Результат интеллектуальной деятельности: ТВЕРДЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ БИОПТЕРИНА, И СПОСОБЫ ПРИМЕНЕНИЯ ТАКИХ КОМПОЗИЦИЙ
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к твердым фармацевтическим композициям, содержащим производные биоптерина, а также способам получения таких твердых фармацевтических композиций. Настоящее изобретение также относится к твердым фармацевтическим композициям по настоящему изобретению для лечения заболеваний.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Биоптерины и их производные являются молекулами, представляющими интерес для фармацевтики. Например, тетрагидробиоптерин (BH4, сапроптерин) недавно был одобрен для лечения гиперфенилаланинемии (HPA) у пациентов взрослой возрастной группы и детской возрастной группы, возрастом 4 года и более, страдающих фенилкетонурией (PKU). Для данной цели тетрагидробиоптерин составляют в коммерческое лекарственное средство (продаваемое под названием куван) в виде растворяемой таблетки с маннитом (E421), безводным гидрофосфатом кальция, кросповидоном типа A, аскорбиновой кислотой (E300), натрия стеарилфумаратом и рибофлавином (E101). Также в данном контексте см. WO 2006/055511.
Другими терапевтически перспективными соединениями биоптерина являются 4-амино-5,6,7,8-тетрагидро-L-биоптерин и 4-амино-7,8-дигидро-L-биоптерин. Было показано, что оба соединения проявляют свойства, отличные от свойств других NO-ингибиторов, что делает данное соединение потенциально более подходящим, чем "классические" аналоги аргинина (Werner et al., (1996). Biochemical Journal 320, 93–6 или патент США 5922713). Было показано, что оба соединения являются эффективными в экспериментальном TBI (см., например, WO 2004/084906, патент США 8222828, европейский патент 0906913 или Terpolilli et al., J Neurotrauma. 2009; 26(11):1963-75). Однако данные производные биоптерина еще не были одобрены для консервативного лечения. Таким образом, существует потребность в обеспечении фармацевтических композиций, которые являются подходящими для терапевтических применений у людей. Теоретически, такую фармацевтическую композицию должно быть легко получить, легко применять и при этом она должна обладать стабильностью – в данном контексте отмечено, что гидрогенизированные производные биоптерина чувствительны к окислению при хранении в течение более длительного периода времени или при получении в виде раствора.
Таким образом, целью настоящего изобретения является обеспечение фармацевтической композиции, содержащей производные биоптерина, которая удовлетворяет данные потребности.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данная проблема решается посредством вариантов осуществления настоящего изобретения, как определено в формуле изобретения, описано в описании и проиллюстрировано в примерах и фигурах.
Настоящее изобретение в одном варианте осуществления относится к твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
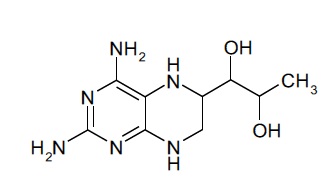 (I),
(I),
и/или соединение, имеющее формулу (II):
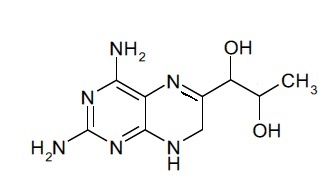 (II),
(II),
и
b) по меньшей мере одну фосфатную соль.
Твердая композиция по настоящему изобретению может быть "приспособлена для внутривенного введения". Это означает, что после смешивания твердой композиции с фармацевтически приемлемым носителем, предпочтительно фармацевтически приемлемой текучей средой, например, водой, или буфером, или какой-либо восстанавливающей текучей средой, получают композицию, которую можно непосредственно использовать для внутривенного применения как таковую. Таким образом, после смешивания твердой композиции с фармацевтически приемлемым носителем получают готовую для применения композицию, которая подходит для внутривенного применения.
В одном варианте осуществления в фармацевтической композиции по настоящему изобретению по меньшей мере одна фосфатная соль представляет собой фосфат натрия, фосфат калия или фосфат аммония. Фосфатная соль может быть выбрана из группы, включающей Na2HPO4 (безводный), Na2HPO4 • 2 H2O, Na2HPO4 • 7 H2O, Na2HPO4 • 12 H2O, NaH2PO4 (безводный), NaH2PO4 • H2O, NaH2PO4 • 2 H2O, K2HPO4 (безводный), K2HPO4 • 3 H2O, KH2PO4 (безводный) и их смеси.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению фосфатная соль представляет собой Na2HPO4 • 2 H2O, и количество Na2HPO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,04 до 0,4.
В дополнительных вариантах осуществления фармацевтической композиции по настоящему изобретению фосфат натрия представляет собой NaH2PO4 • 2 H2O, и количество NaH2PO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение NaH2PO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,01 до 0,09.
В других вариантах осуществления фармацевтическая композиция по настоящему изобретению содержит две различные фосфатные соли натрия. Необязательно, две различные фосфатные соли натрия представляют собой NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количество NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение как NaH2PO4 • 2 H2O, так и Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,02 до 0,5.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количества NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O, присутствующих в композиции, выбраны таким образом, что молярное отношение каждого из NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,02 до 0,5.
В дополнительных вариантах осуществления фармацевтической композиции по настоящему изобретению соединение (I) и/или соединение (II) присутствуют в виде свободного основания.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению фармацевтическая композиция представляет собой лиофилизированную фармацевтическую композицию.
В другом варианте осуществления фармацевтической композиции по настоящему изобретению соединение (I) представляет собой соединение, имеющее формулу (Ia):
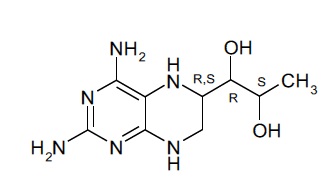 (Ia).
(Ia).
В другом варианте осуществления фармацевтической композиции по настоящему изобретению соединение (II) представляет собой соединение, имеющее формулу (IIa):
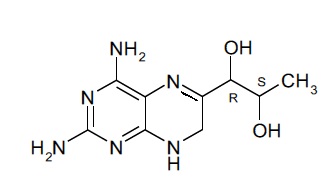 (IIa).
(IIa).
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению фармацевтическая композиция содержит дополнительное фармацевтическое вспомогательное вещество.
В другом варианте осуществления фармацевтической композиции по настоящему изобретению дополнительное фармацевтическое вспомогательное вещество представляет собой неорганическую соль. Неорганическая соль может быть выбрана из MgCl2, CaCl2, NH4Cl, KCl или NaCl, предпочтительно неорганическая соль представляет собой NaCl.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количество NaCl, присутствующего в композиции по настоящему изобретению, выбрано таким образом, что молярное отношение NaCl к соединению (I) или соединению (II) находится в диапазоне от 1,5 до 4, предпочтительно от 1,8 до 3,7.
В некоторых вариантах осуществления фармацевтическая композиция по настоящему изобретению дополнительно содержит кристаллизационную воду.
В других вариантах осуществления фармацевтическая композиция по настоящему изобретению приспособлена для восстановления в воде. "Приспособлена для восстановления в воде" означает, что (дегидратированная или концентрированная) композиция может быть преобразована в жидкое состояния путем добавления воды.
В дополнительных вариантах осуществления фармацевтическая композиция по настоящему изобретению приспособлена для введения посредством инфузии или инъекции.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению соединение (I) представляет собой (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерин.
В дополнительных вариантах осуществления фармацевтической композиции по настоящему изобретению соединение (I) представляет собой (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерин.
В других вариантах осуществления фармацевтической композиции по настоящему изобретению соединение (I) представляет собой диастереомерную смесь, которая содержит больше (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина, чем (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерина.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина и (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерина выбраны таким образом, что отношение количеств (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина к (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерину находится в диапазоне от 0,5 до 2, предпочтительно составляет около 1,3.
В дополнительных вариантах осуществления фармацевтической композиции по настоящему изобретению единичная доза композиции содержит 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 140 ± 30 мг кристаллизационной воды, 70 ± 7 мг дигидрата гидрофосфата динатрия (Na2HPO4 ∙ 2 H2O), 16,5 ± 2 мг дигидрата дигидрофосфата натрия (NaH2PO4 ∙ 2 H2O) и 350 ± 30 мг хлорида натрия (NaCl).
В других вариантах осуществления фармацевтической композиции по настоящему изобретению единичная доза композиции содержит 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 60 ± 50 мг кристаллизационной воды, 70 ± 7 мг дигидрата гидрофосфата динатрия (Na2HPO4 ∙ 2 H2O), 12 ± 2,5 мг дигидрата дигидрофосфата натрия (NaH2PO4 ∙ 2 H2O) и 350 ± 30 мг хлорида натрия (NaCl).
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению композиция содержит 1, 2, 3, 4, 5, 6, 7 или более дополнительных соединений, при этом дополнительные соединения выбраны из группы, включающей одно или более соединений, выбранных из группы, включающей 4-амино-L-биоптерин, (6R,S)-5,6,7,8-тетрагидро-L-биоптерин, 1-[(6R,S)-2,4-диамино-5,6,7,8-тетрагидроптеридин-6-ил]пропанол, 1-[(6R,S)-2,4-диамино-5,6,7,8-тетрагидроптеридин-6-ил]пропан, (1R,2S)-1-[(6R,S)-2-(ацетиламино)-4-амино-5,6,7,8-тетрагидроптерин-6-ил]-1,2-диацетоксипропан, 2,4-диамино-7,8-дигидроптеридин, 2,4-диаминоптеридин.
Настоящее изобретение также относится к применению лиофилизированной фармацевтической композиции по настоящему изобретению для лечения заболевания. В некоторых вариантах осуществления заболевание выбрано из группы, включающей травматическое повреждение головного мозга, нетравматическое повреждение головного мозга, предпочтительно инсульт или менингит, повышенное внутричерепное давление, вторичное повреждение головного мозга.
Кроме того, настоящее изобретение относится к лиофилизированной фармацевтической композиции для применения в лечении заболевания. В некоторых вариантах осуществления заболевание выбрано из группы, включающей травматическое повреждение головного мозга, нетравматическое повреждение головного мозга, предпочтительно инсульт или менингит, повышенное внутричерепное давление, вторичное повреждение головного мозга.
Настоящее изобретение дополнительно относится к способу получения лиофилизированной твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну фосфатную соль, и необязательно NaCl;
при этом способ включает
aa) растворение соединения формулы (III) и/или соединения формулы (II):
 (III)
(III)
в буфере, при этом буфер предпочтительно содержит фосфат;
bb) лиофилизацию раствора, полученного на стадии aa).
В некоторых вариантах осуществления способ по настоящему изобретению дополнительно включает стадию растворения лиофилизата, полученного на стадии bb), в фармацевтически приемлемой текучей среде с получением инъекционного раствора.
Соответственно, настоящее изобретение также относится к способу получения инъекционного раствора, содержащего
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну фосфатную соль, и необязательно NaCl;
при этом способ включает
aa) растворение соединения формулы (III) и/или (II):
(III),
в буфере, при этом буфер предпочтительно содержит фосфат;
bb) лиофилизацию раствора, полученного на стадии aa);
cc) восстановление лиофилизата, полученного на стадии bb), в фармацевтически приемлемой текучей среде с получением инъекционного раствора, при этом лиофилизатом, полученным на стадии bb), заполняют флакон.
В одном варианте осуществления способа по настоящему изобретению лиофилизатом, полученным на стадии bb), заполняют 50 мл флакон,
(i) предпочтительно в количестве приблизительно 1-1,5 г, предпочтительно 1,25 г твердого состава или
(ii) предпочтительно в количестве приблизительно 0,9-1,4 г, предпочтительно 1,15 г твердого состава.
В других вариантах осуществления в способе по настоящему изобретению буфер из стадии aa) представляет собой буфер на основе гидрофосфата натрия, содержащий по меньшей мере одну фосфатную соль.
В других вариантах осуществления в способе по настоящему изобретению буфер из стадии aa) содержит NaOH, буфер на основе гидрофосфата натрия и воду. Необязательно, NaOH представляет собой 5 н. раствор NaOH.
В другом варианте осуществления способа по настоящему изобретению буфер на основе гидрофосфата натрия получают путем раздельного растворения NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O.
В дополнительном варианте осуществления способа по настоящему изобретению буфер на основе гидрофосфата натрия характеризуется значением pH 7,4 в результате добавления раствора NaH2PO4 • 2 H2O к растворенному Na2HPO4 • 2 H2O.
В некоторых вариантах осуществления способа по настоящему изобретению буфер содержит 12-16% (вес./вес.) 5 н. NaOH, 8-12% (вес./вес.) буфера на основе гидрофосфата натрия и 74-78% (вес./вес.) воды для инъекций.
В другом варианте осуществления способа по настоящему изобретению раствор, полученный на стадии aa), стерилизуют фильтрацией, предпочтительно с применением 0,22 мкм фильтра.
В дополнительных вариантах осуществления способа по настоящему изобретению буфер характеризуется значением pH приблизительно 8, 9, 10, 11, 12, 13 или 14.
В еще одном варианте осуществления способа по настоящему изобретению раствор из стадии aa) характеризуется значением pH приблизительно 4, 5, 6, 7, 8, 9, 10 или 11, предпочтительно от 6,5 до 7,6, наиболее предпочтительно 7,4.
В других вариантах осуществления способа по настоящему изобретению лиофилизатом, полученным на стадии bb), заполняют флаконы предпочтительно в количестве приблизительно 1-1,5 г, предпочтительно 1,25 г твердого состава или предпочтительно в количестве приблизительно 0,9-1,4 г, предпочтительно 1,15 г твердого состава.
В дополнительных вариантах осуществления способа по настоящему изобретению буфер получают с дегазированным буфером. В некоторых вариантах осуществления буфер дегазируют азотом до достижения содержания кислорода ˂ 1,0 ppm.
В другом варианте осуществления способа по настоящему изобретению после получения раствора лиофилизацию начинают не позже, чем через 2 часа.
Настоящее изобретение также относится к фармацевтической композиции, получаемой с использованием способа по настоящему изобретению.
Настоящее изобретение дополнительно относится к применению лиофилизированной фармацевтической композиции по настоящему изобретению в изготовлении лекарственного препарата для лечения субъекта с травматическим повреждением головного мозга, таким как закрытая травма черепа, повышенное внутричерепное давление, но также вторичным повреждением головного мозга или нетравматическим повреждением головного мозга, таким как инсульт или менингит.
Кроме того, настоящее изобретение относится к способу лечения заболевания у субъекта, включающему стадию введения лиофилизированной фармацевтической композиции по настоящему изобретению субъекту, нуждающемуся в этом. В некоторых вариантах осуществления в способе лечения заболевания у субъекта максимальная суточная доза составляет 20 мг/кг веса тела в сутки, предпочтительно 17,5, 15,0 или 12,5, 10, 8,5, 7,5, 5,0 или 2,5 мг/кг веса тела в сутки.
Настоящее изобретение также относится к лиофилизированной фармацевтической композиции для применения в лечении заболевания у субъекта, при этом применение предусматривает стадию введения лиофилизированной фармацевтической композиции по настоящему изобретению субъекту, нуждающемуся в этом. В некоторых вариантах осуществления максимальная суточная доза составляет 20 мг/кг веса тела в сутки, предпочтительно 17,5, 15,0 или 12,5, 10, 8,5, 7,5, 5,0 или 2,5 мг/кг веса тела в сутки.
В еще одном варианте осуществления настоящее изобретение относится к твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну неорганическую соль, предпочтительно NaCl.
В таком варианте осуществления количество NaCl, присутствующего в композиции по настоящему изобретению, может быть выбрано так, чтобы молярное отношение NaCl к соединению (I) или соединению (II) находилось в диапазоне от 1,5 до 4, предпочтительно от 1,8 до 3,7, от 1,85 до 3,6, от 1,9 до 3,4, наиболее предпочтительно от 1,9 до 2,5.
В одном варианте осуществления молярное отношение NaCl к соединению (I) или соединению (II) составляет приблизительно 2,2.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг.1 показано получение лекарственного вещества дигидрата 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина дигидрохлорида (VAS203).
На фиг. 2 показана технология изготовления лекарственного продукта VAS203 в соответствии с настоящим изобретением.
На фиг. 3 показаны данные о стабильности 1 г флаконов с VAS203 (№ партии 928606), хранящихся при 2-8°C (таблица 5).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения заключалась в обеспечении фармацевтических композиций. Кроме того, данные композиции должны были быть удобными в применении и стабильными. В данном документе было обнаружено, что при применении твердых фосфатных солей (отдельно или совместно с неорганической солью, такой как NaCl или KCl) совместно с твердыми производными биоптерина, такими как соединения формулы (I) или (II) в результате получают фармацевтические композиции (составы), которые очень хорошо приспособлены для достижения значения pH или диапазона pH, а также осмоляльности, которая при растворении непосредственно пригодна для терапевтического введения. Твердые фармацевтические композиции являются особенно хорошо подходящими и удобными в применении, поскольку они уже приспособлены для внутривенного введения, благодаря чему они также обеспечивают возможность безопасного применения. Кроме того, проблема окисления производных биоптерина в жидкостях была решена путем обеспечения твердых композиций.
Таким образом, настоящее изобретение относится к твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну фосфатную соль.
Кроме того, настоящее изобретение также относится к твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну неорганическую соль, в частности, NaCl.
Термин "твердое вещество" или "твердая композиция", применяемый в данном документе, относится к частицам (ионам, атомам, или молекулам, или соединениям), которые плотно соединенные между собой. Силы между частицами являются такими прочными, что частицы не могут свободно перемещаться, а могут только колебаться. В результате этого твердое вещество имеет стабильную определенную форму и определенный объем. Твердые вещества могут изменять свою форму только под действием силы, например, при дроблении или нарезании. В кристаллических твердых веществах частицы (атомы, молекулы или ионы) соединены в правильно упорядоченную повторяющуюся кристаллическую решетку. Существует множество различных кристаллических структур, при этом одно и то же вещество может иметь более одной структуры (или твердой фазы). Термин "твердое вещество" также охватывает аморфные или некристаллические композиции/вещества/твердые вещества. Как правило, агрегатное состояние определяют при комнатной температуре и давлении окружающей среды. Комнатная температура обычно обозначает небольшой диапазон температур, при которых воздух не чувствуется ни горячим, ни холодным, при этом часто указывается как диапазон от 20 до 23,5°C со средним значением 21°C (70°F). Под давлением окружающей среды подразумевают давление от 900 до 1200 гПа (гектопаскаль), предпочтительно, приблизительно 1000 гПа. Твердые вещества могут быть преобразованы в жидкости посредством плавления, а жидкости могут быть преобразованы в твердые вещества посредством замораживания. Твердые вещества также могут преобразовываться непосредственно в газы путем процесса сублимации.
Твердую композицию по настоящему изобретению можно вводить (обычно при растворении в жидкости, такой как вода) человеку ("введение"). Это обеспечивает введение субъекту терапевтически эффективной дозы твердой композиции по настоящему изобретению.
"Терапевтически эффективное количество" представляет собой дозу соединения формулы (I) и/или соединения формулы (II), которая обеспечивает эффекты, с целью которых ее вводят. Точная доза будет зависеть от цели лечения и будет установлена специалистом в данной области с применением известных методик. Как известно из уровня техники и описано выше, может быть необходимо регулирование относительно системной или локализованной доставки, возраста, веса тела, общего состояния здоровья, пола, режима питания, времени введения, взаимодействия лекарственных средств и тяжести состояния, и оно будет очевидным специалисту в данной области в результате проведения обычных экспериментов. В данном контексте снова отмечено, что оба соединения − 4-амино-5,6,7,8-тетрагидро-L-биоптерин и 4-амино-7,8-дигидро-L-биоптерин − являются фармацевтически активными. Также отмечено, что 4-амино-7,8-дигидро-L-биоптерин может быть получен путем окисления (также самопроизвольного окисления) из 4-амино-5,6,7,8-тетрагидро-L-биоптерина. Таким образом, композиция по настоящему изобретению может содержать либо только 4-амино-5,6,7,8-тетрагидро-L-биоптерин, либо только 4-амино-7,8-дигидро-L-биоптерин, либо смесь двух данных соединений в любом соотношении.
Твердые композиции по настоящему изобретению применимы как для лечения людей, так и для ветеринарных способов применения. Соединения, описанные в данном документе, обладающие необходимой терапевтической активностью, можно вводить в фармацевтически приемлемом носителе пациенту/субъекту, как описано в данном документе. В зависимости от способа введения соединения могут быть составлены различными способами, как этом обсуждается ниже. Твердую композицию по настоящему изобретению можно вводить отдельно или в комбинации с другими терапевтическими средствами.
Твердая композиция по настоящему изобретению может быть дополнительно "приспособлена для внутривенного введения". Это означает, что после смешивания твердой композиции, предпочтительно, с фармацевтически приемлемым носителем, предпочтительно фармацевтически приемлемой текучей средой, например, водой, или буфером, или любой восстанавливающей текучей средой, описанной ниже, получают композицию, которую можно непосредственно использовать для внутривенного применения как таковую. Таким образом, после смешивания твердой композиции с фармацевтически приемлемым носителем получают готовую для применения композицию, которая подходит для внутривенного применения. Внутривенное введение представляет собой инфузию или инъекцию жидких веществ непосредственно в вену, обычно с помощью шприца и полой иглы, которой прокалывают кожу на достаточную глубину для доставки материала в организм субъекта.
Таким образом, в дополнительных вариантах осуществления фармацевтическая композиция по настоящему изобретению приспособлена для введения посредством инфузии или инъекции. Под "инфузией" подразумевают непрерывное введение в течение определенного периода времени. Например, такое введение может занимать от 10 минут до 4 дней. Таким образом, оно может занимать около 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 32, 40, 48, 56, 68, 72, 86 или 96 часов. "Инъекция" означает кратковременный инфузионный способ введения текучей среды в организм субъекта. Обычно такое введение занимает менее 10 минут. Однако инъекцию можно повторять несколько раз в сутки. Например, инъекцию можно осуществлять 1, 2, 3, 4, 5, 6, 7, 8 или 9 раз в сутки. Кроме того, инъекцию(и) можно осуществлять в течение 1, 2, 3 или 4 дней. Однако введение посредством инъекции или инфузии при необходимости также можно осуществлять в течение более длительного периода. Очевидным является то, что точная длительность зависит от множества факторов.
Как правило, композиции для внутривенного введения представляют собой твердую фармацевтическую композицию по настоящему изобретению, смешанную с фармацевтически приемлемым носителем, например, со стерильным изотоническим водным буфером, с получением фармацевтического раствора. При необходимости, данные композиция/раствор также могут содержать солюбилизирующее средство и местное обезболивающее средство, такое как лигнокаин, для ослабления боли в месте инъекции. Обычно ингредиенты поставляются либо раздельно, либо смешанными друг с другом в единичной дозированной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметично закрытом контейнере, таком как флакон, ампула или саше, с указанием количества соединения (I) и/или соединения (II). Если композиция/раствор предназначены для введения посредством инфузии, их можно разводить в инфузионном флаконе, содержащем стерильную воду фармацевтической степени чистоты или солевой раствор. Если композицию вводят посредством инъекции или инфузии, может быть представлена ампула со стерильной водой для инъекций или солевым раствором, так что ингредиенты могут быть смешаны перед введением.
Термин "фармацевтически приемлемый" означает одобренный регулирующим органом или другой общепризнанной фармакопеей для применения животными и, более конкретно, людьми. Обычно "фармацевтически приемлемый" также означает "физиологически приемлемый", что означает, что текучая среда/носитель соответствует нормальному функционированию живого субъекта или является характерным для него и не вызывает токсичность или какие-либо другие нежелательные эффекты. Такие текучая среда/носитель в норме характеризуются приблизительно тем же значением pH и/или осмоляльности, что и, например, текучие среды, такие как кровь животного.
"Фармацевтически приемлемая текучая среда" может быть одной из следующего неограничивающего списка неводных или водных растворителей. Неводными растворителями являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и инъекционные сложные органические эфиры, такие как этилолеат. Водные растворители включают воду, спиртовые/водные растворы, эмульсии или суспензии, в том числе солевой раствор и забуференные среды, раствор ионов натрия, раствор Рингера с декстрозой, декстрозой и ионами натрия, лактатный раствор Рингера или нелетучие масла. Среды для внутривенного введения включают текучую среду и питательные добавки, электролитные добавки (такие как добавки на основе раствора Рингера с декстрозой) и т. п. Предпочтительной фармацевтически приемлемой текучей средой является водный растворитель, например, стерильная вода для инъекций.
В некоторых вариантах осуществления твердую фармацевтическую композицию по настоящему изобретению восстанавливают перед введением. В других вариантах осуществления фармацевтическая композиция по настоящему изобретению приспособлена для восстановления в воде. "Восстановление в воде" означает возвращение (дегидратированной или концентрированной) композиции в жидкое состояние путем добавления фармацевтически приемлемой текучей среды, описанной выше. Предпочтительно, твердая композиция по настоящему изобретению восстановлена в воде.
Настоящее изобретение может охватывать различные твердые композиции. Например, твердая композиция может содержать соединение (I) и/или соединение (II) в дополнение к фосфатной соли. Под "фосфатной солью" подразумевают, что можно применять любую известную фосфатную соль. Фосфатные соли относятся к множеству различных комбинаций химического фосфата с солями и минералами. В одном варианте осуществления в фармацевтической композиции по настоящему изобретению по меньшей мере одна фосфатная соль представляет собой фосфат натрия, фосфат калия или фосфат аммония. Фосфатная соль может быть выбрана из группы, включающей Na2HPO4 (безводный), Na2HPO4 • 2 H2O, Na2HPO4 • 7 H2O, Na2HPO4 • 12 H2O, NaH2PO4 (безводный), NaH2PO4 • H2O, NaH2PO4 • 2 H2O, K2HPO4 (безводный), K2HPO4 • 3 H2O, KH2PO4 (безводный) и их смеси.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению фосфатная соль представляет собой Na2HPO4 • 2 H2O, и количество Na2HPO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,04 до 0,4, предпочтительно от 0,05 до 0,35, от 0,075 до 0,25, от 0,09 до 0,2. В одном варианте осуществления молярное отношение Na2HPO4 • 2 H2O к соединению (I) или соединению (II) составляет приблизительно 0,144.
В дополнительных вариантах осуществления фармацевтической композиции по настоящему изобретению фосфат натрия представляет собой NaH2PO4 • 2 H2O, и количество NaH2PO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение NaH2PO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,01 до 0,09, предпочтительно от 0,015 до 0,07, от 0,02 до 0,05, от 0,025 до 0,04, от 0,035 до 0,05. В одном варианте осуществления молярное отношение NaH2PO4 • 2 H2O к соединению (I) или соединению (II) составляет приблизительно 0,038.
В других вариантах осуществления фармацевтическая композиция по настоящему изобретению содержит две различные фосфатные соли натрия. Необязательно, две различные фосфатные соли натрия представляют собой NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количества NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O, присутствующих в композиции, выбраны таким образом, что молярное отношение как NaH2PO4 • 2 H2O, так и Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,03 до 0,45, от 0,04 до 0,3, от 0,05 до 0,25, от 0,06 до 0,2, от 0,07 до 0,15. В одном варианте осуществления молярное отношение как NaH2PO4 • 2 H2O, так и Na2HPO4 • 2 H2O к соединению (I) или соединению (II) составляет приблизительно 0,18.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количества NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O, присутствующих в композиции, выбраны таким образом, что молярное отношение каждого из NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,025 до 0,4, от 0,025 до 0,3, от 0,025 до 0,2, от 0,03 до 0,1. В некоторых вариантах осуществления количество NaH2PO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение NaH2PO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,01 до 0,09, а количество Na2HPO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,04 до 0,4. В данном случае также применяют другие количества применяемых отдельно фосфатных солей Na2HPO4 • 2 H2O и NaH2PO4 • 2 H2O, как описано выше. В некоторых вариантах осуществления твердая фармацевтическая композиция содержит по меньше мере 1, 2, 3, 4, 5, 6 или более фосфатных солей. В одном варианте осуществления твердая фармацевтическая композиция по настоящему изобретению содержит 2 (различные) фосфатные соли.
Используемый в данном документе термин "приблизительно" понимают как то, что может существовать изменение относительного значения или диапазона (например pH, концентрации, процентного содержания, молярности, времени и т. п.), которое может составлять до 5%, до 10%, до 15% или до 20% и включая данное значение от приведенного значения. Например, если состав содержит приблизительно 5 мг/мл соединения, это понимают как то, что состав может иметь от 4 до 6 мг/мл, предпочтительно от 4,25 до 5,75 мг/мл, более предпочтительно от 4,5 до 5,5 мг/мл и еще более предпочтительно от 4,75 до 5,25 мг/мл, при этом наиболее предпочтительное значение составляет 5 мг/мл. То же самое применяется также по отношению к молярности. Например, молярность, равную приблизительно 1, понимают как молярность, равную от 0,8 до 1,2, предпочтительно от 0,85 до 1,15, более предпочтительно от 0,9 до 1,1, еще более предпочтительно от 0,95 до 1,05, при этом наиболее предпочтительное значение составляет 1.
В качестве альтернативы, настоящее изобретение относится к твердым композициям, которые содержат соединение (I) и/или соединение (II) и неорганическую соль, отличную от фосфатной соли. В данных композициях неорганическая соль может быть единственным компонентом помимо соединения (I) или (II) (в данном случае, фосфатная соль не присутствует). В качестве альтернативы, в композиции по настоящему изобретению неорганическая соль может присутствовать в комбинации по меньшей мере с одним фосфатом. Термин "неорганическая соль" при упоминании в данном документе означает каждую подходящую неорганическую соль. Необязательно, неорганическая соль выбрана из MgCl2, CaCl2, NH4Cl, KCl или NaCl. В некоторых предпочтительных вариантах осуществления неорганическая соль представляет собой NaCl. В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению количество NaCl, присутствующего в композиции по настоящему изобретению (отдельно или вместе по меньшей мере с одним фосфатом), выбрано таким образом, что молярное отношение NaCl к соединению (I) или соединению (II) находится в диапазоне от 1,5 до 4, предпочтительно от 1,8 до 3,7, от 1,85 до 3,6, от 1,9 до 3,4, наиболее предпочтительно от 1,9 до 2,5. В одном из таких вариантов осуществления молярное отношение NaCl к соединению (I) или соединению (II) составляет приблизительно 2,2.
В композициях по настоящему изобретению соединение (I) и/или соединение (II) могут присутствовать в виде диастереомерных смесей или смесей из 1, 2, 3, 4, 5, 6, 7 или 8 стереоизомеров соединения (I) и 1, 2, 3, 4, 5 или 6 стереоизомеров соединения (II), предпочтительно, из одного или двух стереоизомеров.
Соединение формулы (I), таким образом, может включать в себя диастереомерные смеси соединения формулы (I) или смеси из одного или более стереоизомеров соединения формулы (I). Соединение (I) может представлять собой (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерин. В дополнительных вариантах осуществления соединение (I) представляет собой (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерин. В других вариантах осуществления соединение (I) представляет собой диастереомерную смесь, которая содержит больше (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина, чем (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерина. В некоторых вариантах осуществления количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина и (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерина выбраны таким образом, что отношение количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина к (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерину находится в диапазоне от 0,5 до 2, предпочтительно от 0,5 до 1,9, от 0,7 до 1,8, от 0,8 до 1,7, от 0,9 до 1,6, от 1 до 1,5, наиболее предпочтительно от 1,1 до 1,4. В одном варианте осуществления количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина и (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерина выбраны таким образом, что отношение количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина к (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерину составляет около 1,3. В некоторых вариантах осуществления фармацевтическая композиция по настоящему изобретению содержит только соединение (I).
Соединение (I) может также представлять собой соединение, имеющее формулу (Ia):
(Ia).
Подобным образом, соединение формулы (II) при упоминании в данном документе может включать в себя стереоизомерные смеси соединения формулы (II) или смеси из одного или более стереоизомеров соединения формулы (II). В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению соединение (II) представляет собой 4-амино-7,8-дигидро-L-биоптерин. В другом варианте осуществления соединение (II) представляет собой соединение, имеющее формулу (IIa):
(IIa).
В некоторых вариантах осуществления фармацевтическая композиция по настоящему изобретению содержит только соединение (II).
В твердых фармацевтических композициях соединение (I) и/или соединение (II) также могут присутствовать в виде свободного основания. "Свободное основание" при упоминании в данном документе относится к чистой основной форме амина, в отличие от его формы соли. Кроме того, данный термин применяют для описания депротонированной аминной формы соединения. Обычный противоион представляет собой ион из неорганической кислоты, такой как отрицательно заряженный хлорид. Например, можно сравнить свободное основание амина (NH2) с гидрохлоридом амина (NH3+ Cl-) при добавлении HCl.
Как упоминалось выше, производные биоптерина чувствительны к окислению в жидкостях. Следовательно, твердая фармацевтическая композиция также может быть представлена в виде лиофилизированной фармацевтической композиции. Термин "лиофилизированный", применяемый в данном документе, означает сушку вымораживанием, которая представляет собой процесс дегидратации. Принцип сушки вымораживанием заключается в замораживании материала, а затем снижении окружающего давления для обеспечения сублимации замороженной воды в материале непосредственно из твердой фазы в газовую фазу. В некоторых вариантах осуществления конечное содержание остаточной воды в лиофилизированном продукте составляет от 0% до 15%, предпочтительно от 0% до 12%, (вес./вес.). Способы осуществления лиофилизации известны специалисту в данной области.
Как очевидно специалисту в данной области, фармацевтические композиции могут дополнительно содержать 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более дополнительных фармацевтических вспомогательных веществ. "Фармацевтическое вспомогательное вещество" или добавки представляют собой соединения, добавленные к соединениям формулы (I) и/или (II). Данные добавки могут выполнять конкретную функцию. Их можно добавлять для увеличения объема, облегчения изготовления, улучшения стабильности, улучшения доставки и нацеливания лекарственного средства и модифицирования безопасности или фармакокинетического профиля лекарственного средства. Ингредиенты, которые можно применять в ходе изготовления лекарственного продукта, но которые могут не присутствовать в твердой композиции по настоящему изобретению, также считаются вспомогательными веществами (примеры включают воду для лиофилизированных продуктов и инертные газы в свободном пространстве контейнеров). Подходящие фармацевтические вспомогательные вещества включают крахмал, глюкозу, лактозу, сахарозу, трегалозу, маннит, сорбит, глицин, гистидин, рафинозу, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, глицеролмоностеарат, тальк, ион натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, декстрозу, декстран, воду, этанол и т. п. Композиции, при необходимости, также могут содержать незначительные количества смачивающих или эмульгирующих средств или pH-буферных средств. Данные композиции могут принимать форму растворов, суспензий, эмульсии и т. п.
Например, если твердая композиция по настоящему изобретению содержит соединение формулы (I) и/или формулы (II) и фосфатную соль, возможным дополнительным фармацевтическим вспомогательным веществом может быть неорганическая соль. Неорганическая соль может представлять собой любую неорганическую соль, описанную выше. С другой стороны, если фармацевтическая композиция по настоящему изобретению содержит соединение формулы (I) и/или формулы (II) и неорганическую соль, дополнительным фармацевтическим вспомогательным веществом может быть фосфатная соль. Фосфатная соль может представлять собой любую фосфатную соль, описанную выше.
Помимо фармацевтических вспомогательных веществ, которые могут присутствовать в твердых фармацевтических композициях по настоящему изобретению, в некоторых вариантах осуществления фармацевтическая композиция по настоящему изобретению может содержать кристаллизационную воду, но также, естественно, могут присутствовать дополнительные фармацевтические вспомогательные вещества. Как используется в данном документе в соответствии с его обычным значением в данной области, "кристаллизационная вода", или "гидратационная вода", или "вода кристаллизации" означает воду, которая образуется внутри кристаллов – в настоящем изобретении, как неорганические соли, такие как Na2HPO4 или NaH2PO4, так и производное биоптерина соединения (I) или соединения (II) могут содержать кристаллизационную воду. Несмотря на то, что в твердых фосфатных солях, таких как Na2HPO4 ∙ 2 H2O или NaH2PO4 ∙ 2 H2O, кристаллизационная вода присутствует в определенных стехиометрических количествах, количество кристаллизационной воды, присутствующей в твердой форме соединения (I) или соединения (II), может варьировать в зависимости от условий синтеза и/или кристаллизации соединения, которое применяют для получения твердого состава по настоящему изобретению. Без ограничения какой-либо теорией, считают, что кристаллизационная вода, которая присутствует в твердой форме соединения (I) или соединения (II) может быть связана, например, посредством водородных связей, с двумя аминогруппами соединения (I) или (II), которые присутствуют в виде свободного основания. В целях иллюстрации, в данном контексте является предпочтительным, чтобы единичная доза композиции содержала 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 140 ± 30 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 16,5 ± 2 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг хлорида натрия (NaCl). В данной единичной дозе две фосфатные соли натрия присутствуют в виде дигидратов, тогда как кристаллизационная вода, которая присутствует в количестве 140 ± 30 мг кристаллизационной воды, относится только к воде, которая присутствует совместно со свободным основанием соединения (I) и/или соединения (II). Другая иллюстративная единичная доза композиции содержит 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 60 ± 50 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 12 ± 2,5 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг хлорида натрия (NaCl).
Как только что было описано, фармацевтическая композиция по настоящему изобретению может быть представлена в единичной дозе. "Единичная доза" твердых фармацевтических композиций по настоящему изобретению означает, что ингредиенты смешивают вместе в единичной дозе, обычно включающей смесь активных лекарственных компонентов, таких как соединение (I) и/или соединение (II), и нелекарственных компонентов (вспомогательных веществ), таких как по меньшей мере одна фосфатная соль и/или по меньшей мере одна неорганическая соль. Кроме того, единичная доза может дополнительно содержать вспомогательные вещества, такие как по меньшей мере одна фосфатная соль или по меньшей мере одна неорганическая соль, и/или совместно другой не подлежащий повторному использованию материал, который не может рассматриваться ни как ингредиент, ни как упаковка (как, например, оболочка капсулы). Термин единичная доза также может охватывать не подлежащую повторному использованию упаковку в той же мере (в особенности, если каждый лекарственный продукт упакован отдельно). Единичная доза также может содержать восстановленные твердые фармацевтические композиции по настоящему изобретению.
Для иллюстративной единичной дозы далее кратко описан расчет молярных отношений ингредиентов в данной единичной дозе.
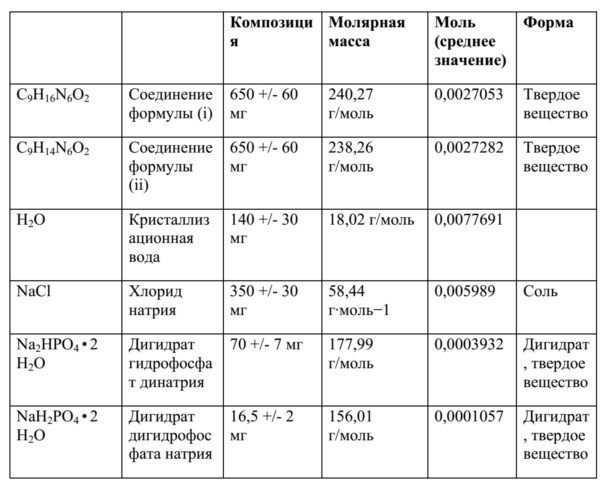
Молярное отношение nNaCl/nсоединение (i) = 0,005989 моль/0,0027053 моль ~ 2,214.
Молярное отношение nNaCl/nсоединение (ii) = 0,005989 моль/0,0027282 моль ~ 2,195.
Молярное отношение nNa2HPO4 • 2 H2O + nNaH2PO4 • 2 H2O/nсоединение (i) = 0,0003932 моль + 0,0001057 моль/0,0027053 моль ~ 0,18.
Молярное отношение nNa2HPO4 • 2 H2O + nNaH2PO4 • 2 H2O/nсоединение (ii) = 0,0003932 моль + 0,0001057 моль/0,0027282 моль ~ 0,18.
Молярное отношение nNa2HPO4 • 2 H2O/nсоединение (i) = 0,0003932 моль/0,0027053 моль ~ 0,145.
Молярное отношение nNa2HPO4 • 2 H2O/nсоединение (ii) = 0,0003932 моль/0,0027282 моль ~ 0,144.
Молярное отношение nNaH2PO4 • 2 H2O/nсоединение (i) = 0,0001057 моль/0,0027053 моль ~ 0,038.
Молярное отношение nNaH2PO4 • 2 H2O/nсоединение (ii) = 0,0001057 моль/0,0027282 моль ~ 0,038.
nсоединение (i) = mсоединение (i)/Mсоединение (i) = 0,710 г/240,27 г∙моль-1 = 0,002955 моль.
nсоединение (i) = mсоединение (i)/Mсоединение (i) = 0,590 г/240,27 г∙моль-1 = 0,0024556 моль.
nсоединение (i) = mсоединение (i)/Mсоединение (i) = 0,650 г/240,27 г∙моль-1 = 0,0027053 моль.
nсоединение (ii) = mсоединение (ii)/Mсоединение (ii) = 0,710 г/238,25 г∙моль-1 = 0,00298 моль.
nсоединение (ii) = mсоединение (ii)/Mсоединение (ii) = 0,590 г/238,25 г∙моль-1 = 0,0024763 моль.
nсоединение (ii) = mсоединение (ii)/Mсоединение (ii) = 0,650 г/238,25 г∙моль-1 =0,0027282 моль.
nNaCl = mNaCl/MNaCl = 0,380 г/58,44 г∙моль-1 = 0,0065023 моль.
nNaCl = mNaCl/MNaCl = 0,320 г/58,44 г∙моль-1 = 0,0054757 моль.
nNaCl = mNaCl/MNaCl = 0,350 г/58,44 г∙моль-1 = 0,005989 моль.
nNa2HPO4 • 2 H2O = mNa2HPO4 • 2 H2O/MNa2HPO4 • 2 H2O = 0,077 г/177,99 г∙моль-1 = 0,0004326 моль.
nNa2HPO4 • 2 H2O = mNa2HPO4 • 2 H2O/MNa2HPO4 • 2 H2O = 0,063 г/177,99 г∙моль-1 = 0,0003539 моль.
nNa2HPO4 • 2 H2O = mNa2HPO4 • 2 H2O/MNa2HPO4 • 2 H2O = 0,07 г/177,99 г∙моль-1 = 0,0003932 моль.
nNaH2PO4 • 2 H2O = mNaH2PO4 • 2 H2O/MNaH2PO4 • 2 H2O = 0,0185 г/156,01 г∙моль-1 = 0,0001185 моль.
nNaH2PO4 • 2 H2O = mNaH2PO4 • 2 H2O/MNaH2PO4 • 2 H2O = 0,0145 г/156,01 г моль-1 = 0,0000929 моль.
nNaH2PO4 • 2 H2O = mNaH2PO4 • 2 H2O/MNaH2PO4 • 2 H2O = 0,0165 г/156,01 г моль-1 = 0,0001057 моль.
nH2O = mH2O/MH2O = 0,17 г/18,02 г моль-1 = 0,0094339 моль.
nH2O = mH2O/MH2O = 0,11 г/18,02 г моль-1 = 0,0061043 моль.
nH2O = mH2O/MH2O = 0,14 г/18,02 г моль-1 = 0,0077691 моль.
В итоге, единичная доза, описанная в данном документе, может содержать 650 ± 60 мг свободного основания 4-амино-7,8-дигидро-L-биоптерина и/или 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 140 ± 30 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 16,5 ± 2 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг NaCl. Также возможно, чтобы единичная доза композиции содержала 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 60 ± 50 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 12 ± 2,5 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг хлорида натрия (NaCl). В типичных вариантах осуществления данная единичная доза относится и упакована для введения в 50 мл флакон, поскольку после восстановления в объеме 50 мл фармацевтически приемлемого носителя, такого как вода ad inject, получают инфузионный или инъекционный раствор, который может быть введен без замедления. Таким образом, например, если применяют отличную от этой единичную дозу, например, в 30 мл флаконе, количества всех компонентов композиции регулируют соответственно. В примере с 30 мл флаконом количество каждого компонента будет уменьшено до 3/5 (60%). Это означает, что если единичная доза в 50 мл флаконе содержит 650 ± 60 мг свободного основания 4-амино-7,8-дигидро-L-биоптерина и/или 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 140 ± 30 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 16,5 ± 2 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг NaCl, то 30 мл единичная доза будет содержать 390 ± 36 мг свободного основания 4-амино-7,8-дигидро-L-биоптерина и/или 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 84 ± 18 мг кристаллизационной воды, 42 ± 4,2 мг Na2HPO4 ∙ 2 H2O, 9,9 ± 1,2 мг NaH2PO4 ∙ 2 H2O и 210 ± 18 мг NaCl. Соответственно, если единичная доза в 50 мл флаконе содержит 650 ± 60 мг свободного основания 4-амино-7,8-дигидро-L-биоптерина и/или 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 60 ± 50 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 12 ± 2,5 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг NaCl, то 30 мл единичная доза будет содержать 390 ± 36 мг свободного основания 4-амино-7,8-дигидро-L-биоптерина и/или 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 36 ± 18 мг кристаллизационной воды, 42 ± 4,2 мг Na2HPO4 ∙ 2 H2O, 7,2 ± 1,2 мг NaH2PO4 ∙ 2 H2O и 210 ± 18 мг NaCl.
Фармацевтическая композиция по настоящему изобретению также может содержать дополнительные молекулы, например, сопутствующие примеси, образованные в ходе способа получения фармацевтической композиции по настоящему изобретению. Таким образом, фармацевтическая композиция по настоящему изобретению может содержать 1, 2, 3, 4, 5, 6, 7 или более дополнительных соединений. Данные дополнительные соединения могут быть выбраны из одного или более соединений, в том числе без ограничения из группы, включающей 4-амино-7,8-дигидро-L-биоптерин (который может быть образован при самопроизвольном окислении 4-амино-5,6,7,8-тетрагидро-L-биоптерина, см. выше), 4-амино-L-биоптерин, (6R,S)-5,6,7,8-тетрагидро-L-биоптерин, 1-[(6R,S)-2,4-диамино-5,6,7,8-тетрагидроптеридин-6-ил]пропанол, 1-[(6R,S)-2,4-диамино-5,6,7,8-тетрагидроптеридин-6-ил]пропан, (1R,2S)-1-[(6R,S)-2-(ацетиламино)-4-амино-5,6,7,8-тетрагидроптерин-6-ил]-1,2-диацетоксипропан, 2,4-диамино-7,8-дигидроптеридин или 2,4-диаминоптеридин, для обозначения некоторых иллюстративных соединений.
Как изложено выше, было показано, что производные птеридина проявляют свойства, отличные от свойств других NO-ингибиторов, что делает данное соединение потенциально более подходящим, чем "классические" аналоги аргинина. Таким образом, настоящее изобретение также относится к применению лиофилизированной фармацевтической композиции по настоящее изобретение для лечения заболевания. В некоторых вариантах осуществления заболевание выбрано из группы, включающей травматическое повреждение головного мозга, нетравматическое повреждение головного мозга, предпочтительно инсульт или менингит, повышенное внутричерепное давление, вторичное повреждение головного мозга.
Термин "травматическое повреждение головного мозга" или "травма головного мозга" возникает, когда внешняя сила травматически повреждает головной мозг. TBI можно классифицировать на основании тяжести, механизма (закрытая или проникающая травма черепа) или других признаков (например, возникновения в определенном месте или в обширной области). Травматическое повреждение головного мозга может возникать вследствие фокусного удара головой, при внезапной акселерации/децелерации в черепе или при сложной комбинации как движения, так и внезапного удара, а также ударных волн или проникновения поражающего элемента. С помощью коматозной шкалы Глазго (GCS), наиболее широко применяемой системы классификации тяжести TBI, оценивают уровень сознания человека по шкале от 3 до15 на основании вербальных, двигательных реакций и реакций открывания глаз на раздражители. В целом, установлено, что TBI со значением по GCS 13 или выше является легким, 9–12 является умеренным и 8 или ниже является тяжелым. Подобные системы существуют для детей младшего возраста. С точки зрения диагностики, есть дополнительные различия между открытыми и закрытыми TBI. Открытое TBI считается повреждением, при котором оболочки головного мозга (твердая оболочка головного мозга) механически разрушены и головной мозг контактирует с окружающей средой через это отверстие. Зачастую открытое TBI связано с выходом ликвора и остатков тканей головного мозга. При закрытом TBI кости черепа или череп остаются неповрежденными, а первичное поражение головного мозга (травма) характеризуется местными очагами поражения, такими как ушиб или гематомы, и/или диффузным поражением тканей головного мозга. Термин "череп" при упоминании в данном документе представляет собой группу, состоящую из мозгового черепа (черепной коробки) и висцерального черепа (черепно-лицевого отдела), существующего костного и хрящевого скелета головы позвоночных животных. "Внутричерепной" означает внутри черепа.
В отличие от этого, "нетравматическое повреждение головного мозга" не включает внешнюю механическую силу для обеспечения повреждения головного мозга. Причины нетравматического повреждения головного мозга могут включать недостаток кислорода, глюкозы или крови. Инфекции могут вызвать энцефалит (набухание головного мозга), менингит (менингеальное набухание) или клеточную токсичность, как, например, обусловленная скоротечной формой заболевания печеночная недостаточность, в случае опухолей или отравляющих веществ. Данные инфекции могут возникать вследствие инсульта, сердечного приступа, обратимого утопления, удушения или диабетической комы, отравления или других связанных с химическими веществами причин, таких как злоупотребление алкоголем или передозировка лекарственными средствами, инфекций или опухолей, а также дегенеративных состояний, таких как болезнь Альцгеймера и болезнь Паркинсона. Острое нейродегенеративное заболевание представлено "инсультом", который относится к потере функции головного мозга вследствие нарушений кровоснабжения головного мозга, в особенности, если он возникает быстро, и часто связан с нарушением мозгового кровообращения. Он может возникнуть вследствие ишемии (отсутствия кровотока), вызванной закупориванием (тромбозом, артериальной эмболией), или кровоизлияния в центральной нервной системе (CNS) или внутричерепных кровеносных сосудах. В результате, пораженная область головного мозга не может функционировать нормально.
Другим заболеванием, которое может подвергаться лечению, является "менингит", который представляет собой острое воспаление оболочек, покрывающих головной мозг и спинной мозг, в общем известных как мягкие оболочки мозга. Воспаление может быть вызвано инфицированием вирусами, бактериями или другими микроорганизмами, и, реже, определенными лекарственными средствами.
Помимо поражения, вызванного в момент повреждения, травма головного мозга (нетравматическое или травматическое повреждение головного мозга) приводит ко "вторичному повреждению" или "вторичному повреждению головного мозга", которое относится к различным явлениям, которые имеют место в течение минут и дней после повреждения. Данные процессы, которые включают изменения мозгового кровотока и давления внутри черепа, способствуют в значительной степени поражению от исходного повреждения. Явления вторичного повреждения могут включать, например, нарушение гематоэнцефалического барьера, высвобождение факторов, которые вызывают воспаление, перенасыщение свободными радикалами, избыточное высвобождение нейромедиатора глутамата (эксайтотоксичность), поступление ионов кальция и натрия в нейроны и дисфункцию митохондрий. Поврежденные аксоны в белом веществе головного мозга могут отделяться от их клеточных тел в результате вторичного повреждения, потенциально уничтожая данные нейроны. Другие факторы при вторичном повреждении представляют собой изменения кровотока в головном мозге; повторяющееся переходное разрушение гематоэнцефалического барьера; ишемию (недостаточный кровоток); гипоксию головного мозга (недостаток кислорода в головном мозге); отек головного мозга (набухание головного мозга) и повышенное внутричерепное давление (давление внутри черепа).
Также возможно, что внутричерепное давление может повышаться вследствие набухания или масс-эффекта от очага, такого как кровоизлияние. В результате церебральное перфузионное давление (давление кровотока в головном мозге) снижается − возникает ишемия. Если давление в черепе поднимается слишком высоко, это может вызвать смерть головного мозга или образование грыжи, в которой части головного мозга сдавливаются структурами в черепе. Термин "внутричерепное давление" (ICP) означает давление внутри черепа и, следовательно, в ткани головного мозга и спинномозговой жидкости (CSF). В организме есть различные механизмы, посредством которых он поддерживает ICP стабильным, при этом значения давления CSF изменяются приблизительно на 1 мм рт. ст. у нормальных взрослых людей посредством сдвигов образования и поглощения CSF. ICP измеряют в миллиметрах ртутного столба (мм рт. ст.) и в состоянии покоя оно составляет в норме 7–15 мм рт. ст. для взрослого, лежащего на спине. Изменения ICP объясняются изменениями объема в одном или более отделений, находящихся в черепе. "Повышенное давление в черепе" или "повышенное внутричерепное давление" означает повышенное давление в черепе субъекта по сравнению с нормальным здоровым субъектом. Поскольку внутричерепное давление в норме составляет от 7 до 15 мм рт. ст.; следовательно, давление в 20–25 мм рт. ст., верхняя граница нормы, уже считается повышенным внутричерепным давлением и может потребоваться лечение для снижения данного давления. Таким образом, повышенным внутричерепным давлением может считаться любое давление выше 20 мм рт. ст. в черепе лежащего на спине субъекта, предпочтительно давление выше 25 мм рт. ст., выше 26 мм рт. ст., выше 27 мм рт. ст., выше 28 мм рт. ст., выше 29 мм рт. ст., выше 30 мм рт. ст., выше 31 мм рт. ст., выше 32 мм рт. ст., выше 33 мм рт. ст., выше 34 мм рт. ст. или выше 35 мм рт. ст.
В этом направлении, настоящее изобретение дополнительно относится к применению лиофилизированной фармацевтической композиции по настоящему изобретению в изготовлении лекарственного препарата для лечения субъекта с закрытой травмой черепа, повышенным внутричерепным давлением и вторичным повреждением головного мозга. "Закрытая травма черепа" при упоминании в данном документе означает тип травматического повреждения головного мозга, при котором череп и твердая оболочка головного мозга остаются неповрежденными (также см. выше).
Кроме того, настоящее изобретение относится к способу лечения заболевания у субъекта, включающему стадию введения лиофилизированной фармацевтической композиции по настоящему изобретению субъекту, нуждающемуся в этом.
Относительно введения лечащий врач и клинические факторы будут определять режим дозирования. Как хорошо известно в области медицины, дозы для любого пациента зависят от множества факторов, в том числе веса пациента, площади поверхности тела, возраста, конкретного вводимого соединения, пола, времени и способа введения, общего состояния здоровья и других лекарственных средств, вводимых одновременно. Типичная суточная доза может находиться, например, в диапазоне от 2,5 мг/кг до 12,5 мг/кг веса тела; однако дозы ниже или выше данного иллюстративного диапазона также предусмотрены, в особенности, с учетом вышеупомянутых факторов.
Дозы предпочтительно вводят один раз в сутки в течение максимально 4 дней, однако в ходе лечения дозы можно вводить в течение более длительных промежутков времени и при необходимости их можно вводить в течение намного более коротких промежутков времени, например, ежедневно. В предпочтительном случае иммунный ответ наблюдают с использованием способов, описанных в данном документе, и дополнительных способов, известных специалисту в данной области, и дозы оптимизируют, например, по времени, количеству и/или композиции. Дозы будут варьировать, но предпочтительная доза для внутривенного введения соединения (I) и/или соединения (II) составляет от примерно 2,5 мг/кг до 12,5 мг/кг веса тела в сутки.
Если режим представляет собой инъекцию или непрерывную кратковременную инфузию, она также должна находиться в диапазоне от приблизительно 1 μг до приблизительно 1 мг единиц на килограмм веса тела в минуту соответственно. Прогресс можно наблюдать посредством периодической оценки.
Анализы in vitro можно необязательно использовать для облегчения определения оптимальных диапазонов доз. Точная доза, используемая в составе, также будет зависеть от способа введения и серьезности заболевания, и она должна быть определена в соответствии с решением практикующего врача и состояний каждого пациента. Эффективные дозы могут быть экстраполированы из кривых зависимости ответа от дозы, полученных из тестовых систем in vitro или животных моделей. Предпочтительно, фармацевтическую композицию вводят непосредственно или в комбинации со вспомогательным средством.
Максимальная суточная доза (DDD) основана на количестве соединения (I) и/или (II), которое обычно применяют в качестве основного показания для субъектов (например, взрослых людей) в сутки. Однако режим дозирования может отличатся между инъекциями и инфузиями или даже несколькими инъекциями и/или инфузиями в сутки. В некоторых вариантах осуществления в способе лечения заболевания у субъекта максимальная суточная доза составляет 12,5 мг/кг веса тела, предпочтительно 12,5, 11,5, 10,5, 9,5, 8,5, 7,5, 6,5, 5,5, 4,5, 3,5, 2,5 мг/кг веса тела. В некоторых вариантах осуществления максимальная суточная доза составляет 10,0, 9,0 или предпочтительно 8,5 мг/кг веса тела, или даже менее, например, 7,5, 5,0, 2,5 мг/кг веса тела.
Как применяется в данном документе, интервал, который определяют как "(от) X до Y", равен интервалу, который определяют как "X-Y". Оба интервала конкретно включают верхнюю границу, а также нижнюю границу. Это означает, что, например, интервал "от 2,5 мг/кг до 12,5 мг/кг" или "2,5-12,5 мг/кг" включает концентрацию 2,5, 3,5, 4,5, 5,5, 6,5, 7,5, 8,5, 9,5, 10,5, 11,5 и 12,5, а также любое приведенное промежуточное значение.
Также предполагается, что фармацевтические композиции используют в подходах совместной терапии, т. е. в совместном введении с другими лекарственными препаратами или лекарственными средствами, например, другими лекарственными средствами для предупреждения, лечения или ослабления заболеваний, таких как травматическое или нетравматическое повреждение головного мозга.
"Субъектами", подлежащими лечению согласно настоящему изобретению, предпочтительно являются млекопитающие, такие как люди, обезьяны, кошки, собаки, лошади, свиньи, крупный рогатый скот, мыши или крысы, при этом люди являются предпочтительными.
Настоящее изобретение также предусматривает способ получения лиофилизированной твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну фосфатную соль, и необязательно NaCl;
при этом способ включает
aa) растворение соединения формулы (III) и/или (II):
(III) или
(II)
в буфере, при этом буфер предпочтительно содержит фосфат; и
bb) лиофилизацию раствора, полученного на стадии aa).
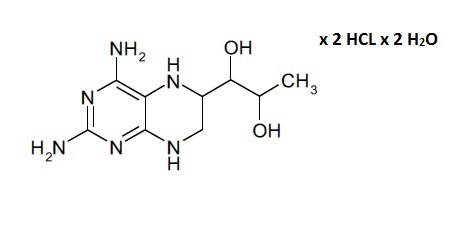
В данном контексте отмечено, что несмотря на то, что соединение формулы (II) является легкодоступным как таковое в результате синтеза, типичными продуктом синтеза соединения формулы (I) является его дигидрат дигидрохлорида, показанный в формуле (III). По этой причине в способе получения твердой композиции по настоящему изобретению, которая содержит соединение формулы (I), 4-амино-5,6,7,8-тетрагидро-L-биоптерин, исходным материалом обычно является дигидрат дигидрохлорида 4-амино-5,6,7,8-тетрагидро-L-биоптерина − соединение формулы (III).
Кроме того, настоящее изобретение дополнительно относится к способу получения лиофилизированной твердой фармацевтической композиции (приспособленной для внутривенного введения), содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну неорганическую кислоту, предпочтительно NaCl;
при этом способ включает
aa) растворение соединения формулы (III) и/или формулы (II):
(III) или
(II)
в буфере;
bb) лиофилизацию раствора, полученного на стадии aa).
Буфер, описанный в стадии aa), применяют для растворения соединения формулы (III) и/или формулы (II) с получением раствора соединение-буфер. Буфер, как правило, представляет собой раствор, который содержит слабую кислоту (или основание) и соль слабой кислоты (или этого основания). Более того, он противостоит изменениям pH при добавлении в него небольших количеств кислоты или щелочи. Различные фосфаты могут образовывать несколько различных комбинаций буферов. Три возможных варианта фосфата натрия могут включать NaH2PO4 - дигидрофосфат натрия, Na2HPO4 - гидрофосфат динатрия, Na3PO4 - фосфат натрия.
Буфер из стадии aa) также может представлять собой буфер на основе гидрофосфата натрия, содержащий по меньшей мере одну фосфатную соль. "Буфер на основе гидрофосфата натрия" представляет собой буфер, содержащий фосфатную соль натрия. В некоторых вариантах осуществления буфер на основе фосфата натрия содержит по меньшей мере одну фосфатную соль натрия, описанную выше. В других вариантах осуществления буфер на основе фосфата натрия содержит 2, 3, 4, 5 или более фосфатных солей натрия, предпочтительно две различные фосфатные соли, описанные выше. В другом варианте осуществления способа по настоящему изобретению буфер на основе гидрофосфата натрия получают путем раздельного растворения NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O.
Термин "раздельное растворение" означает, что растворение каждого фосфата натрия осуществляют на расстоянии друг от друга, например, в разной стеклянной посуде. В дополнительном варианте осуществления способа по настоящему изобретению буфер на основе гидрофосфата натрия характеризуется значением pH 7,4 в результате добавления раствора NaH2PO4 • 2 H2O к растворенному Na2HPO4 • 2 H2O.
Кроме того, буфер из стадии aa) может содержать NaOH, буфер на основе гидрофосфата натрия и воду. Необязательно, NaOH представляет собой 5 н. раствор NaOH. "5 н. раствор NaOH" означает раствор NaOH (гидроксида натрия), который характеризуется нормальностью 5 (5 н.). Если концентрация раствора гидроксида натрия составляет c (NaOH) = 5 моль/л, то его нормальность составляет 5 н.
В некоторых вариантах осуществления способа по настоящему изобретению буфер содержит 12-16% (вес./вес.) 5 н. NaOH, 8-12% (вес./вес.) буфера на основе гидрофосфата натрия и 74-78% (вес./вес.) воды для инъекций.
В дополнительных вариантах осуществления способа по настоящему изобретению буфер характеризуется значением pH приблизительно 8, 9, 10, 11, 12, 13 или 14.
В еще одном варианте осуществления способа по настоящему изобретению раствор из стадии aa) характеризуется значением pH приблизительно 4, 5, 6, 7, 8, 9, 10 или 11, предпочтительно 6,5-7,6, наиболее предпочтительно приблизительно 7,4.
Буфер можно получить различными способами. Например, его можно получать в ходе дегазации с получением дегазированного буфера. "Дегазированный буфер" относится к удалению растворенных газов, например, кислорода, из жидкостей, в особенности, воды или водных растворов. Барботирование раствора инертными газами замещает растворенные вредные реакционноспособные газы, такие как кислород и диоксид азота. Широко применяют азот, аргон, гелий и другие инертные газы. Для завершения замещения раствор необходимо энергично перемешать и барботировать в течение длительного времени. В некоторых вариантах осуществления буфер дегазируют азотом до достижения кислорода ˂ 1,0 ppm (частей на миллион).
В качестве следующей дополнительной стадии способа раствор, полученный на стадии aa), можно стерилизовать фильтрацией, предпочтительно с применением 0,22 мкм фильтра. Стерилизация фильтрацией охватывает любой способ, при котором устраняют (удаляют) или уничтожают все формы микробной жизни, в том числе передающиеся микроорганизмы (такие как грибы, бактерии, вирусы, споровые формы и т. д.), присутствующие на поверхности, содержащиеся в текучей среде, в медикаменте или в композиции (твердой или жидкой). В данном случае, стерилизации достигают путем фильтрации. Стерилизации также можно достигнуть путем применения нагрева, химических средств, излучения, высокого давления или их комбинаций со стадией фильтрации. Фильтр может иметь различные размеры пор. Как правило, фильтр с размером пор 0,2 мкм (микрофильтрация) будет эффективно удалять микроорганизмы. При обработке высокомолекулярных лекарственных средств должны быть удалены или инактивированы вирусы, например, посредством применения нагрева или глутаральдегида или т. п. Применяют нанофильтры с еще меньшим размером пор 20-50 нм (нанофильтрация). Чем меньше размер пор, тем ниже скорость потока. Для достижения более высокой общей пропускной способности или во избежание преждевременного блокирования, можно применять предварительные фильтры для защиты мембранных фильтров с небольшим размером пор.
В другом варианте осуществления способа по настоящему изобретению после получения фармацевтической композиции лиофилизацию начинают не позже, чем через 2 часа, предпочтительно не позже, чем через 1,5 часа, не позже, чем через 1 час, не позже, чем через 30 минут, наиболее предпочтительно не позже, чем через 15 минут.
Следует отметить, что настоящее изобретение также относится к фармацевтической композиции, получаемой посредством способа по настоящему изобретению.
Для лучшей обработки твердой фармацевтической композиции по настоящему изобретению лиофилизатом, полученным на стадии bb), или фармацевтической композицией по настоящему изобретению заполняют флаконы предпочтительно в количестве приблизительно 1-1,5 г, предпочтительно приблизительно 1,25 г твердого состава. В качестве альтернативы, лиофилизатом, полученным на стадии bb), или фармацевтической композицией можно заполнять флаконы в количестве приблизительно 0,9-1,4 г, предпочтительно 1,15 г твердого состава. При использовании в качестве "единичной дозы" количество приблизительно 1-1,5 г или предпочтительно приблизительно 1,25 г твердого состава, или, в качестве альтернативы, количество приблизительно 0,9-1,4 г, предпочтительно 1,15 г твердого состава заполняют в 50 мл флакон. Термин "флакон" относится к (небольшому) стеклянному или пластмассовому сосуду или бутылке, часто применяемым для хранения фармацевтических композиций в виде жидкостей или твердых веществ. В настоящее время флаконы часто изготавливают из пластмассы, такой как полипропилен или полистирол. Естественно, любой другой подходящий контейнер также можно применять для хранения твердой композиции по настоящему изобретению.
Для получения готового к применению инъекционного раствора способ по настоящему изобретению также включает стадию восстановления лиофилизата, полученного на стадии bb) в фармацевтически приемлемой текучей среде с получением инъекционного раствора. Термин "инъекционный раствор" относится к раствору, который может быть использован для внутривенного введения, предпочтительно для инъекции или инфузии (также см. выше раскрытие касательно инъекции). Инъекционный раствор содержит фармацевтически приемлемую текучую среду.
Настоящее изобретение дополнительно характеризуется следующим списком объектов.
Объект 1. Твердая фармацевтическая композиция, приспособленная для внутривенного введения, содержащая
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну фосфатную соль.
Объект 2. Фармацевтическая композиция по п. 1, где по меньшей мере одна фосфатная соль представляет собой фосфат натрия, фосфат калия или фосфат аммония.
Объект 3. Фармацевтическая композиция по п. 2, где фосфатная соль выбрана из группы, включающей Na2HPO4 (безводный), Na2HPO4 • 2 H2O, Na2HPO4 • 7 H2O, Na2HPO4 • 12 H2O, NaH2PO4 (безводный), NaH2PO4 • H2O, NaH2PO4 • 2 H2O, K2HPO4 (безводный), K2HPO4 • 3 H2O, KH2PO4 (безводный) и их смеси.
Объект 4. Фармацевтическая композиция по п. 2, где фосфатная соль представляет собой Na2HPO4 • 2 H2O, и где количество Na2HPO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,04 до 0,4.
Объект 5. Фармацевтическая композиция по п. 2, где фосфат натрия представляет собой NaH2PO4 • 2 H2O, и где количество NaH2PO4 • 2 H2O, присутствующего в композиции, выбрано таким образом, что молярное отношение NaH2PO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,01 до 0,09.
Объект 6. Фармацевтическая композиция по любому из пп. 1-5, содержащая две различные фосфатные соли натрия.
Объект 7. Фармацевтическая композиция по п. 6, где две различные фосфатные соли натрия представляют собой NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O.
Объект 8. Фармацевтическая композиция по п. 7, где количества NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O, присутствующих в композиции, выбраны таким образом, что молярное отношение как NaH2PO4 • 2 H2O, так и Na2HPO4 • 2 H2O к соединению (I) или соединению (II) находится в диапазоне от 0,02 до 0,5.
Объект 9. Фармацевтическая композиция по любому из пп. 1-8, где соединение (I) и/или соединение (II) присутствуют в виде свободного основания.
Объект 10. Фармацевтическая композиция по любому из пп. 1-9, где фармацевтическая композиция представляет собой лиофилизированную фармацевтическую композицию.
Объект 11. Фармацевтическая композиция по любому из пп. 1-10, где соединение (I) представляет собой соединение, имеющее формулу (Ia):
(Ia).
Объект 12. Фармацевтическая композиция по любому из пп. 1-11, где соединение (II) представляет собой соединение, имеющее формулу (IIa):
(IIa).
Объект 13. Фармацевтическая композиция по любому из пп. 1-12, где фармацевтическая композиция содержит дополнительное фармацевтическое вспомогательное вещество.
Объект 14. Фармацевтическая композиция по п. 13, где дополнительное фармацевтическое вспомогательное вещество представляет собой неорганическую соль.
Объект 15. Фармацевтическая композиция по п. 14, где неорганическая соль выбрана из MgCl2, CaCl2, NH4Cl, KCl или NaCl.
Объект 16. Фармацевтическая композиция по п. 15, где неорганическая соль представляет собой NaCl.
Объект 17. Фармацевтическая композиция по п. 16, где количество NaCl, присутствующего в композиции, выбрано таким образом, что молярное отношение NaCl к соединению (I) или соединению (II) находится в диапазоне от 1,5 до 4, предпочтительно от 1,8 до 3,7.
Объект 18. Фармацевтическая композиция по любому из пп. 1-17, где композиция дополнительно содержит кристаллизационную воду.
Объект 19. Фармацевтическая композиция по любому из пп. 1-18, где композиция приспособлена для восстановления в воде.
Объект 20. Фармацевтическая композиция по любому из пп. 1-19, где композиция приспособлена для введения посредством инфузии или инъекции.
Объект 21. Фармацевтическая композиция по любому из пп. 1-20, где соединение (I) представляет собой (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерин.
Объект 22. Фармацевтическая композиция по любому из пп. 1-21, где соединение (I) представляет собой (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерин.
Объект 23. Фармацевтическая композиция по любому из пп. 1-22, где соединение (I) представляет собой диастереомерную смесь, которая содержит больше (6R)-4-амино-5, 6,7, 8-тетрагидро-L-биоптерина, чем (6S)-4-амино-5, 6,7,8-тетрагидро-L-биоптерина.
Объект 24. Фармацевтическая композиция по п. 23, где количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина и (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерина выбраны таким образом, что отношение количества (6R)-4-амино-5,6,7,8-тетрагидро-L-биоптерина к (6S)-4-амино-5,6,7,8-тетрагидро-L-биоптерину находится в диапазоне от 0,5 до 2, предпочтительно составляет около 1,3.
Объект 25. Фармацевтическая композиция по любому из пп. 1-24, где единичная доза композиции содержит 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина, 140 ± 30 мг кристаллизационной воды, 70 ± 7 мг Na2HPO4 ∙ 2 H2O, 16,5 ± 2 мг NaH2PO4 ∙ 2 H2O и 350 ± 30 мг NaCl.
Объект 26. Композиция по любому из пп. 1-25, где композиция содержит 1, 2, 3, 4, 5, 6, 7 или более дополнительных соединений, где дополнительные соединения выбраны из группы, включающей одно или более соединений, выбранных из группы, включающей 4-амино-L-биоптерин, (6R,S)-5,6,7,8-тетрагидро-L-биоптерин, 1-[(6R,S)-2,4-диамино-5,6,7,8-тетрагидроптеридин-6-ил]пропанол, 1-[(6R,S)-2,4-диамино-5,6,7,8-тетрагидроптеридин-6-ил]пропан, (1R,2S)-1-[(6R,S)-2-(ацетиламино)-4-амино-5,6,7,8-тетрагидроптерин-6-ил]-1,2-диацетоксипропан, 2,4-диамино-7,8-дигидроптеридин, 2,4-диаминоптеридин.
Объект 27. Применение лиофилизированной фармацевтической композиции по любому из пп. 1-26 для лечения заболевания.
Объект 28. Применение по п. 27, где заболевание выбрано из группы, включающей травматическое повреждение головного мозга, нетравматическое повреждение головного мозга, предпочтительно инсульт или менингит, повышенное внутричерепное давление, вторичное повреждение головного мозга.
Объект 29. Способ получения лиофилизированной твердой фармацевтической композиции, приспособленной для внутривенного введения, содержащей
a) соединение, имеющее формулу (I):
(I),
и/или соединение, имеющее формулу (II):
(II),
и
b) по меньшей мере одну фосфатную соль;
при этом способ включает
aa) растворение соединения формулы (III) и/или (II):
(III)
в буфере;
bb) лиофилизацию раствора, полученного на стадии aa).
Объект 30. Способ по п. 29, где способ дополнительно включает стадию восстановления лиофилизата, полученного на стадии bb), в фармацевтически приемлемой текучей среде с получением инъекционного раствора.
Объект 31. Способ по п. 29 или п. 30, где буфер из стадии aa) представляет собой буфер на основе гидрофосфата натрия, содержащий по меньшей мере одну фосфатную соль.
Объект 32. Способ по любому из пп. 29-31, где буфер из стадии aa) содержит NaOH, буфер на основе гидрофосфата натрия и воду.
Объект 33. Способ по п. 31, где NaOH представляет собой 5 н. раствор NaOH.
Объект 34. Способ по п. 31, где буфер на основе гидрофосфата натрия получают путем раздельного растворения NaH2PO4 • 2 H2O и Na2HPO4 • 2 H2O.
Объект 35. Способ по п. 33, где буфер на основе гидрофосфата натрия характеризуется значением pH 7,4 в результате добавления раствора NaH2PO4 • 2 H2O к растворенному Na2HPO4 • 2 H2O.
Объект 36. Способ по любому из пп. 29-35, где буфер содержит 12-16% (вес./вес.) 5 н. NaOH, 8-12% (вес./вес.) буфера на основе гидрофосфата натрия и 74-78% (вес./вес.) воды для инъекций.
Объект 37. Способ по любому из пп. 29-36, где раствор, полученный на стадии aa), стерилизуют фильтрацией, предпочтительно с применением 0,22 мкм фильтра.
Объект 38. Способ по любому из пп. 29-37, где буфер характеризуется значением pH приблизительно 8, 9, 10, 11, 12, 13 или 14.
Объект 39. Способ по любому из пп. 29-38, где раствор из стадии aa) характеризуется значением pH приблизительно 4, 5, 6, 7, 8, 9, 10 или 11, предпочтительно от 6,5 до 7,6, наиболее предпочтительно 7,4.
Объект 40. Способ по любому из пп. 29-39, где лиофилизатом, полученным на стадии bb), заполняют флаконы предпочтительно в количестве приблизительно 1-1,5 г, предпочтительно 1,25 г твердого состава.
Объект 41. Способ по любому из пп. 29-40, где буфер получают с дегазированным буфером.
Объект 42. Способ по любому из пп. 29-41, где буфер дегазируют азотом до достижения содержания кислорода ˂ 1,0 ppm.
Объект 43. Способ по любому из пп. 29-42, где после получения раствора лиофилизацию начинают не позже, чем через 2 часа.
Объект 44. Фармацевтическая композиция, получаемая посредством способа по любому из пп. 29-43.
Объект 45. Применение лиофилизированной фармацевтической композиции по любому из пп. 1-28 в изготовлении лекарственного препарата для лечения субъекта с закрытой травмой черепа, повышенным внутричерепным давлением и вторичным повреждением головного мозга.
Объект 46. Способ лечения заболевания у субъекта, включающий стадию введения лиофилизированной фармацевтической композиции по любому из пп. 1-28 субъекту, нуждающемуся в этом.
Объект 47. Способ по п. 46, где максимальная суточная доза составляет 20 мг/кг веса тела, предпочтительно 17,5, 15,0 или 12,5, 10, 8,5, 7,5, 5,0, 2,5 мг/кг веса тела.
ПРИМЕРЫ
Следующие примеры дополнительно иллюстрируют настоящее изобретение. Однако данные примеры не должны рассматриваться как ограничивающие объем настоящего изобретения. Примеры включены в целях иллюстрации, а настоящее изобретение ограничено только формулой изобретения.
Пример 1. Описание способа изготовления
VAS203 получали посредством многостадийного синтеза, начиная из коммерчески доступного L-биоптерина. Схема способа показана на фигуре 1. Синтез VAS203 основан на публикациях W. Pfleiderer et al. в Pteridines 1989, 1, 199-210 и Pteridines 1995, 6, 1-7.
Целью первой фазы разработки было воспроизведение описанных в литературе процедур и оценка их применимости для крупного масштаба. Очистку промежуточных соединений с помощью флэш-хроматографии исключали и замещали стадиями осаждения или кристаллизации. Более того, осуществляли подборы молярных отношений реагентов и условий реакций, так что был доступен стабильный способ вплоть до промежуточного соединения MAE 119 (4-амино-L-биоптерина).
Было проблематично установить стабильный и надежный путь гидрирования MAE 119 до VAS203 с применением PtO2 в качестве катализатора. Тестировали различные растворители и водные среды при различных значениях pH, но только при применении разбавленной хлористоводородной кислоты могли быть достигнуты быстрые и воспроизводимые результаты, в особенности, в отношении диастереомерного соотношения лекарственного вещества. Общее количество остаточного содержания платины может быть существенно снижено с помощью предварительного гидрирования катализатора на основе оксида платины (IV), посредством чего диастереомерное соотношение незначительно изменялось. Доказано, что отработка кислотного раствора VAS203 является затруднительной, поскольку при простом выпаривании воды при пониженном давлении оставался стекловидный остаток. Прорыв был достигнут при применении 2-пропанола в качестве вспомогательного растворителя в процессе выпаривания. В ходе данной процедуры получали твердое вещество, но, к сожалению, 2-пропанол был включен в данное твердое вещество. При обычных условиях высушивания количество остаточного 2-пропанола не могло быть снижено более чем на 5% (вес./вес.). Для решения данной проблемы можно включать конечную стадию лиофилизации с образованием VAS203. Единичные партии могут быть объединены с получением больших количеств.
Сведения структурного определения VAS203 было представлено способом синтеза и подтверждалось элементным анализом, ядерным магнитным резонансом (1H-ЯМР и 13C-ЯМР), УФ- и ИК-спектроскопией (данные не приведены).
Пример 2.
Описание и композиция лекарственного продукта
VAS203 будет поставляться в виде стерильного лиофилизированного порошка от белого до светло-красного или коричневого цвета, загруженного в 50 мл стеклянные флаконы в атмосфере азота в качестве защитной атмосферы. Каждый флакон содержит 650 ± 60 мг свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина и 140 ± 30 мг кристаллизационной воды. Кроме того, флаконы содержат 350 ± 30 мг хлорида натрия (NaCl), 70 ± 7 мг дигидрата гидрофосфата динатрия (Na2HPO4 · 2 H2O) и 16,5 ± 2 мг дигидрата дигидрофосфата натрия (NaH2PO4 · 2 H2O). Пределы допустимых отклонений в композиции лекарственного продукта являлись относительно высокими (± 10%). Причиной этого является изменение содержания гидрохлорида в VAS203. Содержание гидрохлорида в VAS203 варьирует от партии к партии вплоть до 10% (от 2,03 HCl до 2,24 HCl). В ходе получения лекарственного продукта гидрохлорид нейтрализовали в настоящем изобретении путем добавления гидроксида натрия и буфера на основе фосфата натрия с получением изотонического раствора с физиологическим значением pH. Следовательно, содержание молекул, образованных в ходе нейтрализации (хлорид натрия, гидрофосфат динатрия и дигидрофосфат натрия) также варьирует в соответствии с содержанием гидрохлорида в соответствующей партии VAS203. Приведенные пределы допустимых отклонений должны соответствовать спецификациям относительно определяющих качество параметров − pH и осмолярности. Качественная композиция 1 г флаконов с VAS203 приведена в таблице 2.
Таблица 2
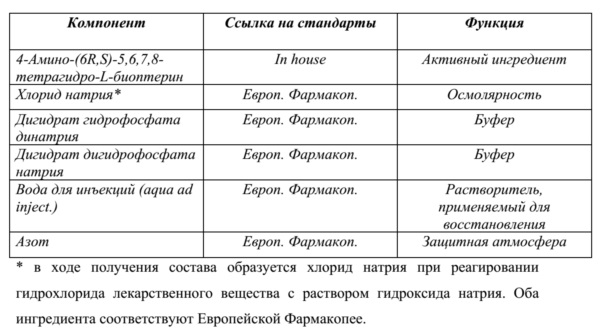
Фармацевтическая разработка
Твердую лиофилизированную дозированную форму разрабатывали в данном случае для VAS203 с получением инфузионного раствора.
1 г VAS 203 в 10 г буферного раствора на основе гидроксида натрия/гидрофосфата натрия с конечным pH 7,4 выбирали для асептической обработки, стерилизовали посредством мембранной фильтрации и заполняли в 50 мл стеклянные флаконы. Далее данный раствор лиофилизировали в соответствии с выбранной программой лиофилизации, в ходе которой получали лиофилизированный продукт с отличной стабильностью. В данной твердой композиции VAS 203 присутствовал в виде свободного основания 4-амино-5,6,7,8-тетрагидро-L-биоптерина. Флаконы закрывали в атмосфере азота, закупоривали лиофилизационными пробками и закрывали белыми вакуумными крышками. Вспомогательные вещества добавляли с целью получения изотонического раствора с физиологическим pH после восстановления с помощью 50 мл воды ad inject. Значение pH конечного изотонического раствора составляло 6,5-7,6. Конечная концентрация лекарственного вещества VAS203 составляла 20 мг/мл.
Описание способа изготовления и способов контроля процесса
Дигидрат дигидрохлорида 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина (VAS203), растворенный в буферном растворе, при pH 7,4 является очень нестабильным и окисляется до его двух метаболитов 4-амино-7,8-дигидро-L-биоптерина и 4-амино-L-биоптерина. Следовательно, важным является, чтобы раствор VAS203 был получен с дегазированным буфером и после получения раствора VAS203 лиофилизация была начата без задержки.
Для обеспечения возможности лиофилизации в 50 мл флаконах необходимо получить концентрированный буферный раствор с VAS203 (1 г VAS203 в 10 г буфера). Буферный раствор получали посредством смешивания базовых растворов:
14% (вес./вес.) 5 н. раствора гидроксида натрия (NaOH)
10% (вес./вес.) раствора буфера на основе гидрофосфата натрия (NaPB)
500 ммоль/л, pH 7,4
76% (вес./вес.) воды для инъекций.
Стерильный буферный раствор затем дегазировали азотом до достижения содержания кислорода <1,0 ppm. Для каждого флакона 9,0 г данного буферного раствора применяли для растворения 1,0 г VAS203. Буферный раствор заполняли в продуваемый азотом сосуд и твердое вещество (лиофилизированное) VAS203 осторожно добавляли в течение 15 минут в атмосфере азота в качестве защитной атмосферы. После проверки значения pH раствор VAS203 стерилизовали фильтрацией с применением 0,22 мкм фильтра Millipak 200. После применения 0,22 мкм фильтры тестировали в отношении целостности. Образец в размере 100 мл отбирали для тестирования биологической нагрузки. Предел биологической нагрузки раствора лекарственного продукта перед стерилизационной фильтрацией определяли как = 10 КОЕ/100 мл. Оставшийся раствор фильтровали второй раз и диспергировали в 50 мл флаконах, 10 г на флакон. Перед заполнением флаконы стерилизовали посредством сухого нагрева в соответствии с разделом 5.1.1. Европейской Фармакопеи. После лиофилизации флаконы закрывали в атмосфере азота, закупоривали лиофилизационными пробками и закрывали белыми вакуумными пробками. Раствор VAS203 получали непосредственно перед лиофилизацией. Способ асептического заполнения валидировали с использованием наполнения средами. Принципиальная схема последовательных стадий способа изготовления с указанием компонентов, применяемых на каждой стадии, показана на фиг. 2. Результатом является твердая композиция, в которой VAS203 присутствует в виде свободного основания 4-амино-(6R,S)-5,6,7,8-тетрагидро-L-биоптерина.
Стабильность
Контроль стабильности проводили в течение 24 месяцев. Образцы хранили в условиях "долгосрочного исследования" при 5°C (препарат с действующим веществом). Ускоренное исследование стабильности проводили при 40°C/75% RH (относительная влажность) в течение 6 месяцев (препарат с действующим веществом). Аналитические процедуры, применяемые в программе исследования стабильности, включали тесты относительно количественного анализа, чистоты и сопутствующих примесей. Применяемые спецификации и использованные способы являлись такими же, как и для выпуска лекарственного продукта.
Контроль стабильности проводили в течение по меньшей мере 60 месяцев. В таблице 3 показаны условия и период хранения для исследования стабильности. Образцы хранили в условиях "долгосрочного исследования" при 5°C (препарат с действующим веществом). Ускоренное исследование стабильности проводили при 40°C/75% RH (относительная влажность) в течение 6 месяцев (препарат с действующим веществом). В таблице 4 приведен график тестирования в разные моменты времени. Аналитические процедуры, применяемые в программе исследования стабильности, включали тесты относительно количественного анализа, чистоты и сопутствующих примесей. Применяемые спецификации и использованные способы являлись такими же, как и для выпуска лекарственного продукта. Иллюстративные результаты исследования стабильности показаны в таблице 5 (которая изображена на фиг. 4).
Таблица 3. Условия для проведения контроля стабильности
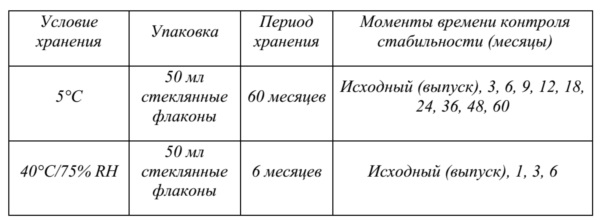
Таблица 4. График тестирования в моменты времени контроля стабильности
* Тесты, подлежащие осуществлению, представляли собой
- A: полная спецификация срока годности
- B: полная спецификация срока годности без пунктов тестирования: "стерильность", "эндотоксины" и "частицы"
Обсуждение
Данные о стабильности доступны для партии, хранящейся при 5°C и при 40°C/75% RH, защищенной от света. Незначительные изменения наблюдали для образцов, хранящихся при обеих температурах в течение периода до 24 месяцев, но эти данные не продемонстрировали устойчивой тенденции, а считались входящими в диапазон вариабельности анализа. Продукты распада не образовывались. Незначительное увеличение может наблюдаться для первого метаболита (продукта окисления) лекарственного вещества (4-амино-7,8-дигидро-L-биоптерина). Через 24 месяца хранения при 5°C в 7 из 10 флаконов было обнаружено в общем количестве 15 видимых частиц.
Данные о стабильности доступны для партии, хранящейся при 5°C и при 40°C/75% RH, защищенной от света. Для образцов, хранящихся при обеих температурах в течение периода до 36 месяцев, не было обнаружено значительных изменений. Продукты распада не образовывались.
Вывод
Флаконы с лекарственным продуктом VAS203 являются стабильными при хранении при 5°C, при защите от света, в течение не менее 36 месяцев. Флаконы с лекарственным продуктом VAS203 являются стабильными при хранении при 40°C/75% RH, при защите от света, в течение не менее 6 месяцев. С учетом вышеизложенного, срок годности длительностью 42 месяца установили для 1 г лиофилизированного порошка VAS203, заполненного в 50 мл стеклянные флаконы в атмосфере азота в качестве защитной атмосферы. В инструкции относительно хранения будет рекомендовано хранить флаконы при 2-8°C, и флаконы должны быть обернуты алюминиевой фольгой и упакованы в картонные коробки для защиты их от света. Все восстановленные лекарственные продукты должны быть визуально проверены на наличие твердых частиц. В инструкции относительно хранения будет рекомендовано хранить флаконы при 2-8°C.
Срок годности может быть продлен, если будут доступными соответствующие данные о долгосрочной и ускоренной стабильности из аналогичного исследования стабильности для флаконов с 1 г VAS203 и 50 мл флаконов плацебо и результаты будут соответствовать действующим спецификациям. Срок годности будет определен в соответствии с принципами, описанными в руководстве ICH Q1E.
Если не обнаружено значительного изменения при ускоренных условиях в течение 6 месяцев и данные долгосрочной стабильности демонстрируют небольшое изменение или его отсутствие в течение периода времени и низкое значение вариабельность, то в качестве продленного срока годности будет использован период в два раза больший охватываемого данными в реальном времени, но срок годности не может превышать продолжительность, доступную из данных долгосрочного исследования, более чем на 12 месяцев.
Пример 3. Стабильность в ходе получения, введения и курса инфузии
Исследование стабильности при комнатной температуре проводили для лекарственного вещества VAS203 для тестирования в отношении любого ухудшения восстановленного раствора в прозрачных 50 мл полипропиленовых (PP) шприцах, которые применяют в клинических исследованиях. В ходе тестирования стабильности PP-шприцы подвергали воздействию нормального дневного света. Содержание, чистоту, сопутствующие примеси и pH раствора контролировали в течение 48 часов (исходный, 6, 24, 48 часов после получения). В таблице 6 приведены результаты тестирования стабильности.
Обсуждение
Содержание раствора (количественный анализ) оставалось постоянным в определенных пределах (650 ± 60 мг) в течение 48 часов. В течение периода тестирования хроматографическая чистота для обоих диастереомеров была удовлетворительной в определенных пределах, значительного изменения не наблюдалось. Относительное содержание сопутствующих примесей оставалось постоянным в определенных пределах в течение 48 часов. Продукты распада не образовывались. Незначительное увеличение может наблюдаться для первого метаболита VAS203 (4-амино-7,8-дигидро-L-биоптерина). Однако данное окисление возможно было вызвано остаточным кислородом в шприце, от 6 до 48 часов изменение было незначительным. Значение pH восстановленного раствора VAS203 оставалось постоянным в определенных пределах в течение 48 часов. Изменение не наблюдалось. Чувствительность к свету не наблюдалась до 48 часов.
Вывод
Проведенные исследования демонстрируют, что восстановленный раствор VAS203 является стабильным в устройстве для его применения (50 мл PP-шприц) при комнатной температуре, при воздействии нормального дневного света в течение 48 часов. Однако срок годности был снижен во избежание микробиологического загрязнения. Срок годности был определен как момент времени получения раствора VAS203 плюс 27 часов.
Таблица 6. Стабильность восстановленного раствора VAS203 в 50 мл PP-шприце (устройство применения), хранящегося при комнатной температуре
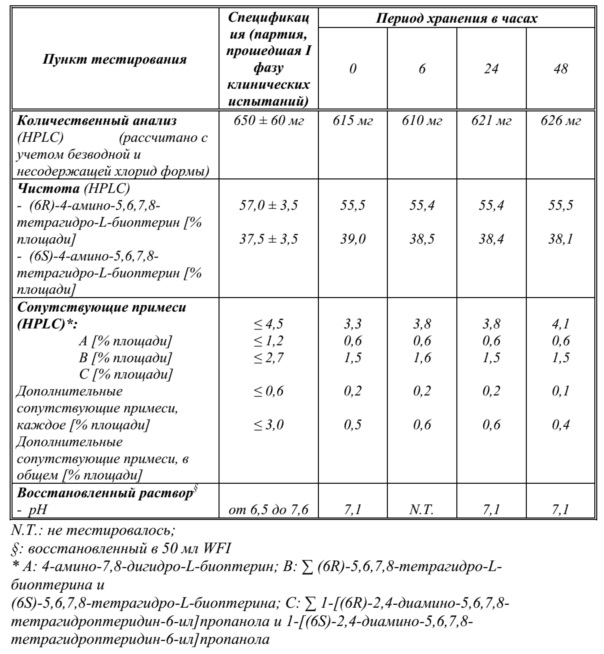
Следует отметить, что используемые в данном документе формы единственного числа включают ссылки на множественное число, если в контексте четко не указано иное. Таким образом, например, ссылка на "реагент" включает один или более таких различных реагентов, а ссылка на "способ" включает ссылку на эквивалентные стадии и способы, известные специалисту средней квалификации в данной области, которые могут быть модифицированы или замещены способами, описанными в данном документе.
Все публикации и патенты, цитируемые в данном раскрытии, включены посредством ссылки во всей своей полноте. В данной степени, в которой материал, включенный посредством ссылки, противоречит или не соответствует данному описанию, описание будет заменять любой такой материал.
Если не указано иное, термин "по меньшей мере", предшествующий ряду элементов, следует понимать как ссылку на каждый элемент в данном ряде. Специалист в данной области поймет или будет способен установить с использованием только обычных экспериментов множество эквивалентов для конкретных вариантов осуществления настоящего изобретения, описанных в данном документе. Такие эквиваленты предназначены для включения в объем настоящего изобретения.
По всему данному описанию и формуле изобретения, которая следует далее, если контекст не требует иного, слово "содержать" и такие вариации, как "содержит" и "содержащий" понимают как означающее включение указанного целого числа или стадии, или группы целых чисел или стадий, но не исключение любого другого целого числа или стадии, или группы целых чисел или стадий. При использовании в данном документе термин "содержащий" может быть заменен термином "состоящий" или в некоторых случаях при использовании в данном документе − термином "имеющий".
При использовании в данном документе термин "состоящий из" исключает любой элемент, стадию или ингредиент, не указанный в заявленном элементе. При использовании в данном документе термин "по сути состоящий из" не исключает материалы или стадии, которые не оказывают существенного влияния на основные и новые характеристики заявленного элемента. В каждом случае в данном документе любой из терминов "содержащий", "по сути состоящий из" и "состоящий из" можно заменить одним из других двух терминов.
Некоторые документы цитируются по всему тексту данного описания. Каждый из документов, цитируемых в данном документе (включая все патенты, заявки на патенты, научные публикации, спецификации производителей, инструкции и т. д.), либо выше, либо ниже, включены в данный документ посредством ссылки во всей своей полноте. Ни одно из приведенного в данном документе не должно истолковываться как признание того, что настоящее изобретение не дает право относить такое раскрытие к более ранней дате посредством предыдущего изобретения.
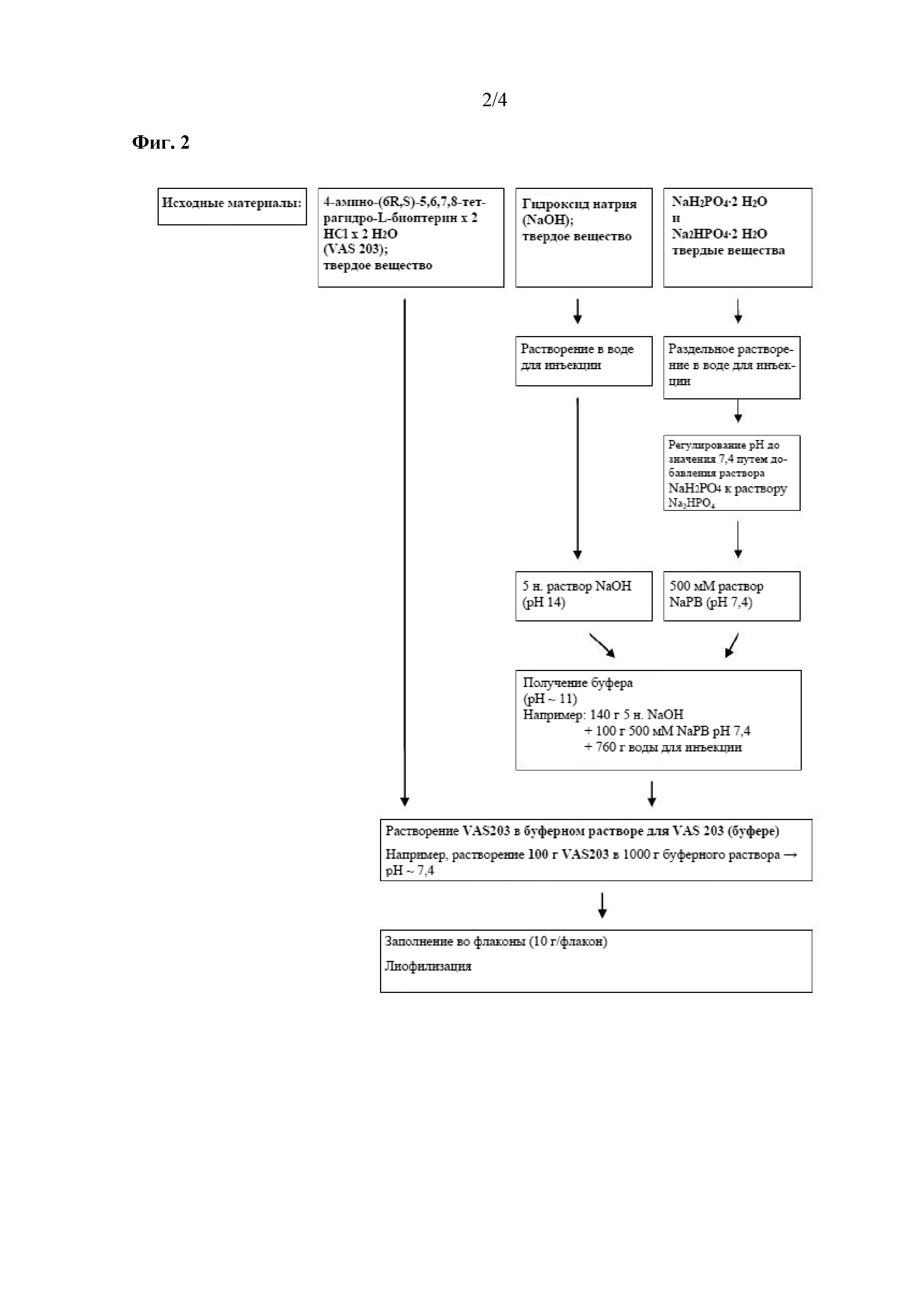
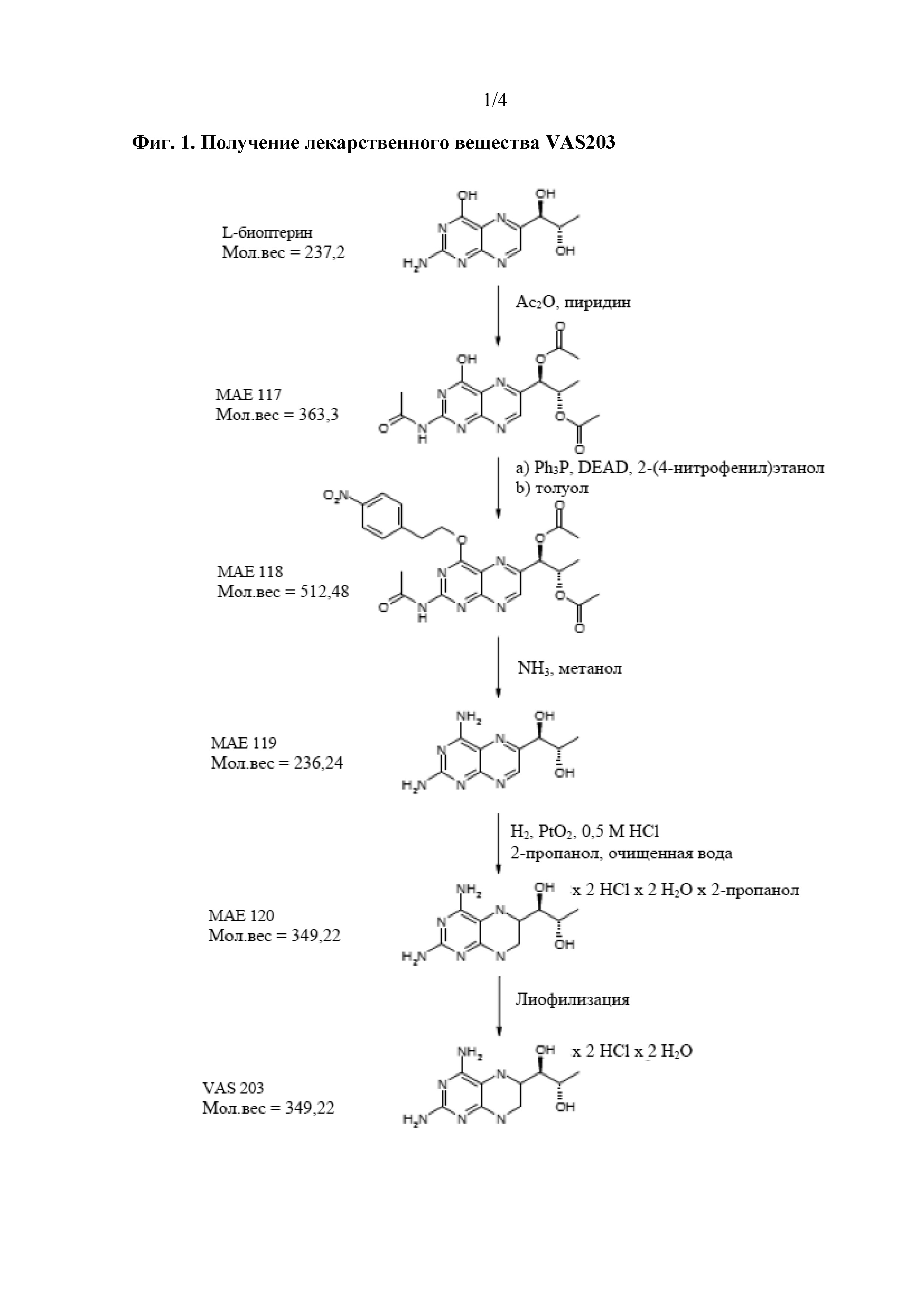
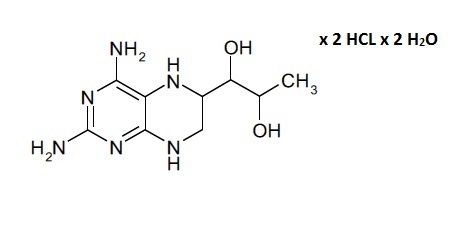
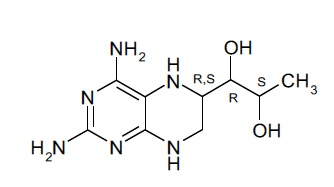
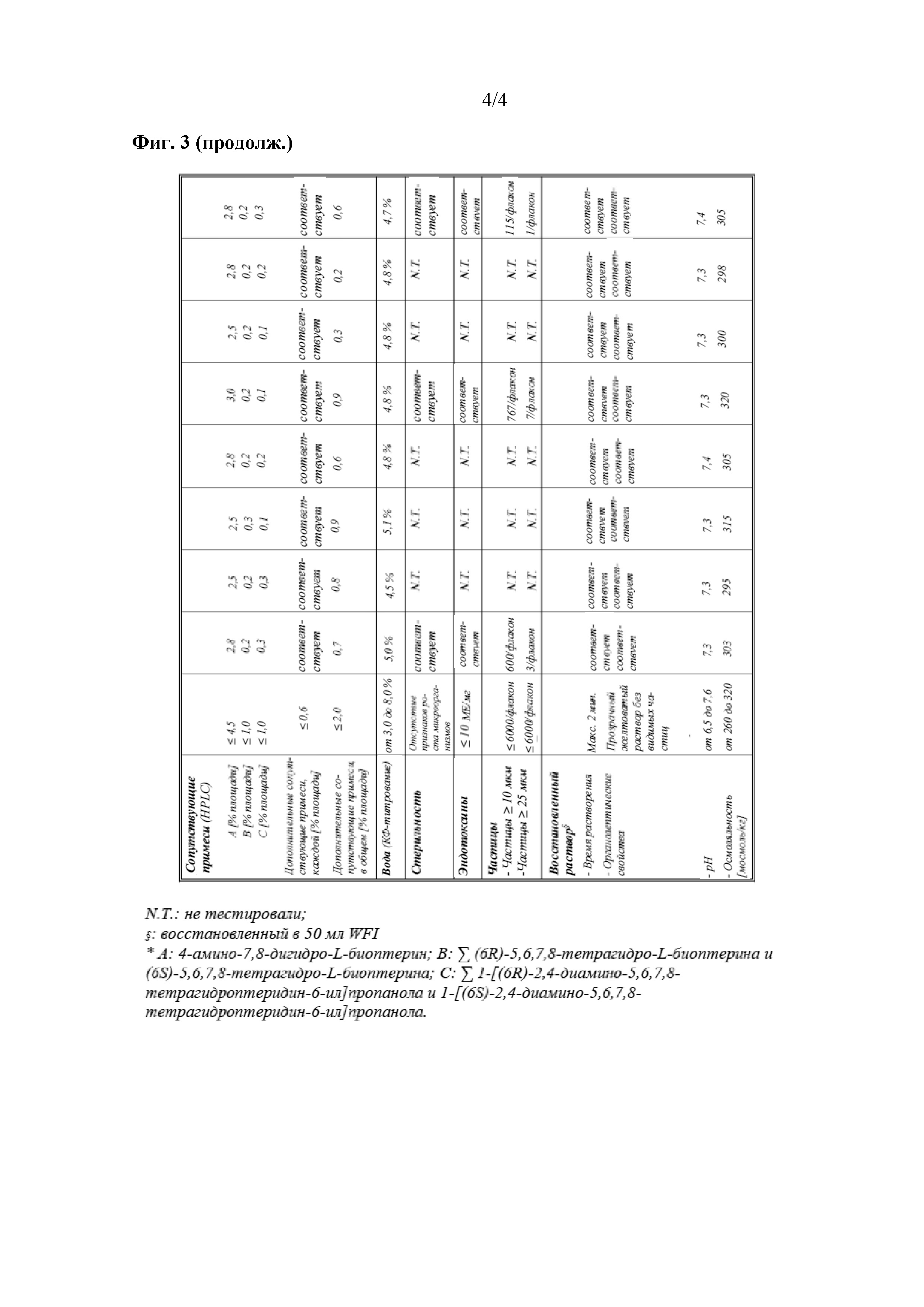
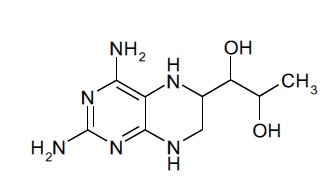
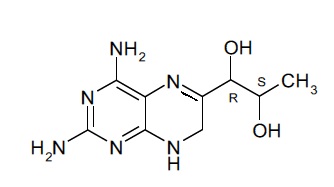
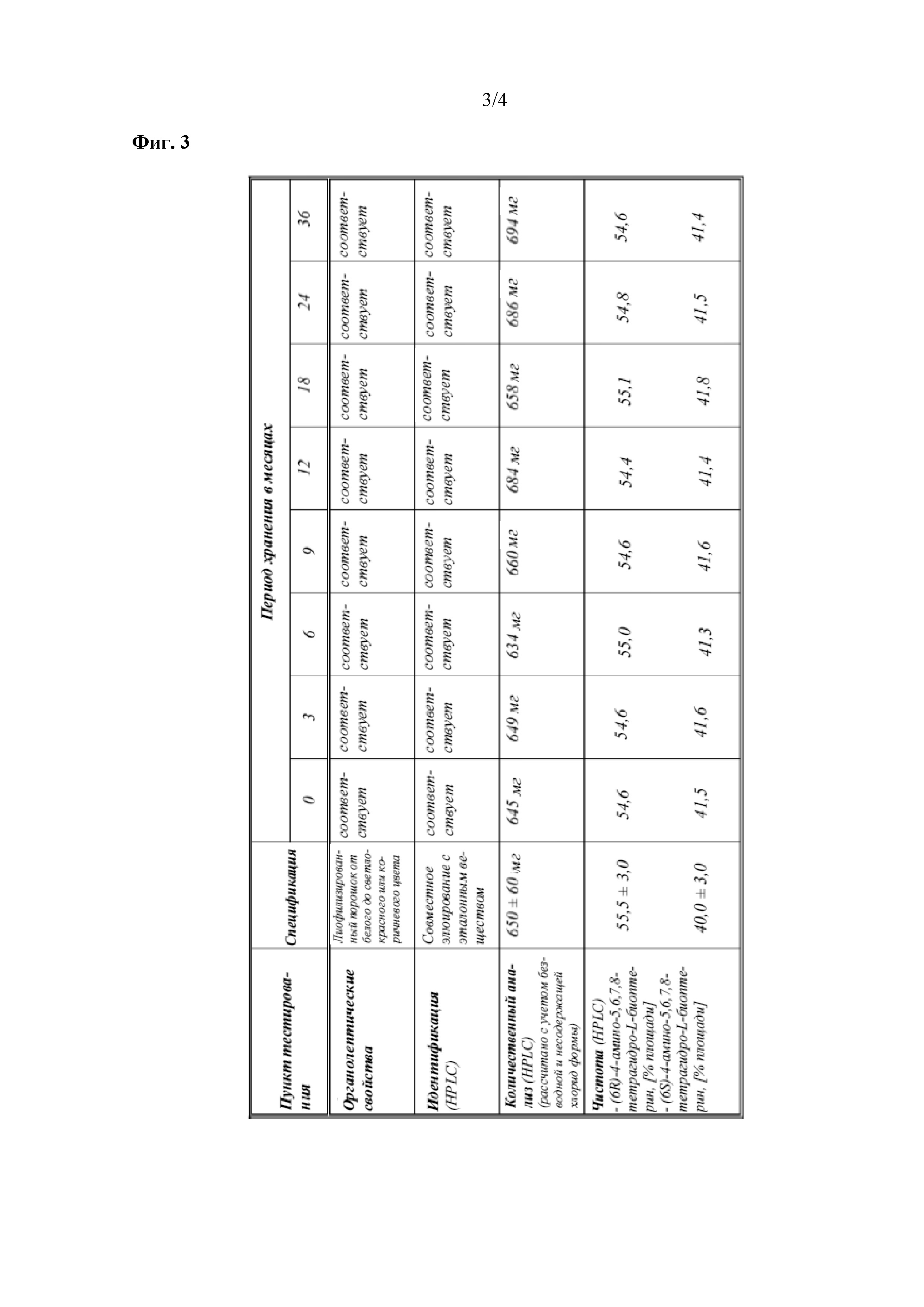
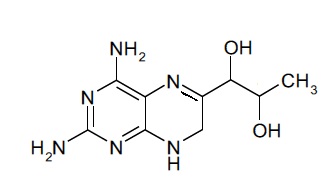
Соединения, способ их получения, применение, фармацевтическая композиция

