Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ И ПРОИЗВОДНЫХ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
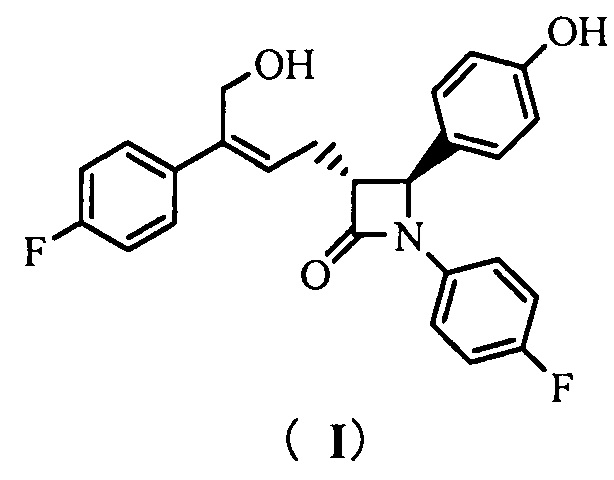
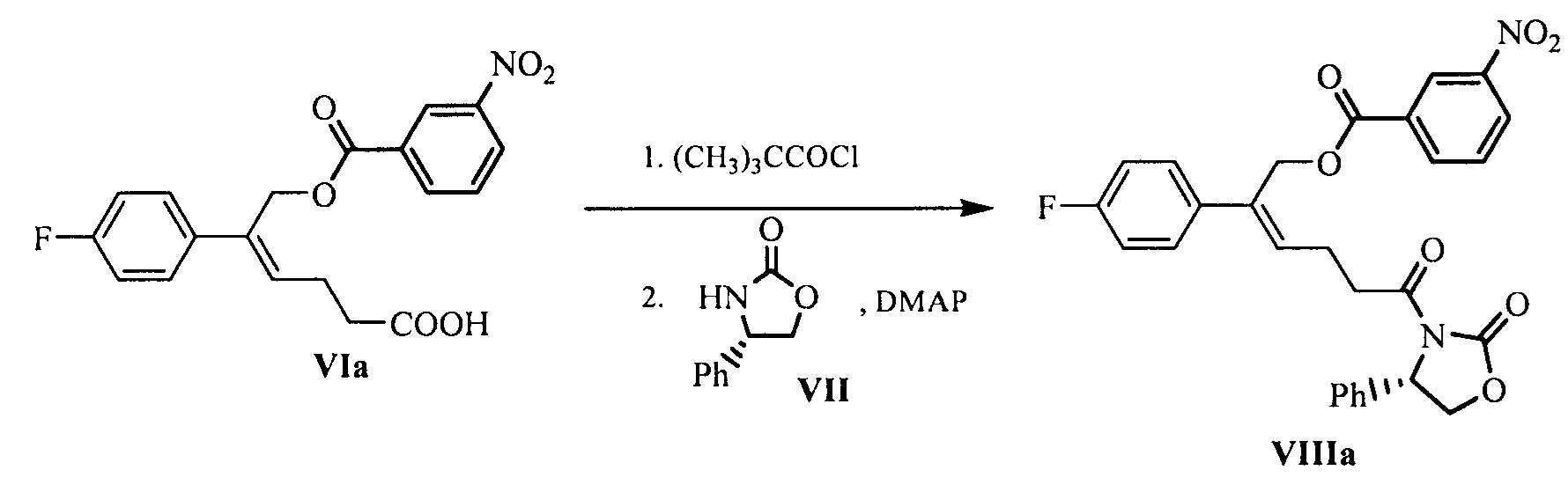
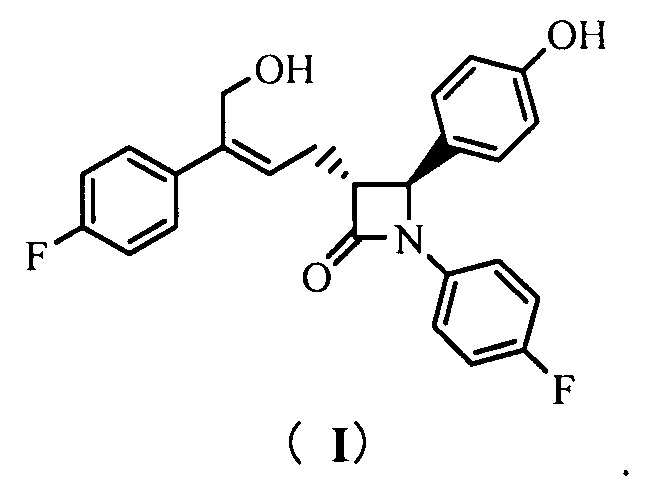
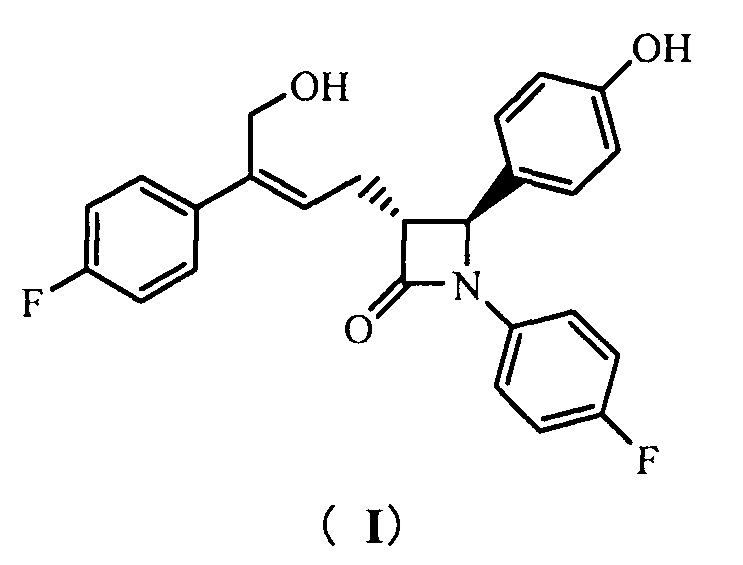
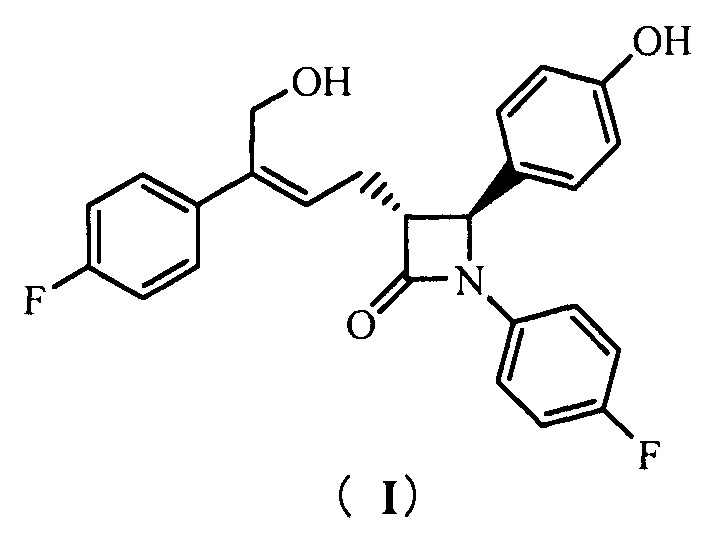
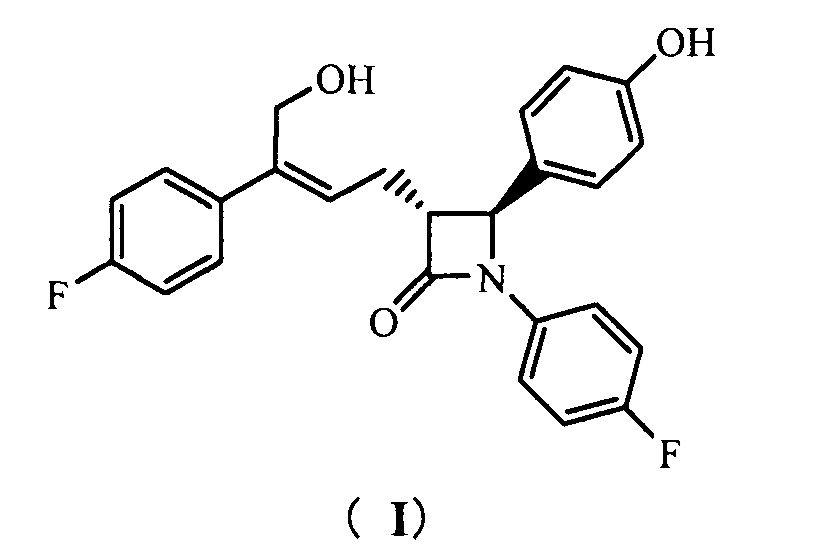
Настоящее изобретение относится к области химии, в частности, к новому способу получения агента, ингибирующего абсорбцию холестерина, соединения формулы (I), т.е., (3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-гидроксибут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она и его синтезированных промежуточных соединений.
УРОВЕНЬ ТЕХНИКИ
В западных странах ишемическая атеросклеротическая болезнь сердца (ишемическая болезнь сердца) является наиболее частой причиной смерти, и холестерин является одним из факторов риска, вызывающих данное заболевание. В настоящее время существует два вида лекарственных средств, которые применяют для снижения уровня холестерина в плазме. Одним из видов являются статины, которые являются ингибиторами ГМГ-КоА редуктазы, и могут эффективно ингибировать биосинтез холестерина in vivo. Другая роль заключается в предотвращении всасывания холестерина в тонком кишечнике, и Эзетимиб является распространенным ингибитором абсорбции холестерина. В патенте США (US 5846966) описан Эзетимиб, при этом химическая структура представляет собой следующую:

Боковая цепь в 3-положении атома углерода азетидинона представляет собой хиральный бензиловый спирт, и хиральный атом углерода имеет конфигурацию S. Взаимосвязь между структурой и активностью показывает, что фармакодинамические свойства конфигурации S лучше, чем конфигурации R, которая указывает, что стереохимия углерода бензила очень важна.
В WO 2011/017907 описан новый вид азетидиноновых соединений, которые также могут эффективно ингибировать абсорбцию холестерина, но боковая цепь в 3-положении атома углерода азетидинона представляет собой не хиральный бензиловый спирт, а ахиральный аллиловый спирт, и фармакодинамические свойства двойных связей конфигурации Z намного лучше, чем двойных связей конфигурации Е. Среди соединений данного типа химическая структура данного соединения с наилучшими фармакодинамическими свойствами, т.е., (3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-гидроксибут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-он, представляет собой следующую:

Поскольку путь синтеза для получения новых азетидиноновых соединений, указанных в WO 2011/017907 слишком долгий, и двойные связи конфигурации Z не могут быть получены стереоселективно, некоторые из стадий не подходят для промышленного производства, поэтому необходимо разработать новый технологический способ. Настоящее изобретение относится к новому способу получения такого типа азетидинона. Сырьевые материалы для нового способа легко получить, и способ включает несколько стадий синтеза, двойная связь конфигурации Z может быть получена стереоселективно, и операция является простой, выход является высоким, стоимость низкой, а способ можно применять для промышленного производства.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
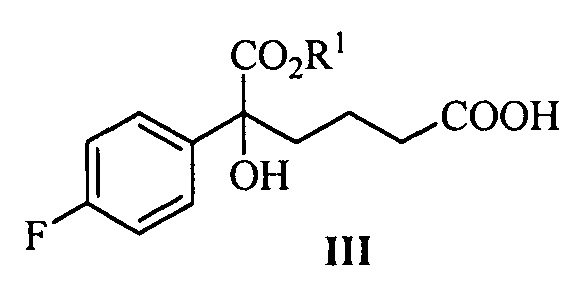
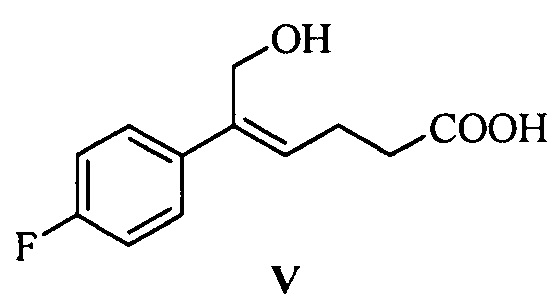
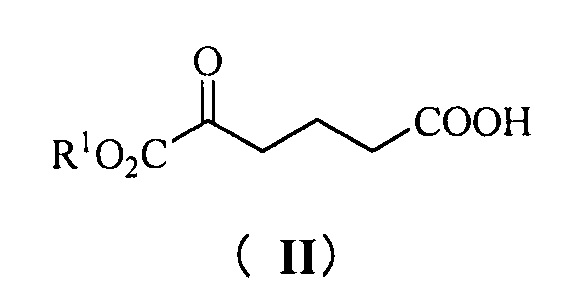
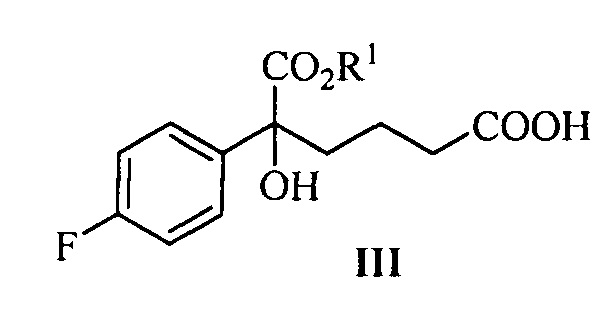
Одна из задач настоящего изобретения заключается в обеспечении новых ключевых промежуточных соединений (соединений формулы III, IV и V) для получения соединения формулы (I) и способов их получения:
 .
.
В одном аспекте настоящего изобретения предложены новые промежуточные соединения формулы III, IV и V, которые можно применять для получения соединения формулы (I):
 ;
;
 ;
;
 ,
,
где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил.
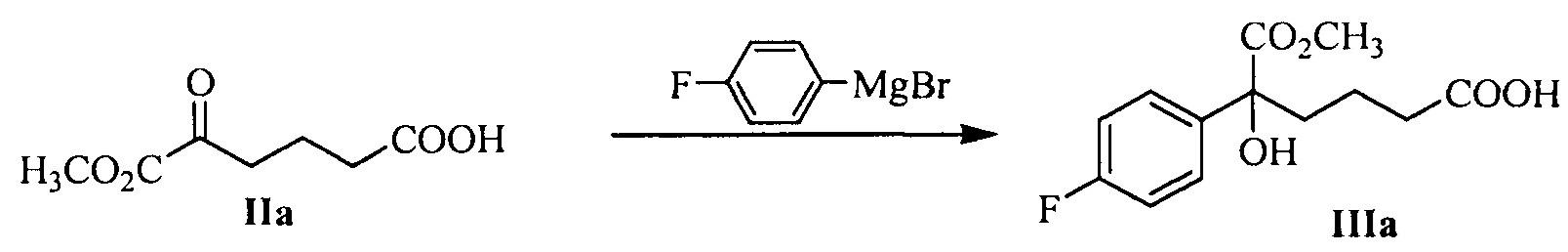
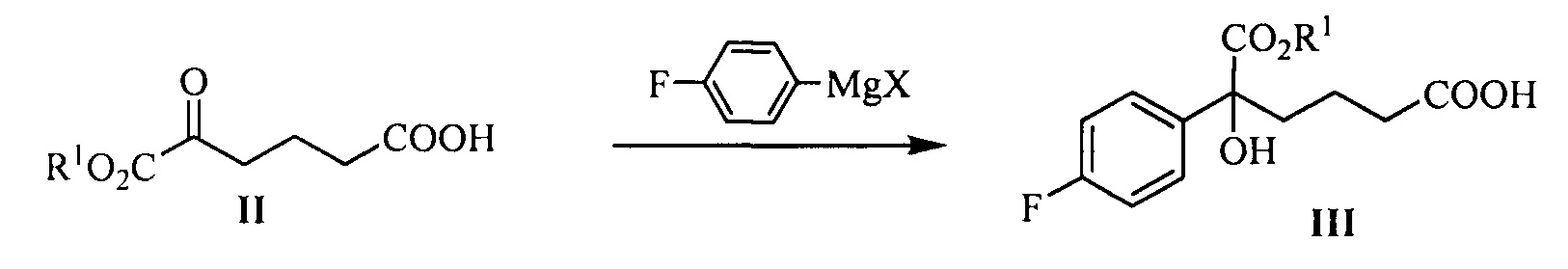
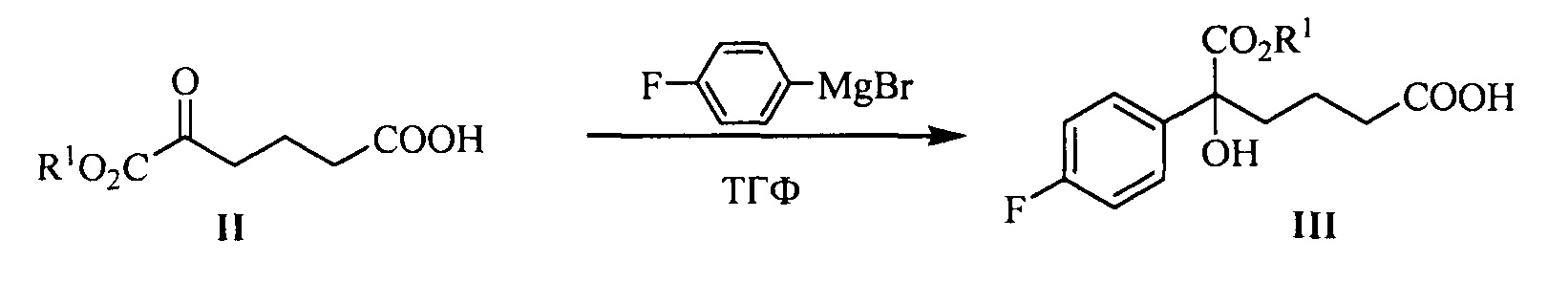
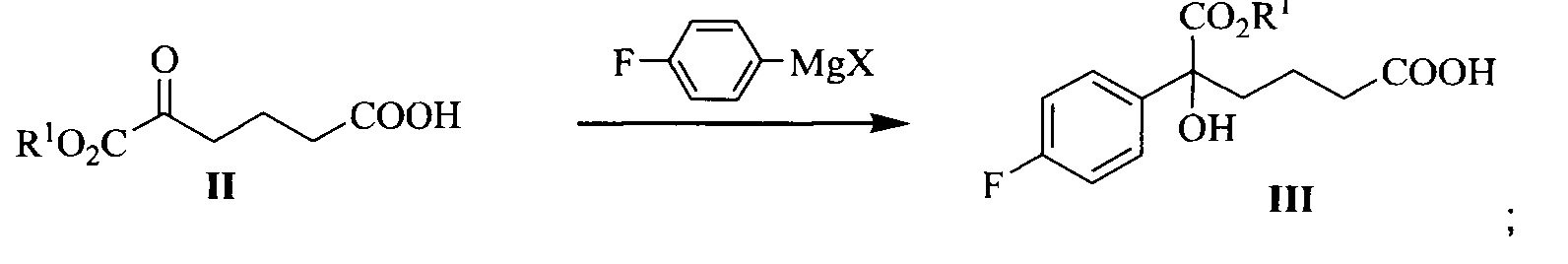
В другом аспекте настоящего изобретения предложен способ получения соединения формулы III, при этом указанный способ включает:
проведение селективного присоединения Гриньяра к кетону формулы II с галогенидом 4-фторфенилмагния, который применяют в качестве реактива Гриньяра, для получения третичного спирта формулы III:
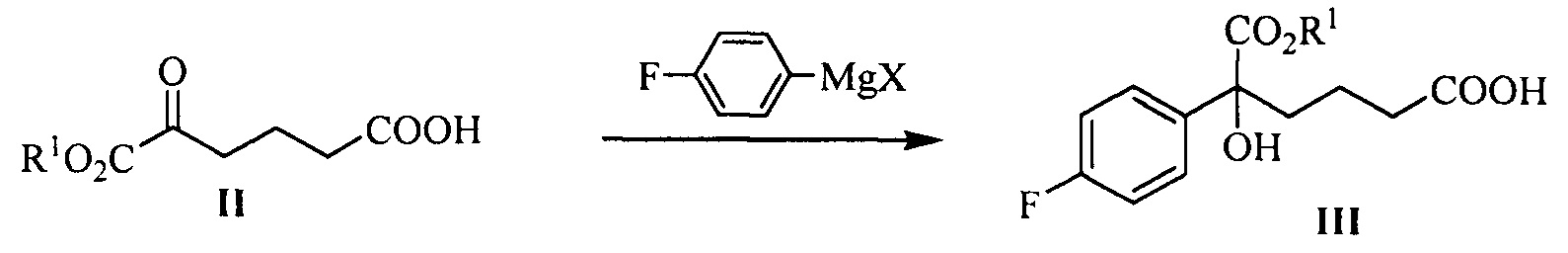
где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил; X представляет собой галоген, предпочтительно хлор, бром или йод.
галогенид 4-фторфенилмагния предпочтительно представляет собой бромид 4-фторфенилмагния.
Молярное соотношение соединения формулы II к галогениду 4-фторфенилмагния в реакции составляет 1:1,0~5,0, предпочтительно 1:1,1~3,0.
Температуру реакции поддерживают между -78°С~-5°С, предпочтительно -50°С~-10°С.
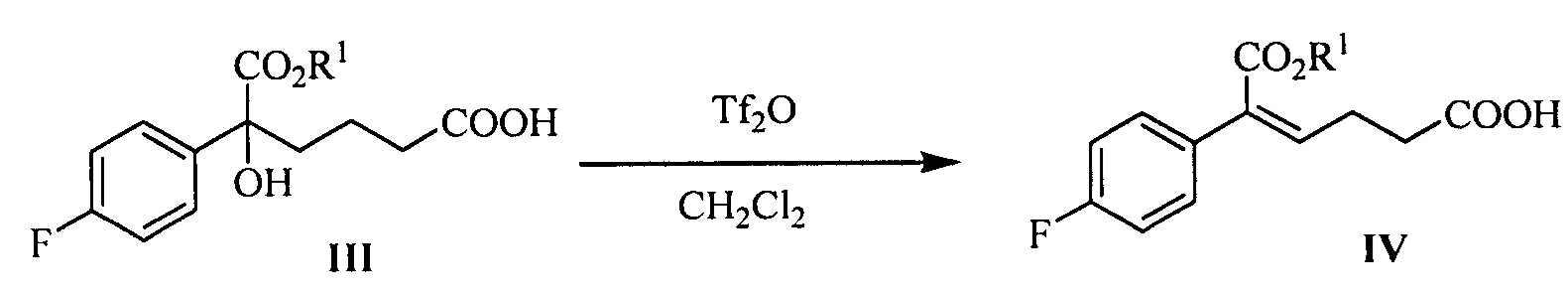
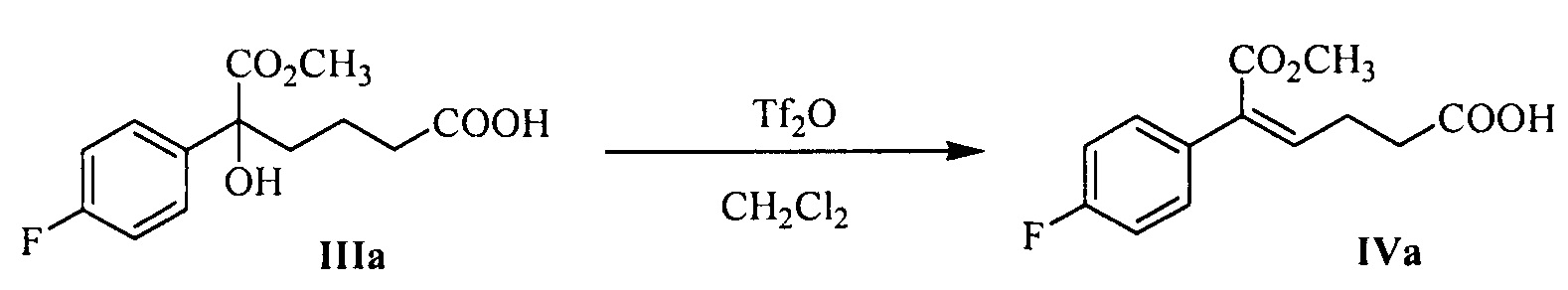
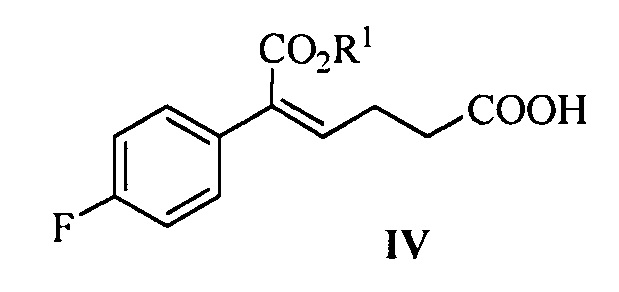
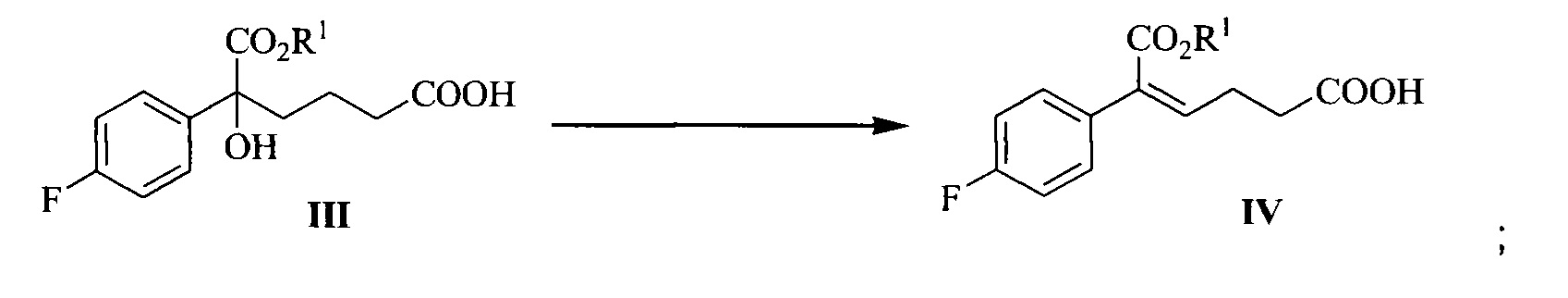
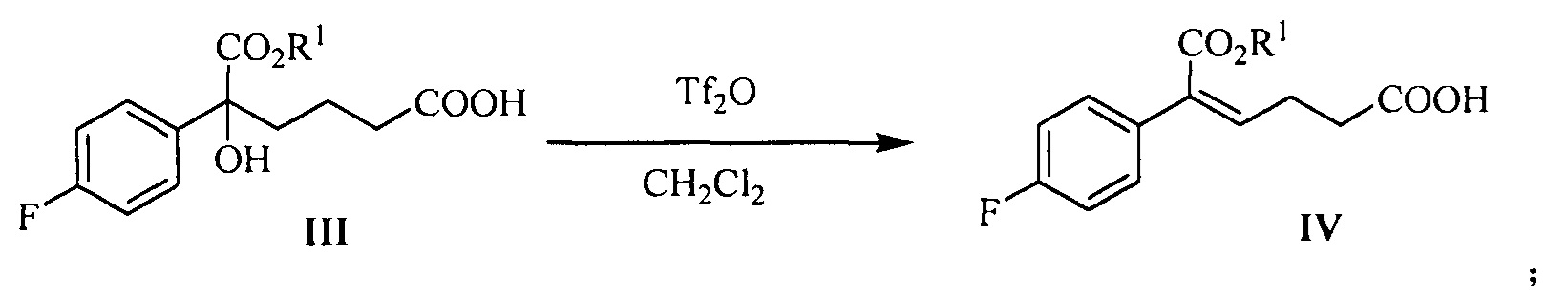
В другом аспекте настоящего изобретения предложен способ получения соединения формулы IV, при этом способ включает следующую стадию: под действием дегидратирующего агента третичный спирт формулы III подвергают стереоселективной дегидратации для получения (Z)-α,β-ненасыщенного сложного эфира формулы IV:

где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил.
В данной реакции указанный дегидратирующий агент выбран из концентрированной серной кислоты, п-толуолсульфоновой кислоты, фосфорной кислоты, трифторметансульфонового ангидрида или метансульфоновой кислоты, предпочтительно трифторметансульфонового ангидрида.
В данной реакции молярное соотношение соединения формулы III к дегидрирующему агенту составляет 1:1,0~3,0, предпочтительно 1:1,0~1,5.
Растворитель для реакции выбран из дихлорметана или толуола, предпочтительно дихлорметана.
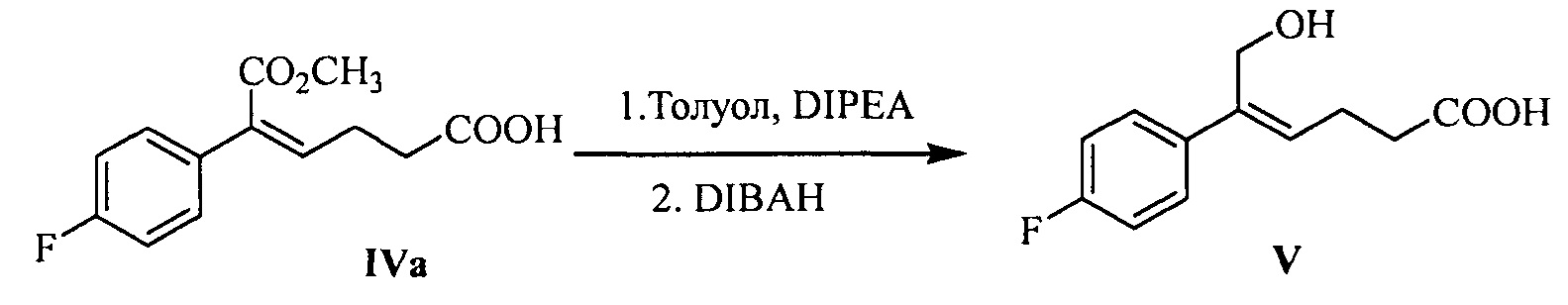
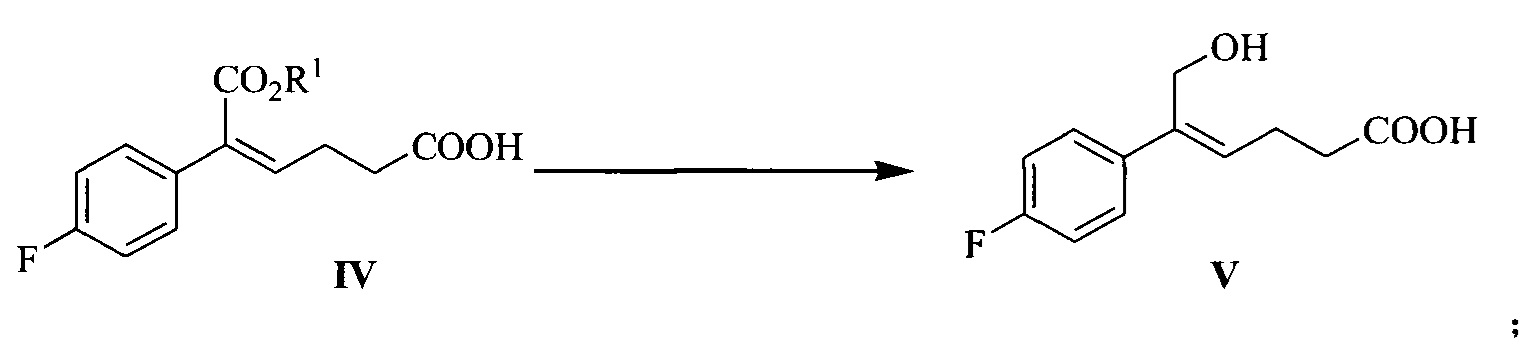
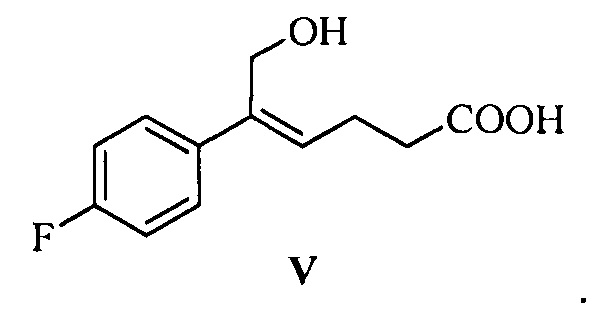
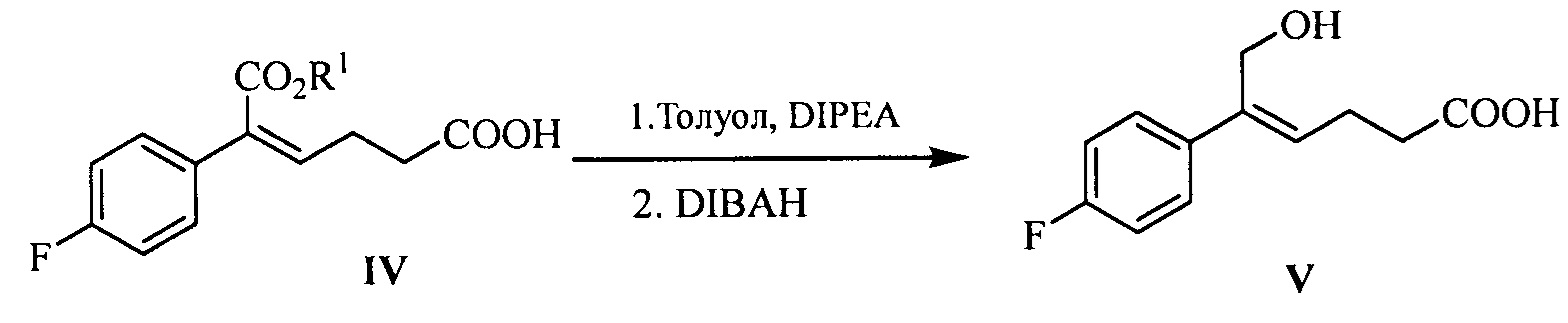
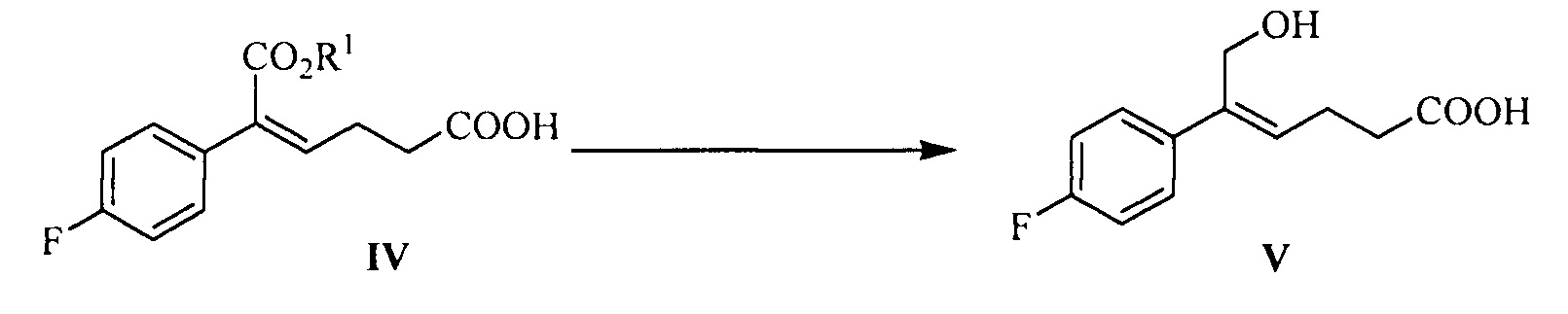
В другом аспекте настоящего изобретения предложен способ получения соединения формулы V, включающий: селективное восстановление сложного эфира формулы IV до спирта формулы V под действием восстанавливающего агента:

где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил.
Восстанавливающий агент реакции предпочтительно представляет собой гидрид диизобутилалюминия (DIBAH).
Растворитель для реакции выбран из дихлорметана, тетрагидрофурана, толуола или диоксана, предпочтительно толуола.
Молярное соотношение соединения формулы IV к восстанавливающему агенту составляет 1:2,5~5,0, предпочтительно 1:3,0~4,0.
Другая задача настоящего изобретения заключается в обеспечении нового способа получения соединения формулы (I) в соответствии с вышеуказанными промежуточными соединениями, чтобы также обеспечить улучшенный и простой способ получения соединения формулы (I) с хорошей селективностью и высоким выходом.
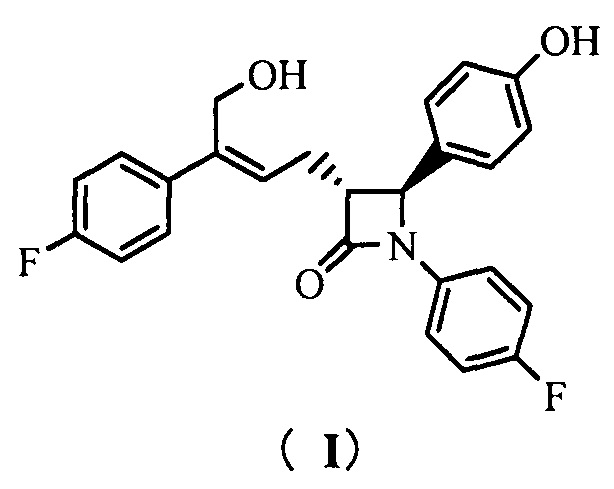
То есть, способ получения соединения (3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-гидроксибут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она, в котором соединение формулы (I) представляет собой новое азетидиноновое соединение, которое может восстанавливать холестерин крови.
При этом способ включает следующие стадии:
(1) Проведение селективного присоединения Гриньяра к кетону формулы II с галогенидом 4-фторфенилмагния, который применяют в качестве реактива Гриньяра, с получением третичного спирта формулы III:

где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил; X представляет собой галоген, предпочтительно хлор, бром или йод.
(2) Под действием дегидратирующего агента третичный спирт формулы III подвергают стереоселективной дегидратации с получением (Z)-α,β-ненасыщенного сложного эфира формулы IV:
 .
.
(3) Под действием восстанавливающего агента сложный эфир формулы IV селективно восстанавливают до спирта формулы V:

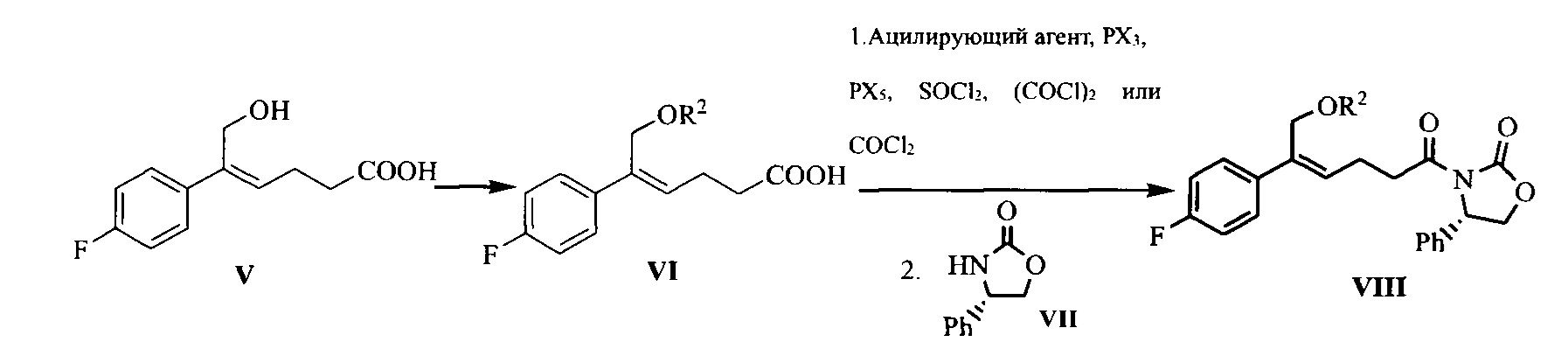
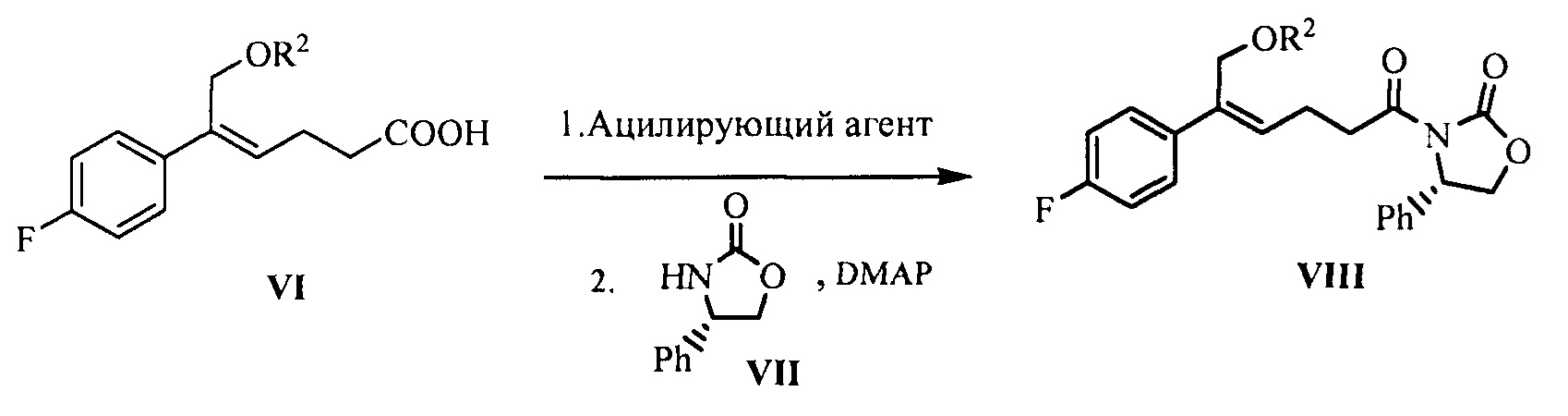
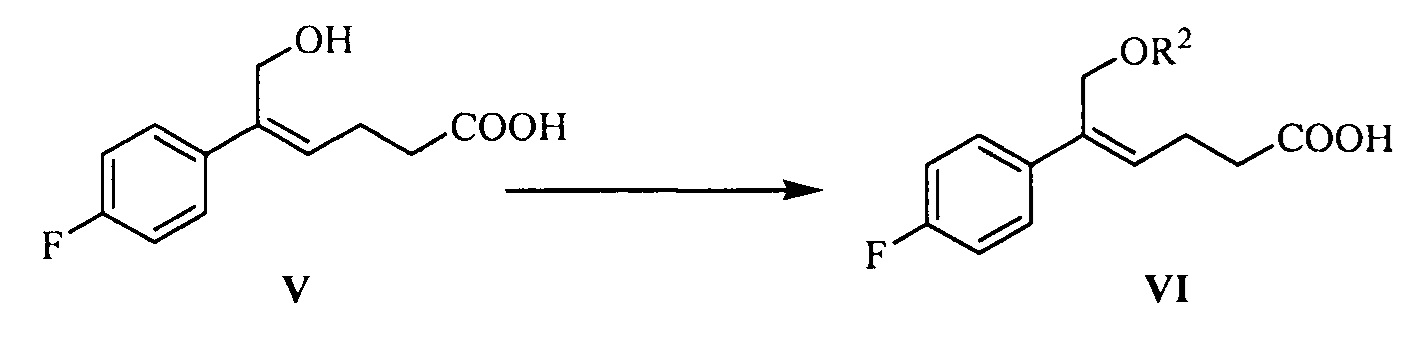
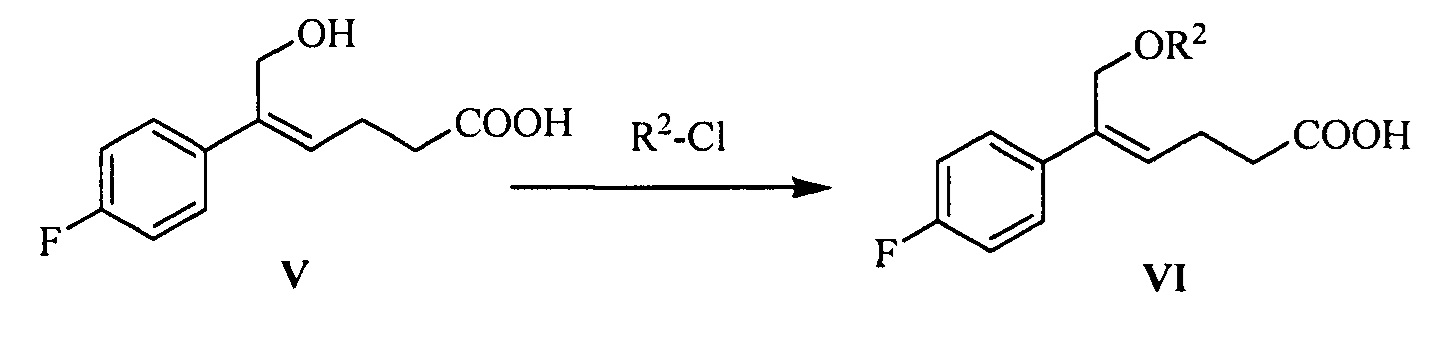
(4) Взаимодействие соединения формулы V с защищающим гидроксил соединением с получением соединения формулы VI:

где R2 представляет собой группу, защищающую гидроксил спирта, такую как: ацетил, замещенный или незамещенный бензоил («замещенный» включает галоген, алкил, нитрозамещенный) и т.д.
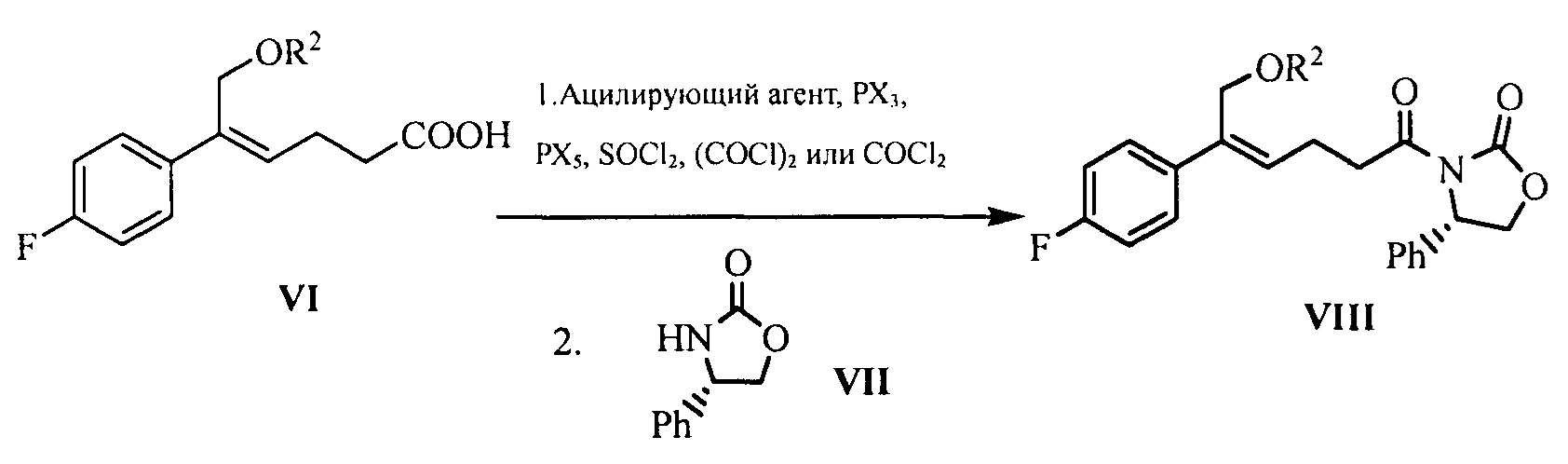
(5) Превращение карбоновой кислоты формулы VI в смешанный ангидрид или ацилгалид, с последующим взаимодействием с (S)-4-фенил-2-оксазолидоном формулы VII, который применяют в качестве хирального вспомогательного вещества с получением производного оксазолидона формулы VIII:
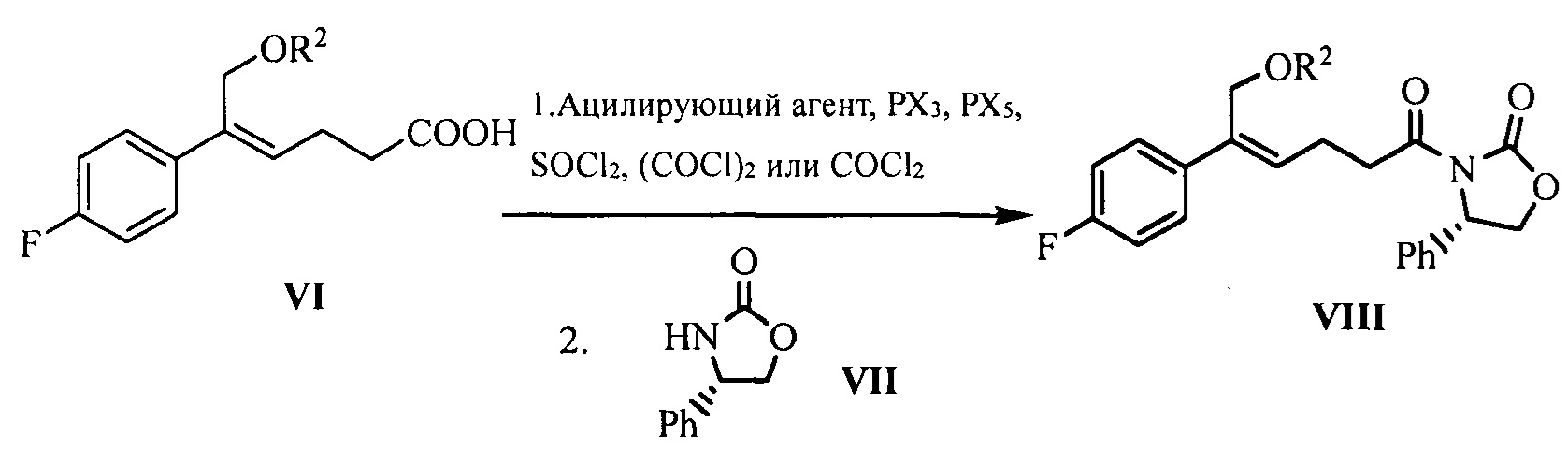
где карбоновую кислоту формулы VI подвергают взаимодействию с ацилирующим агентом с получением смешанного ангидрида; или карбоновую кислоту формулы VI подвергают взаимодействию с тригалогенидом фосфора, пентагалогенидом фосфора, дихлорсульфаном (SOCl2), оксалилхлоридом ((COCl)2) или фосгеном (COCl2) с получением ацилгалогенида; при этом X представляет собой хлор или бром.
Кроме того, стадия (4) и стадия (5) могут быть объединены, т.е. соединение формулы VIII может быть получено из соединения формулы V с помощью однореакторного способа, причем указанный способ включает следующие стадии: защита спиртового гидроксила формулы V в подходящем растворителе с получением соединения формулы VI, последующее превращение карбоновой кислоты формулы VI в смешанный ангидрид или ацилгалогенид без разделения и очистки, затем приведение во взаимодействие с (S)-4-фенил-2-оксазолидоном формулы VII, который применяют в качестве хирального вспомогательного вещества с получением производного оксазолидона формулы VIII:

где карбоновую кислоту формулы VI подвергают взаимодействию с ацилирующим агентом с получением смешанного ангидрида; или карбоновую кислоту формулы VI подвергают взаимодействию с тригалогенидом фосфора, пентагалогенидом фосфора, дихлорсульфаном (SOCl2), оксалилхлоридом ((COCl)2) или фосгеном (COCl2) с получением ацилгалогенида; при этом X представляет собой хлор или бром.
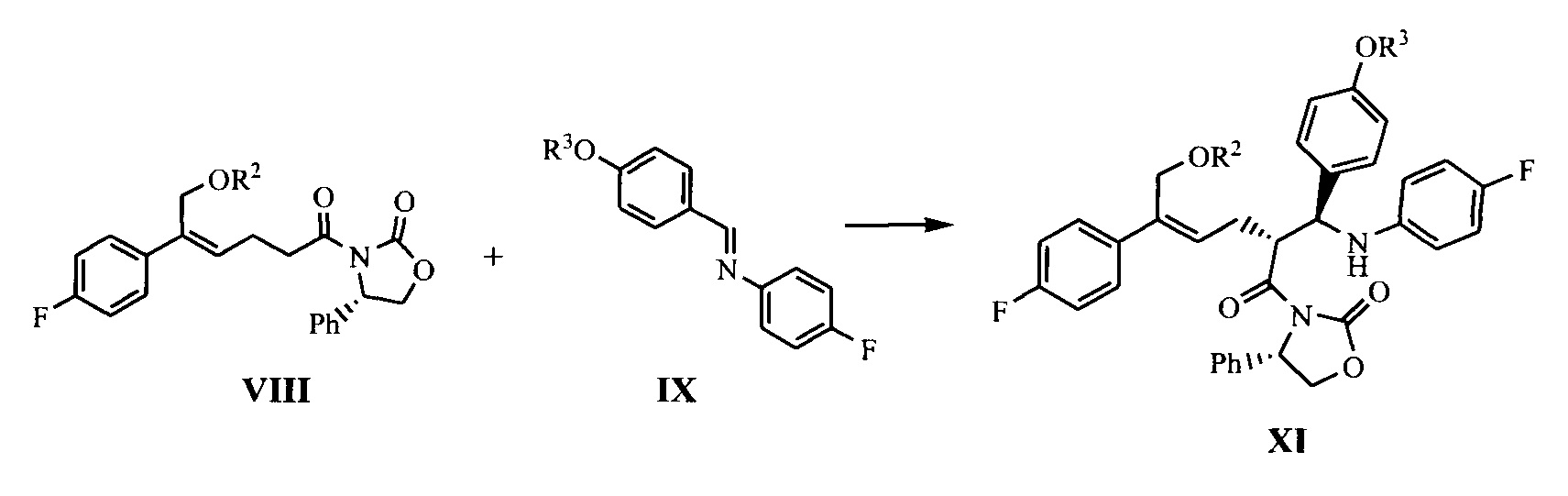
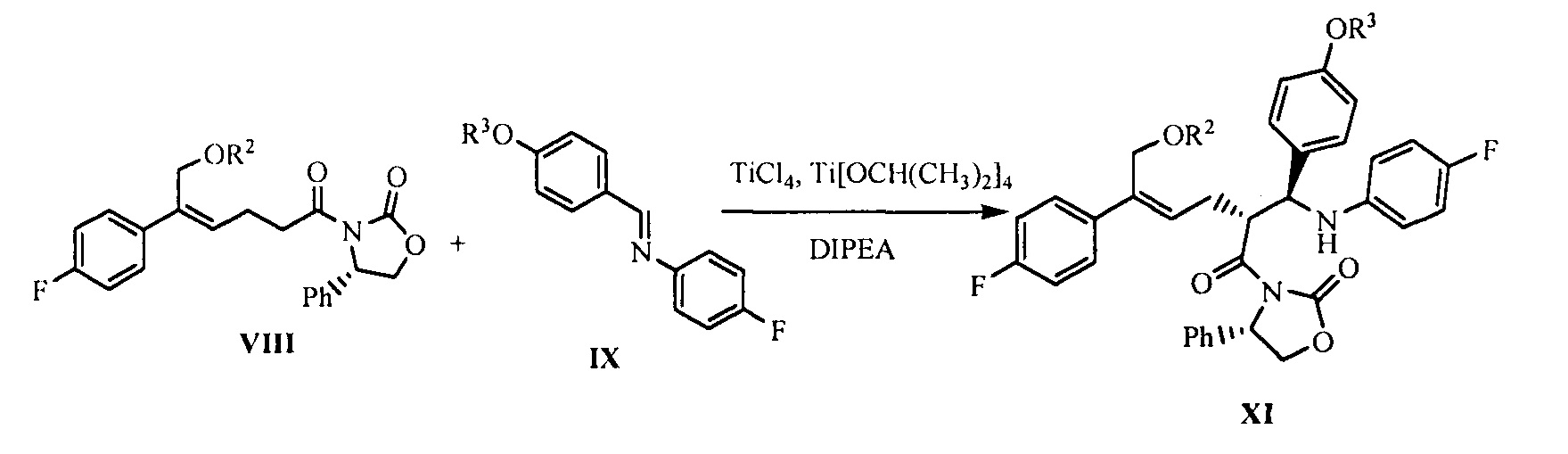
(6) В присутствии кислот Льюиса (тетрахлорид титана (TiCl4) и тетраизопропил титанат) и третичного амина взаимодействие производного оксазолидона формулы VIII с имином формулы IX с получением продукта присоединения формулы XI:
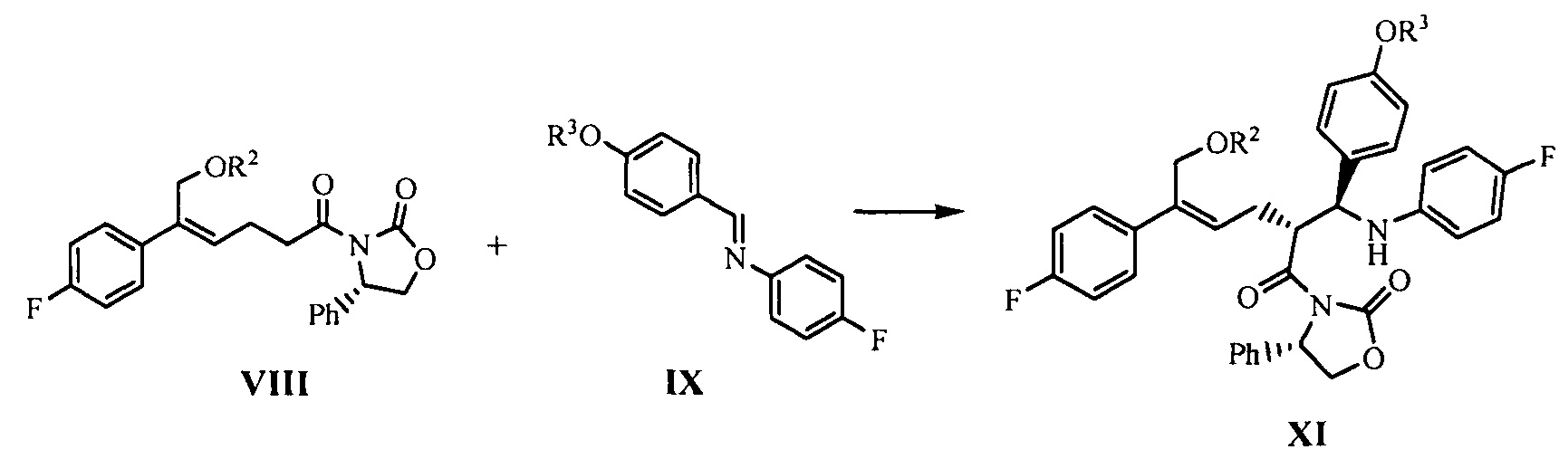
где R2 и R3 представляют собой гидроксилзащищающие группы, такие как: ацетил, замещенный или незамещенный бензоил («замещенный» включает галоген, алкил, нитрозамещенный) и т.д., R2 и R3 могут быть одинаковыми или разными.
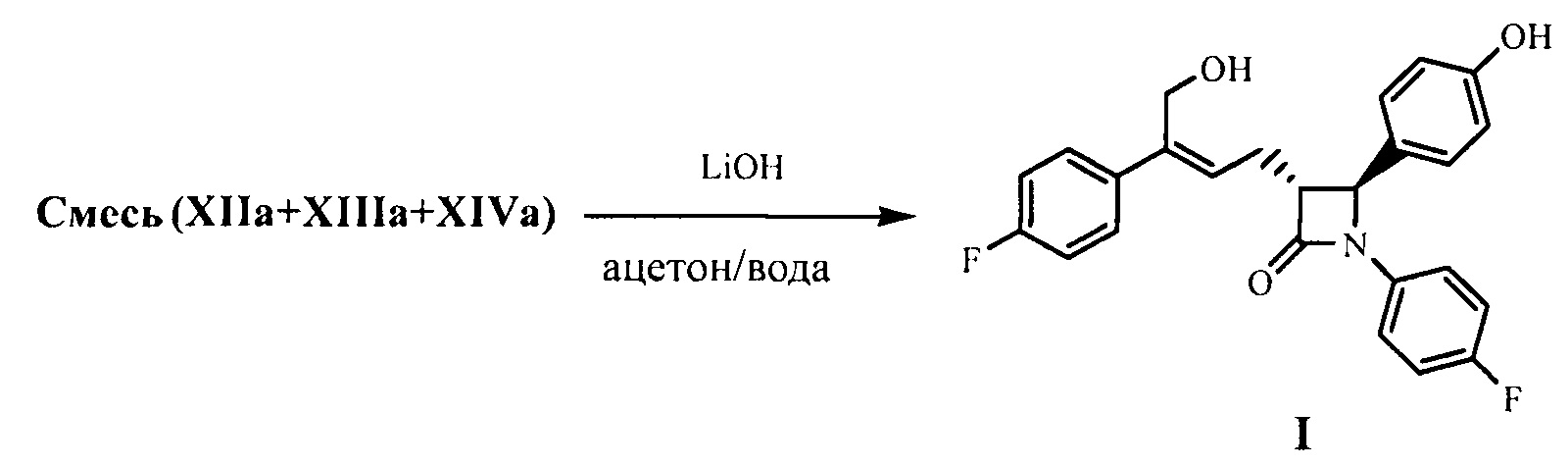
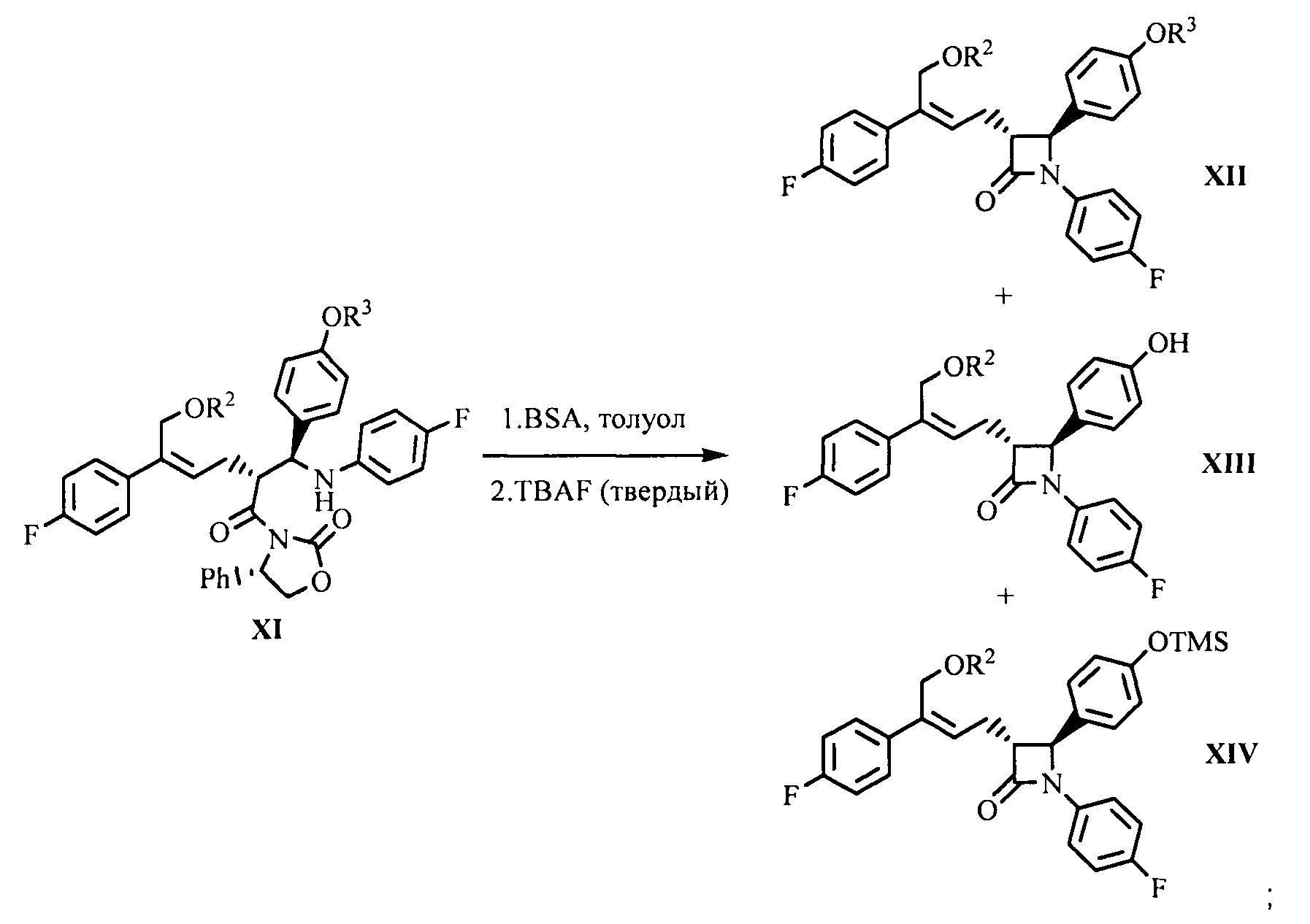
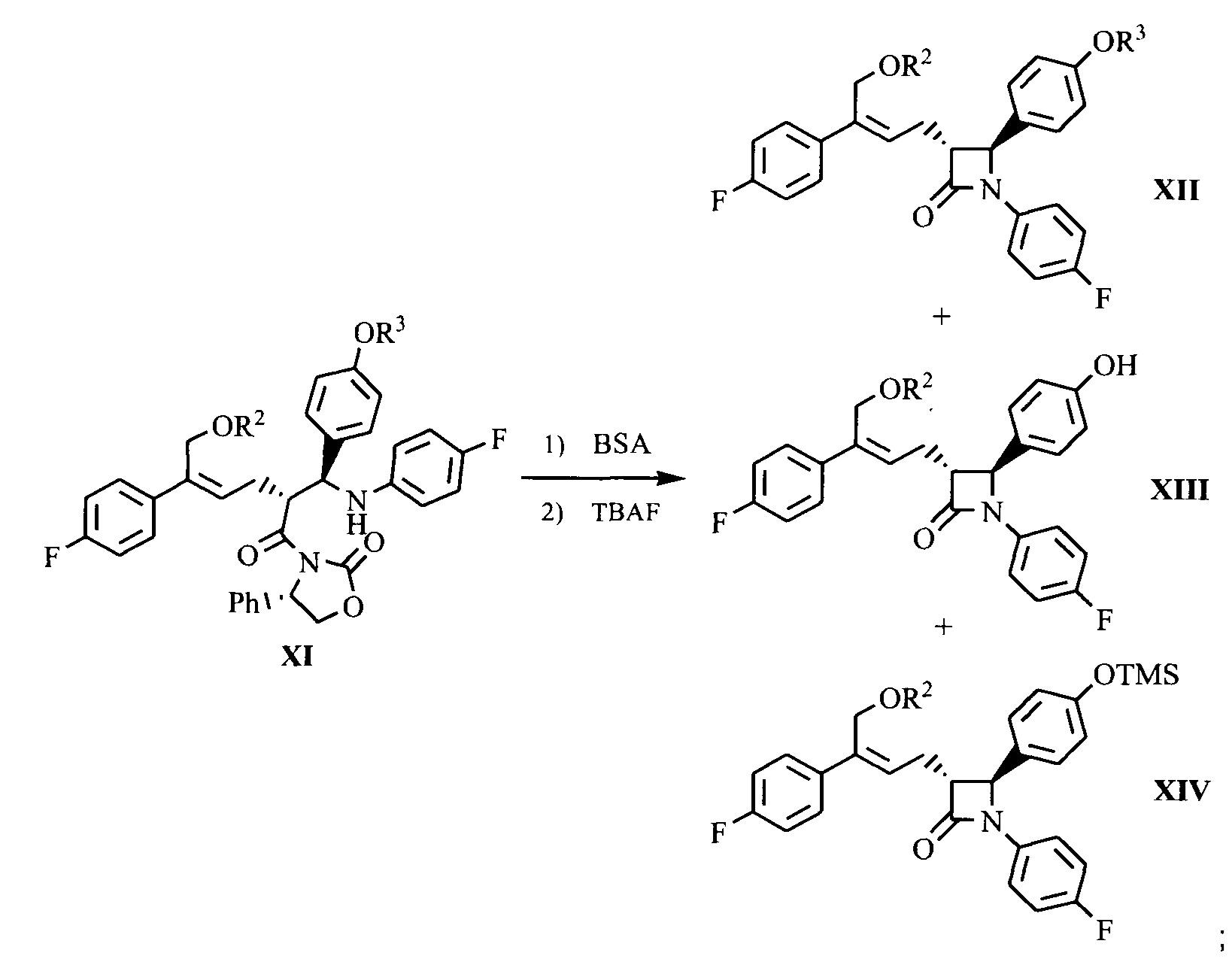
(7) Проведение циклизации для продукта присоединения формулы XI с применением N,O-бис(триметилсилил)ацетамида (BSA) и фторида тетрабутиламмония (TBAF), с получением β-лактамов формул XII, XIII и XIV:
 ;
;
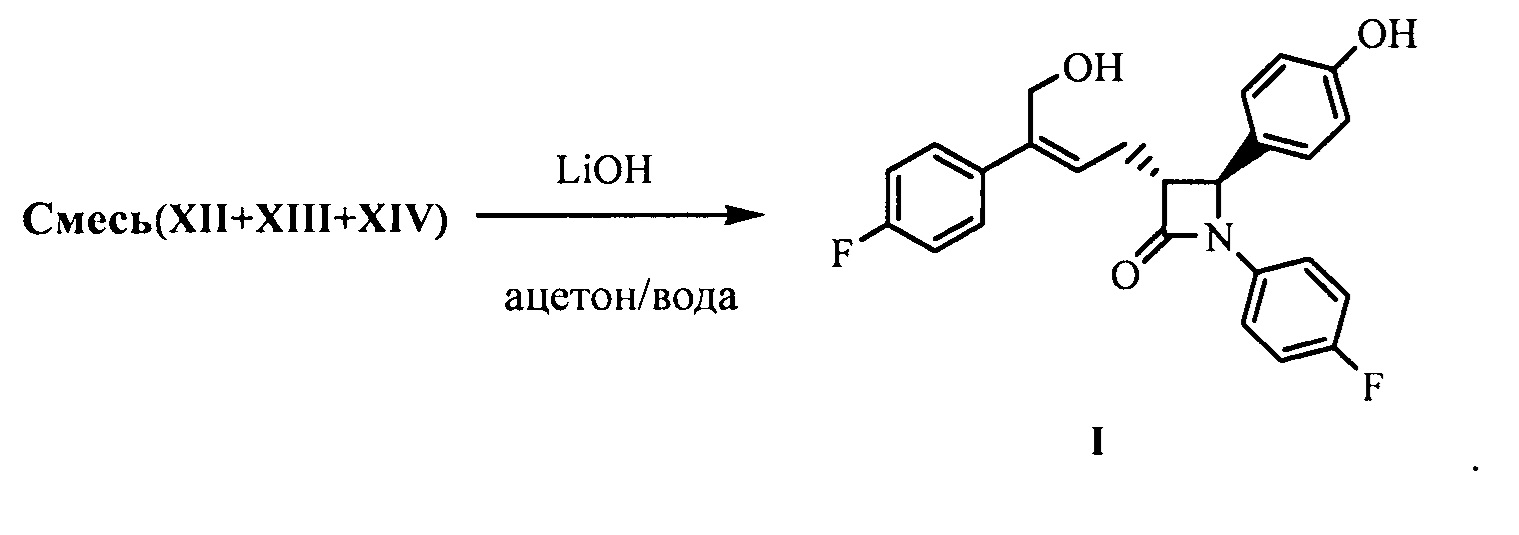
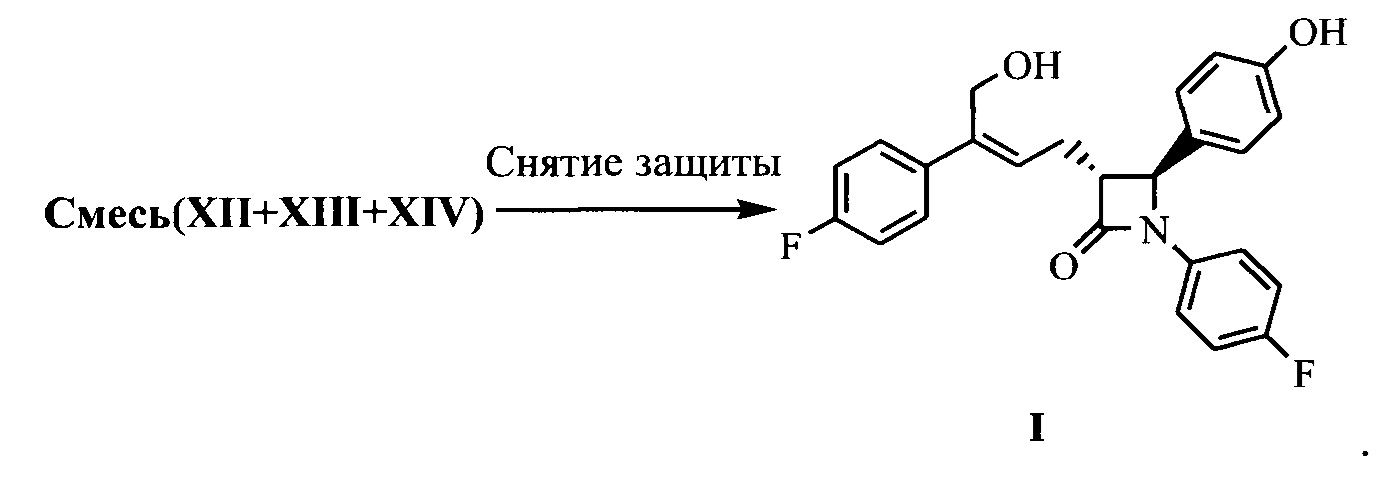
(8) Получение соединения формулы (I) посредством снятия защиты смеси соединений формул XII, XIII и XIV на стадии (7) под действием щелочи:
 .
.
В указанных выше стадиях взаимодействия, где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил; R2 и R3 представляют собой защищающие гидроксил группы, такие как: ацетил, замещенный или незамещенный бензоил («замещенный» включает галоген, алкил, нитрозамещенный) и т.д., R2 и R3 могут быть одинаковыми или разными.
На стадии (1), молярное соотношение соединения формулы II к галогениду 4-фторфенилмагния составляет 1:1,0~5,0, предпочтительно 1:1,1~3,0; галогенид 4-фторфенилмагния предпочтительно представляет собой бромид 4-фторфенилмагния. Температуру реакции поддерживают между -78°С~-5°С, предпочтительно -50°С~-10°С.
На стадии (2), указанный дегидратирующий агент выбран из концентрированной серной кислоты, п-толуолсульфоновой кислоты, фосфорной кислоты, трифторметансульфонового ангидрида или метансульфоновой кислоты, предпочтительно трифторметансульфонового ангидрида. Молярное соотношение соединения формулы III к дегидрирующему агенту составляет 1:1,0~3,0, предпочтительно 1:1,0~1,5. Растворитель для реакции выбран из дихлорметана или толуола, предпочтительно дихлорметана.
На стадии (3), молярное соотношение соединения формулы IV к восстанавливающему агенту составляет 1:2,5~5,0, предпочтительно 1:3,0~4,0. Восстанавливающий агент предпочтительно представляет собой гидрид диизобутилалюминия (DIBAH). Растворитель для реакции выбран из дихлорметана, тетрагидрофурана, толуола или диоксана, предпочтительно толуола.
На стадии (4), группа, защищающая гидроксил спирта, R2 предпочтительно представляет собой замещенный или незамещенный бензоил, более предпочтительно замещенный бензоил, где «замещенный» предпочтительно представляет собой замещенный нитрогруппой, более предпочтительно замещенный нитрогруппой в положении 3. Растворитель для реакции выбран из N,N-диметилформамида (ДМФА), N,N-диметилацетамида (DMA), диметилсульфоксида (ДМСО), 1,3-диметилпропиленмочевины (DMPU) или гексаметилфосфорамида (НМРА), предпочтительно N,N-диметилацетамида (DMA). Молярное соотношение соединения V к гидроксилзащищающему агенту составляет 1:1,0~3,0, предпочтительно 1:1,2~2,3.
На стадии (5), ацилирующий агент выбран из пивалоилхлорида, 3-нитробензоилхлорида или изобутилхлорформиата, предпочтительно пивалоилхлорида или 3-нитробензоилхлорида. Молярное соотношение соединения формулы VI к ацилирующему агенту составляет 1:1,0~2,0, предпочтительно 1:1,1~1,6. Молярное соотношение соединения формулы VI к (S)-4-фенил-2-оксазолидону формулы VII составляет 1:0,5~1,5, предпочтительно 1:0,8~1,1.
Когда стадию (4) объединяют со стадией (5), т.е., соединение формулы VIII получают из соединения формулы V с помощью однореакторного способа, группа, защищающая гидроксил спирта, R2 предпочтительно представляет собой замещенный или незамещенный бензоил, более предпочтительно замещенный бензоил, где «замещенный» предпочтительно представляет собой замещенный нитрогруппой, более предпочтительно замещенный нитрогруппой в положении 3. Растворитель выбран из N,N-диметилформамида (ДМФА), N,N-диметилацетамида (DMA), диметилсульфоксида (ДМСО), 1,3-диметилпропиленмочевины (DMPU) или гексаметилфосфорамида (НМРА), предпочтительно N,N-диметилацетамида (DMA). Молярное соотношение соединения V к агенту, защищающему гидроксильную группу спирта, составляет 1:1,0~3,0, предпочтительно 1:1,0~1,5. Ацилирующий агент выбран из пивалоилхлорида, 3-нитробензоилхлорида или изобутилхлорформиата, предпочтительно пивалоилхлорида или 3-нитробензоилхлорида. Молярное соотношение соединения формулы V к ацилирующему агенту составляет 1:1,0~2,0, предпочтительно 1:1,0~1,5. Молярное соотношение соединения формулы V к (S)-4-фенил-2-оксазолидону составляет 1:0,5~1,5, предпочтительно 1:0,7~1,1.
На стадии (6), группа, защищающая гидроксил фенола R3, предпочтительно представляет собой замещенный или незамещенный бензоил, более предпочтительно замещенный бензоил, где «замещенный» предпочтительно представляет собой замещенный нитрогруппой, более предпочтительно замещенный нитрогруппой в положении 3. Третичный амин предпочтительно представляет собой диизопропилэтиламин (DIPEA). Молярное соотношение соединения формулы VIII к имину (соединение формулы IX) составляет 1:1,0~2,0, предпочтительно 1:1,0~1,2; при этом температуру реакции поддерживают между -90°С~0°С, предпочтительно -80°С~-20°С; при этом спирты, кислоты или смешанные жидкие формы кислот, разбавленных органическими растворителями, можно применять в реакции гашения после обработки; при этом спирты выбраны из метанола, этанола, пропанола, изопропанола, третичного бутанола, предпочтительно изопропанола; при этом кислоты выбраны из неорганических кислот и органических кислот, включая хлористоводородную кислоту, серную кислоту, азотную кислоту, бромистоводородную кислоту, муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, бензойную кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, малеиновую кислоту или винную кислоту, предпочтительно органических кислот, включая муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, бензойную кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, малеиновую кислоту или винную кислоту, более предпочтительно уксусной кислоты или трифторуксусной кислоты.
На стадии (7), растворитель для реакции выбран из ацетонитрила или толуола, предпочтительно толуола. Молярное соотношение соединения формулы XI к N,O-бис(триметилсилил)ацетамиду (BSA) составляет 1:1,0~5,0, предпочтительно 1:2,0~4,0; и молярное соотношение соединения формулы XI к тетрабутиламмония фторида тригидрату (TBAF) составляет 1:0,1~0,5, предпочтительно 1:0,1~0,3.
На стадии (8) растворитель, используемый для снятия защиты с соединений формулы XII, XIII и XIV, предпочтительно представляет собой ацетон, щелочь предпочтительно представляет собой водный раствор гидроксида лития. Молярное соотношение щелочи к соединению XI на стадии (7) составляет 3,0~5,0:1.
В другом аспекте настоящее изобретение также относится к защите промежуточных соединений формулы III, IV и V.
Некоторые термины, используемые в настоящем изобретении, определены следующим образом:
«Галоген» относится к фтору, хлору, брому и йоду.
«Алкил», когда он представляет собой группу или часть группы, относится к линейной или разветвленной алифатической углеводородной группе. Наиболее предпочтительно, он представляет собой C1~С6 алкил, если не обозначено иное, при этом примеры линейного или разветвленного C1~С6 алкила включают, но не ограничиваются ими: метил, этил, н-пропил, 2-пропил, н-бутил, изобутил, третичный бутил, гексил и т.п.
«Комнатная температура» относится к 20~30°С.
Предпочтительные условия реакции настоящего изобретения приведены на следующих схемах
Стадия (1):

Стадия (2):
Стадия (3):

Стадия (4):

Стадия (5):

Стадия (6):

Стадия (7):

Стадия (8):

В указанных выше схемах реакции Tf2O представляет собой трифторметансульфоновый ангидрид, DIBAH представляет собой гидрид диизобутилалюминия, DMAP представляет собой 4-диметиламинопиридин, DIPEA представляет собой диизопропилэтиламин, BSA представляет собой N,O-бис(триметилсилил)ацетамид и TBAF представляет собой фторид тетрабутиламмония, где R1 представляет собой C1-С6 алкил, предпочтительно метил, этил или изопропил, более предпочтительно метил; R2 и R3 представляют собой защищающие гидроксил группы, такие как: ацетил, замещенный или незамещенный бензоил («замещенный» включает галоген, алкил, нитрозамещенный) и т.д., R2 и R3 могут быть одинаковыми или разными.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Способ получения дополнительно проиллюстрирован посредством комбинации указанных выше стадий реакции (1)~(8) ниже.
На стадии (1), спирт формулы III получают посредством реакции присоединения кетокарбонила формулы II и реактива Гриньяра. Способ взаимодействия является следующим: соединение формулы II (1 эквивалент) добавляют к безводному растворителю (такому как тетрагидрофуран или диэтиловый эфир, предпочтительно тетрагидрофуран), температуру понижают до значения между -78°С~-5°С (предпочтительно -50°С~-10°С), добавляют 1,0~5,0 эквивалентов (предпочтительно 1,1~3,0 эквивалентов) реактива Гриньяра (такого как галогенид 4-фторфенилмагния, предпочтительно бромид 4-фторфенилмагния), затем температуру поддерживают для взаимодействия в течение 1~2 часов при перемешивании, взаимодействие прекращают с помощью водного раствора хлорида аммония. Продукт, соединение формулы III, выделяют посредством экстракции и очищают посредством кристаллизации.
В данной реакции сырьевой материал, сложный кетоэфир карбоновой кислоты (соединение формулы II) может быть получен способом синтеза из документа Tetrahedron Letters, 1994, 35, 6089-6092, т.е. в растворителе ацетонитриле, кетон формулы II получают посредством окисления и раскрытия кольца циклопентанон-2-карбоксилата при катализе солью меди (такой как CuCl2⋅2Н2О, CuSO4⋅5H2O, Cu(ОАс)2⋅Н2О или Cu(ClO4)2⋅6H2O). Уравнение представляет собой следующее:
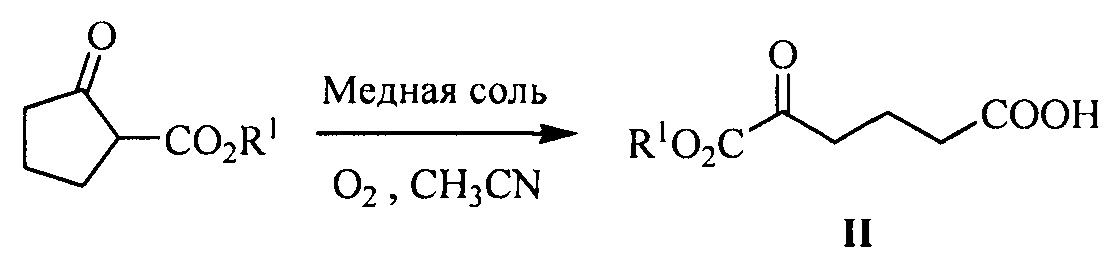 .
.
На стадии (2), (Z)-α,β-ненасыщенный сложный эфир формулы IV получают с помощью стереоселективной дегидратации третичного спирта формулы III. В J.Org.Chem.2006, 71, 5039-5042 описано, что (Z)-α-арил-α,β-ненасыщенный сложный эфир получают с помощью селективной дегидратации α-арил-α-гидроксил сложного эфира под действием дегидратирующего агента (трифторметансульфоновый ангидрид) и щелочи (такой как пиридин или DMAP). Время взаимодействия по литературным данным является долгим (взаимодействие длится 10~12 часов при комнатной температуре), и не указано, является ли ω-карбонил-α-арил-α-гидроксил сложный эфир подходящим для данного взаимодействия. Ранее известное взаимодействие было улучшено способом согласно настоящему изобретению для увеличения эффективности и селективности получения (Z)-ω-карбонил-α-арил-α,β-ненасыщенного сложного эфира. После улучшения время реакции сократилось, селективность улучшилась и необходимости в щелочном катализе (пиридин или DMAP) больше нет. Гидроксильный сложный эфир карбоновой кислоты (соединение формулы III, 1 эквивалент) растворяют в неполярном безводном растворителе (таком как дихлорметан), добавляют 1,0~3,0 эквивалента (предпочтительно 1,0~1,5 эквивалента) дегидратирующего агента (предпочтительно трифторметансульфонового ангидрида) при 5°С~15°С, реакционную смесь нагревают с обратным холодильником в течение 1~2 часов, и затем реакцию останавливают водой. Продукт, соединение формулы IV, разделяют с помощью экстракции.
На стадии (3), сложноэфирную группу соединения формулы IV селективно восстанавливают и сохраняют карбонильную группу. Соединение формулы IV (1 эквивалент) растворяют в подходящем растворителе (предпочтительно толуоле), добавляют щелочь (такую как триэтиламин или диизопропилэтиламин) при комнатной температуре, чтобы позволить карбонильной группе образовать соль, затем температуру понижают, медленно добавляют 2,5~5,0 эквивалентов (предпочтительно 3,0~4,0 эквивалентов) восстанавливающего агента (предпочтительно гидрида диизобутилалюминия) при температуре -30°С~-5°С, реакционную смесь перемешивают в течение 20~60 минут. После окончания реакции реакционный раствор медленно добавляют к щелочи (такой как водный раствор гидроксида калия, гидроксида лития или гидроксида натрия, предпочтительно водный раствор гидроксида натрия) при температуре t<15°C, перемешивают, разделяют на слои, водную фазу экстрагируют подходящим растворителем (например, дихлорметаном) для удаления органических примесей, затем подкисляют кислотой (такой как соляная кислота), затем экстрагируют с помощью подходящего растворителя (например, этилацетата), продукт отделяют и очищают с помощью кристаллизации с получением (Z)-5-(4-фторфенил)-6-гидроксил-гекс-4-еновой кислоты (соединение формулы V).
На стадии (4) гидроксильную группу соединения формулы V селективно защищают и сохраняют карбонильную группу. Соединение формулы V (1 эквивалент) растворяют в подходящем безводном растворителе (предпочтительно N,N-диметилацетамиде), добавляют 1,0~3,0 эквивалента (предпочтительно 1,2~2,3 эквивалента) защищающего гидроксил агента (предпочтительно нитробензоилхлорида, более предпочтительно 3-нитробензоилхлорида) при -5°С~40°С и обеспечивают взаимодействие в течение 5~6 часов. Добавляют подходящую щелочь (такую как пиридин) для гидролиза полученной смеси ангидрида, затем добавляют щелочь (такую как имидазол) для удаления карбоновой кислоты защищающего агента, высвободившейся в процессе гидролиза с образованием соли. Продукт, соединение формулы VI, отделяют с помощью экстракции.
На стадии (5) соединение формулы VI (1 эквивалент) растворяют в безводном инертном растворителе (таком как тетрагидрофуран или дихлорметан, предпочтительно дихлорметан), добавляют 1,0~2,0 эквивалента (предпочтительно 1,1~1,6 эквивалента) ацилирующего агента (такого как пивалоилхлорид, изобутилхлорформиат или 3-нитробензоилхлорид, предпочтительно пивалоилхлорид или 3-нитробензоилхлорид), в то же время обеспечивают взаимодействие смеси в течение 3~4 часов при комнатной температуре в присутствии щелочи (такой как триэтиламин) с получением смеси ангидрида. Затем к полученному раствору смеси ангидрида добавляют 0,5~1,5 эквивалента (предпочтительно 0,8~1,1 эквивалента) (S)-4-фенил-2-оксазолидона формулы VII, добавляют 0,1~0,3 эквивалента подходящего катализатора (такого как 4-диметиламинопиридин) и перемешивают в течение 3-5 часов при комнатной температуре для образования ацилированного производного оксазолидона формулы VIII посредством конденсации. Продукт отделяют с помощью экстракции и очищают кристаллизацией.
Кроме того, стадия (4) может быть объединена со стадией (5), соединение формулы VIII может быть получено из соединения формулы V с помощью однореакторного способа. Соединение формулы V (1 эквивалент) растворяют в подходящем безводном растворителе (предпочтительно N,N-диметилацетамиде), добавляют 1,0~3,0 эквивалента (предпочтительно 1,0~1,5 эквивалента) защищающего гидроксил агента (предпочтительно нитробензоилхлорида, предпочтительно 3-нитробензоилхлорида) при -5°С~40°С, после окончания взаимодействия реакционный раствор добавляют к раствору 1,0~2,0 эквивалентов (предпочтительно 1,0~1,5 эквивалентов) ацилирующего агента (такого как пивалоилхлорид, изобутилхлорформиат или 3-нитробензоилхлорид, предпочтительно пивалоилхлорид или 3-нитробензоилхлорид, более предпочтительно 3-нитробензоилхлорид) и щелочи (такой как триэтиламин), растворенным в безводном инертном растворителе (таком как тетрагидрофуран или дихлорметан, предпочтительно дихлорметан), затем добавляют 0,5~1,5 эквивалентов (предпочтительно 0,7~1,1 эквивалентов) хирального вспомогательного вещества ((S)-4-фенил-2-оксазолидона) формулы VII и 0,1~0,5 эквивалента подходящего катализатора (такого как 4-диметиламинопиридин), температуру поддерживают для взаимодействия в течение 6~7 часов, ацилированное производное оксазолидона формулы VIII получают с помощью конденсации. Продукт отделяют с помощью экстракции и очищают кристаллизацией.
На стадии (6), температуру снижают в присутствии подходящего безводного растворителя (такого как безводный дихлорметан) и добавляют защиту потоком сухого инертного газа (такого как азот), кислоты Льюиса TiCl4 (1,1~1,5 эквивалентов) и тетраизопропил титанат (0,3~0,5 эквивалент) при температуре -5°С~0°С для осуществления взаимодействия при перемешивании в течение 20-40 минут с получением реагента титана, который оставляют для применения. Ацилированное производное оксазолидона формулы VIII (1 эквивалент), защищенное соединение имина формулы IX (1,0~2,0 эквивалентов, предпочтительно 1,0~1,2 эквивалентов) растворяют в безводном растворителе (таком как безводный дихлорметан), добавляют третичный амин (такой как диизопропилэтиламин), перемешивают в течение 10 минут, температуру снижают, медленно по каплям добавляют реагент титана, полученный выше, при температуре -90°С~0°С (предпочтительно -80°С~-20°С), температуру непрерывно поддерживают для осуществления взаимодействия, после окончания реакции добавляют подходящее количество кислоты (предпочтительно уксусной кислоты или трифторуксусной кислоты) для гашения реакции. Между тем, соль титана удаляют путем добавления разбавленной серной кислоты, а затем соединение формулы XI отделяют с помощью экстракции и очищают с помощью кристаллизации.
На стадии (7) соединение формулы XI (1 эквивалент) растворяют в подходящем растворителе (таком как толуол), добавляют 1,0~5,0 эквивалентов (предпочтительно 2,0~4,0 эквивалентов) N,O-бис(триметилсилил)ацетамида (BSA), и подвергают взаимодействию в течение 2-3 часов при температуре 50°С~70°С, затем добавляют 0,1~0,5 эквивалента (предпочтительно 0,1~0,3 эквивалента) тетрабутиламмония фторида (TBAF), подвергают взаимодействию в течение 2~5 часов при температуре, смесь соединений формулы XII, XIII и XIV получают с помощью циклизации.
На стадии (8) смесь соединений формулы XII, XIII и XIV растворяют в подходящем растворителе (предпочтительно ацетоне), добавляют 3-5 эквивалентов (рассчитывают на основании поданного количества соединения XI из стадии 7 как 1 эквивалента) щелочи (предпочтительно водного раствора гидроксида лития) при комнатной температуре для гидролиза гидроксилзащищающей группы, смесь подвергают взаимодействию при перемешивании в течение 2~3 часов, затем подкисляют слабой кислотой (такой как разбавленная серная кислота или разбавленная хлористоводородная кислота), экстрагируют, концентрируют и разделяют посредством колоночной хроматографии с получением соединения формулы I, которое очищают с помощью перекристаллизации.
В настоящем изобретении кетоэфир карбоновой кислоты, представленный формулой (II), применяют в качестве исходного материала и подвергают реакции присоединения Гриньяра, стереоселективной дегидратации, восстановлению сложноэфирной группы, защите гидроксильной группы, присоединению имина после конденсации с хиральным вспомогательным веществом, циклизации и удалению защитной группы с получением соединения, представленного формулой (I). Преимущества настоящего изобретения могут быть объединены следующим образом:
a) По сравнению со способом получения, описанном в WO 2011/017907, количество стадий синтеза значительно уменьшилось с 14 стадий до 8 стадий; в то же время, колоночную хроматографию заменяют на многократную перекристаллизацию, чтобы упростить операцию очистки и снизить стоимость.
b) На стадии 2 настоящего способа Z алкен получают посредством стереоселекции, обеспечивая начало последовательных многостадийных реакций с одного изомера, что облегчает операцию разделения и снижение стоимости.
c) Для защиты гидроксильных групп (включая гидроксильные группы спирта и фенольные гидроксильные группы) применяют нитрозамещенный бензоил, предпочтительно применяют бензоил, нитрозамещенный в положении 3. В одном аспекте способность кристаллизации ключевого промежуточного соединения увеличивается, так что он может быть эффективно очищен с помощью простой операции кристаллизации; в другом аспекте предпочтительно снятие защиты в неагрессивной среде впоследствии и предотвращение возникновения сопутствующих побочных реакций (например, раскрытие кольца β-лактама) в щелочных условиях.
d) Способ согласно настоящему изобретению подходит для промышленного производства, и выход является высоким.
Таким образом, настоящее изобретение имеет преимущества легко доступного сырья, нескольких стадий синтеза, простоты операций, высокого выхода, хорошей стереоселективности и низкой стоимости, а также может быть использовано для промышленного производства.
ВАРИАНТЫ РЕАЛИЗАЦИИ
Следующие примеры предназначены исключительно для иллюстрации настоящего изобретения, настоящее изобретение не ограничивается данными примерами.
Пример 1: Получение сырьевого материала имина IXa

Стадия 1: 12 кг 4-гидроксибензальдегида и 60 л метанола добавляли в 100 л реакционную емкость и растворяли при перемешивании, 12 кг 4-фторанилина по каплям добавляли при комнатной температуре, осуществляли непрерывное взаимодействие указанной смеси в течение 2~3 часов после добавления. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (4-гидроксилбензальдегид), твердое вещество, полученное при взаимодействии, отфильтровывали, сушили и взвешивали 19 кг (выход: 90%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 6,88 (d, 2 Н, J = 8,4 Гц), 7,18-7,27 (m, 4 Н), 7,76 (d, 2 Н, J = 8,4 Гц), 8,46 (s, 1 Н), 10,11 (s, 1 Н).
Стадия 2: Продукт, полученный на стадии 1, и 200 л дихлорметана добавляли в 500 л реакционную емкость и растворяли при перемешивании, при комнатной температуре добавляли 22 кг триэтиламина, 1,8 кг 4-диметиламинопиридина (DMAP), по каплям добавляли 50 л раствора дихлорметана с 20 кг растворенного 3-нитробензоилхлорида, осуществляли непрерывное взаимодействие указанной смеси в течение 2~3 часов после добавления, и контролировали посредством ТСХ до исчезновения пятен сырьевого материала (продукт, полученный на стадии 1). рН доводили до 4~6 посредством 2М хлористоводородной кислоты, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали дихлорметаном (30 л × 2 раза), органические фазы объединяли и затем промывали 1 раз солевым раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении, неочищенный продукт перекристаллизовывали в безводном этаноле, фильтровали и сушили до получения 19 кг имина IXa (выход: 59%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 7,23 (t, 2 Н, J = 8,8 Гц), 7,31-7,35 (m, 2 Н), 7,49 (d, 2 Н, J = 8,4 Гц), 7,90 (t, 1 Н, J = 8,0 Гц), 8,03 (d, 2 Н, J = 8,4 Гц), 8,52-8,58 (m, 2 Н), 8,65 (s, 1 Н), 8,78 (s, 1 Н).
Пример 2: Получение 5-(4-фторфенил)-5-гидрокси-6-метокси-6-оксо-гексановой кислоты (IIIa)

100 г (0,563 моль) 6-метокси-5,6-диоксо-гексановой кислоты (соединение IIa) и 300 мл тетрагидрофурана добавляли в 3 л реакционную колбу, смесь защищали в атмосфере азота и растворяли при перемешивании, температуру снижали до -20°С~-10°С, медленно по каплям добавляли 1М раствор бромида 4-фторфенилмагния в ТГФ (1,4 л, 1,4 моль), температуру поддерживали для взаимодействия в течение 1~2 часов после добавления. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение IIa).
Водный 25% раствор хлорида аммония (60 г хлорида аммония, растворенного в 180 мл воды) добавляли при температуре -20°С~0°С и перемешивали в течение 5 минут, затем рН доводили до 3~5 посредством 4М хлористоводородной кислоты при температуре 0°С ~30°С, затем добавляли 600 мл н-гептана, и перемешивали в течение 5 минут, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали этилацетатом (140 мл × 2 раза), органические фазы объединяли, и затем промывали 2 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении, неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 64,6 г соединения IIIa (ВЭЖХ чистота: 93,2%; выход: 39,6%).
1Н ЯМР (ДМСО-d6): 1,33-1,43 (m, 2 Н), 1,89-1,96 (m, 1 Н), 1,99-2,04 (m, 1 Н), 2,15 (t, 2 Н, J = 7,6 Гц), 3,61 (s, 3 Н), 5,99 (s, 1 Н), 7,12-7,17 (m, 2 Н), 7,47-7,51 (m, 2 Н), 12,02 (s, 1 Н); MS (m/z): 269 [М-Н]-.
Пример 3: Получение (Z)-5-(4-фторфенил)-6-метокси-6-оксо-гекс-4-еновой кислоты (IVa)

64,0 г (0,221 моль) 5-(4-фторфенил)-5-гидрокси-6-метокси-6-оксо-гексановой кислоты (соединение IIIa) и 300 мл дихлорметана добавляли в 500 мл реакционную колбу, смесь растворяли при перемешивании и защищали азотом, температуру снижали, 65,6 г (0,233 моль) трифторметансульфонового ангидрида добавляли при температуре 5°С ~15°С, затем реакционную смесь нагревали с обратным холодильником в течение 1~2 часов и контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение IIIa).
Температуру снижали, 100 мл воды добавляли при температуре 5°С ~15°С для гашения реакции, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали дихлорметаном (60 мл × 2 раза), органические фазы объединяли и затем промывали 3 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении с получением 54,4 г соединения IVa (ВЭЖХ чистота: 95,6%; выход: 93,4%).
1Н ЯМР (ДМСО-d6): 2,42 (t, 2 Н, J = 7,3 Гц), 2,56 (q, 2 Н, J = 7,3 Гц), 3,74 (s, 3 Н), 6,27 (t, 1 Н, J = 7,4 Гц), 7,18 (t, 2 Н, J = 8,8 Гц), 7,32-7,36 (m, 2 Н), 12,19 (s, 1 Н).
Пример 4: Получение (Z)-5-(4-фторфенил)-6-гидрокси-гекс-4-еновой кислоты (V)

54,0 г (0,205 моль) (Z)-5-(4-фторфенил)-6-метокси-6-оксо-гекс-4-еновой кислоты (соединение IVa) и 240 мл толуола добавляли в 1 л реакционную колбу, смесь растворяли при перемешивании и защищали азотом, добавляли 31,0 г (0,240 моль) диизопропилэтиламина и растворяли при перемешивании, температуру снижали до -20°С ~-15°С, медленно по каплям добавляли 382,0 г (0,673 моль) DIBAH в форме раствора в толуоле (25%), температуру поддерживали для взаимодействия в течение 20~40 минут после добавления. Взаимодействие контролировали посредством ТСХ до полного взаимодействия сырьевого материала (соединение Iva).
При температуре ниже 15°С реакционную смесь медленно по каплям добавляли к водному раствору гидроксида натрия (72,2 г гидроксид натрия, растворенный в 300 мл воды), затем раствор перемешивали в течение 30 минут; отстаивали для разделения на слои, водную фазу собирали, водную фазу экстрагировали дихлорметаном (60 мл × 2 раза), дихлорметановую фазу отбрасывали. РН водной фазы доводили до 1~2 с помощью 4М хлористоводородной кислоты при температуре ниже 25°С, добавляли 240 мл этилацетата, раствор перемешивали в течение 5 минут, затем отстаивали для разделения на слои, органическую фазу собирали, и водную фазу экстрагировали этилацетатом (100 мл × 3 раза). Органические фазы объединяли и промывали 2 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении, неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 36,4 г соединения V (ВЭЖХ чистота: 96,3%; выход: 76,4%).
1Н ЯМР (ДМСО-d6): 2,38 (t, 2 Н, J = 7,1 Гц), 2,47 (q, 2 Н, J = 7,0 Гц), 4,34 (s, 2 Н), 4,75 (brs, 1 Н), 5,78 (t, 1 Н, J = 7,2 Гц), 7,13 (t, 2 Н, J = 8,9 Гц), 7,45-7,48 (m, 2 Н), 12,13 (br s, 1 Н).
Пример 5: Получение (Z)-5-(4-фторфенил)-6-(3-нитробензоилокси)гекс-4-еновой кислоты (VIa)

18,0 г (0,077 моль) (Z)-5-(4-фторфенил)-6-гидрокси-гекс-4-еновой кислоты (соединение V) и 60 мл N,N-диметилацетамида добавляли в 250 мл реакционную колбу, смесь растворяли при перемешивании и защищали азотом, температуру снижали, 31,2 г (0,168 моль) 3-нитробензоилхлорида добавляли при температуре -5°С ~5°С, затем обеспечивали взаимодействие реакционной смеси в течение 5~6 часов при температуре -5°С ~5°С, и контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение V).
При температуре 0~10°С добавляли водный раствор пиридина (13,0 г пиридина растворяли в 30 мл воды) и перемешивали в течение 30 минут, затем при температуре 0~10°С, добавляли водный раствор имидазола (22,5 г имидазола растворяли в 50 мл воды), перемешивали в течение 1~2 часов, затем раствор экстрагировали с помощью 120 мл этилацетата и отстаивали для разделения на слои, органическую фазу собирали, и водную фазу экстрагировали этилацетатом (20 мл × 3 раза), органические фазы объединяли, и органическую фазу промывали водой, рН доводили до 3~5 с помощью 2М хлористоводородной кислоты, затем органическую фазу промывали 1 раз насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении с получением 24,0 г соединения VIa (ВЭЖХ чистота: 92,6%; выход: 77,0%).
1Н ЯМР (ДМСО-d6): 2,45 (t, 2 Н, J = 7,1 Гц), 2,59 (q, 2 Н, J = 7,3 Гц), 5,36 (s, 2 Н), 6,09(t, 1 Н, J = 7,4 Гц), 7,18 (t, 2 Н, J = 8,8 Гц), 7,51-7,54 (m, 2 Н), 7,80 (t, 1 Н, J = 7,8 Гц), 8,23(d, 1 Н, J = 7,8 Гц), 8,46-8,48 (m, 2 Н), 12,17 (s, 1 Н).
Пример 6: Получение
[(Z)-2-(4-фторфенил)-6-оксо-6-[(4S)-2-оксо-4-фенил-оксазолидин-3-ил]гекс-2-енил] 3-нитробензоата (VIIIa)
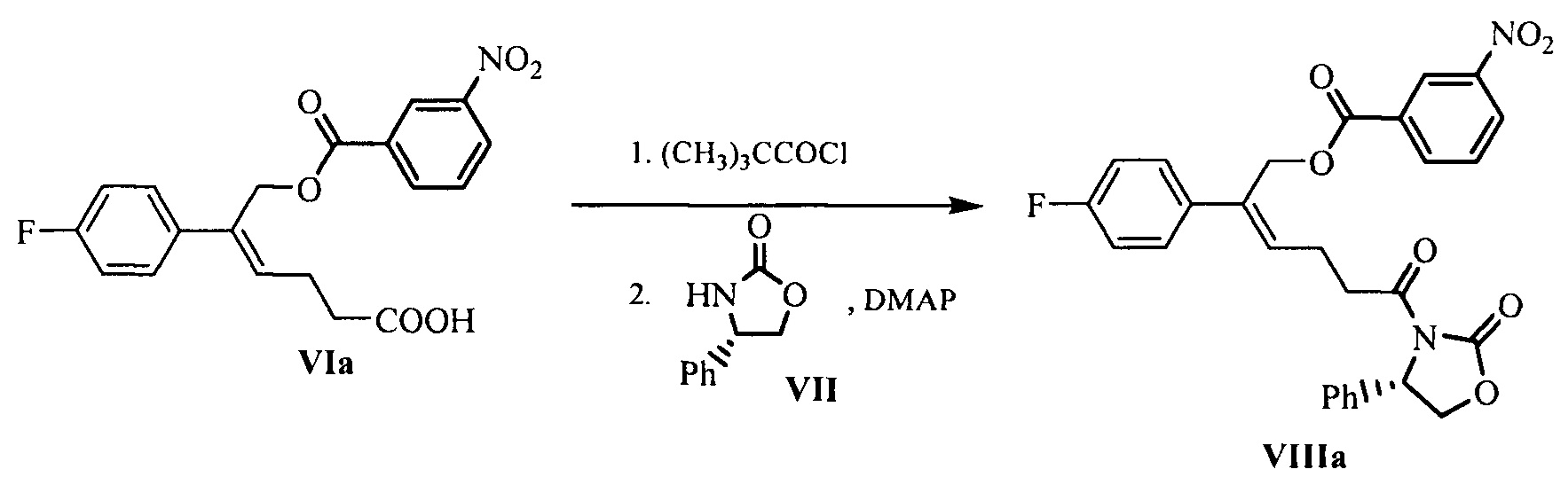
24,0 г (0,060 моль) (Z)-5-(4-фторфенил)-6-(3-нитробензоилокси)-гекс-4-еновой кислоты (соединение VIa) и 100 мл дихлорметана добавляли в 250 мл реакционную колбу, смесь растворяли при перемешивании и защищали азотом. Добавляли 8,9 г (0,074 моль) пивалоилхлорида. При комнатной температуре медленно по каплям добавляли 15,6 г (0,154 моль) триэтиламина, обеспечивали взаимодействие реакционной смеси в течение 3~4 часов при комнатной температуре после добавления. Затем добавляли 7,8 г (0,048 моль) (S)-4-фенил-2-оксазолидинона (соединение VII) и 2,2 г (0,018 моль) 4-диметиламинопиридина и осуществляли взаимодействие при комнатной температуре в течение 3~4 часов. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение VIa).
РН доводили до 4~6 с помощью 2М хлористоводородной кислоты, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, и водную фазу экстрагировали дихлорметаном (30 мл × 2 раза), органические фазы объединяли. Добавляли водный раствор имидазола (11,1 г имидазола растворяли в 30 мл воды) и перемешивали в течение 1~2 часов, затем 1 раз промывали насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили с получением 25,3 г соединения VIIIa (ВЭЖХ чистота: 95,1%; выход: 78,0%).
1Н ЯМР (ДМСО-d6): 2,59 (q, 2 Н, J = 7,2 Гц), 3,00-3,18 (m, 2 Н), 4,15 (dd, 1 Н, J = 8,8, 3,6 Гц), 4,72 (t, 1 Н, J = 8,7 Гц), 5,29 (d, 1 Н, J = 13,2 Гц), 5,32 (d, 1 Н, J = 13,2 Гц), 5,45 (dd, 1 Н, J = 8,6, 3,6 Гц), 6,05 (t, 1 Н, J = 7,5 Гц), 7,17 (t, 2 Н, J = 8,9 Гц), 7,26-7,36 (m, 5 H), 7,46-7,50 (m, 2 H), 7,76-7,80 (m, 1 H), 8,19-8,21 (m, 1 H), 8,45-8,47 (m, 2 H).
Пример 7: Получение соединения формулы VIIIa
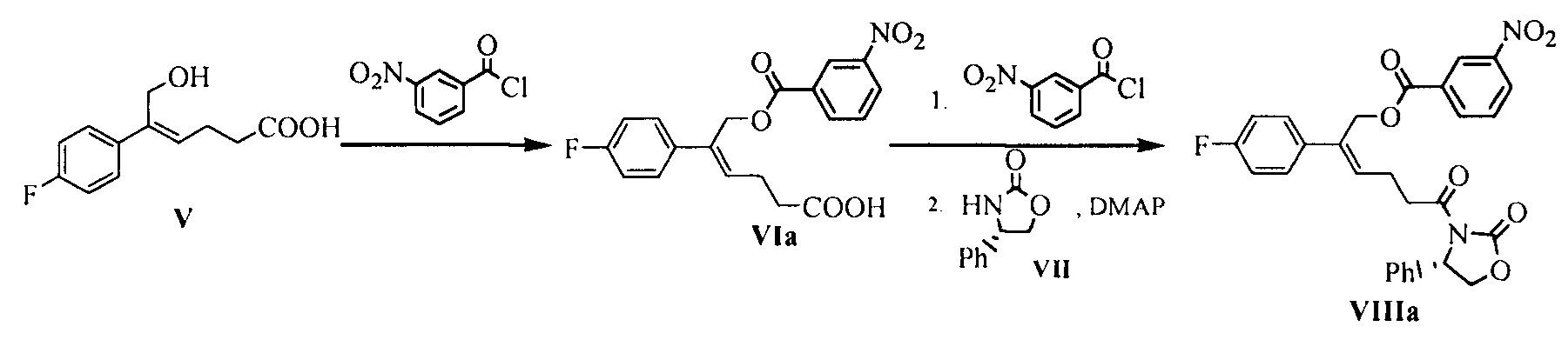
Стадия 1: 18 г (0,080 моль) (Z)-5-(4-фторфенил)-6-гидрокси-гекс-4-еновой кислоты (соединение V) и 90 мл N,N-диметилацетамида добавляли в 250 мл реакционную колбу, смесь растворяли при перемешивании и защищали азотом. 17,8 г (0,096 моль) 3-нитробензоилхлорида добавляли при 25°С~30°С, температуру поддерживали для взаимодействия в течение 2 часов, окончание реакции определяли посредством ВЭЖХ, реакционную смесь оставляли для применения.
Стадия 2: 180 мл дихлорметана и 16,3 г (0,088 моль) 3-нитробензоилхлорида добавляли в 500 мл реакционную колбу и защищали азотом, 32,4 г (0,32 моль) триэтиламина по каплям добавляли при температуре 25°С~30°С, затем реакционную смесь со стадии 1 по каплям добавляли при температуре 25°С~30°С (смесь добавляли в течение 1~2 часов), затем температуру поддерживали для взаимодействия в течение 5 минут после добавления, затем добавляли 11,75 г (0,072 моль) (S)-4-фенил-2-оксазолидинона и 4,4 г (0,036 моль) 4-диметиламинопиридина, температуру поддерживали для взаимодействия в течение 6~7 часов, окончание реакции определяли посредством ВЭЖХ.
90 мл воды добавляли к реакционной смеси, указанный раствор отстаивали для разделения на слои, органическую фазу собирали и водную фазу экстрагировали дихлорметаном (50 мл × 2 раза), органические фазы объединяли. РН органической фазы доводили до 4~6 с помощью 2М хлористоводородной кислоты, затем органическую фазу промывали посредством 90 мл воды до нейтральной реакции, затем добавляли водный раствор имидазола (27 г имидазола растворяли в 50 мл воды), перемешивали в течение 30 минут и отстаивали для разделения на слои, органическую фазу собирали и промывали насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в смеси растворителей этилацетат/петролейный эфир (2/3), фильтровали и сушили до получения 30 г соединения VIIIa (ВЭЖХ чистота: 97,5%; выход: 74,0%).
Пример 8: Получение
[(Z,5R)-5-[(S)-(4-фторанилино)-[4-(3-нитробензоил)оксифенил]метил]-2-(4-фторфенил)-6-оксо-6-[(4S)-2-оксо-4-фенил-оксазолидин-3-ил]гекс-2-енил] 3-нитробензоата (XIa)
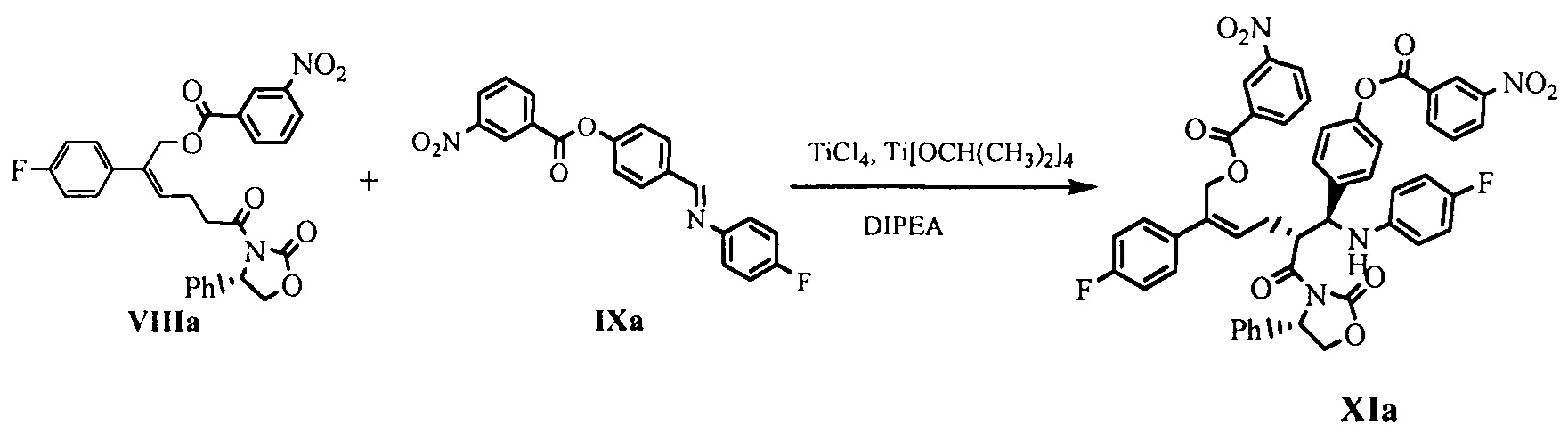
80 мл дихлорметана и 10,5 г (0,055 моль) тетрахлорида титана добавляли в 250 мл реакционную колбу и защищали азотом, температуру снижали, по каплям добавляли 5,2 г (0,018 моль) изопропилата титана при температуре -5°С~0°С, затем раствор перемешивали в течение 30 минут при температуре -5°С~0°С с получением реагента титана. 25,0 г (0,046 моль) соединения формулы VIIIa, 18,3 г (0,050 моль) имина формулы IXa и 350 мл дихлорметана добавляли в 1 л реакционную колбу и растворяли при перемешивании, добавляли 14,3 г (0,111 моль) диизопропилэтиламина и перемешивали, температуру снижали, реагент титана медленно по каплям добавляли при температуре -25°С~-20°С, затем смесь оставляли для взаимодействия в течение 1~2 часов при температуре -25°С~-20°С, взаимодействие контролировали посредством ВЭЖХ до содержания сырьевого материала (соединение VIIIa) <5%.
30 мл уксусной кислоты по каплям добавляли при температуре -25°С~-20°С и перемешивали в течение 5 минут; 150 мл серной кислоты (2М) по каплям добавляли при температуре ниже 10°С и перемешивали в течение 10 минут; указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали дихлорметаном (25 мл × 2 раза), органические фазы объединяли и промывали 3 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 21,4 г соединения XIa (ВЭЖХ чистота: 95,3%; выход: 50,4%).
1Н ЯМР (ДМСО-d6): 2,38-2,45 (m, 1 Н), 2,56-2,64 (m, 1 Н), 4,11 (dd, 1 Н, J = 8,8, 4,7 Гц), 4,62-4,75 (m, 3 Н), 5,15 (s, 2 Н), 5,51 (dd, 1 Н, J = 8,5, 4,6 Гц), 5,98 (t, 1 Н, J = 7,4 Гц), 6,34 (d, 1 Н, J = 9,8 Гц), 6,58-6,62 (m, 2 Н), 6,80 (t, 2 Н, J = 8,9 Гц), 7,13-7,28 (m, 9 Н), 7,45-7,48 (m, 2 Н), 7,54 (d, 2 Н, J = 8,5 Гц), 7,79 (t, 1 Н, J = 7,9 Гц), 7,91 (t, 1 Н, J = 8,0 Гц), 8,18 (d, 1 Н, J = 7,8 Гц), 8,43-8,49 (m, 3 Н), 8,57-8,60 (m, 1 Н), 8,74 (t, 1 Н, J = 1,8 Гц); MS (m/z): 883 [М+Н]+.
Пример 9: Получение соединения формулы XIa
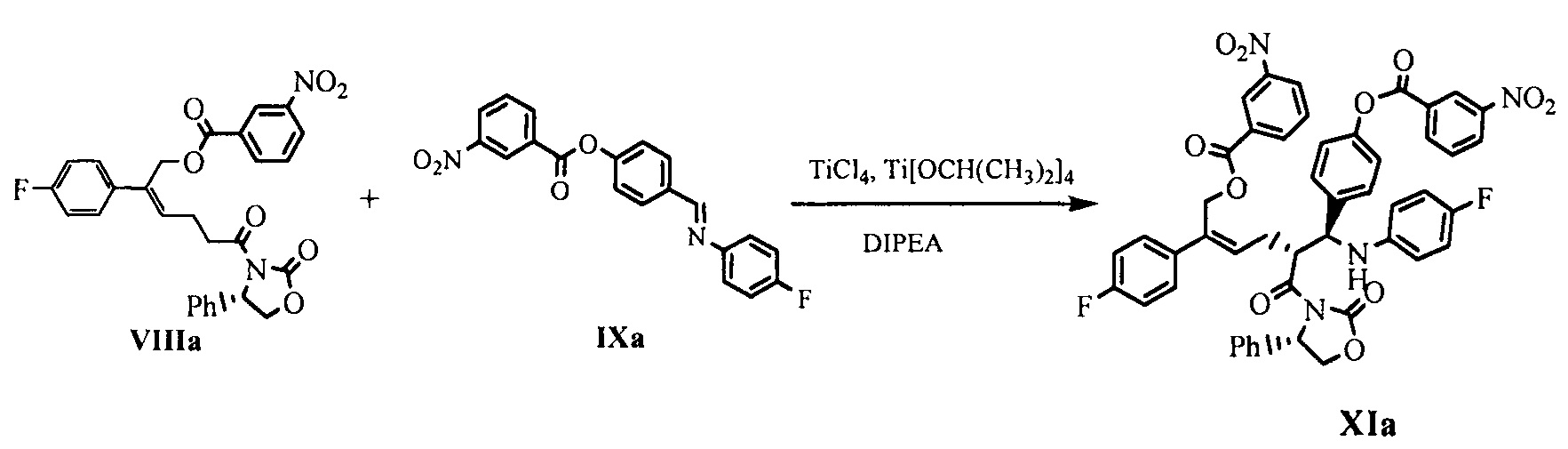
150 мл дихлорметана и 13,2 г (0,069 моль) тетрахлорида титана добавляли в 500 мл реакционную колбу, защищали азотом и перемешивали, температуру снижали, 60 мл раствора дихлорметана с растворенным 6,6 г (0,023 моль) изопропилатом титана по каплям добавляли при температуре -5°С~0°С, затем раствор перемешивали в течение 30 минут при температуре -5°С~0°С с получением реагента титана. 30 г (0,058 моль) соединения формулы VIIIa, 23,2 г (0,064 моль) имина формулы IXa и 900 мл дихлорметана добавляли в 2 л реакционную колбу и защищали азотом и растворяли при перемешивании, добавляли 19,5 г диизопропилэтиламина, температуру снижали, медленно по каплям добавляли реагент титана, полученный выше, при температуре -75°С~-70°С, добавление заканчивали не позднее примерно 2 часов, затем обеспечивали взаимодействие реакционной смеси в течение 5 минут при температуре -75°С~-70°С и контролировали посредством ВЭЖХ до исчезновения пятен сырьевого материала (соединение VIIIa).
Раствор 135 мл 20% трифторуксусной кислоты в дихлорметане быстро добавляли при температуре ниже -70°С и перемешивали в течение 1 минуты; быстро по каплям добавляли 240 мл водного раствора серной кислоты (2М) при температуре ниже -30°С, затем раствор перемешивали и нагревали до комнатной температуры; указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали дихлорметаном (100 мл × 2 раза), органические фазы объединяли и промывали насыщенным солевым водным раствором до нейтральной реакции, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении с получением неочищенного продукта. Неочищенный продукт перекристаллизовывали в смеси растворителей этилацетат/петролейный эфир (1/1), фильтровали и сушили до получения 35 г соединения XIa (ВЭЖХ чистота: 98,9%; выход: 68,7%).
Пример 10: Получение
(3R,4S)-4-[4-(3-нитробензоилокси)фенил]-3-[3-(4-фторфенил)-4-(3-нитробензоилокси)бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (XIIa),
(3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-(3-нитробензоилокси)бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (XIIIa),
(3R,4S)-4-(4-триметилсилилоксифенил)-3-[3-(4-фторфенил)-4-(3-нитробензоилокси)бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (XIVa)

21,0 г (0,025 моль) соединения формулы XIa и 200 мл толуола добавляли в 500 мл реакционную колбу, смесь перемешивали и нагревали, 18,4 г (0,090 моль) N,O-бис(триметилсилил)ацетамида (BSA) добавляли при температуре 50°С~60°С, затем оставляли для взаимодействия в течение 2 часов при данной температуре; затем добавляли 1,0 г (0,003 моль) тетрабутиламмония фторида тригидрата при температуре 50°~60°С и оставляли для взаимодействия в течение 2~3 часов при данной температуре. Взаимодействие контролировали посредством ВЭЖХ до содержания сырьевого материала (соединение XIa) <1,0%.
Температуру снижали ниже 25°С, по каплям добавляли 50 мл ледяной воды и перемешивали в течение 10 минут, затем добавляли 180 мл н-гептана и непрерывно перемешивали в течение 30 минут, твердое вещество выпадало в осадок и его отфильтровывали, и фильтрат отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали толуолом (15 мл × 2 раза), органические фазы объединяли и концентрировали досуха под вакуумом, получали смесь. Некоторое количество смеси отбирали для разделения с получением трех продуктов, т.е. соединений XIIa, XIIIa, XIVa.
Соединение XIIa: 1Н ЯМР (400 МГц, ДМСО-d6): 2,91-3,08 (m, 2 Н), 3,41 (td, 1 Н, J = 8,5,2,1 Гц), 5,14 (d, 1 Н, J = 2,0 Гц), 5,42 (d, 1 Н, J = 13,1 Гц), 5,46 (d, 1 Н, J = 13,1 Гц), 6,17 (t, 1 Н, J = 7,5 Гц), 7,13-7,26 (m, 6 Н), 7,35 (d, 2 Н, J = 8,5 Гц), 7,48-7,53 (m, 4 Н), 7,78 (t, 1 Н, J = 8,0 Гц), 7,91 (t, 1 Н, J = 8,0 Гц), 8,21 (d, 1 Н, J = 7,8 Гц), 8,44-8,47 (m, 2 Н), 8,51 (d, 1 Н, J = 7,9 Гц), 8,56-8,59 (m, 1 Н), 8,76 (t, 1 Н, J = 1,7 Гц); MS (m/z): 720 [М+Н]+, 742 [M+Na]+.
Соединение XIIIa: 1Н ЯМР (400 МГц, ДМСО-d6): 2,85-2,98 (m, 2 Н), 3,30 (td, 1 Н, J = 8,5, 2,2 Гц), 4,92 (d, 1 Н, J = 2,2 Гц), 5,40 (d, 1 Н, J = 13,1 Гц), 5,44 (d, 1 Н, J = 13,1 Гц), 6,13 (t, 1 Н, J = 7,5 Гц), 6,73 (d, 2 Н, J = 8,5 Гц), 7,12 (t, 2 Н, J = 8,8 Гц), 7,16-7,21 (m, 6 Н), 7,47-7,50 (m, 2 Н), 7,79 (td, 1 Н, J = 7,7, 0,9 Гц), 8,21 (d, 1 Н, J = 7,8 Гц), 8,45-8,47 (m, 2 Н), 9,52 (s, 1 Н); MS (m/z): 571 [М+Н]+.
Соединение XIVa: 1Н ЯМР (400 МГц, CDCl3): 0,28 (s, 9 Н), 2,97-3,01 (m, 2 Н), 3,30 (td, 1 Н, J = 7,9, 2,2 Гц), 4,72 (d, 1 Н, J = 2,1 Гц), 5,37 (s, 2 Н), 6,07 (t, 1 Н, J = 7,6 Гц), 6,83 (d, 2 Н, J = 8,5 Гц), 6,94 (t, 2 Н, J = 8,6 Гц), 7,03 (t, 2 Н, J = 8,6 Гц), 7,20 (d, 2 Н, J = 8,5 Гц), 7,24-7,28 (m, 2 Н), 7,35-7,38 (m, 2 Н), 7,61 (t, 1 Н, J = 8,0 Гц), 8,23 (d, 1 Н, J = 7,8 Гц), 8,38-8,41 (m, 1 Н), 8,75 (t, 1 Н, J = 1,7 Гц); MS (m/z): 643 [М+Н]+.
Пример 11: Получение
(3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-гидрокси-бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (I)

Смесь соединений формулы XIIa, XIIIa XIVa, полученных в примере 10, и 90 мл ацетона добавляли в 250 мл реакционную колбу и растворяли при перемешивании, добавляли 23 мл (0,069 моль) водного раствора гидроксида лития (3М) при комнатной температуре, и обеспечивали взаимодействие реакционной смеси в течение 2~3 часов при перемешивании, и контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединений XIIa, XIIIa XIVa).
РН доводили до 4~6 посредством 2М хлористоводородной кислоты при комнатной температуре, затем раствор концентрировали под вакуумом (30~40°С) до малого объема, добавляли 100 мл этилацетата и перемешивали в течение 5 минут, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали этилацетатом (20 мл × 2 раза), органические фазы объединяли, добавляли водный раствор гидрокарбоната натрия (3,8 г гидрокарбоната натрия растворяли в 40 мл воды) и перемешивали в течение 30 минут, указанный раствор отстаивали для разделения на слои. РН органической фазы доводили до примерно 6 с помощью 2М хлористоводородной кислоты, отстаивали для разделения на слои, органическую фазу промывали 1 раз насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали, фильтрат концентрировали досуха при пониженном давлении. Остаток очищали посредством колоночной хроматографии, дважды перекристаллизовывали в смеси растворителей этилацетат и н-гептан, фильтровали и сушили до получения 4,3 г соединения I (ВЭЖХ чистота: 99,2%; выход: 44,7% рассчитан в соответствии с количеством поданного соединения XIa из примера 10).
1Н ЯМР (400 МГц, ДМСО-d6): δ 2,71-2,84 (m, 2 Н), 3,23 (td, 1 Н, J = 6,4, 2,0 Гц), 4,40 (d, 2 Н, J = 5,3 Гц), 4,87 (t, 1 Н, J = 5,3 Гц), 4,94 (d, 1 Н, J = 2,1 Гц), 5,80 (t, 1 H, J = 7,5 Гц), 6,74 (d, 2 Н, J = 8,5 Гц), 7,11-7,17 (m, 4 Н), 7,20-7,25 (m, 4 Н), 7,39-7,43 (m, 2 Н), 9,50 (s, 1 Н); MS (m/z): 422 [М+Н]+.
Пример 12: Получение 5-(4-фторфенил)-5-гидрокси-6-метокси-6-оксо-гексановой кислоты (IIIa)
60 кг (337,9 моль) 6-метокси-5,6-диоксо-гексановой кислоты (соединение IIa) и 180 л тетрагидрофурана добавляли в 2000 л реакционную емкость, смесь защищали азотом и растворяли при перемешивании, температуру снижали до -20°С~-10°С, медленно по каплям добавляли 1М раствор 4-фторфенилмагния бромида в ТГФ (800 л, 800 моль) и температуру поддерживали для взаимодействия в течение 1~2 часов после добавления. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение IIa).
25% водный раствор хлорида аммония (30 кг хлорида аммония, растворенного в 90 л воды) добавляли при температуре -20°С~0°С и перемешивали в течение 5 минут, затем рН доводили до 3~5 с помощью 4М хлористоводородной кислоты при температуре 0°С ~30°С, затем добавляли 400 л н-гептана и перемешивали в течение 5 минут, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали этилацетатом (80 л × 2 раза), органические фазы объединяли и два раза промывали насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 45,6 кг соединения IIIa (ВЭЖХ чистота: 70,8%; выход: 35,4%).
1Н ЯМР (ДМСО-d6): 1,33-1,43 (m, 2 Н), 1,89-1,96 (m, 1 Н), 1,99-2,04 (m, 1 Н), 2,15 (t, 2 Н, J = 7,6 Гц), 3,61 (s, 3 Н), 5,99 (s, 1 Н), 7,12-7,17 (m, 2 Н), 7,47-7,51 (m, 2 Н), 12,02 (s, 1 Н); MS (m/z): 269 [М-Н]-.
Пример 13: Получение (Z)-5-(4-фторфенил)-6-метокси-6-оксо-гекс-4-еновой кислоты (IVa)
40,0 кг (104,9 моль) 5-(4-фторфенил)-5-гидрокси-6-метокси-6-оксо-гексановой кислоты (соединение IIIa) и 200 л дихлорметана добавляли в 300 л реакционную емкость, смесь растворяли при перемешивании и защищали азотом, температуру снижали, 31,2 кг (110,6 моль) трифторметансульфонового ангидрида добавляли при температуре 5~15°С, затем реакционную смесь нагревали с обратным холодильником в течение 1~2 часов и контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение IIIa).
Температуру снижали, реакцию останавливали посредством добавления 50 л воды при температуре 5°С~15°С, перемешивали в течение 5 минут и отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали дихлорметаном (40 л × 2 раза), органические фазы объединяли и промывали 3 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении с получением 25,2 кг соединения IVa (ВЭЖХ чистота: 86,3%; выход: 82,3%).
1Н ЯМР (ДМСО-d6): 2,42 (t, 2 Н, J = 7,3 Гц), 2,56 (q, 2 Н, J = 7,3 Гц), 3,74 (s, 3 Н), 6,27 (t, 1 Н, J = 7,4 Гц), 7,18 (t, 2 Н, J = 8,8 Гц), 7,32-7,36 (m, 2 Н), 12,19 (s, 1 Н).
Пример 14: Получение (Z)-5-(4-фторфенил)-6-гидрокси-гекс-4-еновой кислоты (V)
25,0 кг (85,6 моль) (Z)-5-(4-фторфенил)-6-метокси-6-оксо-гекс-4-еновой кислоты (соединение IVa) и 100 л толуола добавляли в 500 л реакционную емкость, смесь растворяли при перемешивании и защищали азотом, добавляли 13,0 кг (100,8 моль) диизопропилэтиламина, перемешивали в течение 5 минут. Температуру снижали до -20°С~-15°С, медленно по каплям добавляли 159,7 кг (281,2 моль) DIBAH, растворенного в толуоле (25%), и температуру поддерживали для взаимодействия в течение 20~40 минут после добавления. Взаимодействие контролировали посредством ТСХ до полного взаимодействия сырьевого материала (соединение IVa).
Реакционную смесь медленно по каплям добавляли к водному раствору гидроксида натрия (30,2 кг гидроксида натрия растворяли в 140 л воды) при температуре ниже 15°С и перемешивали в течение 30 минут. Указанный раствор отстаивали для разделения на слои, водную фазу собирали и экстрагировали посредством 50 л дихлорметана, фазу дихлорметана отбрасывали, рН водной фазы доводили до 1~2 при температуре ниже 25°С с помощью 6М хлористоводородной кислоты, добавляли 100 л этилацетата и перемешивали в течение 5 минут. Указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали этилацетатом (40 л × 3 раза), органические фазы объединяли и промывали 2 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 15,0 кг соединения V (ВЭЖХ чистота: 92,1%; выход: 72,0%).
1Н ЯМР (ДМСО-d6): 2,38 (t, 2 Н, J = 7,1 Гц), 2,47 (q, 2H, J = 7,0 Гц), 4,34 (s, 2 Н), 4,75 (brs, 1Н), 5,78 (t, 1 Н, J = 7,2 Гц), 7,13 (t, 2 Н, J = 8,9 Гц), 7,45-7,48 (m, 2 Н), 12,13 (br s, 1Н).
Пример 15: Получение (Z)-5-(4-фторфенил)-6-(3-нитробензоилокси)гекс-4-еновой кислоты (VIa)

15,0 кг (61,7 моль) (Z)-5-(4-фторфенил)-6-гидрокси-гекс-4-еновой кислоты (соединение V) и 50 л N,N-диметилацетамида добавляли в 300 л реакционную емкость, смесь растворяли при перемешивании и защищали азотом. Температуру снижали до -5°С~5°С, добавляли 24,9 кг (134,2 моль) 3-нитробензоилхлорида, и температуру поддерживали для взаимодействия в течение 5-6 часов. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение V).
Водный раствор пиридина добавляли (10,4 кг пиридина растворяли в 30 л воды) при температуре 0°С~10°С и перемешивали в течение 30 минут. Затем добавляли водный раствор имидазола (18,0 кг имидазола растворяли в 50 л воды) при температуре 0°С~10°С и перемешивали в течение 1~2 часов, затем добавляли 100 л этилацетата и перемешивали в течение 5 минут. Указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали этилацетатом (20 л × 3 раза), органические фазы объединяли и промывали водой, рН доводили до 3~5 с помощью 2М хлористоводородной кислоты, промывали 1 раз насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении с получением 20,2 кг соединения VIa (ВЭЖХ чистота: 85,6%; выход: 75,2%).
1Н ЯМР (ДМСО-d6): 2,45 (t, 2 Н, J = 7,1 Гц), 2,59 (q, 2 Н, J = 7,3 Гц), 5,36 (s, 2 Н), 6,09(t, 1 Н, J = 7,4 Гц), 7,18 (t, 2 Н, J = 8,8 Гц), 7,51-7,54 (m, 2 Н), 7,80 (t, 1 Н, J = 7,8 Гц), 8,23(d, 1 Н, J = 7,8 Гц), 8,46-8,48 (m, 2 Н), 12,17 (s, 1 Н).
Пример 16: Получение
[(Z)-2-(4-фторфенил)-6-оксо-6-[(45)-2-оксо-4-фенил-оксазолидин-3-ил]гекс-2-енил] 3-нитробензоата (VIIIa)
20,0 кг (45,9 моль) (Z)-5-(4-фторфенил)-6-(3-нитробензоилокси)-гекс-4-еновой кислоты (соединение VIa) и 100 л дихлорметана добавляли в 300 л реакционную емкость, смесь растворяли при перемешивании и защищали азотом. Добавляли 6,8 кг (56,4 моль) пивалоилхлорида. При комнатной температуре медленно по каплям добавляли 12,0 кг (118,8 моль) триэтиламина, обеспечивали взаимодействие реакционной смеси при комнатной температуре в течение 3-4 часов после добавления, затем добавляли 6,0 кг (36,8 моль) (S)-4-фенил-2-оксазолидинона (соединение VII) и 1,7 кг (13,9 моль) 4-диметиламинопиридина и оставляли для взаимодействия при комнатной температуре в течение 4-5 часов. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение VIa).
РН доводили до 4~6 с помощью 2М хлористоводородной кислоты, указанный раствор отстаивали для разделения на слои, органическую фазу собирали, и водную фазу экстрагировали дихлорметаном (25 л × 2 раза), органические фазы объединяли, добавляли водный раствор имидазола (8,6 кг имидазола растворяли в 30 л воды) и перемешивали в течение 2-3 часов, затем 1 раз промывали насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 15,2 кг соединения VIIIa (ВЭЖХ чистота: 91,7%; выход: 58,6%).
1Н ЯМР (ДМСО-d6): 2,59 (q, 2 Н, J = 7,2 Гц), 3,00-3,18 (m, 2 Н), 4,15 (dd, 1 Н, J = 8,8, 3,6 Гц), 4,72 (t, 1 Н, J = 8,7 Гц), 5,29 (d, 1 Н, J = 13,2 Гц), 5,32 (d, 1 Н, J = 13,2 Гц), 5,45 (dd, 1 Н, J = 8,6, 3,6 Гц), 6,05 (t, 1 Н, J = 7,5 Гц), 7,17 (t, 2 Н, J = 8,9 Гц), 7,26-7,36 (m, 5 Н), 7,46-7,50 (m, 2 Н), 7,76-7,80 (m, 1 Н), 8,19-8,21 (m, 1 Н), 8,45-8,47 (m, 2 Н).
Пример 17: Получение
[(Z,5R)-5-[(S)-(4-фторанилино)-[4-(3-нитробензоил)оксифенил]метил]-2-(4-фторфенил)-6-оксо-6-[(4S)-2-оксо-4-фенил-оксазолидин-3-ил]гекс-2-енил] 3-нитробензоата (XIa)

60 л дихлорметана и 6,0 кг (31,6 моль) тетрахлорида титана добавляли в 100 л реакционную емкость и защищали азотом, перемешивали, температуру снижали, добавляли 3,0 кг (10,6 моль) тетраизопропил титаната при температуре -5°С~0°С, затем перемешивали в течение 30 минут при температуре -5°С~0°С с получением реагента титана. 15,0 кг (26,6 моль) соединения формулы VIIIa, 10,6 кг (29,1 моль) имина формулы IXa и 220L дихлорметана добавляли в 500 л реакционную емкость и растворяли при перемешивании, добавляли 8,3 кг (64,3 моль) диизопропилэтиламина и перемешивали в течение 10 минут, температуру снижали, медленно по каплям добавляли реагент титана при температуре -25°С~-20°С, температуру поддерживали для взаимодействия в течение 1~2 часов после добавления. Взаимодействие контролировали посредством ВЭЖХ до содержания сырьевого материала (соединение VIIIa) <5%.
18 л уксусной кислоты добавляли при температуре -25°С~-20°С, затем перемешивали в течение 5 минут; 90 л серной кислоты (2М) добавляли по каплям при температуре ниже 10°С, затем перемешивали в течение 10 минут. Указанный раствор отстаивали для разделения на слои, органическую фазу собирали, и водную фазу экстрагировали посредством 30 л дихлорметана. Органические фазы объединяли и промывали 3 раза насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха при пониженном давлении. Неочищенный продукт перекристаллизовывали в толуоле, фильтровали и сушили до получения 12,3 кг соединения XIa (ВЭЖХ чистота: 92,1%; выход: 48,4%).
1Н ЯМР (ДМСО-d6): 2,38-2,45 (m, 1 Н), 2,56-2,64 (m, 1 Н), 4,11 (dd, 1 Н, J = 8,8, 4,7 Гц), 4,62-4,75 (m, 3 Н), 5,15 (s, 2 Н), 5,51 (dd, 1 Н, J = 8,5, 4,6 Гц), 5,98 (t, 1 Н, J = 7,4 Гц), 6,34 (d, 1 Н, J = 9,8 Гц), 6,58-6,62 (m, 2 Н), 6,80 (t, 2 Н, J = 8,9 Гц), 7,13-7,28 (m, 9 Н), 7,45-7,48 (m, 2 Н), 7,54 (d, 2 Н, J = 8,5 Гц), 7,79 (t, 1 Н, J = 7,9 Гц), 7,91 (t, 1 Н, J = 8,0 Гц), 8,18 (d, 1 Н, J = 7,8 Гц), 8,43-8,49 (m, 3 Н), 8,57-8,60 (m, 1 Н), 8,74 (t, 1 Н, J = 1,8 Гц); MS (m/z): 883 [М+Н]+.
Пример 18: Получение
(3R,4S)-4-[4-(3-нитробензоилокси)фенил]-3-[3-(4-фторфенил)-4-(3-нитробензоилокси)бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (XIIa),
(3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-(3-нитробензоилокси)бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (XIIIa),
(3R,4S)-4-(4-триметилсилилоксифенил)-3-[3-(4-фторфенил)-4-(3-нитробензоилокси)бут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (XIVa)

12,0 кг (12,5 моль) соединения формулы XIa и 120 л толуола добавляли в 300 л реакционную емкость и перемешивали и нагревали, добавляли 10,2 кг (50,0 моль) N,O-бис(триметилсилил)ацетамида (BSA) при 50°С~60°С, оставляли для взаимодействия в течение 2~3 часов при указанной температуре; затем добавляли 0,6 кг (1,9 моль) тетрабутиламмония фторида тригидрата и температуру поддерживали для взаимодействия в течение 2~3 часов. Взаимодействие контролировали посредством ВЭЖХ до содержания сырьевого материала (соединение XIa) <1,0%.
Температуру снижали, 30 л ледяной воды по каплям добавляли при температуре ниже 25°С, раствор перемешивали в течение 10 минут, затем добавляли 100 л н-гептана и перемешивали в течение 30 минут, твердые вещества выпадали в осадок и их отфильтровывали, фильтрат отстаивали для разделения на слои, органическую фазу собирали, и водную фазу экстрагировали толуолом (10 л × 2 раза). Органические фазы объединяли и концентрировали досуха под вакуумом с получением смеси. Некоторое количество смеси отбирали для разделения с получением трех продуктов, т.е. соединений XIIa, XIIIa, XIVa.
Соединение XIIa: 1Н ЯМР (400 МГц, ДМСО-d6): 2,91-3,08 (m, 2 Н), 3,41 (td, 1 Н, J = 8,5, 2,1 Гц), 5,14 (d, 1 Н, J = 2,0 Гц), 5,42 (d, 1 Н, J = 13,1 Гц), 5,46 (d, 1 Н, J = 13,1 Гц), 6,17 (t, 1 Н, J = 7,5 Гц), 7,13-7,26 (m, 6 Н), 7,35 (d, 2 Н, J = 8,5 Гц), 7,48-7,53 (m, 4 Н), 7,78 (t, 1 Н, J = 8,0 Гц), 7,91 (t, 1 Н, J = 8,0 Гц), 8,21 (d, 1 Н, J = 7,8 Гц), 8,44-8,47 (m, 2 Н), 8,51 (d, 1 Н, J = 7,9 Гц), 8,56-8,59 (m, 1 Н), 8,76 (t, 1 Н, J = 1,7 Гц); MS (m/z): 720 [М+Н]+, 742 [M+Na]+.
Соединение XIIIa: 1Н ЯМР (400 МГц, ДМСО-d6): 2,85-2,98 (m, 2 Н), 3,30 (td, 1 Н, J = 8,5, 2,2 Гц), 4,92 (d, 1 H, J = 2,2 Гц), 5,40 (d, 1 Н, J = 13,1 Гц), 5,44 (d, 1 Н, J = 13,1 Гц), 6,13 (t, 1 Н, J = 7,5 Гц), 6,73 (d, 2 Н, J = 8,5 Гц), 7,12 (t, 2 Н, J = 8,8 Гц), 7,16-7,21 (m, 6 Н), 7,47-7,50 (m, 2 Н), 7,79 (td, 1 Н, J = 7,7, 0,9 Гц), 8,21 (d, 1 Н, J = 7,8 Гц), 8,45-8,47 (m, 2 Н), 9,52 (s, 1 Н); MS (m/z): 571 [М+Н]+.
Соединение XIVa: 1Н ЯМР (400 МГц, CDCl3): 0,28 (s, 9 Н), 2,97-3,01 (m, 2 Н), 3,30 (td, 1 Н, J = 7,9, 2,2 Гц), 4,72 (d, 1 Н, J = 2,1 Гц), 5,37 (s, 2 Н), 6,07 (t, 1 Н, J = 7,6 Гц), 6,83 (d, 2 Н, J = 8,5 Гц), 6,94 (t, 2 Н, J = 8,6 Гц), 7,03 (t, 2 Н, J = 8,6 Гц), 7,20 (d, 2 Н, J = 8,5 Гц), 7,24-7,28 (m, 2 Н), 7,35-7,38 (m, 2 Н), 7,61 (t, 1 Н, J = 8,0 Гц), 8,23 (d, 1 Н, J = 7,8 Гц), 8,38-8,41 (m, 1 Н), 8,75 (t, 1 Н, J = 1,7 Гц); MS (m/z): 643 [М+Н]+.
Пример 19: Получение
(3R,4S)-4-(4-гидроксифенил)-3-[3-(4-фторфенил)-4-гидроксибут-2(Z)-енил]-1-(4-фторфенил)азетидин-2-она (I)
Смесь соединений XIIa, XIIIa, XIVa, полученных в примере 18, и 50 л ацетона добавляли в 100 л реакционную емкость и растворяли при перемешивании, добавляли 13 л (39,0 моль) водного раствора гидроксида лития (3М) при комнатной температуре и оставляли для взаимодействия в течение 0,5~1 часа при перемешивании. Взаимодействие контролировали посредством ТСХ до исчезновения пятен сырьевого материала (соединение XIIa, XIIIa, XIVa).
РН доводили до 4~6 с помощью 2М хлористоводородной кислоты при комнатной температуре, затем раствор концентрировали под вакуумом (при 30°С~40°С) до малого объема, добавляли 60 л этилацетата и перемешивали в течение 5 минут, затем указанный раствор отстаивали для разделения на слои, органическую фазу собирали, водную фазу экстрагировали этилацетатом (10 л × 2 раза). Органические фазы объединяли, добавляли водный раствор бикарбоната натрия (2,0 кг бикарбоната натрия растворяли в 20 л воды), перемешивали в течение 30 минут, указанный раствор отстаивали для разделения на слои, рН органической фазы доводили до примерно 6 с помощью 2М хлористоводородной кислоты, указанный раствор отстаивали для разделения на слои, органическую фазу промывали 1 раз насыщенным солевым водным раствором, сушили над безводным сульфатом натрия, фильтровали, фильтрат концентрировали досуха при пониженном давлении. Остаток очищали посредством колоночной хроматографии, дважды кристаллизовали в смеси растворителей этилацетата и н-гептана, фильтровали и сушили до получения 2,1 кг соединения I (ВЭЖХ чистота: 98,9%; выход: 39,4% рассчитан в соответствии с количеством поданного соединения XIa примера 18).
1Н ЯМР (400 МГц, ДМСО-d6): δ 2,71-2,84 (m, 2 Н), 3,23 (td, 1 Н, J = 6,4, 2,0 Гц), 4,40 (d, 2 Н, J = 5,3 Гц), 4,87 (t, 1 Н, J = 5,3 Гц), 4,94 (d, 1 Н, J = 2,1 Гц), 5,80 (t, 1 Н, J = 7,5 Гц), 6,74 (d, 2 Н, J = 8,5 Гц), 7,11-7,17 (m, 4 Н), 7,20-7,25 (m, 4 Н), 7,39-7,43 (m, 2 Н), 9,50 (s, 1 Н); MS (m/z): 422 [М+Н]+.

Соединения, оказывающие возбуждающее действие на рецептор активатора пролиферации пероксисом подтипа б, способ получения и применение указанных соединений
Соединения, обладающие активирующим действием на подтипы рецепторов, активируемых пролифератором пероксисом (ppars), и способ получения и применения указанных соединений
Бензамидные производные, обладающие противораковой активностью, способ их получения и их применение
Способ реализации адаптации контента и сервер для адаптации контента
Способ и устройство для передачи данных полупостоянного планирования
Способ, устройство и система оценки разрешения конфликтов произвольного доступа
Белое светодиодное устройство переменного тока
Соединения, обладающие активирующим действием на подтипы рецепторов, активируемых пролифератором пероксисом (ppars), и способ получения и применения указанных соединений
Система стерильного розлива для поточного добавления частиц
Бензамидные производные, обладающие противораковой активностью, способ их получения и их применение
Горный экскаватор и его опорное устройство поворота и качения
Способ и устройство для передачи информации по восходящей линии связи
Способ, установка, устройство и система для уменьшения влияния помех на канал управления

