Результат интеллектуальной деятельности: Бифункциональные цитотоксические агенты
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым бифункциональным димерам CBI и CPI, полезным для лечения пролиферативных заболеваний. Эти димеры могут функционировать как самостоятельные лекарственные средства, полезные нагрузки в конъюгатах антитело-лекарственное средство (ADC) и соединения-конструкции линкер-полезная нагрузка, полезные в связи с получением или введением таких ADC. Настоящее изобретение также относится к композициям, содержащим вышеупомянутые димеры, конструкции линкер-полезная нагрузка и ADC, и к способам использования этих димеров, конструкций линкер-полезная нагрузка и ADC для лечения патологических состояний, включающих рак.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Мономеры на основе CPI были предметом недавних публикаций. Например, соединения (+)-СС-1065 и дуокармицины являются природными продуктами, выделенными из культурального бульона Streptomyces species, и они, как было показано, проявляют ультрамощную активность против культивированных раковых клеток и у экспериментальных животных. (+)-Ятакемицин (Yatakemycin) был выделен из Streptomyces sp. и представляет собой самый мощный член этого класса природных продуктов. Полагают, что биологическая активность этих природных продуктов связана с селективным в отношении характеристической последовательности ДНК алкилированием N3 аденина в АТ-богатых сайтах по наименее замещенному атому углерода активированного циклопропана. Считается, что связывание этой малой бороздки инициирует каскад клеточных событий, приводящих к апоптозу, как это наблюдалось для дуокармицинов ("Chemical and Biological Explorations of the Family of CC-1065 and the Duocarmycin Natural Products", Current Topics in Medicinal Chemistry, 2009, 9, 1494-1524). Ключевым структурным мотивом в этих и родственных аналогах является структура CPI, которая представляет собой реакционноспособную группу, алкилирующую ДНК:
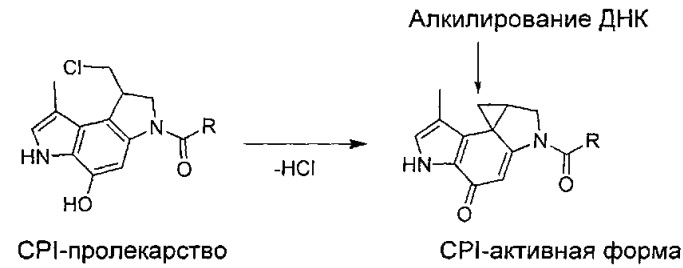
Пролекарственная форма CPI превращается в активное лекарственное вещество в биологической среде в результате реакции межмолекулярной циклизации. (Термин "CPI" происходит из химического названия: 1,2,8,8а-тетрагидроциклопропа[с]пирроло[3,2-е]индол-4(5Н)-он). Итак, CPI-пролекарство превращается в активное лекарственное вещество в результате реакции межмолекулярной циклизации. Фенольные синтетические предшественники (пролекарственная форма) обладают неотличимыми биологическими свойствами (эффективность и селективность алкилирования ДНК, цитотоксические активность в vitro, противоопухолевая активность в vivo) по сравнению с самими циклопропановыми производными (активная форма) ("Design, Synthesis, and Evaluation of Duocarmycin O-Amino Phenol Prodrugs Subject to Tunable Reductive Activation", J. Med. Chem. 2010, 53, 7731-7738). Другими словами, не важно, находится ли CPI головная часть в ее активной циклопропанированной форме или в ее пролекарственной форме. Важно отметить, что в этих соединениях присутствует только один CPI мотив; следовательно, эти соединения действуют как агенты, моноалкилирующие ДНК. Впоследствии были разработаны некоторые другие синтетические аналоги CPI структур, а именно те, которые указаны в "Chemical and Biological Explorations of the Family of CC-1065 and the Duocarmycin Natural Products", Current Topics in Medicinal Chemistry, 2009, 9, 1494-1524. В этом источнике информации указаны синтетические аналоги CBI, Cpzl, CFI, CI и CBQ. Моноалкилирующие аналоги дуокармицина были всесторонне исследованы в предклинических и клинических исследованиях ("Chemical and Biological Explorations of the Family of CC-1065 and the Duocarmycin Natural Products, Current Topics in Medicinal Chemistry, 2009, 9, 1494-1524).
Отдельным, но родственным классом соединений являются бифункциональные аналоги, которые содержат два активных мотива алкилирования ДНК (т.е. CPI). Эти соедринения отличаются от обычных дуокармицинов тем, что у них отсутствует группировка, имеющаяся в дуокармицинах, которая функционирует в качестве мотивов распознавания ДНК. Вместо этого, эти бифункциональные соединения просто содержат два мотива алкилирования (т.е. два CPI мотива), конденсированные вместе. Благодаря наличию двух реакционноспособных мотивов алкилирования эти соединения являются активными ДНК кросс-линкерами, тогда как соединения только с одним мотивом алкилирования (все дуокармицины) только моноалкилируют ДНК.
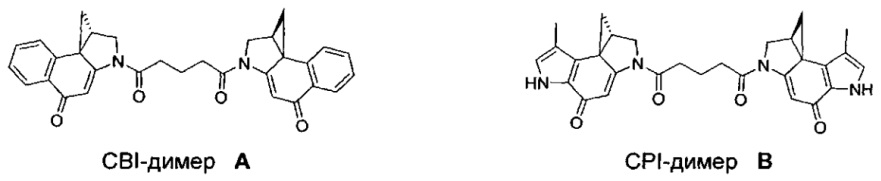
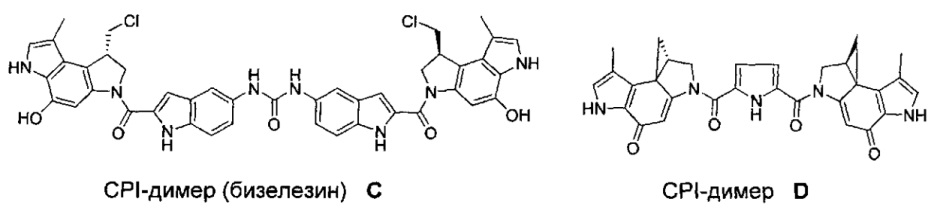
Соединения, изображенные выше, являются репрезентативными примерами из литературы и, как сообщается, являются сильнодействующими цитотоксинами: A ("Glycosidic Prodrugs of Highly Potent Bifunctional Duocarmycin Derivatives for Selective Treatment of Cancer", Angew. Chem. Int. Ed. 2010, 49, 7336-7339; "Duocarmycin Analogues Target Aldehyde Dehydrogenase 1 in Lung Cancer Cells", Angew. Chem. Int. Ed. 2012, 51, 2874-2877; "Bifunctional prodrugs and Drugs", WO 2011/054837, DE 102009051799; "The Two Faces of Potent Antitumor Duocarmycin-Based Drugs: A Structural Dissection Reveals Disparate Motifs for DNA versus Aldehyde Dehydrogenase 1 Affinity", Angew. Chem. Int. Ed. 2013, 52, 1-6; В ("Interstrand DNA Cross-linking with Dimers of the Spirocyclopropyl Alkylating Moiety of CC-1065", J. Am. Chem. SOC. 1989, 111, 6428-6429; "CC-1065 analogs having two CPI subunits useful as antitumor agents and ultraviolet light absorbers", Eur. Pat. Appl. (1990), EP 359454, также для соединений С и D; С ("Synthesis and DNA Cross-Linking by a Rigid CPI Dimer", J. Am. Chem. SOC. 1991, 113, 8994-8995; "Nucleotide Preferences for DNA Interstrand Cross-Linking Induced by the Cyclopropylpyrroloindole Analogue U-77,779", Biochemistry 1993, 32, 2592-2600; "Determination of the Structural Role of the Internal Guanine-Cytosine Base Pair in Recognition of a Seven-Base-Pair Sequence Cross-Linked by Bizelesin", Biochemistry 1995, 34, 11005-11016; "Analysis of the Monoalkylation and Cross-Linking Sequence Specificity of Bizelesin, a Bifunctional Alkylation Agent Related to (+)-CC-1065", J. Am. Chem. SOC. 1993, 115, 5925-5933; "Mapping of DNA Alkylation Sites Induced by Adozelesin and Bizelesin in Human Cells by Ligation-Mediated Polymerase Chain Reaction", Biochemistry 1994, 33, 6024-6030; "DNA Interstrand Cross-Links Induced by the Cyclopropylpyrroloindole Antitumor Agent Bizelesin Are Reversible upon Exposure to Alkali", Biochemistry 1993, 32, 9108-9114; "Replacement of the Bizelesin Ureadiyl Linkage by a Guanidinium Moiety Retards Translocation from Monoalkylation to Cross-Linking Sites on DNA", J. Am. Chem. Soc. 1997, 119, 3434-3442; "DNA interstrand cross-linking, DNA sequence specificity, and induced conformational changes produced by a dimeric analog of (+)-CC-1065", Anti-Cancer Drug Design (1991), 6, 427-452; "A phase I study of bizelesin, a highly potent and selective DNA interactive agent, in patients with advanced solid malignancies", Ann Oncol. 2003 May; 14(5):775-782; "A Phase I study of bizelesin (NSC 615291) in patients with advanced solid tumors", Clin Cancer Res. 2002, 3, 712-717; "Solution conformation of a bizelesin A-tract duplex adduct: DNA-DNA cross-linking of an A-tract straightens out bent DNA", J Mol Biol. 1995, 252, 86-101; "Preclinical pharmacology of bizelesin, a potent bifunctional analog of the DNA-binding antibiotic CC-1065", Cancer Chemother Pharmacol. 1994, 34, 317-322; D ("CC-1065 analogs having two CPI subunits useful as antitumor agents and ultraviolet light absorbers", Eur. Pat. Appl. (1990), EP 359454. Активный мотив алкилирования ДНК может, в принципе, существовать либо в пролекарственной форме, которая превращается в активное лекарственное средство в биологической среде, либо в его активном состоянии, которое не требует дальнейшего превращения. Превращение пролекарства в активное лекарственное средство для бифункциональных кросс-линкеров иллюстрируется примером с использованием CBI димера, как показано ниже:
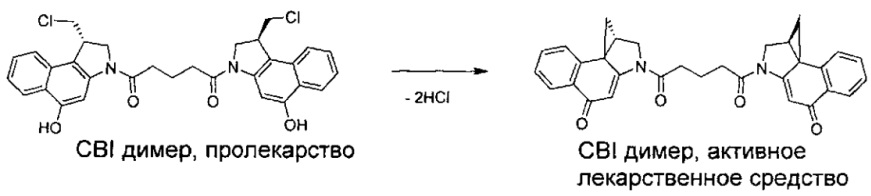
Такое же превращение имеет место для всех бифункциональных кросс-линкеров, которые существуют в их пролекарственном состоянии. Известны другие бифункциональные кросс-линкеры ("Chemical and Biological Explorations of the Family of CC-1065 and the Duocarmycin Natural Products", Current Topics in Medicinal Chemistry, 2009, 9, 1494-1524; "DNA interstrand cross-linking agents and their chemotherapeutic potential", Curr Med Chem. 2012, 19, 364-385; "Design and Synthesis of a Novel DNA-DNA Interstrand Adenine-Guanine Cross-Linking Agent", J. Am. Chem. Soc. 2001, 123, 4865-4866; "Effect of base sequence on the DNA cross-linking properties of pyrrolobenzodiazepine (PBD) dimers", Nucleic Acids Res. 2011, 39, 5800-5812; "Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers", Biochemistry. 2003, 42, 8232-8239; "Bifunctional alkylating agents derived from duocarmycin SA: potent antitumor activity with altered sequence selectivity", Bioorg Med Chem Lett. 2000, 10, 495-498; "Design, Synthesis and Cytotoxicity Evaluation of 1-Chloromethyl-5-hydroxy-1,2-dihydro-3H-benz[e]indole (seco-CBI) Dimers", Bioorganic & Medicinal Chemistry 2000, 8, 1607-1617.
Фосфатная пролекарственная стратегия для мономерных секо-CBI-содержащих цитотоксинов была описана Zhao et al. ("Synthesis and biological evaluation of antibody conjugates of phosphate prodrugs of cytotoxic DNA alkylators for the targeted treatment of cancer", J. Med. Chem. 2012, 55, 766-782) и Zhang et al. ("Immunoconjugates containing phosphate-prodrugged DNA minor groove binding agents, compositions containing them, and methods of making them and their use for treating cancer", WO 2012/162482).
Ни одно из вышеупомянутых соединений, имеющих два CBI- и/или CPI-ядра, связанные вместе с образованием димерных структур (так называемых CBI димеров, CPI димеров или CBI/CPI димеров), не рассматривалось для использования в конъюгатах антитело-лекарственное средство (ADC) в качестве полезной нагрузки.
Конъюгирование лекарственных средств с антителами, либо напрямую, либо через линкеры, предполагает рассмотрение целого ряда факторов, включая идентичность и местоположение химической группы для конъюгирования лекарственного средства, механизм высвобождения лекарственного средства, структурные элементы, обеспечивающие высвобождение лекарственного средства, и структурную модификацию для высвобождаемого свободного лекарственного средства. Кроме того, если лекарственное средство должно высвобождаться после интернализации антитела, то механизм высвобождения лекарственного средства должен быть согласованным с внутриклеточным транспортом конъюгата.
Несмотря на то, что множество разных классов лекарственных средств было опробовано для доставки антителами, только несколько классов лекарственных средств оказались эффективными в форме конъюгатов антитело-лекарственное средство с сохранением подходящего профиля токсичности. Одним таким классом являются ауристатины, производные природного продукта доластатина 10. Репрезентативные ауристатины включают N-метилвалинвалин-долаизолейцин-долапроин-норэфедрин и N-метилвалинвалин-долаизолейцин-долапроин-фенилаланин. Другие родственные тубулин-связывающие агенты включают майтанзины (смотри, например, "Cell-binding agent-maytansinoid conjugates linked via a noncleavable linker, preparation methods, and methods using them for targeting specific cell populations", опубликованный как WO 2005/037992). Другие цитотоксические лекарственные средства, которые были использованы для связывания с антителами, включают ДНК-связывающие лекарственные средства, такие как калихеамицин, который вызывает последовательность-специфическое расщепление двухнитевой ДНК. Еще один класс ДНК-связывающих цитотоксических лекарственных средств, используемых в ADC, включает димерные пирролобензодиазепины (смотри, например, "Preparation of unsymmetrical pyrollobenzodiazepines dimers for inclusion in targeted conjugates", опубликованный как WO 2013/041606). Другим таким классом лекарственных средств, когда были предприняты попытки осуществлять доставку с помощью антитела, являются ДНК-связывающие алкилирующие агенты, такие как аналог дуокармицина СС-1065 (смотри "Preparation of CC-1065 analogs and their conjugates for treatment of cancer", опубликованный как WO 2010/062171) и родственные соединения (смотри "Antibody-grug peptide conjugates for use as cytotoxins in cancer treatment", опубликованный как WO 2007/038658, и "Immunoconjugates containing phosphate-prodrugged DNA minor groove binding agents, compositions containing them, and methods of making them and their use for treating cancer", опубликованный как WO 2012/162482). Однако все эти лекарственные средства имеют ограничения, связанные с показаниями и профилем лечения заболеваний, и поэтому остается потребность в дополнительных лекарственных средствах с улучшенными свойствами, доставляемых посредством конъюгирования с антителом. Соответственно, согласно настоящему изобретению предложены новые ADC с димерами в качестве полезных нагрузок.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение характеризует новые структурные димерные аналоги, которые содержат новые линкерные элементы. Эти новые спейсерные мотивы приводят к соединениям с разными биологическими свойствами, например с улучшенной активностью в анализах пролиферации опухолевых клеток и стабильностью в плазме крови. Данное изобретение также характеризует новые спейсерные элементы для соответствующих CPI димеров и CBI-CPI смешанных структур. Кроме того, согласно изобретению предложено
Кроме того, настоящее изобретение является первым изобретением, раскрывающим такие соединения в связи с ADC модальностью, и включение этих соединений в обеспечивающий направленную доставку ADC является значительным достижением.
Настоящее изобретение относится к цитотоксическим димерам, содержащим субъединицы на основе CBI и/или на основе CPI (включая секо-формы CBI и/или CPI, как подробно изложено в данном описании), к конъюгатам антитело-лекарственное средство, содержащим такие димеры, и к способам использования таковых для лечения рака. Обе CBI и CPI структуры могут быть представлены их секо-формой и могут быть замещены и дериватизированы, как подробно изложено в данном описании.
Таким образом, настоящее изобретение относится к соединениям и фармацевтическим композициям, содержащим их, к их получению и к применению для соединений, в первую очередь, но не исключительно, противораковых агентов. Согласно одному аспекту настоящее изобретение относится к являющемуся "полезной нагрузкой" соединению формулы I:
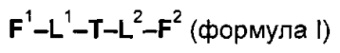
или его фармацевтически приемлемой соли или сольвату, где:
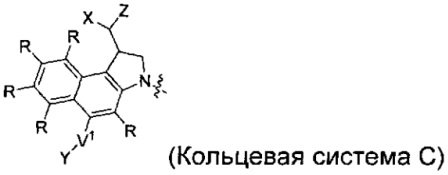
каждый из F1 и F2 независимо выбран из кольцевых систем A, B, C и D:


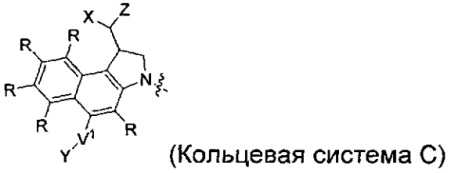

каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C2-C6алкенил, -C2-C6алкинил, -галоген, гидроксил, алкокси, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -NO2, -C6-C14арил и -C6-C14гетероарил, где два или более R возможно соединены с образованием кольца или колец, и где указанные -C6-C14арил и -C6-C14гетероарил возможно замещены 1-5 заместителями, независимо выбранными из групп -C1-C10алкил, -C1-C10алкокси, -галоген, -C1-C10алкилтио, -трифторметил, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -C1-C10алкил-N(C1-C8алкил)2, -C1-C3алкилтио, -NO2 или -C1-C10гетероциклил, для каждой кольцевой системы, в которой имеется R;
каждый V1 независимо представляет собой связь, O, N(R) или S для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O, N(R) или S для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -фенил, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 или -C(O)N(R)2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо представляет собой -OH, -O-ацил, азидо, галоген, цианат, тиоцианат, изоцианат, тиоизоцианат или 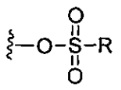 для каждой кольцевой системы, в которой имеется X;
для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из группы, состоящей из H и групп -C1-C6алкил-RA, -C(O)RA, -C(S)RA, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(RA)2, гликозил, -NO2 и -PO(ORA)2, для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил и -C1-C20алкилN(R)2, где указанные -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил и -C1-C20алкилN(R)2 возможно замещены 1-3 заместителями, независимо выбранными из R;
каждый Z независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -С(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)галоген, и где каждый указанный C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -С(O)OC1-C8алкил, -С(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)галоген возможно замещен 1-3 заместителями, независимо выбранными из R, для каждой кольцевой системы, в которой имеется Z;
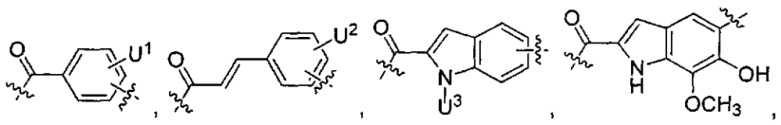
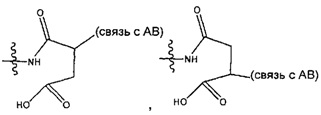
каждый из L1 и L2 независимо выбран из прямой связи, карбонильной или карбонилацильной группы, связанной с F1 или F2 по ацильной группировке, где карбонилацильная группа выбрана из группы, состоящей из:
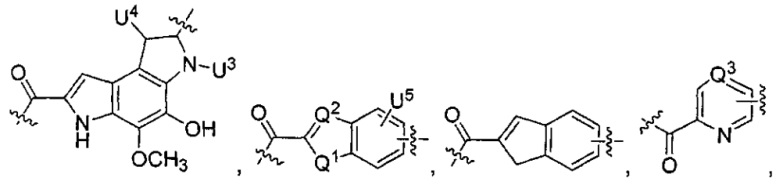
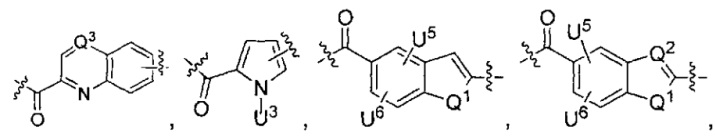
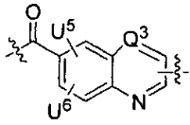 и
и 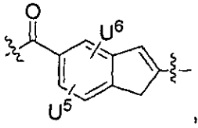 где
где
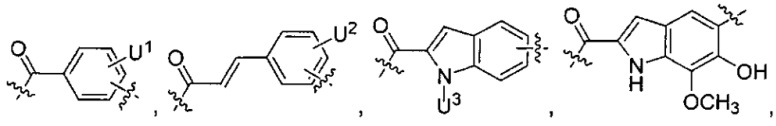
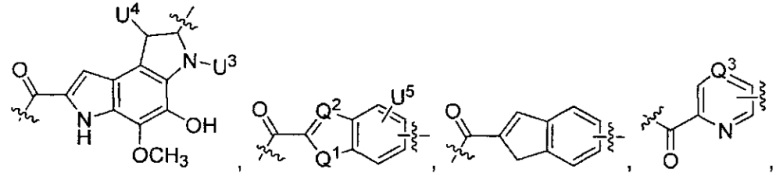
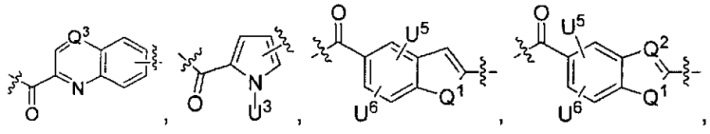
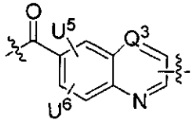
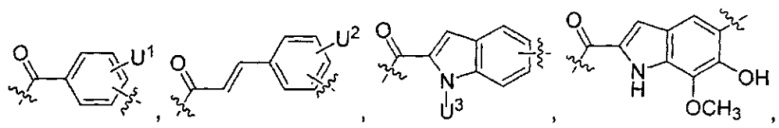
U1 выбран из H и групп -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)фенил или -галоген,
U2 представляет собой H, -OH или -OCH3,
U3 представляет собой H, -CH3 или -C2H5,
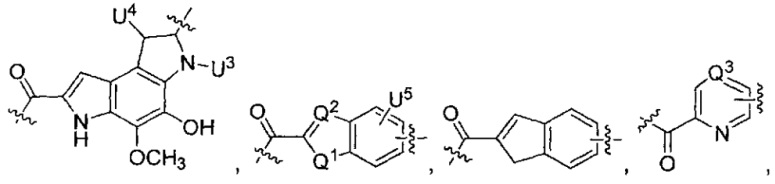
U4 представляет собой H или CH3S-,
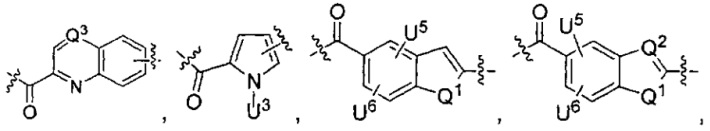
каждый из U5 и U6 независимо выбран из H и групп -галоген, -C1-C4алкил, -C1-C3алкокси, -C1-C6диалкиламино, -NO2, -NHC(O)C1-C10алкил, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 или -NHC(O)фенил,
Q1 представляет собой -O-, -S- или -NH-, и
каждый из Q2 и Q3 независимо представляет собой -CH- или -N-;
T выбран из:
-NHC(O)-,
-C(O)NH-,
-C(O)O-,
-OC(O)-,
-NRB-T1-NRC-, где каждый из RB и RC независимо представляет собой H или -C1-C8алкил, или RB и RC соединены вместе с образованием кольца и вместе представляют собой (CH2)2-3, где T1 выбран из -C(O)-, -C(O)(CH2)nC(O)-, где n означает целое число от 0 до 50, -C(O)PhC(O)-, где Ph представляет собой 1,3- или 1,4-фенилен,
-C(O)hetC(O)-, где het представляет собой моно-, би- или трициклический гетероарил из 5-12 членов, содержащий один, два или три гетероатома, независимо выбранных из O, N, S, P и B, где het возможно замещен 1-8 заместителями, каждый из которых независимо выбран из группы, состоящей из групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -NH2, -NHRD и -NO2, и указанные возможные заместители на het возможно замещены RE, где по меньшей мере один из F1 и F2 выбран из группы, состоящей из Кольцевой системы C и Кольцевой системы D, когда T представляет собой -C(O)hetC(O)-,
где каждый RD независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C(O)-C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2 и -C(O)-галоген, возможно замещенных RE,
где каждый RE независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -С(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2 и -C(O)-галоген, и где каждый RE возможно замещен 1-3 заместителями, независимо выбранными из R,
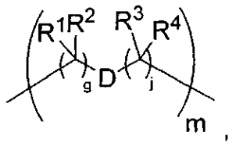
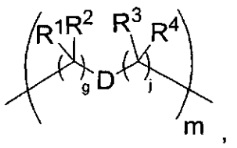
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
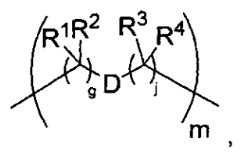
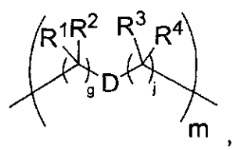
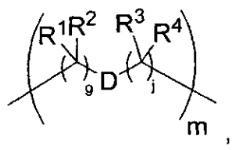
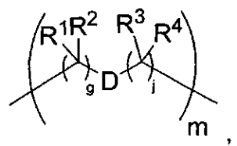
где каждый X1 независимо представляет собой связь, -NRE-, -O- или -S-, где каждый из A1 и B1 независимо представляет собой =O или =S, где каждый из R1, R2, R3 и R4 независимо представляет собой RE, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, или каждый из R1, R2, R3 и R4 связан с разными атомами углерода на D, где каждый из g и j независимо означает целое число от 0 до 50, и m означает целое число от 1 до 50, и где D представляет собой связь или выбран из группы, состоящей из групп -S-, -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло возможно замещены -RE, -C(O)RE, -C(O)ORE, -N(RE)2, -N(R)C(O)RE или -N(R)C(O)ORE, и D дополнительно возможно замещен 1-2 R при условии, что если g означает 0, j означает 0, и T2 представляет собой -C1-C8алкилен-, то один из F1 и F2 выбран из группы, состоящей из Кольцевой системы A и Кольцевой системы B, а другой из F1 и F2 выбран из группы, состоящей из Кольцевой системы C и Кольцевой системы D, и
-G1-T2-G2-, где каждый из G1 и G2 независимо представляет собой -S(O)X1- или -S(O)2X1-.
В воплощениях изобретения переменная n означает число от 0 до 50, предпочтительно от 0 до 25, предпочтительно от 0 до 10 и предпочтительно 1-5. Предпочтительно, переменная n может быть равна 0, 1, 2, 3, 4 или 5.
В других воплощениях изобретения переменная -Y- представляет собой C(O)N(RA)2 или C(S)N(RA)2, где один RA представляет собой водород или -C1-C20алкил, а другой RA представляет собой -C1-C20алкил-N(R)2, так что образуется структура:
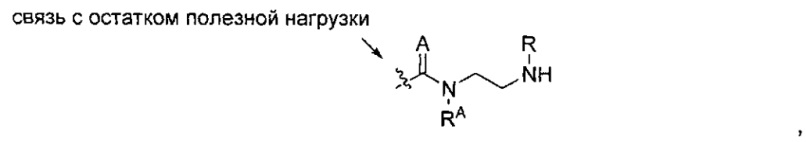
где A представляет собой кислород или серу.
Как указано выше, воплощения настоящего изобретения включают воплощения, где каждый из R1, R2, R3 и R4 представляет собой связь с разными атомами углерода на D. Когда D представляет собой 6-членное карбоциклическое кольцо (ниже изображено жирными линиями), тогда это воплощение может принимать форму кубана:
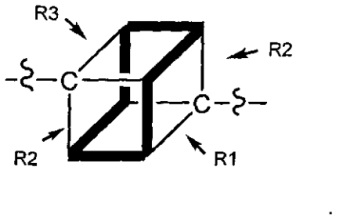
Другие формы кубанов (например, замещенные формы, как изложено в данном документе) и не являющиеся кубанами также возможны и входят в объем изобретения.
Согласно другому аспекту изобретения предложено соединение, представляющее собой конструкцию "линкер-полезная нагрузка", формулы IIA:
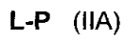
или его фармацевтически приемлемые соль или сольват, где:
P представляет собой:
F1-L1-T-L2-F2,
где:
каждый из F1 и F2 независимо выбран из кольцевых систем A, B, C и D:


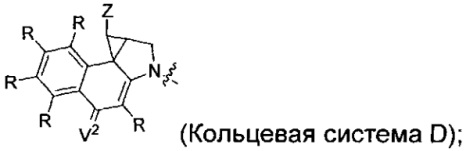
каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C2-C6алкенил, -C2-C6алкинил, -галоген, гидроксил, алкокси, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -NO2, -C6-C14арил и -C6-C14гетероарил, где два или более R возможно соединены с образованием кольца или колец, и где указанные -C6-C14арил и -C6-C14гетероарил возможно замещены 1-5 заместителями, независимо выбранными из групп -C1-C10алкил, -C1-C10алкокси, -галоген, -C1-C10алкилтио, -трифторметил, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -C1-C10алкил-N(C1-C8алкил)2, -C1-C3алкилтио, -NO2 или -C1-C10гетероциклил, для каждой кольцевой системы, в которой имеется R;
каждый V1 независимо представляет собой связь, O, N(R) или S для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O, N(R) или S для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -фенил, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 или -C(O)N(R)2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо выбран из групп -OH, -O-ацил, азидо, галоген, цианат, тиоцианат, изоцианат, тиоизоцианат или 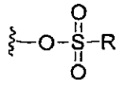 для каждой кольцевой системы, в которой имеется X;
для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из связи, H и групп -C(O)RA, -C(S)RA, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(RA)2, гликозил, -NO2 и -P(O)(ORA)2 для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C1-C20алкилN(R)2, -C1-C20алкилен, -C1-C8гетероалкилен, -C6-C14арилен, аралкилен, -C1-C10гетероцикло, -C3-C8карбоцикло и -C1-C20aлкилN(R)- и RF, где указанный RA возможно замещен 1-3 заместителями, независимо выбранными из R, и где один Y является двухвалентным и связан с L,
RF представляет собой -N(R6)QN(R5)C(O)- и связан с L по карбонилу, расположенному рядом с N(R5), где каждый из R5 и R6 независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил и -C3-C8карбоциклил, или R5 или R6 соединены с замещенным атомом углерода на Q с образованием -C1-C10гетероциклического или -C6-C14гетероарильного кольца, или R5 и R6 соединены вместе с образованием -C1-C10гетероциклической или -C6-C14гетероарильной кольцевой системы, и где Q представляет собой -C1-C8алкилен-, -C1-C8гетероалкилен-, -C6-C14арилен-, -аралкилен-, -C1-C10гетероцикло- или -C3-C8карбоцикло-, где каждый из Q, R5 и R6 независимо возможно замещен 1-3 заместителями, независимо выбранными из R;
каждый Z независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -С(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, и где указанные C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, каждый, возможно замещены 1-3 заместителями, независимо выбранными из R, для каждой кольцевой системы, в которой имеется Z;
каждый из L1 и L2 независимо выбран из прямой связи, карбонильной или карбонилацильной группы, связанной с F1 или F2 по ацильной группировке, где карбонилацильная группа выбрана из группы, состоящей из:
 и
и 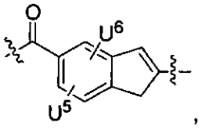 где
где
U1 выбран из H и групп -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)фенил или -галоген,
U2 представляет собой H, -OH или -OCH3,
U3 представляет собой H, -CH3 или -C2H5,
U4 представляет собой H или CH3S-,
каждый из U5 и U6 независимо выбран из H и групп -галоген, -C1-C4алкил, -C1-C3алкокси, -C1-C6диалкиламино, -NO2, -NHC(O)C1-C10алкил, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 или -NHC(O)фенил,
Q1 представляет собой -O-, -S- или -NH-, и
каждый из Q2 и Q3 независимо представляет собой -CH- или -N-;
T выбран из:
-NHC(O)-,
-C(O)NH-,
-C(O)O-,
-OC(O)-,
-NRB-T1-NRC-, где каждый из RB и RC независимо представляет собой H или -C1-C8алкил, или RB и RC соединены вместе с образованием кольца и вместе представляют собой (CH2)2-3, где T1 выбран из -C(O)-, -C(O)(CH2)nC(O)-, где n означает целое число от 0 до 50, -C(O)PhC(O)-, где Ph представляет собой 1,3- или 1,4-фенилен,
-C(O)hetC(O)-, где het представляет собой моно-, би- или трициклический гетероарил из 5-12 членов, содержащий один, два или три гетероатома, независимо выбранных из O, N, S, P и B, где het возможно замещен 1-8 заместителями, каждый из которых независимо выбран из группы, состоящей из групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -NH2, -NHRD и -NO2, и указанные возможные заместители на het возможно замещены RE, где по меньшей мере один из F1 и F2 выбран из группы, состоящей из Кольцевой системы C и Кольцевой системы D, когда T представляет собой -C(O)hetC(O)-,
где каждый RD независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C(O)-C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -С(O)OC1-C8алкил, -С(O)N(C1-C8алкил)2 и -C(O)-галоген, возможно замещенных RE,
где каждый RE независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2 и -C(O)-галоген, и где каждый RE возможно замещен 1-3 заместителями, независимо выбранными из R,
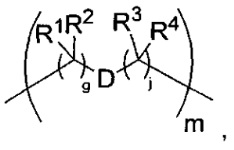
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
где каждый X1 независимо представляет собой связь, -NRE-, -O- или -S-, где каждый из A1 и B1 независимо представляет собой =O или =S, где каждый из R1, R2, R3 и R4 независимо представляет собой RE, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, или каждый из R1, R2, R3 и R4 связан с разными атомами углерода на D, где каждый из g и j независимо означает целое число от 0 до 50, и m означает целое число от 1 до 50, и где D представляет собой связь или выбран из группы, состоящей из групп -S-, -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло возможно замещены -RE, -C(O)RE, -C(O)ORE, -N(RE)2, -N(R)C(O)RE или -N(R)C(O)ORE, и D дополнительно возможно замещен 1-2 R, и
-G1-Т2-G2-, где каждый из G1 и G2 независимо представляет собой -S(O)X1- или -S(O)2X1-;
L представляет собой LA-LB-(LC)1-3, где LA выбран из группы, состоящей из групп -галоген, -N(R)2, -CON(R)2, -S-арил, возможно замещенный -NO2 или -CON(R)2, -S-гетероарил, возможно замещенный -NO2, алкил-SO2-гетероарил, арил-SO2-гетероарил-,
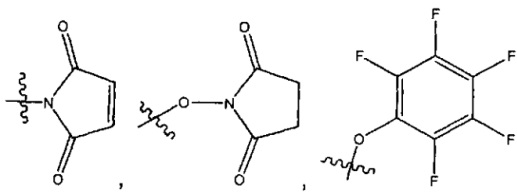 и
и 
LB представляет собой LB1-LB2-LB3, где LB1 отсутствует или представляет собой один или более компонентов, выбранных из группы, состоящей из групп -C(O)-, -C(S)-, -C(O)NR-, -C(O)C1-C6алкил-, -C(O)NRC1-C6алкил-, -C1-C6алкил(OCH2CH2)1-6-, -C(O)C1-C6aлкилNRC(O)-, -С(O)C1-C6алкил(OCH2CH2)1-6-, -C1-C6алкил(OCH2CH2)1-6-C(O)-, -C1-C6алкил-S-S-C1-C6алкилNRС(O)CH2-, -C1-C6алкил(OCH2CH2)1-6NRC(O)CH2-, -C(O)C1-C6алкил-NRC(O)C1-6алкил-, -N=CR-фенил-O-C1-C6алкил-, -N=CR-фенил-O-C1-C6алкил-C(O)-, -C(O)-C1-C6алкил(OCH2CH2)1-6NRC(O)-, -C(O)C1-C6алкил-фенил(NR-C(O)C1-C6алкил)1-4-, -С(O)C1-C6алкил(OCH2CH2)1-6-NRC(O)C1-C6алкил-, -C1-C6алкил-, -S-, -C(O)-CH(NR-C(O)C1-C6алкил)-C1-C6алкил- и (-CH2-CH2-O-)1-20,
где LB2 представляет собой AA0-12, где AA представляет собой природную аминокислоту, неприродную аминокислоту или -(CR15)o-S-S-(CR15)p, где каждый из o и p независимо означает целое число от 1 до 20,
LB3 представляет собой -PABA-, -PABC- или отсутствует;
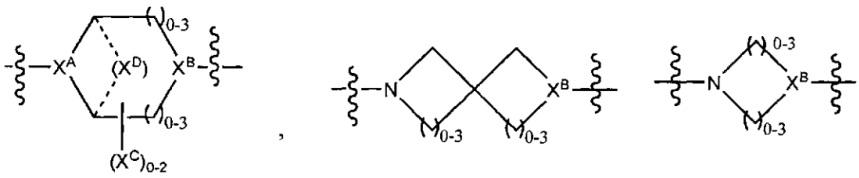
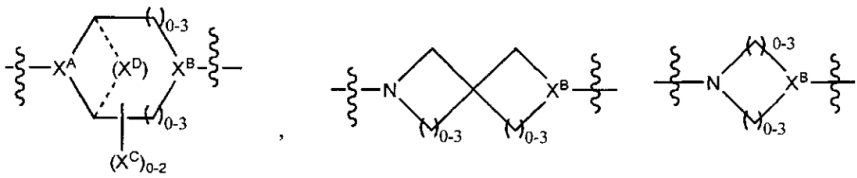
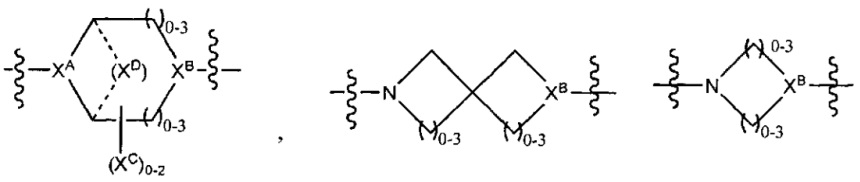
LC отсутствует или независимо выбран из группы, состоящей из групп -C1-C6алкилен-, -NRC3-C8-гетероциклилNR-, -NRC3-C8-карбоциклилNR-, -NRC1-C6алкилNR-, -NRC1-C6алкилен-, -S-, -NR-, -NRNR-, -O(CR2)1-4S-S(CR2)1-4N(R)-, -NRC1-C6-алкиленфениленNR-, -NRC1-C6aлкилeнфeнилeнSO2NR-, -OC1-C6aлкилS-SC1-C6aлкилC(COOR)NR-, -NRC(COOR)C1-C6aлкилS-SC1-C6aлкилO-,
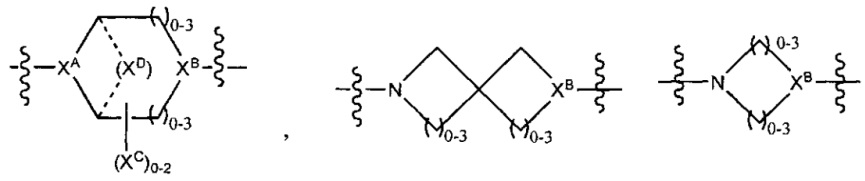
и 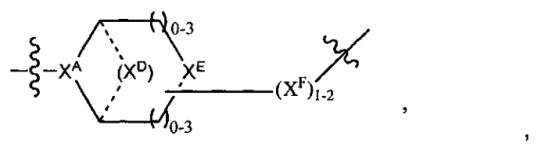
где
XA представляет собой CR или N,
XB представляет собой CH, CR(C(R)2)1-3NR, CR(C(R)2)1-3O, CR(C(R)2)1-3C(O)NR, CR-(C(R)2)1-3C(O)NRNR, CR(C(R)2)1-3SO2NR, CR(C(R)2)1-3NRNR, CR(C(R)2)1-3NRC(O) или N,
каждый XC представляет собой R,
каждый XD представляет собой -(CH2)1-5- или отсутствует;
XE представляет собой O, S, C(R)2, C(R)(C(R)2)1-3-NR2 или NR, и
каждый XF представляет собой (C(R)2)1-3-NR или C(R)2-(C(R)2)1-3-O.
В других воплощениях изобретения переменная -Y- представляет собой C(O)N(RA)2 или C(S)N(RA)2, где один RA представляет собой водород или -C1-C20алкил, а другой RA представляет собой -C1-C20алкил-N(R)-, так что образуется структура:
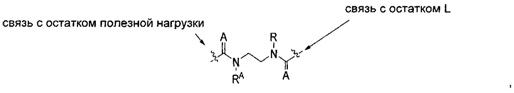
где каждый A независимо представляет собой кислород или серу.
Согласно еще одному аспекту изобретения предложено соединение, представляющее собой конъюгат антитело-лекарственное средство, формулы IIIA:
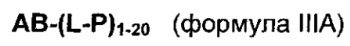
или его фармацевтически приемлемые соль или сольват, где:
AB представляет собой представляет собой антитело;
P представляет собой:
F1-L1-T-L2-F2,
где:
каждый из F1 и F2 независимо выбран из кольцевых систем A, B, C и D:

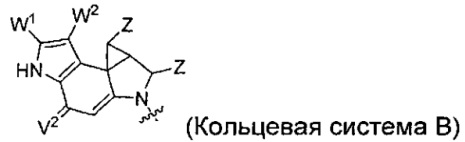
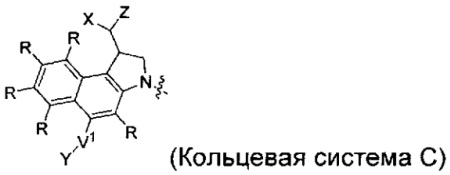
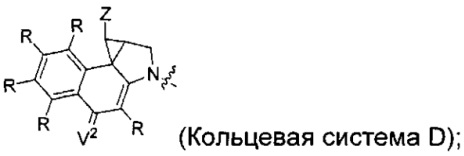
каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C2-C6алкенил, -C2-C6алкинил, -галоген, гидроксил, алкокси, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -NO2, -C6-C14арил и -C6-C14гетероарил, где два или более R возможно соединены с образованием кольца или колец, и где указанные -C6-C14арил и -C6-C14гетероарил возможно замещены 1-5 заместителями, независимо выбранными из групп -C1-C10алкил, -C1-C10алкокси, -галоген, -C1-C10алкилтио, -трифторметил, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -C1-C10алкил-N(C1-C8алкил)2, -C1-C3алкилтио, -NO2 или -C1-C10гетероциклил, для каждой кольцевой системы, в которой имеется R;
каждый V1 независимо представляет собой связь, O, N(R) или S для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O, N(R) или S для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -фенил, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 или -C(O)N(R)2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо выбран из групп -OH, -О-ацил, азидо, галоген, цианат, тиоцианат, изоцианат, тиоизоцианат или 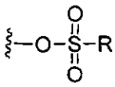 для каждой кольцевой системы, в которой имеется X;
для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из связи, H и групп -C(O)RA, -C(S)RA, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(RA)2) гликозил, -NO2 и -P(O)(ORA)2 для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C1-C20aлкилN(R)2, -C1-C20алкилен, -C1-C8гетероалкилен, -C6-C14арилен, аралкилен, -C1-C10гетероцикло, -C3-C8карбоцикло и -C1-C20алкилN(R)- и RF, где указанный RA возможно замещен 1-3 заместителями, независимо выбранными из R, и где один Y является двухвалентным и связан с L,
RF представляет собой -N(R6)QN(R5)C(O)- и связан с L по карбонилу, расположенному рядом с N(R5), где каждый из R5 и R6 независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил и -C3-C8карбоциклил, или R5 или R6 соединены с замещенным атомом углерода на Q с образованием -C1-C10гетероциклического или -C6-C14гетероарильного кольца, или R5 и R6 соединены вместе с образованием -C1-C10гетероциклической или -C6-C14гетероарильной кольцевой системы, и где Q представляет собой -C1-C8алкилен-, -C1-C8гетероалкилен-, -C6-C14арилен-, -аралкилен-, -C1-C10гетероцикло- или -C3-C8карбоцикло-, где каждый из Q, R5 и R6 независимо возможно замещен 1-3 заместителями, независимо выбранными из R;
каждый Z независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -С(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, и где указанные C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, каждый, возможно замещены 1-3 заместителями, независимо выбранными из R, для каждой кольцевой системы, в которой имеется Z;
каждый из L1 и L2 независимо выбран из прямой связи, карбонильной или карбонилацильной группы, связанной с F1 или F2 по ацильной группировке, где карбонилацильная группа выбрана из группы, состоящей из:
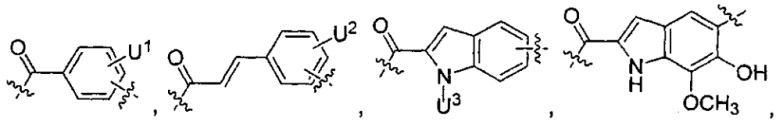
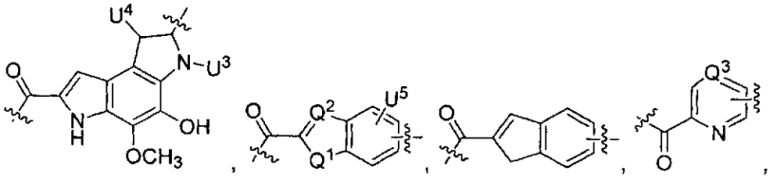
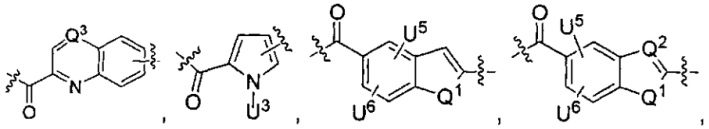
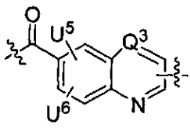 и
и 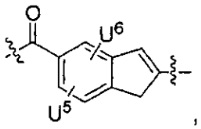 где
где
U1 выбран из H и групп -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)фенил или -галоген,
U2 представляет собой H, -OH или -OCH3,
U3 представляет собой H, -CH3 или -C2H5,
U4 представляет собой H или CH3S-,
каждый из U5 и U6 независимо выбран из H и групп -галоген, -C1-C4алкил, -C1-C3алкокси, -C1-C6диалкиламино, -NO2, -NHC(O)C1-C10алкил, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 или -NHC(O)фенил,
Q1 представляет собой -O-, -S- или -NH-, и
каждый из Q2 и Q3 независимо представляет собой -CH- или -N-;
T выбран из:
-NHC(O)-,
-C(O)NH-,
-C(O)O-,
-OC(O)-,
-NRB-T1-NRC-, где каждый из RB и RC независимо представляет собой H или -C1-C8алкил, или RB и RC соединены вместе с образованием кольца и вместе представляют собой (CH2)2-3, где T1 выбран из -C(O)-, -C(O)(CH2)nC(O)-, где n означает целое число от 0 до 50, -C(O)PhC(O)-, где Ph представляет собой 1,3- или 1,4-фенилен,
-C(O)hetC(O)-, где het представляет собой моно-, би- или трициклический гетероарил из 5-12 членов, содержащий один, два или три гетероатома, независимо выбранных из O, N, S, P и B, где het возможно замещен 1-8 заместителями, каждый из которых независимо выбран из группы, состоящей из групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -NH2, -NHRD и -NO2, и указанные возможные заместители на het возможно замещены RE, где по меньшей мере один из F1 и F2 выбран из группы, состоящей из Кольцевой системы C и Кольцевой системы D, когда T представляет собой -C(O)hetC(O)-,
где каждый RD независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C(O)-C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -С(O)OC1-C8алкил, -C(O)N(C1-Cалкил)2 и -C(O)-галоген, возможно замещенных RE,
где каждый RE независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2 и -C(O)-галоген, и где каждый RE возможно замещен 1-3 заместителями, независимо выбранными из R,
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
где каждый X1 независимо представляет собой связь, -NRE-, -O- или -S-, где каждый из A1 и B1 независимо представляет собой =O или =S, где каждый из R1, R2, R3 и R4 независимо представляет собой RE, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, или каждый из R1, R2, R3 и R4 связан с разными атомами углерода на D, где каждый из g и j независимо означает целое число от 0 до 50, и m означает целое число от 1 до 50, и где D представляет собой связь или выбран из группы, состоящей из групп -S-, -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8 гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло возможно замещены -RE, -C(O)RE, -C(O)ORE, -N(RE)2, -N(R)C(O)RE или -N(R)C(O)ORE, и D дополнительно возможно замещен 1-2 R, и
-G1-Т2-G2-, где каждый из G1 и G2 независимо представляет собой -S(O)X1- или -S(O)2X1-;
L представляет собой LA-LB-(LC)1-3;
LA выбран из связи с AB, группы -N(R)-(связь с AB), алкил-SO2-гетероарил, арил-SO2-гетероарил-,
 и
и 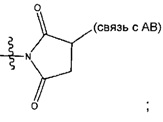
LB представляет собой LB1-LB2-LB3,
где LB1 отсутствует или представляет собой один или более компонентов, выбранных из группы, состоящей из групп -C(O)-, -C(S)-, -C(O)NR-, -C(O)C1-C6алкил-, -C(O)NRC1-C6алкил-, -C1-C6алкил(OCH2CH2)1-6-, -C(O)C1-C6алкилNRC(O)-, -C(O)C1-C6алкил(OCH2CH2)1-6-, -C1-C6алкил(OCH2CH2)1-6-C(O)-, -C1-C6алкил-S-S-C1-C6алкилNRC(O)CH2-, -C1-C6алкил(OCH2CH2)1-6NRC(O)CH2-, -C(O)C1-C6алкил-NRC(O)C1-6алкил-, -N=CR-фенил-O-C1-C6алкил-, -N=CR-фенил-O-C1-C6алкил-C(O)-, -C(O)-C1-C6алкил(OCH2CH2)1-6NRC(О)-, -C(O)C1-C6алкил-фенил(NR-C(O)C1-C6алкил)1-4-, -C(O)C1-C6алкил(OCH2CH2)1-6-NRC(O)C1-C6алкил-, -C1-C6алкил-, -S-, -C(O)-СН(NR-С(O)C1-C6алкил)-C1-C6алкил- и (-CH2-CH2-O-)1-20,
LB2 представляет собой AA0-12, где AA представляет собой природную аминокислоту, неприродную аминокислоту или -(CR15)o-S-S-(CR15)p, где каждый из o и p независимо означает целое число от 1 до 20,
LB3 представляет собой -PABA-, -PABC- или отсутствует;
LC отсутствует или независимо выбран из группы, состоящей из групп -C1-C6алкилен-, -NRC3-C8-гетероциклилNR-, -NRC3-C8-карбоциклилNR-, -NRC1-C6алкилNR-, -NRC1-C6алкилен-, -S-, -NR-, -NRNR-, -O(CR2)1-4S-S(CR2)1-4N(R)-, -NRC1-C6-алкиленфениленNR-, -NRC1-C6алкиленфениленSO2NR-, -OC1-C6алкилS-SC1-C6алкилC(COOR)NR-, -NRC(COOR)C1-C6алкилS-SC1-C6алкилO-,
и 
где
XA представляет собой CR или N,
XB представляет собой CH, CR(C(R)2)1-3NR, CR(C(R)2)1-3O, CR(C(R)2)1-3C(O)NR, CR-(C(R)2)1-3C(O)NRNR, CR(C(R)2)1-3SO2NR, CR(C(R)2)1-3NRNR, CR(C(R)2)1-3NRC(O) или N,
каждый XC представляет собой R,
каждый XD представляет собой -(CH2)1-5- или отсутствует;
XE представляет собой O, S, C(R)2, C(R)(C(R)2)1-3-NR2 или NR, и
каждый XF представляет собой (C(R)2)1-3-NR или C(R)2-(C(R)2)1-3-O.
Согласно другому аспекту изобретения предложено соединение, представляющее собой конструкцию "линкер-полезная нагрузка", формулы IIB:
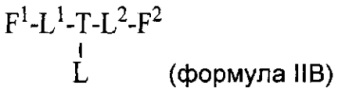
или его фармацевтически приемлемые соль или сольват, где:
каждый из F1 и F2 независимо выбран из кольцевых систем A, B, C и D:


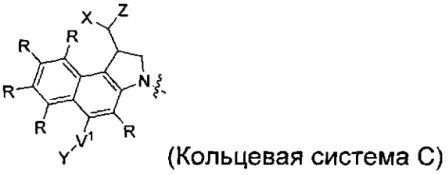

каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C2-C6алкенил, -C2-C6алкинил, -галоген, гидроксил, алкокси, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -NO2, -C6-C14арил и -C6-C14гетероарил, где два или более R возможно соединены с образованием кольца или колец, и где указанные -C6-C14арил и -C6-C14гетероарил возможно замещены 1-5 заместителями, независимо выбранными из групп -C1-C10алкил, -C1-C10алкокси, -галоген, -C1-C10алкилтио, -трифторметил, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -C1-C10алкил-N(C1-C8алкил)2, -C1-C3алкилтио, -NO2 или -C1-C10гетероциклил, для каждой кольцевой системы, в которой имеется R;
каждый V1 независимо представляет собой связь, O, N(R) или S для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O, N(R) или S для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -фенил, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 или -C(O)N(R)2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо выбран из групп -OH, -О-ацил, азидо, галоген, цианат, тиоцианат, изоцианат, тиоизоцианат или 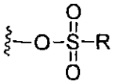 для каждой кольцевой системы, в которой имеется X;
для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из группы, состоящей из H и групп -C1-C6алкил-RA, -C(O)RA, -C(S)RA, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(RA)2, гликозил, -NO2 и -P(O)(ORA)2, для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил и -C1-C20алкилN(R)2, где указанные -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил и -C1-C20алкилN(R)2 возможно замещены 1-3 заместителями, независимо выбранными из R;
каждый Z независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, и где указанные C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, каждый, возможно замещены 1-3 заместителями, независимо выбранными из R, для каждой кольцевой системы, в которой имеется Z;
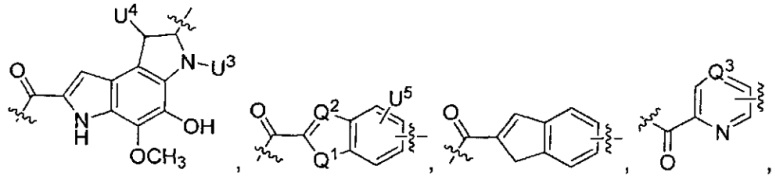
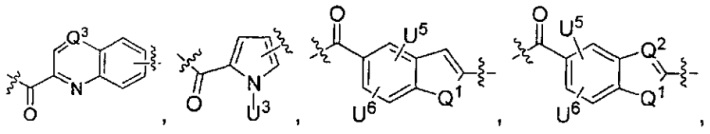
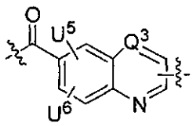
каждый из L1 и L2 независимо выбран из прямой связи, карбонильной или карбонилацильной группы, связанной с F1 или F2 по ацильной группировке, где карбонилацильная группа выбрана из группы, состоящей из:
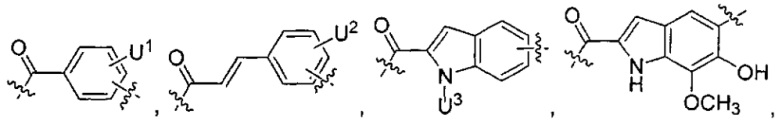
 и
и 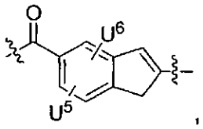 где
где
U1 выбран из H и групп -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)фенил или -галоген,
U2 представляет собой H, -OH или -OCH3,
U3 представляет собой H, -CH3 или -C2H5,
U4 представляет собой H или CH3S-,
каждый из U5 и U6 независимо выбран из H и групп -галоген, -C1-C4алкил, -C1-C3алкокси, -C1-C6диалкиламино, -NO2, -NHC(O)C1-C10алкил, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 или -NHC(O)фенил,
Q1 представляет собой -O-, -S- или -NH-, и
каждый из Q2 и Q3 независимо представляет собой -CH- или -N-;
T выбран из:
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
где каждый X1 независимо представляет собой связь, -NRE-, -O- или -S-, где каждый из A1 и B1 независимо представляет собой =O или =S, где каждый из R1, R2, R3 и R4 независимо представляет собой RE, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, или каждый из R1, R2, R3 и R4 связан с разными атомами углерода на D, где каждый из g и j независимо означает целое число от 0 до 50, и m означает целое число от 1 до 50, и где D выбран из группы, состоящей из групп -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло замещены одним членом группы, выбранным из N(RE)C(O)-, где карбонил связан с L, и -C(O)-, где карбонил связан с L, и дополнительно возможно замещены 1-2 R;
где каждый RE независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2 и -C(O)-галоген, и где каждый RE возможно замещен 1-3 заместителями, независимо выбранными из R;
L представляет собой LA-LB-(LC)1-3;
LA выбран из групп -галоген, -N(R)2, -CON(R)2, -S-арил, возможно замещенный -NO2 или -CON(R)2, -S-гетероарил, возможно замещенный -NO2, алкил-SO2-гетероарил, арилSO2-гетероарил-,
 и
и 
LB представляет собой LB1-LB2-LB3,
где LB1 отсутствует или представляет собой один или более компонентов, выбранных из группы, состоящей из групп -C(O)-, -C(S)-, -C(O)NR-, -C(O)C1-C6алкил-, -C(O)NRC1-C6алкил-, -C1-C6алкил(OCH2CH2)1-6-, -C(O)C1-C6алкилNRC(O)-, -С(O)C1-C6алкил(OCH2CH2)1-6-, -C1-C6алкил(OCH2CH2)1-6-C(O)-, -C1-C6алкил-S-S-C1-C6алкилNRC(O)CH2-, -C1-C6алкил(OCH2CH2)1-6NRC(O)CH2-, -С(O)C1-C6алкил-NRC(O)C1-6алкил-, -N=CR-фенил-O-C1-C6алкил-, -N=CR-фенил-O-C1-C6алкил-C(O)-, -C(O)-C1-C6алкил(OCH2CH2)1-6NRC(O)-, -C(O)C1-C6алкил-фенил(NR-C(O)C1-C6алкил)1-4-, -C(O)C1-C6алкил(OCH2CH2)1-6-NRC(O)C1-C6алкил-, -C1-C6алкил-, -S-, -C(O)-CH(NR-C(O)C1-C6алкил)-C1-C6алкил- и (-CH2-CH2-O-)1-20,
LB2 представляет собой AA0-12, где AA представляет собой природную аминокислоту, неприродную аминокислоту или -(CR15)o-S-S-(CR15)p, где каждый из o и p независимо означает целое число от 1 до 20,
LB3 представляет собой -PABA-, -PABC- или отсутствует;
LC отсутствует или независимо выбран из группы, состоящей из -C1-C6алкилен-, -NRC3-C8-гетероциклилNR-, -NRC3-C8-карбоциклилNR-, -NRC1-C6алкилNR-, -NRC1-C6алкилен-, -S-, -NR-, -NRNR-, -O(CR2)1-4S-S(CR2)1-4N(R)-, -NRC1-C6-алкиленфениленNR-, -NRC1-C6aлкилeнфeнилeнSO2NR-, -OC1-C6алкилS-SC1-C6алкилC(COOR)NR-, -NRC(COOR)C1-C6aлкилS-SC1-C6aлкилO-,
и 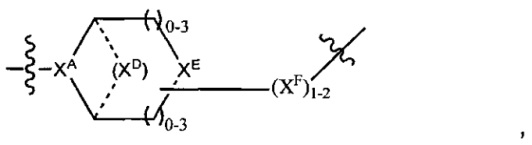
где
XA представляет собой CR или N,
XB представляет собой CH, CR(C(R)2)1-3NR, CR(C(R)2)1-3O, CR(C(R)2)1-3C(O)NR, CR-(C(R)2)1-3C(O)NRNR, CR(C(R)2)1-3SO2NR, CR(C(R)2)1-3NRNR, CR(C(R)2)1-3NRC(O) или N,
каждый XC представляет собой R,
каждый XD представляет собой -(CH2)1-5- или отсутствует;
XE представляет собой O, S, C(R)2, C(R)(C(R)2)1-3-NR2 или NR, и
каждый XF представляет собой (C(R)2)1-3-NR или C(R)2-(C(R)2)1-3-O.
Согласно еще одному другому аспекту изобретения предложено соединение, представляющее собой конъюгат антитело-лекарственное средство, формулы IIIB:
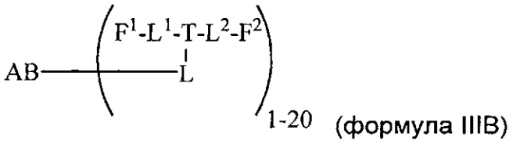
или его фармацевтически приемлемые соль или сольват, где:
AB представляет собой антитело;
каждый из F1 и F2 независимо выбран из кольцевых систем A, B, C и D:


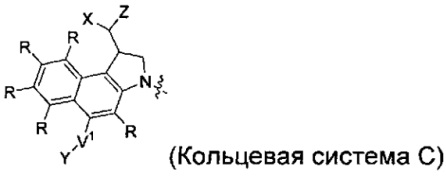
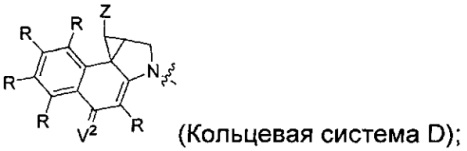
каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C2-C6алкенил, -C2-C6алкинил, -галоген, гидроксил, алкокси, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -NO2, -C6-C14арил и -C6-C14гетероарил, где два или более R возможно соединены с образованием кольца или колец, и где указанные -C6-C14арил и -C6-C14гетероарил возможно замещены 1-5 заместителями, независимо выбранными из групп -C1-C10алкил, -C1-C10алкокси, -галоген, -C1-C10алкилтио, -трифторметил, -NH2, -NH(C1-C8алкил), -N(C1-C8алкил)2, -C1-C10алкил-N(C1-C8алкил)2, -C1-C3алкилтио, -NO2 или -C1-C10гетероциклил, для каждой кольцевой системы, в которой имеется R;
каждый V1 независимо представляет собой связь, O, N(R) или S для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O, N(R) или S для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -фенил, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 или -C(O)N(R)2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо выбран из групп -OH, -O-ацил, азидо, галоген, цианат, тиоцианат, изоцианат, тиоизоцианат или 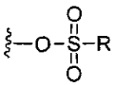 для каждой кольцевой системы, в которой имеется X;
для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из группы, состоящей из H и групп -C1-C6алкил-RA, -C(O)RA, -C(S)RA, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(RA)2, гликозил, -NO2 и -P(O)(ORA)2, для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил и -C1-C20алкилN(R)2, где указанные -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил и -C1-C20aлкилN(R)2 возможно замещены 1-3 заместителями, независимо выбранными из R;
каждый Z независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, и где указанные C1-C8алкил, -C1-C8гетероалкил, -C6-C14арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2, -C(O)OH, -C(O)NHNH2 и -C(O)-галоген, каждый, возможно замещены 1-3 заместителями, независимо выбранными из R, для каждой кольцевой системы, в которой имеется Z;
каждый из L1 и L2 независимо выбран из прямой связи, карбонильной или карбонилацильной группы, связанной с F1 или F2 по ацильной группировке, где карбонилацильная группа выбрана из группы, состоящей из:
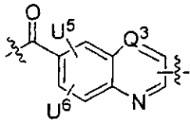 и
и 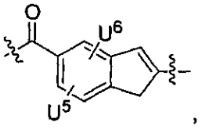 где
где
U1 выбран из H и групп -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)фенил или -галоген,
U2 представляет собой H, -OH или -OCH3,
U3 представляет собой H, -CH3 или -C2H5,
U4 представляет собой H или CH3S-,
каждый из U5 и U6 независимо выбран из H и групп -галоген, -C1-C4алкил, -C1-C3алкокси, -C1-C6диалкиламино, -NO2, -NHC(O)C1-C10алкил, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 или -NHC(O)фенил,
Q1 представляет собой -O-, -S- или -NH-, и
каждый из Q2 и Q3 независимо представляет собой -CH- или -N-;
T выбран из:
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
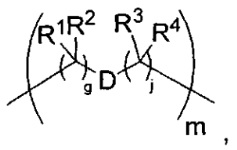
где каждый X1 независимо представляет собой связь, -NRE-, -O- или -S-, где каждый из A1 и B1 независимо представляет собой =O или =S, где каждый из R1, R2, R3 и R4 независимо представляет собой RE, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, или каждый из R1, R2, R3 и R4 связан с разными атомами углерода на D, где каждый из g и j независимо означает целое число от 0 до 50, и m означает целое число от 1 до 50, и где D выбран из группы, состоящей из групп -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C8гетероалкилен-, -аралкилен, -C1-C10гетероцикло и -C3-C8карбоцикло замещены одним членом группы, выбранным из -N(RE)C(O)-, где карбонил связан с L, и -C(O)-, где карбонил связан с L, и дополнительно возможно замещены 1-2 R, и
где каждый RE независимо выбран из группы, состоящей из H и групп -C1-C8алкил, -C1-C8гетероалкил, -арил, -аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C(O)OC1-C8алкил, -C(O)N(C1-C8алкил)2 и -C(O)-галоген, и где каждый RE возможно замещен 1-3 заместителями, независимо выбранными из R;
L представляет собой LA-LB-(LC)1-3;
LA выбран из связи с AB, групп -N(R)-(связь с AB), алкил-SO2-гетероарил, арилSO2-гетероарил-,
 и
и 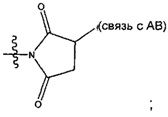
LB представляет собой LB1-LB2-LB3,
где LB1 отсутствует или представляет собой один или более компонентов, выбранных из группы, состоящей из групп -C(O)-, -C(S)-, -C(O)NR-, -C(O)C1-C6алкил-, -C(O)NRC1-C6aлкил-, -C1-C6алкил(OCH2CH2)1-6-, -C(O)C1-C6алкилNRC(O)-, -C(O)C1-C6алкил(OCH2CH2)1-6-, -C1-C6алкил(OCH2CH2)1-6-C(O)-, -C1-C6алкил-S-S-C1-C6алкилNRC(O)CH2-, -C1-C6алкил(OCH2CH2)1-6NRC(O)CH2-, -C(O)C1-C6алкил-NRC(O)C1-6алкил-, -N=CR-фенил-O-C1-C6алкил-, -N=CR-фенил-O-C1-C6алкил-C(O)-, -C(O)-C1-C6алкил(OCH2CH2)1-6NRC(O)-, -C(O)C1-C6алкил-фенил(NR-C(O)C1-C6алкил)1-4-, -C(O)C1-C6алкил(OCH2CH2)1-6-NRC(O)C1-C6алкил-, -C1-C6алкил-, -S-, -C(O)-CH(NR-C(O)C1-C6алкил)-C1-C6алкил- и (-CH2-CH2-O-)1-20,
LB2 представляет собой AA0-12, где AA представляет собой природную аминокислоту, неприродную аминокислоту или -(CR15)o-S-S-(CR15)p, где каждый из o и p независимо означает целое число от 1 до 20,
LB3 представляет собой -PABA-, -PABC- или отсутствует;
LC отсутствует или независимо выбран из группы, состоящей из групп -C1-C6алкилен-, -NRC3-C8-гетероциклилNR-, -NRC3-C8-карбоциклилNR-, -NRC1-C6алкилNR-, -NRC1-C6алкилен-, -S-, -NR-, -NRNR-, -O(CR2)1-4S-S(CR2)1-4N(R)-, -NRC1-C6-алкиленфениленNR-, -NRC1-C6алкиленфениленSO2NR-, -OC1-C6алкилS-SC1-C6алкилC(COOR)NR-, -NRC(COOR)C1-C6алкилS-SC1-C6алкилO-,
и 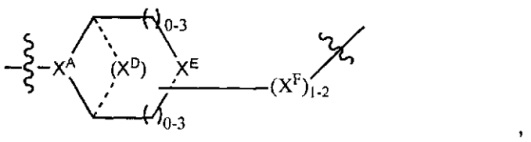
где
XA представляет собой CR или N,
XB представляет собой CH, CR(C(R)2)1-3NR, CR(C(R)2)1-3O, CR(C(R)2)1-3C(O)NR, CR-(C(R)2)1-3C(O)NRNR, CR(C(R)2)1-3SO2NR, CR(C(R)2)1-3NRNR, CR(C(R)2)1-3NRC(O) или N,
каждый XC представляет собой R,
каждый XD представляет собой -(CH2)1-5- или отсутствует;
XE представляет собой O, S, C(R)2, C(R)(C(R)2)1-3-NR2 или NR, и
каждый XF представляет собой (C(R)2)1-3-NR или C(R)2-(C(R)2)1-3-O.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где
каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил и -NH2;
каждый V1 независимо представляет собой O или N(R) для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O или N(R) для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -C(O)OR или -C(O)NR2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо представляет собой галоген для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из группы, состоящей из H и групп -C(O)RA, -C(O)N(RA)2, гликозил, -NO2 и -PO(ORA)2 для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C3-C8карбоциклил и -C1-C20алкилN(R)2, где указанные -C1-C20алкил, -C1-C8гетероалкил, -C3-C8карбоциклил и -C1-C20алкилN(R)2 возможно замещены 1-3 заместителями, независимо выбранными из R;
L1 и L2 каждый из независимо выбран из прямой связи и карбонила; и
T выбран из:
-NRB-T1-NRC-, где каждый из RB и RC независимо представляет собой H или -C1-C8алкил,
-C(O)hetC(O)-, где het представляет собой моноциклический гетероарил из 5-12 членов, содержащий один или два гетероатома, независимо выбранных из O, N и S, где het возможно замещен 1-8 заместителями, каждый из которых независимо выбран из группы, состоящей из групп -C1-C8алкил, -NH2 и -NH2, и указанные возможные заместители на het возможно замещены группой -C1-C8алкил, где по меньшей мере один из F1 и F2 выбран из группы, состоящей из Кольцевой системы C и Кольцевой системы D, когда T представляет собой -C(O)hetC(O)-, и
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
где каждый X1 представляет собой связь, где каждый из A1 и B1 независимо представляет собой =O, где каждый из R1, R2, R3 и R4 независимо представляет собой H, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, и где D представляет собой связь или выбран из группы, состоящей из групп -S-, -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C10гетероцикло и -C3-C8карбоцикло возможно замещены -NH2, -N(R)C(O)H или -N(R)C(O)OH.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где два или более R возможно соединены с образованием кольца или колец.
Дополнительные аспекты изобретения включают такие соединения, как соединения, упомянутые в данном документе, где
каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил и -NH2;
каждый V1 независимо представляет собой O или N(R) для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O или N(R) для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -C(O)OR или -C(O)NR2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо представляет собой галоген для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из связи, H и групп -C(O)RA, -C(S)RA, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(RA)2, гликозил, -NO2 и -P(O)(ORA)2 для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C6-C14арил, аралкил, -C1-C10гетероциклил, -C3-C8карбоциклил, -C1-C20алкилN(R)2, -C1-C20алкилен, -C1-C8гетероалкилен, -C6-C14арилен, аралкилен, -C1-C10гетероцикло, -C3-C8карбоцикло и -C1-C20алкилN(R)- и RF, где указанный RA возможно замещен 1-3 заместителями, независимо выбранными из R, и где один Y является двухвалентным и связан с L,
RF представляет собой -N(R6)QN(R5)C(O)- и связан с L по карбонилу, расположенному рядом с N(R5), где каждый из R5 и R6 независимо выбран из группы, состоящей из H и групп -C1-C8алкил и -C1-C8гетероалкил, или R5 или R6 соединены с замещенным атомом углерода на Q с образованием -C1-C10гетероциклического или -C6-C14гетероарильного кольца, или R5 и R6 соединены вместе с образованием -C1-C10гетероциклической или -C6-C14гетероарильной кольцевой системы, и где Q представляет собой -C1-C8алкилен-, -C6-C14арилен- или -C3-C8карбоцикло-, где каждый из Q, R5 и R6 независимо возможно замещен 1-3 заместителями, независимо выбранными из R;
каждый из L1 и L2 независимо выбран из прямой связи и карбонила; и
T выбран из:
-NRB-T1-NRC-, где каждый из RB и RC независимо представляет собой H или -C1-C8алкил,
-C(O)hetC(O)-, где het представляет собой моноциклический гетероарил из 5-12 членов, содержащий один или два гетероатома, независимо выбранных из O, N и S, где het возможно замещен 1-8 заместителями, каждый из которых независимо выбран из группы, состоящей из групп -C1-C8алкил, -NH2 и -NH2, и указанные возможные заместители на het возможно замещены группой -C1-C8алкил, где по меньшей мере один из F1 и F2 выбран из группы, состоящей из Кольцевой системы C и Кольцевой системы D, когда T представляет собой -C(O)hetC(O)-, и
-C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
где каждый X1 представляет собой связь, где каждый из A1 и B1 независимо представляет собой =O, где каждый из R1, R2, R3 и R4 независимо представляет собой H, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, и где D представляет собой связь или выбран из группы, состоящей из групп -S-, -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C10гетероцикло и -C3-C8карбоцикло возможно замещены -NH2, -N(R)C(O)H или -N(R)C(O)OH.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где
каждый R независимо выбран из группы, состоящей из H и групп -C1-C20алкил и -NH2;
каждый V1 независимо представляет собой O или N(R) для каждой кольцевой системы, в которой имеется V1;
каждый V2 независимо представляет собой O или N(R) для каждой кольцевой системы, в которой имеется V2;
каждый из W1 и W2 независимо представляет собой H, -C1-C5алкил, -C(O)OR или -C(O)NR2 для каждой кольцевой системы, в которой имеются W1 и W2;
каждый X независимо представляет собой галоген для каждой кольцевой системы, в которой имеется X;
каждый Y независимо выбран из группы, состоящей из H и групп -C(O)RA, -C(O)N(RA)2, гликозил, -NO2 и -PO(ORA)2, для каждой кольцевой системы, в которой имеется Y, где каждый RA независимо выбран из группы, состоящей из H и групп -C1-C20алкил, -C1-C8гетероалкил, -C3-C8карбоциклил и -C1-C20алкилN(R)2, где указанные -C1-C20алкил, -C1-C8гетероалкил, -C3-C8карбоциклил и -C1-C20алкилN(R)2 возможно замещены 1-3 заместителями, независимо выбранными из R;
каждый из L1 и L2 независимо выбран из прямой связи и карбонила; и
T представляет собой -C(A1)X1-T2-X1C(B1)-, где T2 представляет собой:
где каждый X1 представляет собой связь, где каждый из A1 и B1 независимо представляет собой =O, где каждый из R1, R2, R3 и R4 независимо представляет собой H, или R1 и R2 образуют кольцевую систему, или R3 и R4 образуют кольцевую систему, или обе пары R1 и R2 и R3 и R4, каждая независимо, образуют кольцевые системы, или R1 и R3 образуют кольцевую систему, или R2 и R4 образуют кольцевую систему, или обе пары R1 и R3 и R2 и R4, каждая независимо, образуют кольцевые системы, где указанные кольцевые системы независимо выбраны из кольцевых систем -C1-C10гетероциклил или -C3-C8карбоциклил, и где D представляет собой связь или выбран из группы, состоящей из групп -S-, -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C10гетероцикло и -C3-C8карбоцикло, где указанные -C1-C8алкилен-, -C6-C14арилен-, -C6-C14гетероарилен-, -C1-C10гетероцикло и -C3-C8карбоцикло возможно замещены -NH2, -N(R)C(O)H или -N(R)C(O)OH.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где
LA выбран из группы, состоящей из групп -галоген, -N(R)2, -CON(R)2, -S-арил, возможно замещенный -NO2 или -CON(R)2, -S-гетероарил, возможно замещенный -NO2, алкил-SO2-гетероарил, арилSO2-гетероарил-, и
LB представляет собой LB1-LB2-LB3, где LB1 отсутствует или представляет собой один или более компонентов, выбранных из группы, состоящей из групп -C(O)-, -C(S)-, -C(O)NR-, -С(O)C1-C6алкил-, -С(O)NRC1-C6алкил-, -C1-C6алкил(OCH2CH2)1-6-, -C(O)C1-C6алкилNRC(O)-, -C(O)C1-C6алкил(OCH2CH2)1-6-, -C1-C6алкил(OCH2CH2)1-6-C(O)-, -C1-C6алкил-S-S-C1-C6алкилNRC(O)CH2-C1-C6алкил(OCH2CH2)1-6NRC(O)CH2-, -C(O)C1-C6алкил-NRC(O)C1-6алкил-, -C(О)-C1-C6алкил(OCH2CH2)1-6NRC(O)-, -C(O)C1-C6алкил-фенил(NR-C(O)C1-C6алкил)1-4-, -С(O)C1-C6алкил(OCH2CH2)1-6-NRC(O)C1-C6алкил-, -C1-C6алкил-, -S-, -С(О)-CH(NR-C(O)C1-C6алкил)-C1-C6алкил- и (-CH2-CH2-O-)1-20, где LB2 представляет собой AA0-12, где AA представляет собой природную аминокислоту, неприродную аминокислоту или -(CR15)o-S-S-(CR15)p, где каждый из o и p независимо означает целое число от 1 до 20, и LB3 представляет собой -PABA-, -PABC- или отсутствует; и
LC отсутствует.
Дополнительные аспекты изобретения включают конъюгаты антитело-лекарственное средство, такие как те, которые упомянуты в данном документе, где LA выбран из связи с AB и групп -NR-(связь с AB), алкил-SO2-гетероарил, арилSO2-гетероарил-,
 и
и 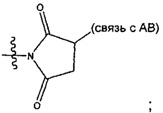
LB представляет собой LB1-LB2-LB3, где LB1 отсутствует или представляет собой один или более компонентов, выбранных из группы, состоящей из групп -C(O)-, -C(S)-, -C(O)NR-, -С(O)C1-C6алкил-, -C(O)NRC1-C6алкил-, -C1-C6алкил(OCH2CH2)1-6-, -C(O)C1-C6алкилNRC(O)-, -C(O)C1-C6алкил(OCH2CH2)1-6-, -C1-C6алкил(OCH2CH2)1-6-C(O)-, -C1-C6алкил-S-S-C1-C6алкилNRC(О)CH2-C1-C6алкил(OCH2CH2)1-6NRC(O)CH2-, -C(O)C1-C6aлкил-NRC(O)C1-6aлкил-, -C(O)-C1-C6алкил(OCH2CH2)1-6NRC(О)-, -С(O)C1-C6алкил-фенил(NR-C(O)C1-C6алкил)1-4-, -C(O)C1-C6алкил(OCH2CH2)1-6-NRC(O)C1-C6алкил-, -C1-C6алкил-, -S-, -C(O)-CH(NR-C(O)C1-C6алкил)-C1-C6алкил- и (-CH2-CH2-О-)1-20, где LB2 представляет собой AA0-12, где AA представляет собой природную аминокислоту, неприродную аминокислоту или -(CR15)o-S-S-(CR15)p где каждый из o и p независимо означает целое число от 1 до 20, и LB3 представляет собой -PABA-, -PABC- или отсутствует; и
LC отсутствует.
Дополнительные аспекты изобретения включают соединения, такие как соединения, которые упомянуты в данном документе, где RF выбран из:
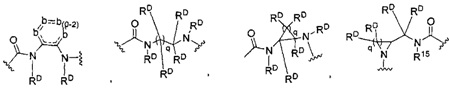 и
и 
где q означает 1-10, и каждый b независимо представляет собой CRD, N, NRD, O или S.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где один или более W представляют собой C1-C3алкил.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где X представляет собой хлор.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где один Y представляет собой H или -C(O)C1-C10алкил.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где один или более Z представляют собой H.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где T выбран из амида или группировки амино-соединительная группа-амино формулы -NH-C(O)-NH- или -NH-C(O)-het-C(O)-NH-.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где амид представляет собой -C(O)NH- или -NHC(O)-.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где het представляет собой гетероарил, выбранный из групп пиррол-2-,5-диил-, фур-2,5-диил-, индол-2,5-диил, бензофуран-2,5-диил и 3,6-дигидробензо[1,2-b:4,3-b]дипиррол-2,7-диил.
Дополнительные аспекты изобретения включают соединения, такие как соединения, упомянутые в данном документе, где L1 и L2 выбраны из карбонила, 2-карбонилиндол-5-ила, 2-карбонил-6-гидрокси-7-метоксииндол-5-ила, 2-карбонил-1,2,3,6-тетрагидробензо[1,2-b:4,3-b]дипиррол-7-ила, 2-карбонил-4-гидрокси-5-метокси-1,2,3,6-тетрагидробензо[1,2-b:4,3-b']дипиррол-7-ила и 2-карбонил-4-гидрокси-5-метокси-1,2,3,6-тетрагидробензо[1,2-b:4,3-b']дипиррол-7-ила.
Дополнительными аспектами изобретения являются соединения, перечисленные в данном документе, где применимо одно или более из следующего: W представляет собой метил; X представляет собой галоген; Y представляет собой водород или -COR, где R представляет собой C1-C10алкил; и Z представляет собой водород.
Изобретение охватывает также соединение, как оно охарактеризовано в данном документе, где T выбран из амида (т.е. -C(O)NH- или -NHC(O)-) или группировки амино-соединительная группа-амино формулы -NH-T'-NH, где T' представляет собой карбонил или -C-(O)-het-C(O)-. Если T представляет собой группировку амино-соединительная группа-амино формулы NH-T'-NH, то T' может представлять собой карбонил (т.е. -C-(O)-) или -C(O)-het-C(O)-, где het представляет собой гетероарил, выбранный из групп пиррол-2-,5-диил-, фур-2,5-диил-, индол-2,5-диил, бензофуран-2,5-диил или 3,6-дигидробензо[1,2-b:4,3-b]дипиррол-2,7-диил.
Также воплощениями изобретения охвачены соединения, которые описаны в данном документе, где L1 и L2 выбраны из 2-карбонилиндол-5-ила, 2-карбонил-6-гидрокси-7-метоксииндол-5-ила, 2-карбонил-1,2,3,6-тетрагидробензо[1,2-b:4,3-b]дипиррол-7-ила, 2-карбонил-4-гидрокси-5-метокси-1,2,3,6-тетрагидробензо[1,2-b:4,3-b']дипиррол-7-ила и 2-карбонил-4-гидрокси-5-метокси-1,2,3,6-тетрагидробензо[1,2-b:4,3-b']дипиррол-7-ила.
Другой аспект изобретения охватывает соединения, которые описаны в данном документе, где LA представляет собой 
Изобретение охватывает также конструкции линкер-полезная нагрузка или конъюгаты антитело-лекарственное средство, содержащие радикал соединений, представляющих собой полезные нагрузки, описанных в данном документе.
Важно, что изобретение охватывает фармацевтические композиции соединений и любых их фармацевтически приемлемых солей или сольватов, описанных в данном документе, где фармацевтическая композиция содержит фармацевтически приемлемый эксципиент.
Изобретение также относится к способам лечения рака, включающим введение нуждающемуся в этом пациенту терапевтически эффективного количества одного или более соединений, описанных в данном документе, или фармацевтической композиции или композиций, содержащих одно или более этих соединений.
Некоторые соединения, включая полезные нагрузки, конструкции линкер-полезная нагрузка и ADC, изображенные в данном документе, показаны в конкретной стереоизомерной форме. Тем не менее, изобретение охватывает все стереоизомерные формы этих соединений. Например, соединение с двумя стереоизомерными центрами может быть изображено как R,S форма соединения, но изобретение предусматривает все стереоизомерные формы, например R,R; R,S; S,R и S,S.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к цитотоксическим бифункциональным соединениям, к конъюгатам антитело-лекарственное средство (ADC), содержащим указанные цитотоксические бифункциональные соединения, и к способам использования таковых для лечения рака и других патологических состояний. Изобретение также относится к способам использования таких соединений и/или конъюгатов in vitro, in situ и in vivo для обнаружения, диагностики или лечения клеток млекопитающего или ассоциированных патологических состояний.
Определения и сокращения
Если конкретно не утверждается иное, приведенные ниже термины и фразы, использованные в данном документе, имеют указанные значения. При использовании товарных наименований в данном документе товарное наименование включает готовый препарат, непатентованное лекарственное средство и активный(ые) фармацевтический(ие) ингредиент(ы) продукта под товарным наименованием, если из контекста не следует иное.
Термин "антитело" (или "Ab") в данном документе использован в самом широком смысле и конкретно охватывает интактные моноклональные антитела, поликлональные антитела, моноспецифические антитела, мультиспецифические антитела (например, биспецифические антитела) и фрагменты антител, которые проявляют желаемую биологическую активность. Интактное антитело имеет в основном две области: вариабельную область и константную область. Вариабельная область связывается и взаимодействует с антигеном-мишенью. Вариабельная область включает в себя участок, определяющий комплементарность (CDR), который распознает и связывается с сайтом специфического связывания на конкретном антигене. Константная область может распознаваться иммунной системой и взаимодействовать с иммунной системой (смотри, например, Janeway et al., 2001, Immuno. Biology, 5th Ed., Garland Publishing, New York). Антитело может быть любого типа или класса (например, IgG, IgE, IgM, IgD и IgA) или подкласса (например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2). Антитело может иметь происхождение из любых подходящих видов. В некоторых воплощениях антитело имеет человеческое или мышиное происхождение. Антитело может быть, например, человеческим, гуманизированным или химерным.
Термины "специфически связывается" и "специфическое связывание" относятся к антителу, связывающемуся с предопределенным антигеном. Обычно антитело связывается с аффинностью по меньшей мере примерно 1×107 М-1 и связывается с предопределенным антигеном с аффинностью, которая по меньшей мере в два раза больше, чем его аффинность к связыванию с неспецифическим антигеном (например, бычьим сывороточным альбумином (BSA), казеином), иным, чем предопределенный антиген или близкородственный антиген.
Термин "моноклональное антитело" в данном документе относится к антителу, полученному из популяции по существу гомогенных антител, т.е. индивидуальные антитела, составляющие популяцию, являются идентичными, за исключением возможных встречающихся в природе мутаций, которые могут присутствовать в незначительных количествах. Моноклональное антитела являются высокоспецифическими, будучи направленными против единственного антигенного сайта. Определение "моноклональное" указывает на характер антитела как полученного из по существу гомогенной популяции антител, и его не следует трактовать как требующее продуцирования антитела любым конкретным способом.
Термин "моноклональные антитела" конкретно охватывает "химерные" антитела, в которых часть тяжелой и/или легкой цепи идентична или гомологична соответствующей последовательности антител, имеющих происхождение из конкретных видов или принадлежащих конкретному классу или подклассу антител, в то время как остальная часть цепи(ей) идентична или гомологична соответствующим последовательностям антител, имеющих происхождение из других видов или принадлежащих другому классу или подклассу антител, а также фрагментов таких антител, при условии, что они проявляют желаемую биологическую активность.
В данном документе "H(C)-" относится к трастузумабу (торговое наименование HERCEPTIN®), который является моноклональным антителом, которое блокирует рецептор HER2/neu, связываясь через один из его цистиновых остатков с соединением по изобретению. В данном документе "H(K)-" относится к трастузумабу, который является моноклональным антителом, которое блокирует рецептор HER2/neu, связываясь через один из его лизиновых остатков с соединением по изобретению.
"Интактное антитело" представляет собой антитело, которое содержит антигенсвязывающую вариабельную область, а также константный домен легкой цепи (CL) и константные домены тяжелой цепи, CH1, CH2, CH3 и CH4, в зависимости от класса антител. Константные домены могут представлять собой константные домены нативной последовательности (например, константные домены человеческой нативной последовательности) или их варианты по аминокислотной последовательности.
Интактное антитело может иметь одну или более "эффекторных функций", которые относятся к биологическим активностям, свойственным Fc-области (например, нативной последовательности Fc-области или варианта аминокислотной последовательности Fc-области) антитела. Примеры эффекторных функций антитела включают комплемент-зависимую цитотоксичность, антитело-зависимую клеточно-опосредованную цитотоксичность (ADCC) и антитело-зависимый клеточно-опосредованный фагоцитоз.
"Фрагмент антитела" содержит часть интактного антитела, предпочтительно содержащую его антигенсвязывающую или вариабельную область. Примеры фрагментов антитела включают фрагменты Fab, Fab', F(ab')2 и Fv, диатела, триатела, тетратела, линейные антитела, одноцепочечные молекулы антител, scFv, scFv-Fc, мультиспецифические фрагменты антитела, образованные из фрагмента(ов) антитела, фрагмент(ы), продуцируемые Fab экспрессионной библиотекой, или эпитоп-связывающие фрагменты любого из вышеуказанных, которые иммуноспецифически связываются с антигеном-мишенью (например, антигеном раковых клеток, вирусным антигеном или микробным антигеном).
Термин "вариабельный" в контексте антитела относится к определенным участкам вариабельных доменов антитела, которые сильно отличаются по последовательности и используются в связывании и специфичности каждого конкретного антитела в отношении его конкретного антигена. Эта вариабельность сконцентрирована в трех сегментах, называемых "гипервариабельными участками" в вариабельных доменах легкой и тяжелой цепи. Более высококонсервативные участки вариабельных доменов называют каркасными областями (FR). Вариабельные домены нативных тяжелой и легкой цепей, каждый, содержат четыре FR, соединенные тремя гипервариабельными участками.
Термин "гипервариабельный участок" в данном документе относится к аминокислотным остаткам антитела, которые ответственны за связывание антигена. Гипервариабельный участок обычно содержит аминокислотные остатки из "участка, определяющего комплементарность" или "CDR" (например, остатки 24-34 (L1), 50-56 (L2) и 89-97 (L3) в вариабельном домене легкой цепи и 31-35 (H1), 50-65 (H2) и 95-102 (L3) в вариабельном домене тяжелой цепи (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)) и/или остатки из "гипервариабельной петли" (например, остатки 26-32 (L1), 50-52 (L2) и 91-96 (L3) в вариабельном домене легкой цепи и 26-32 (Н1), 53-55 (142) и 96-101 (Н3) в вариабельном домене тяжелой цепи (Chothia and Lesk, 1987, J. Mol. Biol. 196:901-917). FR остатки представляют собой те остатки в вариабельном домене, которые являются иными, чем остатки гипервариабельного участка, как определено в данном документе.
"Одноцепочечный фрагмент антитела Fv" или "scFv" содержит V.sub.H и V.sub.L домены антитела, где эти домены присутствуют в единичной полипептидной цепи. Обычно Fv полипептид дополнительно содержит полипептидный линкер между V.sub.H и V.sub.L доменами, который дает возможность scFv образовывать желаемую структуру для связывания антигена. Обзор по scFv смотри в публикации Pluckthun в The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994).
Термин "диатело" относится к небольшим фрагментам антитела с двумя антигенсвязывающими сайтами, и эти фрагменты содержат вариабельный домен тяжелой цепи (VH), соединенный с вариабельным доменом легкой цепи (VL) в одной и той же полипептидной цепи. При использовании линкера, который является слишком коротким, чтобы осуществлялось спаривание двух доменов на одной и той же цепи, домены вынуждены спариваться с комплементарными доменами другой цепи и создавать два антигенсвязывающих сайта. Диатела описаны более полно, например, в EP 0404097; WO 93/11161; и Hollinger et al., 1993, Proc. Natl. Acad. Sci. USA 90:6444-6448.
"Гуманизированные" формы не являющихся человеческими антител (например, грызуна) являются химерными антителами, которые содержат минимальную последовательность, имеющую происхождение из иммуноглобулина, не являющегося человеческим. В большинстве случаев гуманизированные антитела являются человеческими иммуноглобулинами (реципиентное антитело), в которых остатки из гипервариабельного участка реципиента заменены остатками из гипервариабельного участка не являющихся человеческими видов (донорное антитело), таких как мышь, крыса, кролик или не являющийся человеком примат, имеющих желаемую специфичность, аффинность и функциональную способность. В некоторых случаях остатки каркасной области (FR) человеческого иммуноглобулина заменены соответствующими остатками не являющихся человеческими видов. Кроме того, гуманизированные антитела могут содержать остатки, которые не обнаруживаются в реципиентном антителе или в донорном антителе. Эти модификации создают для дополнительного улучшения эффективности антитела. В общем, гуманизированное антитело содержит по существу все из по меньшей мере одного и, как правило, двух вариабельных доменов, в которых все или по существу все гипервариабельные петли соответствуют петлям из иммуноглобулина, не являющегося человеческим, и все или по существу все FR являются FR из последовательности человеческого иммуноглобулина. Гуманизированное антитело возможно также будет содержать по меньшей мере участок константной области иммуноглобулина (Fc), как правило человеческого иммуноглобулина. Дополнительные подробности смотри в Jones et al., 1986, Nature 321:522-525; Riechmann et al., 1988, Nature 332:323-329; и Presta, 1992, Curr. Op. Struct. Biol. 2:593-596.
В данном документе "выделенный" означает отделенный от других компонентов (а) природного источника, такого как растение или клетка или клеточная культура животного, или (б) синтетической органической химической реакционной смеси. В данном документе "очищенный" означает, что выделенный изолят содержит по меньшей мере 95%, а в другом аспекте по меньшей мере 98% соединения (например, конъюгата) от массы изолята.
"Выделенное" антитело является антителом, которое было идентифицировано и выделено и/или извлечено из компонента его естественной среды. Загрязняющие компоненты его естественной среды представляют собой вещества, которые могут препятствовать диагностическому или терапевтическому применению антитела и могут включать ферменты, гормоны и другие белковые или небелковые растворенные вещества. В предпочтительных воплощениях антитело будут очищать (1) до более чем 95% масс. антитела по результатам определения методом Лоури и наиболее предпочтительно более чем 99% масс., (2) до степени, достаточной для получения по меньшей мере 15 остатков N-концевой или внутренней аминокислотной последовательности с использованием секвенатора с вращающимся стаканом, или (3) до гомогенности по результатам анализа методом SDS-PAGE (электрофорез на полиакриламиде с додецилсульфатом натрия) в восстановительных или невосстановительных условиях с использованием красителя кумасси синего или, предпочтительно, серебрянки. Выделенное антитело включает антитело in situ в рекомбинантных клетках, поскольку по меньшей мере один компонент естественной среды антитела не будет присутствовать. Однако обычно выделенное антитело будет получено в результате осуществления по меньшей мере одной стадии очистки.
Антитело, которое "индуцирует апоптоз", представляет собой антитело, которое индуцирует запрограммированную клеточную гибель, что определяют посредством связывания аннексина V, фрагментации ДНК, сжатия клетки, расширения эндоплазматического ретикулума, клеточной фрагментации и/или образования мембранных везикул (называемых апоптотическими тельцами). Клетка представляет собой опухолевую клетку, например молочной железы, яичника, желудка, эндометрия, слюнной железы, легкого, почки, толстой кишки, щитовидной железы, поджелудочной железы или мочевого пузыря. Для оценки клеточных событий, ассоциированных с апоптозом, доступны различные методы. Например, перенос фосфатидилсерина (PS) может быть измерен посредством связывания аннексина; фрагментация ДНК может быть оценена посредством электрофоретического расщепления ДНК; и конденсация ядра/хроматина наряду с фрагментацией ДНК может быть оценена по любому увеличению гиподиплоидных клеток.
Термин "терапевтически эффективное количество" относится к количеству лекарственного средства, эффективному для лечения заболевания или расстройства у млекопитающего. В случае рака терапевтически эффективное количество лекарственного средства может сокращать число раковых клеток; сокращать размер опухоли; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) метастазирование опухоли; ингибировать до некоторой степени рост опухоли; и/или ослаблять до некоторой степени один или более симптомов, ассоциированных с раком. В той степени, в какой лекарственное средство может ингибировать рост существующих раковых клеток и/или уничтожать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим. Для терапии рака эффективность может быть измерена, например, путем оценки времени прогрессирования заболевания (TTP) и/или определения скорости ответной реакции (RR).
Термин "значительное количество" относится к большей части, т.е. больше 50% популяции, смеси или образца.
Термин "внутриклеточный метаболит" относится к соединению, образующемуся в результате метаболического процесса или реакции внутри клетки на конъюгате антитело-лекарственное средство (ADC). Метаболический процесс или реакция может представлять собой ферментативный процесс, такой как протеолитическое расщепление пептидного линкера ADC. Внутриклеточные метаболиты включают, без ограничения, антитела и свободное лекарственное средство, которые подверглись внутриклеточному расщеплению после вхождения, диффузии, захвата или транспорта в клетку.
Термины "внутриклеточное расщепленное" и "внутриклеточное расщепление" относится к метаболическому процессу или реакции внутри клетки на ADC или тому подобному, в результате которого(ой) ковалентное присоединение, например линкера между группировкой лекарственного средства и антителом, разрывается с образованием в результате свободного лекарственного средства или другого метаболита конъюгата, диссоциированного из антитела внутри клетки. Расщепленные группировки ADC являются, таким образом, внутриклеточными метаболитами.
Термин "биодоступность" относится к системной доступности (т.е. к уровням в крови/плазме крови) данного количества лекарственного средства, введенного пациенту. Биодоступность является абсолютным термином, который означает показатель как времени (скорость), так и общего количества (степень) лекарственного средства, которое поступает в общий (системный) кровоток из введенной лекарственной формы.
Термин "цитотоксическая активность" относится к уничтожению клеток, цитостатическому или антипролиферативному эффекту ADC или внутриклеточного метаболита указанного ADC. Цитотоксическая активность может быть выражена в виде значения IC50, которое представляет собой концентрацию (молярную или массовую) на единицу объема, при которой выживает половина клеток.
"Расстройство" представляет собой любое состояние, которое может получать пользу от лечения лекарственным средством или конъюгатом антитело-лекарственное средство. Этот термин охватывает хронические и острые расстройства или заболевания, включая те патологические состояния, которые предрасполагают животного к рассматриваемому расстройству. Не являющиеся ограничивающими примеры расстройств, которые лечат, в данном документе включают доброкачественные и злокачественные новообразования; лейкоз и лимфоидные злокачественности, нейрональные, глиальные, астроцитальные, гипоталамические и другие грандулярные, макрофагальные, эпителиальные, стромальные и бластоцелевые расстройства; и воспалительные, ангиогенные и иммунологические расстройства.
Термины "рак" и "раковый" означают или описывают физиологическое состояние или расстройство у млекопитающих, которое обычно характеризуется нерегулируемым ростом клеток. "Опухоль" содержит одну или более раковых клеток.
Примеры "пациента" включают, без ограничения, человека, крысу, мышь, морскую свинку, обезьяну, свинью, козу, корову, лошадь, собаку, кошку, птицу и дичь. В типичном воплощении пациентом является человек.
Термины "лечить" или "лечение", если из контекста не следует иное, относятся к терапевтическому лечению и профилактическим мерам предупреждения рецидива, целью которых является ингибирование или замедление (уменьшение) нежелательного физиологического изменения или расстройства, такого как развитие или распространение рака. Для целей этого изобретения полезные или желаемые клинические результаты включают, без ограничения, ослабление симптомов, уменьшение степени заболевания, стабилизацию (т.е. не ухудшение) состояния заболевания, задержку или замедление прогрессирования заболевания, ослабление или временное облегчение болезненного состояния и ремиссию (частичную или полную), обнаружимые или не обнаружимые. "Лечение" может также означать продление жизни по сравнению с ожидаемым сроком жизни в отсутствие лечения. Лица, нуждающиеся в лечении, включают те лица, которые уже имеют состояние или расстройство, а также те лица, которые предрасположены к тому, чтобы иметь состояние или расстройство.
В контексте рака термин "лечение" охватывает любое или все из следующего: ингибирование роста опухолевых клеток, раковых клеток или опухоли, ингибирование репликации опухолевых клеток или раковых клеток, сокращение общей опухолевой нагрузки или снижение количества раковых клеток и ослабление одного или более симптомов, связанных с заболеванием.
В контексте аутоиммунного заболевания термин "лечение" охватывает любое или все из следующего: ингибирование репликации клеток, связанной с аутоиммунным заболеванием, включая, без ограничения, клетки, которые продуцируют аутоиммунное антитело, сокращение аутоиммунного-антительного бремени и ослабление одного или более симптомов аутоиммунного заболевания.
В контексте инфекционного заболевания термин "лечение" охватывает любое или все из следующего: ингибирование роста, размножения или репликации патогена, который вызывает инфекционное заболевание, и ослабление одного или более симптомов инфекционного заболевания.
Использованный термин "листовка-вкладыш в упаковке" относится к инструкциям, которые обычно вложены в промышленные упаковки терапевтических продуктов, содержащим информацию о показании (показаниях), использованию, дозировке, введению, противопоказаниям и/или предостережениям, относящимся к применению таких терапевтических продуктов.
Использованные в данном документе термины "клетка", "линия клеток" и "культура клеток" использованы взаимозаменяемым образом, и все такие выражения включают потомство. Слова "трансформанты" и "трансформированные клетки" включают подвергнутую воздействию первичную клетку и культуры или потомство, получаемые из них, независимо от количества пересевов. Понятно также, что все потомство может не быть абсолютно идентичным по содержанию ДНК из-за преднамеренных или случайных мутаций. Мутантное потомство, которое имеет такую же функцию или биологическую активность, проверенную в результате скрининга у первоначально трансформированной клетки, также охвачено. Из контекста будет ясно, если предполагаются особые указания.
В данном документе CBI относится к 1,2,9,9а-тетрагидро-4H-бензо[е]циклопропа[с]индол-4-ону или его замещенной или дериватизированной форме. CBI может также относится к секо-форме CBI, или секо-CBI, которая также известна как 1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ол, или к ее замещенной или дериватизированной форме (или формам).
В данном документе CPI относится к 1,2,8,8а-тетрагидроциклопропа[с]пирроло[3,2-е]индол-4(5H)-ону или его замещенной или дериватизированной форме. CPI может также относится к секо-форме CPI, или секо-CPI, которая также известна как 8-(хлорметил)-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ол, или к его замещенной или дериватизированной форме (или формам).
Если не указано иное, термин "алкил", сам по себе или как часть другого термина, относится к прямоцепочечному или разветвленному насыщенному углеводороду, имеющему указанное количество атомов углерода (например, "C1-C8алкил" относится алкильной группе, имеющей от 1 до 8 атомов углерода). Алкильные группы обычно содержат от 1 до 20 атомов углерода, предпочтительно от 1 до 8 атомов углерода и более предпочтительно от 1 до 4 атомов углерода. Когда количество атомов углерода не указано, тогда алкильная группа имеет от 1 до 8 атомов углерода. Репрезентативные прямоцепочечные C1-C8алкилы включают, без ограничения, метил, этил, н-пропил, н-бутил, н-пентил, н-гексил, н-гептил и н-октил; разветвленные C1-C8алкилы включают, без ограничения, -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил и -2-метилбутил; незамещенные C2-C8алкилы включают, без ограничения, винил, аллил, 1-бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил, 1-гексил, 2-гексил, 3-гексил, ацетиленил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил и 3-метил-1-бутинил. Ссылка на "алкил" в данном документе относится к незамещенным и замещенным группировкам, которые описаны выше.
Если не указано иное, "алкилен", сам по себе или как часть другого термина, относится к насыщенному разветвленному или прямоцепочечному циклическому углеводородному радикалу с указанным количеством атомов углерода, обычно 1-18 атомов углерода, имеющему два центра одновалентных радикалов, получаемых в результате удаления двух атомов водорода с одного и того же или двух разных атомов углерода родительского алкана. Алкиленовые группы обычно содержат от 1 до 18 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 8 атомов углерода и наиболее предпочтительно от 1 до 4 атомов углерода. Типичные алкиленовые радикалы включают, без ограничения, метилен (-CH2-), 1,2-этилен (-CH2CH2-), 1,3-пропилен (-CH2CH2CH2-), 1,4-бутилен (-CH2CH2CH2CH2-) и т.п. "C1-C10" прямоцепочечный алкилен представляет собой прямоцепочечную насыщенную углеводородную группу формулы -(CH2)1-10-. Примеры C1-C10алкилена включают метилен, этилен, пропилен, бутилен, пентилен, гексилен, гептилен, октилен, нонилен и декален. Ссылка на "алкилен" в данном документе относится к незамещенным и замещенным группировкам, которые описаны выше.
Если не указано иное, термин "гетероалкил", сам по себе или в комбинации с другим термином, означает, если не указано иное, стабильный углеводород с прямой или разветвленной цепью, или его комбинации, полностью насыщенный или содержащий от 1 до 3 степеней ненасыщения, состоящий из указанного количества атомов углерода и одного-трех гетероатомов, выбранных из группы, состоящей из O, N, Si и S, и где атомы азота и серы возможно могут быть окисленными, и гетероатом азота возможно может быть кватернизированным. Гетероатом(ы) O, N и S могут располагаться в любом внутреннем положении гетероалкильной группы. Гетероатом Si может располагаться в любом внутреннем положении гетероалкильной группы, включая положение, по которому алкильная группа присоединена к остальной части молекулы. Вплоть до двух гетероатомов могут следовать друг за другом. Гетероалкильные группы обычно содержат от 1 до 15 атомов углерода, предпочтительно от 1 до 12 атомов углерода, более предпочтительно от 1 до 8 атомов углерода и наиболее предпочтительно от 1 до 4 атомов углерода. Ссылка на "гетероалкил" в данном документе относится к незамещенным и замещенным группировкам, которые описаны выше.
Если не указано иное, термин "гетероалкилен", сам по себе или как часть другого заместителя, означает двухвалентную группу, производную от гетероалкила (который описан выше). В гетероалкиленовых группах гетероатомы могут также занимать любой или оба конца цепи. Ссылка на "гетероалкилен" в данном документе относится к незамещенным и замещенным группировкам, которые описаны выше.
Если не указано иное, "арил", сам по себе или как часто другого термина, означает замещенный или незамещенный одновалентный карбоциклический ароматический углеводородный радикал из 5-20, предпочтительно 5-14 или 6-14 атомов углерода, получаемый в результате удаления одного атома водорода с одного атома углерода родительской ароматической кольцевой системы. Типичные арильные группы включают, без ограничения, радикалы бензола, замещенного бензола, нафталина, антрацена, бифенила и т.п. Замещенная карбоцикличекая ароматическая группа (например, арильная группа) может быть замещена одной или более, предпочтительно 1-5, следующими группами: C1-C8алкил, -O-(C1-C8алкил), -C(O)R9, -OC(O)R9, -C(O)OR9, -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(O)2R', -S(O)R', -OH, галоген, -N3, -NH2, -NH(R9), -N(R9)2 и -CN; где каждый R9 независимо выбран из -H, C1-C8алкила и незамещенного арила. В некоторых воплощениях замещенная карбоциклическая ароматическая группа дополнительно может включать одну или более из следующих групп: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R9 и -SR9. "Арилен" представляет собой соответствующую двухвалентную группировку.
"Замещенный алкил" (или "замещенный алкилен", "замещенный гетероалкил" или "замещенный гетероалкилен") означает релевантную алкильную алкил-содержащую группу или радикал, которые описаны выше, в которой(ом) один или более атомов водорода, каждый независимо, замещены заместителем. Типичные заместители включают, без ограничения, -X, -R10, -O-, -OR10, -SR10, -S-, -NR102, -NR103, =NR10, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NR10C(=O)R10R10, -C(=O)NR102, -SO3-, -SO3H, -S(=O)2R10, -OS(=O)2OR10, -S(=O)2NR10, -S(=O)R10, -OP(=O)(OR10)2, -P(=O)(OR10)2, -PO32-, PO3H2, -AsO2H2, -C(=O)R10, -C(=O)X, -C(=S)R10, -CO2R10, -CO2-, -C(=S)OR10, -C(=O)SR10, -C(=S)SR10, -C(=O)NR102, -C(=S)NR102 или -C(=NR10)NR102, где каждый X независимо представляет собой галоген: -F, -Cl, -Br или -I; и каждый R10 независимо представляет собой -H, C1-C20алкил, C1-C20гетероалкил, C6-C20арил, C1-C10гетероциклил, защитную группу или пролекарственную группировку. Арильные, алкиленовые и гетероалкиленовые группы, которые описаны выше, также могут быть замещены подобным образом.
Если не указано иное, "аралкил", сам по себе или как часть другого термина, означает алкильную группу, которая определена выше, замещенную арильной группой, которая определена выше.
Если не указано иное, "C3-C10гетероциклил", сам или как часть другого термина, относится к одновалентной замещенной или незамещенной ароматической или неароматической моноциклической, бициклической или трициклической кольцевой системе, имеющей от 2 до 10, от 2 до 14 или 2-20 атомов углерода, предпочтительно от 3 до 8 атомов углерода (упоминаемых также как члены кольца) и от одного до четырех гетероатомов в качестве членов кольца, независимо выбранных из N, O, P или S, и получаемый в результате удаления одного атома водорода с кольцевого атома родительской кольцевой системы. Один или более атомов N, C или S в гетероциклиле могут быть окисленными. Кольцо, которое содержит гетероатомы, может быть ароматическим или неароматическим. Ароматические гетероциклы иногда упоминаются в данном документе как гетероарилы. Если не указанное иное, гетероциклил присоединен к его боковой группе по любому гетероатому или атому углерода, который обеспечивает образование стабильной структуры. Репрезентативные примеры C2-C10гетероциклила включают, без ограничения, тетрагидрофуранил, оксетанил, пиранил, пирролидинил, пиперидинил, пиперазинил, бензофуранил, бензотиофен, бензотиазолил, индолил, бензопиразолил, пирролил, тиофенил (тиофен), фуранил, тиазолил, имидазолил, пиразолил, триазолил, хинолинил, включая такие группировки, как 1,2,3,4-тетрагидро-хинолинил, пиримидинил, пиридинил, пиридонил, пиразинил, пиридазинил, изотиазолил, изоксазолил, тетразолил, эпоксид, оксетан и BODIPY (бор-дипиррометен) (замещенный или незамещенный). C2-C10гетероциклил может быть замещен группами в количестве вплоть до семи, включающими, без ограничения, C1-C8алкил, C1C8гетероалкил, -OR11, арил, -C(O)R11, -OC(O)R11, -C(O)OR11, -C(O)NH2, -C(O)NHR11, -C(O)N(R11)2, -NHC(O)R11, -S(=O)2R11, -S(O)R11, галоген, -N3, -NH2, -NH(R11), -N(R11)2 и -CN; где каждый R11 независимо выбран из -H, C1-C8алкила, C1-C8гетероалкила и арила. В некоторых воплощениях замещенный гетероциклил может также охватывать одну или более из следующих групп: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R11 и -SR11. Гетероцикло или C2-C10гетероцикло представляет собой соответствующую двухвалентную группировку. Двухвалентные ароматические гетероциклы иногда упоминаются в данном документе как гетероарилен или C2-C10гетероарилен.
Как указано выше, ароматические гетероциклы иногда упоминаются в данном документе как гетероарилы и предпочтительно содержат 5-14, 6-14 или 6-20 атомов углерода в дополнение к гетероатомам. Гетероарилы могут представлять собой моноциклические, бициклические или трициклические кольцевые системы. Репрезентативные гетероарилы включают, без ограничения, триазолил, тетразолил, оксадиазолил, пиридил, фурил, бензофуранил, тиофенил, бензотиофенил, хинолинил, пирролил, индолил, оксазолил, бензоксазолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изоксазолил, пиразолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, хиназолинил, пиримидил, азепинил, оксепинил и хиноксалинил. Гетероарилы возможно замещены. Типичные заместители включают, без ограничения, -X, -Rh, -O-, -ORh, -SRh, -S-, -NRh2, -NRh3, =NRh, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NRhC(=O)Rh, -C(=O)NRh2, -SO3-, -SO3H, -S(=O)2Rh, -OS(=O)2ORh, -S(=O)2NRh, -S(=O)Rh, -OP(=O)(ORh)2, -P(=O)(ORh)2, -PO32-, PO3H2, -AsO2H2, -C(=O)Rh, -C(=O)X, -C(=S)Rh, -CO2Rh, -CO2-, -C(=S)ORh, -C(=O)SRh, -C(=S)SRh, -C(=O)NRh2, -C(=S)NRh2, -C(=NR)NRh2, C1-C20-гетероалкил, C6-C20арил, C3-C8гетероциклил защитную группу или пролекарственную группировку, где каждый X независимо представляет собой галоген: -F, -Cl, -Br или -I; и каждый Rh независимо представляет собой -H или C1-C6алкил. Двухвалентные ароматические гетероциклы иногда упоминаются в данном документе как гетероарилены или C1-C10гетероарилены.
Если не указано иное, "гетероаралкил", сам по себе или как часть другого термина, означает алкильную группу, которая определена выше, замещенную ароматической гетероциклильной группой, которая определена выше. Гетероаралкило представляет собой соответствующую двухвалентную группировку.
Если не указано иное, "C3-C8карбоциклил", сам по себе или как часть другого термина, представляет собой 3-, 4-, 5-, 6-, 7- или 8-членное одновалентное, замещенное или незамещенное, насыщенное или ненасыщенное неароматическое моноциклическое или бициклическое карбоциклическое кольцо, получаемое в результате удаления одного атома водорода с кольцевого атома родительской кольцевой системы. Репрезентативный C3-C8карбоциклил включает, без ограничения, циклопропил, циклобутил, циклопентил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, 1,4-циклогексадиенил, циклогептил, 1,3-циклогептадиенил, 1,3,5-циклогептатриенил, циклооктил, циклооктадиенил, бицикло(1.1.1.)пентан и бицикло(2.2.2.)октан. C3-C8карбоциклильная группа может быть незамещенной или может быть замещенной группами в количестве вплоть до семи, включающими, без ограничения, C1-C8алкил, C1-C8гетероалкил, -OR11, арил, -C(O)R11, -OC(O)R11, -C(O)OR11, -C(O)NH2, -C(O)NHR11, -C(O)N(R11)2, -NHC(O)R11, -S(=O)2R11, -S(=O)R11, -OH, -галоген, -N3, -NH2, -NH(R11), -N(R11)2 и -CN; где каждый R11 независимо выбран из -H, C1-C8алкила, C1-C8гетероалкила и арила. "C3-C8карбоцикло" представляет собой соответствующую двухвалентную группировку.
В данном документе заместитель "азидо" относится к -N=N=N; заместитель "цианат" относится к -O-CN; заместитель "тиоцианат" относится к -S-CN; заместитель "изоцианат" относится к -N=C=O; и заместитель "тиоизоцианат" относится к -S-N=C=O.
Термин "хиральный" относится к молекулам, которые обладают свойством несовмещения со своим зеркальным отражением, тогда как термин "ахиральный" относится к молекулам, которые совмещаются со своим зеркальным отражением.
Термин "стереоизомеры" относится к соединениям, которые имеют идентичное химическое строение, но отличаются расположением атомов или групп в пространстве.
"Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, и его молекулы не являются зеркальными отражениями друг друга. Диастереомеры имеют разные физические свойства, например точки плавления, точки кипения, спектральные свойства и реакционную способность. Смеси диастереомеров могут быть разделены аналитическими методами высокого разрешения, такими как электрофорез и хроматография.
"Гликозил" относится к структуре:
 или ее замещенным формам, включая, например, указанную структуру, замещенную с образованием таких структур, как:
или ее замещенным формам, включая, например, указанную структуру, замещенную с образованием таких структур, как:
 и
и  и многие другие.
и многие другие.
Стереохимические определения и условные обозначения, использованные в данном документе, как правило, соответствуют изложенным в S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms, McGraw-Hill Book Company, New York (1984); и Eliel and Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York (1994). Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоско-поляризованного света. При описании оптически активного соединения префиксы D и L или R и S используют для обозначения абсолютной конфигурации молекулы вокруг ее хирального(ых) центра(ов). Префиксы d и l или (+) и (-) используют для обозначения знака вращения плоско-поляризованного света соединением, причем (-) или l означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры идентичны, за исключением того, что они являются зеркальными отражениями друг друга. Конкретный стереоизомер может также называться энантиомером, и смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров 50:50 называется рацемической смесью или рацематом, которая(ый) может существовать в отсутствии стереоселективности или стереоспецифичности в химической реакции или химическом процессе. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомеров, не имеющей оптической активности.
В данном документе "-PABA-" или "PABA" относится к пара-аминобензойной кислоте и производным от нее группировкам, например к структуре:
или ее вариантам.
В данном документе "-PABC-" или "PABC" относится к пара-аминобензилкарбонилу и производным от него группировкам, например к структуре:
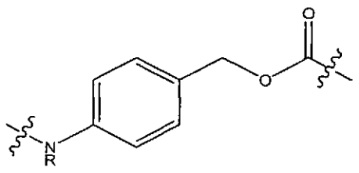
или ее вариантам.
"Производное" аминокислоты охватывает аминокислоту, имеющую заместители или модификации в результате ковалентного присоединения родительской аминокислоты, например в результате алкилирования, гликозилирования, ацетилирования, фосфорилирования и т.п. В определение "производного" также входят, например, один или более аналогов аминокислоты с замещенными связями, а также другие модификации, известные в данной области.
"Природная аминокислота" относится к аргинину, глутамину, фенилаланину, тирозину, триптофану, лизину, глицину, аланину, гистидину, серину, пролину, глутаминовой кислоте, аспарагиновой кислоте, треонину, цистеину, метионину, лейцину, аспарагину, изолейцину и валину, если из контекста не следует иное.
"Защитная группа" относится к группировке, которая, когда она присоединена к реакционноспособной группе в молекуле, маскирует, снижает или предотвращает эту реакционную способность. Примеры защитных групп можно найти в Т.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York, 1999, и в Harrison and Harrison et al., Compendium of Synthetic Organic Methods, Vols. 1-8 (John Wiley and Sons, 1971-1996), которые во всей их полноте включены в данное описание посредством ссылки. Репрезентативные защитные группы гидроксила включают ацильные группы, бензиловые и тритиловые эфиры, тетрагидропираниловые эфиры, триалкилсилиловые эфиры и аллиловые эфиры. Репрезентативные защитные группы аминогруппы включают формил, ацетил, трифторацетил, бензил, бензилоксикарбонил (CBZ), трет-бутоксикарбонил (Boc), триметилсилил (TMS), 2-триметилсилил-этансульфонил (SES), тритил и замещенные тритильные группы, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (FMOC), нитро-вератрилоксикарбонил (NVOC) и тому подобное.
Примеры "защитной группы гидроксила" включают, без ограничения, метоксиметиловый эфир, 2-метоксиэтоксиметиловый эфир, тетрагидропираниловый эфир, бензиловый эфир, пара-метоксибензиловый эфир, триметилсилиловый эфир, триэтилсилиловый эфир, триизопропилсилиловый эфир, трет-бутилдиметилсилиловый эфир, трифенилметилсилиловый эфир, эфир ацетат, замещенные ацетатные эфиры, пивалоат, бензоат, метансульфонат и пара-толуолсульфонат.
"Уходящая группа" относится к функциональной группе, которая может быть замещена другой функциональной группой. Такие уходящие группы общеизвестны в данной области, и примеры включают, без ограничения, галогенид (например, хлорид, бромид, йодид), метансульфонил (мезил), пара-толуолсульфонил (тозил), трифторметилсульфонил (трифлат) и трифторметилсульфонат.
Фраза "фармацевтически приемлемая соль", используемая в данном документе, относится к фармацевтически приемлемым органическим или неорганическим солям соединения. Соединение обычно содержит по меньшей мере одну аминогруппу, и, соответственно, с этой аминогруппой могут быть образованы соли присоединения кислоты. Типичные соли включают, без ограничения, соли сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, малат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может заключать в себе включение другой молекулы, такой как ацетатный ион, сукцинатный ион или другой противоион. Противоион может представлять собой органическую или неорганическую группировку, которая стабилизирует заряд на родительском соединении. Кроме того, фармацевтически приемлемая соль может иметь более чем один заряженный атом в его структуре. В случаях, когда множественные заряженные атомы являются частью фармацевтически приемлемой соли, соль может иметь множественные противоионы. Следовательно, фармацевтически приемлемая соль может иметь один или более заряженных атомов и/или один или более противоионов.
Термины "фармацевтически приемлемый сольват" или "сольват" относятся к ассоциации одной или более молекул растворителя и соединения или конъюгата по изобретению. Примеры растворителей, которые образуют фармацевтически приемлемые сольваты, включают, без ограничения, воду, изопропанол, этанол, метанол, DMSO, этилацетат, уксусную кислоту и этаноламин.
Термины "нагрузка", или "лекарственная нагрузка", или "полезная нагрузка" означают или относятся к среднему количеству полезных нагрузок (в данном документе "полезная нагрузка" и "полезные нагрузки" использованы взаимозаменяемым образом с "лекарственным средством" и "лекарственными средствами") на антитело в молекуле ADC. Лекарственная нагрузка может находиться в диапазоне от 1 до 20 лекарственных средств на антитело. Это иногда упоминается как DAR, или отношение лекарственного средства к антителу. Композиции ADC, описанные в данном документе, обычно имеют DAR от 1 до 20 и в некоторых воплощениях от 1 до 8, от 2 до 8, от 2 до 6, от 2 до 5 и от 2 до 4. Типичные значения DAR составляют 2, 4, 6 и 8. Среднее количество лекарственных средств на антитело, или значение DAR, может быть определено стандартными методами, такими как спектроскопия в УФ/видимой области, масс-спектрометрия, ELISA (твердофазный иммуноферментный анализ) и ВЭЖХ (высокоэффективная жидкостная хроматография). Количественный показатель DAR также может быть определен. В некоторых случаях разделение, очистка и определение характеристик гомогенных ADC, имеющих конкретное значение DAR, может быть осуществлено такими методами, как обращенно-фазная ВЭЖХ или электрофорез. Значение DAR может быть ограничено количеством сайтов присоединения на антителе. Например, в тех случаях, когда сайтом присоединения является цистеиновый тиол, тогда антитело может иметь только одну или несколько цистеиновых тиольных групп, или может иметь только одну или несколько достаточно реакционноспособных тиольных групп, через которые может быть присоединена линкерная единица. В некоторых воплощениях цистеиновый тиол представляет собой тиольную группу цистеинового остатка, которая образует межцепочечную дисульфидную связь. В некоторых воплощениях цистеиновый тиол представляет собой тиольную группу цистеинового остатка, которая не образует межцепочечную дисульфидную связь. В типичных случаях, во время реакции конъюгирования с антителом конъюгируется меньше чем теоретический максимум группировок лекарственных средств. Антитело может содержать, например, много остатков лизина, которые не взаимодействуют с линкером или линкерным промежуточным соединением. Только наиболее реакционноспособные группы лизина могут взаимодействовать с реакционноспособным линкерным реагентом.
Как правило, антитела не содержат много, если и содержат, свободных реакционноспособных цистеиновых тиолов, которые могут быть связаны с лекарственным средством посредством линкера. Большинство тиольных групп цистеиновых остатков в антителах существуют в виде дисульфидных мостиков и должны быть восстановлены восстановителем, таким как дитиотрейтол (DTT). Антитело может быть подвергнуто воздействию денатурирующих условий для высвобождения реакционноспособных нуклеофильных групп, таких как лизин или цистеин. Нагрузку (соотношение лекарственное средство/антитело) ADC можно контролировать несколькими разными способами, включая: (1) ограничение молярного избытка конструкции лекарственное средство-линкер относительно антитела, (2) ограничение времени или температуры реакции конъюгирования, и (3) частичные или ограничивающие восстановительные условия для модификации тиольной группы цистеина. Когда более чем одна нуклеофильная группа взаимодействует с конструкцией лекарственное средство-линкер, тогда полученный продукт представляет собой смесь ADC с распределением одна или более группировок лекарственных средств на антитело. Среднее количество лекарственных средств на антитело может быть рассчитано из смеси, например двойным ELISA-антитело анализом, специфичным в отношении антитела и специфичным в отношении лекарственного средства. Индивидуальные ADC могут быть идентифицированы в смеси посредством масс-спектроскопии и разделены посредством ВЭЖХ, например хроматографии гидрофобного взаимодействия.
Ниже приведен список сокращений и определений, которые не могут быть иным образом определены или описаны в этой заявке: DMSO (означает диметилсульфоксид), МСВР (означает масс-спектрометрия высокого разрешения), ДДМ (означает детектор на диодной матрице), TFA (означает 2,2,2-трифторуксусная кислота или трифторуксусная кислота), TFF (означает фильтрация в тангенциальном потоке), EtOH (означает этанол), М.м. (означает молекулярная масса), ВЭЖХ (означает высокоэффективная жидкостная хроматография), преп. ВЭЖХ (означает препаративная высокоэффективная жидкостная хроматография), т.д. (означает и так далее), тритил (означает 1,1',1'-этан-1,1,1-триилтрибензол), THF (означает тетрагидрофуран), NHS (означает 1-гидрокси-2,5-пирролидиндион), Cbz (означает карбоксибензил), экв. (означает эквивалент), n-BuLi (означает н-бутиллитий), OAc (означает ацетат), MeOH (означает метанол), i-Pr (означает изопропил или пропан-2-ил), NMM (означает 4-метилморфолин), и "-" в таблице означает, что на данный момент нет данных.
Двухвалентные группировки и заместители, использованные в данном документе, относятся к указанным группировкам или заместителям, связанным или соединенным в любом направлении или в обоих направлениях. Например, группировка -C(O)NR- (в определении LB1 и где-либо в другом месте) означает как -C(O)NR-, так и -NRC(O)-; группировка -C(O)C1-C6алкил- означает как -С(O)C1-C6алкил-, так и -C1-C6алкилC(O)-, и так далее. В более общем смысле, описание несимметрической двухвалентной группировки, связанной по ее "левой" и "правой" сторонам, означает обе группировки, которые указаны (левая сторона группировки, связанная по левой стороне, как написано, правая сторона группировки, связанная по правой стороне, как написано), и в обратном направлении группировки, которые указаны (левая сторона группировки, связанная по правой стороне, как написано, правая сторона группировки, связанная по левой стороне, как написано).
Термины "связь" и "отсутствует" оба использованы в данном документе для описания переменной, которая не имеет атома или атомов. Так, если указано, что двухвалентная переменная "отсутствует", то понятно, что это означает, что соседние группировки связаны друг с другом. Например, если LB2 отсутствует, то это означает, что LB1 может быть связан с LB3; или если LB1 и LB2 оба отсутствуют, то имеется в виду, что LA может быть связан с LB3. Аналогично, если двухвалентная переменная определена как представляющая собой "связь", то имеется в виду, что нет присутствующих атомов, и соседние группировки связаны друг с другом. Так, например, если переменная "D" определена как представляющая собой связь, то понятно, что атомы углерода, соседние с D (в структуре, определяющей T2), связаны друг с другом. Имеется в виду, что отсутствующая одновалентная переменная представляет собой водород или электронную пару, способную к дальнейшему ковалентному связыванию.
Антительная единица (A, Ab или AB)
Как указано выше, термин "антитело" (или "A", "Ab" или "AB") в данном документе использован в самом широком смысле и конкретно охватывает интактные моноклональное антитела, поликлональные антитела, моноспецифические антитела, мультиспецифические антитела (например, биспецифические антитела) и фрагменты антител, которые проявляют желаемую биологическая активность. Кроме того, несмотря на то, что некоторые аспекты изобретения, описанные в данном документе, относятся к конъюгатам антитело-лекарственное средство, предусматривается также, что антительная часть конъюгата может быть заменена чем-либо, что специфически связывается или реакционно ассоциируется или образует комплекс с рецептором, антигеном или другой рецептивной группировкой, ассоциированной с данной популяцией клеток-мишеней. Например, вместо антитела конъюгаты по изобретению могут содержать обеспечивающую направленную доставку молекулу, которая связывается, образует комплекс или взаимодействует с рецептором, антигеном или другой рецептивной группировкой популяции клеток, которая стремится быть терапевтически или иным образом биологически модифицированной. Примеры таких молекул включают белки меньшей молекулярной массы, полипептиды или пептиды, лектины, гликопротеины, непептиды, витамины, молекулы, осуществляющие транспорт питательных веществ (такие как, без ограничения, трансферрин), или любые другие связывающие клетки молекулы или вещества. В некоторых аспектах антитело или другая такая обеспечивающая направленную доставку молекула действует, доставляя лекарственное средство в конкретную популяцию клеток-мишеней, с которой антитело или другая обеспечивающая направленную доставку молекула взаимодействует.
В другом аспекте настоящее изобретение относится к соединению, представляющему собой конъюгат антитело-лекарственное средство, формулы IIIA или IIIB, где антитело AB выбрано из трастузумаба, мутантов трастузумаба (например, мутантов трастузумаба, раскрытых в данном документе или в международной патентной заявке PCT/IB2012/056234), ореговомаба, эдреколомаба, цетуксимаба, гуманизированного моноклонального антитела к рецептору витронектина (αvβ3), алемтузумаба, анти-HLA-DR антитела, включая гуманизированное анти-HLA-DR антитело для лечения неходжкинской лимфомы, 131I Lym-1, анти-HLA-Dr10 антитела, включая мышиное анти-HLA-Dr10 антитело для лечения неходжкинской лимфомы, анти-cd33 антитела, анти-cd22 антитела, включая гуманизированное анти-CD22 mAb для лечения болезни Ходжкина или неходжкинской лимфомы, лабетузумаба, бевацизумаба, ибритумомаба тиуксетана, офатумумаба, панитумумаба, ритуксимаба, тозитумомаба, ипилимумаба и гемтузумаба.
Гетероатомы, которые могут присутствовать на антительной единице, включают серу (в одном воплощении из сульфгидрильной группы антитела), кислород (в одном воплощении из карбонильной, карбоксильной или гидроксильной группы антитела) и азот (в одном воплощении из первичной или вторичной аминогруппы антитела). Эти гетероатомы могут присутствовать на антителе в природном состоянии антитела, например в существующем в природе антителе, или могут быть введены в антитело посредством химического модифицирования.
В одном воплощении антительная единица имеет сульфгидрильную группу, и антительная единица связывается через атом серы сульфгидрильной группу.
В другом воплощении антитело имеет остатки лизина, которые могут взаимодействовать с активированными сложными эфирами (такие сложные эфиры включают, без ограничения, сложные эфиры N-гидроксисукцинимида, пентафторфенила и пара-нитрофенила) и поэтому образуют амидную связь, состоящую из атома азота антительной единицы и карбонила.
В еще одном воплощении антительная единица имеет один или более остатков лизина, которые могут быть химически модифицированы для введения одной или более сульфгидрильных групп. Реагенты, которые могут быть использованы для модификации лизинов, включают, без ограничения, N-сукцинимидил-S-ацетилтиоацетат (SATA) и 2-иминотиолата гидрохлорид (реагент Траута (Traut)).
В другом воплощении антительная единица может иметь одну или более углеводных групп, которые могут быть химически модифицированы, чтобы иметь одну или более сульфгидрильных групп.
В еще одном другом воплощении антительная единица может иметь одну или более углеводных групп, которые могут быть окислены с образованием альдегидной группы (смотри, например, Laguzza, et al., 1989, J. Med. Chem. 32(3):548-55). Соответствующий альдегид может образовывать связь с реакционноспособным сайтом, таким как, например, гидразин и гидроксиламин. Другие протоколы модификации белков для присоединения или ассоциации лекарственных средств описаны в Coligan et al., Current Protocols in Protein Science, vol. 2, John Wiley & Sons (2002) (включен в данное описание посредством ссылки).
Когда конъюгаты содержат не являющиеся иммунореакционноспособными белковые, полипептидные или пептидные единицы вместо антитела, тогда полезные не являющиеся иммунореакционноспособными белковые, полипептидные или пептидные единицы включают, без ограничения, трансферрин, эпидермальные факторы роста ("EGF"), бомбезин, гастрин, гастрин-высвобождающий пептид, тромбоцитарный фактор роста, IL-2, IL-6, трансформирующие факторы роста ("TOP"), такие как TGF-α и TGF-β, фактор роста вируса коровьей оспы ("VGF"), инсулин и инсулиноподобные факторы роста I и II, соматостатин, лектины и апопротеин из липопротеина низкой плотности.
Полезные поликлональные антитела представляют собой гетерогенные популяции молекул антител из сывороток иммунизированных животных. Полезные моноклональное антитела представляют собой гомогенные популяции антител к конкретной антигенной детерминанте (например, антигену раковых клеток, вирусному антигену, микробному антигену, белку, пептиду, углеводу, химической, нуклеиновой кислоте или их фрагментам). Моноклональное антитело (mAb) к интересующему антигену может быть получено любым известным в данной области методом, который предусматривает продуцирование молекул антитела линиями непрерывных клеток в культуре.
Полезные моноклональное антитела включают, без ограничения, моноклональные антитела человека, гуманизированные моноклональные антитела, фрагменты антител или химерные моноклональное антитела. Моноклональные антитела человека могут быть получены любыми многочисленными методами, известными в данной области (например, Teng et al., 1983, Proc. Natl. Acad. Sci. USA. 80:7308-7312; Kozbor et al., 1983, Immunology Today 4:72-79; и Olsson et al., 1982, Meth. Enzymol. 92:3-16).
Антитело может представлять собой биспецифическое антитело. Способы получения биспецифических антител известны в данной области и обсуждаются ниже.
Антитело может представлять собой функционально активный фрагмент, производное или аналог антитела, которое(ый) иммуноспецифически связывается с клетками-мишенями (например, антигенами раковых клеток, вирусными антигенами или микробным антигенами) или другими антителами, которые связываются с клетками опухоли или матриксом. В этом отношении "функционально активный" означает, что фрагмент, производное или аналог способен индуцировать антитела против антиидиотипических антител, которые распознают тот же самый антиген, который распознает антитело, из которого фрагмент, производное или аналог происходят. Конкретно, в иллюстративном воплощении антигенность идиотипа молекулы иммуноглобулина может быть усилена путем делеции каркасной области и последовательностей CDR, которые являются C-концевыми по отношению к последовательности CDR, которая специфически распознает антиген. Чтобы определить, какие последовательности CDR связываются с антигеном, синтетические пептиды, содержащие последовательности CDR, могут быть использованы в анализах связывания с антигеном любым методом анализа связывания, известным в данной области (например, анализом BIAcore) (информацию о локализации последовательностей CDR смотри, например, в Kabat et al., 1991, Sequences of Proteins of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, Md.; Kabat E et al., 1980, J. Immunology 125(3):961-969).
Другие полезные антитела включают фрагменты антител, такие как, без ограничения, F(ab')2 фрагменты, Fab фрагменты, Fvs, одноцепочечные антитела, диатела, триатела, тетратела, scFv, scFv-FV или любая другая молекула с такой же специфичностью, как и у антитела.
Дополнительно, рекомбинантные антитела, такие как химерные и гуманизированные моноклональные антитела, содержащие как человеческие участки, так и участки, не являющиеся человеческими, которые могут быть получены стандартными методами, основанными на рекомбинантной ДНК, являются полезными антителами. Химерное антитело представляет собой молекулу, в которой разные участки имеют происхождение из разных видов животных, такие как, например, имеющие вариабельную область из константных областей мышиного моноклонального и человеческого иммуноглобулина (смотри, например, патент США №4816567 и патент США №4816397, полное содержание которых включено в данное описание посредством ссылки). Гуманизированные антитела представляют собой молекулы антител из видов, не являющихся человеческими, имеющие один или более участков, определяющих комплементарность (CDR), из вида, не являющегося человеческим, и каркасную область из молекулы иммуноглобулина человека (смотри, например, патент США №5585089, полное содержание которого включено в данное описание посредством ссылки). Такие химерные и гуманизированные моноклональные антитела могут быть получены известными в данной области методами, основанными на рекомбинантной ДНК, например с использованием методов, описанных в международной публикации WO 87/02671; публикации Европейского патента №0184187; публикации Европейского патента №0171496; публикации Европейского патента №0173494; международной публикации WO 86/01533; патенте США №4816567; публикации Европейского патента №012023; Berter et al., 1988, Science 240:1041-1043; Liu et al., 1987, Proc. Natl. Acad. Sci. USA 84:3439-3443; Liu et al., 1987, J. Immunol. 139:3521-3526; Sun et al., 1987, Proc. Natl. Acad. Sci. USA 84:214-218; Nishimura et al., 1987, Cancer. Res. 47:999-1005; Wood et al., 1985, Nature 314:446-449; и Shaw et al., 1988, J. Natl. Cancer Inst. 80:1553-1559; Morrisoon, 1985, Science 229:1202-1207; Oi et al., 1986, BioTechniques 4:214; патенте США №5225539; Jones et al., 1986, Nature 321:552-525; Verhoeyan et al., 1988, Science 239:1534; и Beidler et al., 1988, J. Immunol. 141:4053-4060; полное содержание которых включено в данное описание посредством ссылки.
Полностью человеческие антитела особенно желательны и могут быть получены с использованием трансгенных мышей, которые неспособны экспрессировать гены тяжелой и легкой цепей эндогенного иммуноглобулина, но которые могут экспрессировать человеческие гены тяжелой и легкой цепей. Трансгенных мышей иммунизируют обычным способом выбранным антигеном, например всем или частью полипептида по изобретению. Моноклональные антитела, направленные против антигена, могут быть получены с использованием стандартной гибридомной технологии. Трансгены человеческого иммуноглобулина, переносимые трансгенными мышами, перегруппировываются во время B-клеточной дифференцировки, и затем подвергаются механизму переключения классов и соматической мутации. Так, используя такой метод, можно получать терапевтически полезные IgG, IgA, IgM и IgE антитела. Обзор по этой технологии получения человеческих антител смотри в Lonberg and Huszar, 1995, Int. Rev. Immunol. 13:65-93. Подробное обсуждение этой технологии получения человеческих антител и человеческих моноклональных антител и протоколы для получения таких антител смотри, например, в патентах США №№5625126, 5633425, 5569825, 5661016, 5545806, каждый из которых во всей его полноте включен в данное описание посредством ссылки. Другие человеческие антитела могут быть приобретены коммерческим путем, например у Abgenix, Inc. (теперь Amgen, Freemont, Calif.) и Medarex (Princeton, N.J.).
Полностью человеческие антитела, которые распознают выбранный эпитоп, могут быть созданы с использованием метода, имещего название "управляемая селекция". В этом подходе выбранное моноклональное антитело, не являющегося человеческим, например мышиное антитело, используют для управляемой селекции полностью человеческого антитела, распознающего такой же эпитоп (смотри, например, Jespers et al., 1994, Biotechnology 12:899-903). Человеческие антитела могут быть также получены с использованием различных методов, известных в данной области, включая библиотеки фагового дисплея (смотри, например, Hoogenboom and Winter, 1991, J. Mol. Biol. 227:381; Marks et al., 1991, J. Mol. Biol. 222:581; Quan and Carter, 2002, The rise of monoclonal antibodies as therapeutics, in Anti-IgE and Allergic Disease, Jardieu and Fick, eds., Marcel Dekker, New York, N.Y., Chapter 20, pp. 427-469).
В других воплощениях антитело представляет собой слитый белок антитела или его функционально активного фрагмента, например в котором антитело подвергнуто слиянию посредством ковалентной связи (например, пептидной связи) либо по N-концу, либо по С-концу с аминокислотной последовательностью другого белка (или его части, предпочтительно части белка из по меньшей мере 10, 20 или 50 аминокислот), который не образует антитело. Предпочтительно, антитело или его фрагмент ковалентно связано с другим белком по N-концу константного домена.
Антитела включают аналоги и производные, которые либо модифицированы, например ковалентным присоединением молекулы любого типа, поскольку такое ковалентное присоединение дает возможность сохранить антигенсвязвающую иммуноспецифичность антитела. Например, но без всякого ограничения, производные и аналоги антитела включают те из них, которые были дополнительно модифицированы, например гликозилированием, ацетилированием, пэгилированием, фосфорилированим, амидированием, дериватизацией известными защитными/блокирующими группами, протеолитическим расщеплением, связью с клеточной антительной единицей или другим белком и т.д. Любые многочисленные химические модификации могут быть осуществлены известными методами, включая, без ограничения, специфическое химическое расщепление, ацетилирование, формилирование, метаболический синтез в присутствии туникамицина и т.д. Дополнительно, аналог или производное может содержать одну или более неприродных аминокислот.
Антитела могут иметь модификации (например, замены, делеции или добавления) аминокислотных остатков, которые взаимодействуют с Fc рецепторами. В частности, антитела могут иметь модификации аминокислотных остатков, идентифицированных как участвующих во взаимодействии между анти-Fc доменом и FcRn рецептором (смотри, например, международную публикацию WO 97/34631, которая во всей ее полноте включена в данное описание посредством ссылки).
Антитела, иммуноспецифические к антигену раковых клеток, могут быть приобретены коммерческим путем или могут быть получены любым методом, известным специалисту в данной области, таким как, например, методы химического синтеза или рекомбинантные экспрессионные методы. Нуклеотидная последовательность, кодирующая антитела, иммуноспецифические к антигену раковых клеток, могут быть получены, например, из базы данных GenBank или подобной ей базы данных, литературных публикаций или путем клонирования и секвенирования.
В конкретном воплощении могут быть использованы известные антитела для лечения рака. Антитела, иммуноспецифические к антигену раковых клеток, могут быть приобретены коммерческим путем или могут быть получены любым методом, известным специалисту в данной области, таким как, например, рекомбинантные экспрессионные методы. Нуклеотидная последовательность, кодирующая антитела, иммуноспецифические к антигену раковых клеток, могут быть получены, например, из базы данных GenBank или подобной ей базы данных, литературных публикаций или путем клонирования и секвенирования. Примеры антител, доступных для лечения рака, включают, без ограничения, OVAREX, которое представляет собой мышиное антитело для лечения рака яичника; PANOREX (Glaxo Wellcome, NC), которое представляет собой мышиное IgG2a антитело для лечения колоректального рака; Cetuximab ERBITUX (Imclone Systems Inc., NY), которое представляет собой анти-EGFR IgG химерные антитело для лечения эпидермальный фактор роста-положительных видов рака, таких как рак в области головы и шеи; Vitaxin (MedImmune, Inc., MD), которое представляет собой гуманизированное антитело для лечения саркомы; CAMPATH I/H (Leukosite, MA), которое представляет собой гуманизированное IgG1 антитело для лечения хронического лимфоцитарного лейкоза (CLL); SMART ID10 (Protein Design Labs, Inc., CA), которое представляет собой гуманизированное анти-HLA-DR антитело для лечения неходжкинской лимфомы; ONCOLYM (Techniclone, Inc., CA), которое представляет собой радиомеченое мышиное анти-HLA-Dr10 антитело для лечения неходжкинской лимфомы; ALLOMUNE (BioTransplant, CA), которое представляет собой гуманизированное анти-CD2 mAb для лечения болезни Ходжкина или неходжкинской лимфомы; и CEACIDE (Immunomedics, NJ), которое представляет собой гуманизированное анти-CEA антитело для лечения колоректального рака.
Стремясь открыть эффективные клеточные мишени для диагностики и терапии рака, исследователи старались идентифицировать трансмембранные или иные ассоциированные с опухолями полипептиды, которые специфически экспрессируются на поверхности раковых клеток одного или более конкретного(ых) типа(ов) по сравнению с одной или более нормальной(ыми) нераковой(ыми) клеткой(ами). Часто такие ассоциированные с опухолями полипептиды в большей степени повсеместно экспрессируются на поверхности раковых клеток по сравнению с экспрессией на поверхности нераковых клеток. Идентификация таких ассоциированных с опухолью поверхностных клеточных антигенных полипептидов дала возможность специфически направленно воздействовать на раковые клетки с целью их разрушения основанными на антителах терапевтическими средствами.
Линкерая единица (L)
Линкер (иногда упоминаемый в данном документе как "[линкер]") представляет собой бифункциональное соединение, которое может быть использовано для связывания лекарственного средства и антитела с образованием конъюгата антитело-лекарственное средство (ADC). Такие конъюгаты полезны, например, в образовании иммуноконъюгатов, направленных против ассоциированных с опухолями антигенов. Такие конъюгаты обеспечивают избирательную доставку цитотоксических лекарственных средств в клетки опухоли.
В ADC линкер служит для присоединения полезной нагрузки к антителу.
В одном аспекте вводят второй участок линкерной единицы, который имеет второй реакционноспособный сайт, например электрофильную группу, которая способна взаимодействовать с нуклеофильной группой, присутствующей на антительной единице (например, на антителе). Полезные нуклеофильные группы на антителе включают, без ограничения, сульфгидрильную группу, гидроксильную группу и аминогруппу. Гетероатом нуклеофильной группы антитела взаимодействует с электрофильной группой на линкерной единице и образует ковалентную связь с линкерной единицей. Полезные электрофильные группы включают, без ограничения, малеимидные и галогенацетамидные группы. Электрофильная группа предоставляет ковалентный сайт для присоединения антитела.
В другом воплощении линкерная единица имеет реакционноспособный сайт, который имеет нуклеофильную группу, которая взаимодействует с электрофильной группой, присутствующей на антителе. Полезные электрофильные группы включают, без ограничения, альдегидные и кетоновые карбонильные группы. Гетероатом нуклеофильной группы линкерной единицы может взаимодействовать с электрофильной группой на антителе и образовывать ковалентную связь с антителом. Полезные нуклеофильные группы на линкерной единице включают, без ограничения, гидразид, оксим, амино, гидразин, тиосемикарбазон, гидразин-карбоксилат и арилгидразид. Электрофильная группа на антителе предоставляет удобный сайт для присоединения линкерной единицы.
Функциональные аминогруппы также являются полезными реакционноспособными сайтами для линкерной единицы, поскольку они могут взаимодействовать с карбоновой кислотой или активированными сложными эфирами соединения с образованием амидной связи. Обычно пептидные соединения по изобретению могут быть получены в результате образования пептидной связи между двумя или более аминокислотами и/или пептидными фрагментами. Такие пептидные связи могут быть образованы, например, методом жидкофазного синтеза (смотри, например, Schroder and Lubke, "The Peptides", volume 1, pp 76-136, 1965, Academic Press), который известен в области пептидной химии.
В контексте данного изобретения, в частности без ограничения линкерными компонентами, такими как L1, L2 (включая L2A, L2B и L2C) и L3, выражение "выбранный из одного или более" или "один или более" указывает на то, что множественные компоненты, которые могут быть одинаковыми или разными, расположены или могут быть расположены последовательно. Так, например, L3 может представлять собой -C1-6алкил-, -NR- или другие индивидуально перечисленные компоненты, а также -C1-6алкил-NR- или любую другую комбинацию 2 или более перечисленных компонентов.
В другом воплощении линкерная единица имеет реакционноспособный сайт, который может взаимодействовать с нуклеофилами антитела, такими как цистеины. Реакционноспособный сайт состоит из гетероцикла, который замещен сульфоном. Сульфон затем замещается нуклеофилом антитела (т.е. цистеином), и вновь образованная связь между антителом и гетероциклом соединяет антитело с линкером (смотри WO 2014/144878).
Синтез соединений и конъюгатов антитело-лекарственное средство
Соединения и конъюгаты по изобретению могут быть получены по методикам синтеза, в общих чертах изложенных ниже в разделе "Иллюстративные примеры". Как описано более подробно ниже, соединения и конъюгаты по изобретению могут быть получены с использованием участка линкерной единицы, имеющего реакционноспособный сайт для связывания с соединением. В одном аспекте вводят второй участок линкерной единицы, который имеет второй реакционноспособный сайт, например электрофильную группу, которая взаимодействует с нуклеофильной группой, присутствующей на антительной единице (например, антителе). Полезные нуклеофильные группы на антителе включают, без ограничения, сульфгидрильные, гидроксильные и аминогруппы. Гетероатом нуклеофильной группы антитела взаимодействует с электрофильной группой на линкерной единице и образует ковалентную связь с линкерной единицей. Полезные электрофильные группы включают, без ограничения, малеимидные и галогенацетамидные группы. Электрофильная группа предоставляет удобный сайт для присоединения антитела.
В другом воплощении линкерная единица имеет реакционноспособный сайт, который имеет нуклеофильную группу, которая взаимодействует с электрофильной группой, присутствующей на антителе. Полезные электрофильные группы на антителе включают, без ограничения, альдегидные и кетоновые карбонильные группы. Гетероатом нуклеофильной группы линкерной единицы может взаимодействовать с электрофильной группой на антителе и образовывать ковалентную связь с антителом. Полезные нуклеофильные группы на линкерной единице включают, без ограничения, гидразид, оксим, амино, гидразин, тиосемикарбазон, гидразин-карбоксилат и арилгидразид. Электрофильная группа на антителе предоставляет удобный сайт для присоединения линкерной единицы.
Функциональные аминогруппы также являются полезными реакционноспособными сайтами для линкерной единицы, поскольку они могут взаимодействовать с карбоновой кислотой или активированными сложными эфирами соединения с образованием амидной связи. Обычно пептидные соединения по изобретению могут быть получены в результате образования пептидной связи между двумя или более аминокислотами и/или пептидными фрагментами. Такие пептидные связи могут быть получены, например, методом жидкофазного синтеза (смотри, например, Schroder and Lubke, "The Peptides", volume 1, pp 76-136, 1965, Academic Press), который общеизвестен в области пептидной химии.
Как описано более подробно ниже, конъюгаты могут быть получены с использованием участка линкера, имеющего реакционноспособный сайт для связывания с соединением по изобретению и введения другого участка линкерной единицы, имеющего реакционноспособный сайт для антитела. В одном аспекте линкерная единица имеет реакционноспособный сайт, который имеет электрофильную группу, которая взаимодействует с нуклеофильной группой, присутствующей на антительной единице, такой как антитело. Электрофильная группа предоставляет удобный сайт для присоединения антитела. Полезные нуклеофильные группы на антителе включают, без ограничения, сульфгидрильные, гидроксильные и аминогруппы. Гетероатом нуклеофильной группы антитела взаимодействует с электрофильной группой на линкерной единице и образует ковалентную связь с линкерной единицей. Полезные электрофильные группы включают, без ограничения, малеиимидные и галогенацетамидные группы.
В другом воплощении линкерная единица имеет реакционноспособный сайт, который имеет нуклеофильную группу, которая взаимодействует с электрофильной группой, присутствующей на антительной единице. Электрофильная группа на антителе предоставляет удобный сайт для присоединения линкерной единицы. Полезные электрофильные группы на антителе включают, без ограничения, альдегидные и кетоновые карбонильные группы. Гетероатом нуклеофильной группы линкерной единицы может взаимодействовать с электрофильной группой на антителе и образовывать ковалентную связь с антителом. Полезные нуклеофильные группы на линкерной единице включают, без ограничения, гидразид, оксим, амино, гидразин, тиосемикарбазон, гидразин-карбоксилат и арилгидразид.
Конъюгирование с трансглутаминазой
В некоторых воплощениях соединение по изобретению может быть ковалентно поперечно связано с Fc-содержащим или Fab-содержащим полипептидом, сконструированным с ацил-донорной глутамин-содержащей меткой (например, Gln-содержащими пептидными метками или Q-метками), или эндогенным глутамином, сделанным реакционноспособным (т.е. способным образовывать ковалентную связь в качестве ацил-донора в присутствии амина и трансглутаминазы) посредством полипептидной инженерии (например, посредством делеции, вставки, замены, мутации аминокислоты или любой их комбинации на полипептиде) в присутствии трансглутаминазы, при условии, что соединение по изобретению содержит амин-донорный агент (например, небольшую молекулу, содержащую реакционноспособный амин, или молекулу, присоединенную к реакционноспособному амину), тем самым образуя стабильную и гомогенную популяцию сконструированного Fc-содержащего полипептидного конъюгата, причем амин-донорный агент сайт-специфическим образом конъюгирован с Fc-содержащим или Fab-содержащим полипептидом через ацил-донорную глутамин-содержащую метку или экспонированный/доступный/реакционноспособный эндогенный глутамин. Например, соединения по изобретению могут быть конъюгированы так, как описано в Международной патентной заявке серийный № PCT/IB2011/054899, полное содержание которой включено в данное описание посредством ссылки. В некоторых воплощениях для облегчения конъюгирования соединения по изобретению с Fc-содержащим или Fab-содержащим полипептидом, сконструированным с ацил-донорной глутамин-содержащей меткой или эндогенным глутамином, сделанным реакционноспособным посредством полипептидной инженерии в присутствии трансглутаминазы, Z представляет собой NH2.
Конъюгирование с константной областью каппа домена легкой цепи человека
В некоторых воплощениях соединение по изобретению может быть ковалентно присоединено к боковой цепи K188 константной области каппа домена легкой цепи человека  (нумерация полноразмерной цепи согласно Kabat). Например, соединения по изобретению могут быть конъюгированы так, как описано в заявке на патент США серийный номер 13/180,204, полное содержание которой включено в данное описание посредством ссылки. В некоторых воплощениях для облегчения конъюгирования с K188
(нумерация полноразмерной цепи согласно Kabat). Например, соединения по изобретению могут быть конъюгированы так, как описано в заявке на патент США серийный номер 13/180,204, полное содержание которой включено в данное описание посредством ссылки. В некоторых воплощениях для облегчения конъюгирования с K188  Z представляет собой
Z представляет собой 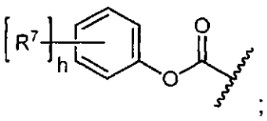 R7 независимо выбран для каждого случая из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3; и h означает 1, 2, 3, 4 или 5.
R7 независимо выбран для каждого случая из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3; и h означает 1, 2, 3, 4 или 5.
В некоторых воплощениях согласно изобретению предложена композиция, содержащая соединение по изобретению, ковалентно конъюгированное с антителом (или его антигенсвязывающим участком), где по меньшей мере примерно 50%, или по меньшей мере примерно 60%, или по меньшей мере примерно 70%, или по меньшей мере примерно 80%, или по меньшей мере примерно 90% соединения по изобретению в композиции конъюгировано с антителом или его антигенсвязывающиим участком по K188 .
В некоторых воплощениях соединения по изобретению могут быть конъюгированы с объединенным сайтом каталитического антитела, такого как антитела к альдолазе, или его антигенсвязывающим участком. Антитела к альдолазе содержат участки объединенного сайта, которые, когда они не обременены (например конъюгированием), катализируют реакцию альдольного присоединения между алифатическом кетонным донором и альдегидным акцептором. Содержание публикации заявки на патент США № US 2006/205670 включено в данное описание посредством ссылки, в частности страницы 78-118, где описаны линкеры, и абзацы [0153]-[0233], где описаны антитела, их полезные фрагменты, варианты и модификации, h38C2, объединенные сайты и участки, определяющие комплементарность (CDR), и связанная с ними технология антител. Термин "объединенный сайт" охватывает CDR и соседние каркасные остатки, которые вовлечены в связывание антигена.
Композиции и способы введения
В других воплощениях еще один аспект изобретения относится к фармацевтическим композициям, содержащим эффективное количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство и фармацевтически приемлемый носитель или наполнитель. В некоторых воплощениях композиции подходят для ветеринарного введения или введения человеку.
Настоящие фармацевтические композиции могут находиться в любой форме, которая дает возможность вводить композицию пациенту. Например, композиция может быть в форме твердого вещества или жидкости. Типичные пути введения включают, без ограничения, парентеральное введение, введение в глаз и введение в опухоль. Парентеральное введение включает подкожные инъекции, внутривенное, внутримышечное или интрастернальное инъецирование или инфузионные способы. В одном аспекте композиции вводят парентерально. В конкретном воплощении композиции вводят внутривенно.
Фармацевтические композиции могут быть приготовлены таким образом, чтобы была обеспечена биодоступность соединения по изобретению и/или его конъюгата антитело-лекарственное средство после введения композиции пациенту. Композиции могут принимать форму одной или более стандартных лекарственных форм, где, например, таблетка может представлять собой однократную лекарственную форму, и контейнер соединения по изобретению и/или его конъюгата антитело-лекарственное средство в жидкой форме может содержать множество стандартных лекарственных форм.
Вещества, используемые в приготовлении фармацевтических композиций, могут быть нетоксичными в используемых количествах. Специалисту в данной области понятно, что оптимальная дозировка активного(ых) ингредиента(ов) в фармацевтической композиции будет зависеть от целого ряда различных факторов. Релевантные факторы включают, без ограничения, тип животного (например, человек), конкретную форму соединения по изобретению и/или его конъюгата антитело-лекарственное средство, способ введения и используемую композицию.
Фармацевтически приемлемый носитель или наполнитель может быть твердым или в виде частиц, и тогда композиции находятся, например, в форме таблетки или в форме порошка. Носитель(и) может быть жидким. Кроме того, носитель(и) может быть в виде частиц.
Композиция может находиться в форме жидкости, например раствора, эмульсии или суспензии. В состав композиции для введения инъекциями также могут быть входить одно или более из следующих веществ: поверхностно-активное вещество, консервант, увлажняющий агент, диспергирующий агент, суспендирующий агент, буфер, стабилизатор и изотонический агент.
Жидкие композиции, представляющие собой растворы, суспензии или другую подобную форму, могут также содержать одно или более из следующего: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический раствор, раствор Рингера, изотонический хлорид натрия, нелетучие масла, такие как синтетические моно- или диглицериды, которые могут служить в качестве растворителя или суспендирующей среды, полиэтиленгликоли, глицерин, циклодекстрин, пропилен гликоль или другие растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты, фосфаты или аминокислоты; и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Парентеральная композиция может быть заключена в ампуле, одноразовом шприце или многодозовом флаконе, изготовленном из стекла, пластика или другого материала. Физиологический раствор представляет собой типичное вспомогательное вещество. Инъецируемая композиция предпочтительно является стерильной.
Количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство, которое является эффективным в лечении конкретного расстройства или состояния, будет зависеть от природы этого расстройства или состояния и может быть определено стандартными клиническими методами. Кроме того, для идентификации оптимальных диапазонов дозировки возможно могут быть использованы анализы in vitro или in vivo. Точная доза, которую нужно использовать в композициях, также будет зависеть от пути введения и серьезности заболевания или расстройства, и ее следует определять в соответствии с суждением практикующего специалиста и обстоятельствами каждого пациента.
Композиции содержат эффективное количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство, исходя из условия, что будет достигаться подходящая дозировка. Обычно это количество составляет по меньшей мере примерно 0,01% соединения по изобретению и/или его конъюгата антитело-лекарственное средство от массы композиции. В типичном воплощении фармацевтические композиции получают таким образом, чтобы парентеральная стандартная лекарственная форма содержала от примерно 0,01% до примерно 2% масс. соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
Для внутривенного введения композиция может содержать от примерно 0,01 до примерно 100 мг соединения по изобретению и/или его конъюгата антитело-лекарственное средство на кг массы тела пациента. В одном аспекте композиция может содержать от примерно 1 до примерно 100 мг соединения по изобретению и/или его конъюгата антитело-лекарственное средство на кг массы тела пациента. В другом аспекте вводимое количество будет находиться в диапазоне от примерно 0,1 до примерно 25 мг/кг массы тела соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
В общем, дозировка соединения по изобретению и/или его конъюгата антитело-лекарственное средство, вводимая пациенту, составляет от примерно 0,01 мг/кг до примерно 20 мг/кг массы тела пациента. В одном аспекте дозировка, вводимая пациенту, составляет от примерно 0,01 мг/кг до примерно 10 мг/кг массы тела пациента. В другом аспекте дозировка, вводимая пациенту, составляет от примерно 0,1 мг/кг до примерно 10 мг/кг массы тела пациента. В еще одном воплощении дозировка, вводимая пациенту, составляет от примерно 0,1 мг/кг до примерно 5 мг/кг массы тела пациента. В еще одном воплощении вводимая дозировка составляет от примерно 0,1 мг/кг до примерно 3 мг/кг массы тела пациента. В еще одном воплощении вводимая дозировка составляет от примерно 1 мг/кг до примерно 3 мг/кг массы тела пациента.
Соединение по изобретению и/или его конъюгат антитело-лекарственное средство можно вводить любым удобным путем, например инфузией или болюсной инъекцией. Введение может быть системным или местным. Известны различные системы доставки, например инкапсулирование в липосомах, микрочастицах, микрокапсулах, капсулах и т.д., и они могут быть использованы для введения соединения по изобретению и/или его конъюгата антитело-лекарственное средство. В некоторых воплощениях пациенту вводят более одного соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
В конкретных воплощениях может быть желательным введение одного или более соединений по изобретению и/или их конъюгатов антитело-лекарственное средство местно в область, нуждающуюся в лечении. Этого можно достичь, например и без ограничения, местной инфузией во время хирургического вмешательства; местным нанесением, например в сочетании с наложением повязки на рану после хирургической операции; инъекциями; с помощью катетера; или посредством имплантата, где имплантат представляет собой пористый, непористый или гелеобразный материал, включая мембраны, такие как силастиковые мембраны или волокна. В одном воплощении введение может представлять собой прямую инъекцию в участок (или бывший участок) раковой, опухолевой или неопластической или пренеопластической ткани. В другом воплощении введение может представлять собой прямую инъекцию в участок (или бывший участок) проявления аутоиммунного заболевания.
В еще одном другом воплощении соединение по изобретению и/или его конъюгат антитело-лекарственное средство можно доставлять в системе контролируемого высвобождения, такой как, без ограничения, насос, или могут быть использованы различные полимерные материалы. В еще одном другом воплощении система контролируемого высвобождения может быть размещена в непосредственной близости от мишени соединения по изобретению и/или его конъюгата антитело-лекарственное средство, например печени, и в этом случае требуется только часть системной дозы (смотри, например, Goodson, Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)). Могут быть использованы другие системы контролируемого высвобождения, которые обсуждаются в обзоре Langer (Science 249:1527-1533 (1990)).
Термин "носитель" относится к разбавителю, вспомогательному веществу или эксципиенту, вместе с которым вводят соединение или его конъюгат антитело-лекарственное средство. Такие фармацевтические носители могут представлять собой жидкости, такие как вода и масла, в том числе нефтяного, животного, растительного или синтетического происхождения. Носителями могут быть физиологический раствор и т.п. Кроме того, могут быть использованы вспомогательные, стабилизирующие и другие агенты. В одном воплощении для введения пациенту соединение или конъюгат и фармацевтически приемлемые носители являются стерильными. Вода является примером носителя для внутривенного введения соединения или конъюгата. Солевые растворы и водные растворы декстрозы и глицерина также могут быть использованы в качестве жидких носителей, особенно для инъецируемых растворов. Композиции по настоящему изобретению, если желательно, могут также содержать незначительные количества увлажняющих или эмульгирующих агентов или pH буферных агентов.
Композиции по настоящему изобретению могут принимать форму растворов, таблеток, порошков, препаратов длительного высвобождения или любую другую форму, подходящую для применения. Другие примеры подходящих фармацевтических носителей описаны в "Remington's Pharmaceutical Sciences" by E.W. Martin.
В одном воплощении соединение по изобретению и/или его конъюгат антитело-лекарственное средство готовят в соответствии с рутинными методиками в виде фармацевтической композиции, пригодной для внутривенного введения животным, в частности людям. Обычно носители или наполнители для внутривенного введения представляют собой стерильные изотонические водные буферные растворы. Если необходимо, композиции могут также содержать солюбилизирующий агент. Композиции для внутривенного введения возможно могут содержать местный анестетик, такой как лигнокаин, для облегчения боли в месте инъекции. Как правило, ингредиенты поставляются либо по отдельности, либо смешанными вместе в стандартной лекарственной форме, например в виде сухого лиофилизированного порошка или безводного концентрата в герметично закупоренном контейнере, таком как ампула или саше, с указанием количества активного агента. Если соединение по изобретению и/или его конъюгат антитело-лекарственное средство предназначено для введения инфузией, то его можно дозировать, например, с использованием инфузионного флакона, содержащего стерильную воду фармацевтического сорта или физиологический раствор. Если соединение по изобретению и/или его конъюгат антитело-лекарственное средство вводят инъекцией, то может быть предоставлена ампула со стерильной водой для инъекций или физиологическим раствором, чтобы ингредиенты можно было смешать перед введением.
Композиция может содержать различные вещества, которые модифицируют физическую форму твердой или жидкой стандартной лекарственной формы. Например, композиция может содержать вещества, которые образуют оболочку, покрывающую активные ингредиенты. Вещества, которые образуют покрывающую оболочку, типично являются инертными и могут быть выбраны из, например, сахара, шеллака и других энтеросолюбильных покровных агентов. Альтернативно, активные ингредиенты могут быть инкапсулированы в желатиновой капсуле.
Композиции по настоящему изобретению в твердой или в жидкой форме могут содержать фармакологический агент, используемый в лечении рака.
Терапевтические применения соединений и их конъюгатов антитело-лекарственное средство
Еще один аспект изобретения относится к способу использования соединений по изобретению и их конъюгатов антитело-лекарственное средство для лечения рака.
Соединения по изобретению и/или их конъюгаты антитело-лекарственное средство полезны для ингибирования размножения опухолевой клетки или раковой клетки, вызывая апоптоз в опухолевой или раковой клетке, или для лечения рака у пациента. Соединения по изобретению и/или их конъюгаты антитело-лекарственное средство можно применять соответственно в различных местах действия для лечения рака у животных. Указанные конъюгаты можно использовать для доставки соединения по изобретению в опухолевую клетку или раковую клетку. Без какой-либо связи с теорией, в одном воплощении антитело конъюгата связывается или ассоциируется с антигеном, ассоциированным раковой клеткой или опухолевой клеткой, и конъюгат может быть захвачен (интернализирован) внутрь опухолевой клетки или раковой клетки посредством рецептор-опосредованного эндоцитоза или по другому механизму интернализации. Антиген может прикрепляться к опухолевой клетке или раковой клетке или может представлять собой внеклеточный матриксный белок, ассоциированный с опухолевой клеткой или раковой клеткой. В некоторых воплощениях сразу после попадания внутрь клетки одна или более специфических пептидных последовательностей ферментативно или гидролитически расщепляется одной или более ассоциированными с опухолевыми клетками или раковыми клетками протеазами, в результате чего соединение по изобретению высвобождается из конъюгата. Высвободившееся соединение по изобретению затем свободно мигрирует в клетке и индуцирует цитотоксическую или цитостатическую активность. Конъюгат также может расщепляться внутриклеточной протеазой с высвобождением соединения по изобретению. В альтернативном воплощении соединение по изобретению отщепляется от конъюгата за пределами опухолевой клетки или раковой клетки, и соединение по изобретению затем проникает в клетку.
В некоторых воплощениях конъюгаты обеспечивают конъюгат-специфическую направленную доставку противоопухолевого или противоракового лекарственного средства, снижая тем самым общую токсичность соединения по изобретению.
В другом воплощении антительная единица связывается с опухолевой клеткой или раковой клеткой.
В другом воплощении антительная единица связывается с антигеном опухолевой клетки или раковой клетки, который находится на поверхности опухолевой клетки или раковой клетки.
В другом воплощении антительная единица связывается с антигеном опухолевой клетки или раковой клетки, который представляет собой внеклеточный матриксный белок, ассоциированный с опухолевой клеткой или раковой клеткой.
Специфичность антительной единицы в отношении конкретной опухолевой клетки или раковой клетки может быть важна для определения тех опухолей или видов рака, которые наиболее эффективно лечатся.
Конкретные типы раковых заболеваний, которые можно лечить соединением по изобретению и/или его конъюгатом антитело-лекарственное средство, включают, без ограничения, карциномы мочевого пузыря, рак молочной железы, шейки матки, ободочной кишки, эндометрия, почки, легкого, пищевода, яичника, предстательной железы, поджелудочной железы, кожи, желудка и яичек; и раковые заболевания крови, в том числе, без ограничения, лейкозы и лимфомы.
Комплексная терапия рака. Раковые заболевания, включая, без ограничения, опухолевое заболевание, метастазы или другое заболевание или расстройство, характеризующееся неконтролируемым ростом клеток, можно лечить или подавлять путем введения соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
В других воплощениях предложены способы лечения рака, включающие введение нуждающемуся в этом пациенту эффективного количества соединения по изобретению и/или его конъюгата антитело-лекарственное средство и химиотерапевтического агента. В одном воплощении химиотерапевтический агент представляет собой агент, с использованием которого, как было установлено, лечение рака не было невосприимчивым. В другом воплощении химиотерапевтический агент представляет собой агент, с использованием которого, как было установлено, лечение рака было невосприимчивым. Соединение по изобретению и/или его конъюгат антитело-лекарственное средство можно вводить пациенту, который также перенес хирургическую операцию в качестве лечения рака.
В некоторых воплощениях пациент также получает дополнительное лечение, такое как лучевая терапия. В конкретном воплощении соединение по изобретению и/или его конъюгат антитело-лекарственное средство вводят параллельно с химиотерапевтическим агентом или с лучевой терапией. В другом конкретном воплощении химиотерапевтический агент вводят или лучевую терапию проводят до или после введения соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
Химиотерапевтический агент можно вводить серией курсов. Можно вводить любой один химиотерапевтический агент или комбинацию химиотерапевтических агентов, например стандартный(ые) химиотерапевтический(ие) агент(ы).
Дополнительно, предложены способы лечения рака соединением по изобретению и/или его конъюгатом антитело-лекарственное средство в качестве альтернативы химиотерапии или лучевой терапии в тех случаях, когда доказано или может оказаться, что химиотерапия или лучевая терапия слишком токсична для субъекта, которого лечат, например вызывает неприемлемые или непереносимые побочные эффекты. Подлежащего лечению пациента возможно можно лечить другим способом лечения рака, таким как хирургическое вмешательство, лучевая терапия или химиотерапия, в зависимости от того, какое лечение является приемлемым или терпимым.
Соединения по изобретению и/или их конъюгаты антитело-лекарственное средство могут быть также использованы в моделях в vitro или ex vivo, таких как для лечения некоторых видов рака, включая, без ограничения, лейкозы и лимфомы, причем такое лечение включает в себя аутологические трансплантаты стволовых клеток. Это лечение может заключать в себе многостадийный процесс, при котором аутологические гематопоэтические стволовые клетки животного собирают и очищают от всех раковых клеток, остаточную популяцию костномозговую клеток животного затем уничтожают путем введения высокой дозы соединения по изобретению и/или его конъюгата антитело-лекарственное средство с применением или без применения большой дозы лучевой терапии, и трансплантат стволовых клеток внедряют назад животному. Затем проводят поддерживающее лечение, при этом функция костного мозга восстанавливается, и пациент выздоравливает.
Изобретение дополнительно описано в приведенных ниже примерах, которые не ограничивают объем изобретения.
Иллюстративные примеры полезных нагрузок и конструкций линкер-полезная нагрузка
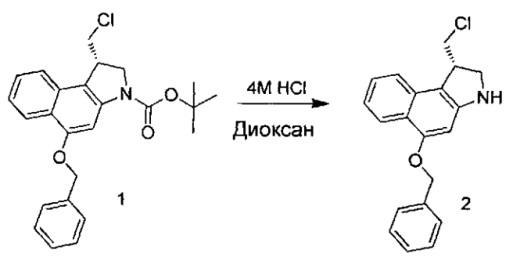
Соединение 2 является коммерчески известным соединением (смотри международную заявку PCT 2005112919, 1 декабря 2005 года).
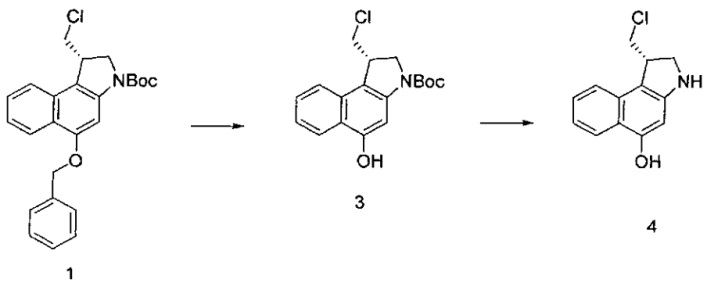
Получение трет-бутил-(S)-1-(хлорметил)-5-гидрокси-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата (3): В раствор трет-бутил-(S)-5-(бензилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата (4 г, 9 ммоль) в THF (250 мл) добавляли Pd-C (0,7 г) при 40°C. Затем порциями добавляли водный HCOONH4 (9,5 мл, 25%), и эту реакционную смесь перемешивали при 40°C в течение 1 часа. Реакционную смесь фильтровали, и фильтрат концентрировали досуха. Полученный остаток растворяли в этилацетате (250 мл) и промывали H2O (20 мл), сушили над Na2SO4 и концентрировали досуха с получением соединения 3 в виде серого твердого вещества (2,9 г, 90%).
Получение (S)-1-(хлорметил)-2,3-дигидро-1Н-бензо[е]индол-5-ола (4): В круглодонную колбу, содержащую соединение 3 (820 мг, 2,46 ммоль), добавляли 4М HCl в диоксане (36 мл, 140 ммоль). Эту реакционную смесь перемешивали при комнатной температуре. Реакционную смесь концентрировали и затем помещали под вакуум (насос с ременным приводом) с получением соединения 4 (684 мг, 100%) в виде серого твердого вещества.
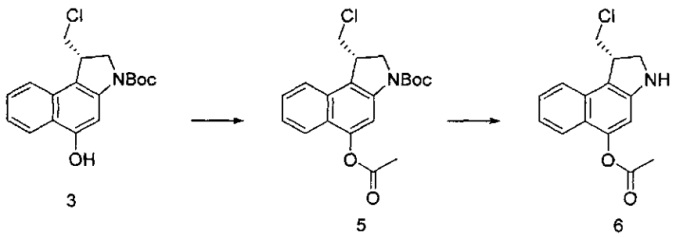
Получение трет-бутил-(S)-5-ацетокси-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата (5): Ацетилхлорид (0,1 мл, 1,4 ммоль) добавляли в раствор (S)-трет-бутил-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-карбоксилата [3] (230 мг, 0,7 ммоль) в CH2Cl2 (6 мл) при 0°C, затем добавляли пиридин (0,11 мл, 1,4 ммоль). Эту смесь перемешивали при 0°C в течение 2 минут и затем при комнатной температуре в течение 1 ч. Смесь концентрировали, и остаток обрабатывали EtOAc и водой и экстрагировали EtOAc. Объединенные органические фазы промывали водой и рассолом и сушили над MgSO4. Растворитель удаляли под вакуумом с получением соединения 5 в виде светло-желтого твердого вещества (235 мг, 91%). ЖХ/МС (Протокол В): m/z 398,3 [M+H].
Получение (S)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата (6): В круглодонную колбу, содержащую (S)-трет-бутил-5-ацетокси-1-(хлорметил)-1H-бензо[е]индол-3(2Н)-карбоксилат (375 мг, 0,998 мМ), добавляли 10 мл 4М HCl в диоксане (40 мМ). Эту реакционную смесь перемешивали при комнатной температуре, и затем растворитель удаляли в вакууме с получением соединения 6 (312 мг, 100%).
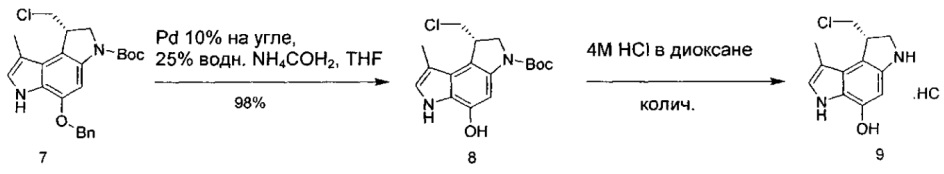
Получение трет-бутил-(1S)-1-(хлорметил)-5-гидрокси-8-метил-1,6-дигидропирроло[3,2-е]индол-3(2Н)-карбоксилата (8): В перемешиваемый раствор соединения 7 (смотри J. Am. Chem. Soc. 1987, 109, 6837-6838) (12,2 г, 28,6 ммоль) в 200 мл THF при 0°C добавляли 10%-ный (масс.) палладий на угле (4 г), затем медленно по каплям добавляли 30 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение примерно 90 мин. Реакционную смесь разбавляли диэтиловым эфиром, после чего добавляли сульфат натрия. Реакционную смесь фильтровали через тонкий слой целита, который затем промывали дважды диэтиловым эфиром. Органические фазы объединяли и затем концентрировали, после чего помещали под вакуум с получением соединения 7 (9,65 г, количественный выход) в виде светло-серого твердого вещества. ЖХ/МС (Протокол В): m/z 337,2 [M+H]+, время удерживания = 1,81 мин.
Получение (S)-8-(хлорметил)-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ола
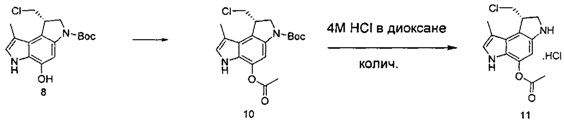
Стадия 1: Синтез трет-бутил-(1S)-5-(ацетилокси)-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3(2Н)-карбоксилата (188): В перемешиваемый раствор соединения 43 (1,99 г, 5,91 ммоль) в 30 мл дихлорметана при 0°C добавляли ацетилхлорид (0,462 мл, 6,50 ммоль), затем сразу добавляли пиридин (0,714 мл, 8,86 ммоль). Эту реакционную смесь перемешивали при 0°C в течение примерно 10 мин. Реакционную смесь наносили на диоксид кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-15% ацетона в гептанах). Содержимое соответствующих пробирок концентрировали и помещали в глубокий вакуум с получением соединения 10 (2,13 г, 95%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол В): m/z 401,1 [M+Na]+, время удерживания = 1,93 мин.
Стадия 2: Синтез соли (8S)-8-(хлорметил)-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил-ацетата с соляной кислотой (189): В круглодонную колбу, содержащую соединение 10 (606 мг, 1,60 ммоль), добавляли 4М HCl в диоксане (24 мл, 96 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Реакционную смесь концентрировали и затем помещали в глубокий вакуум с получением соединения 11 (589 мг, количественный выход) в виде светло-зеленого твердого вещества. ЖХ/МС (Протокол В): m/z 279,1 [M+H]+, время удерживания = 0,72 мин.
Общая методика A: В перемешиваемый раствор моно- или дикислоты в THF, дихлорметане или смеси обоих растворителей при 0°C добавляли оксалилхлорид (1-2,5 зкв.), затем добавляли каталитическое количество DMF. Реакционную смесь перемешивали при 0°C в течение нескольких минут, после чего оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение периода времени от 30 минут до нескольких часов. Реакционную смесь затем концентрировали в вакууме. В некоторых случаях неочищенное вещество затем подвергали азеотропной перегонке от одного до нескольких раз с гептаном или другим релевантным растворителем или растворителями. Неочищенное вещество затем сушили в глубоком вакууме, после чего использовали на следующей стадии.
Общая методика B: В перемешиваемый раствор амина (2-2,5 экв.) в THF, дихлорметане или смеси обоих растворителей при 0°C (или в некоторых случаях в другом релевантном растворителе или растворителях) добавляли хлорангидрид кислоты или хлорангидрид дикислоты, после чего добавляли пиридин (3-6 экв.), триэтиламин (3-6 экв.) или другое релевантное основание (3-6 экв.). Эту реакционную смесь перемешивали при 0°C в течение периода времени от нескольких секунд до нескольких минут, после чего оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение периода времени от 10 минут до нескольких часов. Реакционную смесь затем концентрировали в вакууме. В некоторых случаях неочищенное вещество затем подвергали азеотропной перегонке несколько раз с гептаном или другим релевантным растворителем или растворителями. В большинстве случаев неочищенное вещество затем очищали описанным методом, таким как хроматография на диоксиде кремния или обращенно-фазная С18 хроматография среднего давления.
Получение (S)-фуран-2,5-диилбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2Н)-ил)метанона) (13)
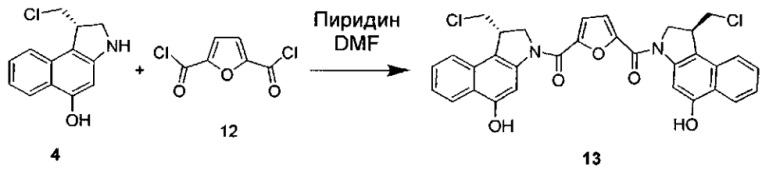
Соединение 4 растворяли в DMF (0,75 мл), добавляли пиридин (13 мкл) и раствор соединения 12 (8 мг) в DMF (0,2 мл, 0,168 ммоль), и полученный раствор перемешивали при комнатной температуре в течение ночи. Смесь разбавляли DCM и промывали водой и рассолом и сушили над MgSO4. Неочищенное вещество очищали флэш-хроматографией на силикагеле (DCM/MOH = 0-10%) с получением продукта 13 в виде зеленого твердого вещества (8 мг, 30%). ЖХ/МС: m/z 587,4 [M+H], время удерживания = 1,0 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.50 (s), 8.14 (d), 7.95 (s), 7.88 (d), 7.55 (t), 7.50 (s), 7.40 (t), 4.78 (m), 4.58 (d), 4.25 (s), 4.02 (d), 3.92 (m).
Получение (S)-((1R,3S)-циклогексан-1,3-диил)бис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2Н)-ил)метанона) (16)
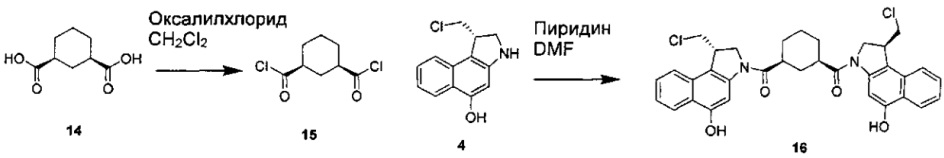
Стадия 1: цис-Циклогексан-1,3-дикарбоновую кислоту (14, 10 мг, 0,058 ммоль) растворяли в THF (2 мл) и добавляли оксалилхлорид (2М в CH2Cl2, 0,09 мл, 0,17 ммоль) и DMF (2 капли) при 0°C. Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 1 ч и концентрировали в вакууме с получением соответствующего хлорангидрида кислоты 15 в виде не совсем белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2: Вышеуказанное соединение 15 растворяли в DMF (2 мл) при 0°C и добавляли HCl-соль (S)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ола (4, 25 мг, 0,093 ммоль), затем добавляли пиридин (0,029 мл, 0,36 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. DMF удаляли при пониженном давлении, и остаток очищали методом ISCO (химическая очистка in situ) с использованием MeOH/DCM (0-20%) с получением продукта 16 в виде темно-синего твердого вещества (8,5 мг, 31%). ЖХ/МС: m/z 603,4 [M+H], время удерживания = 1,03 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.36 (s), 8.09 (d), 8.03 (s), 7.80 (t), 7.53 (t), 7.33 (t), 4.44 (m), 4.33 (d), 4.18 (s), 4.02 (m), 3.85 (m), 2.88 (m), 2.04-1.90 (m), 1.74 (q), 1.52-1.45 (m).
Получение (S)-пиридин-2,6-диилбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)метанона) (18)

(S)-1-(Хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ол (4) (13,5 мг, HCl-соль, 0,05 ммоль) растворяли в DMF (2 мл) и добавляли пиридин (8 мг, 0,10 ммоль), затем добавляли 2,6-пиридиндикарбонилдихлорид (8,5 мг, 0,025 ммоль). Эту смесь перемешивали при комнатной температуре в течение 2 ч. Неочищенное вещество очищали методом ISCO с использованием MeOH/DCM (0-10%) с получением продукта в виде зеленого твердого вещества, который промывали MeOH с получением продукта 18 в виде серого твердого вещества (10 мг, 67%). ЖХ/МС: m/z 598,1 [M+H], время удерживания = 1,0 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.51 (s), 8.29 (t), 8.13 (d), 8.02 (s), 7.82 (d), 7.52 (t), 7.38 (t), 4.63 (s), 4.19-4.10 (m), 3.96 (m), 3.84 (m).
Получение (S)-1,3-фениленбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2Н)-ил)метанона) [20]
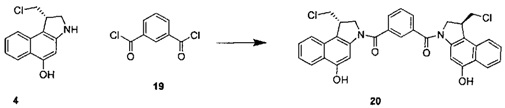
(S)-1-(Хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ол (4) (27 мг, HCl-соль, 0,1 ммоль) растворяли в DMF (2 мл) и добавляли пиридин (0,024 мл, 0,29 ммоль), после чего добавляли хлорангидрид изофталевой кислоты (19, 10 мг, 0,05 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и остаток очищали методом ISCO с использованием MeOH/DCM (0-10%) с получением продукта 20 в виде серого твердого вещества (20 мг, 68%). ЖХ/МС (Протокол В): m/z 597,2 [M+H], время удерживания = 0,99 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.47 (s), 8.13 (d), 7.96 (s), 7.84 (d), 7.72 (t), 7.52 (t), 7.37 (t), 4.44 (s), 4.08 (s), 3.97 (s), 3.86 (s).
Получение (S)-3,3'-тиобис(1-((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)пропан-1-она) (23)
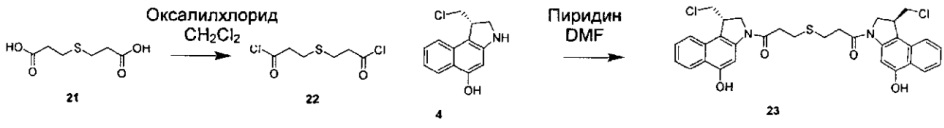
Стадия 1: 3,3'-Тиодипропановую кислоту (21, 8 мг, 0,04 ммоль) растворяли в THF (2 мл) и добавляли оксалилхлорид (2М в CH2Cl2, 0,4 мл, 0,2 ммоль) и DMF (2 капли) при 0°C. Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты 22, который использовали на следующей стадии без дополнительной очистки.
Стадия 2: Вышеуказанное соединение 22 растворяли в DMF (2 мл) при 0°C, добавляли HCl-соль (S)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ола (6) (25 мг, 0,09 ммоль), после чего добавляли пиридин (0,022 мл, 0,27 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. DMF удаляли при пониженном давлении, и остаток очищали методом ISCO с использованием MeOH/DCM (0-10%) с получением продукта 23 в виде не совсем белого твердого вещества (15 мг, 50%). ЖХ/МС (Протокол В): m/z 609,1 [M+H], время удерживания = 1,0 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.36 (s), 8.09 (d), 7.99 (s), 7.79 (d), 7.50 (t), 7.33 (t), 4.37 (m), 4.19 (m), 3.99 (d), 3.82 (m), 2.90-2.82 (m).
Получение (S)-пиридин-3,5-диилбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2Н)-ил)метанона) (26)
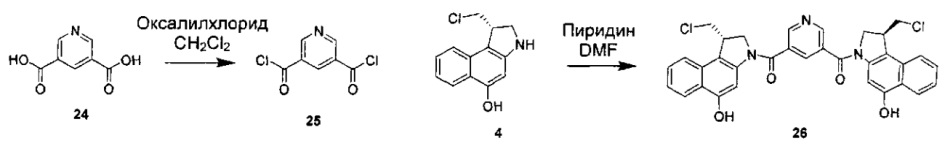
Стадия 1: К пиридин-3,5-дикарбоновой кислоте (24, 7 мг, 0,04 ммоль) добавляли 2 мл DCM, после чего добавляли 2М оксалилхлорид (0,2 мл, 0,4 ммоль) и DMF (2 капли). Прозрачный раствор перемешивали при комнатной температуре в течение 2 ч и концентрировали с получением соответствующего хлорангидрида кислоты 25 в виде желтого твердого вещества.
Стадия 2: Вышеуказанное твердое вещество 25 растворяли в DMF (0,2 мл), и этот раствор добавляли в раствор HCl-соли (S)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ола (4) (25 мг, 0,09 ммоль) в DMF (1 мл), после чего добавляли пиридин (0,02 мл, 0,25 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме, и остаток очищали методом ISCO (MeOH/DCM=0-10%) с получением продукта 26 в виде серого твердого вещества (20 мг, 80%). ЖХ/МС (Протокол В): m/z 598,1 [M+H], время удерживания = 0,95 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.55 (s), 9.00 (s), 8.2 (s), 7.97 (s), 7.84 (d), 7.53 (t), 7.36 (t), 4.50 (s), 4.10 (s), 3.98 (s), 3.86 (s).
Получение (S)-тиофен-2,5-диилбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)метанона) (29) и (S)-5-(1-(хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-карбоновой кислоты (30)
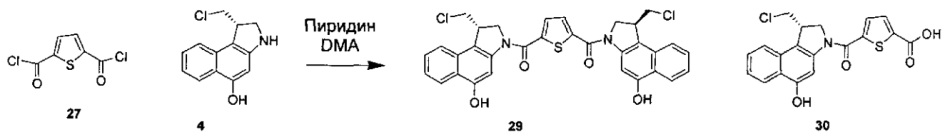
(S)-1-(Хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ол [4] (102 мг, HCl-соль, 0,38 ммоль) растворяли в DMA (2 мл) и добавляли пиридин (0,061 мл, 0,76 ммоль), после чего добавляли тиофен-2,5-дикарбонилдихлорид (27, 40 мг, 0,19 ммоль). Эту смесь перемешивали при комнатной температуре в течение 2 ч. Неочищенное вещество очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением двух продуктов:
(S)-Тиофен-2,5-диилбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)метанон) (29) в виде желтого твердого вещества (60 мг, 52%). ЖХ/МС (Протокол В): m/z 603,0 [M+H], время удерживания = 1,99 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.48 (s), 8.14 (d), 7.86 (m), 7.55 (t), 7.40 (t), 4.78 (t), 4.44 (d), 4.23 (s), 4.03 (d), 3.91 (m).
(S)-5-(1-(Хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбоновая кислота (30) в виде зеленого твердого вещества (23 мг, 31%). ЖХ/МС (Протокол В): m/z 388,1 [M+H], время удерживания = 0,82 мин. 1H ЯМР (400 МГц, MeOD-d4), δ 8.23 (d), 7.82 (m), 7.71 (s), 7.55 (t), 7.40 (t), 4.64 (m), 4.53 (d), 4.15 (t), 4.01 (dd), 3.74 (m).
Получение (S)-(1H-пиррол-2,5-диил)бис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)метанона) (32)
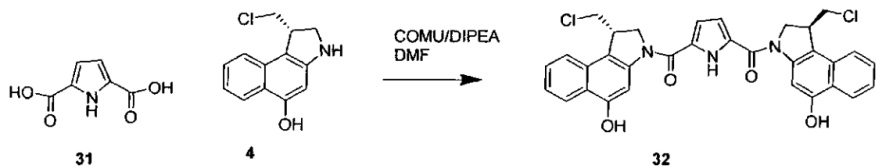
DIPEA (диизопропилэтиламин) (33 мг, 0,25 ммоль) добавляли в раствор 1H-пиррол-2,5-дикарбоновой кислоты (31, 10 мг, 0,064 ммоль) в DMF (1,5 мл), после чего добавляли (S)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ол [4] (38 мг, HCl-соль, 0,14 ммоль) и COMU (гексафторфосфат (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбения) (82 мг, 0,19 ммоль), и эту смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество очищали на системе ВЭЖХ Gilson (ACN (ацетонитрил)/вода, 0,02% TFA) с получением продукта 32 в виде желтого твердого вещества (5 мг, 10%). ЖХ/МС (Протокол В): m/z 586,3 [M+H], время удерживания = 2,04 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 11.66 (s), 10.44 (s), 8.13 (d), 7.92 (s), 7.86 (d), 7.55 (t), 7.38 (t), 5.76 (s), 4.71 (t), 4.44 (d), 4.22 (s), 4.03 (d), 3.88 (m).
Получение (S)-тиофен-2,4-диилбис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)метанона) (35)
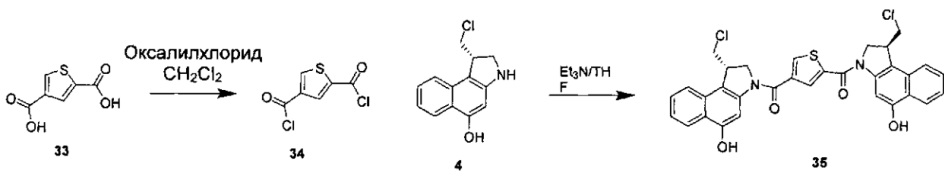
Стадия 1: 2,4-Тиофендикарбоновую кислоту (33, 100 мг, 0,58 ммоль) растворяли в THF (5 мл) и охлаждали до 0°C в ледяной бане. Добавляли оксалилхлорид (0,75 мл, 2М в CH2Cl2, 1,5 ммоль), после чего добавляли 2 капли DMF. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. В этот период времени можно было наблюдать образование белого осадка. Смесь концентрировали в вакууме с получением тиофен-2,4-дикарбонилдихлорида (34) в виде не совсем белого твердого вещества (122 мг, 100%).
Стадия 2: (S)-1-(Хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ол [4] (81 мг, HCl-соль, 0,3 ммоль) растворяли в THF (3 мл) и добавляли Et3N (0,125 мл, 0,9 ммоль) при 0°C, после чего добавляли раствор тиофен-2,4-дикарбонилдихлорида (24, 31,4 мг, 0,15 ммоль) в CH2Cl2 (1 мл). Эту смесь перемешивали при 0°C в течение 5 минут и затем перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали, и остаток обрабатывали MeOH, и полученное желтое твердое вещество собирали фильтрованием с получением неочищенного продукта. Этот неочищенный продукт очищали на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением продукта 35 в виде желтого твердого вещества (40 мг, 44%). ЖХ/МС (Протокол В): m/z 603,3 [M+H], время удерживания = 1,96 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.46 (d), 8.41 (s), 8.13 (d), 8.05 (s), 7.87 (t), 7.54 (t), 7.39 (m), 4.81 (t), 4.61 (s), 4.46 (d), 4.21 (m), 4.18 (m), 4.00 (m), 3.98-3.86 (m).
Получение (S)-(1-метил-1H-пиррол-2,5-диил)бис(((S)-1-(хлорметил)-5-гидрокси-1H-бензо[е]индол-3(2H)-ил)метанона) (38)
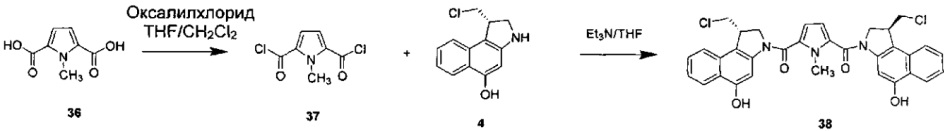
Стадия 1: 1-Метил-1H-пиррол-2,5-дикарбоновую кислоту (36, 20 мг, 0,12 ммоль) растворяли в THF (2 мл) и добавляли оксалилхлорид (2М в CH2Cl2, 0,18 мл, 0,35 ммоль) и DMF (2 капли) при 0°C. Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты 37 в виде не совсем белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2: Вышеуказанное соединение 37 растворяли в THF (2 мл) при 0°C, добавляли соль (S)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ола с HCl [4] (65 мг, 0.24 ммоль), после чего добавляли Et3N (0,1 мл, 0,71 ммоль). Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 38 в виде не совсем белого твердого вещества (31 мг, 44%). ЖХ/МС: m/z 600,5 [M+H], время удерживания = 1,04 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.44 (s), 8.13 (d), 7.84 (d), 7.75 (s), 7.53 (t), 7.38 (t), 6.78 (s), 4.60 (t), 4.30 (d), 4.08 (s), 4.02 (d), 3.9 (s), 3.87 (d).
Получение 3-амино-1,5-бис-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-пентан-1,5-диона (40).
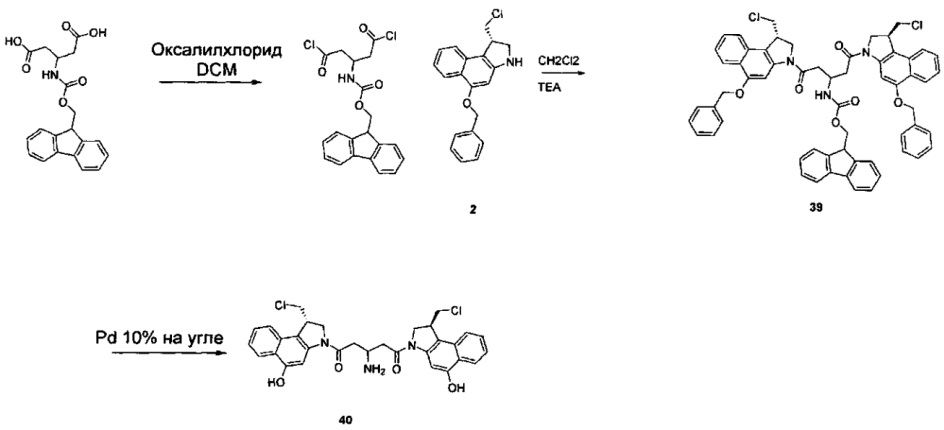
Стадия 1: В продуваемую N2 круглодонную колбу, содержащую 3-(9H-флуорен-9-илметоксикарбониламино)-пентандикислоту (918 мг, 2,48 ммоль) в 20 мл безводного дихлорметана добавляли оксалилхлорид (5,22 ммоль, 0,469 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме до неочищенного остатка. Этот остаток переносили в дихлорметан (10 мл) и по каплям добавляли в круглодонную колбу, содержащую соединение (2) (1610 мг, 4,97 ммоль) в 25 мл дихлорметана и триэтиламина (2,08 мл). Неочищенную реакционную смесь концентрировали в вакууме и обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (39) (2,103 г, 86%) в виде палево-белого твердого вещества. ЖХ/МС (Протокол В): m/z 982 [М+Н+], время удерживания = 2,81 мин.
Стадия 2: Перемешиваемый раствор соединения 39, 9H-флуорен-9-илметилового эфира {3-((S)-5-бензилокси-1-хлорметил-1,2-дигидро-бензо[е]индол-3-ил)-1-[2-((S)-5-бензилокси-1-хлорметил-1,2-дигидро-бензо[е]индол-3-ил)-2-оксо-этил]-3-оксо-пропил}-карбаминовой кислоты (92 мг, 0,104 ммоль), в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (16 мг, 0,15 ммоль), после чего медленно по каплям добавляли 1 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан и промывали водой. Дихлорметановый слой концентрировали, добавляли 2 мл 1М HCl (водн.) и концентрировали. Остаток переносили в этилацетат, и твердое вещество отфильтровывали с получением соединения 40 в виде белого твердого вещества (52 мг, 51%). ЖХ/МС (Протокол В): m/z 578 [M+H+], время удерживания = 1,42 мин.
Получение 3-(3-Амино-фенил)-1,5-бис-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-пентан-1,5-диона (44)
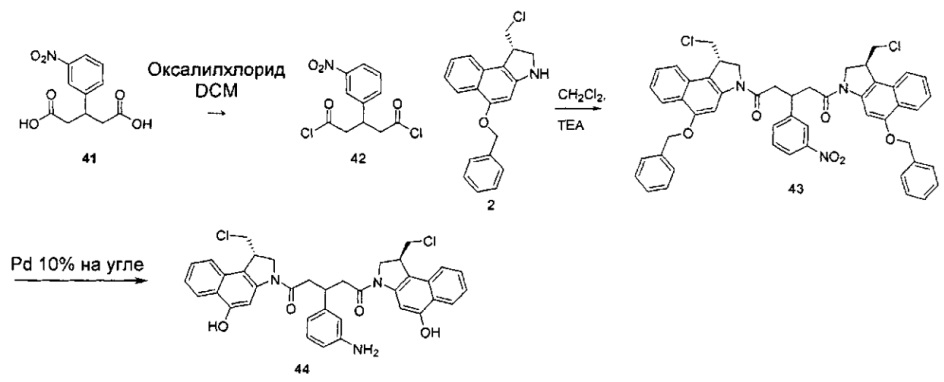
Стадия 1: В продуваемую N2 круглодонную колбу, содержащую 3-(3-нитро-фенил)-пентандикислоту (3,330 мг, 1,30 ммоль) в 15 мл безводного дихлорметана добавляли оксалилхлорид (2,6 ммоль, 0,24 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме с получением соединения 42 в виде белого твердого вещества (378 мг, 1,30 ммоль, количественный выход).
Стадия 2: В круглодонную колбу, содержащую соединение 2 (124 мг, 0,344 ммоль) в 15 мл дихлорметана, добавляли 3-(3-нитро-фенил)-пентандиоилдихлорид (42) (42 мг, 0,172 ммоль). Затем добавляли триэтиламин (0,08 мл), и систему перемешивали в течение 1 часа при комнатной температуре. Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Это неочищенное твердое вещество переносили в 10% MeOH в EtOAc, и белое твердое вещество отфильтровывали с получением целевого продукта 43 (120 мг, 0,172 ммоль, 80%). ЖХ/МС (Протокол В): m/z 864 [M+H+], время удерживания = 2,75 мин.
Стадия 3: Перемешиваемый раствор соединения 43 (85 мг, 0,098 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (16 мг, 0,15 ммоль), после чего медленно по каплям добавляли 2 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан и промывали водой. Дихлорметановый слой концентрировали, добавляли 2 мл 1М HCl (водн.) и концентрировали. Остаток переносили в этилацетат, и твердое вещество отфильтровывали с получением соединения (44) в виде белого твердого вещества (35 мг, 52%). ЖХ/МС: m/z 654 [M+H+], время удерживания = 1,93 мин.
Получение 3-(4-амино-фенил)-1,5-бис-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-пентан-1,5-диона 48
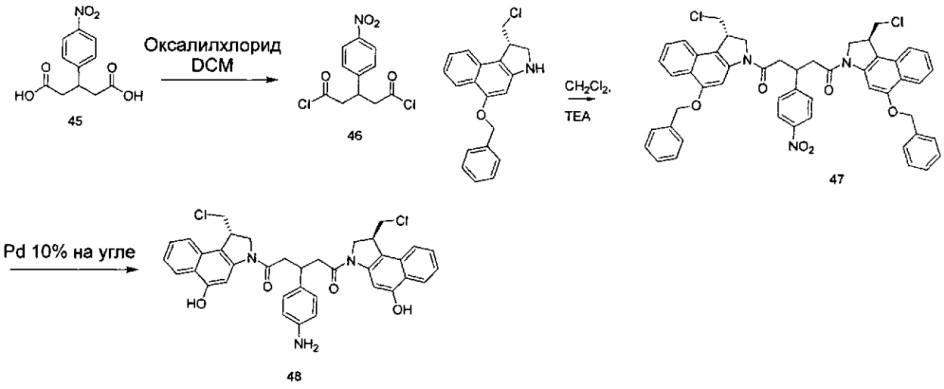
Стадия 1: В продуваемую N2 круглодонную колбу, содержащую 3-(4-нитро-фенил)-пентандикислоту (45, 110 мг, 0,434 ммоль) в 5 мл безводного DCM добавляли оксалилхлорид (0,911 ммоль, 0,082 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме с получением соединения (46) в виде белого твердого вещества (125 мг, 0,434 ммоль, количественный выход). ЖХ/МС, в метаноле: m/z 282,0 [M+H+, для бис-продукта метанолиза], время удерживания = 1,38 мин. (7) (известное коммерчески и в литературе: Тетраhedron, 63(39), 9741-9745; 2007
Стадия 2: В круглодонную колбу, содержащую соединение 2 (111 мг, 0,344 ммоль) в 15 мл дихлорметана, добавляли 3-(4-нитро-фенил)-пентандиоилдихлорид (46) (50 мг, 0,172 ммоль). Затем добавляли триэтиламин (0,144 мл), и систему перемешивали в течение 1 часа при комнатной температуре. Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2x). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Это неочищенное твердое вещество переносили в 10% МеОН в EtOAc, и белое твердое вещество отфильтровывали с получением целевого продукта (47) (101 мг, 0,115 ммоль, 68%). ЖХ/МС: m/z 864 [M+H+], время удерживания = 2,72 мин.
Стадия 3: (10). Перемешиваемый раствор соединения (47), 3-(4-нитро-фенил)-1,5-бис-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-пентан-1,5-диона (90 мг, 0,1 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (17 мг, 0,16 ммоль), после чего медленно по каплям добавляли 1 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан и промывали водой. Дихлорметановый слой концентрировали, добавляли 2 мл 1М HCl (водн.) и концентрировали. Остаток переносили в этилацетат, и твердое вещество отфильтровывали с получением соединения 48 в виде белого твердого вещества. (44 мг, 61%). ЖХ/МС: m/z 654 [M+H+], время удерживания = 1,73 мин.
Получение (S)-3-{2-[2-((S)-5-ацетокси-1-хлорметил-1,2-дигидро-бензо[е]индол-3-ил)-2-оксо-этиламино]-ацетил}-1-хлорметил-2,3-дигидро-1H-бензо[е]индол-5-илового эфира уксусной кислоты (53)
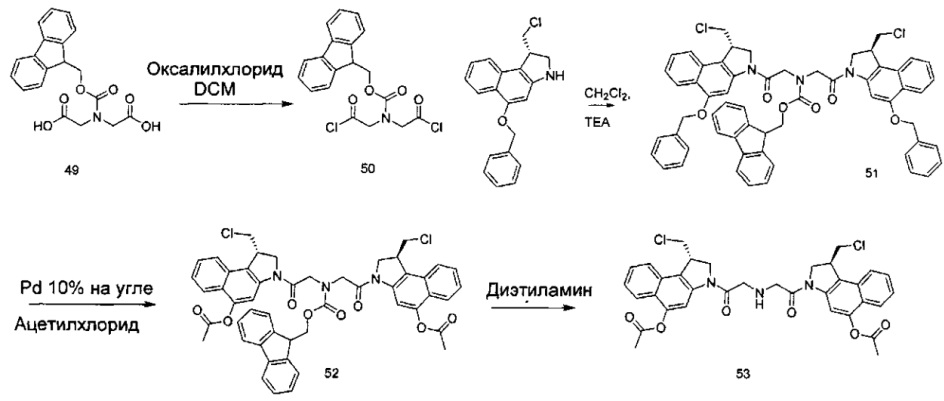
Стадия 1: В продуваемую N2 круглодонную колбу, содержащую 3-[карбоксиметил-(9H-флуорен-9-илметоксикарбонил)-амино]-уксусную кислоту (49, 300 мг, 0,844 ммоль) в 5 мл безводного DCM, добавляли оксалилхлорид (1,94 ммоль, 0,175 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме с получением соединения (50) в виде белого твердого вещества (330 мг, 0,844 ммоль, количественный выход). ЖХ/МС, в метаноле: m/z 384,0 [M+H+, для бис-продукта метанолиза]. Время удерживания = 1,91 мин.
Стадия 2: В круглодонную колбу, содержащую соединение 2 (76 мг, 0,21 ммоль) в 5 мл дихлорметана, добавляли соединение 50 (41 мг, 0,105 ммоль). Затем добавляли триэтиламин (0,088 мл), и эту систему перемешивали в течение 1 часа при комнатной температуре. Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2x). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-75% этилацетата в гептанах) с получением соединения (51) (91 мг, 90%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 966 [M+H+], время удерживания = 2,91 мин.
Стадия 3: Перемешиваемый раствор соединения 51 (40 мг, 0,041 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (10 мг, 0,09 ммоль), после чего медленно по каплям добавляли 1 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан, добавляли ацетилхлорид (1 мл), и реакционную смесь затем концентрировали в вакууме. Остаток обратно переносили в 15 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2x). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (52) (27 мг, 76%) в виде белого твердого вещества. ЖХ/МС: m/z 870 [M+H+], время удерживания = 2,51 мин.
Стадия 4: В оснащенную мешалкой круглодонную колбу, содержащую соединение 52 (25 мг, 0,29 ммоль), добавляли 5 мл дихлорметана и 5 мл диэтиламина. Этот раствор перемешивали в течение 3 часов. Реакционную смесь концентрировали в вакууме и переносили в смесь 50% дихлорметана и гептана и снова концентрировали в вакууме. Эту процедуру повторяли 3 раза. Неочищенное твердое вещество переносили в смесь 50% тетрагидрофурана и 1М HCl (водн.). Белое твердое вещество переносили в эфир и отфильтровывали с получением соединения (15) в виде белого твердого вещества (14 мг, 70%). ЖХ/МС: m/z 648 [M+H+], время удерживания = 1,78 мин.
Получение 3-(4-амино-фенил)-N,N-бис-[2-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-2-оксо-этил]-пропионамида (56)
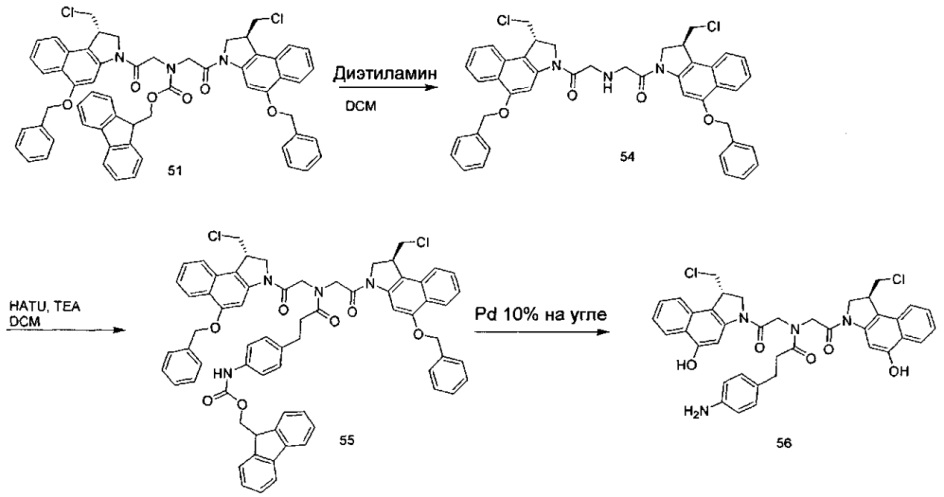
Стадия 1: В оснащенную мешалкой круглодонную колбу, содержащую соединение 51 (300 мг, 0,310 ммоль), добавляли 5 мл дихлорметана и 5 мл диэтиламина. Этот раствор перемешивали в течение 3 часов. Реакционную смесь концентрировали в вакууме и переносили в смесь 50% дихлорметана и гептана и снова концентрировали в вакууме. Эту процедуру повторяли 3 раза с получением соединения (54) в виде белого твердого вещества. (216 мг, 93%). ЖХ/МС: m/z 744 [M+H+], время удерживания = 2,26 мин.
Стадия 2: В продуваемую N2 круглодонную колбу, содержащую соединение 54 (100 мг, 0,134 ммоль) в 5 мл безводного дихлорметана, добавляли 3-[4-(9H-флуорен-9-илметоксикарбониламино)-фенил]-пропионовую кислоту (52 мг, 0,134 ммоль). В этот раствор добавляли гексафторфосфат(диметиламино)-N,N-диметил(3H-[1,2,3]триазоло[4,5-b]пиридин-3-илокси)метанаминия (52 мг, 0,134 ммоль) и триэтиламин (0,05 мл). Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме до неочищенного остатка. Этот остаток обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2x). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (55) (130 мг, 87%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 1113 [M+H+], время удерживания = 2,771 мин.
Стадия 3: Перемешиваемый раствор соединения 55 (115 мг, 0,103 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (10 мг, 0,1 ммоль), после чего медленно по каплям добавляли 1 мл 25%-ного формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан и промывали водой. Дихлорметановый слой концентрировали, добавляли 2 мл 1М HCl (водн.) и концентрировали. Остаток переносили в этилацетат, и твердое вещество отфильтровывали с получением соединения (56) в виде белого твердого вещества. (26 мг, 34%). ЖХ/МС: m/z 711 [M+H+], время удерживания = 1,6 мин.
Получение 9H-флуорен-9-илметилового эфира [(S)-1-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-карбонил)-4-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-4-оксо-бутил]-карбаминовой кислоты 60
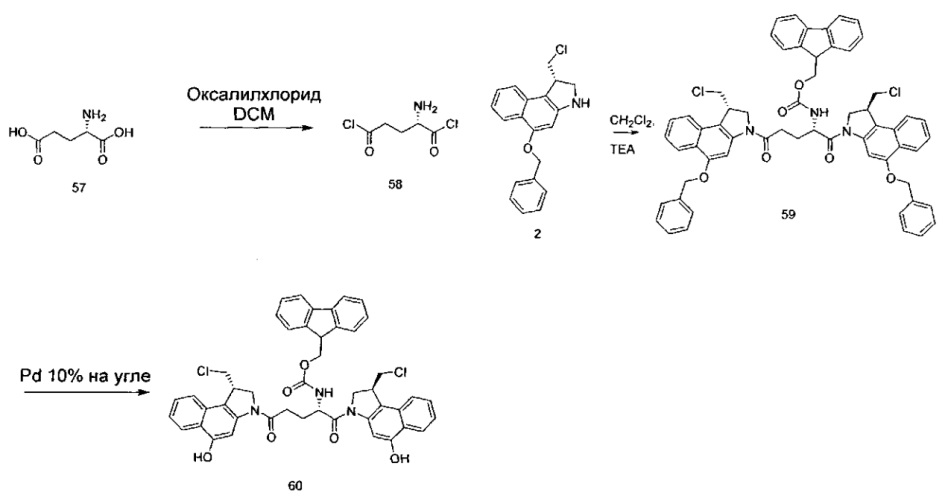
Стадия 1: В продуваемую N2 круглодонную колбу, содержащую (S)-2-(9H-флуорен-9-илметоксикарбониламино)-пентандикислоту 57 (400 мг, 1,08 ммоль) в 15 мл безводного дихлорметана, добавляли оксалилхлорид (2,27 ммоль, 0,205 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме до неочищенного остатка 58. Этот остаток переносили в дихлорметан (10 мл) и по каплям добавляли в круглодонную колбу, содержащую соединение 2 (700 мг, 2,17 ммоль) в 10 мл дихлорметана и триэтиламине (0,905 мл). Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (59) (260 мг, 24%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 980 [M+H+], время удерживания = 2,84 мин.
Стадия 2: Перемешиваемый раствор соединения (59), (250 мг, 0,255 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (64 мг, 12,8 ммоль), после чего медленно по каплям добавляли 2,1 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 30 мин. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан и промывали водой. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (60) (121 мг, 59%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 800 [M+H+], время удерживания = 2,25 мин.
Получение [3-((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-3-оксо-пропил]-амида (S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-карбоновой кислоты (31). Перемешиваемый раствор соединения (65)
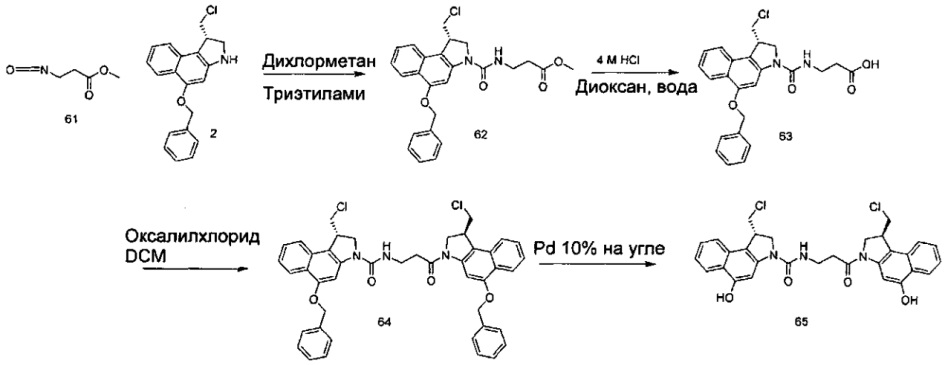
Стадия 1: В круглодонную колбу, содержащую соединение (2) (200 мг, 0,555 ммоль) в дихлорметане (10 мл), по каплям добавляли метиловый эфир 3-изоцианато-пропионовой кислоты 61 (79 мг, 0,555 ммоль) и триэтиламин (0,5 мл). Эту реакционную смесь перемешивали в течение 3 часов. Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (62) (0,231 мг, 89%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 467 [M+H+], время удерживания = 2,11 мин. ЯМР yes
Стадия 2: В оснащенную мешалкой круглодонную колбу, содержащую соединение 62 (230 мг, 0,493 ммоль), добавляли 5 мл 1М HCl (водн.) в 5 мл тетрагидрофурана. Этот раствор перемешивали в течение 3 часов при 70°C. Реакционную смесь концентрировали в вакууме и переносили в 50% дихлорметан в гептане и концентрировали в вакууме. Эту процедуру повторяли 3 раза с получением соединения (63) (180 мг, 83%) в виде белого твердого вещества после концентрирования. ЖХ/МС: m/z 439 [M+H+], время удерживания = 1,83 мин.
Стадия 3: В продуваемую N2 круглодонную колбу, содержащую соединение (63) (110 мг, 0,250 ммоль) в 5 мл безводного DCM, добавляли оксалилхлорид (0,250 ммоль, 0,02 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме до неочищенного остатка. Этот остаток переносили в дихлорметан (10 мл) и по каплям добавляли в круглодонную колбу, содержащую соединение 2 (90 мг, 0,250 ммоль) в 10 мл дихлорметана и триэтиламине (0,5 мл). Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 15 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2x). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (64) (80 г, 43%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 744 [M+H+], время удерживания = 2,60 мин.
Стадия 4:, Соединение 64 (75 мг, 0,100 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (25 мг, 0,24 ммоль), после чего медленно по каплям добавляли 1 мл 25%-ного раствора формиата аммония в воде. Эту реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Неочищенный остаток переносили в дихлорметан и промывали водой. Дихлорметановый слой концентрировали, добавляли 2 мл 1М HCl (водн.) и концентрировали. Остаток переносили в этилацетат, и твердое вещество отфильтровывали с получением соединения (65) в виде белого твердого вещества (15 мг, 26%). ЖХ/МС: m/z 564 [M+H+], время удерживания = 1,88 мин.
Получение (1S,1'S)-3,3'(1H-пиррол-2,5-дикарбонил)бис(1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5,3-диил)диацетата [68]
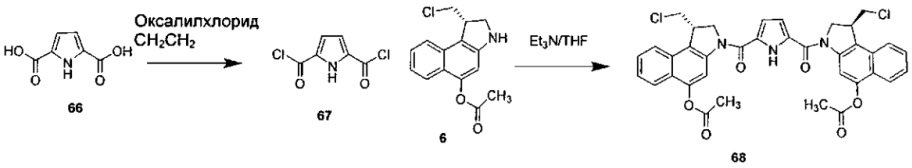
Стадия 1: 1H-пиррол-2,5-дикарбоновую кислоту (32, 50 мг, 0,3 ммоль) растворяли в THF (5 мл) при 0°C, добавляли оксалилхлорид (0,4 мл, 2М в CH2Cl2, 0,8 ммоль), после чего добавляли 2 капли DMF. Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 2 ч. Смесь концентрировали в вакууме с получением 1H-пиррол-2,5-дикарбонилдихлорида (33) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2: Его растворяли в THF (12 мл) при 0°C, добавляли 1H-пиррол-2,5-дикарбонилдихлорид (33, со стадии 2), после чего добавляли Et3N (0,28 мл). Эту смесь перемешивали при 0°C в течение 5 минут и при комнатной температуре в течение 3 ч. Смесь концентрировали в вакууме, и остаток обрабатывали MeOH с получением серого твердого вещества. Это твердое вещество собирали фильтрованием с получением неочищенного продукта в виде серого твердого вещества. Этот неочищенный продукт очищали на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением чистого продукта, (1S,1'S)-3,3'-(1H-пиррол-2,5-дикарбонил)бис(1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5,3-диил)диацетата в виде не совсем белого твердого вещества (34, 60 мг, 30%). ЖХ/МС: m/z 670,4 [M+H], время удерживания = 2,20 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 11.77 (s), 8.17 (s), 8.06 (d), 7.92 (d), 7.65 (t), 7.52 (t), 4.80 (t), 4.5 (d), 4.41 (s), 4.10 (d), 4.02 (m), 2.10 (s).
Получение (1S,1'S)-3,3'-(тиазол-2,5-дикарбонил)бис(1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5,3-диил)диацетата [71]
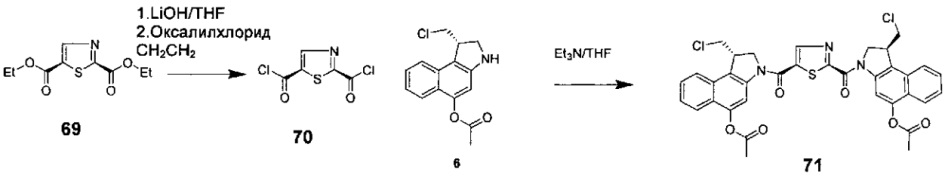
Стадия 1: Диэтилтиазол-2,5-дикарбоксилат (35, 348 мг, 1,5 ммоль) растворяли в THF (10 мл) и добавляли раствор LiOH⋅H2O (383 мг, 9,0 ммоль) в воде (5 мл) при 0°C. Эту смесь перемешивали при 0°C в течение 30 минут, затем при комнатной температуре в течение 4 ч. Смесь концентрировали в вакууме до удаления THF, и остаток подкисляли добавлением 1М водного раствора HCl до pH примерно 4-5. Полученное твердое вещество собирали фильтрованием с получением тиазол-2,5-дикарбоновой кислоты в виде белого твердого вещества (63 мг, 24%). Тиазол-2,5-дикарбоновую кислоту (20 мг, 0,12 ммоль) растворяли в THF (2 мл), добавляли оксалилхлорид (0,18 мл, 2М в DCM) при 0°C, после чего добавляли 2 капли DMF. Смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты 70 в виде белого твердого вещества.
Стадия 2: Желтое твердое вещество 5 суспендировали в THF (3 мл), добавляли хлорангидрид кислоты со стадии 2, после чего добавляли Et3N (0,05 мл, 0,4 ммоль) при 0°C. Эту смесь перемешивали при 0°C в течение 5 минут и затем при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson с получением целевого соединения 71 в виде желтого твердого вещества (3,6 мг, 3,9%). ЖХ/МС: m/z 688,5 [M+H], время удерживания = 2,27 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 8.81 (s), 8.41 (s), 8.27 (s), 8.16 (m), 8.04 (m), 7.74 (m), 7.64 (m), 5.25 (d), 4.94 (q), 4.53 (m), 4.21-4.08 (m), 2.63 (s).
Получение (S)-3-[5-((S)-5-ацетокси-1-хлорметил-1,2-дигидро-бензо[е]индол-3-карбонил)-1-метил-1H-пиразол-3-карбонил]-1-хлорметил-2,3-дигидро-1H-бензо[е]индол-5-илового эфира уксусной кислоты (74)
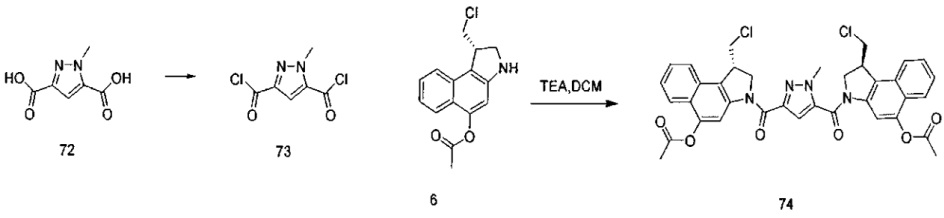
В продуваемую N2 круглодонную колбу, содержащую 1-метил-1H-пиразол-3,5-дикарбоновую кислоту 38 (20 мг, 0,12 ммоль) в 5 мл безводного дихлорметана, добавляли оксалилхлорид (0,248 ммоль, 0,022 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Эту реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме до неочищенного остатка соединения 73. Соединение 73 переносили в дихлорметан (10 мл) и по каплям добавляли в круглодонную колбу, содержащую (S)-1-хлорметил-2,3-дигидро-1H-бензо[е]индол-5-иловый эфир уксусной кислоты (5, 73 мг, 0,236 ммоль) в 5 мл дихлорметана и триэтиламине (2,08 мл). Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2x). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения (74) (12 мг, 15%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 6852 [M+H+], время удерживания = 2,21 мин.
Получение 7-азабицикло[2.2.1]гептан-1,4-диилбис[карбонил(1S)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3,5-диил]-диацетата 79
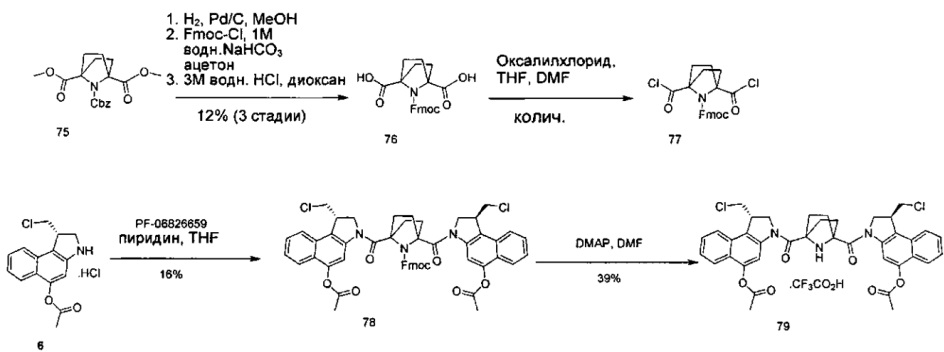
Стадия 1: Смесь 7-бензил-1,4-диметил-7-азабицикло[2.2.1]гептан-1,4,7-трикарбоксилата (3,20 г, 9,21 ммоль) [получен, как описано в Chem. Eur. J. 2012, 18, 1127-1141] в присутствии Pd/C (10%, 1000 мг) гидрировали под давлением баллона при комнатной температуре в течение примерно 2 часов. Реакционную смесь фильтровали через слой целита, и остаток на фильтре промывали раствором 40 мл метанола и 40 мл дихлорметана. Органические фазы объединяли и концентрировали в вакууме с получением светло-желтого твердого вещества. В перемешиваемый раствор этого неочищенного твердого вещества в 40 мл ацетона при 0°C добавляли водн. NaHCO3 (1М, 65 мл, 64,6 ммоль), после чего по каплям добавляли Fmoc-Cl (3,34 г, 12,9 ммоль) в виде раствора в 40 мл ацетона. Реакционную смесь разбавляли 100 мл воды и экстрагировали этилацетатом (100 мл, 3x). Органические фазы объединяли и промывали водой, рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Затем осуществляли хроматографию на силикагеле (градиент: от 12,5% до 17% этилацетата в петролейном эфире). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением белого твердого вещества. Это неочищенное вещество затем суспендировали в водн. HCl (3М, 60 мл) и 80 мл диоксана. Реакционную смесь нагревали до образования флегмы и затем перемешивали при температуре дефлегмации в течение примерно 16 часов. Реакционную смесь затем концентрировали в вакууме до удаления большей части диоксана. Водную фазу затем экстрагировали этилацетатом (100 мл, 2x). Органические фазы объединяли, промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Затем осуществляли хроматографию на силикагеле (градиент: от 8,3% до 25% метанола в дихлорметане). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме и затем снова очищали препаративной ВЭЖХ (метод M, используя от 50% В до 80% В за 30 минут, затем 95% в течение 5 минут) с получением соединения 76 (400 мг, 12%, за 3 стадии) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6): δ 7.81-7.79 (d, 2Н), 7.72-7.71 (d, 2Н), 7.42-7.38 (m, 2Н), 7.34-7.31 (m, 2Н), 4.35-4.33-7.33 (d, 2Н), 4.22-4.19 (m, 1Н), 2.28-2.26 (d, 4Н), 1.93-1.91 (d, 2Н).
Стадия 2: Следуя общей методике A, используя соединение 76 (90 мг, 0,40 ммоль), оксалилхлорид (0,033 мл, 0,39 ммоль), THF (8 мл) и 1 каплю DMF, было получено соединение 77 в виде не совсем белого твердого вещества (79 мг, количественный выход). Неочищенное соединение 77 как есть сразу использовали на следующей стадии.
Стадия 3: Следуя общей методике B, используя соединение 6 (103 мг, 0,331 ммоль), соединение 77 (70 мг, 0,16 ммоль), пиридин (0,051 мл, 0,63 ммоль) и THF (12 мл), было получено неочищенное вещество. Эту реакционную смесь концентрировали в вакууме, растворяли в DMSO и инжектировали в предколонку 12 г C18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 30% до 95% ацетонитрила в воде с 0,02% TFA в каждой фазе). Содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 78 (23 мг, 16%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 922,0 [M+H]+, время удерживания = 2,59 мин.
Стадия 4: В перемешиваемый раствор соединения 78 (17,9 мг, 0,019 ммоль) в 1,0 мл DMF добавляли DMAP (4-(диметиламино)пиридин) (47,4 мг, 0,388 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 60 мин. Неочищенную реакционную смесь инжектировали в предколонку 5 г C18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 30% до 95% ацетонитрила в воде с 0,02% TFA в каждой фазе). Содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 79 (6,1 мг, 39%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол В): m/z 700,1 [M+H]+, время удерживания = 1,47 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 10.26-10.17 (m, 2Н), 8.28-8.24 (m, 2Н), 8.11-8.06 (d, 2Н), 8.00-7.95 (d, 2Н), 7.71-7.64 (t, 2Н), 7.60-7.53 (t, 2Н), 4.56-4.37 (m, 6Н), 4.18-4.05 (m, 4Н), 2.83-2.59 (m, 8Н), 2.49-2.37 (m, 6Н).
Получение (1S,4S)-бицикло[2.1.1]гексан-1,4-диилбис[карбонил(1S)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3,5-диил]-диацетата 82
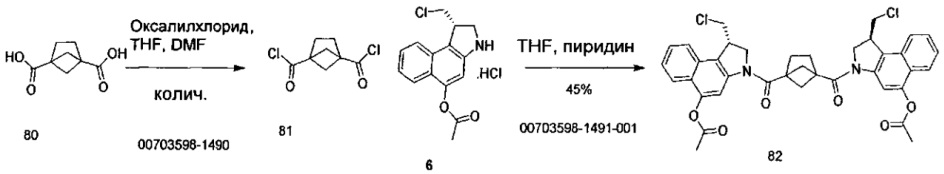
Стадия 1: Следуя общей методике A, используя бицикло[2.1.1]гексан-1,4-дикарбоновую кислоту 80 (30 мг, 0,18 ммоль), оксалилхлорид (0,0303 мл, 0,353 ммоль), THF (4 мл) и 1 каплю DMF, было получено соединение 81 в виде не совсем белого твердого вещества (39 мг, количественный выход). Неочищенное соединение 81 как есть сразу использовали на следующей стадии.
Стадия 2: Следуя общей методике B, используя соединение 6 (106 мг, 0,338 ммоль), соединение 81 (35 мг, 0,17 ммоль), пиридин (0,0545 мл, 0,676 ммоль) и THF (8 мл) и очистку обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 75% ацетонитрила в воде с 0,02% TFA в каждой фазе), было получено соединение 82 (52 мг, 45%) в виде белого твердого вещества. ЖХ/МС (Протокол В): m/z 685,2 [M+H]+, время удерживания = 2,16 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 8.23 (s, 2Н), 8.03-7.99 (d, 2Н), 7.92-7.87 (d, 2Н), 7.63-7.57 (t, 2Н), 7.51-7.44 (t, 2Н), 4.47-4.25 (m, 6Н), 4.13-3.98 (m, 4Н), 2.47 (s, 6Н), 2.27-2.07 (m, 8Н).
Получение бицикло[2.2.2]октан-1,4-диилбис[карбонил(1S)-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3,5(2H)-диил]-диацетата 85
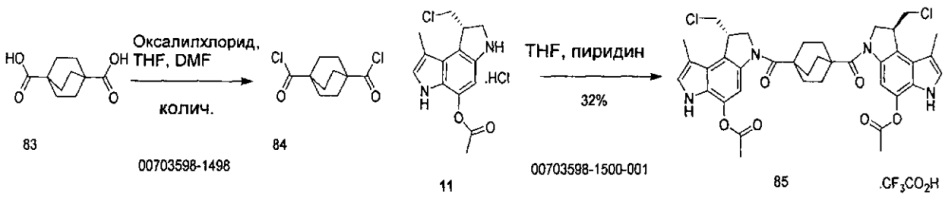
Стадия 1: Следуя общей методике A, используя бицикло[2.2.2]октан-1,4-дикарбоновую кислоту 83 (16 мг, 0,081 ммоль), оксалилхлорид (0,015 мл, 0,17 ммоль), THF (5 мл) и 1 каплю DMF, было получено соединение 84 в виде не совсем белого твердого вещества (19 мг, количественный выход). Неочищенное соединение 84 как есть сразу использовали на следующей стадии.
Стадия 2: Следуя общей методике B, используя соединение 189 (50,9 мг, 0,145 ммоль), соединение 84 (17,0 мг, 0,0723 ммоль), пиридин (0,0233 мл, 0,289 ммоль) и THF (4 мл) и очистку обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 75% ацетонитрила в воде с 0,02% TFA в каждой фазе), было получено соединение 85 (21,6 мг, 32%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 719,3 [M+H]+, время удерживания = 2,27 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 11.00 (s, 2Н), 7.79 (s, 2Н), 7.19 (s, 2Н), 4.68-4.62 (m, 2Н), 4.27-4.19 (m, 2Н), 4.06-3.94 (m, 4Н), 3.65-3.57 (m, 2Н), 2.42-2.32 (m, 12Н), 2.12-1.96 (m, 12Н).
Получение бицикло[2.2.1]гептан-1,4-диилбис[карбонил(1S)-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3,5(2Н)-диил]-диацетата 88
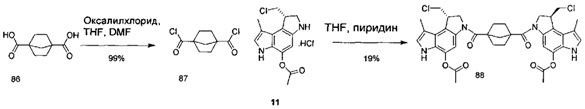
Стадия 1: Следуя общей методике A, используя бицикло[2.2.1]гептан-1,4-дикарбоновую кислоту 86 (16 мг, 0,087 ммоль), оксалилхлорид (0,016 мл, 0,18 ммоль), THF (5 мл) и 1 каплю DMF, было получено соединение 87 в виде не совсем белого твердого вещества (19 мг, 99%). Неочищенное соединение 87 как есть сразу использовали на следующей стадии.
Стадия 2: Следуя общей методике B, используя соединение 189 (54,1 мг, 0,154 ммоль), соединение 87 (17,0 мг, 0,0769 ммоль), пиридин (0,0248 мл, 0,308 ммоль) и THF (4 мл) и очистку обращенно-фазной C18 хроматографией среднего давления (градиент: от 10% до 75% ацетонитрила в воде с 0,02% TFA в каждой фазе), было получено соединение 88 (13,6 мг, 19%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 705,3 [M+H]+, время удерживания = 2,32 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 11.02 (s, 2Н), 7.82 (s, 2Н), 7.19 (s, 2Н), 4.50-4.45 (d, 2Н), 4.26-4.16 (m, 2Н), 4.10-4.02 (m, 2Н), 3.98-3.92 (m, 2Н), 3.65-3.58 (m, 2Н), 2.41-2.33 (m, 12Н), 2.22-2.03 (m, 10Н).
Получение бицикло[1.1.1]пентан-1,3-диилбис[карбонил(1S)-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3,5(2H)-диил]-диацетата 91
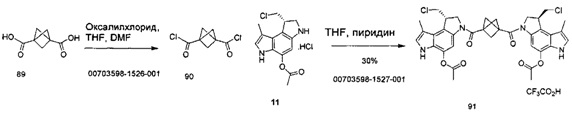
Стадия 1: Следуя общей методике A, используя бицикло[1.1.1]пентан-1,3-дикарбоновую кислоту 89 (31 мг, 0,20 ммоль), оксалилхлорид (0,025 мл, 0,40 ммоль), THF (8 мл) и 1 каплю DMF, было получено соединение 90 в виде не совсем белого твердого вещества (40 мг, количественный выход). Неочищенное соединение 90 как есть сразу использовали на следующей стадии.
Стадия 2: Следуя общей методике B, используя соединение 189 (142 мг, 0,404 ммоль), соединение 90 (39 мг, 0,20 ммоль), пиридин (0,065 мл, 0,81 ммоль) и THF (12 мл) и очистку обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 75% ацетонитрила в воде с 0,02% TFA в каждой фазе), было получено соединение 91 (45,5 мг, 30%) в виде светло-серого твердого вещества. ЖХ/МС (Протокол B): m/z 677,2 [M+H]+, время удерживания = 1,89 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 11.04 (s, 2Н), 7.78 (s, 2Н), 7.20 (s, 2Н), 4.47-4.39 (m, 2Н), 4.36-4.26 (m, 2Н), 4.18-4.08 (m, 2Н), 4.03-3.94 (m, 2Н), 3.77-3.66 (m, 2Н), 2.56 (s, 6Н), 2.41-2.31 (m, 12Н).
Получение (8S)-6-[(3-{[(1S)-5-(ацетилокси)-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)ацетил]-8-(хлорметил)-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил-ацетата 97
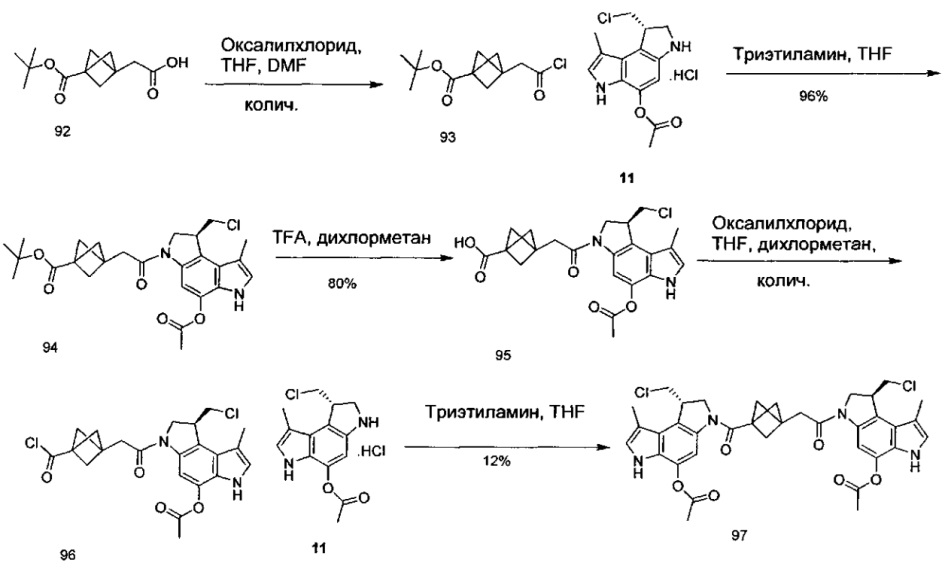
Стадия 1: Следуя общей методике A, используя 3-(2-трет-бутокси-2-оксоэтил)бицикло[1.1.1]пентан-1-карбоновую кислоту 92 [получена, как описано в Bioorg. Med. Chem. 2009, 17, 242-250.] (90 мг, 0,40 ммоль), оксалилхлорид (0,041 мл, 0,477 ммоль), THF (8 мл) и 1 каплю DMF, было получено соединение 93 в виде не совсем белого твердого вещества (103 мг, количественный выход). Неочищенное соединение 93 как есть сразу использовали на следующей стадии.
Стадия 2: Следуя общей методике B, используя соединение 11 (141 мг, 0,40 ммоль), соединение 93 (98 мг, 0,40 ммоль), триэтиламин (0,168 мл, 1,20 ммоль) и THF (30 мл) и очистку хроматографией на силикагеле (градиент: от 0% до 35% ацетона в гептане), было получено соединение 94 (188 мг, 96%) в виде не совсем белого твердого вещества. ЖХ/МС (Протокол B): m/z 487,2 [M+H]+, время удерживания = 2,04 мин.
Стадия 3: В перемешиваемый раствор соединения 94 (184 мг, 0,378 ммоль) в 8 мл дихлорметана добавляли TFA (4,0 мл, 52 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 45 мин. Реакционную смесь концентрировали в вакууме и помещали в глубокий вакуум с получением соединения 95 (164 мг, 80%) в виде светло-серого твердого вещества, которое использовали на следующей стадии без очистки. ЖХ/МС (Протокол В): m/z 431,7 [M+H]+, время удерживания = 1,39 мин.
Стадия 4: Следуя общей методике A, используя соединение 95 (55 мг, 0,101 ммоль), оксалилхлорид (0,0104 мл, 0,121 ммоль), THF (3 мл), дихлорметан (1 мл) и 1 каплю DMF, было получено соединение 96 в виде не совсем белого твердого вещества (46 мг, количественный выход). Неочищенное соединение 96 как есть сразу использовали на следующей стадии.
Стадия 5: Следуя общей методике B, используя соединение 11 (31,3 мг, 0,089 ммоль), соединение 96 (40 мг, 0,089 ммоль), пиридин (0,0215 мл, 0,267 ммоль) и THF (8,0 мл) и очистку обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 70% ацетонитрила в воде с 0,02% TFA в каждой фазе), было получено соединение 97 (10,1 мг, 12%) в виде светло-серого твердого вещества. ЖХ/МС (Протокол B): m/z 691,3 [M+H]+, время удерживания = 1,93 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 11.02 (s, 2Н), 7.86-7.72 (d, 2Н), 7.19 (s, 2Н), 4.43-4.36 (m, 1Н), 4.28-4.14 (m, 3Н), 4.13-4.05 (m, 2Н), 3.96-3.89 (m, 2Н), 3.68-3.60 (m, 2Н), 2.89-2.82 (m, 2Н), 2.73-2.66 (m, 2Н), 2.40-2.30 (m, 12Н), 2.24-2.15 (m, 6Н).
Получение трет-бутил-(1S)-8-амино-5-(бензилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата 99 и трет-бутил-(1R)-8-амино-5-(бензилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата 98 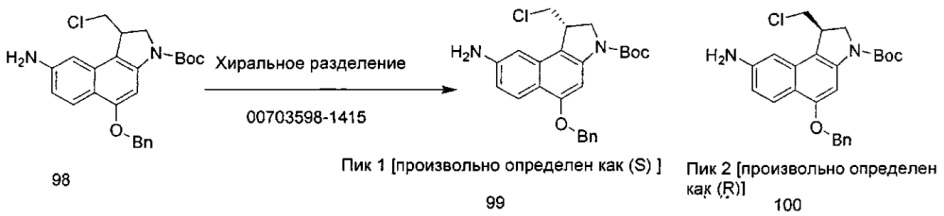
трет-Бутил-8-амино-5-(бензилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилат 98 [полученный с использованием химии, описанной в J. Med. Chem. 2012, 55, 5878-5886] разделяли, используя суперкритическую флюидную хроматографию (метод L1). Пик 1 концентрировали в вакууме с получением соединения 99 (385 мг), которому было произвольно присвоено обозначение (S). ЖХ/МС (Протокол В): m/z 439,1 [M+H]+, время удерживания = 2,34 мин. Пик 2 концентрировали в вакууме с получением соединения 100 (401 мг), которому произвольно было присвоено обозначение (R). ЖХ/МС (Протокол В): m/z 439,1 [M+H]+, время удерживания = 2,34 мин.
Получение трет-бутил-(1R)-8-(ацетиламино)-5-(ацетилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата 102
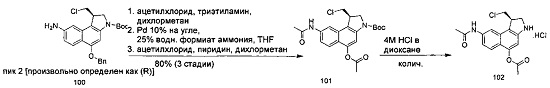
Стадия 1: В перемешиваемый раствор соединения 99 (60 мг, 0,14 ммоль) в 6 мл дихлорметана при 0°C добавляли ацетилхлорид (0,015 мл, 0,206 ммоль), после чего добавляли триэтиламин (0,029 мл, 0,206 ммоль). Эту реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 25 мин. Реакционную смесь разбавляли дихлорметаном и затем переносили в делительную воронку. Органический слой отделяли и затем промывали 1 н. HCl и затем водой. Органический слой сушили над сульфатом натрия, фильтровали и затем концентрировали в вакууме с получением оранжевого твердого вещества. В перемешиваемый раствор неочищенного вещества в 4 мл THF при 0°C добавляли 10% Pd на угле (45 мг), после чего добавляли 25%-ный водный раствор формиата аммония (0,3 мл). Реакционную смесь перемешивали при 0°C в течение примерно 4 часов. Реакционную смесь разбавляли THF и диэтиловым эфиром. Добавляли сульфат натрия, и реакционную смесь фильтровали через тонкий слой целита. Органические фазы концентрировали в вакууме и помещали в глубокий вакуум с получением светло-коричневого твердого вещества. В перемешиваемый раствор неочищенного вещества в 6 мл дихлорметана при 0°C добавляли ацетилхлорид (0,015 мл, 0,211 ммоль), после чего добавляли пиридин (0,017 мл, 0,211 ммоль). Реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 25 мин. Реакционную смесь концентрировали в вакууме. Затем осуществляли хроматографию на силикагеле (градиент: от 0% до 45% ацетона в гептане). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением соединения 101 (49 мг, 80%, за 3 стадии) в виде не совсем белого твердого вещества. ЖХ/МС (Протокол В): m/z 455,9 [M+Na]+23, время удерживания = 2,05 мин.
Стадия 2: В круглодонную колбу, содержащую соединение 101 (45 мг, 0,10 ммоль), добавляли 4М HCl в диоксане (6,0 мл, 24 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 2 часов. Реакционную смесь концентрировали в вакууме и помещали в глубокий вакуум с получением соединения 102 (42 мг, количественный выход) в виде темно-коричневого твердого вещества. ЖХ/МС (Протокол В): m/z 333,0 [M+H]+, время удерживания = 1,65 мин.
Получение трет-бутил-(1S)-8-(ацетиламино)-5-(ацетилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-карбоксилата 103
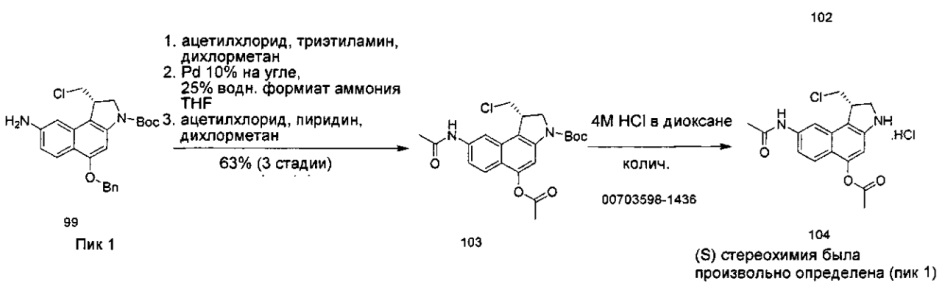
Стадия 1: В перемешиваемый раствор соединения 99 (65 мг, 0,15 ммоль) в 6 мл дихлорметана при 0°C добавляли ацетилхлорид (0,016 мл, 0,22 ммоль), после чего добавляли триэтиламин (0,031 мл, 0,22 ммоль). Эту реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 25 мин. Реакционную смесь разбавляли дихлорметаном и затем переносили в делительную воронку. Органический слой отделяли и затем промывали 1 н. HCl и затем водой. Органический слой сушили над сульфатом натрия, фильтровали и затем концентрировали в вакууме с получением оранжевого твердого вещества. В перемешиваемый раствор неочищенного вещества в 4 мл THF при 0°C добавляли 10% Pd на угле (45 мг), после чего добавляли 25%-ный водный раствор формиата аммония (0,5 мл). Реакционную смесь перемешивали при 0°C в течение примерно 4 часов. Реакционную смесь разбавляли THF и диэтиловым эфир. Добавляли сульфат натрия, и реакционную смесь фильтровали через тонкий слой целита. Органические фазы концентрировали в вакууме и помещали в глубокий вакуум с получением светло-коричневого твердого вещества. В перемешиваемый раствор неочищенного вещества в 8 мл дихлорметана при 0°C добавляли ацетилхлорид (0,015 мл, 0,21 ммоль), после чего добавляли пиридин (0,017 мл, 0,21 ммоль). Реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 25 мин. Реакционную смесь концентрировали в вакууме. Затем осуществляли хроматографию на силикагеле (градиент: от 0% до 25% ацетона в гептане). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением соединения 103 (39,1 мг, 63%, за 3 стадии) в виде белого твердого вещества. ЖХ/МС (Протокол В): m/z 455,0 [M+Na]+23, время удерживания = 2,00 минуты.
Стадия 2: В круглодонную колбу, содержащую соединение 103 (37 мг, 0,085 ммоль), добавляли 4М HCl в диоксане (4,0 мл, 16 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 2 часов. Реакционную смесь концентрировали в вакууме и помещали в глубокий вакуум с получением соединения 104 (34 мг, количественный выход) в виде зеленого твердого вещества. ЖХ/МС (Протокол В): m/z 333,0 [M+H]+, время удерживания = 1,41 мин.
Получение (1S)-3-{[3-(хлоркарбонил)бицикло[1.1.1]пент-1-ил]карбонил}-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ила 107
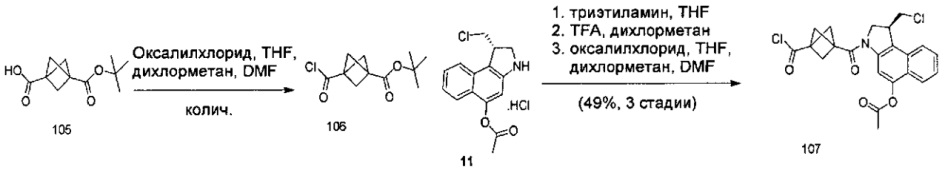
Стадия 1: Следуя общей методике A, используя 3-(трет-бутоксикарбонил)бицикло[1.1.1]пентан-1-карбоновую кислоту 105 (212 мг, 1,0 ммоль), оксалилхлорид (0,094 мл, 1,10 ммоль), THF (3 мл), дихлорметан (6 мл) и 1 каплю DMF, было получено соединение 105 в виде не совсем белого твердого вещества (235 мг, количественный выход). Это неочищенное соединение 105 как есть сразу использовали на следующей стадии.
Стадия 2: Следовали общей методике B, используя соединение 11 (311 мг, 0,997 ммоль), соединение 105 (230 мг, 0,997 ммоль), триэтиламин (0,292 мл, 2,09 ммоль) и THF (20 мл) и очистку хроматографией на силикагеле (градиент: от 10% до 75% ацетона в гептане). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением белого твердого вещества. В перемешиваемый раствор этого неочищенного вещества в 10 мл дихлорметана добавляли TFA (5,0 мл, 65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 90 мин. Реакционную смесь концентрировали в вакууме. Вещество растворяли дихлорметаном, переносили в делительную воронку и затем промывали 1 н. HCl водн., рассолом и водой. Органический слой сушили над сульфатом натрия, фильтровали и затем концентрировали в вакууме, после чего помещали в глубокий вакуум с получением белого твердого вещества. Используя это неочищенное вещество и следуя общей методике A с использованием оксалилхлорида (0,010 мл, 0,121 ммоль), THF (4 мл), дихлорметана (2 мл) и 1 капли DMF, было получено соединение 107 в виде белого твердого вещества (52 мг, 49%, за 3 стадии). Неочищенное соединение 107 как есть сразу использовали на следующей стадии.
Получение (1R)-8-(ацетиламино)-3-[(3-{[(1S)-5-(ацетилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-5-ил-ацетата 108

Следуя общей методике B, используя соединение 107 (21 мг, 0,057 ммоль), соединение 102 (24,6 мг, 0,057 ммоль), триэтиламин (0,024 мл, 0,171 ммоль) и THF (6 мл) и очистку препаративной ВЭЖХ (метод H1), было получено соединение 108 (5,8 мг, 14%) в виде не совсем белого твердого вещества. ЖХ/МС (Протокол В): m/z 728,1 [M+H]+, время удерживания = 2,12 мин.
Получение (1S)-3-[(3-{[(1S)-8-(ацетиламино)-5-(ацетилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата 109

Следуя общей методике B, используя соединение 107 (29,4 мг, 0,068 ммоль), соединение 104 (25 мг, 0,068 ммоль), триэтиламин (0,028 мл, 0,028 ммоль) и THF (8 мл) и очистку обращенно-фазной C18 хроматографией среднего давления (градиент: от 10% до 75% ацетонитрила в воде с 0,02% TFA в каждой фазе), было получено соединение 109 (11,8 мг, 24%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 728,0 [M+H]+, время удерживания = 2,13 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 10.25 (s, 1Н), 8.27 (s, 1Н), 8.20 (s, 1Н), 8.06 (s, 1Н), 8.04-7.98 (m, 1Н), 7.92-7.80 (m, 2Н), 7.63-7.55 (m, 2Н), 7.50-7.45 (m, 1Н), 4.56-4.33 (m, 5Н), 4.29-4.17 (m, 1H), 4.16-3.94 (m, 4Н), 2.62 (s, 6Н), 2.48-2.43 (m, 6Н), 2.11 (s, 3H).
Получение (S)-3-[5-((S)-5-амино-1-хлорметил-1,2-дигидро-бензо[е]индол-3-карбонил)-тиофен-2-карбонил]-1-хлорметил-2,3-дигидро-1H-бензо[е]индол-5-иловый эфир уксусной кислоты 115
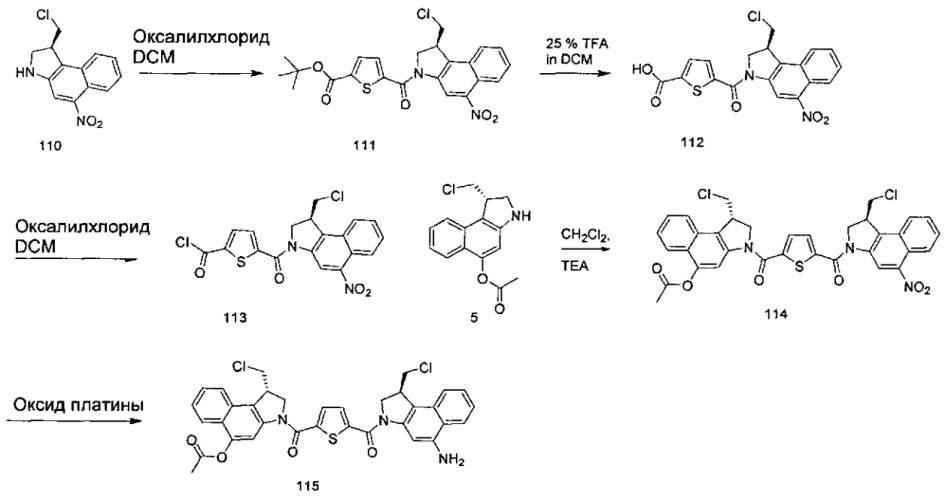
Стадия 1: В продуваемую N2 круглодонную колбу, содержащую моно-трет-бутиловый эфир тиофен-2,5-дикарбоновой кислоты (152 мг, 0,66 ммоль) в 5 мл безводного DCM, добавляли оксалилхлорид (0,66 ммоль, 0,066 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме до неочищенного остатка. Этот остаток затем добавляли в круглодонную колбу, содержащую соединение 110 (200 мг, 0,66 ммоль) в 15 мл безводного дихлорметана. Реакционную смесь перемешивали в течение 2 часов. Остаток разбавляли 15 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения 111 (185 мг, 58%) в виде желтого твердого вещества. ЖХ/МС: m/z 473 [M+H+], время удерживания = 2,25 мин.
Стадия 2: К соединению 111 добавляли 10 мл 25%-ной трифторуксусной кислоты в дихлорметане. Эту реакционную смесь перемешивали в течение 30 мин. Неочищенную реакционную смесь концентрировали в вакууме с получением соединения 112 в виде желтого твердого вещества. ЖХ/МС: m/z 416 [M+H+], время удерживания = 1,65 мин.
Стадия 3: В продуваемую N2 круглодонную колбу, содержащую соединение 112 (100 мг, 0,24 ммоль) в 5 мл безводного DCM, добавляли оксалилхлорид (0,24 ммоль, 0,02 мл). В этот раствор добавляли 1 каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов и концентрировали в вакууме с получением соединения 113 в виде желтого твердого вещества (100 мг, 0,24 ммоль, количественный выход). ЖХ/МС, в метаноле: m/z 282,0 [M+H+, для продукта метанолиза], время удерживания = 1,95 мин.
Стадия 4: В круглодонную колбу, содержащую соединение 5 (28 мг, 0,092 ммоль) в 5 мл дихлорметана, добавляли соединение 113 (40 мг, 0,092 ммоль). Затем добавляли триэтиламин (0,088 мл), и эту систему перемешивали в течение 1 часа при комнатной температуре. Неочищенную реакционную смесь концентрировали в вакууме, обратно переносили в 25 мл дихлорметана и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения 114 (40 мг, 64%) в виде желтого твердого вещества. ЖХ/МС: m/z 674 [M+H+], время удерживания = 2,25 мин.
Стадия 5: В колбу Парра, содержащую соединение 114 (30 мг, 0,044 ммоль) в 15 мл безводного тетрагидрофурана, добавляли оксид платины (5 мг, 0,02 ммоль). Систему закрывали резиновой пробкой и осуществляли гидрирование под давлением H2 50 фунт/кв.дюйм (345 кПа) в течение 3 часов. Через 3 часа колбу Парра продували N2, и неочищенную реакционную смесь фильтровали через слой целита, используя этилацетат. Фильтрат, содержащий целевой неочищенный продукт, затем концентрировали. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения 115 (15 мг, 50%) в виде желтого твердого вещества. ЖХ/МС: m/z 644 [M+H+], время удерживания = 2,06 мин.
Получение ((S)-1-хлорметил-5-гидрокси-1,2-дигидро-бензо[е]индол-3-ил)-[5-((S)-1-хлорметил-5-гидрокси-8-метил-1,6-дигидро-2H-пирроло[3,2-е]индол-3-карбонил)-тиофен-2-ил]-метанона 117
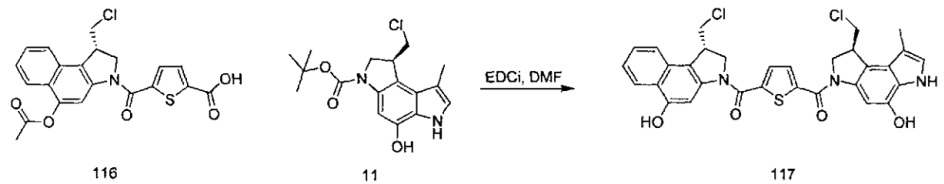
В оснащенной мешалкой круглодонной колбе, содержащей соединение 11 (34 мг, 0,1 ммоль), переносили в N,N-диметилформамид (5 мл) и по каплям добавляли в круглодонную колбу, содержащую 5-((S)-5-ацетокси-1-хлорметил-1,2-дигидро-бензо[е]индол-3-карбонил)-тиофен-2-карбоновую кислоту (42, 44 мг, 0,1 ммоль), 3-(этилиминометиленамино)-N,N-диметилпропан-1-амин (59 мг, 0,3 ммоль) и бикарбонат натрия (36 мг, 0,4 ммоль) в 5 мл N,N-диметилформамида). Эту реакционную смесь перемешивали в течение 30 мин. Добавляли 3 мл 1М HCl (водн.), и неочищенную реакционную смесь концентрировали в вакууме. Затем осуществляли обращенно-фазную хроматографию (градиент: 0%-65% ацетонитрила в воде) с получением соединения 117 (15 мг, 24%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 604 [M-H+], время удерживания = 1,93 минут
Получение (S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-илметил-карбоната 119
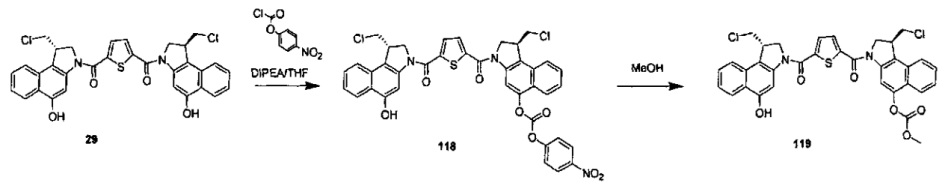
Раствор 4-нитрофенилхлорформиата (11 мг, 0,054 ммоль) в THF (1 мл) добавляли к раствору соединения 29 (27 мг, 0,045 ммоль) в THF (3 мл) и DIPEA (0,032 мл, 0,18 ммоль) при 0°C. Эту смесь перемешивали при 0°C в течение 2 ч и перемешивали при комнатной температуре в течение ночи. ЖХ/МС показала, что образовался соответствующий моно-PNP (пара-нитрофенил)-карбонат 118. В реакционную смесь добавляли метанол (1 мл). После перемешивания в течение 5 минут смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением продукта 119 в виде не совсем белого твердого вещества (5 мг, 20%). ЖХ/МС: m/z 660,7 [M+H], время удерживания = 1,06 мин. 1H ЯМР (400 МГц, CDCl3), δ 8.53 (d), 7.80 (m), 7.72 (d), 7.45 (m), 7.34 (m), 4.72 (m), 4.62 (d), 4.30 (m), 4.11 (t), 4.04 (s), 3.86 (d), 3.71 (d), 3.47 (t), 3.24 (m).
Получение (S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил-(2-(диметиламино)этил)карбамата 123
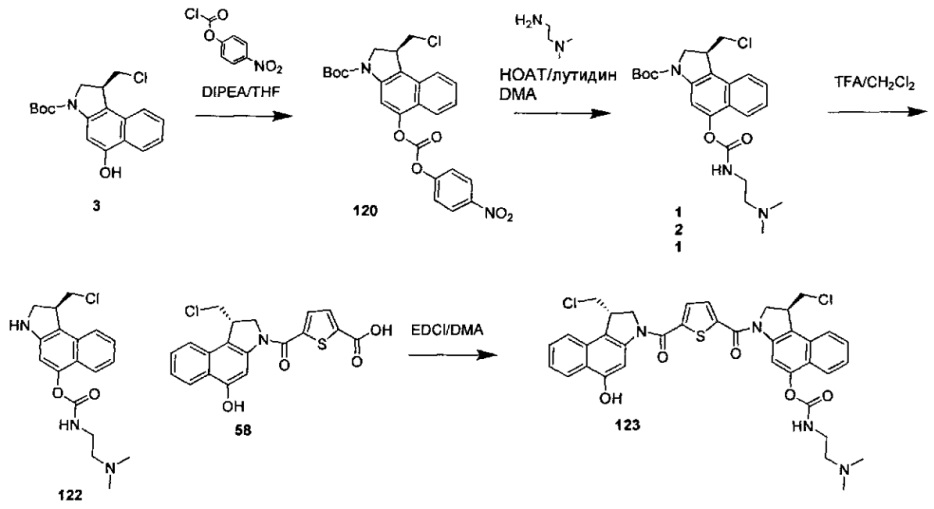
Стадия 1: Раствор 4-нитрофенилхлорформиата (164 мг, 0,78 ммоль) в THF (1 мл) добавляли к раствору соединения 3 (200 мг, 0,60 ммоль) в THF (6 мл) и DIPEA (0,315 мл, 1,8 ммоль) при 0°C, и эту смесь перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали, и остаток обрабатывали EA и водой, экстрагировали EA, промывали водой и рассолом, сушили над MgSO4, и растворитель удаляли в вакууме с получением PNP-карбоната 120 в виде желтой формы (300 мг, 100%). ЖХ/МС: m/z 399,0 [M+H], время удерживания = 2,37 мин.
Стадия 2: N,N-Диметилэтилендиамин (35 мг, 0,4 ммоль) добавляли к раствору вышеуказанного PNP-карбоната 120 (100 мг, 0,2 ммоль) в DMA (3 мл), после чего добавляли лутидин (0,07 мл, 0,6 ммоль) и HOAt (1-гидрокси-7-азабензотриазол) (14 мг, 0,1 ммоль). Эту смесь перемешивали при комнатной температуре в течение 4 ч. Смесь подвергали очистке на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением карбамата (S)-трет-бутил-1-(хлорметил)-5-(((2-(диметиламино)этил)карбамоил)окси)-1H-бензо[е]индол-3(2H)-карбоксилата 121 в виде желтого стекла (86 мг, 77%). ЖХ/МС: m/z 448,1 [M+H], время удерживания = 0,70 мин.
Стадия 3: Вышеуказанное соединение 121 (38 мг, 0,067 ммоль) обрабатывали TFA (0,5 мл) и CH2Cl2 (2 мл) в течение 2 ч, затем концентрировали в вакууме с получением соответствующего, лишенного защиты амина 122, который растворяли в DMA (3 мл). В этот раствор добавляли (S)-5-(1-(хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбоновую кислоту [58] (26 мг, 0,067 ммоль), после чего добавляли EDCI (1-этил-3-(3-диметиламинопропил)-карбодиимид) (27 мг, 0,14 ммоль), и эту смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество очищали на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением соединения 123 (4,5 мг, 8%). ЖХ/МС: m/z 717,4 [M+H], время удерживания = 1,38 мин. 1H ЯМР (400 МГц, CDCl3), δ 8.24 (d), 8.0 (d), 7.75 (d), 7.64 (s), 7.55-7.34 (m), 4.62 (m), 4.13 (t), 4.05 (t), 3.94 (t), 3.64 (t), 3.57-3.45 (m), 3.33 (s), 3.25 (s), 2.89 (s).
Получение (S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил-метил(2-(метиламино)этил)карбамата 126

Стадия 1: В вышеуказанный раствор соединения 120 добавляли N,N,N-триметилэтилендиамин (222 мг, 0,28 ммоль), после чего добавляли лутидин (0,37 мл, 3,2 ммоль) и HOAt (29 мг, 0,2 ммоль). Эту смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали, и остаток разбавляли этилацетатом, промывали рассолом, сушили над MgSO4. Неочищенный реакционный продукт очищали методом ISCO с использованием MeOH/DCM (0-20%) с получением соединения 124 в виде белой пены (245 мг, 50%). ЖХ/МС: m/z 462,2 [M+H], время удерживания = 1,45 мин.
Стадия 2: Вышеуказанное соединение 124 (40 мг, 0,087 ммоль) обрабатывали предварительно охлажденной TFA (1 мл) при 0°C в течение 10 мин. TFA удаляли под вакуумом с получением соответствующего, лишенного защиты амина 125, который растворяли в DMF (3 мл). В этот раствор добавляли (S)-5-(1-(хлорметил)-5-гидрокси-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбоновую кислоту [58] (34 мг, 0,087 ммоль), после чего добавляли EDCI (35 мг, 0,17 ммоль), и эту смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество очищали на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением продукта 126 в виде не совсем белого твердого вещества (25 мг, 39%). ЖХ/МС: m/z 731,1 [M+H], время удерживания = 1,71 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 10.49 (s), 8.26 (s), 8.14 (d), 7.98 (d), 7.88 (d), 7.66 (t), 7.77 (t), 7.40 (t), 4.89 (t), 4.78 (t), 4.55 (d), 4.43 (d), 4.23 (s), 4.08-3.91 (m), 3.73 (s), 3.50 (s), 3.40 (s), 3.26 (s), 2.89 (m).
Получение бицикло[1.1.1]пентан-1,3-диилбис{[(1S)-5-амино-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]метанона} 130
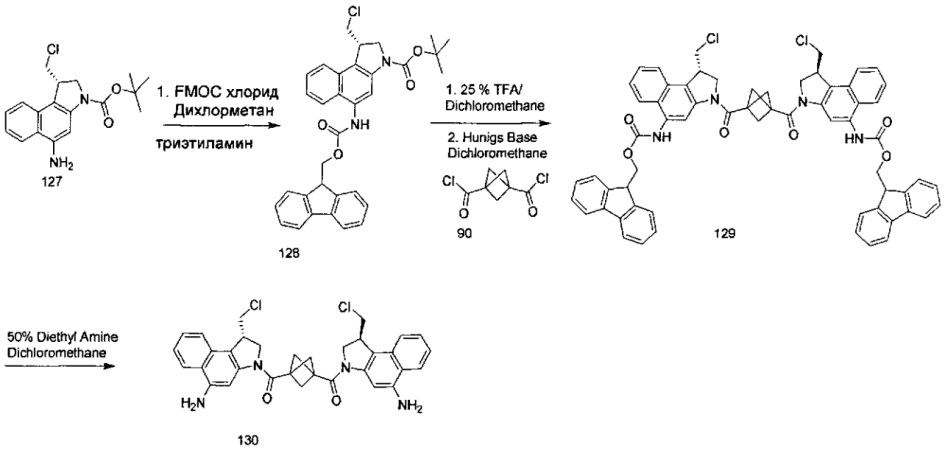
Стадия 1: В оснащенную мешалкой круглодонную колбу, содержащую флуоренилметилоксикарбонилхлорид (560 мг, 2,1 ммоль), добавляли 5 мл безводного DCM, и систему продували азотом. Добавляли соединение 127 (800 мг, 2,1 ммоль), после чего добавляли TEA (0,3 мл, 2,1 ммоль). Систему перемешивали в течение 5 часов. Неочищенную реакционную смесь переносили в этилацетат и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x), бикарбонатом натрия и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного остатка. Неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением соединения 128 в виде желтого твердого вещества (1,096 г, 91%). ЖХ/МС (Протокол B): m/z 455 [M-Boc]+, время удерживания = 2,58 мин.
Стадия 2: В оснащенную мешалкой круглодонную колбу, содержащую соединение 128 (1000 мг, 1,96 ммоль), добавляли 15 мл 25%-ного раствора TFA в DCM. Этот раствор перемешивали в течение 30 мин. Реакционную смесь концентрировали под вакуумом и переносили в смесь 50% DCM и гептана и концентрировали под вакуумом. Эту процедуру повторяли 3 раза (до удаления избытка TFA) с получением белого твердого вещества после концентрирования. Это белое твердое вещество добавляли в перемешиваемый раствор бицикло[1.1.1]пентан-1,3-дикарбонилдихлорида 90 в 10 мл безводного DCM. Реакционную смесь перемешивали в течение 1 часа и концентрировали до неочищенного стеклообразного вещества. Неочищенную реакционную смесь переносили в этилацетат и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x), бикарбонатом натрия и рассолом (2х). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали до неочищенного остатка. Неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением соединения 129 в виде желтого твердого вещества (250 мг, 12%). ЖХ/МС (Протокол B): m/z 1030,7 [M-H]-, время удерживания = 2,29 мин.
Стадия 3: В оснащенную мешалкой круглодонную колбу, содержащую бис(9H-флуорен-9-илметил)-(бицикло[1.1.1]пентан-1,3-диилбис{карбонил[(1S)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3,5-диил]})бискарбамат 129 (20 мг, 0,19 ммоль), добавляли 10 мл смеси 1:1 DCM и DEA. Этот раствор перемешивали в течение 3 часов. Реакционную смесь концентрировали под вакуумом и переносили в 50% DCM в гептане и концентрировали под вакуумом. Эту процедуру повторяли 3 раза (до удаления избытка DEA) с получением белого твердого вещества после концентрирования. Этот неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 10% метанола в DCM) с получением соединения 130 в виде желтого твердого вещества (4 мг, 30%). ЖХ/МС (Протокол B): m/z 585,1 [M+H]+, время удерживания = 1,99 мин.
Получение (1S)-1-(хлорметил)-3-[(4-{[(1S)-1-(хлорметил)-5-гидрокси-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[2.2.1]гепт-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата 134
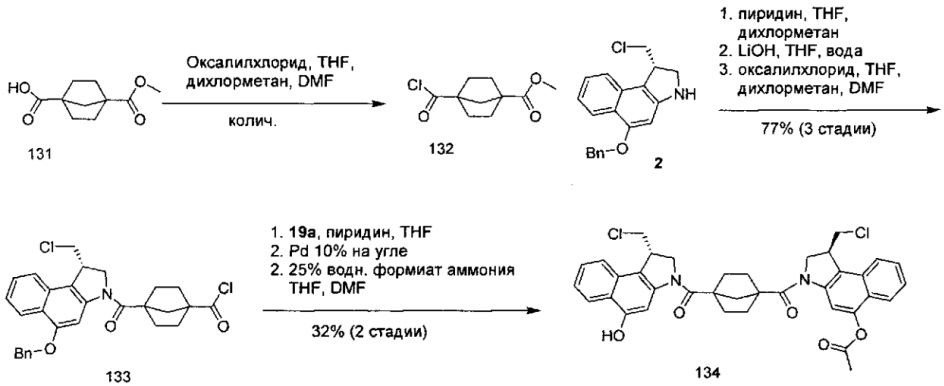
Стадия 1: Следуя общей методике А, используя 4-(метоксикарбонил)бицикло[2.2.1]гептан-1-карбоновую кислоту 131 (75 мг, 0,38 ммоль), оксалилхлорид (0,032 мл, 0,378 ммоль), THF (1,5 мл), дихлорметан (1,5) и 1 каплю DMF, было получено соединение 132 в виде смеси белого масла и твердого вещества (85 мг, количественный выход). Неочищенное соединение 132 как есть сразу использовали на следующей стадии.
Стадия 2: Следуя общей методике B, используя соединение 2 (125 мг, 0,346 ммоль), соединение 132 (75 мг, 0,35 ммоль), пиридин (0,112 мл, 1,38 ммоль), дихлорметан (2 мл) и THF (6 мл) и очистку хроматографией на силикагеле (градиент: от 0% до 25% ацетона в гептане), содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением белого твердого вещества. В перемешиваемый раствор неочищенного вещества в 6 мл THF, добавляли гидроксид лития (52,9 мг, 2,21 ммоль), растворенный в 1,5 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение примерно 3,5 часов. Реакционную смесь концентрировали до меньшего объема, переносили в делительную воронку и разбавляли дихлорметаном. Реакционную смесь промывали 1 н. HCl. Водный слой промывали один раз дихлорметаном. Органические слои объединяли, промывали рассолом, водой, сушили над сульфатом натрия, фильтровали и затем концентрировали в вакууме, после чего помещали в глубокий вакуум. Следуя общей методике A, используя неочищенное вещество, оксалилхлорид (0,024 мл, 0,281 ммоль), THF (4,0 мл), дихлорметан (4,0 мл) и 1 каплю DMF, было получено соединение 133 в виде белого масла и твердой смеси (85 мг, количественный выход). Неочищенное соединение 133 как есть сразу использовали на следующей стадии.
Стадия 3: Следуя общей методике B, используя соединение 19a (79,9 мг, 0,256 ммоль), соединение 133 (130 мг, 0,256 ммоль), пиридин (0,103 мл, 1,28 ммоль) и THF (6 мл), после концентрирования этой реакционной смеси в вакууме было получено неочищенное соединение в виде светло-розового твердого вещества. В перемешиваемый раствор этого неочищенного вещества в 3 мл DMF и 1 мл THF при 0°C добавляли 10% Pd на угле (100 мг), после чего добавляли 25%-ный водный раствор формиата аммония (0,4 мл). Реакционную смесь перемешивали при 0°C в течение примерно 90 мин. Реакционную смесь фильтровали через слой С18, который промывали 70%/30% раствором ацетонитрила и воды с 0,02% TFA в каждой фазе. Вещество концентрировали, используя испаритель Genevac, с получением соединения 134 (54 мг, 32%, за 2 стадии) в виде светло-серого твердого вещества. ЖХ/МС (Протокол B): m/z 657,1 [M+H]+, время удерживания = 2,10 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 10.32 (s, 1Н), 8.24 (s, 1Н), 8.11-8.07 (s, 1Н), 8.02-7.96 (m, 2Н), 7.91-7.86 (d, 1Н), 7.83-7.78 (d, 1Н), 7.63-7.57 (m, 1Н), 7.52-7.45 (m, 2Н), 7.36-7.30 (m, 1Н), 4.54-4.38 (m, 3H), 4.35-4.27 (m, 2Н), 4.16-4.05 (m, 2Н), 4.02-3.90 (m, 2Н), 3.80-3.73 (m, 1Н), 2.47 (s, 3H), 2.26-2.03 (m, 10H).
Получение (3bR,4aS,3b'R,4a'S)-6,6'-(бицикло[1.1.1]пентан-1,3-диилдикарбонил)бис(3-метил-4,4а,5,6-тетрагидроциклоргора[с]пирроло[3,2-е]индол-8(1H)-она)

В смесь соединения 109 (21 мг, 0,026 ммоль) в 2 мл ацетонитрила добавляли триэтиламин (0,40 мл, 2,9 ммоль), после чего добавляли 0,4 мл воды. Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Реакционную смесь концентрировали в вакууме. Затем осуществляли хроматографию на силикагеле (градиент: от 0% до 10% метанола в дихлорметане). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением соединения 135 (1,6 мг, 12%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 521,3 [M+H]+, время удерживания = 1,28 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 11.47 (s, 2Н), 6.85 (s, 4Н), 4.29-4.22 (m, 2Н), 4.17-4.10 (m, 2Н), 3.19-3.09 (m, 2Н), 1.97 (s, 6Н), 1.93-1.87 (m, 2Н), 1.27-1.22 (m, 2Н).
Получение (1aS,9bR,1a'S,9b'R)-3,3'-(тиен-2,5-диилдикарбонил)бис(1,1а,2,3-тетрагидро-5H-бензо[е]циклоргора[с]индол-5-она)

В смесь соединения 57 (44 мг, 0,073 ммоль) в 3 мл ацетонитрила добавляли триэтиламин (0,40 мл, 2,9 ммоль), после чего добавляли 0,4 мл воды. Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Реакционную смесь концентрировали в вакууме. Затем осуществляли хроматографию на силикагеле (градиент: от 0% до 5% метанола в дихлорметане). Содержимое соответствующих пробирок объединяли и концентрировали в вакууме с получением соединения 136 (16,3 мг, 39%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 531,1 [M+H]+, время удерживания = 1,55 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 8.05-8.01 (d, 2Н), 7.80 (s, 2Н), 7.65-7.59 (t, 2Н), 7.48-7.43 (t, 2Н), 7.28-7.23 (d, 2Н), 6.76 (s, 2Н), 4.57-4.51 (m, 2Н), 4.34-4.26 (m, 2Н), 1.85-1.76 (m, 4Н).
Получение (1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-гидрокси-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил-метил-2,3,4-три-O-ацетил-бета-D-глюкопиранозидуроната 141
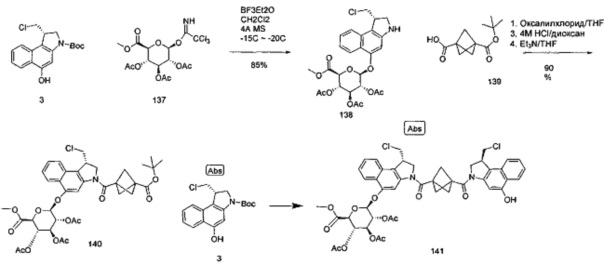
Соединение 140 (464 мг, 0,6 ммоль) растворяли в DCM (4 мл), добавляли TFA (2 мл), и эту смесь перемешивали в течение 2 ч. Смесь концентрировали в вакууме с получением соответствующей кислоты. ЖХ/МС (Протокол В): 688,0 [M+H]+, время удерживания 0,98 мин. Ее растворяли в THF (8 мл), охлаждали до 0°C, добавляли оксалилхлорид (0,9 мл, 2М в DCM), после чего добавляли 2 капли DMF. Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 50 мин. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты.
ЖХ/МС: 702,1 (1,05 мин по Larry, пик соответствующего Me эфира);
Соединение 4 растворяли в THF (10 мл), охлаждали до 0°C, добавляли вышеуказанный хлорангидрид кислоты, после чего добавляли Et3N (0,5 мл, 4,0 ммоль). Эту смесь перемешивали при 0°C в течение 30 мин. Смесь разбавляли EA, промывали водой и рассолом, сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток обрабатывали MeOH. Полученное твердое вещество собирали фильтрованием с получением соединения 141 в виде зеленого твердого вещества (414 мг, 73.5%). ЖХ/МС (Протокол B): 903,2 [M+H]+, время удерживания 1,11 мин.
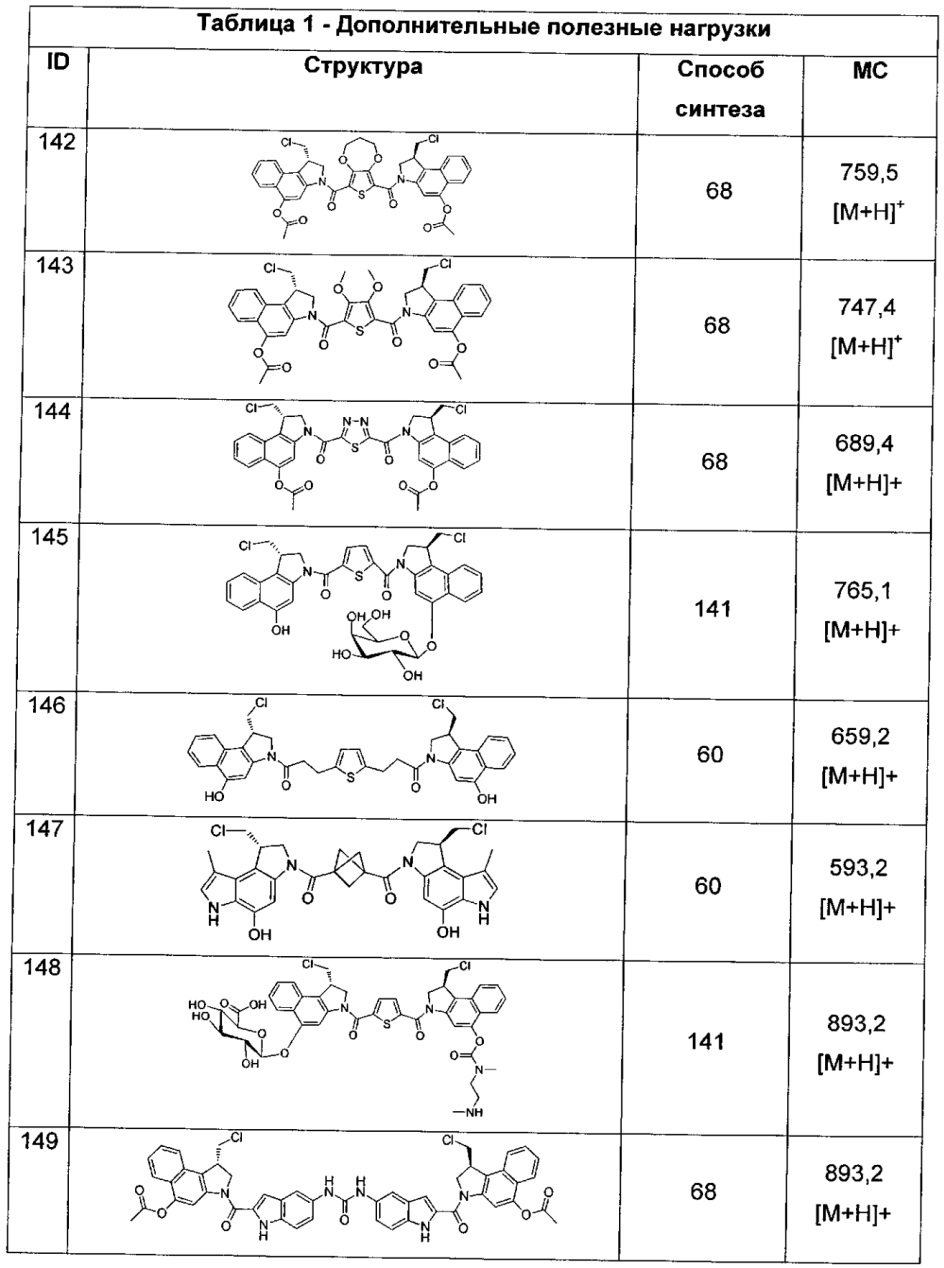
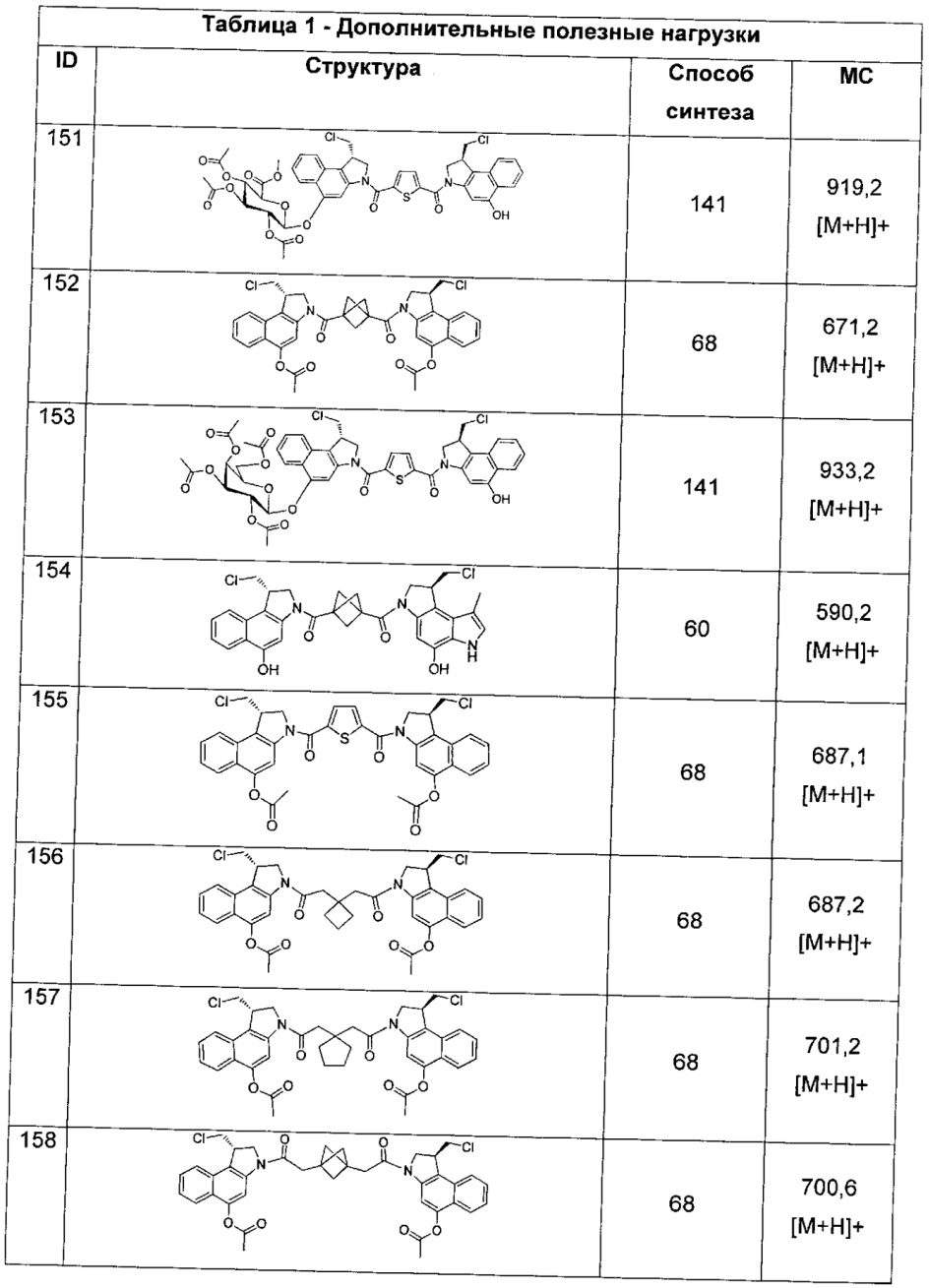
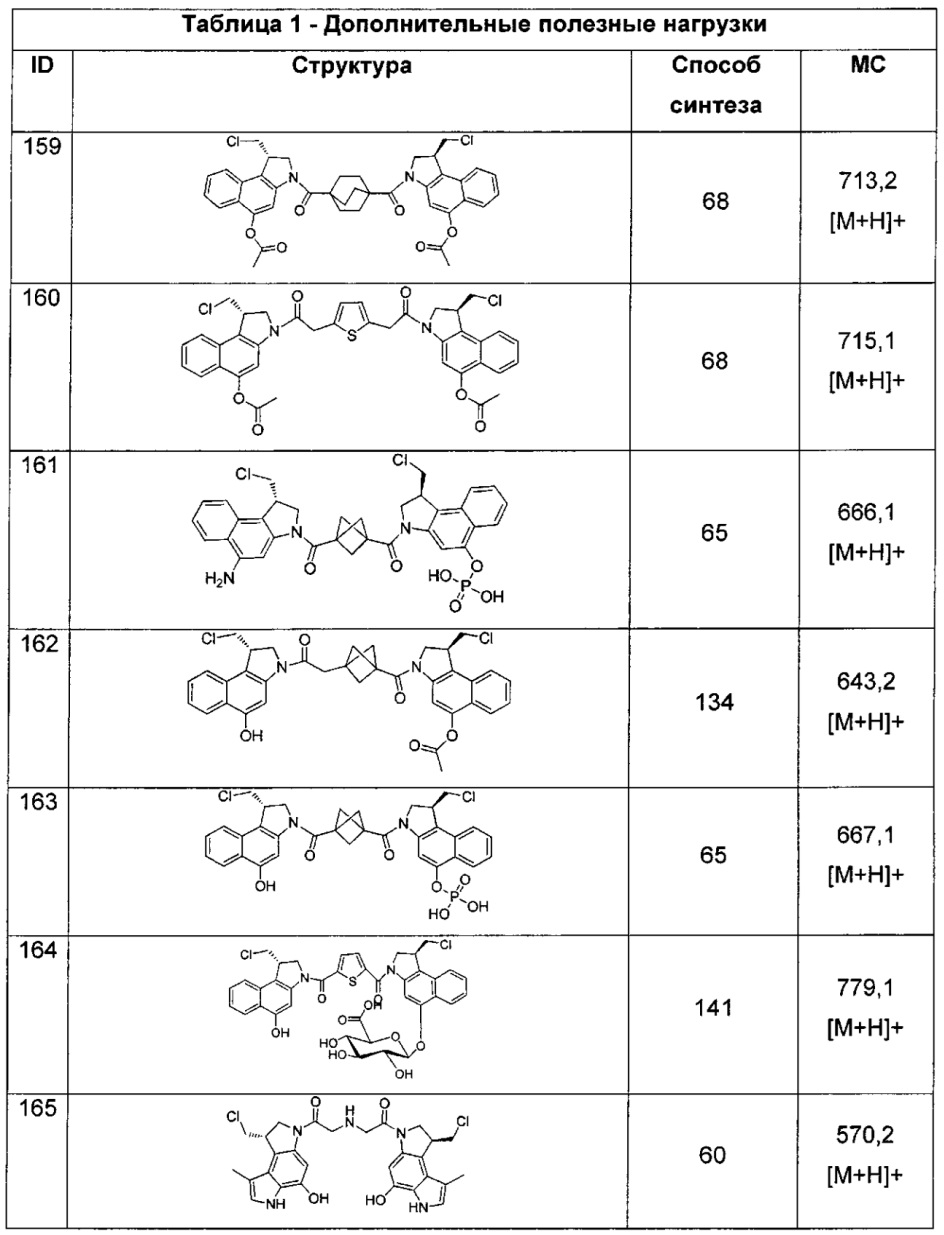
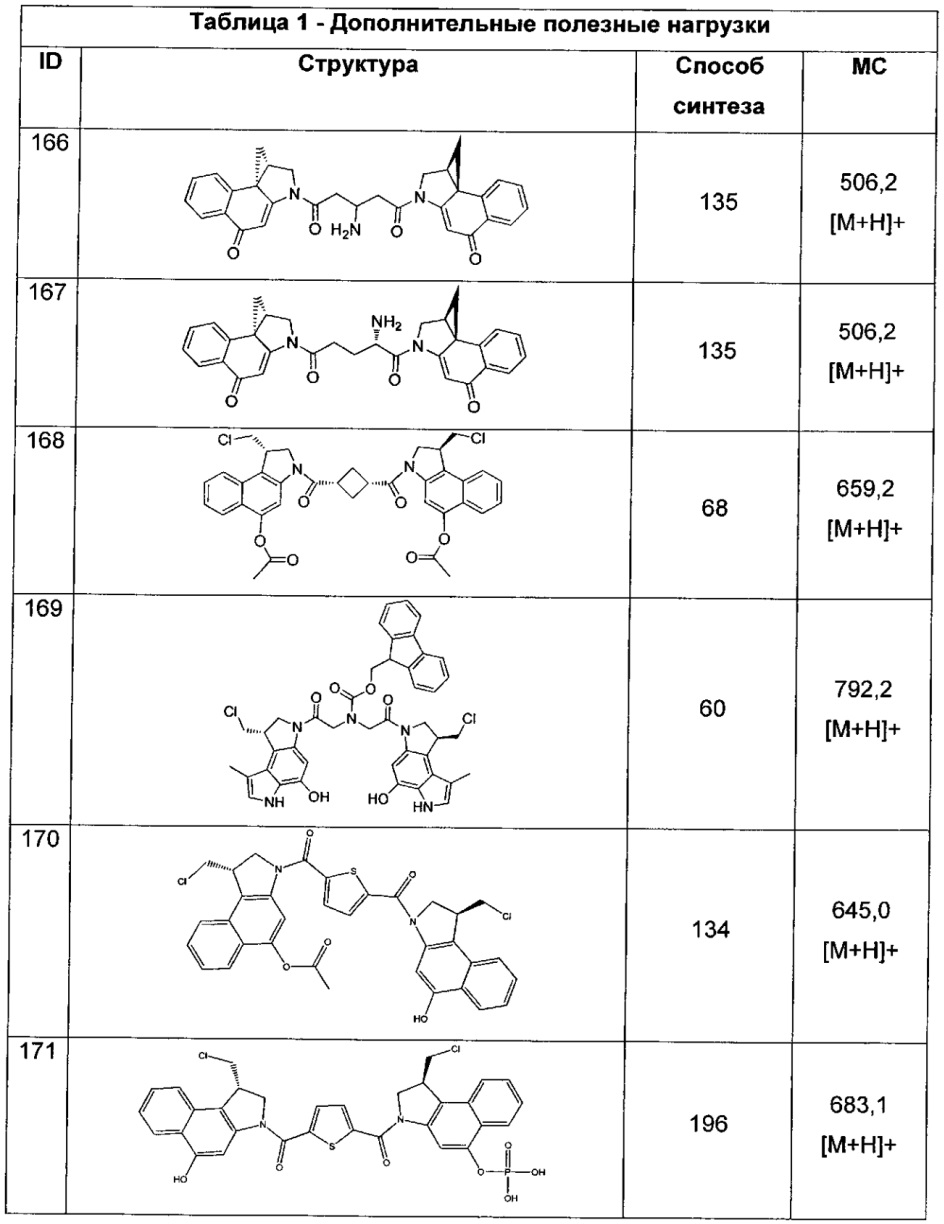
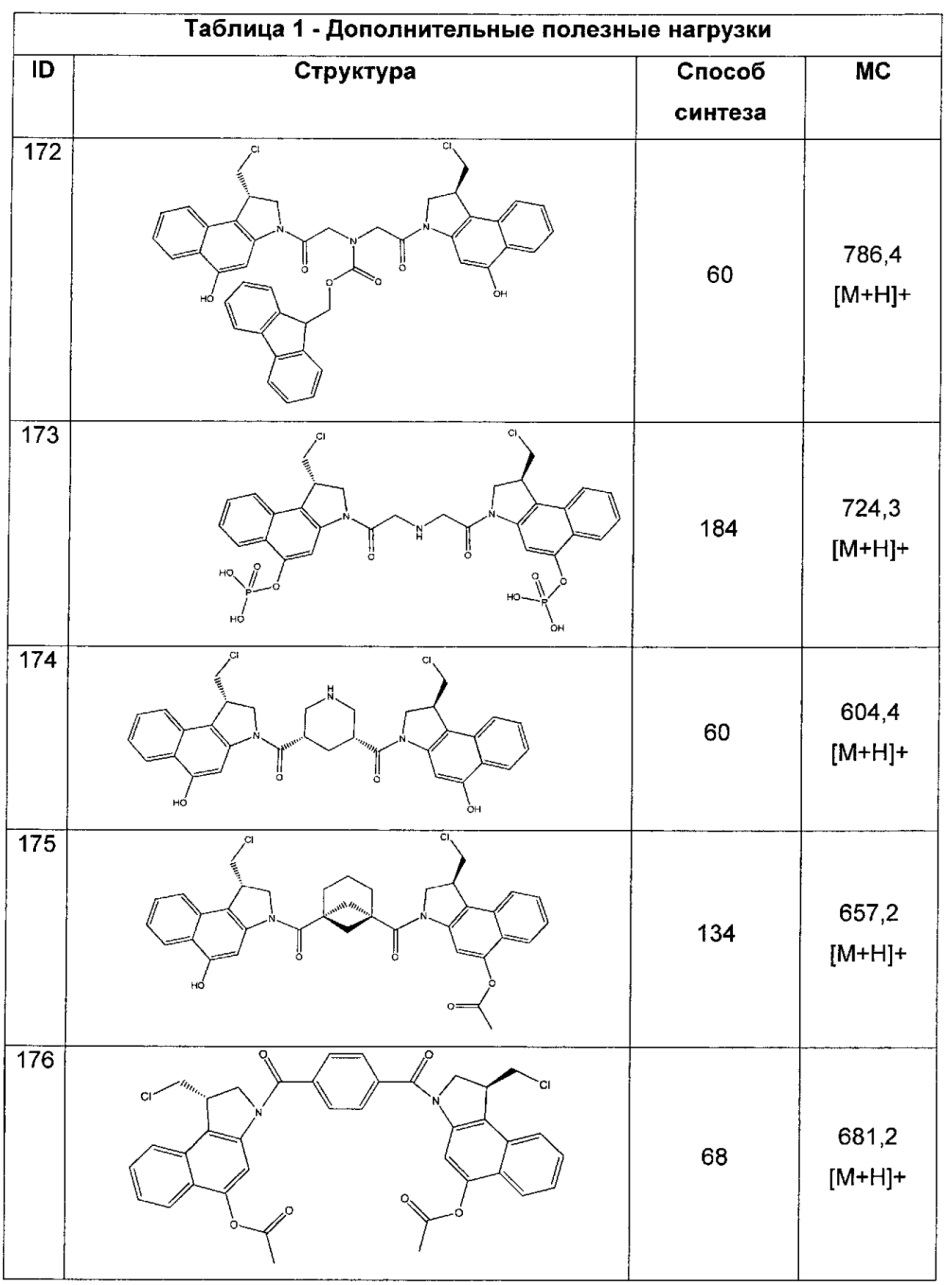
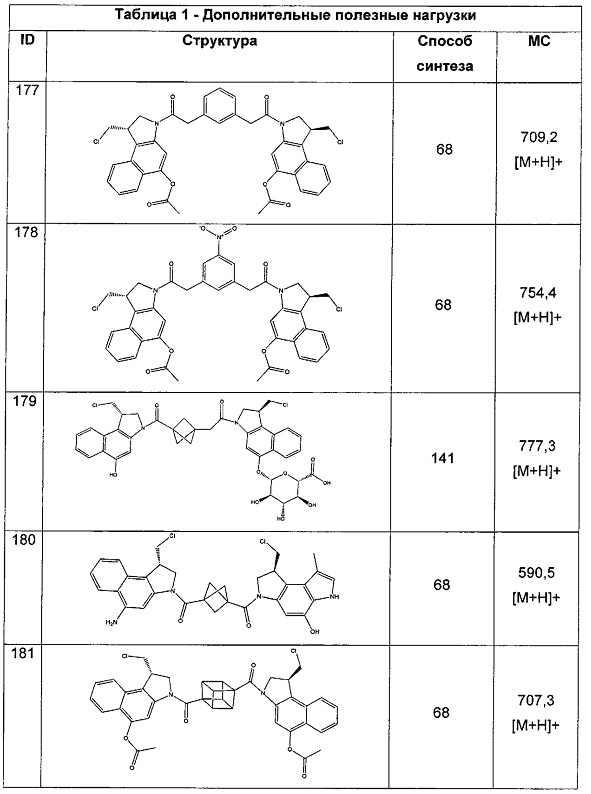
Названия представленных в Таблице 1 соединений приведены ниже:




Получение 4-((23S,26S)-1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-23-изопропил-21,24-диоксо-26-(3-уреидопропил)-3,6,9,12,15,18-гексаокса-22,25-диазагептакозанамидо)бензил-бис(2-((S)-1-(хлорметил)-5-(фосфоноокси)-1H-бензо[е]индол-3(2H)-ил)-2-оксоэтил)карбамата (186)
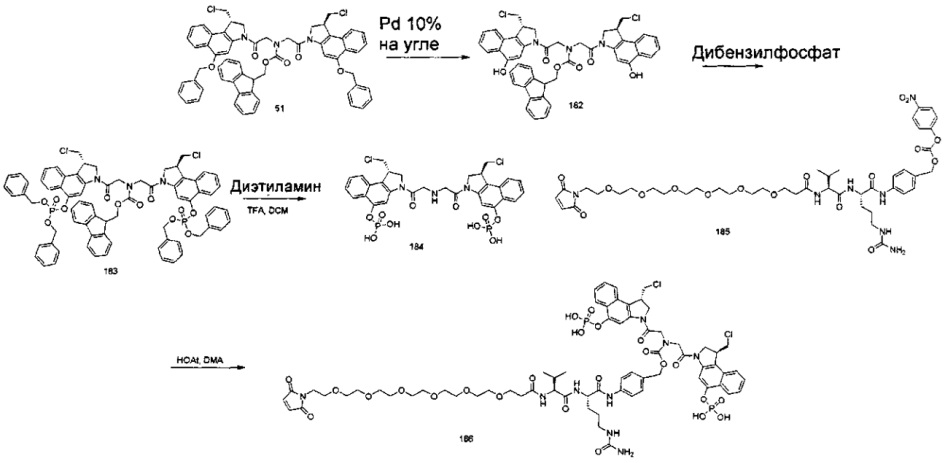
Стадия 1: Перемешиваемый раствор соединения 51 (120 мг, 0,124 ммоль) в 10 мл тетрагидрофурана под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (106 мг, 0,298 ммоль), после чего медленно по каплям добавляли 1 мл 25%-ного формиата аммония в воде. Эту реакционную смесь перемешивали при 0°C в течение 5 часов. Реакционную смесь затем фильтровали через слой целита, и фильтрат затем концентрировали в вакууме. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения 182 (35 мг, 36%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 786 [M+H+], время удерживания = 2,22 мин.
Стадия 2: В перемешиваемый раствор соединения 182 (274 мг, 0,348 ммоль) в 10 мл THF и 10 мл ацетонитрила, добавляли тетрахлорид углерода (2,04 мл, 21,0 ммоль) и основание Хюнига (1,12 мл, 6,45 ммоль), дибензилфосфит (0,9 мл, 4,32 ммоль) и DMAP (каталитическое количество). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Неочищенную реакционную смесь концентрировали в вакууме и затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения 183 (239 мг, 52%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 1308 [M+H+], время удерживания = 2,70 мин.
Стадия 3: В оснащенную мешалкой круглодонную колбу, содержащую соединение 183 (200 мг, 0,153 ммоль), добавляли 5 мл дихлорметана и 5 мл диэтиламина. Этот раствор перемешивали в течение 3 часов. Реакционную смесь концентрировали в вакууме и переносили в смесь 50% дихлорметана и гептана и снова концентрировали в вакууме. Эту процедуру повторяли 3 раза. Неочищенный остаток переносили в 10 мл 25%-ный раствор трифторуксусной кислоты в дихлорметане, после чего добавляли тиофенол (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение двух суток. Неочищенную реакционную смесь концентрировали в вакууме и затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% этилацетата в гептанах) с получением соединения 184 (60 мг, 47%) в виде палево-белого твердого вещества. ЖХ/МС: m/z 724 [M+H+], время удерживания = 1,02 мин.
Стадия 4: В круглодонную колбу, содержащую соединение 184 (75 мг, 0,1 ммоль), добавляли 10 мл DMA, и систему продували N2. В этот раствор при перемешивании добавляли соединение 185 (99 мг, 0,104 ммоль), после чего добавляли HOAt (416 мг, 0,104 ммоль) и основание Хюнига (1 каплю). Эту систему перемешивали при 45°C в течение 3 часов. Неочищенную реакционную смесь концентрировали в вакууме и затем осуществляли обращенно-фазную хроматографию с получением соединения 186 (34 мг, 21%) в виде белого твердого вещества. ЖХ/МС: m/z 1546 [M+H+], время удерживания = 1,23 мин.
Получение (S)-3-(5-(хлоркарбонил)тиофен-2-карбонил)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата 191
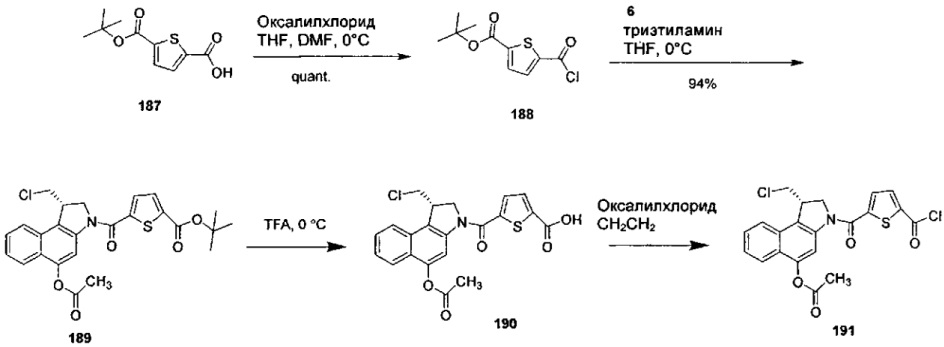
Стадия 1: В перемешиваемый раствор 5-(трет-бутоксикарбонил)тиофен-2-карбоновой кислоты (187) в 20 мл THF при 0°C добавляли оксалилхлорид (0,677 мл, 7,88 ммоль), после чего добавляли 1 каплю DMF. Эту реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 90 мин. Реакционную смесь концентрировали и помещали в глубокий вакуум с получением соединения 188 (1,67 г, количественный выход) в виде белого твердого вещества. Это неочищенное вещество сразу использовали на следующей стадии.
Стадия 2: В перемешиваемый раствор смеси соединения 6 (1,54 г, 4,93 ммоль) в 25 мл THF при 0°C добавляли триэтиламин (1,38 мл, 9,87 ммоль), и затем сразу добавляли соединение 188 (1,46 г, 5,92 ммоль), растворенное в 25 мл THF. Реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь затем перемешивали при комнатной температуре в течение примерно 45 мин. Реакционную смесь наносили на диоксид кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-100% ацетона в гептанах). Содержимое соответствующих пробирок концентрировали и помещали в глубокий вакуум с получением соединения 189 (2,24 г, 94%) в виде коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 486,3 [M+H]+, время удерживания = 2,19 мин.
Стадия 3: Соединение 189 (144 мг, 0,3 ммоль) обрабатывали предварительно охлажденной TFA (3 мл) при 0°C в течение 30 минут, затем концентрировали в вакууме с получением соответствующей кислоты 190. ЖХ/МС: m/z 430,3 [M+H], время удерживания = 1,59 мин. Соединение 190 растворяли в THF (3 мл), добавляли оксалилхлорид (0,2 мл, 2М в CH2Cl2, 0,4 ммоль) при 0°C, после чего добавляли 2 капли DMF (каталитическое количество), и эту смесь перемешивали при 0°C в течение 5 минут и затем при комнатной температуре в течение 2 ч. Смесь концентрировали в вакууме с получением соединения 191 в виде желтого твердого вещества.
Получение (S)-дибензил-(1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил)фосфата (193)
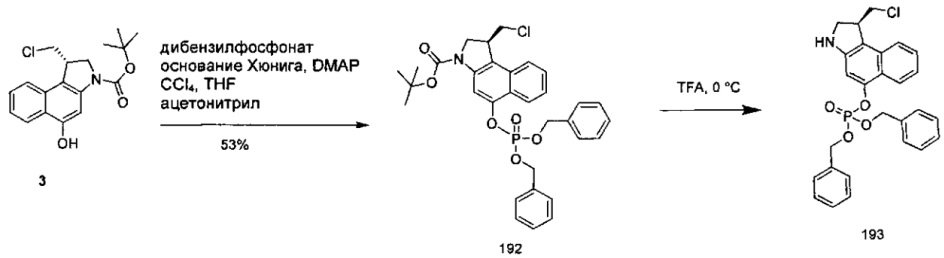
Стадия 1: В перемешиваемый раствор соединения 3 (889 мг, 2,66 ммоль) в 20 мл THF и 20 мл ацетонитрила добавляли тетрахлорид углерода (3,61 мл, 37,3 ммоль), после чего добавляли основание Хюнига (2,0 мл, 11,5 ммоль), дибензилфосфонат (3,65 мл, 16,5 ммоль) и DMAP (65,1 мг, 0,533 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Реакционную смесь концентрировали до меньшего объема, разбавляли несколькими мл DMSO и затем инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 85% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 192 (839 мг, 53%) в виде смеси светло-коричневого масла и твердого вещества. ЖХ/МС (Протокол B): m/z 595,3 [M+2H]+, время удерживания = 2,47 мин.
Стадия 2: В перемешиваемый раствор соединения 192 (834 мг, 1,40 ммоль) в 16 мл дихлорметана добавляли TFA (16 мл, 210 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение 1 минуты и затем сразу концентрировали, после чего помещали в глубокий вакуум с получением соединения 193 (701 мг, количественный выход) в виде смеси зеленого масла и твердого вещества. ЖХ/МС (Протокол B): m/z 494,2 [M+H]+, время удерживания = 2,17 мин.
Получение (1S)-3-(5-((1S)-5-(((бензилокси)(гидрокси)фосфорил)окси)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата [194] и (S)-3-(5-((S)-5-((бис(бензилокси)фосфорил)окси)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата [195]
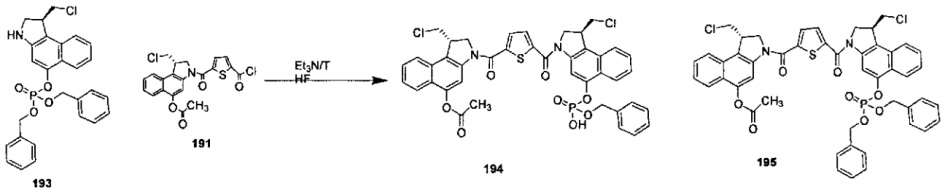
Соединение 193 растворяли в THF (3 мл) при 0°C, добавляли Et3N (0,165 мл, 1,2 ммоль), после чего добавляли раствор соединения 191 в THF (2 мл). Эту смесь перемешивали при 0°C в течение 5 минут и при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson (ACN/вода, 0,02% TFA) с получением двух продуктов: соединения 194 в виде желтого твердого вещества (50 мг, 21%); ЖХ/МС: m/z 815,4 [M+H], время удерживания = 0,96 мин; 1H ЯМР (400 МГц, DMSO-d6), δ 8.42 (s), 8.16 (s), 8.07 (d), 8.02 (d), 7.94 (d), 7.90 (s), 7.64 (q), 7.54 (q), 7.10-7.29 (m), 5.14 (d), 4.86 (q), 4.52 (t), 4.42 (m), 4.11-4.00 (m); и соединения 195 в виде зеленого твердого вещества (50 мг, 19%). ЖХ/МС: m/z 905,4 [M+H], время удерживания = 2,43 мин.
Получение (1S)-1-(хлорметил)-3-[(5-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}тиофен-2-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил-4-нитрофенилкарбоната (196)

В перемешиваемую смесь соединения 195 (200 мг, 0,221 ммоль) в 8 мл метанола добавляли 4М HCl в диоксане (8,0 мл, 230 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Реакционную смесь концентрировали. Неочищенное вещество переносили в 8 мл THF и 8 мл дихлорметана. В этот перемешиваемый раствор при 0°C добавляли 4-нитрофенилкарбонохлоридат (86,3 мг, 0,428 ммоль), после чего добавляли триэтиламин (0,179 мл, 1,28 ммоль). Реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Реакционную смесь концентрировали и затем помещали в глубокий вакуум. В перемешиваемую смесь неочищенного вещества в 10 мл дихлорметана добавляли раствор TFA (5 мл, 70 ммоль) в 10 мл дихлорметана, после чего добавляли тиофенол (0,107 мл, 1,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 6-7 часов. Реакционную смесь концентрировали до меньшего объема, разбавляли несколькими мл DMSO и затем инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 196 (71 мг, 40%) в виде желтого твердого вещества. ЖХ/МС (Протокол B): m/z 848,3 [M+H]+, время удерживания = 1,78 мин. 1H ЯМР (400 МГц, DMSO-d6) δ 8.51 (bra s), 8.35-8.41 (m), 8.09-8.15 (m), 8.00, 7.97-8.02 (d), 7.87-7.93 (m), 7.80-7.86 (m), 7.67-7.73 (m), 7.58-7.65 (m), 7.50-7.55 (m), 4.80-4.93 (m), 4.42-4.58 (m), 4.31-4.37 (m), 3.96-4.15 (m).
Получение 4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил-(2-(((((S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(фосфоноокси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил)окси)карбонил)(2-метоксиэтил)амино)этил)(метил)карбамата [198]
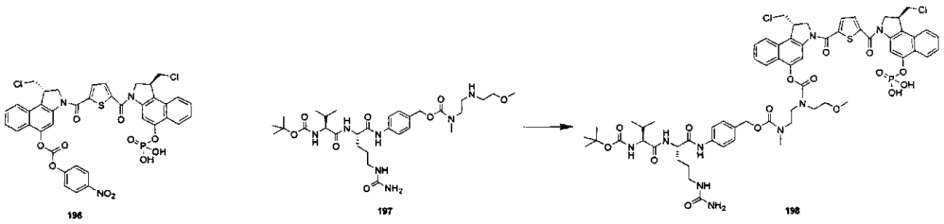
Соединение 196 (15 мг, 0,018 ммоль) растворяли в DMF (1 мл), добавляли раствор соединения 197 (17 мг, 0,023 ммоль) в DMF (1 мл), после чего добавляли DIPEA (0,013 мл, 0,072 ммоль) и лутидин (0,008 мл, 0,072 ммоль), затем HOAt (2,6 мг). Эту смесь перемешивали при комнатной температуре в течение 30 мин. По результатам ЖХ/МС реакция закончилась через 30 мин. Неочищенное вещество очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 198 в виде светло-желтого твердого вещества (13 мг, 53%). ЖХ/МС: m/z 1346,8 [M+H], время удерживания = 1,77 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 8.39 (s), 8.15 (d), 8.01 (m), 7.89 (m), 7.63 (m), 7.53 (m), 7.45-7.23 (m), 6.73 (d), 5.98 (s), 5.07-4.95 (m), 4.84 (t), 4.51 (m), 4.49-4.60 (m), 4.08-3.95 (m), 3.84-3.63 (m), 3.00-2.89 (m), 1.68-1.59 (m), 0.85 (m).
Получение 4-((26S,29S)-1-бром-26-изопропил-2,24,27-триоксо-29-(3-уреидопропил)-6,9,12,15,18,21-гексаокса-3,25,28-триазатриаконтанамидо)бензил-(2-(((((S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(фосфоноокси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил)окси)карбонил)(2-метоксиэтил)амино)этил)(метил)карбамата [201]
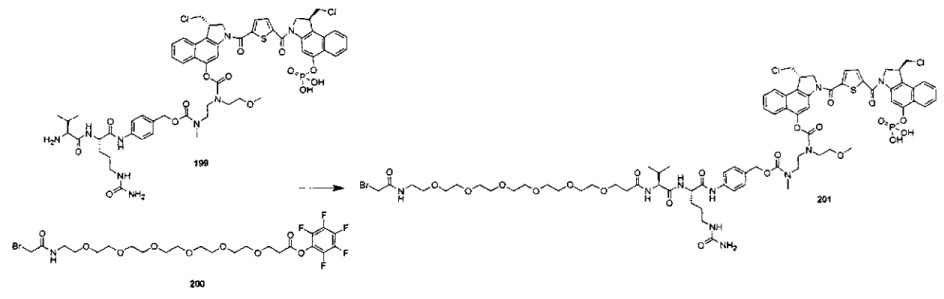
Стадия 1: Соединение 198 (13 мг, 0,01 ммоль) обрабатывали предварительно охлажденной TFA (0°C, 2 мл) в течение 2 минут и концентрировали в вакууме с получением продукта 199 в виде желтого твердого вещества (14 мг, TFA соль, 100%). ЖХ/МС: m/z 1247,9 [M+H], время удерживания = 1,57 мин. 1H ЯМР (400 МГц, DMF-d7), δ 10.13 (s), 8.65 (d), 8.45 (s), 8.17 (d), 7.95-7.85 (m), 7.65-7.22 (m), 5.04-4.97 (m), 4.81 (dd), 4.56 (s), 4.33 (d), 4.07-3.94 (m), 3.73-3.64 (m), 3.50 (s), 3.55-3.09 (m), 2.95-2.85 (m), 2.21 (dd), 1.76 (m), 1.62 (m), 1.46 (s), 0.99 (m).
Стадия 2: Соединение 199 (5 мг, 0,004 ммоль) добавляли в раствор перфторфенил-1-бром-2-оксо-6,9,12,15,18,21-гексаокса-3-азатетракозан-24-оата 200 (3,8 мг, 0,006 ммоль) в DMF (0,5 мл), после чего добавляли DIPEA (0,003 мл, 0,016 ммоль). Эту смесь перемешивали при комнатной температуре в течение 1 ч. Неочищенное вещество очищали на системе ВЭЖХ Gilson, используя ACN/вода (0,02% TFA), с получением продукта 201 в виде желтого твердого вещества (3 мг, 40%). ЖХ/МС: m/z 1704,0 [M+H], время удерживания = 1,61 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 9.88 (s), 8.30 (s), 8.24 (s), 8.06 (m), 7.91 (m), 7.81 (m), 7.54 (m), 7.47 (m), 7.43-7.13 (m), 5.91 (s), 4.98-4.85 (m), 4.76 (m), 4.43 (m), 4.30 (s), 4.14 (m), 4.00-3.90 (m), 3.52 (m), 3.16 (m), 2.92-2.86 (m), 2.31-2.25 (m), 1.90 (s), 1.52 (s), 1.34 (s), 1.32 (m), 0.78 (m).
Получение 4-((23S,26S)-1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-23-изопропил-21,24-диоксо-26-(3-уреидопропил)-3,6,9,12,15,18-гексаокса-22,25-диазагептакозанамидо)бензил-(2-(((((S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(фосфоноокси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил)окси)карбонил)(2-метоксиэтил)амино)этил)(метил)карбамата [206]
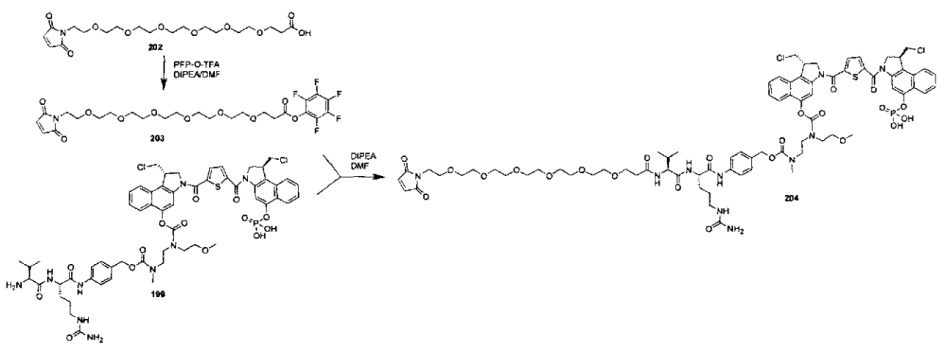
Стадия 1: Соединение 202 (227 мг, 0,52 ммоль) растворяли в CH2Cl2 (2 мл) и DMF (2 мл), добавляли PFP-O-TFA (0,19 мл, 1,05 ммоль) и DIPEA (0,275 мл, 1,57 ммоль). Эту смесь перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением соответствующего PFP-эфира 203 в виде желтого масла (34 мг, 11%). ЖХ/МС: m/z 623,4 [M+Na], время удерживания = 0,92 мин.
Стадия 2: Соединение 203 (3 мг, 0,005 ммоль) добавляли в раствор соединения 199 (7 мг, 0,005 ммоль) в DMF (0,3 мл), после чего добавляли DIPEA (0,005 мл, 0,03 ммоль). Эту смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь подвергали разделению на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 204 в виде желтого твердого вещества (4,6 мг, 60%). ЖХ/МС: m/z 1664,1 [M+H], время удерживания = 1,63 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 8.39 (s), 8.14 (m), 8.10-7.99 (m), 7.63 (m), 7.55-7.5 (m), 7.48 (s), 7.02 (s), 6.52 (s), 5.99 (s), 5.07-4.95 (m), 4.84 (t), 4.52 (t), 4.38 (s), 4.24 (t), 4.08-3.99 (m), 3.61-3.48 (m), 3.00-2.89 (m), 2.68 (s), 2.34 (s), 0.86 (dd).
Получение 4-((23S,26S)-1-амино-23-изопропил-21,24-диоксо-26-(3-уреидопропил)-3,6,9,12,15,18-гексаокса-22,25-диазагептакозанамидо)бензил-(2-(((((S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(фосфоноокси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил)окси)карбонил)(2-метоксиэтил)амино)этил)(метил)карбамата [208]
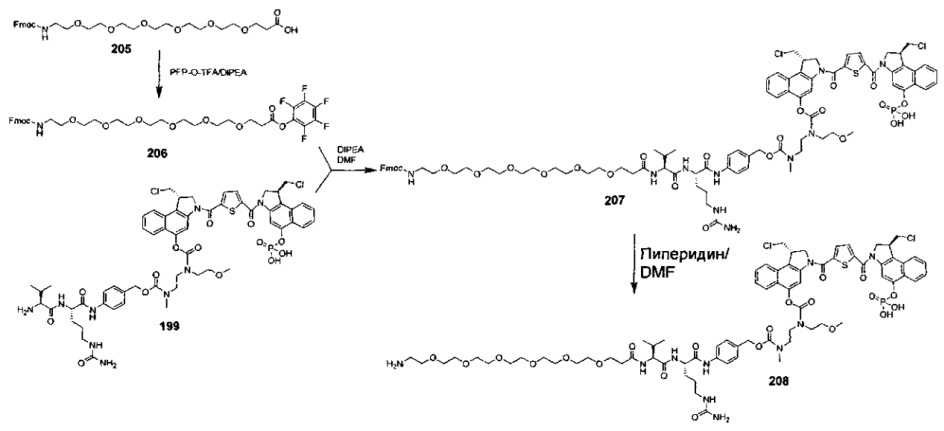
Стадия 1: Соединение 205 (43 мг, 0,07 ммоль) растворяли в DMF (2 мл), добавляли PFP-O-TFA (0,026 мл, 0,14 ммоль), после чего добавляли DIPEA (0,038 мл, 0,21 ммоль). Эту смесь перемешивали при комнатной температуре в течение 2 ч. Неочищенное вещество очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 206 в виде бесцветного масла (39 мг, 72%). ЖХ/МС: m/z 742,2 [M+H], время удерживания = 2,17 мин.
Стадия 2: Соединение 199 (7 мг, 0,005 ммоль) растворяли в DMF (0,6 мл), добавляли раствор вышеуказанного PFP-эфира 206 (3,7 мг, 0,005 ммоль) в DCM (0,1 мл), после чего добавляли DIPEA (0,005 мл, 0,03 ммоль). Эту смесь перемешивали при комнатной температуре в течение 1 ч. Неочищенный продукт 207: ЖХ/МС: m/z 1805,3 [M+H], время удерживания = 1,97 мин.
Стадия 3: В вышеуказанную реакционную смесь добавляли соединение 207, пиперидин (0,02 мл, 0,2 ммоль), и эту смесь перемешивали при комнатной температуре в течение 30 мин. Смесь концентрировали в вакууме, и неочищенное вещество очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 208 в виде желтого твердого вещества (4,2 мг, TFA-соль, 50%). ЖХ/МС: m/z 1584,0 [M+H], время удерживания = 1,54 мин. 1H ЯМР (400 МГц, DMSO-d6), δ 9.98 (s), 8.38 (s), 8.14 (m), 7.98 (m), 7.88 (m), 7.70 (s), 7.62 (m), 7.54 (m), 7.47 (m), 7.27 (m), 6.01 (s), 5.06-5.00 (m), 4.84 (m), 4.51 (m), 4.37 (m), 4.25 (m), 4.08 (m), 4.02 (m), 3.59 (m), 3.25 (m), 2.98 (m), 2.37 (m), 1.97 (s), 1.69 (s), 1.59 9s), 1.39 (m), 0.86 (dd).
Получение (1S)-1-(хлорметил)-3-[(5-{[(1S)-1-(хлорметил)-5-{[(4-нитрофенокси)карбонил]окси}-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}тиофен-2-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил-ацетата (211)
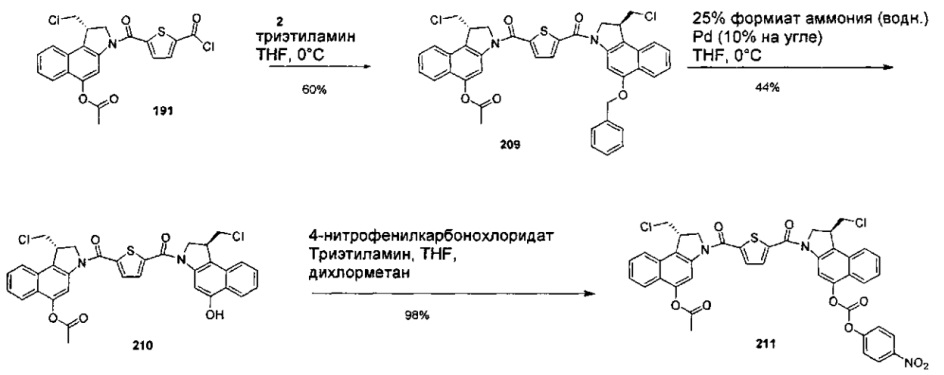
Стадия 1: В перемешиваемую смесь соединения 2 (425 мг, 1,31 ммоль) в 5 мл THF под азотом при 0°C добавляли триэтиламин (0,333 мл, 2,39 ммоль), после чего немедленно добавляли соединение 191 (535 мг, 1,19 ммоль), растворенное в 5 мл THF. Эту реакционную смесь перемешивали при 0°C в течение 5 минут и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Реакционную смесь затем наносили на диоксид кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 5%-80% ацетона в гептанах). Содержимое соответствующих пробирок концентрировали и помещали в глубокий вакуум с получением соединения 209 (530 мг, 60%) в виде желтого твердого вещества. ЖХ/МС (Протокол B): m/z 735,1 [M+H]+, время удерживания = 2,48 мин.
Стадия 6: Перемешиваемый раствор соединения 209 (610 мг, 0,829 ммоль) в 15 мл THF под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (203 мг), после чего медленно по каплям добавляли 2 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 12-24 часов. Реакционную смесь разбавляли диэтиловым эфиром, после чего добавляли сульфат натрия. Реакционную смесь фильтровали через целит, и целит промывали дважды диэтиловым эфиром. Органические фазы объединяли и затем концентрировали. Остаток очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 80% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 210 (206 мг, 44%) в виде желтого твердого вещества. ЖХ/МС (Протокол B): m/z 645,0 [M+H]+, время удерживания = 2,08 мин. 1H ЯМР (400 МГц, DMSO) δ 10.49 (br s), 8.13-8.18 (d), 8.05-8.10 (d), 7.93-7.97 (d), 7.83-7.91 (m), 7.63-7.69 (t), 7.53-7.58 (m), 7.38-7.43 (m), 4.83-4.92 (m), 4.74-4.82 (m), 4.50-4.55 (d), 4.39-4.47 (m), 4.20-4.27 (m), 4.01-4.15, 3.88-3.96 (m), 3.57-3.68 (m), 1.74-1.80, 1.36-1.39 (m).
Стадия 7: В перемешиваемый раствор соединения 210 (195 мг, 0,302 ммоль) в 12 мл дихлорметана и 8 мл THF при 0°C добавляли 4-нитрофенилкарбонохлоридат (122 мг, 0,604 ммоль), после чего добавляли триэтиламин (0,168 мл, 1,21 ммоль). Эту реакционную смесь перемешивали при 0°C в течение 5 минут и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Реакционную смесь концентрировали. Остаток очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 85% ацетонитрила в воде with 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 211 (240 мг, 98%) в виде желтого твердого вещества. ЖХ/МС (Протокол B): m/z 810,3 [M+H]+, время удерживания = 2,35 мин.
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N-[4-({[{2-[({[(1S)-3-[(5-{[(1S)-5-(ацетилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}тиофен-2-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-N~5~-карбамоил-L-орнитинамида (215)
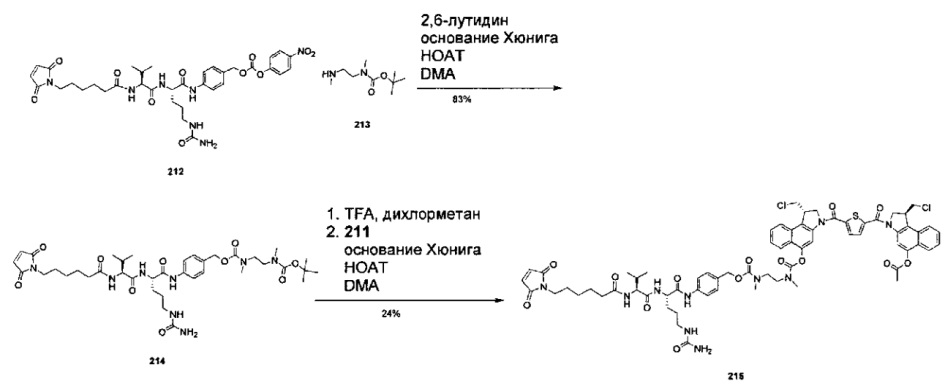
Стадия 1: В перемешиваемый раствор соединения 212 (750 мг, 1,02 ммоль) и соединения 213 добавляли трет-бутил-метил[2-(метиламино)этил]карбамат (192 мг, 1,02 ммоль) в 6 мл DMA, 2,6-лутидин (0,236 мл, 2,03 ммоль), после чего добавляли основание Хюнига (0,354 мл, 2,03 ммоль) и HOAT (69,1 мг, 0,5 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Реакционную смесь инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе) и затем очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 45% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 214 (663 мг, 83%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 787,3 [M+H]+, время удерживания = 1,45 мин.
Стадия 2: В перемешиваемую смесь соединения 214 (40,9 мг, 0,052 ммоль) в 2 мл дихлорметана добавляли TFA (1 мл, 10 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Реакционную смесь концентрировали и затем помещали в глубокий вакуум. Неочищенное вещество переносили в 2 мл DMA, и в этот перемешиваемый раствор добавляли основание Хюнига (0,03 мл, 0,17 ммоль), после чего добавляли 2,6-лутидин (0,02 мл, 0,17 ммоль), HOAT (5,9 мг, 0,043 ммоль) и затем соединение 211 (35 мг, 0,043 ммоль), растворенное в 1 мл DMA. Реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Реакционную смесь инжектировали в предколонку 5 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе) и затем очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 215 (14,1 мг, 24%) в виде желтого твердого вещества. ЖХ/МС (Протокол B): m/z 1359,3 [M+3H]+, время удерживания = 2,01 мин. MCBP: m/z 1359,4549 [M+3H]+.
Получение N-[1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-21-оксо-3,6,9,12,15,18-гексаоксагеникозан-21-ил]-L-валил-N~1~5~-карбамоил-N-{4-[({метил[2-(метиламино)этил]карбамоил}окси)метил]фенил}-L-орнитинамида (215)
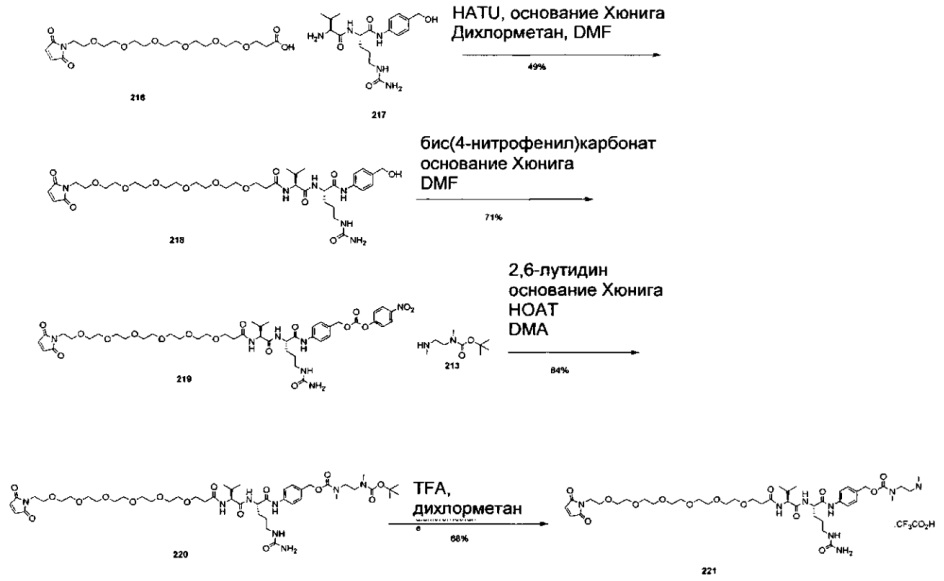
Стадия 1: В круглодонную колбу, содержащую 1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-3,6,9,12,15,18-гексаоксагеникозан-21-овую кислоту, добавляли соединение 216 (628 мг, 1,45 ммоль), 20 мл дихлорметана, 2 мл DMF, HATU (501 мг, 1,32 ммоль) и основание Хюнига (0,92 мл, 5,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 минут, после чего добавляли L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамид, соединение 217 (500 мг, 1,32 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 90 минут, затем гасили добавлением TFA. Реакционную смесь концентрировали до меньшего объема, разбавляли несколькими мл DMSO и затем инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 40% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 218 (514 мг, 49%) в виде светлого твердого вещества. ЖХ/МС (Протокол B): m/z 795,5 [M+H]+, время удерживания = 1,01 мин.
Стадия 2: В перемешиваемый раствор соединения 218 (210 мг, 0,264 ммоль) и бис(4-нитрофенил)карбоната (161 мг, 0,528 ммоль) в 4 мл DMF добавляли основание Хюнига (0,096 мл, 0,554 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 2 часов. Реакционную смесь инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 55% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 219 (180 мг, 71%) в виде твердого вещества. ЖХ/МС (Протокол B): m/z 960,5 [M+H]+, время удерживания = 1,48 мин.
Стадия 3: В перемешиваемый раствор соединения 219 (640 мг, 0,667 ммоль) и соединения 213 [полученного, как описано в J. Med. Chem. 1992, 33, 559-567] (127 мг, 0,674 ммоль) в 6 мл DMA добавляли 2,6-лутидин (0,154 мл, 1,33 ммоль), после чего добавляли основание Хюнига (0,232 мл, 1,33 ммоль) и HOAT (9,1 мг, 0,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 15 мин. Реакционную смесь инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 40% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 220 (564 мг, 84%) в виде воскообразного белого твердого вещества. ЖХ/МС (Протокол B): m/z 1009,7 [M+H]+, время удерживания = 1,43 мин.
Стадия 4: В перемешиваемую смесь соединения 220 (470 мг, 0,466 ммоль) в 6 мл дихлорметана добавляли TFA (3,0 мл, 40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Реакционную смесь концентрировали. Остаток очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 30% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 221 (326 мг, 68%) в виде белого масла/твердой смеси. ЖХ/МС (Протокол B): m/z 909,8 [M+H]+, время удерживания = 0,91 мин.
Получение N-[1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-21-оксо-3,6,9,12,15,18-гексаоксагеникозан-21-ил]-L-валил-N-[4-({[{2-[({[(1S)-3-[(5-{[(1S)-5-(ацетилокси)-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}тиофен-2-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-N~5~-карбамоил-L-орнитинамида (222)
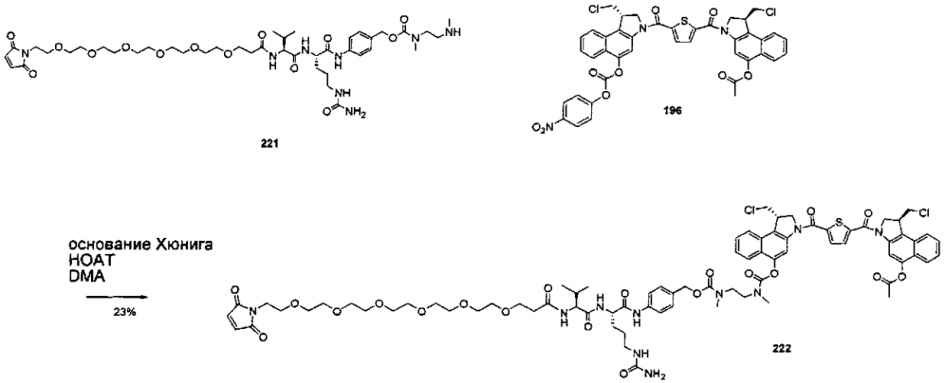
В перемешиваемую смесь соединения 221 (50,1 мг, 0,05 ммоль) в 1 мл DMA, и в этот перемешиваемый раствор добавляли основание Хюнига (0,03 мл, 0,172 ммоль), после чего добавляли 2,6-лутидин (0,02 мл, 0,172 ммоль), HOAT (5,9 мг, 0,043 ммоль) и соединение 211 (35 мг, 0,043 ммоль), растворенное в 1 мл DMA. Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Реакционную смесь инжектировали в предколонку 5 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 222 (15.4 мг, 23%) в виде желтого/белого твердого вещества. ЖХ/МС (Протокол B): m/z 1580,4 [M+2H]+, время удерживания = 1,95 мин. MCBP: m/z 790,7923 [M+2H]+.
Получение N-[1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-21-оксо-3,6,9,12,15,18-гексаоксагеникозан-21-ил]-L-валил-N~5~-карбамоил-N-[4-({[(2-{[({(1S)-1-(хлорметил)-3-[(5-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}тиофен-2-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-L-орнитинамида (223)
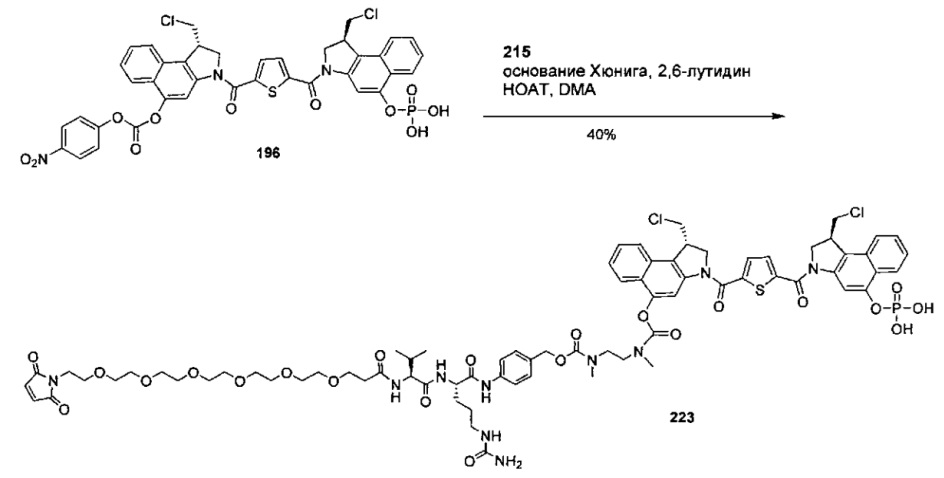
Стадия 1: В перемешиваемый раствор соединения 196 (29,8 мг, 0,035 ммоль) в 0,5 мл DMA добавляли соединение 221 (17,3 мг, 0,019 ммоль) в виде раствора в 1,5 мл DMA, после чего добавляли основание Хюнига (0,024 мл, 0,14 ммоль), 2,6-лутидин (0,016 мл, 0,14 ммоль) и HOAT (4,8 мг, 0,035 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Реакционную смесь инжектировали в предколонку 5 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 75% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего подвергали очистке препаративной ВЭЖХ (метод B), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 222 (22,6 мг, 40%) в виде желтого твердого вещества. ЖХ/МС (Протокол B): m/z 1619,9 [M+3H]+, время удерживания = 1,62 мин. HPLC (Протокол D): время удерживания = 9,339 мин.
Получение метил-3-(хлоркарбонил)бицикло[1.1.1]пентан-1-карбоксилата (225)
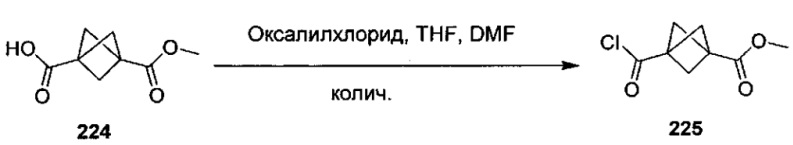
В перемешиваемый раствор соединения 224 в 12 мл THF при 0°C добавляли оксалилхлорид (0,381 мл, 4,44 ммоль), после чего добавляли 1 каплю DMF. Реакционную смесь перемешивали при 0°C в течение примерно 1 минуты и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Реакционную смесь концентрировали и затем помещали в глубокий вакуум с получением соединения 225 (701 мг, количественный выход) в виде белого твердого вещества.
Получение соли (8S)-8-(хлорметил)-6-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил 4-нитрофенилкарбоната с трифторуксусной кислотой 230
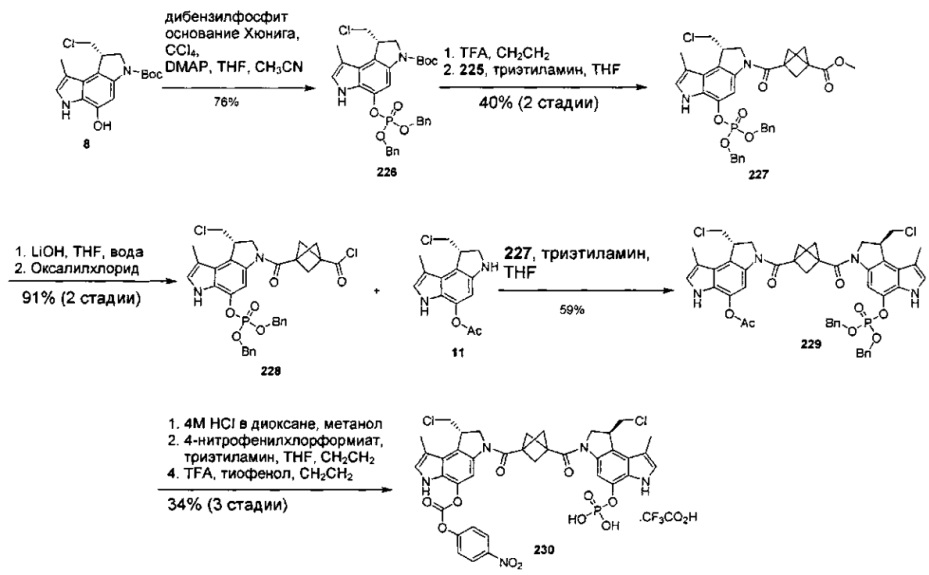
Стадия 1: В перемешиваемый раствор соединения 8 (4,5 г, 13,4 ммоль) в 80 мл THF и 80 мл ацетонитрила добавляли тетрахлорид углерода (18,1 мл, 187 ммоль), после чего добавляли основание Хюнига (9,31 мл, 53,4 ммоль), дибензилфосфит (17,7 мл, 80,2 ммоль) и DMAP (326 мг, 2,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Реакционную смесь наносили на диоксид кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-20% ацетона в гептанах). Содержимое соответствующих пробирок концентрировали и помещали в глубокий вакуум с получением соединения 226 (6,04 г, 76%) в виде светло-желтого твердого вещества. ЖХ/МС (Протокол B): m/z 614,3 [M+NH4]+, время удерживания = 2,38 мин.
Стадия 2: В перемешиваемый раствор соединения 226 (2,15 г, 3,60 ммоль) в 24 мл дихлорметана добавляли TFA (24 мл, 310 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 60 секунд, немедленно концентрировали и затем помещали под вакуум (насос с ременным приводом). В перемешиваемый раствор неочищенного вещества (2,59 г, 3,57 ммоль) в 15 мл THF при 0°C добавляли триэтиламин (1,49 мл, 10,7 ммоль), затем сразу добавляли соединение 225 (674 мг, 3,57 ммоль), растворенное в 15 мл THF. Реакционную смесь перемешивали при 0°C в течение примерно 5 минут и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Реакционную смесь наносили на диоксид кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-30% ацетона в гептанах). Содержимое соответствующих пробирок концентрировали и помещали в глубокий вакуум с получением соединения 227 (920 мг, 40% за 2 стадии) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 649,2 [M+H]+, время удерживания = 2,04 мин.
Стадия 3: В перемешиваемый раствор соединения 227 (895 мг, 1,38 ммоль) в 16 мл THF добавляли гидроксид лития (330 мг, 13,8 ммоль), растворенный в 4 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение примерно 90 мин. Добавляли дихлорметан, после чего добавляли 1 н. водный раствор HCl. Вещество переносили в делительную воронку. Органический слой отделяли, и водный слой промывали дважды дихлорметаном. Органические слои объединяли, промывали однократно рассолом, водой, сушили над сульфатом натрия, фильтровали и затем концентрировали и помещали в глубокий вакуум. Неочищенное вещество переносили в 15 мл THF и 5 мл дихлорметана, затем охлаждали до 0°C. В этот перемешиваемый раствор при 0°C добавляли оксалилхлорид (0,140 мл, 1,63 ммоль), после чего добавляли 1 каплю DMF. Реакционную смесь оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение примерно 60 мин. Реакционную смесь концентрировали и затем помещали в глубокий вакуум 228 (820 мг, 91% за 2 стадии) в виде светло-коричневого твердого вещества. Это неочищенное вещество использовали как есть на следующей стадии.
Стадия 4: В перемешиваемый раствор соединения 11 (527 мг, 1,50 ммоль) в 12 мл THF при 0°C добавляли триэтиламин (0,348 мл, 2,50 ммоль), затем немедленно добавляли соединение 228 (816 мг, 1,25 ммоль), растворенное в 12 мл THF. Реакционную смесь перемешивали при 0°C в течение примерно 5 минут, после чего оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Реакционную смесь наносили на диоксид кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-45% ацетона в гептанах). Содержимое соответствующих пробирок концентрировали и помещали в глубокий вакуум с получением соединения 229 (660 мг, 59%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 895,3 [M+H]+, время удерживания = 2,21 мин.
Стадия 5: В перемешиваемый раствор соединения 229 (652 мг, 0,728 ммоль) в 20 мл метанола добавляли 4М HCl в диоксане (20 мл, 80 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 24 мин. Реакционную смесь концентрировали и затем помещали в глубокий вакуум. В перемешиваемый раствор неочищенного вещества в 16 мл дихлорметана и 16 мл THF при 0°C добавляли пара-нитрофенил-хлорформиат (191 мг, 0,946 ммоль), затем немедленно добавляли триэтиламин (0,508 мл, 3,64 ммоль). Реакционную смесь перемешивали при 0°C в течение примерно 5 минут и затем оставляли нагреваться до комнатной температуры при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Реакционную смесь концентрировали. В перемешиваемый раствор неочищенного вещества в 12 мл дихлорметана добавляли раствор TFA (12 мл, 160 ммоль) в 12 мл дихлорметана, после чего добавляли тиофенол (0,745 мл, 7,28 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 6 часов. Реакционную смесь концентрировали. Неочищенное вещество разбавляли несколькими миллилитрами DMSO и затем инжектировали в предколонку 25 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 15% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 230 (267 мг, 34% за 3 стадии) в виде светло-желтого твердого вещества. ЖХ/МС (Протокол B): m/z 838,3 [M+H]+, время удерживания = 1,68 мин.
Получение соли N-[1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-21-оксо-3,6,9,12,15,18-гексаоксагеникозан-21-ил]-L-валил-N~5~-карбамоил-N-[4-({[(2-{[({(8S)-8-(хлорметил)-6-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-L-орнитинамида с трифторуксусной кислотой (231)
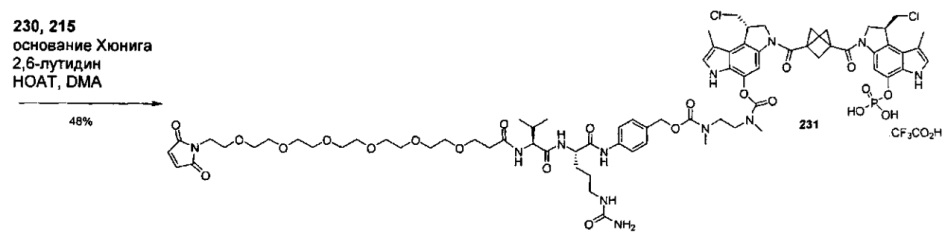
Стадия 1: В сосуд вместимостью 2 драхмы (7,776 г), содержащий соединение 230 (90 мг, 0,11 ммоль) и соединение 215 (121 мг, 0,118 ммоль), добавляли 3,0 мл DMA, после чего добавляли основание Хюнига (0,0748 мл, 0,429 ммоль), 2,6-лутидин (0,0497 мл, 0,429 ммоль) и HOAT (14,7 мг, 0,108 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 15 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 45% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего осуществляли вторую очистку методом H, и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 231 (117 мг, 60%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 1607,8 [M+H]+, время удерживания = 1,60 мин.
Получение соли N~2~-ацетил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N~5~-карбамоил-N-{4-[({метил[2-(метиламино)этил]карбамоил}окси)метил]фенил}-L-орнитинамида с трифторуксусной кислотой (236)
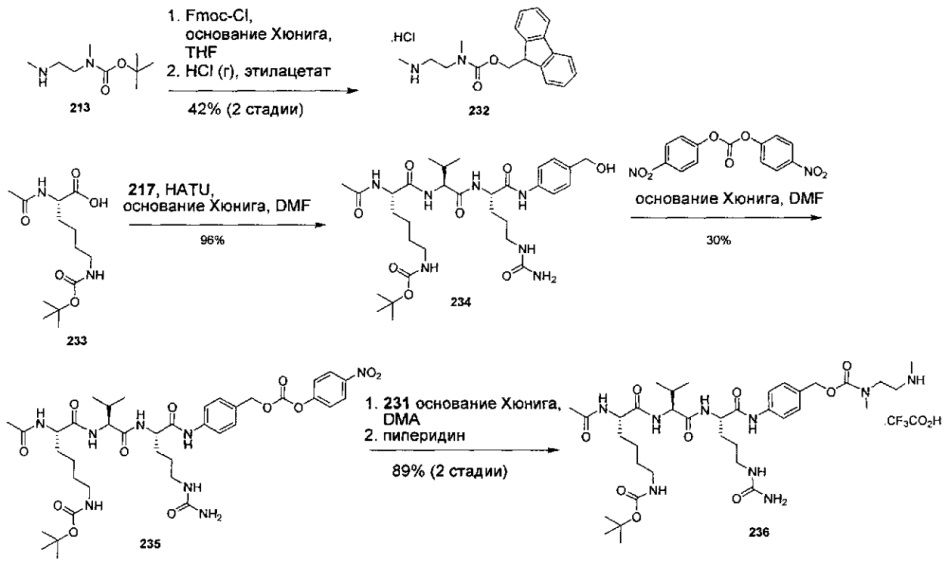
Стадия 1: В перемешиваемый раствор соединения 213 (16,0 г, 85,0 ммоль) и основания Хюнига (23 г, 178 ммоль) в 450 мл THF при 0°C по каплям добавляли Fmoc-Cl (22 г, 85,0 ммоль) в виде раствора в 450 мл THF. Эту смесь перемешивали при 0°C в течение 10 мин. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и затем промывали NH4Cl (водн.) и рассолом. Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографией на силикагеле (градиент: 2,5%-50% этилацетата в петролейном эфире). Содержимое соответствующих пробирок концентрировали. Вещество растворяли в 150 мл этилацетата, после чего добавляли 150 мл HCl в этилацетате. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и добавляли 300 мл MTBE. Полученный осадок собирали фильтрованием с получением соединения 232 (10.4 г, 42% за 2 стадии) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ 8.89 (br, 2Н), 7.91 (d, 2Н), 7.66 (d, 2Н) 7.42 (m, 2Н), 7.36 (m, 2Н), 4.34 (m, 3H), 3.51 (m, 1Н), 3.04 (m, 1Н), 2.85 (s, 3H), 2.72 (m, 1Н), 2.32 (m, 1Н).
Стадия 2: В раствор соединения 217 (481 мг, 1,27 ммоль) в 10 мл DMF добавляли соединение 233 (366 мг, 1,27 ммоль), HATU (660 мг, 1,65 ммоль) и основание Хюнига (0,302 мл, 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Реакционную смесь разбавляли этилацетатом, что вызывало распад твердого вещества. Эту суспензию перемешивали в течение примерно 30 мин. Твердое вещество собирали фильтрованием, промывали свежим этилацетатом и сушили в глубоком вакууме с получением соединения 234 (797 мг, 97%) в виде коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 650,3 [M+H]+, время удерживания = 0,64 минуты.
Стадия 3: В раствор соединения 234 (18,5 г, 28,5 ммоль) в DMF (500 мл) добавляли бис(4-нитрофенил)карбонат (9,54 г, 31,4 ммоль), после чего добавляли основание Хюнига (5,5 г, 42,8 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали. Остаток очищали хроматографией на диоксиде кремния (градиент: 1%-10% метанола в дихлорметане) с получением соединения 235 (6,9 г, 29,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ 8.30 (d, 2Н), 8.12 (d, 1Н), 8.01 (d, 1Н), 7.70 (d, 1Н), 7.64 (d, 2Н), 7.56 (d, 2Н), 7.40 (d, 2Н), 6.78 (m, 1Н), 5.98 (m, 1Н), 5.43 (s, 2Н), 5.24 (s, 2Н), 4.49 (m, 1Н), 4.19 (m, 2Н), 2.86 (m, 4Н), 1.99 (m, 1Н), 1.60 (m, 3H), 1.36 (m, 16Н), 0.82 (m, 6Н).
Стадия 4: В перемешиваемый раствор соединения 235 (500 мг, 0,605 ммоль) и соединения 232 (210 мг, 0,605 ммоль) в 3,0 мл DMA добавляли основание Хюнига (0,316 мл, 1,82 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Затем в реакционную смесь добавляли пиперидин (0,598 мл, 6,05 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение еще примерно 15 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 35% ацетонитрила в воде с 0,02% TFA в каждой фазе). Содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 236 (475 мг, 89% за 2 стадии) в виде прозрачного белого твердого вещества. ЖХ/МС (Протокол B): m/z 764,4 [M+H]+, время удерживания = 1,03 мин.
Получение соли N~2~-ацетил-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(2-{[({(8S)-8-(хлорметил)-6-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-L-орнитинамида с трифторуксусной кислотой (237)
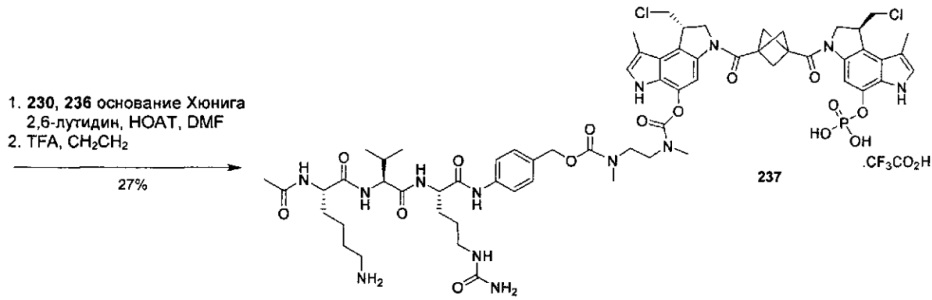
В сосуд вместимостью 2 драхмы (7,776 г), содержащий соединение 230 (100 мг, 0,119 ммоль) и соединение 236 (115 мг, 0,131 ммоль), добавляли DMF (2,0 мл), после чего добавляли основание Хюнига (0,0831 мл, 0,477 ммоль), 2,6-лутидин (0,0552 мл, 0,477 ммоль) и HOAT (16,2 мг, 0,119 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Реакционную смесь концентрировали. К неочищенному образцу добавляли дихлорметан (2 мл). В эту перемешиваемую смесь добавляли TFA (1,0 мл, 13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 30 мин. Реакционную смесь концентрировали. Неочищенное вещество растворяли в DMSO и инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 50% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего осуществляли вторую очистку методом G, и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 237 (55,8 мг, 27%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 1362,8 [M+H]+, время удерживания = 1,44 мин. 1H ЯМР (400 МГц, DMSO-d6): δ 10.96-10.83 (m), 10.06-9.97 (m), 8.16-7.97 (m), 7.87-7.66 (m), 7.59-7.47 (m), 7.37-6.97 (m), 6.54 (s), 6.05 (s), 5.47 (s), 5.12-4.96 (m), 4.45-3.91 (m), 3.74-2.83 (m), 2.76-2.68 (m), 2.59-2.52 (m), 2.39-2.32 (m), 2.02-1.93 (m), 1.83 (s), 1.71-1.21 (m), 0.88-0.77 (m).
Получение 3-{[2-({[(2-{[({(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-дисульфанил}-N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-аланина (244)
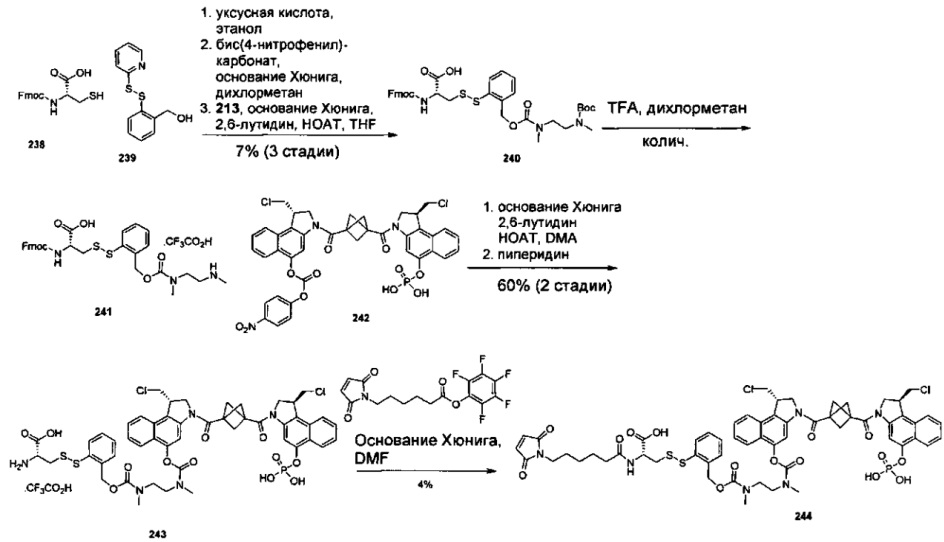
Стадия 1: В перемешиваемую смесь N-[(9H-флуорен-9-илметокси)карбонил]-L-цистеина 238 (17,9 г, 52,1 ммоль) в сухом этаноле (360 мл) при 0°C добавляли уксусную кислоту (2,41 г, 40,1 ммоль). Затем в реакционную смесь при 0°C добавляли раствор [2-(пиридин-2-илдисульфанил)фенил]метанола 239 (10 г, 40,104 ммоль) в сухом этаноле (200 мл). Эту смесь перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь концентрировали в вакууме с получением желтого масла. Остаток очищали препаративой HPLC (метод M) с получением желтой смолы (3,5 г). В перемешиваемый раствор этого неочищенного вещества (2,5 г, 5,191 ммоль) в сухом дихлорметане (100 мл) при 0°C добавляли бис(4-нитрофенил)карбонат (1,9 г, 6,23 ммоль), после чего добавляли основание Хюнига (805 мг, 6,23 ммоль). Эту смесь перемешивали при 0°C в течение  часа и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 23 часов. Реакционную смесь нагревали до 30°C и перемешивали при 30°C в течение примерно 18 часов. Реакционную смесь нагревали до 40°C и перемешивали при 40°C в течение примерно 6 часов. Реакционную смесь промывали 1М HCl (20 мл x 2), рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением остатка (3,89 г) в виде желтого масла. Этот остаток очищали хроматографией на силикагеле (градиент: от 0% до 4% метанол в дихлорметане) с получением желтого твердого вещества (2,48 г). В перемешиваемый раствор этого неочищенного вещества в THF (35 мл) при 0°C добавляли соединение 213 (635 мг, 3,37 ммоль), после чего добавляли основание Хюнига (793 мг, 6,14 ммоль), 2,6-лутидин (657 мг, 6,14 ммоль) и HOAT (41,8 мг, 0,307 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение 40 мин. Реакционную смесь разбавляли этилацетатом (200 мл) и промывали 1М HCl (30 мл, x2) и рассолом. Органические фазы сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта (3,6 г) в виде желтого масла. Этот неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 4% метанола в дихлорметане) с получением продукта (2,35 г) в виде желтой смолы. Продукт затем очищали препаративной ВЭЖХ (метод M, используя градиент от 50% B до 80% B за 30 минут, затем 95% в течение 5 минут). Смесь концентрировали в вакууме и экстрагировали этилацетатом (100 мл, x3). Органические слои объединяли, промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 240 (1,45 г, 7% за 3 стадии) в виде желтой смолы. 1H ЯМР (400 МГц, DMSO-d6): δ 7.91-7.89 (m, 3H), 7.74-7.72 (m, 3H), 7.44-7.31 (m, 7Н), 5.14 (s, 2Н), 4.34-4.24 (m, 4Н), 3.31-3.29 (m, 3H), 3.10-3.09 (m, 1Н), 3.04-3.02 (m, 1Н), 2.86-2.82 (d, 3H), 2.75-2.73 (m, 2Н), 2.67-2.50 (m, 2Н), 1.38-1.31 (m, 9Н).
часа и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 23 часов. Реакционную смесь нагревали до 30°C и перемешивали при 30°C в течение примерно 18 часов. Реакционную смесь нагревали до 40°C и перемешивали при 40°C в течение примерно 6 часов. Реакционную смесь промывали 1М HCl (20 мл x 2), рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением остатка (3,89 г) в виде желтого масла. Этот остаток очищали хроматографией на силикагеле (градиент: от 0% до 4% метанол в дихлорметане) с получением желтого твердого вещества (2,48 г). В перемешиваемый раствор этого неочищенного вещества в THF (35 мл) при 0°C добавляли соединение 213 (635 мг, 3,37 ммоль), после чего добавляли основание Хюнига (793 мг, 6,14 ммоль), 2,6-лутидин (657 мг, 6,14 ммоль) и HOAT (41,8 мг, 0,307 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение 40 мин. Реакционную смесь разбавляли этилацетатом (200 мл) и промывали 1М HCl (30 мл, x2) и рассолом. Органические фазы сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта (3,6 г) в виде желтого масла. Этот неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 4% метанола в дихлорметане) с получением продукта (2,35 г) в виде желтой смолы. Продукт затем очищали препаративной ВЭЖХ (метод M, используя градиент от 50% B до 80% B за 30 минут, затем 95% в течение 5 минут). Смесь концентрировали в вакууме и экстрагировали этилацетатом (100 мл, x3). Органические слои объединяли, промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 240 (1,45 г, 7% за 3 стадии) в виде желтой смолы. 1H ЯМР (400 МГц, DMSO-d6): δ 7.91-7.89 (m, 3H), 7.74-7.72 (m, 3H), 7.44-7.31 (m, 7Н), 5.14 (s, 2Н), 4.34-4.24 (m, 4Н), 3.31-3.29 (m, 3H), 3.10-3.09 (m, 1Н), 3.04-3.02 (m, 1Н), 2.86-2.82 (d, 3H), 2.75-2.73 (m, 2Н), 2.67-2.50 (m, 2Н), 1.38-1.31 (m, 9Н).
Стадия 2: В перемешиваемый раствор соединения 240 (35 мг, 0,050 ммоль) в 4 мл дихлорметана добавляли TFA (2 мл, 30 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Реакционную смесь концентрировали в вакууме и помещали в глубокий вакуум с получением соединения 241 в виде белого твердого вещества (40 мг, количественный выход). ЖХ/МС (Протокол B): m/z 596,5 [M+H]+, время удерживания = 1,38 мин.
Стадия 3: В сосуд, содержащий соединение 241 (29,8 мг, 0,042 ммоль) и соединение 242 (1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил-4-нитрофенил-карбонат [полученный с использованием химического процесса, описанного для получения соединения 229] (35,0 мг, 0,042 ммоль), добавляли 2,0 мл DMA, затем немедленно добавляли основание Хюнига (0,0293 мл, 0,168 ммоль), 2,6-лутидин (0,0195 мл, 0,168 ммоль) и HOAT (5,72 мг, 0,042 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Затем в реакционную смесь добавляли пиперидин (0,30 мл, 3 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 65% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 243 (30 мг, 60%) в виде серого твердого вещества. ЖХ/МС (Протокол B): m/z 838,3 [M+2H]+, время удерживания = 1,55 мин.
Стадия 4: В перемешиваемый раствор соединения 243 (20 мг, 0,017 ммоль) и пентафторфенил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноата (7,03 мг, 0,0186 ммоль) в 1,5 мл DMF добавляли основание Хюнига (0,0118 мл, 0,0677 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 15 мин. Неочищенную реакционную смесь инжектировали в предколонку 5 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 20% до 70% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего осуществляли очистку препаративной HPLC (метод И), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 244 (0,8 мг, 4%) в виде серого твердого вещества. ЖХ/МС (Протокол D): m/z 630,8 [1/2 M+1H]+, время удерживания = 10,786 мин.
Получение 3-{[4-({[(2-{[({(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-дисульфанил}-N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-аланина 250
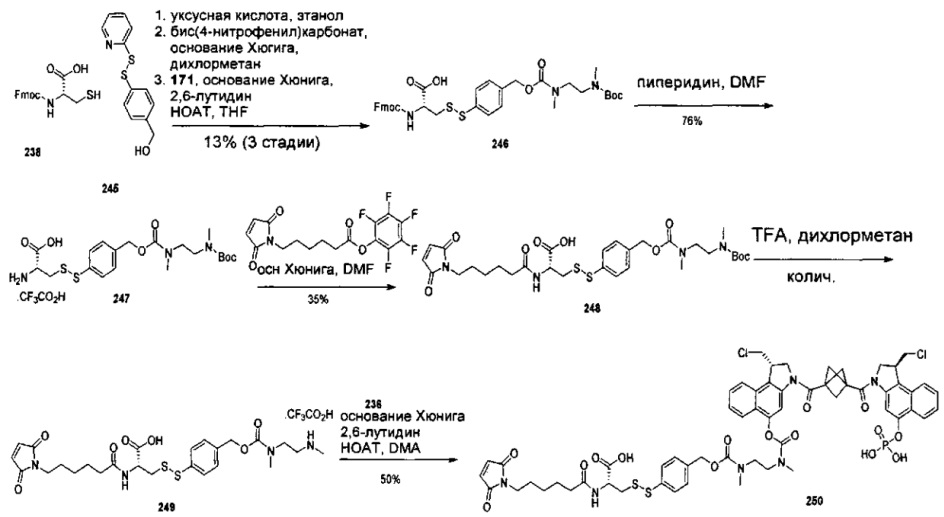
Стадия 1: В перемешиваемую смесь N-[(9H-флуорен-9-илметокси)карбонил]-L-цистеина 238 (11,6 г, 33,7 ммоль) в сухом этаноле (230 мл) при 0°C добавляли уксусную кислоту (1,93 г, 32,1 ммоль). Затем в реакционную смесь при 0°C добавляли раствор [4-(пиридин-2-илдисульфанил)фенил]метанола 245 (10 г, 40,104 ммоль) в сухом этаноле (160 мл). Эту смесь оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали в вакууме с получением желтого масла. Этот остаток очищали препаративной ВЭЖХ (метод M, используя градиент от 45% B до 75% B за 30 минут, затем 95% в течение 5 минут) с получением желтой смолы (8,5 г). В перемешиваемый раствор этого неочищенного вещества (8,0 г, 16,61 ммоль) в сухом дихлорметане (320 мл) при 0°C добавляли бис(4-нитрофенил)карбонат (6,06 г, 19,9 ммоль), после чего добавляли основание Хюнига (2,58 г, 19,9 ммоль). Смесь перемешивали при 0°C в течение 10 минут и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение примерно 15 часов. Затем в реакционную смесь дополнительно добавляли бис(4-нитрофенил)карбонат (1,52 г, 4,98 ммоль) и основание Хюнига (644 мг, 4,98 ммоль, 0,3 экв.). Реакционную смесь перемешивали при комнатной температуре в течение еще 2 часов. Реакционную смесь промывали 1М HCl (50 мл x 2), рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением остатка (17,1 г) в виде желтого масла. Этот остаток очищали хроматографией на силикагеле (градиент: от 0% до 7% метанола в дихлорметане) с получением желтого масла. В перемешиваемый раствор этого неочищенного вещества в THF (103 мл) при 0°C добавляли соединение 171 (1,89 г, 10,0 ммоль), после чего добавляли основание Хюнига (2,36 г, 18,2 ммоль), 2,6-лутидин (1,96 г, 18,2 ммоль) и HOAT (124 мг, 0,912 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение 60 мин. Реакционную смесь разбавляли этилацетатом (200 мл), промывали 1М HCl (30 мл, x2) и рассолом. Органические фазы сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта (7,5 г) в виде желтого масла. Этот неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 4% метанола в дихлорметане) с получением продукта (4,0 г) в виде желтой смолы. Этот продукт затем очищали (метод M, используя градиент от 50% B до 80% B за 30 минут, затем 95% в течение 5 минут). Смесь концентрировали в вакууме и экстрагировали этилацетатом (100 мл, x3). Органические слои объединяли, промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 246 (3,0 г, 13% за 3 стадии) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6): δ 7.89-7.87 (d, 2Н), 7.71-7.70 (d, 2Н), 7.55-7.52 (m, 2Н), 7.50-7.41 (m, 2Н), 7.39-7.30 (m, 4Н), 4.97 (s, 2Н), 4.30-4.22 (m, 4Н), 3.29 (br, 4Н), 3.10-3.01 (m, 2Н), 2.82-2.80 (d, 3H), 2.73 (s, 1Н), 2.66 (s, 2Н), 1.32-1.30 (d, 9Н).
Стадия 2: В перемешиваемый раствор соединения 246 (499 мг, 0,717 ммоль) в 4,0 DMF добавляли пиперидин (1,13 мл, 11,5 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 5 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 50% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением 3-{[4-(4,7,10,10-тетраметил-3,8-диоксо-2,9-диокса-4,7-диазаундец1-ил)фенил]дисульфанил}-L-аланина 247 (320 мг, 76%) в виде серого твердого вещества. ЖХ/МС (Протокол B): m/z 474,5 [M+H]+, время удерживания = 1,19 мин.
Стадия 3: В перемешиваемый раствор соединения 247 (140 мг, 0,238 ммоль) и пентафторфенил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноата (98,9 мг, 0,262 ммоль) добавляли 2 мл DMF, затем немедленно добавляли основание Хюнига (0,124 мл, 0,715 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 5 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 70% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-3-{[4-(4,7,10,10-тетраметил-3,8-диоксо-2,9-диокса-4,7-диазаундец1-ил)фенил]дисульфанил}-L-аланина 248 (56 мг, 35%) в виде прозрачного твердого вещества. ЖХ/МС (Протокол B): m/z 667,3 [M+H]+, время удерживания = 1,71 мин.
Стадия 4: В перемешиваемый раствор соединения 248 (35 мг, 0,050 ммоль) в 4 мл дихлорметана добавляли TFA (2 мл, 30 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Реакционную смесь концентрировали в вакууме и помещали в глубокий вакуум с получением соединения 249 в виде белого твердого вещества (40 мг, количественный выход).
Стадия 4: В сосуд, содержащий соединение 249 (18,0 мг, 0,0264 ммоль) и соединение 242 (22,0 мг, 0,0264 ммоль), добавляли 1,6 мл DMA, затем немедленно добавляли основание Хюнига (0,0184 мл, 0,106 ммоль), 2,6-лутидин (0,0123 мл, 0,106 ммоль) и HOAT (3,60 мг, 0,0264 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего осуществляли очистку препаративной ВЭЖХ (метод 12), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 250 (16,7 мг, 50%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 1261,4 [M+3H]+, время удерживания = 1,71 мин.
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N~5~-карбамоил-N-[4-({[(2-{[({(8S)-8-(хлорметил)-6-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-L-орнитинамида 255
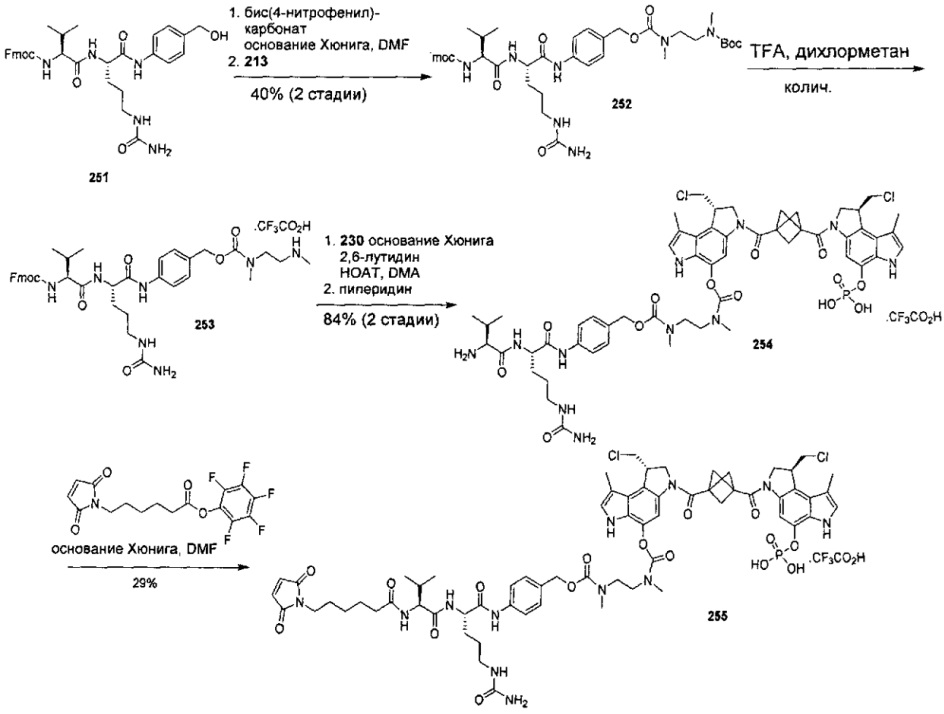
Стадия 1: N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамид 251 (725 мг, 1,2 ммоль) растворяли в 6 мл DMF, после чего осуществляли обработку ультразвуком в течение примерно 10 мин. Затем включали мешалку, и этот раствор перемешивали при комнатной температуре. Затем добавляли бис(4-нитрофенил)карбонат (403 мг, 1,33 ммоль), после чего добавляли основание Хюнига (0,44 мл, 2,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение примерно 5 часов. Добавляли соединение 213 (227 мг, 1,2 ммоль), растворенное в 1 мл DMF. Реакционную смесь перемешивали при комнатной температуре в течение примерно 1 минуты. Неочищенную реакционную смесь инжектировали в предколонку 24 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N~5~-карбамоил-N-[4-(4,7,10,10-тетраметил-3,8-диоксо-2,9-диокса-4,7-диазаундец1-ил)фенил]-L-орнитинамида 252 (395 мг, 40% за 2 стадии) в виде коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 816,7 [M+H]+, время удерживания = 1,88 мин.
Стадия 2: В перемешиваемую смесь соединения 252 (197 мг, 0,241 ммоль) в 6 мл дихлорметана добавляли TFA (2 мл, 30 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 20 мин. Реакционную смесь концентрировали в вакууме и помещали в глубокий вакуум с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N~5~-карбамоил-N-[4-(4,7,10,10-тетраметил-3,8-диоксо-2,9-диокса-4,7-диазаундец-1-ил)фенил]-L-орнитинамида 253 (210 мг, количественный выход) в виде белого и светло-коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 716,7 [M+H]+, время удерживания = 1,27 мин.
Стадия 3: В сосуд, содержащий соединение 230 (48 мг, 0,053 ммоль) и соединение 253 (52,4 мг, 0,063 ммоль), добавляли 2,0 мл DMA, затем немедленно добавляли основание Хюнига (0,036 мл, 0,211 ммоль), 2,6-лутидин (0,024 мл, 0,211 ммоль) и HOAT (7,1 мг, 0,0525 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Затем добавляли пиперидин (0,30 мл, 3 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Неочищенную реакционную смесь инжектировали в предколонку 12 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 10% до 50% ацетонитрила в воде с 0,02% TFA в каждой фазе) и содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 254 (68 мг, 84% за 2 стадии) в виде светло-серого твердого вещества. ЖХ/МС (Протокол B): m/z 1193,5 [M+2H]+, время удерживания = 1,46 мин.
Стадия 4: В перемешиваемый раствор соединения 254 (30 мг, 0,020 ммоль) и пентафторфенил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноата (8,11 мг, 0,0215 ммоль) в 2,0 мл DMF добавляли основание Хюнига (0,0136 мл, 0,0782 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 10 мин. Неочищенную реакционную смесь инжектировали в предколонку 5 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 50% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего осуществляли вторую очистку препаративной HPLC (метод J1). Содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 255 (9,1 мг, 29%) в виде светло-коричневого твердого вещества. ЖХ/МС (Протокол B): m/z 1386,9 [M+2H]+, время удерживания = 1,60 мин.
Получение N-(24-бром-23-оксо-4,7,10,13,16,19-гексаокса-22-азатетракозан-1-оил)-L-валил-N~5~-карбамоил-N-[4-({[(2-{[({(8S)-8-(хлорметил)-6-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-метил-3,6,7,8-тетрагидропирроло[3,2-е]индол-4-ил}окси)карбонил](метил)амино}этил)(метил)карбамоил]окси}метил)фенил]-L-орнитинамида 257
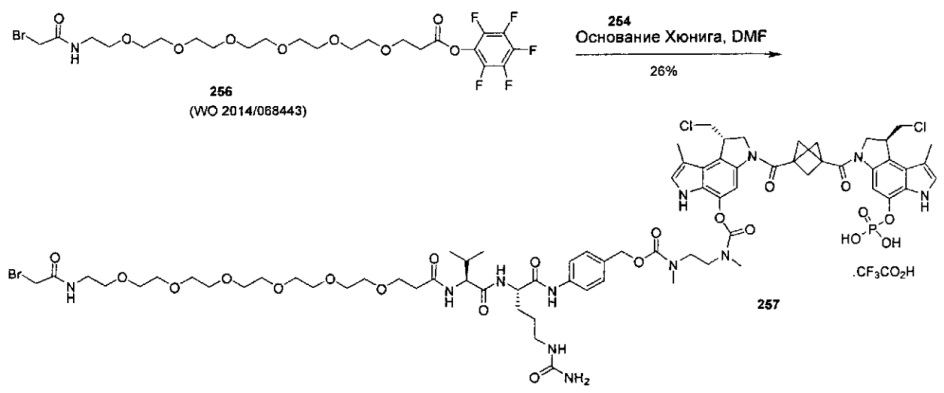
В перемешиваемый раствор соединения 254 (30 мг, 0,020 ммоль) и пентафторфенил-1-бром-2-оксо-6,9,12,15,18,21-гексаокса-3-азатетракозан-24-оата 2556 (13,8 мг, 0,0215 ммоль) [полученного, как описано в WO 2014/068443] в 2,0 мл DMF добавляли основание Хюнига (0,0136 мл, 0,0782 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение примерно 40 мин. Неочищенную реакционную смесь инжектировали в предколонку 5 г С18 (которая была предварительно уравновешена ацетонитрилом и затем водой, с 0,02% TFA в каждой фазе). Вещество очищали обращенно-фазной С18 хроматографией среднего давления (градиент: от 5% до 50% ацетонитрила в воде с 0,02% TFA в каждой фазе), после чего осуществляли вторую очистку препаративной ВЭЖХ (метод K1). Содержимое соответствующих пробирок концентрировали, используя испаритель Genevac, с получением соединения 257 (10,8 мг, 26%) в виде белого твердого вещества. ЖХ/МС (Протокол B): m/z 1649,7 [M+3H]+, время удерживания = 1,53 мин.
Получение N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-[(1S)-3-[(3-карбоксибицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]-L-аланинамида 261
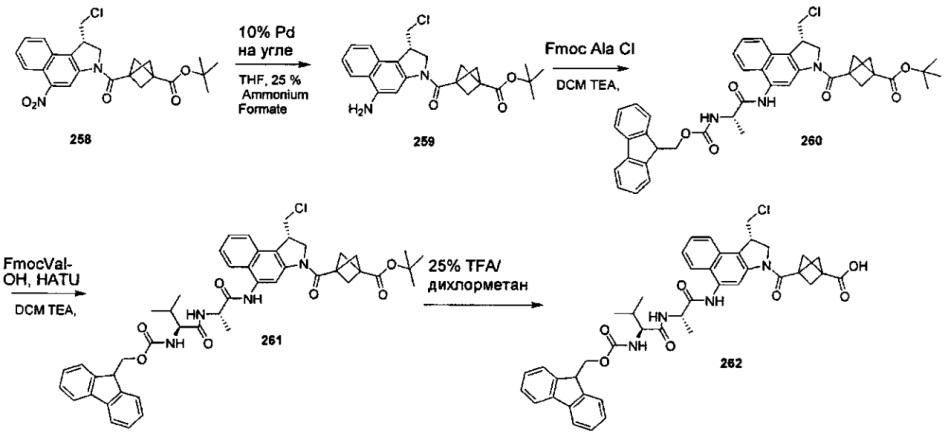
Стадия 1: Перемешиваемый раствор трет-бутил-3-{[(1S)-1-(хлорметил)-5-нитро-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пентан-1-карбоксилата 258 (полученного аналогично соединению 189) (980 мг, 2,14 ммоль) в 7 мл THF под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (30 мг), после чего медленно по каплям добавляли 2 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 3 часов. После окончания реакции реакционную смесь фильтровали через слой целита, и фильтрат концентрировали под вакуумом. Неочищенные продукты очищали хроматографией на силикагеле (градиент: от 0% до 10% MeOH в DCM) с получением трет-бутил-3-{[(1S)-5-амино-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пентан-1-карбоксилата 259 в виде желтого твердого вещества (905 мг, 98%). ЖХ/МС (Протокол B): m/z 427 [M+H]+, время удерживания = 1,92 мин.
Стадия 2. В перемешиваемый раствор соединения 259 (900, 2,11 ммоль) в 5 мл безводного DCM добавляли (9H-флуорен-9-ил)метил-(S)-(1-хлор-1-оксопропан-2-ил)карбамат (695 мг, 2.11 ммоль), после чего по каплям добавляли TEA (0,5 мл). Эту реакционную смесь перемешивали в течение 2 часов. По завершении перемешивания реакционную смесь концентрировали под вакуумом. Неочищенные продукты очищали хроматографией на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением трет-бутил-3-{[(1S)-1-(хлорметил)-5-({N-[(9H-флуорен-9-илметокси)карбонил]-L-аланил}амино)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пентан-1-карбоксилата 260 в виде белого твердого вещества (1,102 г, 73%). ЖХ/МС (Протокол B): m/z 720 [M+H]+, время удерживания = 2,32 мин.
Стадия 3: В оснащенную мешалкой круглодонную колбу, содержащую соединение 260 (1000 мг, 1,388 ммоль), добавляли 15 мл смеси 1:1 DCM в DEA. Этот раствор перемешивали в течение 3 часов. Реакционную смесь концентрировали под вакуумом и переносили в 50% DCM в гептане и снова концентрировали под вакуумом. Эту процедуру повторяли 3 раза (до удаления избытка DEA) с получением неочищенного соединения в виде белого твердого вещества после концентрирования. Это неочищенное белое твердое вещество добавляли в круглодонную колбу, содержащую (((9H-флуорен-9-ил)метокси)карбонил)-L-валин (471 мг, 1,38 ммоль) и HATU (350 мг, 1,38 ммоль) в 10 мл безводного DCM. Затем добавляли TEA (0,5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. По завершении реакционную смесь концентрировали под вакуумом. Неочищенные продукты очищали хроматографией на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-[(1S)-3-{[3-(трет-бутоксикарбонил)бицикло[1.1.1]пент-1-ил]карбонил}-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]-L-аланинамида 261 в виде белого твердого вещества (1,005 г, 88%). ЖХ/МС (Протокол B): m/z 819 [M+H]+, время удерживания = 2,31 мин.
Стадия 4: 10 мл 25% TFA в DCM добавляли в круглодонную колбу, содержащую соединение 261 (1000 мг, 1.22 ммоль). Эту реакционную смесь перемешивали в течение 3 часов. Раствор перемешивали в течение 3 часов. Реакционную смесь концентрировали под вакуумом и переносили в смесь 50% DCM и гептана и концентрировали под вакуумом. Эту процедуру повторяли 3 раза (до удаления избытка TFA) с получением после концентрирования соединения 262 в виде белого твердого вещества (920 мг, 98%). ЖХ/МС (Протокол B): m/z 763 [M+H]+, время удерживания = 1,88 мин.
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N-{(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}-L-аланинамида 266
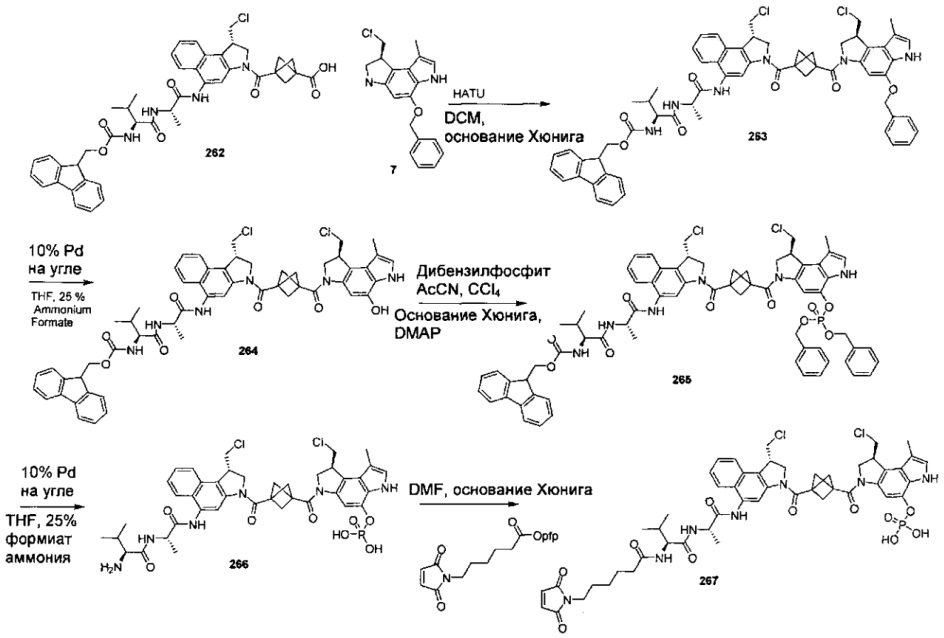
Стадия 1: В круглодонную колбу, содержащую N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-[(1S)-3-[(3-карбоксибицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]-L-аланинамид 262 (580 мг, 0,76 ммоль) в 5 мл THF, добавляли HATU (298 мг, 0,76 ммоль). Эту смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли (1S)-5-(бензилокси)-1-(хлорметил)-8-метил-1,2,3,6-тетрагидропирроло[3,2-е]индол 7, после чего добавляли 0,3 мл основания Хюнига. Реакционную смесь перемешивали в течение 1 часа и концентрировали до неочищенного стеклообразного вещества. Неочищенную реакционную смесь очищали хроматографией на силикагеле (градиент: от 0% до 10% MeOH в DCM) с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-[(1S)-3-[(3-{[(1S)-5-{[бис(бензилокси)фосфорил]окси}-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]-L-аланинамида 263 в виде белого твердого вещества (723 мг, 98%). ЖХ/МС (Протокол B): m/z 1071 [M+H]+, время удерживания = 2,45 мин.
Стадия 2: Перемешиваемый раствор соединения 263 (100 мг, 0,932 ммоль) в 7 мл THF под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (10 мг), после чего медленно по каплям добавляли 0,5 мл 25%-ного формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 1 часа. По завершении перемешивания реакционную смесь фильтровали через слой целита, и фильтрат концентрировали под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 10% MeOH в DCM) с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-{(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-гидрокси-8-метил-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}-L-аланинамида 264 в виде желтого твердого вещества (821 мг, 89%). %). ЖХ/МС (Протокол B): m/z 981 [M+H]+, время удерживания = 2,16 мин.
Стадия 3: В перемешиваемый раствор соединения 264 (650 мг, 0,66 ммоль) в 10 мл THF и 10 мл AcCN добавляли тетрахлорид углерода (2,04 мл, 21,0 ммоль), после чего добавляли основание Хюнига (1,12 мл, 6,45 ммоль), дибензилфосфит (694 мг, 2,65 ммоль) и DMAP (каталитическое количество). Эту реакционную смесь перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь концентрировали до неочищенного стеклообразного вещества. Эту неочищенную реакционную смесь очищали хроматографией на силикагеле (градиент: от 0% до 10% MeOH в DCM) с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-[(1S)-3-[(3-{[(1S)-5-{[бис(бензилокси)фосфорил]окси}-1-(хлорметил)-8-метил-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]-L-аланинамида 265 в виде белого стекла (502 мг, 66%). ЖХ/МС (Протокол B): m/z 1243 [M+H]+, время удерживания = 2,46 мин.
Стадия 4: Перемешиваемый раствор соединения 264 (100 мг, 0,932 ммоль) в 7 мл THF под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (10 мг), после чего медленно по каплям добавляли 0,5 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 1 часа. По завершении перемешивания реакционную смесь фильтровали через слой целита, и фильтрат концентрировали под вакуумом. Неочищенные продукты очищали хроматографией на силикагеле (градиент: от 0% до 10% MeOH в DCM) с получением L-валил-N-{(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-8-метил-5-(фосфоноокси)-1,6-дигидропирроло[3,2-е]индол-3(2H)-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}-L-аланинамида 265 в виде желтого твердого вещества (25 мг, 18%). ЖХ/МС (Протокол B): m/z 839 [M+H]+, время удерживания = 1,54 мин.
Стадия 5: В оснащенную мешалкой круглодонную колбу, содержащую пентафторфенил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноат (18 мг, 0,046 ммоль), добавляли 5 мл безводного DCM, и систему продували N2. В этот раствор добавляли соединение 265 (40 мг, 0,046 ммоль) и TEA (0,05 мл). Систему перемешивали в течение 1 часа. Реакционную смесь концентрировали под вакуумом и очищали с получением соединения 267 (20% 9 мг, Метод N), время удерживания = 15,462 мин. ЖХ/МС (Протокол B): m/z 1032 [M+H]+, время удерживания = 1,55 мин. 1H ЯМР (400 МГц, DMSO-d6) δ 11.34 (s, 1Н), 9.89 (s, 1Н), 8.43 (s, 1Н), 8.20 (d, J=6.8 Гц, 2Н), 7.91 (dd, J=14.4, 8.4 Гц, 3H), 7.85-7.74 (m, 2Н), 7.49 (t, J=7.7 Гц, 1Н), 7.35 (t, J=7.8 Гц, 1Н), 6.96 (d, J=25.4 Гц, 4Н), 4.52 (t, J=7.1 Гц, 1Н), 4.37 (dq, J=22.0, 10.7 Гц, 4Н), 4.18 (dt, J=19.7, 8.5 Гц, 2Н), 4.07-3.85 (m, 4Н), 3.58 (t, J=9.8 Гц, 1Н), 3.43-3.12 (m, 34Н), 2.71 (d, J=8.2 Гц, 1Н), 2.62-2.37 (m, 49Н), 2.28 (s, 3H), 2.09 (qt, J=14.0, 7.1 Гц, 3H), 1.98-1.86 (m, 1Н), 1.39 (dt, J=22.2, 7.2 Гц, 11Н), 1.22-1.05 (m, 6Н), 0.78 (dd, J=9.7, 6.7 Гц, 10H).
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N-{(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}-L-аланинамида 270
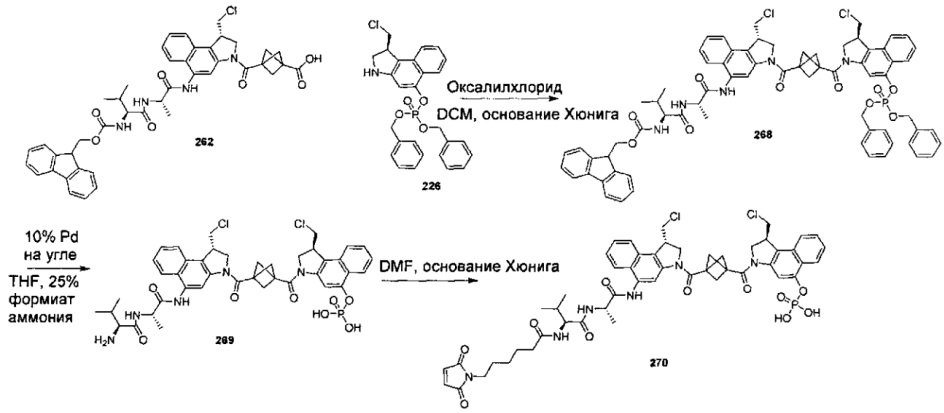
Стадия 1: Соединение 226 (214 мг, 0,36 ммоль) переносили в CH2Cl2 (2 мл) и добавляли TFA (0,5 мл), и после окончания удаления защитной группы растворитель удаляли. В продуваемую N2 круглодонную колбу, содержащую 262 (200 мг, 0,26 ммоль) в 5 мл безводного DCM, добавляли оксалилхлорид (0,024 мл, 0,26 ммоль). В этот раствор добавляли 1 каплю DMF, и эту систему перемешивали в течение 3 часов. Реакционную смесь концентрировали под вакуумом. Остаток переносили в DCM и добавляли в круглодонную колбу, содержащую лишенную защитной группы соединение 226 в 15 мл DCM и TEA (0,144 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Неочищенную реакционную смесь концентрировали под вакуумом, переносили в 25 мл DCM и переносили в делительную воронку. Органический слой промывали 1М HCl (3x), водой (3x) и рассолом (2х). Органический слой сушили над Na2SO4, фильтровали, и фильтрат концентрировали до неочищенного твердого вещества. Неочищенный продукт очищали хроматографией на силикагеле (градиент: от 0% до 10% MeOH в DCM) с получением N-[(9H-флуорен-9-илметокси)карбонил]-L-валил-N-[(1S)-3-[(3-{[(1S)-5-{[бис(бензилокси)фосфорил]окси}-1-(хлорметил)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил]-L-аланинамида 268 в виде желтого твердого вещества (75 мг, 23%). ЖХ/МС (Протокол В): m/z 1238 [M+H]+, время удерживания = 2,53 мин.
Стадия 2: Перемешиваемый раствор соединения 268 (75 мг, 0,061 ммоль) в 5 мл THF под азотом охлаждали до 0°C, используя ледяную баню. Затем добавляли 10%-ный палладий на активированном угле (5 мг), после чего медленно по каплям добавляли 0,5 мл 25%-ного раствора формиата аммония в воде. Реакционную смесь перемешивали при 0°C в течение 3 часов. По завершении перемешивания реакционную смесь фильтровали через слой целита, и фильтрат концентрировали под вакуумом. Неочищенный продукт переносили в этилацетат, и твердое вещество отфильтровывали с получением L-валил-N-{(1S)-1-(хлорметил)-3-[(3-{[(1S)-1-(хлорметил)-5-(фосфоноокси)-1,2-дигидро-3H-бензо[е]индол-3-ил]карбонил}бицикло[1.1.1]пент-1-ил)карбонил]-2,3-дигидро-1H-бензо[е]индол-5-ил}-L-аланина 269 в виде светло-желтого твердого вещества (20 мг, 30%). ЖХ/МС (Протокол B): m/z 838 [M+H]+, время удерживания = 1,27 мин.
Стадия 3: В оснащенную мешалкой круглодонную колбу, содержащую пентафторфенил 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноат (9,0 мг, 0,024 ммоль), добавляли 5 мл безводного DCM, и систему продували N2. В этот раствор добавляли соединение 269 (20 мг, 0,024 ммоль) и TEA (0.05 мл). Систему перемешивали в течение 1 часа. Реакционную смесь концентрировали под вакуумом и очищали посредством ВЭЖХ, Метод N, с получением соединения 270 (5 мг, 20%). Время удерживания = 10,734 мин. ЖХ/МС (Протокол B): m/z 1031 [M+H]+, время удерживания = 1,54 мин.
Получение (2S,3S,4S,5R,6S)-6-(((S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(((2-((((4-((23S,26S)-1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-23-изопропил-21,24-диоксо-26-(3-уреидопропил)-3,6,9,12,15,18-гексаокса-22,25-диазагептакозан-27-амидо)бензил)окси)карбонил)(метил)амино)этил)(метил)карбамоил)окси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил)окси)-3,4,5-тригидрокситетрагидро-2H-пиран-2-карбоновой кислоты 278
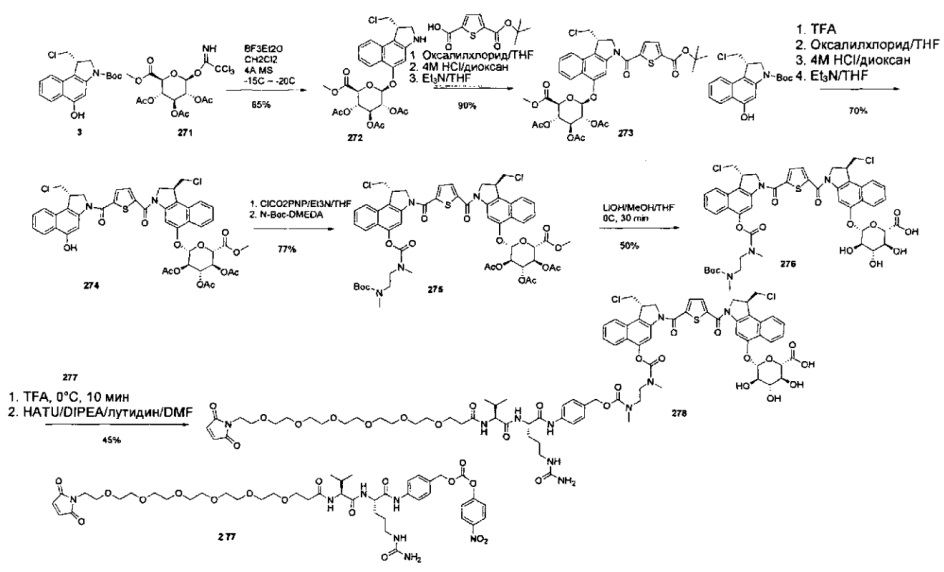
Стадия 1: трет-Бутил-(1S)-1-(хлорметил)-5-гидрокси-1,2-дигидро-3H-бензо[е]индол-3-карбоксилат 3 (683 мг, 2,05 ммоль) растворяли в DCM (70 мл), добавляли молекулярные сита  (3,8 г, порошок, <5 микрон, активированные), и эту смесь перемешивали при комнатной температуре в течение 30 мин. В реакционную смесь добавляли альфа-D-глюкуронида метиловый эфир 2,3,4-триацетат 1-2,2,2-трихлорэтанимидат 271 (1178 мг, 2,45 ммоль), и смесь охлаждали до -15°C. Медленно добавляли раствор BF3⋅Et2O (0,13 мл, 1,02 ммоль) в DCM (10 мл), и реакционную смесь перемешивали при температуре ниже -20°C в течение 1 ч. В смесь добавляли раствор BF3⋅Et2O (0,76 мл, 6 ммоль) в DCM (10 мл) для удаления Boc-группы, и реакционную смесь оставляли нагреваться до комнатной температуры в течение 2 ч. Смесь фильтровали через слой целита, и фильтрат концентрировали с получением зеленой пены (липкой). В нее добавляли 4М HCl (2 мл) и снова концентрировали с получением зеленой пены в виде неочищенного продукта 272, 1130 мг (94%), который использовали на следующей стадии без дополнительной очистки.
(3,8 г, порошок, <5 микрон, активированные), и эту смесь перемешивали при комнатной температуре в течение 30 мин. В реакционную смесь добавляли альфа-D-глюкуронида метиловый эфир 2,3,4-триацетат 1-2,2,2-трихлорэтанимидат 271 (1178 мг, 2,45 ммоль), и смесь охлаждали до -15°C. Медленно добавляли раствор BF3⋅Et2O (0,13 мл, 1,02 ммоль) в DCM (10 мл), и реакционную смесь перемешивали при температуре ниже -20°C в течение 1 ч. В смесь добавляли раствор BF3⋅Et2O (0,76 мл, 6 ммоль) в DCM (10 мл) для удаления Boc-группы, и реакционную смесь оставляли нагреваться до комнатной температуры в течение 2 ч. Смесь фильтровали через слой целита, и фильтрат концентрировали с получением зеленой пены (липкой). В нее добавляли 4М HCl (2 мл) и снова концентрировали с получением зеленой пены в виде неочищенного продукта 272, 1130 мг (94%), который использовали на следующей стадии без дополнительной очистки.
Стадия 2: Моно-tBu эфир тиофендикислоты 187 (189 мг, 0,83 ммоль) растворяли в THF (10 мл), охлаждали до 0°C и добавляли оксалилхлорид (2М в DCM, 0,8 мл, 1,6 ммоль), после чего добавляли DMF (2 капли). Эту смесь перемешивали при 0°C в течение 5 минут и затем при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты в виде не совсем белого твердого вещества. Вышеуказанное твердое вещество смешивали с соединением 272 (246 мг, 0,42 ммоль) и обрабатывали THF (10 мл) при 0°C, после чего добавляли Et3N (0,29 мл, 2 ммоль). Смесь перемешивали при 0°C в течение 5 минут и при комнатной температуре в течение 30 мин. Смесь концентрировали, и остаток очищали колоночной хроматографией на силикагеле, используя смесь EA/Hep (этилацетат/гептан) (50/50), с получением продукта 273 в виде желтого твердого вещества (302 мг, 90%) ЖХ/МС: 760,1.
Стадия 3: Соединение 273 (790 мг, 1,04 ммоль) обрабатывали TFA (2 мл) и DCM (4 мл) при комнатной температуре в течение 1 ч. Смесь концентрировали с получением желтого твердого вещества. Это твердое вещество растворяли в THF (10 мл), охлаждали до 0°C, добавляли оксалилхлорид (2М в DCM, 1 мл, 2 ммоль), после чего добавляли DMF (1 каплю). Смесь перемешивали при 0°C в течение 5 минут и затем при комнатной температуре в течение 1 ч. Смесь концентрировали с получением хлорангидрида кислоты в виде желтого твердого вещества. Соединение 3 (118 мг, 1,56 ммоль) обрабатывали 4М HCl (4 мл) в течение 1 ч и концентрировали в вакууме с получением лишенного Boc-группы соединения в виде зеленого твердого вещества. Его растворяли в THF (10 мл), добавляли раствор вышеуказанного хлорангидрида кислоты в THF (10 мл) при 0°C, после чего добавляли Et3N (0,58 мл, 4,16 ммоль), и эту смесь перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли EA, промывали водой и рассолом и сушили над MgSO4. Смесь концентрировали в вакууме, и остаток обрабатывали MeOH. Полученное твердое вещество собирали фильтрованием с получением продукта в виде желтого твердого вещества 274 (668 мг, 70%). ЖХ/МС: 919,1
Стадия 4: Соединение 274 (576 мг, 0,63 ммоль) растворяли в THF (20 мл), охлаждали до 0°C, добавляли раствор пара-нитрофенилхлорформиата (263 мг, 1,26 ммоль) в DCM (2 мл), после чего добавляли Et3N (0,52 мл, 3,76 ммоль). Эту смесь перемешивали при 0°C в течение 5 минут и затем при комнатной температуре в течение 2 ч. ЖХ/МС показала окончание образования карбоната. Соединение 213 (354 мг, 1,88 ммоль) в THF (2 мл) добавляли в вышеуказанную смесь, и ее перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли EtOAc, промывали водой и рассолом и сушили над MgSO4. Смесь концентрировали в вакууме с получением твердого остатка, который обрабатывали MeOH до образования осадка. Полученное твердое вещество собирали фильтрованием с получением продукта 275 в виде желтого твердого вещества (550 мг, 77%).
Стадия 5: Соединение 275 (550 мг, 0,48 ммоль) растворяли в смеси THF/MeOH (1/1, 10 мл), охлаждали до 0°C, добавляли раствор LiOH⋅H2O (206 мг, 4,8 ммоль) в воде (3 мл), и эту смесь перемешивали при 0°C в течение 1 ч. HOAc (300 мг) добавляли для нейтрализации вышеуказанного раствора, и его концентрировали в вакууме. Остаток очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 276 в виде желтого твердого вещества (243 мг, 50%).
Стадия 6: Соединение 276 (50 мг, 0,05 ммоль) обрабатывали предварительно охлажденной TFA (2 мл) при 0°C в течение 5 минут и концентрировали в вакууме с получением лишенного Boc-группы соединения в виде желтого твердого вещества. Вышеуказанное твердое вещество растворяли в DMF (2 мл), добавляли соединение 277 (48 мг, 0,05 ммоль), после чего добавляли лутидин (0,035 мл, 0,3 ммоль), DIPEA (0,052 мл, 0,3 ммоль) и HOAt (7 мг, 0,05 ммоль). Эту смесь перемешивали при 30°C в течение 7 ч. Неочищенное соединение подвергали очистке на системе ВЭЖХ Gislon (0,02% TFA) с получением продукта 278 в виде желтого твердого вещества, 39 мг (45%). ЖХ/МС: 1715,8/1737,8 (1,71 мин по Larry); 1713,7(-).
Получение (2S,3S,4S,5R,6S)-6-(((S)-3-(5-((S)-5-(((2-((((4-((S)-2-((S)-2-((S)-2-ацетамидо-6-аминогексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)этил)(метил)-карбамоил)окси)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-1-(хлорметил)-2,3-дигидро-1H-бензо[е]индол-5-ил)окси)-3,4,5-тригидрокситетрагидро-2H-пиран-2-карбоновой кислоты 280
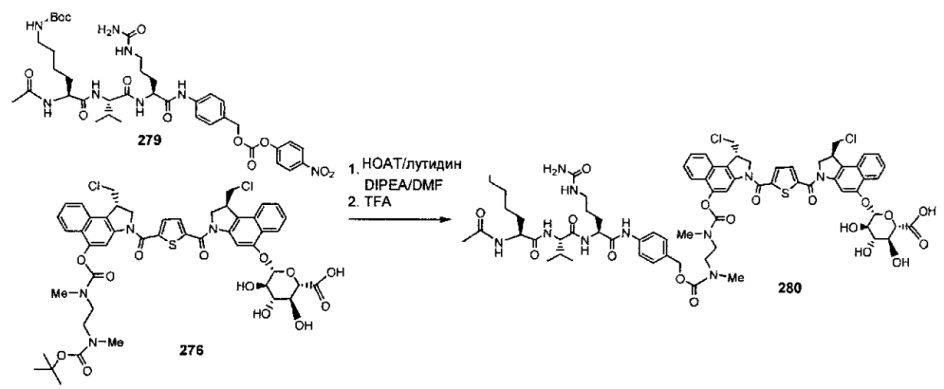
Соединение 276 обрабатывали TFA (2 мл) при 0°C в течение 1 ч. Его концентрировали в вакууме с получением его растворяли в DMF (2 мл), добавляли соединение 279 (59 мг, 0,07 ммоль), после чего добавляли лутидин (0,033 мл, 0,29 ммоль), DIPEA (0,051 мл, 0,29 ммоль) и HOAt (7 мг, 0,05 ммоль). Эту смесь перемешивали при 30°C в течение 4 ч. Смесь концентрировали, и остаток очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта в виде желтого твердого вещества, 48 мг (62%). Его обрабатывали предварительно охлажденной TFA (1,5 мл) в течение 5 минут, затем концентрировали в вакууме с получением неочищенного вещества в виде желтого твердого вещества. Это неочищенное вещество очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением, после сублимационной сушки, продукта 280 в виде желтого порошка (21 мг, 43%). ЖХ/МС: 1470,6
Получение (S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(((2S,3R,4S,5R,6R)-3,4,5-тригидрокси-6-(гидроксиметил)тетрагидро-2H-пиран-2-ил)окси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил-(4-((23S,26S)-1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-23-изопропил-21,24-диоксо-26-(3-уреидопропил)-3,6,9,12,15,18-гексаокса-22,25-диазагептакозан-27-амидо)бензил)этан-1,2-диилбис(метилкарбамата)
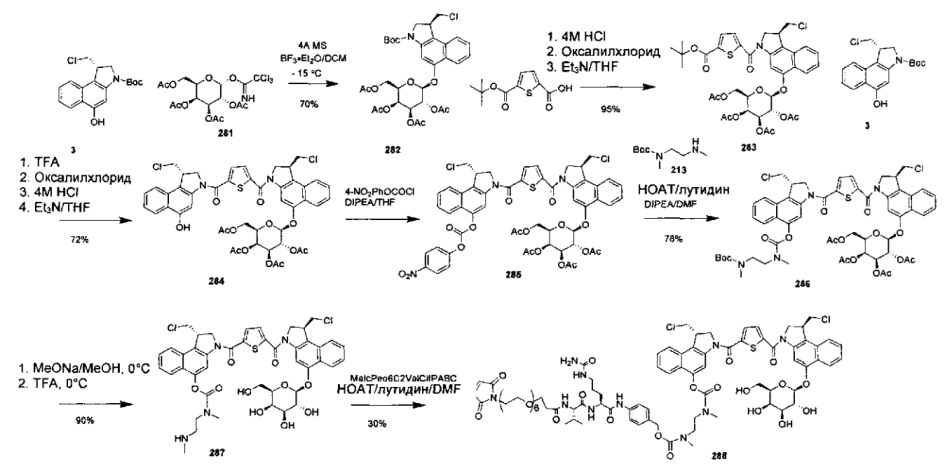
Стадия 1: Соединение 3 (775 мг, 2,3 ммоль) растворяли в DCM (80 мл), добавляли молекулярные сита 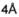 (6,2 г, порошок, <5 микрон, активированные), и эту смесь перемешивали при комнатной температуре в течение 30 мин. В реакционную смесь добавляли альфа-D-галактопонозы 2,3,4,6-тетраацетат 1-2,2,2-трихлорэтанимидат 281 (1260 мг, 2,3 ммоль) и охлаждали ее до -15°C. Медленно добавляли раствор BF3⋅Et2O (0,144 мл, 1,2 ммоль) в DCM (10 мл), и реакционную смесь перемешивали при температуре от -15°C до -20°C в течение 1 ч. Реакционную смесь фильтровали через слой целита, и фильтрат концентрировали. Неочищенное вещество очищали методом ISCO с использованием MeOH/DCM (0-20%) с получением продукта 282 в виде зеленого твердого вещества (1400 мг, 91%).
(6,2 г, порошок, <5 микрон, активированные), и эту смесь перемешивали при комнатной температуре в течение 30 мин. В реакционную смесь добавляли альфа-D-галактопонозы 2,3,4,6-тетраацетат 1-2,2,2-трихлорэтанимидат 281 (1260 мг, 2,3 ммоль) и охлаждали ее до -15°C. Медленно добавляли раствор BF3⋅Et2O (0,144 мл, 1,2 ммоль) в DCM (10 мл), и реакционную смесь перемешивали при температуре от -15°C до -20°C в течение 1 ч. Реакционную смесь фильтровали через слой целита, и фильтрат концентрировали. Неочищенное вещество очищали методом ISCO с использованием MeOH/DCM (0-20%) с получением продукта 282 в виде зеленого твердого вещества (1400 мг, 91%).
Стадия 2: Моно-tBu эфир тиофендикислоты 187 (300 мг, 1,3 ммоль) растворяли в THF (10 мл), охлаждали до 0°C и добавляли оксалилхлорид (2М в DCM, 1 мл, 2 ммоль), после чего добавляли DMF (2 капли). Эту смесь перемешивали при 0°C в течение 5 минут и затем при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты в виде белого твердого вещества. Соединение 282 (664 мг, 1 ммоль) обрабатывали 4М HCl (4 мл) в течение 1 ч при комнатной температуре. Его концентрировали в вакууме с получением лишенного Boc-группы амина в виде зеленого твердого вещества. Вышеуказанное твердое вещество смешивали с THF (10 мл) при 0°C, добавляли Et3N (0,83 мл, 6 ммоль). Смесь перемешивали при 0°C в течение 5 минут и при комнатной температуре в течение 30 мин. Смесь разбавляли EtOAc, промывали водой и рассолом и сушили над MgSO4. Смесь концентрировали в вакууме, остаток обрабатывали MeOH и снова концентрировали с получением твердого остатка, который подвергали перекристаллизации из MeOH. Полученное желтое твердое вещество собирали фильтрованием с получением продукта 283 в виде желтого твердого вещества (500 мг, 65%).
Стадия 3: Соединение 283 (200 мг, 0,26 ммоль) растворяли в THF (6 мл), добавляли оксалилхлорид (0,64 мл, 2М в DCM) при 0°C, после чего добавляли DMF (2 капли). Эту смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 0,5 ч. Смесь концентрировали в вакууме с получением соответствующего хлорангидрида кислоты в виде желтого твердого вещества. Соединение 3 (138 мг, 0,41 ммоль) обрабатывали 4М HCl (1 мл в диоксане) в течение 2 ч. Смесь концентрировали в вакууме с получением лишенного Boc-группы амина в виде зеленой пены. Эту пену растворяли в THF (5 мл), добавляли вышеуказанный хлорангидрид кислоты в THF (5 мл) при 0°C, после чего добавляли Et3N (0,23 мл, 1,55 ммоль). Смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 1 ч. Смесь разбавляли EtOAc, промывали водой и рассолом, сушили над MgSO4 и концентрировали в вакууме с получением твердого остатка, который обрабатывали МеОН. Полученное твердое вещество собирали фильтрованием и промывали диэтиловым эфиром с получением продукта в виде желтого твердого вещества. Фильтрат концентрировали и очищали на системе ВЭЖХ Gilson, используя смесь ACN/вода (0,02% TFA), с получением продукта в виде желтого твердого вещества 284 (200 мг, 83%).
Стадия 4: Соединение 284 (68 мг, 0,073 ммоль) растворяли в THF (3 мл), охлаждали до 0°C, добавляли раствор 4-нитрофенилхлорформиата (46 мг, 0,22 ммоль) в DCM (0,6 мл), после чего добавляли Et3N (0,061 мл, 0,44 ммоль). Эту смесь перемешивали при 0°C в течение 5 минут и при комнатной температуре в течение 1 ч с получением соединения 285. В вышеуказанную реакционную смесь добавляли N-Boc-DMEDA (55 мг, 0,29 ммоль), и смесь перемешивали при комнатной температуре в течение еще 1 ч. Смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson с получением продукта в виде желтого твердого вещества 286 (65 мг, 78%).
Стадия 5: Соединение 286 (10 мг, 0,009 ммоль) растворяли в MeOH (1 мл) при 0°C, добавляли MeONa (0,054 мл, 0,5М в MeOH, 0,027 ммоль), и эту смесь перемешивали при 0°C в течение 5 мин. Смесь нейтрализовали HOAc (0,4 мл, 0,1М в MeOH) и концентрировали в вакууме с получением продукта в виде желтого твердого вещества. Его обрабатывали предварительно охлажденной TFA (0,8 мл) в течение 2 минут и концентрировали в вакууме с получением лишенного Boc-группы соединения в виде желтого твердого вещества 287 (8,3 мг, 90%).
Стадия 6: Соединение 287 (8,3 мг, 0,008 ммоль) растворяли в DMF (1 мл), добавляли Malc-Peg6C2ValCitPABC (9,6 мг, 0,01 ммоль), после чего добавляли лутидин (0,004 мл), DIPEA (0,006 мл) и HOAt (1,1 мг, 0,008 ммоль). Эту смесь перемешивали при комнатной температуре в течение 4 ч. Неочищенное вещество очищали на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта 288 в виде желтого твердого вещества (4 мг, 30%). 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ=8.42 (d, J=8.2 Гц, 1Н), 8.15 (d, J=7.4 Гц, 1Н), 7.95 (d, J=8.2 Гц, 1Н), 7.91 (d, J=8.2 Гц, 1Н), 7.84 (d, J=8.6 Гц, 1Н), 7.80-7.66 (m, 2Н), 7.63-7.48 (m, 4Н), 7.43 (br. s., 3H), 7.23 (d, J=7.8 Гц, 1H), 6.81 (s, 2Н), 5.26-5.12 (m, 2Н), 5.09 (d, J=8.6 Гц, 1H), 4.71-4.54 (m, 4Н), 4.49 (br. s., 1H), 4.33-4.15 (m, 3H), 4.10-3.95 (m, 4H), 3.93-3.77 (m, 6H), 3.77-3.64 (m, 8H), 3.64-3.54 (m, 24H), 3.51 (br. s., 1H), 3.23-3.03 (m, 5H), 3.03-2.95 (m, 2H), 2.60-2.51 (m, 2H), 2.13 (d, J=7.0 Гц, 1H), 1.90 (br. s., 1H), 1.72 (br. s., 1H), 1.57 (br. s., 2H), 0.99 (t, J=6.4 Гц, 6H). ЖХ/МС: 1702,3/829,9/748,7.
Получение 4-((23S,26S)-1-амино-23-изопропил-21,24-диоксо-26-(3-уреидопропил)-3,6,9,12,15,18-гексаокса-22,25-диазагептакозан-27-амидо)бензил((S)-1-(хлорметил)-3-(5-((S)-1-(хлорметил)-5-(((2S,3R,4S,5R,6R)-3,4,5-тригидрокси-6-(гидроксиметил)тетрагидро-2H-пиран-2-ил)окси)-2,3-дигидро-1H-бензо[е]индол-3-карбонил)тиофен-2-карбонил)-2,3-дигидро-1H-бензо[е]индол-5-ил)этан-1,2-диилбис(метилкарбамата) 289
Стадия 1: BocValCitPABC 287 (30,9 мг, 0,048 ммоль) добавляли в раствор соединения 286 (32 мг, 0,032 ммоль) в DMF (2 мл), после чего добавляли лутидин (0,015 мл), DIPEA (0,022 мл) и HOAt (4,4 мг). Эту смесь перемешивали при комнатной температуре в течение 5 ч. Неочищенное соединение подвергали разделению на системе ВЭЖХ Gilson (0,02% TFA) с получением продукта в виде желтого твердого вещества 288 (32 мг, 72%).
Стадия 2: Соединение 288 (16 мг, 0,012 ммоль) обрабатывали предварительно охлажденной TFA (1 мл) в течение 5 минут и концентрировали в вакууме с получением лишенного Boc-группы соединения в виде желтого твердого вещества. Вышеуказанное твердое вещество растворяли в DMF (0,5 мл), добавляли DIPEA (0,013 мл), после чего добавляли раствор 206 (12 мг, 0,016 ммоль) в DCM (0,1 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. В вышеуказанный раствор добавляли пиперидин (0,2 мл), и смесь перемешивали в течение 30 мин. Смесь концентрировали в вакууме, и остаток очищали на системе ВЭЖХ Gilson, используя смесь ACN/вода (0,02% TFA), с получением продукта 289 в виде желтого твердого вещества (8 мг, 40%). 1H ЯМР (400 МГц, DMSO-d6) δ=9.89 (br. s., 1Н), 8.35-8.21 (m, 1Н), 8.15-8.00 (m, 2Н), 7.96 (d, J=7.8 Гц, 1Н), 7.91-7.84 (m, 1Н), 7.84-7.72 (m, 4Н), 7.63 (br. s., 2Н), 7.58-7.50 (m, 3H), 7.50-7.44 (m, 2Н), 7.40 (d, J=6.2 Гц, 2Н), 7.18 (br. s., 2Н), 5.90 (br. s., 1Н), 5.06-4.90 (m, 2Н), 4.87 (d, J=7.4 Гц, 1Н), 4.83-4.68 (m, 2Н), 4.42 (t, J=12.3 Гц, 2Н), 4.32 (br. s., 2Н), 4.23 (br. s., 1Н), 4.19-4.11 (m, 1Н), 4.10-3.95 (m, 3H), 3.95-3.80 (m, 2Н), 3.77-3.62 (m, 3H), 3.60-3.46 (m, 15Н), 3.15 (br. s., 2H), 3.06 (br. s., 1H), 2.89 (d, J=5.1 Гц, 3H), 2.92 (d, J=5.1 Гц, 3H), 2.86-2.74 (m, 3H), 2.35-2.21 (m, 1H), 1.96-1.82 (m, 1H), 1.61 (br. s., 1H), 1.52 (br. s., 1H), 1.43-1.21 (m, 2H), 0.76 (d, J=6.6 Гц, 3H), 0.79 (d, J=6.2 Гц, 3H); 1622,2 [M+H]+;
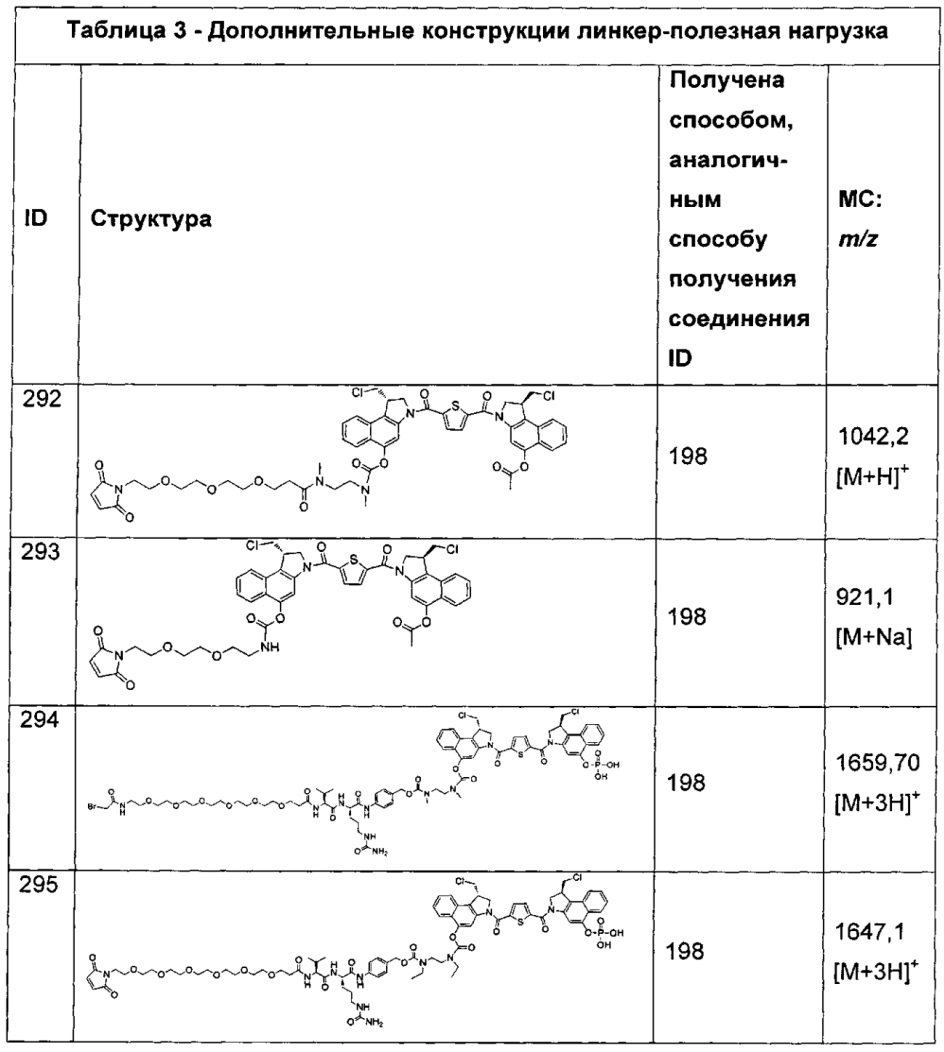
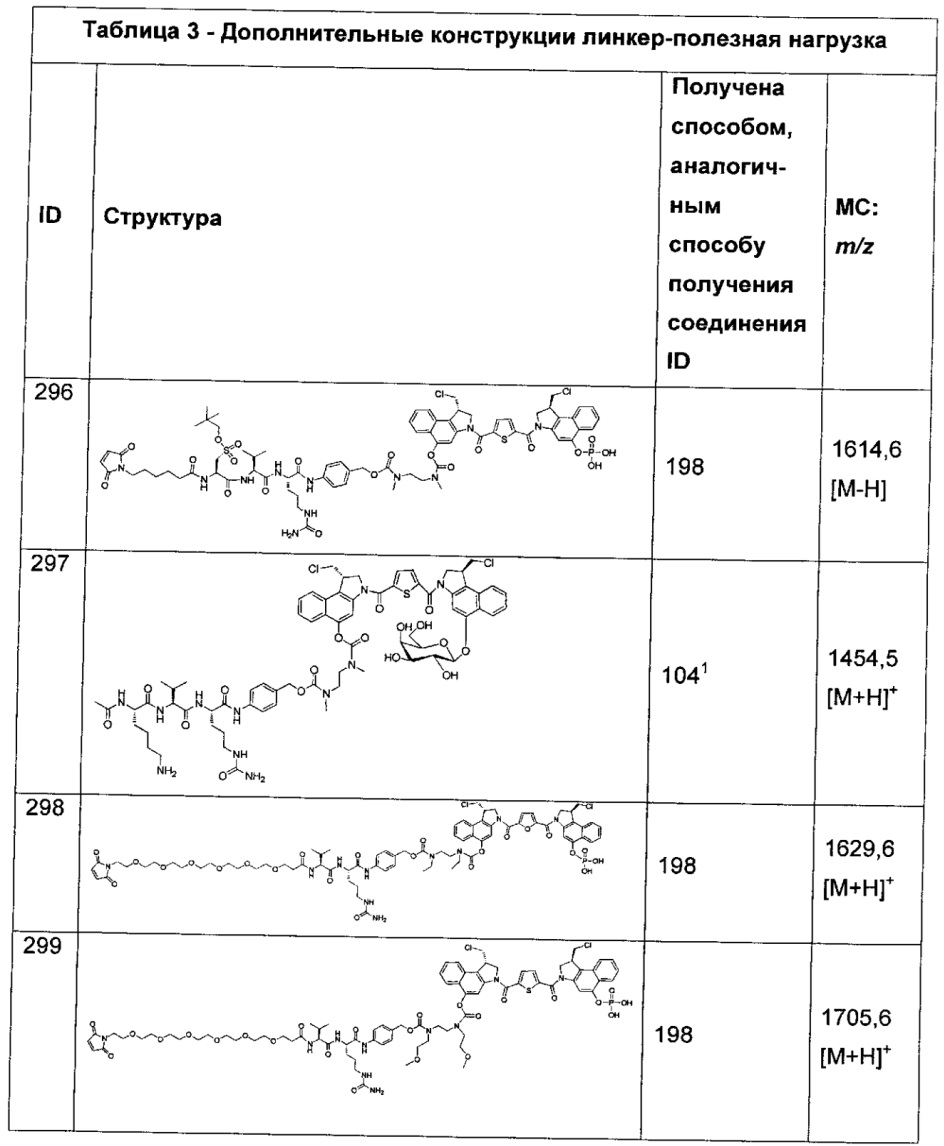
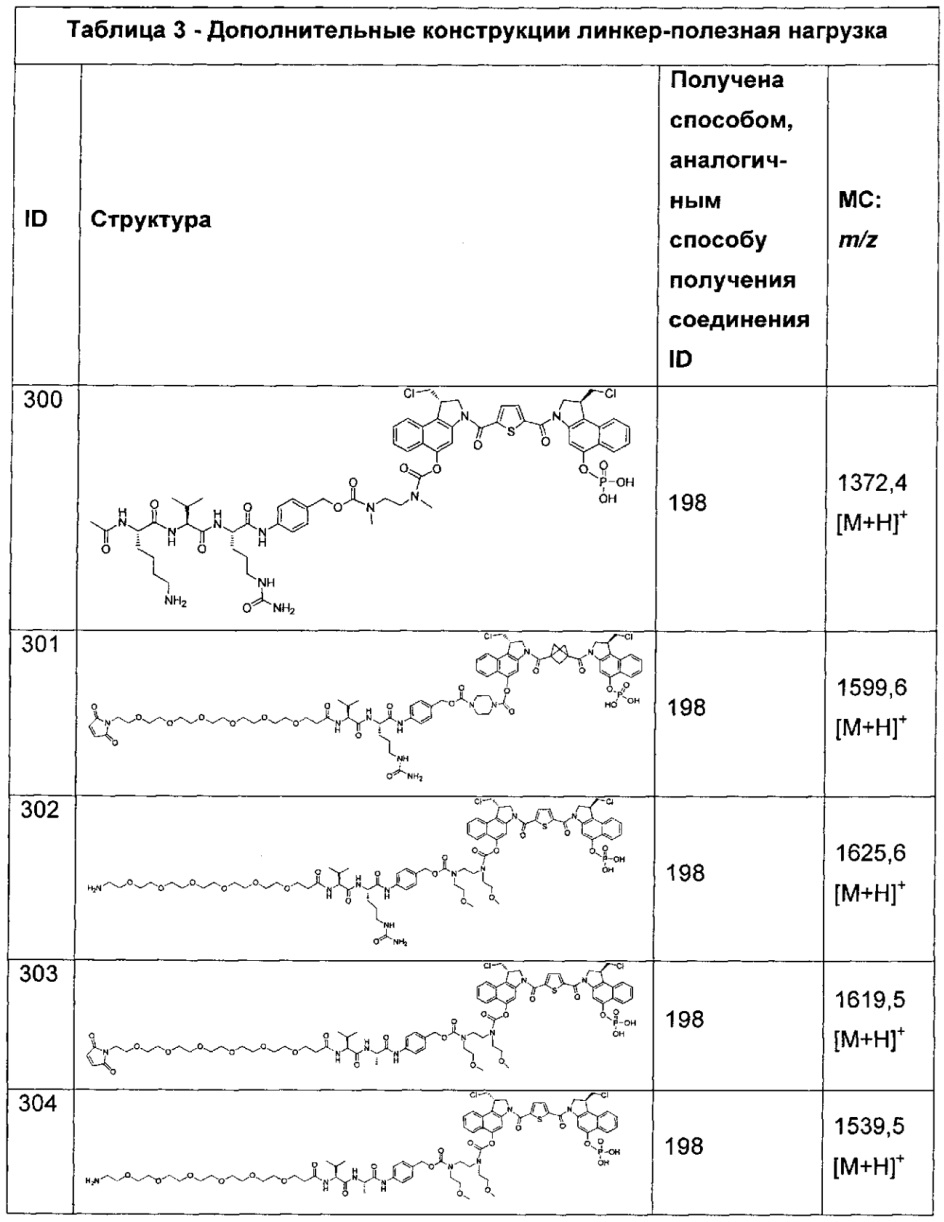
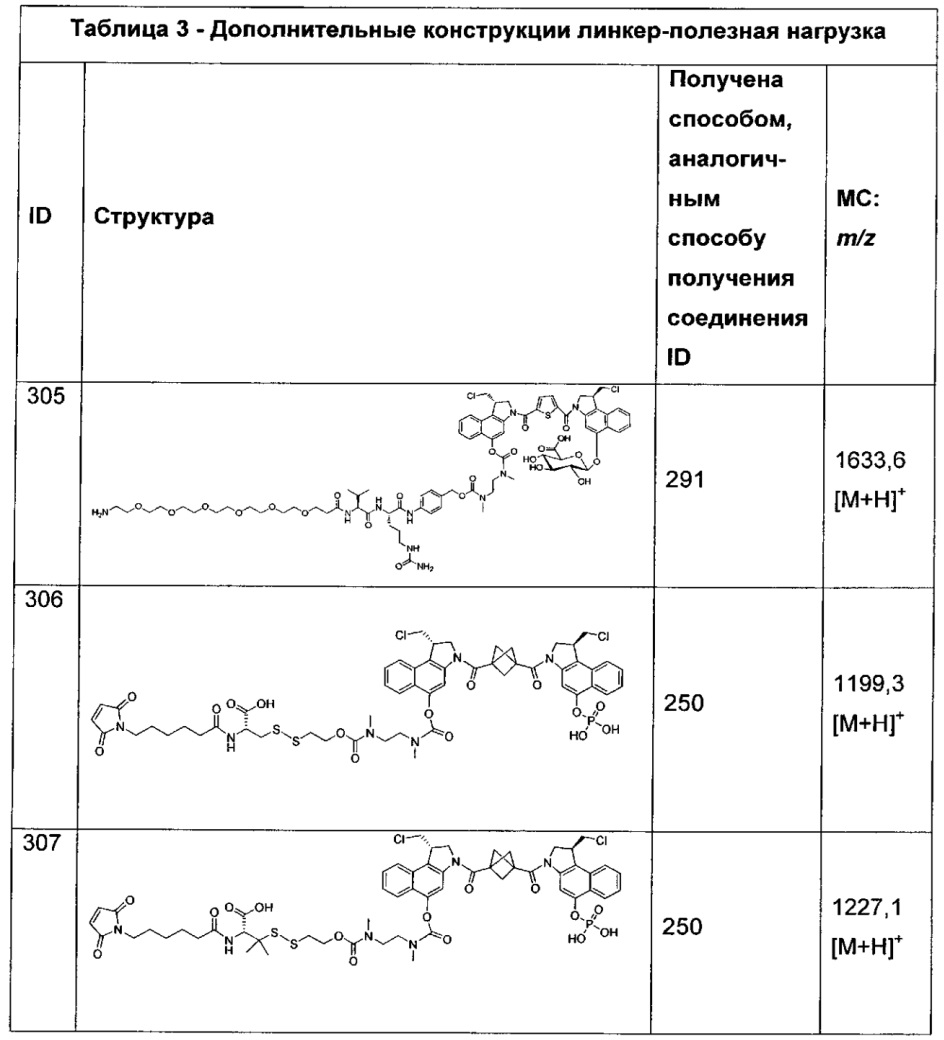
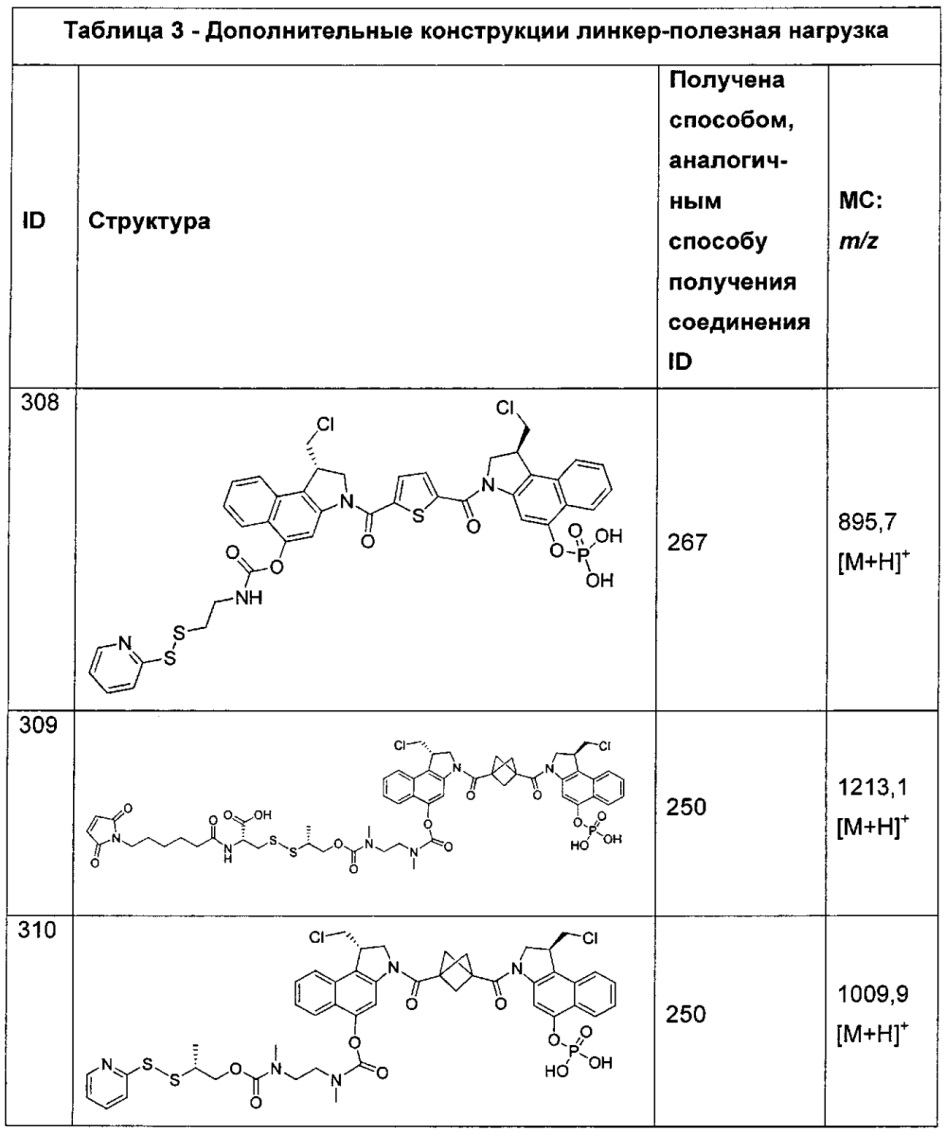
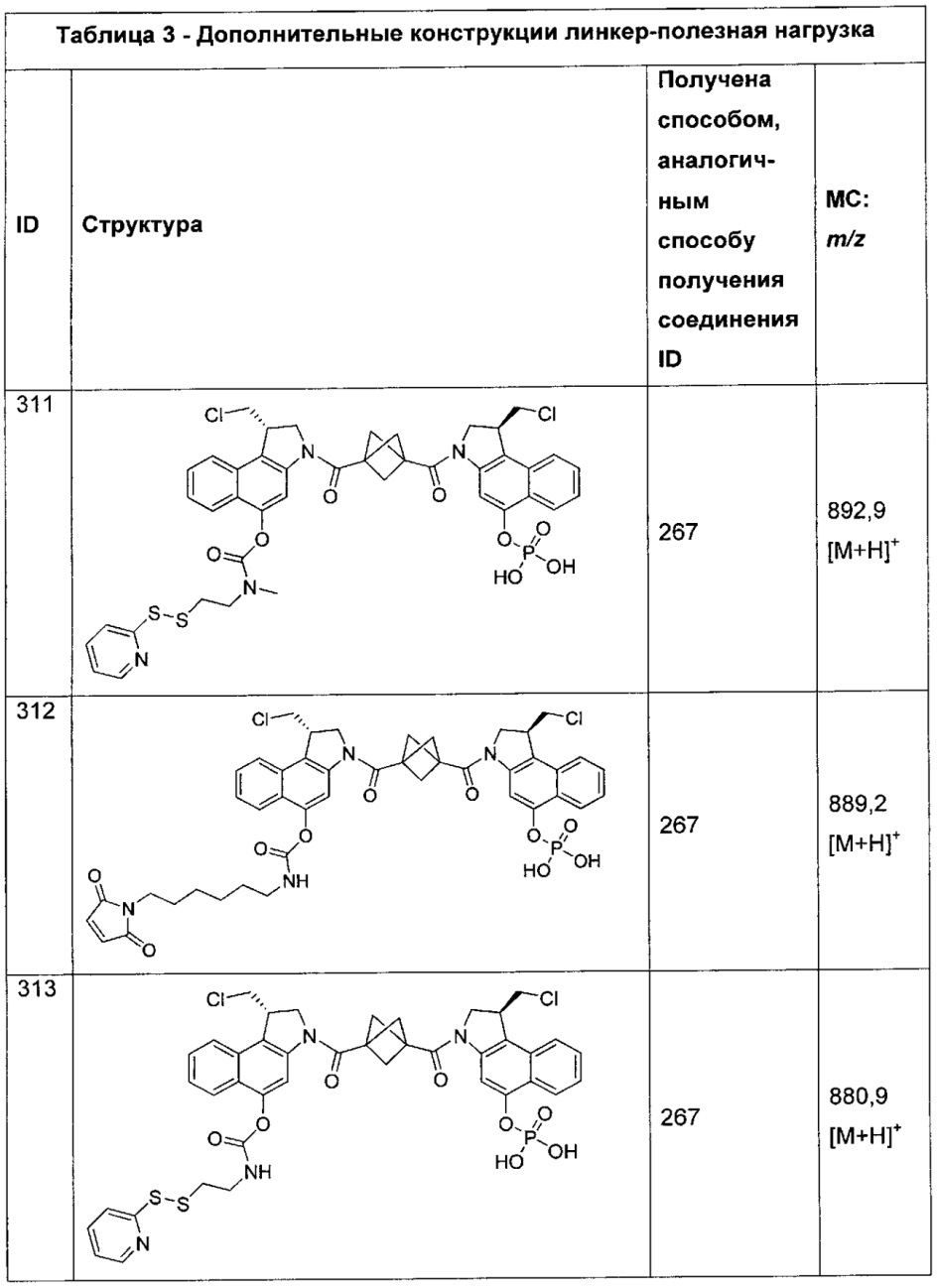
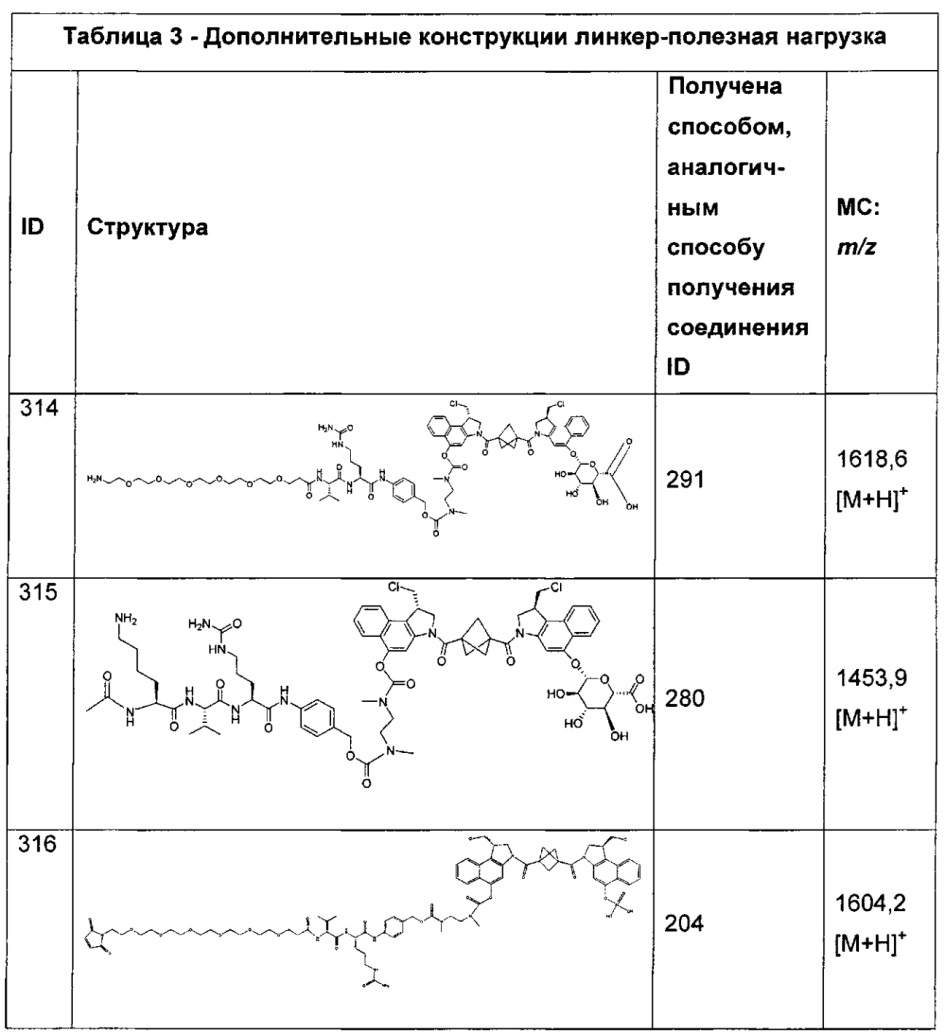





Согласно изобретению предложены также соединения, представленные в Таблицах 5A и 5B.


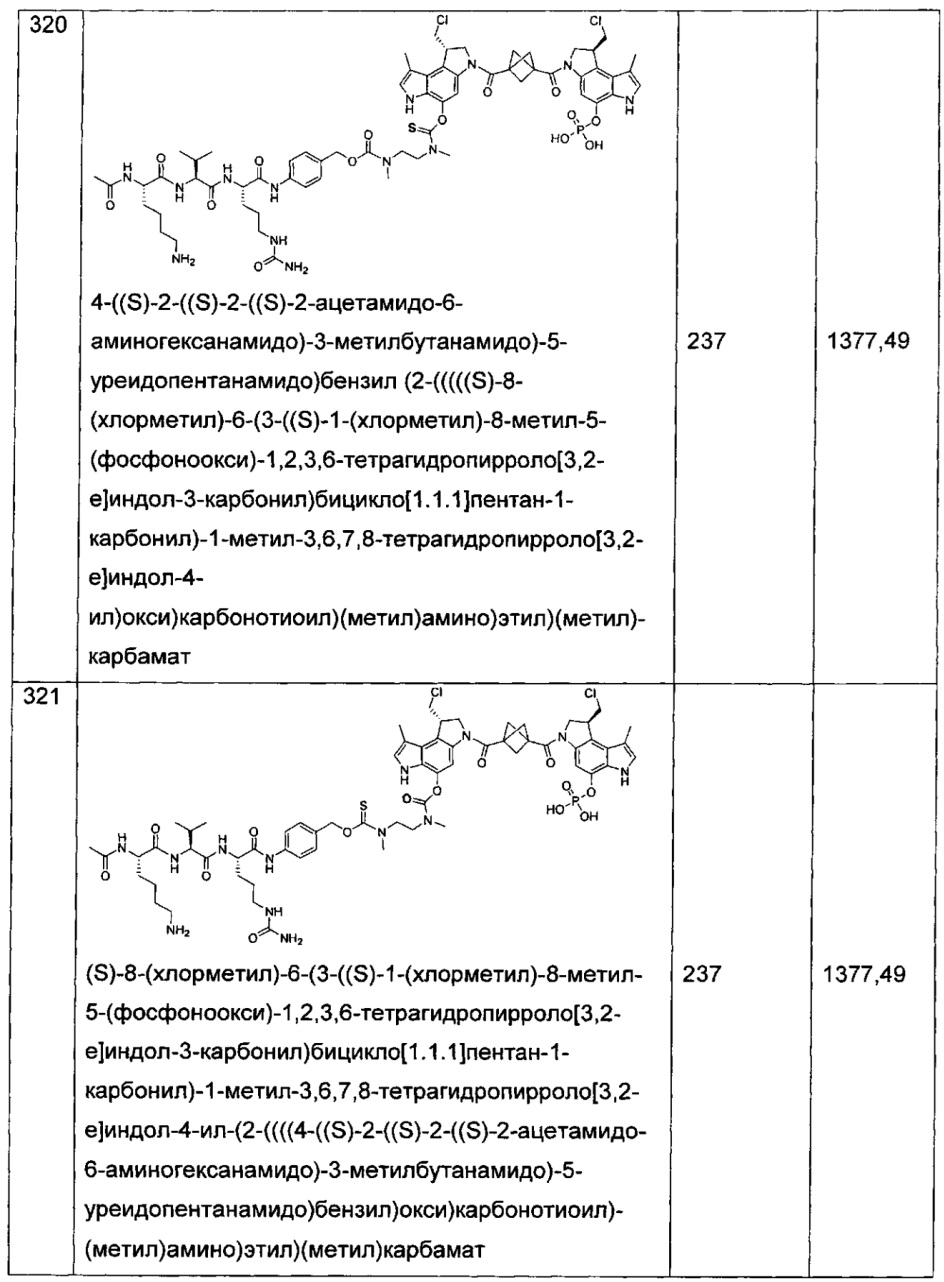
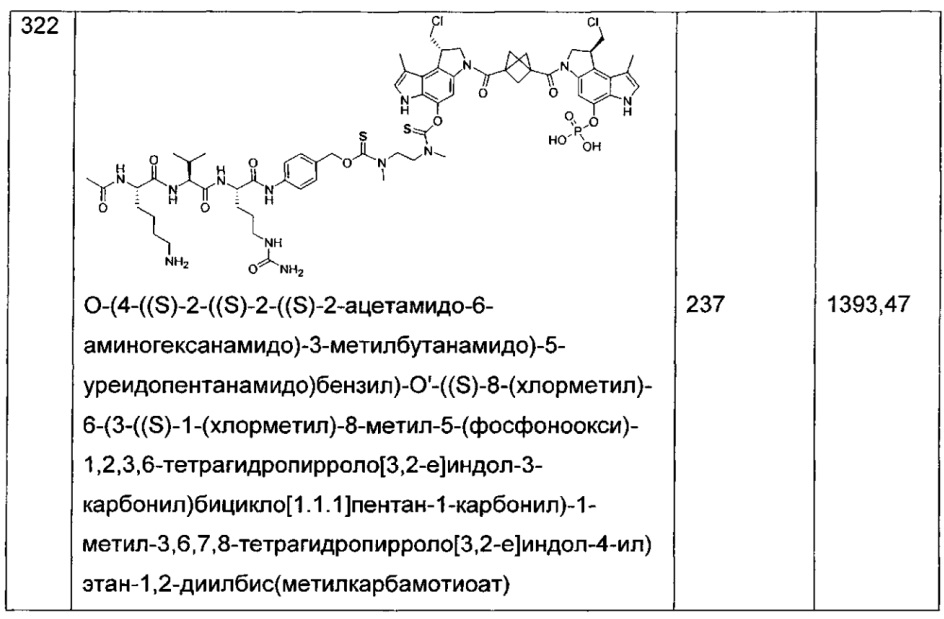
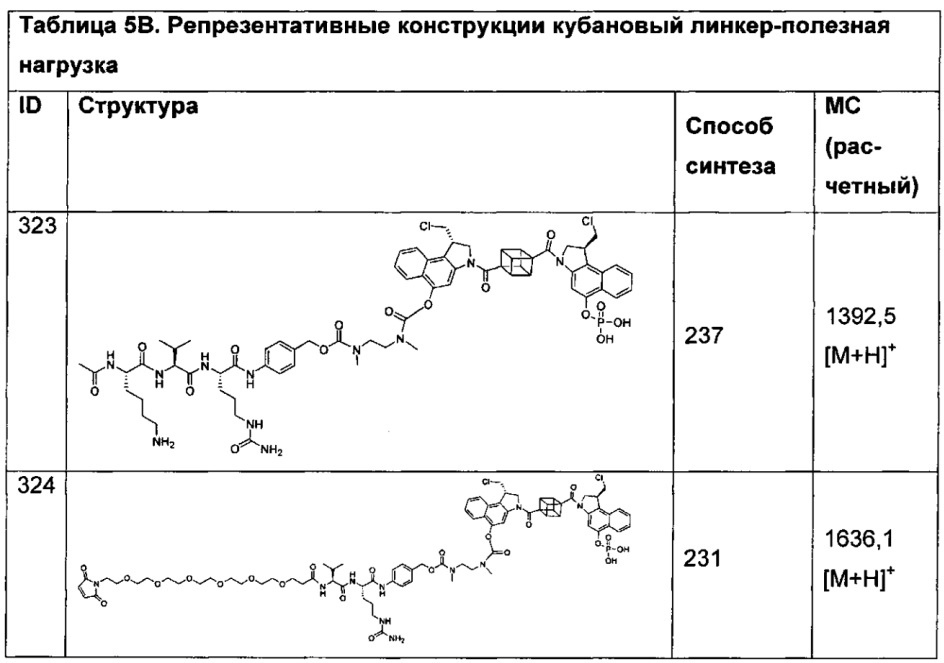
Условия ВЭЖХ и ЖХ/МС, использованные для анализа
Протокол A: колонка: Waters Acquity UPLC HSS T3, 2,1 мм×50 мм, С18, 1,7 мкм; подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: 5% B в течение 0,1 минуты, от 5% до 95% B за 0,9 минуты, 95% B в течение 0,1 минуты; скорость потока: 1,25 мл в минуту; температура: 60°C; детектирование: 200-450 нм; МС (+) диапазон 100-2000 дальтон; инжектируемый объем: 5 мл; прибор: Waters Acquity.
Протокол B: колонка: Waters Acquity UPLC HSS T3, 2,1 мм×50 мм, С18, 1,7 мкм; подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: 5% B в течение 0,1 минуты, от 5% до 95% B за 2,5 минуты, 95% B в течение 0,35 минуты; скорость потока: 1,25 мл в минуту; температура: 60°C; детектирование: 200-450 нм; МС (+) диапазон 100-2000 дальтон; инжектируемый объем: 5 мл; прибор: Waters Acquity.
Протокол C: колонка: Phenomenex Luna С18 (2), 150×3,0 мм, 5 мкм; подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: 50% B в течение 1,5 минуты, от 50% до 100% B за 6,5 минут, затем 100% B в течение 3 минут; скорость потока: 0,75 мл в минуту; температура: 45°C; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; инжектируемый объем: 10 мкл; прибор: Agilent 1200 LCMS.
Протокол D: колонка: Phenomenex Luna С18 PFP(2), 150×3,0 мм, 5 мкм; подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: от 0% до 5% B за 1,5 минуты, от 5% до 100% B заг 8,5 минут, затем 100% B в течение 2 минут; скорость потока: 0,75 мл в минуту; температура: не контролировали; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; инжектируемый объем: 10 мкл; прибор: Agilent 1200 LCMS.
Протокол E: колонка: Phenomenex Luna С18 PFP(2), 150×3,0 мм, 5 мкм; подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: 5% B в течение 1,5 минуты, от 5% до 100% B за 8,5 минут, затем 100% B в течение 2 минут; скорость потока: 0,75 мл в минуту; температура: не контролировали; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; инжектируемый объем: 10 мкл; прибор: Agilent 1200 LCMS.
Протокол F: колонка: Xtimate С18, 30×2,1 мм, 3 мкм; подвижная фаза A: 0,037% TFA в воде (об./об.); подвижная фаза B: 0,037% TFA в ацетонитриле (об./об.); градиент: 10% B в течение 0,1 минуты, от 10% до 80% B за 3 минуты, затем 80% B в течение 0,1 минуты; скорость потока: 1,5 мл в минуту; температура: 40°C; детектирование: ДДМ 220 нм; МС (+) диапазон 100-1000 дальтон; инжектируемый объем: 3 мкл; прибор: Shimadzu.
Протокол G: колонка: Xtimate С18, 30×2,1 мм, 3 мкм; подвижная фаза A: 0,037% TFA в воде (об./об.); подвижная фаза B: 0,037% TFA в ацетонитриле (об./об.); градиент: 10% B в течение 0,1 минуты, от 10% до 80% B за 3 минуты, затем 80% B в течение 0,1 минуты; скорость потока: 1,5 мл в минуту; температура: 40°C; детектирование: ДДМ 220 нм; МС (+) диапазон 100-1000 дальтон; инжектируемый объем: 3 мкл; прибор: Shimadzu.
Протокол H: колонка: Xtimate С18, 30×2,1 мм, 3 мкм; подвижная фаза A: 0,037% TFA в воде (об./об.); подвижная фаза B: 0,037% TFA в ацетонитриле (об./об.); градиент: 0% B в течение 0,1 минуты, от 0% до 60% B за 2 минуты, затем 60% B в течение 0,1 минуты; скорость потока: 1,5 мл в минуту; температура: 40°C; детектирование: ДДМ 220 нм; МС (+) диапазон 100-1000 дальтон; инжектируемый объем: 2 мкл; прибор: Shimadzu.
Условия ВЭЖХ, использованные для очистки
Метод A: колонка: Phenomenex Luna С18(2), 150×21,2 мм, 5 мкм; подвижная фаза A: 0,02% муравьиной кислоты в воде; подвижная фаза B: 0,02% муравьиной кислоты в ацетонитриле; градиент: 40% B в течение 1,5 минут, от 40% до 100% B за 8,5 минут, 100% B в течение 0,5 минуты; скорость потока: 27 мл в минуту; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; прибор: Waters FractionLynx.
Метод B: колонка: Phenomenex Luna PFP (2), 150×21,2 мм, 5 мкм; подвижная фаза A: 0,02% муравьиной кислоты в воде; подвижная фаза B: 0,02% муравьиной кислоты в ацетонитриле; градиент: 30% B в течение 1,5 минут, от 30% до 60% B за 8,5 минут, от 60% B до 100% B за 0,5 минуты, 100% B в течение 2 минут; скорость потока: 27 мл в минуту; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; прибор: Waters FractionLynx.
Метод C: колонка: Phenomenex Synergi Polar RP, 150×21,2 мм, 4 мкм; подвижная фаза A: 0,02% муравьиной кислоты в воде; подвижная фаза B: 0,02% муравьиной кислоты в ацетонитриле; градиент: 20% B в течение 1,5 минут, от 20% до 50% B за 8,5 минут, от 50% B до 100% B за 0,5 минуты, 100% B в течение 2 минут; скорость потока: 27 мл в минуту; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: Waters FractionLynx.
Метод D: колонка: Xtimate С18, 30×2,1 мм, 3 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 25% B в течение 1,5 минут, от 25% до 50% B за 25 минут, затем 100% B в течение 5,0 минут; скорость потока: 90 мл в минуту; температура: не контролировали; детектирование: ДДМ 220 нм; МС (+) диапазон 100-1000 дальтон; прибор: Shimadzu.
Метод E: колонка: LUNA С18, 250×50 мм, 10 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 25% B в течение 1,5 минут, от 25% до 55% B за 25 минут, затем 100% B в течение 5,0 минут; скорость потока: 90 мл в минуту; температура: не контролировали; детектирование: ДДМ 220 нм; МС (+) диапазон 100-1000 дальтон; прибор: Shimadzu.
Метод F: колонка: Phenomenex Luna С18(2), 250×50 мм, 10 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: от 35% до 65% B за 30 минут, затем 100% B в течение 5,0 минут; скорость потока: 90 мл в минуту; температура: не контролировали; детектирование: ДДМ 220 нм; МС (+) диапазон 100-1000 дальтон; прибор: Shimadzu.
Метод G: колонка: Phenomenex Luna С18(2), 250×50 мм, 10 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 10% B в течение 1,5 минут, от 10% B до 55% B за 8,5 минут, от 55% B до 100% B за 0,5 минут, затем выдержка при 100% B в течение 1,5 минут; скорость потока: 27 мл в минуту; температура: не контролировали; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 RP Waters Fractional Lynx LCMS
Метод H: колонка: Phenomenex Luna С18(2), 150×21,2 мм, 5 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 10% B в течение 1,5 минут, от 10% B до 75% B за 8,5 минут, затем от 75% B до 100% B за 2,0 минуты; скорость потока: 27 мл в минуту; температура: не контролировали; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 RP Waters Fractional Lynx LCMS.
Метод H1: колонка: Phenomenex Luna С18(2), 150×21,2 мм, 5 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 1% B в течение 1,5 минут, от 1% B до 100% B за 8,5 минут, затем 100% B в течение 2,0 минут; скорость потока: 27 мл в минуту; температура: не контролировали; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 RP Waters Fractional Lynx LCMS.
Метод I1: колонка: Phenomenex Luna PFP (2), 150×21,2 мм, 5 мкм; подвижная фаза A: 0,02% TFA в воде; подвижная фаза B: 0,02% TFA в ацетонитриле; градиент: 40% B в течение 1,5 минут, от 40% до 100% B за 8,5 минут, 100% B в течение 2,0 минут; скорость потока: 27 мл в минуту; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 Waters FractionLynx LCMS.
Метод I2: колонка: Phenomenex Luna PFP (2), 150×21,2 мм, 5 мкм; подвижная фаза A: 0,02% TFA в воде; подвижная фаза B: 0,02% TFA в ацетонитриле; градиент: 1% B в течение 1,5 минут, от 1% до 100% B за 8,5 минут, 100% B в течение 2,0 минут; скорость потока: 27 мл в минуту; детектирование: ДДМ 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 Waters FractionLynx LCMS.
Метод J1: колонка: Phenomenex Synergi Polar RP, 150×21,2 мм, 4 мкм; подвижная фаза A: 0,02% TFA в воде; подвижная фаза B: 0,02% TFA в ацетонитриле; градиент: 10% B в течение 1,5 минут, от 10% до 75% B за 8,5 минут, от 75% B до 100% B за 0,5 минут, 100% B в течение 2 минут; скорость потока: 27 мл в минуту; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: Waters FractionLynx.
Метод K1: колонка: Phenomenex Luna С18(2), 250×50 мм, 10 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 1% B в течение 1,5 минут, от 1% B до 75% B за 8,5 минут, от 75% B до 100% B за 0,5 минуты, затем выдержка при 100% B в течение 1,5 минут; скорость потока: 27 мл в минуту; температура: не контролировали; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 RP Waters Fractional Lynx LCMS.
Метод L1: колонка: ChiralTech AD-H, 500×21,5 мм, 5 мкм; подвижная фаза A: CO2 (об./об.); подвижная фаза В: метанол (об./об.); градиент: изократические условия 60% CO2, 40% метанола; скорость потока: 36 мл в минуту CO2, 24 мл в минуту метанола; противодавление: 100 бар (100 кПа); детектирование: ДДМ 210; прибор: Thar 80 (Waters).
Метод M: колонка: Phenomenex Synergi, 250×50 мм, 10 мкм; подвижная фаза A: 0,1% TFA в воде; подвижная фаза B: 0,1% TFA в ацетонитриле; градиент: от 40% до 70% B за 30 минут, затем 95% B в течение 5,0 минут; скорость потока: 80 мл в минуту; детектирование: ДДМ 220, 254 нм; МС (+) диапазон 100-1000 дальтон; прибор: Shimadzu LC-20AP.
Метод N: колонка: Phenomenex Luna Phenylhexyl 150×21,2 мм, 5 мкм; подвижная фаза A: 0,2% TFA в воде (об./об.); подвижная фаза B: 0,2% TFA в ацетонитриле (об./об.); градиент: 35% B в течение 1,5 минут, от 35% B до 100% B за 18,5 минут, затем 100%) В в течение 2,0 минут; скорость потока: 27 мл в минуту: температура: не контролировали; детектирование: ДДМ 210-360 нм; МС (+) диапазон 150-2000 дальтон; прибор: 305 RP Waters Fractional Lynx LCMS.
Иллюстративные примеры конъюгатов антитело-лекарственное средство
Протокол A: Общая методика конъюгирования антитела с конструкцией линкер-положительная нагрузка через внутренние дисульфиды
IL13Rα2-AB08-v1.0/1.0-человеческое IgG1 антитело [Pfizer, раствор 12-13 мг/мл в забуференном фосфатами физиологическом растворе Дульбекко (DPBS, Lonza, pH 7,4)] или VEGFR-1121В-человеческое IgG1 антитело [Pfizer, раствор 19,3 мг/мл в забуференном фосфатами физиологическом растворе Дульбекко (DPBS, Lonza, pH 7,4)] восстанавливали добавлением 2,9-3 эквивалентов трис(2-карбоксиэтил)фосфина гидрохлорида (TCEP, 5 мМ раствор в DPBS). Реакционную смесь инкубировали при 37°C в течение 1-1,25 ч и затем оставляли охлаждаться до температуры окружающей среды. Конъюгирование осуществляли путем добавления 7 эквивалентов конструкции линкер-полезная нагрузка [10 мМ раствор в N,N-диметилацетамиде (DMA)]. Дополнительное количество DMA добавляли в реакционную смесь до достижения в сумме 10-15% (об./об.) компонента, представляющего собой органический растворитель, в конечной реакционной смеси. Реакционную смесь инкубировали в течение 1 ч при температуре окружающей среды. Для ADC 1-5 после 1 ч при температуре окружающей среды избыток конструкции линкер-полезная нагрузка гасили добавлением 10 эквивалентов цистеина (20 мМ раствор в DPBS). Погашенную реакционную смесь выдерживали при температуре окружающей среды в течение 15 минут и затем хранили при 4°C до проведения очистки. Для ADC 6-14 после 1 ч при температуре окружающей среды реакционную смесь десольватировали на десольватирующих колонках GE Sephadex gel с использованием DPBS (pH 7,4) в качестве элюента и затем хранили при 4°C до проведения очистки. Неочищенное вещество очищали эксклюзионной хроматографией (ЭХ), используя систему GE AKTA Explorer с колонкой GE Superdex 200 (10/300 GL) и DPBS (pH 7,4) в качестве элюента.
Протокол B: колонка: Agilent Poroshell 300SB-C8, 75×2,1 мм, 2,6 мкм; подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: начальные условия: от 20% B до 45% B за 4 минуты; скорость потока: 1,0 мл в минуту; температура: 60°C; детектирование: 220 нм; МС (+) диапазон 400-2000 Да; инжектируемый объем: 10 мкл; прибор: Agilent 1100 LC, Waters MicromassZQ MS. Деконволюцию осуществляли, используя MaxEnt1.
Протокол C: колонка: GE Superdex 200 (5/150 GL); подвижная фаза: забуференный фосфатами физиологический раствор (PBS, 1X, pH 7,4) с 2% ацетонитрила; изократические условия; скорость потока: 0,25 мл в минуту; температура: комнатная температура; инжектируемый объем: 10 мкл; прибор: Agilent 1100 HPLC.
Протокол D: Получение ADC с трансглутаминазой, проиллюстрированное для конструкции линкер-полезная нагрузка AcLys-vc-MMAD ("Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates", Chem Biol. 2013, 20, 161-7). Для конъюгирования C16-HC и C16-LC с AcLys-vcMMAD антитело доводили до 5 мг/мл в буфере, содержащем 25 мМ Tris-HCl при pH 8,0 и 150 мМ NaCl. AcLys-vc-MMAD добавляли либо в 5-кратном (С16-НС), либо в 10-кратном (C16-LC) молярном избытке относительно антитела, и ферментативную реакцию инициировали добавлением 1% (масс./об.) (С16-НС) или 2% (масс./об.) (С16-LC) бактериальной трансглутаминазы (Ajinomoto Activa TI, Japan). После инкубирования при умеренном встряхивании при 22°C (С16-НС) или при 37°C (C16-LC) в течение 16 часов ADC очищали, используя MabSelect SuRe (GE Healthcare, Inc), по стандартным методикам.
Согласно изобретению предложены также соединения, представленные в Таблицах 6A и 6B.
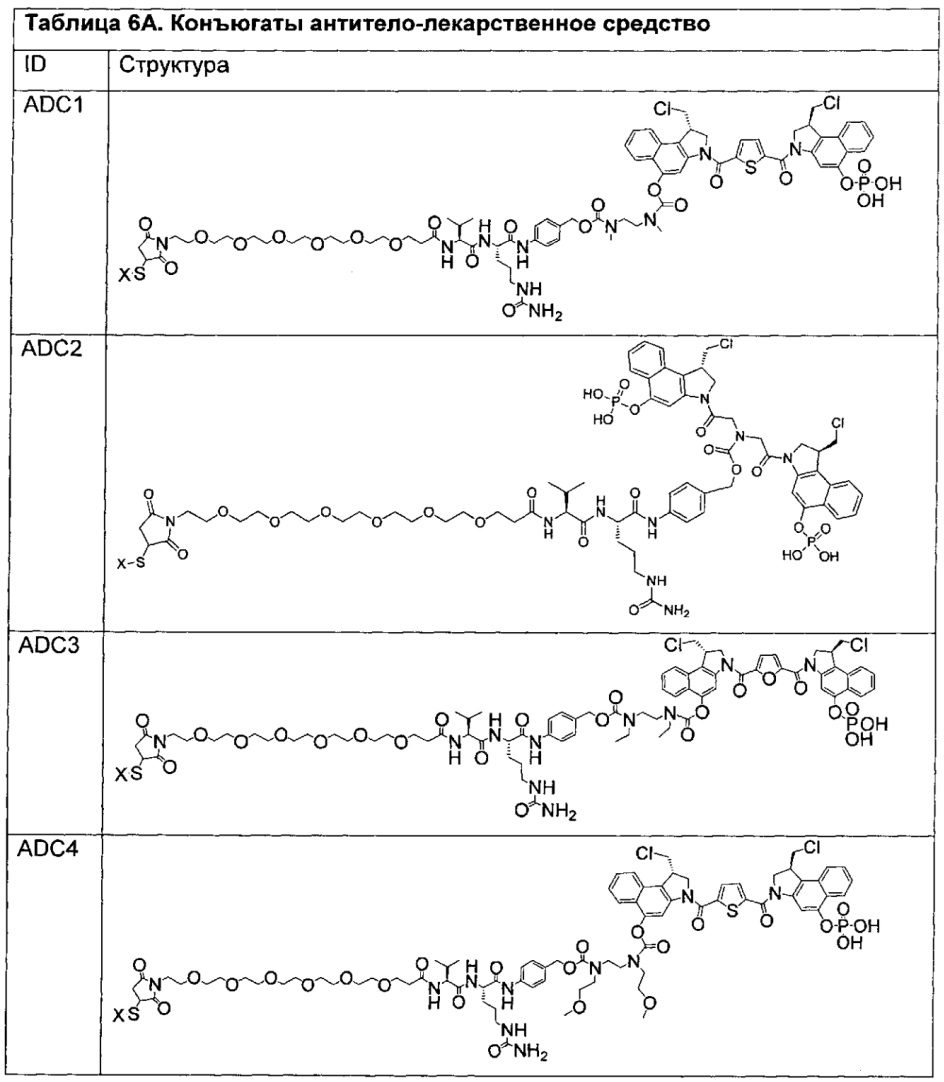
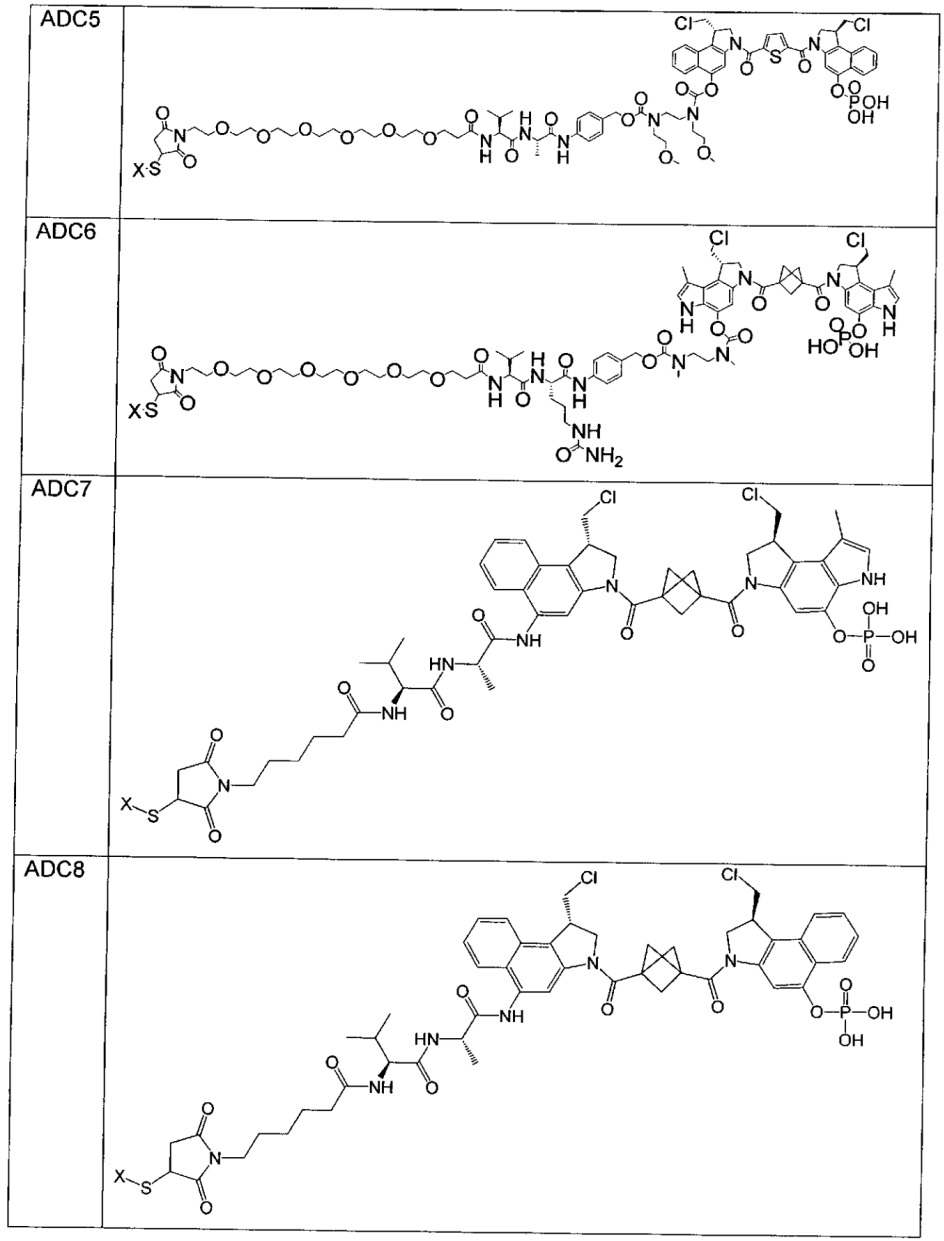
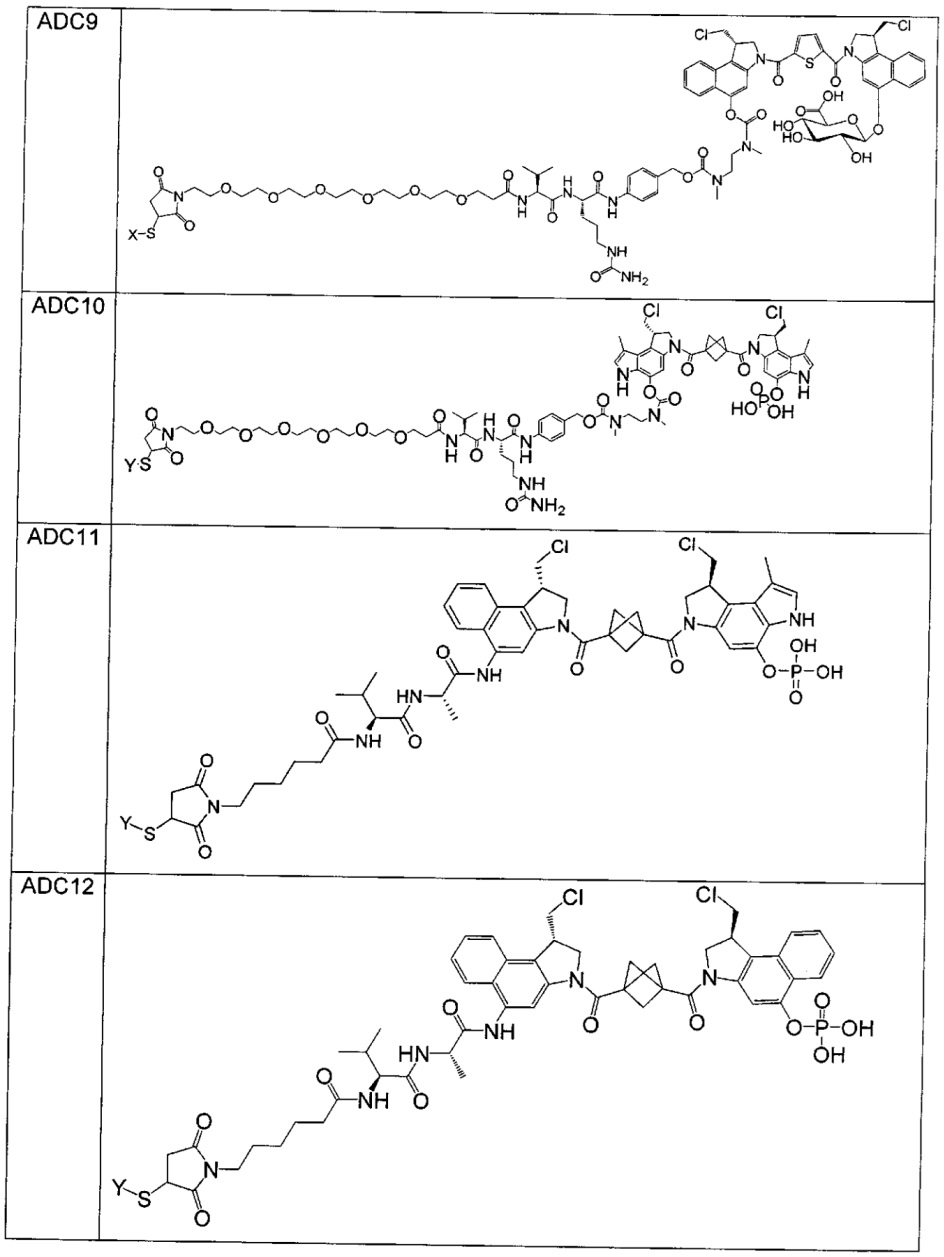
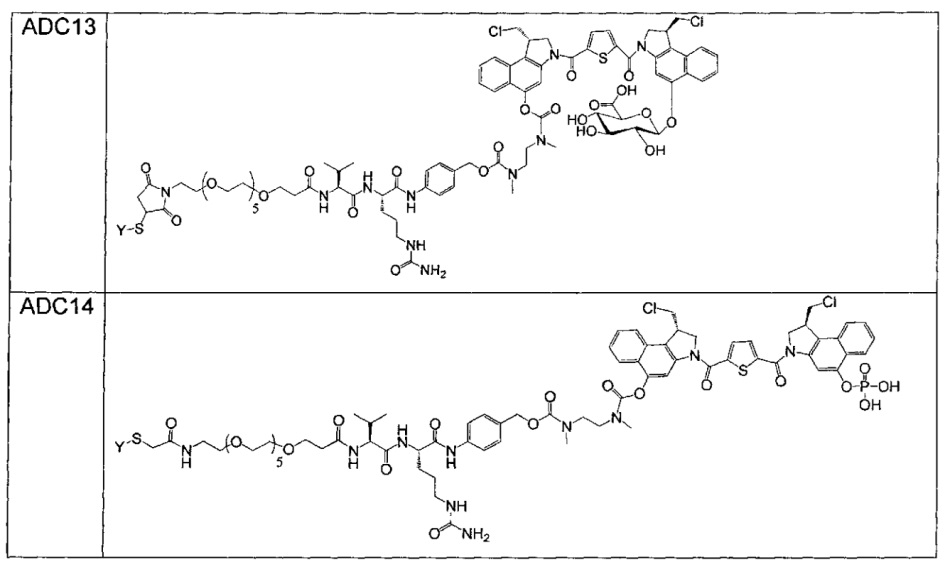
В приведенной выше Таблице "X и Y" означают антитело. Проиллюстрированные ADC были конъюгированы с антителом к IL13 (IL13Rα2-AB08-v1.0/1.0-человеческое IgG1 антитело), которое обозначено буквой X, и антителом к VEGF VEGFR-1121B-hG1, которое обозначено буквой Y.
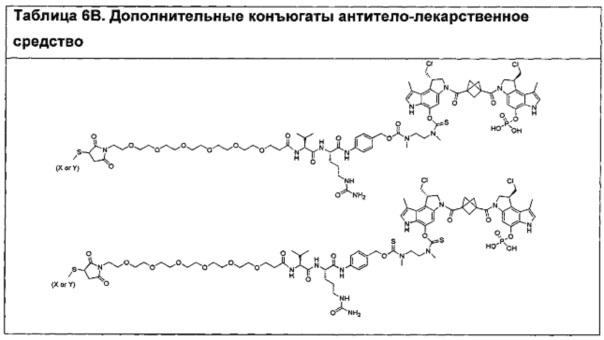
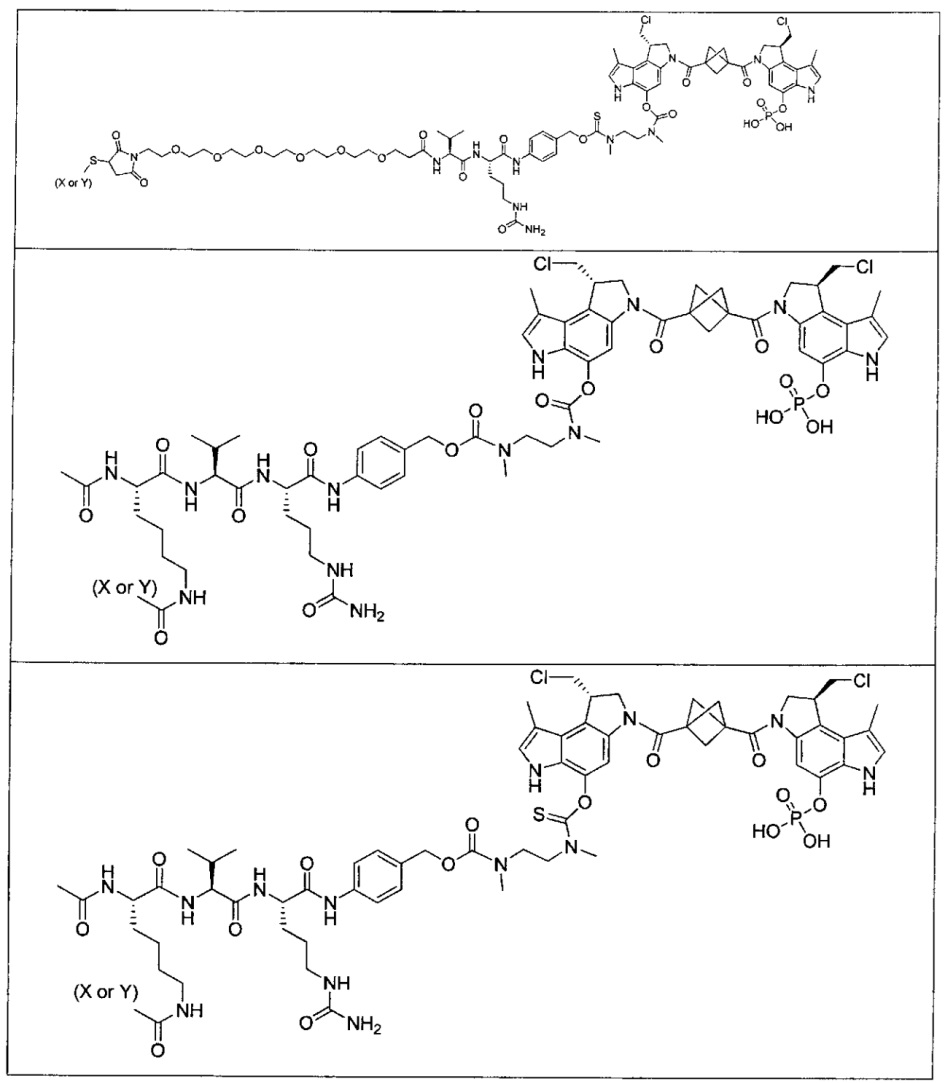
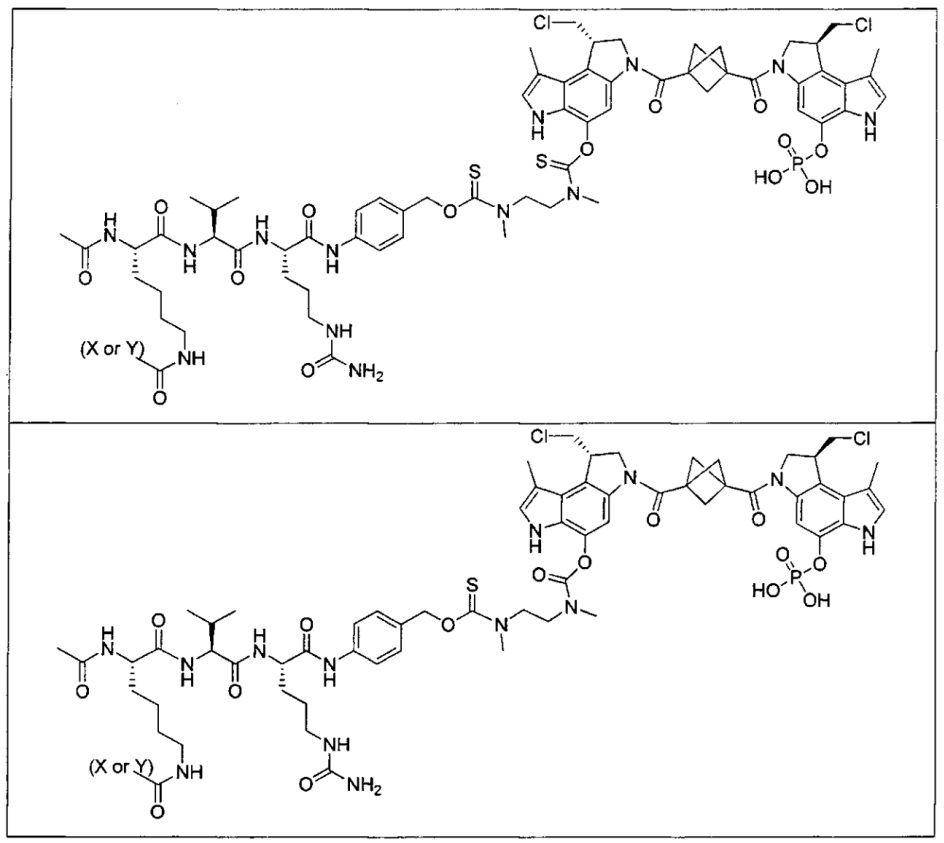
В приведенной выше Таблице "X и Y" означают антитело. Проиллюстрированные ADC конъюгированы с антителом к IL13 (IL13Rα2-AB08-v1.0/1.0-человеческое IgG1 антитело), которое обозначено буквой X, и антителом к VEGF VEGFR-1121B-hG1, которое обозначено буквой Y.
Аналитические данные для проиллюстрированных ADC
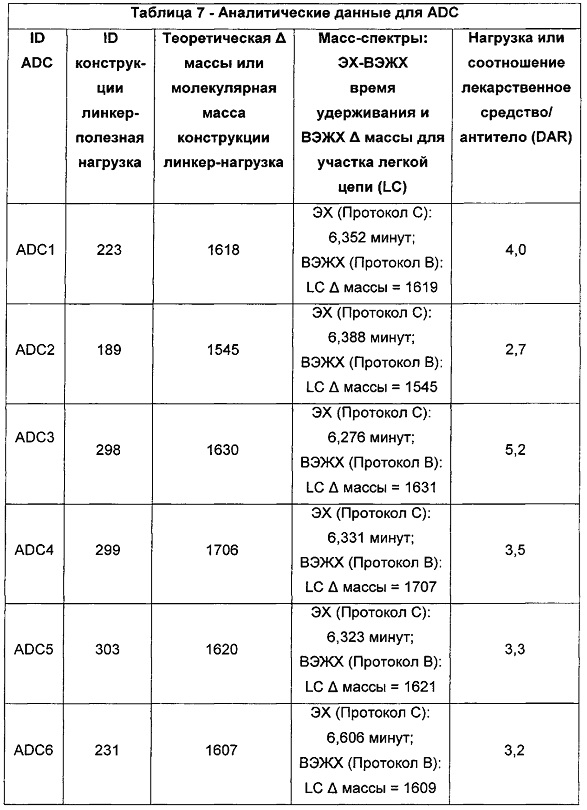
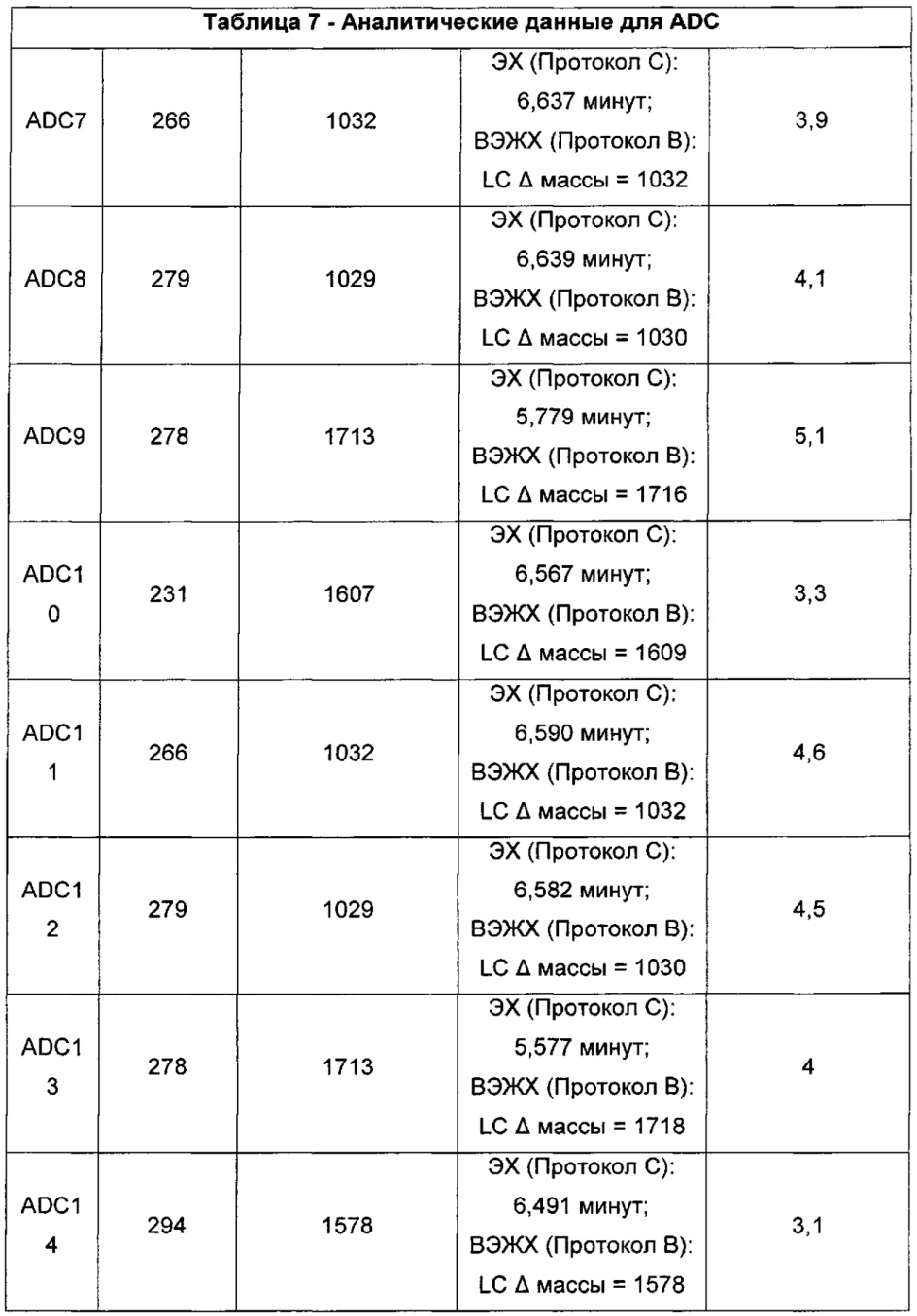
Экспериментальные методики для биологической оценки полезных нагрузок и конъюгатов антитело-лекарственное средство
Линии клеток
Линии раковых клеток были получены из АТСС (Американская коллекция типовых культур) (Manassas, VA), N87 (желудочная карцинома человека, имеющая происхождение из метастатического участка печени), HL60 (лейкоз), А375 (меланома) и HUVEC (эндотелиальные клетки пупочной вены человека) выращивали в средах RPMI 1640. Все среды были дополнены 10% сыворотки плода коровы, 1% пирувата натрия и 1% L-глутамина (Invitrogen, Grand Island, NY). Эндотелиальные клетки пупочной вены человека (HUVEC) были получены от Lonza (Allendale, NJ) и поддерживались в среде EGM2, дополненной EGM-2 SingleQuots (Lonza # СС-4176). Все клетки держали в увлажненном инкубаторе (37°C, 5% CO2).
Методика анализа цитотоксичности для полезных нагрузок
Клетки в 100 мкл среды культивировали в 96-луночном планшете. Линии раковых клеток обрабатывали указанными соединениями путем добавления 50 мкл 3X исходных растворов в двух повторах в 10 концентрациях. Клетки инкубировали с соединениями в течение четырех суток, затем к клеткам добавляли 30 мкл раствора CellTiter® 96 Aqueous One MTS Solution (Promega Cat # G3582), инкубировали в течение 1,5 ч при 37°C, затем измеряли поглощение при 490 нм на планшет-ридере Victor (Perkin Elmer, Waltham, MA). Относительную жизнеспособность клеток определяли в процентах от лунок с необработанными клетками. Значения IC50 вычисляли, используя четырехпараметрическую логистическую модель #203 с XLfit v4.2 (IDBS, Guildford, Surry, UK).
Методика анализа цитотоксичности для ADC
День 0: посев клеток в 100 мкл полной среды в 96-луночном черном планшете с плоским прозрачным дном и культивирование в течение ночи. День 1: добавление 50 мкл 3X титрованных тестируемых соединений до достижения конечного объема 150 мкл и культивирование в течение 72 часов при 37°C, 5% CO2. День 4: добавление 50 мкл Cell TiterGlo во все лунки, вортекс в течение 20-30 минут, считывание на Victor 3 в соответствии с люминесцентной программой. Анализ данных: % выживания вычисляют как 100 x (показания каждой точки данных - среднее значение BKG) среднее значение только для контрольных клеток - среднее значение BKG.
В Таблице, приведенной ниже, представлены данные по IC50 для выбранных полезных нагрузок по настоящему изобретению.
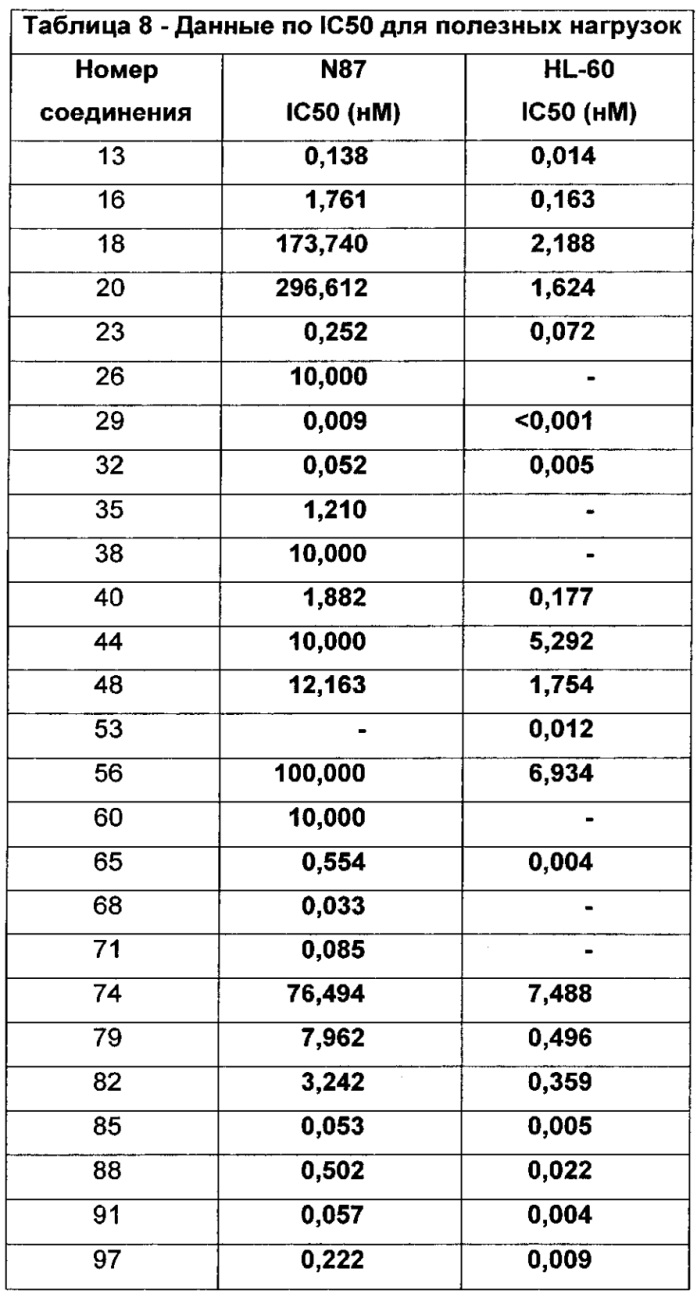
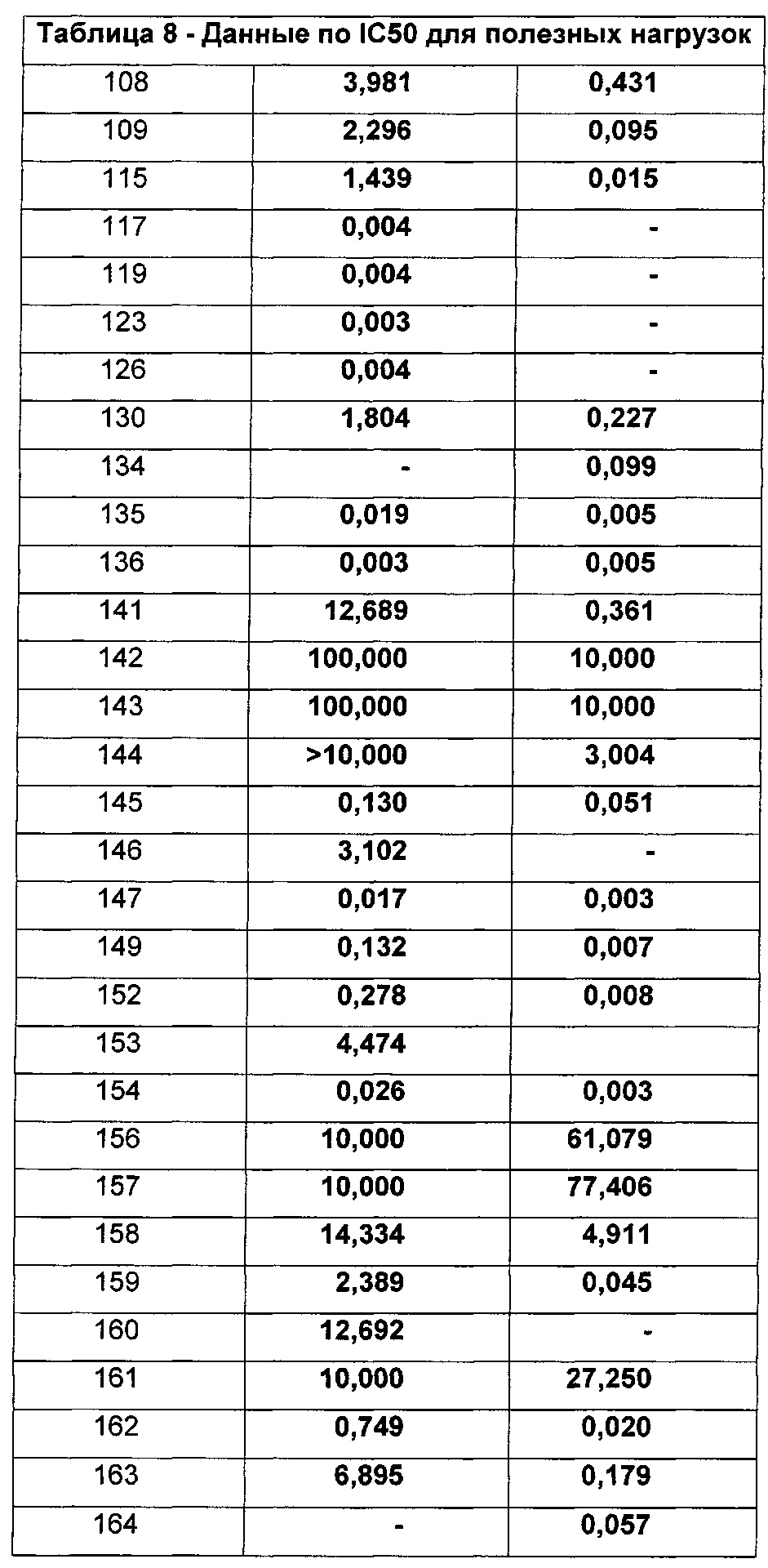
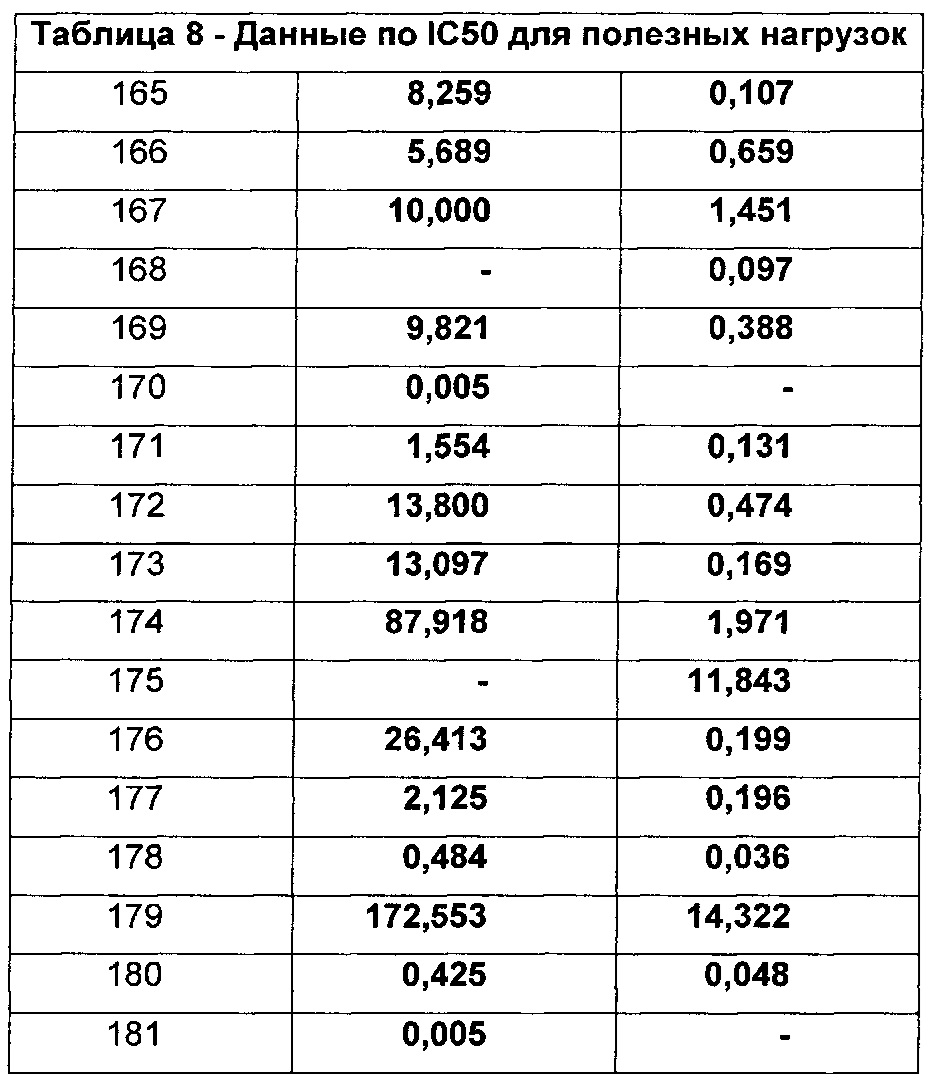
В приведенной ниже Таблице представлены данные по IC50 для выбранных ADC по настоящему изобретению.
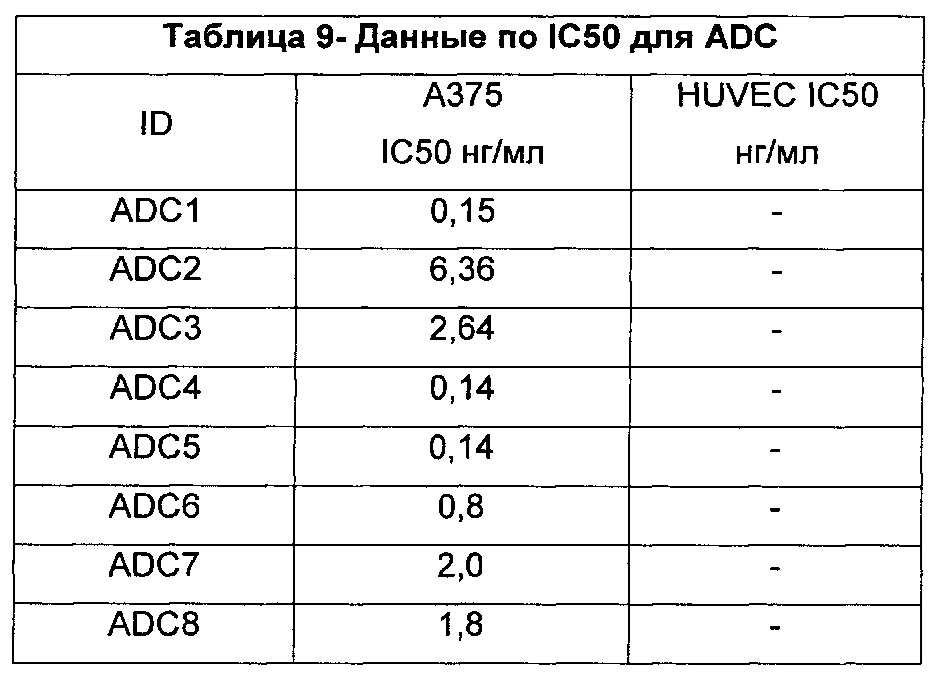
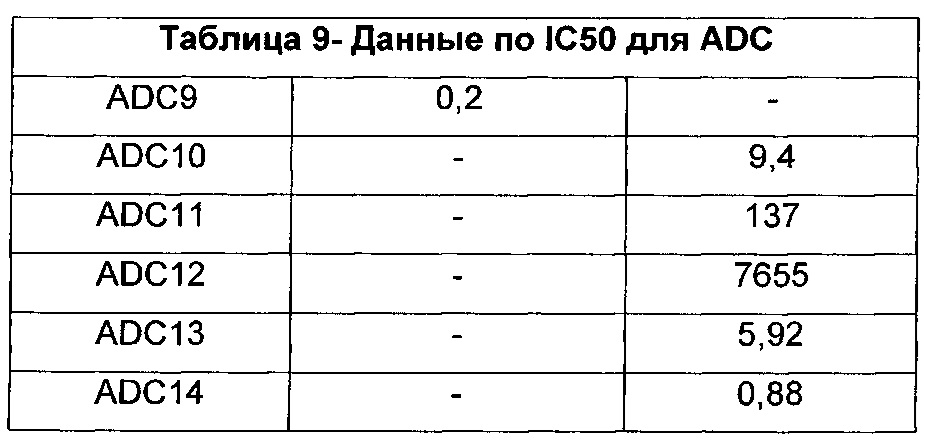
Приведенная ниже схема иллюстрирует, как полезная нагрузка высвобождается при введении пациенту и после отщепления линкера от ADC на примере с одним типом линкера. После высвобождения линкера образуются различные соединения, которые взаимопревращаются в биологической среде. Все образующиеся соединения заявлены как часть данного изобретения и относятся к общей формуле F1-L1-T-L2-F2.
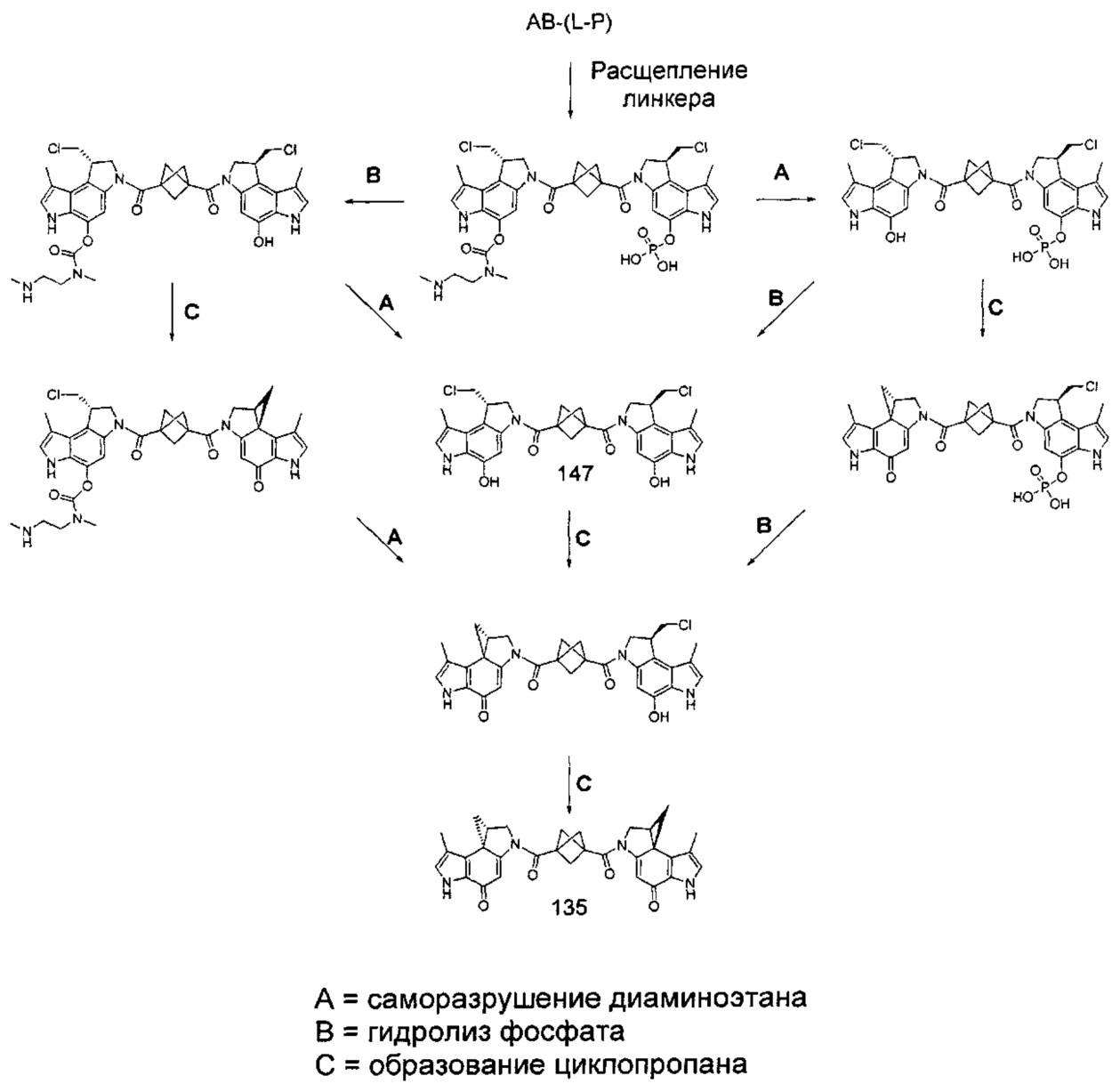
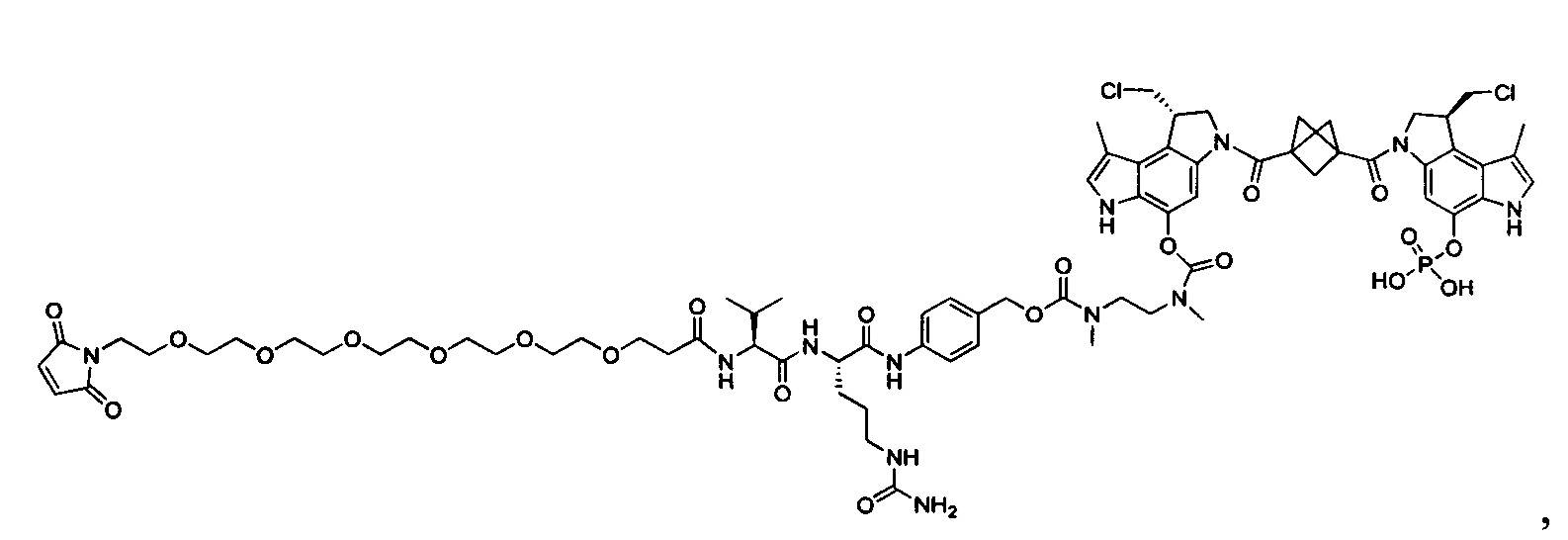
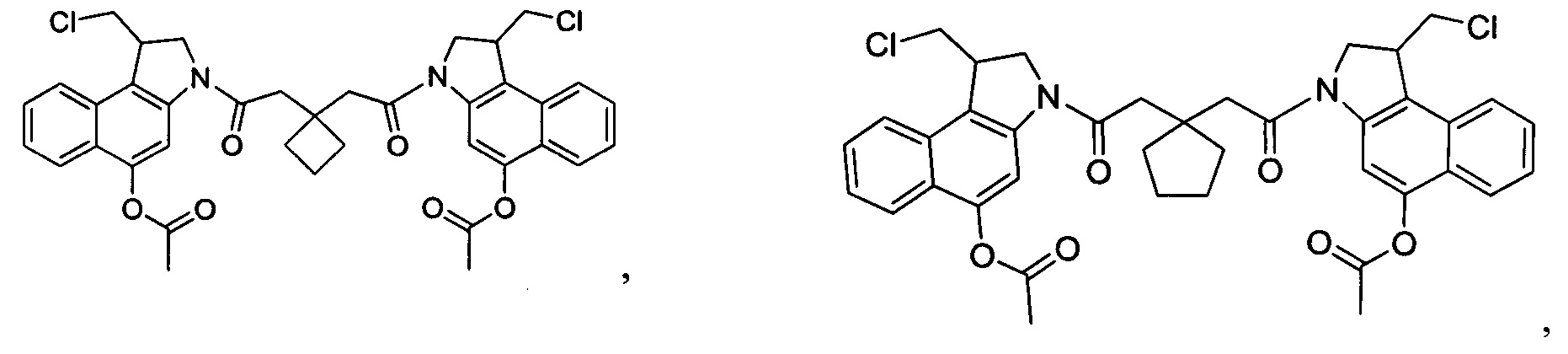
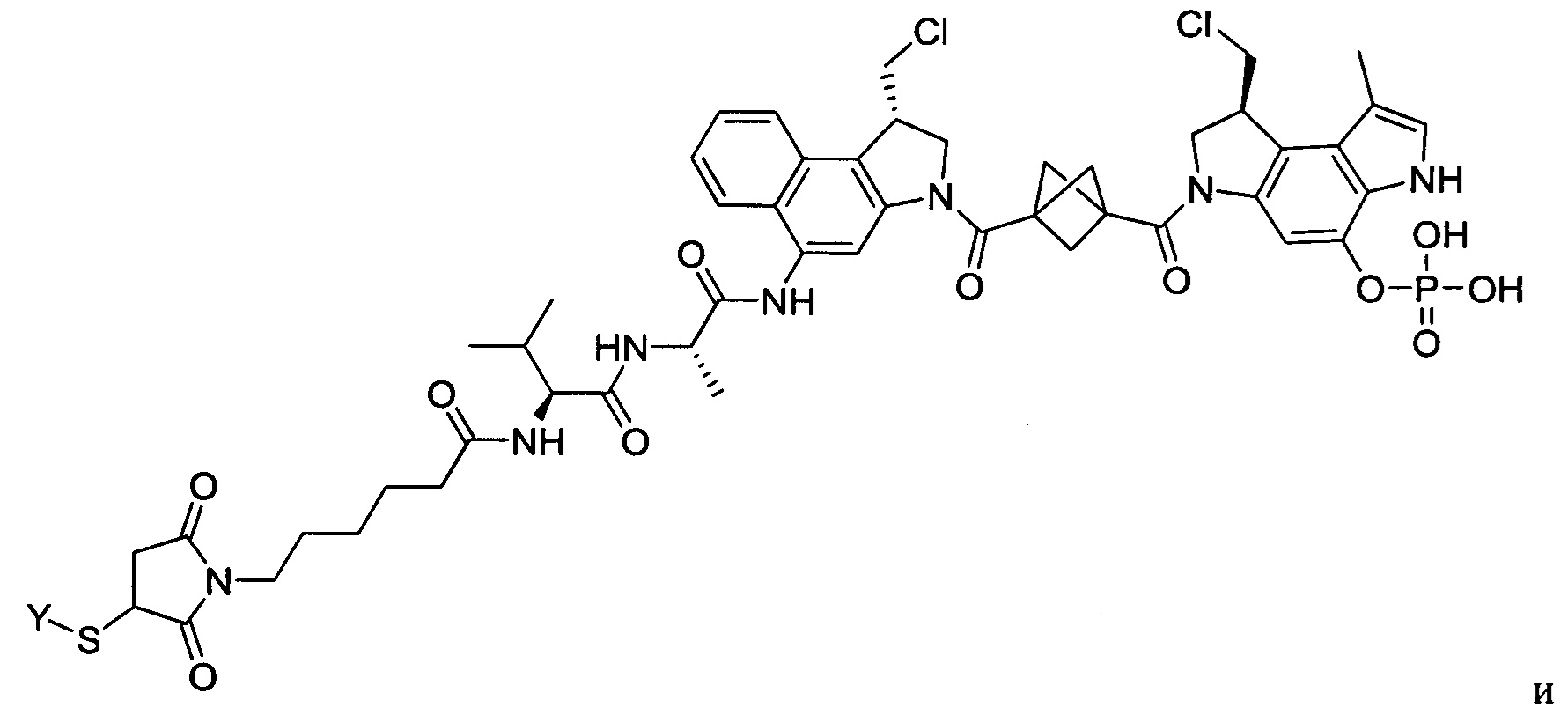
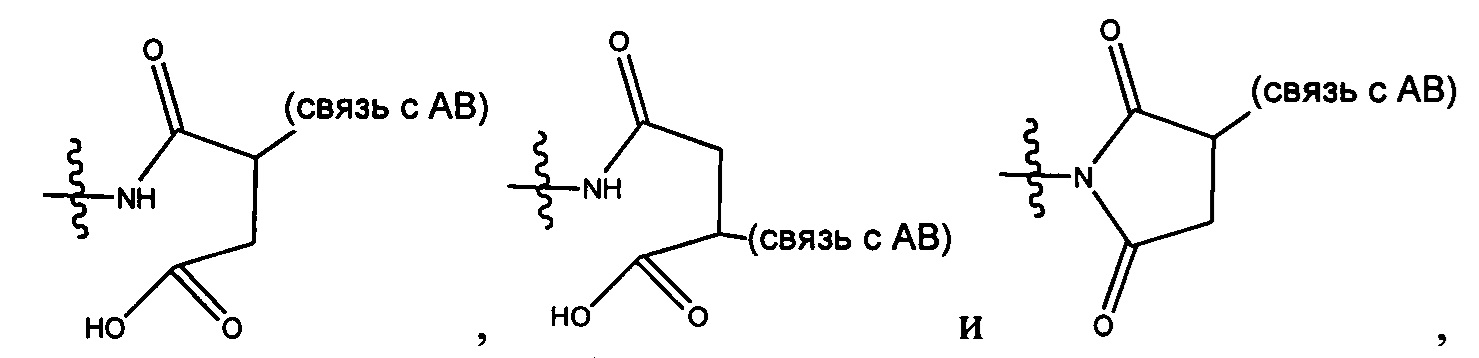
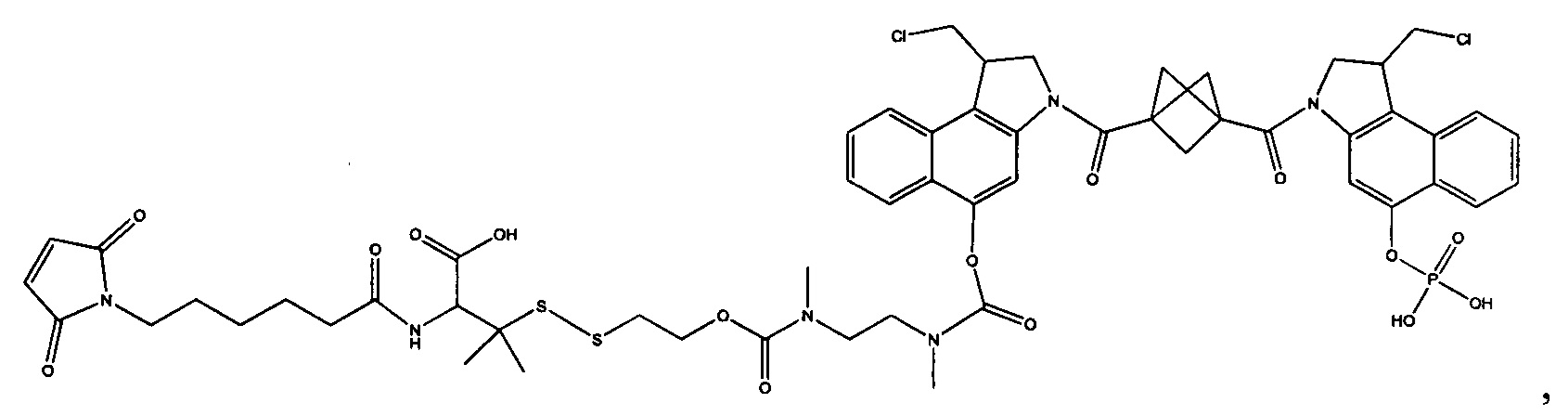

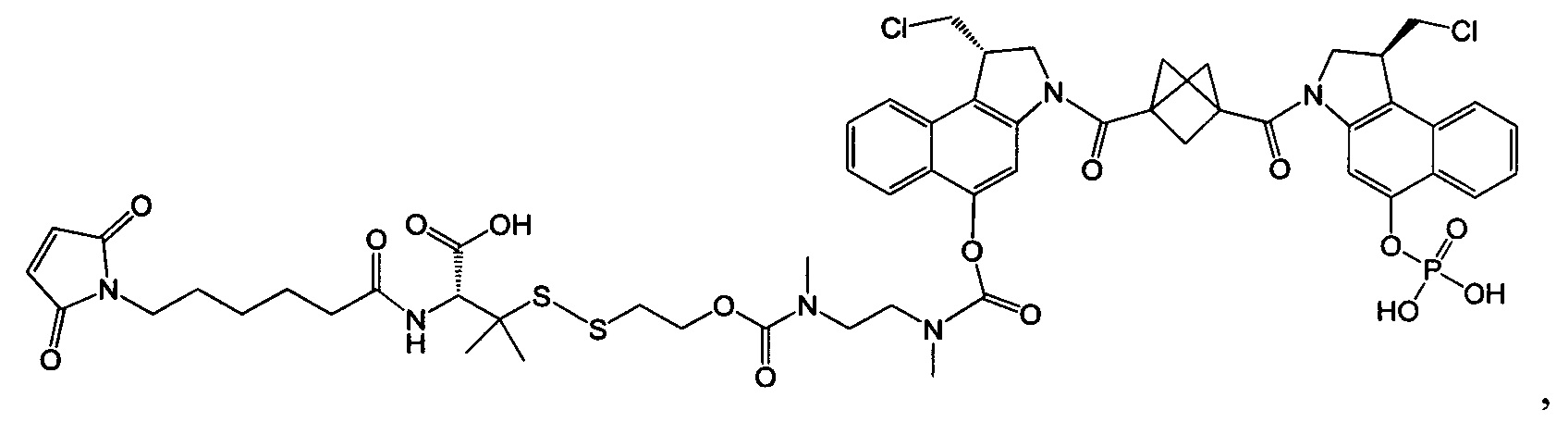
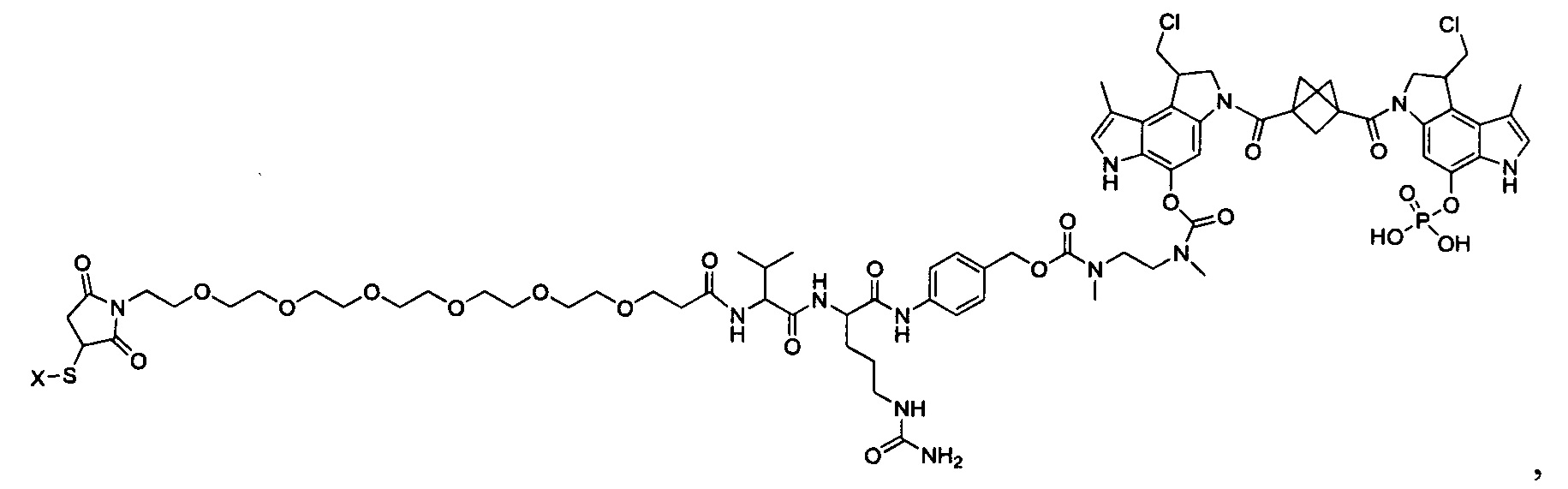
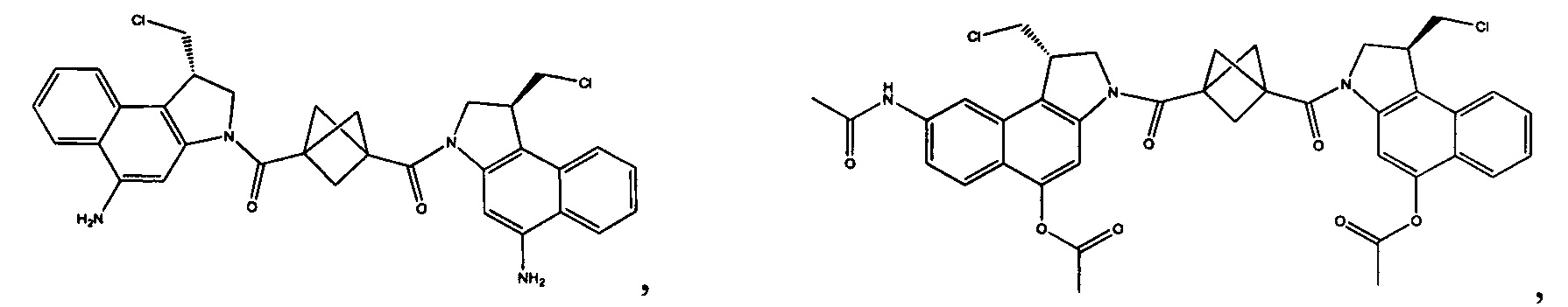
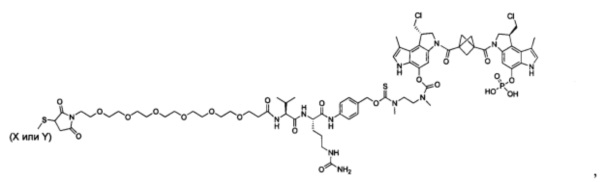
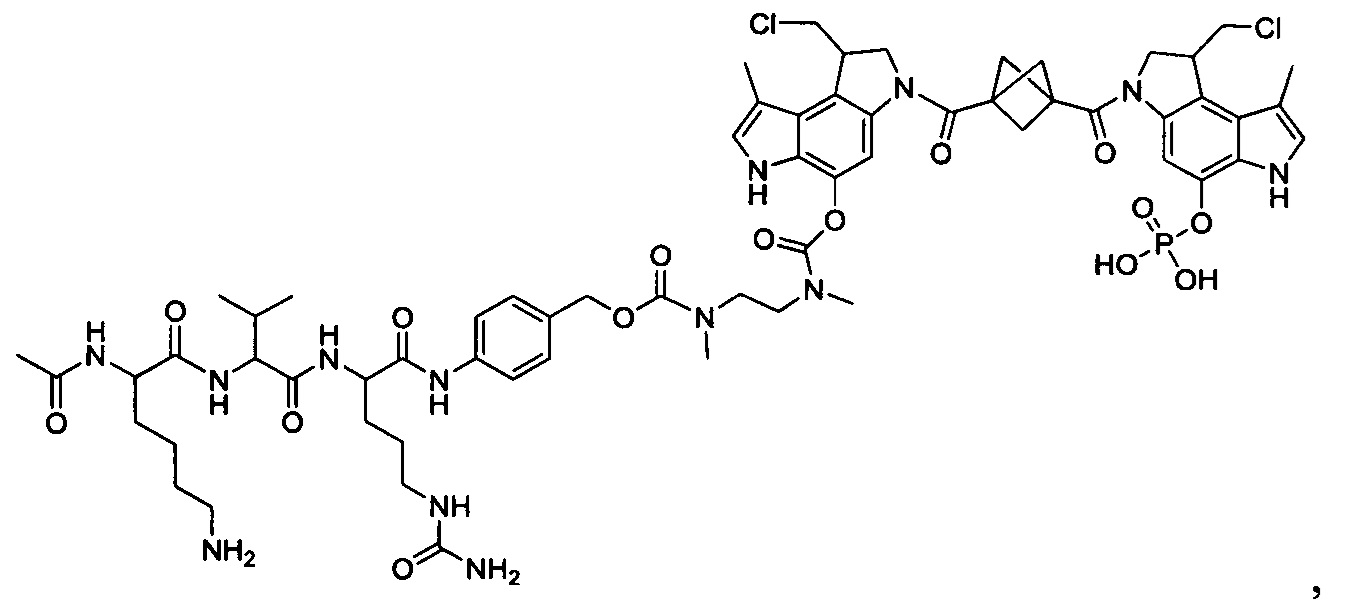
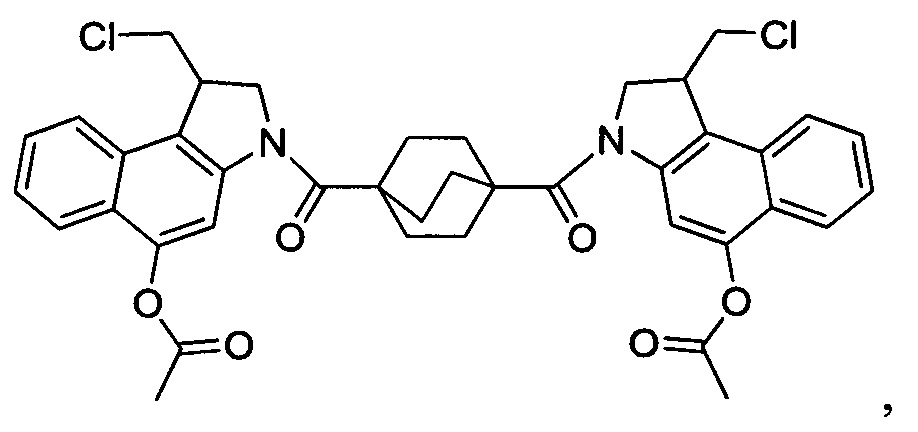
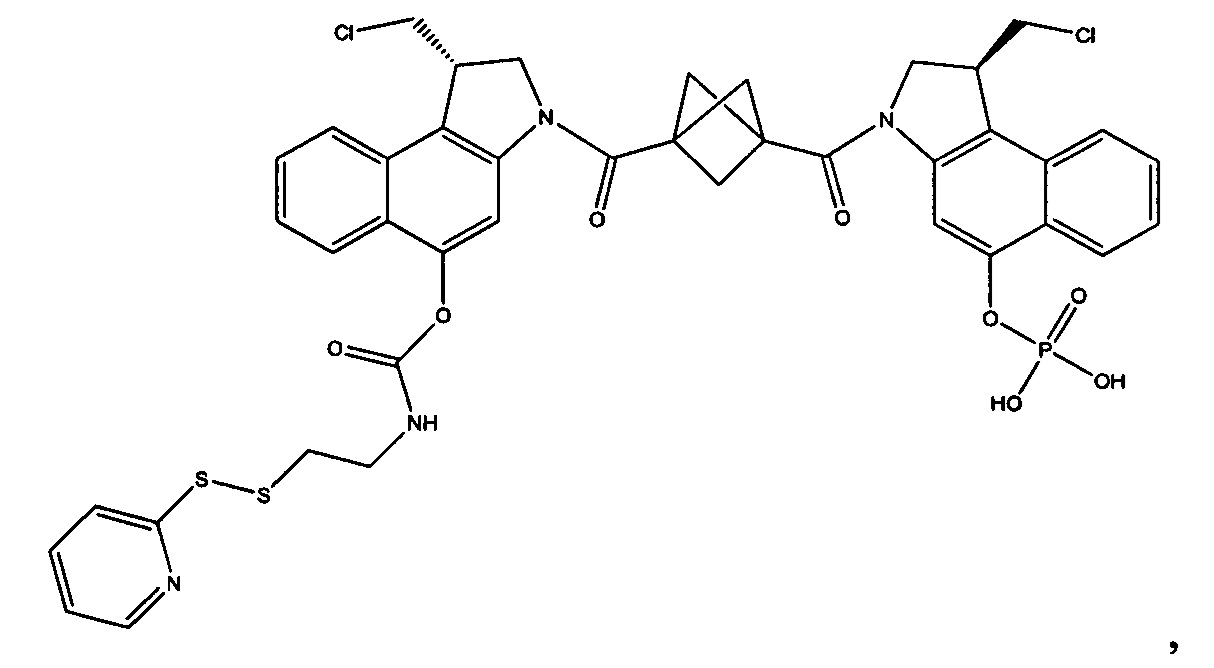
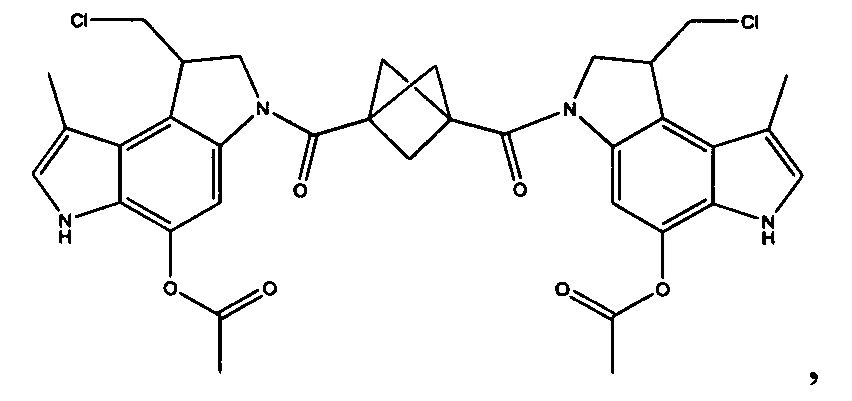
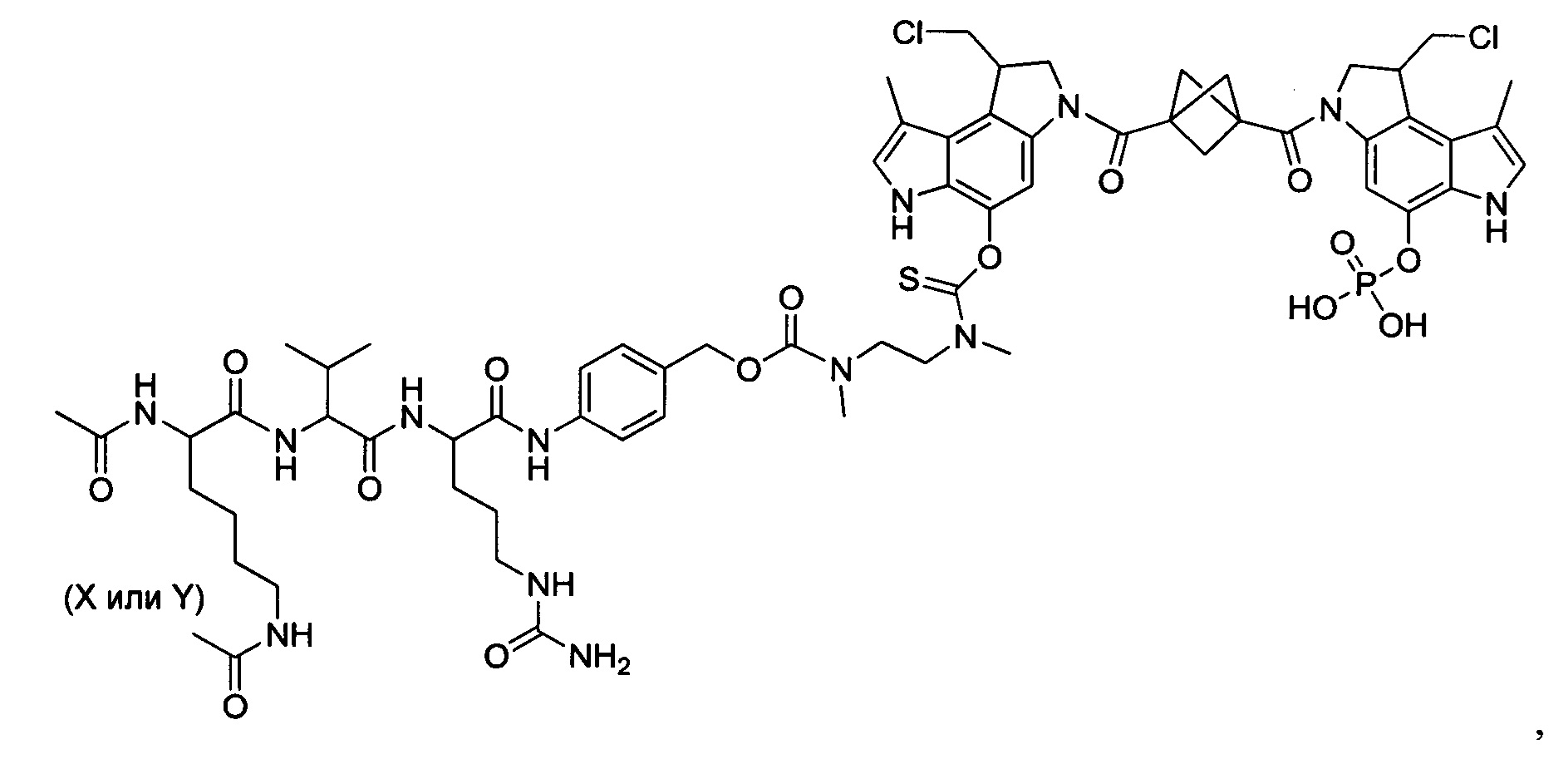
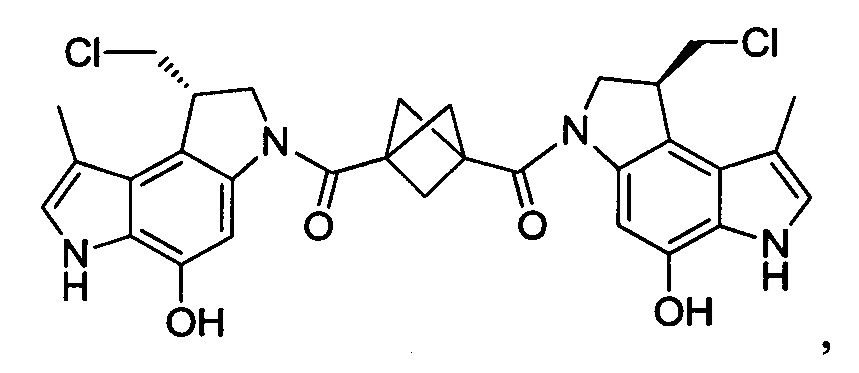
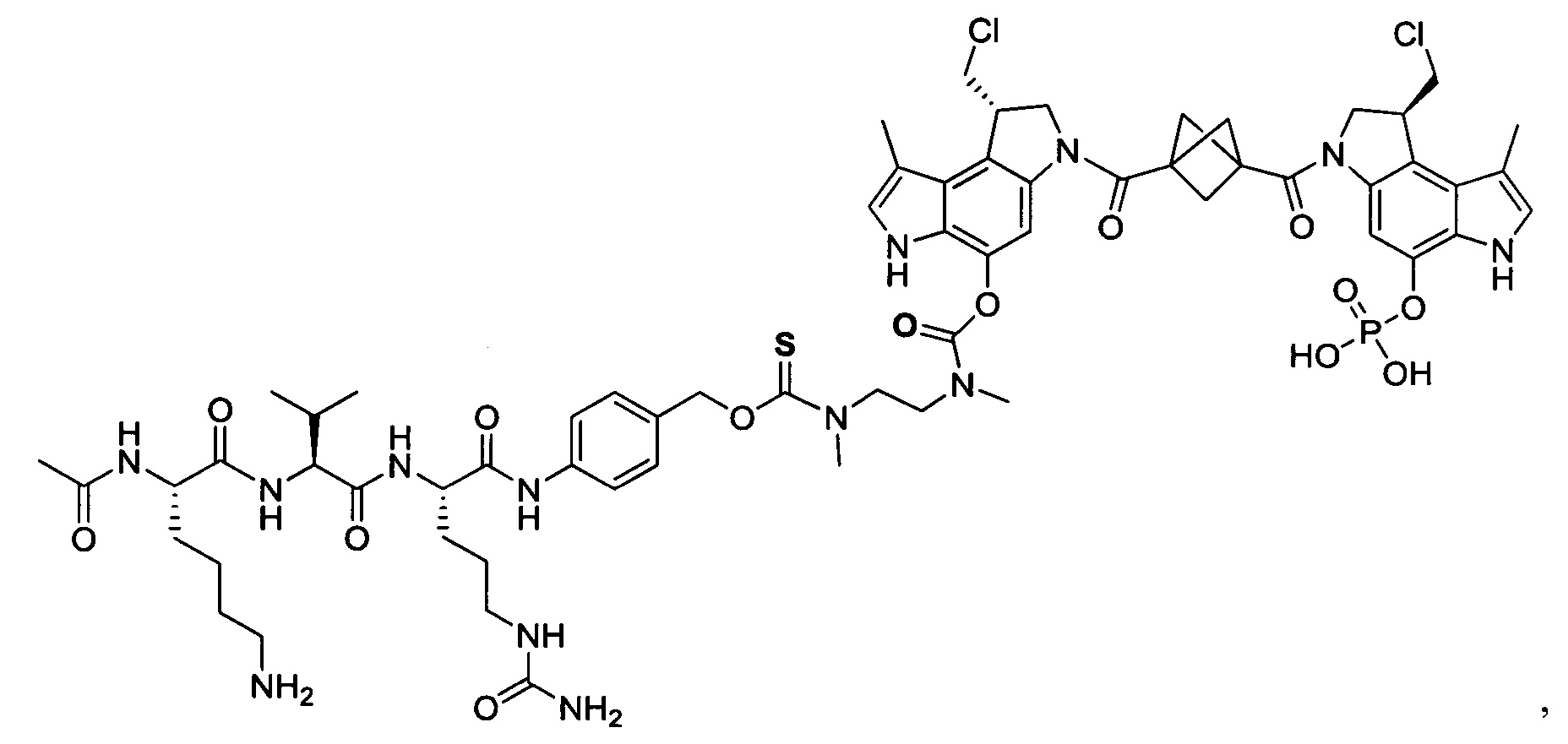
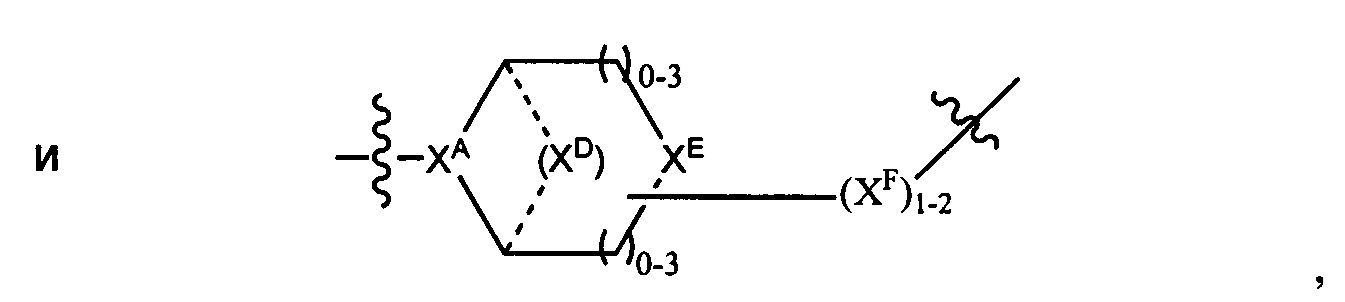
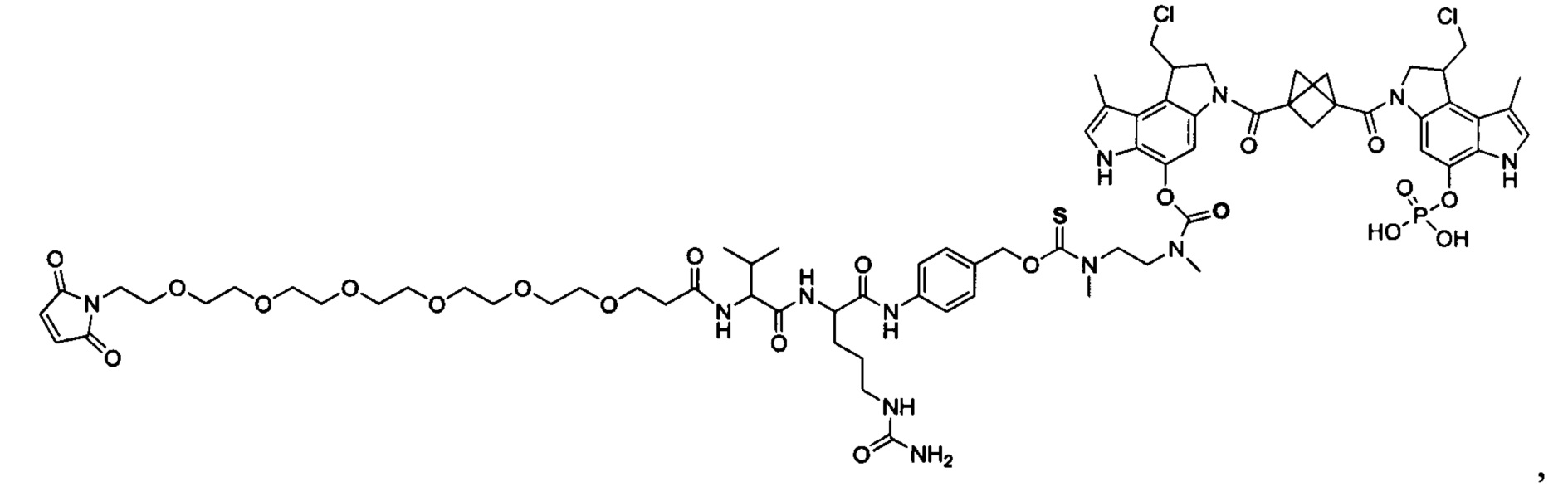
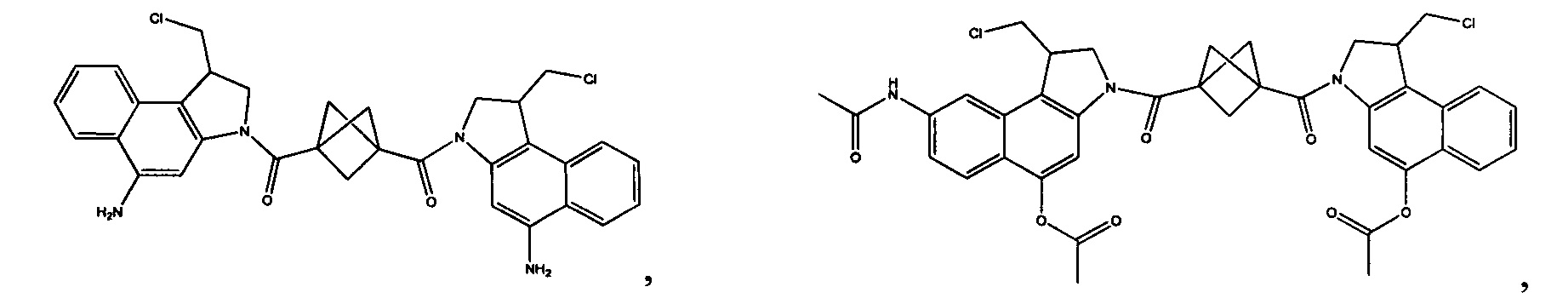
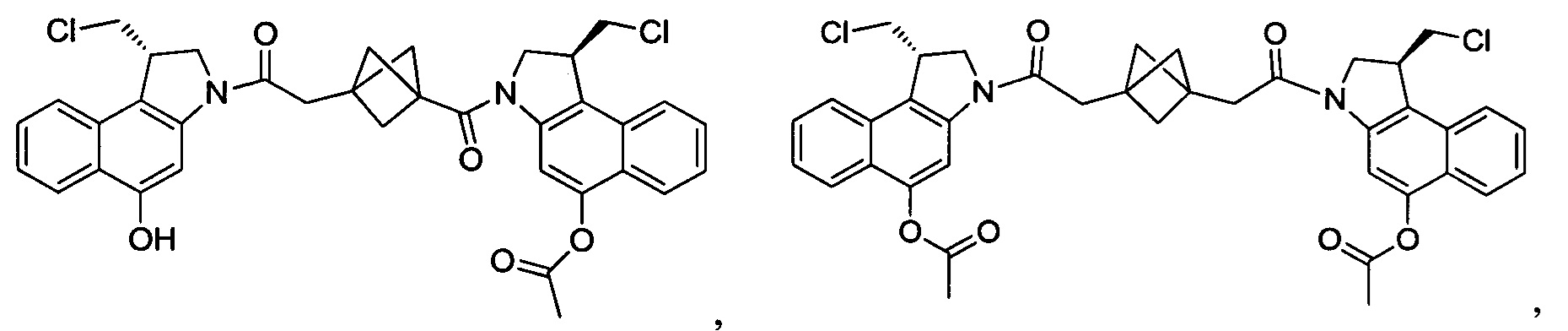
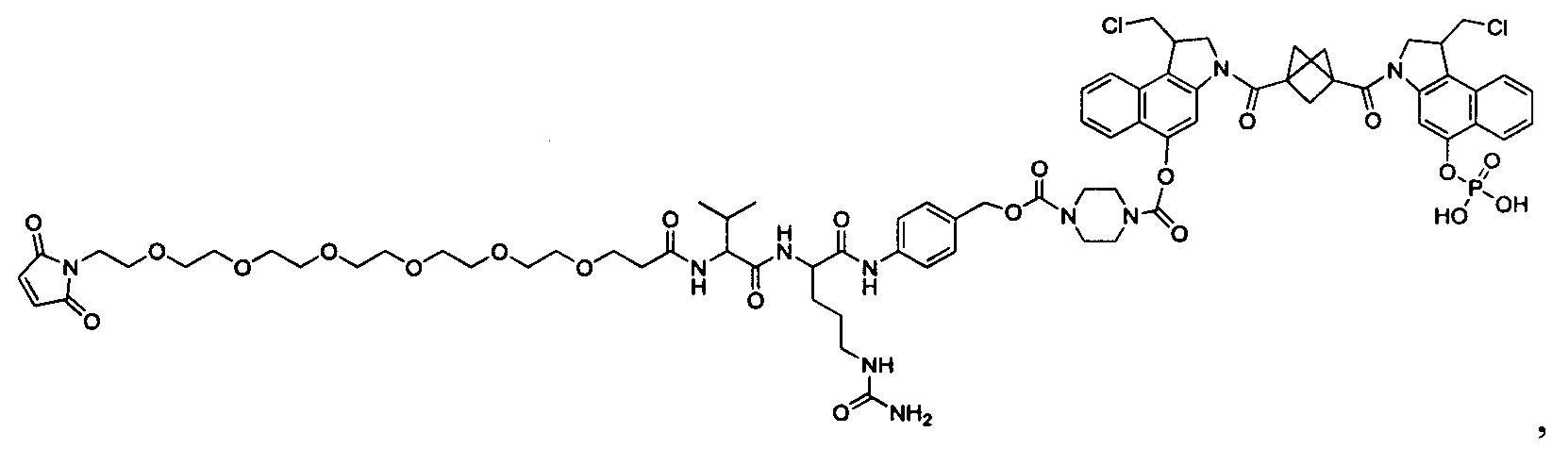
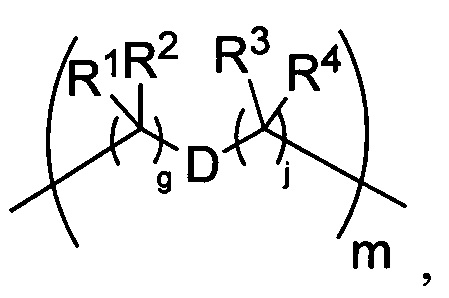
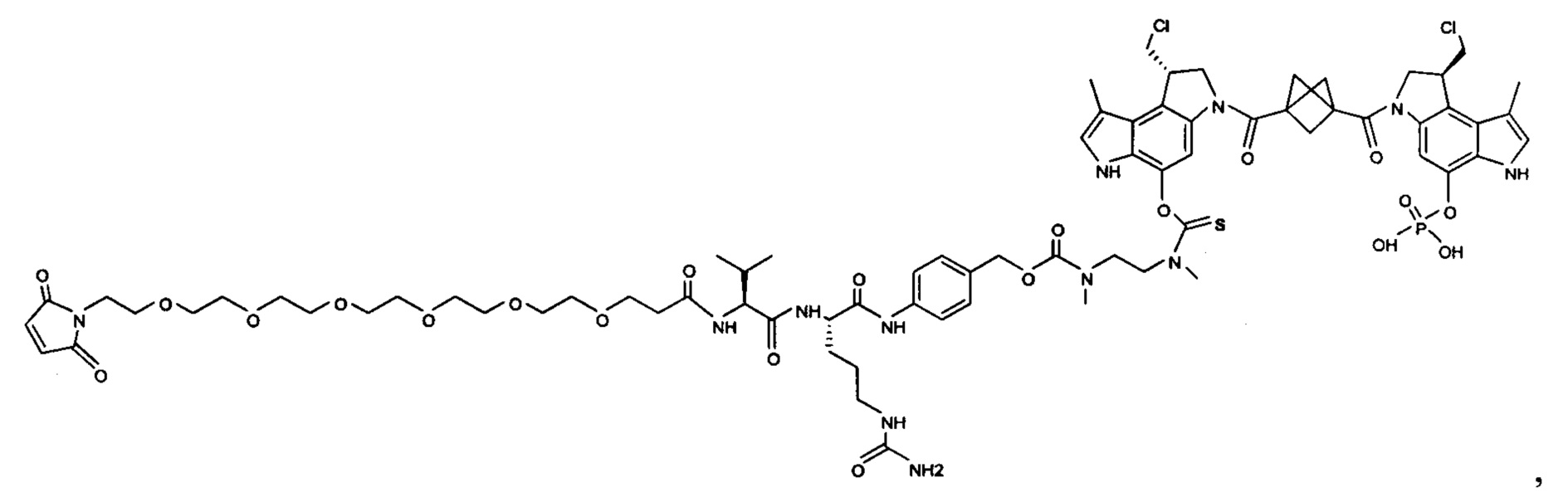
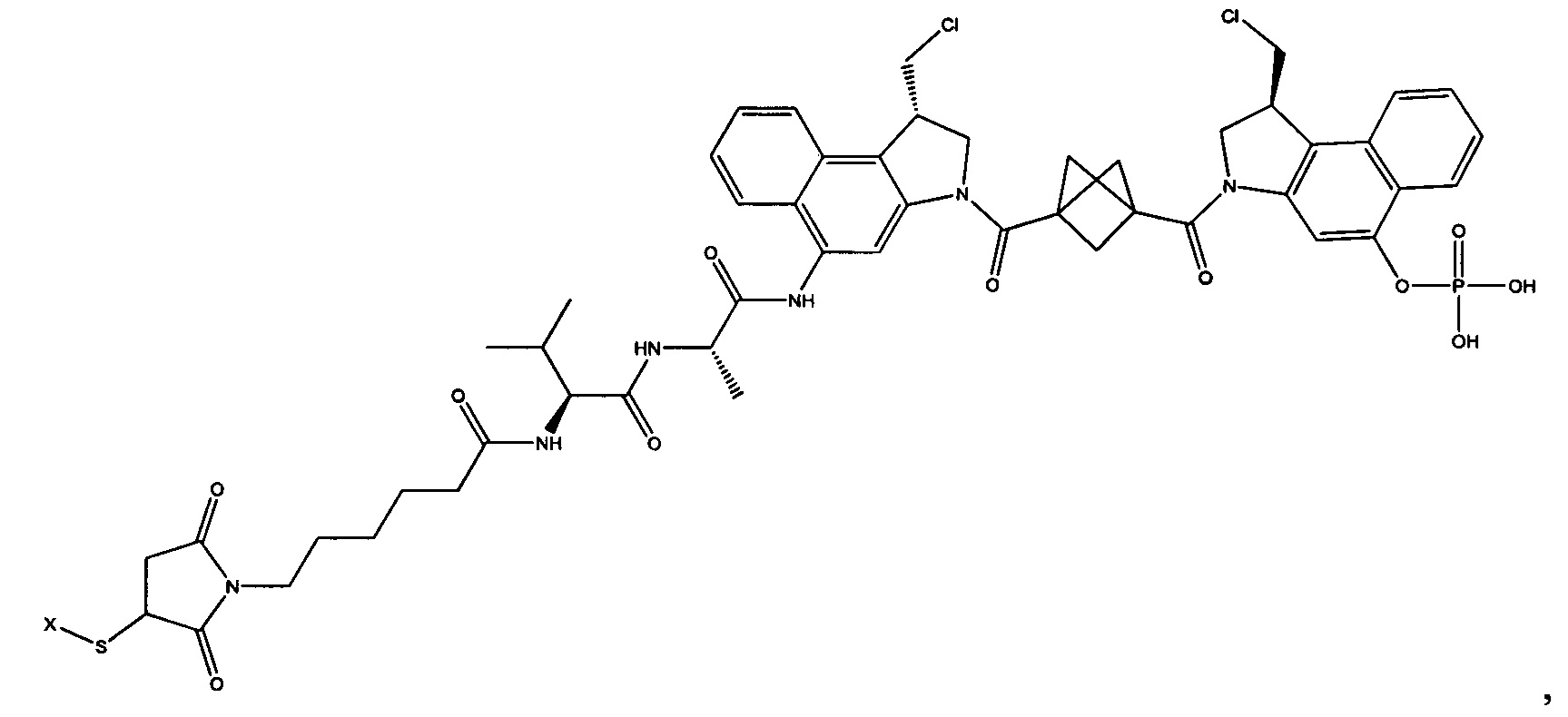
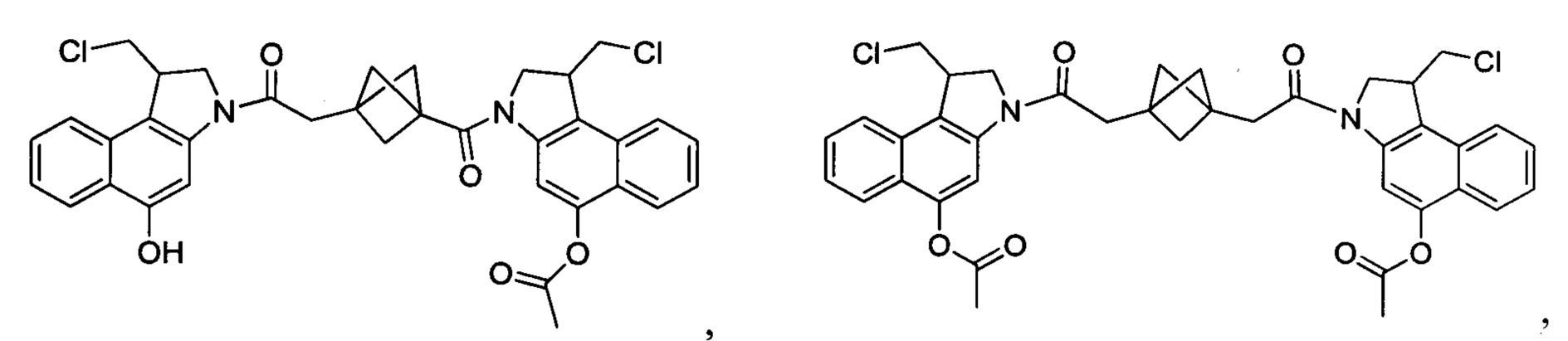
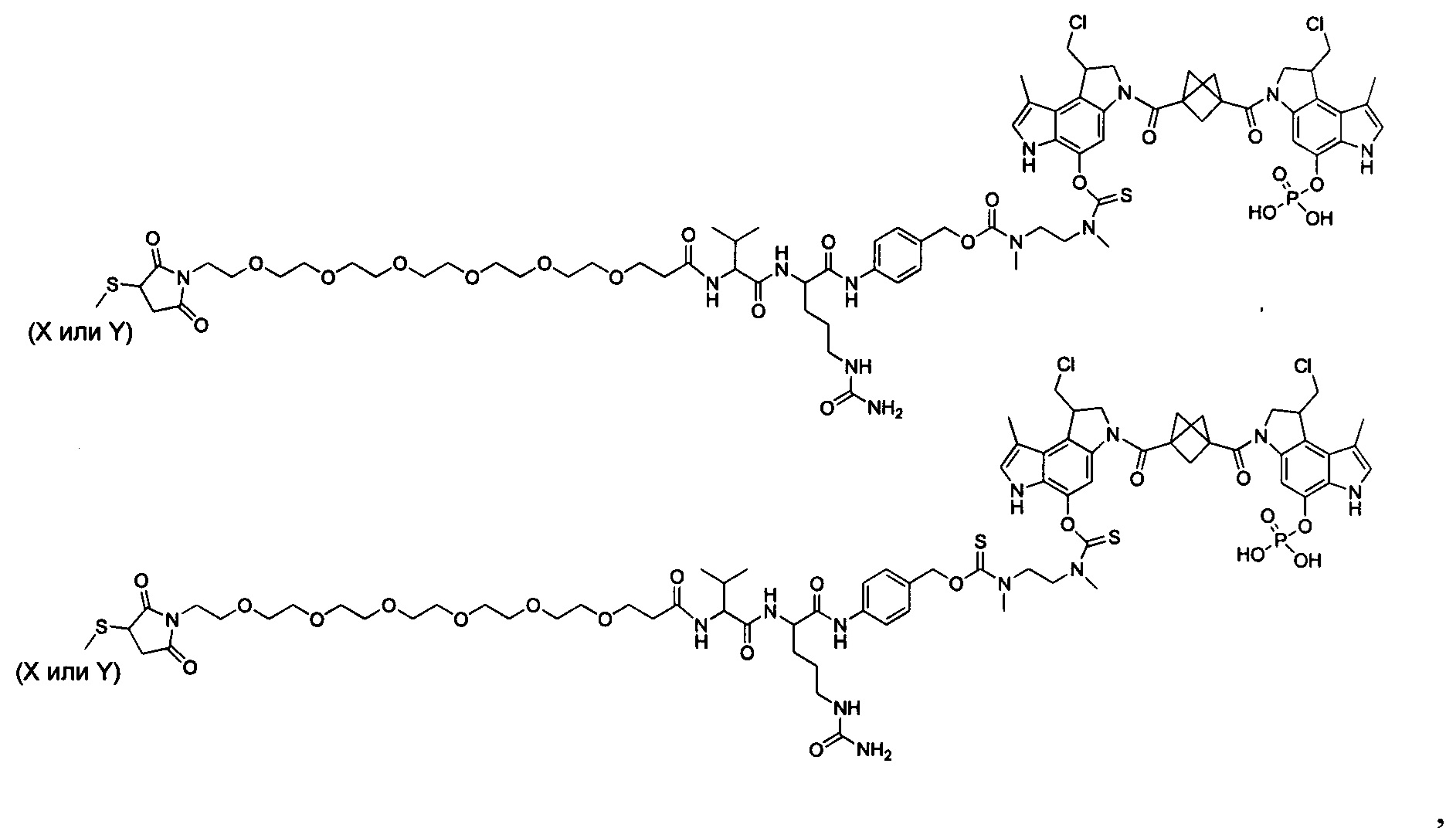
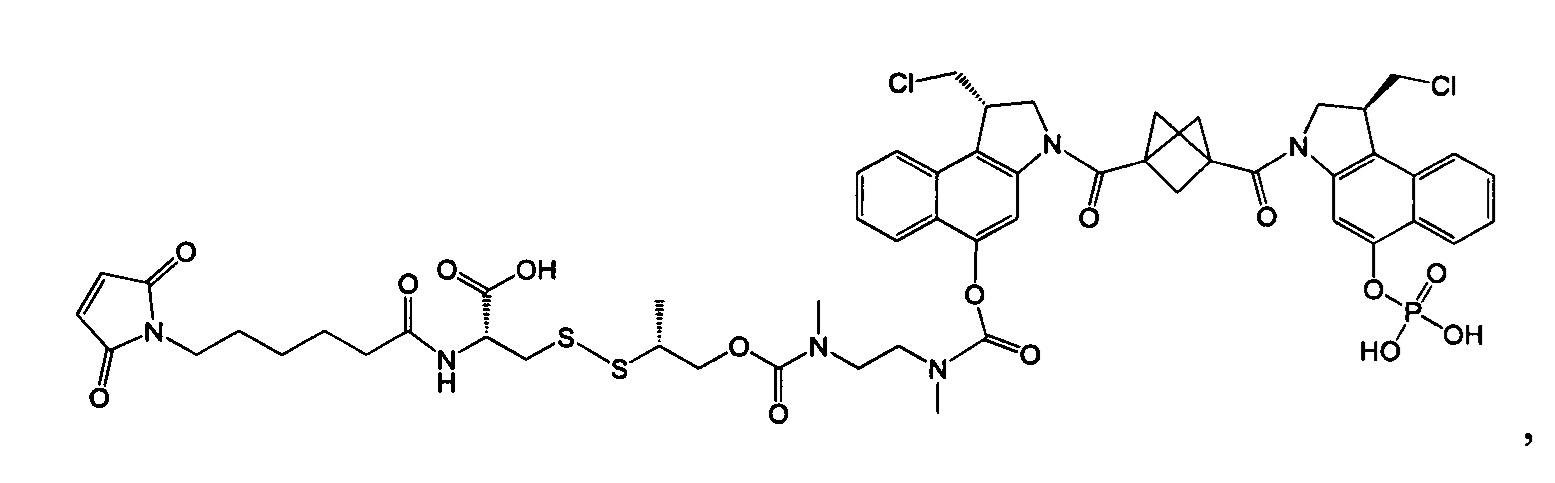
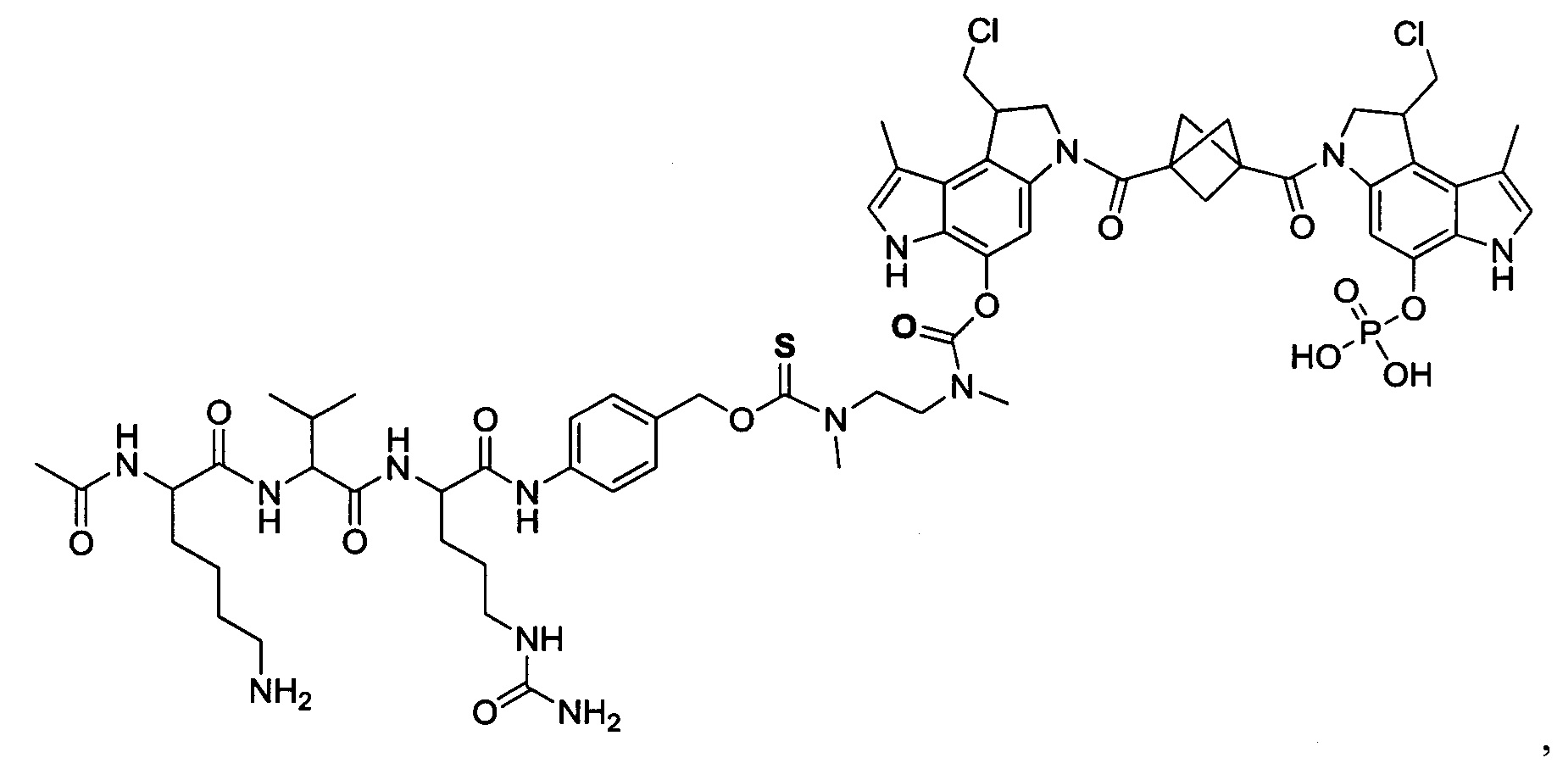
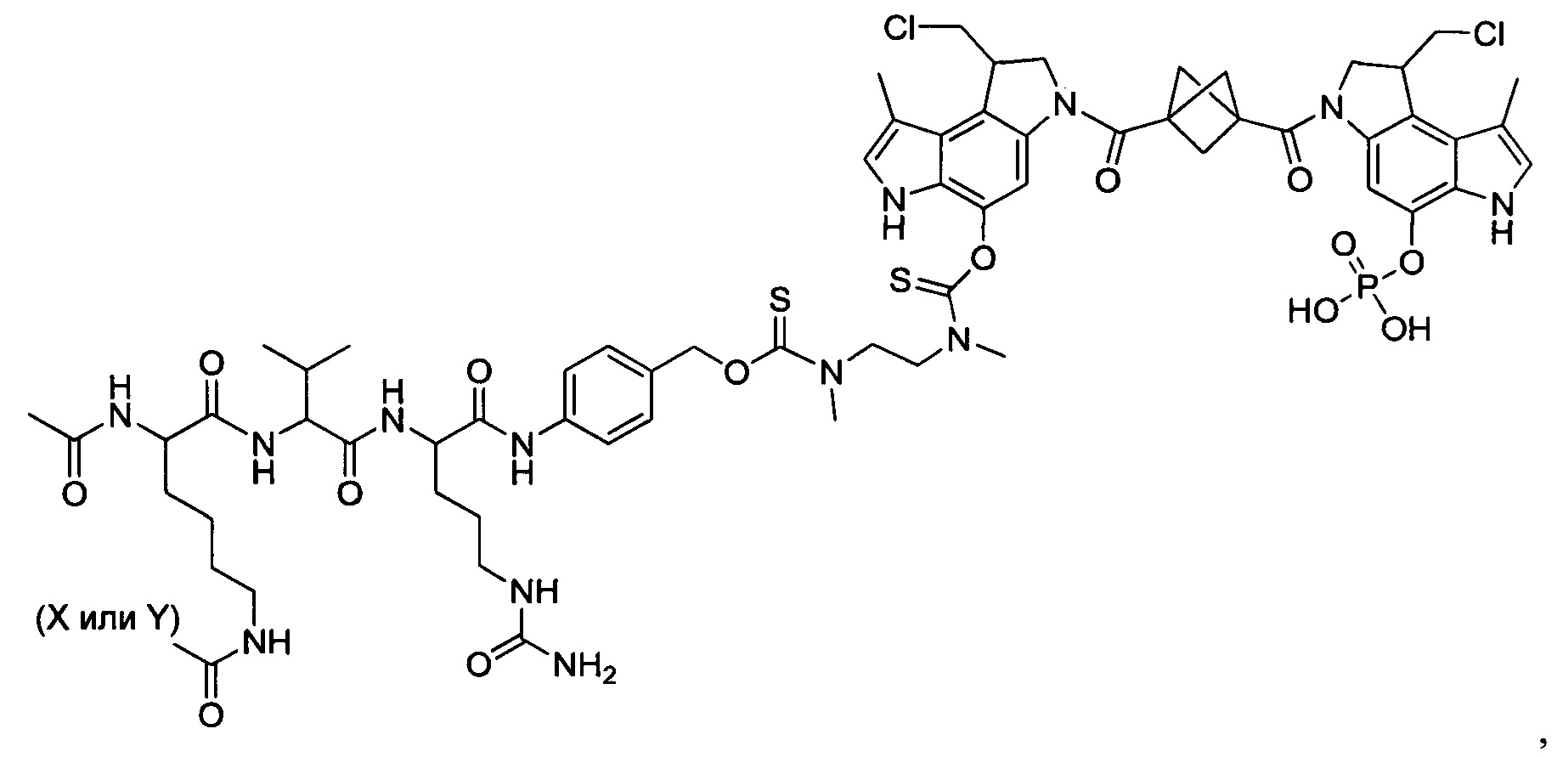
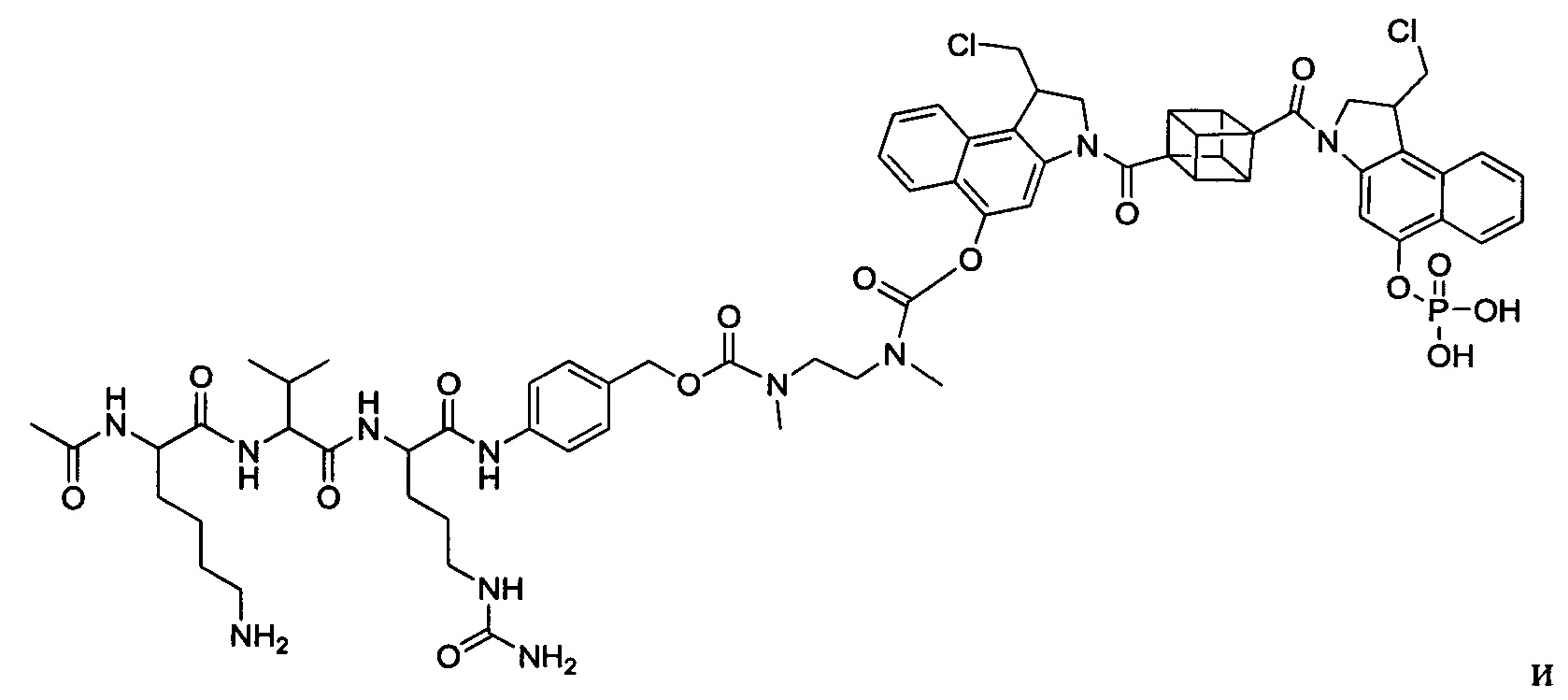
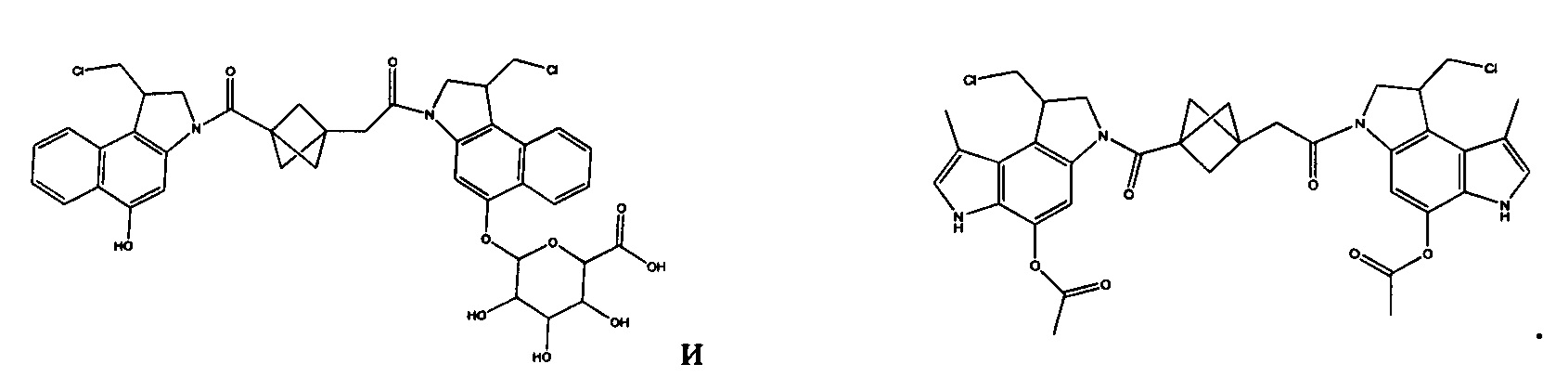
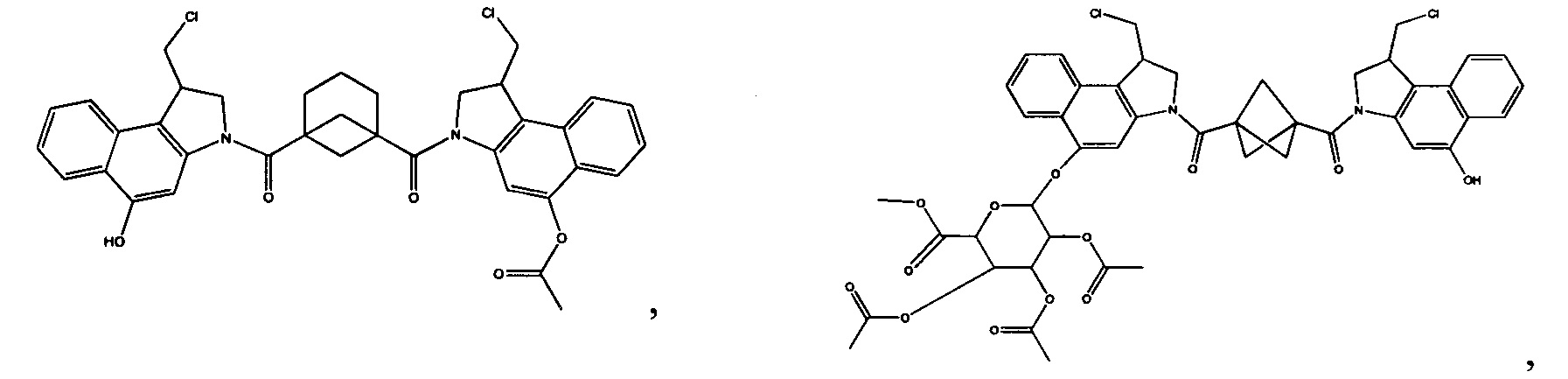
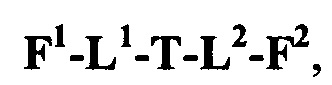
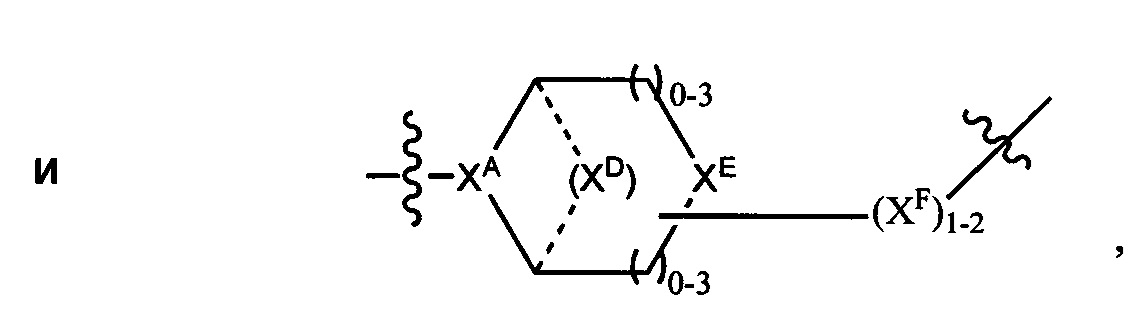
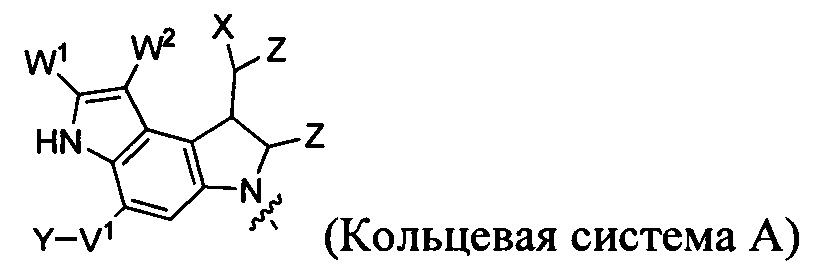
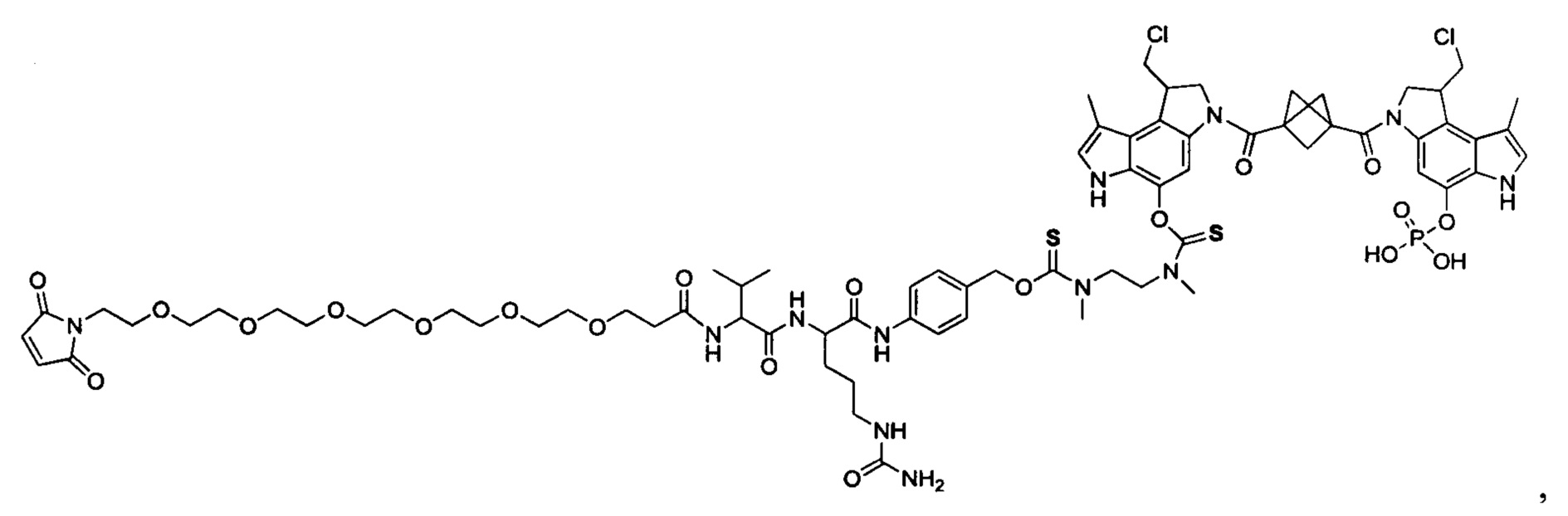
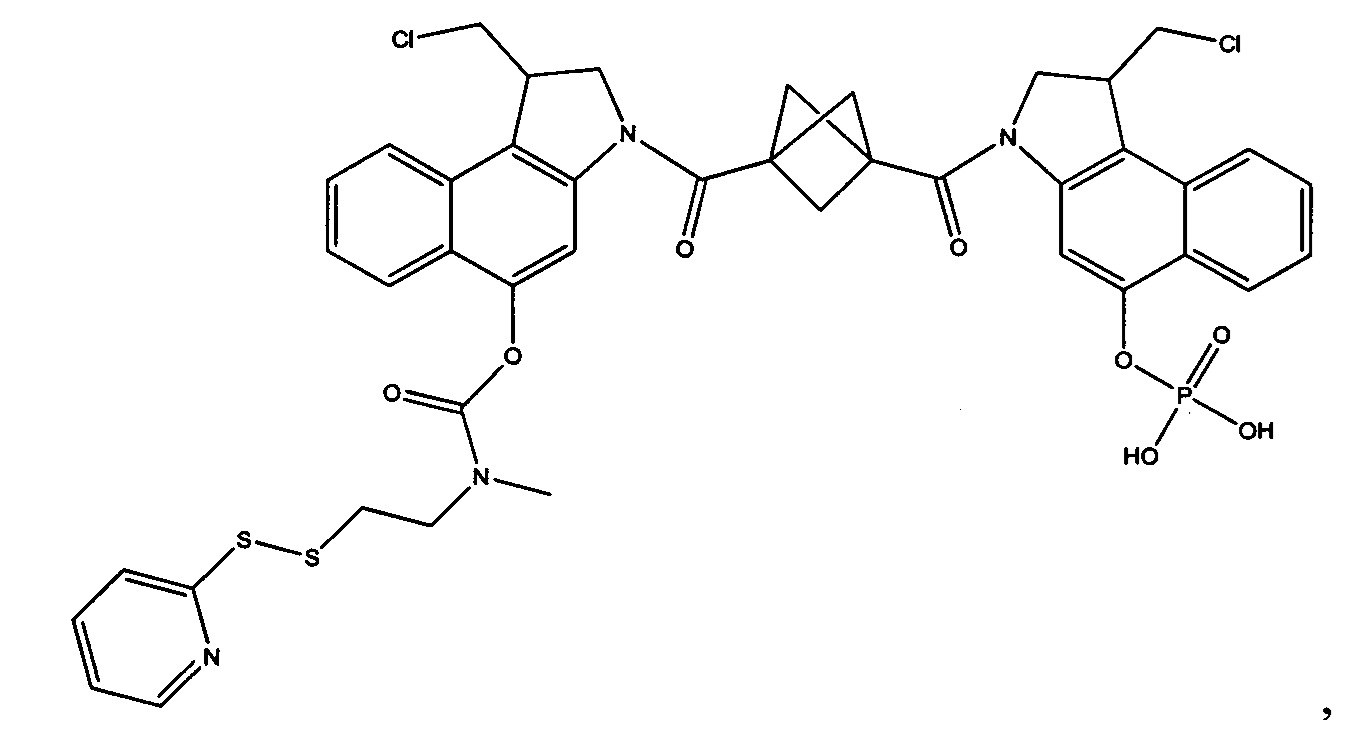
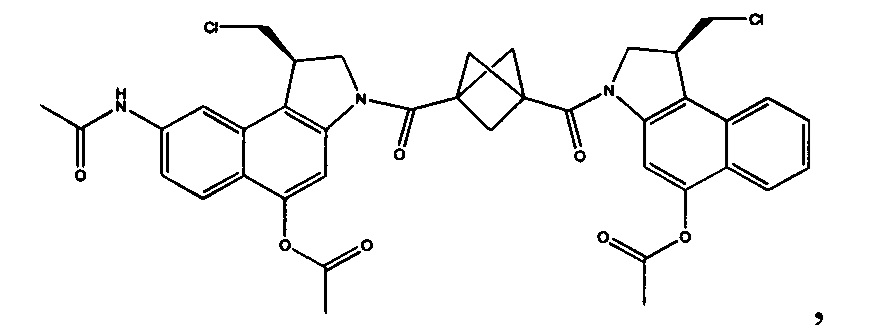
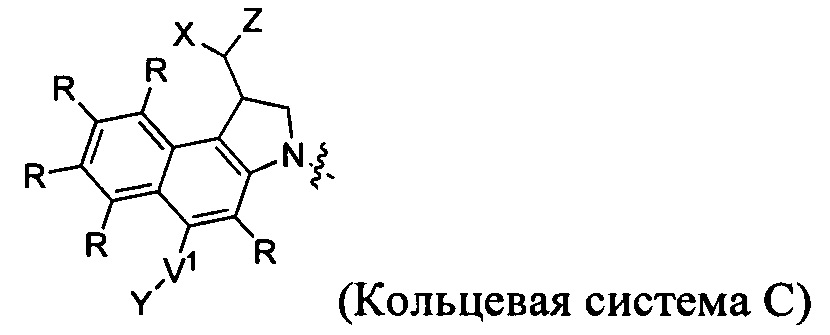
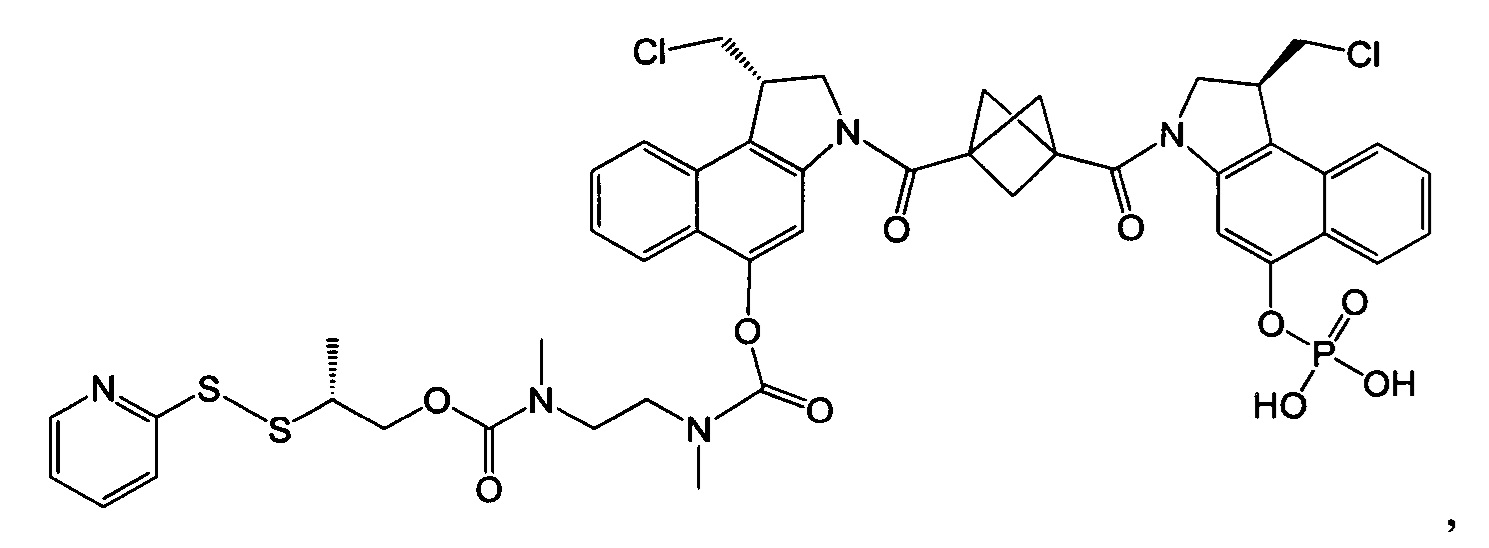
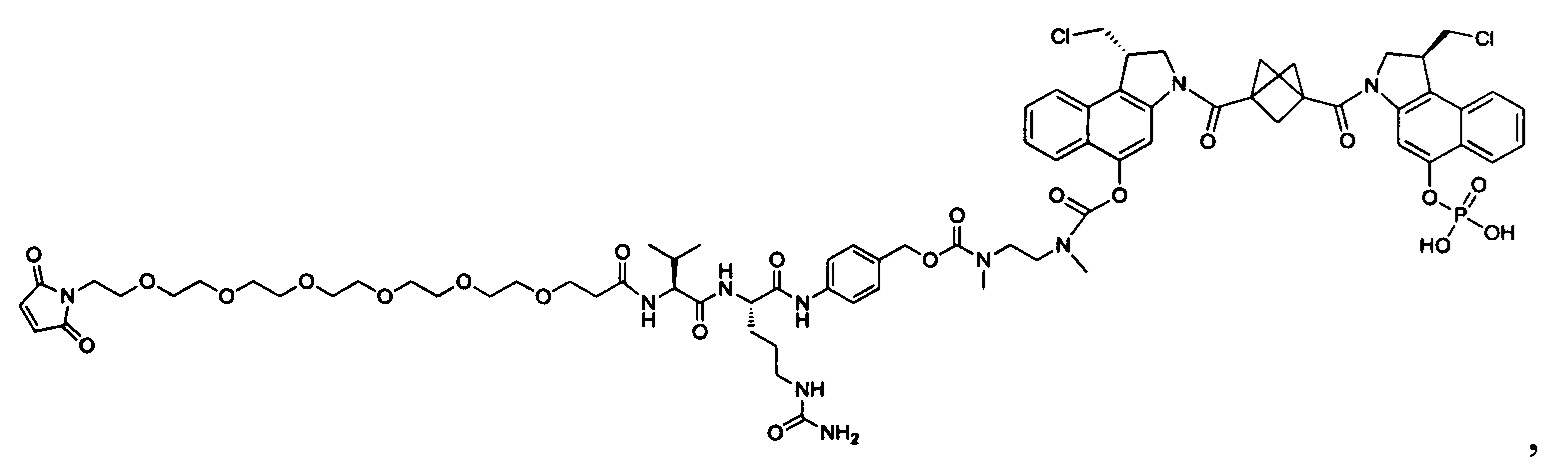
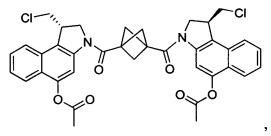
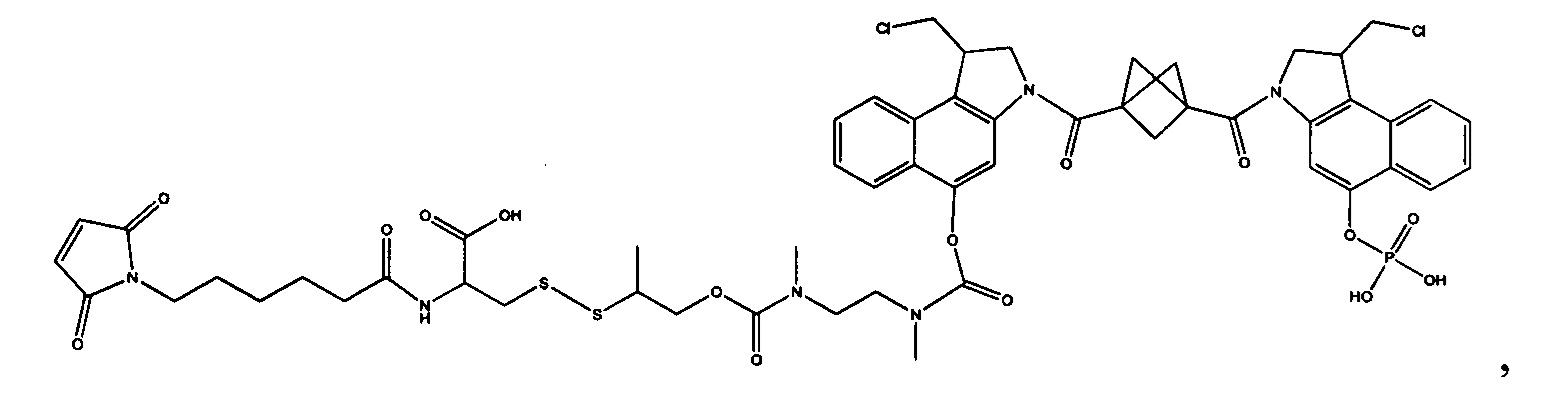
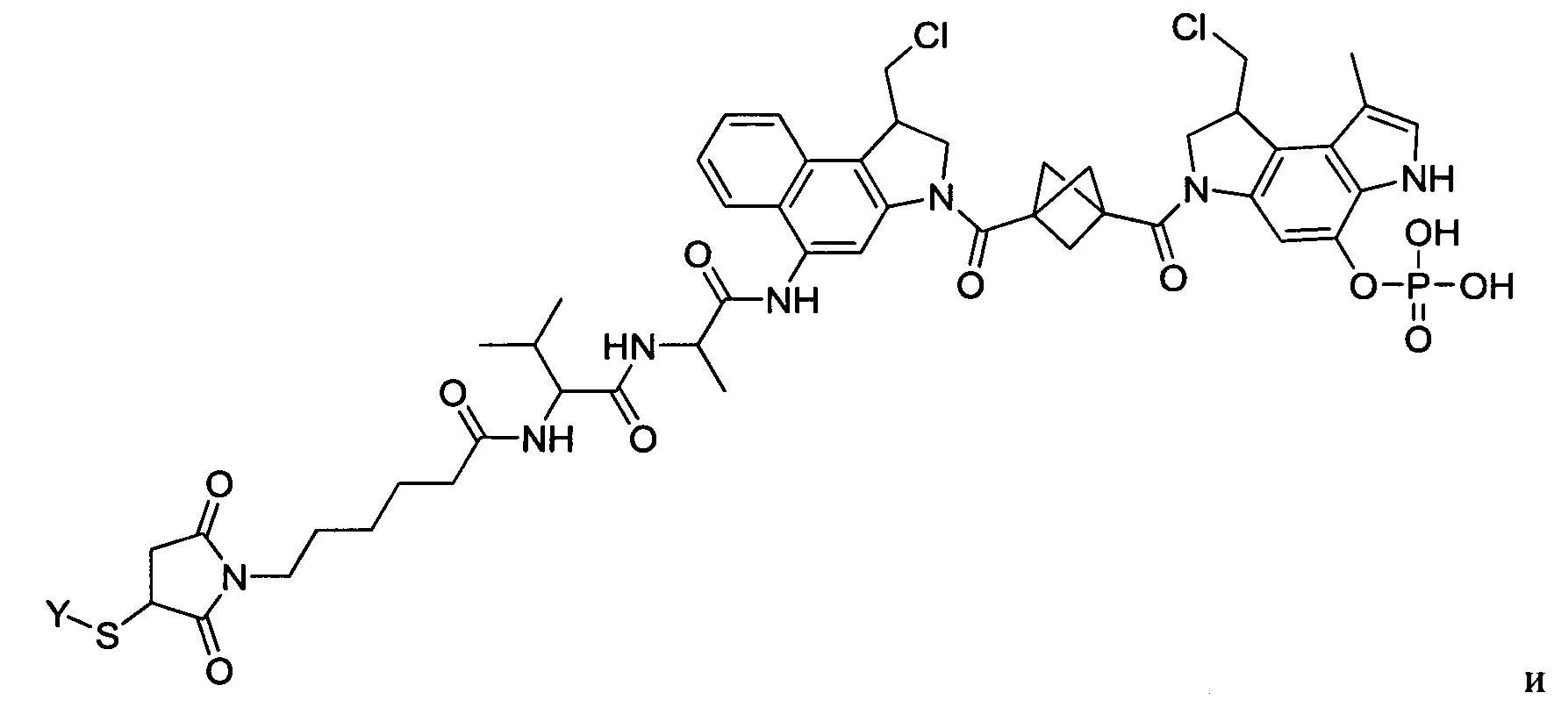
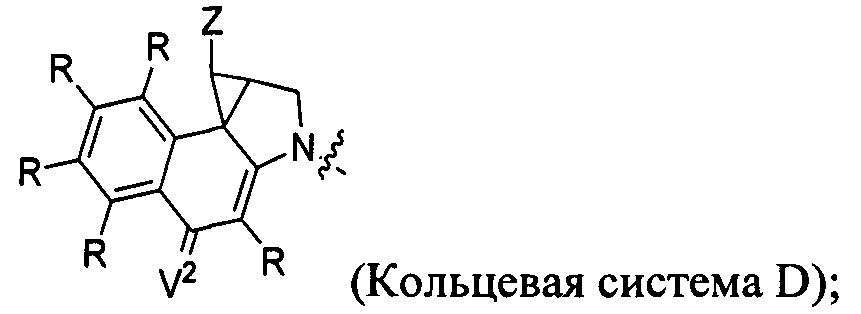
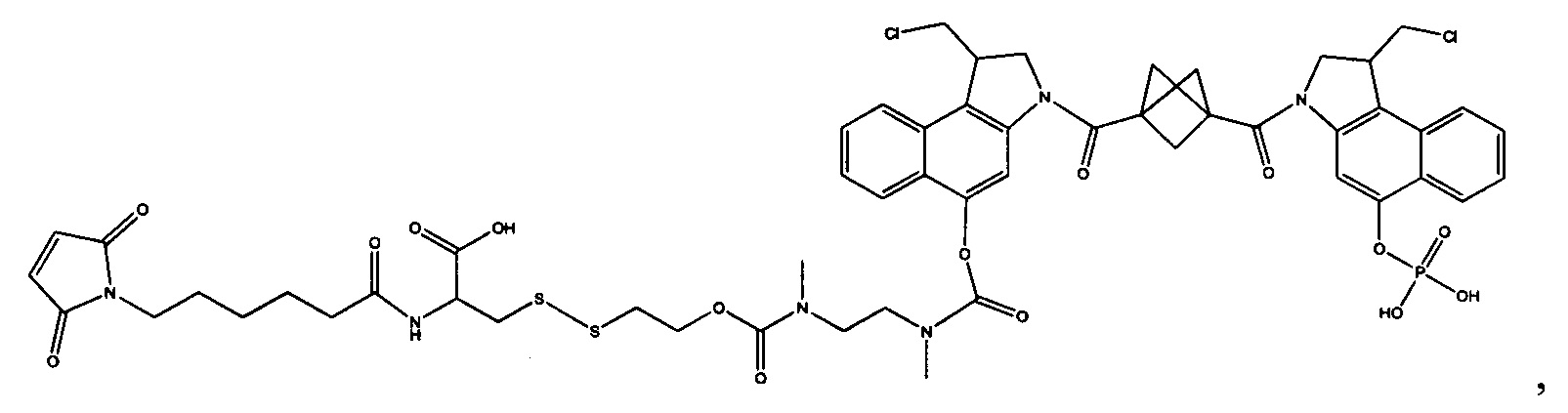
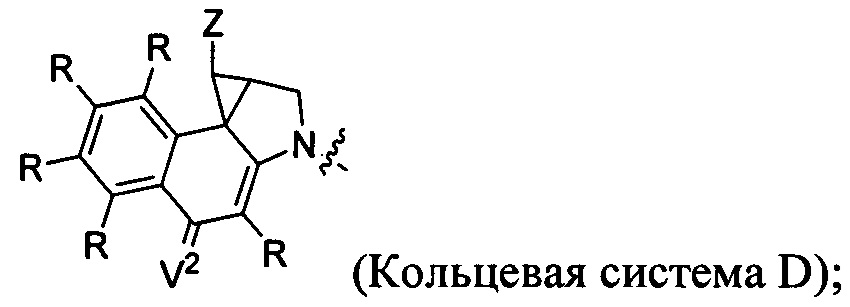
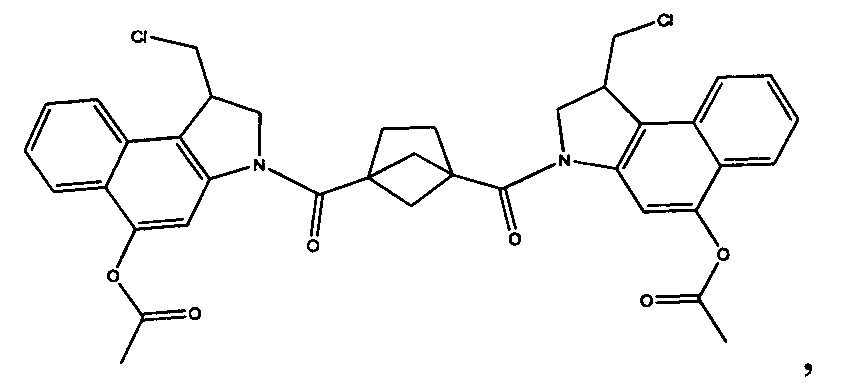
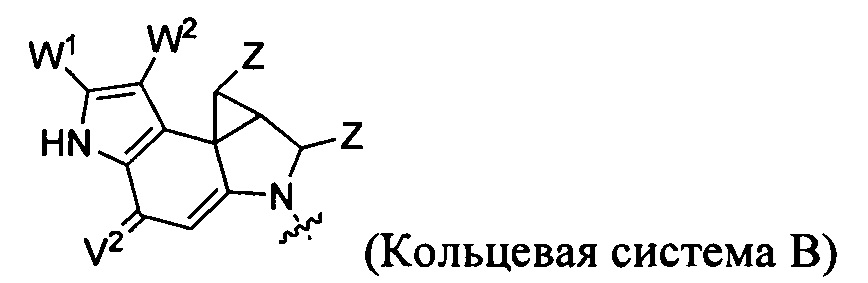
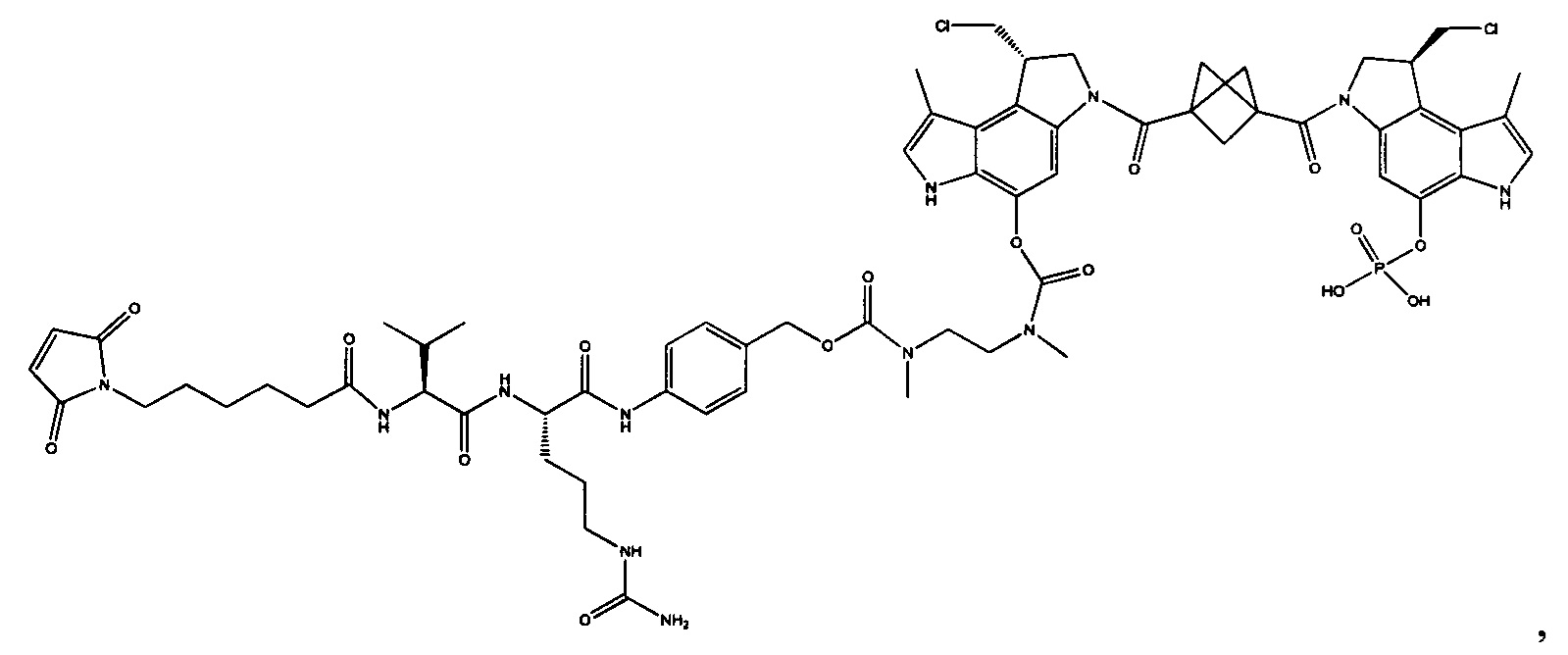
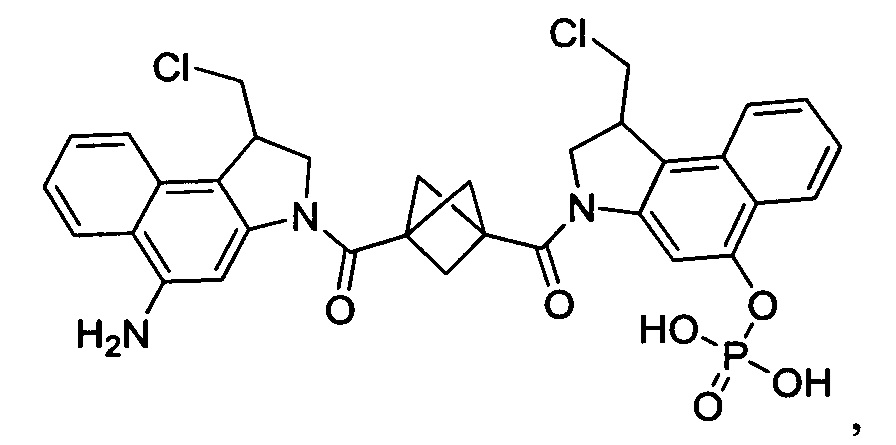
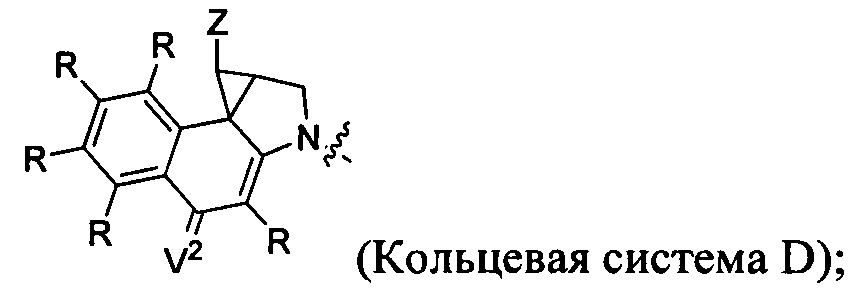
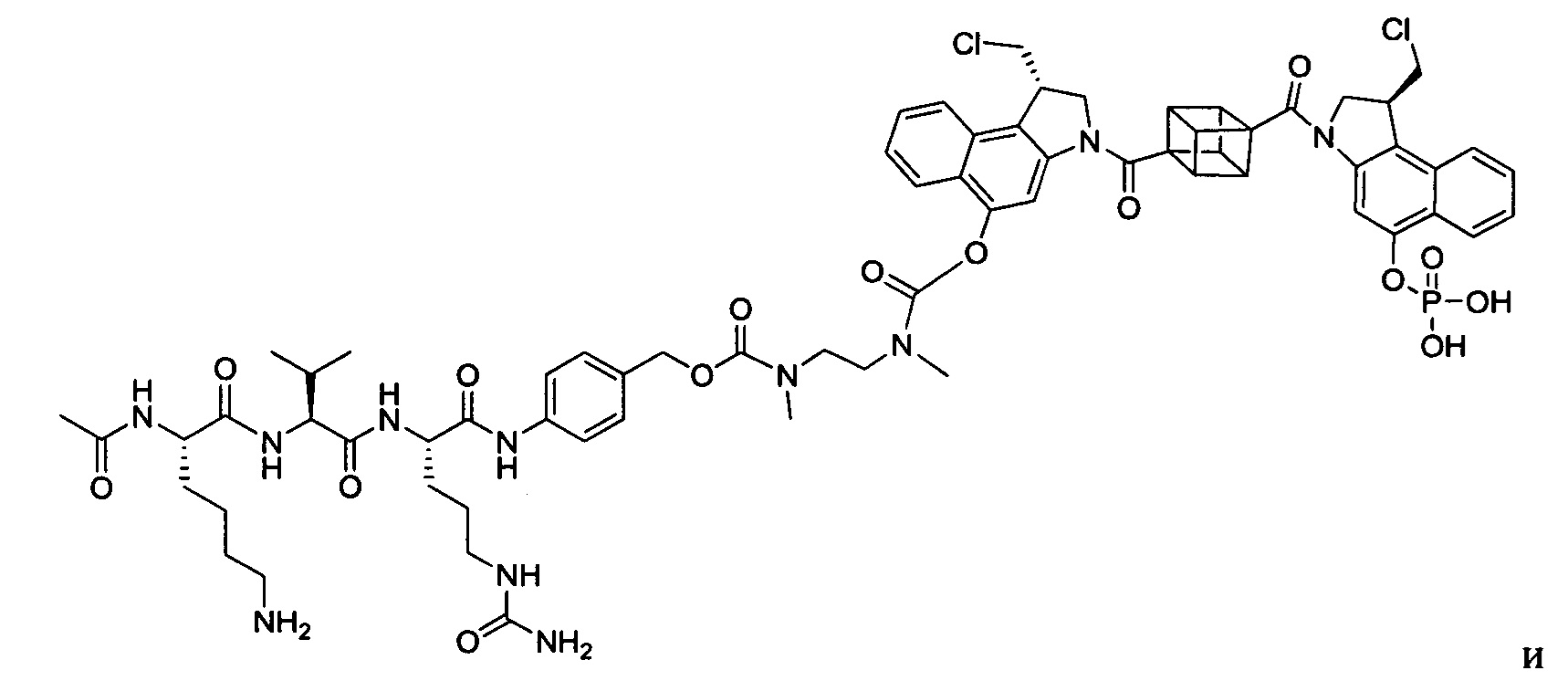
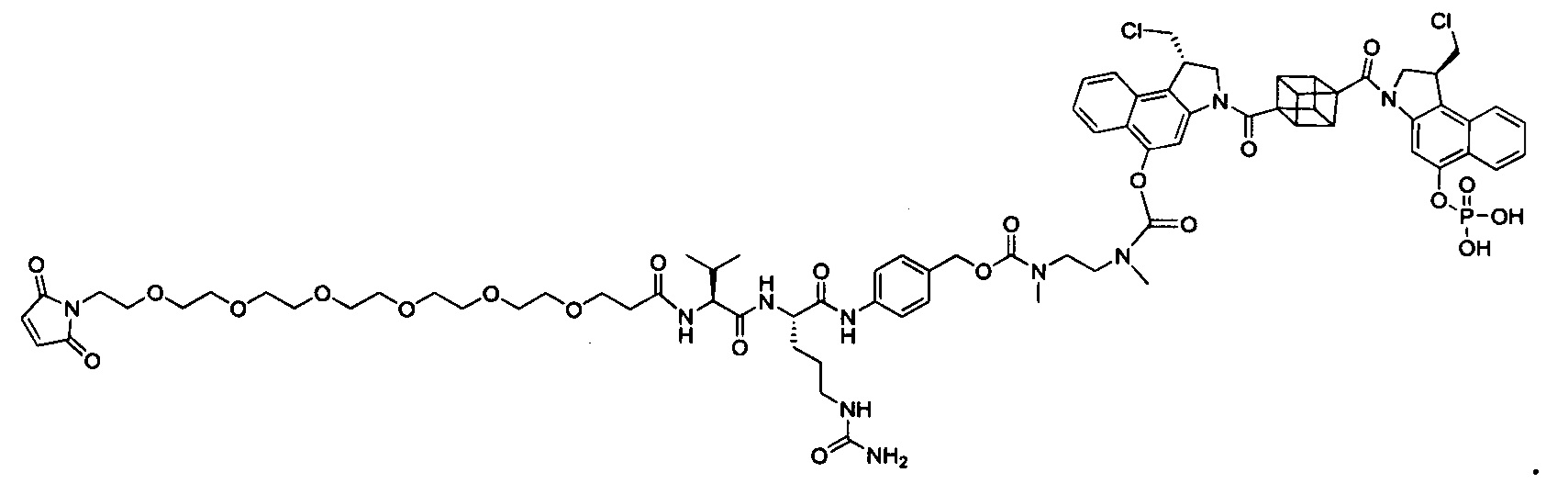
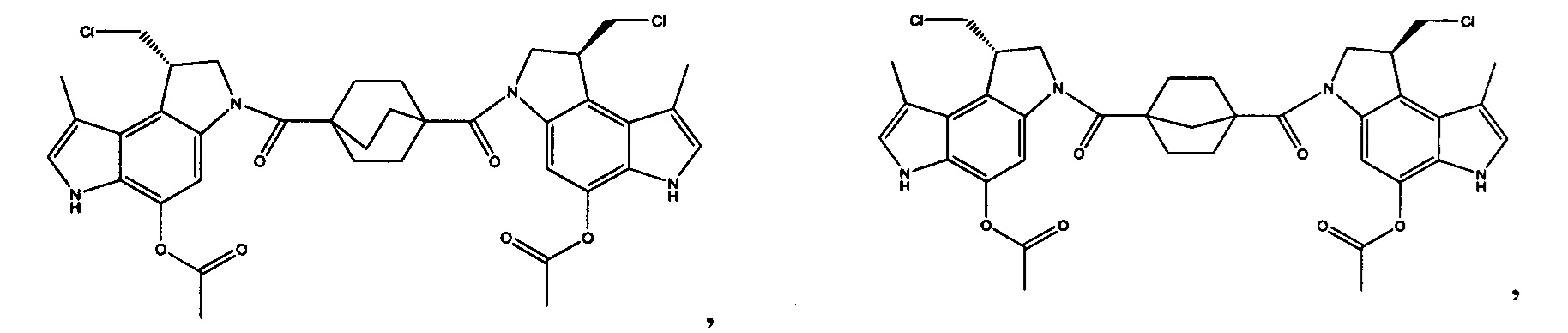
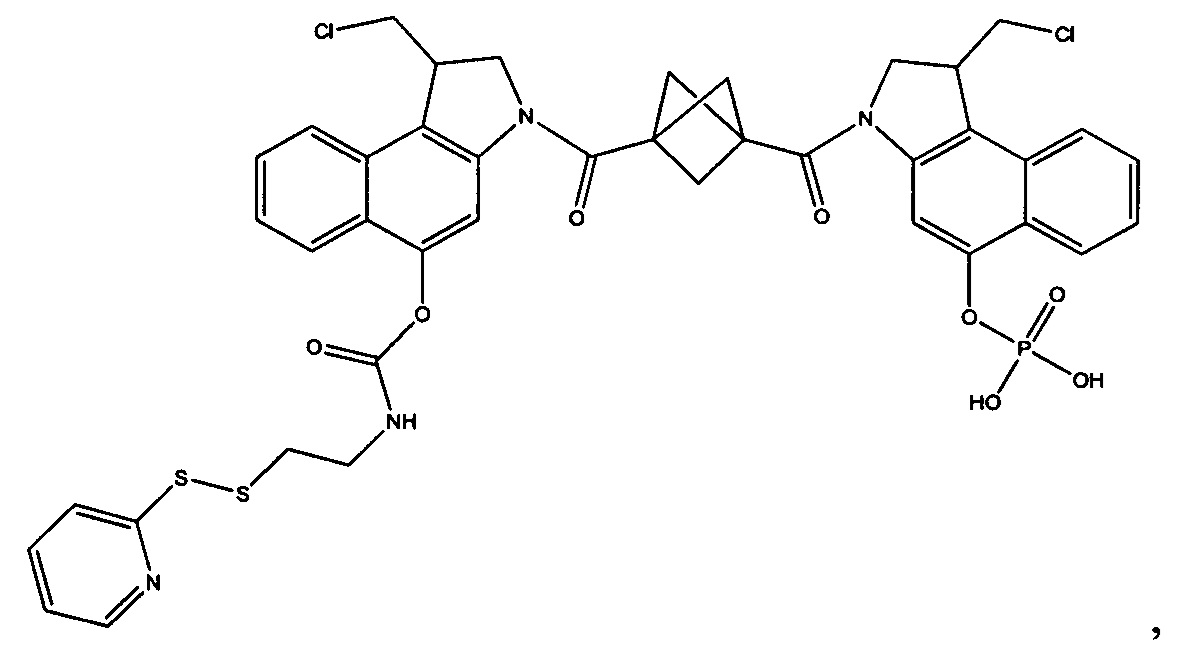
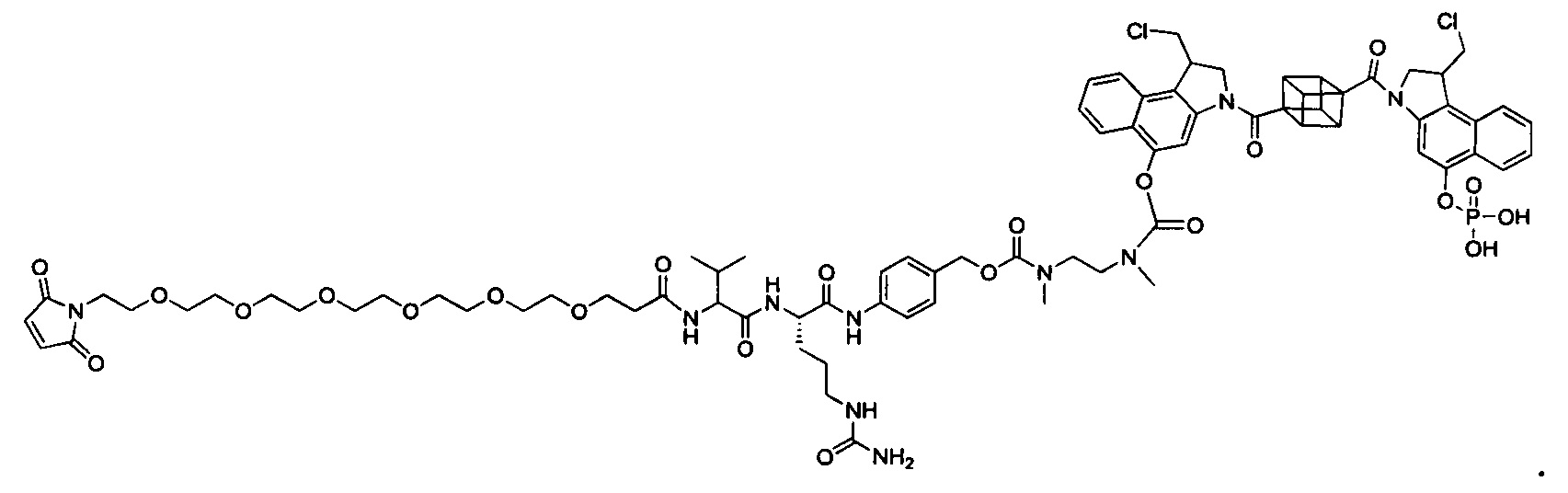
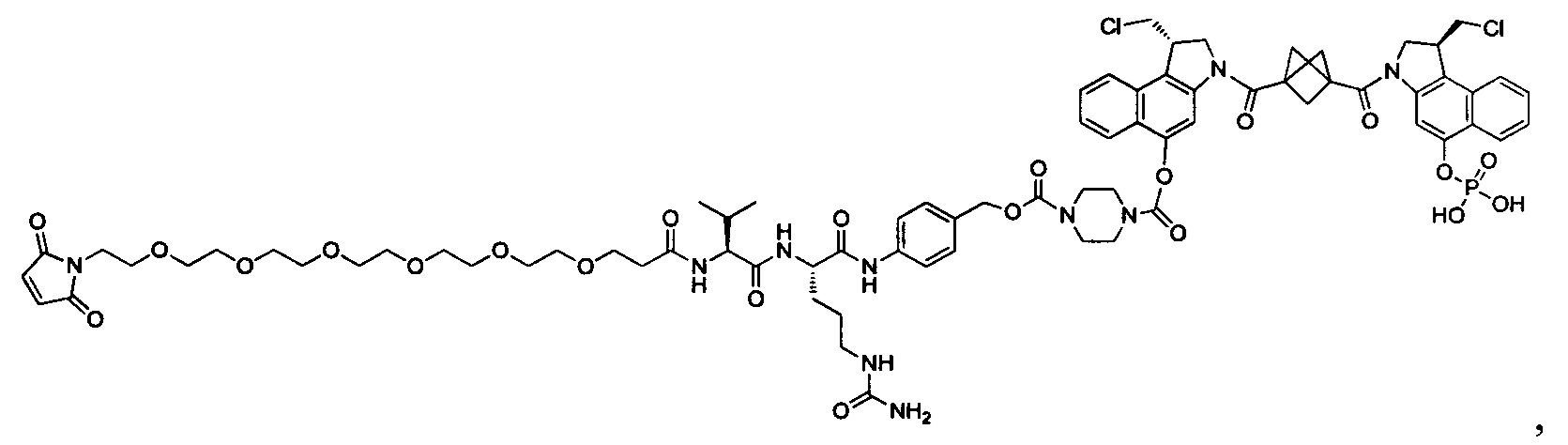
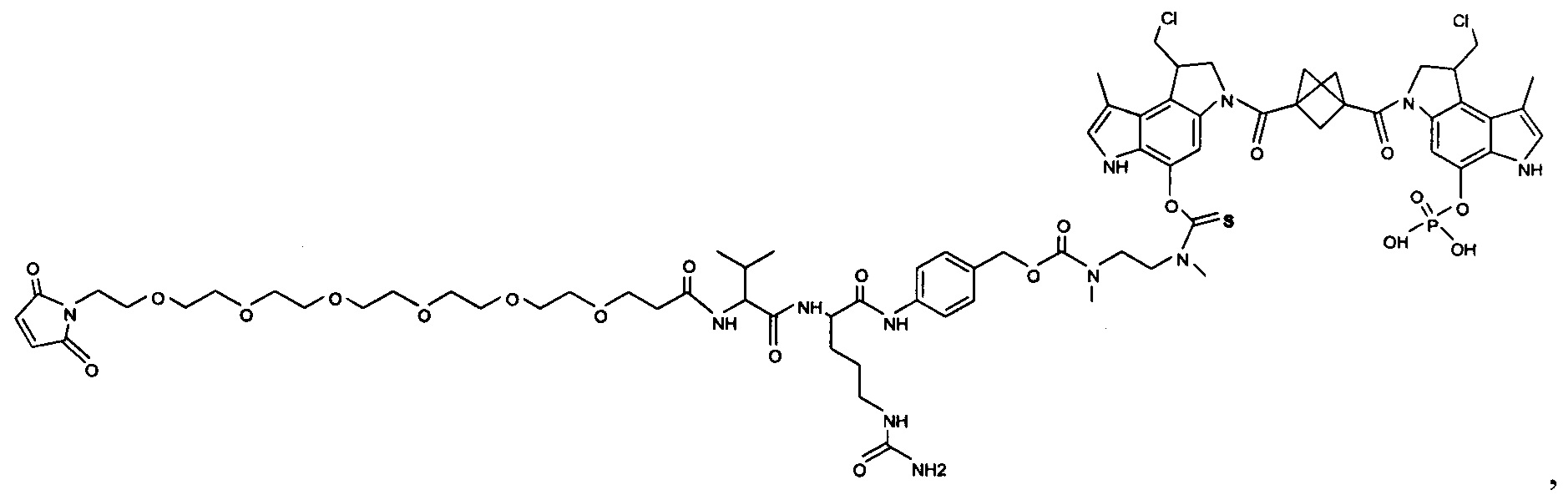
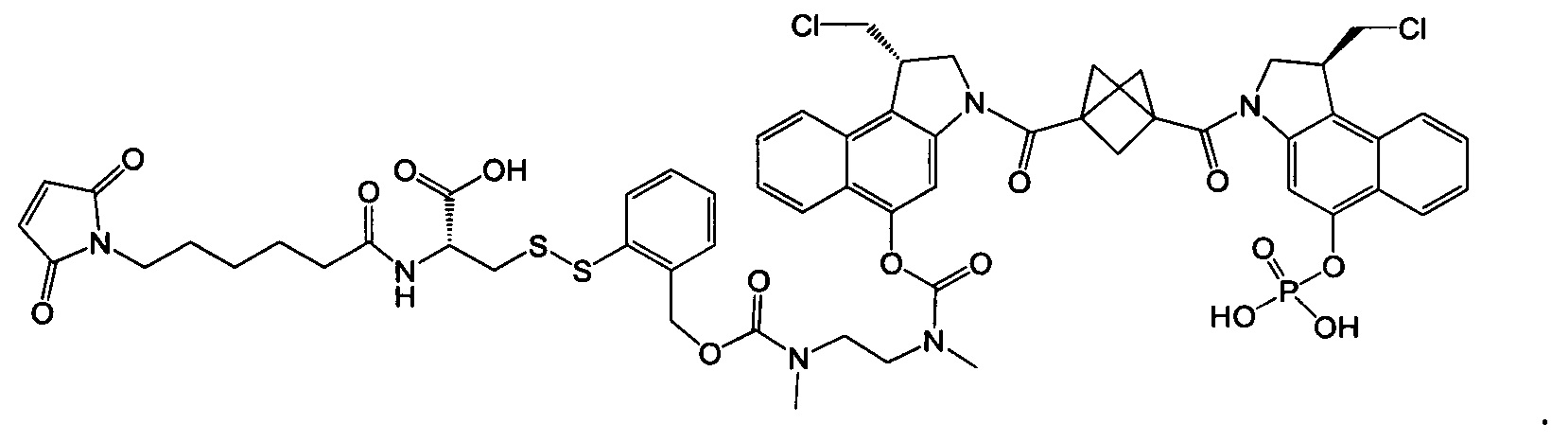
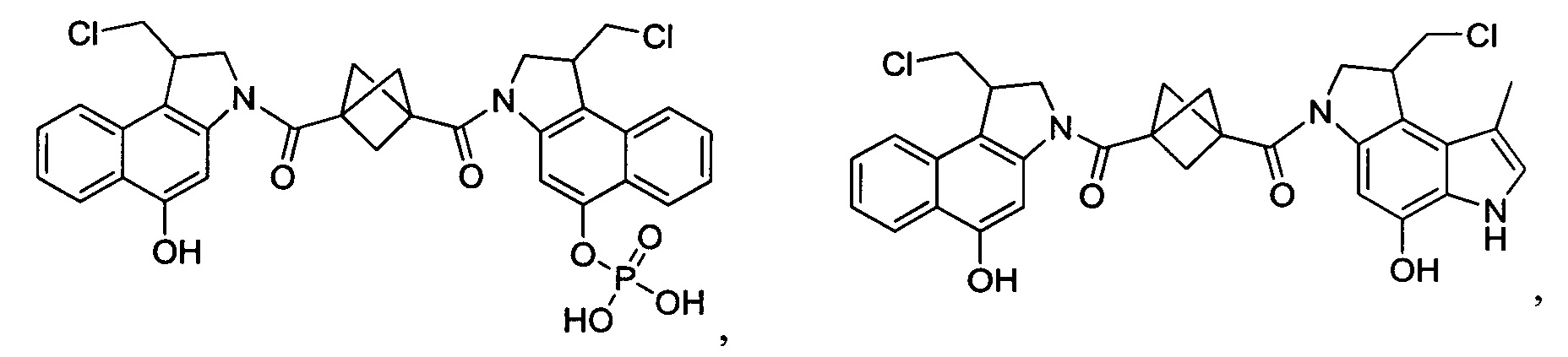
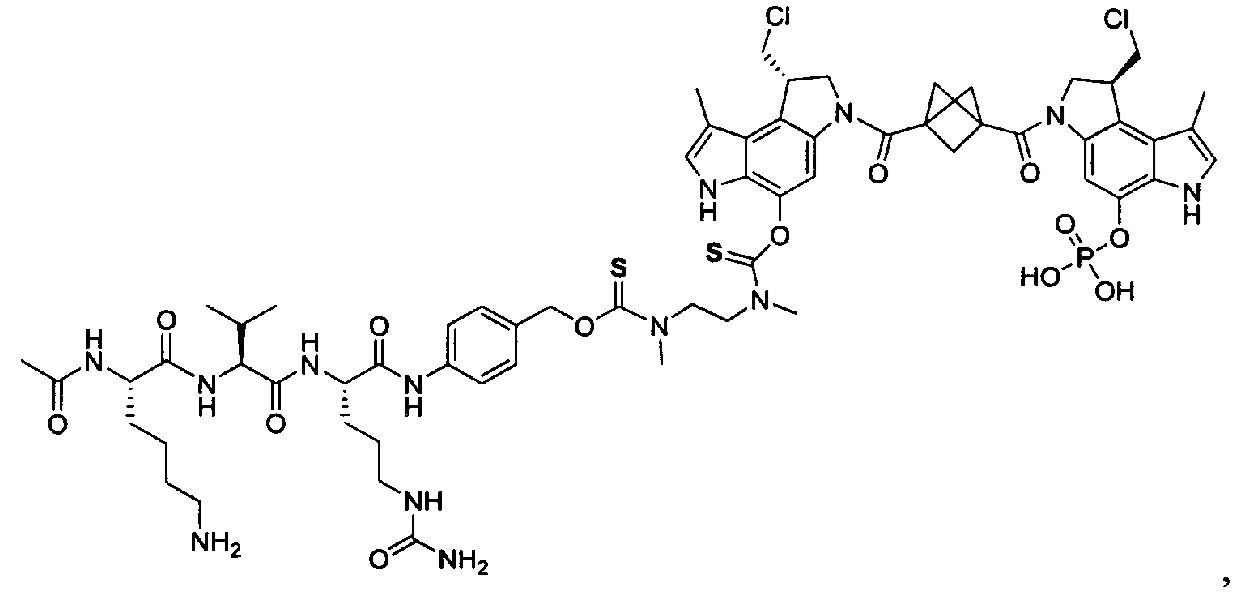
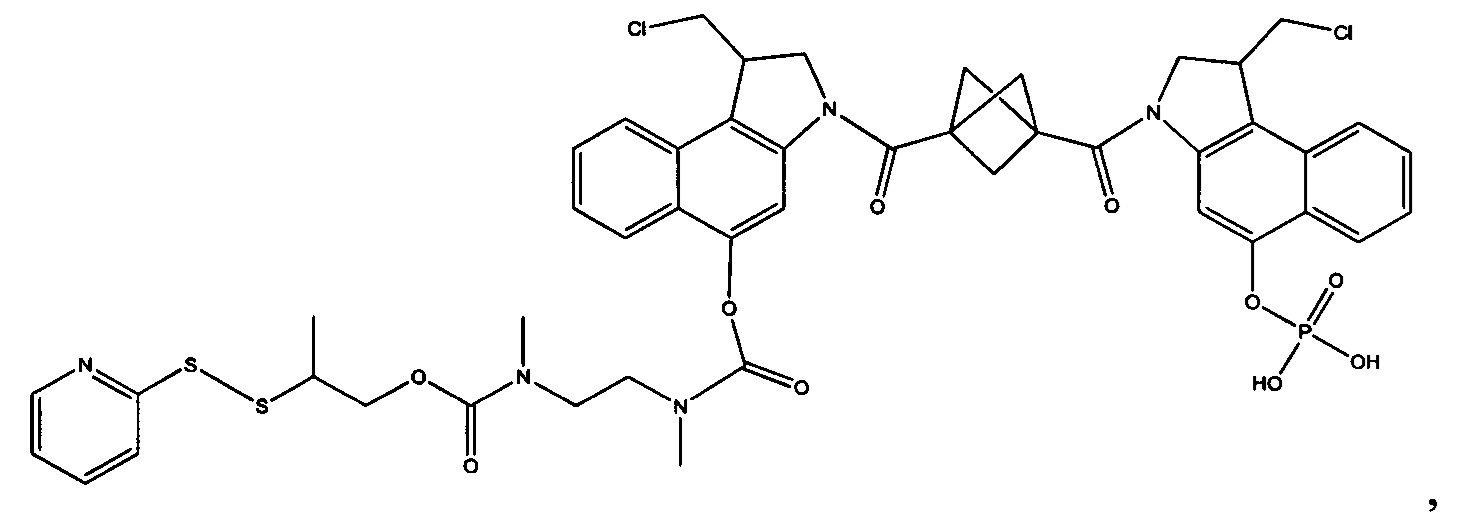
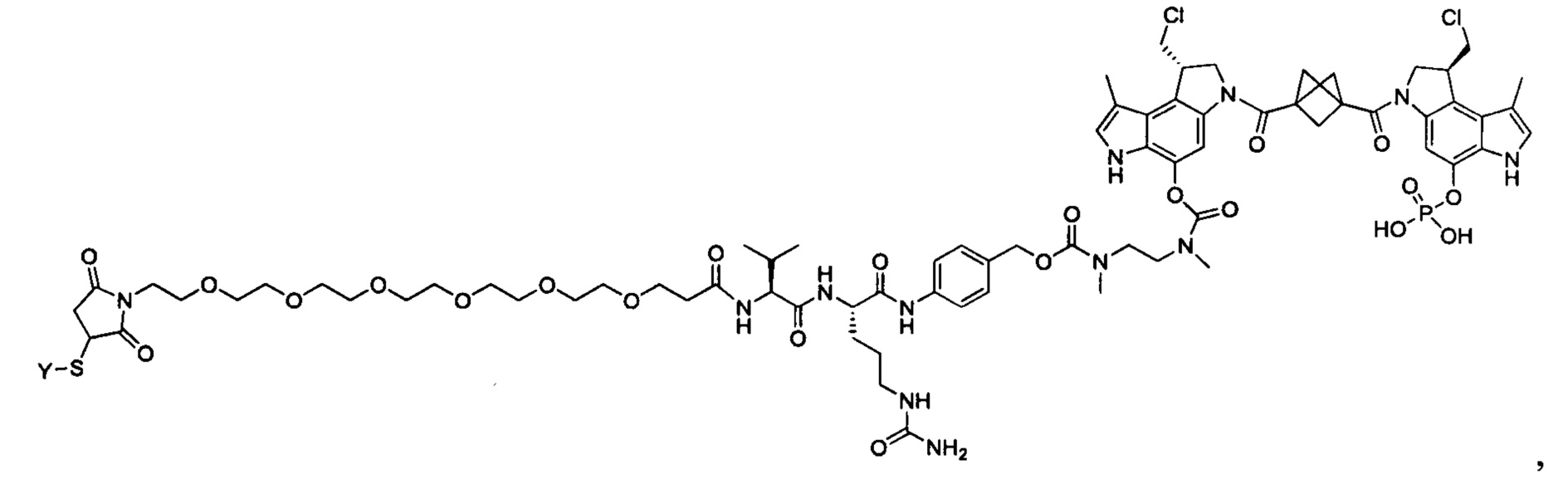
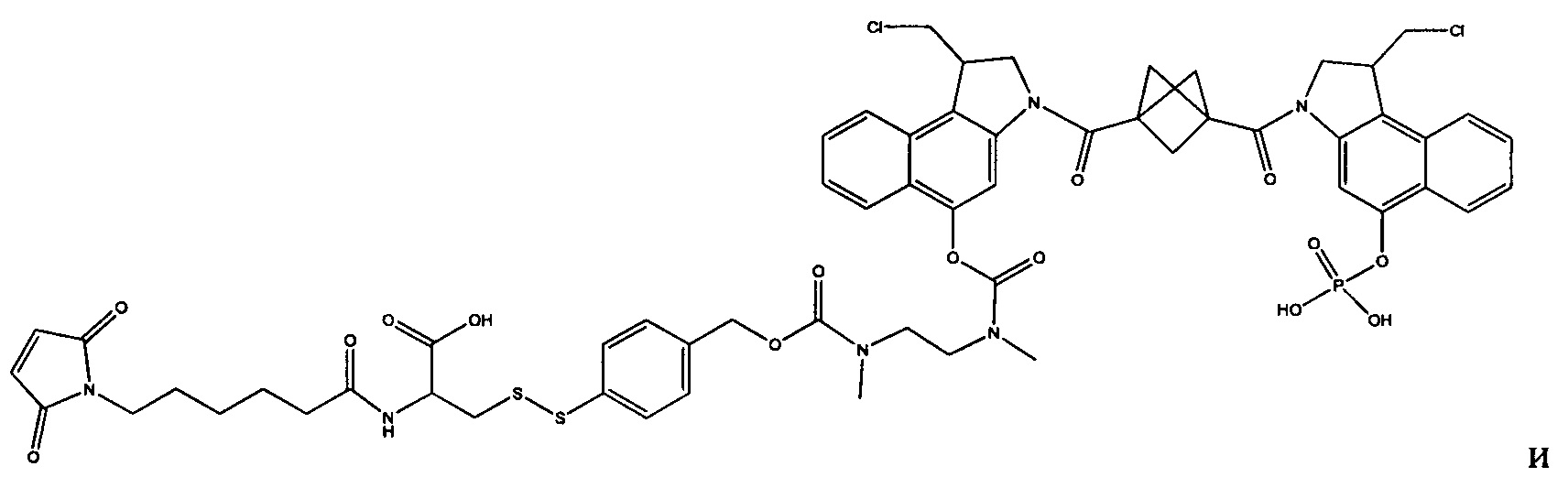
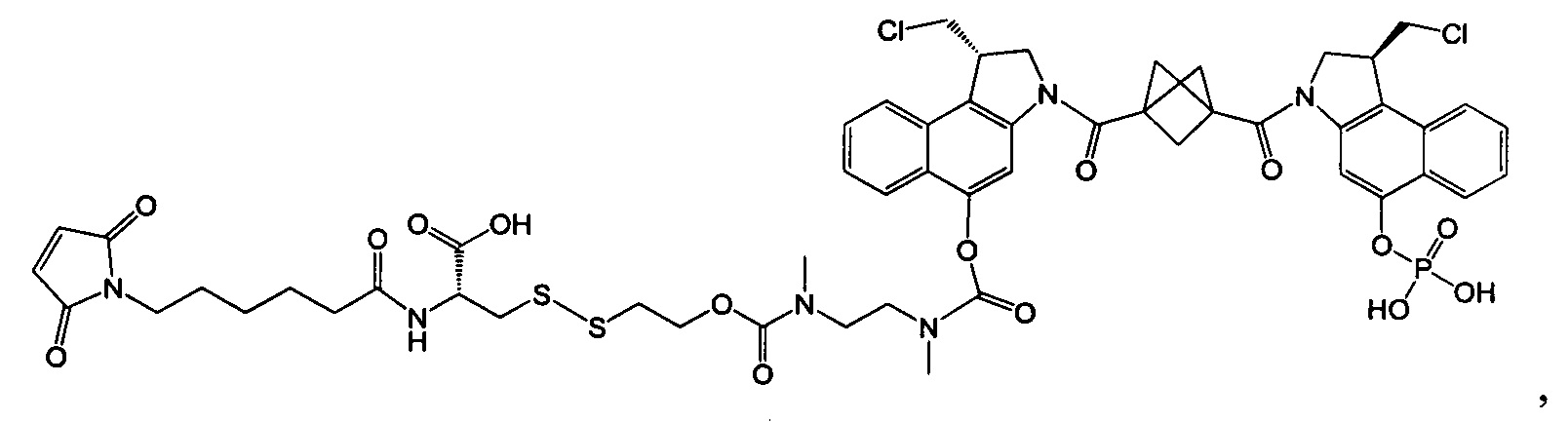
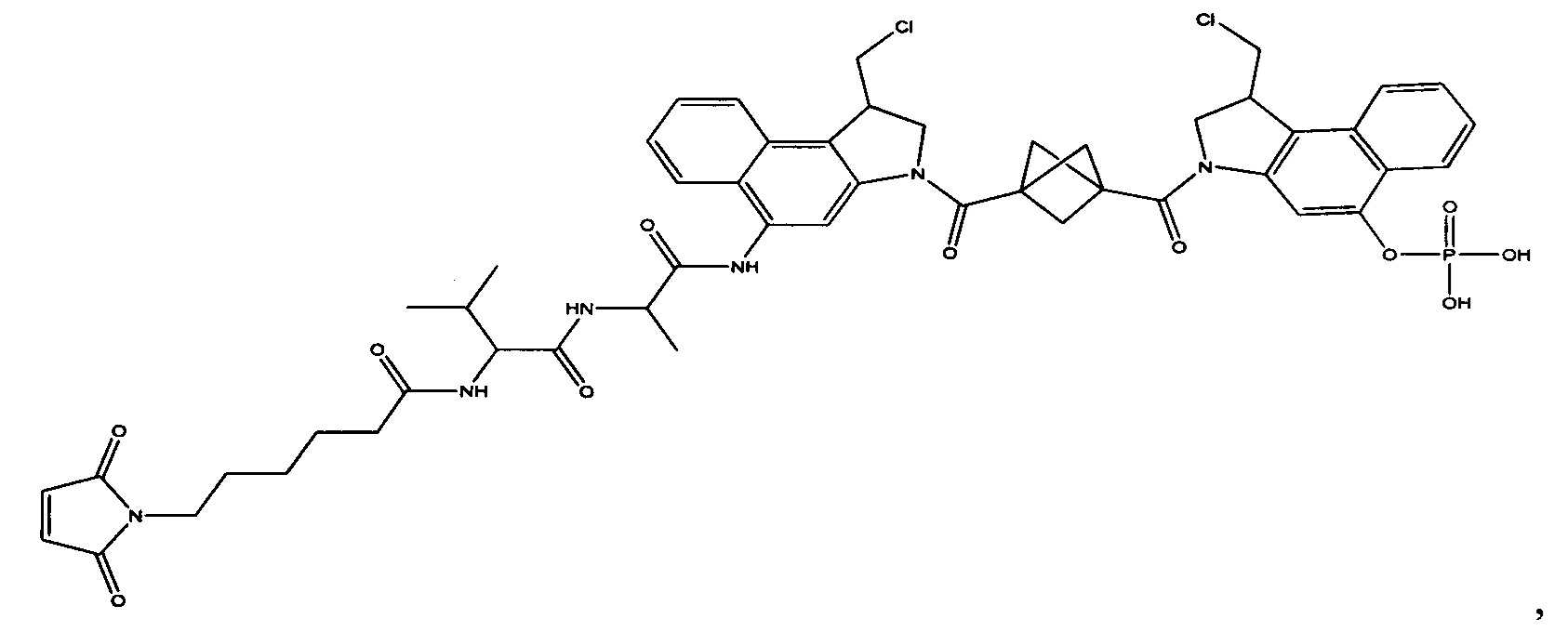
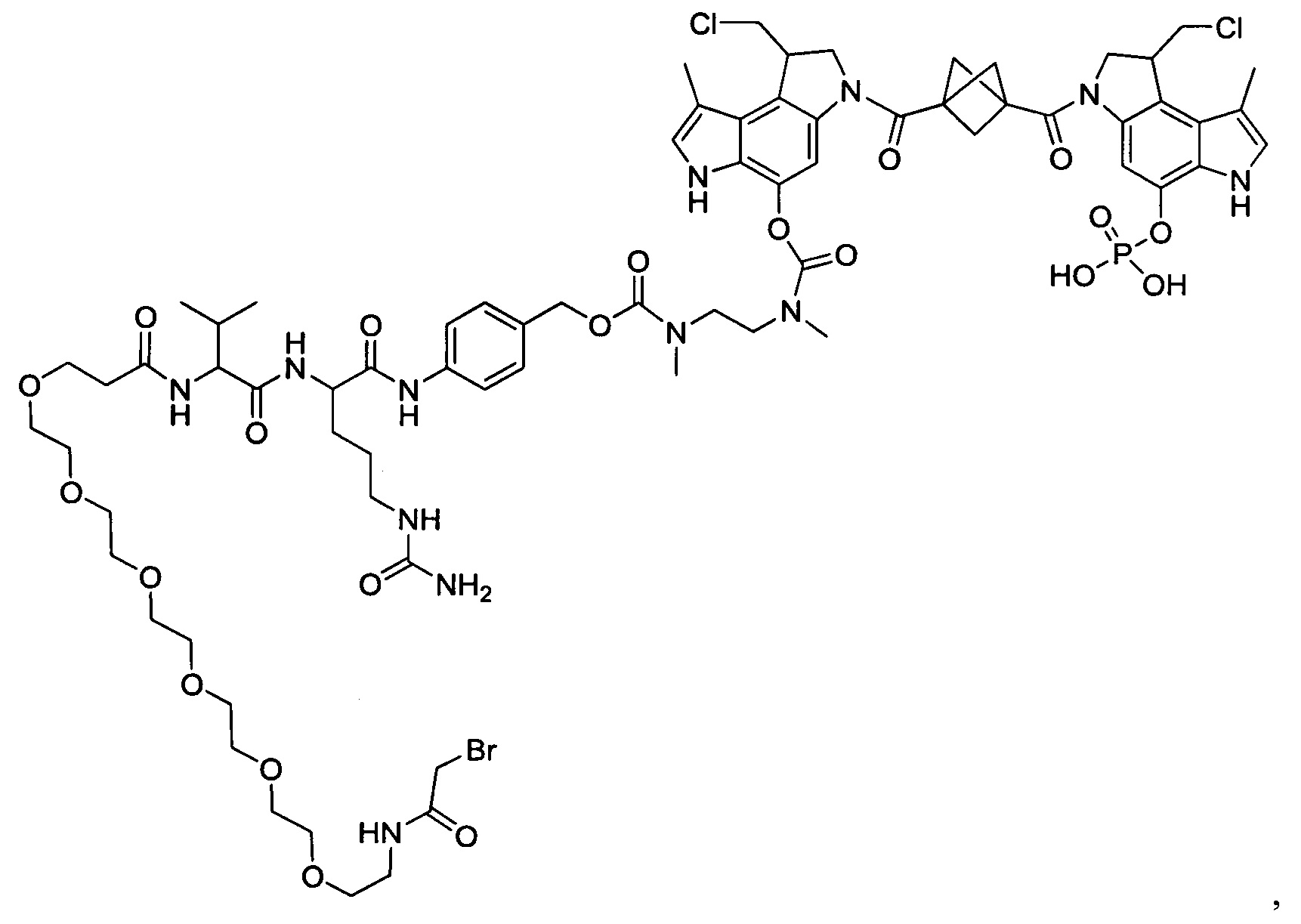
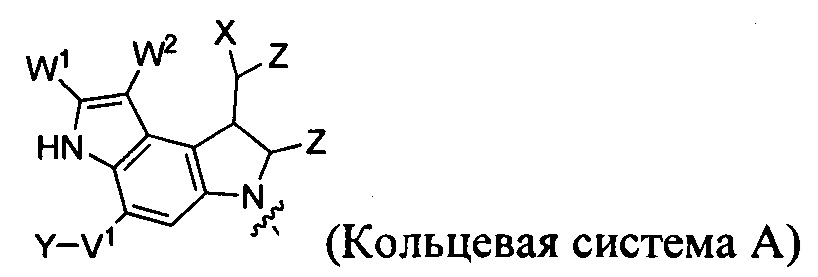
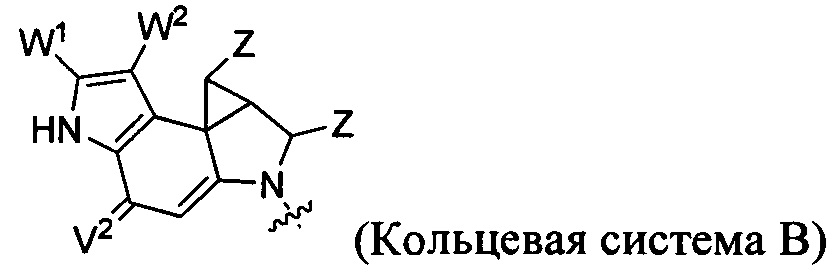
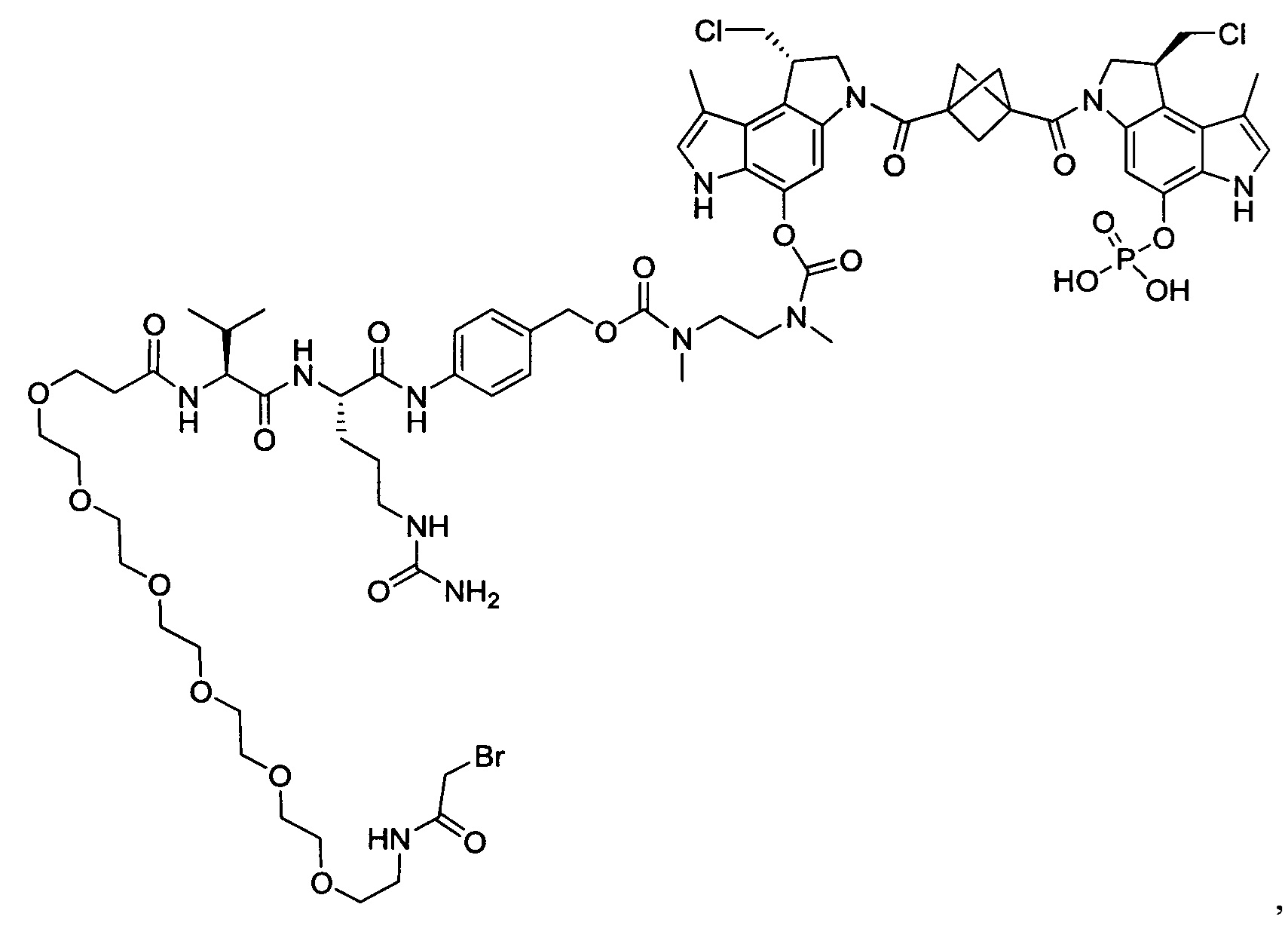
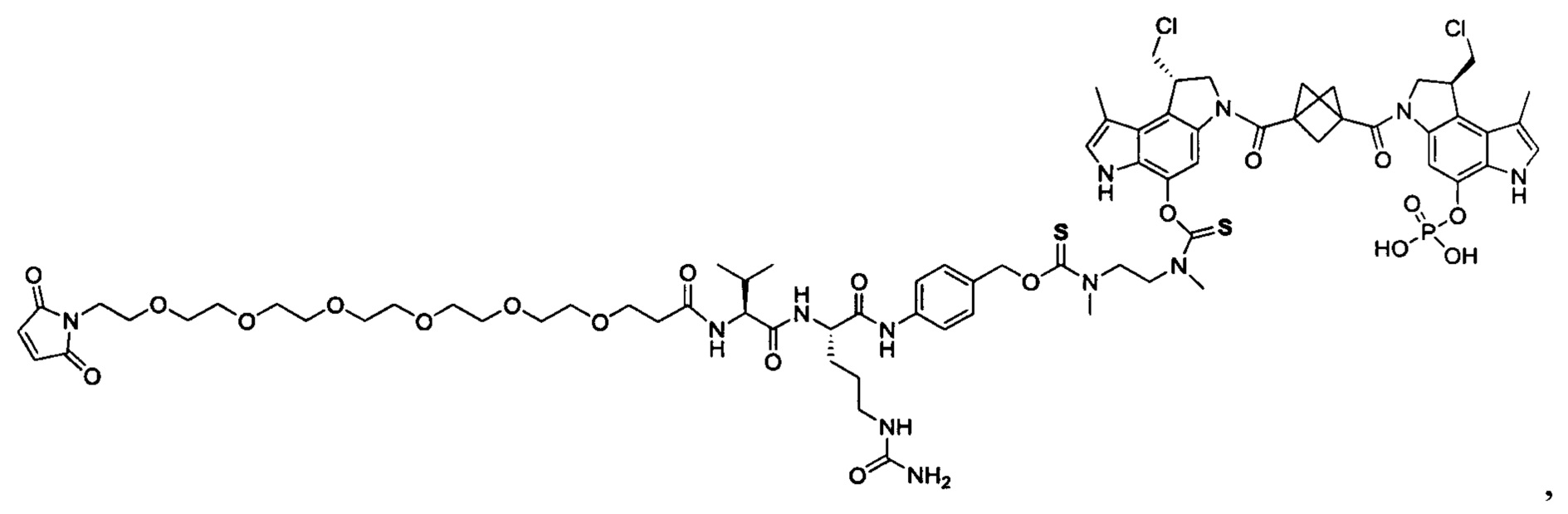
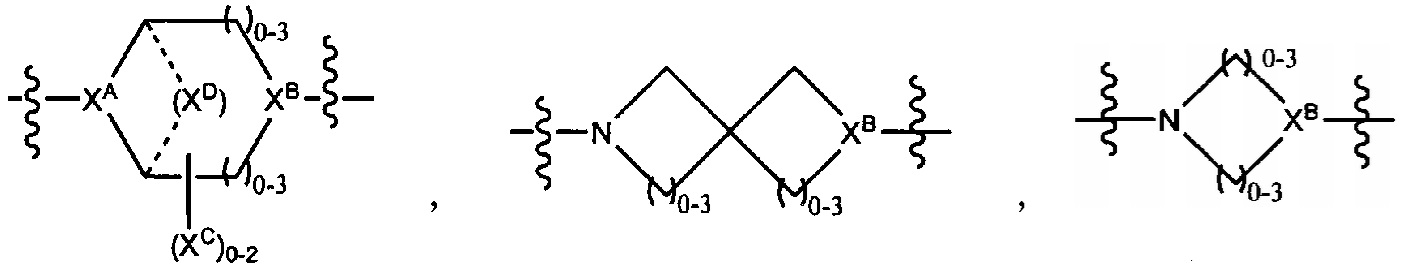
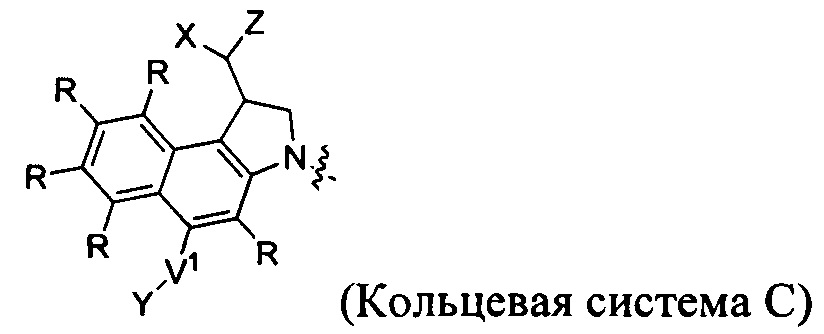
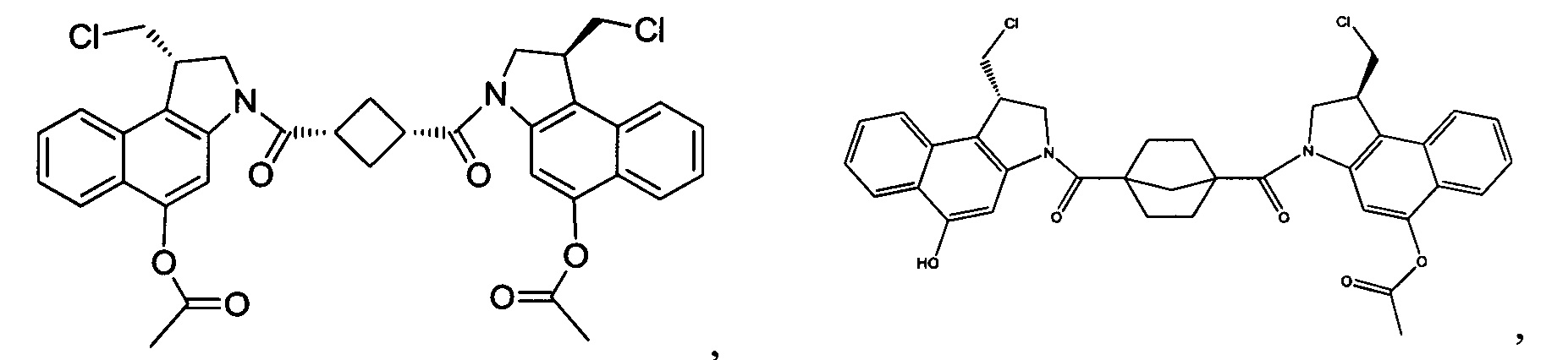
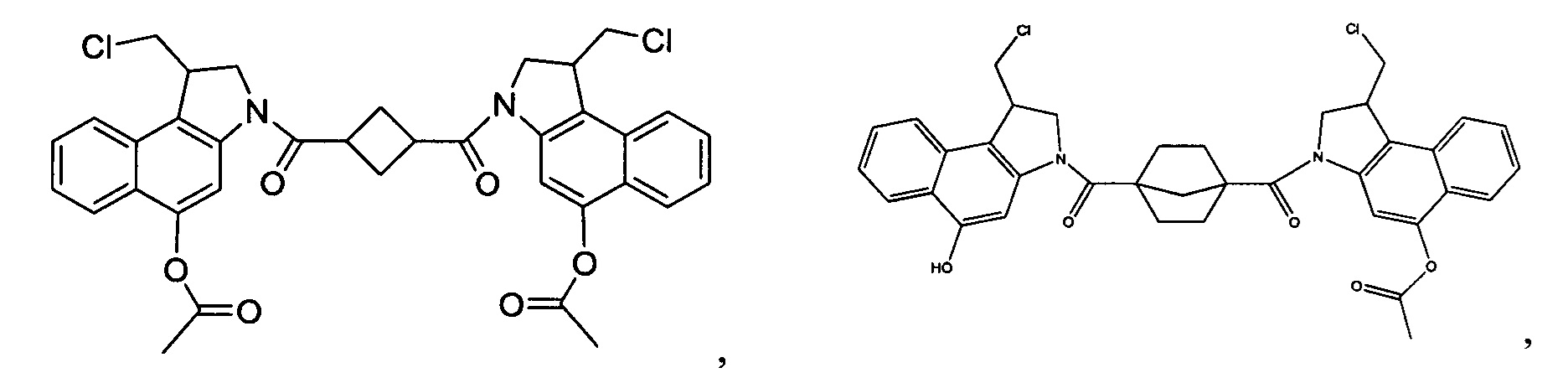
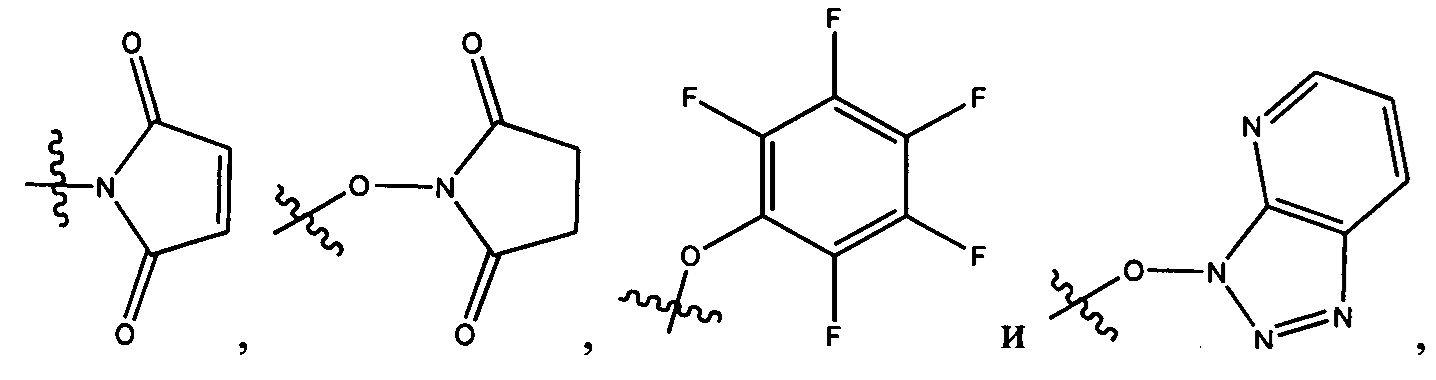
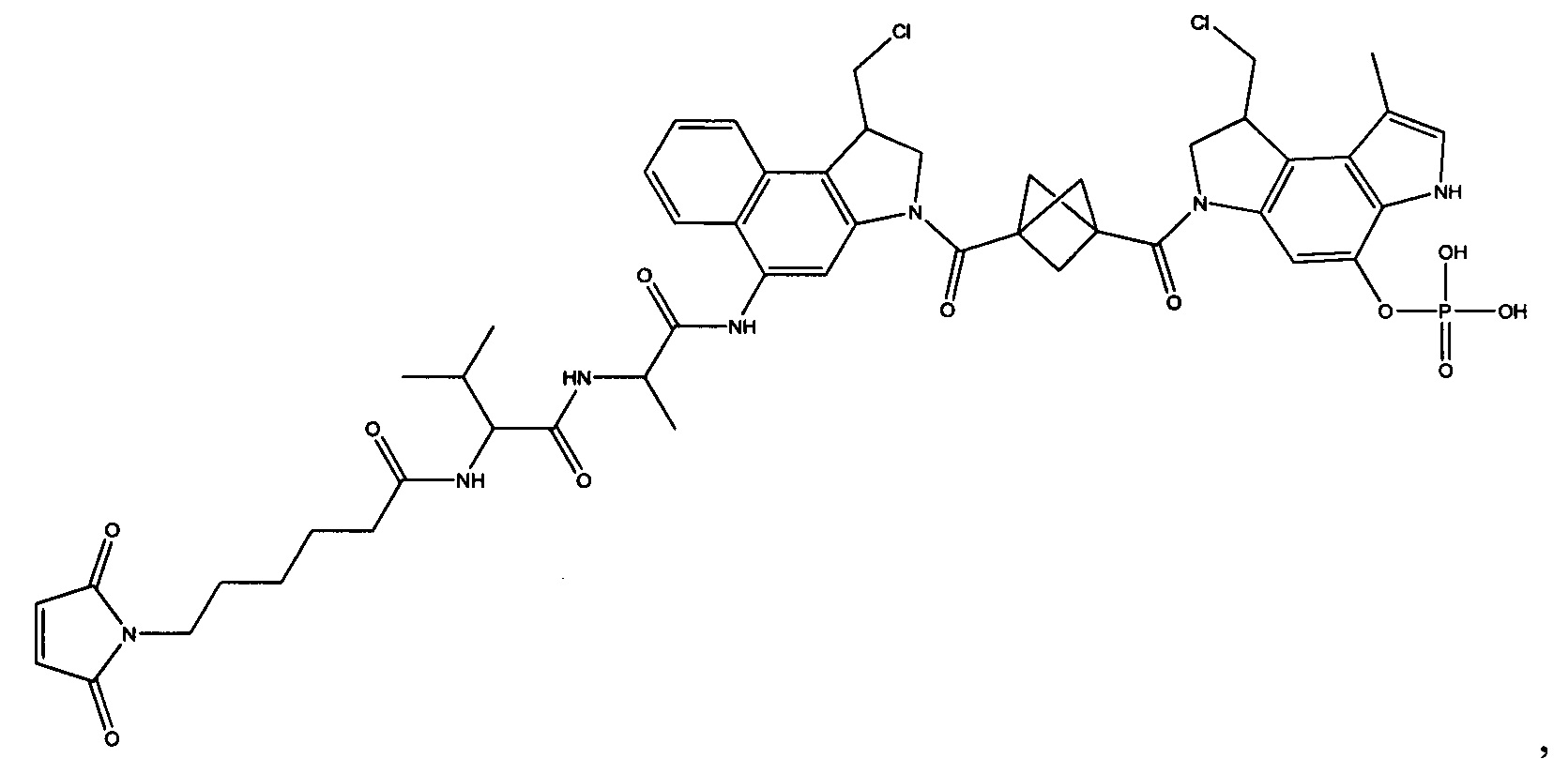
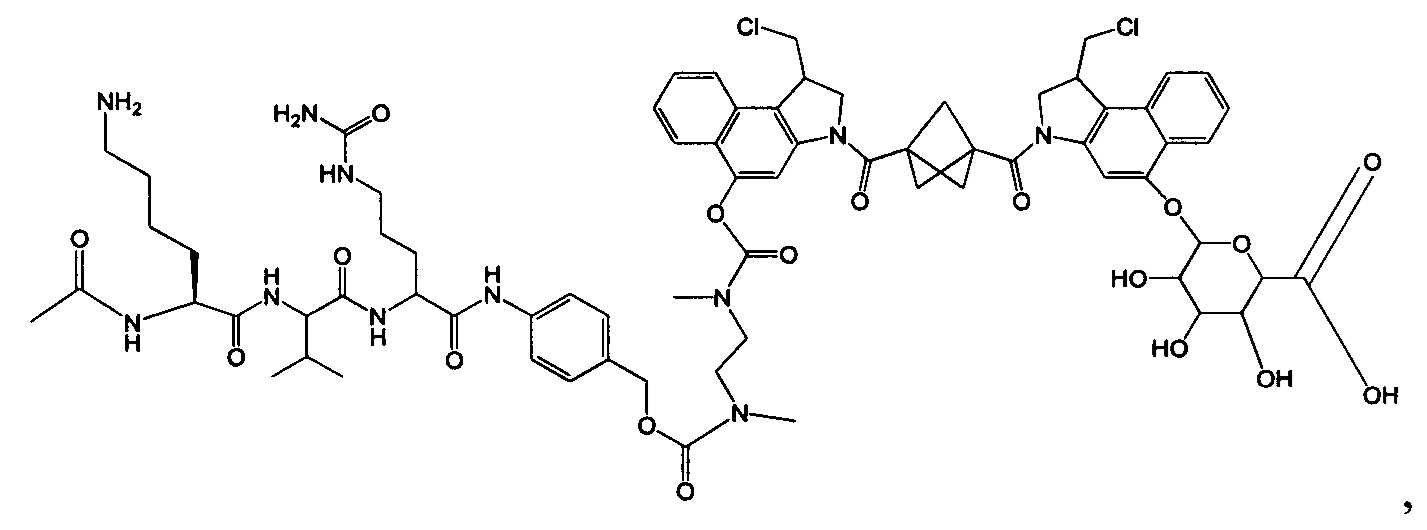
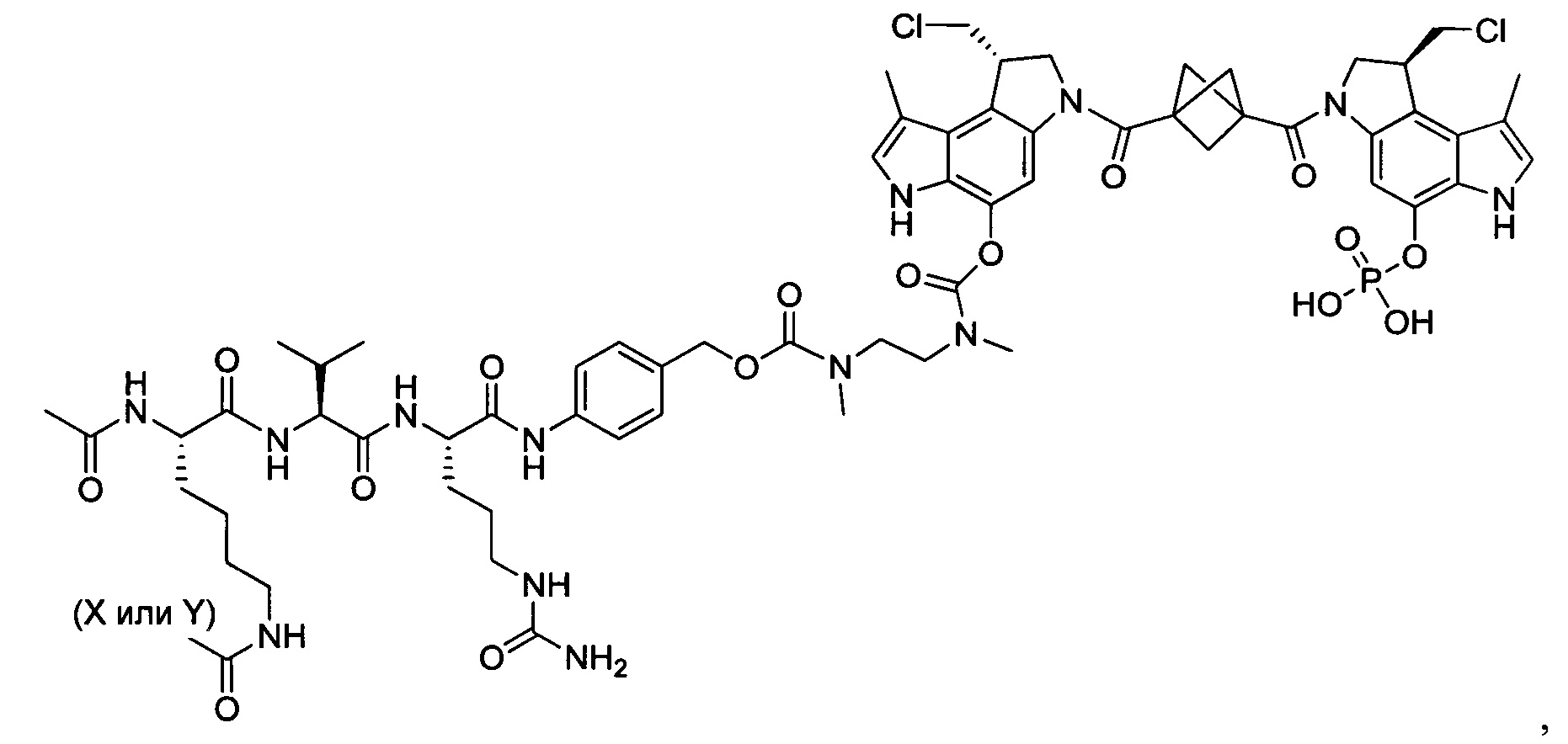
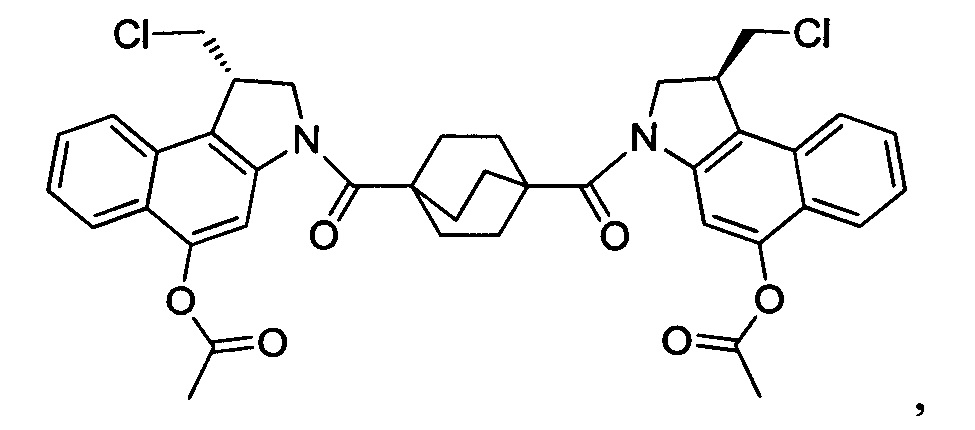
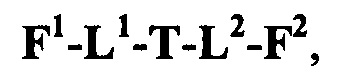
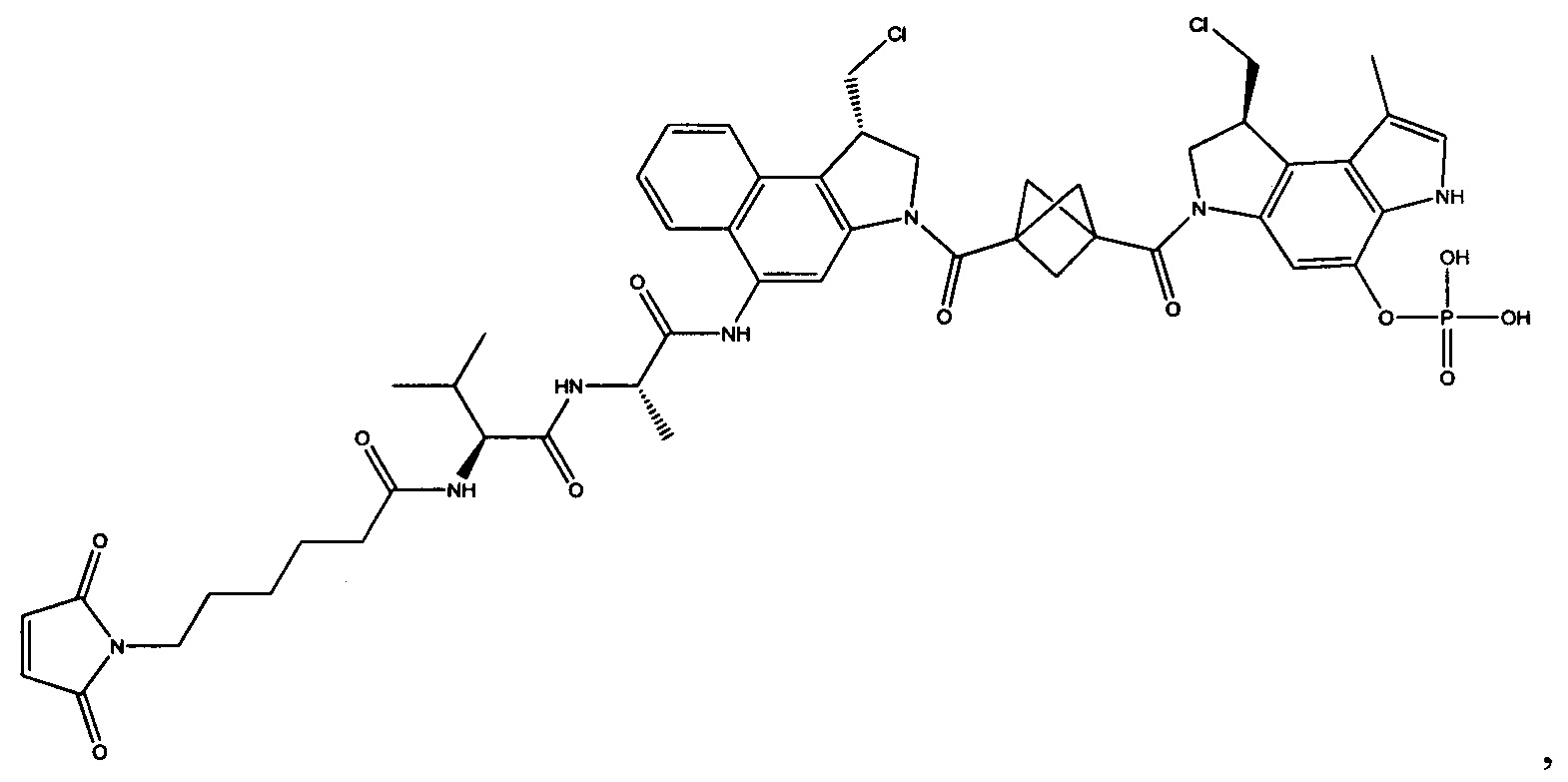
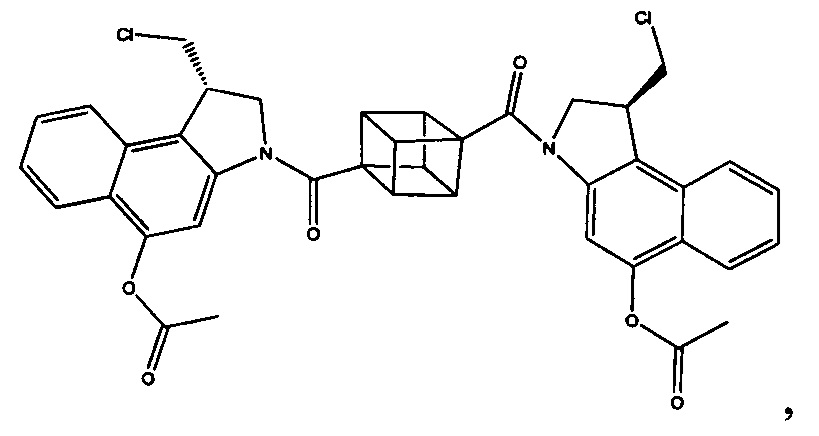
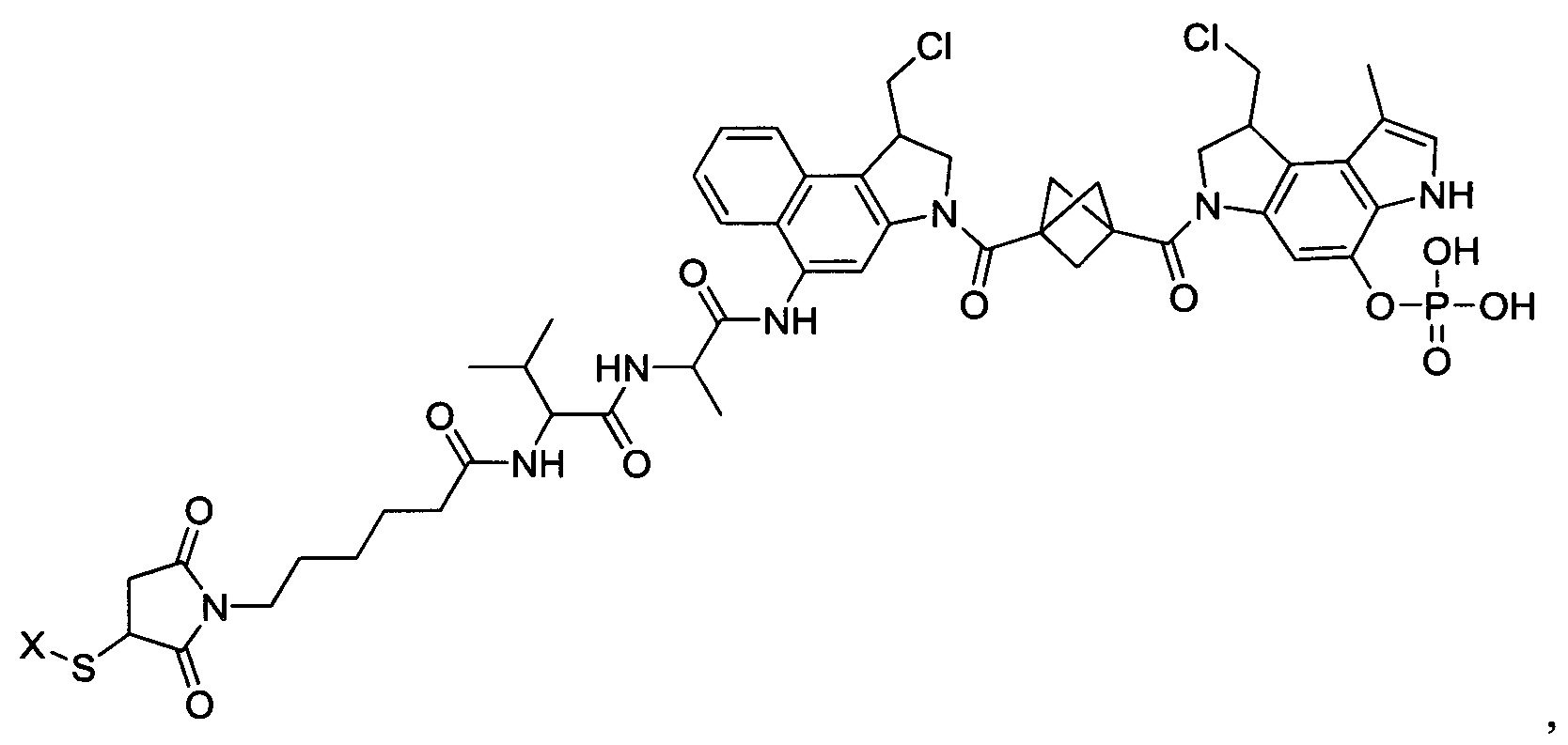
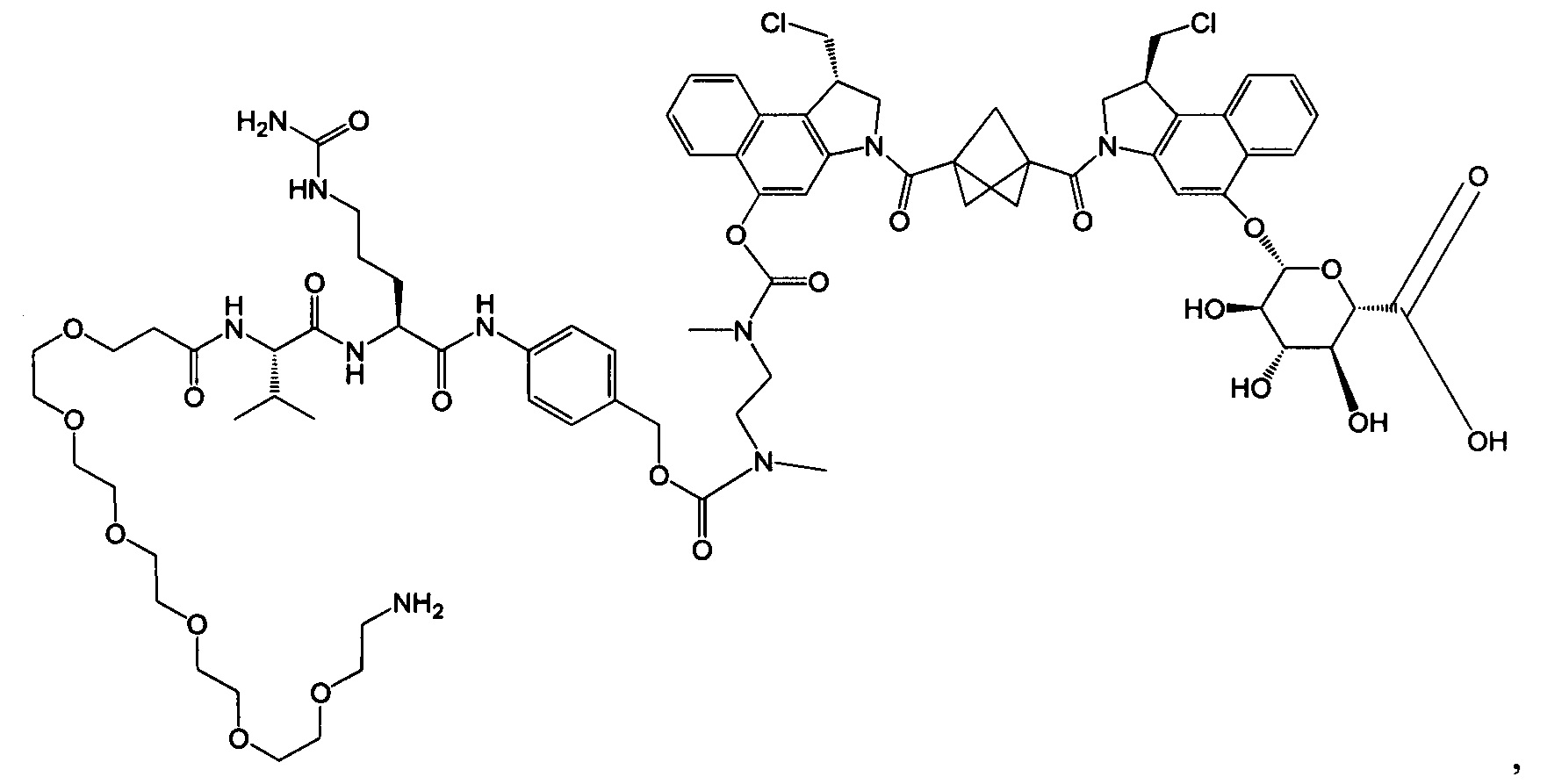
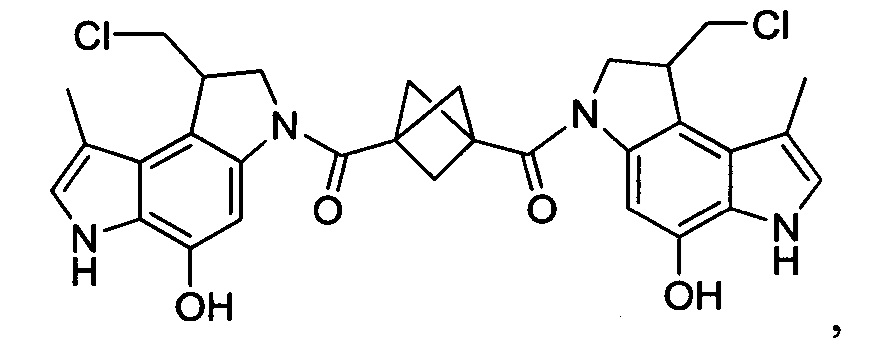
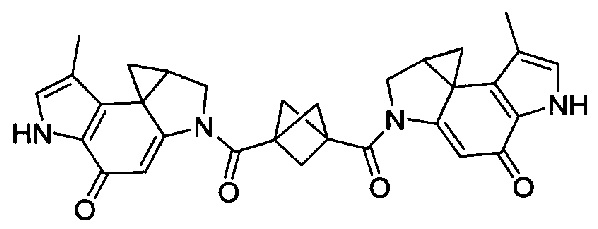
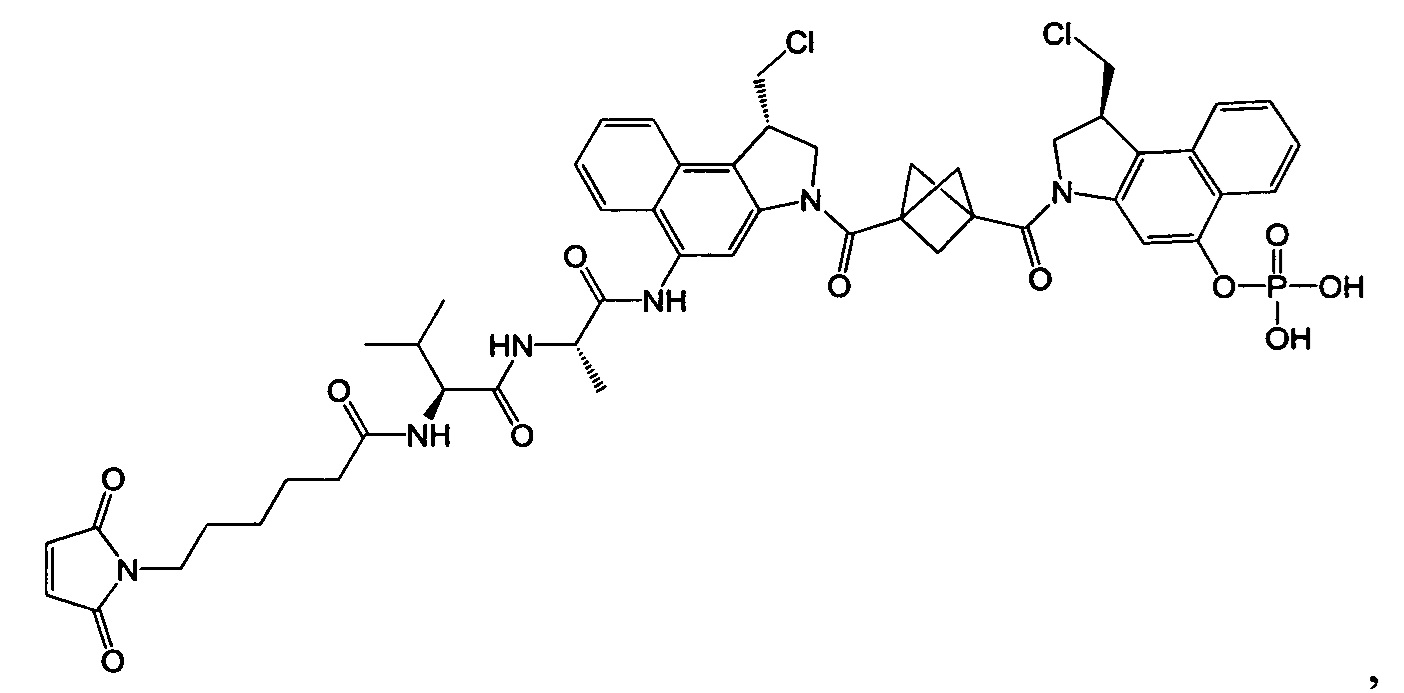
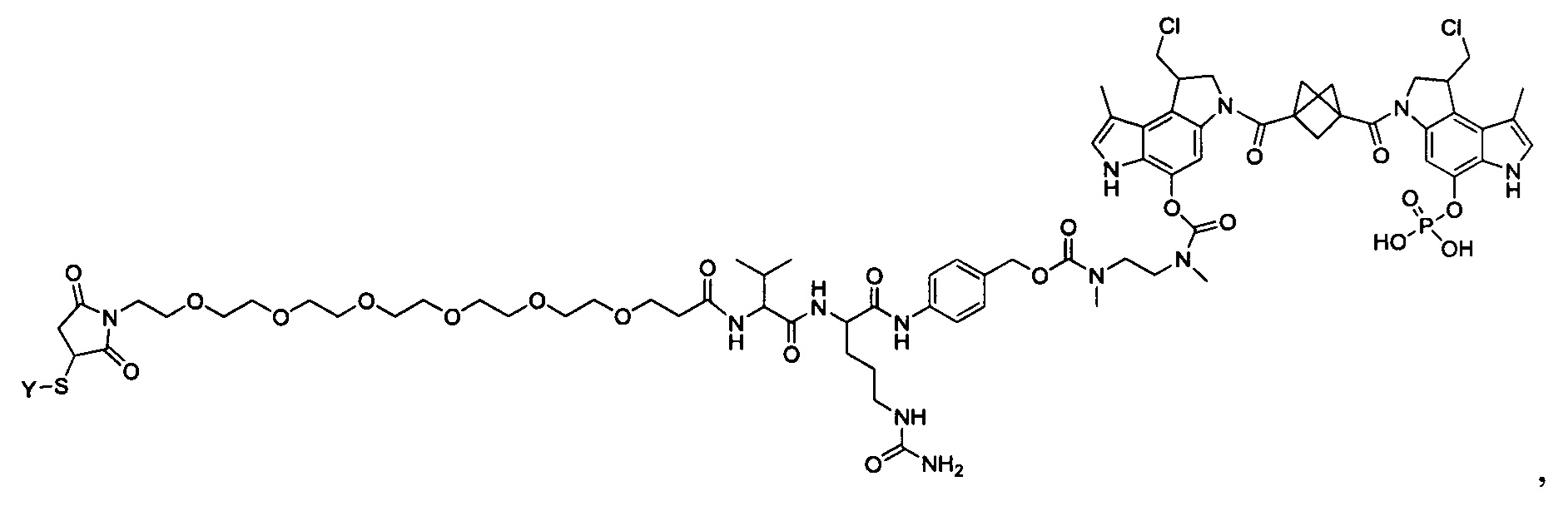
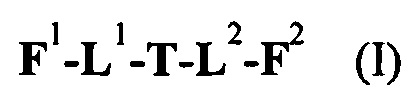
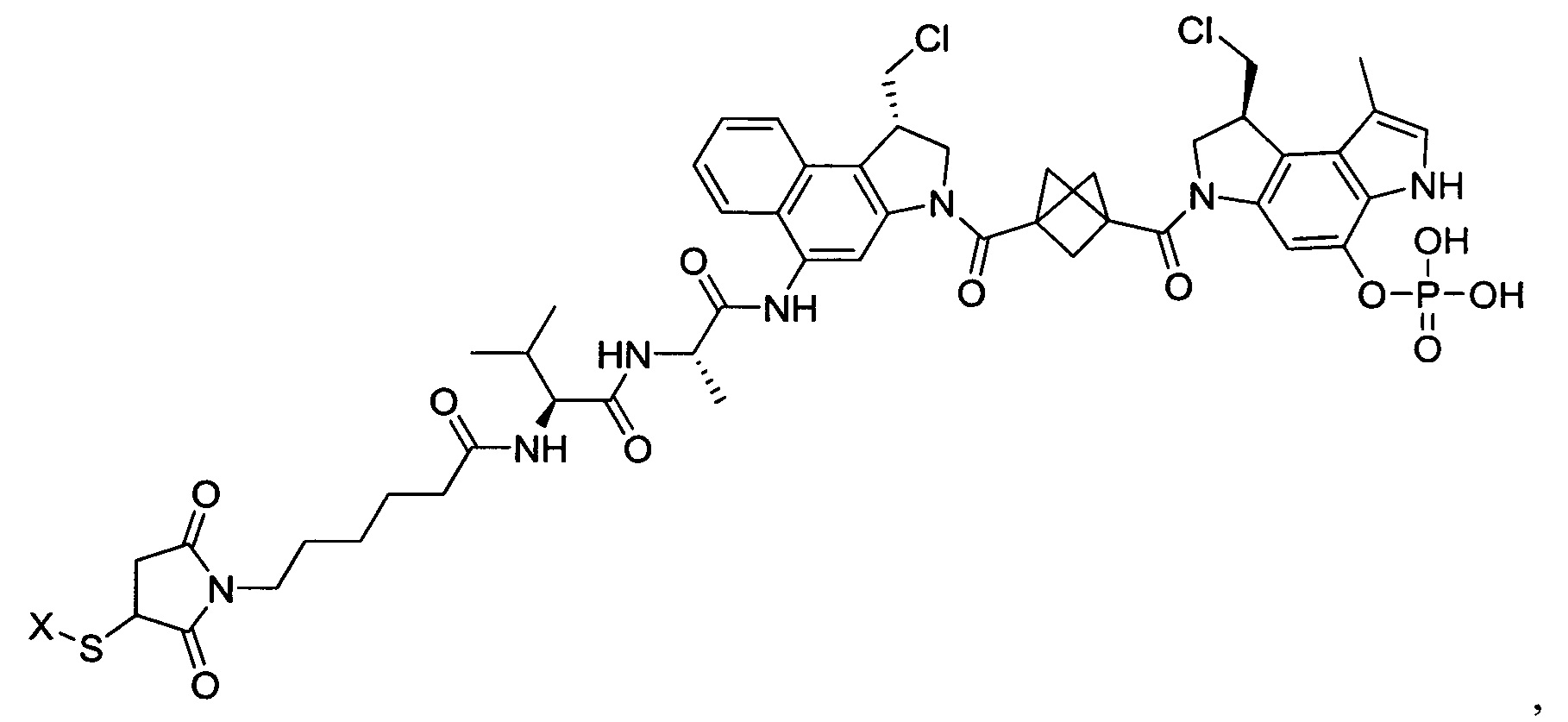
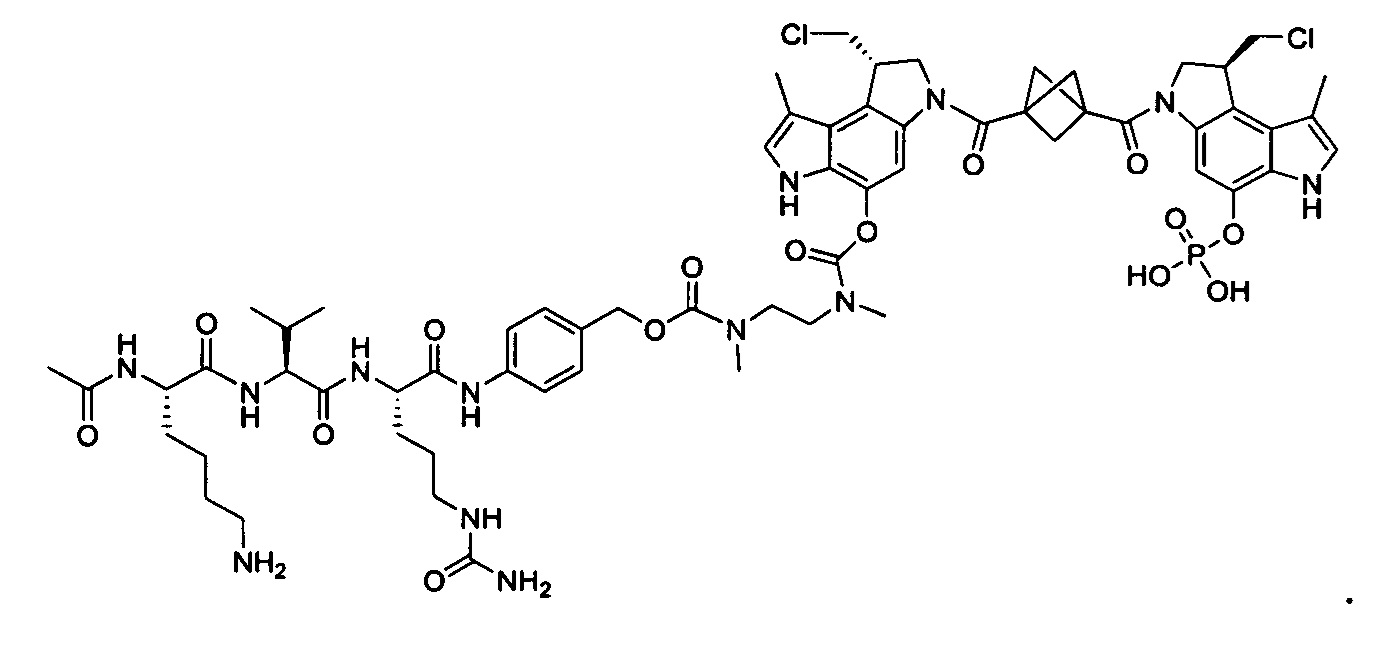
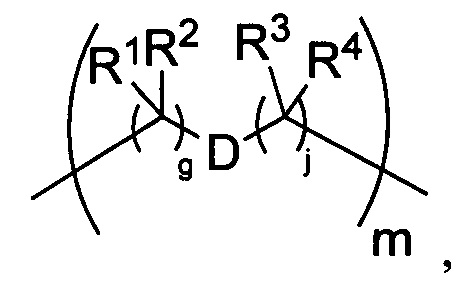
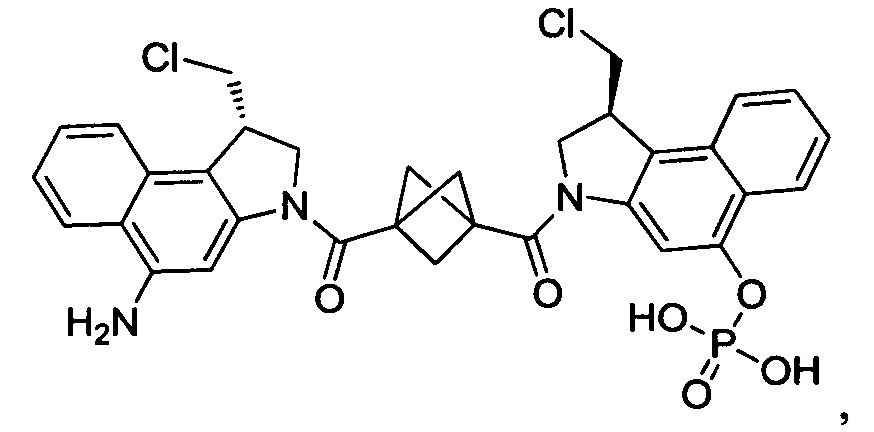
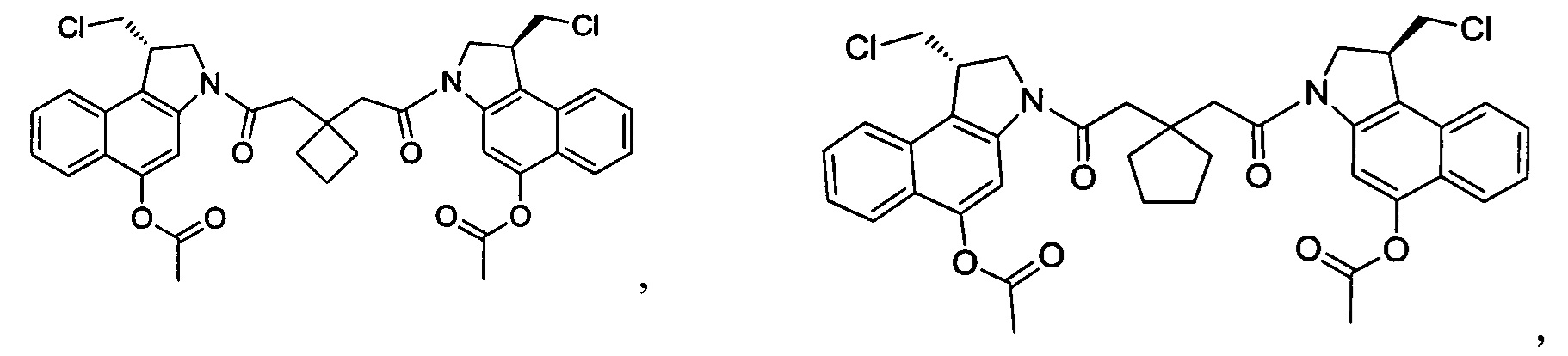
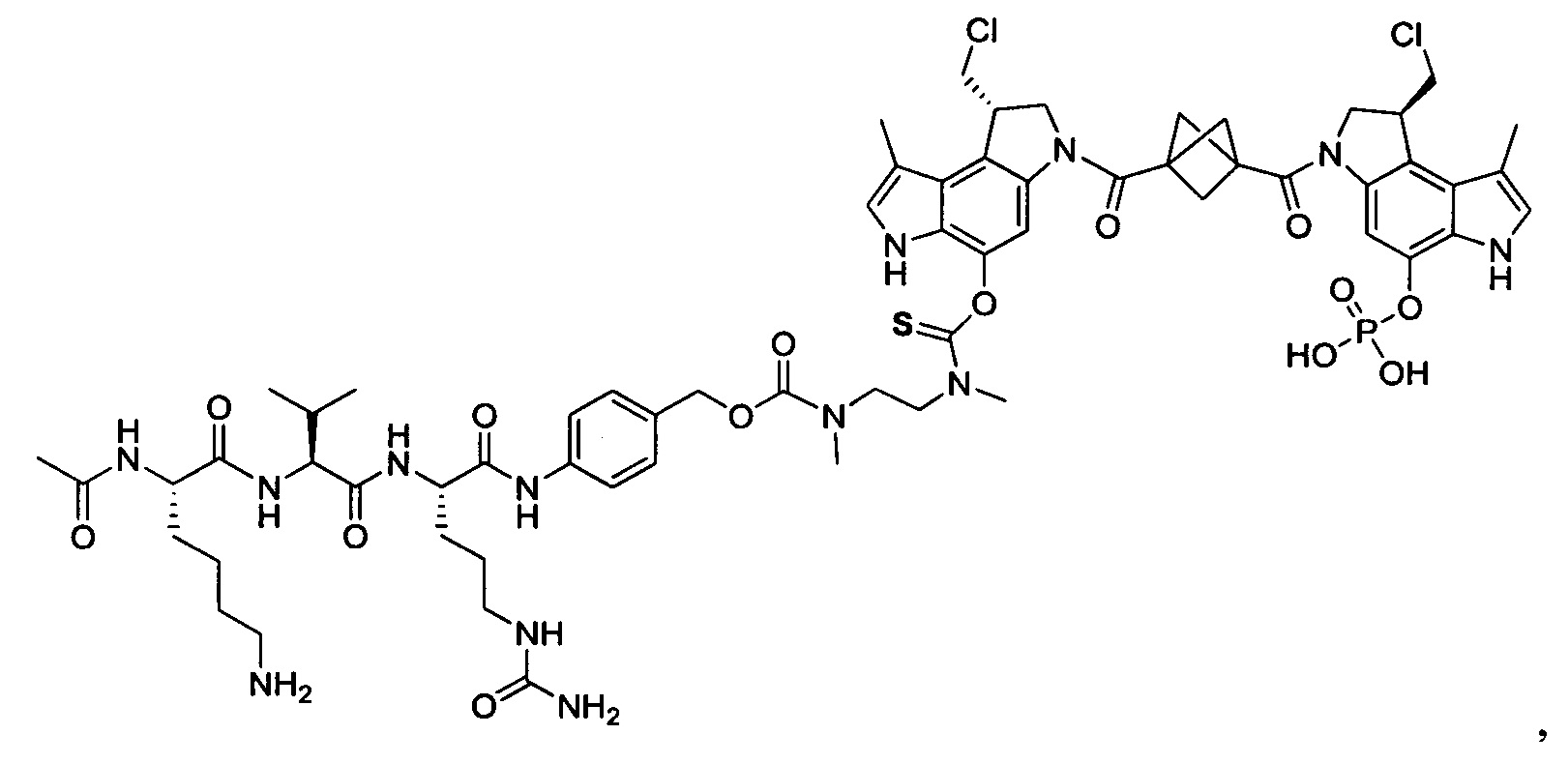
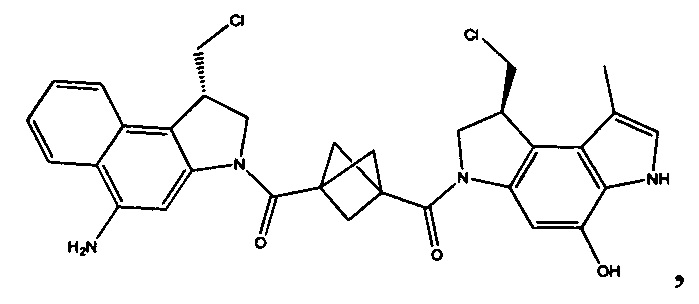
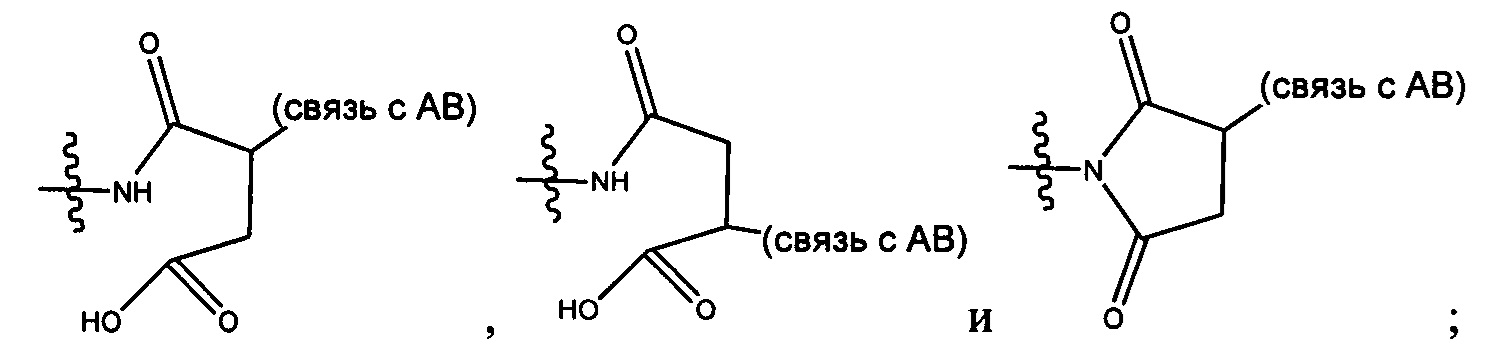
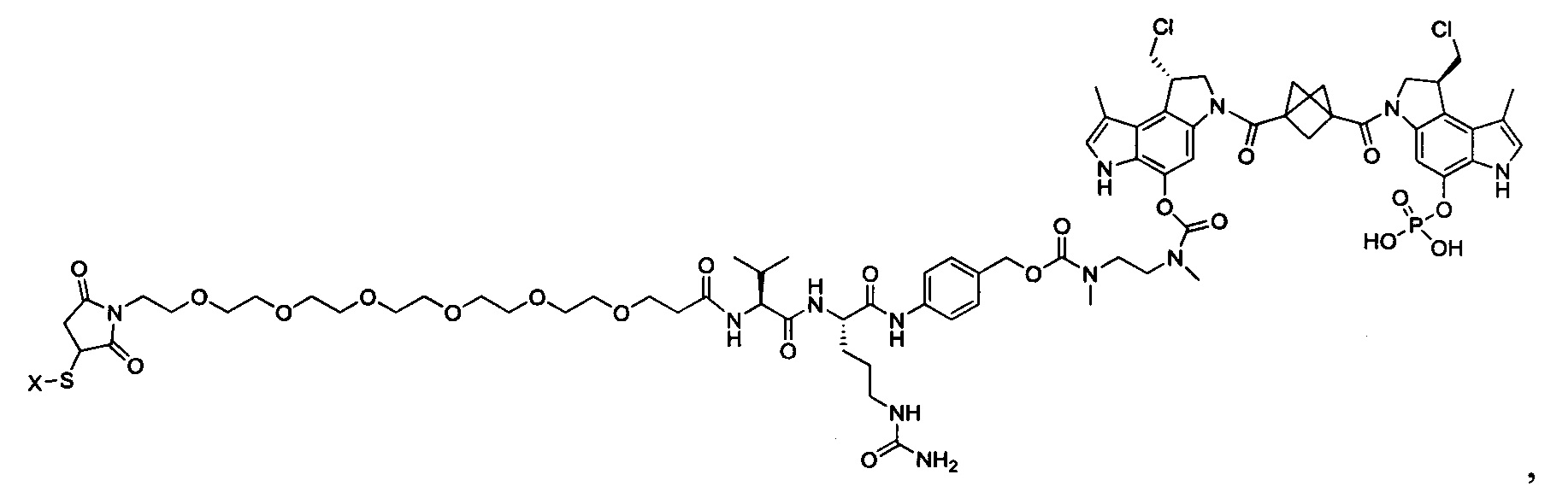
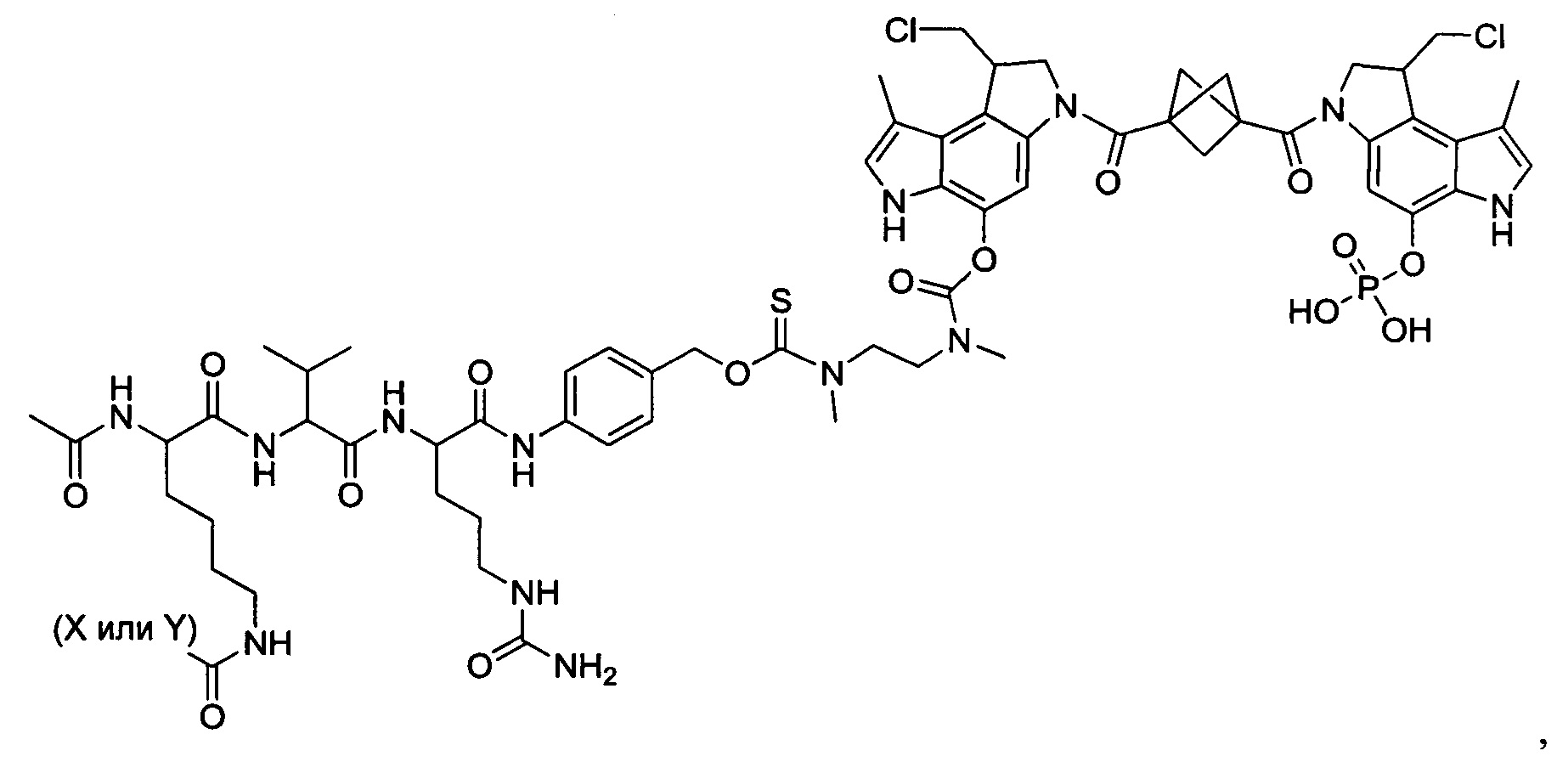
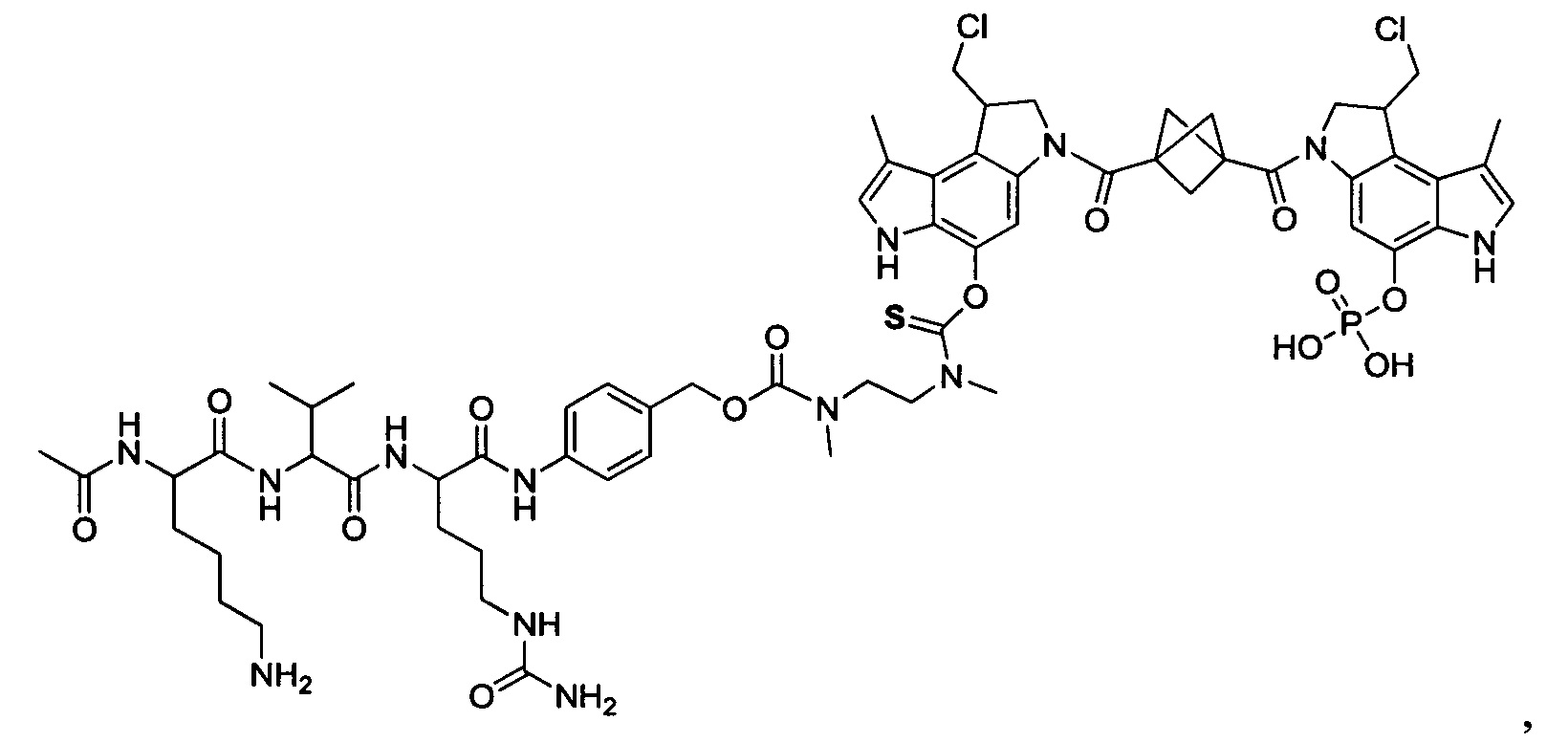
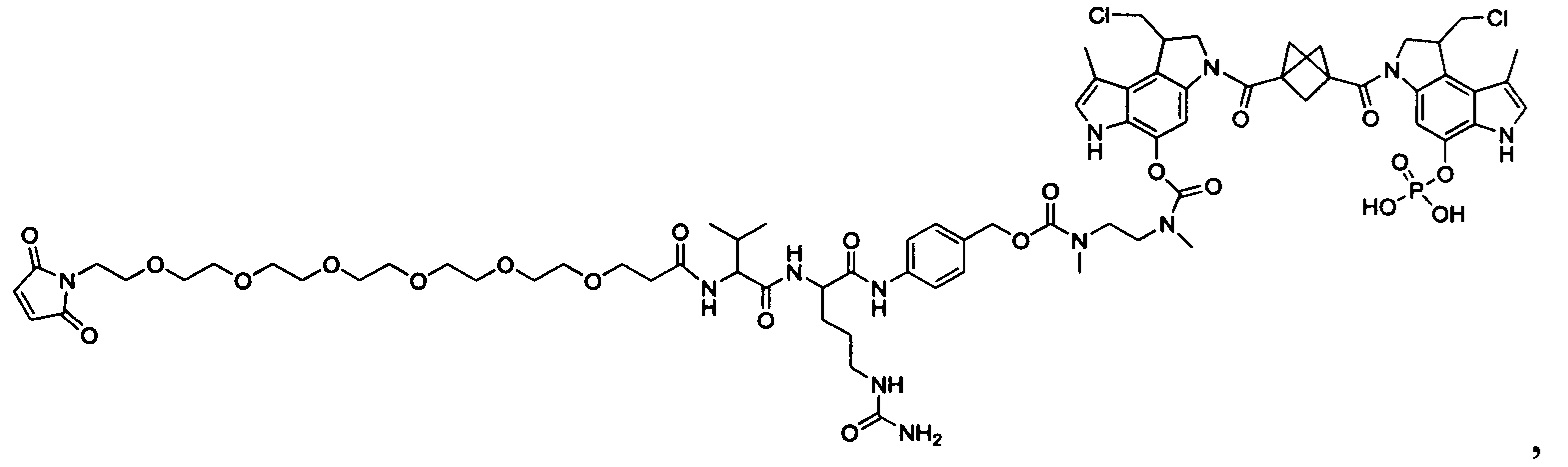
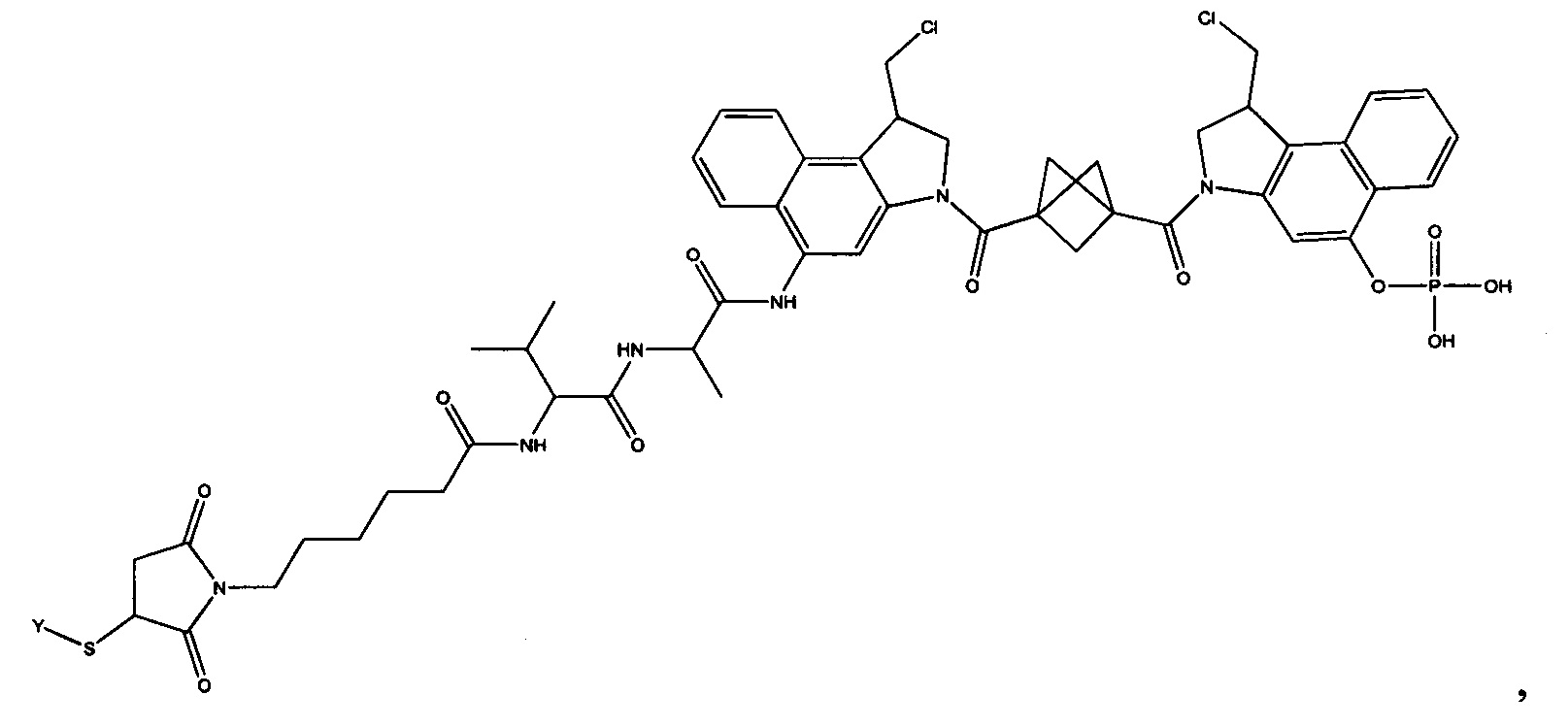
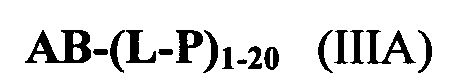
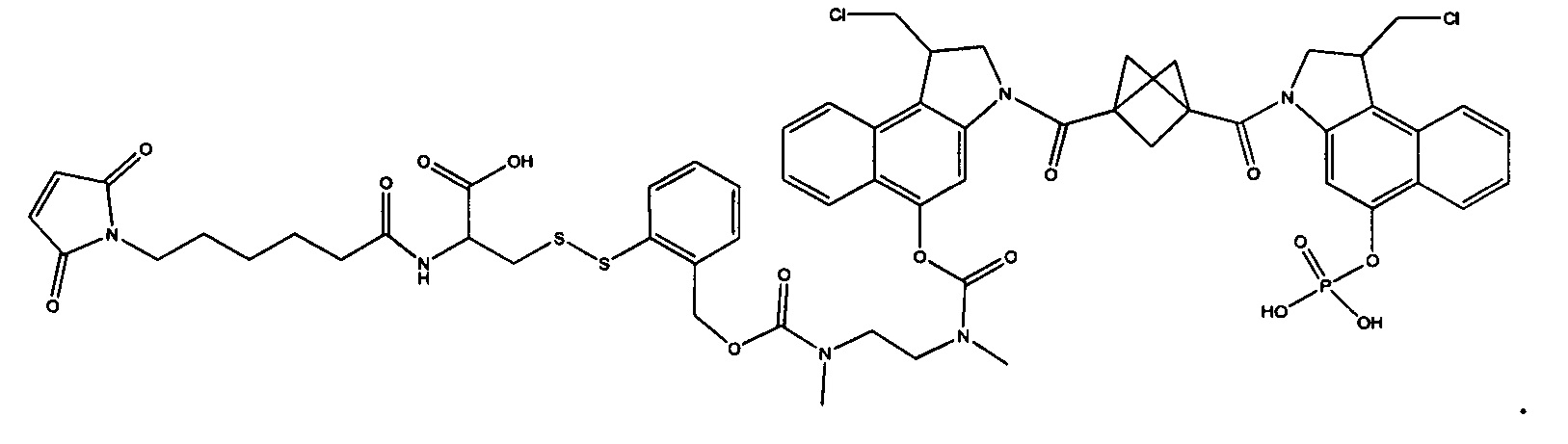
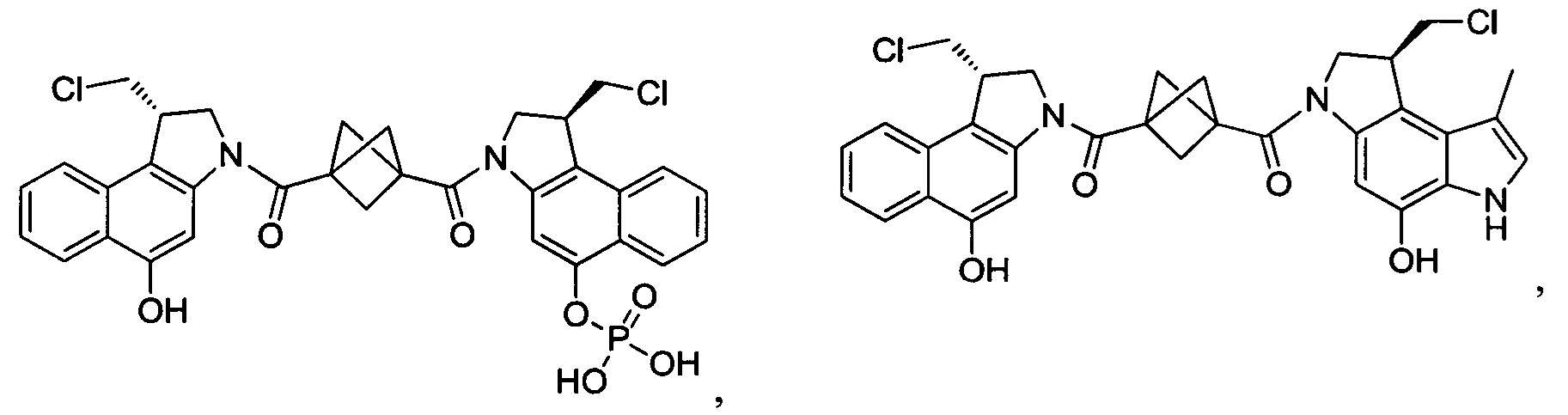
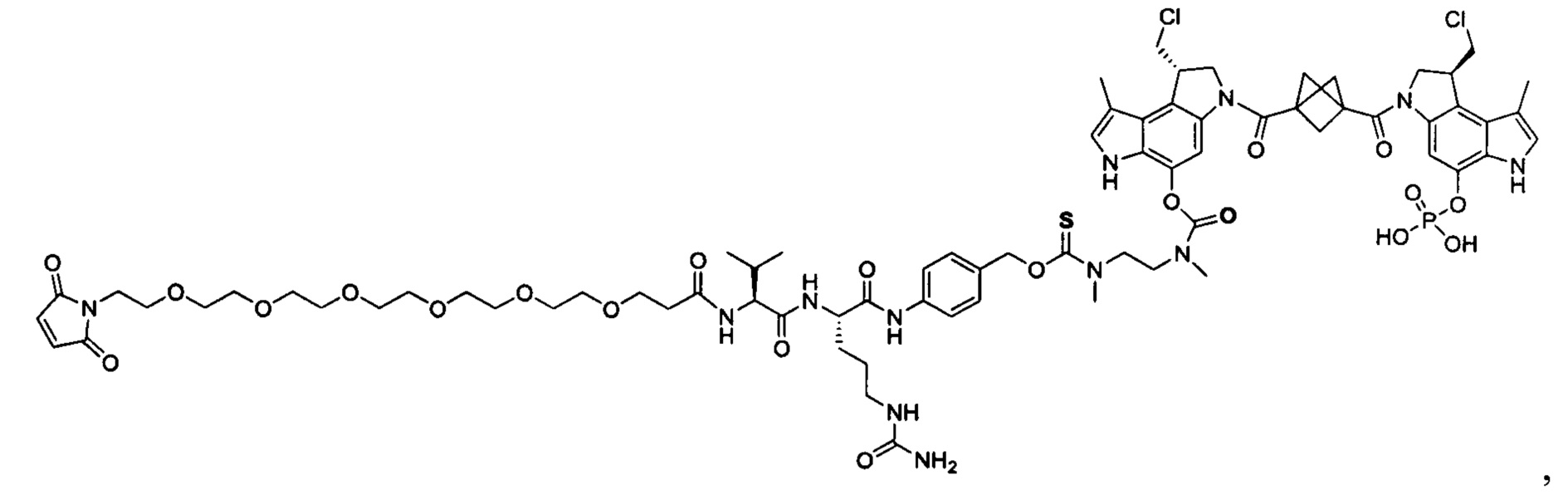
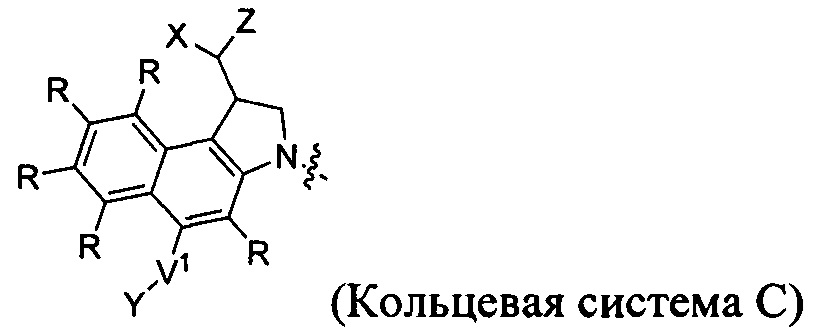
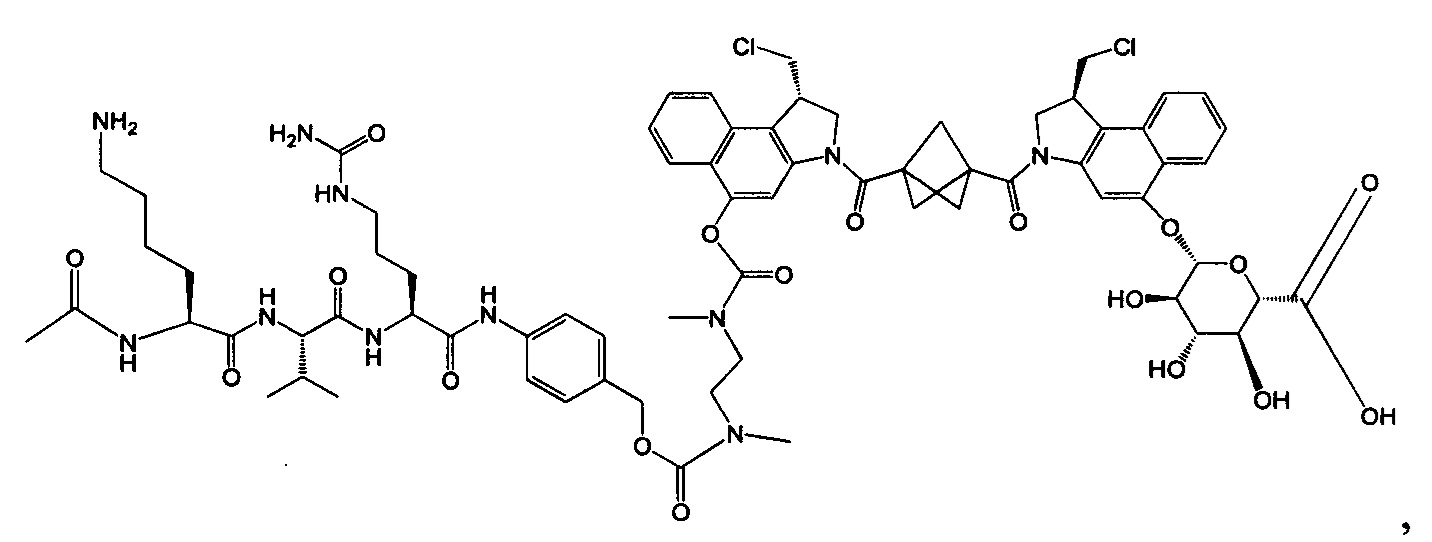
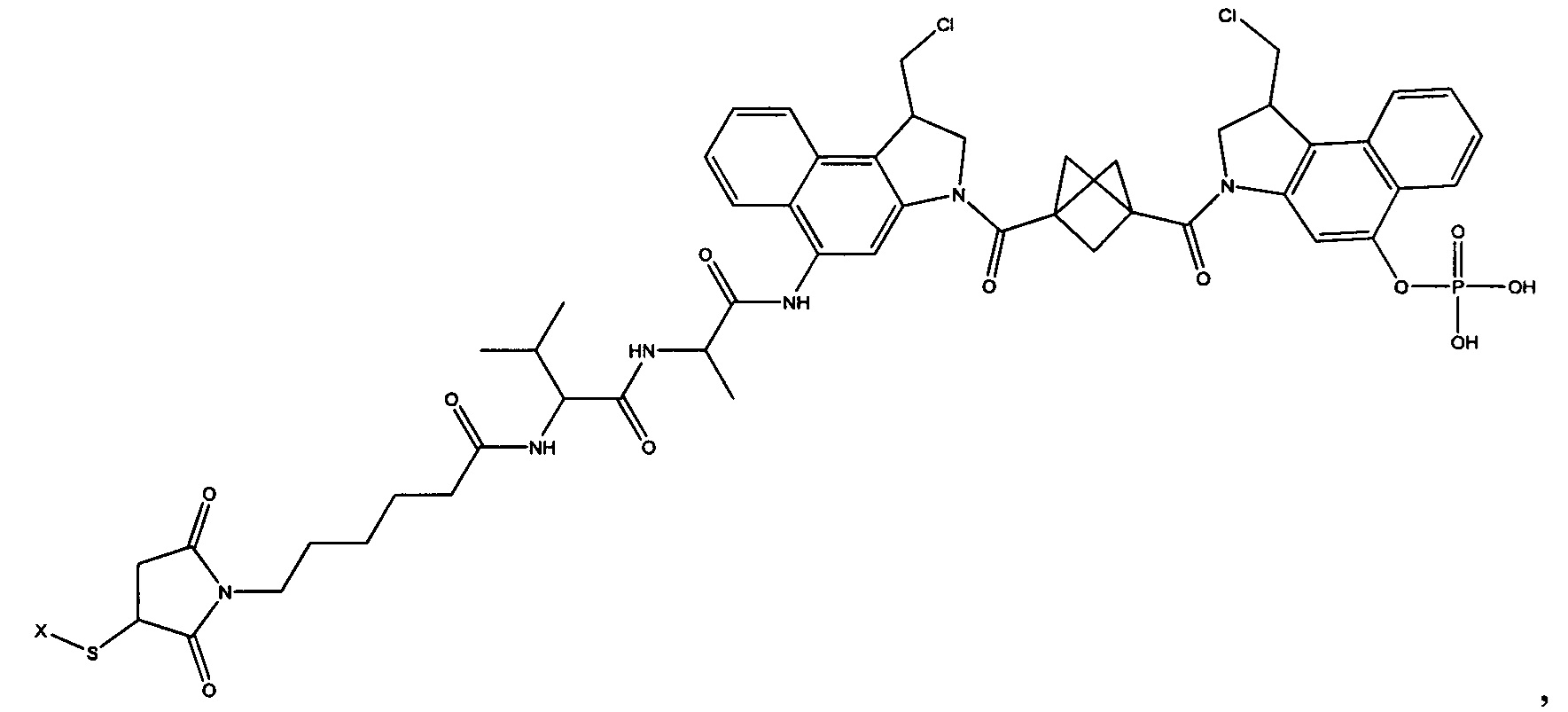
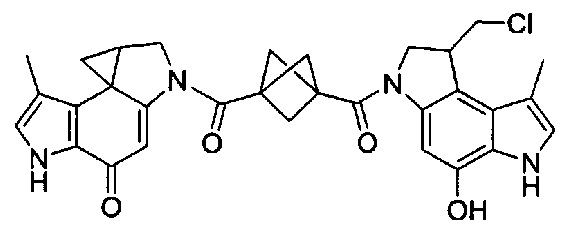
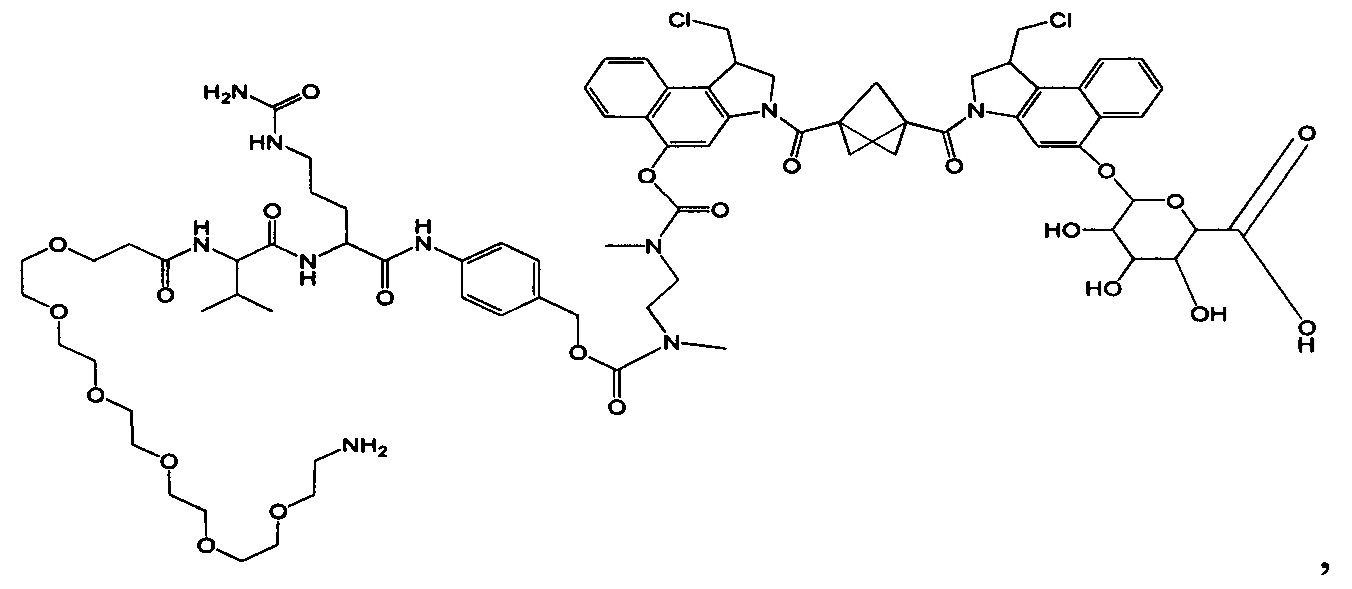
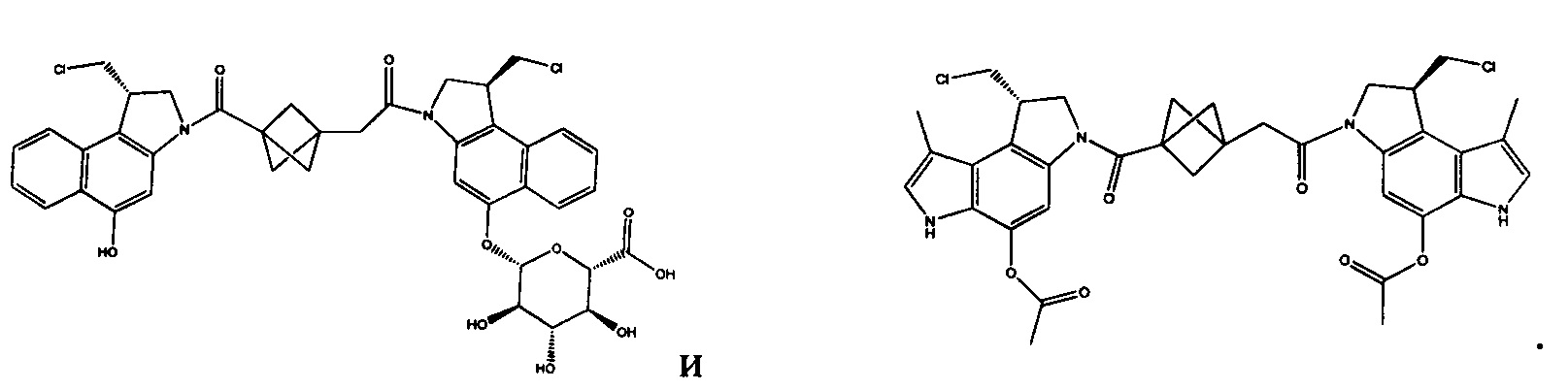
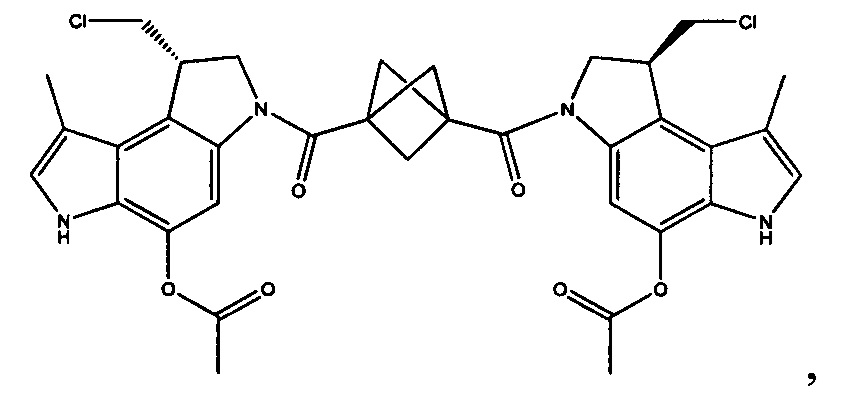
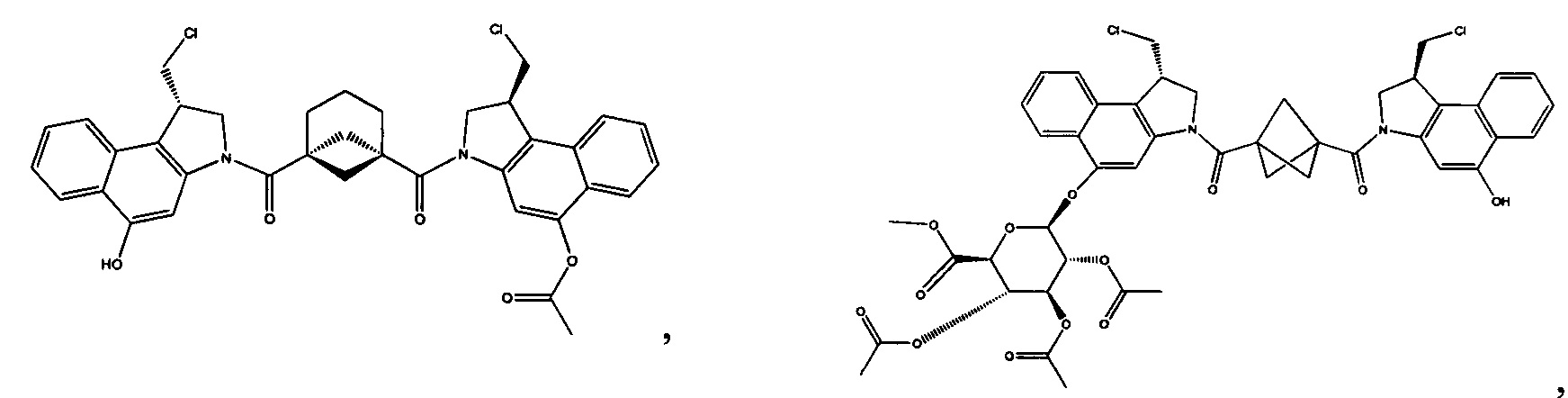
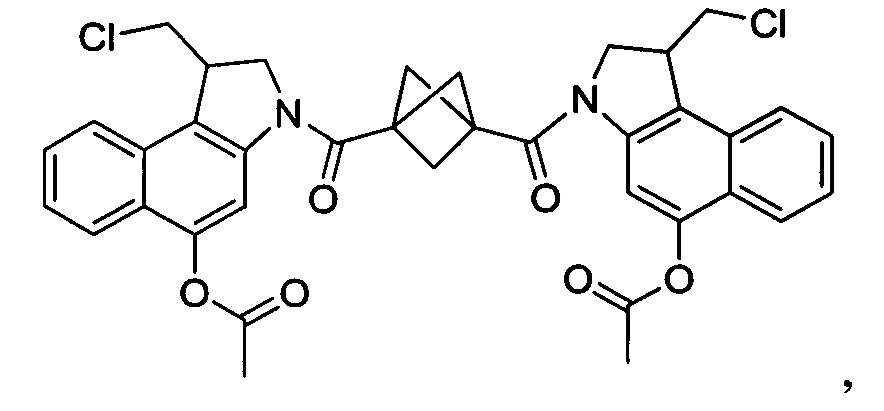
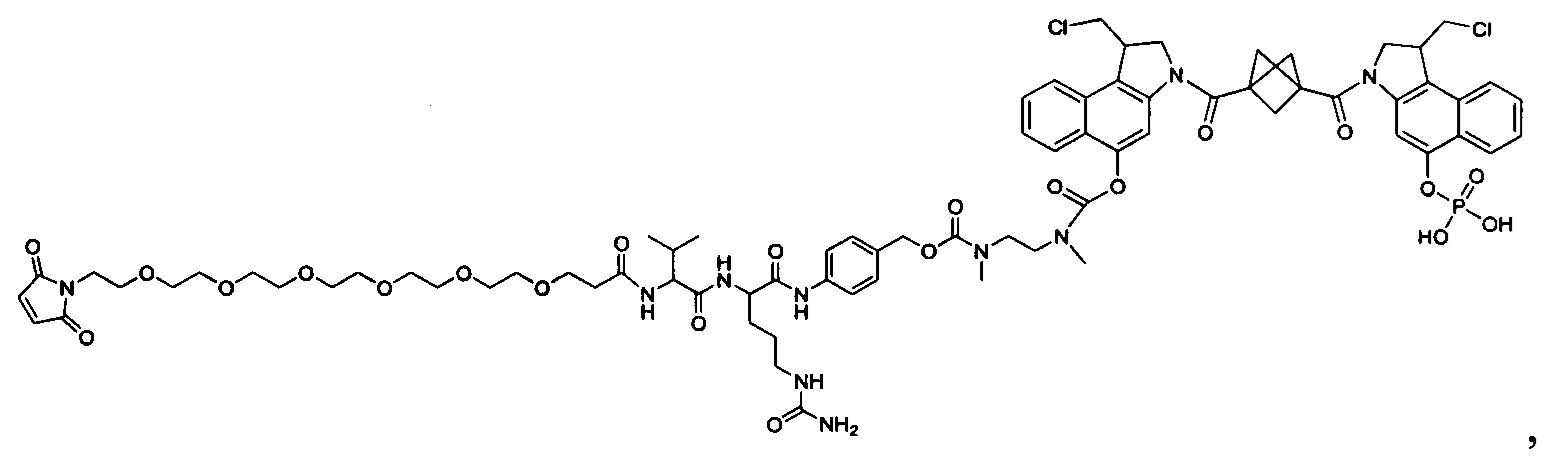
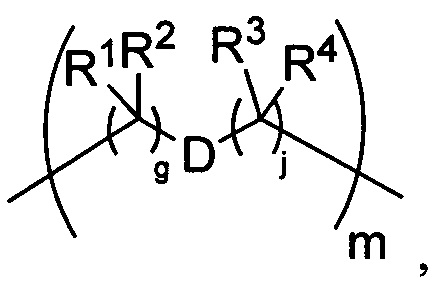
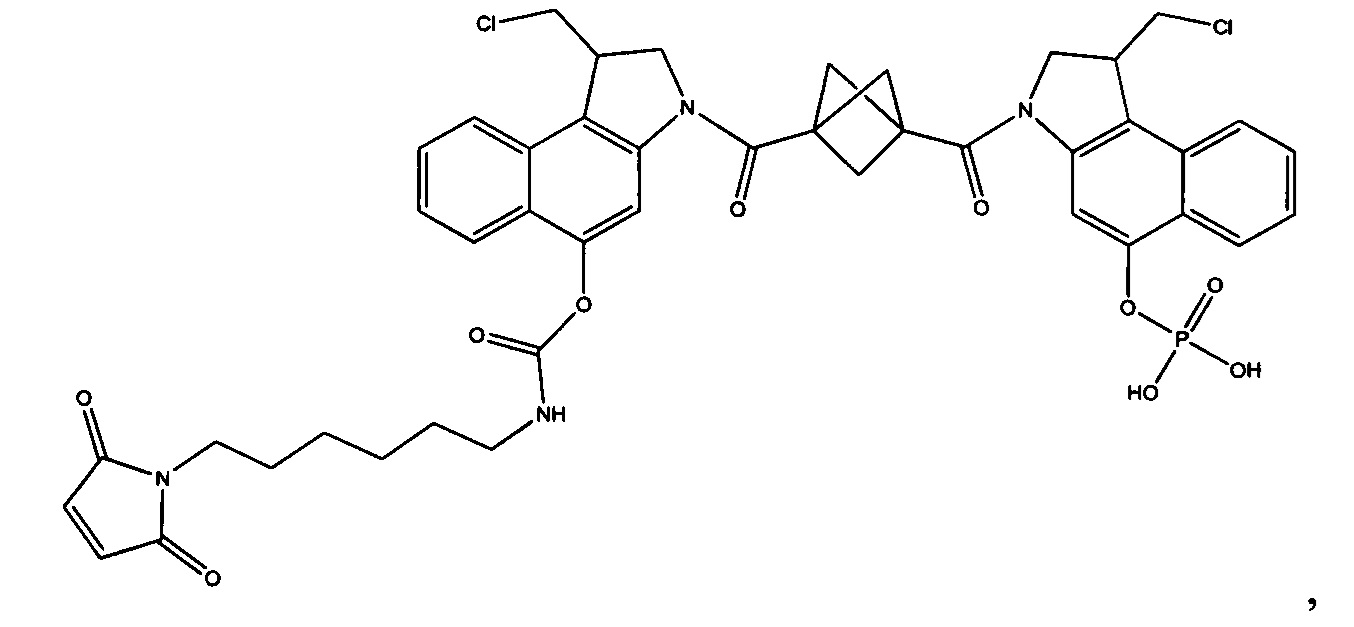
Комбинированная терапия туберкулеза
Новые адъювантные композиции
Стабильный жидкий препарат антитела
Производные 2,3-дигидро-1н-инден-1-ил-2,7-диазаспиро[3.5]нонана и их применение в качестве антагонистов или обратных агонистов грелинового рецептора
Вирус диареи крупного рогатого скота с модифицированным белком e
N1/n2-лактамные ингибиторы ацетил-коа-карбоксилаз
Блокирующие антитела против dkk-1 и их применения
Молекулы, связывающиеся с 4-1вв
Антитела с рн-зависимым связыванием антигена
Лечение антителами против pcsk9
Цитотоксические пептиды и их конъюгаты антитело-лекарственное средство
Аналоги сплицеостатина
Новые 3-индол замещенные производные, фармацевтические композиции и способы применения
Бифункциональные цитотоксические агенты, содержащие cti фармакофор

