Результат интеллектуальной деятельности: НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ И ВЫСОКОСЕЛЕКТИВНЫХ ЛИГАНДОВ АСИАЛОГЛИКОПРОТЕИНОВОГО РЕЦЕПТОРА ДЛЯ ТЕРАПИИ ОНКОЛОГИЧЕСКИХ ПАТОЛОГИЙ ПЕЧЕНИ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к области органической и медицинской химии, а также молекулярной биологии и касается способа получения нового класса биологически активных веществ - низкомолекулярных ковалентно связанных конъюгатов противоопухолевых соединений с высокоселективными лигандами асиалогликопротеинового рецептора (ASGP-R), которые могут быть использованы в качестве лекарственных средств для лечения гепатоцеллюлярной карциномы (ГЦК).
Уровень техники
Гепатоцеллюлярная карцинома занимает 5-е по распространенности и 3-е по смертности среди злокачественных опухолей человека. Ежегодно в мире диагностируют более 600000 новых случаев заболевания [Kumar Y. et al. Transarterial therapies for hepatocellular carcinoma: a comprehensive review with current updates and future directions // Asian Рас J Cancer Prev. - 2016. - T. 17. - C. 473-478]. В связи с широким спектром побочных эффектов и высокой общей токсичностью противоопухолевых препаратов, актуальной задачей является поиск систем для их адресной доставки в пораженные ткани. Адресная доставка - перспективный метод модификации лекарственных агентов, позволяющий улучшить фармакологический профиль, локализовать терапевтические агенты в целевой ткани, органе или клетке, повысить терапевтический индекс и эффективность лекарственных препаратов за счет увеличения его действенной концентрации и снижения общего содержания в организме [Maklakova S. Yu. et al. A new approach to the synthesis of ligands of asialoglycoprotein receptor for targeted delivery of oligonucleotides to hepatocytes // Russian Chemical Bulletin. - 2015. - T. 64. - №. 7. - C. 1655-1662]. Асиалогликопротеиновый рецептор (ASGP-R) - одна из наиболее удобных мишеней для адресной доставки лекарственных агентов в клетки печени. Это обусловлено такими факторами, как: 1) высокая селективность ASGP-R по отношению к производным галактозы, 2) расположение ASGP-R только на поверхности гепатоцитов и 3) высокая концентрация (более 500000 рецепторов на каждой клетке) [Spiess М. The asialoglycoprotein receptor: a model for endocytic transport receptors // Biochemistry. - 1990. - T. 29. - №. 43. - C. 10009-10018; Stockert R.J. The asialoglycoprotein receptor: relationships between structure, function, and expression // Physiological reviews. - 1995. - T. 75. - №. 3. - C. 591-609]. Важным преимуществом ASGP-R является механизм рецептор-опосредованного эндоцитоза, за счет которого лекарственный агент попадает в гепатоцит и освобождается от молекулы-доставщика, а рецептор в течение часа возвращается на поверхность мембраны [Bareford L.М., Swaan P.W. Endocytic mechanisms for targeted drug delivery // Advanced drug delivery reviews. - 2007. - T. 59. - №. 8. - C. 748-758]. Характерной особенностью ASGP-R является его повышенная экспрессия в клетке в присутствии биотина [Collins J.С. et al. Biotin-dependent expression of the asialoglycoprotein receptor in HepG2 // J. Biol. Chem. 1988. V. 263. №. 23. P. 11280-11283]. В связи с этим возможна направленная доставка терапевтических молекул в печень с помощью адресных систем, включающих в себя тканеспецифический лиганд на основе N-ацетилгалактозамина и его производных [Maklakova S. Yu. et al. A new approach to the synthesis of ligands of asialoglycoprotein receptor for targeted delivery of oligonucleotides to hepatocytes // Russian Chemical Bulletin. - 2015. - T. 64. - №. 7. - C. 1655-1662].
Существует ряд примеров систем для направленного транспорта противоопухолевых препаратов в клетки ГЦК с помощью лигандов ASGP-R [Ivanenkov Y.A. et al. Modern approaches for treatment of liver diseases via targeted drug delivery strategies // Russ Chem Rev. - 2017. - T. 86]. В подавляющем большинстве это сополимеры, наночастицы, липосомы или мицеллы, модифицированные остатками галактозы или N-ацетилгалактозамина. Например, полимерный носитель PK2, разработанный Pfizer и исследовавшийся в фазе I/II на пациентах с первичным или метастатическим раком печени [Seymour L.W. et al. Hepatic drug targeting: phase I evaluation of polymer-bound doxorubicin // Journal of clinical oncology. - 2002. - T. 20. - №. 6. - C. 1668-1676]. Доксорубицин (Dox) связывался через расщепляемую тетрапептидную последовательность с N-(2-гидроксипропил)метакриламидным сополимером, содержащим фрагменты N-ацетилгалактозамина. Было продемонстрировано, что таким образом можно обеспечить селективный транспорт доксорубицина в клетки, и препарат был рекомендован для дальнейшего исследования (фазы II), которое тем не менее в результате было приостановлено. Основными недостатками высокомолекулярных систем направленного транспорта (мицеллы, наночастицы, липосомы и т.п.), являются: 1) низкая проницаемость через клеточную мембрану и стенки кровеносных сосудов [Ivanenkov Y.A. et al. Modern approaches for treatment of liver diseases via targeted drug delivery strategies // Russ Chem Rev. - 2017. - T. 86; Goodman Т.Т., Olive P.L., Pun S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids // International journal of nanomedicine. - 2007. - T. 2. - №. 2. - C. 265]; 2) низкая стабильность в плазме крови [ R. et al. Assessment of the integrity of poly (caprolactone)-b-poly (ethylene oxide) micelles under biological conditions: a fluorogenic-based approach // Langmuir. - 2006. - T. 22. - №. 8. - C. 3570-3578; R., Eisenberg A., Maysinger D. Block copolymer micelles as delivery vehicles of hydrophobic drugs: micelle-cell interactions // Journal of drug targeting. - 2006. - T. 14. - №. 6. - C. 343-355; Burt H.M. et al. Development of copolymers of poly (D,L-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel // Colloids and Surfaces B: Biointerfaces. - 1999. - T. 16. - №. 1. - C. 161-171]; 3) сравнительно короткое время полувыведения [Hong М. et al. Efficient tumor targeting of hydroxycamptothecin loaded PEGylated niosomes modified with transferrin // Journal of Controlled Release. - 2009. - T. 133. - №. 2. - C. 96-102].
R. et al. Assessment of the integrity of poly (caprolactone)-b-poly (ethylene oxide) micelles under biological conditions: a fluorogenic-based approach // Langmuir. - 2006. - T. 22. - №. 8. - C. 3570-3578; R., Eisenberg A., Maysinger D. Block copolymer micelles as delivery vehicles of hydrophobic drugs: micelle-cell interactions // Journal of drug targeting. - 2006. - T. 14. - №. 6. - C. 343-355; Burt H.M. et al. Development of copolymers of poly (D,L-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel // Colloids and Surfaces B: Biointerfaces. - 1999. - T. 16. - №. 1. - C. 161-171]; 3) сравнительно короткое время полувыведения [Hong М. et al. Efficient tumor targeting of hydroxycamptothecin loaded PEGylated niosomes modified with transferrin // Journal of Controlled Release. - 2009. - T. 133. - №. 2. - C. 96-102].
В отличие от высокомолекулярных систем доставки, в литературе практически не описаны низкомолекулярные конъюгаты лигандов ASGP-R с противоопухолевыми препаратами, в которых векторный фрагмент и действующее вещество соединены с помощью сравнительно небольшого биоразлагаемого фрагмента (линкера). Хотя добавление галактозного фрагмента в структуру низкомолекулярных конъюгатов достаточно распространенно [Ma Y. et al. Galactose as Broad Ligand for Multiple Tumor Imaging and Therapy // Journal of Cancer. - 2015. - T. 6. - №.7. - C. 658-670; Melisi D. et al. D-Galactose as a vector for prodrug design // Current topics in medicinal chemistry. - 2011. - T. 11. - №. 18. - C. 2288-2298], данные работы не рассматривают его в качестве лиганда ASGP-R, а в частности предполагают увеличение растворимости в водных растворах и сродство к другому рецептору. Заявляемое изобретение представляет собой первый случай, описывающий конъюгаты противоопухолевых лекарственных соединений, связанных с ASGP-R-специфическим лигандом через относительно короткий биодеградируемый линкер. Был произведен синтез новых лигандов асиалогликопротеинового рецептора на основе производных N-ацетилгалактозамина, а также синтез их ковалентно связанных низкомолекулярных конъюгатов с противоопухолевыми лекарственными средствами и биологические испытания полученных конъюгатов на клеточных линиях HepG2, Hek293.
Раскрытие изобретения
Задачей настоящего изобретения является создание новых низкомолекулярных ковалентно связанных конъюгатов противоопухолевых лекарственных соединений с лигандами асиалогликопротеинового рецептора на основе производных N-ацетилгалактозамина для терапии онкологических заболеваний печени.
Техническим результатом является получение новых соединений, обладающих выраженной аффинностью к асиалогликопротеиновому рецептору и токсичностью по отношению к клеточным линиям гепатоцеллюлярной карциномы, низкой токсичностью по отношению к другим тканям и улучшенной биодоступностью действующего противопухолевого соединения.
Поставленная задача решается низкомолекулярными конъюгатами противоопухолевых соединений с лигандами асиалогликопротеинового рецептора на основе производных N-ацетилгалактозамина, общей структурной формулы:
где лиганд представляет собой производное N-ацетилгалактозамина структурных формул 1, 2, 3:

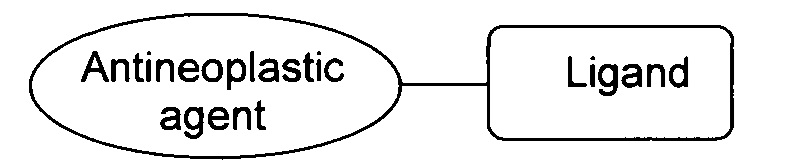
a «Antineoplastic agent» (противоопухолевое соединение) представляет собой соединение, содержащее свободную гидроксильную или амино группу, образующую ковалентную связь с лигандом и обладающее противоопухолевым действием для терапии онкологических заболеваний печени. При этом в качестве противоопухолевого соединения, содержащего свободную гидроксильную предпочтительно использовать паклитаксел, или гемцитабин, или цитарабин, или азацитидин, а в качестве противоопухолевого соединения, содержащего свободную амино группу предпочтительно использовать доксорубицин (Dox*HCl) или даунорубицин. Лиганды формул 1, 2 и 3 характеризуются аффинностью к асиалогликопротеиновому рецептору и предназначены для доставки противоопухолевых соединений, используемых при терапии гепатоцеллюлярной карциномы.
Способ получения низкомолекулярных конъюгатов, заключается в проведении двух последовательных реакций, сначала реакции карбодиимидного синтеза путем смешения противоопухолевого соединения и алкиниловой кислоты с терминальной тройной связью, взятых из расчета, что на 1 мольный эквивалент противоопухолевого соединения берут не менее 1.0 эквивалента алкиниловой кислоты с терминальной тройной связью, в присутствии активатора карбоксильной группы, взятом из расчета на 1 мольный эквивалент алкиниловой кислоты берут 1 эквивалент активатора карбоксильной группы, в среде, обеспечивающей протекание реакции карбодиимидного синтеза, и выделение промежуточного продукта путем колоночной хроматографии - противоопухолевого соединения, модифицированного алкинильной кислотой с теминальной тройной связью, а затем реакции [3+2] азид-алкинового циклоприсоединения посредством смешения промежуточного продукта и компонента, содержащего азидо-группу, в присутствии катализатора - соединения одновалентной меди, в среде, обеспечивающей протекание реакции циклоприсоединения, при этом на 1 мольный эквивалент промежуточного продукта используют по меньшей мере 1.0 эквивалент компонента, содержащего азидо-группу, и по меньшей мере 0.2 эквивалент катализатора, после прохождения реакции осуществляют удаление среды с последующим выделением конечного продукта при помощи колоночной хроматографии. Предпочтительно в качестве противоопухолевого соединения использовать паклитаксел, или гемцитабин, или цитарабин, или азацитидин, или доксорубицин (Dox*HCl), или даунорубицин, при этом в качестве алкиниловой кислоты с терминальной тройной связью использовать проп-2-иновую кислота, бут-3-иновую кислота, пент-4-иновую кислота, гекс-5-иновую кислота, гепт-6-иновую кислоту, а в качестве среды, обеспечивающей протекание реакции карбодиимидного синтеза, использовать ДМФА (диметилформамид), CH2Cl2 (дихлорметан), CHCl3 (хлороформ), тетрогидрофуран, ClCH2CH2Cl (1,2-дихлорэтан). В качестве активатора карбоксильной группы предпочтительно использовать растворимые в среде, обеспечивающей протекание реакции карбодиимидного синтеза, карбодиимиды, а именно 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), N,N'-диизопропилкарбодиимид (DIC), N,N'-дициклогексилкарбодиимид (DCC). Выделение промежуточного продукта предпочтительно проводить с использованием прямой колоночной хроматографии в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. Для проведения реакции [3+2] азид-алкинового циклоприсоединения в качестве компонента, содержащего азидо-группу предпочтительно использовать производное N-ацетилгалактозамина, выбранное из группы, включающей 1-Азидо-1-дезокси-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-(2'-азидоэтил)-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-О-Аллил-2-ацетамидо-2-дезокси-6-азидо-6-дезокси-β-D-галактопиранозы. При этом в качестве соединения одновалентной одновалентной меди, предпочтительно использовать одновалентные соли меди растворимые в среде, обеспечивающей прохождение реакции циклоприсоединения, а именно иодид меди (CuI), бромид меди (CuBr), хлорид меди (CuCl), а в качестве среды, обеспечивающей протекание реакции [3+2] азид-алкинового циклоприсоединения, предпочтительно использовать ДМФА, смесь вода-тетрогидрофуран в объемном соотношении 1:1, диметилсульфоксид (ДМСО). Для выделения конечного продукта - низкомолекулярного конъюгата предпочтительно использовать обращено-фазовую колоночную хроматографию в системе Н2О-MeCN.
Для получения низкомолекулярного конъюгата с противоопухолевым соединением, включающим доступную для модификации аминогруппу, в качестве которых предпочтительно использовать доксорубицин или даунорубицин, сначала получают лиганд формул 1, 2 и 3 путем проведения реакции [3+2] азид-алкинового циклоприсоединения посредством смешения соединения, содержащего терминальный алкильный фрагмент, и соединения, содержащего азидо-группу, взятых в мольном соотношении 1:1, в присутствии катализатора - соли одновалентной меди, взятой из расчета на 1 мольный эквивалент компонента, содержащего азидо-группу, по меньшей мере 0.25 эквивалента катализатора в среде обеспечивающей протекание реакции циклоприсоединения, после прохождения реакции удаляют среду и выделяют лиганд при помощи колоночной хроматографии, с последующим проведением реакции карбодиимидного синтеза между полученным лигандом и противоопухолевым соединением с доступной для модификации аминогруппой, взятых в мольном соотношении 1:1, в присутствии активатора карбоксильной группы, взятом из расчета на 1 мольный эквивалент лиганда по п. 1 не менее 1 эквивалента активатора, в ДМФА, после прохождения реакции удаляют ДМФА и выделяют конъюгат при помощи колоночной хроматографии. При этом в качестве среды, обеспечивающей протекание реакции [3+2] азид-алкинового присоединения, используют ДМФА, смесь вода-тетрогидрофуран в объемном соотношении 1:1, ДМСО. В качестве соединения, содержащего терминальный алкиновый фрагмент, предпочтительно использовать алкиниловые кислоты с терминальной тройной связью, а именно проп-2-иновую кислоту, бут-3-иновую кислоту, пент-4-иновую кислоту, гекс-5-иновую кислоту, гепт-6-иновую кислоту, а в качестве соединения, содержащего азидо-группу предпочтительно использовать производные N-ацетилгалактозамина, а именно 1-Азидо-1-дезокси-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-(2'-азидоэтил)-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-аллил-2-ацетамидо-2-дезокси-6-азидо-6-дезокси-β-D-галактопиранозу. Предпочтительно в качестве солей одновалентной меди использовать растворимые в среде, обеспечивающей прохождение реакции циклоприсоединения, соли меди, а именно CuI, CuBr, CuCl. Пр этом для выделения полученного лиганда предпочтительно использовать прямую колоночную хроматографию системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. При проведении реакции карбодиимидного синтеза в качестве активатора карбоксильной группы предпочтительно использовать растворимые в ДМФА карбодиимиды, а именно 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), N,N'-диизопропилкарбодиимид (DIC), N,N'-дициклогексилкарбодиимид (DCC). Для выделения полученного конъюгата предпочтительно использовать две последовательные колоночные хроматографии прямую колоночную хроматографию системе с градиентом концентраций CH2Cl2:МеОН (20:1) → МеОН и обращено-фазовую колоночную хроматографию в системе Н2О-MeCN.
Краткое описание чертежей
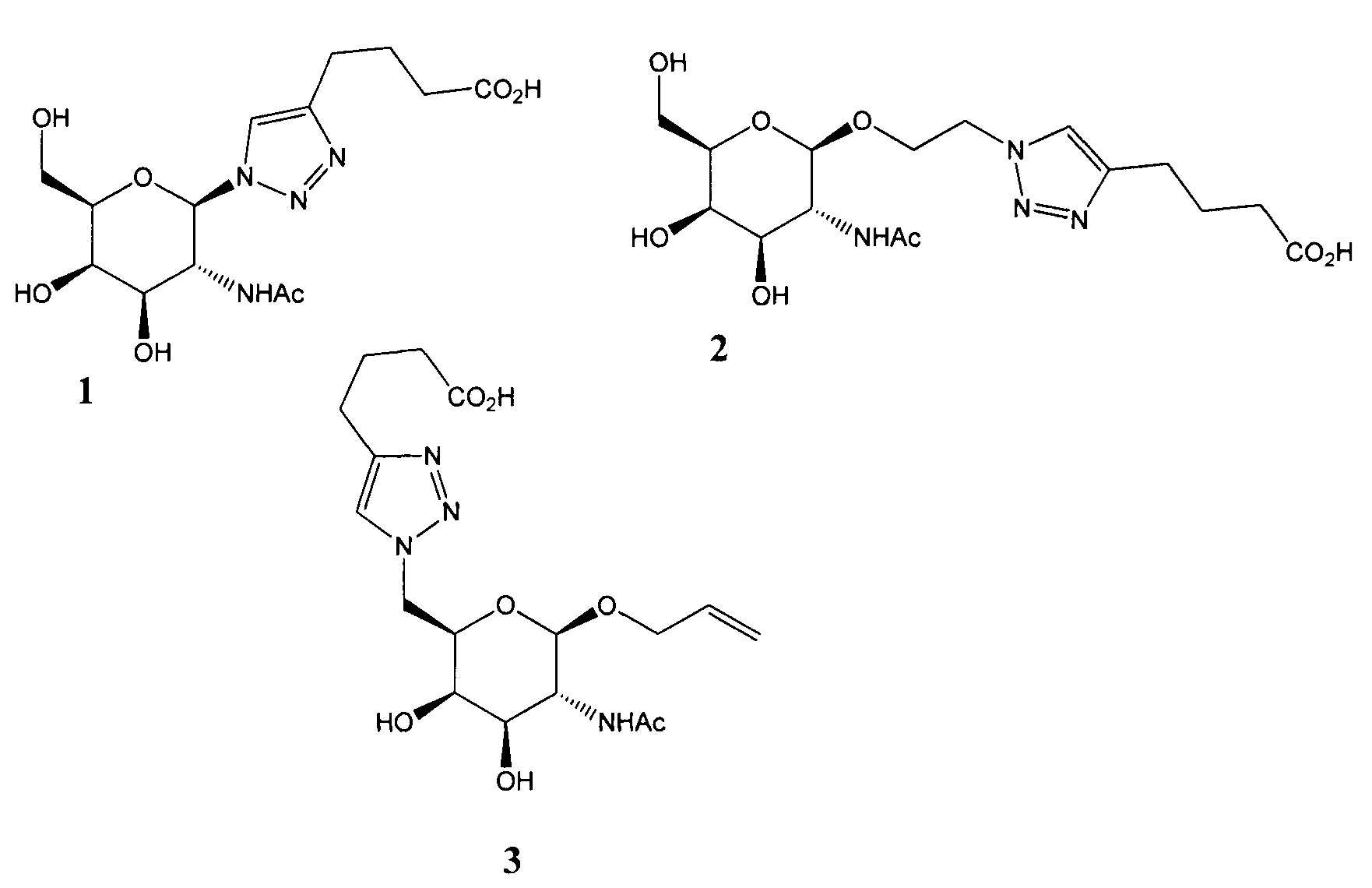
На фиг. 1 представлены снимки, сделанные при помощи микроскопа, конъюгатов (4) - А, (5) - В, (6) - С. Время инкубирования - 24 часа.
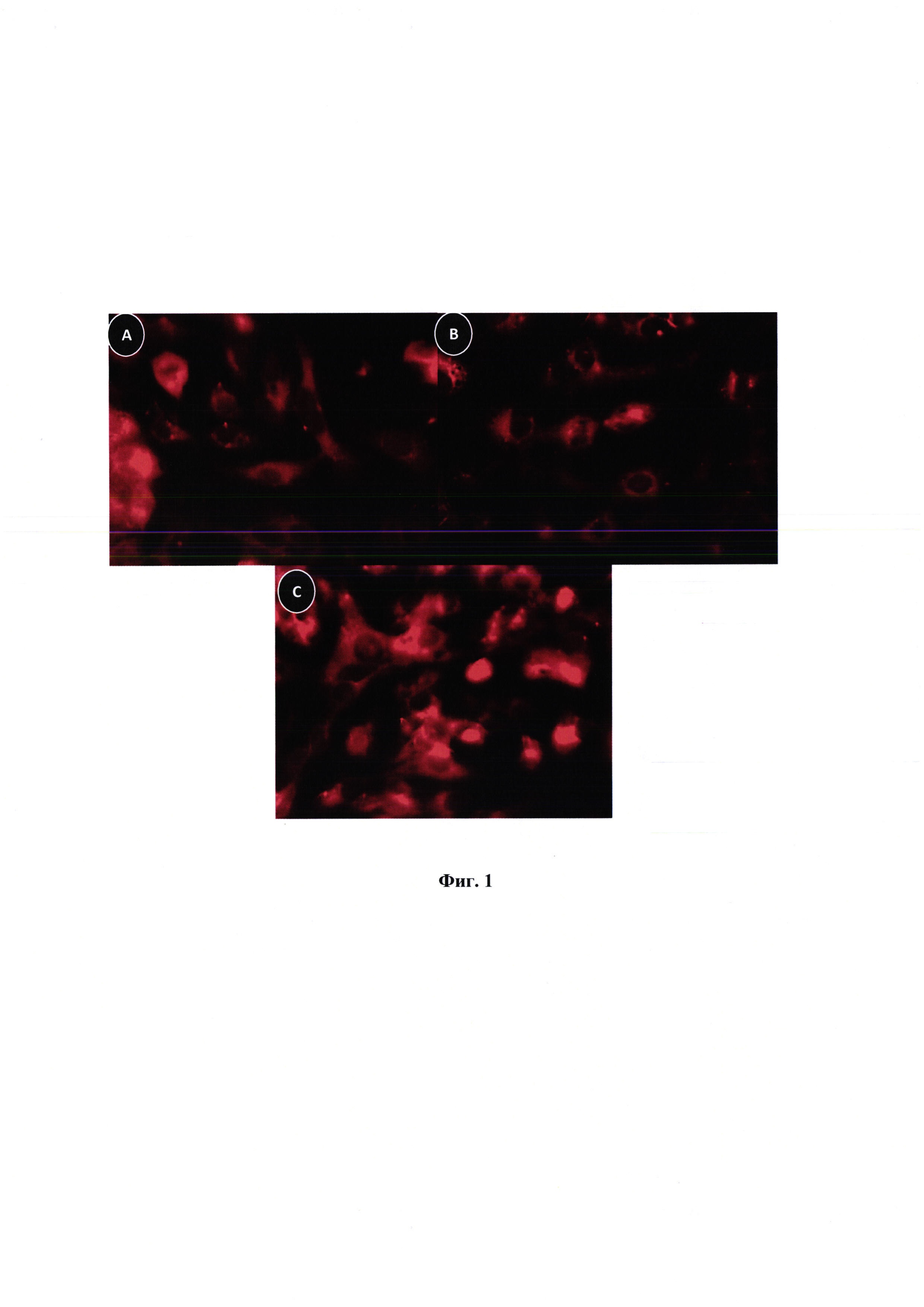
На фиг. 2 представлены снимки необработанных клеток Hepg2, которые фиксировали, пермеабилизировали и иммунизировали антителом против α-тубулина
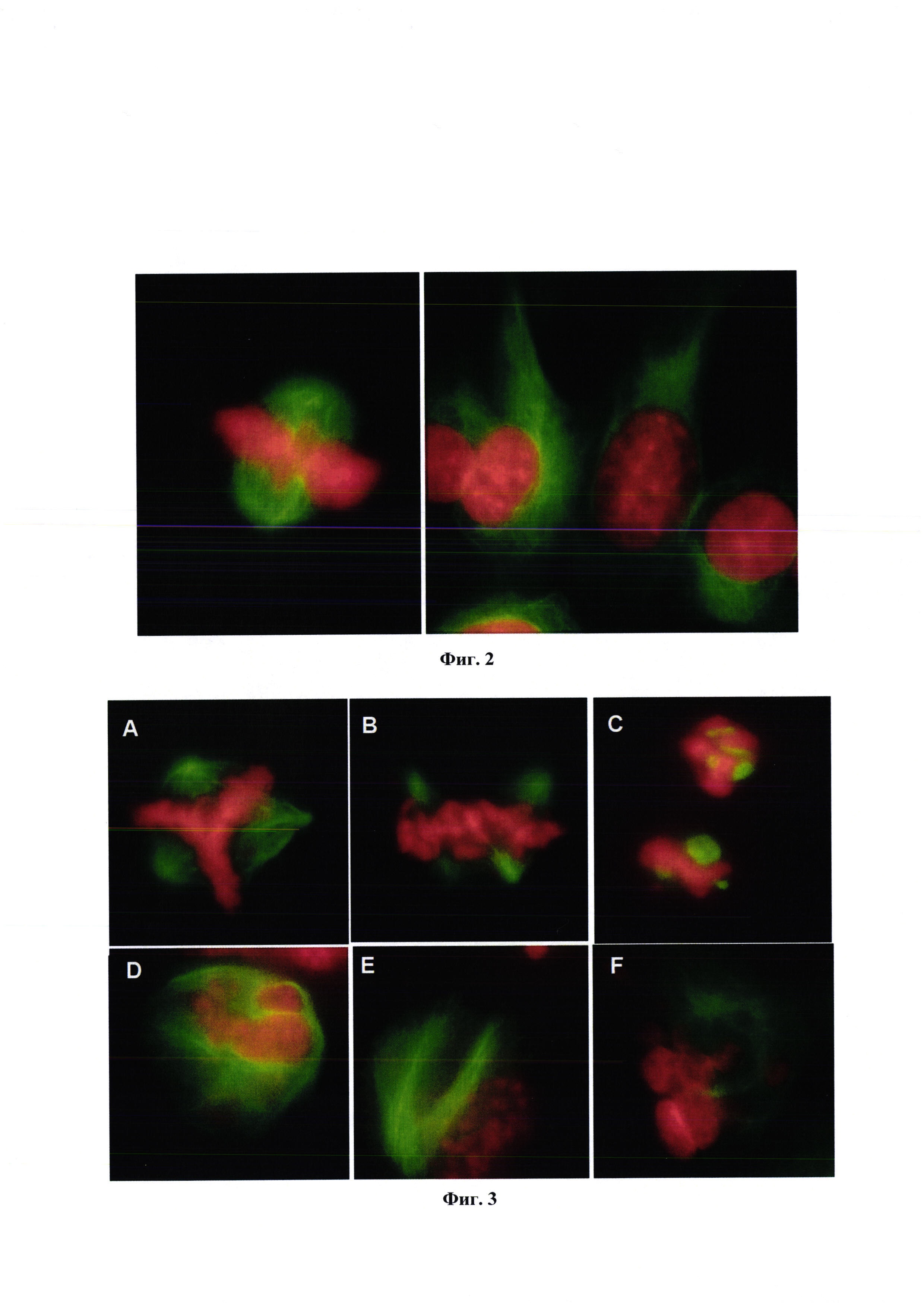
На фиг. 3 представлены снимки клеточной линии Hepg2, обработанных конъюгатами (8-10). Клетки фиксировали, пермеабилизировали и иммунизировали антителом против α-тубулина. На снимках: А - клетки после обработки конъюгатом 8 (6 часов), В - клетки после обработки конъюгатом 8 (39 часов), С - клетки после обработки конъюгатом 9 (3 часа), D - клетки после обработки конъюгатом 9 (17 часа), Е - клетки после обработки конъюгатом 10 (24 часа), F - клетки после обработки конъюгатом 9 (17 часов).
Осуществление изобретения
Ниже приведены определения терминов, которые используются в описании настоящего изобретения.
«Асиалогликопротеиновый рецептор (ASGP-R)» - трансмембранный белок, преимущественно экспрессируемый гепатоцитами, селективно распознающий производные D-галактозы и D-галактозамина и участвующий в их транспорте в клетку.
«Конъюгат» - комплекс, формирующийся при помощи ковалентных связей между противоопухолевым соединенем и его лигандом-носителем.
«Лиганд» - химическое соединение, которое образует нековалентный комплекс с той или иной биомолекулой (чаще всего белком, например клеточным рецептором, но иногда, например, с ДНК) и производит, вследствие такого связывания, те или иные биохимические, физиологические или фармакологические эффекты.
«Противоопухолевое лекарственное соединение (агент)» - вещество, используемое в качестве активного компонента в препаратах для лечения онкологических заболеваний. Примеры используемых противоопухолевых агентов и их ковалентная связь с заявляемым лигандом приведены на схеме (I)
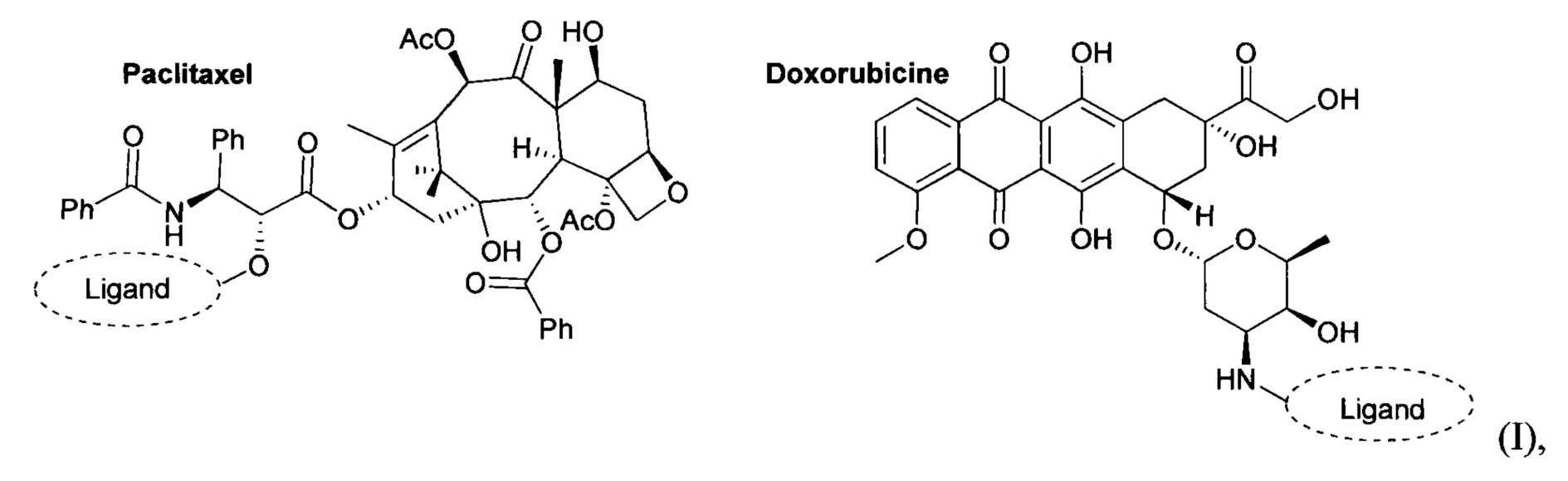
где «Ligand» (лиганд) - это выше описанные производные N-ацетилгалактозамина с структурными формулами 1, 2, 3.
Все используемые реагенты являются коммерчески доступными, выпаривание растворителя осуществляли с использованием роторного испарителя, при пониженном давлении при температуре бани примерно 50°С; контроль за ходом реакции осуществляли при помощи тонкослойной хроматографии (ТСХ), и время реакции указано только для иллюстрации; структуру и чистоту всех выделенных соединений подтверждали, по меньшей мере, одним из следующих методов: ТСХ (пластины для ТСХ с предварительно нанесенным силикагелем 60 F254 Merck), масс-спектрометрия или ядерный магнитный резонанс (ЯМР). Выход продукта приведен только для иллюстрации. Колоночную флэш-хроматографию осуществляли, используя Merck силикагель 60 (230-400 меш ASTM). Масс-спектры высокого разрешения (HRMS) зарегистрированы на спектрометре Bruker microTOF II. Спектры ЯМР регистрировали на приборах Bruker Avance-400 (рабочая частота 400.1 и 100.6 МГц для 1Н и 13С, соответственно) и Agilent 400-MR (рабочая частота 400.0 и 100.6 МГц для 1Н и 13С, соответственно), используя дейтерированный хлороформ (99,8% D) или метанол (99.9%) или ДМСО (99,9% D) в качестве растворителя, если не указано иное, относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, миллионных долях (м.д.); обычные используемые сокращения следующие: с - синглет, д - дублет, т - триплет, кв - квартет, м - мультиплет, шир. - широкий и так далее.
Способ получения конъюгатов противоопухолевых лекарственных соединений с лигандами асиалогликопротеинового рецептора на основе производных N-ацетилгалактозамина для терапии онкологических заболеваний печени проводят по реакции карбодиимидного синтеза с последующей реакцией [3+2] азид-алкинового циклоприсоединения. Для противоопухолевых соединений с доступной для модификации аминогруппой возможен путь синтеза с первоначальным получением лиганда путем реакции [3+2] азид-алкинового циклоприсоединения и последующим проведением реакции карбодиимидного синтеза между лигандом и противоопухолевым соединением.
Образование конъюгата лиганда асиалогликопротеинового рецептора на основе производных N-ацетилгалактозамина с противоопухолевыми соединениями, содержащими амино и гидроксильную группы, доступные для ацилирования алкиновой кислотой с терминальной тройной связью, заключается двух последовательных реакциях. Сначала проводят модификацию противоопухолевого соединения алкиновой кислотой с терминальной тройной связью при помощи реакции карбодиимидного синтеза, которая осуществляется посредством смешения противоопухолевого соединения и алкиновой кислоты с терминальной тройной связью в присутствии активатора карбоксильной группы в среде, обеспечивающей протекание реакции карбодиимидного синтеза, при этом на 1 мольный эквивалент противоопухолевого соединения берут не менее 1 эквивалента кислоты и не менее 1.0 эквивалента активатора карбоксильной группы. После чего проводят выделение противоопухолевого соединения, модифицированного алкиновой кислотой с терминальной тройной связью, путем прямой колоночной хроматографии в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. Затем проводят реакцию [3+2] азид-алкинового циклоприсоединения посредством смешения модифицированного противоопухолевого соединения, содержащего терминальный алкильный фрагмент, и соединения, содержащего азидо-группу в мольном соотношении из расчета, что на 1 мольный эквивалент соединения, содержащего азидо-группу, используют по меньшей мере 1.0 эквивалента соединения, содержащего терминальный алкильный фрагмент. Реакцию проводят в присутствии катализатора - соединения одновалентной меди, взятого из расчета на 1 мольный эквивалент соединения, содержащего азидо-группу берут по меньшей мере 0.2 эквивалент катализатора, при этом реакцию проводят в среде, обеспечивающей растворение соединений. После прохождения реакции осуществляют удаление среды с последующим выделением продукта при помощи колоночной хроматографии.
В качестве противоопухолевого соединения используют паклитаксел, или гемцитабин, или цитарабин, или азацитидин, или доксорубицин, или даунорубицин.
В качестве алкиниловой кислоты с терминальной тройной связью используют проп-2-иновую кислота, бут-3-иновую кислота, пент-4-иновую кислота, гекс-5-иновую кислота, гепт-6-иновую кислоту.
В качестве среды, обеспечивающей протекание реакции карбодиимидного синтеза, используют диметилформамид, дихлорметан, хлороформ, тетрогидрофуран, 1,2-дихлорэтан.
В качестве активатора карбоксильной группы используют растворимые в среде, обеспечивающей протекание реакции карбодиимидного синтеза, карбодиимиды: 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), N,N'-диизопропилкарбодиимид (DIC), N,N'-дициклогексилкарбодиимид (DCC).
Для выделения промежуточного продукта используют прямую колоночную хроматографию в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН.
В качестве среды, обеспечивающей протекание реакции [3+2] азид-алкинового циклоприсоединения, используют ДМФА, смесь вода-тетрогидрофуран в объемном соотношении 1:1, ДМСО или любой другой растворитель, обеспечивающий растворение и инертный к компонентам смеси.
В качестве компонента, содержащего азидо-группу - производные N-ацетилгалактозамина: 1-Азидо-1-дезокси-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-(2'-азидоэтил)-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-аллил-2-ацетамидо-2-дезокси-6-азидо-6-дезокси-β-D-галактопиранозу.
В качестве катализатора - соединений одновалентной меди используют любую растворимую в среде, обеспечивающей прохождение реакции циклоприсоединения, соль одновалентной меди. Например, CuI, CuBr, CuCl и т.п.
Для выделения полученного продукта используют обращено-фазовую колоночную хроматографию в системе Н2О-MeCN.
Верхняя граница содержания используемых реагентов не ограничивается, т.к. избыток какого-либо реагента не уменьшает выходов реакций, однако при большом избытке может понадобиться дополнительная очистка продуктов реакций.
Для противоопухолевых соединений с доступной для модификации аминогруппой присоединение лиганда проводят следующим образом.
На первом этапе получают заявляемые лиганды путем проведения реакции [3+2] азид-алкинового циклоприсоединения посредством смешения соединения, содержащего терминальный алкильный фрагмент, и соединения, содержащего азидо-группу, при этом на 1.0 мольный эквивалент соединения, содержащего азидо-группу, используют по меньшей мере 1.0 эквивалента соединения, содержащего терминальный алкильный фрагмент. Реакцию проводят в присутствии катализатора - соединения одновалентной меди, взятую в количестве из расчета по меньшей мере 0.25 эквивалент катализатора на 1.0 мольный эквивалент соединения, содержащего азидо-группу. Реакцию проводят в среде, обеспечивающей растворение компонентов. При этом в качестве среды, обеспечивающей растворение соединений, используют ДМФА, смесь вода-тетрогидрофуран в объемном соотношении 1:1, ДМСО или любой другой растворитель, обеспечивающий растворение и инертный к компонентам смеси. После прохождения реакции осуществляют удаление среды с последующим выделением продукта при помощи колоночной хроматографии.
В качестве соединений, содержащих терминальный алкиновый фрагмент, используют алкиниловые кислоты с терминальной тройной связью. Длина углеродного фрагмента кислоты не имеет принципиального значения, в качестве алкиниловой кислоты с терминальной тройной связью может выступать: проп-2-иновая кислота, бут-3-иновая кислота, пент-4-иновая кислота, гекс-5-иновая кислота, гепт-6-иновая и т.д.
В качестве компонента, содержащего азидо-группу, используют производные N-ацетилгалактозамина: 1-Азидо-1-дезокси-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-(2'-азидоэтил)-2-ацетамидо-2-дезокси-β-D-галактопиранозу, 1-O-Аллил-2-ацетамидо-2-дезокси-6-азидо-6-дезокси-β-D-галактопиранозу.
В качестве катализатора - соединения одновалентной меди используют любую растворимую в среде, обеспечивающей прохождение реакции циклоприсоединения, соль одновалентной меди. Например, CuI, CuBr, CuCl и т.д.
Для выделения полученного продукта используют прямую колоночную хроматографию системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН.
Образование конъюгата полученного лиганда асиалогликопротеинового рецептора на основе производных N-ацетилгалактозамина с противоопухолевыми соединениями с доступной для модификации аминогруппой заключается в проведении реакции карбодиимидного синтеза посредством смешения соединения, содержащего карбоксильную группу - лиганда формулы 1, или 2, или 3, соответственно, и противоопухолевого соединения, включающего аминогруппу, например доксорубицина или даунорубицина, в мольном соотношении 1:1, в присутствии активатора карбоксильной группы. При этом количество активатора берут из расчета на 1,0 мольный эквивалент противоопухолевого агента по меньшей мере 1.1 эквивалент активатора. Реакцию карбодиимидного синтеза проводят в среде, обеспечивающей растворение соединений и инертной к компонентам смеси. Предпочтительно использовать ДМФА. После прохождения реакции осуществляют удаление среды с последующим выделением продукта при помощи колоночной хроматографии.
В качестве активаторов карбоксильной группы используют любой активатор карбоксильной группы карбодиимидного ряда, например: 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), N,N'-диизопропилкарбодиимид (DIC), N,N'-дициклогексилкарбодиимид (DCC) и т.п
Для выделения полученного продукта используют две последовательные колоночные хроматографии прямую колоночную хроматографию системе с градиентом концентраций CH2Cl2:МеОН (20:1) → МеОН и обращено-фазовую колоночную хроматографию в системе Н2О-MeCN.
Представленные ниже примеры иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Получение 4-(1-(3-ацетамидо-4,5-дигидрокси-6-(гидроксиметил)тетрагидро-2Н-пиран-2-ил)-1Н-1,2,3-триазол-4-ил)бутановой кислоты (1).
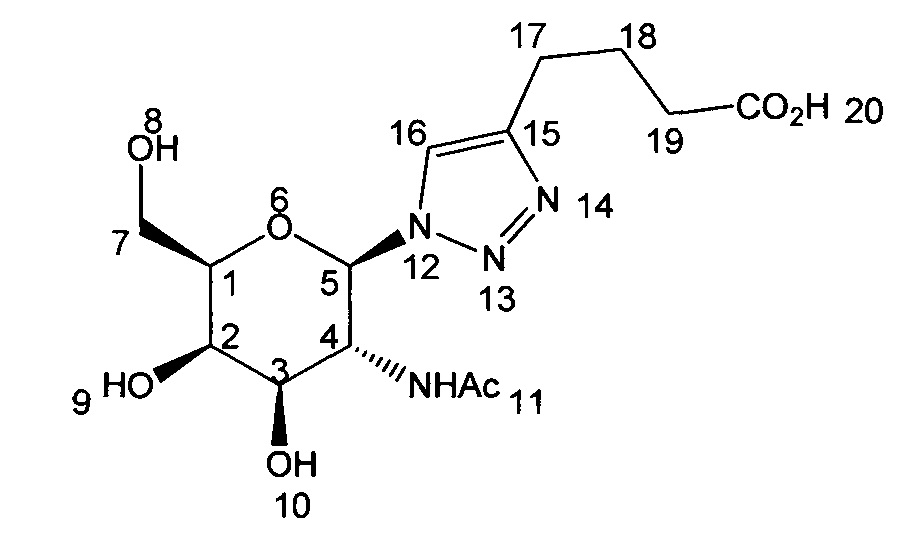
К раствору 108 мг (0,44 ммоль) 1-Азидо-1-дезокси-2-ацетамидо-2-дезокси-β-D-галактопиранозы, полученной согласно методике, описанной в работе [ N. et al. Iron (III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu (0) for the subsequent click chemistry // Chemical Communications. - 2011. - T. 47. - №. 37. - C. 10440-10442], в 15 мл сухого ДМФА добавили 60 мг (0,528 ммоль, 1,2 экв.) гекс-5-иновой кислоты и 21 мг (0,11 ммоль, 0,25 экв.) CuI. Полученную смесь перемешивали в течение 48 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом колоночной хроматографии в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. В результате получили соединение 1 в виде светло-желтого аморфного вещества 125 мг (79%).
N. et al. Iron (III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu (0) for the subsequent click chemistry // Chemical Communications. - 2011. - T. 47. - №. 37. - C. 10440-10442], в 15 мл сухого ДМФА добавили 60 мг (0,528 ммоль, 1,2 экв.) гекс-5-иновой кислоты и 21 мг (0,11 ммоль, 0,25 экв.) CuI. Полученную смесь перемешивали в течение 48 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом колоночной хроматографии в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. В результате получили соединение 1 в виде светло-желтого аморфного вещества 125 мг (79%).
1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 8.03 (с, 1Н, Н-16); 5.75 (д, 1H, J=9.8 Гц, Н-5); 4.51-4.63 (м, 1Н, Н-1); 4.06 (д, 1Н, J=3.1 Гц, Н-2); 4.21-4.30 (м, 1Н, Н-3); 3.73-3.97 (м, 4Н, Н-3,4,7); 2.68-2.80 (м, 2Н, Н-17); 2.20-2.32 (м, 2Н, Н-19); 1.88-2.01 (м, 2Н, Н-18); 1.79 (с, 1H, -NHAc).
МСВР (m/z) для C14H22N4O7: [М+Н]+ 359.1561 (найдено), 359.1562 (рассч.); [M+Na]+ 381.1381 (найдено), 381.1381 (рассч.).
Пример 2. Получение 4-(1-(2-((3-ацетамидо-4,5-дигидрокси-6-(гидроксиметил)тетрагидро-2Н-пиран-2-ил)окси)этил)-1Н-1,2,3-триазол-4-ил)бутановой кислоты (2).

К раствору 30 мг (0,103 ммоль) 1-O-(2'-азидоэтил)-2-ацетамидо-2-дезокси-β-D-галактопиранозы, полученной согласно методике, описанной в работе [Hasegawa A. et al. Studies on the thioglycosides of N-acetylneuraminic acid 10: Synthesis of S-(α-sialosyl)-(2→6)-O-2-acetamido-2-deoxy-β-d-hexopyranosyl ceramide and its related compounds // Journal of carbohydrate chemistry. - 1992. - T. 11. - №. 3. - C. 319-331], в 15 мл сухого ДМФА добавили 13,9 мг (0,124 ммоль, 1,2 экв.) гекс-5-иновой кислоты и 4,9 мг (0,026 ммоль, 0,25 экв.) CuI. Полученную смесь перемешивали в течение 48 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом колоночной хроматографии в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. В результате получили соединение (2) в виде светло-желтого аморфного вещества 31 мг (75%).
1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 8.21 (с, 1H, Н-19); 4.67-4.77 (м, 2Н, Н-1,2); 4.40 (д, 1Н, J=8.4 Гц, Н-5); 4.21-4.30 (м, 1H, Н-3); 3.90-4.03 (м, 2Н, Н-4, 13); 3.80-3.90 (м, 1Н, Н-13); 3.48-3.80 (м, 4Н, Н-7,14); 2.86 (т, 2Н, J=7.4 Гц, Н-20); 2.45 (т, 2Н, J=7.4 Гц, Н-22); 1.92-2.08 (м, 5Н, H-21, -NHAc).
МСВР (m/z) для C16H26N4O8: [М+Н]+ 403.1819 (найдено), 403.1823 (рассч.).
Пример 3. Получение 4-(1-((5-ацетамидо-6-(аллилокси)-3,4-дигидрокситетрагидро-2Н-пиран-2-ил)метил)-1Н-1,2,3-триазол-4-ил)бутановой кислоты (3).

К раствору 30 мг (0,105 ммоль) 1-O-Аллил-2-ацетамидо-2-дезокси-6-азидо-6-дезокси-β-D-галактопиранозы, полученной согласно ранее описанной методике [Wong С.Н. et al. A library approach to the discovery of small molecules that recognize RNA: use of a 1, 3-hydroxyamine motif as core // Journal of the American Chemical Society. - 1998. - T. 120. - №. 33. - C. 8319-8327] в 15 мл сухого ДМФА добавили 14,1 мг (0,125 ммоль, 1,2 экв.) гекс-5-иновой кислоты и 5 мг (0,026 ммоль, 0,25 экв.) CuI. Полученную смесь перемешивали в течение 48 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом колоночной хроматографии в системе с градиентом концентраций CH2Cl2:МеОН (10:1) → МеОН. В результате получили соединение (3) в виде светло-желтого аморфного вещества (28 мг, 67%).
1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 7.99 (с, 1Н, Н-19); 5.65-5.78 (м, 1Н, Н-14); 5.04-5.19 (м, 2Н, Н-15); 4.81 (д, 1H, J=3.6 Гц, Н-2); 4.63-4.71 (м, 2Н, Н-1,5); 4.30 (дд, 1Н, J=3.5 и 11.0 Гц, Н-3); 4.19-4.24 (м, 1Н, Н-4), 3.95 (д, 1Н, J=2.6 Гц, Н-7), 3.84 (дд, 1H, J=3.1 и 11.0 Гц, Н-7), 3.63-3.75 (м, 2Н, Н-13), 2.74-2.86 (м, 2Н, Н-20), 2.32-2.42 (м, 2Н, Н-22), 1.91-2.05 (м, 5Н, Н-21, -NHAc).
МСВР (m/z) для C17H26N4O7: [М+Н]+ 399.1876 (найдено), 399.1874 (рассч.); [M+Na]+ 421.1692 (найдено), 421.1694 (рассч.).
Пример 4. Получение соединения (4).
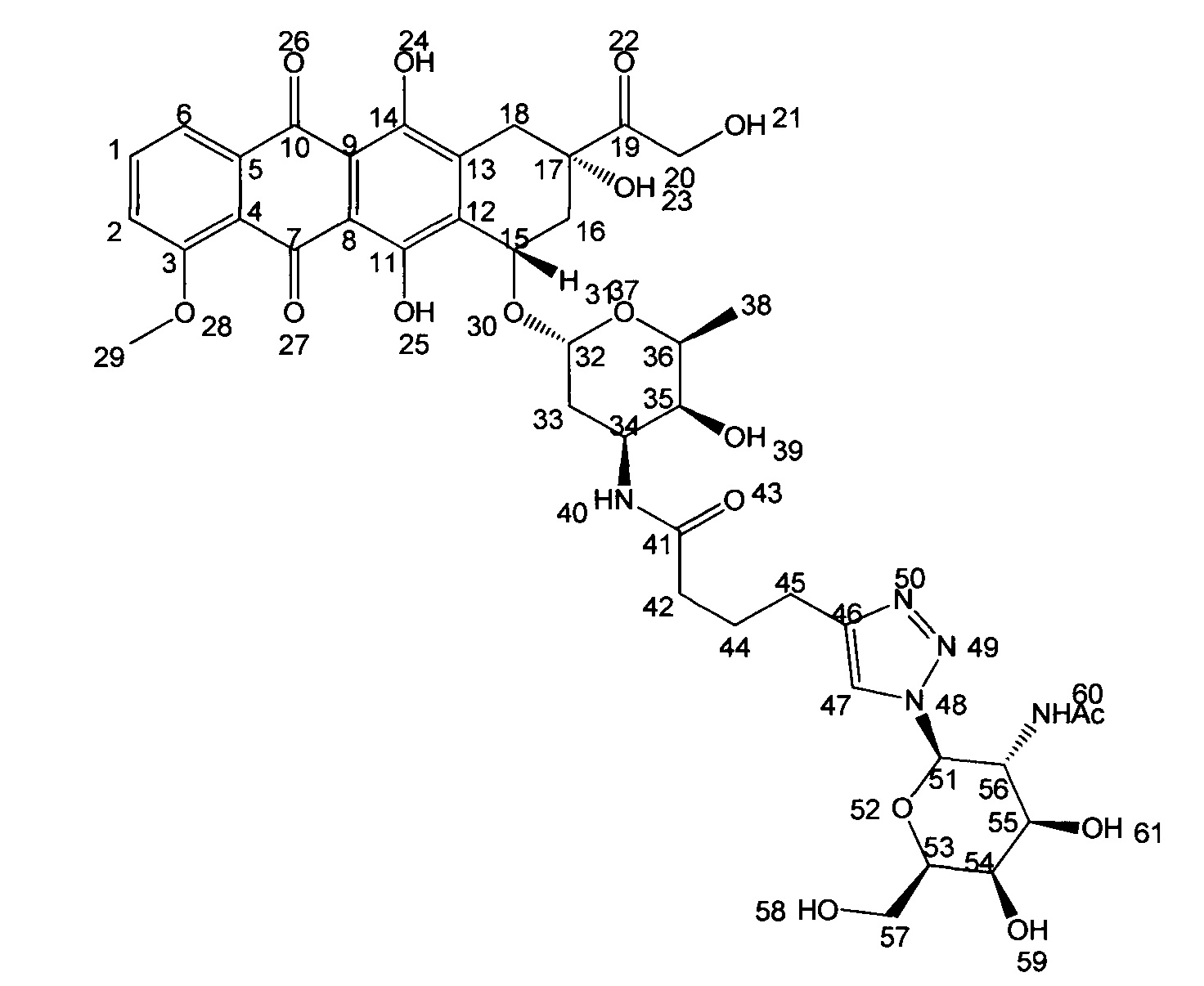
К раствору 6 мг (0,01 ммоль) доксорубицина гидрохлорида в 6 мл сухого ДМФА добавили 3,95 мг (0,01 ммоль, 1 экв.) соединения (1) в присутствии 1,15 мг (0,011 ммоль) Et3N, 1,31 мг (0,011 ммоль, 1,1 экв.) NHS, 2,11 мг (0,011 ммоль, 1,1 экв.) EDC в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали двумя последовательными хроматографированиями: 1) колоночная хроматография на силикагеле (градиентное элюирование CH2Cl2:МеОН (20:1) → МеОН), 2) ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (4) в виде темно-красного порошка (7.3 мг, 80%).
1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 7.72-8.04 (м, 3Н, Н-1,6, 61); 7.56 (д, 1Н, J=7.6 Гц, Н-2); 5.61 (д, 2Н, J=9.2 Гц, Н-51); 5.09-5.22 (м, 1H, Н-32); 4.65 (с, 2Н, Н-20); 4.46 (т, 1Н, J=10.0 Гц, Н-53); 4.09-4.33 (м, 3Н, Н-54,55,56); 4.04 (с, 5Н, Н-29,57); 3.69-3.87 (м, 5Н, Н-31,34,35,36, ОН); 3.62 (уш.с, 1Н, ОН); 2.91-3.18 (м, 2Н, Н-45); 2.27-2.44 (м, 2Н, Н-16); 2.06-2.22 (м, 2Н, Н-42); 1.84-2.03 (м, 4Н, Н-44,33); 1.77 (с, 3H, -NHAc); 1.20-1.34 (м, 5Н, Н-18,38).
МСВР (m/z) для C41H49N5O17: [М+Н]+ 884.3196 (найдено), 884.3196 (рассч.); [M+Na]+ 906.3025 (найдено), 906.3016 (рассч.).
Пример 5. Получение соединения (5).

К раствору 6 мг (0,01 ммоль) Dox*HCl в 6 мл сухого ДМФА добавили 4,16 мг (0,01 ммоль, 1 экв.) соединения (2) в присутствии 1,15 мг (0,011 ммоль, 1,1 экв.) Et3N, 1,31 мг (0,011 ммоль, 1,1 экв.) NHS, 2,11 мг (0,011 ммоль, 1,1 экв.) EDC в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали двумя последовательными хроматографированиями: 1) колоночная хроматография на силикагеле (градиентное элюирование CH2Cl2:МеОН (20:1) → МеОН), 2) ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (5) в виде темно-красного порошка (6,7 мг, 70%).
1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 8.00 (д, 1H, J=7.5 Гц, Н-6); 7.85 (т, J=7.5 Гц, Н-1); 7.74 (с, 1Н, Н-61); 7.58 (д, 1H, J=7.5 Гц, Н-2); 5.43 (м, 1Н, ОН); 5.19 (м, 1Н, Н-32); 4.74 (м, 2Н, Н-55); 4.61 (с, 2Н, Н-20); 4.51 (т, 2Н, J=6.2 Гц, Н-56); 4.33 (д, 1H, J=8.4 Гц, Н-47); 4.28 (д, 1Н, J=6.6 Гц, Н-43); 4.08-4.22 (м, 2Н, Н-44,45); 4.04 (с, 3Н, Н-29); 3.66-3.96 (м, 6Н, Н-31,34,35,36,46, ОН); 3.62 (уш.с, 1Н, ОН); 3.51-3.57 (м, 1Н, Н-49); 3.44-3.50 (м, 1Н, Н-49); 3.00-3.18 (м, 2Н, Н-62); 2.37 (м, 2Н, Н-16); 2.23 (т, 2Н, J=7.8 Гц, Н-64); 1.85-2.05 (м, 7Н, -NHAc, Н-33,63); 1.24-1.34 (м, 5Н, Н-18,38).
МСВР (m/z) для C43H53N5O18: [М+Н]+ 928,3458 (найдено), 928,3456 (рассч.); [M+Na]+ 950,3277 (найдено), 950,3270 (рассч.).
Пример 6. Получение соединения (6).

К раствору 10 мг (0,017 ммоль) Dox*HCl в 10 мл сухого ДМФА добавили 6,87 мг (0,017 ммоль, 1 экв.) соединения (3) в присутствии 1,92 мг (0,019 ммоль, 1,1 экв.) Et3N, 2,18 мг (0,019 ммоль, 1,1 экв.) NHS, 3,64 мг (0,019 ммоль, 1,1 экв.) EDC в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали двумя последовательными хроматографированиями: 1) колоночная хроматография на силикагеле (градиентное элюирование CH2Cl2:МеОН (20:1) → МеОН), 2) ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (6) в виде темно-красного порошка (6 мг, 38%). 1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 7.94 (д, 1Н, J=7.5 Гц, Н-6); 7.83 (т, J=7.5 Гц, Н-1); 7.73 (с, 1H, Н-59); 7.56 (д, 1H, J=7.5 Гц, Н-2); 5.56-5.72 (м, 1H, Н-54); 5.42 (м, 1H, ОН); 4.96-5.23 (м, 3Н, Н-32,55); 4.75 (м, 2Н, Н-53); 4.48-4.56 (м, 2Н, Н-20); 4.20-4.33 (м, 2Н, Н-41,45); 4.08-4.19 (м, 1H, Н-42,43); 4.02 (с, 3Н, Н-29); 3.88 (с, 1H, ОН); 3.74-3.84 (м, 1Н, Н-44) 3.46-3.70 (м, 5Н, Н-31,34,35,36,47); 2.95-3.18 (м, 2Н, Н-64); 2.11-2.42 (м, 4Н, Н-16, 61); 1.81-2.07 (м, 5Н, -NHAc, Н-63); 1.54-1.76 (м, 2Н, Н-33).
МСВР (m/z) для C44H53N5O17: [М+Н]+ 924,3509 (найдено), 924,3543 (рассч.); [M+Na]+ 946,3340 (найдено), 946,3329 (рассч.).
Пример 7. Синтез моноацилированного гекс-5-иновой кислотой паклитаксела (7). [[Pilkington-Miksa М. et al. Design, synthesis, and biological evaluation of novel cRGD-paclitaxel conjugates for integrin-assisted drug delivery // Bioconjugate chemistry. - 2012. - T. 23. - №. 8. - C. 1610-1622.]

К раствору 40,9 мг (47,87 мкмоль) паклитаксела в 20 мл сухого CH2Cl2 добавили 5,9 мг (52,66 мкмоль, 1,1 экв.) гекс-5-иновой кислоты и 8,2 мг (52,66 мкмоль, 1,1 экв.) EDC, а также каталитическое количество DMAP в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов при комнатной температуре, затем CH2Cl2 удалили на роторном испарителе. Полученное вещество очищали методом обращенно-фазовой колоночной хроматографии на приборе INTERCHIM PURIFLASH 430 на обращенно-фазовой колонке PURIFLASH С18-НР 15UM F0012 в системе с градиентом концентраций H2O:MeCN (1:0) → H2O:MeCN (0:1). В результате получили соединение (7) в виде белого порошка (41,3 мг, 91%).
1Н ЯМР (400 МГц, CDCl3, δ, м.д.): 8.15 (д, 2Н, J=7.2 Гц, Н-орто-(Ar1)); 7.75 (д, 2Н, J=7.3 Гц, Н-орто-(Ar3)); 7.62 (т, 1Н, J=7.4 Гц, H-пара-(Ar1)); 7.57-7.48 (м, 3Н, Ароматика); 7.47-7.32 (м, 7Н, Ароматика); 6.90 (д, 1Н, J=9.2 Гц, NH); 6.23-6.32 (м, 2Н, Н-10, 13); 5.97 (дд, 1Н, J=2.9 и 9.2 Гц, H-3'); 5.69 (д, 1Н, J=7.1 Гц, Н-2); 5.51 (д, 1H, J=3.1 Гц, Н-2'); 4.98 (дд, 1Н, J=1.8 и 9.5 Гц, Н-5); 4.45 (дд, 1Н, J=6.7 и 10.8 Гц, Н-7); 4.33 (д, 1H, J=8.4 Гц, Н-20); 4.21 (д, 1H, J=8.6 Гц, Н-20); 3.82 (д, 1Н, J=7.1 Гц, Н-3); 2.48-2.70 (м, 3Н, Н-6, 4'',); 2.47 (с, 3Н, 4-ОАс); 2.13-2.44 (м, 8Н, Н-14, 6'', 10-ОАс, Н-2''); 2.00-1.78 (м, 6Н, Н-6,18, 3''); 1.69 (с, 3Н, Н-19); 1.24 (с, 3Н, Н-17); 1.14 (с, 3Н, Н-16).
Пример 8. Синтез конъюгата паклитаксела (8).
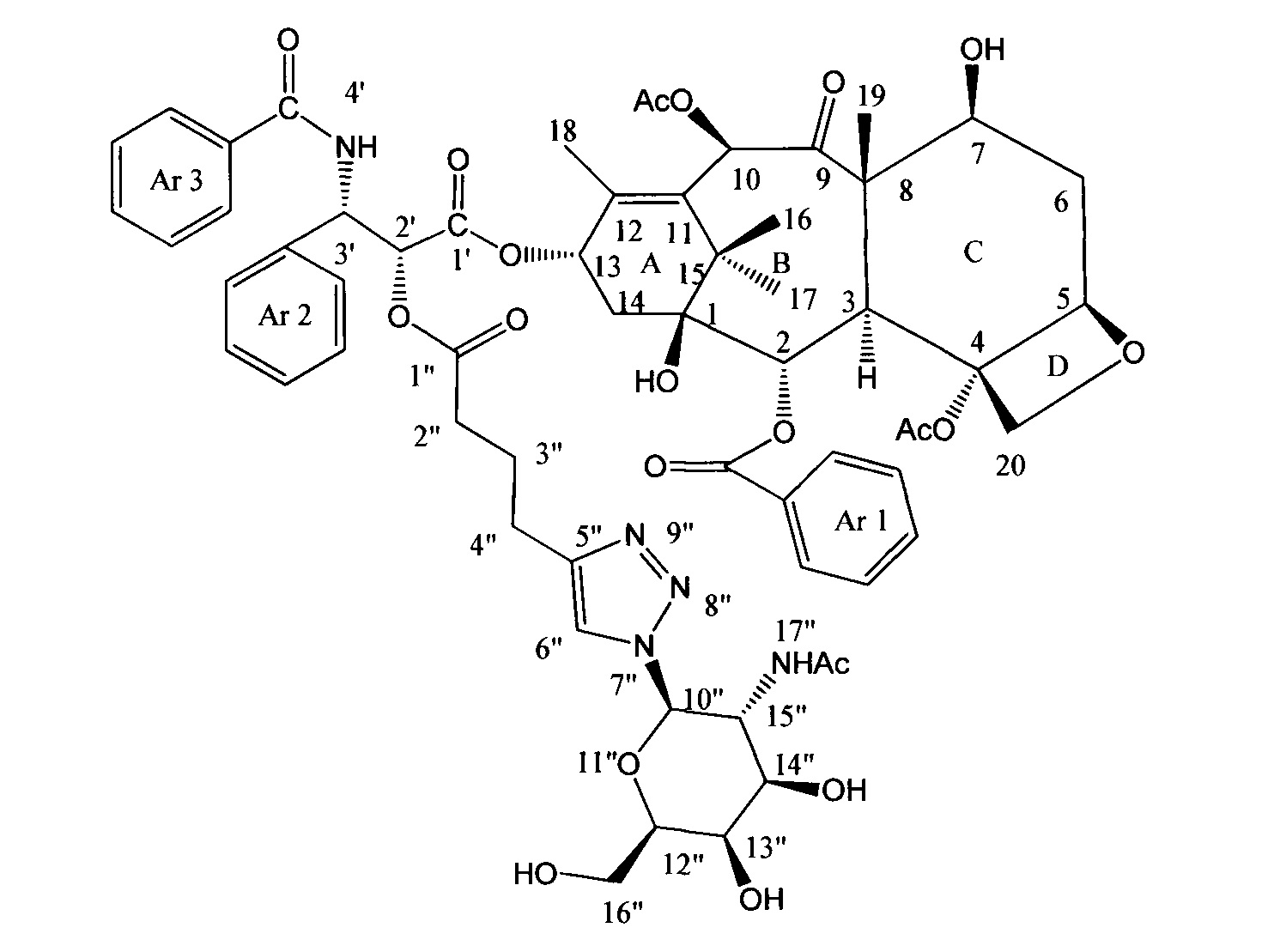
К раствору 10 мг (10,54 мкмоль) соединения (7) в 7 мл сухого ДМФА добавили 2,6 мг (10,54 мкмоль, 1 экв.) 1-Азидо-2-ацетамидо-2-дезокси-3,4,6-три-O-ацетил-β-D-галактопиранозы, полученной согласно методике, описанной в работе [ N. et al. Iron (III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu (0) for the subsequent click chemistry // Chemical Communications. - 2011. - T. 47. - №. 37. - С. 10440-10442] в присутствии 0,4 мг (2,11 мкмоль, 0,2 экв.) CuI и 0,2 мг (2,11 мкмоль, 0,2 экв.) Et3N в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (8) в виде белого порошка (10 мг, 79%).
N. et al. Iron (III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu (0) for the subsequent click chemistry // Chemical Communications. - 2011. - T. 47. - №. 37. - С. 10440-10442] в присутствии 0,4 мг (2,11 мкмоль, 0,2 экв.) CuI и 0,2 мг (2,11 мкмоль, 0,2 экв.) Et3N в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (8) в виде белого порошка (10 мг, 79%).
1Н ЯМР (400 МГц, CDCl3, δ, м.д.): 8.04 (д, 2Н, J=7.8 Гц, Н-орто-(Ar1)); 7.99 (м, 1H, -NH-); 7.82 (с, 1H, Н-6''); 7.69 (д, 2Н, J=6.5 Гц, Н-орто-(Ar3)); 7.29-7.57 (м, 11Н, Ароматика); 6.26 (с, 1H, Н-10); 6.08 (т, 1Н, J=8.7 Гц, Н-13); 5.84-5.91 (м, 1H, Н-3'); 5.60-5.68 (м, 2Н, Н-2, Н-10''); 5.34-5.42 (м, 1Н, Н-2'); 4.91 (д, 1Н, J=9.8 Гц, Н-5); 4.12-4.42 (м, 4Н, Н-7,20, 12''); 3.93-4.00 (м, 1Н, Н-13''); 2.53-2.73 (м, 3Н, Н-6,4''); 2.30-2.50 (м, 5Н, 4-Ас, 2''); 2.10-2.27 (м, 5Н, Н-14, 10-Ас); 1.75-2.06 (м, 6Н, Н-6,3'', -NHAc); 1.65 (с, 3Н, Н-18); 1.60 (с, 3Н, Н-19); 1.11 (с, 3Н, Н-17); 1.07 (с, 3Н, Н-16).
МСВР (m/z) для C61H71N5O20: [М+Н]+ 1194.4771 (найдено), 1194.4765 (рассч.); [M+Na]+ 1216.4585 (найдено), 1216.4585 (рассч.).
Пример 9. Синтез конъюгата паклитаксела (9).
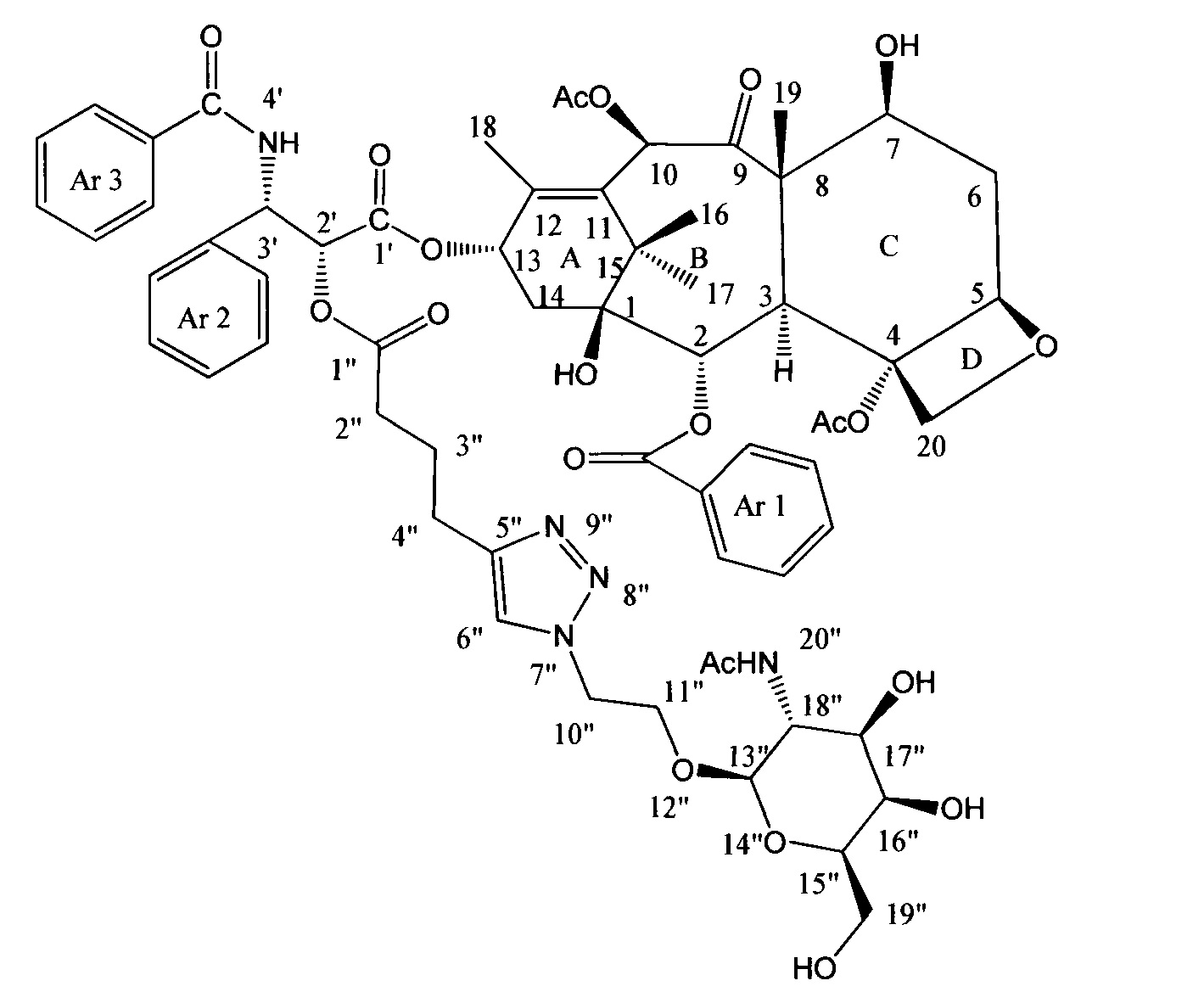
К раствору 10 мг (10,54 мкмоль) соединения (7) в 7 мл сухого ДМФА добавили 3,1 мг (10,54 мкмоль, 1 экв.) 1-O-(2'-азидоэтил)-2-ацетамидо-2-дезокси-β-D-галактопиранозы, полученной согласно методике, описанной в работе [Hasegawa A. et al. Studies on the thioglycosides of N-acetylneuraminic acid 10: Synthesis of S-(α-sialosyl)-(2→6)-O-2-acetamido-2-deoxy-β-d-hexopyranosyl ceramide and its related compounds // Journal of carbohydrate chemistry. - 1992. - T. 11. - №. 3. - C. 319-331], в присутствии 0,4 мг (2,11 мкмоль, 0,2 экв.) CuI и 0,2 мг (2,11 мкмоль, 0,2 экв.) Et3N в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (9) в виде белого порошка (7 мг, 54%).
1Н ЯМР (400 МГц, CDCl3, δ, м.д.): 8.07 (д, 2Н, J=7.5 Гц, Н-орто-(Ar1)); 8.00 (м, 2Н, -NH-, Н-6''); 7.71 (д, 2Н, J=7.0 Гц, Н-орто-(Ar3)); 7.30-7.60 (м, 12Н, Ароматика, -NH-); 6.27 (с, 1Н, Н-10); 6.09 (т, 1H, J=8.7 Гц, Н-13); 5.87 (д, 1Н, J=4.4 Гц, Н-3'); 5.63 (д, 1Н, J=7.0 Гц, Н-2); 5.33-5.42 (м, 1Н, Н-2'); 4.93 (д, 1H, J=9.3 Гц, Н-5); 4.08-4.56 (м, 9Н, Н-7,20, 11'', 13'',15'',16'', +ОН); 3.67-3.98 (м, 7Н, Н-3, 10'', 17'', 18'', 19''); 2.62-2.77 (м, 3Н, Н-6,4''); 2.31-2.52 (м, 5Н, 4-Ас, 2''); 2.09-2.26 (м, 5Н, Н-14, 10-Ас); 1.76-1.99 (м, 9Н, H-6,18,3'', NHAc); 1.62 (с, 1H, Н-19); 1.19 (с, 3Н, Н-17); 1.14 (с, 3Н, Н-16).
МСВР (m/z) для C63H75N5O21: [М+Н]+ 1238.5035 (найдено), 1238.5027 (рассч.); [M+Na]+ 1260.4848 (найдено), 1260.4847 (рассч.).
Пример 10. Синтез конъюгата паклитаксела (10).
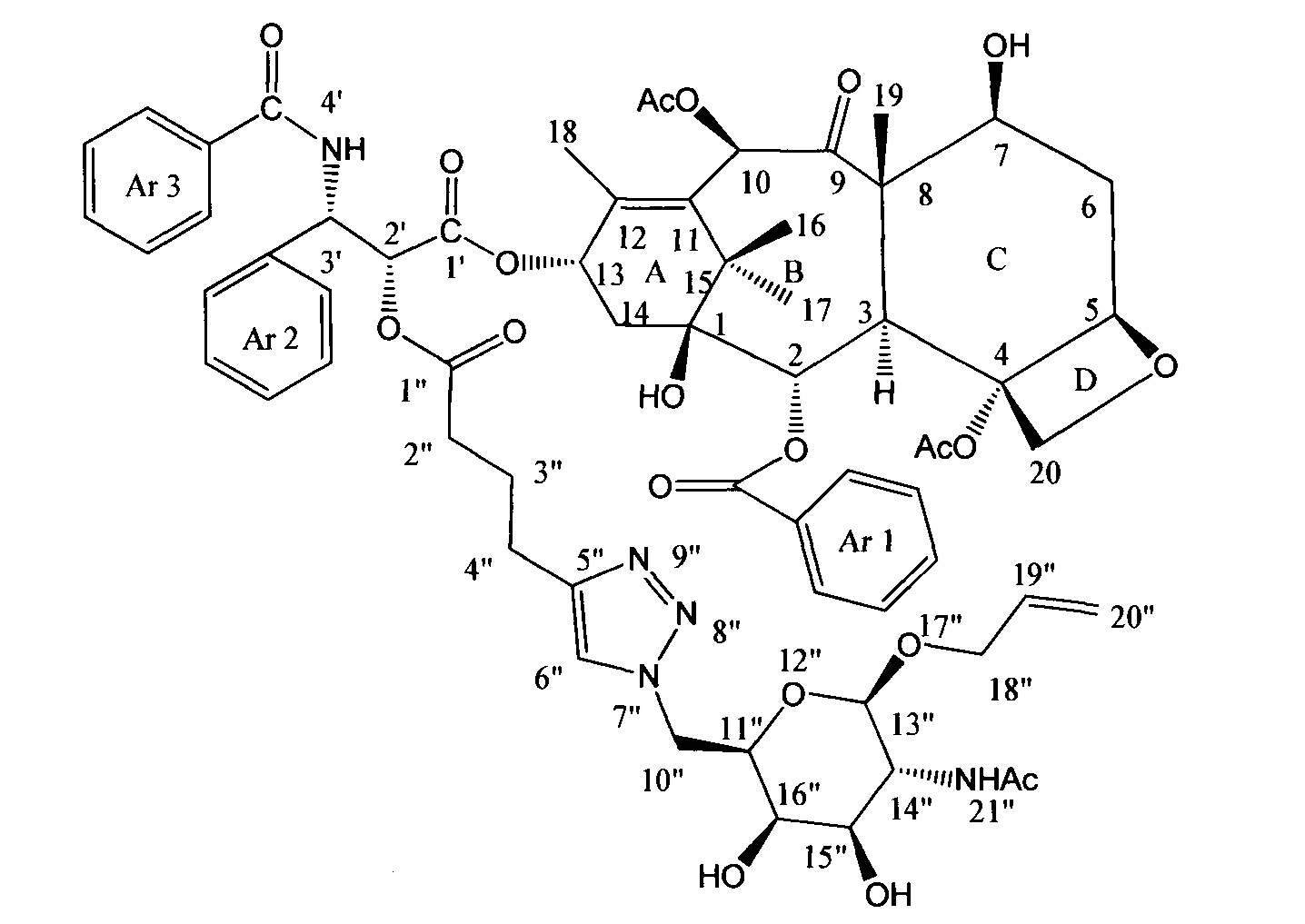
К раствору 6,5 мг (6,85 мкмоль) соединения (7) в 7 мл сухого ДМФА добавили 2,2 мг (6,85 мкмоль, 1 экв.) 1-O-Аллил-2-ацетамидо-2-дезокси-6-азидо-6-дезокси-β-D-галактопиранозы, полученной согласно ранее описанной методике [Wong С.Н. et al. А library approach to the discovery of small molecules that recognize RNA: use of a 1, 3-hydroxyamine motif as core // Journal of the American Chemical Society. - 1998. - T. 120. - №. 33. - C. 8319-8327] в присутствии 0,3 мг (1,58 мкмоль, 0,2 экв.) CuI и 0,2 мг (1,58 мкмоль, 0,2 экв.) Et3N в инертной атмосфере. Полученную смесь перемешивали в течение 24 часов, затем ДМФА удалили на роторном испарителе. Полученное вещество очищали методом ВЭЖХ в обращено-фазовой системе (Н2О - AcCN). В результате получили конъюгат (31) в виде белого порошка (6,5 мг, 77%).
1Н ЯМР (400 МГц, CD3OD, δ, м.д.): 8.11 (д, 2Н, J=7.2 Гц, Н-орто-(Ar1)); 7.80 (д, 2Н, J=7.2 Гц, Н-орто-(Ar3)); 7.77 (с, 1Н, Н-6''); 7.62-7.70 (м, 2Н, -NH-, H-пара-(Ar1)); 7.38-7.60 (м, 9Н, Ароматика); 7.27 (т, 1Н, J=7.1 Гц, Н-пара-(Ar2)); 6.43 (с, 1Н, Н-10); 6.08 (т, 1Н, J=9.1 Гц, Н-13); 5.84 (д, 1H, J=6.1 Гц, Н-3'); 5.70-5.81 (м, 1Н, Н-19''); 5.65 (д, 1Н, J=7.2 Гц, Н-2); 5.46 (д, 1H, J=6.0 Гц, Н-2'); 4.95-5.15 (м, 3Н, Н-5,20''); 4.59-4.66 (м, 1H, Н-11''); 4.17-4.39 (м, 4Н, Н-7, 13'',20); 4.09 (дд, 1Н, J=4.7, 13.3 Гц, Н-14''); 3.76-4.00 (м, 5Н, Н-3, 15'',16'',18''); 3.60 (дд, 1H, J=3.0, 10.7 Гц, Н-10''); 3.42-3.50 (м, 1H, Н-10''); 2.68-2.79 (м, 2Н, Н-4''); 2.44-2.54 (м, 3Н, Н-2'',6); 2.40 (с, 3Н, 4-ОАс); 2.11-2.23 (м, 5Н, Н-14, 10-ОАс); 1.76-2.06 (м, 7Н, Н-6,3'', 18, -NHAc); 1.66 (с, 3Н, Н-19); 1.14 (с, 6Н, Н-16, 17).
МСВР (m/z) для C64H75N5O20: [М+Н]+ 1234.5084 (найдено), 1234.5078 (рассч.); [M+Na]+ 1256.4896 (найдено), 1256.4898 (рассч.).
Пример 11. Измерение константы диссоциации комплекса лиганд-ASGP-R
Эксперимент проводился на приборе Biacore X100 (Biacore АВ, Уппсала, Швеция) с использованием чипа-носителя СМ5, состоящего из золотой пластины, покрытой слоем карбоксиметилированного декстрана. Поверхность чипа включает в себя две проточных ячейки: на одной иммобилизуется анализируемый белок, вторая ячейка - ячейка сравнения.
Использовалась буферная смесь, содержащая 150 mM NaCl, 50 mM CaCl2, 50 mM Tris, рН 7.4. Обе ячейки были активированы смесью, содержащей 0,05 М N-гидроксисукцинимид (NHS) и 0,2 М N-этил-N'-диметиламинопропил карбодиимид (EDC) в течении 7 минут, скорость потока смеси 10 мкл/мин. Поверхность ячейки 1 не модифицировалась. На поверхности ячейки 2 был иммобилизован фермент ASGRP печени кролика, растворенный в буфере: 10 mM Tris-HCl, рН 7.0. Раствор фермента подавался в течении 15 минут со скоростью 10 мкл/мин. Обе ячейки были обработаны 0.1 М раствором этаноламина (рН 8.5) для деактивации. Количество иммобилизированного белка ASGPR - 2000 RU.
Отбор и подготовка проб
Лиганды формул 1, 2, 3, исследуемые на аффинность к белку ASGP-R, были растворены в рабочей буферной смеси (150 mM NaCl, 50 mM CaCl2, 50 mM Tris, рН 7.4). К образцам с низкой растворимостью был добавлен диметилсульфоксид (ДМСО), вплоть до объема, равного половине объема использованной буферной смеси. Более высокие концентрации ДМСО не были использованы ввиду непереносимости белковой структуры экстремальных условий и опасностью необратимой денатурации.
Каждый лиганд был представлен в широком диапазоне концентраций, от 10-2 М до 5*10-11 М. Скорость потока 20 мкл/мин, лиганд подавался в течение 180 с (время связывания), и далее в течение 60 с изучалась диссоциация комплекса. Для восстановления чип-носитель обрабатывался 20 мкл 20 мМ раствором ЭДТА. Эксперименты проводились при 25°С. Все буферные растворы были дегазированы и профильтрованы.
Результаты приведены в таблице 1.


Полученные данные свидетельствуют о большей аффинности к ASGP-R полученных лигандов по отношению к нативному лиганду - N-ацетилгалактозамину.
Пример 12. Изучение токсичности конъюгатов по отношению клеточным линиям гепатоцеллюлярной карциномы.
Результаты исследования представлены в таблице 1. Экспериментальные данные цитоксичности самого паклитаксела на клеточной линии HepG2 представлены в работе Луо и соавторов [Luo D. et al. Effects of Bcl-2 and Bcl-XL protein levels on chemoresistance of hepatoblastoma HepG2 cell line // Biochemistry and Cell Biology. - 2000. - T. 78. - №. 2. - С. 119-126.], а так же получены в настоящей работе.

Для полученных конъюгатов измеренные значения цитотоксичности (СС50) на клеточной линии гепатоцеллюлярной карциномы сравнимы со значением цитотоксичности для противоопухолевого соединения.
Пример 13. Исследование устойчивости конъюгатов 8-10 в моделях биологических сред при рН 5.0 и 7.4.
Были приготовлены буферные растворы (НСООН, NH4OH) с рН 5.0 и 7.4. После растворения 200 мкг конъюгата в 2 мл буфера было отобрано по 200 мкл полученного раствора в заданные временные точки (0; 0.5; 1; 2; 4; 8; 12; 24 часа). Затем образцы подвергали заморозке при -20°С. После чего образцы были исследованы методом ВЭЖХ-МС на приборе Shimadzu Prominence LC-20 с одиночным квадрупольным масс-спектрометром Shimadzu LCMS-2020 с двойным источником ионизации DUIS-ESI-APCI. Аналитической и препаративной колонкой была Phenomenex Luna 3u C18 100A.
Для всех исследованных конъюгатов площадь пика вещества с течением времени оставалась одинаковой в пределах погрешности, из чего следует, что полученные конъюгаты не подвергаются деградации и являются устойчивыми в этих биологических средах.
Пример 14. Исследования накопления доксорубицина и конъюгатов 4-6 в клеточной культуре HepG2 методом флуоресцентной микроскопии.
Флуоресцентная микроскопия осуществлена с использованием системы для визуализации флюоресценции EVOS FL Cell Imaging System. С 40 - кратным увеличением. На фиг. 1 представлены снимки, сделанные при помощи микроскопа, конъюгатов (4) - А, (5) - В, (6) - С. Время инкубирования - 24 часа. Как свидетельствуют представленные, снимки конъюгаты накапливаются в цитоплазме и, следовательно, доставляются лигандами внутрь клетки.
Пример 15. Анализ морфологии ядра и цитоскелета клеточной культуры линии гепатоцеллюлярной карциномы (HepG2) при воздействии на них конъюгатов 8-10 при помощи иммунно-флуоресцентной микроскопии.
Клетки HepG2 выращивали на покрытых поли-L-лизином покровных стеклах в течение 24 часов с последующим воздействием паклитаксела и конъюгатов в концентрации 1 мкмоль. После 24-часового лечения клетки фиксировали в 4% параформальдегиде (Sigma-Aldrich) в PBS в течение 10 минут, три раза промывали в PBS, а затем инкубировали в течение часа с блокирующим раствором (1% бычьего сывороточного альбумина (BSA), 1% Triton Х-100 в PBS). Затем клетки инкубировали в течение ночи с альфа-тубулином Alexa Fluor 488 Mouse Monoclonal Antibody (Invitrogen # 322588) при 2 мкг/мл. Клетки дважды промывали PBS, а ядра окрашивали 4', 6-диамидино-2-фенилиндолом (DAPI) (Invitrogen). Клетки наблюдали с помощью микроскопа Zeiss Axiovert 200 М.
Необработанные клетки показали типичные ядерные и цитоскелетные структуры с образованием нормальных митотических веретен. Микротрубочки, организованные как диффузные в цитоплазме (фиг. 2).
После обработки клеток с помощью паклитаксела характерны изменения морфологии: митозы с более чем двумя полюсами; многие аномальные ядра (микроядра или многоядерные), измененная структура микротрубочек (более толстые и более плотные пучки микротрубочек). Подобные клеточные морфологические изменения наблюдались для обработанных паклитакселем конъюгатов (фиг. 3).
Способ дифференциальной диагностики тяжелой бронхиальной астмы
Способ модификации моноцитов периферической крови для повышения их паракринной активности при аутологической трансплантации
Способ прогнозирования индивидуального риска развития бронхиальной астмы в регионах с высокой и низкой распространенностью гельминтных инфекций
Способ прогнозирования тяжести течения хронической обструктивной болезни легких
Способ получения наногибридного функционального сепарационного материала на основе модифицированного носителя и модифицированных наночастиц металла
Наногибридный функциональный сепарационный материал на основе модифицированного носителя и модифицированных наночастиц металла
Устройство для исследования воздействия низкочастотного магнитного поля на кинетику биохимических процессов в биологических системах, содержащих магнитные наночастицы
Способ покрытия наночастиц магнетита слоем золота
Композиция, ингибирующая теломеразу
Новые диспиро-индолиноны, ингибиторы mdm2/p53 взаимодействия, способ получения и применения

