Результат интеллектуальной деятельности: УЛУЧШЕННЫЕ СМЕШАННЫЕ МЕТАЛЛОКСИДНЫЕ КАТАЛИЗАТОРЫ АММОКСИДИРОВАНИЯ
Вид РИД
Изобретение
Уровень техники
Область техники
Настоящее изобретение относится к улучшенному катализатору для использования при аммоксидировании ненасыщенного углеводорода в соответственный ненасыщенный нитрил. В частности, настоящее изобретение направлено на улучшенную каталитическую композицию для аммоксидирования пропилена и/или изобутилена в акрилонитрил и/или метакрилонитрил, соответственно, причем указанный катализатор содержит комплекс оксидов металлов, содержащий рубидий, висмут, церий, молибден, железо и другие промоторы, и причем указанный катализатор характеризуется соотношением рубидия к висмуту и церию, содержащимся в катализаторе.
Описание предшествующего уровня техники
Катализаторы, содержащие оксиды железа, висмута и молибдена, промотированные подходящими элементами, долго использовались для конверсии пропилена и/или изобутилена при повышенных температурах в присутствии аммиака и кислорода (обычно в виде воздуха) для производства акрилонитрила и/или метакрилонитрила. В частности, каждый из патента Великобритании №1436475; патентов США №4766232; №4377534; №4040978; №4168246; №5223469 и №4863891 направлен на висмут-молибден-железные катализаторы, которые могут быть промотированы элементами II группы, для получения акрилонитрила. Кроме того, патенты США №5093299, №5212137, №5658842, №5834394, №8153546 и документ CN103418400 направлены на висмутом-молибденовые промотированные катализаторы, дающие высокие выходы акрилонитрила.
Отчасти настоящее изобретение относится к висмут-молибден-железным катализаторам, промотированным церием. Было обнаружено, что путем регулирования относительного соотношения висмута к церию влияют на работу катализатора.
Сущность изобретения
Настоящее изобретение направлено на улучшенную смешанную металлоксидную каталитическую композицию для аммоксидирования пропилена и/или изобутилена.
Согласно одному варианту осуществления каталитическая композиция содержит комплекс оксидов металлов, причем относительные соотношения перечисленных элементов в указанном катализаторе представлены следующей формулой:
Mom Bia Feb Ac Dd Ee Ff Gg Ceh Rbn Ox,
где А представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия и цезия; и
D представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, свинца и германия;
G представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
a, b, с, d, е, f, g, h, m, n и x, соответственно, представляют собой атомные отношения висмута (Bi), железа (Fe), A, D, Е, F, G, церия (Се), рубидия (Rb) и кислорода (О) относительно «m» атомов молибдена (Мо), где
а составляет больше чем 0-7,
b составляет 0,1-7,
с составляет больше чем 0-5,
d составляет 0,1-12,
е составляет 0-5,
f составляет 0-5,
g составляет 0-0,2,
h составляет 0,01-5,
m составляет 10-15,
n составляет больше чем 0-5, и
х представляет собой число атомов кислорода, необходимое для удовлетворения валентных требований других присутствующих составляющих элементов;
причем 0,3≤(a+h)/d и
0<(n+c)/(a+h)≤0,2.
Согласно одному варианту осуществления каталитической композиции 0,3≤(a+h)/d, 1,2≤h/b≤5 и 0<(n+c)/(a+h)≤0,2.
Согласно одному варианту осуществления каталитической композиции 0,3≤(a+h)/d, 1,2≤h/b≤5, 0<a/h<1,5 и 0<(n+c)/(a+h)≤0,2.
Согласно одному варианту осуществления каталитической композиции 0,15≤(a+h)/d≤0,4; 0<(n+c)/(a+h)≤0,2 и 0,8≤h/b≤5.
Согласно одному варианту осуществления каталитическая композиция не содержит калий.
Согласно одному варианту осуществления каталитическая композиция при использовании для аммоксидирования пропилена в акрилонитрил обеспечивает выход акрилонитрила больше 80%.
Настоящее изобретение также направлено на способы конверсии олефина, выбранного из группы, состоящей из пропилена и изобутилена или их смесей, в акрилонитрил и/или метакрилонитрил, и другие побочные нитрилы (т.е. соединения с функциональной группой «-CN», такие как ацетонитрил и цианистый водород), и их смеси путем реакции в паровой фазе при повышенной температуре и давлении указанного олефина с содержащим молекулярный кислород газом и аммиаком в присутствии смешанного металлоксидного катализатора, описанного выше.
Настоящее изобретение также направлено на способ конверсии олефина, выбранного из группы, состоящей из пропилена, изобутилена и их смесей, в акролеин и/или акриловую кислоту, метакролеин и/или метакриловую кислоту и их смеси, соответственно, путем реакции в паровой фазе при повышенной температуре и давлении указанного олефина с содержащим молекулярный кислород газом и аммиаком в присутствии смешанного металлоксидного катализатора, описанного выше.
В вышеуказанных соотношениях и при использовании в настоящем раскрытии символ «/» является символом деления, эквивалентным «+» и означает «разделенный на».
Подробное описание изобретения
Настоящее изобретение направлено на улучшенный смешанный металлоксидный катализатор для использования в процессах аммоксидирования пропилена и/или изобутилена. В этих процессах этот улучшенный катализатор обеспечивает большую общую конверсию пропилена и/или изобутилена в нитрилы (т.е. соединения с функциональной группой «-CN», такие как акрилонитрил, метакрилонитрил, ацетонитрил и цианистый водород), больший выход цианистого водорода и больший коэффициент использования аммиака. Эти улучшенные смешанные металлоксидные катализаторы также пригодны в процессах окисления пропилена и/или изобутилена в акролеин и/или акриловую кислоту, метакролеин и/или метакриловую кислоту и их смеси.
Катализатор
Настоящее изобретение направлено на многокомпонентную смешанную металлоксидную каталитическую композицию для аммоксидирования, содержащую комплекс каталитических оксидов, где перечисленные элементы и относительные соотношения перечисленных элементов в указанной каталитической композиции представлены следующей формулой:

где А представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из лития, натрия, калия и цезия; и
D представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, свинца и германия;
G представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а, b, с, d, е, f, g, h, m, n и x, соответственно, представляют собой атомные отношения висмута (Bi), железа (Fe), A, D, Е, F, G, церия (Се), рубидия (Rb) и кислорода (О) относительно «m» атомов молибдена (Мо),
где а составляет больше чем 0-7,
b составляет 0,1-7,
с составляет больше чем 0-5,
d составляет 0,1-12,
е составляет 0-5,
f составляет 0-5,
g составляет 0-0,2,
h составляет 0,01-5,
m составляет 10-15,
n составляет больше чем 0-5, и
х представляет собой число атомов кислорода, необходимое для удовлетворения валентных требований других присутствующих составляющих элементов.
Согласно одному варианту осуществления вышеуказанной каталитической композиции 0,3≤(a+h)/d, и 0<(n+c)/(a+h)≤0,2.
Согласно одному варианту осуществления вышеуказанной каталитической композиции 0,3≤(a+h)/d, 1,2≤h/b≤5 и 0<(n+c)/(a+h)≤0,2.
Согласно одному варианту осуществления каталитической композиции 0,3≤(a+h)/d, 1,2≤h/b≤5, 0<а/h<1,5 и 0<(n+c)/(a+h)≤0,2.
Согласно одному варианту осуществления вышеуказанной каталитической композиции 0,15≤(a+h)/d≤0,4; 0<(n+c)/(a+h)≤0,2 и 0,8≤h/b≤5.
Согласно одному варианту осуществления А представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из лития, натрия и цезия. Согласно одному варианту осуществления каталитическая композиция не содержит калий.
Отчасти каталитическая композиция может характеризоваться соотношением (a+h)/d, где «а» представляет собой относительное количество висмута в катализаторе, «h» представляет собой относительное количество церия в катализаторе, a «d» представляет собой относительные количества никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария в катализаторе. Эти относительные количества представляют собой нижние индексы элементов в формуле катализатора, или в случае «d» сумму нижних индексов из формулы катализатора для любого из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария, присутствующих в катализаторе. Согласно одному варианту осуществления 0,3≤(a+h)/d. Согласно другому независимому варианту осуществления 0,15≤(a+h)/d≤0,4. Другие независимые варианты осуществления представляют собой (каждая строчка ниже является вариантом осуществления):
0,3≤(a+h)/d≤1,
0,3≤(a+h)/d≤0,8,
0,3≤(a+h)/d≤0,6,
0,3≤(a+h)/d≤0,4.
0,15≤(a+h)/d
(a+h)/d≤1,
(a+h)/d≤0,8,
(a+h)/d≤0,6 и
(a+h)/d≤0,4.
Отчасти каталитическую композицию можно охарактеризовать соотношением (n+c)/(a+h), где «n» представляет собой относительное количество рубидия в катализаторе, «с» представляет собой относительное количество любого из лития, натрия, калия и цезия в катализаторе, «а» представляет собой относительное количество висмута в катализаторе, a «h» представляет собой относительное количество церия в катализаторе. Эти относительные количества представляют собой нижние индексы элементов в формуле катализатора, или в случае «с» - сумму нижних индексов из формулы катализатора для любого из лития, натрия, калия и цезия, присутствующих в катализаторе. Согласно одному варианту осуществления 0<(n+c)/(a+h)≤0,2. Другие независимые варианты осуществления представляют собой (каждая строчка ниже является вариантом осуществления):
0<(n+c)/(a+h),
0,02<(n+c)/(a+h),
0,04<(n+c)/(a+h),
0,06<(n+c)/(a+h),
(n+c)/(a+h)≤0,2,
(n+c)/(a+h)≤0,15,
(n+c)/(a+h)≤0,12,
(n+c)/(a+h)≤0,1,
0,02<(n+c)/(a+h)≤0,2,
0,04<(n+c)/(a+h)≤0,15 и
0,06<(n+c)/(a+h)≤0,12,
Отчасти каталитическую композицию можно охарактеризовать соотношением h/b, где «h» представляет собой относительное количество церия в катализаторе, а «b» представляет собой относительное количество железа в катализаторе. Эти относительные количества представляют собой нижние индексы элементов в формуле катализатора. Согласно одному варианту осуществления 0,8≤h/b≤5. Другие независимые варианты осуществления представляют собой (каждая строчка ниже является вариантом осуществления):
1,2≤h/b≤5,
1,5≤h/b≤5,
1,2≤h/b,
1,5≤h/b,
0,8≤h/b и
h/b≤5.
Было обнаружено, что катализаторы, описанные в диапазоне, описанном 0,8≤h/b≤5, как правило, сильнее в том отношении, что они имеют более низкую потерю от истирания, определенную тестом на истирание с помощью затопленной струи.
Отчасти каталитическую композицию можно охарактеризовать соотношением (a/h), где «а» представляет собой относительное количество висмута в катализаторе, «h» представляет собой относительное количество церия в катализаторе. Эти относительные количества представляют собой нижние индексы элементов в формуле катализатора. Согласно одному варианту осуществления 0<a/h≤1,5. Другие независимые варианты осуществления представляют собой (каждая строчка ниже является вариантом осуществления):
0,2≤a/h≤1,5,
0,3≤a/h≤1,5,
0,4≤a/h≤1,5,
0,45≤a/h≤1,5,
0,2≤a/h,
0,3≤a/h,
0,4≤a/h,
0,45≤a/h,
0,65≤a/h,
0,7≤a/h,
0,8≤a/h,
0,90≤a/h,
a/h≤1,2 и
a/h≤1,5.
Согласно другому варианту осуществления каталитическая композиция содержит комплекс оксидов металлов, причем относительные соотношения перечисленных элементов в указанном катализаторе представлены следующей формулой:
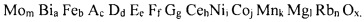
где А представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из лития, натрия, калия и цезия; и
D представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из цинка, кальция, стронция, кадмия и бария;
Е представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а, b, с, d, е, f, g, h, i, j, k, l, m, n и х соответственно, представляют собой атомные отношения висмута (Bi), железа (Fe), A, D, Е, F, G, церия (Се), никеля (Ni), кобальта (Со), марганца (Mn), магния (Mg), рубидия (Rb) и кислорода (О) относительно «m» атомов молибдена (Мо),
где а составляет больше чем 0-7,
b составляет 0,1-7,
с составляет 0,01-5,
d составляет 0,1-12,
е составляет 0-5,
f составляет 0-5,
g составляет 0-0,2,
h составляет 0,01-5,
i составляет 0,1-12,
j составляет 0,1-12,
k составляет 0,1-12,
l составляет 0,1-12,
m составляет 10-15,
n составляет более чем 0-5,
х представляет собой число атомов кислорода, необходимое для удовлетворения валентных требований других присутствующих составляющих элементов.
Согласно одному варианту осуществления вышеуказанной каталитической композиции z=d+i+j+k+l; 0,3≤(a+h)/z; 1,2≤h/b≤5; 0<a/h<1,5; 0,2<i/(i+j+k+l) и 0<(n+c)/(a+h)<0,2. Кроме того, соотношения, ранее описанные выше для h/b, a/h и (n+c)/(a+h), в равной степени относятся к вышеуказанному варианту осуществления каталитической композиции.
Кроме того, отчасти для этого варианта осуществления каталитическую композицию можно охарактеризовать соотношением (a+h)/z, где z=d+i+j+k+l. Независимо, отчасти для этого варианта осуществления каталитическую композицию можно охарактеризовать соотношением i/(i+j+k+l). Для этих соотношений «а» представляет собой относительное количество висмута в катализаторе, «h» представляет собой относительное количество церия в катализаторе, «d» представляет собой относительные количества никеля, цинка, кальция, стронция, кадмия и бария в катализаторе, «i» представляет собой относительное количество никеля в катализаторе, «j» представляет собой относительное количество кобальта в катализаторе, «k» представляет собой относительное количество марганца в катализаторе, «l» представляет собой относительное количество магния в катализаторе. Эти относительные количества представляют собой нижние индексы элементов в формуле катализатора, или в случае «d» - сумму нижних индексов из формулы катализатора для любого из цинка, кальция, стронция, кадмия и бария, присутствующих в катализаторе. Согласно одному варианту осуществления 0,3≤(a+h)/z. Согласно другому независимому варианту осуществления 0,15≤(a+h)/z≤0,4. Согласно другому независимому варианту осуществления 0,2<i/(i+j+k+l).
Согласно другим вариантам осуществления каталитических композиций, описанных в настоящем документе (каждая строка ниже является вариантом осуществления),
0,45≤a/h<1,5 и 0,3≤(a+h)/d,
0,65≤a/h<1,5 и 0,3≤(a+h)/d,
0,70≤a/h<1,5 и 0,3≤(a+h)/d,
0,80≤a/h<1,5 и 0,3≤(a+h)/d,
0,90≤a/h<1,5 и 0,3≤(a+h)/d,
0,45≤a/h<1,5, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,65≤a/h<1,5, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,70≤a/h<1,5, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,80≤a/h<1,5, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,90≤a/h<1,5, 0,3<(a+h)/d и 0,8≤h/b≤5,
0,90≤a/h≤1,2 и 0,3≤(a+h)/d, и
0,45≤a/h≤1,2, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,65≤a/h≤1,2, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,70≤a/h≤1,2, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,80≤a/h≤1,2, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,90≤a/h≤1,2, 0,3≤(a+h)/d и 0,8≤h/b≤5,
0,45≤a/h≤1,5 и 0,3≤(a+h)/d≤1,
0,65≤a/h<1,5 и 0,3≤(a+h)/d≤1,
0,70≤a/h<1,5 и 0,3≤(a+h)/d≤1,
0,80≤a/h<1,5 и 0,3≤(a+h)/d≤1,
0,90≤a/h<1,5 и 0,3≤(a+h)/d≤1,
0,45≤a/h<1,5, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,65≤a/h<1,5, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,70≤a/h<1,5, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,80≤a/h<1,5, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,90≤a/h<1,5, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,90≤a/h≤1,2 и 0,3≤(a+h)/d≤1, и
0,45≤a/h≤1,2, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,65≤a/h≤1,2, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,70≤a/h≤1,2, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,80≤a/h≤1,2, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5,
0,90≤a/h≤1,2, 0,3≤(a+h)/d≤1 и 0,8≤h/b≤5.
Согласно одному варианту осуществления каталитические композиции, описанные в настоящем документе, содержат теллур (Те). Согласно одному варианту осуществления катализатор не содержит сурьму. Согласно одному варианту осуществления каталитические композиции, описанные в настоящем документе, не содержат селен. Согласно другому варианту осуществления компоненты или элементы, обозначенные как «Е» в формулах композиций, описанных в настоящем документе, могут также включать теллур (Те) и/или сурьму (Sb). Согласно одному варианту осуществления компонент или элемент, обозначенный как «Е» в формулах композиций, описанных в настоящем документе, представляет собой хром (Cr). Согласно другому варианту осуществления компоненты или элементы, обозначенные как «Е» в формулах композиций, описанных в настоящем документе, представляют собой хром (Cr) и по меньшей мере один другой из компонентов или элементов, обозначенных как «Е». Согласно одному варианту осуществления компоненты или элементы, обозначенные как «Е» в вышеуказанных формулах композиций, описанных в настоящем документе, представляет собой хром. Согласно одному варианту осуществления композиций, описанных в настоящем документе, «F» может дополнительно включать свинец (Pb). Согласно одному варианту осуществления композиций, описанных в настоящем документе, «F» может дополнительно включать менее чем приблизительно 10 частей на миллион свинца (Pb). Согласно другому варианту осуществления «F» не содержит свинец (Pb). Согласно одному варианту осуществления композиций, описанных в настоящем документе, h составляет от 0,01 до 5. Согласно одному варианту осуществления композиций, описанных в настоящем документе, «m» равняется 12.
При использовании в настоящем документе «каталитическая композиция» и «катализатор» являются синонимами и используются взаимозаменяемо. При использовании в настоящем документе «редкоземельный элемент» означает по меньшей мере один из лантана, церия, празеодима, неодима, прометия, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, скандия и иттрия (хотя церий является редкоземельным элементом, он исключен из данного списка, поскольку церий является отдельно указанным компонентом катализатора, описанного в настоящем документе).
Катализатор настоящего изобретения можно использовать или на носителе, или без носителя (т.е. катализатор может содержать носитель). Подходящие носители представляют собой диоксид кремния, оксид алюминия, цирконий, диоксид титана или их смеси. Носитель обычно служит в качестве связующего для катализатора и дает более сильный (т.е. более устойчивый к истиранию) катализатор. Однако, для промышленных применений подходящая смесь как активной фазы (т.е. комплекса каталитических оксидов, описанного выше), так и носителя является важной для получения приемлемой активности и твердости (устойчивости к истиранию) для катализатора. Обычно носитель составляет от 40 до 60 масс. % катализатора на носителе. Согласно одному варианту осуществления настоящего изобретения носитель может составлять всего лишь приблизительно 30 масс. % катализатора на носителе. Согласно другому варианту осуществления настоящего изобретения носитель может составлять вплоть до приблизительно 70 масс. % катализатора на носителе.
Согласно одному варианту осуществления носитель катализатора представляет собой золь диоксида кремния. Обычно золи диоксида кремния содержат некоторое количество натрия. Согласно одному варианту осуществления золь диоксида кремния содержит менее 600 частей на миллион натрия. Согласно другому варианту осуществления золь диоксида кремния содержит менее 200 частей на миллион натрия. Обычно средний диаметр коллоидных частиц золя диоксида кремния составляет от приблизительно 15 нм до приблизительно 50 нм. Согласно одному варианту осуществления настоящего изобретения средний диаметр коллоидных частиц золя диоксида кремния составляет приблизительно 10 нм и может быть всего лишь приблизительно 4 нм. Согласно другому варианту осуществления настоящего изобретения средний диаметр коллоидных частиц золя диоксида кремния составляет приблизительно 100 нм. Согласно другому варианту осуществления настоящего изобретения средний диаметр коллоидных частиц золя диоксида кремния составляет приблизительно 20 нм. Согласно другому варианту осуществления настоящего изобретения средний диаметр коллоидных частиц золя диоксида кремния составляет приблизительно 40 нм.
Получение катализатора
Катализатор можно получать любым из ряда способов получения катализаторов, которые известны специалистам в данной области техники. Обычный способ получения будет начинаться с получения смеси воды, соединения-источника молибдена и материала носителя (например, золя диоксида кремния). Отдельно соединения-источники остальных элементов в катализаторе объединяют в воде с получением второй смеси. Эти две смеси затем объединяют при перемешивании при незначительно повышенной температуре (приблизительно 40°С) с получением суспензии предшественника катализатора. Суспензию предшественника катализатора затем сушат и денитрифицируют, а затем прокаливают, как описано ниже.
Согласно одному варианту осуществления элементы в определенной выше каталитической композиции объединяют вместе в водной суспензии предшественника катализатора, полученную таким образом водную суспензию предшественника сушат с получением предшественника катализатора и предшественник катализатор прокаливают с получением катализатора.
Согласно другому варианту осуществления определенную выше каталитическую композицию получают путем:
(i) объединения в водном растворе соединений-источников висмута и церия и необязательно одного или нескольких из натрия, калия, рубидия, цезия, кальция, лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, свинца и вольфрама с получением смеси (т.е. первой смеси),
(ii) добавления соединения-источника молибдена в смесь (т.е. первую смесь) для реакции со смесью и получения суспензии осадка и
(iii) объединения суспензии осадка с соединениями-источниками остальных элементов и остального молибдена в катализаторе с получением водной суспензии предшественника катализатора.
При использовании в настоящем документе «соединения-источники» представляют собой соединения, которые содержат и/или обеспечивают один или несколько металлов для смешанной металлоксидной каталитической композиции. При использовании в настоящем документе «остальные элементы» или «остальные элементы в катализаторе» относятся к элементам и количеству этих элементов, которые представлены «А», «D», «Е», «F» и «G» в вышеуказанной формуле, которые не были включены в первую смесь. Согласно одному варианту осуществления некоторые элементы могут быть частью как первой, так и второй смеси. Кроме того, при использовании в настоящем документе «остальной молибден» или «остальной молибден в катализаторе» относится к тому количеству молибдена, которое требуется в готовом катализаторе, которое не присутствовало (т.е. не включено при получении) в суспензии осадка. Наконец, сумма количеств молибдена, обеспечиваемых в соединениях-источниках молибдена, добавленных на (ii) и (iii), равно общему количеству молибдена, находящемуся в катализаторе.
При вышеуказанном получении катализатора соединения-источники остальных элементов и остального молибдена, которые объединяют с суспензией осадка, можно объединять в любом порядке или с любой комбинацией таких остальных элементов и остального молибдена. Согласно одному варианту осуществления смесь соединений-источников остальных элементов и остального молибдена объединяют с суспензией осадка с получением водной суспензии предшественника катализатора. Согласно другому варианту осуществления (i) смесь соединений-источников остальных элементов объединяют с суспензией осадка и (ii) соединения-источники остального молибдена отдельно добавляют в суспензию осадка с получением водной суспензии предшественника катализатора. Согласно другому варианту осуществления соединения-источники остальных элементов и остального молибдена добавляют отдельно (т.е. по одному за раз) в суспензию осадка. Согласно другому варианту осуществления множество (т.е. более одной) смесей соединений-источников остальных элементов и остального молибдена, где каждая смесь содержит одно или несколько соединений-источников остальных элементов или остального молибдена, отдельно добавляют (т.е. одну смесь за раз или множество смесей, добавляемых одновременно) в суспензию осадка с получением водной суспензии предшественника катализатора. Согласно еще одному варианту осуществления смесь соединений-источников остальных элементов объединяют с соединением-источником молибдена и полученную смесь затем добавляют в суспензию осадка с получением суспензии предшественника катализатора. Согласно еще одному варианту осуществления носитель представляет собой диоксид кремния (SiO2), и диоксид кремния объединяют с соединением-источником остального молибдена перед объединением остального молибдена с суспензией осадка (т.е. диоксид кремния и соединение-источник для остального молибдена объединяют с получением смеси, а затем эту смесь добавляют в суспензию осадка, отдельно или в комбинации с одним или несколькими соединениями-источниками остальных элементов).
При вышеуказанном получении катализатора молибден добавляют как при получении суспензии осадка, так и при получении водной суспензии предшественника катализатора. На атомном уровне минимальное количество молибдена, добавленное для получения суспензии осадка, определяется следующим соотношением
Mo=1,5(Bi+Ce)+0,5(Rb+Na+K+Cs)+(Са)+1,5 (сумма числа атомов лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, лютеция, скандия и иттрия) + (Pb) - (W).
Причем в вышеуказанном соотношении «Мо» представляет собой число атомов молибдена, которые необходимо добавить в первую смесь, a «Bi», «Се», «Rb», «Na», «К», «Cs», «Са», «Pb» и «W» представляют собой число атомов висмута, церия, рубидия, натрия, калия, цезия, кальция, свинца и вольфрама, соответственно, находящиеся в первой смеси.
При вышеуказанном получении катализатора обычно количество молибдена, добавленное в первую смесь для получения суспензии осадка, составляет приблизительно 20-35% всего молибдена в готовом катализаторе. Согласно одному варианту осуществления соединение-источник остального молибдена, присутствующего в катализаторе, добавляют в смесь соединения-источника остальных элементов (т.е. вторую смесь) перед объединением смеси остальных элементов с суспензией осадка с получением суспензии предшественника катализатора. Согласно другим вариантам осуществления соединение-источник молибден, содержащий остальной молибден, присутствующий в катализаторе, добавляют в суспензию осадка или перед, или после, или одновременно со смесью соединений-источников остальных элементов (т.е. второй смесью) для получения суспензии предшественника катализатора.
При вышеуказанном получении соединения-источники Bi и Се и необязательно одного или нескольких из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb и W объединяют в водном растворе с получением смеси. Согласно одному варианту осуществления нитрат висмута и необязательно нитраты других металлов (т.е. нитраты Na, К, Rb, Cs, Са, редкоземельного элемента и/или Pb) растворяют в водном растворе цериевого нитрата аммония. Если добавляют вольфрам, соединение-источник обычно представляет собой паравольфрамат аммония, (NH4)10H2(W2O7)6.
Добавленным в смесь, содержащую висмут и церий (и необязательно один или несколько из Na, К, Rb, Cs, Са, редкоземельный элемент, Pb и/или W), является соединение-источник молибдена. Согласно одному варианту осуществления это соединение-источник молибдена представляет собой гептамолибдат аммония, растворенный в воде. При добавлении соединения-источника молибдена в смесь, содержащую висмут и церий, будет происходить реакция, которая будет давать осадок, и полученная смесь представляет собой суспензию осадка.
Суспензию осадка затем объединяют со смесью соединения-источника остальных элементов катализатора и соединением-источником молибдена с получением водной суспензии предшественника катализатора. Смесь соединений-источников остальных элементов и соединения-источника молибдена можно получать объединением соединений-источников остальных элементов в водном растворе (например, соединения-источники объединяют в воде), а затем добавления соединения-источника молибдена. Согласно одному варианту осуществления это соединение-источник молибдена представляет собой гептамолибдат аммония, растворенный в воде. При объединении суспензии осадка со смесью остальных элементов/молибдена порядок добавления не важен, т.е. суспензию осадка можно добавлять в смесь остальных элементов/молибдена или смесь остальных элементов/молибдена можно добавлять в суспензию осадка. Водную суспензию предшественника катализатора поддерживают при повышенной температуре.
Количество водного растворителя в каждой из вышеописанных водных смесей и суспензий может изменяться из-за растворимостей соединений-источников, объединенных с получением конкретного смешанного оксида металлов. Количество водного растворителя должно быть, по меньшей мере, достаточным для получения суспензии или смеси твердых веществ или жидкостей, которую можно перемешивать.
В любом случае соединения-источники предпочтительно объединяют и/или приводят в реакцию при помощи протокола, который включает смешивание соединений-источников на стадии объединения и/или реакции. Конкретный механизм смешивания не важен и может включать, например, смешивание (например, перемешивание или встряхивание) компонентов при реакции любым эффективным способом. Такие способы включают, например, встряхивание содержимого емкости, например, при помощи вибрации, опрокидывания или качания содержащей компоненты емкости. Такие способы также включают, например, перемешивание при помощи перемешивающего элемента, расположенного, по меньшей мере, частично в реакционной емкости, и движущей силы, соединенной с перемешивающим элементом или с реакционной емкостью для получения взаимного перемещения между перемешивающим элементом и реакционной емкостью. Перемешивающий элемент может быть приводным и/или опирающимся на вал перемешивающим элементом. Движущая сила может быть непосредственно соединена с перемешивающим элементом или может быть косвенно соединена с перемешивающим элементом (например, посредством магнитного взаимодействия). Смешивание в общем предпочтительно является достаточным для обеспечения смешивания компонентов для достаточной реакции между компонентами реакционной среды с получением более однородной реакционной среды (например, и получения более однородного предшественника смешанного оксида металлов) по сравнению с неперемешанной реакцией. Это дает более эффективный расход исходных материалов и более однородный продукционный смешанный оксид металлов. Смешивание суспензии осадка на стадии реакции также вызывает образование осадка в растворе, а не на стенках реакционной емкости. Более предпочтительно образование осадка в растворе обеспечивает рост частиц на всех поверхностях частицы, а не ограниченных наружных сторонах, когда рост происходит от стенки реакционной емкости.
Соединение-источник молибдена может содержать оксид молибдена (VI) (МоО3), гептамолибдат аммония или молибденовую кислоту. Соединение-источник молибдена можно вводить из любого оксида молибдена, такого как диоксид, триоксид, пентоксид или гептаоксид. Однако, предпочтительно использовать гидролизуемую или разлагаемую соль молибдена в качестве соединения-источника молибдена.
Обычные соединения-источники для висмута, церия и остальных элементов катализатора представляют собой нитратные соли металлов. Такие нитратные соли легко доступны и легко растворимы.
Соединение-источник висмута может содержать оксид или соль, которая при прокаливании будет давать оксид. Предпочтительны водорастворимые соли, которые легко диспергируются, но образуют стабильные оксиды при тепловой обработке. Согласно одному варианту осуществления соединение-источник висмута представляет собой нитрат висмута, Bi(NO3)3⋅5H2O.
Соединение-источник церия может содержать оксид или соль, которая при прокаливании будет давать оксид. Предпочтительны водорастворимые соли, которые легко диспергируются, но образуют стабильные оксиды при тепловой обработке. Согласно одному варианту осуществления соединение-источник церия представляет собой цериевый нитрат аммония, (NH4)2Ce(NO3)6.
Соединение-источник железа можно получать из любого соединения железа, которое при прокаливании будет давать оксид. Как и с другими элементами, водорастворимые соли предпочтительны для простоты, с которой их можно равномерно диспергировать в катализаторе. Наиболее предпочтительным является нитрат железа.
Соединения-источники для остальных элементов можно получать из любого подходящего источника. Например, кобальт, никель и магний можно вводить в катализатор при помощи нитратных солей. Кроме того, магний можно вводить в катализатор в виде нерастворимого карбоната или гидроксида, который при тепловой обработке дает оксид. Фосфор можно вводить в катализатор в виде соли щелочного металла, или соли щелочноземельного металла, или аммониевой соли, но предпочтительно вводят в виде фосфорной кислоты.
Соединения-источники для щелочных компонентов катализатора можно вводить в катализатор в виде оксида или в виде соли, которая при прокаливании будет давать оксид.
Растворители, помимо воды, которые можно использовать для получения смешанных оксидов металлов согласно настоящему изобретению, включают, помимо прочего, спирты, такие как метанол, этанол, пропанол, диолы (например, этиленгликоль, пропиленгликоль и пр.), органические кислоты, такие как уксусная кислота, а также другие полярные растворители, известные в данной области техники. Соединения-источники металлов, по меньшей мере, частично растворимы в растворителе.
Как указано выше, катализатор настоящего изобретения можно использовать или на носителе, или без носителя (т.е. катализатор может содержать носитель). Подходящие носители представляют собой диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана или их смеси. Носитель можно добавлять в любое время перед сушкой суспензии предшественника катализатора. Носитель можно добавлять в любое время при получении любой смеси элементов, суспензии осадка или суспензии предшественника катализатора или после него. Кроме того, носитель не обязательно добавлять в один момент или на одной стадии (т.е. носитель можно добавлять во множестве моментов при получении). Согласно одному варианту осуществления носитель объединяют с другими ингредиентами при получении водной суспензии предшественника катализатора. Согласно одному варианту осуществления носитель добавляют в суспензию осадка (т.е. после получения суспензии осадка). Согласно одному варианту осуществления носитель объединяют с соединением-источником молибдена перед объединением соединения-источника молибдена с соединениями-источниками остальных элементов в катализаторе с получением «второй смеси», указанной выше.
Суспензию предшественника катализатора сушат и денитрифицируют (т.е. удаляют нитраты) с получением предшественника катализатора. Согласно одному варианту осуществления суспензию предшественника катализатора сушат с получением частиц катализатора. Согласно одному варианту осуществления суспензию предшественника катализатора сушат распылением в микросферические частицы катализатора. Согласно одному варианту осуществления температура на выходе распылительной сушилки составляет от 110°С до 350°С, предпочтительно от 110°С до 250°С, наиболее предпочтительно от 110°С до 180°С.Согласно одному варианту осуществления распылительная сушилка представляет собой распылительную сушилку с параллельными потоками (т.е. частицы распыляются параллельно потоку газа). Согласно другому варианту осуществления распылительная сушилка представляет собой противоточную распылительную сушилку (т.е. частицы распыляются противотоком потоку газа). Согласно другому варианту осуществления распылительная сушилка представляет собой распылительную сушилку с соплами высокого давления. В таких процессах сушки распылением содержащие воду твердые частицы распыляют в контакте с горячим газом (обычно воздухом) так, чтобы испарить воду. Сушку регулируют температурой газа и расстоянием, которое проходят частицы в контакте с газом. Обычно нежелательно регулировать эти параметры для получения слишком быстрой сушки, поскольку это приводит к тенденции образования сухих оболочек на частично высушенных частицах твердой фазы, которые затем растрескиваются, когда вода, поглощенная в частицах, испаряется и стремится улетучиваться. Кроме того, желательно обеспечивать катализатор в форме с насколько возможно небольшим количеством поглощенной воды. Таким образом, если следует использовать реактор с псевдоожиженным слоем, и желательны микросферические частицы, целесообразно выбирать условия сушки распылением с учетом достижения полной сушки без растрескивания частиц. Высушенный материал катализатора затем нагревают для удаления любых оставшихся нитратов. Температура денитрификации может находиться в диапазоне от 100°С до 500°С, предпочтительно от 250°С до 450°С.
Наконец, высушенные и денитрифицированные предшественники катализатора прокаливают для получения готового катализатора. Согласно одному варианту осуществления прокаливание проводят в воздухе. Согласно другому варианту осуществления прокаливание проводят в инертной атмосфере. Согласно одному варианту осуществления предшественник катализатора прокаливают в азоте. Условия прокаливания включают температуры в диапазоне от приблизительно 300°С до приблизительно 700°С, более предпочтительно от приблизительно 350°С до приблизительно 650°С, и согласно некоторым вариантам осуществления прокаливание может происходить при приблизительно 600°С.Согласно одному варианту осуществления прокаливание можно осуществлять на множестве стадий с повышающимися температурами. Согласно одному варианту осуществления первую стадию прокаливания проводят при температуре в диапазоне от приблизительно 300°С до приблизительно 450°С с последующей второй стадией прокаливания, проводимой при температуре в диапазоне от приблизительно 500°С до приблизительно 650°С.
Процесс аммоксидирования
Катализаторы настоящего изобретения пригодны в процессах аммоксидирования для конверсии олефина, выбранного из группы, состоящей из пропилена, изобутилена или их смесей, в акрилонитрил, метакрилонитрил и их смеси, соответственно, путем реакции в паровой фазе при повышенной температуре и давлении указанного олефина с содержащим молекулярный кислород газом и аммиаком в присутствии катализатора. Катализаторы настоящего изобретения также пригодны для аммоксидирования метанола в цианистый водород и аммоксидирования этанола в ацетонитрил. Согласно одному варианту осуществления, используя катализаторы, описанные в настоящем документе, метанол и/или этанол можно совместно подавать в процесс для аммоксидирования пропилена, изобутилена или их смесей в акрилонитрил, метакрилонитрил или их смеси для повышения выхода побочных продуктов - цианистого водорода и/или ацетонитрила, получаемых в таком процессе.
Предпочтительно реакцию аммоксидирования проводят в реакторе с псевдоожиженным слоем, хотя предусматриваются другие типы реакторов, такие как реакторы конвейерного типа. Реакторы с псевдоожиженным слоем для получения акрилонитрила хорошо известны в предшествующем уровне техники. Например, является подходящей конструкция реактора, указанная в патенте США №3230246, включенная в настоящий документ ссылкой.
Условия для проведения реакции аммоксидирования также хорошо известны в предшествующем уровне техники, что показано в патентах США №5093299; №4863891; №4767878 и №4503001; включенных в настоящий документ ссылкой. Обычно процесс аммоксидирования проводят путем контакта пропилена или изобутилена в присутствии аммиака и кислорода с псевдоожиженым слоем катализатора при повышенной температуре с получением акрилонитрила или метакрилонитрила. Можно использовать любой источник кислорода. По экономическим причинам, однако, предпочтительно использовать воздух. Обычное мольное отношение кислорода к олефину в сырье будет находиться в диапазоне от 0,5:1 до 4:1, предпочтительно от 1:1 до 3:1.
Мольное соотношение аммиака к олефину в сырье при реакции может изменяться от 0,5:1 до 2:1. В действительности нет верхнего предела для соотношения аммиак-олефин, но обычно нет причины превышать соотношение 2:1 по экономическим соображениям. Подходящие соотношения в сырье для использования с катализатором настоящего изобретения для получения акрилонитрила из пропилена представляют собой соотношение аммиака к пропилену в диапазоне от 0,9:1 до 1,3:1, а соотношение воздуха к пропилену от 8,0:1 до 12,0:1. Катализатор настоящего изобретения может давать высокие выходы акрилонитрила при относительно низких соотношениях аммиака к пропилену в сырье от приблизительно 1:1 до приблизительно 1,05:1. Эти «условия низкого содержания аммиака» способствуют снижению количества непрореагировавшего аммиака в выходящем потоке реактора, состояния, известного как «проскок аммиака», что следовательно способствует снижению производственных отходов. В частности, непрореагировавший аммиак следует удалять из выходящего потока реактора перед извлечением акрилонитрила. Непрореагировавший аммиак обычно удаляют путем контакта выходящего потока реактора с серной кислотой с получением сульфата аммония или путем контакта выходящего потока реактора с акриловой кислотой с получением акрилата аммония, что в обоих случаях дает поток производственных отходов, который необходимо обрабатывать и/или утилизировать.
Реакцию проводят при температуре в диапазоне от приблизительно 260°С до 600°С, предпочтительно в диапазонах от 310°С до 500°С, в частности предпочтительно от 350°С до 480°С. Время контакта, хотя и не критично, обычно находится в диапазоне от 0,1 до 50 секунд, причем предпочтение отдают времени контакта от 1 до 15 секунд.
Продукты реакции можно извлекать и очищать любым из способов, известных специалистам в данной области техники. Один такой способ предусматривает промывку выходящих газов реактора при помощи холодной воды или подходящего растворителя для удаления продуктов реакции, а затем очистку продукта реакции дистилляцией.
Основная польза катализатора, полученного способом настоящего изобретения, заключается в аммоксидировании пропилена в акрилонитрил. Другая польза включает аммоксидирование пропана в акрилонитрил и аммоксидирование глицерина в акрилонитрил. Катализатор, полученный способом настоящего изобретения, можно также использовать для окисления пропилена в акролеин и/или акриловую кислоту. Такие способы представляют собой обычно двухстадийные способы, причем пропилен превращается в присутствии катализатора главным образом в акролеин на первой стадии, а акролеин превращается в присутствии катализатора главным образом в акриловую кислоту на второй стадии. Катализатор, описанный в настоящем документе, подходит для применения на первой стадии для окисления пропилена в акролеин.
Конкретные варианты осуществления
Для иллюстрации настоящего изобретения катализатор, полученный согласно настоящему изобретению, оценивали и сравнивали при аналогичных условиях реакции с аналогичными катализаторами, полученными способами уровня техники вне объема настоящего изобретения. Эти примеры представлены только с целями иллюстрации. Каталитические композиции для каждого примера являются такими, как показано после номера примера. Все каталитические препараты получали с 39±2 нм золя диоксида кремния. Сравнительные примеры обозначены как таковые, а также обозначены при помощи «С» в таблицах.
Сравнительный пример 1: Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,72Ce1,76Mo12,502Ox+50 масс. % SiO2 (51,3 части на миллион Na, 38,1 нм)
Реакционную смесь А готовили нагреванием 10309 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (9372 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 1829 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (2222 г), Ni(NO3)2⋅6H2O (7108 г), Mg(NO3)2⋅6H2O (4701 г) и Cr(NO3)3⋅9H2O (122,3 г).
Реакционную смесь С готовили нагреванием 2264 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (2059 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 5896 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (1067 г) и RbNO3 (86,5 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (41588 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали путем тепловой обработки в воздухе во вращающейся печи при 343°С в течение 50 минут с последующим прокаливанием в воздухе во вращающейся печи при 570°С в течение 50 минут.
Сравнительный пример 2: K0,2Ni4Mg3Fe0,9Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,4 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (143,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,8 г), Ni(NO3)2⋅6H2O (101,7 г), Mg(NO3)2⋅6H2O (67,2 г) и Cr(NO3)3⋅9H2O (1,75 г).
Реакционную смесь С готовили нагреванием 64,8 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,9 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,7 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (30,5 г) и KNO3 (1,77 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 580°С.
Сравнительный пример 3: Cs0,072K0,12Ni4Mg3Fe0,9Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,7 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,2 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,2 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 64,8 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,9 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), CsNO3 (1,21 г) и KNO3 (1,05 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 4: Cs0,1K0,1Ni4Mg3Fe0,9Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 156,9 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (142,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,7 г), Ni(NO3)2⋅6H2O (101,3 г), Mg(NO3)2⋅6H2O (67,0 г) и Cr(NO3)3⋅9H2O (1,74 г).
Реакционную смесь С готовили нагреванием 64,6 мл деионизированной воды до 65°С а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,75 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,1 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (30,4 г), CsNO3 (1,70 г) и KNO3 (0,88 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 5: K0,1Ni4Mg3Na0,1Fe0,9Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 143,2 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (157,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,8 г), Ni(NO3)2⋅6H2O (101,7 г), Mg(NO3)2⋅6H2O (67,3 г) и Cr(NO3)3⋅9H2O (1,75 г).
Реакционную смесь С готовили нагреванием 64,9 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (59,0 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,8 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательнее добавляя Bi(NO3)3⋅5H2O (30,5 г), NaNO3 (0,74 г) и KNO3 (0,88 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 6: Cs0,1Ni4Mg3Na0,1Fe0,9Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,0 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (142,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,7 г), Ni(NO3)2⋅6H2O (101,4 г), Mg(NO3)2⋅6H2O (67,0 г) и Cr(NO3)3⋅9H2O (1,74 г).
Реакционную смесь С готовили нагреванием 64,7 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,8 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,2 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательнее добавляя Bi(NO3)3⋅5H2O (30,4 г), NaNO3 (0,74 г) и CsNO3 (1,70 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С.Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 7: Ni4Mg3Na0,2Fe0,9Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,6 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (143,3 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,8 г), Ni(NO3)2⋅6H2O (101,8 г), Mg(NO3)2⋅6H2O (67,3 г) и Cr(NO3)3⋅9H2O (1,75 г).
Реакционную смесь С готовили нагреванием 64,9 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (59,0 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,85 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательнео добавляя Bi(NO3)3⋅5H2O (30,6 г) и NaNO3 (1,49 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 620°С.
Сравнительный пример 8: K0,2Ni4Mg3Fe0,9Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,4 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (143,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,8 г), Ni(NO3)2⋅6H2O (101,7 г), Mg(NO3)2⋅6H2O (67,2 г) и Cr(NO3)3⋅9H2O (1,75 г).
Реакционную смесь С готовили нагреванием 64,8 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,9 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,7 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательнео добавляя Bi(NO3)3⋅5H2O (30,5 г) и KNO3 (0,74 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 9: Cs0,144Ni4Mg3Fe0,9Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,8 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,0 г), Mg(NO3)2⋅6H2O (66,2 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 63,3 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (57,6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 116,9 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,7 г) и CsNO3 (2,41 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 10: Cs0,072K0,12Ni4Mg3Fe0,9Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,7 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,3 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 63,8 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,0 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), KNO3 (1,05 г) и CsNO3 (1,21 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 11: K0,24Ni4Mg3Fe0,9Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,5 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,4 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,4 г), Ni(NO3)2⋅6H2O (100,4 г), Mg(NO3)2⋅6H2O (66,4 г) и Cr(NO3)3⋅9H2O (1,73 г).
Реакционную смесь С готовили нагреванием 64,3 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,9 г) и KNO3 (2,09 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 12: Cs0,1K0,1Li0,3Fe0,9Ni4,2Co4,2Bi0,62Ce0,8Cr0,12Mo13,174Ox+50 масс. % SiO2 (39 нм)
Реакционную смесь А готовили нагреванием 242 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдатаа аммония (200,2 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 25,0 мл деионизированной воды до 55°С, а затем последовательным добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (105,1 г), Co(NO3)2⋅6H2O (105,2 г), Cr(NO3)3⋅9H2O (4,13 г), Bi(NO3)3⋅5H2O (25,9 г), LiNO3 (1,78 г), CsNO3 (1,68 г) и KNO3 (0,87 г).
Реакционную смесь С готовили нагреванием 75,5 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С.
Реакционную смесь D готовили добавлением реакционной смеси С в реакционную смесь В.
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А.
Реакционную смесь F готовили добавлением реакционной смеси D в реакционную смесь Е, что давало осаждение твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 60 минут, в течение которых температуре позволяли снизиться до 40°С с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора затем гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 13: Ni4Mg3La0,76Fe1,4Rb0,192Cr0,05Bi0,72Ce1Mo12,791Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 152,5 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (138,6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 31,3 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (49,5 г), Ni(NO3)2⋅6H2O (101,7 г), Mg(NO3)2⋅6H2O (67,3 г) и Cr(NO3)3⋅9H2O (1,75 г).
Реакционную смесь С готовили нагреванием 64,8 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,9 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 95,9 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (30,5 г), La(NO3)3⋅6H2O (28,8 г) и RbNO3 (2,48 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С.
Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 14: Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,85Ce0,35Mo10,496Ox+50 масс. %, 27 части на миллион Na, 39 нм SiO2
Реакционную смесь А готовили нагреванием 184 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (166,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 34 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (39,9 г), Ni(NO3)2⋅6H2O (127,6 г), Mg(NO3)2⋅6H2O (84,4 г) и Cr(NO3)3⋅9H2O (2,19 г).
Реакционную смесь С готовили нагреванием 41 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (36,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 42,1 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (45,2 г) и RbNO3 (3,11 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С.Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 15: Ni6,5Mg2,6Fe1,35Rb0,1Bi0,35Ce0,23Mo12,2Ox+50 масс. % SiO2 (27 частей на миллион Na, 39 нм)
Реакционную смесь А готовили нагреванием 212,5 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (192,2 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 35,6 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9Н2О (52,9 г), Ni(NO3)2⋅6H2O (183,1 г), Mg(NO3)2⋅6H2O (64,6 г) и Cr(NO3)3⋅9H2O (1,94 г).
Реакционную смесь С готовили нагреванием 17,3 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (15,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 24,4 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (16,5 г) и RbNO3 (1,43 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 16: Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,28Ce0,683Mo10,307Ox+50 масс. % SiO2 (27 частей на миллион Na, 39 нм)
Реакционную смесь А готовили нагреванием 196 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (178,0 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 32 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (41,8 г), Ni(NO3)2⋅6H2O (133,8 г), Mg(NO3)2⋅6H2O (88,5 г) и Cr(NO3)3⋅9H2O (2,30 г).
Реакционную смесь С готовили нагреванием 34 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (31,3 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 86,1 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (15,6 г) и RbNO3 (3,26 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси D в реакционную смесь С, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 17: Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,200Ce0,488Mo9,797Ox+50 масс. % SiO2 (27 частей на миллион Na, 39 нм)
Реакционную смесь А готовили нагреванием 206 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (186,6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 33,5 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (44,4 г), Ni(NO3)2⋅6H2O (141,9 г), Mg(NO3)2⋅6H2O (93,8 г) и Cr(NO3)3⋅9H2O (2,24 г).
Реакционную смесь С готовили нагреванием 27 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (24,3 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 65,3 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (11,8 г) и RbNO3 (3,45 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси D в реакционную смесь С, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 18: Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,150Ce0,366Mo9,447Ox+50 масс. % SiO2 (27 частей на миллион Na, 39 нм)
Реакционную смесь А готовили нагреванием 212 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (192,6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 35 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (46,1 г), Ni(NO3)2⋅6H2O (147,4 г), Mg(NO3)2⋅6H2O (97,5 г) и Cr(NO3)3⋅9H2O (2,54 г).
Реакционную смесь С готовили нагреванием 22 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (19,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 50,9 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, 4 последовательно добавляя Bi(NO3)3⋅5H2O (9,22 г) и RbNO3 (3,59 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси D в реакционную смесь С, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Сравнительный пример 19: Ni4Mg3Fe0,9Rb0,22Cr0,05Bi0,72Ce1,76Mo12,501Ox+50 масс. % SiO2 (31 частей на миллион Na, 39 нм)
Реакционную смесь А готовили нагреванием 151 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (137,4 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 27 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (32,6 г), Ni(NO3)2⋅6H2O (104,4 г), Mg(NO3)2⋅6H2O (69,0 г) и Cr(NO3)з⋅9H2O (1,79 г).
Реакционную смесь С готовили нагреванием 66,8 мл деионизированной воды до 55°С, а затем добавлением при перемешивании гептамолибдата аммония (60,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 173,2 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (31,3 г) и RbNO3 (2,91 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением по каплям реакционной смеси D в реакционную смесь С, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 1: Cs0,024K0,16Ni4Mg3Fe0,9Rb0,032Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,6 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,4 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,3 г), Mg(NO3)2⋅6H2O (66,3 г) и Cr(NO3)3⋅9H2O (1,725 г).
Реакционную смесь С готовили нагреванием 64,1 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,3 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,2 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,85 г), KNO3 (1,39 г) и CsNO3 (0,40 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С.
Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С.Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 2: Cs0,072Ni4Mg3Fe0,9Rb0,096Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,7 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,1 г), Mg(NO3)2⋅6H2O (66,2 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 63,6 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (57,8 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,0 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), RbNO3 (1,22 г) и CsNO3 (1,21 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С.
Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 3: K0,1Ni4Mg3Na0,05Fe0,9Rb0,1Cr0,05Bi1,24Ce1,24Mo13,12Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,2 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,3 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 64,3 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), RbNO3 (1,27 г), KNO3 (0,87 г) и NaNO3 (0,37 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 4: K0,1Ni4Mg3Na0,05Fe0,9Rb0,1Cr0,05Bi1,24Ce1,24Mo13,12Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,2 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,3 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 64,3 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), RbNO3 (1,27 г), KNO3 (0,87 г) и NaNO3 (0,37 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 5: K0,12Ni4Mg3Fe0,9Rb0,096Cr0,05Bi1,24Ce1,24Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,5 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,4 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,3 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 64,1 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,2 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,2 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), RbNO3 (1,22 г) и KNO3 (1,05 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С.
Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 6: K0,1Ni4Mg3Fe0,9Rb0,1Cr0,05Bi0,72Ce1,76Mo13,095Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,2 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (142,9 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9Н2О (31,7 г), Ni(NO3)2⋅6H2O (101,5 г), Mg(NO3)2⋅6H2O (67,1 г) и Cr(NO3)3⋅9H2O (1,75 г).
Реакционную смесь С готовили нагреванием 64,7 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,85 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,4 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (30,5 г), RbNO3 (1,29 г) и KNO3 (0,88 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 7: Cs0,072Ni4Mg3Fe0,9Rb0,096Cr0,05Bi0,72Ce1,76Mo13,121Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,4 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (143,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,0 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,6 г), Ni(NO3)2⋅6H2O (101,2 г), Mg(NO3)2⋅6H2O (66,9 г) и Cr(NO3)3⋅9H2O (1,74 г).
Реакционную смесь С готовили нагреванием 64,3 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,4 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 167,9 г 50 масс. % водного раствора (NH4)2Ce(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (30,4 г), RbNO3 (1,23 г) и CsNO3 (1,22 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 8: Cs0,025K0,025Ni4Mg3Na0,03Fe0,9Rb0,125Cr0,05Bi0,72Ce1,76Мо13,095Ох+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 157,0 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (142,7 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 26,1 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,7 г), Ni(NO3)2⋅6H2O (101,4 г), Mg(NO3)2⋅6H2O (67,0 г) и Cr(NO3)3⋅9H2O (1,74 г).
Реакционную смесь С готовили нагреванием 64,7 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,8 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 168,2 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (30,4 г), RbNO3 (1,61 г), KNO3 (0,22 г), NaNO3 (0,22 г) и CsNO3 (0,42 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 9: Cs0,025Ni4Mg3Na0,05Fe0,9Rb0,12Cr0,05Bi1,24Ce1,24Mo13,12Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,6 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,25 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 63,4 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), RbNO3 (1,52 г), KNO3 (0,22 г), NaNO3 (0,37 г) и CsNO3 (0,42 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С.Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С. Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Пример 10: Ni4Mg3Na0,05Fe0,9Rb0,15Cr0,05Bi1,24Ce1,24Mo13,12Ox+50 масс. % SiO2 (31 часть на миллион Na, 38,2 нм)
Реакционную смесь А готовили нагреванием 155,6 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (141,5 г) с получением прозрачного бесцветного раствора.
Реакционную смесь В готовили нагреванием 28,2 мл деионизированной воды до 55°С, а затем добавлением при перемешивании Fe(NO3)3⋅9H2O (31,3 г), Ni(NO3)2⋅6H2O (100,2 г), Mg(NO3)2⋅6H2O (66,27 г) и Cr(NO3)3⋅9H2O (1,72 г).
Реакционную смесь С готовили нагреванием 63,9 мл деионизированной воды до 65°С, а затем добавлением при перемешивании более 30 минут гептамолибдата аммония (58,1 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили (i) нагреванием 117,1 г 50 масс. % водного раствора (NH4)2Се(NO3)6 до 55°С, (ii) в то же время раствор перемешивали и нагревали, последовательно добавляя Bi(NO3)3⋅5H2O (51,8 г), RbNO3 (1,91 г) и NaNO3 (0,37 г).
Реакционную смесь Е готовили добавлением при перемешивании золя диоксида кремния (610 г, 41 масс. % диоксида кремния) в реакционную смесь А с последующим добавлением реакционной смеси В.
Реакционную смесь F готовили добавлением реакционной смеси С в реакционную смесь D, что давало осаждение оранжевого твердого вещества (эта полученная смесь представляла собой суспензию осадка). Перемешивание суспензии осадка продолжали в течение 15 минут, в то же время температуру поддерживали в диапазоне 50-55°С. Реакционную смесь F затем добавляли в реакционную смесь Е с получением готовой суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение одного часа, в то же время ее охлаждали до приблизительно 40°С.Затем ее гомогенизировали в смесителе в течение 3 минут при 5000 об/мин. Суспензию затем сушили распылением в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали тепловой обработкой в течение 3 часов в воздухе при 290°С с последующими дополнительными 3 часами при 425°С. Порошок затем прокаливали в воздухе в течение 3 часов при 560°С.
Тестирование катализатора
Весь катализатор тестировали в лабораторном реакторе для аммоксидирования пропилена в акрилонитрил. Все тестирование проводили в 40 см3 реакторе с псевдоожиженным слоем. Пропилен подавали в реактор со скоростями, показанными в таблице X, от 0,06 до 0,09 WWH (т.е. масса пропилена/масса катализатора/час). Давление внутри реактора поддерживали на 10 фунтах/кв. дюйм. Температура реакции составляла 430°С. Образцы продуктов реакции собирали через несколько дней тестирования (от приблизительно 140 до приблизительно 190 часов работы). Выходящий поток реактора собирали в пузырьковых скрубберах, содержащих холодный раствор HCl. Скорость отходящего газа измеряли при помощи пленочного расходомера, а состав отходящего газа определяли в конце цикла с помощью газового хроматографа, оборудованного газовым анализатором с раздельными колонками. В конце цикла извлечения всю жидкость скруббера разбавляли до приблизительно 200 грамм дистиллированной водой. Взвешенное количество 2-бутанона использовали в качестве внутреннего стандарта в ~50-граммовой аликвоте разбавленного раствора. 2 мкл образец анализировали в GC, оборудованном пламенно-ионизационным детектором и колонкой Carbowax™. Количество NH3 определяли титрованием избытка свободной HCl при помощи раствора NaOH.



Примечания для таблиц 1, 2 и 3 (где применимо):
1. «WWH» представляет массу пропилена на массу катализатора в час в сырье.
2. «% конверсия С3=» представляет мольные проценты однократной конверсии пропилена во все продукты.
3. «Выход AN» представляет процентный выход акрилонитрила.
4. «Селективность к AN» представляет процентную селективность к акрилонитрилу.
5. «а/h» представляет атомное отношение висмута к церию.
6. «(a+h)/d» или «(a+h)/z» представляет отношение (атомов висмута плюс атомов церия) к атомам элементов D в катализаторе (смотрите формулу в п. 1).
7. «h/b» представляет атомное отношение церия к железу.
8. Сравнительные примеры обозначены как «С» перед номером примера.
Данные в таблице 3 ясно показывают преимущество настоящего изобретения. Примеры 1-10 (т.е. примеры в объеме заявляемого изобретения) показывают больший выход акрилонитрила и/или более высокую селективность к акрилонитрилу, чем катализаторы из С1-С19 и С19-С21 (т.е. сравнительных примеров, которые находятся вне объема заявляемого изобретения).
Хотя вышеуказанное описание и вышеуказанные варианты осуществления являются обычными для осуществления на практике настоящего изобретения, очевидно, что много альтернатив, модификаций и вариантов будут очевидны специалистам в данной области техники в свете настоящего описания. Следовательно, предполагается, что все такие альтернативы, модификации и варианты охвачены и попадают в сущность и широкий объем приложенной формулы изобретения.
Улучшенные селективные катализаторы аммоксидирования
Улучшенные селективные катализаторы аммоксидирования
Контроль температуры реактора аммоксидирования
Испарительная установка для технологического потока
Улучшенная конструкция циклонов
Испарительная установка для технологического потока
Регулирование ph закалочной колонны
Управление колонной выделения
Колонна выделения
Циркуляционное орошение колонны головного погона
Способ получения смешанного оксидного катализатора для получения акрилонитрила или метакрилонитрила (варианты)
Способ приготовления смешанных металлоксидных катализаторов окислительного аммонолиза и/или окисления низших алканов
Способ получения усовершенствованных катализаторов аммоксидирования на основе смешанных оксидов металлов
Усовершенствованные катализаторы аммоксидирования на основе смешанных оксидов металлов
Высокоэффективный способ аммоксидирования и катализаторы на основе смешанных оксидов металлов
Износостойкие катализаторы аммоксидирования на основе смешанных оксидов металлов
Смешанные металлооксидные катализаторы
Добавки к катализаторам аммоксидирования на основе смесей оксидов металлов, вводимые перед кальцинированием
Улучшенные селективные катализаторы аммоксидирования
Улучшенные селективные катализаторы аммоксидирования

