Результат интеллектуальной деятельности: ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Изобретение относится к фармацевтическим композициям, содержащим 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту (Соединение 1), методам производства таких препаратов и методам применения содержащих их фармацевтических композиций.
ПРЕДПОСЫЛКИ
CFTR представляет собой анионный канал, опосредованный цАМФ/АТФ и экспрессирующийся в клетках различных типов, включая всасывающий и секреторный эпителий, где он регулирует поток анионов через мембрану, а также активность других ионных каналов и белков. В эпителиальных клетках нормальное функционирование CFTR имеет решающее значение для транспорта электролитов во всем организме, в том числе в тканях дыхательной и пищеварительной систем. CFTR состоит приблизительно из 1480 аминокислот, образующих белок, состоящий из тандемных повторов трансмембранных доменов, каждый из которых содержит шесть трансмембранных спиралей, а также домена, связывающего нуклеотиды. Два трансмембранных домена связаны крупным, полярным, регуляторным (R)-доменом с множеством участков фосфорилирования, регулирующих активность канала и ионные потоки в клетках.
Ген, кодирующий CFTR, определен и секвенирован (см. Gregory, R. J. et al. (1990) Nature 347:382-386; Rich, D. P. et al. (1990) Nature 347:358-362), (Riordan, J. R. et al. (1989) Science 245:1066-1073). Дефект этого гена становится причиной мутаций в CFTR, приводящих к муковисцидозу (МВ), самому распространенному смертельному генетическому заболеванию человека. В США муковисцидоз поражает примерно 1 из 2500 младенцев ежегодно. В популяции США до 10 млн человек являются носителями одиночной копии дефектного гена без очевидных проявлений болезни. И наоборот, индивиды, несущие две копии гена, вызывающего МВ, страдают от инвалидизирующих и приводящих к смерти проявлений МВ, включая хроническое поражение легких.
Мутации в гене CFTR, экспрессирующемся эндогенно в дыхательном эпителии, приводят к снижению секреции анионов в апикальной мембране и дисбалансу транспорта ионов и жидкости у больных муковисцидозом. Обусловленное этим снижение транспорта анионов способствует усиленному накоплению слизи в легких и сопутствующей микробной инфекции, что в конечном итоге приводит к смерти больных МВ. В дополнение к дыхательным нарушениям, больные МВ обычно страдают от желудочно-кишечных проблем и недостаточности поджелудочной железы, при отсутствии лечения приводящей к смерти. Кроме того, большинство мужчин с муковисцидозом бесплодны, а у женщин с муковисцидозом снижена способность к оплодотворению. В противоположность больным, несущим две копии гена МВ и страдающим от тяжелых явлений, у людей, имеющих единственную копию этого гена, отмечается повышенная устойчивость к холере и обезвоживанию в результате диареи - что, вероятно, объясняет относительно высокую распространенность этого гена в популяции.
Анализ последовательности гена CFTR в хромосомах больных МВ выявил ряд мутаций, вызывающих болезнь (Cutting, G. R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61:863:870; and Kerem, B-S. et al. (1989) Science 245:1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). В настоящее время известно более 1000 мутаций в гене МВ, вызывающих заболевание, согласно опубликованным в научной и медицинской литературе данным. Наиболее распространенной мутацией является делеция фенилаланина в положении 508 аминокислотной последовательности CFTR, которую часто обозначаютF508del-CFTR. Эта мутация обнаруживается приблизительно в 70% случаев муковисцидоза и сопровождается тяжелым заболеванием. Прочие мутации включают R117H и G551D.
Делеция остатка 508 в F508del-CFTR препятствует правильному сворачиванию образующегося белка. Это нарушает выход мутантного белка из эндоплазматической сети и движение к плазматической мембране. В результате число каналов в мембране намного ниже, чем в клетках, экспрессирующих CFTR исходного типа. В дополнение к нарушениям ионных потоков, мутации приводят к дефектам воротного механизма канала. Снижение количества каналов в мембране в сочетании с дефектом воротного механизма приводит к снижению транспорта анионов через эпителий и, следовательно, нарушению транспорта ионов и жидкости. (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). Однако исследования показали, что хотя число каналов F508del-CFTR в мембране снижается, они остаются функциональными, но в меньшей степени, чем CFTR исходного типа. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Denning et al., supra; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). Кроме F508del-CFTR, существуют другие нарушения, сопровождающиеся мутациями CFTR и приводящие к нарушению ионных потоков, синтеза и/или воротного механизма каналов, которые могут активироваться или ослабевать, изменяя секрецию анионов и прогресс и/или тяжесть болезни.
Хотя CFTR транспортирует различные молекулы, в дополнение к анионам, очевидно, что эта роль (транспорт анионов) представляет собой один из элементов важного механизма транспорта ионов и воды через эпителий. Остальные элементы включают эпителиальные Na+ каналы, ENaC, совместный транспортер Na+/2Cl-/K+, Na+-K+-АТФазный насос и K+ каналы, ответственные за поступление хлоридов в клетку.
Эти элементы действуют вместе, обеспечивая направленный транспорт через эпителий за счет их избирательной экспрессии и местоположения в клетке. Всасывание хлоридов происходит за счет координированной активности ENaC и CFTR, присутствующих на апикальной мембране, а также Na+-K+-АТФазного насоса и Cl- каналов, экспрессирующихся на базолатеральной поверхности клетки. Вторичный активный транспорт хлоридов со стороны просвета ведет к накоплению хлоридов внутри клетки, которые затем могут пассивно покидать клетку через Cl-каналы, то есть происходит векторный перенос. Конфигурация котранспортера Na+/2Cl-/K+, Na+-K+-АТФазного насоса и K+ каналов базолатеральной мембраны на базолатеральной поверхности CFTR со стороны просвета координирует секрецию хлорида через CFTR со стороны просвета. Поскольку вероятно, что транспорт воды никогда не бывает активным, ее поток через эпителий зависит от трансэпителиальных осмотических градиентов, создаваемых общим потоком ионов натрия и хлорид-ионов.
Как обсуждалось выше, полагают, что делеция остатка 508 при F508del-CFTR препятствует правильному сворачиванию образующегося белка, делая этот мутантный белок неспособным к выходу из ЭС и перемещению к плазматической мембране. Как результат, количество зрелого белка на плазматической мембране оказывается недостаточным и транспорт хлорид-ионов в эпителиальной ткани значительно снижается. Фактически, показано, что этот клеточный феномен дефекта обработки транспортеров АТФ-связывающей кассеты (АСК) в эндоплазматической сети (ЭС) лежит в основе не только МВ, но и большого разнообразия других изолированных и наследственных заболеваний. Возможны два механизма нарушения функции аппарата ЭС: либо за счет нарушения сопряжения с экспортом белков в ЭС, ведущего к деградации, либо за счет накопления этих дефектных/неправильно свернутых белков в ЭС [Aridor M, et al., Nature Med., 5(7), pp. 745-751 (1999); Shastry, B.S., et al., Neurochem. International, 43, pp. 1-7 (2003); Rutishauser, J., et al., Swiss Med Wkly, 132, pp. 211-222 (2002); Morello, JP et al., TIPS, 21, pp. 466-469 (2000); Bross P., et al., Human Mut., 14, pp. 186-198 (1999)].
Соединение 1 в форме соли описано в международной публикации PCT WO 2007056341 в качестве модулятора активности CFTR и, следовательно, эффективного средства лечения CFTR-опосредованных заболеваний, таких как муковисцидоз. Соединение 1 формы I, по существу кристаллическое и не содержащее соли, описывается в патентной заявке США US 20090170905, поданной 4 декабря 2008 г. Соединение 1 формы II и соединение 1 в форме соли-гидрохлорида A раскрываются в опубликованной патентной заявке США US 20110263654, поданной 7 апреля 2011 г. Все заявки включены в настоящей документ полностью посредством ссылки.
Соединение 1, как часть комбинации с ивакафтором (N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид), признано Управлением по надзору за продуктами питания и лекарствами США (FDA) как средство терапии прорыва муковисцидоза, это один из двух подобных препаратов на момент подачи этой заявки (второй - ивакафтор). Это указывает на значительную неудовлетворенную потребность в эффективном лечении, которое было бы направлено на причину муковисцидоза, в отличие от симптоматического лечения. Кроме того, распространенной проблемой является то, что препараты, одобренные FDA, не всегда доступны нуждающимся в них пациентам. Таким образом, существует значительная неудовлетворенная потребность в лекарственных формах, содержащих описанное ранее Соединение 1, и способах их получения непрерывным и контролируемым способом.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение относится к фармацевтическим композициям, фармацевтическим препаратам и твердым лекарственным формам, содержащим 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойную кислоту (Соединение 1), структура которой показана ниже:
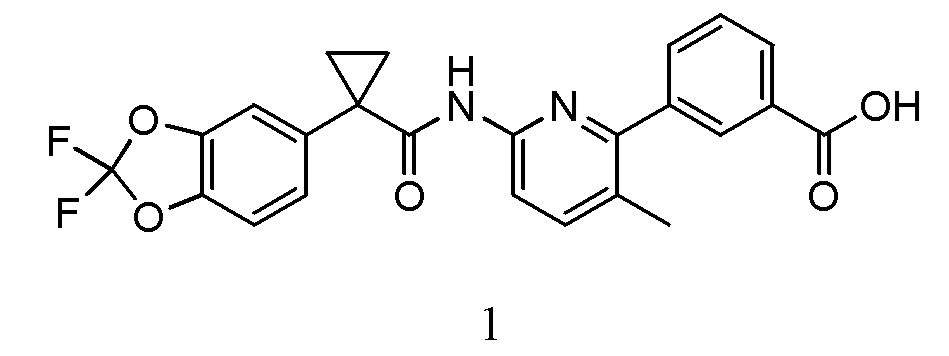
В одном аспекте, изобретение относится к фармацевтической композиции, содержащей:
a. Соединение 1;
b. наполнитель;
c. разрыхлитель;
d. поверхностно-активное вещество;
e. скользящее вещество; и
f. регулятор сыпучести или связующее вещество.
В других вариантах осуществления изобретения Соединение 1 по существу является одной из кристаллических твердых форм. В одном варианте осуществления изобретения Соединение 1 по существу является кристаллической формой I (Соединение 1 формы I). В одном варианте осуществления изобретения Соединение 1 по существу является кристаллической формой II (Соединение 1 формы II). В одном варианте осуществления изобретения Соединение 1 по существу является кристаллической солью HCl (Соединение 1 в форме соли HCl A). Понятно, что термин «Соединение 1», использующийся в этом документе, включает в себя, помимо прочих форм, в том числе некристаллических форм, следующие твердые формы: Соединение 1 формы I, Соединение 1 формы II и/или Соединение 1 в форме соли HCl A.
В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит от 25 мг до 400 мг. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 25 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 50 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 100 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 125 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 150 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 200 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 250 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 300 мг Соединения 1. В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит 400 мг Соединения 1.
В одном аспекте изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В одном аспекте изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом аспекте изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей следующие компоненты:
|
|
В другом аспекте, это изобретение относится к фармацевтической композиции в форме таблеток, которые содержат Соединение 1, а также один или более фармацевтически приемлемых вспомогательных компонентов, например наполнитель, разрыхлитель, поверхностно-активное вещество, разбавитель, связующее вещество, регулятор сыпучести и скользящее вещество или любые их сочетания, при этом за время приблизительно 30 минут таблетка растворяется по меньшей мере на 50%. В другом варианте осуществления скорость растворения составляет по меньшей мере около 75% приблизительно за 30 минут. В другом варианте осуществления скорость растворения составляет по меньшей мере около 90% приблизительно за 30 минут.
В другом аспекте, изобретение относится к фармацевтической композиции, состоящей из таблеток и содержащей порошковую или гранулированную смесь, которая содержит Соединение 1; и один и более фармацевтически приемлемых вспомогательных компонентов, например наполнитель, разрыхлитель, поверхностно-активное вещество, разбавитель, связующее вещество, регулятор сыпучести и скользящее вещество, при этом твердость таблетки составляет по меньшей мере около 5 килофунтов (kP); 1 kP = ~9,8 Н). В другом варианте осуществления расчетная истираемость таблетки менее 1,0% после 400 вращений. В другом аспекте, изобретение относится к фармацевтической композиции, состоящей из таблеток и содержащий порошковую или гранулированную смесь, которая содержит Соединение 1 в форме II, Соединение 1 и один и более фармацевтически приемлемых вспомогательных компонентов, например наполнитель, разрыхлитель, поверхностно-активное вещество, разбавитель, связующее вещество, регулятор сыпучести и скользящее вещество, при этом твердость таблетки составляет по меньшей мере около 5 килофунтов (kP); 1 kP = ~9,8 Н). В другом варианте осуществления расчетная истираемость таблетки менее 1,0% после 400 вращений.
В другом аспекте, изобретение относится к фармацевтической композиции, как описано в настоящем документе, также содержащей дополнительный терапевтический агент. В некоторых вариантах осуществления дополнительный терапевтический агент представляет собой N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид.
В другом аспекте, изобретение относится к способу лечения CFTR-опосредованного заболевания у млекопитающих, включающему введение млекопитающего эффективного количества описанной здесь фармацевтической композиции. В некоторых вариантах осуществления изобретения CFTR-опосредованным заболеванием является муковисцидоз, эмфизема, ХОЗЛ или остеопороз. В других вариантах осуществления изобретения CFTR-опосредованным заболеванием является муковисцидоз. Этот способ может дополнительно включать в себя введение дополнительного терапевтического агента, при этом в некоторых вариантах осуществления дополнительный терапевтический агент выбирают из муколитического агента, бронхорасширяющего средства, антибиотика, противоинфекционного агента, противовоспалительного агента, препарата, потенцирующего CFTR или питательного агента. В другом варианте осуществления дополнительный терапевтический агент представляет собой N-(5-гидрокси-2,4-дитретбутилфенил)-4-оксо-1H-хинолин-3-карбоксамид. В другом варианте осуществления у пациента имеется мутация F508del-CFTR. В другом варианте осуществления пациент гомозиготен по F508del. В другом варианте осуществления пациент гетерозиготен по F508del.
В другом аспекте, изобретение относится к набору, содержащему таблетку в соответствии с настоящим изобретением и отдельный терапевтический агент или содержащую его фармацевтическую композицию. В другом варианте осуществления Соединение 1 в виде таблетки находится в форме I. В другом варианте осуществления терапевтический агент представляет собой препарат для лечения муковисцидоза, не относящийся к Соединению 1. В другом варианте осуществления терапевтический агент представляет собой потенциатор для лечения муковисцидоза. В другом варианте изобретения терапевтический агент представляет собой N-(5-гидрокси-2,4-дитретбутилфенил)-4-оксо-1H-хинолин-3-карбоксамид. В другом варианте осуществления изобретения таблетка и терапевтический агент находятся в отдельных контейнерах. В другом варианте осуществления отдельные контейнеры представляют собой бутылки. В другом варианте осуществления отдельные контейнеры представляют собой флаконы. В другом варианте осуществления отдельные контейнеры представляют собой контурные ячейковые упаковки.
В другом аспекте, изобретение относится к способу получения описанных здесь фармацевтических композиций с помощью роликового пресса, включающему стадии просеивания и взвешивания Соединения 1 и вспомогательных веществ; смешивания Соединения 1 и вспомогательных веществ за приемлемый промежуток времени; вальцевания смеси в форме лент и размола лент на гранулы; смешивания гранул с вспомогательными компонентами, не входящими в состав гранул, за приемлемый промежуток времени; прессования смеси для получения таблеток; нанесения покрытия на таблетки; и, по желанию, печати монограммы на одной или обеих сторонах таблетки.
В другом аспекте, изобретение относится к способу получения описанных здесь фармацевтических композиций путем гранулирования с высоким усилием сдвига, включающего стадии просеивания и взвешивания Соединения 1 и вспомогательных веществ; смешивания Соединения 1 и вспомогательных веществ при одновременном добавлении жидкости для гранулирования, содержащей поверхностно-активное вещество и связующее вещество, при подходящей скорости перемешивания в течение приемлемого времени, и измельчения смеси для получения гранул; сушки гранул; смешивания гранул с вспомогательными компонентами, не входящими в состав гранул, за приемлемый промежуток времени; прессования смеси для получения таблеток; нанесения покрытия на таблетки; и, по желанию, печати монограммы на одной или обеих сторонах таблетки.
В другом аспекте, изобретение относится к непрерывным или полунепрерывным способам получения описанных здесь фармацевтических композиций путем влажного гранулирования с использованием двухшнекового аппарата, включающим в себя стадии просеивания и взвешивания Соединения 1 и вспомогательных веществ; смешивания Соединения 1 и вспомогательных веществ в смесителе и подачи смеси в установку для непрерывного гранулирования при одновременном добавлении жидкости для гранулирования, содержащей поверхностно-активное и связующее вещество, с подходящей скоростью за приемлемый промежуток времени, и измельчения смеси для получения гранул; сушки гранул; смешивания гранул с вспомогательными компонентами, не входящими в состав гранул, за приемлемый промежуток времени; прессования смеси для получения таблеток; нанесения покрытия на таблетки; и, по желанию, печати монограммы на одной или обеих сторонах таблетки.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1 показывает рентгенодифракционную картину, вычисленную на основе структуры одного кристалла Соединения 1 формы I.
Фиг. 2 показывает фактическую рентгенодифракционную картину Соединения 1 формы I.
Фиг. 3 показывает рентгенодифракционную картину Соединения 1 формы II.
Фиг. 4 показывает рентгенодифракционные картины Соединения 1 формы II, выбранных из:
1) Соединения 1 формы II, сольват с метанолом;
2) Соединения 1 формы II, сольват с этанолом;
3) Соединения 1 формы II, сольват с ацетоном;
4) Соединения 1 формы II, сольват с 2-пропанолом;
5) Соединения 1 формы II, сольват с ацетонитрилом;
6) Соединения 1 формы II, сольват с тетрагидрофураном;
7) Соединения 1 формы II, сольват с метиловым эфиром уксусной кислоты;
8) Соединения 1 формы II, сольват с 2-бутаноном;
9) Соединения 1 формы II, сольват с этиловым эфиром муравьиной кислоты; и
10) Соединения 1 формы II, сольват с 2-метилтетрагидрофураном.
Фиг. 5 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с метанолом.
Фиг. 6 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с этанолом.
Фиг. 7 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с ацетоном.
Фиг. 8 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с 2-пропанолом.
Фиг. 9 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с ацетонитрилом.
Фиг. 10 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с тетрагидрофураном.
Фиг. 11 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с метиловым эфиром уксусной кислоты.
Фиг. 12 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с 2-бутаноном.
Фиг. 13 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с этиловым эфиром муравьиной кислоты.
Фиг. 14 показывает рентгенодифракционную картину Соединения 1 формы II, сольват с 2-метилтетрагидрофураном.
Фиг. 15 показывает результат дифференциальной сканирующей калориметрии (ДСК) следов Соединения 1 формы II, сольват с ацетоном.
Фиг. 16 показывает график термогравиметрического анализа (ТГА) Соединения 1 формы II, сольват с ацетоном.
Фиг. 17 показывает конформацию Соединения 1 формы II, сольват с ацетоном, на основании рентгенодифракционного анализа одного кристалла.
Фиг. 18 показывает конформацию димера Соединения 1 в форме соли HCl A.
Фиг. 19 показывает картину рентгеновской дифракции Соединения 1 в форме соли HCl А, вычисленную по кристаллической структуре.
Фиг. 20 показывает 1H ЯМР-спектр Соединения 1.
Фиг. 21 показывает 1H ЯМР-спектр Соединения 1 в форме соли HCl.
Фиг. 22 показывает результат дифференциальной сканирующей калориметрии (ДСК) следов Соединения 1 формы I.
Фиг. 23 показывает конформацию Соединения 1 формы I на основании рентгенодифракционного анализа одного кристалла.
Фиг. 24 показывает конформацию Соединения 1 формы II, сольват с ацетоном, на основании рентгенодифракционного анализа одного кристалла.
Фиг. 25 показывает 13C ЯМР-спектр твердого вещества (вращение 15,0 кГц) Соединения 1 формы II, сольват с ацетоном.
Фиг. 26 показывает 19F ЯМР-спектр твердого вещества (вращение 12,5 кГц) Соединения 1 формы II, сольват с ацетоном.
Фиг. 27 показывает картину рентгеновской дифракции Соединения 1 в форме соли HCl А, вычисленную по кристаллической структуре.
Фиг. 28 показывает графики растворения Соединения 1 в градиенте pH для таблеток, изготовленных путем гранулирования с высоким усилием сдвига (ГВУС) и влажного гранулирования с помощью двухшнекового гранулятора (ВГДГ) (ПМВ означает потерю массы после высушивания, мера содержания воды в порошке/гранулах).
ПОДРОБНОЕ ОПИСАНИЕ
ОПРЕДЕЛЕНИЯ
В настоящем документе «CFTR» означает регулятор трансмембранной проводимости при муковисцидозе.
В настоящем документе обозначения «ΔF508» или «F508del» относятся к специфической мутации в белке CFTR. Эта мутация представляет собой делецию трех нуклеотидов, составляющих кодон аминокислоты фенилаланина в положении 508, что приводит к образованию молекулы белка CFTR без этого остатка фенилаланина.
В настоящем документе пациентом, «гомозиготным» по конкретной мутации, например F508del, называется пациент с той же мутацией в обоих аллелях.
В настоящем документе пациентом, «гетерозиготным» по конкретной мутации, например F508del, называется пациент, имеющий эту мутацию в одном аллеле и другую мутацию в другом аллеле.
В настоящем документе термин «корректор CFTR» относится к соединению, усиливающему или индуцирующему увеличение количества функционального белка CFTR на поверхности клетки, приводя к повышению функциональной активности.
В настоящем документе термин «потенциатор CFTR» относится к соединению, усиливающему или индуцирующему активность канала белка CFTR на поверхности клетки, приводя к повышению функциональной активности.
Термин «активный фармацевтический компонент» или «АФК» в настоящем документе относится к биологически активному компоненту. Примеры АФК включают в себя 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту (Соединение 1).
Термины «твердая форма», «твердые формы» и связанные термины, при использовании применительно к 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил) бензойной кислоте (Соединению 1), относятся к твердой форме, например, кристаллам и т.п., Соединения 1, которое находится преимущественно не в жидком или газообразном состоянии.
Термин «по существу аморфный» в настоящем документе относится к твердому материалу, не имеющему или почти не имеющему дальнего порядка в расположении молекул. Например, по существу аморфный материал имеет степень кристаллизации менее приблизительно 15% (например, степень кристаллизации менее примерно 10% или менее примерно 5%). Также отмечено, что термин «по существу аморфный» включает в себя ключевое слово «аморфный», относящееся к материалу без кристаллической структуры (0%).
В настоящем документе термин «по существу кристаллический» (например, во фразе «по существу кристаллическое Соединение 1 формы I, Соединение 1 формы II или Соединение 1 в форме соли HCl A») относится к твердому материалу, преимущественно имеющему дальний порядок в расположении молекул. Например, по существу кристаллический материал имеет степень кристаллизации более приблизительно 85% (например, степень кристаллизации более примерно 90% или более примерно 95%). Также отмечено, что термин «по существу кристаллический» включает в себя ключевое слово «кристаллический», относящееся к материалу кристаллической структуры 100%.
Термин «кристаллический» и связанные термины в настоящем документе, использующиеся для описания вещества, компонента, продукта или формы, означают, что вещество, компонент или продукт имеют по существу кристаллическую структуру по данным рентгенодифракционного анализа. (См., напр., Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins, Baltimore, Md. (2003); The United States Pharmacopeia, 23rd ed., 1843-1844 (1995)).
В настоящем документе термин «композиция» в целом относится к композиции двух или более компонентов, обычно одного или более лекарственных препаратов (например, Соединения 1 формы I, Соединения 1 формы II или Соединения 1 в форме соли HCl A) и одного или более вспомогательных фармацевтических компонентов.
В настоящем документе термин «твердая лекарственная форма» в целом относится к фармацевтической композиции, включающей, при использовании для приема внутрь, капсулы, таблетки, пилюли, порошки и гранулы. В таких лекарственных формах активное соединение смешано с по меньшей мере одним инертным фармацевтически приемлемым вспомогательным компонентом или носителем.
В настоящем документе «вспомогательное вещество» означает функциональные или нефункциональные компоненты в фармацевтической композиции.
В настоящем документе термин «разрыхлитель» означает вспомогательное вещество, увлажняющее фармацевтическую композицию и способствующую распаду таблеток. В настоящем документе термин «разбавитель» или «наполнитель» означает вспомогательное вещество, увеличивающее объем фармацевтической композиции.
Термин «поверхностно-активное вещество» в настоящем документе означает вспомогательное вещество, придающее фармацевтической композиции большую растворимость и/или смачиваемость.
Термин «связующее вещество» в настоящем документе означает вспомогательное вещество, придающее фармацевтической композиции способность к усиленной когезии или прочности на растяжение (т.е. твердость).
Термин «регулятор сыпучести» в настоящем документе означает вспомогательное вещество, придающее фармацевтической композиции усиленную сыпучесть.
Термин «краситель» в настоящем документе означает вспомогательное вещество, придающее фармацевтической композиции желаемый цвет. Примеры красителей включают готовые имеющиеся в продаже пигменты, например, алюминиевый лак FD&C синий № 1, FD&C синий №2, прочие синие красители FD&C, диоксид титана, оксид железа и/или их сочетания. В одном варианте осуществления изобретения фармацевтическая композиция в соответствии с изобретением имеет пурпурный цвет.
В настоящем документе термин «скользящее вещество» обозначает вспомогательное вещество, добавляемое к фармацевтическим композициям, прессующимся в таблетки. Скользящее вещество помогает спрессовыванию гранул в таблетки и выталкиванию таблетки фармацевтической композиции из штамповочного пресса.
Термины «кубический сантиметр» и «куб. см» в настоящем документе взаимозаменяемы и обозначают единицу объема. Обратите внимание, что 1 куб. см = 1 мл.
Термины «килофунт» и «kP» в настоящем документе взаимозаменяемы и обозначают меру силы, где kP = приблизительно 9,8 Ньютонов.
Термин «стойкость к истиранию» в настоящем документе относится к способности таблетки оставаться целой и сохранять свою форму, несмотря на воздействие внешнего давления. Стойкость к истиранию можно определить количественно по математическому выражению, представленному в уравнении 1:
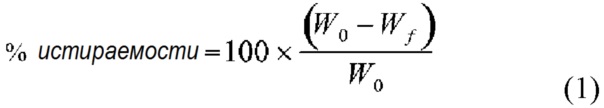
где W0 - исходный вес таблетки, а Wf - конечный вес таблетки после пропускания через фриабилятор. Стойкость к истиранию измеряют с помощью стандартного испытательного аппарата Фарм. США, выполняющего 100-400 вращений испытываемых таблеток. Некоторые таблетки в соответствии с настоящим изобретением характеризуются истираемостью менее 5,0%. В другом варианте осуществления истираемость составляет менее 2,0%. В другом варианте осуществления расчетная истираемость составляет менее 1,0% после 400 вращений.
В настоящем документе термин «средний диаметр частиц» обозначает средний диаметр частиц, измеренный такими методами, как рассеяние лазерного излучения, анализ изображений или анализ с помощью сит. В одном варианте осуществления изобретения гранулы, использующиеся для приготовления фармацевтических композиций в соответствии с изобретением, имеют средний диаметр частиц менее 1,0 мм.
В настоящем документе термин «насыпная плотность» обозначает массу частиц материала, деленную на полный объем, занимаемый частицами. Полный объем включает в себя объем частиц, объем пустот между частицами и объем внутренних пор. Насыпная плотность не является свойством, присущим материалу; она может изменяться в зависимости от способа обработки материала. В одном варианте осуществления изобретения гранулы, использующиеся для изготовления фармацевтических композиций в соответствии с изобретением, имеют насыпную плотность около 0,5-0,7 г/куб. см.
Эффективное количество или «терапевтически эффективное количество» лекарственного вещества в соответствии с настоящим изобретением может варьировать в зависимости от таких факторов, как течение заболевания, возраст и вес пациента, а также способности соединения в соответствии с настоящим изобретением вызывать желаемый ответ у пациента. Дозы можно регулировать для получения оптимального терапевтического ответа. Эффективное количество - также количество, при котором какие-либо токсические или вредоносные эффекты (т.е. побочные эффекты) соединения в соответствии с изобретением перевешиваются терапевтически благоприятными эффектами.
В настоящем документе, если не указано иное, термины «терапевтически эффективное количество» и «эффективное количество» соединения означают достаточное количество для благоприятного терапевтического действия при лечении заболевания или нарушения, или для замедления развития или максимального уменьшения одного или более симптомов, связанных с заболеванием или нарушением. «Терапевтически эффективное количество» и «эффективное количество» соединения означает количество терапевтического препарата, отдельно или в сочетании с одним или более препаратов, обеспечивающее благоприятное терапевтическое действие при лечении заболевания или нарушения. Термин «терапевтически эффективное количество» и «эффективное количество» может включать количество, улучшающее общее лечение, снижающее или предотвращающее симптомы или причины заболевания или нарушения, или повышающее терапевтическую эффективность другого терапевтического препарата.
«По существу чистый» во фразе «по существу чистое соединение 1 формы I, соединение 1 формы II или соединение 1 в форме соли HCl A,» означает чистоту выше, чем приблизительно 90%. В другом варианте осуществления термин «по существу чистый» означает чистоту выше, чем приблизительно 95%. В другом варианте осуществления термин «по существу чистый» означает чистоту выше чем приблизительно 98%. В другом варианте осуществления термин «по существу чистый» означает чистоту выше чем приблизительно 99%.
Применительно к Соединению 1 (т.е. Соединению 1 формы I, Соединению 1 формы II, Соединению 1 в форме соли HCl A), термины «около» и «приблизительно» при использовании в связи с дозами, количествами или массовыми процентами компонентов композиции или лекарственной формы означают дозу, количество или массовый процент, которые, как известно среднему специалисту в этой области, обеспечивают фармакологический эффект, эквивалентный эффекту конкретной дозы, количества или массового процента. В особенности, термин «около» или «приблизительно» означает допустимое отклонение от конкретного значения, по определению среднего специалиста в этой области, которое частично зависит от того, каким образом измеряется или определяется значение. В определенных вариантах осуществления термин «около» или «приблизительно» означает значение в пределах 1, 2, 3 или 4 стандартных отклонений. В определенных вариантах осуществления термин «около» или «приблизительно» означает значение в пределах 30%, 25%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,1% или 0,05% от данного значения или диапазона.
Если не указано иное, термин «Соединение 1» включает в себя, без ограничений, твердые формы Соединения 1, как описано в настоящем документе, в частности, Соединение 1 формы I, Соединение 1 формы II или Соединение 1 в форме соли HCl A, а также их сочетания.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Изобретение относится к фармацевтическим композициям, фармацевтическим составам и твердым лекарственным формам, содержащим Соединение 1, которое может иметь по существу кристаллическую форму. В некоторых вариантах осуществления Соединение 1 находится в кристаллической форме I (Соединение 1 формы I). В некоторых вариантах осуществления Соединение 1 находится в кристаллической форме II (Соединение 1 формы II). В некоторых вариантах осуществления Соединение 1 находится в кристаллической форме в виде соли HCl (Соединение 1 в форме соли HCl A). В некоторых вариантах осуществления этого аспекта количество Соединения 1, присутствующего в фармацевтической композиции, составляет 25 мг, 50 мг, 75 мг, 100 мг, 125 мг, 150 мг, 200 мг, 250 мг или 400 мг. В некоторых вариантах осуществления этого аспекта относительный массовый процент Соединения 1, присутствующего в фармацевтической композиции, от 10 до 75 процентов. В этих и других вариантах осуществления изобретения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойная кислота присутствует как по существу чистое Соединение 1. «По существу чистое» означает чистоту более 90%; предпочтительно, чистоту более 95%; еще предпочтительнее, чистоту более 99,5% (т.е. без примеси других кристаллических форм Соединения 1).
Таким образом, в одном аспекте, изобретение относится к фармацевтической композиции, включающей:
a. Соединение 1;
b. наполнитель;
c. разрыхлитель;
d. поверхностно-активное вещество;
e. разбавитель;
f. скользящее вещество; и
g. регулятор сыпучести или связующее вещество.
В одном варианте осуществления этого аспекта фармацевтическая композиция содержит 25 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 50 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 100 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 125 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 150 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 200 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 250 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 300 мг Соединения 1. В другом варианте осуществления этого аспекта фармацевтическая композиция содержит 400 мг Соединения 1.
В некоторых вариантах осуществления фармацевтические композиции содержат Соединение 1, при этом Соединение 1 присутствует в количестве по меньшей мере 15 массовых % (например, по меньшей мере 20 массовых %, по меньшей мере 30 массовых %, по меньшей мере 40 массовых %, по меньшей мере 50 массовых %, по меньшей мере 60 массовых % или по меньшей мере 70 массовых %) от массы композиции.
В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит Соединение 1, наполнитель, разбавитель, разрыхлитель, поверхностно-активное вещество, регулятор сыпучести и скользящее вещество. В этом варианте осуществления композиция содержит от приблизительно 20 массовых % до приблизительно 50 массовых % (например, приблизительно 25-35 массовых %) Соединения 1 от массы композиции, и, более типично, от 25 массовых % до приблизительно 45 массовых % (например, приблизительно 28-32 массовых %) Соединения 1 от массы композиции.
В некоторых вариантах осуществления изобретения фармацевтическая композиция содержит Соединение 1, наполнитель, разбавитель, разрыхлитель, поверхностно-активное вещество, связующее вещество и скользящее вещество. В этом варианте осуществления композиция содержит от приблизительно 30 массовых % до приблизительно 60 массовых % (например, приблизительно 40-55 массовых %) Соединения 1 от массы композиции и, более типично, от 35 массовых % до приблизительно 70 массовых % (например, приблизительно 45-55 массовых %) Соединения 1 от массы композиции.
Концентрация Соединения 1 в композиции зависит от нескольких факторов, например, количества фармацевтической композиции, необходимого для получения нужного количества Соединения 1, и желаемого графика растворения фармацевтической композиции.
В другом варианте осуществления изобретения фармацевтическая композиция содержит Соединение 1, при этом Соединение 1 находится в твердой форме и имеет средний диаметр частиц, измеренный методом рассеяния света (например, с помощью прибора «Malvern Mastersizer» производства Malvern Instruments, Англия) от 0,1 до 10 микрон. В другом варианте осуществления изобретения размер частиц Соединения составляет 1 от 1 до 5 микрон. В другом варианте осуществления Соединение 1 имеет размер частиц D50 2,0 микрон.
Как показано, в некоторых вариантах осуществления изобретения фармацевтические композиции, предназначенные для приема внутрь, в дополнение к Соединению 1 содержат один или более вспомогательных веществ, таких как наполнители, разрыхлители, поверхностно-активные вещества, разбавители, связующие вещества, регуляторы сыпучести, скользящие вещества, красители или отдушки, а также любые их сочетания.
Наполнители, подходящие для данного изобретения, сравнимы с компонентами фармацевтической композиции, т.е. они существенно не снижают растворимость, твердость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. К примерам наполнителей относятся: целлюлозы, модифицированные целлюлозы (например, натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза, гидроксиметилцеллюлоза, гидроксипропилцеллюлоза), ацетат целлюлозы, микрокристаллическая целлюлоза, фосфаты кальция, двухосновный фосфат кальция, крахмалы (например, кукурузный крахмал, картофельный крахмал), подсластители (например, сорбит, лактоза, сахароза и т.п.), либо любые их сочетания.
Таким образом, в одном варианте осуществления фармацевтическая композиция содержит по меньшей мере один наполнитель в количестве по меньшей мере 5 массовых % (например, по меньшей мере приблизительно 20 массовых %, по меньшей мере приблизительно 30 массовых % или по меньшей мере приблизительно 40 массовых %) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 10 массовых % до приблизительно 60 массовых % (например, от приблизительно 20 массовых % до приблизительно 55 массовых %, от приблизительно 25 массовых % до приблизительно 50 массовых %, или от приблизительно 27 массовых % до приблизительно 45 массовых %) наполнителя от массы композиции. В другом примере фармацевтическая композиция содержит по меньшей мере приблизительно 20 массовых % (т.е. по меньшей мере 30 массовых % или по меньшей мере 40 массовых %) микрокристаллической целлюлозы, например, МКЦ Avicel PH102, от массы композиции. Еще в одном примере фармацевтическая композиция содержит от приблизительно 10 массовых % до приблизительно 60 массовых % (например, от приблизительно 20 массовых % до приблизительно 55 массовых % или от приблизительно 25 массовых % до приблизительно 45 массовых %) микрокристаллической целлюлозы от массы композиции.
Разрыхлители, подходящие для данного изобретения, сравнимы с компонентами фармацевтической композиции, т.е. они значительно не снижают химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. Примеры разрыхлителей включают натриевую соль кросскармеллозы, натрия крахмалгликолят или их сочетания.
Таким образом, в одном варианте осуществления фармацевтическая композиция содержит разрыхлитель в количестве приблизительно 10 массовых % или меньше (например, приблизительно 7 массовых % или меньше, приблизительно 6 массовых % или меньше, или приблизительно 5 массовых % или меньше) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 1 массовых % до приблизительно 10 массовых % (например, от приблизительно 1,5 массовых % до приблизительно 7,5 массовых % или от приблизительно 2,5 массовых % до приблизительно 6 массовых %) разрыхлителя от массы композиции. В другом варианте осуществления фармацевтическая композиция содержит приблизительно 10 массовых % или меньше (например, 7 массовых % или меньше, 6 массовых % или меньше или 5 массовых % или меньше) натриевой соли кросскармелозы от массы композиции. Еще в одном примере фармацевтическая композиция содержит от приблизительно 1 массовых % до приблизительно 10 массовых % (например, от приблизительно 1,5 массовых % до приблизительно 7,5 массовых % или от приблизительно 2,5 массовых % до приблизительно 6 массовых %) натриевой соли кросскармеллозы от массы композиции. В некоторых вариантах осуществления фармацевтическая композиция содержит от приблизительно 0,1 массовых % до приблизительно 10 массовых % (например, от приблизительно 0,5 массовых % до приблизительно 7,5 массовых % или от приблизительно 1,5 массовых % до приблизительно 6 массовых %) разрыхлителя от массы композиции. В прочих вариантах осуществления фармацевтическая композиция содержит от приблизительно 0,5 массовых % до приблизительно 10 массовых % (например, от приблизительно 1,5 массовых % до приблизительно 7,5 массовых % или от приблизительно 2,5 массовых % до приблизительно 6 массовых %) разрыхлителя от массы композиции.
Поверхностно-активные вещества, подходящие для данного изобретения, сравнимы с компонентами фармацевтической композиции, т.е. они значительно не снижают химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. К примерам поверхностно-активных веществ относятся лаурилсульфат натрия (SLS), стеарилфумарат натрия (SSF), полиоксиэтилена 20 сорбитанмоноолеат (т.е. Твин™), любые их сочетания и т.п.
Таким образом, в одном варианте осуществления фармацевтическая композиция содержит поверхностно-активное вещество в количестве приблизительно 10 массовых % или меньше (например, приблизительно 5 массовых % или меньше, приблизительно 2 массовых % или меньше, приблизительно 1 массовых % или меньше, приблизительно 0,8 массовых % или меньше или приблизительно 0,6 массовых % или меньше) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 10 массовых % до приблизительно 0,1 массовых % (например, от приблизительно 5 массовых % до приблизительно 0,2 массовых % или от приблизительно 2 массовых % до приблизительно 0,3 массовых %) поверхностно-активного вещества от массы композиции. В другом примере фармацевтическая композиция содержит 10 массовых % или меньше (например, приблизительно 5 массовых % или меньше, приблизительно 2 массовых % или меньше, приблизительно 1 массовых % или меньше, приблизительно 0,8 массовых % или меньше, или приблизительно 0,6 массовых % или меньше) натрия лаурилсульфата от массы композиции. Еще в одном примере фармацевтическая композиция содержит от приблизительно 10 массовых % до приблизительно 0,1 массовых % (например, от приблизительно 5 массовых % до приблизительно 0,2 массовых % или от приблизительно 2 массовых % до приблизительно 0,3 массовых %) лаурилсульфата натрия от массы композиции.
Связующие вещества, подходящие для данного изобретения, повышают прочность таблетки и сравнимы с компонентами фармацевтической композиции, т.е. существенно не снижают химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. К примерам связующих веществ относятся поливинилпирролидон, двухосновный фосфат кальция, сахароза, кукурузный (маисовый) крахмал, модифицированная целлюлоза (например, гидроксиметилцеллюлоза) или любые их сочетания.
Таким образом, в одном варианте осуществления фармацевтическая композиция содержит по меньшей мере приблизительно 0,1 массового % (например, по меньшей мере приблизительно 1 массовый %, по меньшей мере приблизительно 3 массовых %, по меньшей мере приблизительно 4 массовых % или по меньшей мере приблизительно 5 массовых %) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 0,1 массового % до приблизительно 10 массовых % (например, от приблизительно 1 массового % до приблизительно 10 массовых % или от приблизительно 2 массовых % до приблизительно 7 массовых %) связующего вещества от массы композиции. В другом примере фармацевтическая композиция содержит по меньшей мере приблизительно 0,1 массового % (т.е. по меньшей мере приблизительно 1 массовый %, по меньшей мере приблизительно 2 массовых %, по меньшей мере приблизительно 3 массовых % или по меньшей мере приблизительно 4 массовых %) поливинилпирролидона от массы композиции. В еще одном примере фармацевтическая композиция содержит скользящее вещество в количестве от приблизительно 0,1 массового % до приблизительно 10 массовых % (например, от приблизительно 1 массового % до приблизительно 8 массовых % или от приблизительно 2 массовых % до приблизительно 5 массовых %) поливинилпирролидона от массы композиции.
Разбавители, подходящие для данного изобретения, могут добавлять необходимый объем составу для изготовления таблеток желаемого размера и обычно совместимы с компонентами фармацевтической композиции, т.е. они значительно не снижают растворимость, твердость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Примеры разбавителей включают: сахара, например, кондитерский сахар, сжимаемый сахар, декстраны, декстрин, декстрозу, лактозу, маннит, сорбит, целлюлозу и модифицированные целлюлозы, например, порошковую целлюлозу, тальк, фосфат кальция, крахмал или любые их сочетания.
Таким образом, в одном варианте осуществления фармацевтическая композиция содержит разбавитель в количестве приблизительно 40 массовых % или меньше (например, 35 массовых % или меньше, 30 массовых % или меньше, 25 массовых % или меньше, 20 массовых % или меньше, 15 массовых % или меньше или 10 массовых % или меньше) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 40 массовых % до приблизительно 1 массового % (например, от приблизительно 35 массовых % до приблизительно 5 массовых % или от приблизительно 30 массовых % до приблизительно 7 массовых %, от приблизительно 25 массовых % до приблизительно 10 массовых %, от приблизительно 20 массовых % до приблизительно 15 массовых %) разбавителя от массы композиции. В другом варианте осуществления фармацевтическая композиция содержит приблизительно 40 массовых % или меньше (например, 35 массовых % или меньше, 25 массовых % или меньше или 15 массовых % или меньше) маннита от массы композиции. Еще в одном примере фармацевтическая композиция содержит от приблизительно 35 массовых % до приблизительно 1 массового % (например, от приблизительно 30 массовых % до приблизительно 5 массовых % или от приблизительно 25 массовых % до приблизительно 10 массовых %) маннита от массы композиции.
Регуляторы сыпучести, подходящие для данного изобретения и улучшающие сыпучесть фармацевтической композиции, сравнимы с компонентами фармацевтической композиции, т.е. они значительно не снижают растворимость, твердость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Примеры регуляторов сыпучести включают коллоидный оксид кремния, тальк или их сочетания.
Таким образом, в одном варианте осуществления фармацевтическая композиция содержит регулятор сыпучести в количестве приблизительно 2 массовых % или меньше (например, приблизительно 1,75 массовых % или меньше, приблизительно 1,25 массовых % или меньше, или приблизительно 1,00 массовый % или меньше) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 2 массовых % до приблизительно 0,05 массового % (например, от приблизительно 1,5 массового % до приблизительно 0,07 массового % или от приблизительно 1,0 массового % до приблизительно 0,09 массового %) регулятора сыпучести от массы композиции. В другом примере фармацевтическая композиция содержит приблизительно 2 массовых % или меньше (например, 1,75 массового % или меньше, 1,25 массового % или меньше или 1,00 массового % или меньше) коллоидного диоксида кремния от массы композиции. Еще в одном примере фармацевтическая композиция содержит от приблизительно 2 массовых % до приблизительно 0,05 массового % (например, от приблизительно 1,5 массового % до приблизительно 0,07 массового % или от приблизительно 1,0 массовых % до приблизительно 0,09 массового %) коллоидного диоксида кремния от массы композиции.
В некоторых вариантах осуществления фармацевтическая композиция может содержать скользящее вещество, способное препятствовать адгезии смеси гранул к поверхности (например, поверхности чаши для смешивания, пресса и/или пробойника). Кроме того, скользящее вещество способно уменьшить трение между частицами гранулята и способствовать прессованию или выталкиванию прессованных фармацевтических композиций из пресса. Кроме того, скользящее вещество сравнимо с компонентами фармацевтической композиции, т.е. оно существенно не снижает растворимость, твердость или биологическую активность фармацевтической композиции. Примеры скользящих веществ включают стеарат магния, стеарат кальция, стеарат цинка, стеарат натрия, стеариновую кислоту, стеарат алюминия, лейцин, глицерилбегенат, гидрогенизированное растительное масло или сочетание этих компонентов. В одном варианте осуществления фармацевтическая композиция содержит скользящее вещество в количестве 5 массовых % или меньше (например, 4,75 массовых %, 4,0 массовых % или меньше, 3,00 массовых % или меньше, или 2,0 массовых % или меньше) от массы композиции. Например, фармацевтическая композиция содержит от приблизительно 5 массовых % до приблизительно 0,10 массовых % (например, от приблизительно 4,5 массовых % до приблизительно 0,5 массовых % или от приблизительно 3 массовых % до приблизительно 1 массового %) скользящего вещества от массы композиции. В другом примере фармацевтическая композиция содержит приблизительно 5 массовых % или меньше (например, 4,0 массовых % или меньше, 3,0 массовых % или меньше, 2,0 массовых % или меньше, 1,0 массовый % или меньше) стеарата магния от массы композиции. Еще в одном примере фармацевтическая композиция содержит от приблизительно 5 массовых % до приблизительно 0,10 массового % (например, от приблизительно 4,5 массовых % до приблизительно 0,15 массового % или от приблизительно 3,0 массовых % до приблизительно 0,50 массового %) стеарата магния от массы композиции.
Фармацевтические композиции в соответствии с настоящим изобретением могут дополнительно содержать один или более красителей, ароматизаторов и/или отдушек для улучшения внешнего вида, вкуса и/или запаха композиции. Подходящие красители, ароматизаторы или отдушки сравнимы с компонентами фармацевтической композиции, т.е. они существенно не снижают растворимость, химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. В одном варианте осуществления изобретения фармацевтическая композиция содержит краситель, ароматизатор и/или отдушку. В одном варианте осуществления изобретения фармацевтическая композиция в соответствии с настоящим изобретением имеет пурпурный цвет.
В некоторых вариантах осуществления фармацевтическая композиция включает таблетки или может быть изготовлена в форме таблеток, при этом таблетки могут быть покрыты красителем и дополнительно маркированы логотипом, другим изображением и/или текстом с помощью подходящей печатной краски. В других вариантах осуществления фармацевтическая композиция включает таблетки или может быть изготовлена в форме таблеток, при этом таблетки могут быть покрыты красителем, воском, а также дополнительно маркированы логотипом, другим изображением и/или текстом с помощью подходящей печатной краски. Подходящие красители и печатные краски сравнимы с компонентами фармацевтической композиции, т.е. они существенно не снижают растворимость, химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. Подходящие красители и печатные краски могут быть любого цвета и изготавливаться на основе воды или органических растворителей. В одном варианте осуществления изобретения таблетки, изготовленные из фармацевтической композиции, покрыты красящим веществом и затем маркированы логотипом, другим изображением и/или текстом с помощью подходящей печатной краски. Например, таблетки, содержащие описанную здесь фармацевтическую композицию, могут быть покрыты пленкой, составляющей около 3 массовых % (например, меньше чем приблизительно 6 массовых % или меньше чем приблизительно 4 массовых %) и содержащей краситель. Окрашенные таблетки могут быть маркированы логотипом и текстом, показывающим содержание действующего вещества в таблетке, с помощью подходящей печатной краски. В другом примере, таблетки, содержащие описанную здесь фармацевтическую композицию, могут быть покрыты пленкой, составляющей около 3 массовых % (например, меньше чем приблизительно 6 массовых % или меньше чем приблизительно 4 массовых %) и содержащей краситель.
В другом варианте осуществления изобретения таблетки, изготовленные из фармацевтической композиции, покрыты красящим веществом, воском, а затем маркированы логотипом, другим изображением и/или текстом с помощью подходящей печатной краски. Например, таблетки, содержащие описанную здесь фармацевтическую композицию, могут быть покрыты пленкой, составляющей около 3 массовых % (например, меньше чем приблизительно 6 массовых % или меньше чем приблизительно 4 массовых %) и содержащей краситель. Для покрытия окрашенных таблеток воском можно применять порошковый карнаубский воск в массовом соотношении приблизительно 0,01% от начальной массы сердцевины таблетки. Покрытые воском таблетки могут быть маркированы логотипом и текстом, показывающим содержание действующего вещества в таблетке, с помощью подходящей печатной краски. В другом примере таблетки, содержащие описанную здесь фармацевтическую композицию, могут быть покрыты оболочкой, составляющей примерно 3 массовых % (например, менее чем приблизительно 6 массовых % или менее чем приблизительно 4 массовых %), содержащей краситель. Окрашенные таблетки можно покрыть порошкообразным карнаубским воском в весовом соотношении приблизительно 0,01% от начальной массы сердцевины таблетки. Покрытые воском таблетки могут быть маркированы логотипом и текстом, указывающим содержание действующего вещества в таблетке, с использованием печатных красок для фармацевтической промышленности, например, черной краски (например, Opacode® S-1-17823, краска на основе органического растворителя, поставщик - компания Colorcon, Inc., Вест Пойнт, Пенсильвания.).
Одним из примеров фармацевтической композиции является композиция, содержащая от приблизительно 15 массовых % до приблизительно 70 массовых % (например, от приблизительно 15 массовых % до приблизительно 60 массовых %, от приблизительно 15 массовых % до приблизительно 50 массовых %, от приблизительно 15 массовых % до приблизительно 40 массовых %, от приблизительно 20 массовых % до приблизительно 70 массовых %, от приблизительно 30 массовых % до приблизительно 70 массовых %, от приблизительно 40 массовых % до приблизительно 70 массовых %, от приблизительно 50 массовых % до приблизительно 70 массовых %) Соединения 1 от массы композиции. Вышеупомянутые композиции могут также содержать один или более фармацевтически приемлемых вспомогательных веществ, например, от приблизительно 20 массовых % до приблизительно 50 массовых % наполнителя; от приблизительно 1 массового % до приблизительно 5 массовых % разрыхлителя; от приблизительно 2 массовых % до приблизительно 0,3 массового % скользящего вещества; от приблизительно 0,1 массового % до приблизительно 5 массовых % связующего вещества; от приблизительно 1 массового % до приблизительно 30 массовых % разбавителя; от приблизительно 2 массовых % до приблизительно 0,05 массового % регулятора сыпучести; и от приблизительно 5 массовых % до приблизительно 0,1 массового % скользящего вещества. Либо фармацевтическая композиция включает в себя композицию, содержащую от приблизительно 15 массовых % до приблизительно 70 массовых % (например, от приблизительно 20 массовых % до приблизительно 40 массовых %, от приблизительно 25 массовых % до приблизительно 60 массовых % или от приблизительно 30 массовых % до приблизительно 55 массовых %) Соединения 1 от массы композиции; и один или более вспомогательных веществ, например, от приблизительно 20 массовых % до приблизительно 50 массовых % наполнителя; от приблизительно 1 массового % до приблизительно 5 массовых % разрыхлителя; от приблизительно 2 массовых % до приблизительно 0,3 массового % скользящего вещества; от приблизительно 0,1 массового % до приблизительно 5 массовых % связующего вещества; от приблизительно 1 массового % до приблизительно 30 массовых % разбавителя; от приблизительно 2 массовых % до приблизительно 0,05 массовых % регулятора сыпучести; и от приблизительно 5 массовых % до приблизительно 0,1 массового % скользящего вещества.
Еще один пример фармацевтической композиции содержит от приблизительно 15 массовых % до приблизительно 70 массовых % (например, от приблизительно 15 массовых % до приблизительно 60 массовых %, от приблизительно 15 массовых % до приблизительно 50 массовых %, от приблизительно 15 массовых % до приблизительно 40 массовых %, от приблизительно 20 массовых % до приблизительно 70 массовых %, от приблизительно 30 массовых % до приблизительно 70 массовых %, от приблизительно 40 массовых % до приблизительно 70 массовых % или от приблизительно 50 массовых % до приблизительно 70 массовых %) Соединения 1 от массы композиции и один или более вспомогательных веществ, например, от приблизительно 20 массовых % до приблизительно 50 массовых % наполнителя; от приблизительно 1 массового % до приблизительно 5 массовых % разрыхлителя; от приблизительно 2 массовых % до приблизительно 0,3 массового % скользящего вещества; от приблизительно 0,1 массового % до приблизительно 5 массовых % связующего вещества; от приблизительно 1 массового % до приблизительно 30 массовых % разбавителя; от приблизительно 2 массовых % до приблизительно 0,05 массового % регулятора сыпучести; и от приблизительно 2 массовых % до приблизительно 0,1 массового % скользящего вещества.
Еще один пример фармацевтической композиции содержит от приблизительно 15 массовых % до приблизительно 70 массовых % (например, от приблизительно 15 массовых % до приблизительно 60 массовых %, от приблизительно 15 массовых % до приблизительно 50 массовых %, от приблизительно 15 массовых % до приблизительно 40 массовых %, от приблизительно 20 массовых % до приблизительно 70 массовых %, от приблизительно 30 массовых % до приблизительно 70 массовых %, от приблизительно 40 массовых % до приблизительно 70 массовых % или от приблизительно 50 массовых % до приблизительно 70 массовых %) Соединения 1 от массы композиции и один или более вспомогательных веществ, например, от приблизительно 20 массовых % до приблизительно 50 массовых % наполнителя; от приблизительно 1 массового % до приблизительно 5 массовых % разрыхлителя; от приблизительно 2 массовых % до приблизительно 0,3 массового % скользящего вещества; от приблизительно 0,1 массового % до приблизительно 5 массовых % связующего вещества; от приблизительно 1 массового % до приблизительно 30 массовых % разбавителя; от приблизительно 2 массовых % до приблизительно 0,05 массового % регулятора сыпучести; и от приблизительно 2 массовых % до приблизительно 0,1 массового % скользящего вещества.
Еще один пример фармацевтической композиции содержит от приблизительно 15 массовых % до приблизительно 70 массовых % (например, от приблизительно 15 массовых % до приблизительно 60 массовых %, от приблизительно 15 массовых % до приблизительно 50 массовых %, от приблизительно 15 массовых % до приблизительно 40 массовых %, от приблизительно 20 массовых % до приблизительно 70 массовых %, от приблизительно 30 массовых % до приблизительно 70 массовых %, от приблизительно 40 массовых % до приблизительно 70 массовых % или от приблизительно 50 массовых % до приблизительно 70 массовых %) Соединения 1 и один или более вспомогательных веществ, например, от приблизительно 20 массовых % до приблизительно 50 массовых % наполнителя; от приблизительно 1 массового % до приблизительно 5 массовых % разрыхлителя; от приблизительно 2 массовых % до приблизительно 0,3 массового % скользящего вещества; от приблизительно 0,1 массового % до приблизительно 5 массовых % связующего вещества; от приблизительно 1 массового % до приблизительно 30 массовых % разбавителя; от приблизительно 2 массовых % до приблизительно 0,05 массового % регулятора сыпучести; и от приблизительно 2 массовых % до приблизительно 0,1 массового % скользящего вещества.
В одном варианте осуществления изобретения фармацевтическая композиция в гранулированной форме содержит:
a. приблизительно 30 массовых % Соединения 1 от массы композиции;
b. приблизительно 42 массовых % микрокристаллической целлюлозы от массы композиции;
c. приблизительно 21 массовых % маннита от массы композиции;
d. приблизительно 3 массовых % натриевой соли кросскармелозы от массы композиции;
e. приблизительно 1 массового % лаурилсульфата натрия от массы композиции;
f. приблизительно 2 массовых % стеарата магния от массы композиции; и
g. приблизительно 0,5 массового % коллоидного оксида кремния от массы композиции.
Еще одна гранулированная композиция в виде лекарственной формы для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 50 массовых % Соединения 1;
b. приблизительно 30 массовых % микрокристаллической целлюлозы от массы композиции;
c. приблизительно 13 массовых % маннита от массы композиции;
d. приблизительно 2 массовых % натриевой соли кросскармеллозы от массы композиции;
e. приблизительно 4 массовых % поливинилпирролидона от массы композиции; и
f. приблизительно 1 массовый % лаурилсульфата натрия от массы композиции.
В одном варианте осуществления изобретения фармацевтическая композиция для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 30 массовых % Соединения 1 от массы композиции;
b. приблизительно 42 массовых % микрокристаллической целлюлозы от массы композиции;
c. приблизительно 21 массовый % маннита от массы композиции;
d. приблизительно 3 массовых % натриевой соли кросскармелозы от массы композиции;
e. приблизительно 1 массовый % лаурилсульфата натрия от массы композиции;
f. приблизительно 2,5 массовых % стеарата магния от массы композиции; и
g. приблизительно 0,5 массового % коллоидного оксида кремния от массы композиции.
Еще одна лекарственная форма для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 50 массовых % Соединения 1 от массы композиции;
b. приблизительно 30 массовых % микрокристаллической целлюлозы от массы композиции;
c. приблизительно 13 массовых % маннита от массы композиции;
d. приблизительно 4 массовых % натриевой соли кросскармеллозы от массы композиции;
e. приблизительно 4 массовых % поливинилпирролидона от массы композиции
f. приблизительно 1 массовый % лаурилсульфата натрия от массы композиции; и
g. приблизительно 0,5 массового % сульфата магния от массы композиции.
Еще одна лекарственная форма для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 60 массовых % Соединения 1 от массы композиции;
b. приблизительно 20 массовых % микрокристаллической целлюлозы от массы композиции;
c. приблизительно 13 массовых % маннита от массы композиции;
d. приблизительно 4 массовых % натриевой соли кросскармеллозы от массы композиции;
e. приблизительно 4 массовых % поливинилпирролидона от массы композиции;
f. приблизительно 1 массовый % лаурилсульфата натрия от массы композиции; и
g. приблизительно 0,5 массового % сульфата магния от массы композиции.
Еще одна лекарственная форма для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно от 150 до 250 мг Соединения 1;
b. приблизительно от 40 до 50 мг маннита;
c. приблизительно от 120 до 130 мг микрокристаллической целлюлозы;
d. приблизителен от 10 до 20 мг натриевой соли кросскармелозы;
e. приблизительно от 10 до 20 мг поливинилпирролидона;
f. приблизительно от 1 до 5 мг лаурилсульфата натрия; и
g. приблизительно от 1 до 5 мг стерата магния.
Еще одна лекарственная форма для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 200 мг Соединения 1;
b. приблизительно 43 мг маннита;
c. приблизительно 123 мг микрокристаллической целлюлозы;
d. приблизительно 15 мг натриевой соли кросскармелозы;
e. приблизительно 13 мг поливинилпирролидона;
f. приблизительно 3 мг лаурилсульфата натрия; и
g. приблизительно 4 мг стеарата магния.
Еще одна лекарственная форма для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 200 мг Соединения 1;
b. приблизительно 45 мг маннита;
c. приблизительно 123 мг микрокристаллической целлюлозы;
d. приблизительно 15 мг натриевой соли кросскармелозы;
e. приблизительно 10,4 мг поливинилпирролидона;
f. приблизительно 26 мг лаурилсульфата натрия; и
g. приблизительно 4 мг стеарата магния.
Еще одна лекарственная форма для приема внутрь в соответствии с настоящим изобретением содержит:
a. приблизительно 70 массовых % Соединения 1 от массы композиции;
b. приблизительно 12 массовых % микрокристаллической целлюлозы от массы композиции;
c. приблизительно 11 массовых % маннита от массы композиции;
d. приблизительно 4 массовых % натриевой соли кросскармеллозы от массы композиции;
e. приблизительно 4 массовых % поливинилпирролидона от массы композиции
f. приблизительно 1 массовый % лаурилсульфата натрия от массы композиции; и
g. приблизительно 0,5 массового % сульфата магния от массы композиции.
Фармацевтические композиции в соответствии с настоящим изобретением можно изготавливать в форме таблеток, капсул, пастилок или других твердых форм, подходящих для приема внутрь. Таким образом, в некоторых вариантах осуществления фармацевтические композиции изготовлены в форме таблеток.
Еще в одном варианте осуществления изобретения фармацевтическая композиция в форме таблеток имеет начальную твердость 5-21 kP ± 20% и содержит: приблизительно 30 массовых % Соединения 1; приблизительно 42 массовых % микрокристаллической целлюлозы от массы композиции; приблизительно 21 массовый % маннита от массы композиции; приблизительно 3 массовых % натриевой соли кросскармелозы от массы композиции; приблизительно 1 массовый % лаурилсульфата натрия от массы композиции; приблизительно 2,5 массовых % стеарата магния от массы композиции; и приблизительно 0,5 массового % коллоидного оксида кремния от массы композиции. При этом количество Соединения 1 в таблетированной фармацевтической композиции составляет от приблизительно 25 мг до приблизительно 250 мг, например, 50 мг, 75, 100 мг 150 мг, 200 мг или 250 мг Соединения 1 на таблетку.
Еще в одном варианте осуществления изобретения фармацевтическая композиция в форме таблеток имеет начальную твердость 5-21 kP ± 20% и содержит: приблизительно 49 массовых % Соединения 1; приблизительно 29 массовых % микрокристаллической целлюлозы от массы композиции; приблизительно 12,6 массовых % маннита от массы композиции; приблизительно 4 массовых % натриевой соли кросскармелозы от массы композиции; приблизительно 4 массовых % поливинилпирролидона от массы композиции; приблизительно 1 массовый % лаурилсульфата натрия от массы композиции; и приблизительно 0,5 массового % стеарата магния от массы композиции. Количество Соединения 1 в фармацевтической композиции в форме таблеток варьируется от приблизительно 25 мг до приблизительно 250 мг, например, 50 мг, 75 мг, 100 мг, 150 мг, 200 мг или 250 мг Соединения 1 на таблетку.
В определенных вариантах осуществления фармацевтическая композиция в форме таблеток содержит приблизительно 100 мг Соединения 1. В определенных вариантах осуществления фармацевтическая композиция в форме таблеток содержит приблизительно 200 мг Соединения 1.
Другой аспект настоящего изобретения относится к фармацевтической композиции в форме таблеток или капсул, содержащей Соединение 1 и другие вспомогательные вещества (например, наполнитель, разрыхлитель, поверхностно-активное вещество, связующее вещество, регулятор сыпучести, краситель, скользящее вещество или любое их сочетание), каждое из которых описано выше и в приведенных ниже примерах, при этом таблетка растворяется по меньшей мере на приблизительно 50% (например, по меньшей мере приблизительно на 60%, по меньшей мере приблизительно на 70%, по меньшей мере приблизительно на 80%, по меньшей мере приблизительно на 90% или по меньшей мере приблизительно на 99%) примерно за 30 минут. В одном примере фармацевтическая композиция представляет собой таблетку, содержащую Соединение 1 в количестве от 25 до 250 мг, например, 25 мг, 50 мг, 75 мг, 100 мг, 150 мг, 200 мг или 250 мг, и одно или более вспомогательных веществ (например, наполнитель, разрыхлитель, поверхностно-активное вещество, связующее вещество, регулятор сыпучести, краситель, скользящее вещество или любое их сочетание), каждое из которых описано выше и в приведенных ниже примерах, при этом таблетка растворяется приблизительно на 50-100% (например, от приблизительно 55% до приблизительно 95% или от приблизительно 60% до приблизительно 90%) примерно за 30 минут. В другом примере фармацевтическая композиция представляет собой таблетку, которая содержит композицию, содержащую Соединение 1; и одно или более вспомогательных веществ из следующей группы: наполнитель, разбавитель, разрыхлитель, поверхностно-активное вещество, связующее вещество, регулятор сыпучести и скользящее вещество, при этом таблетка растворяется по меньшей мере приблизительно на 50% (например, по меньшей мере приблизительно на 60%, по меньшей мере приблизительно на 70%, по меньшей мере приблизительно на 80%, по меньшей мере приблизительно на 90% или по меньшей мере приблизительно на 99%) примерно за 30 минут.
В одном варианте осуществления таблетка содержит композицию, содержащую по меньшей мере приблизительно 25 мг (например, по меньшей мере приблизительно 30 мг, по меньшей мере приблизительно 40 мг или по меньшей мере приблизительно 50 мг) Соединения 1; и одно или более вспомогательных веществ из следующей группы: наполнитель, разбавитель, разрыхлитель, поверхностно-активное вещество, связующее вещество, регулятор сыпучести и скользящее вещество. В другом варианте осуществления изобретения таблетка содержит фармацевтическую композицию, содержащую по меньшей мере приблизительно 25 мг (например, по меньшей мере приблизительно 30 мг, по меньшей мере приблизительно 40 мг, по меньшей мере приблизительно 50 мг, по меньшей мере приблизительно 100 мг или по меньшей мере 150 мг) Соединения 1 и одно или более из следующих вспомогательных веществ: наполнитель, разбавитель, разрыхлитель, поверхностно-активное вещество, связующее вещество, регулятор сыпучести и скользящее вещество.
Растворение можно измерить с помощью стандартного аппарата Фарм. США II типа, в котором в качестве среды для растворения используется 0,1% ЦТАБ, растворенный в 900 мл деионизованной воды, забуференной при pH 6,8 одноосновным фосфатом калия, при перемешивании со скоростью 50-75 об/мин и температуре приблизительно 37°C. В каждом сосуде аппарата испытывается одна экспериментальная таблетка. Кроме того, растворение можно измерить с помощью стандартного аппарата Фарм. США II типа, в котором в качестве среды для растворения используется 0,7% лаурилсульфат натрия, растворенный в 900 мл 50 мМ фосфатного буфера (pH 6,8), при перемешивании на скорости около 65 об/мин. и температуре приблизительно 37°C. В каждом сосуде аппарата испытывается одна экспериментальная таблетка. Кроме того, растворение можно измерить с помощью стандартного аппарата Фарм. США II типа, в котором в качестве среды для растворения используется 0,5% лаурилсульфат натрия, растворенный в 900 мл 50 мМ фосфатного буфера (pH 5), при перемешивании на скорости около 65 об/мин. и температуре приблизительно 37°C. В каждом сосуде аппарата испытывается одна экспериментальная таблетка.
СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ 1, СОЕДИНЕНИЯ 1 ФОРМЫ I, СОЕДИНЕНИЯ 1 ФОРМЫ II, СОЕДИНЕНИЯ 1 В ФОРМЕ СОЛИ HCl A
Соединение 1
Соединение 1 используется в качестве исходного вещества для получения других твердых форм и может изготовляться путем соединения функциональной группы кислоты с функциональной группой амина в соответствии со схемами 1-4.
Схема 1. Синтез функциональной группы хлорангидрида.
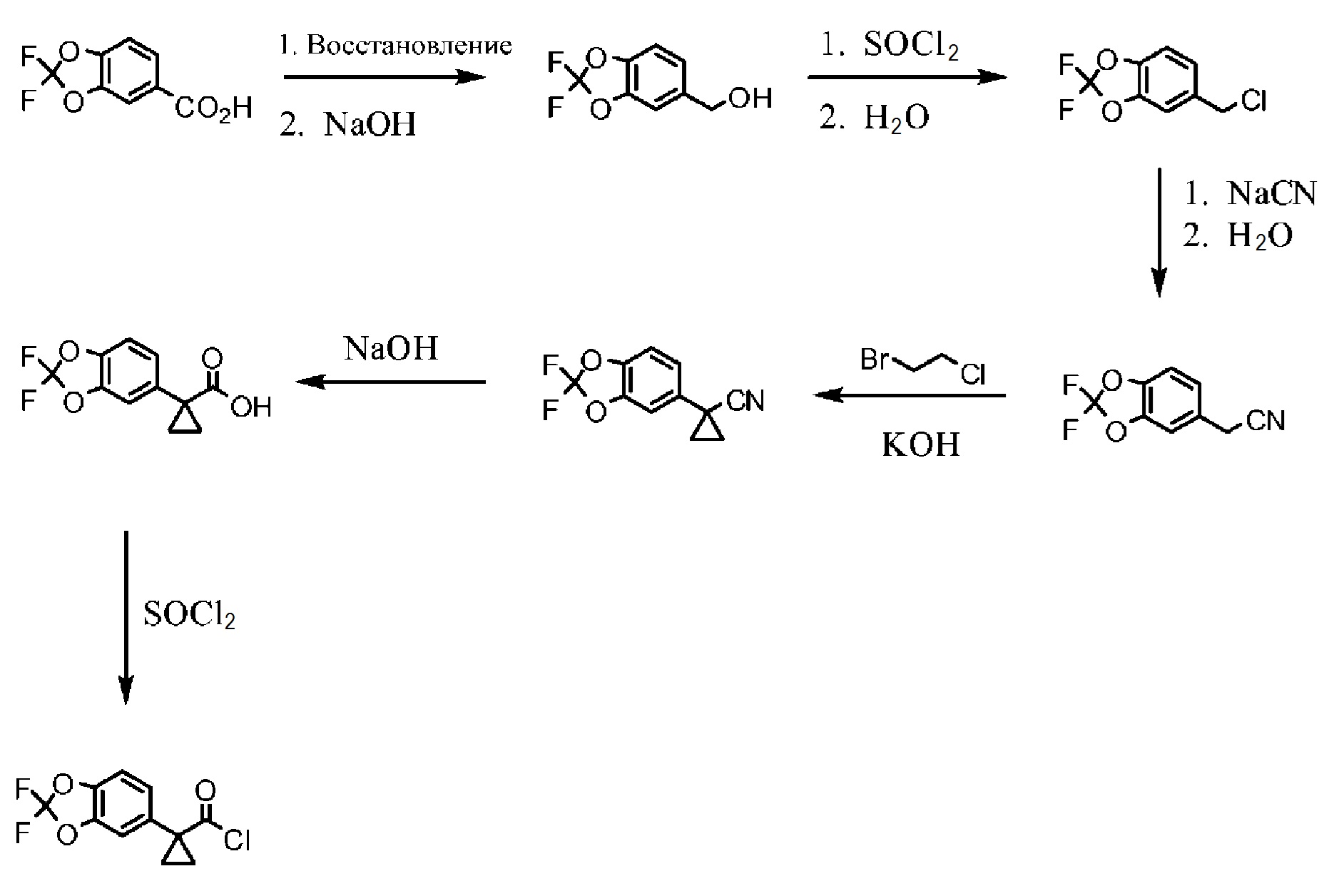
На схеме 1 изображено получение 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлорида, который применяется на Схеме 3 для получения амидной связи Соединения 1.
Исходный материал, 2,2-дифторбензо[d][1,3]диоксол-5-карбоновую кислоту, поставляет компания Saltigo (филиал Lanxess Corporation). Восстановление функциональной группы карбоновой кислоты 2,2-дифторбензо[d][1,3]диоксол-5-карбоновой кислоты до первичного спирта с последующим превращением в соответствующий хлорид путем реакции с тионилхлоридом (SOCl2) дает 5-(хлорметил)-2,2-дифторбензо[d][1,3]диоксол, который в последующем превращается в 2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)ацетонитрил путем реакции с цианидом натрия. Обработка 2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)ацетонитрила основанием и 1-бром-2-хлорэтаном дает 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонитрил. Нитрильная функциональная группа в 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонитриле переводится в карбоновую кислоту с помощью основания, при этом получается 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоновая кислота, которая превращается в требуемый хлорид путем реакции с тионилхлоридом.
Схема 2. Альтернативный синтез функциональной группы хлорангидрида.
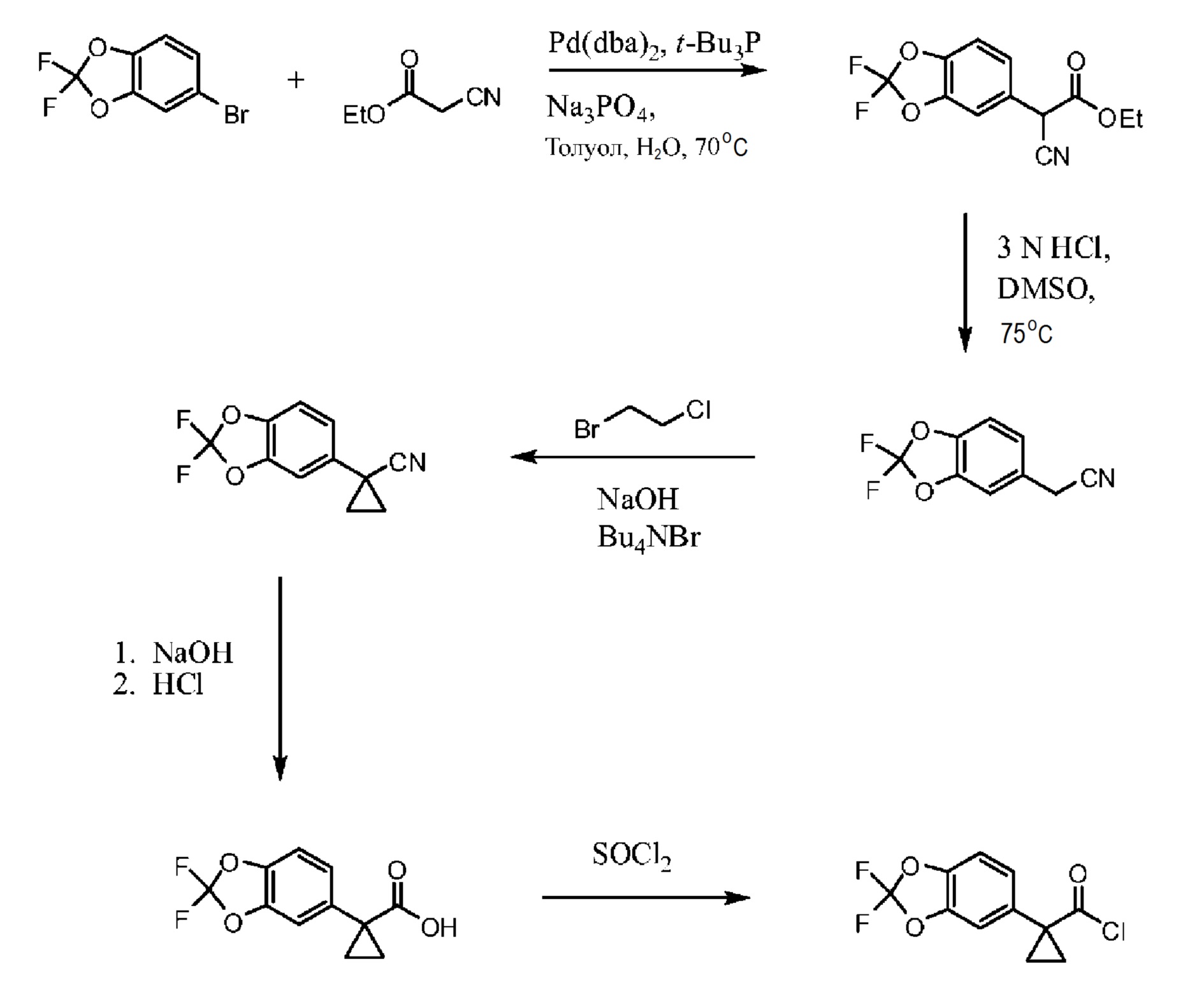
На Схеме 2 изображен альтернативный способ синтеза требуемого хлорангидрида. 5-бромметил-2,2-дифтор-1,3-бензодиоксол соединяется с этилцианоацетатом в присутствии палладиевого катализатора для образования соответствующего альфа-цианоэтилового эфира. Омыление функциональной группы эфира для получения карбоновой кислоты дает цианоэтиловое соединение. Алкилирование цианоэтилового соединения 1-бром-2-хлорэтаном в присутствии основания дает цианоциклопропил. Обработка цианоциклопропила основанием дает соль-карбоксилат, которую превращают в карбоновую кислоту путем обработки кислотой. Затем следует превращение карбоновой кислоты в хлорангидрид с помощью хлорирующего агента, например, тионилхлорида и т.п.
Схема 3.Синтез аминогруппы.
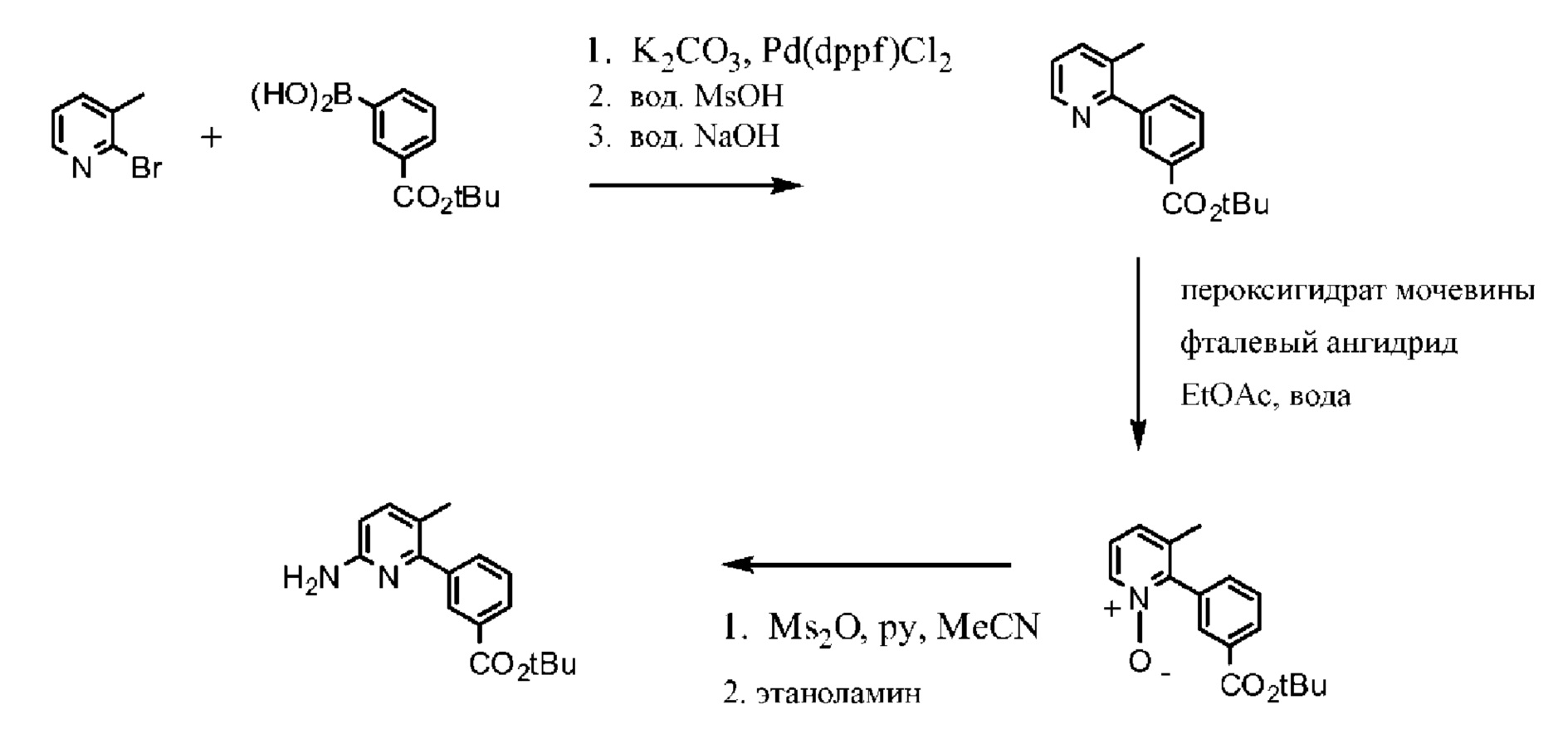
На Схеме 3 изображено получение требуемого трет-бутил 3-(6-амино-3-метилпиридин-2-ил)бензоата, который соединяется с 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлоридом на Схеме 3 для получения Соединения 1. Соединение 2-бром-3-метилпиридина с 3-(трет-бутоксикарбонил)фенилбороновой кислоты в присутствии палладия в качестве катализатора дает трет-бутил 3-(3-метилпиридин-2-ил)бензоат, который в последующем превращается в желаемое соединение.
Схема 4. Образование соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты.
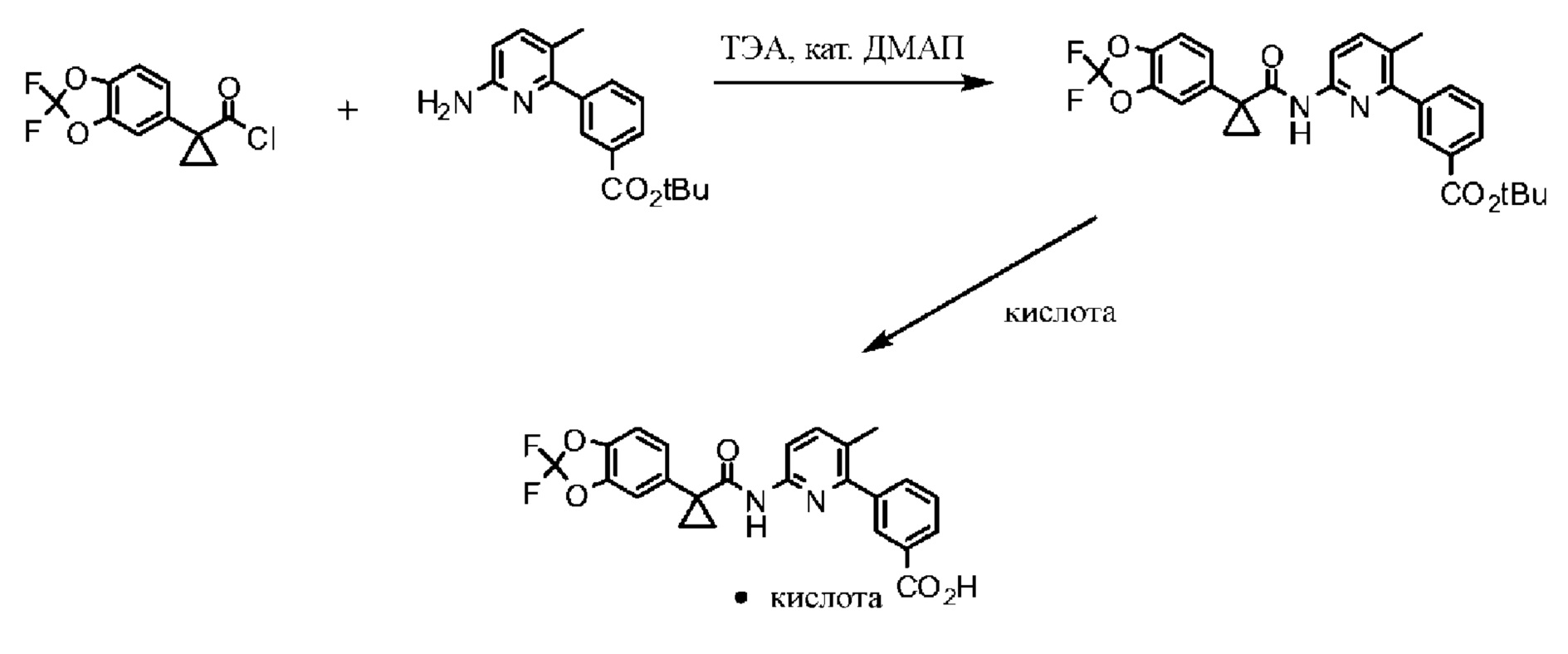
На Схеме 4 изображено соединение 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлорида с трет-бутил 3-(6-амино-3-метилпиридин-2-ил)бензоатом с использованием триэтиламина и 4-диметиламинопиридина для изначального получения трет-бутилового эфира Соединения 1.
Соединение 1 формы I
Соединение 1 формы I получают путем диспергирования или растворения соли, например, соли HCl, Соединения 1 в подходящем растворителе в течение эффективного периода времени. Обработка трет-бутилового эфира кислотой, например HCl, дает соответствующую соль Соединения 1, обычно представляющая собой твердое кристаллическое вещество. Соединение 1 также может быть получено непосредственно из предшественника трет-бутилового эфира путем обработки соответствующей кислотой, например, муравьиной кислотой.
Соль HCl 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты можно использовать для получения Формы I путем диспергирования или растворения соли HCl 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты в подходящем растворители в течение эффективного периода времени. Также могут использоваться прочие соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты, например, соли других неорганических или органических кислот. Другие соли образуются в результате кислотного гидролиза функциональной группы трет-бутилового эфира. Соли-производные других кислот могут включать, например, азотную, серную, фосфорную, борную, уксусную, бензойную и малоновую. Эти соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты могут быть или не быть растворимыми, в зависимости от используемого растворителя, однако нерастворимость не препятствует образованию Соединения I. Например, в одном варианте осуществления подходящим растворителем может быть вода или смесь спирта и воды, например, смесь 50% метанола/воды, несмотря на то, что соль HCl 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты слаборастворима в воде. В одном варианте осуществления подходящим растворителем является вода.
Эффективное время образования Формы I из соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты может быть любым временем от 2 до 24 и более часов. Известно, что необходимый период времени обратно пропорционален температуре. То есть, чем выше температура, тем меньше времени требуется для диссоциации кислоты и образования Формы I. Когда растворителем является вода, перемешивание дисперсной смеси в течение приблизительно 24 ч при комнатной температуре дает приблизительно 98% выход Формы I. Если для способа желателен раствор соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты, можно использовать более высокие температуры. После перемешивания раствора в течение эффективного периода времени при повышенной температуре перекристаллизация при охлаждении дает по существу чистую Форму I. В одном варианте осуществления термин «по существу чистый» означает чистоту выше, чем приблизительно 90%. В другом варианте осуществления термин «по существу чистый» означает чистоту выше, чем приблизительно 95%. В другом варианте осуществления термин «по существу чистый» означает чистоту выше, чем приблизительно 98%. В другом варианте осуществления термин «по существу чистый» означает чистоту выше, чем приблизительно 99%. Выбранная температура частично зависит от используемого растворителя и находится в пределах, которые возможно измерить обычными способами. В одном варианте осуществления температура лежит в пределах от комнатной температуры до приблизительно 80°C. В другом варианте осуществления температура лежит в пределах от комнатной температуры до приблизительно 40°C. В другом варианте осуществления температура лежит в пределах от приблизительно 40°C до приблизительно 60°C. В другом варианте осуществления температура лежит в пределах от приблизительно 60°C до приблизительно 80°C.
Соединение 1 формы I также может быть получено непосредственно из 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата (см. Схему 3), который является предшественником соли Соединения 1. Таким образом, 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоат может вступать в реакцию с соответствующей кислотой, например, с муравьиной кислотой в условиях реакции, подходящих для получения Соединения 1 формы I.
Соединение 1 формы I может быть далее очищено путем перекристаллизации из органического растворителя. Примеры органических растворителей включают, без ограничений, толуол, кумол, анизол, 1-бутанол, изопропиловый эфир уксусной кислоты, бутиловый эфир уксусной кислоты, изобутиловый эфир уксусной кислоты, метил-t-бутиловый эфир, метилизобутилкетон и смеси 1-пропанола и воды. Температуры могут быть такими, как описано выше. Например, Форма I растворяется в 1-бутаноле при 75°C до полного растворения. При охлаждении раствора до 10°C со скоростью 0,2°C/мин образуются кристаллы Формы I, которые можно отфильтровать.
В одном варианте осуществления Соединение 1 формы I характеризуется одним или более пиками на 15,2-15,6 градусов и 14,3-14,7 градусов при рентгенодифракционном анализе порошка с помощью источника Cu Kα-излучения. В другом варианте осуществления Соединение 1 формы I характеризуется одним или более пиками на 15,4, 16,3 и 14,5 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 14,6-15,0 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 14,8 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 17,6-18,0 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 17,8 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 16,4-16,8 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 16,4-16,8 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 16,6 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 7,6-8,0 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 7,8 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 25,8-26,2 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 26,0 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 21,4-21,8 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 21,6 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 23,1-23,5 градусов. В другом варианте осуществления Соединение 1 формы I дополнительно характеризуется пиком на 23,3 градусов. В некоторых вариантах осуществления Соединение 1 формы I характеризуется дифракционной картиной, по существу сходной с показанной на Фиг. 1. В некоторых вариантах осуществления Соединение 1 формы I характеризуется дифракционной картиной, по существу сходной с показанной на Фиг. 2.
В некоторых вариантах осуществления распределение частиц Соединения 1 формы I по размеру D50 составляет около 30 мкм или меньше.
Соединение 1 формы II
Соединение 1 формы II получают путем суспендирования Соединения 1 формы I в подходящем органическом растворителе при достаточной концентрации в течение достаточного времени. Затем суспензию фильтруют с помощью центрифуги или под вакуумом и сушат при комнатных условиях достаточное время для получения Соединения II.
В некоторых вариантах осуществления изобретения приблизительно 20-40 мг Соединения 1 формы I суспендируют приблизительно в 400-600 мкл подходящего растворителя. В другом варианте осуществления изобретения приблизительно 25-35 мг Соединения 1 формы I суспендируют приблизительно в 450-550 мкл подходящего растворителя. В другом варианте осуществления изобретения приблизительно 30 мг Соединения 1 формы I суспендируют приблизительно в 500 мкл подходящего растворителя.
В некоторых вариантах осуществления время, в течение которого Соединение 1 формы I суспендируется в растворителе, составляет от 1 ч до 4 дней. Конкретнее, время, в течение которого Соединение 1 суспендируется в растворителе, составляет от 1 до 3 дней. Конкретнее, это время составляет 2 дня.
В некоторых вариантах осуществления подходящий растворитель выбирается из органического растворителя с молекулами достаточного размера, чтобы заполнить пустоты в кристаллической решетке Соединения 1 формы II. В других вариантах осуществления молекулы сольвента имеют достаточный размер, чтобы уместиться в пустоты размером около 100 Å3.
В других вариантах осуществления растворитель выбирается из группы, состоящей из метанола, этанола, ацетона, 2-пропанола, ацетонитрила, тетрагидрофурана, метилового эфира уксусной кислоты, 2-бутанона, этилового эфира муравьиной кислоты и 2-метилтетрагидрофурана.
В других вариантах осуществления для получения Соединения 1 формы II можно использовать смесь двух или более из этих растворителей. Альтернативно, Соединение 1 формы II может быть получено из смеси, содержащей один или более из этих растворителей и воду.
В некоторых вариантах осуществления эффективное время сушки Соединения 1 формы II составляет от 1 до 24 ч. Конкретнее, это время составляет от 6 до 18 ч. Конкретнее, это время составляет около 12 ч.
В другом варианте осуществления Соединение 1 формы II получают путем диспергирования или растворения соли Соединения 1, например гидрохлорида Соединения 1, в подходящем растворителе в течение эффективного периода времени.
Описанное здесь Соединение 1 формы II содержит кристаллическую решетку Соединения 1, в которой пустоты кристаллической решетки свободны, либо заполнены, либо частично заполнены одной или более молекулами подходящего растворителя. Подходящие растворители включают, без ограничений, метанол, этанол, ацетон, 2-пропанол, ацетонитрил, тетрагидрофуран, метиловый эфир уксусной кислоты, 2-бутанон, этиловый эфир муравьиной кислоты и 2-метилтетрагидрофуран. Определенные физические свойства изоструктурных сольватных форм Соединения 1, например, картина рентгеновской дифракции порошка, точка плавления и свойства при дифференциальной сканирующей калориметрии, значительно не изменяются в зависимости от молекулы растворителя.
В одном варианте осуществления Соединение 1 формы II характеризуется одним или более пиками на 21,50-21,90 градусов и 8,80-9,20 градусов при рентгенодифракционном анализе порошка с помощью Cu трубки в качестве источника Kα излучения. В другом варианте осуществления Соединение 1 формы II характеризуется одним или более пиками на 21,50-21,90 градусов, 8,80-9,20 градусов, 18,00-18,40 градусов и 22,90-23,30 градусов при рентгенодифракционном анализе порошка с помощью Cu трубки в качестве источника Kα излучения. В другом варианте осуществления Соединение 1 формы II характеризуется одним или более пиками на 21,70, 8,98 и 11,04 градусов. В другом варианте осуществления Соединение 1 формы II характеризуется одним или более пиками на 21,70, 8,98, 11,04, 18,16 и 23,06 градусов. В другом варианте осуществления Соединение 1 формы II характеризуется пиком на 21,50-21,90 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 21,70 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 8,80-9,20 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 8,98 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 10,80-11,20 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 11,04 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 18,00-18,40 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 18,16 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 22,90 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 23,06 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 20,40-20,80 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 20,63 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 22,00-22,40 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 22,22 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 18,40-18,80 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 18,57 градусов. В другом варианте осуществления изобретения Соединение 1 формы II дополнительно характеризуется пиком на 16,50-16,90 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 16,66 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 19,70-20,10 градусов. В другом варианте осуществления Соединение 1 формы II дополнительно характеризуется пиком на 19,86 градусов.
В некоторых вариантах осуществления Соединение 1 формы II характеризуется дифракционной картиной, по существу сходной с показанной на Фиг. 3. В некоторых вариантах осуществления Соединение 1 формы II характеризуется дифракционными картинами, по существу сходными с показанной на Фиг. 4.
В другом варианте осуществления изобретения сольват, образующий форму II Соединения 1, выбирается из группы, состоящей из метанола, этанола, ацетона, 2-пропанола, ацетонитрила, тетрагидрофурана, метилового эфира уксусной кислоты, 2-бутанона, этилового эфира муравьиной кислоты и 2-метилтетрагидрофурана. Дифракционные картины представлены для следующих сольватов Соединения 1 формы II: метанол (Фиг. 5), этанол (Фиг. 6), ацетон (Фиг. 7), 2-пропанол (Фиг. 8) ацетонитрил (Фиг. 9), тетрагидрофуран (Фиг. 10), метиловый эфир уксусной кислоты (Фиг. 11), 2-бутанон (Фиг. 12), этиловый эфир муравьиной кислоты (Фиг. 13) и 2-метилтетрагидрофуран (Фиг. 14).
В другом варианте осуществления изобретение относится к Соединению 1 формы II, демонстрирующему 2 или более фазовых переходов, определенных методом дифференциальной сканирующей калориметрии или сходным аналитическим методом, известным специалистам в этой области. В некоторых вариантах осуществления изобретения результаты ДСК Соединения 1 формы II по существу сходны с результатами ДСК, показанными на Фиг. 15. В другом варианте осуществления этого аспекта ДСК показывает два фазовых перехода. В другом варианте осуществления ДСК показывает три фазовых перехода. В другом варианте осуществления один из фазовых переходов происходит между 200 и 207°C. В другом варианте осуществления один из фазовых переходов происходит между 204 и 206°C. В другом варианте осуществления один из фазовых переходов происходит между 183 и 190°C. В другом варианте осуществления один из фазовых переходов происходит между 185 и 187°C. В другом варианте осуществления точка плавления Соединения 1, сольватной формы А лежит в пределах от 183°C до 190°C. В другом варианте осуществления точка плавления Соединения 1, сольватной формы А лежит в пределах от 185°C до 187°C.
В другом варианте осуществления изобретения Соединение 1 формы II содержит от 1 до 10 массовых процентов (массовых %) сольвата по результатам термогравиметрического анализа (ТГА). В некоторых вариантах осуществления изобретения результаты ТГА Соединения 1 формы II по существу сходны с результатами ТГА, показанными на Фиг. 16. В другом варианте осуществления изобретения Соединение 1 формы II содержит от 2 до 5 массовых % сольвата по результатам ТГА или сходного аналитического метода, известного специалисту в этой области.
В другом варианте осуществления изобретения конформация Соединения 1 формы II в виде сольвата с ацетоном по существу сходна с изображенной на Фиг. 17, определенной с помощью рентгеноструктурного анализа одного кристалла.
В другом варианте осуществления изобретения Соединение 1 формы II в виде сольвата с ацетоном имеет группу симметрии кристаллической решетки P21/n и следующие размеры ячейки:
a=16,5235 (10) Å α = 90°
b=12,7425 (8) Å β = 103,736 (4)°
c=20,5512 (13) Å γ = 90°.
Соединение 1 в форме соли HCl A
Соединение 1 в форме соли HCl А можно получить из соли HCl Соединения 1 путем растворения соли HCl Соединения 1 в минимальном количестве растворителя и удаления растворителя путем медленного выпаривания. В другом варианте осуществления растворитель является спиртом. В другом варианте осуществления растворитель является этанолом. Медленное выпаривание обычно выполняется способами, затрудняющими испарение растворителя. Например, в одном варианте осуществления медленное выпаривание включает в себя растворение соли HCl Соединения 1 во флаконе с пленкой «парафильм», в которой проделано отверстие.
В одном варианте осуществления Соединение 1 в форме соли HCl A характеризуется одним или более пиками на 8,80-9,20 градусов и 18,20-18,60 градусов при рентгенодифракционном анализе порошка с помощью Cu трубки в качестве источника Kα излучения. В другом варианте осуществления Соединение 1 в форме соли HCl A характеризуется одним или более пиками на 8,80-9,20 градусов, 17,30-17,70 градусов, 18,20-18,60 градусов, 10,10-10,50 градусов и 15,80-16,20 градусов при рентгенодифракционном анализе порошка с помощью Cu трубки в качестве источника Kα излучения. В другом варианте осуществления изобретения Соединение 1 в форме соли HCl А характеризуется одним или более пиками при 8,96, 17,51 и 18,45 градусов. В другом варианте осуществления изобретения Соединение 1 в форме соли HCl А характеризуется одним или более пиками при 8,96, 17,51, 18,45, 10,33 и 16,01 градусов. В другом варианте осуществление Соединение 1 в форме соли HCl A характеризуется пиком на 8,80-9,20 градусов. В другом варианте осуществление Соединение 1 в форме соли HCl A характеризуется пиком на 8,96 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 17,30-17,70 градусов. В другом варианте осуществление Соединение 1 в форме соли HCl A характеризуется пиком на 17,51 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 18,20-18,60 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 18,45 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 10,10-10,50 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 10,33 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 15,80-16,20 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 16,01 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 11,70-12,10 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 11,94 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком при 7,90-8,30 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 8,14 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 9,90-10,30 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 10,10 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 16,40-16,80 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 16,55 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 9,30-9,70 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 9,54 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 16,40-16,80 градусов. В другом варианте осуществления Соединение 1 в форме соли HCl A дополнительно характеризуется пиком на 16,55 градусов. В некоторых вариантах осуществления изобретения Соединение 1 в форме соли HCl A представляет собой димер, изображенный на Фиг. 18.
В некоторых вариантах осуществления Соединение 1 в форме соли HCl A характеризуется дифракционной картиной, по существу сходной с показанной на Фиг. 19.
В другом варианте осуществления изобретения Соединение 1 в форме соли HCl A имеет кристаллическую структуру, принадлежит к пространственной группе кристаллической решетки P-1 и имеет следующие размеры ячейки:
a=10,2702 (2) Å α = 67,0270 (10)°
b=10,8782 (2) Å β = 66,1810 (10)°
c=12,4821 (3) Å γ = 72,4760 (10)°.
СПОСОБЫ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
Лекарственные формы в соответствии с настоящим изобретением могут быть получены путем компактирования или прессования смеси или композиции, например, порошка или гранул, под давлением для получения стабильной трехмерной формы (например, таблетки). В настоящем документе термин «таблетка» включает в себя прессованную фармацевтическую лекарственную форму любых форм и размеров, с покрытием или без.
Выражение «лекарственная форма» при использовании в настоящем документе означает физически отдельную единицу препарата, подходящего пациенту, проходящему лечение. В целом, компактированная смесь имеет плотность больше, чем у смеси до компактирования. Лекарственная форма в соответствии с настоящим изобретением может иметь почти любую форму, в том числе вогнутые или выпуклые поверхности, закругленные или не закругленные углы, а также округлую или прямолинейную форму. В некоторых вариантах осуществления изобретения прессованные лекарственные формы в соответствии с настоящим изобретением включают округлую таблетку с плоскими поверхностями. Твердые лекарственные формы в соответствии с настоящим изобретением могут быть изготовлены с помощью любого способа компактирования и прессования, известного специалистам в этой области и подходящего для получения прессованных твердых лекарственных форм. В конкретных вариантах осуществления изобретения описанные здесь составы могут быть изготовлены с помощью традиционных методов, известных специалистам в области изготовления фармацевтических препаратов, как описано, например, в соответствующих учебниках. См., например, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins, Baltimore, Md. (2003); Ansel et al., Pharmaceutical Dosage Forms And Drug Delivery Systems, 7th Edition, Lippincott Williams & Wilkins, (1999); The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); Gibson, Pharmaceutical Preformulation And Formulation, CRC Press (2001), которые включены в настоящий документ посредством ссылки.
Гранулирование и прессование
В некоторых вариантах осуществления твердые формы, включая порошки, содержащие действующее вещество (Соединение 1) и фармацевтически приемлемые вспомогательные вещества (например, наполнитель, разрыхлитель, поверхностно-активное вещество, регулятор сыпучести, связующее вещество, скользящее вещество или любое их сочетание), могут быть подвергнуты способу сухого гранулирования. В способе сухого гранулирования порошки слипаются в более крупные частицы с размером, подходящим для последующей обработки. Сухое гранулирование может улучшить сыпучесть смеси, чтобы из нее можно было изготовить таблетки, соответствующие требованиям к однородности по массе или однородности содержимого.
Описанные здесь лекарственные формы могут быть произведены с помощью одной или более стадий перемешивания и гранулирования. Порядок и количество стадий смешивания и гранулирования не имеет решающего значения. Однако по меньшей мере один из вспомогательных компонентов или Соединение 1 могут быть подвергнуты способу сухого гранулирования или влажного гранулирования с большим усилием сдвига перед прессованием в таблетки. Сухое гранулирование Соединения 1 и вспомогательных веществ вместе перед прессованием таблеток представляется простым, недорогим и эффективным способом достижения тесного физического контакта между ингредиентами композиций и составов в соответствии с настоящим изобретением, кроме того, при этом получаются таблетки с хорошей стабильностью. Сухое гранулирование возможно с помощью механического способа, передающего энергию к смеси без использования каких-либо жидких веществ (в форме водных растворов, растворов в органических растворителей или их смесей), в противоположность способам влажного гранулирования, которые также предусмотрены. В целом, механический способ требует компактирования, например, обеспечиваемого вальцеванием. Примером альтернативного способа сухого гранулирования является брикетирование.
В некоторых вариантах осуществления изобретения способ вальцевания включает высокоэффективное механическое компактирование одного или более веществ. В некоторых вариантах осуществления изобретения фармацевтическая композиция, содержащая смесь порошков, подвергается прессованию, в частности, вальцеванию, между двумя роликами, вращающимися в противоположных направлениях, для получения твердого листа, который в последующем дробится в сите для получения частиц. В этом материале, состоящем из частиц, можно добиться тесного механического контакта между компонентами. Примером оборудования для вальцевания является «Minipactor® a Gerteis 3W-Polygran» производства Gerteis Maschinen+Processengineering AG.
В некоторых вариантах осуществления прессование таблеток в соответствии с настоящим изобретением возможно без использования каких-либо жидких веществ (в форме растворов в воде, органических растворителях или их смесей), т.е. с помощью способа сухого гранулирования. В типичном варианте осуществления образующаяся сердцевина таблетки имеет прочность на сжатие в диапазоне от 1 до 15 кПа; например, от 1,5 до 12,5 кПа, предпочтительно в диапазоне от 2 до 10 кПа.
Краткое описание процедуры производства
В некоторых вариантах осуществления компоненты взвешиваются в соответствии с приведенной здесь формулой. Далее, все компоненты, которые должны входить в состав гранул, просеиваются и хорошо перемешиваются. Для смазки к компонентам можно добавить подходящее скользящее вещество, например, стеарат магния. На следующей стадии возможно компактирование/брикетирование смеси порошка и компонентов определенного размера. Затем компактированные или брикетированные смеси перемалываются для получения гранул и просеиваются до нужного размера. Далее к гранулам можно дополнительно добавить скользящее вещество, например, стеарат магния. В последующем композицию в форме гранул в соответствии с настоящим изобретением можно прессовать с помощью подходящих прессов для получения различных лекарственных форм в соответствии с настоящим изобретением. Дополнительно таблетки можно покрыть пленкой, красителем или другим покрытием.
Другой аспект настоящего изобретения относится к способу получения фармацевтической композиции, включающему в себя предварительное смешивание композиции, содержащей Соединение 1 и один или более вспомогательных веществ, выбранных из следующих: наполнитель, разбавитель, связующее вещество, регулятор сыпучести, поверхностно-активное вещество, скользящее вещество, разрыхлитель, и прессование композиции в таблетку, которая растворяется по меньшей мере приблизительно на 50% примерно за 30 минут.
В другом варианте осуществления изобретения используется способ влажного гранулирования для получения фармацевтического состава в соответствии с настоящим изобретением из смеси порошкообразных и жидких компонентов. Например, фармацевтической композиции, содержащей смесь Соединения 1 и одного или более вспомогательных веществ, выбранных из следующих: наполнитель, разбавитель, связующее вещество, регулятор сыпучести, поверхностно-активное вещество, скользящее вещество, разрыхлитель, которые взвешиваются в соответствии с указанной здесь формулой. Далее, один или более компонентов, входящих в состав гранул, просеиваются и перемешиваются с помощью гранулятора с высоким или низким усилием сдвига или двухшнекового гранулятора с использованием воды, или воды с поверхностно-активным веществом, или воды со связующим веществом, или воды с поверхностно-активным и связующим веществом для гранулирования порошкообразной смеси. Для гранулирования порошкообразной смеси могут использоваться другие жидкости, кроме воды, с добавлением или без добавления поверхностно-активного вещества и/или связующего вещества. Далее, влажные гранулы могут быть дополнительно перемолоты с помощью подходящей мельницы. Далее, воду можно по желанию удалить из смеси путем сушки компонентов подходящим способом. Затем высушенные гранулы можно дополнительно измельчить до требуемого размера. Затем можно добавить дополнительные гранулированные вспомогательные вещества путем смешивания (например, наполнитель, разбавитель и регулятор сыпучести). Затем к гранулам нужного размера можно снова добавить скользящее вещество стеарат магния и регулятор сыпучести, например, натриевую соль кросскармеллозы. В последующем композицию в форме гранул в соответствии с настоящим изобретением можно прессовать с помощью подходящих прессов для получения различных лекарственных форм в соответствии с настоящим изобретением. Дополнительно можно покрыть таблетки пленкой, красителем или другим покрытием.
В особенно желательном варианте осуществления изобретения фармацевтические композиции в соответствии с настоящим изобретением изготовляются путем непрерывного влажного гранулирования с помощью двухшнекового гранулятора. При непрерывном производстве с мониторингом и контролем на протяжении процесса получается высококачественный продукт с высоким постоянством свойств. Кроме того, непрерывное производство способствует качеству за счет совершенствования проекта с проектным полем, «богатым данными», и лучшему пониманию влияния переменных на более ранних стадиях процесса на более поздние стадии процесса и качество готового продукта. Кроме того, окончательное производство фармацевтических композиций в соответствии с настоящим изобретением можно начать быстро с помощью оборудования коммерческого масштаба, что позволяет избежать рисков, связанных с увеличением объема производства, и изменений состава не более поздних стадиях разработки. И наконец, непрерывное производство имеет преимущество при коммерческом производстве, например, лучший контроль процесса, меньшая необходимость в манипуляциях с продуктом и эффективность за счет оперативного выпуска. Общий результат - более устойчивый, контролируемый процесс с возможностью изменения масштаба с меньшим количеством подлежащих проверке параметров, благодаря чему повышается качество продукта и, следовательно, безопасность пациентов.
Например, гранулирование с высоким усилием сдвига (ГВУС), распространенная техника гранулирования, связана с хорошо известным риском чрезмерного уплотнения гранул и недостаточного контроля процесса. Увеличение масштаба такого способа очень сложно и связано со значительным риском. Переход со способа ГВУС на способ гранулирования с помощью двухшнекового гранулятора (ГДГ) позволяет увеличить масштаб способа с использованием того же оборудования, чтобы производить партии другого размера за счет увеличения длительности процесса. Это исключает риск, связанный с увеличением масштаба производства, часто свойственный процессам влажного гранулирования. Кроме того, было обнаружено, что процесс ГДГ более устойчив и менее чувствителен к чрезмерному уплотнению гранул. Как видно на Фиг. 28 для таблетки Соединения 1, при способе ГВУС показано значительное замедление растворения при повышении содержания воды, в то время как при способе ГДГ изменений при добавлении сходного количества воды не показано. Удивительно, но при использовании способа влажного гранулирования с помощью двухшнекового аппарата не было обнаружено изменений в производительности способа получения таблеток, содержащих 45-55% или 60-70% Соединения 1 в массовом соотношении. Это не относится к способу ГВУС. Кроме того, этот непрерывный и способствующий повышению качества продукта способ помогает решить распространенную проблему, на которую указывает FDA - недостаточная доступность лекарств для нуждающихся в них пациентов.
В одном варианте осуществления непрерывный способ начинается с подачи вспомогательных компонентов по отдельности и Соединения 1 во встроенный смеситель непрерывного действия через весовой питатель непрерывного действия. Из этого смесителя материал непрерывно подается на конвейер и обрабатывается путем влажного гранулирования в двухшнековом аппарате, сушки, измельчения, добавления вспомогательных веществ, не входящих в состав гранул, смешивания, прессования и нанесения пленочного покрытия.
Например, в одном варианте осуществления таблетки, содержащие Соединение 1, могут изготавливаться непрерывно в соответствии с показанной ниже схемой.
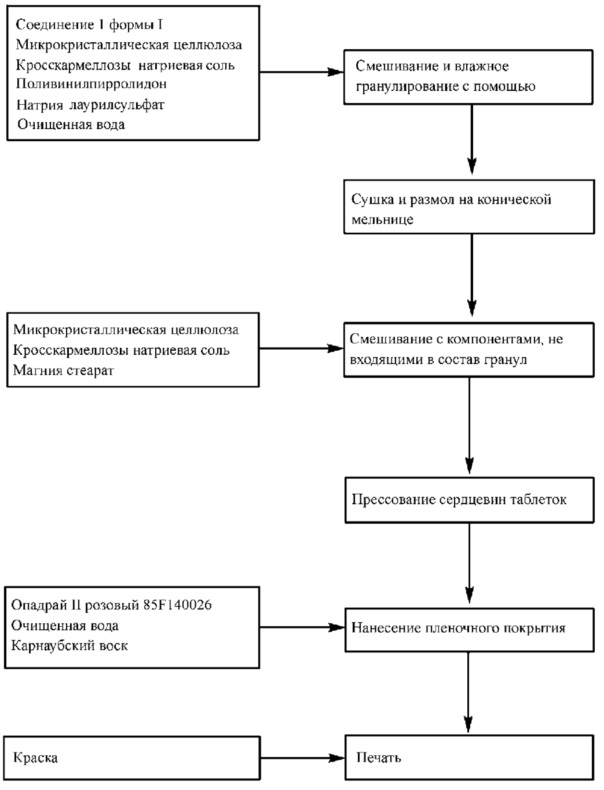
Каждый из компонентов смеси из этого примера описан выше и в примерах ниже. Кроме того, смесь может содержать дополнительные добавки, например, один или более красителей, один или более ароматизаторов или одну или более отдушек, как описано в примерах ниже. В некоторых вариантах осуществления изобретения относительные концентрации (т.е. массовые %) каждого из этих компонентов (и любых дополнительных добавок) в смеси также представлены выше и в примерах ниже. Компоненты, составляющие смесь, могут добавляться последовательно или порциями в любых сочетаниях; и компоненты или сочетания компонентов могут добавляться в любом порядке. В одном варианте осуществления скользящее вещество - последний компонент, добавляемый в смесь.
В другом варианте осуществления смесь содержит композицию Соединения 1 и одного или более вспомогательных веществ; связующее вещество, регулятор сыпучести, поверхностно-активное вещество, разбавитель, скользящее вещество, разрыхлитель и наполнитель, при этом каждый из этих компонентов добавляется в форме порошка (например, с частицами среднего диаметра, измеренного с помощью рассеяния света, 250 мкм или меньше (например, 150 мкм или меньше, 100 мкм или меньше, 50 мкм или меньше, 45 мкм или меньше, 40 мкм или меньше или 35 мкм или меньше). Например, смесь, содержащая композицию Соединения 1, разбавителя, регулятора сыпучести, поверхностно-активного вещества, скользящего вещества, разрыхлителя и наполнителя, при этом каждый из этих компонентов добавляется в форме порошка (например, с частицами среднего диаметра, измеренного с помощью рассеяния света, 250 мкм или меньше (например, 150 мкм или меньше, 100 мкм или меньше, 50 мкм или меньше, 45 мкм или меньше, 40 мкм или меньше или 35 мкм или меньше)). В другом варианте осуществления изобретения смесь содержит композицию Соединения 1, разбавителя, связующего вещества, поверхностно-активного вещества, скользящего вещества, разрыхлителя и наполнителя, при этом каждый из этих компонентов добавляется в форме порошка (например, с частицами среднего диаметра, измеренного с помощью рассеяния света, 250 мкм или меньше (например, 150 мкм или меньше, 100 мкм или меньше, 50 мкм или меньше, 45 мкм или меньше, 40 мкм или меньше или 35 мкм или меньше)).
В другом варианте осуществления смесь содержит композицию Соединения 1 и любую комбинацию следующих веществ: связующее вещество, регулятор сыпучести, разбавитель, поверхностно-активное вещество, скользящее вещество, разрыхлитель и наполнитель, при этом каждое из этих веществ по существу свободны от воды. Каждый из компонентов содержит менее 5 массовых % (например, менее 2 массовых %, менее 1 массовый %, менее 0,75 массовых %, менее 0,5 массовых %, или менее 0,25 массовых %) воды от массы компонента. Например, смесь содержит композицию Соединения 1, разбавитель, регулятор сыпучести, скользящее вещество, разрыхлитель и наполнитель, при этом каждый из этих компонентов по существу свободен от воды. В некоторых вариантах осуществления каждый из компонентов содержит менее 5 массовых % (например, менее 2 массовых %, менее 1 массового %, менее 0,75 массового %, менее 0,5 массового %, или менее 0,25 массового %) воды от массы компонента.
В другом варианте осуществления прессование смеси в таблетки осуществляется путем заполнения формы (например, формы для прессования) смесью и приложения давления. Этого можно достичь с помощью пресса или другого сходного аппарата. В некоторых вариантах осуществления смесь Соединения 1 и вспомогательных компонентов может быть сначала обработана для получения гранулированной формы. Затем гранулы измельчают до получения нужного размера и прессуют в таблетки, либо подготавливаются к инкапсуляции способами, известными в фармацевтике. Кроме того, следует отметить, что приложение давления к смеси в форме можно повторять, при этом давление может быть одинаковым при каждом этапе прессования, либо может использоваться разное давление. В другом примере смесь порошкообразных компонентов или гранул может прессоваться с помощью штамповочного пресса, создающего достаточное давление для формирования таблетки, растворяющейся на 50% или более примерно за 30 минут (например, примерно на 55% или более примерно за 30 минут, или примерно на 60% или более примерно за 30 минут). Например, смесь прессуется с помощью штамповочного пресса для получения таблеток с твердостью по меньшей мере приблизительно 5 кПа (по меньшей мере приблизительно 5,5 кПа, по меньшей мере приблизительно 6 кПа, по меньшей мере приблизительно 7 кПа, по меньшей мере приблизительно 10 кПа или по меньшей мере 15 кПа). В некоторых случаях смесь прессуется для получения таблеток с твердостью от 5 до 20 кПа.
В некоторых вариантах осуществления таблетки, содержащие фармацевтическую композицию, описанную в настоящем документе, могут быть покрыты пленкой, составляющей примерно 3,0 массовых % от массы таблетки и содержащей краситель. В определенных случаях суспензия или раствор красителя, использующиеся для покрытия таблеток, содержит приблизительно 20 массовых % твердых веществ от массы суспензии или раствора красителя. В других случаях покрытые таблетки могут быть маркированы логотипом, другим изображением или текстом.
В других вариантах осуществления способ производства фармацевтических композиций включает использование смеси твердых форм, например, смеси порошкообразных и/или жидких компонентов, смеси, содержащей Соединение 1 и один или более вспомогательных веществ, выбранных из следующих: связующее вещество, регулятор сыпучести, разбавитель, поверхностно-активное вещество, скользящее вещество, разрыхлитель и наполнитель; перемешивание этой смеси до по существу однородного состояния и прессование или компактирование для получения гранулированной формы. Затем гранулированную композицию, содержащую Соединение 1, можно прессовать в таблетки или подготавливать для производства капсул, как описано выше или в примерах ниже. Альтернативно, способы производства фармацевтической композиции включают изготовление смеси Соединения 1 и одного или более вспомогательных веществ, например связующего вещества, регулятора сыпучести, разбавителя, поверхностно-активного вещества, разрыхлителя и наполнителя; перемешивание смеси до по существу однородного состояния и прессование/компактирование смеси для получения гранулированной формы с помощью роликового пресса, при этом используется композиция для сухого гранулирования, описанная в примерах ниже или, альтернативно, прессование/компактирование в гранулы с помощью способа компактирования с высоким усилием сдвига, как описано в примерах ниже. Лекарственные формы, например таблетки, описанные в настоящем документе, могут изготавливаться из гранул, полученных путем добавления Соединения 1 в дополнение к выбранным вспомогательным веществам, описанным в настоящем документе.
В некоторых вариантах осуществления смесь перемешивается с помощью вихревого движения, смешения, встряхивания и т.п. вручную, с помощью мешалки, смесителя, любых их сочетаний и т.п. При последовательном добавлении компонентов или сочетаний компонентов перемешивание может производиться между последовательными добавлениями, непрерывно во время добавления компонентов, после добавления всех компонентов или сочетаний компонентов, или с помощью любого сочетания этих способов. Смесь перемешивается до по существу однородного состава.
В другом варианте осуществления настоящее изобретение включает размол Соединения 1, Соединения 1 формы I, Соединения 1 формы II, Соединения 1 в форме соли HCl A с помощью подходящей традиционной струйной мельницы, в которой используется давление воздуха, подходящее для получения частиц со значительной фракцией размера от 0,1 микрон до 50 микрон. В другом варианте осуществления размер частиц составляет от 0,1 микрон до 20 микрон. В другом варианте осуществления изобретения размер частиц составляет от 0,1 микрон до 10 микрон. В другом варианте осуществления размер частиц составляет от 1,0 микрон до 5 микрон. В другом варианте осуществления Соединение 1, Соединение 1 формы I, Соединение 1 формы II, Соединение 1 в форме соли HCl A имеет размер частиц D50 2,0 микрон.
В различных вариантах осуществления изобретения второй терапевтический агент может входить в состав вместе с Соединением 1, образуя единую или одиночную лекарственную форму, например, таблетку или капсулу.
Растворение лекарственных форм, изготовленных как описано выше, можно исследовать in vitro по методу 711 «Растворение» Фармакопеи США, издание 29, Фармакопейная конвенция США, Инк., Rockville, Md., 2005 (Фарм. США), чтобы определить скорость высвобождения действующего вещества из лекарственной формы. Содержание действующего вещества и примесей традиционно определяется такими методами, как высокоэффективная жидкостная хроматография (ВЭЖХ).
В некоторых вариантах осуществления изобретение включает использование упаковочных материалов, таких как контейнеры и крышки из полиэтилена высокой плотности (ПЭВП), полиэтилена низкой плотности (ПЭНП) и/или полипропилена и/или стекла, лощеной бумаги, алюминиевые пакеты и контурные ячейковые упаковки или полоски, состоящие из алюминия или поливинилхлорида (ПВХ) высокой плотности, возможно, с влагопоглотителем, полиэтиленом (ПЭ), поливинилиденхлоридом (ПВДХ), ПВХ/ПЭ/ПВДХ и т.п. Эти упаковочные материалы могут использоваться для хранения различных фармацевтических композиций и лекарственных форм с сохранением стерильности после соответствующей стерилизации упаковки и ее содержимого химическими или физическими методами, часто применяющимися в фармацевтической промышленности.
СПОСОБЫ ВВЕДЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
В одном аспекте, фармацевтические композиции настоящего изобретения могут вводиться пациенту раз в сутки или примерно раз в 24 часа. Альтернативно, фармацевтические композиции настоящего изобретения могут вводиться пациенту дважды в сутки или примерно раз в 12 ч. Эти фармацевтические композиции вводятся в виде лекарственных форм для приема внутрь, содержащих приблизительно 25 мг, 50 мг, 100 мг, 125 мг, 150 мг, 200 мг, 250 мг или 400 мг Соединения 1. В этом аспекте, в дополнение к Соединению 1 фармацевтические композиции содержат наполнитель; разбавитель; разрыхлитель; поверхностно-активное вещество; по меньшей мере одно из следующего: связующее вещество и регулятор сыпучести; и скользящее вещество. Например, доза 400 мг Соединения 1 может содержать 200 мг Соединения 1, или 4 таблетки в соответствии с изобретением, каждая из которых содержит 100 мг Соединения 1.
Также следует понимать, что соединение и фармацевтически приемлемые композиции и формы в соответствии с настоящим изобретением могут использоваться для комбинированной терапии; то есть, Соединение 1 и его фармацевтически приемлемые композиции могут вводиться одновременно, до или после введения одного или более желаемых лекарств или медицинских процедур. При конкретном сочетании терапий (препаратов или процедур) для использования при комбинированном режиме должна учитываться совместимость желаемых терапевтических препаратов и/или процедур и желаемый терапевтический эффект. Кроме того, следует понимать, что используемые терапии могут давать желаемый эффект при одном нарушении (например, соединение в соответствии с изобретением может вводиться одновременно с другим препаратом, использующимся для лечения этого же заболевания), либо они могут оказывать разные эффекты (например, контроль каких-либо нежелательных явлений). В настоящем документе дополнительные терапевтические агенты, обычно применяющиеся для лечения или профилактики конкретного заболевания, например, CFTR-опосредованного заболевания или состояния, известны как «подходящие для лечения заболевания или состояния».
В одном варианте осуществления дополнительный терапевтический агент выбирается из муколитического агента, бронхорасширяющего средства, антибиотика, противоинфекционного агента или противовоспалительного агента, модулятора CFTR, иного, чем Соединение 1 в соответствии с настоящим изобретением, или питательного вещества.
В одном варианте осуществления дополнительный агент представляет собой (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид. В другом варианте осуществления изобретения дополнительный агент представляет собой N-(5-гидрокси-2,4-дитрет-бутилфенил)-4-оксо-1H-хинолон-3-карбоксамид. В другом варианте осуществления дополнительный агент выбирается из Таблицы 1:
В других вариантах осуществления дополнительный агент представляет собой любое сочетание вышеперечисленных агентов. Например, композиция может содержать Соединение 1, (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид и N-(5-гидрокси-2,4-дитрет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид. В другом варианте осуществления композиция может содержать Соединение 1, N-(5-гидрокси-2,4-дитрет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид, а также любое из соединений из Таблицы 1, т.е. соединения 1-14 из Таблицы 1, или любое их сочетание.
В одном варианте осуществления изобретения дополнительным терапевтическим агентом является антибиотик. Примеры подходящих антибиотиков включают тобрамицин, в том числе торбамицин в форме порошка для ингаляций, азитромицин, азтреонам, включая аэрозольную форму азтреонама, амикацин, включая липосомные формы, ципрофлоксацин, включая формы, подходящие для ингаляционного введения, левофлаксацин, включая аэрозольные формы, а также сочетания двух антибиотиков, например, фосфомицина и тобрамицина.
В другом варианте осуществления изобретения дополнительный агент является муколитическим средством. К примерам подходящих муколитических средств относится Пульмозим®.
В другом варианте осуществления изобретения дополнительный агент является бронхорасширяющим средством. Примеры бронхорасширяющих средств включают альбутерол, метапротенерола сульфат, пирбутерола ацетат, сальметерол или тетрабулина сульфат.
В другом варианте осуществления изобретения дополнительный агент эффективно восстанавливает жидкость, покрывающую поверхность дыхательных путей. Такие агенты улучшают транспорт солей в клетки и из клеток, способствуя увлажнению слизи в дыхательных путях легких и, следовательно, облегчая ее отделение. Примеры таких агентов включают гипертонический раствор хлорида натрия, денуфосол в форме тетранатриевой соли ([[(3S,5R)-5-(4-амино-2-оксопиримидин-1-ил)-3-гидроксиоксолан-2-ил]метокси-гидроксифосфорил][[[(2R,3S,4R,5R)-5-(2,4-диоксопиримидин-1-ил)-3,4-дигидроксиоксолан-2-ил]метокси-гидроксифосфорил]окси-гидроксифосфорил]гидрофосфат) или бронхитол (ингаляционная форма маннита).
В другом варианте осуществления дополнительный агент представляет собой противовоспалительный агент, т.е. агент, способный уменьшать воспаление в легких. Примеры подходящих агентов включают ибупрофен, докозагексановую кислоту (ДГК), силденафил, ингаляционный глутатион, пиоглитазон, гидроксихлорохин или симвастатин.
В другом варианте осуществления дополнительный агент представляет собой модулятор CFTR, кроме Соединения 1, т.е. агент, изменяющий активность CFTR. Примеры таких агентов включают аталурен («PTC124®»; 3-[5-(2-фторфенил)-1,2,4-оксадиазол-3-ил]бензойную кислоту), синапултид, ланковутид, депелестат (человеческий рекомбинантный ингибитор эластазы нейтрофилов) и кабипростон (7-{(2R, 4aR, 5R, 7aR)-2-[(3S)-1,1-дифтор-3-метилпентил]-2-гидрокси-6-оксооктагидроциклопента[b]пиран-5-ил}гептановую кислоту).
В другом варианте осуществления изобретения дополнительный агент представляет собой агент для улучшения питания. Примеры агентов для улучшения питания включают панкреатическую липазу (заместительная терапия ферментом поджелудочной железы), в том числе Pancrease®, Pancreacarb®, Ultrase® или Creon®, Liprotomase® (ранее Trizytek®), Aquadeks® или ингаляционный глутатион. В одном варианте осуществления дополнительный агент для улучшения питания представляет собой панкреатическую липазу.
В другом варианте осуществления дополнительный агент представляет собой соединение, выбираемое из гентамицина, куркумина, циклофосфамида, 4-фенилбутирата, миглустата, фелодипина, нимодипина, филоксина В, генистеина, апигенина, модуляторов цАМФ/цГМФ, например ролипрама, силденафила, милринона, тадалафила, амринона, изопротеренола, альбутерола и алметерола, дезоксиспергуалина, ингибиторов HSP 90, ингибиторов HSP 70, ингибиторов протеосома, например эпоксомицина, лактацистина и т.п.
В другом варианте осуществления изобретения дополнительный агент представляет собой соединение, выбранное из следующих: 3-амино-6-(4-фтор-фенил)-5-трифторметилпиридин-2-карбоновая кислота (3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 5-амино-6′-метил-3-трифторметил-[2,3]бипиридинил-6-карбоновая кислота (3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-циклопропил-N-(3,3,3-трифтор-2-гидрокси-2-метилпропил)-5-(трифторметил)пиколинамид; 3-амино-6-метокси-N-(3,3,3-трифтор-2-гидрокси-2-(трифторметил)пропил)-5-(трифторметил)пиколинамид; 3-амино-6-(4-фтор-фенил)-5-трифторметил-пиридин-2-карбоновая кислота ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-метокси-5-трифторметилпиридин-2-карбоновая кислота ((S-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-метокси-5-трифторметил-пиридин-2-карбоновая кислота ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-(2,4-дихлорфенил)-5-трифторметил-пиридин-2-карбоновая кислота ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-(2,4-дихлорфенил)-5-трифторметилпиридин-2-карбоновая кислота ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-(4-фторфенил)-5-трифторметилпиридин-2-карбоновая кислота (2-гидрокси-2-метилпропил)-амид; 3-амино-5,6-бис-трифторметилпиридин-2-карбоновая кислота ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-5,6-бис-трифторметил-пиридин-2-карбоновая кислота ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; (S)-3-амино-6-этокси-N-(3,3,3-трифтор-2-гидрокси-2-метилпропил)-5-(трифторметил)пиколинамид; 3-амино-6-метокси-5-трифторметилпиридин-2-карбоновая кислота ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-метокси-5-трифторметилпиридин-2-карбоновая кислота ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-6-(4-фторфенил)-5-трифторметилпиридин-2-карбоновая кислота (3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-5,6-бис-трифторметилпиридин-2-карбоновая кислота ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид; 3-амино-5,6-бис-трифторметил-пиридин-2-карбоновая кислота ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амид, или их фармацевтически приемлемые соли. В другом варианте осуществления изобретения дополнительный агент представляет собой соединение, раскрывающееся в патенте США № 8247436 и международной публикации PCT WO 2011113894, каждый из которых полностью включен в настоящую заявку посредством ссылок.
В одном варианте осуществления дополнительный агент представляет собой триметилангелицин. В другом варианте осуществления изобретения дополнительный агент представляет собой соединение, раскрывающееся в патенте WO 2012171954, который включен в настоящий документ посредством ссылки.
В других вариантах осуществления дополнительный агент представляет собой соединение, раскрывающееся в WO 2004028480, WO 2004110352, WO 2005094374, WO 2005120497, или WO 2006101740. В другом варианте осуществления дополнительный агент представляет собой производное бензо[с]хинолизина, обладающее CFTR-модулирующей активностью, или производное бензопирана, обладающее CFTR-модулирующей активностью.
В других вариантах осуществления дополнительный агент представляет собой соединение, раскрывающееся в патентах США №№ 7202262, патентах США №№ 6992096, US 20060148864, US 20060148863, US 20060035943, US 20050164973, WO 2006110483, WO 2006044456, WO 2006044682, WO 2006044505, WO 2006044503, WO 2006044502 или WO 2004091502. В другом варианте осуществления дополнительный агент представляет собой соединение, раскрывающееся в WO 2004080972, WO 2004111014, WO 2005035514, WO 2005049018, WO 2006099256, WO 2006127588 или WO 2007044560. В другом варианте осуществления изобретения дополнительный агент представляет собой N-(5-гидрокси-2,4-ди-трет-бутилфенилфенил)-4-оксо-1H-хинолин-3-карбоксамид.
В одном варианте осуществления изобретения 600 мг Соединения 1 может вводиться нуждающемуся в этом пациенту после одновременного введения 250 мг N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамида (Соединения 2). В этих вариантах осуществления необходимые дозы могут быть получены приемом одной или более таблеток в соответствии с настоящим изобретением. Например, введение 600 мг Соединения 1 может быть достигнуто приемом трех таблеток, каждая из которых содержит 200 мг Соединения 1, четырех таблеток, каждая из которых содержит 150 мг Соединения 1, или одной таблетки, содержащей 400 мг Соединения 1, и одной таблетки, содержащей 200 мг Соединения 1. Соединение 2 может вводиться в форме фармацевтической композиции, содержащей Соединение 2 и фармацевтически приемлемый носитель. Введение может продолжаться до облегчения симптомов заболевания, либо в течение периода, рекомендованного лечащим врачом пациента, например, длительность введения может быть менее 1 недели, 1 неделя, 2 недели, 3 недели, месяц или больше. Периоду одновременного введения может предшествовать период введения одного Соединения 1. Например, возможно введение 600 мг Соединения 1 в течение 2 недель, а затем одновременное введение 250 мг Соединения 2 в течение еще одной недели. В другом варианте осуществления Соединение 1 в дозе 600 мг может вводиться дважды в сутки в течение 28 дней, а затем может вводиться Соединение 2 в дозе 250 мг дважды в сутки на протяжении 28 дней. В другом варианте осуществления Соединение 1 в дозе 600 мг может вводиться раз в сутки в течение 28 дней, а затем может вводиться Соединение 2 в дозе 250 мг раз в сутки на протяжении 28 дней. В другом варианте осуществления Соединение 1 в дозе 600 мг может вводиться раз в сутки на протяжении 28 дней, с последующим совместным введением Соединения 1 в дозе 600 мг раз в сутки и Соединения 2 в дозе 250 мг раз в 12 ч на протяжении 28 дней. В другом варианте осуществления Соединение 1 может вводиться в дозе 600 мг раз в сутки, и Соединение 2 может вводиться в дозе 250 мг раз в сутки.
В одном варианте осуществления изобретения Соединение 1 может вводиться нуждающемуся в этом пациенту в дозе 600 мг, а в последующем к нему может быть добавлено 450 мг N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамида (Соединения 2). В этих вариантах осуществления необходимые дозы могут быть получены приемом одной или более таблеток в соответствии с настоящим изобретением. Например, введение 600 мг Соединения 1 может быть достигнуто приемом трех таблеток, каждая из которых содержит 200 мг Соединения 1, или четырех таблеток, каждая из которых содержит 150 мг Соединения 1. Соединение 2 может вводиться в форме фармацевтической композиции, содержащей Соединение 2 и фармацевтически приемлемый носитель. Введение может продолжаться до облегчения симптомов заболевания, либо в течение периода, рекомендованного лечащим врачом пациента, например, длительность введения может быть менее 1 недели, 1 неделя, 2 недели, 3 недели, месяц или больше. Периоду одновременного введения может предшествовать период введения одного Соединения 1. Например, возможно введение 600 мг Соединения 1 в течение 2 недель, а затем добавление 450 мг Соединения 2 в течение еще одной недели. В другом варианте осуществления Соединение 1 в дозе 600 мг может вводиться дважды в сутки в течение 28 дней, а затем может вводиться Соединение 2 в дозе 450 мг дважды в сутки на протяжении 28 дней.
В одном варианте осуществления изобретения нуждающемуся в этом пациенту может вводиться Соединение 1 в дозе 400 мг, а затем к нему может быть добавлен N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид (Соединение 2) в дозе 350 мг. В этих вариантах осуществления необходимые дозы могут быть получены приемом одной или более таблеток в соответствии с настоящим изобретением. Например, введение 400 мг Соединения 1 может быть достигнуто приемом двух таблеток, каждая из которых содержит 200 мг Соединения 1, или четырех таблеток, каждая из которых содержит 100 мг Соединения 1. Соединение 2 может вводиться в форме фармацевтической композиции, содержащей Соединение 2 и фармацевтически приемлемый носитель. Введение может продолжаться до облегчения симптомов заболевания, либо в течение периода, рекомендованного лечащим врачом пациента, например, длительность введения может быть менее 1 недели, 1 неделя, 2 недели, 3 недели, месяц или больше. Периоду одновременного введения может предшествовать период введения одного Соединения 1. Например, возможно введение 400 мг Соединения 1 в течение 2 недель, а затем одновременное введение 350 мг Соединения 2 в течение еще одной недели. В другом варианте осуществления Соединение 1 в дозе 400 мг может вводиться раз в 8 ч на протяжении 28 дней, а затем может вводиться Соединение 2 в дозе 350 раз в 8 ч на протяжении 28 дней.
В одном варианте осуществления изобретения нуждающемуся в этом пациенту может вводиться Соединение 1 в дозе 400 мг, а затем к нему может быть добавлен N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид (Соединение 2) в дозе 250 мг. В этих вариантах осуществления необходимые дозы могут быть получены приемом одной или более таблеток в соответствии с настоящим изобретением. Например, введение 400 мг Соединения 1 может быть достигнуто приемом двух таблеток, каждая из которых содержит 200 мг Соединения 1, или четырех таблеток, каждая из которых содержит 100 мг Соединения 1. Соединение 2 может вводиться в форме фармацевтической композиции, содержащей Соединение 2 и фармацевтически приемлемый носитель. Введение может продолжаться до облегчения симптомов заболевания, либо в течение периода, рекомендованного лечащим врачом пациента, например, длительность введения может быть менее 1 недели, 1 неделя, 2 недели, 3 недели, месяц или больше. Периоду одновременного введения может предшествовать период введения одного Соединения 1. Например, возможно введение 400 мг Соединения 1 в течение 2 недель, а затем добавление 150 мг или 250 мг Соединения 2 в течение еще одной недели. В другом варианте осуществления Соединение 1 в дозе 400 мг может вводиться дважды в сутки в течение 28 дней, после чего может вводиться Соединение 2 в дозе 250 мг дважды в сутки на протяжении 28 дней. В другом варианте осуществления Соединение 1 в дозе 400 мг может вводиться дважды в сутки в течение 28 дней, после чего может вводиться Соединение 2 в дозе 250 мг раз в сутки на протяжении 28 дней. В другом варианте осуществления Соединение 1 в дозе 400 мг может вводиться раз в сутки на протяжении 28 дней, а затем Соединение 1 в дозе 400 мг раз в сутки может вводиться совместно с Соединением 2 в дозе 250 мг раз в 12 ч на протяжении 28 дней. В другом варианте осуществления Соединение 1 может вводиться в дозе 400 мг дважды в сутки в сочетании с Соединением 2 в дозе 250 мг раз в сутки.
В одном варианте осуществления изобретения нуждающемуся в этом пациенту может вводиться Соединение 1 в дозе 400 мг раз в сутки с последующим добавлением 150 мг Соединения 2 раз в сутки. В этих вариантах осуществления необходимые дозы могут быть получены приемом одной или более таблеток в соответствии с настоящим изобретением. Например, введение 400 мг Соединения 1 может быть достигнуто приемом двух таблеток, каждая из которых содержит 200 мг Соединения 1, или четырех таблеток, каждая из которых содержит 100 мг Соединения 1. Соединение 2 может вводиться в форме фармацевтической композиции, содержащей Соединение 2 и фармацевтически приемлемый носитель. Введение может продолжаться до облегчения симптомов заболевания, либо в течение периода, рекомендованного лечащим врачом пациента, например, длительность введения может быть менее 1 недели, 1 неделя, 2 недели, 3 недели, месяц или больше. Периоду одновременного введения может предшествовать период введения одного Соединения 1. Например, возможно введение 400 мг Соединения 1 в течение 2 недель, а затем одновременное введение 150 мг или 250 мг Соединения 2 в течение еще одной недели.
В одном варианте осуществления изобретения нуждающемуся в этом пациенту может вводиться Соединение 1 в дозе 400 мг раз в сутки с последующим добавлением 150 мг Соединения 2 раз в 12 ч. В другом варианте осуществления изобретения нуждающемуся в этом пациенту может вводиться Соединение 1 в дозе 400 мг раз в сутки с последующим добавлением 250 мг Соединения 2 раз в 12 ч. В этих вариантах осуществления необходимые дозы могут быть получены приемом одной или более таблеток в соответствии с настоящим изобретением. Например, введение 400 мг Соединения 1 может быть достигнуто приемом двух таблеток, каждая из которых содержит 200 мг Соединения 1, или четырех таблеток, каждая из которых содержит 100 мг Соединения 1. Соединение 2 может вводиться в форме фармацевтической композиции, содержащей Соединение 2 и фармацевтически приемлемый носитель. Введение может продолжаться до облегчения симптомов заболевания, либо в течение периода, рекомендованного лечащим врачом пациента, например, длительность введения может быть менее 1 недели, 1 неделя, 2 недели, 3 недели, месяц или больше. Периоду одновременного введения может предшествовать период введения одного Соединения 1. Например, возможно введение 400 мг Соединения 1 в течение 2 недель, а затем одновременное введение 150 мг или 250 мг Соединения 2 в течение еще одной недели.
В другом варианте осуществления Соединение 1 в дозе 200 мг может вводиться раз в сутки на протяжении 28 дней, с последующим совместным введением Соединения 1 в дозе 200 мг раз в сутки и Соединения 2 в дозе 250 мг раз в 12 ч на протяжении 28 дней.
В одном варианте осуществления изобретения таблетки Соединения 1 100 мг, 200 мг и 300 мг могут комбинироваться для получения ряда разных доз. Например, дозы Соединения 1 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 700 мг, 800 мг, 900 мг, 1000 мг, 1100 мг или 1200 мг могут вводиться с использованием таблеток 100 мг, 200 мг и 300 мг и их сочетаний. Например, доза Соединения 1 900 мг может быть введена в форме трех таблеток Соединения 1 300 мг. Доза Соединения 1 600 мг может быть введена в форме трех таблеток 200 мг Соединения 1 или двух таблеток 300 мг Соединения 1. Любые из указанных ранее в этом разделе доз могут быть введены с дозами Соединения 2 и/или по графику приема, описанных в трех предшествующих абзацах.
Эти комбинации применяются для лечения описанных здесь заболеваний, в том числе муковисцидоза. Эти комбинации также используются в наборах, описанных в настоящем документе.
Количество дополнительного терапевтического агента, присутствующего в композициях в соответствии с настоящим изобретением, должно быть не больше, чем было бы введено в обычной ситуации в составе композиции, содержащей терапевтический агент в качестве единственного действующего вещества. Предпочтительно, когда количество дополнительного терапевтического агента в композициях в соответствии с настоящим изобретением варьирует от приблизительно 50% до 100% от количества, обычно присутствующего в композициях, содержащих этот агент в качестве единственного действующего вещества.
В другом аспекте, изобретение относится к набору, содержащему таблетку в соответствии с настоящим изобретением и отдельный терапевтический агент или содержащую его фармацевтическую композицию. В другом варианте осуществления Соединение 1 в виде таблетки представляет собой Форму I. В другом варианте осуществления терапевтический агент представляет собой препарат для лечения муковисцидоза, не относящийся к Соединению 1. В другом варианте осуществления терапевтический агент представляет собой потенциатор для лечения муковисцидоза. В другом варианте изобретения терапевтический агент представляет собой N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид. В другом варианте осуществления изобретения таблетка и терапевтический агент находятся в отдельных контейнерах. В другом варианте осуществления отдельные контейнеры представляют собой бутылки. В другом варианте осуществления отдельные контейнеры представляют собой флаконы. В другом варианте осуществления отдельные контейнеры представляют собой контурные ячейковые упаковки.
ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ КОМПОЗИЦИИ
В одном аспекте, изобретение также относится к способу лечения, уменьшения тяжести или симптоматического лечения заболевания у пациента, при этом способ включает введение пациенту эффективного количества фармацевтической композиции в соответствии с настоящим изобретением, при этом заболевание выбирается из следующих: муковисцидоз, астма, индуцированное дымом ХОЗЛ, хронический бронхит, риносинусит, запор, панкреатит, недостаточность поджелудочной железы, мужское бесплодие, вызванное врожденным двухсторонним отсутствием семявыносящего протока, легкое заболевание легких, идиопатический панкреатит, аллергический бронхолегочный аспергиллез (АБЛА), заболевание печени, наследственная эмфизема, наследственный гемохроматоз, недостаточность свертывания-фибринолиза, например, недостаточность белка C, наследственный ангионевротический отек 1 типа, недостаточность обмена жиров, например, наследственная гиперхолестеринемия, хиломикронемия 1 типа, абеталипопротеинемия, лизосомные болезни накопления, например, болезнь клеточных включений/псевдо-синдром Гурлер, мукополисахаридозы, болезнь Сандхофа/Тея-Сакса, синдром Криглера-Найяра II типа, полиэндокринопатия/гиперинсулинемия, сахарный диабет, карликовость Ларона, недостаточность миелопероксидазы, первичный гипопаратиреоидизм, меланома, гликаноз CDG 1 типа, врожденный гиперпаратиреоидизм, несовершенный остеогенез, наследственная гипофибриногенемия, недостаточность АКТГ, несахарный диабет (НД), нейрофизиологический НД, нефрогенный НД, синдром Шарко-Мари-Тута, болезнь Пелицеуса-Мерцбахера, нейродегенеративные заболевания, например, болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий надъядерный паралич, болезнь Пика, несколько полиглутаминовых неврологических расстройств, например, болезнь Хантингтона, спиномозжечковая атаксия I типа, атрофия мышц спины и глазного яблока, дентато-рубро-паллидо-льюисова атрофия и миотоническая дистрофия, а также губкообразные энцефалопатии, например, наследственная болезнь Кройтцфельда-Якоба (за счет дефекта обработки прионного белка), болезнь Фабри, синдром Штреусслера-Шейнкера, ХОЗЛ, синдром сухого глаза или болезнь Шергена, остеопороз, остеопения, заживление и рост костей (включая восстановление кости, регенерацию кости, снижение резорбции кости и усиление отложения костной ткани), синдром Горэма, нарушения хлоридных каналов, например, врожденная миотония (формы Томсона и Бекера), синдром Барттера III типа, болезнь Дента, гиперэкплексия, эпилепсия, лизосомная болезнь накопления, синдром Эйнджелмена и первичная цилиарная дискинезия (ПЦД), термин для обозначения наследственных нарушений структуры и/или функции ресничек, включая ПЦД с зеркальным расположением органов (также известным как синдром Картагенера), ПЦД без зеркального расположения органов и аплазия ресничек.
Соединение 1, как часть комбинации с ивакафтором (N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид), признан Управлением по надзору за продуктами питания и лекарствами США (FDA) средством терапии прорыва муковисцидоза, это один из двух подобных препаратов на момент подачи этой заявки (второй - ивакафтор). Это показывает значительную неудовлетворенную потребность в эффективном лечении, направленном на причину муковисцидоза, в отличие от симптоматического лечения. Кроме того, распространенной проблемой является то, что препараты, одобренные FDA, не всегда доступны нуждающимся в них пациентам. Таким образом, существует значительная неудовлетворенная потребность в лекарственных формах, содержащих описанное ранее Соединение 1, и способах их получения непрерывным и контролируемым способом.
В одном аспекте изобретение также относится к способу лечения, уменьшения тяжести или симптоматического лечения заболевания у пациента, включающему введение пациенту эффективного количества фармацевтической композиции в соответствии с настоящим изобретением, при этом заболевание выбирается из следующих: генерализованная эпилепсия с фебрильными судорогами плюс(GEFS+), генерализованная эпилепсия с фебрильными и афебрильными судорогами, миотония, парамиотония, врожденная парамиотония, миотония, усиливающаяся под действием калия, гиперкалиемический периодический паралич, синдром удлиненного интервала QT, синдром удлиненного интервала QT/синдром Бругады, аутосомно-доминантный синдром удлиненного интервала QT с глухотой, аутосомно-рецессивный синдром удлиненного интервала QT с характеристиками дизморфии, врожденный или приобретенный синдром удлиненного интервала QT, синдром Тимоти, персистирующая гиперинсулинемическая гипогликемия новорожденных, дилятационная кардиомиопатия, аутосомно-доминантный синдром удлиненного интервала QT, болезнь Дента, остеопетроз, синдром Барттера III типа, болезнь центральных волокон, злокачественная гипертермия и катехоламинергическая полиморфная тахикардия.
В одном аспекте настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения муковисцидоза, включающему введение пациенту эффективного количества фармацевтической композиции в соответствии с настоящим изобретением, при этом у пациента имеется мутация CFTR N1303K, ΔI507 или R560T.
В одном аспекте настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения муковисцидоза, включающему введение пациенту эффективного количества фармацевтической композиции в соответствии с настоящим изобретением, при этом у пациента имеется мутация CFTR или G551D. В другом варианте осуществления пациент гомозиготен по G551D. В другом варианте осуществления изобретения пациент гетерозиготен по G551D, при этом другая мутация CFTR является одной из следующих: F508del, G542X, N1303K, W1282X, R117H, R553X, 1717-1G->A, 621+1G->T, 2789+5G->A, 3849+10kbC->T, R1162X, G85E, 3120+1G->A, ΔI507, 1898+1G->A, 3659delC, R347P, R560T, R334W, A455E, 2184delA или 711+1G->T.
В одном аспекте настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения муковисцидоза, включающему введение пациенту эффективного количества фармацевтической композиции в соответствии с настоящим изобретением, при этом у пациента имеется мутация CFTR F508del. В другом варианте осуществления изобретения пациент гомозиготен по F508del. В другом варианте осуществления изобретения пациент гетерозиготен по F508del, при этом другая мутация CFTR является одной из следующих: G551D, G542X, N1303K, W1282X, R117H, R553X, 1717-1G->A, 621+1G->T, 2789+5G->A, 3849+10kbC->T, R1162X, G85E, 3120+1G->A, ΔI507, 1898+1G->A, 3659delC, R347P, R560T, R334W, A455E, 2184delA или 711+1G->T.
В определенных вариантах осуществления изобретения фармацевтически приемлемые композиции в соответствии с настоящим изобретением, содержащие Соединение 1, эффективны для лечения, ослабления тяжести или симптоматического лечения муковисцидоза у пациентов с остаточной активностью CFTR в апикальной мембране дыхательного и не дыхательного эпителия. Присутствие остаточной активности CFTR на поверхности эпителия можно легко обнаружить известными методами, например, стандартными электрофизиологическими, биохимическими или гистохимическими методами. Такие методы определяют активность CFTR с помощью электрофизиологических техник in vivo или ex vivo, измерения концентрации Cl- в поте или слюне, либо биохимическими или гистохимическими методами ex vivo, позволяющими определить плотность поверхности клеток. С помощью таких методов можно легко определить остаточную активность CFTR у пациентов, гетерозиготных или гомозиготных по ряду мутаций, в том числе у пациентов, гомозиготных или гетерозиготных по большинству распространенных мутаций, F508del, а также других мутаций, например, мутации G551D или мутации R117H. В определенных вариантах осуществления фармацевтические композиции, содержащие Соединение 1, эффективны для лечения, уменьшения тяжести или симптоматического лечения муковисцидоза у пациентов с незначительной или отсутствующей остаточной активностью CFTR. В определенных вариантах осуществления изобретения фармацевтические композиции, содержащие Соединение 1, эффективны для лечения, ослабления тяжести или симптоматического лечения муковисцидоза у пациентов со слабой или отсутствующей остаточной активностью CFTR в апикальной мембране дыхательного эпителия.
В другом варианте осуществления изобретения соединения и композиции в соответствии с настоящим изобретением эффективны для лечения или ослабления тяжести муковисцидоза у пациентов, у которых остаточная активность CFTR индуцирована или усилена. Такая индукция или усиление активности CFTR может быть достигнуто фармакологическими методами. В другом варианте осуществления изобретения соединения и композиции в соответствии с настоящим изобретением эффективны для лечения или ослабления тяжести муковисцидоза у пациентов, у которых остаточная активность CFTR индуцирована или усилена с помощью генной терапии. Такие способы увеличивают число CFTR, присутствующих на клеточной поверхности, тем самым индуцируя прежде отсутствующую активность CFTR у пациента или усиливая существующую степень остаточной активности CFTR у пациента.
В одном варианте осуществления изобретения фармацевтическая композиция в соответствии с настоящим изобретением, содержащая Соединение 1, как описано в настоящем документе, эффективны для лечения или ослабления тяжести муковисцидоза у пациентов с определенными генотипами, имеющих остаточную активность CFTR, т.е. мутациями I класса (отсутствие синтеза), мутациями II класса (неправильное сворачивание), мутациями III класса (нарушение регуляции или воротного механизма каналов), мутациями IV класса (изменение проводимости) или мутациями V класса (снижение синтеза).
В одном варианте осуществления фармацевтические композиции в соответствии с настоящим изобретением, содержащие Соединение 1, как описано в настоящем документе, эффективны для лечения или ослабления тяжести муковисцидоза у пациентов с определенными клиническими фенотипами, например, умеренным или легким клиническим фенотипом, который обычно коррелирует со степенью остаточной активности CFTR в апикальной мембране эпителия. Такие фенотипы включают пациентов с недостаточностью поджелудочной железы.
В одном варианте настоящего изобретения фармацевтические композиции в соответствии с настоящим изобретением, содержащие Соединение 1, как описано в настоящем документе, эффективны для лечения, ослабления тяжести или симптоматического лечения недостаточности поджелудочной железы, идиопатического панкреатита и врожденного двухстороннего отсутствия семявыносящего протока, или легкого легочного заболевания у пациентов с остаточной активностью CFTR.
В одном варианте настоящего изобретения фармацевтические композиции в соответствии с настоящим изобретением, содержащие Соединение 1, как описано в настоящем документе, эффективны для лечения, ослабления тяжести или симптоматического лечения недостаточности поджелудочной железы, идиопатического панкреатита и врожденного двухстороннего отсутствия семявыносящего протока, или легкого легочного заболевания у пациентов с исходным типом CFTR.
Кроме муковисцидоза, модулирование активности CFTR может быть полезно при других заболеваниях, не вызванных непосредственно мутациями в CFTR, например, секреторных заболеваниях и других заболеваниях, связанных с нарушением сворачивания белков и опосредованными CFTR. Они включают, без ограничений, хроническое обструктивное заболевание легких (ХОЗЛ), синдром сухого глаза и синдром Шергена. ХОЗЛ характеризуется ограничением воздушного потока, который прогрессирует и не обратим полностью. Ограничение воздушного потока обусловлено гиперсекрецией слизи, эмфиземой и бронхиолитом. Активация мутантных или немутантных CFTR является потенциальным способом лечения гиперсекреции слизи и нарушения мукоцилиарного клиренса, распространенных при ХОЗЛ. В частности, усиление секреции анионов через CFTR может способствовать транспорту жидкости в жидкость, покрывающую поверхность дыхательных путей, для увлажнения слизи и оптимизации вязкости жидкости вокруг ресничек. Это может улучшить мукоцилиарный клиренс и снизить симптомы, связанные с ХОЗЛ. Синдром сухого глаза характеризуется уменьшением образования слезной жидкости и аномальным составом липидов, белков и муцина в слезной пленке. Существует много причин сухости глаз, некоторые из которых - возраст, лазерная операция на глазах, артрит, лекарственные препараты, химические/термические ожоги, аллергии и заболевания, такие как муковисцидоз и синдром Шергена. Усиление секреции анионов через CFTR должно ускорить транспорт жидкости из эндотелиальных клеток роговицы и секреторных клеток желез, окружающих глаз, и повысить влажность роговицы. Это должно способствовать облегчению симптомов, связанных с синдромом сухого глаза. Синдром Шергена - аутоиммунное заболевание, при котором иммунная система атакует железы, выделяющие жидкость, во всем организме, включая глаза, ротовую полость, кожу, ткань дыхательных органов, печень, влагалище и кишечник. Симптомы включают сухость глаз, рта и влагалища, а также заболевания легких. Заболевание также связано с ревматоидным артритом, системной волчанкой, системным склерозом и полимиозитом/дерматомиозитом. Полагают, что причиной заболевания является нарушение транспорта белков, возможности лечения при котором ограничены. Вещества, усиливающие или индуцирующие активность CFTR, могут способствовать увлажнению различных органов, пораженных болезнью, и облегчать связанные с ней симптомы.
В одном варианте осуществления изобретение относится к способу усиления или индукции активности анионных каналов in vitro или in vivo, включающему контроль функции канала с помощью фармацевтической композиции в соответствии с настоящим изобретением. В другом варианте осуществления изобретения анионный канал является хлоридным каналом или бикарбонатным каналом. В другом варианте осуществления изобретения анионный канал является хлоридным каналом.
Точное количество соединения, необходимое для лечения, будет разным для разных субъектов, в зависимости от вида, возраста, общего состояния субъекта, тяжести инфекции, конкретного агента, способа его введения и т.п. Соединения по настоящему изобретению предпочтительно изготавливают в виде стандартных лекарственных форм для упрощения введения и обеспечения однородности дозирования. Выражение «стандартная лекарственная форма», при использовании в настоящем документе, означает физически отдельную единицу средства, подходящую пациенту, проходящему лечение. Тем не менее, следует понимать, что решение об общей суточной дозе соединений и композиций по настоящему изобретению должен принимать наблюдающий врач по результатам обоснованной клинической оценки. Точный уровень эффективной дозы для каждого конкретного пациента или организма зависит от ряда факторов, включая вид расстройства, в связи с которым проводится лечение, и степень его тяжести; активности конкретного используемого соединения; конкретной используемой композиции; возраст, массу тела, общее состояние здоровья, пол и режим питания пациента; время введения, способ введения и скорость выведения определенного применяемого соединения; продолжительность лечения; лекарственные препараты, применяемые в сочетании или одновременно со специальным применяемым соединением, а также другие подобные факторы, хорошо известные специалистам в области медицины. Используемый в настоящем документе термин «пациент» означает животное, предпочтительно млекопитающее, наиболее предпочтительно человека.
Если где-либо в настоящей заявке название соединения не отражает правильно структуры соединения, структура имеет приоритет над названием.
Описание примеров
ПРД (порошковая рентгеновская дифрактометрия)
Данные о рентгеновской дифракции (РД) Соединения 1, Соединения 1 формы I, Соединения 1 формы II или Соединения 1 в форме соли HCl A получали с помощью дифрактометра порошка Bruker D8 DISCOVER с 2-мерным детектором HI-STAR 2 и плоским графитовым монохроматором. При этом использовалась запаянная Cu трубка с Kα излучением при 40 кВ, 35 мA. Образцы помещали на силиконовые подложки с нулевым фоновым сигналом при 25°C. При анализе каждого образца получали 2 набора данных за 120 секунд каждый при двух разных углах θ2: 8° и 26°. Данные интегрировали с помощью программы GADDS и объединяли с помощью программы DIFFRACTplusEVA. Неопределенность указанных положений пиков составляет ±0,2 градуса.
Описание размола на струйной мельнице
Неизмельченное Соединение 1, Соединение 1 формы I, Соединение 1 формы II или Соединение 1 в форме соли HCl А просеивали для разрушения комков перед загрузкой в струйную мельницу. Все сита были одноразовыми и протирались перед использованием. Неизмельченное Соединение 1, Соединение 1 формы I, Соединение 1 формы II или Соединение 1 в форме соли HCl А добавляется в загрузочную воронку струйной мельницы с контролируемой скоростью подачи, при этом используется сжатый азот. Давление газа лежит в пределах 40-45/45-70 PSI (трубка Вентури/мельница), а скорость подачи составляет от 0,5 до 1,6 кг/час. Соединение 1, Соединение 1 формы I, Соединение 1 формы II или Соединение 1 в форме соли HCl А измельчается в мельнице за счет столкновений частиц со стенками и друг с другом, а затем Соединение 1 формы II или Соединение 1 в форме соли HCl А ссыпается в контейнеры для измельченного продукта. Полагают, что средний специалист в этой области также может получить необходимый размер частиц Соединения 1, Соединения 1 формы I, Соединения 1 формы II или Соединения 1 в форме соли HCl А также путем измельчения на штифтовой мельнице, частично в условиях, описанных выше.
Дифференциальная сканирующая калориметрия (ДСК)
Данные дифференциальной сканирующей калориметрии (ДСК) Соединения 1, Соединения 1 формы I, Соединения 1 формы II или Соединения 1 в форме соли HCl А получали с помощью прибора для ДСК Q100 V9,6 Build 290 (TA Instruments, Нью-Касл, Делавэр). Для калибровки температуры использовали индий, а для калибровки теплопроводности - сапфир. Навеску образцов 3-6 мг помещали в алюминиевые тигли, которые закрывали крышками с одним точечным отверстием. Образцы сканировали при температуре от 25°C до 350°C со скоростью нагрева 1,0°C/мин и при пропускании азота со скоростью 50 мл/мин. Данные собирали с помощью программного обеспечения Thermal Advantage Q SeriesTM версии 2.2.0.248 и анализировали с помощью универсальной программы для анализа 4.1D (TA Instruments, Нью-Касл, Делавэр). Указанные значения относятся к отдельным анализам.
Определение структуры одного кристалла Соединения 1 формы I, Соединения 1 формы II и Соединения 1 в форме соли HCl A
Данные рентгеновской дифракции получали с помощью дифрактометра «Bruker Apex II» с запаянной Cu трубкой в качестве источника Kα излучения и ПЗС-детектором «Apex II». Структуру устанавливали с помощью программы SHELX (Sheldrick, G.M., Acta Cryst, (2008) A64, 112-122). На основании статистики систематических погасаний и интенсивностей была определена структура, которая отнесена к группе симметрии кристаллической решетки P21/n.
Витрид® (натрия бис(2-метоксиэтокси)алюмогидрид [или NaAlH2(OCH2CH2OCH3)2], 65% (по массе) раствор в толуоле) был закуплен в компании Aldrich Chemicals.
2,2-дифтор-1,3-бензодиоксол-5-карбоновая кислота была закуплена в компании Saltigo (филиал Lanxess Corporation).
Приготовление (2,2-дифтор-1,3-бензодиоксол-5-ил)-метанола.

Имеющуюся в продаже 2,2-дифтор-1,3-бензодиоксол-5-карбоновую кислоту (1,0 экв.) суспендировали в толуоле (10 объемов). Vitride® (2 экв.) добавляли через капельную воронку с такой скоростью, чтобы температура оставалась в пределах 15-25°C. В конце периода добавления температуру повышали до 40°C на 2 ч, затем осторожно добавляли 10% (по массе) водный раствор NaOH (4,0 экв.) через капельную воронку, поддерживая температуру 40-50°C. После перемешивания в течение дополнительных 30 минут слоям позволяли разделиться при 40°C. Органическую фазу охлаждали до 20°C, затем промывали водой (2×1,5 объемов), высушивали (Na2SO4), фильтровали и концентрировали для получения неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)-метанола, который использовали непосредственно на следующей стадии.
Приготовление 5-хлорметил-2,2-дифтор-1,3-бензодиоксола.

(2,2-дифтор-1,3-бензодиоксол-5-ил)-метанол (1,0 экв.) растворяли в МТБЭ (5 объемов). Затем добавляли каталитическое количество 4-(N,N-диметил)аминопиридина (ДМАП) (1 молярный %) и вносили SOCl2 (1,2 экв.) в капельную воронку. SOCl2 добавляли с такой скоростью, чтобы поддерживать температуру реакционной смеси 15-25°C. Температуру повышали до 30°C на 1 ч, а затем охлаждали до 20°C. Через капельную воронку добавляли воду (4 объема) при поддержании температуры менее 30°C. После перемешивания в течение дополнительных 30 минут слоям позволяли разделиться. Органический слой перемешивали и добавляли 10% (масс./об.) водный раствор NaOH (4,4 объема). После перемешивания в течение дополнительных 15-20 минут слоям позволяли разделиться. Затем органическую фазу высушивали (Na2SO4), фильтровали и концентрировали для получения неочищенного 5-хлорметил-2,2-дифтор-1,3-бензодиоксола, который использовали непосредственно на следующей стадии.
Приготовление (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила.

Раствор 5-хлорметил-2,2-дифтор-1,3-бензодиоксола (1 экв.) в ДМСО (1,25 объема) добавляли к суспензии NaCN (1,4 экв.) в ДМСО (3 объема) при поддержании температуры в пределах 30-40°C. Смесь перемешивали в течение 1 часа, а затем добавляли воду (6 объемов) и метил трет-бутиловый эфир (МТБЭ) (4 объема). После перемешивания в течение 30 минут слои разделяли. Водный слой экстрагировали МТБЭ (1,8 объема). Комбинированные органические слои промывали водой (1,8 объема), высушивали (Na2SO4), фильтровали и концентрировали для получения неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила, который использовали непосредственно на следующей стадии.
Синтез (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетат-ацетонитрила
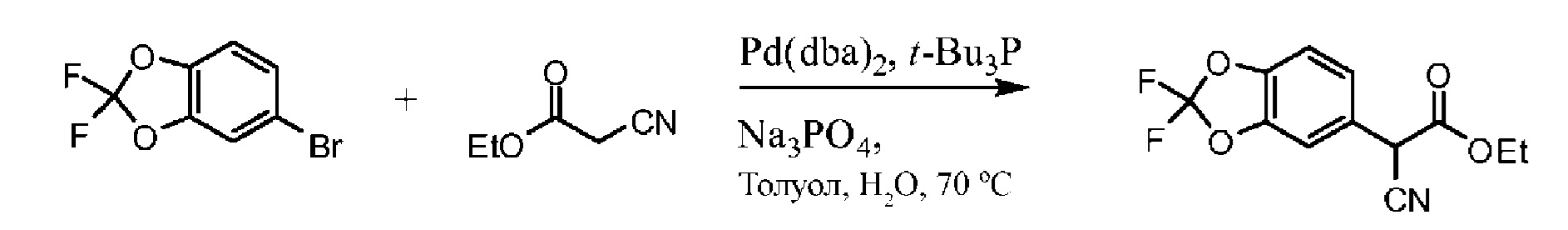
Реакционный сосуд продували азотом и заполняли 900 мл толуола. Растворитель дегазировали путем пропускания азота не менее 16 ч. Затем в реакционный сосуд вносили Na3PO4 (155,7 г, 949,5 ммоль), а затем бис(дибензилиденацетон)Pd (0) (7,28 г, 12,66 ммоль). 10% (массовых) раствор трет-бутилфосфина в гексане (51,23 г, 25,32 ммоль) добавляли в течение 10 минут при 23°C через капельную воронку, продуваемую азотом. Смесь оставляли для перемешивания на 50 минут, а затем добавляли 5-бром-2,2-дифтор-1,3-бензодиоксол (75 г, 316,5 ммоль) в течение 1 минуты. После перемешивания в течение дополнительных 50 минут в смесь вносили этилцианоацетат (71,6 г, 633,0 ммоль) в течение 5 минут, а затем воду (4,5 мл) одной порцией. Смесь нагревали до 70°C в течение 40 минут и анализировали методом ВЭЖХ каждые 1-2 ч для определения процента превращения реактива в продукт. После полного превращения (обычно 100% превращение происходит через 5-8 ч) смесь охлаждали до 20-25°C и фильтровали через целитовый фильтр. Целитовый фильтр ополаскивали толуолом (2×450 мл), а затем комбинированную органическую фазу концентрировали до 300 мл под вакуумом при температуре 60-65°C. К концентрату добавляли 225 мл ДМСО и концентрировали под вакуумом при 70-80°C до прекращения активной отгонки растворителя. Раствор охлаждали до 20-25°C и разводили до 900 мл ДМСО для подготовки к стадии 2. 1H ЯМР (500 мГц, CDCl3) δ 7,16-7,10 (м, 2H), 7,03 (д, J=8,2 Гц, 1H), 4,63 (с, 1H), 4,19 (м, 2H), 1,23 (т, J=7,1 Гц, 3H).
Синтез (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила.
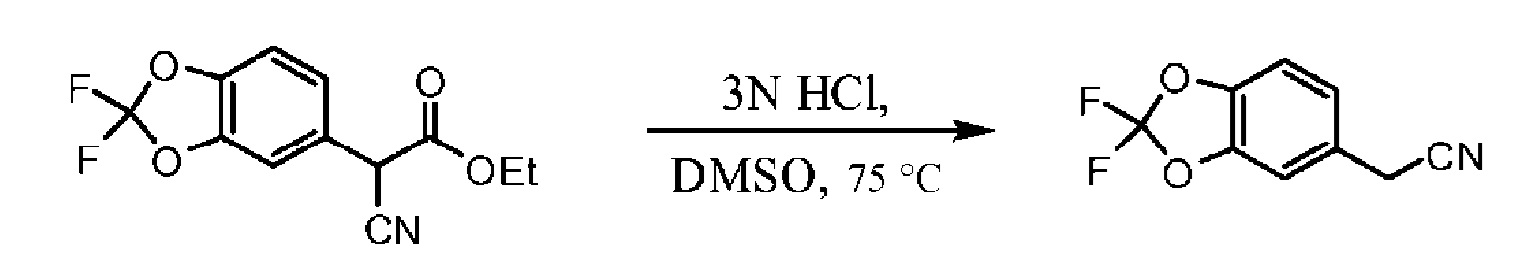
К раствору (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетата-ацетонитрила в ДМСО, описанному выше, добавляли 3 Н HCl (617,3 мл, 1,85 моль) в течение 20 минут при поддержании температуры смеси < 40°C. Затем смесь нагревали до 75°C в течение 1 часа и анализировали методом ВЭЖХ каждые 1-2 ч для определения процента превращения. После превращения > на 99% (обычно через 5-6 ч) реакционную смесь охлаждали до 20-25°C и экстрагировали МТБЭ (2×525 мл) в течение достаточного времени, чтобы фазы могли полностью разделиться. Комбинированную органическую фазу промывали 5% NaCl (2Ч375 мл). Затем раствор переносили в оборудование, подходящее для перегонки под вакуумом 0,19-0,33 кПа (1,5-2,5 Торр), с охлаждаемой колбой-приемником. Раствор концентрировали под вакуумом при < 60°C для удаления растворителей. Затем (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрил отгоняли от полученного маслянистого вещества при 125-130°C (температура термостата) и 0,19-0,27 кПа (1,5-2,0 Торр). (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрил выделяли в форме прозрачного маслянистого вещества с выходом 66% из 5-бром-2,2-дифтор-1,3-бензодиоксола (2 стадии) и чистотой 91,5% AUC по результатам ВЭЖХ (что соответствует количественному содержанию 95% по массе). 1H ЯМР (500 мГц, ДМСО) δ 7,44 (шир. с, 1H), 7,43 (д, J=8,4 Гц, 1H), 7,22 (дд, J=8,2, 1,8 Гц, 1H), 4,07 (с, 2H).
Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонитрила.

Смесь (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила (1,0 экв.), 50 массовых % водного KOH (5,0 экв.) 1-бром-2-хлорэтана (1,5 экв.) и Oct4NBr (0,02 экв.) нагревали до 70°C в течение 1 часа. Реакционную смесь охлаждали, а затем обрабатывали МТБЭ и водой. Органическую фазу промывали водой и насыщенным солевым раствором. Растворитель удаляли для получения (2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонитрила.
Получение 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбоновой кислоты.

(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонитрил гидролизовали с помощью 6 M NaOH (8 экв.) в этаноле (5 объемов) при 80°C в течение ночи. Затем смесь охлаждали до комнатной температуры и выпаривали этанол под вакуумом. Затем остаток поглощали водой с МТБЭ, добавляли 1 М HCl и разделяли слои. После этого слой МТБЭ обрабатывали дициклогексамином (ДЦГА) (0,97 экв.). Суспензию охлаждали до 0°C, фильтровали и промывали гептаном для получения соответствующей соли ДЦГА. Соль поглощали МТБЭ и 10% лимонной кислотой и перемешивали до растворения всех твердых веществ. Слои разделяли и промывали слой МТБЭ водой и насыщенным солевым раствором. После замены растворителя на гептан и последующего титрования получалась 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбоновая кислота, которую высушивали в вакуумном термостате при 50°C в течение ночи.
Получение 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонилхлорида.
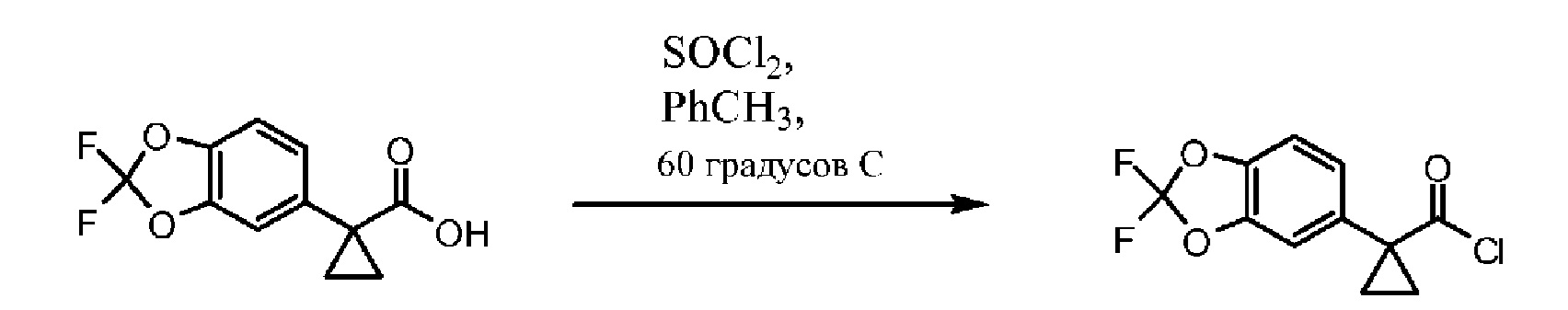
1-(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбоновую кислоту (1,2 экв.) суспендировали в толуоле (2,5 объема) и нагревали смесь до 60°C. Через капельную воронку добавляли SOCl2 (1,4 экв.). Через 30 минут из реакционной смеси отгоняли толуол и SOCl2. К полученной смеси добавляли дополнительный объем толуола (2,5 объемов) и повторяли отгонку для получения хлорида продукта в форме маслянистой жидкости, которую использовали без дальнейшей очистки.
Получение трет-бутил-3-(3-метилпиридин-2-ил)бензоата.
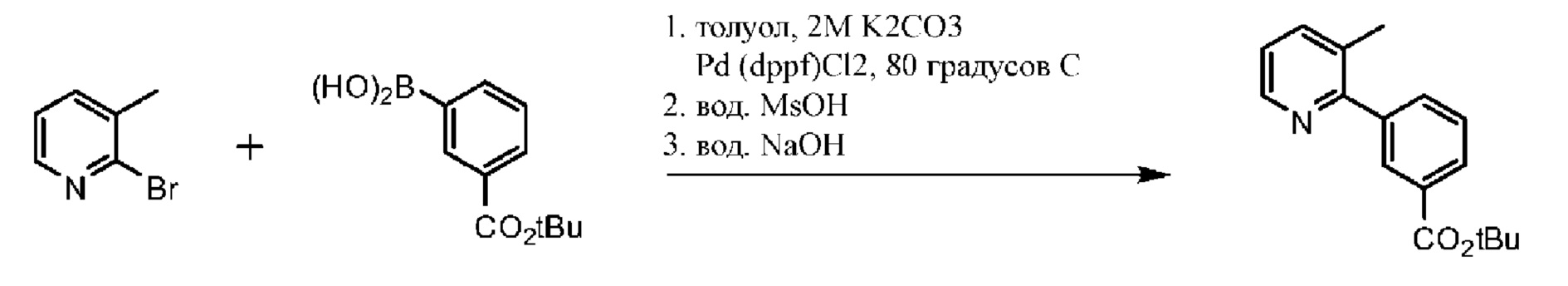
2-бром-3-метилпиридин (1,0 экв.) растворяли в толуоле (12 объемов). Добавляли K2CO3 (4,8 экв.), а затем воду (3,5 объема). Полученную смесь нагревали до 65°C под струей N2 в течение часа. Затем добавляли 3-(трет-бутоксикарбонил)фенилбороновую кислоту (1,05 экв.) и Pd(dppf)Cl2⋅CH2Cl2 (0,015 экв.) и нагревали смесь до 80°C. Через 2 ч нагрев отключали, добавляли воду (3,5 объемов) и позволяли слоям разделиться. Затем органическую фазу промывали водой (3,5 объемов) и экстрагировали 10% водным раствором метансульфоновой кислоты (2 экв. MsOH, 7,7 объемов). Водную фазу подщелачивали добавлением 50% водного раствора NaOH (2 экв.) и экстрагировали EtOAc (8 объемов). Органический слой концентрировали для получения неочищенного трет-бутил-3-(3-метилпиридин-2-ил)бензоата (82%), который использовали непосредственно на следующей стадии.
Получение 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида.

трет-бутил-3-(3-метилпиридин-2-ил)бензоат (1,0 экв.) растворяли в EtOAc (6 объемов). Добавляли воду (0,3 объема), затем пероксигидрат мочевины (3 экв.). После этого к смеси добавляли твердый фталевый ангидрид (3 экв.) порциями с такой скоростью, чтобы температура реакционной смеси оставалась ниже 45°C. После завершения добавления фталевого ангидрида смесь нагревали до 45°C. После перемешивания на протяжении еще 4 часов нагрев отключали. Через капельную воронку добавляли 10% (по массе) водный раствор Na2SO3 (1,5 экв.). После завершения добавления Na2SO3 смесь перемешивали еще 30 минут и разделяли слои. Органический слой перемешивали и добавляли 10% (по массе) водный раствор Na2CO3 (2 экв.). После перемешивания в течение дополнительных 30 минут слоям позволяли разделиться. Органическую фазу промывали 13% (масса/объем) NaCl. Затем органическую фазу отфильтровывали и концентрировали для получения неочищенного 2-(3-(трет-бутоксифенил)фенил)-3-метилпиридин-1-оксида (95%), который использовали непосредственно на следующей стадии.
Получение трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата.
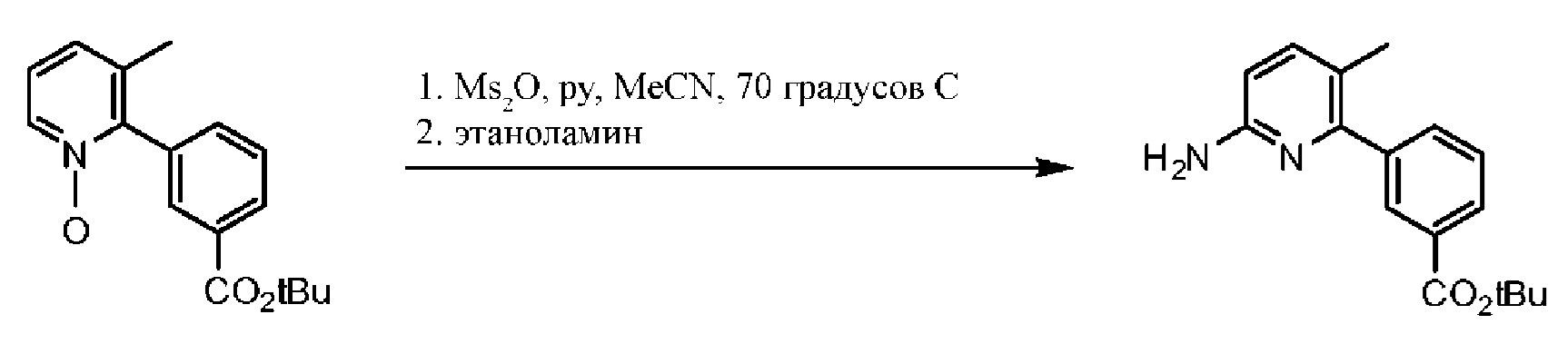
Раствор 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида (1 экв.) и пиридина (4 экв.) в ацетонитриле (8 объемов) нагревали до 70°C. Раствор метансульфонового ангидрида (1,5 экв.) в MeCN (2 объема) добавляли в течение 50 минут через капельную воронку при поддержании температуры 75°C. После завершения добавления смесь перемешивали еще 0,5 ч. Затем смеси давали остыть до комнатной температуры. Через капельную воронку добавляли этаноламид (10 экв.). После перемешивания в течение 2 ч добавляли воду (6 объемов) и смесь охлаждали до 10°C. После перемешивания в течение 3 ч твердое вещество собирали путем фильтрации и промывали водой (3 объема), смесью ацетонитрила/воды 2:1 и ацетонитрилом (2×1,5 объемов). Твердое вещество высушивали до постоянного веса (разница <1%) под вакуумом при температуре 50°C при медленном пропускании N2 для получения трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата в форме красно-желтого твердого вещества (выход 53%).
Получение 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)- циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата.
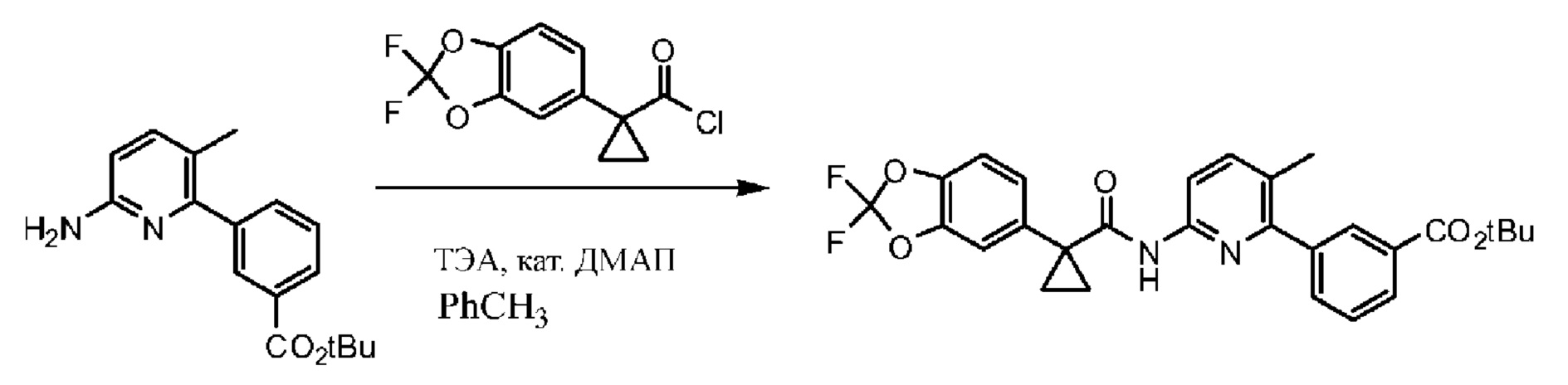
Неочищенный хлорангидрид, описанный выше, растворяли в толуоле (2,5 объемов по объему хлорангидрида) и добавляли через капельную воронку к смеси трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата (1 экв.), ДМАП, (0,02 экв.) и триэтиламина (3,0 экв.) в толуоле (4 объема по объему трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата). Через 2 часа к реакционной смеси добавляли воду (4 объема по объему трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата). После перемешивания в течение 30 минут слои разделяли. Затем органическую фазу отфильтровывали и концентрировали для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата в форме густого маслянистого вещества (количественный выход неочищенного вещества). Затем добавляли ацетонитрил (3 объема от объема неочищенного продукта) и отгоняли до кристаллизации. После этого добавляли воду (2 объема от объема неочищенного продукта) и перемешивали смесь 2 часа. Твердое вещество собирали путем фильтрования, промывали смесью ацетонитрила/воды 1:1 (по объему) (2×1 объема от объема неочищенного продукта) и частично высушивали на фильтре под вакуумом. Затем твердое вещество высушивали до постоянного веса (различие Ѕ <1%) под вакуумом при 60°C при медленном пропускании N2 для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата в форме твердого вещества коричневого цвета.
Получение 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты в форме соли HCl.
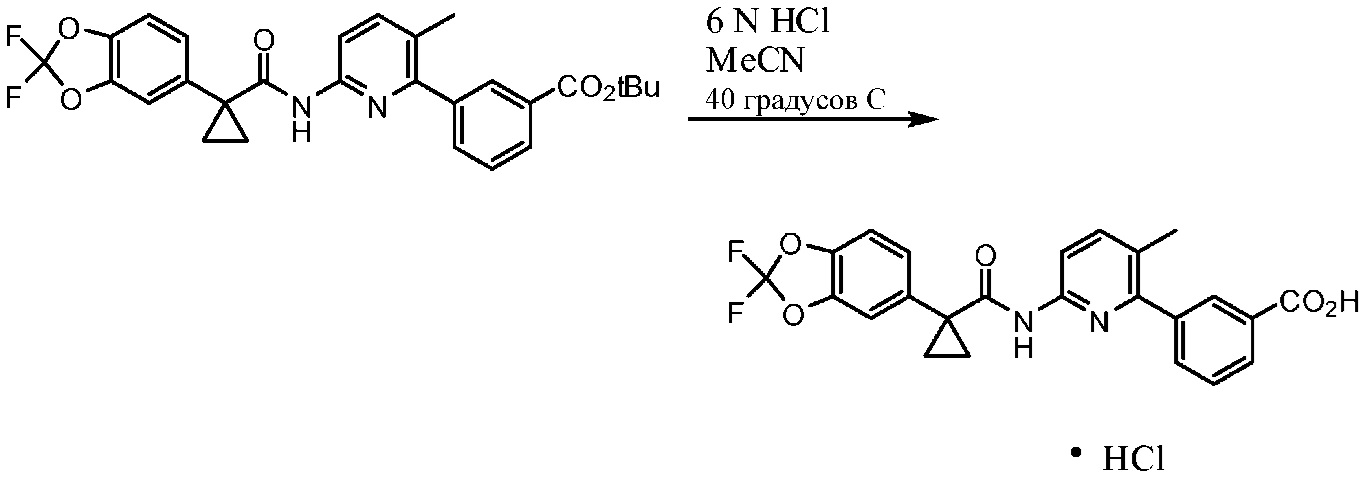
К суспензии 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата (1,0 экв.) в MeCN (3,0 объема) добавляли воду (0,83 объема) и концентрированный водный раствор HCl (0,83 объема). Смесь нагревали до 45±5°C. После перемешивания в течение 24-48 ч реакция завершалась, и смеси давали остыть до комнатной температуры. Добавляли воду (1,33 объема) и перемешивали смесь. Твердое вещество собирали путем фильтрации, промывали водой (2×0,3 объема) и частично высушивали на фильтре под вакуумом. Затем твердое вещество высушивали до постоянного веса (различие <1%) под вакуумом при 60°C при медленном пропускании N2 для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты⋅HCl в форме грязно-белого твердого вещества.
1H ЯМР спектр Соединения 1 показан на Фиг. 20, а на Фиг. 21 показан 1H ЯМР спектр Соединения 1 в форме соли HCl.
В таблице 2 ниже показаны данные 1H ЯМР Соединения I.
|
Получение Соединения 1 формы I, метод A. 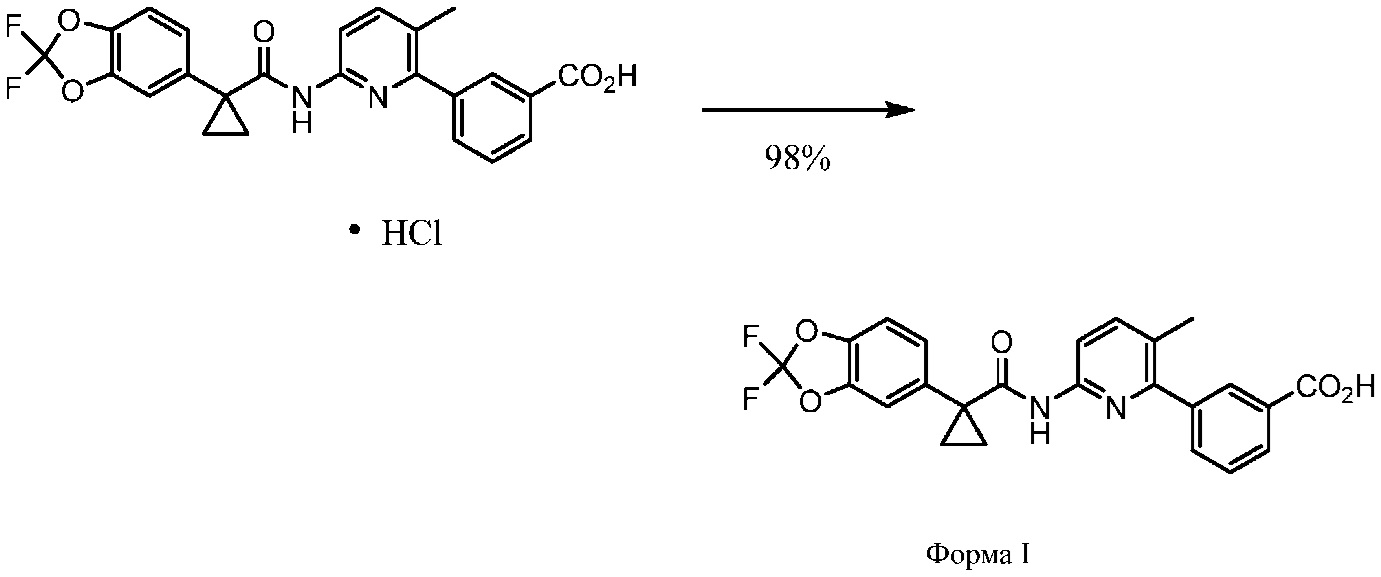
Суспензию 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамид)-3-метилпиридин-2-ил)бензойной кислоты⋅HCl (1 экв.) в воде (10 объемов) перемешивали при комнатной температуре. После перемешивания в течение 24 ч отбирали пробу. Пробу фильтровали и промывали твердое вещество водой (дважды). Твердое вещество анализировали методом ДСК. Когда анализ методом ДСК показывал полное превращение в Форму I, твердое вещество отфильтровывали, промывали водой (2×1,0 объема) и частично высушивали на фильтре под вакуумом. Твердое вещество высушивали до постоянного веса (различие <1%) под вакуумом при 60°C с медленным пропусканием N2 для получения Соединения 1 формы I в виде грязно-белого твердого вещества (выход 98%). 1H ЯМР (400 мГц, ДМСО-d6) 9,14 (с, 1H), 7,99-7,93 (м, 3H), 7,80-7,78 (м, 1H), 7,74-7,72 (м, 1H), 7,60-7,55 (м, 2H), 7,41-7,33 (м, 2H), 2,24 (с, 3H), 1,53-1,51 (м, 2H), 1,19-1,17 (м, 2H).
Получение Соединения 1 формы I, метод B.
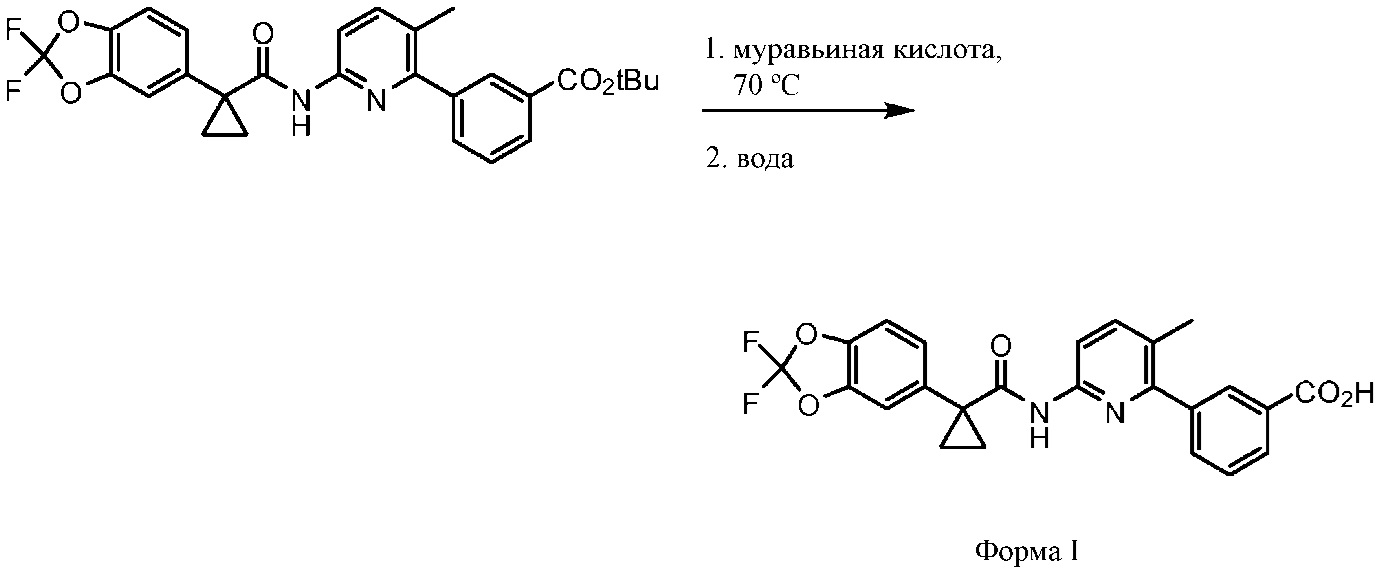
Раствор 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата (1,0 экв.) в муравьиной кислоте (3,0 объема) нагревали при перемешивании до 70±10°C в течение 8 ч. Реакцию считали завершенной после того, как анализ хроматографическими методами показывал остаточное содержание 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата) менее 1,0% AUC. Смеси давали остыть до комнатной температуры. Раствор добавляли к воде (6 объемов), нагревали до 50°C и перемешивали смесь. Затем смесь нагревали до 70±10°C до тех пор, пока содержание 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамид)-3-метилпиридин-2-ил)-трет-бутилбензоата не становилось не выше 0,8% (AUC). Твердое вещество отфильтровывали, промывали водой (2×3 объема) и частично высушивали на фильтре под вакуумом. Твердое вещество высушивали до постоянного веса (различие <1%) под вакуумом при 60°C с медленным пропусканием N2 для получения Соединения 1 формы I в виде грязно-белого твердого вещества.
Результаты ДСК Соединения 1 формы 1 показаны на Фиг. 22. Плавление Соединения 1 формы I происходит при температуре примерно 204°C.
Картину рентгеновской дифракции вычисляли по результатам определения структуры одного кристалла Соединения 1 формы I; она показана на Фиг. 1. В таблице 3 перечислены вычисленные пики, показанные на Фиг. 1.
|
Фактическая картина рентгеновской дифракции Соединения 1 формы I показана на Фиг. 2. В таблице 4 перечислены фактические пики, показанные на Фиг. 2.
|
Бесцветные кристаллы Соединения 1 формы I получали путем охлаждения концентрированного раствора в 1-бутаноле с 75°C до 10°C со скоростью 0,2°C/мин. Выбирали кристаллы с размерами 0,50×0,08×0,03 мм, очищали минеральным маслом, помещали в держатель MicroMount и центрировали с помощью системы Bruker APEX II. Получали три группы по 40 кадров, разделенных в обратном пространстве для обеспечения ориентационной матрицы и получения начальных параметров ячейки. Конечные параметры ячейки получали и уточняли после получения полного набора данных.
Набор дифракционных данных в обратном пространстве получали с разрешением 0,82 Å использованием шагов 0,5° с выдержкой 30 с для каждого кадра. Данные собирали при 100 (2) K. Интеграцию интенсивностей и уточнение параметров ячейки проводили с помощью программы APEXII. Исследование кристалла после сбора данных не показало признаков разрушения.
На Фиг. 23 показана конформация Соединения 1 формы I на основании рентгенодифракционного анализа одного кристалла. Соединение 1 формы I представляет собой моноклинные кристаллы, P21/n, со следующими размерами ячейки: a=4,9626(7) Å, b=12,299(2) Å, c=33,075(4) Å, β=93,938(9)°, V=2014,0 Å3, Z=4. Плотность Соединения 1 формы I, вычисленная по данным о структуре, составляет 1,492 г/см3 при 100 K.
Получение Соединения 1 формы II из Соединения 1 формы I.
Соединение 1 формы I (приблизительно 30 мг) суспендировали в 500 мкл подходящего растворителя (например метанола, этанола, ацетона, 2-пропанола, ацетонитрила, тетрагидрофурана, метилового эфира уксусной кислоты, 2-бутанона, этилового эфира муравьиной кислоты и метилтетрагидрофурана) в течение 2 дней. Затем суспензию фильтровали с помощью центрифуги или под вакуумом и высушивали при комнатной температуре в течение ночи для получения Соединения 1 формы II.
Результаты ДСК Соединения 1 формы II в форме сольвата с ацетоном показаны на Фиг. 15, на которой видно 2 фазовых перехода. Точка плавления Соединения 1 формы II в виде сольвата с ацетоном лежит в пределах от 188°C до 205°C.
Фактическая картина рентгеновской дифракции Соединения 1 формы II показана на Фиг. 3. В таблице 5 перечислены фактические пики, показанные на Фиг. 3, в нисходящем порядке относительной интенсивности.
|
Изображения конформации Соединения 1 формы II в виде сольвата с ацетоном на основании рентгенодифракционного анализа одного кристалла показаны на Фиг. 24. Стехиометрическое соотношение между Соединением 1 формы II и ацетоном составляет приблизительно 4,4:1 (4,48:1 на основании вычисления по данным 1H ЯМР; 4,38:1 по данным рентгенодифракционного анализа). Анализ кристаллической структуры показал упаковку молекул с двумя полостями или карманами на элементарную ячейку, или 1 полостью на молекулу исходного вещества. В сольвате с ацетоном приблизительно 92% пустот заполнено молекулами ацетона. Соединение 1 формы II относится к моноклинной группе симметрии P21/n со следующими размерами ячейки: a=16,5235(10) Å, b=12,7425(8) Å, c=20,5512(13) Å, α=90°, β=103,736(4)°, γ=90°, V=4203,3(5) Å3, = 4. Плотность Соединения 1 в Соединении 1 формы II, вычисленная по данным о структуре, составляет 1,430/см3 при 100 K.
Спектр 13C ЯМР твердого Соединения 1 формы II в форме сольвата с ацетоном показан на Фиг. 25. В таблице 6 показаны химические сдвиги соответствующих пиков.
|
|
Спектр 19F ЯМР твердого Соединения 1 формы II в форме сольвата с ацетоном показан на Фиг. 26. Пики, помеченные звездочкой, соответствуют боковым полосам от вращения. В таблице 7 показаны химические сдвиги соответствующих пиков.
|
Получение Соединения 1 в форме соли HCl A.
Бесцветные кристаллы Соединения 1 в форме соли HCl А получали путем медленного выпаривания концентрированного раствора соли HCl Соединения 1 в этаноле. Выбирали кристалл с размерами 0,30×1/5×0,15 мм, очищали с помощью минерального масла, помещали в держатель MicroMount и центрировали в дифрактометре Bruker APEXII. Получали три группы по 40 кадров, разделенных в обратном пространстве для обеспечения ориентационной матрицы и получения начальных параметров ячейки. Конечные параметры ячейки получали и уточняли после получения полного набора данных.
На Фиг. 18 показано изображение конформации Соединения 1 в форме соли HCl А в виде димера, определенной на основании анализа одного кристалла. Картина рентгеновской дифракции Соединения 1 в форме соли HCl А, вычисленная по кристаллической структуре, показана на Фиг. 27. В таблице 8 указаны фактические пики, показанные на Фиг. 27, в нисходящем порядке относительной интенсивности.
|
Примеры лекарственных форм для приема внутрь, содержащих Соединение 1
Была изготовлена таблетка с компонентами и количествами, указанными в таблице 9 с составом примерной таблетки 1А, содержащей 100 мг действующего вещества, т.е. Соединения 1 формы I. Примерная таблетка 1А (содержащая 100 мг Соединения 1) изготавливается с помощью аппарата для сухого вальцевания. В Таблице 9 были указаны следующие типы/торговые марки: микрокристаллическая целлюлоза: Avicel PH102; маннит: Pearlitol SD 100; Кросскармеллозы натриевая соль: Acdisol; и коллоидный оксид кремния: Cabosil.
|
Была изготовлена таблетка с компонентами и количествами, указанными в таблице 10, показывающей состав примерной таблетки 1B, содержащей 100 мг действующего вещества, т.е. Соединения 1 формы I. Примерная таблетка 1B (содержащая 100 мг Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 10 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в составе композиции для таблетки - кросскармелозы натриевая соль: Acdisol.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 11, показывающей состав примерной таблетки 1C, содержащая 100 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1C (содержащая 100 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 11 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для таблетки - кросскармелозы натриевая соль: Acdisol.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 12, показывающей состав примерной таблетки 1D, содержащая 200 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1D (содержащая 200 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 12 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 13, показывающей состав примерной таблетки 1E, содержащая 200 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1E (содержащая 200 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 13 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712; и в пленочном покрытии - пленочное покрытие: Опадрай II; Воск: карнаубский.
|
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 14, показывающей состав примерной таблетки 1F, содержащая 200 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1F (содержащая 200 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 14 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712; и в пленочном покрытии - пленочное покрытие: Опадрай II; Воск: карнаубский.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 15, показывающей состав примерной таблетки 1G, содержащая 100 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1G (содержащая 100 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 15 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в составе таблетки - кросскармелозы натриевая соль: Acdisol.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 16, показывающей состав примерной таблетки 1H, содержащая 100 мг действующего вещества, т.е. кристаллического Соединения 1 формы I или формы II Примерная таблетка 1H (содержащая 100 мг кристаллического Соединения 1 формы I или формы II) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 16 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712.
|
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 17, показывающей состав примерной таблетки 1I, содержащая 100 мг действующего вещества, т.е. кристаллического Соединения 1 формы I или формы II Примерная таблетка 1I (содержащая 100 мг кристаллического Соединения 1 формы I или формы II) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 17 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712.
|
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 18, показывающей состав примерной таблетки 1J, содержащая 300 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1J (содержащая 300 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 18 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712; и в пленочном покрытии - пленочное покрытие: Опадрай II; Воск: карнаубский.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 19, показывающей состав примерной таблетки 1K, содержащая 300 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1K (содержащая 300 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с высоким усилием сдвига. В таблице 19 указаны следующие типы/торговые марки. Гранулированная смесь, полученная путем гранулирования с высоким усилием сдвига - микрокристаллическая целлюлоза: Avicel PH101; маннит: Pearlitol C50; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712; и в пленочном покрытии - пленочное покрытие: Опадрай II; Воск: карнаубский.
|
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 20, показывающей состав примерной таблетки 1L, содержащая 200 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1L (содержащая 200 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с использованием двухшнекового аппарата. В таблице 20 указаны следующие типы/торговые марки. Гранулированная смесь, полученная с помощью двухшнекового гранулятора - микрокристаллическая целлюлоза: Avicel PH101; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712; и в пленочном покрытии - пленочное покрытие: Опадрай II; Воск: карнаубский.
|
Была изготовлена таблетка с компонентами и количествами, перечисленными в таблице 21, показывающей состав примерной таблетки 1M, содержащая 400 мг действующего вещества, т.е. кристаллического Соединения 1 формы I. Примерная таблетка 1M (содержащая 400 мг кристаллического Соединения 1 формы I) изготавливается с использованием способа влажного гранулирования с использованием двухшнекового аппарата. В таблице 21 указаны следующие типы/торговые марки. Гранулированная смесь, полученная с помощью двухшнекового гранулятора - микрокристаллическая целлюлоза: Avicel PH101; Кросскармеллозы натриевая соль: Acdisol; поливинилпирролидон: Kollidon PVP K30; и в композиции для сердцевин таблеток - микрокристаллическая целлюлоза: Avicel PH200; Кросскармеллозы натриевая соль: Acdisol; и магния стеарат: 5712; и в пленочном покрытии - пленочное покрытие: Опадрай II; Воск: карнаубский.
|
|
Изготовление таблеток из гранулированной композиции, полученной путем вальцевания
Оборудование/ способ
Оборудование
Роликовые прессы: Alexanderwerk WP 120, Vector TF-Mini, или Vector TF-Labo.
Просеивание/взвешивание
Соединение 1 и вспомогательные вещества могут просеиваться до или после взвешивания. Подходящий размер сита - 20, 40 или 60. Для упрощения просеивания Соединение 1 можно предварительно смешать с одним или более вспомогательных веществ.
Смешивание
Соединение 1 и вспомогательные вещества могут добавляться в смеситель в разном порядке. Смешивание может проводиться в смесителе «Turbula» или V-образном смесителе. Компоненты могут смешиваться в течение 10 минут без скользящего вещества, а затем смешиваться еще 3 минуты после добавления скользящего вещества.
Вальцевание
Смесь может подвергаться вальцеванию в форме лент и размалываться на гранулы с помощью аппарата Alexanderwerk WP 120. Использующиеся ролики могут быть роликами 25 мм с давлением сжатия от 18 до 50 бар, скоростью вращения от 3 до 12 об/мин и скоростью винтового питателя 20-80 об/мин. Размеры сита встроенной мельницы могут быть 2 мм (верхнее сито) и 0,8 мм (нижнее сито).
Смешивание
Гранулы, полученные после вальцевания, могут быть смешаны с вспомогательными веществами, не входящими в состав гранул, например, с наполнителями или скользящим веществом, с помощью V-образного смесителя. Время смешивания может быть 5, 3 или 1 минута.
Прессование
Смесь для прессования может быть спрессована в таблетки с помощью отдельного пресса «Riva MiniPress» с оснасткой 10 мм. Вес таблеток, содержащих дозу 100 мг, может быть 200, 250 или 300 мг.
Пленочное покрытие
На таблетки может наноситься пленочное покрытие с использованием дражировочного котла, например, «O'Hara Labcoat».
Печать
На одной или обеих сторонах таблеток, покрытых оболочкой, можно напечатать монограмму, например, с помощью принтера «Hartnett Delta».
Изготовление таблеток из гранулированной композиции, полученной с помощью гранулятора с высоким усилием сдвига
Оборудование/ способ
Оборудование
Гранулятор: «Procept MiPro» с чашей 250 мл или 1 л.
Просеивание/ взвешивание
Соединение 1 и вспомогательные вещества могут просеиваться до или после взвешивания. Возможный размер сита - 20, 40 или 60. Для упрощения просеивания Соединение 1 можно предварительно смешать с одним или более вспомогательных веществ.
Операция гранулирования
Жидкость для гранулирования - лаурилсульфат натрия и связующее вещество, которые добавляются в очищенную воду и перемешиваются до растворения. Подходящее соотношение - 2,5% масс./масс. лаурилсульфата натрия и 10,0% масс./масс. ПВП K30 в воде.
Гранулирование - вспомогательные вещества и соединение 1 вносятся в чашу гранулятора. Порядок добавления может быть следующим: Соединение 1, разрыхлитель, разбавитель и наполнитель. Компоненты могут перемешиваться в чаше 250 мл в течение 1 минуты при скорости мешалки 1000 об/мин. и скорости ножа 1000 об/мин. Гранулирование может проводиться при скорости мешалки 2000 об/мин и скорости ножа 4000 об/мин при добавлении жидкости для гранулирования с помощью шприцевого насоса на скорости 1,5-4,5 г/мин. Время добавления жидкости может быть от 4 до 12 минут. После добавления требуемого количества жидкости гранулы могут уплотняться в течение примерно 10 секунд - 1 минуты. Одним из преимуществ настоящего способа гранулирования с высоким усилием сдвига, которое следует отметить, является использование жидкости для гранулирования, содержащей как поверхностно-активное вещество, так и связующее вещество для улучшения гранулирования за счет повышения смачиваемости. В одном варианте осуществления изобретения поверхностно-активным веществом является лаурилсульфат натрия.
Размол
Уменьшение размера гранул возможно с помощью конической мельницы или мельницы с ситами.
Сушка
Гранулы могут высушиваться в вакуумном сушильном шкафу, полочной сушилке, двухконусной сушилке или сушилке с псевдоожиженным слоем. Гранулы высушивались с помощью вакуумного сушильного шкафа с продувкой азотом.
Смешивание
Гранулы могут смешиваться с вспомогательными веществами, не входящими в состав гранул. Гранулы смешивали с разрыхлителем, разбавителем, наполнителем и скользящим веществом, не входящими в состав гранул. Гранулы смешивали с помощью смесителя «Turbula» в течение 3 минут до добавления скользящего вещества и 1 минуты после добавления скользящего вещества. Можно использовать более крупные смесители, например, 4-квартовый V-образный смеситель.
Прессование
Смесь для прессования может быть спрессована в таблетки с помощью отдельного пресса «Riva MiniPress» с оснасткой 8 или 10 мм. Вес таблеток, содержащих дозу 100 мг, может быть приблизительно 160, 200 или 250 мг.
Пленочное покрытие
На таблетки может наноситься пленочное покрытие с использованием дражировочного котла, например, «O'Hara Labcoat».
Печать
На одной или обеих сторонах таблеток, покрытых оболочкой, можно напечатать монограмму, например, с помощью принтера «Hartnett Delta».
Изготовление таблеток с использованием способа непрерывного влажного гранулирования с помощью двухшнекового аппарата.
Оборудование/способ
Оборудование
Гранулятор: двухшнековый гранулятор «ConsiGma» или «Leistritz» или «Thermo Fisher».
Просеивание/взвешивание
Соединение 1 и вспомогательные вещества могут просеиваться до или после взвешивания. Возможный размер сита - 20, 40 или 60. Для упрощения просеивания Соединение 1 можно предварительно смешать с одним или более вспомогательных веществ.
Смешивание
Соединение 1 и вспомогательные вещества могут добавляться в смеситель в разном порядке. Смешивание может производиться в смесителе «Turbula», в v-образном смесителе, бункерном смесителе или смесителе непрерывного действия. Компоненты могут перемешиваться в течение 10 минут в порционных смесителях или непрерывно в смесителе непрерывного действия.
Операция гранулирования
Жидкость для гранулирования - лаурилсульфат натрия и связующее вещество, которые добавляются в очищенную воду и перемешиваются до растворения. Подходящее соотношение - 2,5% масс./масс. лаурилсульфата натрия и 10,0% масс./масс. ПВП K30 в воде.
Гранулирование - смесь, содержащая Соединение 1 и вспомогательные вещества, может вноситься в двухшнековый гранулятор с помощью весового питателя непрерывного действия со скоростью 10 кг/час. Жидкость для гранулирования может добавляться с помощью перистальтического насоса со скоростью 3,5 кг/час. Гранулятор может работать на скорости 400 об/мин. Заметным преимуществом настоящего способа гранулирования с помощью двухшнекового гранулятора является использование жидкости для гранулирования, содержащей как поверхностно-активное вещество, так и связующее вещество для улучшения гранулирования за счет повышения смачиваемости. В одном варианте осуществления изобретения поверхностно-активным веществом является лаурилсульфат натрия. Другим заметным преимуществом является непрерывность способа, таким образом, в каждый момент времени обрабатывается лишь ограниченный объем материала, что позволяет хорошо контролировать способ и получать продукт высокого качества.
Размол
Уменьшение размера гранул возможно с помощью мельницы с ситами или конической мельницы.
Сушка
Гранулы могут высушиваться в вакуумном сушильном шкафу, полочной сушилке, двухконусной сушилке или сушилке с псевдоожиженным слоем.
Смешивание
Гранулы могут смешиваться с вспомогательными веществами, не входящими в состав гранул. Гранулы смешивали с помощью бункерного смесителя объемом 300 л в течение 60 оборотов.
Прессование
Смесь была спрессована в таблетки с помощью ротационного пресса «Courtoy Modul P».
Пленочное покрытие
На таблетки может наноситься пленочное покрытие с использованием дражировочного котла, например, «O'Hara Labcoat».
Печать
На одной или обеих сторонах таблеток, покрытых оболочкой, можно напечатать монограмму, например, с помощью принтера «Hartnett Delta».
График приема препарата
В другом аспекте настоящее изобретение относится к способу лечения CFTR-опосредованного заболевания у пациента, включающему введение нуждающемуся в этом пациенту эффективного количества фармацевтической композиции в соответствии с настоящим изобретением. В другом варианте осуществления фармацевтическая композиция вводится пациенту раз в две недели. В другом варианте осуществления фармацевтическая композиция вводится пациенту раз неделю. В другом варианте осуществления фармацевтическая композиция вводится пациенту раз в три дня. В другом варианте осуществления фармацевтическая композиция вводится пациенту раз в день. В одном варианте осуществления, если фармацевтическая композиция представляет собой таблетку в соответствии с табл. 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 или 19, она вводится раз в день.
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Методы анализа для обнаружения и измерения способности соединений к коррекции F508del-CFTR
Оптические методы измерения мембранного потенциала для анализа F508del-CFTR-модулирующих свойств соединений.
Оптический метод измерения мембранного потенциала с помощью потенциал-чувствительного датчика резонансного переноса энергии флуоресценции, описанного Gonzalez and Tsien (см. Gonzalez, J. E. и R. Y. Tsien (1995) «Voltage sensing by fluorescence resonance energy transfer in single cells» Biophys J 69(4): 1272-80, и Gonzalez, J. E. and R. Y. Tsien (1997) «Improved indicators of cell membrane potential that use fluorescence resonance energy transfer» Chem Biol 4(4): 269-77) в сочетании с оборудованием для измерения изменений флуоресценции, например, ионного зонда/зонда для измерения потенциала (VIPR) (см. Gonzalez, J. E., K. Oades, et al. (1999) «Cell-based assays and instrumentation for screening ion-channel targets» Drug Discov Today 4(9): 431-439).
Эти потенциал-чувствительные методы основаны на изменении резонансного переноса энергии флуоресценции (РПЭФ) между мембранорастворимым потенциал-чувствительным красителем DiSBAC2(3) и флуоресцентным фосфолипидом CC2-DMPE, который присоединяется к внешнему листку плазматической мембраны и действует как донор РПЭФ. Изменения мембранного потенциала (Вм) приводят к перераспределению отрицательно заряженного DiSBAC2(3) в плазматической мембране и соответствующему изменению переноса энергии от CC2-DMPE. Изменения флуоресценции измеряли с помощью VIPR™ II, встроенного устройства для работы с жидкостями, и флуоресцентного детектора, разработанного для проведения клеточных анализов на микротитрационных 96- или 384-луночных планшетах.
1. Идентификация корректирующих соединений
Для идентификации мелких молекул, корректирующих дефекты транспорта, связанных с F508del-CFTR, был разработан формат метода высокопроизводительного анализа с однократным добавлением реактивов. Лунки инкубировали с бессывороточной средой в течение 16 ч при 37°C в присутствии или отсутствии (отрицательный контроль) исследуемого соединения. В качестве положительного контроля использовали клетки, внесенные в 384-луночные планшеты, которые инкубировали 16 ч при 27°C для «температурной коррекции» F508del-CFTR. В последующем клетки промывали 3Х раствором Кребса-Рингера и вносили потенциал-чувствительные красители. Для активации F508del-CFTR в каждую лунку вносили 10 мкM форсколина и потенциатор CFTR генистеин (20 мкM) вместе со средой, не содержащей Cl-. Добавление среды, не содержащей Cl-, способствовало утечке Cl- в ответ на активацию F508del-CFTR, а возникающую в результате деполяризацию мембраны измеряли оптическим методом с помощью потенциал-чувствительных красителей на основе РПЭФ.
2. Определение потенцирующих соединений
Для идентификации потенциаторов F508del-CFTR был разработан формат метода высокопроизводительного анализа с двукратным добавлением реактивов. Сначала в каждую лунку добавляли среду, не содержащую Cl-, с исследуемым соединением или без. Через 22 секунды снова добавляли не содержащую Cl--среду с 2-10мкМ форсколина для активации F508del-CFTR. Внеклеточная концентрация Cl- после обоих добавлений составила 28 мM, что способствовало оттоку Cl- в ответ на активацию F508del-CFTR, а возникающую в результате деполяризацию мембраны измеряли оптическим методом с помощью потенциал-чувствительных красителей на основе РПЭФ.
3. Растворы
Раствор для бани № 1: (в мM) NaCl 160, KCl 4,5, CaCl2 2, MgCl2 1, ГЭПЭС 10, pH 7,4 с NaOH.
Раствор для бани, не содержащий хлоридов: Хлориды в растворе для бани № 1 заменяются глюконатами.
CC2-ДМФЭ: Готовиться в форме маточного раствора 10 мМ в ДМСО и хранится при -20°C.
DiSBAC2(3): Готовиться в форме маточного раствора 10 мМ в ДМСО и хранится при -20°C.
4. Культура клеток
Для оптического измерения мембранного потенциала используются мышиные фибробласты NIH3T3 со стабильной экспрессией F508del-CFTR. Клетки инкубируют при 37°C в атмосфере с 5% CO2 и при относительной влажности 90% в среде Игла, модифицированной Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1Х добавки аминокислот NEAA, β-ME, 1Х пенициллина/стрептомицина и 25 мM ГЭПЭС в культуральных колбах 175 см2. Для всех оптических методов анализа клетки вносили в лунки 384-луночных планшетов, покрытых матригелем, в количестве 30000 на лунку, и инкубировали 2 ч при 37°C перед инкубированием при 27°C в течение 24 ч для количественного определения потенциатора. Для количественного определения корректирующего действия клетки культивировали при 27°C пли 37°C с соединениями или без в течение 16-24 ч.
Электрофизиологические методы анализа F508del-CFTR-модулирующих свойств соединений
1. Исследование в камере Уссинга
Эксперименты в камере Уссинга проводили на поляризованных эпителиальных клетках, экспрессирующих F508del-CFTR, для дополнительного определения свойств модуляторов F508del-CFTR, определенных с помощью оптических методов. Эпителиальные клетки FRTΔF508-CFTR, выращенные на вставках «Costar Snapwell» для культуры клеток, помещали в камеру Уссинга (Physiologic Instruments, Inc., Сан Диего, Калифорния) и создавали постоянное короткое замыкание между монослоями с помощью системы зажимов (факультет биоинженерии, Университет Айовы, Айова, и компания Physiologic Instruments, Inc., Сан Диего, Калифорния). Трансэпителиальное сопротивление измеряли с помощью импульса 2 мВ. В этих условиях измерения сопротивление эпителия щитовидной железы крыс линии Фишер составило 4 KΩ/см2 или выше. Растворы поддерживали при температуре 27°C и пропускали через них воздух в виде пузырьков. Поправку на смещение потенциала электрода и сопротивление жидкости вычисляли с помощью вставки без клеток. В этих условиях электрический ток отражает поток Cl- через F508del-CFTR, экспрессирующиеся в апикальной мембране. Ток короткого замыкания определяли цифровым способом с помощью интерфейса MP100A-CE и программы AcqKnowledge (v3,2,6; BIOPAC Systems, Санта Барбара, Калифорния).
2. Идентификация корректирующих соединений
В типичной методике используется градиент концентрации Cl- между базолатеральной и апикальной мембраной. Для создания такого градиента использовался нормальный раствор Рингера со стороны базолатеральной мембраны, в то время как со стороны апикальной мембраны эквимолярный раствор NaCl был заменен эквимолярным раствором глюконата натрия (с pH, доведенным до 7,4 путем титрования NaOH), таким образом, был получен значительный градиент концентрации Cl- по обеим сторонам эпителия. Все эксперименты проводили с использованием цельных монослоев. Для полной активации F508del-CFTR вносили форсколин (10 мкM) и ингибитор ФДЭ, IBMX (100 мкM), а затем добавляли потенциатор CFTR генистеин (50 мкM).
Как и в других типах клеток, инкубация клеток эпителия щитовидной железы крыс линии Фишер со стабильной экспрессией F508del-CFTR повышала функциональную плотность CFTR в плазматической мембране. Для определения активности корректирующих соединений клетки инкубировали с 10 мкM исследуемого соединения в течение 24 ч при 37°C и перед записью промывали 3X. Ток короткого замыкания, опосредованный цАМФ и генистеином в ячейках, содержащих соединение, нормировали относительно контрольных значений при 27°C и 37°C и выражали как процентную активность. Предварительная инкубация ячеек с корректирующим соединением значительно повышала ток короткого замыкания, опосредованный цАМФ и генистеином, по сравнению с контролями при 37°C.
3. Определение потенцирующих соединений
В типичной методике используется градиент концентрации Cl- между базолатеральной и апикальной мембраной. Для создания такого градиента использовали нормальный раствор Рингера со стороны базолатеральной мембраны, проницаемость которой повышали при помощи нистатина (360 мкг/мл), в то время как со стороны апикальной мембраны NaCl заменяли эквимолярным раствором глюконата натрия (с рН, доведенным до 7,4 титрованием NaOH), таким образом, достигался значительный градиент Cl- по обеим сторонам эпителия. Все эксперименты проводили через 30 минут после внесения нистатина для повышения проницаемости. Форсколин (10 мкM) и все исследуемые соединения вносили с обеих сторон вкладок для культуры клеток. Эффективность предполагаемых потенциаторов F508del-CFTR сравнивали с активностью известного потенциатора, генистеина.
4. Растворы
Раствор с базолатеральной стороны (в мM): NaCl (135), CaCl2 (1,2), MgCl2 (1,2), K2HPO4 (2,4), KHPO4 (0,6), N-2-гидроксиэтилпиперазин-N’-2-этансульфоновая кислота (ГЭПЭС) (10) и декстроза (10). pH этого раствора доводили до 7,4 титрованием NaOH.
Раствор с апикальной стороны (в мM): такой же, как для базолатеральной стороны, но NaCl заменяли глюконатом Na (135).
5. Культура клеток
Для исследования предполагаемых модуляторов F508del-CFTR, определенных при помощи оптических методов, применяли эксперименты с эпителиальными клетками крыс линии Фишер, экспрессирующими F508del-CFTR (FRTΔF508-CFTR), в камере Уссинга. Клетки культивировали во вкладках для культуры клеток «Costar Snapwell» в течение 5 суток при 37°C и 5% CO2 в среде Хэма F-12 в модификации Куна с 5% фетальной бычьей сыворотки, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Перед использованием для определения потенцирующей активности соединений клетки инкубировали при 27°C в течение 16-48 ч для поправки на F508del-CFTR. Для определения активности корректирующих соединений клетки инкубировали при 27°C или 37°C с соединениями или без, в течение 24 ч.
6. Запись данных токов при исследовании цельных клеток
Макроскопический ток F508del-CFTR (IΔF508) в клетках NIH3T3 со стабильной экспрессией F508del-CFTR после поправки на температуру и исследуемые соединения измеряли с помощью метода локальной фиксации потенциала с перфорацией участка мембраны на цельной клетке. Коротко, фиксацию потенциала IΔF508 проводили при комнатной температуре с помощью усилителя для фиксации потенциала «Axopatch 200B» (Axon Instruments Inc., Фостер Сити, Калифорния). При всех записях использовали частоту 10 кГц с фильтром нижних частот 1 кГц. Пипетки имели сопротивление 5-6 MΩΩ при заполнении внутриклеточным раствором. В этих условиях записи вычисленный потенциал реверсии для Cl- (ECl) при комнатной температуре составил -28 мВ. При всех записях сопротивление уплотнения составило > 20 гΩ, а добавочное сопротивление - < 15 MΩ. Для генерации импульсов, регистрации данных и анализа использовали компьютер с а/ц интерфейсом «Digidata 1320» в сочетании с «Clampex 8» (Axon Instruments Inc.). Баня содержала < 250 мкл физиологического раствора и в ней поддерживалась непрерывная перфузия со скоростью 2 мл/мин с помощью гравитационной перфузионной системы.
7. Идентификация корректирующих соединений
Для определения активности корректирующих соединений для повышения плотности функциональных F508del-CFTR в плазматической мембране мы использовали вышеописанные техники локальной фиксации потенциала для измерения плотности тока после 24-часового воздействия корректирующих соединений. Для полной активации F508del-CFTR в ячейки добавляли 10 мкM форсоколина и 20 мкM генистеина. В наших условиях записи плотность тока после 24-часовой инкубации при 27°C была выше, чем после 24-часовой инкубации при 37°C. Эти результаты согласуются с известным влиянием низкотемпературной инкубации на плотность F508del-CFTR в плазматической мембране. Для определения влияния корректирующих соединений на плотность тока CFTR клетки инкубировали с 10 мкM исследуемого соединения в течение 24 ч при 37°C и сравнивали плотность тока с плотностью тока контролей при 27°C и 37°C (% активность). Перед записью клетки промывали 3Х внеклеточной средой для записи потенциала с целью удаления остатков исследуемого соединения. Предварительная инкубация с 10 мкM корректирующих соединений значительно повышала цАМФ- и генистеин-зависимый ток по сравнению с контролями при 37°C.
8. Идентификация потенцирующих соединений
Способность потенциаторов F508del-CFTR повышать макроскопический ток F508del-CFTR Cl- (IΔF508) в клетках NIH3T3 со стабильной экспрессией F508del-CFTR также исследовали с помощью техники локальной фиксации потенциала с помощью перфорированного фрагмента мембраны. Потенциаторы, выявленные оптическими методами, вызывали дозо-зависимое повышение IΔF508 с силой и эффективностью, сходными с наблюдаемыми в оптических методах. В исследованных клетках потенциал реверсии до и после внесения потенциатора составил около -30 мВ, что соответствует вычисленному ECl (-28 мВ).
9. Растворы
Внутриклеточный раствор (в мM): Cs-аспартат (90), CsCl (50), MgCl2 (1), ГЭПЭС (10) и 240 мкг/мл амфотерцина-B (pH доводили до 7,35 с помощью CsOH).
Внеклеточный раствор (в мM): N-метил-D-глюкамин (NMDG)-Cl (150), MgCl2 (2), CaCl2 (2), ГЭПЭС (10) (pH доводили до 7,35 с помощью HCl).
10. Культура клеток
Для записи потенциала цельных клеток использовали мышиные фибробласты NIH3T3 со стабильной экспрессией F508del-CFTR. Клетки инкубируют при 37°C в атмосфере с 5% CO2 и при относительной влажности 90% в среде Игла, модифицированной Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1Х добавки аминокислот NEAA, β-ME, 1Х пенициллина/стрептомицина и 25 мM ГЭПЭС в культуральных колбах 175 см2. Для записи потенциалов цельной клетки 2500-5000 клеток осаждали на стеклянных покровных стеклах, покрытых поли-L-лизином, культивировали 24-48 ч при 27°C перед использованием для проверки активности потенциаторов, и инкубировали с корректирующим соединением или без, при 37°C для измерения активности корректирующих соединений.
11. Запись потенциалов в одном канале
Активность одиночного канала F508del-CFTR, стабильно экспрессирующегося в клетках NIH3T3, с поправкой на температуру, а также активности потенциаторов определяли методом фиксации потенциала отделенного фрагмента мембраны, перевернутого внутренней стороной наружу. Коротко, запись активности одного канала путем локальной фиксации потенциала проводили при комнатной температуре с помощью усилителя «Axopatch 200B» для локальной фиксации потенциала (Axon Instruments Inc.). При всех записях использовали частоту 10 кГц с фильтром нижних частот 400 Гц. Пипетки для фиксации потенциала были изготовлены из кальций-борофосфатного стекла «Kovar Sealing» №7052 (World Precision Instruments, Inc., Сарасота, Флорида) и имели сопротивление 5-8 MΩ при заполнении внеклеточным раствором. F508del-CFTR активировали после отделения фрагмента мембраны добавлением 1 мM Mg-АТФ и 75 нM цАМФ-зависимой протеинкиназы, каталитическая субъединица (PKA; Promega Corp. Мэдисон, Висконсин). После стабилизации активности канала поддерживали перфузию фрагмента с помощью гравитационной микроперфузионной системы. Трубка для притока жидкости располагалась рядом с фрагментом мембраны, таким образом, полный обмен раствора происходил за 1-2 секунды. Для сохранения активности F508del-CFTR во время быстрой перфузии в омывающий раствор добавляли неспецифический ингибитор фосфатазы F- (10 мM NaF). При этих условиях записи активность канала оставалась постоянной на протяжении всего процесса записи (до 60 минут). Токи, создаваемые перемещением положительно заряженных частиц из внутриклеточного раствора во внеклеточный (анионы перемещались в противоположном направлении), показаны как положительные токи. Потенциал пипетки (Vp) поддерживали на уровне 80 мВ.
Активность канала анализировали с помощью фрагментов мембраны, содержащих ≤2 активных каналов. Максимальное число отверстий одновременно определяло число активных каналов в ходе эксперимента. Для определения амплитуды тока одного канала данные, записанные со 120 секунды активности F508del-CFTR, фильтровали "в автономном режиме" при 100 Гц, а затем использовали для построения диаграмм амплитуды по всем точкам, которые затем приводили в соответствие многомерным Гауссовым функциям с помощью программы Bio-Patch Analysis (Bio-Logic Comp. Франция). Полный микроскопический ток и вероятность открытого состояния канала (Po) определяли после 120 секунд активности канала. Po определяли с помощью программного обеспечения Bio-Patch или по соотношению Po = I/i(N), где I = средний ток, i = амплитуда тока одного канала и N = число активных каналов во фрагменте мембраны.
12. Растворы
Внеклеточный раствор (в мM): NMDG (150), аспарагиновая кислота (150), CaCl2 (5), MgCl2 (2) и ГЭПЭС (10) (pH доводили до 7,35 с помощью трис-основания).
Внутриклеточный раствор (в мM): NMDG-Cl (150), MgCl2 (2), ЭГТА (5), ТЭС (10) и трис-основание (14) (pH доводили до 7,35 с помощью HCl).
13. Культура клеток
Для локальной фиксации потенциала фрагмента мембраны использовали мышиные фибробласты NIH3T3 со стабильной экспрессией F508del-CFTR. Клетки инкубируют при 37°C в атмосфере с 5% CO2 и при относительной влажности 90% в среде Игла, модифицированной Дульбекко с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1Х добавки аминокислот NEAA, β-ME, 1Х пенициллина/стрептомицина и 25 мM ГЭПЭС в культуральных колбах 175 см2. Для записи потенциалов одного канала 2500-5000 клеток осаждали на стеклянных покровных стеклах, покрытых поли-L-лизином, и культивировали 24-48 ч при 27°C перед использованием в эксперименте.
Была измерена активность, т.е. ЭК50 Соединения 1, которая показана в таблице 22.
Таблица 22.
|
ПРОЧИЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Все публикации и патенты, на которые ссылается эта заявка, включены в нее посредством ссылки в той же степени, как если бы каждая отдельная публикация или патентная заявка была специально указана как включенная посредством ссылки. Если значение терминов в каких-либо патентах или публикациях, на которые приводится ссылка, противоречат значению терминов в настоящей заявке, значение терминов в настоящей заявке имеет преимущественную силу. Кроме того, в вышеприведенном обсуждении раскрываются и описываются просто примерные варианты осуществления изобретения. Специалисту в этой области будет понятно из описания и сопровождающих его фигур и пунктов, что в изобретение могут вноситься различные изменения, модификации и вариации без отклонения от сущности и объема изобретения, как описано в следующих пунктах формулы изобретения.
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/938359d05abc69cdc094d1364b4476f3.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/c97a52f889f00f442938645e5dd46d47.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/92d7a4e5e29bfcaf389a0c1576014aa5.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/7275f3098d6b0b8de6adfa06cfdc6297.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/e72e6b36cae94c7518a37df8052a2351.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/4dbe7671753679b0be1f53792d5575d7.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/7145fa6afdcafd7a08976e05e53a52ab.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/9f1e16e97e928c0e096d00929a62af05.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/8ebc742955ca0278f2b2f39fe30d5a35.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/65128f74c892e4c5955f46dbcca50bc7.jpg)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/2cff49722dc484488c788bf424b5087b.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/b0c8378a4f7af3b1fd1b1135e2ca02a7.jpg)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/d87fce7042c0cb5ccc08f48e20e7f711.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/145cb86f65e6743d540eb176ece95fb1.jpg)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/01a4c68c9f356e8ff59995f51f3a27d3.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/54110d85ab383e94e32ed663966b70aa.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/8bcaf49d20ef7e54d6e7c4046b7b76b2.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/8f8fe89a216881a68f60071acb37817e.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/78b33ee246b280d50ecd6535be090853.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/2894a0fdac6fc8ade6e1e2685b455bf9.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/a3396100645fdc185b7f432d5409f1f7.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/8ec5ccfee5795809c8b72ba55142178d.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/6afbf06c6658b023a7ed931e4affc987.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/0761201117ec21f9b72112836724776e.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/fd9ed5ee15f73f960f5c1f49a058f8e5.jpg)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/76ec2f76aeb76bc2a14694535382f271.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/3bfdbc92ca5ba8f5c2c42dc7dbd6a88b.png)
![ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/ae/81/d5/22106bb43827869fbc1812665cb82f7f.png)
Дигидродиазепины, которые можно использовать в качестве ингибиторов протеинкиназ
Способы и промежуточные соединения для получения стерических соединений
Модуляторы атф-связывающих кассетных транспортеров
Твердые формы n-(7-азабицикло[2.2.1]гептан-7-ил-)-2-(трифторметил)фенил)-4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоксамида
Производные 3-карбоксамида-4-оксохинолина, полезные в качестве модуляторов регулятора трансмембранной проводимости кистозного фиброза
Способ модуляции транспортеров атф-связывающей кассеты
Модуляторы транспортеров атф-связывающей кассеты
Способы и промежуточные соединения
Способ получения азабициклических соединений
Способ получения модуляторов регулятора трансмембранной проводимости кистозного фиброза
Дигидродиазепины, которые можно использовать в качестве ингибиторов протеинкиназ
Способы и промежуточные соединения для получения стерических соединений
Модуляторы атф-связывающих кассетных транспортеров
Твердые формы n-(7-азабицикло[2.2.1]гептан-7-ил-)-2-(трифторметил)фенил)-4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоксамида
Производные 3-карбоксамида-4-оксохинолина, полезные в качестве модуляторов регулятора трансмембранной проводимости кистозного фиброза
Способ модуляции транспортеров атф-связывающей кассеты
Модуляторы транспортеров атф-связывающей кассеты
Способы и промежуточные соединения
Способ получения азабициклических соединений
Способ получения модуляторов регулятора трансмембранной проводимости кистозного фиброза

