АНАЛОГИ ГАММА-АМИНОМАСЛЯНОЙ КИСЛОТЫ (ГАМК) ДЛЯ ЛЕЧЕНИЯ БОЛИ И ДРУГИХ ЗАБОЛЕВАНИЙ
Вид РИД
Изобретение
Настоящее изобретение относится к новым терапевтическим агентам, в частности к производным гамма-аминомасляной кислоты, их фармацевтическим композициям, способам их получения, а также к их терапевтической активности при лечении боли.
Предшествующий уровень техники
Потенциал-зависимые кальциевые каналы формируются за счет комбинаций порообразующей субъединицы он и вспомогательных белков α2δ, β и γ (Caterall (2000) Annu Rev. Cell Dev Biol. 16: 521-555). Известно, что белок α2δ регулирует как плотность кальциевых каналов, так и потенциал-зависимую кинетику этих каналов (Felix et al (1997) J. Neuroscience 17: 6884-6891; Klugbauer et al (1999) J. Neuroscience 19: 684-691; Hobom et al (2000) Eur J. Neuroscience 12: 1217-1226, и Qin et al (2002) Mol. Pharmacol. 62: 485-496).
Габапентин (GBP) представляет собой анти-эпилептическое, анти-гипералгезическое и анксиолитическое лекарственное средство, обладающее высокой аффинностью к двум подтипам α2δ субъединицы кальциевого канала α2δ1 и α2δ2. GBP был первоначально предназначен для лечения эпилепсии и также нашел применение в лечении боли и тревожного расстройства (Taylor et al (1998) Epilepsy Res 29: 223-249). Механизм, лежащий в основе действия GBP до сих пор слабо изучен. GBP был первоначально разработан как липофильный аналог гамма-аминомасляной кислоты (ГАМК), но впоследствии было показано, что нет взаимодействия ни с каким-либо из ферментов пути метаболизма ГАМК, ни непосредственно с рецепторами ГАМКА или ГАМКВ. Тем не менее, он способен эффективно преодолевать гематоэнцефалический барьер, с помощью транспортеров аминокислот системы L.
Прегабалин (PGB) представляет собой второе поколение, более сильнодействующее, является преемником GBP для лечения тех же самых состояний, что перечислены выше. GBP (структура G, ниже) и PGB (структура P, ниже) связываются с субъединицей α2δ-1 со значениями IC50 140 нмоль/л и 80 нмоль/л соответственно (Dolphin (2013) Biochim Biophys Acta 1828: 1541-1549).
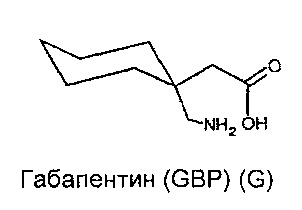
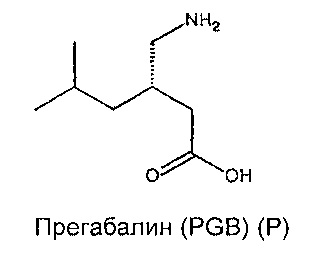
GBP показывает несколько, если таковые имеются, токсических побочных эффектов при клинически значимых дозах. Однако, он обладает сравнительно коротким периодом полураспада, и выводится из организма без изменений, возможно, из-за очень высокой растворимости в воде и очевидного отсутствия связывания белков in vivo. Мягкий седативный эффект, головокружение и атаксия являются основными дозолимитирующими побочными эффектами, и они, как полагают, центрально-опосредованы.
GBP и PGB, в отличие от многих других лекарственных средств центрального действия, являются гидрофильными и двухзарядными при нейтральном pH, что делает их нерастворимыми в липидах, таких как клеточные мембраны. Тем не менее, оба соединения по всей видимости, преодолевают мембранные барьеры кишечника, гематоэнцефалический барьер и клеточные мембраны с помощью специализированной системы транспортеров (система L), которая также транспортирует эндогенные аминокислоты, такие как L-лейцин, L-изолейцин и L-валин (Su et al (2005), J. Pharmacol. Exp. Ther. 313, 1-10).
У млекопитающих существует четыре родственных подтипа белка α2δ, кодируемые разными генами. Каждый подтип белка имеет молекулярную массу приблизительно 150 кДа и состоит из 997-1150 аминокислотных остатков. Только подтипы 1 и 2 α2δ связывают PGB с высокой аффинностью; подтипы 3 и 4, лишены значительного связывания данного лекарственного средства (Fink et al (2002) Neuropharmacology, 42, 229-236). Аффинность связывания PGB аналогична для рекомбинантного белка α2δ 1 и 2 типа, демонстрируя, что PGB не является подтип-селективным (Piechan et al (2004) Soc. Neuroscience Abstr., 111 (program №115)).
Европейская патентная заявка 2192109 A относится к бициклическим гамма-аминокислотным соединениям формулы A,
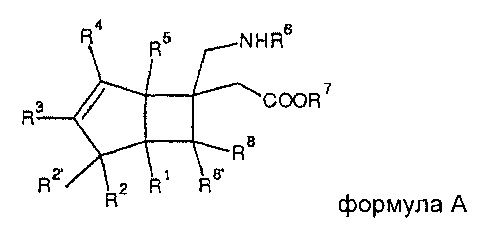
где каждый из R1, R2, R2' и R4-R8 и R8' независимо представляет собой атом водорода, атом галогена или С1-С6 алкильную группу, или R2 и R2' вместе с атомом углерода, к которому они присоединены, образуют С3-С7 циклоалкильную группу; и
R3 представляет собой атом водорода, атом галогена, С1-С6 алкильную группу, С1-С6 алкилгалогенидную группу, гидрокси-С1-С6 алкильную группу, сульфанил-С1-С6 алкильную группу, С1-С6 алкокси-С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу, С1-С6 алкокси группу, С1-С6 алкилсульфанильную группу, С1-С6 алкилсульфанил-С1-С6 алкильную группу, С2-С7 ацилтио С1-С6 алкильную группу, С2-С7-ацилокси С1-С6 алкильную группу или С3-С7 циклоалкильную группу.
Указанные производные обладают активностью в качестве лигандов α2δ и эффективны при лечении боли или расстройств центральной нервной системы. US 2012071685 раскрывает препаративные методы получения некоторых из указанных бициклических производных гамма-аминокислоты.
Краткое описание изобретения
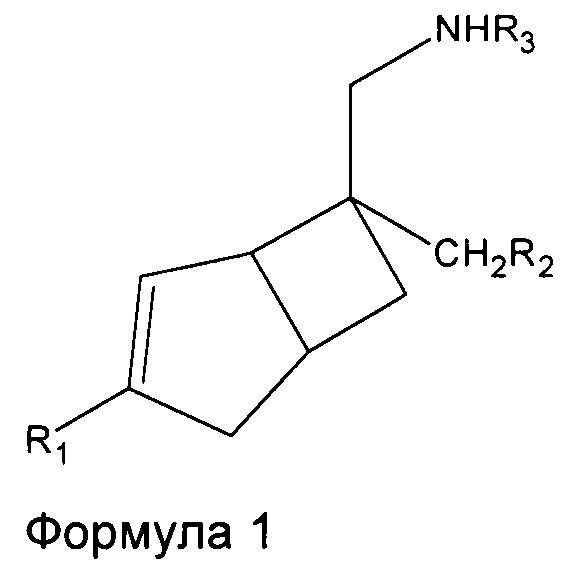
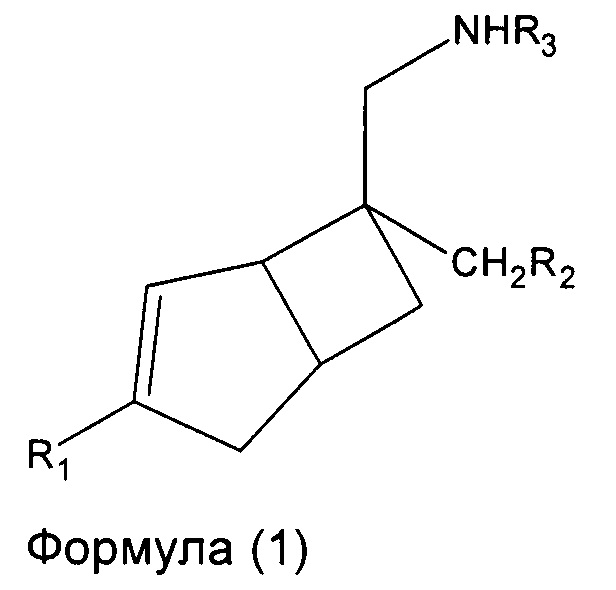
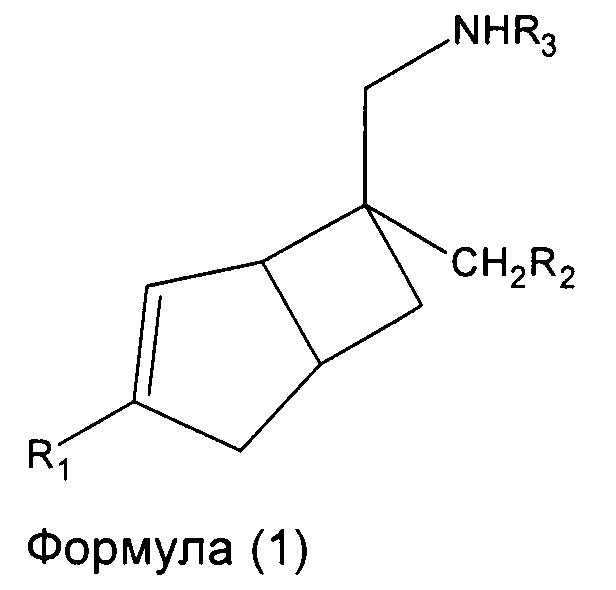
В одном аспекте, настоящее изобретение относится к соединениям формулы 1,
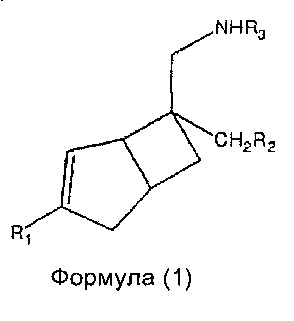
где R1 представляет собой водород, галоген, С1-4 алкильную группу, C1-4 алкилгалогенидную группу, группу (C1-4 алкокси)(C2-4 алкил), C2-4 алкенильную группу, C2-4 алкинильную группу или C3-7 циклоалкильную группу,
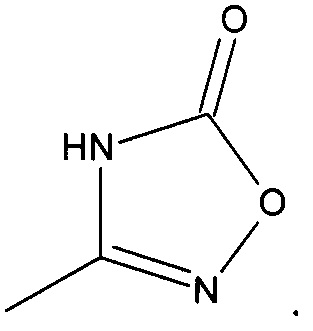
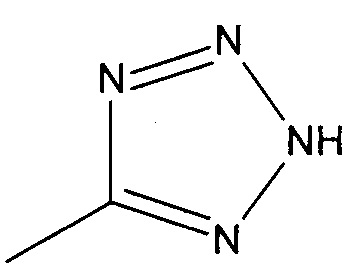
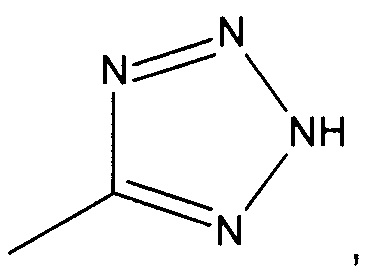
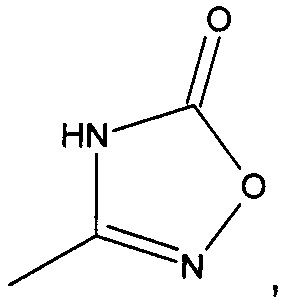
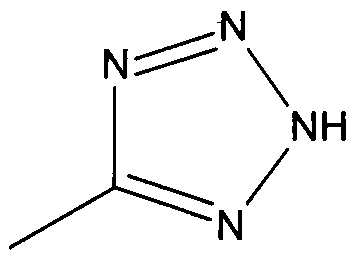
R2 представляет собой 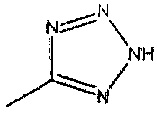 или
или 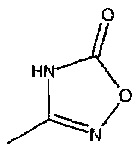 или его таутомер, и
или его таутомер, и
R3 представляет собой водород, С1-4 алкильную группу, группу (C1-4 алкокси)(C2-4 алкил) или C3-7 циклоалкильную группу,
или его фармацевтически приемлемой соли или сольвату. В другом аспекте, настоящее изобретение относится к способу лечения нейропатической боли у субъекта, включающий введение терапевтически эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли или сольвата, указанному субъекту.
В еще одном аспекте настоящее изобретение относится к способу лечения расстройства центральной нервной системы у субъекта, включающий введение терапевтически эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли или сольвата, указанному субъекту.
Настоящее изобретение относится к соединениям, которые связываются с высокой аффинностью с субъединицей α2δ-1 потенциал-зависимых кальциевых каналов.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединения, раскрытые в настоящем документе вместе с подходящим наполнителем или подходящим жидким косметическим средством. Фармацевтическая композиция может быть в любой удобной форме для введения субъекту, например, в виде капсулы, каплеты, таблетки или в виде мази для местного применения.
Подробное описание изобретения
Настоящее изобретение относится к соединениям формулы 1,
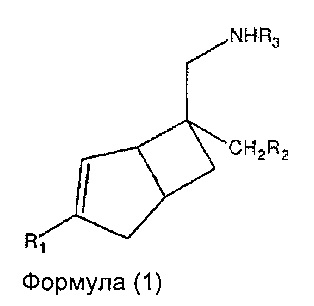
где R1 представляет собой водород, галоген, C1-4 алкильную группу, C1-4 алкилгалогенидную группу, группу (C1-4 алкокси)(C2-4 алкил), C2-4 алкенильную группу, C2-4 алкинильную группу или C3-7 циклоалкильную группу,
R2 представляет собой  или
или 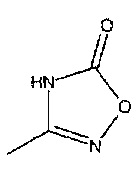 или его таутомер, и
или его таутомер, и
R3 представляет собой водород, C1-4 алкильную группу, группу (C1-4 алкокси)(C2-4 алкил) или С3-7 циклоалкильную группу,
или его фармацевтически приемлемой соли или сольвату.
Следующие термины должны быть поняты в следующих значениях:
Термин "C1-4 алкил" означает линейную или разветвленную алифатическую углеводородную цепь, имеющую от 1 до 4 атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил.
Термин "C1-4 алкил галогенид" означает C1-4 алкильную группу, замещенную одним или более атомами галогена.
Термин "(C1-4 алкокси) (C2-4 алкил)" означает C2-4 алкильную группу, замещенную одной или более C1-4 алкокси группами.
Термин "C2-4 алкенил" означает линейную или разветвленную алифатическую углеводородную цепь, имеющую от 2 до 4 атомов углерода и содержащую углерод-углеродную двойную связь, например, этенил, пропенил, н-бутенил и изобутенил.
Термин "C2-4 алкинил" означает линейную или разветвленную алифатическую углеводородную цепь, имеющую от 2 до 4 атомов углерода и содержащую углерод-углеродную тройную связь, например, этинил, пропинил, н-бутинил и изобутинил.
Термин "C3-7 циклоалкил" означает неароматическую моно- или мультициклическую кольцевую систему, содержащую от 3 до 7 атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "галоген" означает фтор, хлор, бром или йод.
Используемый в настоящем описании термин "соединения формулы 1" включают фармацевтически приемлемые соли и их сольваты. Ссылки на промежуточные соединения также включают соли и их сольваты. Фармацевтически приемлемые соли соединений по настоящему изобретению могут включать в себя соли присоединения основания. Такие соли могут быть образованы с неорганическим основанием, которое дает фармацевтически приемлемый катион, например, соль щелочного металла, такая как соль натрия или калия, или соль щелочноземельного металла, такая как соль кальция или магния. Фармацевтически приемлемые соли по изобретению могут также включать в себя соли присоединения кислоты. Такие соли могут быть образованы с неорганической или органической кислотой, которая дает фармацевтически приемлемый анион, например, соль гидрогалогенида, такая как хлористоводородная или бромистая соль, соль серной кислоты или фосфорной кислоты, или соль органической кислоты, например, соли с ацетатом, фумаратом, малеатом, тартратом, лактатом, цитратом, пируватом, сукцинатом, оксалатом, метансульфонатом или п-толуолсульфонатом. Термин "сольват" относится к соединению формулы 1 в твердом состоянии, в котором молекулы подходящего растворителя включены в кристаллическую решетку. Подходящий растворитель для терапевтического введения является физиологически приемлемым при дозируемом введении. Примерами подходящих растворителей для терапевтического введения являются этанол и вода. Когда вода является растворителем, сольват упоминается как гидрат. В общем, сольваты образуются путем растворения соединения в соответствующем растворителе и выделения сольвата путем охлаждения или с использованием антирастворителя. Сольват, как правило, сушат или, подвергают азеотропной перегонке в условиях окружающей среды. Типичные сольваты включают гидраты, такие как моногидрат, дигидрат или тригидрат.
Если соединения, полученные в соответствии с настоящим изобретением, включают хиральный центр, то соединения существуют в двух энантиомерных формах, соответственно соединения по настоящему изобретению включают рацемат, единичные энантиомеры или их смеси. Энантиомеры могут быть разделены методами, известными специалистам в данной области техники, например, хиральной высокоэффективной жидкостной хроматографией (ВЭЖХ), например, двуокись кремния с присоединенным хиральным лигандом. Энантиомеры также могут быть разделены с помощью методов, включающих образование солей с хиральными кислотами или основаниями, с образованием диастереомерных солей с последующей кристаллизацией. Соединения, полученные в соответствии с настоящим изобретением, согласно изобретению, могут также включать в себя единичные диастереоизомеры или их смеси. Диастереоизомеры могут быть разделены способами, известными специалистам в данной области техники, например, путем образования солей с кислотами или основаниями с последующей кристаллизацией.
Все таутомерные формы также включены в настоящее описание.
Термин "терапевтически эффективное количество" описывает количество соединения в соответствии с настоящим изобретением, которое должен быть использовано человеческими субъектами, с целью достижения желаемого терапевтического эффекта. Это количество может варьироваться в зависимости от субъекта и зависит от таких параметров, как возраст, вес, рост, физическое состояние и история болезни. Соединение в соответствии с настоящим изобретением, может быть эффективным для уменьшения, ингибирования или ослабления нежелательных симптомов у пациента.
В одном из вариантов осуществления соединения в соответствии с настоящим изобретением, R1 представляет собой водород, C1-4 алкильную группу или галоген, предпочтительно водород или C1-4 алкильную группу, более предпочтительно C1-4 алкильную группу, например, метил, этил или пропил. В других предпочтительных соединениях формулы 1, R1 представляет собой этил.
В одном из вариантов осуществления соединений в соответствии с настоящим изобретением, R3 представляет собой водород или С1-С4 алкильную группу, например, метил, этил или пропил. В предпочтительных соединениях формулы 1, R3 представляет собой водород или этил. В других предпочтительных соединениях формулы 1, R3 представляет собой водород.
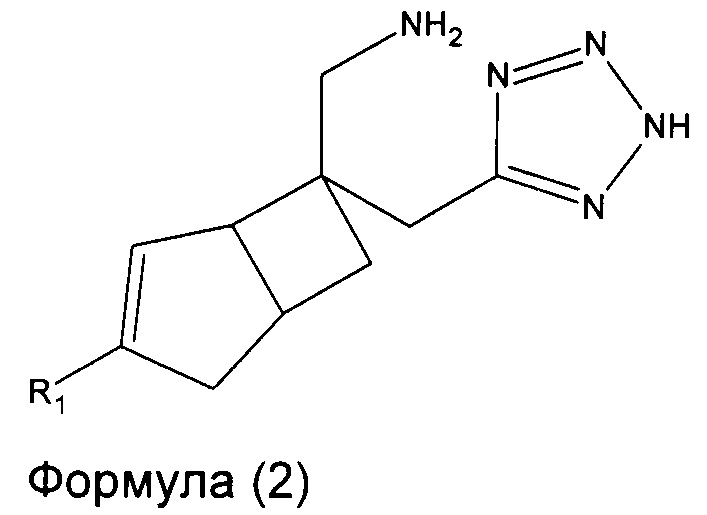
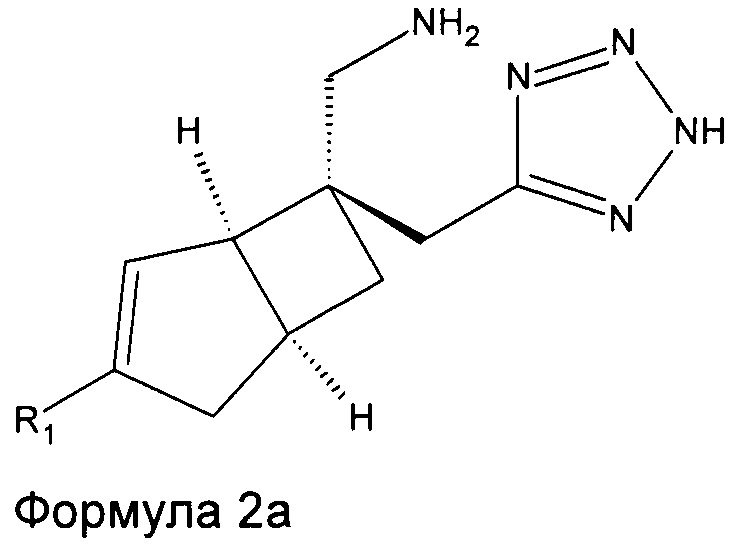
В одном варианте осуществления, настоящее изобретение представляет собой соединение формулы 2:
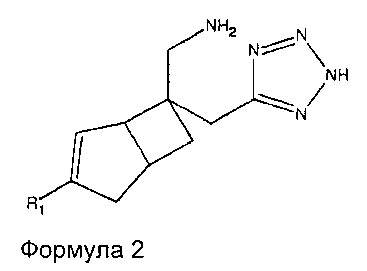
где R1 представляет собой водород, галоген, C1-4 алкильную группу, C1-4 алкилгалогенидную группу, группу (C1-4 алкокси)(C2-4 алкил), C2-4 алкенильную группу, C2-4 алкинильную группу или C3-7 циклоалкильную группу,
или его фармацевтически приемлемую соль или сольват.
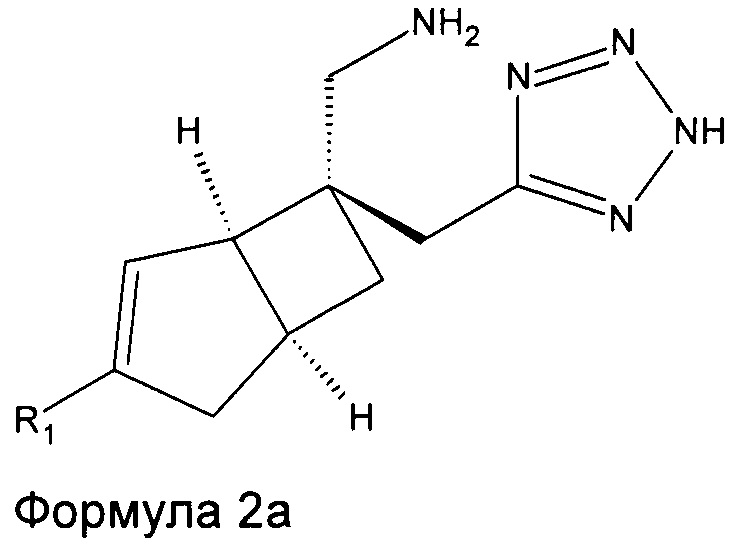
Соединение формулы 2, может иметь определенную стереохимию, как показано в соединении формулы 2a.
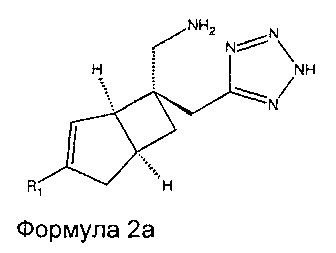
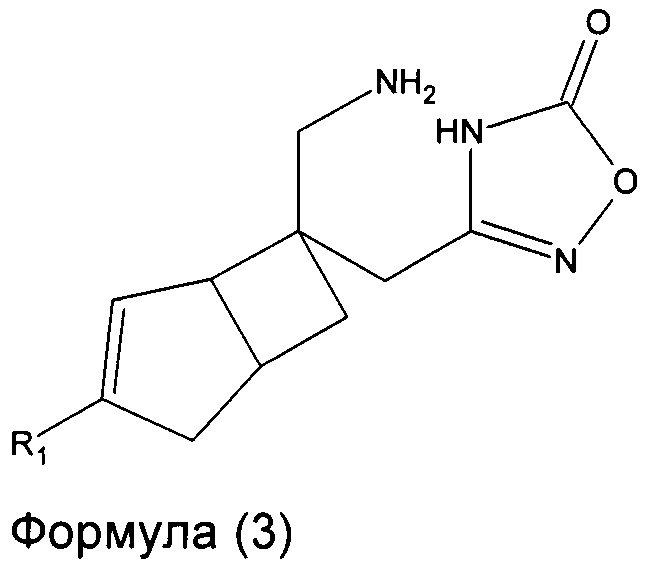
В другом варианте осуществления, настоящее изобретение представляет собой соединение формулы 3:
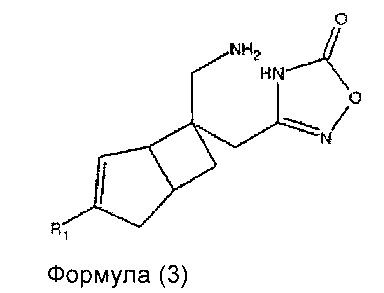
где R1 представляет собой водород, галоген, C1-4 алкильную группу, C1-4 алкилгалогенидную группу, группу (C1-4 алкокси)(C2-4 алкил), C2-4 алкенильную группу, C2-4 алкинильную группу или C3-7 циклоалкильную группу,
или его фармацевтически приемлемую соль или сольват.
Соединение формулы 3, может иметь определенную стереохимию, как показано в соединении формулы 3a.
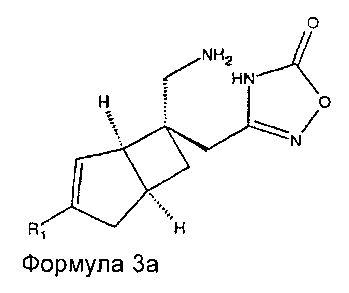
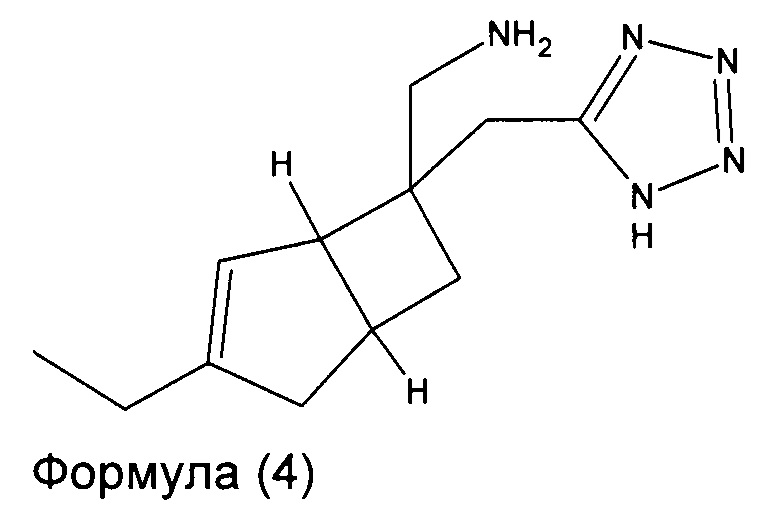
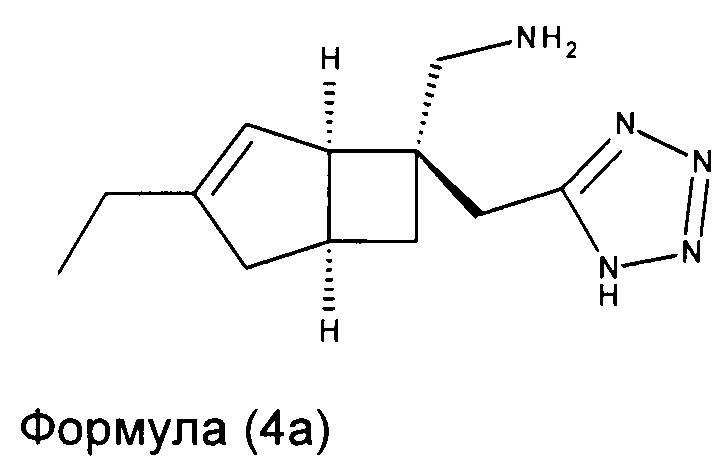
В другом варианте осуществления, настоящее изобретение представляет собой соединение формулы 4:
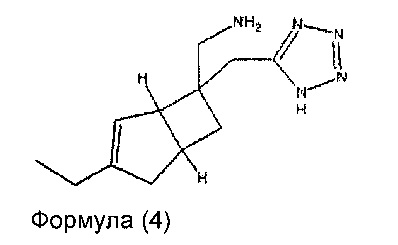
Соединение формулы 4, может иметь определенную стереохимию, как показано в соединении формулы 4а:
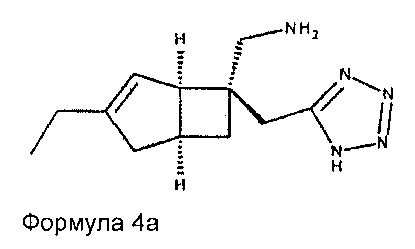
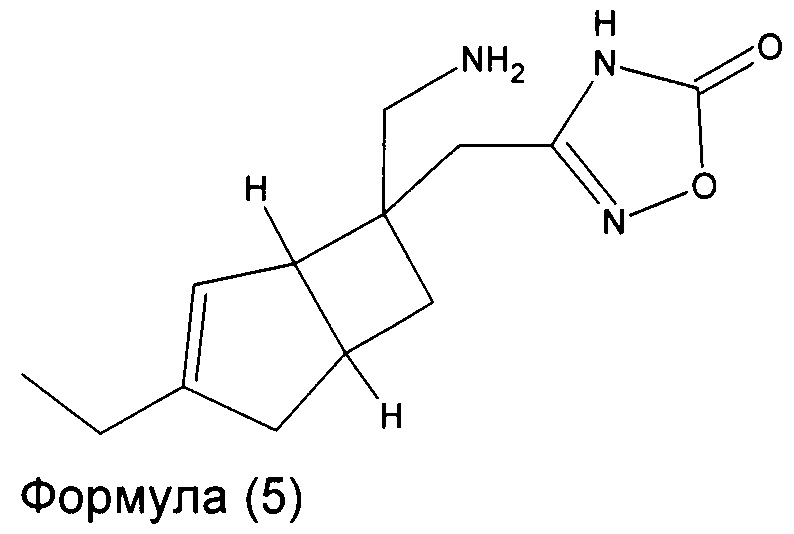
В другом варианте осуществления, настоящее изобретение представляет собой соединение формулы 5:
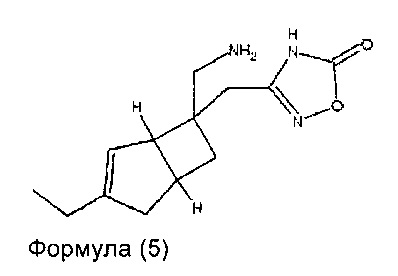
Соединение формулы 5, может иметь определенную стереохимию, как показано в соединении формулы 5a:
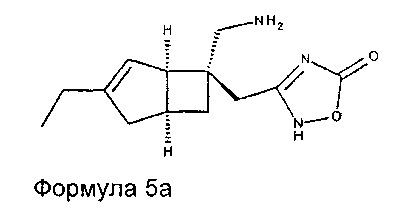
Конкретные соединения формулы 1 представляют собой:
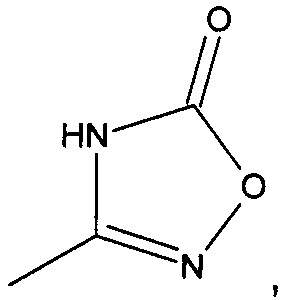
3-(((1R,5S,6S)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-он
3-(((1S,5R,6R)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-он
Рацемический 3-(((1R,5S,6S)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-он
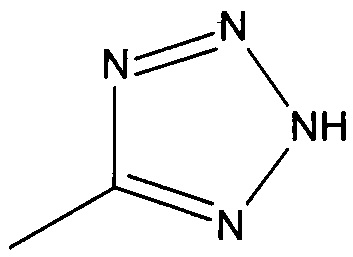
((1R,5S,6S)-6-((1Н-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метанамин
((1S,5R,6R)-6-((1Н-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метанамин
Рацемический ((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метанамин
N-(((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)этанамин
N-(((1S,5R,6R)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)этанамин
Рацемический N-(((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)этанамин
Специалисту будет понятно, что хиральные молекулы могут быть повернуты, но сохраняют то же относительное стереохимическое расположение. Например, соединения Формул 4а и 5а могут быть изображены как показано ниже.
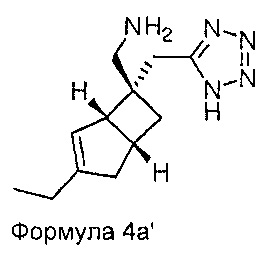
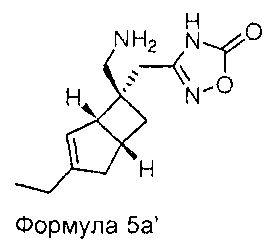
Настоящее изобретение также относится к фармацевтическим композициям, содержащим терапевтически эффективное количество соединения формулы 1 в сочетании с фармацевтически приемлемым носителем. Соединение формулы 1 используют в количестве, эффективном для лечения, уменьшения или ослабления нейропатической боли у субъекта, особенно человека, страдающего от болезненного состояния. Такое лечение боли может быть связано, а может не быть связано с расстройством центральной нервной системой (ЦНС) или периферической нервной системы (ПНС). Соединение формулы 1 также является эффективным для лечения, уменьшения или ослабления любых других, не связанных с болью заболеваний ЦНС. Композиции по настоящему изобретению, содержат терапевтически эффективное количество соединения формулы 1, которое, как правило, находится в диапазоне от 0,1 до 95 масс. % соединения формулы 1, но зависит от точной природы активного вещества и способа введения. Как правило, доза активного вещества находится в диапазоне от 0,1 до 500 мг в виде одной или нескольких доз, в зависимости от точной природы активного вещества и способа введения.
При терапевтическом применении, соединения формулы 1 могут быть введены перорально, ректально, парентерально или местно. Фармацевтические композиции в соответствии с настоящим изобретением могут иметь любую форму в виде композиции для перорального, ректального, парентерального или местного применения, известной специалистам в данной области техники, с использованием носителей, хорошо известных в области фармацевтики. Такие композиции обычно получают в виде стандартной лекарственной формы. Композиции для перорального введения могут включать твердые лекарственные формы, такие как таблетки, капсулы или каплеты, или жидкие лекарственные формы, такие как сиропы и водные или масляные суспензии. Твердые лекарственные формы, такие как таблетки и каплеты, могут быть получены путем смешивания соединения формулы 1 с инертным разбавителем в присутствии дезинтегрирующих агентов и других лекарственных добавок, таких как смазывающие вещества. Капсулы могут быть в форме твердых желатиновых капсул, например, твердые желатиновые капсулы, или мягкие капсулы, которые получают обычными способами, в которых активное вещество заключено в носитель и инкапсулировано. Необязательно, такие лекарственные формы могут включать энтеросолюбильное покрытие, получаемое в соответствии с обычными способами, которые могут быть использованы для изменения скорости высвобождения, или наполнитель, который задерживает высвобождение, чтобы обеспечить замедленное высвобождение, или композиции с замедленным высвобождением. Жидкие лекарственные формы могут быть получены путем растворения активного вещества в приемлемом жидком носителе, таком как вода или масляный наполнитель, необязательно в присутствии одного или нескольких агентов растворения, поверхностно-активных веществ и/или суспендирующих вспомогательных добавок. Композиции для ректального введения являются известными фармацевтическими формами для такого введения, например, суппозитории с восковой или полиэтиленгликолевой основой. Композиции для парентерального введения также являются известными фармацевтическими формами для такого введения, например, стерильные растворы или суспензии в подходящей системе растворителей. Композиции для местного применения могут включать кремы, лосьоны, мази, гели или другие такие лекарственные формы, которые могут быть введены путем нанесения композиции непосредственно на пораженный участок или путем нанесения композиции на основу, такую как трансдермальный пластырь, или в виде композиции, содержащиеся внутри проницаемой мембраны, для применения к болезненной области. Обычные водные и неводные носители, такие как минеральные масла и воски, могут быть использованы отдельно или в комбинации для получения кремов, лосьонов или мазей. Гели могут быть получены путем смешивания соединения формулы 1 с основой для местного применения, содержащей гелеобразующий агент, например, Карбомер, в присутствии воды. Необязательно дополнительно могут быть также включены лекарственные добавки, такие как трансдермальные ускорители, загустители.
В другом варианте осуществления, соединение по настоящему изобретению может быть использовано в сочетании с подходящим фармацевтическим наполнителем для местного лечения боли в спине. Сочетание соединения и фармацевтического наполнителя может быть в форме геля, гель формируют и адаптируют для нанесения на кожу субъекта при боли. В другом варианте осуществления изобретения, комбинация соединения и фармацевтического наполнителя, может быть включена в ткань пластыря, при этом пластырь формируют и адаптируют для размещения на и/или адгезии к коже субъекта при боли. В более предпочтительном варианте осуществления настоящего изобретения, соединение высвобождается с медленной скоростью из фармацевтического наполнителя, помещенного в ткань пластыря.
Соединения формулы 1 включены в фармацевтические композиции в соответствии с настоящим изобретением, которые применяют при условиях, перечисленных ниже.
Изобретение предполагает, что соединения формулы 1 могут быть применены в клинических условиях для лечения нейропатической боли. В другом варианте осуществления изобретения, соединения могут быть применены для лечения боли, связанной с центральной нервной системой (ЦНС). В другом варианте осуществления, соединения по данному изобретению могут быть применены для лечения боли, не связанной с центральной нервной системой. В дополнительном варианте осуществления, соединения по изобретению могут быть применены для лечения боли, не связанной с ПНС. В еще одном варианте осуществления, соединения по изобретению могут быть использованы для лечения заболеваний ЦНС. В одном из вариантов осуществления, заболевание ЦНС выбирают из группы, состоящей из эпилепсии, ишемического цереброваскулярного заболевания, инсульта, церебральных новообразований, болезни Альцгеймера, болезни Пика, болезни Хантингтона, деменции, болезни Паркинсона и других экстрапирамидных расстройств, бокового амиотрофического склероза и других заболеваний двигательных нейронов, прогрессирующей нейронной мышечной атрофии, пигментной дистрофии сетчатки, наследственной атаксии, рассеянного склероза и других демиелинизирующих заболеваний, бактериального и вирусного менингитов, абсцесса мозга, субдурального абсцесса, эпидурального абсцесса, гнойного внутричерепного тромбофлебита, миелита и радикулита, вирусного заболевания центральной нервной системы, прионного заболевания, включая Куру, болезнь Крейтцфельда-Якоба и синдром Герстманна-Штреусслера-Шейнкера, фатальной семейной бессонницы, нарушения питания и обмена веществ нервной системы, нейрофиброматоза, туберозного склероза, мозжечково-ретинального гемангиобластоматоза, энцефалотригеминального синдрома, умственной отсталости и других нарушений развития центральной нервной системы, включая синдром Дауна, церебрального паралича, заболеваний внутреннего скелета, вегетативных расстройств нервной системы, черепно-мозговых нервных расстройств, заболеваний спинного мозга, мышечной дистрофии и других нервно-мышечных расстройств, заболеваний периферической нервной системы, дерматомиозита и полимиозита, наследуемой, метаболической, эндокринной и токсической миопатии, тяжелой миастении, периодического паралича, психических расстройств, включая расстройство настроения, тревожное расстройство и шизофреническое расстройство, сезонного аффективного расстройства (SAD), акатизии, амнезии, кататонии, диабетической нейропатии, поздней дискинезии, дистонии, параноидальных психозов, постгерпетической невралгии, болезни Туретта, прогрессивного надъядерного паралича, кортикобазальной дегенерации и семейной лобно-височной деменции. В другом варианте осуществления, соединения по данному изобретению могут быть применены при лечении боли, связанной с ЦНС, такой как, но не ограничиваясь ими, головная боль и мигрень.
В другом варианте осуществления, соединения по изобретению могут быть применены в сочетании с подходящим лосьоном в фармацевтическом препарате для местного лечения боли в спине. В другом варианте осуществления, соединения по изобретению могут быть применены для местного лечения боли в суставах.
Далее будут описаны способы получения соединений по настоящему изобретению. Эти способы образуют еще один аспект настоящего изобретения.
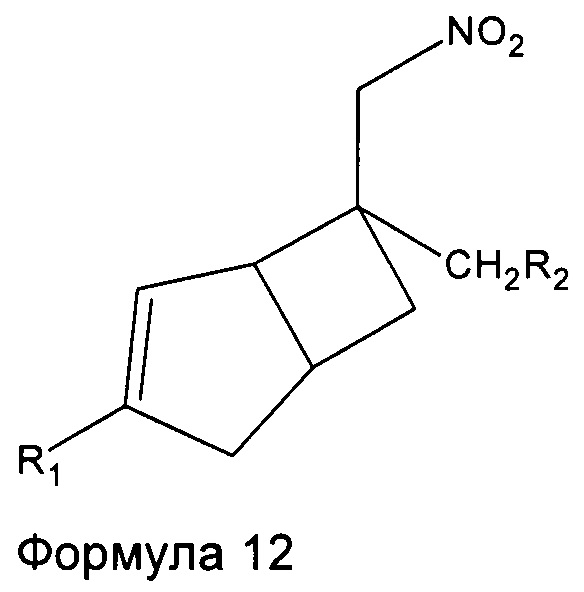
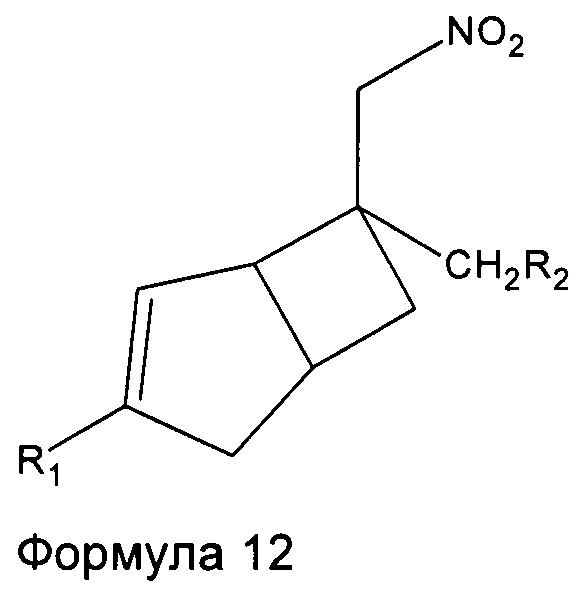
Соединения формулы 1 могут быть получены восстановлением соединений формулы 12
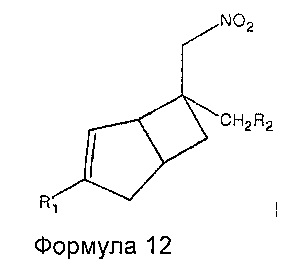
где R1 и R2 являются такими, как определено выше, с подходящим восстанавливающим агентом.
Например, соединения формулы 1, где R1 является таким, как определено в данном описании,
R2 представляет собой 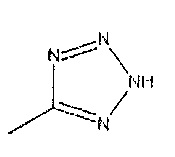
и R3 представляет собой C1-4 алкильную группу, группу (C1-4 алкокси)(C2-4 алкил) или C3-7 циклоалкильную группу, могут быть получены восстановлением соединений формулы 12 с подходящим восстанавливающим агентом с последующим восстановительным аминированием с подходящим альдегидом или кетоном. Подходящие реагенты включают порошок железа с хлоридом аммония, в подходящем растворителе, например, водном спирте, таком как этанол, при подходящей температуре, например, от температуры окружающей среды до температуры нагревания с обратным холодильником. Дополнительно восстановительное аминирование в присутствии подходящего альдегида или кетона с соответствующим восстанавливающим агентом, например, включает использование боргидрида натрия в подходящем хлорированном или эфирном растворителе, таком как дихлорэтан, при температуре окружающей среды.
Соединения формулы 1, где R1 является таким, как определено в данном описании, R2 представляет собой 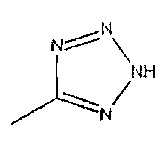 и R3 представляет собой водород, могут быть получены восстановлением соединений формулы 12 с подходящим восстанавливающим агентом, таким как порошок железа с хлоридом аммония, в подходящем растворителе, таком как водный этанол, при температуре, например, от температуры окружающей среды до температуры нагревания с обратным холодильником. Добавление защитной группы к сырому продукту с получением промежуточного соединения формулы 13
и R3 представляет собой водород, могут быть получены восстановлением соединений формулы 12 с подходящим восстанавливающим агентом, таким как порошок железа с хлоридом аммония, в подходящем растворителе, таком как водный этанол, при температуре, например, от температуры окружающей среды до температуры нагревания с обратным холодильником. Добавление защитной группы к сырому продукту с получением промежуточного соединения формулы 13
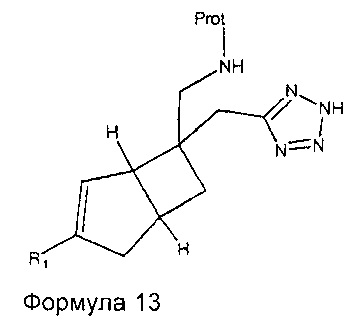
может быть использовано для облегчения очистки с последующим удалением защитной группы в кислых условиях. Подходящие защитные группы включают трет-бутоксикарбонильную группу, полученную путем реакции с ди-трет-бутилдикарбонатом в присутствии подходящего основания и растворителя. Примеры подходящего основания включают N,N-диизопропилэтиламин или триэтиламин, в подходящем растворителе, таком как водный эфирный растворитель, например, водный тетрагидрофуран, при температуре окружающей среды до температуры нагревания с обратным холодильником. Удаление защитной группы может быть осуществлено сильными кислотами, такими как соляная кислота или трифторуксусная кислота в подходящем растворителе, например, трифторуксусная кислота в дихлорметане.
Соединения формулы 1, где R1 и R3 являются такими, как определено в данном описании, и R2 представляет собой 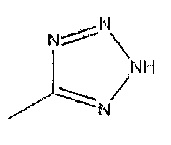 могут быть получены путем взаимодействия соединений формулы 12 с подходящим восстанавливающим агентом, например боргидридом натрия в присутствии никель (II) хлорид гексагидрата, в подходящем растворителе, например метаноле, при температуре от 0°C до температуры окружающей среды.
могут быть получены путем взаимодействия соединений формулы 12 с подходящим восстанавливающим агентом, например боргидридом натрия в присутствии никель (II) хлорид гексагидрата, в подходящем растворителе, например метаноле, при температуре от 0°C до температуры окружающей среды.
Соединения формулы 1, где R3 представляет собой алкил или циклоалкил, могут быть, как правило, получены путем восстановительного аминирования соединений формулы 1, где R3 представляет собой водород, например, путем обработки подходящим соединением альдегида или кетона вместе с подходящим восстанавливающим агентом, таким как боргидрид натрия, цианоборгидрид натрия или триацетоксиборгидрид натрия. Например, при использовании ацетальдегида получают соединения, где R3 представляет собой этильную группу. В качестве альтернативы при использовании циклопентанона получают соединения, где R3 представляет собой циклопентильную группу.
Соединения формулы 1 могут быть также получены путем удаления защитной группы из соединений формулы 1, где R3 замещен защитной группой, например, СООС(CH3)3, которая может быть удалена путем взаимодействия с подходящей кислотой, например, соляной кислотой или трифторуксусной кислотой.
Энантиомеры соединений формулы 1 могут быть получены путем разделения соответствующего рацемата или смеси диастереоизомеров, например, путем образования диастереомерных солей с подходящими хиральными кислотами или основаниями. Примеры подходящих хиральных кислот включают миндальную кислоту, α-метоксифенилуксусную кислоту, винную кислоту, напроксен или кислоту Мошера. Примеры подходящих хиральных оснований включают α-метилбензиламин, 4-хлор-α-метилбензиламин или эфедрин.
Соединения формулы 12, где R1 является таким, как определено в данном описании, и R2 представляет собой 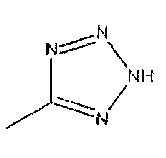 могут быть получены из соединений формулы 6,
могут быть получены из соединений формулы 6,
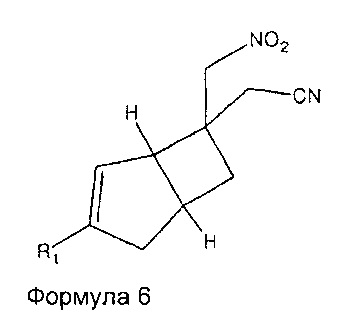
где R1 является таким, как определено в данном описании, с помощью реакции образования циклической структуры с соответствующим азидом, например, натрий- или триметилсилил-азидом, вместе с общеизвестными подходящим катализатором, таким как оксид дибутилолова, пиридин гидрохлорид или хлорид аммония в растворителе, таком как диметилформамид (ДМФ), N-метилпирролидинон (NMP) или толуол, при повышенной температуре от 60°C до 120°C.
Соединения формулы 12, где R2 представляет собой 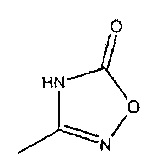
могут быть получены путем взаимодействия соединения формулы 6 с подходящими реагентами двухстадийным способом с получением фрагмента оксадиазолона. Например, первая стадия может включать использование гидроксиламина для получения промежуточного соединения формулы 9
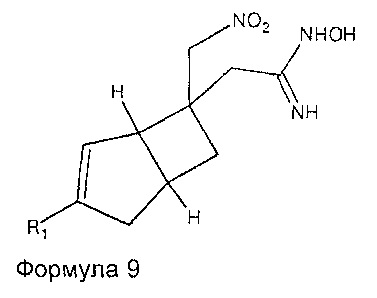
где R1 является таким, как определено в данном описании. Вторая стадия образования циклической структуры может включать взаимодействие промежуточного соединения 9 с подходящим реагентом циклизации, который включает карбонильную группу. Например, подходящие реагенты включают карбонилдиимидазол или фосген. Обработка с помощью карбонилдиимидазола может происходить в подходящем эфирном растворителе, таком как 1,4-диоксан, при температуре нагревания с обратным холодильником.
Соединения формулы 9 могут быть получены путем обработки соединений формулы 6 гидроксиламином в подходящем растворителе, таком как водный этанол, при повышенных температурах, например, в микроволновой печи при 100 Вт.
Соединения формулы 6 могут быть получены путем обработки соединений формулы 7,
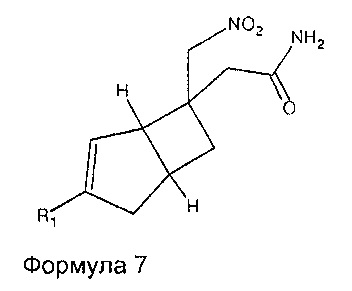
где R1 является таким, как определено в данном описании, подходящим дегидратирующим агентом, например, реагентом Бургесса в подходящем растворителе, таком как дихлорметан, при температуре окружающей среды.
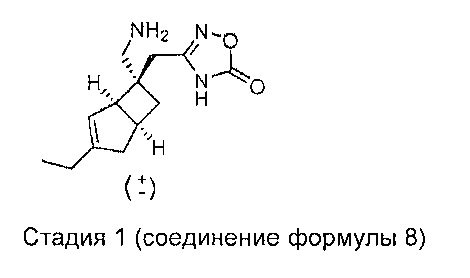
Соединения формулы 7 могут быть получены путем обработки соединений формулы 8,
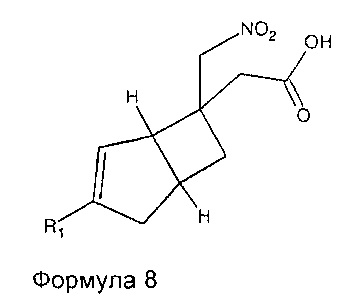
где R1 является таким, как определено в данном описании, подходящим связующим агентом с последующим добавлением концентрированного 0,88 водного аммиака. Подходящий связующий агент может представлять собой 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридин 3-оксидгексафторфосфат (HATU), в присутствии подходящего основания, например N,N-диизопропилэтиламина (основание Хюнига) или триэтиламина, и растворителя, такого как диметилформамид (ДМФ) или N-метилпирролидинон (NMP).
Соединения формулы 8 могут быть получены путем обработки соединения трет-бутил 2-(3-алкил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетата подходящей сильной кислотой, например соляной кислотой или трифторуксусной кислотой, в подходящем растворителе, таком как дихлорметан или 1,4-диоксан.
Соединения трет-бутил 2-(3-алкил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетата могут быть получены, как описано в патенте US 2012/0071685.
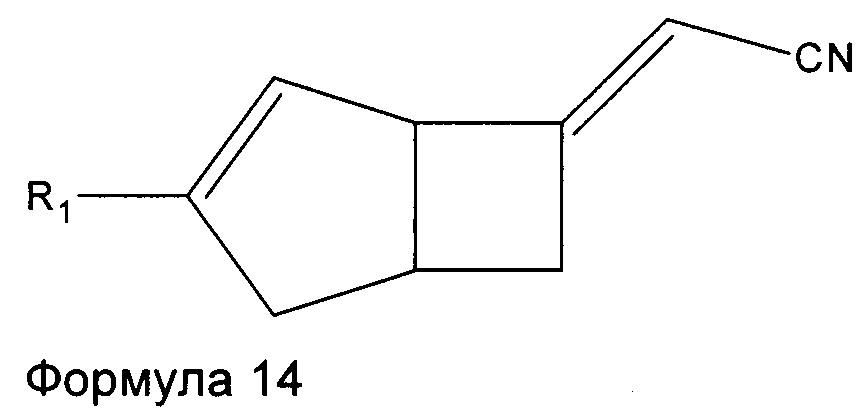
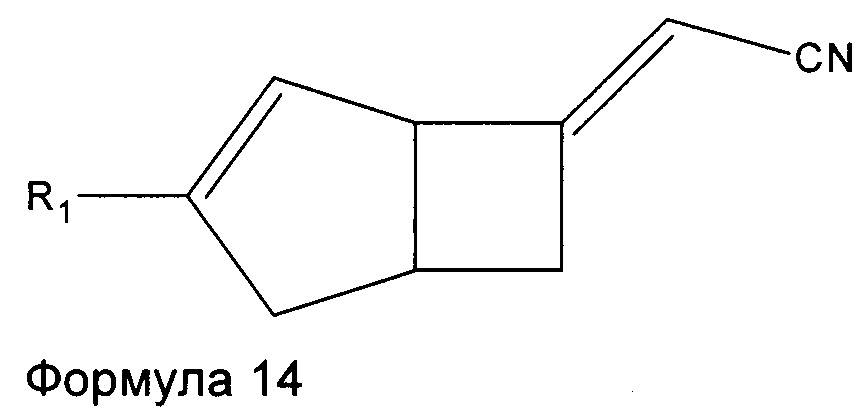
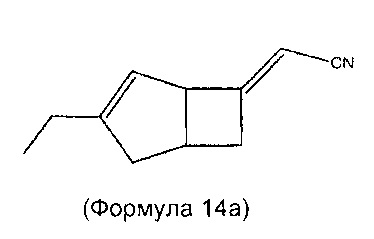
Соединения формулы 6 могут быть получены также путем взаимодействия соединений формулы 14
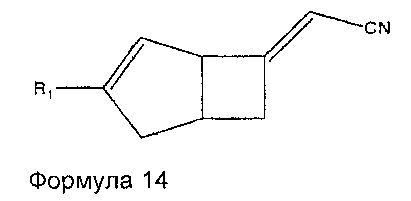
где R1 является таким, как определено в данном описании, с нитрометаном в присутствии подходящего основания, например, 1,8-диазабицикло[5.4.0]ундец-7-ена (ДБУ), и растворителя, такого как дихлорметан.
Соединения формулы 14 могут быть получены путем обработки соединений формулы 15
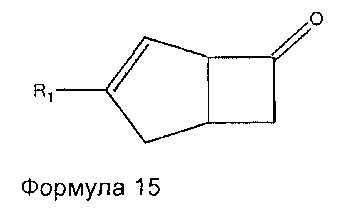
где R1 является таким, как определено в данном описании, подходящим реагентом, образующим двойную связь, например диэтил цианометилфосфатом, в присутствии подходящего основания, такого как трет-бутоксид калия, в подходящем растворителе, например в тетрагидрофуране, при температуре в диапазоне от 0°C до температуры окружающей среды.
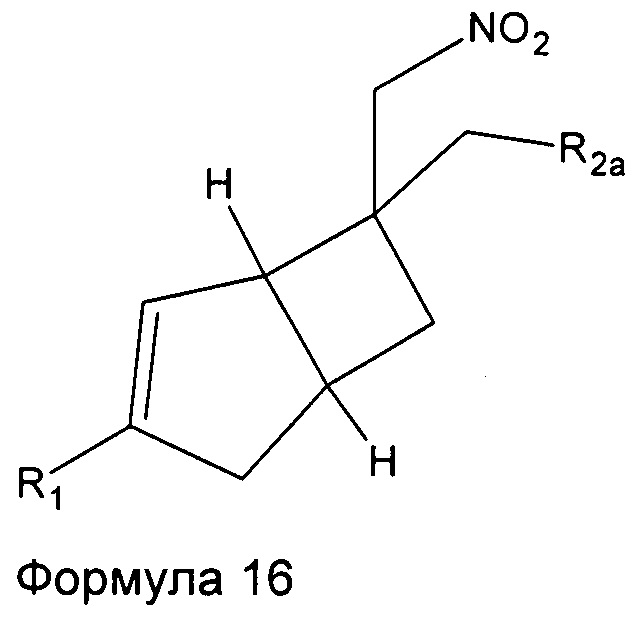
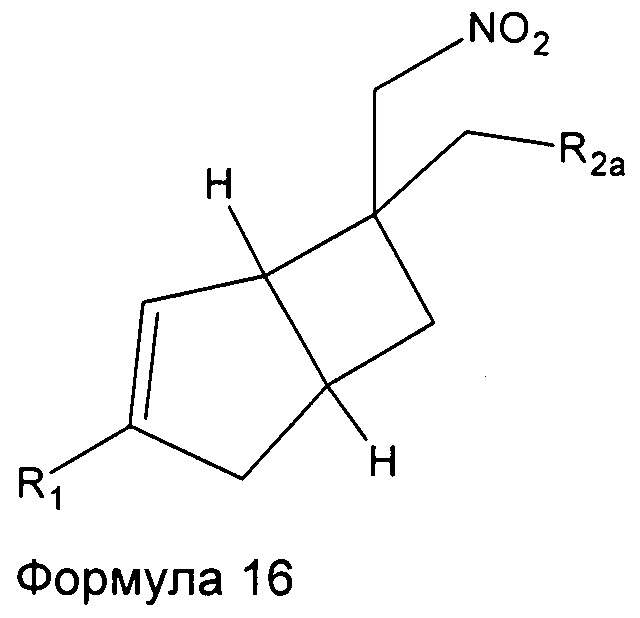
Промежуточные соединения нитрометила 6, 7, 8, 9, 10, 11 и 12 могут быть включены в промежуточную формулу 16
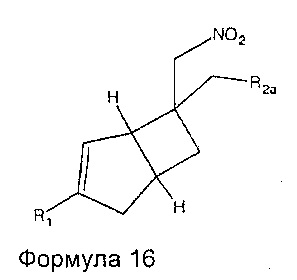
где R1 представляет собой водород, галоген, C1-4 алкильную группу, C1-4 алкилгалогенидную группу, группу (C1-4 алкокси)(C2-4 алкил), C2-4 алкенильную группу, C2-4 алкинильную группу или C3-7 циклоалкильную группу,
R2a представляет собой -R2, -CN, -CONH2, -COOH, -(C=NH)NHOH, и
R2 представляет собой 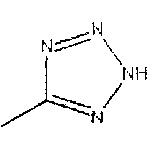 или
или 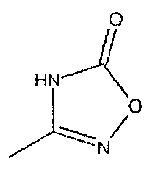 или его таутомер.
или его таутомер.
Подробная информация о предпочтительных стадиях способа получения конкретных соединений формулы 1 представлена в схемах 1-3:
Синтез соединения формулы 4а осуществляют с использованием нитро-нитрильного промежуточного соединения формулы 6: (схема 1)
Схема 1
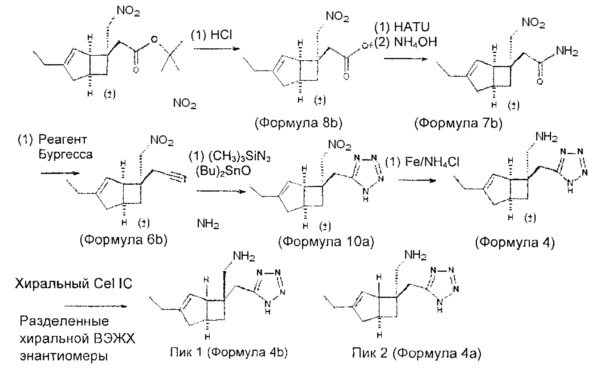
Синтез соединения формулы 5 осуществляют с использованием нитро-нитрильного промежуточного соединения формулы 6: (схема 2)
Схема 2
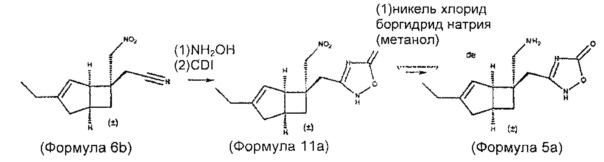
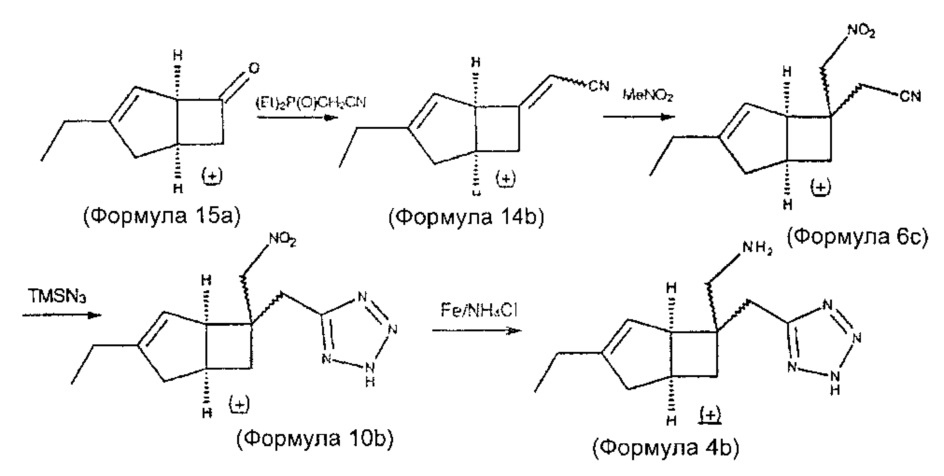
Синтез соединения формулы 4 (показано как 4b) и его энантиомеров может быть выполнен по схеме 3
Схема 3
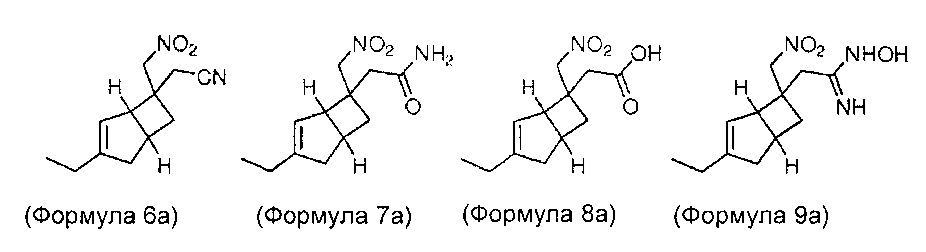
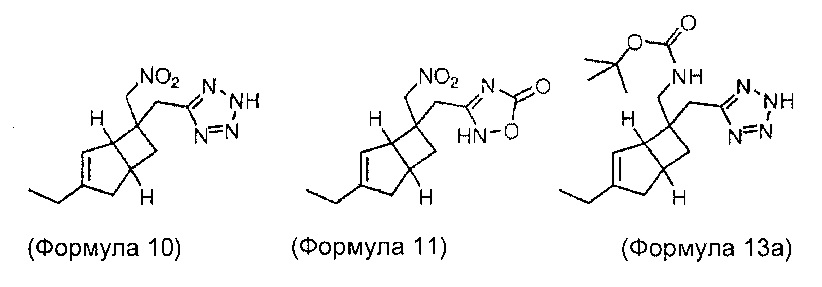
Некоторые промежуточные соединения и их стереоизомеры формул 6, 7, 8, 9, 10, 11, 12, 13, 14 и 16, как полагают, представляют собой новые соединения. Изобретение также охватывает все стереоизомеры промежуточных соединений, содержащих химическую структуру, как показано в формуле 6, формуле 7, формуле 8, формуле 9, формуле 12, формуле 13, формуле 14 и формуле 16 и конкретные соединения, описанные ниже.
Все новые соединения заявлены в качестве дополнительного аспекта настоящего изобретения.
Изобретение иллюстрируется следующими не ограничивающими примерами.
Пример 1: Синтез соединения формулы 3
Синтез рацемического 3-(((1R,5S,6S)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-она
Рацемическая 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) уксусная кислота
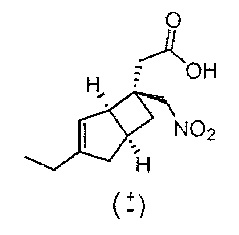
К раствору трет-бутил 2-(3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетата (1,8 г, 6,09 ммоль) (получение раскрыто в US 2012/0071685 A1, пример 5-d) в дихлорметане (20 мл) добавляли трифторуксусную кислоту (20 мл) и смесь оставляли стоять в течение 1 часа. Растворитель удаляли и осадок снова растворяли в толуоле (100 мл) и выпаривали досуха, эта процедура повторялась пятикратно с получением 2-(3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) уксусной кислоты (1,4 г, 5,62 ммоль, выход 92%) в виде бесцветного масла.
ЖХ-МС (жидкостная хромотография с масс-спектрометрией) (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиной кислоты) способ 4 мин, от 5 до 95% ацетонитрил/вода): m/z; 238(M-H)- (ES-), при 2,21 мин, 93% чистота @ 215 нм.
1H ЯМР (ядерный магнитный резонанс) (400 МГц, ДМСО (диметилсульфоксид)-d6): δ 12,25 (1Н, s), 5,26 (1Н, d, J=2,0), 4,87 (2Н, d, J=1,6), 3,13 (1Н, br. s), 2,85 (1H, quin, J=7,5), 2,46-2,32 (3H, m), 2,21 (1H, ddd, J=12,5, 8,8, 2,5), 2,11 (2H, q, J=7,4), 2,03 (1H, br. d, J=16,5), 1,46 (1H, dd, J=11,3, 7,3), 1,04 (3H, t, J=7,4) млн-1. (Толуол также присутствует: 7,26-7,23 (0,3Н, m), 7,18-7,12 (0,45Н, m), 2,30 (0,45Н, m).
Стадия 2 (соединение формулы 7)
Рацемический 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетамид
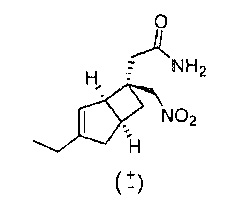
К раствору 2-(3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) уксусной кислоты (2,5 г, 10,45 ммоль) в сухом диметилформамиде (10 мл) добавляли основание Хюнига (2,74 мл, 15,67 ммоль), с последующим добавлением HATU (4,37 г, 11,49 ммоль). Смесь перемешивали в течение 10 минут при комнатной температуре, а затем охлаждали в ледяной воде. К этому охлажденному перемешанному раствору добавляли 0,88 водный раствор аммиака (6,46 мл, 104 ммоль). Реакционную смесь затем оставляли нагреваться до комнатной температуры и перемешивали дополнительно в течение 1 часа. Растворитель удаляли на роторном испарителе и осадок переносили в этилацетат и промывали водой, сушили над сульфатом натрия. Сырой продукт очищали с помощью хроматографии на силикагеле (40 г колонка с диоксидом кремния, градиент растворителя 20-100% простой эфир : изогексан) с получением 2-(3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетамида (2,3 г, 9,46 ммоль, выход 91%) в виде прозрачной бесцветной смолы.
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиной кислоты) способ 4 мин, от 5 до 95% ацетонитрил/вода): m/z; 239(М+Н)+ (ES+); 237 (M-H) - (ES-), при 1,949 мин.
1H ЯМР (400 МГц, ДМСО-d6): δ 7,28, (1Н, br. s), 6,77 (1Н, br. s), 5,27 (1Н, d, J=2,2), 4,88 (2H, dd, J=22,3, 12,3), 3,12 (1H, br. s), 2,85-2,78 (1H, m), 2,46-2,39 (1H, m), 2,24 (1H, br. d, J=1,4), 2,19 (1H, ddd, J=12,4, 8,8, 2,6), 2,11 (2H, q, J=7,5), 2,00 (1H, br. d, J=16,8), 1,42 (1H, dd, J=12,5, 7,4), 1,05 (3H, t, J=7,4) млн-1, (дихлорметан также присутствует: 5,87 (0,9 Н, s))
Стадия 3 (соединение формулы 6)
Рацемический 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрил
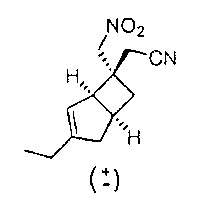
К охлажденному льдом раствору рацемического 2-((1R,5S,6S)-3-этил-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетамида (0,8 г, 3,36 ммоль) в сухом дихлорметане (20 мл) добавляли порциями в течение 20 мин реагент Бургесса (0,880 г, 3,69 ммоль) и смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 3-х часов. Реакционную смесь упаривали до половины объема и наносили на силикагелевый картридж и очищали с помощью хроматографии на Companion (колонка 40 г, 0-60% простой эфир : изогексан) с получением рацемического 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрила (501 мг, 2,23 ммоль, выход 66,4%) в виде прозрачного бесцветного масла.
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиная кислота) способ 4 мин, 5-95% ацетонитрил/вода): m/z 221(М+Н)+ (ES+); 219 (M-H) - (ES-), при 2,34.
1H ЯМР (400 МГц, ДМСО-d6): δ 5,33 (1Н, d, J=2,0), 4,87 (2Н, d, J=1,9), 3,17 (1Н, br. s), 2,93-2,85 (1H, m), 2,65 (2H, br. s), 2,49-2,43 (1H, m), 2,23 (1H, ddd, J=12,5, 8,8, 2,5), 2,16-2,05 (3H, m), 1,56 (1H, dd, J=12,6, 7,2), 1,07 (3H, t, J=7,4) млн-1.
Стадия 4 (соединение формулы 9)
Рацемический 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)-N-гидроксиацетимидамид
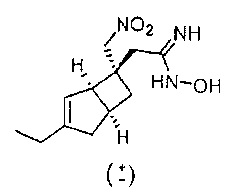
Смесь гидроксиламина в воде (50%-ный водный раствор) (543 мкл, 8,85 ммоль), рацемического 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрила (650 мг, 2,95 ммоль) в этаноле (10 мл) нагревали при температуре 85 градусов в микроволновой печи СЕМ при 100 Вт в течение 2-х часов.
Реакционную смесь упаривали досуха и осадок переносили в этилацетат и промывали водой и сушили над сульфатом натрия. После фильтрации и выпаривания получали рацемический 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)-N-гидроксиацетимидамид (530 мг, 1,883 ммоль, выход 63,8%) в виде бесцветного масла, которое использовали без дополнительной очистки.
ЖХ-МС (Agilent, X-Select, Waters X-Select С18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиная кислота) способ 4 мин, 5-95% ацетонитрил/вода): m/z 254 (M+H)+(ES+); при 1,25 мин.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,92 (1Н, s), 5,36-5,35 (1Н, m), 5,26 (2Н, br. s), 4,96 (1Н, d, J=12,8), 4,83 (1Н, d, J=12,8), 3,13 (1H, br. s), 2,77 (1H, quin, J=7,5), 2,41 (1H, dd, J=16,4, 8,2), 2,20-2,09 (5H, m), 2,02 (1H, d, J=16,0), 1,53 (1H, dd, J=12,4, 7,4), 1,06 (3H, t, J=7,4) млн-1.
Стадия 5 (соединение формулы 11)
Рацемический 3-(((1R,5S,6S)-3-зтил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-он
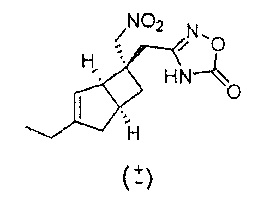
Смесь рацемического 2-((1R,5S,6S)-3-эил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)-N-гидроксиацетимидамида (520 мг, 2,053 ммоль) и карбонилдиимидазола (666 мг, 4,11 ммоль) в диоксане (30 мл) нагревали с обратным холодильником в течение 2-х часов.
Реакционную смесь упаривали досуха и осадок переносили в воду (50 мл) и раствор осторожно подкисляли 1Н соляной кислотой (HCl). Водную смесь экстрагировали эфиром и сушили над сульфатом натрия.
Сырой продукт очищали с помощью хроматографии на силикагеле (колонка 12 г, растворитель : простой эфир) с получением рацемического 3-(((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-она (195 мг, 0,684 ммоль, выход 33,3%) в виде бесцветной смолы.
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, основный (0,1% бикарбонат аммония) способ 4 мин, 5-95% ацетонитрил/вода): 278(M-H)- (ES-), при 1,58 мин, чистота 98% @ 215 нм.
1H ЯМР (400 МГц, ДМСО-d6): δ 12,12 (1Н, s), 5,35 (1Н, q, J=1,9), 4,88 (2Н, q, J=13,5), 3,16 (1Н, br. s), 2,87 91H, quin, J=7,5), 2,66 (2H, d, J=1,4), 2,44 (1H, dd, J=16,6, 7,8), 2,17-2,09 (3H, m), 2,06-2,02 (1H, m), 1,62 (1H, dd, J=12,5, 7,4), 1,06 (3H, t, J=7,5). Эфир присутствует - совпадает с сигналом при 1,06 + сигнал воды при 3,4 млн-1
13C ЯМР (100 МГц, ДМСО-d6): δ 159,44, 157,15, 150,69, 120,63, 80,09, 52,17, 42,75, 41,80, 35,38, 30,19, 28,46, 23,87, 12,25 млн-1.
Стадия 6 (Соединение формулы 3)
Рацемический 3-(((1R,5S,6S)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-он
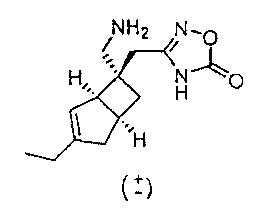
К охлажденному льдом раствору рацемического 3-(((1R,5S,6S)-3-3Wi-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-она (286 мг, 1,024 ммоль) и никель (II) хлорида, 6H2O (24,34 мг, 0,102 ммоль) в метаноле (40 мл) добавляли порциями боргидрид натрия (387 мг, 10,24 ммоль) в течение 15 минут. После завершения добавления реакционную смесь оставляли нагреваться до комнатной температуры.
Реакционную смесь осторожно гасили добавлением уксусной кислоты (1 мл) и полученную смесь упаривали досуха. Осадок растворяли в метаноле и пропускали через слой силикагеля и элюат упаривали досуха с получением почти белого твердого вещества. Сырой продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) (Waters, кислый (0,1% муравьиная кислота), Acidic, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 25-70% ацетонитрила в воде) с получением после лиофилизации рацемического 3-(((1R,5S,6S)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил)-1,2,4-оксадиазол-5(4Н)-она (8,0 мг, 0,031 ммоль, выход 3,07%) в виде бесцветного твердого вещества.
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиная кислота) способ 4 мин, 5-95% ацетонитрил/вода): m/z 250 (М+Н)+ (ES+); 248 (M-H)- (ES-), при 1,143 мин, чистота 100% @ 215 нм.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,27 (0,5Н, s), 5,33 (1Н, d, J=1,5), 3,11 (1Н, d, J=13,1), 3,04 (1Н, J=13,1), 2,97 (1Н, br. s), 2,76 (1H, quin, J=7,5), 2,44-2,34 (3Н, m), 2,12 (2Н, q, J=7,4), 2,03-1,97 (2H, m), 1,31 (1H, dd, J=12,2, 7,4), 1,05 (3H, t, J=7,4) млн-1.
13C ЯМР (100 МГц, ДМСО-d6): δ 170,67, 165,86, 149,09, 121,20, 51,92, 46,98, 42,62, 41,81, 34,69, 32,98, 30,35, 24,11, 12,64 млн-1.
Пример 2: Синтез соединения формулы 2
Синтез рацемического ((1R,5S,6S)-6-((1Н-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамина и энантиомеров
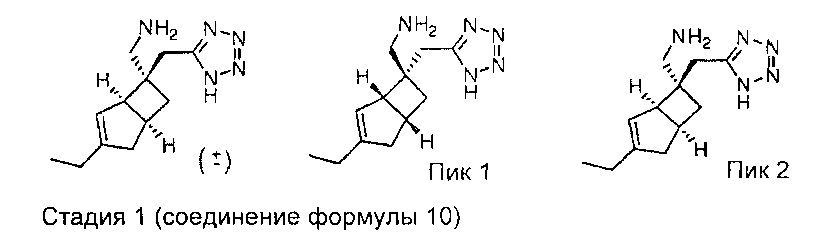
Рацемический 5-(((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)метил)-1H-тетразол
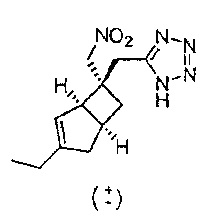
К раствору рацемического 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрила (280 мг, 1,271 ммоль) (получение описано на стадии 3, соединения формулы 3) в сухом толуоле (5 мл) добавляли азидотриметилсилан (675 мкл, 5,08 ммоль) и оксид дибутилолова (63,3 мг, 0,254 ммоль) и смесь нагревали в микроволновой печи СЕМ: мощность 100 Вт, температура 110 градусов в течение 1 часа.
Вышеуказанный процесс был повторен одиннадцать раз и исходные реакционные смеси объединяли. Объединенную реакционную смесь упаривали досуха и осадок переносили в 0,1Н раствор гидроксида натрия (10 мл) и промывали простым эфиром (2×20 мл), водный слой отделяли и подкисляли 1Н соляной кислотой. Сырой продукт затем повторно экстрагировали обратно эфиром и сушили над сульфатом натрия. После фильтрации и выпаривания получали твердый продукт, который очищали с помощью хроматографии на Companion (колонка 4 г, градиент растворителей: 10-70% простой эфир : изогексан) с получением рацемического 5-(((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)метил)-1H-тетразола (1,81 г, 5,50 ммоль, выход 39%) в виде бесцветного твердого вещества.
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиной кислоты) способ 4 мин, 5-95% ацетонитрил/вода): m/z 264 (М+Н)+ (ES+); 262 (M-H)- (ES-), при 2,05 мин, чистота 98% @ 210 нм.
1H ЯМР (400 МГц, ДМСО-d6): δ 16,12 (1Н, br. s), 5,37 (1H, d, J=1,0), 4,80 (2Н, s), 3,22 (1Н, br. s), 3,02 (2H, s), 2,92-2,84 (1H, m), 2,50-2,44 (1H, m), 2,18-2,05 (4H, m), 1,64 (1H, dd, J=12,4, 7,4), 1,06 (3H, t, J=7,4) млн-1.
Стадия 2 (соединение формулы 13)
Рацемический трет-бутил (((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил) карбамат
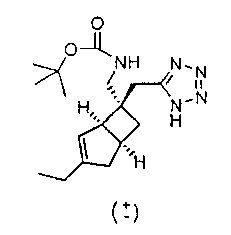
К раствору рацемического 5-(((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил)метил)-1H-тетразола (400 мг, 1,519 ммоль) в растворителе, состоящем из этанола (50 мл) и воды (15 мл) добавляли порошок железа (848 мг, 15,19 ммоль) и хлорид аммония (488 мг, 9,12 ммоль). Смесь перемешивали и нагревали с обратным холодильником в течение 1 часа. Реакционную смесь оставляли охлаждаться и фильтровали через слой целита и тщательно промывали этанолом, а фильтрат упаривали досуха.
Вышеуказанный осадок растворяли в смеси воды (20 мл) и тетрагидрофурана (60 мл) к полученному раствору добавляли ди-трет-бутилдикарбонат (2,8 г, 12,86 ммоль) и триэтиламин (1,79 мл, 12,86 ммоль) и затем нагревали и перемешивали при 40 градусах в течение 1 часа.
Реакционную смесь упаривали до половины исходного объема, и остаток подкисляли добавлением 10%-ного водного раствора лимонной кислоты. Сырой продукт затем экстрагировали этилацетатом, органический слой отделяли, промывали водой и сушили над сульфатом натрия. Сырой продукт очищали с помощью хроматографии на силикагеле (колонка 12 г, градиент растворителя 0-70% простой эфир : изогексан) с получением рацемического трет-бутил (((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил) карбамата (380 мг, 1,140 ммоль, выход 89%) в виде прозрачной бесцветной смолы.
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиная кислота) способ 4 мин, 5-95% ацетонитрил/вода): m/z 334 (М+Н)+ (ES+); 332 (M-H)- (ES-), при 2,34 мин, чистота 98% @ 210 нм.
ЖХ-МС (Agilent, X-Select, Waters X-Bridge C18, 2,5 мкм, 4,6×30 мм, основный (0,1% бикарбонат аммония) способ 4 мин, 5-95% ацетонитрил/вода): m/z 334 (М+Н)+ (ES+); 332 (M-H)- (ES-), при 1,51 мин, чистота 98% @ 215 нм.
1H ЯМР (400 МГц, CD3OD): δ 6,96 (1Н, t, J=5,8), 5,43 (1Н, br. s), 3,2-3,15 (2Н, m), 3,11 (1Н, br. s), 2,99 (1H, d, J=14,9), 2,89 (1H, d, J=14,9), 2,84-2,76 (1H, m), 2,51 (1H, dd, J=16,4, 7,8), 2,17 (2H, q, J=7,4), 2,07 (1H, br. d, J=16,4), 1,90 (1H, ddd, J=11,8, 8,7, 2,6), 1,56 (1H, dd, J=12,1, 7,3), 1,46 (7H, s, tBu (главный ротамер)), 1,44 (2H, s, tBU (второстепенный ротамер)), 1,11 (3H, t, J=7,5) млн-1, (дихлорметан также присутствует: 5,49 (0,5Н, s)).
Стадия 3 (соединение формулы 2)
Рацемический ((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамин
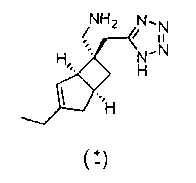
К раствору рацемического трет-бутил (((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил) карбамата (1,3 г, 3,90 ммоль) в дихлорметане (50 мл) добавляли трифторуксусную кислоту (30 мл, 389 ммоль) и смесь оставляли стоять при комнатной температуре в течение 30 минут.
Смесь выпаривали досуха и осадок растворяли в толуоле (60 мл) и выпаривали досуха, эту процедуру повторяли три раза. Осадок растворяли в смеси 1:1 метанола и воды (30 м). Этот раствор наносили на Dowex® 50WX8 водородная форма 100-200 меш с ионообменной смолой (10 г). Смолу промывали и элюировали водой до тех пор, пока элюант не становился нейтральным. Затем продукт элюировали с использованием 2N метанольного раствора аммиака с получением после выпаривания бесцветной смолы. Этот осадок растирали с ацетонитрилом (13 мл) с получением рацемического ((1R,5S,6S)-6-((1Н-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамина в виде бесцветного твердого вещества (365 мг, 1,50 ммоль, 38,5%).
ЖХ-МС (Agilent, X-Select, Waters X-Select C18, 2,5 мкм, 4,6×30 мм, кислый (0,1% муравьиная кислота) способ 4 мин, 5-95% ацетонитрил/вода): m/z 234 (М+Н)+ (ES+); 232 (M-H)- (ES-), при 1,01 мин.
ЖХ-МС (Agilent, X-Select, Waters X-Bridge C18, 2,5 мкм, 4,6×30 мм, основный (0,1% бикарбонат аммония) способ 4 мин, 5-95% ацетонитрил/вода): m/z 234 (М+Н)+ (ES+); 232 (M-H)- (ES-), при 1,21 мин.
1H ЯМР (400 МГц, CD3OD): δ 5,40 (1Н, d, J=1,8), 3,15 (3H, m), 3,05 (1Н, d, J=15,3), 2,94 (1Н, d, J=15,3), 2,83 (1H, m), 2,52 (1H, br. dd, J=16,4, 7,8), 2,19 (2H, q, J=7,8), 2,10 (1H, br. d), 1,93 (1H, ddd, J=12,4, 8,7, 2,7), 1,64 (1H, dd, J=12,6, 7,5), 1,13 (3H, t, 7=7,4) млн-1.
13C ЯМР (100 МГц, CD3OD): δ 159,86, 151,60, 122,57, 53,69, 47,96, 44,73, 43,03, 36,87, 30,21, 25,40, 12,84 млн-1.
Стадия 4
Хиральное разделение энантиомеров рацемического ((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамина
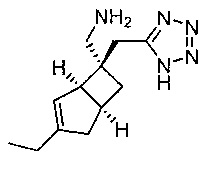

Рацемический ((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамин (155 мг) растворяют используя Diacel Chiralpak IC, 5 мкм, 20×250 мм, 15 мл/мин, 50% этанол:50% изогексан. Для получения после испарения фракций время удерживания пик 1 9,91 мин (32 мг) и время удерживания пик 2 18,91 мин (28 мг).
Аналитическая хиральная хроматография: Diacel Chiralpak IC, 5 мкм, 4,6×250 мм, способ 30 мин, 1,5 мл/мин, 30% этанол:70% изогексан, время удерживания пик 1 9,49 мин %, время удерживания пик 2 19,41 мин при 215 нм.
Пример 3: Синтез соединения формулы 2
Синтез рацемического N-((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил) этанамина
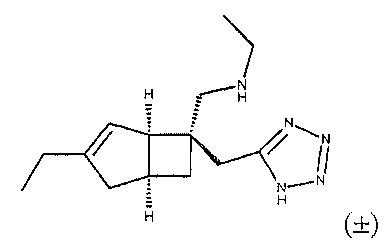
К суспензии рацемического ((1R,5S,6S)-6-((1Н-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамина (соединение формулы 2) (50 мг, 0,214 ммоль) в сухом дихлорэтане (10 мл) добавляли ацетальдегид (121 мкл, 2,143 ммоль) и полученную смесь перемешивали в течение 20 минут, в течение этого времени раствор становился прозрачным.
Растворитель удаляли на роторном испарителе, и осадок переносили в этанол (10 мл) и к этому раствору добавляли боргидрид натрия (81 мг, 2,143 ммоль) и полученную смесь перемешивали в течение 20 мин.
Реакционную смесь подкисляли до pH 1 путем добавления по каплям 1N соляной кислоты и полученную смесь упаривали досуха.
Сырой продукт очищали с помощью препаративной ВЭЖХ (Waters, основный (0,1% бикарбонат аммония), Basic, Waters X-Bridge Prep-C18, 5 мкм, 19×50 мм колонка, 20-50% ацетонитрил в воде) с получением N-(((1R,5S,6S)-6-((1H-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил)метил) этанамина (24 мг, 0,090 ммоль, выход 42,0%) в виде бесцветного твердого вещества.
ЖХ-МС (Agilent, Basic, Waters X-Bridge C18, 2,5 мкм, 4,6×30 мм, основный (0,1% Бикарбонат аммония) способ 4 мин, 5-95% ацетонитрил/вода): m/z 262 (М+Н)+ (ES+); 260 (M-H)- (ES-), при 1,32 мин, чистота 98% @ 215 нм.
1H ЯМР (400 МГц, CD3OD): δ 5,35 (1Н, d, J=1,8), 3,25 (2Н, dd, J=16,0, 13,0), 3,18-3,12 (3H, m), 3,08 (1Н, d, J=15,5), 2,99 (1H, d, J=15,5), 2,88 (1H, quin, J=7,4), 2,52 (1H, dd, J,=16,4, 7,8), 2,21-2,16 (2H, m), 2,10 (1H, d, J=16,4), 1,99 (1H, ddd, J=12,4, 8,7, 2,7), 1,60 (1H, dd, J=12,4, 7,5), 1,40 (3H, t, J=7,3), 1,12 (3H, t, J=7,4) млн-1.
13С ЯМР (100 МГц, CD3OD): δ 160,10, 151,80, 122,38, 56,59, 53,89, 45,05, 44,50, 42,99, 37,25, 31,93, 31,08, 25,39, 12,80, 11,53 млн-1.
Пример 4: Альтернативный путь получения соединения 10
Стадия 1 (соединение формулы 14)
Рацемический (2Е/Z)-2-((1R,5S)-3-этил-6-бицикло[3.2.0]гепт-3-енилиден) ацетонитрил
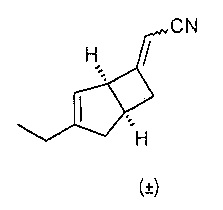
К раствору 1,78М трет-бутоксида калия в тетрагидрофуране (64 мл, 113,9 ммоль), разбавленного в тетрагидрофуране (45 мл) и охлажденного до 0°C добавляли диэтил цианометилфосфат (21,16 г, 119 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 минут и давали нагреться до комнатной температуры и перемешивали еще в течение 30 минут. Смесь переносили в капельную воронку с уравновешенным давлением и добавляли по каплям к раствору рацемического (1R,5S)-3-этилбицикло[3.2.0]гепт-3-ен-6-она (14,8 г, 109 ммоль) в тетрагидрофуране (140 мл) при 0°C.
Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Смесь разбавляли насыщенным водным раствором хлорида аммония (100 мл) и этилацетатом (200 мл) и слои разделяли. Водный слой экстрагировали этилацетатом (50 мл), а объединенные органические слои промывали насыщенным солевым раствором (50 мл) и сушили над сульфатом магния. Осадок после фильтрования очищали с помощью хроматографии на силикагеле (2,5% этилацетат : изогексан) с получением рацемического (2Е)-2-((1R,5S)-3-этил-6-бицикло[3.2.0]гепт-3-енилиден) ацетонитрила в виде смеси E/Z изомеров (14,45 г, 84%).
ЖХ-МС (Agilent, Waters SunFire С18, 4,6×30 мм, муравьиная кислота, ацетонитрил/вода): m/z 160,2 (М+Н)+ (ES+) при 2,88 мин.
1H ЯМР (400 МГц, CDC13): приблизительно 60:40 смесь изомеров алкена δ 5,43 (0,4Н, m), 5,23 (0,6Н, m), 5,09 (0,6Н, m), 4,98 (0,4Н, m), 4,12 (0,4Н, br s), 3,93 (0,6H, br s), 3,19-2,90 (2H, m), 2,74-2,46 (2H, m), 2,29-2,07 (2H, m), 1,14-1,06 (3H, m).
Стадия 2 (соединение формулы 6)
Рацемический 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрил
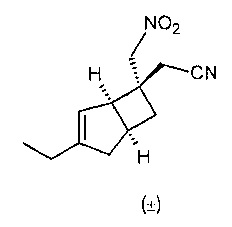
К раствору (1R,5S)-3-этилбицикло[3.2.0]гепт-3-ен-6-она (9,59 г, 60,3 ммоль) в нитрометане (75 мл, 84,6 г, 1,38 моль) в атмосфере азота добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (10 мл, 10,2 г, 66,9 ммоль) и полученную смесь перемешивали в течение 18 ч при комнатной температуре.
Реакционную смесь выливали в 5%-ный водный раствор дигидрофосфата калия ортофосфата (400 мл) и добавляли этилацетат (300 мл). Слои разделяли, и водный слой дополнительно экстрагировали этилацетатом (2×150 мл). Объединенные органические слои сушили над сульфатом магния и упаривали с получением сырого продукта, который был совмещен с сырым продуктом предыдущей реакции, осуществленной наполовину. Осадок очищали с помощью хроматографии на силикагеле (5-10% этилацетат : изогексан) с получением рацемического 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрила (16,5 г, 75 ммоль, выход 83%) в виде приблизительно 2:1 смеси диастереомеров. Данные для основного диастереомера.
ЖХ-МС (Agilent, Waters SunFire C18, 4,6×30 мм, кислый (0,05% муравьиная кислота, способ 6 мин, 3-97% ацетонитрил/вода): m/z 221 (M+H)+ (ES+) при 2,79 мин.
1H ЯМР (400 МГц, ДМСО-d6): δ 5,32 (1Н, d, J=2,1), 4,87 (2Н, s), 3,16 (1Н, br. s), 2,97-2,82 (1Н, m), 2,65 (2Н, s), 2,48-2,40 (1Н, m), 2,23 (1Н, ddd, J=12,4, 8,8,2,5), 2,16 - 2,02 (3H, m), 1,56 (1H, dd, J=12,5, 7,2), 1,07 (3H, t, J=7,5) млн-1.
Стадия 3 (соединение формулы 10)
Рацемический 5-(((1R,5S,6S)-3-этил-6-(нитрометил)-6-бицикло[3.2.0]гепт-3-енил)метил)-1H-тетразол
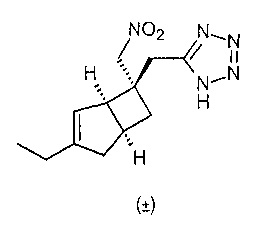
К раствору рацемического 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрила (200 мг, 0,909 ммоль) в сухом толуоле (4 мл) добавляли азидотриметилсилан (590 мкл, 4,44 ммоль) и оксид дибутилолова (113 мг, 0,45 ммоль). Сосуд герметизировали и нагревали до 110°C в течение 18 часов. Смесь охлаждали до комнатной температуры и распределяли между водой (20 мл) и этилацетатом (20 мл). Органический слой обрабатывали 2М раствором гидроксида натрия (20 мл) и водный слой отделяли, а затем подкисляли концентрированной соляной кислотой до приблизительно pH 1. Кислый водный слой повторно экстрагировали этилацетатом (2×20 мл) и объединенные органические слои сушили над сульфатом магния. После фильтрации и выпаривания получали сырой продукт, который очищали с помощью хроматографии на силикагеле (50% простой эфир:изогексан) с получением рацемического 5-(((1R,5S,6S)-3-этил-6-(нитрометил)-6-бицикло[3.2.0]гепт-3-енил)метил)-1H-тетразола (10 мг, 0,038 ммоль, выход 4%).
ЖХ-МС (Agilent, Waters SunFire C18, 4,6×30 мм, кислый (0,05% муравьиной кислоты, способ 6 мин, 3-97% ацетонитрил/вода): m/z 264 (М+Н) + (ES+); 262 (M-H) - (ES-), при 2,35 мин.
1H ЯМР (400 МГц, ДМСО-d6): δ 11,09 (1Н, br. s), 5,37 (1Н, d, J=1,2), 4,80 (2Н, s), 3,22 (1Н, br. s), 3,01 (2H, s), 2,93 - 2,81 (1H, m), 2,50-2,40 (1H, m), 2,19-2,05 (4H, m), 1,63 (1H, dd, J=12,5, 7,5), 1,05 (3H, t, J=7,6) млн-1.
Альтернативные условия для стадии 3 (соединение формулы 10)
Рацемический 5-(((1R,5S,6S)-3-этил-6-(нитрометил)-6-бицикло[3.2.0]гепт-3-енил)метил)-1H-тетразол
К раствору рацемического 2-((1R,5S,6S)-3-этил-6-(нитрометил)бицикло[3.2.0]гепт-3-ен-6-ил) ацетонитрила (170 мг, 0,772 ммоль) в 1-метил-2-пирролидиноне (2,7 мл) добавляли гидрохлорид пиридина (180 мг, 1,57 ммоль) и азид натрия (263 мг, 4,04 ммоль). Колбу нагревали в атмосфере азота до 100°C в течение 18 часов. Температуру колбы повышали до 117-120°C в течение еще 4 ч, после чего ее оставляли охлаждаться до комнатной температуры. Смесь выливали в воду (20 мл) и осторожно подкисляли с помощью водной 2М соляной кислоты. Водный слой экстрагировали этилацетатом (2×20 мл), а затем органический слой взбалтывали с раствором 2М гидроксида натрия (1×20 мл, 1×10 мл). Объединенные водные слои затем подкисляли концентрированной соляной кислотой до приблизительно pH1 и повторно экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали водой (10 мл) и сушили над сульфатом магния. После фильтрации и выпаривания получали сырой продукт, который очищали с помощью хроматографии на силикагеле (7 г диоксида кремния, диэтиловый эфир : изогексан : уксусная кислота 200:300:8) с получением рацемического 5-(((1R,5S,6S)-3-этил-6-(нитрометил)-6-бицикло[3.2.0]гепт-3-енил)метил)-1Н-тетразола (81 мг, 0,304 ммоль, выход 40%).
ЖХ-МС (Agilent, Waters SunFire С18, 4,6×30 мм, кислый (0,05% муравьиной кислоты, способ 6 мин, 3-97% ацетонитрил/вода): m/z 264 (М+Н)+ (ES+); 262 (M-H)- (ES-), при 2,35 мин.
1H ЯМР (400 МГц, ДМСО-d6): δ 16,10 (1Н, br. s), 5,34 (1Н, d, J=1,4), 4,80 (2Н, s), 3,22 (1Н, br. s), 3,02 (2H, s), 2,94-2,81 (1H, m), 2,48-2,40 (1H, m), 2,19-2,05 (4H, m), 1,64 (1H, dd, J=12,5, 7,4), 1,05 (3H, t, J=7,4) млн-1.
Пример 5
Терапевтическая активность соединений по настоящему изобретению, продемонстрирована с помощью анализа аффинного связывания α2δ-1. Это испытание проводили следующим образом.
Анализ связывания субъединиц α2δ-1 кальциевого канала
В данном разделе раскрыт сцинтилляционный проксимальный анализ (SPA) для измерения [3H] габапентина ([3H]GBP) связывающегося с мембранами, содержащими α2δ-1, и его использование для профилирования соединений (Calvo et al (2012) J. Biomol. Screen. 17: 1041-1049).
Мембраны с кальциевыми каналами человека Cav1,2/β3/α2δ-1 (тест Chan) размораживали на льду, аликвотили и хранили при -80°C для последующего использования. Мембраны разводили до 200 мкг/мл (3 мкг/лунку конечной концентрации для анализа (FAC)) в буфере для анализа (10 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) (Sigma) (pH 7,4)). Стоковый раствор [3H]GBP (фирма Perkin Elmer) хранили при -20°C. [3H]GBP разбавляли до 40 нМ (10 нМ FAC) в буфере для анализа. SPA гранулы (Perkin Elmer) повторно суспендировали при 100 мг/мл в 10 мМ HEPES (pH 7,4). Гранулы разбавляли до 40 мг/мл (0,6 мг/лунку FAC) в буфере для анализа. Неспецифическое связывание (NSB) вызывали с использованием избытка прегабалина (Tocris). Прегабалин солюбилизировали в Milli-Q H2O при 10 мМ. 10 мМ прегабалин разбавляли до 400 мкМ (100 мкМ FAC) в буфере для анализа.
Соединения разбавляли до 100 мкМ затем половину протокола разбавляли. Затем разводили в соотношении 1:100 в буфере для анализа до концентрации для анализа  (1 мкМ FAC верхнее разведение).
(1 мкМ FAC верхнее разведение).
SPA гранулы 15 мкл; мембраны 15 мкл; прегабалин или буфер для анализа/испытуемое соединение 15 мкл и [3H]GBP 15 мкл были добавлены в белый 96 луночный планшет, (Perkin Elmer). Планшет для анализа герметизировали и перемешивали в течение 10 секунд на планшетном шейкере, затем помещали на подставку и вставляли в считывающее устройство. Планшет инкубировали в течение ночи (20 часов), а затем считывали на приборе 1450 MicroBeta TriLux Microplate Scintillation and Luminescence Counter при комнатной температуре окружающей среды (RT).
Анализ данных
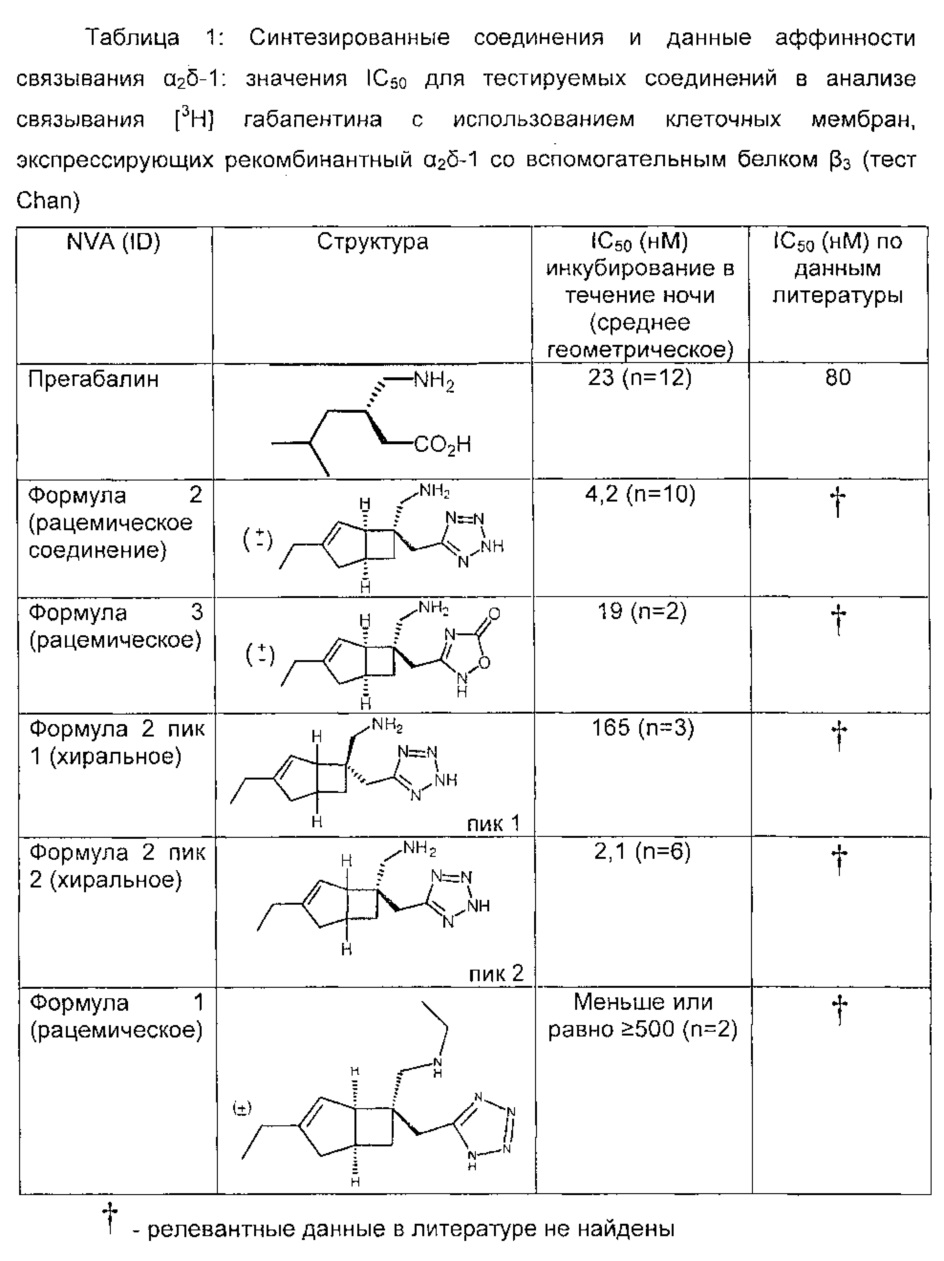
Значения NSB, полученные путем добавления 100 мкМ прегабалина вычитали из значений, полученных для соединений для генерирования значения специфического связывания. Специфическое связывание в импульсах в минуту (cpm) наносили в виде зависимости от концентрации соединения (M) и устанавливали с использованием 4 параметрического логистического уравнения. Составные величины IC50 были вычислены, где концентрация соединения дает 50% ингибирование специфического связывания. Результаты испытаний соединений представлены в таблице 1.
Пример 6
Метод определения кинетических параметров связывания (ассоциации и диссоциации) и аффинности (КД) аналогов ГАМК к субъединицам α2δ-1.
Хорошо известно в данной области техники, что аффинность связывания может быть выражена как КД или равновесная константа диссоциации, где увеличение аффинности связывания коррелирует с уменьшением КД, которое может быть вычислено из кинетических констант связывания. Кинетический анализ связывания аналогов ГАМК с субъединицами α2δ-1 можно определить методом поверхностного плазмонного резонанса, с применением приборов Biacore™ SPR (Biacore, GE Healthcare, Uppsala). Получают кинетические скорости ассоциации (ka, k-on) и скорости диссоциации (kd; k-off). Значения константы равновесия диссоциации (KD; аффинность) рассчитывают как k-off/k-on.
В предшествующем уровне техники и в научных публикациях короткий регион в пределах полной длины субъединицы α2δ-1 потенциалзависимых кальциевых каналов, упоминается как сайт связывания габапентина и прегабалина (Wang et al (1999) Biochem. J. 342, 313-320; Field et al (2006) Proc. Natl: Acad. Sci. USA, 14, 103, 17537-17542).
В этом примере описано, что аналоги ГАМК ((1R,5S,6S)-6-((1Н-тетразол-5-ил)метил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил) метанамин ("тестируемое соединение": получено, как описано в примере 2) и (1R,5S,6S)-6-(аминометил)-3-этилбицикло[3.2.0]гепт-3-ен-6-ил] уксусная кислота ("контрольное соединение": получено, как описано в WO 2010/079668 и US 2012/0071685 A1) способны связываться с рекомбинантным фрагментом субъединицы потенциал-зависимого кальциевого канала человека (α2δ-1, CACNA2D1). Кроме того, сравнительный анализ кинетического связывания этих двух соединений проводили для того, чтобы выявить и охарактеризовать потенциально различные связывающие свойства этих двух соединений для их мишени (CACNA2D1).
Человеческий рекомбинантный фрагмент CACNA2D1 ковалентно иммобилизовали при высокой плотности (26000 RU) на поверхности оптического сенсорного чипа Biacore CM5 в качестве целевого лиганда с использованием соединительной химии тиола, в соответствии с инструкциями изготовителя (Biacore Thiol Coupling Kit, Order Code: Br-10057; GE-Healthcare, Uppsala). Бычий сывороточный альбумин (BSA) иммобилизовали в контрольную проточную кювету для того, чтобы компенсировать неспецифический фон связывания. Повышающие концентрации тестируемого соединения и контрольного соединения, растворенных в буфере Biacore HBS-P (Order Code: BR-100368; GE-Healthcare, Uppsala), пропускали через проточные кюветы при скорости потока 30 мкл/мин. Поверхности сенсорного чипа восстанавливали после каждого прогона 10 мМ соляной кислотой (скорость потока равна 30 мкл/мин), чтобы удалить предварительно связанный материал и заново сформировать сайты связывания активных соединений. Затем формировали субтрактивные сенсограммы (CACNA2D1 рекомбинантный белок минус контрольный BSA).
Кинетический анализ связывания впоследствии проводили с помощью подгонки математической одиночной сенсограммы каждой субтрактивной сенсограммы связывания с использованием алгоритма взаимодействия Langmuir 1:1, программы Biaevaluation 4.0.
Экспериментальные результаты, описанные в данном примере, показали, что оба соединения, при их исследовании, были способны специфически связываться с человеческим рекомбинантным белком CACNA2D1. Кроме того, данные, описанные в данном примере, позволяют разделить свойства связывания тестируемого соединения и контрольного соединения. Эти различия позволяют прогнозировать повышенную фармакологическую активность тестируемого соединения через контрольное соединение.
Следует понимать, что приведенные выше примеры представлены в качестве иллюстрации настоящего изобретения и не предназначены для ограничения его каким-либо образом. Объем настоящего изобретения должен, следовательно, быть определен со ссылкой на приложенную формулу изобретения, наряду с полным объемом эквивалентов, к которым данная формула относится.