Результат интеллектуальной деятельности: СПОСОБ СИНТЕЗА N-(ФОСФОНОМЕТИЛ)ГЛИЦИНА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
[0001] Настоящее изобретение относится к новому способу синтеза N-(фосфонометил)глицина или его производных.
Уровень техники
[0002] N-(фосфонометил)глицин, известный в области агрохимикатов как глифосат, представляет собой высокоэффективный и коммерчески значимый фитотоксикант широкого спектра, применимый при контроле роста прорастающих семян, появляющихся всходов, укоренившейся и созревающей древесной и травянистой растительности, а также водных растений. Глифосат применяют в качестве послевсходового гербицида системного действия для контроля роста широкого разнообразия однолетней и многолетней травы, а также видов широколиственной сорной травы на обрабатываемых посевных площадях, в том числе при производстве хлопка.
[0003] Глифосат и его соли традиционно применяют в водных гербицидных составах, обычно содержащих одно или несколько поверхностно-активных веществ, по отношению к тканям листа (т.е., листве или другим фотосинтезирующим органам) целевого растения. После применения глифосат поглощается тканями листа и перемещается по растению. Глифосат неконкурентно блокирует важный биохимический путь, который является общим практически для всех растений. Более конкретно, глифосат ингибирует путь шикимовой кислоты, что приводит к биосинтезу ароматических аминокислот. Глифосат ингибирует превращение фосфоенолпировиноградной кислоты и 3-фосфошикимовой кислоты в 5-енолпирувил-3-фосфошикимовую кислоту посредством ингибирования фермента синтазы 5-енолпирувил-3-фосфошикимовой кислоты (EPSP синтазы или EPSPS), находящегося в растениях.
[0004] Хорошо известны несколько путей получения посредством которых может быть получен глифосат, например, пути предложенные в патентах US 3969398; СА 1039739; US 3799758; US 3927080; US 4237065 и US 4065491, но все эти пути предусматривают несколько недостатков, включая потери продукта, проблемы защиты окружающей среды и ко всему прочему неприемлемые результаты с экономической точки зрения.
[0005] В патенте СН 620223 раскрывают способ получения N-(фосфонометил)глицина с хорошим выходом посредством осуществления гидролиза бис-N,N'-[O,O'-[ди-низший-алкил]-фосфонометил]-2,5-дикетопиперазина (= соединение I) с помощью водной галогенводородной кислоты. Соединение I получают посредством осуществления реакции N,N'-бисхлорметил-2,5-дикетопиперазина с три-низший-алкил-фосфитом или посредством осуществления реакции двунатриевой соли 2,5-дикетопиперазина с хлорметилфосфоновой кислотой.
[0006] В патенте ES 525399 раскрывают способ получения N-(фосфонометил)глицина с хорошим выходом посредством гидролиза в щелочных или кислотных условиях бис-N,N'-[O,O'-[диметил]фосфонометил]-2,5-дикетопиперазина (соединения II). Соединение II получают в результате реакции 2,5-дикетопиперазина с формальдегидом с образованием N,N'-диметилол-2,5-дикетопиперазина, который в дополнительной реакции с тионилхлоридом превращают в N,N'-бисхлорметил-2,5-дикетопиперазин, который на следующей стадии превращают в соединение II посредством реакции с триметилфосфитом.
[0007] В патентной заявке WO 98/35930 раскрывают способ получения N-ациламинокарбоновой кислоты, которая легко превращается в N-фосфонометил)глицин или его соль или сложный эфир при реакции фосфонометилирования. В одном варианте осуществления реакция фосфонометилирования в результате приводит к замене N-ацил-заместителя N-ациламинокарбоновой кислоты N-фосфонометильной группой. В другом варианте осуществления 2,5-дикетопиперазин, образованный посредством деацилирования N-ациламинокарбоновой кислоты, фосфонометилируют с помощью трихлорида фосфора, фосфористой кислоты или источника фосфористой кислоты в присутствии источника формальдегида.
[0008] В патенте US 4804499 раскрывают способ получения N-замещенной аминометилфосфоновой кислоты, включающий осуществление реакции соединения на основе 2,5-дикетопиперазина с фосфористой кислотой и формальдегидом в кислой среде. Пример 4 иллюстрирует получение N-(фосфонометил)глицина с выходом 52%.
[0009] В патенте US 4400330 раскрывают способ получения N-(фосфонометил)глицина, который включает стадии (1) первого осуществления реакции 2,5-дикетопиперазина с параформальдегидом в ледяной уксусной кислоте, затем добавления галоген-замещенного фосфорсодержащего соединения, все в присутствии растворителя на основе низкомолекулярной карбоновой кислоты, с образованием промежуточного соединения N,N'-бисфосфонометил-2,5-дикетопиперазина, (2) выделения указанного промежуточного соединения, (3) затем осуществления реакции указанного промежуточного соединения N,N'-бисфосфонометил-2,5-дикетопиперазина с гидролизующим средством, выбранным из щелочного или щелочноземельного основания, с образованием соли N-(фосфонометил)глицина и (4) после этого подкисления указанной соли с помощью неорганической кислоты с образованием конечного продукта, N-(фосфонометил)глицина. Стадия (1) в патенте US 4400330 состоит из реакции N,N'-диметилол-2,5-дикетопиперазина и галоген-замещенного фосфорсодержащего соединения, приводящей в результате к образованию N,N'-дигалогенметил-2,5-дикетопиперазина и фосфористой кислоты. Фосфористая кислота затем реагирует с N,N'-дигалогенметил-2,5-дикетопиперазином с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина и хлористоводородной кислоты.
[0010] Вследствие важности N-(фосфонометил)глицина в качестве гербицида имеет место постоянный поиск других способов получения таких соединений.
Цели изобретения
[0011] Настоящее изобретение нацелено на обеспечение улучшенного, в частности, эффективного и благоприятного для окружающей среды, способа получения N-(фосфонометил)глицина или его производных, в котором отсутствуют недостатки способов предшествующего уровня техники.
Краткое описание изобретения
[0012] В настоящем изобретении раскрывают способ синтеза N-(фосфонометил)глицина или одного из его производных, выбранных из группы, состоящий из его солей, его фосфонатных сложных эфиров и солей его фосфонатного сложного эфира, включающий стадии
а) образования в присутствии кислотного катализатора реакционной смеси, содержащей 2,5-дикетопиперазин, формальдегид и соединение, содержащее один или несколько ангидридных фрагментов Р-О-Р, причем указанные фрагменты содержат один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина, его моно- - тетра-фосфонатных сложных эфиров, дегидратированных форм N,N'-бисфосфонометил-2,5-дикетопиперазина и фосфонатных сложных эфиров его дегидратированных форм;
b) осуществления гидролиза указанных N,N'-бисфосфонометил-2,5-дикетопиперазина, его дегидратированных форм или их фосфонатных сложных эфиров с получением N-(фосфонометил)глицина или одного из его производных, выбранных из группы, состоящей из его солей, его фосфонатных сложных эфиров и солей его фосфонатного сложного эфира.
[0013] В предпочтительных вариантах осуществления настоящего изобретения раскрывают один или несколько из следующих признаков:
- молярное отношение формальдегида к 2,5-дикетопиперазину находится в диапазоне от 2 до 8, предпочтительно от 2 до 3 и более предпочтительно от 2,4 до 2,8;
- эквивалентное соотношение фрагмента N-H 2,5-дикетопиперазина к ангидридному фрагменту Р-О-Р находится в диапазоне от 0,2 до 2,5, предпочтительно от 0,3 до 2,0 и более предпочтительно от 0,5 до 1,5;
- соотношение молей кислотного катализатора к эквивалентам N-H 2,5-дикетопиперазина составляет 2
или более и предпочтительно находится в диапазоне от 4 до 10;
- содержимое реактора со стадии а) перемешивают при практически постоянной температуре в диапазоне от 20°С до 120°С, предпочтительно от 40°С до 100°С, в течение периода времени в диапазоне от 30 минут до 24 часов и предпочтительно от 1 часа до 20 часов;
- соединение, содержащее ангидридные фрагменты Р-О-Р, выбрано из группы, состоящей из тетрафосфорного гексаоксида, тетраэтилпирофосфита, и при этом соединения, содержащие ангидридные фрагменты Р-О-Р, получены в результате объединения одного или нескольких соединений, содержащих:
- один или несколько фрагментов Р-ОН с одним или несколькими соединениями, содержащими один или несколько ангидридных фрагментов Р-О-Р или один или несколько фрагментов Р-Х, где атом Р из одного или нескольких соединений находится в степени окисления (+III);
- один или несколько фрагментов Р-Х и воду, где атом Р в содержащем фрагмент Р-Х соединении находится в степени окисления (+III);
- два или более фрагментов Р-О-Р и воду, где содержащее фрагмент Р-О-Р соединение содержит атом Р в степени окисления (+III) и атом Р в степени окисления (+III) или (+V);
где соединения, содержащие один или несколько фрагментов Р-ОН, могут поддаваться таутомеризации фрагмента >Р(=O)Н,
где X представляет собой галогенид, выбранный из группы, состоящей из хлора, брома и йода, и
где уровень галогена в содержащем ангидридный фрагмент Р-О-Р соединении составляет 1000 ppm или менее, предпочтительно 500 ppm или менее и более предпочтительно 200 ppm или менее;
- соединение, содержащее ангидридные фрагменты Р-О-Р, выбрано из группы, состоящий из тетрафосфорного гексаоксида, тетраэтилпирофосфита, и при этом содержащее ангидридные фрагменты Р-О-Р соединение получено в результате объединения фосфористой кислоты и тетрафосфорного гексаоксида, фосфористой кислоты и тетрафосфорного декаоксида, фосфористой кислоты и трихлорида фосфора, диметилфосфита и тетрафосфорного декаоксида, трихлорида фосфора и воды, а также тетрафосфорного гексаоксида и воды;
- соединение, содержащее ангидридные фрагменты Р-О-Р, представляет собой тетрафосфорный гексаоксид;
- стадия а) разделена на две отдельные стадии, включающие
а1) осуществление реакции 2,5-дикетопиперазина с формальдегидом с образованием N,N'-диметилол-2,5-дикетопиперазина;
а2) осуществление реакции указанного N,N'-диметилол-2,5-дикетопиперазина с соединением, содержащим один или несколько ангидридных фрагментов Р-О-Р, причем указанные фрагменты содержат атом P в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), в присутствии кислотного катализатора с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина, его дегидратированных форм или их производных;
- молярное отношение формальдегида к 2,5-дикетопиперазину находится в диапазоне от 2,0 до 8,0, предпочтительно от 2,0 до 3,0 и более предпочтительно от 2,4 до 2,8;
- реакцию 2,5-дикетопиперазина с формальдегидом осуществляют в водных щелочных условиях при pH выше 7,0, предпочтительно в диапазоне от 7,1 до 11,0, более предпочтительно от 7,5 до 10,0 и наиболее предпочтительно от 8,0 до 9,0;
- стадию а1) реакции осуществляют посредством нагревания при температуре в диапазоне от 60°С до 100°С и предпочтительно от 70°С до 90°С в течение периода времени в диапазоне от 20 минут до 120 минут;
- выделяют N,N'-диметилол-2,5-дикетопиперазин, образованный на стадии а1);
- стадия а2) включает растворитель, выбранный из группы, состоящей из 1,4-диоксана, толуола, этилацетата, ацетонитрила, уксусной кислоты, сульфолана, 1-этил-3-метил-имидазолий-бис(трифторметилсульфонил)имида или их смесей;
- содержащее ангидридную группу Р-О-Р соединение и N,N'-диметилол-2,5-дикетопиперазин в присутствии кислотного катализатора и необязательно растворителя постепенно смешивают при поддержании температуры реакции ниже 120°С;
- N,N'-диметилол-2,5-дикетопиперазин в виде твердого вещества или в растворе постепенно добавляют к содержащему ангидридный фрагмент Р-О-Р соединению, включая кислотный катализатор и необязательно растворитель, при поддержании температуры реакции ниже 120°С;
- эквивалентное соотношение N,N'-диметилол-2,5-дикетопиперазина к ангидридному фрагменту Р-О-Р на стадии а2) находится в диапазоне от 0,2 до 2,5, предпочтительно от 0,3 до 2,0 и более предпочтительно от 0,5 до 1,5;
- молярное отношение N,N'-диметилол-2,5-дикетопиперазина к тетрафосфорному гексаоксиду на стадии а2) находится в диапазоне от 0,5 до 5,0, предпочтительно от 0,6 до 4,0 и более предпочтительно от 1,0 и 3,0;
- на стадии а2) после завершения добавления содержащего ангидридный фрагмент Р-О-Р соединения его необязательно нагревают до температуры в диапазоне от 20°С до 120°С, предпочтительно от 40°С до 100°С, и поддерживают при указанной температуре в течение периода времени в диапазоне от 1 часа до 20 часов;
- выделяют N,N'-бисфосфонометил-2,5-дикетопиперазин или его производные образованные, на стадии а) или а2);
- кислотный катализатор представляет собой гомогенный катализатор на основе кислоты Брэнстеда, предпочтительно выбранный из группы, состоящей из метансульфоновой кислоты, трифторметансульфоновой кислоты, трифторуксусной кислоты, п-толуолсульфоновой кислоты, хлористоводородной кислоты, фосфористой кислоты, фосфорной кислоты и их смесей;
- кислотный катализатор представляет собой гетерогенную кислоту Брэнстеда, предпочтительно выбранную из группы, состоящей из:
(i) комбинаций твердых кислотных оксидов металлов как таковых или на подложке из вещества-носителя;
(ii) катионообменных смол, выбранных из группы, включающей сополимеры стирола, этилвинилбензола и дивинилбензола, функционализированные таким образом, что обеспечивается прививка фрагментов SO3H на ароматическую группу и перфорированные смолы, несущие группы карбоновой и/или сульфоновой кислоты;
(iii) органических сульфоновых, карбоновых и фосфоновых кислот Брэнстеда, которые являются практически несмешиваемыми в реакционной среде при температуре реакции;
(iv) кислотного катализатора, полученного:
- в результате взаимодействия твердой подложки, содержащей неподеленную пару электронов, на которую осаждена органическая кислота Брэнстеда; или
- в результате взаимодействия твердой подложки, содержащей неподеленную пару электронов, на которую осаждено соединение, содержащее Льюисовский кислотный центр; или
- из гетерогенных твердых веществ, функционализированных посредством химической прививки группы кислоты Брэнстеда или ее предшественника; и
(v) гетерогенных гетерополикислот общей формулы HxPMyOz, где P выбран из фосфора и кремния, а М выбран из вольфрама и молибдена и их комбинаций;
- кислотный катализатор представляет собой гомогенную кислоту Льюиса, предпочтительно выбранную из группы, состоящей из LiN(CF3SO2)2, Mg(OCF3SO2)2, Al(OCF3SO2)3, Bi(OCF3SO2)3, Sc(OCF3SO2)3;
- кислотный катализатор представляет собой гетерогенную кислоту Льюиса, полученную в результате взаимодействия гомогенного катализатора на основе кислоты Льюиса и органического или неорганического полимерного соединения;
- гидролиз на стадии b) проводят в кислых условиях, предпочтительно достигаемых посредством летучей кислоты, предпочтительно посредством хлористоводородной кислоты;
- летучую кислоту извлекают в конце стадии b), необязательно очищают и повторно применяют;
- гидролиз на стадии b) проводят в щелочных условиях;
- гидролиз на стадии b) проводят в нейтральных условиях, предпочтительно в присутствии ферментного катализатора, предпочтительно амидазы;
- гидролиз на стадии b) осуществляют при температуре в диапазоне от 25°С до 250°С в течение периода времени в диапазоне от 1 часа до 100 часов;
- производные выбраны из группы, состоящей из солей N-(фосфонометил)глицина, фосфонатных сложных эфиров N-(фосфонометил)глицина и солей фосфонатных сложных эфиров N-(фосфонометил)глицина, и при этом катион соли выбран из группы, состоящей из аммония, изопропиламмония, этаноламмония, диметиламмония, триметилсульфония, натрия и калия;
- N-(фосфонометил)глицин получают периодическим или непрерывным способом;
[0014] Формула для реакций, изложенных выше, может быть представлена следующим образом:
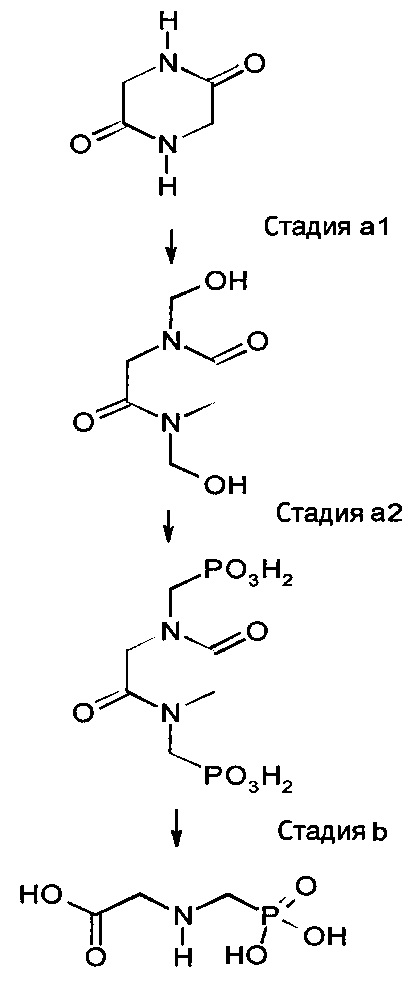
Вышеуказанные промежуточные реакции, как предполагается, представляют собой одну из нескольких возможностей получения N-(фосфонометил)глицина или его производных.
Подробное описание изобретения
[0015] Настоящее изобретение предусматривает эффективный, экономически целесообразный и предпочтительно благоприятный для окружающей среды способ получения N-(фосфонометил)глицина или его производных.
[0016] Под производными по настоящему изобретению подразумевают соли и фосфонатные сложные эфиры N-(фосфонометил)глицина, где
- соли N-(фосфонометил)глицина содержат карбоксилат- и/или (ди)фосфонат-анион и агрономически приемлемый катион или аммоний-катион N-(фосфонометил)глицина и агрономически приемлемый анион.
Предпочтительные соли представляют собой соли аммония, изопропиламмония, этаноламмония, диметиламмония, триметилсульфония, натрия и калия, где соотношение катиона к аниону N-(фосфонометил)глицина находится в диапазоне от 0,1 до 3,0.
При проведении гидролиза в основных условиях соль содержит карбоксилат- и/или фосфонат-анион N-(фосфонометил)глицина и щелочной металл, щелочноземельный металл или аммоний-катион; иначе, при проведении гидролиза в кислых условиях образованная соль содержит аммоний-катион N-(фосфонометил)глицина и анион, переходящий из применяемой для гидролиза кислоты. В данном конкретном последнем случае анион, например, представляет собой хлорид-анион, переходящий из хлористоводородной кислоты, или сульфат-анион, переходящий из серной кислоты.
- фосфонатные сложные эфиры содержат один или несколько замещенных или незамещенных гидрокарбильных групп, которые могут быть разветвленными или неразветвленными, насыщенными или ненасыщенными и могут содержать одно или несколько колец. Подходящие гидрокарбилы включают алкильные, алкенильные, алкинильные и арильные фрагменты. Они также включают алкильные, алкенильные, алкинильные и арильные фрагменты, замещенные другими алифатическими или циклическими гидрокарбильными группами, такими как алкарильная, алкенарильная и алкинарильная.
Замещенный гидрокарбил определяется как гидрокарбил, в котором по меньшей мере один атом водорода был замещен атомом, отличным от водорода, таким как атом галогена, атом кислорода с образованием, например, эфира или сложного эфира, атом азота с образованием амидной или нитрильной группы или атом серы с образованием, например, тиоэфирной группы.
[0017] Способ по настоящему изобретению включает стадии:
a) осуществления реакции 2,5-дикетопиперазина, формальдегида и соединения, содержащего один или несколько ангидридных фрагментов Р-О-Р, причем указанные фрагменты содержат один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), в присутствии кислотного катализатора и необязательно растворителя с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина, его дегидратированных форм или их производных;
b) осуществления гидролиза образованного N,N'-бисфосфонометил-2,5-дикетопиперазина, его дегидратированных форм или их производных с образованием N-(фосфонометил)глицина или одного из его производных.
[0018] Под производными N,N'-бисфосфонометил-2,5-дикетопиперазина и его дегидратированными формами в настоящем изобретении подразумевают моно- - тетра-фосфонатные сложные эфиры N,N'-бисфосфонометил-2,5-дикетопиперазина или фосфонатные сложные эфиры его дегидратированных форм. Фосфонатные сложные эфиры обычно получают посредством применения соединения, содержащего один или несколько ангидридных фрагментов Р-О-Р, замещенных соответствующим гидрокарбильным заместителем.
[0019] Производные N-(фосфонометил)глицина предпочтительно получают как результат реакции гидролиза стадии b), или могут быть получены посредством дополнительной обработки N-фосфонометилглицина. Под производными по настоящему изобретению подразумевают соли, фосфонатные сложные эфиры или соли фосфонатного сложного эфира N-(фосфонометил)глицина. В настоящем изобретении следует понимать, что выражение N-(фосфонометил)глицин включает все производные.
[0020] Молярное отношение формальдегида к 2,5-дикетопиперазину находится в диапазоне от приблизительно 2 до приблизительно 8, предпочтительно от приблизительно 2 до приблизительно 3 и более предпочтительно от приблизительно 2,4 до приблизительно 2,8.
[0021] Эквивалентное соотношение фрагмента N-H 2,5-дикетопиперазина к ангидридному фрагменту Р-О-Р находится в диапазоне от приблизительно 0,2 до приблизительно 2,5, предпочтительно от приблизительно 0,3 до приблизительно 2,0 и более предпочтительно от приблизительно 0,5 до приблизительно 1,5.
[0022] Соотношение молей кислотного катализатора к эквивалентам N-H 2,5-дикетопиперазина составляет по меньшей мере приблизительно 2 и предпочтительно в находится в диапазоне от приблизительно 4 до приблизительно 10.
[0023] Содержимое реактора со стадии а) перемешивают при практически постоянной температуре в диапазоне от приблизительно 20°С до приблизительно 120°С, предпочтительно от приблизительно 40°С до приблизительно 100°С, в течение периода времени в диапазоне от приблизительно 30 минут до приблизительно 24 часов и предпочтительно от приблизительно 1 часа до приблизительно 20 часов.
[0024] 2,5-Дикетопиперазин может быть получен в результате циклодимеризации глицина, как раскрыто, например, в заявке на патент США 2004/0024180, где глицин нагревают в органическом растворителе при температуре от приблизительно 50°С до приблизительно 200°С, в частности, при температуре в диапазоне от приблизительно 80°С до приблизительно 150°С. Диапазон pH, при котором происходит циклодимеризация, предпочтительно составляет от приблизительно 2 до приблизительно 9 и предпочтительно от приблизительно 3 до приблизительно 7.
[0025] Обычно растворители, которые могут применяться для реакции циклодимеризации глицина, предпочтительно представляют собой таковые, способные к образованию низкокипящей азеотропной смеси с водой. Такие растворители представляют собой, например, ацетонитрил, аллиловый спирт, бензол, бензиловый спирт, н-бутанол, 2-бутанол, трет-бутанол, бутиловый сложный эфир уксусной кислоты, тетрахлорид углерода, хлорбензол, хлороформ, циклогексан, 1,2-дихлорэтан, диэтилацеталь, диметилацеталь, этиловый сложный эфир уксусной кислоты, гептан, метилизобутилкетон, 3-пентанол, толуол и ксилол.
[0026] Формальдегид, известный как оксиметилен, характеризующийся формулой CH2O, получают и реализуют в виде водных растворов, содержащих различные, часто незначительные, например, 0,3-3%, количества метанола и, как правило, представляют собой 37% растворы на основе формальдегида, несмотря на то, что могут применяться различные концентрации. Формальдегидные растворы находятся в виде смеси олигомеров. Такие предшественники формальдегида могут, например, быть представлены параформальдегидом, смесью в твердом состоянии линейных поли(оксиметиленгликолей) обычно с достаточно короткой, n=8-100, длиной цепи, и циклическим тримером формальдегида, называемым выражением 1,3,5-триоксан. Концентрации жидкого формальдегида выше приблизительно 37% требуют хранения при температуре выше комнатной температуры во избежание осаждения формальдегидных полимеров. Температура, необходимая для поддержания прозрачного раствора и во избежание отделения твердого полимера, повышается от комнатной температуры по мере того, как концентрация раствора повышается выше приблизительно 37%.
[0027] Несмотря на то, что формальдегид обычно применяют в виде 37% по весу раствора в воде, известного как формалин, его также могут добавлять в виде водного раствора с концентрацией формальдегида, отличной от 37% по весу, либо в виде твердого вещества, такого как, например, параформальдегид или 1,3,5-триоксан.
[0028] Для случая, когда 2,5-дикетопиперазин, формальдегид и содержащее ангидридный фрагмент Р-О-Р соединение, содержащее один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), образуют реакционную смесь, само собой разумеется, что количество воды, присутствующее в водном растворе формальдегида, находится в соответствии с одним требованием, необходимым для преобразования первого содержащего ангидридный фрагмент Р-О-Р соединения в содержащее модифицированный Р-О-Р- ангидридный фрагмент соединение посредством частичного гидролиза указанного первого содержащего ангидридный фрагмент Р-О-Р соединения, после чего будут осуществлять реакцию указанного содержащего модифицированный ангидридный фрагмент Р-О-Р соединения с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина, его дегидратированных форм или их производных. Для этого конкретного случая формальдегид предпочтительно добавляют в безводных условиях, т.е. формальдегид предпочтительно добавляют в виде безводного твердого вещества.
[0029] В случае, когда содержащее ангидридный фрагмент Р-О-Р соединение предпочтительно выбрано из группы, состоящей из тетрафосфорного гексаоксида и частично гидролизованных соединений на основе тетрафосфорного гексаоксида, полученных посредством реакции 1 моля тетрафосфорного гексаоксида с 1, 2, 3, 4 и 5 молями воды, соответственно, следует понимать, что все соединения, содержащие по меньшей мере один ангидридный фрагмент Р-О-Р, где один атом Р находится в степени окисления (+III) и другой атом Р находится в степени окисления (+III) или (+V), могут применяться для целей настоящего изобретения.
[0030] Подходящие содержащие ангидридный фрагмент Р-О-Р соединения могут либо собственно содержать ангидридный фрагмент Р-О-Р в соединении (например, P4O6 или пирофосфиты (RO)2P-O-P(OR)2), либо могут быть получены in situ посредством объединения реагентов, которые будут образовывать необходимый ангидридный фрагмент Р-О-Р.
Подходящие комбинации реагентов представляют собой:
a) соединения, содержащие по меньшей мере один фрагмент Р-ОН (также поддающийся таутомеризации фрагмент >Р(=O)Н в >P(LP)OH (где LP представляет собой неподеленную пару электронов)), такой как, например, что и в случае для диметилфосфита (MeO)2Р(=O)Н), а также соединения, содержащие по меньшей мере один ангидридный фрагмент Р-О-Р, например, P2O5 или P4O6;
b) соединения, содержащие по меньшей мере один фрагмент Р-ОН, и соединения, содержащие по меньшей мере один фрагмент Р-Х (X=Cl, Br, I);
c) соединения, содержащие по меньшей мере один фрагмент Р-Х и H2O;
d) соединения, содержащие ангидридные фрагменты Р-О-Р и H2O, для частичного гидролиза.
В случае a) и b) обязательным является то, что по меньшей мере в одном из используемых соединений атом Р находится в степени окисления (+III), при этом в случае с) необходимо, чтобы атом Р находился в степени окисления (+III), а в случае d) фрагменты Р-О-Р содержали атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V) с целью образования содержащего ангидридный фрагмент Р-О-Р соединения, содержащего атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V).
[0031] Содержащие ангидридный фрагмент Р-О-Р соединения, где ангидридный фрагмент Р-О-Р уже присутствует, представляют собой оксиды фосфора с формулой P4On с n=6-9, пирофосфиты с общей формулой (RO)2P-O-P(OR)2, где R представляет собой алкильную или арильную группу, пирофосфористую кислоту (H4P2O5) и изогипофосфорную кислоту (Н)(НО)Р(O)-O-Р(O)(ОН)2.
[0032] Комбинации, описанные в а), получают посредством осуществления реакции, например, оксидов фосфора с формулой P4On при n=6-10, алкил-замещенных пирофосфитов, пирофосфористой кислоты, изогипофосфорной кислоты, метафосфорной кислоты или полифосфорной кислоты с фосфористой кислотой, фосфорной кислотой, моно- или дизамещенными фосфитами с формулой (RO)PO2H2 или (RO)2POH, где R представляет собой алкильную или арильную группу, фосфатными сложными эфирами (RO)PO3H2 или (RO)2PO2H, фосфоновыми кислотами RPO3H2 или его сложным моноэфиром RPO2H(OR), при условии, что такие комбинации будут приводить к содержащим ангидридный фрагмент Р-О-Р соединениям, содержащим один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V).
[0033] Комбинации, описанные в b), получают посредством объединения PCl3, PBr3, POCl3 или моно- или дихлорфосфитов типа (RO)2PCl и (RO)PCl2 с фосфористой кислотой, фосфорной кислотой или моно- или дизамещенными фосфитами с формулой (RO)PO2H2 или (RO)2POH, при условии, что такие комбинации будут приводить к содержащим ангидридный фрагмент Р-О-Р соединениям, содержащим один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V).
[0034] Комбинации, описанные в с), получают посредством объединения PCl3, PBr3 или моно- или дихлорфосфитов типа (RO)2PCl и (RO)PCl2 с H2O.
[0035] С целью получения содержащих ангидридный фрагмент Р-О-Р соединений, не содержащих функциональные группы Р-Х, оставшиеся функциональные группы Р-Х гидролизируют водой. Оставшиеся ангидридные фрагменты Р-О-Р могут быть гидролизованы, поскольку необходимый ангидридный фрагмент Р-О-Р, где один атом Р находится в степени окисления (+III) и другой атом Р находится в степени окисления (+III) или (+V), остается.
[0036] Наиболее предпочтительные представляют собой тетрафосфорный гексаоксид, тетраэтилпирофосфит и комбинации из фосфористой кислоты и тетрафосфорного гексаоксида, из фосфористой кислоты и тетрафосфорного декаоксида, из фосфористой кислоты и трихлорида фосфора, из диметилфосфита и тетрафосфорного декаоксида, из трихлорида фосфора и воды, а также из тетрафосфорного гексаоксида и воды.
[0037] Количество "реакционно-способных" атомов Р(+III), которые могут быть превращены в фосфоновые кислоты, согласно настоящему изобретению определяют по количеству атомов Р(+III) и количеству ангидридных фрагментов Р-О-Р. Если ангидридных фрагментов Р-О-Р больше, чем атомов Р(+III), то все атомы Р(+III) превращают в фосфоновые кислоты. Если ангидридных фрагментов Р-О-Р меньше, чем атомов Р(+III), то только часть атомов Р(+III), эквивалентную количеству ангидридных фрагментов Р-О-Р, превращают в фосфоновые кислоты.
[0038] В случае, если применяют галоген-содержащие исходные вещества, например, PCl3, POCl3 или PBr3, то уровень галогена в содержащем ангидридный фрагмент Р-О-Р соединении следует поддерживать ниже 1000 ppm, обычно ниже 500 ppm, предпочтительно ниже 200 ppm в пересчете на вещество на основе Р-О-Р, составляющее 100%. Следовательно, все избыточные функциональные группы Р-Х гидролизируют перед реакциями с субстратом посредством добавления одной молекулы H2O на избыточную функциональную группу Р-Х. Образованный НХ удаляют посредством, например, продувки сухим инертным газом, таким как азот или гелий, через раствор.
[0039] Тетрафосфорный гексаоксид, предпочтительно применяемый в объеме настоящего изобретения, может быть представлен практически чистым соединением, содержащим по меньшей мере 85%, предпочтительно более 90%, более предпочтительно по меньшей мере 95% и в одном конкретном исполнении по меньшей мере 97% P4O6. Несмотря на то, что тетрафосфорный гексаоксид, подходящий для применения в контексте настоящего изобретения, может быть получен по любой известной технологии, в предпочтительных вариантах осуществления его получают согласно способу, описанному в патентных заявках WO 2009/068636 и/или WO 2010/055056 в разделе под названием "Способ получения P4O6 с улучшенным выходом" ("Process for the manufacture of P4O6 with improved yield"). Более подробно, кислород или смесь кислорода и инертного газа, а также газообразный или жидкий фосфор преимущественно реагируют в стехиометрических количествах в реакционном блоке при температуре в диапазоне от 1600 до 2000 K с отводом тепла, образованного при экзотермической реакции фосфора и кислорода, при поддержании предпочтительного времени пребывания от 0,5 до 60 секунд с последующим гашением продукта реакции при температуре ниже 700 K и очисткой неочищенного продукта реакции посредством дистилляции. Тетрафосфорный гексаоксид, полученный таким образом, представляет собой чистый продукт, обычно содержащий по меньшей мере 97% оксида. Полученный таким образом P4O6 обычно представлен в виде жидкого вещества с высокой степенью чистоты, содержащего, в частности, низкие уровни элементарного фосфора, Р4, предпочтительно ниже 1000 ppm в пересчете на Р4О6, составляющий 100%. Предпочтительное время пребывания составляет от 5 до 30 секунд, более предпочтительно от 8 до 30 секунд. Продукт реакции в одном предпочтительном исполнении может быть охлажден до температуры ниже 350 K.
[0040] Предполагается, что P4O6, участвующий в реакции при температуре от 24°С (t° плавления) до 120°С, обязательно должен быть жидким или газообразным, несмотря на это, твердые вещества могут, теоретически, применяться для получения реакционной среды.
[0041] С целью удобства и оперативного знания, тетрафосфорный гексаоксид, представленный P4O6, является высокочистым и содержит очень низкие уровни примесей, в частности, элементарного фосфора, Р4, при уровне ниже 1000 ppm, обычно ниже 500 ppm и предпочтительно не более 200 ppm в пересчете на P4O6, составляющий 100%.
[0042] Кислотный катализатор, применяемый в объеме настоящего изобретения, предпочтительно представляет собой гомогенный катализатор на основе кислоты Брэнстеда необязательно в присутствии растворителя, или гетерогенный катализатор на основе кислоты Брэнстеда в присутствии растворителя, или катализатор на основе кислоты Льюиса в присутствии растворителя.
[0043] Гомогенная кислота Брэнстеда предпочтительно выбрана из группы, состоящей из метансульфоновой кислоты, фторметансульфоновой кислоты, трихлорметансульфоновой кислоты, трифторметансульфоновой кислоты, трифторуксусной кислоты, трет-бутилсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты, 2,4,6-триметилбензолсульфоновой кислоты, перфтор- или перхлор-алкилсульфоновых кислот, перфтор- или перхлор-алкилкарбоновых кислот, хлористоводородной кислоты, бромистоводородной кислоты, йодистоводородной кислоты, фосфористой кислоты, фосфорной кислоты и их смесей. Гомогенная кислота Брэнстеда предпочтительно представляет собой метансульфоновую кислоту.
[0044] Гетерогенная кислота Брэнстеда предпочтительно выбрана из группы, состоящей из:
(i) комбинаций твердых кислотных оксидов металлов как таковых или на подложке из вещества-носителя;
(ii) катионообменных смол, выбранных из группы, включающей сополимеры стирола, этилвинилбензола и дивинилбензола, функционализированные таким образом, что обеспечивается прививка фрагментов SO3H на ароматическую группу и перфорированные смолы, несущие группы карбоновой и/или сульфоновой кислоты;
(iii) органических сульфоновых, карбоновых и фосфоновых кислот Брэнстеда, которые являются практически несмешиваемыми в реакционной среде при температуре реакции;
(iv) кислотного катализатора, полученного:
- в результате взаимодействия твердой подложки, содержащей неподеленную пару электронов, на которую осаждена органическая кислота Брэнстеда; или
- в результате взаимодействия твердой подложки, содержащей неподеленную пару электронов, на которую осаждено соединение, содержащее Льюисовский кислотный центр; или
- из гетерогенных твердых веществ, функционализированных посредством химической прививки группы кислоты Брэнстеда или ее предшественника; и
(v) гетерогенных гетерополикислот общей формулы HxPMyOz, где Р выбран из фосфора и кремния, а М выбран из вольфрама и молибдена и их комбинаций.
[0045] Предпочтительные гомогенные кислоты Льюиса могут быть выбраны из солей металла, характеризующихся общей формулой,
MXn,
где М представляет собой элемент основной группы или переходный металл, такой как Li, В, Mg, Al, Bi, Fe, Zn, La, Sc, Yb или Pd; X в MXn, как правило, представляет собой анион кислоты или производное кислоты типа Cl, OTf или NTf2, где Tf представляет собой CF3SO2 и n равняется степени окисления М, которая может составлять от 1 до 5. Например, возможны комбинации LiNTf2, Mg(OTf)2, MgCl2, ZnCl2, PdCl2, Fe(OTf)3, Al(OTf)3, AlCl3, Bi(OTf)3, BiCl3, Sc(OTf)3, Ln(OTf)3, Yb(OTf)3. Предпочтительно применяют комбинации жесткого металла или металла на границе между жестким и мягким согласно принципа HSAB (жестких и мягких кислот и оснований), такого как Li, Mg, Al, Sc, Zn, Bi, и слабо координирующих анионов, таких как OTf или NTf2. Примеры таких предпочтительных комбинаций представляют собой LiNTf2, Mg(OTf)2, Al(OTf)3, Bi(OTf)3.
[0046] Предпочтительные гетерогенные кислоты Льюиса могут быть представлены веществами из дискретно выбранных подклассов, образованными при взаимодействии/связывании гомогенных кислот Льюиса, например, комплексных соединений металлов, солей металлов или металлорганических веществ, с полимерными органическими или неорганическими главными цепями. Пример такого подкласса представляет собой полистироловую матрицу со связанными группами Sc(OTf)2. Такой катализатор может быть получен, например, посредством взаимодействия смолы на основе полистиролсульфоновой кислоты, например, Amberlyst 15, с Sc(OTf)3. Число эквивалентов функциональных групп кислоты Льюиса может быть определено в данном случае различными способами, например, посредством кислотно-основного определения непрореагировавших групп сульфоновой кислоты, посредством количественного определения высвобожденной трифторметансульфоновой кислоты, а также посредством измерения с применением ICP количества Sc в смоле.
[0047] В способе по настоящему изобретению необязательно применяют растворители. Типичные примеры подходящих растворителей представляют собой анизол; уксусную кислоту; хлорированные и фторированные углеводороды, такие как фторбензол, хлорбензол, тетрахлорэтан, тетрахлорэтилен, дихлорэтан, дихлорметан; полярные растворители, такие как диглим, глим, дифенилоксид, производные полиалкиленгликоля с защищенными OH-группами, такими как OR***, где R*** представляет собой низшую алкильную или ацильную группу; алифатические углеводороды, такие как гексан, гептан, циклогексан; ациклические эфиры, такие как дибутиловый эфир, диэтиловый эфир, диизопропиловый эфир, дипентиловый эфир и бутилметиловый эфир; циклические эфиры, такие как тетрагидрофуран, диоксан и тетрагидропиран; смешанные циклические/ациклические эфиры, такие как циклопентилметиловый эфир; циклические и ациклические сульфоны, такие как сульфолан; ароматические растворители, такие как толуол, бензол, ксилол; органические ацетаты, такие как этилацетат; органические нитрилы, такие как ацетонитрил, бензонитрил; жидкости на основе кремния, такие как полиметилфенилсилоксан или его смеси; нереакционно-способные ионогенные жидкости, такие как 1-н-бутил-имидазолий-трифторметансульфонат, и 1-этил-3-метил-имидазолий-бис(трифторметилсульфонил)имид; или их смеси.
[0048] В конкретном варианте осуществления настоящего изобретения кислотный катализатор выступает в качестве катализатора и в качестве растворителя.
[0049] В предпочтительном варианте осуществления настоящего изобретения способ включает стадии:
а1) осуществление реакции 2,5-дикетопиперазина с формальдегидом с образованием N,N'-диметилол-2,5-дикетопиперазина;
а2) осуществления реакции N,N'-диметилол-2,5-дикетопиперазина с соединением, содержащим один или несколько ангидридных фрагментов Р-О-Р, причем указанные фрагменты содержат один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), в присутствии кислотного катализатора и необязательно растворителя с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина, или его дегидратированных форм, или их производных, в зависимости от содержащего ангидридный фрагмент Р-О-Р соединения, применяемого для осуществления реакции с N,N'-диметилол-2,5-дикетопиперазином;
b) осуществления гидролиза образованного N,N'-бисфосфонометил-2,5-дикетопиперазина, или его дегидратированных форм, или их производных с образованием N-(фосфонометил)глицина или одного из его производных.
[0050] На стадии а1) реакцию 2,5-дикетопиперазина с формальдегидом осуществляют предпочтительно в водных щелочных условиях, достигаемых посредством растворения основания, где основание предпочтительно выбрано из группы, состоящей из карбоната натрия, карбоната калия, карбоната кальция, бикарбоната натрия, бикарбоната калия, ацетата натрия, ацетата калия, гидроксида натрия, гидроксида калия или их смесей.
[0051] На стадии а1) молярное отношение формальдегида к 2,5-дикетопиперазину находится в диапазоне от приблизительно 2 до приблизительно 8, предпочтительно от приблизительно 2 до приблизительно 3 и более предпочтительно от приблизительно 2,4 до приблизительно 2,8.
[0052] Способ по настоящему изобретению начинают с получения суспензии 2,5-дикетопиперазина в воде и регуляции pH до значения выше приблизительно 7, предпочтительно в диапазоне от приблизительно 7,1 до приблизительно 11,0, более предпочтительно от приблизительно 7,5 до приблизительно 10,0 и наиболее предпочтительно от приблизительно 8,0 до приблизительно 9,0 посредством добавления водного раствора основания, где получаемая в результате суспензия содержит от приблизительно 5% до приблизительно 50% по весу, предпочтительно от приблизительно 10% до приблизительно 40% по весу и более предпочтительно от приблизительно 15% до приблизительно 35% по весу 2,5-дикетопиперазина.
[0053] Формальдегид, предпочтительно водный раствор формальдегида, добавляют в водную суспензию, содержащую 2,5-дикетопиперазин, при перемешивании. Предпочтительно добавление формальдегида в водный раствор 2,5-дикетопиперазина проводят при комнатной температуре, т.е. при температуре от приблизительно 15°С до приблизительно 30°С, после чего получаемую в результате суспензию нагревают до температуры в диапазоне от приблизительно 60°С до приблизительно 100°С.
[0054] Предпочтительно реакцию 2,5-дикетопиперазина с формальдегидом проводят при перемешивании при температуре в диапазоне от приблизительно 60°С до приблизительно 100°С, предпочтительно от приблизительно 70°С до приблизительно 90°С, более предпочтительно при приблизительно 85°С в течение периода времени в диапазоне от приблизительно 20 минут до приблизительно 120 минут, предпочтительно приблизительно 60 минут.
[0055] После завершения реакции 2,5-дикетопиперазина с формальдегидом образованный N,N'-диметилол-2,5-дикетопиперазин выделяют посредством осаждения и фильтрации. Таким образом, содержимое реактора охлаждают до температуры ниже приблизительно 60°С, предпочтительно до температуры в диапазоне от приблизительно 10°С до приблизительно 40°С и более предпочтительно от приблизительно 15°С до приблизительно 30°С, после чего начинается осаждение N,N'-диметилол-2,5-дикетопиперазина. Дополнительное охлаждение до температуры в диапазоне от приблизительно 2°С до приблизительно 10°С в течение периода времени в диапазоне от приблизительно 4 часов до приблизительно 20 часов обеспечивает завершение процесса осаждения.
[0056] Преципитат N,N'-диметилол-2,5-дикетопиперазина выделяют с применением традиционных способов фильтрации. Преципитат N,N'-диметилол-2,5-дикетопиперазина промывают холодной водой и необязательно полярным растворителем, таким как этанол, и в конце высушивают на воздухе или с помощью способов принудительной сушки, таких как, например, конвекция.
[0057] В предпочтительном варианте осуществления настоящего изобретения фильтрат, также называемый маточным щелоком, предпочтительно повторно применяют при последующем получении N,N'-диметилол-2,5-дикетопиперазина с заменой воды, применяемой для получения суспензии. Для данного случая количество формалина, подлежащее введению на стадию а1), может быть снижено с учетом содержания формальдегида маточного щелока.
[0058] Выход при превращении 2,5-дикетопиперазина в N,N'-диметилол-2,5-дикетопиперазин предпочтительно составляет по меньшей мере приблизительно 85%.
[0059] На стадии а2) N,N'-диметилол-2,5-дикетопиперазин суспендируют в кислотном катализаторе или в растворителе, содержащем кислотный катализатор, где получаемая в результате суспензия содержит от приблизительно 2,5% до приблизительно 25% по весу и предпочтительно от приблизительно 5% до приблизительно 20% по весу N,N'-диметилол-2,5-дикетопиперазина.
[0060] Суспензию предпочтительно нагревают до температуры в диапазоне от приблизительно 40°С до приблизительно 80°С, более предпочтительно от приблизительно 50°С до приблизительно 70°С при перемешивании или поддерживают ниже приблизительно 40°C с применением ультразвуковых колебаний с целью преобразования суспензии в раствор. Растворитель необходим для данного конкретного случая, когда гомогенный кислотный катализатор не растворяет N,N'-диметилол-2,5-дикетопиперазин.
[0061] Как только N,N'-диметилол-2,5-дикетопиперазин растворяется в кислотном катализаторе или в растворителе, содержащем кислотный катализатор, содержащее ангидридный фрагмент Р-О-Р соединение, предпочтительно тетрафосфорный гексаоксид, и раствор, содержащий N,N'-диметилол-2,5-дикетопиперазин, устанавливаемый при температуре в диапазоне от приблизительно 0°С до приблизительно 60°С, постепенно смешивают при перемешивании, таким образом, чтобы в ходе смешивания температура реакционной смеси не превышала приблизительно 120°С, предпочтительно приблизительно 90°С и более предпочтительно приблизительно 60°С.
[0062] На стадии а2) соотношение молей кислотного катализатора к эквивалентам ангидридного фрагмента Р-О-Р, содержащего атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), составляет по меньшей мере приблизительно 1 и предпочтительно находится в диапазоне от приблизительно 2 до приблизительно 10.
[0063] На стадии а2) соотношение молей кислотного катализатора к эквивалентам гидроксила N,N'-диметилол-2,5-дикетопиперазина составляет по меньшей мере приблизительно 2 и предпочтительно находится в диапазоне от приблизительно 4 до приблизительно 10.
[0064] Эквивалентное соотношение N,N'-диметилол-2,5-дикетопиперазина к ангидридному фрагменту Р-О-Р на стадии а2) находится в диапазоне от приблизительно 0,2 до приблизительно 2,5, предпочтительно от приблизительно 0,3 до приблизительно 2,0 и более предпочтительно от приблизительно 0,5 до приблизительно 1,5.
[0065] Содержащее ангидридный фрагмент Р-О-Р соединение представляет собой тетрафосфорный гексаоксид. Молярное отношение N,N'-диметилол-2,5-дикетопиперазина к тетрафосфорному гексаоксиду на стадии а2) находится в диапазоне от приблизительно 0,4 до приблизительно 5,0, предпочтительно от приблизительно 0,6 до приблизительно 4,0 и более предпочтительно от приблизительно 1,0 до приблизительно 3,0.
[0066] Постепенное смешивание содержащего ангидридный фрагмент Р-О-Р соединения, предпочтительно тетрафосфорного гексаоксида, и раствора, содержащего N,N'-диметилол-2,5-дикетопиперазин, проводят при оптимальных условиях смешивания. Как только добавление содержащего ангидридный фрагмент Р-О-Р соединения, предпочтительно тетрафосфорного гексаоксида, завершено, содержимое реактора перемешивают при практически постоянной температуре в диапазоне от приблизительно 20°C до приблизительно 120°C, предпочтительно от приблизительно 40°С до приблизительно 100°С в течение периода времени в диапазоне от приблизительно 1 часа до приблизительно 20 часов и предпочтительно от приблизительно 4 часов до приблизительно 16 часов.
[0067] В предпочтительном варианте осуществления настоящего изобретения N,N'-диметилол-2,5-дикетопиперазин в виде твердого вещества или растворенный в кислотном катализаторе и/или в растворителе предпочтительно постепенно добавляют при оптимальных условиях смешивания в смесь, содержащую содержащее ангидридный фрагмент Р-О-Р соединение, необязательно кислотный катализатор и необязательно растворитель.
[0068] В другом варианте осуществления настоящего изобретения стадию а1) и стадию а2) проводят последовательно, т.е. стадию а2) начинают и завершают без выделения N,N'-диметилол-2,5-дикетопиперазина в конце стадии а1).
Для данного конкретного варианта осуществления стадию а1) проводят в кислых условиях, предпочтительно достигаемых посредством метансульфоновой кислоты, в силу чего формальдегид предпочтительно добавляют в виде безводного твердого вещества.
[0069] В предпочтительном варианте осуществления настоящего изобретения N,N'-бисфосфонометил-2,5-дикетопиперазин, или его дегидратированные формы, или их производное выделяют после завершения стадии а) или а2). Следовательно, содержимое реактора со стадии а) или а2) гасят посредством добавления воды; при этом количество добавляемой воды находится в диапазоне от приблизительно 1 до приблизительно 40 эквивалентов на моль исходного 2,5-дикетопиперазина или N,N'-диметилол-2,5-дикетопиперазина. Предпочтительно количество добавляемой воды находится в диапазоне от приблизительно 6 до приблизительно 30 эквивалентов на моль исходного 2,5-дикетопиперазина или N,N'-диметилол-2,5-дикетопиперазина.
[0070] Преципитат N,N'-бисфосфонометил-2,5-дикетопиперазина или его производных, образованный при гашении, выделяют с применением традиционных способов фильтрации, промывают холодной водой и необязательно высушивают на воздухе или с применением способов принудительной сушки, таких как, например, конвекция.
[0071] Выход при превращении N,N'-диметилол-2,5-дикетопиперазина в N,N'-бисфосфонометил-2,5-дикетопиперазин или его производное, достигаемый согласно способу по настоящему изобретению, предпочтительно составляет по меньшей мере приблизительно 75%.
[0072] Водный раствор кислотного катализатора, предпочтительно водный раствор метансульфоновой кислоты, полученный в качестве фильтрата, после выделения N,N'-бисфосфонометил-2,5-дикетопиперазина или одного из его производных содержит от приблизительно 5% до приблизительно 35% воды по весу. Для повторного применения кислотного катализатора в другом цикле стадии а) или а2) его могут очищать с помощью способов, известных из уровня техники, таких как, например, тонкопленочное испарение или испарение падающей пленки, для удаления избыточной воды.
[0073] После завершения стадии а) или а2) содержимое реактора со стадии а) или а2) гидролизируют на стадии b) и выделяют глифосат в форме свободного основания или соли с применением процедур, известных из литературы.
[0074] Во время гидролиза N,N'-бисфосфонометил-2,5-дикетопиперазин или одно из его производных превращается в N-(фосфонометил)глицин.
[0075] Гидролиз проводят в кислых, нейтральных или щелочных условиях периодическом или непрерывным способом.
[0076] При проведении гидролиза в нейтральных условиях добавление ферментного катализатора является необязательным. Ферментативный гидролиз предпочтительно проводят в присутствии амидазы.
[0077] При проведении гидролиза в кислых или щелочных условиях молярное отношение кислоты или основания к N,N'-бисфосфонометил-2,5-дикетопиперазину или одному из его производных находится в диапазоне от приблизительно 1 до приблизительно 20 и предпочтительно от приблизительно 2 до приблизительно 15.
[0078] Щелочной водный раствор, применяемый для гидролиза N,N'-бисфосфонометил-2,5-дикетопиперазина или одного из его производных, предпочтительно получают из основания, выбранного из группы, состоящей из гидроксидов щелочных металлов, гидроксидов щелочно-земельных металлов, аммиака и первичных алифатических аминов; предпочтительно указанное основание представляет собой гидроксид натрия или гидроксид калия.
[0079] Кислотный водный раствор, применяемый для гидролиза N,N'-бисфосфонометил-2,5-дикетопиперазина или одного из его производных, предпочтительно получают из неорганической кислоты; предпочтительно указанная неорганическая кислота является летучей и наиболее предпочтительно данная кислота представляет собой хлористоводородную кислоту.
[0080] Предпочтительно гидролиз проводят в кислых условиях; более предпочтительно кислые условия достигаются посредством водного раствора хлористоводородной кислоты.
[0081] Гидролиз проводят на стадии b) посредством добавления кислого, нейтрального или щелочного водного раствора к N,N'-бисфосфонометил-2,5-дикетопиперазину или его производным со стадии а) или а2) после завершения стадии а) или а2).
[0082] N,N'-бисфосфонометил-2,5-дикетопиперазин или его производное суспендируют в кислом, нейтральном или щелочном водном растворе, где получаемая в результате суспензия содержит от приблизительно 10% до приблизительно 50% по весу и предпочтительно от приблизительно 20% до приблизительно 40% по весу N,N'-бисфосфонометил-2,5-дикетопиперазина или одного из его производных.
[0083] Гидролиз на стадии b) в кислых или щелочных условиях проводят при температуре в диапазоне от приблизительно 90°С до приблизительно 230°С и предпочтительно от приблизительно 110°С до приблизительно 190°С в герметичном реакторе при аутогенном давлении в течение периода времени в диапазоне от приблизительно 1 часа до приблизительно 50 часов и предпочтительно от приблизительно 5 часов до приблизительно 40 часов.
[0084] Гидролиз на стадии b) в нейтральных условиях предпочтительно проводят при температуре в диапазоне от приблизительно 180°С до приблизительно 250°С в течение периода времени в диапазоне от приблизительно 4 часов до приблизительно 80 часов.
[0085] Гидролиз на стадии b) в нейтральных условиях предпочтительно проводят в присутствии ферментного катализатора, предпочтительно амидазы, при температуре в диапазоне от приблизительно 25°С до приблизительно 80°С в течение периода времени в диапазоне от приблизительно 2 часов до приблизительно 100 часов.
[0086] Выход при превращении N,N'-бисфосфонометил-2,5-дикетопиперазина или одного из его производных в N-(фосфонометил)глицин или одно из его производных согласно способу по настоящему изобретению предпочтительно составляет по меньшей мере приблизительно 95%.
[0087] Для стадии b), осуществляемой в кислых условиях, в частности, при применении летучих кислот, таких как, например хлористоводородная кислота, кислоту могут извлекать в конце стадии b), необязательно очищать и повторно применять. Избыток HCl может быть удален посредством дистилляции при атмосферном или пониженном давлении. Кроме того, удаление летучей кислоты подразумевает, что N-фосфонометилглицин может быть получен как таковой.
[0088] Для стадии b), осуществляемой в основных условиях, при применении летучих основных реагентов, таких как, например, аммиак, основание могут извлекать в конце стадии b), необязательно очищать и повторно применять. Избыток аммиака может быть удален посредством дистилляции при атмосферном или пониженном давлении.
[0089] С другой стороны, проведение стадии b) в присутствии нелетучих кислот или оснований приводит в результате к образованию солей N-(фосфонометил)глицина, которые могут быть дополнительно обработаны, соответственно, основанием или кислотой с целью превращения указанных солей N-(фосфонометил)глицина в N-(фосфонометил)глицин.
[0090] В конкретном варианте осуществления настоящего изобретения стадию а) и стадию b) или стадию а2) и стадию b) проводят последовательно, т.е. стадию b) начинают и завершают без выделения N,N'-бисфосфонометил-2,5-дикетопиперазина или его производных в конце стадии а) или стадии а2). Как стадию а) или стадию а2), так и стадию b) предпочтительно проводят в кислых условиях.
[0091] В другом варианте осуществления настоящего изобретения стадию а1), стадию а2) и стадию b) проводят последовательно, т.е. стадию b) начинают и завершают без выделения N,N'-бисфосфонометил-2,5-дикетопиперазина или его производных в конце стадии а2), при этом стадию а2) начинают и завершают без выделения N,N'-диметилол-2,5-дикетопиперазина в конце стадии а1). Все стадии а1), а2) и b) предпочтительно проводят в кислых условиях. Предпочтительно применяют метансульфоновую кислоту и/или хлористоводородную кислоту.
[0092] Для конкретного случая стадию а) и стадию b) или стадию а1), стадию а2) и стадию b) проводят последовательно, причем стадию а) или стадию а1) и стадию а2) следует проводить в безводных условиях, при этом стадию b) проводят в водных условиях.
Примеры
[0093] Следующие примеры иллюстрируют настоящее изобретение; при этом они предназначены только для иллюстрации настоящего изобретения, но не предназначены для ограничения или в противном случае определения объема настоящего изобретения.
[0094] - Примеры с 1 по 3 иллюстрируют стадию а1) настоящего изобретения, т.е. осуществление реакции 2,5-дикетопиперазина и формальдегида с образованием N,N'-диметилол-2,5-дикетопиперазина.
- Примеры с 4 по 21 иллюстрируют стадию а2) настоящего изобретения, т.е. осуществление реакции N,N'-диметилол-2,5-дикетопиперазина с содержащим ангидридный фрагмент Р-О-Р соединением, содержащим один атом Р в степени окисления (+III) и другой атом Р в степени окисления (+III) или (+V), в присутствии кислотного катализатора с образованием N,N'-бисфосфонометил-2,5-дикетопиперазина. Для примеров с 4 по 17 содержащее ангидридный фрагмент Р-О-Р соединение представляет собой тетрафосфорный гексаоксид.
- В примере 22 иллюстрируют стадию а1) и стадию а2), последовательно осуществляемые без выделения продукта со стадии a) (N,N'-диметилол-2,5-дикетопиперазина).
- В примерах 23-37 иллюстрируют стадию b), т.е. гидролиз.
- В примере 38 иллюстрируют стадию а2) и стадию b), последовательно осуществляемые без выделения продукта со стадии а2) (N,N'-бисфосфонометил-2,5-дикетопиперазина).
- В примере 39 иллюстрируют стадию а1), стадию а2) и стадию b), последовательно осуществляемые без выделения продукта со стадии а1) (N,N'-диметилол-2,5-дикетопиперазина) и без выделения продукта со стадии а2) (N,N'-бисфосфонометил-2,5-дикетопиперазина).
- В примере 40 иллюстрируют стадию а), где тетрафосфорный гексаоксид добавляют в смесь 2,5-дикетопиперазина и 1,3,5-триоксана в присутствии метансульфоновой кислоты при 40°С.
- В примерах 41 и 42 иллюстрируют состав соли глифосата после завершения стадии b).
Пример 1 (стадия а)
[0095] В круглодонной колбе, оснащенной мешалкой, конденсатором с водяным охлаждением и термопарой, присоединенной к терморегулятору, суспендировали 11,41 г (0,1 моль) 2,5-дикетопиперазина в 40 мл воды путем перемешивания. Регулировали pH до значения приблизительно 8 посредством добавления нескольких капель 50% вес/вес водного раствора карбоната калия. После того добавляли 20,5 мл 37% вес/вес формалина при перемешивании и получаемую в результате суспензию нагревали до температуры 85°С и выдерживали при данной температуре в течение 1 часа.
Суспензию затем охлаждали до комнатной температуры, после чего начиналось осаждение белого твердого вещества. Через 10 часов при 4°С осаждение завершали и преципитат фильтровали с помощью воронки Бюхнера. Фильтрат, также называемый маточным щелоком, извлекали и повторно применяли в синтезе из примера 2.
Преципитат затем промывали холодной водой и этанолом и в конце высушивали с выходом 15,12 г (0,087 моль) N,N'-диметилол-2,5-дикетопиперазина (выход = 87%) с чистотой 98%.
Пример 2 (стадия а1)
[0096] С применением оснащения из примера 1 11,41 г (0,1 моль) 2,5-дикетопиперазина суспендировали в маточном щелоке из примера 1 путем перемешивания. Регулировали pH до величины приблизительно 8 посредством добавления нескольких капель 50% вес/вес водного раствора карбоната калия. После того добавляли 18 мл формалина при перемешивании и получаемую в результате суспензию нагревали до температуры 85°С, и поддерживали при данной температуре в течение 1 часа.
Суспензию затем дополнительно обрабатывали как в примере 1 с получением 16,83 г N,N'-диметилол-2,5-дикетопиперазина (выход = 97%) с чистотой 98%.
Пример 3 (стадия а1)
[0097] Фильтрат (= маточный щелок из примера 2) дополнительно применяли в последующем синтезе N,N'-диметилол-2,5-дикетопиперазина согласно способу действия, как в примере 2, где маточный щелок из примера 1 заменяли маточным щелоком из примера 2. N,N'-диметилол-2,5-дикетопиперазин получали с выходом 94% и чистотой 98%.
Пример 4 (стадия а2)
[0098] С применением оснащения из примера 1 1,74 г (10,0 ммоль) N,N'-диметилол-2,5-дикетопиперазина суспендировали в 13 мл (146,9 ммоль) метансульфоновой кислоты путем перемешивания. Затем суспензию нагревали до 60°C с обеспечением растворения N,N'-диметилол-2,5-дикетопиперазина в метансульфоновой кислоте. Полученный таким образом раствор охлаждали в ванне со льдом до температуры 0°С. После того 1,14 г (5,2 ммоль) тетрафосфорный гексаоксид постепенно добавляли при перемешивании таким образом, чтобы температура реакционной смеси не превышала 60°С. Реакционную смесь затем поддерживали при 60°С в течение 16 часов и затем гасили посредством добавления 5 мл воды. Получаемый в результате белый преципитат фильтровали с помощью воронки Бюхнера.
Преципитат затем промывали холодной водой и в конце высушивали с выходом 2,48 г (8,2 ммоль) чистого N,N'-бисфосфонометил-2,5-дикетопиперазина (выход = 82%).
Пример 5 (стадия а2)
[0099] С применением оснащения из примера 1 5,23 г (30,0 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 16 мл (246,4 ммоль) метансульфоновой кислоты. Затем реакционную смесь нагревали до 60°C с растворением N,N'-диметилол-2,5-дикетопиперазина. После охлаждения до 0°С 3,43 г (15,6 ммоль) P4O6 медленно добавляли и среду перемешивали в течение ночи при 60°С. Добавляли 5 мл воды и получаемый в результате белый преципитат отфильтровывали, промывали холодной водой и высушивали с выходом 7,26 г белого твердого вещества, состоящего из 98,5 мольных % N,N'-бис(фосфонометил)-2,5-дикетопиперазина. Общий выход N,N'-бис(фосфонометил)-2,5-дикетопиперазина составлял 79,1%.
Пример 6 (стадия а2)
[00100] С применением оснащения из примера 1 1,74 г (10,0 моль) N,N'-диметилол-2,5-дикетопиперазина суспендировали в смеси 10 мл 1,4-диоксана и 1,3 мл (20 ммоль) метансульфоновой кислоты. Медленно добавляли 1,14 г (5,2 ммоль) Р4O6 и смесь перемешивали в течение ночи при 100°С. Среду на основе реакционной смеси охлаждали до комнатной температуры, добавляли 5 мл воды и продолжали перемешивание в течение 1 часа при 100°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 13,5 мольных %.
Пример 7 (стадия а2)
[00101] С применением оснащения из примера 1 1,74 г (10,0 ммоль) N,N'-диметилол-2,5-дикетопиперазина суспендировали в смеси 15 мл уксусной кислоты и 1,3 мл (20 ммоль) метансульфоновой кислоты. Медленно добавляли 1,14 г (5,2 ммоль) Р4O6 и смесь перемешивали в течение ночи при 100°С. Реакционную смесь охлаждали до комнатной температуры, добавляли 5 мл воды и продолжали перемешивание в течение 1 часа при 100°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 23,0 мольных %.
Пример 8 (стадия а2)
[00102] С применением оснащения из примера 1 1,74 г (10,0 ммоль) N,N'-диметилол-2,5-дикетопиперазина суспендировали в 15 мл уксусной кислоты. Затем 1,14 г (5,2 ммоль) Р4O6 медленно добавляли и смесь перемешивали при 90°С в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли 0,9 мл (20,0 ммоль) трифторметансульфоновой кислоты. Получаемый в результате раствор перемешивали в течение ночи при 90°С. Добавляли 5 мл воды и продолжали перемешивание в течение 1 часа при 90°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 24,6 мольных %.
Пример 9 (стадия а2)
[00103] С применением оснащения из примера 1 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 15 мл 1,4-диоксана в атмосфере N2. Медленно добавляли 1,14 г (5 ммоль) P4O6. Затем реакционную смесь нагревали до 80°С в течение 2 часов и охлаждали до 40°С. Добавляли 0,47 г (1 ммоль) трифторметансульфоната алюминия и смесь нагревали до 80°С в течение ночи. Затем добавляли 2 мл воды и смесь нагревали в течение 2 часов до 80°С. Все летучие вещества удаляли под вакуумом и остаточные твердые вещества растворяли водой и доводили до pH 5,4 посредством добавления гидроксида натрия. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 11,3% вес/вес, как определяли с помощью 31Р-ЯМР спектроскопии в H2O/D2O.
Пример 10 (стадия а2)
[00104] С применением оснащения из примера 1 0,77 г (3,5 ммоль) P4O6 медленно добавляли к смеси 10 мл 1,4-диоксана и 0,9 мл метансульфоновой кислоты в атмосфере N2. Затем температуру поднимали до 60°С. Небольшими порциями добавляли 1,22 г (7 ммоль) N,N'-диметилол-2,5-дикетопиперазина. Затем реакционную смесь нагревали до 100°С в течение 4 часов. После охлаждения до 40°С добавляли 2 мл воды и смесь нагревали в течение 2 часов до 100°С. Все летучие вещества удаляли под вакуумом и остаточные твердые вещества растворяли водой и доводили до pH 5,4 посредством добавления гидроксида натрия. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 14% вес/вес, как определяли с помощью 31Р-ЯМР спектроскопии в H2O/D2O.
Пример 11 (стадия а2)
[00105] С применением оснащения из примера 1 1,22 г (7 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 8 мл трифторуксусной кислоты и нагревали до 40°С в атмосфере N2. Медленно добавляли 0,80 г (3,6 ммоль) P4O6. Затем реакционную смесь нагревали до 80°С в течение ночи. Затем добавляли 2 мл воды и смесь нагревали в течение 2 часов до 80°С. Раствор разбавляли водой и доводили до pH 5,4 посредством добавления гидроксида натрия. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 61,0% вес/вес, как определяли с помощью 31Р-ЯМР спектроскопии в H2O/D2O.
Пример 12 (стадия а2)
[00106] С применением оснащения из примера 1 1,40 г (8 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 5 мл метансульфоновой кислоты и нагревали до 40°С в атмосфере N2. Медленно добавляли 0,92 г (4,2 ммоль) P4O6. Затем реакционную смесь нагревали до 80°С в течение ночи. Затем добавляли 2 мл воды и смесь нагревали в течение 2 часов до 105°С. Образованное белое твердое вещество отфильтровывали и последовательно прополаскивали водой и 2 н. HCl перед сублимационной сушкой. Выделяли 2,06 г белого твердого вещества, состоящего из N,N'-бисфосфонометил-2,5-дикетопиперазина при 95 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 85,2%.
Пример 13 (стадия а2)
[00107] С применением оснащения из примера 1 4,35 г (25 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 13 мл метансульфоновой кислоты и нагревали до 40°С в атмосфере N2. Медленно добавляли 2,86 г (13 ммоль) P4O6. Затем реакционную смесь нагревали до 80°С в течение ночи. Затем добавляли 2 мл воды и смесь нагревали в течение 2 часов до 100°С. Образованное белое твердое вещество отфильтровывали и последовательно прополаскивали этанолом и водой перед высушиванием. Выделяли 5,6 г белого твердого вещества, состоящего из N,N'-бисфосфонометил-2,5-дикетопиперазина при 95 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии в DMSO-d6. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 74,1%.
Пример 14 (стадия а2)
[00108] С применением оснащения из примера 1 8,2 г (100 ммоль) фосфористой кислоты и 0,6 мл (20,8 ммоль) P4O6 предварительно смешивали в течение 20 мин. при 85°С. Затем добавляли 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина и реакционную смесь нагревали до 85°С в течение ночи. Затем добавляли 5 мл воды и смесь перемешивали при 85°С в течение 1 часа. Выход N,N'-бисфосфонометил-2,5-дикетопиперазина в неочищенной реакционной смеси составлял 0,6 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 15 (стадия а2)
[00109] С применением оснащения из примера 1 6 мл (92,4 ммоль) метансульфоновой кислоты, 1,8 г (22 ммоль) фосфористой кислоты и 0,3 мл (2,6 ммоль) P4O6 предварительно смешивали в течение 20 мин. при 85°С. Затем добавляли 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина и реакционную смесь нагревали до 85°С в течение ночи. Затем добавляли 5 мл воды и смесь перемешивали при 85°С в течение 1 часа. Выход N,N'-бисфосфонометил-2,5-дикетопиперазина в неочищенной реакционной смеси составлял 60,7%, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 16 (стадия а2)
[00110] С применением оснащения из примера 1 0,82 г (10 ммоль) фосфористой кислоты смешивали с 5 мл (78 ммоль) метансульфоновой кислоты. Медленно добавляли 1,37 г (10 ммоль) PCl3, затем 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина. Затем реакционную смесь перемешивали в течение 6 часов при 60°С. При температуре окружающей среды добавляли 0,5 мл воды и 20 мл ацетона и смесь выдерживали. Через 1 час образованное белое твердое вещество фильтровали и высушивали. Маточный щелок выдерживали в течение ночи при 4°С, что вызывало образование дополнительного белого твердого вещества, которое собирали и высушивали. Всего получали 1,4 г объединенных белых твердых веществ. Твердое вещество состояло из N,N'-бисфосфонометил-2,5-дикетопиперазина при 82 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 40,8%.
Пример 17 (стадия а2)
[00111] С применением оснащения из примера 1 1,39 г (8 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 5 мл (78 ммоль) метансульфоновой кислоты. Медленно добавляли 4,20 г (16 ммоль) тетраэтилпирофосфита. Затем реакционную смесь нагревали до 60°С в течение 8 часов. Затем добавляли 5 мл воды и все летучие вещества удаляли в вакууме. Остаток суспендировали в 10 мл ацетона и белое твердое вещество отфильтровывали, и высушивали с выходом 1,8 г. Твердое вещество состояло из N,N'-бисфосфонометил-2,5-дикетопиперазина при 72 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 66,0%.
Пример 18 (стадия а2)
[00112] С применением оснащения из примера 1 10 мл (154 ммоль) метансульфоновой кислоты, 1,64 г (20 ммоль) фосфористой кислоты и 2,80 г (20 ммоль) P2O5 смешивали в течение 1 часа при температуре выше 50°С. Затем добавляли 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина и реакционную смесь нагревали до 85°С в течение ночи. Затем добавляли 6 мл воды и смесь перемешивали при 85°С в течение 1 часа. Выход в неочищенной реакционной смеси представлял собой количественно оцениваемый, как определяли с помощью 31Р-ЯМР спектроскопии. Осажденный N,N'-бисфосфонометил-2,5-дикетопиперазин отфильтровывали и высушивали. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 2,56 г (85,0%).
Пример 19 (стадия а2)
[00113] С применением оснащения из примера 1 10 мл (154 ммоль) метансульфоновой кислоты, 1,8 мл (22 ммоль) диметилфосфита и 2,8 г (20 ммоль) P2O5 смешивали в течение 1 часа при температуре выше 50°С. Затем добавляли 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина и реакционную смесь нагревали до 85°С в течение ночи. Затем добавляли 6 мл воды и смесь перемешивали при 85°С в течение 1 часа. Выход N,N'-бисфосфонометил-2,5-дикетопиперазина в неочищенной реакционной смеси составлял 97 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии. Осажденный N,N'-бисфосфонометил-2,5-дикетопиперазин отфильтровывали и высушивали. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 2,53 г (84,0%).
Пример 20 (стадия а2)
[00114] С применением оснащения из примера 1 6 мл метансульфоновой кислоты, 0,97 г (8,8 ммоль) диметилфосфита и 0,85 г (6 ммоль) P2O5 смешивали в течение 20 минут при температуре выше 85°С. Затем добавляли 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина и реакционную смесь нагревали до 85°С в течение ночи. Затем добавляли 5 мл воды и смесь перемешивали при 85°С в течение 1 часа. Выход N,N'-бисфосфонометил-2,5-дикетопиперазина в неочищенной реакционной смеси составлял 3,6 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 21 (стадия а2)
[00115] В круглодонной колбе, оснащенной механической мешалкой, термометром и воронкой для твердой добавки, в атмосфере N2 4,64 г (21,1 ммоль) P4O6 разводили в 24 мл метансульфоновой кислоты при постоянной обработке ультразвуком. Постепенно добавляли 6,96 г (40 ммоль) N,N'-диметилол-2,5-дикетопиперазина в реакционную смесь в течение периода 1,5 часа при постоянной обработке ультразвуком. После добавления обработку ультразвуком поддерживали в течение 1 часа и затем прекращали. В неочищенный реакционной смеси осуществляли реакцию при комнатной температуре в течение 24 часов. Затем постепенно добавляли 8 мл воды в смесь при поддержании температуры ниже 40°С. Выход N,N'-бисфосфонометил-2,5-дикетопиперазина в неочищенной реакционной смеси составлял 93 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 22 (стадия а1 + стадия а2)
[00116] В круглодонной колбе, оснащенной механической мешалкой, термометром и конденсатором, 4,52 г (40 ммоль) 2,5-дикетопиперазина и 2,41 г (80 ммоль) параформальдегида смешивали с 40 мл метансульфоновой кислоты в атмосфере N2. Смесь нагревали до 85°С в течение 2 ч. до растворения твердого вещества. Затем смесь охлаждали до 40°С и медленно добавляли 4,40 г (20 ммоль) P4O6. Затем реакционную смесь нагревали до 85°С в течение ночи. Затем добавляли 10 мл воды и смесь нагревали в течение 2 ч. при 85°С. Выход N,N'-бисфосфонометил-2,5-дикетопиперазина в неочищенной реакционной смеси составлял 17,0%, как определяли с помощью 31Р-ЯМР спектроскопии. Осажденный N,N'-бисфосфонометил-2,5-дикетопиперазин отфильтровывали и высушивали. Общий выход N,N'-бисфосфонометил-2,5-дикетопиперазина составлял 0,97 г (8,0%).
Пример 23 (стадия b)
[00117] В устойчивой к давлению закупоренной пробирке 0,75 г (2,5 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 1,35 мл 30% вес/вес водного раствора HCl (12,41 ммоль HCl, 5 экв.). Получаемую в результате суспензию перемешивали в течение 40 часов при 120°С, что приводило в результате к полному растворению всех твердых веществ. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицина гидрохлорид выявляли при 99,3 мольных %.
Пример 24 (стадия b)
[00118] В устойчивой к давлению закупоренной пробирке 0,70 г (2,3 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 1,8 мл 36% вес/вес водного раствора HCl (34,5 ммоль HCl, 15 экв.). Получаемую в результате суспензию выдерживали в течение 18 часов при 115°С, что приводило в результате к полному растворению всех твердых веществ. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. Наблюдали полное превращение N-(фосфонометил)глицина гидрохлорида.
Пример 25 (стадия b)
[00119] В устойчивой к давлению закупоренной пробирке 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды вместе с 2 эквивалентами метансульфоновой кислоты. Получаемую в результате суспензию выдерживали в течение 20 часов при 150°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-фосфонометил)глицина метансульфонат выявляли при 82,6 мольных %.
Пример 26 (стадия b)
[00120] В устойчивой к давлению закупоренной пробирке 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды вместе с 2 эквивалентами 36% вес/вес HCl. Получаемую в результате суспензию выдерживали в течение 20 часов при 150°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицина гидрохлорид выявляли при 84,3 мольных %.
Пример 27 (стадия b)
[00121] В устойчивой к давлению закупоренной пробирке 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды вместе с 4 эквивалентами 36% вес/вес HCl. Получаемую в результате суспензию выдерживали в течение 20 часов при 165°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицина гидрохлорид выявляли при 93,0 мольных %.
Пример 28 (стадия b)
[00122] В устойчивой к давлению закупоренной пробирке 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды вместе с 2 эквивалентами 98% серной кислоты. Получаемую в результате суспензию выдерживали в течение 20 часов при 165°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицина сульфат выявляли при 93,3 мольных %.
Пример 29 (стадия b)
[00123] В устойчивой к давлению закупоренной пробирке 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 2 мл воды вместе с 2 эквивалентами 98% серной кислоты. Получаемую в результате суспензию выдерживали в течение 20 часов при 165°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицина сульфат выявляли при 93 мольных %.
Пример 30 (стадия b)
[00124] В устойчивой к давлению закупоренной пробирке 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды вместе с 4 эквивалентами 98% серной кислоты. Получаемую в результате суспензию выдерживали в течение 20 часов при 165°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицина сульфат выявляли при 96,9 мольных %.
Пример 31 (стадия b)
[00125] В устойчивой к давлению закупоренной пробирке 1 г (3,2 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 6 мл воды. Получаемую в результате суспензию выдерживали в течение 72 часов при 150°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицин выявляли при 54,9 мольных %.
Пример 32 (стадия b)
[00126] В устойчивой к давлению закупоренной пробирке 0,52 г (0,8 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды. Получаемую в результате суспензию выдерживали в течение 25 часов при 150°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицин выявляли при 34,8 мольных %.
Пример 33 (стадия b)
[00127] В устойчивой к давлению пробирке во внутренней части автоклава Парра 0,50 г (1,6 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 3 мл воды и устанавливали давление N2 1 бар. Получаемую в результате суспензию перемешивали в течение 3 часов при 240°С. Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицин выявляли при 41,0 мольных %.
Пример 34 (стадия b)
[00128] Эксперимент проводили непрерывным способом с применением системы для проточной химии Vapourtec R серии. Взвесь 0,25 г (0,8 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина в воде (1 мл) загружали в пробоотборную петлю. С применением воды в качестве растворителя взвесь элюировали через высокотемпературный трубчатый реактор (10 мл), нагретый до 200°С, при скорости потока 0,1 мл. мин-1 в течение периода 120 минут. Регулятор обратного давления при 150 фунтах на кв. дюйм присоединяли в линию между реактором и сборником, из которого полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. N-(фосфонометил)глицин выявляли при 8,8 мольных %.
Пример 35 (стадия b)
[00129] В устойчивой к давлению закупоренной пробирке 1,0 г (3,3 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 6 мл воды вместе с 10 эквивалентами 36% вес/вес HCl. Получаемую в результате суспензию выдерживали в течение 6 часов при 165°С. Избыток HCl затем отгоняли из раствора посредством дистилляции. Извлекали 0,98 г свободного глифосата посредством кристаллизации из водного остаточного раствора (выделяли 92%) с чистотой приблизительно 99%, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 36 (стадия b)
[00130] В устойчивой к давлению закупоренной пробирке 1,0 г (3,3 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 6 мл воды вместе с 6 эквивалентами 50% вес/вес NaOH. Получаемую в результате суспензию выдерживали в течение 6-7 часов при 165°С. Тринатриевую соль глифосата выявляли в растворе при 98%, как определяли с помощью 31Р-ЯМР спектроскопии. Раствор выпаривали с выходом белого твердого вещества.
Пример 37 (стадия b)
[00131] В устойчивой к давлению закупоренной пробирке 1,0 г (3,3 ммоль) N,N'-бисфосфонометил-2,5-дикетопиперазина суспендировали в 6 мл 29 вес. % водного раствора аммиака (49,6 ммоль). Получаемую в результате суспензию выдерживали в течение 6 часов при 165°С.
Полученный раствор анализировали с помощью 31Р-ЯМР спектроскопии. Аммонийную соль N-(фосфонометил)глицина выявляли при 9 мольных %.
Пример 38 (стадия а2 + стадия b)
[00132] С применением оснащения из примера 1 1,74 г (10 ммоль) N,N'-диметилол-2,5-дикетопиперазина смешивали с 6 мл метансульфоновой кислоты и нагревали до 40°С в атмосфере N2. Медленно добавляли 1,14 г (5,2 ммоль) P4O6. Затем реакционную смесь нагревали до 70°С в течение 6 часов. Затем по каплям добавляли 2 мл воды при комнатной температуре и полученную суспензию хранили при 4°С в течение ночи. Получаемую в результате суспензию разбавляли 9 мл воды, перемешивали до однородности и нагревали в течение 7 часов при 150°С в устойчивой к давлению закупоренной пробирке. При охлаждении добавляли 2 мл пропанола и раствор оставляли для медленного испарения. Выделяли 0,408 г бело-желтоватого твердого вещества, состоящего из мезилатной соли глифосата при 98 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 39 (стадия а1 + стадия а2 + стадия b)
[00133] В круглодонной колбе, оснащенной механической мешалкой, термометром и конденсатором, 2,28 г (20 ммоль) 2,5-дикетопиперазина и 1,20 г (40 ммоль) параформальдегида смешивали с 10 мл уксусной кислоты и 1,3 мл (20 ммоль) метансульфоновой кислоты. Смесь нагревали до 80°С в течение 6 часов до растворения всех твердых веществ. Затем смесь охлаждали до температуры окружающей среды и медленно добавляли 2,20 г (10 ммоль) P4O6. Затем реакционную смесь нагревали в течение 6 часов до 80°С. Затем добавляли 10 мл воды и смесь нагревали в трубке под давлением в течение 8 часов при 150°С. Выход N-(фосфонометил)глицина определяли с помощью 31Р-ЯМР спектроскопии при 11,5% вес/вес.
Пример 40 (стадия а)
[00134] С применением оснащения из примера 1 1,71 г (15 ммоль) 2,5-дикетопиперазина и 0,90 г (10 ммоль) 1,3,5-триоксана смешивали с 10 мл метансульфоновой кислоты и нагревали до 40°С в атмосфере N2. Затем добавляли 1,65 г (0,0075 ммоль) P4O6. Затем реакционную смесь нагревали до 80°С в течение ночи. Затем добавляли 2 мл воды и анализировали полученную смесь. N,N'-бисфосфонометил-2,5-дикетопиперазин выявляли при 5,4 мольных %, как определяли с помощью 31Р-ЯМР спектроскопии.
Пример 41 (стадия b и состав)
[00135] Смешивали 0,5 г свободного глифосата с 1 эквивалентом (2,95 ммоль) 97% изопропиламина в 3 мл воды и получаемую в результате суспензию перемешивали при 80°С в течение 30 минут. Моноизопропиламмонийную соль глифосата выявляли при >99%, как определяли с помощью 31Р-ЯМР спектроскопии. Раствор выпаривали с выходом твердого вещества.
Пример 42 (стадия b и состав)
[00136] Последовательно смешивали 0,3 г свободного глифосата (1,77 ммоль), суспендированного в 2 мл воды, сначала с 3 эквивалентами (5,31 ммоль) 50% вес/вес NaOH в течение 10 минут, а затем с 2 эквивалентами (3,54 ммоль) 36% вес/вес HCl в течение 10 минут. Затем 1 эквивалент триметилсульфония йодида (1,77 ммоль, 0,361 г) добавляли в раствор, который перемешивали при комнатной температуре в течение 2 часов. Монотриметилсульфониевую соль глифосата выявляли при 99%, как определяли с помощью 31Р-ЯМР спектроскопии. Раствор выпаривали с выходом белого твердого вещества.
Способ синтеза альфа-аминоалкиленфосфоновой кислоты
Способ синтеза n-(фосфонометил)глицина
Способ синтеза аминоалкиленфосфоновой кислоты
Способ обработки воды
Способ вторичного извлечения нефти
Способ производства концентрированной фосфористой кислоты
Способ производства аминоалкиленфосфоновой кислоты
Композиция для обработки поверхности, содержащая соединения фосфоновых кислот
Способ получения диалкилфосфитов
Способ получения фосфоноалкилиминодиуксусной кислоты
Способ получения аминогидроксидифосфоновых кислот
Способ синтеза альфа-аминоалкиленфосфоновой кислоты
Способ синтеза n-(фосфонометил)глицина

