РЕКОМБИНАНТНЫЙ МИКРООРГАНИЗМ ДЛЯ ПОЛУЧЕНИЯ ПОЛЕЗНЫХ МЕТАБОЛИТОВ
Вид РИД
Изобретение
Настоящее изобретение относится к рекомбинантному микроорганизму, который характеризуется наличием фосфокетолазной активности и имеет сниженный или инактивированный путь Эмбдена-Мейерхофа-Парнаса (ЕМРР) путем инактивации гена(ов), который(е) кодирует(ют) фосфофруктокиназу, или путем снижения фосфофруктокиназной активности по сравнению с немодифицированным микроорганизмом или таким, который не обладает фосфофруктокиназной активностью, и имеет сниженное или инактивированное окислительное ответвление пентозафосфатного пути (РРР) путем инактивации гена(ов), которые кодируют глюкозо-6-фосфатдегидрогеназу, или путем снижения глюкозо-6-фосфатдегидрогеназной активности по сравнению с немодифицированным микроорганизм или таким, который не обладает глюкозо-6-фосфатдегидрогеназной активностью. Такой микроорганизм может использоваться для продукции полезных метаболитов, таких, как ацетон, изобутен или пропен.
На протяжении нескольких последних десятилетий специалисты-практики в области метаболического инжиниринга прилагали усилия, чтобы обеспечить биологические решения для осуществления продукции химических соединений, обеспечивая, таким образом, альтернативы для более традиционных химических процессов. В общем случае, биологически решения позволяют использовать возобновляемые источники сырья (например, сахара) и конкурировать с существующими процессами на основе нефтехимии. Многоэтапное биологическое решение для получения химического вещества типично включает микроорганизм в качестве катализатора для превращения источника сырья до целевой молекулы. Полный набор ферментативных реакций для продукции определенной целевой молекулы может быть разделен на те, которые относятся к путям метаболизма центрального атома углерода, и те, которые относятся к пути, специфическому для продукта. Реакции, которые относятся к центральному атому углерода, и пути, специфические для продукта, являются связанными тем, что окислительно-восстановительные (типично, НАД(Ф)Н) и энергетические (типично, АТФ) компоненты каждого из них и каждая ферментативная реакция должны обеспечиваться в общем балансе, который осуществляет свой вклад в конкурентность этого процесса. Исторически пути центрального атома углерода гетеротрофов, которые выращиваются на сахарах, были описаны как путь Эмбдена-Мейерхофа-Парнаса (ЕМРР), пентозафосфатный путь (РРР), путь Энтнера-Дудорова (EDP), и фосфокетолазный путь (РКР) (смотри Gottschalk (1986), Bacterial Metabolism, 2ое издание, Springer- Verlag, New York). Каждый центральный путь или комбинация центральных путей обладает преимуществами и недостатками в отношении их специфической целевой молекулы. Для того чтобы обеспечить конкурентные биопроцессы, были описаны рекомбинантные микроорганизмы с модификациями, которые вовлекают ЕМРР, РРР и EDP (M. Emmerling и др., Metab. Eng. 1: 117 (1999); L. 0. Ingram и др., Appl. Environ. Microbiol. 53: 2420 (1987); С.Т. Trinh и др., Appl. Environ. Microbiol. 74: 3634 (2008)). Совсем недавно были описаны рекомбинантные микроорганизмы с модификациями, которые вовлекают РКР (смотри Sonderegger и др. Appl. Environ. Microbiol. 70 (2004), 2892-2897, патент США №7,253,001, Chinen и др. J. Biosci. Bioeng. 103 (2007), 262-269, патент США №7,785,858; Fleige и др., Appl. Microbiol. Cell Physiol.91 (2011), 769-776).
EMPP превращает 1 моль глюкозы в 2 моля пирувата (PYR). В том случае, когда ацетил-СоА является желательным, 1 моль PYR может превращаться в 1 моль ацетил-СоА при сопутствующем образовании 1 моля С02 и 1 моля НАДФ. Подытоженные реакции являются приведенными в уравнении 1.
глюкоза+2АДФ+2H3PO4+2СоА+4НАД+→
2 ацетил-СоА+2CO2+2АТФ+2H2O+4НАДФ+4H+
(Уравнение 1)
РРР обеспечивает средства для превращения 1 моля глюкозы в 1 моль CO2 и 2 моля НАДФН при сопутствующем образовании 0,67 моля фруктоза-6-фосфата (F6P) и 0,33 моля глицеральдегид-3-фосфата (GAP). F6P и GAP, образованные таким образом, должны подвергаться метаболизму с помощью реакций других путей, например, с помощью EMPP. EDP превращает 1 моль глюкозы в 1 моль GAP и 1 моль PYR при сопутствующем образовании 1 моля НАДФН. Как и с помощью РРР, GAP, образованный таким образом, должны подвергаться метаболизму с помощью реакций других путей. РКР обеспечивает средства для превращения 1 моля глюкозы в 1 моль GAP и 1,5 моля ацетилфосфата (АсР). Когда ацетил-СоА является желательным, 1 эквивалент АсР плюс 1 эквивалент коэнзима А (СоА) может превращаться в 1 эквивалент ацетил-СоА и 1 эквивалент неорганического фосфата (Pi) при действии фосфотрансацетилазы.
Для специфических целевых молекул, имеющих происхождение от остатков АсСоА, полученных путем РКР, и приблизительно нейтральный окислительно-восстановительный потенциал к АсСоА остаткам, существует дефицит в отношении общего энергетического баланса. РКР (и, подобно ему, РРР и EDP) не образует АТФ для превращения глюкозы в глюкоза-6-фосфат. В случае зависимого от фосфоенолпирувата (PEP) потребления глюкозы PEP должен образовываться с помощью других способов, например, посредством EMPP. Повторное использование GAP с помощью РКР усугубляет вопрос, в частности, тогда, когда специфический для продукта путь обеспечивает незначительное количество АТФ.
Sonderegger (в приводившейся выше ссылке) и патент США №7,253,001 раскрывают рекомбинантные штаммы Saccharomyces cerevisiae, которые включают нативную или сверхэкспрессированную фосфокетолазную активность вместе со сверхэкспрессированной фосфотрансацетилазой для повышения выхода в процессе превращения смесей глюкоза/ксилоза в этанол. Эти штаммы демонстрируют независимое от PEP потребление глюкозы как для механизма ЕМРР, так и РРР.
Chinen (в приводившейся выше ссылке) и патент США №7,785,858 раскрывают рекомбинантную бактерию, выбранную из группы, которая состоит из семейства Enterobacteriaceae, бактерий Coryneform, бактерий Bacillus, которые имеют повышенную фосфокетолазную активность для превращения глюкозы в целевые молекулы, которые продуцируются посредством промежуточного ацетил-СоА, включая группу, которая состоит из L-глутаминовой кислоты, L-глутамина, L-пролина, L-аргинина, L-лейцина, L-цистеина, сукцината и полигидроксибутирата. Эти штаммы демонстрируют зависимое от РЕР-потребление глюкозы при использовании механизма ЕМРР. Примечательным является то, что активность фосфофруктокиназы в бактерии в соответствии с патентом США №7,785,858 является сниженной по сравнению с таковой для штамма дикого типа или немодифицированного типа (смотри страницу 33).
Вне зависимости от того, использует ли частный микроорганизм независимое от PEP потребление глюкозы или зависимое от PEP потребление глюкозы, это влияет на общий энергетический баланс процесса. Например, штаммы S. cerevisiae естественным образом используют независимое от PEP потребление глюкозы, в то время как штаммы Escherichia coli естественным образом используют зависимое от PEP потребление глюкозы. Были раскрыты штаммы Е. coli, в которых зависимое от PEP потребление глюкозы заменялось независимым от PEP потреблением глюкозы. Flores и др. (Metabolic Engineering (2005) 7, 70-87 и патент США №7,371,558. В частности, патент США №7,371,558 раскрывает модификацию потребления глюкозы для повышения выхода превращения глюкозы в 1,3-пропандиол. Эти штаммы демонстрируют независимое от PEP потребление глюкозы как с механизмом ЕМРР, так и РРР, в частности, при отсутствии фосфокетолазной активности.
Существует потребность в развитии рекомбинантных микроорганизмов, которые включают центральный атом углерода и пути, специфические для продукта, что максимизирует превращение источника сырья в продукт с помощью наилучшего приспособления окислительно-восстановительных условий и энергетических ограничений ферментативных реакций. Заявители решили эту необходимость путем обеспечения воплощений, как определено в пунктах формулы изобретения.
Таким образом, настоящее изобретение относится к рекомбинантному микроорганизму, который характеризуется:
a) наличием фосфокетолазной активности;
b) (i) сниженным или инактивированным путем Эмбдена-Мейерхофа-Парнаса (ЕМРР) посредством инактивации гена(ов), который(е) кодирует(ют) фосфофруктокиназу, или путем снижения фосфофруктокиназной активности по сравнению с немодифицированным микроорганизмом; или
(ii) отсутствием активности фосфофруктокиназы,
и
c) (i) наличием сниженного или инактивированного окислительного ответвления пентозафосфатного пути (РРР) путем инактивации гена(ов), который(е) кодирует(ют) глюкозо-6-фосфатдегидрогеназу, или путем снижения глюкозо-6-фосфатдегидрогеназной активности по сравнению с немодифицированным микроорганизмом; или
(ii) отсутствием активности глюкозо-6-фосфатдегидрогеназы.
Микроорганизм в соответствии с настоящим изобретением характеризуется наличием фосфокетолазной активности, так, что это повышает поток продуцируемого ацетил-СоА. Обычно, микроорганизм превращает глюкозу с помощью пути Эмбдена-Мейерхофа-Парнаса в пируват, который потом может превращаться в ацетил-СоА с помощью фермента пируватдегидрогеназы. Однако такое превращение сопровождается высвобождением СО2 и, таким образом, теряется один атом углерода, который мог бы использоваться в продукции полезных метаболитов. Для того чтобы увеличить количество ацетил-СоА в микроорганизме, является, таким образом, желательным, чтобы ацетил-СоА образовывался с помощью отличного пути для того, чтобы избежать потери атомов углерода. Путем использования микроорганизма, обладающего фосфокетолазной активностью, фосфат и фруктоза-6-фосфат превращаются в эритроза-4-фосфат и ацетилфосфат, а фосфотрансацетилазы далее превращают ацетилфосфат в ацетил-СоА без потери атома углерода. Таким образом, в конце процесса выход ацетил-СоА может повышаться путем применения микроорганизма, обладающего фосфокетолазной активностью. Такой микроорганизм является способным к превращению глюкозы в ацетил-СоА без потери атома углерода. Рекомбинантные микроорганизмы, в которых фосфокетолаза экспрессируется естественным образом или гетерологично, раскрываются патентах США №7,785,858 и 7,253,001.
Термин "фосфокетолазная активность", как используется в настоящем изобретении, означает ферментативную активность, которая является способной к превращению
D-ксилулоза-5-фосфата в D-глицеральдегид-3-фосфат в соответствии со следующей реакцией:
D-ксилулоза-5-фосфат+фосфат→D-глицеральдегид-3-фосфат+ацетилфосфат+вода,
или такой, которая является способной катализировать представленную выше реакцию, а также является способной превращать D-фруктоза-6-фосфат в D-эритроза-4-фосфат в соответствии со следующей реакцией:
D-фруктоза-6-фосфат+фосфат→ацетилфосфат+D-эритроза-4-фосфат+вода.
Бывшие фосфокетолазы обычно классифицируются в соответствии с ЕС 4.1.2.9, а более поздние - ЕС 4.1.2.22. Оба типа фосфокетолаз могут использоваться в рамках данного изобретения. Фигура 1 показывает схемы для общих реакций при использовании двух возможностей фосфокетолазы, как описывается в данной заявке.
Такая ферментативная активность может быть измерена с помощью анализов, которые являются известными в области техники. Пример такого анализа является приведенным в разделе Примеры, представленном ниже.
В контексте настоящего изобретения микроорганизм, который обладает фосфокетолазной активностью, может, например, представлять собой микроорганизм, который естественным образом обладает фосфокетолазной активностью, или микроорганизм, который природным образом не обладает фосфокетолазной активностью и был генетически модифицирован для экспрессии фосфокетолазы, или микроорганизм, который естественным образом обладает фосфокетолазной активностью и который был генетически модифицирован, например, трансформирован с помощью нуклеиновой кислоты, например, вектора, который кодирует фосфокетолазу, для того, чтобы повысить фосфокетолазную активность у указанного микроорганизма.
Микроорганизмы, которые наследственно, то есть природным образом, обладают фосфокетолазной активностью, являются известными в области техники, и любой из них может использоваться в контексте настоящего изобретения.
Является также возможным в контексте настоящего изобретение, чтобы микроорганизм представлял собой микроорганизм, который природным образом обладает фосфокетолазной активностью, но который является генетически модифицированным так, что включает нуклеотидную последовательность, которая позволяет осуществлять экспрессию фосфокетолазы. Подобно этому, микроорганизм может также представлять собой микроорганизм, который естественным образом обладает фосфокетолазной активностью, но который является генетически модифицированным так, что фосфокетолазная активность повышается, например, путем введения экзогенной нуклеотидной последовательности, кодирующей фосфокетолазу.
Генетическая модификация микроорганизмов для экспрессии фермента, который представляет интерес, будет подробно описана ниже.
Фосфокетолаза в соответствии с данным изобретением, которая экспрессируется в микроорганизме, может представлять собой любую фосфокетолазу, в частности, фосфокетолазу из прокариотических или эукариотических организмов. Описывают, например, прокариотические фосфокетолазы из Lactococcus lactis, и такой пример является приведенным в разделе Примеры.
В предпочтительном воплощении настоящего изобретения фосфокетолаза представляет собой фермент, который включает аминокислотную последовательность, как кодируется с помощью SQ0005, показанной в разделе Примеры, или последовательность, которая является, по крайней мере, на n% идентичной такой аминокислотной последовательности и имеющая активность фосфокетолазы, где n представляет собой целое число от 10 до 100, предпочтительно 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99.
Предпочтительно, когда степень идентичности определяется путем сравнения соответствующей последовательности с аминокислотной последовательностью какой-либо одной из упомянутых выше SEQ ID NO. Тогда, когда последовательности, которые сравнивают, не имеют одинаковую длину, степень идентичности предпочтительно либо относится к проценту аминокислотных остатков в более короткой последовательности, которые являются идентичными аминокислотным остаткам в более длинной последовательности, или к проценту аминокислотных остатков в более длинной последовательности, которые являются идентичными аминокислотным остаткам в более короткой последовательности. Степень идентичности последовательности может быть определена в соответствии со способами, известными в данной области техники при использовании предпочтительно приемлемых компьютерных алгоритмов, таких как CLUSTAL.
При использовании метода Clustal анализа для определения, является ли частная последовательность, например, на 80% идентичной эталонной последовательности, могут использоваться параметры по умолчанию или такие параметры могут предпочтительно быть такими, как описано ниже: Матрица: матрица блоков аминокислотных замен 30; штраф за внесение пробела: 10,0; штраф за продление пробела: 0,05; задержка отклонение: 40; протяженность зоны пробела: 8 для сравнения аминокислотных последовательностей. Для сравнения нуклеотидных последовательностей штраф за продление пробела предпочтительно устанавливают на уровне предпочтительно 5,0. Предпочтительно, когда степень идентичности подсчитывается на протяжении полной длины последовательности.
Фосфокетолаза, которая экспрессируется в микроорганизме в соответствии с изобретением, может представлять собой существующую в природе фосфокетолазу или она может быть фосфокетолазой, которая получена из существующей в природе фосфокетолазы, например, путем введения мутаций или других изменений, которые, например, изменяют или улучшают ферментативную активность.
Способы модификации и/или улучшения желательных ферментативных активностей белков являются хорошо известными квалифицированному специалисту в данной области техники и включают, например, случайный мутагенез или сайт-направленный мутагенез и последующую селекцию ферментов, обладающих желательными свойствами, или подходы так называемой "направленной эволюции".
Например, для генетической модификации в прокариотической клетке, молекула нуклеиновой кислоты, которая кодирует фосфокетолазу, может быть введена в плазмиды, которые позволяют осуществлять мутагенез или модификацию последовательности с помощью рекомбинации последовательностей ДНК. Стандартные способы (смотри Sambrook и Russell (2001), Molecular Cloning: A Laboratory Manual, CSH Press, Cold Spring Harbor, NY, USA) позволяют осуществлять замены оснований или прибавлять существующие в природе или синтетические последовательности. ДНК фрагменты могут быть лигированы при использовании адаптеров и линкеров, комплементарных этим фрагментам. Кроме того, могут использоваться средства инжиниринга, которые обеспечивают приемлемые сайты рестрикции или удаление избыточной ДНК или рестрикционных сайтов. В тех случаях, когда инсерции, делеции или замены являются возможными, могут использоваться in vitro мутагенез, "восстановление праймера", рестрикция или лигирование. В общем случае, анализ последовательности, рестрикционный анализ и другие способы биохимии и молекулярной биологии осуществляются как способы анализа. Полученные варианты фосфокетолазы потом подвергают анализу на желаемую активность, например, ферментативную активность, с помощью анализа так, как описано выше, и, в частности, на их повышенную ферментативную активность.
Как описано выше, микроорганизм в соответствии с изобретением может представлять собой микроорганизм, который был генетически модифицирован путем введения молекулы нуклеиновой кислоты, которая кодирует фосфокетолазу. Таким образом, в предпочтительном воплощении рекомбинантный микроорганизм представляет собой рекомбинантный микроорганизм, который был генетически модифицирован для наличия повышенной фосфокетолазной активности. Этого можно достичь, например, путем трансформации микроорганизма с помощью нуклеиновой кислоты, которая кодирует фосфокетолазу. Подробное описание генетической модификации микроорганизмов будет дополнительно приведено ниже. Предпочтительно, когда молекула нуклеиновой кислоты, которая встраивается в микроорганизм, представляет собой молекулу нуклеиновой кислоты, которая является гетерологичной по отношению к микроорганизму, то есть, она в природе не существует в указанном микроорганизме.
В контексте настоящего изобретения "повышенная активность" означает, что экспрессия и/или активность фермента, в частности, фосфокетолазы в генетически модифицированном микроорганизме, составляет, по крайней мере, 10%, предпочтительно, по крайней мере, на 20%, более предпочтительно, по крайней мере, 30% или 50%, даже более предпочтительно, по крайней мере, 70% или 80% и особенно предпочтительно, по крайней мере, 90% или 100% выше, чем в немодифицированном микроорганизме. В более предпочтительных воплощениях повышение экспрессии и/или активности может составлять, по крайней мере, 150%, по крайней мере, 200% или, по крайней мере, 500%. В особенно предпочтительных воплощениях экспрессия является, по крайней мере, 10-кратной, более предпочтительно, по крайней мере, в 100 раз и даже более предпочтительно, по крайней мере, в 1000 раз более высокой, чем в соответствующем немодифицированном микроорганизме.
Термин "повышенная" экспрессия/активность также относится к ситуации, в которой немодифицированный микроорганизм не экспрессирует соответствующий фермент, например, фосфокетолазу, так, что соответствующая экспрессия/активность в немодифицированном микроорганизме равна нулю.
Способы для измерения уровня экспрессии данного белка в клетке являются хорошо известными квалифицированному специалисту в данной области техники. В одном воплощении измерение уровня экспрессии осуществляют путем измерения количества соответствующего белка. Соответствующие способы являются хорошо известными квалифицированному специалисту в данной области техники и включают Вестер-блоттинг, ELISA и т.д. В другом воплощении измерение уровня экспрессии осуществляют путем измерения количества соответствующей РНК. Соответствующие способы являются хорошо известными квалифицированному специалисту в данной области техники и включают, например, Нозерн-блоттинг.
Способы измерения ферментативной активности фосфокетолазы являются хорошо известными в области техники и уже были описаны выше.
Микроорганизм в соответствии с настоящим изобретением дополнительно характеризуется наличием сниженного или инактивированного пути Эмбдена-Мейерхофа-Парнаса (ЕМРР) путем инактивации гена(ов), который(ые) кодирует(ют) фосфофруктокиназу, или путем снижения фосфофруктокиназной активности по сравнению с немодифицированным микроорганизмом или такими, которые не обладают фосфофруктокиназной активностью. Таким образом, микроорганизм представляет собой либо микроорганизм, который естественным образом обладает ЕМРР, включая фосфофруктокиназную активность, но который был модифицирован, в частности, генетически модифицирован, так, что фосфофруктокиназная активность является либо полностью устраненной, либо сниженной по сравнению с соответствующим немодифицированным микроорганизмом, или микроорганизм представляет собой микроорганизм, который естественным образом не обладает фосфофруктокиназной активностью.
Как уже было упомянуто выше, тогда, когда глюкоза подвергается процессингу в помощью ЕМРР до ацетил-СоА, один атом углерода теряется путем высвобождения CO2 на последнем этапе. Путем введения фосфокетолазы этой потере можно избежать. Поскольку фруктоза-6-фосфат представляет собой субстрат для фосфокетолазы, является желательным, чтобы пул фруктоза-6-фосфата поддерживался на высоком уровне в микроорганизме для того, чтобы повысить выход ацетил-СоА. Поскольку фруктоза-6-фосфат представляет собой субстрат для фермента пути Эмбдена-Мейерхофа-Парнаса, то есть, для фосфофруктокиназы, рекомбинантный микроорганизм в соответствии с настоящим изобретением имеет сниженную фосфофруктокиназную активность по сравнению с немодифицированным микроорганизмом, или ген(ы), который(ые) кодирует(ют) фосфофруктокиназу был(были) инактивирован(ы). Это обеспечивает то, что поток фруктоза-6-фосфата направляется на фосфокетолазу и продукцию ацетил-СоА без потери CO2, поскольку фруктоза-6-фосфат или большая часть фруктоза-6-фосфата не может более подвергаться процессингу при использовании пути Эмбдена-Мейерхофа-Парнаса. Рекомбинантные микроорганизмы, в которых фосфокетолаза естественным образом или гетерологично экспрессируется и которые имеют сниженную фосфофруктокиназную активность, описываются в патенте США №7,785,858.
"Фосфофруктокиназная активность" означает ферментативную активность, которая превращает АТФ и фруктоза-6-фосфат в АДФ и фруктоза-1,6-бисфосфат (ЕС 2.7.1.11). Эта ферментативная активность может быть измерена с помощью анализов, которые являются известными в области техники, как, например, описывается Kotlarz и др. (Methods Enzymol. (1982) 90, 60-70).
Термин "микроорганизм, который характеризуется наличием сниженного или инактивированного пути Эмбдена-Мейерхофа-Парнаса (ЕМРР) путем инактивации гена(ов), которые(ые) кодирует(ют) фосфофруктокиназу или путем снижения фосфофруктокиназной активности по сравнению с немодифицированным микроорганизмом" предпочтительно относится к микроорганизму, в котором инактивация гена(ов), которые(ые) кодирует(ют) фосфофруктокиназу или путем снижения фосфофруктокиназной активности по сравнению с немодифицированным микроорганизмом, достигается путем генетической модификации микроорганизма, что ведет к указанной инактивации или снижению.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой рекомбинантный микроорганизм, который имеет инактивированный путь Эмбдена-Мейерхофа-Парнаса (ЕМРР) путем инактивации гена(ов), которые(ые) кодирует(ют) фосфофруктокиназу. Инактивация гена(ов), которые(ые) кодирует(ют) фосфофруктокиназу в контексте настоящего изобретения означает, что ген(ы), которые(ые) кодирует(ют) фосфофруктокиназу, и которые присутствуют в микроорганизме, является(ются) инактивированным(и), так, что они уже не экспрессируют и/или не приводят к синтезу функциональной фосфофруктокиназы. Инактивация может достигаться с помощью различных путей, известных в уровне техники. Инактивация может, например, достигаться путем разрыва гена(ов), который(е) кодирует(ют) фосфофруктокиназу, или путем делеции указанного(ых) гена(ов) путем введения селективного маркера. Альтернативно, промотор гена(ов), который(ые) кодирует(ют) фосфофруктокиназу, может подвергаться мутации так, что ген уже не транскрибируется в мРНК. Другие пути инактивации гена(ов), который(ые) кодируют фосфофруктокиназу, известных в уровне техники, представляют собой: экспрессию полинуклеотида, который кодирует РНК, имеющую нуклеотидную последовательность, комплементарную транскрипту гена(ов) фосфофруктокиназы, так что мРНК может уже не транслироваться в белок, экспрессию полинуклеотида, который кодирует РНК, которая супрессирует экспрессию указанного(ых) гена(ов) с помощью эффекта РНКи; экспрессию полинуклеотида, который кодирует РНК, обладающую активностью специфического расщепления транскрипта указанного(ых) гена(ов); или экспрессию полинуклеотида, который кодирует РНК, которая супрессирует экспрессию указанного(ых) гена(ов) с помощью эффекта косупрессии. Такие полинуклеотиды могут быть встроены в вектор, который может быть введен в микроорганизм путем трансформации, для достижения инактивации гена(ов), который(ые) кодирует(ют) фосфофруктокиназу.
Термин "инактивация" в контексте настоящего изобретения предпочтительно означает полную инактивацию, то есть, означает то, что микроорганизм не демонстрирует фосфофруктокиназной активности. Это означает, в частности, что микроорганизм не демонстрирует фосфофруктокиназной активности, независимой от условий выращивания.
Предпочтительно, "инактивация" означает, что ген(ы), который(ые) кодирует(ют) фосфофруктокиназу, который(ые) присутствует(ют) в микроорганизме, являются генетически модифицированными так, что предотвращают экспрессию фермента. Этого можно достичь, например, с помощью делеции гена или его частей, где делеция его частей предотвращает экспрессию фермента, или путем нарушения гена либо в его кодирующем участке, либо в промоторном участке, где нарушение оказывает эффект, который заключается в том, что белок не экспрессируется или экспрессируется дисфункциональный белок.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой рекомбинантный микроорганизм, который имеет сниженный путь Эмбдена-Мейерхофа-Парнаса (ЕМРР) путем уменьшения фосфофруктокиназной активности по сравнению с немодифицированным микроорганизмом. Предпочтительно, такое снижение достигается путем генетической модификации микроорганизма. Этого можно достичь, например, с помощью случайного мутагенеза или сайт-направленного мутагенеза промотора и/или фермента и последующей селекции промоторов и/или ферментов, которые обладают желательными свойствами, или с помощью комплементарных нуклеотидных последовательностей или эффекта РНКи, как описано выше. Подробное описание генетической модификации микроорганизмов будет представлено дополнительно ниже.
В контексте настоящего изобретения "сниженная активность" означает, что экспрессия и/или активность фермента, в частности, фосфофруктокиназы, в генетически модифицированном микроорганизме является, по крайней мере, на 10%, предпочтительно, по крайней мере, 20%, более предпочтительно, по крайней мере, 30% или 50%, даже более предпочтительно, по крайней мере, 70% или 80% и особенно предпочтительно, по крайней мере, 90% или 100% ниже, чем в соответствующем немодифицированном микроорганизме. Способы для измерения уровня экспрессии данного белка в клетке являются хорошо известными квалифицированному специалисту в данной области техники. Анализы для измерения сниженной ферментативной активности фосфофруктокиназы являются известными в уровне техники.
В другом воплощении микроорганизм в соответствии с настоящим изобретением представляет собой микроорганизм, который не обладает фосфофруктокиназной активностью. Это предпочтительно означает, что такой микроорганизм в природе не обладает фосфофруктокиназной активностью. Это означает, что такой микроорганизм в природе не содержит в своем геноме нуклеотидной последовательности, которая кодирует фермент с фосфофруктокиназной активностью. Примеры таких микроорганизмов представляют собой Zymomonas mobilis (J.S. Suo и др., Nat. Biotechnol. 23:63 (2005)) и Ralstonia eutropha (C. Fleige и др., Appl. Microb. Cell Physiol. 91:769 (2011)).
Микроорганизм в соответствии с настоящим изобретением дополнительно характеризуется наличием сниженного или инактивированного окислительного ответвления пентозафосфатного пути (РРР) путем инактивации гена(ов), который(ые) кодирует(ют) глюкозо-6-фосфатдегидрогеназу, или путем снижения глюкозо-6-фосфатдегидрогеназной активности по сравнению с немодифицированным микроорганизмом или таким, который не обладает глюкозо-6-фосфатдегидрогеназной активностью. Таким образом, микроорганизм представляет собой либо микроорганизм, который естественным образом обладает РРР, включая глюкозо-6-фосфатдегидрогеназную активность, но который был модифицирован, в частности, генетически модифицирован, так, что глюкозо-6-фосфатдегидрогеназная активность является либо полностью отсутствующей, либо такой, которая является сниженной по сравнению с соответствующим немодифицированным микроорганизмом, или микроорганизм представляет собой микроорганизм, который в природе не обладает глюкозо-6-фосфатдегидрогеназной активностью.
Снижение или инактивация окислительного ответвления пентозафосфатного пути дополнительно повышает выход ацетил-СоА, поскольку глюкоза-6-фосфат не будет более прокачиваться через пентозафосфатный цикл. Весь или почти весь глюкоза-6-фосфат в микроорганизме будет превращаться в фруктоза-6-фосфат, который будет потом превращаться в ацетил-СоА.
"Глюкозо-6-фосфатдегидрогеназная активность" означает ферментативную активность, которая превращает глюкоза-6-фосфат и НАДФ+ в 6-фосфоглюконо-δ-лактон и НАДФН (ЕС 1.1.1.49). Эта ферментативная активность может быть измерена с помощью анализов, которые являются известными в области техники, как, например, те, которые описаны Noltmann и др. (J. Biol. Chem. (1961) 236, 1225-1230).
Термин "микроорганизм, который характеризуется наличием сниженного или инактивированного окислительного ответвления пентозафосфатного пути (РРР) путем инактивации гена(ов), который(ые) кодирует(ют) глюкозо-6-фосфатдегидрогеназу, или путем снижения глюкозо-6-фосфатдегидрогеназной активности по сравнению с немодифицированным микроорганизмом" предпочтительно относится к микроорганизму, в котором инактивация гена(ов), который(ые) кодирует(ют) глюкозо-6-фосфатдегидрогеназу или снижение глюкозо-6-фосфатдегидрогеназной активности по сравнению с немодифицированным микроорганизмом достигается путем генетической модификации микроорганизма, что ведет к указанной инактивации или снижению.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой рекомбинантный микроорганизм, который обладает инактивированным окислительным ответвлением пентозафосфатного пути (РРР) путем инактивации гена(ов), который(ые) кодирует(ют) глюкоза-6-фосфат дегидрогеназу. Инактивация гена(ов), который(ые) кодирует(ют) глюкозо-6-фосфатдегидрогеназу в контексте настоящего изобретения означает, что ген(ы), который(ые) кодирует(ют) глюкозо-6-фосфатдегидрогеназу, присутствующий(ие) в микроорганизме, является(ются) инактивированным(и) так, что они больше не экспрессируют и/или не приводят к синтезу функциональной глюкоза-6-фосфатдегидрогеназы. Инактивация может достигаться путем множества различных способов, известных в уровне техники. Инактивация может, например, достигаться путем нарушения гена(ов), который(е) кодирует(ют) глюкозо-6-фосфатдегидрогеназу, или делеции указанного(ых) гена(ов) с помощью введения селективного маркера. Альтернативно, промотор гена(ов), который(е) кодирует(ют) глюкозо-6-фосфатдегидрогеназу, может подвергаться мутации так, что этот(эти) ген(ы) больше не транскрибируется(ются) в мРНК. Другие пути для инактивации гена(ов), который(е) кодирует(ют) фосфофруктокиназу, известные в уровне техники, представляют собой: экспрессию полинуклеотида, который кодирует РНК, имеющую нуклеотидную последовательность, комплементарную транскрипту гена(ов) глюкозо-6-фосфатдегидрогеназы так, что мРНК не может более транслироваться в белок, экспрессию полинуклеотида, который кодирует РНК, супрессирующую экспрессию указанного(ых) гена(ов) посредством эффекта РНКи; экспрессию полинуклеотида, который кодирует РНК, имеющую активность специфического расщепления транскрипта указанного(ых) гена(ов); или экспрессию полинуклеотида, который кодирует РНК, которая супрессирует экспрессию указанного(ых) гена(ов) посредством эффекта косупрессии. Эти полинуклеотиды могут быть введены в вектор, который может быть введен в микроорганизм путем трансформации для достижения инактивации гена(ов), кодирующих глюкозо-6-фосфатдегидрогеназу.
Термин "инактивация" в контексте настоящего изобретения предпочтительно означает полную инактивацию, то есть, тот факт, что микроорганизм не демонстрирует активности глюкозо-6-фосфатдегидрогеназы. Это означает, в частности, то, что микроорганизм не демонстрирует активности глюкозо-6-фосфатдегидрогеназы, независимой от условий выращивания.
Предпочтительно, "инактивация" означает, что ген(ы), который(ые) кодирует(ют) глюкозо-6-фосфатдегидрогеназу которая присутствует в микроорганизме, является(ются) генетически модифицированными так, что предотвращают экспрессию фермента. Этого можно достичь, например, путем делеции гена или его частей, где делеция его частей предотвращает экспрессию фермента, или путем нарушения либо в кодирующем участке, либо в промоторном участке, где нарушение имеет эффект, который заключается в том, что белок не экспрессируется или экспрессируется дисфункциональный белок.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой рекомбинантный микроорганизм, который имеет сниженное окислительное ответвление пентозафосфатного пути (РРР) путем снижения активности глюкозо-6-фосфатдегидрогеназы по сравнению с немодифицированным микроорганизмом. Предпочтительно, такое снижение достигается путем генетической модификации микроорганизма. Этого можно достичь, например, путем случайного мутагенеза или сайт-направленного мутагенеза промотора и/или фермента, и последующей селекции промоторов и/или ферментов, которые обладают желательными свойствами или с помощью комплементарных нуклеотидных последовательностей или эффекта РНКи, как описано выше. Подробное описание генетической модификации микроорганизмов будет приведено ниже.
В контексте настоящего изобретения "сниженная активность" означает, что экспрессия и/или активность фермента, в частности, глюкоза-6-фосфатдегидрогеназы, в генетически модифицированном микроорганизме является, по крайней мере, на 10%, предпочтительно, по крайней мере, 20%, более предпочтительно, по крайней мере, 30% или 50%, даже более предпочтительно, по крайней мере, 70% или 80% и особенно предпочтительно, по крайней мере, на 90% или 100% ниже, чем в соответствующем немодифицированном микроорганизме. Способы для измерения уровня экспрессии данного белка в клетке являются хорошо известными квалифицированному специалисту в данной области техники. Анализы для измерения сниженной ферментативной активности глюкозо-6-фосфатдегидрогеназы являются известными в уровне техники.
В другом воплощении микроорганизм в соответствии с настоящим изобретением представляет собой микроорганизм, который не обладает активностью глюкозо-6-фосфатдегидрогеназы. Это предпочтительно означает, что такой микроорганизм естественным образом не обладает глюкозо-6-фосфатдегидрогеназной активностью. Это означает, что такой микроорганизм естественным образом не содержит в своем геноме нуклеотидной последовательности, которая кодирует фермент с глюкозо-6-фосфатдегидрогеназной активностью. Примеры таких микроорганизмов представляют собой Acinetobacter baylyi (Barbe и др., Nucl. Acids Res. 32 (2004), 5766-5779), гипертермофильный филум архейской эры, такой, как Sulfolobus solfataricus (Nunn и др., J. Biol. Chem. 285 (2010), 33701-33709), Thermoproteus tenax, Thermoplasma acidophilum и Picrophilus torridus (Reher и Schonheit, FEBS Lett. 580 (2006), 1198-1204).
В дополнительном воплощении микроорганизм в соответствии с настоящим изобретением дополнительно характеризуется наличием активности Фруктоза-1,6-бисфосфат фосфатазы, предпочтительно при выращивании на глюкозе. Фруктоза-1,6-бисфосфат фосфатаза представляет собой фермент, который участвует в глюконеогенезе, гидролизующем фруктоза-1,6-бисфосфат в фруктоза-6-фосфат и свободный фосфат. Однако, как правило, во всех организмах в присутствии глюкозы фруктоза-1,6-бисфосфат фосфатазная активность репрессируется, и глюкоза подвергается обработке путем ЕМРР (гликолиза). Микроорганизм в соответствии с настоящим изобретением, который обладает фосфокетолазной активностью и который не имеет фосфофруктокиназной активности или в котором фосфофруктокиназная активность является сниженной, или ген, который кодируют фосфофруктокиназу является инактивированным, выход ацетил-СоА путем конверсии фруктоза-6-фосфата с помощью фосфокетолазы (ЕС 4.1.2.9 или ЕС 4.1.2.22) может быть повышен с помощью обеспечения присутствия Фруктоза-1,6-бисфосфат фосфатазной активности, например, путем дерепрессии фруктоза-1,6-бисфосфат фосфатазы в присутствии глюкозы. Присутствие активности фруктоза-1,6-бисфосфат фосфатазы приводит к рециркуляции фруктоза-1,6-бисфосфата, вырабатываемого с помощью фруктоза-1,6-бисфосфат альдолазы, в фруктоза-6-фосфат, который может потом снова превращаться с помощью фосфокетолазного пути в ацетил-СоА. Действительно, продукт фосфокетолазы - ацетилфосфат взаимопревращается в ацетил-СоА посредством действия фермента фосфатацетилтрансферазы ЕС 2.3.1.8. Таким образом, рекомбинантный микроорганизм в соответствии с настоящим изобретением является способным к выработке ацетил-СоА из глюкозы при стехиометрическом составе, приближающемся к 3:1. Суммарные реакции являются представленными в уравнении 2:
глюкоза+АТФ+3 СоА→3 ацетил-СоА+АДФ+H3PO4+2H2O (Уравнение 2)
Термин "активность фруктоза-1,6-бисфосфат фосфатазы" означает ферментативную активность, которая превращает фруктоза-1,6-бисфосфат и H2O в фруктоза-6-фосфат и фосфат (ЕС 3.1.3.11). Эта ферментативная активность может быть измерена с помощью анализов, которые являются известными в области техники, как, например, описывается Hines и др. (J. Biol. Chem. (2007) 282, 11696-11704). Термины "активность фруктоза-1,6-бисфосфат фосфатазы" и "фруктоза-1,6-бисфосфат фосфатаза" также охватывают ферменты, которые являются бифункциональными в том, что они демонстрируют активность фруктоза-1-6-бисфосфат альдолазы/фосфатазы. Такие бифункциональные ферменты экспрессируются в большинстве простейших и сильно разветвленных бактериальных генеалогиях и в большинстве случаев являются термостабильными. Такие ферменты представляют собой, например, такие, которые описаны для Thermococcus kodakaraensis, Sulfolobus tokodaii, Ignicoccus hospitalis, Cenarchaeum symbiosum, Sulfolobus solfataricas, Thermus thermophilus, Thermoproteus neutrophilus, Moorella thermoacetica и много других (смотри, например, Say и Fuchs (Nature 464 (2010), 1077); Fushinobu и др. (Nature 478 (2011), 538; Du и др. (Nature 478 (2011), 534).
Термин "активность фруктоза-1,6-бисфосфат фосфатазы при выращивании на глюкозе" означает, что микроорганизм экспрессирует фермент с фруктоза-1,6-бисфосфат фосфатазной активностью тогда, когда микроорганизм выращивается на глюкозе. "Выращенный на глюкозе" означает, что микроорганизм выращивается в среде, которая содержит, среди прочих, глюкозу в качестве источника углерода. Предпочтительно, этот термин означает, что микроорганизм выращивается в среде, содержащей глюкозу в качестве единственного источника углерода.
В контексте настоящего изобретения микроорганизм, который обладает активностью фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, может, например, представлять собой микроорганизм, который естественным образом обладает активностью фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, или микроорганизм, который естественным образом не обладает активностью фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, но тот, который был генетически модифицирован для экспрессии фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе. Он может также представлять собой микроорганизм, который естественным образом обладает активностью фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, и который был генетически модифицирован, например, трансформирован с помощью нуклеиновой кислоты, например, вектора, который кодирует фруктоза-1,6-бисфосфат фосфатазу, для того, чтобы повысить фосфокетолазную активность в указанном микроорганизме.
Микроорганизмы которые наследственно, то есть, естественным образом обладают активностью фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, являются хорошо известными в области техники и любой из них может использоваться в контексте настоящего изобретения.
Также является возможным в контексте настоящего изобретения, что микроорганизм представляет собой микроорганизм, который естественным образом не обладает фруктоза-1,6-бисфосфат фосфатазной активностью, в частности, при выращивании на глюкозе, но который является генетически модифицированным так, что способен экспрессировать фруктоза-1,6-бисфосфат фосфатазу, в частности, при выращивании на глюкозе. Этого можно достичь, например, путем мутации промотора гена, который кодирует Фруктоза-1,6-бисфосфат фосфатазу так, что этот ген не является более репрессированным, когда микроорганизм выращивается на глюкозе, или промотор может быть заменен другим промотором, например, конститутивным промотором, который не подвергается регуляции, когда микроорганизм выращивается на глюкозе.
Подобно этому, микроорганизм может также представлять собой микроорганизм, который естественным образом обладает активностью фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, но который является генетически модифицированным так, что улучшает/повышает активность фруктоза-1,6-бисфосфат фосфатазы, в частности, при выращивании на глюкозе, например, путем введения экзогенной нуклеотидной последовательности, которая кодирует фруктоза-1,6-бисфосфат фосфатазу.
Генетическая модификация микроорганизмов для экспрессии фермента, который представляет интерес, будет подробно описана ниже.
Фруктоза-1,6-бисфосфат фосфатаза в соответствии с данным изобретением может представлять собой существующую в природе фруктоза-1,6-бисфосфат фосфатазу, или она может являться фруктоза-1,6-бисфосфат фосфатазой, которая получена из существующей в природе фруктоза-1,6-бисфосфат фосфатазы, например, путем введения мутаций или других изменений, которые, например, изменяют или улучшают ферментативную активность, стабильность и т.д. Способы модификации и/или улучшения желательных ферментативных активностей белков являются хорошо известными квалифицированному специалисту в данной области техники и были описаны выше. Полученные варианты фруктоза-1,6-бисфосфат фосфатазы потом подвергают анализу на их свойства, например, ферментативную активность или регуляцию. Анализы для измерения активности фермента фруктоза-1,6-бисфосфат фосфатазы являются известными в уровне техники. В одном воплощении фруктоза-1,6-бисфосфат фосфатаза представляет собой фермент, который не подвергается регуляции с помощью ингибирования по типу обратной связи.
В предпочтительном воплощении рекомбинантный микроорганизм был генетически модифицирован для того, чтобы иметь повышенную активность фруктоза-1,6-бисфосфат фосфатазы. Этого можно достичь, например, путем трансформации микроорганизма с помощью нуклеиновой кислоты, которая кодирует фруктоза-1,6-бисфосфат фосфатазу. Подробное описание генетической модификации микроорганизмов будет приведено ниже.
В контексте настоящего изобретения "повышенная активность" означает, что экспрессия и/или активность фермента, в частности, фруктоза-1,6-бисфосфат фосфатазы, в генетически модифицированном микроорганизме при выращивании на глюкозе является, по крайней мере, на 10%, предпочтительно, по крайней мере, 20%, более предпочтительно, по крайней мере, 30% или 50%, даже более предпочтительно, по крайней мере, 70% или 80% и особенно предпочтительно, по крайней мере, на 90% или 100% выше, чем в соответствующем немодифицированном микроорганизме при выращивании на глюкозе. В более предпочтительных воплощениях повышение экспрессии и/или активности может составлять, по крайней мере, 150%, по крайней мере, 200% или, по крайней мере, 500%. В частности, в предпочтительных воплощениях экспрессия является, по крайней мере, в 10 раз, более предпочтительно, по крайней мере, в 100 раз и даже более предпочтительно, по крайней мере, в 1000 раз выше, чем в соответствующем немодифицированном микроорганизме, в частности, при выращивании на глюкозе.
Способы для измерения уровня экспрессия данного белка в клетке являются хорошо известными квалифицированному специалисту в данной области техники. В одном воплощении измерение уровня экспрессии осуществляют путем измерения количества соответствующего белка. Соответствующие способы являются хорошо известными квалифицированному специалисту в данной области техники и включают Вестерн-блоттинг, ELISA и т.д. В другом воплощении измерение уровня экспрессии осуществляют путем измерения количества соответствующей РНК. Соответствующие способы являются хорошо известными квалифицированному специалисту в данной области техники и включают, например, Нозерн-блоттинг.
Способы для измерения ферментативной активности фруктоза-1,6-бисфосфата являются известными в уровне техники.
В другом воплощении микроорганизм в соответствии с настоящим изобретением дополнительно характеризуется тем, что ЕМРР является дополнительно сниженным или инактивированным путем инактивации гена(ов), который(ые)е кодирует(ют) глицеральдегид 3-фосфат дегидрогеназу, или путем снижения активности глицеральдегид 3-фосфат дегидрогеназы по сравнению с немодифицированным микроорганизмом. Дополнительное снижение ЕМРР на расположенном ниже этапе путем снижения или инактивации глицеральдегид 3-фосфат дегидрогеназы обеспечивает то, что весь или почти весь глицеральдегид 3-фосфат, который может вырабатываться в микроорганизме, будет подвергаться гликолизу до ацетил-СоА, с помощью чего один атом углерода будет теряться при высвобождении CO2 на последнем этапе, который катализируется пируватдегидрогеназой. Таким образом, блокирование ЕМРР путем снижения или инактивации активности глицеральдегид 3-фосфат дегидрогеназы дополнительно обеспечивает то, что поток направляется к фосфокетолазе.
"Активность глицеральдегид 3-фосфат дегидрогеназы" означает ферментативную активность, которая превращает глицеральдегид 3-фосфат, фосфат и НАД+ в 3-фосфо-О-глицероил фосфат и НАДФ+Н+ (ЕС 1.2.1.12). Эта активность может быть измерена с помощью способов, известных в области техники, например, так, как описывается D'Alessio и др. (J. Biol. Chem. (1971) 246, 4326-4333).
Термин "микроорганизм, который характеризуется наличием дополнительно сниженного или инактивированного пути Эмбдена-Мейерхофа-Парнаса (ЕМРР) путем инактивации гена(ов), который(ые) кодирует(ют) глицеральдегид 3-фосфат дегидрогеназу, или путем снижения активности глицеральдегид 3-фосфат дегидрогеназы по сравнению с немодифицированным микроорганизмом" предпочтительно относится к микроорганизму, в котором инактивация гена(ов), который(ые) кодирует(ют) глицеральдегид 3-фосфат дегидрогеназу, или снижение активности глицеральдегид 3-фосфат дегидрогеназы по сравнению с немодифицированным микроорганизмом достигается путем генетической модификации микроорганизма, что ведет к указанной инактивации или снижению.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой рекомбинантный микроорганизм, в котором ЕМРР является дополнительно сниженным или инактивированным путем инактивации гена(ов), который(ые) кодирует(ют) глицеральдегид 3-фосфатдегидрогеназу, или путем снижения активности глицеральдегид 3-фосфатдегидрогеназы по сравнению с немодифицированным микроорганизмом. Инактивация гена(ов), который(ые) кодирует(ют) глицеральдегид 3-фосфатдегидрогеназу в контексте настоящего изобретения означает, что ген(ы), который(ые) кодирует(ют) глицеральдегид 3-фосфатдегидрогеназу, присутствующий(ие) в микроорганизме, являются инактивированными так, что они уже не экспрессируют и/или не приводят к синтезу функциональной глицеральдегид 3-фосфатдегидрогеназы. Инактивация может достигаться с помощью различных путей, известных в уровне техники. Инактивация может, например, достигаться путем нарушения гена(ов), который(ые) кодирует(ют) глицеральдегид 3-фосфатдегидрогеназу или путем делеции указанного(ых) гена(ов) посредством введения селективного маркера. Альтернативно, промотор гена, который кодирует глицеральдегид 3-фосфатдегидрогеназу, может подвергаться мутации таким образом, что ген(ы) уже не транскрибируются в мРНК. Другие пути для инактивации гена(ов), которые кодируют глицеральдегид 3-фосфатдегидрогеназу, известные в уровне техники, представляют собой: экспрессию полинуклеотида, который кодирует РНК, которая имеет нуклеотидную последовательность, комплементарную транскрипту гена(ов) глицеральдегид 3-фосфатдегидрогеназы так, что мРНК не может более транслироваться в белок, экспрессию полинуклеотида, который кодирует РНК, супрессирующую экспрессию указанного(ых) гена(ов), с помощью эффекта РНКи; экспрессию полинуклеотида, который кодирует РНК, имеющую активность специфического расщепления транскрипта указанного(ых) гена(ов); или экспрессию полинуклеотида, который кодирует РНК, супрессирующую экспрессию указанного(ых) гена(ов) с помощью эффекта косупрессии. Эти полинуклеотиды могут быть встроены в вектор, который может быть введен в микроорганизм путем трансформации, для достижения инактивация гена(ов), который(ые) кодирует(ют) глицеральдегид 3-фосфатдегидрогеназу.
Термин "инактивация" в контексте настоящего изобретения предпочтительно означает полную инактивация, то, есть то, что микроорганизм не демонстрирует активности глицеральдегид 3-фосфатдегидрогеназы. Это означает, в частности, что микроорганизм не демонстрирует активности глицеральдегид 3-фосфатдегидрогеназы, независимой от условий выращивания.
Предпочтительно, "инактивация" означает, что ген(ы), который(ые) кодирует(ют) глицеральдегид 3-фосфатдегидрогеназу, который(ые) присутствует(ют) в микроорганизме, являются генетически модифицированным(и) так, что предотвращает(ют) экспрессию фермента. Этого можно достичь, например, с помощью делеции гена или его частей, где делеция его частей предотвращает экспрессию фермента, или путем нарушения гена, либо в его кодирующем участке, либо в промоторном участке, где нарушение оказывает эффект, который заключается в том, что белок не экспрессируется или экспрессируется дисфункциональный белок.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой рекомбинантный микроорганизм, который имеет сниженный ЕМРР посредством снижения активности глицеральдегид 3-фосфатдегидрогеназы по сравнению с немодифицированным микроорганизмом. Предпочтительно, такое снижение достигается путем генетической модификации микроорганизма. Этого можно достичь, например, путем случайного мутагенеза или сайт-направленного мутагенеза промотора и/или фермента и последующей селекции промоторов и/или ферментов, которые обладают желательными свойствами, или с помощью комплементарных нуклеотидных последовательностей или эффекта РНКи, как описано выше. Подробное описание генетической модификации микроорганизмов будет приведено ниже.
В контексте настоящего изобретения "сниженная активность" означает, что экспрессия и/или активность фермента, в частности, глицеральдегид 3-фосфатдегидрогеназы, в генетически модифицированном микроорганизме является, по крайней мере, на 10%, предпочтительно, по крайней мере, 20%, более предпочтительно, по крайней мере, 30% или 50%, даже более предпочтительно, по крайней мере, 70% или 80% и особенно предпочтительно, по крайней мере, на 90% или 100% ниже, чем в соответствующем немодифицированном микроорганизме. Способы для измерения уровня экспрессия данного белка в клетке являются хорошо известными квалифицированному специалисту в данной области техники. Анализы для измерения сниженной активности фермента глицеральдегид 3-фосфатдегидрогеназы являются известными в уровне техники.
Термин "микроорганизм" в контексте настоящего изобретения относится к бактерии, а также к грибам, таким, как дрожжи, а также к водорослям и простейшим. В одном предпочтительном воплощении микроорганизм представляет собой бактерию. В принципе может использоваться любая бактерия. Предпочтительные бактерии, которые используются в процессе в соответствии с изобретением, представляет собой бактерии рода Bacillus, Clostridium, Corynebacterium, Pseudomonas, Zymomonas или Escherichia. В особенно предпочтительном воплощении бактерия относится к роду Escherichia и даже более предпочтительно к виду Escherichia coli. В другом предпочтительном воплощении бактерия относится к виду Pseudomonas putida или виду Zymomonas mobilis или виду Corynebacterium glutamicum.
В другом предпочтительном воплощении микроорганизм представляет собой грибы, более предпочтительно грибы рода Saccharomyces, Schizosaccharomyces, Aspergillus, Trichoderma, Kluyveromyces или Pichia и даже более предпочтительно такие виды, как Saccharomyces cerevisiae, Schizosaccharomyces pombe, Aspergillus niger, Trichoderma reesei, Kluyveromyces marxianus, Kluyveromyces lactis, Pichia pastoris, Pichia torula или Pichia utilis.
В более предпочтительном воплощении, где рекомбинантный микроорганизм представляет собой бактерию, в которой ген(ы) который(ые) кодирует(ют) PTS транспортер, зависимый от PEP, является(ются) инактивированным(и). В контексте настоящего изобретения инактивация означает, что ген(ы), который(ые) кодирует(ют) PTS транспортер, зависимый от PEP, который(ые) является(ются) присутствующим(и) в микроорганизме является (являются) инактивированным(и), так, что он(они) больше не экспрессируют и/или не приводят к синтезу функционального PTS транспортера, зависимого от PEP. Инактивация гена(ов), который(ые) кодирует(ют) PTS транспортер, зависимый от PEP, должна быть такой, что бактерия больше не является способной к транспорту глюкозы посредством PTS транспортера, зависимого от PEP.
PTS транспортеры, зависимые от PEP (например, из Е. coli, В. subtilis), являются известными в области техники. Пример для инактивации PTS транспортера, зависимого от PEP, представлен в разделе Примеры, приведенном ниже.
Инактивация может достигаться при использовании множества различных путей, известных в уровне техники. Инактивация может, например, достигаться путем нарушения гена(ов), который(ые) кодирует(ют) PTS транспортер, зависимый от PEP, или путем делеции указанного(ых) гена(ов) с помощью введения селективного маркера. Альтернативно, промотор гена(ов), который(ые) кодирует(ют) PTS транспортер, зависимый от PEP, может подвергаться мутации таким образом, что ген(ы) больше не транскрибируются в мРНК. Другие пути для инактивации гена(ов), который(ые) кодирует(ют) PTS транспортер, зависимый от PEP, известные в области техники, представляют собой: экспрессию полинуклеотида, который кодирует РНК, имеющую нуклеотидную последовательность, комплементарную транскрипту гена(ов) транспортера, зависимого от PEP, так, что мРНК не может более транслироваться в белок, экспрессию полинуклеотида, который кодирует РНК, супрессирующую экспрессию указанного(ых) гена(ов), с помощью эффекта РНКи; экспрессию полинуклеотида, который кодирует РНК, имеющую активность специфического расщепления транскрипта указанного(ых) гена(ов); или экспрессию полинуклеотида, который кодирует РНК, супрессирующую экспрессию указанного(ых) гена(ов) с помощью эффекта косупрессии. Эти полинуклеотиды могут быть встроены в вектор, который может быть введен в микроорганизм путем трансформации, для достижения инактивация гена(ов), который(ые) кодирует(ют) транспортер, зависимый от PEP.
Термин "рекомбинантный" означает, что микроорганизм в соответствии с настоящим изобретением является генетически модифицированным так, что содержит молекулу нуклеиновой кислоты, которая кодирует фермент, как определено выше, в отличие от микроорганизма дикого типа или немодифицированного микроорганизма, или что ген, который кодирует фермент, как определено выше, был делегирован по сравнению с микроорганизмом дикого типа или немодифицированным микроорганизмом.
Молекула нуклеиновой кислоты, которая кодирует фермент, как определено выше, может использоваться самостоятельно или как часть вектора.
Молекулы нуклеиновой кислоты могут дополнительно включать последовательности контроля экспрессия, оперативно связанные с полинуклеотидом, входящим в состав молекулы нуклеиновой кислоты. Термин "оперативно связанный" или "функционально связанный", как используется в описании к данной заявке, относится к связи между одной или более последовательностями контроля экспрессия и кодирующим участком в полинуклеотиде, который экспрессируется таким образом, что экспрессия достигается при условиях, совместимых с последовательностью контроля экспрессии.
Экспрессия включает транкрипцию гетерологичной последовательности ДНК, предпочтительно в способную к трансляции мРНК. Регуляторные элементы, которые обеспечивают экспрессию в грибах, а также бактериях, являются хорошо известными среднему специалисту в данной области техники. Они охватывают промоторы, энхансеры, сигналы терминации, нацеливающие сигналы и тому подобное. Примеры приводятся в данной заявке ниже в связи с объяснение, которое касается векторов.
Промоторы для использования в связи с молекулой нуклеиновой кислоты могут быть гомологичными или гетерологичными в отношении своего происхождения и/или в отношении гена, который подвергается экспрессии. Приемлемые промоторы, например, представляют собой промоторы, которые предоставляются для конститутивной экспрессии. Однако промоторы, которые являются инактивированными только в определенной точке времени, определяемой внешними факторами, также могут использоваться. Искусственные и/или химически индуцируемые промоторы могут использоваться в этом контексте.
Векторы могут дополнительно включать последовательности контроля экспрессии, оперативно связанные с указанными полинуклеотидами, которые содержаться в векторах. Такие последовательности контроля экспрессии могут быть приемлемыми для обеспечения транкрипция и синтеза способной к трансляции РНК в бактериях или грибах.
Кроме того, является возможным встраивать различные мутации в полинуклеотиды с помощью обычных для молекулярной биологии способов (смотри, например, Sambrook и Russell (2001), Molecular Cloning: A Laboratory Manual, CSH Press, Cold Spring Harbor, NY, USA), что приводит к синтезу полипептидов, которые возможно обладают модифицированными биологическими свойствами. Встраивание точечных мутаций является возможным в положениях, в которых модификация аминокислотной последовательности, например, оказывает влияние на биологическую активность или регуляцию полипептида.
Кроме того, могут быть получены мутанты, обладающие модифицированной активностью в отношении субстрата или продукта. Предпочтительно, такие мутанты демонстрируют повышенную активность. Альтернативно, могут быть получены мутанты, каталитическая активность которых устраняется без потери активности связывания субстрата.
Более того, введение мутаций в полинуклеотиды, которые кодируют фермент, как определено выше, позволяют снижать или повышать скорость генной экспрессии и/или активности ферментов, которые кодируются указанными полинуклеотидами.
Для генетически модифицированных бактерий или грибов полинуклеотиды, которые кодируют фермент, как определено выше, или части таких молекул, могут быть введены в плазмиды, что позволяет осуществлять мутагенез или модификацию последовательностей путем рекомбинации последовательностей ДНК. Стандартные способы (смотри Sambrook и Russell (2001), Molecular Cloning: A Laboratory Manual, CSH Press, Cold Spring Harbor, NY, USA) позволяют осуществлять замену оснований или прибавлять природные синтетические последовательности. ДНК фрагменты могут быть соединены друг с другом путем применения адаптеров и линкеров к этим фрагментам. Кроме того, могут использоваться средства инжиниринга, которые обеспечивают приемлемые сайты рестрикции или удаление избыточной ДНК или рестрикционных сайтов. В тех случаях, когда инсерции, делеции или замены являются возможными, могут использоваться in vitro мутагенез, "восстановление примера", рестрикция или лигирование. В общем случае, анализ последовательностей, рестрикционный анализ и другие способы биохимии и молекулярной биологии осуществляются в качестве аналитических способов.
Таким образом, в соответствии с настоящим изобретением рекомбинантный микроорганизм может быть получен путем генетической модификации грибов или бактерий, которая включает введение описанных выше полинуклеотидов, молекул нуклеиновой кислоты или векторов в грибы или бактерии.
Изобретение относится к рекомбинантным микроорганизмам, в частности, к бактериям и грибам, генетически модифицированным с помощью описанных выше полинуклеотидов, молекул нуклеиновой кислоты или векторов, или получаемых с помощью описанного выше способа для получения генетически модифицированных бактерий или грибов, и к клеткам, которые имеют происхождение от таких трансформированных бактерий или грибов и содержат полинуклеотид, молекулу нуклеиновой кислоты или вектор, как определено выше. В предпочтительном воплощении клетка-хозяин является генетически модифицированной таким образом, что она содержит полинуклеотид, стабильно интегрированный в геном.
Полинуклеотид экспрессируется так, что это приводит к получению полипептида, который обладает активностями, описанными выше. Обзор различных систем экспрессии, например, содержится в Methods in Enzymology 153 (1987), 385-516, in Bitter и др. (Methods in Enzymology 153 (1987), 516-544) и у Sawers и др. (Applied Microbiology and Biotechnology 46 (1996), 1-9), Billman-Jacobe (Current Opinion in Biotechnology 7 (1996), 500-4), Hockney (Trends in Biotechnology 12 (1994), 456-463), Griffiths и др., (Methods in Molecular Biology 75 (1997), 427-440). Обзор экспрессионных систем грибов, например, является представленным Hensing и др. (Antonie van Leuwenhoek 67 (1995), 261-279), Bussineau и др. (Developments in Biological Standardization 83 (1994), 13-19), Gellissen и др. (Antonie van Leuwenhoek 62 (1992), 79-93, Fleer (Current Opinion in Biotechnology 3 (1992), 486-496), Vedvick (Current Opinion in Biotechnology 2 (1991), 742-745) и Buckholz (Bionechnology 9 (1991), 1067-1072).
Экспрессия векторов была широко описана в литературе. Как правило векторы содержат не только ген селективного маркера и сайт начала репликации, которые обеспечивают репликацию в выбранной клетке-хозяине, а также бактериальный или вирусный промотор, и в большинстве случаев сигнал терминации транскрипции. Между промотором и сигналом терминации в общем случае существует, по крайней мере, один сайт рестрикции или полилинкер, который позволяет осуществлять инсерцию кодирующей последовательности ДНК. ДНК последовательность, которая в природе осуществляет со-контроль транкрипции соответствующего гена, может использоваться в качестве промоторной последовательности, если она является активной в выбранном организме хозяина. Однако эта последовательность может быть заменена на другие промоторные последовательности. Является возможным использовать промоторы, которые обеспечивают конститутивную экспрессию гена, и индуцибельные промоторы, которые позволяют осуществлять спланированный контроль экспрессии гена. Бактериальные и вирусные промоторные последовательности, обладающие этими свойствами, подробно описываются в литературе. Регуляторные последовательности для экспрессии в микроорганизмах (например, Е. coli, S. cerevisiae) являются в достаточной мере описанными в литературе. Промоторы, которые позволяют осуществлять особенно высокую экспрессию размещенных ниже последовательностей, представляют собой, например, Т7 промотор (Studier и др., Methods in Enzymology 185 (1990), 60-89), lacUV5, trp, trp-lacUVS (DeBoer и др., в Rodriguez и Chamberlin (Eds), Promoter, Structure и Function; Praeger, New York, (1982), 462-481; DeBoer и др., Proc. Natl. Acad. Sci. USA (1983), 21-25), Ip1, rac (Boros и др., Gene 42 (1986), 97-100). Индуцибельные промоторы предпочтительно используются для синтеза полипептидов. Эти промоторы часто приводят к более высоким выходам полипептидов, чем конститутивные промоторы. Для того чтобы получить оптимальное количество полипептида, часто используют двухэтапный процесс.Сначала хозяйские клетки культивируют при оптимальных условиях до относительно высокой плотности клеток. На втором этапе индуцируют транкрипцию в зависимости от типа используемого промотора. В этой связи tac промотор, который может индуцироваться лактозой или IPTG (=изопропил-β-D-тиогалактопиранозид), является особенно приемлемым (deBoer и др., Proc. Natl. Acad. Sci. USA 80 (1983), 21-25). Сигналы терминации транкрипции также являются описанными в литературе.
Трансформация хозяйской клетки с помощью полинуклеотида или вектора в соответствии с изобретением может осуществляться с помощью стандартных способов, таких, которые, например, описываются у Sambrook и Russell (2001), Molecular Cloning: A Laboratory Manual, CSH Press, Cold Spring Harbor, NY, USA; Methods in Yeast Genetics, A Laboratory Course Manual, Cold Spring Harbor Laboratory Press, 1990. Клетка-хозяин культивируется в питательной среде, которая соответствует требованиям конкретной клетки-хозяина, используемой, в частности, в отношении значения рН, температуры, концентрации соли, аэрации, антибиотиков, витаминов, микроэлементов и т.д.
В другом аспекте в соответствии с настоящим изобретением рекомбинантный микроорганизм дополнительно характеризуется тем, что является способным к превращению ацетил-СоА в ацетон. Способы для обеспечения такого рекомбинантного микроорганизма являются, например, раскрытыми в ЕР 2295593. Термин "который является способным к превращению ацетил-СоА в ацетон" в контексте настоящего изобретения означает, что организм/микроорганизм имеет способность вырабатывать ацетон в клетке благодаря присутствию ферментов, обеспечивающих ферментативные активности, которые позволяют осуществлять продукцию ацетона из ацетил-СоА.
Ацетон вырабатывается определенными микроорганизмами, такими как Clostridium acetobutylicum, Clostridium beijerinckii, Clostridium cellulolyticum, Bacillus polymyxa и Pseudomonas putida. Синтез ацетона наилучшим образом характеризуется у Clostridium acetobutylicum. Он начинается с реакции (реакционный этап 1), в которой две молекулы ацетил-СоА конденсируются с образованием ацетоацетил-СоА. Эта реакция катализируется с помощью ацетил-СоА ацетилтрансферазы (ЕС 2.3.1.9). Ацетоацетил-СоА потом превращается в ацетоацетат с помощью реакции с уксусной кислотой или масляной кислотой, что также приводит к продукции ацетил-СоА или бутирил-СоА (реакционный этап 2). Эта реакция катализируется с помощью, например, ацетоацетилСоА трансферазы (ЕС 2.8.3.8). АцетоацетилСоА трансфераза является известной из различных организмов, например, из Е. coli, где она кодируется, например, с помощью гена atoAD, или из Clostridium acetobutylicum, где она кодируется, например, с помощью гена ctfAB. Однако другие ферменты также могут катализировать эту реакцию, например, 3-оксокислота СоА трансферазы (ЕС 2.8.3.5) или сукцинат СоАлигазы (ЕС 6.2.1.5).
В завершение, ацетоацетат превращается в ацетон с помощью этапа декарбоксилирования (реакционный этап 3), который катализируется с помощью ацетоацетат декарбоксилазы (ЕС 4.1.1.4).
Описанные выше реакционные этапы 1 и 2 и ферменты, которые катализируют их являются характерными не только для синтеза ацетона, но и могут быть обнаружены в различных организмах. В противовес этому, реакционный этап 3, который катализируется с помощью ацетоацетат декарбоксилазы (ЕС 4.1.1.4), обнаруживается только в тех организмах, которые являются способными к выработке ацетона.
В предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой микроорганизм, который естественным образом обладает способностью продуцировать ацетон. Таким образом, предпочтительные микроорганизмы относятся к роду Clostridium, Bacillus или Pseudomonas, более предпочтительно к видам Clostridium acetobutylicum, Clostridium beijerinckii, Clostridium cellulolyticum, Bacillus polymyxa или Pseudomonas putida.
В другом предпочтительном воплощении рекомбинантный микроорганизм в соответствии с настоящим изобретением представляет собой микроорганизм, который имеет происхождение от организма/микроорганизма, который в природе не продуцирует ацетон, но который был генетически модифицирован для того, чтобы вырабатывать ацетон, то есть, путем введения гена(ов), необходимого(ых) для того, чтобы позволить осуществлять продукцию ацетона в микроорганизме. В принципе любой микроорганизм может быть генетически модифицирован таким образом. Ферменты, которые отвечают за синтез ацетона, были описаны выше. Гены, которые кодируют соответствующие ферменты, являются хорошо известными в области техники и могут использоваться для генетической модификации данного микроорганизма для того, чтобы получать ацетон. Как описано выше, реакционные этапы 1 и 2 синтеза ацетона существуют в природе у большинства микроорганизмов. Однако реакционный этап 3 является отличительным свойством и ключевой характеристикой для синтеза ацетона. Таким образом, в предпочтительном воплощении генетически модифицированный микроорганизм, который получен из микроорганизма, который в природе не вырабатывает ацетон, является модифицированным так, что содержит нуклеотидную последовательность, которая кодирует фермент, катализирующий превращение ацетоацетата в ацетон путем декарбоксилирования, например, с помощью ацетоацетат декарбоксилазы (ЕС 4.1.1.4). Нуклеотидные последовательности из нескольких организмов, которые кодируют этот фермент, являются известными в области техники, например, adc ген из Clostridium acetobutylicum (номера доступа Uniprot Р23670 и Р23673), Clostridium beijerinckii (Clostridium MP; Q9RPK1), Clostridium pasteurianum (номер доступа Uniprot P81336), Bradyrhizobium sp.(штамм BTAi1/ATCC BAA-1182; номер доступа Uniprot A5EBU7), Burkholderia mallei (ATCC 10399 A9LBSO), Burkholderia mallei (номер доступа Uniprot A3MAE3), Burkholderia mallei FMH A5XJB2, Burkholderia cenocepacia (номер доступа Uniprot AOB471), Burkholderia ambifaria (номер доступа Uniprot QOb5P1), Burkholderia phytofirmans (номер доступа Uniprot B2T319), Burkholderia spec. (номер доступа Uniprot Q38ZUO), Clostridium botulinum (номер доступа Uniprot B2TLN8), Ralstonia pickettii (номер доступа Uniprot B2UIG7), Streptomyces nogalater (номер доступа Uniprot Q9EYI7), Streptomyces avermitilis (номер доступа Uniprot Q82NF4), Legionella pneumophila (номер доступа Uniprot Q5ZXQ9), Lactobacillus salivarius (номер доступа Uniprot Q1WVG5), Rhodococcus spec. (номер доступа Uniprot QOS7W4), Lactobacillus plantarum (номер доступа Uniprot Q890GO), Rhizobium leguminosarum (номер доступа Uniprot Q1M911), Lactobacillus casei (номер доступа Uniprot Q03B66), Francisella tularensis (номер доступа Uniprot QOBLC9), Saccharopolyspora erythreae (номер доступа Uniprot A4FKR9), Korarchaeum cryptofilum (номер доступа Uniprot B1L3N6), Bacillus amyloliquefaciens (номер доступа Uniprot A7Z8K8), Cochliobolus heterostrophus (номер доступа Uniprot Q8NJQ3), Sulfolobus islandicus (номер доступа Uniprot C3ML22) и Francisella tularensis subsp.holarctica (штамм OSU18).
Более предпочтительно, когда микроорганизм является генетически модифицированным, с тем, чтобы быть трансформированным с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 2 синтеза ацетона, то есть, превращение ацетоацетил СоА в ацетоацетат.
Даже более предпочтительно, когда микроорганизм является генетически модифицированным с тем, чтобы быть трансформированным с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 1 синтеза ацетона, то есть, конденсацию двух молекул ацетил СоА в ацетоацетил СоА.
В особенно предпочтительном воплощении микроорганизм является генетически модифицированным с тем, чтобы быть трансформированным с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 1 синтеза ацетона, и с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 2 синтеза ацетона, или с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 1 синтеза ацетона, и с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 3 синтеза ацетона, или с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 2 синтеза ацетона, и с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 3 синтеза ацетона, или с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 1 синтеза ацетона, и с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 2 синтеза ацетона, и с помощью молекулы нуклеиновой кислоты, которая кодирует фермент, способный катализировать упомянутый выше реакционный этап 3 синтеза ацетона.
Способы получения упомянутых выше генетически модифицированных микроорганизмов являются хорошо известными в области техники. Таким образом, в общем случае, микроорганизм является трансформированным с помощью конструкции ДНК, которая позволяет осуществлять экспрессию соответствующего фермента в микроорганизме. Такая конструкция обычно включает кодирующую последовательность, которая представляет интерес, связанную с регуляторными последовательностями, которые позволяют осуществлять транкрипцию и трансляцию в соответствующей хозяйской клетке, например, промотор и энхансер, и/или терминатор транкрипции и/или сайт связывания рибосомы и т.д. В уровне техники описаны микроорганизмы, которые были генетически модифицированы так, чтобы быть способными вырабатывать ацетон. В частности, гены, например, из Clostridium acetobutylicum, были введены в Е. coli, что позволяло, таким образом, осуществлять синтез ацетона в Е. coli, бактерии, которая естественным образом не продуцирует ацетон (Bermejo и др., Appl. Environ. Microbiol. 64 (1998); 1079-1085; Hanai и др., Appl. Environ. Microbiol. 73 (2007), 7814-7818). В частности, Hanai и др. (в процитированном месте) показывает, что является достаточным ввести последовательность нуклеиновой кислоты, которая кодирует ацетоацетатдекарбоксилазу (например, такую из Clostridium acetobutylicum) для того, чтобы достичь продукции ацетона в Е. coli, что свидетельствует о том что эндогенные ферменты в Е. coli, которые катализируют упомянутые выше реакционные этапы 1 и 2 (то есть, экспрессию продуктов генов atoB и atoAD Е. coli), являются достаточными для обеспечения субстрата для продукции ацетона.
В другом аспекте в соответствии с настоящим изобретением рекомбинантный микроорганизм дополнительно характеризуется тем, что является способным к превращению ацетил-СоА в ацетон и превращению ацетона в изобутен. Способы обеспечения такого рекомбинантного микроорганизма являются например, раскрытыми в ЕР-А 2295593 (ЕР 09170312), WO 2010/001078 и ЕР 10188001.
В другом аспекте в соответствии с настоящим изобретением рекомбинантный микроорганизм характеризуется тем, что является способным к превращению ацетил-СоА в ацетон и превращению ацетона в пропен. Способы получения такого рекомбинантного микроорганизма представляют собой, например, такие, которые раскрыты у Hanai и др., Appl. Environ. Microbiol. 73 (2007), 7814-7818.
Специалист в данной области техники сможет признать, что дополнительные генетические модификации микроорганизмов в соответствии с настоящим изобретением могут приводить к улучшениям в отношении эффективности, с которой микроорганизмы в соответствии с настоящим изобретением превращают источник сырья в продукт. Например, природные микроорганизмы обычно вырабатывают продукты, такие как формиат, ацетат, лактат, сукцинат, этанол, глицерол, 2,3-бутандиол, метилглиоксал и водород; которые все могут быть вредными для продукции, например, ацетона, изобутена или пропена из сахаров. Устранение или существенное снижение таких нежелательных побочных продуктов может достигаться путем устранения или уменьшения активности ферментов, которые приводят к их выработке. Такие активности включают, но без ограничения таковыми, группу, состоящую из:
- ацетил-СоА+формиат=СоА+пируват (например, такой, который катализируется с помощью формиат С-ацетилтрансферазы, также является известным как пируват формиатлиаза (ЕС 2.3.1.54); для Е. coli-pfIB, NCBI-GenelD: 945514);
- АТФ+ацетат=АДФ+ацетилфосфат (например, такой, который катализируется с помощью ацетаткиназы (ЕС 2.7.2.1); для Е. coli-ackA, NCBI-GenelD: 946775);
- (Р)-лактат+НАД*=пируват+НАДФ+Н* (например, такой, который катализируется с помощью L-лактатдегидрогеназы (ЕС 1.1.1.28); для Е. coli - IdhA, NCBI-GenelD: 946315);
- фосфат+оксалоацетат=фосфоенолпируват+НСО3- (например, такой, который катализируется с помощью фосфоенолпируват карбоксилазы (ЕС 4.1.1.31); для Е. coli - ррс, NCBI-GenelD: 948457);
- АТФ+оксалоацетат=АДФ+фосфоенолпируват+СО2 (например, такой, который катализируется с помощью фосфоенолпируват карбоксикиназы (АТФ) (ЕС 4.1.1.49); для Е. coli - pck, NCBI-GenelD: 945667);
- сукцинат+акцептор=фумарат+восстановленный акцептор (например, такой, который катализируется с помощью сукцинатдегидрогеназы (ЕС 1.3.99.1); для Е. coli -, которая влючает frdA и frdB, NCBI-GenelD: 948667 и 948666, соответственно);
- 2-оксокарбоксилат (например, пируват)=альдегид (например, ацетальдегид+СО2 (например, такой, который катализируется с помощью пируватдекарбоксилазы (ЕС 4.1.1.1));
- ацетальдегид+СоА+НАД+=ацетил-СоА+НАДФ+H+ (например, такой, который катализируется с помощью ацетальдегиддегидрогеназы (ацетилирование) (ЕС 1.2.1.10); для Е. coli - adhE, NCBI-GenelD: 945837);
- sn-глицерин 3-фосфат+НАД(Ф)+=глицерон фосфат+НАД(Ф)Н+H+ (например, такой, который катализируется с помощью глицерин-3-фосфат дегидрогеназы [НАД(Р)+] (ЕС 1.1.1.94); для E. coli - gpsA, NCBI-GenelD: 948125);
- 2 пируват=2-ацетолактат+СО2 (например, такой, который катализируется с помощью ацетолактатсинтазы (ЕС 2.2.1.6); для E. coli - ilvH и ilvl, NCBI-GenelD: 947267 и 948793, соответственно);
- глицерон фосфат=метилглиоксал+фосфат (например, такой, который катализируется с помощью метилглиоксалсинтазы (ЕС 4.2.3.3); для Е. coli - mgsA, NCBI-GenelD: 945574); и
- формиат+H+=CO2+Н2 (например, такой, который катализируется с помощью формиатгидрогеназы (ЕС 1.2.1.2 вместе с ЕС 1.12.1.2); для Е. coli - fdhF (EC 1.2.1.2), NCBI-GenelD: 948584).
Таким образом, в предпочтительном воплощении микроорганизм в соответствии с изобретение характеризуется тем, что одна или более из приведенных выше ферментативных активностей устраняются или снижаются.
Специалист в данной области техники сможет признать, что генетические модификации регуляторных элементов в микроорганизмах в соответствии с настоящим изобретением могут приводить к улучшениям в отношении эффективности, с которой микроорганизмы в соответствии с настоящим изобретением превращают источник сырья в продукт. В отношении Е. coli такие генетические модификации включают, но без ограничения таковыми, группу, которая состоит из:
- делеции fnr гена (NCBI-GenelD: 945908), глобального регулятора анаэробного роста;
- делеции rpoS гена (NCBI-GenelD: 947210), РНК полимеразы, сигма S (сигма 38) фактора.
Таким образом, в другом предпочтительном воплощении микроорганизм в соответствии с изобретение демонстрирует, по крайней мере, одну из этих делеции.
Дополнительный аспект в соответствии с настоящим изобретением заключается в применении рекомбинантного микроорганизма в соответствии с настоящим изобретением для превращения глюкозы в ацетил-СоА. Ацетил СоА (также является известным как ацетил коэнзим А) по химической структуре представляет собой тиоэстер между коэнзимом А (тиол) и уксусной кислотой и является молекулой важного предшественника для продукции полезных метаболитов. Ацетил-СоА может в дальнейшем превращаться с помощью рекомбинантного микроорганизма в полезные метаболиты, такие как L-глутаминовая кислота, L-глутамин, L-пролин, L-аргинин, L-лейцин, сукцинат и полигидроксибутират.
Другой аспект в соответствии с настоящим изобретением представляет собой применение рекомбинантного микроорганизма в соответствии с настоящим изобретением, который является способным к превращению ацетил-СоА в ацетон для превращения глюкозы в ацетон.
Дополнительный аспект в соответствии с настоящим изобретением представляет собой применение рекомбинантного микроорганизма в соответствии с настоящим изобретением, который является способным к превращению ацетил-СоА в ацетон и ацетона в изобутен для превращения глюкозы в изобутен.
Еще один аспект в соответствии с настоящим изобретением представляет собой применение рекомбинантного микроорганизма в соответствии с настоящим изобретением, который является способным к превращению ацетил-СоА в ацетон и ацетона в пропен для превращения глюкозы в пропен.
В соответствии с этим настоящее изобретение также относится к способу производства ацетона и/или изобутена, и/или пропена из глюкозы, в котором упомянутый выше рекомбинантный микроорганизм культивируют при условиях, которые позволяют осуществлять продукцию ацетона и/или изобутена, и/или пропена и в котором ацетон и/или изобутен, и/или пропен изолируют. Микроорганизмы культивируют при приемлемых условиях культивирования, которые позволяют осуществлять ферментативную(ые) реакцию(и). Специфические условия культивирования зависят от используемого микроорганизма, но являются хорошо известными квалифицированному специалисту в данной области техники. Условия культивирования в общем случае выбирают таким образом, что они позволяют осуществлять экспрессию генов, которые кодируют ферменты для соответствующих реакций. Различные способы являются известными квалифицированному специалисту в данной области техники для того, что улучшить и усовершенствовать экспрессию определенных генов на определенных этапах культивирования, и представляют собой, например, индукцию генной экспрессии с помощью химических индукторов или сдвига температуры.
В другом предпочтительном воплощении способ в соответствии с изобретением также включает этап сбора газообразных продуктов, в частности, изобутена или пропена, удаление газа из реакции, то есть, восстановление продуктов, которые дегазированы, например, из культуры. Таким образом, в предпочтительном воплощении способ осуществляется в присутствии системы для сбора изобутена или пропена в газообразной форме в процессе реакции.
В сущности, короткие алкены, такие как изобутен и пропен, приобретают газообразное состояние при комнатной температуры и атмосферном давлении. Способ в соответствии с изобретение, таким образом, не требует экстракции продукта жидкой культуральной среды, этап, который является весьма дорогим при осуществлении в промышленном масштабе. Откачивание и хранение газообразных углеводородов и их возможное последующее физическое отделение и химическое превращение может осуществляться в соответствии с любым способом, известным специалисту в данной области техники.
Настоящее изобретение дополнительно описывается со ссылкой на следующие неограничивающие фигуры и примеры.
Фигура 1 показывает две схемы для продукции ацетил-СоА из глюкоза-6-фосфата с помощью фосфокетолазного пути при использовании либо одной, либо обоих активностей фосфокетолазы ЕС 4.1.2.9 и ЕС 4.1.2.22.
ПРИМЕРЫ
ОБЩИЕ СПОСОБЫ И МАТЕРИАЛЫ
Процедуры для лигирования и трансформации являются хорошо известными в области техники. Методики, приемлемые для использования в следующих примерах, могут быть найдены у Sambrook J., и др., Molecular Cloning: A Laboratory Manual, 2-ое изд., Cold Spring Harbor, N.Y., 1989, и Sambrook J., выше.
Материалы и способы, приемлемые для поддержания и роста бактериальных культур, являются хорошо известными в области техники. Методики, приемлемые для использования в следующих примерах, могут быть найдены в Manual of Methods for General Bacteriology (Philipp Gerhardt, R.G.E. Murray, Ralph N. Costilow, Eugene W. Nester, Willis A. Wood, Noel R. Krieg и G. Briggs Philips, eds).
Все реагенты и материалы, используемые для роста и поддержания бактериальных клеток, являются полученными от Sigma-Aldrich Company (St. Louis, МО), если не указано иное.
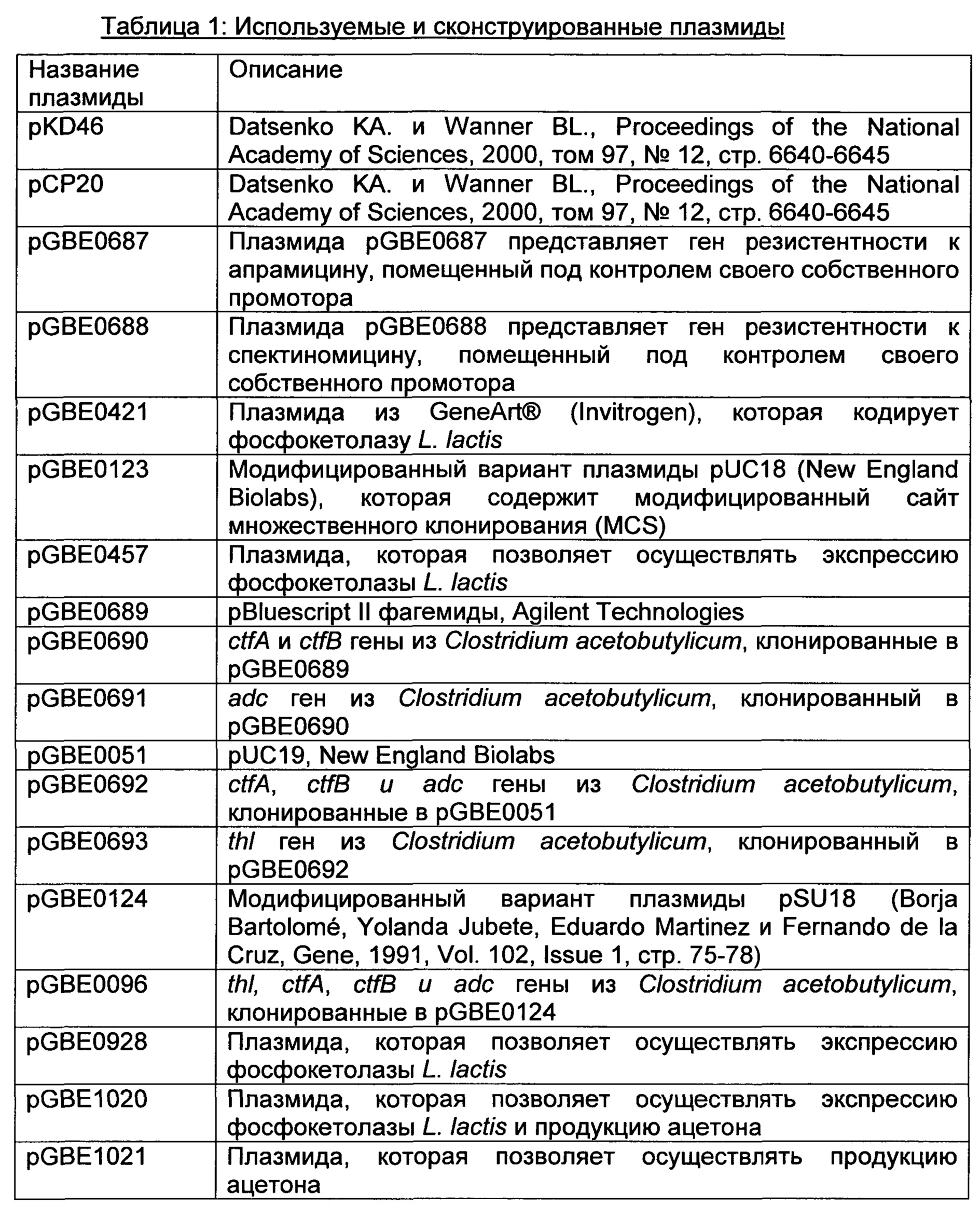
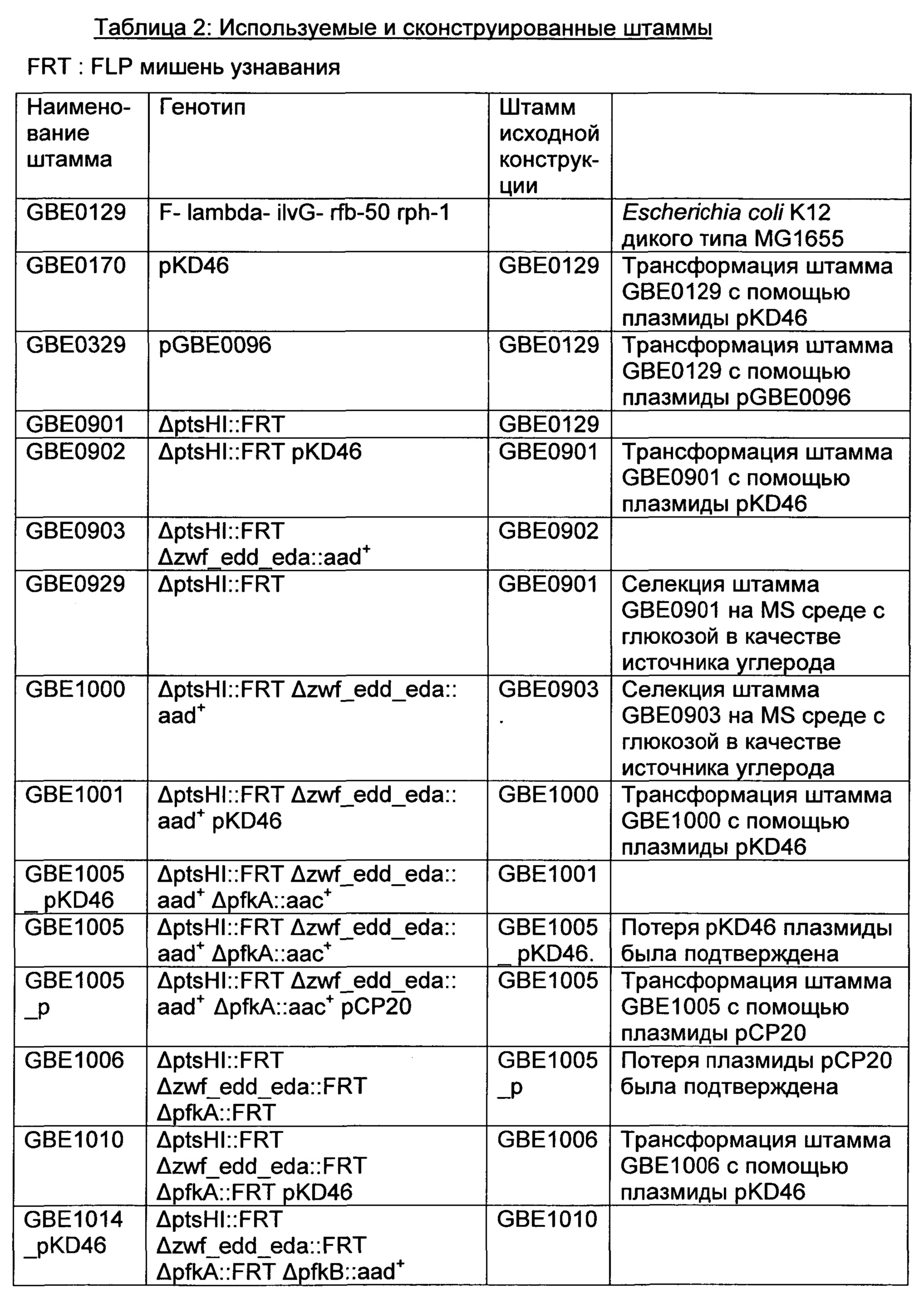
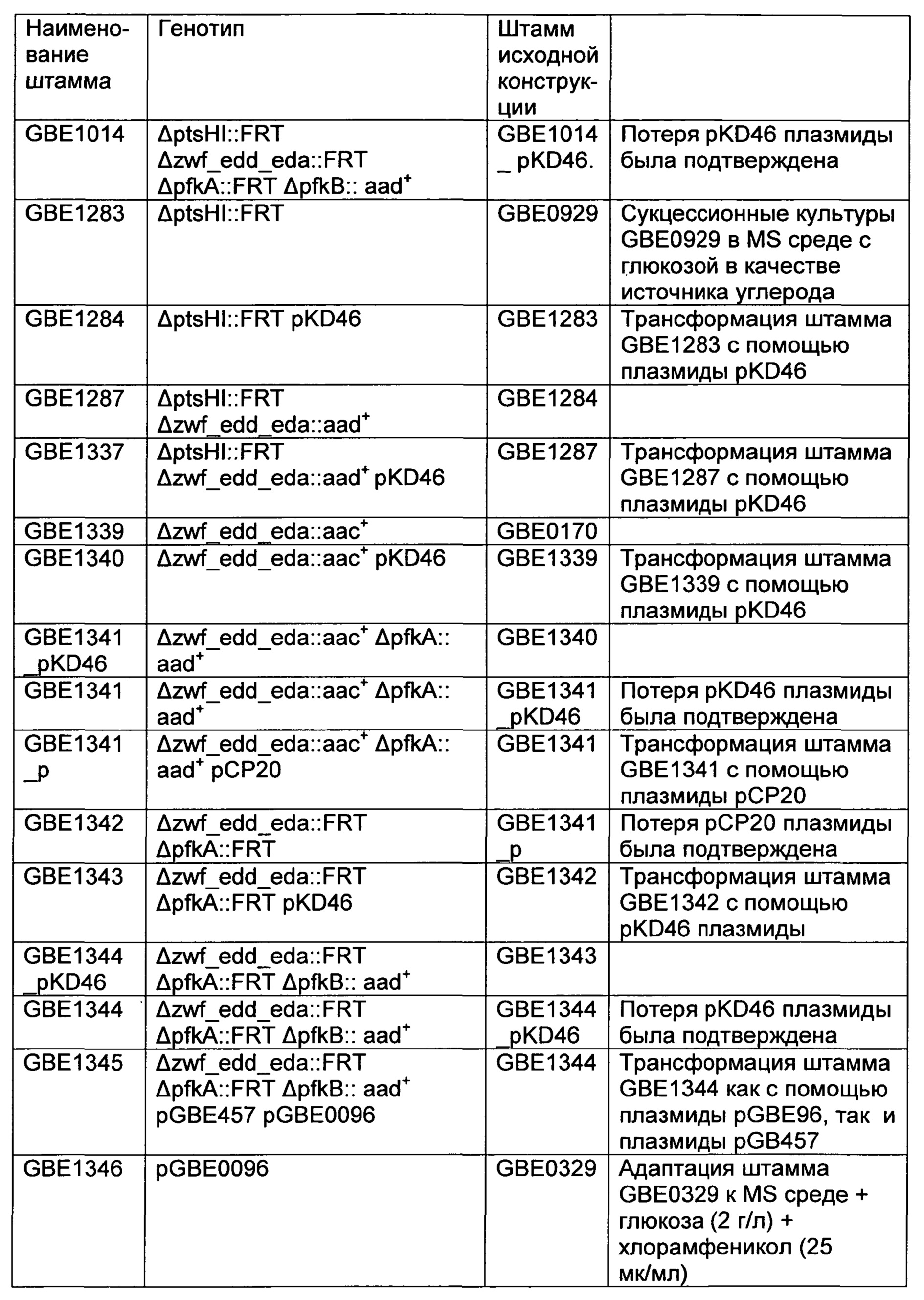
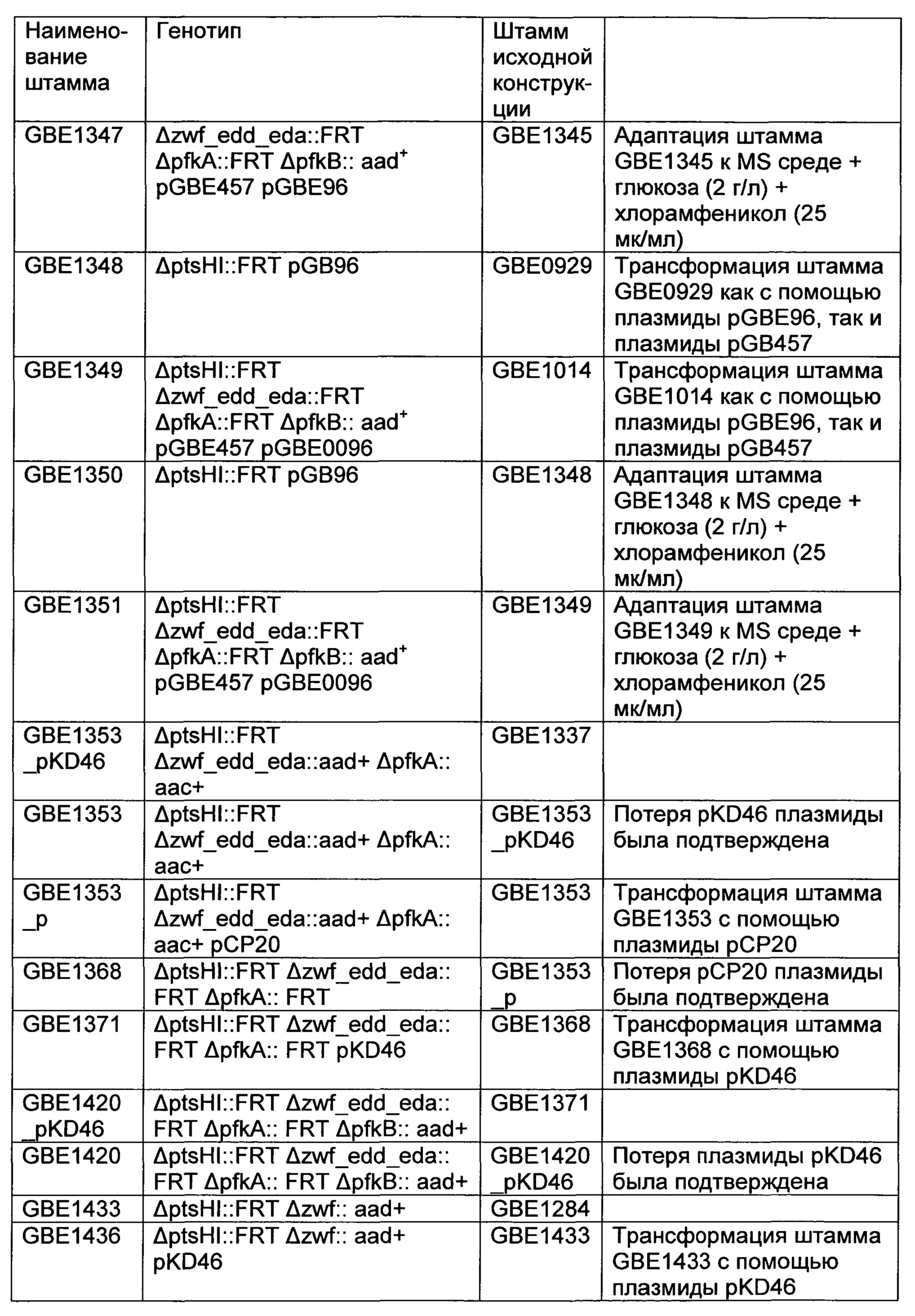
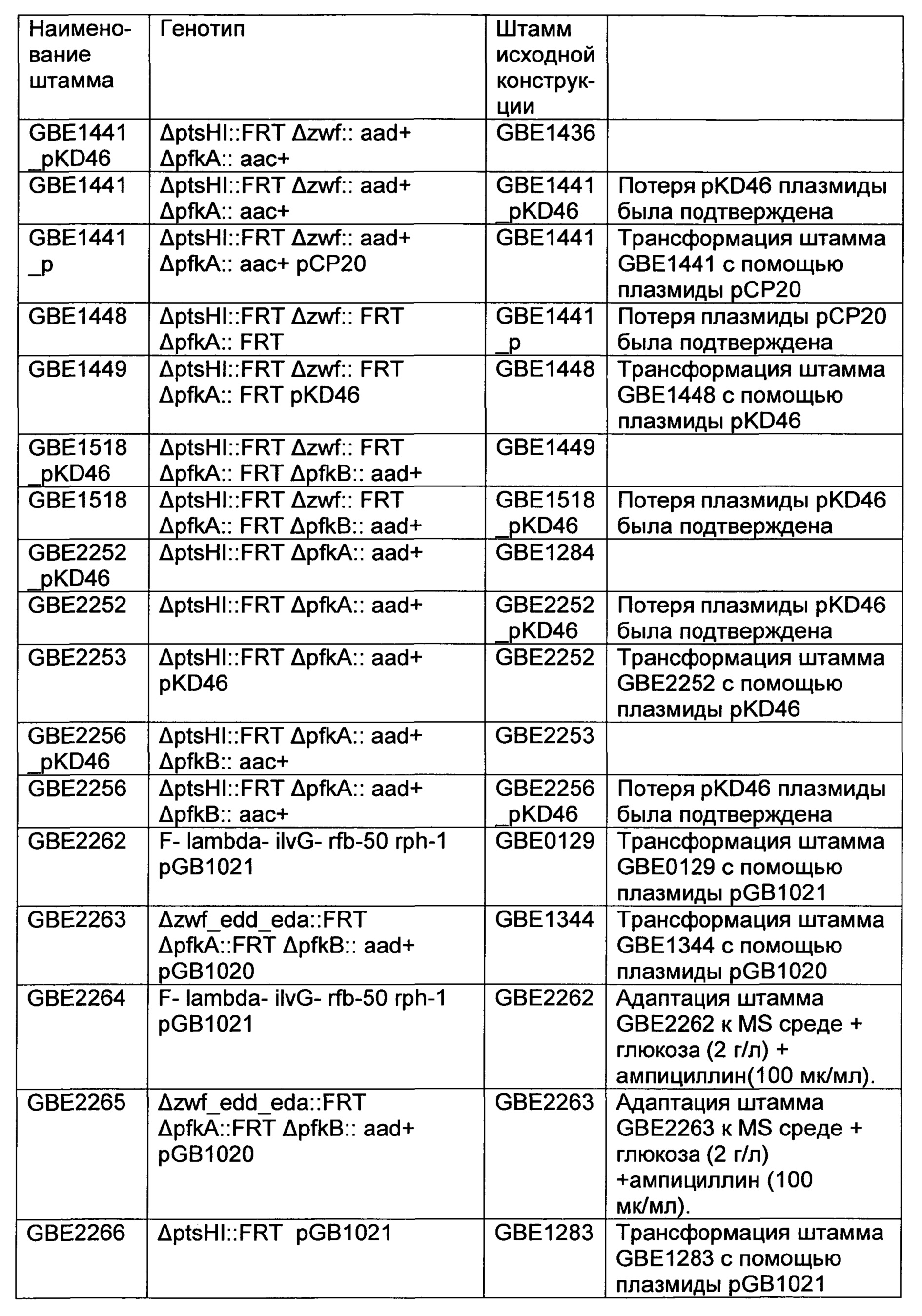
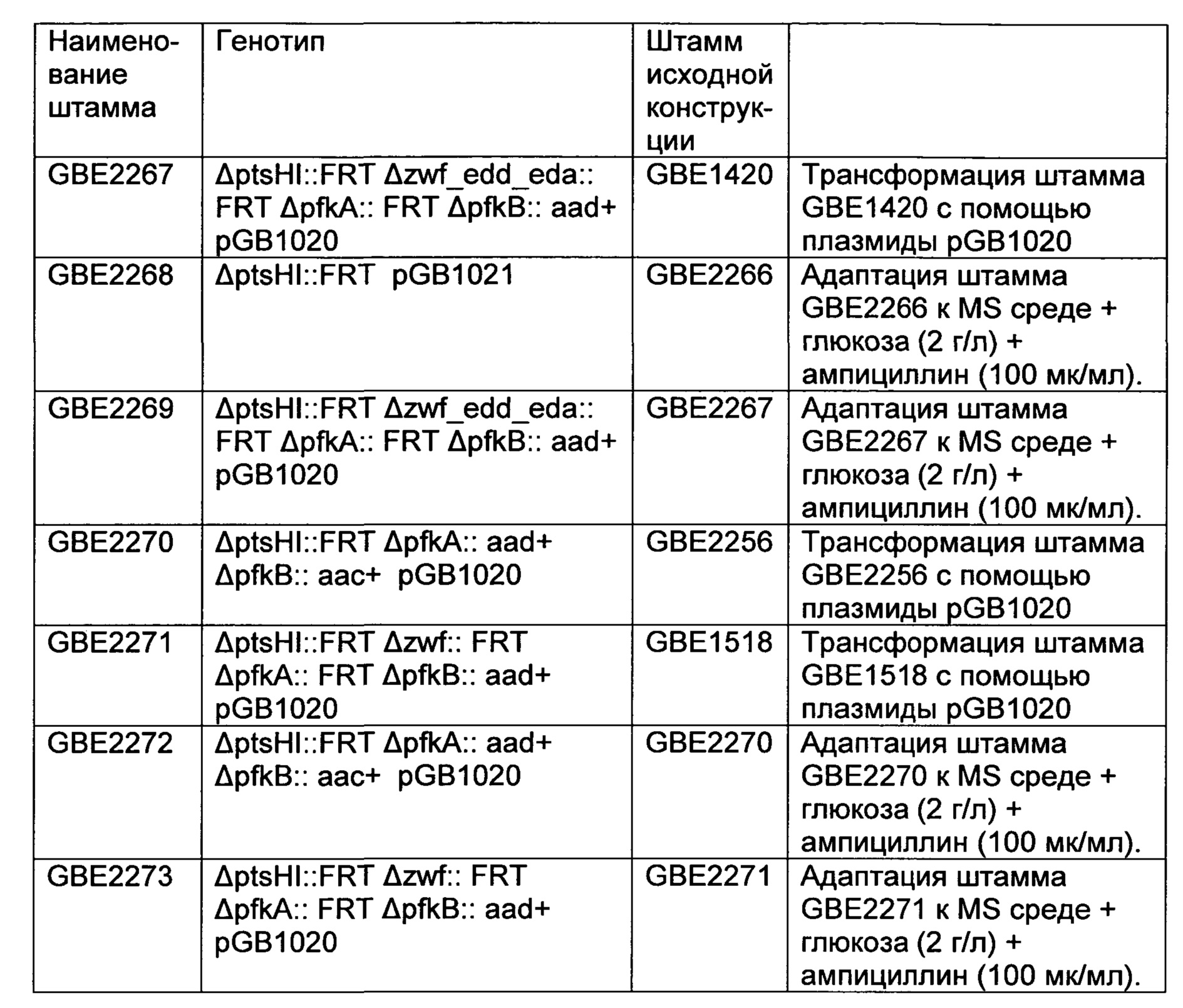
Таблица 3: Последовательности бактериальных хромосомных участков, используемые гены, плазмидные участки.


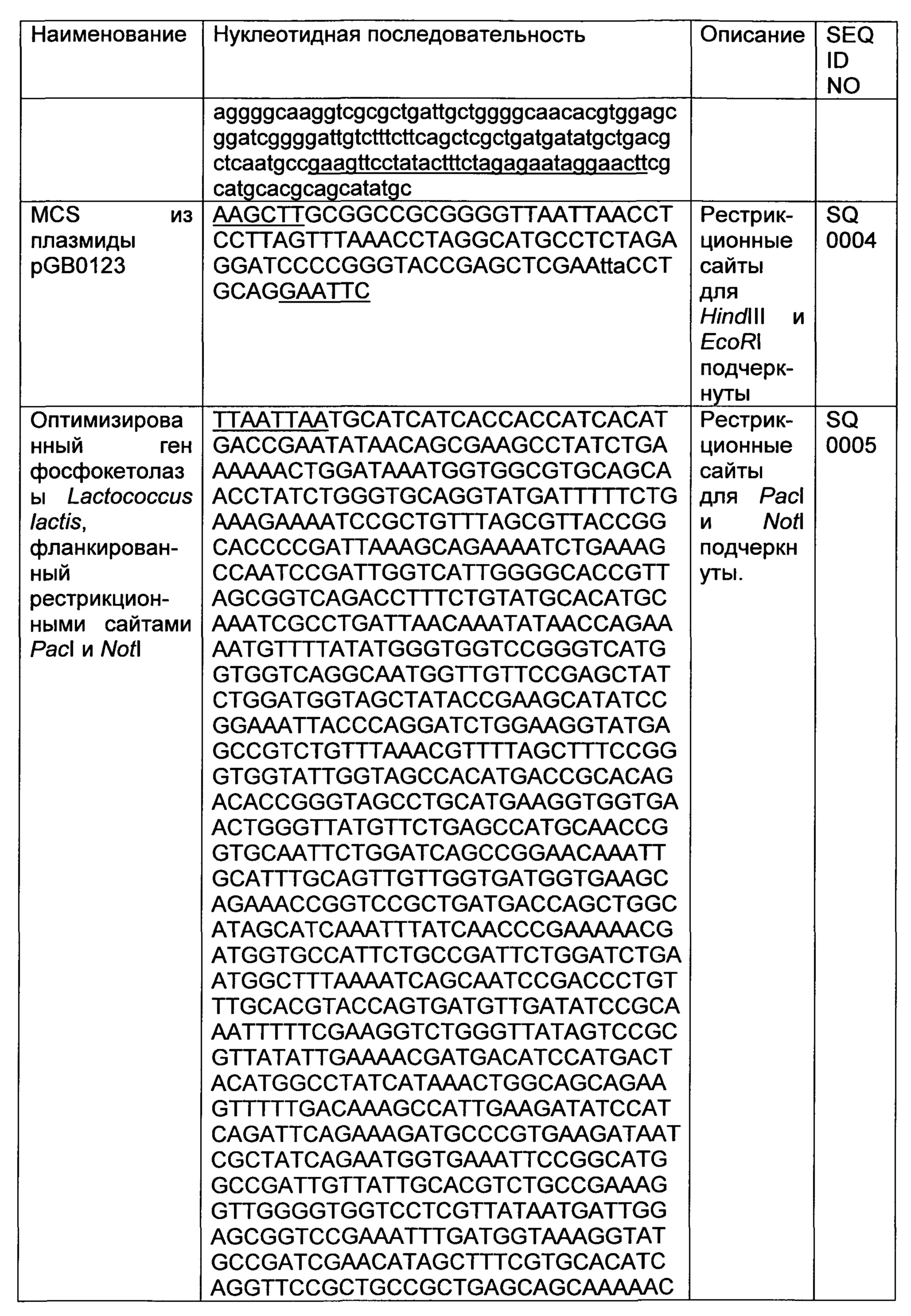


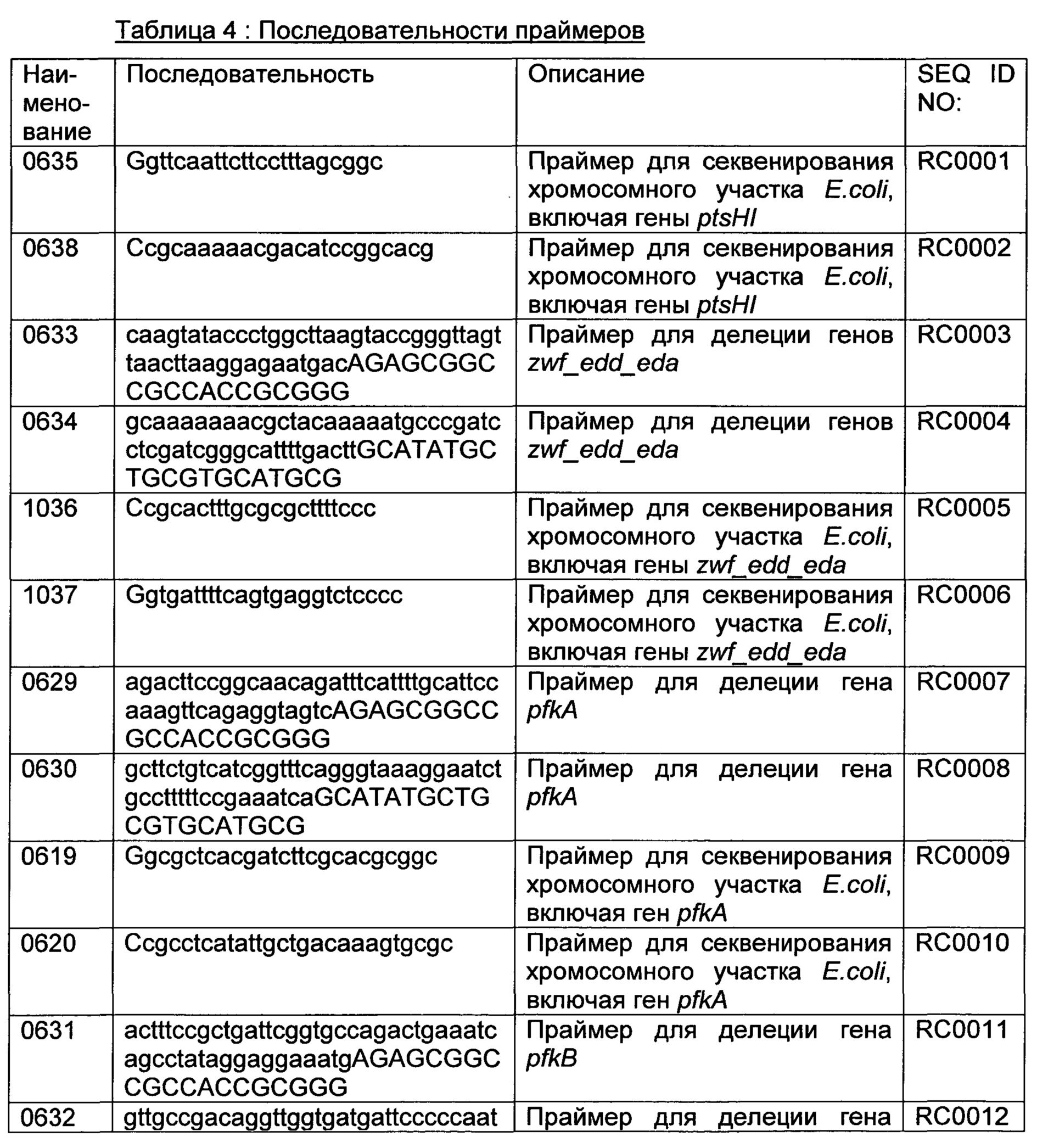
Хромосомная интеграция для генного нокаута.
Для интеграции ДНК в специфический участок хромосомы является необходимой гомология встраиваемой ДНК и хромосомного сайта-мишени, а также селективного маркера. Является предпочтительным, если маркер будет легко удаляться после интеграции. Система FRT/Flp рекомбиназа обеспечивает механизм удаления маркера. Сайты FRT представляют собой сайты узнавания для Flp рекомбиназ. Flp представляет собой сайт специфической рекомбиназы, которая вырезает промежуточную ДНК из непосредственно повторяемых сайтов узнавания.
Интеграционную кассету, которая содержит гомологичные плечи к хромосомному сайту-мишени и которая кодирует селективный маркер, фланкированный FRT (Datsenko КА. и Wanner BL, Proceedings of the National Academy of Sciences, 2000, Vol.97, №12, стр.6640-6645) сайтами, трансформировали в хозяйские клетки, которые несут pKD46 (Datsenko КА. И Wanner BL., Proceedings of the National Academy of Sciences, 2000, том 97, №12, стр.6640-6645). Успешные интегранты отбирали на основе роста клеток в присутствии антибиотика. Таким образом, pKD46 извлекали из клеток и рекомбиназную плазмиду потом вводили в интегранты для удаления гена антибиотика. Штаммы, которые содержат FRT кассету, трансформировали с помощью рСР20 плазмиды, которая кодирует Flp рекомбиназу (Datsenko КА. И Wanner BL., Proceedings of the National Academy of Sciences, 2000, том 97, №12, стр.6640-6645). После удаления интегрированного маркера рекомбиназные плазмиды извлекали из штамма.
ПРИМЕР 1: Конструирование штамма GBE1014
Цель селекции заключалась в том, чтобы описать конструкцию штамма Escherichia coli, названного GBE1014, для которого зависимое от PEP потребление глюкозы является инактивированным путем делеции PTS транспортных генов, зависимое от АТФ потребление глюкозы является невозможным, путь Эмбдена-Мейерхофа-Парнаса (ЕМРР) является инактивированным путем делеции генов фосфофруктокиназы, а пентозафосфатный путь (РРР) является инактивированным путем делеции гена глюкозо-6-фосфатдегидрогеназы.
Конструирование начинали со штамма GBE0901. GBE0901 представляет собой Escherichia coli (Migula) Castellani и Chalmers, штамм MG1655 (АТСС #700926), в котором исходную нуклеотидную последовательность от bp 2531736 до 2533865 (геномная база данных NCBI), содержащую гены ptsH и ptsl, заменяли SEQ SQ0001. Делеция оказывает влияние на зависимую от PEP систему фосфотрансферазы (PTS), что приводит к зависимому от PEP потреблению глюкозы, инактивированному в штамме GBE0901. Делецию генов ptsHI подтверждали с помощью ПЦР, и олигонуклеотиды 0635 и 0638 (представленные как SEQ RC0001 и RC0002, соответственно) использовали в качестве праймеров. Полученный продукт ПЦР размером 0,4 кб подвергали секвенированию при использовании тех же праймеров.
Штамм GBE0901 культивировали в LB среде, и GBE0901 клетки получали как электрокомпетентные. Электрокомпетентные GBE0901 клетки трансформировали с помощью плазмиды pKD46 (Datsenko KA. и Wanner BL, Proceedings of the National Academy of Sciences, 2000, том 97, №12, стр.6640-6645) и потом высаживали на LB планшеты, содержащие ампициллин (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С. Трансформация GBE0901 клеток с помощью плазмиды pKD46 позволяла получить штамм GBE0902.
Плазмида pGBE0688 представляет ген резистентности к спектиномицину, помещенный под контролем своего собственного промотора. Последовательность этой кассеты резистентности указана в Таблице 3 (SEQ SQ0002).
Плазмиду pGBE0688 использовали в качестве матрицы с праймерами 0633 и 0634 (представлены как SEQ RC0003 и RC0004, соответственно) для получения продукта ПЦР размером 1,3 кб. Этот продукта ПЦР размером 1,3 кб трансформировали в электрокомпетентную бактерию GBE0902, и трансформационную смесь потом высаживали на LB планшеты, содержащие спектиномицин (50 мк/мл), и инкубировали в течение ночи при 37°С для получения штамма GBE0903. В штамме GBE0903 ДНК последовательность, которая состоит из генов zwf, edd и eda, была делетирована. Эти гены соответственно кодируют глюкоза-6-фосфатдегидрогеназу, 6-фосфоглюконатдегидратазу и 2-кето-3-дезокси-6-фосфоглюконат альдолазу. Эта делетированная ДНК последовательность, включая гены zwf, edd и eda, была заменена на кассету резистентности к спектиномицину. Для того чтобы проверить эффективную делецию генов zwf, edd и eda, осуществляли ПЦР амплификацию при использовании праймеров 1036 и 1037 (представлены как SEQ RC0005 и RC0006, соответственно). Получали заключительный ПЦР продукт размером 1,9 кб. Этот ПЦР продукт размером 1,9 кб подвергали секвенированию с помощью тех же праймеров 1036 и 1037.
Штамм GBE0903 потом высаживали на LB планшеты, инкубировали при 37°С, и изолированные колонии подвергали скринингу на планшетах с MS (Richaud С., Mengin-Leucreulx D., Pochet S., Johnson EJ., Cohen GN. и Marliere P; The Journal of Biological Chemistry; 1993; том 268; №36; стр.26827-26835) при использовании с глюкозой в качестве источника углерода (2 г/л). Спустя 48 часов инкубации при 37°С колонии становились видимыми и их переносили на жидкую среду MS с добавлением глюкозы (2 г/л). Эта инкубация в течение ночи при 37°С индуцировала потерю плазмиды pKD46. Один изолят имел увеличенное вдвое время 7 часов, его называли GBE1000.
Штамм GBE1000 делали электрокомпетентным. GBE1000 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46 и потом высаживали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С. Трансформация GBE1000 клеток с помощью плазмиды pKD46 обеспечивала получение штамма GBE1001.
Плазмида pGBE0687 представляет ген резистентности к апрамицину, помещенный под контролем своего собственного промотора. Последовательность этой кассеты резистентности представлена в Таблице 3 (SEQ SQ0003).
Плазмиду pGBE0687 использовали в качестве матрицы вместе с праймерами 0629 и 0630 (представлены как SEQ RC0007 и RC0008, соответственно) для получения продукта ПЦР размером 1,2 кб. Полученный продукт ПЦР размером 1,2 кб трансформировали в электрокомпетентные GBE1001 бактерии, и трансформационную смесь высаживали на LB планшеты, содержащие апрамицин (50 мк/мл). Планшеты потом инкубировали в течение ночи при 37°С для получения нового штамма под названием GBE1005_pKD46. В штамме GB1005_pKD46 ген pfkA фосфофруктокиназы делегировали и заменяли на кассету резистентности к апрамицину. Для проверки того, что была проведена делеция гена pfkA, осуществляли ПЦР амплификацию при использовании праймеров 0619 и 0620 (представлены как SEQ RC0009 и RC0010, соответственно). Этот продукт ПЦР размером 1,7 кб подвергали секвенированию с помощью тех же праймеров 0619 и 0620. Для того, чтобы проверить потерю плазмиды pKD46, штамм GBE1005_pKD46 высевали на LB планшеты и инкубировали в течение ночи при 42°С. Потерю плазмиды pKD46 подтверждали путем высевания изолированных колоний на LB планшеты, содержащие ампициллин (100 мк/мл), инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, которые инкубировали при 37°С, и называли GBE1005. GBE1005 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
Спектиномициновую кассету помещали в соответствующие локусы генов zwf_edd_eda, а апрамициновую кассету помещали в соответствующие локусы гена pfkA. Для того чтобы вырезать кассеты резистентности, содержащие гены резистентности к спектиномицину и апрамицину, штамм GBE1005 трансформировали с помощью плазмиды рСР20 (Datsenko KA. и Wanner BL., Proceedings of the National Academy of Sciences, 2000, том 97, №12, стр.6640-6645) для получения штамма GBE1005_p. После инкубации в течение ночи на LB планшетах, содержащих ампициллин (100 мк/мл), при 30°С, изолированные колонии повторно высевали штрихом на LB планшеты с добавлением ампициллина (100 мк/мл) и инкубировали в течение ночи при 30°С.Изолированные колонии потом высевали на LB планшеты и инкубировали в течение ночи при 42°С, что вызывало потерю рСР20 плазмиды. Потом для того, чтобы проверить эффективное вырезание двух кассет устойчивости и потерю плазмиды рСР20, изолированные колонии высевали штрихом на LB планшеты, содержащие спектиномицин (50 мк/мл), инкубировали в течение ночи при 37°С на LB планшеты, содержащие апрамицин (50 мк/мл), которые инкубировали в течение ночи при 37°С, на LB планшеты, содержащие ампициллин (100 мк/мл), которые инкубировали в течение ночи при 30°С, на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, которые инкубировали при 37°С, и называли GBE1006. GBE1006 клетки не росли на LB планшетах, содержащих спектиномицин (50 мк/мл), на LB планшетах, содержащих апрамицин (50 мк/мл), и на LB планшетах с добавлением ампициллина (100 мк/мл).
Штамм GBE1006 делали электрокомпетентным, и GBE1006 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46. Трансформированные клетки потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С до получения нового штамма под названием GBE1010. Продукт ПЦР получали при использовании плазмиды pGBE0688 в качестве матрицы и олигонуклеотидов 0631 и 0632 (представлены как SEQ RC0011 и RC0012, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентные GBE1010 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие спектиномицин (50 мк/мл). Планшеты инкубировали в течение ночи при 37°С для получения штамма GBE1014_pKD46. В штамме GBE1014_ pKD46 ген фосфофруктокиназы pfkB был делетирован, и делетированную ДНК последовательность заменяли кассетой, содержащей ген устойивости к спектиномицину. Для проверки того, что произошла делеция гена pfkB, осуществляли ПЦР амплификацию при использовании праймеров 0621 и 0622 (представлены как SEQ RC0013 и RC0014, соответственно). Заключительный продукт ПЦР размером 2,2 кб подвергали секвенированию при использовании тех же праймеров 0621 и 0622.
Для индукции потери плазмиды pKD46 штамм GBE1014_pKD46 высевали на LB планшеты, и планшеты инкубировали в течение ночи при 42°С.Потерю плазмиды pKD46 проверяли путем высевания изолированных колоний на LB планшеты с добавлением ампициллина (100 мк/мл), инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, растущий на LB планшетах, инкубировали при 37°С и называли GBE1014. GBE1014 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
ПРИМЕР 2: Конструирование штамма GBE0929
Штамм GBE0901 высевали на LB планшеты при 37°С, и изолированные колонии подвергали скринингу на MS планшетах с глюкозой в качестве источника углерода (2 г/л). Спустя 48 часов инкубации при 37°С колонии становились видимыми. Один изолят имел двойное время 5 часов, его называли GBE0929.
ПРИМЕР 3: Конструирование штамма GBE1344
Конструирование начинали с использования штамма под названием GBE0129. GBE0129 представляет собой бактерию MG1655 Escherichia coli (АТСС #700926).
Штамм GBE0129 культивировали в LB среде, и GBE0129 клетки делали электрокомпетентными. Электрокомпетентные GBE0129 клетки трансформировали с помощью плазмиды pKD46, и потом трансформанты высаживали на LB планшеты, содержащие ампициллин (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С для получения нового штамма под названием GBE0170.
Продукт ПЦР получали при использовании плазмиды pGBE0687 в качестве матрицы и олигонуклеотидов 0633 и 0634 (представлены как SEQ RC0003 и RC0004, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,2 кб трансформировали в электрокомпетентные GBE0170 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие апрамицин (50 мк/мл). Планшеты инкубировали в течение ночи при 37°С. Такая инкубация запускала потерю pKD46 плазмиды и приводила к созданию нового штамма под названием GBE1339. В штамме GBE1339 глюкозо-6-фосфатдегидрогеназа, кодируемая геном zwf, 6-фосфоглюконат дегидратаза, кодируемая геном edd, и 2-кето-3-дезокси-6-фосфоглюконат альдолаза, кодируемая геном ее/а, были неактивными. После этого гены zwf, edd и eda делетировали и заменяли кассетой, содержащей ген устойчивости к апрамицину. Для проверки того, что делеция генов zwf, edd и eda была эффективной, проводили ПЦР амплификацию при использовании праймеров 1036 и 1037 (представлены как SEQ RC0005 и RC0006, соответственно). Этот продукт ПЦР размером 1,8 кб подвергали секвенированию с помощью тех же праймеров 1036 и 1037.
Штамм GBE1339 делали электрокомпетентным, и GBE1339 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46. Потом трансформанты высевали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С для получения штамма GBE1340. Получение ПЦР продукта осуществляли при использовании плазмиды pGBE0688 в качестве матрицы и олигонуклеотидов 0629 и 0630 (представлены как SEQ RC0007 и RC0008, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентную бактерию GBE1340, и трансформационную смесь высевали на LB планшеты, содержащие спектиномицин (50 мк/мл). Планшеты инкубировали в течение ночи при 37°С для получения штамма GBE1341_pKD46. В штамме GB1341_pKD46 ген pfka, кодирующий фосфофруктокиназу, заменяли геном устойчивости к спектиномицину. Для проверки того, что делеция гена pfkA была эффективной, осуществляли ПЦР амплификацию при использовании праймеров 0619 и 0620 (представлены как SEQ RC0009 и RC0010, соответственно). Этот продукт ПЦР размером 1,8 кб подвергали секвенированию с помощью тех же праймеров 0619 и 0620. Для индукции потери плазмиды pKD46 для штамма GBE1341_pKD46 клетки GBE1341_pKD46 высевали на LB планшеты и инкубировали при 42°С. Потерю плазмиды pKD46 подтверждали путем высевания изолированных колоний на LB планшеты с добавлением ампициллина (100 мк/мл), инкубировали в течение ночи при 30°С, на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, растущий на LB планшетах, инкубировали при 37°С, он получал название GBE1341. GBE1341 не рос на LB планшетах с добавлением ампициллина (100 мк/мл).
Для того чтобы вырезать кассеты резистентности, содержащие ген устойчивости к апрамицину и спектиномицину, которые были соответственно расположены в бывшем локусе генов zwf_edd_eda и pfkA, штамм GBE1341 трансформировали с помощью плазмиды рСР20 для получения нового штамма под названием GBE1341_p. После инкубации в течение ночи на LB планшетах, содержащих ампициллин (100 мк/мл) при 30°С, изолированные колонии повторно высевали штрихом на LB планшеты с добавлением ампициллина (100 мк/мл) для еще одной инкубации в течение ночи при 30°С. Изолированные колонии потом высевали на LB планшеты и инкубировали в течение ночи при 42°С. Эта инкубация при 42°С запускала потерю плазмиды рСР20. Впоследствии, для того, чтобы проверить вырезание и потерю плазмиды рСР20, изолированные колонии высевали штрихом на LB планшеты, содержащие спектиномицин (50 мк/мл), и инкубировали в течение ночи при 37°С, на LB планшеты с добавлением апрамицина (50 мк/мл), которые инкубировали в течение ночи при 37°С, на LB планшеты, содержащие ампициллин (100 мк/мл), которые инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, растущий на LB планшетах, инкубировали при 37°С, его называли GBE1342. GBE1342 клетки не росли на LB планшетах с добавлением спектиномицина (50 мк/мл), на LB планшетах с добавлением апрамицина (50 мк/мл) и на LB планшетах, содержащих ампициллин (100 мк/мл).
Штамм GBE1342 был сделан электрокомпетентным, и GBE1342 клетки трансформировали при использовании плазмиды pKD46. Потом трансформанты высевали на LB планшеты с добавлением ампициллина (100 мк/мл) и инкубировали в течение ночи при 30°С для получения штамма GBE1343. Получали продукт ПЦР при использовании плазмиды pGBE0688 в качестве матрицы и олигонуклеотидов 0631 и 0632 (представлены как SEQ RC0011 и RC0012, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентную бактерию GBE1343, и трансформационную смесь высевали на LB планшеты, содержащие спектиномицин (50 мк/мл), после чего осуществляли инкубацию в течение ночи при 37°С.Получали новый штамм под названием GBE1344_pKD46. В штамме GBE1344_pKD46 ген pfkb, кодирующий фосфофруктокиназу, был делетирован и заменен кассетлй резистентности к спектиномицину. Для проверки того, что делеция гена pfkB была эффективной, осуществляли ПЦР амплификацию при использовании праймеров 0621 и 0622 (представлены как SEQ RC0013 и RC0014, соответственно). Полученный продукт ПЦР размером 2,2 кб подвергали секвенированию с помощью тех же праймеров 0621 и 0622. Для индукции потери плазмиды pKD46 штамм GBE1344_pKD46 высевали на LB планшеты и инкубировали в течение ночи при 42°С. Потерю плазмиды pKD46 проверяли путем высевания изолированных колоний LB планшеты, содержащие ампициллин (100 мк/мл), и инкубировали в течение ночи при 30°С, а также на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, растущий на LB планшетах, инкубировали при 37°С и называли GBE1344. GBE1344 клетки не росли на LB планшетах, содержащих ампициллин (100 мк/мл).
ПРИМЕР 4: Конструирование плазмиды pGBE0457
Целью этого раздела является описать конструирование плазмиды, которая позволяет осуществлять экспрессию фосфокетолазы YP_003354041.1 из Lactococcus lactis в штаммах Е. coli.
Плазмида pGBE0123 представляет собой модифицированный вариант плазмиды pUC18 (New England Biolabs) и содержит модифицированный сайт множественного клонирования (MCS). Исходный MCS из pUC18 (от сайта рестрикции HindIII до рестрикционного сайта EcoRI) был заменен последовательностью SQ0004 (Таблица 3). Плазмида pGB0123 позволяет осуществлять экспрессию двух рекомбинантных белков под контролем промотора Plac.

Ген фосфокетолазы L. lactis был оптимизирован по кодонам GeneArt® (Invitrogen) для осуществления оптимальной экспрессии в Escherichia coli. Кроме того, прибавляли His-метку на 5' конце гена, а также прибавляли дополнительный стоп-кодон на 3' конце (SQ0005). Конструкцию гена фланкировали с помощью рестрикционных сайтов PacI и NotI и обеспечивали в составе плазмиды pGBE0421.
Для эксперимента клонирования продукты ПЦР и рестрикционные фрагменты очищали с помощью геля при использовании набора для экстракции QIAquick gel (Qiagen). Рестрикционные ферменты и Т4 ДНК лигазу (New England Biolabs, Beverly, MA) использовали в соответствии с инструкциями производителя.
Плазмиду pGBE0421 переваривали с помощью рестрикционных ферментов PacI и NotI для создания продукта размером 2,6 кб. Плазмиду pGB0123 переваривали также с помощью рестрикционных ферментов PacI и NotI и лигировали с рестрикционным фрагментом размером 2,6 кб. Полученную плазмиду (pGBE0457) подвергали секвенированию с помощью праймеров 1061, 1062, 1063, 1064 и 1065 (представлены как SEQ RC0015, RC0016, RC0017, RC0018 и RC0019, соответственно).
Экспрессию фосфокетолазы из Lactococcus lactis проверяли на белковом геле, после очистки рекомбинантного белка с использованием His ловушки (набор Protino Ni-IDA 1000, Macherey Nagel). Очистку осуществляли в соответствии с рекомендациями производителя. Ферментативный анализ с очищенным ферментом также осуществляли для того, чтобы определить фосфокетолазную активность на двух различных субстратах: ксилулоза-5-фосфате и фруктоза-6-фосфате. Экспериментальная процедура была такой же, что и та, которую использовали Leo Meile и др., (Journal of Bacteriology, May 2001, стр.2929-2936), за исключением того, что значение рН раствора составляло 7,5, и осуществляли прибавление 1 мМ MgCl2. Для осуществления этого ферментативного анализа 10 мкг очищенного белка прибавляли к 75 мкл реакционной смеси. Специфическая активность (мкмоль образовавшегося ацетил-Р/мин/мг белка) составляла 2815 мкмоль/мин/мг белка и 1941 мкмоль/мин/мг белка для D-ксилулоза-5-фосфата и D-фруктоза-6-фосфата, соответственно.
ПРИМЕР 5: Конструирование плазмиды pGBE0096
Конструирование плазмиды, которая отвечает за продукцию ацетона в E. coli, основывалось на конструировании плазмиды, описанной Bermejo LL, Welker NE. и Papoutsakis ET., Applied и Environmental Microbiology, 1998, том 64, №3, стр.1076-1085.
Заказывали штамм Clostridium acetobutylicum (ATCC 824). Геномную ДНК из этого штамма получали из бактериальной хромосомы, полученная плазмида получила наименование d pSOL1.
Гены ctfA и ctfB подвергались ПЦР амплификации из pSOU плазмиды с помощью праймеров 0070 и 0071 (представлены как SEQ RC0020 и RC0021, соответственно). Вводили BamHI сайт рестрикции на 5' конце продукта ПЦР и EcoRV сайт рестрикции на 3' конце продукта ПЦР. Полученный продукт ПЦР размером 1,3 кб переваривали с помощью рестрикционных ферментов BamHI и EcoRV, потом лигировали с pGB0689 (фагемида pBluescript II, Agilent Technologies), которую переваривали также с помощью рестрикционных ферментов, BamHI и EcoRV. Полученную плазмиду (pGBE0690) подвергали секвенированию с помощью праймеров 1066 и 1067 (представлены как SEQ RC0022 и RC0023, соответственно).
Ген adc и ген-терминатор подвергались ПЦР амплификации из pSOL1 плазмиды с помощью праймеров 0072 и 0073 (представлены как SEQ RC0024 и RC0025, соответственно). ПЦР амплификация позволяла осуществить встраивание EcoRV сайта рестрикции на 5' конце и SalI сайта рестрикции на 3' конце. Полученный продукт ПЦР размером 0,8 кб переваривали с помощью рестрикционных ферментов EcoRV и SalI. Плазмиду pGBE0690 переваривали также с помощью рестрикционных ферментов, EcoRV и SalI и потом лигировали с продуктом ПЦР размером 0,8 кб. Полученную плазмиду (pGBE0691) подвергали секвенированию с помощью праймеров 1066, 1067, 1068 и 1069 (представлены как SEQ RC0022, RC0023, RC0026 и RC0027, соответственно).
Плазмиду pGBE0691 переваривали с помощью рестрикционных ферментов BamHI и SalI для создания продукта размером 2,2 кб. Рестрикционный фрагмент размером 2,2 кб содержал гены ctfA, ctfB и adc. Плазмиду pGBE0051 (pUC19, New England Biolabs) переваривали также с помощью рестрикционных ферментов BamHI и SalI и потом лигировали с рестрикционным фрагментом размером 2,2 кб. Полученную плазмиду (pGBE0692) подвергали секвенированию с помощью праймеров 1066, 1067, 1068 и 1069 (представлены как SEQ RC0022, RC0023, RC0026 и RC0027, соответственно).
Геномная ДНК гена thl и его соответствующий промотор thl из Clostridium acetobutylicunn (ATCC 824) подвергались ПЦР амплификации с помощью праймеров 0074 и 0075 (представлены как SEQ RC0028 и RC0029, соответственно). ПЦР амплификация позволяла осуществить встраивание KpnI сайта рестрикции на 5' конце и BamHI сайта рестрикции на 3' конце. Полученный продукт ПЦР размером 1,4 кб переваривали с помощью рестрикционных ферментов KpnI и BamHI, и аналогично для плазмиды pGBE0692. Переваренную pGBE0692 плазмиду лигировали с продуктом ПЦР размером 1,4 кб. Полученную плазмиду pGBE0693 подвергали секвенированию с помощью праймеров 1066, 1067, 1068, 1069, 1070, 1071, 1072, 1073 и 1074 (представлены как SEQ RC0022, RC0023, RC0026 RC0027, RC0030, RC0031, RC0032, RC0033 и RC0034, соответственно).
Плазмида pGBE0124 представляет собой модифицированный вариант плазмиды pSU18 (Borja Bartolome, Yolanda Jubete, Eduardo Martinez и Fernando de la Cruz, Gene, 1991, том 102, выпуск 1, стр.75-78), она содержала модифицированный сайт множественного клонирования (MCS). Исходный MCS из pSU18 (от EcoRI сайта рестрикции до HindM сайта рестрикции) был заменен последовательностью SEQ SQ0006 (Таблица 3). Плазмида pGB0124 позволяет осуществлять экспрессию двух рекомбинантных белков под контролем Plac промотора.
Плазмиду pGBE0693 переваривали с помощью рестрикционных ферментов KpnI и SalI для создания продукта размером 3,5 кб. Плазмиду pGBE0124 переваривали также с помощью рестрикционных ферментов KpnI и SalI и потом лигировали с рестрикционным фрагментом размером 3,5 кб. Полученную плазмиду (pGBE0096) подвергали секвенированию с помощью праймеров 1066, 1067, 1068, 1069, 1070, 1071, 1072, 1073 и 1074 (представлены как SEQ RC0022, RC0023, RC0026 RC0027, RC0030, RC0031, RC0032, RC0033 и RC0034, соответственно).
ПРИМЕР 6: Получение ацетона с помощью штаммов GBE1346 и GBE1347.
Описание трансформации плазмиды в релевантные штаммы
Штамм GBE0129 делали электрокомпетентным, и GBE0129 электрокомпетентные клетки трансформировали с помощью плазмиды pGBE0096. Трансформанты потом высевали на LB планшеты, содержащие хлорамфеникол (25 мк/мл), и планшеты инкубировали в течение ночи при 30°С для получения штамма GBE0329.
Штамм GBE1344 делали электрокомпетентным, и GBE1344 электрокомпетентные клетки трансформировали с помощью двух плазмид pGBE0457 и pGBE0096. Трансформанты потом высевали на LB планшеты с добавлением ампициллина (100 мк/мл) и хлорамфеникола (25 мк/мл). Планшеты инкубировали в течение ночи при 30°С для получения штамма GBE1345.
Изолированные колонии из штаммов GBE0329 и GBE1345 подвергали скринингу на MS планшетах, содержащих глюкозу в качестве источника углерода (2 г/л) и хлорамфеникол (25 мк/мл). Эти планшеты инкубировали при 30°С для получения штаммов GBE1346 и GBE1347, соответственно. Через 4 дня инкубации при 30°С колонии переносили на MS жидкую среду, содержащую глюкозу (2 г/л) и хлорамфеникол (25 мк/мл), и инкубировали 3 дня при 30°С.
Описание условий культивирования флаконов
Для осуществления экспериментов по ферментации MS среду с 200 мМ фосфата дикалия использовали вместо 50 мМ фосфата дикалия. Полученная среда имела название MSP.
400 мл MSP жидкой среды, содержащей глюкозу (10 г/л) и хлорамфеникол (25 мк/мл), инокулировали либо с помощью прекультуры штамма GBE1346, либо с помощью прекультуры штамма GBE1347. Исходное значение OD600 составляло 0,1. 400 мл культуры инкубировали в бутылках на 500 мл, закрытых завинчивающимися крышками, при 30°С, при скорости 170 об/мин. Аликвоты 2 мл отбирали через 1 день, 2 дня, 3 дня, 6 дней, 7 дней и 8 дней. Для взятия аликвоты каждого образца бутылки открывали на 10 секунд.
Описание аналитических способов
Аликвоты фильтровали и концентрацию глюкозы определяли с помощью аналитического набора для определения глюкозы (НК) (GAHK20-1KT, Sigma) в соответствии с рекомендациями производителя. Кнцентрацию ацетона определяли с помощью газовой хроматографии при использовании газового хроматографа 450-GC (Bruker) и следующей программы:
- Колонка: DB-WAX (123-7033, Agilent Technologies)
- Инжектор с расщеплением/без расщепления: Т°=250°С
- Печь:
- 80°С в течение 6 минут
- 10°С в минуту до 220°С
- 220°С в течение 7 минут
- Струя, вытекающая из колонки: 1,5 мл/минута (Nitrogen)
- Детектор FID: T°=300°С
Результаты
Соотношение полученный [ацетон] (мМ)/потребленная [глюкоза] (мМ) было выше для штамма GBE1347, чем для штамма GBE1346.
ПРИМЕР 7: Получение ацетона с помощью штаммов GBE1350 и GBE1351.
Описание трансформации плазмиды в релевантные штаммы
Штамм GBE0929 делали электрокомпетентным, и GBE0929 электрокомпетентные клетки трансформировали с помощью плазмиды под названием pGBE0096. Трансформанты потом высевали на LB планшеты с добавлением хлорамфеникола (25 мк/мл), и планшеты инкубировали в течение ночи при 30°С для получения штамма GBE1348.
Штамм GBE1014 делали электрокомпетентным, подвергали трансформации с помощью двух плазмид pGBE0457 и pGBE0096. Трансформанты потом высевали на LB планшеты с добавлением ампициллина (100 мк/мл) и хлорамфеникола (25 мк/мл), и планшеты инкубировали в течение ночи при 30°С для получения штамм GBE1349.
Изолированные колонии из штаммов GBE1348 и GBE1349 подвергали скринингу на MS планшетах, содержащих глюкозу в качестве источника углерода (2 г/л) и хлорамфеникол (25 мк/мл). Эти планшеты инкубировали в течение 4 дней при 30°С для получения штаммов GBE1350 и GBE1351, соответственно. Изолированные колонии потом переносили на MS жидкую среду, содержащцю глюкозу (2 г/л) и хлорамфеникол (25 мк/мл). Клетки GBE1350 и GBE1351 инкубировали при 30°С.
Описание условий культивирования флаконов
400 мл MSP среды, содержащей глюкозу (10 г/л) и хлорамфеникол (25 мк/мл), инокулировали либо с помощью прекультуры штамма GBE1350, либо с помощью прекультуры штамма GBE1351. Исходное значение OD600 составляло 0,1. 400 мл кульутры инкубировали в бутылках на 500 мл, закрытых завинчивающимися крышками, при 30°С, при скорости 170 об/мин. Аликвоты 2 мл отбирали через 1 день, 2 дня, 3 дня, 6 дней, 7 дней и 8 дней. Для взятия аликвоты каждого образца бутылки открывали на 10 секунд.
Описание аналитических способов
Аликвоты фильтровали и концентрацию глюкозы определяли с помощью аналитического набора для определения глюкозы (НК) (GAHK20-1KT, Sigma) в соответствии с рекомендациями производителя. Концентрацию ацетона определяли с помощью газовой хроматографии при использовании газового хроматографа 450-GC (Bruker) и следующей программы:
- Колонка: DB-WAX (123-7033, Agilent Technologies)
- Инжектора с расщеплением/без расщепления: Т°=250°С
- Печь:
- 80°С в течение 6 минут
- 10°С в минуту до 220°С
- 220°С в течение 7 минут
- Струя, вытекающая из колонки: 1,5 мл/минута (Nitrogen)
- Детектор FID: T°=300°С
Результаты
Соотношение полученный [ацетон] (мМ)/потребленная [глюкоза] (мМ) было выше для штамма GBE1351, чем для штамма GBE1350GBE.
ПРИМЕР 8: Конструирование плазмиды pGBE1020
Целью этого раздела является описать конструирование плазмиды, которая позволяет осуществлять экспрессию фосфокетолазы YP_003354041.1 из Lactococcus lactis, а также позволяет осуществлять продукцию ацетона в штаммах E. coli.
Ген фосфокетолазы L. lactis был амплифицирован с помощью ПЦР из pGBE0421 плазмиды с помощью праймеров 1516 и 1517 (представлены как SEQ RC0035 и RC0036, соответственно).
ПЦР амплификация позволяла осуществить встраивание EcoRI сайта рестрикции и сайта связывания рибосомы (RBS) на 5' конце и KpnI сайта рестрикции на 3' конце. Полученный продукт ПЦР размером 2,5 кб переваривали с помощью рестрикционных ферментов EcoRI и KpnI. pGBE0123 плазмиду переваривали также с помощью рестрикционных ферментов EcoRI и KpnI и потом лигировали с продуктом ПЦР размером 2,5 кб. Полученную плазмиду (pGBE0928) подвергали секвенированию с помощью праймеров 1061, 1062, 1063, 1064 и 1065 (представлены как SEQ RC0015, RC0016, RC0017, RC0018 и RC0019, соответственно).
Плазмиду pGBE0096 переваривали с помощью рестрикционных ферментов KpnI и NotI для создания продукта размером 3,6 кб. pGBE0928 плазмиду переваривали также с помощью рестрикционных ферментов KpnI и NotI и лигировали с рестрикционным фрагментом размером 3,6 кб. Полученную плазмиду (pGBE1020) подвергали секвенированию с помощью праймеров 1994, 1995, 1996, 1997, 1998, 1999, 2000, 2001, 2002 и 2003 (представлены как SEQ RC0037, RC0038, RC0039, RC0040, RC0041, RC0042, RC0043, RC0044, RC0045 и RC0046, соответственно).
ПРИМЕР 9: Конструирование плазмиды pGBE1021
Целью этого раздела является описать конструирование плазмиды, которая позволяет осуществлять экспрессию продукцию ацетона в штаммах E. coli.
Плазмиду pGBE0096 переваривали с помощью рестрикционных ферментов KpnI и NotI для создания продукта размером 3,6 кб.
Плазмиду pGBE0123 переваривали также с помощью рестрикционных ферментов KpnI и NotI и потом лигировали с продуктом ПЦР размером 3,6 кб. Полученную плазмиду (pGBE1021) подвергали секвенированию с помощью праймеров 1994, 1995, 1996, 1997, 1998, 1999, 2000, 2001, 2002 и 2003 (представлены как SEQ RC0037, RC0038, RC0039, RC0040, RC0041, RC0042, RC0043, RC0044, RC0045 и RC0046, соответственно).
ПРИМЕР 10: Продукция ацетона штаммами GBE2264 и GBE2265.
Описание трансформации плазмиды в релевантные штаммы
Штамм GBE0129 делали электрокомпетентным, и GBE0129 электрокомпетентные клетки трансформировали с помощью плазмиды pGB1021. Трансформанты потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С для получения штамма GBE2262.
Штамм GBE1344 делали электрокомпетентным, и GBE1344 электрокомпетентные клетки трансформировали с помощью плазмиды pGBE1020. Трансформанты потом высевали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С для получения штамма GBE2263.
Изолированные колонии из штаммов GBE2262 и GBE2263 подвергали скринингу на MS планшетах, содержащих глюкозу в качестве источника углерода (2 г/л) и ампициллин (100 мк/мл). Эти планшеты инкубировали при 30°С для получения штаммов GBE2264 и GBE2265, соответственно. Через 4 дня инкубации при 30°С колонии переносили на MS жидкую среду, содержащую глюкозу (2 г/л), дрожжевой экстракт (0,1 г/л) и ампициллин (100 мк/мл), и инкубировали 3 дня при 30°С.
Описание условий культивирования флаконов
Жидкую среду MSP (200 мл), содержащую глюкозу (10 г/л), дрожжевой экстракт (0,1 г/л) и ампициллин (100 мк/мл), инокулировали либо с помощью прекультуры штамма GBE2264, либо с помощью прекультуры штамма GBE2265. Исходное значение ODeoo составляло 0,1. 200 мл культуры инкубировали в бутылках на 250 мл, закрытых завинчивающимися крышками, при 30°С, при скорости 170 об/мин Аликвоты (2 мл) отбирали через 1 день, 2 дня, 4 дня, 5 дней и 6 дней. Для взятия аликвоты каждого образца бутылки открывали на 10 секунд.
Описание аналитических способов
Аликвоты фильтровали, и концентрацию глюкозы определяли с помощью анализа ВЭЖХ при использовании Agilent ВЭЖХ (1260 Infinity) и Hi-Plex колонки (Agilent, PL1170-6830) с предохранительной колонкой (Agilent, PL Hi-Plex H предохранительная колонка, PL1170-1830).
- Объем введения: 20 мкл
- Состав растворителя: [H2SO4]: 5,5 мМ
- Температура колонок: 65°С
- RID (G1362A): установка температуры: 35°С
Ацетон экстрагировали из отфильтрованных аликвот путем смешивания с метилацетатом (1 объем метилацетата для 2 объемов отфильтрованной аликвоты). Концентрацию ацетона определяли с помощью газовой хроматографии при использовании газового хроматографа 450-GC (Bruker) и следующей программы:
- Колонка: DB-WAX (123-7033, Agilent Technologies)
- Инжектор:
- Соотношение расщепления: 10
- Т°=250°С
- Печь:
- 50°С в течение 9 минут
- 20°С в минуту до 180°С
- 180°С в течение 5 минут
- Поток из колонки: 1,5 мл/минута (Nitrogen)
- Детектор FID: Т°=300°С
Результаты
Соотношение полученный [ацетон] (мМ)/потребленная [глюкоза] (мМ) было выше для штамма GBE2265, чем для штамма GBE2264.
ПРИМЕР 11: Конструирование штамма GBE1283
Штамм GBE0929 высевали на MS планшеты, содержащие глюкозу в качестве источника углерода (2 г/л). Изолированную колонию переносили на жидкую среду MS, содержащую глюкозу (2 г/л), и инкубировали в течение 3 дней при 30°С. Жидкую среду MS (100 мл), содержащую глюкозу (2 г/л), инокулировали при использовании прекультуры штамма GBE0929. Исходное значение OD600 составляло 0,1. 100 мл культуры инкубировали в колбах Эрленмейера на 1 л при 30°С и при скорости 170 об/мин. Тогда, когда значение OD600 стало превышать 1, брали аликвоту культуры и использовали в качестве инокулята для свежей культуры (100 мл культуры инкубировали в колбах Эрленмейера на 1 л при температуре 30°С и скорости 170 об/мин). Штамм GBE0929 подвергали субкультивированию 10 раз для получения штамма GBE1283.
ПРИМЕР 12: Конструирование штамма GBE2256.
Штамм GBE1283 культивировали в LB среде, и GBE1283 клетки делали электрокомпетентными. Электрокомпетентные GBE1283 клетки трансформировали с помощью pKD46 плазмиды и потом переносили на LB планшеты, содержащие ампициллин (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С. Трансформация GBE1283 клеток с помощью плазмиды pKD46 обеспечивала получение штамма GBE1284.
Плазмиду pGBE0688 использовали в качестве матрицы вместе с праймерами 0629 и 0630 (представлены как SEQ RC0007 и RC0008, соответственно) для получения продукта ПЦР размером 1,2 кб. Полученный продукт ПЦР размером 1,2 кб трансформировали в электрокомпетентные GBE1284 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие спектиномицин (50 мк/мл). Потом планшеты инкубировали в течение ночи при 37°С для получения нового штамма под названием GBE2252_pKD46. В штамме GB2252_pKD46 ген pfkA фосфофруктокиназы был делегирован и заменен кассетой резистентности к спектиномицину. Для проверки того, что произошла делеция гена pfkA, осуществляли ПЦР амплификацию при использовании праймеров 0619 и 0620 (представлены как SEQ RC0009 и RC0010, соответственно). Этот продукт ПЦР размером 1,7 кб подвергали секвенированию с помощью тех же праймеров 0619 и 0620. Для того, чтобы проверить потерю плазмиды pKD46, штамм GBE2252_pKD46 высевали на LB планшеты и инкубировали в течение ночи при 42°С. Потерю плазмиды pKD46 подтверждали путем высевания изолированных колоний на LB планшеты, содержащие ампициллин (100 мк/мл), инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, инкубировали при 37°С и называли GBE2252. GBE2252 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
Штамм GBE2252 делали электрокомпетентным, и GBE2252 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46. Трансформированные клетки потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С до получения нового штамма под названием GBE2253.
Продукт ПЦР получали при использовании плазмиды pGBE0687 в качестве матрицы и олигонуклеотидов 0631 и 0632 (представлены как SEQ RC0011 и RC0012, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентные GBE2253 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие апрамицин (50 мк/мл). Планшеты инкубировали в течение ночи при 37°С для получения штамма GBE2256_pKD46. В штамме GBE2256_pKD46 ген pfkB фосфофруктокиназы был делетирован, и делегированную ДНК последовательность заменяли кассетой, содержащей ген устойчивости к апрамицину. Для проверки того, что произошла делеция pfkB гена, осуществляли ПЦР амплификацию при использовании праймеров 0621 и 0622 (представлены как SEQ RC0013 и RC0014, соответственно). Этот заключительный продукт ПЦР размером 2,2 кб подвергали секвенированию при использовании тех же праймеров 0621 и 0622.
Для индукции потери плазмиды pKD46 штамм GBE2256_pKD46 высевали на LB планшеты и планшеты инкубировали в течение ночи при 42°С. Потерю плазмиды pKD46 проверяли путем высевания изолированных колоний на LB планшеты с добавлением ампициллина (100 мк/мл), инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, выращиваемый на LB планшетах, инкубировали при 37°С и называли GBE2256. GBE2256 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
ПРИМЕР 13: Конструирование штамма GBE1518
Плазмиду pGBE0688 использовали в качестве матрицы с праймерами 0633 и 1109 (представлены как SEQ RC0003 и RC0047, соответственно) для получения продукта ПЦР размером 1,3 кб. Этот продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентные GBE1284 бактерии, и трансформационную смесь потом высаживали на LB планшеты, содержащие спектиномицин (50 мк/мл), и инкубировали в течение ночи при 37°С для получения штамма GBE1433. В штамме GBE1433 ДНК последовательность, которая состояла из гена zwf, делетировали. Эту делегированную ДНК последовательность, включая ген zwf, заменяли кассетой резистентности к спектиномицину. Для того, чтобы проверить эффективную делецию гена zwf, осуществляли ПЦР амплификацию при использовании праймеров 1036 и 1110 (представлены как SEQ RC0005 и RC0048, соответственно). Получали заключительный продукт ПЦР размером 1,5 кб. Этот продукт ПЦР размером 1,5 кб подвергали секвенированию с помощью тех же праймеров 1036 и 1110.
Штамм GBE1433 делали электрокомпетентным. GBE1433 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46 и потом высаживали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С. Трансформация GBE1433 клеток с помощью плазмиды pKD46 обеспечивала получение штамма GBE1436.
Плазмиду pGBE0687 использовали в качестве матрицы вместе с праймерами 0629 и 0630 (представлены как SEQ RC0007 и RC0008, соответственно) для получения продукта ПЦР размером 1,2 кб. Полученный продукт ПЦР размером 1,2 кб трансформировали в электрокомпетентные GBE1436 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие апрамицин (50 мк/мл). Потом планшеты инкубировали в течение ночи при 37°С для получения нового штамма под названием GBE1441_pKD46. В штамме GB1441_pKD46 ген pfkA фосфофруктокиназы был делегирован и заменен кассетой резистентности к апрамицину. Для проверки того, что произошла делеция гена pfkA, осуществляли ПЦР амплификацию при использовании праймеров 0619 и 0620 (представлены как SEQ RC0009 и RC0010, соответственно). Этот продукт ПЦР размером 1,7 кб подвергали секвенированию с помощью тех же праймеров 0619 и 0620. Для того, чтобы проверить потерю плазмиды pKD46, штамм GBE1441_pKD46 высевали на LB планшеты и инкубировали в течение ночи при 42°С.Потерю плазмиды pKD46 подтверждали путем высевания изолированных колоний на LB планшеты, содержащие ампициллин (100 мк/мл), инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, которые инкубировали при 37°С, и называли GBE1441. GBE1441 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
Спектиномициновую кассету помещали в соответствующие локусы zwf гена, а апрамициновую кассету помещали в соответствующие локусы pfkA гена. Для того, чтобы вырезать кассеты резистентности, содержащие ген устойчивости к спектиномицину и апрамицину, штамм GBE1441 трансформировали с помощью плазмиды рСР20 для получения штамма GBE1441_p. После инкубации в течение ночи на LB планшетах, содержащих ампициллин (100 мк/мл), при 30°С изолированные колонии повторно высевали штрихом на LB планшеты с добавлением ампициллина (100 мк/мл) и инкубировали в течение ночи при 30°С. Изолированные колонии потом высевали на LB планшеты и инкубировали в течение ночи при 42°С, что вызывало потерю рСР20 плазмиды. Потом для того, чтобы проверить эффективное вырезание двух кассет устойчивости и потерю рСР20 плазмиды, изолированные колонии высевали штрихом на LB планшеты, содержащие спектиномицин (50 мк/мл), которые инкубировали в течение ночи при 37°С, на LB планшеты, содержащие апрамицин (50 мк/мл), которые инкубировали в течение ночи при 37°С, на LB планшеты, содержащие ампициллин (100 мк/мл), которые инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, которые инкубировали при 37°С, и штамм называли GBE1448. GBE1448 клетки не росли на LB планшетах, содержащих спектиномицин (50 мк/мл), на LB планшетах, содержащих апрамицин (50 мк/мл), и на LB планшетах с добавлением ампициллина (100 мк/мл).
Штамм GBE1448 делали электрокомпетентным, и GBE1448 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46. Трансформированные клетки потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С до получения нового штамма под названием GBE1449. Продукт ПЦР получали при использовании плазмиды pGBE0688 в качестве матрицы и олигонуклеотидов 0631 и 0632 (представлены как SEQ RC0011 и RC0012, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентные GBE1449 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие спектиномицин (50 мк/мл). Планшеты инкубировали в течение ночи при 37°С для получения штамма GBE1518_pKD46. В штамме GBE1518_pKD46 ген pfkB фосфофруктокиназы был делегирован, и делетированную ДНК последовательность заменяли кассетой, содержащей ген устойивости к спектиномицину. Для проверки того, что произошла делеция гена pfkB, осуществляли ПЦР амплификацию при использовании праймеров 0621 и 0622 (представлены как SEQ RC0013 и RC0014, соответственно). Заключительный продукт ПЦР размером 2.2 кб подвергали секвенированию при использовании тех же праймеров 0621 и 0622.
Для индукции потери плазмиды pKD46 штамм GBE1518_ pKD46 высевали на LB планшеты и планшеты инкубировали в течение ночи при 42°С. Потерю плазмиды pKD46 проверяли путем высевания изолированных колоний на LB планшеты с добавлением ампициллина (100 мк/мл), которые инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, растущий на LB планшетах, инкубировали при 37°С и называли GBE1518. GBE1518 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
ПРИМЕР 14: Конструирование штамма GBE1420
Плазмиду pGBE0688 использовали в качестве матрицы с праймерами 0633 и 0634 (представлены как SEQ RC0003 и RC0004, соответственно) для получения продукта ПЦР размером 1,3 кб. Этот продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентные GBE1284 бактерии, и трансформационную смесь потом высаживали на LB планшеты, содержащие спектиномицин (50 мк/мл), и инкубировали в течение ночи при 37°С для получения штамма GBE1287. В штамме GBE1287 ДНК последовательность, состоящая из генов zwf, edd и eda, была делетирована. Эту делетированную ДНК последовательность, включающую гены zwf, edd и eda, заменяли кассетой резистентности к спектиномицину. Для того, чтобы проверить эффективную делецию генов zwf, edd и eda, осуществляли ПЦР амплификацию при использовании праймеров 1036 и 1037 (представлены как SEQ RC0005 и RC0006, соответственно). Получали заключительный продукт ПЦР размером 1,9 кб. Этот продукт ПЦР размером 1,9 кб подвергали секвенированию с помощью тех же праймеров 1036 и 1037.
Штамм GBE1287 делали электрокомпетентным. GBE1287 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46 и потом высаживали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С. Трансформация GBE1287 клеток при использовании плазмиды pKD46 обеспечивала получение штамма GBE1337.
Плазмиду pGBE0687 использовали в качестве матрицы вместе с праймерами 0629 и 0630 (представлены как SEQ RC0007 и RC0008, соответственно) для получения продукта ПЦР размером 1,2 кб. Полученный продукт ПЦР размером 1,2 кб трансформировали в электрокомпетентные GBE1337 бактерии, и трансформационную смесь высевали на LB планшеты, содержащие апрамицин (50 мк/мл). Планшеты потом инкубировали в течение ночи при 37°С для получения нового штамма под названием GBE1353_pKD46. В штамме GB1353_pKD46 ген pfkA фосфофруктокиназы был делетирован и заменен на кассету резистентности к апрамицину. Для проверки того, что произошла делеция гена pfkA, осуществляли ПЦР амплификацию при использовании праймеров 0619 и 0620 (представлены как SEQ RC0009 и RC0010, соответственно). Этот продукт ПЦР размером 1,7 кб подвергали секвенированию с помощью тех же праймеров 0619 и 0620. Для того, чтобы проверить потерю плазмиды pKD46, штамм GBE1353_pKD46 высевали на LB планшеты и инкубировали в течение ночи при 42°С.Потерю плазмиды pKD46 подтверждали путем высевания изолированных колоний на LB планшеты, содержащие ампициллин (100 мк/мл), которые инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, которые инкубировали при 37°С, и называли GBE1353. GBE1353 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
Спектиномициновую кассету помещали в соответствующие локусы генов zwf_edd_eda, а апрамициновую кассету помещали в соответствующие локус гена pfkA. Для того, чтобы вырезать кассеты резистентности, содержащие ген устойчивости к спектиномицину и апрамицину, штамм GBE1353 трансформировали с помощью плазмиды рСР20 для получения штамма GBE1353_p.После инкубации в течение ночи на LB планшетах, содержащих ампициллин (100 мк/мл), при 30°С, изолированные колонии повторно высевали штрихом на LB планшеты с добавлением ампициллина (100 мк/мл) и инкубировали в течение ночи при 30°С. Изолированные колонии потом высевали на LB планшеты и инкубировали в течение ночи при 42°С, что вызывало потерю рСР20 плазмиды. Потом для того, чтобы проверить эффективное вырезание двух кассет устойчивости и потерю плазмиды рСР20, изолированные колонии высевали штрихом на LB планшеты, содержащие спектиномицин (50 мк/мл), которые инкубировали в течение ночи при 37°С, на LB планшеты, содержащие апрамицин (50 мк/мл), которые инкубировали в течение ночи при 37°С, на LB планшеты, содержащие ампициллин (100 мк/мл), которые инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм выращивали на LB планшетах, которые инкубировали при 37°С, и называли GBE1368. GBE1368 клетки не росли на LB планшетах, содержащих спектиномицин (50 мк/мл), на LB планшетах, содержащих апрамицин (50 мк/мл), и на LB планшетах с добавлением ампициллина (100 мк/мл).
Штамм GBE1368 делали электрокомпетентным, и GBE1368 электрокомпетентные клетки трансформировали с помощью плазмиды pKD46. Трансформированные клетки потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С до получения нового штамма под названием GBE1371. Продукт ПЦР получали при использовании плазмиды pGBE0688 в качестве матрицы и олигонуклеотидов 0631 и 0632 (представлены как SEQ RC0011 и RC0012, соответственно) в качестве праймеров. Полученный продукт ПЦР размером 1,3 кб трансформировали в электрокомпетентные GBE1371 бактерии и трансформационную смесь высевали на LB планшеты, содержащие спектиномицин (50 мк/мл). Планшеты инкубировали в течение ночи при 37°С для получения штамма GBE1420_pKD46. В штамме GBE1420_pKD46 ген pfkB фосфофруктокиназы был делетирован, и делетированную ДНК последовательность заменяли кассетой, содержащей ген устойивости к спектиномицину. Для проверки того, что произошла делеция pfkB гена, осуществляли ПЦР амплификацию при использовании праймеров 0621 и 0622 (представлены как SEQ RC0013 и RC0014, соответственно). Этот заключительный продукт ПЦР размером 2,2 кб подвергали секвенированию при использовании тех же праймеров 0621 и 0622.
Для индукции потери плазмиды pKD46 штамм GBE1420_pKD46 высевали на LB планшеты, и планшеты инкубировали в течение ночи при 42°С. Потерю плазмиды pKD46 проверяли путем высевания изолированных колоний на LB планшеты с добавлением ампициллина (100 мк/мл), которые инкубировали в течение ночи при 30°С, и на LB планшеты, которые инкубировали в течение ночи при 37°С. Полученный штамм, растущий на LB планшетах, инкубировали при 37°С и называли GBE1420. GBE1420 клетки не росли на LB планшетах с добавлением ампициллина (100 мк/мл).
ПРИМЕР 15: Продукция ацетона штаммами GBE2268 и GBE2269.
Описание трансформации плазмиды в релевантные штаммы
Штамм GBE1283 делали электрокомпетентным, и GBE1283 электрокомпетентные клетки трансформировали с помощью плазмиды pGB1021. Трансформанты потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С для получения штамма GBE2266.
Штамм GBE1420 делали электрокомпетентным, и GBE1420 электрокомпетентные клетки трансформировали с помощью плазмиды pGBE1020. Трансформанты потом высевали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С для получения штамма GBE2267.
Изолированные колонии из штаммов GBE2266 и GBE2267 подвергали скринингу на MS планшетах, содержащих глюкозу в качестве источника углерода (2 г/л), и ампициллин (100 мк/мл). Эти планшеты инкубировали при 30°С для получения штаммов GBE2268 и GBE2269, соответственно. Через 4 дня инкубации при 30°С колонии переносили на MS жидкую среду, содержащую глюкозу (2 г/л), дрожжевой экстракт (0,1 г/л) и ампициллин (100 мк/мл), и инкубировали в течение 3 дней при 30°С.
Описание условий культивирования флаконов
Жидкую среду MSP (200 мл), содержащую глюкозу (10 г/л), дрожжевой экстракт (0,1 г/л) и ампициллин (100 мк/мл), инокулировали либо с помощью прекультуры штамма GBE2268, либо с помощью прекультуры штамма GBE2269. Исходное значение OD600 составляло 0,1. 200 мл культуры инкубировали в бутылках на 250 мл, закрытых завинчивающимися крышками, при 30°С, при скорости 170 об/мин. Аликвоты (2 мл) отбирали через 1 день, 2 дня, 4 дня, 5 дней, 6 дней, 7 дней и 8 дней. Для взятия аликвоты каждого образца бутылки открывали на 10 секунд.
Описание аналитических способов
Аликвоты фильтровали и концентрацию глюкозы определяли с помощью анализа ВЭЖХ при использовании Agilent ВЭЖХ (1260 Infinity) и Hi-Plex колонки (Agilent, PL1170-6830) с предохранительной колонкой (Agilent, PL Hi-Plex H предохранительная колонка, PL1170-1830).
- Объем введения: 20 мкл
- Состав растворителя: [H2SO4]: 5,5 мМ
- Температура колонок: 65°С
- RID (G1362A): установка температуры: 35°С
Ацетон экстрагировали из отфильтрованных аликвот путем смешивания с метилацетатом (1 объем метилацетата для 2 объемов отфильтрованной аликвоты). Концентрацию ацетона определяли с помощью газовой хроматографии при использовании газового хроматографа 450-GC (Bruker) и следующей программы:
- Колонка: DB-WAX (123-7033, Agilent Technologies)
- Инжектор:
- Соотношение расщепления: 10
- Т°=250°С
- Печь:
- 50°С в течение 9 минут
- 20°С в минуту до 180°С
- 180°С в течение 5 минут
- Струя из колонки: 1,5 мл/минута (Nitrogen)
- Детектор FID: Т°=300°С
Результаты
Соотношение полученный [ацетон] (мМ)/потребленная [глюкоза] (мМ) было выше для штамма GBE2269, чем для штамма GBE2268.
ПРИМЕР 16: Продукция ацетона штаммами GBE2272 и GBE2273.
Описание трансформации плазмиды в релевантные штаммы
Штамм GBE2256 делали электрокомпетентным, и GBE2256 электрокомпетентные клетки трансформировали с помощью плазмиды pGB1020. Трансформанты потом высевали на LB планшеты, содержащие ампициллин (100 мк/мл), и планшеты инкубировали в течение ночи при 30°С для получения штамма GBE2270.
Штамм GBE1518 делали электрокомпетентным, и GBE1518 электрокомпетентные клетки трансформировали с помощью плазмиды pGBE1020. Трансформанты потом высевали на LB планшеты с добавлением ампициллина (100 мк/мл). Планшеты инкубировали в течение ночи при 30°С для получения штамма GBE2271.
Изолированные колонии из штаммов GBE2270 и GBE2271 подвергали скринингу на MS планшетах, содержащих глюкозу в качестве источника углерода (2 г/л) и ампициллин (100 мк/мл). Эти планшеты инкубировали при 30°С для получения штаммов GBE2272 и GBE2273, соответственно. Через 4 дня инкубации при 30°С, колонии переносили на MS жидкую среду, содержащую глюкозу (2 г/л), дрожжевой экстракт (0,1 г/л) и ампициллин (100 мк/мл), и инкубировали в течение 3 дней при 30°С.
Описание условий культивирования флаконов
Жидкую среду MSP (200 мл), содержащую глюкозу (10 г/л), дрожжевой экстракт (0,1 г/л) и ампициллин (100 мк/мл), инокулировали либо с помощью прекультуры штамма GBE2272, либо с помощью прекультуры штамма GBE2273. Исходное значение OD600 составляло 0,1. 200 мл культуры инкубировали в бутылках на 250 мл, закрытых завинчивающимися крышками, при 30°С, при скорости 170 об/мин. Аликвоты (2 мл) отбирали через 1 день, 2 дня, 4 дня, 5 дней, 6 дней, 7 дней и 8 дней. Для взятия аликвоты каждого образца бутылки открывали на 10 секунд.
Описание аналитических способов
Аликвоты фильтровали и концентрацию глюкозы определяли с помощью анализа ВЭЖХ при использовании Agilent ВЭЖХ (1260 Infinity) и Hi-Plex колонки (Agilent, PL1170-6830) с предохранительной колонкой (Agilent, PL Hi-Plex H предохранительная колонка, PL1170-1830).
- Объем введения: 20 мкл
- Состав растворителя: [H2SO4]: 5,5 мМ
- Температура колонок: 65°С
- RID (G1362A): установка температуры: 35°С Ацетон экстрагировали из отфильтрованных аликвот путем смешивания с метилацетатом (1 объем метилацетата для 2 объемов отфильтрованной аликвоты). Концентрацию ацетона определяли с помощью газовой хроматографии при использовании газового хроматографа 450-GC (Bruker) и следующей программы:
- Колонка: DB-WAX (123-7033, Agilent Technologies)
- Инжектор:
- Соотношение расщепления: 10
- Т°=250°С
- Печь:
- 50°С в течение 9 минут
- 20°С в минуту до 180°С
- 180°С в течение 5 минут
- Струя из колонки: 1,5 мл/минута (Nitrogen)
- Детектор FID: Т°=300°С
Результаты
Соотношение полученный [ацетон] (мМ)/потребленная [глюкоза] (мМ) было выше для штамма GBE2273, чем для штамма GBE2272


















Способ получения алкенов путем комбинированного ферментативного превращения 3-гидроксиалкановых кислот
Получение летучих диенов с использованием ферментативной дегидратации низших алкенолов
Способ получения 3-гидрокси-3-метилмасляной кислоты из ацетона и ацетил-соа
Способ получения алкенов путем комбинированного ферментативного превращения 3-гидроксиалкановых кислот
Получение летучих диенов с использованием ферментативной дегидратации низших алкенолов

