Результат интеллектуальной деятельности: АНАЛОГ БЕНЗОФУРАНА В КАЧЕСТВЕ ИНГИБИТОРА NS4B
Вид РИД
Изобретение
Область техники, к которой относится изобретение
[1] Настоящее изобретение относится к новому классу аналогов бензофурана в качестве ингибиторов NS4B, в частности, относится к соединению, имеющему структурную формулу (I), или к его фармацевтически приемлемой соли.
Предшествующий уровень техники
[2] HCV представляет собой один из основных патогенов человека и согласно оценкам во всем мире насчитывается приблизительно 0,17 миллиарда носителей хронической HCV-инфекции, что в 5 раз превышает количество носителей инфекции, вызываемой вирусом иммунодефицита человека 1 типа. Хронические HCV-инфекции могут перерастать в тяжелые прогрессирующие заболевания печени, в том числе в цирроз печени и гепатоцеллюлярную карциному. Таким образом, хроническая HCV-инфекция является основной причиной смерти, связанной с заболеваниями печени, в мире.
[3] В настоящее время стандартную терапию хронической HCV-инфекции осуществляют посредством α-интерферона, рибавирина и противовирусного лекарственного совместного введения средства прямого действия (DAA), которое представляет собой одно из лекарственных средств, разрешенных к применению за последние два года. Хотя лечебный эффект значительно α-интерферона и рибавирина, терапия улучшен по сравнению с совместным введением является неэффективной для некоторых носителей хронической HCV-инфекции и вирус α-интерферон может становиться устойчивым к лекарственному средству. Кроме того, и рибавирин характеризуются очевидными нежелательными реакциями. Следовательно, крайне необходимым является новое и эффективное лекарственное средство для лечения хронической HCV-инфекции.
[4] HCV является вирусом с одноцепочечной РНК, который принадлежит к отдельному роду семейства Flaviviridae. Все представители семейства Flaviviridae являются оболочечными вирусными частицами, содержащими геном, представленный цепью РНК, который кодирует все известные вирус-специфические белки посредством трансляции одной непрерывной открытой рамки считывания (ORF).
[5] Существует значительная неоднородность среди нуклеотидов генома HCV и кодируемых аминокислотных последовательностей. Было определено, что существует по меньшей мере 6 главных генотипов и более 50 подтипов. Распределение основных генотипов HCV варьируется по всему миру. Несмотря на большое количество исследований роли генотипов в патогенезе и лечении, клиническая важность генетической неоднородности HCV остается неясной.
[6] Геном HCV, представленный РНК, содержит приблизительно 9500 нуклеотидов с одной открытой рамкой считывания, кодирующей один полипротеин из приблизительно 3000 аминокислот. В инфицированных клетках полипротеин расщепляется клеточными протеазами и вирусными протеазами в нескольких участках с получением структурных и неструктурных (NS) белков. Что касается HCV, образование зрелых неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A и NS5B) достигалось с помощью двух типов вирусных протеаз. Обычно считают, что первым типом (NS2) является металлопротеаза, расщепляющая в участке соединения NS2-NS3; вторая протеаза представляет собой сериновую протеазу, содержащуюся в N-концевой области NS3 (также называемую в данном документе протеазой NS3), которая опосредует все последующие расщепления ниже NS3, расщепление по цис-типу в участке соединения NS3-NS4A и расщепления по транс-типу в участках соединения NS4A-NS4B, NS4B-NS5A и NS5A-NA5B. Белок NS4A, по-видимому, имеет ряд функций, например, является кофактором протеазы NS3 и, возможно, содействует NS3 и другим ферментативным компонентам, необходимым для репликации вируса, в осуществлении локализации в мембране. Белок NS3 также демонстрирует нуклеозидтрифосфатазную и РНК-хеликазную активности. Функции двух белков NS4B и NS5A не вполне ясны, но они играют важную роль в репликации HCV. NS4B представляет собой трансмембранный белок, участвующий в образовании репликативного комплекса вируса. NS5A представляет собой фосфорилированный белок, участвующий в репликации РНК вируса и образовании вирусных частиц. NS5B (также известный как полимераза HCV) представляет собой РНК-зависимую РНК-полимеразу, участвующую в репликации РНК генома HCV.
[7] В WO2013095275, WO2012122716, CN102863428A и т. д. соответственно сообщается о ряде соединений в качестве ингибиторов HCV, эффекты которых в аспектах активности, растворимости и т. п. нуждаются в дальнейшем улучшении.
Описание изобретения
[8] Целью настоящего изобретения является обеспечение соединения, имеющего структурную формулу (I),
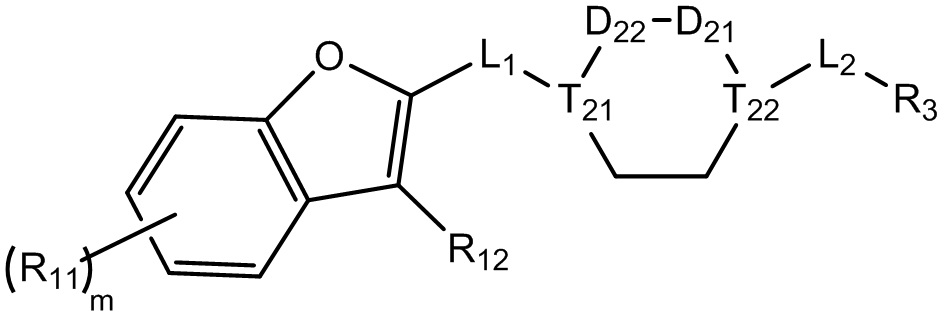
(I),
[9] или его фармацевтически приемлемой соли, где
[10] компонент 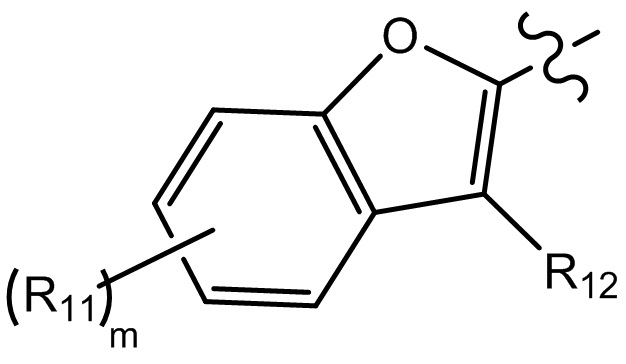 может быть заменен на
может быть заменен на 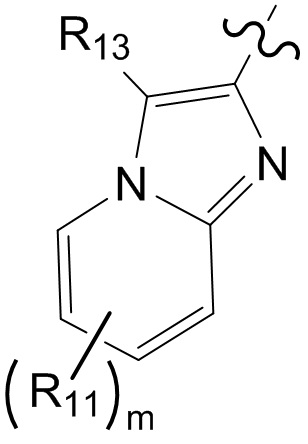 или
или 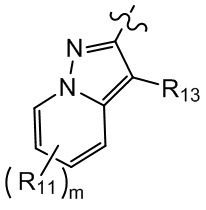 ;
;
[11] ноль, один или два из T21-22 выбраны из N, при этом остальные из них выбраны из C(Rt);
[12] каждый из D21-22, L1-2 независимо выбран из группы, состоящей из -[C(Rd1)(Rd2)]0-2-, -C(=O)-, -C(=O)N(Rd3)-, -N(Rd4)-, -C(=NRd5)-, -S(=O)2N(Rd6)-, -S(=O)N(Rd7)-, -O-, -S-, -C(=O)O-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(Rd8)C(=O)N(Rd9)-;
[13] m выбрано из группы, состоящей из 1, 2, 3 и 4;
[14] R3 выбран из группы, состоящей из H, F, Cl, Br, I, CN, OH, SH, NH2, CHO, COOH, C(=O)NH2, S(=O)NH2, S(=O)2NH2, или выбран из группы, состоящей из C1-10-алкила или гетероалкила, C3-10-циклогидрокарбила или гетероциклогидрокарбила, C1-10-алкила или гетероалкила, замещенного C3-10-циклогидрокарбилом или гетероциклогидрокарбилом, необязательно замещенных нолем, одним, двумя или тремя Rt;
[15] каждый из R11-13, Rt, Rd1, Rd2 независимо выбран из группы, состоящей из H, F, Cl, Br, I, CN, OH, SH, NH2, CHO, COOH, C(=O)NH2, или выбран из группы, состоящей из C1-10-алкила или гетероалкила, необязательно замещенного R01, C3-10-циклогидрокарбила или гетероциклогидрокарбила, C1-10-алкила или гетероалкила, замещенного C3-10-циклогидрокарбилом или гетероциклогидрокарбилом;
[16] R01 выбран из группы, состоящей из F, Cl, Br, I, CN, OH, SH, NH2 и R02;
[17] R02 выбран из группы, состоящей из C1-10-алкила, C1-10-алкиламино, N,N-бис(C1-10-алкил)амино, C1-10-алкоксила, C1-10-алканоила, C1-10-алкоксикарбонила, C1-10-алкилсульфонила, C1-10-алкилсульфинила, C3-10-циклоалкила, C3-10-циклоалкиламино, C3-10-гетероциклоалкиламино, C3-10-циклоалкоксила, C3-10-циклоалканоила, C3-10-циклоалкоксикарбонила, C3-10-циклоалкилсульфонила и C3-10-циклоалкилсульфинила;
[18] "гетеро" представляет собой гетероатом или гетероатомную группу, выбранную из группы, состоящей из -C(=O)N(Rd3)-, -N(Rd4)-, -C(=NRd5)-, -S(=O)2N(Rd6)-, -S(=O)N(Rd7)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и/или -N(Rd8)C(=O)N(Rd9)-;
[19] каждый из Rd3-d9 независимо выбран из группы, состоящей из H, OH, NH2 и R02;
[20] R02 необязательно замещен R001;
[21] R001 выбран из группы, состоящей из F, Cl, Br, I, CN, OH, N(CH3)2, NH(CH3), NH2, CHO, COOH, C(=O)NH2, тригалогенметила, дигалогенметила, моногалогенметила, аминометила, гидроксиметила, метила, метиламино, формила, метоксикарбонила, метилсульфонила и метилсульфинила;
[22] количество R01, R001, гетероатомов или гетероатомных групп независимо выбрано из группы, состоящей из 0, 1, 2 и 3;
[23] необязательно существует другая соединяющая связь (CH2)1-3 между T21 и T22.
[24] В некоторых вариантах осуществления настоящего изобретения каждый из D21-22, L1-2 независимо выбран из группы, состоящей из (CH2)0-2, -C(=O)-, -C(=O)NH- и -C(=O)N(Me)-.
[25] В некоторых вариантах осуществления настоящего изобретения компонент выбран из 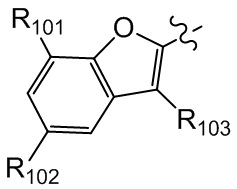 , каждый из R101-103 независимо выбран из группы, состоящей из H, F, Cl, Br, I, CN, OH, SH, NH2, CHO, COOH, C(=O)NH2, или выбран из группы, состоящей из C1-10-алкила или гетероалкила, C3-10-циклогидрокарбила или гетероциклогидрокарбила и C1-10-алкила или гетероалкила, замещенного C3-10-циклогидрокарбилом или гетероциклогидрокарбилом, необязательно замещенных нолем, одним, двумя или тремя R01, при этом R01 определен в формуле изобретения.
, каждый из R101-103 независимо выбран из группы, состоящей из H, F, Cl, Br, I, CN, OH, SH, NH2, CHO, COOH, C(=O)NH2, или выбран из группы, состоящей из C1-10-алкила или гетероалкила, C3-10-циклогидрокарбила или гетероциклогидрокарбила и C1-10-алкила или гетероалкила, замещенного C3-10-циклогидрокарбилом или гетероциклогидрокарбилом, необязательно замещенных нолем, одним, двумя или тремя R01, при этом R01 определен в формуле изобретения.
[26] В некоторых вариантах осуществления настоящего изобретения каждый из R101-103 независимо выбран из группы, состоящей из F, Cl, Br, -CF3, -CHF2, CN, Me, этила, пропила, циклопропила и изопропила.
[27] В некоторых вариантах осуществления настоящего изобретения компонент выбран из группы, состоящей из 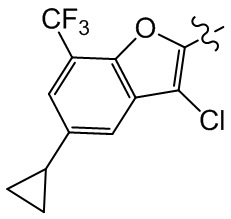 ,
, 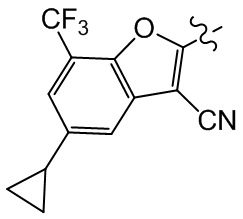 ,
, 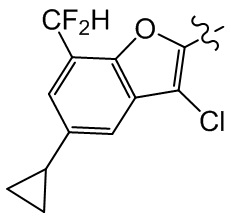 ,
, 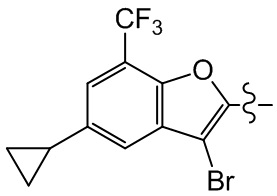 ,
, 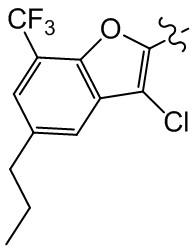 ,
, 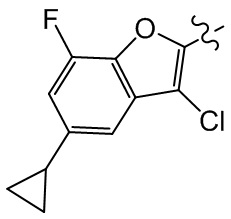 ,
, 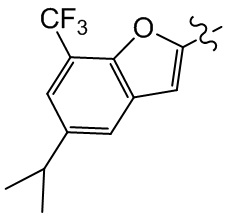 ,
, 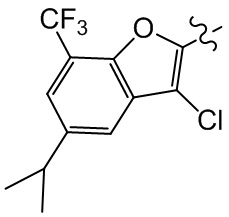 и
и 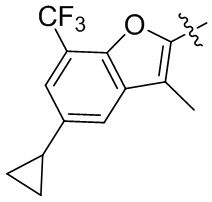 ;
;
или компонент выбран из 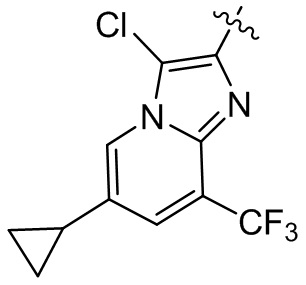 ;
;
или компонент выбран из 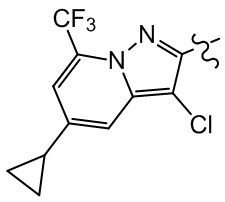 .
.
[28] В некоторых вариантах осуществления настоящего изобретения компонент 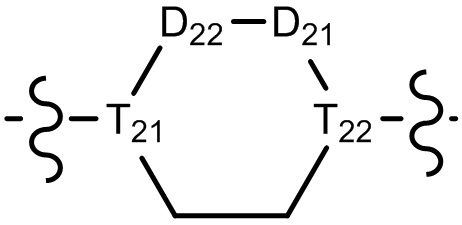 выбран из группы, состоящей из
выбран из группы, состоящей из 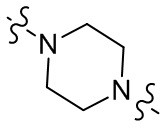 ,
, 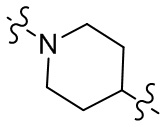 ,
, 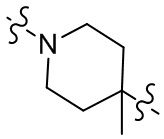 ,
, 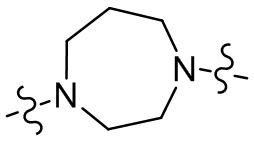 ,
, 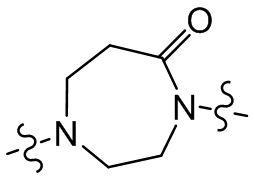 ,
, 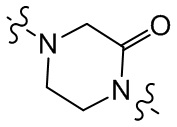 ,
, 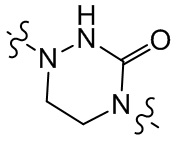 и
и 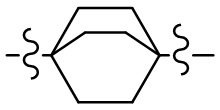 .
.
[29] В некоторых вариантах осуществления настоящего изобретения R3 выбран из группы, состоящей из 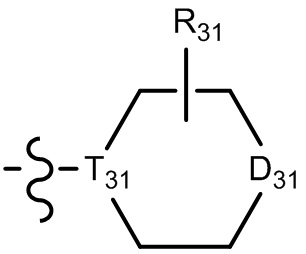 ,
, 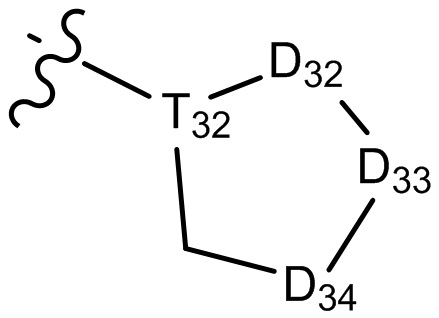 ,
, 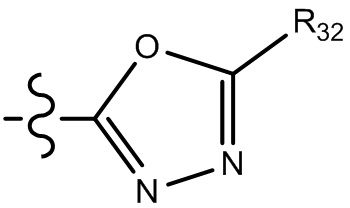 ,
, 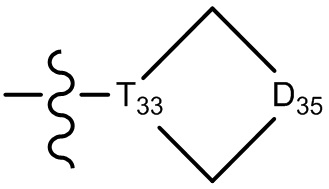 , -N(R33)(R34) и O(R35), или выбран из C1-3-алкила, циклопропила, фенила или пиридила, замещенного R001, где
, -N(R33)(R34) и O(R35), или выбран из C1-3-алкила, циклопропила, фенила или пиридила, замещенного R001, где
[30] каждый из T31-33 независимо выбран из группы, состоящей из N и C(Rt);
[31] каждый из D31-35 независимо выбран из группы, состоящей из -[C(Rd1)(Rd2)]0-2-, -C(=O)-, -C(=O)N(Rd3)-, -N(Rd4)-, -C(=NRd5)-, -S(=O)2N(Rd6)-, -S(=O)N(Rd7)-, -O-, -S-, -C(=O)O-, -C(=S)-, -S(=O)- и -S(=O)2-;
[32] каждый из R31-35 независимо выбран из группы, состоящей из H, F, Cl, Br, I, CN, OH, SH, NH2, CHO, COOH и C(=O)NH2, или выбран из группы, состоящей из C1-10-алкила или гетероалкила, необязательно замещенного R01, C3-10-циклогидрокарбила или гетероциклогидрокарбила, C1-10-алкила или гетероалкила, замещенного C3-10-циклогидрокарбилом или гетероциклогидрокарбилом;
[33] определения Rt, Rd1-d7, R01 соответствуют таковым в пункте 1 формулы изобретения;
[34] необязательно существует другая соединяющая связь (CH2)1-3 между T31 и D31, D33 и D34, T33 и D35.
[35] В некоторых вариантах осуществления настоящего изобретения каждый из D31-35 независимо выбран из группы, состоящей из -C(=O)-, -O-, метилена, -N(CH3)-, -CH(OH)- и -CF2-; каждый из R31-34 независимо выбран из H, метила, этила, н-пропила, изопропила.
[36] В некоторых вариантах осуществления настоящего изобретения R3 выбран из группы, состоящей из 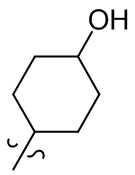 ,
, 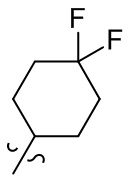 ,
, 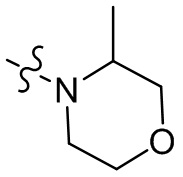 ,
, 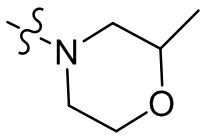 ,
, 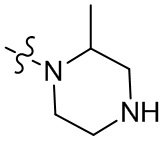 ,
, 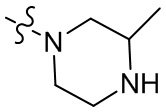 ,
, 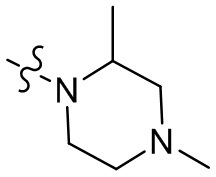 ,
, 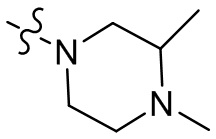 ,
, 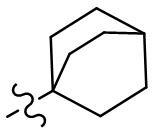 ,
, 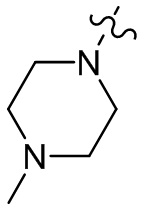 ;
; 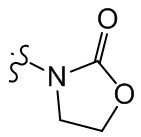 ,
, 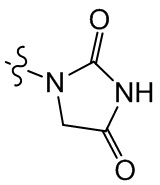 ,
, 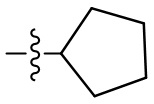 ,
, 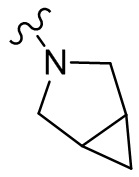 ;
; 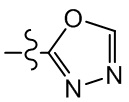 ,
,  ;
;  ,
,  ,
,  ,
, 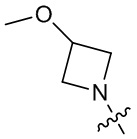 ,
, 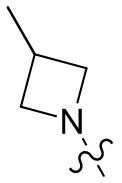 ,
, 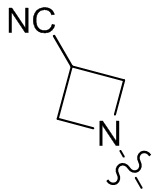 ,
, 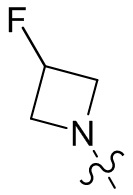 ,
, 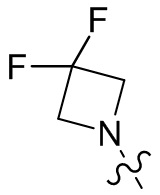 ,
, 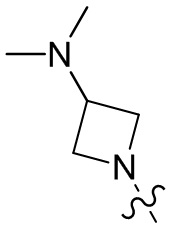 ,
, 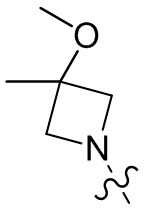 ,
, 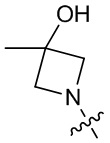 ,
, 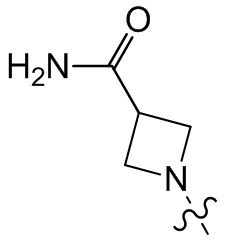 ,
, 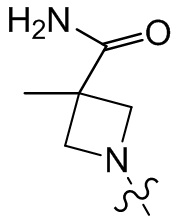 ,
, 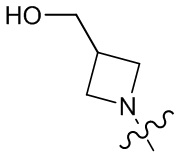 ,
, 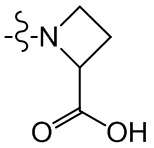 ,
,  ,
, 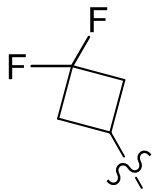 ;
;  ,
,  ,
, 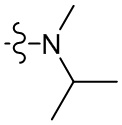 ,
, 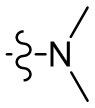 ,
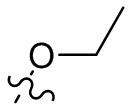
, 
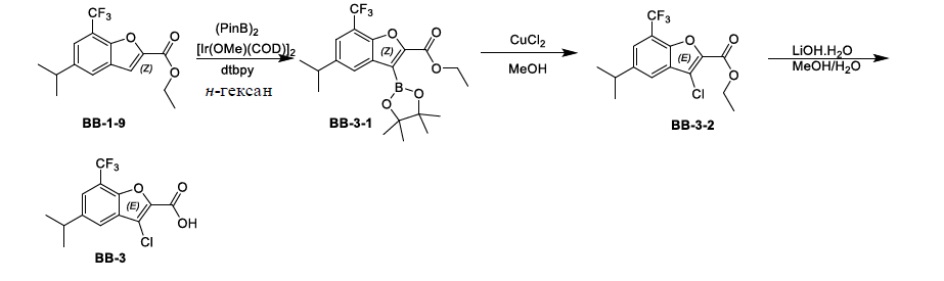
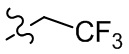 ,
, 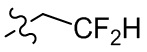 ,
, 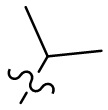 ,
, 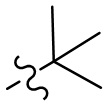 ,
, 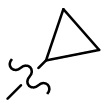 ,
,  и
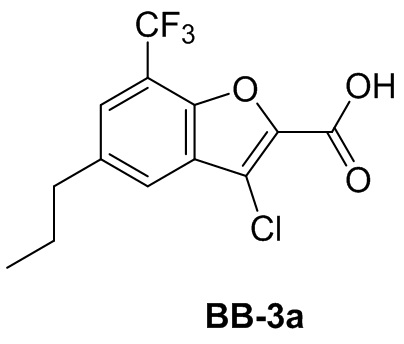
и  .
.
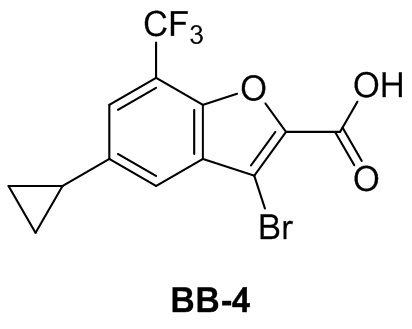
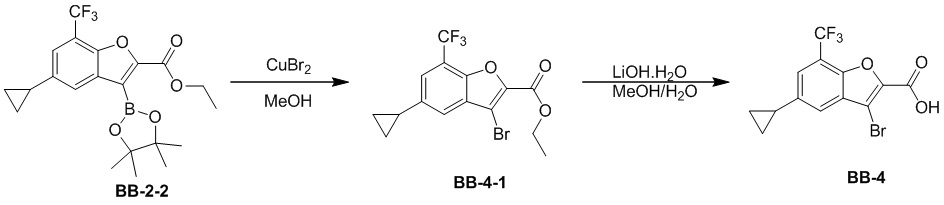
[37] В некоторых вариантах осуществления настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из группы, состоящей из
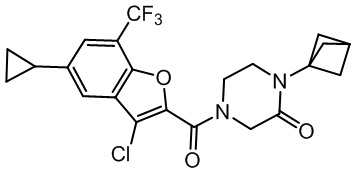
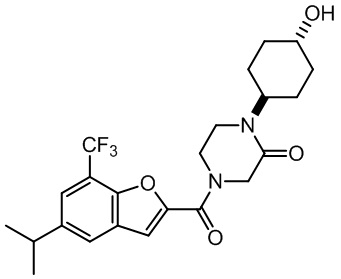
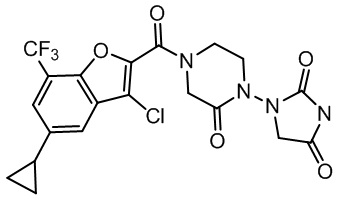
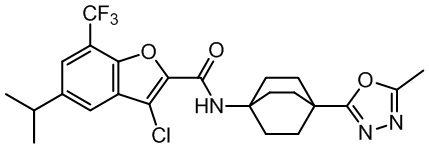
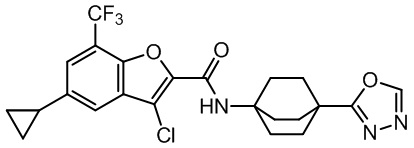
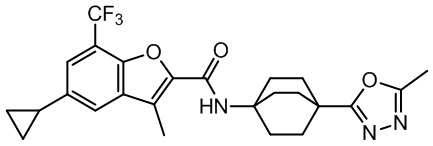
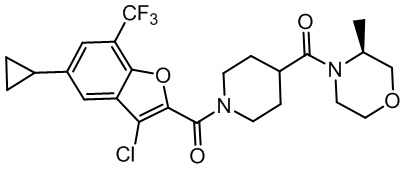
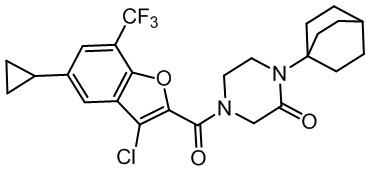
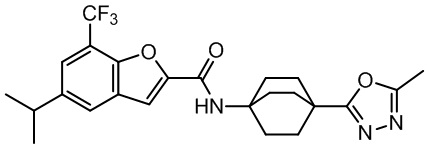
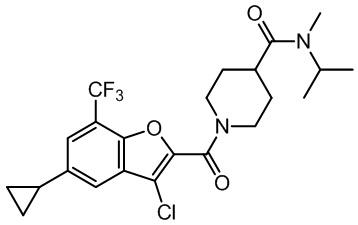
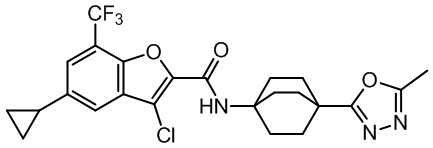
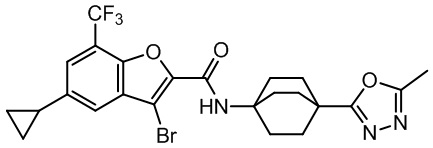
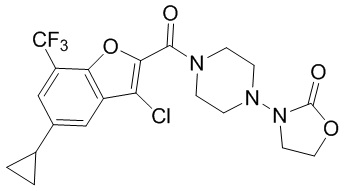
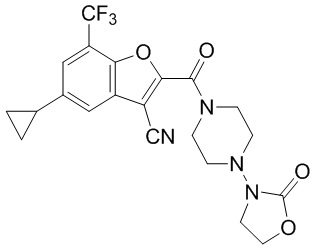
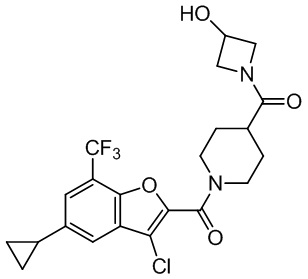
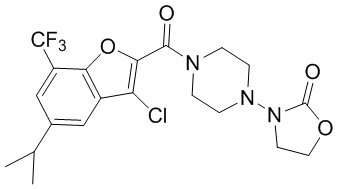
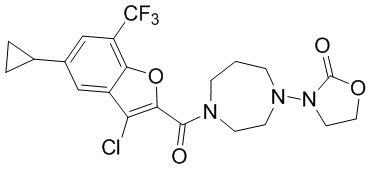
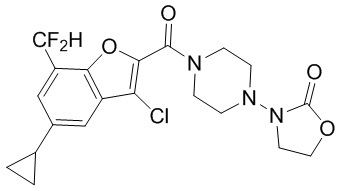
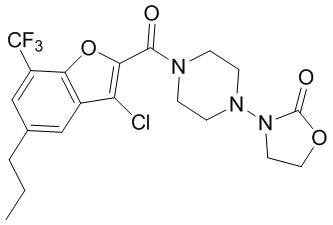
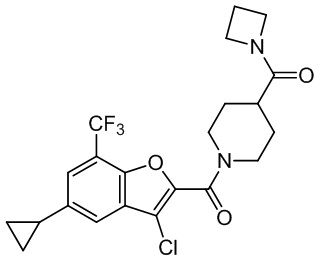
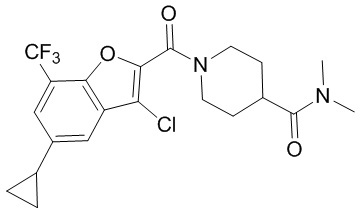
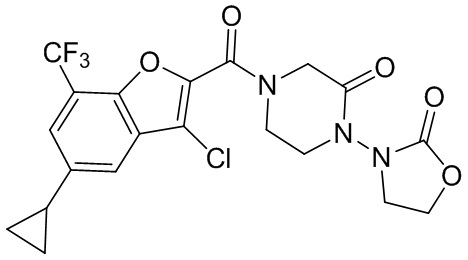
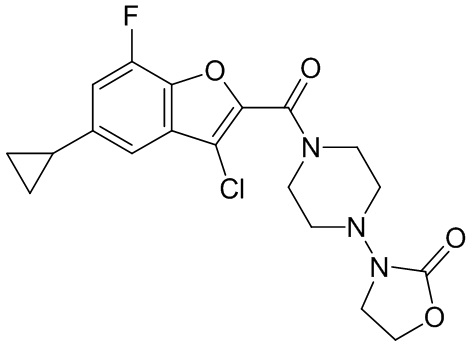
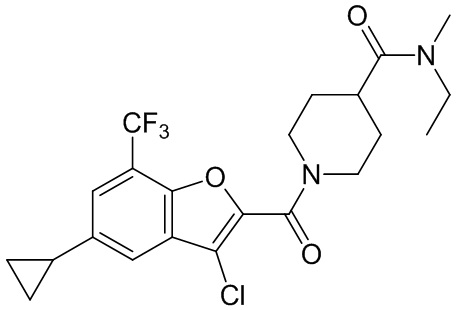
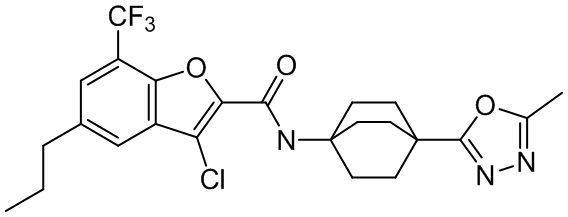
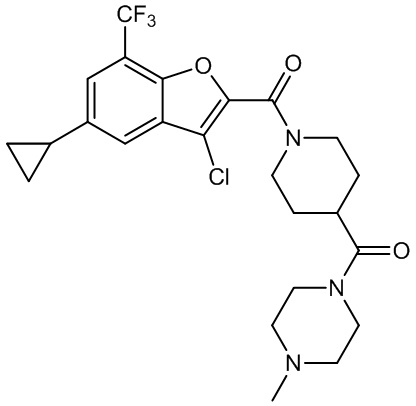
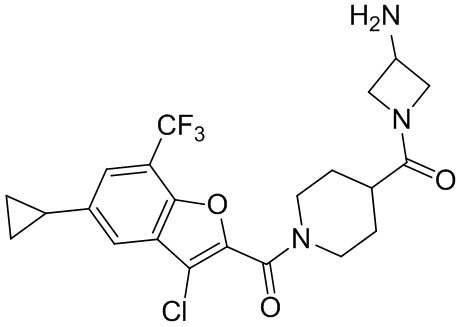
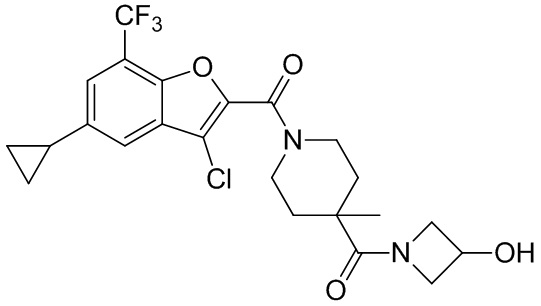
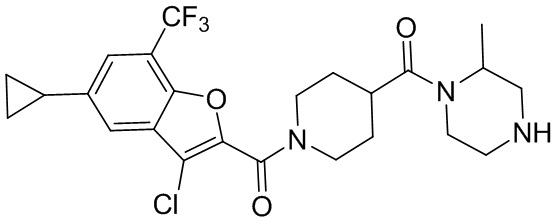
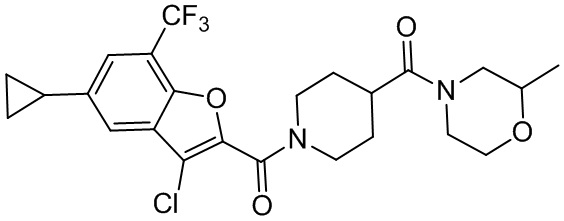
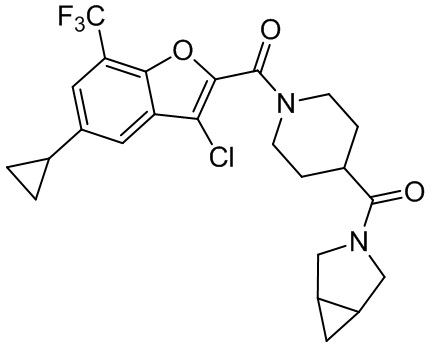
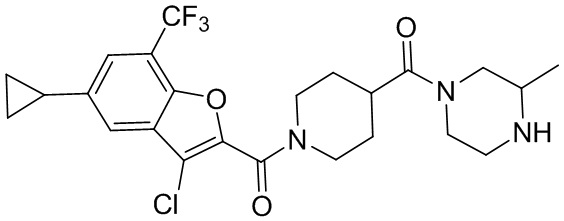
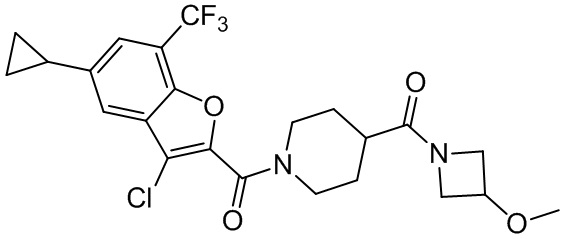
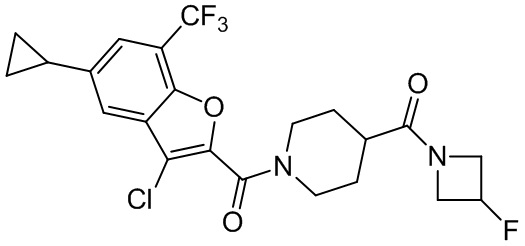
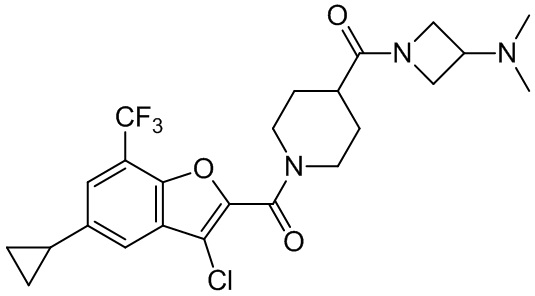
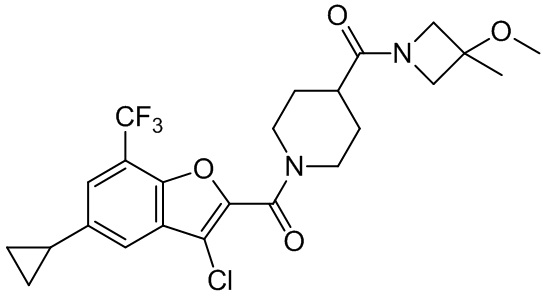
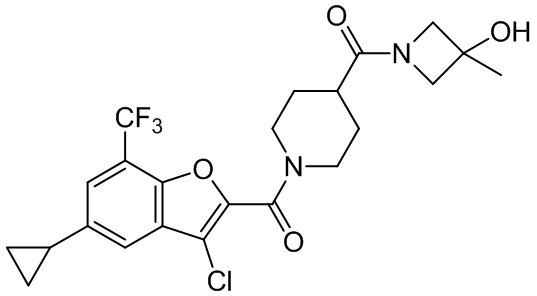
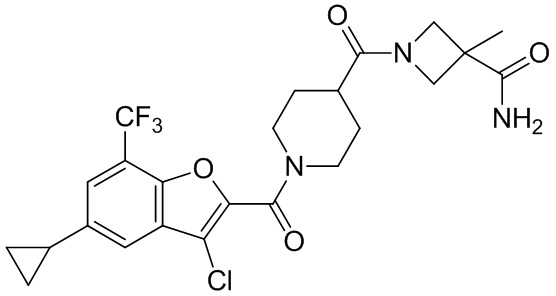
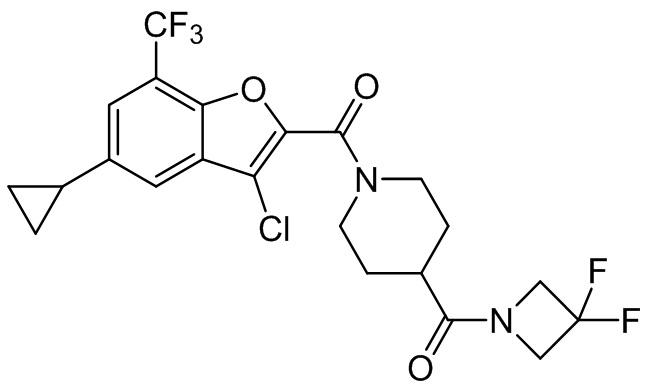
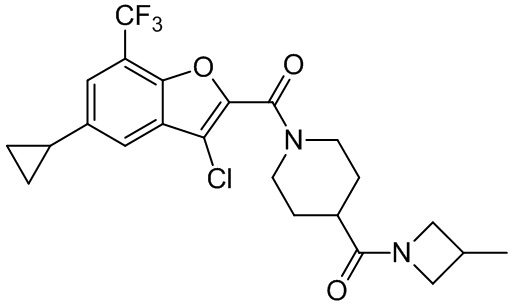
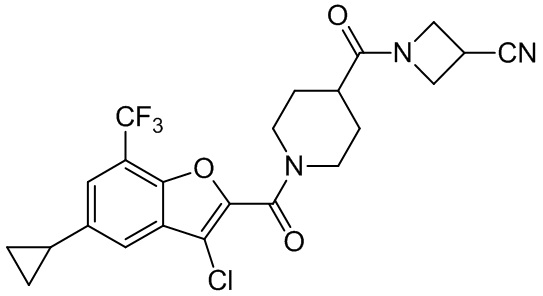
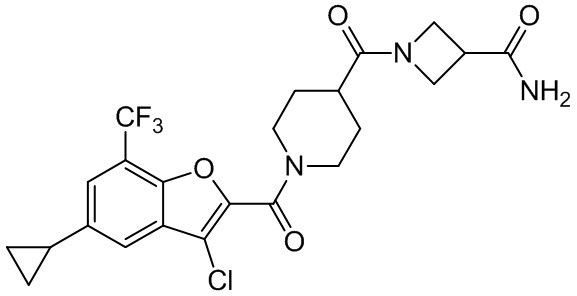
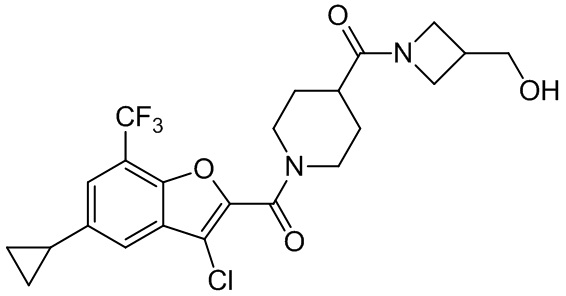
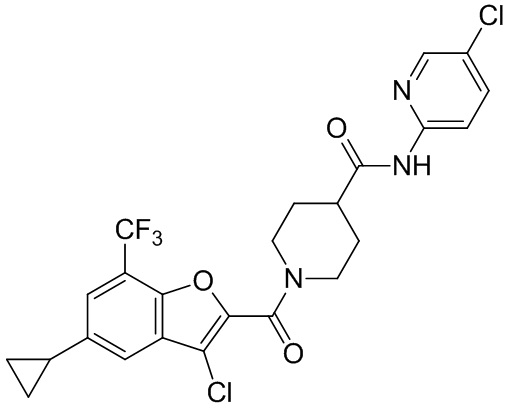
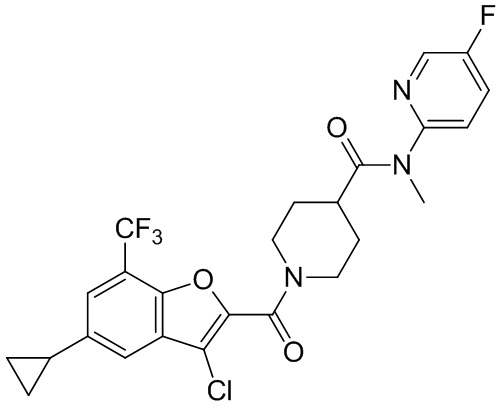
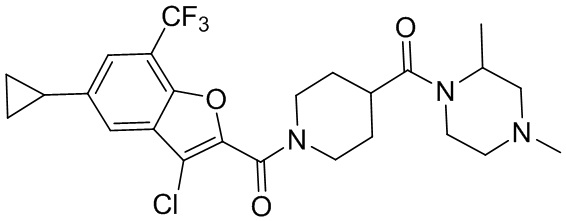
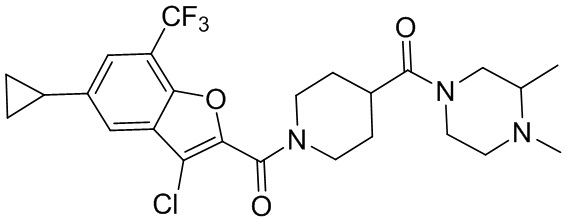
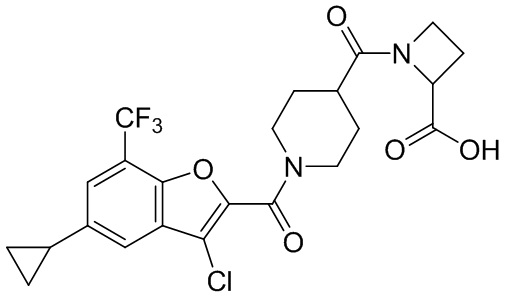
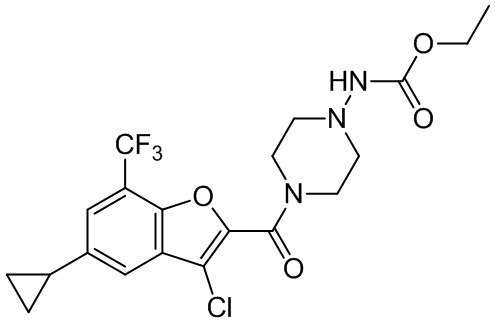
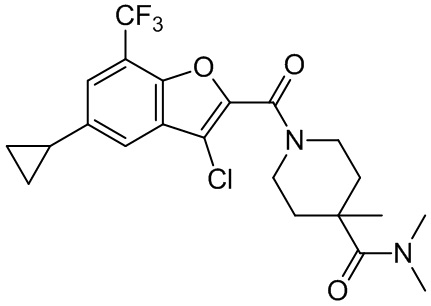
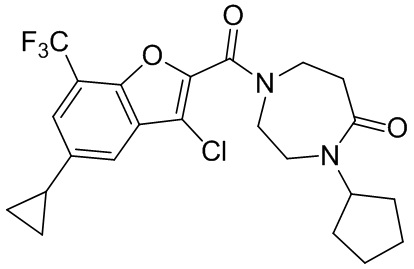
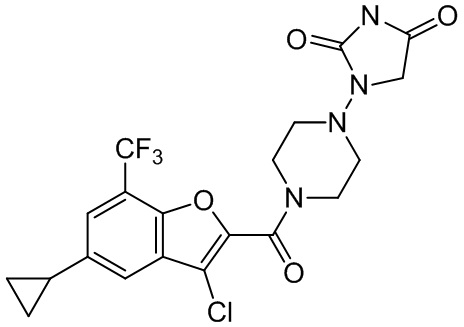
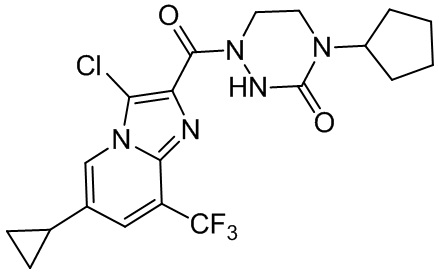
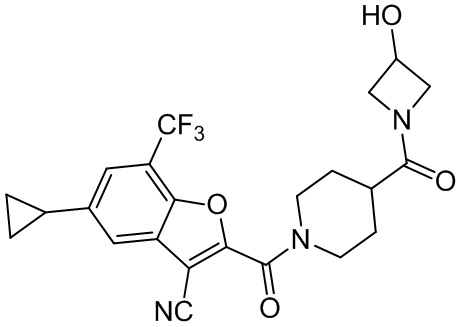
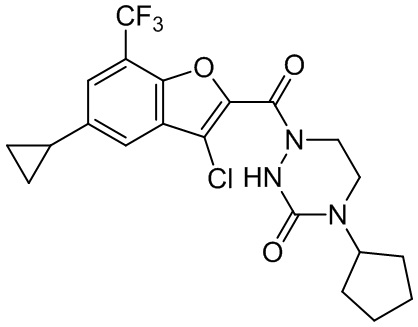
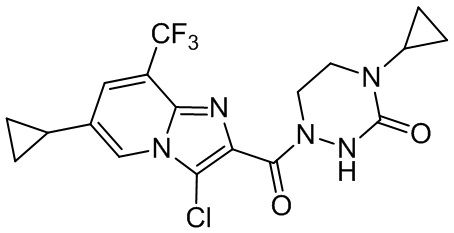
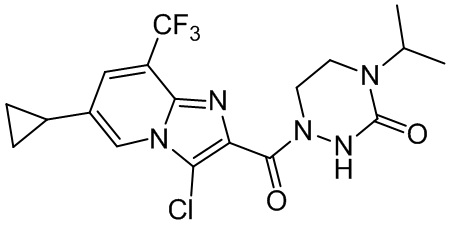
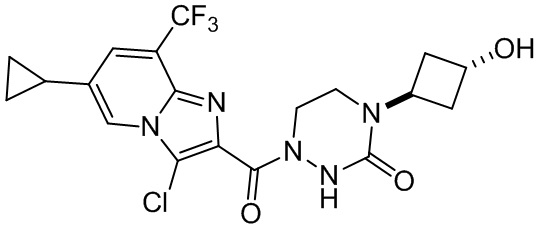
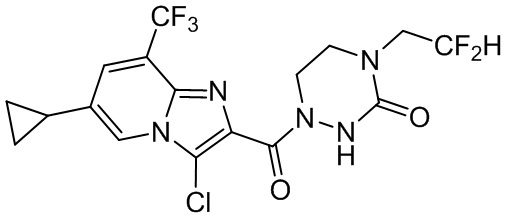
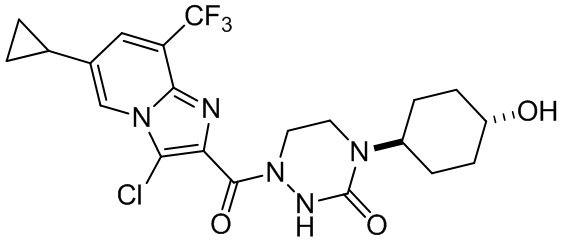
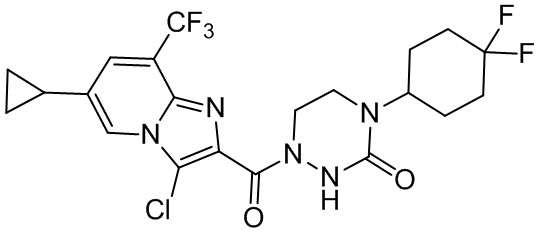
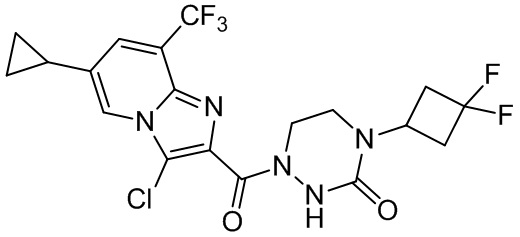
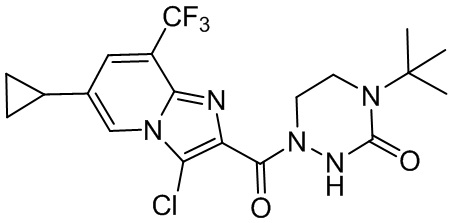
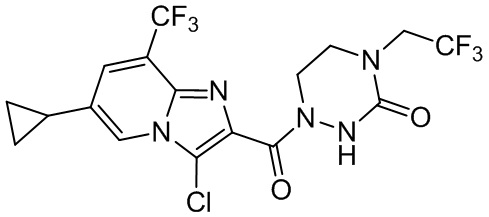
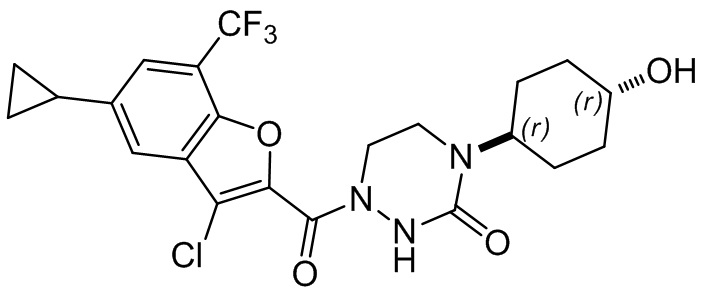
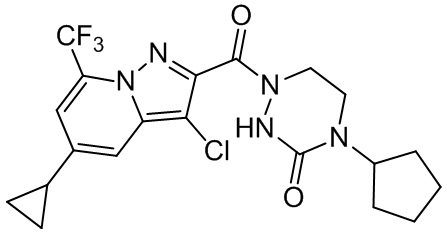
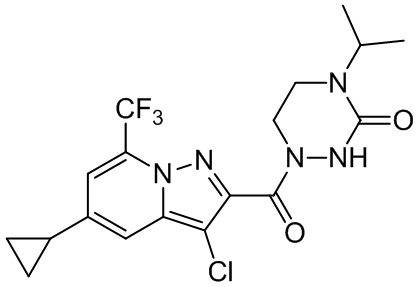 и
и 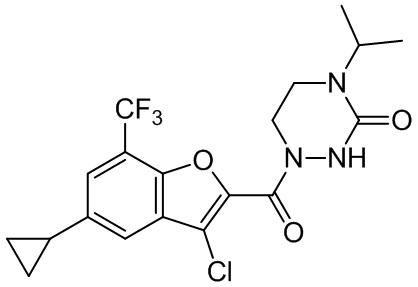 .
.
[38] Соответствующие определения
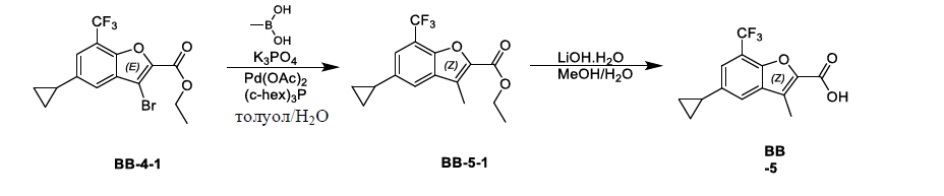
[39] Если не указано иное, предусматривается, что следующие термины и фразы, используемые в данном документе, имеют следующие значения. Конкретный термин или фраза не должны рассматриваться как неопределенные или неясные в отсутствие конкретного определения и в то же время должны быть понятными в соответствии с обычным значением. В случае появления в данном документе торгового наименования оно относится к соответствующему продукту или его активному ингредиенту.
[40] C1-10 выбран из C1, C2, C3, C4, C5, C6, C7, C8, C9 и C10; C3-10 выбран из C3, C4, C5, C6, C7, C8, C9 и C10.
[41] C1-10-алкил или гетероалкил, циклический или гетероциклический C3-10-гидрокарбонил, C1-10-алкил или гетероалкил, замещенный циклическим C3-10-гидрокарбонилом или гетероциклическим C3-10-гидрокарбонилом, включают без ограничения:
[42] C1-10-алкил, C1-10-алкиламино, N,N-бис(C1-10-алкил)амино, C1-10-алкоксил, C1-10-алкилацил, C1-10-алкоксикарбонил, C1-10-алкилсульфонил, C1-10-алкилсульфинил, C3-10-циклоалкил, C3-10-циклоалкиламино, C3-10-гетероциклоалкиламино, C3-10-циклоалкокси, C3-10-циклоалкилацил, C3-10-циклоалкоксикарбонил, C3-10-циклоалкилсульфонил, C3-10-циклоалкилсульфинил;
[43] метил, этил, н-пропил, изопропил, -CH2C(CH3)(CH3)(OH), циклопропил, циклобутил, пропилметилен, циклопропилкарбонил, бензилокси, трифторметил, аминометил, гидроксиметил, метокси, формил, метоксикарбонил, метилсульфонил, метилсульфинил, этокси, ацетил, этилсульфонил, этоксикарбонил, диметиламино, диэтиламино, диметиламинокарбонил, диэтиламинокарбонил;
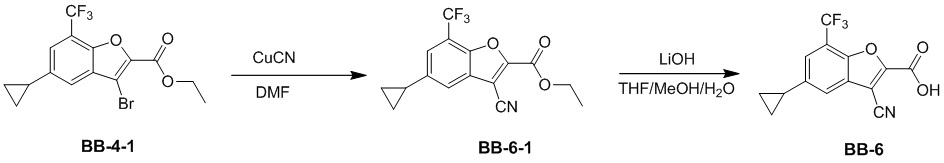
[44] N(CH3)2, NH(CH3), -CH2CF3, -CH2CH2CF3, -CH2CH2F, -CH2CH2S(=O)2CH3, -CH2CH2CN, 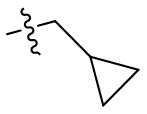 ,
, 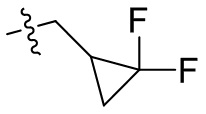 ,
, 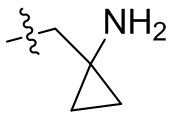 ,
, 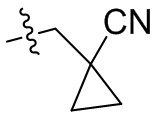 ,
, 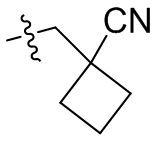 ,
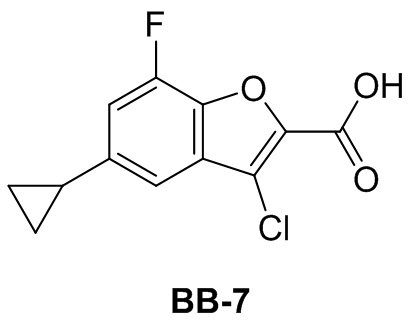
, 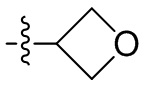 , -CH2CH(OH)(CH3)2, -CH2CH(F)(CH3)2, -CH2CH2F, -CH2CF3, -CH2CH2CF3, -CH2CH2NH2, -CH2CH2OH, -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2N(CH3)2, -S(=O)2CH3, -CH2CH2S(=O)2CH3,
, -CH2CH(OH)(CH3)2, -CH2CH(F)(CH3)2, -CH2CH2F, -CH2CF3, -CH2CH2CF3, -CH2CH2NH2, -CH2CH2OH, -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2N(CH3)2, -S(=O)2CH3, -CH2CH2S(=O)2CH3, 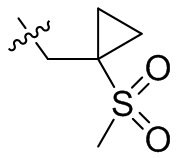 ,
, 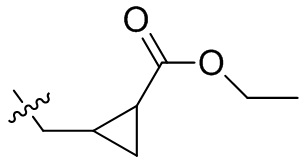 ,
, 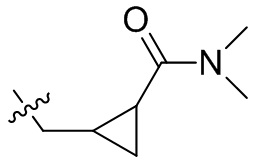 ,
, 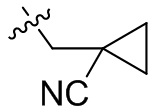 ,
, 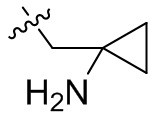 ,
, 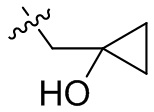 ,
, 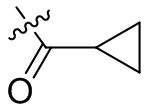 ,
, 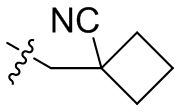 ,
, 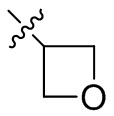 ,
, 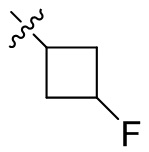 ,
, 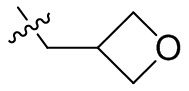 ,
, 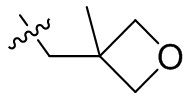 ,
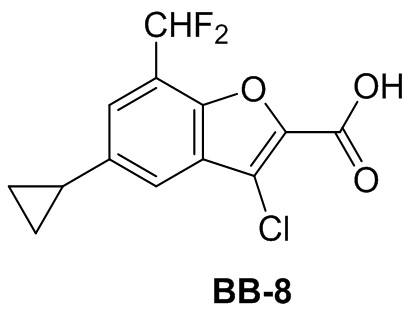
,  ,
,  ,
,  ,
,  ,
, 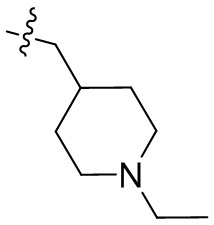 ,
, 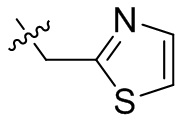 ,
, 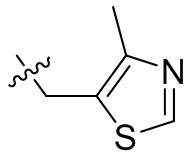 ,
, 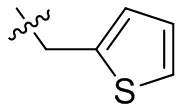 ,
, 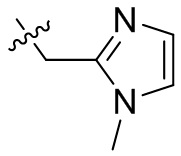 ,
, 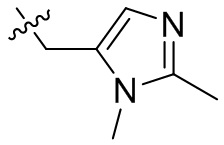 ,
, 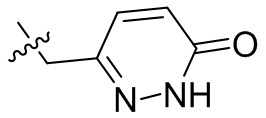 ;
; 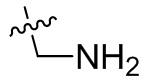 ,
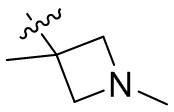
,  ,
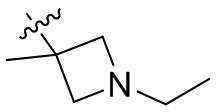
,  ,
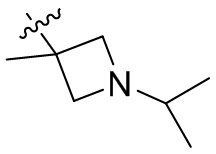
,  ,
,  ,
, 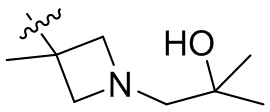 ,
, 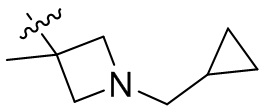 ,
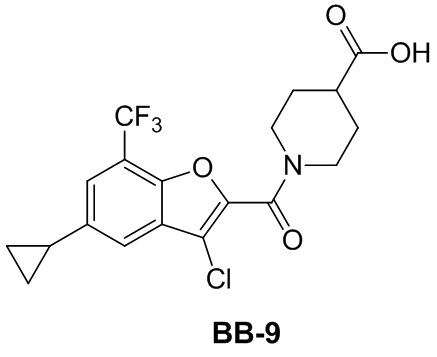
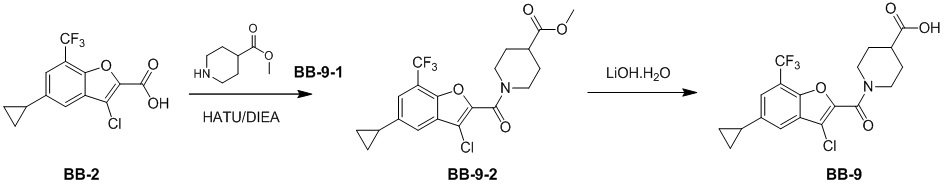
, 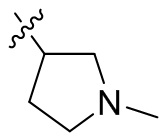 ,
, 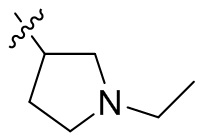 ,
, 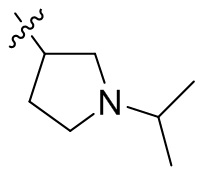 ,
, 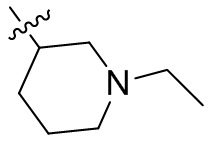 ,
, 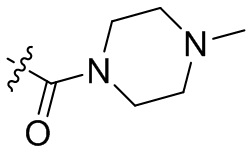 ,
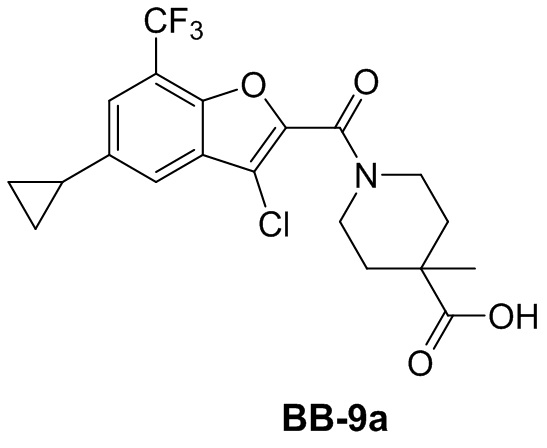
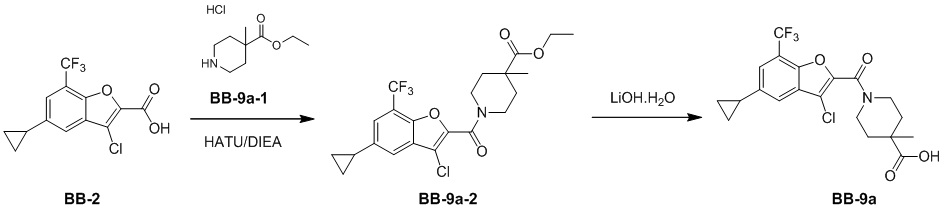
, 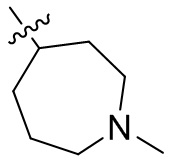 ,
, 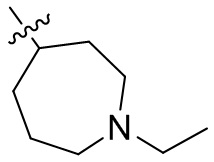 ,
, 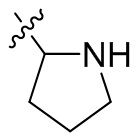 ,
, 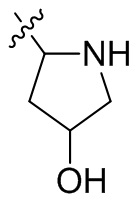 ,
, 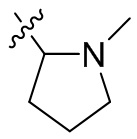 ,
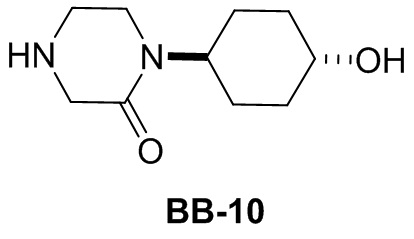
, 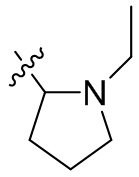 ,
, 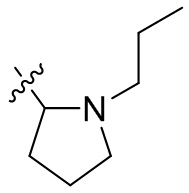 ,
, 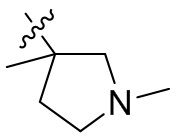 ,
, 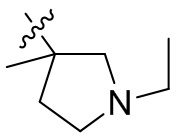 ,
, 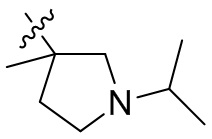 ,
, 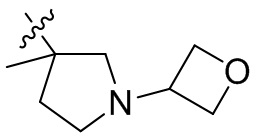 ,
, 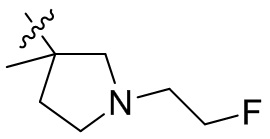 ,
, 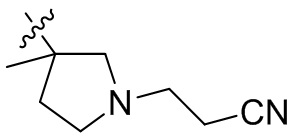 ,
, 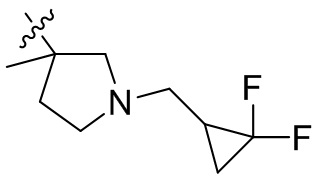 ,
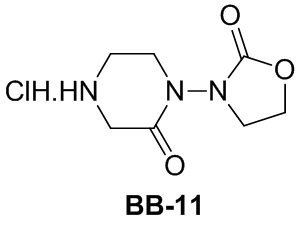
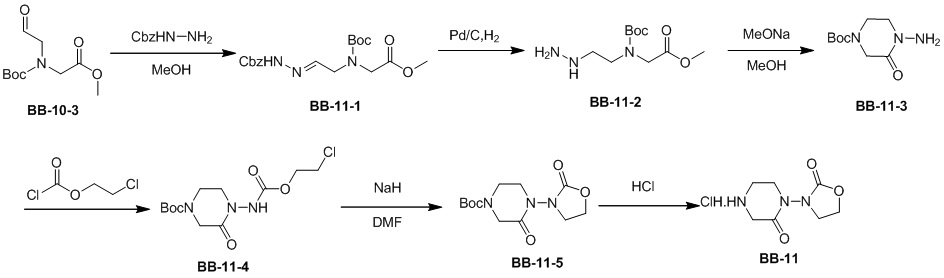
, 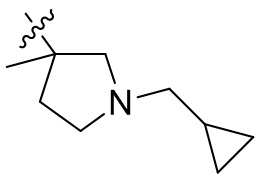 ,
, 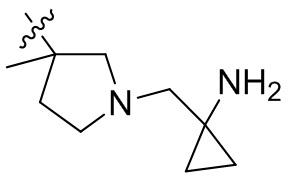 ,
, 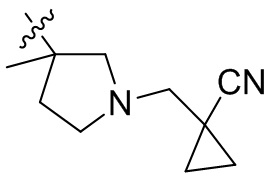 ,
, 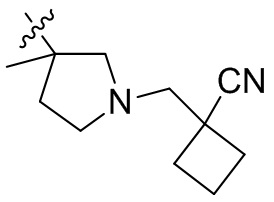 ,
, 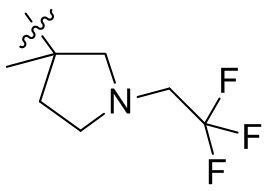 ,
, 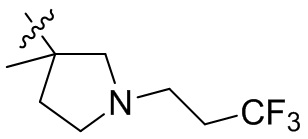 ,
, 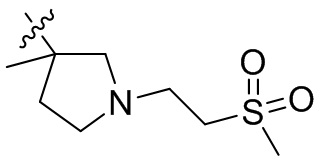 ,
, 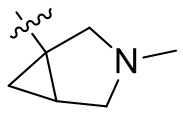 ,
, 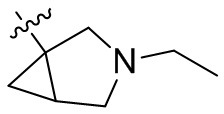 ,
, 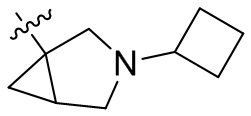 ,
, 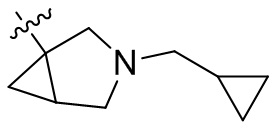 ,
,  ,
,  ,
,  ,
,  ,
, 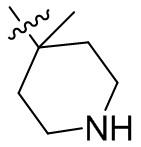 ,
, 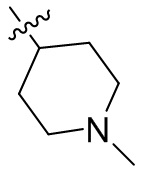 ,
, 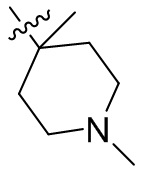 ,
, 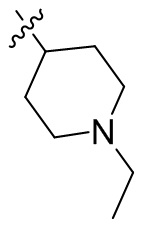 ,
,  ,
,  ,
,  ,
,  ,
, 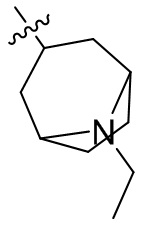 ,
, 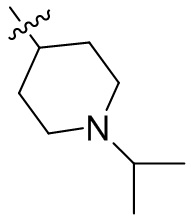 ,
, 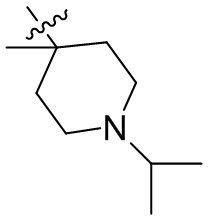 ,
, 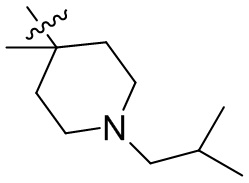 ,
, 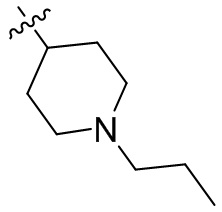 ,
, 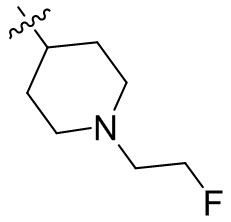 ,
, 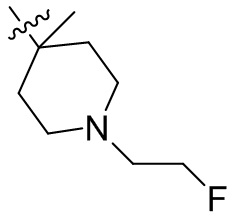 ,
, 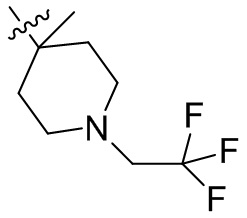 ,
, 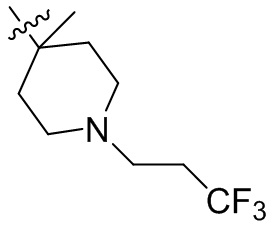 ,
, 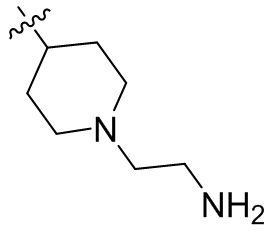 ,
, 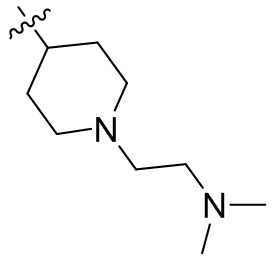 ,
, 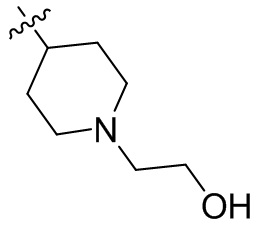 ,
,  ,
,  ,
,  ,
,  ,
, 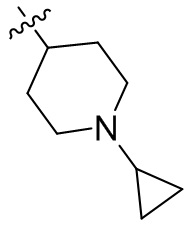 ,
, 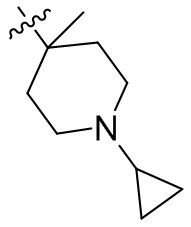 ,
, 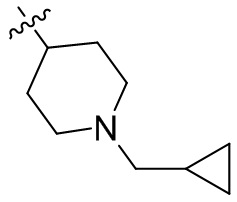 ,
, 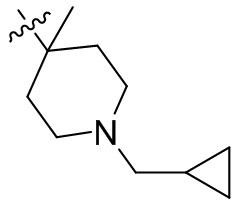 ,
, 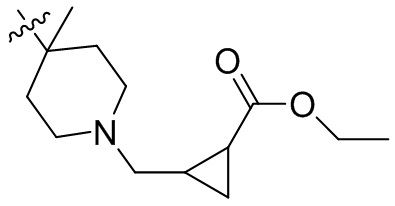 ,
, 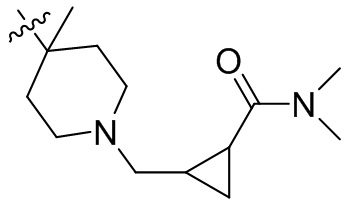 ,
, 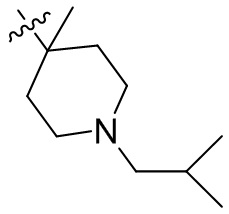 ,
, 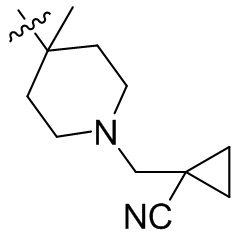 ,
,  ,
,  ,
,  ,
,  ,
, 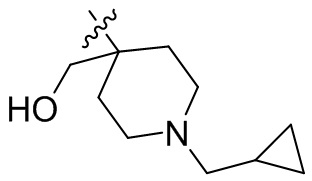 ,
, 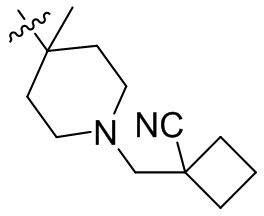 ,
, 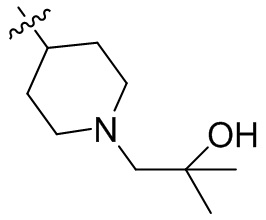 ,
, 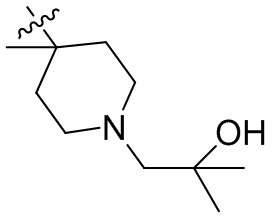 ,
, 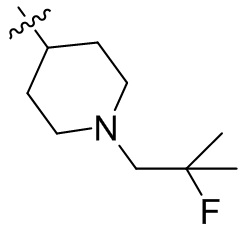 ,
, 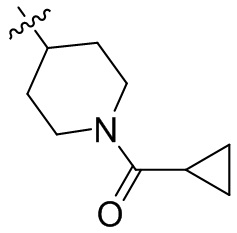 ,
, 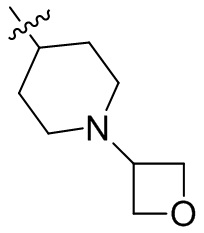 ,
, 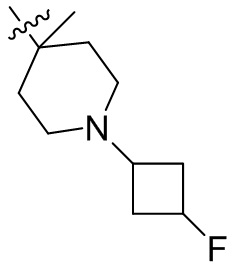 ,
, 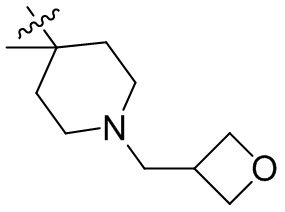 ,
, 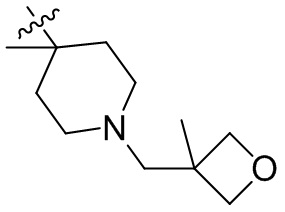 ,
, 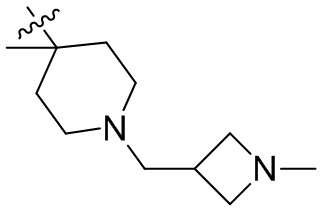 ,
, 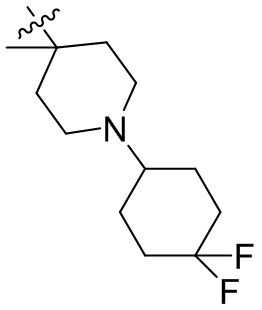 ,
, 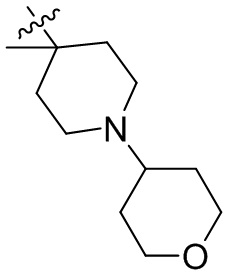 ,
, 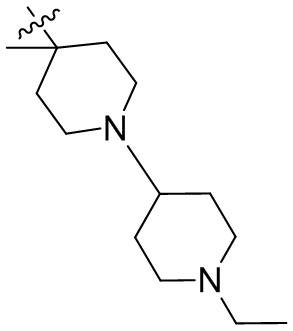 ,
, 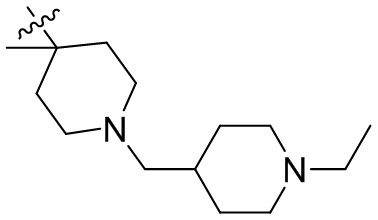 ,
, 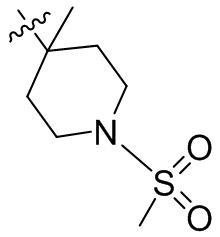 ,
, 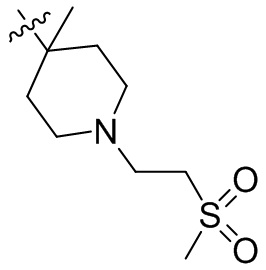 ,
, 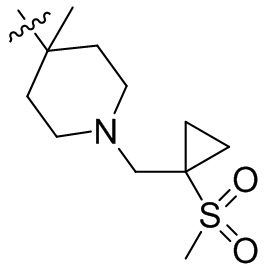 ,
, 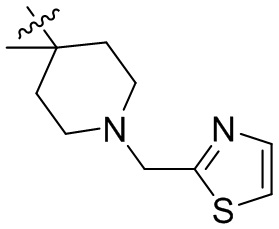 ,
, 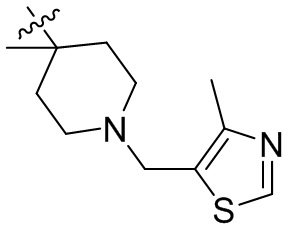 ,
,  ,
,  ,
,  ,
,  ,
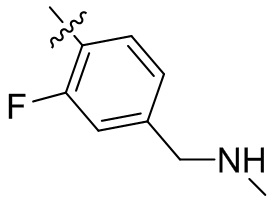
,  ,
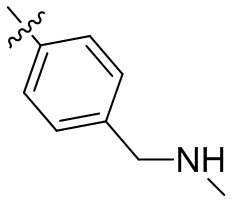
,  ,
,  ,
,  ,
,  ,
,  и
и
[45] фенил, тиазолил, бифенил, нафтил, циклопентил, фурил, 3-пирролинил, пирролидинил, 1,3-диоксоланил, пиразолил, 2-пиразолинил, пиразолидинил, имидазолил, оксазолил, тиазолил, 1,2,3-азолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,3,4-тиадиазолил, 4H-пиранил, пиридил, пиперидил, 1,4-диоксанил, морфолинил, пиридазинил, пиримидинил, пиразинил, пиперазинил, 1,3,5-тритианил, 1,3,5-триазинил, бензофуранил, бензотиофенил, индолил, бензимидазолил, бензoтиазолил, пуринил, хинолинил, изохинолинил, циннолинил или хиноксалинил;
[46] C6-12-арил, C6-12-аралкил, C6-12-гетероароматические радикалы или C6-12-гетероаралкил;  ,
,  ,
, 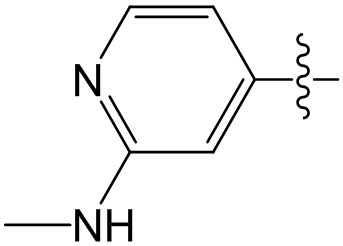 ;
; 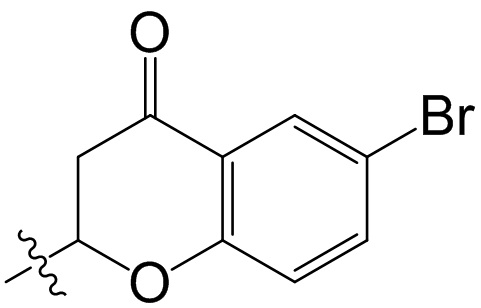 ,
, 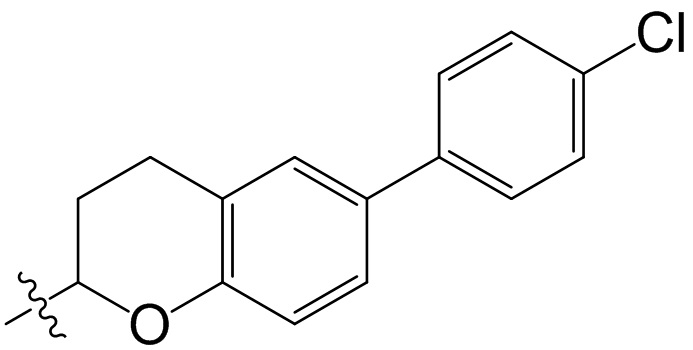 ,
, 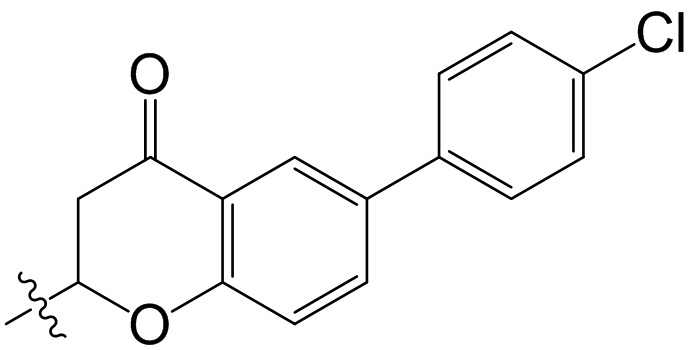 ,
, 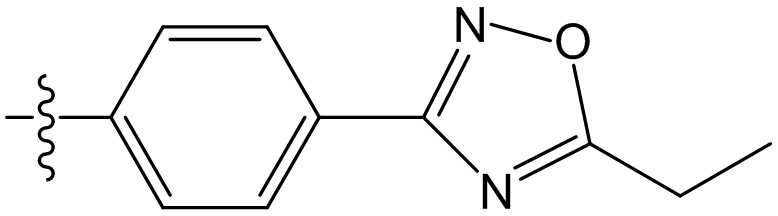 ,
, 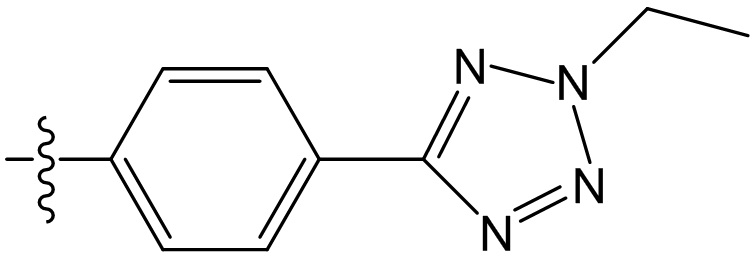 ,
, 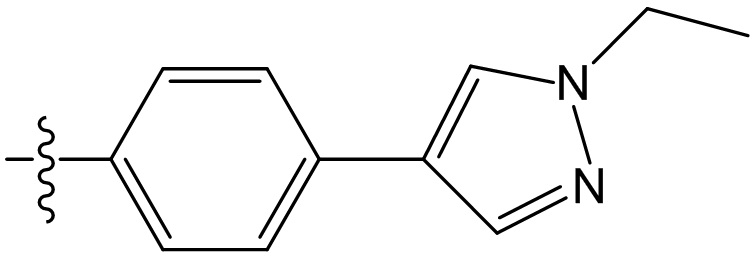 ,
, 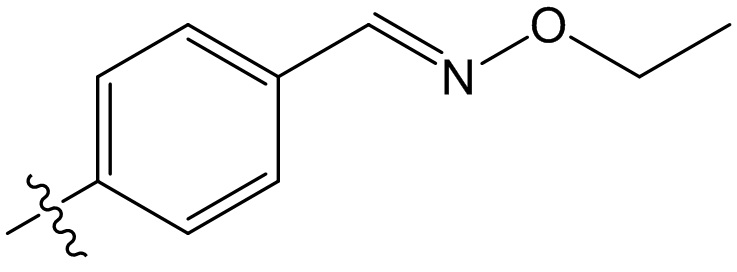 ,
, 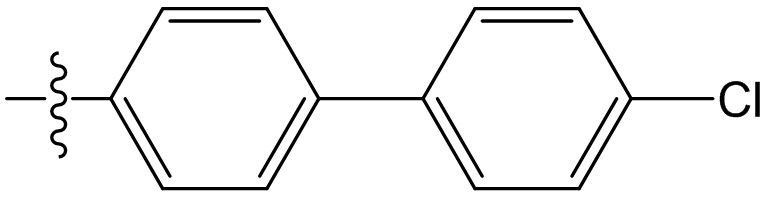 ,
, 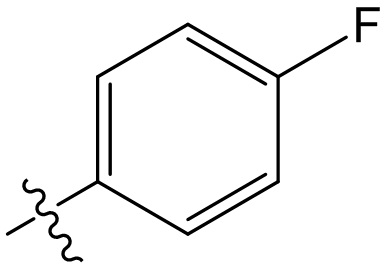 ,
, 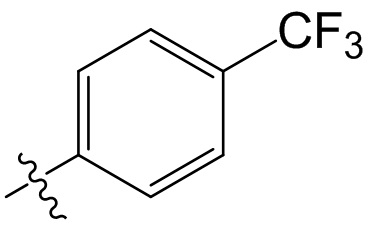 ,
, 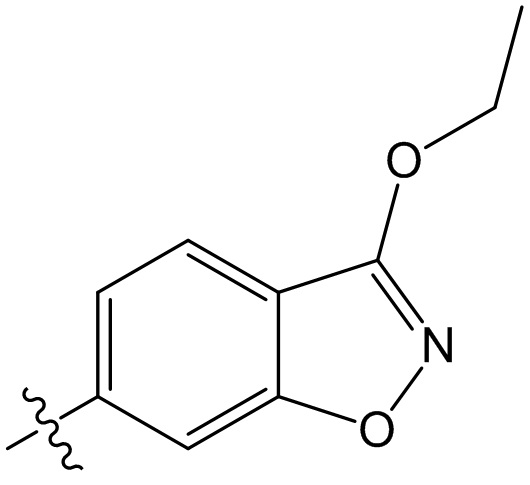 ,
, 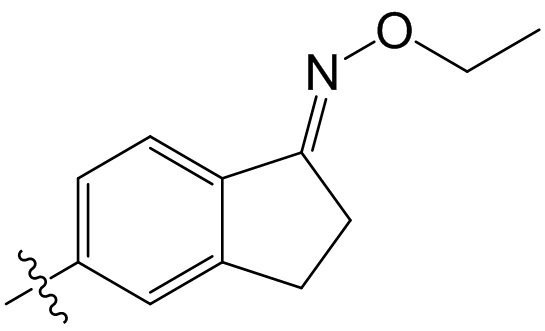 ,
, 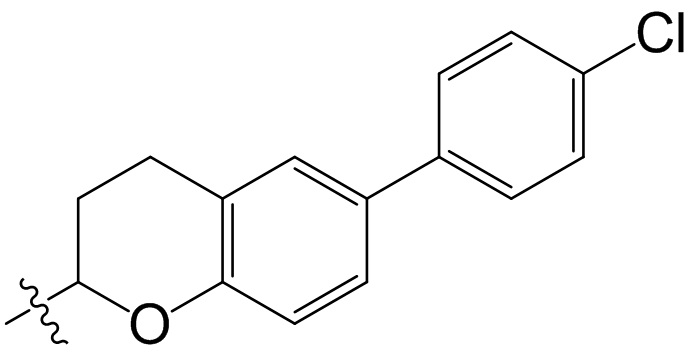 ,
, 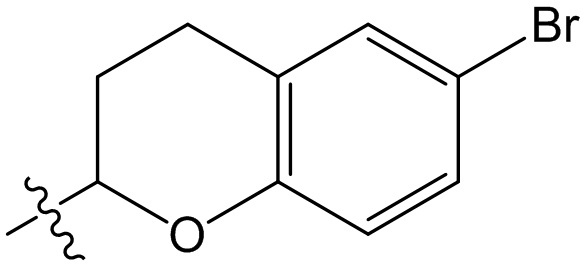 ,
, 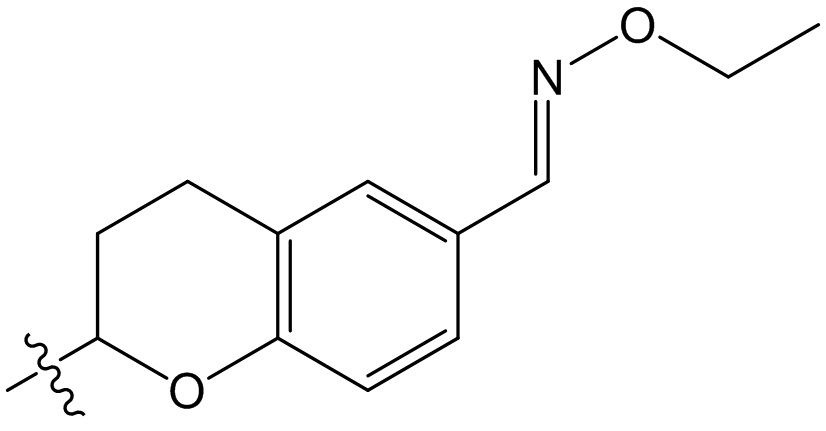 ;
;
 ,
,  ,
,  ,
,  ,
, 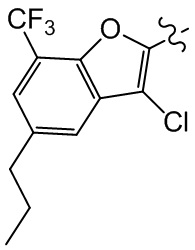 , ,
, , 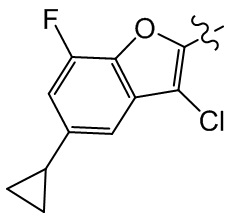 ,
, 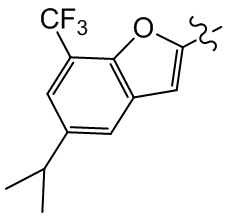 ,
, 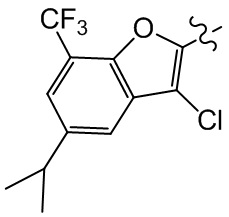 , ,
, ,  ;
;
 ,
,  , , ,
, , ,  ,
, 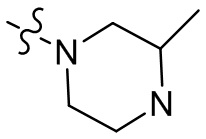 , , , , ,
, , , , , 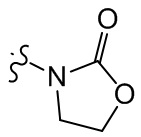 ,
, 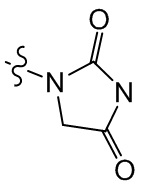 , , ,
, , , 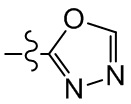 ,
, 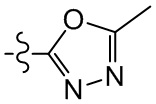 , , , , , , , , , , ,
, , , , , , , , , , , 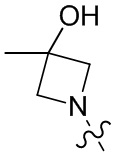 , , ,
, , , 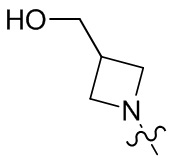 ,
, 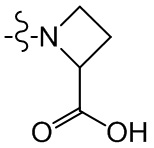 , , ,
, , , 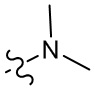 ,
, 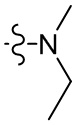 ,
, 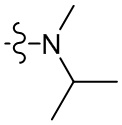 , , ,
, , , 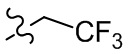 ,
, 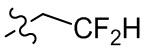 , , , , , .
, , , , , .
[47] В данном документе термин “фармацевтически приемлемый” распространяется на те соединения, материалы, композиции и/или лекарственные формы, которые входят в объем объективных медицинских представлений и пригодны для применения в контакте с человеческой и животной тканью, но без избыточных токсичности, раздражения, аллергических реакций или других проблем или осложнений, а также удовлетворяют приемлемому соотношению польза/риск.
[48] Термин “фармацевтически приемлемая соль” относится к соли соединения по настоящему изобретению, которую получают с использованием соединения с конкретным заместителем, раскрытым в настоящем изобретении, и относительно нетоксичных кислоты или щелочи. Если соединение по настоящему изобретению содержит относительно кислую функциональную группу, то можно получить соль присоединения щелочи путем приведения соединения в нейтральной форме в контакт с достаточным количеством щелочи в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения щелочи включает соль натрия, калия, кальция, аммония, органического аммония или магния и т. п. Если соединение по настоящему изобретению содержит относительно щелочную функциональную группу, то можно получить соль присоединения кислоты путем приведения соединения в нейтральной форме в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, при этом неорганическая кислота включает такие соединения, как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, бикарбонат, фосфорная кислота, гидрофосфат, дигидрофосфат, серная кислота, гидросульфат, йодистоводородная кислота, фосфористая кислота и т. д.; и соль органической кислоты, при этом органическая кислота включает такие соединения, как уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, субериновая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, фенилсульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота, винная кислота, метилсульфоновая кислота и т. п.; а также включают соль аминокислоты (например, аргинина и т. д.) и соль органической кислоты, такой как глюкуроновая кислота и т. п. (см. Berge et al., “Pharmaceutical Salts”, Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторое конкретное соединение по настоящему изобретению содержит как щелочную, так и кислую функциональные группы, так что оно может быть преобразовано в любую соль присоединения щелочи или присоединения кислоты.
[49] Нейтральную форму соединения предпочтительно восстанавливают путем приведения соли в контакт с основанием или кислотой традиционным образом и затем отделения исходного соединения. Различие между исходной формой соединения и различными формами солей заключается в некоторых физических свойствах, таких как различная растворимость в полярном растворителе.
[50] “Фармацевтически приемлемая соль” в настоящем изобретении является производным соединения по настоящему изобретению, где исходное соединение модифицировано путем образования соли с кислотой или щелочью. Примеры фармацевтически приемлемой соли включают без ограничения соль присоединения неорганической кислоты или органической кислоты щелочи, такой как амин, соль щелочного металла или органическую соль радикала кислоты, такой как карбоновая кислота, и т. д. Фармацевтически приемлемая соль включает обычно нетоксичные соли или соли четвертичного аммония исходного соединения, такие как соль, образованная нетоксичной неорганической кислотой или органической кислотой. Обычно нетоксичная соль включает без ограничения соли, полученные из неорганических кислот и органических кислот, при этом неорганические кислоты или органические кислоты выбраны из 2-ацетоксибензойной кислоты, 2-изэтионовой кислоты, уксусной кислоты, аскорбиновой кислоты, фенилсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, эдетовой кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидройодата, гидроксила, гидроксинафтойной, изэтионовой кислоты, молочной кислоты, лактозы, додекансульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактуронана, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, фолиновой кислоты, янтарной кислоты, аминосульфоновой кислоты, сульфаниловой кислоты, серной кислоты, дубильной кислоты, винной кислоты и п-толуолсульфоновой кислоты.
[51] Фармацевтически приемлемую соль по настоящему изобретению можно получить традиционным способом с использованием исходного соединения, содержащего кислую или щелочную группу. Как правило, способ получения соли предусматривает осуществление реакции данных соединений в формах свободных кислот или щелочей со стехиометрическим количеством соответствующих щелочей или кислот в воде или органическом растворителе или смеси воды и органического растворителя. Как правило, предпочтительным выбором является неводная среда, такая как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил и т. д.
[52] Помимо формы соли, для соединения в настоящем изобретении существует форма пролекарства. Пролекарство соединения, описанное в настоящем изобретении, легко преобразуется в соединение по настоящему изобретению посредством химических изменений в физиологических условиях. Кроме того, пролекарство может быть преобразовано в соединение по настоящему изобретению посредством химического или биохимического способа в условиях in vivo.
[53] Некоторые соединения по настоящему изобретению могут существовать в формах, отличных от сольватов, или в форме сольватов, в том числе в форме гидратов. Как правило, форма сольвата аналогична форме, отличной от сольвата, обе из которых включены в объем настоящего изобретения. Некоторые соединения по настоящему изобретению могут содержать асимметрические атомы углерода (оптические центры) или двойные связи. В объем настоящего изобретения включены рацемические изомеры, диастереомеры, геометрические изомеры и отдельные изомеры.
[54] Схематическое изображение рацемического изомера, амбискалемического и скалемического или энантиомерно чистого соединения по настоящему изобретению приведено из Maehr, J. Chem. Ed. 1985, 62: 114-120. Если не указано иное, абсолютная конфигурация стереоцентра представлена клиновидными и пунктирными линиями. Если соединение по настоящему изобретению содержит винильную двойную связь или другой центр геометрической асимметрии, то, если не указано иное, включены геометрические E- и Z-изомеры. Аналогично, все таутомерные формы находятся в объеме настоящего изобретения.
[55] Соединение по настоящему изобретению может существовать в виде конкретного геометрического или стереоизомерного изомера. Настоящее изобретение предусматривает все данные классы соединений, в том числе цис- и транс-изомеры, (-)- и (+)-антимеры, (R)- и (S)-антимеры, диастереомеры, (D)-изомеры, (L)-изомеры, а также их рацемические смеси и другие, такие как смеси, обогащенные энантиомерами или диастереоизомерами, при этом все данные смеси находятся в объеме настоящего изобретения. Другие асимметрические атомы углерода могут находиться в заместителях, таких как алкил. Все данные изомеры и их смеси включены в объем настоящего изобретения.
[56] Оптически активные (R)- и (S)-изомеры, (D)- и (L)-изомеры можно получать с помощью асимметрического синтеза или хиральных реагентов или других традиционных методик. Если желаемым является энантиомер соединения по настоящему изобретению, в получении можно использовать асимметрический синтез или дериватизирующее действие хиральных вспомогательных реагентов, при котором получаемые в результате смеси диастереомеров выделяют, и вспомогательные группы отщепляют с получением чистого необходимого энантиомера. Или же, если молекула содержит щелочную функциональную группу (такую как аминогруппа) или кислые функциональные группы (такие как карбоксильная), при взаимодействии с соответствующей оптически активной кислотой или щелочью образуется соль диастереомера, и затем чистый энантиомер можно повторно использовать после отделения от соли диастереомера с помощью общепринятых способов, известных из уровня техники. Кроме того, разделение энантиомера и диастереомера обычно осуществляют с помощью хроматографического способа, при этом в хроматографическом способе используется хиральная неподвижная фаза, и необязательно в комбинации со способом химической дериватизации (например, с получением карбамата из амина).
[57] Один или более одного атома, составляющих соединение по настоящему изобретению, могут содержать неестественную долю изотопов атомов. Например, соединение может быть меченным радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). Все варианты изотопного состава соединения, раскрытого в настоящем изобретении, независимо от того, являются они радиоактивными или нет, включены в объем настоящего изобретения.
[58] Термин “фармацевтически приемлемый носитель” относится к любому составу или несущей среде, способным доставлять эффективное количество активного вещества, раскрытого в настоящем изобретении, которые не препятствуют биологической активности активного вещества и не проявляют токсических побочных эффектов в отношении реципиента или пациента, при этом типичный носитель включает воду, растительные и минеральные масла, основу для крема, матрицу для лосьона, матрицу для мази и т. д. Матрица содержит суспензию, средство для повышения вязкости, вещества, способствующие проникновению через кожу, и т. д. Их состав хорошо известен специалисту в области косметологии или лекарственных средств местного действия. Другая информация относительно носителя может быть упомянута в Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которого включено в данный документ во всей своей полноте.
[59] Термин “наполнитель”, как правило, относится к носителю, разбавителю и/или среде, необходимым для получения эффективной фармацевтической композиции.
[60] Что касается лекарственного средства или фармакологически активного средства, термин “эффективное количество” или “терапевтически эффективное количество” относится к достаточному количеству лекарственного средства или состава, с помощью которого можно достигать желаемых эффектов, но которое не проявляет токсичность. Что касается состава для перорального применения по настоящему изобретению, “эффективное количество” одного активного вещества в композиции представляет собой количество, необходимое для достижения желаемых эффектов в комбинации с другим активным веществом в композиции. Определение эффективного количества варьируется между людьми, и оно зависит от возраста и общего состояния пациента, его получающего, а также от конкретного активного вещества. В одном случае соответствующее эффективное количество может определять специалист в данной области в соответствии со стандартными испытаниями.
[61] Термин “активный ингредиент”, “терапевтическое средство”, “активное вещество” или “активное средство” относится к химическому объекту, который может обеспечивать эффективное лечение нарушений, болезней или состояний целевого субъекта.
[62] Термин “замещенный” относится к одному или более атомам водорода у конкретного атома, необязательно замещенным заместителем, в том числе дейтерием и вариантом водорода, при условии, что валентное состояние конкретного атома является нормальным и соединение, полученное после замещения, является стабильным. Если заместитель представляет собой кетонную группу (т. e. =O), это означает, что два атома водорода замещены. Замещение кетонной группой не происходит в ариле. Термин “необязательно замещенный” означает то, что объект может быть замещенным или не быть замещенным, и при этом, если не указано иное, тип и количество заместителей может быть произвольным в рамках предположений о стабильности, существующих в химии.
[63] Если какой-либо параметр (например, R) встречается более одного раза в композиции или структуре соединения, то определение для каждого случая встречаемости является независимым. Следовательно, например, если группа замещена 0-2 R, группа может быть необязательно замещена не более чем двумя R, и в каждом случае R имеет независимые возможные варианты. Кроме того, комбинация заместителей и/или их вариантов допускается лишь в случаях, когда такая комбинация будет приводить к получению стабильного соединения.
[64] Если количество соединяющих групп равно 0, как, например, в -(CRR)0-, это означает, что соединяющая группа представляет собой одинарную связь.
[65] Если один из параметров выбран из одинарной связи, это означает, что две группы, к которым он присоединен, соединены непосредственно, например, если L в A-L-Z представляет собой одинарную связь, это означает, что структура в действительности представляет собой A-Z.
[66] Если заместитель одной связью может быть перекрестно соединен с двумя атомами кольца, то заместитель может быть связан с произвольными атомами в кольце. Если не указано, посредством какого атома указанный заместитель соединен с общей структурной формулой, включая соединение, которое не упомянуто конкретно, то заместитель может быть связан с любым из его атомов. Комбинация заместителей и/или их вариантов допускается лишь в случаях, когда такая комбинация будет приводить к получению стабильного соединения. Например, структурная единица 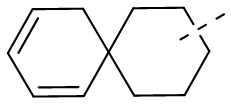 или
или 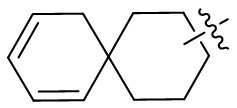 означает, что может иметь место соединение с любым атомом циклогексила или циклогексадиена.
означает, что может иметь место соединение с любым атомом циклогексила или циклогексадиена.
[67] Если не указано иное, термины “галогенированный” или “галоген” сами по себе или в качестве части другого заместителя относятся к атому фтора, хлора, брома или йода. Кроме того, подразумевается, что термин “галогенированный алкил” включает моногалогенированный алкил и полигалогенированный алкил. Например, подразумевается, что термин “галогенированный (C1-C4)-алкил” включает без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил и 3-бромпропил и т. д.
[68] Примеры галогенированного алкила включают без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил. “Алкокси” означает, что алкильная группа с конкретным количеством атомов углерода соединена кислородным мостиком. C1-6-алкокси включает C1-, C2-, C3-, C4-, C5- и C6-алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентилокси. “Циклоалкил” включает насыщенную циклическую группу, такую как циклопропил, циклобутил или циклопентил. 3-7-членный циклоалкил включает C3-, C4-, C5-, C6- и C7-циклоалкил. “Алкенил” включает линейную или разветвленную углеводородную цепь, где в любых стабильных участках цепи присутствует одна или более двойных связей C-C, как, например, в случае винила и пропенила.
[69] Термин “галоген” или “радикал галогена” относится к фтору, хлору, брому и йоду.
[70] Если не указано иное, термин “гетеро” относится к гетероатому или гетероатомной группе (т. e. группе, содержащей гетероатом), в том числе другим атомам помимо углерода (C) и водорода (H), и группам, содержащим данные гетероатомы, таким как, в том числе, кислород (O), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2- и необязательно замещенные -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- или -S(=O)N(H)-.
[71] Если не указано иное, “кольцо” относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Кольцо включает отдельное кольцо, сочлененное кольцо, спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком. Количество атомов в кольце, как правило, определяют как количество элементов кольца, например, “5-7-членное кольцо” представляет собой кольцо, образованное 5-7 атомами. Если не указано иное, кольцо необязательно содержит 1-3 гетероатома. Следовательно, “5-7-членное кольцо” включает, например, фенилпиридин и пиперидинил; с другой стороны, термин “5-7-членный гетероциклоалкил” включает пиридил и пиперидинил, но не включает фенил. Термин “кольцо” также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо соответствует вышеприведенному определению.
[72] Если не указано иное, термин “гетероцикл” или “гетероциклил” относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом и гетероатомную группу, при этом они могут быть насыщенными, частично ненасыщенными или ненасыщенными (ароматическими), при этом они содержат атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, которые независимо выбраны из группы, состоящей из N, O и S, где любой гетероцикл может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Атомы азота и серы необязательно могут быть окисленными (т. e. NO и S(O)p, p равно 1 или 2). Атом азота может быть замещенным или незамещенным (т. e. N или NR, где R представляет собой H или другой заместитель, который был определен в данном документе). Гетероцикл может быть присоединенным к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если образованное соединение является стабильным, то гетероцикл, описанный в данном документе, может быть замещен по его атому углерода или азота. Атом азота в гетероцикле необязательно является кватернизированным. В качестве предпочтительного варианта осуществления настоящего изобретения, если общее количество атомов S и O, содержащихся в гетероцикле, превышает 1, данные гетероатомы не являются смежными друг с другом. В качестве другого предпочтительного варианта настоящего изобретения, общее количество атомов S и O в гетероцикле составляет не более 1. Используемый в данном документе термин “ароматическая гетероциклическая группа” или “гетероарил” относится к стабильному 5-, 6-, 7-членному моноциклу или бициклу или 7-, 8-, 9- или 10-членному бициклическому гетероароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, которые независимо выбраны из группы, состоящей из N, O и S. Атом азота может быть замещенным или незамещенным (т. e. N или NR, где R представляет собой H или другой заместитель, который был определен в данном документе). Атомы азота и серы необязательно могут быть окисленными (т. e. NO и S(O)p, p равно 1 или 2). Следует отметить, что общее количество атомов S и O в гетероароматическом кольце составляет не более 1. Кольца с внутренним мостиком также включены в определение гетероцикла. Если один или более атомов (т. e. C, O, N или S) соединены с двумя несмежными атомами углерода или атомами азота, то образуется кольцо с внутренним мостиком. Предпочтительное кольцо с внутренним мостиком включает без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну углерод-азотную группу. Следует отметить, что мостик всегда преобразует моноциклическое кольцо в трициклическое кольцо. В кольце с внутренним мостиком заместитель в кольце может также быть расположен на мостике.
[73] Примеры гетероциклических соединений включают без ограничения: акридинил, азоцинил, бензимидазолил, бензофуранил, бензoмеркаптофуранил, бензoмеркаптофенил, бензоксазолил, бензоксазолинил, бензoтиазолил, бензoтриазолил, бензoтетразолил, бензoизоксазолил, бензoизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоалкенил, индолинил, индолизинил, индолил, 3H-индолил, изатиновую группу, изобензофуранил, изоиндолил, изоиндолинил, изохинолил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндил, пиримидил, фенантридинил, фенантролинил, феназин, фенотиазин, бензoпуринил, феноксазинил, фталазинил, пиперазинил, пиперидил, оксопиперидинил, 4-оксопиперидинил, пиперонил, птеридил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, оксазолопиридин, пиридиноимидазол, пиридинотиазол, пиридил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазил, изотиазолилтиенил, тиенил, тиофеноксазолил, тиофенотиазолил, тиофеноимидазолил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включены конденсированные кольцевые и спирокольцевые соединения.
[74] Если не указано иное, термин “гидрокарбонил” или его конкретное представление (такое как алкил, алкенил, алкинил, фенил и т. д.) само по себе или в качестве части другого заместителя представляет собой линейный, разветвленный или циклический гидрокарбонил или их комбинацию, которые могут быть полностью насыщенными, моноциклическими или полициклическими ненасыщенными, могут быть монозамещенными, дизамещенными или полизамещенными, могут быть одновалентными (такими как метил), двухвалентными (такими как метилен) или многовалентными (такими как метенил), могут включать двухвалентные или многовалентные группы атомов с указанным количеством атомов углерода (как, например, C1-C10 относится к группе, имеющей 1-10 атомов углерода). Термин “алкил” включает без ограничения алифатический гидрокарбонил и ароматический гидрокарбонил, при этом алифатический гидрокарбонил включает линейную и циклическую структуры, в частности, включает без ограничения алкил, алкенил и алкинил, при этом ароматический гидрокарбонил включает без ограничения 6-12-членный ароматический гидрокарбонил, такой как бензол, нафталин и т. п. В некоторых вариантах осуществления термин “гидрокарбонил” относится к линейной или разветвленной группам или их комбинации, которые могут быть полностью насыщенными, моноциклическими или полициклическими ненасыщенными, могут включать двухвалентные и поливалентные группы. Примеры насыщенного гидрокарбонила включают без ограничения гомологи или изомеры метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила, втор-бутила, изобутила, циклогексила, (циклогексил)метила, циклопропилметила и н-амила, н-гексила, н-гептила, н-октила и т. п. Ненасыщенный алкил содержит одну или более двойных или тройных связей, и его примеры включают без ограничения винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-бутадиенил, 2,4-(пентадиенил), 3-(1,4-пентадиенил), ацетенил, 1- и 3-пропинил, 3-бутинил и более сложные гомологи и изомеры.
[75] Если не указано иное, термин “гетерогидрокарбонил” или его конкретные представления (такие как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т. д.) сами по себе или термин в комбинации с другим термином относятся к стабильному линейному, разветвленному или циклическому гидрокарбонилу или их комбинациям, которые состоят из определенного количества атомов углерода и по меньшей мере одного гетероатома. В некоторых вариантах осуществления термин “гетерогидрокарбонил” сам по себе или термин в комбинации с другим термином относятся к стабильному линейному, разветвленному гидрокарбонилу или их комбинациям, которые состоят из определенного количества атомов углерода и по меньшей мере одного гетероатома. В типичном варианте осуществления гетероатом выбран из группы, состоящей из B, O, N и S, в которой атомы азота и серы необязательно являются окисленными, и атом азота необязательно является кватернизированным. Гетероатомы B, O, N и S могут располагаться в любом внутреннем положении гетерогидрокарбонила (включая положение, в котором гидрокарбонил присоединен к остальной части молекулы). Примеры включают без ограничения -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Не более чем два гетероатома являются смежными, как, например, в -CH2-NH-OCH3.
[76] Термины “алкокси”, “алкиламино” и “алкилтио” (или тиоалкокси) являются идиоматическими выражениями, которые относятся к алкильной группе, присоединенной к остальной части молекулы посредством кислорода, аминогруппы или атома серы соответственно.
[77] Если не указано иное, термин “циклогидрокарбонил”, “гетероциклогидрокарбонил” или его конкретные представления (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероцикловинил, циклоалкинил, гетероциклоалкинил и т. д.) сами по себе или термин в комбинации с другими терминами соответственно относятся к циклическому “гидрокарбонилу”, “гетерогидрокарбонилу”. Кроме того, что касается гетерогидрокарбонила или гетероциклогидрокарбонила (такого как гетероалкил, гетероциклоалкил), гетероатомы могут занимать положение, в котором гетероциклическое кольцо присоединено к остальной части молекулы. Примеры циклоалкила включают без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т. д. Неограниченные примеры гетероциклила включают 1-(1,2,5,6-тетрагидропиридинил), 1-пиперидил, 2-пиперидил, 3-пиперидил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуранилиндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[78] Если не указано иное, термин “арил” относится к полиненасыщенному ароматическому углеводородному заместителю, который может быть монозамещенным, дизамещенным или полизамещенным, может быть одновалентным, двухвалентным или многовалентным. Он может быть моноциклическим или полициклическим (предпочтительно 1-3 кольца). Они конденсированы или соединены с помощью ковалентной связи. Термин “гетероарил” относится к арилу (или кольцу), содержащему 1-4 гетероатома. В иллюстративном варианте осуществления гетероатом выбран из группы, состоящей из B, O, N и S, в которой атомы азота и серы необязательно являются окисленными, и атом азота необязательно является кватернизированным. Гетероарильная группа может быть соединена с остальной частью молекулы посредством гетероатома. Неограниченные примеры арила или гетероарила включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-бензoтиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалил, 5-хиноксалил, 3-хинолил и 6-хинолил. Любой из заместителей в арильной и гетероарильной кольцевых системах выбран из приемлемых заместителей, описанных ниже.
[79] Для простоты при использовании в комбинации с другими терминами (например, арилокси, арилтио, аралкил) арил включает определение арильного и гетероарильного колец, определенных выше. Следовательно, подразумевается, что термин “аралкил” включает группы, в которых арил присоединен к алкилу (например, бензил, фенилэтил, пиридилметил), в том числе те алкилы, где атомы углерода (как, например, метиленовые) были заменены такими атомами, как атомы кислорода, как, например, в феноксиметиле, 2-пиридилоксиметил-3-(1-нафтокси)пропиле и т. д.
[80] Термин “уходящая группа” относится к функциональной группе или атому, которые могут быть заменены другой функциональной группой или атомом посредством реакции замещения (например, реакции нуклеофильного замещения). Например, типичные уходящие группы включают трифлат; хлор, бром, йод; сульфонат, такой как мезилат, тозилат, п-бромбензолсульфонат, п-тозилат и т. д.; ацилокси, такой как ацетокси, трифторацетокси и т. д.
[81] Термин “защитная группа” включает без ограничения “защитную группу для аминогруппы”, “защитную группу для гидроксила” или “защитную группу для меркаптогруппы”. Термин “защитная группа для аминогруппы” относится к защитной группе, которая подходит для предотвращения побочных реакций, происходящих по атому азота аминогруппы. Типичная защитная группа для аминогруппы включает без ограничения: формил; ацил, такой как алканоил (такой как ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), трифенилметил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. д. Термин “защитная группа для гидроксила” относится к защитной группе, которая подходит для предотвращения побочных реакций по гидроксильной группе. Типичная защитная группа для гидроксила включает без ограничения: алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (такой как ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (дифенилметил, DPM); силил, такой как триметилсилил (TMS), и трет-бутилдиметилсилил (TBS), и т. д.
[82] Соединение по настоящему изобретению может быть получено посредством многих способов синтеза, которые хорошо известны специалисту в данной области, включая конкретные варианты осуществления, перечисленные ниже, и их комбинацию с другими способами химического синтеза и эквивалентными альтернативными способами, которые известны специалисту в данной области, при этом предпочтительные варианты осуществления включают без ограничения варианты осуществления настоящего изобретения.
[83] Применяемые в настоящем изобретении растворители являются коммерчески доступными и их можно применять без дополнительной очистки. Реакции осуществляют в атмосфере инертного газообразного азота с безводными растворителями. Данные о ЯМР-спектрах регистрируют с помощью спектрометра Bruker Avance III 400 (400 МГц), и химический сдвиг (ppm) рассчитывают на основе эталонного сигнала тетраметилсилана в слабом поле. Масс-спектрометрические показатели измеряют с помощью Agilent серии 1200 и 6110 (&1956A). Прибор для LC/MS или MS Shimadzu содержит DAD: SPD-M20A (LC) и микромасс-спектрометрический детектор Shimadzu 2020. Масс-спектрометр оснащен источником ионизации электрораспылением (ESI), который может функционировать в режиме положительной ионизации или отрицательной ионизации.
[84] В настоящем изобретении приняты следующие сокращения: водн. означает водный; HATU означает гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; EDC означает гидрохлорид N-(3-диметиламинопропил)-N’-этилкарбодиимида; м-CPBA означает м-хлорпербензойную кислоту; экв. означает эквивалент, количественно равный; CDI означает карбонилдиимидазол; DCM означает дихлорметан; PE означает петролейный эфир; DIAD означает диизопропилазодикарбоксилат; DMF означает N,N-диметилформамид; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; MeOH означает метанол; Cbz означает бензилоксикарбонил, защитную группу для аминогруппы; Boc означает трет-бутоксикарбонил, защитную группу для амина; HOAc означает уксусную кислоту; NaCNBH3 означает цианоборогидрид натрия; к. т. означает комнатную температуру; в теч. ночи означает в течение ночи; THF означает тетрагидрофуран; Boc2O означает ди-трет-бутилдикарбонат; TFA означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; SOCl2 означает тионилхлорид; CS2 означает дисульфид углерода; TsOH означает п-толуолсульфоновую кислоту; NFSI означает N-фторбензолсульфонимид; NCS означает N-хлорсукцинимид; н-Bu4NF означает фторид тетрабутиламмония; iPrOH означает 2-пропанол; т. пл. означает температуру плавления; LDA означает диизопропиламид лития; Pd(dppf)2Cl2 означает 1,1’-[бис(дифенилфосфино)ферроцен]дихлорпалладий; (c-hex)3P означает трициклогексилфосфат; (PinB)2 означает бис(пинаколато)дибор; Ir(OMe)(COD)]2 означает бис(1,5-циклооктадиен)ди-μ-метоксидииридий (I); dtbpy означает 4,4'-ди-трет-бутил-2,2'-дипиридин; NBS означает N-бромсукцинимид; DAST означает трифторид диэтиламиносеры; DIEA означает N,N-диизопропилэтиламин; DPPA означает дифенилфосфорилазид; TsCl означает п-толуолсульфонилхлорид; HOBT означает 1-гидроксибензотриазол; EDCI означает гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида; PhSH означает тиофенол; DMAD означает диметилацетилендикарбоксилат.
[85] Соединениям даны названия вручную или с помощью программного обеспечения ChemDraw®, коммерчески доступным соединениям даны названия в соответствии с каталогом поставщика.
Подробное описание предпочтительного варианта осуществления
[86] Следующие примеры дополнительно иллюстрируют настоящее изобретение, но настоящее изобретение не ограничено ими.
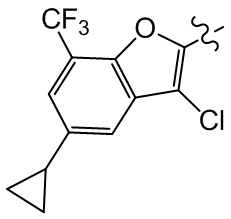
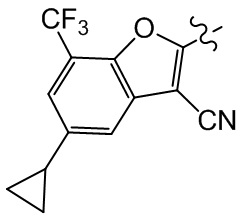
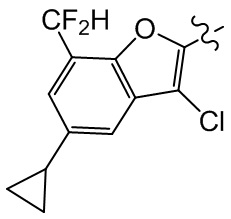
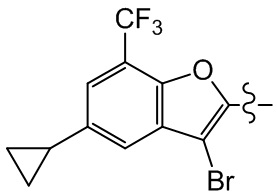
Сравнительный пример 1: фрагмент BB-1
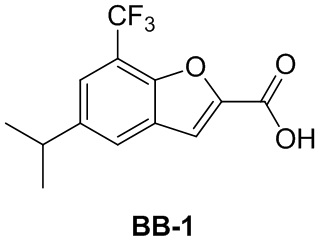
[87] Путь синтеза:
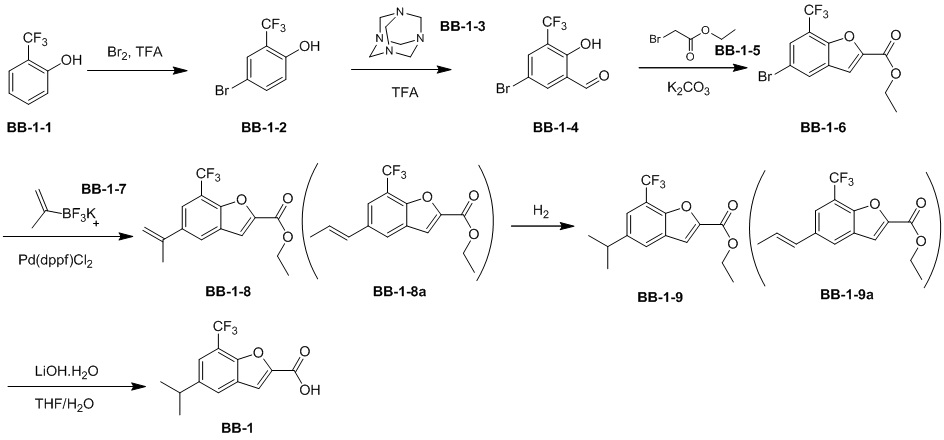
[88] Этап 1: синтез соединения BB-1-2
[89] Соединение BB-1-1 (6,0 г, 37 ммоль) растворяли в TFA (30 мл), затем по каплям медленно добавляли жидкий бром (5,91 г, 37 ммоль), и реагирующие вещества перемешивали при к. т. в течение 2 часов. После завершения реакции добавляли EA (200 мл), органические фазы промывали солевым раствором (50 мл × 2), насыщенным карбонатом натрия (50 мл × 2), солевым раствором (50 мл), высушивали над сульфатом натрия, концентрировали с получением целевого соединения BB-1-2 (серое твердое вещество, 8,9 г, выход: 100%). 1H-ЯМР (400 МГц, CDCl3): δ 7,63 (s, 1H), 7,52 (d, J = 8,8 Гц, 1H), 6,88 (d, J = 8,8 Гц, 1H), 6,09 (s, 1H).
[90] Этап 2: синтез соединения BB-1-4
[91] Соединение BB-1-2 (1 г, 4,13 ммоль) растворяли в TFA (5 мл), затем добавляли метенамин BB-1-3 (2,33 г, 16,6 ммоль). Реагирующие вещества нагревали до 90°C в непроветриваемом контейнере и перемешивали в течение ночи. После охлаждения добавляли воду (20 мл) и 50% серную кислоту (7 мл), смесь дополнительно перемешивали в течение 1 часа, фильтровали, твердое вещество промывали водой, высушивали с получением целевого соединения BB-1-4 (серое твердое вещество, 0,60 г, выход 53,75%). 1H-ЯМР (400 МГц, CDCl3): δ 11,62 (s, 1H), 9,93 (s, 1H), 7,92 (s, 1H), 7,88 (s, 1H).
[92] Этап 3: синтез соединения BB-1-6
[93] Реагирующее вещество BB-1-4 (0,60 г, 2,23 ммоль) растворяли в DMF (8 мл), затем добавляли K2CO3 (0,925 г, 6,69 ммоль), соединение BB-1-5 (0,41 г, 2,45 ммоль). Реагирующие вещества нагревали до 100°C и перемешивали в течение 2 часов. Добавляли EA (50 мл), смесь промывали солевым раствором (20 мл), высушивали, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/3) с получением целевого соединения BB-1-6 (желтое твердое вещество, 0,35 г, выход 46,5%). MS (ESI): масса/заряд 337 [M+H]+, 339 [M+H+2]+.
[94] Этап 4: синтез соединений BB-1-8 и BB-1-8a
[95] Соединение BB-1-6 (0,34 г, 1,01 ммоль) растворяли в изопропаноле (5 мл), затем добавляли изопропенилтрифторборат калия BB-1-7 (0,194 г, 1,31 ммоль), TEA (0,510 г, 5,04 ммоль), 1,1’-[бис(дифенилфосфино)ферроцен]дихлорпалладий (0,074 г, 0,101 ммоль). В реакционной смеси обеспечивали протекание реакции в течение 3 часов в атмосфере газообразного азота. После охлаждения добавляли EA (100 мл), смесь промывали солевым раствором (20 мл), высушивали над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/5) с получением целевых соединений BB-1-8 и BB-1-8a (желтое масло, 0,295 г, выход 98%). MS (ESI): масса/заряд 299 [M+H]+.
[96] Этап 5: синтез соединений BB-1-9 и BB-1-9a
[97] Соединения BB-1-8 и BB-1-8a (0,295 г, 0,989 ммоль) растворяли в EA (15 мл), добавляли 10% Pd/C (0,03 г), реагирующие вещества перемешивали в атмосфере газообразного водорода при давлении 20 фунтов/кв. дюйм в течение 4 часов при к. т. Реакционную смесь выпаривали досуха с получением целевых соединений BB-1-9 и BB-1-9a, которые непосредственно использовали на следующем этапе (серое твердое вещество, 0,295 г, выход 99%). MS (ESI): масса/заряд 301 [M+H]+.
[98] Этап 6: синтез соединения BB-1
[99] Соединение BB-1-9 (0,105 г, 0,35 ммоль) растворяли в THF/H2O (3 мл, 2:1), добавляли гидрат гидроксида лития (0,073 г, 1,75 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции pH доводили до 1 с помощью 2M хлористоводородной кислоты, затем смесь экстрагировали с помощью EA (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали, концентрировали с получением целевого соединения BB-1 (серое твердое вещество, 0,093 г, выход 98%). MS (ESI): масса/заряд 273 [M+H]+.
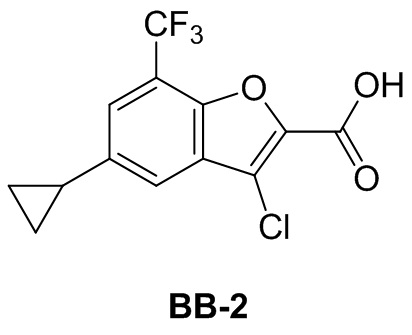
Сравнительный пример 2: фрагмент BB-2
[100] Путь синтеза:
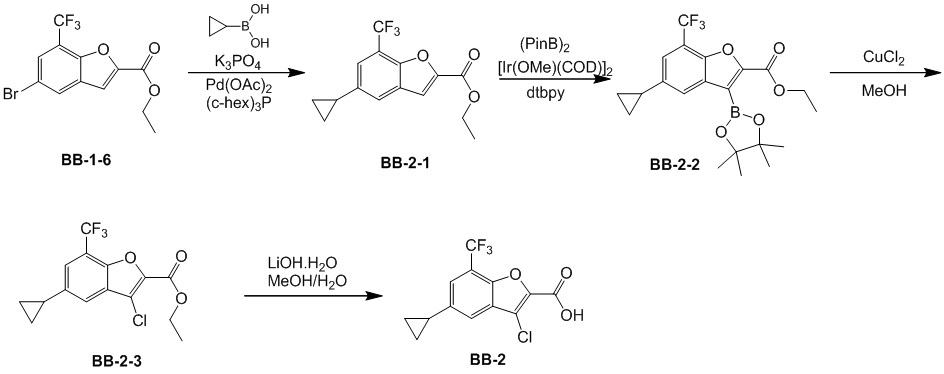
[101] Этап 1: синтез соединения BB-2-1
[102] Соединение BB-1-6 (2 г, 5,93 ммоль) растворяли в смеси толуол/H2O (30 мл, 2:1), затем последовательно добавляли циклопропилбороновую кислоту (1,02 г, 11,86 ммоль), Pd(OAC)2 (0,133 г, 0,593 ммоль), трициклогексилфосфат (0,183 г, 0,652 ммоль), K3PO4 (3,78 г, 17,79 ммоль). Реакционную смесь перемешивали при 100°C в течение 3 часов. После охлаждения добавляли EA (100 мл), органическую фазу промывали солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/10) с получением целевого соединения BB-2-1 (желтое масло, 1,6 г, выход 90,4%). MS (ESI): масса/заряд 299 [M+H]+.
[103] Этап 2: синтез соединения BB-2-2
[104] Соединение BB-2-1 (1,6 г, 5,36 ммоль) растворяли в н-гексане (30 мл), затем последовательно добавляли бис(пинаколато)дибор (1,497 г, 5,896 ммоль), ди-μ-метоксобис(1,5-циклооктадиен)дииридий (I) (0,216 г, 0,32 ммоль), 4,4'-ди-трет-бутил-2,2'-дипиридин (0,144 г, 0,536 ммоль). Реагирующие вещества кипятили с обратным холодильником в течение 2 часов в атмосфере газообразного азота. Реакционную смесь выпаривали досуха, затем очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/10) с получением целевого соединения BB-2-2 (белое твердое вещество, 2,1 г, выход 92%). MS (ESI): масса/заряд 343 [M-82+H]+.
[105] Этап 3: синтез соединения BB-2-3
[106] Соединение BB-2-2 (0,50 г, 1,18 ммоль) растворяли в метаноле (8 мл), затем добавляли CuCl2 (0,317 г, 2,36 ммоль). Реакционную смесь нагревали до 50°C, и в ней обеспечивали протекание реакции в течение 16 ч. После охлаждения реакционную смесь выпаривали досуха, затем очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/10) с получением целевого соединения BB-2-3 (белое твердое вещество, 0,310 г, выход 79%). MS (ESI): масса/заряд 333 [M+H]+.
[107] Этап 4: синтез соединения BB-2
[108] Соединение BB-2-3 (0,31 г, 0,932 ммоль) растворяли в смеси метанол/H2O (6 мл, 2:1), затем добавляли гидрат гидроксида лития (0,156 г, 3,73 ммоль). После завершения реакции pH доводили до 1 с помощью 2 н. хлористоводородной кислоты, затем смесь экстрагировали с помощью DCM, объединенную органическую фазу промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали, концентрировали с получением целевого соединения BB-2, которое непосредственно использовали на следующем этапе (белое твердое вещество, 0,270 г, выход 95%). MS (ESI): масса/заряд 305 [M+H]+.
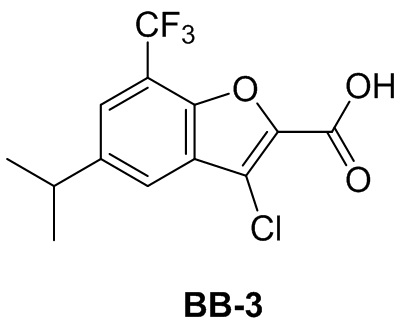
Сравнительный пример 3: фрагмент BB-3
[109] Путь синтеза:
[110] Этап 1: синтез соединения BB-3-1
[111] Соединение BB-1-9 (0,20 г, 0,666 ммоль) растворяли в н-гексане (3 мл), затем последовательно добавляли бис(пинаколато)дибор (0,186 г, 0,763 ммоль), ди-μ-метоксобис(1,5-циклооктадиен)дииридий (I) (0,028 г, 0,042 ммоль), 4,4'-ди-трет-бутил-2,2'-дипиридин (0,018 г, 0,067 ммоль), реагирующие вещества кипятили с обратным холодильником в течение 16 часов в атмосфере газообразного азота. После выпаривания досуха смесь очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/5) с получением целевого соединения BB-3-1 (белое твердое вещество, 0,20 г, выход 70%). MS (ESI): масса/заряд 345 [M-82+H]+.
[112] Этап 2: синтез соединения BB-3-2
[113] Соединение BB-3-1 (0,20 г, 0,469 ммоль) растворяли в метаноле (4 мл), затем добавляли CuCl2 (0,126 г, 0,938 ммоль), реакционную смесь нагревали до 50°C, и в ней обеспечивали протекание реакции в течение 16 часов. После охлаждения реакционную смесь выпаривали досуха, затем очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/5) с получением целевого соединения BB-3-2 (желтое твердое вещество, 0,140 г, выход 89%). MS (ESI): масса/заряд 335 [M+H]+.
[114] Этап 3: синтез соединения BB-3
[115] Соединение BB-3-2 (0,130 г, 0,388 ммоль) растворяли в смеси метанол/H2O (5 мл, 2:1), затем добавляли гидрат гидроксида лития (0,081 г, 1,94 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции pH доводили до 1 с помощью 2 н. хлористоводородной кислоты. А затем смесь экстрагировали с помощью EA (50 мл × 2), объединенную органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали, концентрировали с получением соединения BB-3 (белое твердое вещество, неочищенный продукт, 0,119 г, выход 100%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 307 [M+H]+.
Сравнительный пример 3a: фрагмент BB-3a
[116] Сравнительный пример 3a (фрагмент BB-3a) синтезировали в соответствии со способом получения фрагмента BB-3 в сравнительном примере 3, но исходным материалом было соединение BB-1-9a. MS (ESI): масса/заряд 307 [M+H]+.
Сравнительный пример 4: фрагмент BB-4
[117] Путь синтеза:
[118] Этап 1: синтез соединения BB-4-1
[119] Соединение BB-2-2 (0,30 г, 0,707 ммоль) растворяли в метаноле (8 мл), затем добавляли CuBr2 (0,316 г, 1,41 ммоль), реакционную смесь нагревали до 50°C, и в ней обеспечивали протекание реакции в течение 20 часов. После охлаждения реакционную смесь выпаривали досуха с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/10) с получением целевого соединения BB-4-1 (серое твердое вещество, 0,22 г, выход 82%). MS (ESI): масса/заряд 377 [M+H]+, 379 [M+H+2]+.
[120] Этап 2: синтез соединения BB-4
[121] К смеси метанол/H2O (5 мл, 2:1) добавляли соединение BB-4-1 (0,120 г, 0,318 ммоль), затем добавляли моногидрат гидроксида лития (0,089 г, 1,59 ммоль). После осуществления реакции в течение 2 часов при к. т. реакционную смесь выпаривали при пониженном давлении с последующим доведением pH до 1 с помощью 2 н. хлористоводородной кислоты, затем смесь экстрагировали с помощью EA (25 мл × 3), органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении с получением целевого соединения BB-4 (серое твердое вещество, 0,105 г, выход 95%). MS (ESI): масса/заряд 349 [M+H]+, 351 [M+H+2]+.
Сравнительный пример 5: фрагмент BB-5
[122] Путь синтеза:
[123] Этап 1: синтез соединения BB-5-1
[124] Соединение BB-4-1 (0,100 г, 0,265 ммоль) растворяли в смеси толуол/H2O (3 мл, 2:1), затем добавляли метилбороновую кислоту (0,032 г, 0,53 ммоль), Pd(OAc)2 (0,006 г, 0,0265 ммоль), трициклогексилфосфат (0,008 г, 0,029 ммоль), K3PO4 (0,169 г, 0,795 ммоль). Реагирующие вещества нагревали до 100°C и перемешивали в течение 16 часов. Реакционную смесь охлаждали, и добавляли EA (100 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью пластины с тонким слоем силикагеля (проявитель EA/PE = 1/6) с получением целевого соединения BB-5-1 (серое твердое вещество, 0,075 г, выход 90,6%). MS (ESI): масса/заряд 313 [M+H]+.
[125] Этап 2: синтез соединения BB-5
[126] Соединение BB-5-1 (0,075 г, 0,24 ммоль) растворяли в смеси метанол/H2O (3 мл, 2:1), затем добавляли гидрат гидроксида лития (0,040 г, 0,96 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции pH доводили до 1 с помощью 2 н. хлористоводородной кислоты, затем смесь экстрагировали с помощью DCM (50 мл × 2), объединенную органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-5 (серое твердое вещество, 0,067 г, выход 98%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 285 [M+H]+.
Сравнительный пример 6: фрагмент BB-6
[127] Путь синтеза:
[128] Этап 1: синтез соединения BB-6-1
[129] Соединение BB-4-1 (0,160 г, 0,424 ммоль) растворяли в DMF (3 мл), затем добавляли CuCN (0,152 г, 1,7 ммоль), в реакционной смеси обеспечивали протекание реакции при 160°C в течение 4 часов. После охлаждения смеси добавляли EA (50 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью пластины с тонким слоем силикагеля (проявитель EA/PE = 1/3) с получением целевого соединения BB-6-1 (серое твердое вещество, 0,040 г, выход 29%). MS (ESI): масса/заряд 324 [M+H]+.
[130] Этап 2: синтез соединения BB-6
[131] Соединение BB-6-1 (0,040 г, 0,124 ммоль) растворяли в смеси THF/MeOH/H2O (3 мл, 1:1:1), затем добавляли гидрат гидроксида лития (0,021 г, 0,495 ммоль). Реакционную смесь перемешивали при к. т. с обеспечением протекания в ней реакции в течение 2 часов. После завершения реакции pH доводили до 1 с помощью 6 н. хлористоводородной кислоты, затем смесь экстрагировали с помощью DCM (50 мл × 2), объединенную органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-6 (серое твердое вещество, 0,036 г, выход 98,6%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 296 [M+H]+.
Сравнительный пример 7: фрагмент BB-7
[132] Путь синтеза:
[133] Этап 1: синтез соединения BB-7-2
[134] Соединение BB-7-1 (5 г, 26,18 ммоль) растворяли в TFA (5 мл), затем в атмосфере газообразного азота порциями добавляли соединение BB-1-3 (7,34 г, 52,36 ммоль). Реагирующие вещества перемешивали при к. т. в течение 20 минут, затем нагревали до 90°C и обеспечивали протекание реакции между ними в течение ночи. После охлаждения добавляли H2O (30 мл) и 50% серную кислоту (15 мл), смесь перемешивали в течение 2 часов, фильтровали и высушивали с получением целевого соединения BB-7-2 (коричневое твердое вещество, 3,5 г, выход 61%). 1H-ЯМР (400 МГц, CDCl3): δ 10,85 (br. s., 1H), 9,84 - 9,89 (m, 1H), 7,45 - 7,53 (m, 2H).
[135] Этап 2: синтез соединения BB-7-3
[136] Соединение BB-7-2 (1 г, 4,57 ммоль) растворяли в DMF (12 мл), затем добавляли K2CO3 (1,89 г, 13,7 ммоль), этилбромацетат (0,838 г, 5,02 ммоль). Реакционную смесь нагревали до 100°C, и в ней обеспечивали протекание реакции в течение 2 часов. После охлаждения добавляли EA (100 мл). Органическую фазу промывали солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/3) с получением целевого соединения BB-7-3 (желтое твердое вещество, 0,400 г, выход 30%). 1H-ЯМР (400 МГц, CDCl3): δ 7,61 (s, 1H), 7,48 (d, J = 3,01 Гц, 1H), 7,34 (dd, J = 9,54, 1,51 Гц, 1H), 4,46 (q, J = 7,03 Гц, 2H), 1,43 (t, J = 7,03 Гц, 3H).
[137] Этап 3: синтез соединения BB-7-4
[138] Соединение BB-7-3 (0,287 г, 1 ммоль) растворяли в смеси толуол/H2O (5 мл, 2:1), затем добавляли циклопропилбороновую кислоту (0,172 г, 2 ммоль), Pd(OAc)2 (0,022 г, 0,1 ммоль), трициклогексилфосфат (0,028 г, 0,1 ммоль), K3PO4 (0,637 г, 3 ммоль). Реагирующие вещества нагревали до 100°C, и обеспечивали протекание реакции между ними в течение 4 часов. После охлаждения добавляли EA (100 мл), органическую фазу промывали солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-7-4 (серое твердое вещество, 0,240 г, выход 96,7%). MS (ESI): масса/заряд 249 [M+H]+.
[139] Этап 4: синтез соединения BB-7-5
[140] Соединение BB-7-4 (0,080 г, 0,322 ммоль) растворяли в н-гексане (3 мл), затем последовательно добавляли (PinB)2 (0,106 г, 0,419 ммоль), ди-μ-метоксобис(1,5-циклооктадиен)дииридий (I) (0,015 г, 0,022 ммоль), 4,4’-ди-трет-2,2’-бипиридин (0,009 г, 0,032 ммоль), реагирующие вещества кипятили с обратным холодильником в атмосфере газообразного азота в течение 16 часов. Реакционную смесь выпаривали досуха, затем очищали с помощью пластины с тонким слоем силикагеля (проявитель EA/PE = 1/3) с получением целевого соединения BB-7-5 (белое твердое вещество, 0,070 г, выход 58%). MS (ESI): масса/заряд 293 [M-82+H]+.
[141] Этап 5: синтез соединения BB-7-6
[142] Соединение BB-7-5 (0,070 г, 0,187 ммоль) растворяли в метаноле (3 мл), затем добавляли CuCl2 (0,05 г, 0,374 ммоль), реакционную смесь нагревали до 50°C, и в ней обеспечивали протекание реакции в течение 20 часов. После охлаждения реакционную смесь выпаривали досуха, затем добавляли EA (50 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-7-6 (серое твердое вещество, 0,052 г, выход 98%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 283 [M+H]+.
[143] Этап 6. Синтез соединения BB-7
[144] Соединение BB-7-6 (0,052 г, 0,184 ммоль) растворяли в смеси THF/MeOH/H2O (4 мл, 2:1:1), затем добавляли гидрат гидроксида лития (0,039 г, 0,92 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции pH доводили до 1 с помощью 2 н. хлористоводородной кислоты, затем смесь экстрагировали с помощью EA (50 мл × 2), объединенную органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-7 (серое твердое вещество, 0,045 г, выход 96%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 255 [M+H]+.
Сравнительный пример 8: фрагмент BB-8
[145] Путь синтеза:
[146] Этап 1: синтез соединения BB-8-2
[147] Соединение BB-8-1 (5 г, 36,72 ммоль) растворяли в HOAc (30 мл), смесь охлаждали до 0°C, затем по каплям добавляли жидкий бром (6,75 г, 42,23 ммоль), и в полученной в результате смеси обеспечивали протекание реакции при 0°C в течение 2 часов. После завершения реакции добавляли H2O (100 мл), смесь фильтровали, твердое вещество промывали с помощью H2O и высушивали с получением целевого соединения BB-8-2 (желтое твердое вещество, 7,5 г, выход 95%). 1H-ЯМР (400 МГц, CDCl3): δ 11,18 (s, 1H), 9,81 (s, 1H), 7,51 - 7,47 (m, 2H), 2,25 (s, 3H).
[148] Этап 2: синтез соединения BB-8-3
[149] Соединение BB-8-2 (5,4 г, 25,11 ммоль) растворяли в DMF (70 мл), затем добавляли K2CO3 (6,94 г, 50,22 ммоль), этилбромацетат (5,03 г, 30,13 ммоль). В реакционной смеси обеспечивали протекание реакции при к. т. в течение 1 часа, затем ее нагревали до 80°C, и в ней обеспечивали протекание реакции в течение 16 часов. После охлаждения добавляли EA (50 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-8-3 (белое твердое вещество, 4,2 г, выход 59%). MS (ESI): масса/заряд 283 [M+H]+, 285 [M+H+2]+.
[150] Этап 3: синтез соединения BB-8-4
[151] Соединение BB-8-3 (0,50 г, 1,77 ммоль) растворяли в CCl4 (7 мл), затем добавляли NBS (0,377 г, 2,12 ммоль), бензоилпероксид (0,043 г, 0,177 ммоль). В реакционной смеси обеспечивали протекание реакции при 80°C в течение 16 ч. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-8-4 (серое твердое вещество, 0,660 г, неочищенный продукт). MS (ESI): масса/заряд 363 [M+H]+, 361 [M+H-2]+, 365 [M+H+2]+.
[152] Этап 4: синтез соединения BB-8-5
[153] Соединение BB-8-4 (0,30 г, 0,829 ммоль) растворяли в ацетонитриле (10 мл), затем добавляли N-метилморфолин-N-оксид (0,970 г, 8,29 ммоль). Реакционную смесь перемешивали при к. т. в течение 4 часов, затем выпаривали досуха. Добавляли EA (50 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/5) с получением целевого соединения BB-8-5 (белое твердое вещество, 0,180 г, 73,11%). MS (ESI): масса/заряд 297 [M+H]+, 299 [M+H+2]+.
[154] Этап 5: синтез соединения BB-8-6
[155] Соединение BB-8-5 (0,170 г, 0,572 ммоль) растворяли в толуоле (2 мл), затем добавляли трифторид диэтиламиносеры (0,185 г, 1,15 ммоль), реагирующие вещества перемешивали при 25°C в течение 16 часов в атмосфере газообразного азота. После завершения реакции добавляли EA (50 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-8-6 (серое твердое вещество, 0,175 г, 95,84%). MS (ESI): масса/заряд 319 [M+H]+, 321 [M+H+2]+.
[156] Этап 6: синтез соединения BB-8-7
[157] Соединение BB-8-6 (0,170 г, 0,533 ммоль) растворяли в толуоле (3 мл) и H2O (1 мл), затем добавляли трициклогексилфосфат (0,015 г, 0,053 ммоль), циклопропилбороновую кислоту (0,10 г, 1,16 ммоль), Pd(OAc)2 (0,012 г, 0,053 ммоль), реагирующие вещества нагревали до 110°C и перемешивали в течение 5 часов в атмосфере газообразного азота. После охлаждения добавляли EA (50 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/5) с получением целевого соединения BB-8-7 (желтое твердое вещество, 0,132 г, 88,40%). MS (ESI): масса/заряд 281 [M+H]+.
[158] Этап 7: синтез соединения BB-8-8
[159] Соединение BB-8-7 (0,132 г, 0,471 ммоль) растворяли в н-гексане (3 мл), затем последовательно добавляли (PinB)2 (0,145 г, 0,571 ммоль), 4,4’-ди-трет-2,2’-бипиридин (0,013 г, 0,048 ммоль), ди-μ-метоксобис(1,5-циклооктадиен)дииридий (I) (0,019 г, 0,029 ммоль), реагирующие вещества кипятили с обратным холодильником в атмосфере газообразного азота в течение 16 часов. Реакционную смесь выпаривали досуха, очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/5) с получением целевого соединения BB-8-8 (белое твердое вещество, 0,10 г, 52,27%). MS (ESI): масса/заряд 325 [M-82+H]+.
[160] Этап 8: синтез соединения BB-8-9
[161] Соединение BB-8-8 (0,095 г, 0,234 ммоль) растворяли в метаноле (4 мл), затем добавляли CuCl2 (0,063 г, 0,468 ммоль), реакционную смесь нагревали до 50°C, и в ней обеспечивали протекание реакции в течение 16 часов. После охлаждения реакционную смесь выпаривали досуха, затем добавляли EA (50 мл). Органическую фазу промывали солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-8-9 (белое твердое вещество, 0,073 г, 99,19%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 315 [M+H]+.
[162] Этап 9: синтез соединения BB-8
[163] Соединение BB-8-9 (0,070 г, 0,222 ммоль) растворяли в метаноле (2 мл) и H2O (2 мл), затем добавляли гидрат гидроксида лития (0,050 г, 1,19 ммоль), реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции pH доводили до 1 с помощью 3 н. хлористоводородной кислоты, затем смесь экстрагировали с помощью EA (50 мл × 2), объединенную органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-8 (белое твердое вещество, 0,063 г, 98,81%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 287 [M+H]+.
Сравнительный пример 9: фрагмент BB-9
[164] Путь синтеза:
[165] Этап 1: синтез соединения BB-9-2
[166] Соединение BB-2 (0,30 г, 0,98 ммоль), соединение BB-9-1 (0,141 г, 0,98 ммоль), HATU (0,449 г, 1,18 ммоль) и DIPEA (0,382 г, 2,95 ммоль) растворяли в безводном DMF (3 мл), перемешивали при 20°C в течение 6 часов, добавляли H2O, смесь экстрагировали с помощью EA (20 мл × 2). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении с получением соединения BB-9-2 (желтое масло, 0,460 г, выход 100%), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 430,2 [M+H]+.
[167] Этап 2: синтез соединения BB-9
[168] Соединение BB-9-2 (0,460 г, 1,07 ммоль) растворяли в смеси THF:метанол: H2O = 2:2:1 (10 мл), добавляли моногидрат гидроксида лития (0,135 г, 3,21 ммоль). Реакционную смесь перемешивали при 18°C в течение 2 часов, растворитель удаляли при пониженном давлении, остаток вновь растворяли в H2O, pH доводили до приблизительно 2-3 с помощью 2 н. водного раствора хлористоводородной кислоты, собирали твердое вещество с получением соединения BB-9 (белое твердое вещество, 0,380 г, выход 85,4%), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 416 [M+H]+.
Сравнительный пример 9a: фрагмент BB-9a
[169] Путь синтеза:
[170] Этап 1: синтез соединения BB-9a-2
[171] Соединение BB-2 (0,147 г, 0,483 ммоль), соединение BB-9a-1 (0,100 г, 0,483 ммоль) растворяли в DMF (1 мл), добавляли DIPEA (0,187 г, 1,45 ммоль) и HATU (0,275 г, 0,724 ммоль). Реакционную смесь перемешивали при 18°C в течение 2 часов, добавляли H2O (3 мл), смесь экстрагировали с помощью EA (20 мл × 3). Органические фазы объединяли, промывали с помощью H2O (20 мл × 2) и насыщенного солевого раствора (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения BB-9a-2 (желтое твердое вещество, 0,220 г, выход 99,58%). MS (ESI): масса/заряд 458 [M+H]+.
[172] Этап 2: синтез соединения BB-9a
[173] Соединение BB-9a-2 (0,220 г, 0,480 ммоль) растворяли в метаноле (5 мл), добавляли моногидрат гидроксида лития (0,101 г, 2,40 ммоль) и H2O (1 мл). Реакционную смесь перемешивали при 25°C в течение 16 часов, затем растворитель удаляли при пониженном давлении, остаток вновь растворяли в H2O, pH доводили до 3 с помощью конц. хлористоводородной кислоты. Смесь фильтровали, осадок на фильтре растворяли в DCM (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения BB-9a (желтое твердое вещество, 0,150 г, выход 72,63%). MS (ESI): масса/заряд 430 [M+H]+.
Сравнительный пример 10: фрагмент BB-10
[174] Путь синтеза:
[175] Этап 1: синтез соединения BB-10-2
[176] Соединение BB-10-1 (10,0 г, 52,85 ммоль) растворяли в DMF (120 мл), добавляли аллилбромид (7,67 г, 63,42 ммоль), и затем порциями добавляли NaH (3,17 г, 79,28 ммоль, 60%) при 0°C, реакционную смесь перемешивали при 0°C в течение 2 часов. После завершения реакции ее гасили H2O. Смесь экстрагировали с помощью EA (50 мл × 2), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-10-2 (серое твердое вещество, 9,3 г, выход 76,7%). MS (ESI): масса/заряд 230 [M+H]+.
[177] Этап 2: синтез соединения BB-10-3
[178] Соединение BB-10-2 (9 г, 39,25 ммоль) растворяли в метаноле (200 мл), охлаждали до -78°C, вводили O3 до посинения реакционной смеси, затем вводили N2 до обесцвечивания реакционной смеси, затем добавляли Me2S (15 мл, 203 ммоль), в смеси обеспечивали протекание реакции при к. т. в течение ночи. После завершения реакции смесь концентрировали, добавляли EA (150 мл), органическую фазу промывали солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-10-3 (желтое масло, 9,1 г, выход 100%). MS (ESI): масса/заряд 232 [M+H]+.
[179] Этап 3: синтез соединения BB-10-5
[180] Соединение BB-10-3 (6,35 г, 27,46 ммоль) растворяли в метаноле (100 мл), затем добавляли BB-10-4 (5,0 г, 32,95 ммоль), DIPEA (4,26 г, 32,95 ммоль), Na2SO4 (20 г), реакционную смесь перемешивали при к. т. в течение 1 часа, затем добавляли NaBH4 (1,25 г, 32,95 ммоль), реакционную смесь перемешивали при к. т. в течение 2 часов. Затем порциями добавляли NaH (2,2 г, 54,92 ммоль, 60%), реакционную смесь перемешивали при к. т. в течение 1 часа. Реакционную смесь концентрировали, добавляли EA (200 мл), органическую фазу промывали солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который затем перекристаллизовывали (EA/PE = 1/5) с получением целевого соединения BB-10-5 (желтое твердое вещество, 5,58 г, выход 67%). MS (ESI): масса/заряд 299 [M+H]+.
[181] Этап 4: синтез соединения BB-10
[182] Соединение BB-10-5 (5,5 г, 18,43 ммоль) растворяли в DCM (20 мл), затем добавляли раствор хлористоводородная кислота/диоксан (50 мл, 4 н.). Реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции смесь концентрировали с получением целевого соединения BB-10 (желтое твердое вещество, 4,4 г, выход 100%). MS (ESI): масса/заряд 199 [M+H]+.
Сравнительный пример 11: фрагмент BB-11
[183] Путь синтеза:
[184] Этап 1: синтез соединения BB-11-1
[185] Соединение BB-10-3 (0,450 г, 1,95 ммоль) растворяли в метаноле (5 мл), затем добавляли бензилкарбазат (0,356 г, 2,14 ммоль), Na2SO4 (0,50 г), реагирующие вещества перемешивали при к. т. в течение 2 часов. Реакционную смесь фильтровали, фильтрат концентрировали с получением целевого соединения BB-11-1 (серое твердое вещество, 0,740 г, выход 100%). MS (ESI): масса/заряд 402 [M+Na]+.
[186] Этап 2: синтез соединения BB-11-2
[187] Соединение BB-11-1 (0,22 г, 0,58 ммоль) растворяли в метаноле (10 мл), затем добавляли Pd(OH)2/C (0,020 г), реагирующие вещества перемешивали при к. т. в течение 3 часов в атмосфере газообразного водорода (15 фунтов/кв. дюйм). Реакционную смесь фильтровали, фильтрат концентрировали с получением целевого соединения BB-11-2 (серое твердое вещество, 0,110 г, выход 76,7%).
[188] Этап 3: синтез соединения BB-11-3
[189] Соединение BB-11-2 (0,110 г, 0,445 ммоль) растворяли в метаноле (3 мл), затем добавляли MeONa (0,048 г, 0,889 ммоль), реагирующие вещества перемешивали при к. т. в течение 2 часов. После завершения реакции ее гасили путем добавления H2O по каплям, смесь экстрагировали с помощью EA (50 мл × 2), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-11-3 (желтое масло, 0,090 г, выход 94%). MS (ESI): масса/заряд 160 [M-tBu+H]+.
[190] Этап 4: синтез соединения BB-11-4
[191] Соединение BB-11-3 (0,090 г, 0,418 ммоль) растворяли в DCM (3 мл), затем добавляли 2-хлорэтилхлорформиат (0,072 г, 0,502 ммоль), DIPEA (0,162 г, 1,25 ммоль), реагирующие вещества перемешивали при 0°C в течение 3 часов. После завершения реакции добавляли DCM (100 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-11-4 (желтое твердое вещество, 0,110 г, выход 74,3%). MS (ESI): масса/заряд 322 [M+H]+.
[192] Этап 5: синтез соединения BB-11-5
[193] Соединение BB-11-4 (0,100 г, 0,31 ммоль) растворяли в DMF (3 мл), затем добавляли NaH (0,025 г, 0,62 ммоль), реагирующие вещества перемешивали при к. т. в течение 2 часов. После завершения реакции ее гасили путем добавления H2O по каплям, смесь экстрагировали с помощью EA (50 мл × 2), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-11-5 (желтое масло, 0,080 г, выход 90,2%). MS (ESI): масса/заряд 308 [M+Na]+.
[194] Этап 6: синтез соединения BB-11
[195] Соединение BB-11-5 (0,080 г, 0,28 ммоль) растворяли в DCM (2 мл), затем добавляли раствор хлористоводородная кислота/диоксан (3 мл, 4 н.), реагирующие вещества перемешивали при к. т. в течение 2 часов. Реакционную смесь концентрировали с получением целевого соединения BB-11, которое непосредственно использовали на следующем этапе (серое твердое вещество, 0,064 г, выход 100%). MS (ESI): масса/заряд 186 [M+H]+.
Сравнительный пример 12: фрагмент BB-12
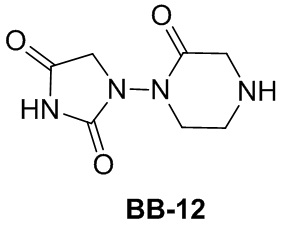
[196] Путь синтеза:
[197] Этап 1: синтез соединения BB-12-2
[198] Соединение BB-12-1 (15,0 г, 67,20 ммоль) растворяли в DMF (170 мл), добавляли аллилбромид (9,76 г, 80,64 ммоль), и затем при 0°C порциями добавляли NaH (3,23 г, 80,64 ммоль, 60%), реакционную смесь перемешивали при 0°C в течение 2 часов. После завершения реакции ее гасили путем добавления H2O, смесь экстрагировали с помощью EA (150 мл × 2), органическую фазу промывали солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-12-2 (серое твердое вещество, 12,0 г, выход 67,83%). MS (ESI): масса/заряд 264 [M+H]+.
[199] Этап 2: синтез соединения BB-12-3
[200] Соединение BB-12-2 (12 г, 45,58 ммоль) растворяли в метаноле (250 мл), охлаждали до -78°C, вводили O3 до посинения реакционной смеси, затем вводили N2 до обесцвечивания реакционной смеси, затем добавляли Me2S (15 мл, 203 ммоль), реакционную смесь перемешивали в течение ночи. После завершения реакции смесь концентрировали, добавляли EA (250 мл), органическую фазу промывали солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-12-3 (желтое масло, 12 г, выход 99,26%). MS (ESI): масса/заряд 266 [M+H]+.
[201] Этап 3: синтез соединения BB-12-5
[202] Соединение BB-12-4 (поставщик: TCI, 0,1 г, 0,66 ммоль) и соединение BB-12-3 (0,175 г, 0,66 ммоль) растворяли в безводном метаноле (10 мл), реагирующие вещества перемешивали при к. т. в течение ночи, затем растворитель удаляли при пониженном давлении с получением соединения BB-12-5 (желтое масло, 0,239 г, выход 100%), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 363,0 [M+H]+.
[203] Этап 4: синтез соединения BB-12-6
[204] Соединение BB-12-5 (0,239 г, 0,66 ммоль) растворяли в смеси HOAc: H2O = 1:1 (3 мл), затем добавляли NaBH3CN (0,041 г, 0,66 ммоль). Реакционную смесь перемешивали при к. т. в течение 3 часов, pH доводили до 6-7 с помощью 1 н. водного раствора NaOH, и затем смесь экстрагировали с помощью EA (20 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении с получением соединения BB-12-6 (бледно-желтое масло, 0,240 г, выход 100%), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 365,1 [M+H]+.
[205] Этап 5: синтез соединения BB-12-7
[206] Соединение BB-12-6 (0,190 г, 0,52 ммоль) растворяли в смеси THF:метанол: H2O = 2:2:1 (5 мл), добавляли моногидрат гидроксида лития (0,109 г, 2,61 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов, растворитель удаляли при пониженном давлении, остаток вновь растворяли в H2O, pH доводили до приблизительно 2-3 с помощью 2 н. водного раствора хлористоводородной кислоты, смесь экстрагировали смесью DCM:метанол = 10:1 (10 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении с получением соединения BB-12-7 (желтое масло, 0,120 г, выход 65,7%), которое непосредственно использовали на следующем этапе без очистки.
[207] Этап 6: синтез соединения BB-12-8
[208] Соединение BB-12-7 (0,183 г, 0,52 ммоль), HATU (0,238 г, 0,63 ммоль) и DIPEA (0,338 г, 2,61 ммоль) растворяли в сухом DMF (3 мл). Реакционную смесь перемешивали при к. т. в течение ночи, очищали с помощью препаративной хроматографии (колонка: Agela DuraShell C18, 150*25*5 мкм, элюирующее средство: MeCN + 0,075% TFA, H2O + 0,075% TFA) с получением целевого соединения BB-12-8 (бледно-желтое масло, 0,040 г, выход 23%). 1H-ЯМР (400 МГц, CD3OD): δ 7,42 - 7,35 (m, 5H), 5,19 (s, 2H), 4,28 - 3,72 (m, 8H).
[209] Этап 7: синтез соединения BB-12
[210] Соединение BB-12-8 (0,040 г, 0,12 ммоль) растворяли в сухом метаноле (5 мл), добавляли Pd/C (0,010 г, 10%), реакционную смесь перемешивали при к. т. в течение 2 часов в атмосфере газообразного водорода (15 фунтов/кв. дюйм), фильтровали, фильтрат выпаривали при пониженном давлении с получением соединения BB-12 (белое твердое вещество, 0,030 г, выход 100%), которое непосредственно использовали на следующем этапе без очистки.
Сравнительный пример 13: фрагмент BB-13
[211] Путь синтеза:
[212] Этап 1: синтез соединения BB-13-2
[213] Соединение BB-13-1 (1,00 г, 4,71 ммоль) растворяли в трет-BuOH (15 мл), затем добавляли TEA (0,572 г, 5,65 ммоль), ди-трет-бутилдикарбонат (3,70 г, 17,0 ммоль), затем по каплям медленно добавляли DPPA (1,56 г, 5,65 ммоль). Реагирующие вещества перемешивали при 28°C в течение 5 часов, затем нагревали до температуры дефлегмирования в течение 16 часов. После завершения реакции растворитель выпаривали, остаток растворяли в MTBE (50 мл), промывали насыщенным раствором NaHCO3 (20 мл), водную фазу экстрагировали с помощью MTBE (50 мл × 2), объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/6) с получением целевого соединения BB-13-2 (белое твердое вещество, 0,67 г, выход 50%). 1H-ЯМР (400 МГц, CDCl3): δ 3,65 (s, 3H), 1,89 (s, 12H), 1,41 (s, 9H).
[214] Этап 2: синтез соединения BB-13-3
[215] Соединение BB-13-2 (0,056 г, 0,2 ммоль) растворяли в метаноле (2,0 мл), затем добавляли гидроксид лития (2 н., 2 мл), pH реагирующих веществ доводили до 4-5 с помощью 1 н. хлористоводородной кислоты, смесь экстрагировали с помощью EA (20 мл × 3), промывали с помощью H2O, высушивали над сульфатом натрия и выпаривали с получением целевого соединения BB-13-3 (белое твердое вещество, 0,040 г, выход 80%). MS (ESI): масса/заряд 214 [M+H-56]+.
[216] Этап 3: синтез соединения BB-13-4
[217] Реагирующее вещество BB-13-3 (2,00 г, 7,43 ммоль) растворяли в DMF (20 мл), затем добавляли бензилкарбазат (1,23 г, 7,43 ммоль), DIPEA (2,88 г, 22,3 ммоль), HATU (4,24 г, 11,1 ммоль). Реагирующие вещества перемешивали при 26°C в течение 16 часов. Добавляли H2O (40 мл), фильтровали, осадок на фильтре растворяли в DCM (50 мл), высушивали, фильтровали и концентрировали с получением целевого соединения BB-13-4 (белое твердое вещество, 2,5 г, выход 81%). 1H-ЯМР (400 МГц, CDCl3): δ 7,55 (m, 5H), 5,16 (s, 2H), 1,69 (s, 12H), 1,43 (s, 9H).
[218] Этап 4: синтез соединения BB-13-5
[219] Соединение BB-13-4 (1,00 г, 2,40 ммоль) растворяли в метаноле (20 мл), затем добавляли влажный Pd/C (0,2 г). Реакционную смесь перемешивали при 26°C в течение 16 ч. в атмосфере газообразного азота (15 фунтов/кв. дюйм). Реакционную смесь фильтровали с помощью целита, промывали метанолом (50 мл), фильтрат концентрировали с получением целевого соединения BB-13-5 (серое твердое вещество, 0,665 г, выход 98%). 1H-ЯМР (400 МГц, CDCl3): δ 4,37 (br, 1H), 1,90 - 1,80 (m, 12H), 1,42 (s, 9H).
[220] Этап 5: синтез соединения BB-13-6
[221] Соединение BB-13-5 (0,665 г, 2,35 ммоль) растворяли в триэтилортоформиате (5 мл), добавляли п-TsOH (0,040 г, 0,235 ммоль), реагирующие вещества перемешивали при 90°C в течение 2 часов. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/2) с получением целевого соединения BB-13-6 (белое твердое вещество, 0,560 г, выход 81%). 1H-ЯМР (400 МГц, CDCl3): δ 8,31 (s, 1H), 4,40 (br, 1H), 2,10 - 1,96 (m, 12H), 1,44 (s, 9H).
[222] Этап 6: синтез соединения BB-13
[223] Соединение BB-13-6 (0,560 г, 1,91 ммоль) растворяли в DCM (20 мл), добавляли TFA (4 мл). Реакционную смесь перемешивали при 26°C в течение 2 часов. После завершения реакции смесь промывали с помощью H2O (30 мл), pH водной фазы доводили до 9 с помощью Na2CO3, затем смесь экстрагировали с помощью DCM (20 мл × 5), объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-13 (белое твердое вещество, 0,200 г, выход 54%). 1H-ЯМР (400 МГц, CDCl3): δ 8,31 (s, 1H), 2,08 - 2,04 (br, 6H), 1,71 - 1,68 (m, 6H).
Сравнительный пример 14: фрагмент BB-14
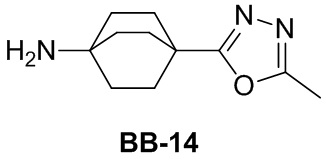
[224] Путь синтеза:
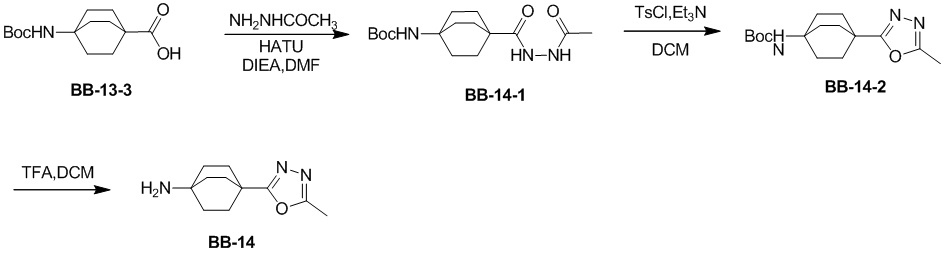
[225] Этап 1: синтез соединения BB-14-1
[226] Соединение BB-13-3 (1,65 г, 6,13 ммоль) растворяли в DMF (20 мл), затем добавляли гидразид уксусной кислоты (0,499 г, 6,74 ммоль), DIPEA (2,37 г, 18,38 ммоль), HATU (3,03 г, 7,96 ммоль), реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции добавляли H2O, затем смесь экстрагировали с помощью EA (50 мл × 3), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 4/1) с получением целевого соединения BB-14-1 (белое твердое вещество, 1,6 г, выход 80%). MS (ESI): масса/заряд 326 [M+H]+.
[227] Этап 2: синтез соединения BB-14-2
[228] Соединение BB-14-1 (0,500 г, 1,54 ммоль) растворяли в DCM (8 мл), затем добавляли TEA (0,311 г, 3,07 ммоль), п-TsCl (0,469 г, 2,46 ммоль), реакционную смесь перемешивали при к. т. в течение 4 часов. После завершения реакции добавляли DCM (100 мл), органическую фазу промывали солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/1) с получением целевого соединения BB-14-2 (белое твердое вещество, 0,330 г, выход 70%). MS (ESI): масса/заряд 308 [M+H]+.
[229] Этап 3: синтез соединения BB-14
[230] Соединение BB-14-2 (0,320 г, 1,04 ммоль) растворяли в DCM (4 мл), затем добавляли TFA (1 мл), реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции добавляли DCM (100 мл), органическую фазу промывали насыщенным водным раствором Na2CO3, солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-14 (белое твердое вещество, 0,205 г, выход 95%). MS (ESI): масса/заряд 208 [M+H]+.
Сравнительный пример 15: фрагмент BB-15
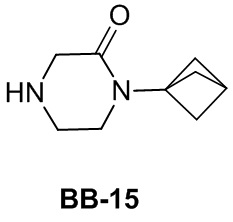
[231] Путь синтеза:
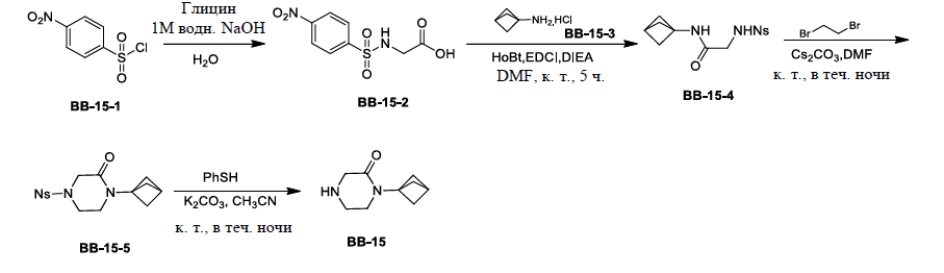
[232] Этап 1: синтез соединения BB-15-2
[233] Глицин (1,51 г, 20,1 ммоль) суспендировали в H2O (5 мл), по каплям добавляли водный раствор NaOH (1M, 10 мл), добавляли соединение BB-15-1 (6,20 г, 28,0 ммоль), по каплям порциями добавляли NaOH (1M, 20 мл), pH поддерживали при значении более 9. После добавления реагирующие вещества перемешивали при к. т. в течение дополнительных 30 минут. Реакционную смесь фильтровали, фильтрат подкисляли водным раствором хлористоводородной кислоты (5M), и выпавшее в осадок твердое вещество отфильтровывали с получением целевого соединения BB-15-2 (желтое твердое вещество, 2,4 г, выход 33%). 1H-ЯМР (400 МГц, CDCl3): δ 8,31 (d, J = 8,8 Гц, 2H), 8,03 (d, J = 8,8 Гц, 2H), 3,78 (s, 2H).
[234] Этап 2: синтез соединения BB-15-4
[235] Соединение BB-15-3 (поставщик: Aroma Circle Corp., 0,050 г, 0,417 ммоль) добавляли к 1,5 мл DMF, затем добавляли соединение BB-15-2 (0,108 г, 0,417 ммоль), HOBt (0,062 г, 0,459 ммоль), DIPEA (0,161 г, 1,25 ммоль), EDC-HCl (0,088 г, 0,459 ммоль). После перемешивания при к. т. в течение 5 часов органическую фазу вливали в H2O, экстрагировали с помощью EA (25 мл), органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении, полученный в результате остаток очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/3) с получением целевого соединения BB-15-4 (желтое твердое вещество, 0,075 г, выход 55,6%). MS (ESI): масса/заряд 326 [M+H]+.
[236] Этап 3: синтез соединения BB-15-5
[237] Соединение BB-15-4 (0,100 г, 0,308 ммоль) добавляли к 3 мл DMF, затем добавляли дибромэтан (0,348 г, 1,85 ммоль), CsCO3 (0,401 г, 1,23 ммоль). После перемешивания при к. т. в течение 16 часов реакцию гасили H2O, проводили высушивание над безводным сульфатом натрия, смесь выпаривали при пониженном давлении с получением целевого соединения BB-15-5 (желтое твердое вещество, 0,080 г, выход 80%). MS (ESI): масса/заряд 352 [M+H]+.
[238] Этап 4: синтез соединения BB-15
[239] Соединение BB-15-5 (0,080 г, 0,228 ммоль) добавляли к 2 мл MeCN, затем добавляли PhSH (0,075 г, 0,684 ммоль). После перемешивания при к. т. в течение 16 часов в атмосфере газообразного азота органическую фазу вливали в DCM, фильтровали, смесь выпаривали при пониженном давлении с получением целевого соединения BB-15 (желтое масло, 0,025 г, выход 65,8%). MS (ESI): масса/заряд 167 [M+H]+.
Сравнительный пример 16: фрагмент BB-16
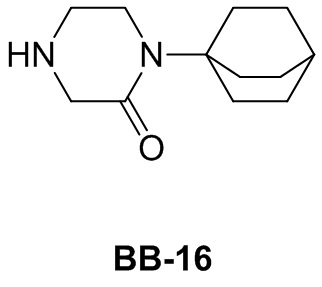
[240] Путь синтеза:
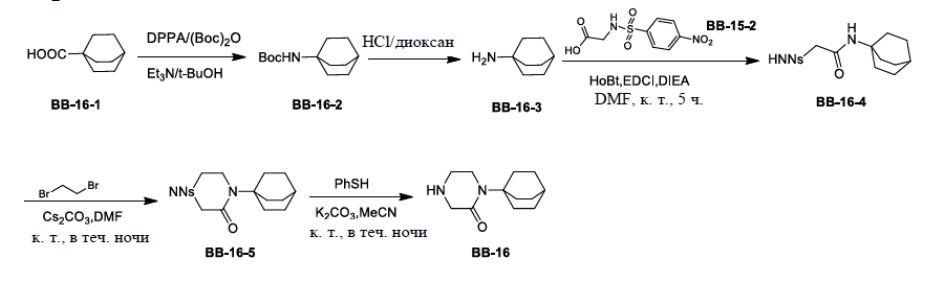 Этап 1: синтез соединения BB-16-2
Этап 1: синтез соединения BB-16-2
[241] Соединение BB-16-1 (поставщик: Shanghai SHUYA, 0,50 г, 3,24 ммоль), ди-трет-бутилдикарбонат (2,5 г, 11,67 ммоль) и TEA (0,393 г, 3,89 ммоль) растворяли в трет-бутаноле (20 мл), медленно добавляли DPPA (1,07 г, 3,89 ммоль) в атмосфере газообразного азота, затем реакционную смесь перемешивали при к. т. в течение приблизительно 8 часов с последующим нагреванием до температуры дефлегмирования в течение 16 часов, растворитель удаляли при пониженном давлении, остаток экстрагировали с помощью MTBE (150 мл × 3), высушивали над безводным сульфатом натрия, смесь выпаривали при пониженном давлении, полученный в результате остаток очищали с помощью препаративной флэш-хроматографии (элюирующее средство: PE/EA = 30:1) с получением целевого соединения BB-16-2 (белое твердое вещество, 0,30 г, выход 41%), которое непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 4,27 - 4,13 (br, 1H), 1,77 - 1,63 (m, 6H), 1,62 - 1,51 (m, 6H), 1,50 - 1,45 (m, 1H), 1,35 (s, 9H).
[242] Этап 2: синтез соединения BB-16-3
[243] Соединение BB-16-2 (0,300 г, 1,33 ммоль) добавляли к 5 мл DCM, затем добавляли раствор хлористоводородная кислота/диоксан (5 мл). После перемешивания при к. т. в течение 16 часов смесь выпаривали при пониженном давлении с получением целевого соединения BB-16-3 (белое твердое вещество, 0,200 г, выход 100%). MS (ESI): масса/заряд 126 [M+H]+.
[244] Этап 3: синтез соединения BB-16-4
[245] Соединение BB-16-3 (0,200 г, 1,242 ммоль) добавляли к 5 мл DMF, затем добавляли соединение BB-15-2 (0,324 г, 1,242 ммоль), HOBt (0,184 г, 1,366 ммоль), EDC-HCl (0,262 г, 1,366 ммоль), DIPEA (0,480 г, 3,72 ммоль). После перемешивания при к. т. в течение 5 часов органическую фазу вливали в H2O, смесь экстрагировали с помощью EA (50 мл × 2), органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении, полученный в результате остаток очищали с помощью колоночной флэш-хроматографии (элюирующее средство EA/PE = 1/3) с получением целевого соединения BB-16-4 (желтое масло, 0,200 г, выход 43,8%). MS (ESI): масса/заряд 368 [M+H]+.
[246] Этап 4: синтез соединения BB-16-5
[247] Соединение BB-16-4 (0,150 г, 0,408 ммоль) добавляли к 5 мл DMF, затем добавляли соединение 1,2-дибромэтан (0,462 г, 2,451 ммоль), CsCO3 (0,531 г, 1,632 ммоль). После перемешивания при 60°C в течение 16 часов реакцию гасили с помощью H2O, смесь высушивали над безводным сульфатом натрия, выпаривали при пониженном давлении с получением целевого соединения BB-16-5 (желтое твердое вещество, 0,100 г, выход 80%). MS (ESI): масса/заряд 394 [M+H]+.
[248] Этап 5: синтез соединения BB-16
[249] Соединение BB-16-5 (0,100 г, 0,487 ммоль) добавляли к 5 мл MeCN, затем добавляли PhSH (0,429 г, 3,9 ммоль), K2CO3 (0,269 г, 1,95 ммоль). После перемешивания при к. т. в течение 16 часов в атмосфере газообразного азота органическую фазу вливали в DCM, фильтровали, смесь выпаривали при пониженном давлении с получением целевого соединения BB-16 (желтое масло, 0,025 г, выход 24,6%). MS (ESI): масса/заряд 209 [M+H]+.
Сравнительный пример 17: фрагмент BB-17
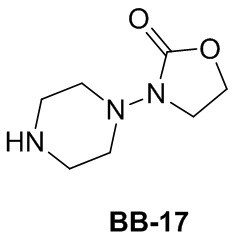
[250] Путь синтеза:
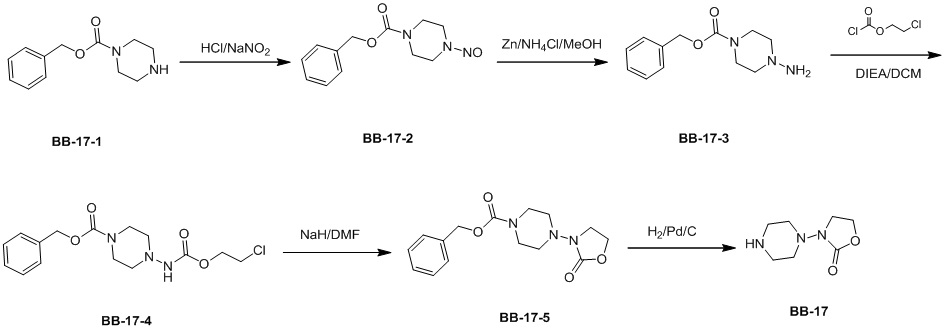
[251] Этап 1: синтез соединения BB-17-2
[252] Соединение BB-17-1 (7,0 г, 31,78 ммоль) растворяли в разбавленной хлористоводородной кислоте (6M, 150 мл), и смесь перемешивали до однородности, по каплям медленно добавляли раствор NaNO2 (4 н., 30 мл) в атмосфере газообразного азота, затем смесь перемешивали при к. т. в течение приблизительно 1 часа, добавляли 100 мл H2O, смесь экстрагировали с помощью EA (100 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении с получением целевого соединения BB-17-2 (7,5 г, выход 66%), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 250 [M+H]+.
[253] Этап 2: синтез соединения BB-17-3
[254] Соединение BB-17-2 (4,0 г, 16 ммоль), порошкообразный цинк (6,2 г, 96 ммоль) и NH4Cl (10 г, 192 ммоль) растворяли в метаноле (80 мл), смесь перемешивали при к. т. в течение 10 минут, затем нагревали до 45°C и перемешивали в течение 2 часов, фильтровали при пониженном давлении, осадок на фильтре промывали метанолом, фильтрат концентрировали досуха при пониженном давлении, к остатку добавляли DCM, и смесь перемешивали в течение 1 часа, фильтрат концентрировали досуха при пониженном давлении с получением целевого соединения BB-17-3 (3,5 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 236 [M+H]+.
[255] Этап 3: синтез соединения BB-17-4
[256] Соединение BB-17-3 (3,5 г неочищенного продукта) и DIPEA (3,81 г, 29,6 ммоль) растворяли в DCM (100 мл), в бане со льдом по каплям медленно добавляли 2-хлорэтилхлорформиат (2,55 г, 18 ммоль) в атмосфере газообразного азота, затем реакционную смесь перемешивали при к. т. в течение приблизительно 2 часов с последующим разбавлением с помощью H2O (80 мл), смесь экстрагировали с помощью DCM (100 мл) (60 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении с получением соединения BB-17-4 (5,0 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 342 [M+H]+.
[257] Этап 4: синтез соединения BB-17-5
[258] Соединение BB-17-4 (5,0 г неочищенного продукта) растворяли в DMF (100 мл), в бане со льдом добавляли NaH (60%, 2,0 г) в атмосфере газообразного азота, затем смесь перемешивали при к. т. в течение приблизительно 2 часов, реакцию гасили путем добавления H2O (100 мл), смесь экстрагировали с помощью EA (300 мл), органическую фазу промывали насыщенным солевым раствором (30 мл × 5), высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении, остаток очищали с помощью препаративной флэш-хроматографии (элюирующее средство: PE/EA = 1:10) с получением целевого соединения BB-17-5 (2,4 г). MS (ESI): масса/заряд 306 [M+H]+.
[259] Этап 4: синтез соединения BB-17
[260] Соединение BB-17-5 (2,4 г, 7,86 ммоль) растворяли в метаноле (80 мл), затем добавляли Pd/C (0,400 г), реакционную смесь перемешивали при к. т. в течение ночи в атмосфере газообразного водорода (15 фунтов/кв. дюйм), и затем Pd/C отфильтровывали, фильтрат концентрировали досуха при пониженном давлении с получением целевого соединения BB-17 (1,2 г, выход 89%), которое непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 4,30 (t, J = 7,8 Гц, 2H), 3,62 (t, J = 7,8 Гц, 2H), 3,03 - 2,87 (m, 8H).
Сравнительный пример 18: фрагмент BB-18
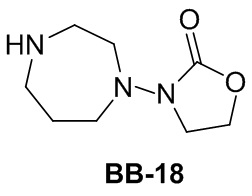
[261] Путь синтеза:
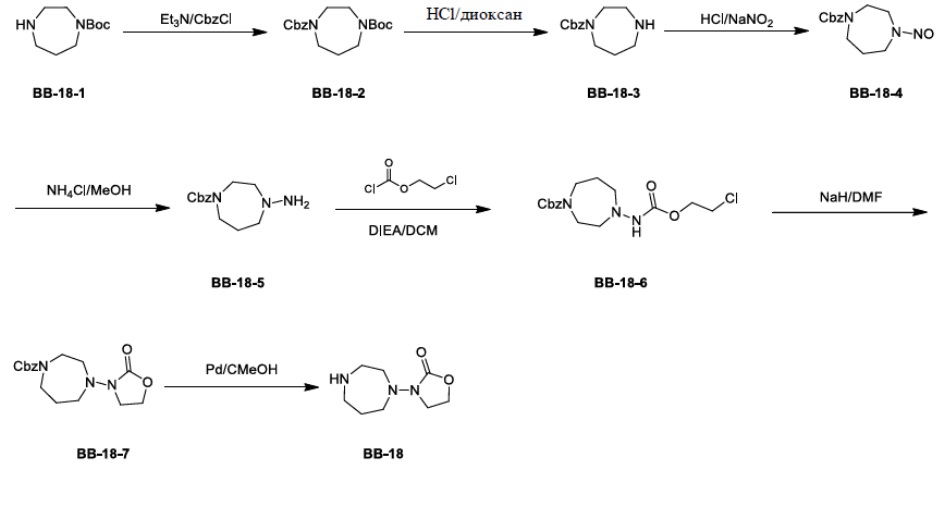
[262] Этап 1: синтез соединения BB-18-2
[263] Соединение BB-18-1 (2,0 г, 10 ммоль) и TEA (2,02 г, 20 ммоль) растворяли в DCM (40 мл), в бане со льдом по каплям медленно добавляли бензилхлорформиат (2,0 г, 11 ммоль) в атмосфере газообразного азота, затем смесь перемешивали при к. т. в течение 24 часов, добавляли разбавленную хлористоводородную кислоту (1 н., 30 мл), смесь экстрагировали с помощью EA (80 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении с получением целевого соединения BB-18-2 (2,6 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 279 [M+H-56]+.
[264] Этап 2: синтез соединения BB-18-3
[265] Соединение BB-18-2 (2,6 г неочищенного продукта) растворяли в растворе HCl/диоксан (4M, 25 мл), и смесь перемешивали до однородности, затем реакционную смесь перемешивали при к. т. в течение 2 часов, затем концентрировали с получением целевого соединения BB-18-3 (1,8 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 235 [M+H]+.
[266] Этап 3: синтез соединения BB-18-4
[267] Соединение BB-18-3 (1,8 г неочищенного продукта) растворяли в разбавленной хлористоводородной кислоте (6M, 40 мл), и смесь перемешивали до однородности, по каплям медленно добавляли раствор NaNO2 (2 н., 17 мл) в атмосфере газообразного азота, затем реакционную смесь перемешивали при к. т. в течение 1 часа, добавляли 50 мл H2O, смесь экстрагировали с помощью EA (60 мл × 3). Органические фазы объединяли и промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия и концентрировали для удаления растворителя с получением таким образом целевого соединения BB-18-4 (2 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 264 [M+H]+.
[268] Этап 4: синтез соединения BB-18-5
[269] Соединение BB-18-4 (2 г неочищенного продукта), порошкообразный цинк (2,73 г, 42 ммоль) и NH4Cl (4,45 г, 84 ммоль) растворяли в метаноле (50 мл), смесь перемешивали при к. т. в течение 10 минут, затем нагревали до 45°C и перемешивали в течение 2 часов, смесь фильтровали при пониженном давлении, осадок на фильтре промывали метанолом, фильтрат концентрировали досуха при пониженном давлении, к остатку добавляли DCM (100 мл), и смесь перемешивали в течение 1 часа, нерастворимые вещества отфильтровывали, фильтрат концентрировали досуха при пониженном давлении с получением целевого соединения BB-18-5 (1,8 г неочищенного продукта), которое использовали на следующем этапе без очистки. MS (ESI): масса/заряд 250 [M+H]+.
[270] Этап 5: синтез соединения BB-18-6
[271] Соединение BB-18-5 (1,8 г неочищенного продукта) и DIPEA (1,85 г, 14,4 ммоль) растворяли в DCM (40 мл), в бане со льдом по каплям медленно добавляли соединение 2-хлорэтилхлорформиат (1,36 г, 9,6 ммоль) в атмосфере газообразного азота, затем смесь перемешивали при к. т. в течение 2 часов, разбавляли с помощью H2O (50 мл), экстрагировали с помощью DCM (50 мл × 3). Органические фазы объединяли и промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия и концентрировали для удаления растворителя с получением таким образом целевого соединения BB-18-6 (2,5 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 356 [M+H]+.
[272] Этап 6: синтез соединения BB-18-7
[273] Соединение BB-18-6 (0,710 г неочищенного продукта) растворяли в DMF (15 мл), в бане со льдом добавляли NaH (60%, 0,320 г) в атмосфере газообразного азота, затем смесь перемешивали при к. т. в течение приблизительно 2 часов, затем проводили гашение путем добавления H2O (50 мл), смесь экстрагировали с помощью EA (200 мл), органическую фазу промывали насыщенным солевым раствором (20 мл × 5), высушивали над безводным сульфатом натрия, выпаривали при пониженном давлении, полученный в результате остаток очищали с помощью препаративной флэш-хроматографии (элюирующее средство: PE/EA = 1:10) с получением целевого соединения BB-18-7 (0,300 г, выход 47%). MS (ESI): масса/заряд 320 [M+H]+.
[274] Этап 7: синтез соединения BB-18
[275] Соединение BB-18-7 (0,300 г, 0,94 ммоль) растворяли в метаноле (5 мл), затем добавляли Pd/C (0,020 г), смесь перемешивали при к. т. в течение ночи в атмосфере газообразного водорода (15 фунтов/кв. дюйм), Pd/C отфильтровывали, фильтрат концентрировали досуха при пониженном давлении с получением целевого соединения BB-18 (0,170 г, выход 97,7%), которое непосредственно использовали на следующем этапе.
Сравнительный пример 19: фрагмент BB-19
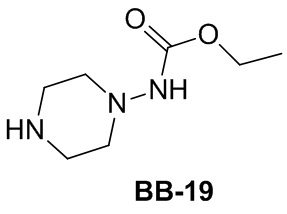
[276] Путь синтеза:
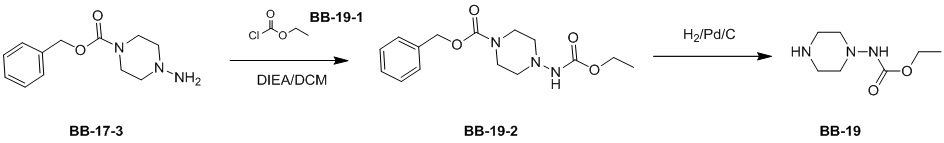
[277] Этап 1: синтез соединения BB-19-2
[278] Соединение BB-17-3 (1,0 г, 4,2 ммоль) и TEA (0,860 г, 8,5 ммоль) растворяли в DCM (20 мл), по каплям медленно добавляли соединение BB-19-1 (0,680 г, 6,3 ммоль) при -10°C в атмосфере газообразного азота, затем смесь перемешивали при к. т. в течение 2 часов, разбавляли путем добавления H2O (50 мл), экстрагировали с помощью DCM (50 мл × 3). Органические фазы объединяли и промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия и концентрировали для удаления растворителя с получением таким образом целевого соединения BB-19-2 (1,3 г неочищенного продукта), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 308 [M+H]+.
[279] Этап 2: синтез соединения BB-19
[280] Соединение BB-19-2 (0,100 г, 0,32 ммоль) растворяли в метаноле (10 мл), затем добавляли Pd/C (0,020 г), смесь перемешивали при к. т. в течение ночи в защитной атмосфере газообразного водорода из баллона (15 фунтов/кв. дюйм), Pd/C отфильтровывали, фильтрат концентрировали досуха при пониженном давлении с получением целевого соединения BB-19 (0,056 г, выход 99,4%), которое непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 174 [M+H]+.
Сравнительный пример 20: фрагмент BB-20
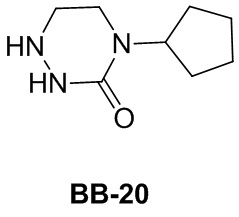
[281] Путь синтеза:
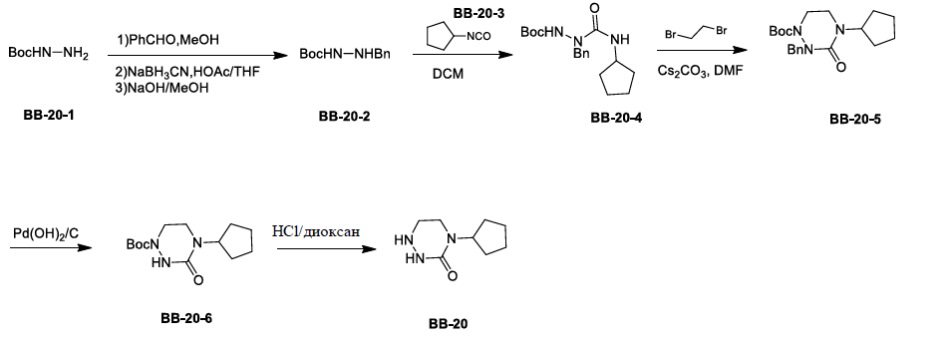
Этап 1: синтез соединения BB-20-2
[282] Соединение BB-20-1 (1 г, 7,6 ммоль) растворяли в THF (10 мл), добавляли бензальдегид (0,77 мл, 7,6 ммоль). Реакционную смесь перемешивали при к. т. в течение 4 часов, затем концентрировали. Полученное в результате твердое вещество (1,3 г, 7,6 ммоль) затем вновь растворяли в THF (15 мл), добавляли AcOH (9 мл) и NaBH3CN (1,05 г, 16,7 ммоль), реакционную смесь перемешивали при к. т. в течение ночи. pH доводили до 9-10 с помощью 1M водного раствора NaOH, смесь экстрагировали с помощью EA (50 мл × 3). Органическую фазу промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением 1,5 г твердого вещества. Затем полученное в результате твердое вещество (1,5 г, 7,6 ммоль) вновь растворяли в метаноле (10 мл), добавляли 1M водный раствор NaOH (10 мл). Реакционную смесь перемешивали при к. т. в течение 2 часов, концентрировали и экстрагировали с помощью DCM (50 мл × 3). Органическую фазу промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта BB-20-2 (белое твердое вещество, 1,37 г, 81,45%), который непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 7,28 - 7,33 (m, 4H), 4,02 (s, 2H), 1,45 (s, 9H). MS (ESI): масса/заряд 167,0 [M+H-56]+.
[283] Этап 2: синтез соединения BB-20-4
[284] Соединение BB-20-3 (0,833 г, 7,5 ммоль) растворяли в DCM (20 мл), добавляли соединение BB-20-2 (1,1 г, 5 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов, затем гасили путем добавления H2O, экстрагировали с помощью DCM (100 мл × 3). Органические фазы объединяли, промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали, неочищенный продукт очищали с помощью препаративной ВЭЖХ (колонка: Phenomenex Gemini C18, 250*50 мм*5 мкм, элюирующее средство: MeCN + 0,05% NH3.H2O, H2O + 0,05% NH3.H2O) с получением BB-20-4 (желтое твердое вещество, 0,730 г, выход 49%). 1H-ЯМР (400 МГц, DMSO-d6): δ 7,24 - 7,29 (m, 5H), 6,10 (br. s., 5H), 3,92 (s, 2H), 1,73 - 1,77 (m, 8H), 1,45 (s, 9H). MS (ESI): масса/заряд 304,0 [M+H]+.
[285] Этап 3: синтез соединения BB-20-5
[286] Соединение BB-20-4 (0,600 г, 1,80 ммоль) и 1,2-дибромэтан (0,507 г, 2,70 ммоль) растворяли в DMF (10 мл), добавляли CsCO3 (1,76 г, 5,40 ммоль). Реакционную смесь перемешивали при к. т. в течение 10 минут в атмосфере газообразного азота, затем нагревали до 40°C, перемешивали в течение 48 часов. Реакционную смесь охлаждали до к. т. и вливали в 50 мл H2O. Водную фазу экстрагировали с помощью EA (100 мл × 3). Органические фазы объединяли, промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной флэш-хроматографии на силикагеле (элюирующее средство: EA/PE = 1/1) с получением BB-20-5 (желтое масло, 0,140 г, выход 22%). MS (ESI): масса/заряд 382,2 [M+Na]+.
[287] Этап 4: синтез соединения BB-20-6
[288] Соединение BB-20-5 (0,200 г, 0,556 ммоль) растворяли в метаноле (20 мл), добавляли Pd(OH)2/C (20%, 0,020 г) в атмосфере газообразного аргона. Реакционную смесь продували газообразным водородом 3 раза с последующей регулировкой давления газообразного водорода (50 фунтов/кв. дюйм) при 50°C, реакционную смесь перемешивали в течение 48 часов, затем фильтровали и концентрировали с получением неочищенного продукта BB-20-6 (серое твердое вещество, 0,180 г, неочищенный продукт), который непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 270 [M+H]+.
[289] Этап 5: синтез соединения BB-20
[290] Соединение BB-20-6 (0,180 г, неочищенный продукт) растворяли в растворе HCl/диоксан (3 мл, 4M), реакционную смесь перемешивали при к. т. в течение 1 часа, затем концентрировали с получением гидрохлорида соединения BB-20 (желтое твердое вещество, 0,140 г, 100%) в виде неочищенного продукта, который непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 170 [M+H]+.
Сравнительный пример 21: фрагмент BB-21
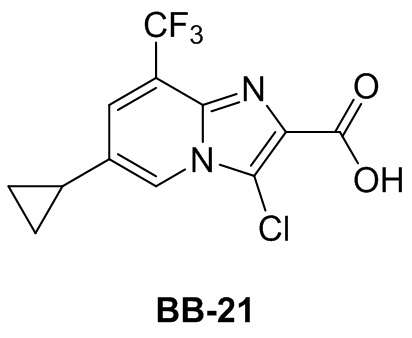
[291] Путь синтеза:
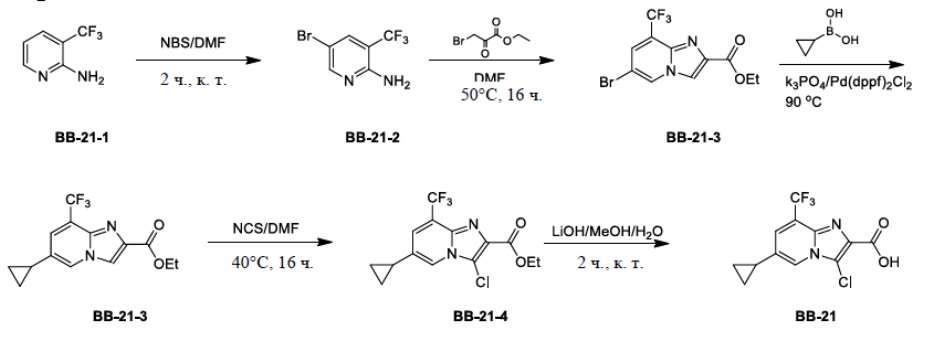
[292] Этап 1: синтез соединения BB-21-2
[293] Соединение BB-21-1 (42,0 г, 259 ммоль) растворяли в DMF (400 мл), затем добавляли NBS (46,1 г, 259 ммоль). Реагирующие вещества перемешивали при 20°C в течение 2 часов. Реакционную смесь вливали в смесь 5% NaHSO3 и H2O (10:1) при перемешивании, осадок отфильтровывали, промывали с помощью H2O, растворяли в DCM (500 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-20-2 (желтое твердое вещество, 51,0 г, выход 89,7%). 1H-ЯМР (400 МГц, CDCl3): δ 8,27 (s, 1H), 7,80 (s, 1H), 5,02 (br, 2H). MS (ESI): масса/заряд 241 [M+H]+, 243 [M+H+2]+.
[294] Этап 2: синтез соединения BB-21-3
[295] Соединение BB-21-2 (51,0 г, 212 ммоль) растворяли в DMF (500 мл), затем добавляли этилбромпируват (82,5 г, 423 ммоль). Реагирующие вещества перемешивали при 50°C в течение 16 часов. Реакционную смесь охлаждали до к. т., вливали в смесь льда и воды (1 л) и перемешивали в течение 20 минут, осадок отфильтровывали, осадок на фильтре растворяли в EA (500 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-21-3 (желтое твердое вещество, 71,0 г, выход 99,5%). 1H-ЯМР (400 МГц, CDCl3): δ 8,49 (s, 1H), 8,25 (s, 1H), 7,69 (s, 1H), 4,47 (q, J = 6,8 Гц, 2H), 1,43 (t, J = 6,8 Гц, 3H). MS (ESI): масса/заряд 337 [M+H]+, 339 [M+H+2]+.
[296] Этап 3: синтез соединения BB-21-4
[297] Соединение BB-21-3 (55,0 г, 163 ммоль) и циклопропилбороновую кислоту (15,4 г, 179 ммоль) растворяли в диоксане (500 мл), затем добавляли K3PO4 (103,9 г, 489 ммоль) и комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроценпалладия (II) и дихлорметана (2,70 г, 3,26 ммоль) в атмосфере газообразного азота. Реагирующие вещества перемешивали при 90°C в течение 16 часов. Реакционную смесь охлаждали до к. т., фильтровали с помощью целита, фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство: EA/PE = 1/4) с получением целевого соединения BB-21-4 (желтое твердое вещество, 24,0 г, выход 49,3%). 1H-ЯМР (400 МГц, CDCl3): δ 8,18 (s, 1H), 8,06 (s, 1H), 7,38 (s, 1H), 4,46 (q, J = 7,2 Гц, 2H), 1,98 (m, 1H), 1,43 (t, J = 7,2 Гц, 3H), 1,07 (m, 2H), 0,76 (m, 2H). MS (ESI): масса/заряд 299 [M+H]+.
[298] Этап 4: синтез соединения BB-21-5
[299] Соединение BB-21-4 (28,0 г, 93,9 ммоль) растворяли в DMF (250 мл), добавляли NCS (12,5 г, 93,9 ммоль). Реагирующие вещества перемешивали при 40°C в течение 16 часов. Реакционную смесь охлаждали до к. т. и вливали в H2O (500 мл), перемешивали в течение 20 минут, осадок отфильтровывали, высушивали с получением целевого соединения BB-21-5 (белое твердое вещество, 31,0 г, выход 99,3%). MS (ESI): масса/заряд 333 [M+H]+.
[300] Этап 5: синтез соединения BB-21-6
[301] Соединение BB-21-5 (31,0 г, 93,2 ммоль) растворяли в метаноле (400 мл), добавляли соединение моногидрат гидроксида лития (19,6 г, 466 ммоль) и H2O (100 мл). Реагирующие вещества перемешивали при 20°C в течение 2 часов. Реакционную смесь концентрировали, pH остатка доводили до значения ниже 3 с помощью конц. хлористоводородной кислоты, смесь экстрагировали с помощью EA (400 мл × 3). Объединенную органическую фазу промывали насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-21-6 (белое твердое вещество, 27,3 г, выход 89,6%). 1H-ЯМР (400 МГц, CDCl3): δ 8,05 (s, 1H), 7,42 (s, 1H), 2,02 (m, 1H), 1,08 (m, 2H), 0,76 (m, 2H). MS (ESI): масса/заряд 305 [M+H]+.
Сравнительный пример 22: фрагмент BB-22
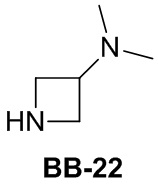
[302] Путь синтеза:
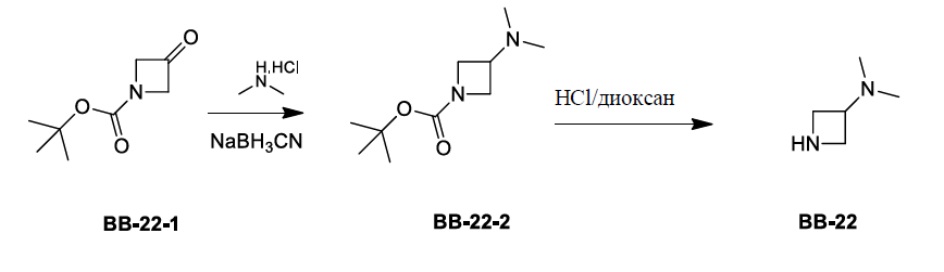
[303] Этап 1: синтез соединения BB-22-2
[304] Соединение BB-22-1 (0,1 г, 0,584 ммоль) растворяли в DCM (1 мл), добавляли гидрохлорид диметиламина (0,072 г, 0,876 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов, добавляли NaBH3CN (0,055 г, 0,876 ммоль), затем смесь перемешивали при к. т. в течение ночи. После завершения реакции реакционную смесь вливали в 20 мл H2O, экстрагировали смесью DCM/метанол (10/1) (30 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-22-2 (желтое масло, 0,12 г) в виде неочищенного продукта. 1H-ЯМР (400 МГц, CDCl3): δ 4,10 - 4,21 (m, 2H), 3,87 - 3,95 (m, 2H), 3,81 (d, J = 4,41 Гц, 1H), 2,16 (s, 6H), 1,43 - 1,44 (m, 9H).
[305] Этап 2: синтез соединения BB-22
[306] Соединение BB-22-2 (0,11 г, 0,549 ммоль) растворяли в DCM (2 мл), добавляли раствор HCl/диоксан (1 мл, 4M), реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции смесь концентрировали с получением целевого соединения гидрохлорида BB-22 (желтое твердое вещество, 0,075 г, неочищенный продукт), которое непосредственно использовали на следующем этапе без очистки.
Сравнительный пример 23: фрагмент BB-23
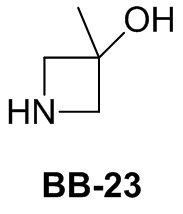
[307] Путь синтеза:
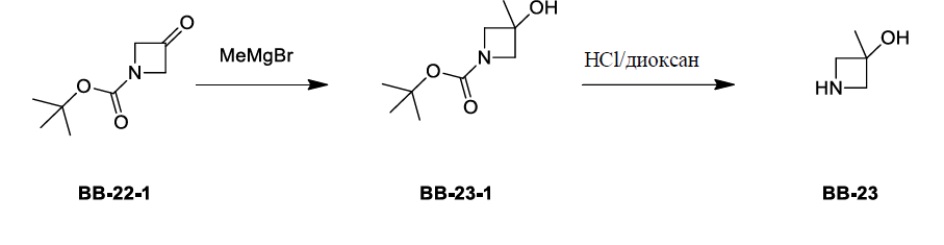
[308] Этап 1: синтез соединения BB-23-1
[309] Соединение BB-22-1 (0,4 г, 2,34 ммоль) растворяли в безводном THF (5 мл), реакционную смесь охлаждали до 0°C, и по каплям добавляли раствор бромметилмагния в THF (3M, 1,00 мл) в атмосфере газообразного азота. Затем реакционную смесь нагревали до к. т., перемешивали в течение 1 часа, добавляли 1M водный раствор NH4Cl (10 мл). Реакционную смесь экстрагировали с помощью EA (30 мл × 3), органические фазы объединяли и промывали насыщенным солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-23-1 (желтое твердое вещество, 0,47 г) в виде неочищенного продукта. 1H-ЯМР (400 МГц, CDCl3): δ 4,07 - 4,17 (m, 1H), 3,79 - 3,88 (m, 4H), 1,51 (s, 3H), 1,43 (s, 9H).
[310] Этап 2: синтез соединения BB-23
[311] Соединение BB-23-1 (0,15 г, 0,80 ммоль) растворяли в DCM (2 мл), добавляли раствор HCl/диоксан (1 мл, 4M), и реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции смесь концентрировали с получением целевого соединения гидрохлорида BB-23 (желтое твердое вещество, 0,099 г, неочищенный продукт), которое непосредственно использовали на следующем этапе без очистки.
Сравнительный пример 24: фрагмент BB-24
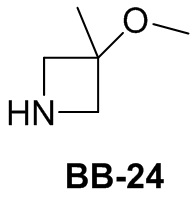
[312] Путь синтеза:
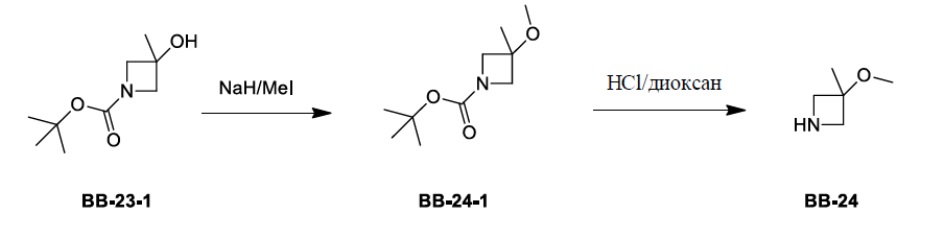
[313] Этап 1: синтез соединения BB-24-1
[314] Соединение BB-23-1 (0,2 г, 1,07 ммоль) растворяли в безводном THF (5 мл), добавляли NaH (0,17 г, 4,30 ммоль, 60%) при к. т. Реакционную смесь перемешивали в течение 10 мин., добавляли MeI (1,14 г, 8,03 ммоль) и реакционную смесь перемешивали при к. т. в течение ночи. После завершения реакции ее гасили путем добавления 20 мл H2O, смесь экстрагировали с помощью EA (50 мл × 3), органические фазы объединяли и промывали насыщенным солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-23-1 (желтое масло, 0,26 г) в виде неочищенного продукта. 1H-ЯМР (400 МГц, CDCl3): δ 3,90 (d, J = 9,04 Гц, 2H), 3,66 (d, J = 9,04 Гц, 2H), 3,19 - 3,29 (m, 3H), 1,45 (s, 3H), 1,44 (s, 9H).
[315] Этап 2: синтез соединения BB-24
[316] Соединение BB-23-1 (0,14 г, 0,70 ммоль) растворяли в DCM (2 мл), добавляли раствор HCl/диоксан (1 мл, 4M), реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции смесь концентрировали с получением целевого соединения гидрохлорида BB-24 (белое твердое вещество, 0,095 г), неочищенный продукт непосредственно использовали на следующем этапе без очистки.
Сравнительный пример 25: фрагмент BB-25
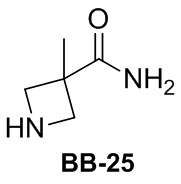
[317] Путь синтеза:
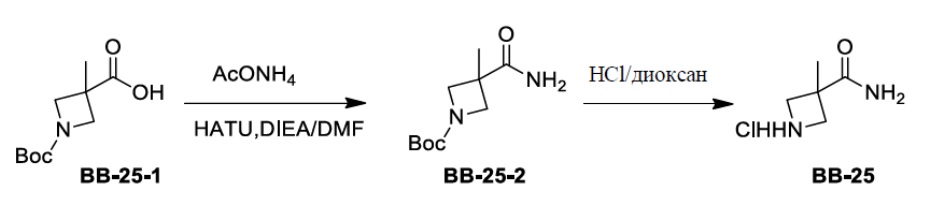
[318] Этап 1: синтез соединения BB-25-2
[319] Соединение BB-25-1 (0,1 г, 0,46 ммоль) и NH4OAc (0,035 г, 0,046 ммоль) растворяли в DMF (2 мл), затем добавляли HATU (0,265 г, 0,697 ммоль) и DIPEA (0,18 г, 1,39 ммоль), реагирующие вещества перемешивали при к. т. в течение 16 часов. После завершения реакции добавляли EA, органическую фазу промывали водным раствором NaHCO3 и насыщенным солевым раствором, высушивали над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали для удаления растворителя с получением таким образом целевого соединения BB-25-2 (белое твердое вещество, 0,05 г, неочищенный продукт), неочищенный продукт непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 237 [M+Na]+.
[320] Этап 2: синтез соединения BB-25-3
[321] Соединение BB-25-2 (0,05 г, неочищенный продукт) растворяли в THF (1,5 мл), затем добавляли раствор HCl/диоксан (1,5 мл, 4M), реагирующие вещества перемешивали при к. т. в течение 1,5 часа, затем смесь концентрировали при пониженном давлении для удаления растворителя с получением таким образом целевого соединения BB-25 (белое твердое вещество, 0,025 г, неочищенный продукт).
Сравнительный пример 26: фрагмент BB-26
[322] Путь синтеза:
[323] Этап 1: синтез соединения BB-26
[324] Соединение BB-26-1 (0,100 г, 0,534 ммоль) растворяли в DCM (2 мл), затем добавляли раствор HCl/диоксан (1 мл, 4M). Реакционную смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь концентрировали с получением целевого соединения BB-26 (белое твердое вещество, 0,060 г, выход 90,91%).
Сравнительный пример 27: фрагмент BB-27
[325] Путь синтеза:
[326] Этап 1: синтез соединения BB-27-2
[327] Соединение BB-27-1 (0,10 г, 0,411 ммоль) растворяли в DMF (3 мл), затем добавляли гидрохлорид диметиламина (0,070 г, 0,858 ммоль), DIPEA (0,210 г, 1,62 ммоль), HATU (0,184 г, 0,484 ммоль), реагирующие вещества перемешивали при к. т. в течение 2 часов. После завершения реакции добавляли EA, органическую фазу промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении для удаления растворителя с получением таким образом целевого соединения BB-27-2 (светло-желтое масло, 0,111 г, выход 99,89%). MS (ESI): масса/заряд 293 [M+Na]+.
[328] Этап 2: синтез соединения BB-27-3
[329] Соединение BB-27-2 (0,111 г, 0,411 ммоль) растворяли в DCM (3 мл), затем добавляли раствор HCl/диоксан (3 мл, 4M, 12,00 ммоль), реагирующие вещества перемешивали при к. т. в течение 2 часов. После завершения реакции смесь концентрировали с получением непосредственно целевого соединения BB-27, которое использовали на следующем этапе (желтое твердое вещество, 0,085 г, неочищенный продукт).
Сравнительный пример 28: фрагмент BB-28
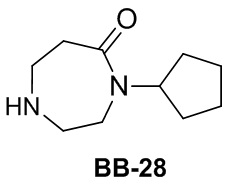
[330] Путь синтеза:
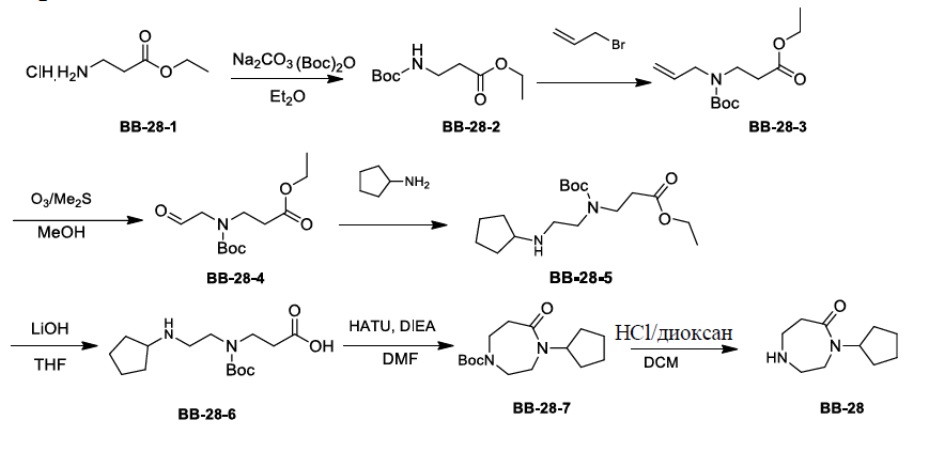
[331] Этап 1. Синтез соединения BB-28-2
[332] Соединение BB-28-1 (10 г, 85,36 ммоль), ди-трет-бутилдикарбонат (18,6 г, 85,36 ммоль) растворяли в Et2O (100 мл), затем составляли насыщенный раствор Na2CO3 (18,1 г, 170,73 ммоль) и добавляли в реакционную смесь, реагирующие вещества перемешивали при 25°C в течение ночи. После завершения реакции твердое вещество удаляли путем фильтрации, фильтрат концентрировали с получением целевого соединения BB-28-2 (желтое твердое вещество, 14 г, неочищенный продукт). MS (ESI): масса/заряд 240 [M+Na]+.
[333] Этап 2: синтез соединения BB-28-3
[334] Соединение BB-28-1 (6 г, 27,6 ммоль) и аллилбромид (4 г, 33,1 ммоль) растворяли в DMF (60 мл), затем порциями добавляли NaH (1,7 г, 41,4 ммоль, 60%) при 0°C, реакционную смесь перемешивали при 0°C в течение 3 часов. После завершения реакции реакцию гасили путем добавления H2O, смесь экстрагировали с помощью EA, органическую фазу промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство: EA/PE = 1/10) с получением целевого соединения BB-28-3 (желтое масло, 2 г, неочищенный продукт). MS (ESI): масса/заряд 158 [M-Boc+H]+.
[335] Этап 3: синтез соединения BB-28-4
[336] Соединение BB-28-3 (2 г, 7,8 ммоль) растворяли в метаноле (20 мл), смесь охлаждали до -78°C, вводили O3 до посинения реакционной смеси, затем вводили N2 до обесцвечивания реакционной смеси, затем добавляли Me2S (12,4 г, 38,8 ммоль), обеспечивали протекание реакции при 25°C в течение ночи. После завершения реакции смесь концентрировали, добавляли EA, органическую фазу промывали солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-28-4 (желтое масло, 1,8 г, неочищенный продукт). MS (ESI): масса/заряд 160 [M-Boc+H]+.
[337] Этап 4: синтез соединения BB-28-5
[338] Соединение BB-28-4 (0,5 г, 1,93 ммоль), циклопентиламин (0,197 г, 2,31 ммоль), DIPEA (0,29 г, 1,93 ммоль) и безводный Na2SO4 (0,197 г, 2,31 ммоль) растворяли в сухом метаноле (5 мл), реагирующие вещества перемешивали при к. т. в течение 2 часов, медленно добавляли NaBH4 (0,088 г, 2,31 ммоль), затем смесь перемешивали при к. т. в течение 2 часов. Реакцию гасили водным раствором NH4Cl, смесь экстрагировали с помощью EA, промывали с помощью H2O, высушивали над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной флэш-хроматографии (элюирующее средство: метанол/DCM = 1/10) с получением целевого соединения BB-28-5 (желтое масло, 0,25 г, неочищенный продукт). MS (ESI): масса/заряд 329 [M+H]+.
[339] Этап 5: синтез соединения BB-28-6
[340] Соединение BB-28-5 (0,1 г, 0,3 ммоль) растворяли в THF (0,5 мл), затем добавляли водный раствор LiOH (1 мл, 2M). Реакционную смесь перемешивали при 30°C в течение 4 часов, затем концентрировали при пониженном давлении для удаления THF с последующей сублимационной сушкой с получением соединения BB-28-6 (желтое масло, 0,09 г, неочищенный продукт), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 301 [M+H]+.
[341] Этап 6: синтез соединения BB-28-7
[342] Соединение BB-12-7 (0,09 г, 0,3 ммоль), HATU (0,172 г, 0,45 ммоль) и DIPEA (0,116 г, 0,9 ммоль) растворяли в сухом DMF (2 мл). Реакционную смесь перемешивали при 20°C в течение 4 часов, после завершения реакции реакционную смесь вливали в EA. Смесь промывали солевым раствором, высушивали над сульфатом натрия и концентрировали с получением неочищенного продукта, который выделяли и очищали с помощью пластины с тонким слоем силикагеля (проявитель: EA/PE = 1/3) с получением соединения BB-28-6 (желтое масло, 0,05 г, 55%). MS (ESI): масса/заряд 227 [M-56+H]+.
[343] Этап 7: синтез соединения BB-28
[344] Соединение BB-28-7 (0,05 г, 0,18 ммоль) растворяли в DCM (1 мл), добавляли раствор HCl/диоксан (1 мл, 4M), реакционную смесь перемешивали при 20°C в течение 1,5 часа, затем концентрировали при пониженном давлении с получением соединения BB-28 (белое твердое вещество, 0,030 г, неочищенный продукт), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 183 [M+H]+.
Сравнительный пример 29: фрагмент BB-29
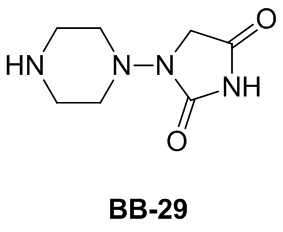
[345] Путь синтеза:
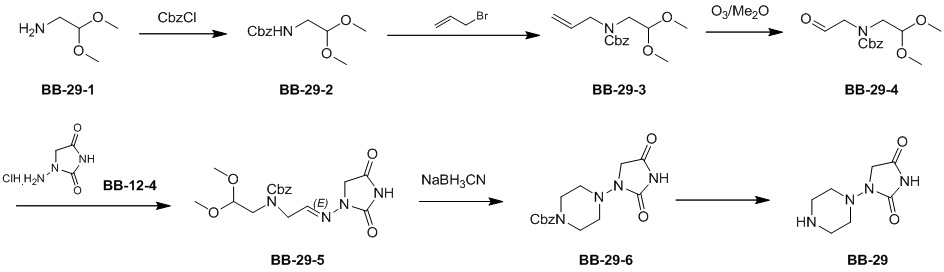
[346] Этап 1: синтез соединения BB-29-2
[347] Соединение BB-29-1 (4,56 г, 43,37 ммоль) растворяли в толуоле (24 мл), NaOH (2,19 г, 54,65 ммоль) растворяли в H2O (12 мл), затем к смеси медленно добавляли водный раствор NaOH. К реакционной смеси по каплям медленно добавляли бензилхлорформиат (6,96 г, 40,77 ммоль), температуру реакционного раствора поддерживали при 10-20°C. После добавления реагирующие вещества перемешивали при к. т. в течение 16 часов. После завершения реакции органическую фазу отделяли, промывали солевым раствором (50 мл × 2), высушивали над сульфатом натрия, фильтровали и концентрировали с получением целевого соединения BB-29-2 (желтое масло, 11 г, неочищенный продукт), которое непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 7,38 - 7,27 (m, 5H), 5,1 (s, 2H), 4,38 (t, J = 5,4 Гц, 1H), 3,38 (s, 6H), 3,35 - 3,32 (m, 2H).
[348] Этап 2: синтез соединения BB-29-3
[349] Соединение BB-29-2 (5,62 г, 23,49 ммоль), KOH (2,64 г, 46,98 ммоль) и BTMA (0,088 г, 0,474 ммоль) суспендировали в толуоле (20 мл), затем медленно добавляли раствор 3-аллилбромида (5,68 г, 46,98 ммоль) в толуоле (6 мл), температуру реагирующих веществ поддерживали при 20-30°C. Реагирующие вещества перемешивали при к. т. в течение 16 часов, затем при 30°C в течение 24 часов. После завершения реакции добавляли 20 мл H2O, органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого соединения BB-29-3 (желтое масло, 5,20 г, неочищенный продукт). 1H-ЯМР (400 МГц, CDCl3): δ 7,35 - 7,29 (m, 5H), 5,79 - 5,73 (m, 1H), 5,17 - 5,14 (m, 3H), 4,53 - 4,07 (m, 1H), 4,00 - 3,96 (m, 2H), 3,40 - 3,31 (m, 8H).
[350] Этап 3: синтез соединения BB-29-4
[351] Реагирующее вещество BB-29-3 (2,00 г, 7,16 ммоль) растворяли в смеси метанол/DCM (3:1, 40 мл), смесь охлаждали до -78°C, вводили O2 в течение 5 минут, затем вводили O3 до тех пор, пока раствор не приобретал сплошную синюю окраску, затем вводили O2 до обесцвечивания раствора. К реакционной смеси добавляли Me2S (0,344 г, 7,16 ммоль), и смесь медленно нагревали до к. т., затем перемешивали при к. т. в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения BB-29-4 (желтое масло, 2,70 г, неочищенный продукт), которое непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 7,37 - 7,28 (m, 5H), 5,18 - 5,12 (m, 2H), 4,45 - 4,34 (m, 1H), 3,47 - 3,45 (m, 2H), 3,42 - 3,40 (m, 2H), 3,39 - 3,30 (m, 3H), 3,09 (s, 1H).
[352] Этап 4: синтез соединения BB-29-5
[353] Соединение BB-29-4 (0,500 г, 1,32 ммоль) растворяли в метаноле (5 мл), затем добавляли соединение BB-12-4 (TCI, 0,199 г, 1,32 ммоль), реакционную смесь перемешивали при к. т. в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения BB-29-5 (оранжевое масло, 0,550 г, неочищенный продукт), которое непосредственно использовали на следующем этапе.
[354] Этап 5: синтез соединения BB-29-6
[355] Соединение BB-29-5 (0,450 г, 1,19 ммоль) суспендировали в муравьиной кислоте (5 мл), медленно добавляли NaBH3CN (0,150 г, 2,38 ммоль), реагирующие вещества перемешивали при к. т. в течение 20 часов в атмосфере газообразного азота. Реакционную смесь концентрировали при пониженном давлении, остаток выделяли и очищали с помощью ВЭЖХ (колонка: Phenomenex Gemini C18, 150*25 мм*5 мкм, элюирующее средство: MeCN, H2O) с получением целевого соединения BB-29-6 (желтое масло, 0,040 г, выход 10%). MS (ESI): масса/заряд 301 [M+H]+.
[356] Этап 6: синтез соединения BB-29
[357] Соединение BB-29-6 (0,050 г, 0,157 ммоль) растворяли в метаноле (5 мл), затем добавляли Pd/C (0,010 г), реагирующие вещества перемешивали при к. т. в течение 1 часа в атмосфере газообразного водорода (15 фунтов/кв. дюйм). Реакционную смесь фильтровали, фильтрат концентрировали при пониженном давлении с получением целевого соединения BB-29 (желтое масло, 0,030 г, неочищенный продукт). MS (ESI): масса/заряд 185 [M+H]+.
Сравнительный пример 30: фрагменты BB-30 и BB-30a
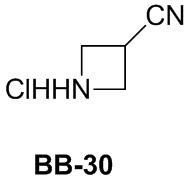
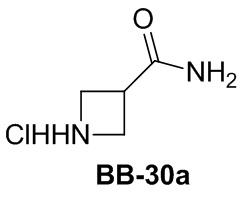
[358] Путь синтеза:
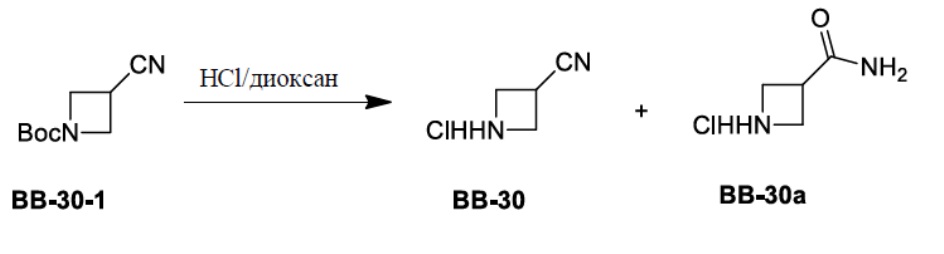
[359] Этап 1: синтез соединений BB-30 и BB-30a
[360] Соединение BB-30-1 (0,200 г, 1,10 ммоль) растворяли в DCM (2 мл), затем добавляли раствор HCl/диоксан (1 мл, 4M). Реакционную смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь концентрировали с получением целевых соединений BB-30 и BB-30a (белое твердое вещество, 0,130 г, выход 99%), которые не выделяли и непосредственно использовали на следующем этапе. MS (ESI): масса/заряд 83 [M+H]+. MS (ESI): масса/заряд 101 [M+H]+.
Сравнительный пример 31: фрагмент BB-31
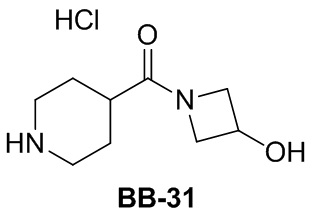
[361] Путь синтеза:
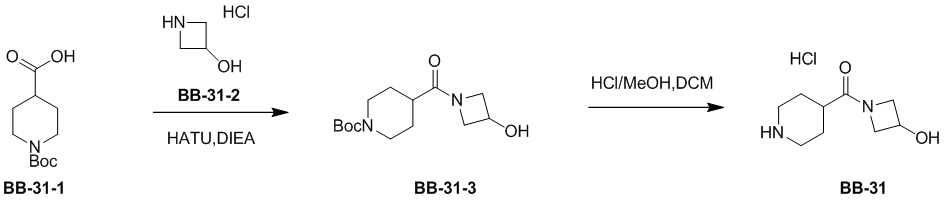
[362] Этап 1: синтез соединения BB-31-3
[363] Соединение BB-31-1 (0,5 г, 2,18 ммоль) и BB-31-2 (0,239 г, 2,18 ммоль) растворяли в DMF (5 мл), затем добавляли DIPEA (0,845 г, 6,54 ммоль) и HATU (1,24 г, 3,27 ммоль). Реагирующие вещества перемешивали при 20°C в течение 3 часов. Добавляли H2O (10 мл), смесь экстрагировали с помощью EA (20 мл × 3). Объединенную органическую фазу промывали с помощью H2O (20 мл × 2), солевого раствора (20 мл), высушивали над сульфатом натрия. Смесь фильтровали и концентрировали с получением целевого соединения BB-31-3 (желтое масло, 0,6 г, выход 97%). MS (ESI): масса/заряд 307 [M+Na]+.
[364] Этап 2: синтез соединения BB-31
[365] Соединение BB-31-3 (0,1 г, 0,352 ммоль) растворяли в DCM (2 мл), затем добавляли раствор HCl/MeOH (1 мл, 4M). Реакционную смесь перемешивали при 20°C в течение 1 часа. Реакционную смесь концентрировали с получением целевого соединения BB-31 (белое масло, 0,077 г, выход 99%). MS (ESI): масса/заряд 185 [M+H]+.
Сравнительный пример 32: фрагмент BB-32
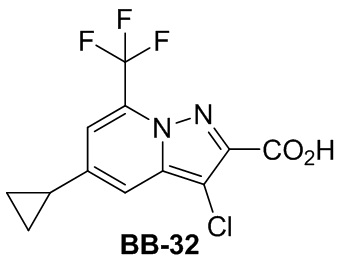
[366] Путь синтеза:
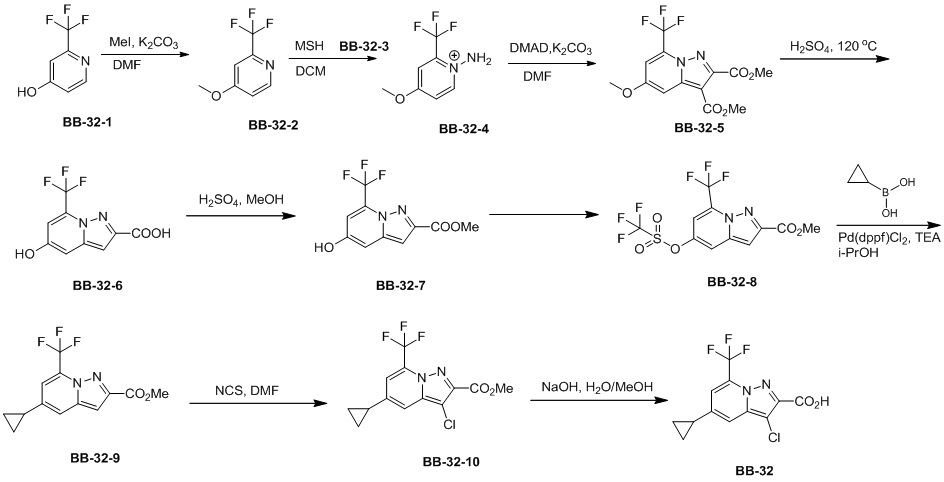
[367] Этап 1: синтез соединения BB-32-2
[368] Соединение BB-32-1 (22 г, 134,9 ммоль) и безводный K2CO3 (23 г, 161,9 ммоль) растворяли в безводном DMF (250 мл), затем медленно добавляли MeI (19 мл, 296,8 ммоль). Реакционную смесь нагревали до 65°C в течение 2 часов. Затем реакционную смесь разбавляли с помощью H2O, экстрагировали с помощью смеси PE/DCM 9:1 (3 × 200 мл). Органические фазы объединяли, промывали с помощью H2O (50 мл), насыщенного солевого раствора (50 мл), высушивали над безводным сульфатом натрия и концентрировали с получением целевого соединения BB-32-2 (желтое масло, 20 г, неочищенный продукт), которое непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 8,50 (d, 1H), 7,30 (s, 1H), 6,90 (d, 1H). MS (ESI): масса/заряд 178,1 [M+H]+.
[369] Этап 2: синтез соединения BB-32-4
[370] Этил-O-метилсульфонилацетогидроксамат (50 г, 175,4 ммоль) растворяли в диоксане (48 мл, 561,3 ммоль), реакционную смесь охлаждали до 0°C в атмосфере газообразного азота. Затем по каплям добавляли HClO4 (22 мл, 368,3 ммоль) в течение 30 минут, реакционную смесь перемешивали при к. т. в течение 2 часов, добавляли 100 мл смеси льда и воды. Реакционную смесь экстрагировали с помощью DCM (3 × 200 мл), органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), высушивали над безводным твердым K2CO3 и концентрировали при 30°C с получением 200 мл раствора соединения BB-32-3 в DCM. MS (ESI): масса/заряд 216,1 [M+H]+. Смешивали соединение BB-32-2 (20 г, 112,9 ммоль) и раствор соединения BB-32-3 в DCM. Реакционную смесь перемешивали при к. т. в течение 15 часов и концентрировали с получением неочищенного продукта, который затем растворяли в 200 мл MTBE и перемешивали в течение 10 минут, и смесь фильтровали с получением соединения BB-32-4 (светло-желтое твердое вещество, 15 г, выход 69%).
[371] Этап 3: синтез соединения BB-32-5
[372] Соединение BB-32-4 (25 г, 129 ммоль) и безводный K2CO3 (37,6 г, 272 ммоль) растворяли в DMF (250 мл), по каплям медленно добавляли DMAD (28,9 мл, 229,3 ммоль) при 0°C. Реакционную смесь перемешивали при к. т. в течение 48 часов, затем вливали в 300 мл H2O, большое количество твердого вещества осаждали и фильтровали, осадок на фильтре сохраняли. Фильтрат экстрагировали с помощью PE/EA (3 × 300 мл), органические фазы объединяли, промывали с помощью H2O (50 мл), насыщенного солевого раствора (50 мл), высушивали над безводным сульфатом натрия и концентрировали. Полученный в результате неочищенный продукт вместе с полученным осадком на фильтре очищали с помощью прибора для колоночной флэш-хроматографии (элюирующее средство: DCM/PE = 1/1) с получением целевого соединения BB-32-5 (желтое твердое вещество, 4,8 г, выход 10%). 1H-ЯМР (400 МГц, CDCl3): δ 7,60 (s, 1H), 7,13 (s, 1H), 3,98-4,00 (s, 6H), 3,90-3,94 (s, 3H). MS (ESI): масса/заряд 333,0 [M+H]+.
[373] Этап 4: синтез соединения BB-32-6
[374] Соединение BB-32-5 (4,8 г, 14,5 ммоль) растворяли в смешанном растворе конц. серной кислоты (25 мл, 450 ммоль) и H2O (10 мл), реакционную смесь нагревали до 120°C и перемешивали в течение 16 часов. Реакционную смесь вливали в 200 мл ледяной воды, большое количество твердого вещества осаждали и фильтровали. Фильтрат экстрагировали с помощью EA (100 мл × 4). Органические фазы объединяли, непосредственно высушивали над безводным сульфатом натрия и концентрировали для удаления растворителя. Остаток вместе с полученным осадком на фильтре высушивали с получением целевого соединения BB-32-6 (желтое твердое вещество, 4,2 г, неочищенный продукт), которое непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 247,0 [M+H]+.
[375] Этап 5: синтез соединения BB-32-7
[376] Соединение BB-32-6 (4,2 г, 17 ммоль) растворяли в метаноле (50 мл), по каплям медленно добавляли конц. серную кислоту (5 мл), и реакционную смесь кипятили с обратным холодильником при 90°C в течение 2 часов. Реакционную смесь концентрировали, остаток разбавляли с помощью H2O (50 мл), затем экстрагировали с помощью EA (3 × 50 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и концентрировали. Полученное в результате твердое вещество выделяли и очищали с помощью колонки для экспресс-хроматографии (элюирующее средство: метанол/DCM = 1/10) с получением целевого соединения BB-32-7 (желтое твердое вещество, 2,7 г, выход 61%). MS (ESI): масса/заряд 261,0 [M+H]+.
[377] Этап 6: синтез соединения BB-32-8
[378] Соединение BB-32-7 (1,2 г, 4,6 ммоль) растворяли в DCM (15 мл), последовательно добавляли DIPEA (1,19 г, 9,2 ммоль) и N-(трифторметил)бензолсульфонамид (3,2 г, 8,98 ммоль) при к. т. Реакционную смесь перемешивали при к. т. в течение 3 часов, затем дополнительно добавляли N-(трифторметил)бензолсульфонамид (1 г, 2,8 ммоль) и концентрировали для удаления растворителя. Полученное в результате твердое вещество выделяли и очищали с помощью колонки для экспресс-хроматографии (элюирующее средство: PE/DCM = 10/1) с получением целевого соединения BB-32-8 (желтое твердое вещество, 1,2 г, выход 67%). 1H-ЯМР (400 МГц, CDCl3): δ 7,76 (s, 1H), 7,35 (s, 1H), 7,26 - 7,28 (m, 1H).
[379] Этап 7: синтез соединения BB-32-9
[380] Соединение BB-32-8 (0,5 г, 1,27 ммоль) растворяли в диоксане (5 мл), последовательно добавляли циклопропилбороновую кислоту (0,219 г, 2,54 ммоль), K3PO4 (0,812 г, 3,82 ммоль) и комплекс дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (II) и дихлорметана (0,05 г, 0,06 ммоль). Реакционную смесь перемешивали при 90°C и кипятили с обратным холодильником в течение 3 часов в атмосфере газообразного азота, затем охлаждали до к. т., фильтровали для удаления нерастворимых веществ. Фильтрат вливали в смесь EA (50 мл) и H2O (20 мл) и распределяли на неподвижной фазе, органическую фазу высушивали над безводным сульфатом натрия и концентрировали. Полученное в результате твердое вещество выделяли и очищали с помощью пластины для препаративной хроматографии (проявитель: PE/EA = 5/1) с получением целевого соединения BB-32-9 (желтое масло, 0,041 г, выход 11%). MS (ESI): масса/заряд 285,0 [M+H]+.
[381] Этап 8: синтез соединения BB-32-10
[382] Соединение BB-32-9 (0,04 г, 0,14 ммоль) растворяли в DMF (0,5 мл), добавляли NCS (0,028 г, 0,21 ммоль). Реакционную смесь перемешивали при 60°C в течение 1 часа, затем разбавляли с помощью H2O, экстрагировали с помощью PE/DCM (3 × 20 мл). Органические фазы объединяли, промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Полученное в результате твердое вещество выделяли и очищали с помощью пластины для препаративной хроматографии (элюирующее средство: PE/EA = 10/1) с получением целевого соединения BB-32-10 (желтое масло, 0,046 г, выход 100%). MS (ESI): масса/заряд 319,0 [M+H]+.
[383] Этап 9: синтез соединения BB-32
[384] Соединение BB-32-10 (0,046 г, 0,15 ммоль) растворяли в смешанном растворе метанола (2 мл) и H2O (0,4 мл), добавляли моногидрат гидроксида лития (0,037 г, 0,75 ммоль). Реакционную смесь перемешивали при к. т. в течение ночи, концентрировали для удаления метанола, затем pH реакционной смеси доводили до 1 с помощью 1M разбавленной хлористоводородной кислоты, смесь экстрагировали с помощью DCM (3 × 20 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и концентрировали с получением целевого соединения BB-32 (серое твердое вещество, 0,028 г, выход 62%) в виде неочищенного продукта, который непосредственно использовали на следующем этапе без очистки. MS (ESI): масса/заряд 305,0 [M+H]+.
[385] Сравнительные примеры 33-41: фрагменты BB-33-41
[386] Сравнительные примеры в нижеследующей таблице синтезировали согласно этапам 1-5 в сравнительном примере 20 (фрагмент BB-20).
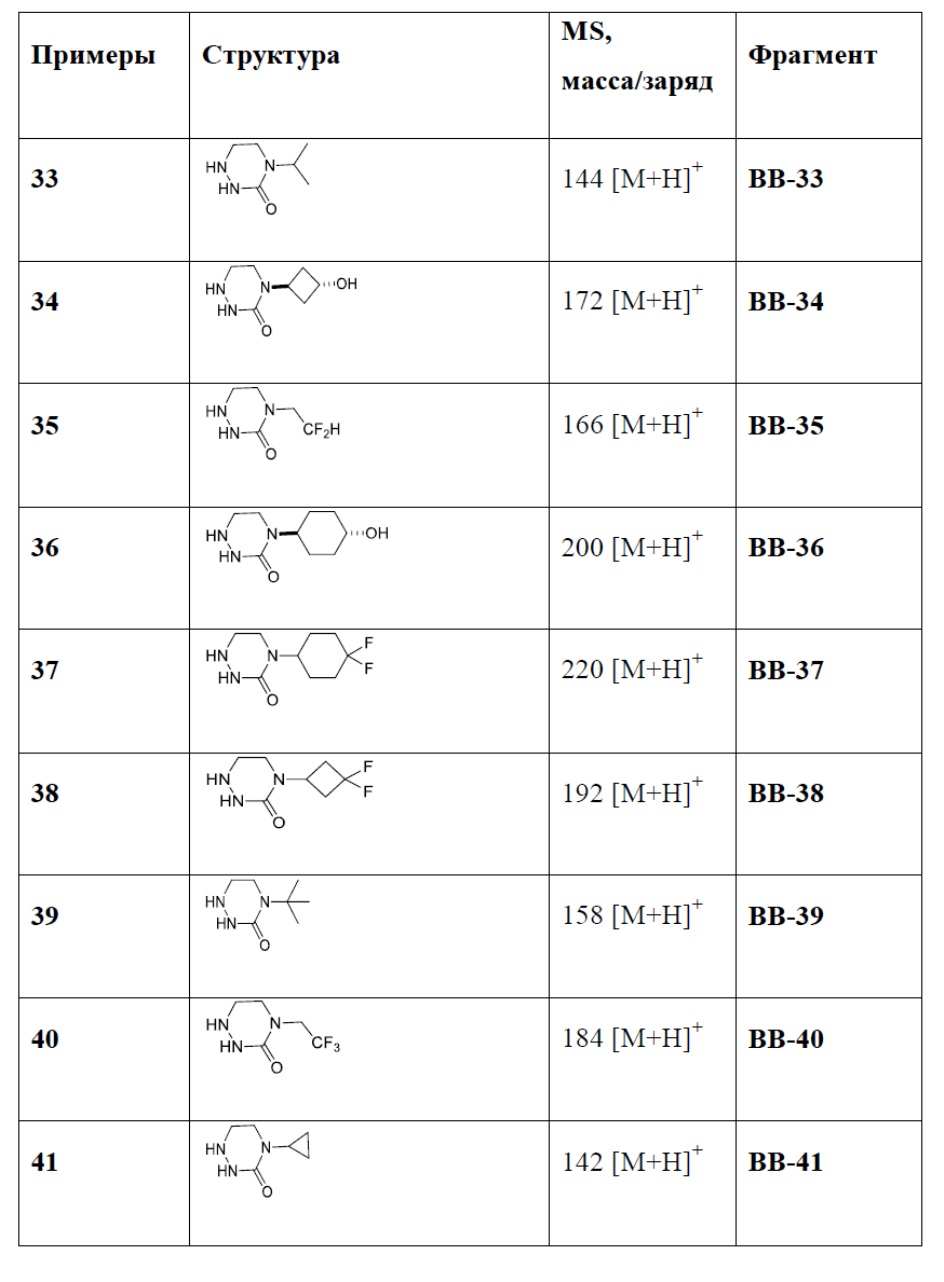
Вариант осуществления 1: соединение YD_0346
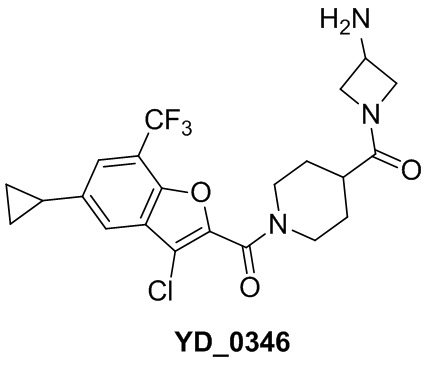
[387] Путь синтеза:
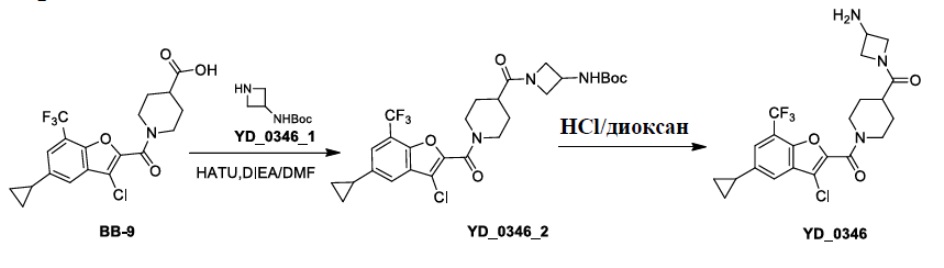
[388] Этап 1: синтез соединения YD_0346_2
[389] Соединение BB-9 (0,035 г, 0,084 ммоль) и соединение YD_0346_1 (0,019 г, 0,093 ммоль) растворяли в DMF (2 мл), затем добавляли HATU (0,048 г, 0,126 ммоль) и DIPEA (0,032 г, 0,253 ммоль), реакционную смесь перемешивали при к. т. в течение 3 часов. После завершения реакции добавляли EA, органическую фазу промывали с помощью H2O и насыщенного солевого раствора, высушивали над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали при пониженном давлении для удаления растворителя с получением таким образом неочищенного продукта, неочищенный продукт выделяли и очищали с помощью пластины с тонким слоем силикагеля (проявитель: EA/PE = 1/2) с получением целевого соединения YD_0346_2 (белое твердое вещество, 0,040 г, выход 83%). MS (ESI): масса/заряд 570 [M+H]+, 541 [M-56+H]+.
[390] Этап 2: синтез соединения YD_0346
[391] Соединение YD_0346_2 растворяли в THF (1 мл), затем добавляли раствор HCl/1,4-диоксан (4M, 2 мл), реакционную смесь перемешивали при 20°C в течение 2 часов. Затем смесь концентрировали при пониженном давлении для удаления растворителя, остаток выделяли и очищали с помощью препаративной ВЭЖХ (колонка: Gemini, 150*25 мм*5 мкм, элюирующее средство: MeCN + 0,075% HCl, H2O + 0,075% HCl) с последующей сублимационной сушкой с получением целевого соединения YD_0346 (белое твердое вещество, 0,0155 г, выход 43,6%). 1H-ЯМР (400 МГц, MeOD) = 7,68 - 7,60 (m, 1H), 7,60 - 7,54 (m, 1H), 4,74 - 4,54 (m, 2H), 4,33 (d, J = 8,0 Гц, 2H), 4,23 - 4,12 (m, 1H), 4,00 (d, J = 4,0 Гц, 2H), 3,86 - 3,78 (m, 1H), 3,76 - 3,68 (m, 1H), 3,64 - 3,35 (m, 1H), 3,20 - 2,99 (m, 1H), 2,80 - 2,61 (m, 1H), 2,25 - 2,14 (m, 1H), 2,06 - 1,66 (m, 4H), 1,19 - 1,07 (m, 2H), 0,89 - 0,77 (m, 2H). MS (ESI): масса/заряд 470 [M+H]+.
[392] Варианты осуществления в нижеследующей таблице синтезировали согласно этапам 1-2 в варианте осуществления 1 (соединение YD_0346)
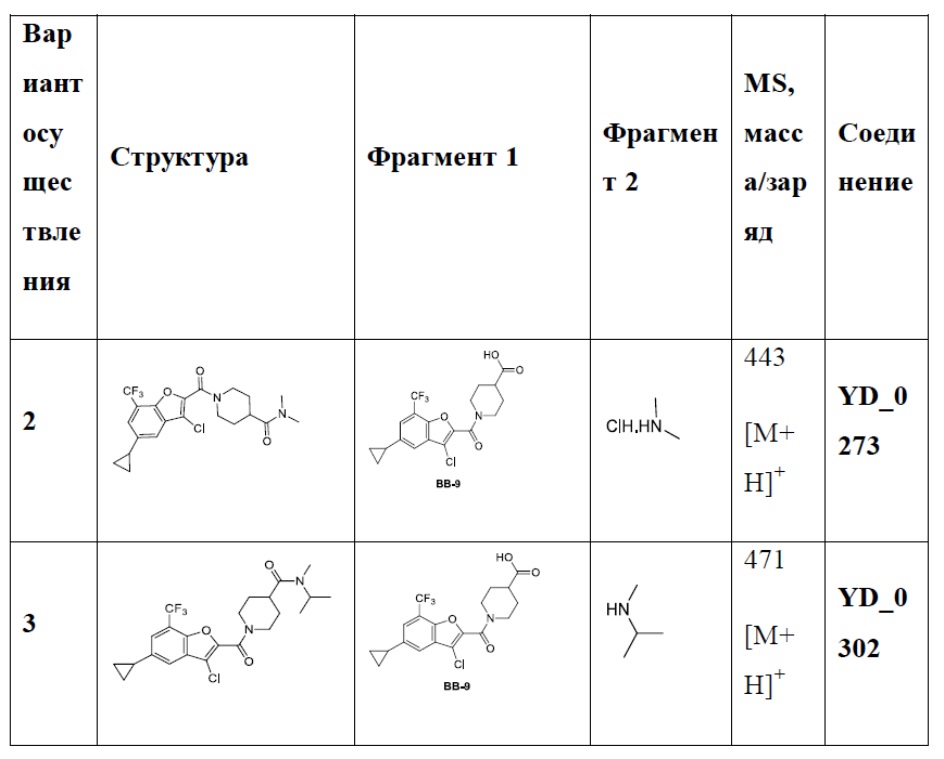
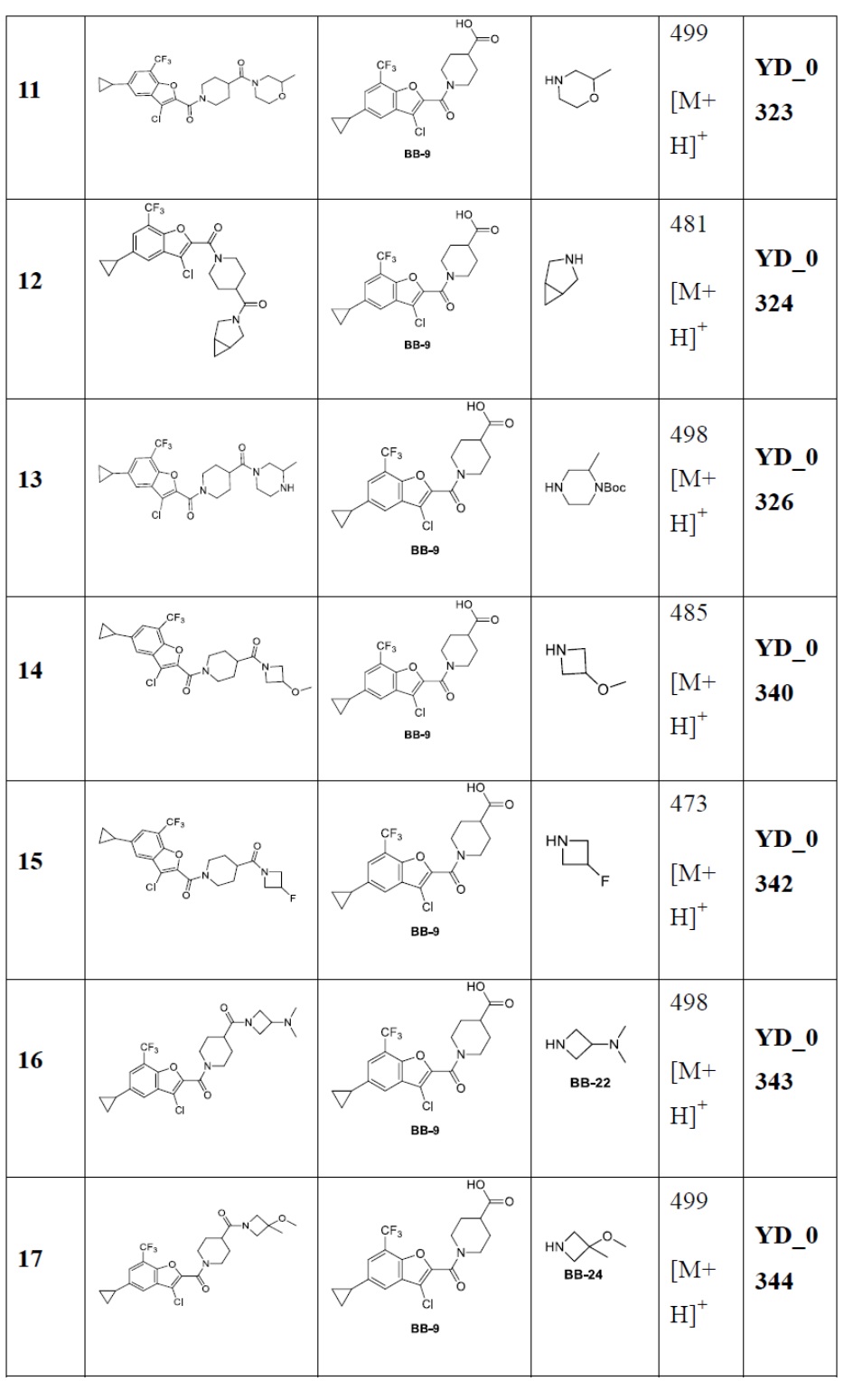
Вариант осуществления 25: YD_0327
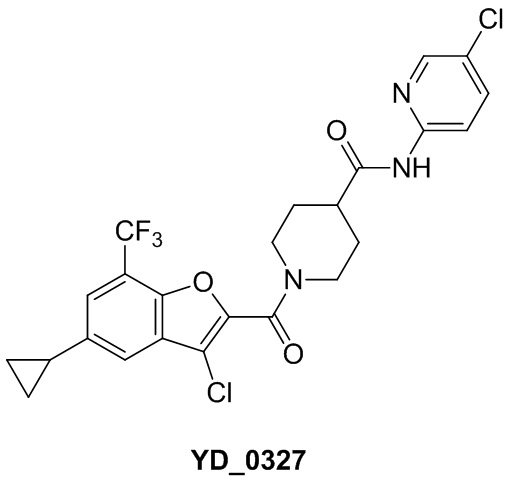
[393] Путь синтеза:
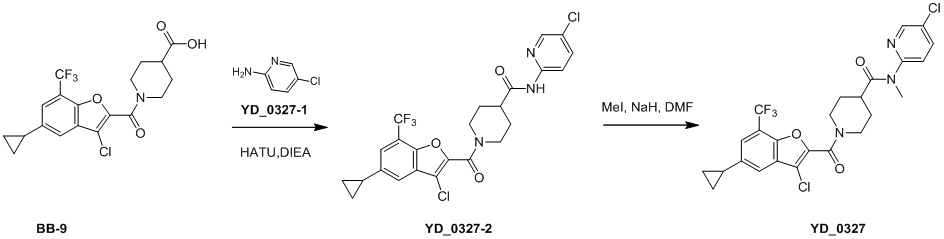
[394] Этап 1: синтез соединения YD_0327-2
[395] Соединение BB-9 (0,100 г, 0,241 ммоль), соединение YD_0327-1 (0,046 г, 0,361 ммоль) растворяли в DMF (1 мл), добавляли DIPEA (0,093 г, 0,722 ммоль) и HATU (0,137 г, 0,361 ммоль), реакционную смесь перемешивали при 25°C в течение 5 часов, добавляли H2O (2 мл). Твердое вещество отфильтровывали и собирали, осадок на фильтре очищали с помощью пластины для тонкослойной хроматографии (проявитель EA/PE = 1/2) с получением соединения YD_0327-2 (желтое твердое вещество, 0,040 г, выход 31,60%). MS (ESI): масса/заряд 527 [M+H]+.
[396] Этап 2: синтез соединения YD_0327
[397] Соединение YD_0327-2 (0,040 г, 0,076 ммоль) растворяли в DMF (1 мл), добавляли NaH (0,006 г, 0,152 ммоль, 60%), реакционную смесь перемешивали при 25°C в течение 10 минут, добавляли MeI (0,054 г, 0,380 ммоль). Реакционную смесь перемешивали при 25°C в течение 2 часов, затем гасили с помощью H2O (0,5 мл), и проводили выделение и очистку с помощью препаративной ВЭЖХ (колонка: Phenomenex Gemini C18, 150*25 мм*5 мкм, элюирующее средство: MeCN + 0,05% NH3.H2O, H2O + 0,05% NH3.H2O) с получением целевого соединения YD_0327 (желтое твердое вещество, 0,004 г, выход 9,74%). 1H-ЯМР (400 МГц, CD3OD): δ 8,19 (s, 1H), 7,98 (d, J = 9,2 Гц, 1H), 7,72 (d, J = 9,2 Гц, 1H), 7,61 (s, 1H), 7,53 (s, 1H), 4,51 (d, J = 13,2 Гц, 1H), 3,92 (d, J = 13,2 Гц, 1H), 3,80 (s, 3H), 3,40 - 3,30 (m, 1H), 3,15 - 3,10 (m, 1H), 2,70 - 2,65 (m, 1H), 2,15 - 2,05 (m, 3H), 1,95 - 1,80 (m, 2H), 1,10 - 1,05 (m, 2H), 0,85 - 0,75 (m, 2H). MS (ESI): масса/заряд 540 [M+H]+.
Вариант осуществления 26: YD_0329
[398] Этап 1: синтез соединения YD_0329
[399] Соединение YD_0329 синтезировали согласно этапам 1-2 в варианте осуществления 25 (соединение YD_0327) за исключением того, что исходными материалами были соединение BB-9 и 2-амино-5-фторпиридин. 1H-ЯМР (400 МГц, CD3OD): δ 8,17 (s, 1H), 8,10 - 8,05 (m, 1H), 7,80 - 7,75 (m, 1H), 7,62 (s, 1H), 7,54 (s, 1H), 4,52 (d, J = 13,2 Гц, 1H), 3,92 (d, J = 13,2 Гц, 1H), 3,83 (s, 3H), 3,40 - 3,30 (m, 1H), 3,15 - 3,10 (m, 1H), 2,70 - 2,65 (m, 1H), 2,15 - 2,05 (m, 3H), 1,95 - 1,80 (m, 2H), 1,10 - 1,05 (m, 2H), 0,85 - 0,75 (m, 2H). MS (ESI): масса/заряд 524 [M+H]+.
Вариант осуществления 27: YD_0321
[400] Путь синтеза:
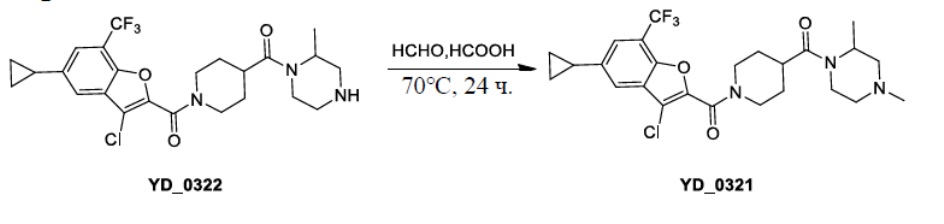
[401] Этап 1: синтез соединения YD_0321
[402] Соединение YD_0322 (0,065 г, 0,130 ммоль) добавляли к 2 мл MeOH, затем добавляли водный раствор формальдегида (0,008 г, 0,266 ммоль), муравьиную кислоту (22 мг, 0,458 ммоль). Реакционную смесь перемешивали при 70°C в течение 24 часов. Реакционную смесь выпаривали досуха при пониженном давлении, затем проводили выделение с помощью препаративной ВЭЖХ (колонка: Phenomenex Gemini C18, 150*25 мм*5 мкм, элюирующее средство: MeCN + 0,05% NH3.H2O, H2O + 0,05% NH3.H2O) с получением соединения YD_0321 (0,008 г, выход 12%). MS-ESI: масса/заряд 512 [M+H]+. 1H-ЯМР (400 МГц, CDCl3): δ 7,49 (m, 1H), 7,42 (m, 1H), 4,61 - 4,75 (m, 2H), 4,03 - 4,09 (m, 1H), 3,45 - 3,61 (m, 2H), 3,20 - 3,27 (m, 1H), 2,91 - 3,09 (m, 2H), 2,68 - 2,83 (m, 3H), 2,26 (s, 3H), 2,01 - 2,09 (m, 2H), 1,86 - 2,04 (m, 3H), 1,39 - 1,40 (m, 1H), 1,22 - 1,26 (m, 2H), 1,05 - 1,10 (m, 2H), 0,77 - 0,79 (m, 2H).
Вариант осуществления 28: YD_0325
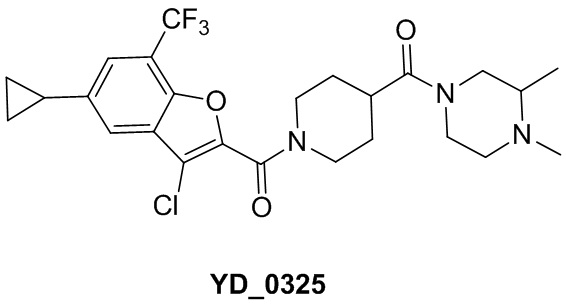
[403] Этап 1: синтез соединения YD_0325
[404] Соединение YD_0325 синтезировали согласно этапу 1 в варианте осуществления 27 (соединение YD_0321) за исключением того, что исходным материалом было соединение YD_0326. 1H-ЯМР (400 МГц, CDCl3): δ 7,62 (m, 1H), 7,49 (m, 1H), 4,55 - 4,63 (m, 1H), 4,31 - 4,46 (m, 1H), 4,02 - 4,11 (m, 1H), 3,58 - 3,80 (m, 1H), 3,12 - 3,43 (m, 2H), 2,79 - 3,10 (m, 4H), 2,49 - 2,56 (m, 1H), 2,31 (s, 3H), 2,01 - 2,11 (m, 2H), 1,72 - 1,96 (m, 4H), 1,12 - 1,21 (m, 2H), 0,77 - 0,79 (m, 2H). MS-ESI: масса/заряд 512 [M+H]+.
Вариант осуществления 29: YD_0351
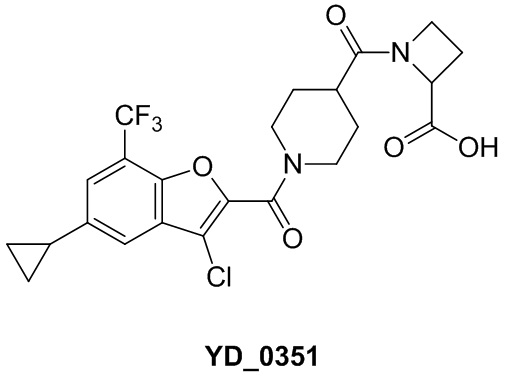
[405] Путь синтеза:
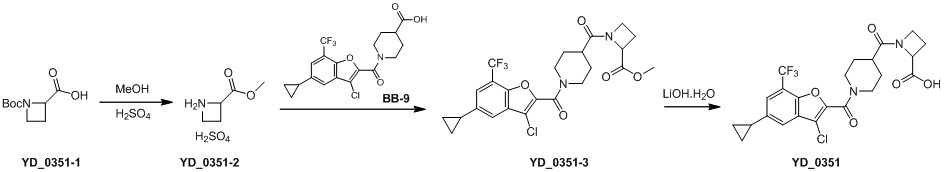
[406] Этап 1: синтез соединения YD_0351-2
[407] Соединение YD_0351-1 (0,090 г, 0,449 ммоль) растворяли в MeOH (5 мл), затем добавляли конц. серную кислоту (0,008 г, 0,081 ммоль), реакционную смесь перемешивали с обратным холодильником в течение 5 часов. После завершения реакции растворитель удаляли путем концентрирования при пониженном давлении с получением неочищенного продукта YD_0351-2 (желтое масло, 0,090 г, неочищенный продукт), который непосредственно использовали на следующем этапе. 1H-ЯМР (400 МГц, CDCl3): δ 5,19 - 5,14 (m, 1H), 4,18 - 4,12 (m, 1H), 3,99 - 3,94 (m, 1H), 3,71 (s, 3H), 2,86 - 2,82 (m, 1H), 2,75 - 2,69 (m, 1H).
[408] Этап 2: синтез соединения YD_0351-3
[409] Соединение YD_0351-2 (0,031 г, 0,144 ммоль) растворяли в DMF (2 мл), затем добавляли соединение BB-9 (0,060 г, 0,144 ммоль), DIPEA (0,093 г, 0,722 ммоль) и HATU (0,066 г, 0,173 ммоль), реакционную смесь перемешивали при к. т. в течение 6 часов. После завершения реакции добавляли H2O (5 мл), смесь экстрагировали с помощью EA (10 мл × 2). Органические фазы объединяли и высушивали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта YD_0351-3 (желтое масло, 0,130 г, неочищенный продукт). MS (ESI): масса/заряд 513 [M+H]+.
[410] Этап 3: синтез соединения YD_0351
[411] Соединение YD_0351-3 (0,130 г, 0,253 ммоль) растворяли в MeOH (3 мл) и H2O (1 мл), затем добавляли моногидрат гидроксида лития (0,032 г, 0,760 ммоль), реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции растворитель удаляли путем концентрирования при пониженном давлении, полученный в результате остаток вновь растворяли в H2O, и pH доводили до приблизительно 2-3 с помощью 2 н. водного раствора хлористоводородной кислоты. Твердое вещество собирали с получением неочищенного продукта, который выделяли и очищали с помощью препаративной ВЭЖХ (колонка: Synergy, 150*30 мм, элюирующее средство: MeCN + 0,075% TFA, H2O + 0,075% TFA) с получением YD_0351 (белое твердое вещество, 0,030 г, выход 24%). 1H-ЯМР (400 МГц, CD3OD): δ 7,64 (s, 1H), 7,56 (s, 1H), 5,15 - 5,11 (m, 0,5H), 4,71 - 4,75 (m, 0,5H), 4,61 - 4,57 (m, 1H), 4,36 - 4,29 (m, 1H), 4,01 - 3,96 (m, 2H), 3,39 - 3,34 (m, 0,5H), 3,14 - 3,03 (m, 0,5H), 3,02 - 2,85 (m, 0,5H), 2,75 - 2,73 (m, 2H), 2,59 - 2,52 (m, 0,5H), 2,21 - 2,16 (m, 2H), 1,83 - 1,80 (m, 4H), 1,14 - 1,09 (m, 2H), 0,84 - 0,82 (m, 2H). MS (ESI): масса/заряд 499 [M+H]+.
Вариант осуществления 30: YD_0019
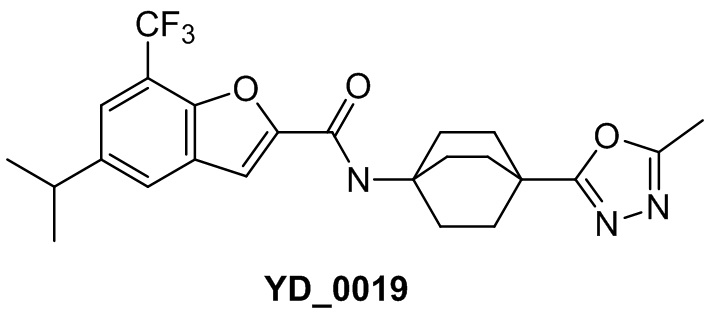
[412] Путь синтеза:
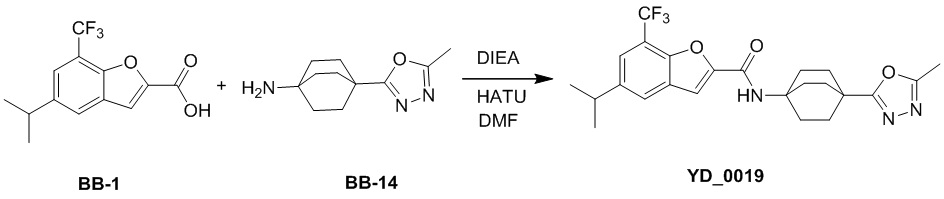
[413] Этап 1: синтез соединения YD_0019
[414] Соединение BB-1 (0,035 г, 0,128 ммоль) растворяли в DMF (2 мл), затем добавляли соединение BB-14 (0,027 г, 0,128 ммоль), DIPEA (0,050 г, 0,386 ммоль) и HATU (0,063 г, 0,167 ммоль). Реакционную смесь перемешивали при к. т. в течение 2 часов. После завершения реакции добавляли EA (100 мл), затем смесь промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который выделяли с помощью пластины с толстым слоем силикагеля (проявитель: PE/EA = 1/1) с получением целевого соединения YD_0019 (белое твердое вещество, 0,050 г, выход 84%). MS (ESI): масса/заряд 462 [M+H]+. 1H-ЯМР (400 МГц, MeOD): δ 7,87 (s, 1H), 7,62 (s, 1H), 7,55 (s, 1H), 3,15 - 3,11 (m, 1H), 2,52 (s, 3H), 2,13 - 2,26 (m, 12H), 1,36 (d, J = 7,2 Гц, 6H).
[415] Варианты осуществления в нижеследующей таблице синтезировали согласно этапу 1 в варианте осуществления 30 (соединение YD_0019).
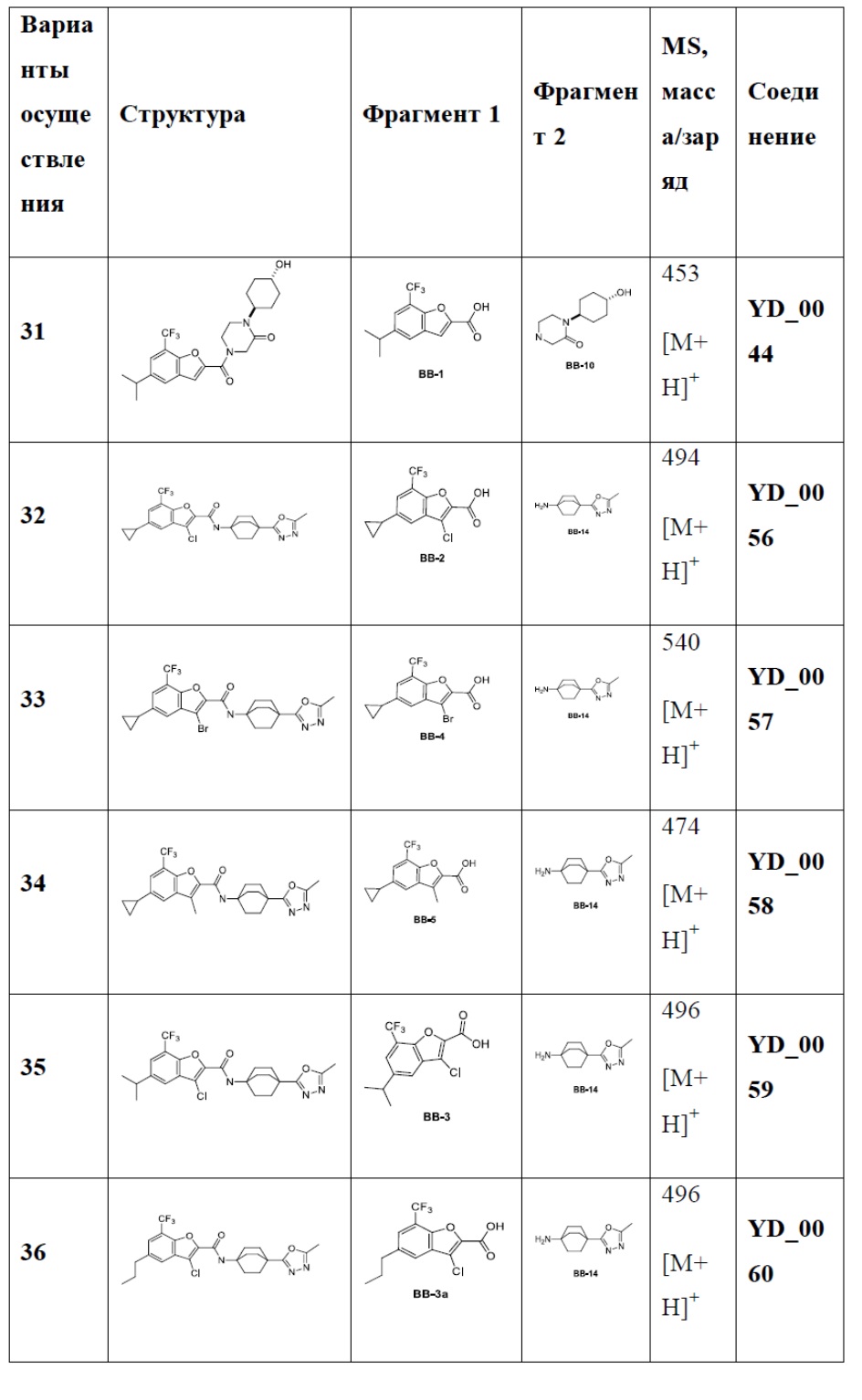
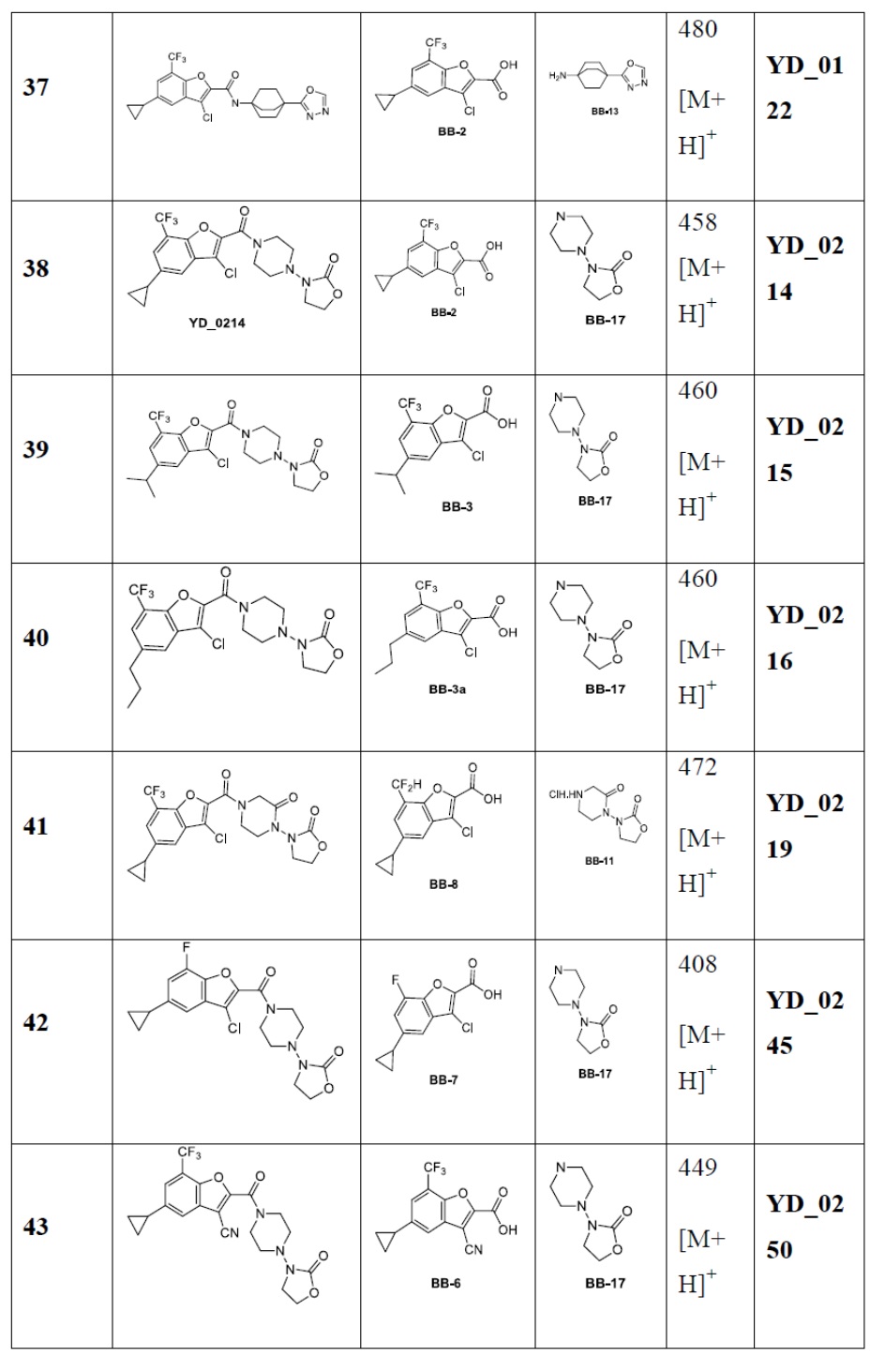
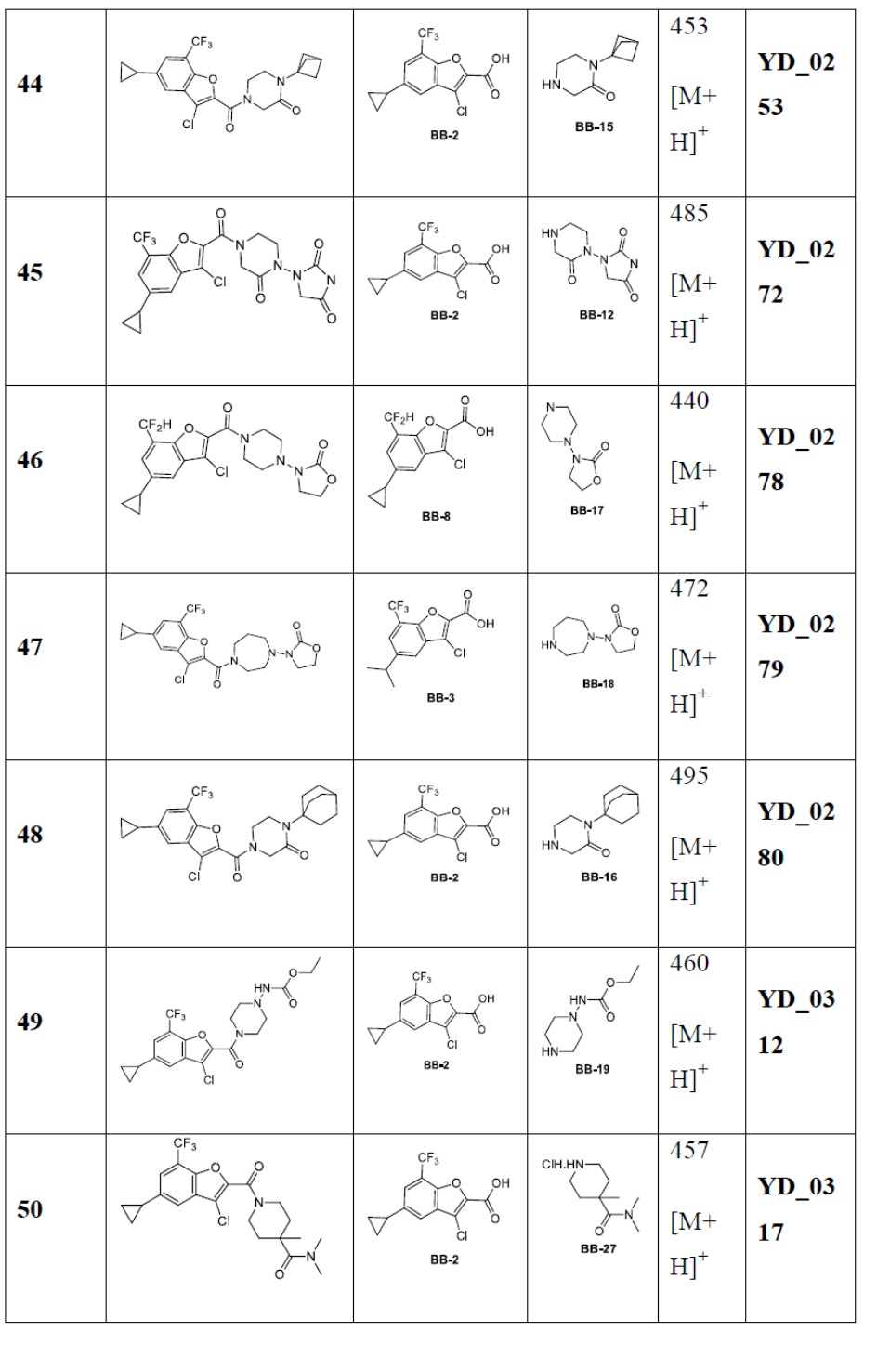
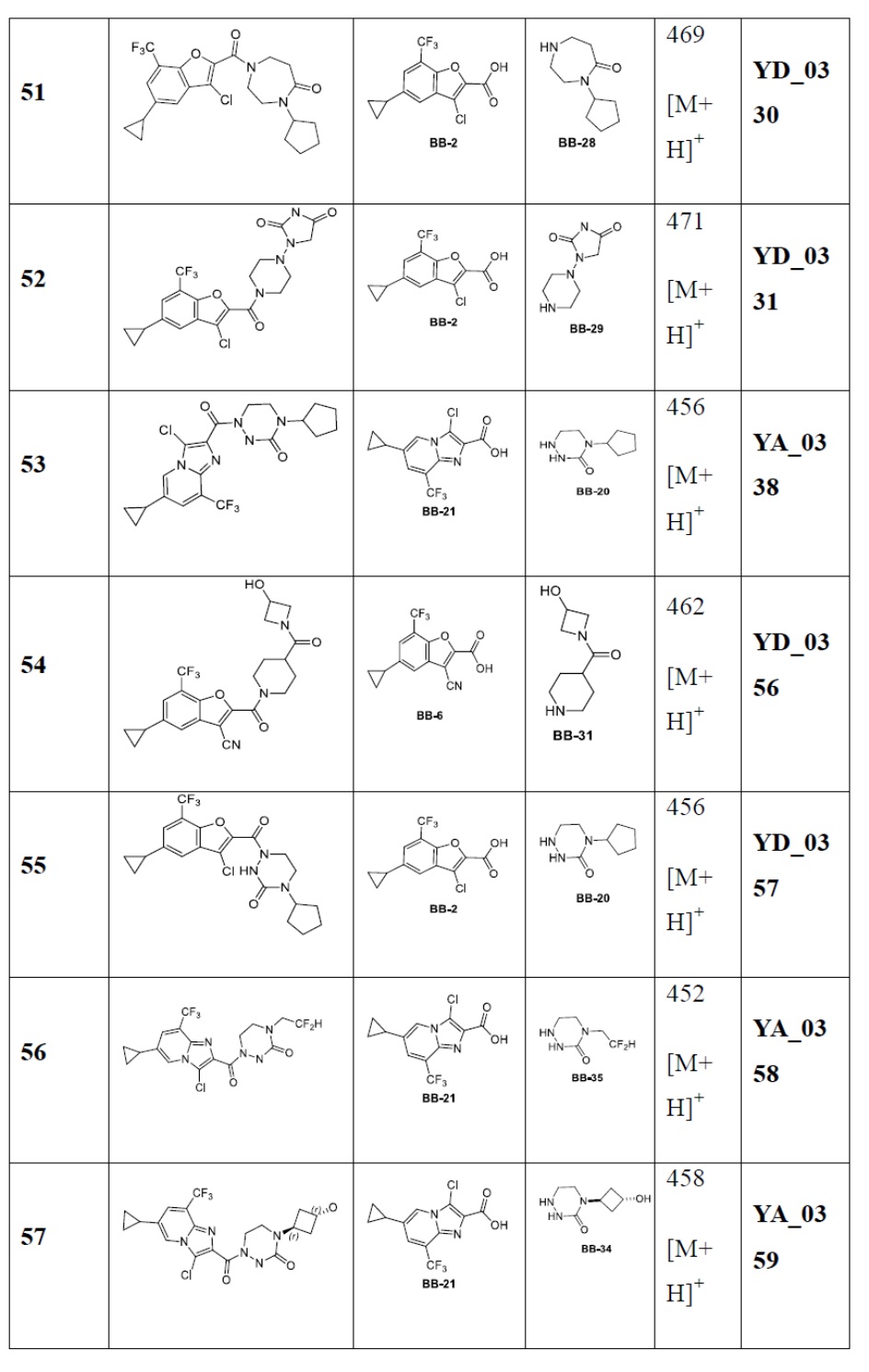
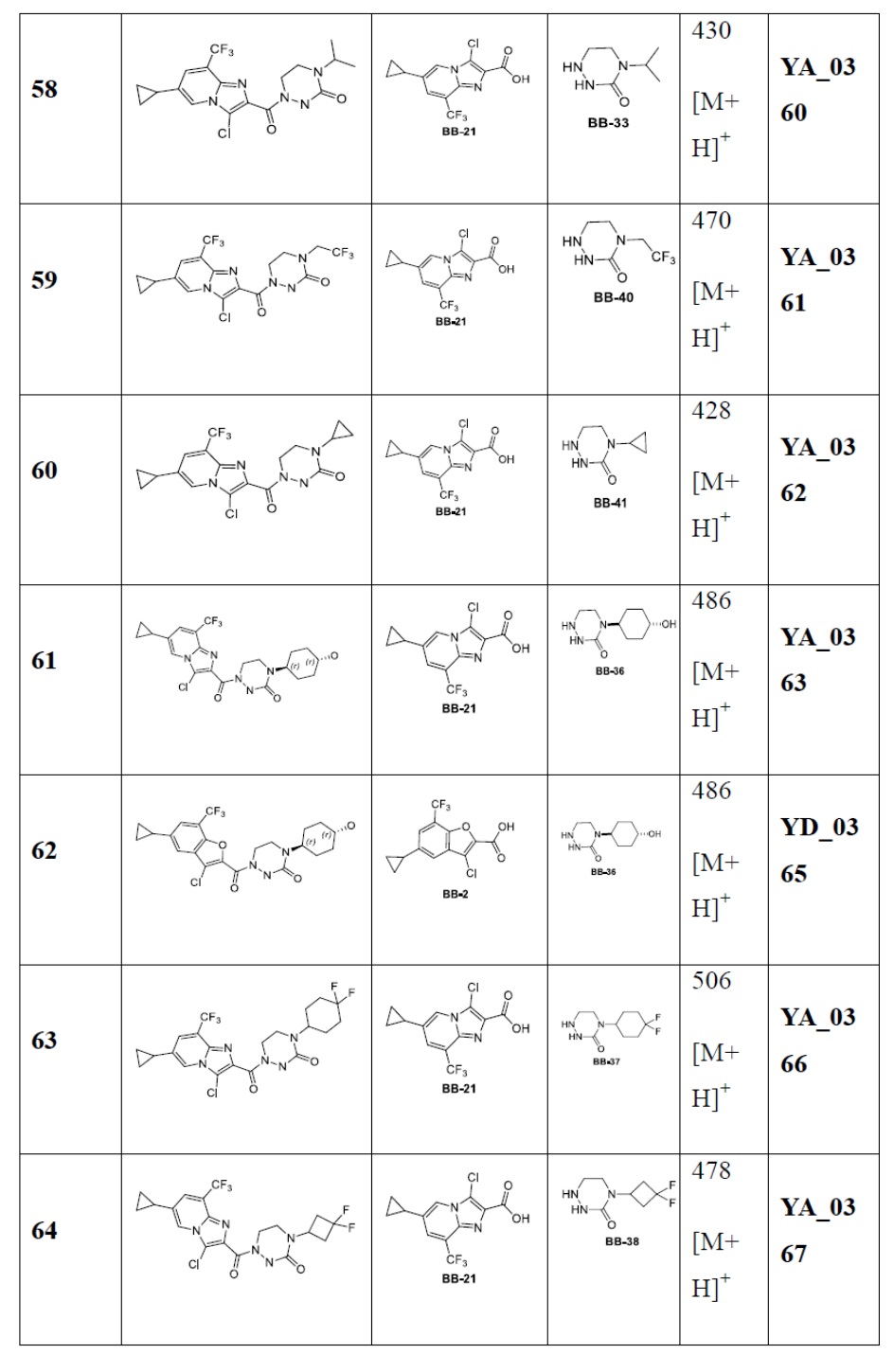
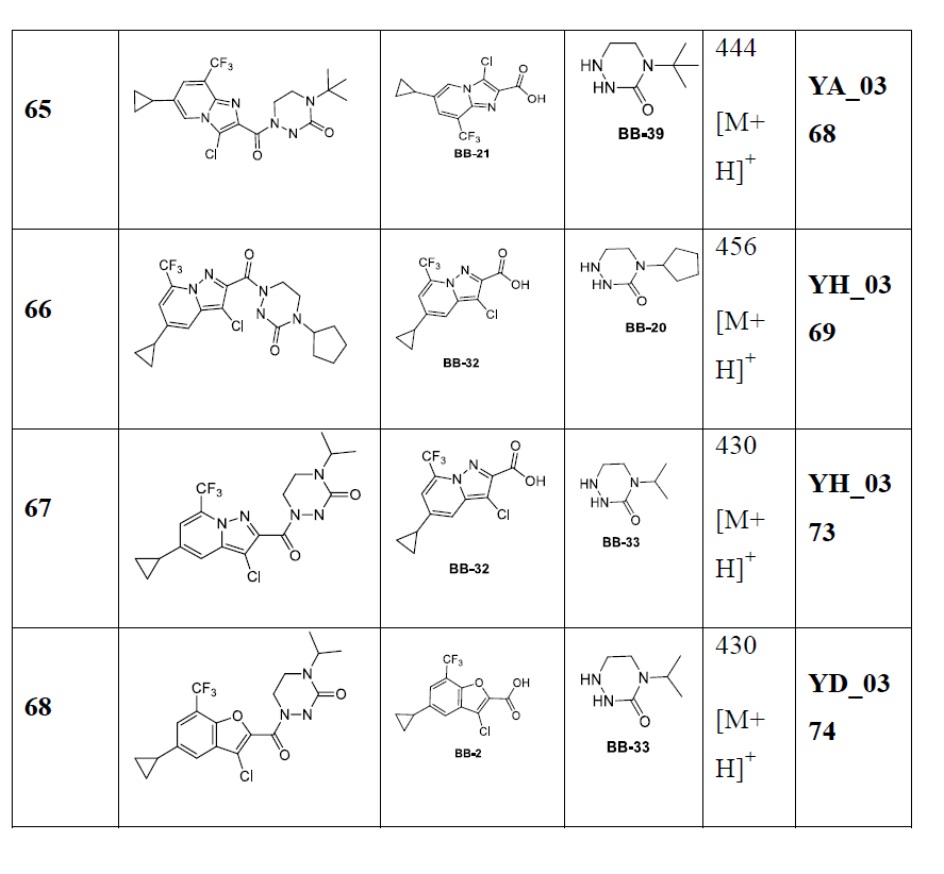
[416] Эксперимент 1. Анализ in vitro
[417] Цель эксперимента
[418] Определяли значения EC50 и CC50 для соединений, противодействующих HCV, с использованием клеток, стабильно трансфицированных репликонами (характеризующихся репликацией) HCV генотипов 1a (HCV-1a) и 1b (HCV-1b). Источником репликона генотипа 1а являются клоны H77, содержащие адаптивные мутации K1691R, K2040R и S2204I. Источником репликона генотипа 1b являются клоны Con1, содержащие адаптивные мутации E1202G, T1280I и K1846T.
[419] Вводная справочная информация
Система репликона HCV генетического подтипа 1a (HCV-1a) и 1b (HCV-1b) содержит ген неструктурного белка HCV соответствующего генетического подтипа, ген NEO устойчивости к G418 и ген люциферазы, что обуславливает возможность стабильной экспрессии белка, ассоциированного с HCV, и люциферазы в клетках. Путем выявления уровня экспрессии гена люциферазы можно определять уровень репликации HCV. Следовательно, данная система используется в качестве модели для скрининга активности соединения, противодействующего HCV, in vitro.
[420] Материалы для эксперимента
Линии клеток с репликоном HCV: клетки с HCV-1a и HCV-1b
Среда для культивирования клеток: среда DMEM (Invitrogen, № по каталогу 11960077) с добавлением 10% фетальной бычьей сыворотки (FBS, Sigma, № по каталогу 12003C) и 1% пенициллина-стрептомицина (пенициллин - 5000 МЕ/мл, стрептомицин - 10 мг/мл, Hyclone, № по каталогу SV30010)
Трипсин (Invitrogen, № по каталогу 25200072)
PBS (Invitrogen, № по каталогу 10010023)
Трипановый синий (Invitrogen, № по каталогу 15250061)
CellTiter-Fluor (Promega, № по каталогу G6082)
Bright-Glo (Promega, № по каталогу E2650)
CO2-инкубатор, Thermo 240 I
Multidrop, Thermo
Сборочная установка для планшетов POD 810, Labcyte
Ручной автоматический счетчик клеток Scepter, Millipore
Спектрофотометр для микропланшетов, Molecular Device
[421] Процедура и способ проведения эксперимента
a) Получение, разбавление и добавление раствора соединения
Порошкообразное соединение растворяли в 100% DMSO. Затем соединение подвергали 5-кратному разбавлению с 6 точками и добавляли в планшет для культивирования клеток с помощью дозатора жидкостей Echo. Убеждались в том, что конечная концентрация DMSO составляла 0,5%. Каждое соединение тестировали в двух повторностях.
b) Культивирование клеток (клетки с репликоном HCV-1a или HCV-1b)
1) Поглощение надосадочной жидкости культуры из культуры клеток и промывание клеток с помощью 10 мл PBS.
2) Добавление предварительно нагретого трипсина в колбы с промытыми культурами клеток, вращение культурального сосуда для обеспечения равномерного покрытия дна культурального сосуда трипсином, затем помещение в инкубатор при 37°C, 5% CO2 для расщепления.
3) Суспендирование клеток в каждой колбе для культивирования тканей T150 с 10-15 мл среды для культивирования, поглощение 0,1 мл жидкости и разбавление в 2 раза раствором трипанового синего согласно подсчету.
4) Разбавление клеток до 8×104 клеток/мл средой для культивирования, добавление разбавленных клеток в 96-луночный планшет, содержащий соединение (Greiner, № по каталогу 655090) (100 мкл/лунка, 8000 клеток/лунка), с помощью автоматического отделителя жидкости (Thermo Scientific). Затем помещение в инкубатор при 37°C, 5% CO2 на 3 дня. Контрольная лунка с клетками: без соединения, содержит только 0,5% DMSO.
5) Добавление флуоресцентного субстрата Cell Titer-fluor в лунку с клетками после инкубирования в течение 30 минут, выявление сигнала с помощью считывающего устройства для микропланшетов EnVision (возбуждение при 405 нм и считывание при 515 нм). Эффект соединений в отношении цитотоксичности для клеток с репликоном HCV анализировали в соответствии с данными флуоресценции, которые применяли для расчета значений CC50.
6) Затем добавление субстрата для люциферазы Bright-Glo после инкубирования в течение 5 минут, выявление активности люциферазы с помощью считывающего устройства для микропланшетов EnVision (длина волны > 700 нм); анализ ингибирующей активности соединений, противодействующих HCV, в соответствии с данными люминесценции, которые применяли для расчета значений EC50.
c) Обработка и анализ данных
Значения EC50 или CC50 получали с помощью аппроксимации данных о процентном значении ингибирования (% ингиб.) нелинейной регрессией с использованием программного обеспечения GraphPad Prism.
Результаты экспериментов показаны в таблице 1.
Таблица 1. Результаты экспериментов для EC50/CC50 в отношении клеток с репликоном HCV
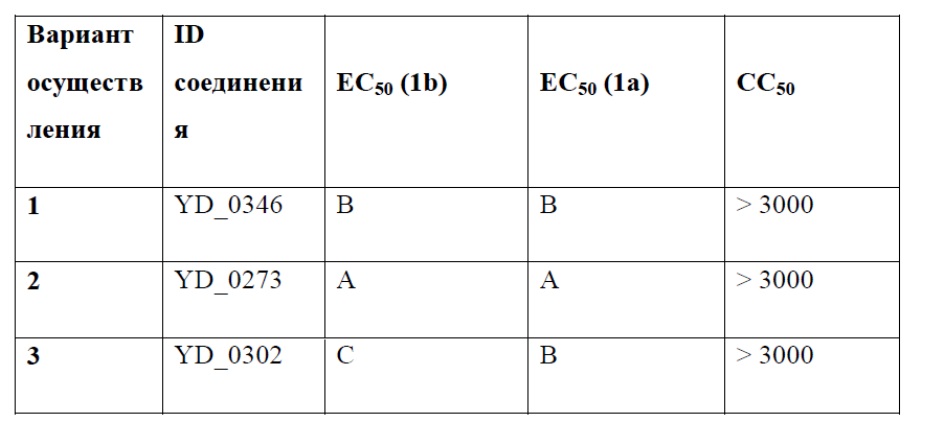
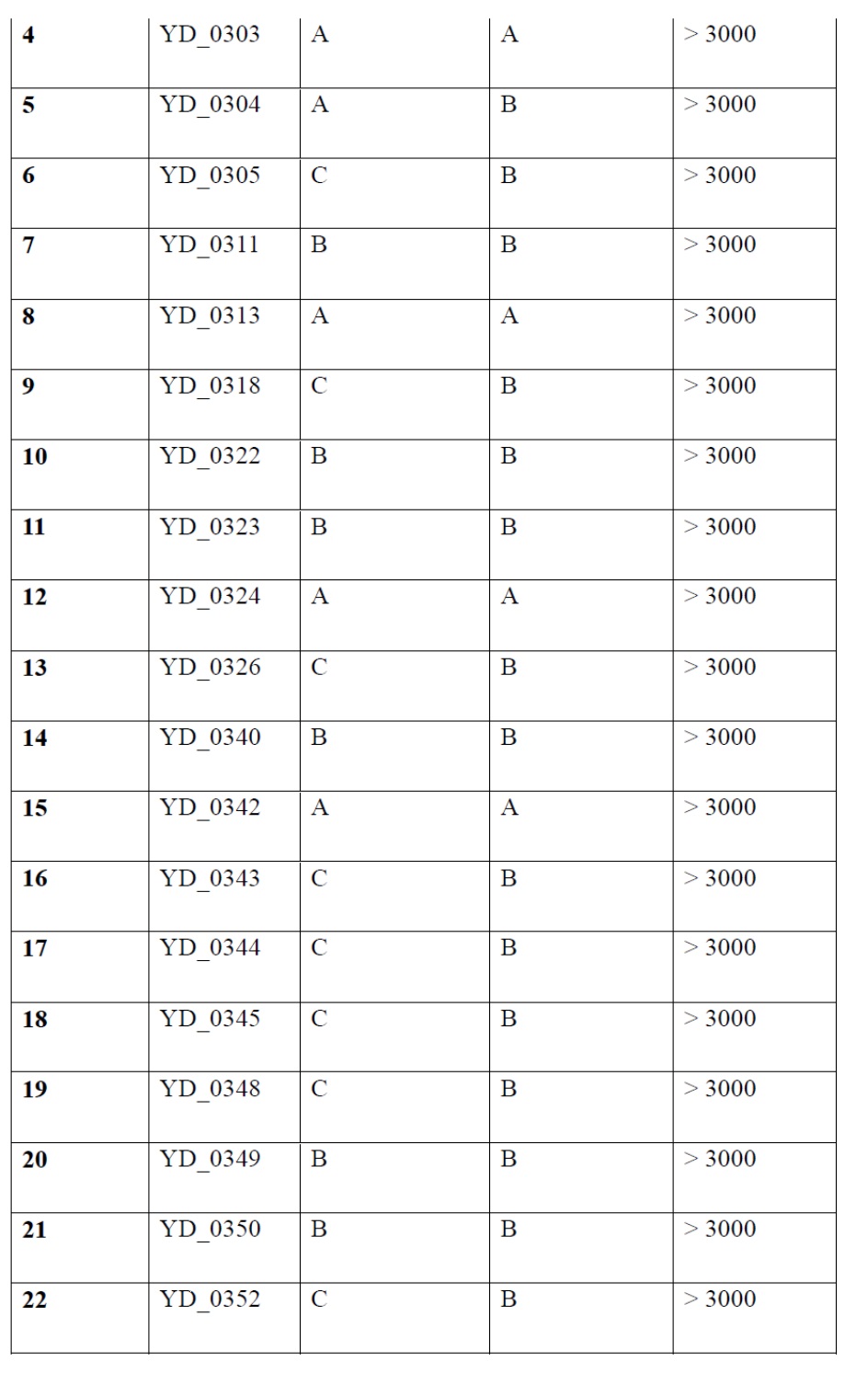
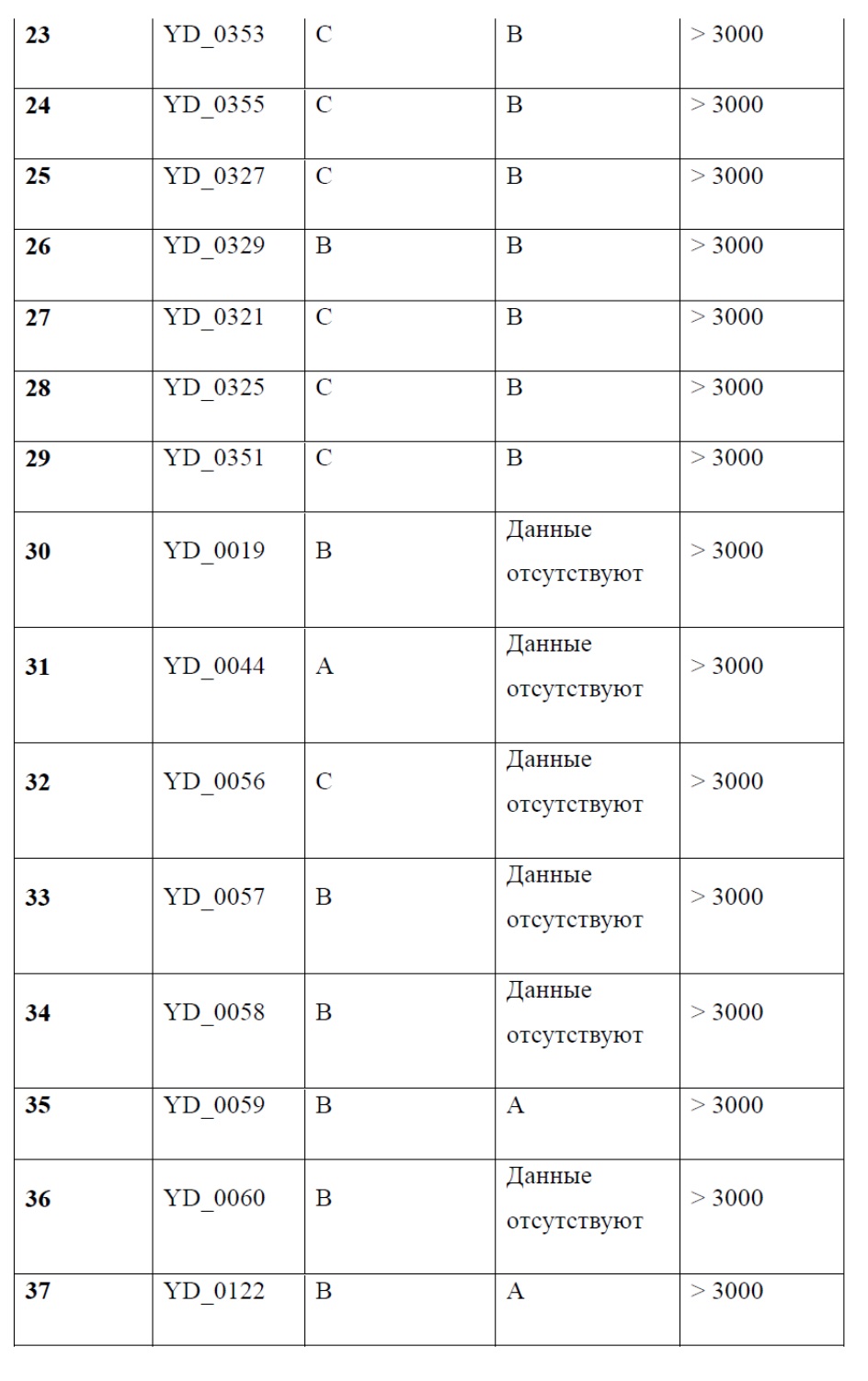
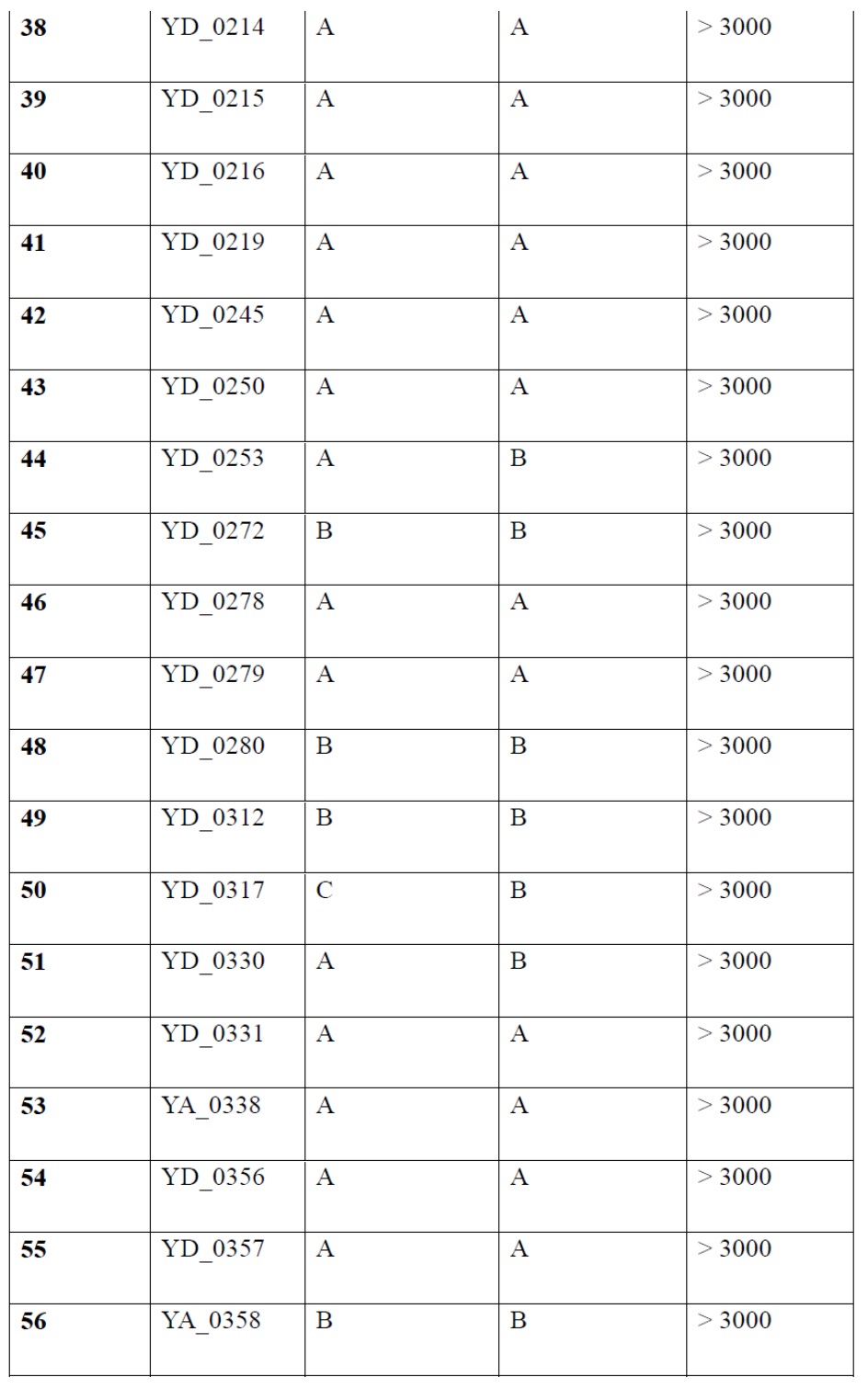
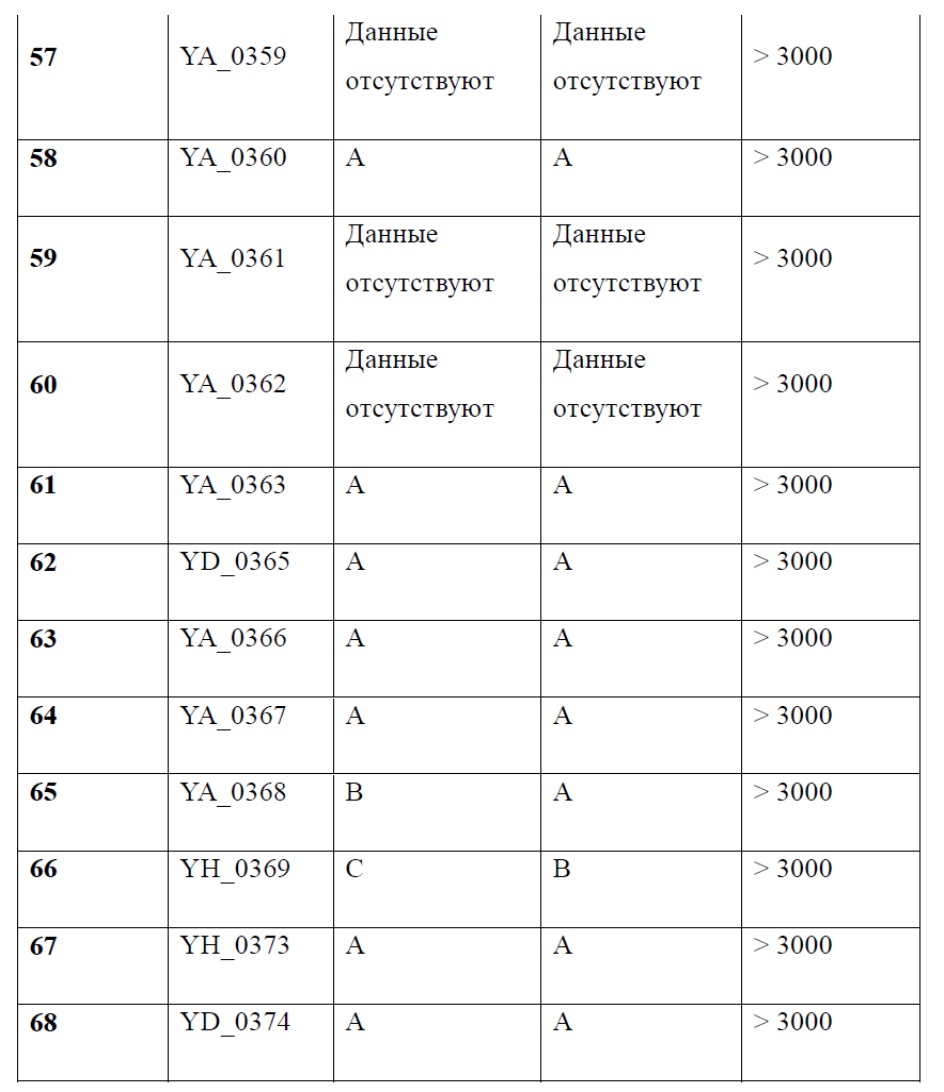
[422] Примечание: A (0,1 нМ - 100 нМ); B (100,01 нМ - 1000 нМ); C (1000,01 нМ - 5000 нМ).
Вывод: соединения по настоящему изобретению обладают превосходной активностью против HCV in vitro.
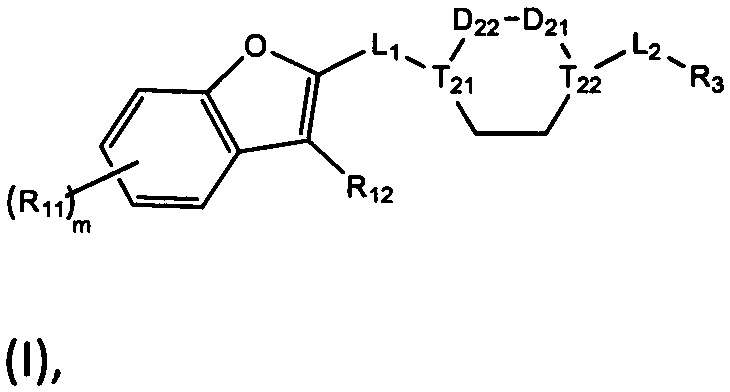
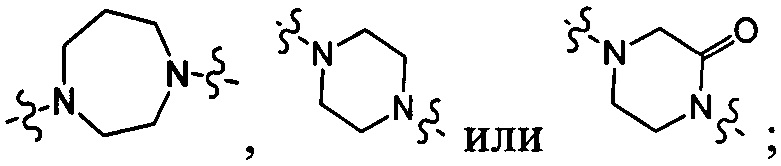
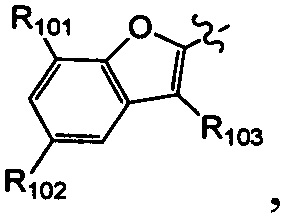
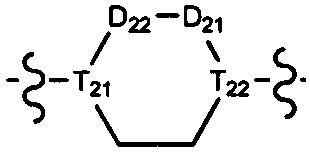
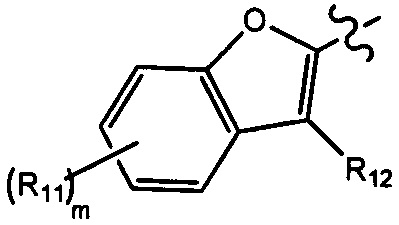
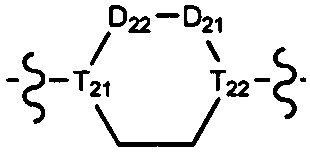
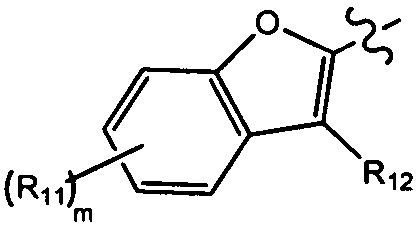
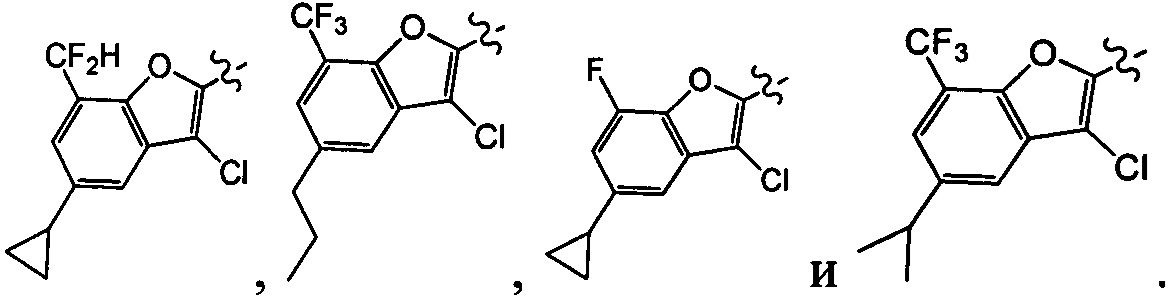
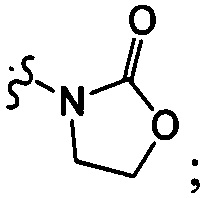
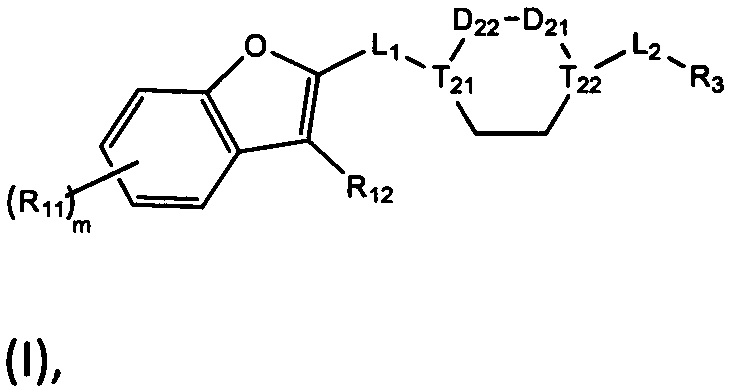
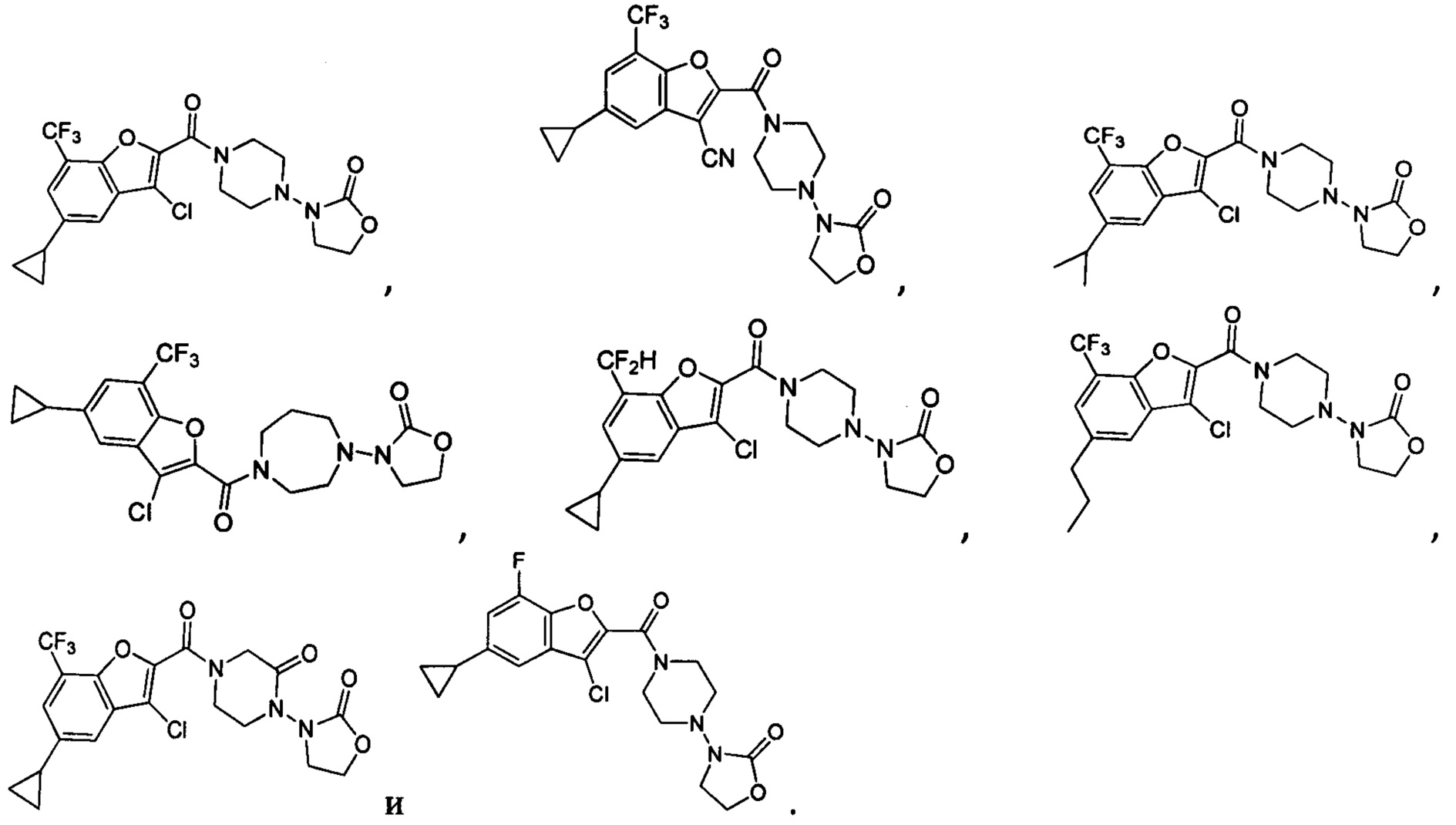
Устройство для получения композитной пленки из многоэлементного сплава
Внутренний блок кондиционера воздуха
Настенный кондиционер воздуха
Внутренний блок кондиционера
Внутренний блок кондиционера воздуха
Просеивающее твердый материал устройство для подземной разработки с закладкой
Новый способ пцр-секвенирования и его применение в генотипировании hla
Устройство заслонки
Производные пиперазинил пиримидина, способ их получения и их применение
Способ, устройство и система управления полномочиями
Соединения тиенил[3, 2-d]пиримидин-4-он, способ получения, фармацевтические композиции и их применение

