АНАЛОГИ КОРТИСТАТИНА ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ И/ИЛИ ИММУНОПАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к аналогам кортистатина и их применениям. Соединения по настоящему изобретению представляют собой пептидные лиганды с потенциалом применения в диагностике, предупреждении или лечении таких патологий, при которых экспрессируются рецепторы, способные связываться с кортистатином.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
Кортистатин (CST) представляет собой природный эндогенный пептид из 14 аминокислот, открытый у крыс в 1996 году (CST-14) [de Lecea et al., Nature, 1996, 381, 242-245], и позднее, в 1997 году, обнаруженный у человека в виде удлиненной формы из 17 аминокислот (CST-17) [Fukusimi et al., Biochem. Biophys. Res. Commun, 1997, 232, 157-163]. Кортистатин, фактически, существует в двух биологически активных формах, поскольку его предшественник (препро-CST) приводит к образованию CST-14 и CST-29 у грызунов и к CST-17, и CST-29 у человека.
Кортистатин имеет высокую гомологию с другим эндогенным пептидом, соматостатином (SST), который является высококонсервативным и обнаружен у млекопитающих в форме соматостатина-14 (SST-14) и соматостатина-28 (SST-28):
Последовательности кортистатина и соматостатина:
H2N-Pc[CKNFFWKTFSSC]K-OH кортистатин-14 (крыса/мышь),
H2N-DRMPс[CRNFFWKTFSSC]K-OH кортистатин-17 (человек),
H2N-AGc[CKNFFWKTFTSC]-OH соматостатин-14 (человек/крыса/мышь).
Известно, что кортистатин взаимодействует с 5 G-белок-связанными мембранными рецепторами, описанными для соматостатина, sstr1-sstr5 [a) Spier et al., Brain Research Reviews 2000, 33, 228-241; b) Patel et al., Endocrinology 1994, 135, 2814-2817]. Но кортистатин не является соматостатином [Gonzalez-Rey et al., Mol. Cell. Endocrinol. 2008, 286 (1-2), 135-140], и, таким образом, в дополнение к своей наномолярной аффинности к рецепторам соматостатина, кортистатин также взаимодействует с рецептором грелина (GHSR). Кроме того, при поиске специфического рецептора для кортистатина орфанный рецептор MrgX2 был описан как первый специфический рецептор человека для кортистатина [Robas et al., J. Biol. Chem. 2003, 278, 44400-44404]. В последующем отсутствие этого рецептора в клетках иммунной системы и его высокая аффинность к другим нейропептидам, таким как проадреномедуллин, привели к тому, что в настоящее время его не рассматривают в качестве специфического рецептора кортистатина [van Hagen et al., Mol. Cell. Endocrinol. 2008, 286(1-2), 141-147], и получение характеристик специфического рецептора кортистатина является вопросом, который остается нерешенным.
Иммуномодулирующая активность кортистатина была широко продемонстрирована на экспериментальных моделях заболеваний, которые протекают с воспалительным или аутоиммунным ответами, таких как летальный эндотоксический шок, болезнь Крона и ревматоидный артрит [a) Gonzalez-Rey et al., J. Exp. Med. 2006, 203(3), 563-571; b) Gonzalez-Rey et al., Proc. Natl. Acad. Sci. USA 2006, 103, 4228-4233; c) Gonzalez-Rey et al., Ann. Rheum. Dis. 2007, 66 (5), 582-588; d) WO 2007/082980 А1]. Указанное иммунорегуляторное действие может быть связано с его экспрессией в лимфоцитах, моноцитах, макрофагах и дендритных клетках и клетках иммунной системы [a) Dalm V.A. et al., Am. J. Physiol. Endocrinol. Metab. 2003, 285, E344-353; b) Dalim V.A. et al., J. Clin. Endocrinol. Metab. 2003, 88, 270-276]. Экспрессия кортистатина и его рецепторов в иммунной системе человека и при патологиях иммунной системы была недавно рассмотрена [van Hagen et al., Mol. Cell. Endocrinol. 2008, 286(1-2), 141-147].
В упомянутых выше научных исследованиях, которые показали эффективность кортистатина при заболеваниях с воспалительным и иммунным компонентом, использовали CST-29. CST-29 представляет собой длинный эндогенный пептид с высокой сложностью синтеза и, следовательно, с низкой промышленной рентабельностью для его промышленного использования в фармацевтическом секторе. Его фармацевтическое применение также представляет дополнительную проблему: он характеризуется низкой стабильностью в сыворотке.
Другие исследуемые предложения доказывают эффективность эндогенного пептида CST-17 в сочетании с нейропептидом EI в лечении воспалительных и аутоиммунных заболеваний [WO 2009/043523 А2], преимущество которого в отношении промышленного использования заключается в низкой сложности синтеза. Однако он по-прежнему обладает недостатком, который заключается в низкой стабильности в сыворотке из-за его нативной структуры из L-аминокислот.
В целом, лекарственные средства на основе пептидов являются предпочтительными, поскольку пептиды по существу являются нетоксичными, их эффективность в низких дозах гарантирует, что они не вызывают значительных побочных эффектов по сравнению с другими лекарственными средствами на основе малых молекул или антител, но они должны быть модифицированы для улучшения их биодоступности и времени полужизни. Включение неприродных аминокислот в природную последовательность является одной из стратегий, известных из предшествующего уровня техники, для повышения стабильности эндогенного пептида. Например, были описаны модификации соматостатина галогенированными аминокислотами п-хлор-Phe и пентафтор-Phe в положениях 6, 7 и 11 [WO 2007/081792 А2; Meyers С.A. et al., Digestion 1981, 21(1), 21-4]. Те же положения 6, 7 и 11 в исходном соматостатине также были модифицированы мезитилаланином и мезитилглицином, давая в результате аналоги соматостатина, которые являются более стабильными [WO 2010/128098 А1]. Однако эти стабилизирующие модификации могут нарушать функциональность исходной молекулы. Данный случай относится к октреотиду, аналогу соматостатина для клинического применения, который является намного более стабильным, чем исходная молекула, сохраняет связывание с рецептором sstr2, но полностью утрачивает аффинность к рецепторам sstr1 и sstr4. [Patel et al., Endocrinology 1994, 135, 2814-2817].
Время полужизни в крови эндогенных пептидов, таких как соматостатин и кортистатин, является чрезвычайно коротким, с трудом достигая лишь нескольких минут [Skamene et al., Clin. Endocrinol. 1984, 20, 555-564]. Таким образом, существует необходимость поиска новых синтетических аналогов кортистатина для лечения тех патологий, при которых экспрессируются специфические рецепторы кортистатина и рецепторы, которые используются совместно с другими молекулами, такими как соматостатин (sstr1, sstr2, sstr3, sstr4 и/или sstr5) и/или грелин (GHSR), являющихся, кроме того, более стабильными в крови, чем кортистатин.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение раскрывает новые пептиды, аналоги кортистатина, с противовоспалительным и/или иммунорегуляторным действием, аналогичным такому действию у природного пептида. Определенные модификации с неприродными аминокислотами, такими как мезитилаланин и/или дигалогенфенилаланины, плюс включение жирных кислот или ПЭГилирований, сохраняют и даже улучшают противовоспалительное и противоаутоиммунное действие природной молекулы in vitro и in vivo. Кроме того, основное преимущество новых аналогов кортистатина заключается в том, что полученные пептиды, имеющие одну или несколько модификаций, обладают существенно более длительным временем полужизни в сыворотке, чем у эндогенной молекулы. Синтез новых аналогов кортистатина является экономически целесообразным (с последовательностями предпочтительно от 13 до 17 аминокислот), что представляет собой один из аспектов, гарантирующий их полезность в фармацевтической промышленности. Соединения по данному изобретению являются новыми соединениями и функционально эквивалентны кортистатину, поскольку все они обладают противовоспалительным и/или иммунорегуляторным эффектом, который аналогичен кортистатину, in vitro и/или in vivo.
Определения
В данном документе включены значения некоторых терминов и выражений, как они используются в контексте настоящего изобретения, с целью оказания содействия в его понимании.
Термин "аналог кортистатина" относится к соединению, которое взаимодействует по меньшей мере с одним из рецепторов кортистатина, известных и используемых совместно с другими молекулами, такими как 5 рецепторов соматостатина (sstr1, sstr2, sstr3, sstr4 и/или sstr5) или рецептор грелина, и/или специфический рецептор кортистатина, по-прежнему нуждающийся в идентификации. Из этого следует, что он представляет собой лиганд указанных рецепторов кортистатина и может быть функциональным эквивалентом (или агонистом) кортистатина с активностью, аналогичной таковой активности кортистатина.
Термин "функциональный эквивалент (или агонист)" относится к соединению, которое демонстрирует аффинность к некоторым рецепторам исходной молекулы и с точки зрения качества данного соединения вызывает такие же эффекты, как и эндогенный лиганд рецептора.
Термин "аналог соматостатина" относится к соединению, которое взаимодействует с одним или несколькими рецепторами соматостатина (sstr) и также известно как лиганд указанных рецепторов. Это определение введено Bevan et al. [в J. Clin. Endocrinol. Metabolism. 2005, 90, 1856-1863]. В ЕР 1040837 A2 термину аналог соматостатина также дают определение в отношении всех модифицированных производных нативного соматостатина, которые проявляют связанную с соматостатином активность, такую как взаимодействие по меньшей мере с одним из рецепторов соматостатина (sstr1, sstr2, sstr3, sstr4 или sstr5)".
В данном описании аббревиатуры, используемые для аминокислот, соответствуют правилам Комитета по биохимической номенклатуре IUPAC IUB, изложенным в Eur. J. Biochem, 1984, 138, 9-37 (фигура 1).
Фигура 1: аминокислоты (стереохимия не указана, во всех случаях это может быть L-, D- или DL-)
Ala (А): аланин;
Asn (N): аспарагин;
Asp (D): аспарагиновая кислота;
Arg (R): аргинин;
Cys (С): цистеин;
Gly (G): глицин;
Lys (K): лизин;
Met (М): метионин;
Phe (F): фенилаланин;
Pro (Р): пролин;
Ser (S): серин;
Thr (T): треонин;
Trp (W): триптофан;
Phg: фенилглицин;
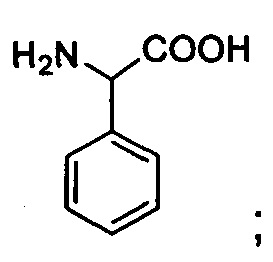
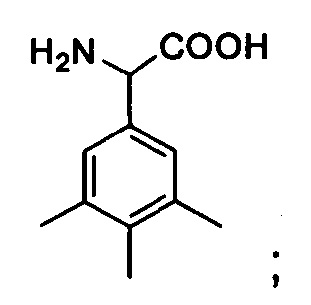
Msa: 2,4,6-триметилфенилаланин или 3-мезитилаланин,
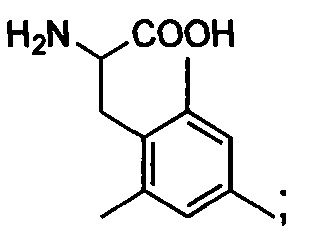
Tmp: 3,4,5-триметилфенилаланин,
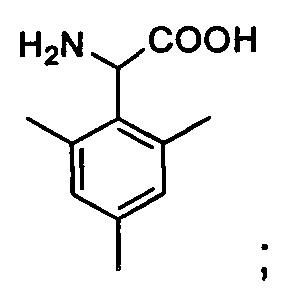
Msg: 2,4,6-триметилфенилглицин или 2-мезитилглицин,
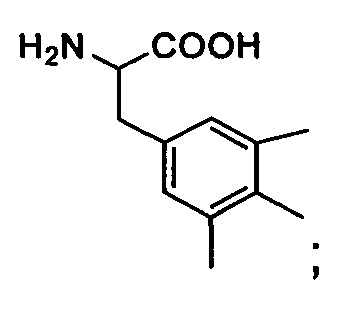
3,4,5-триметилфенилглицин,
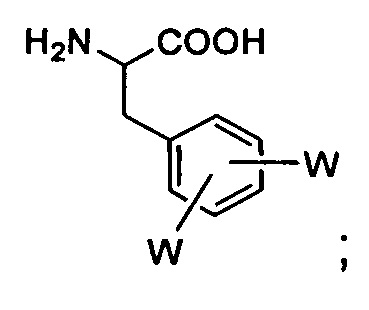
DiW-Phe (где W представляет собой F, Cl, Br или I): дигалогенфенилаланин, которiй является фенилаланином, в котором фенильная группа замещена двумя атомами галогена,
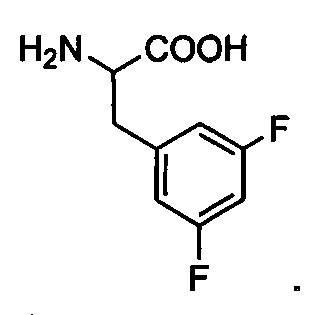
Dfp: 3,5-дифторфенилаланин,
Сокращение "Ас-" используется в данном описании для идентификации ацетильной группы (СН3-СО-), сокращение "Palm-" используется для идентификации пальмитоильной группы (CH3-(CH2)14-CO-) и сокращение "Myr-" используется для идентификации миристоильной группы (СН3-(СН2)12-СО-).
Термин "нециклическая алифатическая группа" используется в данном изобретении для охвата неразветвленных или разветвленных алкильных, алкенильных и алкинильных групп.
Термин "алкильная группа" относится к насыщенной, неразветвленной или разветвленной группе, имеющей от 1 до 24, предпочтительно от 1 до 16, более предпочтительно от 1 до 14, еще более предпочтительно от 1 до 12, еще более предпочтительно 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода, и которая связана с остальной частью молекулы одинарной связью, включая, например и без ограничения: метильную, этильную, изопропильную, изобутильную, трет-бутильную, гептильную, октильную, децильную, додецильную, лаурильную, гексадецильную, октадецильную, амильную, 2-этилгексильную, 2-метилбутильную, 5-метилгексильную группу и подобные.
Термин "алкенильная группа" относится к неразветвленной или разветвленной группе, имеющей от 2 до 24, предпочтительно от 2 до 16, более предпочтительно от 2 до 14, еще более предпочтительно от 2 до 12, еще более предпочтительно 2, 3, 4, 5 или 6 атомов углерода, с одной или несколькими углерод-углеродными двойными связями, предпочтительно с 1, 2 или 3 углерод-углеродными двойными связями, конъюгированными или неконъюгированными, которая связана с остальной частью молекулы одинарной связью, включая, например и без ограничения: винильную(-СН2=СН2), аллильную (-СН2-СН=СН2), олеильную, линолеильную группы и подобные.
Термин "алкинильная группа" относится к неразветвленной или разветвленной группе, имеющей от 2 до 24, предпочтительно от 2 до 16, более предпочтительно от 2 до 14, еще более предпочтительно от 2 до 12, еще более предпочтительно 2, 3, 4, 5 или 6 атомов углерода, с одной или несколькими углерод-углеродными тройными связями, предпочтительно с 1, 2 или 3 углерод-углеродными тройными связями, конъюгированными или неконъюгированными, которая связана с остальной частью молекулы одинарной связью, включая, например и без ограничения: этинильную группу, 1-пропинильную, 2-пропинильную, 1-бутинильную, 2-бутинильную, 3-бутинильную, пентинильную, такую как 1-пентинильная, и подобную. Алкинильные группы также могут содержать одну или несколько углерод-углеродных двойных связей, включая, например и без ограничения: бут-1-ен-3-инильную, пент-4-ен-1-инильную группу и подобные.
Термин "алициклическая группа" используется в данном изобретении для охвата, например и без ограничения: циклоалкильных, или циклоалкенильных, или циклоалкинильных групп.
Термин "циклоалкил" относится к насыщенной моно- или полициклической алифатической группе, имеющей от 3 до 24, предпочтительно от 3 до 16, более предпочтительно от 3 до 14, еще более предпочтительно от 3 до 12, еще более предпочтительно 3, 4, 5 или 6 атомов углерода, и которая связана с остальной частью молекулы одинарной связью, включая, например и без ограничения: циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклогептильную, метилциклогексильную, диметилциклогексильную, октагидроиндольную, декагидронафталиновую, додекагидрофеналеновую группу и подобные.
Термин "циклоалкенил" относится к неароматической моно- или полициклической алифатической группе, имеющей от 5 до 24, предпочтительно от 5 до 16, более предпочтительно от 5 до 14, еще более предпочтительно от 5 до 12, еще более предпочтительно 5 или 6 атомов углерода, с одной или несколькими углерод-углеродными двойными связями, предпочтительно с 1, 2 или 3 углерод-углеродными двойными связями, конъюгированными или неконъюгированными, которая связана с остальной частью молекулы одинарной связью, включая, например и без ограничения: циклопент-1-ен-1-ильную группу и подобные.
Термин "циклоалкинил" относится к неароматической моно- или полициклической алифатической группе, имеющей от 8 до 24, предпочтительно от 8 до 16, более предпочтительно от 8 до 14, еще более предпочтительно от 8 до 12, еще более предпочтительно 8 или 9 атомов углерода, с одной или несколькими углерод-углеродными тройными связями, предпочтительно с 1, 2 или 3 углерод-углеродными тройными связями, конъюгированными или неконъюгированными, которая связана с остальной частью молекулы одинарной связью, включая, например и без ограничения: циклоокт-2-ин-1-ильную группу и подобную. Циклоалкинильные группы также могут содержать одну или несколько углерод-углеродных двойных связей, включая, например и без ограничения: циклоокт-4-ен-2-инильную группу и подобные.
Термин "арильная группа" относится к ароматической группе, имеющей от 6 до 30, предпочтительно от 6 до 18, более предпочтительно от 6 до 10, еще более предпочтительно 6 или 10 атомов углерода, которая содержит 1, 2, 3 или 4 ароматических кольца, связанных углерод-углеродной связью, или которые конденсированы, включая, например и без ограничения: фенильную, нафтильную, дифенильную, инденильную, фенантрильную или антранильную, среди прочего; или аралкильную группу.
Термин "аралкильная группа" относится к алкильной группе, замещенной ароматической группой, с от 7 до 24 атомов углерода и включая, например и без ограничения: -(СН2)1-6-фенил, -(СН2)1-6-(1-нафтил), -(CH2)1-6-(2-нафтил), -(СН2)1-6-СН(фенил)2 и подобные.
Термин "гетероциклильная группа" относится к углеводородному кольцу из 3-10 членов, в котором один или несколько атом в кольце, предпочтительно 1, 2 или 3 атома в кольце, представляет собой элемент, отличающийся от углерода, такой как азот, кислород или сера, и которое может быть насыщенным или ненасыщенным. Для целей настоящего изобретения гетероцикл может быть циклической, моноциклической, бициклической или трициклической системой, которая может включать системы конденсированных колец; а атомы азота, углерода или серы в гетероциклильном радикале могут быть необязательно окислены; атом азота может быть необязательно кватернизован; и гетероциклильный радикал может быть частично или полностью насыщенным или ароматическим. Наиболее предпочтительно термин гетероциклил относится к кольцу из 5 или 6 членов. Примерами насыщенных гетероциклильных групп являются диоксан, пиперидин, пиперазин, пирролидин, морфолин и тиоморфолин. Примерами ароматических гетероциклильных групп, также известных как гетероароматические группы, являются пиридин, пиррол, фуран, тиофен, бензофуран, имидазолин, хинолеин, хинолин, пиридазин и нафтиридин.
Термин "гетероарилалкильная группа" относится к алкильной группе, замещенной с помощью замещенной или незамещенной ароматической гетероциклильной группы, где алкильная группа имеет от 1 до 6 атомов углерода, а ароматическая гетероциклильная группа от 2 до 24 атомов углерода и от 1 до 3 атомов, отличных от углерода, включая, например и без ограничения: -(СН2)1-6-имидазолил, -(СН2)1-6-триазолил, -(CH2)1-6-тиенил, -(СН2)1-6-фурил, -(СН2)1-6-пирролидинил и подобные.
Как понятно из данной области техники, в определенных выше группах может быть соответствующая степень замещения. Следовательно, замещение может присутствовать в группах по данному изобретению, где это точно указано. Упоминания в данном документе замещенных групп в группах по данному изобретению указывают, что определенный радикал может быть замещен в одном или нескольких доступных положениях одним или несколькими заместителями, предпочтительно в 1, 2 или 3 положениях, более предпочтительно в 1 или 2 положениях, еще более предпочтительно в 1 положении. Эти заместители включают, например и без ограничения: С1-С4алкил; гидроксил; С1-С4алкоксил; амино; С1-С4аминоалкил; С1-С4карбонилоксил; С1-С4оксикарбонил; галоген, такой как фтор, хлор, бром и йод; циано; нитро; азид; С1-С4алкилсульфонил; тиол; С1-С4алкилтио; арилоксил, такой как феноксил; NRb(C=NRb)NRbRc; где Rb и Rc независимо выбраны из группы, состоящей из Н, С1-С4алкила, С2-С4алкенила, С2-С4алкинила, С3-С10циклоалкила, С6-С18арила, С7-С17аралкила, гетероциклила из 3-10 членов или защитной группы для аминогруппы.
Соединения по настоящему изобретению
Первый аспект данного изобретения относится к соединению, определенному формулой (I),
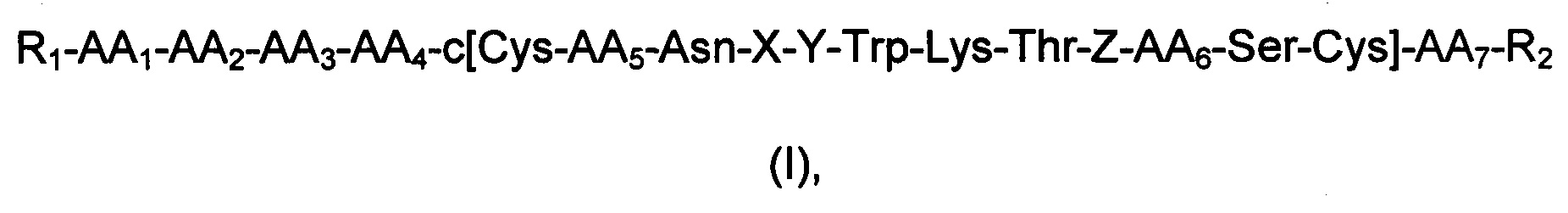
его стереоизомерам, их смесям и/или его фармацевтически приемлемым солям, где
AA1 представляет собой Asp или связь,
АА2 представляет собой Arg или связь,
АА3 представляет собой Met, или Ala, или связь,
АА4 представляет собой Pro или Gly,
АА5 представляет собой Lys или Arg,
АА6 представляет собой Ser или Thr,
АА7 представляет собой Lys или связь,
X, Y, Z представляют собой аминокислоты Phe, Phg, Msa, 3,4,5-триметилфенилаланин, Msg, 3,4,5-триметилфенилглицин и/или дигалогенфенилаланин, diW-Phe;
W выбран из группы, состоящей из F, Cl, Br и I;
R1 выбран из группы, состоящей из Н, нециклической замещенной или незамещенной алифатической группы, замещенной или незамещенной эпициклической группы, замещенного или незамещенного гетероциклила, замещенного или незамещенного гетероарилалкила, замещенного или незамещенного арила, замещенного или незамещенного аралкила, полимера, полученного из полиэтиленгликоля, хелатообразующего средства и R5-CO-;
R2 выбран из группы, состоящей из -NR3R4, -OR3 и -SR3;
R3 и R4 независимо выбраны из группы, состоящей из Н, нециклической замещенной или незамещенной алифатической группы, замещенного или незамещенного алициклила, замещенного или незамещенного гетероциклила, замещенного или незамещенного гетероарилалкила, замещенного или незамещенного арила и замещенного или незамещенного аралкила, и полимера;
R5 выбран из группы, состоящей из Н, нециклической замещенной или незамещенной алифатической группы, замещенного или незамещенного алициклила, замещенного или незамещенного арила, замещенного или незамещенного аралкила, замещенного или незамещенного гетероциклила и замещенного или незамещенного гетероарилалкила;
и при условии, что:
- по меньшей мере одна из аминокислот X, Y или Z представляет собой Msa,
3,4,5-триметилфенилаланин, Msg, 3,4,5-триметилфенилглицин и/или дигалогенфенилаланин, diW-Phe;
- если AA1 и АА2 представляют собой связи, АА3 представляет собой Ala, АА4 представляет собой Gly, АА5 представляет собой Lys, АА6 представляет собой Thr и АА7 представляет собой связь, то по меньшей мере одна из аминокислот X, Y или Z представляет собой дигалогенфенилаланин, diW-Phe.
В предпочтительном варианте осуществления по меньшей мере одна из аминокислот X, Y или Z представляет собой дигалогенфенилаланин, diW-Phe. Предпочтительно, W представляет собой фтор. Более предпочтительно, дигалогенфенилаланин представляет собой 3,5-дифторфенилаланин (Dfp).
В предпочтительном варианте осуществления АА4 представляет собой Pro. В более предпочтительном варианте осуществления АА3 представляет собой Met или связь, а АА4 представляет собой Pro. Предпочтительно, по меньшей мере одна из аминокислот X, Y или Z представляет собой Msa и/или 3,5-дифторфенилаланин (Dfp).
Группы R1 и R2 связаны с аминоконцевым (N-концевым) и карбоксиконцевым (С-концевым) концами пептидных последовательностей, соответственно, и они могут быть аминокислотами.
В соответствии с предпочтительным вариантом осуществления данного изобретения R1 выбран из группы, состоящей из Н, полимера, полученного из полиэтиленгликоля, и R5-CO-, где R5 выбран из группы, состоящей из замещенного или незамещенного С1-С24алкильного радикала, замещенного или незамещенного С2-С24алкенила, замещенного или незамещенного С2-С24алкинила, замещенного или незамещенного С3-С24циклоалкила, замещенного или незамещенного С5-С24циклоалкенила, замещенного или незамещенного С8-С24циклоалкинила, замещенного или незамещенного С6-С30арила, замещенного или незамещенного С7-С24аралкила, замещенного или незамещенного гетероциклильного кольца из 3-10 членов и замещенного или незамещенного гетероарилалкила из 2-24 атомов углерода и 1-3 атомов, отличных от углерода, где алкильная цепь имеет от 1 до 6 атомов углерода. Более предпочтительно, R1 выбран из группы, состоящей из Н, ацетила, трет-бутаноила, фенила, гексаноила, 2-метилгексаноила, циклогексан карбоксила, октаноила, деканоила, лауроила, миристоила, пальмитоила, стеароила, бегенила, олеоила и линолеоила. Еще более предпочтительно, R1 представляет собой Н, ацетил, гексаноил, октаноил, лауроил, миристоил или пальмитоил.
В соответствии с другим предпочтительным вариантом осуществления R1 выбран из полимера, полученного из полиэтиленгликоля с молекулярной массой, составляющей от 200 до 35000 дальтон.
В соответствии с другим предпочтительным вариантом осуществления R2 представляет собой -NR3R4, -OR3 или -SR3, где R3 и R4 независимо выбраны из группы, состоящей из Н, замещенного или незамещенного С1-С24алкила, замещенного или незамещенного С2-С24алкенила, замещенного или незамещенного С2-С24алкинила, замещенного или незамещенного С3-С24циклоалкила, замещенного или незамещенного С5-С24циклоалкенила, замещенного или незамещенного С8-С24циклоалкинила, замещенного или незамещенного С6-С30арила, замещенного или незамещенного С7-С24аралкила, замещенного или незамещенного гетероциклильного кольца из 3-10 членов и замещенного или незамещенного гетероарилалкила из 2-24 атомов углерода и 1-3 атомов, отличных от углерода, где алкильная цепь имеет от 1 до 6 атомов углерода, и полимера, полученного из полиэтиленгликоля. Необязательно, R3 и R4 могут быть связаны насыщенной или ненасыщенной углерод-углеродной связью, образующей цикл с атомом азота. Более предпочтительно, R2 представляет собой -NR3R4 или -OR3, где R3 и R4 независимо выбраны из группы, состоящей из Н, замещенного или незамещенного С1-С24алкила, замещенного или незамещенного С2-С24алкенила, замещенного или незамещенного С2-С24алкинила, замещенного или незамещенного С3-С10циклоалкила, замещенного или незамещенного арила С6-С15, замещенного или незамещенного гетероарилалкильного кольца из 3-10 членов и алкильной цепи из 1-6 атомов углерода, и полимера, полученного из полиэтиленгликоля. Более предпочтительно, R3 и R4 выбраны из группы, состоящей из Н, метила, этила, гексила, додецила или гексадецила. Еще более предпочтительно, R3 представляет собой Н, a R4 выбран из группы, состоящей из Н, метила, этила, гексила, додецила или гексадецила. В соответствии с еще более предпочтительным вариантом осуществления R2 выбран из -ОН и -NH2.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения R1 или R2 представляет собой хелатообразующее средство, необязательно в комплексе с детектируемым или радио-терапевтическим элементом. Хелатообразующее средство относится к группе, которая способна образовывать координационные комплексы с детектируемым или радиотерапевтическим элементом. Предпочтительно, хелатообразующее средство представляет собой группу, способную образовывать комплексы с ионами металлов, более предпочтительно, выбранную из группы, состоящей из DOTA, DTPA, ТЕТА или их производных. Хелатообразующее средство может связываться непосредственно или через линкер.
Детектируемый элемент относится к любому радиоактивному, флуоресцентному элементу или позитивно-контрастному элементу для магнитно-резонансной томографии, предпочтительно, иону металла, который демонстрирует детектируемые свойства в диагностической методике in vivo. Радио-терапевтический элемент понимают как элемент, который испускает α-излучение, β-излучение или γ-излучение.
В конкретном варианте осуществления соединения по настоящему изобретению выбраны из группы последовательностей, описанных ниже:


Специалист в данной области поймет, что аминокислотные последовательности, приведенные в настоящем изобретении, можно химически модифицировать, например, посредством химических модификаций, которые обоснованы с точки зрения физиологии, таких как фосфорилирование, ацетилирование, амидирование, ПЭГилирование, н-октаноилирование или пальмитоилирование, среди прочих.
Соединения по настоящему изобретению могут существовать в виде стереоизомеров или смесей стереоизомеров; например, аминокислоты, образующие их, могут иметь L-, D- конфигурацию, или быть рацемическими независимо одна от другой. Следовательно, можно получить изомерные смеси, а также рацемические смеси или диастереоизомерные смеси, или чистые диастереизомеры или энантиомеры, в зависимости от числа асимметрических атомов углерода, по которым представлены изомеры или смеси изомеров. Предпочтительными структурами пептидов по настоящему изобретению являются чистые изомеры, т.е. один энантиомер или диастереоизомер.
Например, если не указано иное, то подразумевается, что аминокислота представляет собой L- или D-, или их смеси, рацемические или нерацемические. Способы получения, описанные в данном документе, позволяют специалисту в данной области получить каждый из стереоизомеров соединения по настоящему изобретению путем выбора аминокислоты с соответствующей конфигурацией. Например, аминокислота Trp может представлять собой L-Trp или D-Trp.
Более предпочтительно, соединения, включенные в формулу (I), выбраны из группы, состоящей из:


Фармацевтически приемлемые соли соединений, полученных согласно настоящему изобретению, также находятся в пределах области настоящего изобретения. Термин "фармацевтически приемлемые соли" означает соль, одобренную для применения у животных и, более конкретно, у человека, и включает в себя соли, которые используют для образования солей присоединения оснований, независимо от того, являются они неорганическими, например и без ограничения: литий, натрий, калий, кальций, магний, марганец, медь, цинк или алюминий среди прочих, или органическими, например и без ограничения: этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, аргинин, лизин, гистидин или пиперазин среди прочих, или для образования солей присоединения кислот, независимо от того, являются они органическими, например и без ограничения: ацетат, цитрат, лактат, малонат, малеат, тартрат, фумарат, бензоат, аспартат, диаспартат, триаспартат, глутамат, сукцинат, олеат, трифторацетат, оксалат, памоат или глюконат среди прочих, или неорганическими, например и без ограничения: хлорид, сульфат, борат или карбонат среди прочих. Природа соли не является существенно важной, при условии, что она фармацевтически приемлема. Фармацевтически приемлемые соли соединений по настоящему изобретению могут быть получены обычными способами, которые хорошо известны из уровня техники [Berge S.M. et al., J. Pharm. Sci. 1977, 66, 1-19].
Способ получения
Синтез соединений по настоящему изобретению, их стереоизомеров или их фармацевтически приемлемых солей может быть осуществлен в соответствии с обычными способами, известными в данной области.
В одном из вариантов осуществления настоящего изобретения соединения синтезируют с использованием способов твердофазного пептидного синтеза или синтеза в растворе.
Способы твердофазного пептидного синтеза описаны, например, в [Stewart J.M. и Young J.D., 1984, "Solid Phase Peptide Synthesis, 2nd edition" Pierce Chemical Company, Rockford, Illinois; Bodanzsky M. и Bodanzsky A., 1984 "The practice of Peptide Synthesis" Springer Verlag, Berlin; Lloyd-Williams P., Albericio F. and Giralt E. (1997) "Chemical Approaches to the Synthesis of Peptides and Proteins" CRC, Boca Raton, FL, USA]. Способы синтеза в растворе и комбинации способов твердофазного синтеза и синтеза в растворе или ферментативного синтеза описаны в [(Kullmann W. etal., J. Biol. Chem. 1980, 255, 8234-8238].
В одном из вариантов осуществления данного изобретения соединения с формулой (I), их стереоизомеры, их смеси или их косметически или фармацевтически приемлемые соли получают с помощью способа, включающего следующие процедуры:
1. твердофазный синтез;
2. отщепление пептида от полимерного носителя;
3. циклизация пептида в растворе;
4. удаление защитных групп;
или в качестве альтернативы:
1. твердофазный синтез;
2. твердофазная циклизация;
3. отщепление пептида от полимерного носителя и одновременное удаление защитных групп, предпочтительно, с помощью обработки трифторуксусной кислотой.
Предпочтительно, С-конец закрепляют на твердой подложке и процедура, проводимая в твердой фазе, таким образом, предусматривает связывание аминокислоты с защищенным N-концом и свободным С-концом с аминокислотой со свободным N-концом и с С-концом, закрепленным на полимерном носителе; удаление защищенной группы N-конца и повторение этой последовательности столько раз, сколько необходимо для получения в результате пептида, предпочтительно от 13 до 17 аминокислот, с последующим в конечном итоге отщеплением синтезированного пептида от исходной полимерной подложки.
Функциональные группы боковых цепей аминокислот во время синтеза остаются обычно защищенными с помощью временных или постоянных защитных групп и могут быть удалены одновременно или ортогонально в процессе отщепления пептида от полимерной подложки.
В качестве альтернативы, твердофазный синтез можно проводить с использованием конвергентной стратегии путем закрепления пептидного фрагмента на полимерной положке или на пептидном фрагменте, предварительно закрепленном на полимерной подложке. Стратегии конвергентного синтеза широко известны специалистам в данной области и описаны в Lloyd-Williams P. et al., Tetrahedron 1993, 49, 11065-11133.
Способ может включать дополнительные стадии снятия защиты с N-конца и С-конца, и/или отщепления пептида от полимерной подложки обычным образом с помощью стандартных процедур и условий, известных из уровня техники, после чего функциональные группы на указанных концах можно модифицировать. Необязательную модификацию N-конца и С-конца можно проводить с пептидами согласно формуле (I), прикрепленными к полимерному носителю, или сразу после того, как пептид отщепили от полимерной подложки.
Необязательно R1 можно ввести путем реакции N-конца пептида по настоящему изобретению с соединением R1-Z, где R1 имеет вышеуказанное содержание и Z представляет собой уходящую группу, например и без ограничения: тозильную группу, мезильную группу и галогеновую группу среди прочих; посредством реакции нуклеофильного замещения в присутствии подходящего основания и растворителя, где фрагменты имеют функциональные группы, которые не участвуют в образовании связи N-C и обычно защищены с помощью временных или постоянных защитных групп. R1 также можно вводить с помощью реакции N-конца соединения по настоящему изобретению с группой R5COOH или ее сложными эфирами, галогенангидридами или ее ангидридом.
Необязательно и/или дополнительно радикалы R2 можно вводить с помощью реакции соединения HR2, где R2 представляет собой -OR3, -NR3R4 или -SR3, с комплементарным фрагментом, который соответствует пептиду с формулой (I), в котором R2 представляет собой -ОН, в присутствии соответствующего растворителя и основания, такого как, N,N-диизопропилэтиламин (DIEA) или триэтиламин, или добавки, такой как 1-гидроксибензотриазол (HOBt) или 1-гидроксиазабензотриазол (HOAt), и дегидратирующего средства, такого как карбодиимид, соль урония, соль фосфония или соль амидиния среди прочих, с получением таким образом пептида согласно настоящему изобретению с общей формулой (I), где указанные фрагменты имеют функциональные группы, которые не участвуют в образовании связи N-C, О-С или S-C, и соответствующим образом защищены с помощью временных или постоянных защитных групп. В качестве альтернативы, другие радикалы R2 можно встраивать одновременно с процессом отщепления пептида от полимерной подложки.
Специалист в данной области легко поймет, что стадии снятия защиты/отщепления С-конца и N-конца и их последующую дериватизацию можно провести в обычном порядке согласно способам, известным из уровня техники. [Smith М. В. and March J., 1999 "March's Advanced Organic Chemistry Reactions, Mechanisms and Structure", 5th Edition, John Wiley & Sons, 2001].
Термин "защитная группа" относится к группе, которая блокирует органическую функциональную группу и которую можно удалить в контролируемых условиях. Защитные группы, их относительная реакционная способность и условия, при которых они остаются инертными, известны специалисту в данной области.
Примерами типичных защитных групп для аминогруппы являются амиды, такие как амидацетат, амидбензоат, амидпивалат; карбаматы, такие как бензилоксикарбонил (Cbz или Z), 2-хлорбензил (CIZ), пара-нитробензилоксикарбонил (pNZ), трет-бутилоксикарбонил (Boc), 2,2,2-трихлорэтилоксикарбонил (Troc), 2-(триметилсилил)этилоксикарбонил (Teoc), 9-флуоренилметилоксикарбонил (Fmoc) или аллилоксикарбонил (Alloc), тритил (Trt), метокситритил (Mtt), 2,4-динитрофенил (Dnp), N-[1-(4,4-диметил-2,6-диоксоциклогекс-1-илиден)этил] (Dde), 1-(4,4-диметил-2,6-дилксо-циклогексилиден)-3-метилбутил (ivDde), 1-(1-адамантил)-1-метилэтоксикарбонил (Adpoc) среди прочих; предпочтительно Boc или Fmoc.
Примерами типичных защитных групп для карбонильной группы являются сложные эфиры, такие как сложный трет-бутиловый эфир (tBu), сложный аллиловый эфир (АН), сложный трифенилметиловый эфир (сложный тритиловый эфир, Trt), сложный циклогексиловый эфир (cHx), сложный бензиловый эфир (Bzl), сложный орто-нитробензиловый эфир, сложный лара-нитробензиловый эфир, сложный пара-метоксибензиловый эфир, сложный триметилсилилэтиловй эфир, сложный 2-фенилизопропиловый эфир, сложный флуоренилметиловый эфир (Fm), сложный 4-(N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино) бензиловый эфир (Dmab) среди прочих; предпочтительными защитными группами по настоящему изобретению являются сложные эфиры All, tBu, cHex, Bzl и Trt.
Трехфункциональные аминокислоты можно защитить в ходе процесса синтеза с помощью временных или постоянных защитных групп, являющихся ортогональными по отношению к защитным группам N-конца и С-конца. Для защиты аминогруппы боковой цепи лизина используют защитные средства для вышеупомянутой аминогруппы. Боковую цепь триптофана можно защитить с помощью любой из защитных групп вышеупомянутых аминогрупп или можно использовать незащищенной. Боковую цепь треонина и серина можно защитить с помощью сложного трет-бутилового эфира (tBu). Боковую цепь цистеина можно защитить с помощью защитной группы, выбранной из группы, состоящей из тритила и ацетаминометила. Боковую цепь аспарагина можно защитить с помощью защитной группы, выбранной из группы, состоящей из метокситритила, тритила или ксантила, или можно использовать незащищенной. Боковую цепь аргинина защищают с помощью защитной группы, выбранной из группы, состоящей из тозила (Tos), 4-метокси-2,3,6-триметилбензолсульфонила (Mtr), Alloc, нитро, 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонила (Pbf) и 2,2,5,7,8-пентаметилхроман-6-сульфонила (Pmc). Используют боковую цепь метионина, защищенную с помощью сульфоксидной группы, или используют незащищенной. Боковую цепь аспарагиновой кислоты защищают с помощью защитной группы, выбранной из группы, состоящей из Trt, Bzl, сНх, tBu и All. Предпочтительными защитными группами трехфункциональных аминокислот по настоящему изобретению являются сложные эфиры tBu в боковых цепях серина и треонина; Boc в боковых цепях лизина, Trt в боковых цепях цистеина; Pbf в боковых цепях аргинина и Fmoc или Boc в качестве временной защитной группы N-конца.
Примеры этих дополнительных защитных групп, их введение и удаление можно найти в литературе [Greene T.W. и Wuts P.G.M., (1999) "Protective groups in organic synthesis" John Wiley & Sons, New York; Atherton В. и Sheppard R.C. (1989) "Solid Phase Peptide Synthesis: A practical approach" IRL Oxford University Press]. Термин "защитные группы" также включает полимерные подложки, используемые в твердофазном синтезе.
Если синтез проводят полностью или частично в твердой фазе, допустимые для использования в способе по настоящему изобретению твердые носители, могут включать полистирольные носители, полиэтиленгликоль, привитый к полистиролу и им подобные, например и без ограничения: п-метилбензгидриламиновые смолы (МВНА) [Matsueda G.R. et al., Peptides 1981, 2, 45-50], 2-хлортритиловые смолы [Barlos K. et al. 1989 Tetrahedron Lett. 30:3943-3946; Barlos K. et al., 1989 Tetrahedron Lett. 30, 3947-3951], смолы TentaGel® (Rapp Polymere GmbH), смолы ChemMatrix® (Matrix Innovation, Inc) и им подобные, которые могут содержать или не содержать подвижный линкер, такой как 5-(4-аминометил-3,5-диметоксифенокси) валериановая кислота (PAL) [Albericio F. et al., 1990, J. Org. Chem. 55, 3730-3743], 2-[4-аминометил-(2,4-диметоксифенил)] феноксилуксусная кислота (AM) [Rink H., 1987, Tetrahedron Lett. 28, 3787-3790], Wang [Wang S.S., J. Am. Chem. Soc, 1973, 95, 1328-1333] и им подобные, которые способны отщеплять полузащищенный пептид и образовывать цикл в растворе на стадии снятия защиты в растворе или даже твердофазной циклизации и последующим одновременным снятием защиты и отщеплением пептида.
Фармацевтические композиции
Соединения по настоящему изобретению можно вводить любым способом, который приводит к контакту соединений и их участка приложения действия в организме млекопитающего, предпочтительно у человека, и в виде композиции, которая их содержит.
В связи с этим еще одним аспектом настоящего изобретения является фармацевтическая композиция, которая содержит фармацевтически эффективное количество по меньшей мере одного соединения с общей формулой (I), его стереоизомеры, их смеси и/или его фармацевтически приемлемые соли. Фармацевтическая композиция по настоящему изобретению может включать соединение с общей формулой (I), его стереоизомеры, их смеси и/или его твердые фармацевтически приемлемые соли, полученные путем сушки вымораживанием или сушки распылением, и может быть восстановлена в растворителе, подходящем для ее введения.
Фармацевтическая композиция по настоящему изобретению может содержать по меньшей мере один фармацевтически приемлемый наполнитель. Количество и природа фармацевтически приемлемых наполнителей зависит от желаемого способа введения. Фармацевтически приемлемые наполнители хорошо известны экспертам в данной области [Rowe R.C., Sheskey P.J., Quinn, М.Е. (2009) "Handbook of Pharmaceutical Excipients, 6th Edition", Pharmaceutical Press and American Pharmacists Association]. Указанные композиции можно получить с использованием стандартных способов, известных в данной области.
Соединения по настоящему изобретению характеризуются различной растворимостью в воде в соответствии с природой их аминокислотной последовательности или любых возможных модификаций на их N-конце и/или С-конце. Таким образом, соединения по настоящему изобретению можно вводить в состав композиции в виде водного раствора, а те, которые не растворимы в воде, можно растворять в фармацевтически приемлемых стандартных растворителях, таких как и без ограничения: этанол, пропанол, изопропанол, пропиленгликоль, глицерин, диметилсульфоксид, бутиленгликоль или полиэтиленгликоль или любая их комбинация.
Фармацевтически эффективное количество соединений по настоящему изобретению, подлежащее введению, и их дозировка будут зависеть от ряда факторов, в том числе возраста, состояния пациента, природы и тяжести нарушения или заболевания, подлежащего лечению или предупреждению, пути и частоты введения, а также от специфической природы соединений, которые будут использоваться.
"Фармацевтически эффективное количество" необходимо понимать как нетоксичное, но достаточное для обеспечения желаемого эффекта количество соединения по настоящему изобретению. Соединения по настоящему изобретению используют в фармацевтической композиции по настоящему изобретению в фармацевтически эффективных для достижения желаемого эффекта концентрациях; в их предпочтительной форме эффективная суточная доза в организме человека составляет от 0,1 мг до 1000 мг/день, предпочтительно от 0,5 до 100 мг/день и еще более предпочтительно от 1 до 10 мг/день.
Частота введения фармацевтической композиции может быть, например и без ограничения: ежемесячной, раз в две недели, еженедельной, два раза в неделю, три раза в неделю или ежедневной.
Соединения по настоящему изобретению, их стереоизомеры, их смеси и/или их косметически или фармацевтически приемлемые соли также можно включать в системы доставки и/или фармацевтические системы с замедленным высвобождением.
Термин "системы доставки" относится к разбавителю, вспомогательному веществу, наполнителю или носителю, с которыми вводится пептид по настоящему изобретению. Эти фармацевтические носители могут быть жидкостями, такими как вода, масла или поверхностно активные вещества, в том числе получаемые из нефти, животного, растительного или синтетического происхождения, например и без ограничения: арахисовое масло, соевое масло, минеральное масло, кунжутное масло, касторовое масло, полисорбаты, сложные эфиры сорбита, сульфаты эфиров, сульфаты, бетаины, гликозиды, мальтозиды, жирные спирты, ноноксинолы, полоксамеры, полиоксиэтилены, полиэтиленгликоли, декстроза, глицерин, дигитонин и подобные. Специалист в данной области имеет представление о разбавителях, вспомогательных веществах или наполнителях, которые можно использовать в различных системах доставки, в которых можно вводить соединения по настоящему изобретению.
Термин "замедленное высвобождение" используется в обычном смысле, означая систему доставки соединения, которая обеспечивает постепенное высвобождение указанного соединения на протяжении периода времени и предпочтительно, хотя и не обязательно, при относительно постоянных уровнях высвобождения соединения в течение периода времени.
Примеры систем доставки или замедленного высвобождения включают без ограничения липосомы, смешанные липосомы, олеосомы, ниосомы, этосомы, милличастицы, микрочастицы, наночастицы и твердые липидные наночастицы, наноструктурированные липидные носители, губки, циклодекстрины, везикулы, мицеллы, смешанные мицеллы на основе поверхностно-активных веществ, смешанные мицеллы на основе поверхностно-активных веществ-фосфолипидов, миллисферы, микросферы и наносферы, липосферы, милликапсулы, микрокапсулы и нанокапсулы, а также микроэмульсии и наноэмульсии, которые можно добавлять для достижения большей биодоступности активного компонента и/или улучшения его фармакокинетических и фармакодинамических свойств.
Фармацевтические композиции соединений по настоящему изобретению, их стереоизомеров, их смесей и/или их фармацевтически приемлемых солей можно вводить любым подходящим путем, для которого будут включены фармацевтически приемлемые наполнители, необходимые для составления желаемой формы введения, путем местного или системного применения, например и без ограничения: наружный, энтеральный или парентеральный путь. В контексте настоящего изобретения термин "наружный" путь включает кожный и глазной пути, термин "энтеральный" путь включает введение в систему пищеварения, такое как пероральный, трансбуккальный, гастральный, сублингвальный и ректальный пути, а термин "парентеральный" относится к назальному, ушному, глазному, вагинальному путям, подкожным инъекциям, внутрикожному, внутрисосудистому, например, внутривенному, внутримышечному, внутриглазному, интраспинальному, внутричерепному, внутрисуставному, интратекальному и интраперитонеальному путям, а также любой другой подобной методике введения или инфузии. Также рассматривается обработка in vitro, например, в культурах поврежденных клеток и/или стволовых клетках, и обработка ex vivo.
Более конкретно, лечение, предупреждение и/или диагностику с использованием соединений и композиций по настоящему изобретению проводят in vivo, поскольку предпочтительным путем введения является подкожный.
В более конкретном аспекте фармацевтические композиции по настоящему изобретению содержат другие терапевтические средства, например и без ограничения: другие противовоспалительные средства, иммунодепрессивные средства, или метаболические ингибиторы, или ингибиторы ферментов, нестероидные противовоспалительные средства (NSAID), такие как ибупрофен, тенидап, напроксен, мелоксикам, месалазин, пироксикам, диклофенак, индометацин и сульфасалазин, кортикостероиды, такие как преднизолон, гидрокортизон, беклометазон, будесонид; противовоспалительные лекарственные средства, подавляющие цитокины (CSAID), ингибиторы синтеза нуклеотидов, такие как метотрексат и лефлуномид, иммуносупрессоры, такие как циклоспорин, такролимус (FK-506), ингибиторы mTOR, такие как сиролимус (рапамицин) или производные рапамицина, средства-ингибиторы фактора некроза опухолей (TNFα), такие как инфликсимаб, адалимумаб, этанерцепт, цертолизумаб, голимумаб, ингибиторы СОХ-2, такие как целекоксиб, рофекоксиб, вальдекоксиб и их варианты, ингибиторы фосфодиэстеразы, ингибиторы фосфолипазы, такие как аналоги трифторметилкетона, ингибиторы фактора роста эндотелия сосудов или ингибиторы рецепторов фактора роста, ингибиторы ангиогенеза, натализумаб (антитело к интегрину альфа-4), ритуксимаб (антитело к CD20), абатацепт (антитело к CD80 и CD86), фостаматиниб (ингибитор тирозинкиназы селезенки Syk), тоцилизумаб (антитело к IL-6), анакинра (антитело к IL-1), тофацитиниб (ингибитор Janus-киназы), 6-меркаптопурины (6-МР), азатиоприн, балсалазид, сульфасалазин, месалазин, ольсалазин, хлорохин, гидроксихлорохин, пеницилламин, ауранофин, ауротиомалат, азатиоприн, колхицин, агонисты бета-2 адренергических рецепторов, такие как сальбутамол, тербуталин и сальметерол, ксантины, такие как теофиллин и аминофиллин, кромогликат, недокромил, кетотифен, ипратропий, окситропий, микофенолат мофетил, агонисты аденозина, антитромботические средства, пенициллин, ингибиторы комплемента и адренергические средства.
Применения
В связи с другим аспектом настоящее изобретение относится к соединению с общей формулой (I), его стереоизомерам, их смесям и/или его фармацевтически приемлемым солям для применения в медицине.
Другой аспект настоящего изобретения относится к соединению с общей формулой (I), его стереоизомерам, их смесям и/или его фармацевтически приемлемым солям для лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, при которых экспрессируются рецепторы соматостатина sstr1, sstr2, sstr3, sstr4 и/или sstr5, и/или рецептор грелина, и/или специфический рецептор кортистатина, или их комбинации.
В более конкретном аспекте данное изобретение относится к соединению с общей формулой (I), его стереоизомерам, их смесям и/или его фармацевтически приемлемым солям для лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, которые выбраны из группы, состоящей из заболеваний иммунной системы, воспалительных патологий, опухолей, рака, нейродегенеративных заболеваний, глазных болезней, респираторных нарушений, инфекций, боли, заживления ран, регенерации тканей, септических процессов и нарушений, связанных с имплантами/трансплантатами органов или тканей.
В дополнительном конкретном аспекте настоящее изобретение относится к соединению с общей формулой (I), его стереоизомерам, их смесям и/или его фармацевтически приемлемым солям для лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, которые выбраны из группы, состоящей из эндотоксемии, септического шока, синдрома токсического шока, сепсиса, воспалительного заболевания кишечника, болезни Крона, хронического колита, язвенного колита, аутоиммунного гастрита, ревматоидного артрита, остеоартрита, рассеянного склероза, диареи, диареи 3-4 степени, диареи, связанной с лучевой терапией и/или химиотерапией, симптоматического лечения карциноидного синдрома или випомы, эндокринного рака, рака поджелудочной железы, хронического панкреатита, акромегалии, симптоматического лечения гастроэнтеропанкреатических нейроэндокринных опухолей, варикозного расширения вен пищевода, гипертрофической легочной остеоартропатии и тиреотропной аденомы, колоректального рака, рака молочной железы, рака яичников, рака предстательной железы, рака щитовидной железы, рака легкого, рака желудка, гепатоцеллюлярной карциномы, болезни Альцгеймера, артрита, аллергий, волчанки, красной волчанки, лимфопролиферативного расстройства, диабетической ретинопатии, макулярного отека, эндокринологической офтальмопатии, синдрома Кушинга, нейропатической боли, рестеноза, ангиогенеза, гипертиреоидизма, гипотиреоза, гиперинсулинемии, гипокальциемии, болезни Паджета, кахексии и синдрома Золлингера-Эллисона, гангренозной пиодермии, тиреопатии, инсулинзависимого сахарного диабета 1 типа, тиреоидита Хашимото, болезни Грейвса, аутоиммунного гепатита, аллергического энцефаломиелита, увеоретинита, увеита, отторжения имплантата, отторжения трансплантата, реакции трансплантат против хозяина, эндокардита Либмана-Сакса, смешанного заболевания соединительной ткани, склеродермии, дерматополимиозита, гранулематоза Вегенера, синдрома Шегрена, гранулемы, склерозирующего лишая, первичного билиарного цирроза печени, кератита, гломерулонефрита, реактивного артрита, синовиалита, синдрома Рейтера, болезни Лайма, псориатического артрита, индуцированного артрита, анкилозирующего спондилита, тяжелой миастении, васкулита, аллергий, дерматита или экземы, псориаза, фиброзного дерматита, хронического обструктивного заболевания легких (COPD), энцефаломиелита, аутоиммунного тиреоидита, язвы в пожилом возрасте, ирита, конъюнктивита, кератоконъюнктивита, спондилоартропатии, вагинита, проктита, медикаментозного дерматита, обратимых лепрозных реакций, лепрозной эритемы, острой некротической геморрагической энцефалопатии, идиопатической прогрессирующей двусторонней нейросенсорной тугоухости, апластической анемии, эритроцитарной анемии, идиопатической тромбоцитопении, полихондрита, хронического активного гепатита, синдрома Стивенса-Джонсона, идиопатического спру, красного плоского лишая и саркоидоза.
Другой вариант осуществления настоящего изобретения относится к применению соединения с общей формулой (I), его стереоизомеров, их смесей и/или его фармацевтически приемлемых солей в получении фармацевтической композиции для лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, при которых экспрессируются рецепторы соматостатина sstr1, sstr2, sstr3, sstr4 и/или sstr5, и/или рецептор грелина, и/или специфический рецептор кортистатина, или их комбинации.
В более конкретном аспекте данное изобретение относится к применению соединения с общей формулой (I), его стереоизомеров, их смесей и/или его фармацевтически приемлемых солей в получении фармацевтической композиции для лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, которые выбраны из группы, состоящей из заболеваний иммунной системы, воспалительных нарушений, опухолей, рака, нейродегенеративных заболеваний, глазных болезней, респираторных заболеваний, инфекций, боли, заживления ран, регенерации тканей, септических процессов и нарушений, связанных с имплантами/трансплантатами органов или тканей.
В более конкретном аспекте настоящее изобретение относится применению соединения с общей формулой (I), его стереоизомеров, их смесей и/или его фармацевтически приемлемых солей в получении фармацевтической композиции для лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, которые выбраны из группы, состоящей из эндотоксемии, септического шока, синдрома токсического шока, сепсиса, воспалительного заболевания кишечника, болезни Крона, хронического колита, язвенного колита, аутоиммунного гастрита, ревматоидного артрита, остеоартрита, рассеянного склероза, диареи, диареи 3-4 степени, диареи, связанной с лучевой терапией и/или химиотерапией, симптоматического лечения карциноидного синдрома или випомы, эндокринного рака, рака поджелудочной железы, хронического панкреатита, акромегалии, симптоматического лечения гастроэнтеропанкреатических нейроэндокринных опухолей, варикозного расширения вен пищевода, гипертрофической легочной остеоартропатии и тиреотропной аденомы, колоректального рака, рака молочной железы, рака яичников, рака предстательной железы, рака щитовидной железы, рака легкого, рака желудка, гепатоцеллюлярной карциномы, болезни Альцгеймера, артрита, аллергий, волчанки, красной волчанки, лимфопролиферативного расстройства, диабетической ретинопатии, макулярного отека, эндокринологической офтальмопатии, синдрома Кушинга, нейропатической боли, рестеноза, ангиогенеза, гипертиреоидизма, гипотиреоза, гиперинсулинемии, гиперкальциемии, болезни Паджета, кахексии и синдрома Золлингера-Эллисона, гангренозной пиодермии, тиреопатии, инсулинзависимого сахарного диабета 1 типа, тиреоидита Хашимото, болезни Грейвса, аутоиммунного гепатита, аллергического энцефаломиелита, увеоретинита, увеита, отторжения имплантата, отторжения трансплантата, реакции трансплантат против хозяина, эндокардита Либмана-Сакса, смешанного заболевания соединительной ткани, склеродермии, дерматополимиозита, гранулематоза Вегенера, синдрома Шегрена, гранулемы, склерозирующего лишая, первичного билиарного цирроза печени, кератита, гломерулонефрита, реактивного артрита, синовиалита, синдрома Рейтера, болезни Лайма, псориатического артрита, индуцированного артрита, анкилозирующего спондилита, тяжелой миастении, васкулита, аллергий, дерматита или экземы, псориаза, фиброзного дерматита, хронического обструктивного заболевания легких (COPD), энцефаломиелита, аутоиммунного тиреоидита, язвы в пожилом возрасте, ирита, конъюнктивита, кератоконъюнктивита, спондилоартропатии, вагинита, проктита, медикаментозного дерматита, обратимых лепрозных реакций, лепрозной эритемы, острой некротической геморрагической энцефалопатии, идиопатической прогрессирующей двусторонней нейросенсорной тугоухости, апластической анемии, эритроцитарной анемии, идиопатической тромбоцитопении, полихондрита, хронического активного гепатита, синдрома Стивенса-Джонсона, идиопатического спру, красного плоского лишая и саркоидоза.
В дополнительном аспекте настоящее изобретение относится к способу лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, при которых экспрессируются рецепторы соматостатина sstr1, sstr2, sstr3, sstr4 и/или sstr5, и/или рецептор грелина, и/или специфический рецептор кортистатина, или их комбинации, включающему введение фармацевтически эффективного количества по меньшей мере одного соединения с общей формулой (I), его стереоизомеров, их смесей и/или его фармацевтически приемлемых солей.
В другом конкретном аспекте данное изобретение относится к способу лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, которые выбраны из группы, состоящей из заболеваний иммунной системы, воспалительных нарушений, опухолей, рака, нейродегенеративных заболеваний, глазных болезней, респираторных заболеваний, инфекций, боли, заживления ран, регенерации тканей, септических процессов и нарушений, связанных с имплантами/трансплантатами органов или тканей, включающему введение фармацевтически эффективного количества по меньшей мере одного соединения с общей формулой (I), его стереоизомеров, их смесей и/или его фармацевтически приемлемых солей.
В другом конкретном аспекте данное изобретение относится к способу лечения, предупреждения и/или диагностики тех состояний, нарушений и/или патологий, которые выбраны из группы, состоящей из эндотоксемии, септического шока, синдрома токсического шока, сепсиса, воспалительного заболевания кишечника, болезни Крона, хронического колита, язвенного колита, аутоиммунного гастрита, ревматоидного артрита, остеоартрита, рассеянного склероза, диареи, диареи 3-4 степени, диареи, связанной с лучевой терапией и/или химиотерапией, симптоматического лечения карциноидного синдрома или випомы, эндокринного рака, рака поджелудочной железы, хронического панкреатита, акромегалии, симптоматического лечения гастроэнтеропанкреатических нейроэндокринных опухолей, варикозного расширения вен пищевода, гипертрофической легочной остеоартропатии и тиреотропной аденомы, колоректального рака, рака молочной железы, рака яичников, рака предстательной железы, рака щитовидной железы, рака легкого, рака желудка, гепатоцеллюлярной карциномы, болезни Альцгеймера, артрита, аллергий, волчанки, красной волчанки, лимфопролиферативного расстройства, диабетической ретинопатии, макулярного отека, эндокринологической офтальмопатии, синдрома Кушинга, нейропатической боли, рестеноза, ангиогенеза, гипертиреоидизма, гипотиреоза, гиперинсулинемии, гиперкальциемии, болезни Паджета, кахексии и синдрома Золлингера-Эллисона, гангренозной пиодермии, тиреопатии, инсулинзависимого сахарного диабета 1 типа, тиреоидита Хашимото, болезни Грейвса, аутоиммунного гепатита, аллергического энцефаломиелита, увеоретинита, увеита, отторжения имплантата, отторжения трансплантата, реакции трансплантат против хозяина, эндокардита Либмана-Сакса, смешанного заболевания соединительной ткани, склеродермии, дерматополимиозита, гранулематоза Вегенера, синдрома Шегрена, гранулемы, склерозирующего лишая, первичного билиарного цирроза печени, кератита, гломерулонефрита, реактивного артрита, синовиалита, синдрома Рейтера, болезни Лайма, псориатического артрита, индуцированного артрита, анкилозирующего спондилита, тяжелой миастении, васкулита, аллергий, дерматита или экземы, псориаза, фиброзного дерматита, хронического обструктивного заболевания легких (COPD), энцефаломиелита, аутоиммунного тиреоидита, язвы в пожилом возрасте, ирита, конъюнктивита, кератоконъюнктивита, спондилоартропатии, вагинита, проктита, медикаментозного дерматита, обратимых лепрозных реакций, лепрозной эритемы, острой некротической геморрагической энцефалопатии, идиопатической прогрессирующей двусторонней нейросенсорной тугоухости, апластической анемии, эритроцитарной анемии, идиопатической тромбоцитопении, полихондрита, хронического активного гепатита, синдрома Стивенса-Джонсона, идиопатического спру, красного включающему введение фармацевтически эффективного количества по меньшей мере одного соединения с общей формулой (I), его стереоизомеров, их смесей и/или его фармацевтически приемлемых солей.
ПРИМЕРЫ
Следующие конкретные примеры, представленные в данном патентном документе, служат для иллюстрации природы настоящего изобретения. Данные примеры включены лишь в иллюстративных целях и не должны интерпретироваться как ограничивающие изобретение, заявленное в данном документе.
Сокращения
Сокращения, используемые в настоящем описании, имеют следующие значения:
Ac2O, уксусный ангидрид; АсОН, уксусная кислота; Adpoc, 1-(1-адамантил)-1-метилэтокси-карбонил; All, аллил; Alloc, аллилоксикарбонил; Boc, трет-бутилоксикарбонил; Bzl, бензил; Cbz, бензилоксикарбонил; cHx, циклогексил; CIZ, 2-хлорбензил; CST, кортистатин; DCM, дихлорметан; Dde, N-[1-(4,4-диметил-2,6-диоксоциклогекс-1-илиден)этил]; DMEM, модифицированная Дульбекко среда Игла; Dfp, 3,5-дифторфенилаланин; DIEA, N,N'-диизопропилэтиламин; DIPCDI, диизопропилкарбодиимид; Dmab, 4-(N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино)бензил; DMF, N,N-диметилформамид; Dnp, 2,4-динитрофенил; DOTA, 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусная кислота; DTPA, диэтилентриаминпентауксусная кислота; ESI-MS, масс-спектрометрия с ионизацией электрораспылением; Fm, флуоренилметил; Fmoc, 9-флуоренилметилоксикарбонил; HF, фтористоводородная кислота; НОВТ, N-гидроксибензотриазол; HPLC, высокоэффективная жидкостная хроматография; IC50, полумаксимальная ингибирующая концентрация; ivDde, 1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метил-бутил; Ki, константа ингибирования для лекарственного средства; LPS, липополисахарид; М, молекулярная масса; Mtt, метокситритил; мкл, микролитр; мкмоль, микромоль; pNZ, п-нитробензилоксикарбонил; RP-HPLC, обращенно-фазовая HPLC; SST, соматостатин; sstry рецепторы соматостатина; tBu, трет-бутил; Теос, 2-(триметилсилил)этилоксикарбонил; TFA, трифторуксусная кислота; TFE, 2,2,2-трифторэтанол; TIS, триизопропилсилан; tr, время удерживания; Trt, тритил; Troc, 2,2,2-трихлорэтилоксикарбонил; Z, бензилоксикарбонил.
ПРИМЕР 1. Синтез соединения 1
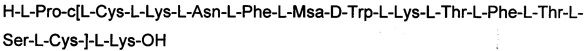
Смолу помещали в реактор для синтеза, оснащенный фильтровальной пластиной и ключом. Встраивание С-концевого остатка проводили на 0,25 г 2-хлортритиловой смолы (1,6 ммоля/г). Первую аминокислоту Fmoc-Lys(Boc)-OH (1 экв.) растворяли в 1,25 мл DCM и 75 мкл DMF. Добавляли DIEA (3 экв.). Раствор с аминокислотой и основанием переносили в реактор и перемешивали в течение 45 минут. После этого добавляли 0,2 мл МеОН и оставляли для прохождения реакции в течение 10 минут. Отфильтровывали и фильтрат удаляли. Смолу промывали с помощью DCM и DMF. При каждой промывке фильтраты отфильтровали и удаляли. Для встраивания следующих аминокислот использовали 2,5 экв. Fmoc-аминокислоты, 2,5 экв. НОВТ и 2,5 экв. DIPCDI. Для реакции сочетания их оставляли для прохождения реакции в течение 40-60 минут, и встраивание аминокислоты контролировали с помощью нингидринового теста. Если нингидриновый тест был положительным, стадию реактивации выполняли в течение 15-30 минут с 0,83 экв. НОВТ и 0,83 экв. DIPCDI. Если нингидриновый тест по-прежнему оставался положительным, выполняли повторное сочетание с 1,25 экв. Fmoc-аминокислоты, НОВТ и DIPCDI. Если нингидриновый тест был отрицательным, синтез проводили со стадией снятия защитной группы Fmoc путем двукратной обработки раствором 20% пиперидина в DMF. Пептидил-смолу промывали 5 раз с помощью DMF, отфильтровывая и удаляя каждый раз фильтры, и затем встраивали следующую аминокислоту. N-концевую аминокислоту встраивали в форме Boc-Pro-ОН. Получали 1,03 г пептид-смолы.
1,03 г (0,3 ммоля) пептидил-смолы помещали в реактор. 9,6 мл раствора AcOH: TFE: DCM добавляли при перемешивании магнитной мешалкой и оставляли для прохождения реакции в течение 2 часов. Его фильтровали в реакторе с фильтровальной пластиной и извлекали фильтрат. Смолу промывали 3 раза с помощью 2,55 мл раствора AcOH: TFE: DCM, и извлекали фильтраты.
Готовили раствор 0,73 г (10 экв.) йода в 3,57 мл раствора AcOH : TFE : DCM. Фильтраты, извлеченные при кислотном гидролизе, переносили в реактор, который содержал раствор йода, и оставляли для прохождения реакции при перемешивании. Готовили раствор 1,52 г (22 экв.) тиосульфата натрия в 6,12 мл воды и добавляли в реактор сразу после завершения окисления, и полное обесцвечивание наблюдали через 5 минут. Перемешивание прекращали и смесь оставляли для остаивания до разделения фаз. Экстрагирование выполняли путем обработки водной фазы с помощью DCM 3 раза и органической фазы с помощью 5% лимонной кислоты : NaCl (объем : вес). Органические фракции выпаривали и осадок сушили в вакууме. Твердый осадок промывали водой в фильтровальной пластине. Получали 0,73 г защищенного окисленного продукта.
6,8 мл реакционной смеси TFA : H2O : DCM : анизол (55:5:30:10) добавляли в реактор. 0,73 г окисленного и защищенного пептида добавляли к предыдущему раствору и оставляли на 4 часа для прохождения реакции. Добавляли гептан (13 мл) и перемешивали в течение 5 минут. Перемешивание прекращали и оставляли для отстаивания. Водную фазу выливали в холодный эфир и оставляли на 15-30 минут. Полученную суспензию фильтровали через фильтровальную пластину и фильтраты удаляли. Осадок промывали эфиром, удаляя фильтраты при каждом промывании. Твердое вещество подвергали сушке вымораживанием и получали 0,56 г неочищенного продукта.
Неочищенный продукт очищали в полупрепаративной системе, оснащенной колонкой NW50, заполненной диоксидом кремния Кромасил с размером частиц 10 микрометров. Пептид суспендировали в 0,1 н. АсОН и добавляли смолу DOWEX, подготовленную в 0,1 н. АсОН. Конечное соединение ацетата извлекали посредством фильтрования и характеризовали с помощью масс-спектрометрии на устройстве для ESI-MS.
Определение характеристик: ESI-MS: теоретическая М=1777,1 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=889,3, [М+3Н]+/3=593,1.
ПРИМЕР 2. Синтез соединения 2
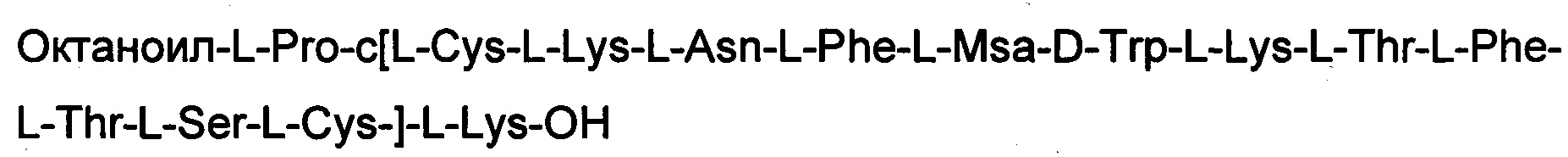
Соединение получали с помощью способа, описанного в Примере 1. В начале использовали 0,25 г смолы и использовали такие же эквивалентные соотношения. N-концевую аминокислоту добавляли в форме Fmoc-Pro-OH. Октаноильную кислоту встраивали в последовательность с использованием 5 экв. кислоты, 5 экв. НОВТ и 5 экв. DIPCDI. Получили 0,5 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1903,35 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=952,4, [М+3Н]+/2=635,2.
ПРИМЕР 3. Синтез соединения 3
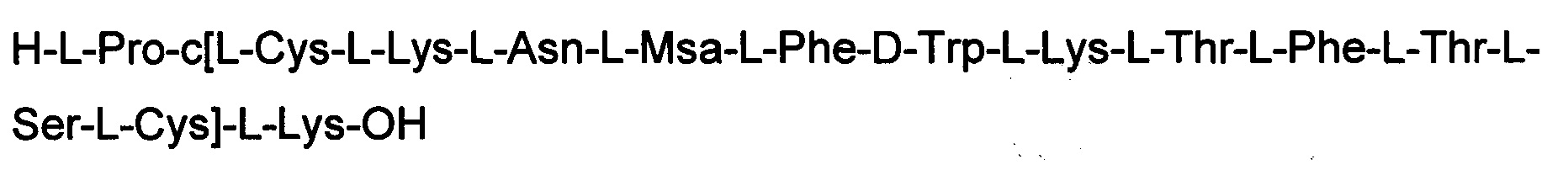
Соединение получали с помощью способа, описанного в Примере 1. Использовали 0,25 г смолы и такие же эквивалентные соотношения. N-концевую аминокислоту добавляли в форме Boc-Pro-ОН. Получали 0,53 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1777,15 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=889,3 [М+3Н]+/2=593,1.
ПРИМЕР 4. Синтез соединения 4
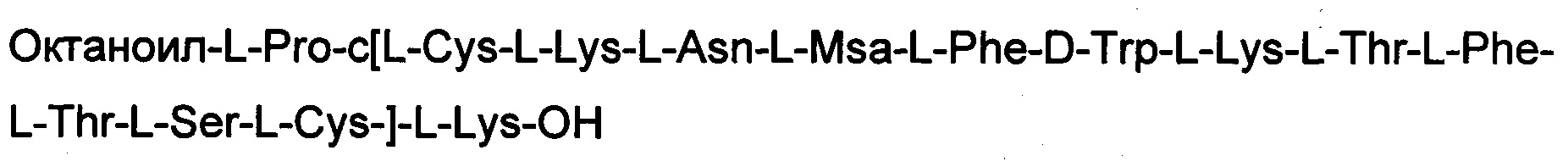
Соединение получили с помощью способа, описанного в Примере 1. Использовали 0,25 г смолы и такие же эквивалентные соотношения. N-концевую аминокислоту добавляли в форме Fmoc-Pro-OH. Октаноильную кислоту встраивали в последовательность с использованием 5 экв. кислоты, 5 экв. НОВТ и 5 экв. DIPCDI. Получали 0,55 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1903,35 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=952,4, [М+3Н]+/2=635,2.
ПРИМЕР 5. Синтез соединения 5
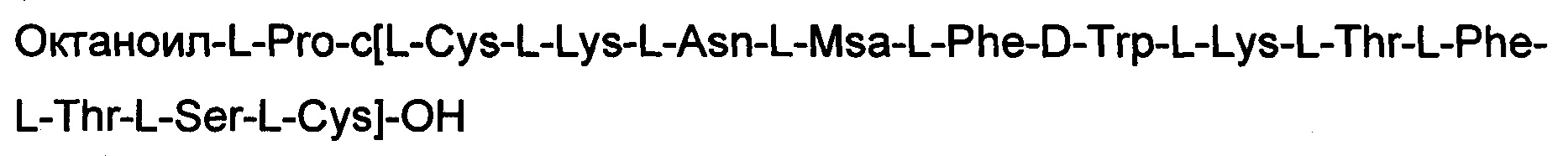
Соединение получили с помощью способа, описанного в Примере 1. Использовали 0,4 г смолы и такие же эквивалентные соотношения. N-концевую аминокислоту добавляли в форме Fmoc-Pro-OH. Октаноильную кислоту встраивали в последовательность с использованием 5 экв. кислоты, 5 экв. НОВТ и 5 экв. DIPCDI. Получали 0,53 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1775,18 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=888,6, [М+3Н]+/2=592,7.
ПРИМЕР 6. Синтез соединения 6
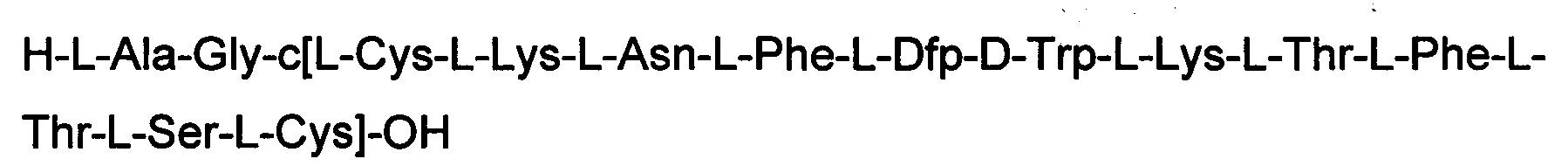
Соединение получили с помощью способа, описанного в Примере 1, и таких же эквивалентных соотношений. N-концевую аминокислоту добавляли в форме Boc-Ala-OH. Получали 0,53 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1673,9 г/моль, экспериментальная М: (масса/заряд): [М+Н]+=1674,8; [М+2Н]+/2=837,9.
ПРИМЕР 7. Синтез 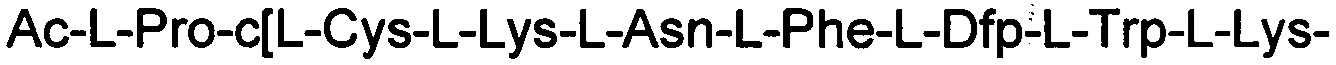
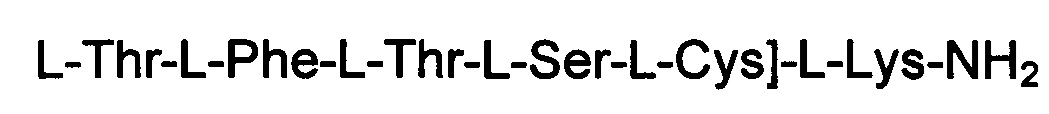 (соединение 7)
(соединение 7)
Соединение получали с помощью способа, описанного в Примере 1, с использованием смолы МВНА и таких же эквивалентных соотношений. N-концевую аминокислоту добавляли в форме Fmoc-Pro-OH. Ацетилирование проводили в твердой фазе с использованием 5 экв. уксусного ангидрида и 10 DIEA. Получали 0,48 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1812,15 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=907,07, [М+3Н]+/3=605,05.
ПРИМЕР 8. Синтез 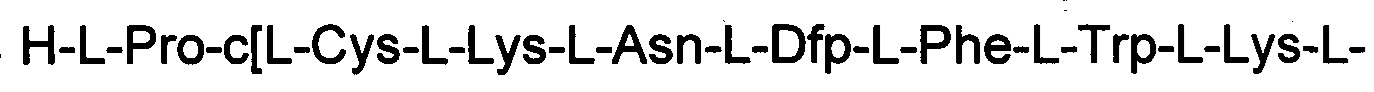
 (соединение 8)
(соединение 8)
Соединение получали с помощью способа, описанного в Примере 1, с использованием смолы МВНА и таких же эквивалентных соотношений. N-концевую аминокислоту добавляли в форме Boc-Pro-ОН. Получали 0,5 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=1798,15 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=900,1, [М+3Н]+/3=600,3.
ПРИМЕР 9. Синтез 
 (соединение 9)
(соединение 9)
Соединение получали с помощью способа, описанного в Примере 1, и таких же эквивалентных соотношений. N-концевую аминокислоту добавляли в форме Fmoc-Asp(OtBu)-OH. После снятия защиты Fmoc и заключительного кислотного гидролиза получали 0,54 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=2207,54 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=1104,7; [М+3Н]+/3=736,8.
ПРИМЕР 10. Синтез 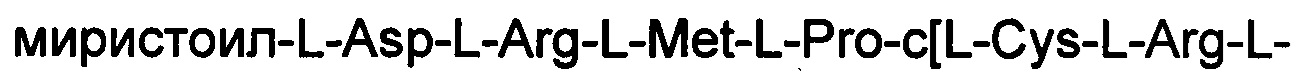
 (соединение 10)
(соединение 10)
Соединение получали с помощью способа, описанного в Примере 1, и таких же эквивалентных соотношений. N-концевую аминокислоту добавляли в форме Fmoc-Asp(OtBu)-OH. Миристиновую кислоту встраивали с использованием 5 экв. кислоты, 5 экв. НОВТ и 5 экв. DIPCDI. Получали 0,52 г неочищенного продукта.
Определение характеристик: ESI-MS: теоретическая М=2411,5 г/моль, экспериментальная М: (масса/заряд): [М+2Н]+/2=1206,8; [М+3Н]+/3=804,8.
ПРИМЕР 11. Значения связывания новых аналогов кортистатина с рецепторами соматостатина (sstr1-sstr5)
Использовали клетки СНО-K1, в которых независимо экспрессировался каждый из 5 рецепторов соматостатина (sstr1-sstr5). Клетки инкубировали в буфере HEPES, рН 7,4, с новыми аналогами кортистатина (соединения 1-10) в диапазоне концентраций от 0,1 нМ до 10 мкМ в течение 2-4 часов и 125I-Tyr11-соматостатин-14 использовали в качестве радиоактивного лиганда, а соматостатин-14 в качестве немеченного лиганда. Радиоактивность, полученную в отсутствие соматостатина-14, рассматривали как общее связывание, а полученную в присутствии 1 мкМ соматостатина-14, рассматривали как неспецифическую связь. Специфическое связывание рассматривали как разница между полным связыванием и неспецифическим связыванием. В тестируемом диапазоне концентраций от 0,1 нМ до 10 мкМ исследованные новые аналоги кортистатина давали значения процента ингибирования специфического связывания, которые составляли более 50%. Указанные значения коррелировали со значениями IC50 для новых аналогов кортистатина в пределах следующих диапазонов, все из которых находятся в наномолярном диапазоне (IC50 (sstr1)=1 нМ - 50 нМ; IC50 (sstr2)=1 нМ -50 нМ; IC50 (sstr3, sstr4 и sstr5)=0,5 нМ - 5 нМ), диапазоне значений, опубликованных для кортистатина [Spier et al., Brain Research Reviews 2000, 33, 228-241] IC50 (sstr1)=1-5 нМ; IC50 (sstr2, sstr3, sstr5)=0,1 нМ - 5 нМ; IC50 (sstr4)=0,1 нМ - 20 нМ). Результаты указывают, что исследованные новые аналоги кортистатина взаимодействуют с рецепторами соматостатина sstr1-sstr 5 при наномолярной аффинности.
ПРИМЕР 12. Влияние новых аналогов кортистатина на воспалительную реакцию in vitro
Клетки Raw 264 культивировали в полной среде DMEM до достижения 80% конфлюентности монослоя. Клетки инкубировали в отсутствии или в присутствии липополисахарида (LPS, 1 мкг/мл, из Е. coli серотипа 055:В5). Клетки, инкубируемые в отсутствие липополисахарида, использовали в качестве эталона (исходный уровень). Новые аналоги кортистатина добавляли при концентрации 100 нМ в начале культивирования. Через 24 часа собирали надосадочные жидкости и измеряли уровни цитокинов и оксида азота. Уровень цитокинов (TNFальфа и IL-6) устанавливали с помощью анализа ELISA. Количество оксида азота (NO) устанавливали с помощью теста Грисса. Равные объемы надосадочных жидкостей из культуры (90 мкл) и реагентов Грисса смешивали и измеряли оптическую плотность при 550 нм. Количество нитрита рассчитывали относительно стандартной кривой NaNO2.
Для сравнения оценивали значения для кортистатина-14 (CST-14), соматостатина-14 (SOM-14) и трех аналогов соматостатина, описанных в WO 2010/128098 А1:

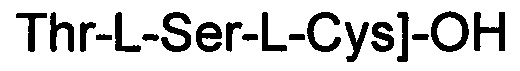 (соединение 11),
(соединение 11),

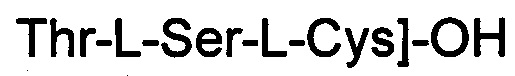 (соединение 12),
(соединение 12),
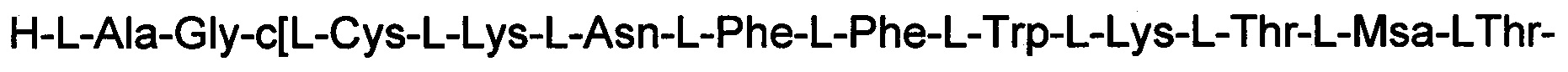
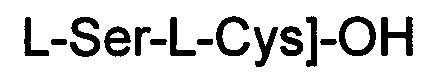 (соединение 13).
(соединение 13).
В качестве максимального измерения воспаления, полученные активированные значения TNFальфа, IL-6 и NO составляли 5,78 нг/мл; 4,48 нг/мл и 5,23 нг/мл, соответственно. Соответствующие значения исходного уровня в отсутствие воспаления составляли 0,48 нг/мл; 0 нг/мл и 0,44 нг/мл. Значения TNFальфа, IL-6 и NO, полученные после обработки нативным пептидом CST-14 составляли 3,17 нг/мл; 3,78 нг/мл и 3,08 нг/мл, соответственно, все из которых ниже максимально измеренных значений воспаления, следовательно, CST-14 показывал противовоспалительную эффективность. Значения, полученные для SST-14, составляли 5,21 нг/мл; 4,2 нг/мл и 4,32 нг/мл, и значения, полученные для исследованных аналогов соматостатина (соединения 11-13), установили в диапазонах 5,18-5,24 нг/мл; 4,19-4,45 нг/мл и 3,4-5,05 нг/мл, соответственно.
Значения, полученные после обработки новыми аналогами кортистатина, установили в диапазонах 2,51-4,9 нг/мл; 2,77-4,23 нг/мл и 3,04-4,49 нг/мл, соответственно, для TNFальфа, IL-6 и NO, демонстрируя большую эффективность указанных методик обработки для уменьшения воспаления in vitro, чем у тестированных аналогов соматостатина (соединения 11-13).
Данные, полученные для CST-14 и SST-14, указывают, что обе молекулы и их аналоги являются эффективными в уменьшении воспаления in vitro.
ПРИМЕР 13. Влияние новых аналогов кортистатина на иммунный ответ in vitro
Спленоциты самцов 8-недельных мышей C57BI/6 получали после механической диссоциации клеток, фильтрации через нейлоновую сетку и лизиса эритроцитов. Спленоциты инкубировали в полной среде DMEM до достижения плотности 106 клеток/мл. Неприкрепившиеся клетки (образованные на 80% Т-клетками) использовали для измерения цитокинов и для анализов пролиферации. Т-клетки культивировали в полной среде DMEM и стимулировали антителами к CD3 (2 мкг/мл) в присутствии разных аналогов кортистатина в концентрации 100 нМ. Через 48 часов выделяли надосадочные жидкости культур и определяли уровни цитокинов (IFNγ и IL-2) с использованием анализа ELISA. Для определения влияния разных аналогов кортистатина на пролиферацию клетки культивировали в течение 72 часов и добавляли 0,5 мкКи (0,0185 МБк)/лунка [3Н]-тимидина на последние 8 часов культивирования, мембраны собирали и измеряли добавленный [3Н]-тимидин с использованием сцинтилляционного счетчика.
Для сравнения определяли значения кортистатина-14 (CST-14), соматостатина-14 (SOM-14) и трех аналогов соматостатина, описанных в WO 2010/128098 А1:
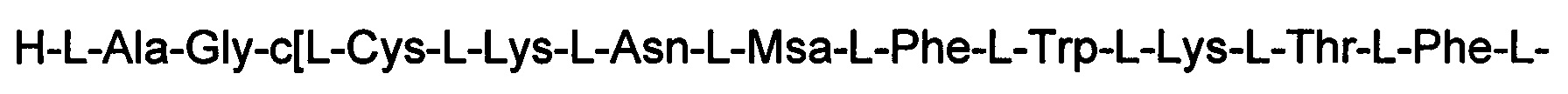
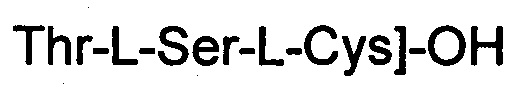 (соединение 11),
(соединение 11),

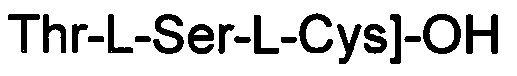 (соединение 12),
(соединение 12),
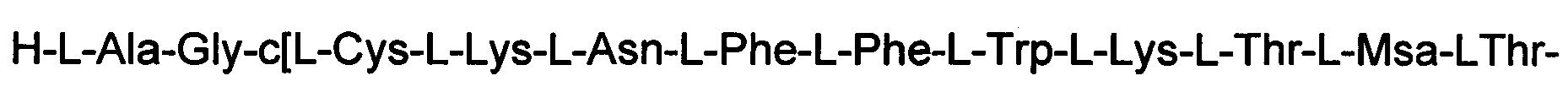
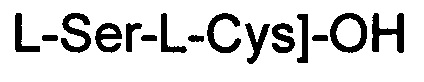 (соединение 13).
(соединение 13).
В качестве максимального показателя иммунного ответа полученные активированные значения пролиферации, INFγ и IL-2, составили 9843; 2,52 нг/мл и 3,22 нг/мл, соответственно. Соответствующие значения исходного уровня, полученные в отсутствие активации иммунного ответа, составили 640; 0 нг/мл и 0,23 нг/мл. Значения пролиферации, INFγ и IL-2, полученные после обработки нативным пептидом CST-14, составляли 5500; 1,13 нг/мл и 1,52 нг/мл, соответственно, все из которых ниже максимальных значений иммунного ответа, указывая на эффективность CST-14 относительно модулирования иммунного ответа. Значения, полученные для SST-14, составляли 9936; 2,31 нг/мл и 3,14 нг/мл, и значения для исследованных аналогов соматостатина (соединения 11-13) установили в диапазонах 10216-10466; 2,51-2,3 нг/мл и 3,23-3,37 нг/мл, соответственно.
Значения, полученные после обработки новыми аналогами кортистатина, обнаружены в диапазонах 5863-9316; 1,33-2,56 нг/мл и 1,8-3,23 нг/мл, соответственно для пролиферации, INFγ и IL-2, демонстрируя эффективность указанных методик обработки для уменьшения иммунного ответа in vitro.
Обработка активированных спленоцитов новыми аналогами кортистатина снижала уровни пролиферации, INFγ и/или IL-2, указывая на эффективность модулирования гиперактивного иммунного ответа.
Сравнительные данные, полученные для CST-14 и его новых аналогов и SST-14 и его аналогов, указывают, что CST-14 и аналоги оказывают большее влияние на уменьшение иммунного ответа in vitro.
ПРИМЕР 14. Стабильность в сыворотке крови новых аналогов кортистатина
Новые соединения инкубировали с 90% сывороткой крови человека при 37°С. Аликвоты экстрагировали в разные моменты времени инкубации. Добавляли метанол для осаждения белков из сыворотки крови, центрифугировали и надосадочную жидкость подвергали хроматографическому анализу с использованием RP-HPLC (градиент: 20-80% В за 30 мин, В=0,07%TFA в ацетонитриле). Исчезновение исходного продукта анализировали с использованием участка, соответствующего исходному продукту, и рассчитывали время полужизни.
Новые соединения имели более длительное время полужизни, чем у кортистатина. В данных экспериментальных условиях время полужизни кортистатина в сыворотке крови составляло 2 минуты. Время полужизни соединений 4, 6 и 5 составляло 21 минуту, 3,8 часа и 35 часов, соответственно. Для CST-14 основным метаболитом является соединение 14. Основным метаболитом соединения 4 является соединение 5. В обоих случаях исходный пептид утрачивает Lys с С-конца, что дает метаболит, который является стабильным в течение ряда часов.
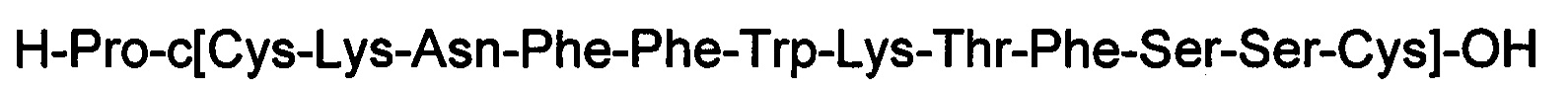 (соединение 14).
(соединение 14).
ПРИМЕР 15. Эффективность соединений 4, 5 и 6 в модели индуцированного коллагеном артрита (CIA) у мышей
Мышам DBA/1 вводили куриный коллаген II типа в полном адъюванте Фрейнда и М. tuberculosis в 0-й и 21-й дни. С 25-го по 29-й день мышам подкожно вводили физиологический раствор (контроль) или 0,4 мг/кг соединений 4, 5 или 6 один раз в день. В качестве контрольной обработки использовали антитело к TNFальфа и вводили внутривенно на 25-й и 32-й дни. Клиническую степень артрита анализировали ежедневно с использованием шкалы от 0 до 10 в соответствии с тяжестью повреждения. Опухание лапы оценивали путем измерения толщины с использованием штангенциркуля каждые пять дней на протяжении всего исследования (дни 20, 25, 30, 35, 40, 45, 50).
Обработка мышей с CIA соединениями 4, 5 и 6 показала существенное улучшение в снижении клинической степени артрита и уменьшении опухания лап, указывая на их эффективность в данной экспериментальной модели болезни с воспалительным и иммунным ответом. Клиническая степень артрита (0-10) у больных мышей, обработанных физиологическим раствором (контроль), достигала максимального значения 8,5 на 21-й день после второй инъекции коллагена. В тот же период обработка этих же мышей с CIA соединениями по настоящему изобретению (соединения 4-6) снизила степень артрита до значений в пределах диапазона от 3 до 5,6. Следует отметить, что эти значения находятся в пределах диапазона, который включает в себя значение степени артрита, полученное для мышей с CIA, обработанных положительным контролем с антителом к TNFальфа (4,9), эквивалентным используемым в клинике методикам лечения ревматоидного артрита.
На 50-й день исследования толщина лапы больных CIA мышей достигала величины 2,83 мм. Контроль с антителом к TNFальфа снижал воспаление в лапах до величины 2,5 мм. Соединения 4-6 также снижали величины воспаления до 2,3-2,5 мм.
ПРИМЕР 16. Эффективность соединений 4, 5 и 6 в экспериментальной модели болезни Крона
Использовали самцов 6-8-недельных мышей BALB/c. Им ректально вводили тринитробензолсульфоновую кислоту (TBNS) в 50% этаноле для индуцирования колита. Контрольные мыши получили 50% этанол. Мышей обработали путем подкожного введения буфера (PBS) или различных аналогов кортистатина (0,4 мг/кг) на 3-й, 4-й и 5-й день, когда заболевание уже сформировалось.
Животных наблюдали ежедневно и контролировали наличие диареи (степень колита) с использованием шкалы от 0 до 4, потерю веса и выживаемость. Кроме того, при вскрытии на 10-й день два независимых исследователя посредством слепого метода оценивали и классифицировали толстый кишечник в соответствии с макроскопическим повреждением, используя шкалу от 0 до 10 на основе критериев, которые отражают воспаление, гиперемию, отек толстого кишечника и распространение язв. Классификацию степени колита в соответствии с консистенцией стула и ректальными кровотечениями также проводили два независимых наблюдателя: 0 = нормальный вид стула, 1 = незначительное изменение консистенции стула; 2 = умеренное изменение консистенции стула; 3 = умеренное изменение консистенции стула и наличие крови в стуле; 4 = тяжелая водянистая диарея и умеренное/значительное количество крови в стуле. Вес мышей, страдающих от колита, индуцированного обработкой TNBS в 50% этаноле, снижался с 1-го дня (22 г) по 10-й день (17 г). Вес здоровых контрольных мышей, обработанных только 50% этанолом, увеличивался с 1-го дня (22 г) по 10-й день (25 г). Обработка больных мышей антителом к TNFальфа в качестве положительного контроля замедляла потерю веса с 1-го дня (21 г) по 10-й день (19 г); то же происходило при обработке соединениями-аналогами кортистатина 4-6, при этом вес на 10-й день составил от 19 до 20 г, значения, которые значительно выше, чем значение веса, полученное для больных мышей (17 г).
Степень колита (0-4), оцененная in vivo на 6-й день исследования, показала значение исходного уровня 0,25 для здоровых контрольных мышей, обработанных 50% этанолом, и максимальное значение 3,6 для больных мышей, обработанных TBNS в 50% этаноле. Разные методики обработки аналогами кортистатина приводили к значениям степени колита от 1,4 до 2,1, аналогично значениям у группы положительного контроля, обработанной антителом к TNFальфа (2,1), показывая эффективность указанных методик обработки.
Степень макроскопических повреждений в толстом кишечнике (0-10) проанализировали при вскрытии. Значение исходного уровня 0,2 получали для здоровых контрольных мышей, обработанных 50% этанолом, и максимальное значение 7,5 для больных мышей, обработанных TBNS в 50% этаноле. Разные методики обработки аналогами кортистатина показали значения степени повреждения от 1,9 до 2,8. Обработка соединением-антителом к TNFальфа дала значение 2,5 в том же диапазоне, что и соединения по настоящему изобретению.
ПРИМЕР 17. Эффективность соединений 4, 5 и 6 в экспериментальной модели язвенного колита
Использовали самцов 7-8-недельных мышей C57BI/6. Им давали без ограничения питьевую воду с 5% раствором натриевой соли сульфата декстрана (DSS) с 0-го дня по 7-й день исследования для индуцирования острого колита. Животным в контрольной группе давали нормальную воду. Мышей обрабатывали путем подкожного введения буфера (PBS) или разными аналогами кортистатина (0,4 мг/кг) на 1-й, 2-й и 3-й день исследования. CST использовали в качестве референтного продукта. Тяжесть колита оценивали ежедневно с использованием шкалы значений от 0 до 4, указывающих клинический индекс активности заболевания с учетом консистенции стула, наличия кровянистого стула и потери веса. Мышей умерщвляли на 8-й день исследования и после вскрытия устанавливали степень макроскопического повреждения толстого кишечника, используя шкалу от 0 до 8. Пероральное введение 5% DSS привело к значительному увеличению индекса активности заболевания (0-4), начиная от значения исходного уровня 0 до значения 3,7 в группе заболевания. У больных мышей, обработанных CST и аналогами кортистатина, получали значения клинического индекса активности заболевания значительно более низкие и подобные, в дипазоне 1-1,5. Кроме того, обработка новыми аналогами кортистатина с 1-го по 3-й день исследования значительно повысила выживаемость больных животных с 50% до 100%. На макроскопическом уровне степень колита у больных животных составяла 6,6; тогда как у больных мышей, обработанных CST, она составляла 0,87, а у больных мышей, обработанных новыми аналогами кортистатина, она составляла 0,74-1,5. Средний вес толстого кишечника больных мышей составил 763 мг, средний вес толстого кишечника больных мышей, обработанных CST, составил 621 мг и средний вес толстого кишечника больных мышей, обработанных новыми аналогами кортистатина, составил 614-621 мг, что указывает на снижение воспаления толстого кишечника. Все эти результаты демонстрируют эффективность новых аналогов кортистатина в данных экспериментальных моделях колита.



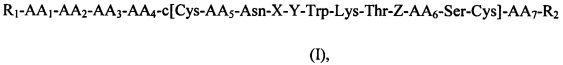







Пептиды, используемые в лечении и/или наблюдении за кожей, слизистыми оболочками и/или волосами, и их применение в косметических или фармацевтических композициях
Композиции для лечения боли и/или воспаления
Пептидные лиганды соматостатиновых рецепторов
Местный глазной пептидный состав
Аналоги бикалутамида или (s)-бикалутамид в качестве активирующих экзоцитоз соединений для применения в лечении расстройства лизосомного накопления или гликогеноза

