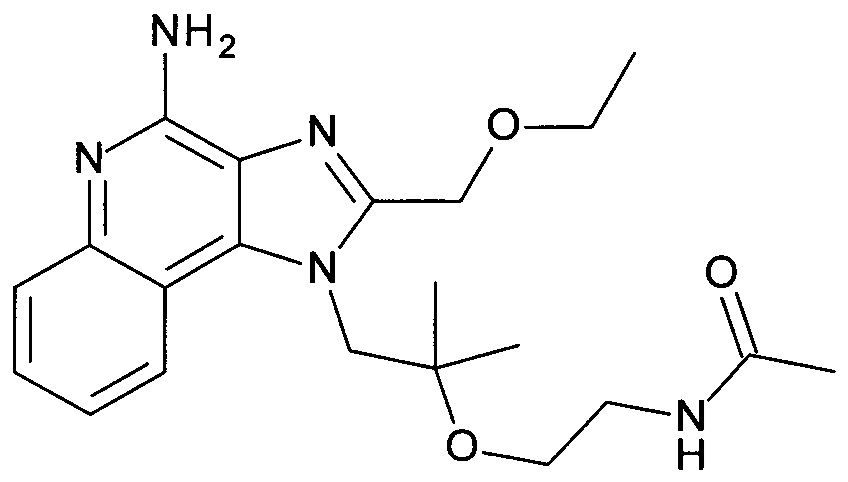
1-(2-(2-Аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин (32 мг, 89.5 мкмоль) объединили с ДХМ (1.0 мл) с получением бесцветного раствора. К этому раствору добавили триэтиламин (25.0 мкл, 179 мкмоль) и ацетилхлорид (7.00 мкл, 98.5 мкмоль) и реакционную смесь перемешивали 18 часов при комнатной температуре. После концентрирования смеси под вакуумом остаток очистили с помощью флеш-хроматографии (силикагель-NH2, 0% - 10% метанол в ДХМ) с получением соединения, указанного в заголовке (19 мг, 53%), в виде белой пены; LC-MS (Площадь УФ пика, ESI) 97%, 400.2348 (МН+).
Пример 5
3-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этиламино)пропан-1-ол
а) 2-(Этоксиметил)-1-[2-метил-2-[2-(3-тритилоксипропиламино)этокси]пропил]-имидазо[4,5-с]хинолин-4-амин
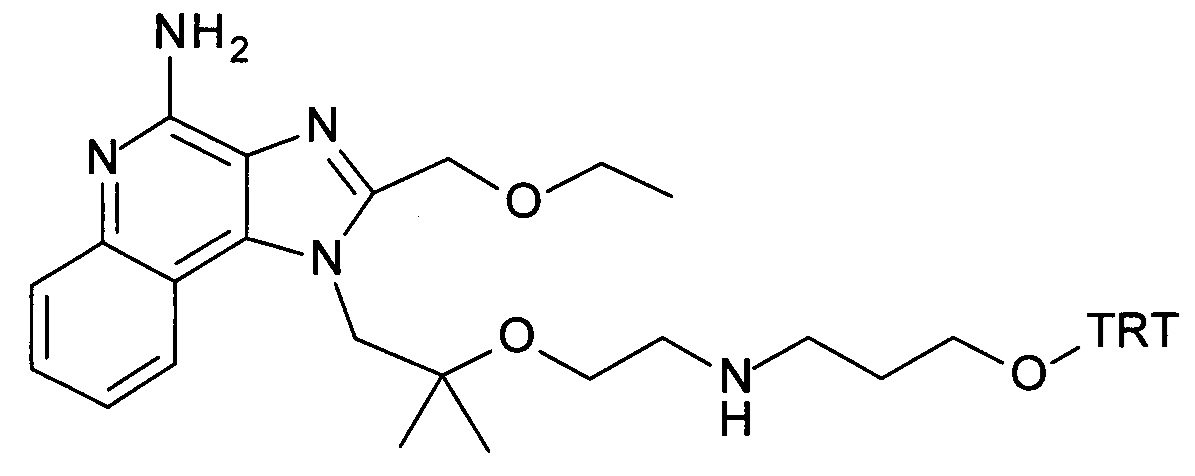
1-(2-(2-Аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин (50 мг, 140 мкмоль) и 3-(трифенилметокси)-пропаналь (CAN 67057-68-5; 44.3 мг, 140 мкмоль) объединили с этанолом (500 мкл) с получением светло-желтой суспензии. Смесь перемешивали в течение 2 часов при комнатной температуре. После этого добавили борогидрид натрия (5.82 мг, 154 мкмоль) и перемешивание при комнатной температуре продолжили в течение ночи. Смесь перемешивали с водой (2 мл) и этилацетатом (5 мл), высушили пропусканием через картридж ChemElut® и сконцентрировали под вакуумом. Неочищенное вещество очистили с помощью флеш-хроматографии (силикагель-NH2, 0% - 100% этилацетат в гептане) с последующей другой флеш-хроматографией (силикагель, 0 - 10% метанол в этилацетате) с получением соединения, указанного в заголовке (10 мг, 11%) в виде желтоватой смолы; LC-MS (Площадь УФ пика, ESI) 89%, 658.5 (МН+).
b) 3-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этиламино)пропан-1-ол
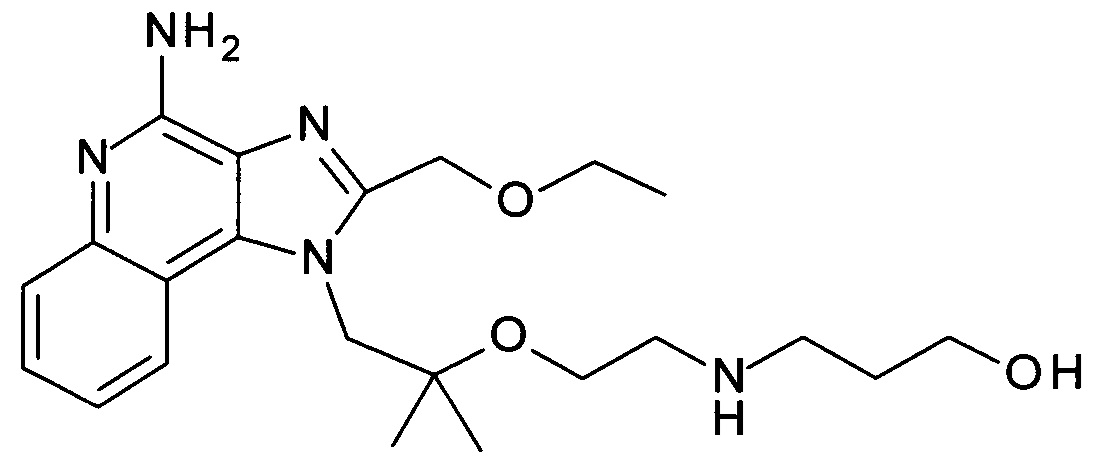
2-(Этоксиметил)-1-[2-метил-2-[2-(3-тритилоксипропиламино)этокси]пропил]-имидазо[4,5-с]хинолин-4-амин (10 мг, 15.2 мкмоль) объединили с ДХМ (1 мл) с получением бесцветного раствора. ТФУ (100 мкл, 1.3 ммоль) добавили и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Смесь сконцентрировали под вакуумом и остаток очистили с помощью флеш-хроматографии (силикагель-NH2, 0% - 30% метанол в этилацетате) с получением соединения, указанного в заголовке (2.3 мг, 36%), в виде бесцветной смолы; LC-MS (Площадь УФ пика, ESI) 95.6%, 416.2661 (МН+).
Пример 6
трет-бутил 6-(2-(1-(4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-илокси)этиламино)-6-оксогексилкарбамат
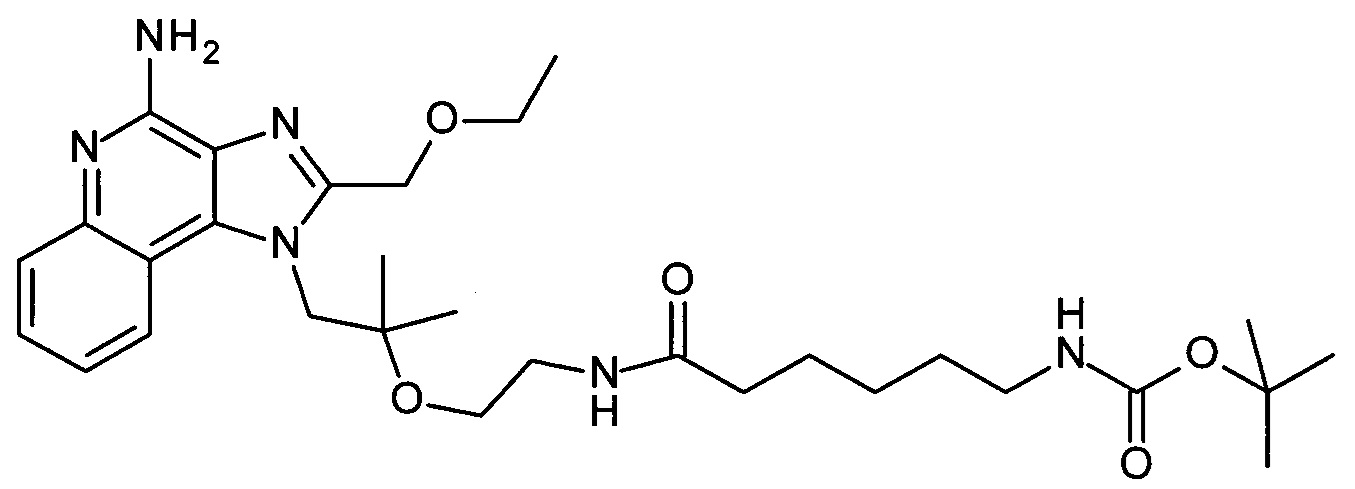
6-(трет-Бутоксикарбониламино)гексановую кислоту (197 мг, 850 мкмоль) объединили с ДМФ (10.9 мл) с получением бесцветного раствора. О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (TBTU, 300 мг, 936 мкмоль) и N,N-диизопропилэтиламин (DIEA, 728 мкл, 4.25 ммоль) добавили при перемешивании в инертной атмосфере. Затем добавили 1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин (Пример 1 с, 304 мг, 850 мкмоль) и смесь перемешивали в течение 2 часов при комнатной температуре. Смесь сконцентрировали под вакуумом, растворили в этилацетате (5 мл), перемешивали в течение 1 мин с холодным раствором гидроксида натрия (1 н) и высушили пропусканием через ChemElut® (10 г). Органическую фазу сконцентрировали под вакуумом снова и остаток очистили с помощью флеш-хроматографии (силикагель, 10% метанол в дихлорметане) с получением соединения, указанного в заголовке (0.135 г, 23%), в виде светло-коричневого масла; LC-MS (Площадь УФ пика, ESI) 83%, 571.5 (МН+).
Пример 7
(Е)-Этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)акрилат
а) 5-амино-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1H-имидазол-4-карбонитрил
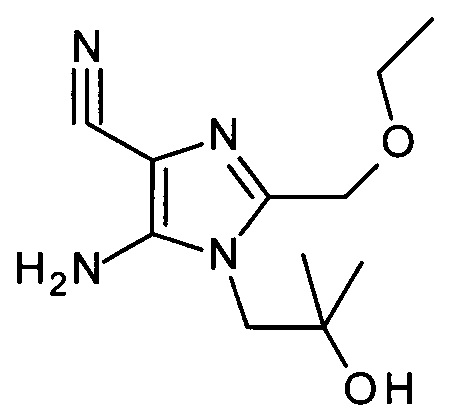
Триэтиламин (5.28 мл, 37.9 ммоль) добавили при перемешивании к суспензии 2-аминомалононитрила 4-метилбензолсульфоната (8 г, 31.6 ммоль) в ТГФ (120 мл) с получением светло-коричневого раствора. 1,1,1,2-тетраэтоксиэтан (7.82 г, 37.9 ммоль) добавили и реакционную смесь перемешивали в атмосфере аргона при температуре кипения. Через 4 ч добавили дополнительное количество 2.3 г 1,1,1,2-тераэтоксиэтана и смесь нагревали в течение дополнительных 2 ч. Смеси дали остыть до комнатной температуры, добавили триэтиламин (5.28 мл, 37.9 ммоль) и 1-амино-2-метилпропан-2-ол (3.63 мл, 37.9 ммоль) и реакционную смесь перемешивали в течение ночи. Смесь сконцентрировали под вакуумом, и остаток разделили между этилацетатом (200 мл, 2×150 мл) и раствором бикарбоната натрия (2 М, 100 мл). Органические фазы объединили, высушили над MgSO4, сконцентрировали под вакуумом и очистили с помощью флеш-хроматографии (силикагель, 0% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке после стадии кристаллизации из гептана (4.47 г, 59%) в виде белого кристаллического осадка; LC-MS (Площадь УФ пика, ESI) 100%, 239.1514 (МН+).
b) 2-(Этоксиметил)-1-(2-гидрокси-2-метилпропил)-5-йод-1Н-имидазол-4-карбонитрил
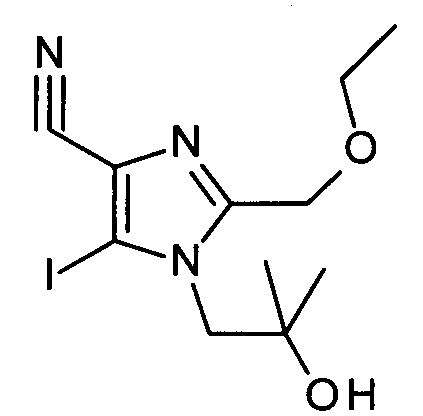
Дийодометан (14.5 мл, 180 ммоль) добавили в атмосфере аргона при перемешивании к суспензии 5-амино-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1Н-имидазол-4-карбонитрила (4.36 г, 18.3 ммоль) в хлороформе (170 мл). Реакционную смесь нагрели до 80°С, раствор изоамилнитрила (9.86 мл, 73.2 ммоль) в хлороформе (30 мл) добавили с помощью шприцевого насоса в течение периода 40 мин и смесь перемешивали в течение дополнительных 30 мин при 80°С. После охлаждения до комнатной температуры смесь сконцентрировали под вакуумом и очистили с помощью флеш-хроматографии (силикагель, 20% - 50% этилацетат в гептане) с получением соединения, указанного в заголовке (3.66 г, 57%), в виде коричневого масла; LC-MS (Площадь УФ пика, ESI) 90%, 350.0374 (МН+).
с) Метил 3-амино-4-[5-циано-2-(этоксиметил)-3-(2-гидрокси-2-метилпропил)-1Н-имидазол-4-ил]бензоат
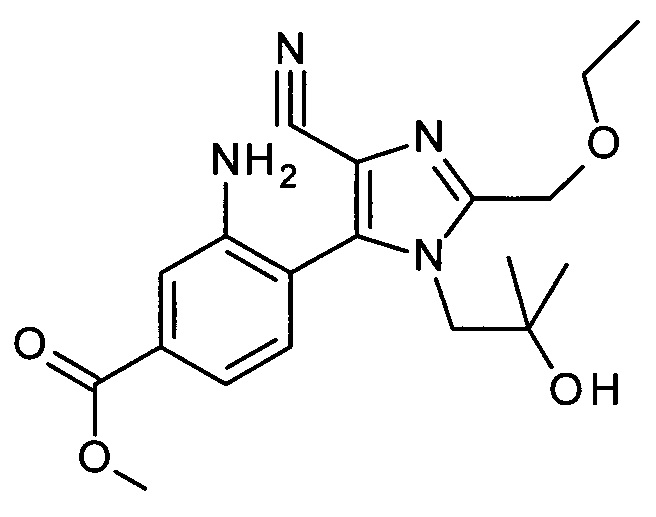
В инертной атмосфере 2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-5-йод-1H-имидазол-4-карбонитрил (3.66 г, 10.5 ммоль) объединили с диметоксиэтаном (43 мл) с получением светло-коричневого раствора. К этому раствору добавили Pd(OAc)2 (118 мг, 524 мкмоль), трифенилфосфин (275 мг, 1.05 ммоль) и метил 3-амино-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (4.36 г, 15.7 ммоль) при перемешивании. В заключение добавили Na2CO3 (2 М, 15.7 мл, 31.4 ммоль). Реакционную смесь нагрели до 100°С и перемешивали в течение 1 часа. После охлаждения до комнатной температуры смесь разделили между этилацетатом (70 мл, 2×50 мл) и водой (70 мл). Органические фазы объединили, высушили над MgSO4, сконцентрировали под вакуумом и очистили с помощью флеш-хроматографии (силикагель, 0% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке (2.79 г, 71%) в виде светло-коричневой пены, которую использовали на следующей стадии без дополнительной очистки.
d) Метил 4-амино-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1Н-имидазо[4,5-с]хинолин-7-карбоксилат
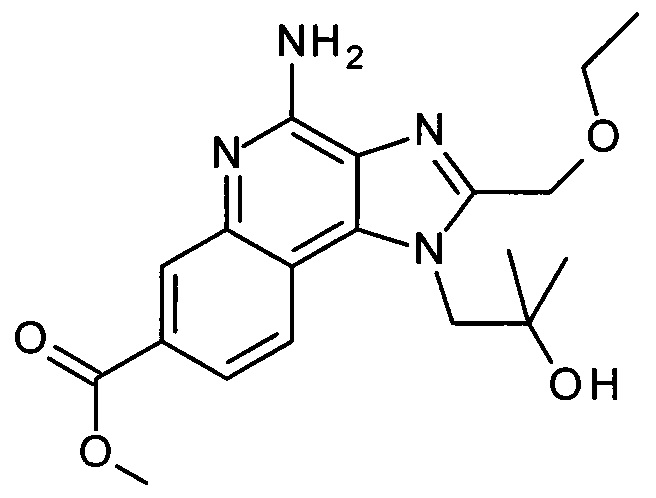
Метил 3-амино-4-(4-циано-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1Н-имидазол-5-ил)бензоат (2.79 г, 7.49 ммоль) объединили с раствором HCl в диоксане (4М, 56.2 мл, 225 ммоль) с получением оранжевого раствора. Реакционную смесь нагрели до 90°С в атмосфере аргона при перемешивании. Через 1 ч при 90°С смесь охладили до комнатной температуры и сконцентрировали под вакуумом с получением бежевого осадка. Осадок растворили в этилацетате (500 мл), промыли смесью воды (100 мл) и насыщенного раствора бикарбоната натрия (250 мл). Водную фазу экстрагировали этилацетатом (2×250 мл), органические фазы объединили, высушили над MgSO4 и сконцентрировали под вакуумом с получением желтого осадка (3.05 г). Твердый материал перекристаллизовали из смеси этилацетат/гептан с получением соединения, указанного в заголовке (2.4 г, 86%), в виде светло-желтого осадка; LC-MS (Площадь УФ пика, ESI) 99%, 373.1884 (МН+).
е) Метил 2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-карбоксилат
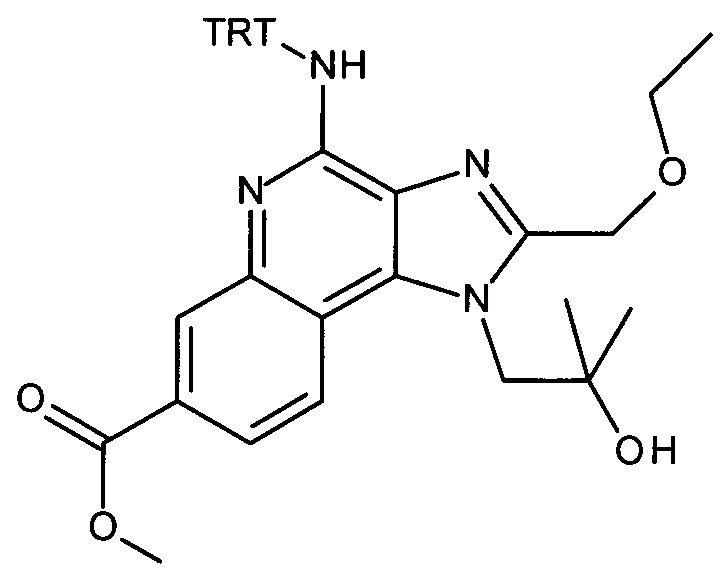
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1а, с использованием метил 4-амино-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1Н-имидазо[4,5-с]хинолин-7-карбоксилата в качестве исходного вещества и выделили (0.77 г, 96%) в виде светло-коричневого осадка; LC-MS (Площадь УФ пика, ESI) 96%, 615.4 (МН+).
f) 2-(Этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-карбоновая кислота
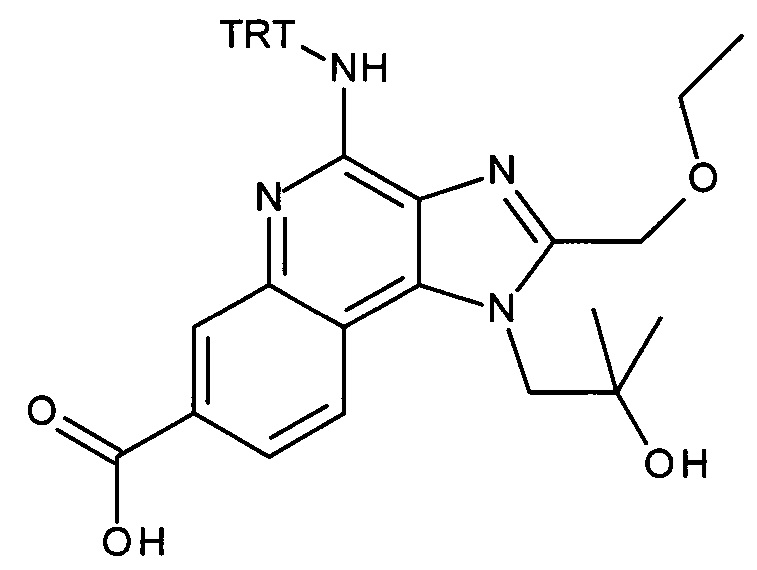
Раствор гидроксида натрия (1 н, 3.51 мл) добавили к метил 2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-карбоксилату (540 мг, 878 мкмоль), растворенному в ТГФ (6.21 мл) и метаноле (621 мкл). Реакционную смесь перемешивали в течение 120 часов при комнатной температуре и сконцентрировали под вакуумом. Остаток разделили между этилацетатом (30 мл) и холодным раствором 1М HCl (5 мл). Водный слой экстрагировали этилацетатом (2×50 мл). Органические слои промыли водой (20 мл) и солевым раствором (20 мл), объединили, высушили над MgSO4, и сконцентрировали под вакуумом с получением соединения, указанного в заголовке (0.499 г, 94%), в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 99%, 601.3 (МН+).
g) 2-(Этоксиметил)-1-(2-гидрокси-2-метилпропил)-N-метокси-N-метил-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-карбоксамид
К раствору 2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1H-имидазо[4,5-с]хинолин-7-карбоновой кислоты (484 мг, 741 мкмоль) в ДМФ (5.19 мл) в инертной атмосфере добавили TBTU (357 мг, 1.11 ммоль), DIEA (388 мкл, 2.22 ммоль) и N,O-диметилгидроксиламина гидрохлорид (108 мг, 1.11 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и после этого смесь сконцентрировали под вакуумом. Остаток разделили между этилацетатом (40 мл, 2×20 мл) и водой (40 мл). Органические фазы промыли водой (2×20 мл), объединили, высушили над MgSO4, сконцентрировали под вакуумом и очистили с помощью флеш-хроматографии (силикагель, 50% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке (0.53 г, колич.), в виде белой пены; LC-MS (Площадь УФ пика, ESI) 92%, 644.4 (МН+).
n) 2-(Этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-карбальдегид
К раствору 2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-N-метокси-N-метил-4-(тритиламино)-1H-имидазо[4,5-с]хинолин-7-карбоксамида (520 мг, 808 мкмоль) в ТГФ (7 мл) в инертной атмосфере добавили при перемешивании при 0°С раствор алюмогидрида лития в ТГФ (1 М, 404 мкл, 404 мкмоль). Смесь перемешивали 1 ч при 0°С, медленно добавили насыщенный раствор хлорида аммония (10 мл) и в заключение добавили воду (20 мл) и этилацетат (30 мл). Фазы разделили, водный слой экстрагировали этилацетатом (30 мл), а органические фазы промыли водой (20 мл), объединили, высушили над MgSO4, и сконцентрировали под вакуумом с получением соединения, указанного в заголовке (0.49 г, колич.), в виде белой пены (LC-MS (Площадь УФ пика, ESI) 83%, 585.4 (МН+)), которую использовали на следующей стадии без дополнительной очистки.
i) Этил (Е)-3-[2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-ил]проп-2-еноат
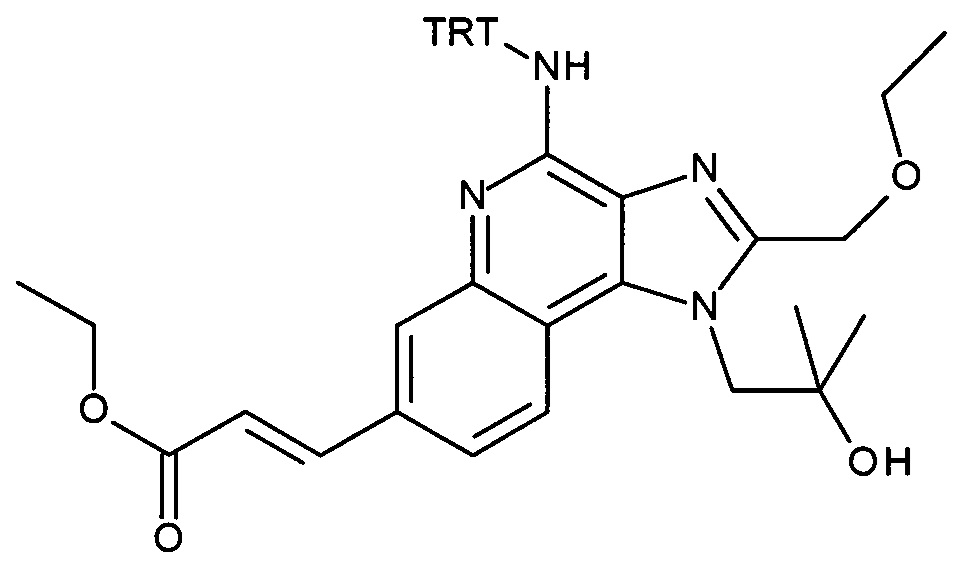
(Карбэтоксиметилен)трифенилфосфоран (422 мг, 1.21 ммоль) добавили в раствор 2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-карбальдегида (472 мг, 808 мкмоль) в дихлорметане (8 мл) в атмосфере аргона. Реакционную смесь перемешивали в течение 20 ч при комнатной температуре и сконцентрировали под вакуумом. Кристаллизация из дихлорметана и диэтилового эфира дала соединение, указанное в заголовке (0.399 г, 75%), в виде белого осадка, LC-MS (Площадь УФ пика, ESI) 89%, 655.4 (МН+).
j) Этил (Е)-3-[1-[2-[2-(трет-бутоксикарбониламино)этокси]-2-метилпропил]-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-ил]проп-2-еноат
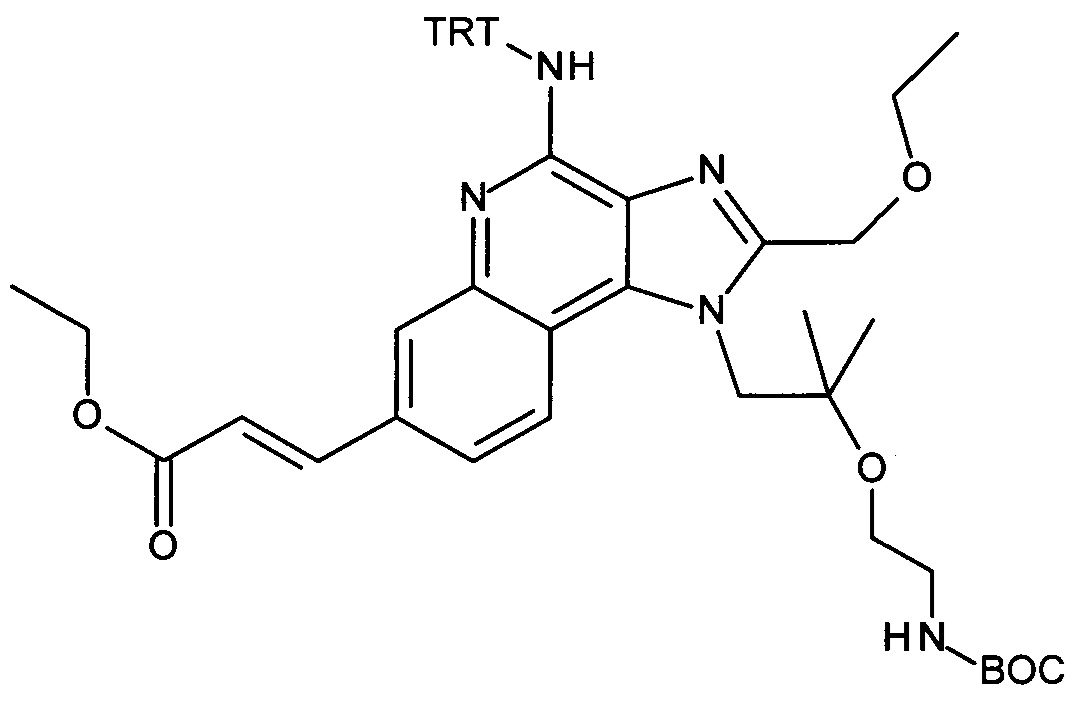
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1b, с использованием этил (Е)-3-[2-(этоксиметил)-1-(2-гидрокси-2-метил пропил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-ил]проп-2-еноата в качестве исходного вещества и выделили (0.28 г, 42%) в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 76%, 798.6 (МН+).
k) Этил (Е)-3-[4-амино-1-[2-(2-аминоэтокси)-2-метилпропил1-2-(этоксиметил)имидазо[4,5-с]хинолин-7-ил]проп-2-еноат
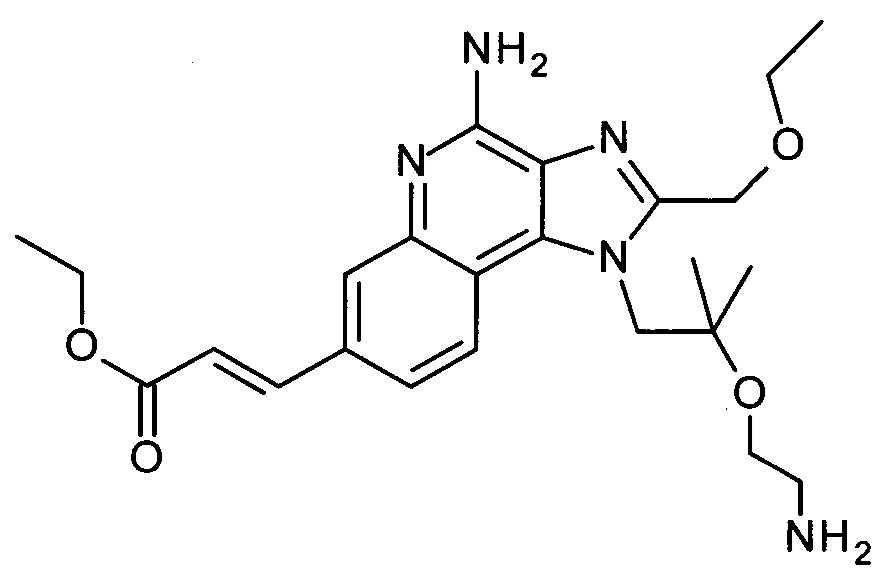
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1с, с использованием этил (Е)-3-[1-[2-[2-(трет-бутоксикарбониламино)этокси]-2-метилпропил]-2-(этоксиметил)-4-(тритиламино)-1H-имидазо[4,5-с]хинолин-7-ил]проп-2-еноата в качестве исходного вещества и выделили (21 мг, 34%) в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 93%, 456.4 (МН+).
Пример 8
Этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноата гидрохлорид
а) Этил 3-[1-[2-[2-(трет-бутоксикарбониламино)этокси]-2-метилпропил]-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-ил]пропаноат
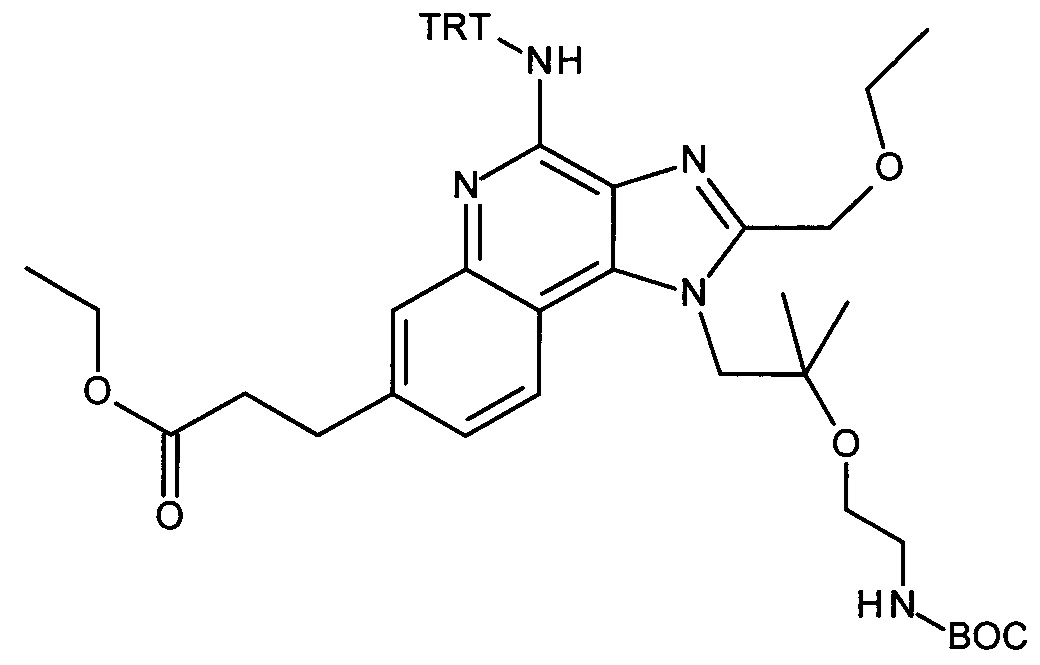
Палладий на угле 10% (100 мг, 940 мкмоль) в этаноле (10 мл) добавили к суспензии (Е)-этил 3-(1-(2-(2-(трет-бутоксикарбониламино)этокси)-2-метилпропил)-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-ил)акрилата (280 мг, 351 мкмоль) в этилацетате (30 мл). Смесь перемешивали в течение 6 ч в атмосфере водорода при комнатной температуре, отфильтровали и растворитель удалили под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель-NH2, 0% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке (0.163 г, 58%) в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 74%, 800.5 (МН+).
b) Этил 3-(4-амино-1-(2-(2-аминоэтокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноата гидрохлорид
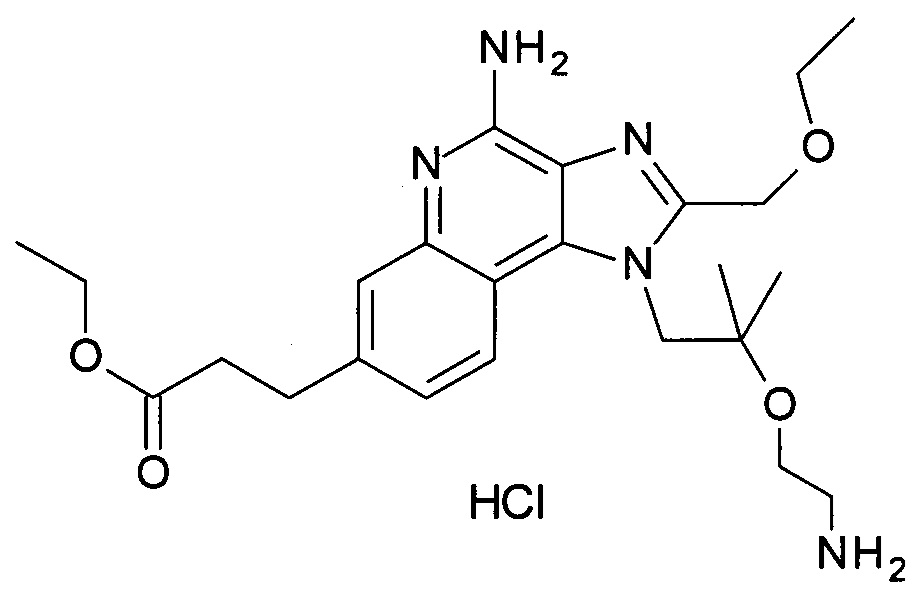
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1с, с использованием этил 3-[1-[2-[2-(трет-бутоксикарбониламино)этокси]-2-метилпропил]-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-7-ил]пропаноата в качестве исходного вещества и выделили (15 мг, 56%) в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 93%, 458.4 (МН+).
Пример 9
Этил 3-(4-амино-1-(2-(2-аминоацетокси)-2-метилпропил)-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-7-ил)пропаноат
а) Этил 3-[4-[бис(бензилоксикарбонил)амино]-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1H-имидазо[4,5-с]хинолин-7-ил]пропаноат
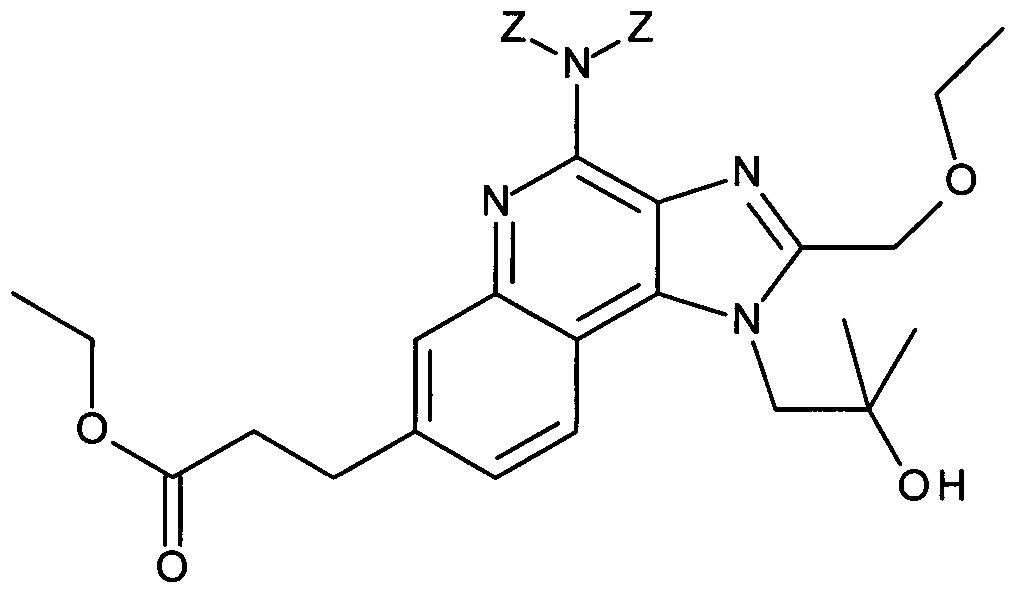
К суспензии этил 3-(4-амино-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1H-имидазо[4,5-с]хинолин-7-ил)пропаноата гидрохлорида (300 мг, 665 мкмоль), DMAP (15 мг, 123 мкмоль) и DIEA (1.86 мл, 10.6 ммоль) в дихлорметане (6 мл) добавили бензилхлорформиат (380 мкл, 2.66 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 ч, дополнительное количество бензилхлорформиата (908 60 мкл, 5.32 ммоль) добавили и перемешивание продолжили в течение дополнительных 6 ч. После этого смесь влили в дихлорметан (50 мл), экстрагировали HCl (1М, 20 мл) и водой (20 мл). Водный слой экстрагировали дихлорметаном (50 мл), органические фазы объединили, высушили над MgSO4 и сконцентрировали под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель, 0% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке (0.459 г, колич.) в виде бесцветного масла; LC-MS (Площадь УФ пика, ESI) 86%, 683.5 (МН+).
b) Этил 3-[4-(бензилоксикарбониламино)-1-[2-[2-(дибензиламино)ацетил]окси-2-метилпропил]-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-7-ил]пропаноат
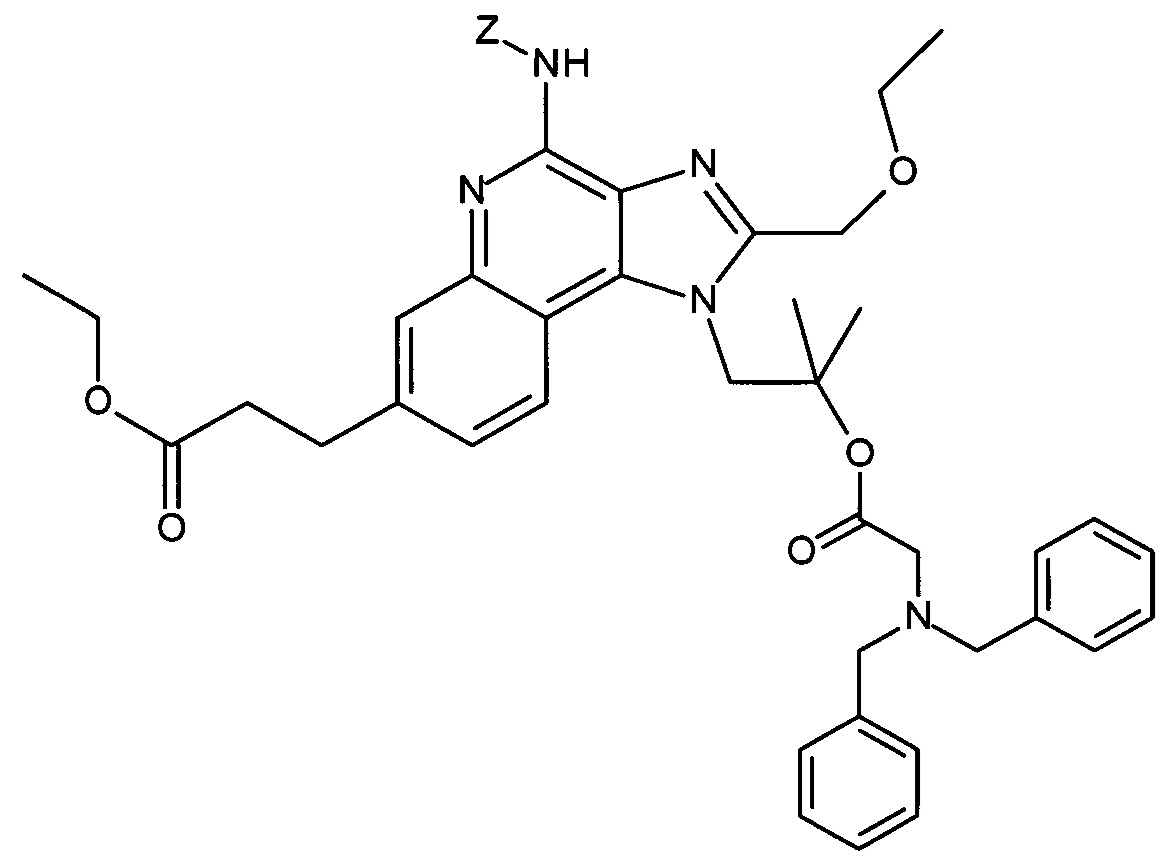
К суспензии этил 3-(4-(бис(бензилоксикарбонил)амино)-2-(этоксиметил)-1-(2-гидрокси-2-метилпропил)-1H-имидазо[4,5-с]хинолин-7-ил)пропаноата (300 мг, 439 мкмоль), 2-(дибензиламино)уксусной кислоты (337 мг, 1.32 ммоль) и DMAP (107 мг, 879 мкмоль) в дихлорметане (4 мл) добавили 1-этил-3-(3-диметиламинопропил)карбодиимид (253 мг, 1.32 ммоль) и молекулярные сита (300 мг). Через 1 ч перемешивания при комнатной температуре исходные соединения растворились, перемешивание при комнатной температуре продолжили в течение 6 ч и смесь оставили на выходные. Остаток разбавили дихлорметаном (50 мл) и промыли HCl (1 М, 20 мл) и водой (20 мл). Водную фазу экстрагировали дихлорметаном (50 мл), органические фазы объединили, высушили над MgSO4 и сконцентрировали под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель, 0% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке (182 мг, 52%), в виде светло-желтого масла; LC-MS (Площадь УФ пика, ESI) 98%, 784.6 (МН+).
с) Этил 3-(4-амино-1-(2-(2-аминоацетокси)-2-метилпропил)-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-7-ил)пропаноат
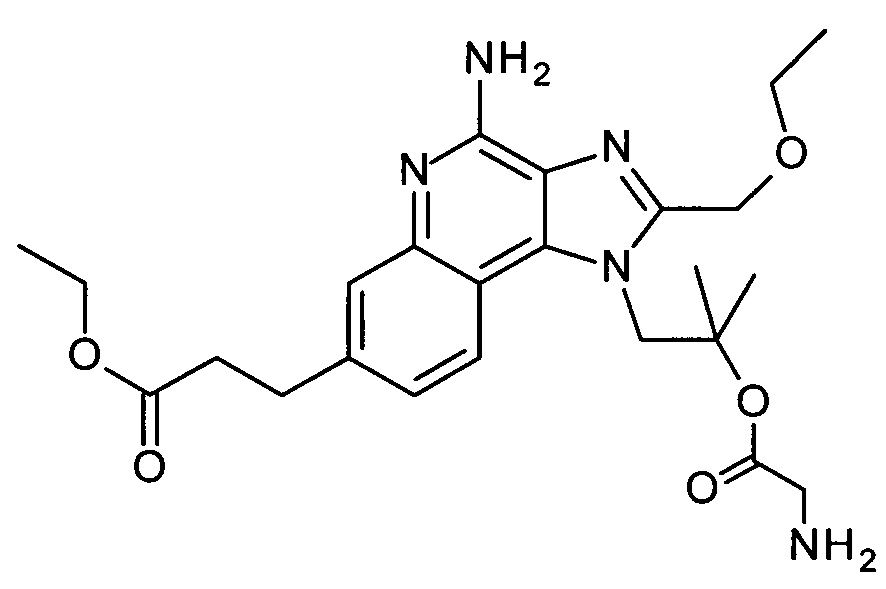
Этил 3-(4-(бензилоксикарбониламино)-1-(2-(2-(дибензиламино)ацетокси)-2-метилпропил)-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-7-ил)пропаноат (182 мг, 232 мкмоль) объединили этилацетатом (20 мл) с получением светло-желтого раствора. Палладий на угле 10% (600 мг, 232 мкмоль) добавили и смесь перемешивали в течение 20 ч при комнатной температуре в атмосфере Н2. Затем смесь отфильтровали через целит® и сконцентрировали под вакуумом. Неочищенное вещество очистили с помощью флеш-хроматографии (силикагель, 20 г, 0% - 30% метанол в ТГФ) до 27 мг загрязненного образца, который дополнительно очистили с помощью препаративной ВЭЖХ с получением соединения, указанного в заголовке (6 мг, 5%), в виде светло-желтого масла; LC-MS (Площадь УФ пика, ESI) 79%, 516.5 (МНСОО-).
Пример 10
1-(2-(2-Аминоэтокси)-2-метилпропил)-2-пентил-1Н-имидазо[4,5-с]хинолин-4-амин
а) 2-метил-1-(2-пентил-1H-имидазо[4,5-с]хинолин-1-ил)пропан-2-ол
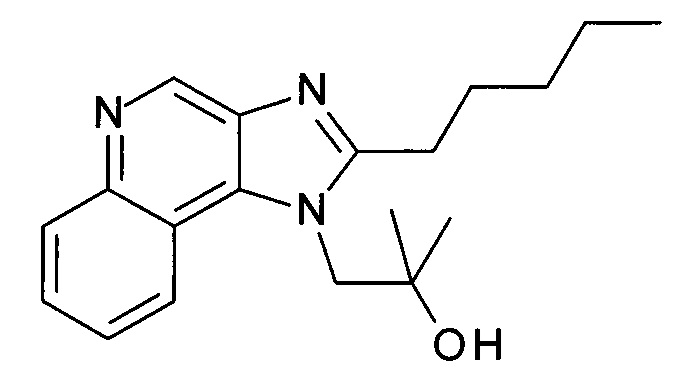
К раствору 1-(3-аминохинолин-4-иламино)-2-метилпропан-2-ола (CAN 129655-59-0, 0.94 г, 4.06 ммоль) в дихлорметане (9.5 мл) в инертной атмосфере добавили по каплям при перемешивании при комнатной температуре в течение периода 6 мин раствор гексаноилхлорида (422 мкл, 4.47 ммоль) в дихлорметане (6.4 мл). Через 3 ч добавили дополнительное количество гексаноилхлорида (192 мкл, 2.03 ммоль) и перемешивание продолжили в течение дополнительного часа и в заключение смесь сконцентрировали под вакуумом. Остаток перерастворили в этаноле (13.5 мл) и, после добавления раствора гидроксида натрия (1 М, 10.8 мл), смесь нагревали при перемешивании в аргоне до 90°С. Через 75 мин смеси дали остыть до комнатной температуры и сконцентрировали под вакуумом. Остаток, смешанный с этилацетатом (30 мл) и водой (5 мл), высушили посредством фильтрации через ChemElut® и этилацетат удалили под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель-NН2, 0% - 100% этилацетат в гептане) с получением соединения, указанного в заголовке (747 мг, 89%) в виде желтого масла; LC-MS (Площадь УФ пика, ESI) 95%, 312.2 (МН+).
b) 2-метил-1-(5-оксидо-2-пентил-1H-имидазо[4,5-с]хинолин-5-ий-1-ил)пропан-2-ол
К раствору 2-метил-1-(2-пентил-1Н-имидазо[4,5-с]хинолин-1-ил)пропан-2-ола (457 мг, 1.36 ммоль) в дихлорметане (21.2 мл) добавили при перемешивании 3-хлорпероксибензойную кислоту (283 мг, 1.64 ммоль) за одну порцию. Реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После этого смесь разделили между холодным дихлорметаном (50 мл) и раствором гидроксида натрия (1 М, 20 мл). Органическую фазу промыли холодной водой (20 мл) и солевым раствором (20 мл) и водные фазы экстрагировали дихлорметаном (2×50 мл). Все органические фазы объединили, высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом, остаток очистили с помощью флеш-хроматографии (силикагель, 0% - 20% метанол в дихлорметане) с получением соединения, указанного в заголовке (360 мг, 80%), в виде светло-желтого осадка; LC-MS (Площадь УФ пика, ESI) 99%, 328.2 (МН+).
с) 1-(4-амино-2-пентил-1H-имидазо[4,5-с]хинолин-1-ил)-2-метил-пропан-2-ол
К раствору 2-метил-1-(5-оксидо-2-пентил-1Н-имидазо[4,5-с]хинолин-5-ий-1-ил)пропан-2-ола (353 мг, 1.08 ммоль) в безводном дихлорметане (47.6 мл) добавили бензоилизоцианат (90%, 286 мкл, 2.05 ммоль) при перемешивании в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивали при температуре кипения в течение 35 мин. Раствор затем сконцентрировали под вакуумом. Остаток объединили с безводным метанолом (17.0 мл) с получением белой суспензии. Добавили раствор метоксида натрия в метаноле (1.7 мл, 9.16 ммоль) и смесь перемешивали в течение 1 ч при 70°С. После этого смесь охладили до комнатной температуры и растворители удалили под вакуумом. Остаток разделили между этилацетатом (30 мл) и водой (15 мл), высушили пропусканием через картридж ChemElut® и сконцентрировали под вакуумом. Остаток очистили с помощью флеш-хроматографии (силикагель, 0% - 10% метанол в дихлорметане) с получением соединения, указанного в заголовке (221 мг, 61%), в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 98%, 327.2185 (МН+).
d) 2-метил-1-[2-пентил-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил]пропан-2-ол
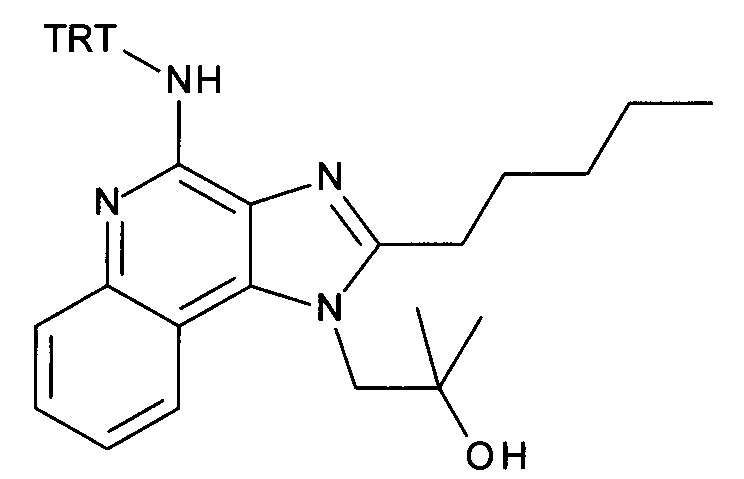
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1а, с использованием 1-(4-амино-2-пентил-1H-имидазо[4,5-с]хинолин-1-ил)-2-метил-пропан-2-ола в качестве исходного вещества и выделили (230 мг, 71%) в виде белого кристаллического осадка; LC-MS (Площадь УФ пика, ESI) 99%, 569.3 (МН+).
f) трет-бутил N-[2-[1,1-диметил-2-[2-пентил-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси]этил]карбамат
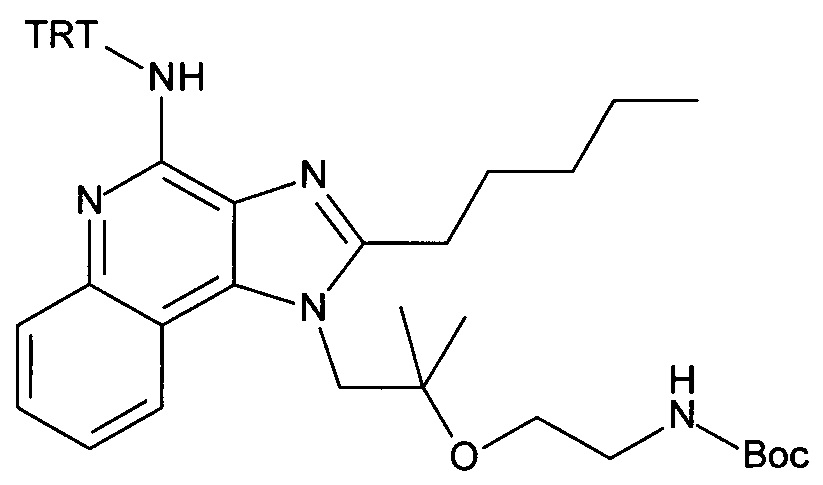
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1b, с использованием 2-метил-1-[2-пентил-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил]пропан-2-ола в качестве исходного вещества и выделили (306 мг, колич.) в виде светло-желтого масла; LC-MS (Площадь УФ пика, ESI) 53%, 712.2 (МН+).
g) 1-(2-(2-Аминоэтокси)-2-метилпропил)-2-пентил-1H-имидазо[4,5-с]хинолин-4-амин
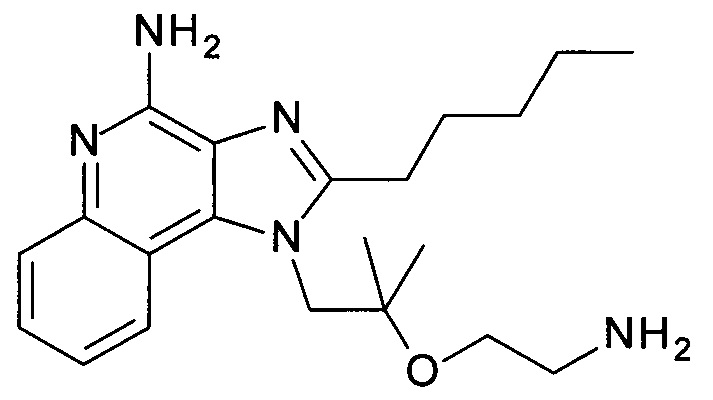
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1с, с использованием трет-бутил N-[2-[1,1-диметил-2-[2-пентил-4-(тритиламино)-1H-имидазо[4,5-с]хинолин-1-ил]этокси]этил]карбамата в качестве исходного вещества и выделили (30 мг, 36%) в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 98%, 370.2622 (МН+).
Пример 11
1-(2-(2-Аминоэтокси)-2-метилпропил)-7-бромо-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-4-амина гидрохлорид
а) 1-[7-бромо-2-(этоксиметил)-4-(тритиламино)-1H-имидазо[4,5-с]хинолин-1-ил]-2-метилпропан-2-ол
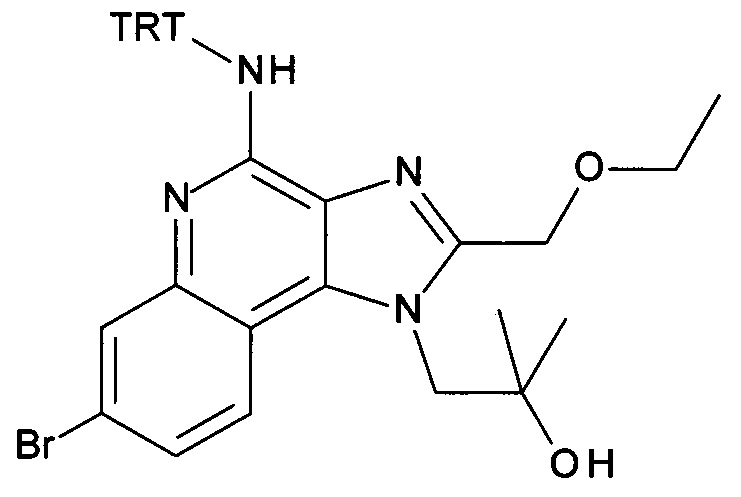
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1а, с использованием 1-(4-амино-7 bromo-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-2-ола в качестве исходного вещества и выделили (100 мг, 88%) в виде светло-коричневого кристаллического осадка; LC-MS (Площадь УФ пика, ESI) 95%, 637.3 (МН+).
b) трет-бутил N-[2-[2-[7-бромо-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил]-1,1-диметил-этокси]этил]карбамат
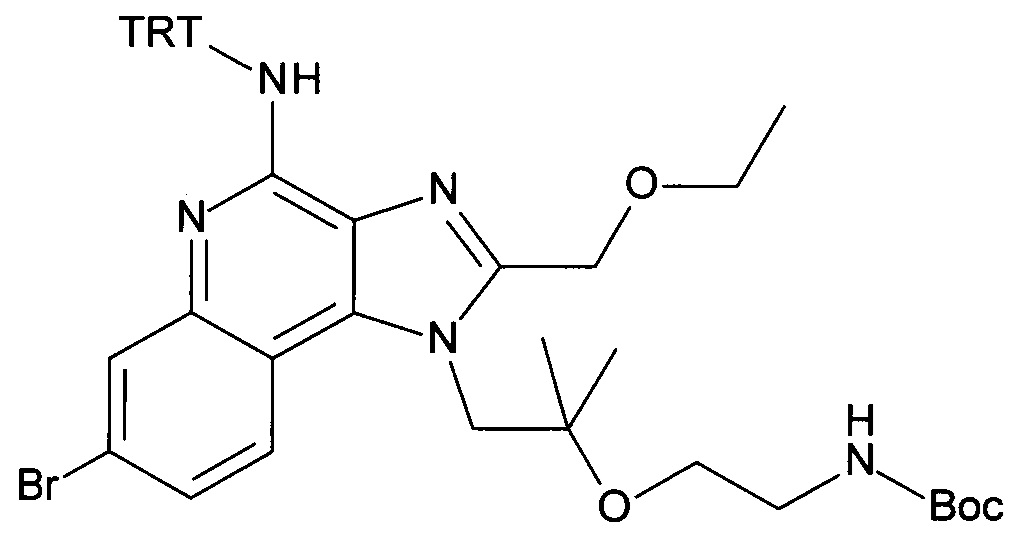
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1b, с использованием 1-[7-бромо-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил]-2-метилпропан-2-ола в качестве исходного вещества и выделили (20 мг, 44%) в виде белого осадка, который использовали без дополнительной очистки на следующей стадии.
с) 1-(2-(2-Аминоэтокси)-2-метилпропил)-7-бромо-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амина гидрохлорид
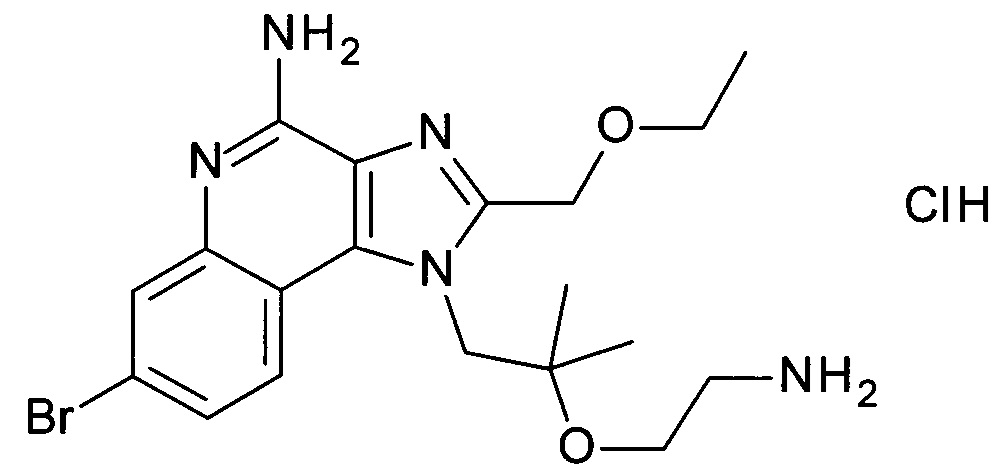
Соединение, указанное в заголовке, синтезировали по аналогии с примером 1с, с использованием трет-бутил N-[2-[2-[7-бромо-2-(этоксиметил)-4-(тритиламино)-1Н-имидазо[4,5-с]хинолин-1-ил]-1,1-диметил-этокси]этил]карбамата в качестве исходного вещества и выделили (7 мг, 58%) в виде белого осадка; LC-MS (Площадь УФ пика, ESI) 99%, 438.2 (МН+).
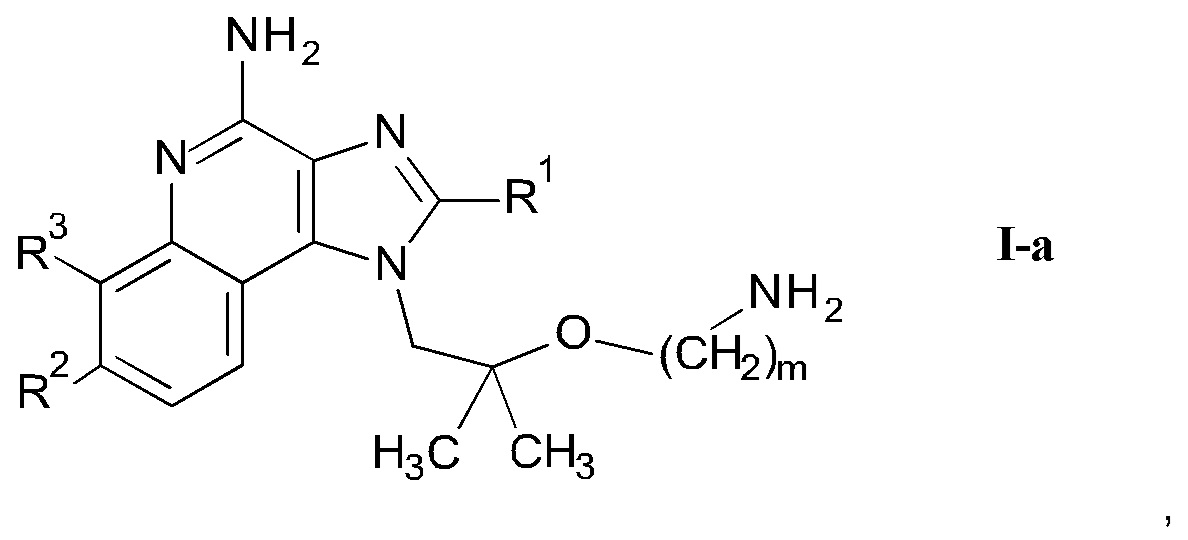
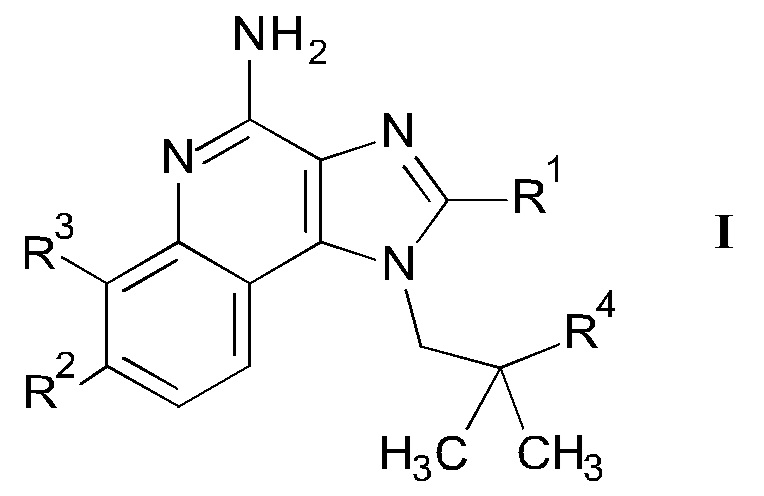
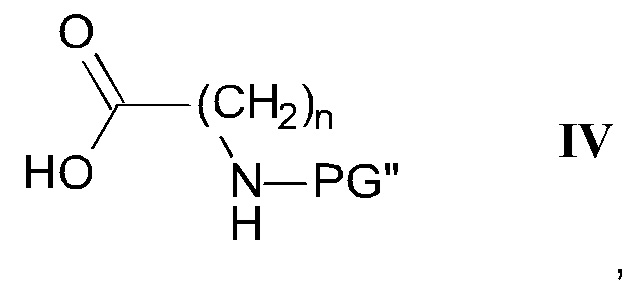
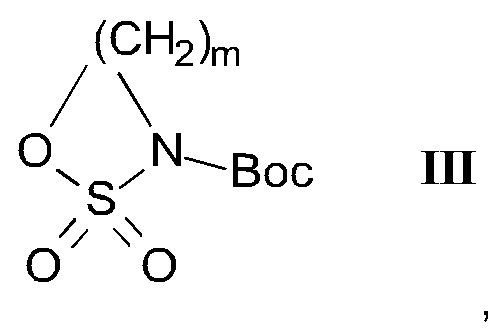
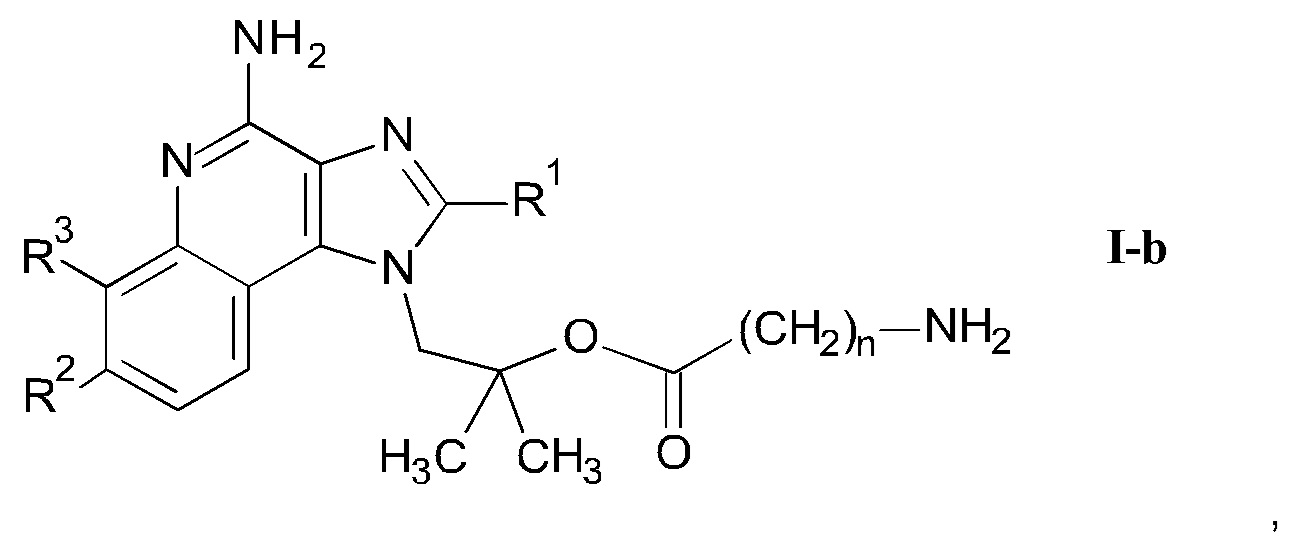
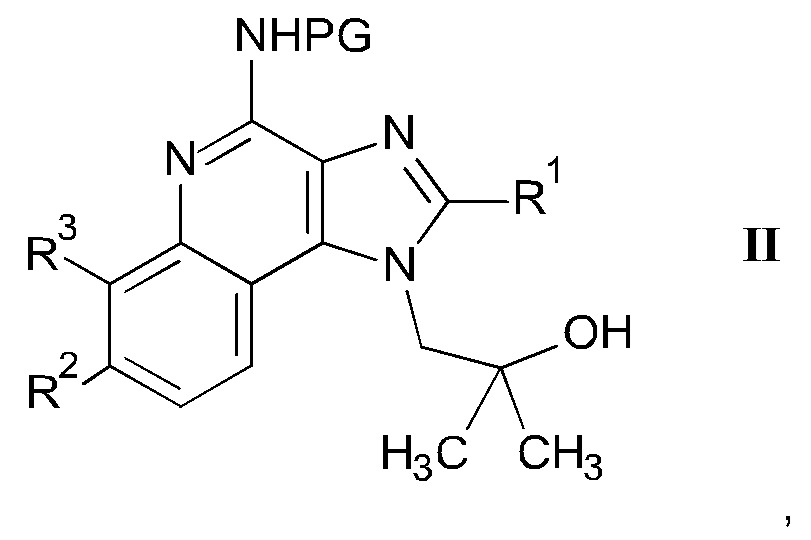
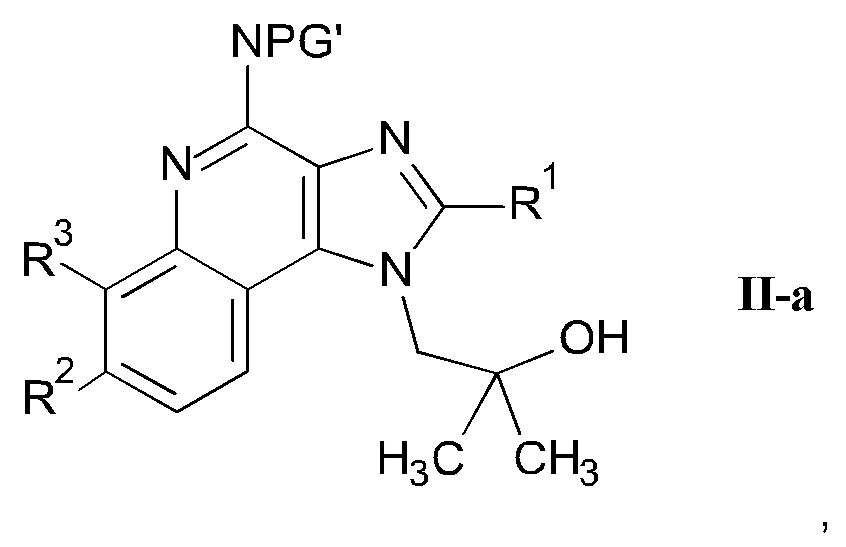
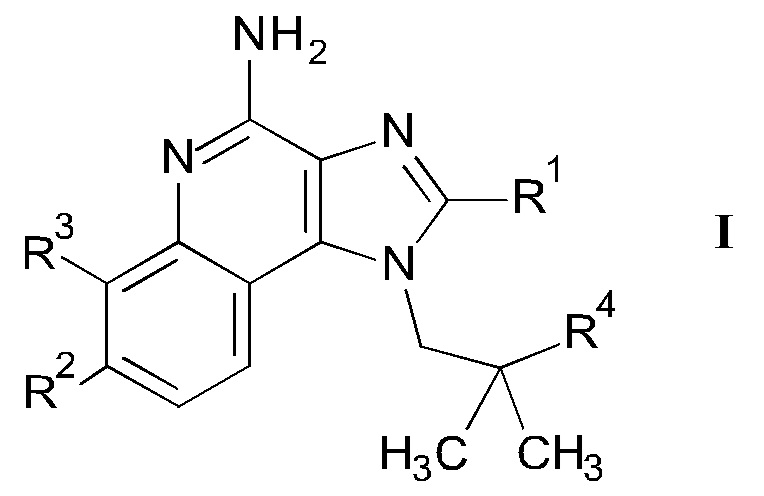
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные изоксазоло-пиридина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Пуриновые соединения, ингибирующие рi3к, и способы применения
Алкилциклогексиловые эфиры дигидротетраазабензоазуленов
Способ синтеза производных амино-метилтетралина
Карбоксил- или гидроксилзамещенные производные бензимидазола
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
N-пиридин-3-ил или n-пиразин-2-ил карбоксамиды в качестве агентов, повышающих уровень холестерина лпвп
Азотосодержащие производные гетероарилов
Производные n-(имидазопиримидин-7-ил)-гетероариламидов и их применение в качестве ингибиторов pde10a
Модуляторы фосфодиэстеразы 10
Циклопентил- и циклогептилпиразолы в качестве модуляторов fxr
Новые производные пиразина
Макроциклические амиды в качестве ингибиторов протеазы
Новые производные азетидина

