Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ТИОФЕНА, ИСПОЛЬЗУЕМЫЕ ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА
Вид РИД
Изобретение
Настоящее изобретение относится к производным тиофена, используемым для лечения патологий, связанных с метаболическим синдромом, в частности, для лечения или предотвращения сахарного диабета.
Сахарный диабет представляет собой весьма гетерогенную группу заболеваний, которые характеризуются рядом общих свойств: гипергликемией, функциональными и количественными отклонениями показателей бета-клеток поджелудочной железы от нормы, резистентностью тканей к инсулину, а также увеличением риска развития в долгосрочной перспективе осложнений, в частности, сердечно-сосудистых осложнений.
Сахарный диабет II типа представляет собой существенную проблему здравоохранения. Распространение данного заболевания резко увеличивается в большинстве промышленно развитых стран, и еще более - в странах с развивающейся экономикой. На сегодняшний день можно говорить об эпидемии данного заболевания, вызывающего серьезные осложнения, которые вполне могут приводить к инвалидности или даже к смертельному исходу, в том числе вследствие почечной недостаточности, инфаркта миокарда или сердечно-сосудистого инсульта.
Недавно установленные количественные показатели сахарного диабета являются следующими (данные ВОЗ (Всемирной организации здравоохранения):
- Во всем мире от сахарного диабета страдают более 220 миллионов человек.
- Сахарный диабет в 3 раза увеличивает риск возникновения инсульта.
- В западном мире сахарный диабет является первой причиной слепоты и почечной недостаточности.
- Согласно оценкам, в 2005 году сахарный диабет стал причиной гибели 1,1 миллиона человек.
- Согласно прогнозам ВОЗ, в период с 2005 по 2030 год количество смертей, вызванных сахарным диабетом, увеличится в два раза. Во Франции уход за пациентами, страдающими от диабета, а также лечение таких пациентов оказывают существенную нагрузку на бюджет государственной системы медицинского страхования. Принимая во внимание тревожные прогнозы количества пациентов в мире, у которых будет выявлен диабет в период с сегодняшнего дня до 2030 года, многие фармацевтические и биотехнологические компании интенсивно производят инвестиции в исследование и разработку в области метаболизма и, более конкретно, в области сахарного диабета II типа с целью выведения на рынок новых альтернативных лекарственных препаратов.
В настоящее время лечение сахарного диабета II типа, способное восстановить нормальное гликемическое равновесие в течение 24 часов и не обладающее побочными действиями, отсутствует. Ни один из существующих вариантов лечения не принимает во внимание всеобъемлющую патологию данного заболевания; кроме того, существующие варианты лечения нацелены исключительно на исправление того или иного недостатка. Средства для лечения сахарного диабета, которые недавно были выведены на рынок, не продемонстрировали какого-либо существенного улучшения гликемического контроля по сравнению с контролем, наблюдавшимся для ранее существующих вариантов лечения, а также оказывали нежелательные побочные действия, что оставляет пространство для новых потенциальных способов лечения.
Вследствие этого существует потребность в новых молекулах, используемых для лечения или предотвращения сахарного диабета или осложнений и/или сопутствующих патологий данного заболевания, предпочтительно сахарного диабета II типа.
Авторы настоящего изобретения неожиданно обнаружили, что определенные производные тиофена обладают активностью ингибирования продукции глюкозы печенью, а также активностью стимулирования секреции инсулина в ответ на поступление глюкозы, и вследствие этого указанные производные тиофена могут использоваться, в частности, в качестве продуктов для фармацевтического применения пациентами, которые нуждаются в таких продуктах, в особенности для предотвращения и/или лечения сахарного диабета и осложнений и/или сопутствующих патологий данного заболевания (ожирение, гипертензия и т.д.), предпочтительно сахарного диабета II типа.
Подробное описание изобретения
Вследствие этого настоящее изобретение относится к производным тиофена следующей общей формулы I:
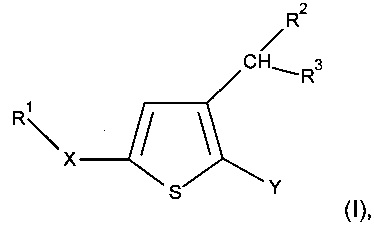 где:
где:
Y представляет собой арильную группу, предпочтительно фенильную (Рh), гетероарильную группу, предпочтительно фурильную, или бензо-1,3-диоксольную группу, причем указанная арильная или гетероарильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -O(С1-С6 алкила), предпочтительно -ОМе, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -OCF3 или -OCHF2, или -О(С1-С6 алкильной) группой, предпочтительно -ОМе; C1-C6 алкила, предпочтительно метила, замещенного одним или несколькими атомами галогена, предпочтительно F, такого как, например, -CF3, или -О(С1-С6 алкильной) группой, предпочтительно -ОМе, такого как, например, -СН2ОМе, или группой -ОН, такого как, например, -СН2ОН; -SО2(C1-C6 алкила), предпочтительно -SО2Me; -CONRaRb, где Ra представляет собой атом водорода или С1-С6 алкильную группу, предпочтительно метил, и Rb представляет собой С1-С6 алкильную группу; или -ОН; примерами необязательно замещенных арильных групп являются Ph, 4-F-Ph, 2,3-(F)2-Ph, 2-F-4-Cl-Ph, 4-Cl-Ph, 3,4-(Cl)2-Ph, 3-NC-Ph, 4-NC-Ph, 3-MeO-Ph, 4-MeO-Ph, 2-MeO-3-F-Ph и 2-OH-3-F-Ph;
X представляет собой группу -SO2 или группу 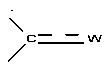 , предпочтительно группу
, предпочтительно группу 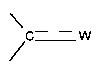 , где
, где  представляет собой связь и W представляет собой атом кислорода или группу -NOR4, где R4 представляет собой атом водорода, С1-С6 алкильную группу или (С1-С6 алкил)арильную группу, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -O(C1-C6 алкила), предпочтительно -ОМе, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, или -O(С1-С6 алкильной) группой, предпочтительно -ОМе; С1-С6 алкила, замещенного одним или несколькими атомами галогена, предпочтительно F, или -О(С1-С6 алкильной) группой, предпочтительно -ОМе или группой -ОН; -SО2(C1-C6 алкила); -CONRa'Rb', где Ra' представляет собой атом водорода или С1-С6 алкильную группу и Rb' представляет собой С1-С6 алкильную группу или -ОН или
представляет собой связь и W представляет собой атом кислорода или группу -NOR4, где R4 представляет собой атом водорода, С1-С6 алкильную группу или (С1-С6 алкил)арильную группу, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -O(C1-C6 алкила), предпочтительно -ОМе, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, или -O(С1-С6 алкильной) группой, предпочтительно -ОМе; С1-С6 алкила, замещенного одним или несколькими атомами галогена, предпочтительно F, или -О(С1-С6 алкильной) группой, предпочтительно -ОМе или группой -ОН; -SО2(C1-C6 алкила); -CONRa'Rb', где Ra' представляет собой атом водорода или С1-С6 алкильную группу и Rb' представляет собой С1-С6 алкильную группу или -ОН или
 отсутствует и W представляет собой -ОН;
отсутствует и W представляет собой -ОН;
R1 представляет собой
- С1-С6 алкильную группу, предпочтительно метильную или этильную, причем указанная алкильная группа необязательно замещена атомом галогена, предпочтительно Cl, такая как, например, -(CH2)2Cl;
- С3-С6 циклоалкильную группу, предпочтительно циклопропильную или циклогексильную;
- (С1-С6 алкил)O(С1-С6 алкильную) группу;
- (С1-С6 алкил)NR(C1-C6 алкильную) группу, где R представляет собой атом водорода или С1-С6 алкильную группу, предпочтительно метильную;
- арильную группу, предпочтительно фенильную (Ph), причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -О(С1-С6 алкила), предпочтительно -ОМе, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -OCF3 или -OCHF2, или -O(С1-С6 алкильной) группой, предпочтительно -ОМе; -SO2(С1-С6 алкила), предпочтительно -SO2Me; -CONRa"Rb", где Ra" представляет собой атом водорода или С1-С6 алкильную группу, предпочтительно метильную, и Rb" представляет собой C1-C6 алкильную группу, предпочтительно метильную; или С1-С6 алкильную группу, предпочтительно метильную, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -CF3, или -O(C1-C6 алкильной) группой, предпочтительно -ОМе, такая как, например, -СН2ОМе, или группой -ОН, такая как, например, -СН2ОН; примерами необязательно замещенной арильной группы являются Ph, 2-F-Ph, 3-F-Ph, 4-F-Ph, 2,3-(F)2-Ph, 2,4-(F)2-Ph, 2,5-(F)2-Ph, 3,5-(F)2-Ph, 3-CI-Ph, 2,4-(CI)2-Ph, 3,4-(CI)2-Ph, 4-NC-Ph, 2-MeO-Ph, 4-MeO-Ph, 3-MeO-Ph, 3-F-4-MeO-Ph и 3-Ме-4-F-Ph;
- (С1-С6 алкил)арильную группу; предпочтительно (C1-C6 алкил)фенильную, в частности, бензильную или (СН2)2фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из F или Cl, в частности, F; -O(С1-С6 алкила), предпочтительно -ОМе; или С1-С6 алкила, предпочтительно метила; примерами необязательно замещенных (С1-С6 алкил)арильных групп являются CH2Ph, CH2-4-F-Ph и (CH2)2Ph;
- -NH-арильную группу, предпочтительно -NH-фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, предпочтительно F или Cl; -О(C1-С6 алкила), предпочтительно -ОМе; или С1-С6 алкила, предпочтительно метила; примерами необязательно замещенных -NH-арильных групп являются NH-4-Br-Ph, NH-3-MeO-Ph и NH-4-MeO-Ph;
-NH(C1-C6 алкил)арильную группу, предпочтительно -ΝΗ(C1-C6 алкил)фенильную, в частности, -NН(СН2)фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена; -O(С1-С6 алкила), предпочтительно -ОМе; или C1-С6 алкила, предпочтительно метила; примерами необязательно замещенных -NН(C1-С6 алкил)арильных групп являются -NHCH2-3-MeO-Ph и NHCH2-4-MeO-Ph;
- гетероарильную группу, предпочтительно фурильную, пиридильную или тиазолильную, необязательно замещенную атомом галогена, в частности, -Cl (предпочтительно указанная группа не замещена);
- группу -ОН;
- морфолиновую группу; или
- N-фенилпиперазиновую группу;
- NH-NH-CO-арильную группу, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из атома галогена, предпочтительно Cl, и -O(С1-С6 алкильной) группы, предпочтительно -ОМе;
NH-NH-CO-гетероарильную группу, предпочтительно NH-NH-CO-пиридильную.
R2 представляет собой атом водорода; С1-С6 алкильную группу, предпочтительно метильную группу; (С1-С6 алкил)арильную группу, предпочтительно (С1-С6 алкил)фенильную группу, в частности, бензильную группу; или (С1-С6 алкил)O(С1-С6 алкильную) группу, предпочтительно группу СН2ОСН3; предпочтительно R2 представляет собой атом водорода;
R3 представляет собой
- группу -COOR5, где R5 представляет собой атом водорода, С1-С6 алкильную группу, такую как, например, метильную, этильную, изопропильную и т-бутильную группы, или глюкопиранозную группу;
- группу -COSR6, где R6 представляет собой атом водорода или С1-С6 алкильную группу;
- группу -CONR7R8, где R7 представляет собой атом водорода или С1-С6 алкильную группу, такую как метильная группа, и R8 представляет собой атом водорода; С1-С6 алкильную группу, предпочтительно этильную или метильную, необязательно замещенную группой -ОН, такую как, например, -(СН2)2ОН; группу -ОН; -O(С1-С6 алкильную) группу, предпочтительно Оэтильную; группу -NH2; группу -(С1-С6 алкил)NR9R10, предпочтительно (CH2)2NR9R10, где R9 и R10 оба представляют собой С1-С6 алкильную группу, предпочтительно метильную или этильную группу; группу -(С1-С6 алкил)СООН, предпочтительно -СН2СООН; группу -(С1-С6алкил)СОО(С1-С6 алкил), предпочтительно -СН2СООэтил; арильную группу, предпочтительно фенильную (Рh); или гетероарильную группу; примерами группы -CONR7R8 являются CONH2, CONHEt, CONHOH, CONHOEt, CONHNH2, CONH(CH2)2OH, CONH(CH2)2NMe2, CONH(CH2)2NEt2, CONMeCH2COOH, CONMeCH2COOEt, CONMeOMe, CONHPh и СONHгетероарил;
- группу -CSNR11R12, где R11 и R12 независимо друг от друга представляют собой атом водорода или С1-С6 алкильную группу, такую как, например, этильная группа, предпочтительно R11 представляет собой атом водорода и R12 представляет собой С1-С6 алкильную группу, такую как, например, этильная группа;
- - группу CN;
- - группу C(=NH)NHOH;
- -СОморфолиновую группу;
- -СОпиролидиновую группу;
- -CON-Ме-пиперазиновую группу;
- -СОгуанидиновую или группу -СОгуанидин-ВОС;
- тетразольную группу; или
- оксадиазолоновую группу;
или энантиомер, диастереоизомер, гидрат, сольват, таутомер, рацемическую смесь или фармацевтически приемлемую соль указанного соединения,
за исключением соединений с (а) по (z1) следующих формул:
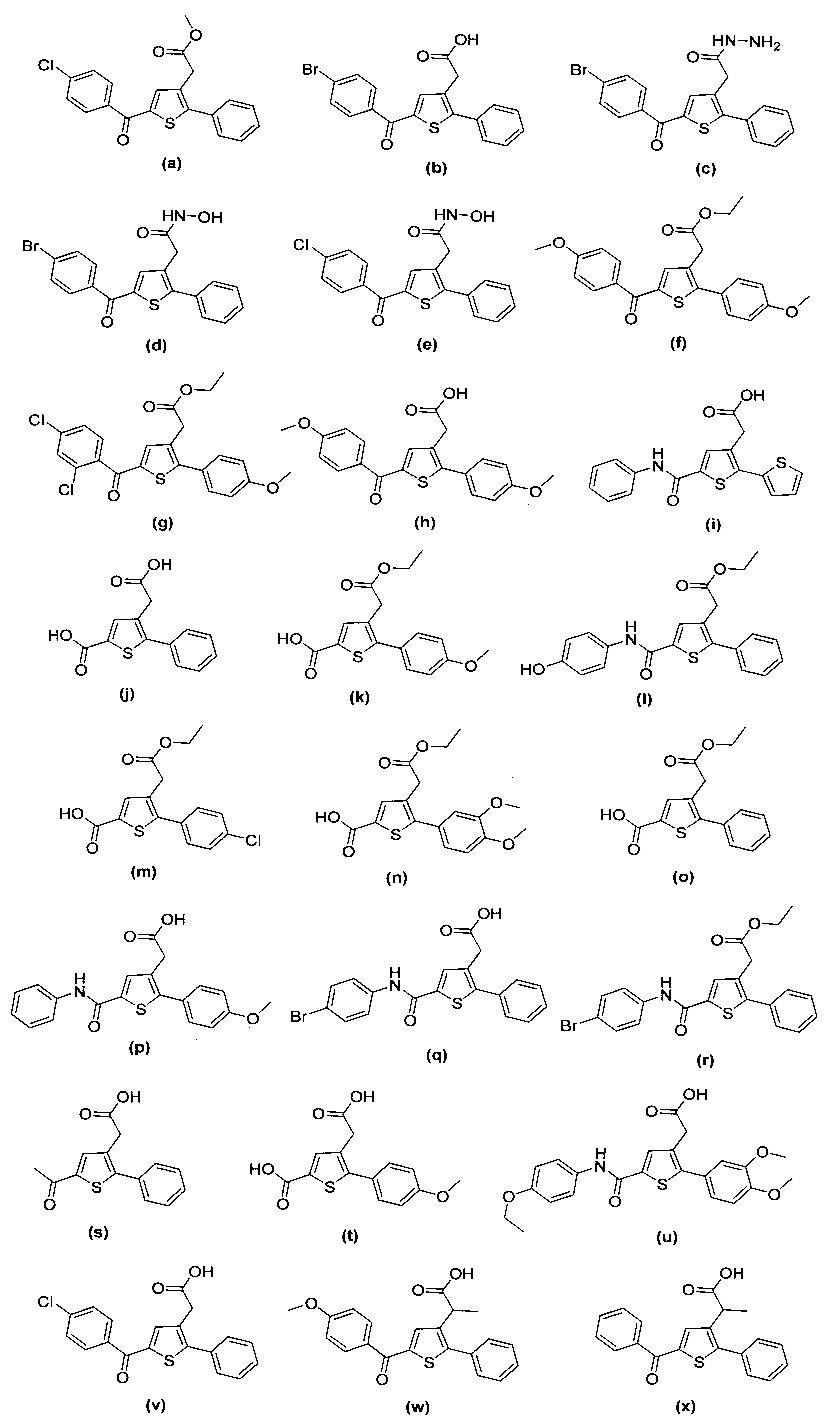
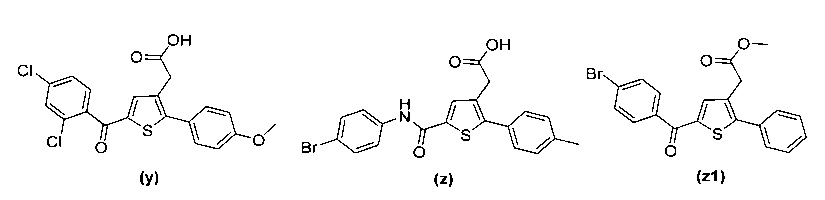
В документах с [1] по [9] раскрыты 11 соединений, структуры которых охватывает общая формула (I) (соединения от (а) до (е), (q), (t), от (v) до (х) и (z1) выше), однако какая-либо противодиабетическая активность, свойственная данным соединениям, не была описана. Вследствие этого данные соединения исключены из продуктов формулы (I), но не из применения данных продуктов для лечения или предотвращения сахарного диабета.
Более того, 16 соединений, структуры которых охватывает общая формула (I) (соединений с (f) по (р), (r), (s), (u), (у) и (z) выше), являются коммерчески доступными, однако какая-либо терапевтическая активность, свойственная данным соединениям, не была раскрыта. Вследствие этого данные соединения исключены из продуктов формулы (I), но не из применения данных продуктов в качестве лекарственного препарата, в частности, для лечения или предотвращения сахарного диабета.
В рамках настоящего изобретения под «арильной группой» понимают ароматическое кольцо, содержащее от 5 до 8 атомов углерода, или несколько сочлененных ароматических колец, содержащих от 5 до 14 атомов углерода. В частности, арильные группы могут представлять собой моноциклические или бициклические группы, предпочтительно фенильные или нафтильные группы. Предпочтительно, арильная группа представляет собой фенильную группу (Ph).
В рамках настоящего изобретения под «гетероарильной группой» понимают любую углеводородную ароматическую группу, содержащую от 3 до 9 атомов и содержащую один или несколько гетероатомов, таких как, например, атомы серы, азота или кислорода. Гетероарильная группа согласно настоящему изобретению может быть образована одним или несколькими сочлененными кольцами. Примерами гетероарильных групп являются фурильная, изоксазильная, пиридильная, тиазолильная, пиримидильная, бензимидазольная, бензоксазольная, бензотиазольная группы. Предпочтительно гетероарильную группу выбирают из фурильной, пиридильной и тиазолильной групп, предпочтительно гетероарильная группа представляет собой фурильную группу.
В рамках настоящего изобретения под «атомом галогена» понимают любой атом галогена, который предпочтительно выбирают из Cl, Br, I или F, в частности, выбирают из F, Cl или Вr, в частности из F или Cl.
В рамках настоящего изобретения под «С1-С6 алкильной группой» понимают любую алкильную группу, содержащую от 1 до 6 атомов углерода, линейную или разветвленную, в частности, метильную, этильную, н-пропильную, изо-пропильную, н-бутильную, изо-бутильную, втор-бутильную, т-бутильную, н-пентильную, н-гексильную группы. Предпочтительно, С1-С6 алкильная группа представляет собой метильную, этильную, изо-пропильную или т-бутильную группу, в частности, метильную или этильную группу, более конкретно метильную группу.
В рамках настоящего изобретения под «С3-С6 циклоалкильной группой» понимают любое насыщенное или углеводородное кольцо, содержащее от 3 до 6 атомов углерода, в частности, циклопропильную, циклобутильную, циклопентильную или циклогексильную группу. Предпочтительно, С3-С6 циклоалкильная группа представляет собой циклопропильную или циклогексильную группу.
В рамках настоящего изобретения под «(С1-С6 алкил)арильной группой» понимают любую арильную группу, как определено выше, связанную с С1-С6 алкильной группой, как определено выше. В частности, примерами (С1-С6 алкил)арильной группы является бензильная группа или -(СН2)2фенильная группа.
В рамках настоящего изобретения под «фармацевтически приемлемым» понимают, что такое вещество можно использовать для приготовления фармацевтической композиции, которая является в общем виде безопасной, нетоксичной и использование которой не является нежелательным ни с биологической, ни с какой-либо другой точки зрения, и которая является приемлемой для ветеринарного применения, а также для приготовления фармацевтических препаратов, применяемых у человека.
В рамках настоящего изобретения под «фармацевтически приемлемыми солями соединения» понимают соли, которые являются фармацевтически приемлемыми, как определено в настоящей заявке, и которые обладают желаемой фармакологической активностью родительского соединения. Такие соли включают:
(1) соли присоединения кислоты, образованные из минеральных кислот, таких как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное; или образованные из органических кислот, таких как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, гидроксинафтоевая кислота, 2-гидроксиэтансульфоновая кислота, молочная кислота, малеиновая кислота, яблочная кислота, миндальная кислота, метансульфоновая кислота, муконовая кислота, 2-нафталинсульфоновая кислота, пропионовая кислота, салициловая кислота, янтарная кислота, дибензоил-L-винная кислота, винная кислота, п-толуолсульфоновая кислота, триметилуксусная кислота, трифторуксусная кислота и тому подобное; или
(2) соли, образованные в случае замещения протона кислоты, присутствующего в родительском соединении, ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо в результате координирования указанного протона кислоты с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин и тому подобное. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия и гидроксид натрия.
В рамках настоящего изобретения под «сольватом соединения» понимают любое соединение, полученное в результате добавления молекулы инертного растворителя к соединению согласно настоящему изобретению; при этом сольват образуется в результате взаимной силы притяжения. Сольваты представляют собой, например, алкоголяты соединения. Гидрат представляет собой сольват, в котором используемым инертным растворителем является вода. Гидрат может представлять собой моно-, ди- или тригидрат.
В рамках настоящего изобретения под «таутомером» понимают любой изомер для получения соединений согласно настоящему изобретению, причем указанные соединения являются взаимнопревращаемыми в результате обратимой химической реакции, которую называют реакцией таутомеризации. В большинстве случаев данная реакция происходит в результате миграции атома водорода, сопровождающейся изменением локализации двойной связи. В растворе соединения, способного к таутомеризации, устанавливается равновесие между 2 таутомерами. Соотношение между таутомерами также зависит от растворителя, от температуры и от значения рН. Вследствие этого таутомеры представляют собой трансформацию функциональной группы в другую группу, наиболее часто в результате сопутствующего смещения атома водорода и π-связи (двойной или тройной связи). Общеизвестные таутомеры представляют собой, например, альдегиды/кетоны - спирты или более конкретные пары енолов; амиды - имидокислоты; лактамы - лактимы; имины - енамины; енамины - енамины. В частности, таутомеры могут включать таутомерию «цикл-цепь», возникающую в случае, если движение протона сопровождается трансформацией открытой структуры в одно кольцо.
Согласно предпочтительному варианту реализации настоящего изобретения  представляет собой связь и W представляет собой атом кислорода или группу -NOR4, где R4 представляет собой атом водорода, С1-С6 алкильную группу, такую как, например, метильная или этильная группа, или (С1-С6 алкил)арильную группу, такую как, например, (С1-С6 алкил)фенильная группа, в частности, бензильная группа, причем указанная арильная группа, предпочтительно фенильная, необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -C1-С6 алкила), предпочтительно -ОМе, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, или -O(С1-С6 алкильной) группой, предпочтительно -ОМе; С1-С6 алкила, замещенного одним или несколькими атомами галогена, предпочтительно F, или -О(C1-С6 алкильной) группой, предпочтительно -ОМе, или группой -ОН; -SО2(С1-С6 алкила); -CONRa'Rb', где Ra' представляет собой атом водорода или C1-С6 алкильную группу и Rb' представляет собой С1-C6 алкильную группу или -ОН.
представляет собой связь и W представляет собой атом кислорода или группу -NOR4, где R4 представляет собой атом водорода, С1-С6 алкильную группу, такую как, например, метильная или этильная группа, или (С1-С6 алкил)арильную группу, такую как, например, (С1-С6 алкил)фенильная группа, в частности, бензильная группа, причем указанная арильная группа, предпочтительно фенильная, необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -C1-С6 алкила), предпочтительно -ОМе, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, или -O(С1-С6 алкильной) группой, предпочтительно -ОМе; С1-С6 алкила, замещенного одним или несколькими атомами галогена, предпочтительно F, или -О(C1-С6 алкильной) группой, предпочтительно -ОМе, или группой -ОН; -SО2(С1-С6 алкила); -CONRa'Rb', где Ra' представляет собой атом водорода или C1-С6 алкильную группу и Rb' представляет собой С1-C6 алкильную группу или -ОН.
Предпочтительно  представляет собой связь и W представляет собой атом кислорода или группу -NOR4, где R4 представляет собой атом водорода или С1-С6 алкильную группу, такую как, например, метильная или этильная группа, еще более предпочтительно W представляет собой атом кислорода.
представляет собой связь и W представляет собой атом кислорода или группу -NOR4, где R4 представляет собой атом водорода или С1-С6 алкильную группу, такую как, например, метильная или этильная группа, еще более предпочтительно W представляет собой атом кислорода.
Согласно другому предпочтительному варианту реализации настоящего изобретения Y представляет собой арильную, предпочтительно фенильную, гетероарильную группу, предпочтительно фурильную, или бензо-1,3-диоксольную группу, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -O(С1-С6 алкила), предпочтительно -ОМе; или -ОН; предпочтительно Y представляет собой арильную группу, предпочтительно фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F, в частности, Cl; или -O(С1-С6 алкила), предпочтительно -ОМе. Предпочтительно заместитель в фенильной группе находится в орто- и/или мета- и/или пара- положении. Еще более предпочтительно Y представляет собой фенильную группу, предпочтительно замещенную в орто- и/или мета- и/или пара- положениях, одним или несколькими атомами галогена, предпочтительно Cl и/или F, в частности, Cl.
Согласно другому предпочтительному варианту реализации настоящего изобретения R1 представляет собой С3-С6 циклоалкильную группу, предпочтительно циклопропильную или циклогексильную; арильную группу, предпочтительно фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -CN; атома галогена, который предпочтительно выбирают из Cl или F; -O(С1-С6 алкила), предпочтительно -ОМе; или С1-С6 алкила, предпочтительно метила; гетероарильную группу, предпочтительно фуранильную, пиридильную или тиазолильную, причем указанная фуранильная группа необязательно замещена атомом галогена, в частности, Cl (предпочтительно указанная группа не замещена); или морфолиновую группу. Предпочтительно R1 представляет собой фенильную группу (Ph), причем указанная фенильная группа необязательно замещена одной или несколькими группами, которые выбирают из атома галогена, который предпочтительно выбирают из Cl или F, в частности, Cl, и -O(С1-С6 алкила), предпочтительно -ОМе, в частности, указанная фенильная группа необязательно замещена одной или несколькими -O(С1-С6 алкильными) группами, предпочтительно -ОМе; или фуранильной, пиридильной или тиазолильной группой, причем указанная фуранильная группа необязательно замещена атомом галогена, в частности, Cl (предпочтительно указанная группа не замещена).
Согласно другому варианту реализации настоящего изобретения R2 представляет собой атом водорода, С1-С6 алкильную группу, предпочтительно метильную группу, или (С1-С6 алкил)O(С1-С6 алкильную) группу, в частности, атом водорода.
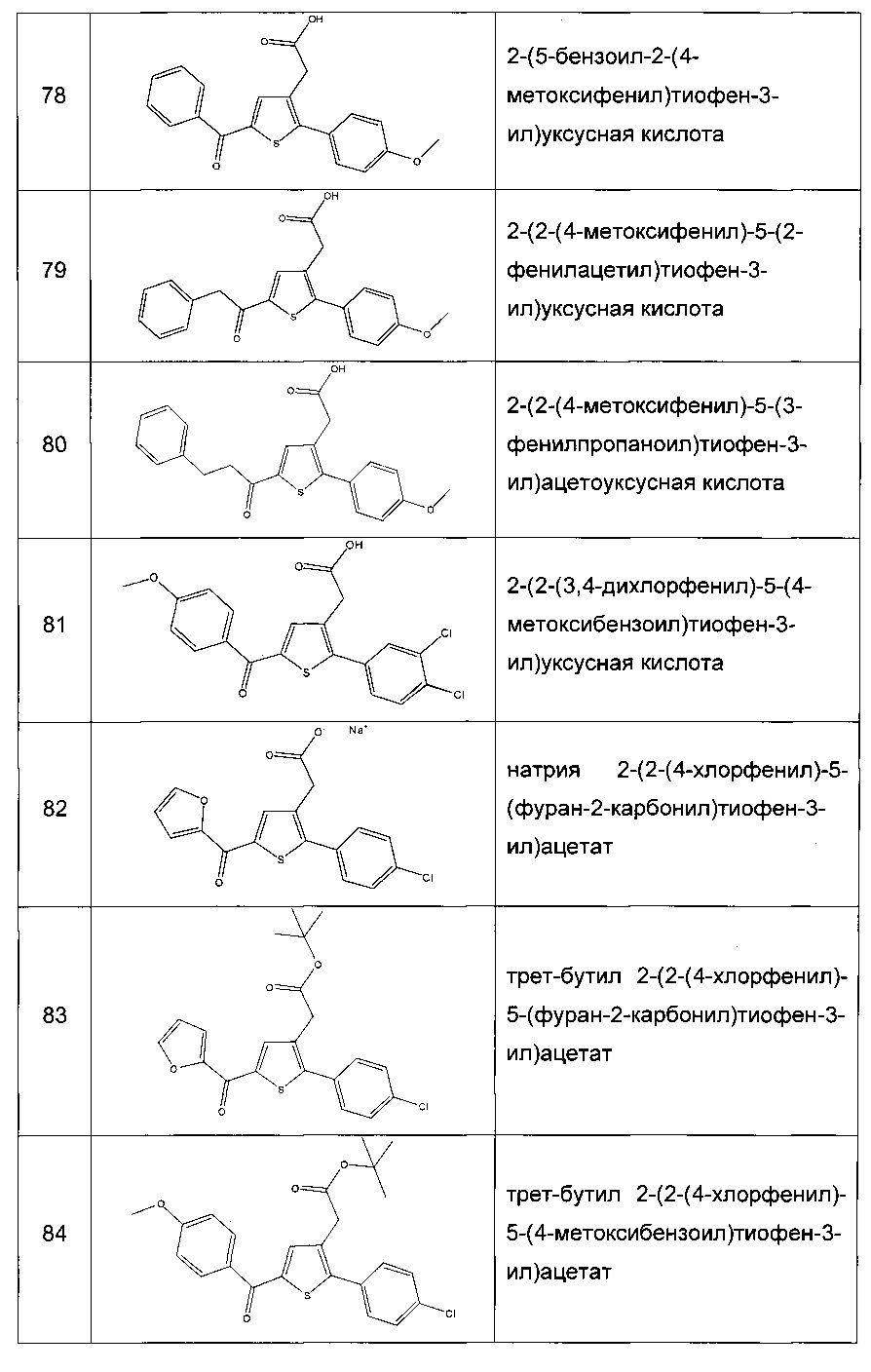
Согласно другому предпочтительному варианту реализации настоящего изобретения R3 представляет собой -СОгуанидиновую группу, группу -COOR5, где R5 представляет собой атом водорода или С1-С6 алкильную группу, такую как, например, метильная, этильная, изопропильная и т-бутильная группа; группу -CONR7R8, где R7 представляет собой атом водорода и R8 представляет собой атом водорода; С1-С6 алкильную группу, предпочтительно этильную или метильную, необязательно замещенную группой -ОН, такую как, например, -(СН2)2ОН; группу -ОН; -O(С1-С6 алкильную) группу, предпочтительно -Оэтильную; или группу -(С1-С6 алкил)NR9R10, где R9 и R10 оба представляют собой С1-С6 алкильную группу, предпочтительно метильную или этильную группу; или -СОморфолиновой группой; предпочтительно, R3 представляет собой группу -CONHOH, -СОгуанидиновую группу или группу -COOR5, где R5 представляет собой атом водорода или С1-С6 алкильную группу, такую как, например, метильная, этильная, изопропильная и т-бутильная группа. Еще более предпочтительно R3 представляет собой группу -COOR5, где R5 представляет собой атом водорода или С1-С6 алкильную группу, такую как, например, метильная, этильная, изопропильная и т-бутильная группа. Более предпочтительно R3 представляет собой группу -СООН или COOEt, в частности, СООН.
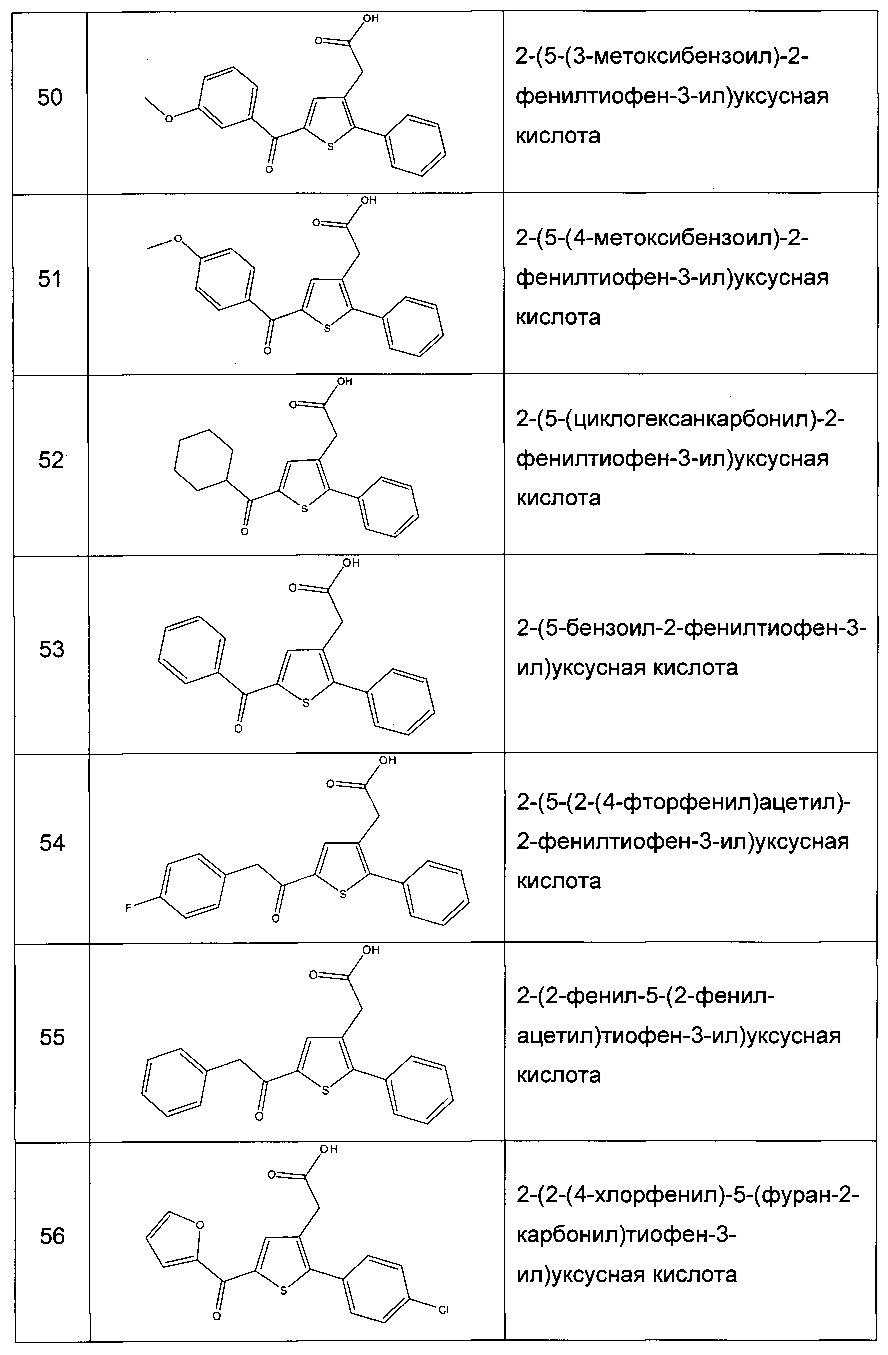
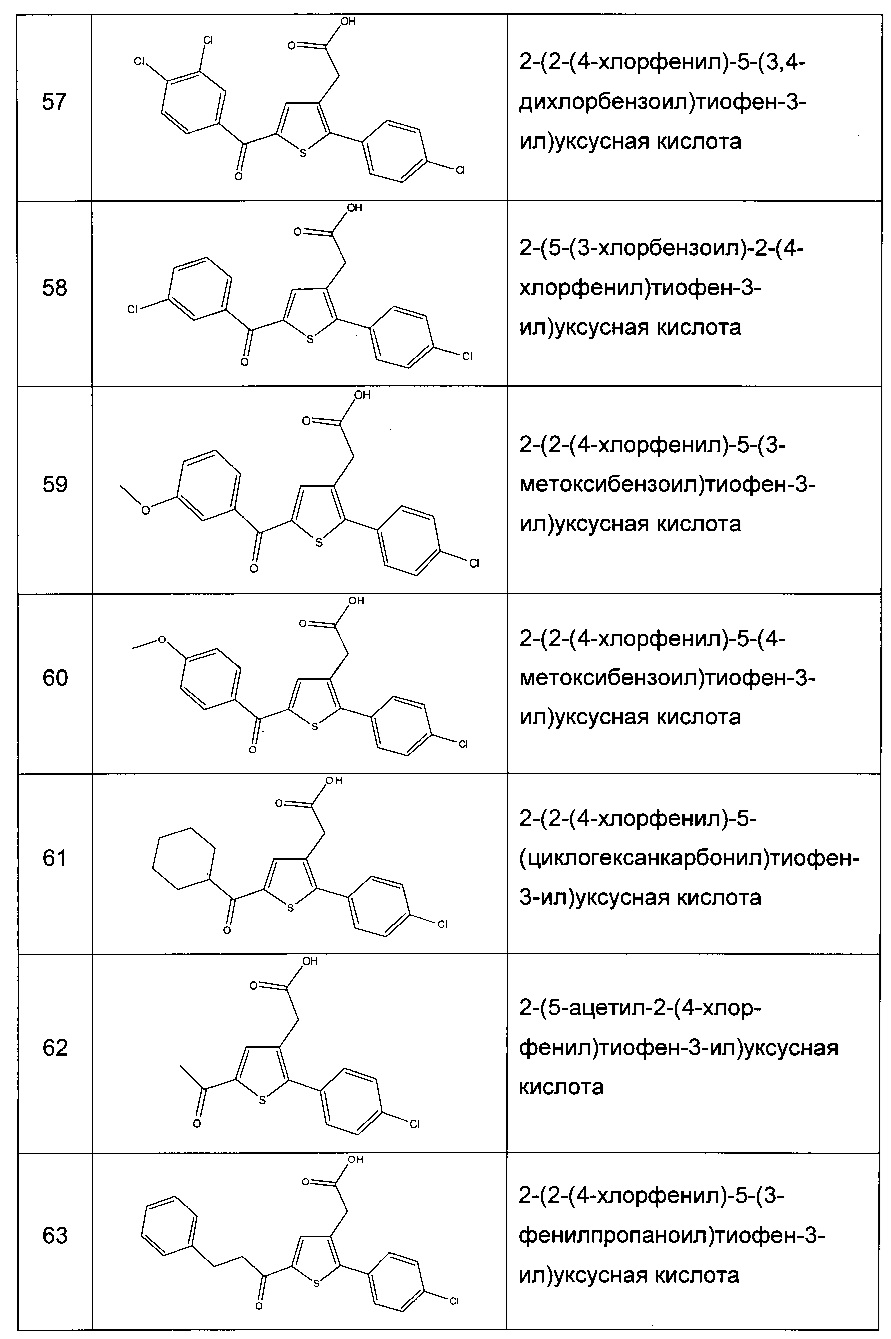
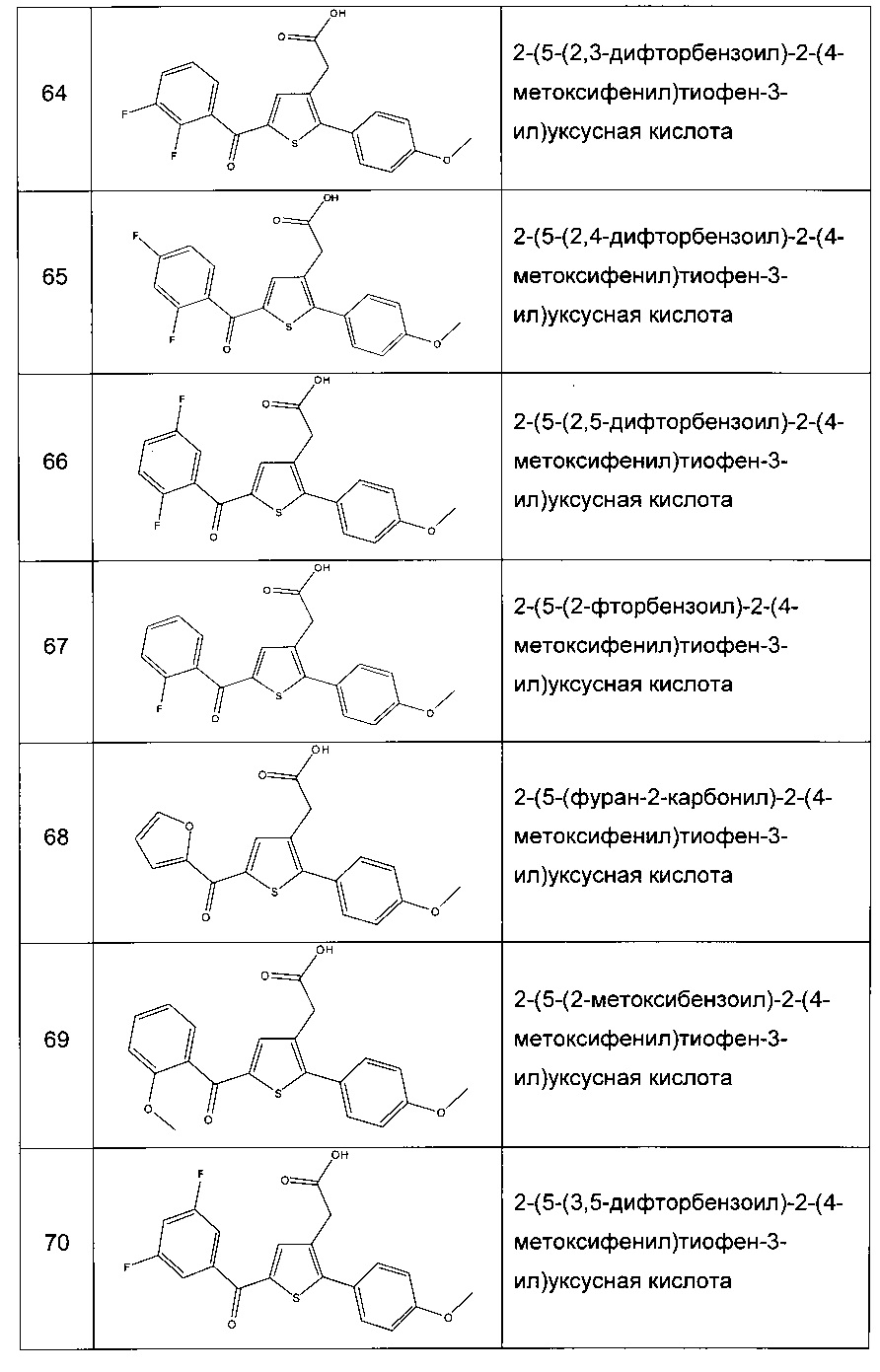
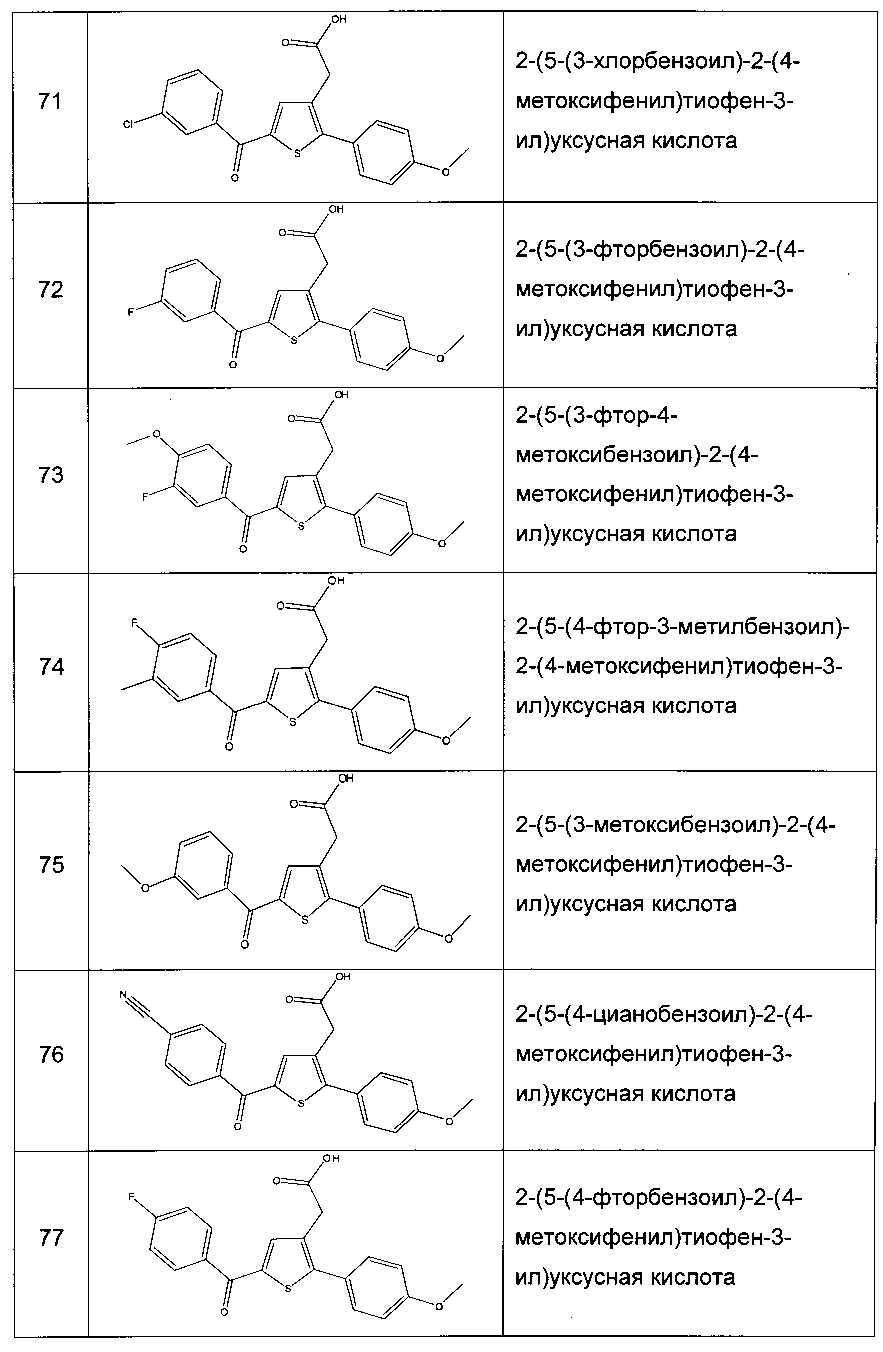
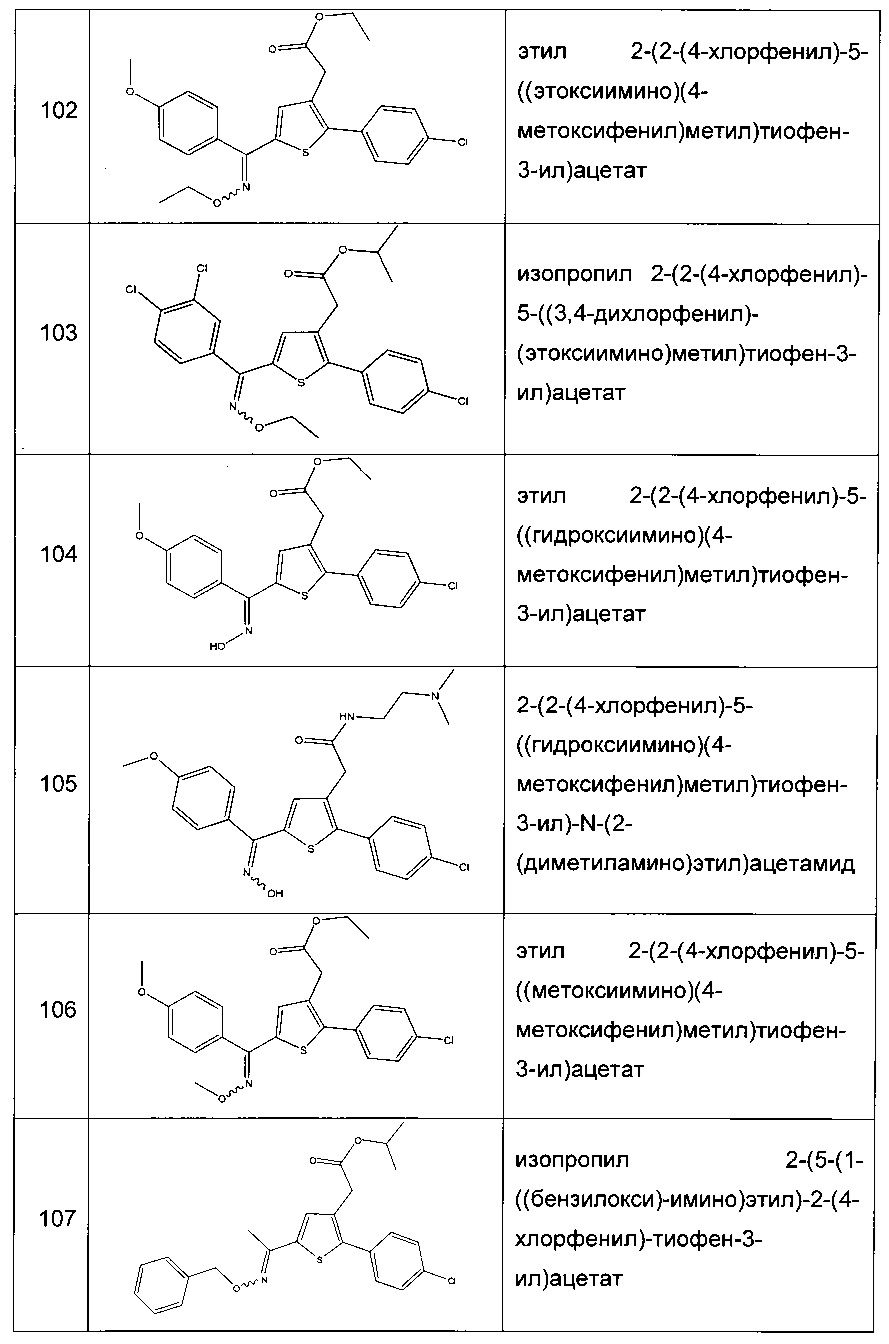
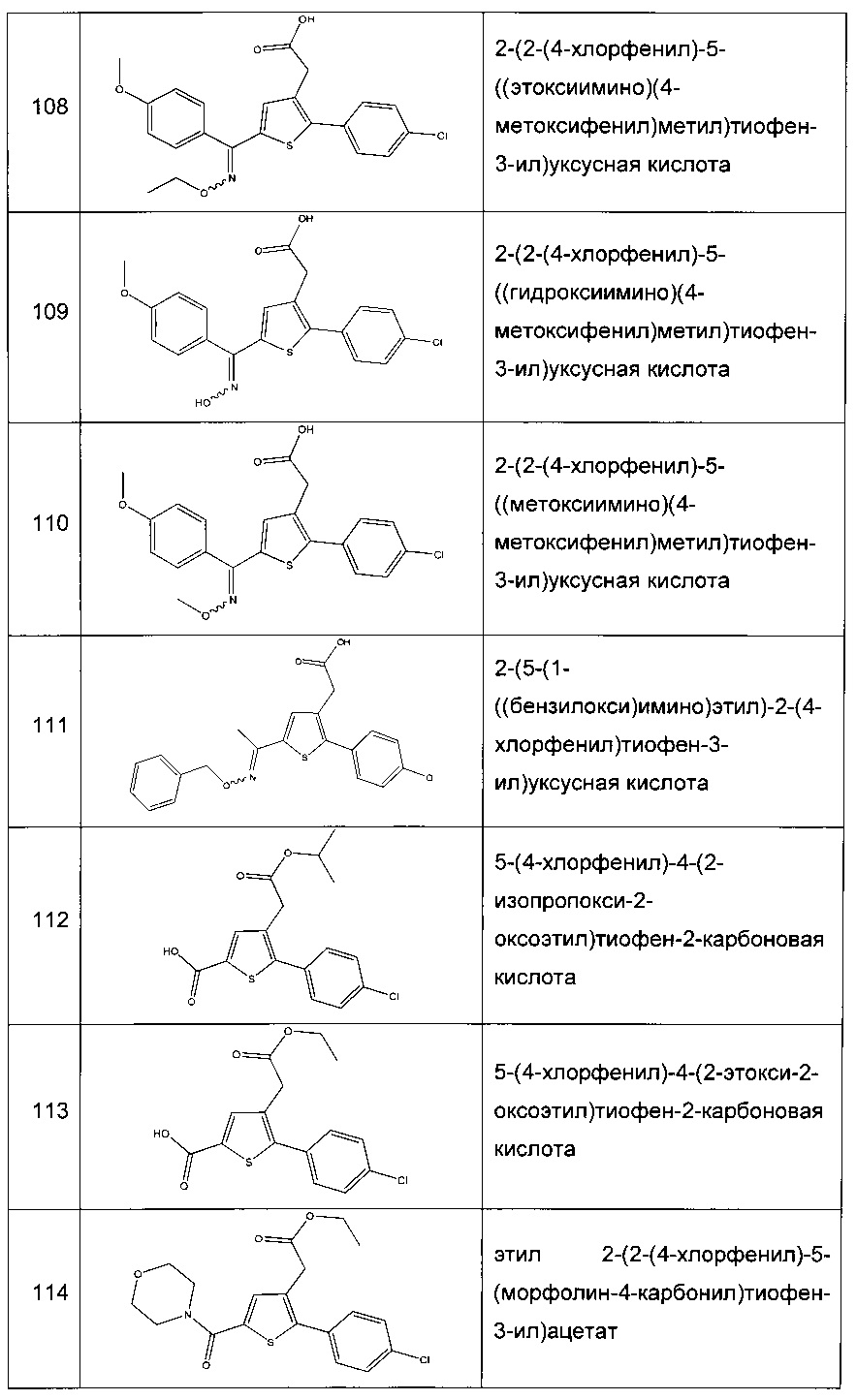
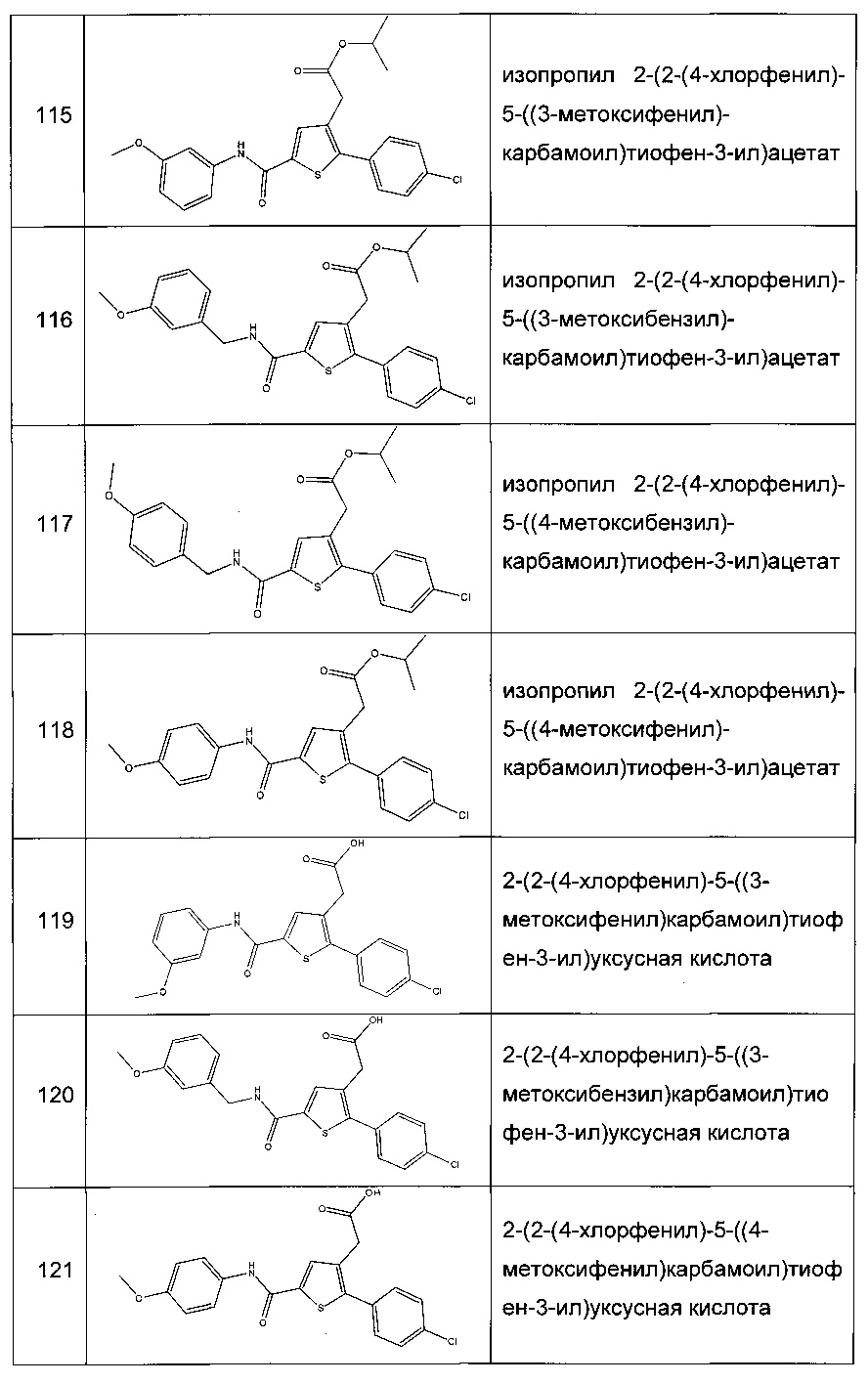
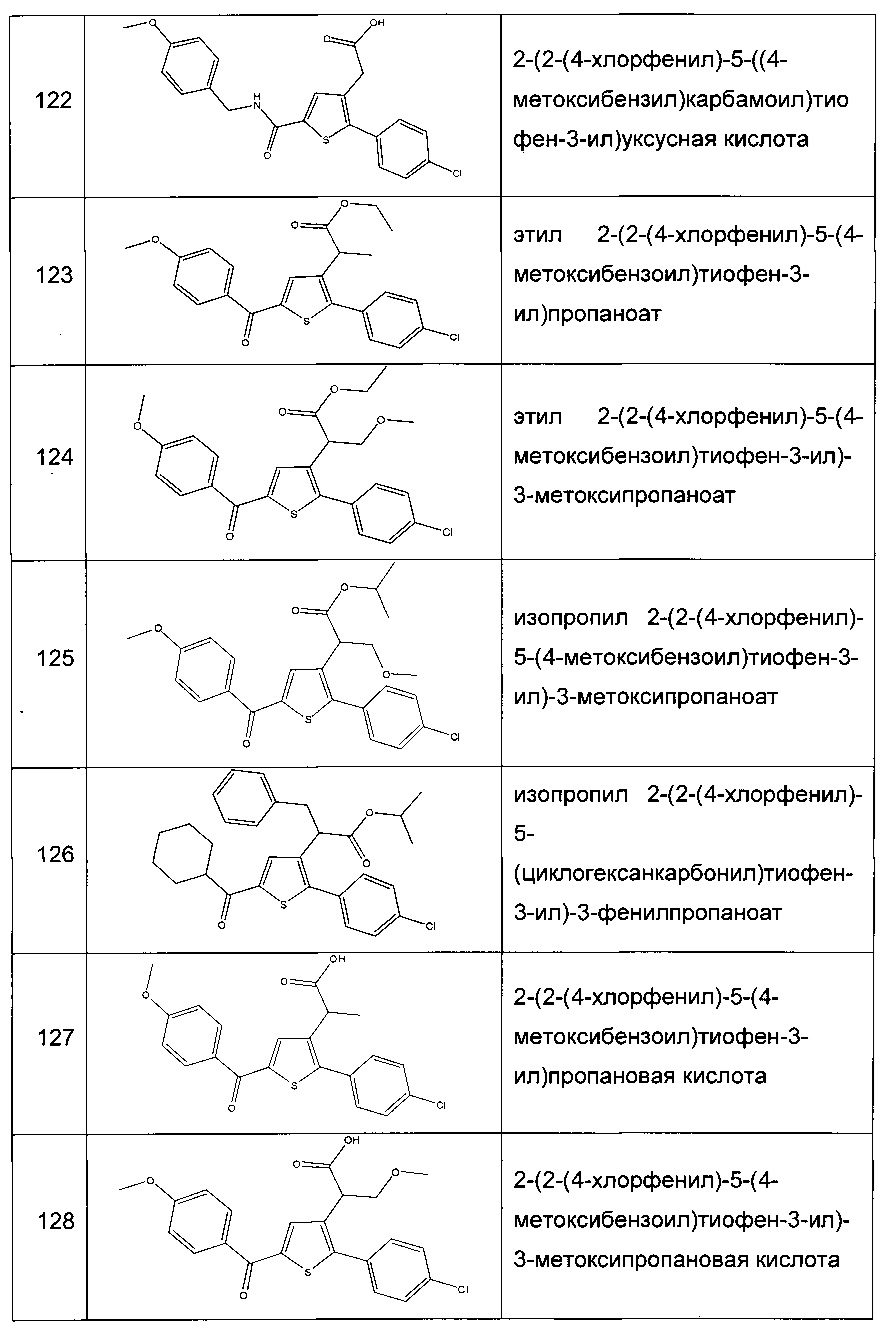
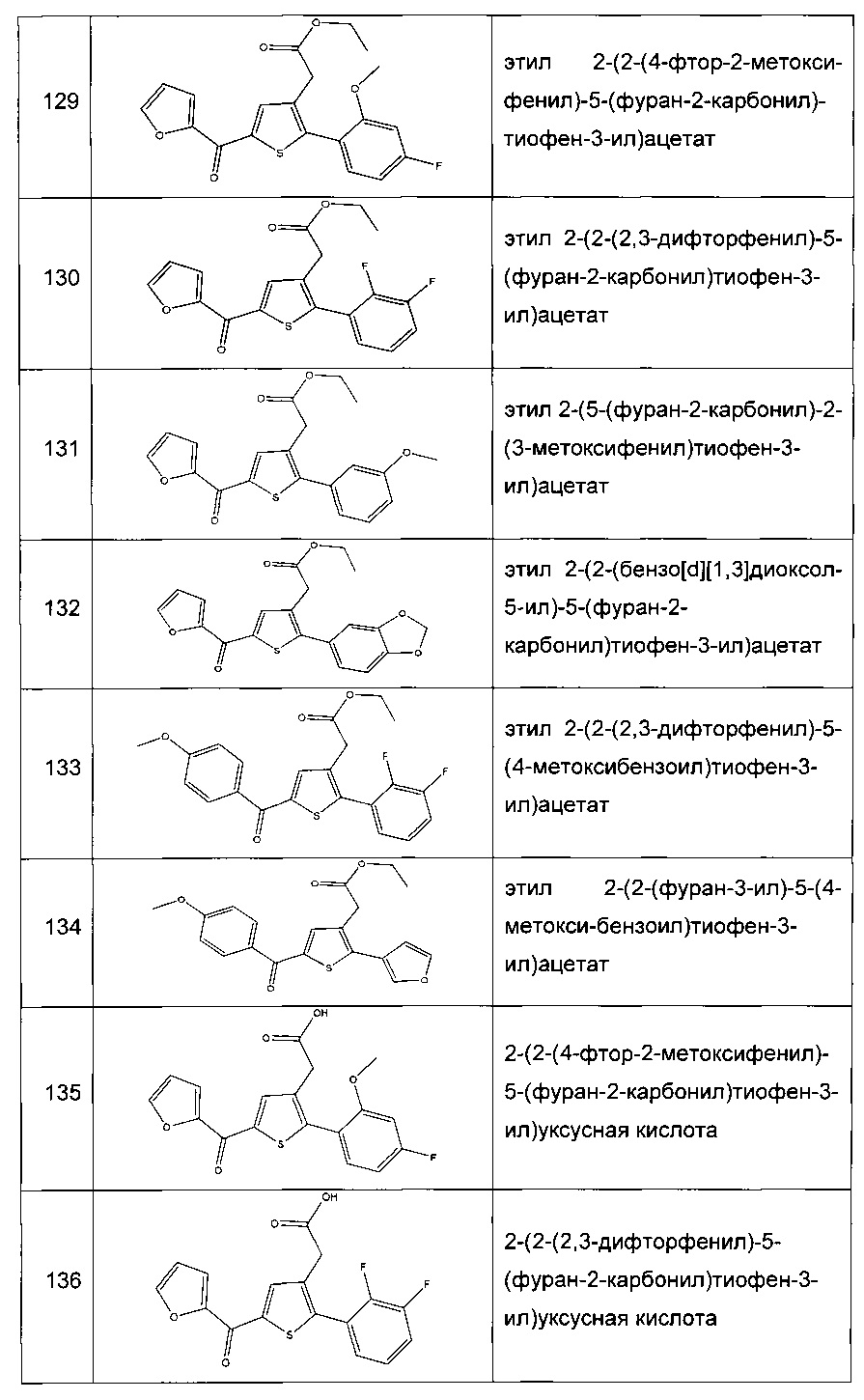
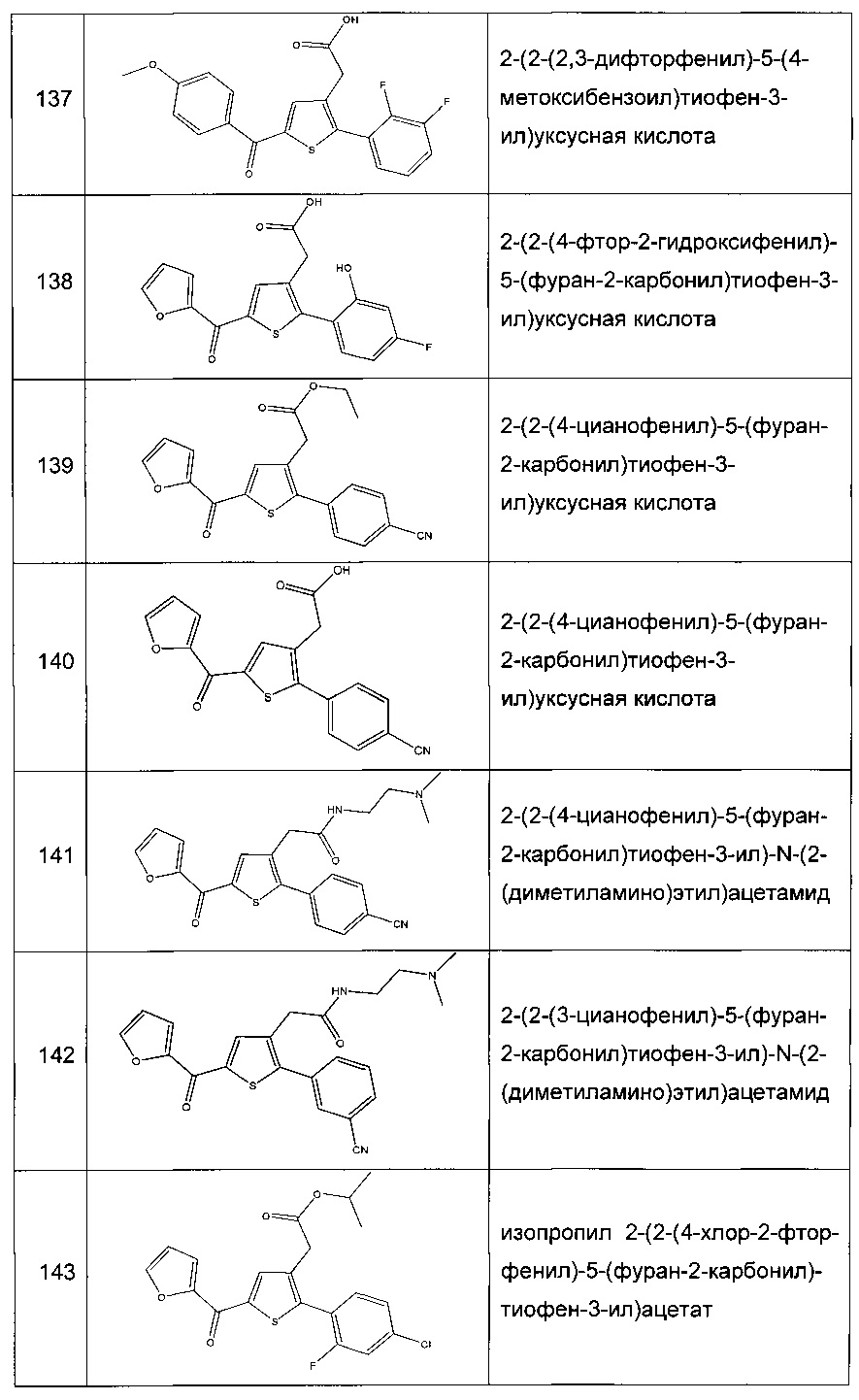
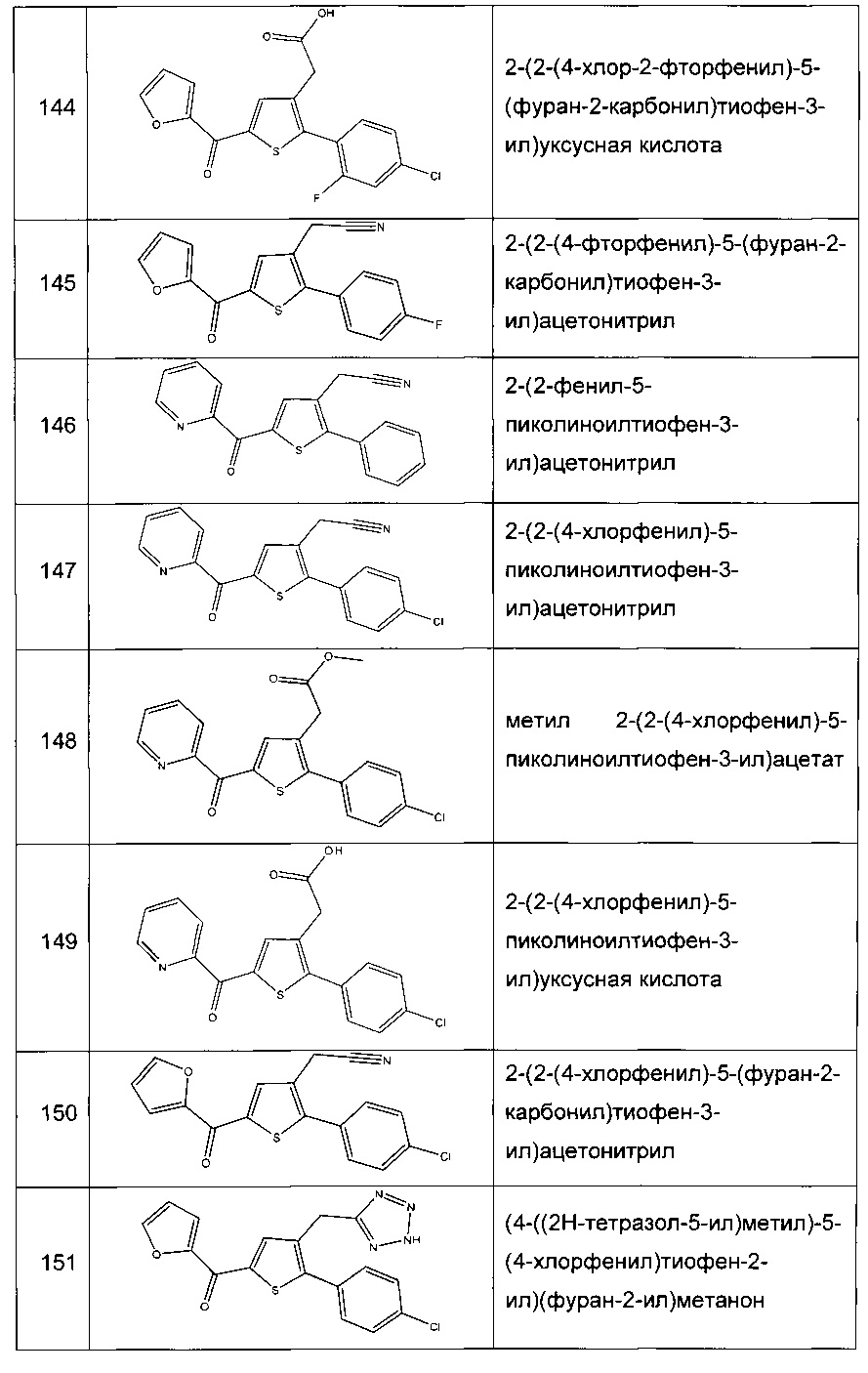
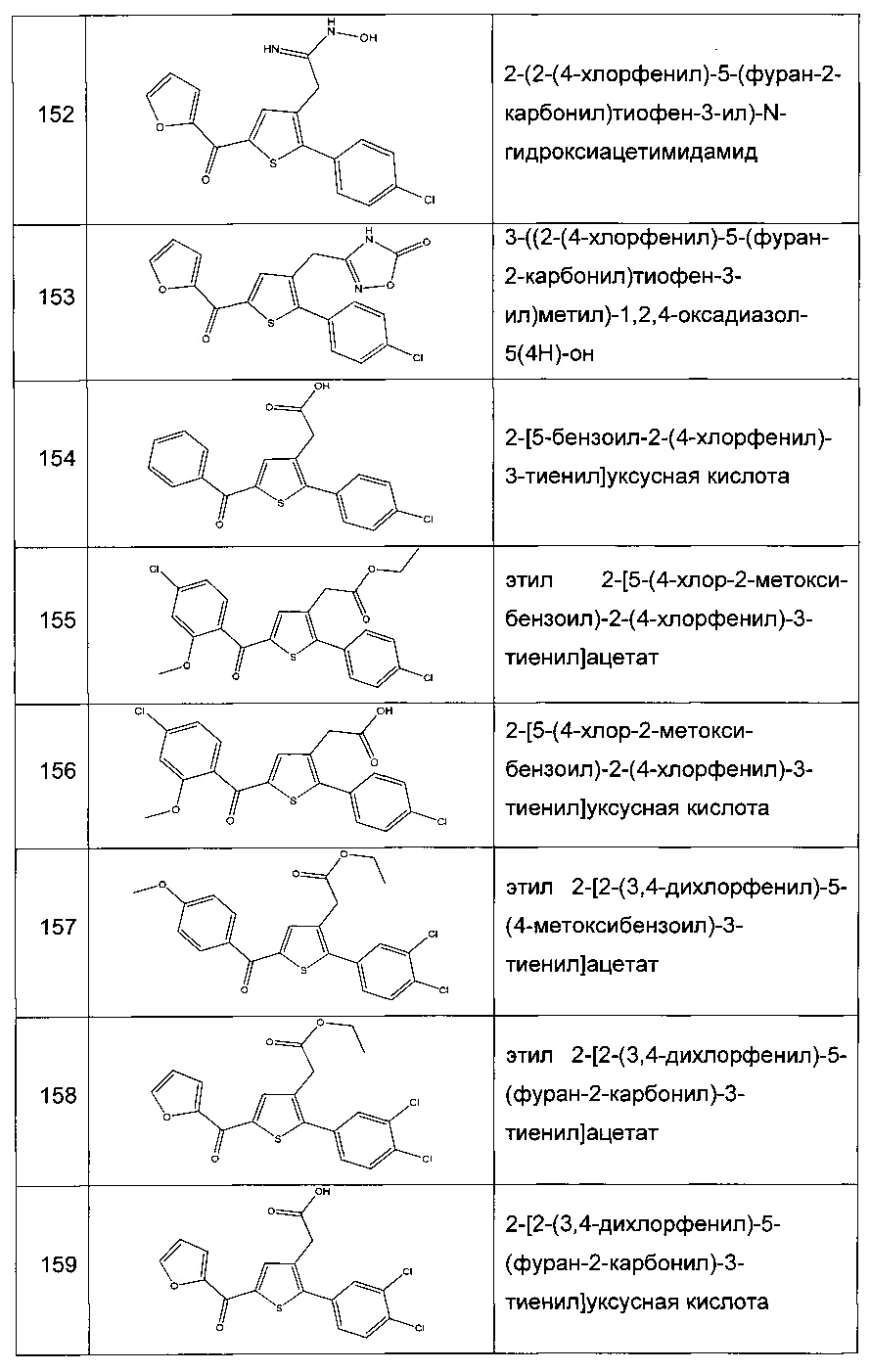
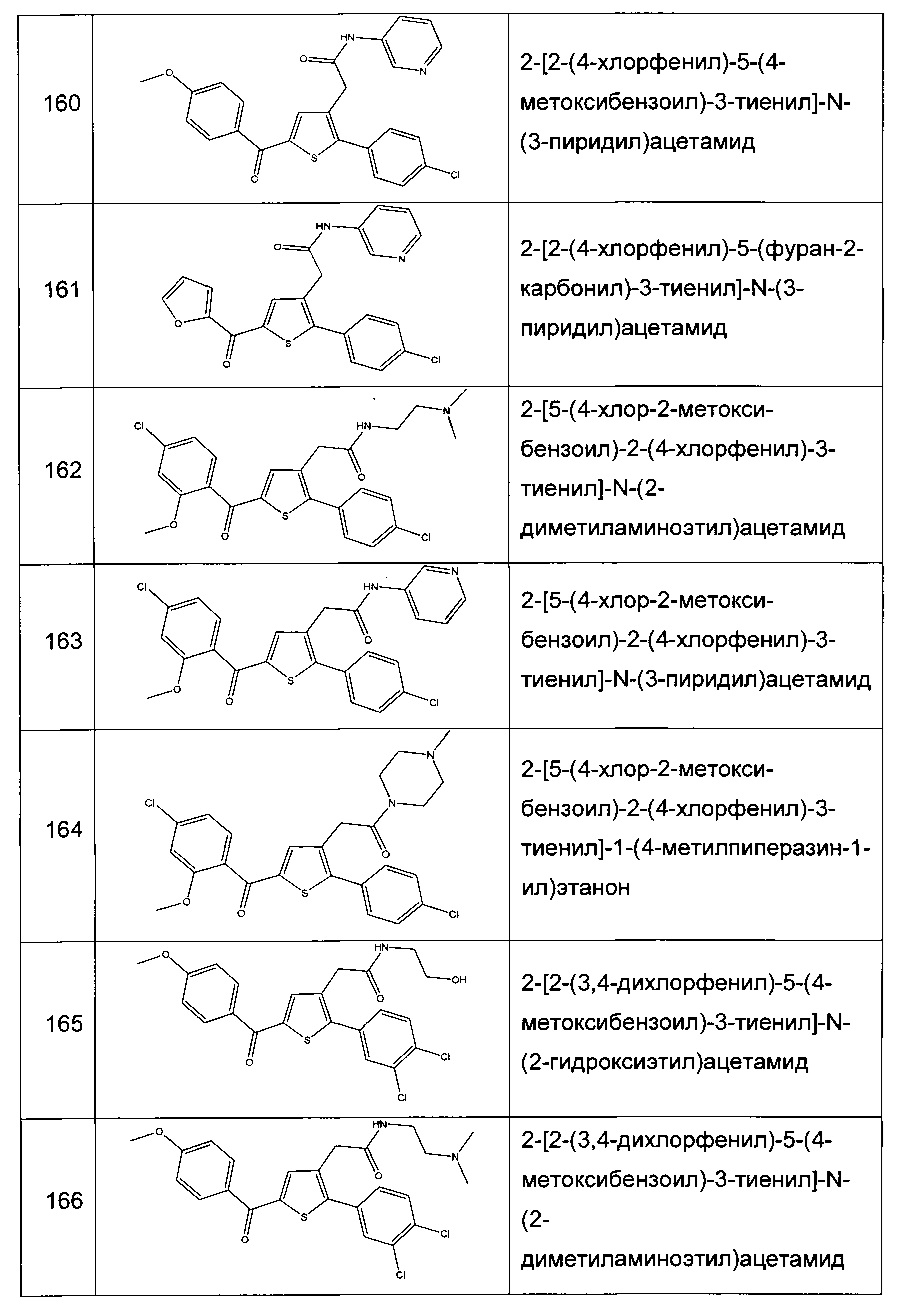
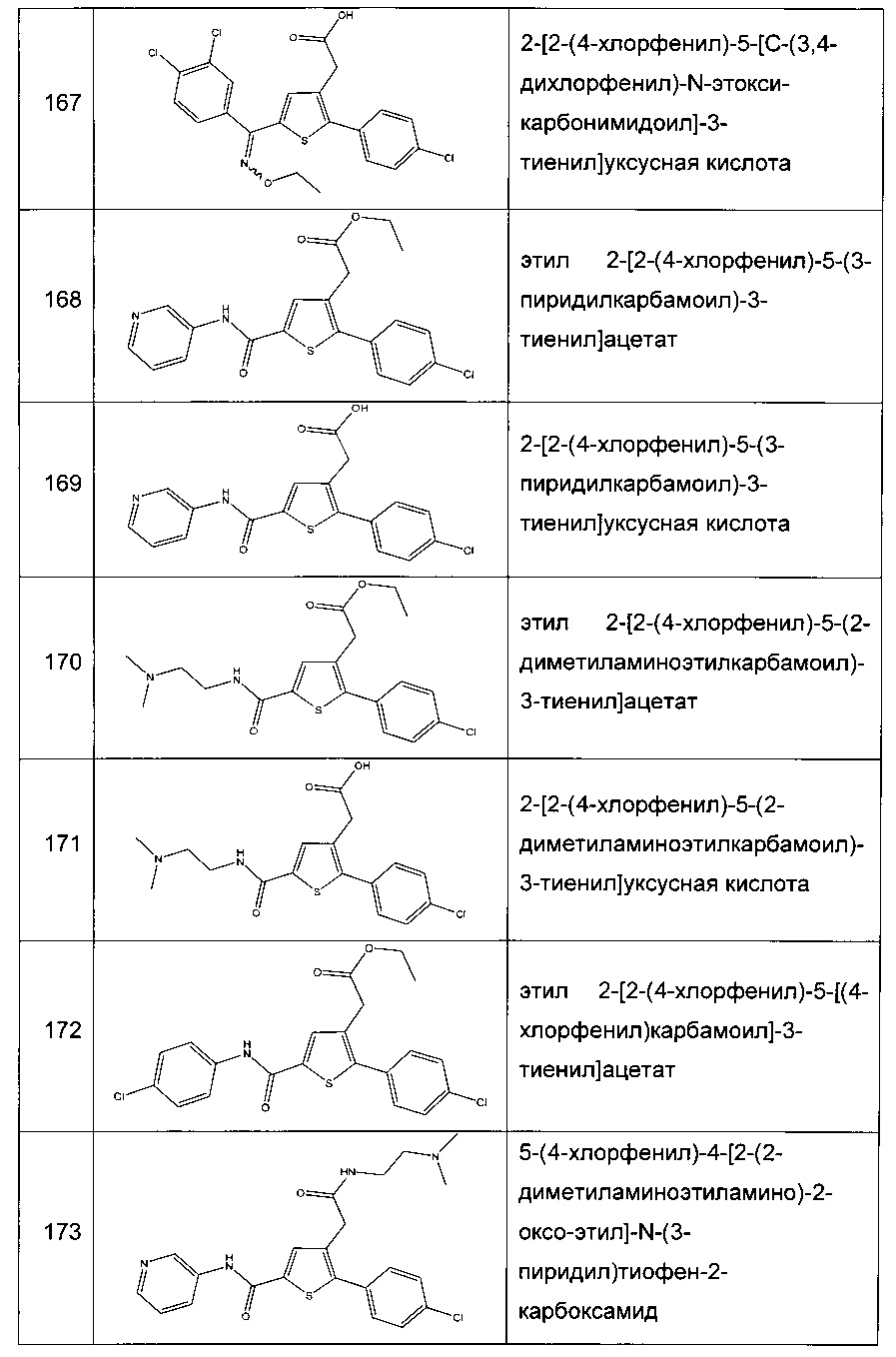
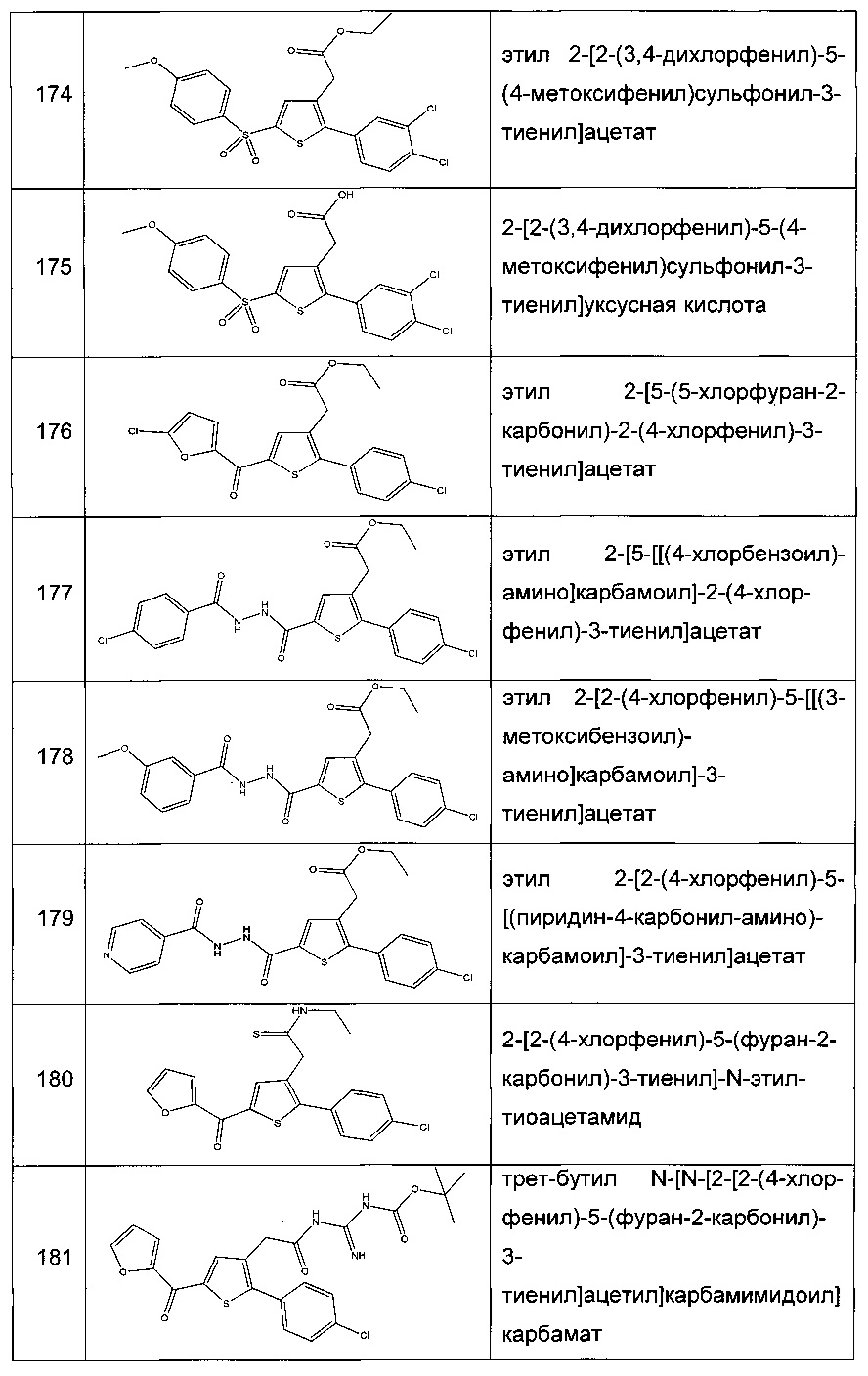
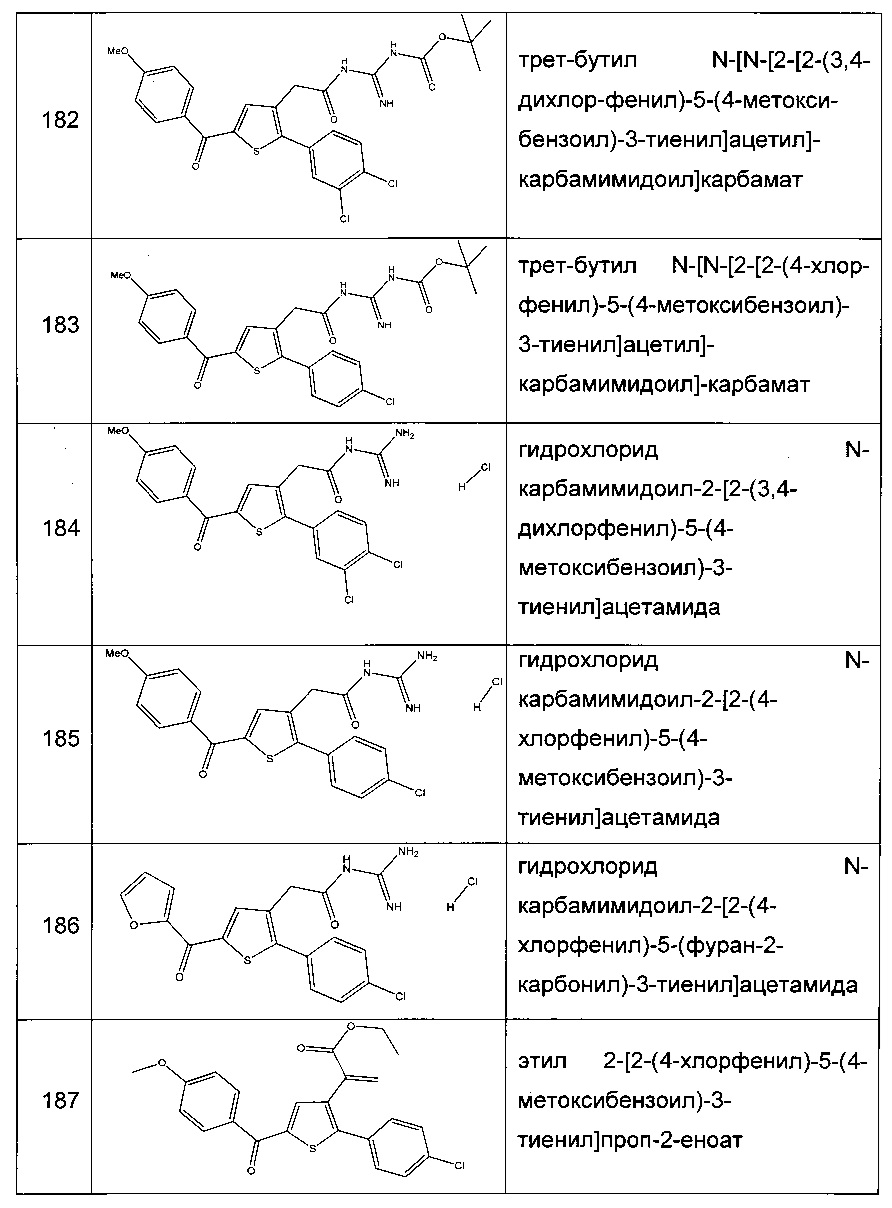
Согласно конкретному варианту реализации настоящего изобретения, вызывающему интерес, производные тиофена выбирают из соединений формул с 1 по 187, приведенных в таблице 1 ниже.
Согласно другому, вызывающему еще больший интерес варианту реализации настоящего изобретения производные тиофена выбирают из 102 соединений, имеющих номера 3, 5, 6, 10-15, 18, 19, 21, 22, 24-27, 29-33, 35, 39, 40, 43, 44, 46, 48-52, 56, 58-61, 63-65, 67, 68, 70-78, 81, 83, 84, 86, 89, 91, 93, 95, 96, 98, 100, 102, 104, 106, 108-110, 114, 123-125, 127, 128, 130, 133, 136, 137, 139, 140, 142-144, 148, 149, 154, 155, 156-159, 165-167, 175, 176 и 182-187 в таблице 1 ниже.
Еще более предпочтительно, что указанные производные тиофена представляют собой соединения 10, 13, 49, 56, 58, 60, 63, 100, 104, 110, 124, 127, 128, 130, 136, 143, 148, 149, 156, 157-159, 167, 175, 176, 184 и 185, приведенные в таблице 1 ниже.
Настоящее изобретение также относится к фармацевтической композиции, содержащей производное тиофена согласно настоящему изобретению и фармацевтически приемлемый эксципиент.
Данные композиции могут быть приготовлены в состав для введения млекопитающим, включая людей. Доза варьирует в зависимости от лечения и в зависимости от соответствующего заболевания. Данные фармацевтические композиции приспособлены для введения посредством любого приемлемого пути, например, перорального (включая буккальный и сублингвальный пути), посредством ректального, назального, местного (включая трансдермальный), вагинального, интраокулярного или парентерального (включая подкожный, внутримышечный или внутривенный) пути. Предпочтительно фармацевтические композиции приспособлены для перорального введения. Данные составы могут быть приготовлены с применением всех способов объединения активных компонентов с подходящими фармацевтически приемлемыми эксципиентами, известных специалисту в данной области техники.
Подходящие единичные лекарственные формы для перорального введения включают таблетки, желатиновые капсулы, порошки, гранулы и растворы для перорального применения или суспензии в водных или неводных жидкостях, съедобные или пищевые пены либо жидкие эмульсии типа «вода в масле» или «масло в воде». В случае если твердая композиция приготовлена в форме таблетки, главный активный компонент предпочтительно смешивают в форме порошка с приемлемым фармацевтическим эксципиентом, таким как желатин, крахмал, лактоза, стеарат магния, тальк, аравийская камедь или тому подобное. Представляется возможным покрыть таблетки сахарозой или другими подходящими материалами, либо таблетки могут быть дополнительно обработаны таким образом, чтобы данные таблетки обладали пролонгированной или отсроченной активностью и непрерывно высвобождали заранее определенное количество активного компонента.
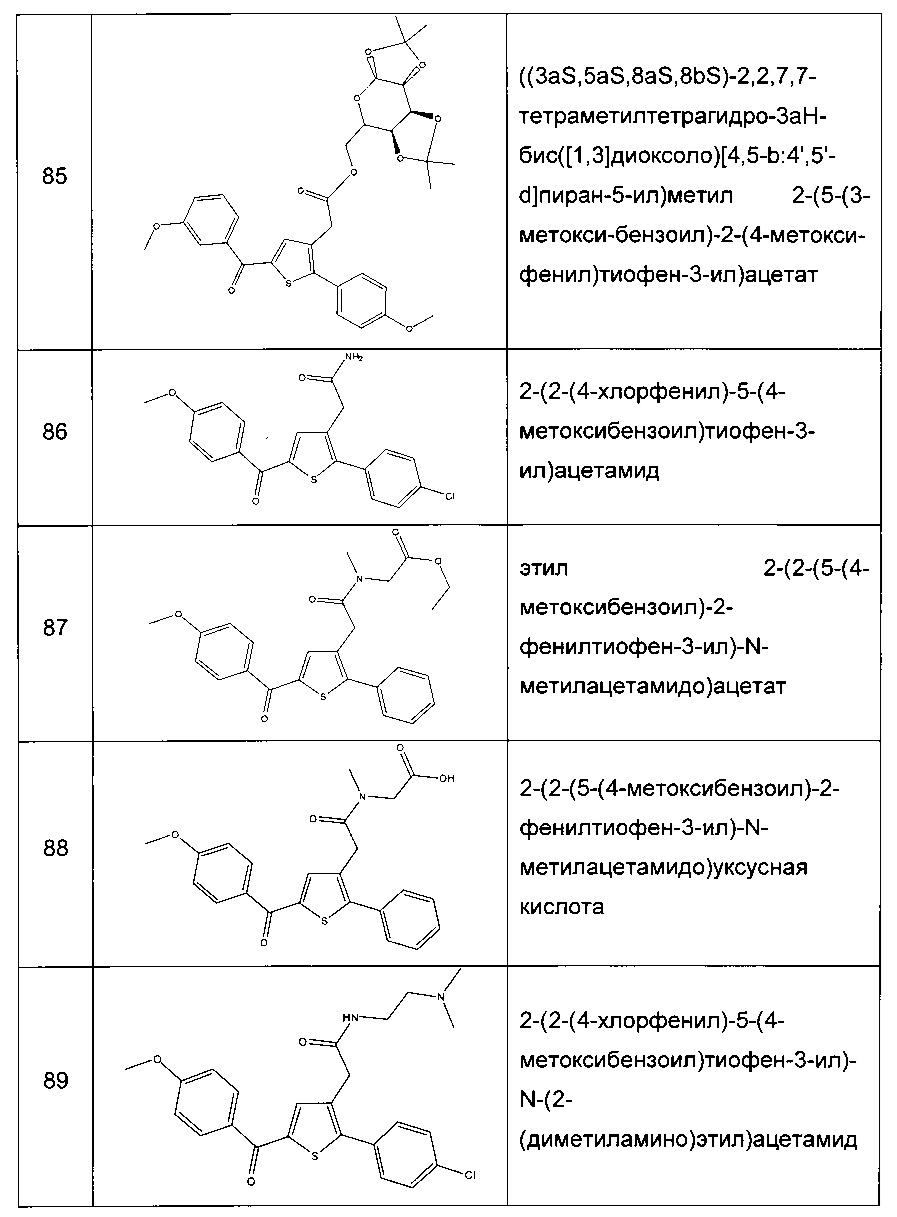
Препарат в желатиновых капсулах получают в результате смешивания активных компонентов, предпочтительно в виде порошка, с разбавителем и в результате розлива полученной смеси в мягкие или твердые желатиновые капсулы, в частности, в желатиновые капсулы. Перед помещением в желатиновые капсулы к композиции могут быть добавлены смягчающие вещества, такие как, например, тальк, стеарат магния, стеарат кальция или полиэтиленгликоль в твердой форме. Также для улучшения доступности лекарственного препарата после приема желатиновой капсулы в состав может быть добавлено разрыхляющее или солюбилизирующее вещество, такое как, например, карбонат кальция или карбонат натрия.
Дополнительно, в случае необходимости, в смесь могут быть добавлены связывающие вещества, смягчающие вещества и подходящие разрыхлители, а также окрашивающие вещества. Подходящие связывающие вещества могут представлять собой, например, крахмал, желатин, естественные сахара, такие как, например, глюкоза или бета-лактоза, подслащивающие вещества, полученные из кукурузы, синтетические или растительные каучуки, такие как, например, камедь или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и тому подобное. Смягчающие вещества, которые могут применяться в данных лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхлители включают крахмал, метил целлюлозу, агар, бентонит, ксантановую камедь и тому подобное. Таблетки могут быть изготовлены, например, путем приготовления смеси порошка, гранулирования или сухого прессования смеси, добавления смягчающего вещества и разрыхляющего вещества и прессования смеси для получения таблеток. Смесь порошка приготавливают в результате смешивания активного компонента, к которому добавляют соответствующий разбавитель или основу и, в некоторых случаях, связывающее вещество, такое как, например, карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, вещество, замедляющее растворение, такое как, например, парафин, вещество, ускоряющее всасывание, такое как, например, четвертичная соль, и/или поглощающее вещество, такое как, например, бентонит, каолин или дикальций фосфат. Смеси порошков могут быть гранулированы путем их смачивания связывающим веществом, таким как, например, сироп, крахмальная паста, мазь на основе камеди или растворы целлюлозы либо полимерные материалы, и продавливания через сито. Гранулы могут быть смазаны в результате добавления стеариновой кислоты, соли стеариновой кислоты, талька или минерального масла для того, чтобы избежать прилипания к формам, в которых осуществляется производство таблеток. Смазанную смесь затем прессуют для получения таблеток. В некоторых случаях на таблетки наносят непрозрачный или прозрачный защитный слой, состоящий из слоя шеллака, слоя сахара или полимерных материалов. К таким покрытиям можно добавить окрашивающие вещества для того, чтобы отличить данные таблетки от других.
Препарат в форме сиропа или эликсира может содержать активный компонент совместно с подслащивающим веществом, антисептическим средством, веществом, придающим вкус, а также подходящим окрашивающим веществом. Как правило, препараты в форме сиропа получают в результате растворения соединения в водном растворе с подходящим веществом, придающим вкус, тогда как эликсиры приготавливают с применением нетоксичного спиртового носителя.
Порошки или гранулы, которые могут быть диспергированы в воде, могут содержать активные компоненты, смешанные с диспергирующим веществом или со смачивающими веществами, или с суспендирующими веществами, такими как, например, этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбитола, а также с усилителями вкуса и аромата или подслащивающими веществами.
Для ректального введения прибегают к применению суппозиториев, которые приготавливают со связывающими веществами, плавящимися при ректальной температуре, например, с маслом какао или полиэтиленгликолями.
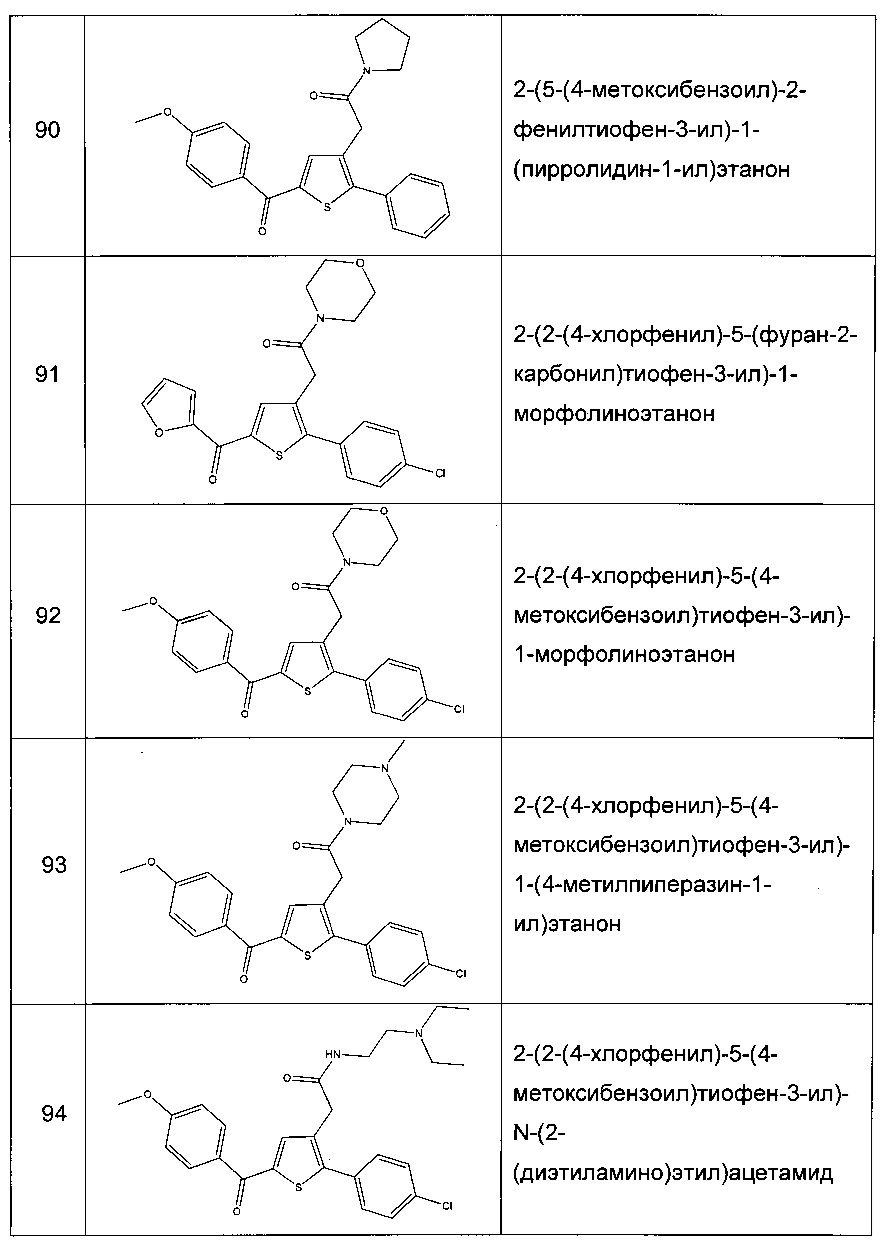
Для парентерального, интраназального или интраокулярного введения применяют водные суспензии, изотонические солевые растворы или стерильные и инъецируемые растворы, которые содержат фармакологически совместимые диспергирующие вещества и/или смачивающие вещества.
Активный компонент может быть также приготовлен в состав в форме микрокапсул, в некоторых случаях с одним или несколькими дополнительными носителями.
Фармацевтические композиции, приспособленные для введения местным путем, могут быть приготовлены в состав в форме крема, мази, суспензии, лосьона, порошка, раствора, пасты, геля, спрея, аэрозолей или масел.
Фармацевтические композиции, приспособленные для введения интраназальным путем, вспомогательный эксципиент в которых находится в твердом состоянии, включают порошки, размер частиц которых находится в диапазоне, например, от 20 до 500 микрон, и которые вводят посредством ингаляции из контейнера, содержащего порошок, причем указанный контейнер располагают напротив носа.
Фармацевтические составы, приспособленные для введения вагинальным путем, могут быть введены в форме буфера, крема, геля, пасты, пены или спрея.
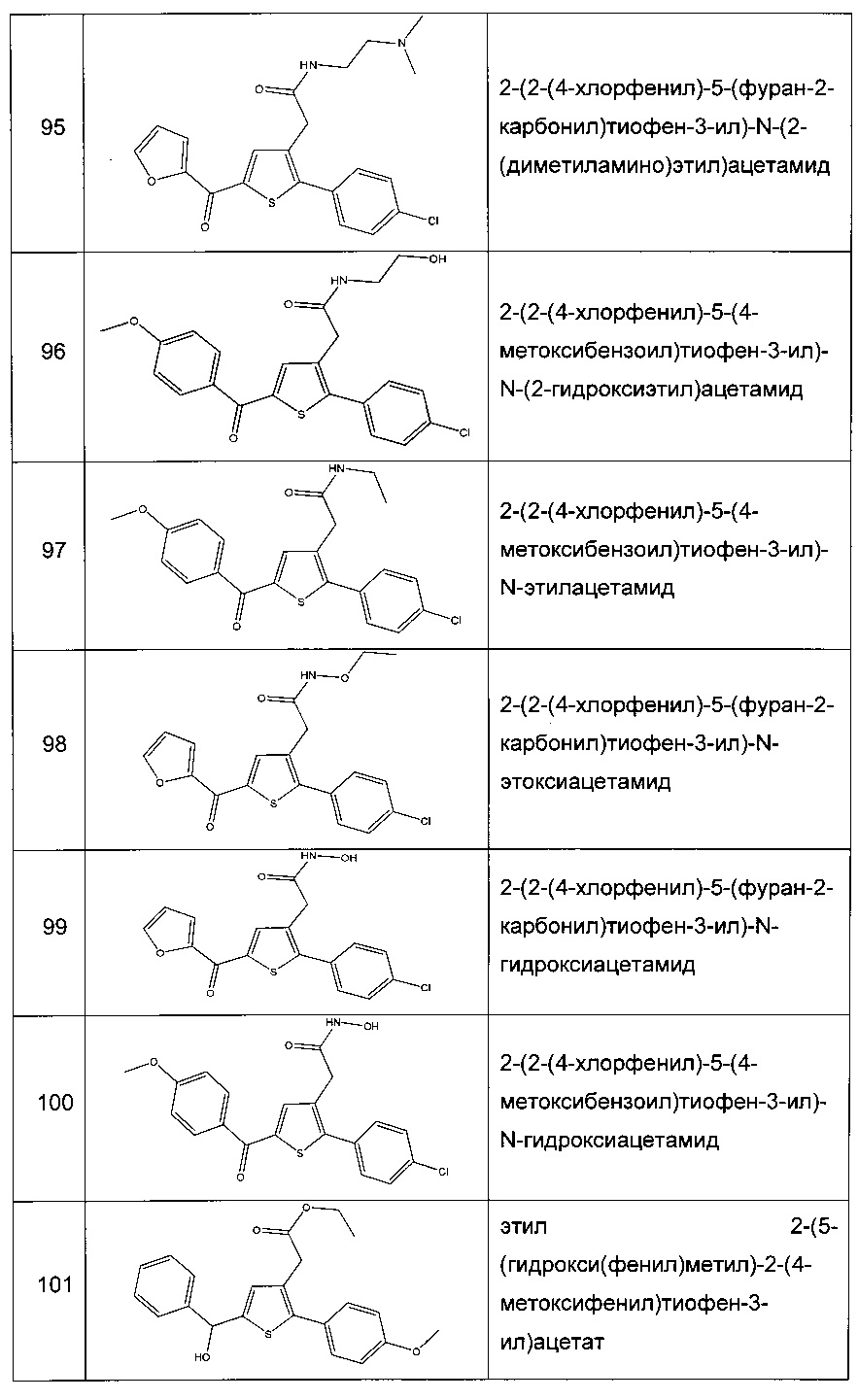
Согласно предпочтительному варианту реализации настоящего изобретения фармацевтическая композиция согласно настоящему изобретению дополнительно содержит другой активный компонент, предпочтительно обладающий комплементарным или синергетическим действием. В частности, данный активный агент представляет собой другой противодиабетический агент, который предпочтительно выбирают из инсулина, сульфонилмочевины, глинидов, бигуанидов, тиазолидиндионов, агонистов GLP-1R (рецептора глюкагоноподобного пептида-1), ингибиторов DPP-IV (дипептидилпептидазы-4), ингибиторов SGLT-2 (натрий-глюкозного котранспортера 2), предпочтительно выбирают из инсулина, глибенкламида, гликлазида, глипизида, глимепирида, репаглинида, натеглинида, метформина, троглитазона, розиглитазона, пиоглитазона, экзенатида, лираглутида, ситаглиптина, вилдаглиптина, саксаглиптина, алоглиптина, дапаглифлозина. Более конкретно, данный активный агент представляет собой метформин. Данный второй активный агент может быть введен в той же фармацевтической композиции, что и производное тиофена согласно настоящему изобретению. Данный второй активный агент может быть также введен отдельно, например, в то же время или способом, протяженным во времени. Предпочтительно, данный второй активный агент вводят пероральным путем.
Настоящее изобретение также относится к производному тиофена согласно настоящему изобретению или к указанному производному тиофена, которое выбирают из соединений формулы с (f) по (р), (r), (s), (u), (у) и (z), как определено выше, для применения в качестве лекарственного препарата. Более того, как указано выше, соединения с (f) по (р), (r), (s), (u), (у) и (z) являются коммерчески доступными, однако сообщения о какой-либо терапевтической активности, которой обладают данные соединения, отсутствуют.
Таким образом, вследствие этого данные соединения никогда не были раскрыты в качестве лекарственных препаратов.
Настоящее изобретение также относится к применению производного тиофена согласно настоящему изобретению или указанного производного тиофена, которое выбирают из соединений формул с (f) по (р), (r), (s), (u), (у) и (z), как определено выше, для приготовления лекарственного препарата.
Согласно настоящему изобретению соединения формулы (I) обладают гипогликемической активностью. Указанные соединения могут вызывать уменьшение гипергликемии, более конкретно, гипергликемии сахарного диабета II типа. В частности, соединения согласно настоящему изобретению обладают гипогликемической активностью и являются вследствие этого пригодными для лечения и/или предотвращения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, таких как, например, патологии, связанные с метаболическим синдромом, предпочтительно сахарного диабета II типа или гипергликемии. Данные лекарственные препараты являются особенно активными у лиц пожилого возраста. Под «лицами пожилого возраста» понимают лиц, мужчин или женщин, возраст которых составляет 65 лет или более.
Термин «резистентность к инсулину» в рамках настоящего изобретения относится к состоянию, при котором нормальное количество инсулина не может вызвать физиологический или нормальный молекулярный ответ.
Вследствие этого, настоящее изобретение относится к производному тиофена согласно настоящему изобретению или к указанному производному тиофена, которое выбирают из соединений формул с (а) по (z1), как определено выше, для применения в качестве лекарственного препарата, предназначенного для лечения и/или предотвращения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, предпочтительно сахарного диабета II типа и гипергликемии.
Более того, как указано выше, соединения формул с (а) по (z1) никогда не были раскрыты в качестве противодиабетических агентов.
Авторы настоящего изобретения обнаружили, что производные согласно настоящему изобретению дают возможность стимулировать секрецию инсулина клетками INS1 и ингибировать продукцию глюкозы печенью на изолированных гепатоцитах крысы.
Предпочтительно сахарный диабет выбирают из раннего, позднего, детского сахарного диабета, диабета лиц пожилого возраста и беременных женщин, в частности, лиц пожилого возраста. Предпочтительно негативные явления сахарного диабета и осложнения и/или патологии, связанные с сахарным диабетом, выбирают из гипергликемии, функциональных и количественных отклонений показателей эндокринных клеток поджелудочной железы от нормы, резистентности к инсулину, диабетической нейропатии, диабетической нефропатии, диабетической ретинопатии, воспаления, ожирения, гипертензии, сердечно-сосудистых, микрососудистых, нейрологических проблем, а также проблем с заживлением ран. Предпочтительно, данные негативные явления представляют собой гипергликемию, функциональные и количественные отклонения показателей эндокринных клеток поджелудочной железы от нормы, резистентность к инсулину и воспаление.
Предпочтительно пациент, который получает лечение, имеет факторы риска, связанные с сахарным диабетом, т.е. заболевания, возникновение которых прямо или опосредованно связано с появлением сахарного диабета. В частности, такие факторы риска включают семейный анамнез, гестационный сахарный диабет, избыточный вес, ожирение, недостаточную физическую нагрузку, гипертензию, высокое содержание триглицеридов, воспаление и гиперлипидемию.
Настоящее изобретение также относится к применению производного тиофена согласно настоящему изобретению или производного тиофена, которое выбирают из соединений формул с (а) по (z1), как определено выше, для приготовления лекарственного препарата, предназначенного для лечения и/или предотвращения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, в особенности сахарного диабета II типа и гипергликемии.
Наконец, настоящее изобретение относится к способу лечения и/или превентивного и/или профилактического лечения и/или лечения, направленного на замедление возникновения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, предпочтительно сахарного диабета II типа и гипергликемии, причем указанный способ включает введение эффективного количества производного тиофена согласно настоящему изобретению или производного тиофена, которое выбирают из соединений формул с (а) по (z1), как определено выше, пациенту, нуждающемуся в указанном соединении.
Эффективное количество можно корректировать в зависимости от природы и тяжести патологии, которую подвергают лечению, от пути введения, а также от веса и возраста пациента. Как правило, единица дозирования будет находиться в диапазоне от 0,5 мг до 2000 мг в сутки, которые принимают за один или несколько приемов, предпочтительно от 1 до 1000 мг.
Производные тиофена согласно настоящему изобретению получены способами, хорошо известными специалисту в данной области техники, и частично способами, описанными ниже.
Настоящее изобретение станет более понятным после прочтения описания и следующих примеров, которые приведены в качестве неограничивающего пояснения.
Описание синтеза и общие схемы
Соединения общей формулы (I) могут быть приготовлены в результате применения или адаптирования любого способа, известного в качестве такового специалисту в данной области техники и/или достижимого специалистом в данной области техники, в особенности способов, описанных автором Larock в руководстве Comprehensive Organic Transformations, VCH Pub., 1989, или в результате применения или адаптирования способов, описанных в нижеследующих процедурах.
Синтез молекул общей формулы (I) близок, иногда идентичен синтезу, который был описан в документах [2], [5], [9], [10], [11] и [12]; при этом приведенный перечень ссылок не следует рассматривать как исчерпывающий.
Различные группы R1-R8 и Y на схемах с 1 по 13 соответствуют определениям, данным ранее.
Схема 1: Образования кольца тиофена можно достичь в результате 3-х этапов, начиная от соответствующим образом замещенной 4-фенил-4-оксо бутановой кислоты; кислотную функцию этерифицируют в стандартных условиях, хлорформилирование проводят на этапе, предшествующем кристаллизации, в присутствии серы. Основным продуктом реакции является промежуточное соединение 1.4, образование которого сопровождается выделением побочного продукта 1.5. Затем 1.4 ацилируют или сульфонилируют в условиях реакции Фриделя-Крафтса; при омылении сложноэфирной функции данный процесс приводит к образованию продуктов типа 1.7 и 1.9, соответственно.
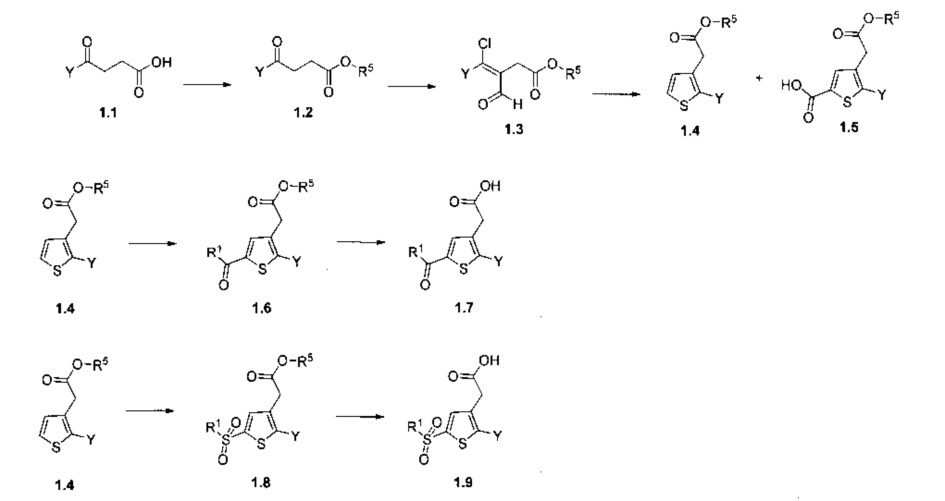
Смешанные сложные эфиры можно приготовить из кислот 1.7 или 1.9 согласно стандартным условиям этерификации или в результате приготовления промежуточного соединения хлорангидрида и последующей реакции ацилирования. Реакции переэтерификации из производных типа 1.6 или 1.8 можно также провести согласно стандартным условиям.
Схема 2: Приготовление амидов типа 2.1 осуществляли из производных 1.7 через промежуточное соединение хлорангидрид в результате проведения реакции ацилирования амина либо в условиях сочетания пептидов, таких как
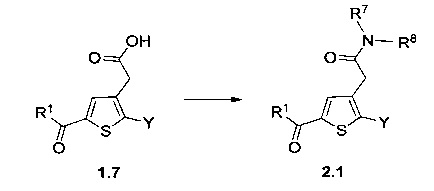
Схема 3: Восстановление кетона проводили в стандартных условиях восстановления, которые, как было доказано, являются селективными.
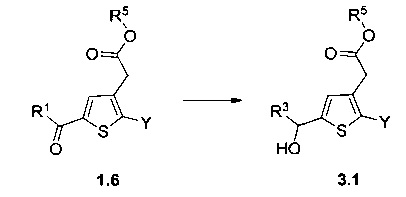
Схема 4: Приготовление оксимов проводили из производных 1.6.
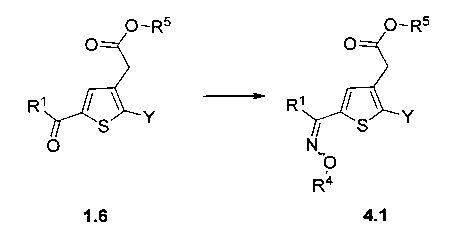
Схема 5: Производные формулы 1.5 использовали для приготовления амидов через этап образования промежуточного соединения хлорангидрида в результате реакции ацилирования амина либо в условиях сочетания пептидов, таких как EDC.HCl, HOBt или, помимо этого, РуВОР. А представляет собой арильную группу, (С1-С6 алкил)арильную группу или, помимо этого, группу арилСОNН.
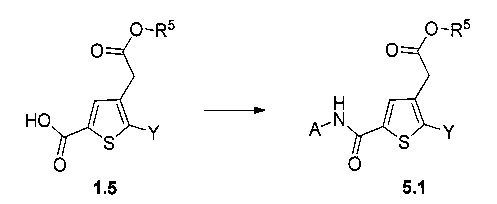
Схема 6: Замещенные по альфа-атому производные кислоты были приготовлены в результате депротонирования в щелочных условиях и последующего добавления электрофильного реактива. В случае, когда электрофильный реактив представляет собой бром(метокси)метан, в зависимости от условий эксперимента, реакция может привести к образованию акрилата типа 6.2.
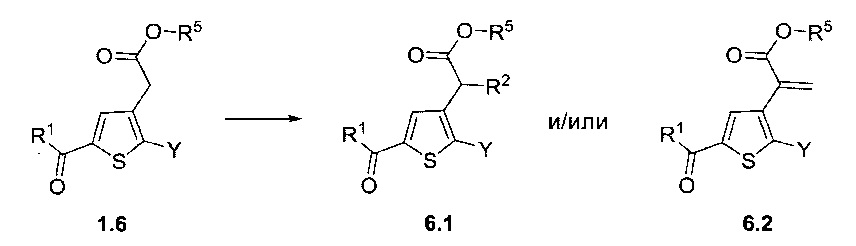
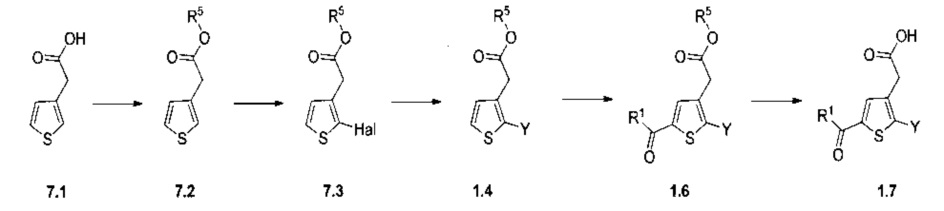
Схема 7: Специфичное введение определенных групп осуществляли в результате применения методов синтеза, альтернативных методу, описанному на схеме 1. Начиная с тиофенуксусной кислоты, проводили реакцию этерификации и последующей галогенизации, введение арильного или гетероарильного заместителей было возможно в результате применения превращения Сузуки для получения производных типа 1.4, которые затем обрабатывали тем же способом, что и ранее, т.е., вовлекали в реакцию ацилирования по типу Фриделя-Крафтса и затем проводили омыление с получением производных 1.7.
Hal представляет собой атом галогена, предпочтительно бром.
Схема 8: Данная процедура может быть в определенных случаях упрощена; производные типа 8.1 приготовили в результате ацилирования производных типа 7.2, после чего полученные производные вовлекали в реакции арилирования, катализируемые палладием, с использованием производных иодфенила с получением производных типа 1.6.
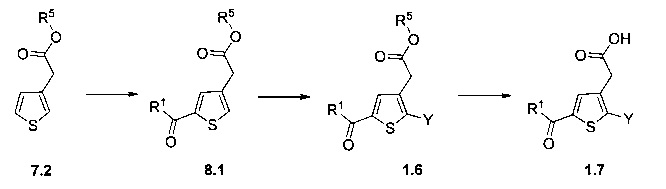
Схема 9: В альтернативном методе приготовления соединений формулы I применяли 2-боронат-3-метилпроизводное тиофена, которое вовлекали в реакции Сузуки. Производные типа 9.2 ацилировали согласно условиям по типу Фриделя-Крафтса. Получение производных 1.6 проводили в результате галогенизации, цианирования и последующего спиртового гидролиза производных 9.3.
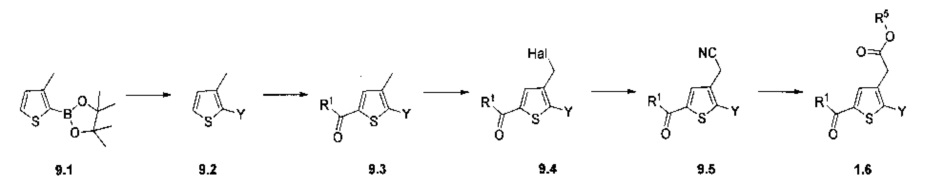
Схема 10: Для введения в соединение конкретных групп был разработан другой метод синтеза, альтернативный методам, описанным ранее. Начиная с метил 5-бром-4-метилтиофен-2-карбоксилата, проводили реакцию омыления с получением соответствующей кислоты для приготовления амида Вайнреба. В присутствии органометаллического соединения типа галоген-магний из производных типа 10.3 получали производные типа 10.4, которые затем вовлекали в реакции Сузуки с последующей галогенизацией и цианированием с получением производных типа 10.7.
Последние вовлекали в реакции спиртового гидролиза с получением сложных эфиров типа 1.6.
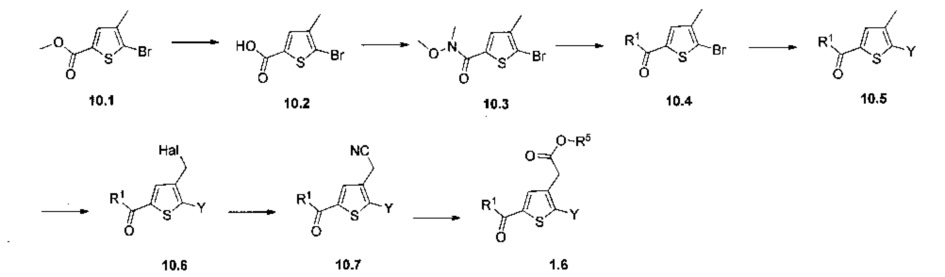
Схема 11: Введение заместителей в кислотную функцию требует другого метода синтеза, в котором в качестве исходного продукта использовали 2-(тиофен-3-ил)ацетонитрил; данный продукт вовлекали в реакции галогенизации и последующего сочетания типа Сузуки, катализируемого палладием, с получением производных типа 11.3. Полученные производные ацилировали согласно условиям реакции Фриделя-Крафтса с получением производных типа 10.7.
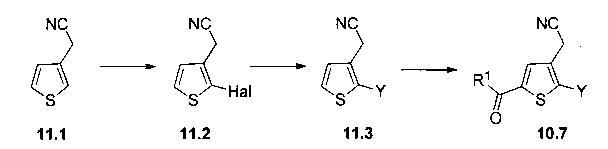
Схема 12: Производные типа 10.7 подвергали обработке азидом с получением тетразольных производных 12.1.
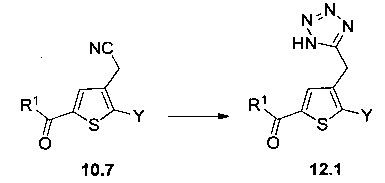
Схема 13: Производные типа 10.7 подвергали обработке гидроксиламином с получением открытых производных типа 13.1, которые затем вовлекали в реакции карбонилирования в присутствии карбонилдиимидазола с получением оксадиазолонов типа 13.2.
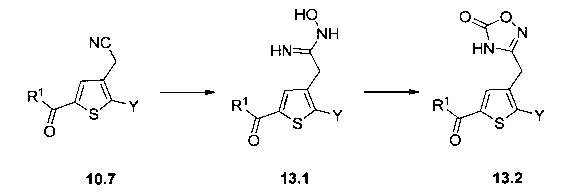
Схема 14: Производные типа 1.4 подвергали гидролизу в щелочных условиях с получением промежуточных соединений - производных кислоты типа 14.1, которые затем вовлекали в реакции сочетания пептидов, в которых могут быть получены производные типа 14.2. Последние обрабатывали реактивом Лавессона из соответствующих тиоамидов 14.3. Наконец, реакция ацилирования по типу Фриделя-Крафтса позволяла получить соединения типа 14.4.
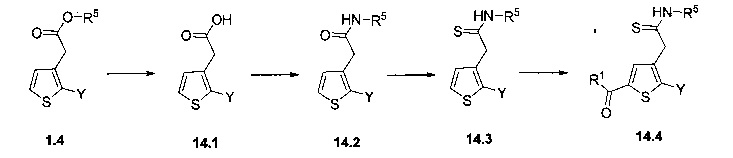
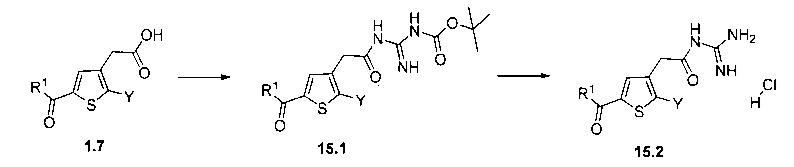
Схема 15: Условия сочетания пептидов в присутствии гуанидин-Вос приводят к образованию из производных 1.7 производных типа 15.1, из которых затем в условиях кислотного гидролиза можно получить незащищенные соединения 15.2.
Примеры:
Оборудование и методы
Спектр протонного (1Н) ядерного магнитного резонанса (ЯМР) получали на приборе Bruker Avance DPX300 (300,16 МГц). Химический сдвиг (δ) измеряли в миллионных долях (м.д.). Спектры калибровали по химическому сдвигу используемого дейтерированного растворителя. Константы взаимодействия (J) выражены в Герцах (Гц); мультиплетность представлена следующим образом: синглет (с), дублет (д), дублет-дублет (дд), триплет (т), триплет-дублет (тд), квадруплет (к), мультиплет (м). Масс-спектры (МС) получали на спектрометре Agilent Technologies MSD типа G1946A, образцы ионизировали с помощью источника «Химической ионизации при атмосферном давлении» (APCl, Atmospheric pressure chemical ionization).
Сокращения

Ряд примеров, приведенных ниже, используется для иллюстрации объема настоящего изобретения, но не для ограничения области применения настоящего изобретения.
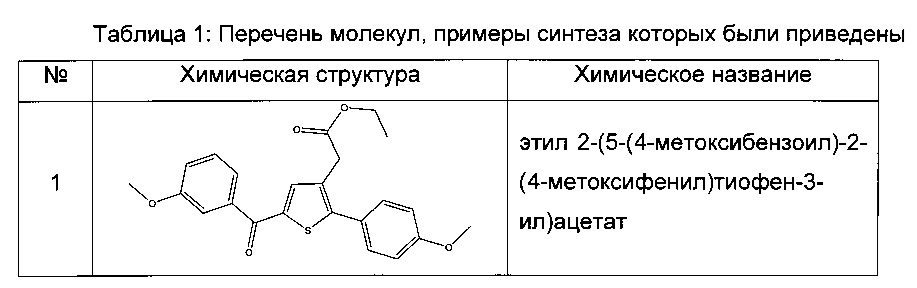
Пример 1: Приготовление производного №1: этил 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетат
Этап 1: Приготовление этил 4-метоксифенил-4-оксобутаноата
 50 г (240 ммоль) 4-метоксифенил-4-оксобутановой кислоты растворяли в 320 мл этанола, к раствору добавляли 0,64 мл (12 ммоль) серной кислоты. Смесь нагревали в колбе с обратным холодильником в течение 16 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан). 54,04 г (выход=95%) этил 4-метоксифенил-4-оксобутаноата получали в виде бесцветного масла. ЖХ/МС (жидкостная хроматография - масс-спектрометрия): m/z=237 (МН+), степень чистоты (УФ-детекция при 254 нм)=84%. 1Н ЯМР (300 МГц, DMSO) δ 7,97 (д, J=8,9 Гц, 2Н), 7,05 (д, J=8,9 Гц, 2Н), 4,05 (к, J=7,1 Гц, 2Н), 3,85 (с, 3Н), 3,30-3,18 (м, 2Н), 2,69-2,56 (м, 2Н), 1,18 (т, J=7,1 Гц, 3Н).
50 г (240 ммоль) 4-метоксифенил-4-оксобутановой кислоты растворяли в 320 мл этанола, к раствору добавляли 0,64 мл (12 ммоль) серной кислоты. Смесь нагревали в колбе с обратным холодильником в течение 16 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан). 54,04 г (выход=95%) этил 4-метоксифенил-4-оксобутаноата получали в виде бесцветного масла. ЖХ/МС (жидкостная хроматография - масс-спектрометрия): m/z=237 (МН+), степень чистоты (УФ-детекция при 254 нм)=84%. 1Н ЯМР (300 МГц, DMSO) δ 7,97 (д, J=8,9 Гц, 2Н), 7,05 (д, J=8,9 Гц, 2Н), 4,05 (к, J=7,1 Гц, 2Н), 3,85 (с, 3Н), 3,30-3,18 (м, 2Н), 2,69-2,56 (м, 2Н), 1,18 (т, J=7,1 Гц, 3Н).
Этап 2: Приготовление этил (Z/Е)-4-хлор-3-формил-4-(4-метоксифенил) бут-3-еноата
 54 8 г (228 ммоль) этил 4-метоксифенил-4-оксобутаноата солюбилизировали в 52,9 мл диметилформамида (683 ммоль), в полученный раствор медленно добавляли 53,1 мл (569 ммоль) фосфорилтрихлорида, реакция была высоко экзотермической. Полученную смесь нагревали до температуры 80°С в течение 3 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь выливали на 1 л смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×200 мл этилацетата. Объединенные органические фазы промывали 300 мл воды, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан). 57 г (выход=75%) этил (Z/Е)-4-хлор-3-формил-4-(4-метоксифенил)бут-3-еноата получали в виде оранжевого масла. ЖХ/МС: m/z=283 (МН+), степень чистоты (УФ-детекция при 254 нм)=93%. 1Н ЯМР (300 МГц, DMSO) δ 9,37 (с, 1Н), 7,50 (д, J=8,8 Гц, 2Н), 7,08 (д, J=8,8 Гц, 2Н), 4,12 (д, J=7,1 Гц, 2Н), 3,84 (с, 3Н), 3,55 (с, 2Н), 1,19 (т, J=7,1 Гц, 3Н).
54 8 г (228 ммоль) этил 4-метоксифенил-4-оксобутаноата солюбилизировали в 52,9 мл диметилформамида (683 ммоль), в полученный раствор медленно добавляли 53,1 мл (569 ммоль) фосфорилтрихлорида, реакция была высоко экзотермической. Полученную смесь нагревали до температуры 80°С в течение 3 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь выливали на 1 л смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×200 мл этилацетата. Объединенные органические фазы промывали 300 мл воды, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан). 57 г (выход=75%) этил (Z/Е)-4-хлор-3-формил-4-(4-метоксифенил)бут-3-еноата получали в виде оранжевого масла. ЖХ/МС: m/z=283 (МН+), степень чистоты (УФ-детекция при 254 нм)=93%. 1Н ЯМР (300 МГц, DMSO) δ 9,37 (с, 1Н), 7,50 (д, J=8,8 Гц, 2Н), 7,08 (д, J=8,8 Гц, 2Н), 4,12 (д, J=7,1 Гц, 2Н), 3,84 (с, 3Н), 3,55 (с, 2Н), 1,19 (т, J=7,1 Гц, 3Н).
Этап 3: Приготовление этил 2-(2-(4-метоксифенил)тиофен-3-ил)ацетата
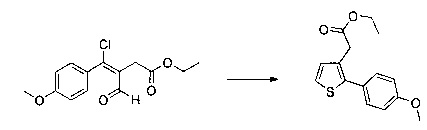 57 г (189 ммоль) этил (Z/Е)-4-хлор-3-формил-4-(4-метоксифенил)бут-3-еноата солюбилизировали в 400 мл тетрагидрофурана. К раствору добавляли 19,71 мл (284 ммоль) 2-меркапто-уксусной кислоты и 79 мл (567 ммоль) триэтиламина. Полученную смесь нагревали в колбе с обратным холодильником в течение 6 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме. Осадок переносили в 200 мл диметилформамида, полученную смесь нагревали до температуры 130°С в течение 2 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь обрабатывали 600 мл воды. Водную фазу экстрагировали 2×200 мл этилацетата. Объединенные органические фазы промывали 2×200 мл воды, 300 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: циклогексан/дихлорметан, 3/1, об./об. (объем/объем)). 29,09 г (выход=53%) этил 2-(2-(4-метоксифенил)тиофен-3-ил)ацетата получали в виде бесцветного масла. ЖХ/МС: m/z=277 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,46 (д, J=5,2 Гц, 1Н), 7,36 (д, J=8,7 Гц, 2Н), 7,03 (т, J=6,5 Гц, 3Н), 4,07 (к, J=7,1 Гц, 2Н), 3,79 (с, 3Н), 3,62 (с, 2Н), 1,16 (т, J=7,1 Гц, 3Н). Обратите внимание: в примере 15 (этап 3) описана обработка, отличная от приведенной выше, которая позволяет выделить побочный продукт типа 1.5, представленный на схеме 1.
57 г (189 ммоль) этил (Z/Е)-4-хлор-3-формил-4-(4-метоксифенил)бут-3-еноата солюбилизировали в 400 мл тетрагидрофурана. К раствору добавляли 19,71 мл (284 ммоль) 2-меркапто-уксусной кислоты и 79 мл (567 ммоль) триэтиламина. Полученную смесь нагревали в колбе с обратным холодильником в течение 6 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме. Осадок переносили в 200 мл диметилформамида, полученную смесь нагревали до температуры 130°С в течение 2 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь обрабатывали 600 мл воды. Водную фазу экстрагировали 2×200 мл этилацетата. Объединенные органические фазы промывали 2×200 мл воды, 300 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: циклогексан/дихлорметан, 3/1, об./об. (объем/объем)). 29,09 г (выход=53%) этил 2-(2-(4-метоксифенил)тиофен-3-ил)ацетата получали в виде бесцветного масла. ЖХ/МС: m/z=277 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,46 (д, J=5,2 Гц, 1Н), 7,36 (д, J=8,7 Гц, 2Н), 7,03 (т, J=6,5 Гц, 3Н), 4,07 (к, J=7,1 Гц, 2Н), 3,79 (с, 3Н), 3,62 (с, 2Н), 1,16 (т, J=7,1 Гц, 3Н). Обратите внимание: в примере 15 (этап 3) описана обработка, отличная от приведенной выше, которая позволяет выделить побочный продукт типа 1.5, представленный на схеме 1.
Этап 4: Приготовление этил 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетата (производное №1)
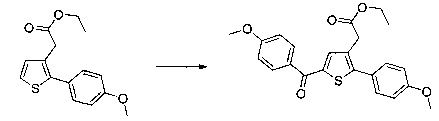 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 5 мл дихлорметана, 0,5 г (1,809 ммоль) этил 2-(2-(4-метоксифенил)тиофен-3-ил)ацетата и 0,367 мл (2,71 ммоль) 4-метоксибензоил хлорида. Затем смесь помещали в условия с температурой 5°С при перемешивании магнитной мешалкой, к смеси добавляли по частям 0,362 г (2,71 ммоль) хлорида алюминия. Полученную смесь перемешивали при к.т. в течение 5 д, после чего выливали на лед и перемешивали в течение 1 ч. Водную фазу экстрагировали 2×20 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент циклогексана/дихлорметана, от 100% до 0% циклогексана, об./об.). 0,652 г (выход=87%) этил 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетата получали в виде бледно-коричневого масла. ЖХ/МС: m/z=411 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 7,87 (д, J 8,8 Гц, 2Н), 7,70 (с, 1Н), 7,46 (д, J=8,8 Гц, 2Н), 7,10 (дд, J=13,7, 8,9 Гц, 4Н), 4,06 (к, J=7,1 Гц, 2Н), 3,87 (с, 3Н), 3,81 (с, 3Н), 3,74 (с, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 5 мл дихлорметана, 0,5 г (1,809 ммоль) этил 2-(2-(4-метоксифенил)тиофен-3-ил)ацетата и 0,367 мл (2,71 ммоль) 4-метоксибензоил хлорида. Затем смесь помещали в условия с температурой 5°С при перемешивании магнитной мешалкой, к смеси добавляли по частям 0,362 г (2,71 ммоль) хлорида алюминия. Полученную смесь перемешивали при к.т. в течение 5 д, после чего выливали на лед и перемешивали в течение 1 ч. Водную фазу экстрагировали 2×20 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент циклогексана/дихлорметана, от 100% до 0% циклогексана, об./об.). 0,652 г (выход=87%) этил 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетата получали в виде бледно-коричневого масла. ЖХ/МС: m/z=411 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 7,87 (д, J 8,8 Гц, 2Н), 7,70 (с, 1Н), 7,46 (д, J=8,8 Гц, 2Н), 7,10 (дд, J=13,7, 8,9 Гц, 4Н), 4,06 (к, J=7,1 Гц, 2Н), 3,87 (с, 3Н), 3,81 (с, 3Н), 3,74 (с, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
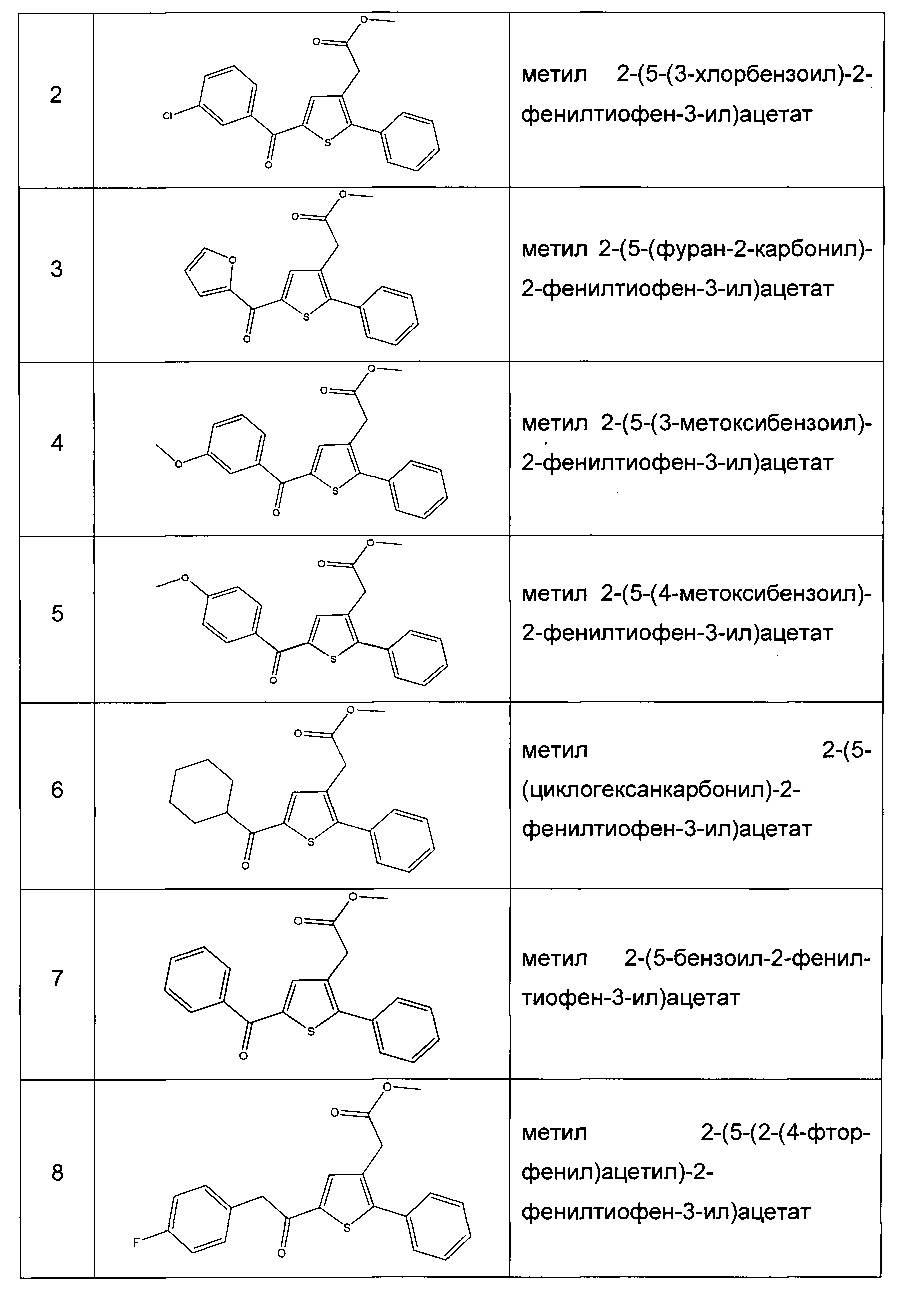
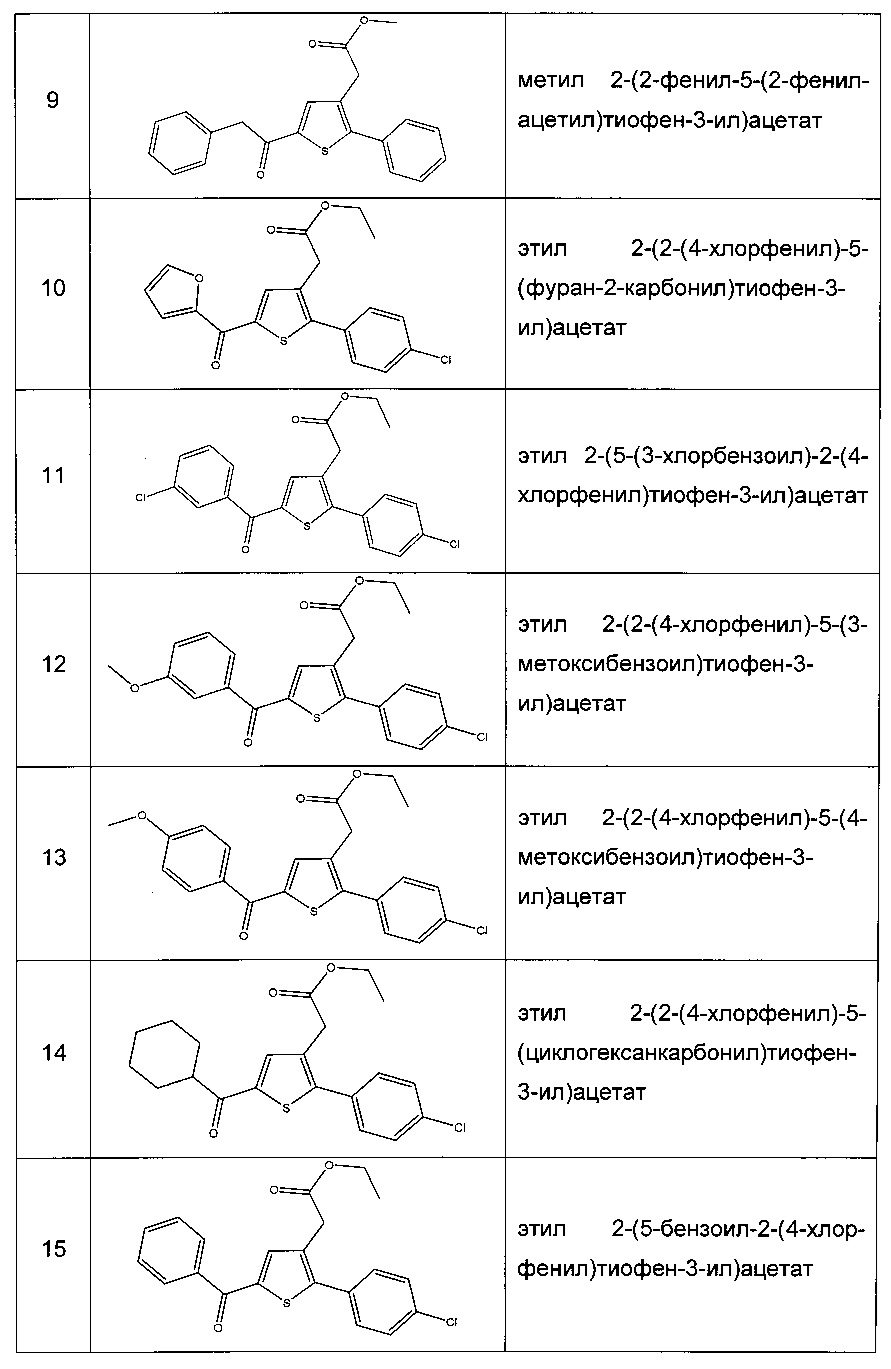
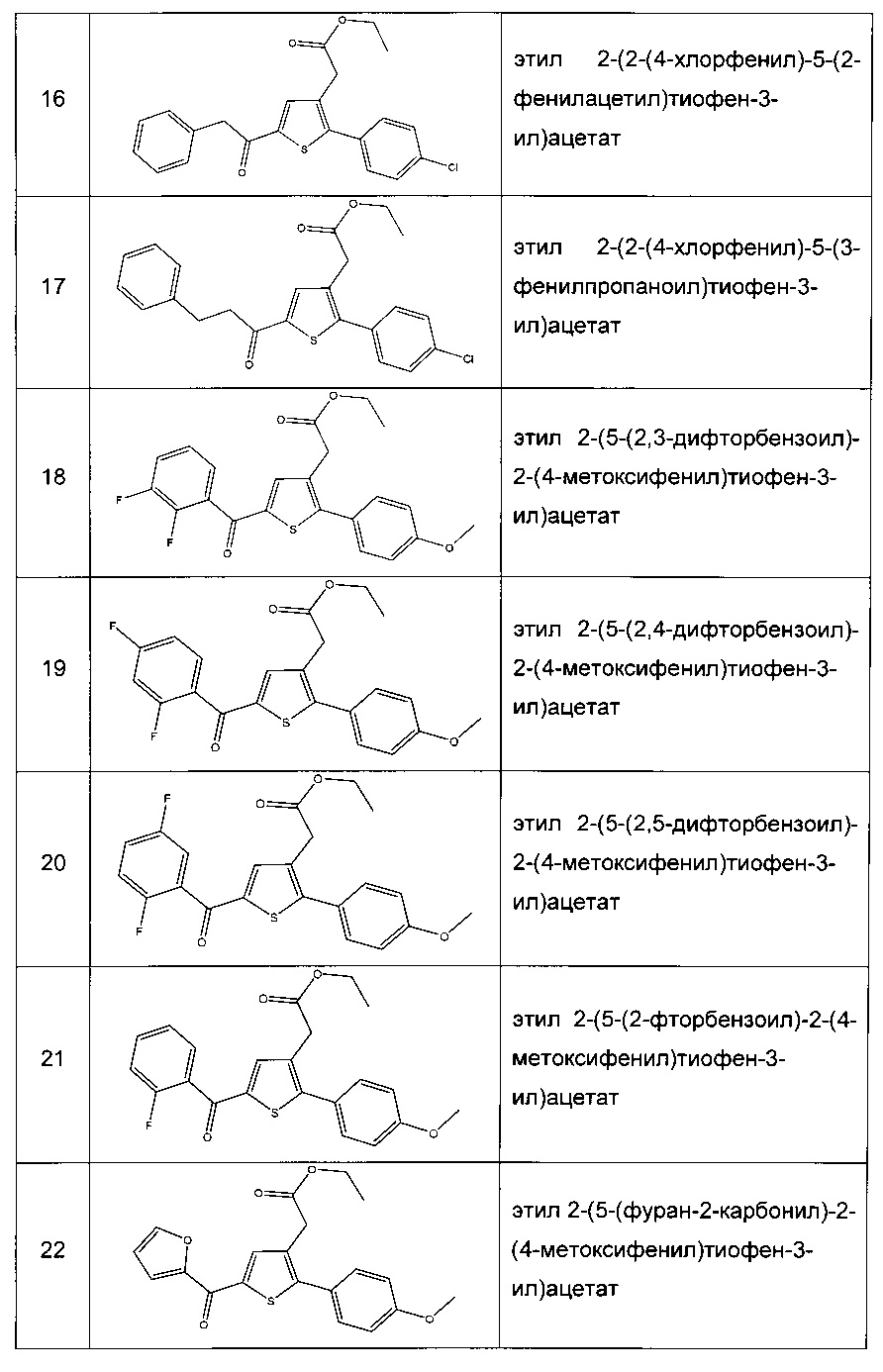
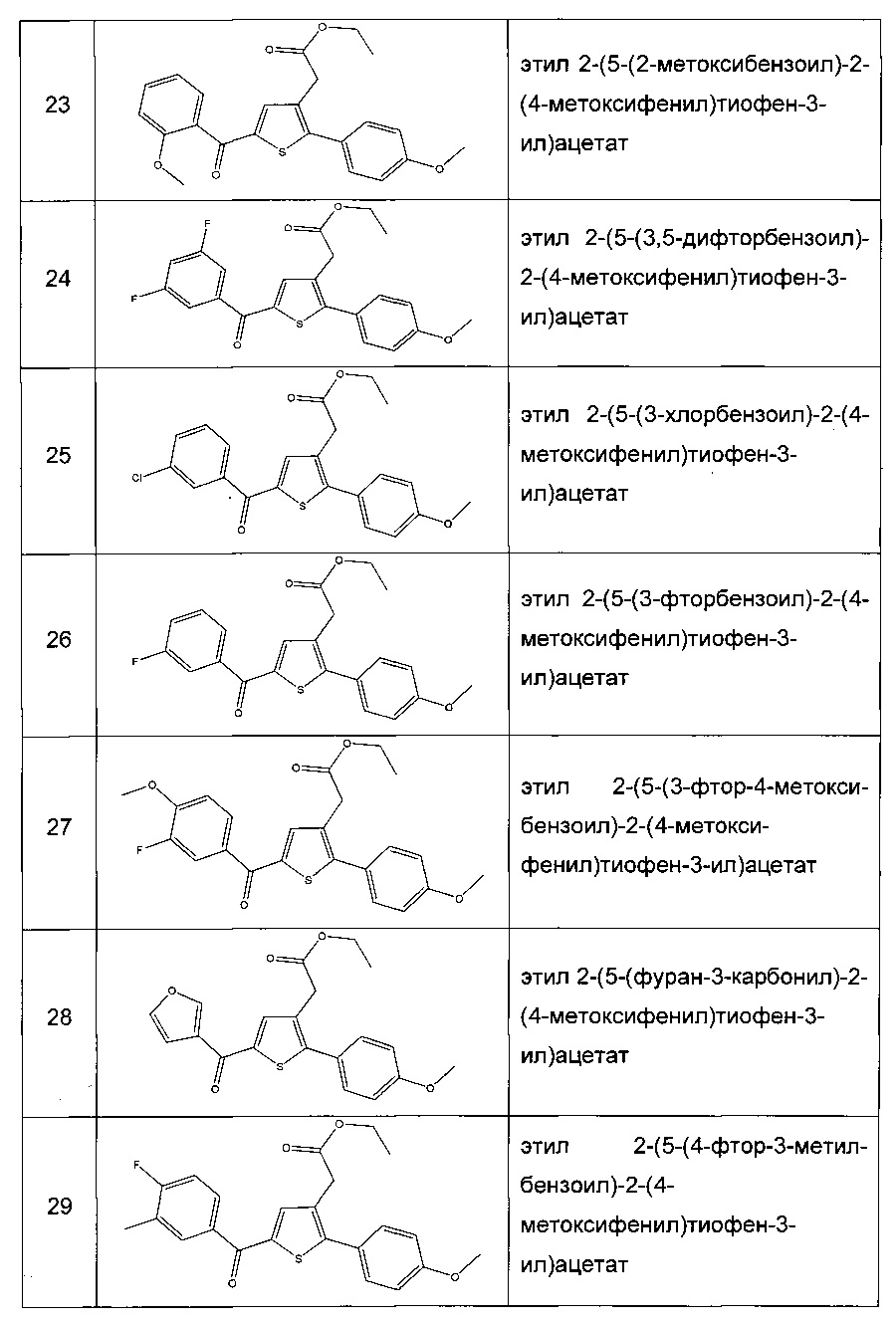
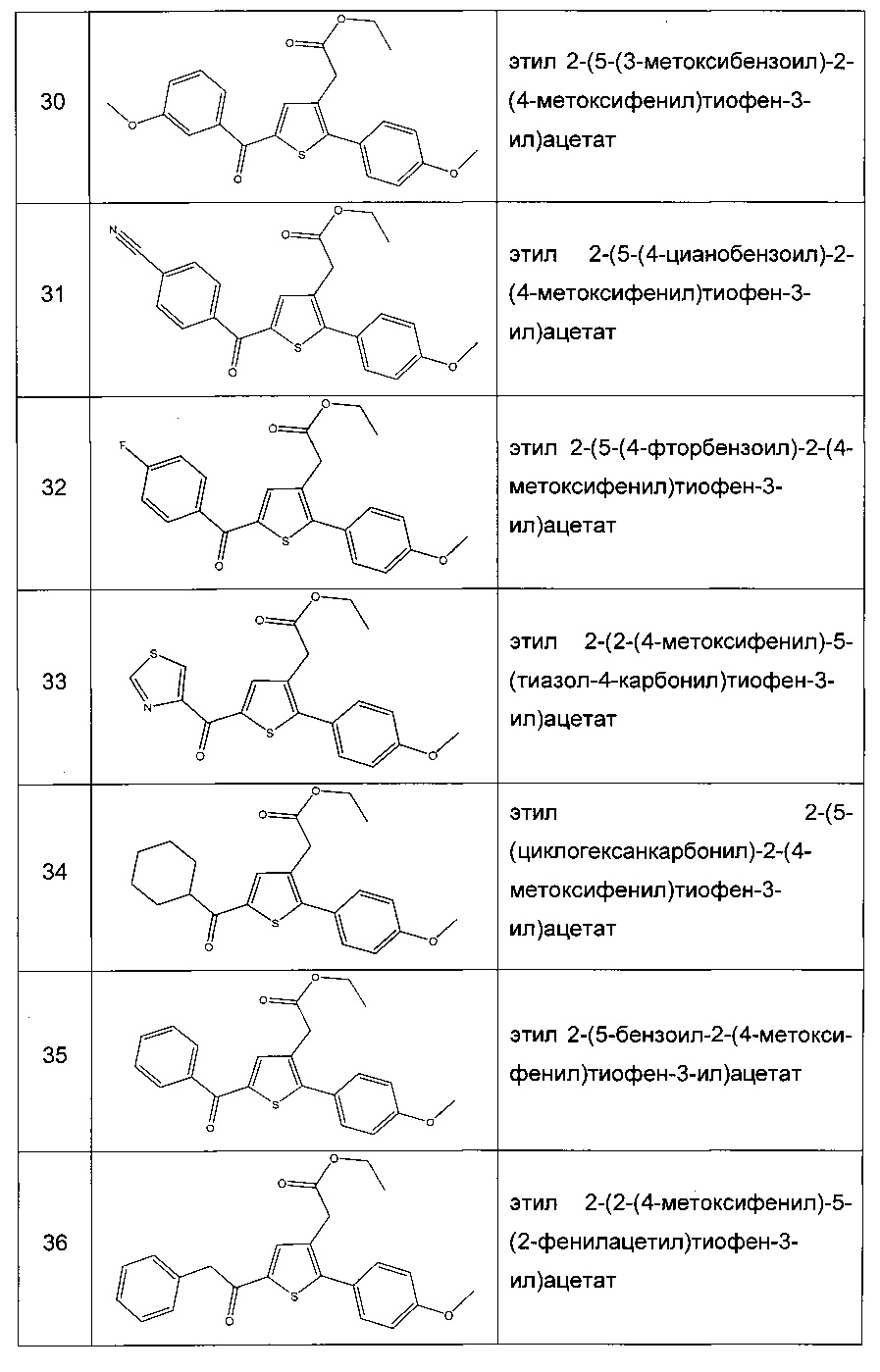
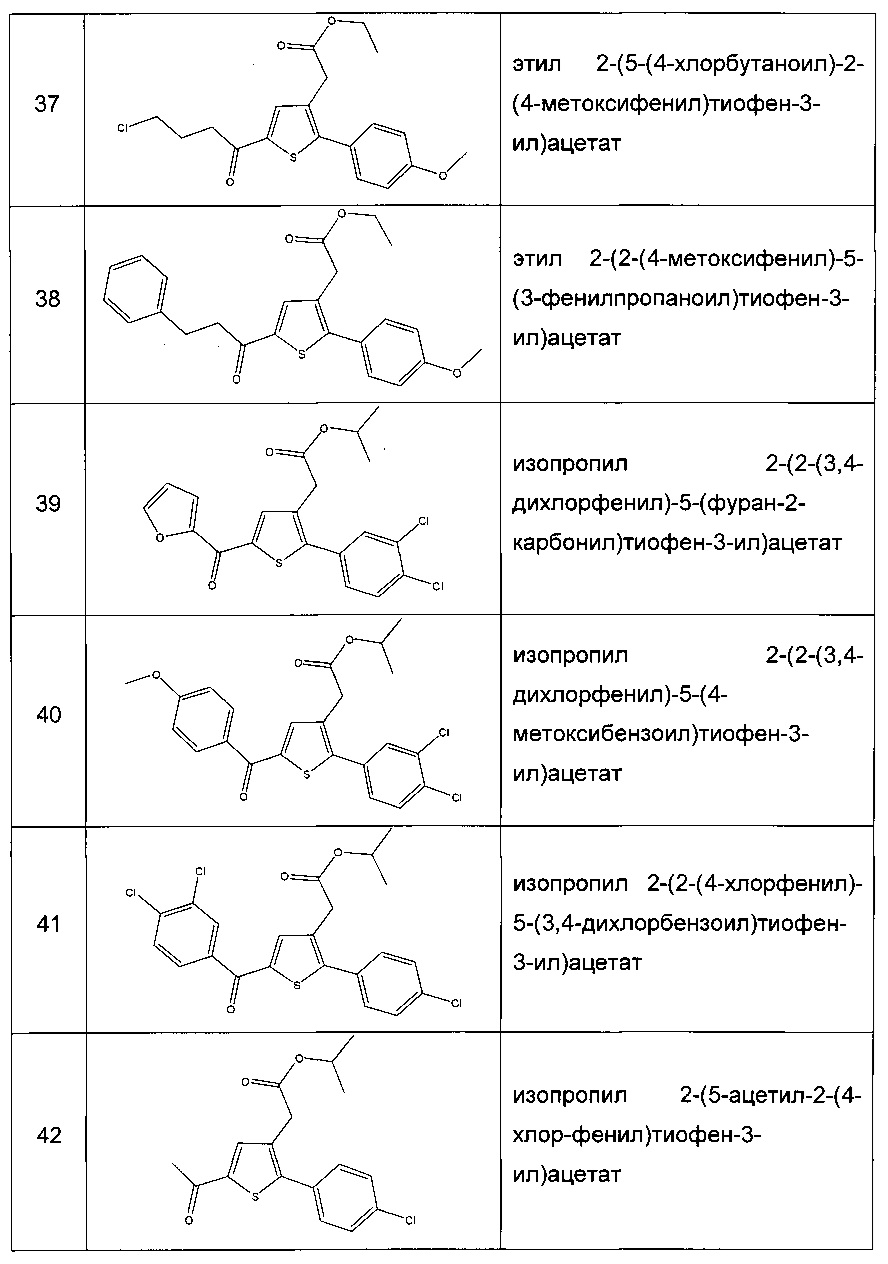
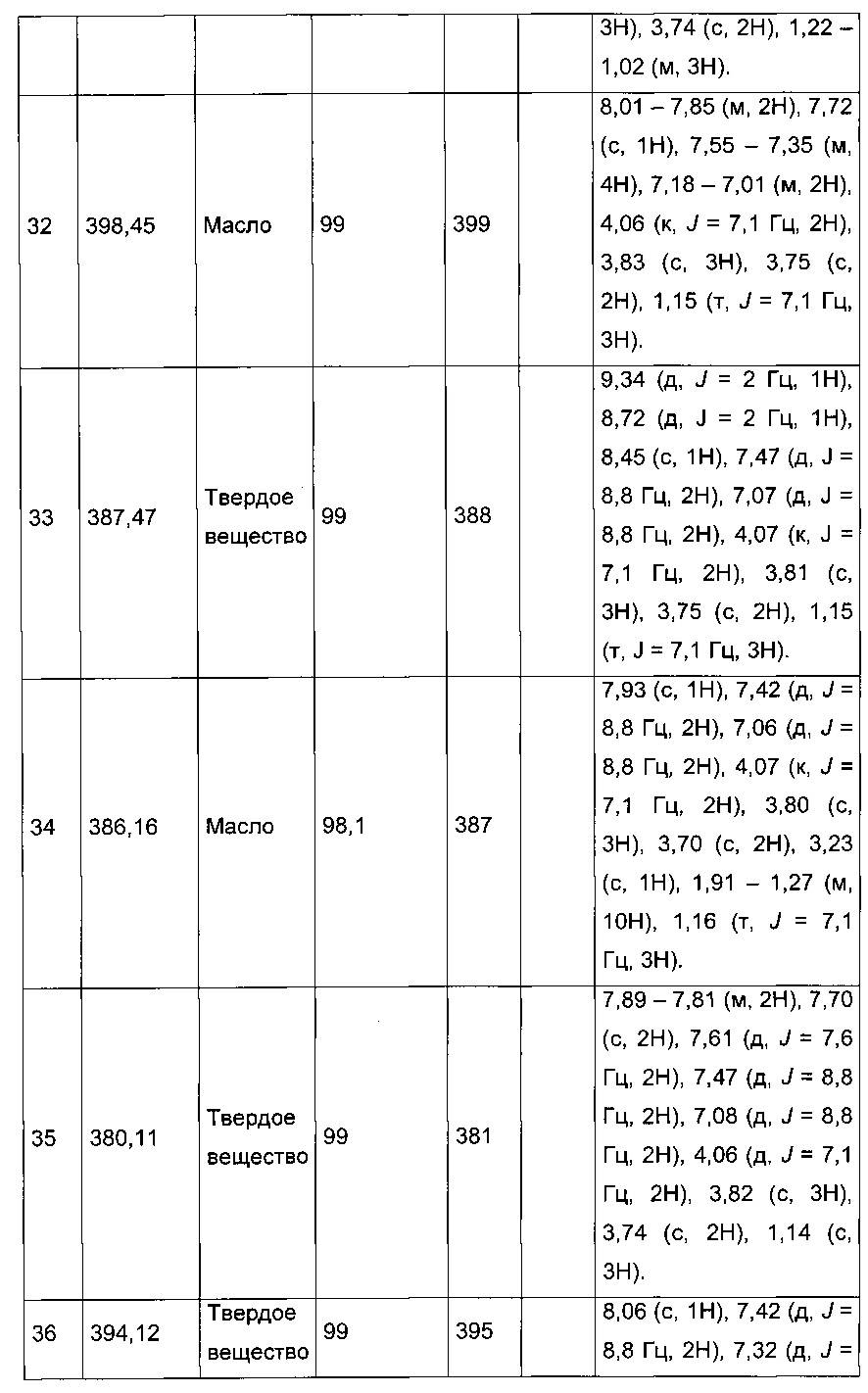
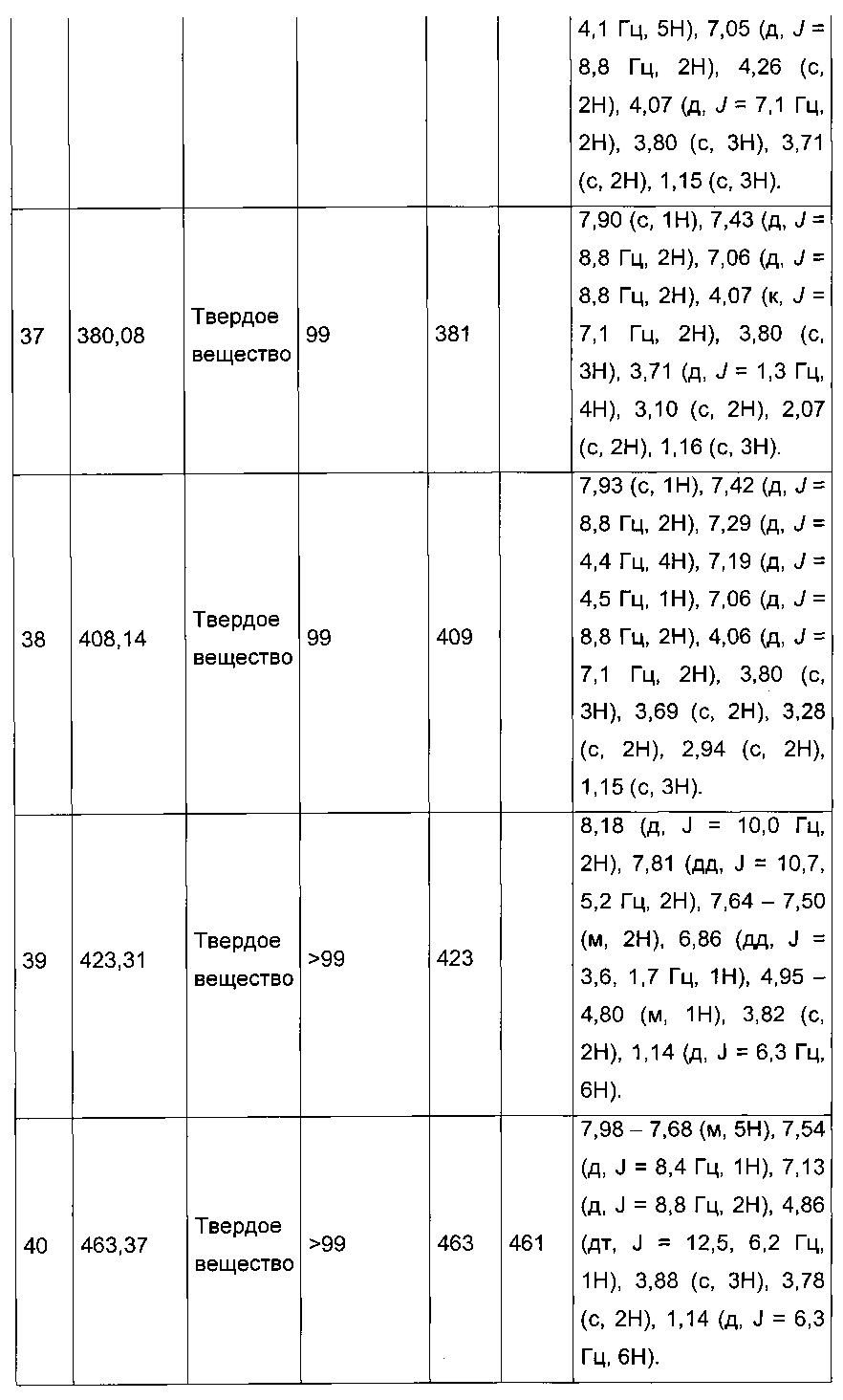
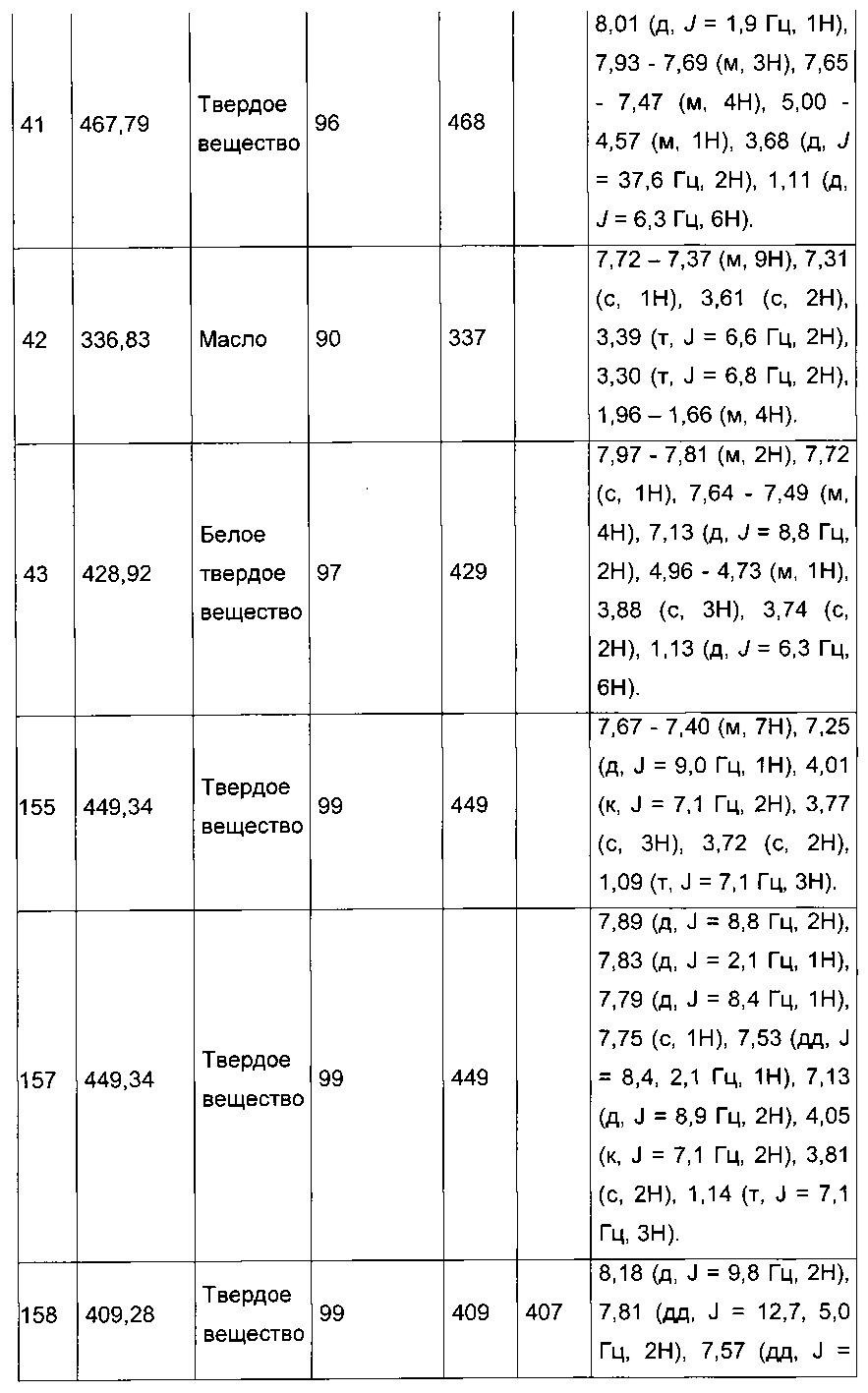
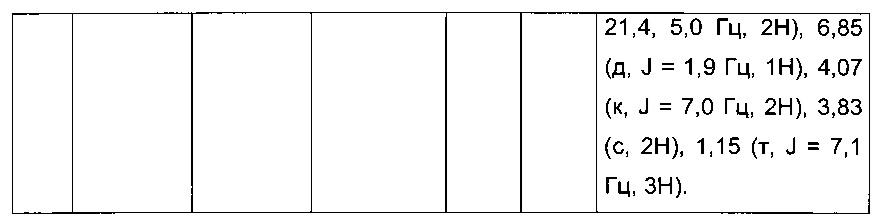
Производные с 2 по 43, 155, 157 и 158 были приготовлены согласно той же последовательности этапов с 1 по 4:
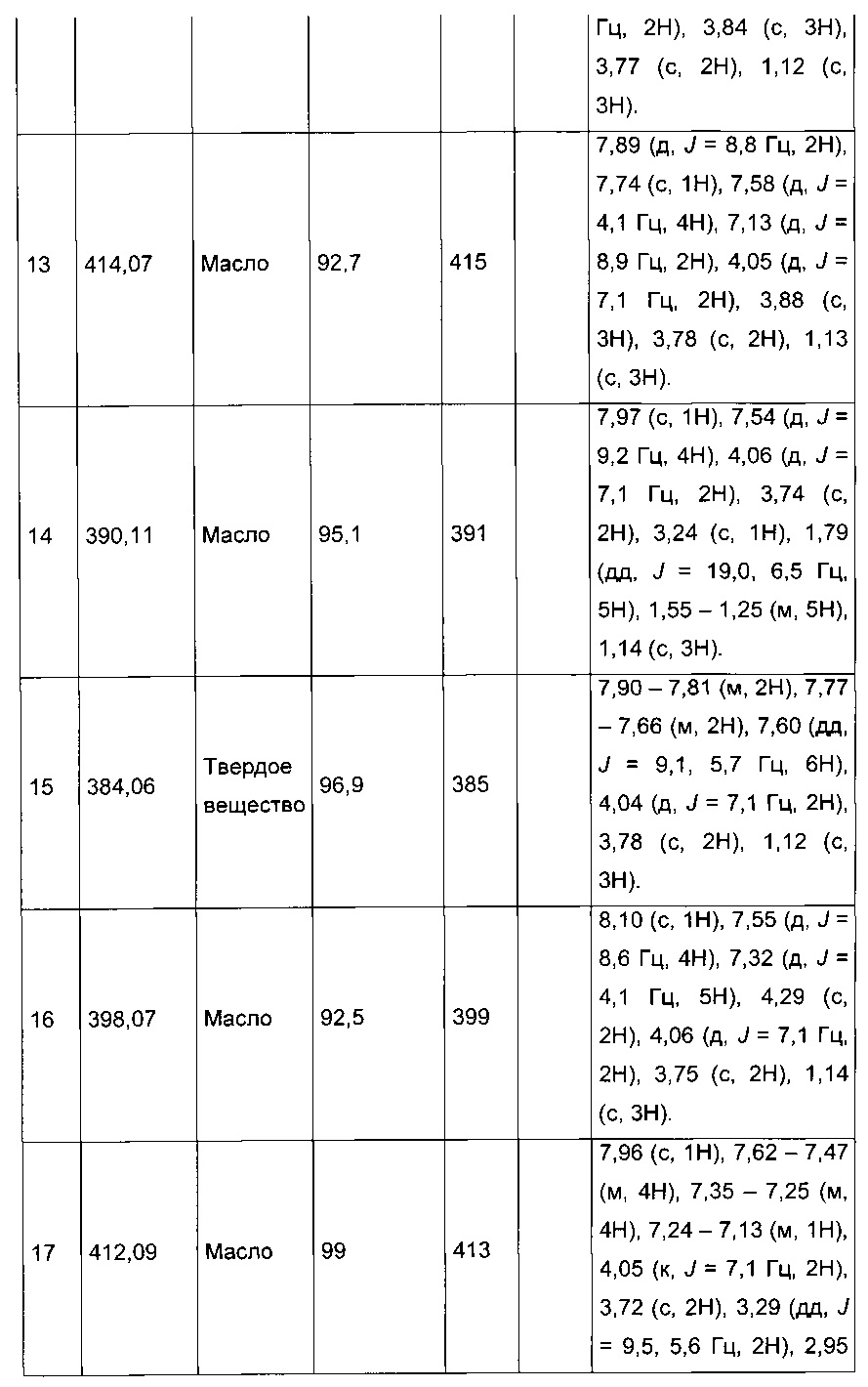
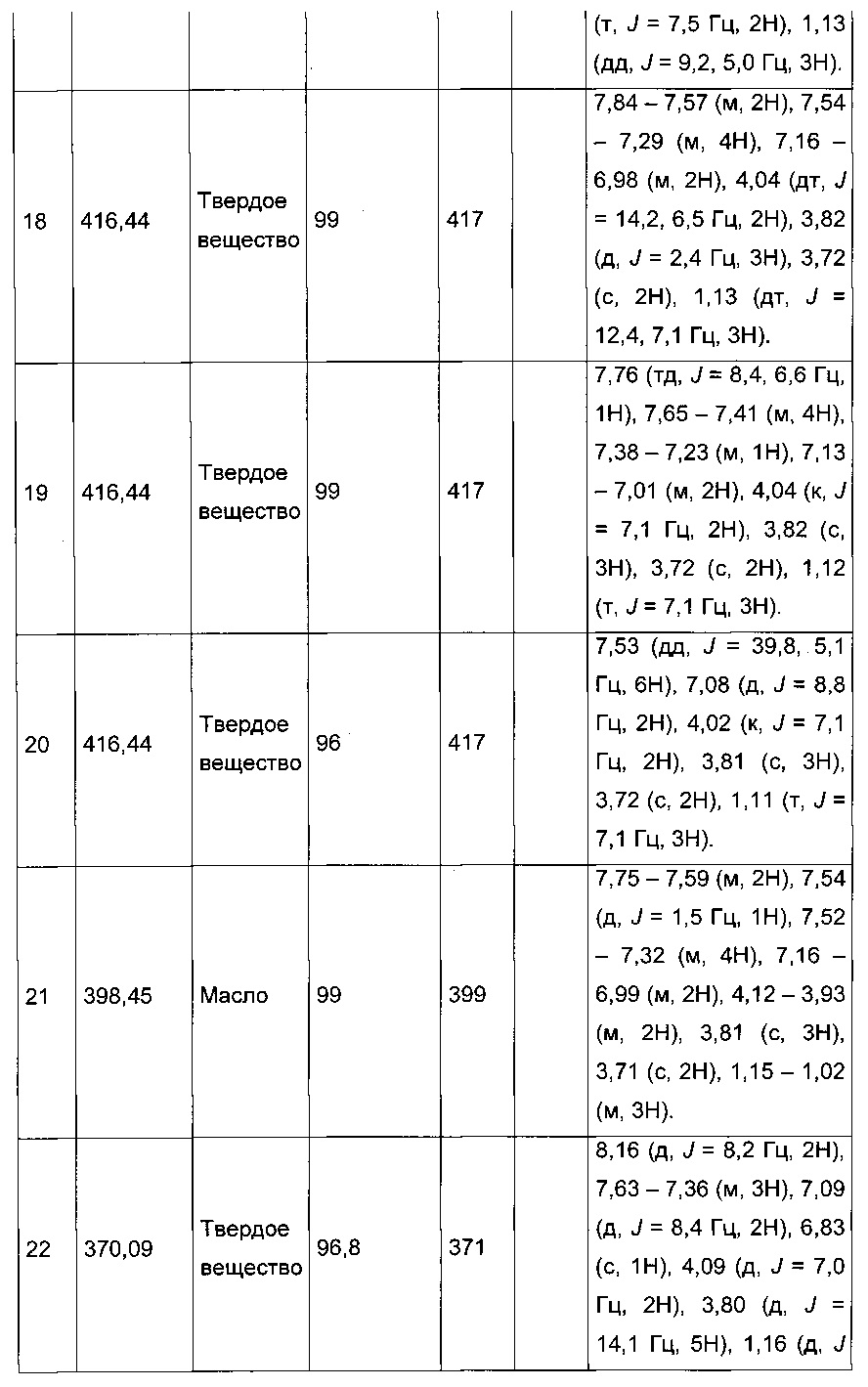
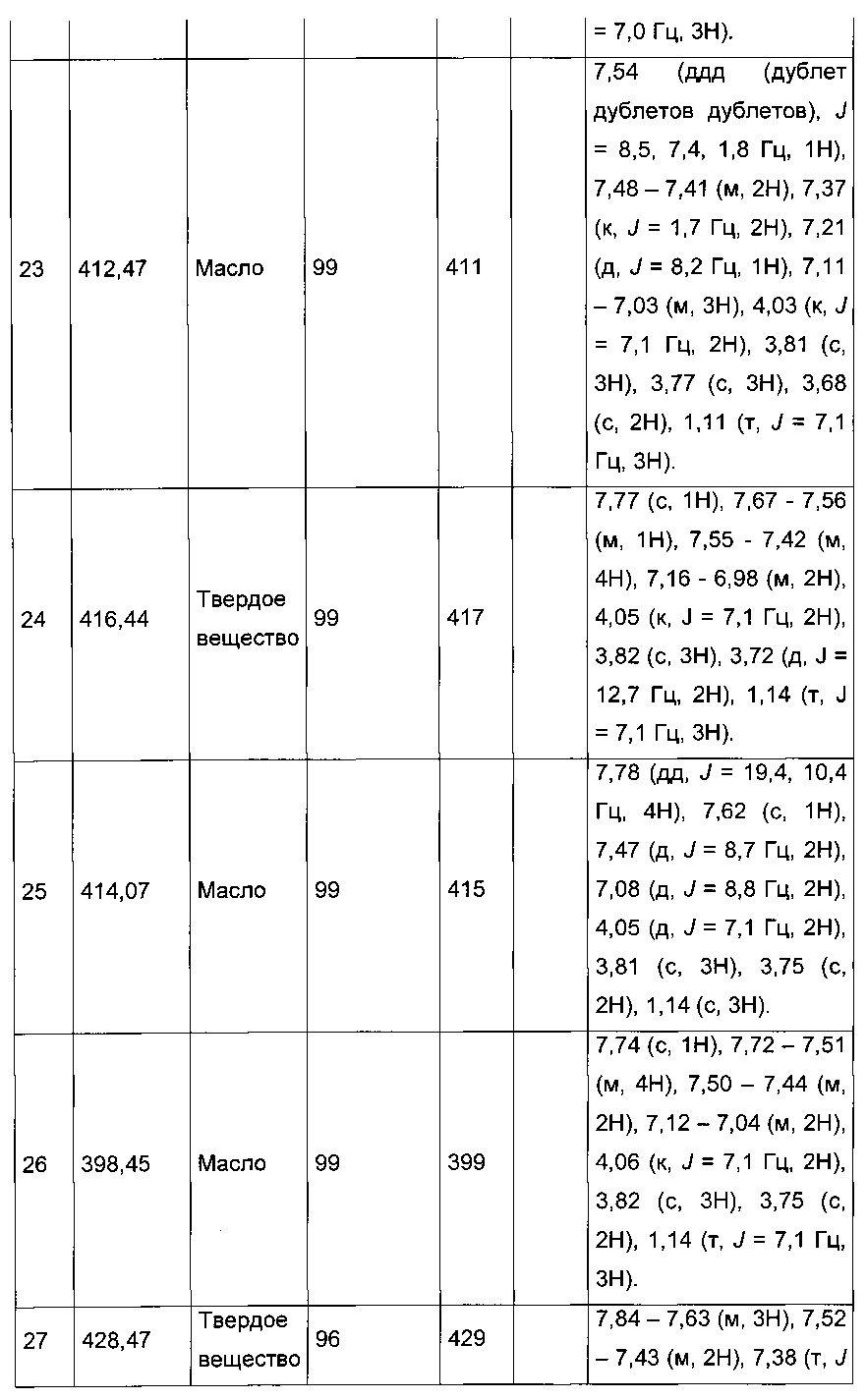
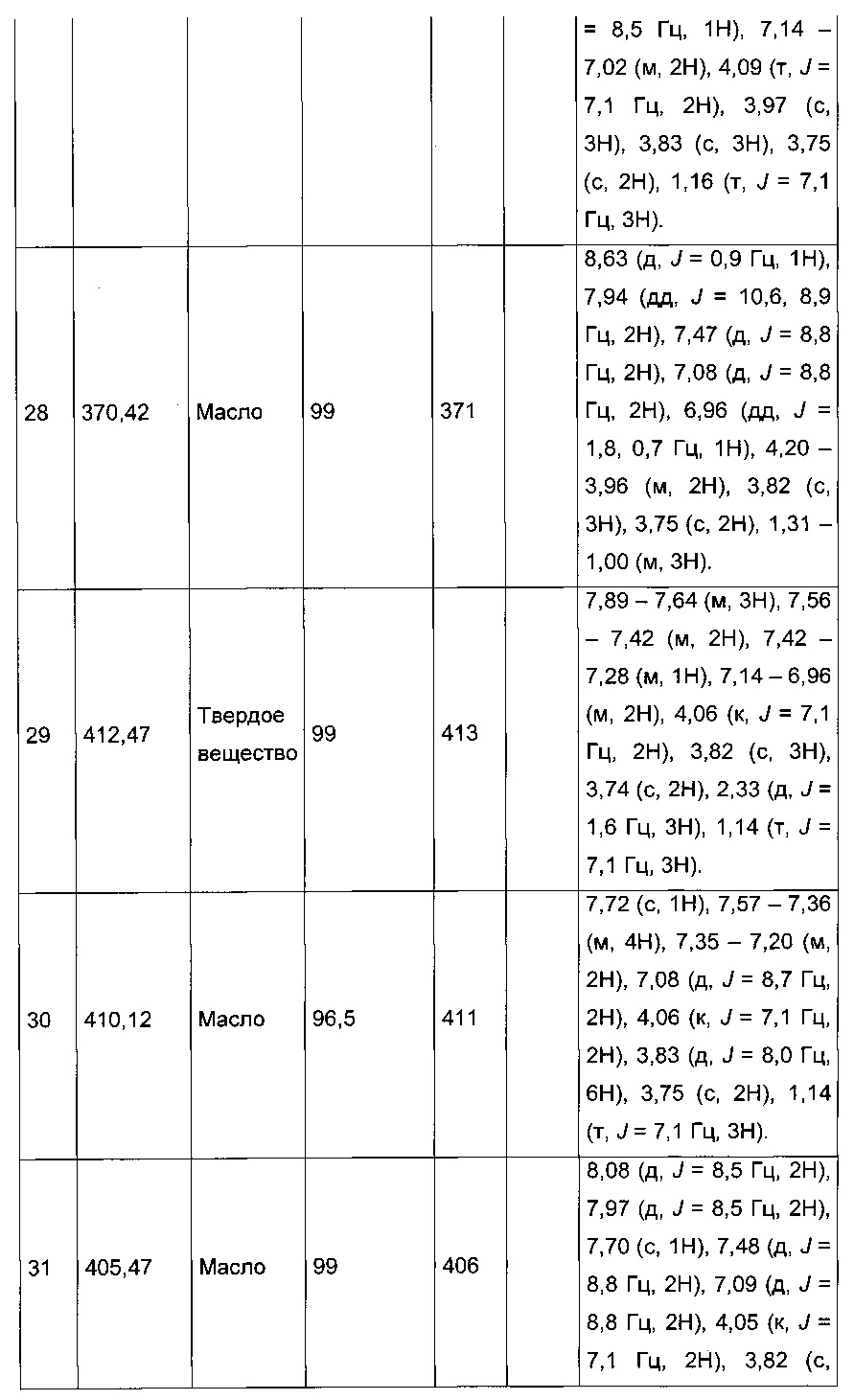
Пример 2: Приготовление производного №44: изопропил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетат
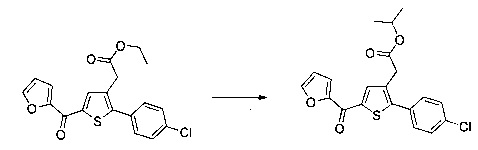 200 мг (0,518 ммоль) этил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 5 ммоль 2-пропанола, добавляли каплю серной кислоты, полученную смесь перемешивали магнитной мешалкой в колбе с обратным холодильником в течение 17 ч. Смеси позволяли остыть до к.т., после чего твердому веществу давали возможность преципитировать в реакционную среду. Твердое вещество выделяли в результате фильтрации и высушивали в вакуумном колпаке с получением 110 мг (выход=54%) изопропил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата в виде белого порошка. ЖХ/МС: m/z=389 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,15 (дд, J=6,5, 5,5 Гц, 2Н), 7,58 (д, J=2,4 Гц, 5Н), 6,84 (дд, J=3,6, 1,7 Гц, 1Н), 5,00-4,76 (м, 1Н), 3,77 (с, 2Н), 1,15 (д, J=6,3 Гц, 6Н).
200 мг (0,518 ммоль) этил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 5 ммоль 2-пропанола, добавляли каплю серной кислоты, полученную смесь перемешивали магнитной мешалкой в колбе с обратным холодильником в течение 17 ч. Смеси позволяли остыть до к.т., после чего твердому веществу давали возможность преципитировать в реакционную среду. Твердое вещество выделяли в результате фильтрации и высушивали в вакуумном колпаке с получением 110 мг (выход=54%) изопропил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата в виде белого порошка. ЖХ/МС: m/z=389 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,15 (дд, J=6,5, 5,5 Гц, 2Н), 7,58 (д, J=2,4 Гц, 5Н), 6,84 (дд, J=3,6, 1,7 Гц, 1Н), 5,00-4,76 (м, 1Н), 3,77 (с, 2Н), 1,15 (д, J=6,3 Гц, 6Н).
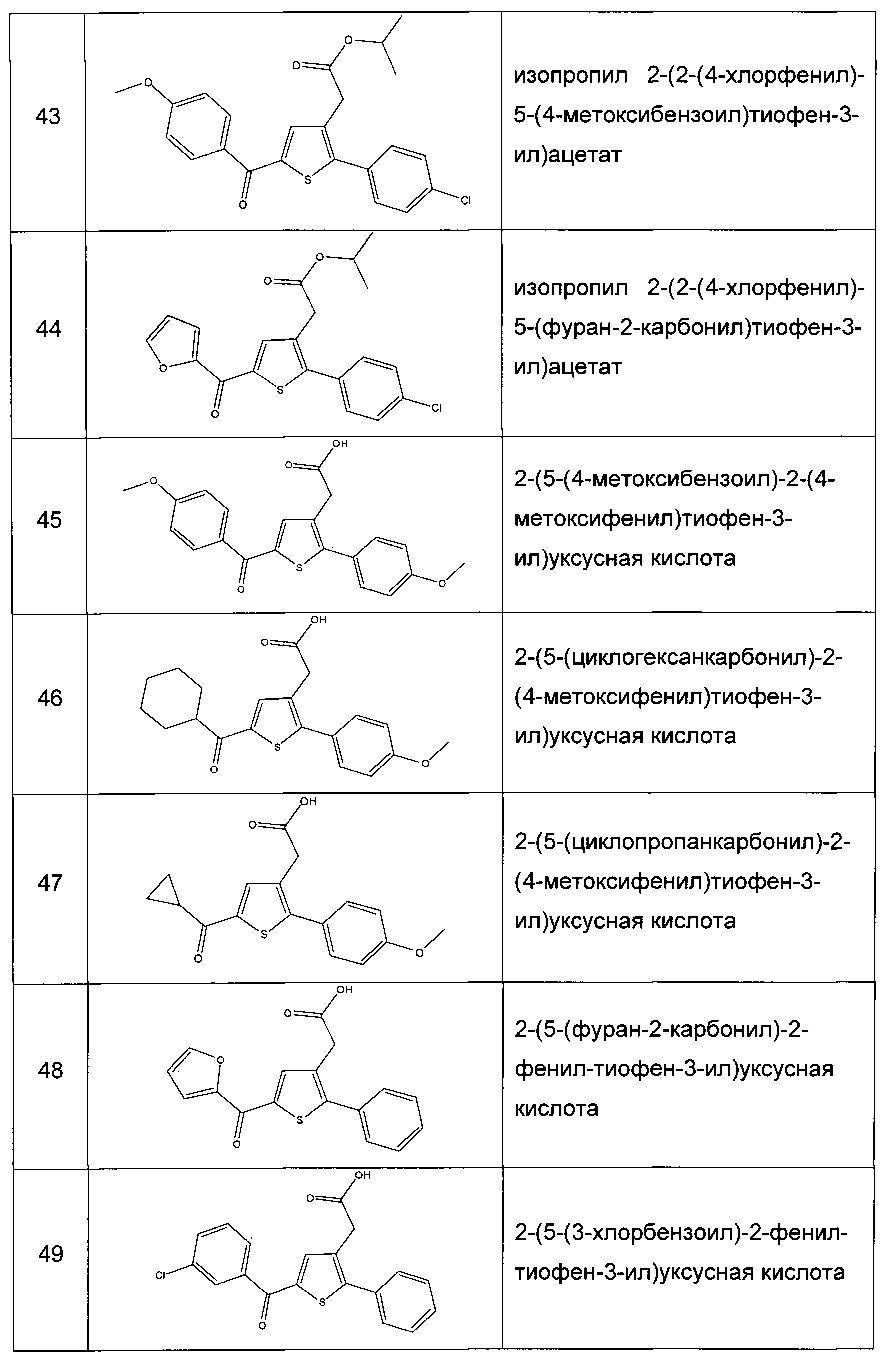
Пример 3: Приготовление производного номер 45: 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)уксусная кислота
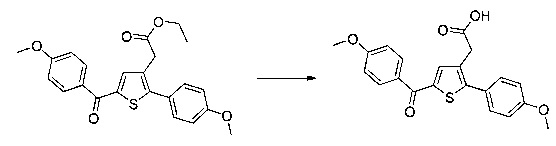
0,616 г (1,503 ммоль) этил 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетата солюбилизировали в 5 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 0,301 мл (3,01 ммоль) 30 масс. % водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу экстрагировали 2×5 мл этилацетата. Затем значение рН водной фазы снижали путем добавления водного раствора 1 н хлористоводородной кислоты до появления преципитата. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и высушивали в вакуумном колпаке с получением 0,536 г (выход=92%) 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=383 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,66 (с, 1Н), 7,87 (д, J=8,8 Гц, 2Н), 7,69 (с, 1Н), 7,48 (д, J=8,7 Гц, 2Н), 7,10 (дд, J=12,0, 8,8 Гц, 4Н), 3,87 (с, 3Н), 3,81 (с, 3Н), 3,64 (с, 2Н).
Производные с 46 по 81, 154, 156 и 159 были приготовлены согласно аналогичной процедуре:

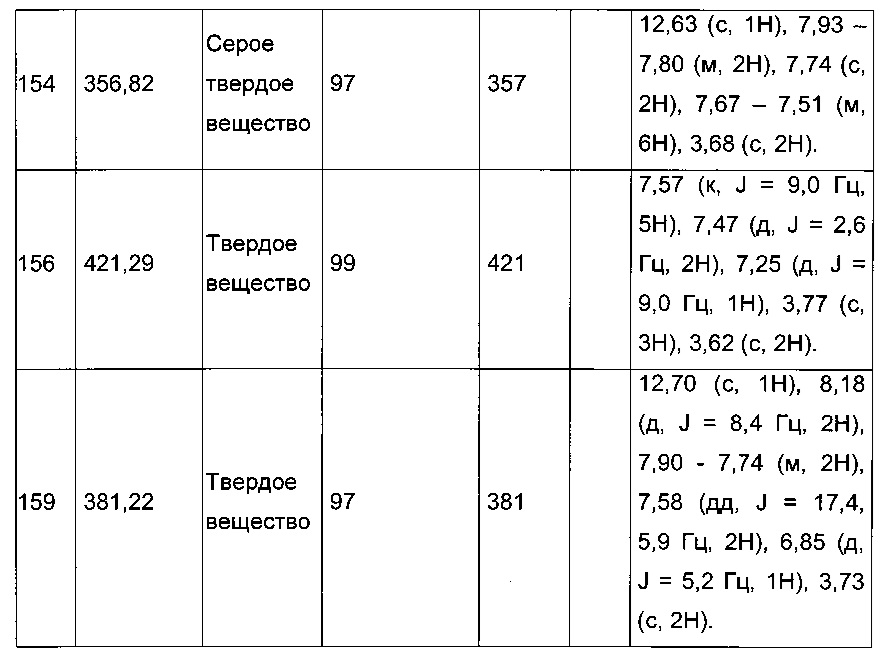
Пример 4: Приготовление производного №82: натрий 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетат
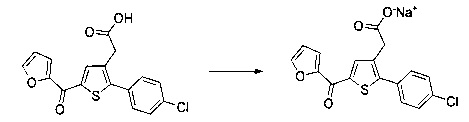 0,21 г (0,606 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в 3 мл метанола, к раствору добавляли при перемешивании магнитной мешалкой 0,112 мл (0,606 ммоль) 30 масс. % раствора метоксида натрия. Полученную смесь перемешивали при к.т. в течение 1 ч. Смесь обрабатывали 10 мл воды, метанол выпаривали в вакууме. Оставшуюся водную фазу лиофилизировали с получением 0,222 г (выход=97%) натрий 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата в виде желтого твердого вещества. ЖХ/МС: m/z=347 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,13 (дд, J=3,4, 2,4 Гц, 2Н), 7,89-7,74 (м, 2Н), 7,58-7,47 (м, 3Н), 6,81 (дд, J=3,6, 1,7 Гц, 1Н), 3,30 (с, 2Н).
0,21 г (0,606 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в 3 мл метанола, к раствору добавляли при перемешивании магнитной мешалкой 0,112 мл (0,606 ммоль) 30 масс. % раствора метоксида натрия. Полученную смесь перемешивали при к.т. в течение 1 ч. Смесь обрабатывали 10 мл воды, метанол выпаривали в вакууме. Оставшуюся водную фазу лиофилизировали с получением 0,222 г (выход=97%) натрий 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата в виде желтого твердого вещества. ЖХ/МС: m/z=347 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,13 (дд, J=3,4, 2,4 Гц, 2Н), 7,89-7,74 (м, 2Н), 7,58-7,47 (м, 3Н), 6,81 (дд, J=3,6, 1,7 Гц, 1Н), 3,30 (с, 2Н).
Пример 5: Приготовление производного №83: трет-бутил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетат
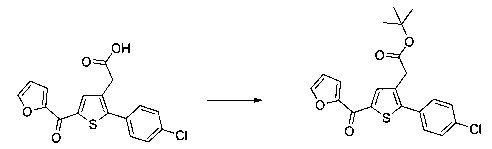 280 мг (0,783 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в 5 мл дихлорметана, к раствору добавляли при перемешивании магнитной мешалкой 0,205 мл (2,35 ммоль) оксалил хлорида и каплю диметилформамида. Смесь перемешивали при к.т. в течение 2 часов, а затем концентрировали в вакууме. Полученный продукт добавляли по каплям при перемешивании магнитной мешалкой к раствору 0,225 мл (2,349 ммоль) трет-бутанола в 5 мл дихлорметана, который находился при температуре 5°С. Смесь выдерживали в течение 15 мин. при температуре 5°С, после чего ледяную баню удаляли, и смесь перемешивали при к.т. в течение 40 ч. Реакционную среду вливали в 30 мл воды, водную фазу экстрагировали 3×20 мл дихлорметана. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaHCO3, 30 мл воды, 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 95% до 90% гептана, об./об.). 0,2 г (выход=62%) трет-бутил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=403 (МН+), степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 8,29-7,99 (м, 2Н), 7,77-7,40 (м, 5Н), 6,84 (дд, J=3,6, 1,7 Гц, 1Н), 3,72 (с, 2Н), 1,35 (с, 9Н).
280 мг (0,783 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в 5 мл дихлорметана, к раствору добавляли при перемешивании магнитной мешалкой 0,205 мл (2,35 ммоль) оксалил хлорида и каплю диметилформамида. Смесь перемешивали при к.т. в течение 2 часов, а затем концентрировали в вакууме. Полученный продукт добавляли по каплям при перемешивании магнитной мешалкой к раствору 0,225 мл (2,349 ммоль) трет-бутанола в 5 мл дихлорметана, который находился при температуре 5°С. Смесь выдерживали в течение 15 мин. при температуре 5°С, после чего ледяную баню удаляли, и смесь перемешивали при к.т. в течение 40 ч. Реакционную среду вливали в 30 мл воды, водную фазу экстрагировали 3×20 мл дихлорметана. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaHCO3, 30 мл воды, 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 95% до 90% гептана, об./об.). 0,2 г (выход=62%) трет-бутил 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=403 (МН+), степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 8,29-7,99 (м, 2Н), 7,77-7,40 (м, 5Н), 6,84 (дд, J=3,6, 1,7 Гц, 1Н), 3,72 (с, 2Н), 1,35 (с, 9Н).
Производное 84 приготовили согласно аналогичной процедуре:

Пример 6: Приготовление производного номер 85: 2-(5-(3-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетат 1,2-3,4-ди-O-изопропилиден-α-~D-галактогалактопиранозы
 0,314 г (0,812 ммоль) 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)уксусной кислоты солюбилизировали в 4 мл дихлорметана, добавляли при перемешивании магнитной мешалкой 0,145 г (0,894 ммоль) карбонилдиимидазола, смесь перемешивали при к.т. в течение 2 часов. Добавляли при перемешивании магнитной мешалкой раствор 211 мг (0,812 ммоль) 1,2-3,4-ди-O-изопропилиден-α-D-галактогалактопиранозы в 4 мл дихлорметана, полученную смесь перемешивали при к.т. в течение 16 ч. Реакционную среду концентрировали в вакууме, неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 9/1, об./об.). 386 мг (выход=75%) 2-(5-(3-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетата 1,2-3,4-ди-О-изопропилиден-α-D-галактогалактопиранозы получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=625 (МН+), степень чистоты (УФ-детекция при 254 нм)=>99%. 1Н ЯМР (300 МГц, DMSO) δ 1Н ЯМР (300 МГц, DMSO) δ 7,71 (с, 26Н), 7,60-7,37 (м, 104Н), 7,37-7,21 (м, 52Н), 7,16-7,03 (м, 51Н), 5,41 (д, J=5,0 Гц, 25Н), 4,58 (дд, J=7,9, 2,4 Гц, 26Н), 4,35 (дд, J=5,0, 2,4 Гц, 26Н), 4,22-3,99 (м, 80Н), 3,89 (д, J=2,8 Гц, 19Н), 3,85 (т, J=7,9 Гц, 160Н), 3,75 (с, 50Н), 1,99 (с, 3Н), 1,33 (с, 78Н), 1,29 (с, 79Н), 1,25 (д, J=4,6 Гц, 160Н), 1,17 (с, 6Н), 0,84 (д, J=6,6 Гц, 5Н).
0,314 г (0,812 ммоль) 2-(5-(4-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)уксусной кислоты солюбилизировали в 4 мл дихлорметана, добавляли при перемешивании магнитной мешалкой 0,145 г (0,894 ммоль) карбонилдиимидазола, смесь перемешивали при к.т. в течение 2 часов. Добавляли при перемешивании магнитной мешалкой раствор 211 мг (0,812 ммоль) 1,2-3,4-ди-O-изопропилиден-α-D-галактогалактопиранозы в 4 мл дихлорметана, полученную смесь перемешивали при к.т. в течение 16 ч. Реакционную среду концентрировали в вакууме, неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 9/1, об./об.). 386 мг (выход=75%) 2-(5-(3-метоксибензоил)-2-(4-метоксифенил)тиофен-3-ил)ацетата 1,2-3,4-ди-О-изопропилиден-α-D-галактогалактопиранозы получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=625 (МН+), степень чистоты (УФ-детекция при 254 нм)=>99%. 1Н ЯМР (300 МГц, DMSO) δ 1Н ЯМР (300 МГц, DMSO) δ 7,71 (с, 26Н), 7,60-7,37 (м, 104Н), 7,37-7,21 (м, 52Н), 7,16-7,03 (м, 51Н), 5,41 (д, J=5,0 Гц, 25Н), 4,58 (дд, J=7,9, 2,4 Гц, 26Н), 4,35 (дд, J=5,0, 2,4 Гц, 26Н), 4,22-3,99 (м, 80Н), 3,89 (д, J=2,8 Гц, 19Н), 3,85 (т, J=7,9 Гц, 160Н), 3,75 (с, 50Н), 1,99 (с, 3Н), 1,33 (с, 78Н), 1,29 (с, 79Н), 1,25 (д, J=4,6 Гц, 160Н), 1,17 (с, 6Н), 0,84 (д, J=6,6 Гц, 5Н).
Пример 7: Приготовление производного номер 86: 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамид
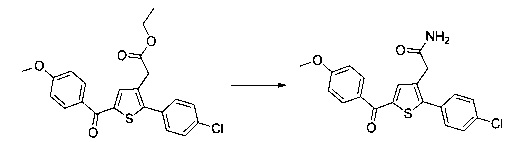 0,25 г (0,603 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата суспендировали в 2,15 мл (15,06 ммоль) 7 М аммиачного раствора метанола. Реакционную среду перемешивали при к.т. магнитной мешалкой в течение 5 д, после чего вливали в 25 мл воды. После перемешивания магнитной мешалкой в течение 10 мин. преципитировало твердое вещество, которое выделяли в результате фильтрации, промывали 2×5 мл воды и рекристаллизовали из этанола. 0,137 г (выход=58%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)-тиофен-3-ил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=386 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,88 (д, J=8,8 Гц, 2Н), 7,75 (с, 1Н), 7,72-7,52 (м, 5Н), 7,13 (д, J=8,9 Гц, 2Н), 7,05 (с, 1Н), 3,88 (с, 3Н), 3,47 (с, 2Н).
0,25 г (0,603 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата суспендировали в 2,15 мл (15,06 ммоль) 7 М аммиачного раствора метанола. Реакционную среду перемешивали при к.т. магнитной мешалкой в течение 5 д, после чего вливали в 25 мл воды. После перемешивания магнитной мешалкой в течение 10 мин. преципитировало твердое вещество, которое выделяли в результате фильтрации, промывали 2×5 мл воды и рекристаллизовали из этанола. 0,137 г (выход=58%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)-тиофен-3-ил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=386 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,88 (д, J=8,8 Гц, 2Н), 7,75 (с, 1Н), 7,72-7,52 (м, 5Н), 7,13 (д, J=8,9 Гц, 2Н), 7,05 (с, 1Н), 3,88 (с, 3Н), 3,47 (с, 2Н).
Пример 8: Приготовление производного №87: этил 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо)ацетат
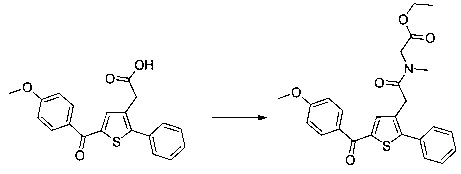 0,091 г (0,593 ммоль) гидрохлорида этилового эфира саркозина солюбилизировали в 3 мл тетрагидрофурана. К полученному раствору добавляли при перемешивании магнитной мешалкой 0,149 г (1,079 ммоль) карбоната калия и 0,2 г (0,593 ммоль) 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)уксусной кислоты. Смесь перемешивали при к.т. в течение 16 ч, после чего вливали в 20 мл воды. Водную фазу экстрагировали 2×20 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/этилацетата, от 100% до 75% дихлорметана, об./об.). 0,133 г (выход=54%) этил 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо)ацетата получали в виде бледно-коричневого масла. ЖХ/МС: m/z=452 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,88 (д, J=8,8 Гц, 2Н), 7,66-7,39 (м, 6Н), 7,12 (д, J=8,8 Гц, 2Н), 4,15 (д, J=40,5 Гц, 4Н), 3,87 (с, 3Н), 3,74 (д, J=35,2 Гц, 2Н), 2,92 (д, J=51,8 Гц, 3Н), 1,15 (дд, J=13,1, 6,0 Гц, 3Н).
0,091 г (0,593 ммоль) гидрохлорида этилового эфира саркозина солюбилизировали в 3 мл тетрагидрофурана. К полученному раствору добавляли при перемешивании магнитной мешалкой 0,149 г (1,079 ммоль) карбоната калия и 0,2 г (0,593 ммоль) 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)уксусной кислоты. Смесь перемешивали при к.т. в течение 16 ч, после чего вливали в 20 мл воды. Водную фазу экстрагировали 2×20 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/этилацетата, от 100% до 75% дихлорметана, об./об.). 0,133 г (выход=54%) этил 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо)ацетата получали в виде бледно-коричневого масла. ЖХ/МС: m/z=452 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,88 (д, J=8,8 Гц, 2Н), 7,66-7,39 (м, 6Н), 7,12 (д, J=8,8 Гц, 2Н), 4,15 (д, J=40,5 Гц, 4Н), 3,87 (с, 3Н), 3,74 (д, J=35,2 Гц, 2Н), 2,92 (д, J=51,8 Гц, 3Н), 1,15 (дд, J=13,1, 6,0 Гц, 3Н).
Пример 9: Приготовление производного №88: 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо)уксусная кислота
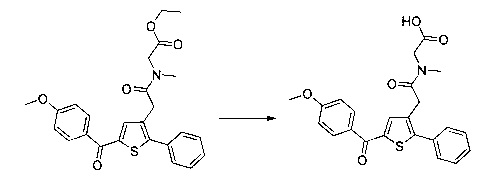 0,099 г (0,203 ммоль) этил 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо) ацетата солюбилизировали в 3 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 0,02 мл (0,203 ммоль) 30 масс. % водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 5 мл воды, водную фазу экстрагировали 5 мл этилацетата. Затем значение рН водной фазы снижали путем добавления раствора 1 н хлористоводородной кислоты до появления преципитата. Твердое вещество выделяли в результате фильтрации, промывали 2×3 мл воды и высушивали в вакуумном колпаке с получением 0,01 г (выход=12%) 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=424 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,85 (с, 1Н), 7,89 (д, J=8,8 Гц, 2Н), 7,70-7,42 (м, 6Н), 7,18-7,08 (м, 2Н), 4,04 (д, J=21,1 Гц, 2Н), 3,87 (с, 3Н), 3,73 (д, J=34,0 Гц, 2Н), 2,91 (д, J=49,4 Гц, 3Н).
0,099 г (0,203 ммоль) этил 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо) ацетата солюбилизировали в 3 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 0,02 мл (0,203 ммоль) 30 масс. % водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 5 мл воды, водную фазу экстрагировали 5 мл этилацетата. Затем значение рН водной фазы снижали путем добавления раствора 1 н хлористоводородной кислоты до появления преципитата. Твердое вещество выделяли в результате фильтрации, промывали 2×3 мл воды и высушивали в вакуумном колпаке с получением 0,01 г (выход=12%) 2-(2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-N-метилацетамидо)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=424 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,85 (с, 1Н), 7,89 (д, J=8,8 Гц, 2Н), 7,70-7,42 (м, 6Н), 7,18-7,08 (м, 2Н), 4,04 (д, J=21,1 Гц, 2Н), 3,87 (с, 3Н), 3,73 (д, J=34,0 Гц, 2Н), 2,91 (д, J=49,4 Гц, 3Н).
Пример 10: Приготовление производного №89: 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамид
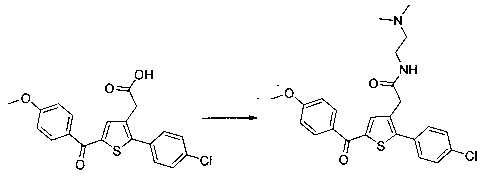 0,1 г (0,258 ммоль) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)уксусной кислоты солюбилизировали в 2 мл дихлорметана. К раствору добавляли при перемешивании магнитной мешалкой 0,059 г (0,310 ммоль) EDC, 0,047 г (0,310 ммоль) HOBt и 0,108 мл (0,775 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 15 мин., затем добавляли 0,034 мл (0,310 ммоль) N,N-диметилэтан-1,2-диамина, полученную смесь перемешивали при к.т. в течение 16 ч. К реакционной смеси добавляли 20 мл дихлорметана. Смесь промывали 2×20 мл воды. Органическую фазу высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, от 100% до 90% дихлорметана, об./об.). 49 мг (выход=39%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=457 (МН+), степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,06 (т, J=5,5 Гц, 1Н), 7,87 (д, J=8,8 Гц, 2Н), 7,78-7,65 (м, 3Н), 7,58 (д, J=8,5 Гц, 2Н), 7,13 (д, J=8,9 Гц, 2Н), 3,88 (с, 3Н), 3,49 (с, 2Н), 3,14 (дд, J=12,3, 6,4 Гц, 2Н), 2,25 (т, J=6,6 Гц, 2Н), 2,11 (с, 6Н).
0,1 г (0,258 ммоль) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)уксусной кислоты солюбилизировали в 2 мл дихлорметана. К раствору добавляли при перемешивании магнитной мешалкой 0,059 г (0,310 ммоль) EDC, 0,047 г (0,310 ммоль) HOBt и 0,108 мл (0,775 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 15 мин., затем добавляли 0,034 мл (0,310 ммоль) N,N-диметилэтан-1,2-диамина, полученную смесь перемешивали при к.т. в течение 16 ч. К реакционной смеси добавляли 20 мл дихлорметана. Смесь промывали 2×20 мл воды. Органическую фазу высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, от 100% до 90% дихлорметана, об./об.). 49 мг (выход=39%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=457 (МН+), степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,06 (т, J=5,5 Гц, 1Н), 7,87 (д, J=8,8 Гц, 2Н), 7,78-7,65 (м, 3Н), 7,58 (д, J=8,5 Гц, 2Н), 7,13 (д, J=8,9 Гц, 2Н), 3,88 (с, 3Н), 3,49 (с, 2Н), 3,14 (дд, J=12,3, 6,4 Гц, 2Н), 2,25 (т, J=6,6 Гц, 2Н), 2,11 (с, 6Н).
Производные с 160 по 166 были приготовлены согласно аналогичной процедуре:
Пример 11: Приготовление производного №90: 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-1-(пирролидин-1-ил)этанон
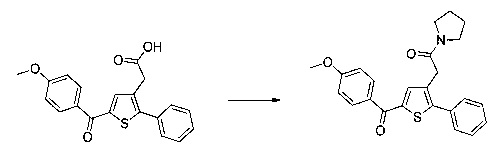 0,095 г (0,270 ммоль) 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)уксусной кислоты солюбилизировали в 3 мл дихлорметана, к раствору добавляли при перемешивании магнитной мешалкой 0,047 мл (0,540 ммоль) оксалил хлорида и каплю диметилформамида. Смесь перемешивали при к.т. в течение 2 часов, а затем концентрировали в вакууме. Полученный продукт солюбилизировали в 2 мл тетрагидрофурана, после чего добавляли по каплям при перемешивании магнитной мешалкой к раствору 0,049 мл (0,593 ммоль) пирролидина в 2 мл тетрагидрофурана. Смесь перемешивали при к.т. в течение 16 ч. Реакционную среду вливали в 20 мл воды, водную фазу экстрагировали 2×10 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: дихлорметан/этилацетат, 3/1, об./об.). 0,077 г (выход=65%) 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-1-(пиррол идин-1 -ил)этанона получали в виде бледно-коричневого масла. ЖХ/МС: m/z=406 (МН+), степень чистоты (УФ-детекция при 254 нм)=93%. 1Н ЯМР (300 МГц, DMSO) δ 7,87 (д, J=8,8 Гц, 2Н), 7,64 (с, 1Н), 7,57-7,42 (м, 5Н), 7,13 (д, J=8,9 Гц, 2Н), 3,87 (с, 3Н), 3,68 (с, 2Н), 3,46-3,23 (м, 4Н), 1,91-1,68 (м, 4Н).
0,095 г (0,270 ммоль) 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)уксусной кислоты солюбилизировали в 3 мл дихлорметана, к раствору добавляли при перемешивании магнитной мешалкой 0,047 мл (0,540 ммоль) оксалил хлорида и каплю диметилформамида. Смесь перемешивали при к.т. в течение 2 часов, а затем концентрировали в вакууме. Полученный продукт солюбилизировали в 2 мл тетрагидрофурана, после чего добавляли по каплям при перемешивании магнитной мешалкой к раствору 0,049 мл (0,593 ммоль) пирролидина в 2 мл тетрагидрофурана. Смесь перемешивали при к.т. в течение 16 ч. Реакционную среду вливали в 20 мл воды, водную фазу экстрагировали 2×10 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: дихлорметан/этилацетат, 3/1, об./об.). 0,077 г (выход=65%) 2-(5-(4-метоксибензоил)-2-фенилтиофен-3-ил)-1-(пиррол идин-1 -ил)этанона получали в виде бледно-коричневого масла. ЖХ/МС: m/z=406 (МН+), степень чистоты (УФ-детекция при 254 нм)=93%. 1Н ЯМР (300 МГц, DMSO) δ 7,87 (д, J=8,8 Гц, 2Н), 7,64 (с, 1Н), 7,57-7,42 (м, 5Н), 7,13 (д, J=8,9 Гц, 2Н), 3,87 (с, 3Н), 3,68 (с, 2Н), 3,46-3,23 (м, 4Н), 1,91-1,68 (м, 4Н).
Производные с 91 по 100 были приготовлены согласно аналогичной процедуре:
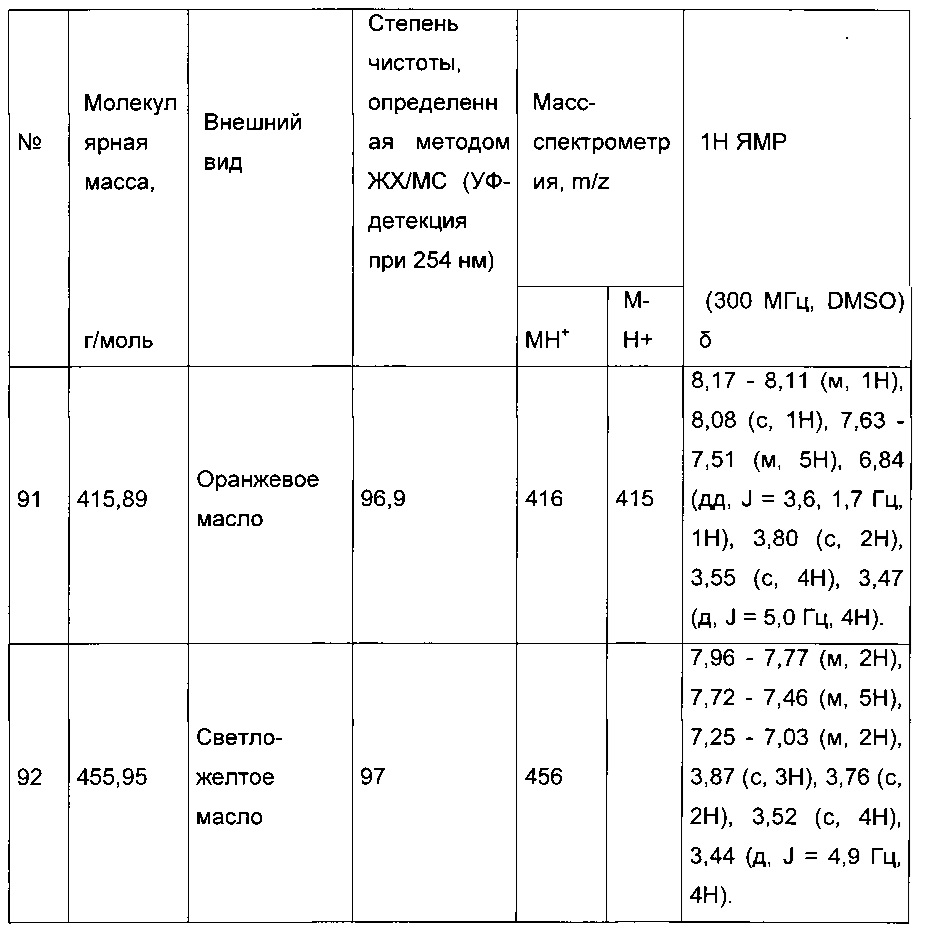
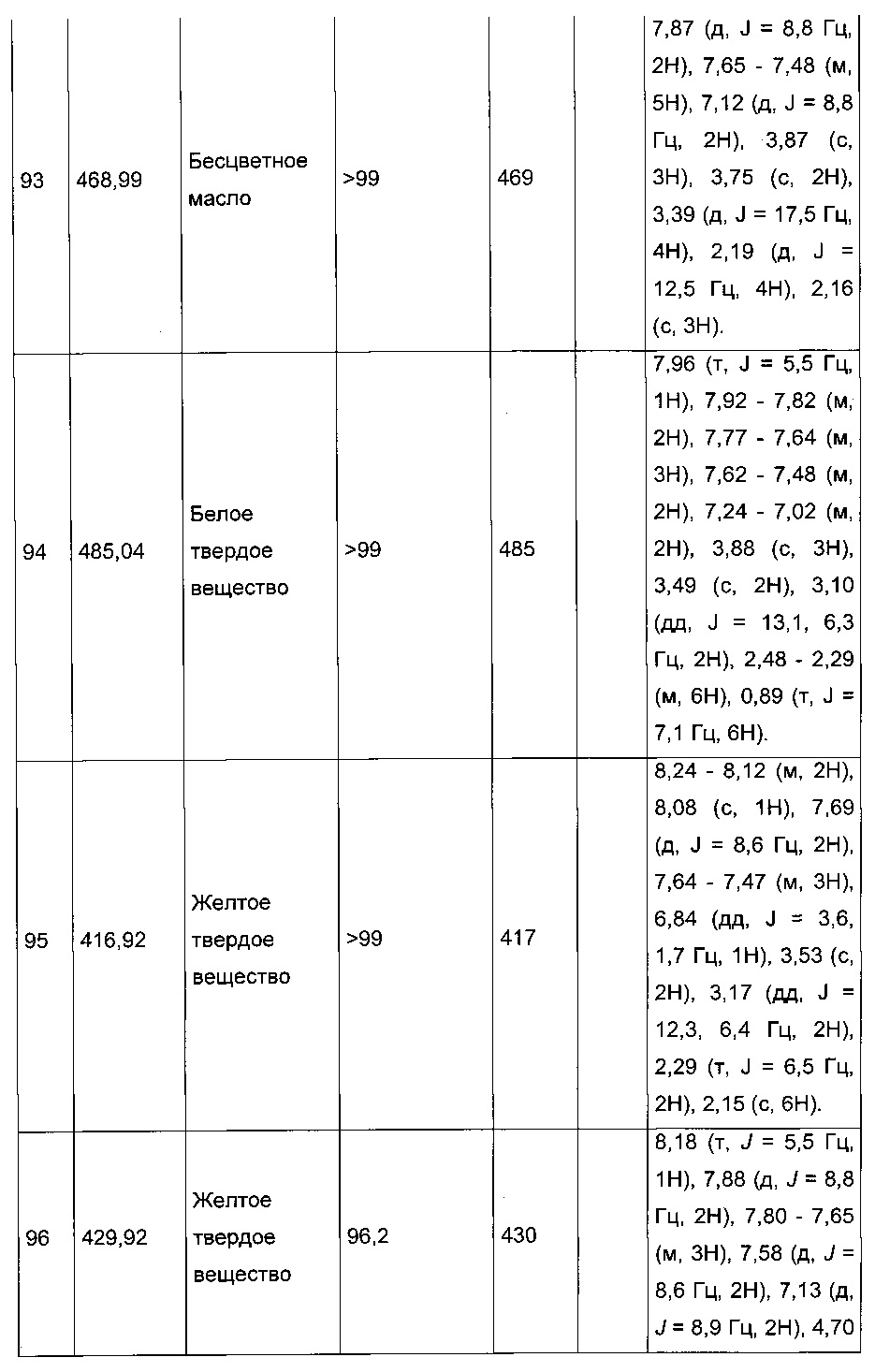
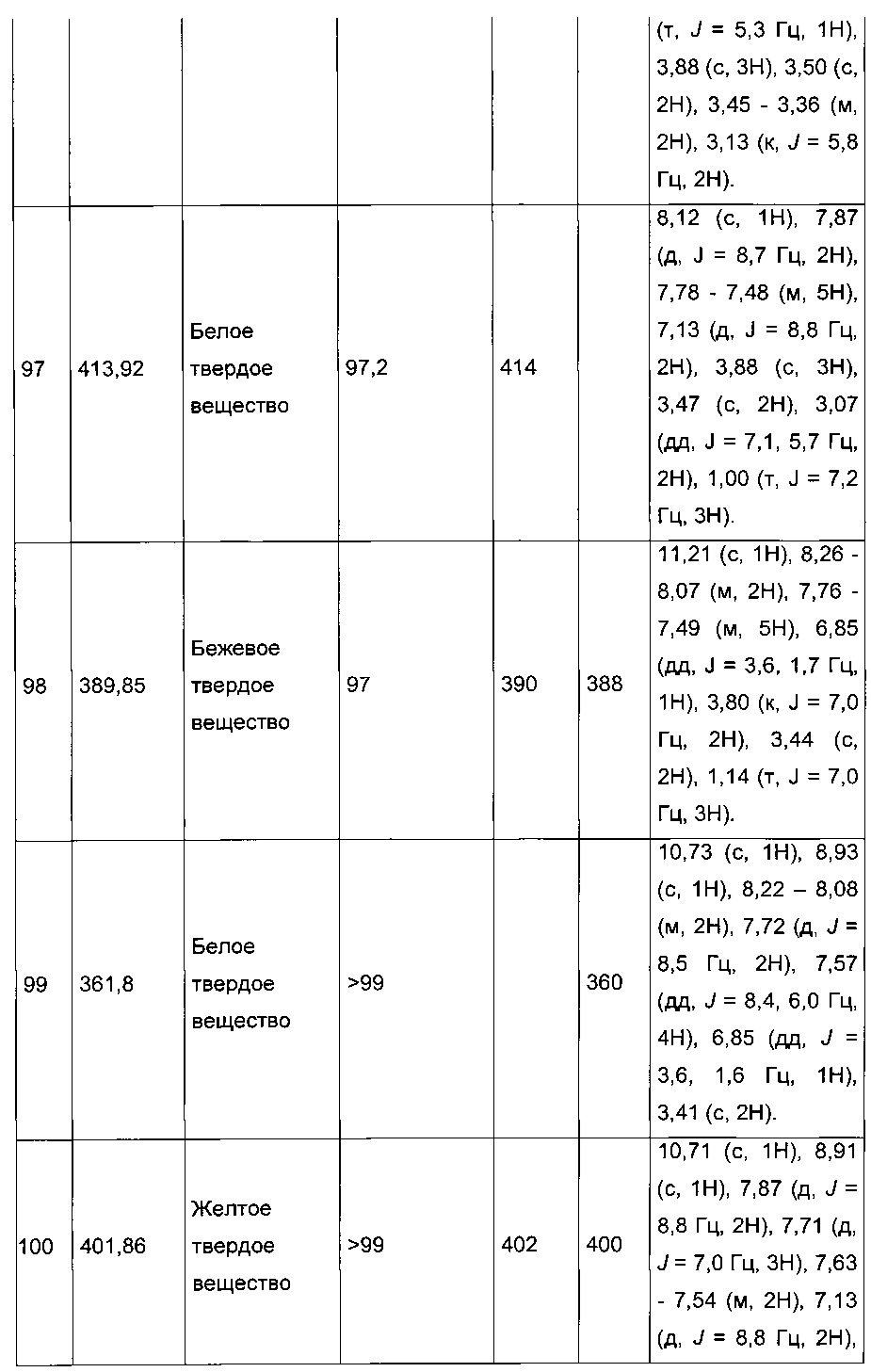
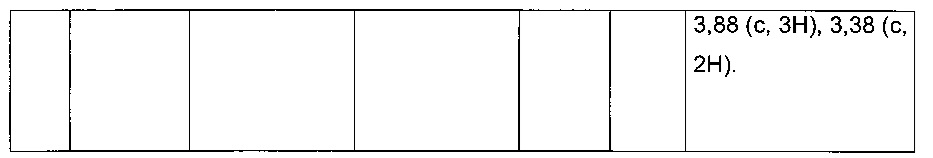
Пример 12: Приготовление производного №101: этил 2-(5-(гидрокси (фенил)метил)-2-(4-метоксифенил)тиофен-3-ил)ацетат
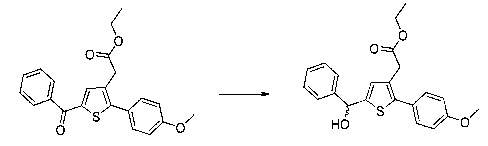 2,8 г (7,36 ммоль) этил 2-(5-бензоил-2-(4-метоксифенил)тиофен-3-ил)ацетата солюбилизировали в 50 мл этанола. Полученную смесь при перемешивании магнитной мешалкой, помещали на ледяную баню при температуре 0°С, после чего добавляли 0,557 г (14,72 ммоль) бороводорода натрия. Смесь выдерживали в течение 15 мин. при температуре 0°С, после чего ледяную баню удаляли, и смесь перемешивали при к.т. в течение 72 ч. Реакционную среду вливали в 400 мл смеси, состоящей из воды и льда, водную фазу экстрагировали 2×200 мл этилацетата. Объединенные органические фазы промывали 2×50 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/ацетона, от 100% до 95% дихлорметана, об./об.). 1,05 г (выход=37%) этил 2-(5-(гидрокси(фенил)метил)-2-(4-метокси фенил)тиофен-3-ил)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 7,40 (д, 2Н), 7,20-7,35 (м, 5Н), 6,80 (д, 2Н), 6,75 (с, 1Н), 5,92 (с, 1Н), 4,07 (к, 2Н), 3,75 (с, 3Н), 3,44 (с, 2Н), 2,45 (с, 1Н), 1,15 (т, 3Н).
2,8 г (7,36 ммоль) этил 2-(5-бензоил-2-(4-метоксифенил)тиофен-3-ил)ацетата солюбилизировали в 50 мл этанола. Полученную смесь при перемешивании магнитной мешалкой, помещали на ледяную баню при температуре 0°С, после чего добавляли 0,557 г (14,72 ммоль) бороводорода натрия. Смесь выдерживали в течение 15 мин. при температуре 0°С, после чего ледяную баню удаляли, и смесь перемешивали при к.т. в течение 72 ч. Реакционную среду вливали в 400 мл смеси, состоящей из воды и льда, водную фазу экстрагировали 2×200 мл этилацетата. Объединенные органические фазы промывали 2×50 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/ацетона, от 100% до 95% дихлорметана, об./об.). 1,05 г (выход=37%) этил 2-(5-(гидрокси(фенил)метил)-2-(4-метокси фенил)тиофен-3-ил)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 7,40 (д, 2Н), 7,20-7,35 (м, 5Н), 6,80 (д, 2Н), 6,75 (с, 1Н), 5,92 (с, 1Н), 4,07 (к, 2Н), 3,75 (с, 3Н), 3,44 (с, 2Н), 2,45 (с, 1Н), 1,15 (т, 3Н).
Пример 13: Приготовление производного номер 102: этил 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)ацетат
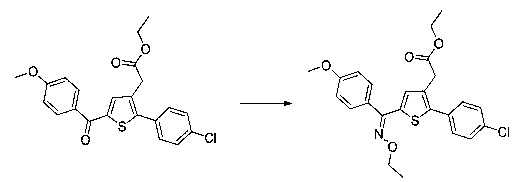 0,3 г (0,701 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата солюбилизировали в 1,5 мл этанола при перемешивании магнитной мешалкой. Затем к полученному раствору добавляли 0,342 г (3,51 ммоль) хлорида О-этилгидроксиламмония и 0,187 мл (2,315 ммоль) пиридина. Смесь перемешивали при температуре 50°С в течение 24 ч. Смеси позволяли остыть до к.т., после чего смесь вливали в 5 мл воды. Водную фазу экстрагировали 2×5 мл этилацетата. Объединенные органические фазы промывали 5 мл насыщенного водного раствора NaHCO3, 5 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 9/1, об./об.). 0,242 г (выход=73%) этил 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)ацетата получали в виде бесцветного масла. ЖХ/МС: m/z=458 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 7,62-7,31 (м, 6Н), 7,12 (с, 1Н), 7,10-6,98 (м, 2Н), 4,30 (к, J=7,0 Гц, 1,6Н), 4,11 (к, J=7,0 Гц, 0,4Н), 4,01 (р, J=7,1 Гц, 2Н), 3,82 (с, 3Н), 3,64 (д, J=12,6 Гц, 2Н), 1,33 (т, J=7,0 Гц, 2,4Н), 1,26-1,15 (м, 0.6Н), 1,09 (т, J=7,1 Гц, 3Н).
0,3 г (0,701 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата солюбилизировали в 1,5 мл этанола при перемешивании магнитной мешалкой. Затем к полученному раствору добавляли 0,342 г (3,51 ммоль) хлорида О-этилгидроксиламмония и 0,187 мл (2,315 ммоль) пиридина. Смесь перемешивали при температуре 50°С в течение 24 ч. Смеси позволяли остыть до к.т., после чего смесь вливали в 5 мл воды. Водную фазу экстрагировали 2×5 мл этилацетата. Объединенные органические фазы промывали 5 мл насыщенного водного раствора NaHCO3, 5 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 9/1, об./об.). 0,242 г (выход=73%) этил 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)ацетата получали в виде бесцветного масла. ЖХ/МС: m/z=458 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 7,62-7,31 (м, 6Н), 7,12 (с, 1Н), 7,10-6,98 (м, 2Н), 4,30 (к, J=7,0 Гц, 1,6Н), 4,11 (к, J=7,0 Гц, 0,4Н), 4,01 (р, J=7,1 Гц, 2Н), 3,82 (с, 3Н), 3,64 (д, J=12,6 Гц, 2Н), 1,33 (т, J=7,0 Гц, 2,4Н), 1,26-1,15 (м, 0.6Н), 1,09 (т, J=7,1 Гц, 3Н).
Производные с 103 по 107 были приготовлены согласно аналогичной процедуре:

Пример 14: Приготовление производного №108: 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)уксусная кислота
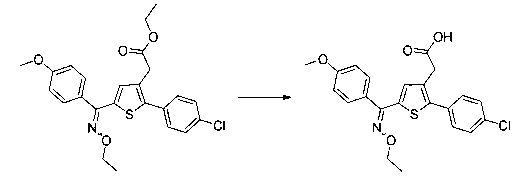 0,238 г (0,505 ммоль) этил 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)ацетата солюбилизировали при перемешивании магнитной мешалкой в 1,1 мл смеси метанол-тетрагидрофуран (1/1, об./об.), добавляли 0,545 мл (0,545 ммоль) 1 М водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 16 ч. Смесь нейтрализовали добавлением 1 н водного раствора хлористоводородной кислоты до появления преципитата. Смесь концентрировали в вакууме, полученный осадок переносили в изопропанол, неорганические соли удаляли в результате фильтрации, полученный фильтрат концентрировали в вакууме, после чего переносили в 2 мл воды и перемешивали магнитной мешалкой при температуре 80°С в течение 1 ч. Смесь концентрировали в вакууме с получением 0,191 г (выход=82%) 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=430 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) (наблюдалась смесь изомеров Z/E) δ 7,65-7,29 (м, 6Н), 7,16-6,97 (м, 3Н), 4,30 (к, J=7,0 Гц, 1,5Н), 4,17-4,04 (м, 0,5Н), 3,82 (с, 3Н), 3,53 (д, J=13,9 Гц, 2Н), 1,40-1,29 (м, 1,5Н), 1,20 (дд, J=13,0, 6,0 Гц, 0,5Н).
0,238 г (0,505 ммоль) этил 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)ацетата солюбилизировали при перемешивании магнитной мешалкой в 1,1 мл смеси метанол-тетрагидрофуран (1/1, об./об.), добавляли 0,545 мл (0,545 ммоль) 1 М водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 16 ч. Смесь нейтрализовали добавлением 1 н водного раствора хлористоводородной кислоты до появления преципитата. Смесь концентрировали в вакууме, полученный осадок переносили в изопропанол, неорганические соли удаляли в результате фильтрации, полученный фильтрат концентрировали в вакууме, после чего переносили в 2 мл воды и перемешивали магнитной мешалкой при температуре 80°С в течение 1 ч. Смесь концентрировали в вакууме с получением 0,191 г (выход=82%) 2-(2-(4-хлорфенил)-5-((этоксиимино)(4-метоксифенил)метил)тиофен-3-ил)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=430 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) (наблюдалась смесь изомеров Z/E) δ 7,65-7,29 (м, 6Н), 7,16-6,97 (м, 3Н), 4,30 (к, J=7,0 Гц, 1,5Н), 4,17-4,04 (м, 0,5Н), 3,82 (с, 3Н), 3,53 (д, J=13,9 Гц, 2Н), 1,40-1,29 (м, 1,5Н), 1,20 (дд, J=13,0, 6,0 Гц, 0,5Н).
Производные с 109 по 111 были приготовлены согласно аналогичной процедуре:
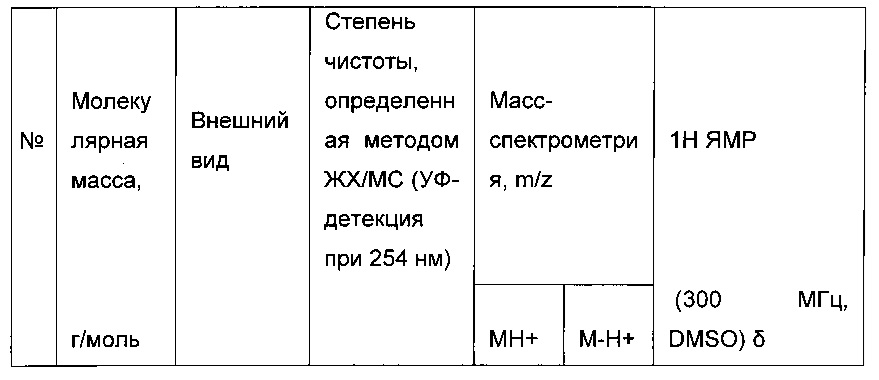
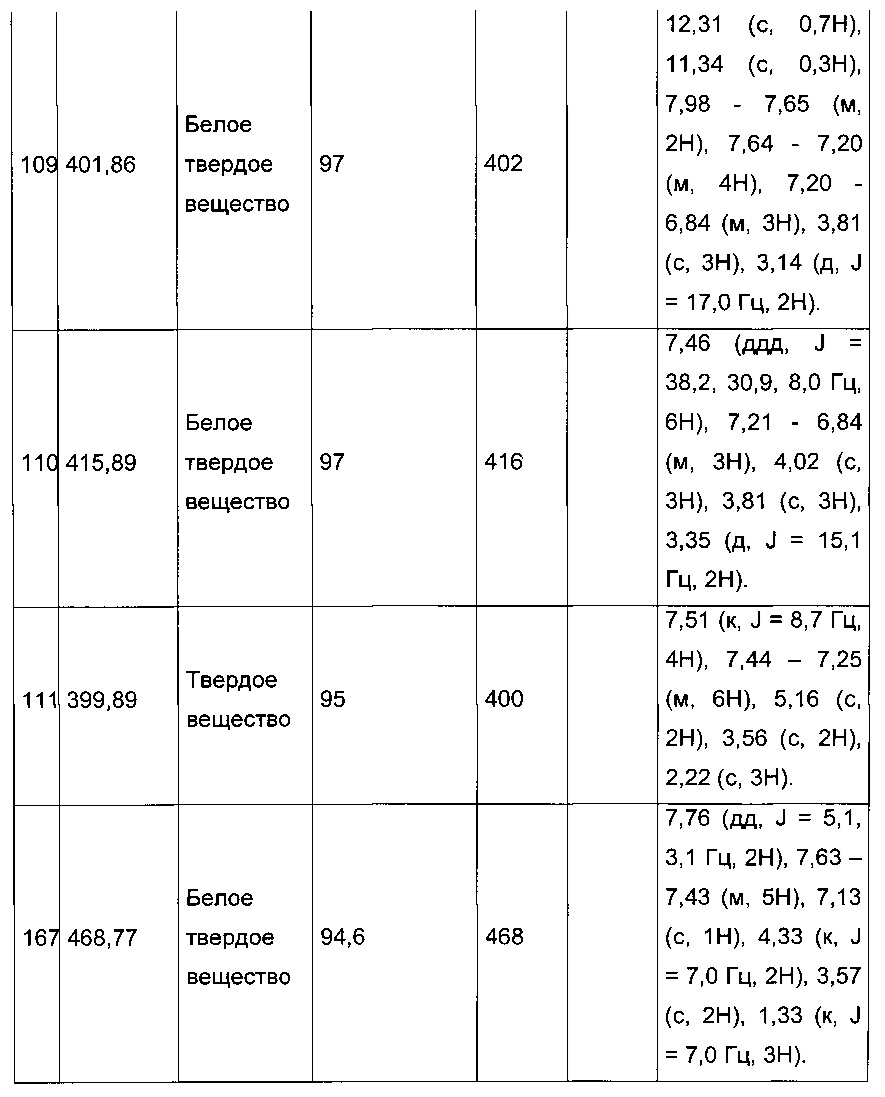
Пример 15: Приготовление производного №112: 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновая кислота
Этап 1: Приготовление изопропил 4-хлорфенил-4-оксобутаноата
 50 г (235 ммоль) 4-хлорфенил-4-оксобутановой кислоты солюбилизировали в 300 мл изопропанола, к раствору добавляли 0,63 мл (11,76 ммоль) серной кислоты. Смесь нагревали в колбе с обратным холодильником в течение 6 д при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан). 59,83 г (выход=>99%) изопропил 4-хлорфенил-4-оксобутаноата получали в виде бесцветного масла. ЖХ/МС: m/z=255 (МН+), степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 8,13-7,85 (м, 2Н), 7,70-7,47 (м, 2Н), 4,86 (дт, J=12,5, 6,3 Гц, 1Н), 3,29-3,22 (м, 2Н), 2,65-2,54 (м, 2Н), 1,16 (д, J=6,3 Гц, 6Н).
50 г (235 ммоль) 4-хлорфенил-4-оксобутановой кислоты солюбилизировали в 300 мл изопропанола, к раствору добавляли 0,63 мл (11,76 ммоль) серной кислоты. Смесь нагревали в колбе с обратным холодильником в течение 6 д при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан). 59,83 г (выход=>99%) изопропил 4-хлорфенил-4-оксобутаноата получали в виде бесцветного масла. ЖХ/МС: m/z=255 (МН+), степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 8,13-7,85 (м, 2Н), 7,70-7,47 (м, 2Н), 4,86 (дт, J=12,5, 6,3 Гц, 1Н), 3,29-3,22 (м, 2Н), 2,65-2,54 (м, 2Н), 1,16 (д, J=6,3 Гц, 6Н).
Этап 2: Приготовление изопропил (Z/Е)-4-хлор-3-формил-4-(4-хлорфенил)бут-3-еноата
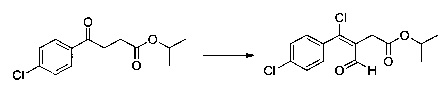 59,8 г (235 ммоль) изопропил 4-хлорфенил-4-оксобутаноата растворяли в 54,5 мл диметилформамида (704 ммоль), в раствор медленно добавляли 54,7 мл (587 ммоль) фосфорилтрихлорида, реакция была высоко экзотермической. Затем полученную смесь нагревали до температуры 80°С в течение 2 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь вливали в 1 л смеси, состоящей из воды и льда, и полученную смесь перемешивали магнитной мешалкой при к.т. в течение 16 ч. Водную фазу экстрагировали 300 мл, а затем 2×150 мл этилацетата. Объединенные органические фазы промывали 2×200 мл воды, 300 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/дихлорметана, от 50 до 100% дихлорметана, об./об.). 47,4 г (выход=62%) изопропил (Z/Е)-4-хлор-3-формил-4-(4-хлорфенил)бут-3-еноата получали в виде оранжевого масла. ЖХ/МС: m/z=301 (МН+), степень чистоты (УФ-детекция при 254 нм)=92%. 1Н ЯМР (300 МГц, DMSO) δ 10,29-9,28 (м, 1Н), 7,71-7,38 (м, 4Н), 4,87 (дк, J=25,0, 6,3 Гц, 1Н), 3,53 (с, 19Н), 3,18-1,16 (м, 2Н).
59,8 г (235 ммоль) изопропил 4-хлорфенил-4-оксобутаноата растворяли в 54,5 мл диметилформамида (704 ммоль), в раствор медленно добавляли 54,7 мл (587 ммоль) фосфорилтрихлорида, реакция была высоко экзотермической. Затем полученную смесь нагревали до температуры 80°С в течение 2 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь вливали в 1 л смеси, состоящей из воды и льда, и полученную смесь перемешивали магнитной мешалкой при к.т. в течение 16 ч. Водную фазу экстрагировали 300 мл, а затем 2×150 мл этилацетата. Объединенные органические фазы промывали 2×200 мл воды, 300 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/дихлорметана, от 50 до 100% дихлорметана, об./об.). 47,4 г (выход=62%) изопропил (Z/Е)-4-хлор-3-формил-4-(4-хлорфенил)бут-3-еноата получали в виде оранжевого масла. ЖХ/МС: m/z=301 (МН+), степень чистоты (УФ-детекция при 254 нм)=92%. 1Н ЯМР (300 МГц, DMSO) δ 10,29-9,28 (м, 1Н), 7,71-7,38 (м, 4Н), 4,87 (дк, J=25,0, 6,3 Гц, 1Н), 3,53 (с, 19Н), 3,18-1,16 (м, 2Н).
Этап 3: Приготовление изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)ацетата и 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновой кислоты (производное №112)
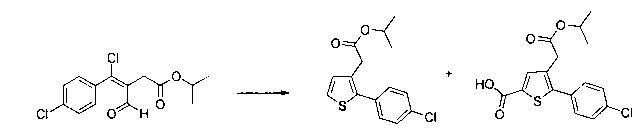 47,4 г (157 ммоль) изопропил (Z/Е)-4-хлор-3-формил-4-(4-хлорфенил)бут-3-еноата солюбилизировали в 250 мл тетрагидрофурана, к раствору добавляли 16,40 мл (236 ммоль) 2-меркапто-уксусной кислоты и 65,8 мл (472 ммоль) триэтиламина. Полученную смесь нагревали в колбе с обратным холодильником в течение 4 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме. Осадок переносили в 175 мл диметилформамида, полученную смесь нагревали до температуры 130°С в течение 2 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь обрабатывали 500 мл воды. Водную фазу экстрагировали 300 мл, а затем 2×150 мл этилацетата. Объединенные органические фазы промывали 2×200 мл воды, 300 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок отбирали и растирали в гептане. Твердое вещество осаждали и выделяли в результате фильтрации с получением 22,8 г (выход=30%) 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновой кислоты. Фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/дихлорметан, 2/1, об./об.). 16,18 г (выход=34%) изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)ацетата получали в виде бледно-коричневого масла. 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновая кислота: ЖХ/МС: неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 13,19 (с, 1Н), 7,69 (с, 1Н), 7,53 (к, J=8,7 Гц, 4Н), 4,86 (дт, J=12,5, 6,2 Гц, 1Н), 3,69 (с, 2Н), 1,13 (д, J=6,3 Гц, 6Н).
47,4 г (157 ммоль) изопропил (Z/Е)-4-хлор-3-формил-4-(4-хлорфенил)бут-3-еноата солюбилизировали в 250 мл тетрагидрофурана, к раствору добавляли 16,40 мл (236 ммоль) 2-меркапто-уксусной кислоты и 65,8 мл (472 ммоль) триэтиламина. Полученную смесь нагревали в колбе с обратным холодильником в течение 4 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме. Осадок переносили в 175 мл диметилформамида, полученную смесь нагревали до температуры 130°С в течение 2 часов при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь обрабатывали 500 мл воды. Водную фазу экстрагировали 300 мл, а затем 2×150 мл этилацетата. Объединенные органические фазы промывали 2×200 мл воды, 300 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок отбирали и растирали в гептане. Твердое вещество осаждали и выделяли в результате фильтрации с получением 22,8 г (выход=30%) 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновой кислоты. Фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/дихлорметан, 2/1, об./об.). 16,18 г (выход=34%) изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)ацетата получали в виде бледно-коричневого масла. 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновая кислота: ЖХ/МС: неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 13,19 (с, 1Н), 7,69 (с, 1Н), 7,53 (к, J=8,7 Гц, 4Н), 4,86 (дт, J=12,5, 6,2 Гц, 1Н), 3,69 (с, 2Н), 1,13 (д, J=6,3 Гц, 6Н).
Изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)ацетат: ЖХ/МС: m/z=294 (М), степень чистоты (УФ-детекция при 254 нм)=96%. 1Н ЯМР (300 МГц, DMSO) δ 7,51 (дт, J=19,3, 6,9 Гц, 5Н), 7,08 (д, J=5,2 Гц, 1Н), 4,87 (дт, J=12,5, 6,3 Гц, 1Н), 3,63 (с, 2Н), 1,15 (д, J=6,3 Гц, 6Н).
Производное номер 113 приготовили согласно той же последовательности этапов с 1 по 3.
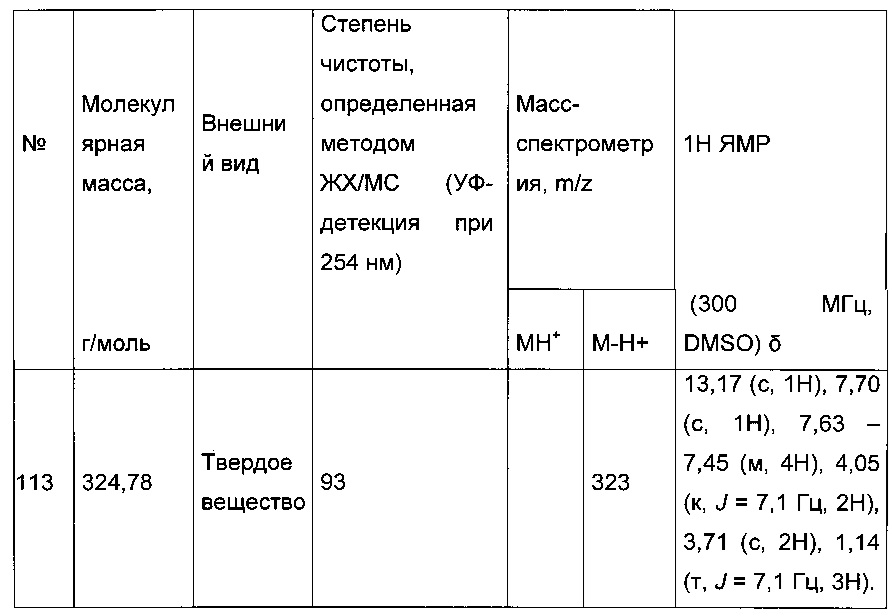
Пример 16: Приготовление производного №114: этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетат
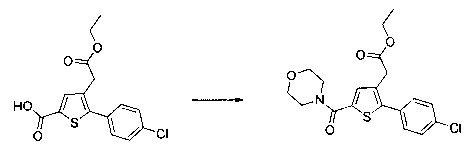 0,44 г (1,244 ммоль) 5-(4-хлорфенил)-4-(2-этокси-2-оксоэтил)тиофен-2-карбоновой кислоты солюбилизировали в 5 мл дихлорметана, к раствору добавляли при перемешивании магнитной мешалкой 0,327 мл (3,73 ммоль) оксалил хлорида и по каплям диметилформамид. Смесь перемешивали при к.т. в течение 2 часов, а затем концентрировали в вакууме. Полученный продукт добавляли по каплям при перемешивании магнитной мешалкой к раствору 0,542 мл (6,22 ммоль) морфолина в 5 мл дихлорметана. Смесь перемешивали при к.т. в течение 16 ч. Реакционную среду вливали в 20 мл воды, водную фазу экстрагировали 3×25 мл дихлорметана. Объединенные органические фазы промывали 20 мл насыщенного водного раствора NaHCO3, 20 мл воды, 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, 98/2, об./об.). 0,359 г (выход=68%) этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетата получали в виде желто-оранжевого масла. ЖХ/МС: m/z=394 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,63-7,44 (м, 4Н), 7,41 (с, 1Н), 4,16-3,95 (м, 2Н), 3,66 (дд, J=11,0, 7,8 Гц, 10Н), 1,14 (т, J=7,1 Гц, 3Н).
0,44 г (1,244 ммоль) 5-(4-хлорфенил)-4-(2-этокси-2-оксоэтил)тиофен-2-карбоновой кислоты солюбилизировали в 5 мл дихлорметана, к раствору добавляли при перемешивании магнитной мешалкой 0,327 мл (3,73 ммоль) оксалил хлорида и по каплям диметилформамид. Смесь перемешивали при к.т. в течение 2 часов, а затем концентрировали в вакууме. Полученный продукт добавляли по каплям при перемешивании магнитной мешалкой к раствору 0,542 мл (6,22 ммоль) морфолина в 5 мл дихлорметана. Смесь перемешивали при к.т. в течение 16 ч. Реакционную среду вливали в 20 мл воды, водную фазу экстрагировали 3×25 мл дихлорметана. Объединенные органические фазы промывали 20 мл насыщенного водного раствора NaHCO3, 20 мл воды, 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, 98/2, об./об.). 0,359 г (выход=68%) этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетата получали в виде желто-оранжевого масла. ЖХ/МС: m/z=394 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,63-7,44 (м, 4Н), 7,41 (с, 1Н), 4,16-3,95 (м, 2Н), 3,66 (дд, J=11,0, 7,8 Гц, 10Н), 1,14 (т, J=7,1 Гц, 3Н).
Пример 17: Приготовление производного №115: этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетат
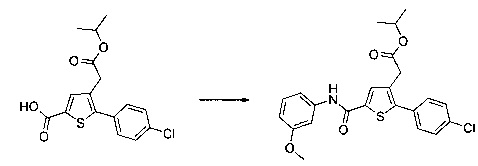 0,5 г (1,476 ммоль) 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновой кислоты солюбилизировали в 10 мл диметилформамида, к раствору добавляли при перемешивании магнитной мешалкой 0,311 г (1,623 ммоль) EDC, 0,249 г (1,623 ммоль) HOBt и 0,411 моль (2,95 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 30 мин., после чего добавляли 0,205 мл (1,77 ммоль) 3-метоксианилина. Затем смесь перемешивали при к.т. в течение 3 часов. Реакционную среду разводили 20 мл этилацетата и промывали 2×20 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 20 мл этилацетата, объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали путем титрования в 10 мл диизопропилового эфира, коричневое твердое вещество выделяли в результате фильтрации с получением 0,529 г (выход=80%) этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетата. ЖХ/МС: m/z=444 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 10,27 (с, 1Н), 8,00 (с, 1Н), 7,55 (к, J=8,6 Гц, 4Н), 7,42 (д, J=2,1 Гц, 1Н), 7,34 (д, J=8,3 Гц, 1Н), 7,26 (т, J=8,1 Гц, 1Н), 6,70 (дд, J=7,8, 1,8 Гц, 1Н), 4,90 (дт, J=12,5, 6,2 Гц, 1Н), 3,76 (с, 3Н), 3,70 (с, 2Н), 1,16 (д, J=6,3 Гц, 6Н).
0,5 г (1,476 ммоль) 5-(4-хлорфенил)-4-(2-изопропокси-2-оксоэтил)тиофен-2-карбоновой кислоты солюбилизировали в 10 мл диметилформамида, к раствору добавляли при перемешивании магнитной мешалкой 0,311 г (1,623 ммоль) EDC, 0,249 г (1,623 ммоль) HOBt и 0,411 моль (2,95 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 30 мин., после чего добавляли 0,205 мл (1,77 ммоль) 3-метоксианилина. Затем смесь перемешивали при к.т. в течение 3 часов. Реакционную среду разводили 20 мл этилацетата и промывали 2×20 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 20 мл этилацетата, объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали путем титрования в 10 мл диизопропилового эфира, коричневое твердое вещество выделяли в результате фильтрации с получением 0,529 г (выход=80%) этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетата. ЖХ/МС: m/z=444 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 10,27 (с, 1Н), 8,00 (с, 1Н), 7,55 (к, J=8,6 Гц, 4Н), 7,42 (д, J=2,1 Гц, 1Н), 7,34 (д, J=8,3 Гц, 1Н), 7,26 (т, J=8,1 Гц, 1Н), 6,70 (дд, J=7,8, 1,8 Гц, 1Н), 4,90 (дт, J=12,5, 6,2 Гц, 1Н), 3,76 (с, 3Н), 3,70 (с, 2Н), 1,16 (д, J=6,3 Гц, 6Н).
Производные под номерами с 116 по 118, 168 и 170 были приготовлены согласно аналогичной процедуре 
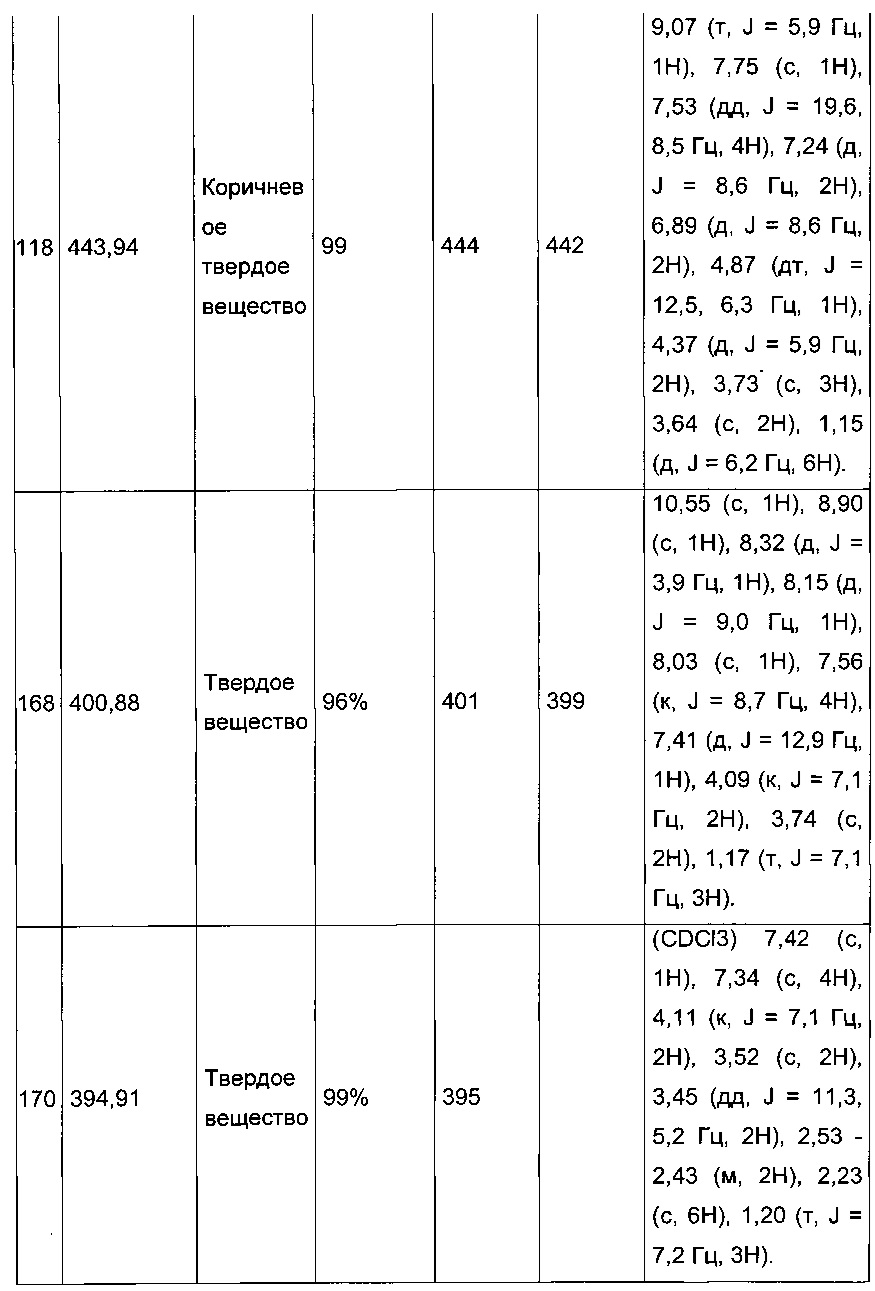
Пример 18: Приготовление производного №119: 2-(2-(4-хлор-фенил)-5-((3-метоксифенил)карбамоил)тиофен-3-ил)уксусная кислота
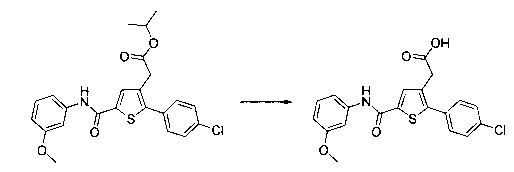 0,1 г (0,225 ммоль) этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетата солюбилизировали при перемешивании магнитной мешалкой в 2 мл метанола, добавляли 0,248 мл (0,248 ммоль) 1 М водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 3 часов. Добавляли 0,01 мл (0,01 ммоль) 1 М водного раствора гидроксида натрия. Смесь снова перемешивали при к.т. в течение 3 часов. Реакционную среду нейтрализовали добавлением раствора 0,019 мл (0,338 ммоль) уксусной кислоты в 5 мл воды. Смесь экстрагировали 2×10 мл этилацетата, объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SО4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 0,082 г (выход=86%) 2-(2-(4-хлорфенил)-5-((3-метоксифенил)карбамоил) тиофен-3-ил)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=402 (МН+); степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 12,69 (с, 1Н), 10,29 (с, 1Н), 8,03 (с, 1Н), 7,66-7,49 (м, 4Н), 7,42 (т, J=2,1 Гц, 1Н), 7,34 (д, J=8,4 Гц, 1Н), 7,25 (т, J=8,1 Гц, 1Н), 6,69 (дд, J=8,0, 2,1 Гц, 1Н), 3,75 (с, 3Н), 3,62 (с, 2Н).
0,1 г (0,225 ммоль) этил 2-(2-(4-хлорфенил)-5-(морфолин-4-карбонил)тиофен-3-ил)ацетата солюбилизировали при перемешивании магнитной мешалкой в 2 мл метанола, добавляли 0,248 мл (0,248 ммоль) 1 М водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 3 часов. Добавляли 0,01 мл (0,01 ммоль) 1 М водного раствора гидроксида натрия. Смесь снова перемешивали при к.т. в течение 3 часов. Реакционную среду нейтрализовали добавлением раствора 0,019 мл (0,338 ммоль) уксусной кислоты в 5 мл воды. Смесь экстрагировали 2×10 мл этилацетата, объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SО4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 0,082 г (выход=86%) 2-(2-(4-хлорфенил)-5-((3-метоксифенил)карбамоил) тиофен-3-ил)уксусной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=402 (МН+); степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 12,69 (с, 1Н), 10,29 (с, 1Н), 8,03 (с, 1Н), 7,66-7,49 (м, 4Н), 7,42 (т, J=2,1 Гц, 1Н), 7,34 (д, J=8,4 Гц, 1Н), 7,25 (т, J=8,1 Гц, 1Н), 6,69 (дд, J=8,0, 2,1 Гц, 1Н), 3,75 (с, 3Н), 3,62 (с, 2Н).
Производные под номерами с 120 по 122, 169, 171 и 172 были приготовлены согласно аналогичной процедуре.
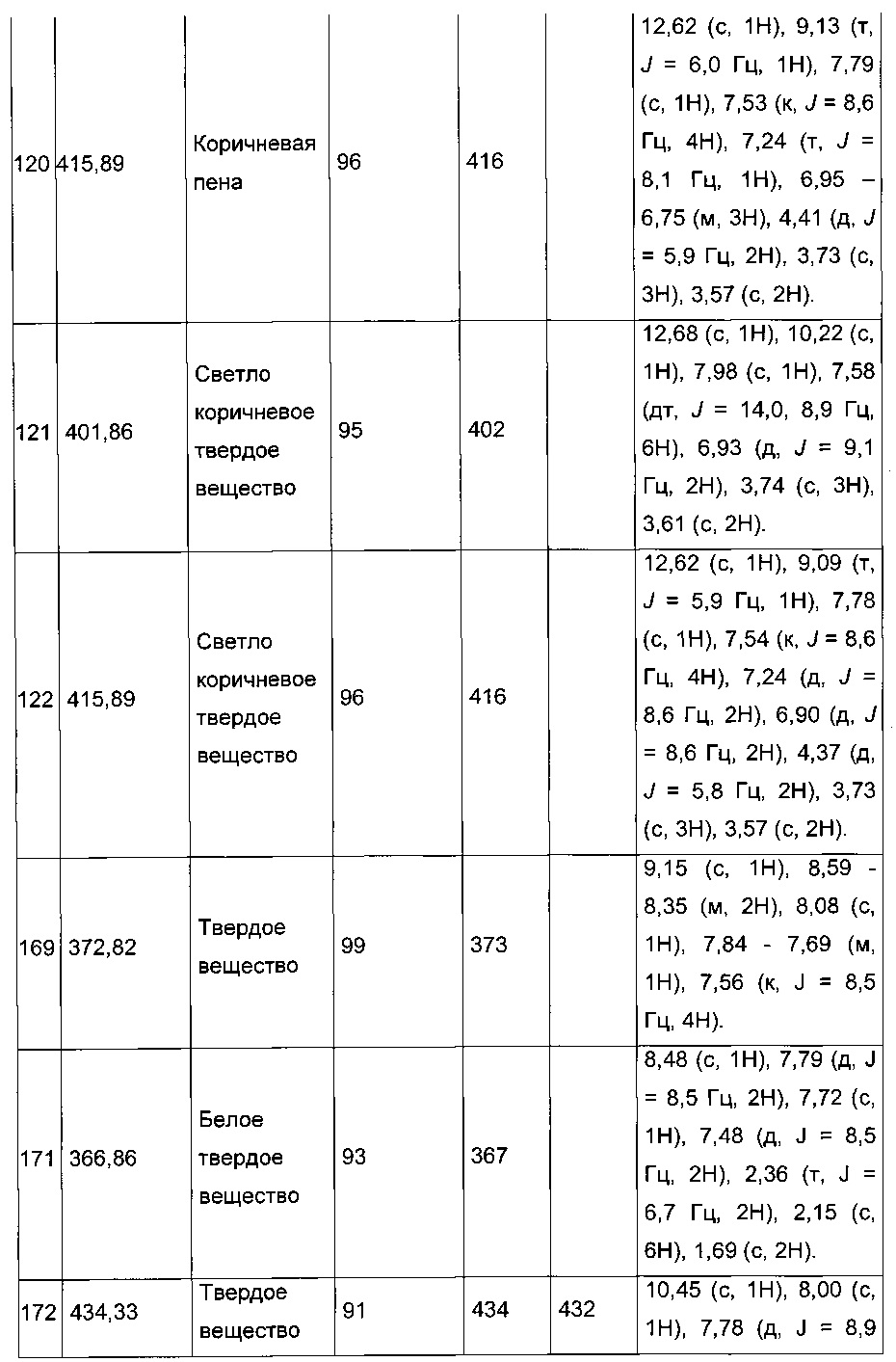
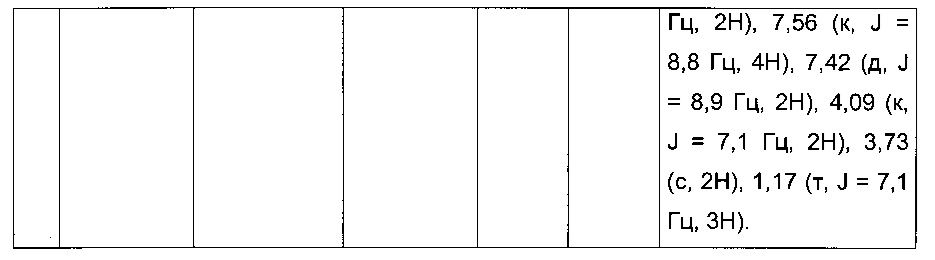
Пример 19: Приготовление производного номер 123: этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропаноат
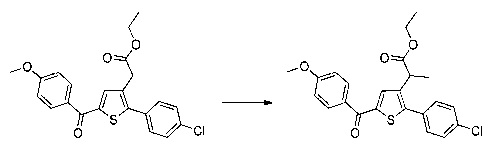 0,25 г (0,597 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата растворяли в 5 мл тетрагидрофурана при перемешивании магнитной мешалкой. Смесь охлаждали до температуры -20°С и добавляли по каплям 0,663 мл (1,193 ммоль) 2 М раствора диизопропиламида лития в тетрагидрофуране. Смесь перемешивали в течение 1 ч при температуре -20°С, после чего добавляли 0,039 мл (0,626 ммоль) метилиодида. Смесь перемешивали еще в течение 1 ч при температуре -20°С. Не дожидаясь нагрева смеси до к.т., смесь вливали в 10 мл воды. Водную фазу экстрагировали 3×10 мл этилацетата, объединенные органические фазы промывали 15 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/петролейного эфира, от 50 до 70% дихлорметана, об./об.). 0,145 г (выход=55%) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)-тиофен-3-ил)пропаноата получали в виде бледно-желтого масла. ЖХ/МС: m/z=429 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 92-7,83 (м, 2Н), 7,68 (с, 1Н), 7,65-7,51 (м, 4Н), 7,19-7,08 (м, 2Н), 4,05 (дт, J=7,2, 3,4 Гц, 2Н), 3,92-3,80 (м, 4Н), 1,43 (д, J=7,1 Гц, 3Н), 1,13 (т, J=7,1 Гц, 3Н).
0,25 г (0,597 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата растворяли в 5 мл тетрагидрофурана при перемешивании магнитной мешалкой. Смесь охлаждали до температуры -20°С и добавляли по каплям 0,663 мл (1,193 ммоль) 2 М раствора диизопропиламида лития в тетрагидрофуране. Смесь перемешивали в течение 1 ч при температуре -20°С, после чего добавляли 0,039 мл (0,626 ммоль) метилиодида. Смесь перемешивали еще в течение 1 ч при температуре -20°С. Не дожидаясь нагрева смеси до к.т., смесь вливали в 10 мл воды. Водную фазу экстрагировали 3×10 мл этилацетата, объединенные органические фазы промывали 15 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/петролейного эфира, от 50 до 70% дихлорметана, об./об.). 0,145 г (выход=55%) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)-тиофен-3-ил)пропаноата получали в виде бледно-желтого масла. ЖХ/МС: m/z=429 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 92-7,83 (м, 2Н), 7,68 (с, 1Н), 7,65-7,51 (м, 4Н), 7,19-7,08 (м, 2Н), 4,05 (дт, J=7,2, 3,4 Гц, 2Н), 3,92-3,80 (м, 4Н), 1,43 (д, J=7,1 Гц, 3Н), 1,13 (т, J=7,1 Гц, 3Н).
Производное номер 124 приготовили согласно аналогичной процедуре.
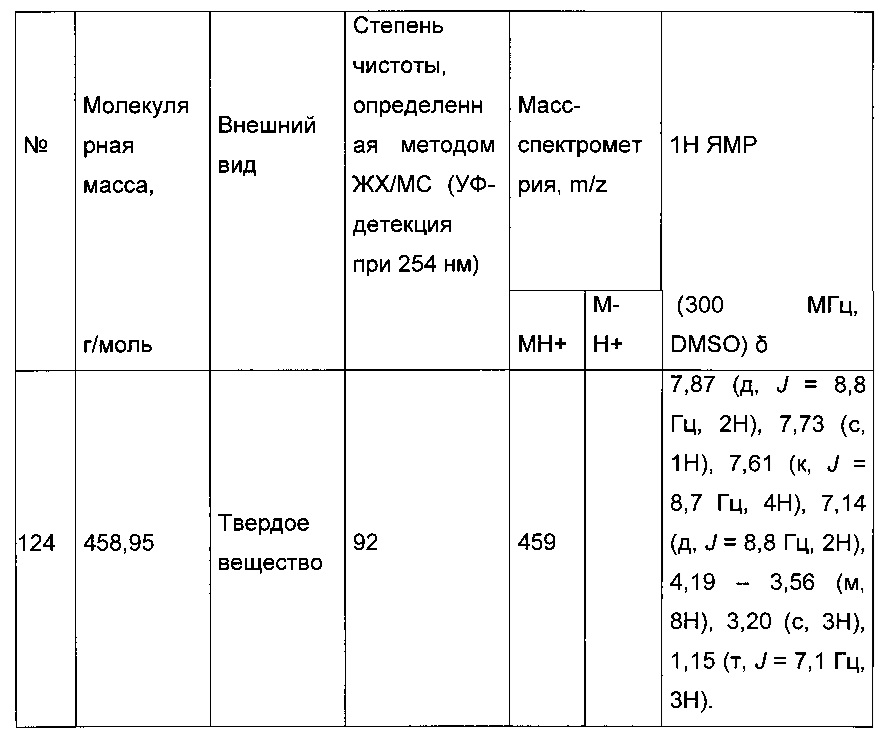
Пример 20: Приготовление производного номер 125: изопропил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропаноат
 0,2 г (0,382 ммоль) изопропил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата солюбилизировали в 3 мл тетрагидрофурана при перемешивании магнитной мешалкой. Смесь охлаждали до температуры -80°С и добавляли по каплям 0,765 мл (0,765 ммоль) 1 н раствора бис-триметилсилиламида лития в тетрагидрофуране. Смесь перемешивали в течение 30 мин. при температуре -50°С, после чего снова охлаждали до температуры -80°С и добавляли 0,058 мл (0,765 ммоль) метоксиметан хлорида. Холодную баню удаляли, смесь перемешивали в течение еще 1 часа, после чего смеси позволяли нагреться до к.т. Реакционную среду вливали в 20 мл насыщенного раствора хлорида аммония. Водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 15 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100% до 0% петролейного эфира, об./об.). 0,062 г (выход=33%) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропаноата получали в виде твердого вещества. ЖХ/МС: m/z=473 (МН+); степень чистоты (УФ-детекция при 254 нм)=96%. 1Н ЯМР (300 МГц, DMSO) δ 7,87 (д, J=8,6 Гц, 2Н), 7,72 (с, 1Н), 7,61 (д, J=8,5 Гц, 4Н), 7,14 (д, J=8,6 Гц, 2Н), 4,90 (дт, J=12,4, 6,1 Гц, 1Н), 4,02-3,76 (м, 5Н), 3,72-3,60 (м, 1Н), 3,20 (с, 3Н), 1,15 (дд, J=14,8, 6,1 Гц, 6Н).
0,2 г (0,382 ммоль) изопропил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата солюбилизировали в 3 мл тетрагидрофурана при перемешивании магнитной мешалкой. Смесь охлаждали до температуры -80°С и добавляли по каплям 0,765 мл (0,765 ммоль) 1 н раствора бис-триметилсилиламида лития в тетрагидрофуране. Смесь перемешивали в течение 30 мин. при температуре -50°С, после чего снова охлаждали до температуры -80°С и добавляли 0,058 мл (0,765 ммоль) метоксиметан хлорида. Холодную баню удаляли, смесь перемешивали в течение еще 1 часа, после чего смеси позволяли нагреться до к.т. Реакционную среду вливали в 20 мл насыщенного раствора хлорида аммония. Водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 15 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100% до 0% петролейного эфира, об./об.). 0,062 г (выход=33%) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропаноата получали в виде твердого вещества. ЖХ/МС: m/z=473 (МН+); степень чистоты (УФ-детекция при 254 нм)=96%. 1Н ЯМР (300 МГц, DMSO) δ 7,87 (д, J=8,6 Гц, 2Н), 7,72 (с, 1Н), 7,61 (д, J=8,5 Гц, 4Н), 7,14 (д, J=8,6 Гц, 2Н), 4,90 (дт, J=12,4, 6,1 Гц, 1Н), 4,02-3,76 (м, 5Н), 3,72-3,60 (м, 1Н), 3,20 (с, 3Н), 1,15 (дд, J=14,8, 6,1 Гц, 6Н).
Пример 21: Приготовление производного номер 126: изопропил 2-(2-(4-хлорфенил)-5-(циклогексанкарбонил)тиофен-3-ил)-3-фенилпропаноат
Этап 1: Приготовление этил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата
 1 г (3,56 ммоль) этил 2-(2-(4-хлорфенил)тиофен-3-ил)ацетата солюбилизировали в атмосфере аргона в 5 мл тетрагидрофурана. Смесь охлаждали до температуры -50°С и добавляли по каплям 0,663 мл (3,56 ммоль) 2 М раствора лития диизопропиламида в тетрагидрофуране при перемешивании магнитной мешалкой. Реакционную смесь перемешивали в течение 5 мин. при температуре -20°С, после чего охлаждали до -50°С. Добавляли по каплям 1,272 мл (10,68 ммоль) бензил бромида. Смесь перемешивали при температуре -50°С в течение 15 мин., после чего холодную баню удаляли, и смесь перемешивали в течение 1 ч, позволяя смеси нагреться до к.т. Реакционную среду вливали в 30 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×20 мл дихлорметана, объединенные органические фазы промывали 30 мл 1 н водного раствора хлористоводородной кислоты, 30 мл воды, после чего высушивали над MgS04, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 1,285 г (выход=94%) этил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенил-пропаноата в виде оранжевого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 7,54 (д, J=5,3 Гц, 1Н), 7,43 (д, J=8,5 Гц, 2Н), 7,24 (д, J=5,3 Гц, 1Н), 7,13 (д, J=7,8 Гц, 6Н), 6,99-6,86 (м, 2Н), 4,04-3,91 (м, 2Н), 3,85 (т, J=7,8 Гц, 1Н), 3,23 (дд, J=13,5, 7,6 Гц, 1Н), 2,95 (дд, J=13,5, 8,0 Гц, 1Н), 1,03 (т, J=7,1 Гц, 3Н).
1 г (3,56 ммоль) этил 2-(2-(4-хлорфенил)тиофен-3-ил)ацетата солюбилизировали в атмосфере аргона в 5 мл тетрагидрофурана. Смесь охлаждали до температуры -50°С и добавляли по каплям 0,663 мл (3,56 ммоль) 2 М раствора лития диизопропиламида в тетрагидрофуране при перемешивании магнитной мешалкой. Реакционную смесь перемешивали в течение 5 мин. при температуре -20°С, после чего охлаждали до -50°С. Добавляли по каплям 1,272 мл (10,68 ммоль) бензил бромида. Смесь перемешивали при температуре -50°С в течение 15 мин., после чего холодную баню удаляли, и смесь перемешивали в течение 1 ч, позволяя смеси нагреться до к.т. Реакционную среду вливали в 30 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×20 мл дихлорметана, объединенные органические фазы промывали 30 мл 1 н водного раствора хлористоводородной кислоты, 30 мл воды, после чего высушивали над MgS04, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 1,285 г (выход=94%) этил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенил-пропаноата в виде оранжевого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 7,54 (д, J=5,3 Гц, 1Н), 7,43 (д, J=8,5 Гц, 2Н), 7,24 (д, J=5,3 Гц, 1Н), 7,13 (д, J=7,8 Гц, 6Н), 6,99-6,86 (м, 2Н), 4,04-3,91 (м, 2Н), 3,85 (т, J=7,8 Гц, 1Н), 3,23 (дд, J=13,5, 7,6 Гц, 1Н), 2,95 (дд, J=13,5, 8,0 Гц, 1Н), 1,03 (т, J=7,1 Гц, 3Н).
Этап 2: Приготовление 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропановой кислоты
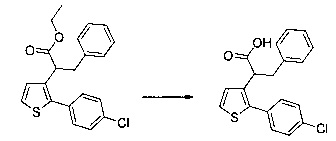 1 г (3,56 ммоль) этил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата солюбилизировали в 10 мл этанола при перемешивании магнитной мешалкой. Добавляли 0,311 мл (3,11 ммоль) 10 М водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 3 д, а затем концентрировали в вакууме. Добавляли 1 н водный раствор хлористоводородной кислоты до появления преципитата, водную фазу экстрагировали 2×25 мл этилацетата. Объединенные органические фазы промывали 40 мл воды, 40 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Осадок очищали путем титрования в 15 мл смеси петролейного эфира и диизопропилового эфира (1/1, об./об.). Твердое вещество выделяли в результате фильтрации, промывали 10 мл петролейного эфира и высушивали в вакуумном колпаке с получением 0,681 г (выход=70%) 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропановой кислоты. ЖХ/МС: m/z=340 (М-Н+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 12,56 (с, 1Н), 7,58 (д, J=5,3 Гц, 1Н), 7,46 (д, J=8,4 Гц, 2Н), 7,27 (д, J=5,3 Гц, 1Н), 7,17 (дд, J=10,7, 7,7 Гц, 5Н), 6,94 (дд, J=7,3, 1,9 Гц, 2Н), 3,83 (т, J=7,7 Гц, 1Н), 2,93 (дд, J=13,6, 8,1 Гц, 1Н).
1 г (3,56 ммоль) этил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата солюбилизировали в 10 мл этанола при перемешивании магнитной мешалкой. Добавляли 0,311 мл (3,11 ммоль) 10 М водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 3 д, а затем концентрировали в вакууме. Добавляли 1 н водный раствор хлористоводородной кислоты до появления преципитата, водную фазу экстрагировали 2×25 мл этилацетата. Объединенные органические фазы промывали 40 мл воды, 40 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Осадок очищали путем титрования в 15 мл смеси петролейного эфира и диизопропилового эфира (1/1, об./об.). Твердое вещество выделяли в результате фильтрации, промывали 10 мл петролейного эфира и высушивали в вакуумном колпаке с получением 0,681 г (выход=70%) 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропановой кислоты. ЖХ/МС: m/z=340 (М-Н+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 12,56 (с, 1Н), 7,58 (д, J=5,3 Гц, 1Н), 7,46 (д, J=8,4 Гц, 2Н), 7,27 (д, J=5,3 Гц, 1Н), 7,17 (дд, J=10,7, 7,7 Гц, 5Н), 6,94 (дд, J=7,3, 1,9 Гц, 2Н), 3,83 (т, J=7,7 Гц, 1Н), 2,93 (дд, J=13,6, 8,1 Гц, 1Н).
Этап 3: Приготовление изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата
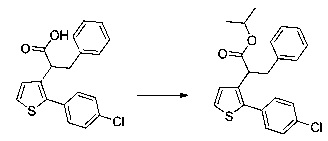 0,55 г (1,604 ммоль) 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропановой кислоты растворяли в 25 мл изопропанола, к полученному раствору добавляли каплю серной кислоты. Смесь нагревали в колбе с обратным холодильником в течение 16 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок переносили в 25 мл, и смесь перемешивали при к.т. в течение 16 ч. Преципитат выделяли в результате фильтрации; промывали смесью, состоящей из 10 мл воды и 0,5 мл петролейного эфира, и высушивали в вакуумном колпаке с получением 0,46 г (выход=71%) изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата в виде белого твердого вещества. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 7,57 (д, J=5,3 Гц, 1Н), 7,46 (д, J=8,5 Гц, 2Н), 7,34-7,05 (м, 6Н), 6,96 (д, J=5,4 Гц, 2Н), 4,78 (дт, J=12,5, 6,2 Гц, 1Н), 3,84 (т, J=7,8 Гц, 1Н), 3,24 (дд, J=13,5, 8,1 Гц, 1Н), 2,96 (дд, J=13,5, 7,6 Гц, 1Н), 1,02 (дд, J=6,2, 3,4 Гц, 6Н).
0,55 г (1,604 ммоль) 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропановой кислоты растворяли в 25 мл изопропанола, к полученному раствору добавляли каплю серной кислоты. Смесь нагревали в колбе с обратным холодильником в течение 16 ч при перемешивании магнитной мешалкой. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок переносили в 25 мл, и смесь перемешивали при к.т. в течение 16 ч. Преципитат выделяли в результате фильтрации; промывали смесью, состоящей из 10 мл воды и 0,5 мл петролейного эфира, и высушивали в вакуумном колпаке с получением 0,46 г (выход=71%) изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата в виде белого твердого вещества. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 7,57 (д, J=5,3 Гц, 1Н), 7,46 (д, J=8,5 Гц, 2Н), 7,34-7,05 (м, 6Н), 6,96 (д, J=5,4 Гц, 2Н), 4,78 (дт, J=12,5, 6,2 Гц, 1Н), 3,84 (т, J=7,8 Гц, 1Н), 3,24 (дд, J=13,5, 8,1 Гц, 1Н), 2,96 (дд, J=13,5, 7,6 Гц, 1Н), 1,02 (дд, J=6,2, 3,4 Гц, 6Н).
Этап 4: Приготовление изопропил 2-(2-(4-хлорфенил)-5-(циклогексан-карбонил)тиофен-3-ил)-3-фенилпропаноата (производное номер 126)
 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 2 мл дихлорметана, 0,285 г (2,14 ммоль) триалюминий хлорида, 0,173 г (1,77 ммоль) циклогексан карбонилхлорида и раствор 0,429 г (1,07 ммоль) изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата в 3 мл дихлорметана. Полученную смесь перемешивали при к.т. в течение 16 ч, после чего вливали в 25 мл смеси, состоящей из воды и льда, и перемешивали в течение 30 мин. Водную фазу экстрагировали 2×15 мл этилацетата. Объединенные органические фазы промывали 15 мл насыщенного водного раствора NaHCO3, 15 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: диизопропиловый эфир/петролейный эфир, 1/9, об./об.). 0,108 г (выход=20%) изопропил 2-(2-(4-хлорфенил)-5-(циклогексанкарбонил)тиофен-3-ил)-3-фенилпропаноата получали в виде твердого вещества. ЖХ/МС: m/z=495 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 8,07 (с, 1Н), 7,51 (д, J=8,3 Гц, 2Н), 7,20 (дд, J=18,0, 7,5 Гц, 5Н), 7,00 (д, J=7,2 Гц, 2Н), 4,81 (дт, J=12,5, 6,2 Гц, 1Н), 3,85 (т, J=7,8 Гц, 1Н), 3,10 (дд, J=13,4, 8,0 Гц, 1Н), 1,76 (т, J=23,5 Гц, 5Н), 1,58-1,11 (м, 6Н), 1,04 (т, J=6,0 Гц, 6Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 2 мл дихлорметана, 0,285 г (2,14 ммоль) триалюминий хлорида, 0,173 г (1,77 ммоль) циклогексан карбонилхлорида и раствор 0,429 г (1,07 ммоль) изопропил 2-(2-(4-хлорфенил)тиофен-3-ил)-3-фенилпропаноата в 3 мл дихлорметана. Полученную смесь перемешивали при к.т. в течение 16 ч, после чего вливали в 25 мл смеси, состоящей из воды и льда, и перемешивали в течение 30 мин. Водную фазу экстрагировали 2×15 мл этилацетата. Объединенные органические фазы промывали 15 мл насыщенного водного раствора NaHCO3, 15 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: диизопропиловый эфир/петролейный эфир, 1/9, об./об.). 0,108 г (выход=20%) изопропил 2-(2-(4-хлорфенил)-5-(циклогексанкарбонил)тиофен-3-ил)-3-фенилпропаноата получали в виде твердого вещества. ЖХ/МС: m/z=495 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 8,07 (с, 1Н), 7,51 (д, J=8,3 Гц, 2Н), 7,20 (дд, J=18,0, 7,5 Гц, 5Н), 7,00 (д, J=7,2 Гц, 2Н), 4,81 (дт, J=12,5, 6,2 Гц, 1Н), 3,85 (т, J=7,8 Гц, 1Н), 3,10 (дд, J=13,4, 8,0 Гц, 1Н), 1,76 (т, J=23,5 Гц, 5Н), 1,58-1,11 (м, 6Н), 1,04 (т, J=6,0 Гц, 6Н).
Пример 22: Приготовление производного №127: 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановая кислота
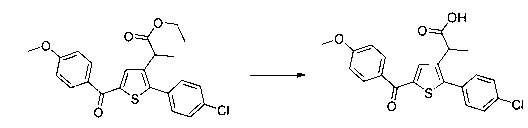 0,087 г (0,197 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)-пропаноата солюбилизировали в 1 мл смеси тетрагидрофурана и этанола (1/1, об./об.), к раствору добавляли при перемешивании магнитной мешалкой 0,197 мл (0,197 ммоль) 1 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу экстрагировали 10 мл метил-трет-бутилового эфира. Водную фазу затем подкисляли до значения рН 3-4 добавлением 1 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 3×10 мл этилацетата, объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 0,077 г (выход=95%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановой кислоты в виде белого твердого вещества. ЖХ/МС: m/z=401 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 7,97-7,79 (м, 4Н), 7,73 (с, 1Н), 7,57 (т, J=10,0 Гц, 2Н), 7,13 (д, J=8,8 Гц, 2Н), 3,88 (с, 3Н), 3,46 (д, J=7,1 Гц, 2Н), 1,25 (д, J=7,0 Гц, 3Н).
0,087 г (0,197 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)-пропаноата солюбилизировали в 1 мл смеси тетрагидрофурана и этанола (1/1, об./об.), к раствору добавляли при перемешивании магнитной мешалкой 0,197 мл (0,197 ммоль) 1 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу экстрагировали 10 мл метил-трет-бутилового эфира. Водную фазу затем подкисляли до значения рН 3-4 добавлением 1 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 3×10 мл этилацетата, объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 0,077 г (выход=95%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановой кислоты в виде белого твердого вещества. ЖХ/МС: m/z=401 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 7,97-7,79 (м, 4Н), 7,73 (с, 1Н), 7,57 (т, J=10,0 Гц, 2Н), 7,13 (д, J=8,8 Гц, 2Н), 3,88 (с, 3Н), 3,46 (д, J=7,1 Гц, 2Н), 1,25 (д, J=7,0 Гц, 3Н).
Производное 128 приготовили согласно аналогичной процедуре:
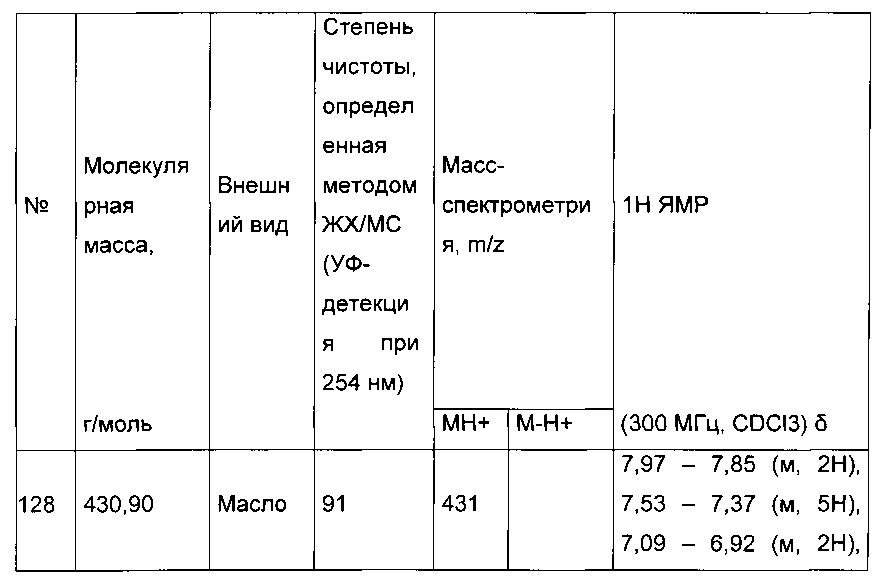
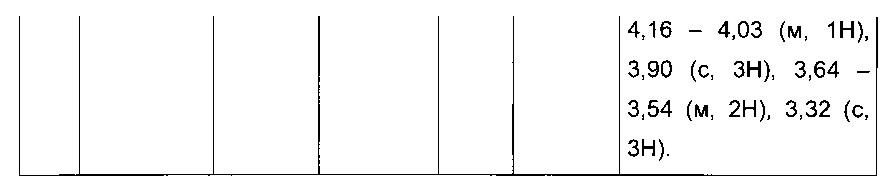
Пример 23: Приготовление производного №129: этил 2-(2-(4-фтор-2-метоксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетат
Этап 1: Приготовление этил 2-(тиофен-3-ил)ацетата
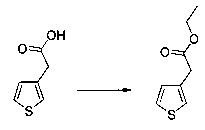 10,75 г (74,1 ммоль) 2-(тиофен-3-ил)уксусной кислоты солюбилизировали в 100 мл этанола, к раствору добавляли 6 мл (72 ммоль) серной кислоты. Смесь нагревали при перемешивании магнитной мешалкой в колбе с обратным холодильником в течение 24 ч. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок обрабатывали 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×100 мл этилацетата. Объединенные органические фазы промывали 100 мл воды, 100 мл насыщенного водного раствора NaHCO3 и 100 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 12,27 г (выход=97%) этил 2-(тиофен-3-ил)ацетата в виде бесцветного масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, CDCl3) δ 7,24 (дд, J=6,0, 3,0 Гц, 1Н), 7,16-7,07 (м, 1Н), 7,02 (дд, J=4,9, 1,1 Гц, 1Н), 4,13 (к, J=7,1 Гц, 2Н), 3,62 (с, 2Н), 1,23 (т, J=7,1 Гц, 3Н).
10,75 г (74,1 ммоль) 2-(тиофен-3-ил)уксусной кислоты солюбилизировали в 100 мл этанола, к раствору добавляли 6 мл (72 ммоль) серной кислоты. Смесь нагревали при перемешивании магнитной мешалкой в колбе с обратным холодильником в течение 24 ч. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок обрабатывали 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×100 мл этилацетата. Объединенные органические фазы промывали 100 мл воды, 100 мл насыщенного водного раствора NaHCO3 и 100 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 12,27 г (выход=97%) этил 2-(тиофен-3-ил)ацетата в виде бесцветного масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, CDCl3) δ 7,24 (дд, J=6,0, 3,0 Гц, 1Н), 7,16-7,07 (м, 1Н), 7,02 (дд, J=4,9, 1,1 Гц, 1Н), 4,13 (к, J=7,1 Гц, 2Н), 3,62 (с, 2Н), 1,23 (т, J=7,1 Гц, 3Н).
Этап 2: Приготовление этил 2-(2-бромтиофен-3-ил)ацетата
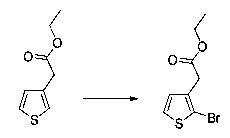 12,2 г (71,7 ммоль) этил 2-(тиофен-3-ил)ацетата солюбилизировали в 100 мл тетрагидрофурана при перемешивании магнитной мешалкой, добавляли 121,7 ммоль N-бромсукцинимида, и смесь перемешивали в колбе с обратным холодильником в течение 3 часов. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок переносили в минимальное количество петролейного эфира, фильтровали на геле гидроксида кремния (элюент: 100% дихлорметан) с получением 18,38 г (выход=88%) этил 2-(2-бром-тиофен-3-ил)ацетата в виде бледно-желтого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, CDCl3) 6 7,27-7,16 (м, 1Н), 7,03-6,86 (м, 1Н), 4,12 (кд, J=7,1, 3,2 Гц, 2Н), 3,64-3,48 (м, 2Н), 1,22 (тд, J=7,1, 1,4 Гц, 3Н).
12,2 г (71,7 ммоль) этил 2-(тиофен-3-ил)ацетата солюбилизировали в 100 мл тетрагидрофурана при перемешивании магнитной мешалкой, добавляли 121,7 ммоль N-бромсукцинимида, и смесь перемешивали в колбе с обратным холодильником в течение 3 часов. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок переносили в минимальное количество петролейного эфира, фильтровали на геле гидроксида кремния (элюент: 100% дихлорметан) с получением 18,38 г (выход=88%) этил 2-(2-бром-тиофен-3-ил)ацетата в виде бледно-желтого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, CDCl3) 6 7,27-7,16 (м, 1Н), 7,03-6,86 (м, 1Н), 4,12 (кд, J=7,1, 3,2 Гц, 2Н), 3,64-3,48 (м, 2Н), 1,22 (тд, J=7,1, 1,4 Гц, 3Н).
Этап 3: Приготовление этил 2-(2-(4-фтор-2-метоксифенил)тиофен-3-ил)ацетата
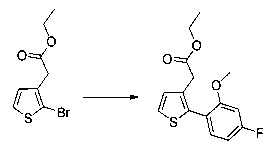 0,7 г (2,53 ммоль) этил 2-(2-бромтиофен-3-ил)ацетата солюбилизировали в атмосфере аргона в 10 мл смеси, состоящей из толуола и этанола (1/1, об./об.), добавляли при перемешивании магнитной мешалкой 0,645 г (3,79 ммоль) 4-фтор-2-метоксифенилбориновой кислоты, 8,85 мл (17,7 ммоль) 2 М водного раствора Na2CO3 и 0,148 г (0,126 ммоль) тетракис(трифенилфосфина) палладия [0]. Смесь перемешивали в колбе с обратным холодильником в течение 3 часов. Смеси позволяли остыть до к.т., после чего смесь выливали, разводили 15 мл воды и 15 мл этилацетата, после чего фильтровали через целит. После сепарации водную фазу экстрагировали 20 мл этилацетата. Объединенные органические фазы промывали 10 мл воды, 10 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/этилацетата, от 100 до 90% петролейного эфира, об./об.). 0,529 г (выход=70%) этил 2-(2-(4-фтор-2-метоксифенил)тиофен-3-ил)ацетата получали в виде светло-желтой пены. ЖХ/МС: m/z=295 (МН+); степень чистоты (УФ-детекция при 254 нм)=83%. 1Н ЯМР (300 МГц, CDCl3) δ 7,51 (д, J=5,2 Гц, 1Н), 7,26 (дд, J=8,4, 6,9 Гц, 1Н), 7,09-7,00 (м, 2Н), 6,84 (тд, J=8,4, 2,5 Гц, 1Н), 4,01 (к, J=7,1 Гц, 2Н), 3,74 (с, 3Н), 3,41 (с, 2Н), 1,13 (т, J=7,1 Гц, 3Н).
0,7 г (2,53 ммоль) этил 2-(2-бромтиофен-3-ил)ацетата солюбилизировали в атмосфере аргона в 10 мл смеси, состоящей из толуола и этанола (1/1, об./об.), добавляли при перемешивании магнитной мешалкой 0,645 г (3,79 ммоль) 4-фтор-2-метоксифенилбориновой кислоты, 8,85 мл (17,7 ммоль) 2 М водного раствора Na2CO3 и 0,148 г (0,126 ммоль) тетракис(трифенилфосфина) палладия [0]. Смесь перемешивали в колбе с обратным холодильником в течение 3 часов. Смеси позволяли остыть до к.т., после чего смесь выливали, разводили 15 мл воды и 15 мл этилацетата, после чего фильтровали через целит. После сепарации водную фазу экстрагировали 20 мл этилацетата. Объединенные органические фазы промывали 10 мл воды, 10 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/этилацетата, от 100 до 90% петролейного эфира, об./об.). 0,529 г (выход=70%) этил 2-(2-(4-фтор-2-метоксифенил)тиофен-3-ил)ацетата получали в виде светло-желтой пены. ЖХ/МС: m/z=295 (МН+); степень чистоты (УФ-детекция при 254 нм)=83%. 1Н ЯМР (300 МГц, CDCl3) δ 7,51 (д, J=5,2 Гц, 1Н), 7,26 (дд, J=8,4, 6,9 Гц, 1Н), 7,09-7,00 (м, 2Н), 6,84 (тд, J=8,4, 2,5 Гц, 1Н), 4,01 (к, J=7,1 Гц, 2Н), 3,74 (с, 3Н), 3,41 (с, 2Н), 1,13 (т, J=7,1 Гц, 3Н).
Этап 4: Приготовление этил 2-(2-(4-фтор-2-метоксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата (производное номер 129)
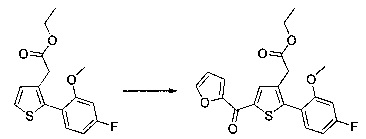 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 5 мл дихлорметана, 0,583 г (4,37 ммоль) триалюминий хлорида, раствор 0,19 мл (1,924 ммоль) 2-фуроил хлорида в 2 мл дихлорметана и раствор 0,52 г (1,749 ммоль) этил 2-(2-(4-фтор-2-метоксифенил)тиофен-3-ил)ацетата в 2 мл дихлорметана. Полученную смесь перемешивали в колбе с обратным холодильником в течение 3 часов и затем при к.т. в течение 16 ч, после чего вливали в 15 мл смеси, состоящей из воды и льда, и перемешивали в течение 30 мин. Добавляли 15 мл дихлорметана, смесь фильтровали через целит. После сепарации водную фазу экстрагировали 20 мл дихлорметана. Объединенные органические фазы промывали 20 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 0% петролейного эфира, об./об.). 0,287 г (выход=41%) этил 2-(2-(4-фтор-2-метокси-фенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде твердого вещества. ЖХ/МС: m/z=389 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,19-8,11 (м, 2Н), 7,56 (д, J=3,6 Гц, 1Н), 7,37 (дд, J=8,5, 6,8 Гц, 1Н), 7,11 (дд, J=11,4, 2,4 Гц, 1Н), 6,91 (тд, J=8,4, 2,5 Гц, 1Н), 6,84 (дд, J=3,6, 1,7 Гц, 1Н), 4,04 (к, J=7,1 Гц, 2Н), 3,78 (с, 3Н), 3,53 (с, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 5 мл дихлорметана, 0,583 г (4,37 ммоль) триалюминий хлорида, раствор 0,19 мл (1,924 ммоль) 2-фуроил хлорида в 2 мл дихлорметана и раствор 0,52 г (1,749 ммоль) этил 2-(2-(4-фтор-2-метоксифенил)тиофен-3-ил)ацетата в 2 мл дихлорметана. Полученную смесь перемешивали в колбе с обратным холодильником в течение 3 часов и затем при к.т. в течение 16 ч, после чего вливали в 15 мл смеси, состоящей из воды и льда, и перемешивали в течение 30 мин. Добавляли 15 мл дихлорметана, смесь фильтровали через целит. После сепарации водную фазу экстрагировали 20 мл дихлорметана. Объединенные органические фазы промывали 20 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 0% петролейного эфира, об./об.). 0,287 г (выход=41%) этил 2-(2-(4-фтор-2-метокси-фенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде твердого вещества. ЖХ/МС: m/z=389 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,19-8,11 (м, 2Н), 7,56 (д, J=3,6 Гц, 1Н), 7,37 (дд, J=8,5, 6,8 Гц, 1Н), 7,11 (дд, J=11,4, 2,4 Гц, 1Н), 6,91 (тд, J=8,4, 2,5 Гц, 1Н), 6,84 (дд, J=3,6, 1,7 Гц, 1Н), 4,04 (к, J=7,1 Гц, 2Н), 3,78 (с, 3Н), 3,53 (с, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
Производные под номерами с 130 по 134 были приготовлены согласно той же последовательности 4 этапов.
Пример 24: Приготовление производного номер 135: 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановая кислота
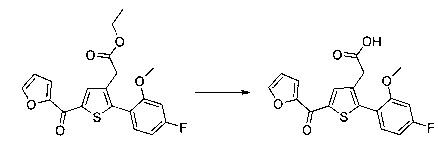 0,207 г (0,522 ммоль) этил 2-(2-(4-фтор-2-метоксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 3 мл смеси, состоящей из тетрагидрофурана и этанола (1/2, об./об.), к полученному раствору добавляли при перемешивании магнитной мешалкой 0,057 мл (0,575 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 3 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу подкисляли до значения рН 2-3 добавлением 12 н водного раствора хлористоводородной кислоты. Добавляли 2 мл петролейного эфира, смесь интенсивно перемешивали в течение 15 мин. Преципитат выделяли в результате фильтрации, промывали 5 мл воды и 5 мл петролейного эфира, после чего высушивали в вакуумном колпаке с получением 0,136 г (выход=70%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановой кислоты. ЖХ/МС: m/z=361 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 12,44 (с, 1Н), 8,15 (с, 2Н), 7,56 (д, J=3,6 Гц, 1Н), 7,38 (дд, J=8,4, 6,8 Гц, 1Н), 7,11 (дд, J=11,4, 2,4 Гц, 1Н), 6,91 (тд, J=8,4, 2,4 Гц, 1Н), 6,86-6,77 (м, 1Н), 3,78 (с, 3Н), 3,44 (с, 2Н).
0,207 г (0,522 ммоль) этил 2-(2-(4-фтор-2-метоксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 3 мл смеси, состоящей из тетрагидрофурана и этанола (1/2, об./об.), к полученному раствору добавляли при перемешивании магнитной мешалкой 0,057 мл (0,575 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 3 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу подкисляли до значения рН 2-3 добавлением 12 н водного раствора хлористоводородной кислоты. Добавляли 2 мл петролейного эфира, смесь интенсивно перемешивали в течение 15 мин. Преципитат выделяли в результате фильтрации, промывали 5 мл воды и 5 мл петролейного эфира, после чего высушивали в вакуумном колпаке с получением 0,136 г (выход=70%) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановой кислоты. ЖХ/МС: m/z=361 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 12,44 (с, 1Н), 8,15 (с, 2Н), 7,56 (д, J=3,6 Гц, 1Н), 7,38 (дд, J=8,4, 6,8 Гц, 1Н), 7,11 (дд, J=11,4, 2,4 Гц, 1Н), 6,91 (тд, J=8,4, 2,4 Гц, 1Н), 6,86-6,77 (м, 1Н), 3,78 (с, 3Н), 3,44 (с, 2Н).
Производные 136 и 137 были приготовлены согласно аналогичной процедуре.

Пример 25: Приготовление производного номер 138: 2-(2-(4-фтор-2-гидроксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусная кислота
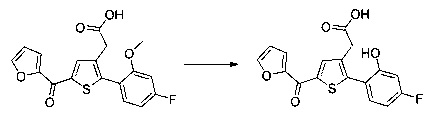 0,122 г (0,816 ммоль) D,L-метионин солюбилизировали в 0,882 мл (13,69 ммоль) метансульфоновой кислоты при перемешивании магнитной мешалкой. К раствору добавляли 0,1 г (0,272 ммоль) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановой кислоты. Смесь нагревали до температуры 80°С в течение 4 д, при этом каждые 24 ч добавляли 0,041 г (0,272 ммоль) D,L-метионина (т.е. в сумме проводили 3 добавления за 4 д). Смеси позволяли остыть до к.т., после чего смесь вливали в 15 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 15 мл изобутанола. Органическую фазу промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgS04, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме и очищали методом препаративной ВЭЖХ/МС с получением 0,02 г (выход=21%) 2-(2-(4-фтор-2-гидроксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты в виде светло-желтого твердого вещества. ЖХ/МС: m/z=347 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 11,66 (с, 1Н), 8,14 (с, 2Н), 7,55 (д, J=4,0 Гц, 1Н), 7,29 (дд, J=8,9, 6,9 Гц, 1Н), 6,83 (дд, J=3,6, 1,7 Гц, 1Н), 6,75 (т, J=8,1 Гц, 2Н), 3,51 (с, 2Н).
0,122 г (0,816 ммоль) D,L-метионин солюбилизировали в 0,882 мл (13,69 ммоль) метансульфоновой кислоты при перемешивании магнитной мешалкой. К раствору добавляли 0,1 г (0,272 ммоль) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)пропановой кислоты. Смесь нагревали до температуры 80°С в течение 4 д, при этом каждые 24 ч добавляли 0,041 г (0,272 ммоль) D,L-метионина (т.е. в сумме проводили 3 добавления за 4 д). Смеси позволяли остыть до к.т., после чего смесь вливали в 15 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 15 мл изобутанола. Органическую фазу промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgS04, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме и очищали методом препаративной ВЭЖХ/МС с получением 0,02 г (выход=21%) 2-(2-(4-фтор-2-гидроксифенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты в виде светло-желтого твердого вещества. ЖХ/МС: m/z=347 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 11,66 (с, 1Н), 8,14 (с, 2Н), 7,55 (д, J=4,0 Гц, 1Н), 7,29 (дд, J=8,9, 6,9 Гц, 1Н), 6,83 (дд, J=3,6, 1,7 Гц, 1Н), 6,75 (т, J=8,1 Гц, 2Н), 3,51 (с, 2Н).
Пример 26: Приготовление производного номер 139: этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетат
Этап 1: Приготовление этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата
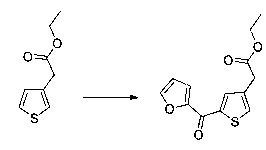 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 25 мл дихлорметана, 7,84 г (0,585 ммоль) триалюминий хлорида, раствор 3,35 мл (32,3 ммоль) 2-фуроил хлорида в 10 мл дихлорметана и раствор 5 г (29,35 ммоль) этил 2-(тиофен-3-ил)ацетата в 10 мл дихлорметана. Полученную смесь перемешивали в колбе с обратным холодильником в течение 1 ч, после чего вливали в 75 мл смеси, состоящей из воды и льда, и перемешивали при к.т. в течение 1 ч. Водную фазу экстрагировали 2×50 мл дихлорметана. Объединенные органические фазы промывали 75 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/этилацетата, от 100 до 80% петролейного эфира, об./об.). 5,9 г (выход=72%) этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде твердого вещества. ЖХ/МС: m/z=265 (МН+); степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 8,12 (с, 2Н), 7,88 (с, 1Н), 7,54 (д, J=3,6 Гц, 1Н), 6,82 (дд, J=3,6, 1,7 Гц, 1Н), 4,11 (к, J=7,1 Гц, 2Н), 3,79 (с, 2Н), 1,21 (т, J=7,1 Гц, 3Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 25 мл дихлорметана, 7,84 г (0,585 ммоль) триалюминий хлорида, раствор 3,35 мл (32,3 ммоль) 2-фуроил хлорида в 10 мл дихлорметана и раствор 5 г (29,35 ммоль) этил 2-(тиофен-3-ил)ацетата в 10 мл дихлорметана. Полученную смесь перемешивали в колбе с обратным холодильником в течение 1 ч, после чего вливали в 75 мл смеси, состоящей из воды и льда, и перемешивали при к.т. в течение 1 ч. Водную фазу экстрагировали 2×50 мл дихлорметана. Объединенные органические фазы промывали 75 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/этилацетата, от 100 до 80% петролейного эфира, об./об.). 5,9 г (выход=72%) этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде твердого вещества. ЖХ/МС: m/z=265 (МН+); степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 8,12 (с, 2Н), 7,88 (с, 1Н), 7,54 (д, J=3,6 Гц, 1Н), 6,82 (дд, J=3,6, 1,7 Гц, 1Н), 4,11 (к, J=7,1 Гц, 2Н), 3,79 (с, 2Н), 1,21 (т, J=7,1 Гц, 3Н).
Этап 2: Приготовление этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата (производное номер 139)
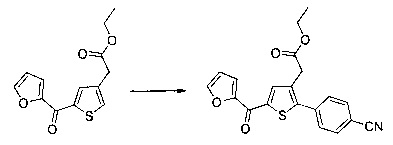 1,189 г (4,27 ммоль) этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата растворяли в 20 мл DMSO при перемешивании магнитной мешалкой, после чего добавляли 0,999 г (4,27 ммоль) 4-иодбензонитрила, 0,497 г (8,55 ммоль) фторида калия, 0,6 г (0,855 ммоль) Pd(Cl)2(PPh3)2 и 1,452 г (8,55 ммоль) нитрата серебра. Смесь перемешивали при температуре 130°С в течение 10 мин. Смеси позволяли остыть до к.т., после чего смесь вливали в 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл воды и 50 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, переносили в 20 мл дихлорметана и фильтровали через гель диоксида кремния (элюент: 100% дихлорметан). Полученный фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 0% петролейного эфира, об./об.). 254 мг (выход=15%) этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=366 (МН+); степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,18 (д, J=15,8 Гц, 2Н), 7,99 (д, J=8,3 Гц, 2Н), 7,75 (д, J=8,3 Гц, 2Н), 7,61 (д, J=3,6 Гц, 1Н), 6,85 (дд, J=3,6, 1,6 Гц, 1Н), 4,06 (к, J=7,1 Гц, 2Н), 3,85 (с, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
1,189 г (4,27 ммоль) этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата растворяли в 20 мл DMSO при перемешивании магнитной мешалкой, после чего добавляли 0,999 г (4,27 ммоль) 4-иодбензонитрила, 0,497 г (8,55 ммоль) фторида калия, 0,6 г (0,855 ммоль) Pd(Cl)2(PPh3)2 и 1,452 г (8,55 ммоль) нитрата серебра. Смесь перемешивали при температуре 130°С в течение 10 мин. Смеси позволяли остыть до к.т., после чего смесь вливали в 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл воды и 50 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, переносили в 20 мл дихлорметана и фильтровали через гель диоксида кремния (элюент: 100% дихлорметан). Полученный фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 0% петролейного эфира, об./об.). 254 мг (выход=15%) этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=366 (МН+); степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,18 (д, J=15,8 Гц, 2Н), 7,99 (д, J=8,3 Гц, 2Н), 7,75 (д, J=8,3 Гц, 2Н), 7,61 (д, J=3,6 Гц, 1Н), 6,85 (дд, J=3,6, 1,6 Гц, 1Н), 4,06 (к, J=7,1 Гц, 2Н), 3,85 (с, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
Пример 27: Приготовление производного номер 140: 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусная кислота
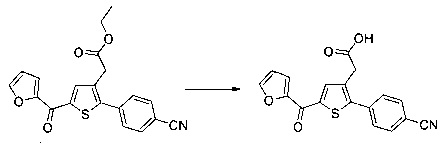 0,189 г (0,491 ммоль) этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 1,5 мл смеси тетрагидрофурана и этанола (1/2, об./об.), к раствору добавляли при перемешивании магнитной мешалкой 0,054 мл (0,541 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 1 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу подкисляли до значения рН 2-3 добавлением 4 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×10 мл этилацетата, объединенные органические фазы промывали 20 мл воды и 20 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Осадок переносили в 10 мл воды, перемешивали при к.т. в течение 1 ч, полученное твердое вещество выделяли в результате фильтрации и промывали водой с получением 166 мг (выход=68%) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 12,69 (с, 1Н), 8,21 (с, 1Н), 8,16 (д, J=0,8 Гц, 1Н), 8,00 (д, J=8,2 Гц, 2Н), 7,76 (д, J=8,3 Гц, 2Н), 7,61 (д, J=3,6 Гц, 1Н), 6,85 (дд, J=3,6, 1,6 Гц, 1Н), 3,75 (с, 2Н).
0,189 г (0,491 ммоль) этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 1,5 мл смеси тетрагидрофурана и этанола (1/2, об./об.), к раствору добавляли при перемешивании магнитной мешалкой 0,054 мл (0,541 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 1 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды, водную фазу подкисляли до значения рН 2-3 добавлением 4 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×10 мл этилацетата, объединенные органические фазы промывали 20 мл воды и 20 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Осадок переносили в 10 мл воды, перемешивали при к.т. в течение 1 ч, полученное твердое вещество выделяли в результате фильтрации и промывали водой с получением 166 мг (выход=68%) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 12,69 (с, 1Н), 8,21 (с, 1Н), 8,16 (д, J=0,8 Гц, 1Н), 8,00 (д, J=8,2 Гц, 2Н), 7,76 (д, J=8,3 Гц, 2Н), 7,61 (д, J=3,6 Гц, 1Н), 6,85 (дд, J=3,6, 1,6 Гц, 1Н), 3,75 (с, 2Н).
Пример 28: Приготовление производного номер 141: 2-(2-(4-цианофенил)-5-(фуpaн-2-кapбoнил)тиoфeн-3-ил)-N-(2-(димeтилaминo)этил)aцeтaмид
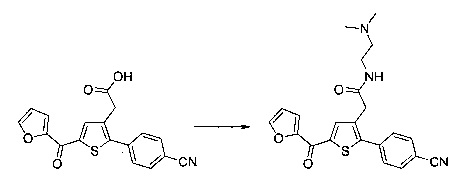 0,125 г (0,371 ммоль) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты растворяли в смеси 5 мл дихлорметана и 0,1 мл диметилформамида. К полученному раствору добавляли при перемешивании магнитной мешалкой при температуре 0°С 0,045 мл (0,408 ммоль) N,N-диметилэтан-1,2-диамина, 0,178 г (0,926 ммоль) EDC.HCl и 0,055 г (0,408 ммоль) HOBt. Смесь перемешивали при к.т. в течение 16 ч. К реакционной смеси добавляли 30 мл воды. Водную фазу экстрагировали 30 мл дихлорметана, органическую фазу промывали 10 мл насыщенного водного раствора NaHCO3, 10 мл воды и 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 0,16 г (выход=30%) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=408 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,20 (с, 1Н), 8,15 (д, J=1,0 Гц, 2Н), 7,98 (д, J=8,4 Гц, 2Н), 7,88 (д, J=8,4 Гц, 2Н), 7,58 (д, J=3,5 Гц, 1Н), 6,86 (дд, J=3,6, 1,7 Гц, 1Н), 3,56 (с, 2Н), 3,16 (дд, J=12,3, 6,4 Гц, 2Н), 2,27 (т, J=6,6 Гц, 2Н), 2,13 (с, 6Н).
0,125 г (0,371 ммоль) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты растворяли в смеси 5 мл дихлорметана и 0,1 мл диметилформамида. К полученному раствору добавляли при перемешивании магнитной мешалкой при температуре 0°С 0,045 мл (0,408 ммоль) N,N-диметилэтан-1,2-диамина, 0,178 г (0,926 ммоль) EDC.HCl и 0,055 г (0,408 ммоль) HOBt. Смесь перемешивали при к.т. в течение 16 ч. К реакционной смеси добавляли 30 мл воды. Водную фазу экстрагировали 30 мл дихлорметана, органическую фазу промывали 10 мл насыщенного водного раствора NaHCO3, 10 мл воды и 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 0,16 г (выход=30%) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=408 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,20 (с, 1Н), 8,15 (д, J=1,0 Гц, 2Н), 7,98 (д, J=8,4 Гц, 2Н), 7,88 (д, J=8,4 Гц, 2Н), 7,58 (д, J=3,5 Гц, 1Н), 6,86 (дд, J=3,6, 1,7 Гц, 1Н), 3,56 (с, 2Н), 3,16 (дд, J=12,3, 6,4 Гц, 2Н), 2,27 (т, J=6,6 Гц, 2Н), 2,13 (с, 6Н).
Пример 29: Приготовление производного номер 142: 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамид
Этап 1: Приготовление 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты
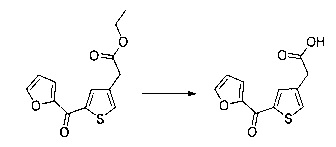 2 г (7,57 ммоль) этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 10 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 0,832 мл (8,32 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 2 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 15 мл воды, водную фазу промывали 2×15 мл этилацетата и затем подкисляли до значения рН 1-2 добавлением 12 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. 1,715 г (выход=96%) 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты получали в виде оранжевого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 12,43 (с, 1Н), 8,11 (с, 2Н), 7,86 (с, 1Н), 7,54 (д, J=3,6 Гц, 1Н), 6,81 (дд, J=3,6, 1,7 Гц, 1Н), 3,70 (с, 2Н).
2 г (7,57 ммоль) этил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 10 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 0,832 мл (8,32 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 2 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 15 мл воды, водную фазу промывали 2×15 мл этилацетата и затем подкисляли до значения рН 1-2 добавлением 12 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. 1,715 г (выход=96%) 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты получали в виде оранжевого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 12,43 (с, 1Н), 8,11 (с, 2Н), 7,86 (с, 1Н), 7,54 (д, J=3,6 Гц, 1Н), 6,81 (дд, J=3,6, 1,7 Гц, 1Н), 3,70 (с, 2Н).
Этап 2: Приготовление N-(2-(диметиламино)этил)-2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетамида
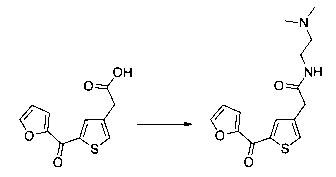 0,416 г (1,761 ммоль) 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в смеси 5 мл дихлорметана и 0,1 мл диметилформамида. К полученному раствору добавляли при перемешивании магнитной мешалкой при температуре 0°С 0,212 мл (1,937 ммоль) N,N-диметилэтан-1,2-диамина, 0,844 г (4,4 ммоль) EDC.HCl и 0,262 г (1,937 ммоль) HOBt. Смесь перемешивали при к.т. в течение 16 ч. К реакционной смеси добавляли 30 мл воды. Водную фазу экстрагировали 30 мл дихлорметана, органическую фазу промывали 10 мл насыщенного водного раствора NaHCO3, 10 мл воды и 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 0,16 г (выход=30%) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 8,07 (дд, J=3,8, 1,2 Гц, 2Н), 7,77 (с, 1Н), 7,51 (д, J=3,2 Гц, 1Н), 6,79 (дд, J=3,6, 1,7 Гц, 1Н), 3,50 (с, 2Н), 3,15 (т, J=6,6 Гц, 2Н), 2,31 (т, J=6,6 Гц, 2Н), 2,13 (с, 6Н).
0,416 г (1,761 ммоль) 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в смеси 5 мл дихлорметана и 0,1 мл диметилформамида. К полученному раствору добавляли при перемешивании магнитной мешалкой при температуре 0°С 0,212 мл (1,937 ммоль) N,N-диметилэтан-1,2-диамина, 0,844 г (4,4 ммоль) EDC.HCl и 0,262 г (1,937 ммоль) HOBt. Смесь перемешивали при к.т. в течение 16 ч. К реакционной смеси добавляли 30 мл воды. Водную фазу экстрагировали 30 мл дихлорметана, органическую фазу промывали 10 мл насыщенного водного раствора NaHCO3, 10 мл воды и 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 0,16 г (выход=30%) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 8,07 (дд, J=3,8, 1,2 Гц, 2Н), 7,77 (с, 1Н), 7,51 (д, J=3,2 Гц, 1Н), 6,79 (дд, J=3,6, 1,7 Гц, 1Н), 3,50 (с, 2Н), 3,15 (т, J=6,6 Гц, 2Н), 2,31 (т, J=6,6 Гц, 2Н), 2,13 (с, 6Н).
Этап 3: Приготовление 2-(2-(3-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида (производное номер 142)
 0,16 г (0,522 ммоль) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида солюбилизировали в 2 мл DMSO при перемешивании магнитной мешалкой, после чего добавляли 0,12 г (0,522 ммоль) 3-иодбензонитрила, 0,061 г (1,044 ммоль) фторида калия, 0,073 г (0,104 ммоль) Pd(Cl)2(PPh3)2 и 0,177 г (1,044 ммоль) нитрата серебра. Смесь перемешивали при температуре 130°С в течение 10 мин. Смеси позволяли остыть до к.т., после чего смесь вливали в 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл воды и 50 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, переносили в 20 мл дихлорметана и фильтровали через гель диоксида кремния (элюент: 100% дихлорметан). Полученный фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 0% петролейного эфира, об./об.). 45 мг (выход=21%) этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=408 (МН+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,18 (с, 1Н), 8,12 (д, J=6,4 Гц, 2Н), 7,94 (т, J=7,7 Гц, 2Н), 7,71 (т, J=7,8 Гц, 1Н), 7,57 (д, J=3,6 Гц, 1Н), 6,83 (дд, J=3,6, 1,7 Гц, 1Н), 3,18 (т, J=6,6 Гц, 2Н), 2,20 (с, 6Н).
0,16 г (0,522 ммоль) 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-(2-(диметиламино)этил)ацетамида солюбилизировали в 2 мл DMSO при перемешивании магнитной мешалкой, после чего добавляли 0,12 г (0,522 ммоль) 3-иодбензонитрила, 0,061 г (1,044 ммоль) фторида калия, 0,073 г (0,104 ммоль) Pd(Cl)2(PPh3)2 и 0,177 г (1,044 ммоль) нитрата серебра. Смесь перемешивали при температуре 130°С в течение 10 мин. Смеси позволяли остыть до к.т., после чего смесь вливали в 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл воды и 50 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, переносили в 20 мл дихлорметана и фильтровали через гель диоксида кремния (элюент: 100% дихлорметан). Полученный фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 0% петролейного эфира, об./об.). 45 мг (выход=21%) этил 2-(2-(4-цианофенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=408 (МН+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,18 (с, 1Н), 8,12 (д, J=6,4 Гц, 2Н), 7,94 (т, J=7,7 Гц, 2Н), 7,71 (т, J=7,8 Гц, 1Н), 7,57 (д, J=3,6 Гц, 1Н), 6,83 (дд, J=3,6, 1,7 Гц, 1Н), 3,18 (т, J=6,6 Гц, 2Н), 2,20 (с, 6Н).
Пример 30: Приготовление производного номер 143: изопропил 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетат
Этап 1: Приготовление изопропил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата
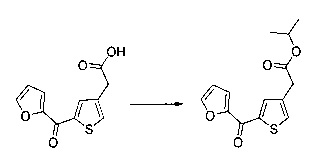 1 г (4,23 ммоль) 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в 10 мл изопропанола, к полученному раствору добавляли 1 каплю серной кислоты. Смесь нагревали при перемешивании магнитной мешалкой в колбе с обратным холодильником в течение 24 ч. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок переносили в 15 мл дихлорметана, органическую фазу промывали 10 мл насыщенного водного раствора NaHCO3 и 10 мл воды, после чего высушивали над MgS04, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 0,667 г (выход=53%) изопропил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата в виде оранжевого масла. ЖХ/МС: m/z=279 (МН+); степень чистоты (УФ-детекция при 254 нм)=94%.
1 г (4,23 ммоль) 2-(5-(фуран-2-карбонил)тиофен-3-ил)уксусной кислоты солюбилизировали в 10 мл изопропанола, к полученному раствору добавляли 1 каплю серной кислоты. Смесь нагревали при перемешивании магнитной мешалкой в колбе с обратным холодильником в течение 24 ч. Смеси позволяли остыть до к.т., после чего смесь концентрировали в вакууме, неочищенный осадок переносили в 15 мл дихлорметана, органическую фазу промывали 10 мл насыщенного водного раствора NaHCO3 и 10 мл воды, после чего высушивали над MgS04, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 0,667 г (выход=53%) изопропил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата в виде оранжевого масла. ЖХ/МС: m/z=279 (МН+); степень чистоты (УФ-детекция при 254 нм)=94%.
Этап 2: Приготовление изопропил 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата (производное номер 143)
 0,66 г (2,29 ммоль) изопропил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 10 мл DMSO при перемешивании магнитной мешалкой, и затем добавляли 0,572 г (2,29 ммоль) 4-хлор-2-фтор-1-иодбензена, 0,259 г (4,46 ммоль) фторида калия, 0,313 г (0,446 ммоль) Pd(Cl)2(PPh3)2 и 0,757 г (4,46 ммоль) нитрата серебра. Смесь перемешивали при температуре 130°С в течение 10 мин. Смеси позволяли остыть до к.т., после чего смесь вливали в 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл воды и 50 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, переносили в 20 мл дихлорметана и фильтровали через гель диоксида кремния (элюент: 100% дихлорметан). Полученный фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/дихлорметана, от 100 до 0% петролейного эфира, об./об.). 135 мг (выход=1%) изопропил 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=407 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 8,18 (д, J=16,2 Гц, 2Н), 7,82-7,29 (м, 5Н), 6,85 (с, 1Н), 4,83 (дт, J=12,2, 6,0 Гц, 1Н), 3,64 (с, 2Н), 1,12 (д, J=6,1 Гц, 6Н).
0,66 г (2,29 ммоль) изопропил 2-(5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 10 мл DMSO при перемешивании магнитной мешалкой, и затем добавляли 0,572 г (2,29 ммоль) 4-хлор-2-фтор-1-иодбензена, 0,259 г (4,46 ммоль) фторида калия, 0,313 г (0,446 ммоль) Pd(Cl)2(PPh3)2 и 0,757 г (4,46 ммоль) нитрата серебра. Смесь перемешивали при температуре 130°С в течение 10 мин. Смеси позволяли остыть до к.т., после чего смесь вливали в 100 мл смеси, состоящей из воды и льда. Водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл воды и 50 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме, переносили в 20 мл дихлорметана и фильтровали через гель диоксида кремния (элюент: 100% дихлорметан). Полученный фильтрат концентрировали в вакууме и очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/дихлорметана, от 100 до 0% петролейного эфира, об./об.). 135 мг (выход=1%) изопропил 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=407 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 8,18 (д, J=16,2 Гц, 2Н), 7,82-7,29 (м, 5Н), 6,85 (с, 1Н), 4,83 (дт, J=12,2, 6,0 Гц, 1Н), 3,64 (с, 2Н), 1,12 (д, J=6,1 Гц, 6Н).
Пример 31: Приготовление производного номер 144: 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)уксусная кислота
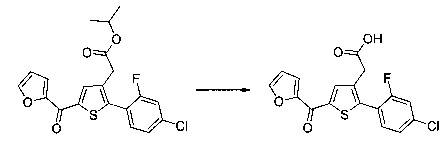 0,115 г (0,283 ммоль) изопропил 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 3 мл изопропанола, к раствору добавляли при перемешивании магнитной мешалкой 0,028 мл (0,283 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 24 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 15 мл воды, водную фазу промывали 15 мл метил-трет-бутилового эфира, а затем подкисляли до значения рН 1-2 добавлением 4 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×10 мл этилацетата, объединенные органические фазы промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Осадок переносили в 10 мл воды, перемешивали при к.т. в течение 1 ч, полученное твердое вещество выделяли в результате фильтрации и промывали водой с получением 71 мг (выход=63%) 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)-тиофен-3-ил)уксусной кислоты. ЖХ/МС: m/z=365 (МН+); степень чистоты (УФ-детекция при 254 нм)=91%. 1Н ЯМР (300 МГц, DMSO) δ 12,54 (с, 1Н), 8,18 (д, J=14,1 Гц, 2Н), 7,68 (д, J=10,0 Гц, 1Н), 7,58 (дд, J=13,0, 5,8 Гц, 2Н), 7,46 (д, J=10,3 Гц, 1Н), 6,91-6,75 (м, 1Н), 3,57 (с, 2Н).
0,115 г (0,283 ммоль) изопропил 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетата солюбилизировали в 3 мл изопропанола, к раствору добавляли при перемешивании магнитной мешалкой 0,028 мл (0,283 ммоль) 10 н водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 24 ч. Смесь концентрировали в вакууме, полученный осадок переносили в 15 мл воды, водную фазу промывали 15 мл метил-трет-бутилового эфира, а затем подкисляли до значения рН 1-2 добавлением 4 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×10 мл этилацетата, объединенные органические фазы промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Осадок переносили в 10 мл воды, перемешивали при к.т. в течение 1 ч, полученное твердое вещество выделяли в результате фильтрации и промывали водой с получением 71 мг (выход=63%) 2-(2-(4-хлор-2-фторфенил)-5-(фуран-2-карбонил)-тиофен-3-ил)уксусной кислоты. ЖХ/МС: m/z=365 (МН+); степень чистоты (УФ-детекция при 254 нм)=91%. 1Н ЯМР (300 МГц, DMSO) δ 12,54 (с, 1Н), 8,18 (д, J=14,1 Гц, 2Н), 7,68 (д, J=10,0 Гц, 1Н), 7,58 (дд, J=13,0, 5,8 Гц, 2Н), 7,46 (д, J=10,3 Гц, 1Н), 6,91-6,75 (м, 1Н), 3,57 (с, 2Н).
Пример 32: Приготовление производного номер 145: 2-(2-(4-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрил
Этап 1: Приготовление 2-(4-фторфенил)-3-метилтиофена
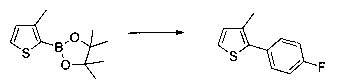 1 г (4,46 ммоль) 4,4,5,5-тетраметил-2-(3-метилтиофен-2-ил)-1,3,2-диоксаборолана солюбилизировали в атмосфере аргона в 15 мл толуола, добавляли при перемешивании магнитной мешалкой 0,781 г (4,46 ммоль) 1-бром-4-фторбензена, 15,62 мл (31,2 ммоль) 2 М водного раствора Na2CO3 и 0,26 г (0,223 ммоль) тетракис(трифенилфосфина) палладия [0]. Смесь перемешивали в колбе с обратным холодильником в течение 3 часов. Смеси позволяли остыть до к.т., после чего смесь фильтровали через целит. Водную фазу экстрагировали 20 мл этилацетата. Органическую фазу промывали 10 мл воды, 10 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% петролейный эфир). 0,509 г (выход=51%) 2-(4-фторфенил)-3-метилтиофена получали в виде масла и непосредственно использовали на следующем этапе.
1 г (4,46 ммоль) 4,4,5,5-тетраметил-2-(3-метилтиофен-2-ил)-1,3,2-диоксаборолана солюбилизировали в атмосфере аргона в 15 мл толуола, добавляли при перемешивании магнитной мешалкой 0,781 г (4,46 ммоль) 1-бром-4-фторбензена, 15,62 мл (31,2 ммоль) 2 М водного раствора Na2CO3 и 0,26 г (0,223 ммоль) тетракис(трифенилфосфина) палладия [0]. Смесь перемешивали в колбе с обратным холодильником в течение 3 часов. Смеси позволяли остыть до к.т., после чего смесь фильтровали через целит. Водную фазу экстрагировали 20 мл этилацетата. Органическую фазу промывали 10 мл воды, 10 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% петролейный эфир). 0,509 г (выход=51%) 2-(4-фторфенил)-3-метилтиофена получали в виде масла и непосредственно использовали на следующем этапе.
Этап 2: Приготовление (5-(4-фторфенил)-4-метилтиофен-2-ил)(фуран-2-ил)метанона
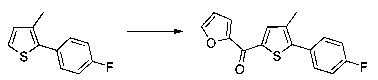 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 20 мл дихлорметана, 0,5 г (2,237 ммоль) 2-(4-фторфенил)-3-метилтиофена, 0,265 мл (2,68 ммоль) 2-фуроил хлорида. Полученную смесь охлаждали до температуры 5°С, после чего добавляли по частям 0,328 г (2,46 ммоль) триалюминий хлорида. Смесь перемешивали при к.т. в течение 16 ч, а затем вливали в 150 мл смеси, состоящей из воды и дихлорметана (2/1, об./об.), и перемешивали при к.т. в течение 30 мин. Смесь фильтровали через целит. После сепарации водную фазу экстрагировали 50 мл дихлорметана, объединенные органические фазы промывали 50 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/этилацетата, от 100 до 75% петролейного эфира, об./об.). 0,58 г (выход=89%) (5-(4-фторфенил)-4-метилтиофен-2-ил)(фуран-2-ил)метанона получали в виде пены. ЖХ/МС: m/z=287 (МН+); степень чистоты (УФ-детекция при 254 нм)=88%. 1Н ЯМР (300 МГц, DMSO) δ 8,13 (дд, J=1,6, 0,7 Гц, 1Н), 8,09 (с, 1Н), 7,62 (ддд, J=6,3, 4,2, 3,0 Гц, 3Н), 7,36 (т, J=8,9 Гц, 2Н), 6,83 (дд, J=3,6, 1,7 Гц, 1Н), 2,34 (с, 3Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 20 мл дихлорметана, 0,5 г (2,237 ммоль) 2-(4-фторфенил)-3-метилтиофена, 0,265 мл (2,68 ммоль) 2-фуроил хлорида. Полученную смесь охлаждали до температуры 5°С, после чего добавляли по частям 0,328 г (2,46 ммоль) триалюминий хлорида. Смесь перемешивали при к.т. в течение 16 ч, а затем вливали в 150 мл смеси, состоящей из воды и дихлорметана (2/1, об./об.), и перемешивали при к.т. в течение 30 мин. Смесь фильтровали через целит. После сепарации водную фазу экстрагировали 50 мл дихлорметана, объединенные органические фазы промывали 50 мл воды, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/этилацетата, от 100 до 75% петролейного эфира, об./об.). 0,58 г (выход=89%) (5-(4-фторфенил)-4-метилтиофен-2-ил)(фуран-2-ил)метанона получали в виде пены. ЖХ/МС: m/z=287 (МН+); степень чистоты (УФ-детекция при 254 нм)=88%. 1Н ЯМР (300 МГц, DMSO) δ 8,13 (дд, J=1,6, 0,7 Гц, 1Н), 8,09 (с, 1Н), 7,62 (ддд, J=6,3, 4,2, 3,0 Гц, 3Н), 7,36 (т, J=8,9 Гц, 2Н), 6,83 (дд, J=3,6, 1,7 Гц, 1Н), 2,34 (с, 3Н).
Этап 3: Приготовление (4-(бромметил)-5-(4-фторфенил)тиофен-2-ил)(фуран-2-ил)метанона
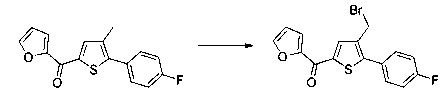 0,56 г (1,741 ммоль) (5-(4-фторфенил)-4-метилтиофен-2-ил)(фуран-2-ил)метанона растворяли в 15 мл четыреххлористого углерода при перемешивании магнитной мешалкой, затем добавляли 0,286 г (1,741 ммоль) AIBN и 0,31 г (1,741 ммоль) N-бромсукцинимида. Смесь перемешивали в колбе с обратным холодильником под освещением 300 ватт в течение 9 ч, при этом в моменты времени 3 и 6 ч добавляли по 0,043 г (0,26 ммоль) AIBN и 0,047 г (0,26 ммоль) N-бромсукцинимида. Реакционную среду концентрировали в вакууме, полученный осадок переносили в 50 мл смеси, состоящей из воды и льда, водную фазу экстрагировали 3×50 мл этилацетата. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 66% петролейного эфира, об./об.). 0,308 г (выход=36%) (4-(бромметил)-5-(4-фторфенил)тиофен-2-ил)(фуран-2-ил)метанона получали в виде твердого вещества. ЖХ/МС: m/z=367 (МН+); степень чистоты (УФ-детекция при 254 нм)=75%. 1Н ЯМР (300 МГц, DMSO) δ 7,98 (с, 1Н), 7,89 (дд, J=12,5, 8,0 Гц, 3Н), 7,74 (д, J=7,7 Гц, 1Н), 7,67 (д, J=5,2 Гц, 1Н), 7,26 (д, J=5,2 Гц, 1Н), 4,70 (с, 2Н), 1,74-1,51 (м, 6Н), 1,43 (с, 3Н).
0,56 г (1,741 ммоль) (5-(4-фторфенил)-4-метилтиофен-2-ил)(фуран-2-ил)метанона растворяли в 15 мл четыреххлористого углерода при перемешивании магнитной мешалкой, затем добавляли 0,286 г (1,741 ммоль) AIBN и 0,31 г (1,741 ммоль) N-бромсукцинимида. Смесь перемешивали в колбе с обратным холодильником под освещением 300 ватт в течение 9 ч, при этом в моменты времени 3 и 6 ч добавляли по 0,043 г (0,26 ммоль) AIBN и 0,047 г (0,26 ммоль) N-бромсукцинимида. Реакционную среду концентрировали в вакууме, полученный осадок переносили в 50 мл смеси, состоящей из воды и льда, водную фазу экстрагировали 3×50 мл этилацетата. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100 до 66% петролейного эфира, об./об.). 0,308 г (выход=36%) (4-(бромметил)-5-(4-фторфенил)тиофен-2-ил)(фуран-2-ил)метанона получали в виде твердого вещества. ЖХ/МС: m/z=367 (МН+); степень чистоты (УФ-детекция при 254 нм)=75%. 1Н ЯМР (300 МГц, DMSO) δ 7,98 (с, 1Н), 7,89 (дд, J=12,5, 8,0 Гц, 3Н), 7,74 (д, J=7,7 Гц, 1Н), 7,67 (д, J=5,2 Гц, 1Н), 7,26 (д, J=5,2 Гц, 1Н), 4,70 (с, 2Н), 1,74-1,51 (м, 6Н), 1,43 (с, 3Н).
Этап 4: Приготовление 2-(2-(4-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила (производное номер 145)
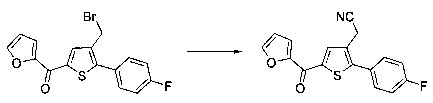 0,3 г (0,616 ммоль) (4-(бром-метил)-5-(4-фторфенил)тиофен-2-ил)(фуран-2-ил)метанона растворяли в 10 мл тетрагидрофурана при перемешивании магнитной мешалкой, затем добавляли 0,044 г (0,678 ммоль) цианида калия и 1 мл воды. Смесь перемешивали в колбе с обратным холодильником в течение 16 ч. Реакционную среду вливали в 15 мл воды, водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом препаративной ВЭЖХ/МС. Получали 0,024 г (выход=11%) 2-(2-(4-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила в виде светло-желтого твердого вещества. ЖХ/МС: m/z=312 (МН+); степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,25 (с, 1Н), 8,18 (с, 1Н), 7,74-7,57 (м, 3Н), 7,41 (т, J=8,8 Гц, 2Н), 6,86 (дд, J=3,6, 1,7 Гц, 1Н), 4,13 (с, 2Н).
0,3 г (0,616 ммоль) (4-(бром-метил)-5-(4-фторфенил)тиофен-2-ил)(фуран-2-ил)метанона растворяли в 10 мл тетрагидрофурана при перемешивании магнитной мешалкой, затем добавляли 0,044 г (0,678 ммоль) цианида калия и 1 мл воды. Смесь перемешивали в колбе с обратным холодильником в течение 16 ч. Реакционную среду вливали в 15 мл воды, водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 15 мл воды и 15 мл насыщенного водного раствора NaCl, после чего высушивали над MgSO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом препаративной ВЭЖХ/МС. Получали 0,024 г (выход=11%) 2-(2-(4-фторфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила в виде светло-желтого твердого вещества. ЖХ/МС: m/z=312 (МН+); степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,25 (с, 1Н), 8,18 (с, 1Н), 7,74-7,57 (м, 3Н), 7,41 (т, J=8,8 Гц, 2Н), 6,86 (дд, J=3,6, 1,7 Гц, 1Н), 4,13 (с, 2Н).
Пример 33: Приготовление производного номер 146: 2-(2-фенил-5-пиколиноилтиофен-3-ил)ацетонитрил
Этап 1: Приготовление 5-бром-4-метилтиофен-2-карбоновой кислоты
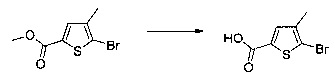 5 г (21,27 ммоль) метил 5-бром-4-метилтиофен-2-карбоксилата солюбилизировали в 250 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 106 мл (106 ммоль) 1 н водного раствора гидроксида натрия. Полученную смесь перемешивали при температуре 80°С в течение 2 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 250 мл воды и добавляли 100 мл 1 н водного раствора хлористоводородной кислоты. Преципитат выделяли в результате фильтрации, промывали минимальным количеством воды и 10 мл пентана. Белое твердое вещество высушивали в вакуумном колпаке и получали 3,63 г (выход=76%) 5-бром-4-метилтиофен-2-карбоновой кислоты. ЖХ/МС: m/z=219 (М-Н+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 13,34 (с, 1Н), 7,53 (с, 1Н), 2,15 (с, 3Н).
5 г (21,27 ммоль) метил 5-бром-4-метилтиофен-2-карбоксилата солюбилизировали в 250 мл этанола, к раствору добавляли при перемешивании магнитной мешалкой 106 мл (106 ммоль) 1 н водного раствора гидроксида натрия. Полученную смесь перемешивали при температуре 80°С в течение 2 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 250 мл воды и добавляли 100 мл 1 н водного раствора хлористоводородной кислоты. Преципитат выделяли в результате фильтрации, промывали минимальным количеством воды и 10 мл пентана. Белое твердое вещество высушивали в вакуумном колпаке и получали 3,63 г (выход=76%) 5-бром-4-метилтиофен-2-карбоновой кислоты. ЖХ/МС: m/z=219 (М-Н+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 13,34 (с, 1Н), 7,53 (с, 1Н), 2,15 (с, 3Н).
Этап 2: Приготовление 5-6ром-N-метокси-N,4-диметилтиофен-2-карбоксамида
 3,63 г (16,26 ммоль) 5-бром-4-метилтиофен-2-карбоновой кислоты солюбилизировали в дихлорметане при перемешивании магнитной мешалкой, затем добавляли 4,27 мл (48,8 ммоль) оксалил хлорида и по каплям диметилформамид. Смесь перемешивали при к.т. в течение 2 часов, после чего концентрировали в вакууме. Осадок переносили в 75 мл смеси дихлорметана/пиридина (2/1, об./об.), а затем добавляли 2,427 г (24,38 ммоль) N,O-диметилгидроксиламина гидрохлорида. Смесь перемешивали при к.т. в течение 3 часов, после чего вливали в 300 мл воды. Водную фазу экстрагировали 3×150 мл этилацетата. Объединенные органические фазы промывали 2×150 мл 1 н водного раствора хлористоводородной кислоты, 150 мл воды и 150 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 4,32 г (выход=100%) 5-бром-N-метокси-N,4-диметилтиофен-2-карбоксамида получали в виде коричневого масла. ЖХ/МС: m/z=265 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 7,64 (с, 1Н), 3,76 (с, 3Н), 3,26 (с, 3Н), 2,17 (с, 3Н).
3,63 г (16,26 ммоль) 5-бром-4-метилтиофен-2-карбоновой кислоты солюбилизировали в дихлорметане при перемешивании магнитной мешалкой, затем добавляли 4,27 мл (48,8 ммоль) оксалил хлорида и по каплям диметилформамид. Смесь перемешивали при к.т. в течение 2 часов, после чего концентрировали в вакууме. Осадок переносили в 75 мл смеси дихлорметана/пиридина (2/1, об./об.), а затем добавляли 2,427 г (24,38 ммоль) N,O-диметилгидроксиламина гидрохлорида. Смесь перемешивали при к.т. в течение 3 часов, после чего вливали в 300 мл воды. Водную фазу экстрагировали 3×150 мл этилацетата. Объединенные органические фазы промывали 2×150 мл 1 н водного раствора хлористоводородной кислоты, 150 мл воды и 150 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 4,32 г (выход=100%) 5-бром-N-метокси-N,4-диметилтиофен-2-карбоксамида получали в виде коричневого масла. ЖХ/МС: m/z=265 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 7,64 (с, 1Н), 3,76 (с, 3Н), 3,26 (с, 3Н), 2,17 (с, 3Н).
Этап 3: Приготовление (5-6ром-4-метилтиофен-2-ил)(пиридин-2-ил)метанона
 В колбу, помещенную в атмосферу аргона, вносили при температуре 0°С: 25 мл тетрагидрофурана, 0,407 мл (3,75 ммоль) 2-иодпиридина и 3,75 мл (3,75 ммоль) этилмагния бромида, ледяную баню убирали, и смесь перемешивали при к.т. в течение 30 мин. Затем добавляли по каплям раствор 1 г (3,75 ммоль) 5-бром-N-метокси-N,4-диметилтиофен-2-карбоксамида в 25 мл тетрагидрофурана. Смесь перемешивали при к.т. в течение 3 часов, после чего вливали в 75 мл водного раствора насыщенного хлорида аммония. Органическую фазу экстрагировали 3×100 мл этилацетата, 100 мл насыщенного водного раствора NaHCO3, 100 мл воды, 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 95% до 75% гептана, об./об.). 0,357 г (выход=33%) (5-бром-4-метилтиофен-2-ил)(пиридин-2-ил)метанона получали в виде белого твердого вещества. ЖХ/МС: m/z=283 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 8,79 (с, 1Н), 8,08 (д, J=26,9 Гц, 3Н), 7,73 (с, 1Н), 2,22 (с, 3Н).
В колбу, помещенную в атмосферу аргона, вносили при температуре 0°С: 25 мл тетрагидрофурана, 0,407 мл (3,75 ммоль) 2-иодпиридина и 3,75 мл (3,75 ммоль) этилмагния бромида, ледяную баню убирали, и смесь перемешивали при к.т. в течение 30 мин. Затем добавляли по каплям раствор 1 г (3,75 ммоль) 5-бром-N-метокси-N,4-диметилтиофен-2-карбоксамида в 25 мл тетрагидрофурана. Смесь перемешивали при к.т. в течение 3 часов, после чего вливали в 75 мл водного раствора насыщенного хлорида аммония. Органическую фазу экстрагировали 3×100 мл этилацетата, 100 мл насыщенного водного раствора NaHCO3, 100 мл воды, 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 95% до 75% гептана, об./об.). 0,357 г (выход=33%) (5-бром-4-метилтиофен-2-ил)(пиридин-2-ил)метанона получали в виде белого твердого вещества. ЖХ/МС: m/z=283 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 8,79 (с, 1Н), 8,08 (д, J=26,9 Гц, 3Н), 7,73 (с, 1Н), 2,22 (с, 3Н).
Этап 4: Приготовление (4-метил-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона
 В реактор, пригодный для обработки микроволнами, вносили при перемешивании магнитной мешалкой: 3 мл смеси, состоящей из диметилового эфира, этанола и воды (1/1/1, об./об./об.), 0,357 г (1,253 ммоль) 5-бром-4-метилтиофен-2-ил)(пиридин-2-ил)метанона, 0,816 г (2,505 ммоль) карбоната цезия и 0,229 г (1,879 ммоль) фенилбориновой кислоты. Смесь облучали при температуре 150°С в течение 15 мин. Смеси позволяли остыть до к.т., после чего реакционную среду экстрагировали 3×10 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 100% до 95% гептана, об./об.). 0,321 г (выход=91%) (4-метил-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=280 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 8,85-8,77 (м, 1Н), 8,19 (с, 1Н), 8,17-8,02 (м, 2Н), 7,78-7,67 (м, 1Н), 7,65-7,39 (м, 5Н), 2,35 (с, 3Н).
В реактор, пригодный для обработки микроволнами, вносили при перемешивании магнитной мешалкой: 3 мл смеси, состоящей из диметилового эфира, этанола и воды (1/1/1, об./об./об.), 0,357 г (1,253 ммоль) 5-бром-4-метилтиофен-2-ил)(пиридин-2-ил)метанона, 0,816 г (2,505 ммоль) карбоната цезия и 0,229 г (1,879 ммоль) фенилбориновой кислоты. Смесь облучали при температуре 150°С в течение 15 мин. Смеси позволяли остыть до к.т., после чего реакционную среду экстрагировали 3×10 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 100% до 95% гептана, об./об.). 0,321 г (выход=91%) (4-метил-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=280 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 8,85-8,77 (м, 1Н), 8,19 (с, 1Н), 8,17-8,02 (м, 2Н), 7,78-7,67 (м, 1Н), 7,65-7,39 (м, 5Н), 2,35 (с, 3Н).
Этап 5: Приготовление (4-(бромметил)-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона
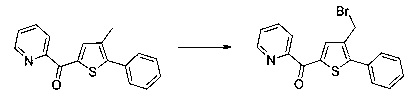 0,157 г (0,556 ммоль) (4-метил-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона солюбилизировали в 4,5 мл четыреххлористого углерода при перемешивании магнитной мешалкой, затем добавляли 0,091 г (0,556 ммоль) AIBN и 0,099 г (0,556 ммоль) N-бромсукцинимида. Смесь перемешивали в колбе с обратным холодильником в течение 6 ч. Реакционную среду вливали в 30 мл смеси, состоящей из воды и льда, водную фазу экстрагировали 3×20 мл этилацетата. Объединенные органические фазы промывали 50 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 95% гептана, об./об.). 0,199 г (выход=56%) (4-(бромметил)-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=359 (МН+); степень чистоты (УФ-детекция при 254 нм)=82%. 1Н ЯМР (300 МГц, DMSO) δ 8,89-8,74 (м, 1Н), 8,23-7,98 (м, 4Н), 7,82-7,34 (м, 5Н), 4,76 (с, 2Н).
0,157 г (0,556 ммоль) (4-метил-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона солюбилизировали в 4,5 мл четыреххлористого углерода при перемешивании магнитной мешалкой, затем добавляли 0,091 г (0,556 ммоль) AIBN и 0,099 г (0,556 ммоль) N-бромсукцинимида. Смесь перемешивали в колбе с обратным холодильником в течение 6 ч. Реакционную среду вливали в 30 мл смеси, состоящей из воды и льда, водную фазу экстрагировали 3×20 мл этилацетата. Объединенные органические фазы промывали 50 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 95% гептана, об./об.). 0,199 г (выход=56%) (4-(бромметил)-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=359 (МН+); степень чистоты (УФ-детекция при 254 нм)=82%. 1Н ЯМР (300 МГц, DMSO) δ 8,89-8,74 (м, 1Н), 8,23-7,98 (м, 4Н), 7,82-7,34 (м, 5Н), 4,76 (с, 2Н).
Этап 6: Приготовление 2-(2-фенил-5-пиколиноилтиофен-3-ил)ацетонитрила (производное номер 146)
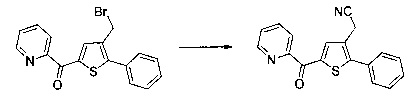 0,135 г (0,309 ммоль) (4-(бромметил)-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона солюбилизировали в 6 мл этанола при перемешивании магнитной мешалкой, затем добавляли 0,024 г (0,361 ммоль) цианида калия и 0,5 мл воды. Смесь перемешивали при к.т. в течение 2 часов, после чего перемешивали при температуре 55°С в течение 90 мин. Реакционную среду концентрировали в вакууме, полученный осадок переносили в 20 мл смеси, состоящей из воды и этилацетата (1/1, об./об.). После сепарации водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 2×15 мл воды, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 95 до 90% гептана, об./об.). 0,033 г (выход=36%) 2-(2-фенил-5-пиколиноилтиофен-3-ил)ацетонитрила получали в виде белого твердого вещества. ЖХ/МС: m/z=305 (МН+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,91-8,76 (м, 1Н), 8,36 (с, 1Н), 8,25-8,03 (м, 2Н), 7,75 (ддд, J=7,2, 4,7, 1,6 Гц, 1Н), 7,66-7,41 (м, 5Н), 4,14 (с, 2Н).
0,135 г (0,309 ммоль) (4-(бромметил)-5-фенилтиофен-2-ил)(пиридин-2-ил)метанона солюбилизировали в 6 мл этанола при перемешивании магнитной мешалкой, затем добавляли 0,024 г (0,361 ммоль) цианида калия и 0,5 мл воды. Смесь перемешивали при к.т. в течение 2 часов, после чего перемешивали при температуре 55°С в течение 90 мин. Реакционную среду концентрировали в вакууме, полученный осадок переносили в 20 мл смеси, состоящей из воды и этилацетата (1/1, об./об.). После сепарации водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 2×15 мл воды, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 95 до 90% гептана, об./об.). 0,033 г (выход=36%) 2-(2-фенил-5-пиколиноилтиофен-3-ил)ацетонитрила получали в виде белого твердого вещества. ЖХ/МС: m/z=305 (МН+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,91-8,76 (м, 1Н), 8,36 (с, 1Н), 8,25-8,03 (м, 2Н), 7,75 (ддд, J=7,2, 4,7, 1,6 Гц, 1Н), 7,66-7,41 (м, 5Н), 4,14 (с, 2Н).
Соединение 147 приготовили согласно той же последовательности этапов с 1 по 6.
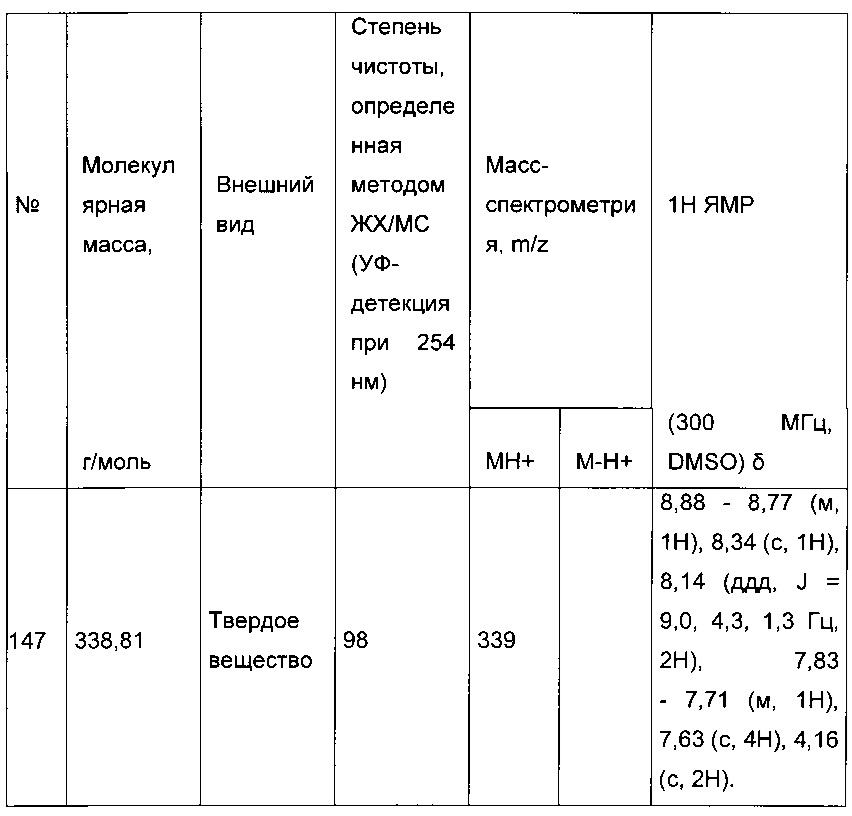
Пример 34: Приготовление производного номер 148: приготовление этил 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетата
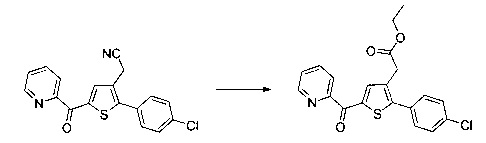 0,2 г (0,578 ммоль) 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетонитрила солюбилизировали при перемешивании магнитной мешалкой в 5 мл метанола, получали беловатую суспензию, к которой добавляли 0,1 мл (1,876 ммоль) серной кислоты. Смесь перемешивали в колбе с обратным холодильником в течение 8 д. Реакционную среду концентрировали в вакууме, полученный осадок переносили в 20 мл смеси, состоящей из воды и этилацетата (1/1, об./об.). После сепарации водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SО4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 85% до 70% гептана, об./об.). 0,215 г (выход=60%) этил 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетата получали в виде белого твердого вещества. ЖХ/МС: m/z=372 (МН+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,86-8,77 (м, 1Н), 8,26 (с, 1Н), 8,19-8,04 (м, 2Н), 7,73 (с, 1Н), 7,58 (д, J=3,0 Гц, 4Н), 3,80 (с, 2Н), 3,61 (с, 3Н).
0,2 г (0,578 ммоль) 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетонитрила солюбилизировали при перемешивании магнитной мешалкой в 5 мл метанола, получали беловатую суспензию, к которой добавляли 0,1 мл (1,876 ммоль) серной кислоты. Смесь перемешивали в колбе с обратным холодильником в течение 8 д. Реакционную среду концентрировали в вакууме, полученный осадок переносили в 20 мл смеси, состоящей из воды и этилацетата (1/1, об./об.). После сепарации водную фазу экстрагировали 2×15 мл этилацетата, объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SО4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 85% до 70% гептана, об./об.). 0,215 г (выход=60%) этил 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетата получали в виде белого твердого вещества. ЖХ/МС: m/z=372 (МН+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,86-8,77 (м, 1Н), 8,26 (с, 1Н), 8,19-8,04 (м, 2Н), 7,73 (с, 1Н), 7,58 (д, J=3,0 Гц, 4Н), 3,80 (с, 2Н), 3,61 (с, 3Н).
Пример 35: Приготовление производного номер 149: приготовление 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)уксусной кислоты
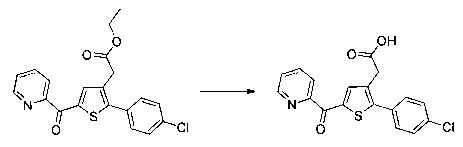 0,099 г (0,264 ммоль) этил 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетата солюбилизировали в 6 мл метанола при перемешивании магнитной мешалкой. К раствору добавляли 0,527 мл (0,527 ммоль) 1 н водного раствора NaOH. Смесь перемешивали при к.т. в течение 16 ч, а затем концентрировали в вакууме. Осадок переносили в 5 мл воды и добавляли 1 н водный раствор хлористоводородной кислоты до появления преципитата, который выделяли в результате фильтрации и высушивали в вакуумном колпаке. Получали 0,07 г (выход=73%) 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)уксусной кислоты. ЖХ/МС: m/z=358 (М-Н+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,57 (с, 1Н), 8,81 (д, J=4,4 Гц, 1Н), 8,27 (с, 1Н), 8,19-8,04 (м, 2Н), 7,73 (м, 1Н), 7,59 (м, 4Н), 3,68 (с, 2Н).
0,099 г (0,264 ммоль) этил 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)ацетата солюбилизировали в 6 мл метанола при перемешивании магнитной мешалкой. К раствору добавляли 0,527 мл (0,527 ммоль) 1 н водного раствора NaOH. Смесь перемешивали при к.т. в течение 16 ч, а затем концентрировали в вакууме. Осадок переносили в 5 мл воды и добавляли 1 н водный раствор хлористоводородной кислоты до появления преципитата, который выделяли в результате фильтрации и высушивали в вакуумном колпаке. Получали 0,07 г (выход=73%) 2-(2-(4-хлорфенил)-5-пиколиноилтиофен-3-ил)уксусной кислоты. ЖХ/МС: m/z=358 (М-Н+); степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,57 (с, 1Н), 8,81 (д, J=4,4 Гц, 1Н), 8,27 (с, 1Н), 8,19-8,04 (м, 2Н), 7,73 (м, 1Н), 7,59 (м, 4Н), 3,68 (с, 2Н).
Пример 36: Приготовление производного номер 150: (2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрил
Этап 1: Приготовление 2-(2-бромтиофен-3-ил)ацетонитрила
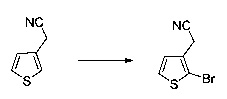 5,9 г (46 ммоль) 2-(тиофен-3-ил)ацетонитрила солюбилизировали в 25 мл четыреххлористого углерода при перемешивании магнитной мешалкой и добавляли 8,27 г (46 ммоль) N-бромсукцинимида. Добавляли при интенсивном перемешивании 0,04 мл (0,460 ммоль) перхлорной кислоты, полученную смесь перемешивали при к.т. в течение 4,5 ч. К реакционной смеси добавляли 0,155 г (1,839 ммоль) бикарбоната натрия. Твердое вещество удаляли в результате фильтрации, фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 9/1, об./об.). 8,47 г (выход=57%) 2-(2-бромтиофен-3-ил)ацетонитрила получали в виде бледно-желтого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 1Н ЯМР (300 МГц, DMSO) δ 7,67 (д, J=5,6 Гц, 1Н), 7,08 (д, J=5,6 Гц, 1Н), 3,93 (с, 2Н).
5,9 г (46 ммоль) 2-(тиофен-3-ил)ацетонитрила солюбилизировали в 25 мл четыреххлористого углерода при перемешивании магнитной мешалкой и добавляли 8,27 г (46 ммоль) N-бромсукцинимида. Добавляли при интенсивном перемешивании 0,04 мл (0,460 ммоль) перхлорной кислоты, полученную смесь перемешивали при к.т. в течение 4,5 ч. К реакционной смеси добавляли 0,155 г (1,839 ммоль) бикарбоната натрия. Твердое вещество удаляли в результате фильтрации, фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 9/1, об./об.). 8,47 г (выход=57%) 2-(2-бромтиофен-3-ил)ацетонитрила получали в виде бледно-желтого масла. ЖХ/МС: m/z=неионизированный. 1Н ЯМР (300 МГц, DMSO) δ 1Н ЯМР (300 МГц, DMSO) δ 7,67 (д, J=5,6 Гц, 1Н), 7,08 (д, J=5,6 Гц, 1Н), 3,93 (с, 2Н).
Этап 2: Приготовление 2-(2-бром-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила
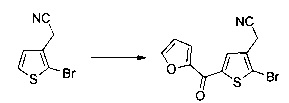 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 4 мл 1,2-дихлорэтана, 0,2 г (0,99 ммоль) 2-(2-бромтиофен-3-ил)ацетонитрила и 0,145 г (1,089 ммоль) триалюминий хлорида. Смесь помещали в условия с температурой 0°С при перемешивании магнитной мешалкой и добавляли по каплям раствор 0,113 мл (1,089 ммоль) 2-фуроил хлорида в 1 мл 1,2-дихлорэтана. Полученную смесь перемешивали при к.т. в течение 4 д, а затем добавляли 0,073 г (0,455 ммоль) триалюминий хлорида и 0,057 мл (0,455 ммоль) 2-фуроил хлорида и продолжали перемешивание при к.т. в течение 24 ч. Реакционную среду вливали в 30 мл воды. Водную фазу экстрагировали 2×25 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaHCO3, 30 мл насыщенного водного раствора NaCl, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 0,211 г (выход=71%) 2-(2-бром-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила получали в виде желтого твердого вещества. ЖХ/МС: m/z=296 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 1Н ЯМР (300 МГц, DMSO) δ 8,17 (д, J=0,9 Гц, 1Н), 8,14 (с, 1Н), 7,61 (д, J=3,7 Гц, 1Н), 6,85 (дд, J=3,6, 1,7 Гц, 1Н), 4,05 (с, 2Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 4 мл 1,2-дихлорэтана, 0,2 г (0,99 ммоль) 2-(2-бромтиофен-3-ил)ацетонитрила и 0,145 г (1,089 ммоль) триалюминий хлорида. Смесь помещали в условия с температурой 0°С при перемешивании магнитной мешалкой и добавляли по каплям раствор 0,113 мл (1,089 ммоль) 2-фуроил хлорида в 1 мл 1,2-дихлорэтана. Полученную смесь перемешивали при к.т. в течение 4 д, а затем добавляли 0,073 г (0,455 ммоль) триалюминий хлорида и 0,057 мл (0,455 ммоль) 2-фуроил хлорида и продолжали перемешивание при к.т. в течение 24 ч. Реакционную среду вливали в 30 мл воды. Водную фазу экстрагировали 2×25 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaHCO3, 30 мл насыщенного водного раствора NaCl, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 0,211 г (выход=71%) 2-(2-бром-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила получали в виде желтого твердого вещества. ЖХ/МС: m/z=296 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 1Н ЯМР (300 МГц, DMSO) δ 8,17 (д, J=0,9 Гц, 1Н), 8,14 (с, 1Н), 7,61 (д, J=3,7 Гц, 1Н), 6,85 (дд, J=3,6, 1,7 Гц, 1Н), 4,05 (с, 2Н).
Этап 3: Приготовление 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила (производное номер 150)
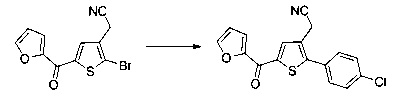 0,207 г (0,699 ммоль) 2-(2-бром-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила солюбилизировали в атмосфере аргона в 6 мл смеси, состоящей из толуола и этанола (7/5, об./об.), добавляли при перемешивании магнитной мешалкой 0,173 г (1,048 ммоль) 4-хлорфенилбориновой кислоты, 2,342 мл (4,68 ммоль) 2 М водного раствора Na2CO3 и 0,040 г (0,035 ммоль) тетракис(трифенилфосфина) палладия [0]. Смесь перемешивали в колбе с обратным холодильником в течение 2,5 ч. Смеси позволяли остыть до к.т., после чего смесь разводили 30 мл воды. Водную фазу экстрагировали 2×30 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 8/2, об./об.). 0,169 г (выход=72%) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=328 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,25 (с, 1Н), 8,19 (с, 1Н), 7,63 (д, J=3,2 Гц, 5Н), 6,86 (дд, J=3,6, 1,7 Гц, 1Н), 4,16 (с, 2Н).
0,207 г (0,699 ммоль) 2-(2-бром-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила солюбилизировали в атмосфере аргона в 6 мл смеси, состоящей из толуола и этанола (7/5, об./об.), добавляли при перемешивании магнитной мешалкой 0,173 г (1,048 ммоль) 4-хлорфенилбориновой кислоты, 2,342 мл (4,68 ммоль) 2 М водного раствора Na2CO3 и 0,040 г (0,035 ммоль) тетракис(трифенилфосфина) палладия [0]. Смесь перемешивали в колбе с обратным холодильником в течение 2,5 ч. Смеси позволяли остыть до к.т., после чего смесь разводили 30 мл воды. Водную фазу экстрагировали 2×30 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 8/2, об./об.). 0,169 г (выход=72%) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=328 (МН+); степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 8,25 (с, 1Н), 8,19 (с, 1Н), 7,63 (д, J=3,2 Гц, 5Н), 6,86 (дд, J=3,6, 1,7 Гц, 1Н), 4,16 (с, 2Н).
Пример 37: Приготовление производного номер 151: приготовление (4-((2Н-тетразол-5-ил)метил)-5-(4-хлорфенил)тиофен-2-ил)(фуран-2-ил)метанона
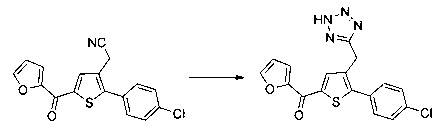 0,149 г (0,45 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила растворяли в 3 мл диметилформамида при перемешивании магнитной мешалкой. К данному раствору добавляли 0,088 г (1,35 ммоль) азида натрия и 0,096 г (1,8 ммоль) хлорида аммония. Смесь перемешивали при температуре 160°С в течение 5 ч. Смеси позволяли остыть до к.т., после чего реакционную среду разводили 5 мл воды, значение рН доводили до 2 добавлением 1 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 3×15 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, от 100 до 95% дихлорметана, об./об.). 0,015 г (выход=8%) (4-((2Н-тетразол-5-ил)метил)-5-(4-хлорфенил)тиофен-2-ил)(фуран-2-ил)метанона получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=370 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 12,12 (с, 1Н), 7,94 (с, 1Н), 7,60 (к, J=8,6 Гц, 4Н), 7,20 (д, J=16,2 Гц, 2Н), 6,42-6,22 (м, 1Н), 4,35 (с, 2Н).
0,149 г (0,45 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила растворяли в 3 мл диметилформамида при перемешивании магнитной мешалкой. К данному раствору добавляли 0,088 г (1,35 ммоль) азида натрия и 0,096 г (1,8 ммоль) хлорида аммония. Смесь перемешивали при температуре 160°С в течение 5 ч. Смеси позволяли остыть до к.т., после чего реакционную среду разводили 5 мл воды, значение рН доводили до 2 добавлением 1 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 3×15 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, от 100 до 95% дихлорметана, об./об.). 0,015 г (выход=8%) (4-((2Н-тетразол-5-ил)метил)-5-(4-хлорфенил)тиофен-2-ил)(фуран-2-ил)метанона получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=370 (МН+); степень чистоты (УФ-детекция при 254 нм)=97%. 1Н ЯМР (300 МГц, DMSO) δ 12,12 (с, 1Н), 7,94 (с, 1Н), 7,60 (к, J=8,6 Гц, 4Н), 7,20 (д, J=16,2 Гц, 2Н), 6,42-6,22 (м, 1Н), 4,35 (с, 2Н).
Пример 38: Приготовление производного номер 152: приготовление 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-гидроксиацетимидамида
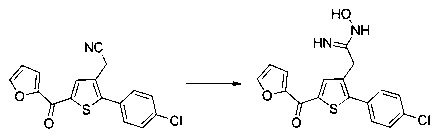 0,1 г (0,299 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила солюбилизировали в 3 мл метанола при перемешивании магнитной мешалкой. К данному раствору добавляли 0,028 г (0,404 ммоль) гидроксиламина гидрохлорида и 0,043 г (0,314 ммоль) карбоната калия. Смесь перемешивали при к.т. в течение 1 ч, а затем нагревали в колбе с обратным холодильником в течение 5 ч. Реакционную среду вливали в 20 мл воды. Водную фазу экстрагировали 2×20 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 4/6, об./об.). 0,023 г (выход=21%) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-гидроксиацетимидамида получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=361 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 9,11 (с, 1Н), 8,19 (с, 1Н), 8,14 (с, 1Н), 7,69 (д, J=8,5 Гц, 2Н), 7,65-7,50 (м, 3Н), 6,84 (дд, J=3,5, 1,6 Гц, 1Н), 5,66 (с, 2Н), 3,39 (с, 2Н).
0,1 г (0,299 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)ацетонитрила солюбилизировали в 3 мл метанола при перемешивании магнитной мешалкой. К данному раствору добавляли 0,028 г (0,404 ммоль) гидроксиламина гидрохлорида и 0,043 г (0,314 ммоль) карбоната калия. Смесь перемешивали при к.т. в течение 1 ч, а затем нагревали в колбе с обратным холодильником в течение 5 ч. Реакционную среду вливали в 20 мл воды. Водную фазу экстрагировали 2×20 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 4/6, об./об.). 0,023 г (выход=21%) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-гидроксиацетимидамида получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=361 (МН+); степень чистоты (УФ-детекция при 254 нм) >99%. 1Н ЯМР (300 МГц, DMSO) δ 9,11 (с, 1Н), 8,19 (с, 1Н), 8,14 (с, 1Н), 7,69 (д, J=8,5 Гц, 2Н), 7,65-7,50 (м, 3Н), 6,84 (дд, J=3,5, 1,6 Гц, 1Н), 5,66 (с, 2Н), 3,39 (с, 2Н).
Пример 39: Приготовление производного номер 153: приготовление 3-((2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)-метил)-1,2,4-оксадиазол-5(2Н)-она
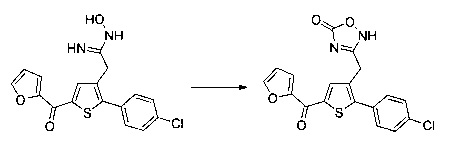 0,05 г (0,139 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-гидроксиацетимидамида солюбилизировали в 1 мл 1,4-диоксана при перемешивании магнитной мешалкой. К раствору добавляли 0,028 г (0,173 ммоль) карбонил диимидазола и 0,023 мл (0,152 мл) DBU (1,8-диаза-бицикло[5,4,0]-ундец-7-цен). Смесь перемешивали при температуре 105°С в течение 30 мин. Реакционную среду вливали в 10 мл воды. Водную фазу промывали 5 мл этилацетата, значение рН водной фазы доводили до 2 добавлением 1 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×10 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. 0,02 г (выход=36%) 3-((2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)метил)-1,2,4-оксадиазол-5(2Н)-она получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=387 (МН+); степень чистоты (УФ-детекция при 254 нм)=96%. 1Н ЯМР (300 МГц, DMSO) δ 12,40 (с, 1Н), 8,19 (с, 1Н), 8,16 (д, J=1,1 Гц, 1Н), 7,66-7,57 (м, 5Н), 6,85 (дд, J=3,6, 1,7 Гц, 1Н), 3,99 (с, 2Н).
0,05 г (0,139 ммоль) 2-(2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)-N-гидроксиацетимидамида солюбилизировали в 1 мл 1,4-диоксана при перемешивании магнитной мешалкой. К раствору добавляли 0,028 г (0,173 ммоль) карбонил диимидазола и 0,023 мл (0,152 мл) DBU (1,8-диаза-бицикло[5,4,0]-ундец-7-цен). Смесь перемешивали при температуре 105°С в течение 30 мин. Реакционную среду вливали в 10 мл воды. Водную фазу промывали 5 мл этилацетата, значение рН водной фазы доводили до 2 добавлением 1 н водного раствора хлористоводородной кислоты. Водную фазу экстрагировали 2×10 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. 0,02 г (выход=36%) 3-((2-(4-хлорфенил)-5-(фуран-2-карбонил)тиофен-3-ил)метил)-1,2,4-оксадиазол-5(2Н)-она получали в виде светло-желтого твердого вещества. ЖХ/МС: m/z=387 (МН+); степень чистоты (УФ-детекция при 254 нм)=96%. 1Н ЯМР (300 МГц, DMSO) δ 12,40 (с, 1Н), 8,19 (с, 1Н), 8,16 (д, J=1,1 Гц, 1Н), 7,66-7,57 (м, 5Н), 6,85 (дд, J=3,6, 1,7 Гц, 1Н), 3,99 (с, 2Н).
Пример 40: Приготовление производного №173: 5-(4-хлорфенил)-4-[2-(2-димeтилaминoэтилaминo)-2-oкco-этил]-N-(3-пиpидил)тиoфeн-2-кapбoкcaмид
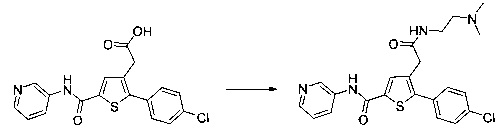 80 мг (0,215 ммоль) 2-[2-(4-хлорфенил)-5-(3-пиридилкарбамоил)-3-тиенил]уксусной кислоты и 0,049 мл (0,429 ммоль) N,N-диметилэтан-1,2-диамина солюбилизировали в 2 мл DMPU. К раствору добавляли при перемешивании магнитной мешалкой 0,045 мл (0,322 моль) триэтиламина и 112 мг (0,215 ммоль) РуВОР. Смесь перемешивали при к.т. в течение 4 часов. Добавляли 10 мл воды, перемешивание продолжали в течение 30 мин., и полученное твердое вещество фильтровали через фритту, промывали водой и высушивали в вакууме. 0,053 г (выход=53%) 5-(4-хлорфенил)-4-[2-(2-димeтилaминoэтилaминo)-2-oкco-этил]-N-(3-пиpидил)тиoфeн-2-кapбoкcaмидa получали в виде твердого вещества. ЖХ/МС: m/z=443 (МН+), степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 10,58 (с, 1Н), 8,90 (с, 1Н), 8,32 (д, J=4,6 Гц, 1Н), 8,16 (д, J=8,5 Гц, 2Н), 8,02 (с, 1Н), 7,59 (к, J=8,6 Гц, 4Н), 7,40 (д, J=12,9 Гц, 1Н), 3,51 (с, 2Н), 2,31 (с, 6Н).
80 мг (0,215 ммоль) 2-[2-(4-хлорфенил)-5-(3-пиридилкарбамоил)-3-тиенил]уксусной кислоты и 0,049 мл (0,429 ммоль) N,N-диметилэтан-1,2-диамина солюбилизировали в 2 мл DMPU. К раствору добавляли при перемешивании магнитной мешалкой 0,045 мл (0,322 моль) триэтиламина и 112 мг (0,215 ммоль) РуВОР. Смесь перемешивали при к.т. в течение 4 часов. Добавляли 10 мл воды, перемешивание продолжали в течение 30 мин., и полученное твердое вещество фильтровали через фритту, промывали водой и высушивали в вакууме. 0,053 г (выход=53%) 5-(4-хлорфенил)-4-[2-(2-димeтилaминoэтилaминo)-2-oкco-этил]-N-(3-пиpидил)тиoфeн-2-кapбoкcaмидa получали в виде твердого вещества. ЖХ/МС: m/z=443 (МН+), степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 10,58 (с, 1Н), 8,90 (с, 1Н), 8,32 (д, J=4,6 Гц, 1Н), 8,16 (д, J=8,5 Гц, 2Н), 8,02 (с, 1Н), 7,59 (к, J=8,6 Гц, 4Н), 7,40 (д, J=12,9 Гц, 1Н), 3,51 (с, 2Н), 2,31 (с, 6Н).
Пример 41: Приготовление производного №174: этил 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]ацетат
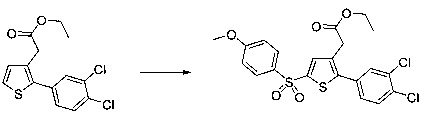 В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 2 мл дихлорметана, 0,1 г (0,317 ммоль) этил 2-(2-(3,4-дихлорфенил)тиофен-3-ил)ацетата и 0,131 г (0,634 ммоль) 4-метоксибензена-1-сульфонилхлорида. Смесь помещали в условия с температурой 5°С при перемешивании магнитной мешалкой и добавляли по частям 0,085 г (0,634 ммоль) хлорида алюминия. Полученную смесь перемешивали при к.т. в течение 24 часов, а затем выливали на лед и перемешивали в течение 1 часа. Водную фазу экстрагировали 2×5 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/дихлорметана, от 100% до 0% петролейного эфира, об./об.). 0,120 г (выход=76%) этил 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]ацетата получали в виде бледно-коричневого масла. ЖХ/МС: m/z=486 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,93 (д, J=8,9 Гц, 2Н), 7,82-7,72 (м, 3Н), 7,45 (дд, J=8,3, 2,1 Гц, 1Н), 7,18 (д, J=9,0 Гц, 2Н), 4,01 (к, J=7,1 Гц, 2Н), 3,85 (с, 3Н), 3,74 (с, 2Н), 1,10 (т, J=7,1 Гц, 3Н).
В колбу, помещенную в атмосферу с потоком аргона, вносили при перемешивании магнитной мешалкой 2 мл дихлорметана, 0,1 г (0,317 ммоль) этил 2-(2-(3,4-дихлорфенил)тиофен-3-ил)ацетата и 0,131 г (0,634 ммоль) 4-метоксибензена-1-сульфонилхлорида. Смесь помещали в условия с температурой 5°С при перемешивании магнитной мешалкой и добавляли по частям 0,085 г (0,634 ммоль) хлорида алюминия. Полученную смесь перемешивали при к.т. в течение 24 часов, а затем выливали на лед и перемешивали в течение 1 часа. Водную фазу экстрагировали 2×5 мл дихлорметана. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/дихлорметана, от 100% до 0% петролейного эфира, об./об.). 0,120 г (выход=76%) этил 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]ацетата получали в виде бледно-коричневого масла. ЖХ/МС: m/z=486 (МН+), степень чистоты (УФ-детекция при 254 нм)=98%. 1Н ЯМР (300 МГц, DMSO) δ 7,93 (д, J=8,9 Гц, 2Н), 7,82-7,72 (м, 3Н), 7,45 (дд, J=8,3, 2,1 Гц, 1Н), 7,18 (д, J=9,0 Гц, 2Н), 4,01 (к, J=7,1 Гц, 2Н), 3,85 (с, 3Н), 3,74 (с, 2Н), 1,10 (т, J=7,1 Гц, 3Н).
Пример 42: Приготовление производного №175: 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]уксусная кислота
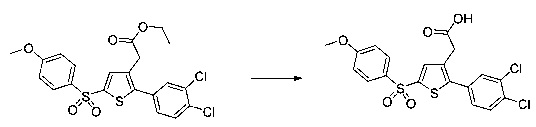 0,090 г (0,185 ммоль) этил 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]-ацетата солюбилизировали в 2 мл этанола и 2 мл THF (тетрагидрофурана), к раствору добавляли при перемешивании магнитной мешалкой 0,020 мл (0,204 ммоль) 30 масс. % водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды. Затем значение рН водной фазы снижали путем добавления 1 н водного раствора хлористоводородной кислоты до появления преципитата. В раствор добавляли 1 мл диизопропилового эфира, после чего полученный раствор перемешивали в течение 2 часов. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и 1 мл диизопропилового эфира и высушивали в вакуумном колпаке с получением 0,072 г (выход=84%) 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]уксусной кислоты в виде белого твердого вещества. Степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,69 (с, 1Н), 7,93 (д, J=8,9 Гц, 2Н), 7,76 (д, J=9,6 Гц, 3Н), 7,46 (дд, J=8,3, 2,1 Гц, 1Н), 7,18 (д, J=8,9 Гц, 2Н), 3,85 (с, 3Н), 3,63 (с, 2Н).
0,090 г (0,185 ммоль) этил 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]-ацетата солюбилизировали в 2 мл этанола и 2 мл THF (тетрагидрофурана), к раствору добавляли при перемешивании магнитной мешалкой 0,020 мл (0,204 ммоль) 30 масс. % водного раствора гидроксида натрия. Полученную смесь перемешивали при к.т. в течение 16 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл воды. Затем значение рН водной фазы снижали путем добавления 1 н водного раствора хлористоводородной кислоты до появления преципитата. В раствор добавляли 1 мл диизопропилового эфира, после чего полученный раствор перемешивали в течение 2 часов. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и 1 мл диизопропилового эфира и высушивали в вакуумном колпаке с получением 0,072 г (выход=84%) 2-[2-(3,4-дихлорфенил)-5-(4-метоксифенил)сульфонил-3-тиенил]уксусной кислоты в виде белого твердого вещества. Степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 12,69 (с, 1Н), 7,93 (д, J=8,9 Гц, 2Н), 7,76 (д, J=9,6 Гц, 3Н), 7,46 (дд, J=8,3, 2,1 Гц, 1Н), 7,18 (д, J=8,9 Гц, 2Н), 3,85 (с, 3Н), 3,63 (с, 2Н).
Пример 43: Приготовление производного №176: этил 2-[5-(5-хлорфуран-2-карбонил)-2-(4-хлорфенил)-3-тиенил]ацетат
 200 мг (0,534 ммоль) этил 2-[2-(4-хлорфенил)-5-(фуран-2-карбонил)-3-тиенил]ацетата растворяли в 3 мл ацетонитрила, после чего добавляли 142,4 мг (1,068 ммоль) N-хлор-сукцинимида. Реакционную смесь нагревали в колбе с обратным холодильником в течение 15 ч. Смесь промывали 5 мл воды, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/дихлорметана, от 100 до 0% гептана, об./об.). 78 мг (выход=31%) этил 2-[5-(5-хлорфуран-2-карбонил)-2-(4-хлорфенил)-3-тиенил]ацетата получали в виде масла без интенсивной окраски. ЖХ/МС: m/z=409 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,14 (с, 1Н), 7,69 (д, J=3,7 Гц, 1Н), 7,64-7,43 (м, 4Н), 6,93 (д, J=3,7 Гц, 1Н), 4,11-4,00 (м, 2Н), 3,82 (д, J=12,5 Гц, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
200 мг (0,534 ммоль) этил 2-[2-(4-хлорфенил)-5-(фуран-2-карбонил)-3-тиенил]ацетата растворяли в 3 мл ацетонитрила, после чего добавляли 142,4 мг (1,068 ммоль) N-хлор-сукцинимида. Реакционную смесь нагревали в колбе с обратным холодильником в течение 15 ч. Смесь промывали 5 мл воды, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/дихлорметана, от 100 до 0% гептана, об./об.). 78 мг (выход=31%) этил 2-[5-(5-хлорфуран-2-карбонил)-2-(4-хлорфенил)-3-тиенил]ацетата получали в виде масла без интенсивной окраски. ЖХ/МС: m/z=409 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 8,14 (с, 1Н), 7,69 (д, J=3,7 Гц, 1Н), 7,64-7,43 (м, 4Н), 6,93 (д, J=3,7 Гц, 1Н), 4,11-4,00 (м, 2Н), 3,82 (д, J=12,5 Гц, 2Н), 1,14 (т, J=7,1 Гц, 3Н).
Пример 44: Приготовление производного №177: этил 2-[5-[[(4-хлорбензоил)амино]карбамоил]-2-(4-хлорфенил)-3-тиенил]ацетат
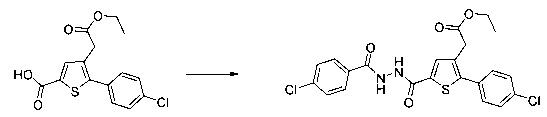 100 мг (0,308 ммоль) 5-(4-хлорфенил)-4-(2-этокси-2-оксо-этил)тиофен-2-карбоновой кислоты, 0,172 мл (0,985 ммоль) триэтиламина и 52,5 мг (0,308 ммоль) 4-хлорбензогидразида погружали в 2 мл DMF. Раствор охлаждали до температуры 0°С и добавляли 240 мг (0,462 ммоль) РуВОР. Смесь перемешивали при к.т. в течение 4 ч. Раствор вливали в 10 мл воды с добавлением 1 мл диизопропилового эфира. После 30 минут интенсивного перемешивания полученное твердое вещество фильтровали, а затем промывали водой и диизопропиловым эфиром. 123 мг (выход=78%) этил 2-[5-[[(4-хлор-бензоил)амино]карбамоил]-2-(4-хлорфенил)-3-тиенил]ацетата получали в виде твердого вещества. ЖХ/МС: m/z=477 (МН+), степень чистоты (УФ-детекция при 254 нм)=93%. 1Н ЯМР (300 МГц, DMSO) δ 10,68 (с, 2Н), 7,93 (д, J=8,5 Гц, 2Н), 7,86 (с, 1Н), 7,57 (дт, J=17,0, 8,6 Гц, 6Н), 4,09 (к, J=7,1 Гц, 2Н), 3,72 (с, 2Н), 1,17 (т, J=7,1 Гц, 3Н).
100 мг (0,308 ммоль) 5-(4-хлорфенил)-4-(2-этокси-2-оксо-этил)тиофен-2-карбоновой кислоты, 0,172 мл (0,985 ммоль) триэтиламина и 52,5 мг (0,308 ммоль) 4-хлорбензогидразида погружали в 2 мл DMF. Раствор охлаждали до температуры 0°С и добавляли 240 мг (0,462 ммоль) РуВОР. Смесь перемешивали при к.т. в течение 4 ч. Раствор вливали в 10 мл воды с добавлением 1 мл диизопропилового эфира. После 30 минут интенсивного перемешивания полученное твердое вещество фильтровали, а затем промывали водой и диизопропиловым эфиром. 123 мг (выход=78%) этил 2-[5-[[(4-хлор-бензоил)амино]карбамоил]-2-(4-хлорфенил)-3-тиенил]ацетата получали в виде твердого вещества. ЖХ/МС: m/z=477 (МН+), степень чистоты (УФ-детекция при 254 нм)=93%. 1Н ЯМР (300 МГц, DMSO) δ 10,68 (с, 2Н), 7,93 (д, J=8,5 Гц, 2Н), 7,86 (с, 1Н), 7,57 (дт, J=17,0, 8,6 Гц, 6Н), 4,09 (к, J=7,1 Гц, 2Н), 3,72 (с, 2Н), 1,17 (т, J=7,1 Гц, 3Н).
Производные 178 и 179 были приготовлены согласно аналогичной процедуре:
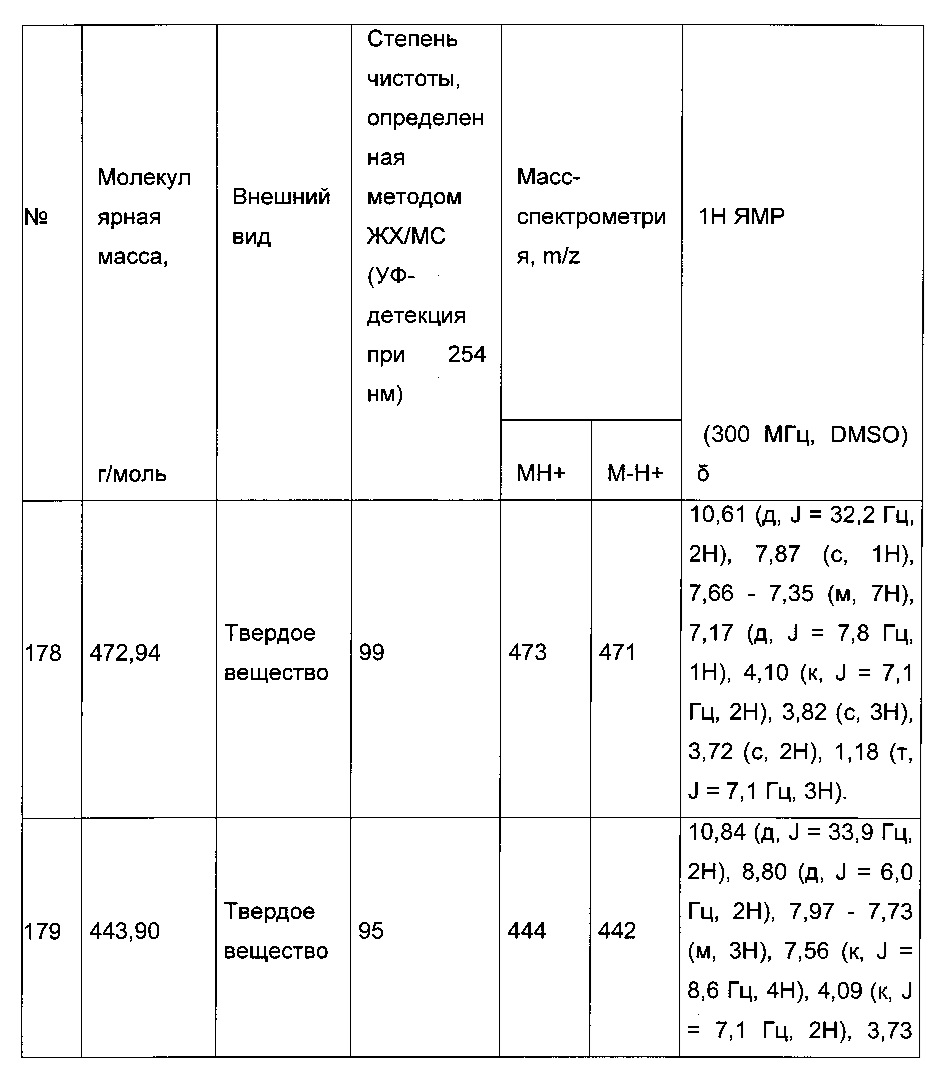
Пример 45: Приготовление производного №180: 2-[2-(4-хлорфенил)-5-(фуран-2-карбонил)-3-тиенил]-N-этил-тиоацетамид
Этап 1: Приготовление 2-[2-(4-хлорфенил)-3-тиенил]уксусной кислоты
 1,5 г (5,34 ммоль) этил 2-[2-(4-хлорфенил)-3-тиенил]ацетата погружали в 8 мл этанола, после чего добавляли 0,588 мл (5,88 ммоль) 30 масс. % водного раствора гидроксида натрия. Раствор перемешивали при к.т. в течение 18 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 15 мл воды. Затем значение рН водной фазы снижали путем добавления 1 н водного раствора хлористоводородной кислоты до появления преципитата. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и высушивали в вакуумном колпаке. 1,227 г (выход=88%) 2-[2-(4-хлорфенил)-3-тиенил]уксусной кислоты получали в виде твердого вещества. ЖХ/МС: m/z=251 (МН-), степень чистоты (УФ-детекция при 254 нм)=93%.
1,5 г (5,34 ммоль) этил 2-[2-(4-хлорфенил)-3-тиенил]ацетата погружали в 8 мл этанола, после чего добавляли 0,588 мл (5,88 ммоль) 30 масс. % водного раствора гидроксида натрия. Раствор перемешивали при к.т. в течение 18 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 15 мл воды. Затем значение рН водной фазы снижали путем добавления 1 н водного раствора хлористоводородной кислоты до появления преципитата. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и высушивали в вакуумном колпаке. 1,227 г (выход=88%) 2-[2-(4-хлорфенил)-3-тиенил]уксусной кислоты получали в виде твердого вещества. ЖХ/МС: m/z=251 (МН-), степень чистоты (УФ-детекция при 254 нм)=93%.
Этап 2: Приготовление 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-ацетамида
 0,6 г (2,374 ммоль) 2-[2-(4-хлорфенил)-3-тиенил]уксусной кислоты, 0,290 г (3,55 ммоль) гидрохлорида этанамина, 1,236 г (2,374 ммоль) РуВОР и 0,827 мл (5,94 ммоль) триэтиламина погружали в 2 мл DMF. Затем раствор перемешивали при к.т. в течение 18 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл 1 н водного раствора хлористоводородной кислоты, после чего перемешивали в течение 1 часа. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и высушивали в вакуумном колпаке. 0,559 г (выход=79%) 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-ацетамида получали в виде твердого вещества. ЖХ/МС: m/z=280 (МН+), степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,11 (с, 1Н), 7,62 (д, J=8,5 Гц, 2Н), 7,57-7,46 (м, 3Н), 7,08 (д, J=5,2 Гц, 1Н), 3,39 (с, 2Н), 3,14-3,02 (м, 2Н), 1,01 (т, J=7,2 Гц, 3Н).
0,6 г (2,374 ммоль) 2-[2-(4-хлорфенил)-3-тиенил]уксусной кислоты, 0,290 г (3,55 ммоль) гидрохлорида этанамина, 1,236 г (2,374 ммоль) РуВОР и 0,827 мл (5,94 ммоль) триэтиламина погружали в 2 мл DMF. Затем раствор перемешивали при к.т. в течение 18 часов. Смесь концентрировали в вакууме, полученный осадок переносили в 10 мл 1 н водного раствора хлористоводородной кислоты, после чего перемешивали в течение 1 часа. Твердое вещество выделяли в результате фильтрации, промывали 2×5 мл воды и высушивали в вакуумном колпаке. 0,559 г (выход=79%) 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-ацетамида получали в виде твердого вещества. ЖХ/МС: m/z=280 (МН+), степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 8,11 (с, 1Н), 7,62 (д, J=8,5 Гц, 2Н), 7,57-7,46 (м, 3Н), 7,08 (д, J=5,2 Гц, 1Н), 3,39 (с, 2Н), 3,14-3,02 (м, 2Н), 1,01 (т, J=7,2 Гц, 3Н).
Этап 3: Приготовление 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-тиоацетамида
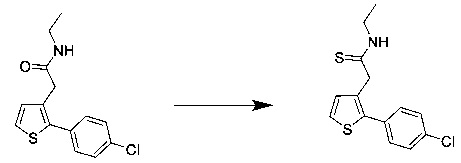 К 5 мл толуола добавляли 559 мг (1,998 ммоль) 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-ацетамида и 808 мг (1,998 ммоль) реактива Лавессона. Раствор перемешивали и нагревали в колбе с обратным холодильником в течение 4 ч. Добавляли 10 мл воды, полученную смесь экстрагировали 2×15 мл этилацетата. Собранные органические фазы промывали 10 мл воды и 10 мл солевого раствора, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100% до 0% петролейного эфира, об./об.). 476 мг (выход=77%) получали в виде масла. ЖХ/МС: m/z=296 (МН+), степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 10,22 (с, 1Н), 7,66 (д, J=8,6 Гц, 2Н), 7,56-7,47 (м, 3Н), 7,02 (д, J=5,2 Гц, 1Н), 3,86 (с, 2Н), 3,59-3,47 (м, 2Н), 1,14 (т, J=7,3 Гц, 3Н).
К 5 мл толуола добавляли 559 мг (1,998 ммоль) 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-ацетамида и 808 мг (1,998 ммоль) реактива Лавессона. Раствор перемешивали и нагревали в колбе с обратным холодильником в течение 4 ч. Добавляли 10 мл воды, полученную смесь экстрагировали 2×15 мл этилацетата. Собранные органические фазы промывали 10 мл воды и 10 мл солевого раствора, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/диизопропилового эфира, от 100% до 0% петролейного эфира, об./об.). 476 мг (выход=77%) получали в виде масла. ЖХ/МС: m/z=296 (МН+), степень чистоты (УФ-детекция при 254 нм)=95%. 1Н ЯМР (300 МГц, DMSO) δ 10,22 (с, 1Н), 7,66 (д, J=8,6 Гц, 2Н), 7,56-7,47 (м, 3Н), 7,02 (д, J=5,2 Гц, 1Н), 3,86 (с, 2Н), 3,59-3,47 (м, 2Н), 1,14 (т, J=7,3 Гц, 3Н).
Этап 4: Приготовление 2-[2-(4-хлорфенил)-5-(фуран-2-карбонил)-3-тиенил]-N-этил-тиоацетамида (производное номер 180)
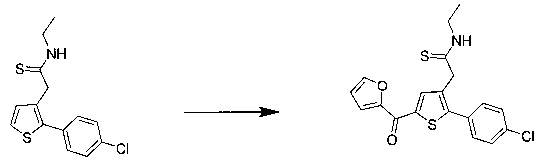 215 мг (0,727 ммоль) 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-тиоацетамида растворяли в 3 мл дихлорметана. При температуре +5°С добавляли 0,107 мл (1,090 ммоль) 2-фуран-карбонил хлорида, а затем, также при температуре +5°С, добавляли 97 мг (0,727 ммоль) хлорида алюминия. Раствор перемешивали при к.т. в течение 18 ч. Смесь вливали в 200 мл воды, перемешивали в течение 3 часов и фильтровали через целит, после чего раствор экстрагировали дважды 15 мл дихлорметана. Собранные органические фазы промывали 10 мл воды, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/дихлорметана, от 100% до 0% петролейного эфира, об./об.). 51 мг (выход=17%) 2-[2-(4-хлорфенил)-5-(фуран-2-карбонил)-3-тиенил]-N-этил-тиоацетамида получали в виде твердого вещества. ЖХ/МС: m/z=390 (МН+), степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 10,27 (с, 1Н), 8,14 (д, J=6,6 Гц, 2Н), 7,70 (д, J=8,6 Гц, 2Н), 7,62-7,49 (м, 3Н), 6,89-6,81 (м, 1Н), 3,94 (с, 2Н), 3,67-3,46 (м, 2Н), 1,15 (т, J=7,2 Гц, 3Н).
215 мг (0,727 ммоль) 2-[2-(4-хлорфенил)-3-тиенил]-N-этил-тиоацетамида растворяли в 3 мл дихлорметана. При температуре +5°С добавляли 0,107 мл (1,090 ммоль) 2-фуран-карбонил хлорида, а затем, также при температуре +5°С, добавляли 97 мг (0,727 ммоль) хлорида алюминия. Раствор перемешивали при к.т. в течение 18 ч. Смесь вливали в 200 мл воды, перемешивали в течение 3 часов и фильтровали через целит, после чего раствор экстрагировали дважды 15 мл дихлорметана. Собранные органические фазы промывали 10 мл воды, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент петролейного эфира/дихлорметана, от 100% до 0% петролейного эфира, об./об.). 51 мг (выход=17%) 2-[2-(4-хлорфенил)-5-(фуран-2-карбонил)-3-тиенил]-N-этил-тиоацетамида получали в виде твердого вещества. ЖХ/МС: m/z=390 (МН+), степень чистоты (УФ-детекция при 254 нм)=94%. 1Н ЯМР (300 МГц, DMSO) δ 10,27 (с, 1Н), 8,14 (д, J=6,6 Гц, 2Н), 7,70 (д, J=8,6 Гц, 2Н), 7,62-7,49 (м, 3Н), 6,89-6,81 (м, 1Н), 3,94 (с, 2Н), 3,67-3,46 (м, 2Н), 1,15 (т, J=7,2 Гц, 3Н).
Пример 46: Приготовление производного №182: N-(N-(трет-бутоксикарбонил)карбамимидоил)-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамид
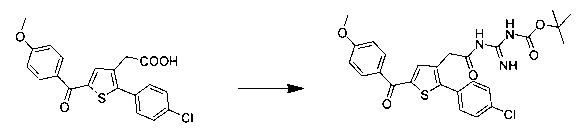 К 1 мл DMF добавляли 142 мг (0,337 ммоль) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)уксусной кислоты, 175 мг (0,337 ммоль) РуВОР и 64,4 мг (0,404 ммоль) Вос-гуанидина. Затем добавляли 0,141 мл (1,011 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 4 ч. Раствор выливали на 10 мл воды и 5 мл диизопропилового эфира при интенсивном перемешивании. Полученное твердое вещество фильтровали, после чего промывали водой и диизопропиловым эфиром. 172 мг (выход=90%) N-(N-(трет-бут-оксикарбонил)карбамимидоил)-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)-тиофен-3-ил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=462 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 11,05 (с, 1Н), 8,70 (с, 2Н), 7,93-7,77 (м, 5Н), 7,57 (д, J=8,4 Гц, 1Н), 7,13 (д, J=8,8 Гц, 2Н), 3,88 (с, 3Н), 3,75 (с, 2Н), 1,39 (с, 9Н).
К 1 мл DMF добавляли 142 мг (0,337 ммоль) 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)уксусной кислоты, 175 мг (0,337 ммоль) РуВОР и 64,4 мг (0,404 ммоль) Вос-гуанидина. Затем добавляли 0,141 мл (1,011 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 4 ч. Раствор выливали на 10 мл воды и 5 мл диизопропилового эфира при интенсивном перемешивании. Полученное твердое вещество фильтровали, после чего промывали водой и диизопропиловым эфиром. 172 мг (выход=90%) N-(N-(трет-бут-оксикарбонил)карбамимидоил)-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)-тиофен-3-ил)ацетамида получали в виде белого твердого вещества. ЖХ/МС: m/z=462 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 11,05 (с, 1Н), 8,70 (с, 2Н), 7,93-7,77 (м, 5Н), 7,57 (д, J=8,4 Гц, 1Н), 7,13 (д, J=8,8 Гц, 2Н), 3,88 (с, 3Н), 3,75 (с, 2Н), 1,39 (с, 9Н).
Производные 181 и 183 были приготовлены согласно аналогичной процедуре.
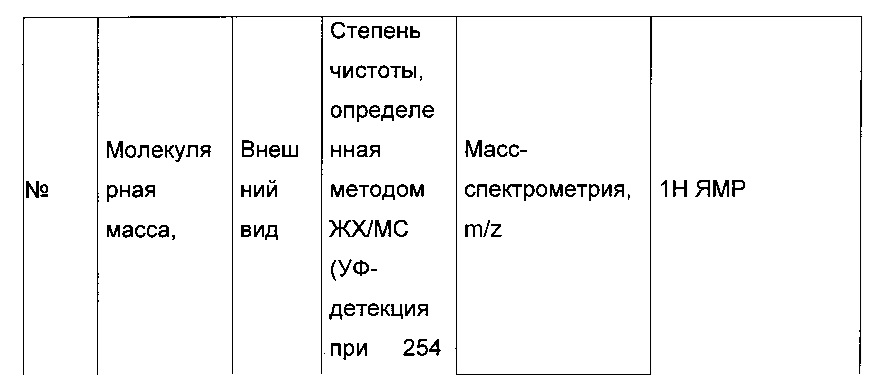
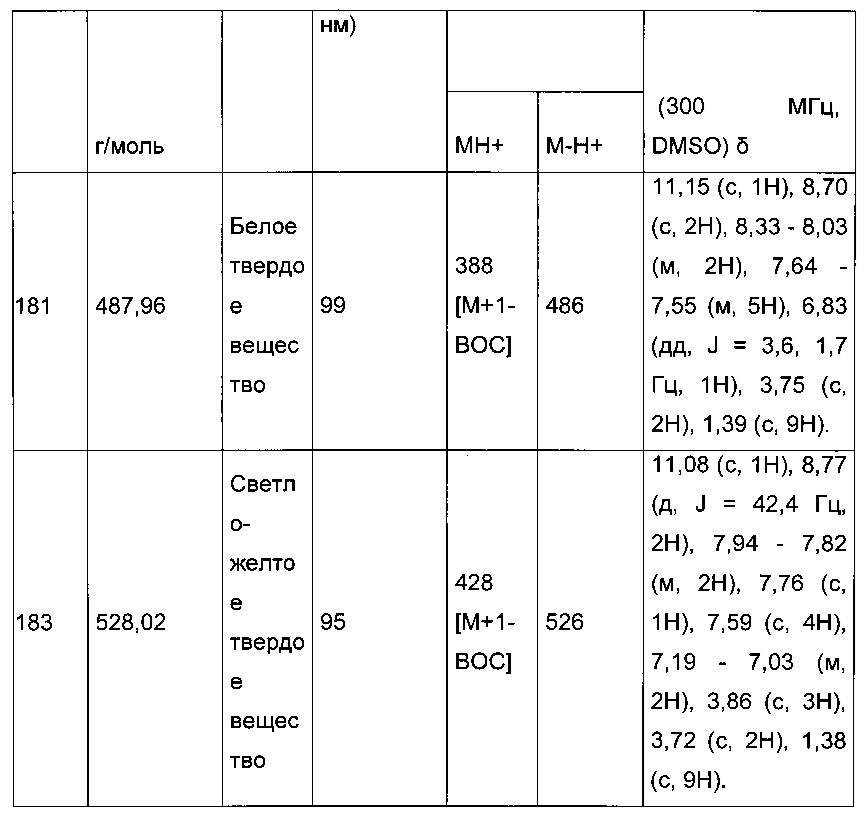
Пример 47: Приготовление производного №185: гидрохлорид N-карбамимидоил-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамида
 481 мг (0,911 ммоль) N-(N-(трет-бутоксикарбонил)карбамимидоил)-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамида растворяли в 2 мл дихлорметана, а затем добавляли 2 мл (26 ммоль) трифторуксусной кислоты. Смесь перемешивали при к.т. в течение 2 часов. Добавляли 0,25 мл (1 ммоль) раствора хлористоводородной кислоты (концентрацией 4 молей на литр) в диоксане, после чего растворители выпаривали в вакууме. Полученное твердое вещество растирали в 5 мл кипящего пропан-2-ола, после охлаждения раствора твердое вещество фильтровали и промывали 1 мл пропан-2-ола, а затем высушивали в вакууме. 421 мг (выход=99%) гидрохлорида N-карбамимидоил-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамида получали в виде желтого твердого вещества. ЖХ/МС: m/z=428 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 11,78 (с, 1Н), 8,36 (с, 4Н), 7,88 (д, J=8,8 Гц, 2Н), 7,81 (с, 1Н), 7,66-7,45 (м, 4Н), 7,12 (д, J=8,9 Гц, 2Н), 3,88 (д, J=7,2 Гц, 5Н).
481 мг (0,911 ммоль) N-(N-(трет-бутоксикарбонил)карбамимидоил)-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамида растворяли в 2 мл дихлорметана, а затем добавляли 2 мл (26 ммоль) трифторуксусной кислоты. Смесь перемешивали при к.т. в течение 2 часов. Добавляли 0,25 мл (1 ммоль) раствора хлористоводородной кислоты (концентрацией 4 молей на литр) в диоксане, после чего растворители выпаривали в вакууме. Полученное твердое вещество растирали в 5 мл кипящего пропан-2-ола, после охлаждения раствора твердое вещество фильтровали и промывали 1 мл пропан-2-ола, а затем высушивали в вакууме. 421 мг (выход=99%) гидрохлорида N-карбамимидоил-2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетамида получали в виде желтого твердого вещества. ЖХ/МС: m/z=428 (МН+), степень чистоты (УФ-детекция при 254 нм)=99%. 1Н ЯМР (300 МГц, DMSO) δ 11,78 (с, 1Н), 8,36 (с, 4Н), 7,88 (д, J=8,8 Гц, 2Н), 7,81 (с, 1Н), 7,66-7,45 (м, 4Н), 7,12 (д, J=8,9 Гц, 2Н), 3,88 (д, J=7,2 Гц, 5Н).
Производные 184 и 186 были приготовлены согласно аналогичной процедуре.
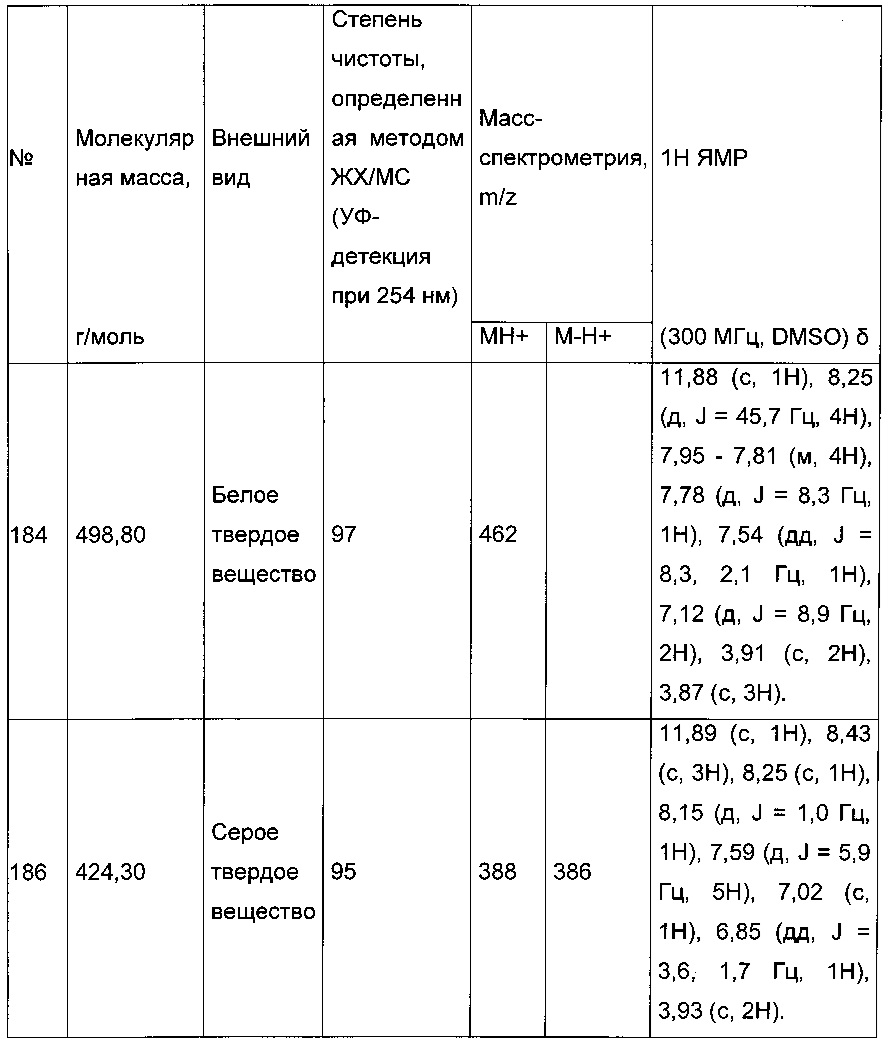
Пример 48: Приготовление производного №187: этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)акрилат
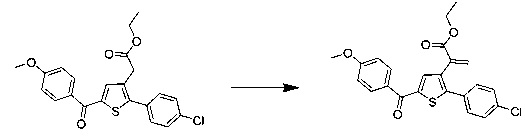 1,5 г (3,62 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата растворяли в 20 мл тетрагидрофурана в атмосфере аргона, а затем охлаждали до температуры -78°С. В полученный раствор вливали по каплям 10,85 мл (10,85 ммоль) раствора бис(триметилсилил)амида лития (концентрацией один моль на литр) в тетрагидрофуране. Раствор перемешивали при температуре -78°С в течение 30 мин., после чего медленно добавляли 0,984 мл (10,85 ммоль) бром(метокси)метана. Смесь перемешивали, смеси позволяли нагреться до к.т. в течение 16 ч. Затем смесь вливали в 50 мл воды, экстрагировали 3 фракциями по 15 мл дихлорметана, собранные органические фазы промывали 20 мл воды, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 100 до 0% гептана, об./об.). 0,696 г (выход=41%) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)акрилата получали в виде твердого вещества. ЖХ/МС: m/z=427 (МН+), степень чистоты (УФ-детекция при 254 нм)=90,6%. 1Н ЯМР (300 МГц, DMSO) δ 7,93 (д, J=8,9 Гц, 2Н), 7,70 (с, 1Н), 7,56-7,51 (м, 2Н), 7,48-7,41 (м, 2Н), 7,11 (д, J=8,9 Гц, 2Н), 6,39 (д, J=0,8 Гц, 1Н), 6,11 (д, J=0,7 Гц, 1Н), 3,93-3,78 (м, 5Н), 0,91 (т, J=7,1 Гц, 3Н).
1,5 г (3,62 ммоль) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)ацетата растворяли в 20 мл тетрагидрофурана в атмосфере аргона, а затем охлаждали до температуры -78°С. В полученный раствор вливали по каплям 10,85 мл (10,85 ммоль) раствора бис(триметилсилил)амида лития (концентрацией один моль на литр) в тетрагидрофуране. Раствор перемешивали при температуре -78°С в течение 30 мин., после чего медленно добавляли 0,984 мл (10,85 ммоль) бром(метокси)метана. Смесь перемешивали, смеси позволяли нагреться до к.т. в течение 16 ч. Затем смесь вливали в 50 мл воды, экстрагировали 3 фракциями по 15 мл дихлорметана, собранные органические фазы промывали 20 мл воды, высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 100 до 0% гептана, об./об.). 0,696 г (выход=41%) этил 2-(2-(4-хлорфенил)-5-(4-метоксибензоил)тиофен-3-ил)акрилата получали в виде твердого вещества. ЖХ/МС: m/z=427 (МН+), степень чистоты (УФ-детекция при 254 нм)=90,6%. 1Н ЯМР (300 МГц, DMSO) δ 7,93 (д, J=8,9 Гц, 2Н), 7,70 (с, 1Н), 7,56-7,51 (м, 2Н), 7,48-7,41 (м, 2Н), 7,11 (д, J=8,9 Гц, 2Н), 6,39 (д, J=0,8 Гц, 1Н), 6,11 (д, J=0,7 Гц, 1Н), 3,93-3,78 (м, 5Н), 0,91 (т, J=7,1 Гц, 3Н).
Тест стимуляции секреции инсулина клетками INS-1
Различные соединения исследовали на клетках INS-1, линии бета-клеток поджелудочной железы, для оценки способности исследуемых соединений потенциировать секрецию инсулина в ответ на поступление глюкозы. Вкратце, клетки культивировали в культуральной среде RPMI 1640 с 10 мМ глюкозы, содержащей 1 мМ пирувата натрия, 50 мкМ 2-меркаптоэтанола, 2 мМ глутамина, 10 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин этансульфоновая кислота), 100 МЕ/мл пенициллина, 100 мкг/мл стрептомицина и 10% инактивированной фетальной телячьей сыворотки (среда описана Asfari et al. [13]). Для проведения теста секреции инсулина клетки INS-1 высевали и культивировали в 96-луночных планшетах. Через 3 дня культивирования при температуре 37°С во влажной атмосфере (95% атмосферного воздуха/5% СO2) среду удаляли, и клетки инкубировали в течение 16 ч в среде, содержащей 5 мМ глюкозы и 1% инактивированной фетальной телячьей сыворотки. В день проведения теста клетки промывали буфером Кребса (рН 7,4), содержащим 0,1% бычьего альбумина, а затем предынкубировали в течение 30 мин. при температуре 37°С в том же буфере, содержащем 2,8 мМ глюкозы. Затем клетки снова промывали буфером Кребса, после чего инкубировали в течение 1 ч в буфере для проведения теста секреции (буфер Кребса, рН 7,4, содержащий 0,1% бычьего альбумина, 3,5 мМ глюкозы и исследуемые молекулы). По окончании теста супернатант клеток отбирали и измеряли количество инсулина, секретируемого клетками, с помощью набора для анализа методом ELISA (Enzyme-Linked Immuno Sorbent Assay, метод твердофазного иммуносорбентного анализа) с применением антител крысы против инсулина (ELISA Alpco, кат. №80-INSRTH-E10). Каждое условие изучали в трех повторах. В качестве положительных контролей при проведении теста использовали 3,5 мМ глюкозы, 10-7 М GLP-1 (глюкагонподобный пептид-1) и смесь 10-7 М Форсколина/10-5 М IBMX (3-изобутил-1-метилксантин). Соединение стимулирует секрецию инсулина, если данный показатель больше или равен 130% от показателя контроля для данной дозы глюкозы.
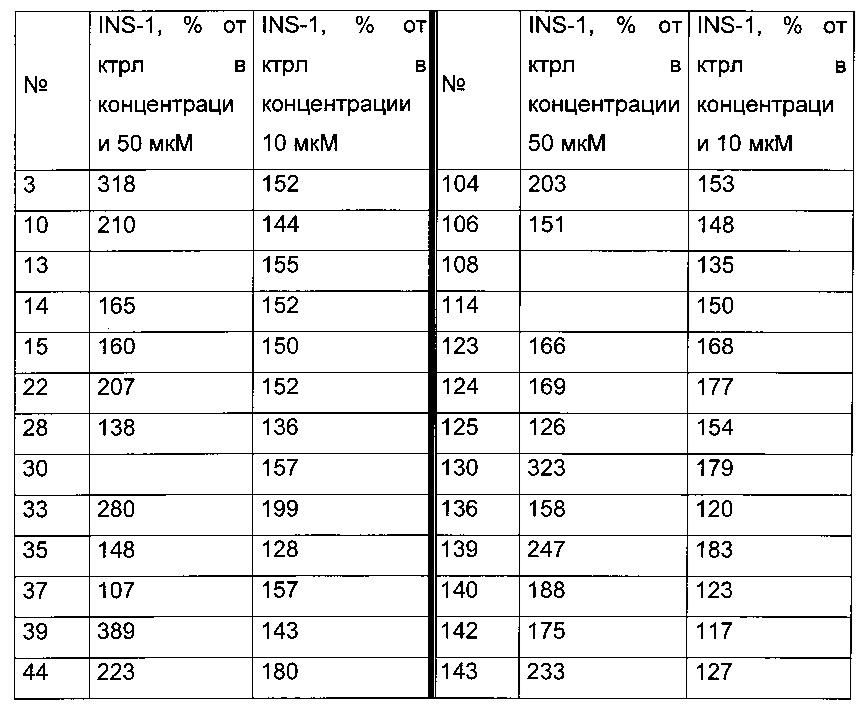
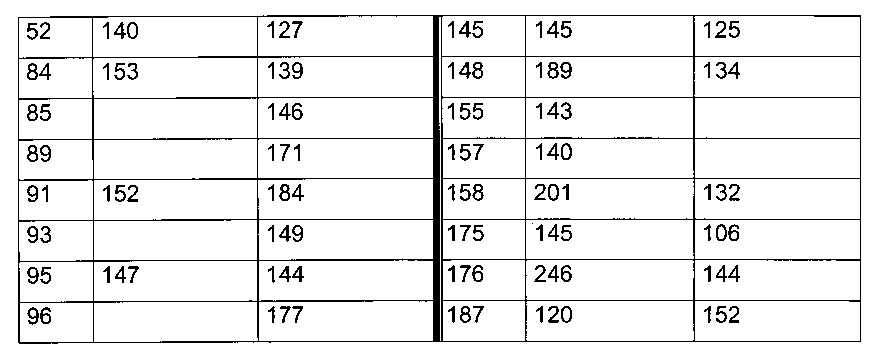
Исходя из полученных результатов, исследуемые производные формулы I оказывают значительное действие на потенциализацию секреции инсулина 8-клетками поджелудочной железы INS-1 в ответ на поступление глюкозы. Полученные значения активации составляют от 132% до свыше 300%, вне зависимости от соответствующей дозы.
Тест ингибирования продукции глюкозы печенью
Гепатоциты выделяли из печени крыс Wistar, не получавших пищу в течение 24 ч, после перфузии коллагеназы в воротную вену. Свежевыделенные гепатоциты высевали на 6-луночных планшетах, покрытых коллагеном и содержащих среду для адгезии (Williams Medium). После адгезии среду заменяли на среду RPMI 1640 без добавления глюкозы, содержащую гидрокортизон (7,10-5 М), и проводили инкубацию в течение от 16 до 18 ч. На следующий день в течение 3 ч проводили тест продукции глюкозы печенью в среде Кребса. В базальных условиях клетки инкубировали исключительно с буфером Кребса, в стимулирующих условиях клетки помещали в буфер Кребса/лактат/пируват, в продуцирующих условиях клетки подвергали воздействию химических соединений в среде буфер Кребса/лактат/пируват. В случае, когда соединения растворяли в DMSO, все условия тестов выполнялись в присутствии конечной концентрации DMSO 0,1%. Положительный контроль при проведении теста представлял собой меркаптопиколинат, который, как известно, оказывает ингибирующее действие на продукцию глюкозы печенью посредством ингибирования фосфоэнолпируваткарбоксикиназы. В случае краткосрочной обработки соединения инкубировали в течение 3 часов. В случае долгосрочной обработки соединения инкубировали в течение 20 ч, пока гепатоциты культивировали в RPMI, а затем добавляли во время проведения теста продукции глюкозы печенью в течение 3 часов. По окончании 3 часов инкубации супернатант отбирали и измеряли содержания глюкозы колориметрическим методом с применением глюкозооксидазы. Клетки лизировали 0,1% водным раствором NaOH и проводили измерение количества белка методом Лоури. Результаты поредставлены в ммолях глюкозы на мг белка. Соединение ингибирует продукцию глюкозы в печени, если данный показатель меньше или равен 75% от показателя контроля для данной дозы глюкозы.
3 ч инкубации
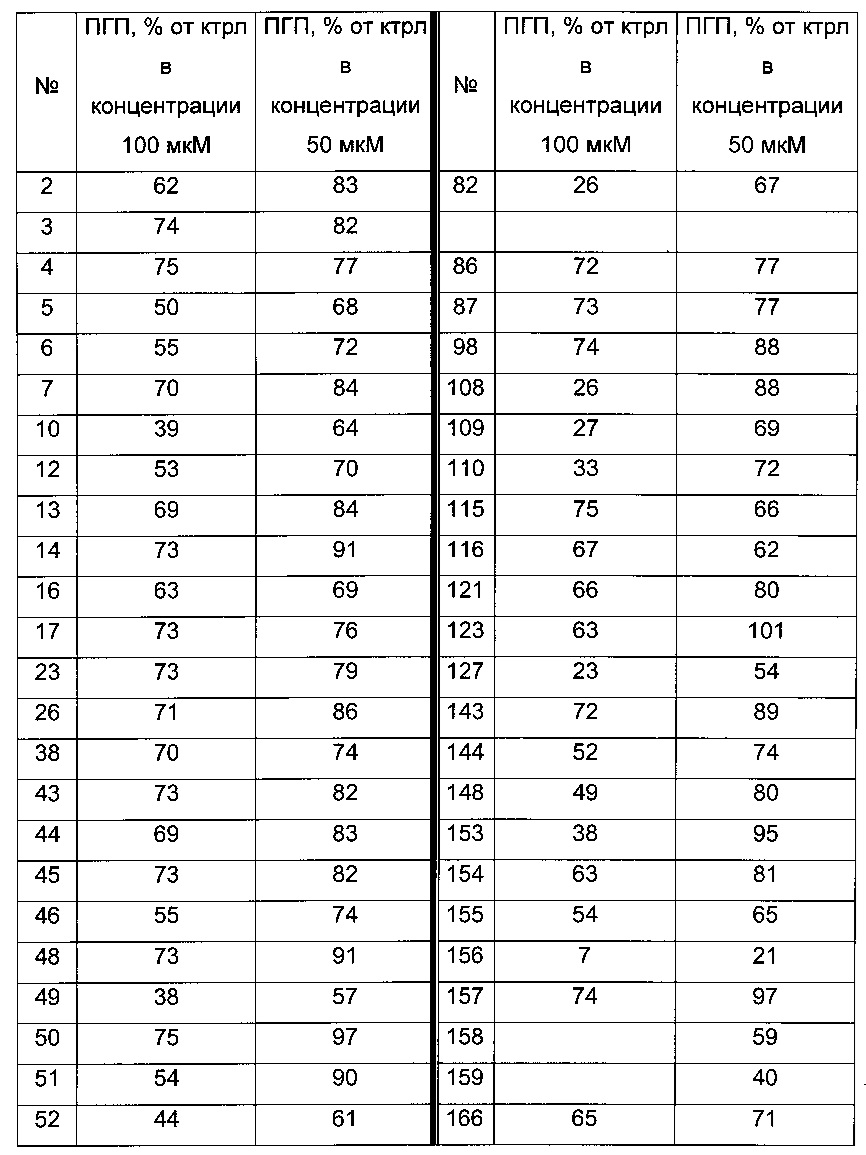
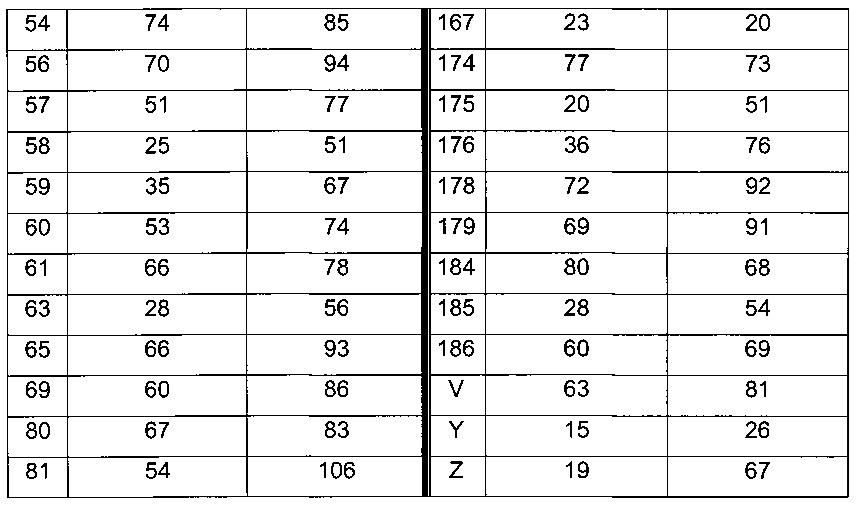
Исследованные производные формулы I оказывают значительное действие на ингибирование продукции глюкозы печенью. Наиболее мощное ингибирование было достигнуто при применении производных 49, 58, 63, 127, 156, 167, 175, 185 и Y в двух дозах и производных 158 и 159 в дозе 50 мкМ.
20 ч инкубирования
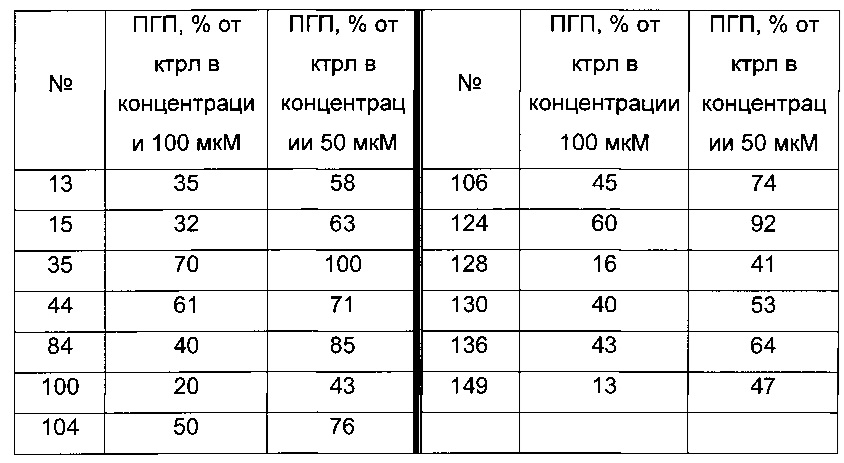
Исследованные производные формулы I оказывают значительное действие на ингибирование продукции глюкозы печенью. Наиболее мощное ингибирование было достигнуто при применении производных 13, 100, 128, 130 и 149 в двух дозах.
Исследование действия соединений на секрецию инсулина в ответ на поступление глюкозы на изолированных перфузируемых поджелудочных железах диабетических крыс N0STZ
Оборудование и методы:
Поджелудочные железы отбирали от крыс, у которых появление диабета было вызвано инъекцией Стрептозотоцина в день рождения [14]; животные получали анестезию Фентобарбиталом (Нембутал©: 45 мг/кг; интраперитонеальное введение). Данные крысы характеризуются специфическим дефицитом инсулина в ответ на поступление глюкозы [15], который наблюдается у человека при сахарном диабете II типа. Изоляцию и перфузирование поджелудочных желез проводили согласно модификации [16] процедуры, описанной Sussman et al. [17]. Действие соединений или веществ-эталонов изучали в течение 35 минут (от t=20 мин. до t=55 мин.) в буфере Кребса при отсутствии глюкозы (GO) или в присутствии глюкозы в концентрации 2,8 мМ (G2,8 мМ), и затем в течение 20 минут (от t=55 мин. до t=75 мин.) в присутствии глюкозы в концентрации 16,5 мМ. Концентрацию инсулина, секретируемого в среду, измеряли методом Elisa (ELISA Alpco, кат. №80-INSRTH-Е10). Результаты выражали в виде среднего значения +/- СОС (стандартная ошибка среднего) результатов нескольких экспериментов.
Таблица результатов:
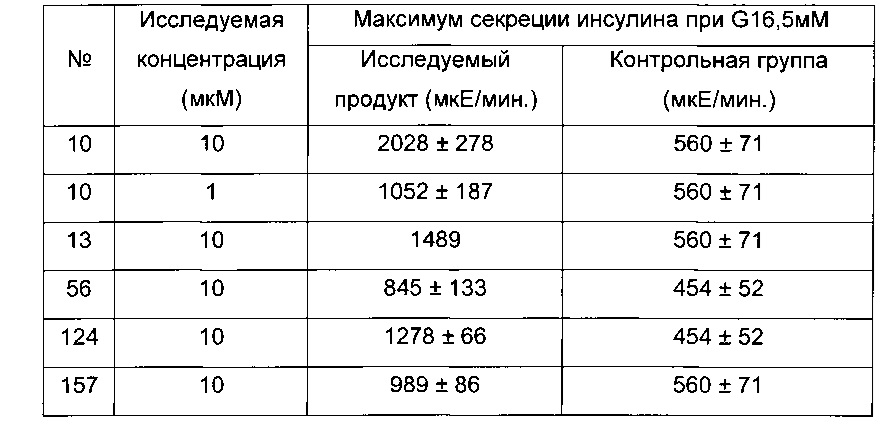
Исследованные производные формулы I оказывают значительное действие на восстановление секреции инсулина в ответ на поступление глюкозы на изолированных перфузируемых поджелудочных железах диабетических крыс N0STZ. Наиболее интенсивная секреция была достигнута при применении соединений 10, 13 и 124.
Исследование противодиабетической активности соединения 10 на крысах GK (крысах линии Гото-Какизаки)
Противодиабетическую активность соединения 10 оценивали на крысах GK-модели сахарного диабета II типа, не сопровождающегося ожирением. Данная модель была получена в результате скрещивания крыс Wistar, выбранных на основании легкого отсутствия толерантности к глюкозе [18]. Данные крысы обладают большинством нарушений функций, которые наблюдаются у человека при сахарном диабете II типа [19]: гипергликемией, отсутствием толерантности к глюкозе, резистентностью к инсулину, а также нарушенной инсулиновой реакцией на глюкозу. Данные животные были выведены в Metabrain и содержались в помещениях для животных с регулируемой температурой (22±2°С) при постоянной влажности (50±20%) с циклом день/ночь 12 ч (световая фаза с 7 ч до 19 ч) и имели неограниченный доступ к пище и воде. Условия содержания и проведения экспериментов соответствовали европейским директивам относительно здоровья и этичного обращения с лабораторными животными (ETS123). Используемые в данном исследовании крысы представляли собой самок крыс GK возрастом 16 недель, которые не получали пищи в течение 2 часов до начала исследования (состояние после всасывания, postabsorptive condition). Тест толерантности к глюкозе проводили при внутривенном введении (ВВТТГ) на 2 группах крыс: одна группа получала перорально соединение 10 в единичной дозе 20 мг/кг, контрольная группа получала перорально носитель. Тест толерантности к глюкозе проводили через 1 ч после перорального введения соединения 10 на животных, которые ранее получили анестезию фенобарбиталом (45 мг/кг, интраперитонеальное введение). Образец крови отбирали в момент времени Т0 непосредственно перед введением нагрузки глюкозы (0,5 г/кг, внутривенное введение), а также в моменты времени × Т5, Т10, Т15, Т20 и Т30 мин. после введения нагрузки глюкозы. Образцы крови центрифугировали, плазму отбирали для определения гликемии. Результаты, представленные выше, выражены
- в виде процента уменьшения гликемии в момент времени Т0 для группы, получавшей соединение 10, по сравнению с контрольной группой.
- в виде процента уменьшения AUC (area under the curve, площадь под кривой гликемии в зависимости от времени) для группы, получавшей соединение 10, по сравнению с контрольной группой.

Полученные результаты свидетельствуют, что соединение 10 при введении в единичной дозе 20 мг/кг способно уменьшить базальную гипергликемию и отсутствие толерантности к глюкозе у животных с диабетом II типа.
Перечень цитируемой литературы
[1] WO 2008051197;
[2] Park et al., Bioorganic & Medicinal Chemistry (2006), 14(2), 395-408;
[3] Shengwu Jishu Tongxun (2007), 18(4), 625-627;
[4] US 20090163545;
[5] Floquet et al., Bioorganic & Medicinal Chemistry Letters (2007), 17(7), 1966-1970;
[6] Khimiko-Farmatsevticheskii Zhurnal (1987), 21(11), 1320-6;
[7] WO 2002095361;
[8] Pang et al., PLoS ONE (2010), 5(4), e10129;
[9] Tang et al., PLoS ONE (2007) 2(8), e761;
[10] US 20120114696;
[11] Merino et al., Bioorganic & Medicinal Chemistry (2006), 14(2), 3583-3591;
[12] Pang et al., PLoS ONE (2009), 4(11), e7730;
[13] Asfari et al., Endocrinology 130: 167-178, 1992;
[14] Portha et al., Diabetes, 23, (1974), 889-895;
[15] Giroix etal., Diabetes, 32, (1983), 445-451;
[16] Assan et al., Nature, 239, (1972), 125-126;
[17] Sussman et al., Diabetes, 15, (1966), 466-472;
[18] Goto et al., Proc. Jpn. Acad. 51, 80-85, 1975;
[19] Portha et al., Mol. Cell. Endocrinol., 297: 73-85, 2009.

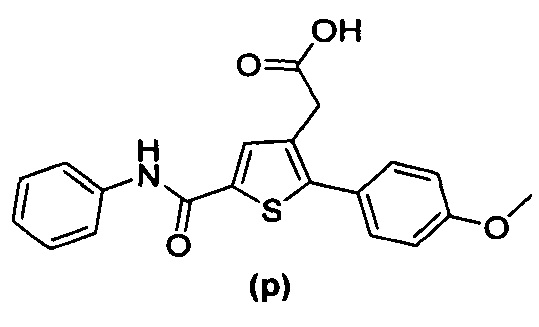
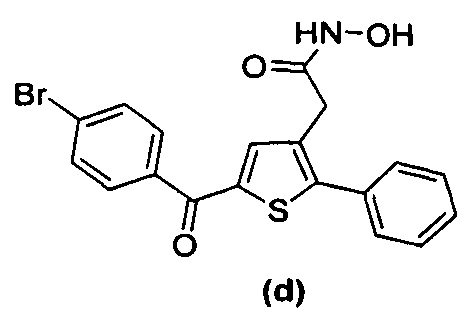
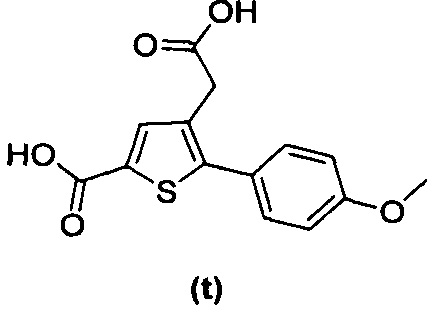
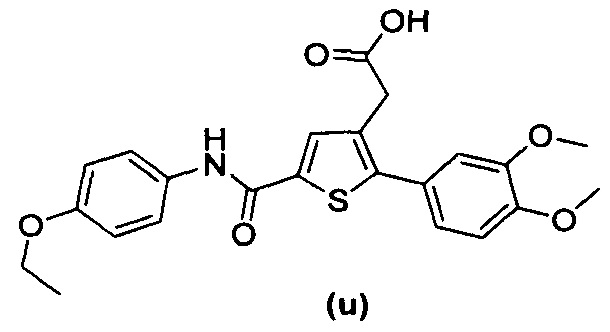
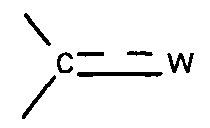
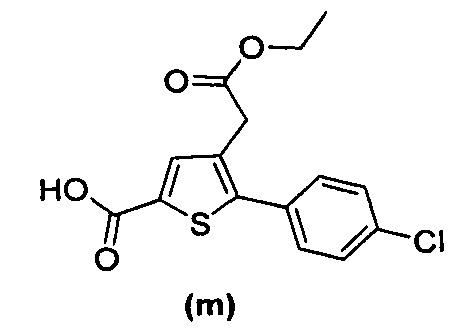
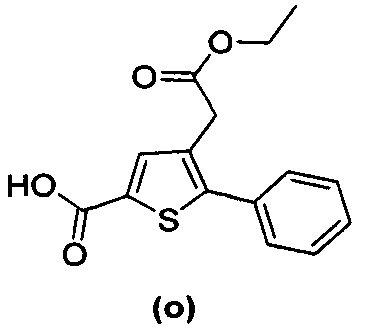
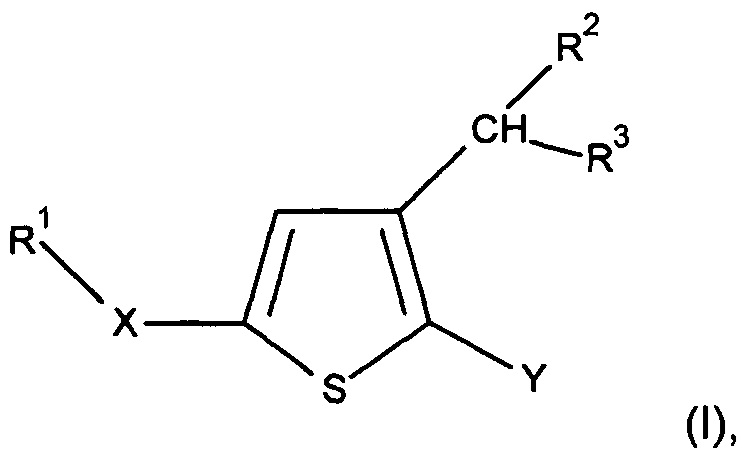
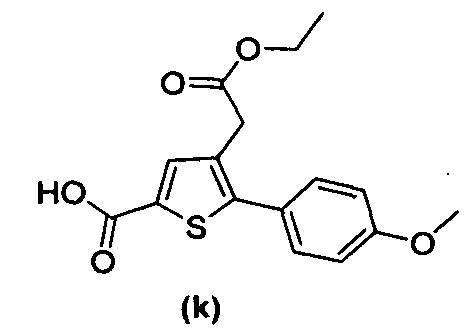
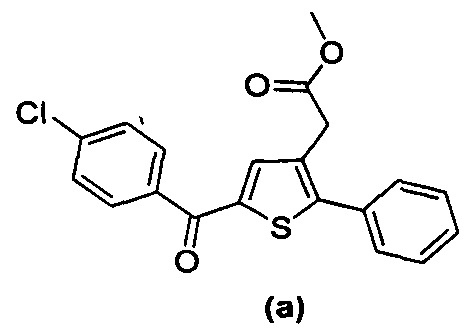
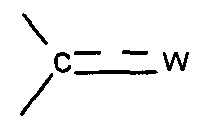
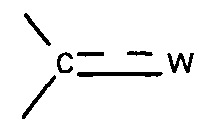
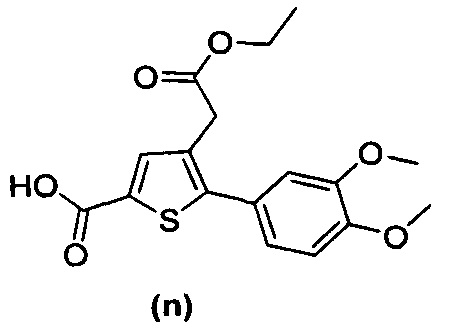

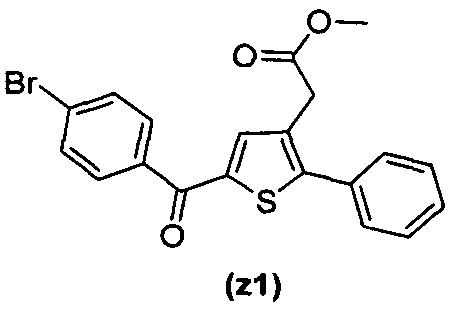
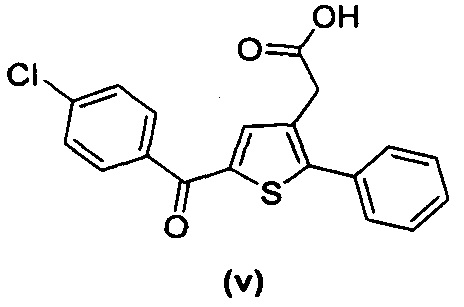

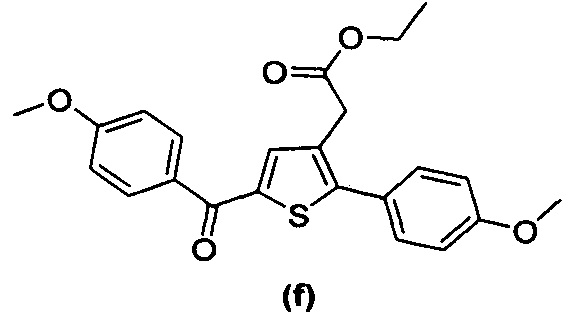
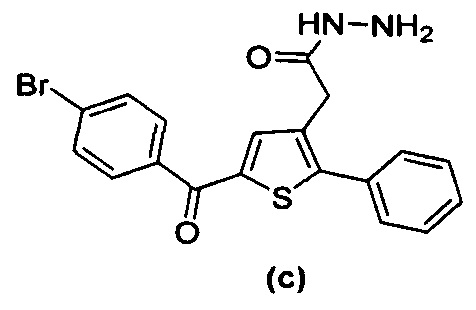
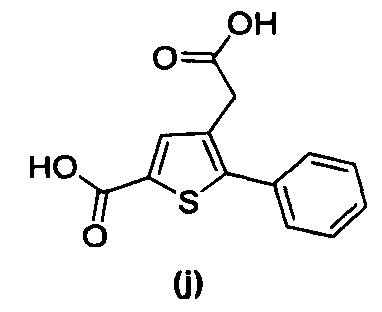
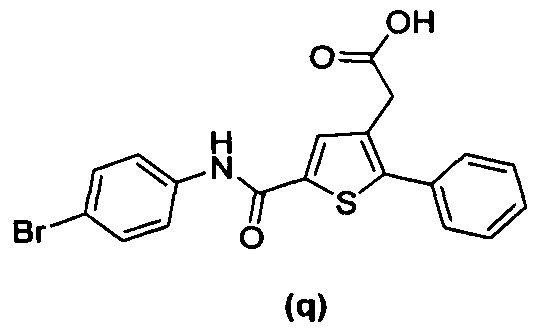

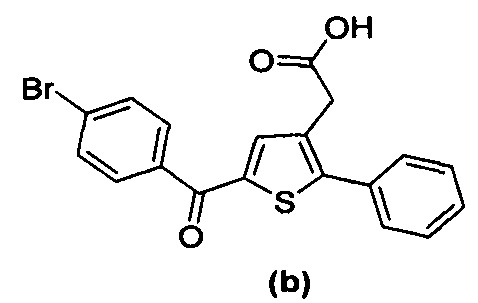
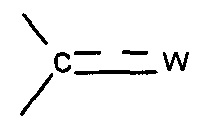
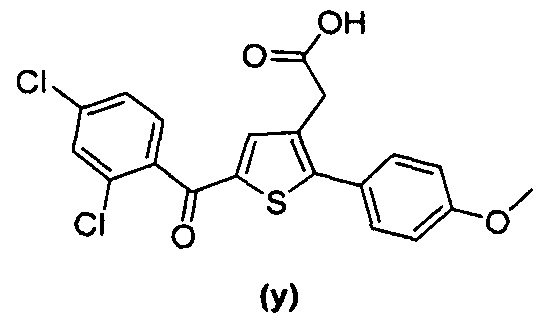

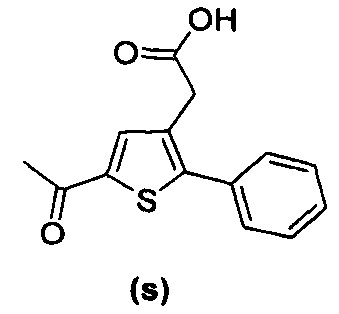
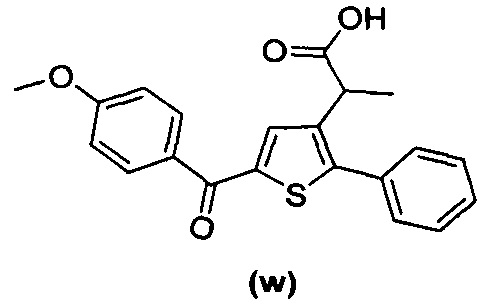
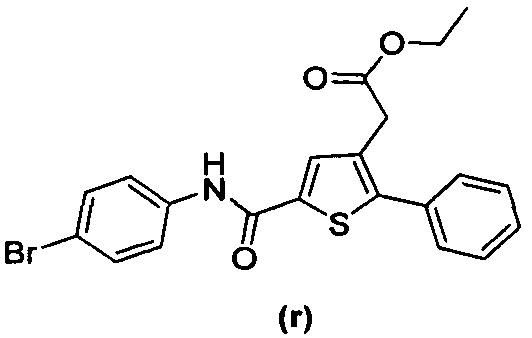
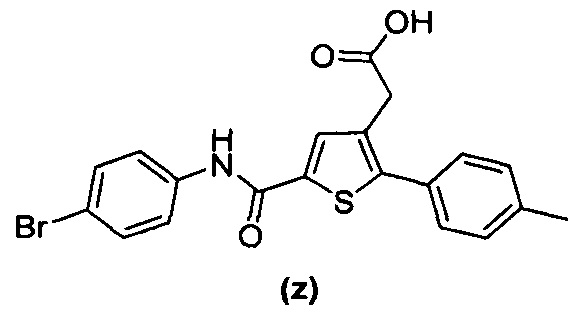
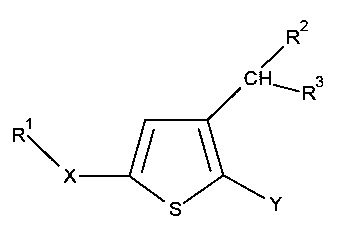
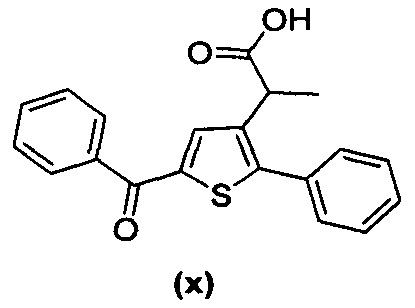
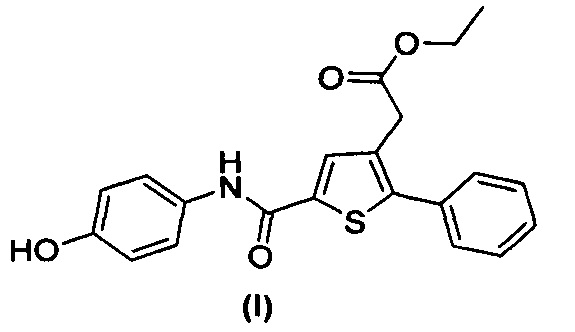
Производное имидазопиридина, используемое для лечения сахарного диабета
Производные, применяемые для лечения мышечной атрофии
Производное имидазопиридина, используемое для лечения сахарного диабета

