Результат интеллектуальной деятельности: СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ
Вид РИД
Изобретение
Область техники
[0001] Настоящее изобретение относится к соли моноциклического производного пиридина и ее кристаллу с ингибирующим действием в отношении FGFR.
Уровень техники
[0002] FGF (фактор роста фибробластов) известен как фактор роста, контролирующий ряд физиологических функций, таких как рост клеток, миграция клеток, клеточная инфильтрация, выживаемость клеток, индукция дифференцировки, заживление ран и ангиогенез.
FGF контролирует различные физиологические функции посредством рецепторов FGF (FGFR: FGFR1, FGFR2, FGFR3 и FGFR4), то есть рецепторных тирозинкиназ. Каждый FGFR включает в себя три типа доменов - внеклеточный домен, трансмембранный домен и внутриклеточный тирозинкиназный домен. При связывании FGF с внеклеточным доменом FGFR образуется димер рецептора. После этого активируется внутриклеточная тирозинкиназа, а затем внутриклеточный сигнал передается главным образом посредством пути MAPK (митоген-активируемой протеинкиназы)/ERK (регулируемой внеклеточным сигналом киназы) или пути PI3K (фосфатидилинозитол-3-киназы)/Akt.
[0003] При этом сообщалось, что различные виды рака, такие как рак молочной железы, рак мочевого пузыря, EMS (8p11 миелопролиферативный синдром), рак желудка, рак эндометрия и рак предстательной железы, обусловлены возникновением нарушения сигнального пути FGF/FGFR, сопровождаемого усилением продуцирования FGF, амплификацией гена FGFR, сверхэкспрессией FGFR, продуцированием гибридного белка FGFR, мутацией FGFR и т. п. (непатентный литературный источник 1). Более того, следующие заболевания были зарегистрированы как виды рака, сопровождаемые нарушением сигнального пути FGF/FGFR: немелкоклеточная карцинома легкого, мелкоклеточная карцинома легкого, рак яичника, саркома, рак толстой кишки, меланома, глиобластома, астроцитома и рак головы и шеи (непатентные литературные источники 2 и 3), рак щитовидной железы (непатентный литературный источник 4), рак поджелудочной железы (непатентные литературные источники 5 и 6), рак печени (непатентный литературный источник 7), рак кожи (непатентный литературный источник 8), рак почки (непатентный литературный источник 9) и плоскоклеточная карцинома легкого и подобные заболевания (непатентные литературные источники 10, 11 и 12).
[0004] Кроме того, сигнальный путь FGF/FGFR является одним из главных сигнальных путей ангиогенеза в эндотелиальных клетках вместе с сигнальным путем VEGF (фактор роста эндотелия сосудов)/KDR (рецептор, имеющий в составе домен, содержащий киназу), и, как сообщалось, вовлекается во взаимодействие раковых стромальных клеток (фибробластов) и раковых клеток (непатентный литературный источник 1).
Следовательно, предполагается, что ингибитор FGFR, нацеливающийся на сигнальный путь FGF/FGFR, функционирует как противоопухолевое лекарственное средство против видов рака, сопровождаемых нарушением сигнального пути FGF/FGFR, на основе его ингибирующего действия в отношении нарушения сигнального пути и его ингибирующего действия в отношении сигнального пути ангиогенеза. Недавно сообщалось про селективный ингибитор FGFR, рассматриваемый как нечувствительный к поражению противостоящим эффектом другого сигнального пути, например, селективный ингибитор FGFR против FGFR1, FGFR2 или FGFR3, который очевидно отличается по структуре от соединения в соответствии с настоящим изобретением. Однако при разработке в качестве противоопухолевого лекарственного средства для людей селективный ингибитор FGFR остается позади противоопухолевого лекарственного средства, одновременно нацеливающегося как на сигнальный путь FGF/FGFR, так и на сигнальный путь VEGF/KDR, и еще не был представлен на рынке (непатентные литературные источники 13 и 14; патентные литературные источники 1 и 2). В патентном литературном источнике 3 раскрыты пиримидиновые производные, но не раскрыто ингибирующее действие в отношении нарушения сигнального пути FGF/FGFR. В патентном литературном источнике 4 раскрыты пиридиновые производные или пиримидиновые производные, которые ингибируют ангиогенез, индуцируемый с помощью VEGF и FGF. Однако ни в одном из этих литературных источников не раскрыты соединения в соответствии с настоящим изобретением.
Список использованной литературы
Патентная литература
[0005]
Патентный литературный источник 1: международная публикация № WO 2008/075068.
Патентный литературный источник 2: международная публикация № WO 2006/000420.
Патентный литературный источник 3: международная публикация № WO 2002/032872.
Патентный литературный источник 4: международная публикация № WO 2004/020434.
Непатентная литература
[0006]
Непатентный литературный источник 1: Nicholas et al., "Fibroblast growth factor signalling: from development to cancer", Nature Reviews Cancer. 2010; 10: 116-129.
Непатентный литературный источник 2: Jorgen WESCHE et al., Fibroblast growth factors and their receptors in cancer, Biochem J. 2011: 437; 199-213.
Непатентный литературный источник 3: Gennaro Daniele et al., FGF Receptor Inhibitors: Role in Cancer Therapy, Curr Oncol Rep. 2012; 14:111-119.
Непатентный литературный источник 4: Rosanne St. Bernard et al., Fibroblast Growth Factor Receptors as Molecular Targets in Thyroid Carcinoma, Endocrinology. 2005; 146: 1145-1153.
Непатентный литературный источник 5: Toshiyuki Ishiwata et al., Enhanced Expression of Fibroblast Growth Factor Receptor 2 IIIc Promotes Human Pancreatic Cancer Cell Proliferation, Am J Pathol. 2012; 180: 1928-1941.
Непатентный литературный источник 6: G Chen et al., Inhibition of endogenous SPARC enhances pancreatic cancer cell growth: modulation by FGFR1-III isoform expression, Br J Cancer. 2010; 102: 188-195.
Непатентный литературный источник 7: Dorothy M. French et al., Targeting FGFR4 Inhibits Hepatocellular Carcinoma in Preclinical Mouse Models, PLoS One. 2012; 7: e36713.
Непатентный литературный источник 8: Armelle Logie et al., Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans, Hum Mol Genet 2005; 14: 1153-1160.
Непатентный литературный источник 9: Tsimafeyeu I et al., Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma, Scand J Urol Nephrol 2011; 45: 190-195.
Непатентный литературный источник 10: Jonathan Weiss et al., Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer, Sci Transl Med. 2010; 2: issue 62 62-93.
Непатентный литературный источник 11: Hidefumi Sasaki et al., Increased FGFR1 copy number in lung squamous cell carcinomas, Mol Med Report. 2012; 5: 725-728.
Непатентный литературный источник 12: The Cancer Genome Atlas Research Network, Comprehensive genomic characterization of squamous cell lung cancers, Nature 2012; 489: 519-525.
Непатентный литературный источник 13: Paul R Gavine et al., AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family, Cancer Res. 2012; 72: 2045-2056.
Непатентный литературный источник 14: Vito Guagnano et al., Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase, J Med Chem. 2011; 54: 7066-7083.
Сущность изобретения
Техническая задача
[0007] Соединение, представленное следующей формулой (I), 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, далее в данном документе называемое соединением (I), обладает ингибирующими действиями в отношении FGFR1, FGFR2 и FGFR3. Как правило, физические свойства соединения, его соли и их кристаллов, применяемых в качестве фармацевтического препарата, сильно влияют на биодоступность лекарственного средства, чистоту активного фармацевтического ингредиента, назначение препарата и т. п. Таким образом, целью настоящего изобретения является обеспечение соли соединения (I) или ее кристалла, которые можно применять в качестве основных материалов для фармацевтических препаратов.
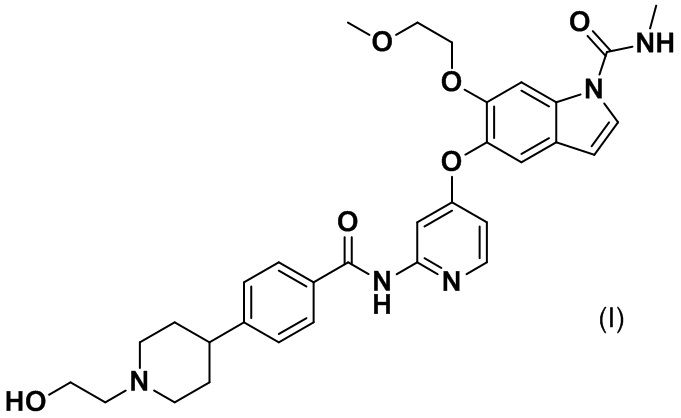
[0008] Авторы настоящего изобретения провели серьезные испытания соединения (I) с учетом вышеуказанных обстоятельств и в результате получили соли соединения (I) или их кристаллы, довершая тем самым настоящее изобретение.
Решение задачи
[0009] В частности, настоящее изобретение предусматривает следующее [1]-[17].
[1] Соль, состоящая из 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I):
и янтарной кислоты или малеиновой кислоты.
[2] Соль согласно вышеприведенному [1], которая представляет собой сукцинатную соль.
[3] Соль согласно вышеприведенному [1], которая представляет собой малеатную соль.
[4] Соль согласно вышеприведенному [2], которая представляет собой 1,5-сукцинатную соль 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида.
[5] Соль согласно вышеприведенному [2], которая представляет собой 0,5-сукцинатную соль 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида.
[6] Соль согласно вышеприведенному [1], которая представляет собой малеатную соль 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида.
[7] Кристалл соли, состоящей из 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I):
и янтарной кислоты или малеиновой кислоты.
[8] Кристалл соли, состоящей из 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I):
и янтарной кислоты.
[9] Кристалл соли, состоящей из 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I):
и малеиновой кислоты.
[10] Кристалл 1,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 22,4° согласно измерению с помощью порошковой рентгеновcкой дифракции.
[11] Кристалл согласно вышеприведенному [10], характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 22,4°, 25,3° и 23,3° согласно измерению с помощью порошковой рентгеновcкой дифракции.
[12] Кристалл (α) 0,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 19,8° согласно измерению с помощью порошковой рентгеновcкой дифракции.
[13] Кристалл мелеатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 20,1° согласно измерению с помощью порошковой рентгеновcкой дифракции.
[14] Кристалл сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, характеризующийся пиками при химических сдвигах (±0,5 ppm) 108,5 ppm, 155,1 ppm и 179,9 ppm в спектре твердотельного 13C-ЯМР.
[15] Кристалл согласно вышеприведенному [14], характеризующийся пиками при химических сдвигах (±0,5 ppm) 27,1 ppm, 34,8 ppm, 108,5 ppm, 155,1 ppm и 179,9 ppm в спектре твердотельного 13C-ЯМР.
[16] Кристалл 1,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, характеризующийся порошковой дифрактограммой, показанной на фигуре 1.
[17] Фармацевтическая композиция, содержащая соль или кристалл согласно вышеприведенным [1] - [16] в качестве активного ингредиента.
Предпочтительные эффекты изобретения
[0010] Соли соединения (I) и их кристаллы, предусмотренные настоящим изобретением, обладают свойствами, показанными в примерах, гигроскопичностью, как показано в нижеописанных испытательных примерах, и потенциалом к применению в качестве лекарственного вещества в фармацевтических препаратах.
Краткое описание графических материалов
[0011] Фигура 1 представляет собой порошковую дифрактограмму кристалла 1,5-сукцинатной соли соединения (I), полученного в примере 1. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 2 представляет собой порошковую дифрактограмму кристалла 0,5-сукцинатной соли соединения (I) (α), полученного в примере 2. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 3 представляет собой порошковую дифрактограмму кристалла малеатной соли соединения (I), полученного в примере 3. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 4 представляет собой спектр твердотельного 13C-ЯМР кристалла 1,5-сукцинатной соли соединения (I), полученного в примере 1.
Фигура 5 представляет собой диаграмму гигроскопичности кристалла 1,5-сукцинатной соли соединения (I), полученного в примере 1. По оси абсцисс показана относительная влажность, а по оси ординат показано изменение веса.
Фигура 6 представляет собой порошковую дифрактограмму кристалла соединения (I) в свободной форме (свободная форма A), полученного в примерах получения 1-15. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 7 представляет собой порошковую дифрактограмму кристалла соединения (I) в свободной форме (свободная форма B), полученного в сравнительном примере 1. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 8 представляет собой порошковую дифрактограмму кристалла соединения (I) в свободной форме (гидрат свободной формы), полученного в сравнительном примере 2. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 9 представляет собой порошковую дифрактограмму кристалла мезилатной соли соединения (I), полученного в сравнительном примере 3. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 10 представляет собой порошковую дифрактограмму кристалла тозилатной соли соединения (I), полученного в сравнительном примере 4. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 11 представляет собой порошковую дифрактограмму кристалла бензоатной соли соединения (I), полученного в сравнительном примере 5. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 12 представляет собой порошковую дифрактограмму кристалла фумаратной соли соединения (I), полученного в сравнительном примере 6. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 13 представляет собой порошковую дифрактограмму кристалла 0,5-сукцинатной соли соединения (I) (α), полученного в примере 4. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 14 представляет собой порошковую дифрактограмму кристалла 0,5-сукцинатной соли соединения (I) (β), полученного в примере 5. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Фигура 15 представляет собой порошковую дифрактограмму кристалла 1,5-сукцинатной соли соединения (I), полученного в примере 6. По оси абсцисс показан угол дифракции (2θ), а по оси ординат показана интенсивность пика.
Описание вариантов осуществления
[0012] Соль соединения (I) по настоящему изобретению, ее кристалл и способы их получения будут описаны более подробно ниже.
[0013] В настоящем описании "соль" относится к химической структурной единице, состоящей из соединения (I) в качестве основного компонента и конкретного числа эквивалентов кислоты по отношению к соединению (I).
[0014] Примеры "соли", как используется в данном документе, включают соли неорганических кислот, соли органических кислот и соли кислых аминокислот, и, в частности, предпочтительными являются фармацевтически приемлемые соли.
[0015] Предпочтительные примеры соли неорганической кислоты включают соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты и т. п, а предпочтительные примеры соли органической кислоты включают соли органических карбоновых кислот, таких как уксусная кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, виннокаменная кислота, яблочная кислота, лимонная кислота, молочная кислота, стеариновая кислота и бензойная кислота, и соли органических сульфоновых кислот, таких как метансульфоновая кислота (мезиловая кислота), этансульфоновая кислота, бензолсульфоновая кислота и п-толуолсульфоновая кислота (тозиловая кислота); в частности, предпочтительными являются янтарная кислота и малеиновая кислота, а особенно предпочтительной - янтарная кислота.
[0016] Предпочтительные примеры соли кислой аминокислоты включают соли аспарагиновой кислоты, глутаминовой кислоты и т. п.
[0017] Соли согласно настоящему изобретению могут быть безводными, гидратами или сольватами. Как используется в данном документе, гидрат или сольват относятся к твердому веществу, образуемому соединением (I) или его солью совместно с молекулами воды или молекулами растворителя, при этом твердое вещество может быть кристаллическим. Примеры растворителей в сольватах включают кетоновые растворители, такие как ацетон, 2-бутанон и циклогексанон; сложноэфирные растворители, такие как метилацетат и этилацетат; эфирные растворители, такие как 1,2-диметоксиэтан и трет-бутилметиловый эфир; спиртовые растворители, такие как метанол, этанол, 1-пропанол и изопропанол; и полярные растворители, такие как N-метил-2-пирролидон, N,N-диметилформамид и диметилсульфоксид. Число молекул воды или молекул растворителя по отношению к соединению (I) или его соли конкретно не ограничено, и их примеры включают одну молекулу или две молекулы.
[0018] Как используется в данном документе, "кристалл" относится к кристаллу соединения (I) или его соли. Следовательно, кристалл 1,5-сукцинатной соли соединения (I), например, означает кристалл соли, образованной между соединением (I) и янтарной кислотой, с 1,5 молекулами янтарной кислоты на 1 молекулу соединения (I).
[0019] Примеры кристаллов, предпочтительных для настоящего изобретения, включают
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 22,4° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 22,4° и 25,3° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 22,4°, 25,3° и 23,3° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 22,4°, 25,3°, 23,3°, 13,2° и 22,0° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 22,4°, 25,3°, 23,3°, 13,2°, 22,0°, 19,3°, 15,7°, 22,7°, 20,6° и 16,0° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (α), характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 19,8° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (α), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 19,8° и 15,7° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (α), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 19,8°, 15,7° и 13,9° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (α), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 19,8°, 15,7°, 13,9°, 21,4° и 25,0° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (α), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 19,8°, 15,7°, 13,9°, 21,4°, 25,0°, 20,6°, 18,2°, 26,8°, 18,8° и 22,4° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (β), характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 16,6° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (β), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 16,6° и 19,7° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (β), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 16,6°, 19,7° и 15,7° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (β), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 16,6°, 19,7°, 15,7°, 9,3° и 14,3° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 0,5-сукцинатной соли соединения (I) (β), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 16,6°, 19,7°, 15,7°, 9,3°, 14,3°, 21,8°, 20,6°, 18,7°, 18,1° и 26,5° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл малеатной соли соединения (I), характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 20,1° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл малеатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 20,1° и 17,0° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл малеатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 20,1°, 17,0° и 16,2° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл малеатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 20,1°, 17,0°, 16,2°, 22,8° и 21,9° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл малеатной соли соединения (I), характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 20,1°, 17,0°, 16,2°, 22,8°, 21,9°, 25,8°, 9,0°, 15,2°, 24,3° и 19,6° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся наличием пиков при химических сдвигах (±0,5 ppm) 108,5 ppm, 155,1 ppm и 179,9 ppm в спектре твердотельного 13C-ЯМР; или
кристалл 1,5-сукцинатной соли соединения (I), характеризующийся наличием пиков при химических сдвигах (±0,5 ppm) 27,1 ppm, 34,8 ppm, 108,5 ppm, 155,1 ppm и 179,9 ppm в спектре твердотельного 13C-ЯМР.
[0020] Пики согласно измерению с помощью порошковой рентгеновcкой дифракции, описанные выше, являются характерными для соответствующих кристаллов и характерными дифракционными пиками для кристаллов 1,5-сукцинатных солей соединения (I), (α) и (β) 0,5-сукцинатных солей соединения (I), малеатных солей соединения (I).
[0021] Как правило, погрешности измерения углов дифракции (2θ) в диапазоне±0,2° могут возникать при измерении с помощью порошковой рентгеновcкой дифракции, и, таким образом, вышеописанные значения углов дифракции необходимо рассматривать как включающие значения в диапазоне примерно±0,2°. Таким образом, в настоящее изобретение включены не только кристаллы определенных солей с пиками при точно таких же углах дифракции согласно измерению с помощью порошковой рентгеновcкой дифракции, но также кристаллы с пиками в пределах диапазона погрешности измерения углов дифракции, составляющего примерно±0,2°.
[0022] Таким образом, как используется в данном документе, "характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 22,4°", например, означает "характеризующийся дифракционным пиком при угле дифракции (2θ) от 22,2° до 22,6°". То же применимо и к другим углам дифракции.
[0023] Как правило, интенсивности пиков и ширина на половине высоты при углах дифракции (2θ) согласно измерению с помощью порошковой рентгеновcкой дифракции различаются при каждом измерении из-за отличий в условиях измерения и разброса размера и формы каждой частицы порошка кристалла и не всегда стабильны, даже если формы кристаллов одинаковые. Таким образом, в случае сравнения порошковой дифрактограммы, когда углы дифракции (2θ) являются одинаковыми, а интенсивности пиков и ширина на половине высоты отличаются, эти отличия не подразумевают, что они получены из-за различий в форме кристалла. Таким образом, кристалл соли, характеризующийся порошковой дифрактограммой, которая имеет вышеуказанные отличия относительно характерных дифракционных пиков определенного кристалла соли согласно настоящему изобретению, означает, что кристалл имеет такую же форму кристалла, что и соль согласно настоящему изобретению. Как используется в данном документе, "характеризующийся порошковой дифрактограммой согласно фиг. 1." означает, что это охватывает не только случай точно такой же дифрактограммы, как показанная на фиг. 1, но также случай с такими же характерными углами дифракции, но отличающимися интенсивностями пиков и шириной на половине высоты. Таким образом, каждый кристалл, характеризующийся такой порошковой дифрактограммой, означает, что кристалл идентичен кристаллу согласно настоящему изобретению.
[0024] Как используется в данном документе, "характеризующийся пиками при химических сдвигах (±0,5 ppm) 27,1 ppm, 34,8 ppm, 108,5 ppm, 155,1 ppm и 179,9 ppm" означает "характеризующийся пиками, каждый из которых фактически эквивалентен пикам при химических сдвигах (±0,05 ppm) 27,1 ppm, 34,8 ppm, 108,5 ppm, 155,1 ppm и 179,9 ppm, при проведении твердотельной 13C-ЯМР спектрометрии при обычных условиях измерения или фактически таких же условиях, как в данном описании".
[0025] При определении того, "характеризуются ли пиками, фактически эквивалентными" или нет, вышеописанные значения химических сдвигов следует рассматривать как включающие значения в диапазоне приблизительно±0,5 ppm, поскольку, как правило, погрешности измерения химических сдвигов (ppm) в диапазоне±0,5 ppm могут возникать в спектре твердотельного 13C-ЯМР. Таким образом, в настоящее изобретение включены не только кристаллы с точно такими же химическими сдвигами в твердотельном 13C-ЯМР спектре, но также кристаллы с химическими сдвигами в пределах диапазона погрешности приблизительно±0,5 ppm. Таким образом, "характеризующийся пиком при химическом сдвиге (±0,5 ppm) 27,1 ppm", как используется в данном документе, например, означает "характеризующийся пиком при химическом сдвиге от 26,6 ppm до 27,6 ppm". То же применимо и к другим химическим сдвигам в спектрах твердотельного 13C ЯМР.
[0026] Способ получения солей соединения (I) или их кристаллов или т. п., который является одним вариантом осуществления согласно настоящему изобретению, будет показан ниже.
[0027]
Получение соединения (I)
Соединение (I) можно синтезировать, как конкретно описано ниже в примере получения 1.
[0028]
Способы получения солей соединения (I)
Соли соединения (I) согласно настоящему изобретению можно получить с помощью традиционных способов получения солей. В частности, их можно получить, например, путем суспендирования или растворения соединения (I) в растворителе, при нагревании в случае необходимости, затем добавления в полученную суспензию или раствор кислоты и перемешивания или отстаивания полученной суспензии или раствора в течение времени от нескольких минут до нескольких дней при комнатной температуре или при охлаждении на ледяной бане. Соли соединения (I) можно получать в виде кристаллов или аморфных веществ согласно способам получения. Аморфное вещество можно получить путем добавления в способы получения операций сублимационной сушки и т. п., в случае необходимости. Примеры растворителей, подлежащих применению в этих способах, включают спиртовые растворители, такие как этанол, 1-пропанол и изопропанол; ацетонитрил; кетоновые растворители, такие как ацетон и 2-бутанон; сложноэфирные растворители, такие как этилацетат; растворители на основе насыщенных углеводородов, такие как гексан и гептан; эфирные растворители, такие как трет-бутилметиловый эфир, или воду. Каждый из этих растворителей можно применять как самостоятельно, так и в смеси из двух или более из них.
[0029]
Способы получения кристаллов соединения (I) или его солей
Кристалл соединения (I) или его соли можно получить с помощью вышеуказанных способов получения соединения (I) или его соли, путем растворения при нагревании соединения (I) или его соли в растворителе и кристаллизации этого посредством охлаждения при перемешивании.
[0030] Соединение (I) или его соль, подлежащие применению при кристаллизации, могут быть в любом виде; это может быть сольват, гидрат, ангидрид, аморфное вещество, кристаллическое вещество (включая вещества, состоящие из множества кристаллических полиморфов) или их комбинация.
[0031] Примеры растворителей, подлежащих применению при кристаллизации, включают спиртовые растворители, такие как метанол, этанол, изопропанол и 1-пропанол; ацетонитрил; амидные растворители, такие как N,N-диметилформамид; сложноэфирные растворители, такие как этилацетат; растворители на основе насыщенных углеводородов, такие как гексан и гептан; кетоновые растворители, такие как ацетон и 2-бутанон; эфирные растворители, такие как трет-бутилметиловый эфир, или воду. Кроме того, каждый из этих растворителей можно применять как самостоятельно, так и в смеси из двух или более из них.
[0032] Количество растворителя, подлежащее применению, можно выбрать подходящим образом при условии, что нижний предел представляет собой количество, при котором соединение (I) или его соль растворяются при нагревании или при котором суспензию можно перемешивать, и что верхний предел представляет собой количество, при котором выход кристалла значительно не уменьшается.
[0033] В ходе кристаллизации можно добавлять затравочный кристалл (например, кристалл требуемого соединения (I) или его соли) или его можно не добавлять. Температура, при которой добавляют затравочный кристалл, конкретно не ограничена, но предпочтительно составляет от 0 до 80°C.
[0034] Хотя температуру, используемую при растворении нагреванием соединения (I) или его соли, то есть при которой растворяются соединение (I) или его соль, можно подходящим образом выбрать в зависимости от растворителя, но она предпочтительно находится в диапазоне от 50°C до температуры, при которой применяемый для перекристаллизации растворитель начинает дефлегмировать, и более предпочтительно от 55 до 80°C.
[0035] Посредством охлаждения в ходе кристаллизации можно получить вещества, содержащие различные формы кристаллов (полиморфизм) в случае быстрого охлаждения. Таким образом, охлаждение необходимо осуществлять при контролировании соответствующим образом скорости охлаждения с учетом ее влияния на качество, размер зерен и другие характеристики кристалла. Например, предпочтительным является осуществление охлаждения при скорости охлаждения от 5 до 40°С/час. Более предпочтительным является осуществление охлаждения при скорости охлаждения, например, от 5 до 25°C/час.
[0036] Кроме того, конечную температуру кристаллизации можно выбрать подходящим образом для получения выхода, качества и подобной характеристики кристалла, но она предпочтительно составляет от -25 до 30°C.
[0037] Целевой кристалл можно получать путем выделения образованного кристалла посредством традиционной процедуры фильтрации, промывания при необходимости отфильтрованного кристалла растворителем и затем его сушки. В качестве растворителя, подлежащего применению для промывки кристалла, можно применять тот же растворитель, что и при кристаллизации. Предпочтительно, он представляет собой, например, этанол, ацетон, 2-бутанон, этилацетат, диэтиловый эфир, трет-бутилметиловый эфир, гексан и т. п. Каждый из этих растворителей можно применять как самостоятельно, так и в смеси из двух или более из них.
[0038] Кристалл, выделенный посредством процедуры фильтрации, можно высушить соответствующим образом, путем выдерживания его в воздухе или в потоке азота или путем нагревания.
[0039] Что касается времени сушки, время, необходимое для снижения количества остаточного растворителя ниже предварительно определенного количества, можно выбрать подходящим образом в зависимости от промышленного масштаба, сушильного устройства, температуры сушки и т. п. Кроме того, сушку можно осуществлять в потоке воздуха или при пониженном давлении. Степень понижения давления можно определить соответствующим образом в зависимости от производственного масштаба, сушильного устройства, температуры сушки и т. п. При необходимости полученный кристалл можно оставить на воздухе после сушки.
[0040] Соли соединения (I) и их кристаллы можно составлять с помощью традиционного способа, и примеры лекарственных форм включают составы для перорального применения (например, таблетки, гранулы, порошки, капсулы и сиропы), инъекционные растворы (для внутривенного введения, внутримышечного введения, подкожного введения и внутрибрюшинного введения) и препараты для наружного применения (например, составы, впитывающиеся через кожу (например, мази и пластыри), глазные препараты, назальные препараты и суппозитории).
[0041] Для получения твердых составов для перорального применения к солям соединения (I) и их кристаллам при необходимости можно добавлять среду, связывающее вещество, разрыхлитель, смазывающее средство, краситель и т. п, при этом согласно традиционному способу можно получить таблетку, гранулу, порошкообразное средство или капсулу. Кроме того, такие таблетку, гранулу, порошкообразное средство, капсулу или подобное при необходимости можно подвергать нанесению покрытия.
Примеры среды включают лактозу, кристаллическую целлюлозу и т. п., примеры связывающего вещества включают гидроксипропилцеллюлозу и т. п., примеры разрыхлителя включают кальция/натрия кроскармеллозу и т. п., примеры смазывающего средства включают стеарат магния и т. п., примеры красителя включают оксид титана и т. п., и примеры средства для нанесения покрытия включают гидроксипропилметилцеллюлозу и т. п., но эти компоненты не ограничиваются вышеупомянутыми примерами.
Твердый состав, такой как таблетка, капсула, гранула или порошок, обычно может содержать любое количество солей соединения (I) и их кристаллов, при условии, что он проявляет эффективность настолько, чтобы считать его применимым в качестве лекарственного препарата.
[0042] Для получения инъекционного раствора (для внутривенного введения, для внутримышечного введения, для подкожного введения, для внутрибрюшинного введения или для других путей введения) к солям соединения (I) и их кристаллам при необходимости добавляют регулятор pH, буферное средство, суспендирующее средство, солюбилизирующее средство, антиоксидант, консервант (антисептик), изотоническое средство и т. п., при этом с помощью традиционного способа можно получить инъекционный препарат. Препараты можно подвергать лиофилизации с получением лиофилизированных препаратов по типу растворяемых непосредственно перед приемом.
В качестве регулятора pH и буферного средства можно применять, например, органическую кислоту или неорганическую кислоту и/или их соль или т. п. В качестве суспендирующего средства можно применять, например, гидроксипропилцеллюлозу или т. п. В качестве солюбилизирующего средства можно применять, например, полисорбат 80 или т. п. В качестве антиоксиданта можно применять, например, α-токоферол или т. п. В качестве консерванта можно применять, например, метилпарагидроксибензоат, этилпарагидроксибензоат или т. п. В качестве изотонического средства можно применять, например, глюкозу или т. п.
Состав для инъекций обычно может содержать любое количество солей соединения (I) и их кристаллов, при условии, что он проявляет эффективность настолько, чтобы считать его применимым в качестве лекарственного препарата.
[0043] Для получения состава для наружного применения основной исходный материал добавляют к солям соединения (I) и их кристаллам и, при необходимости, добавляют, например, консервант, стабилизатор, регулятор pH, антиоксидант, краситель и т. п., как описанные выше, при этом с помощью традиционных способов можно получить, например, препарат для чрескожного применения (мазь, пластырь и т. п.), глазные капли, капли в нос, суппозиторий и т. п.
В качестве подлежащего применению основного исходного материала можно применять различные исходные материалы, обычно применяемые для получения, например, медицинских препаратов, лечебно-профилактической косметики и косметических средств. Конкретные их примеры включают такие исходные материалы, как масла животного и растительного происхождения, минеральные масла, сложноэфирные масла, воски, эмульгаторы, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и очищенная вода.
Препарат для наружного применения обычно может содержать любое количество солей соединения (I) и их кристаллов, при условии, что он проявляет эффективность настолько, чтобы считать его применимым в качестве лекарственного препарата.
[0044] Доза солей соединения (I) и их кристаллов зависит от степени тяжести симптома, возраста, пола и веса пациента, пути введения и типа соли, конкретного вида заболевания и т. п., и особенно не ограничивается, если не превышает максимальную дозу лекарственного препарата, которая может быть принята без неприемлемой побочной реакции, при этом взрослому пациенту их вводят, один раз в сутки или при разделении несколько раз в сутки, в дозе для перорального введения, как правило, составляющей примерно 30 мкг - 10 г, конкретнее 100 мкг - 5 г и более конкретно 100 мкг - 1 г, или в дозе для инъекционного введения, как правило, составляющей примерно 30 мкг - 1 г, конкретнее 100 мкг - 500 мг и более конкретно 100 мкг - 300 мг.
Пример
[0045] Соединения в соответствии с настоящим изобретением можно получить, например, с помощью способов, описанных в примерах получения и примерах, описанных ниже. Однако эти способы являются только лишь примерами, и поэтому соединения в соответствии с настоящим изобретением так или иначе не ограничиваются теми, что получены конкретными примерами, описанными ниже.
[0046] При порошковой рентгеновской дифрактометрии кристаллов, полученных в следующих примерах и сравнительных примерах, полученные кристаллы помещали на предметный столик порошкового рентгеновского дифрактометра и анализировали при следующих условиях. Результаты показаны на фиг. 1-3 и 6-15.
[0047]
Условия измерений
Держатель образца: алюминиевый
Анод: медный
Детектор: сцинтилляционный счетчик
Напряжение в трубке: 50 кВ
Сила тока в трубке: 300 мА
Щель: DS (отклоняющая щель) 0,5 мм (ограничивающая высоту щель 2 мм), SS (рассеивающая щель) открыта, RS (приемная щель) открыта
Скорость сканирования: 10°/мин
Интервал отбора проб: 0,02°
Диапазон сканирования: 5-35°
[0048] Твердотельные 13C-ЯМР спектры кристаллов измеряли при следующих условиях. Результаты показаны на фиг. 4.
Условия измерений
Применяемый прибор: AVANCE400 (производства Bruker Corporation)
Температура при измерениях: комнатная температура (22°C)
Эталонный материал: глицин (внешний стандарт: 176,03 ppm)
Измеряемое ядро: 13C (100,6248425 МГц)
Частота повторения импульсов: 3 секунды
Импульсный режим: измерение TOSS
[0049] В примерах получения Silica gel 60 (Kanto Chemicals) или Presep Silica Gel (WAKO) применяли в качестве силикагеля для очистки, применяемого для колоночной хроматографии на силикагеле, если не указано иное. Кроме того, NH-силикагель (Fuji Silysia Chemical LTD.) или Hi-Flash Column Amino (YAMAZENE CORPORATION) применяли в качестве силикагеля для очистки, применяемого для колоночной хроматографии на NH-силикагеле.
[0050] Спектрометр Varian Mercury 400, Varian Mercury Plus 400, Varian INOVA 500 или Avance 600 МГц (Bruker) применяли для измерения спектров протонного ядерного магнитного резонанса, при этом спектры протонного ядерного магнитного резонанса измеряли при 400 MГц, если не указано иное. Химические сдвиги спектров протонного ядерного магнитного резонанса регистрируются в единицах измерения σ (ppm) относительно тетраметилсилана, а константы взаимодействия регистрируются в единицах измерения Герц (Гц). Сокращения паттернов расщепления являются следующими: s: синглет, d: дуплет, t: триплет; m: мультиплет и brs: широкий синглет.
[0051] В примерах получения, примерах и сравнительных примерах соответственным образом применяли коммерчески доступные продукты в качестве коммерчески доступных соединений.
[0052] [Пример получения 1-1]. N-(4-хлорпиридин-2-ил)ацетамид
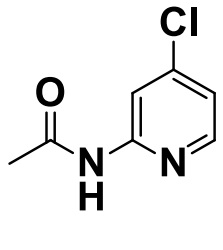
Коммерчески доступный 2-амино-4-хлорпиридин (50 г, 389 ммоль) растворяли в уксусном ангидриде (500 мл), добавляли триэтиламин (271 мл, 1,94 моль) при 20°C и смесь перемешивали при 60°C в течение 12 часов. Смесь охлаждали до комнатной температуры, а затем растворитель выпаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат=от 4: 1 до 1: 1), а затем целевую фракцию концентрировали в вакууме с получением титульного соединения (66 г, 99%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 2,21 (3H, s), 7,05 (1H, dd, J=5,4, 1,9 Гц), 8,15 (1H, d, J=5,4 Гц), 8,30 (2H, brs).
[0053] [Пример получения 1-2]. Фенилметилкарбамат
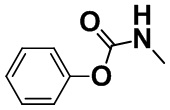
Смесь коммерчески доступного гидрохлорида метиламина (50 г, 0,74 моль), пиридина (124 мл, 1,53 моль) и N,N-диметилформамида (500 мл) перемешивали при 5°C и каплями добавляли коммерчески доступный фенилхлоркарбонат (94 мл, 0,75 моля) на протяжении 2 часов. После завершения покапельного добавления смесь перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. Реакционную смесь добавляли в ледяную воду (2 л) и дважды экстрагировали этилацетатом (1,5 л). Органический слой промывали водой (1 л) и насыщенным солевым раствором (300 мл). Органический слой высушивали над безводным сульфатом магния и затем растворитель выпаривали. К сконцентрированному остатку добавляли н-гептан и этилацетат, осадок собирали фильтрацией и промывали н-гептаном и трет-бутилметиловым эфиром с получением титульного соединения (74,2 г, 66%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 2,90 (3H, d, J=4,9 Гц), 4,95 (1H, brs), 7,08-7,16 (2H, m), 7,16-7,24 (1H, m), 7,31-7,41 (2H, m).
[0054] [Пример получения 1-3]. 1-(4-Фенилпиперидин-1-ил)этанон
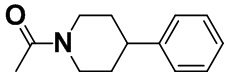
Смесь коммерчески доступного 4-фенилпиперидина (10 г, 62 ммоль), пиридина (5,7 мл, 70,5 ммоль) и тетрагидрофурана (80 мл) перемешивали при 0°C и капали смесь ацетилхлорида (5 мл, 70,3 ммоль) и тетрагидрофурана (20 мл) на протяжении 10 минут. Смесь перемешивали в атмосфере азота при 25°C в течение 14 часов. Этилацетат (100 мл) и воду (100 мл) добавляли в реакционную жидкость для отделения. Водный слой экстрагировали этилацетатом (100 мл), затем объединяли органические слои и полученное промывали насыщенным водным раствором натрия бикарбоната (100 мл), водой (100 мл), а затем насыщенным солевым раствором (50 мл). Органический слой высушивали над безводным сульфатом магния, а затем растворитель выпаривали с получением титульного соединения (12,3 г, 98%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 1,52-1,78 (2H, m), 1,81-1,99 (2H, m), 2,14 (3H, s), 2,63 (1H, td, J=12,9, 2,7 Гц), 2,74 (1H, tt, J=12,1, 3,7 Гц), 3,17 (1H, td, J=13,2, 2,6 Гц), 3,84-4,02 (1H, m), 4,69-4,89 (1H, m), 7,08-7,43 (5H, m).
[0055] [Пример получения 1-4]. 4-(1-Ацетилпиперидин-4-ил)бензойная кислота
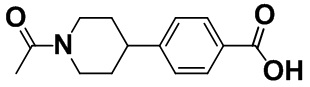
Смесь алюминия хлорида(III) (26 г, 195 ммоль) и дихлорметана (200 мл) перемешивали при 0°C и капали оксалилхлорид (20 мл, 228 ммоль) на протяжении 10 минут. Затем капали смесь 1-(4-фенилпиперидин-1-ил)этанона (12,3 г, 60,5 ммоль), описанного в примере получения 1-3, и дихлорметана (50 мл) на протяжении 30 минут. Смесь перемешивали в атмосфере азота при 25°C в течение 14 часов. Реакционную жидкость выливали на лед и добавляли этилацетат (1 л) и воду (1 л) для отделения. Водный слой дважды экстрагировали этилацетатом (1 л), затем органический слой дважды промывали водой (1 л), а затем насыщенным солевым раствором (500 мл). Органический слой высушивали над безводным сульфатом магния, а затем растворитель выпаривали. К сконцентрированному остатку добавляли этилацетат, продукт собирали фильтрацией и промывали этилацетатом с получением титульного соединения (9,09 г, 61%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 1,49-1,82 (2H, m), 1,92 (2H, t, J=13,2 Гц), 2,15 (3H, s), 2,65 (1H, t, J=11,7 Гц), 2,75-2,94 (1H, m), 3,08-3,30 (1H, m), 3,97 (1H, d, J=13,2 Гц), 4,82 (1H, d, J=12,8 Гц), 7,30 (2H, d, J=8,4 Гц), 8,05 (2H, d, J=8,1 Гц).
[0056] [Пример получения 1-5]. Гидрохлорид 4-(пиперидин-4-ил)бензойной кислоты
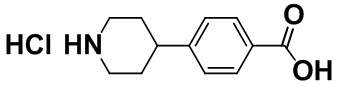
Смесь 4-(1-ацетилпиперидин-4-ил)бензойной кислоты (4,50 г, 18,2 ммоль), описанной в примере получения 1-4, и 5 M хлористоводородной кислоты (50 мл, 250 ммоль) перемешивали в атмосфере азота при 140°C в течение 18 часов. Смесь охлаждали до комнатной температуры, а затем продукт собирали фильтрацией и промывали водой с получением титульного соединения (3,77 г, 86%).
Спектр 1H-ЯМР (DMSO-d6) δ (ppm): 1,60-2,15 (4H, m), 2,76-3,16 (3H, m), 3,27-3,45 (2H, m), 7,36 (2H, d, J=8,1 Гц), 7,92 (2H, d, J=8,1 Гц), 8,65-9,04 (2H, m), 12,89 (1H, brs).
[0057] [Пример получения 1-6]. 4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойная кислота
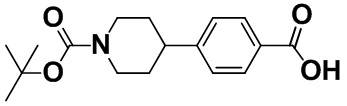
Смесь гидрохлорида 4-(пиперидин-4-ил)бензойной кислоты (2,00 г, 8,27 ммоль), описанного в примере получения 1-5, 1 M раствора натрия гидроксида (25 мл, 25 ммоль) и ацетона (50 мл) перемешивали при 25°C и по каплям добавляли раствор ди-трет-бутилдикарбоната (1,9 г, 8,71 ммоль) в ацетоне (25 мл) на протяжении 10 минут. Смесь перемешивали в атмосфере азота при 25°C в течение 18 часов. Добавляли 1 M хлористоводородную кислоту (17 мл) при охлаждении при 0°C. Смесь дважды экстрагировали этилацетатом (100 мл). Органический слой промывали насыщенным солевым раствором (50 мл). Органический слой высушивали над безводным сульфатом магния, а затем концентрировали в вакууме. К сконцентрированному остатку добавляли н-гептан и трет-бутилметиловый эфир, продукт собирали фильтрацией и промывали н-гептаном с получением титульного соединения (2,30 г, 91%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 1,49 (9H, s), 1,57-1,76 (2H, m), 1,84 (2H, d, J=13,5 Гц), 2,62-2,97 (3H, m), 4,27 (2H, brs), 7,28-7,36 (2H, m), 7,98-8,10 (2H, m).
[0058] [Пример получения 1-7]. 3-Гидрокси-4-(2-метоксиэтокси)бензальдегид
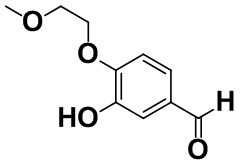
Коммерчески доступный 3,4-дигидроксибензальдегид (39,3 г, 285 ммоль) и карбонат натрия (45,2 г, 427 ммоль) растворяли в N,N-диметилформамиде (400 мл), затем добавляли коммерчески доступный 2-бромэтилметиловый эфир (26,7 мл, 285 ммоль) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 дней. Смесь охлаждали до 0°C, а затем добавляли 2 M хлористоводородную кислоту, этилацетат и воду для разделения. Водный слой экстрагировали этилацетатом, затем объединенный органический слой промывали насыщенным солевым раствором и высушивали над безводным сульфатом магния, а затем фильтровали. Растворитель выпаривали, добавляли дихлорметан, осадок отделяли фильтрацией, а затем полученный фильтрат очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат=от 17:3 до 1:1). Целевую фракцию концентрировали в вакууме с получением титульного соединения (12,9 г, 23%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 3,47 (3H, s), 3,76-3,80 (2H, m), 4,25-4,29 (2H, m), 6,40 (1H, brs), 7,01 (1H, d, J=8,4 Гц), 7,41 (1H, dd, J=8,2, 2,0 Гц), 7,45 (1H, d, J=1,8 Гц), 9,85 (1H, s).
[0059] [Пример получения 1-8]. 3-(Бензилокси)-4-(2-метоксиэтокси)бензальдегид
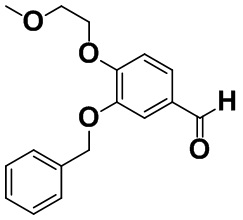
Карбонат калия (11,8 г, 85,7 ммоль) и бензилхлорид (10 мл, 86,9 ммоль) добавляли в жидкую смесь 3-гидрокси-4-(2-метоксиэтокси)бензальдегида (12,9 г, 65,9 ммоль), описанного в примере получения 1-7, в этаноле (130 мл) в атмосфере азота при комнатной температуре и смесь нагревали с обратным холодильником при 90°C в течение 2 часов. Смесь охлаждали до 0°C, а затем добавляли 2 M хлористоводородную кислоту, этилацетат и воду для разделения. Органический слой промывали насыщенным солевым раствором, высушивали над безводным сульфатом магния, а затем фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат=от 9:1 до 1:1). Целевую фракцию концентрировали в вакууме с получением титульного соединения (17,6 г, 93%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 3,46 (3H, s), 3,79-3,85 (2H, m), 4,24-4,30 (2H, m), 5,18 (2H, s), 7,03 (1H, d, J=8,1 Гц), 7,29-7,35 (1H, m), 7,35-7,41 (2H, m), 7,43-7,50 (4H, m), 9,82 (1H, s).
[0060] [Пример получения 1-9]. (E)-2-(бензилокси)-1-(2-метоксиэтокси)-4-(2-нитровинил)бензол
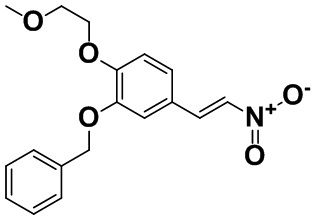
3-(Бензилокси)-4-(2-метоксиэтокси)бензальдегид (17,6 г, 61,5 ммоль), описанный в примере получения 1-8, растворяли в уксусной кислоте (49,3 мл), затем добавляли ацетат аммония (5,69 г, 73,8 ммоль) и нитрометан (8,32 мл, 154 ммоль) в атмосфере азота при комнатной температуре и смесь нагревали с обратным холодильником при 130°C в течение 2 часов. Смесь охлаждали до комнатной температуры, а затем осадок собирали фильтрацией и промывали этанолом с количественным получением титульного соединения.
Спектр 1H-ЯМР (CDCl3) δ (ppm): 3,46 (3H, s), 3,78-3,84 (2H, m), 4,21-4,27 (2H, m), 5,16 (2H, s), 6,97 (1H, d, J=8,4 Гц), 7,06 (1H, d, J=1,8 Гц), 7,16 (1H, dd, J=8,4, 2,2 Гц), 7,30-7,48 (6H, m), 7,91 (1H, d, J=13,5 Гц).
[0061] [Пример получения 1-10]. 6-(2-Метоксиэтокси)-1H-индол-5-ол
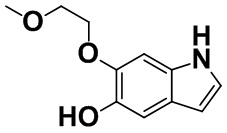
В смесь (E)-2-(бензилокси)-1-(2-метоксиэтокси)-4-(2-нитровинил)бензола (20,2 г, 61,5 ммоль), описанного в примере получения 1-9, и уксусной кислоты (120 мл) добавляли 69% азотную кислоту (15 мл, 233 ммоль) при 25°C и смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь выливали на лед, осадок собирали фильтрацией, а затем промывали водой с получением неочищенного продукта (23,0 г).
Неочищенный продукт (23,0 г) суспендировали в метаноле (500 мл), затем добавляли 10% палладий на угле (содержание воды 50%) (8 г) при комнатной температуре и смесь перемешивали в атмосфере водорода в течение 6 часов. Катализатор отфильтровывали через целит, фильтрат концентрировали в вакууме и полученное очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат=от 2:1 до 1:1). Целевую фракцию концентрировали в вакууме с получением титульного соединения (3,94 г, 31%).
Спектр 1H-ЯМР (CDCl3) δ (ppm): 3,48 (3H, s), 3,69-3,78 (2H, m), 4,16-4,23 (2H, m), 6,24 (1H, s), 6,41 (1H, ddd, J=3,1, 2,1, 0,8 Гц), 6,97 (1H, s), 7,10 (1H, dd, J=3,2, 2,5 Гц), 7,15 (1H, s), 7,94 (1H, brs).
[0062] [Пример получения 1-11]. N-(4-((6-(2-метоксиэтокси)-1H-индол-5-ил)окси)пиридин-2-ил)ацетамид
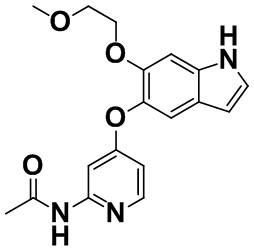
6-(2-Метоксиэтокси)-1H-индол-5-ол (3,94 г, 19,0 ммоль), описанный в примере получения 1-10, и N-(4-хлорпиридин-2-ил)ацетамид (3,25 г, 19,0 ммоль), описанный в примере получения 1-1, растворяли в диметилсульфоксиде (25 мл), затем 97% трет-бутоксид калия (2,20 г, 19,0 ммоль) добавляли при комнатной температуре и смесь нагревали и перемешивали при 150°C в течение 13 часов. Воду и этилацетат добавляли в реакционную жидкость при комнатной температуре для разделения. Водный слой три раза экстрагировали этилацетатом и объединенный органический слой промывали водой. Органический слой высушивали над безводным сульфатом натрия. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и затем полученное очищали с помощью колоночной хроматографии на NH-силикагеле (н-гептан:этилацетат=от 2:3 до 0:1 - этилацетат:метанол=от 49:1 до 9:1). Целевую фракцию концентрировали в вакууме с получением титульного соединения (3,45 г, 53%).
Спектр 1H-ЯМР (500 МГц, CDCl3) δ (ppm): 2,13 (3H, s), 3,27 (3H, s), 3,54-3,58 (2H, m), 4,07-4,11 (2H, m), 6,46-6,50 (1H, m), 6,54 (1H, dd, J=5,8, 1,9 Гц), 7,05 (1H, s), 7,14-7,17 (1H, m), 7,36 (1H, s), 7,75 (1H, brs), 8,02 (1H, d, J=5,8 Гц), 8,10 (1H, brs), 8,19 (1H, brs).
[0063] [Пример получения 1-12]. 4-((6-(2-Метоксиэтокси)-1H-индол-5-ил)окси)пиридин-2-амин
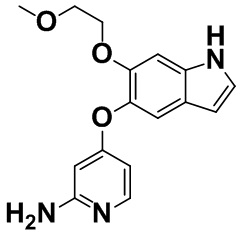
N-(4-((6-(2-метоксиэтокси)-1H-индол-5-ил)окси)пиридин-2-ил)ацетамид (3,45 г, 10,1 ммоль), описанный в примере получения 1-11, растворяли в метаноле (50 мл), добавляли 2 M раствор натрия гидроксида (50 мл) при комнатной температуре и смесь нагревали и перемешивали при 70°C в течение 3 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой три раза экстрагировали этилацетатом и объединенный органический слой высушивали над безводным сульфатом натрия. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и затем полученное очищали с помощью колоночной хроматографии на NH-силикагеле (н-гептан:этилацетат=от 3:7 до 0:1 - этилацетат:метанол=от 49:1 до 24:1). Целевую фракцию и фракцию смеси концентрировали в вакууме отдельно друг от друга, фракцию смеси снова очищали с помощью колоночной хроматографии на силикагеле (этилацетат:метанол=от 1:0 до 9:1), а затем полученное объединяли с вышеупомянутой целевой фракцией с получением титульного соединения (2,60 г, 86%).
Спектр 1H-ЯМР (500 МГц, CDCl3) δ (ppm): 3,31 (3H, s), 3,58-3,63 (2H, m), 4,08-4,11 (2H, m), 4,28 (2H, brs), 5,90 (1H, d, J=2,4 Гц), 6,29 (1H, dd, J=6,1, 2,2 Гц), 6,44-6,52 (1H, m), 7,06 (1H, s), 7,15-7,20 (1H, m), 7,34 (1H, s), 7,88 (1H, d, J=5,8 Гц), 8,22 (1H, brs).
[0064] [Пример получения 1-13]. 5-[(2-Аминопиридин-4-ил)окси]-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид
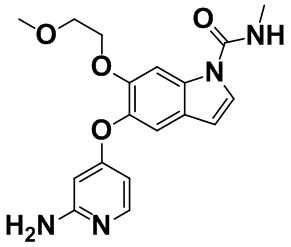
4-((6-(2-Метоксиэтокси)-1H-индол-5-ил)окси)пиридин-2-амин (2,60 г, 8,67 ммоль), описанный в примере получения 1-12, растворяли в N,N-диметилформамиде (50 мл), затем добавляли 50-72% маслянистый гидрид натрия (499 мг) в атмосфере азота при комнатной температуре. Добавляли фенилметилкарбамат (1,97 г, 13,0 ммоль), описанный в примере получения 1-2, и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь охлаждали до 0°C и добавляли этилацетат и воду для разделения. Водный слой дважды экстрагировали этилацетатом, к водному слою добавляли хлорид натрия и полученное три раза экстрагировали этилацетатом. Объединенный органический слой высушивали над безводным сульфатом натрия. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и затем полученное очищали с помощью колоночной хроматографии на NH-силикагеле (н-гептан:этилацетат=от 1:4 до 0:1 - этилацетат:метанол=от 49:1 до 24:1). Целевую фракцию концентрировали в вакууме, добавляли этилацетат, осадок собирали фильтрацией и промывали с получением титульного соединения (2,23 г, 72%).
Спектр 1H-ЯМР (500 МГц, CDCl3) δ (ppm): 3,06 (3H, d, J=4,9 Гц), 3,29 (3H, s), 3,59-3,63 (2H, m), 4,14-4,17 (2H, m), 4,30 (2H, brs), 5,52-5,59 (1H, m), 5,89 (1H, d, J=2,4 Гц), 6,27 (1H, dd, J=5,8, 1,9 Гц), 6,55 (1H, d, J=3,9 Гц), 7,27-7,29 (2H, m), 7,89 (1H, d, J=5,9 Гц), 7,99 (1H, s).
[0065] [Пример получения 1-14]. 6-(2-Метоксиэтокси)-N-метил-5-{[2-({[4-(пиперидин-4-ил)фенил]карбонил}амино)пиридин-4-ил]окси}-1H-индол-1-карбоксамид
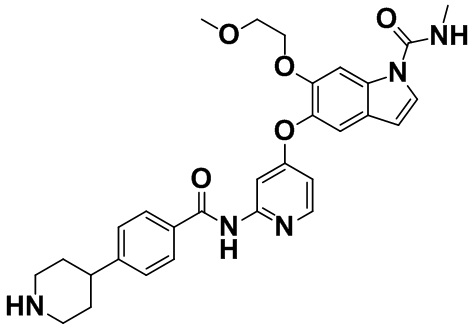
Бензотриазол (609 мг, 5,11 ммоль) растворяли в дихлорметане (25 мл), добавляли тионилхлорид (373 мкл, 5,11 ммоль) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. В реакционную смесь при комнатной температуре добавляли 4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (1,3 г, 4,26 ммоль), описанную в примере получения 1-6, и смесь перемешивали в течение 30 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным сульфатом натрия, и полученное промывали дихлорметаном, фильтрат добавляли в смесь 5-[(2-аминопиридин-4-ил)окси]-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (0,95 г, 2,67 ммоль), описанного в примере получения 1-13, триэтиламина (1,86 мл, 13,3 ммоль) и 4-диметиламинопиридина (16 мг, 0,133 ммоль) в N,N-диметилформамиде (3 мл) и дихлорметане (20 мл) при 0°C на протяжении 5 минут и смесь промывали дихлорметаном (10 мл), а затем перемешивали при той же температуре в течение 5 минут. Смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли 40% водный раствор метиламина (2,3 мл, 26,7 ммоль), а затем смесь перемешивали при комнатной температуре в течение 1,5 часов. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь для разделения и водный слой три раза экстрагировали этилацетатом. Объединенный органический слой высушивали над безводным сульфатом натрия. Осушающее средство отфильтровывали, затем фильтрат концентрировали в вакууме и полученное очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат=от 1:1 до 0:1 - этилацетат:метанол=от 49:1 до 23:2) с получением неочищенного продукта (1,11 г).
Неочищенный продукт (1,11 г) растворяли в дихлорметане (50 мл) и добавляли при комнатной температуре трифторуксусную кислоту (5,0 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, затем полученное концентрировали в вакууме, а затем остаток растворяли в дихлорметане и триэтиламине и полученное концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат:метанол=от 1:0 до 22:3) с получением титульного соединения (829 мг, 57%).
Спектр 1H-ЯМР (500 МГц, CDCl3) δ (ppm): 1,59-1,69 (2H, m), 1,83 (2H, d, J=14,1 Гц), 2,68 (1H, tt, J=12,0, 3,6 Гц), 2,75 (2H, td, J=12,2, 2,4 Гц), 3,04 (3H, d, J=4,9 Гц), 3,17-3,23 (2H, m), 3,26 (3H, s), 3,55-3,61 (2H, m), 4,15-4,21 (2H, m), 5,57-5,65 (1H, m), 6,53 (1H, d, J=3,4 Гц), 6,62 (1H, dd, J=5,8, 2,4 Гц), 7,25 (1H, d, J=3,9 Гц), 7,30-7,34 (3H, m), 7,77-7,82 (2H, m), 7,91 (1H, d, J=2,4 Гц), 8,02 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,50 (1H, brs).
[0066] [Пример получения 1-15]. 5-({2-[({4-[1-(2-Гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид
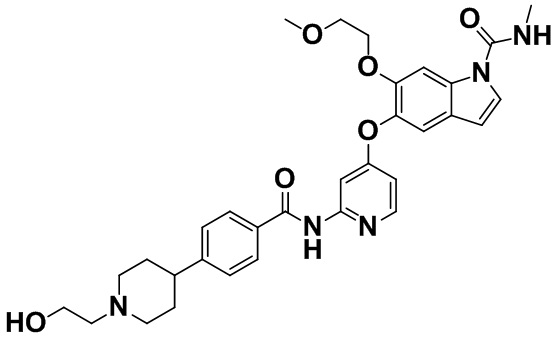
Триацетоксиборгидрид натрия (114 мг, 0,54 ммоль) и коммерчески доступный 2-гидроксиацетальдегид (34,4 мг, 0,57 ммоль) добавляли в смесь 6-(2-метоксиэтокси)-N-метил-5-{[2-({[4-(пиперидин-4-ил)фенил]карбонил}амино)пиридин-4-ил]окси}-1H-индол-1-карбоксамида (100 мг, 0,18 ммоль), описанного в примере получения 1-14, и тетрагидрофурана (4 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор бикарбоната натрия и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой промывали насыщенным солевым раствором, затем высушивали над безводным сульфатом натрия, а затем фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат:метанол=1:0-97:3-9:1). Целевую фракцию концентрировали в вакууме, затем осадок собирали фильтрацией и промывали жидкой смесью диэтилового эфира и н-гексана с получением титульного соединения (90 мг, 83%). Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения (соединения (I) в свободной форме (свободная форма A)) показаны ниже. (2θ±0,2°): 10,4°, 10,9°, 11,4°, 13,5°, 16,1°, 19,7°, 20,4°, 21,5°, 23,3° и 24,3°.
Спектр 1H-ЯМР (CDCl3) δ (ppm): 1,70-1,92 (4H, m), 2,15-2,24 (2H, m), 2,53-2,65 (3H, m), 3,01-3,09 (5H, m), 3,26 (3H, s), 3,56-3,60 (2H, m), 3,64 (2H, t, J=5,2 Гц), 4,15-4,20 (2H, m), 5,49-5,54 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,8, 2,3 Гц), 7,24-7,28 (1H, m), 7,30-7,35 (3H, m), 7,81 (2H, d, J=8,2 Гц), 7,91 (1H, d, J=2,4 Гц), 8,01 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,50 (1H, brs).
[0067] [Пример 1]
Получение кристалла 1,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (другое название: 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид=бутандиоат (2:3))
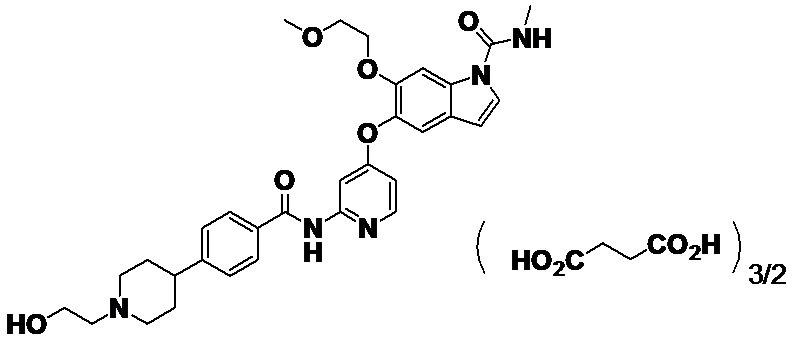
[0068] В колбе для извлечения взвешивали 2,93 г 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, добавляли 60 мл этанола и смесь нагревали и перемешивали при 70°C на масляной бане до растворения. Добавляли янтарную кислоту (1,23 г), затем выключали масляную баню и постепенно охлаждали. Смесь перемешивали при комнатной температуре в течение 2 часов и дополнительно перемешивали при 5°C в течение 1 часа. Твердое вещество собирали фильтрацией с получением титульного соединения (3,70 г).
Спектр 1H-ЯМР (600 МГц, CD3OD) δ (ppm): 1,96-2,10 (4H, m), 2,52 (6H, s), 2,93 (1H, m), 2,96 (3H, s), 3,01 (2H, m), 3,16 (2H, t, J=5,4 Гц), 3,22 (3H, s), 3,56 (2H, t, J=4,7 Гц), 3,61 (2H, m), 3,87 (2H, t, J=5,4 Гц), 4,14 (2H, t, J=4,6 Гц), 6,61 (1H, d, J=3,6 Гц), 6,68 (1H, dd, J=5,8, 2,3 Гц), 7,37 (1H, s), 7,42 (2H, d, J=8,3 Гц), 7,58 (1H, d, J=3,6 Гц), 7,73 (1H, d, J=2,2 Гц), 7,88 (2H, d, J=8,3 Гц), 8,08 (1H, s), 8,15 (1H, d, J=5,8 Гц).
13C-ЯМР(100 МГц, в твердом состоянии) δ (ppm): 27,1, 28,3, 29,7, 34,8, 38,0, 41,3, 54,0, 57,3, 59,7, 60,9, 72,1, 72,5, 103,3, 104,2, 108,5, 116,9, 126,9, 128,6, 134,5, 136,7, 140,7, 149,4, 151,3, 155,1, 169,5, 170,1, 175,6, 179,9, 183,7.
[0069] [Пример 2]
Получение кристалла 0,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (α)
[0070] В 30-мл колбу для извлечения добавляли 117 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, и янтарную кислоту (11,8 мг), добавляли 2 мл раствора изопропанола/воды (8/2, об./об.), осуществляли облучение ультразвуком и смесь перемешивали при комнатной температуре в течение 2-3 часов. Твердое вещество собирали фильтрацией с получением титульного соединения (77,5 мг).
Спектр 1H-ЯМР (600 МГц, CD3OD) δ (ppm): 1,86-2,00 (4H, m), 2,51 (2H, s), 2,62 (2H, m), 2,79 (1H, m), 2,87 (2H, t, J=5,5 Гц), 2,96 (3H, s), 3,22 (3H, s), 3,36 (2H, d, J=11,8 Гц), 3,56 (2H, t, J=4,6 Гц), 3,79 (2H, t, J=5,7 Гц), 4,15 (2H, t, J=4,6 Гц), 6,61 (1H, d, J=3,6 Гц), 6,68 (1H, dd, J=5,7, 2,1 Гц), 7,37 (1H, s), 7,40 (2H, d, J=8,2 Гц), 7,58 (1H, d, J=3,6 Гц), 7,73 (1H, d, J=2,0 Гц), 7,86 (2H, d, J=8,3 Гц), 8,08 (1H, s), 8,14 (1H, d, J=5,8 Гц).
[0071] [Пример 3]
Получение кристалла малеатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида
[0072] Малеиновую кислоту (24,1 мг) и 2 мл ацетона добавляли к 101 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, и смесь перемешивали при комнатной температуре в течение ночи. Твердое вещество собирали фильтрацией с получением титульного соединения (113 мг).
[0073] [Пример 4]
Получение кристалла 0,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (α)
[0074] В пробирку добавляли 550 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, янтарную кислоту (55,3 мг) и воду (5,5 мл) и осуществляли облучение ультразвуком, а затем смесь перемешивали при комнатной температуре в течение ночи. Твердое вещество отфильтровывали в течение ночи. Твердое вещество измельчали в агатовой ступке и хранили в условиях 40°C/75% RH в течение примерно 1,5 часов, а затем получали титульное соединение (620 мг).
Спектр 1H-ЯМР (CD3OD) δ (ppm): 1,86-2,00 (4H, m), 2,51 (2H, s), 2,62 (2H, m), 2,79 (1H, m), 2,87 (2H, brt, J=5,5 Гц), 2,96 (3H, s), 3,22 (3H, s), 3,36 (2H, brd, J=11,8 Гц), 3,56 (2H, brt, J=4,6 Гц), 3,79 (2H, t, J=5,7 Гц), 4,15 (2H, brt, J=4,6 Гц), 6,61 (1H, d, J=3,6 Гц), 6,68 (1H, dd, J=5,7, 2,1 Гц), 7,37 (1H, s), 7,40 (2H, d, J=8,2 Гц), 7,58 (1H, d, J=3,6 Гц), 7,73 (1H, d, J=2,0 Гц), 7,86 (2H, d, J=8,3 Гц), 8,08 (1H, s), 8,14 (1H, d, J=5,8 Гц).
[0075] [Пример 5]
Получение кристалла 0,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (β)
[0076] Образец, полученный в примере 4, высушивали при пониженном давлении в течение 3 дней с получением титульного соединения.
Спектр 1H-ЯМР (CD3OD) δ (ppm): 1,87-2,00 (4H, m), 2,51 (2H, s), 2,65 (2H, m), 2,79 (1H, m), 2,89 (2H, brt, J=5,6 Гц), 2,95 (3H, s), 3,22 (3H, s), 3,38 (2H, brd, J=12,1 Гц), 3,56 (2H, m), 3,80 (2H, t, J=5,6 Гц), 4,14 (2H, m), 6,60 (1H, d, J=3,7 Гц), 6,67 (1H, dd, J=5,8, 2,3 Гц), 7,37 (1H, s), 7,40 (2H, d, J=8,4 Гц), 7,57 (1H, d, J=3,7 Гц), 7,73 (1H, d, J=2,3 Гц), 7,86 (2H, d, J=8,4 Гц), 8,08 (1H, s), 8,14 (1H, d, J=5,8 Гц).
[0077] [Пример 6]
Получение аморфной 1,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида
[0078] 251 мг 1,5-сукцинатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида растворяли в 25 мл 50% водного раствора трет-бутилового спирта. В пробирку добавляли 3 мл раствора образца и раствор образца замораживали в этаноле, охлажденном на сухом льду. Растворитель удаляли в лиофилизаторе с получением титульного соединения (30,8 мг).
Спектр 1H-ЯМР (CD3OD) δ (ppm): 1,96-2,11 (4H, m), 2,53 (6H, s), 2,93 (1H, m), 2,96 (3H, s), 3,00 (2H, m), 3,15 (2H, t, J=5,4 Гц), 3,22 (3H, s), 3,56 (2H, m), 3,60 (2H, brd, J=12,4 Гц), 3,87 (2H, t, J=5,5 Гц), 4,15 (2H, brt, J=4,6 Гц), 6,61 (1H, d, J=3,7 Гц), 6,68 (1H, brd, J=3,8 Гц), 7,37 (1H, s), 7,42 (2H, d, J=8,3 Гц), 7,58 (1H, d, J=3,8 Гц), 7,73 (1H, brs), 7,88 (2H, d, J=8,3 Гц), 8,08 (1H, s), 8,15 (1H, brd, J=5,0 Гц).
[0079] [Сравнительный пример 1]
Получение кристалла 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (свободная форма B)
[0080] В пробирке взвешивали 93,2 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, затем добавляли 2,99 мл изопропанола и 264 мкл воды. Смесь нагревали при 70-100°C на масляной бане до растворения. Полученный раствор перемешивали при -5°C на бане с терморегулированием в течение 16 часов. Осажденное твердое вещество собирали фильтрацией и высушивали при пониженном давлении в течение ночи с получением титульного соединения. Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения показаны ниже. (2θ±0,2°): 7,8°, 10,8°, 13,1°, 14,2°, 17,8°, 21,5°, 21,7°, 23,4°, 24,5° и 29,0°.
[0081] [Сравнительный пример 2]
Получение кристалла 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (гидрат свободной формы)
[0082] 2 мл изопропанола и 2 мл воды добавляли к 208 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15. После осуществления облучения ультразвуком в ледяной воде смесь перемешивали при 5°C в течение 3 дней. Суспендированное твердое вещество собирали фильтрацией с получением титульного соединения (106 мг). Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения показаны ниже. (2θ±0,2°): 8,8°, 9,6°, 15,2°, 16,3°, 20,0°, 20,8°, 21,4°, 22,0°, 23,8° и 27,1°.
[0083] [Сравнительный пример 3]
Получение кристалла мезилатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида
[0084] 2 мл ацетона добавляли к 30,1 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, затем добавляли метансульфоновую кислоту (4,0 мкл) и перемешивали при комнатной температуре в течение 4 дней. Твердое вещество собирали фильтрацией с получением титульного соединения (20,4 мг). Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения показаны ниже. (2θ±0,2°): 11,7°, 13,7°, 15,2°, 16,9°, 18,0°, 18,7°, 19,9°, 21,1°, 22,0° и 24,1°.
[0085] [Сравнительный пример 4]
Получение кристалла тозилатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида
[0086] 2 мл ацетона добавляли к 30,7 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, затем добавляли моногидрат п-толуолсульфоновой кислоты (12,3 мг) и перемешивали при комнатной температуре в течение 4 дней. Твердое вещество собирали фильтрацией с получением титульного соединения (15,1 мг). Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения показаны ниже. (2θ±0,2°): 11,9°, 12,6°, 13,5°, 13,8°, 17,6°, 18,0°, 18,6°, 20,4°, 21,4° и 23,3°.
[0087] [Сравнительный пример 5]
Получение кристалла бензоатной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида
[0088] 0,2 мл этилацетата добавляли в смесь 20,3 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, и бензойной кислоты (8,91 мг) и перемешивали при комнатной температуре. Через 2 часа добавляли 0,1 мл этилацетата и реакционную смесь дополнительно перемешивали в течение ночи. Твердое вещество собирали фильтрацией с получением титульного соединения. Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения показаны ниже. (2θ±0,2°): 9,3°, 13,9°, 14,5°, 15,8°, 18,1°, 19,4°, 20,5°, 21,3°, 22,6° и 26,2°.
[0089] [Сравнительный пример 6]
Получение кристалла фумаратной соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида
[0090] 2 мл ацетона добавляли в смесь 30,6 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, и фумаровой кислоты (7,24 мг) и перемешивали при комнатной температуре в течение ночи. Твердое вещество собирали фильтрацией с получением титульного соединения. Типичные углы дифракции согласно измерению с помощью порошковой рентгеновской дифракции для полученного соединения показаны ниже. (2θ±0,2°): 9,6°, 13,8°, 15,7°, 16,7°, 19,8°, 21,0°, 22,0°, 22,4°, 24,7° и 25,7°.
[0091] [Сравнительный пример 7]
Получение кристалла соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида и хлористоводородной кислоты
[0092] 2 мл ацетона и 6 н. хлористоводородную кислоту (10,0 мкл) добавляли к 29,5 мл 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, и реакционную смесь перемешивали при комнатной температуре. Титульное соединение отделяли от растворителя в масляной форме.
[0093] [Сравнительный пример 8]
Получение кристалла соли 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида и бромистоводородной кислоты
[0094] 2 мл ацетона и бромистоводородную кислоту (7,8 мкл) добавляли к 32,7 мг 5-({2-[({4-[1-(2-гидроксиэтил)пиперидин-4-ил]фенил}карбонил)амино]пиридин-4-ил}окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, описанного в примере получения 1-15, и реакционную смесь перемешивали при комнатной температуре. Титульное соединение отделяли от растворителя в масляной форме.
[0095] [Испытательный пример]
Проводили следующие испытательные примеры и оценивали физические свойства или фармакологические эффекты соединения (I), описанного в примере получения 1-15, или солей соединения (I) и их кристаллов.
[0096] [Испытательный пример 1]. Гигроскопичность
Устройство, анализирующее динамическую сорбцию паров, применяли для оценки гигроскопичности 1,5-сукцинатной соли соединения (I) из примера 1. Температуру на удерживающей образец детали устройства поддерживали при 25°C, а относительную влажность (RH) увеличивали постепенно в диапазоне от 5% до 95%. Относительную влажность регулировали путем корректировки относительных расходов сухого 0% RH и влажного 100% RH азота. Вес образца измеряли каждые 2 минуты при помощи микровесов. Влажность постепенно изменялась, когда величина изменения массы за 5 минут составляла менее 0,01%. Результаты показаны на фиг. 5.
[0097] [Испытательный пример 2]. Внеклеточная ингибирующая активность в отношении киназы
В белый 96-луночный планшет с плоским дном (Sumitomo Bakelite Co., Ltd., MS-8496W) добавляли 10 мкл раствора белка FGFR1 (Carna Biosciences, Inc., 08-133), разведенного до 1 мкг/мл с помощью буфера для анализа (20 мM HEPES-NaOH, 0,01% Triton X-100, 2 мM DTT и 5 мM MgCl2), 10 мкл раствора буфера для анализа, содержащего субстрат CSK-tide (Ana Spec Inc., 63843) с конечной концентрацией 1000 нМ и ATP (Promega Corporation, V9102) с конечной концентрацией 58,3 мкМ, и 5 мкл тестируемого вещества, разведенного с помощью буфера для анализа, и обеспечивали протекание реакции при комнатной температуре в течение 1 часа. Для измерения киназной активности применяли набор для анализа киназной активности ADP-Glo (TM) (Promega Corporation, V9102). После окончания реакции в каждую лунку планшета добавляли 25 мкл реактива ADP-Glo и обеспечивали протекание реакции при комнатной температуре в течение 40 минут с целью остановки киназной реакции и удаления оставшейся ATP. Далее добавляли реактив для обнаружения киназы и обеспечивали протекание реакции при комнатной температуре в течение 40 минут для осуществления превращения ADP в ATP, реакции связывания люциферазы/люциферина и люминесцентной реакции с помощью ATP. Для оценки ферментативной активности интенсивность люминесценции в каждой лунке измеряли с помощью Envision (TM) (PerkinElmer Co., Ltd.). Значения люминесценции в лунках, содержащих белок киназу без добавления тестируемого вещества, определяли как 100%, а значения люминесценции в лунках без добавления как тестового вещества, так и белка киназы определяли как 0%. Затем рассчитывали отношение значений люминесценции в присутствии тестируемого вещества. На основании этого отношения значений люминесценции рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования киназной активности на 50% (т. e. значение IC50).
[0098] Измеряли соответственно внеклеточную ингибирующую активность в отношении киназы FGFR2, внеклеточную ингибирующую активность в отношении киназы FGFR3 и внеклеточную ингибирующую активность в отношении киназы FGFR4 путем применения белка FGFR2 (Carna Biosciences, Inc., 08-134), белка FGFR3 (Carna Biosciences, Inc., 08-135) или белка FGFR4 (Carna Biosciences, Inc., 08-136) таким же образом, как в вышеуказанном случае определения внеклеточной ингибирующей активности в отношении киназы FGFR1. Однако, относительно концентрации ATP внеклеточную ингибирующую активность в отношении киназы оценивали с конечной концентрацией 35 мкM для FGFR2, с конечной концентрацией 16,7 мкM для FGFR3 и с конечной концентрацией 75 мкM для FGFR4. Для FGFR3 и FGFR4 реакцию с тестируемым веществом проводили при комнатной температуре в течение 2 часов. Их результаты показаны в таблице 1.
[0099] <Данные об внеклеточной ингибирующей активности в отношении киназы>
[Таблица 1]
|
[0100] [Испытательный пример 3]. Анализ ингибирования роста SNU-16
Сообщалось, что клеточная линия рака желудка человека SNU-16 (номер в ATCC CRL-5974) характеризуется усиленной экспрессией гена FGFR2 (Cancer Res. 2008. 68: 2340-2348). Клетки SNU-16 поддерживали в среде RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS и пенициллин/стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191), в инкубаторе с 5% CO2 (37°C). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток SNU-16, доведенной до концентрации 1 × 104 клеток/мл с применением среды RPMI-1640, содержащей 10% FBS, и клетки инкубировали в течение ночи в инкубаторе с 5% CO2 (37°C). На следующий день добавляли 50 мкл тестируемого вещества, разведенного с применением среды RPMI-1640, содержащей 10% FBS, и полученное инкубировали в течение 3 дней в инкубаторе с 5% CO2 (37°C). Затем в каждую лунку добавляли 10 мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное инкубировали в течение 1-2 часов в инкубаторе с 5% CO2 (37°C) для осуществления цветной реакции. Значение коэффициента поглощения измеряли при помощи ENVISION (TM) (PerkinElmer Co., Ltd.) при 450 нм. Значение коэффициента поглощения в лунках без добавления тестируемого вещества определяли как 100%, а значение коэффициента поглощения в лунках, не содержащих клетки, определяли как 0%. Затем рассчитывали отношение коэффициентов поглощения в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования роста клеток на 50% (т. е. значение IC50), и она показана в таблице 2.
[0101]
<Данные оценки активности в отношении ингибирования роста SNU-16>
[Таблица 2]
|
[0102] [Испытательный пример 4]. Противоопухолевый эффект на мышиной модели подкожной ксенотрансплантации SNU-16
Концентрацию клеток клеточной линии рака желудка человека SNU-16, которые культивировали в среде RPMI-1640, содержащей 10% FBS и пенициллин/стрептомицин, доводили до 1 × 108 клеток/мл с применением сбалансированного солевого раствора Хенкса (GIBCO № 24020) с получением суспензии клеток, и суспензию смешивали с MATRIGEL (BD Biosciences, № по кат. 354234) в соотношении 1:1 с получением суспензии клеток с концентрацией 5 × 107 клеток/мл. Суспензию клеток в объеме 100 мкл инокулировали безтимусным мышам возрастом 6-7 недель (BALB/cAJcl-nu/nu, самки, Clea Japan Inc.) подкожно в правый бок. Через семь дней после инокуляции у каждой мыши измеряли наименьший диаметр и наибольший диаметр опухоли с применением электронного цифрового штангенциркуля (штангенциркуль Digimatic TM, Mitutoyo Corporation), для того чтобы рассчитать объем опухоли согласно следующей расчетной формуле:
Объем опухоли (мм3)=наибольший диаметр (мм)
× наименьший диаметр (мм) × наименьший диаметр (мм)/2
На основании значений объемов опухолей, полученных в первый день введения, безтимусных мышей распределяли по группам, так что средние значения объемов опухолей были практически равными среди групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 с получением раствора с 10-кратной концентрацией и приготовленный таким образом раствор хранили в холодильнике перед применением. Непосредственно перед введением исходный раствор разбавляли с помощью 5% раствора глюкозы с получением готового раствора для введения (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). Каждый оцениваемый образец вводили перорально группе введения тестируемого вещества в объеме 20 мл/кг один раз в день на протяжении 11 дней, и контрольной группе при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и группы введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляло 0,9 или больше, соответствующую группу введения тестируемого вещества определяли как хорошо переносящую. У группы введения тестируемого вещества, определенной таким образом как хорошо переносящая, рассчитывали соотношение (T/C) (%) объема опухоли, наблюдаемого у группы дозирования тестируемого вещества, к объему опухоли, наблюдаемому у контрольной группы, полученные в последний день, и это показано в таблице 3.
[0103]
<Данные оценки противоопухолевого эффекта на мышиной модели подкожной ксенотрансплантации SNU-16>
[Таблица 3]
|

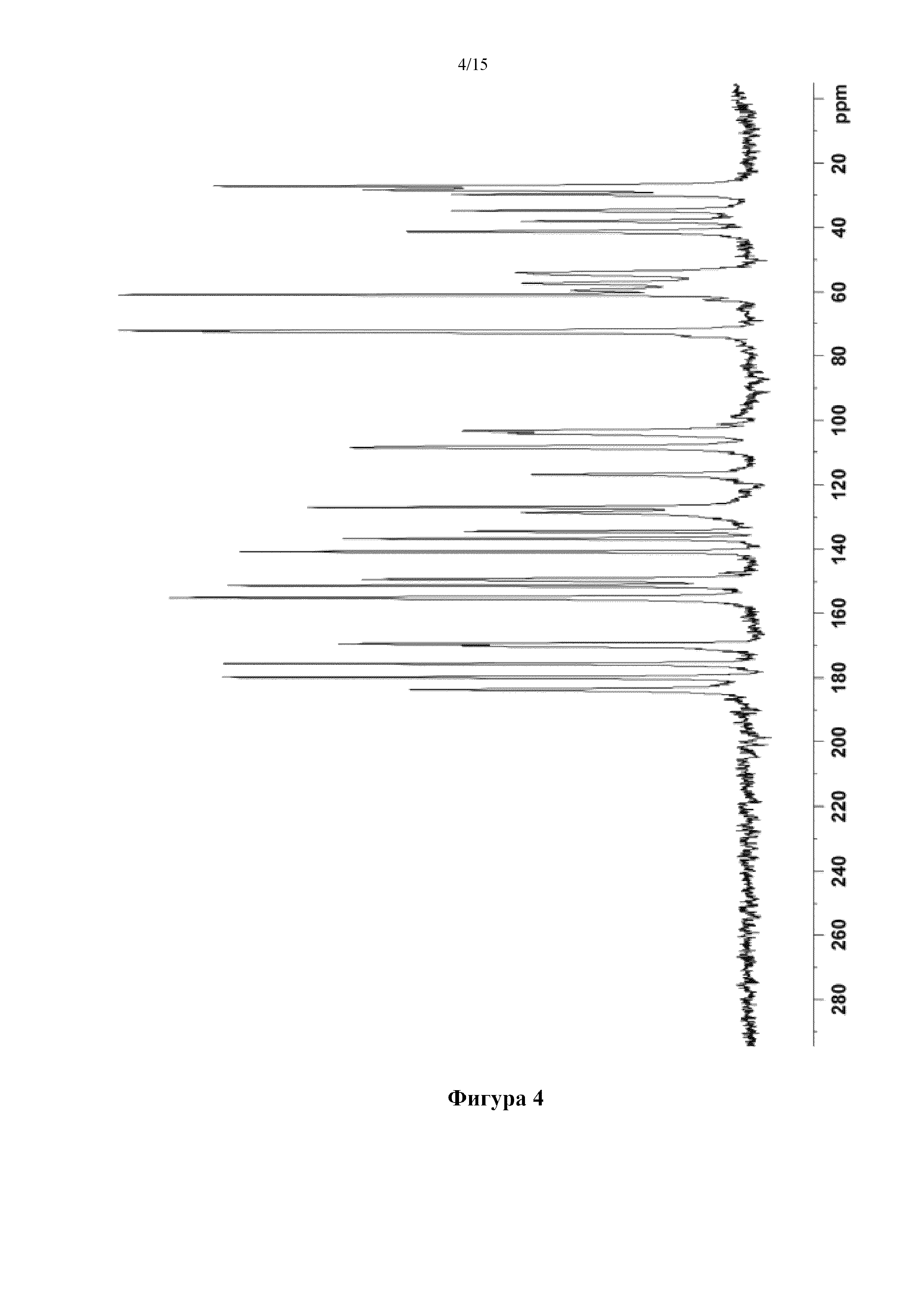
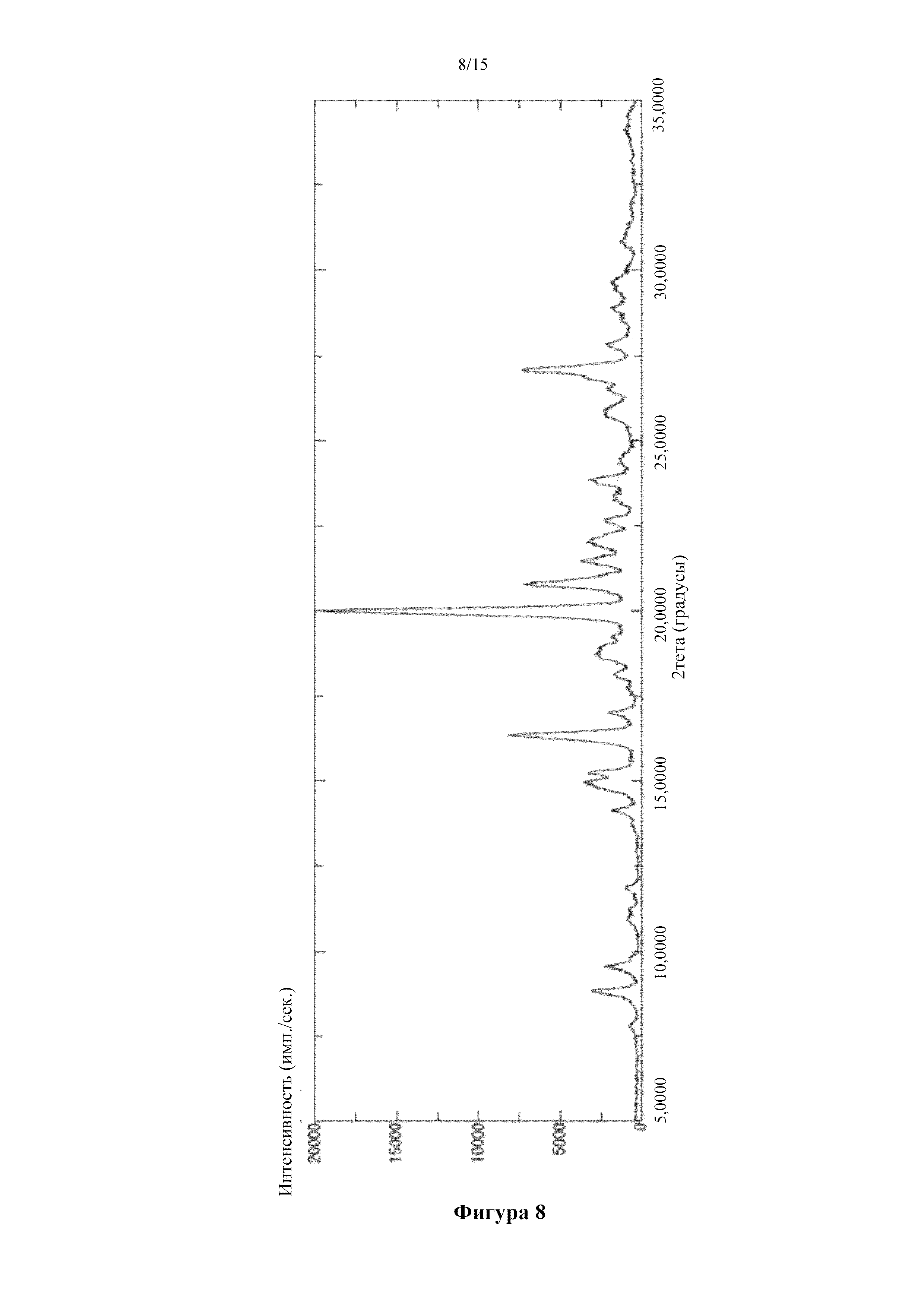
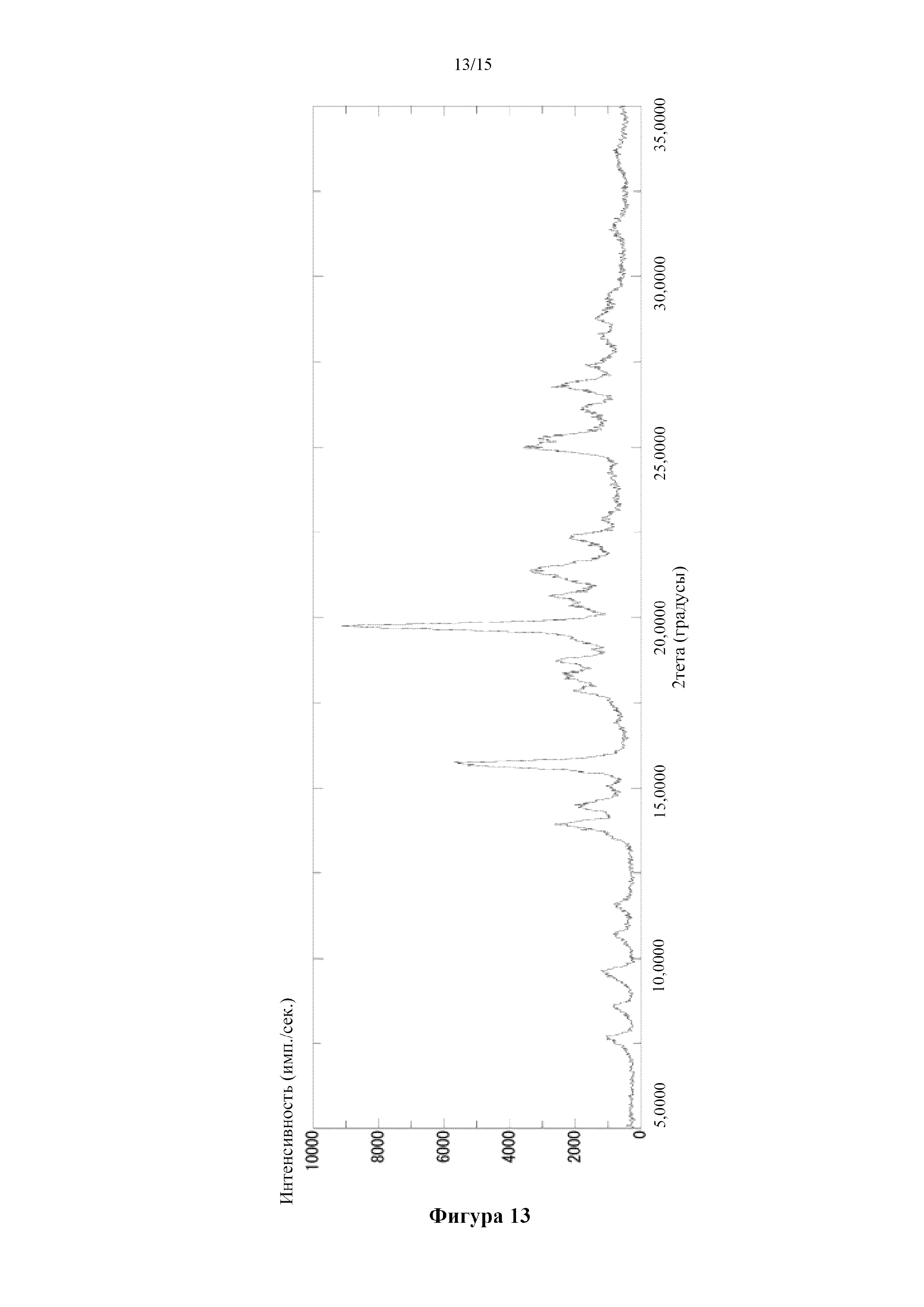
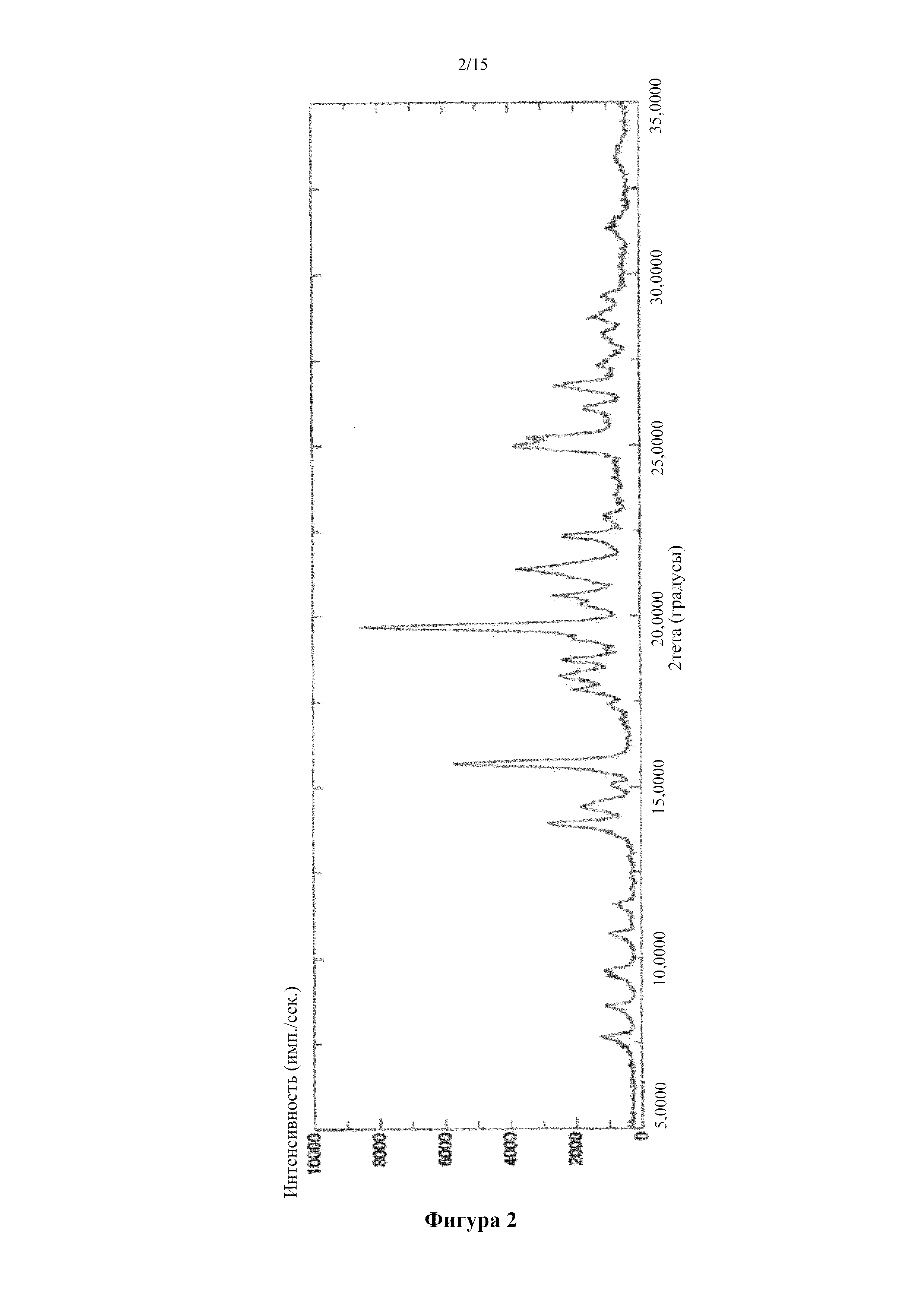
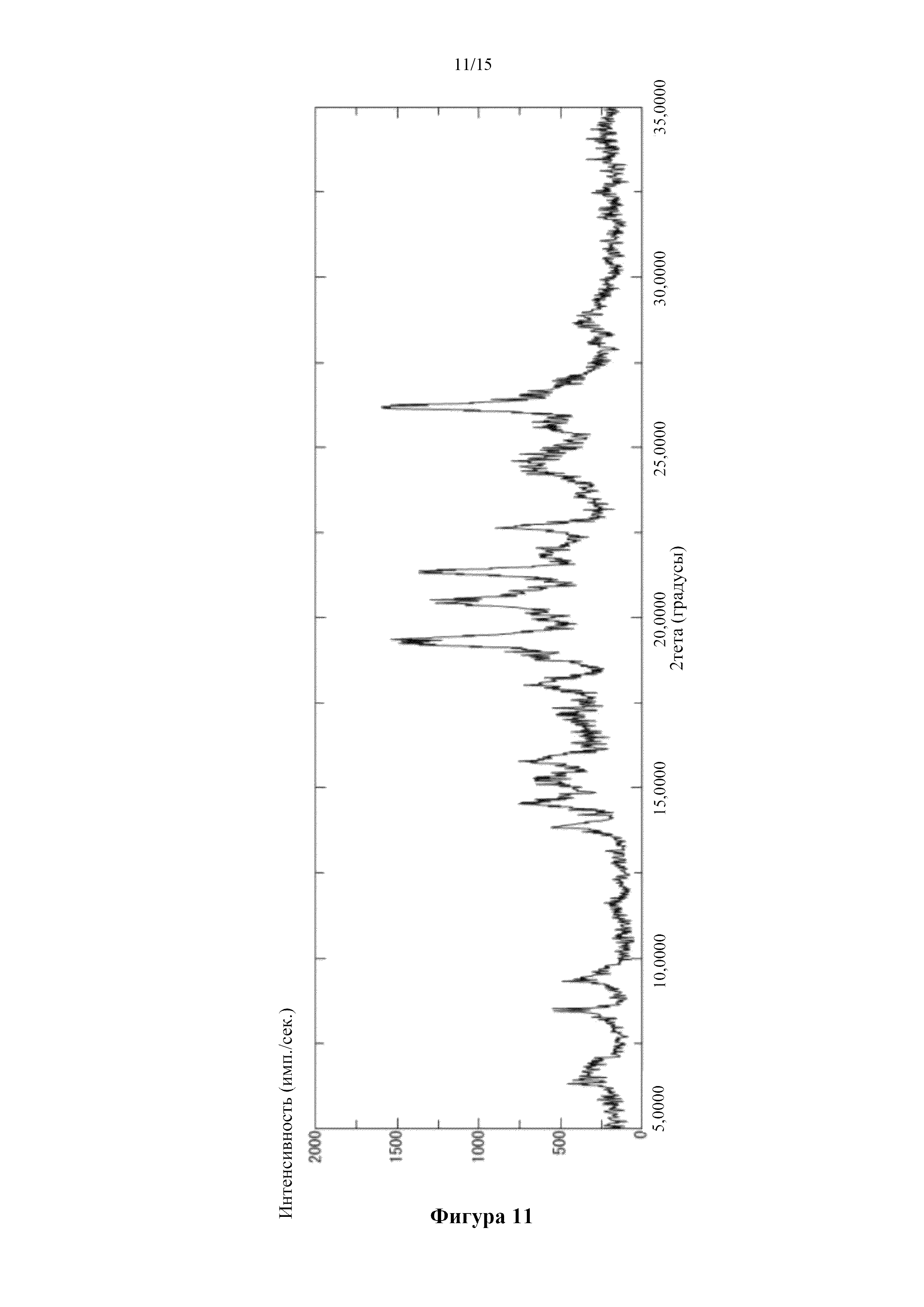




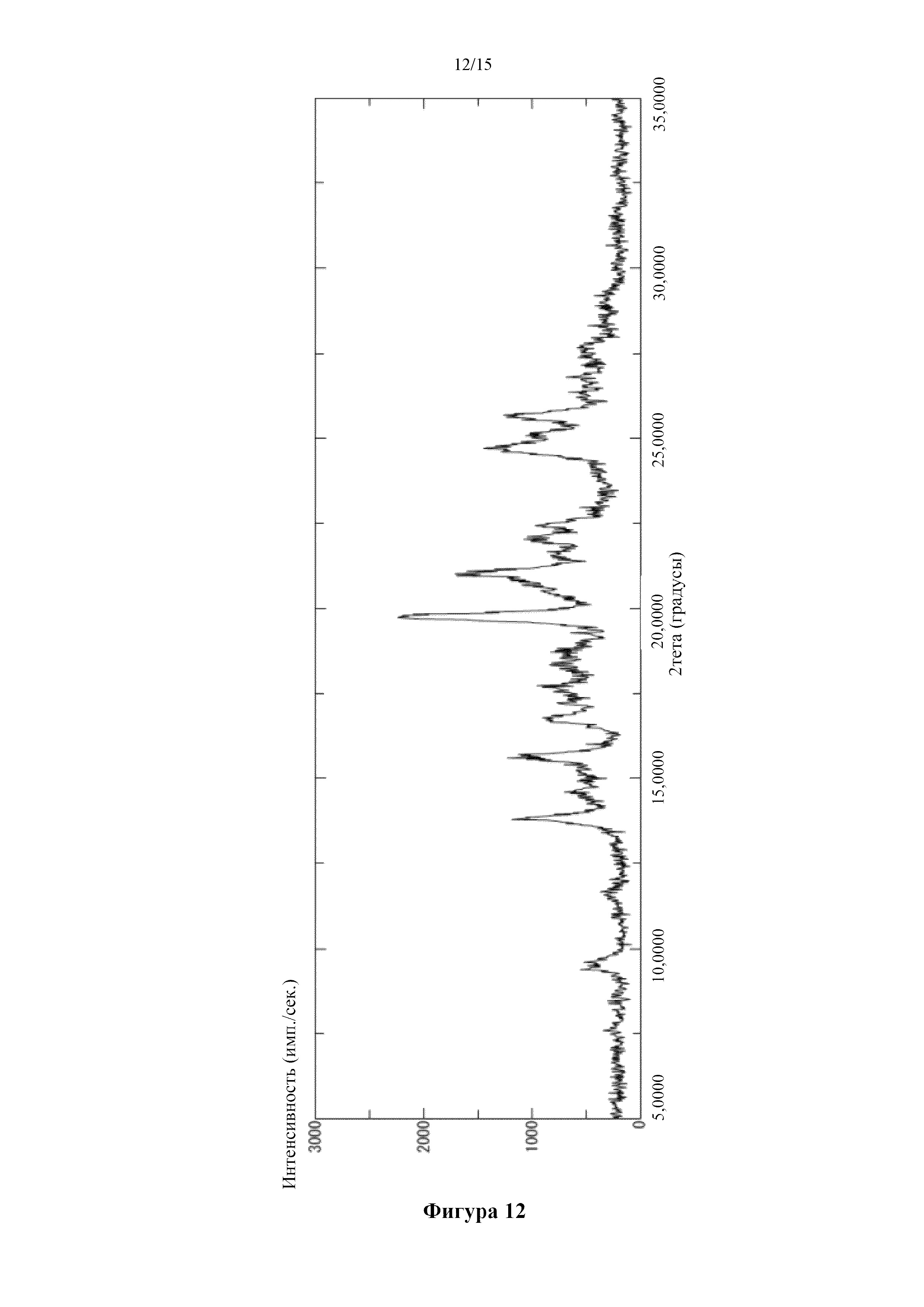
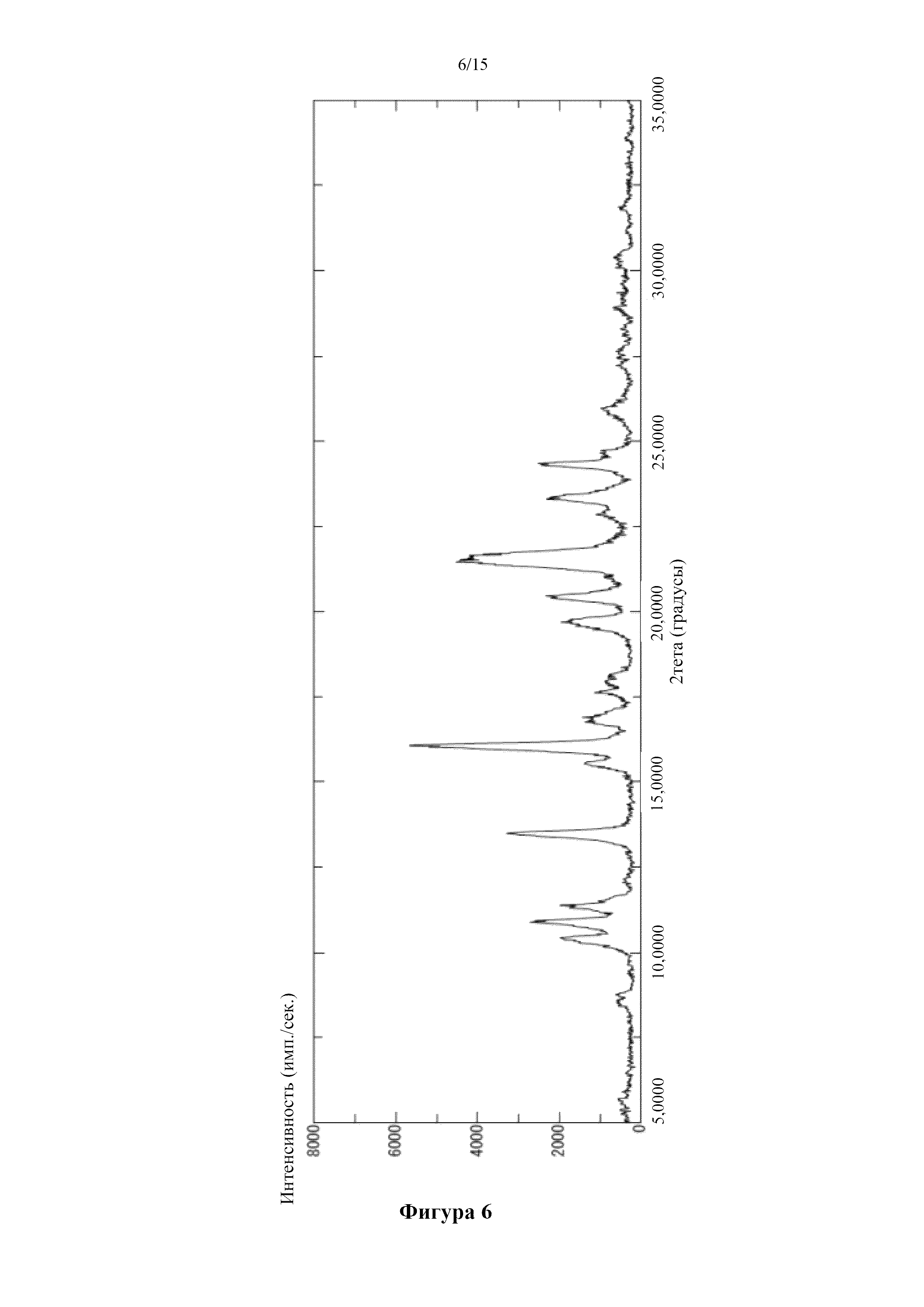

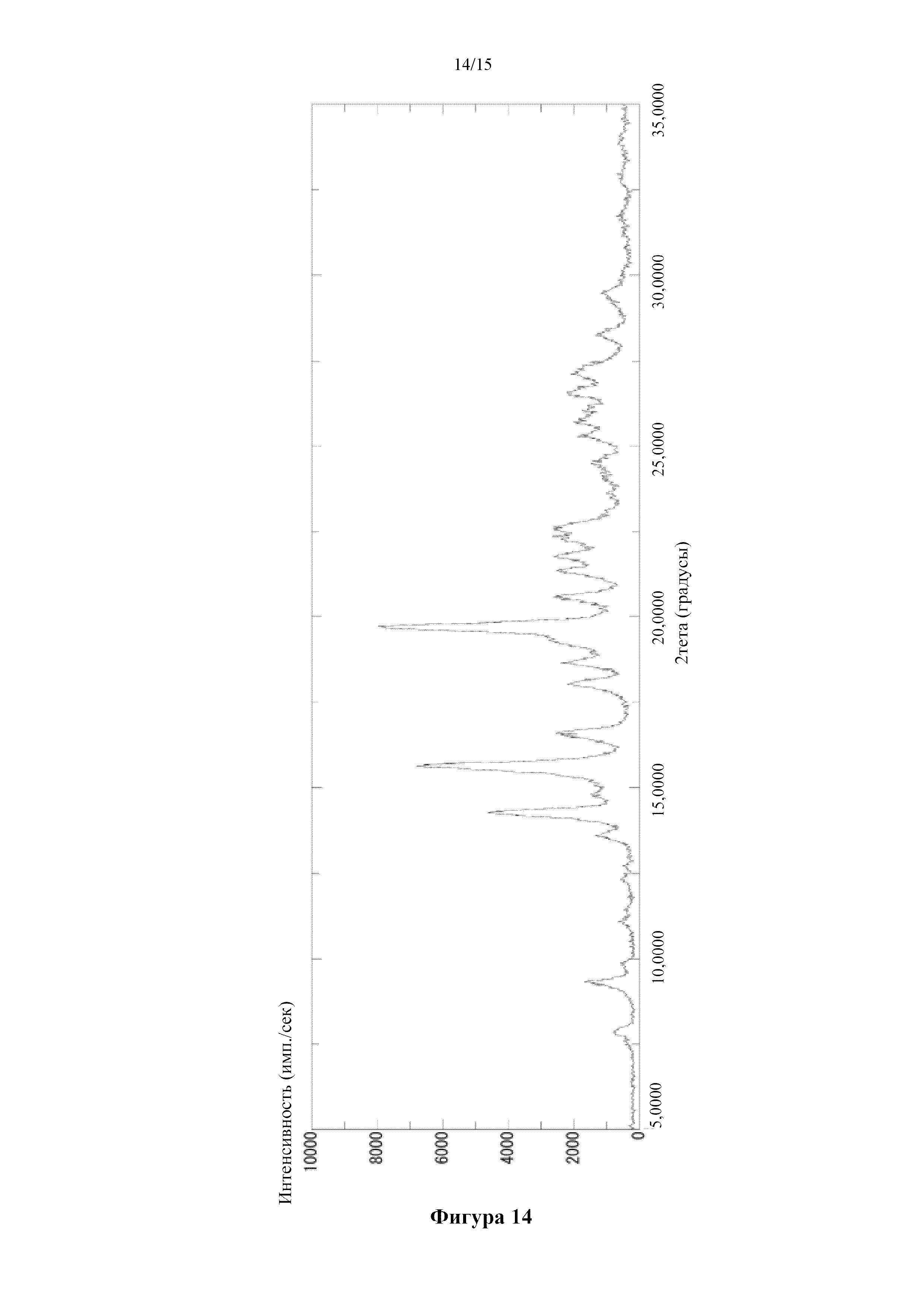
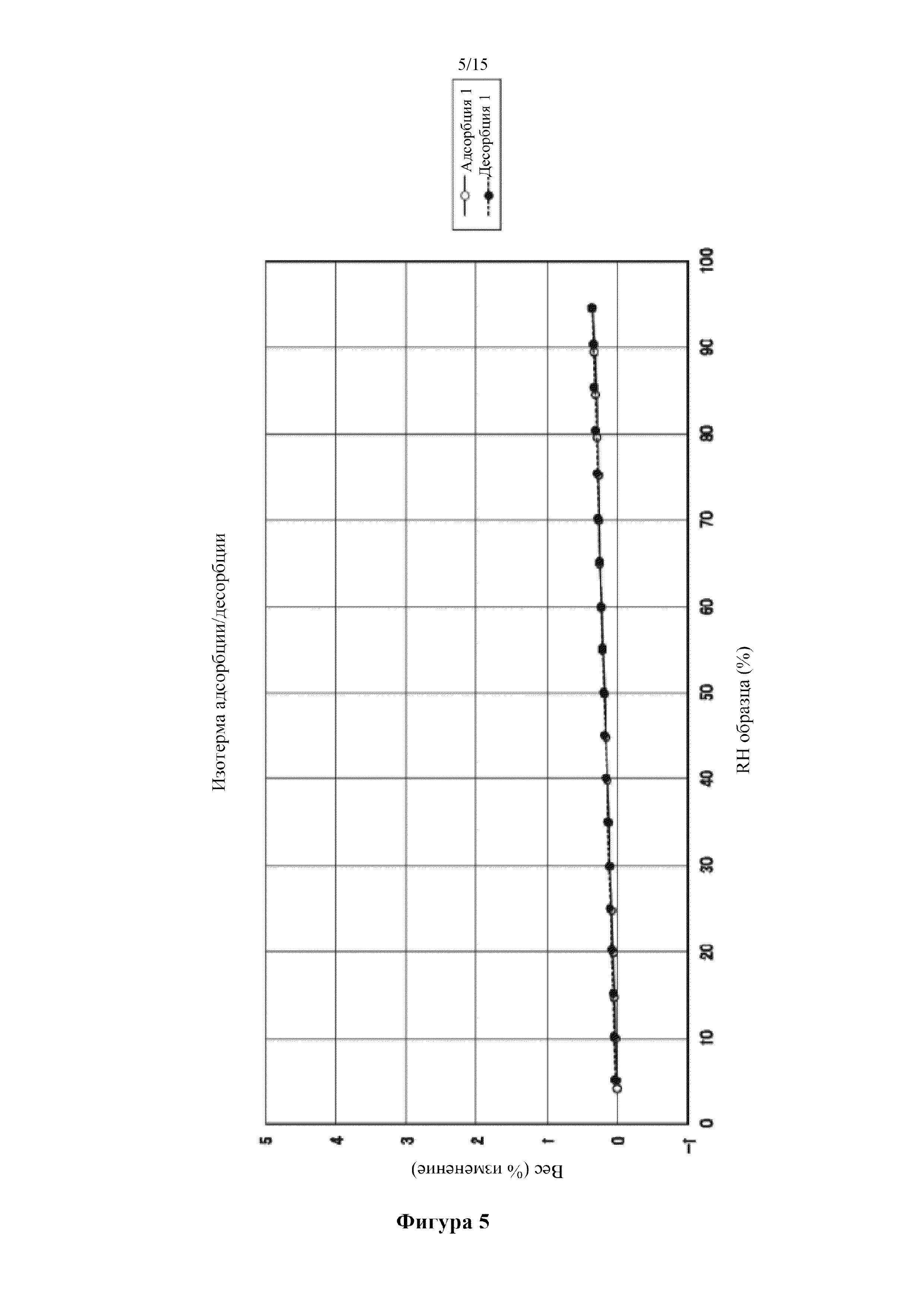
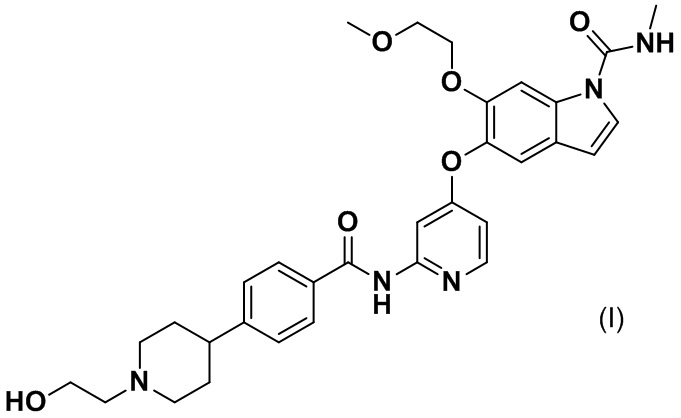
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства
Соль производного пирролидин-3-ил-уксусной кислоты и ее кристаллы
Соль производного пиразолохинолина и ее кристалл

