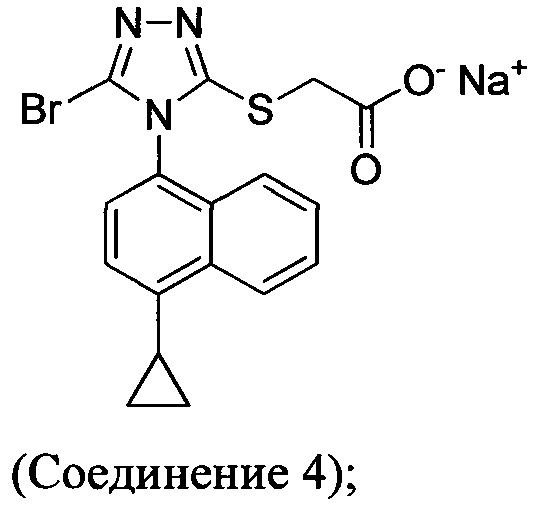
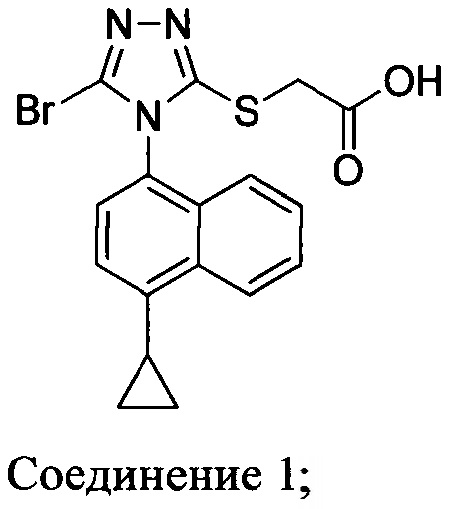
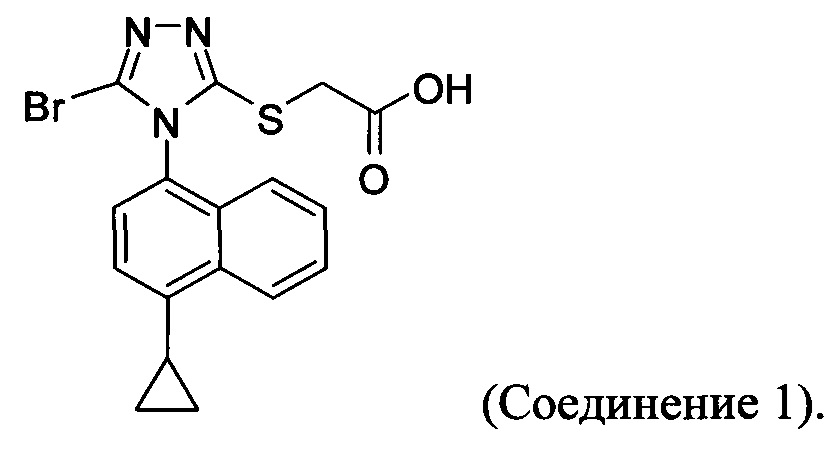
ПОЛУЧЕНИЕ 2-(5-БРОМ-4-(4-ЦИКЛОПРОПИЛНАФТАЛИН-1-ИЛ)-4H-1,2,4-ТРИАЗОЛ-3-ИЛТИО)УКСУСНОЙ КИСЛОТЫ
Вид РИД
Изобретение
Сведения о предшествующем уровне техники
Мочевая кислота образуется в результате окисления ксантина. Нарушения метаболизма мочевой кислоты включают, но без ограничения, полицитемию, миелоидную метаплазию, подагру, рецидивные приступы подагры, подагрический артрит, гиперурикемию, гипертензию, сердечно-сосудистое заболевание, ишемическую болезнь сердца, синдром Леша-Найхана, синдром Келли-Сигмиллера, заболевание почек, камни в почках, почечную недостаточность, воспаление суставов, артрит, уролитиаз, сатурнизм, гиперпаратиреоз, псориаз или саркоидоз.
Краткое описание изобретения
Для получения химических соединений в крупном масштабе на опытной установке часто требуются эффективные методы синтеза. В настоящем документе предлагаются определенные масштабируемые способы и способы для синтеза соединений Формулы (I):
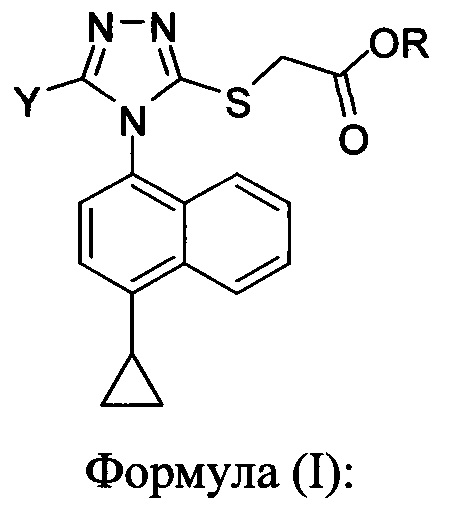
в которой
R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил, -C3-C10 циклоалкенил, или R представляет собой противоион; и
Y представляет собой H, OH, NH2, F, Cl, Br или I.
Соединения Формулы (I), описанные здесь, являются полезными для лечения нарушений обмена мочевой кислоты.
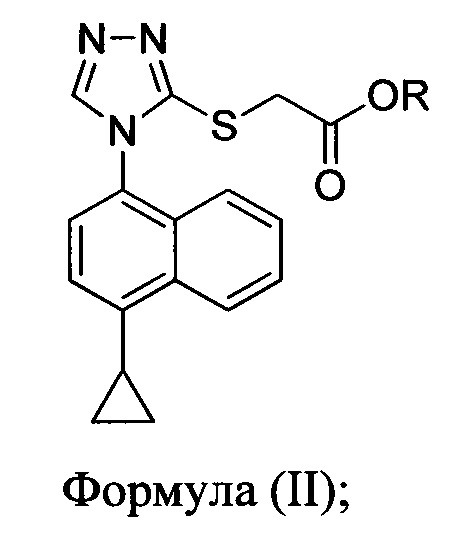
В одном аспекте в настоящем документе предлагается способ (Способ 1) для получения:
включающий приведение в контакт соединения Формулы (II) или его соли,
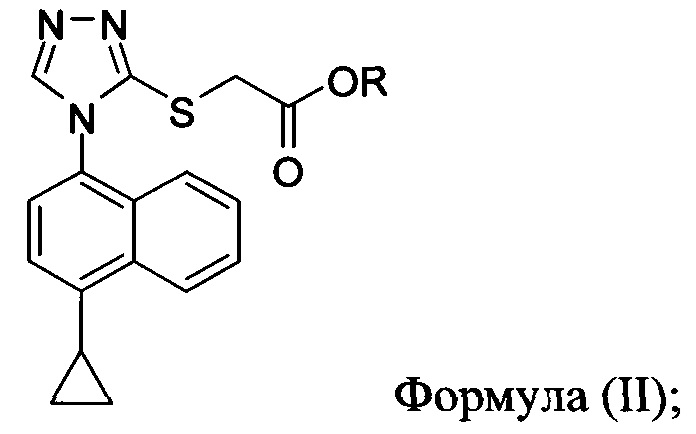
в которой R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил, с N-бромсукцинимидом (NBS) и растворителем для обеспечения соединения структуры:
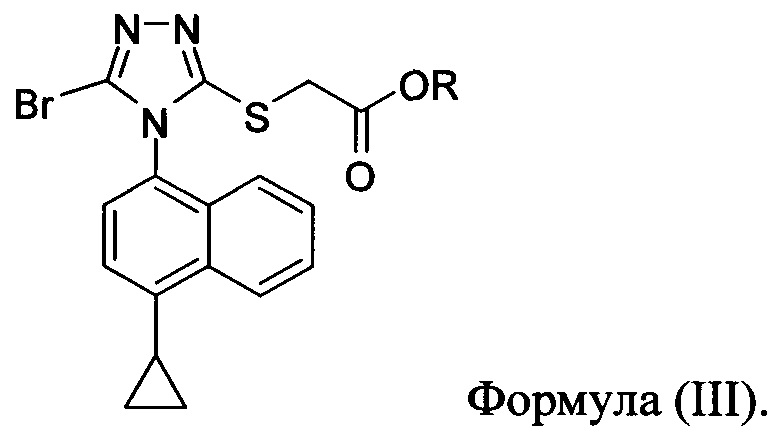
В некоторых вариантах осуществления описанного выше способа R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил. В некоторых вариантах осуществления описанного выше способа R представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изоамил, пентил, гексил, гептил, октил, нонил, терпенил, борнил, аллил, линалил или геранил.
В определенных вариантах осуществления описанного выше способа R представляет собой метил или этил. В некоторых конкретных вариантах осуществления описанного выше способа R представляет собой метил.
В некоторых вариантах осуществления описанного выше способа реакционную смесь перемешивают в течение, по меньшей мере, 12 часов при температуре между примерно комнатной температурой и примерно 32°C.
В некоторых вариантах осуществления описанного выше способа анализ реакционной смеси показывает ≤1,5% площади по данным HPLC соединения Формулы (II).
В некоторых вариантах осуществления описанного выше способа анализ реакционной смеси показывает ≤0,2% площади по данным HPLC соединения Формулы (II).
В одном варианте осуществления описанного выше способа, указанный способ дополнительно включает:
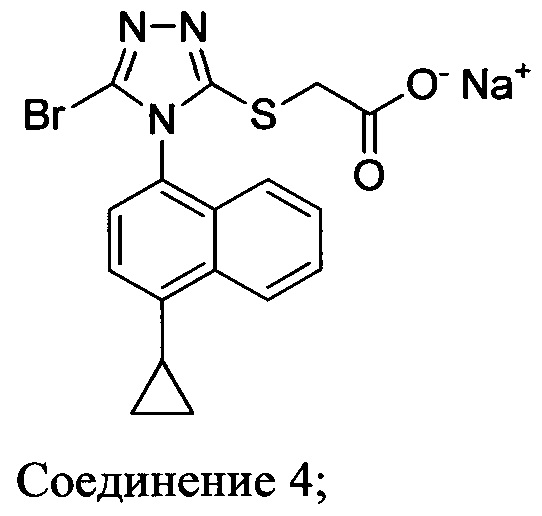
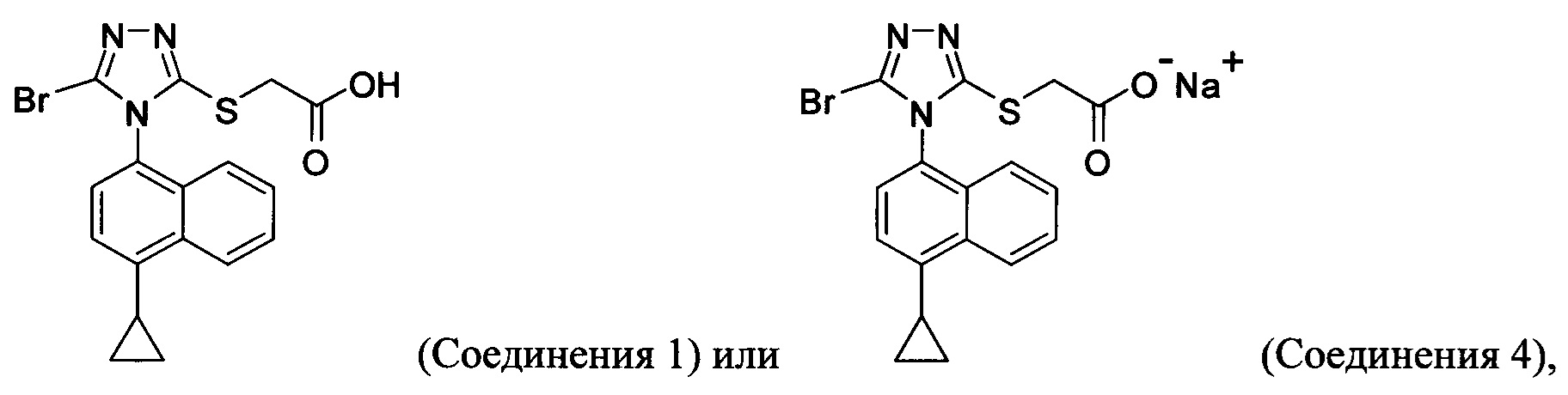
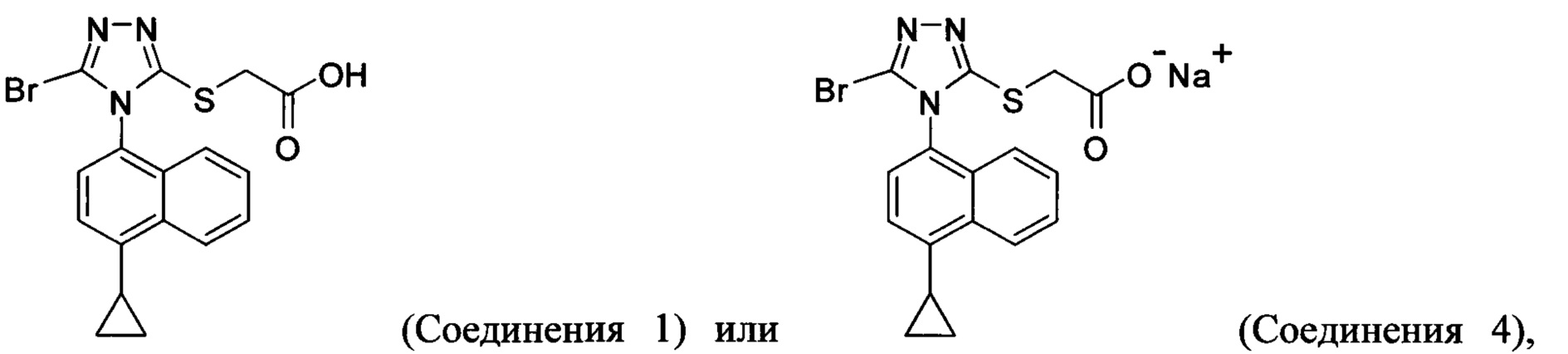
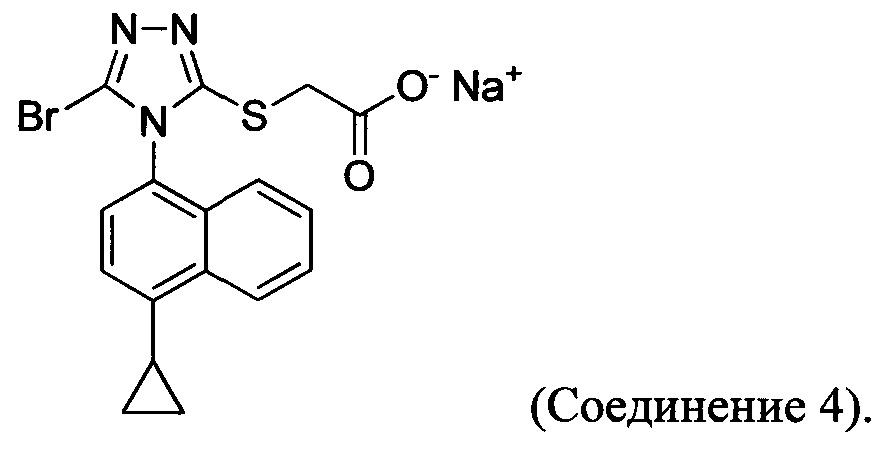
(i) приведение в контакт соединения Формулы (III) с раствором гидроксида натрия для получения Соединения 4; и
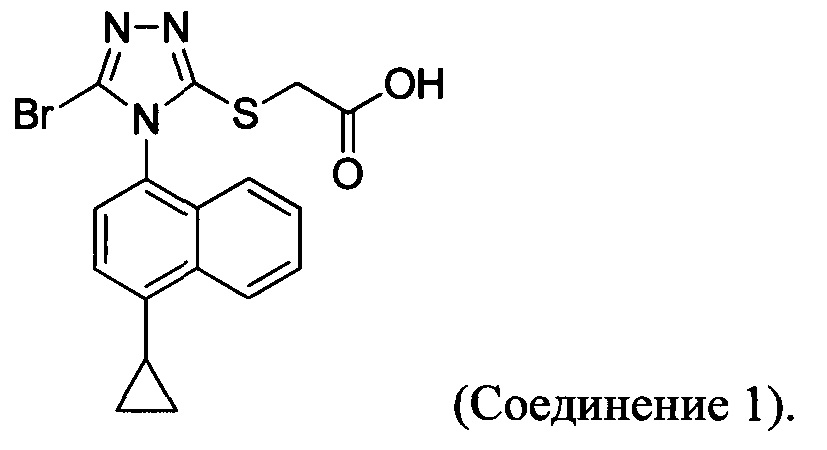
(ii) необязательно приведение в контакт Соединения 4 с кислотой для получения Соединения 1.
В последующих вариантах осуществления описанного выше способа стадия (i) дополнительно включает, необязательно, кристаллизацию Соединения 4 из водного раствора гидроксида натрия.
В некоторых вариантах осуществления описанного выше способа, указанный способ дополнительно включает:
(a) растворение Соединения 4 в воде и добавление в смесь этилацетата;
(b) приведение в контакт бифазной смеси, полученной на стадии (a), с кислотой и отделение органической фазы для получения Соединения 1.
В определенных вариантах осуществления кислота стадии (b) представляет собой соляную кислоту, бромистоводородную кислоту, уксусную кислоту, серную кислоту или фосфорную кислоту. В некоторых конкретных вариантах осуществления кислота стадии (b) представляет собой бромистоводородную кислоту.
В некоторых вариантах осуществления описанного выше способа, указанный способ дополнительно включает перекристаллизацию Соединения 1 из этилацетата. В таком варианте осуществления способ дополнительно включает, необязательно, добавление в смесь н-гептана.
В настоящем документе предлагается Соединение 3, которое может быть получено с помощью описанного выше способа и имеет структуру:
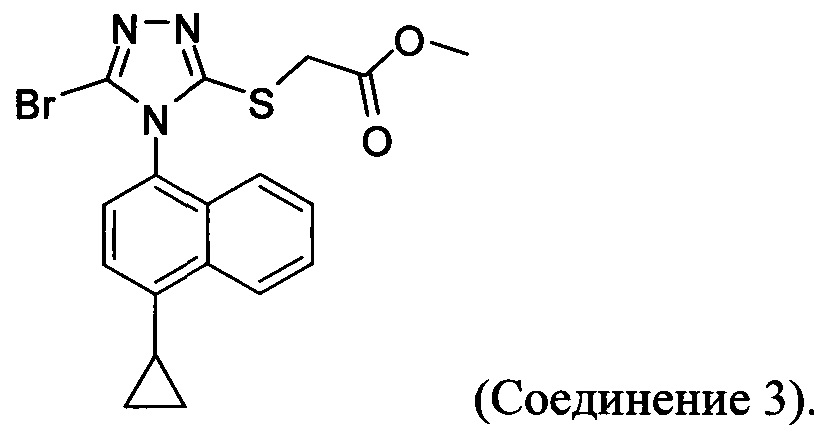
В настоящем документе предлагается Соединение 3-А, которое может быть получено с помощью описанного выше способа и имеет структуру:
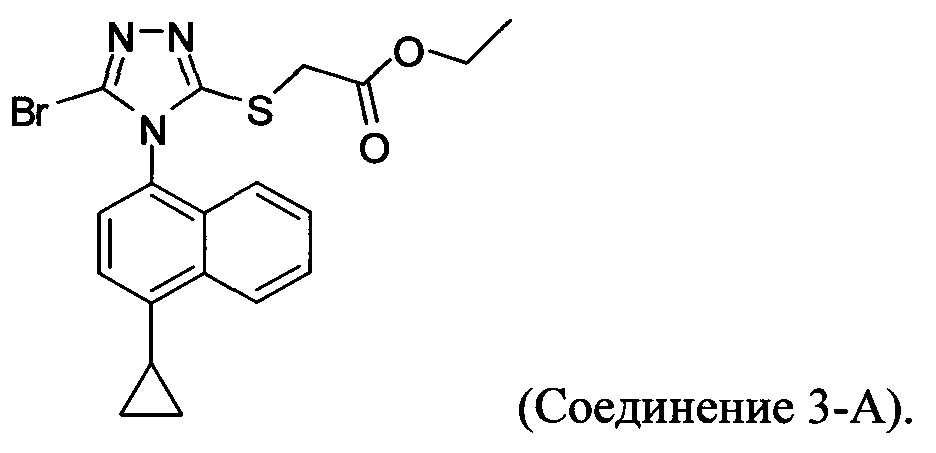
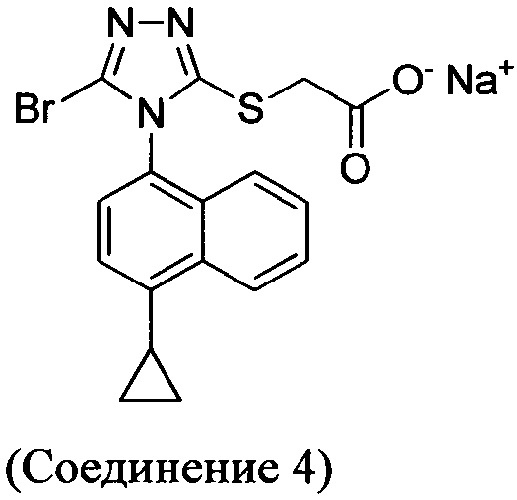
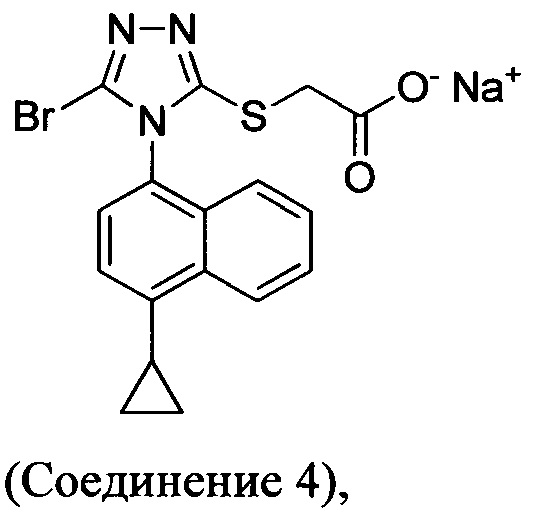
В настоящем документе предлагается Соединение 4, которое может быть получено с помощью способов, описанных выше. В некоторых вариантах осуществления Соединение 4 представляет собой кристаллический полиморф, характеризующийся пиками при 4,90, 9,83 и 25,29 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В некоторых вариантах осуществления Соединение 4 представляет собой кристаллическую полиморфную форму А.
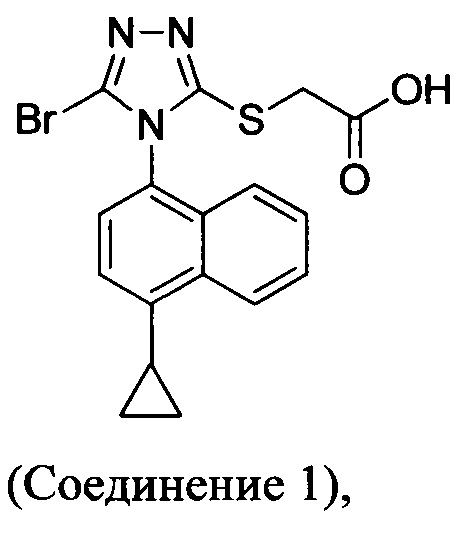
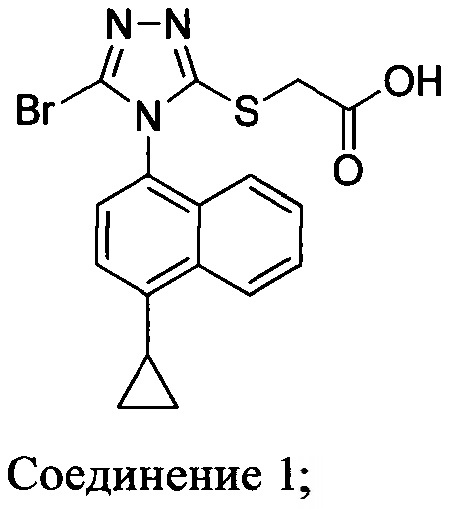
В настоящем документе предлагается Соединение 1, которое может быть получено с помощью описанного выше способа.
В одном аспекте в настоящем документе предлагается Соединение 1, содержащее не более чем 0,1% Соединения 2 по площади согласно данным HPLC анализа
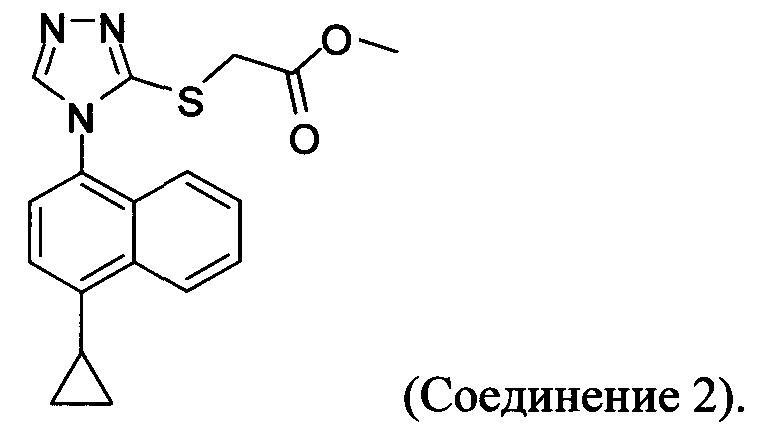
В одном варианте осуществления Соединение 1, содержащее не более чем 0,1% Соединения 2, получено с помощью способа, описанного выше.
В другом аспекте в настоящем документе предлагается Соединение 1, содержащее не более чем 0,1% Соединения 3 по площади согласно данным HPLC анализа.
В одном варианте осуществления Соединение 1, содержащее не более чем 0,1% Соединения 3, получено с помощью способа, описанного выше.
В последующем аспекте в настоящем документе предлагается Соединение 1, содержащее не более чем 0,1% Соединения 2, и не более чем 0,1% Соединения 3 по площади согласно данным HPLC анализа.
В некоторых вариантах осуществления Соединение 1 представляет собой кристаллический полиморф, характеризующийся пиками при 10,32, 18,84 и 20,75 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В определенных вариантах осуществления Соединение 1 представляет собой кристаллическую полиморфную форму 1. В других вариантах осуществления Соединение 1 представляет собой кристаллический полиморф, характеризующийся пиками при 10,46, 18,76 и 19,83 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В определенных вариантах осуществления Соединение 1 представляет собой кристаллическую полиморфную форму 2.
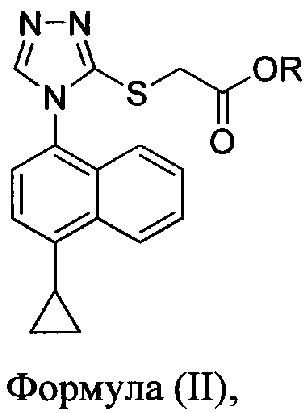
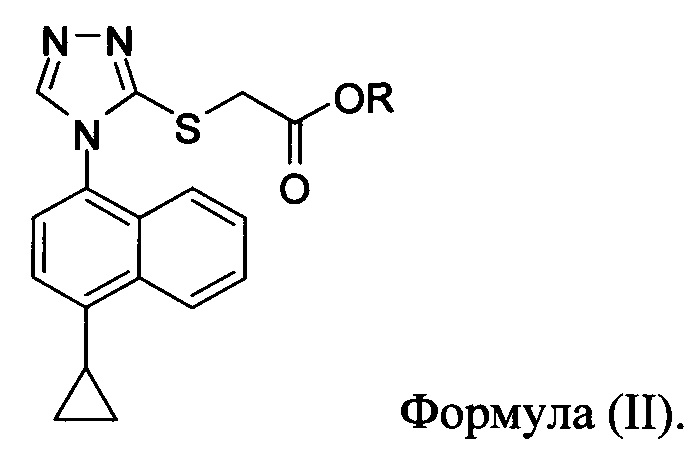
В некоторых вариантах осуществления в настоящем документе предлагается реакционная смесь, содержащая соединение Формулы (II) или его соль
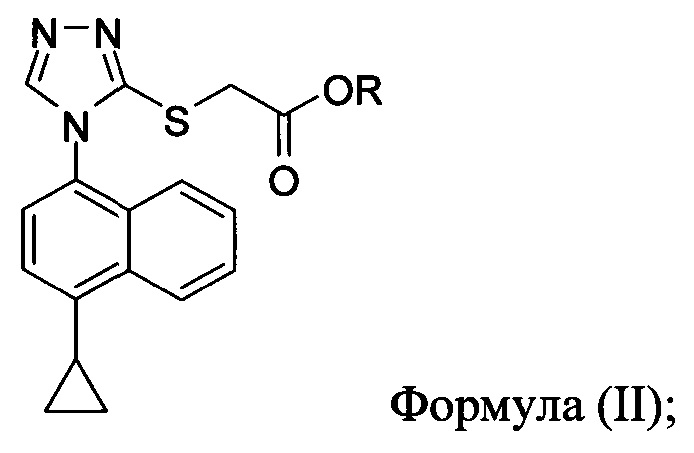
в которой R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил;
бромирующий агент; и растворитель.
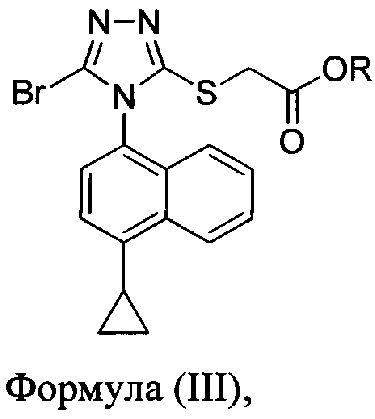
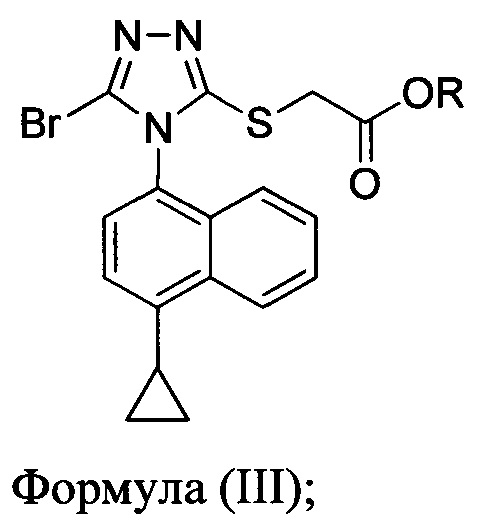
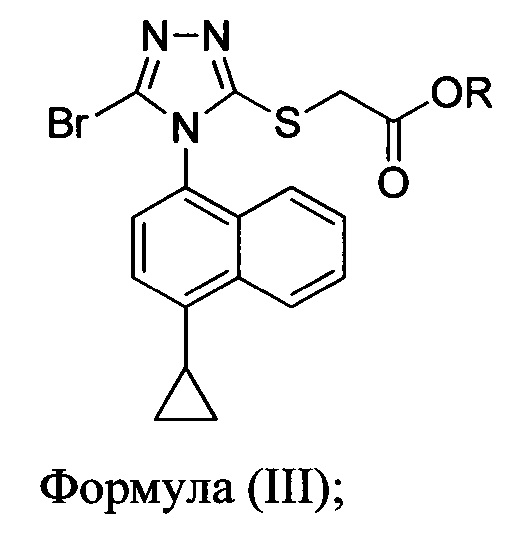
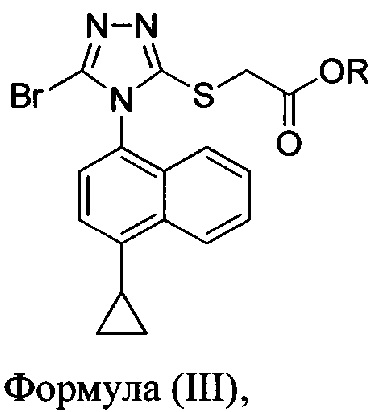
В некоторых вариантах осуществления реакционной смеси, описанной выше, R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил. В определенных вариантах осуществления бромирующий агент представляет собой N-бромсукцинимид (NBS). В некоторых вариантах осуществления растворитель представляет собой THF, DMF, ацетонитрил или МТВЕ. В определенных вариантах осуществления растворитель представляет собой THF. В последующих или дополнительных вариантах осуществления реакционной смеси, описанной выше, получено соединение формулы (III):
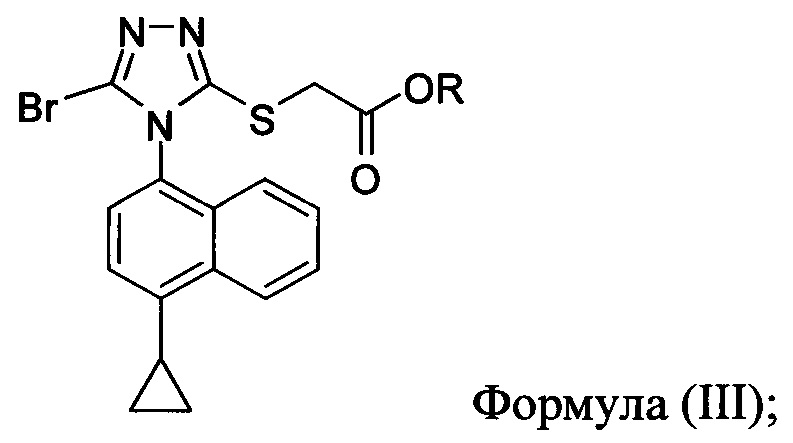
в которой R представляет собой H, -C1-C30 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил; растворитель; и основание.
В некоторых вариантах осуществления получено соединение формулы (III), в которой R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил.
В некоторых вариантах осуществления, предложенных в настоящем документе, описана реакционная смесь, содержащая соединение формулы (III):
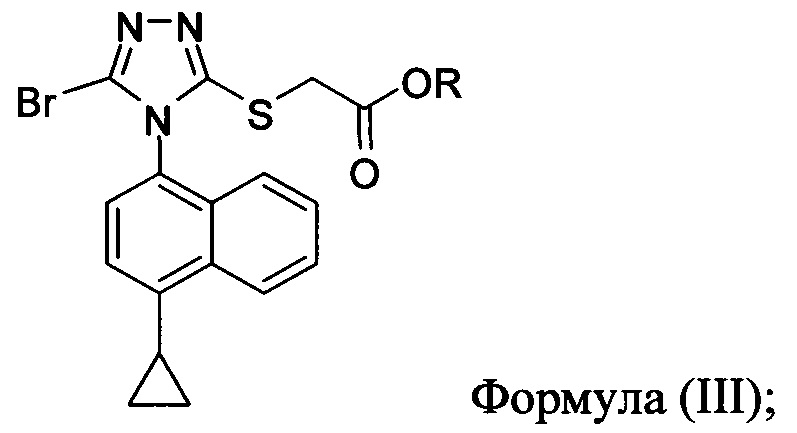
в которой R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил.
В некоторых вариантах осуществления реакционной смеси, описанной выше, R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил. В некоторых вариантах осуществления растворителем является вода. В последующих или дополнительных вариантах осуществления кислота представляет собой бромистоводородную кислоту. В последующих или дополнительных вариантах осуществления реакционной смеси, описанной выше, получено Соединение 4:
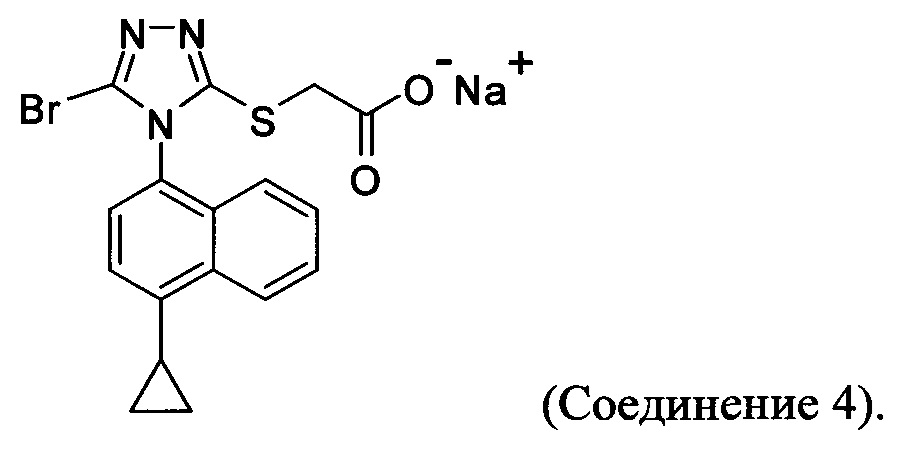
В других вариантах осуществления, предложенных в настоящем документе, описана реакционная смесь, содержащая Соединение 4:
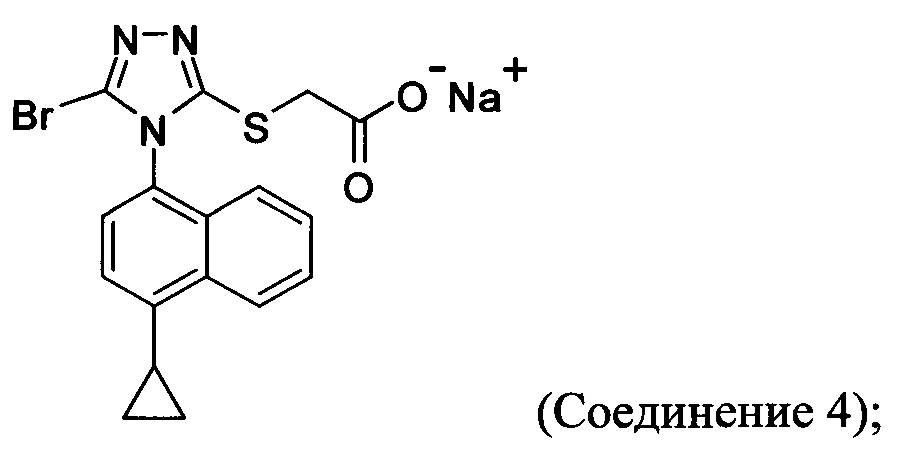
растворитель; и кислоту.
В некоторых вариантах осуществления реакционной смеси, описанной выше, кислота представляет собой бромистоводородную кислоту. В некоторых вариантах осуществления растворителем является вода. В последующих или дополнительных вариантах осуществления реакционной смеси, описанной выше, получено Соединение 1:
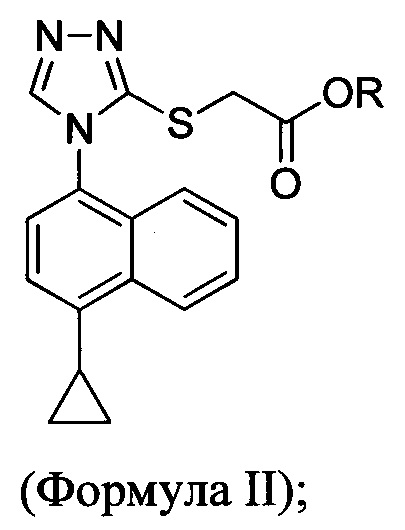
В одном аспекте соединение Формулы (II), используемое в Способе 1 выше, получено с помощью способа (Способ 2), включающего приведение в контакт соединения, имеющего структуру:
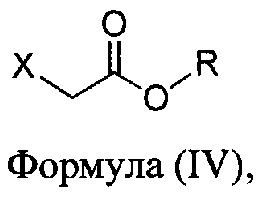
с основанием, растворителем и соединением Формулы (IV):
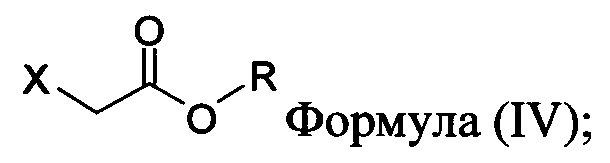
в которой X представляет собой галогено, тозилат, мезилат, трифлат или безилат, и R представляет собой -C1-C10 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил;
для получения соединения Формулы (II):
В некоторых вариантах осуществления способа, описанного выше, соединение Формулы (IV) выбрано из метил-бромацетата, этил-бромацетата, метил-хлорацетата и этил-хлорацетата.
В некоторых вариантах осуществления способа, описанного выше, способ дополнительно включает перемешивание реакционной смеси при температуре между примерно 25°C и примерно 40°C в течение, по меньшей мере, одного часа.
В некоторых вариантах осуществления способа, описанного выше, сырой продукт реакции, содержащий соединение Формулы (II), промывают охлажденной смесью этилацетата (EtOAc) и изопропанола.
В настоящем документе предлагается соединение, имеющее структуру:
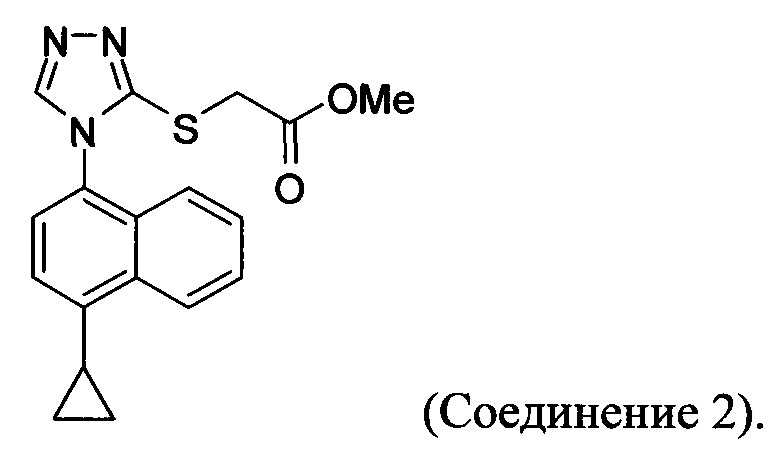
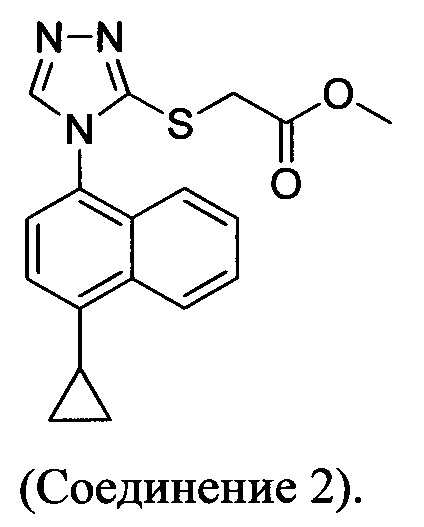
В настоящем документе предлагается Соединение 2, имеющее чистоту, по меньшей мере, 98% по данным HPLC. В настоящем документе предлагается Соединение 2, имеющее чистоту, по меньшей мере, 99% по данным HPLC.
В одном варианте осуществления Соединение 2, а именно Соединение 2, имеющее чистоту, по меньшей мере, 98% по данным HPLC, или Соединение 2, имеющее чистоту, по меньшей мере, 99% по данным HPLC, получено с помощью способов, описанных выше.
В настоящем документе предлагается соединение, имеющее структуру:
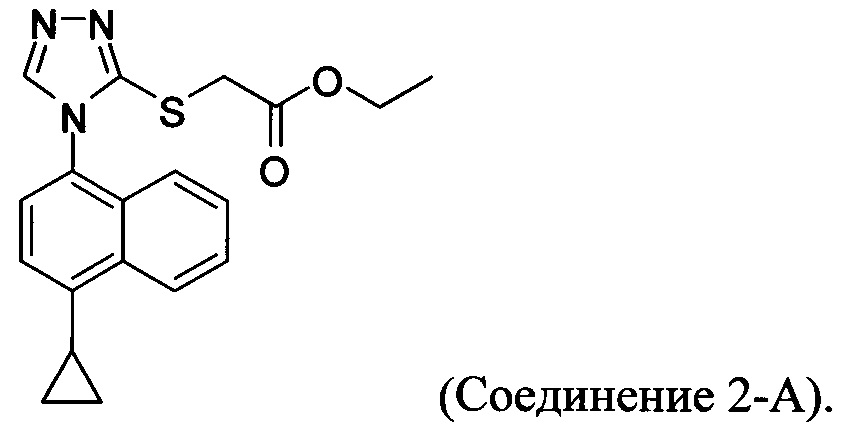
В одном варианте осуществления Соединение 2-А получено с помощью способа, описанного выше.
В настоящем документе в некоторых вариантах осуществления предлагается реакционная смесь, содержащая соединение формулы (IV):
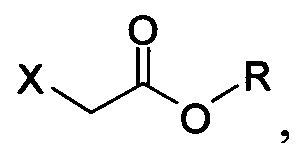
в которой X представляет собой уходящую группу; R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил; Соединение 5; основание; и растворитель. В некоторых вариантах осуществления реакционной смеси, описанной выше, соединение формулы (IV) представляет собой метил-бромацетат, этил-бромацетат, метил-хлорацетат или этил-хлорацетат. В определенных вариантах осуществления соединение формулы (II):

в которой R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил, получено в реакционной смеси, описанной выше. В определенных вариантах осуществления Соединение 2 или 2-А получено в реакционной смеси, описанной выше.
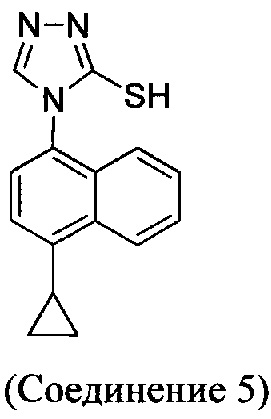
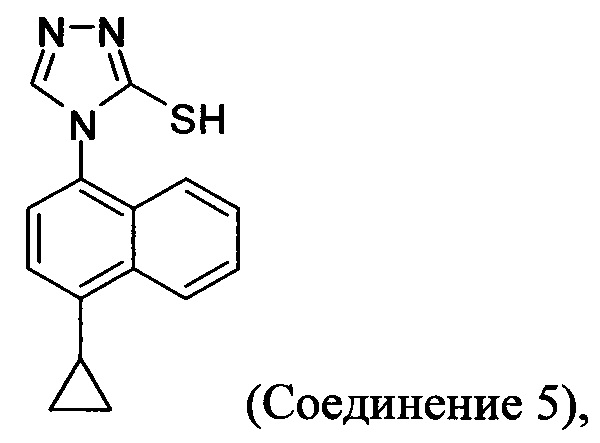
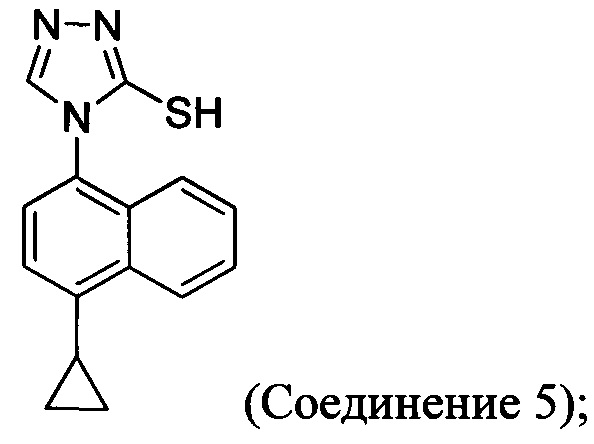
В одном аспекте Соединение 5, используемое в Способе 2, описанном выше, получено с помощью способа (Способ 3), включающего:
(5-i) приведение в контакт соединения, имеющего структуру:
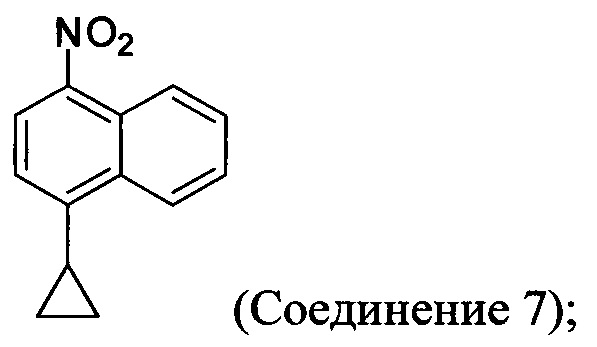
с азотной кислотой, водой и растворителем для получения соединения, имеющего структуру:
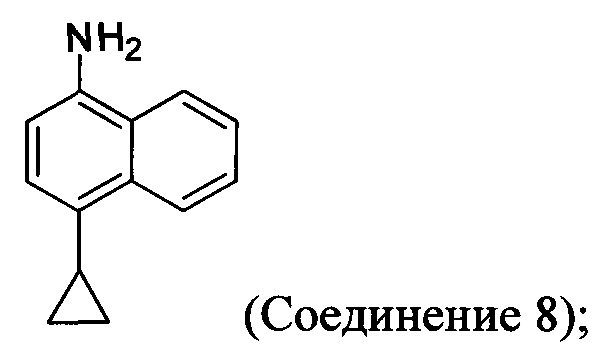
(5-ii) приведение в контакт Соединения 7 с водородом, палладием на угле и одним или несколькими растворителями для получения соединения, имеющего структуру:
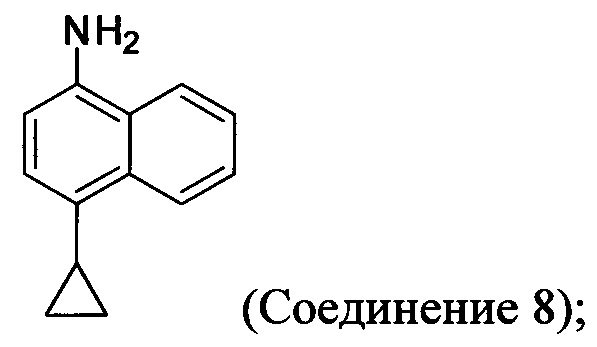
(5-iii) приведение в контакт Соединения 8 с кислотой для получения соли Соединения 8;
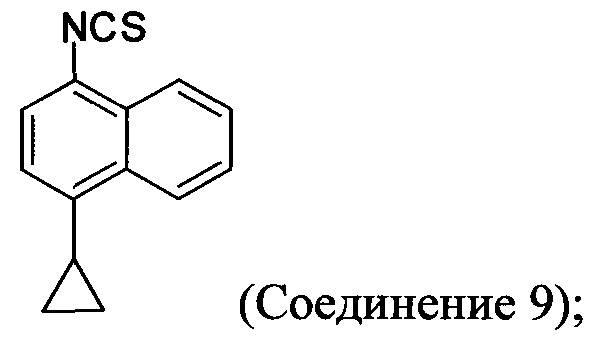
(5-iv) приведение в контакт соли Соединения 8, полученной на стадии (5-iii), с основанием, тиофосгеном и растворителем, и перемешивание смеси примерно при 5°C для получения соединения, имеющего структуру:
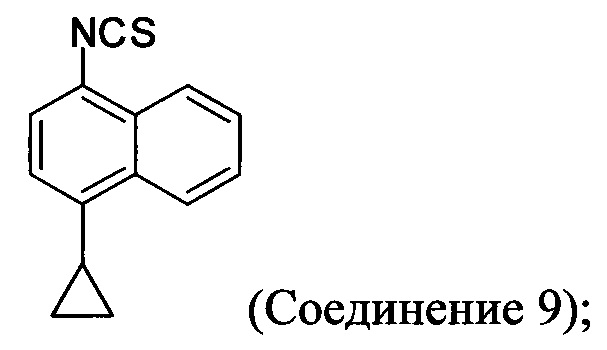
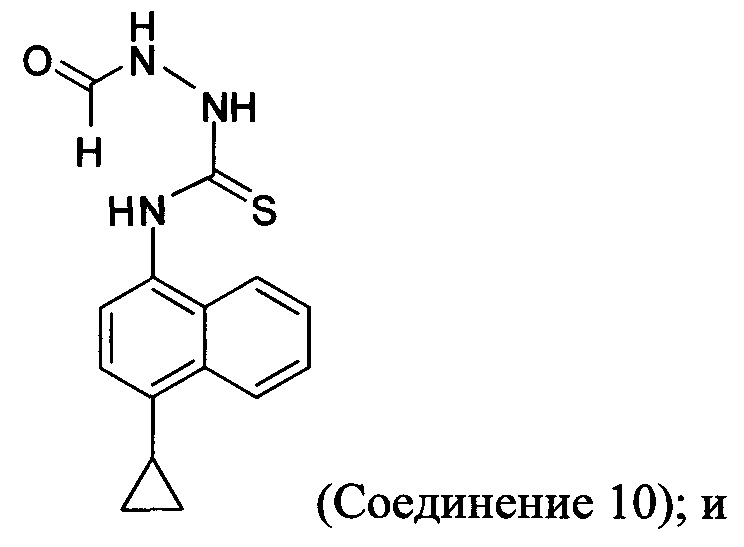
(5-v) приведение в контакт Соединения 9 с формилгидразином и растворителем для получения соединения, имеющего структуру:
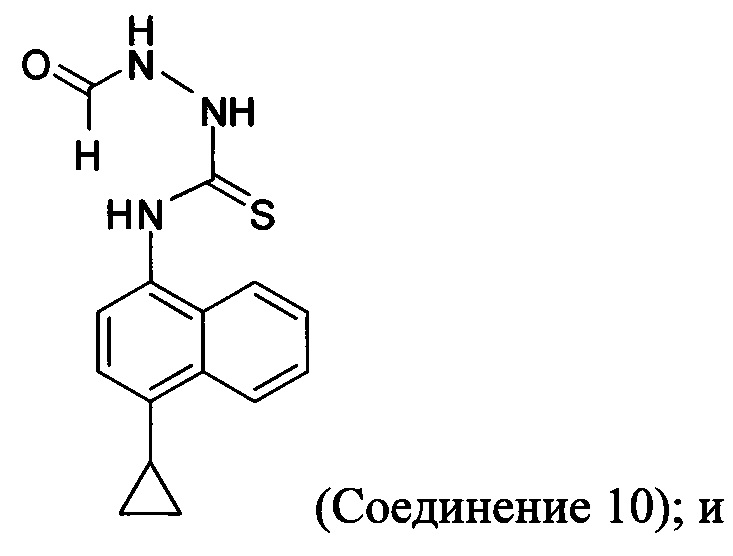
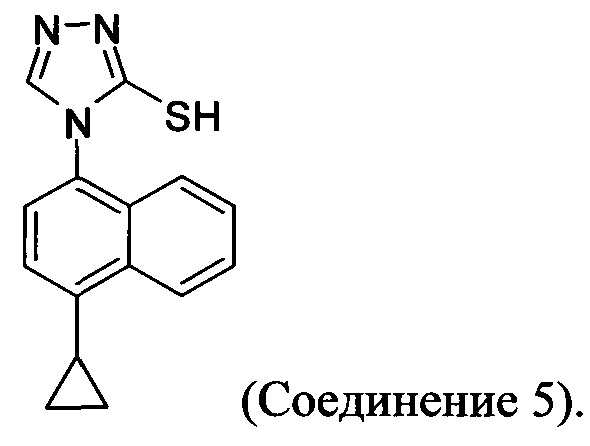
(5-vi) приведение в контакт Соединения 10 с основанием, водой и растворителем для получения Соединения 5:
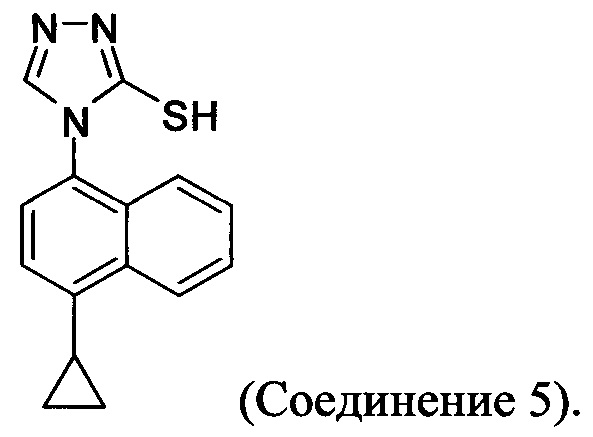
В некоторых вариантах осуществления способа, описанного выше, кислота стадии (5-iii) выбрана из соляной кислоты, щавелевой кислоты и винной кислоты. В конкретных вариантах осуществления способа, описанного выше, кислота стадии (5-iii) представляет собой щавелевую кислоту.
В одном варианте осуществления способа, описанного выше, соль соединения 8 стадии (5-iv) представляет собой оксалат.
В настоящем документе предлагается оксалат Соединения 8, имеющий структуру:
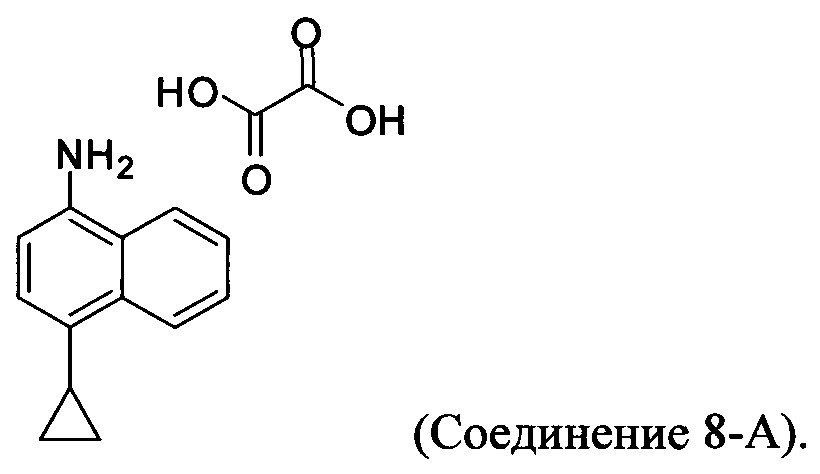
В одном случае Соединение 8-А получено с помощью способов, описанных выше.
В некоторых вариантах осуществления в настоящем документе предлагается реакционная смесь, содержащая Соединение 9, нуклеофил и растворитель.
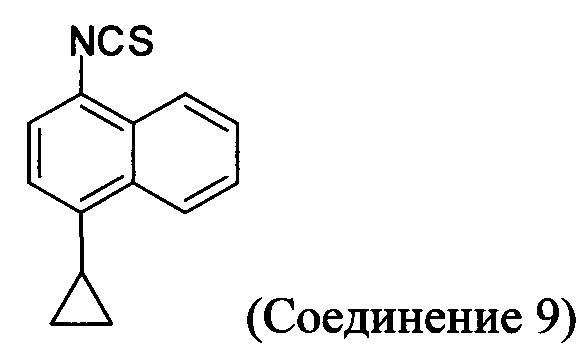
В некоторых вариантах осуществления реакционной смеси, описанной выше, нуклеофил представляет собой формилгидразин. В определенных вариантах осуществления Соединение 10 получено в реакционной смеси, описанной выше.
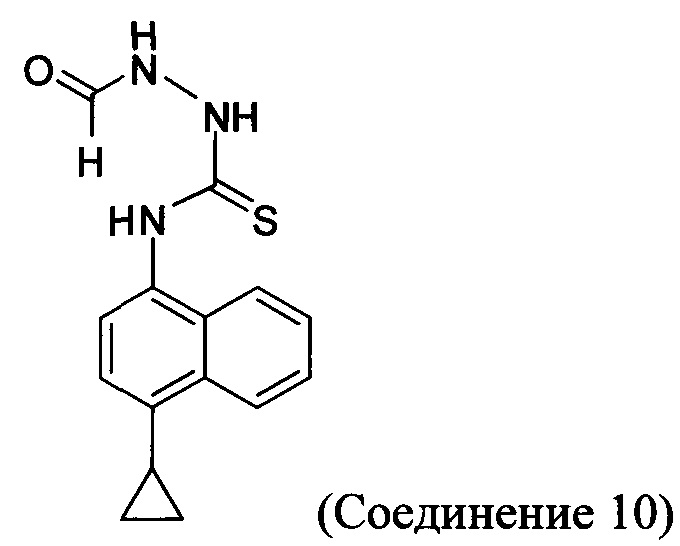
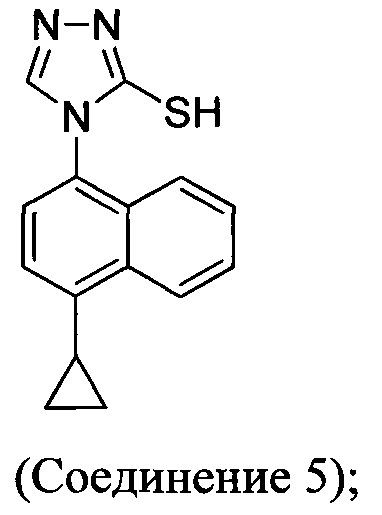
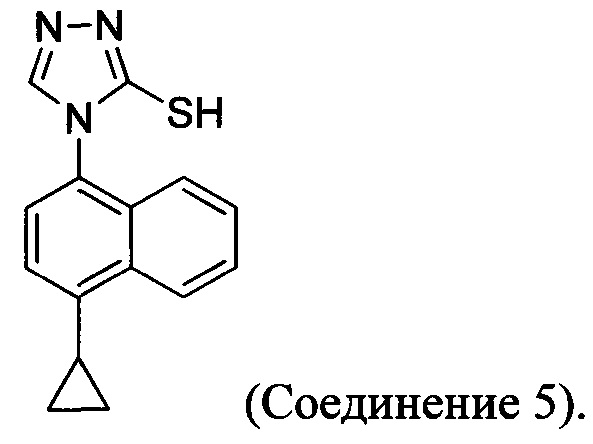
Кроме того, в настоящем документе предлагается реакционная смесь, содержащая Соединение 10, основание и растворитель. В некоторых вариантах осуществления основание представляет собой бикарбонат калия, карбонат калия, бикарбонат натрия, карбонат натрия или карбонат цезия. В определенных вариантах осуществления Соединение 5 получено в реакционной смеси, описанной в настоящем документе.
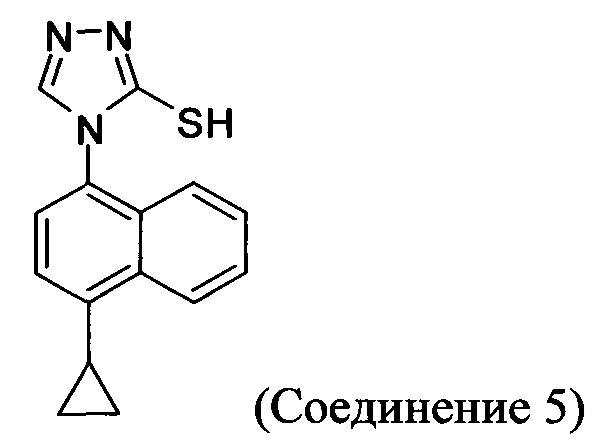
В одном аспекте Соединение 5, используемое в Способе 2, описанном выше, получено с помощью способа (Способ 4), включающего:
(5-i) приведение в контакт соединения, имеющего структуру:
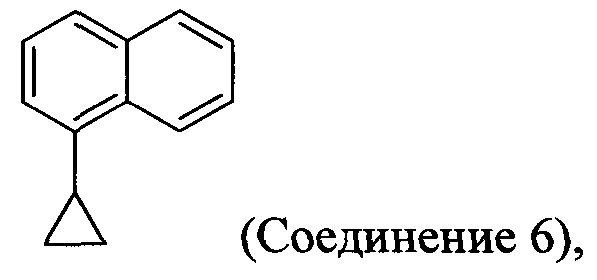
с азотной кислотой, водой и растворителем для получения соединения, имеющего структуру:
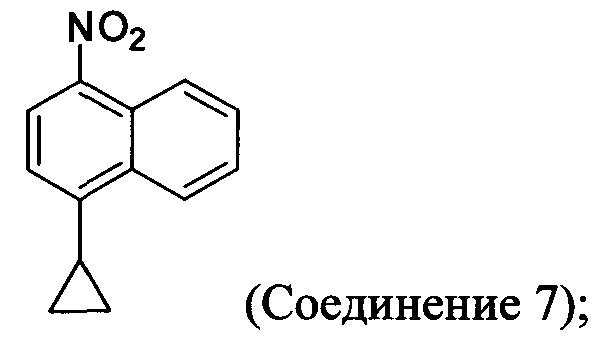
(5-ii) приведение в контакт Соединения 7 с водородом, палладием на угле и одним или несколькими растворителями для получения соединения, имеющего структуру:
(5-iii-A) приведение в контакт Соединения 8 с тиоцианатом натрия, водой и растворителем, и нагрев смеси при температуре, по меньшей мере, 130°C для получения соединения, имеющего структуру:
(5-iv-A) приведение в контакт Соединения 9, полученного на стадии (5-iii-A), с формилгидразином и растворителем для получения соединения, имеющего структуру:
(5-vi) приведение в контакт Соединения 10, полученного на стадии (5-iv-A), с основанием, водой и растворителем для получения Соединения 5:
В настоящем документе предлагается соединение, имеющее структуру:
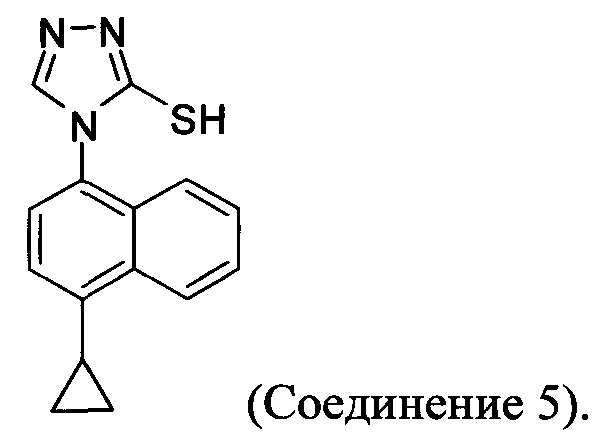
В настоящем документе предлагается Соединение 5, получаемое с помощью Способа 3 или Способа 4, описанного выше.
В настоящем документе предлагается Соединение 9, получаемое с помощью Способа 3 или Способа 4, описанного выше.
В настоящем документе предлагается Соединение 10, получаемое с помощью Способа 3 или Способа 4, описанного выше.
В настоящем документе предлагается Соединение 1, имеющее чистоту, по меньшей мере, 98% площади по данным HPLC-анализа.
В настоящем документе предлагается Соединение 1, имеющее чистоту, по меньшей мере, 98% площади по данным HPLC-анализа, и которое получено с помощью способов, описанных выше. В одном варианте осуществления Соединение 1 получено с использованием Способа 1, Способа 2 и Способа 3, описанных выше. В другом варианте осуществления Соединение 1 получено с использованием Способа 1, Способа 2 и Способа 4, описанных выше.
В одном аспекте в настоящем документе предлагается способ (Способ 5) для получения
включающий:
(i) приведение в контакт соединения, имеющего структуру
с основанием, растворителем и соединением Формулы (IV):
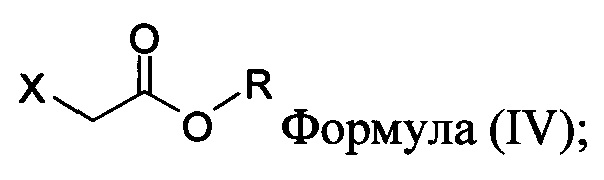
в которой X представляет собой галогено, тозилат, мезилат, трифлат или безилат, и R представляет собой -C1-C20 алкил, -C1-C10 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил;
для получения соединения Формулы (II):
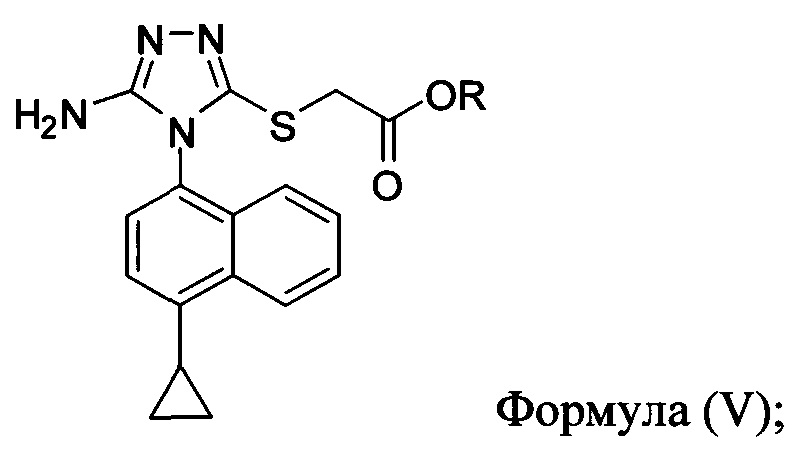
(ii) приведение в контакт соединения Формулы (V) с бромидом меди (II), нитритом калия и растворителем при температуре между примерно 14°C и примерно 22°C для получения соединения, имеющего структуру:
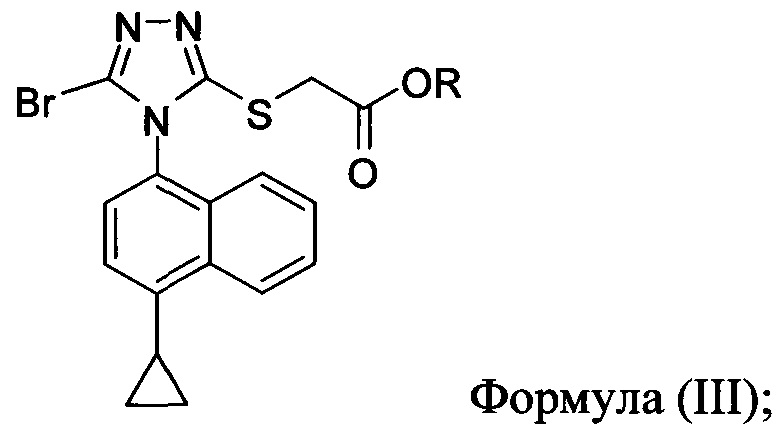
(iii) приведение в контакт раствора соединения Формулы (III), полученного на стадии (ii), в растворителе с водным раствором гидроксида натрия для получения соединения, имеющего структуру:
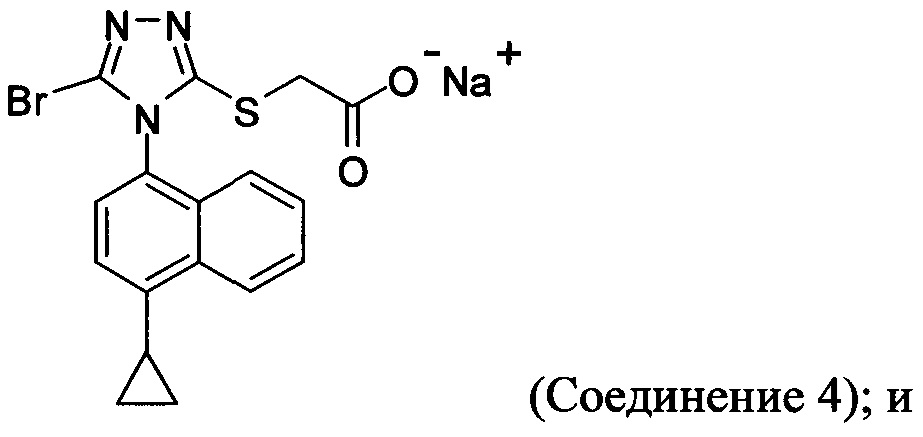
(iv) необязательно, приведение в контакт водного раствора Соединения 4, полученного на стадии (iii), с кислотой для получения смеси, содержащей Соединение 1.
В некоторых вариантах осуществления способа, описанного выше, R представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изоамил, пентил, гексил, гептил, октил, нонил, терпенил, борнил, аллил, линалил или геранил.
В определенных вариантах осуществления способа, описанного выше, R представляет собой метил или этил. В некоторых конкретных вариантах осуществления способа, описанного выше, R представляет собой метил.
В некоторых вариантах осуществления способа, описанного выше, соединение Формулы (IV), полученное на стадии (i), выбрано из метил-бромацетата, этил-бромацетата, метил-хлорацетата и этил-хлорацетата.
В некоторых вариантах осуществления способа, описанного выше, кислота, полученная на стадии (iv), представляет собой соляную кислоту, бромистоводородную кислоту, уксусную кислоту или фосфорную кислоту. В некоторых конкретных вариантах осуществления способа, описанного выше, кислота, полученная на стадии (iv), представляет собой бромистоводородную кислоту.
В некоторых вариантах осуществления способа, описанного выше, указанный способ дополнительно включает фильтрацию смеси, полученной на стадии (iv), для получения Соединения 1 в виде твердого вещества.
В некоторых вариантах осуществления способа, описанного выше, указанный способ дополнительно включает экстракцию смеси, полученной на стадии (iv), этилацетатом и удаление этилацетата для получения Соединения 1 в виде твердого вещества.
В некоторых вариантах осуществления способа, описанного выше, указанный способ дополнительно включает перекристаллизацию Соединения 1 из этилацетата.
В настоящем документе предлагается Соединение 1, получаемое с помощью способа, описанного выше.
В настоящем документе предлагается Соединение 3, получаемое с помощью способа, описанного выше, и имеющее структуру:
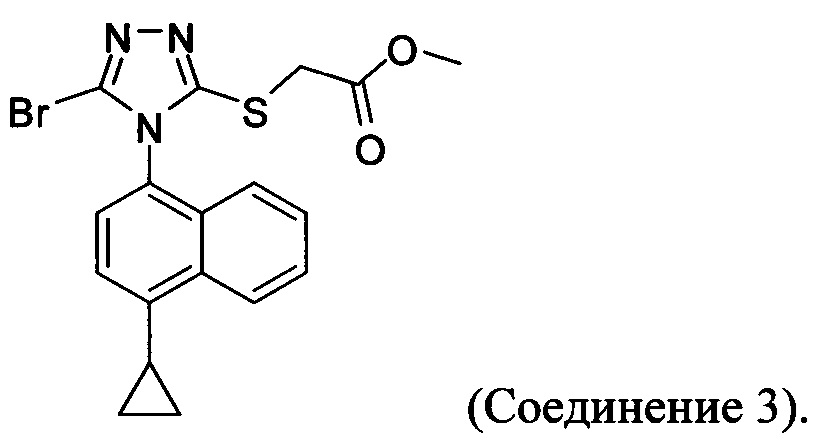
В настоящем документе предлагается Соединение 3-А, получаемое с помощью способа, описанного выше, и имеющее структуру:
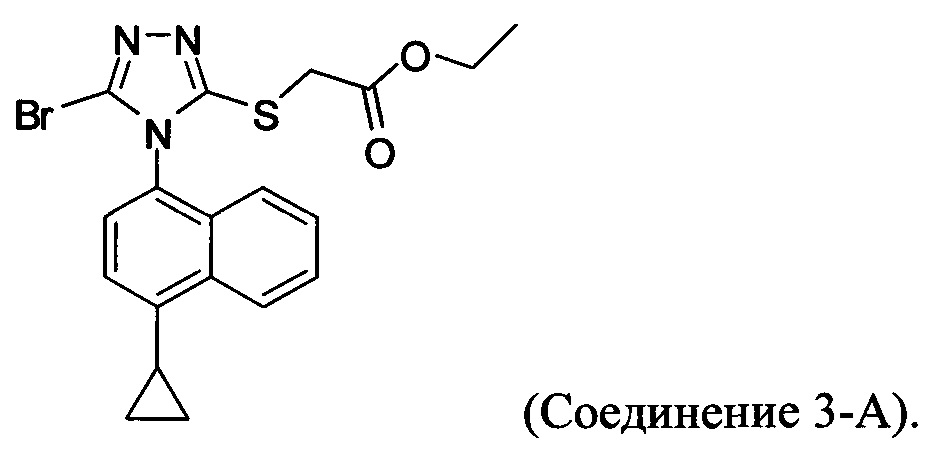
В настоящем документе предлагается Соединение 4, получаемое с помощью способа, описанного выше.
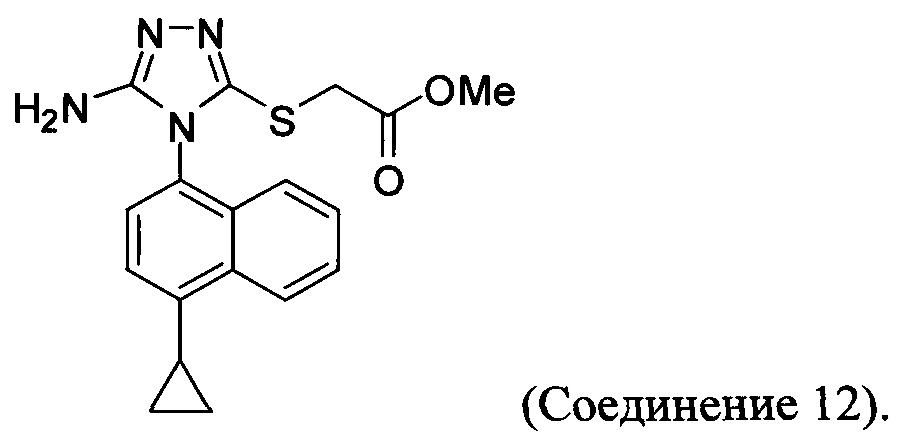
В настоящем документе предлагается Соединение 12, получаемое с помощью способа, описанного выше, и имеющее структуру:
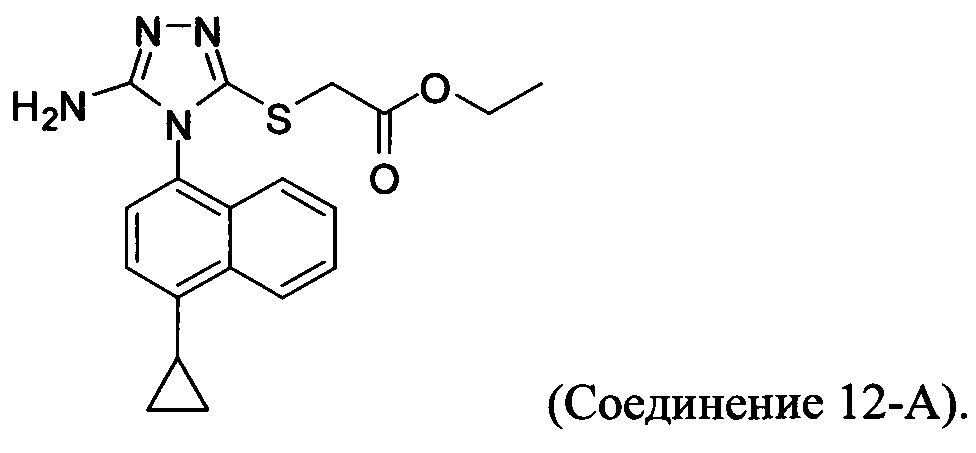
В настоящем документе предлагается Соединение 12-А, получаемое с помощью способа, описанного выше, и имеющее структуру:
В последующем аспекте любой из способов, описанных выше, является подходящим для синтеза любого соединения Формулы (I).
В одном аспекте в настоящем документе предлагается способ (Способ 1а) для получения соединения формулы (III):
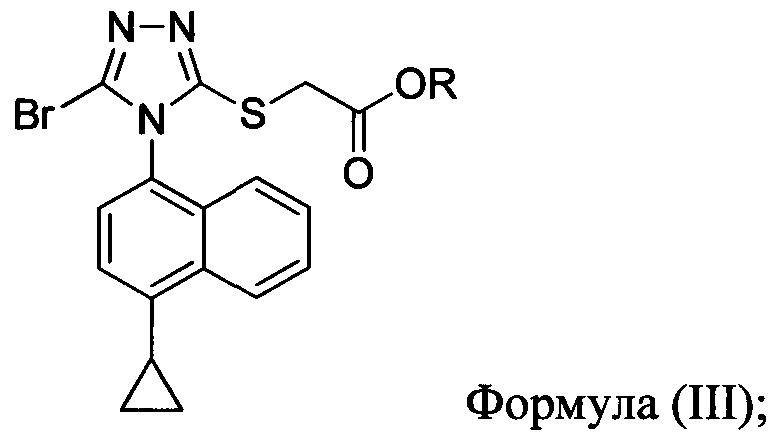
включающий приведение в контакт соединения Формулы (II):
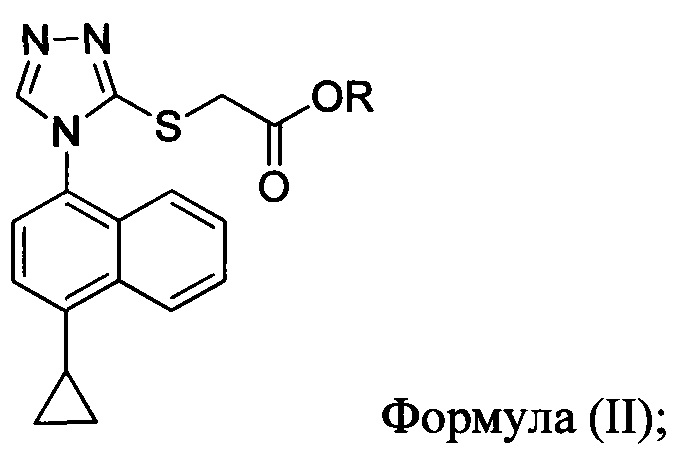
в которой R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил; с N-бромсукцинимидом (NBS) и растворителем. В некоторых вариантах осуществления R представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изоамил, пентил, гексил, гептил, октил, нонил, терпенил, борнил, аллил, линалил или геранил. В определенных вариантах осуществления R представляет собой метил или этил. В некоторых вариантах осуществления соединение Формулы (II), NBS и растворитель перемешивают в течение, по меньшей мере, 12 часов при температуре между примерно комнатной температурой и примерно 32°C.
В некоторых вариантах осуществления Способа 1а, описанного выше, указанный способ дополнительно включает приведение в контакт соединения Формулы (III) с раствором гидроксида натрия для получения Соединения 4:
В определенных вариантах осуществления способ включает кристаллизацию Соединения 4 из водного раствора гидроксида натрия. В альтернативных вариантах осуществления способ дополнительно включает приведение в контакт Соединения 4 с кислотой для получения Соединения 1:
В определенных вариантах осуществления кислота представляет собой бромистоводородную кислоту. В другом альтернативном варианте осуществления способ дополнительно включает: (a) растворение Соединения 4 в воде и добавление этилацетата в смесь; и (b) приведение в контакт бифазной смеси, полученной на стадии (a), с кислотой и отделение органической фазы для получения Соединения 1.
В настоящем документе обеспечено Соединение 3 или Соединение 3-А:
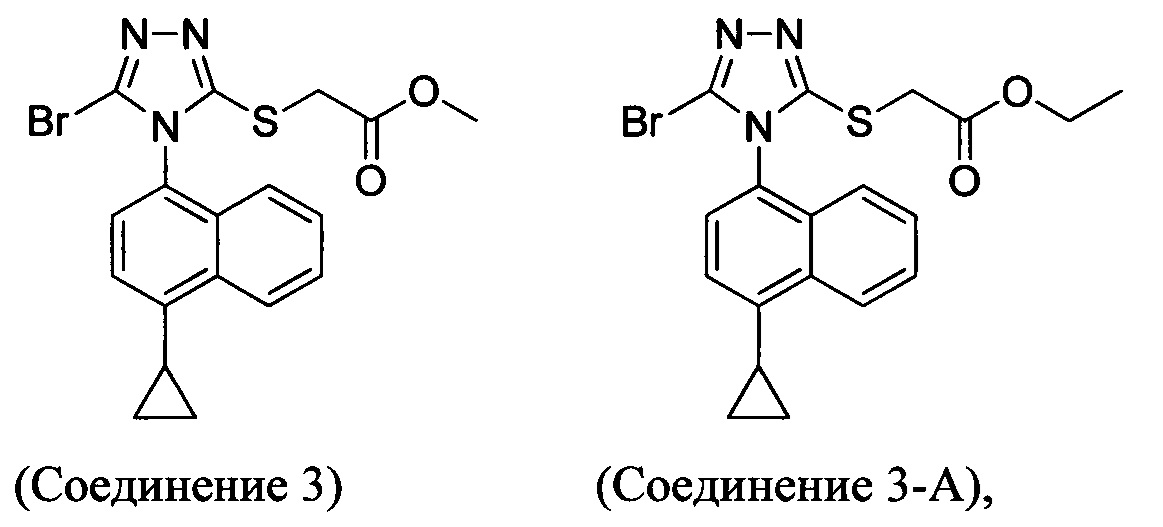
полученное с помощью способов в соответствии с Способом 1а, описанным выше.
В настоящем документе предлагается Соединение 4:
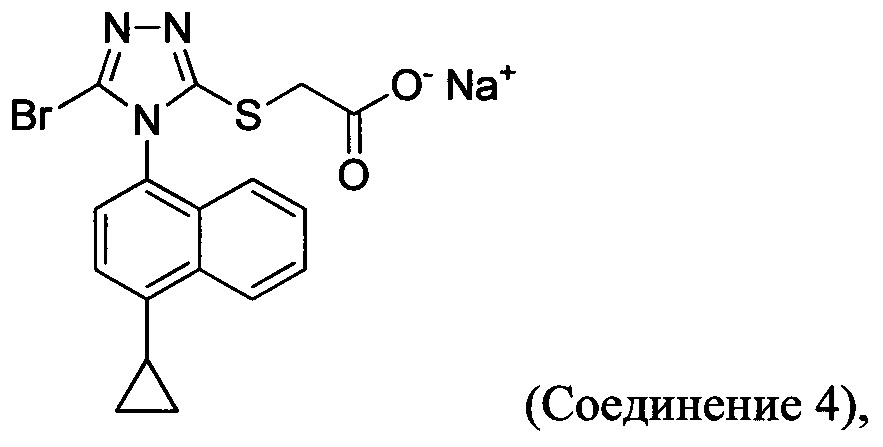
полученное с помощью способов в соответствии с Способом 1а, описанным выше.
В настоящем документе предлагается Соединение 1:
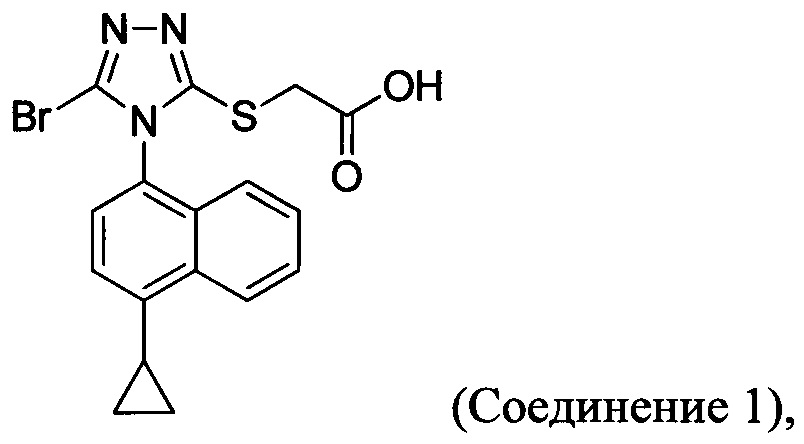
полученное с помощью способов в соответствии с Способом 1а, описанным выше. В некоторых вариантах осуществления Соединение 1 представляет собой кристаллический полиморф, характеризующийся пиками при 10,46, 18,76 и 19,83 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В определенных вариантах осуществления Соединение 1 представляет собой кристаллическую полиморфную форму 2.
В другом аспекте в настоящем документе предлагается реакционная смесь, содержащая соединение Формулы (II):
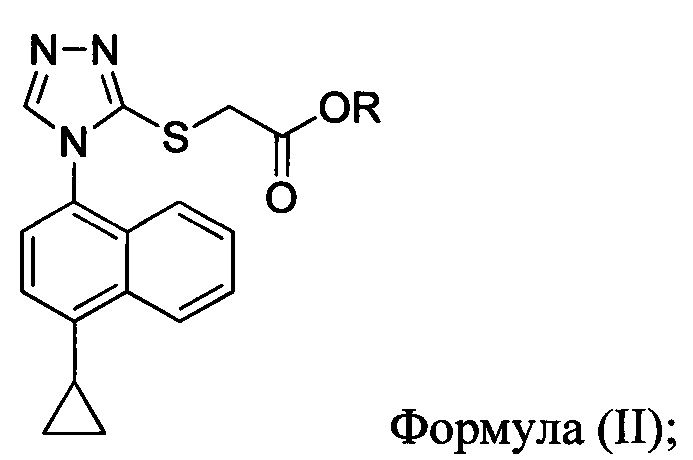
в которой R представляет собой -C1-C10 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил; бромирующий агент; и растворитель. В некоторых вариантах осуществления бромирующий агент представляет собой N-бромсукцинимид (NBS).
В последующем аспекте в настоящем документе предлагается реакционная смесь, содержащая соединение формулы (III)
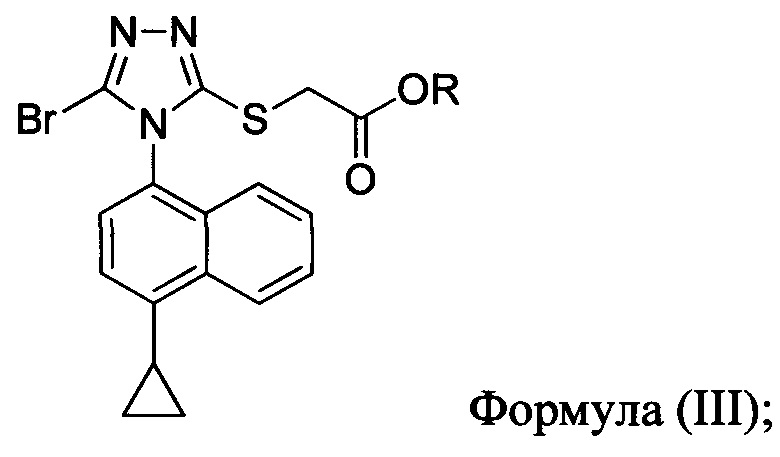
в которой R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил; основание; и растворитель.
В некоторых вариантах осуществления основание представляет собой гидроксид натрия.
В другом аспекте в настоящем документе предлагается реакционная смесь, содержащая Соединение 4:
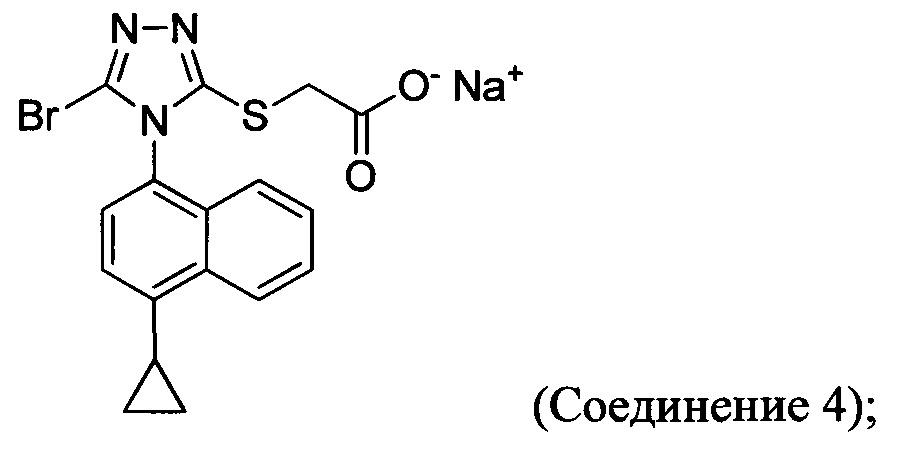
кислоту; и растворитель. В некоторых вариантах осуществления кислота представляет собой бромистоводородную кислоту.
В одном аспекте в настоящем документе предлагается способ (Способ 2а), в котором соединение Формулы (II), используемое в Способе 1а выше, получено с помощью способа, включающего приведение в контакт соединения 5:
с основанием, растворителем и соединением Формулы (IV):
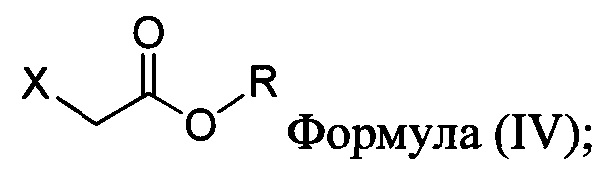
в которой:
X представляет собой галогено, тозилат, мезилат, трифлат или безилат; и
R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил.
В некоторых вариантах осуществления соединение Формулы (IV) выбрано из метил-бромацетата, этил-бромацетата, метил-хлорацетата и этил-хлорацетата. В определенных вариантах осуществления сырой продукт реакции, содержащий соединение Формулы (II), промывают охлажденной смесью этилацетата и изопропанола.
Кроме того, в настоящем документе предлагается Соединение 2 или Соединение 2-А:
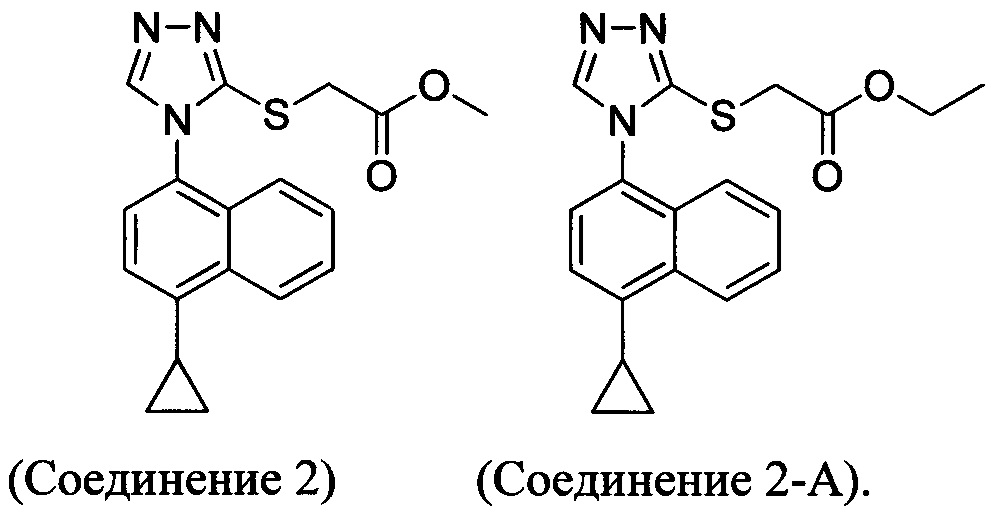
В определенных вариантах осуществления Соединение 2 или Соединение 2-А получено с помощью способов в соответствии с Способом 2а, описанным выше.
В другом аспекте в настоящем документе предлагается реакционная смесь, содержащая Соединение 5:
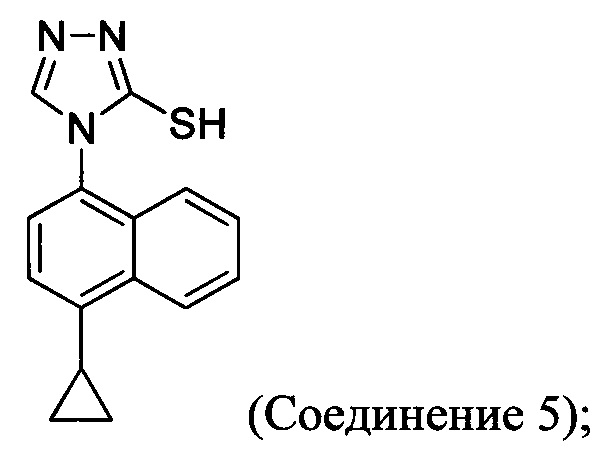
соединение Формулы (IV):
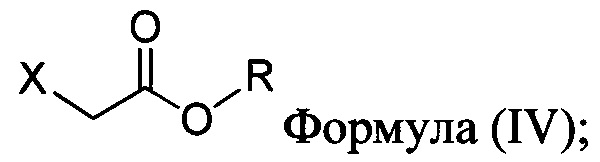
в которой:
X представляет собой уходящую группу; и
R представляет собой -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил или -C3-C10 циклоалкенил; основание; и растворитель. В некоторых вариантах осуществления соединение Формулы (IV) представляет собой метил-бромацетат, этил-бромацетат, метил-хлорацетат или этил-хлорацетат.
В другом аспекте в настоящем документе обеспечен способ (Способ 3а), в котором Соединение 5, используемое в Способе 2а выше, получено с помощью способа, включающего:
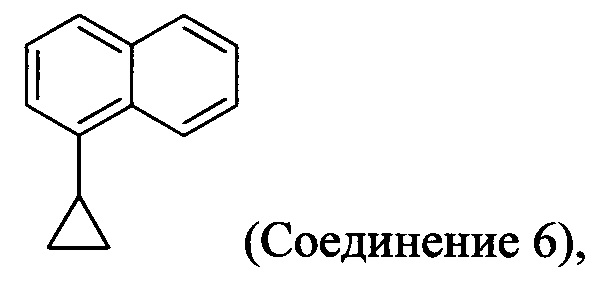
(5-i) приведение в контакт Соединения 6:
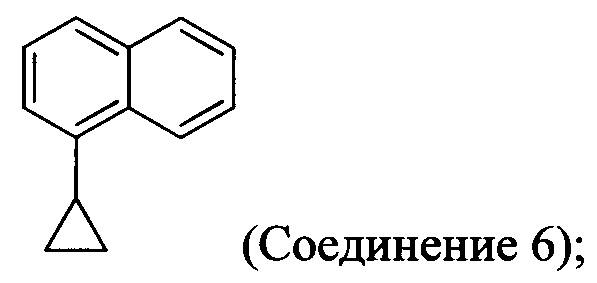
с азотной кислотой, водой и растворителем для получения соединения 7:
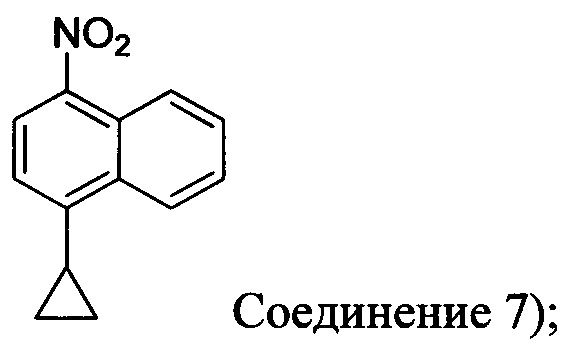
(5-ii) приведение в контакт Соединения 7 с водородом, палладием на угле и одним или несколькими растворителями для получения Соединения 8:

(5-iii) приведение в контакт Соединения 8 с кислотой для получения соли Соединения 8;
(5-iv) приведение в контакт соли Соединения 8, полученной на стадии (5-iii), с основанием, тиофосгеном и растворителем для получения Соединения 9:
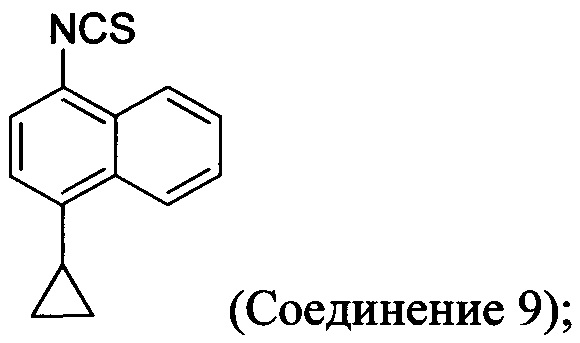
(5-v) приведение в контакт Соединения 9 с формилгидразином и растворителем для получения Соединения 10:
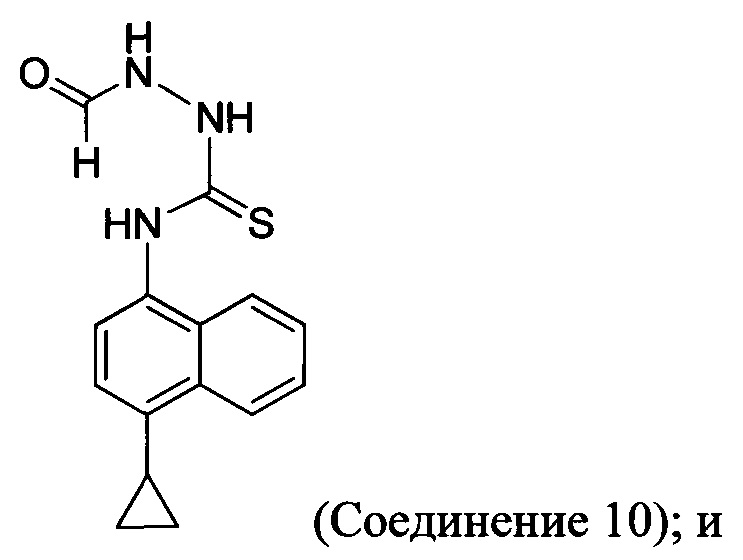
(5-vi) приведение в контакт Соединения 10 с основанием, водой и растворителем для получения Соединения 5:
Кроме того, в настоящем документе предлагается соединение, выбранное из Соединений I-X:
2-(4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение I); 2-(4-(4-циклопропилнафталин-1-ил)-5-гидрокси-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение II); 2-(5-амино-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение III); 2-(5-бром-4-(1-циклопропилнафталин-2-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение IV); 2-(5-бром-4-(4-метилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение V); 2-(5-бром-4-(4-пропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение VI); 2-(5-бром-4-(5-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение VII); 2-(5-бром-4-(нафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение VIII); 2-(5-хлор-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота (Соединение IX); и 4-(5-(карбоксиметилтио)-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-иламино)-4-оксобутановая кислота (Соединение X). В некоторых вариантах осуществления образец 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты содержит меньше, чем примерно 2%, меньше, чем примерно 1,5%, меньше, чем примерно 1,0%, меньше, чем примерно 0,5%, меньше, чем примерно 0,4%, меньше, чем примерно 0,3%, меньше, чем примерно 0,2%, меньше, чем примерно 0,1%, меньше, чем примерно 0,05%, меньше, чем примерно 0,02% любого из Соединений I-X. В предпочтительных вариантах осуществления образец 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты содержит меньше, чем примерно 0,5% любого из Соединений I-X.
Включение посредством отсылки
Все публикации, патенты и патентные заявки, упомянутые в данной заявке, включены в настоящий документ посредством отсылки в той же степени, как если бы каждая отдельная публикация, патент или патентная заявка была конкретно и индивидуально указана как включенная посредством отсылки.
Краткое описание чертежей
Характеризующие новизну отличительные признаки изобретения изложены более подробно в прилагаемой формуле изобретения. Лучшее понимание отличительных признаков и преимуществ настоящего изобретения будет получено при обращении к следующему подробному описанию, в котором изложены иллюстративные варианты осуществления, в которых используются принципы настоящего изобретения, и сопровождающим чертежам, на которых:
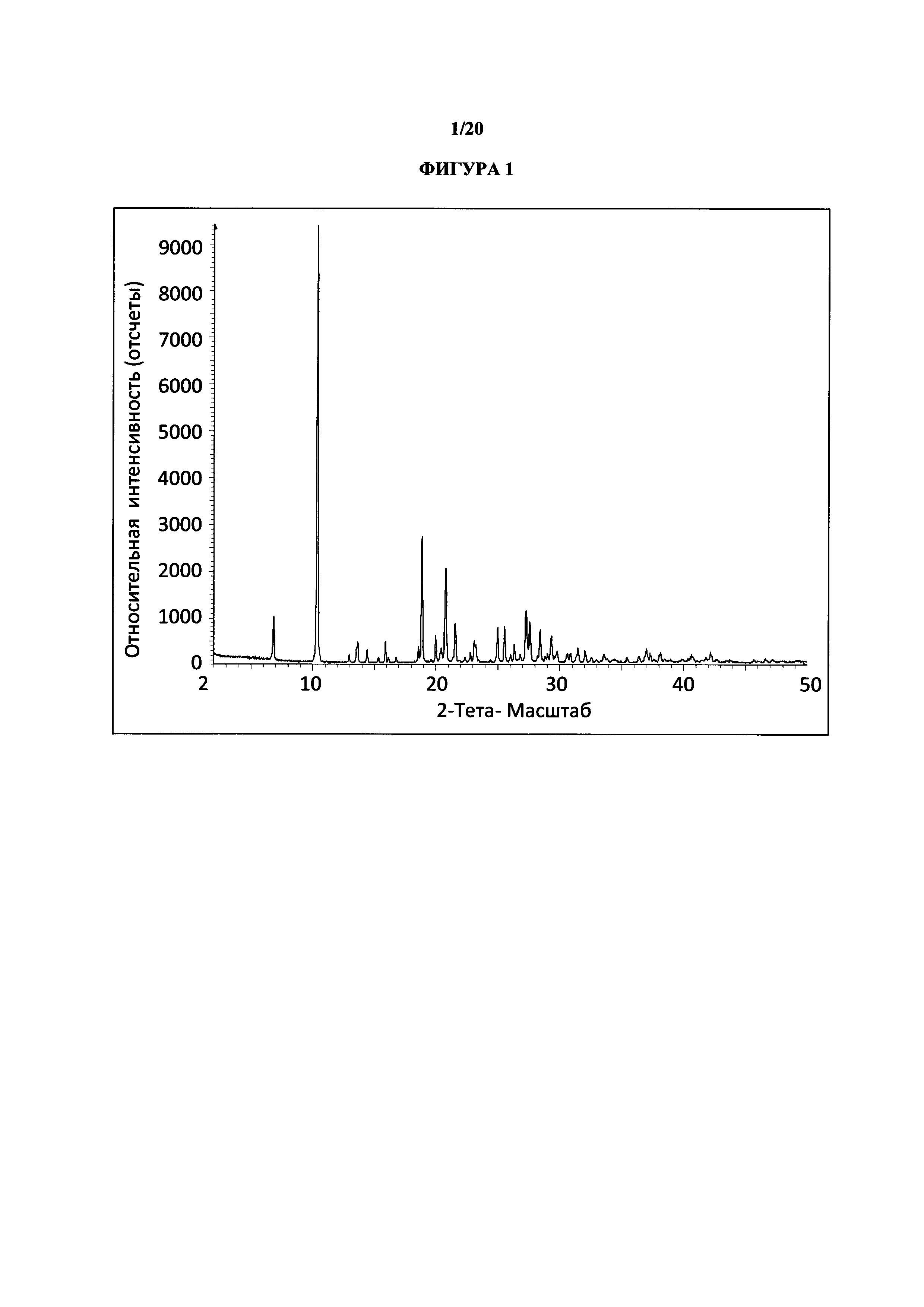
Фигура 1 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы 1 (исходные данные).
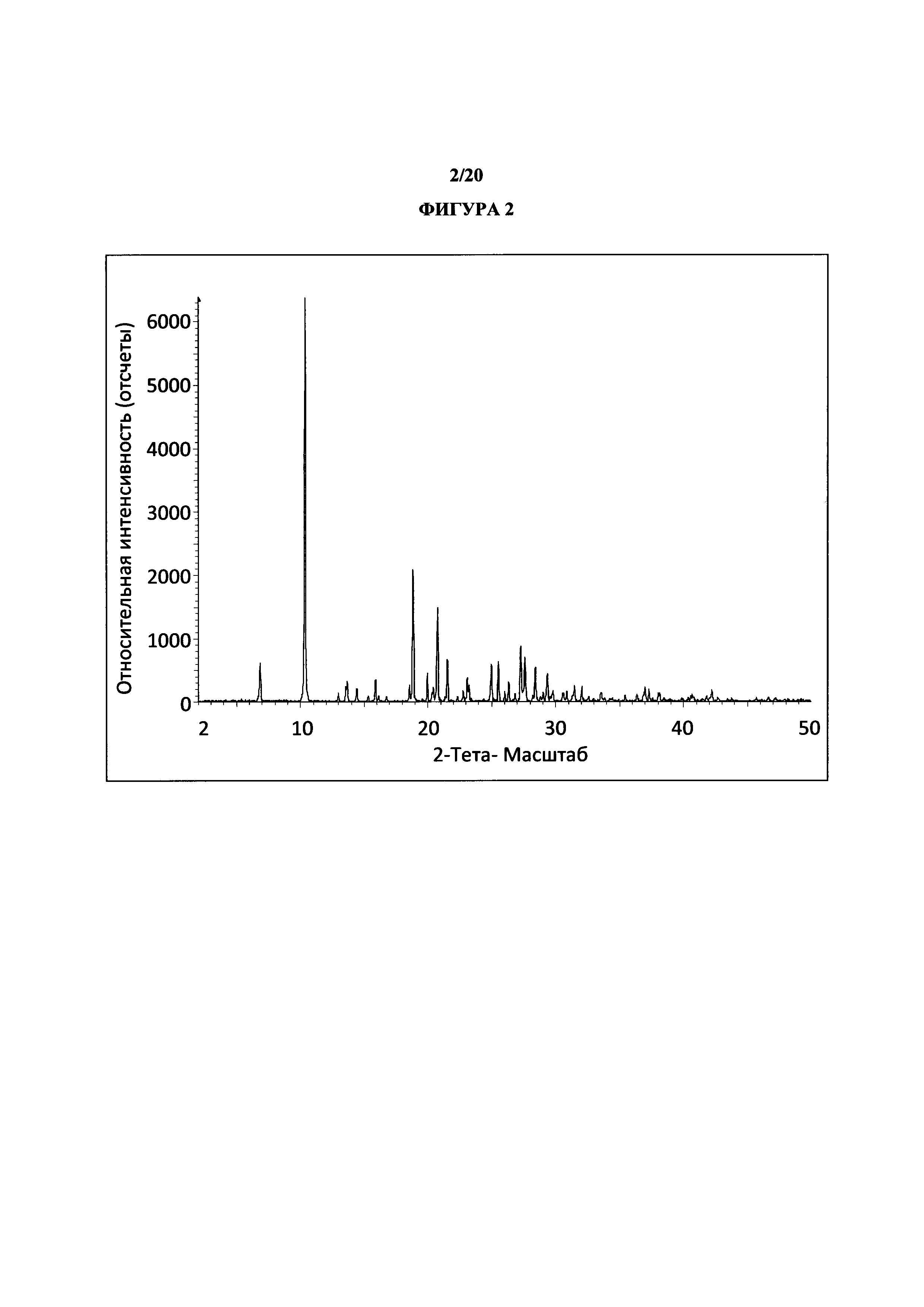
Фигура 2 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы 1 (с удаленным фоновым сигналом и отсеченным Kα2).
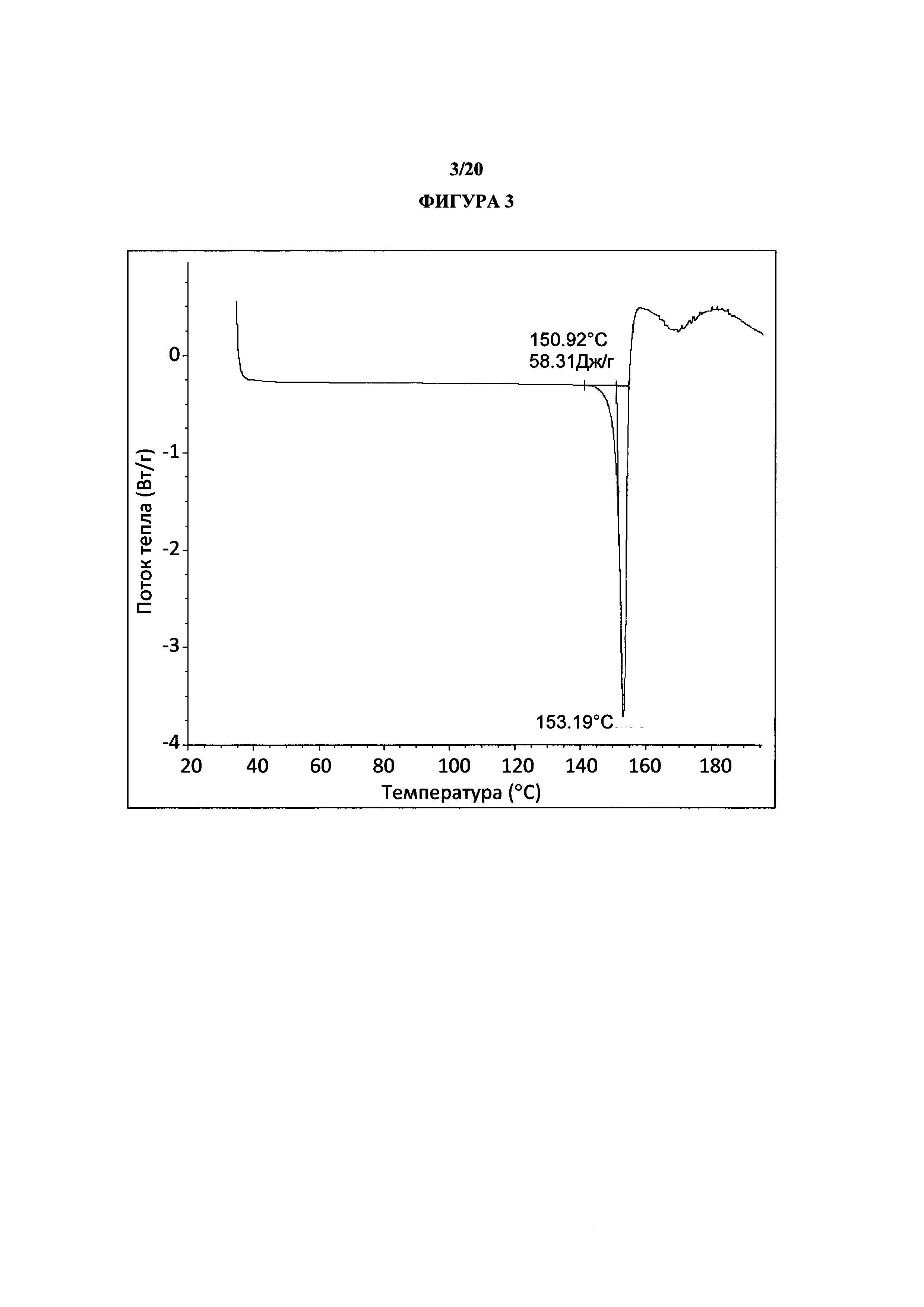
Фигура 3 представляет собой иллюстративную термограмму дифференциальной сканирующей калориметрии полиморфной формы 1.
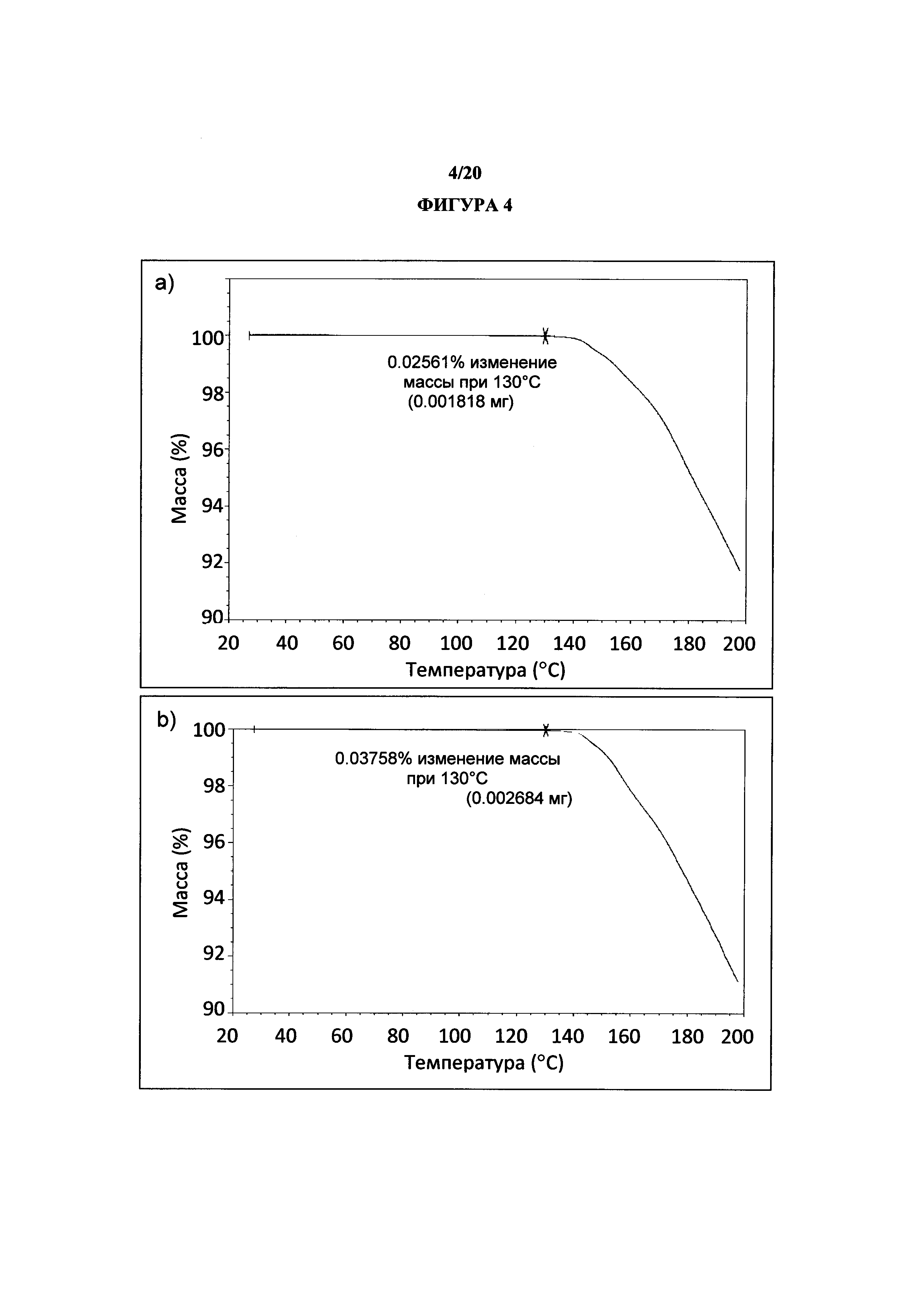
Фигура 4 представляет собой иллюстративные термогравиметрические анализы (a) Rep 1 и (b) Rep 2 полиморфной формы 1.
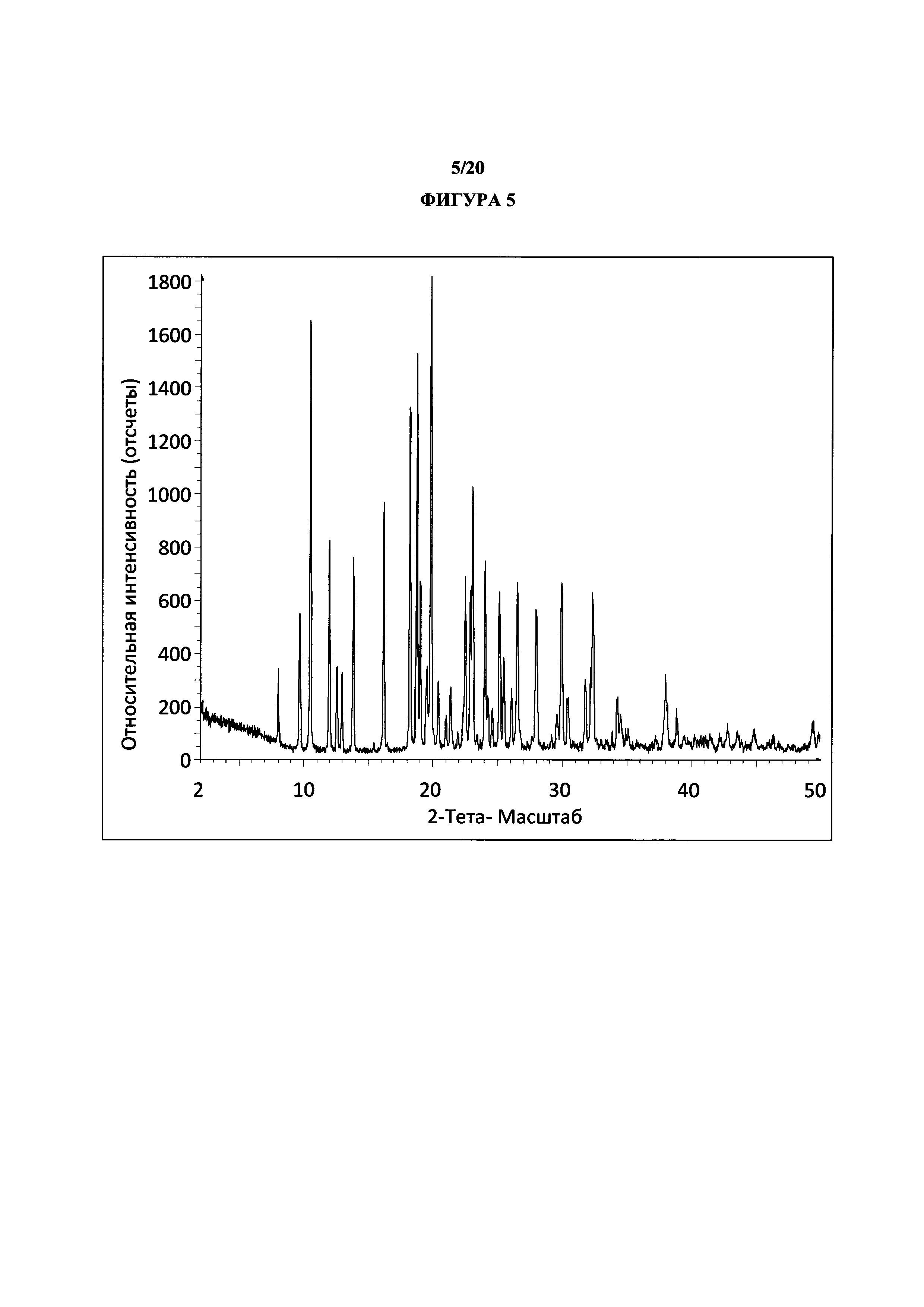
Фигура 5 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы 2 (исходные данные).
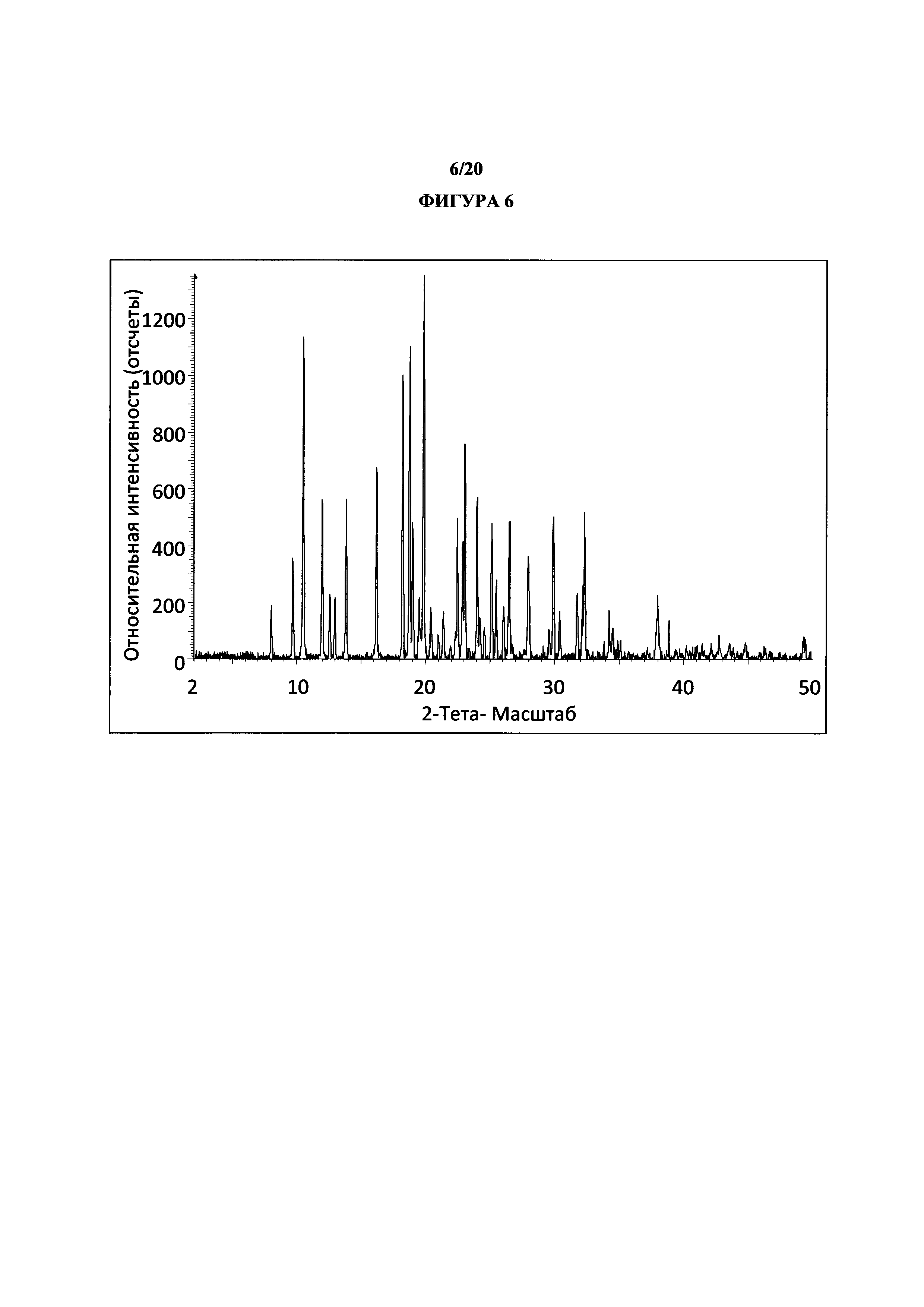
Фигура 6 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы 2 (с удаленным фоновым сигналом и отсеченным Kα2).
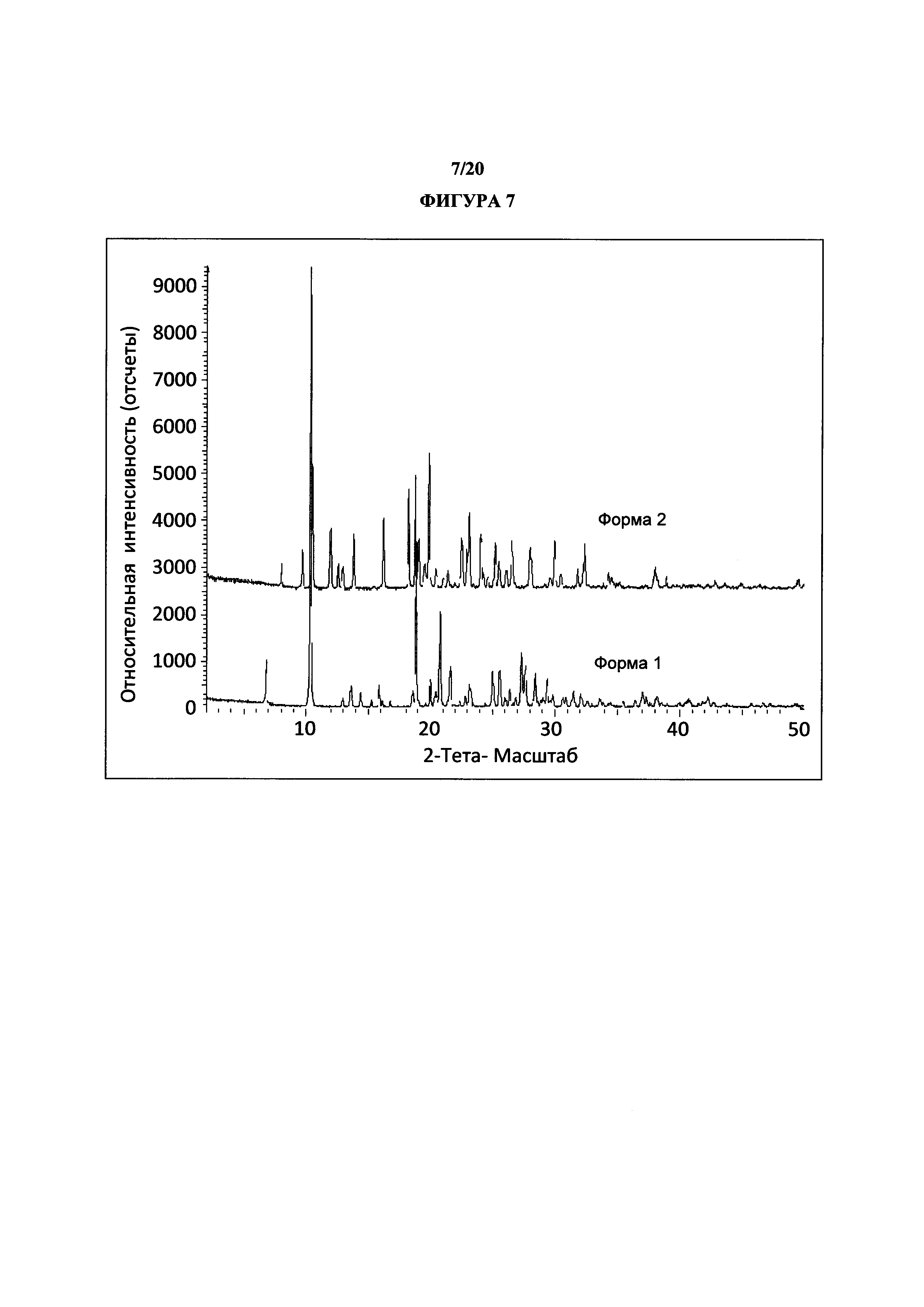
Фигура 7 представляет собой иллюстративное перекрывание порошковых рентгеновских дифрактограмм полиморфной формы 1 (нижняя) и формы 2 (верхняя).
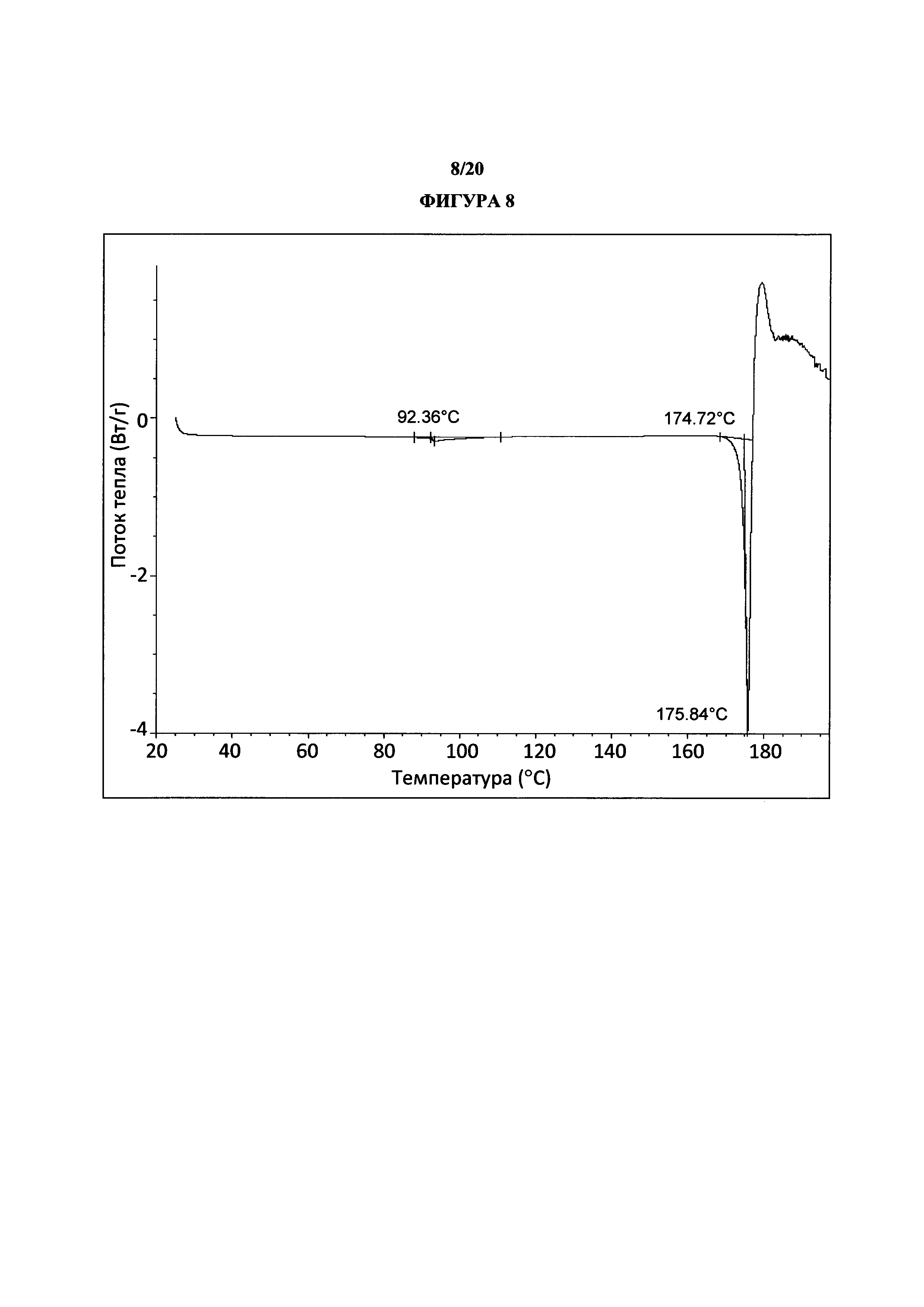
Фигура 8 представляет собой иллюстративную термограмму дифференциальной сканирующей калориметрии полиморфной формы 2.
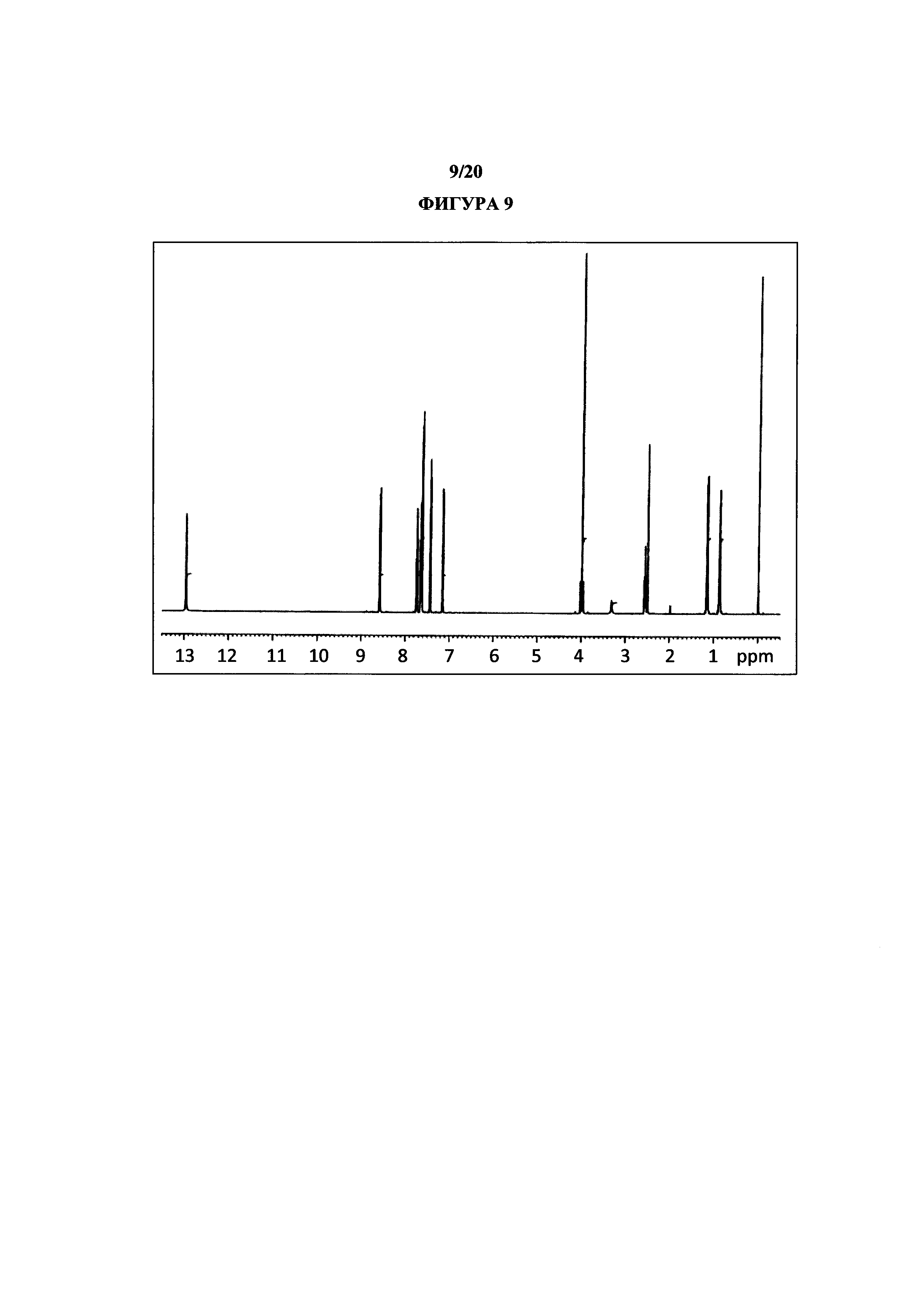
На фигуре 9 показан иллюстративный спектр 1H ЯМР (DMSO-d6) полиморфной формы 2.
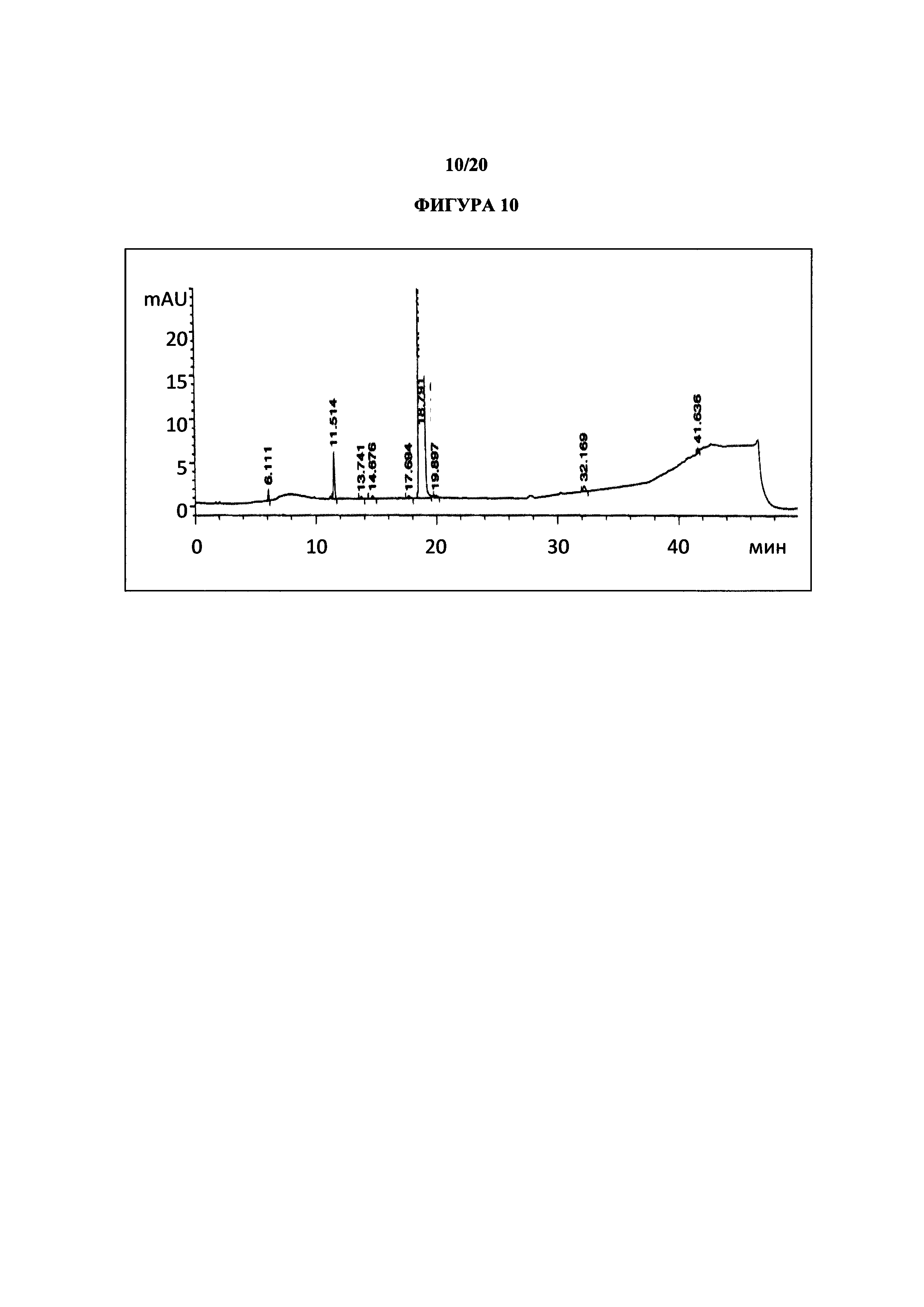
Фигура 10 представляет собой иллюстративную хроматограмму HPLC полиморфной формы 2.
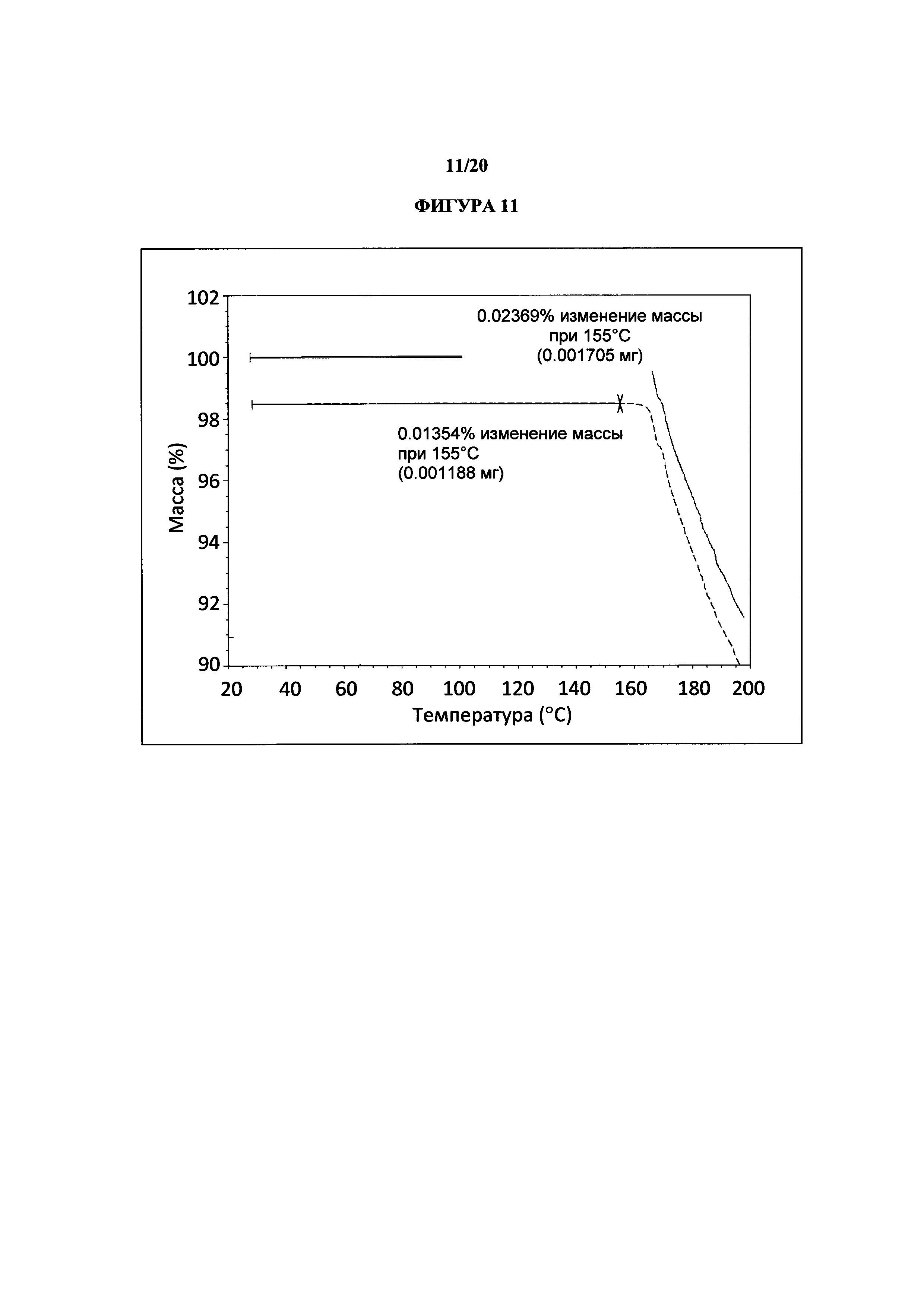
Фигура 11 представляет собой иллюстративную термограмму термогравиметрического анализа полиморфной формы 2.
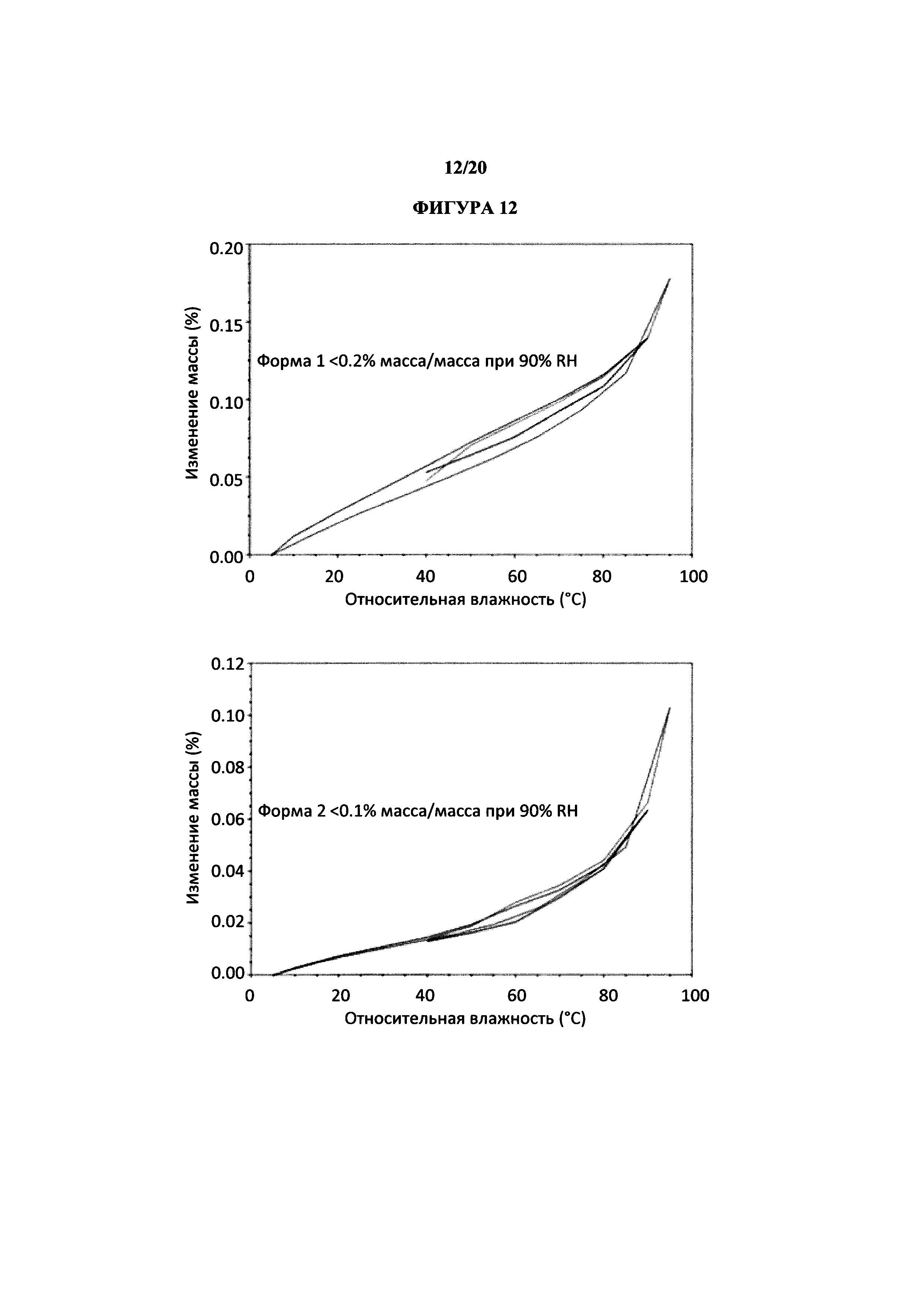
На фигуре 12 представлено иллюстративное исследование гравиметрической паровой сорбции полиморфной формы 1 и 2.
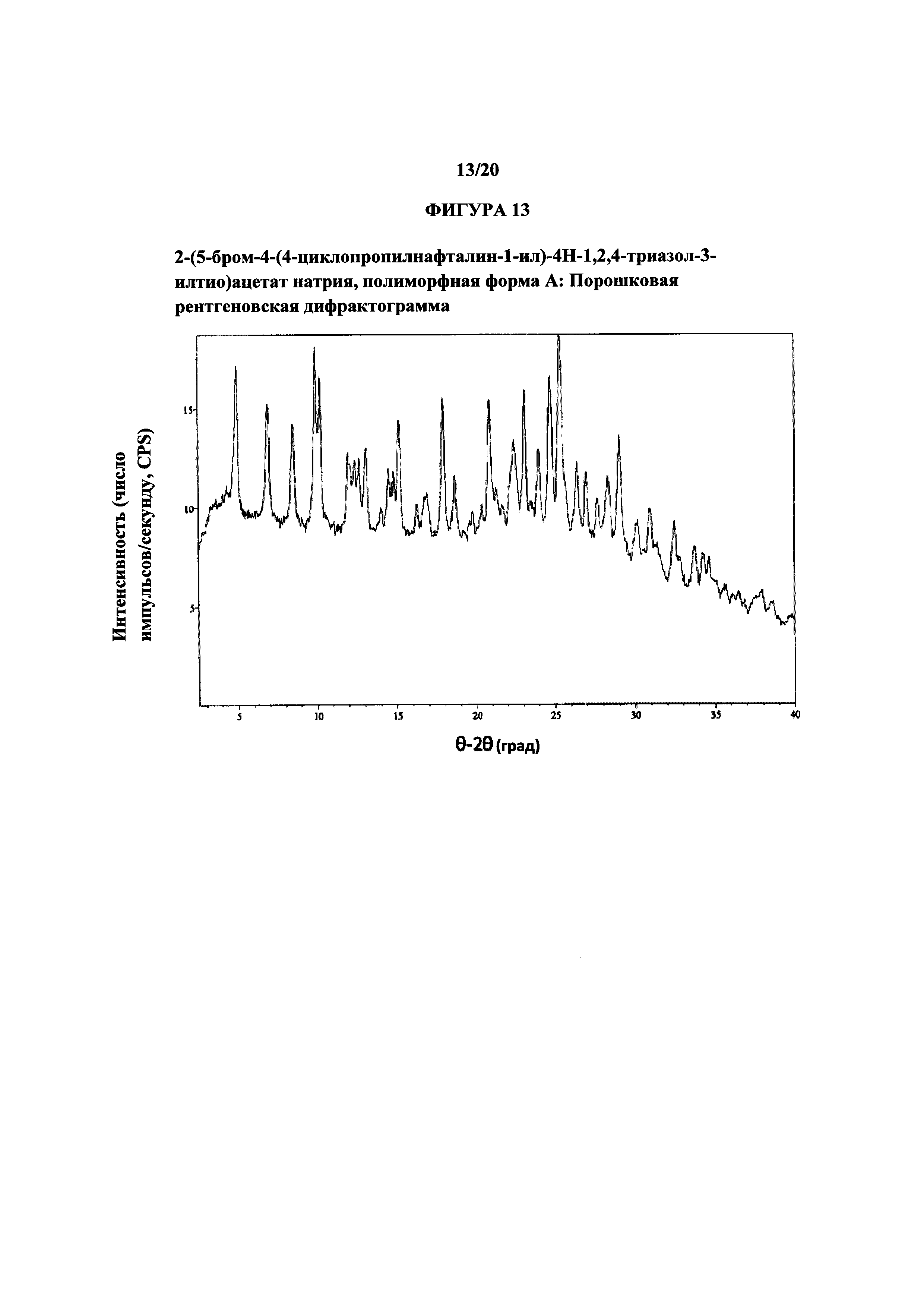
Фигура 13 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы А.
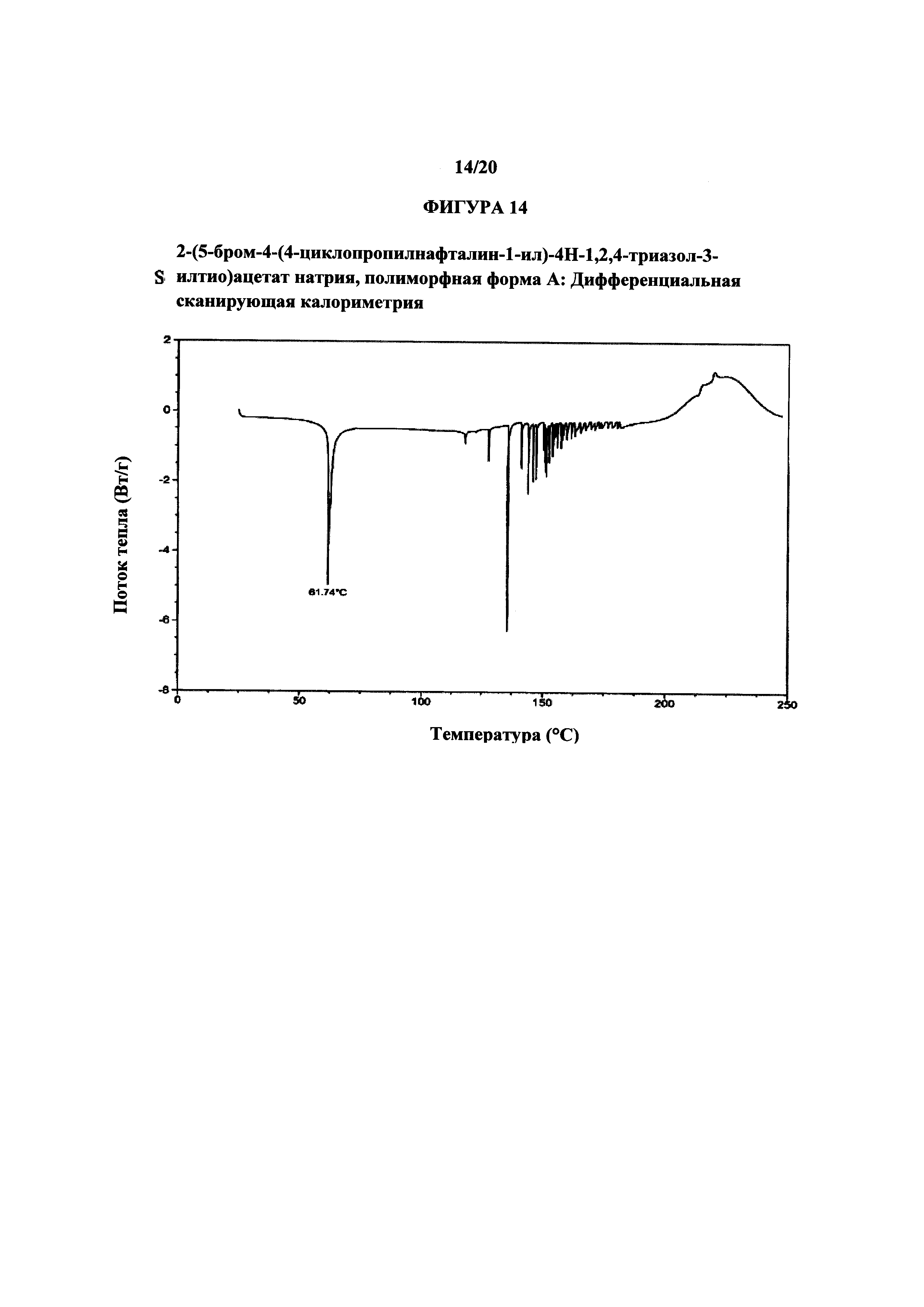
Фигура 14 представляет собой иллюстративную термограмму дифференциальной сканирующей калориметрии полиморфной формы А.
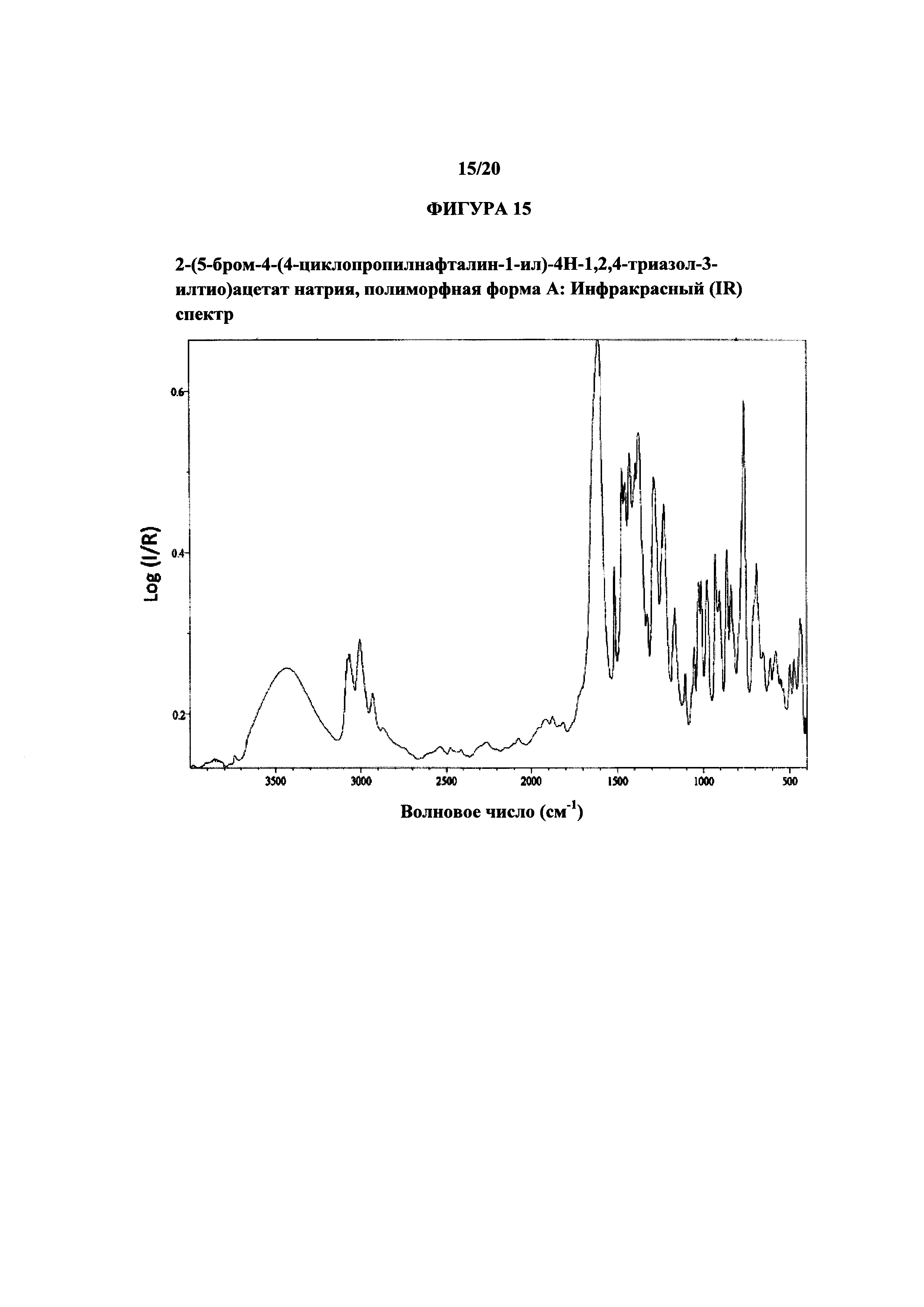
На фигуре 15 показан иллюстративный инфракрасный спектр полиморфной формы А.
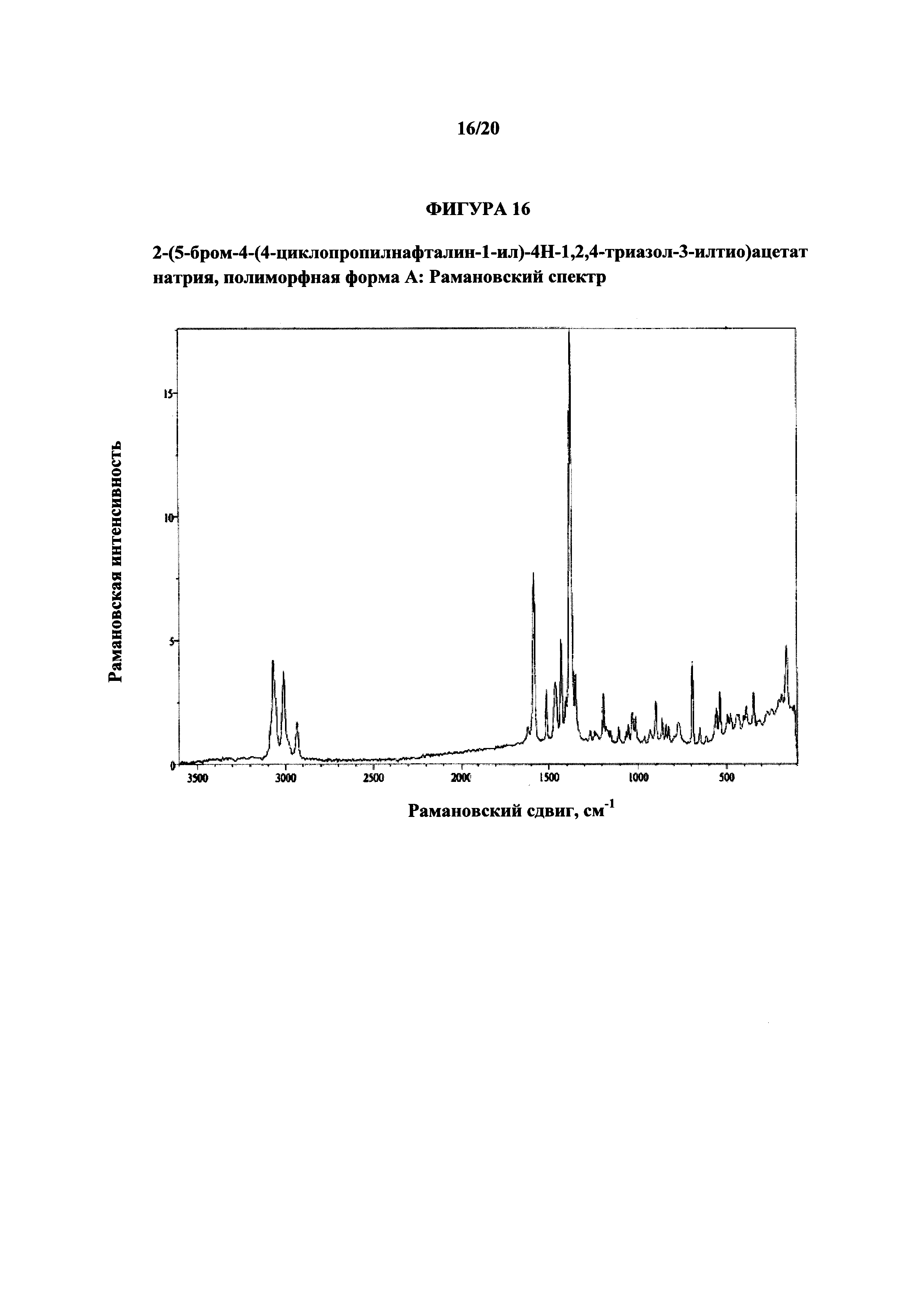
На фигуре 16 показан иллюстративный романовский спектр полиморфной формы А.
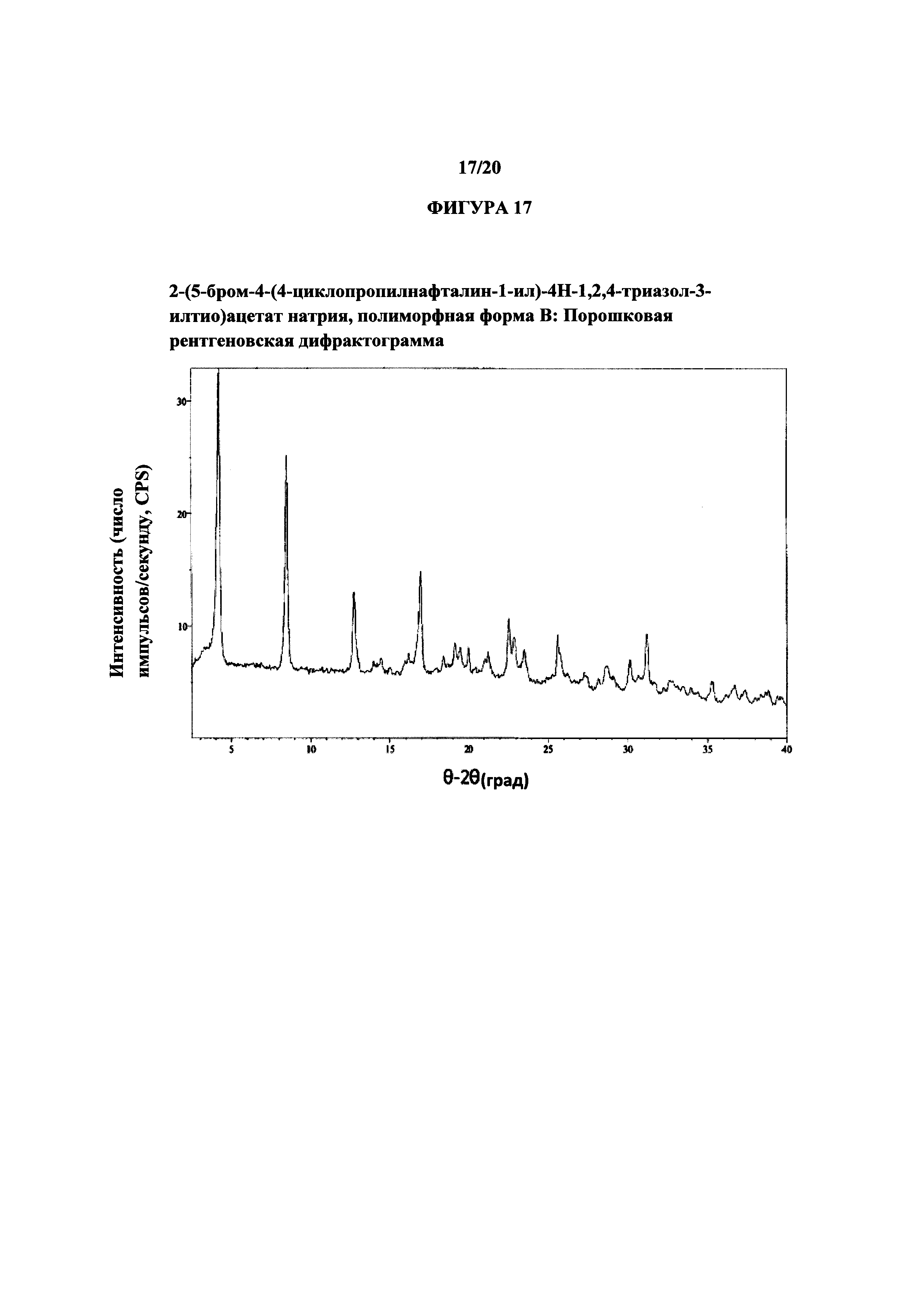
Фигура 17 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы В.
Фигура 18 представляет собой иллюстративную термограмму дифференциальной сканирующей калориметрии полиморфной формы В.
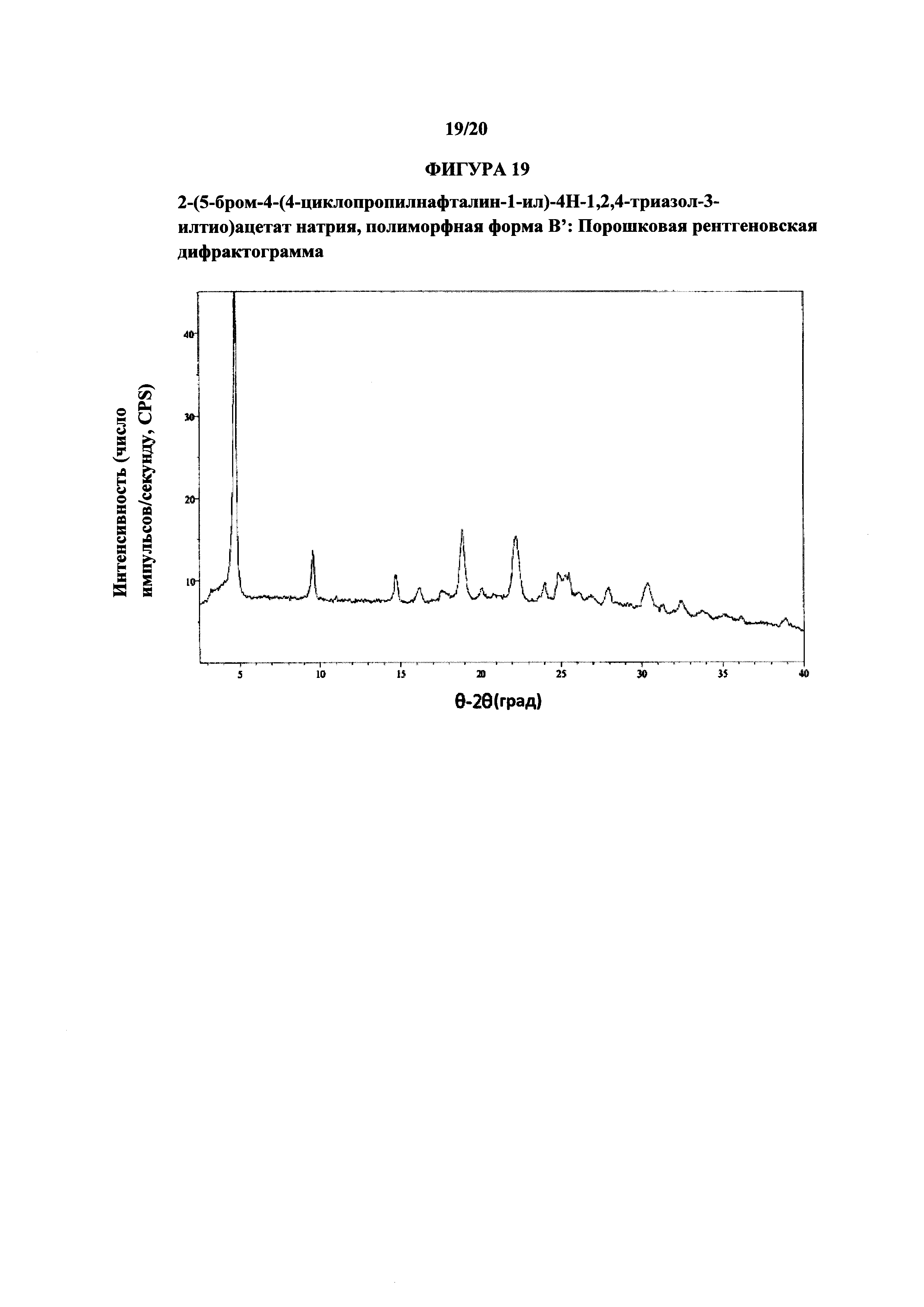
Фигура 19 представляет иллюстративную порошковую рентгеновскую дифрактограмму полиморфной формы В'.
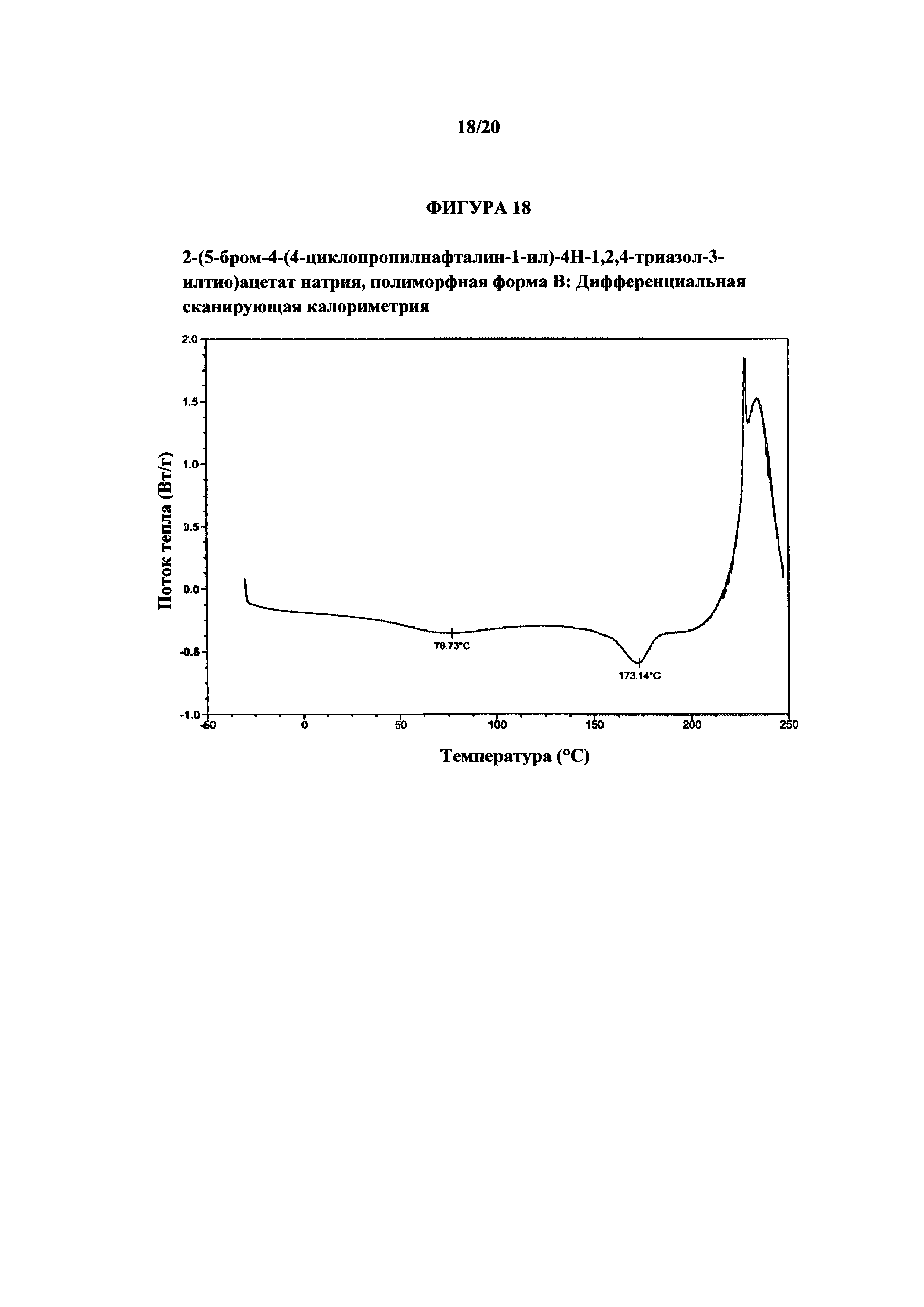
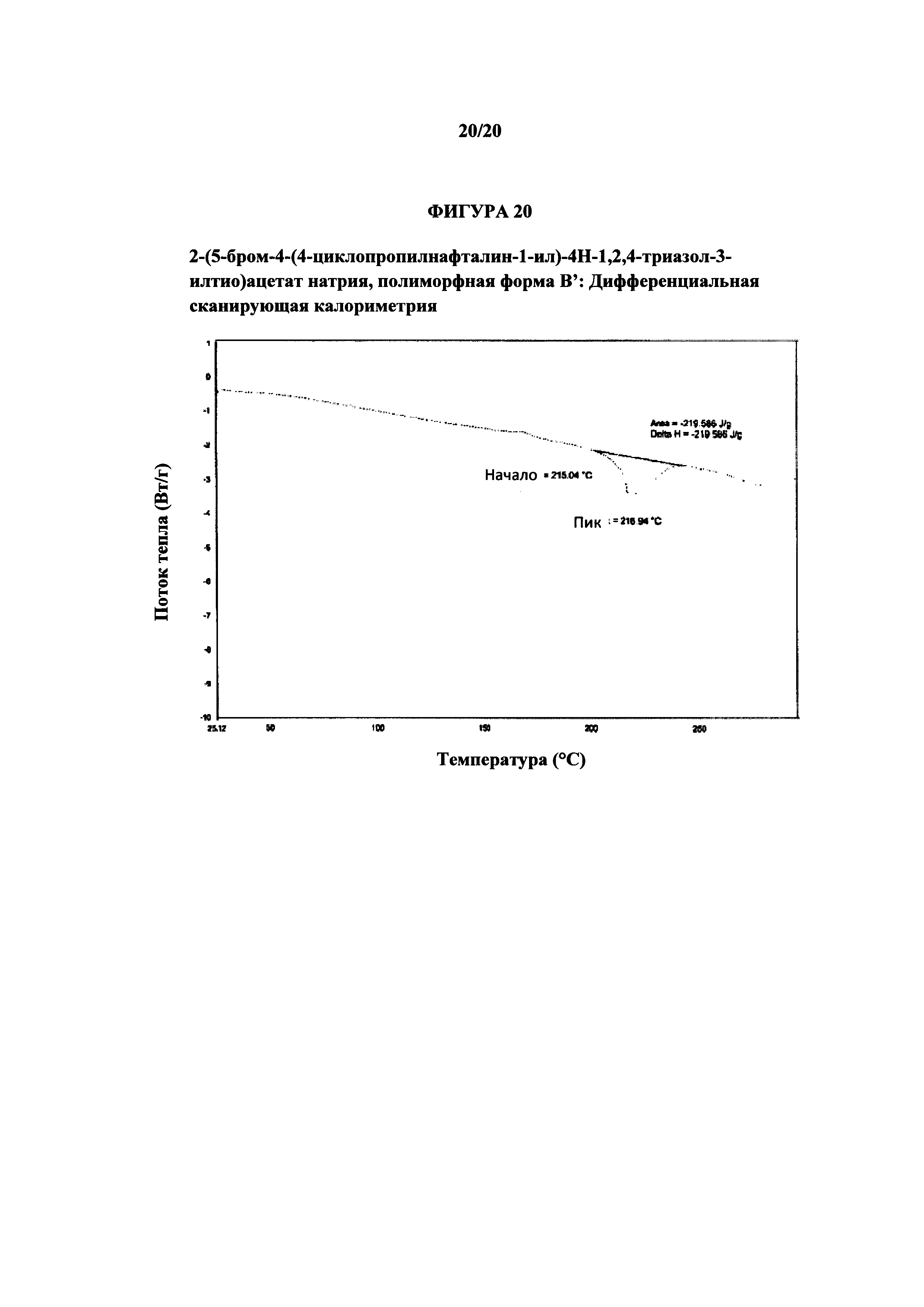
Фигура 20 представляет собой иллюстративную термограмму дифференциальной сканирующей калориметрии полиморфной формы В'.
Подробное описание изобретения
Как правило, производство в крупном масштабе клинически полезных кандидатных лекарственных средств, должно согласовываться с надлежащей производственной практикой. В настоящем документе предлагаются конкретные способы и способы для получения соединений Формулы (I):
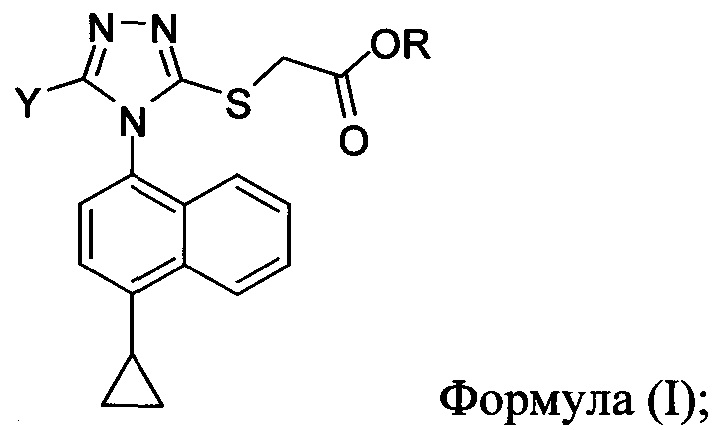
в которой
R представляет собой H, -C1-C20 алкил, -C1-C20 алкенил, -C3-C10 циклоалкил, -C3-C10 циклоалкенил, или R представляет собой противоион; и
Y представляет собой H, OH, NH2, F, Cl, Br или I.
Способы и способы синтеза, предложенные в настоящем документе, ликвидируют некоторые недостатки, связанные с получением, и обеспечивают синтез соединений высокой степени чистоты при одновременном уменьшении отходов и/или побочных продуктов, и уменьшении использования вызывающих коррозию материалов. Усовершенствованные способы и способы синтеза соединений Формулы (I), описанные здесь, обеспечивают получение в крупном масштабе, согласующееся с руководством по надлежащей производственной практике (GMP).
Определенная химическая терминология
Если не указано иное, все употребляемые здесь технические и научные термины имеют значение, обычно воспринимаемое специалистами с обычным уровнем подготовки в области техники, к которой относится заявленный объект изобретения. В случае наличия нескольких определений используемых здесь терминов, преимущественной силой будут обладать приведенные в данном разделе.
Следует понимать, что вышеизложенное общее описание и последующее подробное изложение являются лишь примерами и пояснениями, и не ограничивают ни один из заявленных объектов изобретения. В данной заявке использование единственного числа включает множественное, если специально не указано иное. Следует отметить, что используемое в настоящем описании и прилагаемой формуле изобретения указания на единственное число предполагает указание и на множественное число, если только контекст четко не указывает на обратное. Следует также отметить, что применение «или» означает «и/или», если не указано иное. Кроме того, использование термина «включая», а также других его форм, таких как «включает», «включают» и «включали», не является ограничивающим.
Определение стандартных химических терминов можно найти в работах, на которые сделана ссылка, включая, без ограничения, Carey and Sundberg "Advanced Organic Chemistry 4th Ed." Vols. A (2000) and В (2001), Plenum Press, New York. Если не указано иное, используются стандартные способы масс-спектроскопии, ядерного магнитного резонанса (NMR), высокоэффективной жидкостной хроматографии (HPLC), инфракрасного (IR) спектроскопического анализа и UV/Vis-спектроскопии, и фармакологии, известные в области техники. Если не указаны специальные определения, номенклатура, используемая здесь, является известной в области техники. Стандартные методы могут использоваться для химического синтеза, химических анализов, получения, изготовления и доставки фармацевтических средств и лечения пациентов. Реакции и методы очистки могут быть проведены, например, с применением наборов согласно инструкциям изготовителя, или так, как это обычно выполняется в данной области техники, или как описано здесь. Вышеизложенные методики и процедуры могут в целом выполняться обычными методами, известными в данной области техники, и как описано в различных общих и более конкретных источниках, цитируемых и обсуждаемых в описании данного изобретения. Во всем описании группы и их заместители могут быть выбраны специалистом в данной области таким образом, чтобы получить стабильные фрагменты и соединения.
Если группы заместителей определены их обычными химическими формулами, написанных слева направо, они в равной степени охватывают химически идентичные заместители, которые могут быть получены при записи структуры справа налево. Неограничивающим примером является -CH2O-, что эквивалентно -OCH2-.
Если не указано иное, использование общих химических терминов, таких как, но без ограничения, «алкил», «амин», «арил», эквивалентно их необязательно замещенным формам. Например, используемый здесь термин «алкил» включает необязательно замещенный алкил.
В некоторых вариантах осуществления соединения, представленные здесь, имеют один или несколько стереоцентров. В некоторых вариантах осуществления стереоцентр может существовать в R конфигурации, S конфигурации или их сочетании. В некоторых вариантах осуществления соединения, представленные здесь, имеют одну или несколько двойных связей. В некоторых вариантах осуществления соединения, представленные здесь, имеют одну или несколько двойных связей, и каждая из них существует в E (транс) или Z (цис) конфигурации, или их сочетании. Представление одного конкретного стереоизомера, региоизомера, диастереомера, энантиомера или эпимера следует рассматривать как включающее все возможные стереоизомеры, региоизомеры, диастереомеры, энантиомеры или эпимеры и их смеси. Таким образом, представленные здесь соединения включают все отдельные конфигурационные стереоизомерные, региоизомерные, диастереомерные, энантиомерные и эпимерные формы, а также их соответствующие смеси. Способы обращения или поддержания в неизменной форме определенного стереоцентра, а также разделения смесей стереоизомеров описаны, например, в Fumiss et al. (eds.), VOGEL'S ENCYCLOPEDIA OF PRACTICAL ORGANIC CHEMISTRY 5.sup.TH ED., Longman Scientific and Technical Ltd., Essex, 1991, 809-816; and Heller, Ace. Chem. Res. 1990, 23, 128.
Используемые здесь термины «фрагмент», «химический фрагмент», «группа» и «химическая группа» означают определенную часть или функциональную группу молекулы. Химические фрагменты обычно представляют собой известные химические структурные единицы, входящие в состав или присоединенные к молекуле.
Используемый здесь термин «реагент» означает нуклеофил или электрофил, используемый для создания ковалентных связей.
Термин «связь» или «одинарная связь» означает химическую связь между двумя атомами или двумя фрагментами, если соединенные связью атомы рассматриваются как часть более крупной подструктуры.
Термин «необязательный» или «необязательно» обозначает, что описанное далее событие или обстоятельство может иметь место, а может не иметь, так что описание включает и случаи, когда указанное событие или обстоятельство имеет место, и случаи, когда оно места не имеет. Например, «необязательно замещенный алкил» означает «алкил» или «замещенный алкил», как определено ниже. Кроме того, необязательно замещенная группа может быть незамещенной (например, -CH2CH3), полностью замещенной (например, -CF3CF3), монозамещенной (например, -CH2CH2F) или замещенной на любом уровне между полностью замещенной и монозамещенной (например, -CH2CHF2, -CH2CF3, -CF2CH3, -CFHCHF2, и т.п.). Специалисту в данной области техники будет понятно, что любые группы, содержащие один или несколько заместителей, не должны привносить замещение или паттерн замещения (например, замещенный алкил включает необязательно замещенные циклоалкильные группы, которые, в свою очередь, определены как включающие необязательно замещенные алкильные группы, и так, потенциально, до бесконечности), являющиеся стерически невозможными и/или синтетически нереальными. Таким образом, любые описанные заместители должны в целом восприниматься как имеющие максимальную молекулярную массу приблизительно 1000 дальтон, обычно, до приблизительно 500 дальтон (кроме случаев, когда макромолекулярные заместители явно определены, например, полипептиды, полисахариды, полиэтиленгликоли, ДНК, РНК и т.п.).
Используемый здесь термин C1-Cx включает C1-C2, C1-C3 … C1-Cx. Только в качестве примера, группа, обозначенная как «C1-C4», означает наличие от одного до четырех атомов углерода во фрагменте, т.е. группы, содержащие 1 атом углерода, 2 атома углерода, 3 атома углерода или 4 атома углерода, а также диапазоны C1-C2 и C1-C3. Таким образом, только в качестве примера, «C1-C4 алкил» означает наличие от одного до четырех атомов углерода в алкильной группе, т.е. алкильная группа выбрана из метила, этила, пропила, изо-пропила, н-бутила, изо-бутила, втор-бутила и трет-бутила. При любом ее появлении в настоящем документе числовой диапазон, такой как «от 1 до 10», относится к каждому целому числу в данном диапазоне; например, «от 1 до 10 атомов углерода» означает, что группа может иметь 1 атом углерода, 2 атома углерода, 3 атома углерода, 4 атома углерода, 5 атомов углерода, 6 атомов углерода, 7 атомов углерода, 8 атомов углерода, 9 атомов углерода или 10 атомов углерода.
Используемый здесь термин «алкил», отдельно или в сочетании, относится к необязательно замещенному с прямой цепью или необязательно замещенному с разветвленной цепью насыщенному углеводородному монорадикалу, содержащему от одного до десяти атомов углерода, более предпочтительно от одного до шести атомов углерода. Примеры включают, но без ограничения, метил, этил, н-пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, трет-амил и гексил, а также более длинные алкильные группы, такие как гептил, октил и т.п. При любом их появлении в настоящем документе, числовой диапазон, такой как «C1-C6 алкил» или «C1-6 алкил», означает, что алкильная группа может иметь 1 атом углерода, 2 атома углерода, 3 атома углерода, 4 атома углерода, 5 атомов углерода или 6 атомов углерода, хотя настоящее определение также включает использование термина «алкил» без указания числового диапазона.
Используемый здесь термин «алкилен», отдельно или в сочетании, относится к дирадикалу, производному от указанного выше монорадикала, алкила. Примеры включают, но без ограничения: метилен (-CH2-), этилен (-CH2CH2-), пропилен (-CH2CH2CH2-), изопропилен (-CH(CH3)CH2-) и т.п.
Используемый здесь термин «алкенил», отдельно или в сочетании, относится к необязательно замещенному с прямой цепью или необязательно замещенному с разветвленной цепью углеводородному монорадикалу, содержащему одну или несколько углерод-углеродных двойных связей и от двух до приблизительно десяти атомов углерода, более предпочтительно от двух до шести атомов углерода. Эта группа может быть в цис- или транс-конформации относительно двойной связи(ей) и должна пониматься как охватывающая оба изомера. Примеры включают, но без ограничения, этенил (-CH=CH2), 1-пропенил (-CH2CH=CH2), изопропенил [-C(CH3)=CH2], бутенил, 1,3-бутадиенил и т.п. При любом появлении в настоящем документе числовой диапазон, такой как «C2-C6 алкенил» или «C2-6 алкенил», означает, что алкенильная группа может иметь 2 атома углерода, 3 атома углерода, 4 атома углерода, 5 атомов углерода или 6 атомов углерода, хотя настоящее определение также включает использование термина «алкенил» без указания числового диапазона.
Используемый здесь термин «алкенилен», отдельно или в сочетании, относится к дирадикалу, производному от указанного выше монорадикала, алкенила. Примеры включают, но без ограничения, этенилен (-CH=CH-), пропениленовые изомеры (например, -CH2CH=CH- и -C(CH3)=CH-) и т.п.
Используемый здесь термин «алифатический», отдельно или в сочетании, относится к необязательно замещенному, имеющему прямую или разветвленную цепь, нециклическому, насыщенному, частично ненасыщенному или полностью ненасыщенному неароматическому углеводороду. Таким образом, термин обобщенно включает алкильные, алкенильные и алкинильные группы.
Используемый здесь термин «углеродная цепь», отдельно или в сочетании, относится к любой алкильной, алкенильной, алкинильной, гетероалкильной, гетероалкенильной или гетероалкинильной группе, которая является линейной, циклической или любой их сочетаниями. Если цепь является частью линкера и этот линкер включает одно или несколько колец в составе основного скелета, для целей расчета длины цепи сама «цепь» включает только те атомы углерода, которые составляют верхнюю или нижнюю часть этого кольца, а не то и другое, а если верхняя и нижняя часть кольца(ец) имеют неодинаковую длину, для определения длины цепи используется более короткое расстояние. Если цепь содержит гетероатомы в составе основного скелета, эти атомы не считаются частью длины углеродной цепи.
Используемые здесь термины «цикл», «циклический», «кольцо» и «-членное кольцо», отдельно или в сочетании, относятся к любой ковалентно замкнутой структуре, включая алициклические, гетероциклические, ароматические, гетероароматические и полициклические слитые или неслитые кольцевые системы, описанные здесь. Кольца могут быть необязательно замещенными. Кольца могут образовывать часть слитой кольцевой системы. Термин «-членный» указывает число скелетных атомов, составляющих кольцо. Таким образом, только в качестве примера, циклогексан, пиридин, пиран и пиримидин являются шестичленными кольцами, а циклопентан, пиррол, тетрагидрофуран и тиофен - пятичленными кольцами.
Используемый здесь термин «слитый», отдельно или в сочетании, относится к циклическим структурам, в которых два или больше колец имеют одну или несколько общих связей.
Используемый здесь термин «циклоалкил», отдельно или в сочетании, относится к необязательно замещенному, насыщенному, углеводородному монорадикальному кольцу, содержащему от трех до приблизительно пятнадцати атомов углерода в кольце, или от трех до приблизительно десяти атомов углерода в кольце, хотя оно может включать дополнительные, не входящие в кольцо атомы углерода в качестве заместителей (например, метилциклопропил). При любом появлении в настоящем документе, числовой диапазон, такой как «C3-C6 циклоалкил» или «C3-6 циклоалкил», означает, что циклоалкильная группа может состоять из 3 атомов углерода, 4 атомов углерода, 5 атомов углерода или 6 атомов углерода, то есть представляет собой циклопропил, циклобутил, циклопентил или циклогептил, хотя данное определение также включает использование термина «циклоалкил» без указания числового диапазона. Термин включает слитые, неслитые, мостиковые и спиро радикалы. Слитый циклоалкил может содержать от двух до четырех слитых колец, при этом кольцом прикрепления является циклоалкильное кольцо, а другие отдельные кольца могут быть алициклическими, гетероциклическими, ароматическими, гетероароматическими или любым их сочетанием. Примеры включают, но без ограничения, циклопропил, циклопентил, циклогексил, декалинил и бицикло[2.2.1]гептил и адамантильные кольцевые системы. Иллюстративные примеры включают, но не ограничиваются следующими фрагментами:
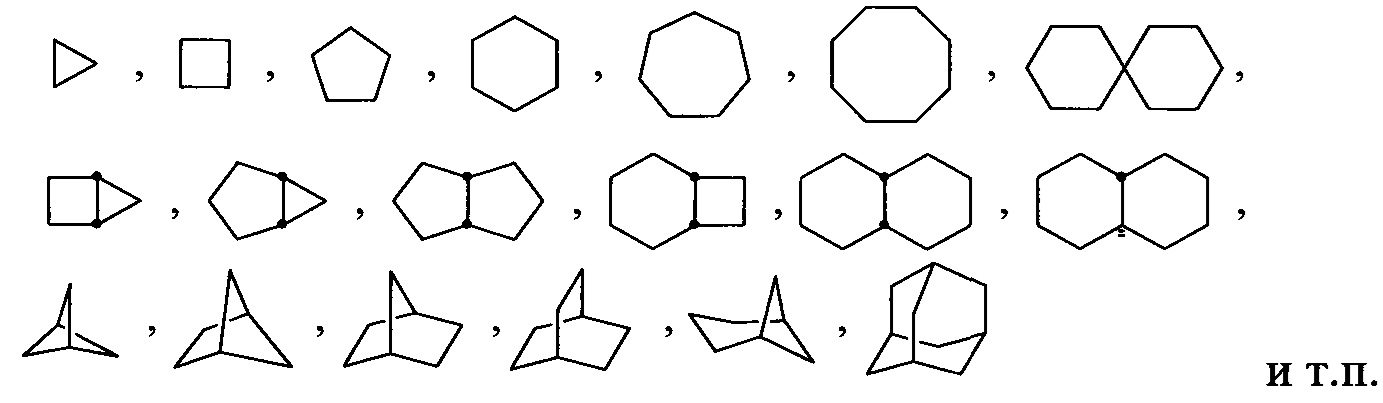
Используемый здесь термин «циклоалкенил», отдельно или в сочетании, относится к необязательно замещенному углеводородному неароматическому монорадикальному кольцу, имеющему одну или несколько двойных углерод-углеродных связей и от трех до приблизительно двадцати атомов углерода в кольце, от трех до приблизительно двенадцати атомов углерода в кольце, или от трех до приблизительно десяти атомов углерода в кольце. Термин включает слитые, неслитые, мостиковые и спиро радикалы. Слитый циклоалкенил может содержать от двух до четырех слитых колец, при этом кольцом прикрепления является циклоалкенильное кольцо, а другие отдельные кольца могут быть алициклическими, гетероциклическими, ароматическими, гетероароматическими или любым их сочетанием. Слитые кольцевые системы могут быть слиты по связи, такой как углерод-углеродная одинарная связь или двойная связь. Примеры циклоалкенилов включают, но без ограничения, циклогексенил, циклопентадиенил и бицикло[2.2.1]гепт-2-еновые кольцевые системы. Иллюстративные примеры включают, но без ограничения, следующие фрагменты:
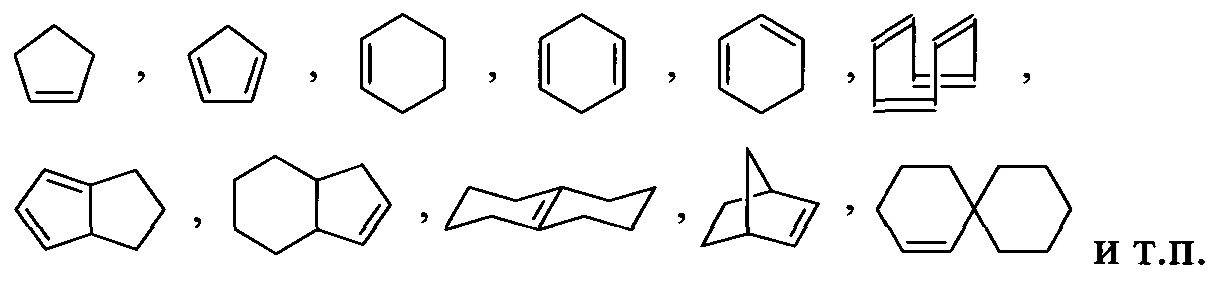
Используемые здесь термины «алициклил» или «алициклический», отдельно или в сочетании, относятся к необязательно замещенным, насыщенным, частично ненасыщенным или полностью ненасыщенным неароматическим углеводородным кольцевым системам, содержащим от трех до приблизительно двадцати атомов углерода в кольце, от трех до приблизительно двенадцати атомов углерода в кольце, или от трех до приблизительно десяти атомов углерода в кольце. Таким образом, эти термины совместно включают циклоалкильные и циклоалкенильные группы.
Используемый здесь термин «карбоциклил», отдельно или в сочетании, означает обобщенно алициклильные или арильные группы; то есть все углеродные, ковалентно замкнутые кольцевые структуры, которые могут быть насыщенными, частично ненасыщенными, полностью ненасыщенными или ароматическими. Карбоциклические кольца могут быть образованы тремя, четырьмя, пятью, шестью, семью, восемью, девятью или более чем девятью атомами углерода. Карбоциклы могут быть необязательно замещенными. Этот термин отличает Карбоциклические от гетероциклических колец, в которых кольцевой скелет содержит по меньшей мере один атом, отличный от углерода.
Используемые здесь термины «галоген», «галогено» или «галогенид», отдельно или в сочетании, означают к фтор, хлор, бром и йод.
Используемый здесь термин «гидрокси», отдельно или в сочетании, означает монорадикал -OH.
Используемые здесь термины «карбокси» или «карбоксил», отдельно или в сочетании, означают фрагмент -C(O)OH, который может быть также записан как -COOH. «Карбоксилатный анион» представляет собой депротонированный карбоксильный фрагмент и записан как -COO или -COO-.
Следует понимать, что в случаях использования двух или большего числа радикалов подряд для определения заместителя, присоединенного к структуре, первый названный радикал считается концевым, а последний названный радикал считается присоединенным к рассматриваемой структуре. Таким образом, например, радикал арилалкил прикреплен к рассматриваемой структуре алкильной группой.
Используемый здесь «3,5-дизамещенный 4-(4-RC-нафталин-1-ил)-4Н-1,2,4-триазол» относится к:
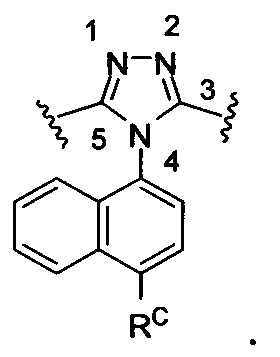
Используемый здесь «3-замещенный-5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол» относится к:
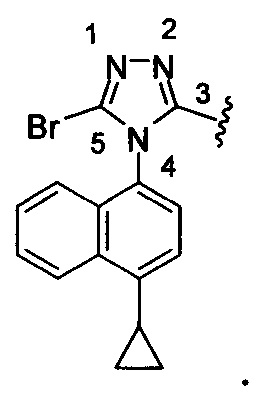
Термин «полиморфная форма 1» относится к кристаллической форме 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоте, которая проявляет порошковую рентгеновскую дифрактограмму по существу такую же, как показанная на фигуре 1 и/или фигуре 2, и/или профиль дифференциальной сканирующей калориметрии по существу такой же, как показанный на фигуре 3.
Термин «полиморфная форма 2» относится к кристаллической форме 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты, которая проявляет порошковую рентгеновскую дифрактограмму по существу такую же, как показанная на фигуре 5 и/или фигуре 6, и/или профиль дифференциальной сканирующей калориметрии по существу такой же, как показанный на фигуре 8.
Термин «полиморфная форма А» относится к кристаллической форме 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, которая проявляет порошковую рентгеновскую дифрактограмму по существу такую же, как показанная на фигуре 13, и/или профиль дифференциальной сканирующей калориметрии по существу такой же, как показанный на фигуре 14.
Термин «полиморфная форма В» относится к кристаллической форме 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, которая проявляет порошковую рентгеновскую дифрактограмму по существу такую же, как показанная на фигуре 17, и/или профиль дифференциальной сканирующей калориметрии по существу такой же, как показанный на фигуре 18.
Термин «полиморфная форма В'» относится к кристаллической форме 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, которая проявляет порошковую рентгеновскую дифрактограмму по существу такую же, как показанная на фигуре 19, и/или профиль дифференциальной сканирующей калориметрии по существу такой же, как показанный на фигуре 20.
Используемый здесь «противоион» представляет собой ион, присутствие которого обеспечивает образование всех нейтрально заряженных соединений. Соответственно, в одном случае противоион представляет собой положительно заряженный ион, например, для соединения Формулы (I), который уравновешивает отрицательный заряд, связанный с карбоксилатным анионом соединения Формулы (I) и обеспечивает образование в целом нейтрально заряженных соединений. Примеры противоионов для соединений Формулы (I) включают, но не ограничиваются, Na+, K+, Li+, Cs+, Mg++, Fe+++, Al+++, или любой другой фармацевтически приемлемый органический или неорганический катион. В случае, когда катион имеет более одного единичного заряда (например, Mg++), следует понимать, что один или несколько карбоксилатных анионных фрагментов (например, карбоксилатные фрагменты соединений Формулы (I)) потребуется для образования в целом нейтрально заряженных соединений. В другом случае, противоион представляет собой отрицательно заряженный ион. Например, для Соединения 8 отрицательно заряженный противоион уравновешивает положительный заряд, связанный с протонированной формой Соединения 8. Примеры таких отрицательно заряженных противоионов включают, но без ограничения, оксалатный, цитратный, тартратный, ацетатный, хлоридный, бромидный, фторидный или любой другой фармацевтически приемлемый органический или неорганический анион.
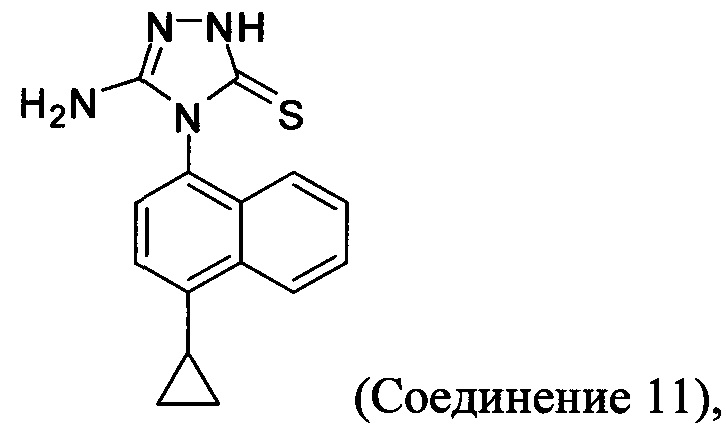
Начальные попытки
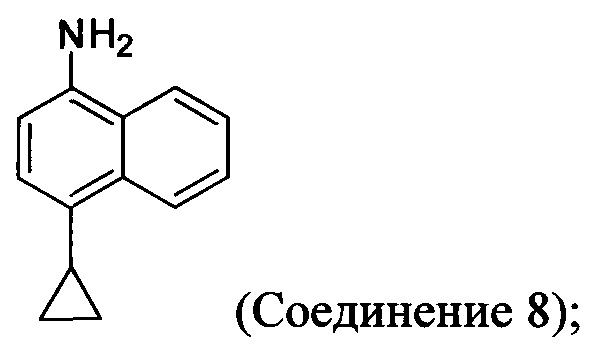
Начальные усилия для идентификации оптимальных условий синтеза соединений Формулы (I), включающих, например. Соединение 1 и Соединение 4, были направлены на использование Соединения 11 (3-амино-4-(4-циклопропилнафталин-1-ил)-1Н-1,2,4-триазол-5(4Н)-тион) в качестве исходного материала.
На Схеме 1 показан иллюстративный синтез Соединения 1 и Соединения 4.

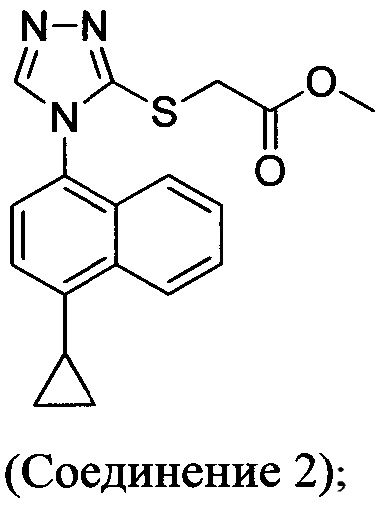
Начиная с Соединения 11, в результате взаимодействия с соединением Формулы (IV) в присутствии DMF в качестве растворителя получают соединение Формулы (V). Можно использовать любой реагент Формулы (IV). В одном варианте осуществления о реакцию Соединения 11 проводят с бром-метилацетатом 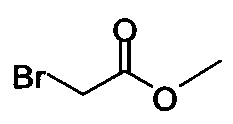 или хлор-метилацетатом
или хлор-метилацетатом 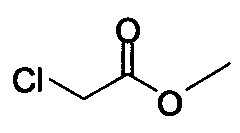 для получения соединения Формулы (V), в которой R представляет собой метил (Соединение 2). В другом варианте осуществления реакцию Соединения 11 проводят с бром-этилацетатом или хлор-этилацетатом для получения соединения Формулы (V), в которой R представляет собой этил (Соединение 2-А). В другом варианте осуществления реакцию Соединения 11 проводят с бромуксусной кислотой
для получения соединения Формулы (V), в которой R представляет собой метил (Соединение 2). В другом варианте осуществления реакцию Соединения 11 проводят с бром-этилацетатом или хлор-этилацетатом для получения соединения Формулы (V), в которой R представляет собой этил (Соединение 2-А). В другом варианте осуществления реакцию Соединения 11 проводят с бромуксусной кислотой 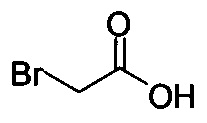 для получения соединения Формулы (V), в которой R представляет собой H. Используют любой подходящий растворитель. В одном варианте осуществления растворитель представляет собой DMF. В альтернативных вариантах осуществления растворитель представляет собой диоксан, ацетонитрил, хлороформ, дихлорметан, тетрагидрофуран (THF), N-метилпирролидон (NMP), диметилсульфоксид (DMSO) и т.п.
для получения соединения Формулы (V), в которой R представляет собой H. Используют любой подходящий растворитель. В одном варианте осуществления растворитель представляет собой DMF. В альтернативных вариантах осуществления растворитель представляет собой диоксан, ацетонитрил, хлороформ, дихлорметан, тетрагидрофуран (THF), N-метилпирролидон (NMP), диметилсульфоксид (DMSO) и т.п.
Соединение Формулы (V) затем подвергают реакции с бромидом меди (II) в присутствии нитрита калия и растворителя, и температуру реакции поддерживают примерно при комнатной температуре или ниже для получения соединения Формулы (III). В некоторых вариантах осуществления соединение формулы (V) подвергают воздействию CuCl2/KNO2, CuCl2/NaNO2, CuBr2/NaNO2, pTsOH/NaNO2/KBr или Br2. В одном варианте осуществления температура реакции находится в интервале от примерно 14°C до примерно 22°C. В другом варианте осуществления температура реакции находится в интервале от примерно 12°C до примерно 25°C. Используют любой подходящий растворитель. В одном варианте осуществления растворитель представляет собой ацетонитрил. В другом варианте осуществления растворитель представляет собой дихлорметан, NMP, диоксан, THF и т.п. В одном варианте осуществления в результате реакции получают соединение Формулы (III), которое представляет собой Соединение 3. В другом варианте осуществления в результате реакции получают соединение Формулы (III), которое представляет собой Соединение 3-А. В другом варианте осуществления, в котором R представляет собой H, в результате реакции получают Соединение 1.
В объеме соединений Формулы (III) предусматриваются соединения сложных эфиров, которые представляют собой пролекарства Соединения 1. Примеры таких сложных эфиров включают и не ограничиваются метиловым, этиловым, пропиловым, изопропиловым, бутиловым, трет-бутиловым, изоамиловым, пентиловым, гексиловым, гептиловым, октиловым, нониловым, терпениловым, борниловым, аллиловым, линалиловым и/или гераниловым сложными эфирами.
Сложноэфирную группу в соединении Формулы (III) гидролизуют с использованием любой подходящей кислоты, включая, но без ограничения, уксусную, трифторуксусную, серную, азотную, фосфорную, соляную или бромистоводородную кислоту, для получения Соединения 1.
Затем Соединение 1 превращают в соль. В одном варианте осуществления Соединение 1 перемешивают в водном растворе гидроксида натрия для получения Соединения 4. В некоторых вариантах осуществления Соединение 4 представляет собой кристаллический полиморф, характеризующийся пиками при 4,90, 9,83 и 25,29 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В некоторых вариантах осуществления Соединение 4 представляет собой кристаллическую полиморфную форму А. В альтернативных вариантах осуществления Соединение 1 перемешивают в водном растворе гидроксида калия, гидроксида лития, гидроксида цезия или любом другом подходящем основном растворе для получения соединения Формулы (I), в которой R представляет собой противоион. В некоторых таких вариантах осуществления спирт (например, метанол, этанол, изопропанол) используют в качестве сорастворителя для стадии реакции, включающей превращение Соединения 1 в соль.
Водный раствор Соединения 4 (или соединения Формулы (I), в которой R представляет собой противоион) подкисляют с использованием подходящей кислоты, такой как бромистоводородная кислота. Другие кислоты, которые являются подходящими для этой стадии, включают, но без ограничения, уксусную, трифторуксусную, серную, азотную, фосфорную, соляную кислоту и т.п. Смесь экстрагируют подходящим органическим растворителем, таким как этилацетат. Другие растворители, подходящие для экстракции, включают, но без ограничения, дихлорметан, трет-бутилметиловый эфир, трет-бутанол и т.п. Соединение 1 затем необязательно перекристаллизуют из EtOAc. В некоторых вариантах осуществления Соединение 1 перекристаллизуют из этилацетата и с использованием н-гептанов в качестве растворителя противоионов. Следует учесть, что для перекристаллизации Соединения 1 можно использовать любой другой подходящий растворитель или комбинацию растворителей.
В некоторых вариантах осуществления Соединение 1 представляет собой кристаллический полиморф, характеризующийся пиками при 10,32, 18,84 и 20,75 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В определенных вариантах осуществления Соединение 1 представляет собой полиморфную форму 1. В других вариантах осуществления Соединение 1 представляет собой кристаллический полиморф, характеризующийся пиками при 10,46, 18,76 и 19,83 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В определенных вариантах осуществления Соединение 1 представляет собой кристаллическую полиморфную форму 2.
Синтез Соединения 11 - первый способ
Синтез Соединения 11, описанный на Схеме 1, был успешно осуществлен в одном иллюстративном варианте осуществления с использованием последовательности реакций, описанных на Схеме 2.
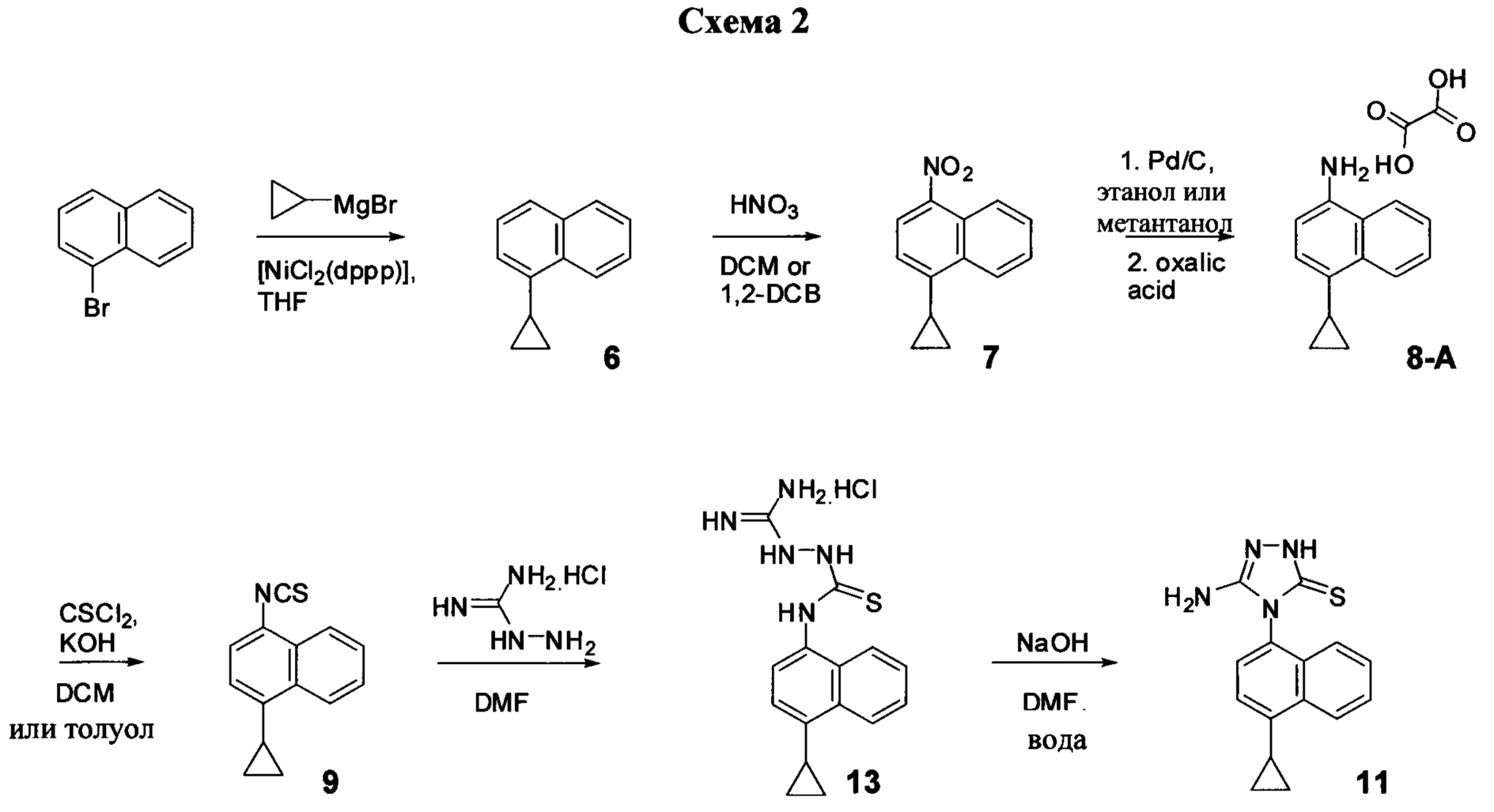
Начиная с 1-бромнафталина, в результате реакции с подходящим реактивом Гриньяра получают Соединения 6.
Затем получают Соединение 11 с помощью способа (Способ 6), включающего:
(11-i) приведение в контакт Соединения 7 с водородом, палладием на угле в одном или нескольких подходящих растворителях для получения соединения, имеющего структуру:
(11-i) приведение в контакт Соединения 8 с щавелевой кислотой для получения оксалата Соединения 8;
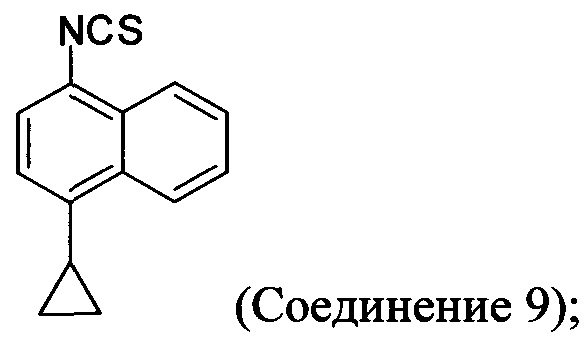
(11-iii) приведение в контакт оксалата Соединения 8, полученного на стадии (11-i), с основанием, тиофосгеном и растворителем, и перемешивание смеси при температуре ниже комнатной для получения соединения структуры:
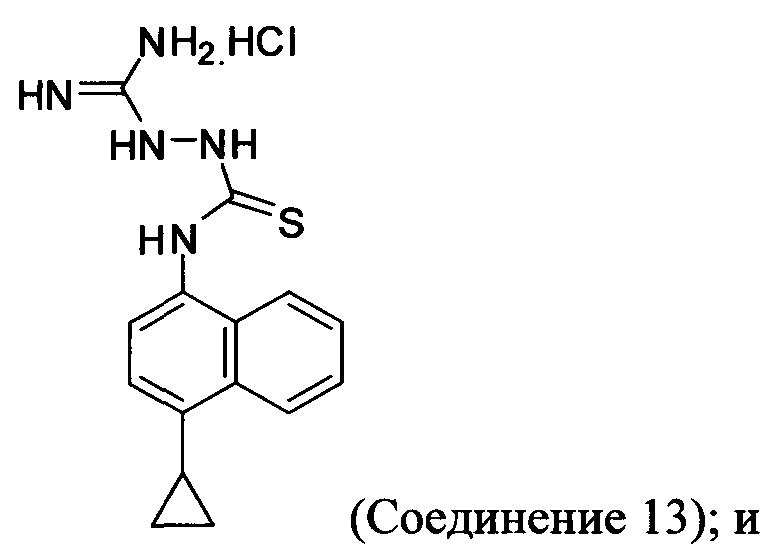
(11-iv) приведение в контакт Соединения 9, полученного на стадии (11-iii), с гидрохлоридом аминогуанидина, основанием и растворителем для получения соединения, имеющего структуру:
(11-v) приведение в контакт Соединения 13, полученного на стадии (11-iv), с основанием, водой и растворителем для получения Соединения 11.
В настоящем документе предлагается Соединение 9, полученное с помощью Способа 6, описанного выше. В настоящем документе предлагается Соединение 11, полученное с помощью Способа 6, описанного выше. В настоящем документе предлагается Соединение 13, полученное с помощью Способа 6, описанного выше.
В одном варианте осуществления растворитель, подходящий для использования на стадии (11-i) Способа 6, описанного выше, представляет собой метанол, этанол или дихлорбензол, или любую их комбинацию. В одном варианте осуществления растворитель, подходящий для использования на стадии (11-i) Способа 6, представляет собой метанол. В другом варианте осуществления растворитель, подходящий для использования на стадии (11-i) Способа 6, представляет собой этанол. Альтернативные растворители, такие как, например, THF, также предусматриваются в объеме вариантов осуществления, представленных в настоящем документе.
В объеме вариантов осуществления, представленных здесь, предусматривается использование других кислот и/или солей кислот на стадии (11-ii) и (11-iii) Способа 6, описанного выше, включая и не ограничиваясь использованием лимонной кислоты, винной кислоты, уксусной кислоты, соляной кислоты и т.п. для получения соответствующих кислых солей Соединения 8.
В одном варианте осуществления растворитель, подходящий для использования на стадии (11-iii) Способа 6, представляет собой толуол. В альтернативных вариантах осуществления растворитель, подходящий для использования на стадии (11-iii) Способа 6, представляет собой дихлорбензол, дихлорметан, ксилолы или любой другой подходящий растворитель. В некоторых случаях реакционную смесь, полученную на стадии (11-iii) Способа 6, перемешивают при температуре между примерно 0°C и примерно 10°C, между примерно 5°C и примерно 15°C, или между примерно 5°C и примерно 25°C. В некоторых случаях реакционную смесь, полученную на стадии (11-iii) Способа 6, перемешивают при примерно 5°C. В некоторых случаях реакционную смесь, полученную на стадии (11-iii) Способа 6, перемешивают при примерно комнатной температуре.
В последующих вариантах осуществления основание, подходящее для реакции на стадии (11-iii) Способа 6, выбирают из гидроксида калия, гидроксида натрия, гидроксида лития, карбоната калия, карбоната цезия, фосфата калия или любого другого подходящего основания. В некоторых вариантах осуществления основание, подходящее для реакции на стадии (11-iii) Способа 6, представляет собой гидроксид калия.
В последующих вариантах осуществления основание, подходящее для реакции на стадии (11-iv) Способа 6, представляет собой органическое или неорганическое основание. Неограничивающие примеры включают триэтиламин, диизопропиламин, диизопропилэтиламин, гидроксид калия, гидроксид натрия, гидроксид лития, карбонат калия, карбонат цезия, фосфат калия или любое другое подходящее основание. В одном варианте осуществления реакцию на стадии (11-iv) Способа 6, описанного выше, проводят в присутствии диизопропилэтиламина (DIEA) или гидроксида натрия. Для реакции на стадии (11-iv) Способа 6 выбирают любой подходящий растворитель, включая, например, DMF, THF, ацетонитрил, диоксан, NMP или т.п. В одном варианте осуществления реакцию на стадии (11-iv) Способа 6 проводят в DMF.
В одном варианте осуществления растворитель, подходящий для использования на стадии (11-v) Способа 6, представляет собой DMF. В альтернативном варианте осуществления растворитель, подходящий для использования на стадии (11-v) Способа 6, представляет собой толуол, дихлорбензол, ксилолы, NMP, ацетонитрил, диоксан или любой другой подходящий растворитель. В последующих вариантах осуществления основание, подходящее для реакции на стадии (11-v) Способа 6, выбирают из гидроксида калия, гидроксида натрия, гидроксида лития, карбоната калия, карбоната цезия, фосфата калия или любого другого подходящего основания. В некоторых вариантах осуществления основание, подходящее для реакции на стадии (11-v) Способа 6, представляет собой гидроксид натрия.
Синтез Соединения 11 - второй способ
В другом иллюстративном варианте осуществления синтез Соединения 11, описанный на Схеме 1, осуществляют с использованием последовательности реакций, описанных на Схеме 3.
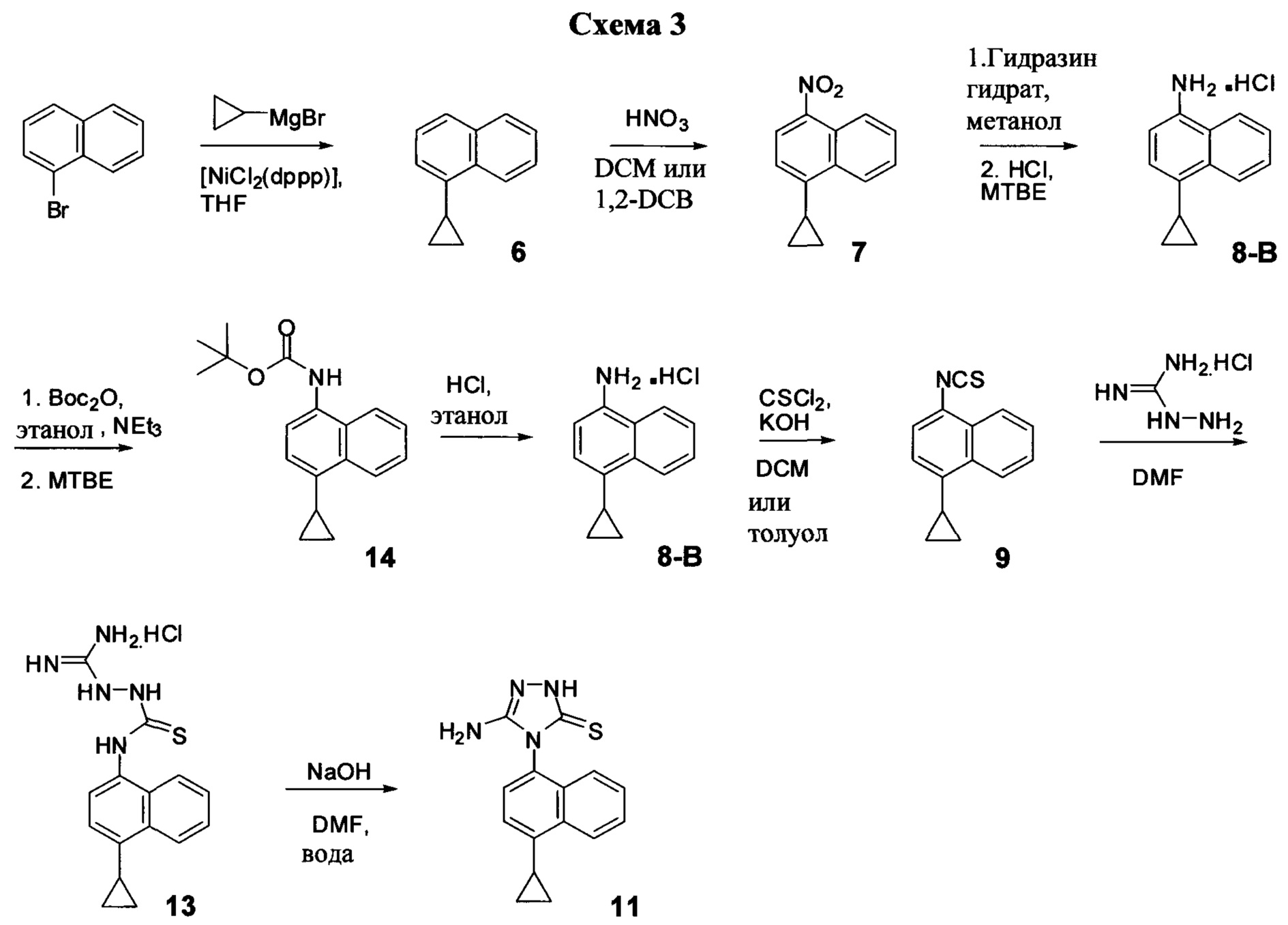
Начиная с 1-бромнафталина, в результате реакции с подходящим реактивом Гриньяра получают Соединение 6.
Соединение 11 затем получают с помощью способа (Способ 7), включающего:
(11-i-A) приведение в контакт Соединения 7 с гидразингидратом и растворителем для получения соединения, имеющего структуру:
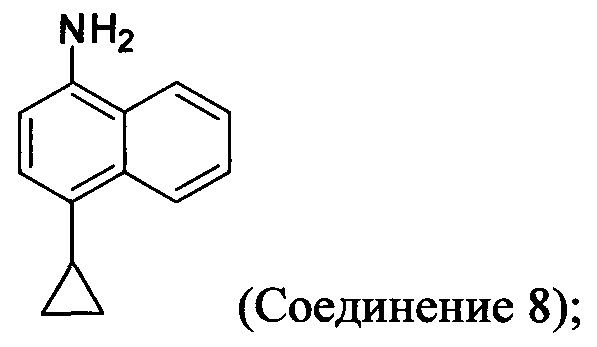
(11-ii-A) приведение в контакт Соединения 8 с соляной кислотой и растворителем для получения хлористоводородной соли Соединения 8;
(11-iii-A) защиту аминогруппы в соли, полученной на стадии (11-ii-A), для получения соединения, имеющего структуру:
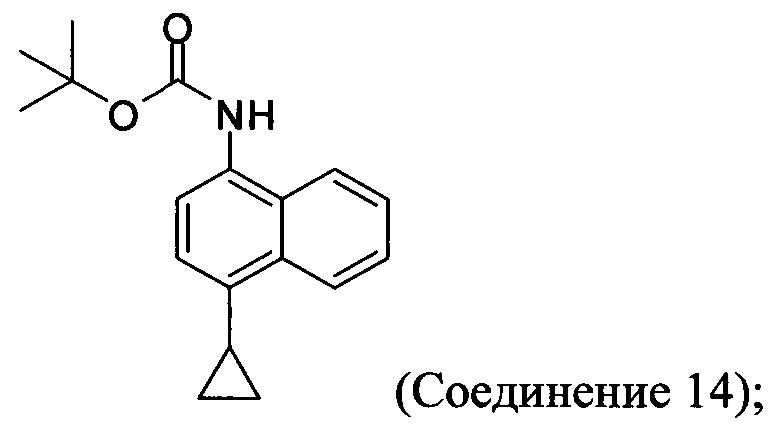
(11-iv-A) приведение в контакт Соединения 14 с соляной кислотой и этанолом для получения хлористоводородной соли Соединения 8;
(11-v-A) приведение в контакт хлористоводородной соли Соединения 8, полученной на стадии (11-iv-A), с основанием, тиофосгеном и растворителем для получения соединения, имеющего структуру:
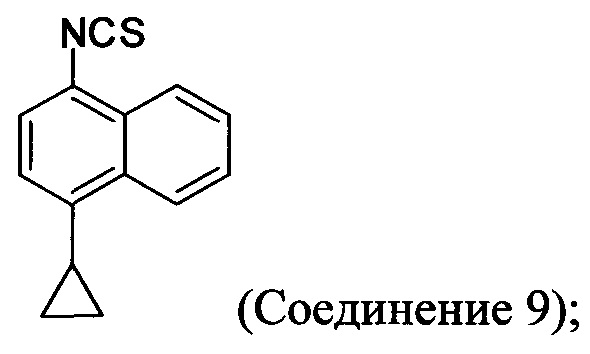
(11-vi-A) приведение в контакт Соединения 9, полученного на стадии (11-v-A), с гидрохлоридом аминогуанидина, основанием и растворителем для получения соединения, имеющего структуру:
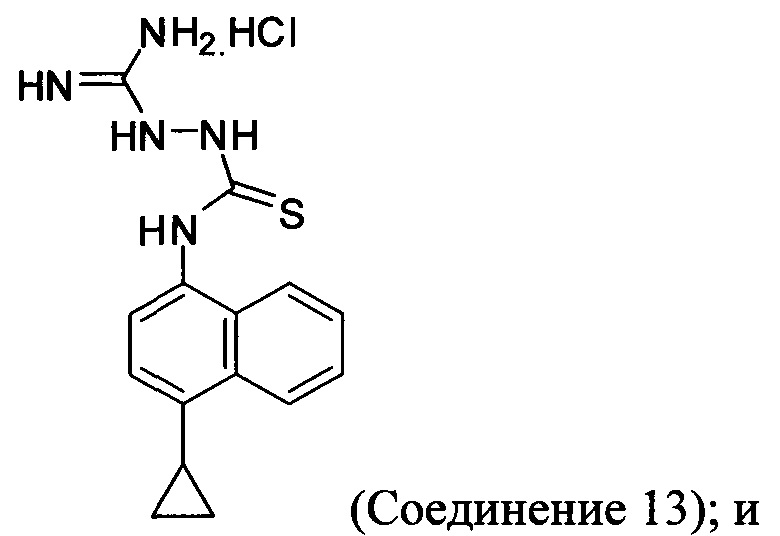
(11-vii-A) приведение в контакт Соединения 13, полученного на стадии (11-vi-A), с основанием, водой и растворителем для получения Соединения 11.
В настоящем документе предлагается Соединение 9, полученное с помощью Способа 7, описанного выше. В настоящем документе предлагается Соединение 11, полученное с помощью Способа 7, описанного выше. В настоящем документе предлагается Соединение 13, полученное с помощью Способа 7, описанного выше. В настоящем документе предлагается Соединение 14, полученное с помощью Способа 7, описанного выше.
В настоящем документе предлагается хлористоводородная соль Соединения 8, получаемая с помощью способа 7, описанного выше, и имеющая структуру:
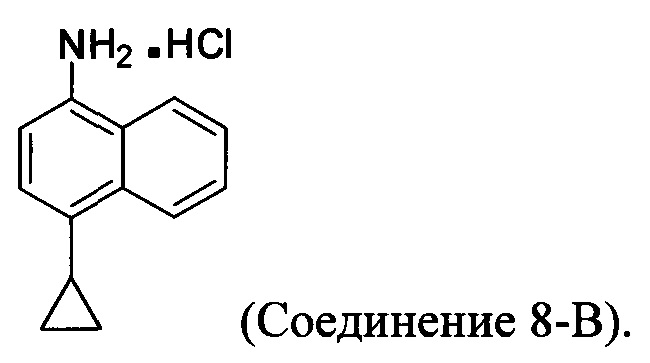
В одном варианте осуществления растворитель, подходящий для использования на стадии (11-i-A) Способа 7, описанного выше, представляет собой метанол, этанол или дихлорбензол, или любую их комбинацию. В одном варианте осуществления растворитель, подходящий для использования на стадии (11-i-A) Способа 7, представляет собой метанол. В другом варианте осуществления растворитель, подходящий для использования на стадии (11-i-A) Способа 7, представляет собой этанол. Альтернативные растворители, такие как, например, THF, также предполагаются в объеме представленных здесь вариантов осуществления.
В объеме представленных здесь вариантов осуществления предполагается использование других кислот и/или солей кислот на стадии (11-ii-A), (11-iii-A) и (11-v-А) Способа 7, описанного выше, включая и не ограничиваясь использованием лимонной кислоты, винной кислоты, уксусной кислоты, соляной кислоты и им подобных для получения соответствующих кислых солей Соединения 8. В объеме представленных здесь вариантов осуществления предполагается использование подходящего растворителя на стадии (11-ii-A) Способа 7, включая THF, диоксан, диэтиловый эфир, метил-трет-бутиловый эфир (МТВЕ) или т.п. В одном варианте осуществления растворитель, используемый на стадии (11-ii-A) Способа 7, представляет собой МТВЕ.
В одном варианте осуществления защитная группа, используемая на стадии (11-iii-A) Способа 7, представляет собой трет-бутилоксикарбонил. Может быть использована любая подходящая защитная группа для аминов. В одном варианте осуществления реакцию на стадии (11-iii-A) Способа 7 проводят в присутствии бутилоксикарбонил-ангидрида, этанола, триэтиламина и МТВЕ для получения Соединения 14.
В одном варианте осуществления растворитель, подходящий для использования на стадии (11-v-A) Способа 7, представляет собой дихлорметан. В альтернативных вариантах осуществления растворитель, подходящий для использования на стадии (11-v-A) Способа 7, представляет собой толуол, дихлорбензол, ксилолы или любой другой подходящий растворитель. В некоторых случаях реакционную смесь, полученную на стадии (11-v-A) Способа 7, перемешивают при температуре между примерно 0°C и примерно 10°C, между примерно 5°C и примерно 15°C, или между примерно 5°C и примерно 25°C. В некоторых случаях реакционную смесь, полученную на стадии (11-v-A) Способа 7, перемешивают при температуре примерно 5°C. В некоторых случаях реакционную смесь, полученную на стадии (11-v-A) Способа 7, перемешивают при примерно комнатной температуре.
В последующих вариантах осуществления основание, подходящее для реакции на стадии (11-v-A) Способа 7, выбирают из гидроксида калия, гидроксида натрия, гидроксида лития, карбоната калия, карбоната цезия, фосфата калия или любого другого подходящего основания. В некоторых вариантах осуществления основание, подходящее для реакции на стадии (11-v-A) Способа 7, представляет собой гидроксид калия.
В последующих вариантах осуществления основание, подходящее для реакции на стадии (11-vi-A) Способа 7, представляет собой органическое или неорганическое основание. Неограничивающие примеры включают триэтиламин, диизопропиламин, диизопропилэтиламин, гидроксид калия, гидроксид натрия, гидроксид лития, карбонат калия, карбонат цезия, фосфат калия или любое другое подходящее основание. В одном варианте осуществления реакцию на стадии (11-vi-A) Способа 7, описанного выше, проводят в присутствии диизопропилэтиламина (DIEA) или гидроксида натрия. Любой подходящий растворитель, выбранный для реакции на стадии (11-vi-A) Способа 7, включает, например, DMF, THF, ацетонитрил, диоксан, NMP и т.п. В одном варианте осуществления реакцию на стадии (11-vi-A) Способа 7 проводят в DMF.
В одном варианте осуществления растворитель, подходящий для использования на стадии (11-vii-A) Способа 7 представляет собой DMF. В альтернативных вариантах осуществления растворитель, подходящий для использования на стадии (11-vii-A) Способа 7, представляет собой толуол, дихлорбензол, ксилолы, NMP, ацетонитрил, диоксан или любой другой подходящий растворитель. В последующих вариантах осуществления основание, подходящее для реакции на стадии (11-vii-A) Способа 7, выбирают из гидроксида калия, гидроксида натрия, гидроксида лития, карбоната калия, карбоната цезия, фосфата калия или любого другого подходящего основания. В некоторых вариантах осуществления основание, подходящее для реакции на стадии (11-vii-A) Способа 7, представляет собой гидроксид натрия.
Недостатки начальных попыток
В последовательности реакций, описанных на Схеме 1, требуется использование бромида меди (II), в результате чего происходит образование отходов и требуются дополнительные затраты на их утилизацию. Дополнительным недостатком этой процедуры является то, что Соединение 1 выделено дважды для достижения желательных уровней чистоты.
Кроме того, для синтеза Соединения 11 требуется слишком длинная последовательность стадий, описанных выше на Схеме 2 и Схеме 3, и использование гидрохлорида аминогуанидина. Для удаления Соединения 13 также требуются дополнительные стадии очистки. Кроме того, как показано на Схеме 3, Соединение 8-В выделено дважды для достижения желательных уровней чистоты для Соединения 11.
Усовершенствованные способы
Для того, чтобы избежать недостатков процедур, описанных выше и в Схемах 1-3, были изучены альтернативные процедуры для синтеза соединения Формулы (I), включая Соединение 1 и Соединение 4. Усовершенствованные процедуры, описанные ниже, имеют определенные преимущества. Усовершенствованные процедуры уменьшают количество стадий, требуемых для получения соединений Формулы (I), включая Соединение 1 или Соединение 4. Усовершенствованные способы обеспечивают более легкую очистку целевых соединений и не требуют повторных способов выделения этого же самого соединения. Усовершенствованные способы позволяют избежать использования гидрохлорида аминогуанидина и последующих дополнительных стадий очистки, требуемых для удаления Соединения 13. Кроме того, усовершенствованные способы, описанные ниже, позволяют избежать использования вызывающих коррозию химических соединений и снижают уровень отходов, таких как отходы, образующиеся в результате реакции с участием бромида меди, описанной на Схеме 1 выше.
Таким образом, разработаны определенные новые способы для синтеза соединений Формулы (I), включая Соединение 1 и Соединение 4, описанные далее и в разделе краткого описания изобретения. Усовершенствованием в новых способах является использование соединения Формулы (II) в качестве промежуточного соединения для синтеза соединений Формулы (I). Дополнительным усовершенствованием в новом способе является использование формилгидразина вместо аминогуанидина для синтеза триазола. Использование формилгидразина позволяет избежать образования семикарбазидов, таких как Соединение 13 в старом способе, и уменьшает количество стадий очистки, требуемых для удаления промежуточных семикарбазидов, таких как Соединение 13 в старом способе.
Синтез соединений Формулы (II)
В одном варианте осуществления на Схеме 4 ниже описан иллюстративный синтез соединений Формулы (II).

В настоящем документе в некоторых вариантах осуществления предлагается способ для синтеза соединений формулы (II). В некоторых вариантах осуществления Соединение 6 превращают в соединение формулы (II) (например. Соединение 2). В некоторых вариантах осуществления Соединение 6 превращают в Соединение 7. В некоторых вариантах осуществления Соединение 7 превращают в Соединение 8. В последующих и дополнительных вариантах осуществления Соединение 8 превращают в Соединение 9. В последующих и дополнительных вариантах осуществления Соединение 9 превращают в Соединение 10. В последующих и дополнительных вариантах осуществления Соединение 10 превращают в Соединение 5. В некоторых вариантах осуществления Соединение 5 превращают в соединение формулы (II).
В некоторых вариантах осуществления Соединение 6 превращают в Соединение 7 в присутствии одного или нескольких нитрирующих реагентов. Неограничивающие примеры подходящих нитрирующих реагентов включают HNO3, HNO3 с кислотой (например, H2SO4), NH4NO3/трифторуксусную кислоту, изоамил нитрат/BF3⋅Et2O, изоамил нитрат/TfOH, Cu(NO3)/TFAA, AgNO3/Tf2O и Hg(NO3)2/HNO3. Для реакции нитрования используют любой подходящий растворитель. В некоторых вариантах осуществления подходящий растворитель, используемый для реакции, представляет собой галогенбензол (например, 1,2-дихлорбензол), толуол, воду, ионные жидкости или их комбинацию.
В некоторых вариантах осуществления Соединение 7 восстанавливают для получения Соединения 8 в присутствии одного или нескольких восстанавливающих агентов. В некоторых вариантах осуществления подходящие восстанавливающие агенты включают палладий (например, палладий на углероде, 5% палладий на углероде, 10% палладий на углероде, палладий на сульфате бария, хлорид палладия на углероде), оксид палладия, никелевый катализатор Ренея, металлическое железо в уксусной кислоте (например, Fe/HCl в водном растворе этанола), FeCl3/HCl, хлорид олова (II) в кислоте, металлический цинк, дитионит натрия, литийалюминийгидрид, гидрид диизобутилалюминия, супер-гидрид, дииодид самария, металлический самарий (например, Sm (4 экв)/NH4Cl в метаноле), сульфид натрия (например, сульфид натрия/ NH4Cl в водном растворе NH4OH), сернистый водород/основание, хлорид титана (III) или любой другой подходящий способ восстановления. Любой подходящий растворитель может быть использован для восстановления. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинацию.
В одном варианте осуществления Соединение 8, содержащее аминогруппу, необязательно превращают в соль кислоты. Неограничивающие примеры кислот, которые могут быть использованы для синтеза кислых солей Соединения 8, включают щавелевую кислоту, винную кислоту, лимонную кислоту, муравьиную кислоту, малоновую кислоту, малеиновую кислоту, адипиновую кислоту, муравьиную кислоту, хлоруксусную кислоту, дихлоруксусную кислоту, трихлоруксусную кислоту, трифторуксусную кислоту, бензойную кислоту, соляную кислоту или любую другую подходящую кислоту. В некоторых вариантах осуществления свободный амин Соединения 8 используется в последующих стадиях.
В некоторых вариантах осуществления Соединение 8 превращают в Соединение 9 в присутствии реагента переноса тионила. В некоторых вариантах осуществления реагент переноса тионила представляет собой тиофосген. В некоторых вариантах осуществления Соединение 8 или его кислую соль превращают в тиоизоцианат в присутствии тиофосгена и подходящего основания. В некоторых вариантах осуществления основание представляет собой бикарбонат калия, карбонат калия, ацетат калия, гидроксид калия, ацетат натрия, бензоат натрия, бикарбонат натрия, карбонат натрия, гидроксид натрия, метасиликат натрия, сесквикарбонат натрия, тринатрия фосфат, карбонат кальция, гидроксид кальция, гидроокись железа, гидроксид лития, гидроксид бария, гидроксид цезия, гидроксид стронция, гидроксид рубидия, карбонат цезия, трет-бутоксид калия или фосфат калия. В определенных вариантах осуществления основание представляет собой бутиллитий, диизопропиламид лития, диэтиламид лития, амид натрия, гидрид натрия, бис(триметилсилил)амид натрия, лития, бис(триметилсилил)амид лития. В других вариантах осуществления основание представляет собой аммиак, триэтиламин, пропиламин, метиламин, диметиламин, триметиламин, метилдиэтиламин, диизопропилэтиламин, анилин, пиперидин, пиридин, 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU) или пирролидин. Любой подходящий растворитель может быть использован для превращения путем нуклеофильного присоединения.
В некоторых вариантах осуществления реагент переноса тионила представляет собой дисульфид углерода, тиоцианат натрия, тиокарбонил-диимидазол, тиоцианат калия, тиоцианат цинка, тиоцианат серебра или тиоцианат аммония. В некоторых случаях Соединение 8 или его соль превращают в тиоизоцианат в присутствии дисульфида углерода (CS2). В последующих или дополнительных вариантах осуществления реакция, кроме того, содержит водный раствор NH4OH, с последующим добавлением нитрата свинца (Pb(NO3)2). В альтернативных вариантах осуществления реакция Соединения 8 или его кислой соли с CS2, кроме того, включает основание и растворитель (например, THF) с последующим TsCl. В альтернативных вариантах осуществления Соединение 8 или его кислую соль (например. Соединение 8-А) превращают в тиоизоцианат в присутствии тиокарбонил-диимидазола и растворителя (например, DMF). В других вариантах осуществления Соединение 8 или его соль превращают в тиоизоцианат в присутствии тиоцианата (например, тиоцианата натрия). В определенных вариантах осуществления реакция Соединения 8 с тиоцианатом (например, тиоцианатом натрия) обеспечивает получение промежуточного соединения тиомочевины. В последующих или дополнительных вариантах осуществления удаление аммиака из промежуточного соединения тиомочевины обеспечивает получение Соединения 9. В определенных вариантах осуществления промежуточное соединение тиомочевины нагревают до высоких температур (например, 100°C, 110°C, 120°C, 130°C, 140°C, 150°C, 160°C, 170°C, 180°C, 190°C или 200°C) для получения Соединения 9. В некоторых вариантах осуществления растворитель, используемый для любого превращения Соединения 8 в Соединение 9, представляет собой ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлобензол, NMP или их комбинацию.
В некоторых вариантах осуществления Соединение 9 превращают в Соединение 10 в присутствии нуклеофила. В некоторых вариантах осуществления нуклеофил представляет собой формилгидразин. Любой подходящий растворитель может быть использован для превращения путем нуклеофильного присоединения. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинацию.
В некоторых вариантах осуществления Соединение 10 циклизируют для получения Соединения 5 в присутствии одного или нескольких оснований. Любое подходящее основание может быть использовано в реакции циклизации. В некоторых вариантах осуществления основание представляет собой бикарбонат калия, карбонат калия, ацетат калия, гидроксид калия, ацетат натрия, бензоат натрия, бикарбонат натрия, карбонат натрия, гидроксид натрия, метасиликат натрия, сесквикарбонат натрия, тринатрия фосфат, карбонат кальция, гидроксид кальция, гидроокись железа, гидроксид лития, гидроксид бария, гидроксид цезия, гидроксид стронция, гидроксид рубидия, карбонат цезия, трет-бутоксид калия или фосфат калия. В определенных вариантах осуществления основание представляет собой бутиллитий, диизопропиламид лития, диэтиламид лития, амид натрия, гидрид натрия, бис(триметилсилил)амид натрия лития, бис(триметилсилил)амид лития. В других вариантах осуществления основание представляет собой аммиак, триэтиламин, пропиламин, метиламин, диметиламин, триметиламин, метилдиэтиламин, диизопропилэтиламин, анилин, пиперидин, пиридин, 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU) или пирролидин. Любой подходящий растворитель может быть использован для превращения путем нуклеофильного присоединения. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетан, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинацию.
В некоторых вариантах осуществления Соединение 5 подвергают нуклеофильному замещению для получения соединения формулы (II) в присутствии электрофила. В некоторых вариантах осуществления электрофил имеет структуру 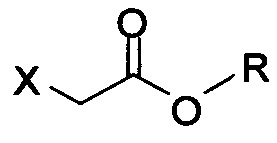 . В некоторых вариантах осуществления X представляет собой уходящую группу. В определенных вариантах осуществления X представляет собой галогено (например, бром, йод или хлор), тозилат, мезилат, безилат, трифлат, нонафлаты, фторсульфонаты или OH. В некоторых вариантах осуществления R представляет собой алкил. В других вариантах осуществления R представляет собой C1-20 алкил, C1-10 алкил, C1-6 алкил или C1-3 алкил. В некоторых вариантах осуществления R представляет собой метиловый, этиловый, пропиловый, изопропиловый, бутиловый, изобутиловый, трет-бутиловый, изоамиловый, пентиловый, гексиловый, гептиловый, октиловый, нониловый, терпениловый, борниловый, аллиловый, линалиловый и/или гераниловый эфиры. В некоторых вариантах осуществления R представляет собой H. Любой подходящий растворитель может быть использован для превращения путем нуклеофильного замещения. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинации.
. В некоторых вариантах осуществления X представляет собой уходящую группу. В определенных вариантах осуществления X представляет собой галогено (например, бром, йод или хлор), тозилат, мезилат, безилат, трифлат, нонафлаты, фторсульфонаты или OH. В некоторых вариантах осуществления R представляет собой алкил. В других вариантах осуществления R представляет собой C1-20 алкил, C1-10 алкил, C1-6 алкил или C1-3 алкил. В некоторых вариантах осуществления R представляет собой метиловый, этиловый, пропиловый, изопропиловый, бутиловый, изобутиловый, трет-бутиловый, изоамиловый, пентиловый, гексиловый, гептиловый, октиловый, нониловый, терпениловый, борниловый, аллиловый, линалиловый и/или гераниловый эфиры. В некоторых вариантах осуществления R представляет собой H. Любой подходящий растворитель может быть использован для превращения путем нуклеофильного замещения. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинации.
В некоторых вариантах осуществления способа, описанного выше (Схема 4), Соединения или промежуточные соединения выделяют и используют в последовательных стадиях синтеза без каких-либо дополнительных стадий очистки. В определенных вариантах осуществления Соединения или промежуточные соединения (например. Соединение 7, 8, 9, 10, 5 или соединение формулы II) используют без промежуточных стадий выделения или очистки. В некоторых вариантах осуществления Соединения или промежуточные соединения очищают перед использованием в последующих стадиях синтеза. В определенных вариантах осуществления Соединения или промежуточные соединения очищают путем кристаллизации. В некоторых вариантах осуществления Соединения или промежуточные соединения очищают путем дистилляции, колоночной хроматографии, обращенно-фазовой хроматографии, препаративной тонкослойной хроматографии или их комбинаций.
В последующих вариантах осуществления на Схеме 5 ниже описан альтернативный иллюстративный синтез соединений Формулы (II).
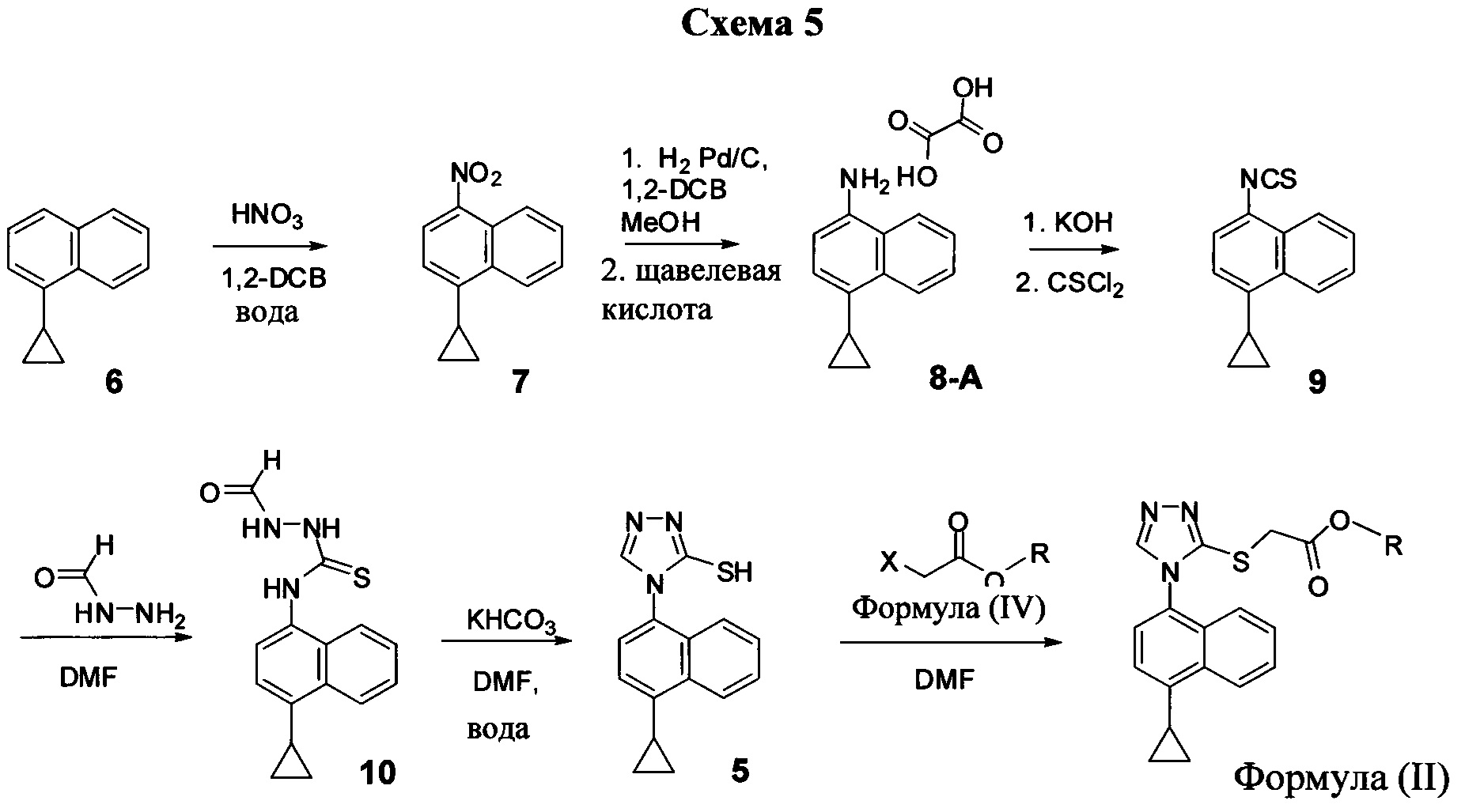
При обращении к Схеме 5, описанной выше, начиная с Соединения 6, в результате реакции нитрования получают Соединение 7. Нитрогруппу в Соединении 7 восстанавливают до амина (Соединение 8) с использованием любого подходящего способа восстановления. В одном варианте осуществления восстановление проводят с использованием газообразного водорода и палладия на угле в присутствии подходящего растворителя. В некоторых вариантах осуществления используют каталитическое количество палладия (например, 0,001%, 0,005%, 0,01%, 0,02%, 0,05%, 0,1%, 0,2%, 0,5%, 1%, 2%, 3%, 4%, 5% или 10%). В некоторых вариантах осуществления используют стехиометрическое количество палладия на угле. В одном варианте осуществления растворитель, подходящий для каталитического гидрирования, выбирают из метанола, этанола, трет-бутанола, THF, дихлорбензола или любой их комбинации.
В одном варианте осуществления Соединение 8, содержащее аминогруппу, превращают в кислую соль (например, оксалат). В одном варианте осуществления кислую соль амина (Соединение 8) используют в последующих стадиях (например, как показано на Схеме 5).
В одном варианте осуществления, как показано на Схеме 5, Соединение 8 или его кислую соль (например. Соединение 8-А) превращают в тиоизоцианат (Соединение 9) в присутствии тиофосгена и подходящего основания. В некоторых из таких вариантов осуществления растворитель, подходящий для синтеза Соединения 9 из Соединения 8 или его соли, представляет собой толуол. В альтернативном варианте осуществления растворитель, подходящий для синтеза Соединения 9 из Соединения 8 или его соли, в соответствии со Схемой 4 представляет собой дихлорбензол, ксилолы, дихлорметан или любой другой подходящий растворитель. В некоторых случаях реакционную смесь перемешивают при температуре между примерно 0°C и примерно 10°C, между примерно 5°C и примерно 15°C, или между примерно 5°C и примерно 25°C. В некоторых случаях реакционную смесь перемешивают при температуре от примерно 5 до примерно 100°C. В некоторых случаях реакционную смесь перемешивают при температуре примерно 5°C. В некоторых случаях реакционную смесь перемешивают при примерно комнатной температуре. В последующих вариантах осуществления основание, подходящее для синтеза Соединения 9 из Соединения 8 или его соли, выбирают из гидроксида калия, гидроксида натрия, гидроксида лития, карбоната калия, карбоната цезия, фосфата калия или любого другого подходящего основания. В некоторых вариантах осуществления основание, подходящее для синтеза Соединения 9 из Соединения 8 или его соли, представляет собой гидроксид калия.
В альтернативном варианте осуществления свободное основание Соединения 8 превращают в тиоизоцианат Соединения 9 в присутствии тиоцианата натрия и воды. В одном варианте осуществления растворитель, используемый для реакции, представляет собой бутанол, этанол, воду, ацетонитрил, диоксан, толуол, ксилол, DCB, DMF, NMP или любой другой подходящий растворитель. В одном варианте осуществления реакционную смесь перемешивают при температуре, по меньшей мере, 100°C, 110°C, 120°C, 130°C, 140°C или 150°C. В некоторых конкретных вариантах осуществления реакцию свободного основания Соединения 8 с тиоцианатом натрия проводят в присутствии воды и ксилолов при температуре, по меньшей мере, 130°C. Также, в объеме представленных здесь вариантов осуществления предусматривается использование других тиоцианатов, таких как тиоцианат калия, тиоцианат цинка, тиоцианат серебра, тиоцианат аммония, или других подходящих реагентов.
Тиоизоцианат Соединения 9 превращают в Соединение 10 в присутствии формилгидразина и подходящего растворителя. В одном варианте осуществления растворитель представляет собой DMF. В альтернативных вариантах осуществления растворитель представляет собой ацетонитрил, THF, диоксан, дихлорметан, дихлорбензол, NMP или любой другой подходящий растворитель. На этой стадии предпочтительно избегать образования Соединения 13, как описано выше на Схеме 2 и Схеме 3.
Соединение 10 циклизуют в Соединение 5 с использованием подходящего основания, воды и растворителя. В одном варианте осуществления основание представляет собой бикарбонат калия. В альтернативном варианте осуществления основание выбирают из гидроксида калия, гидроксида натрия, бикарбоната натрия, гидроксида лития, карбоната калия, карбоната цезия, фосфата калия или любого другого подходящего основания. В одном варианте осуществления растворитель, используемый для превращения Соединения 10 в Соединение 5, представляет собой DMF. В других вариантах осуществления растворитель, используемый для превращения Соединения 10 в Соединение 5, представляет собой THF, ацетонитрил, DCM, DCB, этанол, метанол, диоксан, NMP или любой другой подходящий растворитель.
Соединение 5 превращают в соединение Формулы (II) в присутствии любого подходящего основания и сложного ацетатного эфира Формулы (IV), содержащего уходящую группу. В одном варианте осуществления в результате реакции Соединения 5 с бромметилацетатом 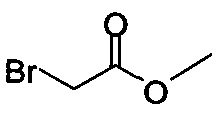 или хлорметилацетатом
или хлорметилацетатом 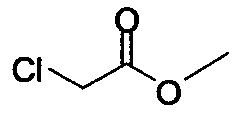 получают соединение Формулы (II), в которой R представляет собой метил (Соединение 2). В другом варианте осуществления реакцию Соединения 5 проводят с бромэтилацетатом
получают соединение Формулы (II), в которой R представляет собой метил (Соединение 2). В другом варианте осуществления реакцию Соединения 5 проводят с бромэтилацетатом 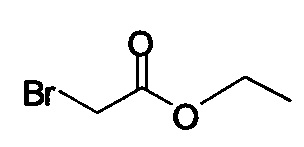 или хлорэтилацетатом
или хлорэтилацетатом 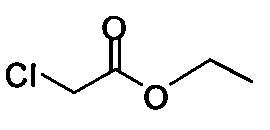 для получения соединения Формулы (II), в которой R представляет собой этил (Соединение 2-А). Используют любой подходящий растворитель. В одном варианте осуществления растворителем является DMF. В альтернативных вариантах осуществления растворителем является диоксан, ацетонитрил, хлороформ, дихлорметан, тетрагидрофуран (THF), N-метилпирролидон (NMP), диметилсульфоксид (DMSO) и т.п. В одном варианте осуществления соединение Формулы (II) выделяют в виде влажного кека, который необязательно промывают охлажденным этилацетатом и изопропанолом, и/или их комбинацией. Синтез соединений Формулы (I) из соединений Формулы (II)
для получения соединения Формулы (II), в которой R представляет собой этил (Соединение 2-А). Используют любой подходящий растворитель. В одном варианте осуществления растворителем является DMF. В альтернативных вариантах осуществления растворителем является диоксан, ацетонитрил, хлороформ, дихлорметан, тетрагидрофуран (THF), N-метилпирролидон (NMP), диметилсульфоксид (DMSO) и т.п. В одном варианте осуществления соединение Формулы (II) выделяют в виде влажного кека, который необязательно промывают охлажденным этилацетатом и изопропанолом, и/или их комбинацией. Синтез соединений Формулы (I) из соединений Формулы (II)
На Схеме 6а описан иллюстративный синтез соединений Формулы (I).
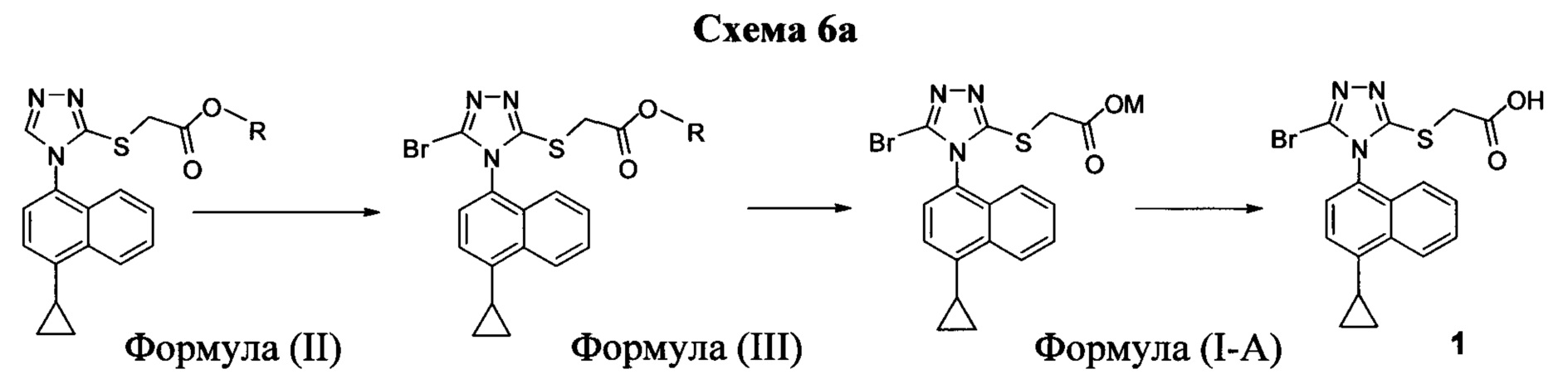
В настоящем документе в некоторых вариантах осуществления предлагается способ для синтеза Соединения 1. Синтез соединений формулы (II) описан выше (например. Схемы 1, 4 и 5). В некоторых вариантах осуществления соединение формулы (II) превращают в Соединение 1. В некоторых вариантах осуществления соединение формулы (II) превращают в соединение формулы (III). В некоторых вариантах осуществления соединение формулы (III) превращают в соединение формулы (I-A). В последующих или дополнительных вариантах осуществления соединение формулы (I-A) превращают в Соединение 1.
В некоторых вариантах осуществления соединение формулы (II) бромируют в присутствии бромирующего агента для получения соединения формулы (III). В некоторых вариантах осуществления бромирование соединения формулы (II), в которой R=H, обеспечивает получение Соединения 1. В определенных вариантах осуществления бромирующий агент представляет собой N-бромсукцинимид (NBS), Br2, BrCl/Br2, тетрабутиламмонийтрибромид, бромид аммония/оксон (в метаноле и/или воде), дибромид селена, Fe2Br3/Br2, AlCl3/Br2, FeCl2/Br2, ZnCl3/Br2, 1,2-дипиридинийтрибромид-этан, NBS/кислота (трифторметансульфоновая кислота и BF3-H2O), NBS/концентрированная серная кислота, NBS/тетрабутиламмония бромид, LiBr/PhI/m-хлорпербензойная кислота/TsOH, AuCl3/NBS, NBS/Pd(OAc)2, N,N,N',N'-тетрабромбензол-1,3-дисульфониламид/поли[N-бромбензол-1,3-дисульфониламид], LiTMP/ZnCl2/Br2, [Ir(COD)(OMe)]2/B2pin2/dtbpy/CuBr2 или [Ir(COD)(OMe)]2/B2pin2/dtbpy/CuCl2. Для бромирования может быть использован любой подходящий растворитель. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинацию.
В некоторых вариантах осуществления соединение формулы (III) необязательно гидролизуют в присутствии основания для получения соединения формулы (I-A), в которой M представляет собой катион. В определенных вариантах осуществления M выбран из Na+, Li+, K+, Cs+, Ba+, Ca+ или любого другого подходящего катиона. В некоторых вариантах осуществления основание представляет собой гидроксид натрия, гидроксид лития, гидроксид калия, гидроксид цезия, гидроксид бария, гидроокись железа, гидроксид кальция, аммиак или любое другое подходящее основание. В определенных вариантах осуществления основание представляет собой бикарбонат калия, карбонат калия, ацетат калия, гидроксид калия, ацетат натрия, бензоат натрия, бикарбонат натрия, карбонат натрия, гидроксид натрия, метасиликат натрия, сесквикарбонат натрия, тринатрия фосфат, карбонат кальция, гидроксид кальция, карбонат цезия или фосфат калия. Для бромирования может быть использован любой подходящий растворитель. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинацию. В некоторых вариантах осуществления растворителем является вода.
В некоторых вариантах осуществления соединение формулы (I-A) необязательно обрабатывают кислотой для получения Соединения 1. В некоторых вариантах осуществления кислота представляет собой бромистоводородную кислоту, серную кислоту, соляную кислоту, йодистоводородную кислоту, азотную кислоту, фосфорную кислоту, фторантимоновую кислоту, фтороборную кислоту, гексафторфосфорную кислоту, трифторуксусную кислоту, уксусную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, трифторметансульфоновую кислоту, лимонную кислоту, муравьиную кислоту, глюконовую кислоту, молочную кислоту, щавелевую кислоту, малоновую кислоту, винную кислоту или любую подходящую кислоту. Любой подходящий растворитель может быть использован для получения свободной кислоты Соединения 1. В некоторых вариантах осуществления растворитель представляет собой воду, ацетонитрил, DMF, THF, толуол, ксилолы, диоксан, бутанол, метанол, этанол, диэтиловый эфир, ацетон, гексан, пентан, гептан, этилацетат, дихлорметан, дихлорэтан, дихлорбензол, NMP или их комбинации. В некоторых вариантах осуществления растворитель представляет собой этилацетат и гептан.
В некоторых вариантах осуществления способа, описанного выше (Схема 6а), соединения (например, соединения формулы (III), (I-A) и Соединение 1) выделяют и используют в последовательных стадиях синтеза без каких-либо дополнительных стадий очистки. В определенных вариантах осуществления соединения (например, соединения формулы (III), (I-A) и Соединение 1) используют без промежуточных стадий выделения или очистки. В некоторых вариантах осуществления соединения (например, соединения формулы (III), (I-A) и Соединение 1) очищают перед использованием в последующих стадиях синтеза. В определенных вариантах осуществления соединения (например, соединения формулы (III), (I-A) и Соединение 1) очищают путем кристаллизации. В некоторых вариантах осуществления соединения (например, соединения формулы (III), (I-A) и Соединение 1) очищают путем дистилляции, колоночной хроматографии, обращенно-фазовой хроматографии, препаративной тонкослойной хроматографии или их комбинаций.
На Схеме 6b описан иллюстративный синтез соединений Формулы (I), включая Соединение 1 и Соединение 4.
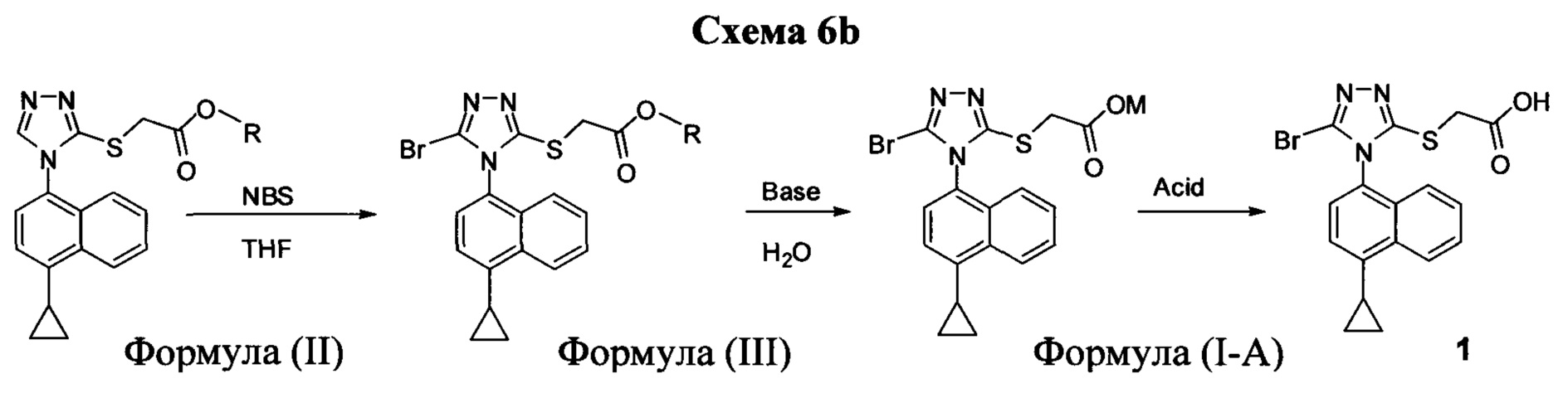
В результате реакции соединения Формулы (II) с N-бромсукцинимидом (NBS) в подходящем растворителе получают соединение Формулы (III). Растворители, подходящие для NBS-опосредованной реакции, включают DMF, ацетонитрил, МТВЕ или любой другой подходящий растворитель. Сложноэфирную группу в соединении Формулы (III) необязательно гидролизуют для получения кислой соли соединения Формулы (I), то есть соединения Формулы (I-A), в которой M выбран из Na+, Li+, K+, Cs+ или любого другого подходящего катиона. Кислую соль необязательно выделяют и/или кристаллизуют. Соединение Формулы (I-A) превращают в Соединение 1 в присутствии кислоты. Соединение 1 необязательно кристаллизуют из подходящего растворителя или смеси растворителей.
В конкретном варианте осуществления на Схеме 7 описан синтез Соединения 1 и Соединения 4.
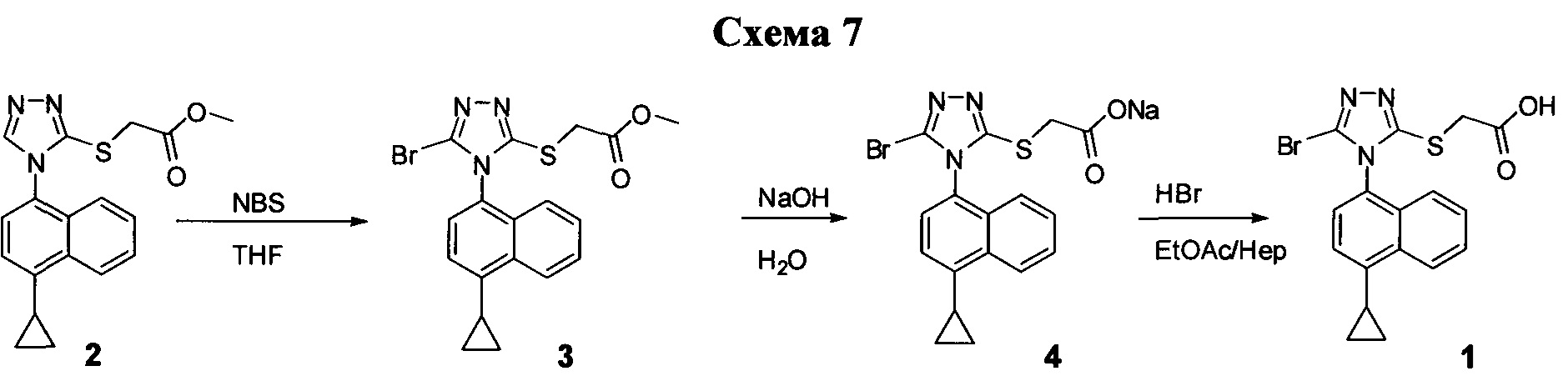
Таким образом, в настоящем документе предлагается способ (Способ 8) для получения Соединения 1, имеющего следующую структуру:
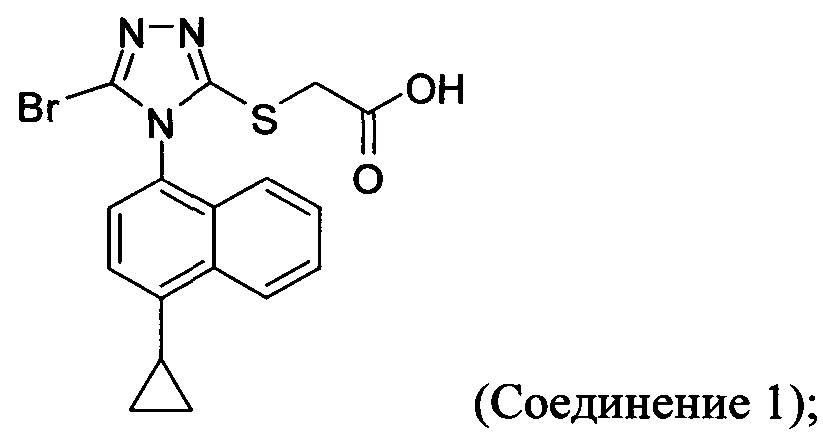
усовершенствованием в способе является приведение в контакт соединения структуры
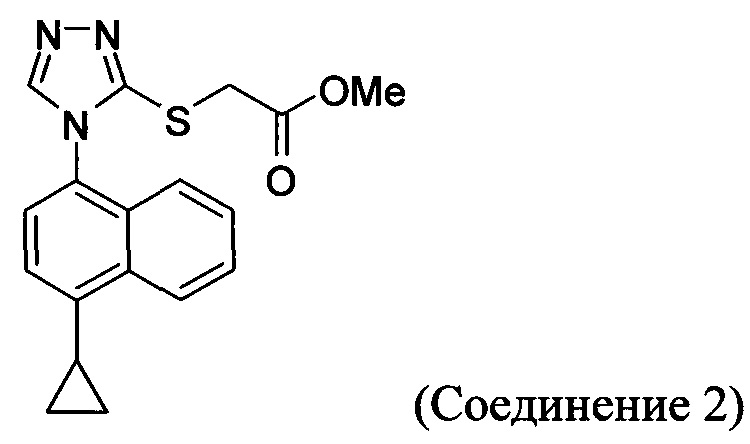
с бромирующим агентом (например, N-бромсукцинимидом (NBS)) и растворителем.
В одном варианте осуществления растворитель представляет собой THF. В альтернативных вариантах осуществления растворитель представляет собой DMF, ацетонитрил или любой другой подходящий растворитель.
В одном случае NBS добавляют в раствор Соединения 2 в THF, при этом раствор THF поддерживают при температуре между примерно комнатной температурой и примерно 32°C. В одном варианте осуществления реакционную смесь затем перемешивают в течение, по меньшей мере, 12 часов при температуре между примерно комнатной температурой и примерно 32°C. В альтернативных вариантах осуществления реакционную смесь перемешивают в течение, по меньшей мере, 2 часов, 4 часов, 6 часов, 8 часов, 10 часов, 12 часов, 14 часов, 20 часов, 24 часов или дольше. В одном случае, если содержание Соединения 2 при анализе реакционной смеси составляет ≥1,5% площади по данным высокоэффективной жидкостной хроматографии (HPLC), в реакционную смесь добавляют дополнительное количество NBS. В одном варианте осуществления после перемешивания реакционной смеси в течение, по меньшей мере, 12 часов и/или, необязательно, добавления дополнительного количества NBS анализ реакционной смеси показывает ≤1,5% площади по данным HPLC Соединения 2. В другом варианте осуществления после перемешивания реакционной смеси в течение, по меньшей мере, 12 часов и/или, необязательно, добавления дополнительного количества NBS анализ реакционной смеси показывает ≤0,2% площади по данным HPLC Соединения 2.
В одном варианте осуществления способ для синтеза Соединения 1 дополнительно включает:
(1-i) экстракцию реакционной смеси растворителем (например, толуолом) при поддержании температуры смеси между примерно 2°C и примерно 7°C;
(1-ii) обратную экстракцию органической фазы, полученной на стадии (1-i), раствором дисульфита натрия один или несколько раз до тех пор, пока с помощью HPLC анализа не будет обнаружен NBS в водной фазе;
(1-iii) промывание органической фазы, полученной на стадии (1-ii), водой;
(1-iv) промывание органической фазы, полученной на стадии (1-iii), раствором бикарбоната натрия один или несколько раз до тех пор, пока водная фаза не будет иметь pH, по меньшей мере, 8; и
(1-v) сбор органической фазы, содержащей соединение, имеющее структуру:
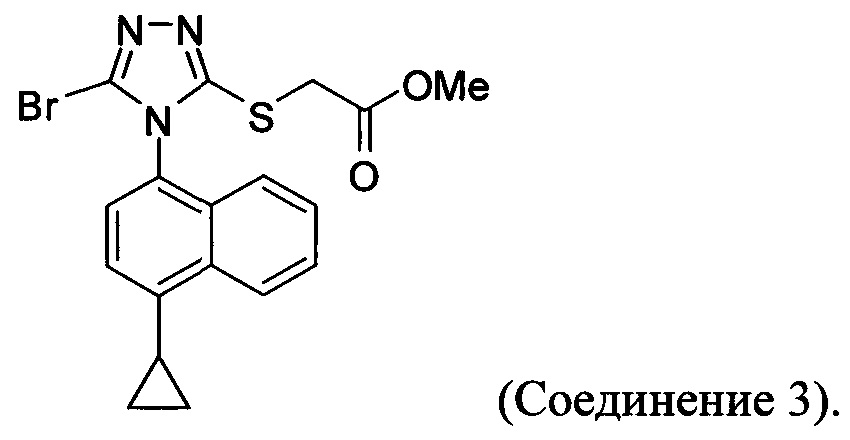
В одном случае способ дополнительно включает, необязательно, концентрированно органической фазы, полученной на стадии (1-v), при пониженном давлении для получения Соединения 3.
В одном случае способ дополнительно включает стадии:
(1-vi) приведение в контакт органической фазы, полученной на стадии (1-v) по пункту 7, с раствором основания (например, гидроксида натрия) до тех пор, пока площадь пика Соединения 3 в органической фазе не будет ниже, чем 50 mAU по данным HPLC анализа; и
(1-vii) сбор водной фазы, содержащей соединение структуры:
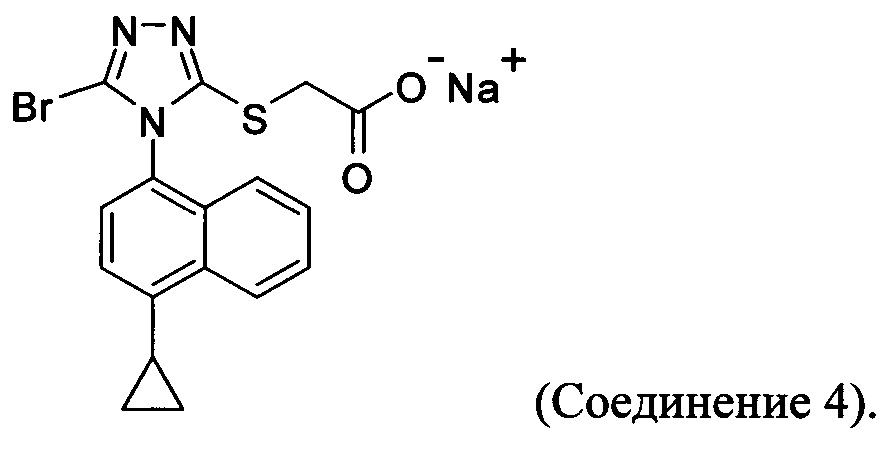
В одном случае способ дополнительно включает стадии:
(1-viii) концентрирование водной фазы, полученной на стадии (1-vii) по пункту 8, при пониженном давлении;
(1-ix) добавление воды для доведения объема смеси, полученной на стадии (1-viii), примерно до 5,5±5% рассчитанной с помощью HPLC анализа массы Соединения 1 в водной фазе стадии (1-viii);
(1-х) охлаждение смеси, полученной на стадии (1-ix), с получением суспензии, содержащей кристаллическое Соединение 4; и
(1-xi) фильтрацию суспензии, полученной на стадии (1-х), с получением влажного кека, содержащего кристаллическое Соединение 4.
В одном случае способ дополнительно включает, необязательно, сушку влажного кека, полученного на стадии (1-xi), для получения Соединения 4.
В одном случае способ дополнительно включает стадии:
(1-xii) растворение влажного кека, содержащего Соединение 4, полученного на стадии (1-xi) по пункту 10, в воде;
(1-xiii) добавление растворителя (например, этилацетата) в раствор, полученный на стадии (1-xii);
(1-xiv) добавление кислоты (например, 24% бромистоводородной кислоты) при поддержании температуры реакционной смеси ниже 35°C и поддержании значения pH реакционной смеси между примерно 2,0 и примерно 4,0; и
(1-xv) отделение органической фазы, содержащей Соединение 1.
В одном случае способ дополнительно включает:
(1-xvi) концентрирование органической фазы, полученной на стадии (1-xv) по пункту 10, при пониженном давлении;
(1-xvii) перемешивание смеси, полученной на стадии (1-xvi), при температуре между примерно 32°C и примерно 38°C в течение, по меньшей мере, 8 часов;
(1-xviii) добавление сорастворителя (например, н-гептана) в смесь, полученную на стадии (1-xvii), и охлаждение смеси; и
(1-xix) фильтрацию суспензии, полученной на стадии (1-xviii), с получением Соединения 1.
В одном варианте осуществления на стадии (1-xix), указанной выше, получают кристаллическое Соединение 1. Кристаллические полиморфы Соединения 1 описаны в международной заявке на патент PCT/US 11/20233 и международной заявке на патент PCT/US 11/67657, и раскрытие полиморфов Соединения 1 и/или Соединения 4 изложено в международной заявке на патент PCT/US 11/20233, международной заявке на патент PCT/US 11/67657, которые включены здесь путем отсылки.
В одном аспекте в настоящем документе предлагается Соединение 1, содержащее не больше, чем 0,1% Соединения 3 по площади по данным HPLC анализа. В одном варианте осуществления Соединение 1, имеющее не больше, чем 0,1% Соединения 3, получено с помощью способов, описанных выше. В последующем аспекте в настоящем документе предлагается Соединение 1, имеющее не больше, чем 0,1% Соединения 2 и не больше, чем 0,1% Соединения 3 по площади по данным HPLC анализа. В еще другом аспекте в настоящем документе предлагается Соединение 1, имеющее чистоту ≥98%. В последующем аспекте в настоящем документе предлагается Соединение 1, имеющее чистоту ≥99%. В любом из вышеизложенных вариантов осуществления чистота определена с помощью HPLC анализа. В любом из вышеизложенных вариантов осуществления Соединение 1 получено с помощью Способа 1, Способа 2, Способа 3, Способа 4, Способа 5, Способа 6, Способа 7 или Способа 8, или любой их комбинации.
Другие формы раскрытых в настоящем документе соединений
Изомеры
В некоторых вариантах осуществления соединения, описанные здесь, существуют в виде геометрических изомеров. В некоторых вариантах осуществления соединения, описанные здесь, обладают одной или несколькими двойными связями. Соединения, представленные здесь, включают все цис, транс, син, анти, энтгеген (Е) и цузаммен (Z) изомеры, а также их соответствующие смеси. В некоторых ситуациях соединения существуют в виде таутомеров. Соединения, описанные здесь, включают все возможные таутомеры в пределах описанных здесь формул. В некоторых ситуациях соединения, описанные здесь, обладают одним или несколькими хиральными центрами, и каждый центр существует в R или S конфигурации. Соединения, описанные здесь, включают все диастереомерные, энантиомерные и эпимерные формы, а также их соответствующие смеси. В дополнительных вариантах осуществления предложенных здесь соединений и способов смеси энантиомеров и/или диастереомеров, получающиеся из одного этапа приготовления, сочетания или взаимного превращения, являются полезными для описанных здесь применений. В некоторых вариантах осуществления описанные здесь соединения получены в виде их индивидуальных стереоизомеров путем реакции рацемической смеси соединения с оптически активным разделяющим агентом с образованием пары диастереоизомерных соединений, разделением диастереомеров и получением оптически чистых энантиомеров. В некоторых вариантах осуществления предпочтительными являются диссоциируемые комплексы (например, кристаллические диастереомерные соли). В некоторых вариантах осуществления диастереомеры имеют различающиеся физические свойства (например, температуры плавления, температуры кипения, растворимости, реакционную способность и т.п.) и разделяются с учетом этих различий. В некоторых вариантах осуществления диастереомеры разделяют хиральной хроматографией или, предпочтительно, методами разделения/расщепления, основанными на различиях в растворимости. В некоторых вариантах осуществления затем получают оптически чистый энантиомер вместе с разделяющим агентом, используя любые практические способы, не приводящие к рацемизацию.
Меченые соединения
В некоторых вариантах осуществления соединения, описанные здесь, существуют в их изотопно-меченых формах. В некоторых вариантах осуществления способы, раскрытые здесь, включают способы лечения заболеваний путем введения таких изотопно-меченых соединений. В некоторых вариантах осуществления способы, раскрытые здесь, включают способы лечения заболеваний путем введения таких изотопно-меченых соединений в виде фармацевтических композиций. Таким образом, в некоторых вариантах осуществления раскрытые здесь соединения включают изотопно-меченые соединения, которые идентичны указанным здесь соединениям, за исключением того, что один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно встречающегося в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Описанные здесь соединения и их метаболиты, фармацевтически приемлемые соли, сложные эфиры, пролекарства, сольваты, гидраты или производные, которые содержат указанные выше изотопы и/или другие изотопы других атомов, входят в объем данного изобретения. Некоторые изотопно-меченые соединения, например те, в которые включены радиоактивные изотопы, такие как 3H и 14C, могут использоваться в анализах распределения лекарственного средства и/или субстрата в ткани. Тритий, то есть изотопы 3H и углерод-14, то есть 14C, особенно предпочтительны с учетом простоты их получения и обнаружения. Кроме того, замещение тяжелыми изотопами, такими как дейтерий, то есть 2H, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный период полужизни или снижение требуемой дозировки. В некоторых вариантах осуществления изотопно-меченые соединения, их фармацевтически приемлемые соли, сложные эфиры, пролекарства, сольваты, гидраты и производные получены любым подходящим способом.
В некоторых вариантах осуществления соединения, описанные здесь, мечены другими средствами, включая, но без ограничения, использование хромофоров или флуоресцентных фрагментов, биолюминесцентных или хемилюминесцентных меток. Фармацевтически приемлемые соли
В некоторых вариантах осуществления соединения, описанные здесь, существуют в виде их фармацевтически приемлемых солей. В некоторых вариантах осуществления способы, описанные здесь, включают способы лечения заболеваний путем введения таких фармацевтически приемлемых солей. В некоторых вариантах осуществления способы, раскрытые здесь, включают способы лечения заболеваний путем введения таких фармацевтически приемлемых солей в виде фармацевтических композиций.
В некоторых вариантах осуществления соединения, описанные здесь, содержат кислотные или основные группы и поэтому могут взаимодействовать с любым числом неорганических или органических оснований, неорганических и органических кислот с образованием фармацевтически приемлемой соли. В некоторых вариантах осуществления эти соли получены in situ во время конечного выделения и очистки соединений по изобретению, или путем отдельной реакции очищенного соединения в его свободной форме с подходящей кислотой или основанием и выделения полученной таким образом соли.
Примеры фармацевтически приемлемых солей включают соли, полученные путем реакции описанных здесь соединений с неорганической, органической кислотой или неорганическим основанием, и такие соли включают ацетат, акрилат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бисульфит, бромид, бутират, бутин-1,4-диоат, камфорат, камфосульфонат, капроат, каприлат, хлорбензоат, хлорид, цитрат, циклопентанпропионат, деканоат, диглюконат, дигидрофосфат, динитробензоат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолят, гемисульфат, гептаноат, гексаноат, гексин-1,6-диоат, гидроксибензоат, гамма-гидроксибутират, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, иодид, изобутират, лактат, малеат, малонат, метансульфонат, манделат, метафосфат, метансульфонат, метоксибензоат, метилбензоат, моногидрофосфат, 1-нафталинсульфонат, 2-нафталинсульфонат, никотинат, нитрат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, пиросульфат, пирофосфат, пропиолат, фталат, фенилацетат, фенилбутират, пропансульфонат, салицилат, сукцинат, сульфат, сульфит, сукцинат, суберат, себакат, сульфонат, тартрат, тиоцианат, тозилат ундеконат и ксилолсульфонат.
Кроме того, описанные здесь соединения могут быть получены в виде фармацевтически приемлемых солей путем реакции свободной основной формы соединения с фармацевтически приемлемой неорганической или органической кислотой, включая, но без ограничения, неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метафосфорная кислота и т.п.; и органические кислоты, такие как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, Q-толуолсульфоновая кислота, винная кислота, трифторуксусная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, арилсульфоновая кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-метилбицикло-[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4'-метиленбис-(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота и муконовая кислота. В некоторых вариантах осуществления другие кислоты, такие как щавелевая кислота, хотя сами по себе не являются фармацевтически приемлемыми, используются при получении солей, применимых в качестве промежуточных соединений для получения соединений по изобретению и их фармацевтически приемлемых солей, образованных путем добавления кислоты.
В некоторых вариантах осуществления описанные здесь соединения, которые содержат свободную кислотную группу, взаимодействуют с подходящим основанием, таким как гидроксид, карбонат, бикарбонат, сульфат, фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Иллюстративные щелочные или щелочноземельные соли включают соли лития, натрия, калия, кальция, магния и алюминия, и т.п. Иллюстративные примеры оснований включают гидроксид натрия, гидроксид калия, гидроксид холина, карбонат натрия, N+(C1-4 алкил)4, и т.п.
Иллюстративные органические амины, используемые для образования солей присоединения основания, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и т.п. Следует понимать, что соединения, описанные здесь, также включают кватернизацию любых основных азотсодержащих групп, которые они содержат. В некоторых вариантах осуществления водо- или маслорастворимые, или диспергируемые продукты получены с помощью такой кватернизации. Соединения, описанные здесь, могут быть получены в виде фармацевтически приемлемых солей, образованных, когда кислотный протон присутствующий в исходном соединении, либо замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо образует координационную связь с органическим основанием. Соли присоединения основания могут быть также получены путем взаимодействия описанных здесь соединений в форме их свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием, включая, но без ограничения, органические основания, такие как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п., и неорганические основания, такие как гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и т.п. Кроме того, формы солей раскрытых соединений могут быть получены с использованием солей исходных материалов или промежуточных соединений.
Сольваты
В некоторых вариантах осуществления соединения, описанные здесь, существуют в виде сольватов. В изобретении предлагаются способы лечения заболеваний путем введения таких сольватов. Кроме того, в изобретении предлагаются способы лечения заболеваний путем введения таких сольватов в виде фармацевтических композиций.
Сольваты содержат стехиометрические или нестехиометрические количества растворителя, и в некоторых вариантах осуществления образованы в способе кристаллизации с применением фармацевтически приемлемых растворителей, таких как вода, этанол и т.п. Гидраты образуются в случае, когда растворителем является вода, или алкоголяты образуются в случае, когда растворителем является спирт. Сольваты описанных здесь соединений могут быть удобно получены или образованы в ходе описанных здесь способов. Только в качестве примера, гидраты описанных здесь соединений могут быть удобно получены путем перекристаллизации из смеси водного/органического растворителя, используя органические растворители, включая, но без ограничения, диоксан, тетрагидрофуран или метанол. Кроме того, предложенные здесь соединения могут существовать в несольватированных и сольватированных формах. В целом, сольватированные формы считаются эквивалентными несольватированным формам в рамках предложенных здесь соединений и способов.
Полиморфы
В некоторых вариантах осуществления соединения, описанные здесь, существуют в виде полиморфов. В изобретении предлагаются способы лечения заболеваний путем введения таких полиморфов. Кроме того, в изобретении предлагаются способы лечения заболеваний путем введения таких полиморфов в виде фармацевтических композиций.
Таким образом, соединения, описанные здесь, включают все их кристаллические формы, известные как полиморфы. Полиморфы включают различные варианты кристаллической структуры при одинаковом элементном составе соединения. В определенных случаях полиморфы имеют различные рентгеновские дифрактограммы, спектры инфракрасного излучения, температуры плавления, плотность, твердость, кристаллическую форму, оптические и электрические свойства, стабильность и растворимость. В определенных случаях различные факторы, такие как растворитель для перекристаллизации, скорость кристаллизации и температура хранения, могут привести к доминированию той или иной кристаллической формы.
Полиморфная форма 1
В одном варианте осуществления полиморфная Форма 1 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата имеет порошковую рентгеновскую дифрактограмму, характеризующуюся дифракционной картиной, приведенной в Таблице 1А или Таблице 1В. В некоторых вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата, содержащий, по меньшей мере, 3 пика (±0.1°2θ) в Таблице 1А или 1В. В определенных вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата, содержащий, по меньшей мере, 4 пика (±0.1°2θ) в Таблице 1А или 1В, по меньшей мере 5 пиков (±0.1°2θ) в Таблице 1А или 1В, по меньшей мере 6 пиков (±0.1°2θ) в Таблице 1А или 1В, по меньшей мере 8 пиков (±0.1°2θ) в Таблице 1А или 1В, по меньшей мере 10 пиков (±0.1°2θ) в Таблице 1А, по меньшей мере 15 пиков (±0.1°2θ) в Таблице 1А, по меньшей мере 20 пиков (±0.1°2θ) в Таблице 1А, по меньшей мере 25 пиков (±0.1°2θ) в Таблице 1А, или по меньшей мере 30 пиков (±0.1°2θ) в Таблице 1А.
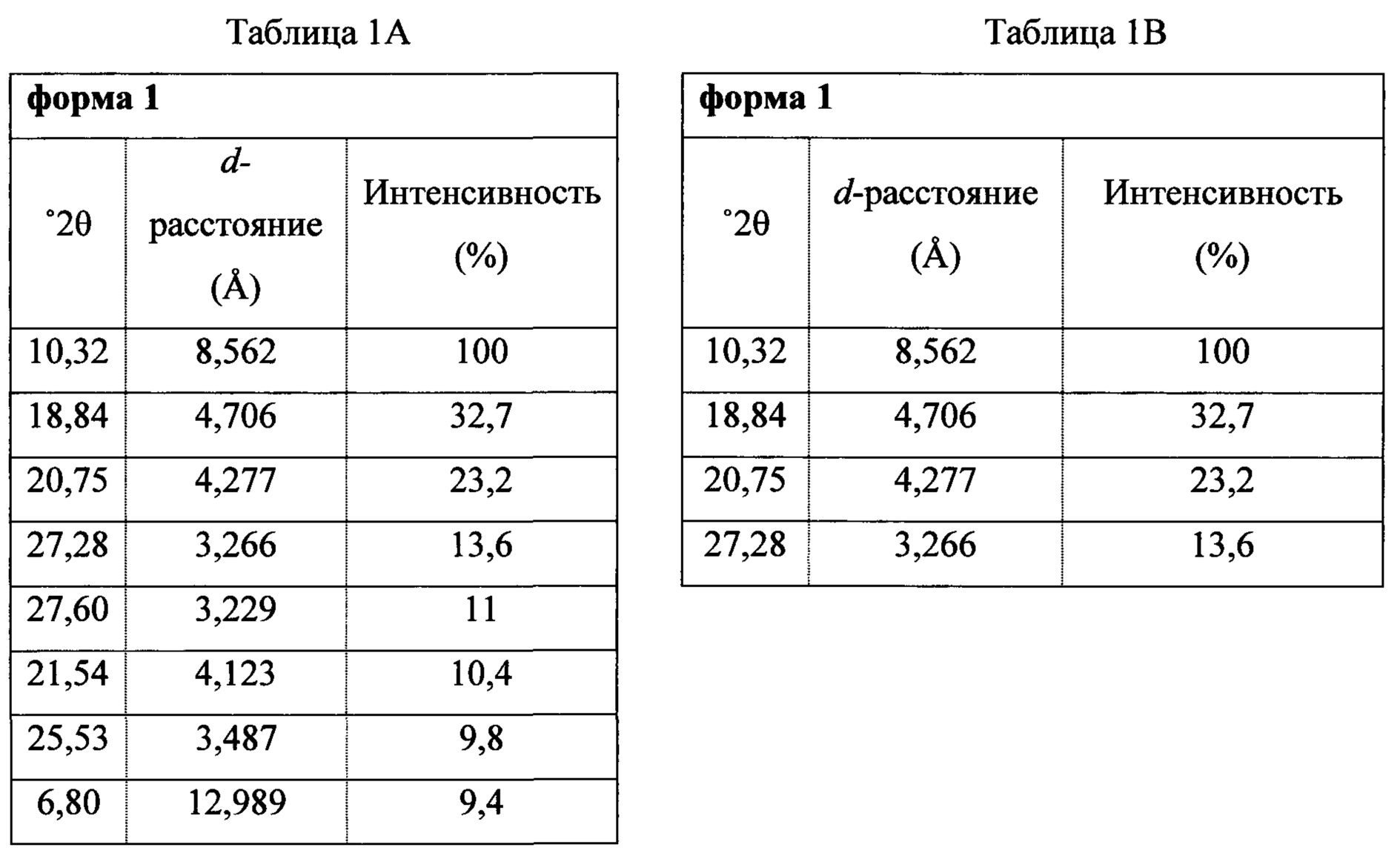

В одном варианте осуществления в настоящем документе предлагается полиморфная форма 1 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата, характеризующаяся пиками на порошковой рентгеновской дифрактограмме при 10,32, 18,84 и 20,75 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В последующих вариантах осуществления полиморфная форма 1 дополнительно характеризуется, по меньшей мере, одним пиком, возникающим при 6,80, 21,54, 24,97, 25,53, 27,28 и 27,60 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В последующих вариантах осуществления полиморфная форма 1 дополнительно характеризуется, по меньшей мере, двумя пиками, возникающими при 6,80, 21,54, 24,97, 25,53, 27,28 и 27,60 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В еще последующих вариантах осуществления полиморф имеет порошковую рентгеновскую дифрактограмму по существу такую же, как порошковая рентгеновская дифрактограмма, показанная на фигуре 1.
Полиморфная форма 2
В одном варианте осуществления полиморфная Форма 2 2-(5-бром-4-(4-циалопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата имеет порошковую рентгеновскую дифрактограмму, характеризующуюся дифракционной картиной, приведенной в Таблице 2А или Таблице 2В. В некоторых вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата, содержащий, по меньшей мере, 3 пика (±0.1°2θ) в Таблице 2А или 2В. В определенных вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата, содержащий, по меньшей мере, 4 пика (±0.1°2θ) в Таблицы 2А или 2В, по меньшей мере 5 пиков (±0.1°2θ) в Таблице 2А или 2В, по меньшей мере 6 пиков (±0.1°2θ) в Таблице 2А или 2В, по меньшей мере 8 пиков (±0.1°2θ) в Таблице 2А или 2В, по меньшей мере 10 пиков (±0.1°2θ) в Таблице 2А, по меньшей мере 15 пиков (±0.1°2θ) в Таблице 2А, по меньшей мере 20 пиков (±0.1°2θ) в Таблице 2А, по меньшей мере 25 пиков (±0.1°2θ) в Таблице 2А, или по меньшей мере 30 пиков (±0.1°2θ) в Таблице 2А.
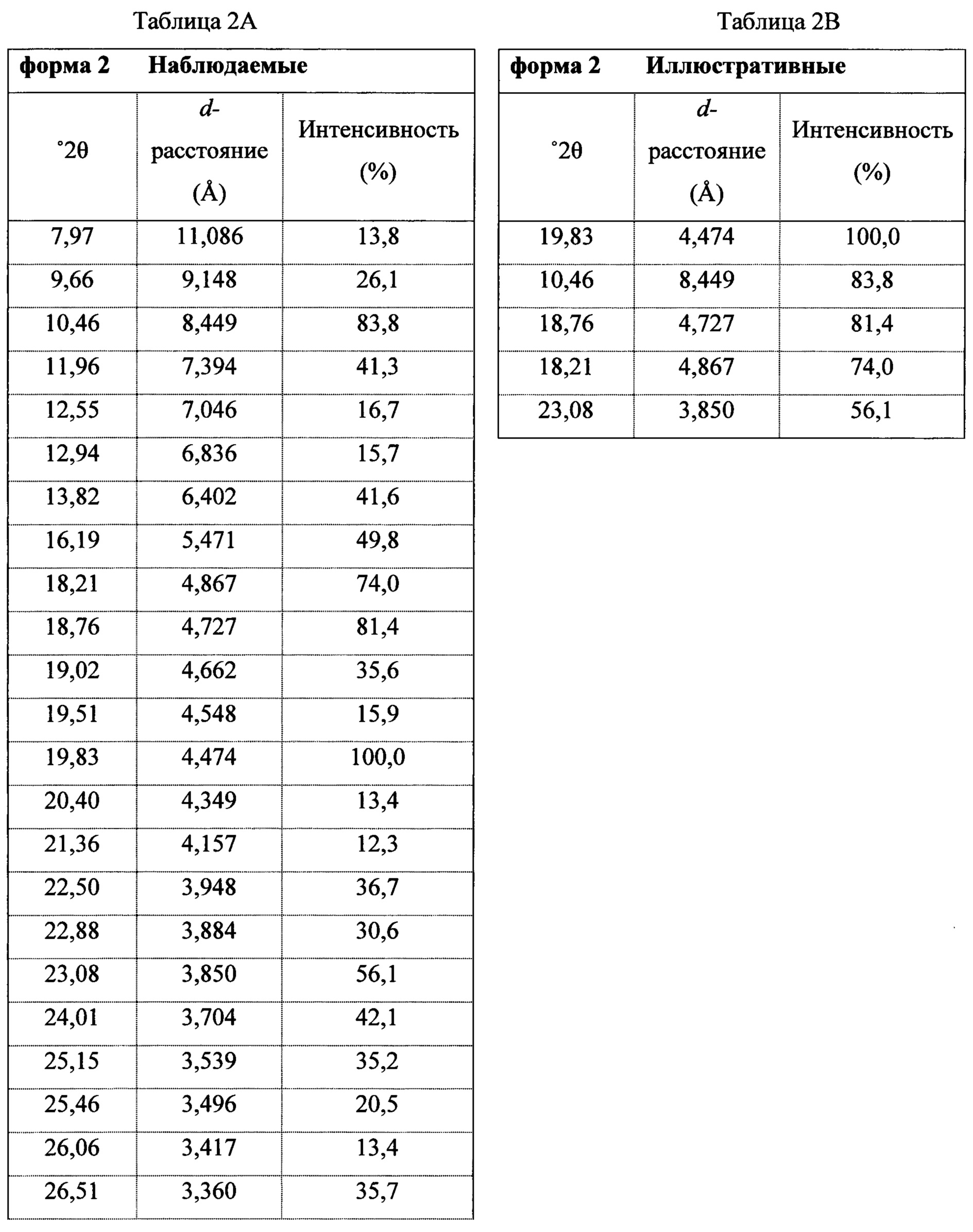
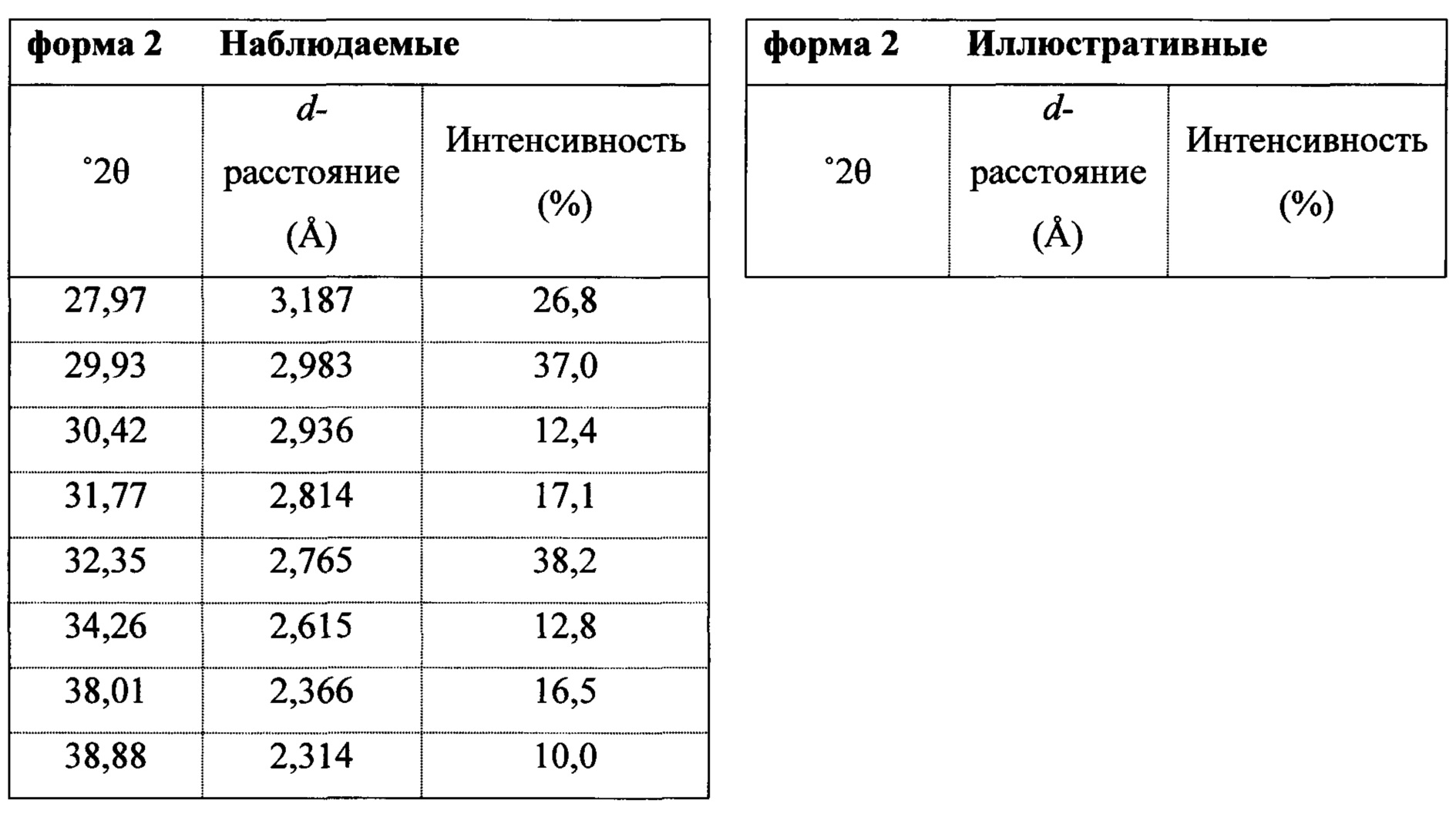
В одном варианте осуществления, предложенном здесь, полиморфная форма 2 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата характеризуется пиками на порошковой рентгеновской дифрактограмме при 10,46, 18,76 и 19,83 градусах два-тета (°26)±0,1 градус два-тета (°2θ). В последующих вариантах осуществления полиморфная форма 2 дополнительно характеризуется, по меньшей мере, один пиком, возникающим при 18,21 или 23,08 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В последующих вариантах осуществления полиморфная форма 2 дополнительно характеризуется, по меньшей мере, двумя пиками, возникающими при 18,21 или 23,08 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В еще последующих вариантах осуществления полиморфная форма 2 имеет порошковую рентгеновскую дифрактограмму по существу такую же, как порошковая рентгеновская дифрактограмма, показанная на фигуре 5.
Было обнаружено, что в определенных случаях кристаллические полиморфы 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата проявляют повышенную стабильность по сравнению с аморфной твердой формой карбоновой кислоты. В некоторых случаях улучшенная стабильность кристаллических полиморфов 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата обеспечивает получение фармацевтических лекарственных форм, проявляющих пониженную изменчивость дозы, присутствующей в заданной лекарственной форме, уменьшение присутствия примесей в препарате готовой лекарственной формы и увеличенный срок хранения изготовленных лекарственных форм по сравнению с фармацевтической лекарственной формой, изготовленной с использованием аморфной твердой формы карбоновой кислоты. В некоторых вариантах осуществления полиморф, описанный здесь (например. Форма 1 или Форма 2), демонстрирует отсутствие деградации (например, меньше чем 0,01%, меньше чем 0,1%, меньше чем 0,5% по массе) в течение по меньшей мере 3 месяцев в условиях ускоренной деградации (например, при 40°C и 75% относительной влажности (RH)), в течение по меньшей мере 4 месяцев в условиях ускоренной деградации (например, при 40°C и 75% относительной влажности (RH)), в течение по меньшей мере 5 месяцев в условиях ускоренной деградации (например, при 40°C и 75% относительной влажности (RH)), в течение по меньшей мере 6 месяцев в условиях ускоренной деградации (например, при 40°C и 75% относительной влажности (RH)), в течение по меньшей мере 9 месяцев в условиях ускоренной деградации (например, при 40°C и 75% относительной влажности (RH)), в течение по меньшей мере 12 месяцев в условиях ускоренной деградации (например, при 40°С и 75% относительной влажности (RH)), и/или (ii) в течение по меньшей мере 12 месяцев в условиях долгосрочного испытания (например, при 25°С и 60% относительной влажности (RH)), в течение по меньшей мере 18 месяцев в условиях долгосрочного испытания (например, при 25°С и 60% относительной влажности (RH)), в течение по меньшей мере 24 месяцев в условиях долгосрочного испытания (например, при 25°С и 60% относительной влажности (RH)).
Кроме того, было обнаружено, что в определенных случаях кристаллические полиморфы 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата проявляют пониженную гигроскопичность по сравнению с другими формами в твердом состоянии по данным результатов исследований гравиметрической паровой сорбции (GVS). На Фигуре 12 показано исследование GVS формы 1 и формы 2. Было обнаружено, что форма 1 поглощает <0,2% масса/масса при высокой влажности, и Форма 2 поглощает <0,1% масса/масса при высокой влажности. Это свойство пониженной гигроскопичности в значительной степени облегчает изготовление твердых фармацевтических лекарственных форм.
Полиморфная форма 4
В одном варианте осуществления полиморфная Форма А 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия имеет порошковую рентгеновскую дифрактограмму, характеризующуюся дифракционной картиной, приведенной в Таблице 1А или Таблице 1В. В некоторых вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, имеющий по меньшей мере 3 пика (±0,1°2θ) в Таблице 1А или 1В. В определенных вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, имеющий по меньшей мере 4 пика (±0,1°2θ) в Таблице 1А или 1В, по меньшей мере 5 пиков (±0,1°2θ) в Таблице 1А или 1В, по меньшей мере 6 пиков (±0,1°2θ) в Таблице 1А или 1В, по меньшей мере 8 пиков (±0,1°2θ) в Таблице 1А или 1В, по меньшей мере 10 пиков (±0,1°2θ) в Таблице 1А, по меньшей мере 15 пиков (±0,1°2θ) в Таблице 1А, по меньшей мере 20 пиков (±0,1°2θ) в Таблице 1А, по меньшей мере 25 пиков (±0,1°2θ) в Таблице 1А, или по меньшей мере 30 пиков (±0,1°2θ) в Таблице 1А.
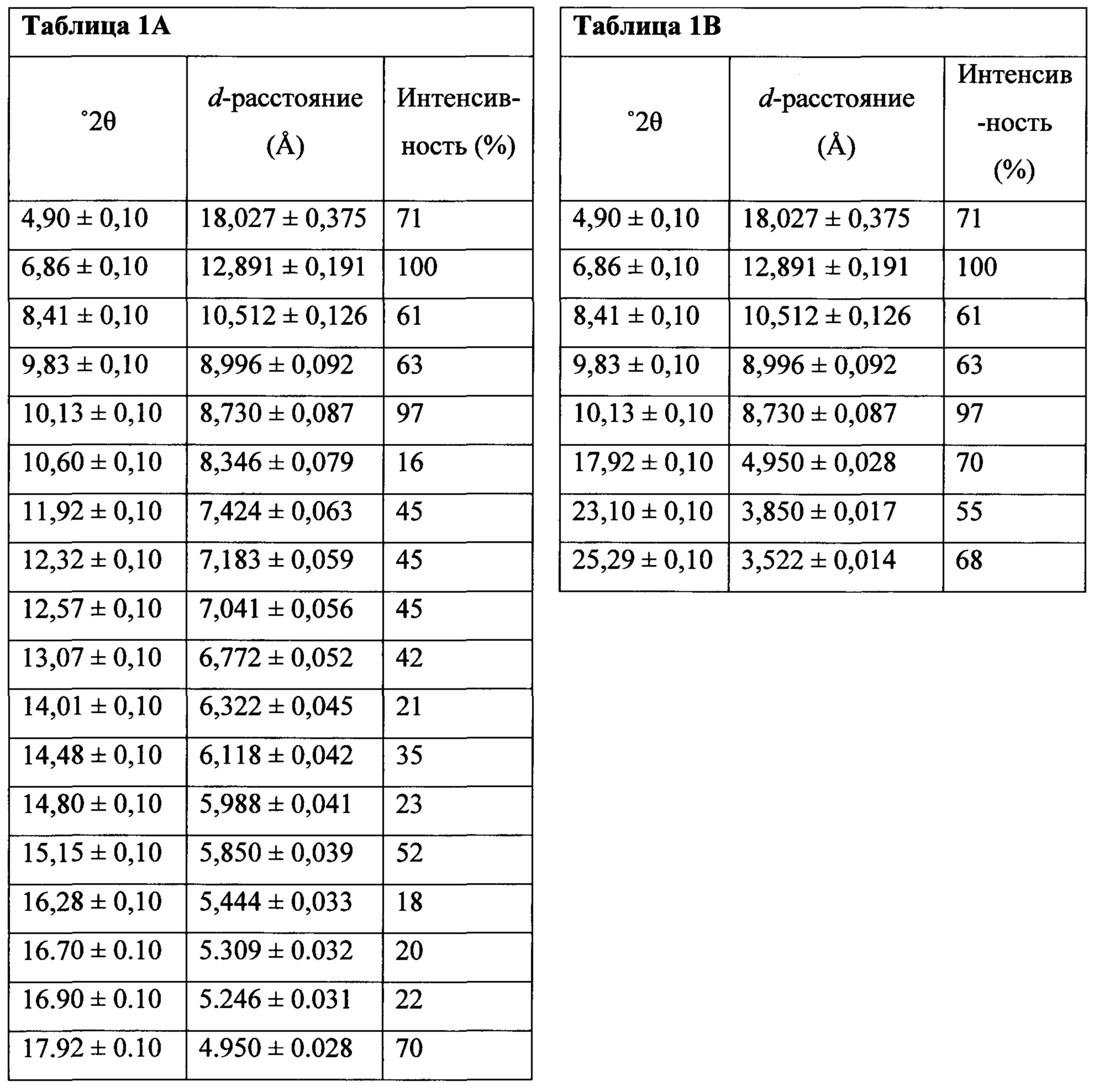
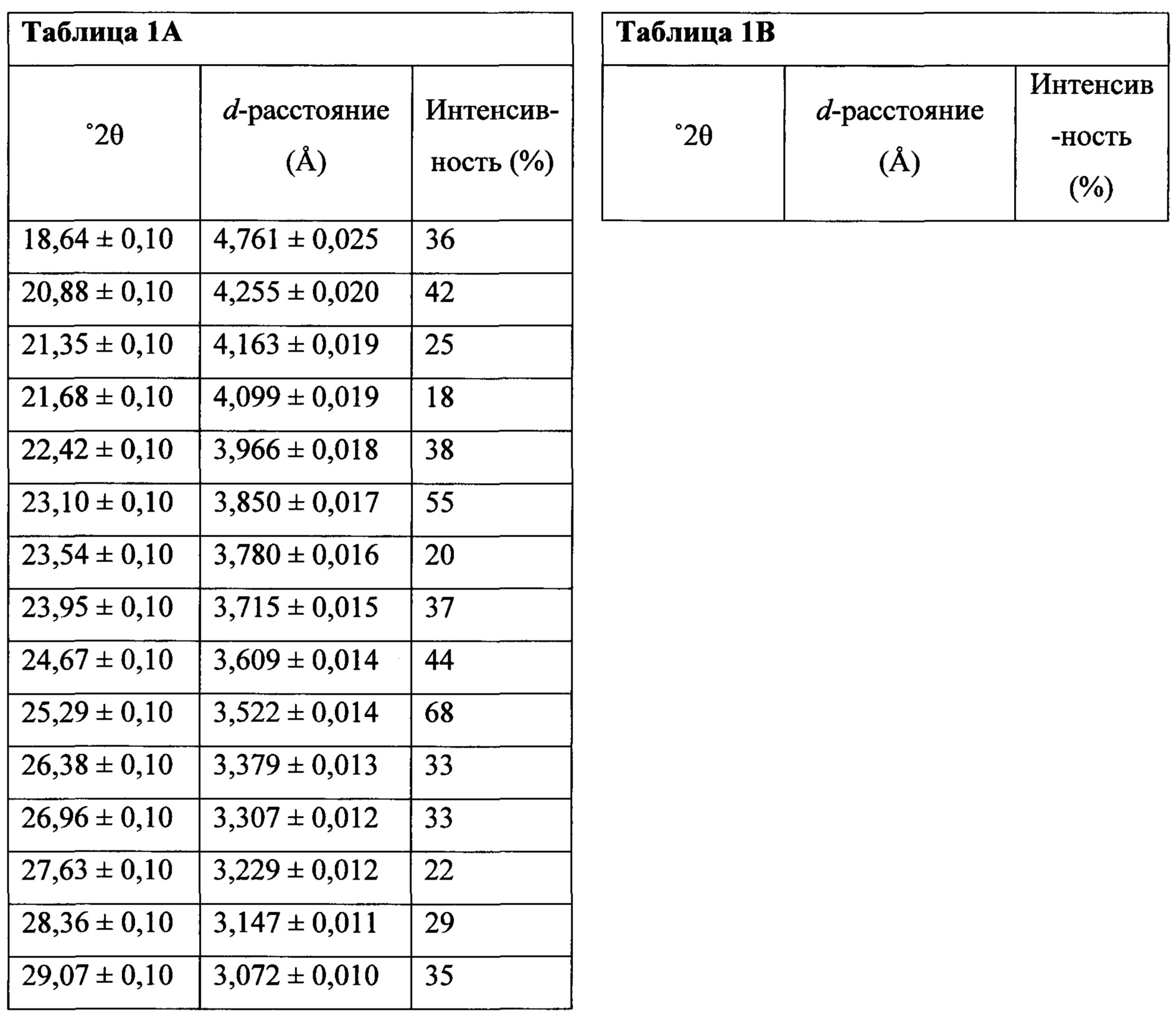
В одном варианте осуществления, предложенном здесь, полиморфная форма А 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия характеризуется пиками на порошковой рентгеновской дифрактограмме при 4,90, 9,83 и 25,29 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В последующих вариантах осуществления полиморфная форма А дополнительно характеризуется, по меньшей мере, одним пиком, возникающим при 6,86, 8,41, 10,13, 17,92 и 23,10 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В последующих вариантах осуществления полиморфная форма А дополнительно характеризуется, по меньшей мере, двумя пиками, возникающими при 6,86, 8,41, 10,13, 17,92 и 23,10 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В еще последующих вариантах осуществления полиморф имеет порошковую рентгеновскую дифрактограмму по существу такую же, как порошковая рентгеновская дифрактограмма, показанная на фигуре 13.
Полиморфная форма В
В одном варианте осуществления полиморфная форма В 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия имеет порошковую рентгеновскую дифрактограмму, характеризующуюся дифракционной картиной, приведенной в таблице 2А или 2В. В некоторых вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, содержащий по меньшей мере 2 пика (±0,1°2θ) в таблице 2А или 2В. В определенных вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, содержащий по меньшей мере 3 пика (±0,1°2θ) в таблице 2А или 2В, по меньшей мере 4 пика (±0,1°2θ) в таблице 2А или 2В, по меньшей мере 5 пиков (±0,1°2θ) в таблице 2А, по меньшей мере 6 пиков (±0,1°2θ) в таблице 2А, по меньшей мере 8 пиков (±0,1°2θ) в таблице 2А, по меньшей мере 10 пиков (±0,1°2θ) в таблице 2А, по меньшей мере 12 пиков (±0,1°2θ) в таблице 2А, по меньшей мере 14 пиков (±0,1°2θ) в таблице 2А, по меньшей мере 16 пиков (±0,1°2θ) в таблице 2А.
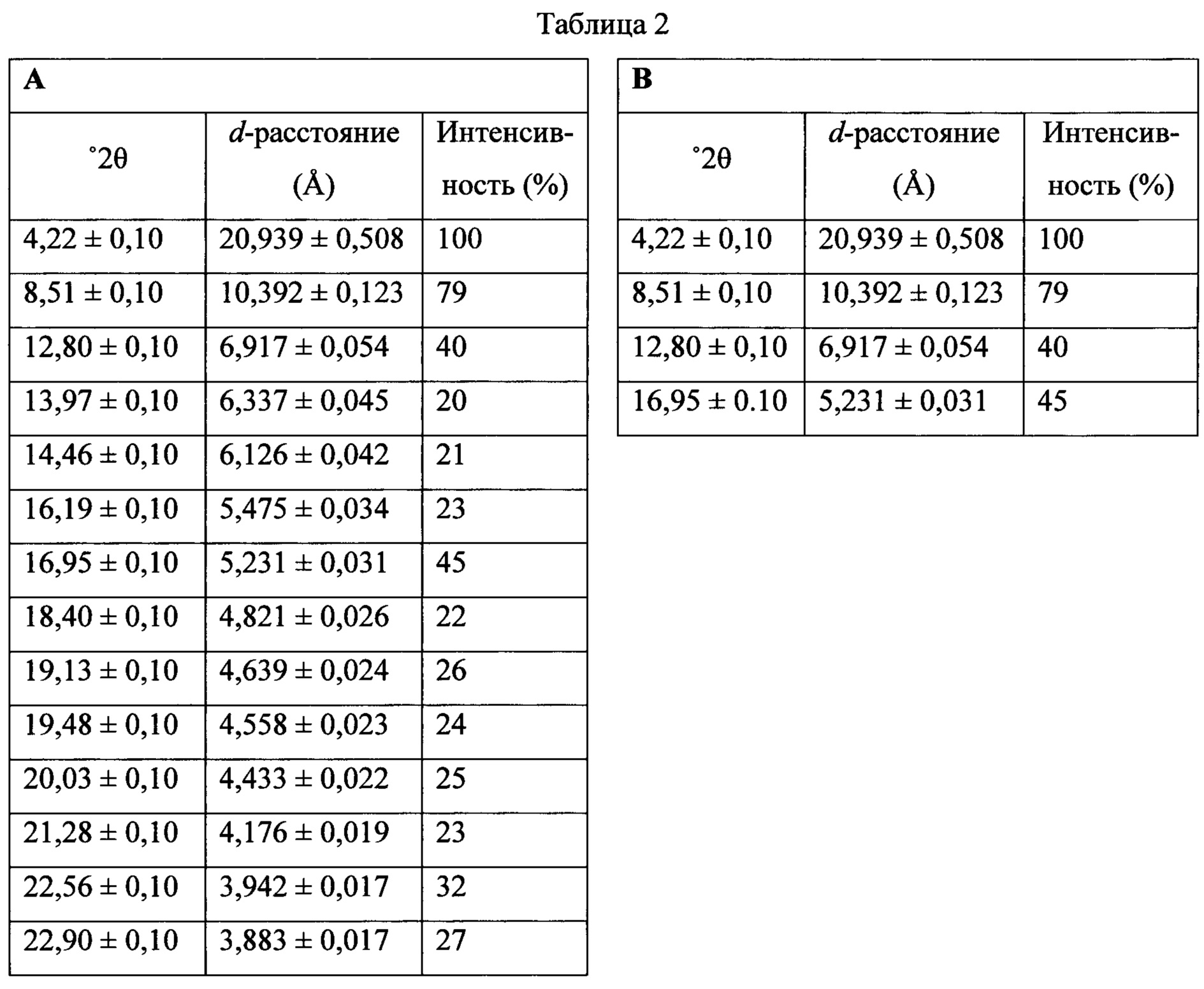

В одном варианте осуществления, предложенном здесь, полиморфная форма В 2-(5-бром-4-(4-циклопропилнафталин-1 -ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия характеризуется пиками на порошковой рентгеновской дифрактограмме при 4,22, 8,51 и 16,95 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В последующем варианте осуществления полиморфная форма В дополнительно характеризуется пиком, возникающим при 12,80 градусах два-тета (°2θ)±0,1 градус два-тета (°2θ). В еще последующих вариантах осуществления полиморф имеет порошковую рентгеновскую дифрактограмму по существу такую же, как порошковая рентгеновская дифрактограмма, показанная на фигуре 17.
Полиморфная форма В'
В одном варианте осуществления полиморфная форма В' 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия имеет порошковую рентгеновскую дифрактограмму, характеризующуюся дифракционной картиной, приведенной на фигуре 19.
Смешивание с аморными формами в твердом состоянии
В определенных вариантах осуществления любой из полиморфов, описанных здесь (например. Форма 1), необязательно содержит (или смешан или в комбинации) определенное количество аморфного 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата. В некоторых вариантах осуществления содержание аморфного компонента полиморфа (например, Формы 1) или полиморфной комбинации составляет менее чем 50 масс. % полиморфа или полиморфной комбинации, менее чем 25 масс. % полиморфа или полиморфной комбинации, менее чем 15 масс. % полиморфа или полиморфной комбинации, менее чем 10 масс. % полиморфа или полиморфной комбинации, или менее чем 5 масс. % полиморфа или полиморфной комбинации.
В определенных вариантах осуществления любой из описанных здесь полиморфов (например. Форма А) необязательно содержит (или смешан или в комбинации) определенное количество аморфного 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия. В некоторых вариантах осуществления содержание аморфного компонента полиморфа (например, Формы А) или полиморфной комбинации составляет менее чем 50 масс. % полиморфа или полиморфной комбинации, менее чем 25 масс. % полиморфа или полиморфной комбинации, менее чем 15 масс. % полиморфа или полиморфной комбинации, менее чем 10 масс. % полиморфа или полиморфной комбинации, или менее чем 5 масс. % полиморфа или полиморфной комбинации.
Размер частиц
В определенных вариантах осуществления в настоящем документе предлагается частица полиморфа 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата (например, кристаллическая или содержащая кристаллический компонент). В некоторых вариантах осуществления в настоящем документе предлагается полиморф 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата (например, кристалличесский или содержащий кристаллический компонент), имеющий размер частиц примерно 5-50 микрон. В некоторых вариантах осуществления средний размер частиц составляет, по меньшей мере, 10 микрон, 15-50 микрон, 15-35 микрон, 35-45 микрон, 35-40 микрон, около 40 микрон и т.п. В некоторых вариантах осуществления частицы 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата (например, кристаллические или содержащие кристаллический компонент, такой как полиморф Формы 1), имеющие средний диаметр больше, чем 5 или 10 микрон, имеют улучшенные параметры стабильности по сравнению с меньшими диаметрами.
Пролекарства
В некоторых вариантах осуществления соединения, описанные здесь, существуют в форме пролекарств. В изобретении предлагаются способы лечения заболеваний путем введения таких пролекарств. Кроме того, в изобретении предлагаются способы лечения заболеваний путем введения таких пролекарств в виде фармацевтических композиций.
Пролекарства обычно являются предшественниками лекарственного средства, которые после введения индивидууму и последующей абсорбции превращаются в активные или более активные единицы посредством способа, такого как превращение метаболическим путем. Некоторые Пролекарства имеют химическую группу, присутствующую в пролекарстве, которая делает их менее активным и/или придает растворимость или некоторые другие свойства лекарственному средству. После отщепления химической группы и/или модифицирования из Пролекарства формируется активный препарат. Пролекарства часто оказываются полезными, потому что в некоторых ситуациях их легче вводить, чем исходное лекарственное средство. Пролекарства являются, например, биодоступными при пероральном введении, тогда как исходное лекарственное средство таковым не является. В определенных случаях пролекарство также имеет лучшую растворимость в фармацевтических композициях по сравнению с исходным лекарственным средством. Примером Пролекарства, без ограничения, является описанное здесь соединение, которое вводят в виде сложного эфира («пролекарство»), чтобы облегчить прохождение через клеточную мембрану, где растворимость в воде вредна для мобильности, но которое затем метаболически гидролизуется в карбоновую кислоту, активную единицу, после поступления в клетку, где растворимость в воде является полезной. Другим примером Пролекарства является короткий пептид (полиаминокислота), связанный с кислотной группой, где пептид метаболизируется с высвобождением действующего вещества. (Смотри, например, Bundgaard, "Design and Application of Prodrugs" in A Textbook of Drug Design and Development, Krosgaard-Larsen and Bundgaard, Ed., 1991, Chapter 5, 113-191, который включен здесь путем отсылки).
В некоторых вариантах осуществления Пролекарства составлены как обратимые производные лекарственного средства для использования в качестве модификаторов с целью улучшения транспорта лекарственного средства в сайт-специфические ткани. До настоящего времени составление пролекарств было направлено на повышение эффективной растворимости в воде терапевтического соединения для нацеливания его на области, в которых вода является основным растворителем.
Кроме того, производные-пролекарства описанных здесь соединений могут быть получены способами, описанными здесь, которые известны в данной области (более подробная информация приведена в Saulnier et al., Bioorganic and Medicinal Chemistry Letters, 1994, 4, 1985). Только в качестве примера, соответствующие Пролекарства могут быть получены путем взаимодействия недериватизированного соединения с подходящим карбамилирующим агентом, таким как, но без ограничения, 1,1-ацилоксиалкилкарбанохлоридат, пара-нитрофенил карбамат или аналогичными. Пролекарственные формы описанных здесь соединений, отличающиеся тем, что пролекарство метаболизируется in vivo с образованием производного, как указано здесь далее, включены в объем формулы изобретения. Действительно, некоторые из описанных здесь соединений могут быть пролекарствами для другого производного или активного соединения.
В некоторых вариантах осуществления пролекарства включают соединения, в которых аминокислотный остаток или полипептидная цепь из двух или более (например, двух, трех или четырех) аминокислотных остатков ковалентно связаны через амидную или сложноэфирную связь со свободной амино, гидрокси или карбоновокислотной группой соединений по настоящему изобретению. Аминокислотные остатки включают, но без ограничения, 20 аминокислот природного происхождения, а также включают 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, орнитин и метионин сульфон. В других вариантах осуществления пролекарства включают соединения, в которых остаток нуклеиновой кислоты или олигонуклеотид из двух или более (например, двух, трех или четырех) остатков нуклеиновой кислоты ковалентно связаны с соединением по настоящему изобретению. В некоторых вариантах осуществления пролекарства включают соединения, содержащие сложный алкиловый эфир. В некоторых таких вариантах осуществления сложный алкиловый эфир отщепляется in vivo с получением Соединения 1. Неограничивающие примеры сложноэфирных пролекарств включают метиловый, этиловый, пропиловый, изопропиловый, бутиловый, изобутиловый, трет-бутиловый, изоамиловый, пентиловый, гексиловый, гептиловый, октиловый, нониловый, терпениловый, борниловый, аллиловый, линалиловый или гераниловый сложные эфиры.
Фармацевтически приемлемые пролекарства описанных здесь соединений также включают, но без ограничения, сложные эфиры, карбонаты, тиокарбонаты, N-ацильные производные, N-ацилоксиалкильные производные, четвертичные производные третичных аминов, основания N-Манниха, основания Шиффа, аминокислотные конъюгаты, сложные фосфатные эфиры, соли металлов и сложные эфиры сульфоновых кислот.Соединения, имеющие свободные амино, амидо, гидрокси или карбоксильные группы, могут быть превращены в пролекарства. Например, свободные карбоксильные группы могут быть дериватизированы как амиды или сложные алкиловые эфиры. В определенных вариантах осуществления все из этих фрагментов пролекарств содержат группы, включающие, но без ограничения, эфирные, аминовые и карбоксильные функциональные группы.
Пролекарства, содержащие гидроксильную группу, включают сложные эфиры, такие как, но без ограничения, сложные ацилоксиалкиловые (например, ацилоксиметиловые, ацилоксиэтиловые) эфиры, сложные алкиловые эфиры, сложные ариловые эфиры, сложные фосфатные эфиры, сложные сульфонатные эфиры, сложные сульфатные эфиры и сложные эфиры, содержащие дисульфид; простые эфиры, амиды, карбаматы, гемисукцинаты, диметиламиноацетаты и фосфорилоксиметилоксикарбонилы, как описано в Advanced Drug Delivery Reviews 1996,79,115.
Пролекарства на основе аминов включают, но без ограничения, следующие группы и комбинации групп:
а также сульфоамиды и фосфонамиды.
Примеры
Предложенные ниже примеры и препараты далее иллюстрируют соединения по настоящему изобретению и способы их получения. Следует понимать, что объем настоящего изобретения не ограничивается каким-либо образом объемом следующих примеров и препаратов.
Пример 1А; Синтез Соединения 2
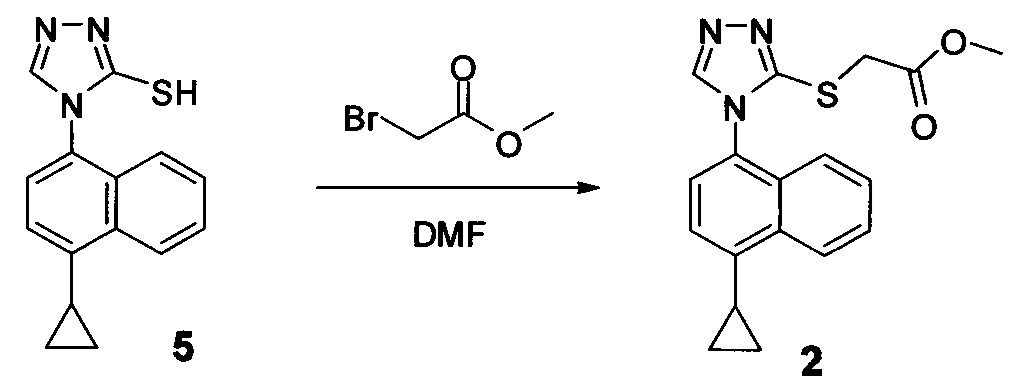
В реактор загружают диметилформамид (2,63 кг ±5%  2,8 л ±5%). Диметилформамид нагревают до температуры между 27°C и 35°C. В этот же реактор порциями загружают Соединение 5 (1,0 кг ±1%) при поддержании температуры между 27°C и 35°C. В реакционную смесь добавляют метилбромацетат (0,6008 / М × 100 кг ±1% 0,3620 / М × 100 л ±1%) при поддержании температуры между 27°C и 35°C, например, между 28°C и 33°C, где М представляет собой чистоту метилбромацетата по GC в % площади. При добавлении метилбромацетата происходит немного экзотермический способ.
2,8 л ±5%). Диметилформамид нагревают до температуры между 27°C и 35°C. В этот же реактор порциями загружают Соединение 5 (1,0 кг ±1%) при поддержании температуры между 27°C и 35°C. В реакционную смесь добавляют метилбромацетат (0,6008 / М × 100 кг ±1% 0,3620 / М × 100 л ±1%) при поддержании температуры между 27°C и 35°C, например, между 28°C и 33°C, где М представляет собой чистоту метилбромацетата по GC в % площади. При добавлении метилбромацетата происходит немного экзотермический способ.
Смесь перемешивают в течение по меньшей мере 10 минут, но не более 20 минут при температуре между 27°C и 35°C, например, между 28°C и 33°C.
В реакционную смесь порциями добавляют бикарбонат натрия (0,314 кг±1%) при поддержании температуры между 27°C и 35°C в течение по меньшей мере 30 минут, но не более 70 минут. Во время добавления порций бикарбоната натрия происходит выделение газообразного диоксида углерода.
Смесь перемешивают в течение по меньшей мере 1 часа, но не более 4 часов при температуре между 27°C и 35°C, например, между 28°C и 33°C. Реакционную смесь охлаждают до температуры между 5°C и 10°C, и затем отбирают образец для анализа методом HPLC.
Реакция считается завершенной, если содержание Соединения 5 составляет меньше, чем 0,50% площади по данным HPLC, предпочтительно меньше, чем 0,20% площади по данным HPLC. Если завершение реакции не достигается после второго образца, реакционную смесь нагревают до температуры между 27°C и 35°C. В реакционную смесь добавляют метилбромацетат (0,0172 / М × 100 кг ±1% 0,0103 / М × 100 л ±1%) при поддержании температуры между 27°C и 35°C, например, между 28°C и 33°C. Смесь перемешивают в течение по меньшей мере 30 минут, но не более 2 часов при температуре между 27°C и 35°C, например, между 28°C и 33°C, затем отбирают образец для анализа методом HPLC.
Если реакция завершена, в реакционную смесь добавляют озонированную деионизированную воду (9,0 л ±5%) в течение, по меньшей мере, 15 минут при поддержании температуры между 5°C и 20°C. Во время добавления происходит выделение газообразного диоксида углерода. При добавлении озонированной деионизированной воды происходит немного экзотермический способ. Смесь перемешивают в течение, по меньшей мере, 30 минут при поддержании температуры между 5°C и 10°C.
В смесь добавляют раствор, предварительно приготовленный путем растворения бикарбоната натрия (0,105 кг ±1%) в озонированной деионизированной воде (1,47 л ±5%), в течение, по меньшей мере, 10 минут при поддержании температуры между 5°C и 20°C до достижения pH смеси между 6,7 и 8,0, например, между 6,9 и 7,3. Суспензию перемешивают в течение, по меньшей мере, 60 минут при поддержании температуры между 5°C и 10°C. Суспензию фильтруют. Влажный фильтрационный осадок дважды промывают озонированной деионизированной водой (2,0 л ±5%), предварительно охлажденной до температуры между 5°C и 10°C.
Влажный фильтрационный осадок дважды промывают смесью этилацетата (0,09 кг ±5% 0,1 л ±5%) и изопропилового спирта (0,79 кг ±5% 1,0 л ±5%), предварительно охлажденной до температуры между 0°C и 5°C.
Влажный фильтрационный осадок сушат в вакууме при температуре ниже 45°C до тех пор, пока содержание воды (по данным анализа по методу Карла Фишера) не составит 0,5% масса/масса, предпочтительно 0,1% масса/масса или меньше, содержание этилацетата по данным GC не составит 200 ppm или меньше, содержание изопропилового спирта по данным GC не составит 600 ppm или меньше, и содержание диметилформамида по данным GC не составит 10000 ppm или меньше.
Пример 1 В: Синтез Соединения 2
Получение в крупном масштабе метил 2-((4-(4-ииклопропшнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата
Размер партии для получения в крупном масштабе метил 2-((4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата составляет 1,0 кг. Рассчитанный стехиометрический выход составил 126,95% масса/масса, при этом предполагаемый выход составил 120±6% масса/масса (95±5 мол. %).
Стадия 1
В реактор загружают диметилформамид (2,86 кг ±5%; 3,05 л ±5%) и нагревают до 15-23°C. В реактор порциями добавляют 4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-тиол (1,0 кг ±1,5%), поддерживая температуру между 15°C и 23°C. В реактор добавляют бикарбонат натрия (0,161 кг ±1%), поддерживая температуру между 15°C и 23°C. В реактор добавляют метилбромацетат (0,6095 кг ±1%; 0,3672 л ±1%), поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C.
Если чистота метилбромацетата составляет <99.0% (по данным GC), то добавляют количество согласно формуле: 0,6065 / М×100 кг ±1%; 0,3654 /М×100 л ±1%, где М представляет собой чистоту метилбромацетата (по GC в % площади). При добавлении метилбромацетата происходит немного экзотермический способ. Линию загрузки споласкивают диметилформамидом (0,19 кг ±5%; 0,2 л ±5%), добавляя промывочную жидкость в реактор, поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C.
Смесь перемешивают в течение по меньшей мере 20 минут, но не более 40 минут, поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C. В реактор порциями добавляют бикарбонат натрия (0,161 кг ±1%) в течение по меньшей мере 20 минут, но не более 120 минут, поддерживая температуру между 15°C и 30°C. Следует учесть, что при добавлении бикарбоната натрия происходит эндотермический способ и выделяется газообразный диоксид углерода. Смесь перемешивают в течение по меньшей мере 1 часа, но не более 8 часов, поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C.
Реакция считается завершенной, когда содержание 4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-тиола составляет меньше, чем 1,0% площади по данным HPLC, предпочтительно меньше, чем 0,50% площади по данным HPLC. В случае завершения реакции переходят к стадии 3. В случае, если реакция не завершалась, берут еще один образец для анализа HPLC; если реакция все еще не завершалась, переходят к стадии 2.
СТАДИЯ 2
В реактор добавляют метилбромацетат (0,01146 кг ±1%; 0,00683 л ±1%), поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C.
Если чистота метилбромацетата составляет <99.0%, (по данным GC), то добавляют количество согласно формуле: 0,0114 / М×100 кг ±1%; 0,0068 / М×100 л ±1%, где М представляет собой чистоту метилбромацетата (по GC в % площади). Линию загрузки споласкивают диметилформамидом (4,7 кг ±5%; 5 л ±5% - фиксированное количество), добавляя промывочную жидкость в реактор, поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C.
Смесь перемешивают в течение по меньшей мере 30 минут, но не более 2 часов, поддерживая температуру между 15°C и 30°C, предпочтительно между 17°C и 21°C. Реакция считается завершенной, когда содержание 4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-тиола составляет меньше, чем 1,0% площади по данным HPLC, предпочтительно меньше, чем 0,50% площади по данным HPLC.
В случае завершения реакции переходят к стадии 3. В случае, если реакция не завершалась, берут еще один образец для анализа методом HPLC. Если реакция все еще не завершалась после второго образца, повторяют стадию 2.
СТАДИЯ 3
В реакционную смесь добавляют озонированную деионизированную воду (0,5 л ±5%) в течение, по меньшей мере, 15 минут, поддерживая температуру между 10°C и 30°C. При добавлении происходит немного экзотермический способ и может выделяться газообразный диоксид углерода. Смесь перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 10°C и 30°C, при этом в этот период времени может происходить образование осадка, представляющее собой немного экзотермический способ.
В реакционную смесь добавляют вторую порцию озонированной деионизированной воды (8,5 л ±5%) в течение, по меньшей мере, 30 минут, поддерживая температуру между 10°C и 30°C, при этом в этот период времени происходит осаждение продукта. При добавлении протекает экзотермический способ и может выделяться диоксид углерода.
Суспензию охлаждают до 5-10°C. Измеряют pH суспензии. При необходимости регулируют pH до значения между 6,3 и 8,3, предпочтительно между 6,9 и 7,3 путем добавления в течение, по меньшей мере, 10 минут предварительно приготовленного раствора бикарбоната натрия (0,105 кг ±1%) в озонированной деионизированной воде (1,47 л ±5%), поддерживая температуру между 5°C и 10°C.
Добавляют озонированную деионизированную воду (1,0 л ±5%) в течение, по меньшей мере, 10 минут, поддерживая температуру между 5°C и 10°C. Суспензию перемешивают в течение, по меньшей мере, 60 минут, поддерживая температуру между 5°C и 10°C.
СТАДИЯ 4
Суспензию фильтруют. Влажный фильтрационный осадок промывают озонированной деионизированной водой (2,0 л ±5%), предварительно охлажденной до температуры между 5°C и 10°C. Промывают влажный фильтрационный осадок озонированной деионизированной водой (2,0 л ±5%), предварительно охлажденной до 5-10°C. Промывают влажный фильтрационный осадок раствором этилацетата (0,09 кг ±5%; 0,1 л ±5%) и изопропиловым спиртом (0,79 кг ±5%; 1,0 л ±5%), предварительно охлажденным до 0-5°C. Влажный фильтрационный осадок промывают раствором этилацетата (0,09 кг ±5%; 0,1 л ±5%) и изопропиловым спиртом (0,79 кг ±5%; 1,0 л ±5%), предварительно охлажденным до 0-5°C.
Влажный фильтрационный осадок сушат в вакууме при температуре ниже 60°C до тех пор, пока содержание воды (по данным анализа по методике Карла Фишера) не составит ≤0.2% масса/масса, предпочтительно ≤0.1% масса/масса; содержание этилацетата (по данным GC) не составит ≤200 ppm; содержание изопропилового спирта (по данным GC) не составит ≤600 ppm; и содержание диметилформамида (по данным GC) не составит ≤10000 ppm.
Пример 2А; Синтез Соединения 1 и Соединения 4
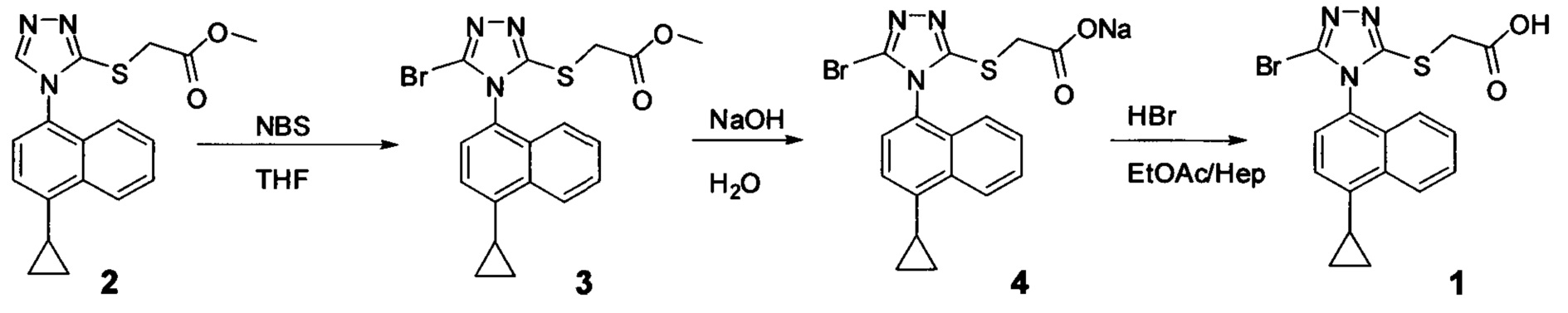
В реактор загружают Соединение 2 (1,0 кг ±1%). В этот же реактор добавляют тетрагидрофуран (6,2 кг ±1% 7,0 л ±1%). Смесь нагревают до температуры между 35°C и 42°C. Смесь перемешивают в течение, по меньшей мере, 10 минут при температуре между 35°C и 42°C для получения прозрачного раствора. Реакционную смесь охлаждают до температуры между 27°C и 32°C.
В реакционную смесь добавляют N-бромсукцинимид (0,734 кг ±1%) при поддержании температуры между 27°C и 32°C, например, между 27°C и 30°C. Смесь перемешивают при температуре между 27°C и 32°C, например, между 27°C и 30°C до завершения реакции.
Реакция считается завершенной, когда содержание Соединения 2 составляет меньше, чем 1,5% площади по данным HPLC, предпочтительно меньше, чем 0,2% площади по данным HPLC.
Отбирают образец для анализа методом HPLC через 20-40 минут перемешивания для определения содержания Соединения 2. На основе анализа HPLC, необязательно, добавляют дополнительное количество N-бромсукцинимида (0,105 кг ±1%) при поддержании температуры между 27°C и 32°C, например, 27°C и 30°C. В противном случае перемешивание продолжают при температуре между 27°C и 32°C, например, между 27°C и 30°C до завершения реакции.
Реакционную смесь охлаждают до температуры между 2°C и 7°C, например, между 2°C и 5°C. В смесь добавляют толуол (4,33 кг ±5%) при поддержании температуры между 2°C и 7°C, например, между 2°C и 5°C.
В реакционную смесь добавляют озонированную деионизированную воду (5,0 л ±5%) в течение, по меньшей мере, 10 минут при поддержании температуры между 2°C и 7°C, например, между 2°C и 5°C. При добавлении озонированной деионизированной воды происходит экзотермическим способ и может возникнуть газовыделение. Смесь перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 2°C и 7°C, например, между 4°C и 6°C.
Перемешивание останавливают и оставляют для разделения слоев, по меньшей мере, на 30 минут. Удаляют водную фазу (нижнюю фазу). В органическую фазу добавляют раствор, предварительно приготовленный путем растворения дисульфита натрия (0,112 кг ±1%) в озонированной деионизированной воде (5,0 л ±5%), в течение, по меньшей мере, 10 минут при поддержании температуры между 2°C и 7°C. При добавлении раствора дисульфита натрия происходит экзотермический способ. Во время добавления может происходить выделение газа.
Суспензию перемешивают в течение, по меньшей мере, 30 минут при поддержании температуры между 2°C и 7°C. Отбирают образец смеси. Если водная фаза образца имеет бледно-желтую окраску, то выполняют еще одну стадию промывки дисульфитом натрия. Если водная фаза образца является бесцветной, то образец отправляют на анализ HPLC. Если пик N-бромсукцинимида обнаруживают с помощью HPLC, то выполняют еще одно промывание дисульфитом натрия и повторяют анализ HPLC до тех пор, пока NBS не будет обнаруживаться с помощью HPLC.
Перемешивание останавливают и оставляли слои для разделения, по меньшей мере, на 15 минут. Удаляют водную фазу (нижнюю фазу) и объединяют с предыдущей водной фазой. Органическую фазу, содержащую Соединение 3, нагревают до температуры между 18°C и 25°C. В органическую фазу добавляют озонированную деионизированную воду (5,0 л ±5%), поддерживая температуру между 18°C и 25°C. Смесь перемешивают в течение, по меньшей мере, 15 минут, поддерживая температуру между 18°C и 25°C. Перемешивание останавливают и оставляют слои для разделения, по меньшей мере, на 15 минут. Водную фазу (нижнюю фазу) удаляют.
В органическую фазу добавляют раствор, предварительно приготовленный путем растворения бикарбоната натрия (0,35 кг ±1%) в озонированной деионизированной воде (5,0 л ±5%), поддерживая температуру между 18°C и 25°C.
Смесь перемешивают в течение, по меньшей мере. 15 минут, поддерживая температуру между 18°C и 25°C. Перемешивание останавливают, и слои оставляют для разделения, по меньшей мере, на 15 минут. Водную фазу (нижнюю фазу) удаляют.
Если pH удаленной водной фазы ниже 8,0, повторяют стадию промывки бикарбонатом натрия до тех пор, пока pH водной фазы не достигнет значения выше 8,0.
В органическую фазу, содержащую Соединение 3, в течение, по меньшей мере, 10 минут, поддерживая температуру между 18°C и 25°C, добавляют раствор, предварительно приготовленный путем растворения гидроксида натрия (чистого) (0,1473 кг ±1%) в озонированной деионизированной воде (3,61 л ±5%).
Смесь перемешивают при температуре между 18°C и 25°C в течение, по меньшей мере, 2 часов до завершения реакции. Реакция считается завершенной, когда площадь пика по данным HPLC Соединения 3 в органической фазе составляет меньше, чем 50 mAU. Если реакция не завершается, то реакционную смесь перемешивают дополнительно в течение 2 часов перед взятием образца. Если завершение реакции не достигается через 6 часов перемешивания, то добавляют дополнительное количество водного раствора гидроксида натрия, и повторно берут образец через 3 часа после добавления. На данном этапе реакционная смесь содержит две фазы. Перемешивание останавливают и оставляют слои для разделения, по меньшей мере, на 30 минут. Водную фазу (нижнюю фазу) отводят в реактор или сборник. Эту стадию повторяют и водные слои объединяют. Органическую фазу (верхнюю фазу) удаляют для утилизации.
Водные фазы концентрируют в вакууме при температуре 40°C или ниже до тех пор, пока дистиллят не прекратит собираться с использованием давления не ниже, чем 75 мбар.
Отбирают образец водной фазы для определения содержания Соединения 4 в концентрированной водной фазе. Регулируют объем концентрированной водной фазы до объема примерно 5,5×W л ±5%, где W представляет собой количество Соединения 1 в кг, рассчитанное из анализируемого образца.
Смесь охлаждают до температуры между 5°C и 0°C в течение, по меньшей мере, 2 часов. Смесь перемешивают в течение, по меньшей мере, 2 часов, поддерживая температуру между 0°C и 5°C. На данном этапе происходит кристаллизация натриевой соли, то есть Соединения 4.
Суспензию нагревают до температуры между 17°C и 19°C. Смесь перемешивают в течение, по меньшей мере, 1 часа, поддерживая температуру между 17°C и 19°C. После этого периода перемешивания натриевая соль Соединения 4 остается в кристаллизованном состоянии.
Смесь охлаждают до температуры между 15°C и 13°C в течение, по меньшей мере, 4 часов. Смесь охлаждают до температуры между 5°C и 0°C в течение, по меньшей мере, 2 часов. Смесь перемешивают в течение, по меньшей мере, 2 часов и, предпочтительно, не более 4 часов, поддерживая температуру между 0°C и 5°C.
Суспензию фильруют. Влажный фильтрационный осадок промывают до 3 раз озонированной деионизированной водой (0,50 л ±5%), предварительно охлажденной до температуры между 0°C и 5°C в потоке азота в сочетании с вакуумом.
Влажный фильтрационный осадок, содержащий Соединение 4, растворяют озонированной деионизированной водой (4,5 л ±5%) при температуре не выше 35°C, и раствор переносят в реактор или сборник. Отбирают образец водного раствора для определения содержания Соединения 1 (W2). W2 представляет собой количество Соединения 1 в кг, рассчитанное из анализируемого образца.
В водный раствор добавляют этилацетат (9,0×W2 кг ±5% 10,0×W2 л ±5%). В смесь добавляют 24% водный раствор бромистоводородной кислоты (переменная величина) при поддержании температуры 35°C или ниже до тех пор, пока pH смеси не достигнет значения между 2,0 и 4,0, например, между 3,0 и 4,0. Водный раствор гидроксида натрия добавляют в смесь, если значение pH опускается ниже 2,0. Смесь перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 30°C и 35°C. Перемешивание останавливают и оставляли слои для разделения, по меньшей мере, на 30 минут.
Органическую фазу (нижнюю фазу) направляют в реактор или сборник и отбирают образец водной фазы для анализа HPLC. Если площадь пика по данным HPLC Соединения 1 выше, чем 500 mAU, промывку бромистоводородной кислотой повторяют, и водные фазы, содержащие Соединение 1, объединяют.
В водную фазу добавляют этилацетат (0,9×W2 кг ±5% 1,0×W2 л ±5%) при поддержании температуры между 30°C и 35°C. Смесь перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 30°C и 35°C. Перемешивание останавливают, и слои оставляют для разделения, по меньшей мере, на 30 минут.
Органическую фазу (верхнюю фазу) направляют в реактор или сборник, и отбирают образец водной фазы для анализа HPLC. Если площадь пика по данным HPLC Соединения 1 выше, чем 500 mAU, то экстракцию этилацетатом повторяют.
Водную фазу (нижнюю фазу) удаляют для утилизации. В объединенную органическую фазу добавляют озонированную деионизированную воду (2,0×W2 л ±5%) при поддержании температуры между 30°C и 35°C. Смесь перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 30°C и 35°C. Перемешивание останавливают, и оставляют слои для разделения, по меньшей мере, на 30 минут. Водную фазу (нижнюю фазу) удаляют для утилизации.
Органическую фазу фильтруют через фильтр с пористостью не выше, чем 1 микрон, и фильтрат переносят в реактор или сборник. Фильтрованную органическую фазу концентрируют в вакууме при температуре 38°C или ниже, например, при температуре между 18°C и 25°C до конечного объема между 7,3×W2 л и 7,7×W2 л. На данном этапе может происходить кристаллизация Соединения 1.
Смесь нагревают до температуры между 35°C и 42°C, например, между 38°C и 40°C, и перемешивают в течение, по меньшей мере, 3 часов, но не более 8 часов, например, между 3 и 5 часами при поддержании температуры.
Необязательно, затравку свободной кислоты Соединения 1 добавляют в виде твердого вещества или суспендированного в н-гептане (0,5×W2 л - фиксированное количество), предварительно пропущенном через фильтр с пористостью не больше, чем 1 микрон.
Фильтрованную органическую фазу концентрируют в вакууме при температуре 38°C или ниже, например, при температуре между 18°C и 25°C до достижения конечного объема между 3,8×W2 л и 4,2×W2 л.
В суспензию добавляют н-гептан (0,68×W2 кг ±5% 1,0×W2 л ±5%), предварительно пропущенный через фильтр с пористостью не больше, чем 1 микрон, при поддержании температуры между 35°C и 42°C, например, между 38°C и 40°C. Смесь перемешивают в течение, по меньшей мере, 1 часа, поддерживая температуру между 35°C и 42°C, предпочтительно между 38°C и 40°C. Смесь охлаждают до температуры между 10°C и 5°C в течение, по меньшей мере, 2 часов. Суспензию фильтруют.
Влажный фильтрационный осадок промывают раствором этилацетата (0,63×W2 кг ±5% 0,7×W2 л ±5%) и н-гептана (0,48×W2 кг ±5% 0,7×W2 л ±5%), предварительно охлажденного до температуры между 5°C и 10°C, и фильтруют через фильтр с пористостью не больше, чем 1 микрон. Влажный фильтрационный осадок сушат в вакууме при температуре 50°C или ниже до тех пор, пока содержание н-гептана по данным GC не составит 5000 ppm или ниже, содержание этилацетата по данным GC не составит 2500 ppm или ниже, и содержание воды (по данным анализа по методу Карла Фишера) не составит 0,5% масса/масса или ниже.
Пример 2 В: Синтез Соединения 1 и Соединения 4
Получение в крупном масштабе 2-(5-бром-4-(4-ииклопропылнафталин-1-ил)-4Н-1,2Л-триазол-3-илтио)ацетата
Размер партии для получения в крупном масштабе 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата составляет 1,0 кг. Рассчитанный стехиометрический выход составляет 119,1% масса/масса, при этом предполагаемый выход составляет 89±24% масса/масса (75±20 мол.%).
Стадия 1
В реактор загружают 2-((4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетат (1,0 кг ±2%). В реактор добавляют тетрагидрофуран (6,0 кг ±1%; 6,8 л ±1%). Смесь нагревают до температуры между 25°C и 35°C, предпочтительно между 29°C и 31°C.
В реактор добавляют N-бромсукцинимид (0,8128 кг ±1%) в течение <3 часов, поддерживая температуру между 25°C и 40°C, предпочтительно между 31°C и 34°C. Линию загрузки споласкивают тетрагидрофураном (0,18 кг ±1%; 0,20 л ±1%), добавляя промывочную жидкость в реактор, поддерживая температуру между 25°C и 40°C, предпочтительно между 31°C и 34°C.
Смесь перемешивают при температуре между 25°C и 40°C, предпочтительно между 31°C и 34°C до завершения реакции. Реакция считается завершенной, когда содержание метил 2-((4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата составляет <1.5% площади по данным HPLC, предпочтительно 0.2% площади по данным HPLC. Если реакция не завершается через 3 часа, добавляют дополнительное количество N-бромсукцинимида (0,052 кг ±1%) при 25-40°C, предпочтительно между 31°C и 34°C, споласкивают линию загрузки тетрагидрофураном (0,18 кг ±1%; 0,20 л ±1%) и добавляют промывочную жидкость в реактор, поддерживая температуру 25-40°C, предпочтительно между 31°C и 34°C. Выдерживают после дополнительной загрузки N-бромсукцинимида перед повторным отбором образца.
СТАДИЯ 2
Реакционную смесь охлаждают до 2-7°C, предпочтительно между 2°C и 5°C. В реактор добавляют толуол (5,20 кг ±5%; 6,0 л ±5%), поддерживая температуру между 2°C и 7°C, предпочтительно между 2°C и 5°C.
В реактор добавляют озонированную деионизированную воду (5,0 л ±5%) в течение, по меньшей мере, 10 минут, поддерживая температуру между 2°C и 7°C, предпочтительно между 2°C и 5°C. При добавлении происходит экзотермический способ и может возникать выделение газа. Перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 2°C и 7°C, предпочтительно между 4°C и 6°C. Перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения слоев. В случае образования эмульсии добавляют толуол (0,43 кг ±5%; 0,5 л ±5%), поддерживая температуру между 2°C и 7°C, предпочтительно между 4°C и 6°C, и смесь перемешивают в течение 15 минут перед повторным отстаиванием фаз. Водную, то есть нижнюю фазу, удаляют.
В органическую фазу добавляют предварительно приготовленный раствор дисульфита натрия (0,154 кг ±1%) в озонированной деонизированной воде (5,0 л ±5%) в течение, по меньшей мере, 10 минут, поддерживая температуру между 2°C и 7°C. При добавлении происходит экзотермический способ и может возникнуть газовыделение. Во время приготовления раствора дисульфита натрия добавляют дополнительное количество дисульфита натрия (0,028 кг ±1%) для каждого дополнительного количества N-бромсукцинимида (0,052 кг ±1%), добавляемого во время реакции бромирования, вплоть до максимального загружаемого количества дисульфита натрия (0,196 кг ±1%).
Суспензию перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 2°C и 7°C. Перемешивание останавливают и оставляют, по меньшей мере, на 15 минут для разделения слоев. Водную фазу, то есть нижнюю фазу, удаляют и объединяют с ранее удаленной водной фазой.
Органическую фазу нагревают до 18-25°C. В органическую фазу добавляют озонированную деионизированную воду (5,0 л ±5%), поддерживая температуру между 18°C и 25°C. Перемешивают в течение, по меньшей мере, 15 минут, поддерживая температуру между 18°C и 25°C. Перемешивание останавливают и оставляют, по меньшей мере, на 15 минут для разделения слоев. Водную фазу, то есть нижнюю фазу, удаляют.
В органическую фазу добавляют приготовленный ранее раствор бикарбоната натрия (0,42 кг ±1%) в озонированной деионизированной воде (6,01±5%), поддерживая температуру между 18°C и 25°C. Во время добавления может происходить выделение газа. Перемешивают в течение, по меньшей мере, 15 минут, поддерживая температуру между 18°C и 25°C. Перемешивание останавливают и оставляют, по меньшей мере, на 15 минут для разделения слоев. Водную фазу, то есть нижнюю фазу, удаляют. Измеряют pH удаленной водной фазы. Если pH ниже 8,0, то переходят к стадии 3. Если pH выше 8,0, то переходят к стадии 4.
СТАДИЯ 3
В органическую фазу добавляют предварительно приготовленный раствор бикарбоната натрия (0,42 кг ±1%) в озонированной деионизированной воде (6,0 л ±5%), поддерживая температуру между 18°C и 25°C. Во время добавления может происходить выделение газа. Перемешивают в течение, по меньшей мере, 15 минут, поддерживая температуру между 18°C и 25°C. Перемешивание останавливают и оставляют, по меньшей мере, на 15 минут для разделения фаз. Водную фазу, то есть нижнюю фазу, удаляют. Измеряют pH удаленной водной фазы. Если pH ниже 8,0, то повторяют стадию 3. Если pH выше 8,0, то переходят к стадии 4.
СТАДИЯ 4
В органическую фазу добавляют предварительно приготовленный раствор чистого гидроксида натрия (0,1473 кг ±1%) в озонированной деионизированной воде (3,61 л ±5%) в течение, по меньшей мере, 10 минут, поддерживая температуру между 20°C и 30°C. Смесь перемешивают при температуре между 20°C и 30°C в течение, по меньшей мере, 2 часов до завершения реакции. Реакция считается завершенной, когда площадь пика по данным HPLC метил 2-((5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата в органической фазе составляет меньше, чем 150 mAU* (при 292 нм). Измеряют pH после 2 часов перемешивания. Если pH>12.0, то образец анализируют методом HPLC в отношении площади пика метил 2-((5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата в органической фазе. Если pH реакционной смеси <12.0, то добавляют предварительно приготовленный раствор чистого гидроксида натрия (0,0118 кг ±1%) в озонированной деионизированной воде (0,29 л ±5%), и повторно отбирают образец для измерения pH и/или HPLC после 2 часов перемешивания. Дополнительное добавление раствора гидроксида натрия повторяют до тех пор, пока площадь пика по данным HPLC метил 2-((5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата не составит ≤12.0.
На данном этапе реакционная смесь содержит две фазы; перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения фаз. Водную, то есть нижнюю фазу, направляют в реактор или сборник.
В органическую фазу добавляют предварительно приготовленный раствор чистого гидроксида натрия (0,002 кг ±1%) в озонированной деионизированной воде (2,0 л ±5%), поддерживая температуру между 18°C и 25°C. Перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 18°C и 25°C. Перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения слоев. Нижнюю водную фазу удаляют и объединяют с полученной ранее водной фазой. Верхнюю органическую фазу удаляют для утилизации.
Отбирают образец объединенных водных фаз для определения содержания 2-((5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)уксусной кислоты. Водные фазы концентрируют в вакууме при ≤50°C, предпочтительно между 35°C и 45°C, до получения конечного объема между 5,0 и 5,6×W л, (где W представляет собой количество 2-((5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)уксусной кислоты («свободной кислоты») в кг, рассчитанное из анализируемого образца. Температура в рубашке реактора не должна превышать 55°C.
СТАДИЯ 5
Охлаждают до 0-5°C в течение, по меньшей мере, 2 часов и перемешивают в течение, по меньшей мере, 2 дополнительных часов, поддерживая температуру между 0°C и 5°C. На данном этапе будет возникать кристаллизация 2-((4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)ацетата натрия. Суспензию нагревают до 17-19°C и перемешивают в течение, по меньшей мере, 1 дополнительного часа, поддерживая температуру между 17°C и 19°C. Соль Na должна оставаться в кристаллизованном состоянии в течение этого периода перемешивания. Охлаждают до 13-15°C в течение, по меньшей мере, 4 часов. Затем охлаждают до 0-5°C в течение, по меньшей мере, 2 часов. Перемешивают в течение, по меньшей мере, 2 часов и предпочтительно не более 4 часов, поддерживая температуру при 0-5°C.
СТАДИЯ 6
Суспензию фильтруют при 0-5°C. Часть маточных растворов можно использовать для ополаскивания продукта со стенок реактора. При необходимости влажный фильтрационный осадок промывают до трех раз озонированной деионизированной водой (0,45×W л ±5%), предварительно охлажденной до 0-5°C. Используют поток азота в сочетании с вакуумом. Непосредственно после переноса материал содержится в фильтре в течение как минимум 30 минут перед фильтрацией.
СТАДИЯ 7
Влажный фильтрационный осадок растворяют в озонированной деионизированной воде (4.0×W л ±5%) при температуре ≤50°C, предпочтительно между 35°C и 45°C, и раствор переносят в реактор или сборник. Температура в рубашке реактора и/или фильтре должна составлять <55°C. Реактор и фильтр промывают озонированной деионизированной водой (0.5×W л ±5%) и объединяют промывочную жидкость с полученным ранее водным раствором. Снова промывают реактор и фильтр озонированной деионизированной водой (0.5×W л ±5%), и объединяют промывочную жидкость с полученным ранее водным раствором.
Отбирают образец раствора натриевой соли для определения содержания 2-((5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-ил)тио)уксусной кислоты («свободной кислоты»; W2). Если W2 на 5% меньше по сравнению с W, то используют W2 вместо W на стадиях 10 и 11.
Добавляют этилацетат (9,9×W кг ±5%; 11,0×W л ±5%). Добавляют 24% водный раствор бромистоводородной кислоты (переменная величина) для достижения pH между 2,0 и 4,0, предпочтительно между 2,5 и 3,5, поддерживая температуру при ≤38°C, предпочтительно между 28°C и 33°C. Предполагаемое требуемое количество составляет примерно 0,80×W кг; 0,66×W л раствора бромистоводородной кислоты. Если pH падает ниже 2,0, то добавляют водный раствор гидроксида натрия.
Перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру при ≤38°C, предпочтительно между 28°C и 33°C. Перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения слоев. Верхнюю органическую фазу направляют в реактор или сборник, и отбирают образец водной фазы для анализа HPLC. Если площадь пика по данным HPLC свободной кислоты составляет >500 mAU* (292 нм), то переходят к стадии 8. Если площадь пика по данным HPLC свободной кислоты составляет <500 mAU* (292 нм), то переходят к стадии 9.
СТАДИЯ 8
В водную фазу добавляют этилацетат (0,9×W2 кг ±5%; 1,0×W2 л ±5%), поддерживая температуру при ≤38°C, предпочтительно между 28°C и 33°C. Смесь перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру при ≤38°C, предпочтительно между 28°C и 33°C. Перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения слоев. Верхнюю органическую фазу удаляют в реактор или сборник, объединяя с полученной ранее органической фазой, и отбирают образец водной фазы для анализа HPLC. Если площадь пика по данным HPLC свободной кислоты составляет >500 mAU* (292 нм), то повторяют стадию 8. Если площадь пика по данным HPLC свободной кислоты составляет >500 mAU* (292 нм), то переходят к стадии 9.
СТАДИЯ 9
В объединенную органическую фазу добавляют озонированную деионизированную воду (2.0×W 1±5%), поддерживая температуру между 30°C и 35°C. Перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 30°C и 35°C. Перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения слоев. Нижнюю водную фазу удаляют для утилизации.
В органическую фазу добавляют озонированную деионизированную воду (2,0×W 1±5%), поддерживая температуру между 30°C и 35°C. Перемешивают в течение, по меньшей мере, 30 минут, поддерживая температуру между 30°C и 35°C. Перемешивание останавливают и оставляют, по меньшей мере, на 30 минут для разделения слоев. Нижнюю водную фазу удаляют для утилизации. Органическую фазу фильтруют (пористость фильтра <1 мкм), и фильтрат переносят в реактор или сборник. Реактор и фильтр промывают этилацетатом (0,45×W кг ±5%; 0.5×W л ±5%) и фильтрат переносят в реактор или сборник.
СТАДИЯ 10
Фильтрованную органическую фазу концентрируют в вакууме при ≤38°C, предпочтительно между 18°C и 25°C (температура рубашки реактора не должна превышать 42°C) до конечного объема 7.3-7.7×W л, при этом может происходить кристаллизация продукта. Нагревают до 35-42°C в течение, по меньшей мере, 3 часов, предпочтительно 3-5 часов. Смесь перемешивают в течение, по меньшей мере, 4 часов, предпочтительно <8 часов, поддерживая температуру между 35°C и 42°C, предпочтительно между 38°C и 40°C. Продукт должен кристаллизоваться в начале этой стадии. Если кристаллизации не наблюдается после 1 часа перемешивания при 35-42°C, в партию можно добавить затравку в виде твердого вещества (переменное количество) или суспендированного в н-гептане (0,5×W L - фиксированное количество), предварительно пропущенном через фильтр (пористость фильтра <1 мкм).
Фильтрованную органическую фазу концентрируют в вакууме при <38°C, предпочтительно при температуре между 18°C и 25°C (температура рубашки <42°C) до конечного объема между 3,5×W л и 3,8×W л. Нагревают до 35-42°C в течение, по меньшей мере, 1 часа, предпочтительно не более 4 часов. В суспензию в течение, по меньшей мере, 1 часа добавляют предварительно фильтрованный (пористость фильтра <1 мкм) н-гептан (0,85×W кг ±5%; 1,25×W л ±5%), поддерживая температуру между 35°C и 42°C, предпочтительно между 38°C и 40°C. Перемешивают в течение, по меньшей мере, 1 часа, поддерживая температуру между 35°C и 42°C, предпочтительно между 38°C и 40°C. Охлаждают до 5-10°C в течение, по меньшей мере, 2 часов. Перемешивают в течение, по меньшей мере, 2 часов при 5-10°C.
СТАДИЯ 11
Суспензию фильтруют. При необходимости используют часть маточных растворов для ополаскивания продукта со стенок реактора перед промывкой н-гептаном. Промывают влажный фильтрационный осадок раствором этилацетата (0,63×W кг ±5%; 0,7×W л ±5%) и н-гептаном (0,48×W кг ±5%; 0,7×W л ±5%), предварительно охлажденным до температуры между -5 и 10°C, предпочтительно 0-5°C, и фильтруют (пористость фильтра <1 мкм). Влажный фильтрационный осадок сушат в вакууме при ≤60°C до тех пор, пока содержание н-нептана по данным GC не составит ≤5000 ppm; содержание этилацетата по данным GC не составит ≤2500 ppm; и содержание воды (по данным анализа по методу Карла Фишера) не составит ≤0.5% масса/масса.
Пример 2С: Синтез Соединения 1 и Соединения 4
Оптимизация стадии бромирования: Бромирование Соединения 2 для получения Соединения 3
Для определения оптимальных условий для стадии бромирования использовали несколько комбинаций бромирующих агентов, растворителей, времени реакции и температуры реакции. Результаты приведены в таблице ниже.
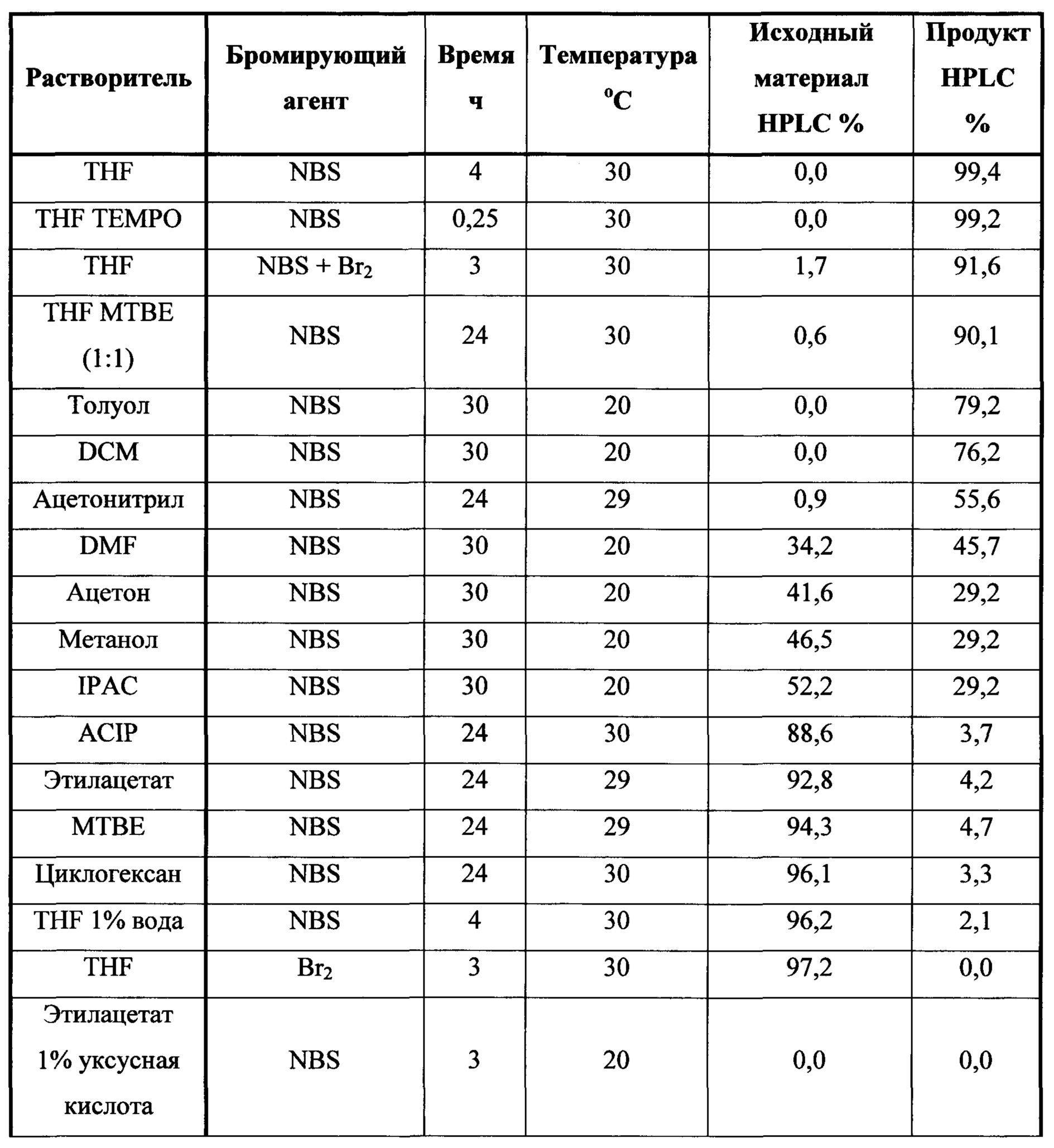
Пример 3: Получение Соединения 5 из Соединения 8 - способ с использованием тиоцианата
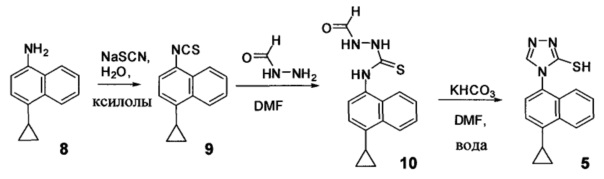
Соединение 8 суспендируют в ксилолах вместе с тиоцианатом натрия и добавляют воду. Смесь нагревают до 90°C до расходования Соединение 8. Смесь затем нагревают до 140°C при отгонке воды до образования изотиоцианатного Соединения 9. Добавляют силикагель и суспензию фильтруют. Твердую фазу промывают ксилолами, и фильтрат экстрагируют дважды водным раствором HCl. Затем раствор концентрируют в вакууме насколько это возможно, и остаток растворяют в DMF. Добавляют раствор формилгидразина в DMF. Смесь перемешивают при 50-55°C и добавляют водный раствор карбоната калия. Затем смесь перемешивают до завершения превращения, охлаждают, и pH доводят до 6-7 путем добавления серной кислоты. Продукт изолируют путем фильтрации, промывают изопропанолом и водой. После сушки продукт Соединение 5 выделяют в виде желтоватого твердого вещества. По желанию. Соединение 5 дополнительно очищают путем растворения в изопропаноле при нагревании с обратным холодильником и обработки углем. Смесь фильтруют и продукт снова кристаллизуют путем концентрирования в вакууме.
Пример 4: Получение Соединения 5 из Соединения 8-А - способ с использованием тиофосгена
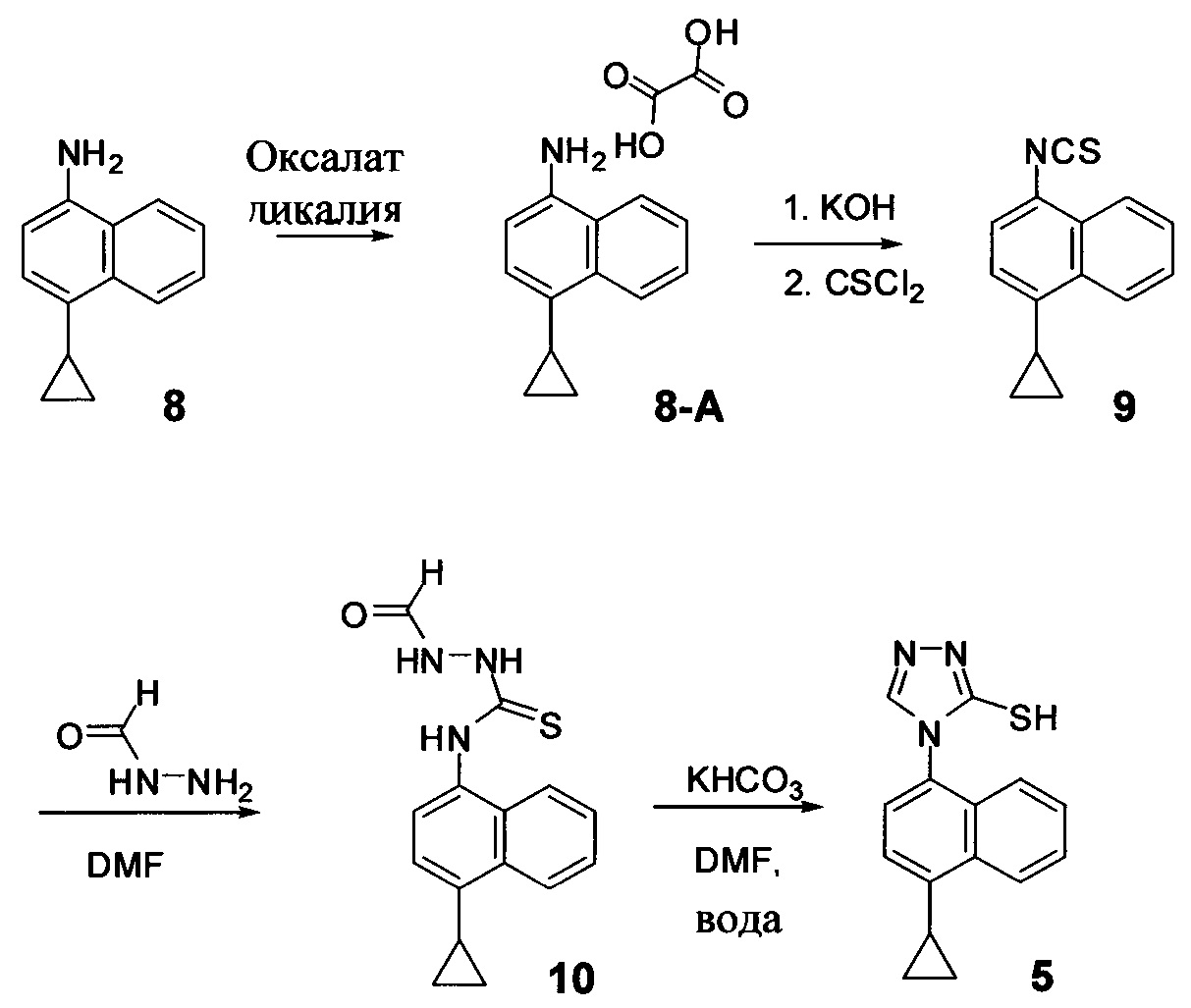
Соединение 8 (44 г), 200 мл толуола и 475 г 20% оксалата дикалия энергично перемешивают, и смесь, содержащую Соединение 8-А, охлаждают до 5°C. Тиофосген (1,5 л) добавляют в течение 1 часа. Смесь фильтруют через целит, и фильтрационный осадок промывают толуолом. Фильтрат собирают, и толуол удаляют в вакууме. Остаток растворяют в смеси 16:1 гексаны:этилацетат, и смесь фильтруют через силикагель. Фильтрат собирают, и растворители удаляют в вакууме с получением Соединения 9 в виде твердого вещества (69 г).
6 г Соединения 9 суспендируют в 12 мл ацетонитрила, и смесь нагревают до 35°С. Добавляют тремя порциями 1,68 г формилгидразида в течение двадцати минут, и смесь перемешивают в течение 2 ч при 35°С, затем охлаждают до 4°С и перемешивают в течение ночи. Реакционную смесь фильтруют, фильтрационный осадок промывают смесью 5:1 гексан:этилацетат и сушат при 40°С с получением 5,85 г Соединения 5.
Пример 5: Синтез Соединения 1 из Соединения 11
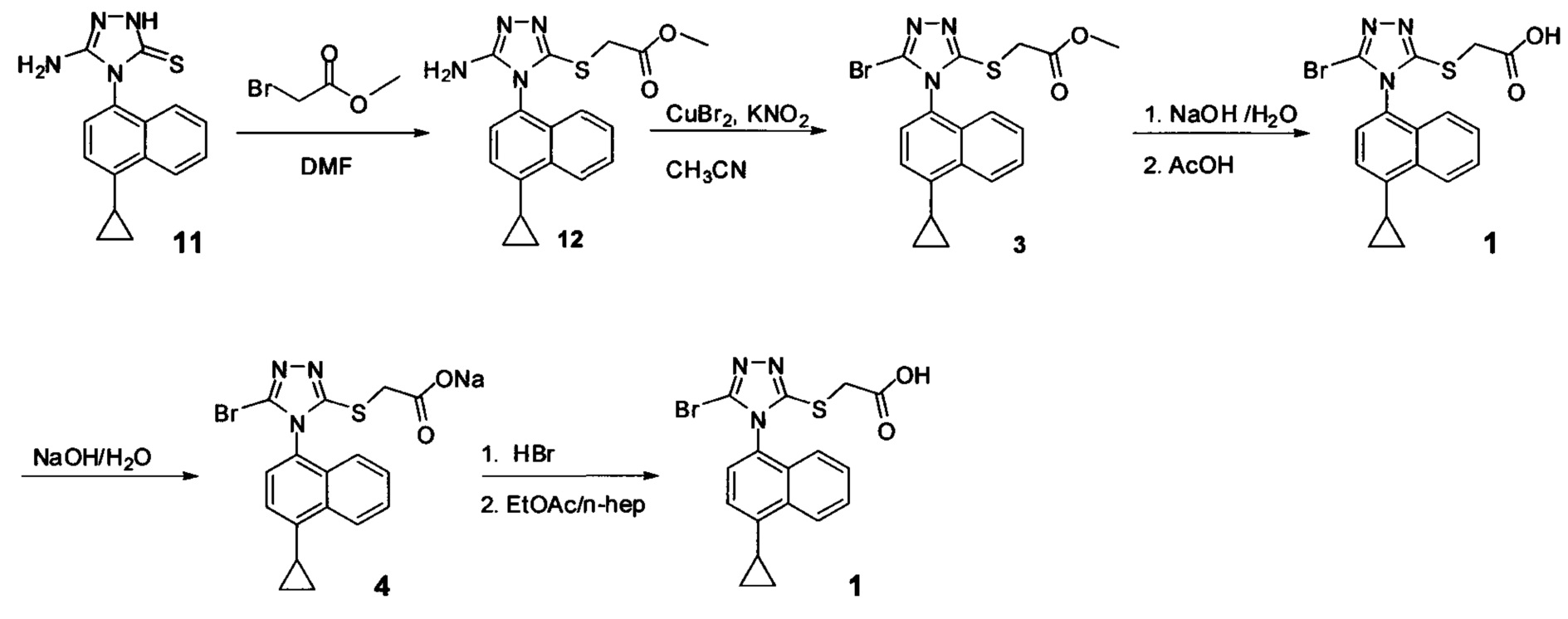
Соединение 11 и метилбромацетат растворяют в DMF и перемешивают при температуре между 14 и 22°С с получением Соединения 12. Продукт выделяют путем охлаждения реакционной смеси до температуры 10-15°С с последующим доведением pH водным раствором бикарбоната натрия. Полученное твердое вещество, Соединение 12, фильтруют и промывают сначала водой, затем охлажденным этилацетатом (0-5°C).
Соединение 12 смешивают с бромидом меди (II) и нитритом калия в ацетонитриле, и перемешивают при температуре между 14 и 20°C до завершения реакции. После добавления в реакционную смесь водного раствора гидроксида натрия и лимонной кислоты продукт. Соединение 3, экстрагируют с использованием толуола, и органический слой промывают несколько раз водными растворами гидроксида аммония и цитрата натрия для удаления солей меди.
Сырое Соединение 3 в растворе подвергают опосредованному основанием гидролизу с добавлением водного раствора гидроксида натрия. Бифазную смесь перемешивают при 18-25°C до завершения гидролиза сложного эфира, затем pH водного слоя доводят до значения между 8 и 9 с использованием водного раствора бромистоводородной кислоты. После разделения двух фаз в водный слой добавляют этилацетат, и pH доводят до значения между 5,15 и 5,35 для экстракции продукта, Соединения 1, в органический слой. Этот способ повторяют несколько раз до экстракции всего Соединения 1. Объединенные органические слои нагревают при 30-35°C, затем циркулируют через слой активированного углерода и затем через фильтр с пористостью меньше, чем 0,5 микрон. Добавляют водный раствор бикарбоната натрия для экстракции Соединения 1 в водный слой, слой концентрируют при пониженном давлении при температуре ниже 40°C, и добавляют уксусную кислоту при поддержании температуры между 40 и 60°C.
Смесь нагревают до температуры между 73 и 77°C, и сырая свободная кислота Соединения 1 кристаллизуется, и в этот момент добавляют воду. Сырую свободную кислоту фильтруют при 7-13°C, промывают холодной смесью уксусной кислоты и воды, и сушат при пониженном давлении при температуре ниже 50°C.
Натриевая соль (Соединение 4) образуется путем добавления эквимолярного водного раствора гидроксида натрия в суспензию сырого Соединения 1 в воде, перемешанную при 18-25°C. Соединение 4, кристаллизованное при охлаждении водной смеси, фильтруют и промывают холодной водой, затем растворяют в теплой воде и фильтруют через фильтр с пористостью не больше, чем 1 микрон. Добавляют этилацетат, бифазную смесь нагревают до 30-35°C, и добавляют водный раствор бромистоводородной кислоты. Соединение 1 экстрагируют в органический слой после добавления этилацетата. Органический слой, содержащий продукт, промывают водой, затем концентрируют при пониженном давлении. Соединение 1 кристаллизуется из раствора. Различное количество н-гептана добавляют для завершения кристаллизации. Кристаллическое Соединение 1 фильтруют и промывают смесью этилацетат и н-гептана, и сушат при пониженном давлении при поддержании температуры сушки ниже 50°C.
Пример 6: Синтез Соединения 11 - способ 1
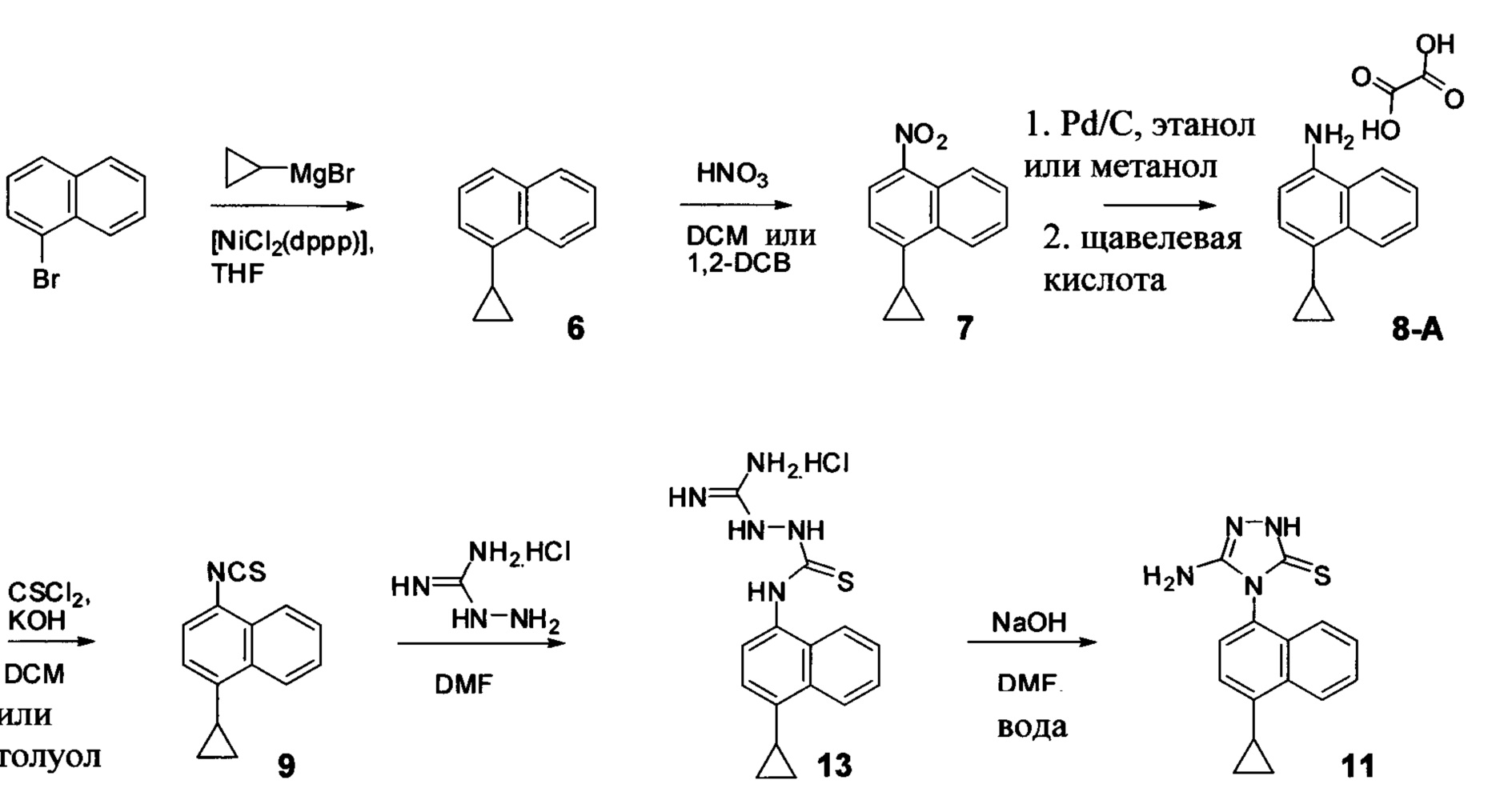
В соответствии с этим способом, циклопропилмагния бромид добавляют в раствор бромнафталина в тетрагидрофуране, перемешанный при 0-5°C в присутствии каталитического количества [1,3-бис(дифенилфосфино)пропан]дихлорникеля(II) с образованием циклопропилнафталина, который разбавляют этилацетатом и промывают.
Циклопропилнафталин (Соединение 6) растворяют в дихлорметане или 1,2-дихлорбензоле, затем добавляют азотную кислоту при 0°C, и реакционную смесь оставляли нагреваться до комнатной температуры. После завершения реакции смесь нейтрализуют бикарбонатом натрия, затем промывают водой, и 1-циклопропил-4-нитронафталин (Соединение 7) используют на следующей стадии без дополнительной очистки.
Соединение 7 растворяют в этаноле или метаноле, и гидролизуют в присутствии палладия на угле. Полученный 1-амино-4-циклопропилнафталин (Соединение 8) кристаллизуют в виде оксалата (Соединения 8-А), затем растворяют в дихлорметане или толуоле, и обрабатывают тиофосгеном в присутствии гидроксида калия для получения соответствующего изотиоцианатного промежуточного соединения (Соединение 9).
В 1-циклопропил-4-изотиоцианатнафталине (Соединение 9) меняют растворитель на DMF и конденсируют с помощью гидрохлорида аминогуанидина с образованием соответствующего замещенного тиосемикарбазида (Соединение 13). Это промежуточное Соединение 13 нагревают в присутствии водного раствора гидроксида натрия с образованием Соединения 11, которое очищают путем кристаллизации из смеси этанола и воды, или смеси метанола, DMF и воды.
Пример 7: Синтез Соединения 11 - способ 2
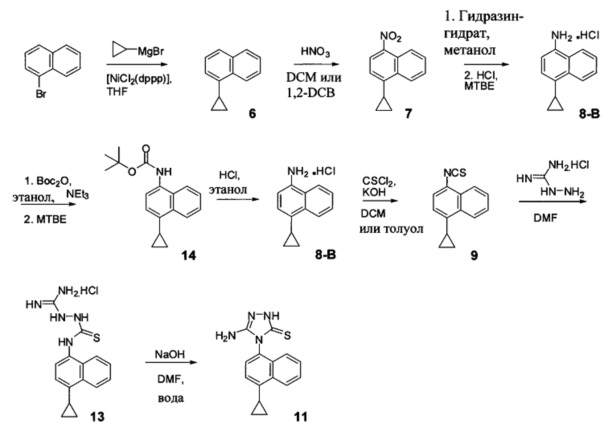
Циклопропилмагния бромид добавляют в раствор бромнафталина в тетрагидрофуране, перемешивают при 0-5°С в присутствии каталитического количества [1,3-бис(дифенилфосфино)пропан]дихлорникеля(II) с образованием циклопропилнафталина, который разбавляют этилацетатом и промывают.
Циклопропилнафталин (Соединение 6) растворяют в дихлорметане, затем добавляют азотную кислоту при 0°C, и реакционную смесь оставляют нагреваться до комнатной температуры. После завершения реакции смесь нейтрализуют бикарбонатом натрия, затем промывают водой, и 1-циклопропил-4-нитронафталин (Соединение 7) используют на следующей стадии без дополнительной очистки.
Соединение 7 растворяют в метаноле и гидролизуют с использованием гидразингидрата при нагревании с обратным холодильником. Сырой 1-амино-4-циклопропилнафталин (Соединение 8) растворяется в этаноле и вступает в реакцию с ди-трет-бутил-дикарбонатом в присутствии триэтиламина с получением трет-бутил-4-циклопропилнафталин-1-илкарбоамата (Соединение 14), которое осаждается из метил-трет-бутилкетона.
Защитную группу в Соединении 14 удаляют соляной кислотой в этаноле с получением амино-4-циклопропилнафталина, который кристаллизуется в виде хлористоводородной соли (соединение 8-В).
4-Циклопропилнафталин-1-амина гидрохлорид (Соединение 8-В) растворяют в дихлорметане и обрабатывают тиофосгеном в присутствии гидроксида натрия с образованием соответствующего изотиоцианатного промежуточного Соединения 9.
У 1-циклопропил-4-изотиоцианатнафталина меняют растворитель на DMF и конденсируют гидрохлоридом аминогуанидина с образованием соответствующего замещенного тиосемикарбазида (Соединение 13). Соединение 13 нагревают в присутствии водного раствора гидроксида натрия с образованием Соединения 11, которое очищают путем кристаллизации из смеси этанола и воды, затем перекристаллизуют из смеси диметилформамида и воды.
Хотя были показаны и описаны предпочтительные варианты осуществления настоящего изобретения, специалисту в данной области будет очевидно, что такие варианты осуществления представлены только в качестве примера. Многочисленные варианты, изменения и замещения будут возникать у специалиста в данной области без отступления от изобретения. Следует понимать, что различные альтернативы вариантам осуществления изобретения, описанным здесь, могут быть использованы при практическом осуществлении изобретения. Подразумевается, что следующая далее формула изобретения определяет объем изобретения, и что тем самым охватываются способы и структуры, находящиеся в пределах объема этой формулы изобретения, и их эквиваленты.
Пример 8: Получение 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты - полиморфной формы 1
2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусную кислоту - форму 1 получают из сырого 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, как описано ниже:
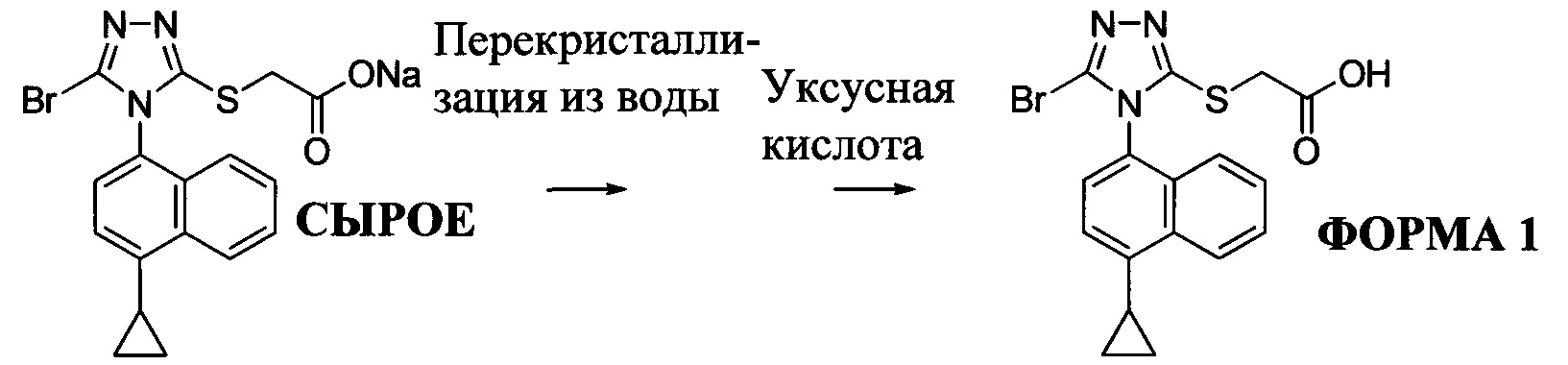
Стадия 1: 2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетат натрия (60 г) и воду (300 мл) перемешивают и недолго нагревают (40-50°C) до растворения всех твердых веществ. Раствор охлаждают и перемешивали на ледяной ванне в течение 1-2 ч, после чего начинает происходить формирование кристаллов (или, если кристаллизация не начинается, в раствор вносят затравку в виде незначительного количества кристаллов 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия). Перемешивание на ледяной ванне продолжают до завершения кристаллизации, и затем твердое вещество изолируют путем фильтрации через воронку со спеченным фильтром (средней пористости) под вакуумом. Фильтрационный осадок промывают ледяной водой (в количестве, достаточном для покрытия фильтрационного осадка), и жидкость полностью пропускают под вакуумом с получением влажного фильтрационного осадка (126,5 г).
Стадия 2: Фильтрационный осадок растворяют в воде (~70 г присутствует в фильтрационном осадке плюс 130 мл; концентрация 200-250 мг/мл) при 60-70°C, и медленно добавляют в уксусную кислоту (200 мл). Раствор уксусная кислота/вода (1:1 объем/объем) охлаждают до комнатной температуры при непрерывном перемешивании и затем дополнительно охлаждают до 0°C, вызывая формирование кристаллов, которые изолируют путем вакуумной фильтрации через воронку со спеченным фильтром средней пористости. Твердые вещества промывают смесью ледяная кислота/вода (1:1 объем/объем) и сушат в вакуумной печи с получением 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты (39,5 г, 78%).
Пример 9: Получение 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты - полиморфной формы 2
2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусную кислоту-форму 2 получают из 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, как описано ниже:
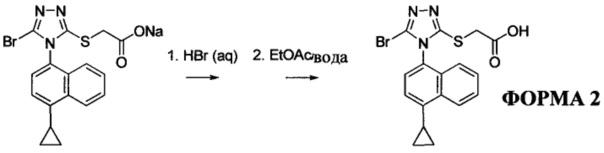
Суспензию 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия (50,0 г сырого образца 97,6% а/а; KF=12,6%; 43,3 г рассчитанная фактическая) и деионизированную воду (217 мл) нагревают (30-35°C) при энергичном перемешивании в течение 10-15 мин, во время которого суспензия растворяется, оставляя только следовые количества твердых веществ. Смесь фильтруют через фильтровальную воронку со средней фриттой, и прозрачный фильтрат охлаждают до 10°C. Приблизительно половину смеси водного раствора бромистого водорода (48 масс. %, 18 г, 106,8 ммоль, 1,05 экв) и деионизированной воды (~13 мл) добавляют в фильтрат в течение 10 мин при 10-15°C, во время которого происходит образование некоторых твердых веществ. Этилацетат (347 мл) добавляют при энергичном перемешивании, что приводит к растворению всех твердых веществ. Оставшийся раствор бромистого водорода добавляют в течение 10 мин при 10°C, и перемешивание продолжают в течение 5-10 мин, во время которого происходит образование мутной суспензии. Перемешивание останавливают, оставляют для разделения фаз и водный слой удаляют. Органический слой промывают деионизированной водой (110 мл) при энергичном перемешивании в течение 5-10 мин, и после разделения фаз водный слой удаляют. Органический слой нагревают до 45-50°C, и растворители удаляют с использованием слабого вакуума, что приводит к образованию суспензии (конечный объем ~200 мл), которую нагревают (45-50°С) при умеренном перемешивании в течение 1 ч, постепенно (3-4 ч) охлаждают до 20-25°C и выдерживают при 20-25°C еще в течение 12 ч, и в заключение охлаждают до 5-10°C и выдерживают в течение 20-30 мин. Суспензию затем фильтруют в вакууме через фильтровальную воронку Бюхнера с использованием фильтровальной бумаги Whatman No. 3. Поскольку присутствуют твердые вещества быстрой фильтрации, маточный раствор циклически пропускают через сосуд для извлечения остаточных твердых веществ, которые собирают вместе с начальной партией. Твердые вещества промывают холодным (5°C) этилацетатом (26 мл) и оставляют для сушки на воронке в течение, по меньшей мере, 10 мин, затем смачивают в н-гептане (30 мл) в течение, по меньшей мере, 10 мин, и повторно применяют вакуум в течение ~6 ч. Твердые вещества переносят в посуду для сушки и сушат в вакуумной печи (25 мм.рт.ст.) в течение, по меньшей мере, 16 ч при 35-40°C при продувании азотом. 2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусную кислоту-форму 2 получали в виде свободнотекучего твердого вещества грязно-белого цвета (28,39 г, 69%), содержащего следовые количества воды (0,16 масс. %) и этилацетата (700 ppm).

Пример 10: Превращение 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты - полиморфной формы 1 в полиморфную форму 2
Способ 1
Этилацетат (200 мл) добавляют в раствор 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты - полиморфной формы 1 (30 г) в ацетоне (200 мл) при 60°C. Часть растворителя (~200 мл) удаляют в низком вакууме и добавляют свежий этилацетат (200 мл), после чего следует еще один цикл дистилляции, во время которого начинается кристаллизация. Температуру водяной ванны медленно повышают до 70°C, и в течение этого периода проводят четыре дополнительных цикла добавления этилацетата/дистилляции до получения конечного объема ~200 мл. Смесь оставляют медленно охлаждаться до комнатной температуры и затем помещают в морозильную камеру на ночь. Твердые вещества изолируют путем фильтрации, промывают ледяным этилацетатом и сушат в вакуумной печи для получения 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты - формы 2.
Способ 2
Раствор 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусной кислоты - форма 1 в одном из растворителей, указанных ниже, медленно выпаривают при комнатной температуре для кристаллизации, замораживают, твердые кристаллы изолируют и промывают растворителем для получения твердой полиморфной формы 2, содержащей следовые количества растворителя и воды, как указано.
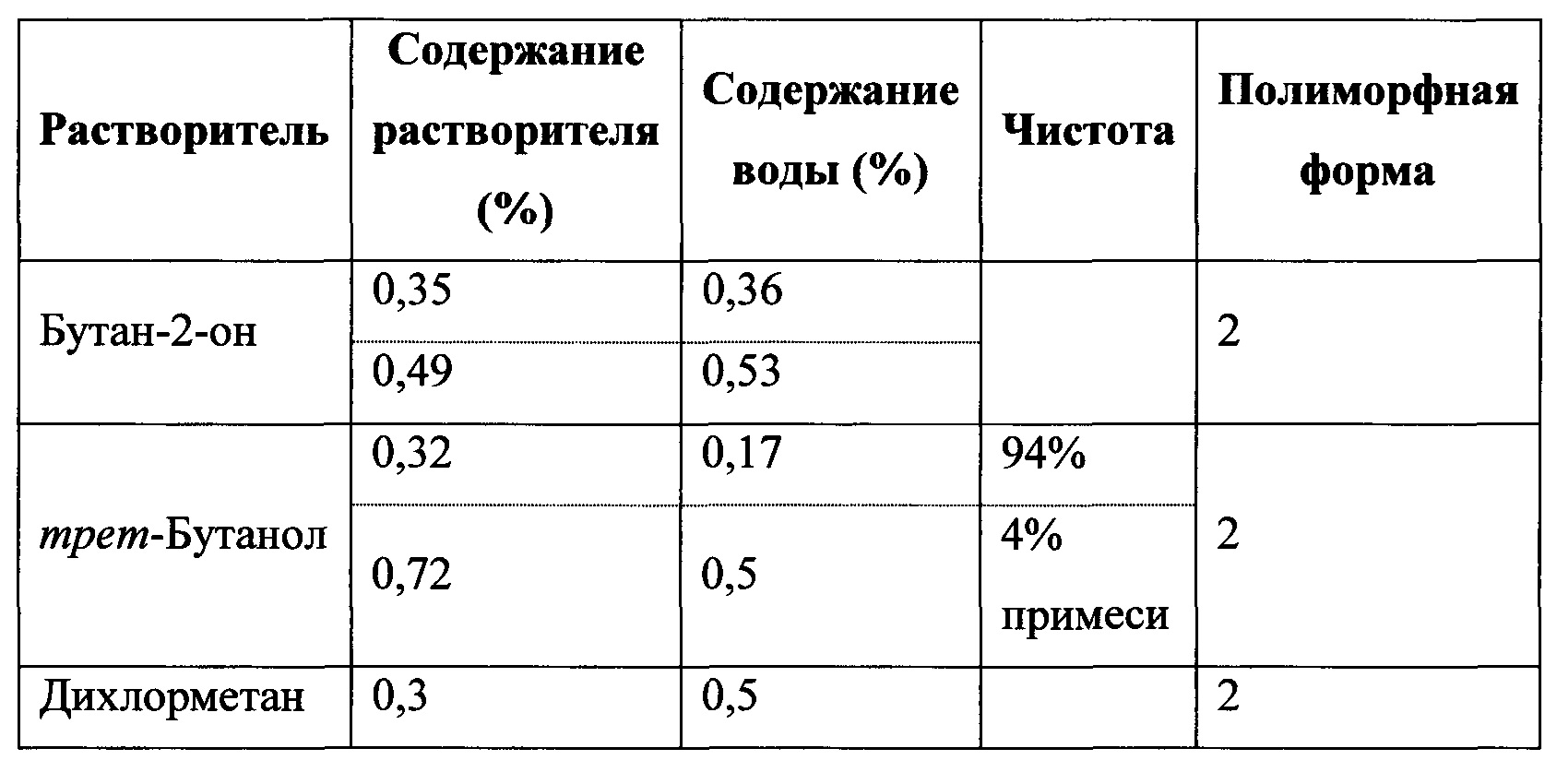
Способ 3
Твердую полиморфную форму 1 выдерживают в равновесном состоянии с ее насыщенным раствором ацетонитрила, этилацетата или толуола при 60°C в течение 6 дней для получения твердой полиморфной формы 2.
Выдерживание твердой полиморфной формы 1 в равновесном состоянии с ее насыщенным раствором ацетона при 60°C в течение 6 дней приводит к разложению.
Способ 4
Твердую полиморфную форму 1 и растворитель (20 мкл) нагревают при 60°C в течение 13 дней для получения твердой полиморфной формы 2.
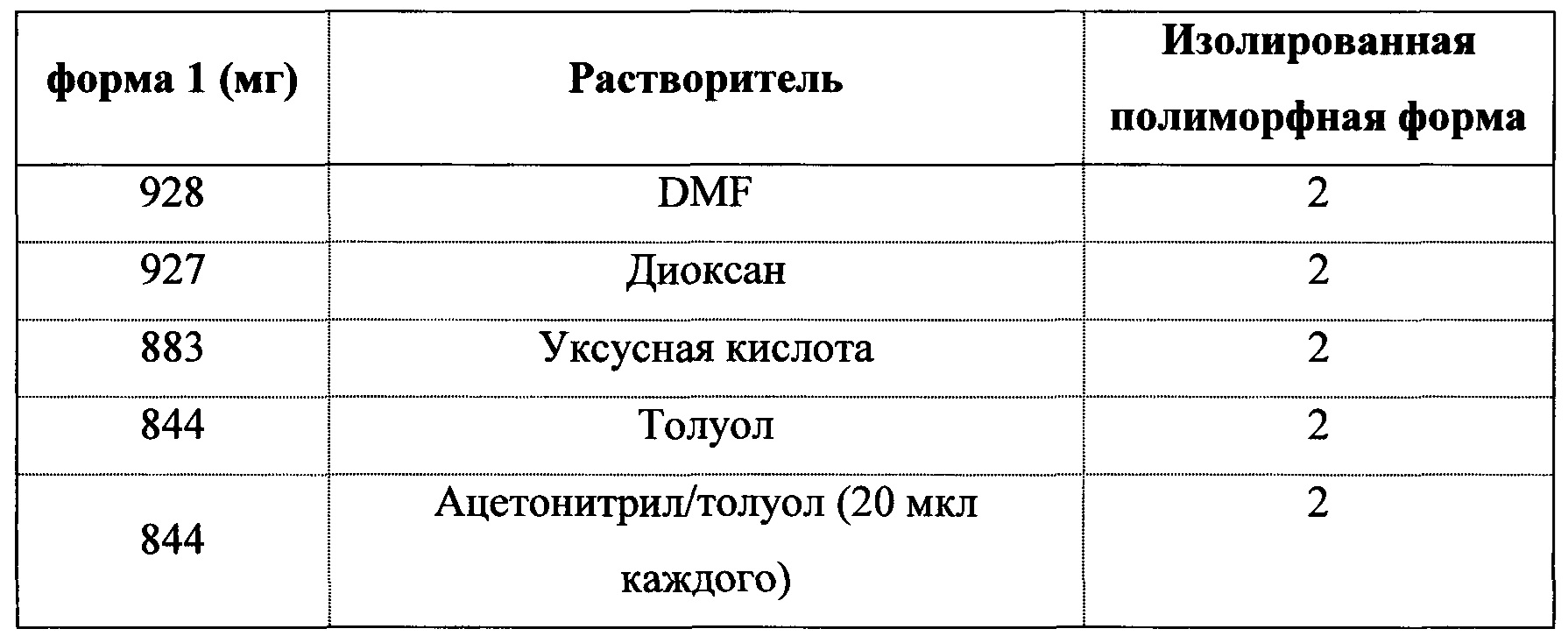

Пример 11А: Анализ кристаллической полиморфной формы 1
Порошковая рентгеновская дифрактография
Порошковая рентгеновская дифрактограмма полиморфной формы 1 показана на фигуре 1 (исходные данные) и 2 (с удалением фонового сигнала и отсечением Kα2); наблюдаемые и репрезентативные пики на картине XRPD показаны в таблицах ниже (получены с корректировкой на фоновый сигнал и отсечением Kα2).
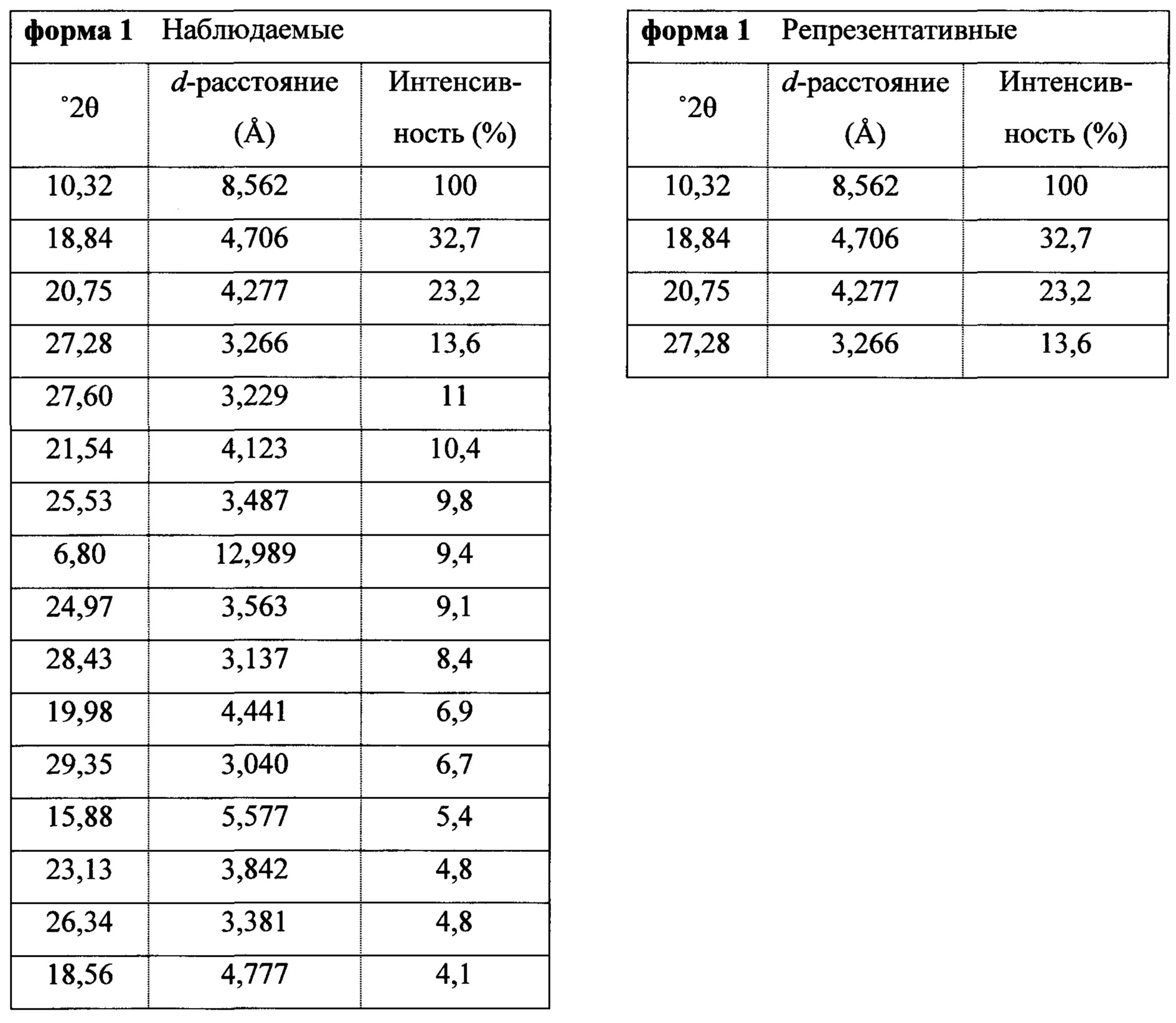
Дифференциальная сканирующая калориметрия (DSC)
Термограмма дифференциальной сканирующей калориметрии для формы 1 показана на фигуре 3; была записана температуре перехода, равная 150,7°C.
Сканирующая электронная микроскопия (SEM)
Анализ SEM показал, что первичные кристаллы формы 1 состоят из агломератов (средний размер ~25 мкм) плоских кристаллов (размер ~5 мкм).
Термогравиметрический анализ (TGA)
Повторные термограммы TGA для формы 1 представлены на фигурах 4(a) и (b), и показывают, что материал не содержит значительных уровней летучих веществ.
Растворимость
Форму 1 (~25 мг) и ацетатный буфер (25 мМ, pH 5, 4 мл), содержащий и не содержащий хлорид натрия (ионная сила доведена до 0,1М), помещали в стеклянную пробирку, которую герметично закрывали и помещали на лабораторный ротатор в инкубатор при 25°C. Через 1, 5 и 7 дней образцы фильтровали и анализировали методом HPLC. Растворимость формы 1 (мг/мл) в различные моменты времени, с хлоридом натрия и без хлорида натрия, показана в таблице ниже:

Пример 11В: Анализ кристаллической полиморфной формы 2
Порошковая рентгеновская дифрактография
Порошковая рентгеновская дифрактограмма полиморфной формы 2 показана на фигурах 5 (исходные данные) и 6 (с удаленным фоновым сигналом и отсечением Kα2); наблюдаемые и репрезентативные пики на картине XRPD приведены в таблице ниже (получены с корректировкой на фоновый сигнал и отсечение Kα2).
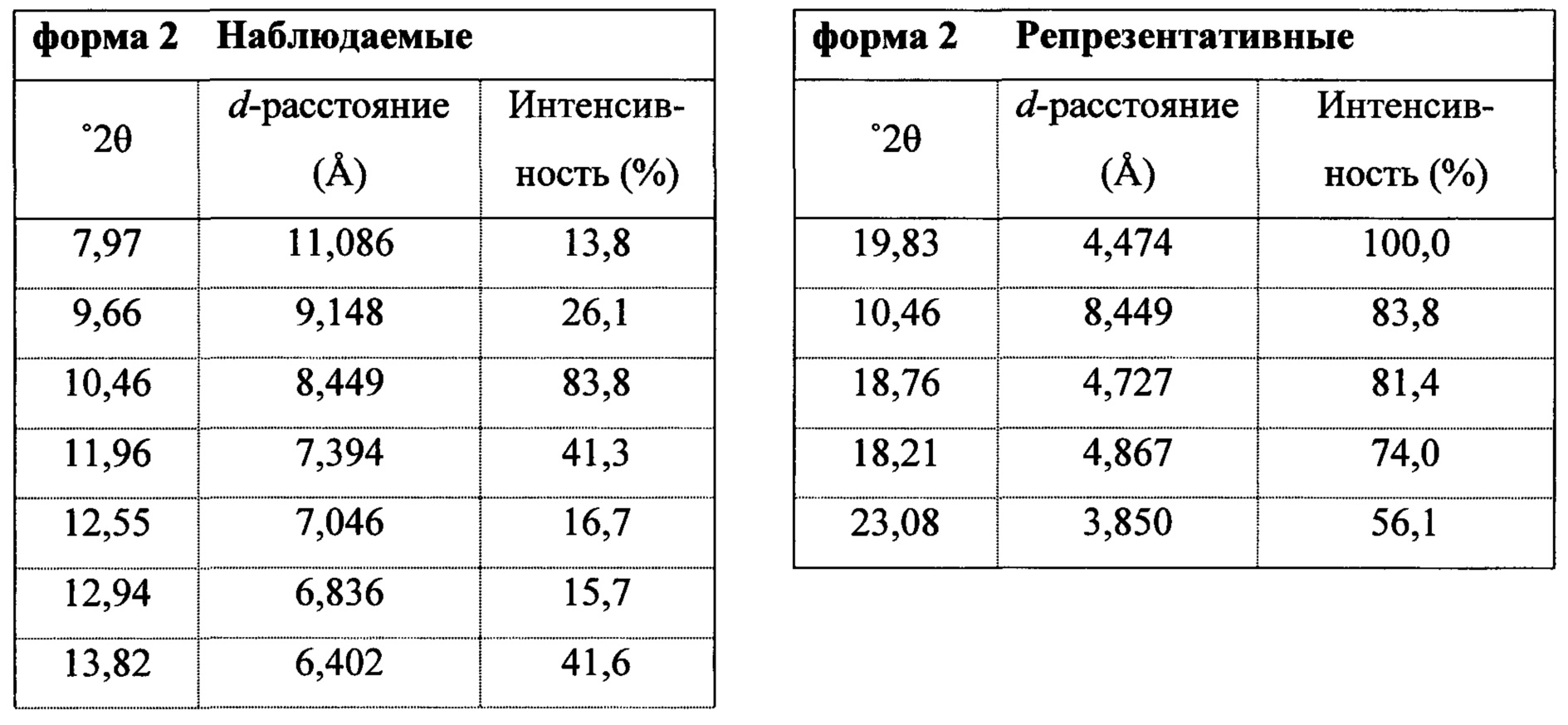
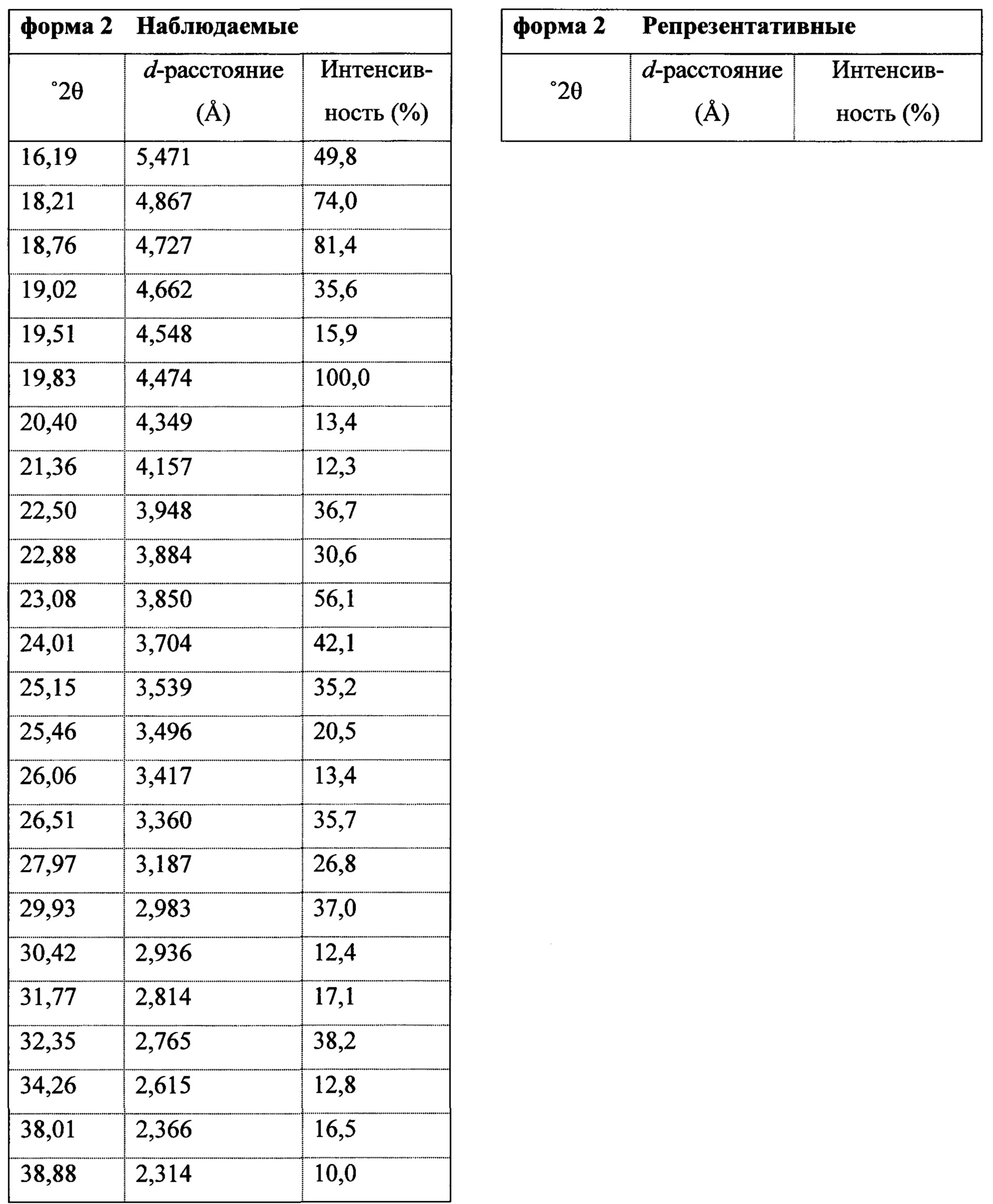
На фигуре 7 показано перекрывание картин XRPD (смещение по оси у) формы 1 (нижняя) и формы 2 (верхняя).
Дифференциальная сканирующая калориметрия (DSC)
Термограмма дифференциальной сканирующей калориметрии для формы 2 показана на фигуре 8, записана температура плавления при 174,7°C.
1Н ЯМР-спектроскопия
1Н ЯМР-спектр, снятый в DMSO-d6, полиморфной формы 2 показан на фигуре 9, и характеристические пики приведены в таблице ниже:
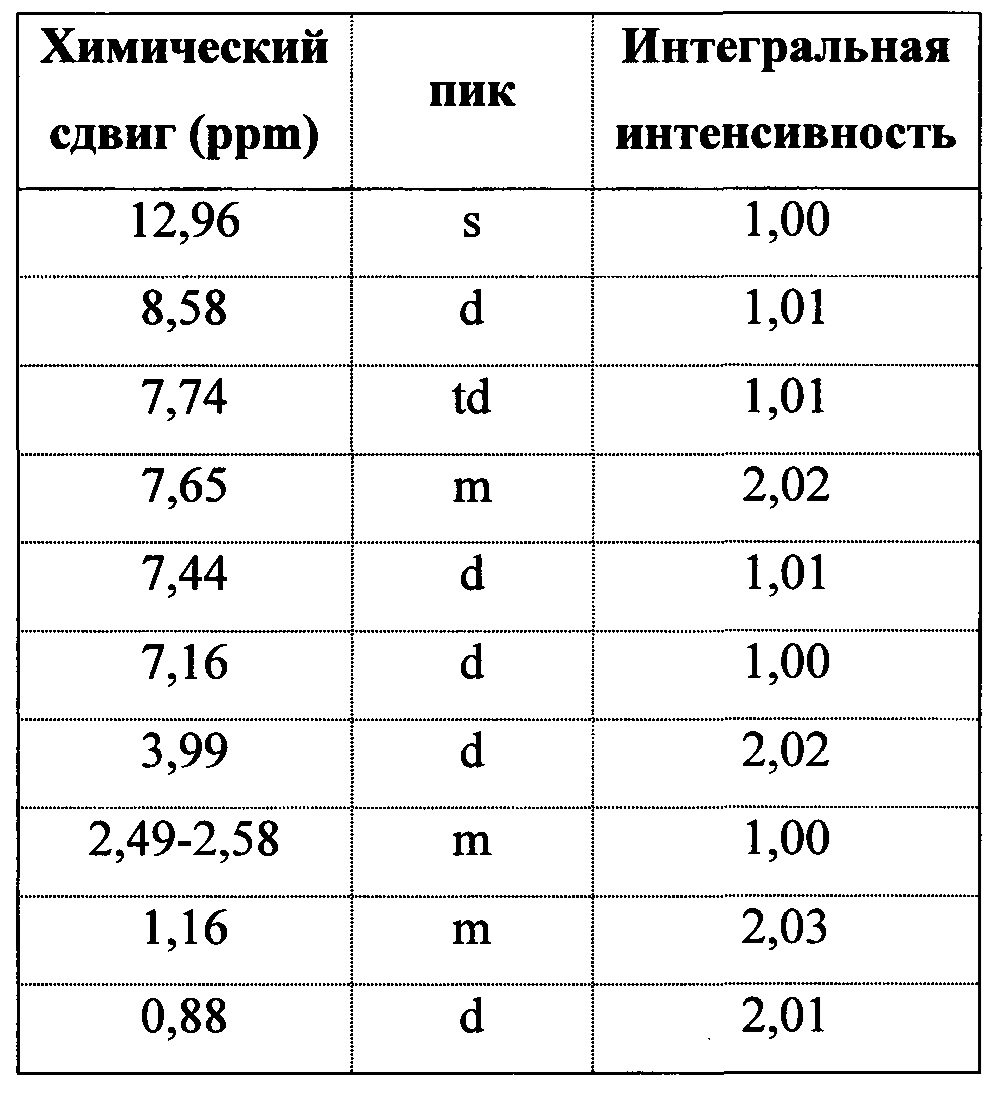
HPLC
Хроматограмма HPLC полиморфной формы 2 показана на фигуре 10. Перечень пиков, присутствующих на хроматограмме, приведен в таблице ниже:
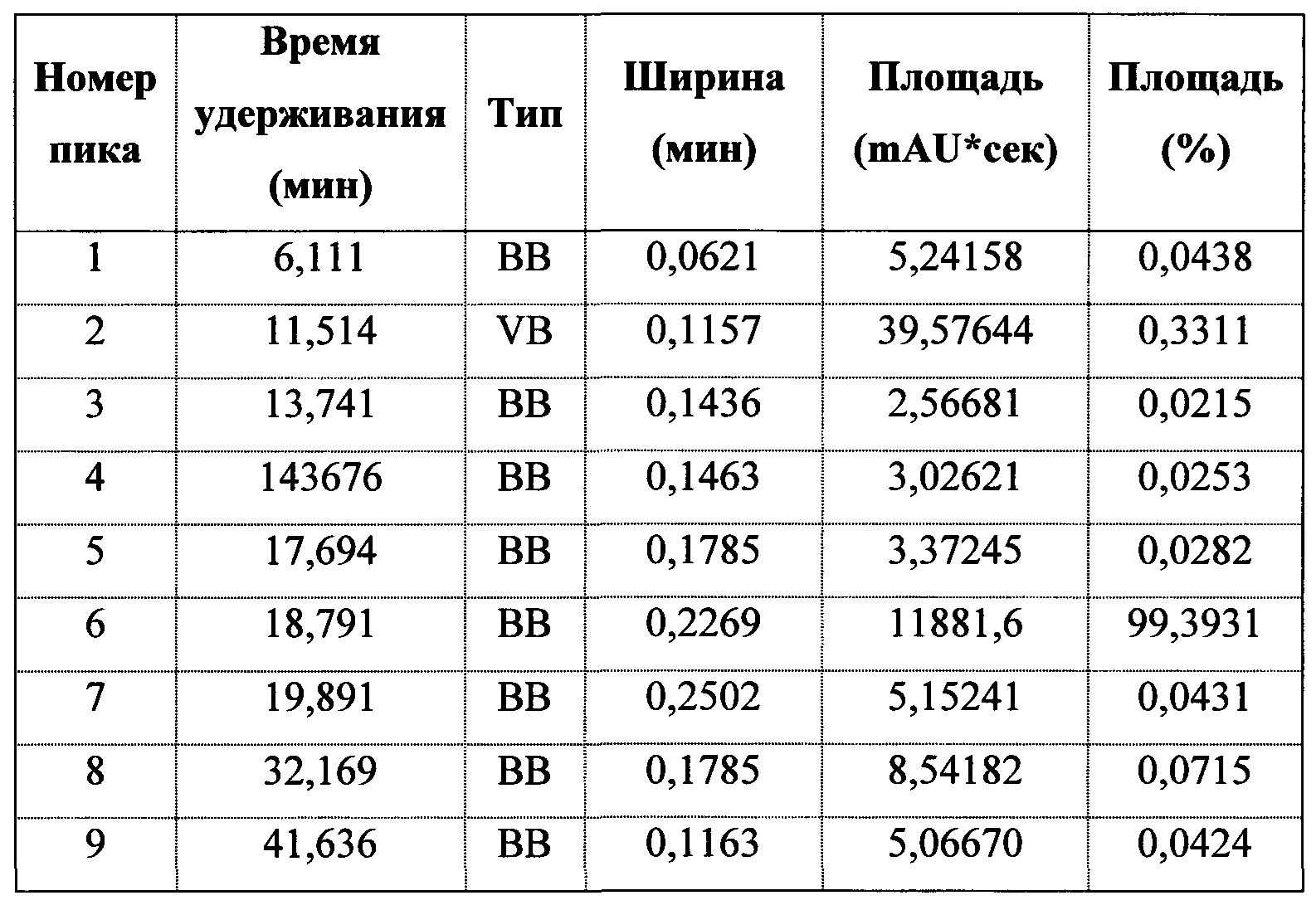
Сканирующая электронная микроскопия (SEM)
Анализ SEM показал первичные кристаллы формы 2, состоящие из агломератов (средний размер ~25 мкм) столбчатых кристаллов (размер ~10 мкм).
Термогравиметрический анализ (TGA)
На фигуре 11 показано перекрывание термограмм TGA для формы 2, которое указывает на то, что материал не содержит значительных уровней летучих веществ.
Растворимость
Форму 2 (~25 мг) и ацетатный буфер (25 мМ, pH 5, 4 мл), содержащий и не содержащий хлорид натрия, (ионная сила доведена до 0,1М), помещают в стеклянную пробирку, которую герметично закрывают и помещают на лабораторный ротатор в инкубатор при 25°C. Через 1, 5 и 7 дней образцы фильтруют и анализируют методом HPLC. Растворимость формы 2 (мг/мл) в различные моменты времени, с хлоридом натрия и без хлорида натрия, приведена в таблице ниже:
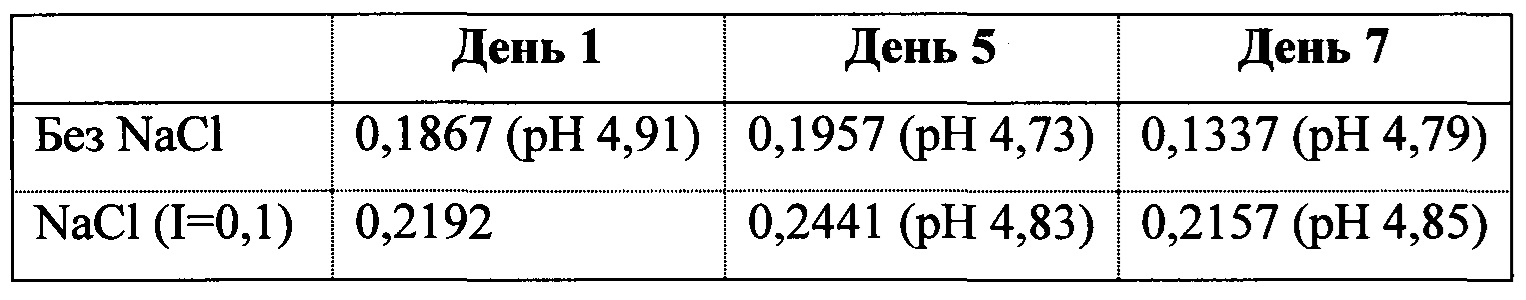
Форму 2 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата тестируют в различных условиях для определения стабильности лекарственного средства. Деградации упакованной Формы 2 не наблюдалось в течение 1 месяца в условиях ускоренной деградации (при 40°C 75% относительной влажности (RH), или при 25°C и 60% относительной влажности (RH)). Упаковка представляет собой двойные пластиковые пакетики из полиэтилена низкой плотности, находящиеся внутри контейнера из полиэтилена высокой плотности (HDPE).
Стабильность кристаллического полиморфа 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата
Было обнаружено, что кристаллические полиморфы 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата проявляют повышенную стабильность по сравнению с аморфными формами в твердом состоянии карбоновой кислоты. Улучшенная стабильность кристаллических полиморфов 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата обеспечивает получение фармацевтических лекарственных форм, проявляющих пониженную изменчивость дозы, присутствующей в заданной лекарственной форме, уменьшение присутствия примесей в препарате готовой лекарственной формы и увеличенный срок хранения изготовленных лекарственных форм по сравнению с фармацевтической лекарственной формой, изготовленной с использованием аморфной твердой формы карбоновой кислоты.
Пример 12А: Порошковая рентгеновская дифрактография (XRPD)
Порошковые рентгеновские дифрактограммы (XRPD) записывают на дифрактометре Bruker D8 Advance, в конфигурации theta/theta Брэгга-Брентано. Падающий пучок рентгеновских лучей получают с использованием CuKα (λ=1,5418Ả) анода (напряжение на трубке=40 кВ, ток=40 мА), изготовленного параллельно с 1,0 мм первичной щелью Соллера со стороны источника и 1,0 мм вторичной щелью Соллера со стороны детектора. Излучение CuKβ удаляют с помощью щели 1,0 мм графитового монохроматора со стороны детектора. Сцинтилляционный детектор (NaI) используют с щелью 0,1 мм. Используют непрерывный режим сканирования с размером шага 0.02°2θ и 5с на шаг в интервале 2-50°2θ. Приблизительно 25 мг материала аккуратно впрессовывают на кремниевую (Si) пластинку с нулевым фоном для обеспечения плоской поверхности материала. Данные регистрируют с использованием программного обеспечения Bruker Diffracplus XRD Commander, версия 2.3. Перечни пиков получают с использованием программного обеспечения Bruker Diffracplus EVA, версия 9.0 с удалением фонового сигнала и отсечением Kα2. Функционирование прибора проверяют по сертифицированному алюминиевому стандарту NIST SRM1976. Параметры прибора для проведения XRPD (Bruker D8 Advance) представлены в таблице ниже:
Пример 12В: Дифференциальная сканирующая калориметрия (DSC)
Дифференциальную сканирующую калориметрию выполняют на дифференциальном сканирующем калориметре ТА Instruments Q2000. Калибровку по температуре выполняют с использованием сертифицированного индиевого стандарта NIST. Дублирующие образцы готовят путем герметичного закрытия приблизительно 2-5 мг (точно записанного) материала в негерметичной чашке ТА Tzero. Негерметичную Tzero чашку/крышку взвешивают и используют на контрольной стороне ячейки. Образцы нагревают со скоростью 10°C/мин от 25°C до 200°C при продувке азота со скоростью 50 мл/мин. Температуру плавления (Tm) и теплоту плавления (ΔHm) измеряют с использованием программного обеспечения ТА Universal Analysis software v4.4.
Пример 12С: Сканирующая электронная микроскопия (SEM)
SEM-изображения получают на сканирующем электронном микроскопе (SEM) JSM-6100 фирмы JEOL. Образец распределяют на столике SEM для установки образца, содержащем двустороннюю угольную ленту, и покрывают золотом методом напыления в течение 60 сек с использованием установки Denton Desk II. Сканирующий электронный микроскоп (SEM) функционирует при ускоряющем напряжении 15 кВ. Изображения получают с использованием программного обеспечения DIPS v2.5 (Digital Imaging Processing System) с медленным сканированием, с установлением разрешения 800×640 пикселей и интегратора при 50 мкс без усреднения. Изображения получают при увеличении в диапазоне от 50Х до 5000Х.
Пример 12D: Термогравиметрический анализ TGA
Термогравиметрический анализ (TGA) выполняют при помощи ТА Instrument Q5000. Калибровку по массе проверяют с использованием сертифицированной массы 50 мг. Дублирующие образцы готовят путем взвешивания и помещения ~5-10 мг материала в Pt ТА тигель. Образцы нагревают со скоростью 10°C/мин до 200°C с продувкой азота со скоростью 25 мл/мин. Потери массы измеряют с использованием программного обеспечения ТА Universal Analysis software v4.4.
Пример 13А: Получение кристаллической полиморфной формы А 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия
Деионизированную воду (0,5 мл) добавляют в перемешанную суспензию аморфного 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия (1,00 г, содержащего 1,8 масс. % воды) и этилацетата (4 мл) с образованием бифазной смеси, которую перемешивают при комнатной температуре в течение 18 часов. Полученную суспензию фильтруют в вакууме, и твердую фазу промывают этилацетатом (2×10 мл). Фильтрационный осадок сушат в вакууме при 18-20°C с продувкой азота в течение 4,5 часов с получением 0,78 г 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, содержащего 13,0 масс. % воды (70,3% извлечение, безводная основа). Изолированное твердое вещество обозначают как Форма А.
Пример 13В: Анализ кристаллической полиморфной формы А 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия Порошковая рентгеновская дифрактография
Анализируют одну картину XRPD с использованием дифрактометра Panalytical и одну картину XRPD с использованием дифрактометра Inel. Воспроизводимость и относительные интенсивности пиков хорошо согласуются между порошковыми рентгеновскими дифрактограммами, что указывает на хорошие статистические показатели частиц и ориентации. Картина XRPD для Формы А показана на Фигуре 13; наблюдаемые и репрезентативные пики на картине XRPD приведены в таблицах ниже:
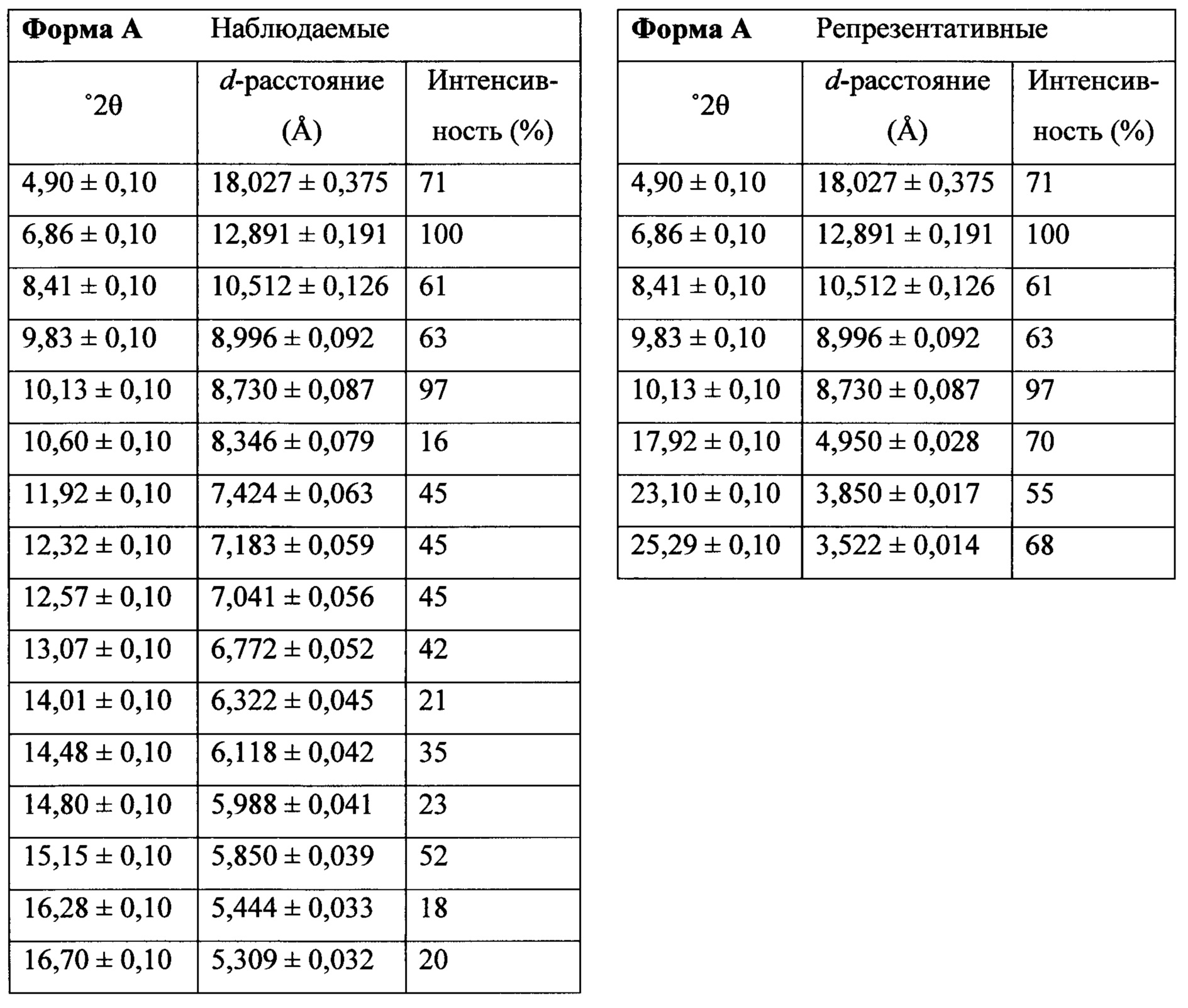
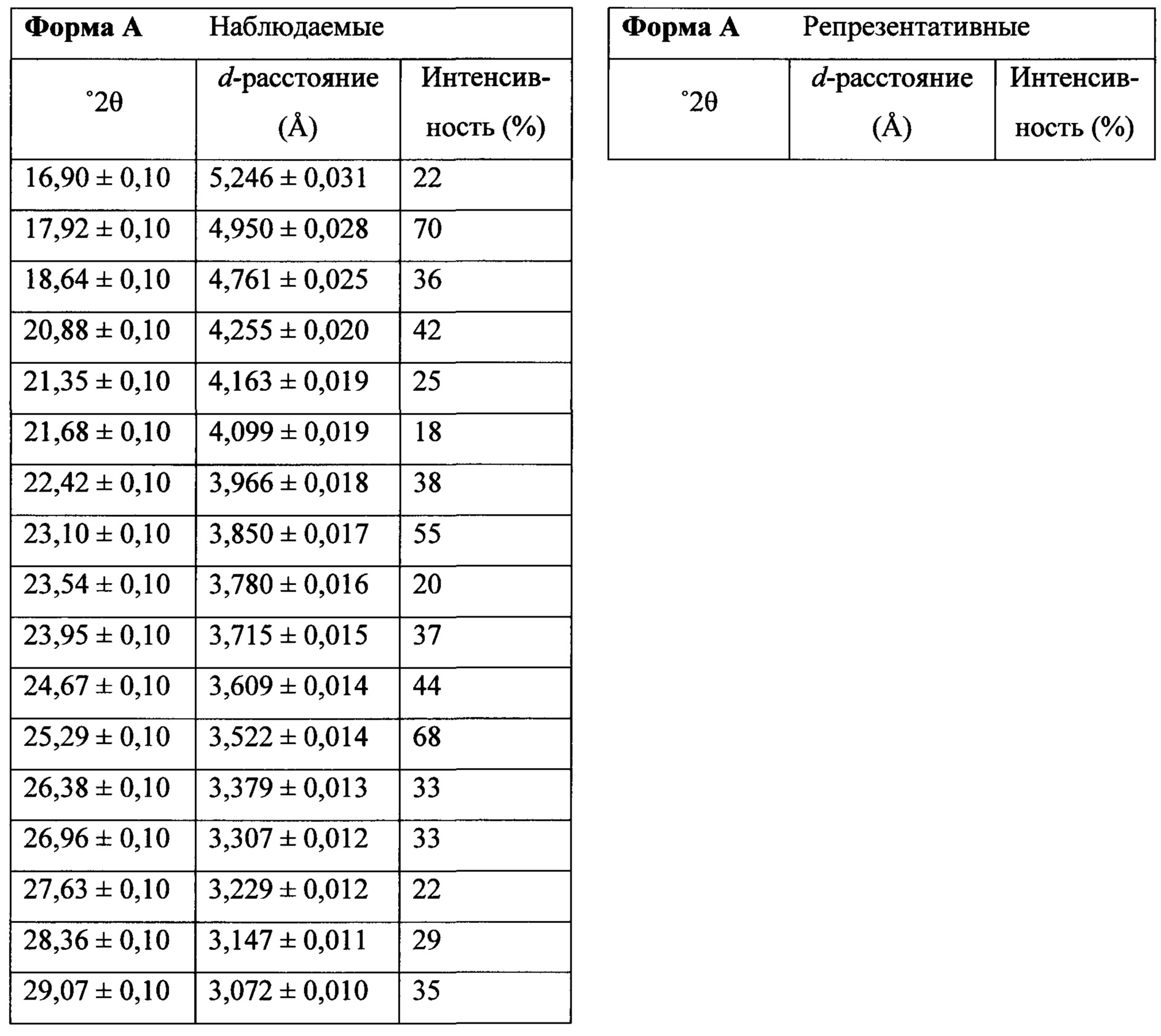
Термограмма дифференциальной сканирующей калориметрии для Формы А показана на Фигуре 14.
Инфракрасный спектр поглощения Формы А показан на Фигуре 15.
Рамановский спектр Формы А показан на Фигуре 16.
Форму А 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия тестируют в различных условиях для определения термодинамической стабильности. Деградации упакованной Формы А не наблюдается в течение 6 месяцев в условиях ускоренной деградации (при 40°С и 75% относительной влажности (RH)). Более того, деградации упакованной Формы А не наблюдается в течение 12 месяцев в условиях долгосрочного испытания (при 25°С и 60% относительной влажности (RH)). Упаковка представляет собой двойные пластиковые пакетики из полиэтилена низкой плотности, находящиеся внутри безводного мешочка из фольги с запаянными швами в контейнере из полиэтилена высокой плотности (HDPE). Результаты стабильности Формы А показали улучшение по сравнению с аморфной свободной кислотой в твердом состоянии.
Пример 14А: Приготовление кристаллической полиморфной Формы В 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия
Приготовление i: Смесь 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия (3,02 г, Форма А) и насыщенного водой этилацетата (6 мл) перемешивают при 45-50°C в течение 16 часов с получением бифазной смеси, которую постепенно охлаждают до комнатной температуры в течение 2 часов и перемешивают дополнительно в течение 21 часа с получением однородной суспензии. Суспензию фильтруют в вакууме, промывают этилацетатом, и фильтрационный осадок сушат в вакууме при 18-20°C при продувке азотом в течение 2 часов с получением 2,77 г 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия, содержащего 12,9 масс. % воды (91,7% извлечение, безводная основа).
Приготовление ii: 2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетат натрия (Форма А) перемешивают при ~50°C в насыщенном водой этилацетате (0,5 мл) в течение ночи, превращая твердые вещества в масло. Масло соскребают с помощью стоматологической зубочистки и оставляют перемешиваться при комнатной температуре. Примерно через 3 дня оптическая микроскопия показала образование кристаллических твердых веществ. Жидкость удаляют путем декантации и твердые вещества изолируют.
Изолированные твердые вещества обозначают как Форма В.
Пример 14В: Анализ кристаллической полиморфной Формы В 2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия
Порошковая рентгеновская дифрактография
Анализируют одну картину XRPD с использованием дифрактометра Panalytical и одну картину XRPD с использованием дифрактометра Inel. Воспроизводимость и относительные интенсивности пиков хорошо согласуются между порошковыми рентгеновскими дифрактограммами, что указывает на хорошие статистические показатели частиц и ориентации. Картина XRPD для Формы В показана на фигуре 17; наблюдаемые и репрезентативные пики на картине XRPD показаны в таблицах ниже:
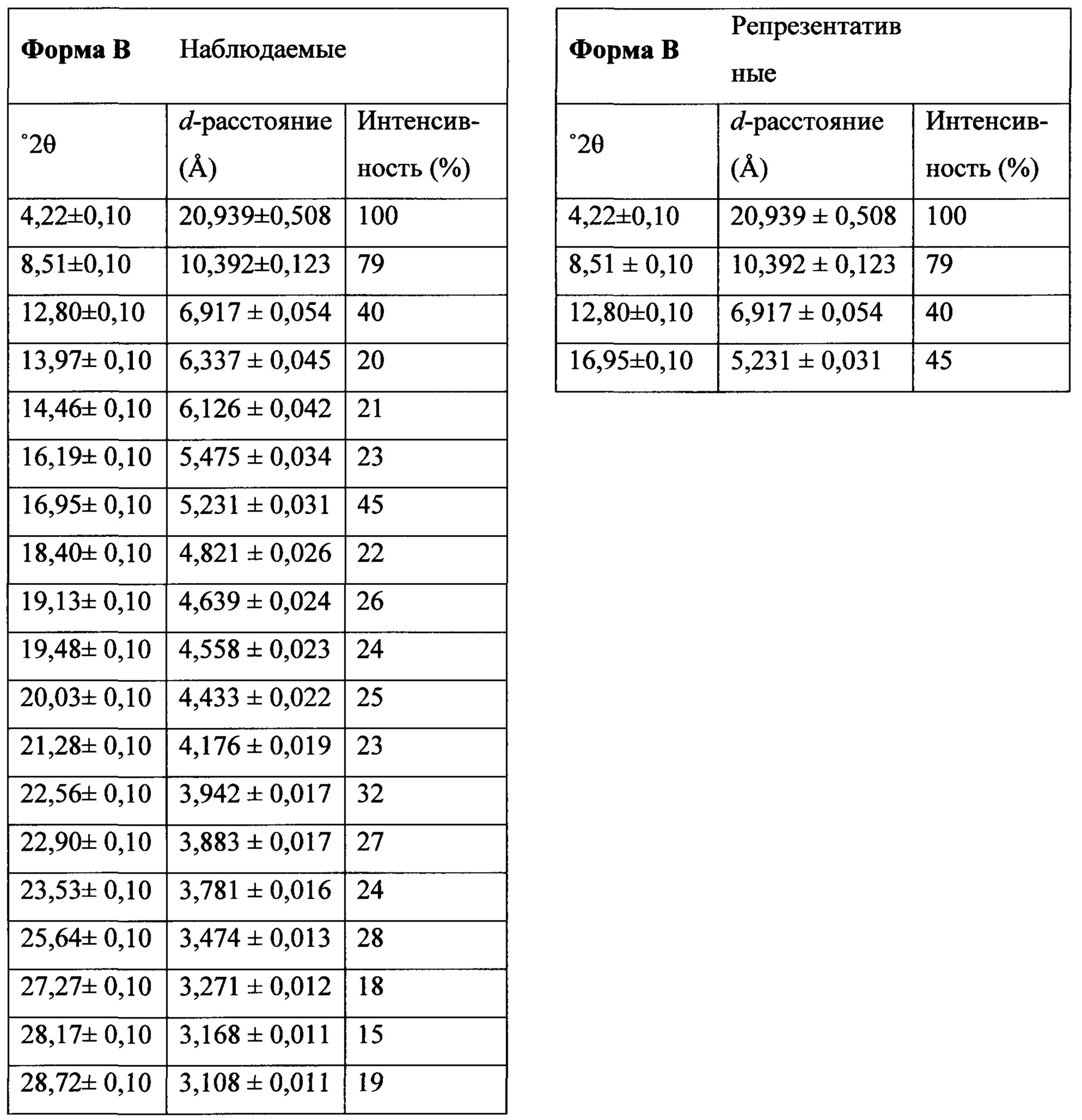
Термограмма дифференциальной сканирующей калориметрии для Формы А показана на Фигуре 18.
Пример 15А: Приготовление кристаллической полиморфной формы В' 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия
2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетат натрия (Форма В, содержащая 12,9 масс. % воды) сушат в вакууме при комнатной температуре в течение 1-3 дней с получением твердого вещества грязно-белого цвета, обозначенного как форма В'.
Пример 15В: Анализ кристаллической полиморфной Формы В' 2-(5-Бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия
Порошковая рентгеновская дифрактограмма для Формы В', показанная на фигуре 19, имеет сходство с порошковой рентгеновской дифрактограммой для Формы В, однако неоднородные сдвиги пиков между картинами указывают на различное сольватационное состояние одного и того же полиморфа. Термограмма дифференциальной сканирующей калориметрии Формы В' показана на фигуре 20.
Стабильность кристаллического полиморфа 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия
Было обнаружено, что кристаллические полиморфы 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия проявляют повышенную стабильность по сравнению с аморфной формой карбоновой кислоты в твердом состоянии. Улучшенная стабильность кристаллических полиморфов 2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)ацетата натрия обеспечивает получение фармацевтических лекарственных форм, проявляющих пониженную изменчивость дозы, присутствующей в заданной лекарственной форме, уменьшение присутствия примесей в препарате готовой лекарственной формы и увеличенный срок хранения изготовленных лекарственных форм по сравнению с фармацевтической лекарственной формой, изготовленной с использованием аморфной твердой формы карбоновой кислоты.
Пример 16: Примеси, идентифицированные в образцах Соединения 1, полученного с использованием описанных здесь способов
Следующие соединения (Соединения 1-Х) были идентифицированы как примеси в образцах Соединения 1, которое было получено с использованием описанных здесь способов, включая Примеры 1 и 2:
2-(4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(4-(4-циклопропилнафталин-1-ил)-5-гидрокси-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-амино-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-бром-4-(1-циклопропилнафталин-2-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-бром-4-(4-метилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-бром-4-(4-пропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-бром-4-(5-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-бром-4-(нафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота;
2-(5-хлор-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-илтио)уксусная кислота; и
4-(5-(карбоксиметилтио)-4-(4-циклопропилнафталин-1-ил)-4Н-1,2,4-триазол-3-иламино)-4-оксобутановая кислота.