Результат интеллектуальной деятельности: АГОНИСТЫ МУСКАРИНОВЫХ РЕЦЕПТОРОВ
Вид РИД
Изобретение
Данное изобретение относится к классу новых мостиковых бициклических соединений, их солям, фармацевтическим композициям, содержащим их, а также их применению в терапии человеческого организма. В частности, данное изобретение нацелено на класс соединений, которые являются агонистами мускаринового рецептора M1 и/или рецептора М4, и, следовательно, применяются в лечении болезни Альцгеймера, шизофрении, когнитивных расстройств и других заболеваний, опосредованных мускариновыми рецепторами М1/М4, а также в лечении или облегчении боли.
УРОВЕНЬ ТЕХНИКИ
Мускариновые ацетилхолиновые рецепторы (mAChR) являются представителями суперсемейства рецепторов, сопряженных с G белком, которые опосредуют работу нейромедиатора ацетилхолина, как в центральной, так и в периферической нервной системе. Было клонировано пять подтипов mAChR, от M1 до М5. mAChR M1 преимущественно постсинаптически экспрессируется в коре головного мозга, гиппокампе, стриатуме и таламусе; mAChR М2 расположены преимущественно в стволе головного мозга и таламусе, но также и в коре головного мозга, гиппокампе и полосатом теле, где они расположены на холинергических синоптических окончаниях (Langmead et al., 2008 Br J Pharmacol). Тем не менее, mAChR M2 также экспрессируются по периферии на ткани сердца (где они опосредуют вагусную иннервацию сердца), а также в гладких мышцах и железах внешней секреции. mAChR М3 экспрессируются на относительно низком уровне в ЦНС, но широко экспрессируются в гладких мышцах и железистых тканях, таких как потовые и слюнные железы (Langmead et al., 2008 Br J Pharmacol).
Мускариновые рецепторы в центральной нервной системе, особенно mAChR M1, играют решающую роль в опосредовании более высокой когнитивной обработки. Заболевания, связанные с когнитивными нарушениями, такие как болезнь Альцгеймера, сопровождаются гибелью холинергических нейронов в базальных отделах переднего мозга (Whitehouse et al., 1982 Science). При шизофрении, которая также характеризуется когнитивными - нарушениями, плотность mAChR снижается в префронтальной коре, гиппокампе и дорсальном полосатом теле субъектов с шизофренией (Dean et al., 2002 Mol Psychiatry). Кроме того, в моделях на животных было показано, что блокада или поражение центральных холинергических путей приводят к значительным когнитивным расстройствам, а неселективные антагонисты mAChR вызывают психотомиметические эффекты у пациентов с психическими расстройствами. Холинергическая заместительная терапия в значительной степени была основана на применении ингибиторов ацетилхолинэстеразы с целью предотвращения распада эндогенного ацетилхолина. Эти соединения демонстрировали эффективность в отношении симптоматического клинически выраженного снижения когнитивных способностей, но приводили к дозолимитирующим побочным эффектам в результате стимуляции периферических mAChR М2 и М3, в том числе к нарушенной двигательной активности желудочно-кишечного тракта, брадикардии, тошноте и рвоте (http://www.drugs.com/pro/donepezil.html; http://www.drugs.com/pro/rivastigmine.html).
Дальнейшие попытки исследования были направлены на идентификацию прямых агонистов mAChR M1 для целевого усиления когнитивной функции. Такие попытки привели к идентификации ряда агонистов, примерами которых являются такие соединения, как ксаномелин, AF267B, сабкомелин, миламелин и цевимелин. Многие из этих соединений продемонстрировали высокую эффективность в доклинических моделях познания как у грызунов, так и/или у не относящихся к человеку приматов. Миламелин продемонстрировал эффективность в отношении скополамин-индуцированного дефицита в кратковременной и пространственной памяти у грызунов; сабкомелин показал эффективность в задаче на различение визуального объекта у мармозеток, а ксаномелин инвертировал индуцированный антагонистом mAChR дефицит когнитивного функционирования в парадигме пассивного избегания.
Болезнь Альцгеймера (БА) является наиболее распространенным нейродегенеративным заболеванием (26,6 миллиона человек во всем мире в 2006 году), которое поражает пожилых людей, приводя к полной потере памяти и когнитивной дисфункции. Этиология данного заболевания сложна, но характеризуется двумя характерными осложнениями на головной мозг: агрегатами амилоидных бляшек, в основном состоящими из β-амилоидного пептида (Аβ), и нейрофибриллярными клубками, образованными гиперфосфорилированными тау-белками. Накопление Аβ считается основным признаком развития БА и, в связи с этим, многие возможные терапии для лечения БА в настоящее время нацелены на ингибирование образования Аβ. Аβ получают в результате протеолитического расщепления мембраносвязанного белка-предшественника амилоида (АРР). Процессинг АРР происходит двумя путями, неамилоидогенным и амилоидогенным. Расщепление АРР γ-секретазой является общим для обоих путей, но в первом АРР расщепляется α-секретазой с образованием растворимого АРРα. Сайт расщепления находится в пределах последовательности Аβ, тем самым препятствуя его образованию. Тем не менее, при амилоидогенном пути АРР расщепляется β-секретазой с образованием растворимого АРРβ, а также Аβ. Исследования in vitro показали, что агонисты mAChR могут способствовать процессингу АРР по растворимому, неамилоидогенному пути. Исследования in vivo показали, что агонист mAChR, AF267B, модифицировал АД-подобную патологию у трансгенной мыши 3×TgAD, модели различных компонентов болезни Альцгеймера (Caccamo et al., 2006 Neuron). И, наконец, агонист mAChR, цевимелин, продемонстрировал небольшое, но существенное снижение уровней Аβ в спинномозговой жидкости у пациентов, страдающих болезнью Альцгеймера, тем самым демонстрируя потенциальную болезнь-модифицирующую эффективность (Nitsch et al., 2000 Neurol).
Кроме того, доклинические исследования свидетельствовали о том, что агонисты mAChR проявляют атипичный антипсихотически-подобный профиль в ряде доклинических парадигм. Агонист mAChR, ксаномелин, инвертирует ряд управляемых дофамином особенностей поведения, в том числе индуцированную амфетамином локомоцию у крыс, индуцированное апоморфином лазание у мышей, управляемое агонистом дофамина вращение у крыс с односторонним поражением 6-OH-DA и индуцированное амфетамином двигательное беспокойство у обезьян (без склонности к EPS). Кроме того, было показано, что он ингибирует возбуждение дофаминовых клеток А10, но не А9, и условный рефлекс избегания, а также индуцирует экспрессию c-fos в префронтальной коре и прилежащем ядре, но не в полосатом теле у крыс. Все эти данные наводят на мысль об атипичном антипсихотически-подобном профиле (Mirza et al., 1999 CNS Drug Rev). Мускариновые рецепторы также вовлечены в нейробиологию наркотической зависимости. Подкрепляющий эффект кокаина и других вызывающих привыкание веществ опосредуется мезолимбической дофаминовой системой, где поведенческие и нейрохимические исследования показали, что холинергические мускариновые подтипы рецепторов играют важную роль в регуляции дофаминергической нейропередачи. Например, мыши М(4) (-/-) демонстрировали значительно повышенную в зависимости от вознаграждения активность поведения в результате воздействия кокаина (Schmidt et al Psychopharmacology (2011) Aug; 216(3): 367-78). Кроме того, ксаномелин демонстрировал блокировку эффектов от кокаина в этих моделях.
Мускариновые рецепторы также участвуют в контроле движений и потенциально представляют новые методы лечения двигательных расстройств, таких как болезнь Паркинсона, СДВГ, болезнь Хантингтона, синдром Туретта и другие синдромы, связанные с дофаминергической дисфункцией в качестве основного патогенетического фактора заболевания.
Каждый из ксаномелина, сабкомелина, миламелина и цевимелина продемонстрировали прогресс на различных стадиях клинических исследований при лечении болезни Альцгеймера и/или шизофрении. Фаза II клинических исследований с ксаномелином продемонстрировала его эффективность в отношении различных доменов когнитивных симптомов, в том числе поведенческих расстройств и галлюцинаций, связанных с болезнью Альцгеймера (Bodick et al., 1997 Arch Neurol). Это соединение также оценивали в небольшом исследовании II фазы шизофреников и обеспечивали значительное снижение позитивных и негативных симптомов по сравнению с контролем с помощью плацебо (Shekhar et al., 2008 Am J Psych). Тем не менее, во всех клинических исследованиях ксаномелин и другие родственные агонисты mAChR проявляли недопустимый предел безопасности в отношении холинергических побочных эффектов, включающих тошноту, боль в области желудочно-кишечного тракта, диарею, диафорез (чрезмерное потоотделение), гиперсаливацию (повышенное слюноотделение), обморок и брадикардию.
Мускариновые рецепторы участвуют в центральной и периферической болях. Боль можно подразделить на три различных вида: острую, воспалительную и нейропатическую. Острая боль имеет важную защитную функцию по защите организма от стимулов, которые могут приводить к повреждениям тканей, тем не менее, купировать послеоперационную боль необходимо. Воспалительная боль может возникать по многим причинам, в том числе из-за повреждения тканей, аутоиммунной реакции и патогенной инвазии, и инициируется под воздействием медиаторов воспаления, таких как нейропептиды и простагландины, которые приводят к нейрональному воспалению и боли. Нейропатическая боль связана с аномальными болевыми реакциями на неболевые стимулы. Нейропатическая боль связана с рядом различных заболеваний/травм, таких как повреждение спинного мозга, рассеянный склероз, сахарный диабет (диабетическая нейропатия), вирусная инфекция (такая как ВИЧ или герпес). Кроме того, она характерна при раке, как в результате заболевания, так и в виде побочного эффекта химиотерапии. Активация мускариновых рецепторов проявила себя в качестве болеутоляющего средства в ряде болевых состояний за счет активации рецепторов в спинном мозге и высших центрах болевой чувствительности в головном мозге. Повышение эндогенных уровней ацетилхолина с помощью ингибиторов ацетилхолинэстеразы, прямой активации мускариновых рецепторов агонистами или аллостерическими модуляторами продемонстрировало наличие анальгетической активности. В противоположность этому, блокада мускариновых рецепторов антагонистами или с использованием нокаутных мышей повышает болевую чувствительность. Данные о роли рецептора M1 в боли рассматриваются D.F. Fiorino и М. Garcia-Guzman, 2012.
В последнее время было идентифицировано небольшое количество соединений, которые проявляют улучшенную селективность в отношении mAChR подтипа M1 по сравнению с экспрессирующимися на периферии подтипами mAChR (Bridges et al., 2008 Bioorg Med Chem Lett; Johnson et al., 2010 Bioorg Med Chem Lett; Budzik et al., 2010 ACS Med Chem Lett). Несмотря на повышенный уровень селективности в отношении mAChR подтипа М3, некоторые из этих соединений сохраняют значительную агонистическую активность как при этом подтипе, так и при mAChR подтипа М2. В данном документе описывается ряд соединений, которые неожиданно проявляют высокие уровни селективности в отношении mAChR M1 и/или М4 по сравнению с рецепторами подтипов М2 и М3.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, обладающим активностью агонистов мускариновых рецепторов M1 или M1 и М4. Более конкретно, данное изобретение относится к соединениям, которые обладают селективностью в отношении рецептора M1 по сравнению с рецепторами подтипов М2 и М3.
Соответственно, в первом варианте реализации изобретения (Вариант реализации 1.1) данное изобретение относится к соединению формулы (1) или формулы (1а):
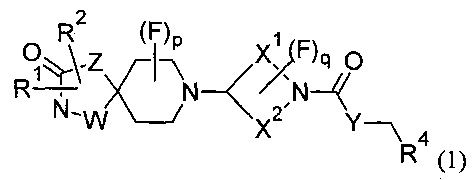
или его соли, где:
р равно 0, 1 или 2;
q равно 0, 1 или 2;
W представляет собой С или N;
Z представляет собой CH2, N, О или S;
Y представляет собой N, О, S или CH2;
X1 и X2 представляют собой насыщенные углеводородные группы, которые вместе содержат в общей сложности от пяти до девяти атомов углерода и которые соединены таким образом, что фрагмент:
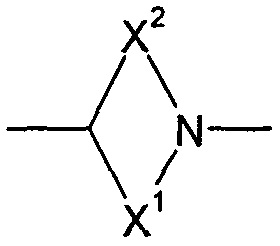
образует мостиковую бициклическую кольцевую систему;
R1 может представлять собой Н, галоген, CN, ОН, C1-3 алкокси, NH2, необязательно замещенный C1-6 алкил, необязательно замещенный С2-6 алкенил, необязательно замещенный С2-6 алкинил, необязательно замещенный С3-6 циклоалкил, необязательно замещенный С3-6 циклоалкенил, CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, NR5R6, COOR5, CONR5R6, NR7CONR5R6, NR7COOR5, OCONR5R6, SR5, SOR5, SO2R5; SO3R5;
R2 может независимо представлять собой Н, галоген, CN, ОН, C1-3 алкокси, NH2, необязательно замещенный C1-6 алкил, необязательно замещенный С2-6 алкенил, необязательно замещенный С2-6 алкинил, необязательно замещенный С3-6 циклоалкил, необязательно замещенный С3-6 циклоалкенил, CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, NR5R6, COOR5, CONR5R6, NR7CONR5R6, NR7COOR5, OCONR5R6, SR5, SOR5, SO2R5; или R1 и R2 вместе образуют необязательно замещенное циклоалкильное или гетероциклоалкильное кольцо;
R4 может представлять собой Н, необязательно замещенный C1-5 алкил, необязательно замещенный C1-5 алкенил, необязательно замещенный C1-5 алкинил, необязательно замещенный С2-6 циклоалкил, необязательно замещенный С2-6 циклоалкенил;
R5, R6 и R7 могут независимо представлять собой Н, C1-6 алкил;
или формулы (1а)
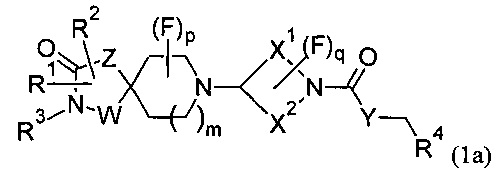
или его соли, где:
m равно 1 или 2;
р равно 0, 1 или 2;
q равно 0, 1 или 2;
W представляет собой С или N;
Z представляет собой CH2, N, О или S;
Y представляет собой N, О, S или CH2;
X1 и X2 представляют собой насыщенные углеводородные группы, которые вместе содержат в общей сложности от пяти до девяти атомов углерода и которые соединены таким образом, что фрагмент:
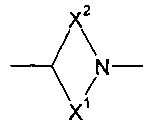
образует мостиковую, бициклическую кольцевую систему;
R1 может представлять собой Н, галоген, CN, ОН, C1-3 алкокси, NH2, необязательно замещенную C1-6 неароматическую углеводородную группу, где один или более атомов углерода необязательно замещены гетероатомом, выбранным из О, N или S, Wa или CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, NR5R6, COOR5, CONR5R6, NR7CONR5R6, NR7COOR5, OCONR5R6, SR5, SOR5, SO2R5; SO3R5;
R2 может независимо представлять собой Н, галоген, CN, ОН, C1-3 алкокси, NH2, необязательно замещенную C1-6 неароматическую углеводородную группу, где один или более атомов углерода необязательно замещены гетероатомом, выбранным из О, N или S, Wa или CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, NR5R6, COOR5, CONR5R6, NR7CONR5R6, NR7COOR5, OCONR5R6, SR5, SOR5, SO2R5; или R1 и R2 или R3 и R2 вместе образуют необязательно замещенное циклоалкильное или гетероциклоалкильное кольцо;
R3 может независимо представлять собой Н, ОН, необязательно замещенную C1-6 неароматическую углеводородную группу, где один или более атомов углерода необязательно замещены гетероатомом, выбранным из О, N или S, Wa или CH2-W8, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо; или R3 и R2 вместе образуют необязательно замещенное циклоалкильное или гетероциклоалкильное кольцо;
R4 может представлять собой Н, необязательно замещенный C1-5 алкил, необязательно замещенный C1-5 алкенил, необязательно замещенный C1-5 алкинил, необязательно замещенный C2-6 циклоалкил, необязательно замещенный C2-6 циклоалкенил;
R5, R6 и R7 могут независимо представлять собой Н, C1-6 алкил.
Конкретные и предпочтительные соединения формулы (1) или формулы (1а) являются такими, как определено в следующих ниже Вариантах реализации изобретения 1.2-1.66:
1.2 Соединение по Варианту реализации 1.1, где R1 представляет собой Н или C1-6 неароматическую углеводородную группу, содержащую 0, 1- или 2-кратные связи углерод-углерод, при этом углеводородная группа необязательно замещена одним-шестью атомами фтора и при этом один или два, но не все атомы углерода углеводородной группы могут быть необязательно замещены гетероатомом, выбранным из О, N и S, и их окисленных форм.
1.3 Соединение по любому из Вариантов реализации 1.1 и 1.2, где R1 выбран из Н; C1-6 алкила; C2-6 алкенила; C2-6 алкинила; и C1-6 неароматических углеводородных групп, состоящих из C3-6 циклоалкильной или C5-6 циклоалкенильной группы либо содержащих ее; каждый из указанных алкила, алкенила, алкинила и неароматических углеводородных групп необязательно замещены одним-шестью атомами фтора, и при этом один или два, но не все атомы углерода каждого из алкила, алкенила, алкинила и неароматических углеводородных групп могут быть необязательно замещены гетероатомом, выбранным из О, N и S, и их окисленных форм.
1.4 Соединение по Варианту реализации 1.1, где R1 представляет собой группу Wa или CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, или R1 и R соединены друг с другом с образованием кольца, которое может быть слитым или спироциклическим.
1.5 Соединение по Варианту реализации 1.1, где R1 представляет собой NR5R6, COOR5, CONR5R6, NR7CONR5R6, NR7COOR5, OCONR5R6, SR5, SOR5, SO2R5, где R5, R6 и R7 могут независимо представлять собой Н, C1-6 алкил.
1.6 Соединение по любому из Вариантов реализации 1.1-1.5, где R1 выбран из:
- Н;
- Галогена;
- Циано;
- ОН;
- C1-3 алкокси;
- NH2;
- C1-6 алкила, необязательно замещенного 1-6 атомами фтора;
- C3-6 алкила, необязательно замещенного 1 гетероатомом, выбранным из О, N или S;
- C2-6 алкенила;
- C2-6 алкинила;
- C3-6 циклоалкила;
- CH2-C3-6 циклоалкила;
- C5-6 циклоалкенила;
- CH2-арила;
- CH2-гетероарила;
- арила;
- гетероарила;
- NR5R6, где R5 и R6 независимо представляют собой Н, C1-6 алкил;
- COOR5, где R5 представляет собой Н, C1-6 алкил;
- CONR5R6, где R5 и R6 независимо представляют собой Н, C1-6 алкил;
- NR7CONR5R6, где R5, R6 и R7 независимо представляют собой Н, C1-6 алкил;
- NR7COOR5, где R5 и R7 независимо представляют собой Н, C1-6 алкил;
- OCONR5R6, где R5 и R6 независимо представляют собой Н, C1-6 алкил;
- SR5, где R5 представляет собой Н, C1-6 алкил;
- SOR5, где R5 представляет собой Н, C1-6 алкил;
- SO2R5, где R5 представляет собой Н, C1-6 алкил;
- SO3R5, где R5 представляет собой Н, C1-6 алкил;
- спироцикла формулы (CH2)n, где n равно 2, 3, 4, 5 или 6.
1.7 Соединение по Варианту реализации 1.6, где R1 представляет собой Н или C1-6 алкил, необязательно замещенный 1-6 атомами фтора.
1.8 Соединение по Варианту реализации 1.5, где R1 представляет собой Н или C1-5 алкил.
1.9 Соединение по любому из Вариантов реализации 1.1-1.8, где R2 представляет собой Н или C1-6 неароматическую углеводородную группу, содержащую 0, 1- или 2-кратные связи углерод-углерод, при этом углеводородная группа необязательно замещена одним-шестью атомами фтора и при этом один или два, но не все атомы углерода углеводородной группы могут быть необязательно замещены гетероатомом, выбранным из О, N и S и их окисленных форм.
1.10 Соединение по любому из Вариантов реализации 1.1-1.9, где R2 выбран из Н; C1-6 алкила; C2-6 алкенила; C2-6 алкинила и C1-6 неароматических углеводородных групп, состоящих из C3-6 циклоалкильной или C5-6 циклоалкенильной группы или содержащих ее; каждый из указанных алкила, алкенила, алкинила и неароматических углеводородных групп необязательно замещены одним-шестью атомами фтора, и при этом один или два, но не все атомы углерода каждого из алкила, алкенила, алкинила и неароматических углеводородных групп могут быть необязательно замещены гетероатомом, выбранным из О, N и S и их окисленных форм.
1.11 Соединение по любому из Вариантов реализации 1.1-1.8, где R2 представляет собой группу CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, или R1 и R2 соединены друг с другом с образованием кольца, которое может быть слитым или спироциклическим.
1.12 Соединение по любому из Вариантов реализации 1.1-1.8, где R2 представляет собой NR5R6, COOR5, CONR5R6, NR7CONR5R6, NR7COOR5, OCONR5R6, SR5, SOR5, SO2R5, где R5, R6 и R7 могут независимо представлять собой Н, C1-6 алкил.
1.13 Соединение по любому из Вариантов реализации 1.1-1.12, где R2 выбран из:
- Н;
- Галогена;
- Циано;
- ОН;
- C1-3 и алкокси;
- NH2;
- C1-6 алкила, необязательно замещенного 1-6 атомами фтора;
- C2-6 алкенила;
- C2-6 алкинила;
- C3-6 циклоалкила;
- C5-6 циклоалкенила;
- CH2-арила;
- CH2-гетероарила;
- NR5R6, где R5 и R6 независимо представляют собой Н, C1-6 алкил;
- COOR5, где R5 представляет собой Н, C1-6 алкил;
- CONR5R6, где R5 и R6 независимо представляют собой Н, C1-6 алкил;
- NR7CONR5R6, где R5, R6 и R7 независимо представляют собой Н, C1-6 алкил;
- NR7COOR5, где R5 и R7 независимо представляют собой Н, C1-6 алкил;
- OCONR5R6, где R5 и R6 независимо представляют собой Н, C1-6 алкил;
- SR5, где R5 представляет собой Н, C1-6 алкил;
- SOR5, где R5 представляет собой Н, C1-6 алкил;
- SO2R5, где R5 представляет собой Н, C1-6 алкил;
- SO3R5, где R5 представляет собой Н, C1-6 алкил.
1.13 Соединение по Варианту реализации 1.12, где R2 представляет собой Н или C1-6 алкил, необязательно замещенный 1-6 атомами фтора.
1.14 Соединение по Варианту реализации 1.13, где R2 представляет собой Н или C1-6 алкил.
1.15 Соединение по любому из Вариантов реализации 1.1-1.14, где R1 и R2 выбран из водорода и C1-6 алкила.
1.16 Соединение по Варианту реализации 1.15, где R1 и R2 независимо представляют собой Н, метил, этил, пропил, изопропил или бензил.
1.17 Соединение по Варианту реализации 1.1, где R1 и R2 вместе или R3 и R2 вместе образуют необязательно замещенное циклоалкильное или гетероциклоалкильное кольцо. Данное кольцо может замещать атом группы R3 азотом. Данное кольцо может быть слитым или спироциклическим.
1.18 Соединение по Варианту реализации 1.17, где R1 и R2 вместе образуют циклоалкильное кольцо, необязательно содержащее не более 2 гетероатомов, выбранных из О, S или N, и необязательно замещенное не более чем 6 атомами F.
1.19 Соединение по Варианту реализации 1.1, где R3 представляет собой Н, ОН или C1-6 неароматическую углеводородную группу, содержащую 0, 1- или 2-кратные связи углерод-углерод, при этом углеводородная группа необязательно замещена одним-шестью атомами фтора и при этом один или два, но не все атомы углерода углеводородной группы могут быть необязательно замещены гетероатомом, выбранным из О, N и S, и их окисленных форм.
1.20 Соединение по Варианту реализации 1.19, где R3 выбран из Н; ОН, C1-6 алкила; C2-5 алкенила; C2-6 алкинила; и C1-6 неароматических углеводородных групп, состоящих из C3-6 циклоалкильной или C5-6 циклоалкенильной группы, или содержащих ее; каждый из указанных алкила, алкенила, алкинила и неароматических углеводородных групп, необязательно замещены одним-шестью атомами фтора, и при этом один или два, но не все атомы углерода каждого из алкила, алкенила, алкинила и неароматических углеводородных групп могут быть необязательно замещены гетероатомом, выбранным из О, N и S, и их окисленных форм.
1.21 Соединение по Варианту реализации 1.19, где R3 представляет собой группу Wa или CH2-Wa, где Wa представляет собой необязательно замещенное 5- или 6-членное циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо, или R1 и R2 соединены друг с другом с образованием кольца, которое может быть слитым или спироциклическим.
1.22 Соединение по любому из Вариантов реализации 1.19-1.21, где R3 выбран из:
- Н;
- ОН;
- C1-6 алкила, необязательно замещенного 1-6 атомами фтора;
- C3-6 алкила, необязательно замещенного 1 гетероатомом, выбранным из О, N или S;
- C2-6 алкенила;
- C2-6 алкинила;
- C3-6 циклоалкила;
- CH2-C3-6 циклоалкила;
- C5-6 циклоалкенила;
- CH2-арила;
- CH2-гетероарила;
- арила;
- гетероарила;
1.23 Соединение по Варианту реализации 1.22, где R3 представляет собой Н или C1-6 алкил, необязательно замещенный 1-6 атомами фтора.
1.24 Соединение по Варианту реализации 1.5, где R3 представляет собой Н или C1-5 алкил.
1.25 Соединение по любому из Вариантов реализации 1.1-1.24, где Z представляет собой CH2, N, О или S.
1.26 Соединение по Варианту реализации 1.25, где Z представляет собой CH2, N или О.
1.27 Соединение по Варианту реализации 1.25, где Z представляет собой CH2.
1.28 Соединение по Варианту реализации 1.25, где Z представляет собой N.
1.29 Соединение по Варианту реализации 1.25, где Z представляет собой О. Если Z представляет собой О, R3 может быть задано как Н. В альтернативном варианте, если Z представляет собой О, m может быть задано как 2. В альтернативном варианте, если Z представляет собой О, либо R3 представляет собой Н, либо m равно 2.
1.30 Соединение по любому из Вариантов реализации 1.1-1.29, где R4 представляет собой Н или ациклическую C1-4 углеводородную группу, необязательно замещенную одним или более атомами фтора.
1.31 Соединение по Варианту реализации 1.30, где R4 представляет собой Н или ациклическую C1-3 углеводородную группу, необязательно замещенную одним или более атомами фтора.
1.32 Соединение по Варианту реализации 1.31, где R4 представляет собой Н или C1-3 алкильную группу, или C1-2 алкинильную группу.
1.33 Соединение по Варианту реализации 1.32, где R4 выбирают из Н, метила, фторметила, этила, этинила и 1-пропинила.
1.34 Соединение по Варианту реализации 1.33, где R4 представляет собой метил.
1.35 Соединение по любому из Вариантов реализации 1.1-1.34, где р равно 0 или 1.
1.36 Соединение по Варианту реализации 1.35, где р равно 0.
1.37 Соединение по Варианту реализации 1.35, где q равно 0 или 1.
1.38 Соединение по любому из Вариантов реализации 1.1-1.37, где m равно 0.
1.39 Соединение по любому из Вариантов реализации 1.1-1.37, где m равно 1.
1.40 Соединение по любому из Вариантов реализации 1.1-1.39, где Y представляет собой N, О или CH2.
1.41 Соединение по Варианту реализации 1.40, где Y представляет собой N.
1.42 Соединение по Варианту реализации 1.40, где Y представляет собой О.
1.43 Соединение по любому из Вариантов реализации 1.1-1.40, где W представляет собой С.
1.44 Соединение по Вариантам реализации 1.1-1.43, где мостиковая бициклическая кольцевая система представляет собой азабицикло-гептановую, азабицикло-октановую или азабицикло-нонановую кольцевую систему.
1.45 Соединение по Варианту реализации 1.44, где мостиковую бициклическую кольцевую систему выбирают из приведенных ниже кольцевых систем ВА-ВН, которые могут быть замещены 0-2 необязательными атомами фтора:

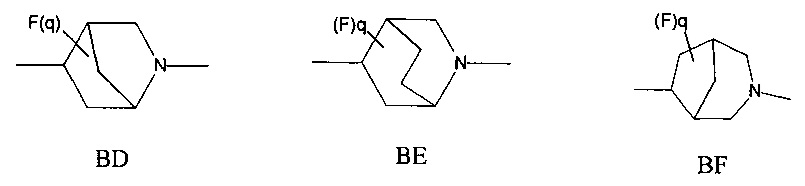
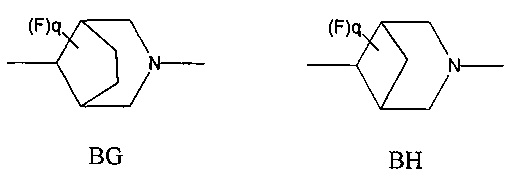
1.46 Соединение по Варианту реализации 1.45, где q равно 0 или 1.
1.47 Соединение по любому из Вариантов реализации 1.1-1.46, где R5 представляет собой Н или C1-6 алкил.
1.48 Соединение по Варианту реализации 1.47, где R5 представляет собой Н.
1.49 Соединение по Варианту реализации 1.47, где R5 представляет собой С1-3 алкил.
1.50 Соединение по любому из Вариантов реализации 1.1-1.49, где R6 представляет собой Н или С1-5 алкил.
1.51 Соединение по Варианту реализации 1.50, где R6 представляет собой Н.
1.52 Соединение по Варианту реализации 1.50, где R6 представляет собой С1-3 алкил.
1.53 Соединение по любому из Вариантов реализации 1.1-1.52, где R7 представляет собой Н или С1-5 алкил.
1.54 Соединение по Варианту реализации 1.53, где R7 представляет собой Н.
1.55 Соединение по Варианту реализации 1.53, где R7 представляет собой С1-3 алкил.
1.56 Соединение по Варианту реализации 1.1, имеющее формулу (2) или формулу (2а):
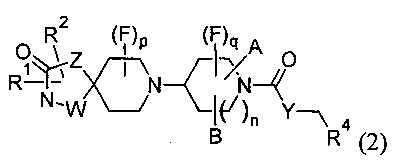
где т равно 1 или 2;
А и В соединены друг с другом с образованием углеродного мостика из 1-3 атомов, где n равно 1, или 1-2 атомов углерода, где n равно 2, а р, q, W, Z, Y, R1, R2 и R4 являются такими, как определено в любом из Вариантов реализации 1.1-1.43; или
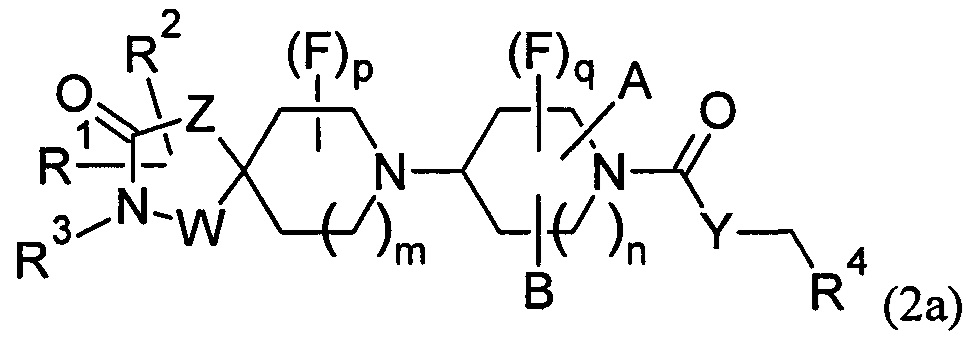
где n равно 1 или 2;
А и В соединены друг с другом с образованием углеродного мостика из 1-3 атомов, где n равно 1, или 1-2 атомов углерода, где n равно 2, a m, р, q, W, Z, Y, R1, R2, R3 и R4 являются такими, как определено в любом из Вариантов реализации 1.1-1.43.
1.57 Соединение по Варианту реализации 1.1, имеющее формулу (3) или формулу (3а):
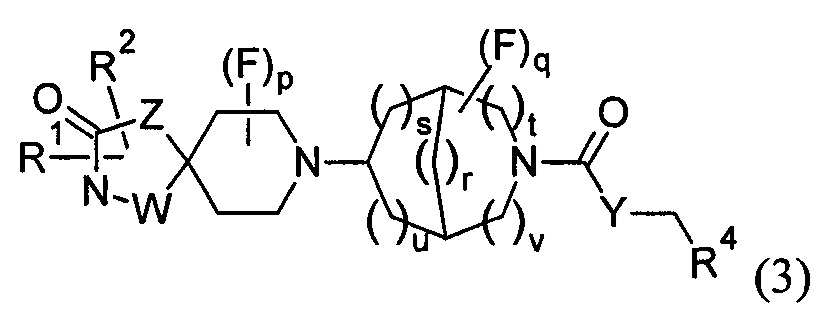
где r равно 1, 2 или 3, и каждое из s, t, u и v равно 0 или 1, при условии, что сумма r, s, t, u и v равна 3, 4 или 5, а р, q, W, Z, Y, R1, R2 и R4 являются такими, как определено в любом из Вариантов реализации 1.1-1.43; или
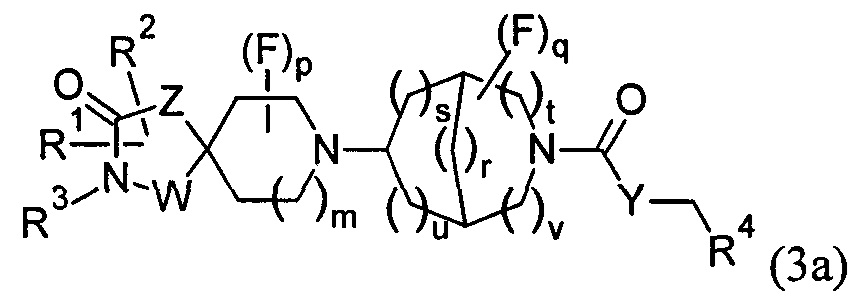
где r равно 1, 2 или 3, и каждое из s, t, u и v равно 0 или 1, при условии, что сумма r, s, t, u и v равна 3, 4 или 5, a m, р, q, W, Z, Y, R1, R2, R3 и R4 являются такими, как определено в любом из Вариантов реализации 1.1-1.43.
1.58 Соединение по Варианту реализации 1.1, имеющее формулу (4) или формулу (4а):
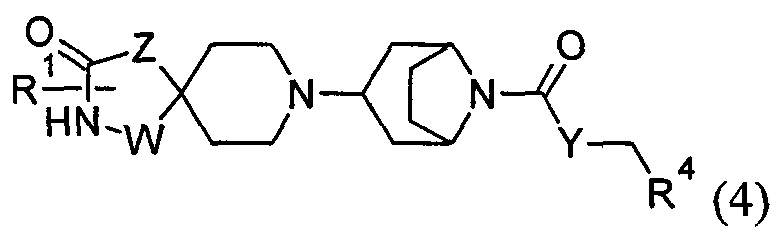
где R1, W, Z, Y и R4 являются такими, как определено в любом из Вариантов реализации 1.1-1.43; или
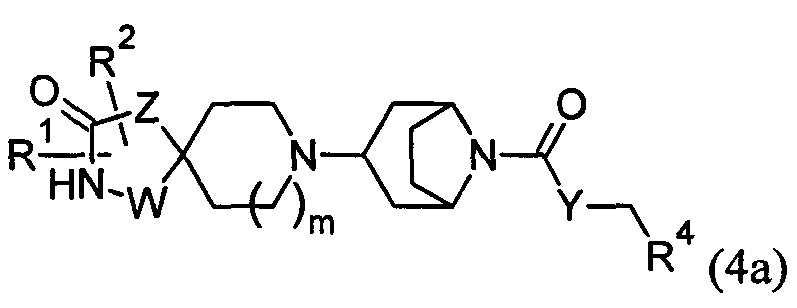
где m, R1, R2, W, Z, Y и R4 являются такими, как определено в любом из Вариантов реализации 1.1-1.43.
1.59 Соединение по любому из Вариантов реализации 1.56-1.58, где Z представляет собой CH2, N или О.
1.60 Соединение по любому из Вариантов реализации 1.56-1.59, где R4 выбран из Н, метила, этила, этинила и 1-пропинила.
1.61 Соединение по Варианту реализации 1.60, где R4 выбран из Н или метила.
1.62 Соединение по Варианту реализации 1.1, которое является таким, как определено в любом из Примеров от 1-1 до 9-2.
1.63 Соединение по любому из Вариантов реализации 1.1-1.61, имеющее молекулярную массу менее чем 550, например менее чем 500 или менее чем 450.
1.64 Соединение по любому из Вариантов реализации 1.1-1.63, которое находится в виде соли.
1.65 Соединение по Варианту реализации 1.64, отличающееся тем, что соль представляет собой кислотно-аддитивную соль.
1.66 Соединение по Варианту реализации 1.64 или Варианту реализации 1.65, отличающееся тем, что соль представляет собой фармацевтически приемлемую соль.
Определения
В этой заявке используются следующие определения, если не указано иное.
Термин "лечение", в отношении применения соединений формулы (1) или формулы (1а), используется для описания любой формы вмешательства, когда соединение вводят субъекту, страдающему от заболевания или расстройства, о котором идет речь, или подверженному риску, или потенциально подверженному риску такого заболевания или расстройства. Таким образом, термин "лечение" охватывает как превентивное (профилактическое) лечение, так и лечение, в котором представлены поддающиеся измерению или обнаружению симптомы заболевания или расстройства.
Термин "терапевтически эффективное количество", при использовании по тексту данного документа (например, в отношении способов лечения заболевания или состояния), относится к количеству соединения, которое является эффективным для получения желаемого терапевтического эффекта. Например, если состояние представляет собой боль, то эффективное терапевтическое количество представляет собой количество, достаточное для обеспечения желаемого уровня облегчения боли. Желаемый уровень облегчения боли может представлять собой, например, полное устранение боли или уменьшение интенсивности боли.
Термин "неароматическая углеводородная группа" (как, например, "C1-5 неароматическая углеводородная группа" или "ациклическая C1-5 неароматическая углеводородная группа") относится к группе, состоящей из атомов углерода и водорода, которая не содержит ароматических колец. Углеводородная группа может быть полностью насыщенной или может содержать одну или более двойных связей углерод-углерод или тройных связей углерод-углерод, или смеси двойных и тройных связей. Углеводородная группа может представлять собой группу с неразветвленной или разветвленной цепью, или может состоять из циклической группы или содержать ее. Таким образом, термин «неароматический углеводород» включает алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкенилалкил CH2-циклоалкил и т.п.
Термины "алкил", "алкенил", "алкинил", "циклоалкил" и "циклоалкенил" используются в их обычном смысле (например, как определено в Золотой книге ИЮПАК), если не указано иное.
Термин "циклоалкил", при использовании по тексту данного документа, если позволяет указанное количество атомов углерода, включает как моноциклические циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, так и бициклические и трициклические группы. Бициклические циклоалкильные группы включают мостиковые кольцевые системы, такие как бициклогептан, бициклооктан и адамантан.
В приведенных выше определениях R1, R2, R3 и R4 указано, что один или два, но не все атомы углерода неароматической углеводородной группы могут быть необязательно замещены гетероатомом, выбранным из О, N и S и их окисленных форм. Следует принимать во внимание, что если атом углерода замещен гетероатомом, низшие валентности гетероатомов по сравнению с углеродом означают, что меньшее количество атомов свяжется с гетероатомами, чем связалось бы с атомом углерода, который был замещен. Так, например, замещение атома углерода (валентность четыре) в группе CH2 кислородом (валентность два) будет означать, что полученная в результате молекула будет содержать на два атома водорода меньше, а замещение атома углерода (валентность четыре) в группе CH2 азотом (валентность три) будет означать, что полученная в результате молекула будет содержать на один атом водорода меньше.
Примеры замещений гетероатомов атомами углерода включают замещение атома углерода в цепи -CH2-CH2-CH2- кислородом или серой с получением простого эфира -CH2-О-CH2- или тиоэфира -CH2-S-CH2-, замещение атома углерода в группе СН2-С≡С-Н азотом с получением нитрильной (циано) группы CH2-C≡N, замещение атома углерода в группе -CH2-CH2-CH2- на С=O с получением кетона -CH2-С(O)-CH2-, замещение атома углерода в группе -CH2-CH2-CH2- на S=O или SO2 с получением сульфоксида -CH2-S(O)-CH2- или сульфона -CH2-S(O)2-CH2-, замещение атома углерода в цепи -CH2-CH2-CH2- на C(O)NH с получением амида -CH2-CH2-C(O)-NH-, замещение атома углерода в цепи -CH2-CH2-CH2- азотом с получением амина -CH2-NH-CH2- и замещение атома углерода в цепи -CH2-CH2-CH2- на С(O)O с получением сложного эфира (или карбоновой кислоты) -СН2-CH2-С(O)-O-. В каждом таком замещении должен оставаться по меньшей мере один атом углерода углеводородной группы.
Соли
Многие соединения формулы (1) или формулы (1а) могут существовать в виде солей, например, кислотно-аддитивные соли или, в некоторых случаях, соли органических и неорганических оснований, такие как карбоксилатные, сульфонатные и фосфатные соли. Все такие соли находятся в пределах объема данного изобретения, а ссылки на соединения формулы (1) или формулы (1а) включают солевые формы соединений, как определено в Вариантах реализации изобретения 1.64-1.66.
Данные соли, как правило, представляют собой кислотно-аддитивные соли.
Соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основный или кислотный фрагмент, обычными химическими способами, такими как способы, описанные в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Как правило, такие соли могут быть получены взаимодействием форм свободных кислот или оснований этих соединений с соответствующим основанием или кислотой в воде или в органическом растворителе, или в смеси обоих; как правило, применяют неводные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Кислотно-аддитивные соли (как определено в Варианте реализации изобретения 1.65) могут образовываться с широким спектром кислот, как неорганических, так и органических. Примеры кислотно-аддитивных солей, подпадающих под Вариант реализации изобретения 1.65, включают моно- или ди-соли, образованные с кислотой, выбранной из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+) камфорной, камфорсульфоновой, ((+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, галогенводородных кислот (например, бромистоводородной, хлористоводородной, йодистоводородной), изэтионовой, молочной (например, (+)-L-молочной, (±)-DL-молочной), лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, пировиноградной, L-пироглутаминовой, салициловой, 4-амино-салициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-винной, тиоциановой, n-толуолсульфоновой, ундециленовой и валериановой кислот, а также ацилированных аминокислот и катионообменных смол.
Если соединения формулы (1) или формулы (1а) содержат аминную функцию, они могут образовывать соли четвертичного аммониевого основания, например, посредством реакции с алкилирующим средством в соответствии со способами, хорошо известными специалистам в данной области техники. Такие четвертичные аммониевые соединения находятся в пределах объема формулы (1) или формулы (1а).
Соединения по данному изобретению могут существовать в виде моно- или ди-солей, в зависимости от pKa кислоты, из которой образуется соль.
Солевые формы соединений по данному изобретению, как правило, представляют собой фармацевтически приемлемые соли, и примеры фармацевтически приемлемых солей описаны в публикации Berge et al., 1977, "Pharmaceutically Acceptable Salts," J. Pharm. Set, Vol. 66, pp. 1-19. Тем не менее, соли, не являющиеся фармацевтически приемлемыми, также могут быть получены в виде промежуточных форм, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие формы солей, не являющихся фармацевтически приемлемыми, которые могут применяться, например, при очистке или разделении соединений по данному изобретению, также составляют часть данного изобретения.
Стереоизомеры
Стереоизомеры представляют собой изомерные молекулы, которые имеют одинаковую молекулярную формулу и последовательность связанных атомов, но отличаются только в трехмерных ориентациях их атомов в пространстве. Эти стереоизомеры могут представлять собой, например, геометрические изомеры и оптические изомеры.
Геометрические изомеры
В геометрических изомерах изомерия обусловлена различными ориентациями атома или группы относительно двойной связи, как в цис-, так и транс- (Z- и Е-) изомерии относительно двойной связи углерод-углерод или цис- и транс-изомерах относительно амидной связи, или син- и анти-изомерии относительно двойной связи углерод-азот (например, в оксимах), или поворотной изомерии относительно связи, где вращение ограничено, или цис- и транс-изомерии относительно кольца, такого как циклоалкановое кольцо.
Соответственно, в другом варианте реализации изобретения (Вариант реализации 1.67) данное изобретение относится к геометрическому изомеру соединения по любому из Вариантов реализации изобретения 1.1-1.66.
Оптические изомеры
Если соединения формулы содержат один или более хиральных центров и могут существовать в виде двух или более оптических изомеров, ссылки на данные соединения включают все их оптические изомерные формы (например, энантиомеры, эпимеры и диастереоизомеры) либо в виде индивидуальных оптических изомеров, либо смесей (например, рацемических смесей), либо двух или более оптических изомеров, если контекст не предусматривает иное.
Соответственно, в другом варианте реализации изобретения (Вариант реализации 1.68) данное изобретение относится к соединению по любому из Вариантов реализации изобретения 1.1-1.67, которое содержит хиральный центр.
Оптические изомеры можно охарактеризовать и идентифицировать по их оптической активности (то есть как изомеры + и - или изомеры d и l), или их можно охарактеризовать с точки зрения их абсолютной стереохимии с помощью номенклатуры "R и S", разработанной Cahn, Ingold и Prelog, см. Advanced Organic Chemistry by Jerry March, 4th Edition, John Wiley & Sons, New York, 1992, pages 109-114, а также см. Cahn, Ingold & Prelog, Angew. Chem. Int. Ed. Engl., 1966, 5, 385-415. Оптические изомеры могут быть разделены с помощью ряда методик, в том числе хиральной хроматографии (хроматографии на хиральной подложке), и такие методики хорошо известны специалистам в данной области техники. В качестве альтернативы хиральной хроматографии, оптические изомеры могут быть разделены посредством образования диастереоизомерных солей с хиральными кислотами, такими как (+)-винная кислота, (-)-пироглутаминовая кислота, (-)-ди-толуоил-L-винная кислота, (+)-миндальная кислота, (-)-яблочная кислота и (-)-камфорсульфоновая кислота, разделения диастереоизомеров с применением избирательной кристаллизации, а затем диссоциации солей с получением индивидуального энантиомера свободного основания.
Если соединения по данному изобретению существуют в виде двух или более оптических изомерных форм, один энантиомер в паре энантиомеров может демонстрировать преимущества по сравнению с другим энантиомером, например, с точки зрения биологической активности. Таким образом, в определенных обстоятельствах, в качестве терапевтического средства желательно применять только один из пары энантиомеров или только один из множества диастереоизомеров.
Соответственно, в другом варианте реализации изобретения (Вариант реализации 1.69) данное изобретение относится к композициям, содержащим соединение по Варианту реализации 1.68, которое имеет один или более хиральных центров, при этом по меньшей мере 55% (например, по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%) соединения по Варианту реализации 1.65 присутствует в виде единственного оптического изомера (например, энантиомера или диастереоизомера).
В одном общем варианте реализации изобретения (Вариант реализации 1.70) 99% или более (например, практически все) от общего количества соединения (или соединения для применения) по Варианту реализации 1.68 присутствует в виде единственного оптического изомера.
Например, в одном варианте реализации изобретения (Вариант реализации 1.71) соединение присутствует в виде единственного энантиомера.
В другом варианте реализации изобретения (Вариант реализации 1.72) соединение присутствует в виде единственного диастереоизомера.
Данное изобретение также относится к смеси оптических изомеров, которая может быть рацемической или нерацемической. Таким образом, данное изобретение предлагает:
1.73 Соединение по Варианту реализации изобретения 1.68, которое находится в виде рацемической смеси оптических изомеров.
1.74 Соединение по Варианту реализации изобретения 1.68, которое находится в виде нерацемической смеси оптических изомеров.
Изотопы
Соединения по данному изобретению, как определено в любом из Вариантов реализации изобретения 1.1-1.74, могут содержать одно или более изотопных замещений, а ссылка на конкретный элемент включает в свой объем все изотопы элемента. Например, ссылка на водород включает в свой объем 1Н, 2Н (D) и 3Н (Т). Аналогичным образом, ссылки на углерод и кислород включают в свой объем, соответственно, 12С, 13С и 14С, а также 16О и 18O.
Аналогичным образом, ссылка на конкретную функциональную группу также включает в свой объем изотопные варианты, если из контекста не следует иное. Например, ссылка на алкильную группу, такую как этильная группа, также охватывает варианты, в которых один или более атомов водорода в группе находятся в виде изотопа дейтерия или трития, например, как в этильной группе, в которой все пять атомов водорода находятся в виде изотопа дейтерия (пердейтероэтильная группа).
Изотопы могут быть радиоактивными или нерадиоактивными. В одном варианте реализации данного изобретения (Вариант реализации 1.75) соединение по любому из Вариантов реализации 1.1-1.74 не содержит радиоактивных изотопов. Такие соединения являются предпочтительными для применения в терапевтических целях. Тем не менее, в другом варианте реализации изобретения (Вариант реализации 1.76) соединение по любому из Вариантов реализации 1.1-1.74 может содержать один или более радиоизотопов. Соединения, содержащие такие радиоизотопы, можно применять в диагностическом контексте.
Сольваты
Соединения формулы (1) или формулы (1а), как определено в любом из Вариантов реализации изобретения 1.1-1.76, могут образовывать сольваты. Предпочтительными сольватами являются сольваты, образованные посредством включения в твердотельную структуру (например, кристаллическую структуру) соединений по данному изобретению молекул нетоксичного фармацевтически приемлемого растворителя (далее по тексту именуемого как сольватирующий растворитель). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены посредством рекристаллизации соединений по данному изобретению растворителем или смесью растворителей, содержащей сольватирующий растворитель. В каждом конкретном случае определить, был ли сольват образован или нет, можно подвергнув кристаллы соединения анализу с применением хорошо известных и стандартных методик, таких как термогравиметрический анализ (ТГА), дифференциальная сканирующая калориметрия (ДСК) и рентгеноструктурная кристаллография. Сольваты могут представлять собой стехиометрические и нестехиометрические сольваты. В частности, предпочтительными сольватами являются гидраты, а примеры гидратов включают полугидраты, моногидраты и дигидраты.
Соответственно, в дополнительных Вариантах реализации изобретения 1.77 и 1.78 данное изобретение предлагает:
1.77 Соединение по любому из Вариантов реализации 1.1-1.76, которое находится в виде сольвата.
1.78 Соединение по Варианту реализации 1.77, отличающееся тем, что сольват представляет собой гидрат.
Для более подробного рассмотрения сольватов и способов, применяемых для их получения и характеристики, см. публикацию Bryn et al., Solid-State Chemistry of Drugs, Second Edition, published by SSCI, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
В альтернативном варианте, вместо того чтобы находиться в виде гидрата, соединение по данному изобретению может быть безводным. Таким образом, в другом варианте реализации изобретения (Вариант реализации 1.79) данное изобретение относится к соединению, как определено в любом из Вариантов реализации 1.1-1.76, в безводной форме (например, безводной кристаллической форме).
Кристаллические и аморфные формы
Соединения по любому из Вариантов реализации изобретения 1.1-1.79 могут существовать в кристаллическом или некристаллическом (например, аморфном) состоянии. Существует ли соединение в кристаллическом состоянии или нет, можно легко определить с помощью стандартных методик, таких как порошковая рентгеновская дифракция (XRPD). Кристаллы и их кристаллические структуры можно обнаружить с помощью ряда методов, в том числе монокристаллической рентгеноструктурной кристаллографии, порошковой рентгеновской дифракции (XRPD), дифференциальной сканирующей калориметрии (ДСК) и инфракрасной спектроскопии, например, инфракрасной спектроскопии с Фурье-преобразованием (FTIR). Поведение кристаллов в условиях различной влажности можно анализировать с помощью гравиметрических исследований сорбции паров, а также с помощью XRPD. Определение кристаллической структуры соединения может быть выполнено с помощью рентгеноструктурной кристаллографии, которая может быть осуществлена в соответствии со стандартными методами, такими как методы, описанные в данном документе, и как описано в публикации Fundamentals of Crystallography, С. Giacovazzo, H.L. Monaco, D. Viterbo, F. Scordari, G. Gilli, G. Zanotti and M. Catti, (International Union of Crystallography/Oxford University Press, 1992 ISBN 0-19-855578-4 (p/b), 0-19-85579-2 (h/b)). Этот метод включает анализ и интерпретацию рентгеновской дифракции монокристалла. Трехмерная структура, которая обычно существует в кристаллической форме, не существует в виде аморфного твердого вещества, а положение молекул относительно друг друга в аморфной форме, по сути, является случайным, см., например, публикацию Hancock et al. J. Pharm. Sci. (1997), 86, 1).
Соответственно, в дополнительных вариантах реализации данного изобретения предлагается:
1.80 Соединение по любому из Вариантов реализации 1.1-1.79, которое находится в кристаллической форме.
1.781 Соединение по любому из Вариантов реализации 1.1-1.79, которое является:
(а) на 50-100% кристаллическим, и, в частности, является по меньшей мере на 50% кристаллическим, или по меньшей мере на 60% кристаллическим, или по меньшей мере на 70% кристаллическим, или по меньшей мере на 80% кристаллическим, или по меньшей мере на 90% кристаллическим, или по меньшей мере на 95% кристаллическим, или по меньшей мере на 98% кристаллическим, или по меньшей мере на 99% кристаллическим, или по меньшей мере на 99,5% кристаллическим, или по меньшей мере на 99,9% кристаллическим, например, на 100% кристаллическим.
1.82 Соединение по любому из Вариантов реализации изобретения 1.1-1.79, которое находится в аморфной форме.
Пролекарства
Соединения формулы (1) или формулы (1а), как определено в любом из Вариантов реализации изобретения 1.1-1.76, могут быть представлены в виде пролекарства. Под термином "пролекарство" подразумевается, например, любое соединение, которое превращается in vivo в биологически активное соединение формулы (1) или формулы (1а), как определено в любом из Вариантов реализации изобретения 1.1-1.76.
Например, некоторые пролекарства представляют собой сложные эфиры активного соединения (например, физиологически приемлемый метаболически лабильный сложный эфир). В процессе метаболизма сложноэфирная группа (-C(=O)OR) расщепляется с образованием активного лекарственного средства. Такие сложные эфиры могут быть получены посредством этерификации, например, любых гидроксильных групп, присутствующих в исходном соединении, при необходимости, с предварительной защитой любых других реакционноспособных групп, присутствующих в исходном соединении, с последующим снятием защиты, если необходимо.
Кроме того, некоторые пролекарства активируются под воздействием ферментов с образованием активного соединения или такого соединения, которое при дальнейшей химической реакции дает активное соединение (например, как в ADEPT, GDEPT, LIDEPT и т.д.). Например, пролекарство может представлять собой производное сахара или другой конъюгат гликозида, или может представлять собой производное сложного эфира аминокислоты.
Соответственно, в другом варианте реализации изобретения (Вариант реализации 1.83) данное изобретение предлагает пролекарство соединения, как определено в любом из Вариантов реализации 1.1-1.76, при этом соединение содержит функциональную группу, которая способна преобразоваться в физиологических условиях с образованием гидроксильной группы или аминогруппы.
Комплексы и клатраты
Также формулой (1) или формулой (1а) в Вариантах реализации изобретения 1.1-1.83 охватываются комплексы (например, комплексы включения или клатраты с соединениями, такие как циклодекстрины или комплексы с металлами) соединений по Вариантам реализации 1.1-1.83.
Соответственно, в другом варианте реализации изобретения (Вариант реализации 1.84) данное изобретение относится к соединению по любому из Вариантов реализации 1.1-1.83 в виде комплекса или клатрата.
Биологическая активность и применение в терапевтических целях
Соединения по настоящему изобретению обладают активностью агонистов мускариновых рецепторов M1. Мускариновая активность соединений может быть определена с помощью анализа фосфо-ERK1/2, описанного ниже в Примере А.
Существенным преимуществом соединений по данному изобретению является то, что они обладают высокой селективностью в отношении рецептора M1 по сравнению с рецепторами подтипов М2 и М3. Соединения по данному изобретению не являются ни агонистами, ни антагонистами подтипов рецепторов М2 и М3. Например, в то время как соединения по данному изобретению, как правило, имеют значения pEC50 по меньшей мере 6 (предпочтительно по меньшей мере 6,5) и значения Emax более 80 (предпочтительно более 95) в отношении рецептора M1 в функциональном анализе, описанном в Примере А, они могут иметь значения pEC50 менее 5 и значения Emax менее 20%, при испытании в отношении подтипов М2 и М3 в функциональном анализе Примера А.
Некоторые соединения по данному изобретению обладают активностью как в отношении рецепторов M1, так и М4.
Соответственно, в Вариантах реализации данного изобретения 2.1-2.9 предлагается:
2.1 Соединение по любому из Вариантов реализации 1.1-1.84 для применения в медицине.
2.2 Соединение по любому из Вариантов реализации 1.1-1.84 для применения в качестве агонистов мускариновых рецепторов M1 или M1 и М4.
2.3 Соединение по любому из Вариантов реализации 1.1-1.84, которое представляет собой агонист мускаринового рецептора M1, имеющий pEC50 более 6,9 и Emax по меньшей мере 80 в отношении рецептора M1 в анализе Примера А в данном документе или анализе, по существу подобном ему.
2.4 Соединение по Варианту реализации 2.3, которое представляет собой агонист мускаринового рецептора M1, имеющий pEC50 более 7,0.
2.5 Соединение по Варианту реализации 2.3 или по Варианту реализации 2.4, имеющее Emax по меньшей мере 90 в отношении рецептора М1.
2.6 Соединение по любому из Вариантов реализации 1.1-1.84, которое представляет собой агонист мускариновых рецепторов M1 и М4, имеющий pEC50 в диапазоне от 6,0 до 8,7 и Emax по меньшей мере 60 в отношении рецептора М4 в анализе Примера А в данном документе или анализе, по существу подобном ему.
2.7 Соединение по любому из Вариантов реализации 1.1-1.84, которое представляет собой агонист мускариновых рецепторов M1 и М4, имеющий pEC50 в диапазоне от 6,0 до 8,1 и Emax по меньшей мере 90 в отношении рецептора М4 в анализе Примера А в данном документе или анализе, по существу подобном ему.
2.8 Соединение по Варианту реализации 2.6, которое представляет собой агонист мускаринового рецептора М4, имеющий pEC50 в диапазоне от 7,5 до 8,7.
2.9 Соединение по Варианту реализации 2.7, которое представляет собой агонист мускаринового рецептора М4, имеющий pEC50 в диапазоне от 6,5 до 7,5.
2.10 Соединение по Варианту реализации 2.6 или по Варианту реализации 2.8, имеющее Emax по меньшей мере 75 в отношении рецептора М4.
2.11 Соединение по Варианту реализации 2.7 или по Варианту реализации 2.9, имеющее Emax по меньшей мере 95 в отношении рецептора М4.
2.12 Соединение по любому из Вариантов реализации 2.3-2.11, которое является селективным в отношении рецепторов M1 и М4 по сравнению с мускариновыми рецепторами М2 и М3.
2.13 Соединение по Варианту реализации 2.12, которое является селективным в отношении рецептора M1 по сравнению с мускариновыми рецепторами М2 и М3.
2.14 Соединение по любому из Вариантов реализации 2.3-2.5, которое является селективным в отношении рецептора M1 по сравнению с мускариновыми рецепторами М2, М3 и М4.
2.15 Соединение по любому из Вариантов реализации 2.3-2.14, которое имеет pEC50 менее 5 и Emax менее 50 в отношении мускариновых рецепторов подтипов М2 и М3.
2.16 Соединение по Варианту реализации 2.15, которое имеет pEC50 менее 4,5 и/или Emax менее 30 в отношении мускариновых рецепторов подтипов М2 и М3.
2.17 Соединение по любому из Вариантов реализации 1.1-1.84 и Вариантов реализации 2.3-2.16 для применения в лечении заболевания или состояния, опосредованного мускариновым рецептором М1.
Благодаря активности агониста мускариновых рецепторов M1 или M1 и М4, соединения по данному изобретению можно применять в лечении болезни Альцгеймера, шизофрении и других психотических расстройств, когнитивных расстройств и других заболеваний, опосредованных мускариновыми рецепторами M1 или M1 и М4, а также можно применять в лечении различных видов боли.
Соответственно, в Вариантах реализации данного изобретения 2.16-2.39 предлагается:
2.18 Соединение по любому из Вариантов реализации изобретения 1.1-1.84 для применения в лечении когнитивного расстройства или психотического расстройства.
2.19 Соединение для применения в соответствии с Вариантом реализации изобретения 2.18, отличающееся тем, что когнитивное расстройство или психотическое расстройство включает, возникает в результате или связано с состоянием, выбранным из когнитивных нарушений, умеренных когнитивных нарушений, лобно-височной деменции, сосудистой деменции, деменции с тельцами Леви, пресенильной деменции, сенильной деменции, атаксии Фридрейха, синдрома Дауна, хореи Хантингтона, гиперкинезии, мании, синдрома Туретта, болезни Альцгеймера, прогрессирующего надъядерного паралича, нарушений когнитивных функций, в том числе нарушения внимания, ориентации, способности к обучению, функции памяти (то есть расстройства памяти, амнезия, амнестические расстройства, синдром транзиторной глобальной амнезии и возрастное нарушение памяти) и речевой функции; когнитивных нарушений в результате инсульта, болезни Хантингтона, болезни Пика, СПИД-ассоциированной деменции или других состояний деменции, таких как мультиинфарктная деменция, алкогольная деменция, гипотиреоз-ассоциированная деменция и деменция, связанная с другими дегенеративными заболеваниями, такими как атрофия мозжечка и боковой амиотрофический склероз; других острых или подострых состояний, которые могут вызвать снижение когнитивных способностей, таких как делирий или депрессивная травма (состояние псевдодеменции), черепно-мозговая травма, возрастное снижение когнитивных способностей, инсульт, нейродегенерация, обусловленные действием наркотических и нейротоксических средств состояния, возрастные когнитивные нарушения, связанные с аутизмом когнитивные нарушения, синдром Дауна, связанное с психозом когнитивное расстройство и связанные с пост-электрошоковой терапией когнитивные расстройства; когнитивных расстройств, вызванных злоупотреблением наркотиками или отменой приема наркотических средств, в том числе никотина, каннабиса, амфетамина, кокаина, синдрома дефицита внимания с гиперактивностью (СДВГ) и дискинетических расстройств, таких как болезнь Паркинсона, вызванный нейролептиками паркинсонизм, а также поздней дискинезии, шизофрении, шизофреноподобного заболевания, психотической депрессии, мании, острой мании, параноидальных, галлюциногенных и бредовых расстройств, расстройств личности, обсессивно-компульсивных расстройств, шизотипических расстройств, бредовых расстройств, психоза на фоне злокачественной опухоли, нарушения обмена веществ, эндокринного заболевания или нарколепсии, психоза на фоне злоупотребления наркотиками или отмены приема наркотических средств, биполярных расстройств и шизоаффективного расстройства.
2.20 Соединение по любому из Вариантов реализации изобретения 1.1-1.84 для применения в лечении болезни Альцгеймера.
2.21 Соединение по любому из Вариантов реализации изобретения 1.1-1.84 для применения в лечении шизофрении.
2.22 Способ лечения когнитивного расстройства у субъекта (например, у млекопитающего пациента, такого как человек, например, у человека, нуждающегося в таком лечении), включающий введение терапевтически эффективной дозы соединения по любому из Вариантов реализации изобретения 1.1-1.84.
2.23 Способ по Варианту реализации изобретения 2.22, отличающийся тем, что когнитивное расстройство включает, возникает в результате или связано с состоянием, которое определено в Варианте реализации изобретения 2.17.
2.24 Способ по Варианту реализации изобретения 2.23, отличающийся тем, что когнитивное расстройство возникает в результате или связано с болезнью Альцгеймера.
2.25 Способ по Варианту реализации изобретения 2.23, отличающийся тем, что когнитивное расстройство представляет собой шизофрению.
2.26 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для изготовления лекарственного средства для лечения когнитивного расстройства.
2.27 Применение по Варианту реализации изобретения 2.26, отличающееся тем, что когнитивное расстройство включает, возникает в результате или связано с состоянием, которое определено в Варианте реализации изобретения 2.19.
2.28 Применение по Варианту реализации изобретения 2.27, отличающееся тем, что когнитивное расстройство возникает в результате или связано с болезнью Альцгеймера.
2.29 Применение по Варианту реализации изобретения 2.28, отличающееся тем, что когнитивное расстройство представляет собой шизофрению.
2.30 Соединение по любому из Вариантов реализации изобретения 1.1-1.84 для лечения или уменьшения интенсивности острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, невралгии тройничного нерва, герпетической невралгии, общих невралгий, висцеральной боли, остеоартритической боли, постгерпетической невралгии, диабетической нейропатии, радикулита, ишиаса, позвоночной боли, головной боли или боли в шее, тяжелой или хронической боли, ноцицептивной боли, прорыва боли, послеоперационной боли или боли, связанной с онкологическим заболеванием.
2.31 Способ лечения или уменьшения интенсивности острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, невралгии тройничного нерва, герпетической невралгии, общих невралгий, висцеральной боли, остеоартритической боли, постгерпетической невралгии, диабетической нейропатии, радикулита, ишиаса, позвоночной боли, головной боли или боли в шее, тяжелой или хронической боли, ноцицептивной боли, прорыва боли, послеоперационной боли или боли, связанной с онкологическим заболеванием, включающий введение терапевтически эффективной дозы соединения согласно любому из Вариантов реализации изобретения 1.1-1.84.
2.32 Соединение по любому из Вариантов реализации изобретения 1.1-1.84 для лечения периферических нарушений, таких как снижение внутриглазного давления при глаукоме, и лечения сухости слизистой оболочки глаз и сухости слизистой оболочки рта, в том числе синдрома Шегрена.
2.33 Способ лечения периферических нарушений, таких как снижение внутриглазного давления при глаукоме, и лечения сухости слизистой оболочки глаз и сухости слизистой оболочки рта, в том числе синдрома Шегрена, включающий введение терапевтически эффективной дозы соединения согласно любому из Вариантов реализации изобретения 1.1-1.84.
2.34 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для изготовления лекарственного средства для лечения или уменьшения интенсивности острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, невралгии тройничного нерва, герпетической невралгии, общих невралгий, висцеральной боли, остеоартритической боли, постгерпетической невралгии, диабетической нейропатии, радикулита, ишиаса, позвоночной боли, головной боли или боли в шее, тяжелой или хронической боли, ноцицептивной боли, прорыва боли, послеоперационной боли или боли, связанной с онкологическим заболеванием, или для лечения периферических нарушений, таких как снижение внутриглазного давления при глаукоме, и лечения сухости слизистой оболочки глаз и сухости слизистой оболочки рта, в том числе синдрома Шегрена.
2.35 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для применения в лечении повреждений кожи, например, вследствие вульгарной пузырчатки, герпетиформного дерматита, пемфигоида и других пузырчатых состояний кожи.
2.36 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для применения в лечении, профилактике, облегчении или реверсии состояний, связанных с измененной функцией и двигательной активностью желудочно-кишечного тракта, таких как функциональная диспепсия, синдром раздраженного кишечника, гастроэзофагеальный кислотный рефлюкс (ГЭР) и пищеводная диспепсия, симптомы пареза желудка и хронической диареи.
2.37 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для применения в лечении обонятельной дисфункции, такой как Босма-Хенкин-Христиансен синдром, химическое отравление (например, селеном и серебром), гипопитуитаризм, синдром Кальмана, переломы костей черепа, терапии опухолей и гипоактивность щитовидной железы.
2.38 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для лечения наркотической зависимости.
2.39 Применение соединения по любому из Вариантов реализации изобретения 1.1-1.84 для лечения двигательных расстройств, таких как болезнь Паркинсона, СДВГ, болезнь Хантингтона, синдром Туретта и другие синдромы, связанные с дофаминергической дисфункцией в качестве основного патогенетического фактора заболевания.
Способы получения соединений формулы (1) или формулы (1а)
Соединения формулы (1) или формулы (1а) могут быть получены в соответствии со способами синтеза, хорошо известными специалистам в данной области техники и описанными в данном документе.
Соответственно, в другом варианте реализации (Вариант реализации 3.1) данное изобретение относится к способу получения соединения, как определено в любом из Вариантов реализации изобретения 1.1-1.84, включающему:
(А) реакцию соединения формулы (10)
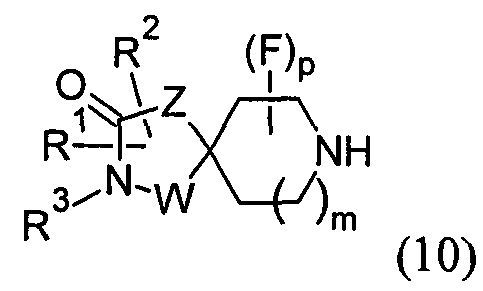
с соединением формулы (11):
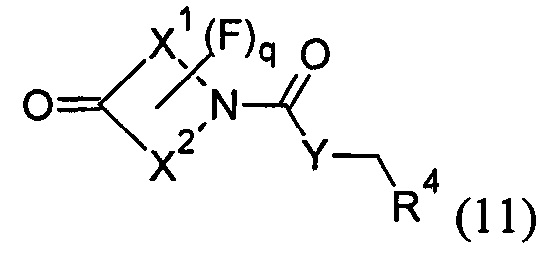
в условиях восстановительного аминирования; где R1, R2, R3, R4, X1, X2, W, Y, Z, m, р и q представляют собой такие, как определено в любом из Вариантов реализации изобретения 1.1-1.84; или
(B) реакцию соединения формулы (12)
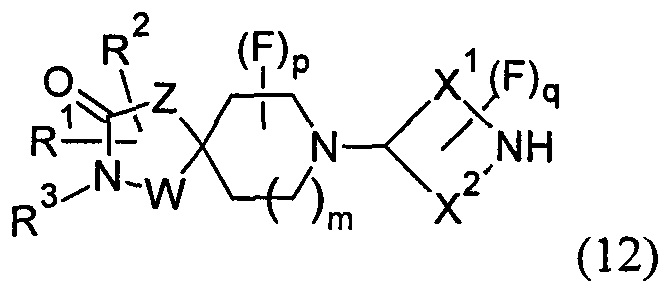
с соединением формулы Cl-C(=O)-CH2-R4, в присутствии основания; или
(C) реакцию соединения формулы (10)
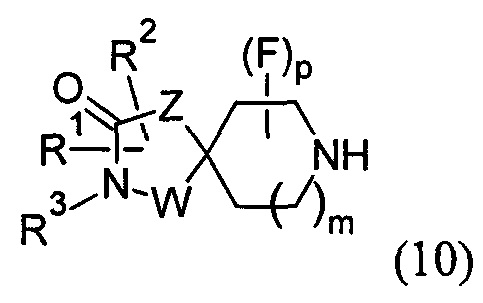
с соединением формулы (13):
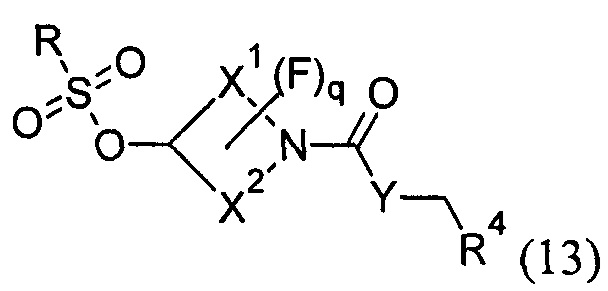
в условиях нуклеофильного замещения; где R1, R2, R3, R4, X1, X2, W, Y, Z, m, р и q представляют собой такие, как определено в любом из Вариантов реализации изобретения 1.1-1.84; и необязательно:
(D) превращение одного соединения формулы (1) или формулы (1а) в другое соединение формулы (1) или формулы (1а).
В варианте (А) способа пиперидиновый гетероцикл (10) подвергают реакции с замещенным кетоном (11) в условиях восстановительного аминирования. Реакцию восстановительного аминирования, как правило, проводят при температуре окружающей среды с применением борогидридного восстановителя, такого как триацетоксиборгидрид натрия, в растворителе, таком как дихлорметан или дихлорэтан, содержащий уксусную кислоту.
В варианте (С) способа пиперидиновый гетероцикл (10) подвергают реакции с сульфоновым эфиром (13, R = метил или 4-метилбензил) в реакции нуклеофильного замещения, которую, как правило, проводят при умеренном нагревании (например, до температуры, составляющей от около 40°С до около 70°С) либо в чистом виде, без растворителя, либо в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил или диметилацетамид.
Промежуточные соединения формулы (12) могут быть получены посредством серии реакций, показанной ниже на Схеме 1.
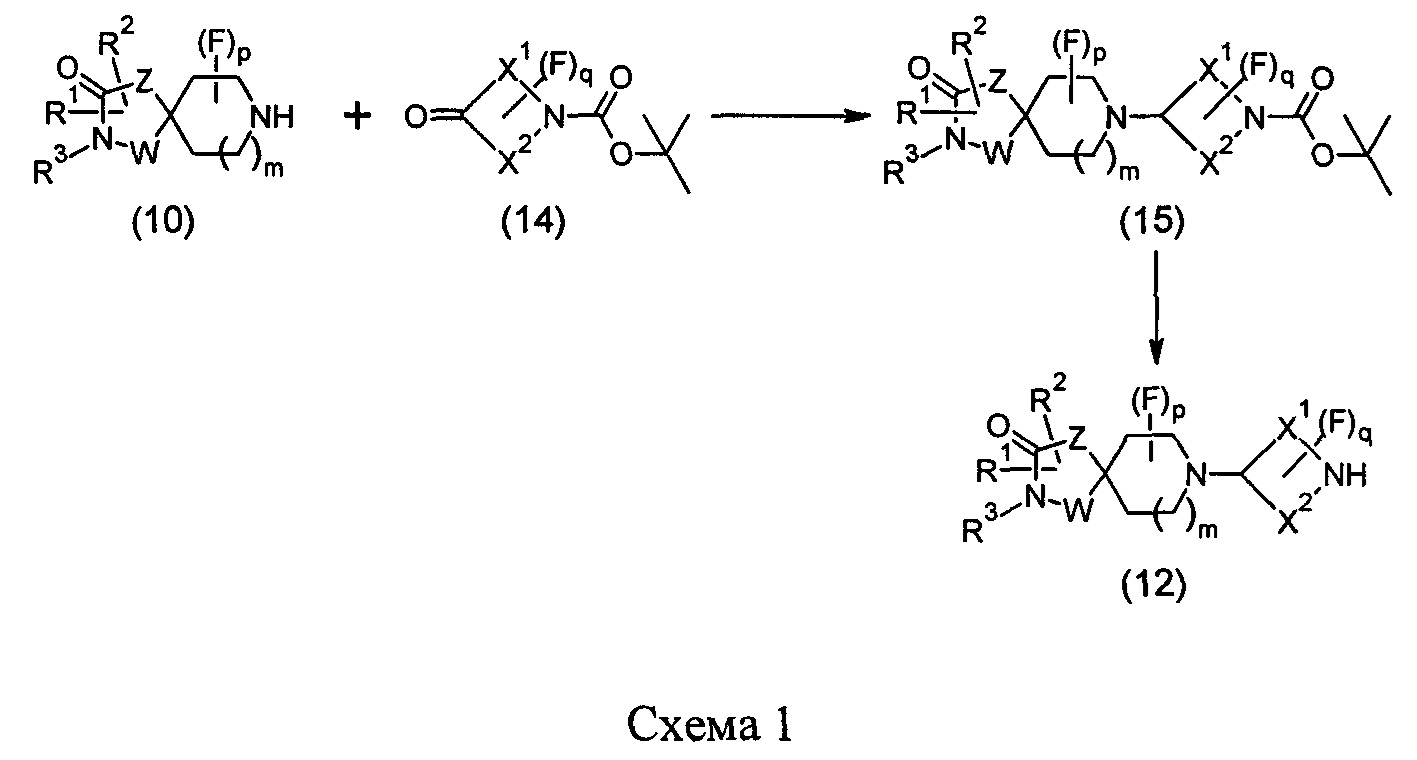
На Схеме 1 реакции пиперидиновый гетероцикл (10) приводят в контакт с Вос-защищенным кетоном (14) в условиях восстановительного аминирования. Реакцию восстановительного аминирования, как правило, проводят при умеренном нагревании (например, до температуры, составляющей от около 40°С до около 70°С) в присутствии либо цианоборогидрида натрия в комбинации с хлоридом цинка, либо триацетоксиборогидрида натрия в комбинации с изопропоксидом титана в растворителе, таком как дихлорметан или дихлорэтан, содержащий уксусную кислоту, с получением промежуточного пиридинового соединения (15), с которого затем снимают защиту путем удаления Вос-группы посредством обработки кислотой (например, трифторуксусной кислотой в дихлорметане) с получением соединения (12).
Соединения формулы (12) также могут быть получены посредством последовательности реакций, показанной ниже на Схеме 2.
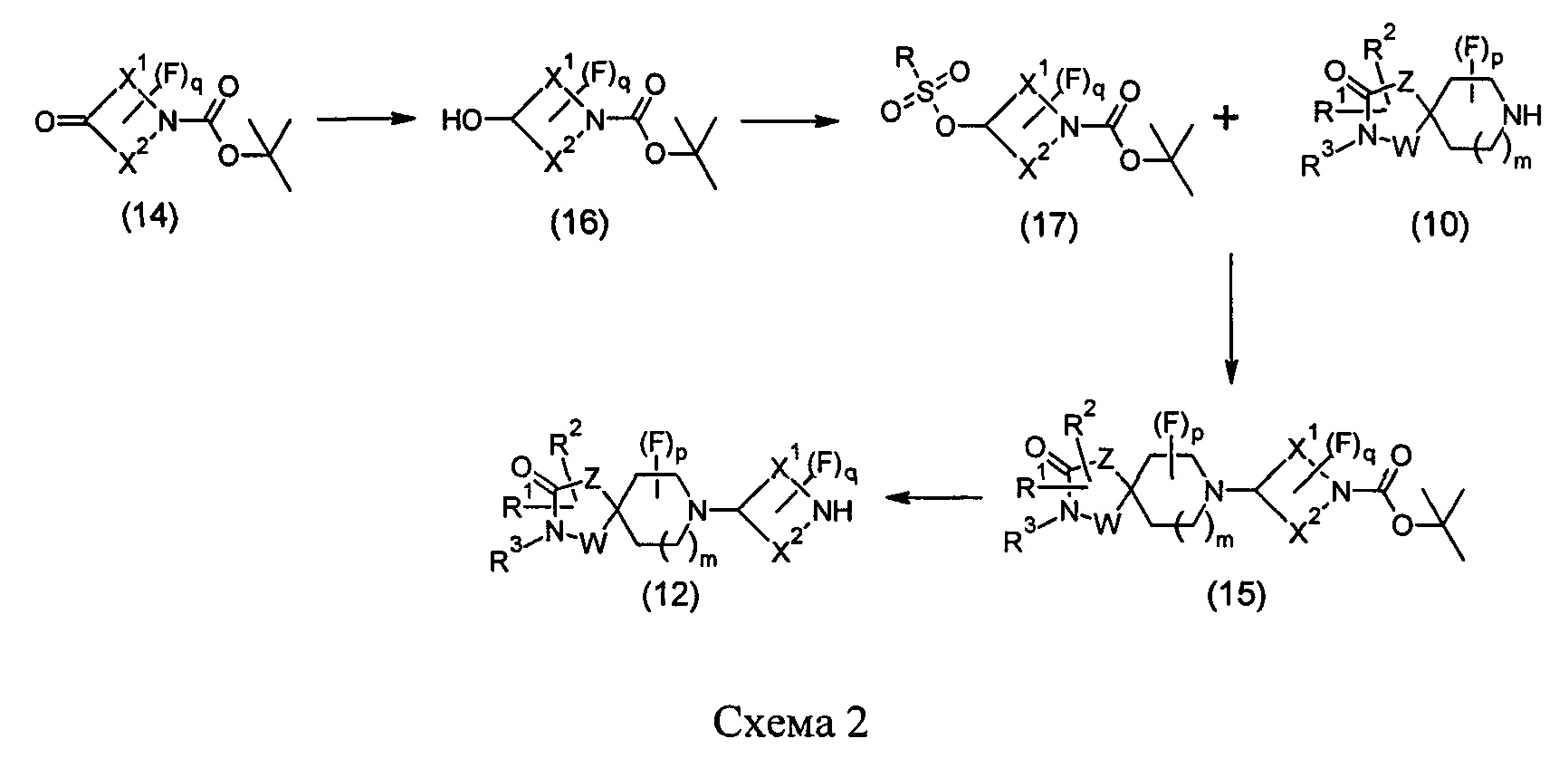
На Схеме 2 Вос-защищенный кетон (14) восстанавливают до спирта (16) с помощью борогидрида натрия в метаноле. Затем спирт (16) активируют как сульфоновый эфир (17, R = метил или 4-метилбензил) с применением соответствующего сульфонилхлорида в дихлорметане в присутствии третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин. Сульфоновый эфир (17) подвергают реакции с пиперидиновым гетероциклом (10) в реакции нуклеофильного замещения, которую, как правило, проводят при умеренном нагревании (например, до температуры, составляющей от около 40°С до около 70°С) либо в чистом виде, без растворителя, либо в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил или диметилацетамид, с получением соединения (15), с которого затем снимают защиту путем удаления Вос-группы посредством обработки кислотой (например, трифторуксусной кислотой в дихлорметане) с получением соединения (12).
После получения одно соединение формулы (1) или формулы (1а), или его защищенное производное, может быть превращено в другое соединение формулы (1) или формулы (1а) с помощью способов, хорошо известных специалистам в данной области техники. Примеры способов синтеза для превращения одной функциональной группы в другую функциональную группу изложены в стандартных текстах, таких как Advanced Organic Chemistry и Organic Syntheses (см. ссылки выше) или Fiesers' Reagents for Organic Synthesis, Volumes 1-17, John Wiley, edited by Mary Fieser (ISBN: 0-471-58283-2).
Во многих из описанных выше реакций может быть необходимо защитить одну или более групп, чтобы предотвратить реакцию в нежелательном положении на молекуле. Примеры защитных групп, а также способы защиты и снятия защиты с функциональных групп можно найти в публикации Protective Groups in Organic Synthesis (Т. Greene and P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Полученные приведенными выше способами соединения могут быть выделены и очищены с помощью любого из множества методов, хорошо известных специалистам в данной области техники, а примеры таких способов включают методы рекристаллизации и хроматографии, такие как колоночная хроматография (например, флэш-хроматография) и ВЭЖХ.
Фармацевтические композиции
Хотя активное соединение можно вводить отдельно, предпочтительно вводить его в виде фармацевтической композиции (например, препарата).
Соответственно, в другом варианте реализации (Вариант реализации 4.1) данного изобретения предлагается фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (1) или формулы (1а), как определено в любом из Вариантов реализации 1.1-1.84, вместе по меньшей мере с одним фармацевтически приемлемым вспомогательным веществом.
В одном варианте реализации изобретения (Вариант реализации 4.2) композиция представляет собой таблетированную композицию.
В другом варианте реализации изобретения (Вариант реализации 4.3) композиция представляет собой капсулированную композицию.
Фармацевтически приемлемое вспомогательное вещество (вещества) может быть выбрано, например, из носителей (например, твердого, жидкого или полутвердого носителя), адъювантов, разбавителей (например, твердых разбавителей, таких как наполнители или объемообразующие препараты; и жидких разбавителей, таких как растворители и сорастворители), гранулирующих средств, связующих веществ, средств для повышения текучести, покрывающих средств, регуляторов скорости высвобождения (например, замедляющих или задерживающих высвобождение полимеров или восков), связывающих веществ, разрыхлителей, буферных веществ, смазывающих веществ, консервантов, антигрибковых и антибактериальных средств, антиоксидантов, буферных веществ, регулирующих тоничность средств, загустителей, ароматизаторов, подсластителей, пигментов, пластификаторов, исправляющих вкус лекарственного средства веществ, стабилизаторов или любых других вспомогательных веществ, обычно применяемых в фармацевтических композициях.
Термин "фармацевтически приемлемый", используемый в данном документе, означает соединения, материалы, композиции и/или лекарственные формы, которые в рамках медицинской оценки подходят для применения в контакте с тканями субъекта (например, субъекта-человека) без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соответствующих приемлемому соотношению польза/риск. Каждое вспомогательное вещество также должно быть "приемлемым" в плане совместимости с другими ингредиентами композиции.
Фармацевтические композиции, содержащие соединения формулы (1) или формулы (1а), могут быть изготовлены в соответствии с известными методиками, см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Фармацевтические композиции могут быть в любой форме, подходящей для перорального, парентерального, местного, интраназального, эндобронхиального, сублингвального, офтальмологического, ушного, ректального, интравагинального или трансдермального введения.
Фармацевтические лекарственные формы, подходящие для перорального введения, включают таблетки (с покрытием или без покрытия), капсулы (с твердой или мягкой оболочкой), каплеты, пилюли, леденцы, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, подъязычные таблетки, пластины или пластыри, такие как щечные пластыри.
Таблетированные композиции могут содержать стандартную дозу активного соединения вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например, лактоза, сахароза, сорбитол или маннитол; и/или несахарный разбавитель, такой как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза, или ее производное, такое как микрокристаллическая целлюлоза (МКЦ), метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, а также крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты, как связующие и гранулирующие средства, такие как поливинилпирролидон, разрыхлители (например, набухающие сшитые полимеры, такие как сшитая карбоксиметилцеллюлоза), смазывающие вещества (например, стеараты), консерванты (например, парабены), антиоксиданты (например, ВНТ), буферные вещества (например, фосфатные или цитратные буферы) и шипучие вещества, такие как смеси цитрат/бикарбонат. Такие вспомогательные вещества хорошо известны, и нет необходимости обсуждать их в данном документе более подробно.
Таблетки могут быть выполнены с возможностью высвобождения лекарственного средства либо при контакте с жидкостями желудка (таблетки с немедленным высвобождением), либо высвобождения контролируемым образом (таблетки с контролируемым высвобождением) в течение продолжительного периода времени, либо при контакте с определенной областью желудочно-кишечного тракта.
Фармацевтические композиции, как правило, содержат от около 1% (масс./масс.) до около 95%, предпочтительно % (масс./масс.) действующего вещества и от 99% (масс./масс.) до 5% (масс./масс.) фармацевтически приемлемого вспомогательного вещества (например, как определено выше), или комбинации таких вспомогательных веществ. Предпочтительно композиции содержат от около 20% (масс./масс.) до около 90% (масс./масс.) действующего вещества и от 80% (масс./масс.) до 10% фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ. Фармацевтические композиции содержат от около 1% до около 95%, предпочтительно от около 20% до около 90% действующего вещества. Фармацевтические композиции по данному изобретению могут быть, например, в виде стандартной дозы, например, в виде ампул, флаконов, суппозиториев, предварительно заполненных шприцов, драже, порошков, таблеток или капсул.
Таблетки и капсулы могут содержать, например, 0-20% разрыхлителя, 0-5% смазывающих веществ, 0-5% средств для повышения текучести и/или 0-99% (масс./масс.) наполнителей, и/или объемообразующих препаратов (в зависимости от дозы лекарственного средства). Они также могут содержать 0-10% (масс./масс.) полимерных связующих, 0-5% (масс./масс.) антиоксидантов, 0-5% (масс./масс.) пигментов. Таблетки с замедленным высвобождением, как правило, дополнительно содержат 0-99% (масс./масс.) контролирующие высвобождение (например, задерживающие) полимеры (в зависимости от дозы). Пленочная оболочка таблетки или капсулы, как правило, содержит 0-10% (масс./масс.) полимеров, 0-3% (масс./масс.) пигментов и/или 0-2% (масс./масс.) пластификаторов.
Композиции для парентерального введения, как правило, содержат 0-20% (масс./масс.) буферов, 0-50% (масс./масс.) сорастворителей и/или 0-99% (масс./масс.) воды для инъекций (ВДИ) (в зависимости от дозы и в случае применения лиофилизата). Композиции для внутримышечных депо также могут содержать 0-99% (масс./масс.) масла.
Фармацевтические композиции могут быть предоставлены пациенту в "упаковках для пациента", содержащих полный курс лечения в одной упаковке, как правило, блистерной упаковке.
Соединения формулы (1) или формулы (1а) будут, как правило, предоставлены в виде стандартной лекарственной формы и в связи с этим будут, как правило, содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, композиция может содержать от 1 нг до 2 г действующего вещества, например, от 1 нг до 2 мг действующего вещества. В пределах этих диапазонов конкретные поддиапазоны соединения составляют от 0,1 мг до 2 г действующего вещества (обычно от 10 мг до 1 г, например от 50 мг до 500 мг) или от 1 мкг до 20 мг (например, от 1 мкг до 10 мг, например от 0,1 мг до 2 мг действующего вещества).
В композициях для перорального введения стандартная лекарственная форма может содержать от 1 мг до 2 г, как правило, от 10 мг до 1 г, например от 50 мг до 1 г, например от 100 мг до 1 г активного соединения.
Активное соединение вводят пациенту, нуждающемуся в этом (например, пациенту человеку или животному) в количестве, достаточном для достижения желаемого терапевтического эффекта (эффективное количество). Точные количества вводимого соединения могут быть определены наблюдающим врачом в соответствии со стандартными методиками.
ПРИМЕРЫ
Теперь данное изобретение будет проиллюстрировано, но не ограничено, ссылкой на конкретные варианты реализации, описанные в приведенных ниже примерах.
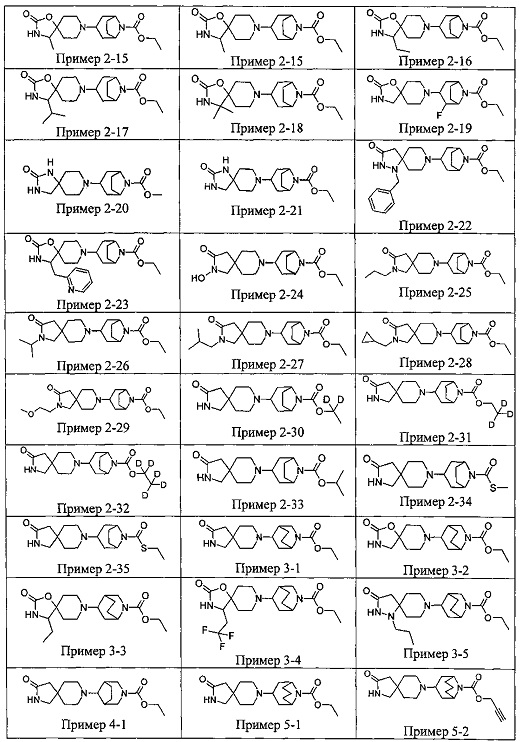
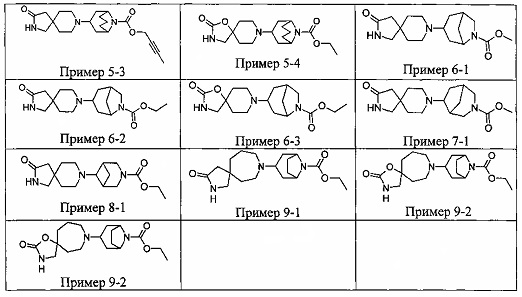
ПРИМЕРЫ ОТ 1-1 ДО 9-2
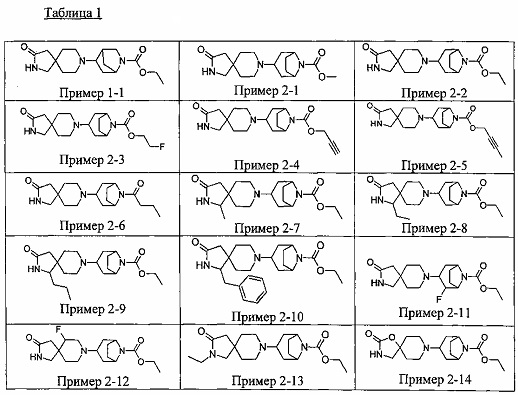
Были получены соединения Примеров от 1-1 до 9-2, приведенных ниже в Таблице 1. Характеристики их ЯМР и ЖХМС, а также способы, применяемые для их получения, приведены в Таблице 3. Исходные материалы для каждого из Примеров приведены в Таблице 2.
Общие методики
Если препаративные пути не включены, соответствующее промежуточное соединение является коммерчески доступным. Коммерческие реагенты использовали без дополнительной очистки. Комнатная температура (кт) означает температуру около 20-27°C. 1Н ЯМР-спектры регистрировали при 300 или 400 МГц либо на приборе Bruker, либо Varian. Значения химических сдвигов, то есть (δ)-значения, выражены в миллионных долях (м.д.). Следующие сокращения используются для мультиплетности ЯМР-сигналов: с = синглет, уш = уширенный, д = дублет, т = триплет, к = квартет, кв = квинтет, тд = триплет дублетов, тт = триплет триплетов, кд = квартет дублетов, ддд = дублет дублета дублетов, ддт = дублет дублета триплетов, м = мультиплет. Константы взаимодействия приведены в виде значений J, измеренных в Гц. Результаты ЯМР и масс-спектроскопии были скорректированы для учета фоновых пиков. Хроматография означает колоночную хроматографию, осуществляемую с применением силикагеля 60-120 меш и в условиях давления азота (флэш-хроматография). ТСХ для контроля реакций означает ТСХ, проводимую с применением указанной подвижной фазы и Silica gel F254 (Merck) в качестве неподвижной фазы. СВЧ-опосредованные реакции проводили в микроволновых реакторах Biotage Initiator или СЕМ Discover.
Эксперименты ЖХМС, как правило, проводили в условиях электрораспылительной ионизации, как определено для каждого соединения, при следующих условиях:
Способы А и В ЖХМС
Приборы: Waters Alliance 2795, PDA детектор Waters 2996, Micromass ZQ; Колонка: Waters X-Bridge C-18, 2,5 микрона, 2,1×20 мм или Phenomenex Gemini-NX C-18, 3 микрона, 2,0×30 мм; Градиент [время (мин)/растворитель D в С (%)]: Способ А: 0,00/2, 0,10/2, 2,50/95, 3,50/95, 3,55/2, 4,00/2 или Способ В: 0,00/2, 0,10/2, 8,40/95, 9,40/95, 9,50/2, 10,00/2; Растворители: растворитель С = 2,5 л H2O + 2,5 мл раствора аммиака; растворитель D = 2,5 л MeCN + 135 мл H2O + 2,5 мл раствора аммиака); Вводимый объем 3 мкл; УФ-детектирование 230-400 нМ; температура колонки 45°С; Скорость потока 1,5 мл/мин.
Способ С ЖХМС
Приборы: ЖХ Agilent 1260 Infinity с диодно-матричным детектором (DAD), одноквадрупольный МС Agilent 6120В Single с источником API-ES; Колонка: Phenomenex Gemini-NX C-18, 3 микрона, 2,0×30 мм; Градиент [время (мин)/растворитель B в А (%)]: Способ: 0,00/5, 2,00/95, 2,50/95, 2,60/5, 3,00/5; Растворители: растворитель А = 2,5 л Н2О + 2,5 мл (28% NH3 в Н2О); растворитель В = 2,5 л MeCN + 129 мл H2O + 2,7 мл (28% NH3 в H2O); Вводимый объем 0,5 мкл; УФ-детектирование 190-400 нМ; температура колонки 40°С; Скорость потока 1,5 мл/мин.
Способы D и Е ЖХМС
Приборы: HP 1100 с DAD G1315A, Micromass ZQ; Колонка: Waters X-Bridge C-18, 2,5 микрона, 2,1×20 мм или Phenomenex Gemini-NX C-18, 3 микрона, 2,0×30 мм; Градиент [время (мин)/растворитель D в С (%)]: Способ D: 0,00/2, 0,10/2, 2,50/95, 3,50/95, 3,55/2, 4.00/2 или Способ Е: 0,00/2, 0,10/2, 8,40/95, 9,40/95, 9,50/2, 10,00/2; Растворители: растворитель С = 2,5 л H2O + 2,5 мл 28% раствора аммиака в Н2О; растворитель D = 2,5 л MeCN + 135 мл Н2О + 2,5 мл 28% раствора аммиака в Н2О); Вводимый объем 1 мкл; УФ-детектирование 230-400 нМ; Масс-детектирование 130-800 а.е.м. (положительное и отрицательное электрораспыление); температура колонки 45°С; Скорость потока 1,5 мл/мин.
Способ F ЖХМС
Приборы: Waters Acquity Н Class, фотодиодная матрица, одноквадрупольный (SQ) детектор; Колонка: ВЕН C18, 1,7 микрон, 2,1×50 мм; Градиент [время (мин)/растворитель B в А (%)]: 0,00/5, 0,40/5, 0,8/35, 1,20/55, 2,50/100, 3,30/100 4,00/5; Растворители: растворитель А=5 мМ ацетата аммония и 0,1% муравьиной кислоты в H2O; растворитель В=0,1% муравьиной кислоты в MeCN; Вводимый объем 2 мкл; УФ-детектирование 200-400 нМ; Масс-детектирование 100-1200 а.е.м. (положительное электрораспыление); колонка при температуре окружающей среды; Скорость потока 0,5 мл/мин.
Способ G ЖХМС
Приборы: Waters 2695, фотодиодная матрица, детектор ZQ-2000; Колонка: X-Bridge C18, 5 микрон, 150×4,6 мм; Градиент [время (мин)/растворитель B в А (%)]: 0,00/10, 5,00/90, 7,00/100, 11,00/100, 11,01/10, 12,00/10; Растворители: растворитель А=0,1% аммиака в H2O; растворитель В=0,1% аммиака в MeCN; Вводимый объем 10 мкл; УФ-детектирование 200-400 нМ; Масс-детектирование 60-1000 а.е.м. (положительное электрораспыление); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
Способ Н ЖХМС
Приборы: Waters 2695, фотодиодная матрица, детектор ZQ-2000; Колонка: X-Bridge C18, 5 микрон, 150×4,6 мм; Градиент [время (мин)/растворитель B в А (%)]: 0,00/100, 7,00/50, 9,00/0, 11,00/0, 11,01/100, 12,00/100; Растворители: растворитель А=0,1% аммиака в H2O; растворитель В=0,1% аммиака в MeCN; Вводимый объем 10 мкл; УФ-детектирование 200-400 нМ; Масс-детектирование 60-1000 а.е.м. (положительное электрораспыление); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
Способ I ЖХМС
Приборы: Waters Acquity UPLC, PDA детектор Waters 3100, SQD; Колонка: Acquity HSS-T3, 1,8 микрон, 2,1×100 мм; Градиент [время (мин)/растворитель B в А (%)]: 0,00/10, 1,00/10, 2,00/15, 4,50/55, 6,00/90, 8,00/90, 9,00/10, 10,00/10; Растворители: растворитель А=0,1% трифторуксусной кислоты в воде; растворитель В = ацетонитрил; Вводимый объем 1 мкл; Длина волны детектирования 214 нм; Температура колонки 30°С; Скорость потока 0,3 мл в мин.
Способ J ЖХМС:
Приборы: Waters 2695, фотодиодная матрица, детектор ZQ-2000; Колонка: X-Bridge C18, 3,5 микрон, 50×4,6 мм; Градиент [время (мин)/растворитель B в А (%)]: 0,01/0, 0,20/0, 5,00/90, 5,80/95, 7,20/95, 7,21/100, 10,00/100; Растворители: растворитель А=0,1% аммиака в Н2О; растворитель В=0,1% аммиака в MeCN; Вводимый объем 10 мкл; УФ-детектирование 200-400 нМ; Масс-детектирование 60-1000 а.е.м. (положительное электрораспыление); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
Данные ЖХМС в экспериментальном разделе приведены в следующем формате: отношение массы иона к заряду, время удерживания, УФ-активность.
Аббревиатуры
д = день (дней)
ДХЭ = дихлорэтан
ДХМ = дихлорметан
DIPEA = диизопропилэтиламин
ДМФА = диметилформамид
ДМСО = диметилсульфоксид
ДФФА = дифенилфосфорилазид
ES = электрораспылительная ионизация
Et3N = триэтиламин
EtOAc = этилацетат
ч = час (часов)
ВЭЖХ = высокоэффективная жидкостная хроматография
ЖХ = жидкостная хроматография
LDA = диизопропиламид лития
LiHMDS = бис(триметилсилил)амид лития
MeCN = ацетонитрил
МеОН = метанол
мин = минута (минут)
МС = масс-спектрометрия
N2 = азот
NaCNBH3 = цианоборогидрид натрия
ЯМР = ядерно-магнитный резонанс
кт = комнатная температура
насыщ. = насыщенный
р-р = раствор
СФХ = сверхкритическая флюидная хроматография
STAB = триацетоксиборогидрид натрия
TBAF = фторид тетрабутиламмония
ТГФ = тетрагидрофуран
ТСХ = тонкослойная хроматография
Префиксы н-, втор-, изо- и трет- имеют свои обычные значения: нормальный, вторичный, изо- и третичный.
Синтез промежуточных соединений:
Путь 1
Типовой способ получения пиперидинов на примере получения Промежуточного соединения 3, 1-этил-2,8-диазаспиро[4,5]декан-3-она гидрохлорида
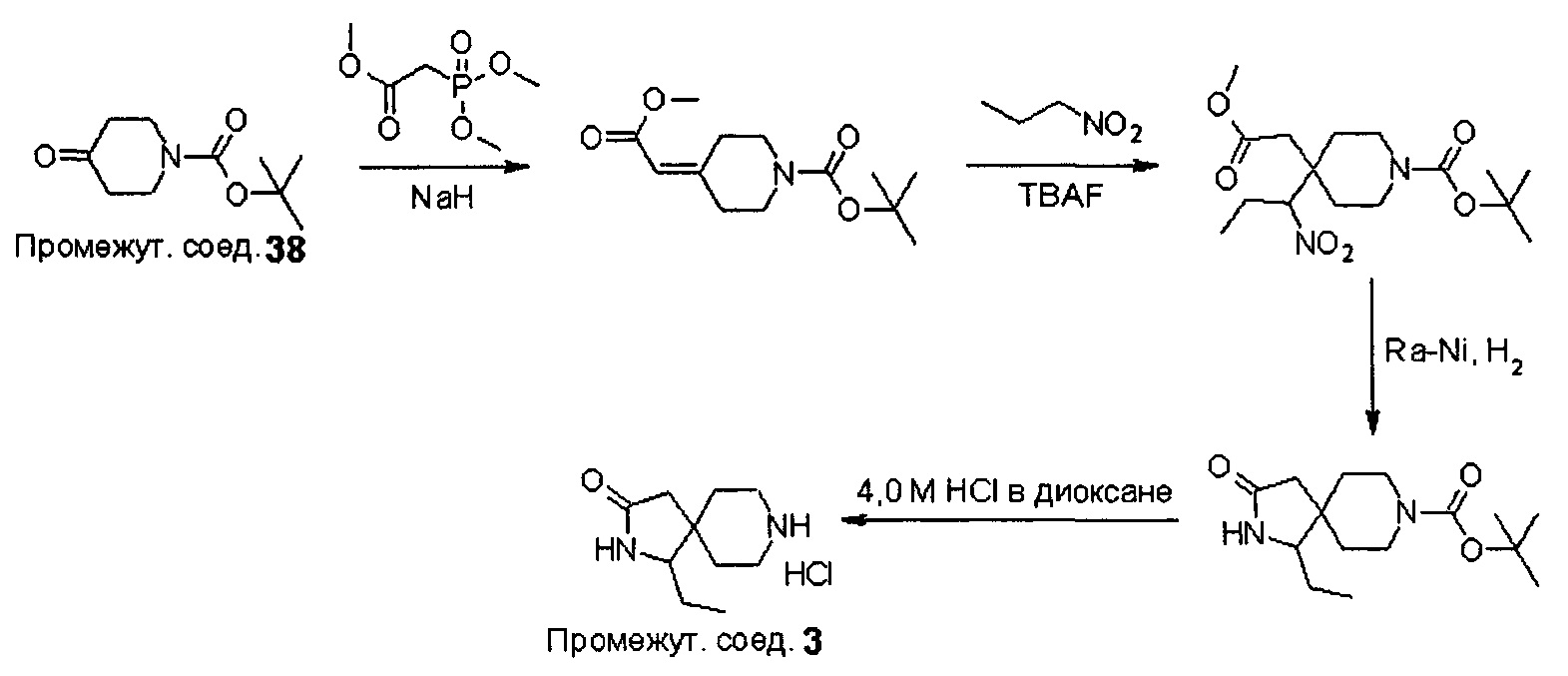
Гидрид натрия в минеральном масле (60%, 11,9 г, 297 ммоль) растворяли в ДМФА (200 мл) и по каплям добавляли метил-2-(диметоксифосфорил)ацетат (52,0 г, 286 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 20 мин, а затем по каплям добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (45,5 г, 228 ммоль) в ДМФ (100 мл) при 0°С. Реакционную смесь перемешивали при кт в течение 2 ч, а затем разбавляли ледяной водой (20 мл), фильтровали и удаляли растворители в вакууме с получением трет-бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (42,5 г, 72,9%) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 256 (М+Н)+ (ES+), при 2,47 мин, УФ-активно
трет-Бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилат (5,0 г, 19,60 ммоль) растворяли в ТГФ (50,0 мл), затем 1,0 М TBAF в ТГФ (25,5 мл, 25,5 ммоль) по каплям добавляли в реакционную смесь с последующим добавлением 1-нитропропана (2,62 г, 29,4 ммоль), реакционную смесь нагревали до 70°С в течение 24 ч. Реакционную смесь выливали в ледяную воду (150 мл), экстрагировали EtOAc (500 мл), водный слой дополнительно экстрагировали EtOAc (2×250 мл), органические слои объединяли, сушили (Na2SO4). Растворители удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, 0-6% EtOAc в гексане) с получением трет-бутил-4-(2-метокси-2-оксоэтил)-4-(1-нитропропил)пиперидин-1-карбоксилата (1,1 г, 40,9%) в виде желтого масла.
ЖХМС (Способ F): m/z 345 (М+Н)+ (ES+), при 2,43 мин, УФ-неактивно
трет-Бутил-4-(2-метокси-2-оксоэтил)-4-(1-нитропропил)пиперидин-1-карбоксилат (0,7 г, 2,03 ммоль) растворяли в МеОН (15 мл) и добавляли Raney®-Nickel (140,0 мг, 20% масс./масс). Реакционную смесь продували Н2, а затем перемешивали при кт в течение 16 ч. Реакционную смесь фильтровали через целит и удаляли растворители в вакууме с получением трет-бутил-1-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилата (0,28 г, 48,0%) в виде белого твердого вещества.
ЖХМС (Способ F): m/z 283 (М+Н)+ (ES+), при 1,95 мин, УФ-неактивно
трет-Бутил-1-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилат (0,27 г, 0,96 ммоль) растворяли 4,0 М HCl в 1,4-диоксане (5,0 мл) при комнатной температуре. Реакционную смесь перемешивали при кт в течение 16 ч и затем удаляли растворители в вакууме. Остаток растирали в порошок с диэтиловым эфиром с получением 1-этил-2,8-диазаспиро[4,5]декан-3-она гидрохлорида, Промежуточного соединения 3 (0,15 г, 84,7%) в виде белого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 2
Типовой способ получения кетонов на примере получения Промежуточного соединения 6, 6-фтор-2,8-диазаспиро[4,5]декан-3-она гидрохлорида
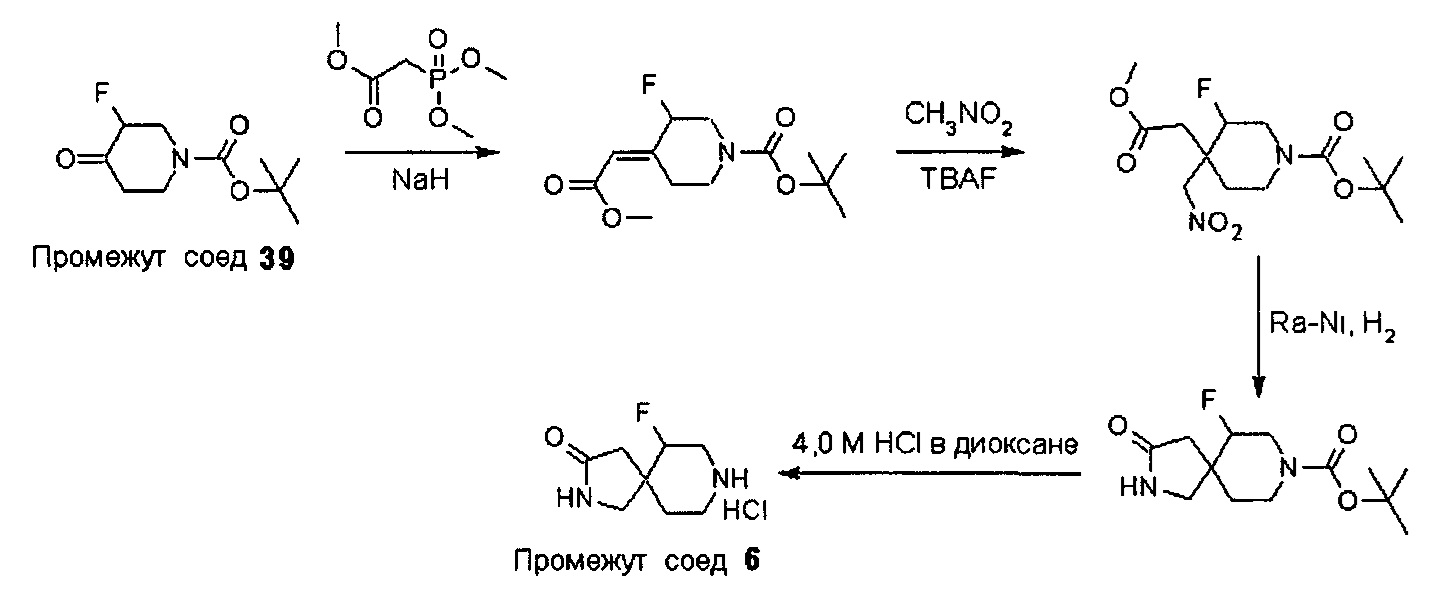
Гидрид натрия в минеральном масле (60%, 0,18 г, 4,6 ммоль) суспендировали в ТГФ (12 мл) и по каплям добавляли метил-2-(диметоксифосфорил)ацетат (0,84 г, 4,6 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч, а затем по каплям добавляли трет-бутил-3-фтор-4-оксо-пиперидин-1-карбоксилат (1,0 г, 4,6 ммоль) в ТГФ (5 мл) при 0°С.Реакционную смесь перемешивали при кт в течение 16 ч, а затем гасили водой (10 мл). Реакционную смесь экстрагировали EtOAc (3×20 мл), органические слои объединяли и промывали насыщ. р-ром NaHCO3 (20 мл) и насыщенным раствором хлорида натрия (20 мл), а затем сушили (Na2SO4). Растворители удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 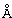 , 25 мл в мин, градиент от 0% до 35% EtOAc в изогексане]) с получением трет-бутил-3-фтор-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (0,94 г, 75%).
, 25 мл в мин, градиент от 0% до 35% EtOAc в изогексане]) с получением трет-бутил-3-фтор-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (0,94 г, 75%).
1H ЯМР: (400 МГц, ДМСО-d6) δ: 1,39 (д, J=2,5 Гц, 9 Н), 2,20-2,35 (м, 1H), 2,74-2,96 (м, 2Н), 3,64 (д, J=2,0 Гц, 3 Н), 4,02-4,20 (м, 1H), 4,22-4,43 (м, 1H), 5,05 (ддд, J=47,5, 4,5, 3,5 Гц, 1H), 5,98 (с, 1H), 6,19 (с, 0,5Н), 6,31 (с, 0,5Н)
трет-Бутил-3-фтор-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилат (0,94 г, 3,5 ммоль) и нитрометан (0,32 г, 5,2 ммоль) растворяли в 1,0 М TBAF в ТГФ (10 мл),, реакционную смесь нагревали при 50°С в атмосфере N2 в течение 2 д. Растворители удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 , 25 мл в мин, градиент от 0% до 40% EtOAc в изогексане]) с получением трет-бутил-3-фтор-4-(2-метокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-карбоксилата (0,47 г, 41%).
1H ЯМР: (400 МГц, ДМСО-d6) δ: 1,37 (с, 9Н), 1,59-1,74 (м, 2Н), 2,62-2,71 (м, 1H), 2,71-2,83 (м, 1H), 2,94-3,08 (м, 1H), 3,16-3,28 (м, 1H), 3,60 (с, 3Н), 3,66-3,84 (м, 1H), 3,94-4,07 (м, 1H), 4,64-4,71 (м, 1H), 4,71-4,86 (м, 2Н)
трет-Бутил-3-фтор-4-(2-метокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-карбоксилат (0,47 г, 1,41 ммоль) растворяли в EtOH (50 мл) и трижды пропускали через H-Cube®, оснащенный системой картриджа с катализатором ThalesNano CatCart®, 70 мм Raney®-Nickel (THS01132), при давлении 40 бар и температуре 50°С. Растворители удаляли в вакууме с получением трет-бутил-6-фтор-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилата (0,35 г, 85%) в виде белого твердого вещества, которое использовали без дополнительной очистки.
1H ЯМР: (400 МГц, ДМСО-d6) δ: 1,37 (с, 9Н), 1,42-1,56 (м, 1H), 1,56-1,74 (м, 1Н), 2,12 (с, 2Н), 2,84-2,92 (м, 1H), 2,94-3,06 (м, 1H), 3,06-3,21 (м, 1H), 3,28 (д, J=9,5 Гц, 1H), 3,71-3,83 (м, 1H), 3,83-4,02 (м, 1H), 4,41-4,60 (м, 1H), 7,58-7,70 (м, 1H)
трети-Бутил-6-фтор-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилат (0,35 г, 1,27 ммоль) суспендировали в 4 М HCl в 1,4-диоксане (10 мл) и перемешивали при кт в течение 16 ч. Растворители удаляли в вакууме с получением 6-фтор-2,8-диазаспиро[4,5]декан-3-она гидрохлорида, Промежуточного соединения 6 (0,27 г, 100% условно) в виде белого твердого вещества, которое использовали без дополнительной очистки.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 3
Для получения Промежуточного соединения 7, 2-этил-2,8-диазаспиро [4,5]декан-3-она гидрохлорида
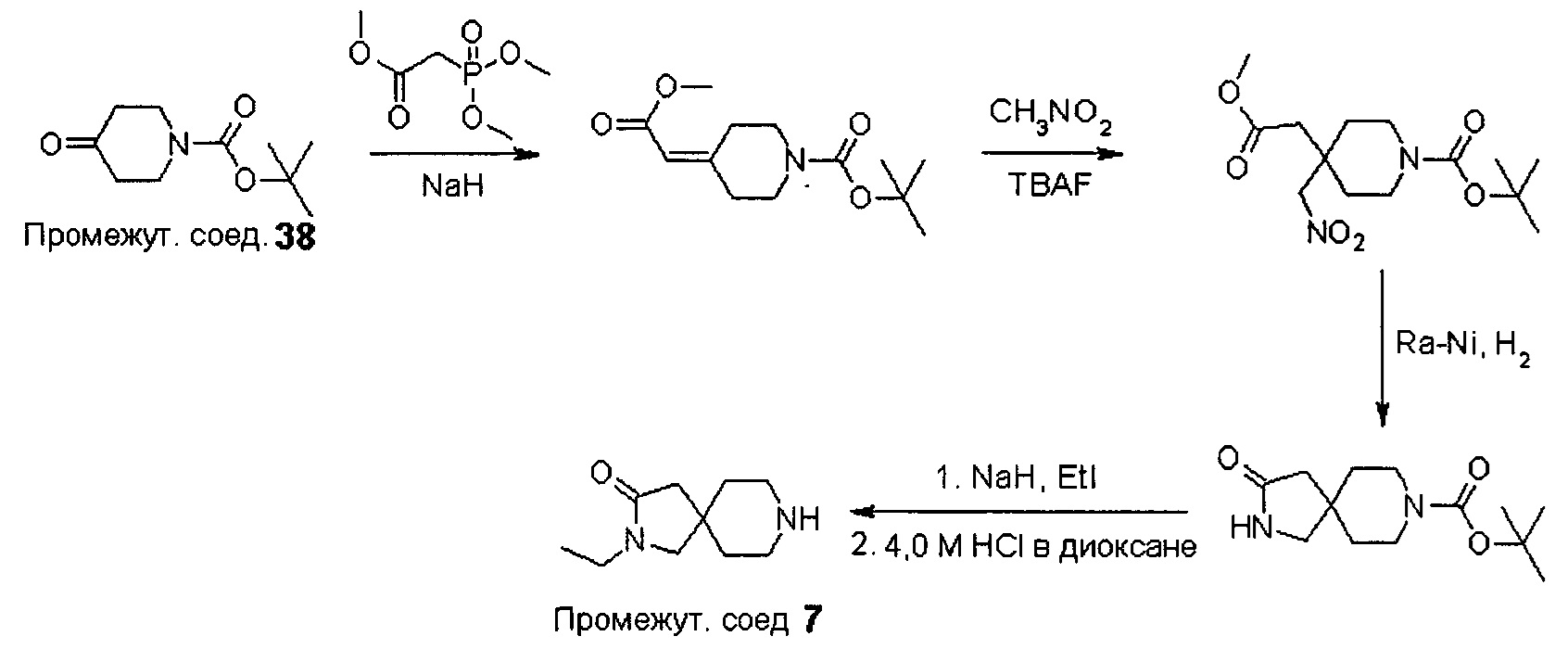
Гидрид натрия в минеральном масле (60%, 11,9 г, 297 ммоль) растворяли в ДМФА (200 мл) и по каплям добавляли метил-2-(диметоксифосфорил)ацетат (52,0 г, 286 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 20 мин, а затем по каплям добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (45,5 г, 228 ммоль) в ДМФА (100 мл) при 0°С. Реакционную смесь перемешивали при кт в течение 2 ч, а затем разбавляли ледяной водой (20 мл), фильтровали и удаляли растворители в вакууме с получением трет-бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (42,5 г, 72,9%) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 256 (М+Н)+ (ES+), при 2,47 мин, УФ-активно
трет-Бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилат (42,5 г, 166 ммоль) и нитрометан (11,2 г, 183 ммоль) растворяли в ТГФ (200 мл), по каплям добавляли 1,0 М раствора TBAF в ТГФ (250 мл, 250 ммоль) при 0°С. Реакционную смесь нагревали с обратным холодильником при 70°С в течение 16 ч. Реакционную смесь распределяли между Н2О (150 мл) и EtOAc (90 мл), водный слой дополнительно экстрагировали EtOAc (2×90 мл); органические слои объединяли и сушили (Na2SO4). Растворитель удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, от 0 до 30% EtOAc в гексанах) с получением трет-бутил-4-(2-метокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-карбоксилата (40,3 г, 76,5%) в виде белого твердого вещества.
ЖХМС (Способ F): m/z 261 (М+Н-56)+ (ES+), при 2,30 мин, УФ-неактивно
трет-Бутил-4-(2-метокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-карбоксилат (40,0 г, 126 ммоль) и Raney-Nickel (40,0 г) растворяли в EtOH (800 мл) и реакционную смесь продували Н2 в течение 16 ч. Реакционную смесь фильтровали через целит, промывали МеОН и растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, 0-4% МеОН в ДХМ) с получением трет-бутил-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилата (22,9 г, 71,2%) в виде белого твердого вещества.
ЖХМС (Способ F): m/z 255 (М+Н)+ (ES+), при 1,81 мин, УФ-неактивно
60% NaH (0,63 г, 15,7 ммоль) растворяли в ДМФА (15,0 мл) и по каплям добавляли трет-бутил-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилат (1,00 г, 3,92 ммоль) в ДМФА (5 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин, затем по каплям добавляли йодистый этил (0,48 мл, 5,88 ммоль) и оставляли реакционную смесь нагреваться до кт и перемешивали в течение 1 ч. Реакционную смесь распределяли между Н2О (30 мл) и EtOAc (25 мл), водный слой дополнительно экстрагировали EtOAc (2×25 мл), органические слои объединяли и сушили (Na2SO4). Растворитель удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, 0-3% МеОН в ДХМ) с получением трет-бутил-2-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилата (1,0 г, 90,3%) в виде белого твердого вещества.
ЖХМС (Способ F): m/z 283 (М+Н)+ (ES+), при 2,00 мин, УФ-неактивно
трет-Бутил-2-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-карбоксилат (1,00 г, 3,54 ммоль) растворяли в 4,0 М HCl в 1,4-диоксане (15,0 мл) и реакционную смесь перемешивали при кт в течение 5 ч. Растворитель удаляли в вакууме и остаток растирали в порошок с ацетоном (3×10 мл) с получением 2-этил-2,8-диазаспиро[4,5] декан-3-она гидрохлорида, Промежуточного соединения 7 (0,55 г, 71,2%) в виде белого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 4
Типовой способ получения пиперидинов на примере получения Промежуточного соединения 12, 4,4-диметил-1-окса-3,8-диазаспиро[4,5]декан-2-она
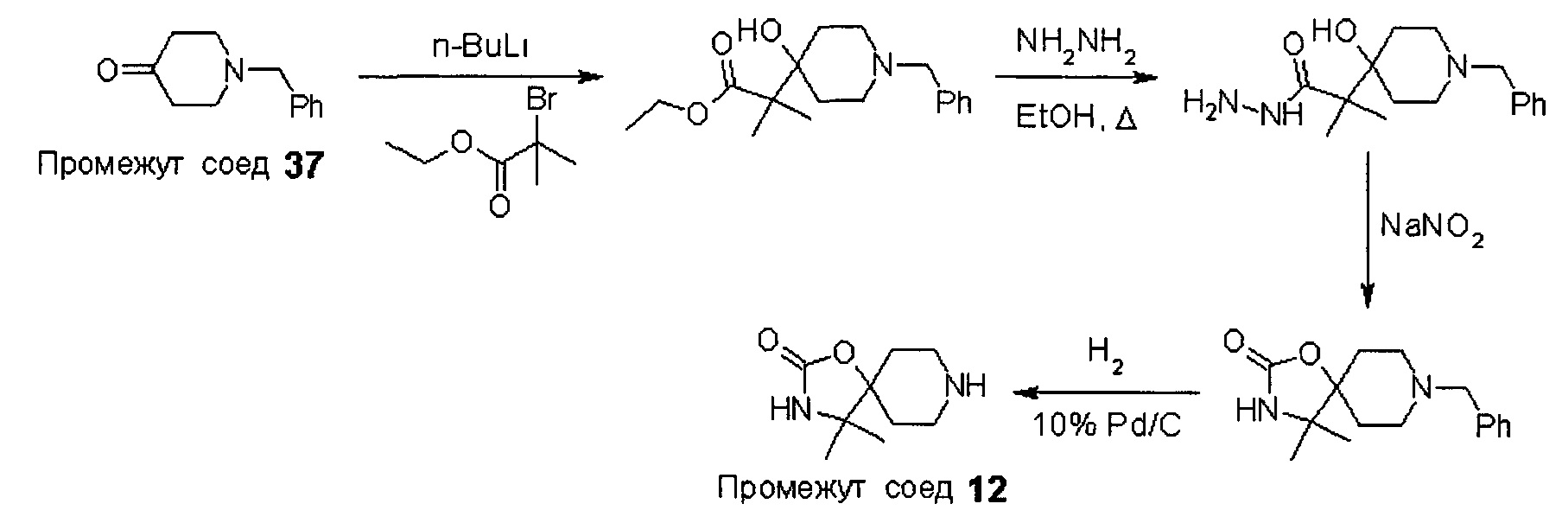
Этиловый эфир 2-бром-2-метилпропионовой кислоты (15,4 г, 79,2 ммоль) растворяли в Et2O (100 мл) и охлаждали до -78°С в атмосфере N2. По каплям добавляли н-бутиллитий (99 мл, 158 ммоль) и реакционную смесь перемешивали при -78°С в течение 1 ч. По каплям добавляли N-бензил-4-пиперидон (10 г, 52,8 ммоль) в Et20 (100 мл) и реакционную смесь перемешивали при -60°С в течение 2 ч. Реакционную смесь гасили насыщ. р-ром NH4Cl (200 мл) и затем разбавляли водой (500 мл). Реакционную смесь экстрагировали EtOAc (3×200 мл), органические слои объединяли и сушили (Na2SO4). Растворитель удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-15% EtOAc в гексане) с получением этил-2-(1-бензил-4-гидроксипиперидин-4 ил)-2-метилпропаноата (12,0 г, 74,3%) в виде желтой смолы.
ЖХМС (Способ F): m/z 306 (М+Н)+ (ES+), при 1,79 мин, УФ-активно
Этил-2-(1-бензил-4-гидроксипиперидин-4-ил)-2-метилпропаноат (12,0 г, 39,3 ммоль) и 85% гидразингидрат (80 мл) растворяли в EtOH (30 мл). Реакционную смесь нагревали с обратным холодильником при 100°С в течение 120 ч. Растворитель удаляли в вакууме с получением 2-(1-бензил-4-гидроксипиперидин-4-ил)-2-метилпропангидразида (15,0 г, 131%) в виде желтой смолы, которую использовали неочищенной на следующей стадии.
ЖХМС (Способ F): m/z 292 (М+Н)+ (ES+), при 1,37 мин, УФ-активно
2-(1-Бензил-4-гидроксипиперидин-4-ил)-2-метилпропангидразид (15 г, 39,3 ммоль условно) растворяли в воде (60 мл) и затем подкисляли концентрированной HCl (5 мл), реакционную смесь охлаждали до 5°С. Добавляли NaNO2 (4,2 г, 61,8 ммоль) в воде (8 мл) при 0°С и реакционную смесь нагревали до 60°С в течение 1 ч. Реакционную смесь подщелачивали 20% раствором NaOH и разбавляли водой (500 мл), экстрагировали EtOAc (3×200 мл), органические слои объединяли и сушили (Na2SO4). Растворитель удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-2% МеОН в ДХМ) с получением 8-бензил-4,4-диметил-1-окса-3,8-диазаспиро[4,5]декан-2-она (5,0 г, 46,4% [за две стадии]) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 275 (М+Н)+ (ES+), при 1,50 мин, УФ-активно
8-Бензил-4,4-диметил-1-окса-3,8-диазаспиро[4,5]декан-2-он (5,0 г, 18,2 ммоль) растворяли в МеОН (30 мл). Добавляли 10% PaVC (0,5 г) и реакционную смесь перемешивали в атмосфере Н2 (1 атм) при 50°С в течение 2 ч. Реакционную смесь фильтровали через целит и удаляли растворитель в вакууме. Остаток растирали в порошок с Et2O с получением 4,4-диметил-1-окса-3,8-диазаспиро[4,5]декан-2-она, Промежуточного соединения 12 (1,5 г, 45,4%) в виде желтого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 5
Типовой способ получения кетонов на примере получения Промежуточного соединения 16, метил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилата
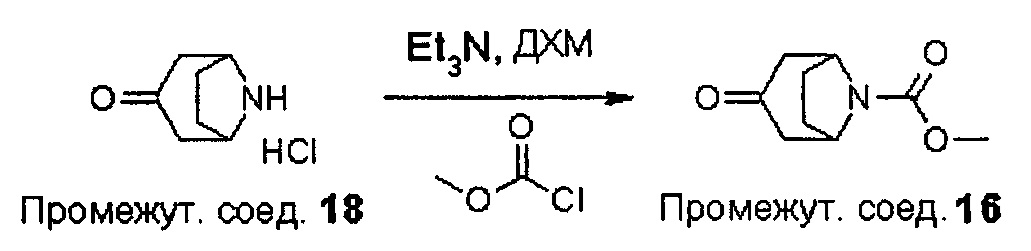
Гидрохлорид нортропинона (1,00 г, 6,1 ммоль) суспендировали в ДХМ (20 мл) и охлаждали до 0°С в атмосфере N2, добавляли триэтиламин (1,25 г, 12,4 ммоль) и метилхлорформиат (0,64 г, 6,8 ммоль), и реакционную смесь перемешивали при кт в течение 16 ч. Реакционную смесь разбавляли ДХМ (20 мл) и промывали насыщ. р-ром NaHCO3 (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем сушили (MgSO4), растворители удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 , 25 мл в минуту, градиент от 0% до 6% МеОН в ДХМ]), с получением метил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилата, Промежуточного соединения 16 (0,88 г, 77,6%) в виде бледно-желтой смолы.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 6
Типовой способ получения кетонов на примере получения Промежуточного соединения 34, этил-3-оксо-6-азабицикло[3,2,1]октан-6-карбоксилата
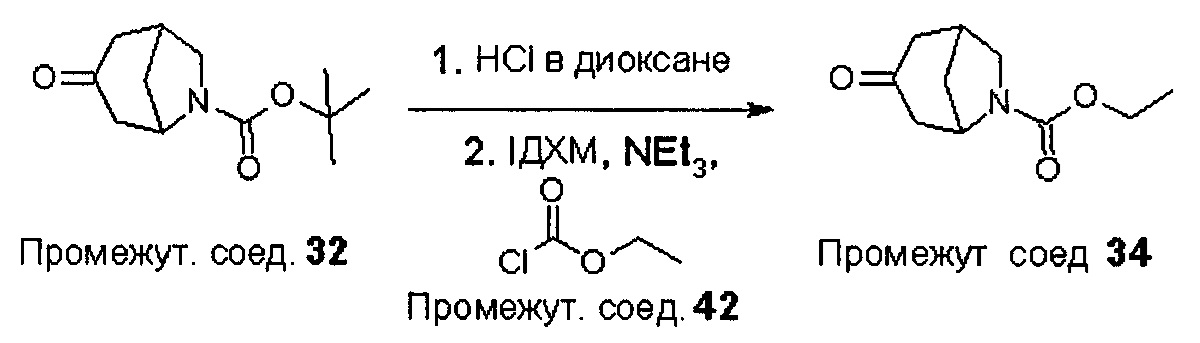
трет-Бутил-3-оксо-6-азабицикло[3,2,1]октан-6-карбоксилат (0,60 г, 2,7 ммоль) добавляли частями к 4,0 М HCl в 1,4-диоксане (10 мл, 40 ммоль), реакционную смесь перемешивали при кт в течение 24 ч и затем удаляли растворители в вакууме. Остаток растворяли в ДХМ (10 мл) и Et3N (0,75 мл, 5,4 ммоль) и охлаждали до 0°С. По каплям добавляли этилхлорформиат (0,28 мл, 3,0 ммоль) и реакционную смесь перемешивали при кт в течение 18 ч. Реакционную смесь распределяли между ДХМ (10 мл) и насыщ. р-ром NaHCO3 (10 мл), водный слой экстрагировали ДХМ (2×10 мл). Органические слои объединяли и промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4 и удаляли растворители в вакууме с получением этил-3-оксо-6-азабицикло[3,2,1]октан-6-карбоксилата, Промежуточного соединения 34 (0,43 г, 81%) в виде желтой смолы.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 7
Типовой способ получения активированных карбаматов на примере получения Промежуточного соединения 58, 4-нитрофенил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
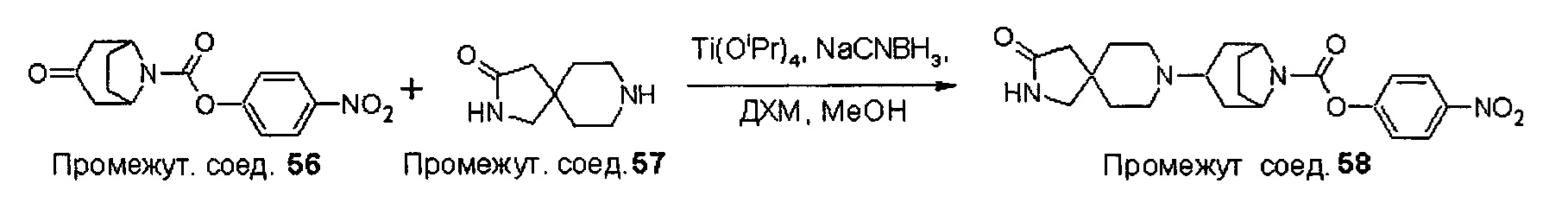
2,8-Диазаспиро[4,5]декан-3-он (1,12 г, 7,24 ммоль) и 4-нитрофенил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилат (2,10 г, 7,24 ммоль) растворяли в ДХМ (10 мл), обрабатывали изопропоксидом (2,57 г, 9,05 ммоль) титана (IV), а затем перемешивали при кт в атмосфере азота в течение ночи. Реакционную смесь разбавляли МеОН (30 мл) и добавляли NaCNBH3 (0,91 г, 14,48 ммоль), и реакционную смесь перемешивали при кт в атмосфере азота в течение ночи. Добавляли воду (10 мл) и ДХМ (10 мл) и раствор пропускали через подушку целита для удаления твердых веществ. Фильтрат разделяли и водную фазу экстрагировали ДХМ (3×25 мл). Органические фазы объединяли и промывали насыщ. р-ром NaHCO3 (25 мл), и сушили пропусканием через картридж для разделения фаз от Biotage. Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 10 г от Biotage, 40-63 мкм, 60 , 10 мл в минуту, градиент от 0% до 10% МеОН в ДХМ]) с получением 4-нитрофенил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Промежуточного соединения 58 в виде смеси диастереоизомеров (204 мг, 6,6%) в виде желтого стекловидного твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 8
Типовой способ получения пиперидинов на примере получения Промежуточного соединения 67, 1-бензил-1,2,8-триазаспиро[4,5]декан-3-она гидрохлорида
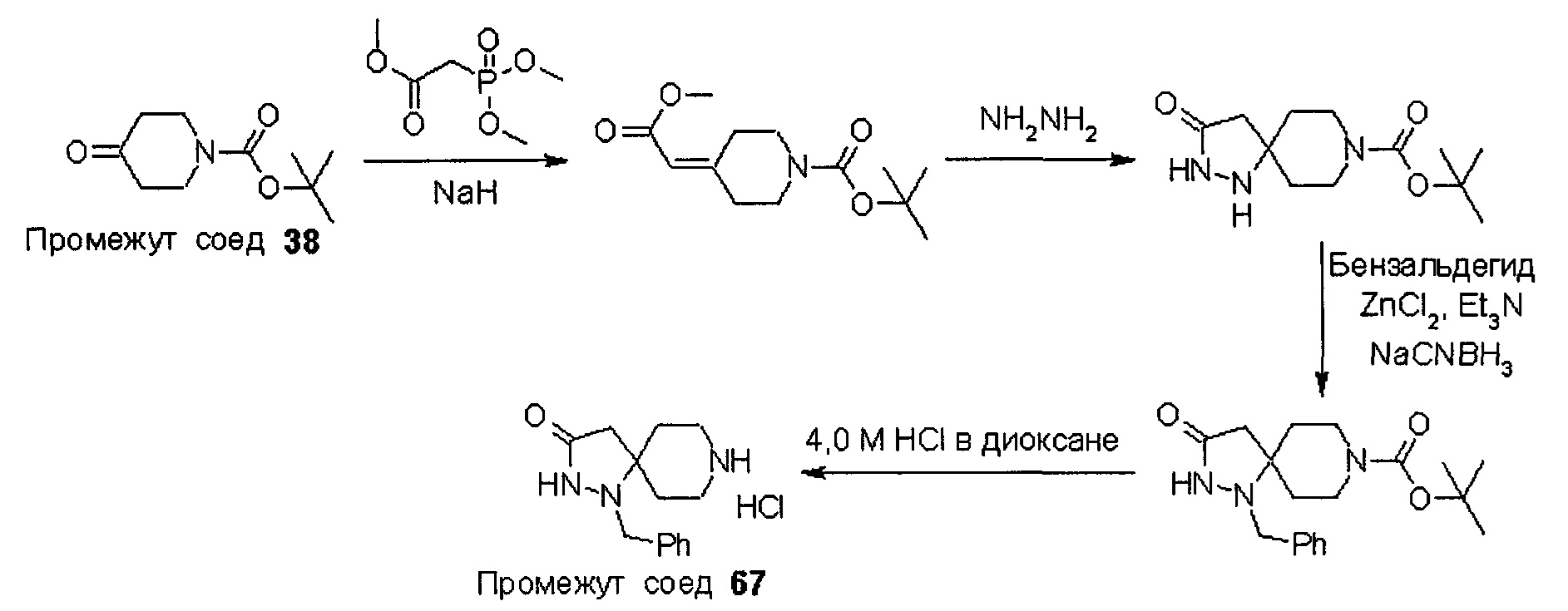
Гидрид натрия в минеральном масле (60%, 11,9 г, 297 ммоль) растворяли в ДМФА (200 мл) и по каплям добавляли метил-2-(диметоксифосфорил) ацетат (52,0 г, 286 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 20 мин, а затем по каплям добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (45,5 г, 228 ммоль) в ДМФ (100 мл) при 0°С. Реакционную смесь перемешивали при кт в течение 2 ч, а затем разбавляли ледяной водой (20 мл), фильтровали и удаляли растворители в вакууме с получением трет-бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (42,5 г, 72,9%) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 256 (М+Н)+ (ES+), при 2,47 мин, УФ-активно
К трет-бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилату (3,0 г, 11,8 ммоль) в EtOH (20 мл) добавляли гидразингидрат (1,1 мл, 23,5 ммоль) и реакционную смесь перемешивали при 80°С в течение 8 ч. Смесь распределяли между водой (150 мл) и EtOAc (120 мл), водный слой дополнительно экстрагировали EtOAc (2×120 мл), а объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл) и сушили (Na2SO4). Растворители удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, размер ячеек сита: 60-120 меш, от 4,0% до 10,0% МеОН в ДХМ) с получением трет-бутил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилата (1,78 г, 59,3%) в виде белого твердого вещества.
ЖХМС (Способ F): m/z 256 (М+Н)+ (ES+), при 1,70 мин, УФ-неактивно
трет-Бутил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилат (0,3 г, 1,18 ммоль), бензальдегид (0,12 мл, 1,29 ммоль), ZnCl2 (8,0 мг, 0,06 ммоль) и Et3N (0,80 мл, 5,89 ммоль) растворяли в МеОН (10 мл) и реакционную смесь перемешивали при 50°С в течение 2 ч. Затем смесь охлаждали до 0°С перед тем, как частями добавить NaCNBH3 (222 мг, 3,52 ммоль) и дополнительно перемешивать при 40°С в течение 30 ч. Смесь распределяли между Н2О (60 мл) и EtOAc (40 мл), и водный слой дополнительно экстрагировали EtOAc (2×40 мл). Объединенные органические слои сушили (Na2SO4), растворитель удаляли в вакууме и неочищенный остаток очищали растиранием в порошок с гексаном (3×3 мл) с получением трет-бутил-1-бензил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилата (320 мг, 79,0%) в виде желтой смолы.
ЖХМС (Способ G): m/z 346 (М+Н)+ (ES+), при 5,91 мин, УФ-активно
трет-Бутил-1-бензил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилат (0,3 г, 0,87 ммоль) растворяли в 1,4-диоксане (2 мл) и по каплям добавляли 4,0 М HCl в 1,4-диоксане (10 мл), реакционную смесь перемешивали при 30°С в течение 16 ч. Растворители удаляли в вакууме и остаток очищали растиранием в порошок с Et2O (3×3 мл) с получением 1-бензил-1,2,8-триазаспиро[4,5]декан-3-она, Промежуточного соединения 67 (0,21 г, 98,6%) в виде не совсем белого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 9
Типовой способ получения кетонов на примере получения Промежуточного соединения 77, 8-этил-3-оксо-8-азабицикло [3,2,1]октан-8-карботиоата
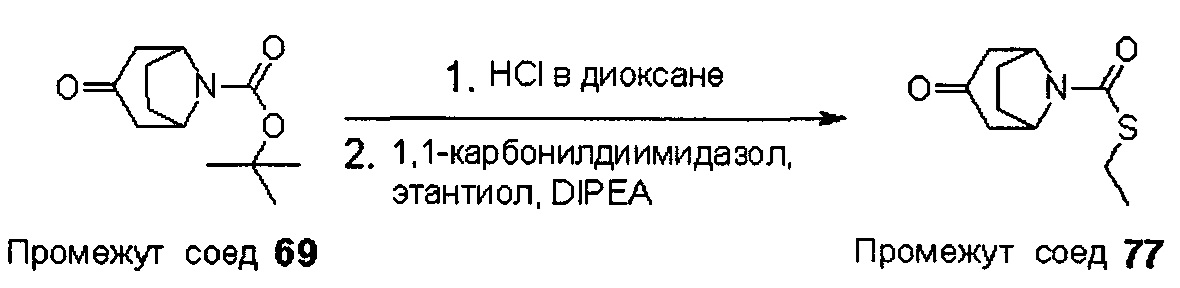
К трет-бутил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилату (1,00 г, 4,4 ммоль) в 1,4-диоксане (3 мл) добавляли 4,0 М HCl в 1,4-диоксане (10 мл, 40 ммоль), реакционную смесь перемешивали при 30°С в течение 7 ч, а затем удаляли растворители в вакууме. К части остатка (0,20 г, 1,3 ммоль) в ДХМ (10 мл) добавляли DIPEA (0,40 мл, 2,5 ммоль), этантиол (0,10 мл, 1,8 ммоль) и 1,1-карбонилдиимидазол (0,29 г, 1,8 ммоль), и полученную смесь перемешивали при кт в течение 18 ч. Реакционную смесь распределяли между Н2О (100 мл) и EtOAc (70 мл), и водный слой дополнительно экстрагировали EtOAc (2×70 мл). Объединенные органические слои сушили над Na2SO4 и растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, размер ячеек сита: 60-120 меш, 20-30% EtOAc в гексане) с получением S-этил-3-оксо-8-азабицикло[3,2,1]октан-8-карботиоата, Промежуточного соединения 77 (120 мг, 45,1%) в виде желтого масла.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 10
Типовой способ получения Промежуточного соединения 80, 1-окса-3,8-диазаспиро [4,6]ундекан-2-она
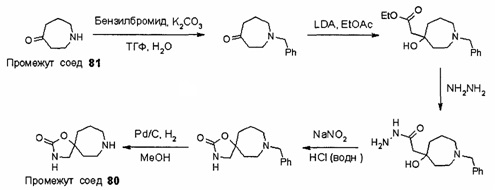
Азепан-4-она гидрохлорид (32 г, 214 ммоль), бензилбромид (40 г, 235 ммоль), K2CO3 (36 г, 257 ммоль) и воду (40 мл) растворяли в ТГФ (160 мл) и перемешивали при 50°С в течение 3 ч. Реакционную смесь разбавляли Н2О (500 мл), экстрагировали EtOAc (3×200 мл), а объединенные органические слои сушили (Na2SO4) и удаляли растворители в вакууме. Неочищенный остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-15% EtOAc и гексана) с получением 1-бензилазепан-4-она (18,0 г, 41,5%) в виде желтой жидкости.
ЖХМС (Способ F): m/z 204 (М+Н)+ (ES+), при 0,91 мин
Диизопропиламин (24,1 мл, 177,3 ммоль) растворяли в ТГФ (100 мл), охлаждали до -78°С в атмосфере N2 и по каплям добавляли 1,6 М раствор н-бутиллития (89,0 мл, 142,0 ммоль) при -78°С. Реакционную смесь перемешивали при 0°С в течение 40 мин перед тем, как добавить EtOAc (9,4 г, 160,4 ммоль) при -78°С и дополнительно перемешивать в течение 10 мин. Затем добавляли 1-бензилазепан-4-он (18 г, 88,6 ммоль) в ТГФ (160 мл) при -78°С и полученную смесь перемешивали при кт в течение 1 ч. Реакционную смесь гасили насыщенным раствором NH4Cl, разбавляли водой (500 мл), экстрагировали EtOAc (3×200 мл), а объединенные органические слои сушили (Na2SO4) и удаляли растворители в вакууме. Неочищенный остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-25% EtOAc в гексане) с получением этил-2-(1-бензил-4-гидроксиазепан-4-ил)ацетата (17,5 г, 67,8%) в виде желтой смолы.
ЖХМС (Способ F): m/z 293 (М+Н)+ (ES+), при 1,60 мин
Этил-2-(1-бензил-4-гидроксиазепан-4-ил)ацетат (17,5 г, 59,9 ммоль) и гидразингидрат (100 мл) перемешивали при 100°С в течение 4 ч. Реакционную смесь выпаривали в вакууме с получением 2-(1-бензил-4-гидроксиазепан-4-ил)ацетогидразида (22 г в неочищенном виде) в виде желтой смолы, которую брали непосредственно для следующей стадии.
ЖХМС (Способ K): m/z 278 (М+Н)+ (ES+), при 3,40 мин
2-(1-Бензил-4-гидроксипиперидин-4-ил)ацетогидразид (22,0 г, 79,0 ммоль) растворяли в Н2О (120 мл) и подкисляли концентрированной HCl при 0°С. К реакционной смеси добавляли NaNO2 (14,0 г, 197,6 ммоль) в Н2О (30 мл) при 0°С и продолжали перемешивание при 60°С в течение 1 ч. Реакционную смесь подщелачивали 20% раствором NaOH, разбавляли Н2О (500 мл), экстрагировали EtOAc (3×200 мл), а объединенные органические слои сушили (Na2SO4) и удаляли растворители в вакууме. Неочищенный продукт очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-8% МеОН в ДХМ) с получением 8-бензил-1-окса-3,8-диазаспиро[4,6]ундекан-2-она (8,5 г, 41,4%) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 261 (М+Н)+ (ES+), при 1,44 мин
К раствору 8-бензил-1-окса-3,8-диазаспиро[4,6]ундекан-2-она (8,5 г, 32,5 ммоль) в МеОН (50 мл) добавляли 10% Pd/C (2,5 г) и суспензию перемешивали при 60°С в течение 2 ч при давлении Н2, равном 1 атм. Реакционную смесь фильтровали через целит и удаляли растворители в вакууме с получением 1-окса-3,8-диазаспиро[4,6]ундекан-2-она, Промежуточного соединения 80 (5,3 г, 94,8%) в виде светло-желтого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 11
Типовой способ получения пиперидинов на примере получения Промежуточного соединения 82, 4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-2-она
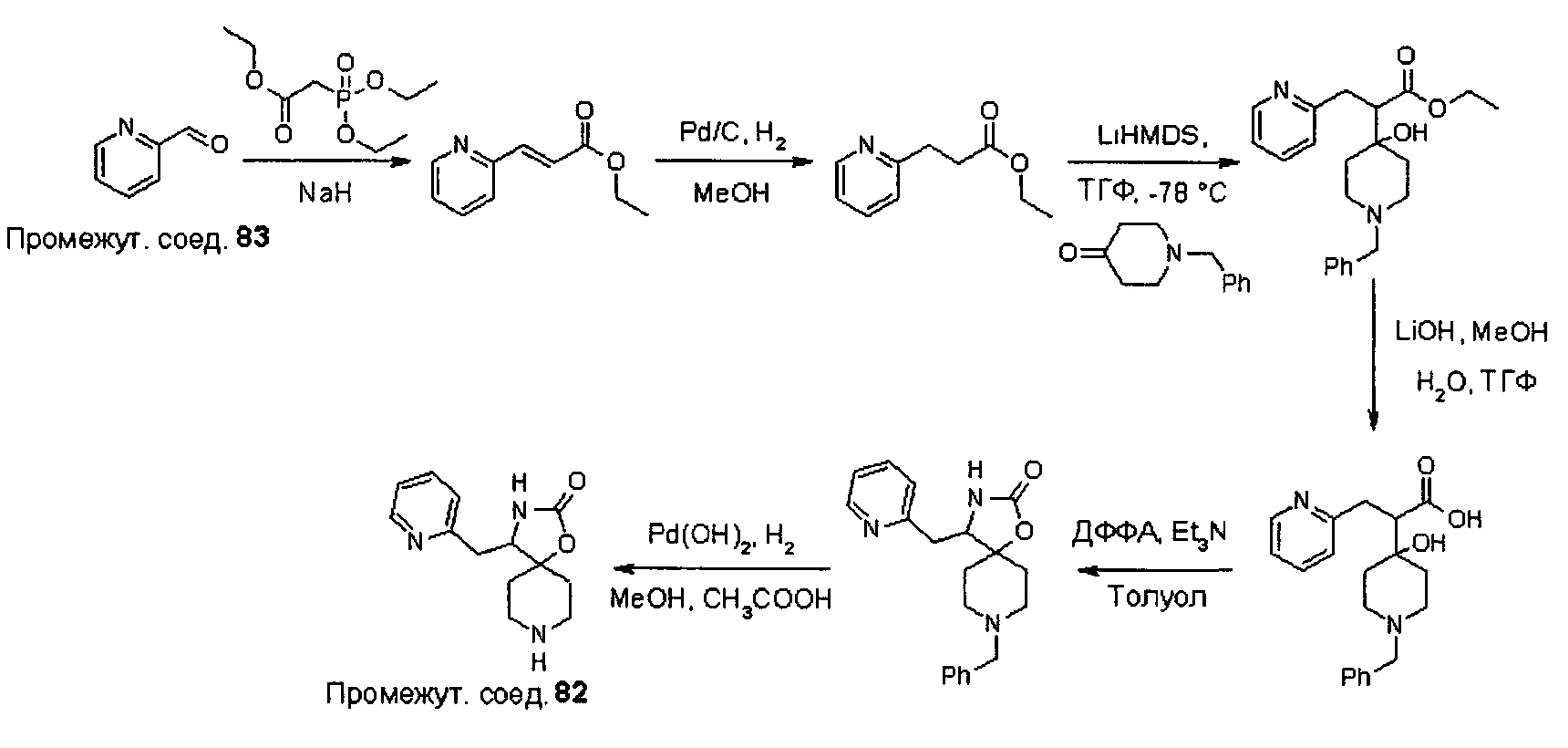
К раствору NaH (8,96 г, 50% в минеральном масле, 186,9 ммоль) в ТГФ (160 мл) добавляли триэтилфосфонацетат (20,5 мл, 102,7 ммоль) при 0°С. После перемешивания в течение 1 ч при 0°С медленно добавляли пиколинальдегид (10,00 г, 93,4 ммоль) при 0°С и реакционную смесь перемешивали при кт в течение 2 ч. Реакционную смесь гасили Н2О (10 мл) и водный слой экстрагировали EtOAc (3×100 мл). Органические слои объединяли, сушили (Na2SO4) и удаляли растворители в вакууме. Неочищенный остаток очищали с помощью колоночной флэш-хроматографии [нормальная фаза, силикагель (100-200 меш), градиент от 10% до 30% EtOAc в гексане] с получением этил-(Е)-3-(пиридин-2-ил)акрилата (7,90 г, 49%) в виде жидкости.
m/z(ES+): 178 (М+Н)+
К раствору этил-(Е)-3-(пиридин-2-ил)акрилата (7,9 г, 23,0 ммоль) в МеОН (100 мл) добавляли 10% Pd/C (0,80 г, 50% влажности) и реакционную смесь перемешивали в атмосфере Н2 (1 атм) при кт в течение 16 ч. Реакционную смесь фильтровали через подушку целита, тщательно промывали МеОН и удаляли растворители в вакууме с получением этил-3-(пиридин-2-ил)пропаноата (7,8 г, 98%) в виде жидкости.
m/z (ES+): 179 (М+Н)+
К раствору этил-3-(пиридин-2-ил)пропаноата (2,90 г, 16,2 ммоль) в ТГФ (60 мл) медленно добавляли LiHMDS (1 М, 48,6 мл, 48,6 ммоль) при -78°С и перемешивали в течение 30 мин с последующим добавлением 1-бензилпиперидин-4-она (3,10 г, 16,2 ммоль) при -78°С, и реакционную смесь перемешивали при -78°С в течение 4 ч. После завершения реакции реакционную смесь гасили насыщ. раствором NH4Cl (30 мл) и водный слой экстрагировали EtOAc (3×30 мл). Органические слои объединяли, сушили (Na2SO4) и удаляли растворители в вакууме. Неочищенный остаток очищали с помощью колоночной флэш-хроматографии [нормальная фаза, силикагель (100-200 меш), градиент от 10% до 30% EtOAc в гексане] с получением этил-2-(1-бензил-4-гидроксипиперидин-4-ил)-3-(пиридин-2-ил)пропаноата (2,80 г, 50%) в виде жидкости.
m/z (ES+): 369 (М+Н)+
К раствору этил-2-(1-бензил-4-гидроксипиперидин-4-ил)-3-(пиридин-2-ил) пропаноата (2,80 г, 7,61 ммоль) в МеОН : ТГФ (1:1, 30 мл) добавляли LiOH.H2O (1,28 г, 30,4 ммоль) в воде (10 мл) при кт и реакционную смесь перемешивали в течение 16 ч. Реакционную смесь подкисляли ледяной уксусной кислотой и экстрагировали EtOAc (3×20 мл). Органические слои объединяли и промывали насыщенным раствором хлорида натрия, сушили (Na2SO4) и удаляли растворители в вакууме с получением 2-(1-бензил-4-гидроксипиперидин-4-ил)-3-(пиридин-2-ил)пропановой кислоты (2,16 г, 84%) в виде бледно-желтого твердого вещества.
m/z (ES+): 339 (М+Н)+
К раствору 2-(1-бензил-4-гидроксипиперидин-4-ил)-3-(пиридин-2-ил)пропановой кислоты (1,70 г, 5,11 ммоль) в толуоле (30 мл) добавляли ДФФА (1,32 мл, 6,13 ммоль) и Et3N (0,84 мл, 6,13 ммоль), и реакционную смесь нагревали при 80°С в течение 16 ч. Реакционную смесь охлаждали до кт и удаляли растворители в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии [нормальная фаза, силикагель (100-200 меш), градиент от 1% до 30% EtOAc в гексане] с получением 8-бензил-4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-2-она (1,25 г, 56%) в виде белого твердого вещества.
m/z (ES+): 338 (М+Н)+
К раствору 8-бензил-4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-2-она (0,80 г, 2,37 ммоль) в МеОН (40 мл), после дегазации в атмосфере N2, добавляли 10% Pd(OH)2 на углероде (0,15 г, 50% влажности). Реакционную смесь перемешивали в атмосфере Н2 (1 атм) при кт в течение 16 ч. После завершения реакции реакционную смесь фильтровали через подушку целита, тщательно промывали МеОН и удаляли растворители в вакууме с получением 4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-2-она, Промежуточного соединения 83 (0,58 г, 98%) в виде жидкости.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 12
Типовой способ получения пиперидинов на примере получения Промежуточного соединения 88, 4-(2,2,2-трифторэтил)-1-окса-3,8-диазаспиро[4,5]декан-2-она
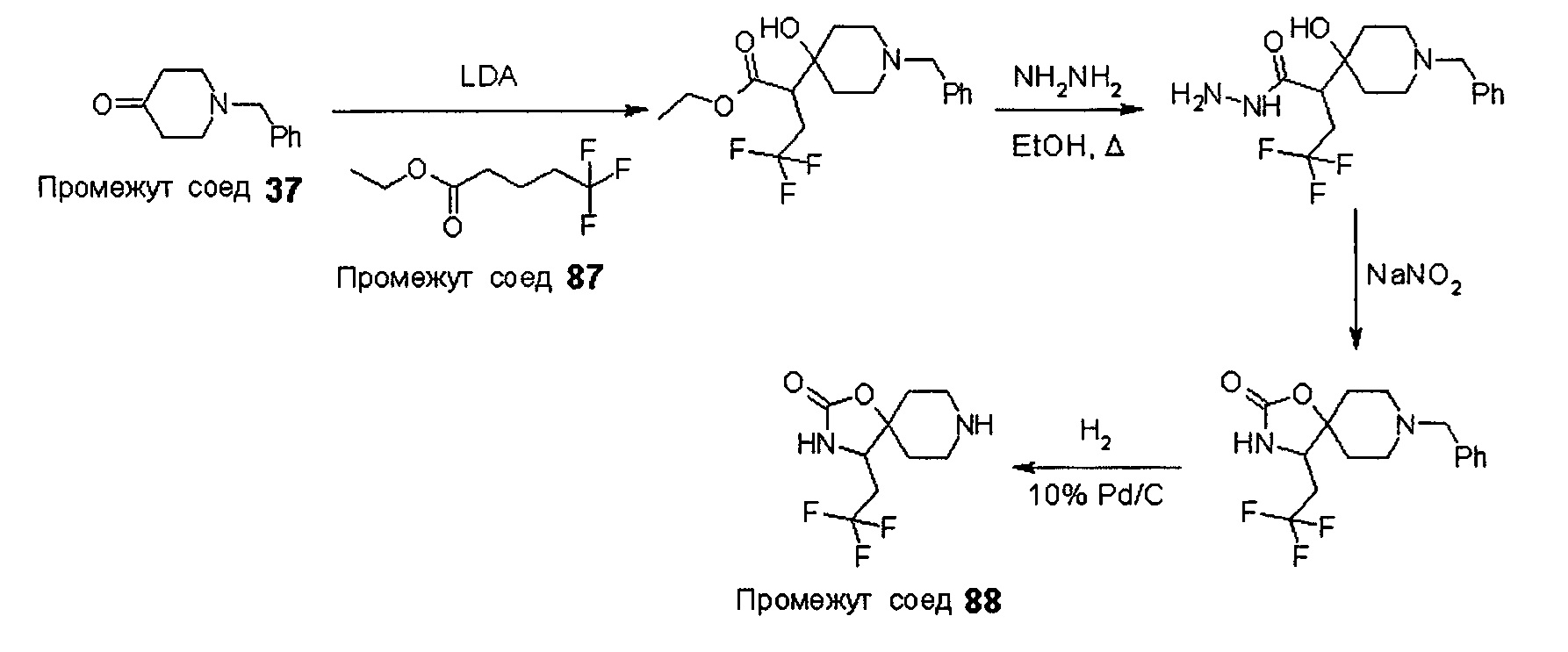
Диизопропиламин (12,8 г, 126,98 ммоль) растворяли в ТГФ (100 мл) и охлаждали до -78°С в атмосфере азота. По каплям добавляли н-бутиллитий (79,3 мл, 126,98 ммоль, 1,6 М в ТГФ) и реакционную смесь перемешивали при -78°С в течение 1 ч. Этил-4,4,4-трифторбутаноат (16,2 г, 95,23 ммоль) добавляли в течение 30 мин, затем реакционную смесь перемешивали при -78°С в течение 1 ч. По каплям добавляли N-бензилпиперидон (15 г, 79,36 ммоль) и реакционную смесь перемешивали при -78°С в течение 30 минут. Реакционную смесь гасили насыщенным раствором NH4Cl (200 мл), разбавляли водой (500 мл) и экстрагировали EtOAc (3×200 мл), а объединенные органические слои сушили (Na2SO4) и удаляли растворители в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-25% EtOAc в гексане) с получением этил-2-(1-бензил-4-гидроксипиперидин-4-ил)-4,4,4-трифторбутаноата (24,0 г, 84,2%) в виде желтой смолы.
ЖХМС (Способ F): m/z 360 (М+Н)+ (ES+), при 1,75 мин, УФ-активно
Этил-2-(1-бензил-4-гидроксипиперидин-4-ил)-4,4,4-трифторбутаноат (24,0 г, 66,85 ммоль) и 85% гидразингидрат (200 мл) растворяли в этаноле (100 мл). Реакционную смесь нагревали с обратным холодильником и оставляли перемешиваться при 100°С в течение 72 ч. Реакционную смесь выпаривали в вакууме с получением неочищенного продукта 2-(1-бензил-4-гидроксипиперидин-4-ил)-4,4,4-трифторбутангидразида (28,0 г) в виде желтой смолы. Неочищенный продукт использовали на следующей стадии без какой-либо очистки.
ЖХМС (Способ F): m/z 346 (М+Н)+ (ES+), при 1,41 мин, УФ-активно
Неочищенный 2-(1-бензил-4-гидроксипиперидин-4-ил)-4,4,4-трифторбутангидразид (28 г, 81,1 ммоль) растворяли в воде (200 мл), подкисляли концентрированной HCl и охлаждали до 0°С. Добавляли NaNO2 (16,7 г, 243,2 ммоль) в воде (50 мл) при 0°С и реакционную смесь оставляли перемешиваться при 60°С в течение 1 ч. Реакционную смесь подщелачивали 20% раствором NaOH, разбавляли водой (500 мл) и экстрагировали EtOAc (3×200 мл), а объединенные органические слои сушили (Na2SO4) и удаляли растворители в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 60-120 меш, 0-3,0% МеОН в дихлорметане) с получением 8-бензил-4-(2,2,2-трифторэтил)-1-окса-3,8-диазаспиро[4,5]декан-2-она (1,2 г, 4,5%) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 329 (М+Н)+ (ES+), при 1,48 мин, УФ-активно
8-Бензил-4-(2,2,2-трифторэтил)-1-окса-3,8-диазаспиро[4,5]декан-2-он (1,2 г, 3,65 ммоль) растворяли в метаноле (30 мл). Добавляли Pd/C (300 мг, 10% Pd/C 50% влажности) и реакционную смесь перемешивали в атмосфере водорода (1 атм) при 50°С в течение 2 ч. Реакционную смесь фильтровали через целит и удаляли растворители в вакууме. Неочищенный продукт растирали в порошок с диэтиловым эфиром с получением 4-(2,2,2-трифторэтил)-1-окса-3,8-диазаспиро[4,5]декан-2-она, Промежуточного продукта 88 (0,75 г, 88,2%) в виде желтого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 13
Типовой способ получения пиперидинов на примере получения Промежуточного соединения 88, 1-пропил-1,2,8-триазаспиро[4,5]декан-3-она гидрохлорида
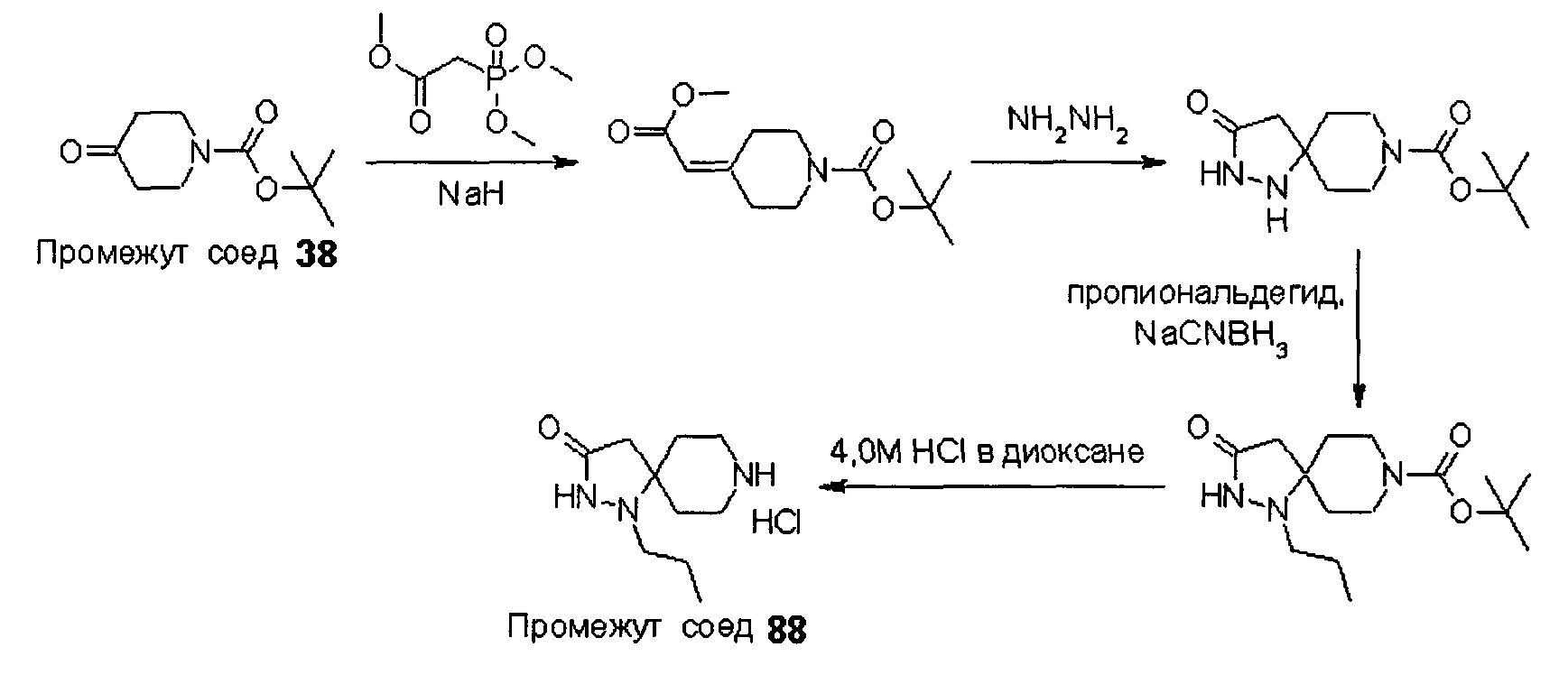
Гидрид натрия в минеральном масле (60%, 11,9 г, 297 ммоль) растворяли в ДМФА (200 мл) и по каплям добавляли метил-2-(диметоксифосфорил)ацетат (52,0 г, 286 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 20 мин, а затем по каплям добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (45,5 г, 228 ммоль) в ДМФ (100 мл) при 0°С. Реакционную смесь перемешивали при кт в течение 2 ч, а затем разбавляли ледяной водой (20 мл), фильтровали и удаляли растворители в вакууме с получением трет-бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (42,5 г, 72,9%) в виде желтого твердого вещества.
ЖХМС (Способ F): m/z 256 (М+Н)+ (ES+), при 2,47 мин, УФ-активно
трет-Бутил-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилат (3,0 г, 11,8 ммоль) растворяли в EtOH (20 мл) и добавляли гидразингидрат (1,1 мл, 23,5 ммоль), и перемешивали реакционную смесь при 80°С в течение 8 ч. Реакционную смесь распределяли между водой (150 мл) и EtOAc (120 мл), водный слой дополнительно экстрагировали EtOAc (2×120 мл), органические слои объединяли, промывали насыщенным раствором хлорида натрия (100 мл) и сушили (Na2SO4). Растворители удаляли в вакууме и остаток очищали с помощью колоночной хроматографии (нормально-фазовый силикагель, размер ячеек сита: 60-120 меш, от 4,0% до 10,0% МеОН в ДХМ) с получением трет-бутил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилата (1,78 г, 59,3%) в виде белого твердого вещества.
ЖХМС (Способ F): m/z 256 (М+Н)+ (ES+), при 1,70 мин, УФ-неактивно
трет-Бутил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилат (500 мг, 1,96 ммоль) растворяли в МеОН (10 мл). Добавляли пропиональдегид (0,2 мл, 2,16 ммоль) и триэтиламин (0,8 мл, 5,88 ммоль), реакционную смесь перемешивали при 45°С в течение 3 ч. Частями добавляли NaCNBH3 (370 г, 5,88 ммоль) и реакционную смесь перемешивали при кт в течение 17 ч. Растворители удаляли в вакууме и остаток распределяли между Н2О (100 мл) и EtOAc (80 мл), водный слой экстрагировали EtOAc (2×80 мл), органические слои объединяли, промывали насыщенным раствором хлорида натрия (100 мл) и сушили (Na2SO4). Растворители удаляли в вакууме и остаток очищали растиранием в порошок с гексаном (3×3 мл) с получением трет-бутил-1-пропил-3-оксо-1,2,8-триазаспиро[4,5]декан-8-карбоксилата (560 мг, 96,2%) в виде желтой смолы.
ЖХМС (Способ Е): m/z 298 (М+Н)+ (ES+), при 3,72 мин, УФ-неактивно
трет-Бутил-1-пропил-3-оксо-1,2,8-триазаспиро [4,5]декан-8-карбоксилат (610 г, 2,05 ммоль) растворяли в 1,4-диоксане (3 мл) и по каплям добавляли 4,0 М HCl в диоксане (5 мл), реакционную смесь перемешивали при 25°С в течение 16 ч. Растворители удаляли в вакууме и остаток очищали растиранием в порошок с Et2O (3×3 мл) с получением 1-пропил-1,2,8-диазаспиро[4,5]декан-3-она гидрохлорида, Промежуточного соединения 88 (470 мг, 98,3%) в виде не совсем белого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Общие способы синтеза:
Путь а
Типовой способ получения пиперидинов через восстановительное аминирование NaCNBH3 на примере получения Примера 2-2, этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
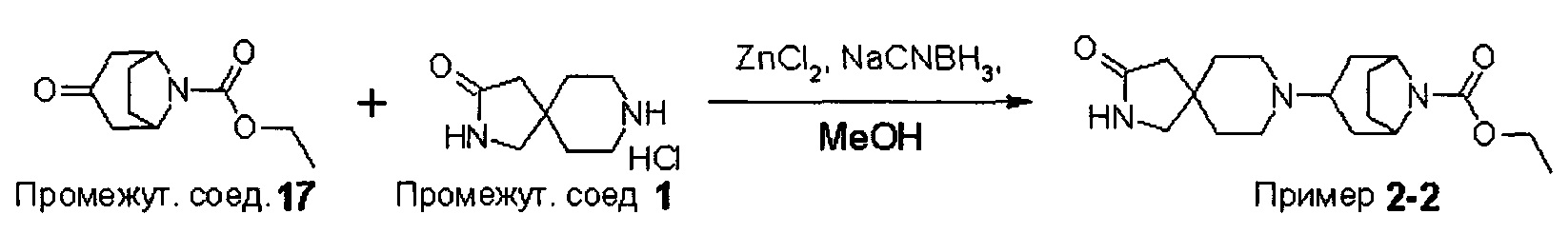
2,8-Диазаспиро[4,5]декан-3-она гидрохлорид (0,40 г, 1,78 ммоль) растворяли в МеОН (3 мл) и обрабатывали K2CO3 (0,49 г, 3,55 ммоль) в минимальном количестве воды, чтобы обессолить. Полученную смесь выпаривали в вакууме. Остаток и этил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилат (0,35 г, 1,78 ммоль) растворяли в МеОН (8 мл) и добавляли хлорид цинка (0,73 г, 5,33 ммоль). Реакционную смесь перемешивали при 50°С, в атмосфере азота, в течение 2 ч, затем охлаждали до кт и добавляли NaCNBH3 (0,23 г, 3,55 ммоль). Реакционную смесь перемешивали при 50°С в атмосфере азота в течение 16 ч. Реакционную смесь охлаждали до кт и обрабатывали насыщ. р-ром NaHCO3, органический растворитель удаляли в вакууме и водный слой экстрагировали ДХМ (2×10 мл), органические слои объединяли и промывали насыщенным раствором хлорида натрия (10 мл) и сушили пропусканием через картридж для разделения фаз от Biotage. Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж PuriFlash Column 15 Silica HP-Silica 15 мкм 40G от Interchim, 30 мл в мин, градиент от 0% до 10% МеОН в ДХМ]), с получением этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 1 (16 мг, 2,5%) Примера 2-2 в виде не совсем белого твердого вещества, и этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 2 (10 мг, 1,7%) Примера 2-2 в виде не совсем белого твердого вещества.
Данные для Изомеров 1 и 2 приведены в Таблице 3.
Путь b
Типовой способ получения пиперидинов через восстановительное аминирование NaCNBH3 на примере получения Примера 2-8, этил-3-(1-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
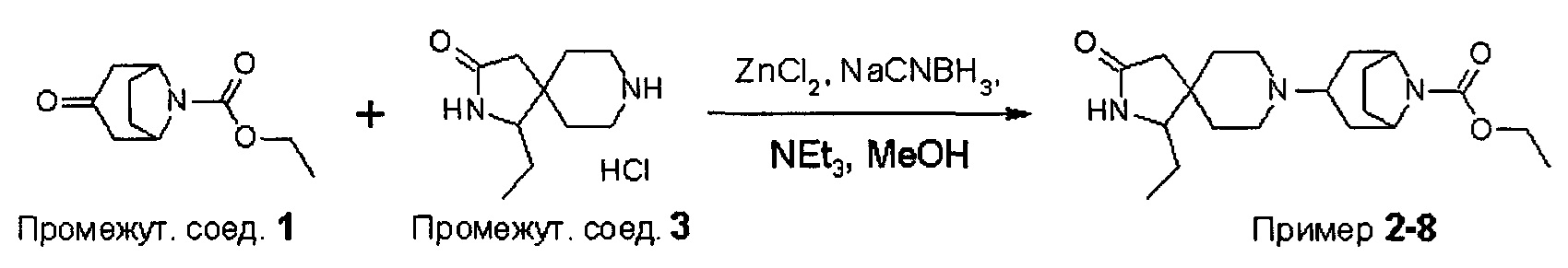
1-Этил-2,8-диазаспиро[4,5]декан-3-она гидрохлорид (0,1 г, 0,55 ммоль), этил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилат (0,2 г, 0,60 ммоль), Et3N (0,38 мл, 2,74 ммоль) и ZnCl2 (0,04 г, 0,03 ммоль) растворяли в МеОН (5 мл) в атмосфере N2 и перемешивали при 60°С в течение 16 ч. Реакционную смесь охлаждали до 0°С и частями добавляли NaCNBH3 (0,17 г, 2,74 ммоль), реакционную смесь перемешивали в атмосфере N2 при 60°С в течение 16 ч. Растворители удаляли в вакууме и остаток распределяли между водой (50 мл) и EtOAc (30 мл), водный слой дополнительно экстрагировали с EtOAc (2×30 мл); органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили (Na2SO4). Растворители удаляли в вакууме и остаток очищали с помощью препаративной обращенно-фазовой ВЭЖХ (Durashell, 250×21,2 мм, 5 мкм, 13 мл в мин, градиент от 30% до 100% (в течение 28 мин), затем 100% (3 мин) ацетонитрил в 50% ацетонитрил/вода (0,1% аммиака)) с получением этил-3-(1-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 1 (0,03 г, 15,1%) Примера 2-8 в виде бесцветного твердого вещества, и этил-3-(1-этил-3-оксо-2,8-диазаспиро[4,5]декан-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 2 (0,006 г, 3,1%) Примера 2-8 в виде бесцветного твердого вещества.
Данные для обоих изомеров приведены в Таблице 3.
Путь с
Типовая процедура получения пиперидинов через восстановительное аминирование триацетоксиборогидрида натрия в ДМФА на примере получения Примера 3-2, этил-5-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло [2,2,2] октан-2-карбоксилата
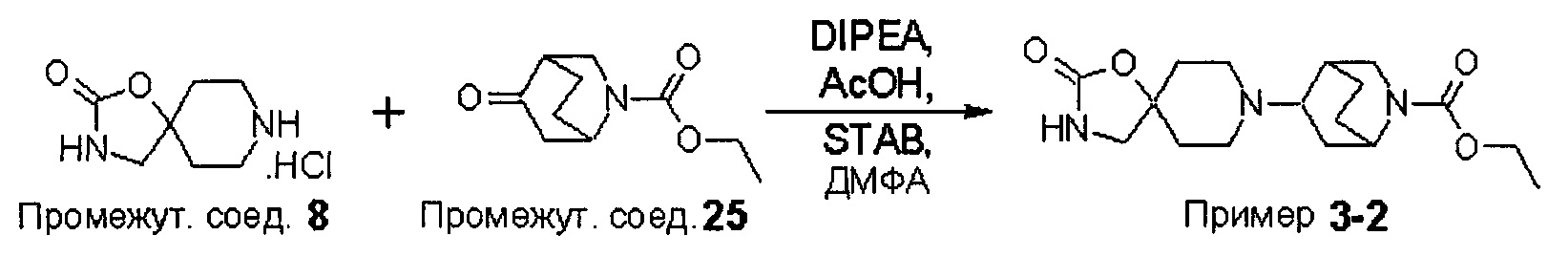
1-Окса-3,8-диазаспиро[4,5]декан-2-она гидрохлорид (0,10 г, 0,52 ммоль) и этил-5-оксо-2-азабицикло[2,2,2]октан-2-карбоксилат (0,10 г, 0,51 ммоль) смешивали в ДМФА (5 мл) при кт. Добавляли DIPEA (0,18 мл, 1,0 ммоль) и АсОН (0,044 мл, 0,77 ммоль) с последующим добавлением STAB (0,32 г, 1,5 ммоль). Реакционную смесь перемешивали в атмосфере азота при 45°С в течение 3 д и при 60°С в течение 1 д, растворители удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 , 30 мл в мин, градиент от 0% до 10% Растворителя А в ДХМ в течение 10 объемов колонки, затем изократическое элюирование 10% Растворителем А в ДХМ в течение 5 объемов колонки, где растворитель А представляет собой 10% (7 М NH3/МеОН) в МеОН]) с получением смеси диастереомеров. Эту смесь очищали с помощью препаративной обращенно-фазовой ВЭЖХ (колонка Gemini-NX 5 мкм C18 110A Axia от Phenomenex, 100×30 мм, элюирование 15-55% градиентом MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где Растворитель В представляет собой 0,2% (28% NH3/H2O) в H2O] и сбор фракций посредством мониторинга при 205 нм) с получением этил-5-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 1 (0,074 г, 43%) Примера 3-2 в виде бесцветного твердого вещества, и этил-5-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 2 (0,023 г, 13%) Примера 3-2 в виде бесцветного твердого вещества.
Данные для Изомеров 1 и 2 приведены в Таблице 3.
Путь d
Типовая процедура получения пиперидинов через восстановительное аминирование триацетоксиборогидрида натрия на примере получения Примера 5-2, проп-2-ин-1-ил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата
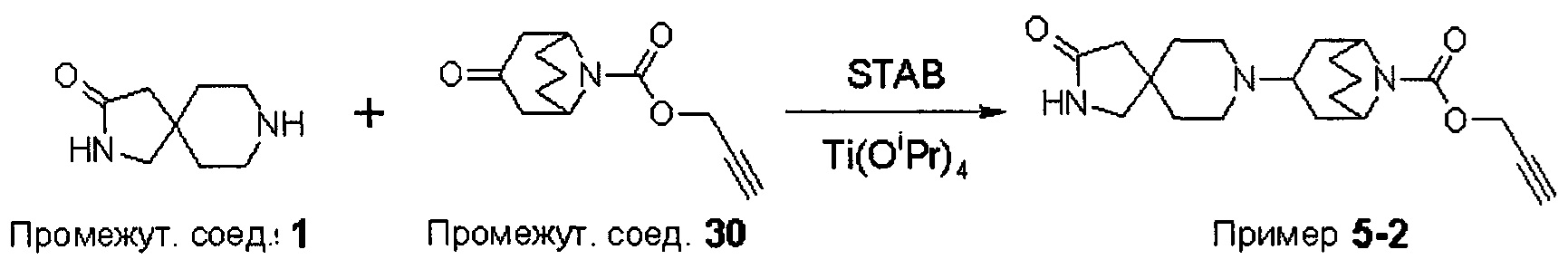
2,8-Диазаспиро[4,5]декан-3-он (0,12 г, 0,75 ммоль) и проп-2-ин-1-ил-3-оксо-9-азабицикло[3,3,1]нонан-9-карбоксилат (0,17 г, 0,75 ммоль) растворяли в ДХЭ (7,5 мл) при кт и добавляли изопропоксид титана (0,66 мл, 2,25 ммоль). Реакционную смесь нагревали с обратным холодильником в атмосфере N2 в течение 16 ч, затем охлаждали до кт. Добавляли STAB (0,80 г, 3,75 ммоль), реакционную смесь снова нагревали до температуры кипения с обратным холодильником в течение 16 ч, затем охлаждали до кт. Реакционную смесь гасили добавлением насыщ. р-ра NaHCO3 (10 мл), разбавляли ДХМ (10 мл), затем фильтровали через подушку целита. Слои разделяли и водный слой экстрагировали ДХМ (4×20 мл). Органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили (MgSO4). Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 , 27 мл в мин, градиент от 1% до 10% МеОН в ДХМ]) с получением неразделимой смеси диастереомеров. Эту смесь очищали с помощью препаративной обращенно-фазовой ВЭЖХ (колонка Gemini-NX 5 мкм C18 110A Axia от Phenomenex, 100×30 мм, элюирование 15-35% градиентом MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель В представляет собой 0,2% (28% NH3/H2O) в Н2О] и сбор фракций посредством мониторинга при 205 нм) с получением проп-2-ин-1-ил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата, Изомера 1 {0,02 г, 7%) Примера 5-2 в виде бесцветного твердого вещества, и проп-2-ин-1-ил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата, Изомера 2 (0,03 г, 11%) Примера 5-2 в виде бесцветного твердого вещества.
Данные для обоих изомеров приведены в Таблице 3.
Путь е
Типовой способ получения пиперидинов через восстановительное аминирование триацетоксиборогидрида натрия, снятие защитной группы Вое и образование этилкарбамата на примере получения Примера 5-1, этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата
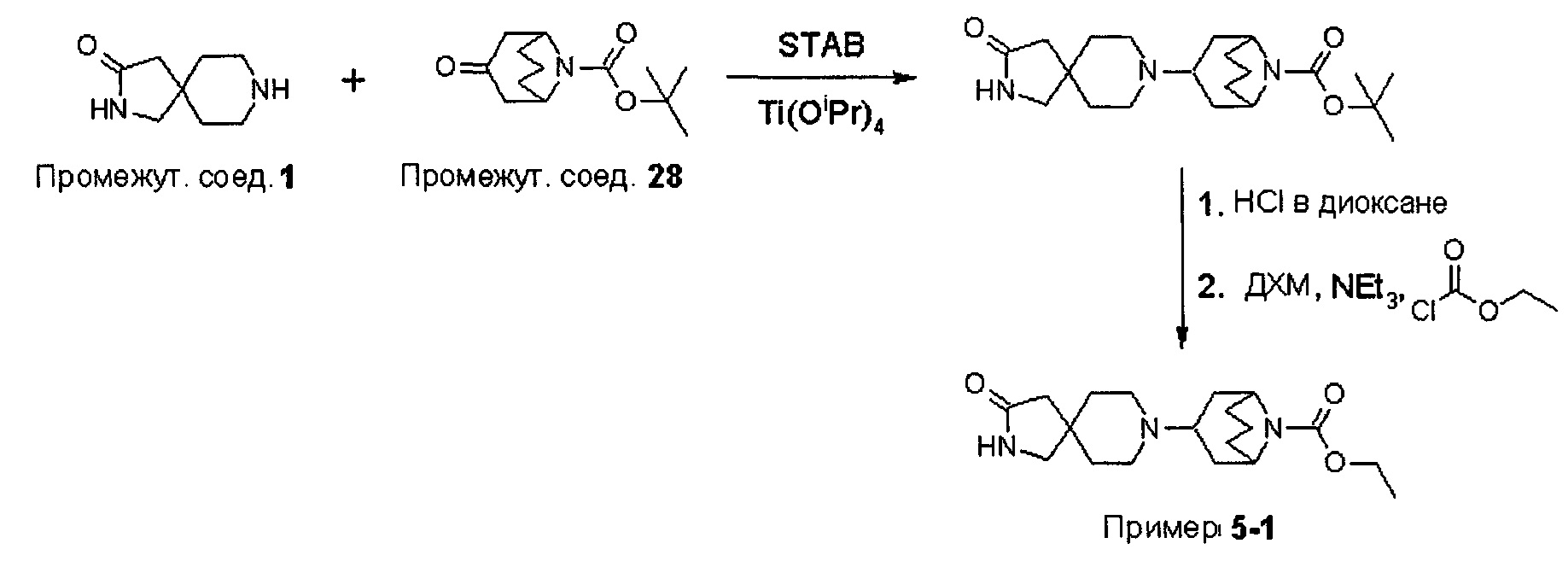
2,8-Диазаспиро[4,5]декан-3-он (0,15 г, 1,0 ммоль) и трет-бутил-3-оксо-9-азабицикло[3,3,1]нонан-9-карбоксилат (0,25 г, 0.1.05 ммоль) растворяли в ДХЭ (10,0 мл) при кт и добавляли изопропоксид титана (0,89 мл, 3,0 ммоль). Реакционную смесь перемешивали в течение ночи с обратным холодильником в атмосфере N2, затем охлаждали до комнатной температуры. Добавляли STAB (1,06 г, 5,0 ммоль), реакционную смесь снова нагревали до температуры кипения с обратным холодильником, поддерживали температуру в течение ночи и затем охлаждали до комнатной температуры. Реакционную смесь гасили добавлением насыщ. р-ра NaHCO3 (10 мл), разбавляли ДХМ (10 мл), затем фильтровали через подушку целита. Слои разделяли и водный слой экстрагировали ДХМ (4×20 мл). Органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили (MgSO4). Растворители удаляли в вакууме с получением неочищенного трет-бутил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата, который использовали без какой-либо очистки.
ЖХМС (Способ С): m/z 378 (М+Н)+ (ES+), при 1,54 мин, УФ-активно
трет-Бутил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилат (0,38 г, 1,0 ммоль условно) растворяли в ДХМ (10 мл), добавляли 4,0 М HCl в 1,4-диоксане (1,25 мл, 5,0 ммоль) и реакционную смесь перемешивали при кт в течение 18 ч. Летучие вещества удаляли в вакууме, остаток растворяли в ДХМ (10 мл), по каплям добавляли Et3N (0,70 мл, 5,0 ммоль) и этилхлорформиат (143 мкл, 1,50 ммоль) и раствор перемешивали при кт в течение 18 ч. Затем смесь выливали в насыщ. р-р NaHCO3 (20 мл), экстрагировали ДХМ (4×20 мл), органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили (MgSO4). Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 , 27 мл в минуту, градиент от 1% до 10% МеОН в ДХМ]) с получением неразделимой смеси диастереомеров. Эту смесь очищали с помощью препаративной обращенно-фазовой ВЭЖХ (колонка Gemini-NX 5 мкм C18 110А Axia от Phenomenex, 100×30 мм, элюирование 20-30% градиентом MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где Растворитель В представляет собой 0,2% (28% NH3/H2O) в Н2О] и сбор фракций посредством мониторинга при 205 нм) с получением этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата, Изомера 1 (0,02 г, 6%) Примера 5-1 в виде бесцветного твердого вещества, и этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-9-азабицикло[3,3,1]нонан-9-карбоксилата, Изомера 2 (0,01 г, 3%) Примера 5-1 в виде бесцветного твердого вещества.
Данные для обоих изомеров приведены в Таблице 3.
Путь f
Типовой способ получения пиперидинов через восстановительное аминирование NaCNBH3, снятие защитной группы Вое и образование этилкарбамата на примере получения Примера 7-1, этил-6-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,2,1]октан-3-карбоксилата
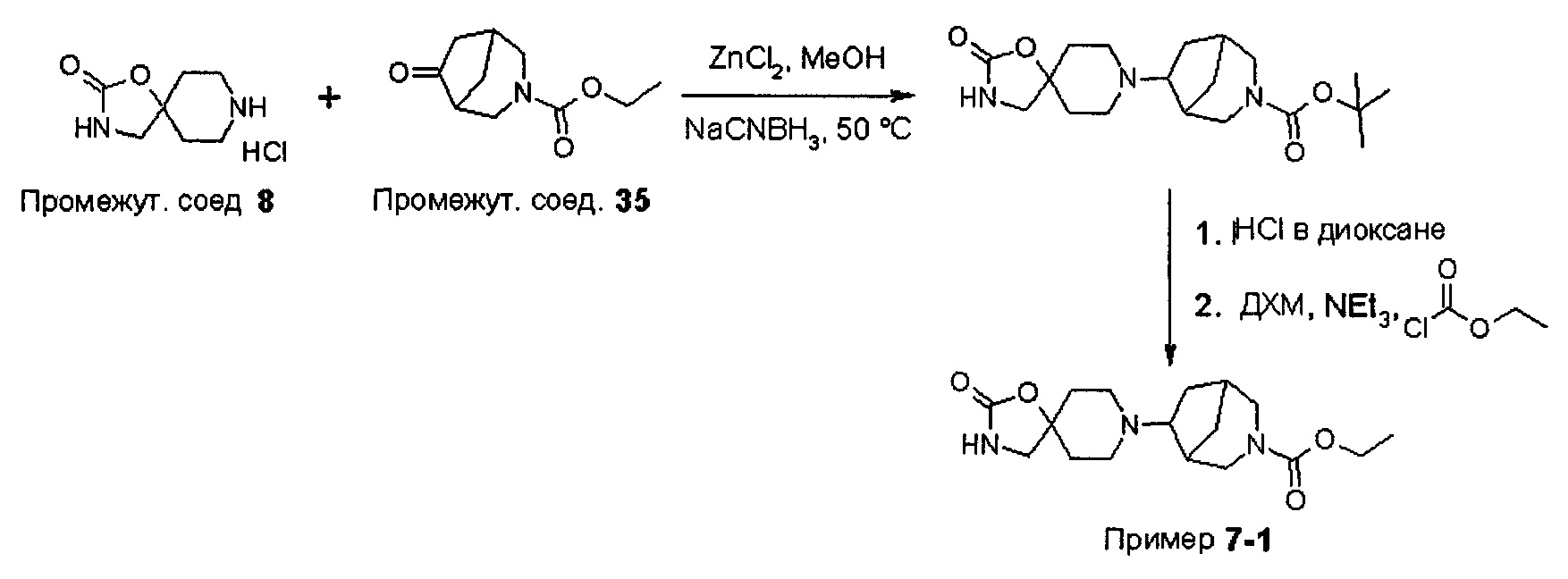
Пример 7-1
1-Окса-3,8-диазаспиро[4,5]декан-2-она гидрохлорид (0,15 г, 0,76 ммоль) растворяли в МеОН (3 мл) и обрабатывали K2CO3 (0,11 г, 0,76 ммоль) в минимальном количестве воды, чтобы обессолить. Растворители удаляли в вакууме, а остаток растворяли в МеОН (8 мл) и добавляли этил-6-оксо-3-азабицикло[3.2.1]октан-3-карбоксилат (0,15 г, 0,76 ммоль) и ZnCl2 (0,31 г, 2,28 ммоль). Реакционную смесь перемешивали при 50°С в атмосфере N2 в течение 2 ч, затем охлаждали до кт и добавляли NaCNBH3 (0,10 г, 1,52 ммоль). Реакционную Смесь перемешивали при 50°С в атмосфере N2 в течение 16 ч, затем охлаждали до кт и гасили насыщ. р-ром NaHCO3 (10 мл). Растворители удаляли в вакууме и водный слой промывали ДХМ (2×10 мл), органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили пропусканием через картридж для разделения фаз от Biotage. Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 10 г от Biotage, 40-63 мкм, 60 , 12 мл в минуту, градиент от 0% до 10% МеОН в ДХМ]) с получением трет-бутил-6-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,2,1]октан-3-карбоксилата (6,0 мг, 2,2%) в виде бесцветной смолы.
ЖХМС (Способ D): m/z 366 (М+Н)+ (ES+), при 1,76 мин, УФ-неактивно
трет-Бутил-6-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,2,1]октан-3-карбоксилат (6,0 мг, 0,016 ммоль) разводили в 4,0 М HCl в 1,4-диоксане (3 мл) и перемешивали при кт в атмосфере N2 в течение 16 ч. Растворители удаляли в вакууме, а остаток растворяли в ДХМ (4 мл) и охлаждали до 0°С в атмосфере N2, добавляли Et3N (5 мг, 0,048 ммоль) и этилхлорформиат (4 мг, 0,032 ммоль) и реакционную смесь перемешивали при кт в атмосфере N2 в течение 16 ч. Реакционную смесь разбавляли ДХМ (10 мл) и промывали насыщ. р-ром NaHCO3 (20 мл), водный слой экстрагировали ДХМ (2×15 мл), органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили пропусканием через картридж для разделения фаз от Biotage. Растворители удаляли в вакууме, а остаток очищали с помощью препаративной обращенно-фазовой ВЭЖХ (колонка Gemini-NX 5 мкм C18 110А Axia от Phenomenex, 100×30 мм, элюирование 20-50% градиентом MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель В представляет собой 0,2% (28% NH3/H2O) в Н2О] и сбор фракций посредством мониторинга при 205 нм) с получением этил-6-(2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,2,1]октан-3-карбоксилата, Изомера 1 (0,84 мг, 15%) Примера 7-1 в виде бесцветной смолы.
Данные для этого соединения приведены в Таблице 3.
Путь g
Типовой способ получения пиперидинов через восстановительное аминирование NaCNBH3 на примере получения Примера 2-23, этил-3-(2-оксо-4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
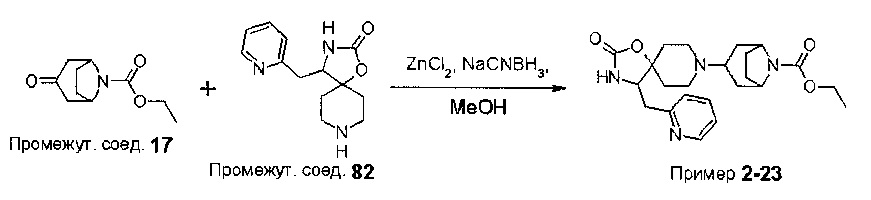
К раствору 4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-2-она (0,10 г, 0,41 ммоль) и этил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилата (0,80 г, 0,41 ммоль) в МеОН (5 мл) добавляли ZnCl2 (0,17 г, 1,21 ммоль). Реакционную смесь перемешивали при 60°С, в атмосфере N2, в течение 6 ч, затем охлаждали до кт и добавляли NaCNBH3 (0,08 г, 1,21 ммоль). Реакционную смесь перемешивали при 60°С в атмосфере азота в течение 16 ч. Реакционную смесь охлаждали до кт, растворитель удаляли в вакууме, а остаток распределяли между насыщ. р-ром NaHCO3 (10 мл) и ДХМ (10 мл), водный слой дополнительно промывали ДХМ (2×10 мл). Органические слои объединяли и промывали насыщенным раствором хлорида натрия, сушили (Na2SO4) и удаляли растворители в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, силикагель (100-200 меш), градиент от 2% до 5% МеОН в ДХМ] с получением этил-3-(2-оксо-4-(пиридин-2-илметил)-1-окса-3,8-диазаспиро[4,5]декан-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата в виде смеси диастереомеров, Примера 2-23 (32 мг, 20%) в виде белого твердого вещества.
Данные для Примера 2-23 приведены в Таблице 3.
Путь h
Способ получения Примера 2-24, этил(3-эндо)-3-(2-гидрокси-3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
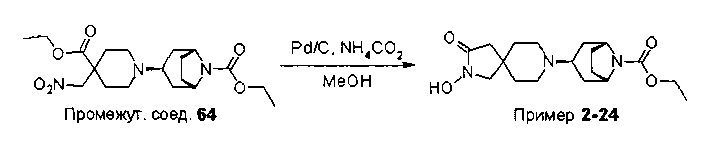
Раствор этил(3-эндо)-3-[4-(2-этокси-2-оксоэтил)-4-(нитрометил)пиперидин-1-ил]-8-азабицикло[3,2,1]октан-8-карбоксилата (2,00 г, 4,87 ммоль) в безводном МеОН (30 мл) разделяли поровну между 4×20 мл микроволновыми виалами. Растворы дегазировали азотом, а затем добавляли 10% палладий на углероде (0,13 мг, 1,21 ммоль) и формиат аммония (1,54 г, 24,20 ммоль). Виалы герметично закрывали и перемешивали при кт в течение 4 ч. Четыре реакционные смеси объединяли и фильтровали через подушку целита в атмосфере азота, а растворители удаляли в вакууме. Остаток очищали помощью препаративной обращенно-фазовой ВЭЖХ [Gemini-NX C18, 5 мкм, 100×30 мм, 30 мл в мин, 5-35% MeCN/Вода + 0,2% аммиака (28% раствор аммиака) с получением Примера 2-24, 8-[8-(этоксикарбонил)-8-азабицикло[3,2,1]окт-3-ил]-3-оксо-2,8-диазаспиро[4,5]декан-2-олата (0,90 г, 52,5%) в виде твердого вещества белого цвета.
Данные для указанного в заголовке соединения приведены в Таблице 3.
Путь i
Типовой способ получения пиперидинов через N-алкилирование на примере получения Примера 2-25, этил-3-(3-оксо-2-пропил-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло [3,2,1] октан-8-карбоксилата
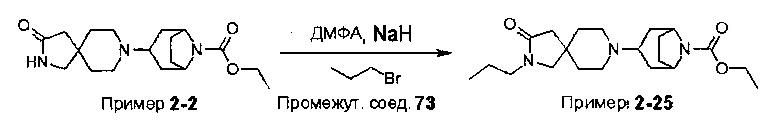
Этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилат (0,20 г, 0,59 ммоль) растворяли в ДМФА (5,0 мл). NaH (60%) (0,071 г, 1,78 ммоль) добавляли при 0°С и реакционную смесь перемешивали при 0°С в течение 30 мин. Добавляли 1-бромпропан (0,11 г, 0,89 ммоль) и реакционную смесь перемешивали при кт в течение 1 ч. Реакционную смесь гасили добавлением воды (20 мл) и экстрагировали EtOAc (150 мл), водный слой дополнительно экстрагировали EtOAc (2×15 мл); органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили (Na2SO4). Растворители удаляли в вакууме, а остаток очищали с помощью препаративной обращенно-фазовой ВЭЖХ [X-Bridge, 150×19 мм, 5 мкм, 12 мл в мин, изократическое элюирование 29% (в течение 20 мин), а затем 100% (4 мин) MeCN в 50% MeCN/вода (0,1% аммиака)] с получением этил-3-(3-оксо-2-пропил-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Примера 2-25 (14,5 мг, 6,5%) в виде бесцветного твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 3.
Путь j
Типовой способ получения пиперидинов через образование карбамата на примере получения Примера 2-30, (1,1-2Н2)этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
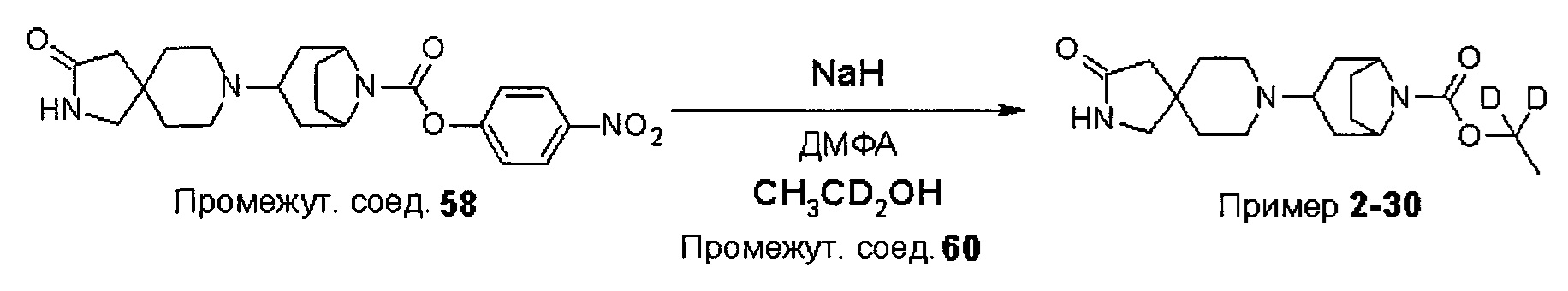
4-Нитрофенил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилат (100 мг, 0,24 ммоль) растворяли в безводном ДМФА (3 мл) и добавляли 60% суспензию гидрида натрия в минеральном масле (47 мг, 1,18 ммоль). Реакционную смесь перемешивали при кт в атмосфере азота в течение 10 мин. Добавляли этанол-1,1-d2 (57 мг, 1,18 ммоль) и реакционную смесь перемешивали в течение ночи при кт в атмосфере азота. К реакционной смеси добавляли воду (1 мл) и удаляли растворители в вакууме. Остаток распределяли между ДХМ (10 мл) и насыщ. водным NaHCO3 (10 мл), водный слой экстрагировали ДХМ (2×10 мл). Объединенные органические слои сушили пропусканием через картридж для разделения фаз от Biotage и удаляли растворители в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 10 г от Biotage, 40-63 мкм, 60 , 12 мл в мин, градиент от 0% до 10% МеОН в ДХМ]) с получением (1,1-2Н2)этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 1 (11 мг, 14%) Примера 2-30 в виде бледно-желтой смолы, и (1,1-2Н2)этил-3-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-8-азабицикло[3,2,1]октан 8-карбоксилата, Изомера 2 (18 мг, 23%) Примера 2-30 в виде бледно-желтой смолы.
Данные для обоих изомеров приведены в Таблице 3.
Путь k
Типовой способ получения пиперидинов через восстановительное аминирование триацетоксиборогидрида натрия в ДМФА на примере получения Примера 3-1, этил-5-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата
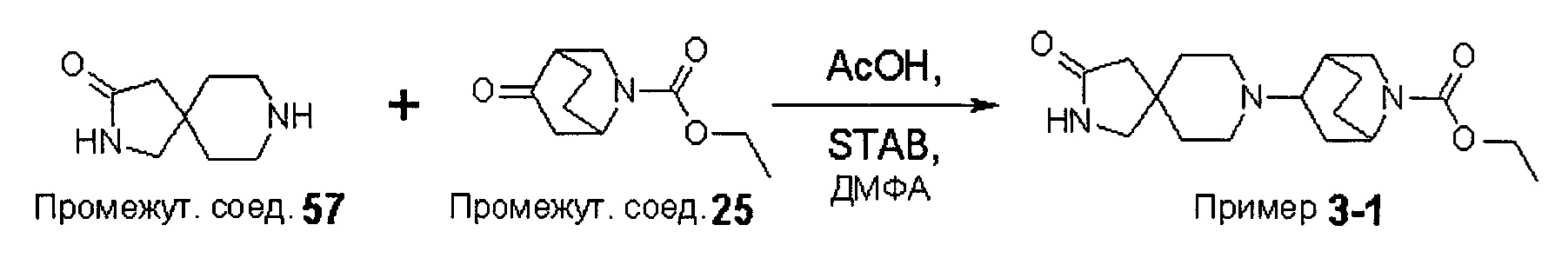
К раствору этил-5-оксо-2-азабицикло[2,2,2]октан-2-карбоксилата (6,70 г, 34 ммоль) и 2,8-диазаспиро[4,5]декан-3-она (5,24 г, 34 ммоль) в ДМФА (30 мл) добавляли НОАс (2,9 мл, 51 ммоль) в атмосфере азота, реакционную смесь перемешивали при кт в течение 20 мин. Добавляли Na(OAc)3BH (21,60 г, 102 ммоль) и реакционную смесь перемешивали при 45°С в течение 3 д. Затем реакционную смесь нагревали до 60°С и перемешивали в течение еще 24 ч. Растворитель удаляли в вакууме, а остаток растворяли в воде (20 мл) и подщелачивали насыщ. р-ром NaHCO3. Водный слой выпаривали досуха и полученное белое твердое вещество разбавляли ДХМ (100 мл). Суспензию перемешивали при кт в течение 30 мин, фильтровали и промывали ДХМ (4×25 мл) осадок на фильтре. Органические слои объединяли и удаляли растворитель в вакууме. Остаток очищали с помощью препаративной обращенно-фазовой ВЭЖХ (Прибор: Gilson, Колонка: XBridge 21,2 250 мм C18, 10 мкм; Подвижная фаза: А: вода (10 ммоль/л NH4HCO3) В: CAN); Скорость потока (мл/мин): 25,00) с получением двух рацемических изомеров этил-5-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, которые дополнительно очищали с помощью хиральной СФХ (Колонка: OJ-H, 4,6 250 мм; Сорастворитель: МеОН (0,1% NH4OH); температура колонки: 40; скорость потока CO2: 2,55) с получением этил-5-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 1 (0,78 г, 6,9%) Примера 3-1 в виде бесцветного твердого вещества, этил-5-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 2 (1,20 г, 10,5%) Примера 3-1 в виде бесцветного твердого вещества, этил-5-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 3 (0,45 г, 3,9%) Примера 3-1 в виде бесцветного твердого вещества, и этил-5-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 4 (1,30 г, 11,4%) Примера 3-1 в виде бесцветного твердого вещества.
Данные для всех четырех изомеров приведены в Таблице 3.
Путь 1
Типовой способ получения пиперидинов через восстановительное аминирование NaCNBH3 на примере получения Примера 3-3, этил-5-(4-этил-2-оксо-1-окса-3,8-диазаспиро [4,5] дец-8-ил)-2-азабицикло [2,2,2] октан-2-карбоксилата
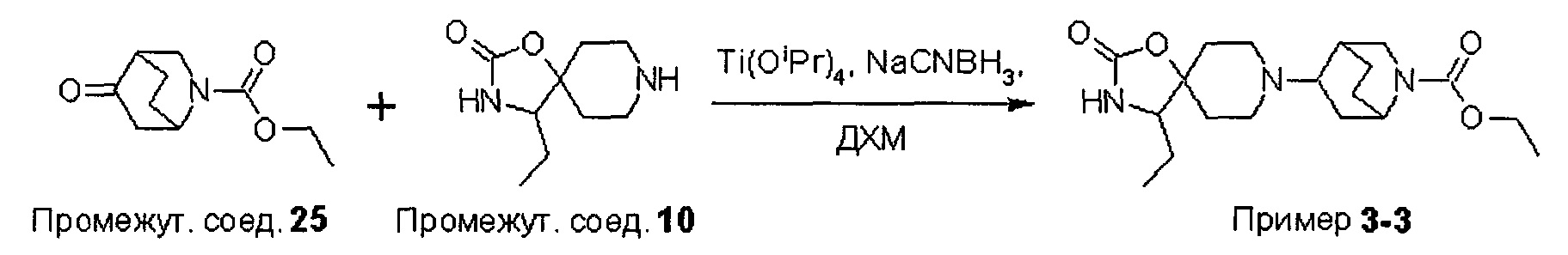
Этил-5-оксо-2-азабицикло[2,2,2]октан-2-карбоксилат (0,077 г, 0,39 ммоль) и 4-этил-1-окса-3,8-диазаспиро[4,5]декан-2-он (0,070 г, 0,39 ммоль) растворяли в ДХМ (3,9 мл). Добавляли изопропоксид (0,35 мл, 1,17 ммоль) титана(IV) и реакционную смесь перемешивали при кт в атмосфере азота в течение 3 ч. Добавляли NaCNBH3 (0,049 г, 0,78 ммоль) и смесь перемешивали при кт в атмосфере азота в течение ночи. Реакционную смесь распределяли между водой (10 мл) и ДХМ (10 мл) и пропускали раствор через подушку целита для удаления твердых веществ. Слои фильтрата разделяли и водную фазу экстрагировали ДХМ (3×25 мл). Органические фазы объединяли и промывали насыщ. р-ром NaHCO3 (25 мл), и сушили пропусканием через картридж для разделения фаз от Biotage. Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 10 г от Biotage, 40-63 мкм, 60 , 10 мл в минуту, градиент от 0% до 10% МеОН в ДХМ]) с получением смеси диастереомеров этил-5-(4-этил-2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата. Эту смесь диастереомеров дополнительно очищали с помощью препаративной обращенно-фазовой ВЭЖХ (колонка Gemini-NX 5 мкм C18 110A Axia от Phenomenex, 100×30 мм, элюирование 20-50% градиентом MeCN/Растворитель B в течение 12,5 мин при 30 мл/мин [где Растворитель В представляет собой 0,2% (28% NH3/H2O) в H2O] и сбор фракций посредством мониторинга при 205 нм) с получением этил-5-(4-этил-2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 1 (0,021 г, 14,8%) Примера 3-3 в виде бесцветного твердого вещества, и этил-5-(4-этил-2-оксо-1-окса-3,8-диазаспиро[4,5]дец-8-ил)-2-азабицикло[2,2,2]октан-2-карбоксилата, Изомера 2 (0,022 г, 15,5%) Примера 3-3 в виде бесцветного твердого вещества.
Данные для Изомера 2 приведены в Таблице 2.
Путь m
Типовой способ получения пиперидинов через восстановительное аминирование NaBH4 на примере получения Примера 8-1, этил-6-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,1,1]гептан-3-карбоксилата
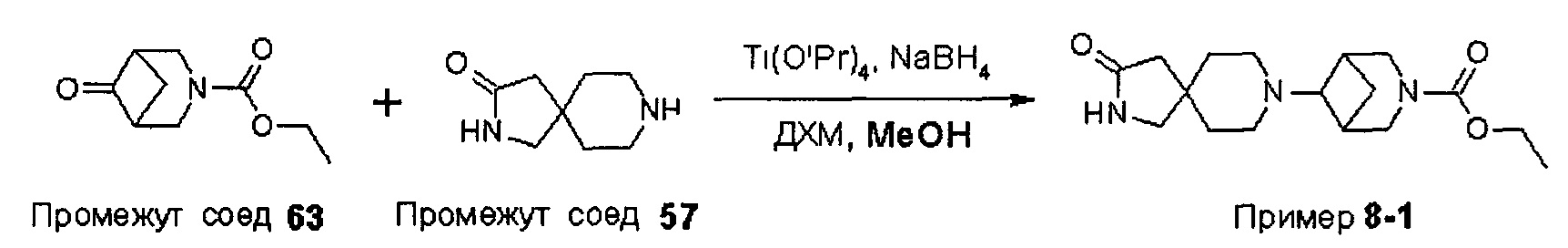
2,8-Диазаспиро[4,5]декан-3-он (0,11 г, 0,72 ммоль) и этил-6-окса-3-азабицикло[3,1,1]гептан-3-карбоксилат (0,12 г, 0,65 ммоль) растворяли в ДХМ (6,5 мл). Добавляли изопропоксид (0,35 мл, 1,17 ммоль) титана(IV) и реакционную смесь перемешивали при кт в атмосфере азота в течение ночи. Реакционную смесь охлаждали до -78°С и добавляли МеОН (15 мл), реакционную смесь перемешивали в течение 15 мин при -78°С. Добавляли NaBH4 (0,18 г, 5,20 ммоль) и реакционную смесь перемешивали при -78°С в течение 1 ч, затем при кт в атмосфере азота в течение ночи. Реакционную смесь обрабатывали NaOH (1 М, 10 мл) и перемешивали в течение 30 мин. Осадок собирали фильтрованием, промывали МеОН (3×20 мл) и удаляли растворители в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 25 г от Biotage, 40-63 мкм, 60 , 25 мл в мин, градиент от 0% до 10% МеОН в ДХМ]) с получением смеси диастереомеров этил-6-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,1,1]гептан-3-карбоксилата. Смесь диастереомеров очищали с помощью препаративной обращенно-фазовой ВЭЖХ (колонка Gemini-NX 5 мкм C18 110A Axia от Phenomenex, 100×30 мм, элюирование 15-50% градиентом MeCN/Растворитель B в течение 12,5 мин при 30 мл/мин [где Растворитель В представляет собой 0,2% (28% NH3/H2O) в H2O] и сбор фракций посредством мониторинга при 205 нм) с получением этил-6-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,1,1]гептан-3-карбоксилата, Изомера 1 (0,006 г, 2,9%) Примера 8-1 в виде бесцветного твердого вещества, и этил-6-(3-оксо-2,8-диазаспиро[4,5]дец-8-ил)-3-азабицикло[3,1,1]гептан-3-карбоксилата, Изомера 2 (0,040 г, 19,2%) Примера 8-1 в виде бесцветного твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь n
Типовая процедура получения азепанов через восстановительное аминирование NaCNBH3 на примере получения Примера 9-1, этил-3-(3-оксо-2,8-диазаспиро [4,6]ундец-8-ил)-8-азабицикл о [3,2,1] октан-8-карбоксилата
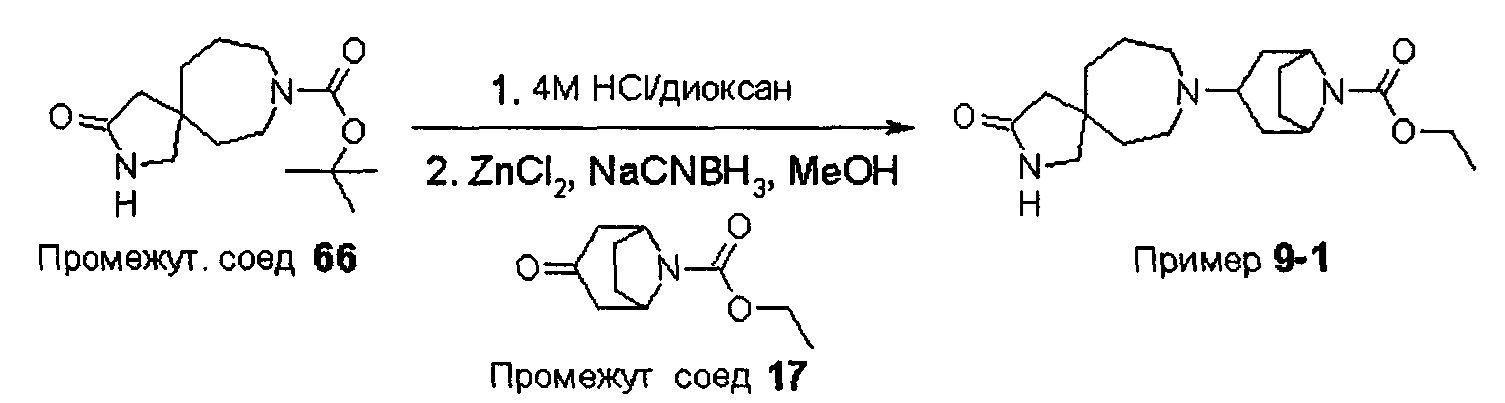
трет-Бутил-3-оксо-2,8-диазаспиро[4,6]ундекан-8-карбоксилат (0,398 г, 1,49 ммоль) добавляли к 4,0 М HCl в 1,4-диоксане (8 мл) и перемешивали при кт в атмосфере N2 в течение 16 ч. Растворители удаляли в вакууме с получением 2,8-диазаспиро[4,6]ундекан-3-она гидрохлорида (0,303 г, 100%), который использовали без дополнительной очистки. Часть 2,8-диазаспиро[4,6]ундекан-3-она гидрохлорида (0,179 г, 0,74 ммоль) растворяли в МеОН (5 мл) и добавляли карбонат калия (0,102 г, 0,74 ммоль), растворенный в минимальном количестве воды, для обессоливания амина. Растворители удаляли в вакууме, а остаток растворяли в МеОН (8 мл) и добавляли этил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилат (0,14 г, 0,74 ммоль) и ZnCl2 (0,30 г, 2,29 ммоль). Реакционную смесь перемешивали при 50°С в атмосфере N2 в течение 2 ч, затем охлаждали до кт и добавляли NaCNBH3 (0,09 г, 1,49 ммоль). Реакционную смесь перемешивали при 50°С в атмосфере N2 в течение 16 ч, затем охлаждали до кт и гасили насыщ. р-ром NaHCO3 (10 мл). Метанол удаляли в вакууме и полученный раствор промывали ДХМ (2×10 мл), органические слои объединяли и промывали насыщенным раствором хлорида натрия, затем сушили пропусканием через картридж для разделения фаз от Biotage. Растворители удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (нормальная фаза, [картридж SNAP KP-sil 10 г от Biotage, 40-63 мкм, 60 , 12 мл в минуту, градиент от 2% до 10% МеОН в ДХМ]) с получением этил-3-(3-оксо-2,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 1 (0,034 г, 13,0%) Примера 9-1 в виде желтой смолы, и этил-3-(3-оксо-2,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 2 (0,02 г, 0,9%) Примера 9-1 в виде желтой смолы.
Данные для этих соединений приведены в Таблице 3.
Путь о
Типовой способ получения азепанов через восстановительное аминирование NaCNBH3 на примере получения Примера 9-2, этил-3-(2-оксо-1-окса-3,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата
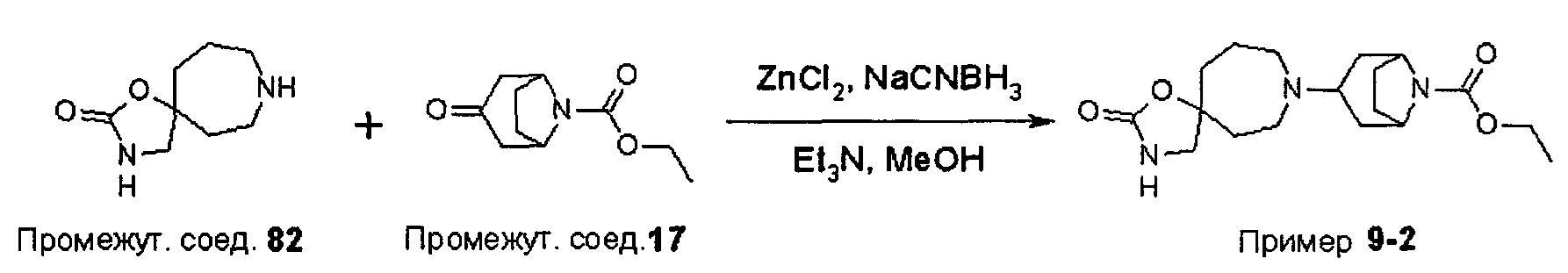
1-Окса-3,8-диазаспиро[4,6]ундекан-2-она гидрохлорид (5,3 г, 31,0 ммоль), этил-3-оксо-8-азабицикло[3,2,1]октан-8-карбоксилат (6,1 г, 31,0 ммоль), Et3N (13,0 мл, 93,0 ммоль), молекулярные сита 4 (2,0 г) и ZnCl2 (1,0 М в диэтиловом эфире, 1,5 мл, 1,5 ммоль) растворяли в МеОН (160 мл) и перемешивали с обратным холодильником в атмосфере N2 в течение 8 ч. Реакционную смесь охлаждали до 0°С и частями добавляли NaCNBH3 (5,8 г, 93,0 ммоль), реакционную смесь перемешивали с обратным холодильником в атмосфере N2 в течение 48 ч. Реакционную смесь фильтровали через целит и удаляли растворители в вакууме. Остаток распределяли между Н2О (500 мл) и EtOAc (150 мл), и водный слой дополнительно экстрагировали EtOAc (2×150 мл). Объединенные органические слои сушили над Na2SO4 и удаляли растворители в вакууме. Остаток очищали с помощью колоночной хроматографии (нормальная фаза, нейтральный силикагель, 100-200 меш, 0-8% МеОН в ДХМ) с получением двух изомеров этил-3-(2-оксо-1-окса-3,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, основного изомера (3,00 г, 27,6%) в виде бесцветной смолы и минорного изомера (0,40 г, 3,7%) в виде бесцветной смолы. Основной изомер (100 мг) дополнительно очищали с помощью хиральной препаративной ВЭЖХ [Chiral PAK IB 5 мкм, 250×20 мм, 13,0 мл/мин с применением метода изократического элюирования в 0,1% ДЭА в н-гексан : IPA : МеОН (19:1:1) в течение 50 мин] с получением этил-3-(2-оксо-1-окса-3,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 1 (45,0 мг, 45,0%) Примера 9-2 в виде бесцветной смолы, этил-3-(2-оксо-1-окса-3,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 2 (40,0 мг, 40,0%) Примера 9-2 в виде бесцветной смолы. Минорный изомер (150 мг) дополнительно очищали с помощью хиральной препаративной ВЭЖХ [СФХ Chiral PAK IC 5 мкм, 250×21 мм, 15,0 мл/мин с применением метода изократического элюирования в 0,1% ДЭА в н-гексан : IPA : ТНР : МеОН (14:1:1:4) в течение 33 мин] с получением этил-3-(2-оксо-1-окса-3,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 3 (50,0 мг, 33,3%) Примера 9-2 в виде бесцветной смолы, и этил-3-(2-оксо-1-окса-3,8-диазаспиро[4,6]ундец-8-ил)-8-азабицикло[3,2,1]октан-8-карбоксилата, Изомера 4 (52,4 мг, 34,9%) Примера 9-2 в виде бесцветной смолы.
Данные для указанного в заголовке соединения приведены в Таблице 3.
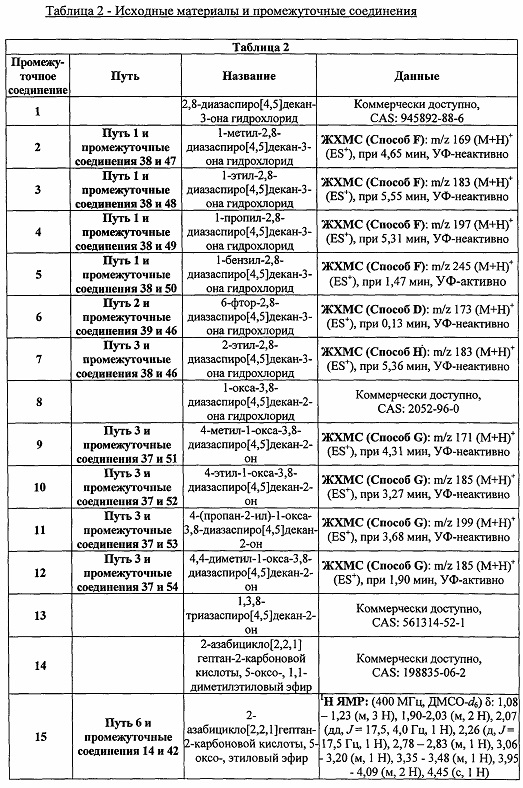
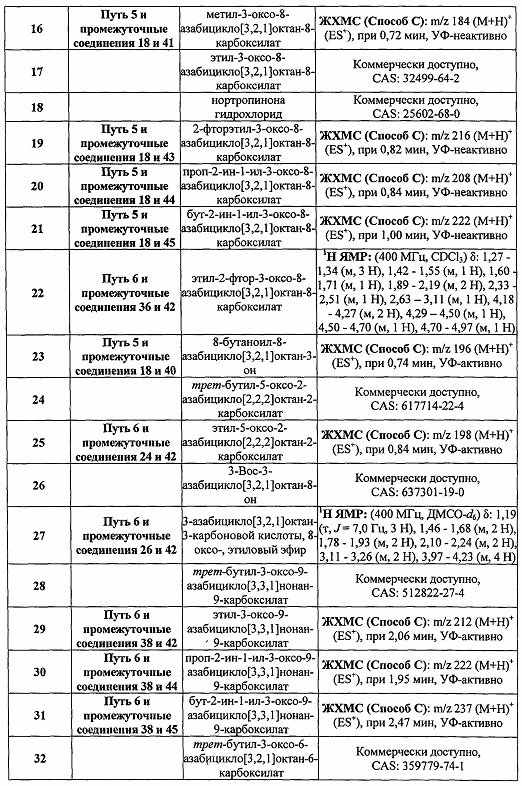
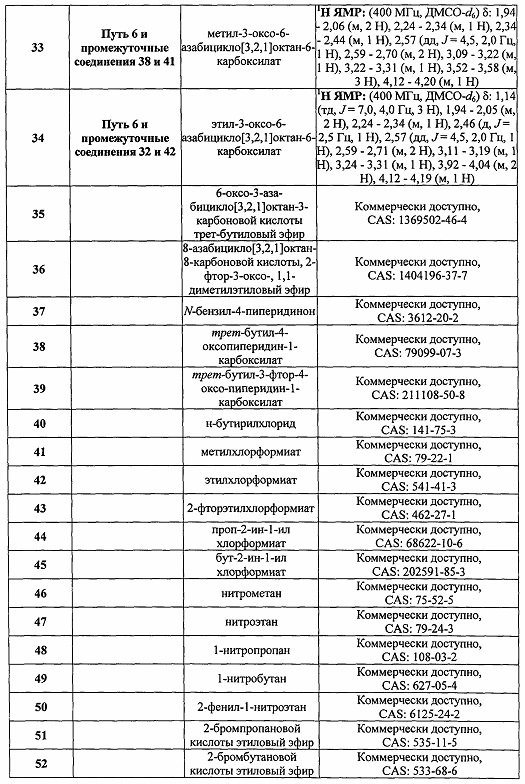
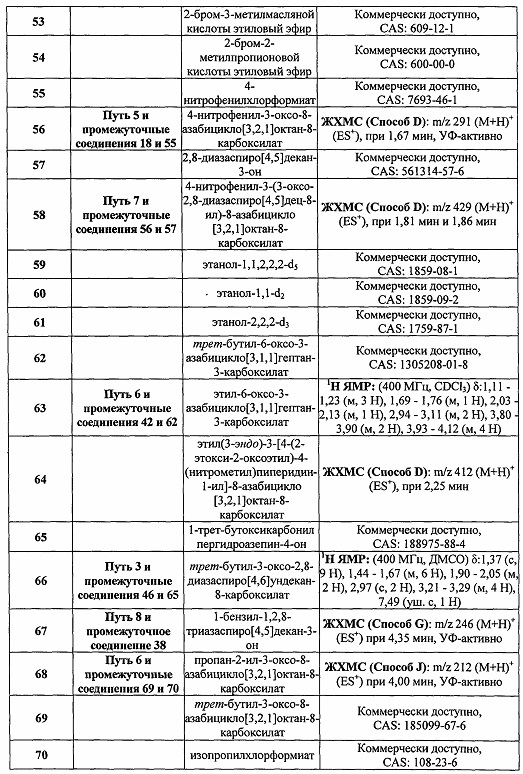
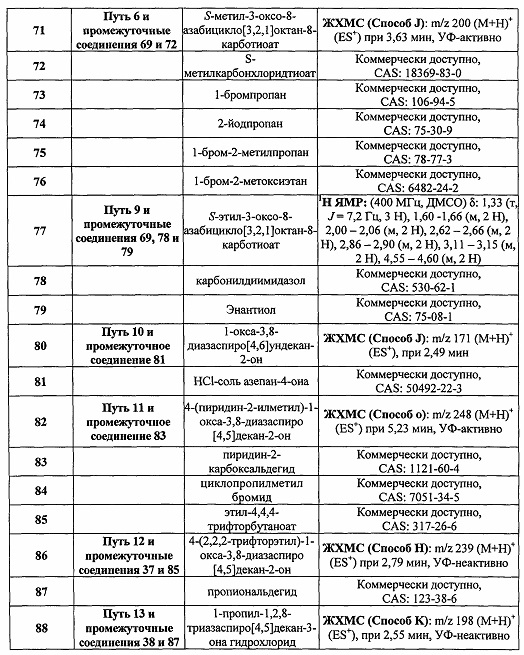
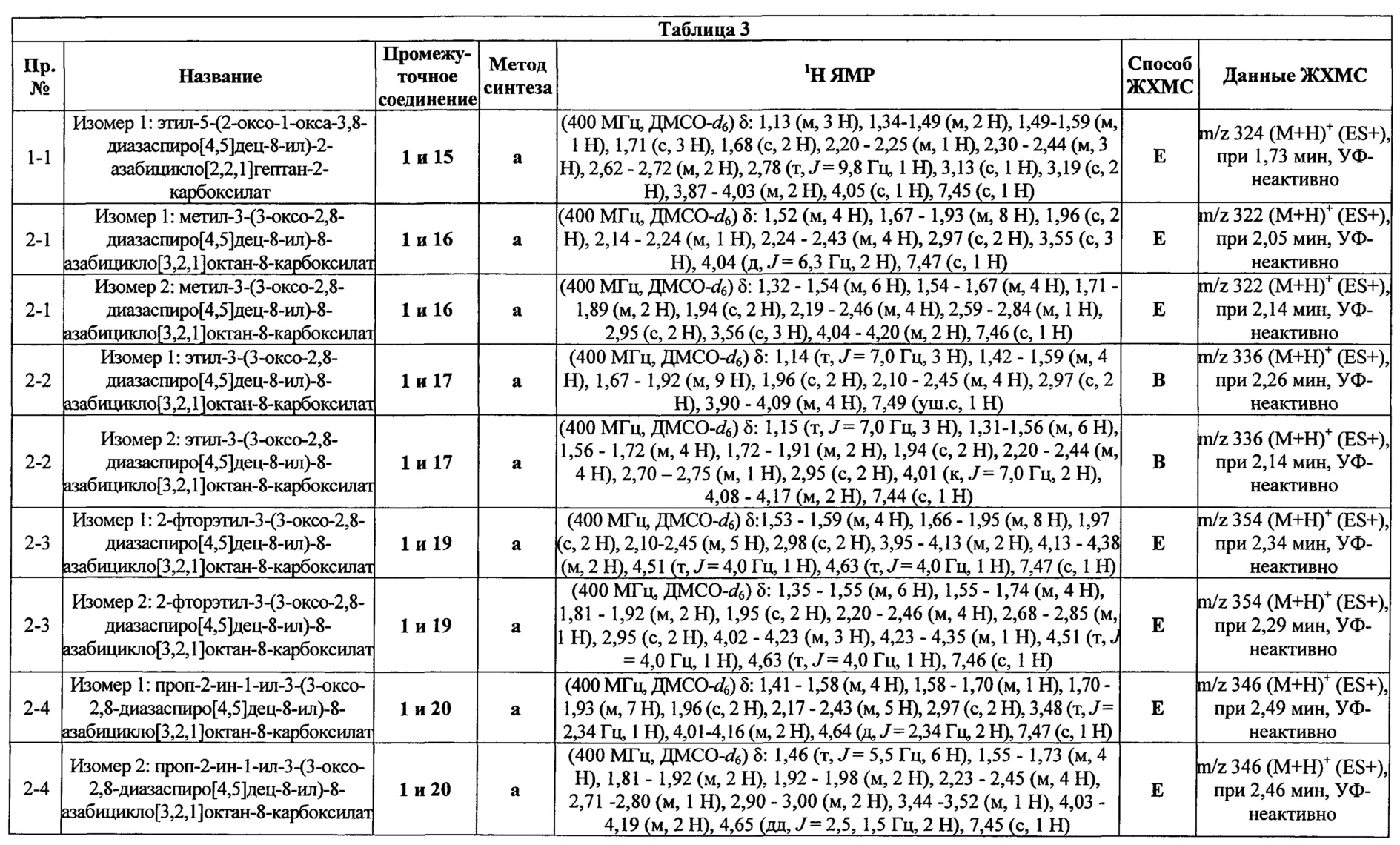
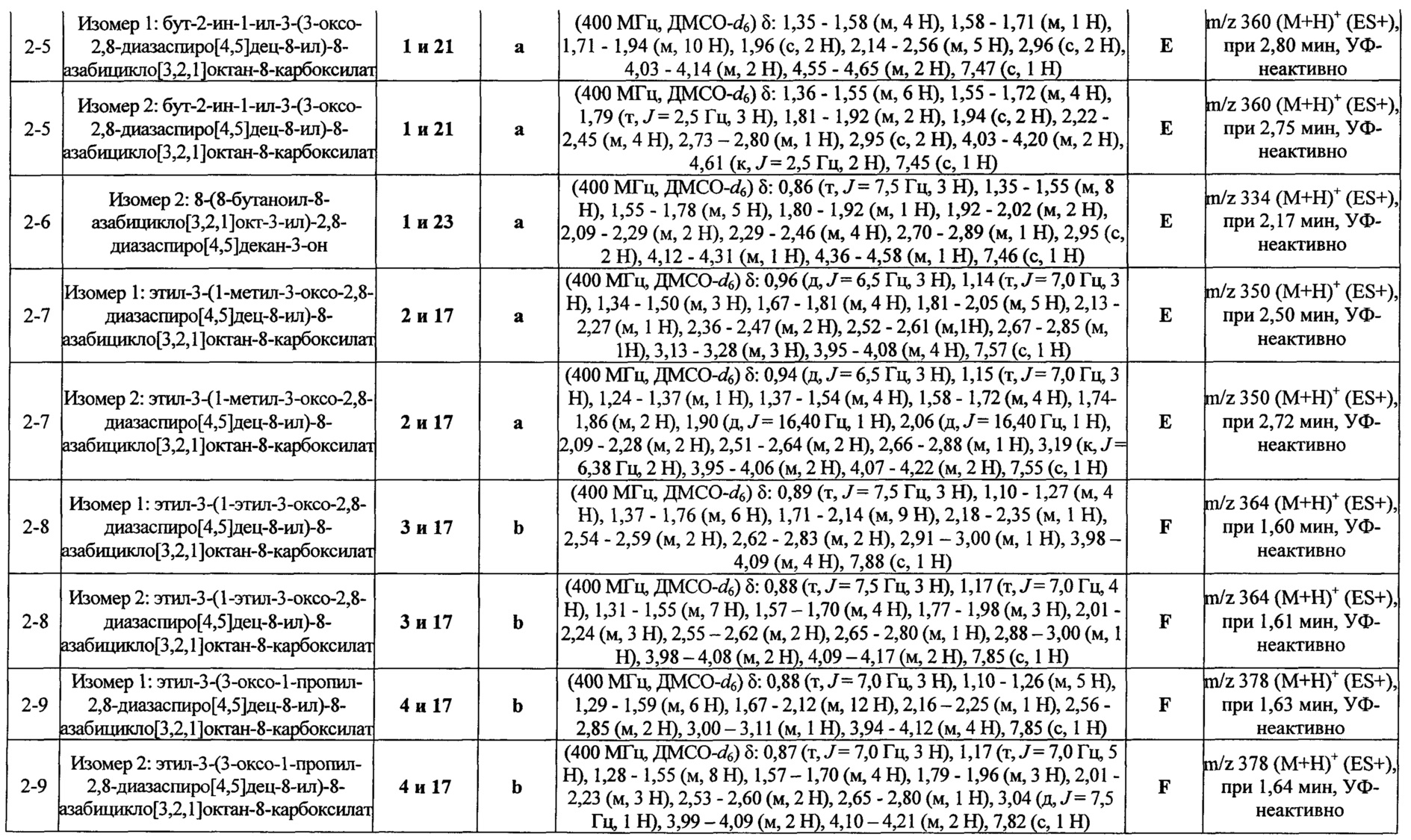
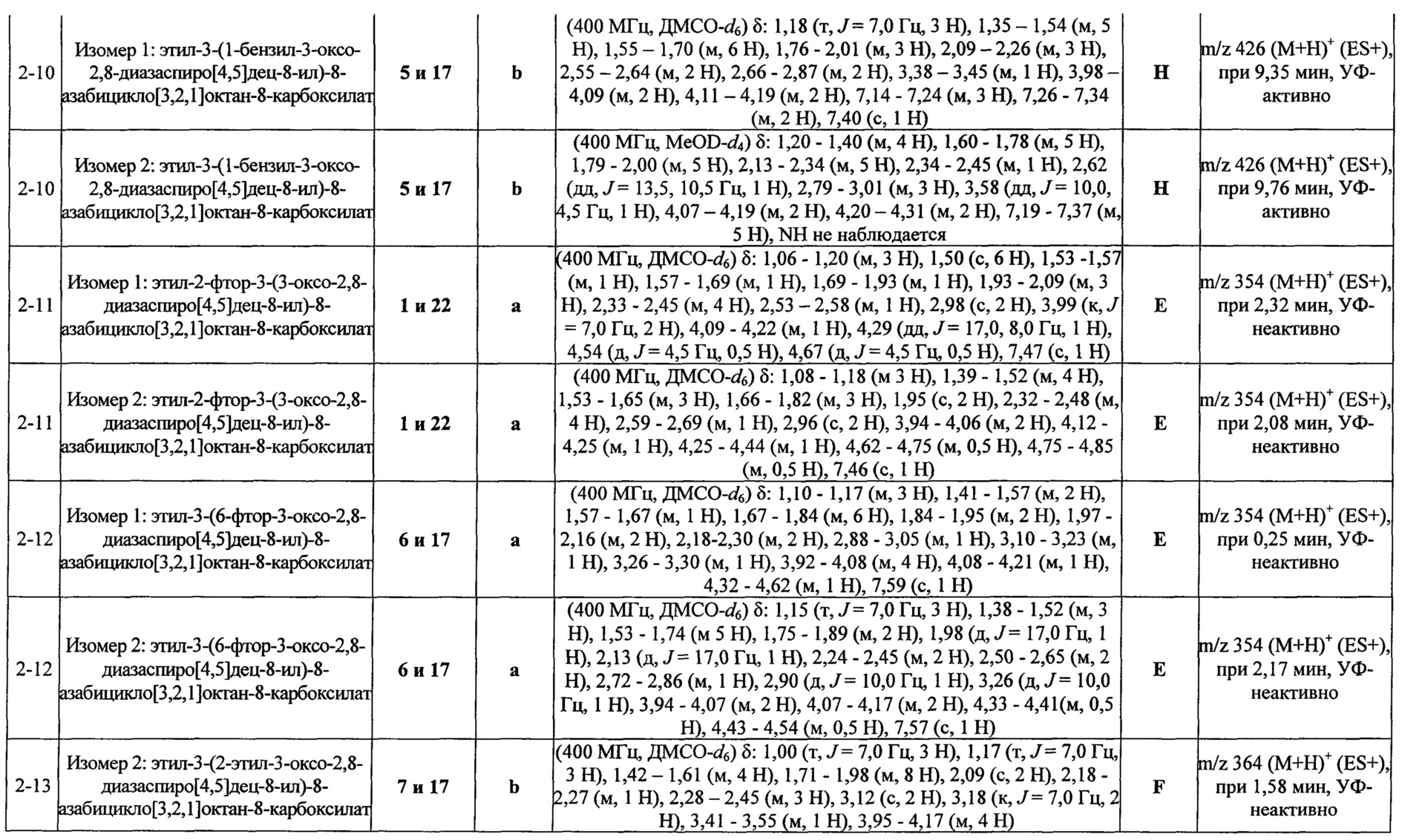
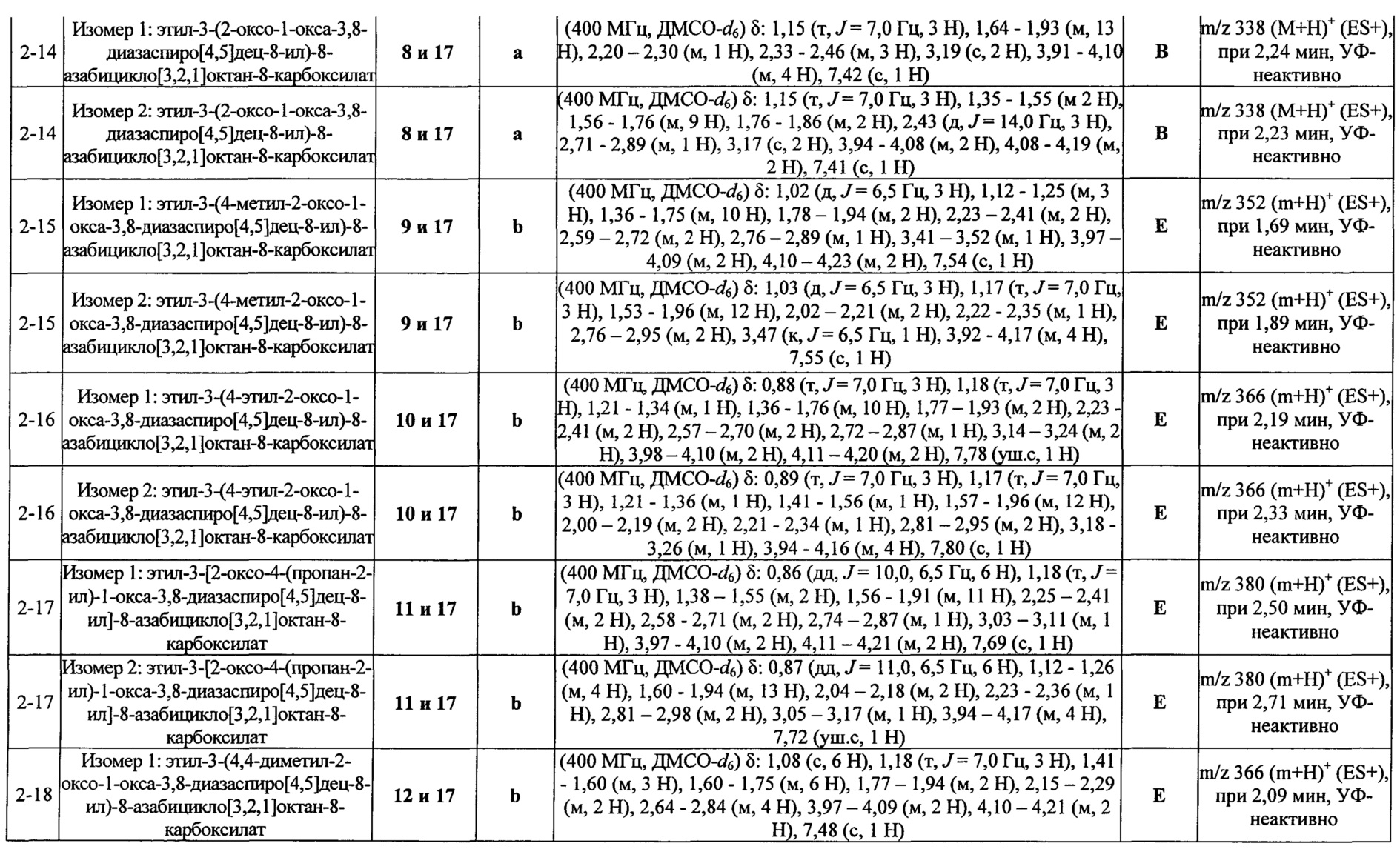
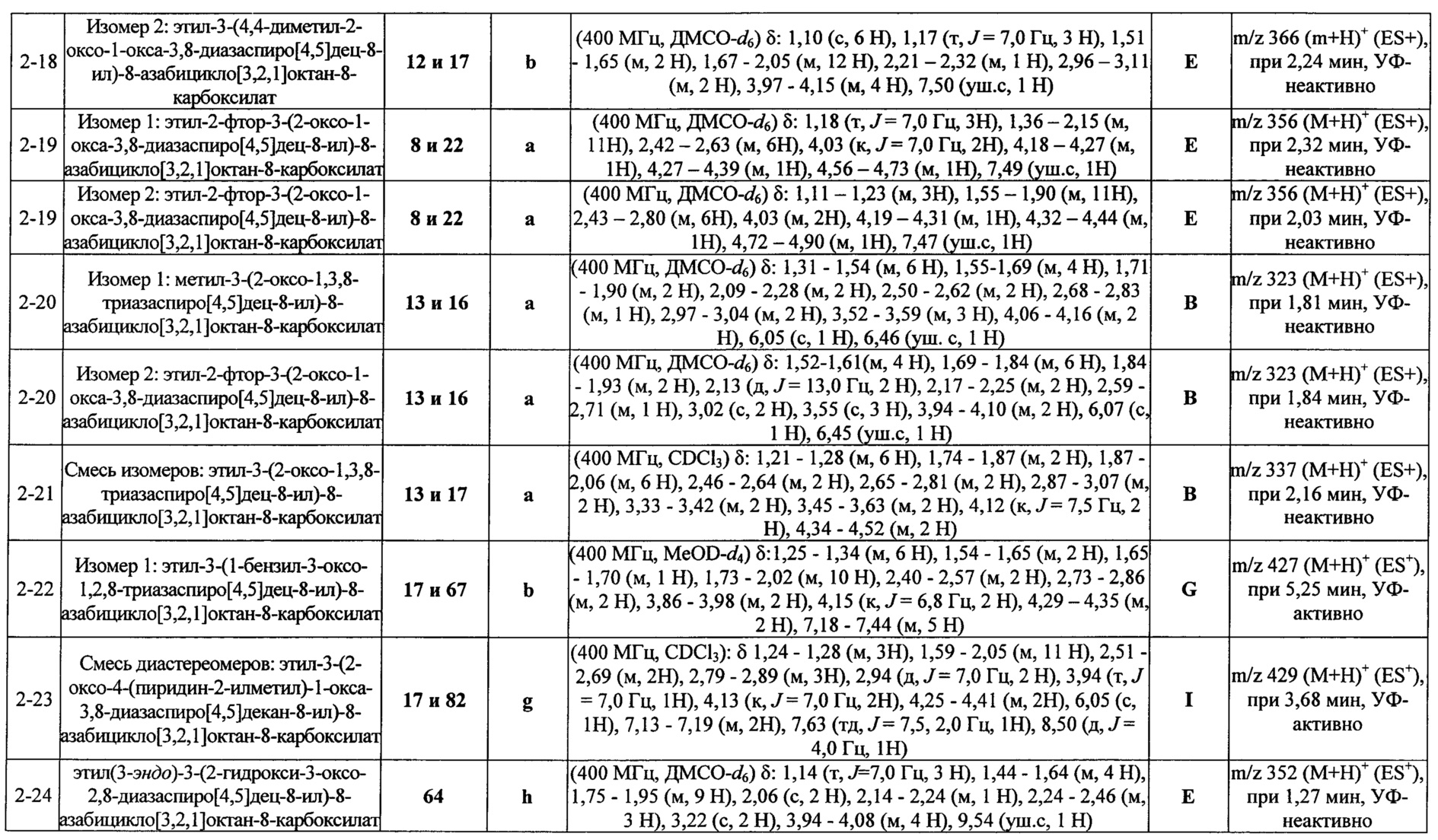
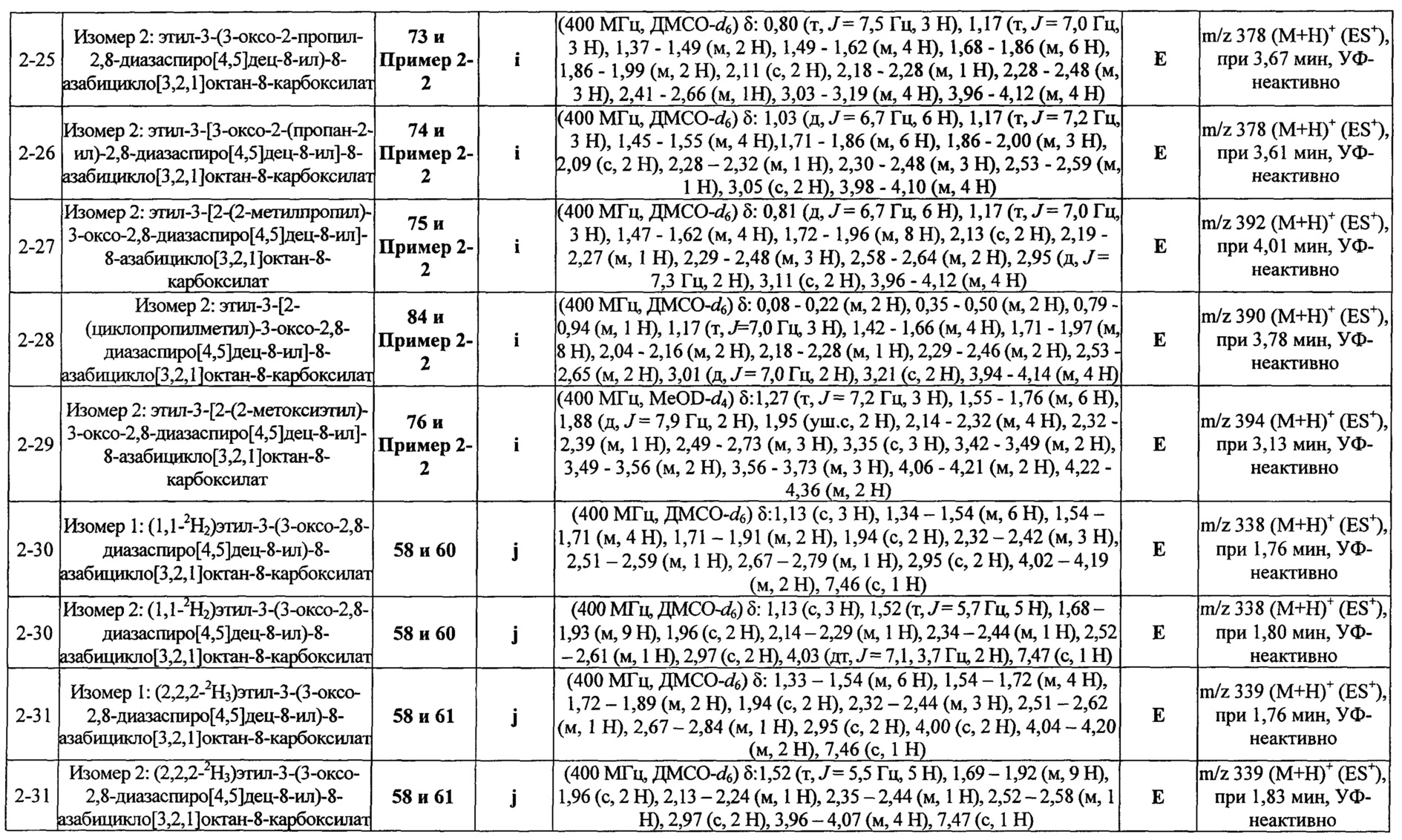
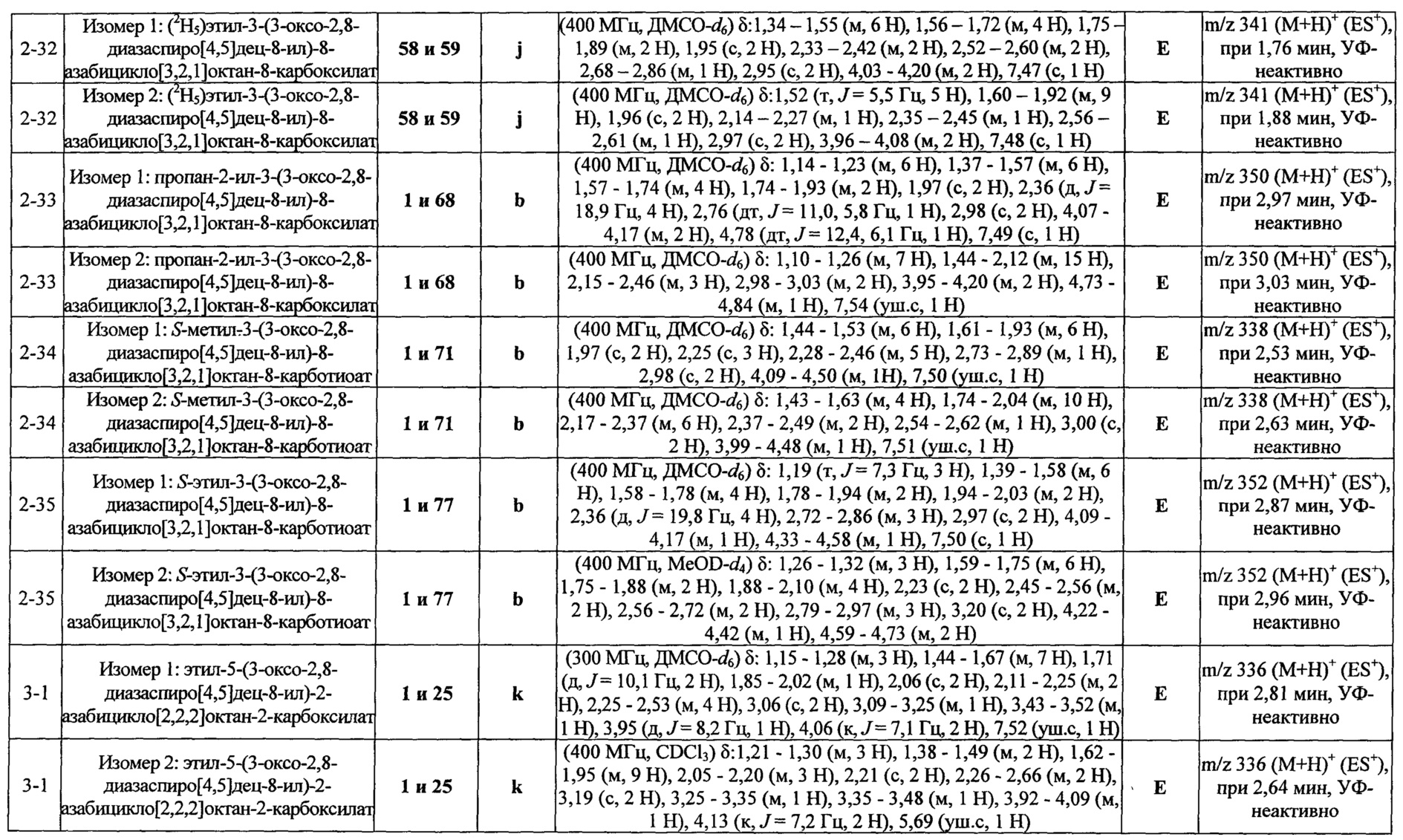
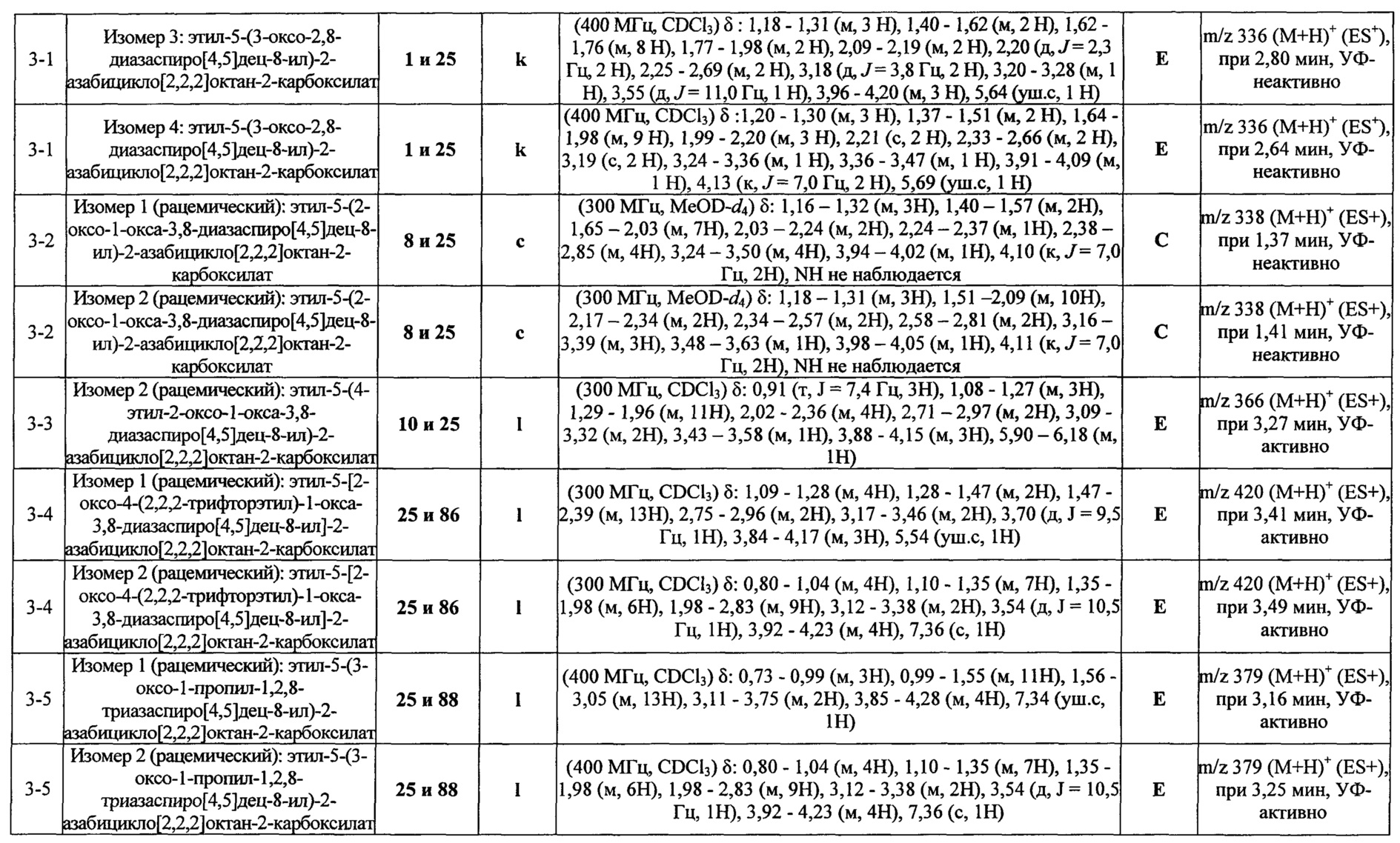
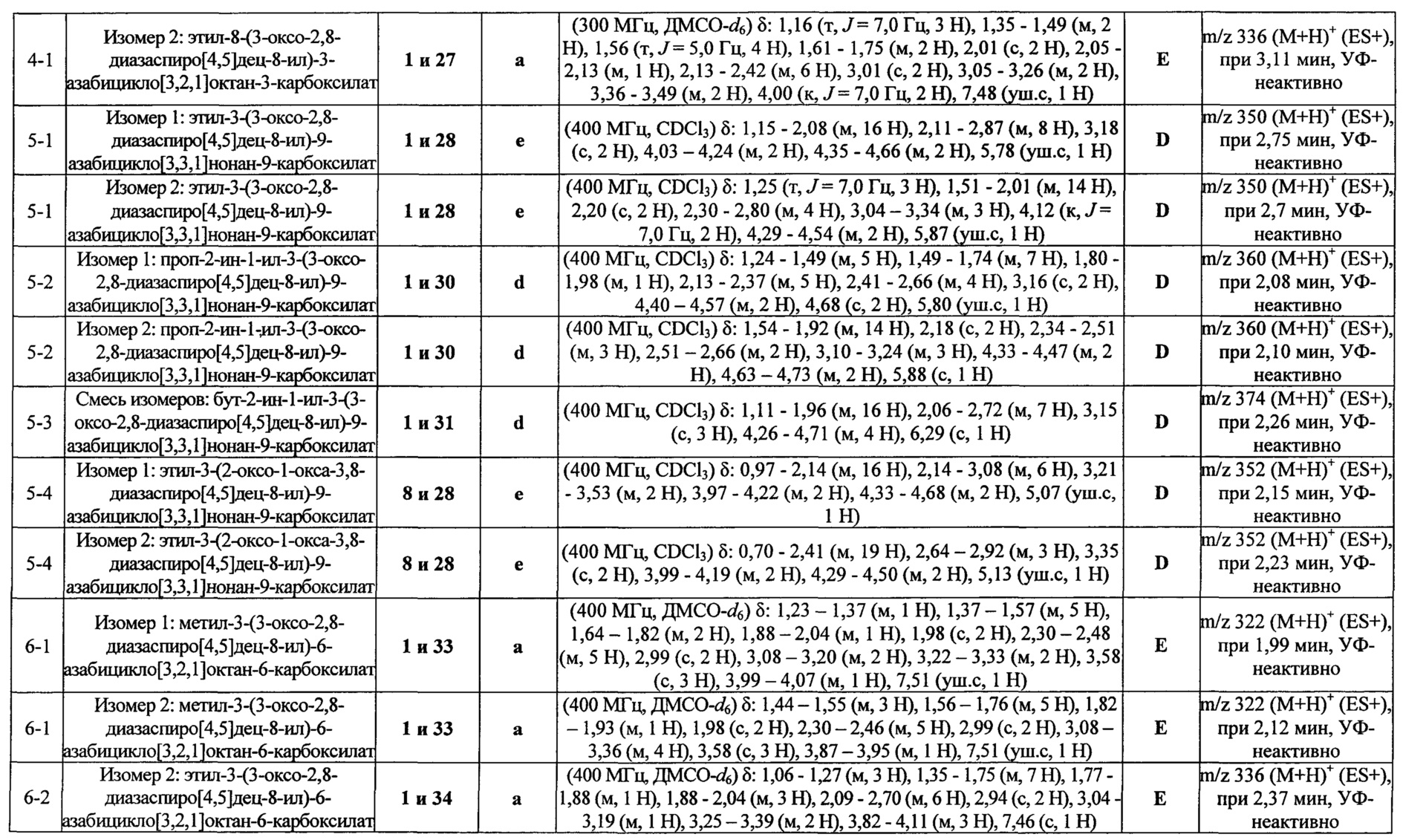
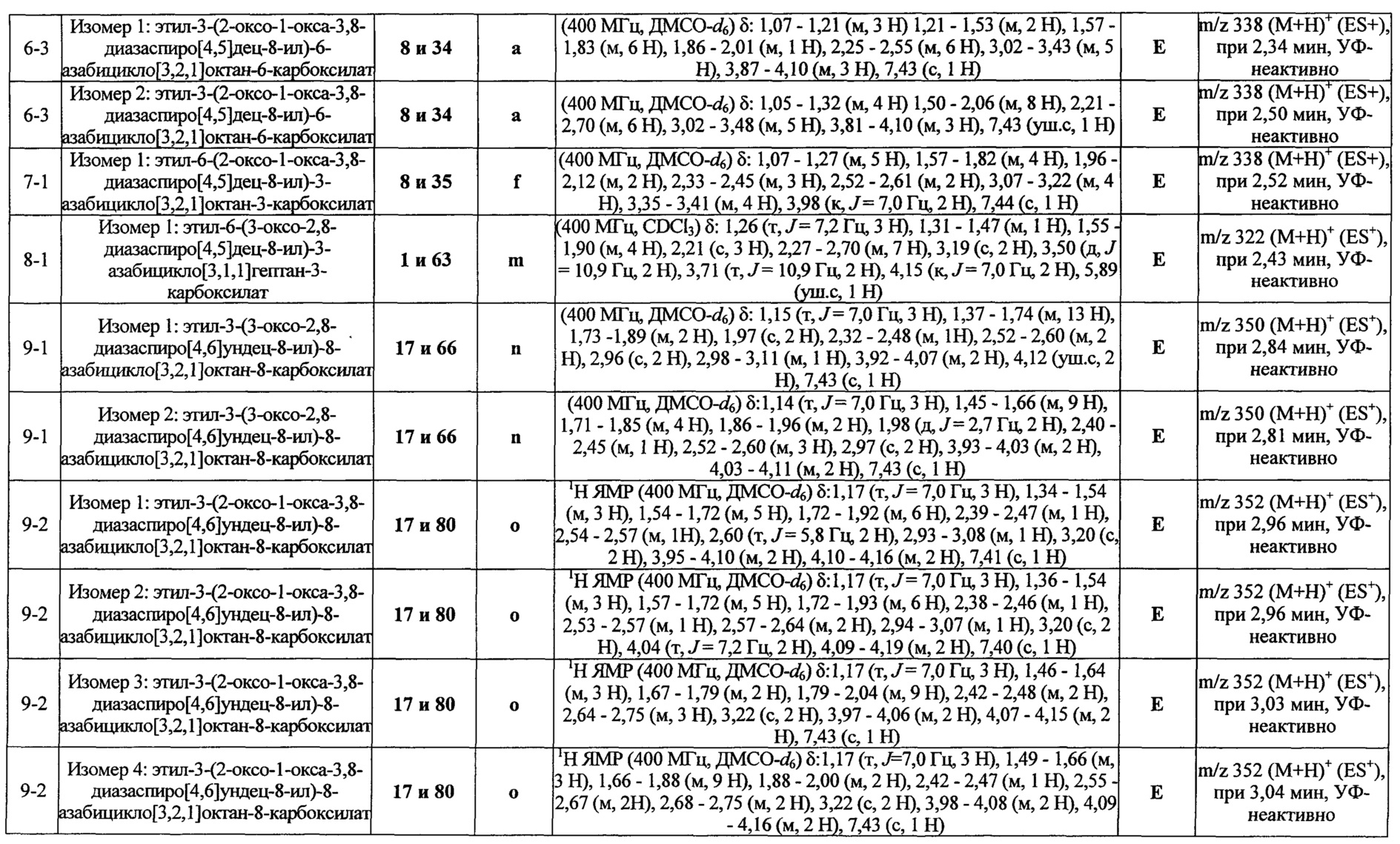
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ ПРИМЕР А
Анализы фосфо-ERK1/2
Функциональные анализы проводили с помощью системы Alphascreen Surefire phospho-ERK1/2 (Crouch & Osmond, Comb. Chem. High Throughput Screen, 2008). Фосфорилирование ERK1/2 является сопутствующим следствием активации рецептора, сопряженного как с Gq/11, так и Gi/o белком, что делает его весьма подходящим для оценки рецепторов M1, М3 (сопряженных с Gq/11) и М2, M4 (сопряженных с Gi/o), вместо использования различных форматов анализа для различных подтипов рецепторов. Клетки СНО, стабильно экспрессирующие мускариновый рецептор M1, М2, М3 или М4 человека, высевали (25 тыс/лунка) на 96-луночные планшеты для тканевых культур в MEM-альфа + 10% диализировали FBS. После прилипания клетки оставляли в бессывороточной среде на протяжении ночи. Стимуляцию агониста осуществляли посредством добавления 5 мкл агониста к клеткам в течение 5 мин (37°С). Среду удаляли и добавляли 50 мкл буфера для лизиса. Через 15 мин 4-микролитровый образец переносили в 384-луночный планшет и добавляли 7 мкл смеси для детектирования. Планшеты инкубировали в течение 2 ч при осторожном перемешивании в темноте и затем считывали на сканирующем спектрофотометре для прочтения планшетов PHERAstar.
Показатели pEC50 и Emax рассчитывали на основе полученных данных для каждого подтипа рецепторов.
Для большинства примеров существуют два диастереомера, которые были разделены, если не указано иное, Аналитические данные для активных изомеров приведены в Таблице 3. В примерах 3-1 и 9-2 были разделены четыре энантиомера, данные представлены для всех изомеров.
Результаты приведены ниже в Таблице 4.
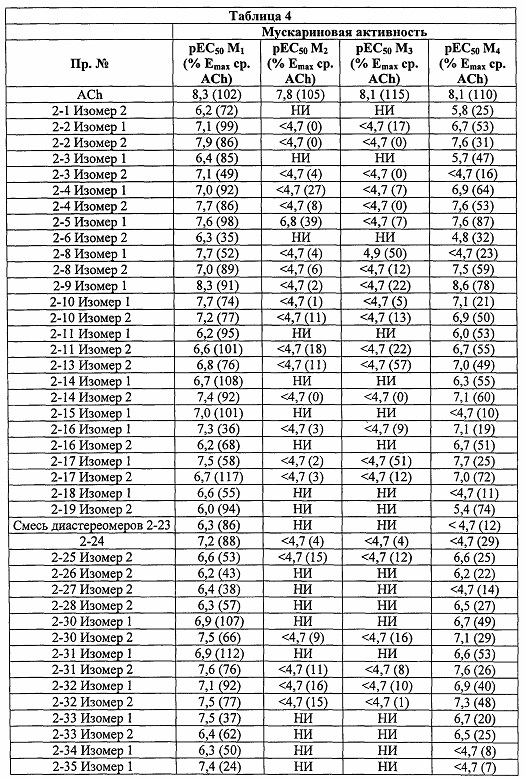
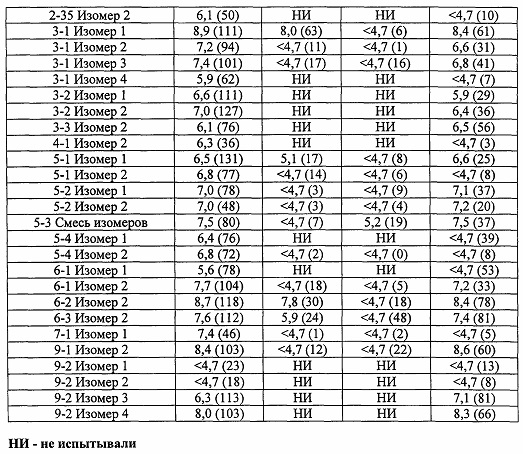
ПРИМЕР В
Пассивное избегание
Исследования проводили, как описано ранее у Foley et al., (2004) Neuropsychopharmacology. В задаче пассивного избегания введение скополамина (1 мг/кг, внутрибрюшинно) через 6 часов после обучения оказывало на животных амнестическое воздействие в отношении парадигмы. Исследовали диапазон доз, составляющий 3, 10 и 30 мг/кг перорально (п/о) свободного основания, вводимого энтерально за 90 минут до начала периода обучения через желудочный зонд.
Было обнаружено, что Изомер 1 из Примера 2-2 инвертирует скополамин-индуцированную амнезию парадигмы в зависимости от дозы, при примерной ED50, составляющей около 10 мг/кг (п/о). Эффект от 30 мг/кг был аналогичным эффекту, произведенному ингибитором холинэстеразы донепезилом (0,1 мг/кг, внутрибрюшинно), который служил в качестве положительного контроля (Фигура 1).
Эквиваленты
Приведенные выше примеры представлены с целью иллюстрации данного изобретения и не должны истолковываться как накладывающие какое-либо ограничение на объем данного изобретения. Очевидно, что возможны многочисленные модификации и изменения в конкретных вариантах реализации данного изобретения, описанных выше и проиллюстрированных на примерах, без отхода от принципов, лежащих в основе данного изобретения. Все такие модификации и изменения предназначены для включения в эту заявку.
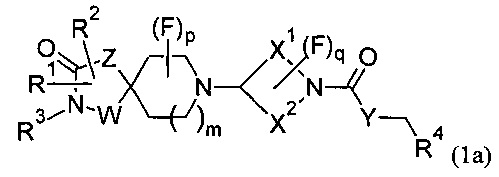
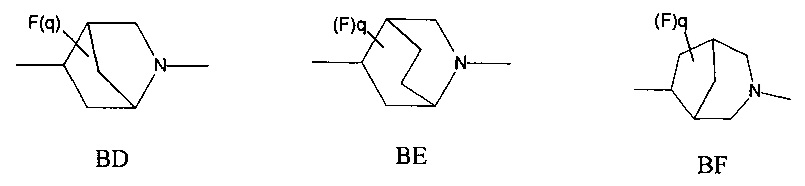
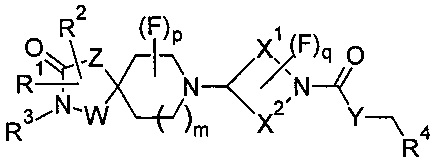
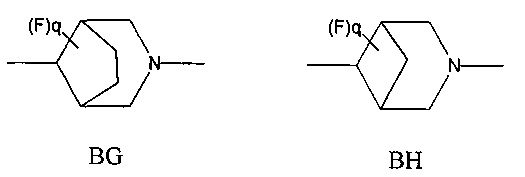
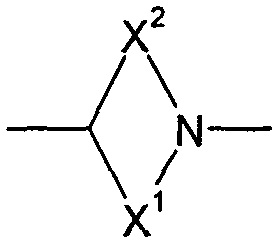
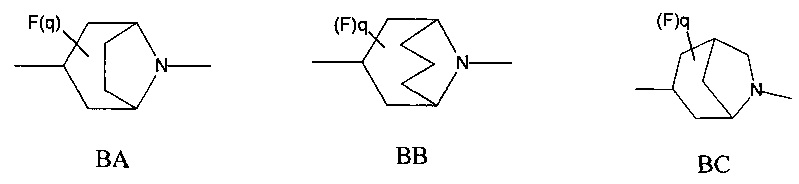
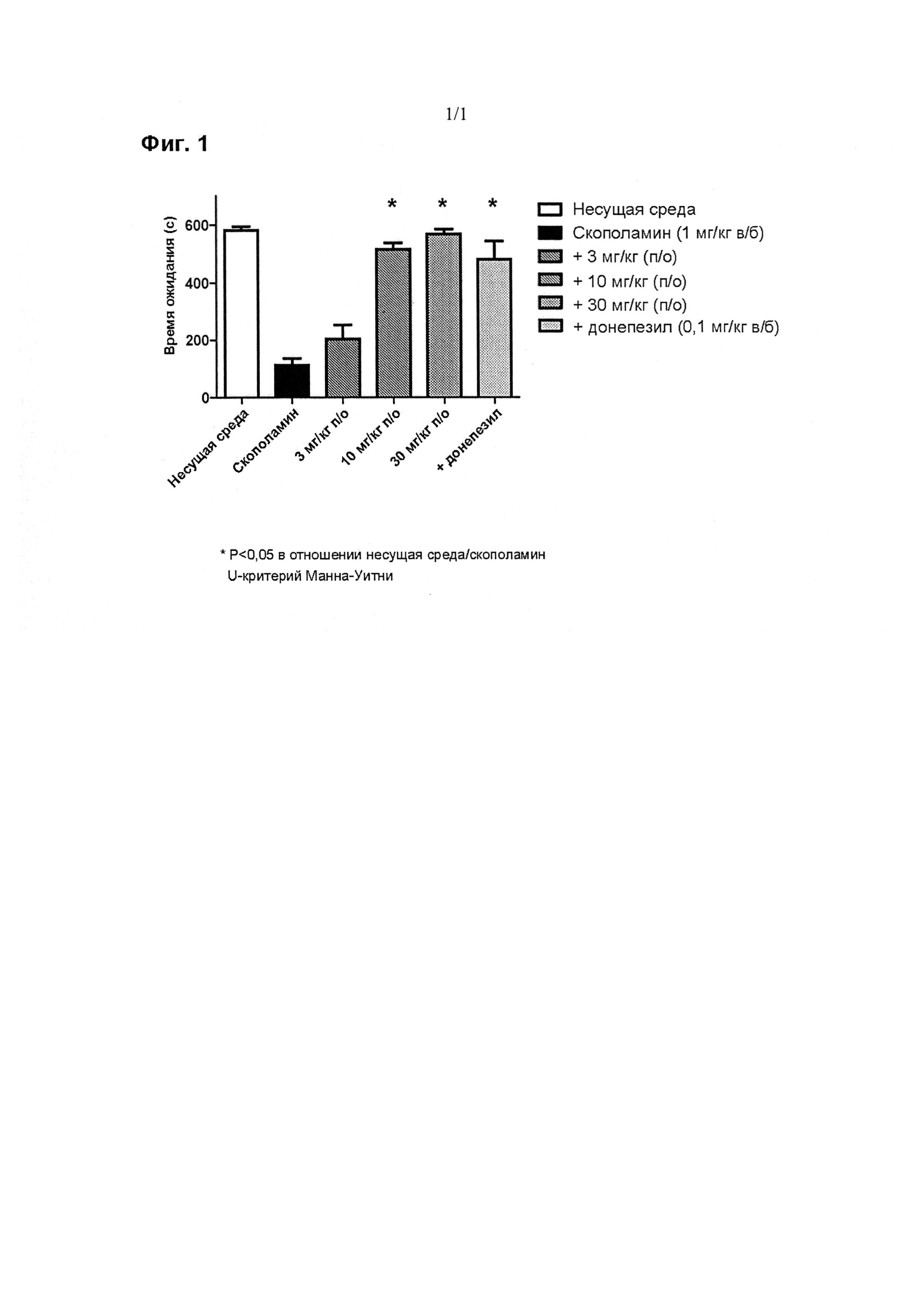
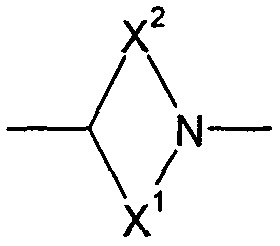
Фармацевтические соединения
Бициклические азотсодержащие соединения как агонисты м1 мускариновых рецепторов
Фармацевтические соединения

