Результат интеллектуальной деятельности: КОМПОЗИЦИИ АНТАГОНИСТОВ НЕЙРОКИНИНА-1 ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтическим композициям антагониста нейрокинина-1 и их пролекарствам для внутривенного введения, получению фармацевтических композиций и их применению.
УРОВЕНЬ ТЕХНИКИ
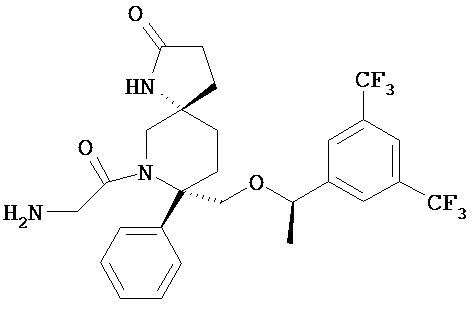
(5S,8S)-8-[{(1R)-1-(3,5-Бис-(трифторметил)фенил)-этокси}-метил]-8-фенил-1,7-диазаспиро[4.5]декан-2-он (соединение, представленное формулой I; также упоминается в настоящем изобретении как Соединение 1) и его соли были описаны в патенте США №7,049,320 (патент ’320), выданном 23 мая 2006 года. Процесс синтеза Соединения 1 подробно описывается в Примере 72а патента ’320 (см. в патенте ’320 колонка 43, со строки 55 до колонки 45, строка 20; колонка 75, со строки 55 колонки 80, строка 21; колонка 90 со строки 35 до строки 63; и колонка 98, строка 1 до колонки 99, строка 24, который включен сюда посредством ссылки. Смотри также WO 2008/11833, Примеры 1-6, которые включены сюда посредством ссылки). WO 2005/063243 раскрывает некоторые фармацевтические композиции, включающие антагонисты NK-1. Композиции, описанные в нем, требуют полианионное производное бета-циклодекстрина, содержащее примерно от одной до семи натрий сульфонатных групп, отделенных от липо-фильной полости по меньшей мере одной спейсерной бутокси группой, т.е. Captisol®. В упомянутом патенте отсутствует ссылка на внутривенные композиции, которые минимизируют гемолиз. Кроме того, различные солевые формы соединения, представленного формулой I, были описаны, например, в опубликованном патенте США 2007/0244142, который также включен посредством ссылки.
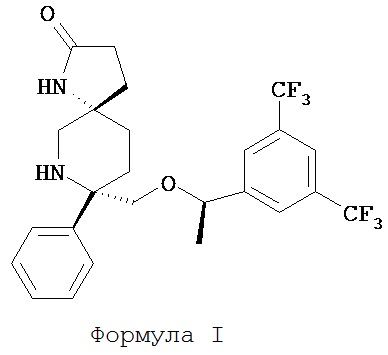
Соединение формулы I классифицируется как соединение тахикинина и является антагонистом рецепторов нейропептида нейрокинина-1 (NK-1). Соединение формулы I может быть в форме свободного основания или в форме фармацевтически приемлемой соли. Свободное основание или соль могут быть в аморфной форме, или фармацевтически приемлемой соли, используемой в настоящем изобретении, может быть в кристаллической форме или в форме кристаллогидрата или сольватной форме. В растворе и в зависимости от pH этого раствора, соединение формулы I может быть в форме смеси свободного амина/соли. Пролекарства соединения формулы I также могут быть использованы в композициях, подходящих для парентерального введения. Пролекарства, в которых либо свободный амин (либо оба амина) в соединении формулы I имеют водород, замещенный группой, выбранной из Y и его солей, где Y выбирается из -Р(O)(ОН)2, -S(O)n1R1, -С(O)(С1-6алкил)Х, -С(O)(С1-6алкил)(арил), -C(O)OR4; X выбирается из NR2R3, -Р(O)(ОН)2 или -S(O)n1R1; R1 является Н или С1-6алкила; R2 является Н или С1-6алкила; R3 является Н или С1-6алкила; R4 является Н или C1-6алкила; значение n1 равное 0-4 подходит для применения согласно настоящему изобретению. Подходящие катионы или дикатионы для ионизированной формы (форм) пролекарств включают соли металлов или катионы органических аминов, в том числе соли меглумина и подобные (N-метил-D-глюкамин). Такие пролекарства могут применяться вместе с описанными парентеральными носителями или без них в подходящей жидкой композиции для лечения пациентов, нуждающихся в таком лечении. Такие пролекарства после парентерального введения пациенту превращаются в непролекарственную форму лекарственного средства (или в его соль). Такие пролекарства могут быть в аморфной форме или в кристаллической форме и/или в форме кристаллосольватов/гидратов.
Было показано, что антагонисты рецепторов NK-1 могут быть полезными терапевтическими средствами, например, при лечении боли, воспаления, мигрени, тошноты (рвоты) и ноцицепции
Сущность изобретения
Настоящее изобретение в широком плане относится к композициям, пригодным для внутривенного введения пациенту, нуждающемся в таком лечении, где названные композиции включают соединение Формулы I и его фармацевтически приемлемые соли, гидраты, сольваты, а также носитель, выбранный из группы, состоящей из водорастворимых органических растворителей, неионных поверхностно-активных веществ, нерастворимых в воде липидов, органических липидов/полутвердых веществ и фосфолипи-дов. Растворимые в воде органические растворители могут быть выбраны из, например, полиэтиленгликоля 300, полиэтиленгликоля 400, этанола, пропиленгликоля, глицерина, N-метил-2-пирролидона, диметилацетамида и диметилсульфоксида. Неионные поверхностно-активные вещества могут быть выбраны из Кремофора EL, Кремофора RH 40, Кремофора RH 60, сукцината d-a-токоферол полиэтиленгликоля 1000, полисорбита 80, солю-тола HS 15, моноолеата сорбитана, полоксамера 407, Labrifil M-1944CS, Labrafil M-2125CS, лабразола, Gellucire 44/14, Softigen 767 и моно- и ди-эфиров жирных кислот с ПЭГ 300, 400 или 1750. Нерастворимые в воде липиды выбираются из касторового масла, кукурузного масла, хлопкового масла, оливкового масла, арахисового масла, масла мяты перечной, подсолнечного масла, кунжутного масла, соевого масла, гидрогенизированного растительного масла, гидрогенизированного соевого масла, среднеце-почечных триглицеридов из кокосового и пальмового масла. Органические жидкости и полутвердые вещества могут быть выбраны из пчелиного воска, D-α-токоферола, олеиновой кислоты и среднецепочечных моно- и диг-лицеридов. Фосфолипиды выбираются из лецитина, гидрогенизированного соевого фосфатидилхолина, дистеароилфосфатидилглицерина, L-α-димиристоилфосфатидилглицерина и L-α-димиристоилфосфатидилгли-церина и других, как описано в настоящем изобретении. Композиции изготавливаются для обеспечения достаточной растворимости и химической стабильности, которая определяется как <5-10% деградации в течение одного года (предпочтительно за два года) при конкретных условиях хранения, которые изменяются в зависимости от конкретной композиции, места хранения и т.д. Предпочтительно, композиции применяются в качестве внутривенных композиций. При необходимости или по предписанию, эти композиции могут иметь широкое назначение как парентеральные композиции, приемлемые для доставки известными специалистам в данной области техники методами, включая внутривенное (ВВ), внутримышечное (ВМ) или подкожное (ПК) введение. Пролекарства могут быть использованы в пероральной форме или в парентеральных композициях, содержащих водные/солевые системы доставки вместе или без дополнительных носителей для доставки, как изложено выше.
Настоящее изобретение описывает и заявляет, в частности, фармацевтические композиции и композиции Соединения 1 и его фармацевтически приемлемых солей для применения в целях лечения тошноты и/или рвоты. Предпочтительной формой Соединения 1, используемой для получения композиций, описываемых в настоящем изобретении, включая внутривенные композиции, является кристаллическая соль моногидрат гидрохлорид. Разработка заявляемых композиций потребовала существенного экспериментирования и усилий, как описано ниже и в Примерах, чтобы преодолеть проблему низкой растворимости лекарственного средства и, в частности, проблему гемолиза (изменение, растворение или разрушение красных кровяных телец приводит к утечке крови в мочу), предположительно вызываемого временными локальными свободными концентрациями некоторых форм Соединения 1 при его внутривенном введении экспериментальным животным в виде болюса. Гемолиз крови не происходит при введении обычных перпероральных форм Соединения 1. Гемолиз в большинстве случаев также не возникает при пероральном или очень медленном введении животным, но имел место в ходе либо медленной инфузии или и/или болюсного введения с некоторыми композициями.
Трудности, первоначально возникшие при разработке композиции Соединения 1 для внутривенного введения, заключались в том, что Соединение 1 имеет низкую растворимость при физиологических значениях pH 7,4 (<4 мкг/мл) и, следовательно, требуется повышение растворимости для достижения терапевтического уровня концентрации в плазме крови в организме и при ожидаемой дозе в 100 мг. Кроме того, было желательно увеличить концентрацию лекарственного средства и дозу в композиции, так как это будет способствовать сокращению объема инфузии, вводимой пациентам.
Для решения проблемы низкой растворимости Соединения 1 при физиологических значениях pH были проведены исследования, чтобы установить, какие системы растворителей позволят повысить растворимость Соединения 1. Captisol® (производное β-циклодекстрина, также упоминаемое в настоящем изобретении как каптизол) и композиции на основе сорастворителей, содержащих пропиленгликоль и этанол, существенно улучшили растворимость Соединения 1. Однако композиции на основе сорастворителей при внутривенном введении неожиданно вызвали гемолиз. Дальнейшие попытки уменьшить/свести к минимуму возникновение гемо-лиза, наблюдаемого при внутривенном введении композиций Соединения 1 на основе каптизола, посредством варьирования концентрации каптизола, объема, скорости введения или добавления буферов, или использования различных комбинаций этанола, пропиленгликоля и полиэтиленгликоля 400, были успешны в отдельных случаях. Например, композиция на основе Каптизола, введенная путем инфузии в течение 15-минутного периода крысам в дозах 10 мг/кг и 5 мг/кг и в объемной дозе 10 мл/кг (концентрационная доза 1 мг/мл до 0,5 мг/мл, соответственно), дала низкое число случаев гемолиза (1/5). Кроме того, болюсное введение композиции на основе Каптизола в дозе 10 мг/кг и в объемной дозе 5 мл/кг (дозовая концентрация 2 мг/мл), вызвала гемолиз у 2/5 от числа крыс. Болюсное введение Каптизола при более низкой концентрации (например, по сравнению с ин-фузионными концентрациями 1 мг/мл и 0,5 мг/мл), вероятно, может привести к еще меньшему проявлению гемолиза. Было сделано предположение, что временная локальная высокая концентрация свободного Соединения 1 в месте инфузии может быть основной причиной гемолиза.
Для того чтобы проверить вышеупомянутую гипотезу, исследования проводились на крысах путем внутривенного введения (медленное ручное болюсное введение, 1-2 минуты) мицеллярной композиции, содержащей макрогол 15-гидроксистеарат (Solutol® HS15, также упоминаемый в настоящем изобретении как солютол) (10 мг/мл лекарственного средства, 22% Solutol HS15, 20 мМ Фосфатного буфера, pH 7.0) и тестированием возникновения гемолиза в различные промежутки времени и с разными дозами (10 мг/кг, 20 мг/кг и 30 мг/кг) после введения лекарственного средства. Наблюдали, что возникновение гемолиза у крыс существенно сокращалось в период 30-60 минут после введения лекарственного средства по сравнению с 15 минутным периодом после введения. Этот результат, в комбинации с тем фактом, что Соединение 1 обладает высокой пероральной доступностью, привело к выводу, что высокая локальная концентрация свободного Соединения 1 была ответственна за временный гемолиз, который происходил в течение первых 30 минут после внутривенного введения.
Чтобы свести к минимуму/уменьшить гемолиз, связанный с Соединением 1, была применена стратегия, которая базировалась на предположении, что медленная скорость введения лекарственного средства может привести к снижению гемолиза. Это включало эксперименты по введению мицеллярных (7,5% солютола, 25 мг/кг, 5 мл/кг, 5 мг/мл) растворов Соединения 1 крысам путем 15 минутной медленной инфузии, а также болюсный метод введения. Высокий уровень возникновения гемолиза наблюдался у крыс, когда мицеллярная композиция вводилась с помощью болюсного метода (5/5), в то время как гемолиз не наблюдался при инфузии (0/10).
Таким образом, снижение концентрации солютола в мицеллярной композиции Соединения 1 с 22%, например до 7,5%, и введение композиции медленной инфузией крысам снижает возникновение гемолиза. Однако, при болюсном методе введения композиции на основе 7,5% солютола (например, низкая концентрация солютола), гемолиз наблюдался, как указано выше. Таким образом, некоторые из заявленных композиций, перечисленных в настоящем изобретении, подходят для медленной инфузии, но не для болюсного метода введения. Другие композиции, как далее описано в настоящем изобретении, подходят как для медленной инфузии, так и для болюсного введения.
Дальнейшее экспериментирование на композиции на основе солютола было проведено для оценки влияния добавления различных типов масел, процента включения масла, а также диапазона pH композиций на основе солютола на возникновение гемолиза у крыс. Считалось, что добавление масла в мицеллярный раствор для формирования микроэмульсий, т.е. загруженных маслом мицелл, должно дополнительно способствовать замедлению высвобождения Соединения 1 из гидрофобного кора и предотвращать быстрое распределение/перенос Соединения 1 в красные кровяные тельца.
Настоящее изобретение относится к парентеральным композициям, включающим, а) соединение формулы I или его фармацевтически приемлемую соль,
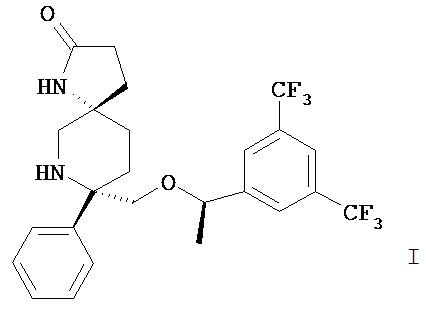
и b) фармацевтически приемлемый носитель.
Термин "фармацевтически приемлемый носитель" означает любой подходящий компонент, который повышает растворимость соединения формулы 1 или его фармацевтически приемлемой соли, чтобы способствовать парентеральной доставке терапевтической концентрации этого соединения или соли в целевой сайт(ы) рецептора NK-1. Носители выбираются из группы, состоящей из кремофоров, эмульсий, микроэмульсий, мицелл, отрицательно заряженных мицелл, загруженных маслом мицелл, интралипидов, ЧСА, липосом и отрицательно и положительно заряженных аминокислот и т.п., как дополнительно описано в настоящем изобретении. Фармацевтически приемлемые носители не включают в себя композиции с β-циклодекстрином. В случае липосом, эмульсий, мицелл и загруженных маслом мицелл, считается, что такие носители должны сохранять лекарственное средство внутри липофильного кора для усиления удерживания и, в то же время, экранировать лекарственное средство в коре. Композиции на основе сывороточного альбумина человека относятся к прочному связыванию ЧСА с Соединением 1 и могли бы свести к минимуму распределение свободного лекарственного средства в красных кровяных тельцах. Такие композиции могут быть сформированы совместно с солютолом, миглиолом и витамином Е. Отрицательно заряженные аминокислоты могут образовывать комплекс и нейтрализовать часть Соединения 1, что сохраняет положительный заряд и тем самым предотвращает распределение Соединения 1 в красные кровяные тельца. Положительно заряженные аминокислоты должны образовывать комплекс с отрицательно заряженной частью Соединения 1 и нейтрализовать его, и уменьшить воздействие соединения на красные кровяные тельца. Отрицательно заряженные мицеллы будут отталкивать отрицательно заряженные эритроциты и предотвращать контакт Соединения 1 с красными кровяными тельцами.
Термин "мицеллярная композиция" означает, что композиция существует в виде мицеллы и является производной или изготовлена из любого компонента, который образует или способен образовать мицеллы в фармацевтически приемлемой системе доставки, такой как вода, физиологический раствор, водный раствор глюкозы и т.п.
Термин "эмульсионная композиция" означает, что композиция существует в виде эмульсии и является производной или изготовлена из любого компонента, который образует или может образовать эмульсию, когда представлена в и/или комбинируются с фармацевтически приемлемой системой доставки, такой как вода, физиологический раствор, водный раствор сахарозы и т.п. Предпочтительные эмульсионные композиции, которые предотвращают любые гемолитические эффекты при болюсном или медленном инфузионным введении, имеют содержание масла около 10% или менее. Концентрация лекарственного средства, варьируемая от около 1 мг/мл до 30 мг/мл, с меньшим объемом и более высокой концентрацией является предпочтительной для внутривенного введения. Фармацевтические композиции могут быть приготовлены так, чтобы увеличить или повысить растворимость антагониста NK-1, а также могут быть существенно разбавленными, чтобы избежать любых возможных гемолитических последствий, однако некоторые объемы разбавления могут оказаться непригодными для введения пациентам, нуждающихся в таком лечении.
Эмульсионная композиция или мицеллярная композиция по настоящему изобретению дополнительно содержит активный фармацевтический ингредиент, выбранный из соединения формулы I или Ia и/или их фармацевтически приемлемых солей, гидратов, полиморфных или физических форм. Такие эмульсионные или мицеллярные композиции, загруженные лекарственное средством, могут дополнительно содержать эксипиенты, которые облегчают доставку и/или полезны для предотвращения или смягчения последствий таких факторов, как гемолиз. Такие дополнительные эксипиенты, могут, таким образом, включать, например, масла или другие компоненты, которые повышают или дополнительно повышают растворимость при смягчении любых потенциальных гемолитических эффектов.
Такие эмульсионные композиции или мицеллярные композиции дополнительно могут быть процессированы для создания более стабильных физических форм или растворов и могут быть дополнительно процессированы, например, для обеспечения стерилизованных парентеральных растворов.
Настоящее изобретение также относится к парентеральным композициям, содержащим соединение формулы 1 или его фармацевтически приемлемую соль в форме наночастицы. Наночастицы соединения формулы 1 или его соли могут быть затем включены в раствор для доставки такой наночастицы внутривенным путем. Наночастицы Соединения 1 и его фармацевтически приемлемой соли могут дополнительно включать фармацевтически приемлемый носитель. Считается, что медленное растворение таких наночастиц (~200 нм), может привести к меньшему гемолизу вследствие уменьшения растворимости лекарственного средства в солюбилизированной фракции.
Настоящее изобретение также относится к способу доставки в организм пациента соединения формулы I и его фармацевтически приемлемой соли, содержащему (а) объединение соединения формулы I или его фармацевтически приемлемой соли с фармацевтически приемлемым носителем для формирования композиции для внутривенного введения, (b) доставку парентеральной композиции в организм пациента, нуждающегося в таком лечении.
После испытаний и значительных усилий изобретателей разрешить вышеупомянутые проблемы, связанные с солютол-содержащими композициями, было обнаружено, что добавление как среднецепочечного, так и длинно-цепочечного масла в определенном соотношении к солютолу дает солюбилизированную, стабильную (химически и физически) микроэмульсионную композицию Соединения 1, которая при внутривенном введении экспериментальным животным показывает минимальный гемолиз. Другой объект настоящего изобретения относится к фармацевтической композиции для парентерального введения, содержащей:
а) соединение формулы I или его фармацевтически приемлемую соль
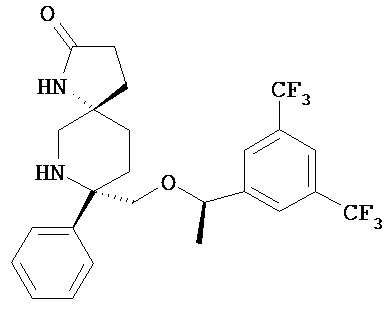
и
b) солюбилизатор, выбранный из группы, состоящей из мицеллы, загруженной маслом, или микроэмульсии.
Предпочтительный вариант осуществления изобретения включает фармацевтическую внутривенную композицию, которая включает:
а) соединение формулы I или его фармацевтически приемлемую соль
и b) эмульгатор.
Настоящее изобретение относится к фармацевтической композиции для внутривенного введения, содержащей соединение формулы 1 или его фармацевтически приемлемую соль и сывороточный альбумин человека (ЧСА).
Настоящее изобретение также относится к композиции для внутривенного введения, содержащей соединение формулы 1 или его фармацевтически приемлемую соль, где соединение или его соль находится в виде наночастицы.
Изобретение дополнительно относится к внутривенной композиции, содержащей соединение формулы 1 или его фармацевтически приемлемую соль и носитель для доставки, выбранный из кремофора.
Кроме того, изобретение относится к внутривенной композиции, содержащей соединение формулы 1 или его фармацевтически приемлемую соль и носитель для доставки, выбранный из мицеллы.
Кроме того, изобретение относится к внутривенной композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль и носитель для доставки, выбранный из липосомы.
Изобретение предпочтительно относится к внутривенной эмульсионной композиции, которая подходит как для болюсного, так и для инфузионного введения.
Предпочтительный вариант осуществления изобретения содержит внутривенную композицию, содержащую соединение формулы I или его фармацевтически приемлемую соль и по меньшей мере один эмульгатор, в котором формируется эмульсия, подлежащая микрофлюидизации для формирования капель, имеющих средний диаметр менее 500 нм, средний диаметр и/или D90 около 600 нм или менее.
Изобретение дополнительно относится к внутривенной композиции, содержащей соединение формулы 1 или его фармацевтически приемлемую соль и отрицательно или положительно заряженную аминокислоту.
Изобретение дополнительно относится к внутривенной композиции, содержащей соединение формулы 1 или его фармацевтически приемлемую соль, которые лиофилизируются.
Изобретение дополнительно относится к внутривенной композиции, содержащей соединение формулы или его фармацевтически приемлемую соль в виде порошка. Порошок растворяется или добавляется к жидкости с образованием жидкой внутривенной композиции, содержащей соединение формулы 1 или его соль, которую вводят пациенту, нуждающемуся в таком лечении. Эмульгаторы, такие как полисорбат 80 (твин 80) и т.п., могут быть добавлены к этой композиции, также как и другие неактивные ингредиенты, такие как регуляторы pH, консерванты (ЭДТА) и т.д.
В каждом из вышеперечисленных примеров осуществления, предпочтительной формой соединения формулы I или его соли, добавляемой в композицию, является форма твердого кристаллического моногидрата гидрохлорида.
В каждом из вышеперечисленных примеров осуществления, альтернативная форма соединения формулы 1 или его соли выбирается из пролекарства соединения формулы I. Такие пролекарства могут вводиться любым методом доставки, включая пероральный путь или внутривенное введение.
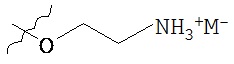
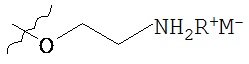
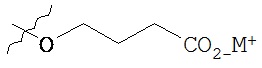
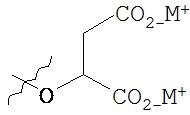
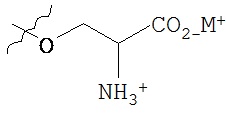
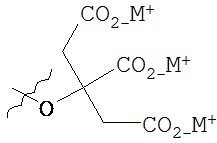
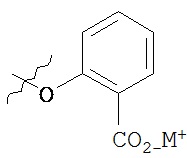
Такие пролекарства могут быть выбраны из соединения формулы 1а и его фармацевтически приемлемых солей:
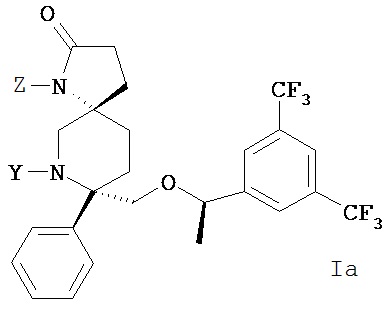
где Z и Y независимо выбираются из группы, состоящей из Н, -РО(ОН)O-M+, -РО(O+)22М+, -PO(O-)2D2+, -[C(R1)(R2)]n-PO(OH)O-M+, -[C(R1)(R2)]n-PO(O-)22M+, -[C(R1)(R2)n-PO(O-)2D2+, -C(O)[C(R1)(R2)]m-OPO(O-)22M+, -C(O)[C(R1)(R2)]oNR1R2, -C(O)[C(R1)(R2)]pCO2-M+, -SO3-М+, -[C(R1)(R2)]qSO3-M+ и -[C(R1)(R2)]rOC(O)OR3, где R3 выбирается из группы
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ;
;
при условии, что Z и Y не могут оба представлять собой Н; М+ выбирается из одновалентного катиона; D+ выбирается из двухвалентных катионов; R1 и R2 независимо выбирают из Н или С1-6алкила, n равно 1-4, m, о и p независимо выбираются из 0-4, и R выбран из C1-6алкила
В предпочтительном варианте осуществления пролекарства, Z выбирается из Н, и Y выбирается из любой одной из вышеуказанных групп для Z и Y, исключая Н. В более предпочтительном варианте выполнения настоящего изобретения такие пролекарства выбираются из:
и
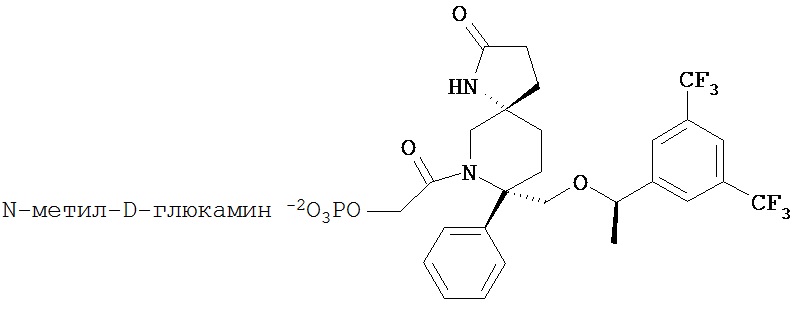
Предпочтительные M+ соли выбирается из, например, солей аммония, солей щелочных металлов, таких как натрий, солей щелочноземельных металлов, таких как кальций и магний, солей органических оснований, таких как N-метил-D-глюкамин или дициклогексиламин, солей аминокислот, таких как аргинин, лизин и т.п.
Пролекарства получают при взаимодействии амина или надлежащим образом защищенного амина с активированной группой Z-X или Y-X, или любым обычным методом формирования варианта Пролекарства соединения формулы I.
Пролекарства по настоящему изобретению обладают улучшенной растворимостью по сравнению с родоначальным лекарственным средством и, таким образом, полезны и удобны для внутривенного введения.
Предпочтительным вариантом осуществления изобретения является фармацевтическая композиция, которая включает:
а) соединение формулы I
или его фармацевтически приемлемую соль;
b) макрогол 15-гидроксистеарат в количестве от 0,50% до 10,0 мас.% от всей композиции;
c) среднецепочечный триглицерид в количестве от 0,10% до 2,5 мас.% от всей композиции;
d) длинноцепочечный триглицерид в количестве от 0,10% до 1,5 мас.% от всей композиции, а также
e) по меньшей мере один буфер, где массовое отношение макрогол 15-гидроксистеарат: среднецепочечный триглицерид: длинноцепочечный триглицерид в композиции составляет около 5-100:1-5:1, и где pH композиции составляет от 6,5 до 8,0.
Приведенная выше композиция может быть в виде загруженной маслом мицеллы или микроэмульсии.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит макрогол 15-гидроксистеарат в количестве от 0,50% до 7,5 мас.% от всей композиции; среднецепочечный триглицерид в количестве от 0,15% до 1,5 мас.% от всей композиции и длинноцепочечный триглицерид в количестве от 0,10% до 1,2 мас.% от всей композиции.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая включает макрогол 15-гидроксистеарат в количестве от 0,88% до 4,84 мас.% от всей композиции, среднецепочечный триглицерид в количестве от 0,20% до 1,20 мас.% от всей композиции, и длинноцепочечный триглицерид в количестве от 0,10% до 0,75 мас.% от всей композиции.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
(а)соединение формулы I
или его фармацевтически приемлемую соль;
(b) макрогол 15-гидроксистеарат в количестве около 4,4 мас.% от всей композиции;
(c) по меньшей мере один среднецепочечный триглицерид в количестве около 1,1 мас.% от всей композиции;
(d) рафинированное соевое масло в количестве около 0,66 мас.% от всей композиции, а также
(e) фосфатный буфер, в котором рН композиции составляет около 7,5.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
(а)соединение формулы 1
или его фармацевтически приемлемую соль;
(b) макрогол 15-гидроксистеарат в количестве около 0,88 мас.% от всей композиции;
(c) по меньшей мере один среднецепочечный триглицерид в количестве около 0,22 мас.% от всей композиции;
(d) рафинированное масло сои в количестве около 0,12 мас.% от всей композиции, а также (е) фосфатный буфер, где рН композиции составляет около 7,5.
Другим объектом настоящего изобретения является способ получения фармацевтической композиции, которая содержит:
a) нагревание (i) расплавленного макрогол 15-гидроксистеарата, (ii) среднецепочечного триглицерида, (iii) длинноцепочечного триглицерида с образованием композиции;
b) добавление воды в композицию с образованием микроэмульсионной композиции;
c) добавление в микроэмульсионную композицию соединения формулы I
или его фармацевтически приемлемой соли и
d) добавление по меньшей мере одного буфера и доведение pH от около 6,5 до 8,0, и формирование фармацевтической композиции, в которой макрогол 15-гидроксистеарат присутствует в количестве от около 0,50% до около 10,0% от общей массы фармацевтической композиции, среднецепочечный триглицерид присутствует в количестве от около 0,10% до около 2,5% от общей массы фармацевтической композиции, и длинноцепочечный триглицерид присутствует в количестве от около 0,10% до около 1,5 мас.% от общей массы фармацевтической композиции, и где массовое отношение макрогол 15-гидроксистеарат: среднецепочечный триглицерид: длинноцепочечный триглицерид в фармацевтической композиции составляет около 5-100:1-5:1.
Другим объектом настоящего изобретения является фармацевтическая композиция, которая содержит:
а) соединение формулы
или его фармацевтически приемлемую соль и
b) пегилированный гидроксистеарат в количестве от около 0,88% до около 5,0 мас.% от общей массы композиции, где пегилированный гидроксистеарат в основном свободен от полиэтиленгликоля, и где pH композиции составляет от около 6,5 до около 8.
Другим объектом настоящего изобретения является способ лечения тошноты и/или рвоты у пациентов, нуждающихся в таком лечении, который содержит инфузионное внутривенное введение пациенту эффективного количества фармацевтической композиции по настоящему изобретению и в котором гемолиз у пациента сводится к минимуму.
Другим объектом настоящего изобретения является способ минимизации гемолиза у пациентов путем внутривенного введения соединения формулы 1 или его фармацевтически приемлемой соли,
Причем способ содержит внутривенное введение с помощью инфузии пациенту эффективного количества фармацевтической композиции по настоящему изобретению.
В другом варианте осуществления настоящего изобретения перечисленные внутривенные композиции могут быть использованы в комбинации с другими лекарственными средствами против рвоты и тошноты; с противовоспалительными или стероидными агентами (например, дексаметазоном) и с химиотерапевтическими агентами. Перечисленные внутривенные композиции могут назначаться пациенту в соответствии с предписанием и режимом, прописанным врачом. Другие такие композиции включают ондан-сетрон и другие известные 5НТ3-антагонисты. Таким образом, соединение формулы 1 и его соли могут быть использованы для инфузии вместе с другими противорвотными агентами для профилактики острой и отсроченной тошноты и рвоты, связанных с начальными и повторными курсами очень высоко вызывающей рвоту химиотерапии рака, включая, например, лечение цисплатином. Соединение формулы 1 и их соли для инфузии могут быть использованы с другими противорвотными агентами для профилактики острой и отсроченной тошноты и рвоты, связанных с начальным и повторным курсом умеренно вызывающей рвоту химиотерапии рака. В дополнение к лечению цисплатином другие противоопухолевые агенты, которые вводятся в этой комбинации в дозированном режиме, включают этопозид, фторурацил, гемцитабин, винорельбин, паклитаксел, циклофосфамид, доксорубицин, доцетаксел и также могут включать темозоломид.
Лечение соединением формулы 1 следует начинать за тридцать минут до начала химиотерапии в 1-й день такой терапии. Внутривенная композиция может медленно вводиться инфузией в течение пятнадцати минут или путем болюсного введения, в зависимости от композиции.
В другом варианте осуществления настоящего изобретения, внутривенную композицию соединения формулы I и его фармацевтически приемлемой соли можно вводить отдельно или в комбинации с другими агентами для лечения и/или профилактики послеоперационной тошноты и рвоты. Такие агенты включают комбинации других противорвотных терапевтических агентов, таких как ондансетрон и других 5НТ3-антагонистов.
Краткое описание чертежей
Фиг.1. Получение микроэмульсионных композиций для внутривенного введения Соединения 1 (схематическая блок-схема).
Фиг.2. Равновесная растворимость Соединения 1 в композиции с 20% Солютола (ромбы) и в композиции с 16% Каптизола (квадраты) в зависимости от pH.
Фиг.3. Схематическое изображение способа производства эмульсионных композиций Соединения 1.
Фиг.4. Распределение частиц по размеру в необработанной композиции, BOL15SO и в обработанной композиции, включая 3 прохода при давлении 2000 фунтов на квадратный дюйм с использованием камеры взаимодействия H30Z.
Фиг.5. Распределение частиц по размерам из необработанной композиции INFWSO и обработанной композиции, включая 3 прохода при давлении 2000 пси с использованием камеры взаимодействия H30Z, и при давлении 4100 пси с использованием камеры взаимодействия H20Y.
Фиг.6. Распределение частиц по размерам из необработанной композиции BOL10WS и обработанной композиции, включая 2 и 3 прохода композиции с помощью камеры взаимодействия H30Z при давлениях 2000 и 4100 пси.
Фиг.7. Оптические микроскопические изображения композиций BOL 15SO и INFWSO в процессе микрофлюидизации.
Фиг.8. Оптические микроскопические изображения композиций BOL 10WSO в процессе микрофлюидизации.
Фиг.9. Оптические микроскопические изображения композиций INFSO в процессе микрофлюидизации.
Фиг.10. Оптические микроскопические изображения композиций BOL10SO в процессе микрофлюидизации.
Фиг.11. Кривая распределения частиц по размеру для INFWSO после стерилизации.
Фиг.12. Кривая распределения частиц по размеру для BOL 10WSO после стерилизации.
Фиг.13. Уровень Соединения 1 в крови в нг/мл при внутривенном введении в различных дозах.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В контексте настоящего изобретения для нижеследующих терминов, пока не указано иное, следует понимать следующие смысловые значения:
Термин "микроэмульсии", используемый в настоящем изобретении, относится к прозрачной, стабильной, изотропной жидкой смеси масла, воды и поверхностно-активного вещества. Этот термин также означает мицеллу, загруженную маслом.
Термин «мицелла» или «мицеллярные», используемый в настоящем изобретении, относится к коллоидным агрегатам амфифильных молекул (сурфактантов), которые возникают при или выше хорошо известной концентрации, называемой критической концентрацией мицеллообразования. Макрогол 15-гидроксистеарат (Solutol® HS15, доступный от BASF Ludvig-shafen, Германия) является конкретным примером поверхностно-активного вещества с критической концентрацией мицеллообразования в диапазоне от 0,005 до 0,02%.
Термин "гемолиз", используемый в настоящем изобретении, означает разрушение красных кровяных телец, что приводит к высвобождению гемоглобина из эритроцитов в плазму крови. Гемолиз может быть измерен методами, хорошо известными специалистам в данной области техники, например, полосками реагента Hemastix® (Bayer Corp, Elkhart, IN), которые детектируют кровь в моче. Полоски реагента Hemastix® меняют окраску от желтой до темно-зеленой, в зависимости от количества гемоглобина, найденного в моче. Шкала Hemastix® выглядит следующим образом: 0=отрицательный, 1=отсутствуют следы гемолиза, 3=следы гемолиза, 4=небольшой +, 5=умеренный++, и 6=большой+++.
Фразы "минимальный гемолиз» и «гемолиз минимизируется", используемые в настоящем изобретении, означают, что при введении экспериментальным животным композиции Соединения 1, либо гемолиз не наблюдался у экспериментальных животных или не более двух экспериментальных животных из десяти экспериментальных млекопитающих обнаруживают следовой уровень гемолиза, определенный, например, как значение три или меньше по шкале Hemastix®.
Термин "стабильный", используемый в настоящем изобретении, относится к химической и физической стабильности.
Физическая стабильность относится к мицеллам и микроэмульсиям, означая, что нет никаких существенных различий в размерах частиц / капель и что не наблюдается разделение фаз.
Химическая стабильность, как используется в настоящем изобретении, относится к сохранению активности Соединения 1 в допустимом диапазоне (более 90% заявленной активности).
Термин "эффективное количество", используемый в настоящем изобретении, относится к количествам Соединения 1 или его фармацевтически приемлемой соли, которые будут препятствовать, улучшать или уменьшать тошноту и/или рвоту у пациента, например, у млекопитающих, таких как человек и приматы или домашнее животное, такие как собака или кошка.
Термины "лечить", "лечение" или "терапия", используемые в настоящем изобретении, относятся к предотвращению или улучшению или уменьшению тошноты и/или рвоты.
Термин "около", используемый в настоящем изобретении, должен пониматься специалистами в данной области техники, как плюс-минус 10% от конкретного значения.
Настоящее изобретение относится к фармацевтическим композициям в виде микроэмульсий для внутривенного введения, содержащим соединение Формулы I (также называемое в настоящем изобретении как Соединение 1),
способам получения фармацевтических композиций и способам лечения тошноты и/или рвоты, используя такие фармацевтические композиции.
В отличие от композиций Соединения 1 на основе сорастворителей, которые при тестировании на экспериментальных животных вызывали гемолиз, проблема гемолиза, вызываемого внутривенным введением Соединения 1 была успешно решена у экспериментальных животных с помощью фармацевтической композиции по настоящему изобретению, которая содержит от около 0,50% до около 10,0% солютола, от около 0,10% до около 2,5% среднецепочечного триглицерида и около от 0,10% до около 1,5% длинноцепочечного триглицерида, в которой массовое отношение макрогол 15-гидроксистеарат:среднецепочечный триглицерид: длинноцепочечный триглицерид составляет около 5-100:1-5:1. Было сделано предположение, не связанное с каким либо конкретным механизмом действия растворяющей системы, что внутривенное введение фармацевтической композиции имеет результатом значительное снижение гемолиза засчет снижения скорости переноса Соединения 1 из мицеллярного кора к красным кровяным тельцам. Предпочтительная концентрация была в пределах 1-15 мг/мл.
Проблема гемолиза, вызванная внутривенным введением Соединения 1, была также решена использованием композиции, содержащей только пегилированный гидроксистеарат в определенном диапазоне концентраций, в котором пегилированный гидроксистеарат был по существу свободен от полиэтиленгликоля.
В другом объекте настоящее изобретение обеспечивает фармацевтическую композицию, содержащую:
а)соединение формулы I
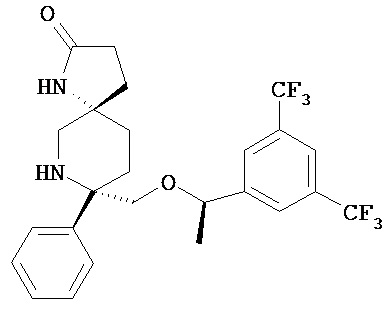
или его фармацевтически приемлемую соль;
b) макрогол 15-гидроксистеарат (Solutol® HS15) в количестве от около 0,50% до около 10,0 мас.% от всей композиции;
c) среднецепочечный триглицерид в количестве от около 0,1% до около 2,5 мас.% от всей композиции;
d) длинноцепочечный триглицерид в количестве от около 0,10% до около 1,5 мас.% от всей композиции, а также
(е) по меньшей мере один буфер, в котором массовое отношение макрогол 15-гидроксистеарат: длинноцепочечный триглицерид составляет около 5-100:1-5:1, и где pH композиции составляет от примерно от около 6,5 до около 8,0.
Композиция по настоящему изобретению удивительно связана с низки гемолизом при внутривенном введении путем инфузии в организм пациента.
Соединение формулы I может образовывать фармацевтически приемлемые соли с органическими и неорганическими кислотами. Примерами подходящих кислот для образования солей являются соляная, серная, фосфорная, уксусная, лимонная, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие минеральные и карбоновые кислоты, хорошо известные специалистам в данной области техники. В некоторых вариантах выполнения соединение формулы I присутствует в виде гидрохлорида. Соли получают взаимодействием свободного основания с достаточным количеством желаемой кислоты для производства соли обычным путем.
В некоторых вариантах осуществления изобретения, соединения формулы I или его фармацевтически приемлемая соль присутствуют в фармацевтической композиции в концентрации от 1 мг/мл до 15 мг/мл, от 2,0 мг/мл до 10 мг/мл или 2,0 мг/мл от всей композиции.
Макрогол 15-гидроксистеарат (Solutol® HS15, доступен от BASF Ludvigshafen, Германия) является поверхностно-активным веществом с критической концентрацией мицеллообразования в диапазоне от 0,005% до 0,02%. Не желая быть ограниченными каким-либо конкретным механизмом действия, изобретатели считают, что концентрация выше критической концентрацией мицеллообразования приводит к формированию мицелл, которые обеспечивают гидрофобное окружение для инкапсуляции Соединения 1 и уменьшение воздействия Соединения 1 на красные кровяные тельца. Solutol® HS15 присутствует в фармацевтической композиции в количестве от около 0,50% до около 10,0 мас.% от всей композиции.
Подходящие среднецепочечные триглицериды, включают, но не ограничены этим, триглицерид каприловой кислоты, триглицерид каприновой кислоты и триглицерид каприловой/каприновой кислот, продаваемый как пропиленгликоль дикаприлат/дикапринат - MIGLYOL® 812 или MIGLYOL® 810, SASOL North America; триглицерид из кокосового масла продается под названием САРТЕХ 300/САРТЕХ 850®, Abitech Corp; триглицерид каприловой/каприловой кислот продается под названием САРТЕХ 355®, Abitech Corp; каприловый/каприловый/лауриновый триглицерид продается под названием САРТЕХ 350®, Abitech Corp; каприловый/каприловый/линолевый триглицерид продается под названием САРТЕХ 810®, Abitech Corp; каприловый/каприловый/стеариновый триглицерид продается под названием САРТЕХ SBE®, Abitech Corp, и комбинации из двух или более из них. Среднецепочечные триглицериды присутствуют в фармацевтической композиции в количестве от около 0,1% до около 2,5 мас.% от всей композиции.
Подходящие длинноцепочечные триглицериды и/или жирные кислоты включают, но не ограничены этим, соевое масло, продаваемый как SUPER-REFINED SOYBEAN OIL USP®, Croda; кукурузное масло продается как SUPER-REFINED CORN OIL NF®, Croda; хлопковое масло, продаваемый как SUPER-REFINED COTTONSEED OIL NF®, Croda оливковое масло, продаваемый как SUPER-REFINED OLIVE OIL NF®, Croda; арахисовое масло, продаваемый как SUPER-REFINED PEANUT OIL BF®, Croda; сафлоровое масло, продаваемый как SUPER-REFINED SAFFLOWER USP®, Croda; кунжутное масло, продаваемый как SUPER-REFINED SESAME NF®, Croda; масло печени акулы, продаваемый как SUPER-REFINED SHARK LIVER®, Croda; этилолеат, продаваемый как Crodamol EO®, Croda; касторовое масло, мононенасыщенные омега-9 жирные кислоты, продаваемый как олеиновая кислота Croda и комбинации из двух или более из них. Длинноцепочечные триглицериды или длинноцепочечные жирные кислоты или то и другое вместе присутствуют в фармацевтической композиции в количестве от около 0,10% до около 1,5 мас.% от всей композиции.
В варианте осуществления изобретение обеспечивает фармацевтическую композицию, которая содержит макрогол 15-гидроксистеарат в количестве от около 0,50% до около 7,5 мас.% от всей композиции, сред нецепочечный триглицерид в количестве от около 0,15% до около 1,5 мас.% от всей композиции и длинноцепочечный триглицерид в количестве от около 0,10% до около 1,2 мас.% от всей композиции.
В другом варианте осуществления изобретение обеспечивает фармацевтическую композицию, которая содержит макрогол 15-гидроксистеарат в количестве от около 0,88% до около 4,84 мас.% от всей композиции, среднецепочечный триглицерид в количестве от около 0,20% до около 1,20 мас.% от всей композиции и длинноцепочечный триглицерид в количестве от около 0,10% до около 0,75 мас.% от всей композиции.
В другом варианте осуществления среднецепочечный триглицерид является каприловый/каприновый триглицерид, продаваемый как пропиленгли-коль дикаприлат/дикапрат-MIGLYOL® 810 или MIGLYOL® 812, SASOL North America (Houston, Texas), и длинноцепочечный триглицерид в виде соевого масла в сверхочищенной форме.
Фармацевтическая композиция также содержит по меньшей мере один буфер. Неограничивающие примеры подходящих буферов, которые могут быть включены в фармацевтическую композицию, включают фосфатные буферы (pH 7-8), такие как фосфат натрия, фосфат калия и буферы фосфата кальция, сукцинатные буферы (pH 4-6), цитратные буферы (pH 2-6), TRIS (pH 7-8) от 5-20 мМ и их смеси.
Количество буфера в фармацевтической композиции может зависеть от типа и концентрации буфера. Например, предположим, 10-20 мМ фосфатный буфер для поддержания pH между 6,5 и 8,0, фосфат может быть добавлен к композиции в количестве от 0,01% до 0,5 мас.%; или около 0,05% до 0,02 мас.% или от 0,4% до 0,2 мас.% от всей композиции. В качестве примера, когда используется система фосфат натрия двухосновный безводный/моноосновный дегидратированный, около 0,2 мас.% моноосновного фосфата натрия и около 0,284 мас.% двухосновного (безводного) фосфата натрия будет добавляться в композицию.
Значение pH композиции в настоящем изобретении регулируется, в случае необходимости, с помощью щелочи, такой как гидроксид натрия, для значений pH от около 6,5 до около 8,0. В некоторых вариантах pH композиции составляет примерно от около 7,0 до около 8,0, pH около pH 7,5 или pH 7,5.
При необходимости, фармацевтическая композиция по данному изобретению может дополнительно содержать другие вспомогательные вещества, например, антиоксиданты (консерванты), антимикробные агенты, хелатирующие агенты, альбумин, регуляторы тоничности, наполнители и др. В качестве примера, антимикробные агенты представлены ВАС (0,1%-0.025%), бензойной кислотой (0,1-0,2%), бензиловым спиртом (0.0001-1.5%); антиоксидантами являются ВНТ, ВНА (0,0001-0,001%), гистидин, метионин, глицин, сульфит/бисульфит натрия (0,01-0,2%) и аскорбиновая кислота (0,02-0,2%); хелатирующими агентами являются ЭДТА (0,01-1,0%), ДТПА (диэтилентриамин пентауксусной кислоты); альбумин (0.05-1.2%); регуляторы тоничности (NaCl, маннит (1-10%), глицерин (0,2-2,5%); а также наполнители маннит (1-10%), сахароза, глицин (0,1%-2,5%), трегалоза и лактоза (0,1-3,0%).
В другом варианте осуществления изобретение обеспечивает фармацевтическую композицию, которая содержит:
(а) соединение формулы 1
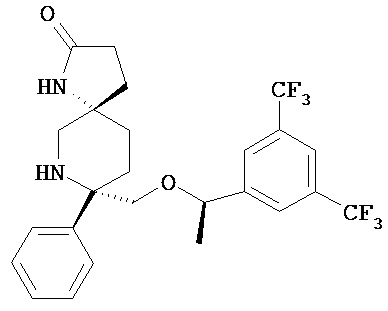
или его фармацевтически приемлемую соль;
(b) макрогол 15-гидроксистеарат (Solutol® HS 15) в количестве около 4,4%, или 4,4 мас.% от всей композиции;
(c) по меньшей мере один среднецепочечный триглицерид в количестве около 1,1%, или 1,1 мас.% от всей композиции;
(a) рафинированное масло сои в количестве около 0,66%, или 0,66 мас.% от всей композиции, а также
(e) фосфатный буфер, в котором pH композиции составляет около 7,5.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
(а) соединение формулы 1
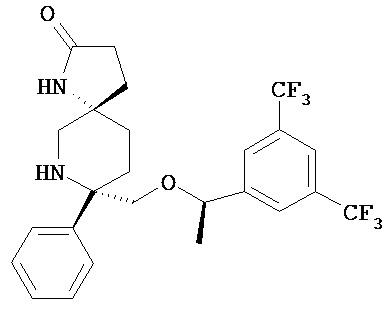
или его фармацевтически приемлемую соль;
(b) макрогол 15-гидроксистеарат (Solutol® HS 15) в количестве около 0,88% и 0,88 мас.% от всей композиции;
(c) по меньшей мере один среднецепочечный триглицерид в количестве около 0,22%, или 0,22 мас.% от всей композиции;
d) рафинированное масло сои в количестве около 0,12%, или 0,12 мас.% от всей композиции, а также
(е) фосфатный буфер, в котором pH композиции составляет около 7,5. Примеры среднецепочечных триглицеридов описаны выше.
Фармацевтическую композицию по настоящему изобретению можно стерилизовать любым из нескольких методик стерилизации, в том числе фильтрующей стерилизацией и стерилизацией в автоклаве. В варианте осуществления, когда фармацевтическая композиция стерилизуется методом фильтрации, например, используют фильтры с размером пор от 0,01 до 0,45 мкм, например, 0,22 мкм. Соответственно, фармацевтическая композиция по настоящему изобретению является химически и физически стабильной в течение от 1 до 6 месяцев, как правило, 3 месяца при температуре от 5°C до 40°C/75% 0 В (относительная влажность).
В другом объекте настоящее изобретение обеспечивает способ получения фармацевтической композиции, который содержит следующие этапы:
a) смешивания и нагревания (I) расплавленного макрогол 15-гидроксистеарата, (ii) среднецепочечного триглицерида (iii) длинноцепо-чечного триглицерида с образованием композиции;
b) добавления воды в композицию для формирования микроэмульсионной композиции;
c) добавления в состав микроэмульсионной композиции соединения формулы I
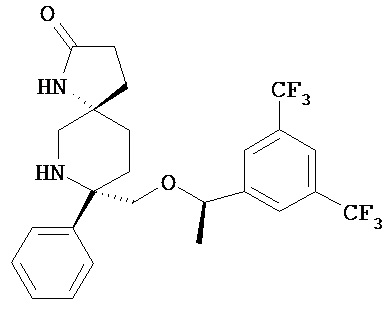
или его фармацевтически приемлемой соли и
d) добавления по меньшей мере одного буфера и регулирование рН от 6,5 до 8, чтобы сформировать фармацевтическую композицию, в которой макрогол 15-гидроксистеарат присутствует в количестве от около 0,50% до около 10,0% от общей массы фармацевтической композиции, среднецепочечный триглицерид присутствуют в количестве от около 0,10% до около 2,5% от общей массы фармацевтической композиции, и длинноцепочечный триглицерид присутствуют в количестве от около 0,10% до около 1,5 мас.% от общей массы фармацевтической композиции, и где массовое отношение макрогол 15-гидроксистеаратсреднецепочечный триглицерид:длинноцепочечный триглицерид в фармацевтической композиции составляет около 5-100:1-5:1.
Примеры среднецепочечных и длинноцепочечных триглицеридов и концентрации Соединения 1 описаны выше.
В качестве примера настоящего способа получения фармацевтической композиции, Solutol® HS15 расплавляют при нагревании Solutol® HS15 при температуре от 60°С до 65°С, или при температуре около 65°C. Расплавленный Solutol® HS15, среднецепочечный и длинноцепочечный триглицериды смешивают при температуре от 50°C до 65°C, или около 60°C, в течение от около 5 минут до около 15 минут, чтобы сформировать композицию. Затем добавляют воду для инфузий, чтобы сформировать микроэмульсионную композицию, с последующим добавлением Соединения 1, чтобы сформировать фармацевтическую композицию. Затем добавляют одноосновный/двухосновный фосфат натрия к фармацевтической композиции. Значение pH фармацевтической композиции доводят до pH, например, 7,5 с использованием гидроксида натрия, и тоничность композиции корректируется тоничным агентом, хлоридом натрия, примерно до около 290 мМ. Затем добавляется вода до необходимого объема с последующей стерилизацией композиции путем фильтрования через 0,22 мкм фильтр (см. ниже пример 5).
Способ получения Соединения 1 описан в ’320 патенте как выполнение по Примеру 72а и он осуществляется через 18 отдельных стадий из коммерчески доступных исходных материалов (описание см. в патенте ’320, со строки 55 колонки 43 до строки 20 колонки 45; со строки 55 колонки 75 до строки 21 колонки 80; со строки 35 колонки 90 до строки 63; и со строки 1 колонки 98 до строки 24 колонки 99, который включен сюда посредством ссылки. Смотри также WO 2008/11833, Примеры 1-6, которые включены сюда посредством ссылки).
В другом варианте осуществления изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение формулы I
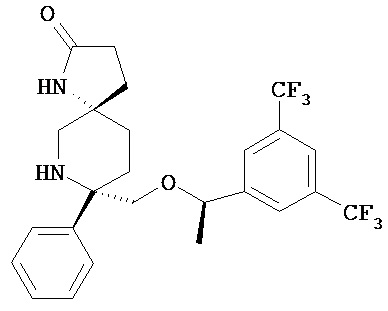
или его фармацевтически приемлемую соль и
b) пегилированный гидроксистеарат в количестве от 0,88% до 5 мас.% от всей композиции, в котором пегилированный гидроксистеарат по существу свободен от полиэтиленгликоля (ПЭГ), и где pH композиции составляет от около 6,5 до около 8,0. Термин "по существу свободный" означает, что композиция с пегилированным гидроксистеаратом содержит не более 2% свободного ПЭГ.
Для получения пегилированного гидроксистеарата, по существу свободного от ПЭГ, свободный ПЭГ, содержащийся в пегилированном гидроксистеарате, например, Solutol® HS15, может быть удален с помощью известных методов, например, диализом (см. ниже пример 5, получение диализировнного солютола в солевом растворе в фосфатном буфере, и Таблицу 9).
Фармацевтические композиции по настоящему изобретению полезны для лечения тошноты и/или рвоты. Соответственно, другим объектом настоящего изобретения является способ лечения тошноты и/или рвоты у пациентов, нуждающихся в таком лечении, например, у человека или приматов, таких как обезьяны, или домашние животные, например, собака или кошка. Способ содержит внутривенное инфузионное введение пациенту эффективного количества фармацевтической композиции по настоящему изобретению, где гемолиз сведен к минимуму благодаря внутривенному введению фармацевтической композиции. Фармацевтические композиции являются полезными при лечении рвоты, задерживая ее наступление, после начала применения химиотерапии, на срок от 24 часов до нескольких дней, как это было экспериментально установлено. Смотрите Gonzales et al, Oncology Special Edition, Vol.5 (2002), p.53-58. Фармацевтические композиции также полезны при лечении острой тошноты и/или рвоты, вызванной химиотерапией, облучением, движением и алкоголем (например, этанолом), высокими дозами антибиотиков, вирусными или бактериальными гастроэнтеритами. Фармацевтические композиции могут быть приготовлены совместно, в целях включения по меньшей мере одно другого средства против рвоты, например, дексаметазон или ондансетрон HCl. Эти фармацевтические композиции также могут быть получены в комбинации с другими средствами против рвоты и тошноты и/или химиотерапевтическими агентами по предписанию врача.
Фармацевтическая композиция может быть разбавлена для введения подходящим водными разбавителем(ями), такими как физиологический раствор, декстроза, декстроза в фильтрованной воде и маннит, чтобы получить любую промежуточную содержание от около 0,88% до около 4,4 мас.% Solutol® HS15 от всей композиции.
Режим дозирования, используемый для фармацевтической композиции по настоящему изобретению, выбирается на основе учета различных факторов, включающий виды, возраст, вес, пол и состояния здоровья пациента, нуждающегося в лечении, а также степени тяжести тошноты и/или рвоты, с которыми сталкивается пациент. Обычный квалифицированный врач может легко определить и прописать эффективное количество Соединения 1 в целях предотвращения, улучшения или уменьшения тошноты и/или рвоты. Например, общая суточная доза соединения формулы I для взрослого человека составляет от 1 мг/кг до 9 мг/кг массы тела пациента или от 1 мг/кг до 3 мг/кг массы тела пациента (предполагаемая доза в 200 мг для пациента весом 70 кг).
Фармацевтическая композиция может быть введена путем внутривенной инфузии в течение от 15 до 90 минут, от 15 до 60 минут, или от 15 до 30 минут. Для некоторых композиций состав можно вводить путем болюсной инъекции.
Другим объектом настоящего изобретения является способ минимизации гемолиза у пациента, например, у млекопитающих, таких, как человек или примат или домашнее животное, например, собака или кошка, путем внутривенного введения соединения формулы I
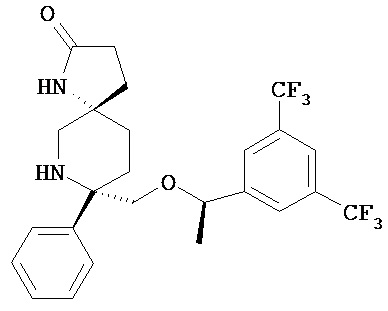
или его фармацевтически приемлемой соли, содержащий внутривенную инфузию пациенту эффективного количества фармацевтической композиции по настоящему изобретению.
Настоящее изобретение относится к способу лечения пациентов, нуждающихся в таком лечении, внутривенной композицией, содержащей соединение Формулы I или его фармацевтически приемлемую соль, где доза такого соединения варьирует от 100 до 200 мг, и композицию вводят пациенту в виде разовой дозы один раз перед химиотерапией или до цикла облучения, или один раз до или после хирургической операции с целью лечения ИХТР (индуцированные химиотерапией тошнота и рвота), ПОТР (послеоперационные тошнота и рвота) или РИТР(радиационно-индуцированные тошнота и рвота).
В дополнение к вышеописанным композициям, содержащим солютол, настоящее изобретение также относится к эмульсионным композициям, которые подходят как для болюсного, так и для медленного внутривенного инфузионного введения. Соединения, полезные для таких композиций, включают соединения формулы I и Ia и их фармацевтически приемлемые соли.
Были разработаны парентеральные эмульсии на основе средне- и длинноцепочечных триглицеридов и с дополнительными эксипиентами, такими как яичные лецитины. Некоторые из этих композиций, полученные с яичным фосфатидилхолином и Lipoid E80S, приводили к чистым (т.е., незначительный гемолиз или его отсутствие) композициям при введении млекопитающим (крысам) как болюсным, так и инфузионым путем. Предпочтительные композиции подвергались микрофлюидизации, приводящей к эмульсиям, имеющим меньший размер капель и относительно узкое распределение частиц по размерам (до 500 нм среднего диаметра и с D90 менее чем 600 нм). Такие композиции были также физически стабильны после стерилизации в автоклаве при 121°C.
Таким образом, в другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а)соединение Формулы
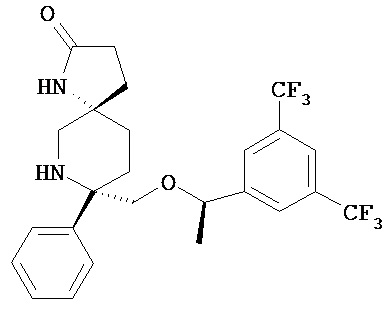
или его фармацевтически приемлемую соль; и
b) фосфолипид.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение Формулы I
или его фармацевтически приемлемую соль; и
b) длинноцепочечную жирную кислоту и/или длинноцепочечный триглицерид.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение Формулы 1
или его фармацевтически приемлемую соль; и b) среднецепочечный триглицерид.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение Формулы I
или его фармацевтически приемлемую соль; и
b) среднецепочечный триглицерид/или длинноцепочечную жирную кислоту; а также
c) фосфолипид.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение Формулы 1
или его фармацевтичевски приемлемую соль; и
b) среднецепочечный триглицерид и/или длинноцепочечную жирную кислоту или триглицерид; а также
c) фосфолипид и
d) глицерин.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение Формулы I
или его фармацевтически приемлемая соль; и
b) среднецепочечный триглицерид и/или длинноцепочечную жирную кислоту или триглицерид; и
c) фосфолипид;
d) глицерин и
e)этанол.
В предпочтительном варианте выполнения настоящего изобретения массовый процент загрузки масла (например, компонент(ы) b) составляет около 10 мас.% относительно общей массы композиции.
В другом варианте осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит:
а) соединение Формулы
или его фармацевтически приемлемую соль; и
b) 10% среднецепочечного триглицерида и/или длинноцепочечной жирной кислоты или триглицерида; а также
c) 1,2% фосфолипида и
d) 2,25% глицерина.
В другом варианте осуществления изобретения, эффективное количество одной или нескольких композиций, перечисленных в настоящем изобретении, может быть использовано в комбинации с другими активными ингредиентами либо в отдельных дозах до или после введения дополнительного активного ингредиента (или одновременно с ним), либо в фиксированной комбинации доз антогониста NK-1 в комбинации с другим таким активным ингредиентом. Композиции по настоящему изобретению можно вводить в комбинации с одним или более селективными ингибиторами обратного захвата серотонина («СИОЗС») для лечения депрессии или тревоги. Представители СИОЗС включают флуоксетин, флувоксамин, пароксетин, сертралин и их фармацевтически приемлемые соли. В другом объекте изобретение относится к способу лечения рвоты или задержки начала рвоты от часов до нескольких дней после получения химиотерапии. Комбинации внутривенных композиций по настоящему изобретению с другим антирвотным агентом, таким как антагонист серотонинового 5-НТ3 рецептора, кортикостероидом или замещенным бензамидом, могут быть использованы для лечения других форм рвоты, в том числе острой рвоты, вызванных химиотерапией, лучевой терапией, движением и/или алкоголем (этанолом), а также послеоперационной тошноты и рвоты. Примеры антагонистов 5-НТ3 включают палосетрон, доласетрон, ондансетрон и гранисетрон или их фармацевтически приемлемые соли. Примером подходящего кортикостероида является дексаметазон. Примером подходящего бензамида является метоклопрамид. Предпочтительные комбинации включают комбинации любых двух из вышеуказанных или любого одного из вышеуказанных с (или непосредственно в составе) внутривенной композицией HK-1.
Настоящее изобретение относится к способу лечения острой и отсроченной тошноты и рвоты, связанных с начальными и повторными курсами высокоэметогенной химиотерапии рака, содержащим введение внутривенной композиции соединения формулы I в комбинации по меньшей мере с одним дополнительным противорвотным агентом.
Настоящее изобретение относится к способу лечения острой и отсроченной тошноты и рвоты, связанных с начальными и повторными курсами умеренноэметогенной химиотерапии рака, содержащим введение внутривенной композиции соединения формулы I в комбинации по меньшей мере с одним дополнительным противорвотным агентом.
Настоящее изобретение относится к способу лечения острой и отсроченной тошноты и рвоты, связанных с начальными и повторными курсами высокоэметогенной химиотерапии рака, содержащим введение внутривенной композиции соединения формулы Ia в комбинации по меньшей мере с одним дополнительным противорвотным агентом.
Настоящее изобретение относится к способу лечения острой и отсроченной тошноты и рвоты, связанных с начальными и повторными курсами умеренноэметогенной химиотерапии рака, содержащим введение внутривенной композиции соединения формулы Ia в комбинации по меньшей мере с одним дополнительным противорвотным агентом.
Следующие ниже примеры изложены в качестве руководства для практикующего врача и не предназначены каким либо путем ограничить объем настоящего изобретения.
ПРИМЕРЫ
Примеры 1-4 описывают разработку определенных внутривенных композиций Соединения 1 или другие исследования, касающиеся гемолиза. Пример 5 обеспечивает описание разработку внутривенной микроэмульсионной (загруженной маслом мицеллы) композиции Соединения 1 настоящего изобретения. Пример 6 обеспечивает дополнительные композиции. Пример 7 описывает эмульсионную композицию, подходящую как для болюсного, так и для медленного инфузионного введения. Пример 8 обеспечивает примеры пролекарств.
Пример 1. Оценка растворимости Соединения 1 в системе на основе циклодекстрина и в других системах сорастворителей.
Растворимость Соединения 1 в 16% Captisol® (сульфобутиловый эфир β-циклодекстрина, доступен от Cydex Inc. of Overland Park, Kansas) и системе сорастворителей пропиленгликоль: этанол (ПГ: EtOH, 40%: 10%) устанавливали путем определения равновесной растворимости Соединения 1 в этих системах растворителей. Концентрацию Соединения 1 определяли методом высокоэффективной жидкостной хроматографии (ВЭЖХ), как описано в примере 5, в различные моменты времени для определения равновесной растворимости, которая была достигнута после достижения плато растворимости (заметно не менялось) с течением времени. Оптимальную растворимость Соединения 1 нашли с 16% Каптизола (данные не представлены).
Пример 2. Исследования на крысах внутривенного введения композиций NK-1. содержащих Captisol® и другие системы сорастворителей.
16% Captisol® и систему сорастворителей ПГ: ЕЮН 40%: 10% вводили внутривенно крысам.
Образцы мочи у крыс оценивали на гемолиз с использованием полосок реагента Hemastix® (№2190, Bayer Corp.Elkhart IN). Наличие крови в моче обнаруживали с использованием полосок реагента Hemastix® (№2190, Bayer Corp.Elkhart IN.).Полоски реагента Hemastix® реагента детектируют пероксидподобную активность гемоглобина. Цвет полосок изменяется от желтого до темно-зеленого, в зависимости от количества гемоглобина, найденного в моче. Шкала Hemastix® выглядит следующим образом: 0=отрицательный, 1=отсутствуют следы гемолиза, 3=следы гемолиза, 4=небольшой+, 5=умеренный++, и 6=большой+++.
Несмотря на то, что в композициях с циклодекстрином и с сорастворителями удалось улучшить растворимость Соединения 1, такие композиции с сорастворителями неожиданно привели к сильному гемолизу (значение 6 на шкале Hemastix®) при введении методом внутривенной инфузии. Введение путем медленной 15 минутной инфузии композиции с каптизолом привело к более низкому уровню появления гемолиза. (Табл. 1).
|
Было сделано предположение, что основной причиной, вызывающей гемолиз, была временная местная высокая концентрация соединения в месте введения. Дополнительные эксперименты на крысах для определения, может ли снизить уровень гемолиза в течении болюсного введения использование Captisol® в различных концентрациях, объемах, скоростях введения или добавлением буферов, не увенчались успехом.
Пример 3. Исследования по оценке появления гемолиза у крыс после болюсного введения композиции, содержащей 22% солютола
Исследования затем выполняли на самцах и самках крыс с целью понижения локальной высокой концентрации свободного Соединения 1 посредством: 1) включения Соединения 1 в гидрофобный кор мицелл, используя Solutol® HS15 (макрогол 15-гидроксистеарат, также называемый в настоящем изобретении как солютол), 2) введения болюсным путем Соединения 1, включенного в солютол, а также 2) тестированием возникновения гемолиза (с помощью полосок реагента Hemastix®) через различные промежутки времени (15-60 минут).
Результаты показали, что, как у самцов, так и у самок крыс, возникновение гемолиза со временем значительно сокращается (Табл. 2), и привели к выводу, что локальная высокая концентрация свободного Соединения 1 была ответственна за транзиторный гемолиз, который возникал в течение первых нескольких минут после внутривенного введения.
|
Одноразовое внутривенное замедленное на 1-2 минуты болюсное введение в хвостовую вену, используя иглу и шприц подходящего размера. Композиция: 10 мг/мл лекарственного средства, 22% Solutol® HS15, 20 мМ фосфатного буфера, pH 7,0.
Пример 4. Исследования по оценке возникновения гемолиза у крыс путем варьирования типа внутривенного введения
Для снижения возникновения гемолиза использовали стратегию, которая включала эксперименты, чтобы проверить, влияет ли на гемолиз скорость введения препарата. Это включало эксперименты по введению крысам Соединения 1 до крыс в мицеллярных растворах (HS Solutol® 15), как инфузионным, так и болюсным путем. Высокий уровень возникновения гемолиза наблюдали у крыс, когда мицеллярный раствор вводили с помощью болюсного способа, в то время как гемолиз не наблюдался в той же дозе через 15 минут при медленной 15 минутной внутривенной инфузии этой же дозы препарата (Табл. 3).
|
Пример 5. Создание внутривенной микроэмульсионной композиции Соединения 1 по настоящему изобретению
МАТЕРИАЛЫ И МЕТОДЫ
Материалы для создания микроэмульсионной композиции
Материалы, использованные в этом исследовании, приведены в Таблице 4.
|
Гемолитическое тестирование на крысах
Методологией данного исследования было введение (в хвостовую вену крысы) каждой композиции в течение 15 минут путем внутривенной инфузии с использованием инфузионного насоса или введением композиции в течение двух минут болюсным путем, используя шприц. Мочу проверяли на наличие свободного гемоглобина с использованием метода объединенного сбора мочи за шестичасовой интервал. Для шестичасового объединенного сбора мочи, крысам вводили дозу и помещали в метаболический отсек. Через шесть часов после введения дозы, крыс подвергали ингаляционному усыплению диоксидом углерода, и отбирали образец мочи. Как минимум пять субъектов протестировали для каждой композиции и образцы мочи проанализировали на гемолиз с использованием полосок реагента Hemastix®, как показано в сравнительном примере 2.
Для 15 минутных образцов и образцов одного часа крысам вводили дозу и возвращали в их обычные индивидуальные домашние отсеки, без подстилки. При любом выделении мочи крысой в отсеке, мочу анализировали полосками реагента Hemastix® для определения присутствия гемоглобинурии. Если крыса не выделяла мочу в отсеке, но выделяла после усыпления, мочу анализировали в усыпляющей камере.
Получение солютола диализом в фосфатно-солевом буферном растворе/дистиллированной воде
Солютол является амфифильной молекулой и содержит около 30% свободного полиэтиленгликоля (ПЭГ). Цель проведения диализа состояла в минимизации/удалении в молекуле гидрофильной составляющей. Чтобы получить солютол, лишенный свободного ПЭГ, готовили 50 мл композиции в виде плацебо (содержащей 20% солютола в фосфатном буфере) и фильтровали через 0,22 мкм фильтр. Диализный мешок смачивали фосфатным буфером с pH 7,0. Записывали вес влажного мешка. Затем диализный мешок заполняли 50 мл вышеупомянутой композицией плацебо и этот мешок помещали в не менее чем 750 мл фосфатного буфера при pH 7,0, который перемешивался мешалкой со средней скоростью. Буфер снаружи диализного мешка заменяли дважды в день, процесс повторяли в течении 72 часов, в течение которых сделали по меньшей мере 6 замен буфера за 72 часа. По прошествии 72 часов измеряли вес диализного мешка одного с раствором плацебо внутри. Плацебо композиция также была проанализирована с помощью ВЭЖХ для подтверждения уровня ПЭГ в растворе.
Аналитический тест-метод для определения содержания свободного полиэтиленгликоля (ПЭГ) в солютоле
Условия ВЭЖХ для тестирования свободного ПЭГ в молекуле солютола состоят в следующем:
Колонка: Nucleosil CIS Sum 150×4,6 мм
Мобильная фаза А: 0,025% раствор фосфорной кислоты
Мобильная фаза Б: ацетонитрил
Температура колонки: 30°C, Температура образца: 20°C, Объем ввода: 20 мкл
Скорость потока: 1 мл/мин. Длина волны УФ: 190 нм. Время выхода: 60 мин
Разбавитель образца: Метанол
Программа градиента:
|
Способ приготовления микроэмульсионной композиции Соединения 1
Ниже приведен процесс получения микроэмульсионной композиции для Соединения 1. Блок-схема способа приготовления микроэмульсионной композиции изображена на Фиг.1.
(i) промывка пустого стеклянного химического стакана;
(ii) взвешивание и загрузка солютола, который предварительно расплавляли в его оригинальном контейнере при температуре 65°C в течение примерно 15 минут в вышеназванный стеклянный стакан;
(iii) взвешивание и загрузка соевого масла в вышеназванный стакан;
(iv) взвешивание и загрузка миглиола в вышеназванный стакан;
(v) тщательное смешивание вышеупомянутых компонентов в течение приблизительно пяти минут с использованием мешалки;
(vi) добавление воды для инъекции в вышеназванный стакан после его тарирования, чтобы сформировать микроэмульсию, и затем добавление Соединения 1 и перемешивание;
(vii) микроэмульсионная композиция перемешивали до гомогенности с помощью магнитной мешалки в течение 15 минут;
(viii) взвешивание и загрузка одноосновного и двухосновного фосфата натрия в вышеназванную композицию;
(ix) проверка pH композиции и доведение pH с помощью 1 Н NaOH до pH 7,5;
(x) проверка осмоляльности раствора и доведение тоничности с помощью насыщенного солевого раствора (NaCl) до прибл. 290 мМ ± 30 мМ;
(xi) вода, в достаточном количестве для получения конечного объема продукта;
(xii) фильтрование раствора через 0,22 мкм фильтр Millipore Durapore-GV в ламинарном шкафу;
(xiii) асептическое укупоривание композиции в 10 или 20 мл стеклянные флаконы для конечного использования.
Аналитический метод испытаний для анализа образцов, содержащих соединения формулы I
Условия ВЭЖХ для анализа Соединения 1:
Колонка: Prodigy ODS (3) 150Х4.6 мм 3 мкм
Мобильная фаза А: 0,1% раствор фосфорной кислоты
Мобильный фаза Б: ацетонитрил
Темп колонки: 30°C. Темп образца: 5°C. Объем ввода: I0 мкл.
Скорость потока: 1 мл/мин. Длина волны УФ: 215 нм. Время выхода: 60 мин
Разбавитель для образцов: Метанол
Программа градиента:
|
А. Оценка растворимости Соединения 1 в солютоле в сравнении с Captisol®
Кривая равновесия растворимости Соединения 1 в 20% солютола строили в зависимости от pH. Как показано на Фиг.2, равновесная растворимость Соединения 1 была существенно выше (~50 мг/мл) в 20% солютола при pH 7,0, по сравнению с <6 мкг/мл, полученной с циклодекстриновой композицией с 16% Captisol®, что ближе к растворимости в воде Соединения 1 при pH 7,0. Следовательно, масштабность повышения растворимости Соединения 1 несомненно была значительнее для мицеллярной композиции солютола, по сравнению с композицией на основе Captisol®.
В. Оценка возникновения гемолиза у крыс при внутривенном введении композиции солютола, содержащей Соединение 1.
Мицеллярный раствор с 22% солютола в дозе 10 мг/мл Соединения 1 при pH 8,0 был использовали в качестве отправной точки для композиции Соединения 1. Показатель pH 8,0 в композиции выбрали, чтобы он был выше значения рКа Соединения 1 равного 6.9, поскольку Соединение 1 должно быть ионизированным для предпочтительного включения в гидрофобный кор мицеллы. Однако когда мицеллярную композицию с 22% солютола испытывали на крысах, получили весьма неожиданные результаты in vivo. Тяжелый гемолиз наблюдался у крыс при введении композиции болюсным вливанием для всех испытанных доз от 10-30 мг/кг Соединения 1 (Таблица 5).
|
Результаты гемолиза после одноразового внутривенного замедленного на 1-2 минуты болюсного введения в хвостовую вену композиции с 22% Solutol® HS15, 10 мг/мл препарата, 20 мМ фосфатного буфера, используя иглу и шприц подходящего размера.
Было сделано предположение, что высокий уровень солютола может быть ответственен за гемолитическую активность в вышеупомянутых мицел-лярных композициях, которые вводят болюсным вливанием. Препарат солютола является амфифильной молекулой, имеющей липофильную часть (12-гидроксистеариновая кислота), которая составляет около 70% препарата, и гидрофильную часть, которая состоит из 30% свободного ПЭГ. Также предполагалось, что присутствие свободного ПЭГ в молекуле солютола повышает общую гидрофильность и облегчает переход из липофильного кора к водной фазе, тем самым увеличивая воздействие препарата на красные кровяные тельца.
Вышеприведенную гипотезу проверяли путем уменьшения содержания солютола в композиции, что тем самым должно было снизить содержание свободного PEG в молекуле. При снижении концентрации солютола количество случаев гемолиза существенно усеньшилось с наибольшего числа 6 случаев на 10 крыс, протестированных с композицией с 22% солютола (доза 20 мг/кг через инфузию) до 0 случаев на 10 крыс, протестированных с композицией с 7,5% солютола, при гораздо более высокой дозе в 25 мг/кг. При 6%-ной концентрации солютола количество случаев гемолиза также значительно уменьшалось до 1/10 и 2/10 случаев возникновения гемолиза в двух отдельных исследованиях (Таблица 6). Возникновение гемолиза, наблюдаемое при самых низких уровнях солютола, может быть связано с фоном и с вариабельностью, которая наблюдалась во время гемолитического тестирования крыс.
|
Для дополнительного подтверждения вышеприведенной гипотезы, свободный ПЭГ удалялся из молекулы солютола путем диализа. Уменьшение содержания свободного ПЭГ с 8,5% до 0% существенно сократило гемоли-тическое проявление. Возникновение случаев гемолиза уменьшалось с 10/10 у крыс, которым вводилась композиция 15% солютола, содержавшая 8,5% свободного ПЭГ, до 1 случая на 10 крыс и до 2 случаев на 10 крыс, получивших соответственно, композиции 6 и 8% солютола, содержащие 2% и 0% свободного ПЭГ (Таблица 7).
|
На основании кривой равновесия растворимости, было установлено, что концентрация 10 мг/мл Соединения 1 может быть достигнута при пятикратном снижении концентрации сурфактанта в текущей лидерной композиции, содержащей 22% солютола, что позволило бы при солюбилизации достичь концентрации 50 мг/мл Соединения 1. Таким образом, мицеллярная композиция с 4,4% солютола (в 5 раз уменьшенный уровень по сравнению с 22% солютола) и с концентрацией 10 мг/мл Соединения 1, pH 8,0, была выбрана в качестве композиции для всех дальнейших испытаний. Однако, когда эта композиция с концентрацией 10 мг/мл Соединения 1 была протестирована на крысах в дозе 20 мг/кг, число случаев гемолиза оставалось еще высоким, приводя к 14 случаям гемолиза из 20 протестированных крыс, разделенных на две группы, по 10 крыс в каждой группе (Таблица 8). Повторное тестирование композиции с 4,4% солютола при pH 8,0 дало аналогичные результаты гемолиза у 7 из 8 субъектов (Таблица 8).
|
С. Оценка случаев гемолиза у крыс при внутривенном введении микроэмульсионных композиций Соединения 1
Вследствие ограниченной возможности самой мицеллы предотвращать возникновение гемолитические случаи при высоких концентрациях в мицеллы загружают масла (с образованием микроэмульсий) для предохранения лекарственного средства внутри гидрофобного кора и замедления распределения Соединения 1 в красные кровяные тельца за счет повышения общей гидрофобности мицелл при включении триглицеридов. Было высказано предположение, что включение триглицеридов также может служить для увеличения срока существования мицеллы, которая в противном случае является высокодинамичной структурой с коротким временем жизни. Мицеллы находятся в равновесии с отдельными молекулами поверхностно-активных веществ/мономеров, которые постоянно обмениваются между основным объемом и мицеллами, кроме того, сами мицеллы непрерывно подвергаются распаду и повторной сборке.
Различные короткоцепочечные, среднецепочечные и длинноцепочечные триглицериды загружались в мицеллы, в том числе capryol, кукурузное масло, соевое масло и MIGLYOL® 812 (также упоминаемый в настоящем изобретении как миглиол), чтобы получить физически стабильные микроэмульсионные композиции с высокой степенью загрузки маслом. Наибольшее количество масла при концентрации 20% солютола может быть загружено с помощью среднецепочечного триглицерида миглиола. Не меньше чем 5% этого среднецепочечного триглицерида цепи может быть загружено в 20% раствор солютола без ущерба для физической стабильности композиции.
Таким образом, композиция, содержащая 20% солютола, 4,4% миглиола и 10 мг/мл Соединения 1, была протестирована на крысах на гемолиз при дозе 20 мг/кг Соединения 1, вводимого путем инфузии. Эта композиция не приводила к гемолитическим случаям у 8 протестированных крыс. Результаты были подобными тем, которые наблюдались со сниженными концентрациями солютола и МСТ, составляющими 4,4% солютола и 1,1% миглиола на 10 мг/мл Соединения 1, приведшие только к 1 случаю гемолиза из 8 обследованных субъектов (Таблица 9).
|
Для подтверждения положительных результатов, полученных с композициями, обогащенных маслами, эти композиции были протестированы снова на крысах в отдельном исследовании для любого случая возникновения гемолиза. Удивительно, но композиция с 20% солютола, 4,4% миглиола привела на этот раз к большой частоте гемолиза с 4 случаями гемолиза в 8 протестированных субъектах. Результаты с 4,4% солютола и 1,1% миглиола были, однако, сопоставимы с предыдущими результатами, вызвав только один случай гемолиза в 7 субъектах, которым была введена эта композиция (Таблица 10).
Для снижения гемолитических проявлений, наблюдавшихся для вышеупомянутых композиций, они были далее модифицированы включением длинноцепочечных триглицеридов (ДЦТ), для дополнительного увеличения загрузки масла в мицеллу и для получения более выраженной гидро-фобности для улучшения удержания лекарственного средства. Соевое масло, длинноцепочечный триглицерид, было использовано для улучшения загрузки масла в композицию солютола-миглиола. Максимальное количество соевого масла, которое может быть загружено в смесь 20% солютола и 5% миглиола, составляет примерно 3%. Соевое масло также использовалось для повышения физической стабильности вышеупомянутой композиции, возможно через перестройку мицеллярной конфигурации путем включения длинной углеводородной цепи в среднецепочечный триглицерид миглиол для формирования более компактной структуры.
Результаты с 20% солютола, 4,4% миглиола и 3% соевого масла при 10 мг/мл Соединения 1 не отличались по сравнению с аналогичной композицией без соевого масла. Вышеназванная композиция привела к гемолизу у 5 из 8 крыс, протестированных с этой композицией. Напротив, 4,4% солютола, 1,1% миглиола и 0,66% соевого масла на 10 мг/мл Соединения 1 дала абсолютно ясные результаты при тестировании на 8 субъектах (Таблица 10). Однако 20% солютола, 4,4% миглиола и 1,5% витамина Е при 10 мг/мл Соединения 1 дала результаты, свободные от гемолиза при тестировании на 8 субъектах в дозе 20 мг/кг путем инфузии (Таблица 10).
Выводы, полученные из вышеприведенных исследований были таковы (а) меньший % солютола дает лучшие результаты, так же как и меньший % свободного ПЭГ в молекуле солютола (b) загрузка масла приводит к усилению удержания препарата в мицеллярном коре и дает лучшие результаты с точки зрения снижения гемолитического проявления по сравнению с простыми мицеллярными растворами и (с) более высокий % солютола дает более вариабельным результатам даже при наличии масел, как это наблюдалось для 20% солютола с миглиолом без масла, так и для комбинации миглиола с соевым маслом, по сравнению с 4,4% солютола в комбинации с вышеуказанными маслами. Добавление масла заметно улучшает некоторые композиции с высоким процентным содержанием солютола (например, композиции с витамином Е).
|
|
Таким образом, для того чтобы обеспечить приемлемый уровень уверенности в обеспечении не гемолитических результатов с обогащенными маслом внутривенных композиций Соединения 1, были намечены три критерия для помощи в выборе лидерной композиции.
(a) Проверка/повторная проверка надежности (композиция должна быть пригодной к проверке на чистоту в >1 исследовании)
(b) Иметь последовательную воспроизводимость по партиям (композиция должна быть пригодной к перекрестной проверке на чистоту через >1 партию)
(c) Должна быть надежной (композиция должна быть пригодной к проверке на чистоту по меньшей мере n=10 субъектов)
Композиции, которые были проверены на основе вышеуказанных принципов, включают 4,4% солютола, 1,1% миглиола и 0,66% соевого масла и 0,88% солютола, 0,2% миглиола и 0,12% соевого масло в дозе 2 мг/мл Соединения 1 при pH 8,0. Уровни солютола уменьшились в 5 раз в предыдущем случае с 4,4% до 0,88%, для дополнительного уменьшения случаев возникновения гемолитических проявлений и для получения воспроизводимых результатов, которые были более последовательными при более низких уровнях солютола как это подтверждено предыдущими данными.
|
4,4% солютола, 1,1% миглиола и 0,66% соевого масла, 2 мг/мл Соединения 1 вводили инфузионно, что привело только к трем случаям гемолиза из 59 крыс, протестированных с этой композицией. Исследование включало результаты, полученные: (а) из четырех отдельных опытов, проведенных на трех различных сайтах, (b) выполненные по меньшей мере с четырьмя различными партиями и (с) проведенные на в большом количестве субъектов, по меньшей мере на 20 крысах (n=20) в каждом из таких опытов (Таблица 11). Таким образом, композиция успешно отвечает вышеуказанным критериям (а-с).
Результаты оказались немного лучше для композиции с 0,88% солютола, 0,2% миглиола, 0,12% соевого масла и 2 мг/мл соединения 1 при pH 8,0; в итоге был только один случай гемолиза из 50 крыс, получивших эту композицию. Эти результаты также включают данные, полученные (а) из трех отдельных опытов, (b) выполненные с тремя различными партиями и (с) проведенные на большом количестве крыс (n=20) в двух таких опытах (Таблица 12).
|
D. Оценка случаев гемолиза у обезьян при внутривенном введении микроэмульсионной композиции Соединения 1
Композиции с уровнями солютола 1,5%, 3,5% и 5,0% с маслами (солютол:миглиол: соевое масло в соотношении 6.67:1.67:1) при pH 7,5 (pH 7,5 выбрана для химической стабильности композиции, также для уменьшения доли ионизированного Соединение 1, рКа 6.9, пригодная для красных кровяных телец) были проверены на 3 обезьянах для выявления любого случая гемолиза. Композиция с 1,5% солютола привела к гемолизу у одной из трех протестированных обезьян. Обезьяна с положительным тестом на гемолиз имела рейтинг +1 по шкале с 6 пунктами, с точки зрения тяжести гемолиза. Кроме того, 2,5% солютола также привел к одному случаю гемолиза из 3 обезьян, которые получили эту композицию (тяжесть+2 у обезьяны, давшей положительный тест на гемолиз) (Таблица 13).Тем не менее, 5,0% солютола дал ясные результаты, без случаев гемолиза у всех 3 обезьян, которым вводилась эта композиция (Таблица 13).
|
Пример 6 - Исследования с дополнительными композициями
В Таблице 14 приводится список других композиций, которые были получены и протестированы на гемолиз, в дополнение к ранее обсужденным композициям с солютолом, сорастворителем и циклодекстрином.
|
Кроме того, несколько других/идентичных композиций были получены и испытаны, как показано ниже. В каждом случае, препарат соединения 1 был в виде моногидрата HCl соли.
Композиция 1:
Соединение 1 + циклодекстрин (Каптизол);
Способ доставки (общая суточная доза) (объем дозы) (концентрация дозы) - результаты гемолиза:
Болюс (ОСД 5 мг/кг) (ОД 5 мл/кг) (КД 1 мг/мл) - 5/5
Медленная инфузия (ОСД 10 мг/кг) (ОД 10 мл/кг) (КД 1 мг/мл) - 1/5
Композиция 2:
Соединение 1+сорастворитель (ПЭГ/EtOH)
В-
МИ (ОСД 2,5 мг/кг) (1,5 мл/кг) (1,5 мг/мл) - 3/5
Композиция 3:
Соединение 1+Полиэтиленгликоль-660-гидростеарат, Solutol® HS 15 (22%), 20 мМ фосфатный буфер, pH 7, солютол HS 15 образует мицеллы
10 мг/мл лекарственного средства; 10, 20 и 30 мг/кг гемолиз у самцов, чаще в начальные периоды медленного болюсной инфузии.
Композиция 4:
Болюс или инфузия Доза (мг/кг) Доза об. (мл/кг) Доза конц. (мг/мл) Частота случаев гемолиза + Число случаев + гемоглобинурии/пол
|
Для следующих композиций дозы вводили в хвостовую вену крыс с помощью инфузии в течение 15 минут. Данные для некоторых композиций из таблицы 14 ниже повторяются в композициях 5-10. Данные приведены как доза (мг/кг); объем дозы (мл/кг); концентрация дозы (мг/мл) и частота случаев гемолиза
Композиция 5:
10 мл интралипид (20% соевого масла, 1,2% яичного фосфолипида, 2,25% глицерина), 100 мкл NK-1/этанол 1 мг/мл, 80 мкл 0,1 н. NaOH, pH 6,07 интралипид 5510/2 (эмульсия)
Композиция 6 (аналогично с интралипидом)
|
Композиция 7; 10 мл ЧСА (20%), 100 мкл NK-1/этанол раствор (1 мг/мл) pH 6,66
|
Композиция 8; 45 г сорбитола 15 HS, 15 г пропиленгликоля (ПГ), 250 мг NK-1, 8 г буфера PBS (1X), разбавленного 2 г раствора NK-1 в солютоле, pH 6,5, или 50 мг/мл лекарственного средства в солютоле: ПГ (3:1 по массе), разбавленного физиологическим раствором до концентрации 1 или 5 мг/мл лекарственного средства перед использованием.
|
Мицелла = менее 25 нм в диаметре Композиция 9:
|
Композиция 10
22% солютола, pH 7, фосфатный буфер 10 мг/мл, гемолиз при медленном болюсном введении в хвостовую вену крысы
Композиция 11
|
Композиция 12
|
Композиция 13
|
Композиция 14
|
Композиция 15
|
Композиция 16*
Обогащенная маслом мицелла (солютол HS 15+миглиол H12N (средне-цепочечный триглицерид масла)/соевое масло)
Главные ненасыщенные жирные кислоты в триглицеридах соевого масла представлены 7%-ми альфа-линоленовой кислоты (С-18:3), 51%-ом лино-левой кислоты (С-18:2) и 23%-ами олеиновой кислоты (С-18:1). Оно также содержит насыщенные жирные кислоты: 4% стеариновой кислоты и 10% пальмитиновой кислоты. 4,4% солютола (1.45% свободного ПЭГ)/1,1% миглиола (pH 8)
|
Композиция 17
|
Композиция 18
|
Композиция 19
|
Композиция 20
|
Композиция 21
|
Композиция 21А
|
Ком позиция 21 В
|
Композиция 22
Соединение 1 2 мг/мл 0.88%Солютола/%0.2 Миглиола/ 0.12%сои 15 минутная инфузия
|
Композиция 23
Соединение 1 2 мг/мл 4.4% Солютола /1% Миглиола/0.6% сои, pH 7,5, 15 минутная инфузия
|
Композиции 24-26;
- Кремофор RH40 1,25%; Доза-20 мг/кг; Конц. препарата: - 2 мг/мл; Гемолиз: - 0/8, 15 мин инфузия
- Кремофор EL 1,25%; Доза-20 мг/кг; Конц. препарата: - 2 мг/мл; Гемолиз: - 1/6, 15 мин медленная инфузия
- Кремофор EL 6% Доза-20 мг/кг; Конц. препарата: - 2 мг/мл; Гемолиз: -4/4;
Болюсное введение
Композиция 27 (смешанная мицелла):
960 мкл ПОФХ/этанол (240 мг ПОФХ)
200 мг Na-соли гликолевой кислоты
15 мг соединения 1 (HCl-моногидрата)
3 мл буфера PBS (1X)
Целевая конц.: 5 мг/мл
pH: 6,6
Встряхнуть энергично перед разбавлением. Разбавить сток-раствор буфером PBS (pH 7,4) до требуемой концентрации. Инфузия должна быть завершена в течение 2 ч после разведения. ПОФХ: пальмитоилолеилфос-фатидилхолин.
Примечание - эта композиция дала высокое число случаев гемолиза при медленном, в течение 15 мин, инфузионном введении 5 мг/кг; 5 мл/кг и при концентрации дозы 1 мг/мл. В отсутствие дополнительной информации, эта композиция не подходит в качестве внутривенной композиции при частном пути введения Длительные инфузии могут быть пригодными и/или необходимыми для минимизации гемолиза.
Композиция 28
6,7% Солютола и 13,3% интралипида, 2 мг/мл лекарственного средства
В предпочтительном варианте осуществления, композиции Соединения 1 вводятся внутривенной инфузией в течение 15-30 минутного периода и могут быть представлены во флаконе, содержащим 100 мг/флакон с 50 мл раствора и концентрацией лекарственного средства 2 мг/ мл. Доза и объем могут быть скорректированы для обеспечения дополнительных целевых доз (например, целевая доза от 100 до 200 мг).
Одно особое преимущество соединения 1 заключается в том, что оно имеет период полураспада (T1/2) 180 часов, что делает его особенно удобным для разовой терапевтической дозы на цикл лечения CINV (тошноты и рвоты, индуцируемых химиотерапией); T1/2 при дозах, изменяющихся в диапазоне от 5 до 200 мг как нг/мл активного фармацевтического ингредиента, по времени при разных дозах: нг/мл на момент непосредственно после введения меняется в диапазоне от 850 нг/мл (200 мг), 430 нг/мл (100 мг), 280 нг/мл (50 мг) с половинной концентрацией, достигаемой для всех доз в пределах 180 ч после введения.
Внутривенные композиции могут быть предварительно смешаны при 2 мг/мл или могут быть в виде концентрата для разбавления (10 мг/мл и 20 мг/мл для разбавления до 2 мг/мл).
Композиция с Intralipid®, содержащая 10 мл Intralipid® (20%) (20% соевого масла, 1,2% фосфолипидов яичного желтка, 2,25% глицерина и воду для инъекции); 100 мкл раствора смеси соединения 1/этанол (1 мг/мл), 80 мкл 0,1 н. NaOH для получения pH 6.07. Композиция интралипида является эмульсией.
Эмульсионная композиция (композиция 6) состоит из пальмитоилолеоил-фосфатидилхолина (ПОФХ), олеиновой кислоты, глицерина и твин 80.
Размеры мицеллярных частиц, как правило, менее 100 нм и входят в диапазон от 15 до 100 нм. Мицеллярная композиция содержит 45 г Солютола HS; 15 г пропиленгликоля; 250 мг соединения 1 (в виде кристаллической соли моногидрата гидрохлорида); 8 г буфера PBS (1X) разбавленного 2 г соединения 1, Солютола 15S и раствор пропиленгликоля; pH 6,5 до 10 мг/ мл; разбавленного перед употреблением или получали как 50 мг/мл раствор с лекарственное средством в Солютола: ПГ (3:1, В:В), разбавленного физиологическим раствором до концентрации препарата 1 или 5 мг/мл перед использованием.
Композиция ЧСА содержала 10 мл 20% ЧСА, 100 мкл раствора Соединение 1/этанол (1 мг/мл), pH 6,66.
Пример 7 - Эмульсионная композиция, удобная для болюсного и медленного инфузионного введения
Также была разработана парентеральная эмульсионная композиция на основе эмульсии из среднецепочечных и длинноцепочечных триглицеридов и подходящего использования эмульгаторов, таких как лецитины из яйца и олеат натрия. Некоторые из этих композиций, как описано ниже, дали ясные результаты при введении как болюсным путем, так и медленной инфузией. Кроме того эмульсии привели к уменьшению гемолиза даже при очень высоких дозах 30 мг/кг. Композиции подвергали микрофлюидизации, в которой условия обработки оптимизировали так, чтобы получить эмульсии с меньшим размером капли и узким распределением частиц по размерам (psd). Все композиции процессировались в камере взаимодействия H30Z (200 микрон) при давлении 2000 пси, что позволило достичь средних диаметров до 500 нм и D90 менее 600 нм. Некоторые композиции были также охарактеризованы как физически стабильные после одного цикла стерилизации в автоклаве при 121°C в течение 20 минут. Такие композиции не приводили к заметному увеличению размера капель после процесса стерилизации. Таким образом, физически стабильная композиция, имеющая соединение 1 (в форме ее кристаллического моногидрата гидрохлорида при приготовлении) была пригодна как для болюсного, так и для медленного инфузионного внутривенного введения без гемолиза в подопытных животных.
Эмульсионные композиции, описанные ниже, обладают преимуществом в виде повышенного удержания лекарственного средства в гидрофобном коре, увеличенного размера капли по сравнению с мицеллярной композицией, что замедляет выход/распределение препарата из масляных капелек в красные кровяные тельца, а также обеспечивают большую гибкость в разработке составов путем использования природных липидов в качестве эмульгаторов, избегая таким образом, например, свободного полиэтиленгликоля, который присутствует в солютоле, и что таким образом предотвращает переход лекарственного средства из гидрофобного окружения в красные кровяные тельца. Парентеральные эмульсии используются для переноса плохо растворимых в воде лекарственных средств и обычно представляют собой маленькие капельки масла в водном растворе. Предпочтительный эмульгатор выбирается из тех, которые считаются безопасными для использования при парентеральном введении. В настоящей разработке лецитин является предпочтительным эмульгатором. Очищенный яичный лецитин является наиболее предпочтительным. Дополнительные стабилизаторы могут быть добавлены в том числе, например, олеиновая кислота или олеат натрия. Среднецепочечные (миглиол) и длинноцепочечные (соевое масло) триглицериды также могут быть добавлены. Яичный фосфатидилхолин может быть использован в качестве стабилизатора также как и Lipoid Е 80 (77,7% ФХ, 7,8% ФЭ), 2,5% ЛФХ, СФМ 3,0%) (ФХ = фосфатидилхолин, ФЭ = фосфатидилэтаноламин, ЛФХ = лизо-ФХ, СФМ = сфингомиелин) или Lipoid E80S. Глицерин может быть использован как регулятор тоничности и этанол может использоваться как сорастворитель/солюбилизатор для лецитина и лекарственного средства (соединение формулы 1 или его соль, например, соединение 1).
Следующие материалы, используемые в этом примере суммированы в Таблице 15.
|
Гемолитическое тестирование на животных
Методологией этого примера являлось введение каждой композиции в течение 15 минут методом внутривенной инфузии с использованием инфузионного насоса или введение композиций в течение 2 минут болюсной инфузией с использованием шприца и доставки композиции в хвостовую вену крысы. Моча тестировали полосками на присутствие свободного гемоглобина использованием метода объединенного сбора мочи в 6-часовом интервале. Как минимум четыре субъекта протестировали для каждой композиции и наблюдали частоту гемолиза у таких животных.
Получение непроцессированной эмульсионной композиции Общая методика получения эмульсии была следующей (Фиг.3):
(1) Взвешивание необходимого количества миглиола и соевого масла в стакане;
(2) Гомогенизация вышеназванного премикса в течение 5 минут для смешивания двух компонентов;
(3) Добавление необходимого количества Lipoid E80® / Lipoid E80S в вышеназванный стакан;
(4) Добавление необходимого количества этанола, содержащего 200-250 мкг/мл соединения 1 в виде моногидрата гидрохлорида;
(5) Взвешивание нужного количества стерильной воды в отдельном стакане;
(6) Добавление определенного количества глицерина в водную фазу и смешивание в течение 2-3 минут;
(7) Добавление определенного количества олеата натрия в водную фазу, если он присутствует в составе;
(8) Раздельное нагревание масляной и водной фазы около 65-70°C в течение 15-20 минут;
(9) Добавление водной фазы в масляную фазу и добавление стерильной воды, сколько необходимо до достижения конечного объема, гомогенизация дисперсии приблизительно 1-2 минуты;
(10) Охлаждение дисперсии до комнатной температуры перед заполнением пробирки.
Получение процессированных эмульсионных композиций с использованием микрофлюидизирования
Необработанная эмульсия, описанная выше или в объеме изобретения, могла быть последовательно пропущена через микрофлюидизатор, что приводило к более стабильной эмульсии с узким распределением капель по размерам. Процесс микрофлюидизирования предполагает использование микрофлюидизатора, который сконструирован для обеспечения необходимого давления на продукт с постоянной скоростью потока продукта. В результате поток продукта разгоняется до высоких скоростей через точно подогнанные геометрически фиксированные микроканалы (камера взаимодействия), создавая сдвиг скоростей в потоке продукта, который значительно выше, чем любые другие традиционные методы обработки. Это приводит к тому, что капли распадаются на меньшие размеры и приводит к образованию более равномерного и узкого распределения капель по размерам для повышения физической стабильности.
Методика для процессирования/характеристики композиций с использованием микрофлюидизатора
Коллоидные эмульсии обрабатывали с использованием либо камеры взаимодействия F20Y (75 микрон) с давлением в диапазоне от 4100 до 10000 пси либо камеры взаимодействия H30Z (200 мкм) при давлении 2000 и 5000 пси. Количество проходов составляло от одного до трех. Использовали водяную баню для камеры взаимодействия, чтобы предотвращать повышение температуры продукта в течение стадии микрофлюи-дизации. Анализ включал использование оптического микроскопа и анализатора размеров частиц. Изображения регистрировали для каждого образца, полученного после обработки. Анализ размеров частиц производили для непроцессированного образца и в конце каждого цикла микрофлюиди-зации.
Стерилизация паром
Композиции помещали и запечатывали в стеклянную пробирку и помещали в автоклав для окончательной стерилизации на один цикл при 121°C в течение 20 минут. Размер частиц измеряли с помощью прибора Malvern Zetasizer Nano-ZS для наблюдения изменений в распределение размера капель после процесса стерилизации. Прибор работает по принципу динамического рассеяния света.
Композиции и результаты
Две эмульсионные композиции были сформированы с яичным ФХ в сочетании со средне- и длинноцепочечными триглицеридами. Эмульсионная композиция не вызывала гемолитических случаев как при инфузии, так и при болюсном введении 4 и 6 крысам, соответственно (Таблица 16). Тем не менее, именно эти композиции не были стабильными в 24 часовой период, если их не встряхивать с приданием сдвига для адекватного диспер-гирования.
|
Эмульсионные композиции для дополнительных испытаний на крысах были сформулированы при 20 мас.% загрузки маслом с использованием в качестве эмульгатора Lipoid E80S для придания физической стабильности. Высокое содержание масла было желательно, чтобы сохранять Соединение 1 в гидрофобном коре. Lipoid E80S имеет высокое содержание фосфа-тидилэтаноламина (ФЭ) и действует как анионный эмульгатор. Данный продукт коммерчески доступен от Lipoid KG of Ludwigshafen, Германия. Содержание ФЭ в Lipoid E80S составляет около 20%, а яичный фосфатидилхолин составляет около 80% вещества. Эти композиции были протестированы в дозе 20 мг/кг соединения 1, как при болюсном, так и при инфузион-ом методе. Количество случаев гемолиза было высоким при загрузке этим маслом, как это видно из Таблицы 17.
|
Снижение загрузки масла в композиции с 20% до 10% дало ясные результаты по гемолизу как для болюсного пути, так и для медленного инфузи-онного пути (Таблица 18).
|
Таким образом, предпочтительный состав эмульсионной композиции содержит 10% загруженного масла. Олеат натрия добавляли как дополнительный ингредиент, но не было получено подходящих гемолитических результатов при всех способах доставки (Таблица 19).
|
Композиции без олеата натрия и с более высокой концентрацией загруженного лекарственного средства (30 мг/кг) также протестировали (Таблица 20).
Таблица 20
|
Пять эмульсионных композиций, перечисленных ниже, обработали с помощью микрофлюидной методики. Окончательный размер частиц (D10, D50 и D90), полученных после обработки этих композиций, а также количество проходов, использованное давление, необходимое для продавливания
жидкости через камеру взаимодействия и тип использованной камеры взаимодействия отражены в Таблице 21.
|
|
INFWSO является таким же составом композиции как в партии Мо.:85266-M/L 10%инф
INFSO имеет тот же состав композиции как в партии No.:85266-M/L10% SO инф
BOL10WSO имеет тот же состав композиции как в партии No.: 85266-M/L10%
BOL10SO имеет тот же состав композиции как в партии No.:85266-M/L10% SO бол
BOL15SO имеет тот же состав композиции как в партии No.: 85266-M/L10% бол 3
Более узкое и более равномерное распределение частиц по размерам (psd) получили для всех процессированных композиций. Как видно на Фиг.4, кривая psd для BOL15SO значительно смещается влево после микро-флюидизации по сравнению с непроцессированной композицией, подвергнутой трем проходам при давлении 2000 пси. Значения D50 и D60 для непроцессированной BOL15SO были 4,505 мкм и 10,745 мкм соответственно, и сдвигаются до 410 нм и 569 нм, соответственно, после трехкратного прохождения композиции с использованием камеры взаимодействия H30Z (200 мкм) при давлении 2000 пси. Микроскопические изображения подтверждают однородность в psd масляных капель, полученных после трехкратной микрофлюидизации композиции при давлении 2000 пси по сравнению с одним проходом при 2000 пси (Фиг.7).
На Фиг.7 показаны изображения в оптическом микроскопе композиций BOL15SO и INFWSO в течение микрофлюидизации. Результаты оптической микроскопии наглядно показывают влияние числа циклов микрофлюидизации на композиции, BOL 15SO (2 верхних изображения) и INFWSO (4 изображения внизу). Наблюдаемые изображения на правой стороне лишены очень крупных масляных глобул с более равномерным и узким распределением по размерам.
Аналогичное наблюдение было сделано по отношению к INFWSO. Значения D50 и D90 изменились с 2,643 мкм и 4,848 мкм для не-процессированной композиции, до 442 нм и 578 нм после трех проходов при 2000 пси в камере взаимодействия H30Z. Кривая psd масленых капель далее смещалась левее и значения D50 и D90 были зафиксированы как 318 и 437 нм, соответственно, используя камеру взаимодействия F20Y с микропористыми каналами в 75 микрон после 3 проходов композиции при давлении 4100 пси (Фиг.5) Микроскопические изображения процессированной композиции после одного прохода при 2000 пси и при 4100 пси показывают большие масляные глобулы и неоднородное psd, которое наглядно становилось более однородным после большего числа проходов композиции (Фиг.7).
Образец BOL10WSO дал аналогичные результаты по процессированию, отраженные на кривой psd для капель масла, значительно сдвинутой влево после микрофлюидизации. Значения D50 и D90 были зафиксированы как 422 мкм и 595 мкм при использовании камеры взаимодействия H30Z при процессировании этих эмульсий при давлении 2000 пси после трех циклов, по сравнению с непроцессированными композициями, которые имели значения D50 и D90 как 3892 мкм и 6873 мкм. Использование более высокого давления в 5000 пси в той же камере взаимодействия и при трех проходах дополнительно сдвигает кривую влево, давая значения D50 и D90 341 нм и 482 нм, соответственно (Фиг.6). Эффект микрофлюидизации в зависимости от числа циклов и более мелкие масляные капли, полученное после обработки композиции при более высоком давлении наблюдается четко в микроскопических изображениях (Фиг.8).
На Фиг.8 показаны изображения в оптическом микроскопе композиций BOL10WSO в течение микрофлюидизации.
Результаты оптической микроскопии четко показывают влияние давления и числа циклов микрофлюидизации на композицию BOL10WSO. На изображениях с правой стороны наблюдается отсутствие очень больших масляных глобул с большей однородностью и узким распределением по размеру. Кроме того, как и ожидалось, капли масла также становятся меньше, когда композиция подвергается микрофлюидизации при более высоком давлении.
Аналогичные наблюдения были сделаны для композиций INFSO и BOL10SO (Фиг.9 и 10). На Фиг.9 показаны изображения в оптическом микроскопе композиций BOL15SO и INFWSO в процессе микрофлюидизации. Результаты оптической микроскопии ясно показывают влияние числа циклов микрофлюидизации на композицию INFSO. Большее число циклов приводит к более равномерному распределению частиц по размерам. На Фиг.10 показаны изображения в оптическом микроскопе композиций BOL10SO в процессе микрофлюидизации.
Таким образом, процессирование микрофлюидизиованием дает физически стабильные эмульсионные композиции, содержащие соединение формулы I и его фармацевтически приемлемые соли. Настоящее изобретение также относится к эмульсионным композициям Формулы I и их солей, имеющих капли со средним диаметром около 500 нм или менее и D90 около 600 или менее. Еще меньшие размеры капель были получены, когда композиции подвергались дополнительным проходам и более высоким давлениям. Далее композиции подвергались автоклавированию для стерилизации композиций для внутривенного введения. Видимых проблем со стабильностью не возникало в результате автоклавирования. INFWSO и BOL10WSO автоклавировали и после визуального осмотра средний размер частиц, полученных после одного цикла был определен как 353,4 нм и 336,7 нм, соответственно (Фиг.11 и 12). Таким образом, результаты с отсутствием гемолиза воспроизводимо достигаются при использовании эмульсионных композиций на основе фосфолипида как для болюсного, так и инфузионного введения.
Как указывалось выше, композиции являются полезными для профилактики и лечения тошноты и рвоты, связанных с лучевой и химиотерапией терапией при лечении рака и послеоперационных проявлений тошноты и рвоты. Курс терапии содержит одну дозу композиции, содержащую соединение Формулы I или его соль, за один день до начала химиотерапии с последующим приемом раз в день во второй и третий дни. Курс лечения также содержит прием единственной дозы в первый день. Внутривенный раствор может быть в форме раствора с концентрацией 2 мг/мл, 50 мл раствора, необходимого для введения 100 мг пациенту, нуждающемуся в таком лечении. Композиция может быть готова к употреблению в виде предварительного смешанного раствора или концентрата, который можно разбавить перед использованием. Лиофилизированные композиции, описанные в настоящем изобретении, могут быть в форме порошка, который смешивается с физиологическим раствором или соответствующим буферным раствором перед использованием.
Пример 8 - Пролекарства
Два Пролекарства, показанных ниже, были получены путем взаимодействия активированного карбонильного соединения с соединением формулы I. Соль глюкаминфосфата, показанная ниже, дала результат свободный от гемолиза в водном растворе глюкозы без какого-либо солюбилизатора при измерении в двух различных точках времени и как в моче, так и плазме.
Пролекарства:
,
Данные гемолиза для Пролекарства - соли глюкамин фосфата в глюкоза/вода:
|
Данные для амина, полученного с носителем (10% Солютола): 0.25 часа, 3/3 гемолиз.

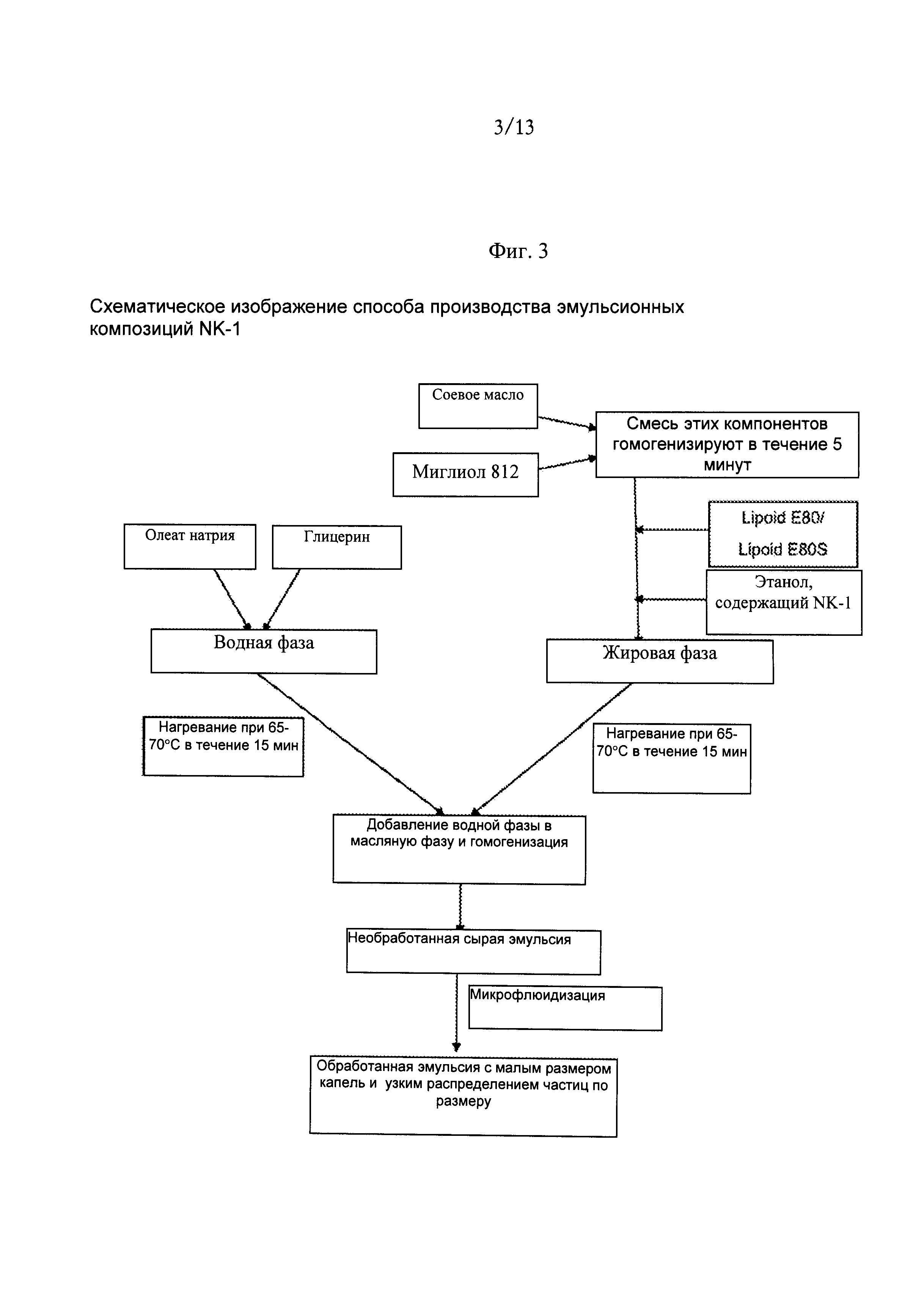
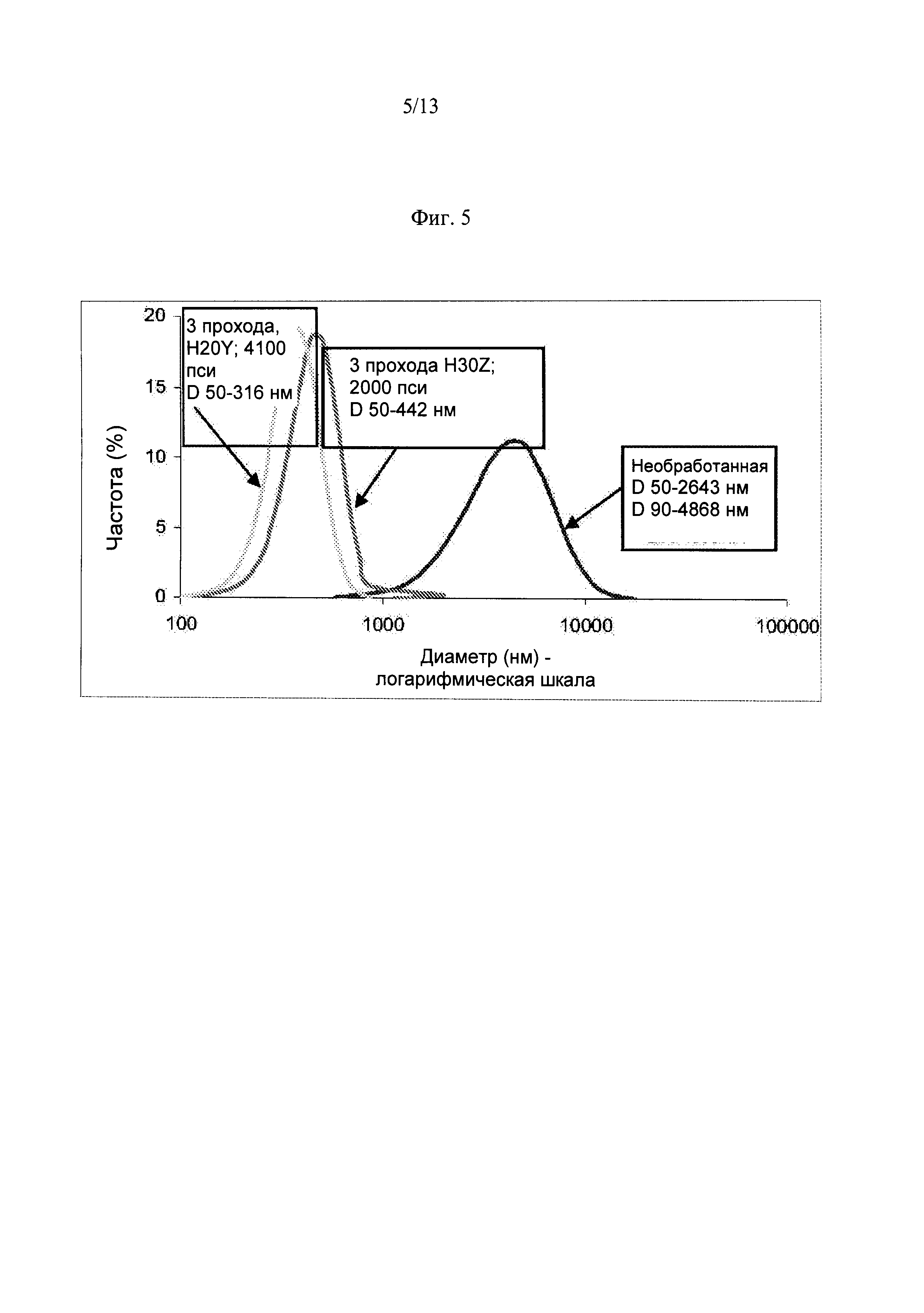
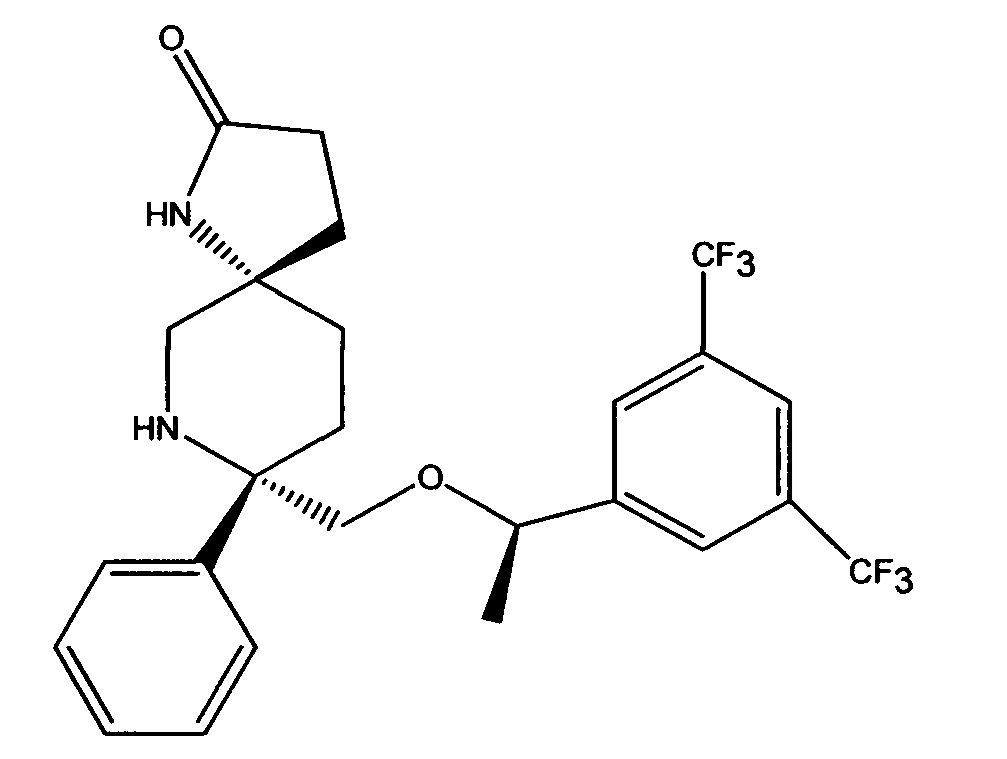
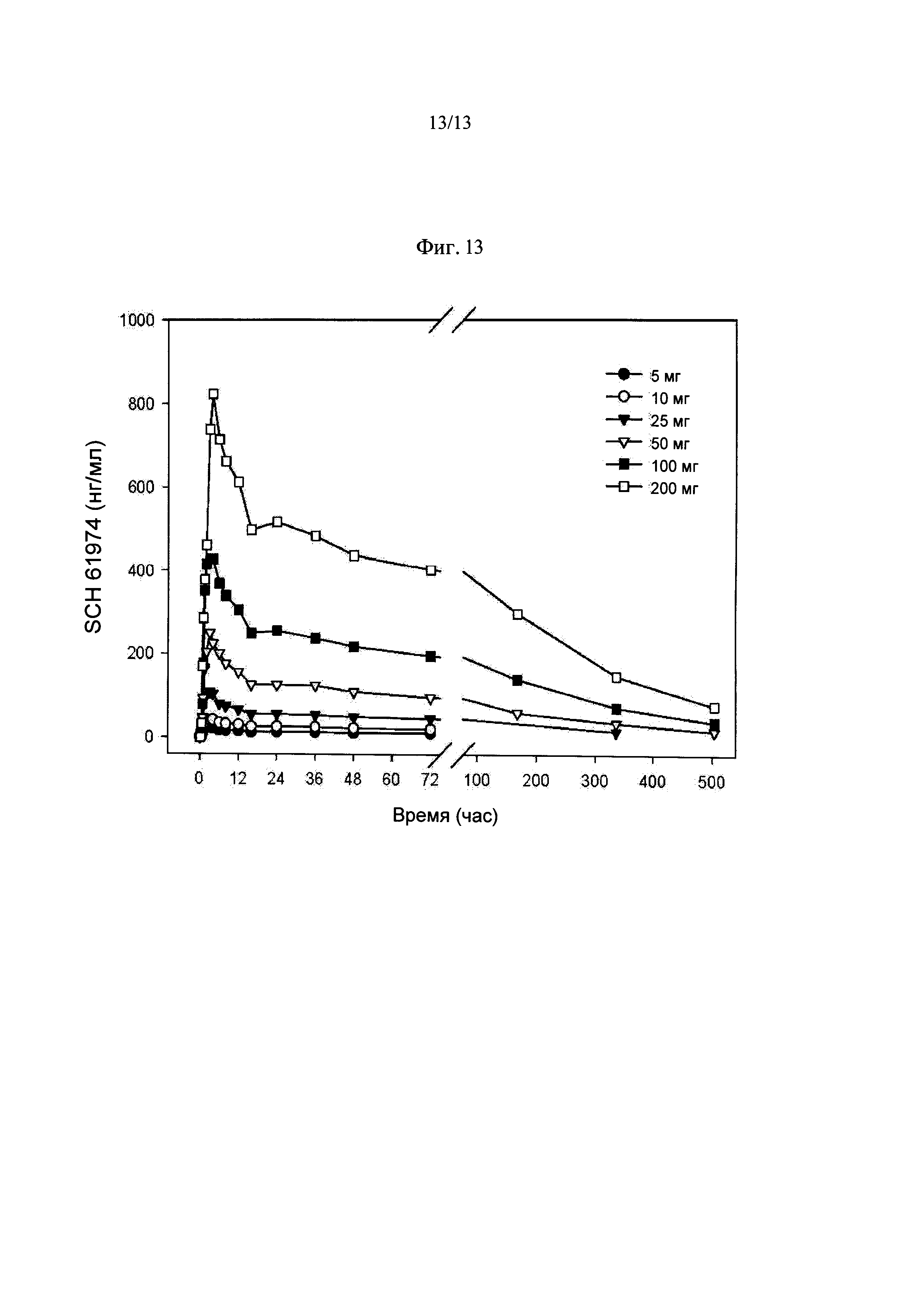
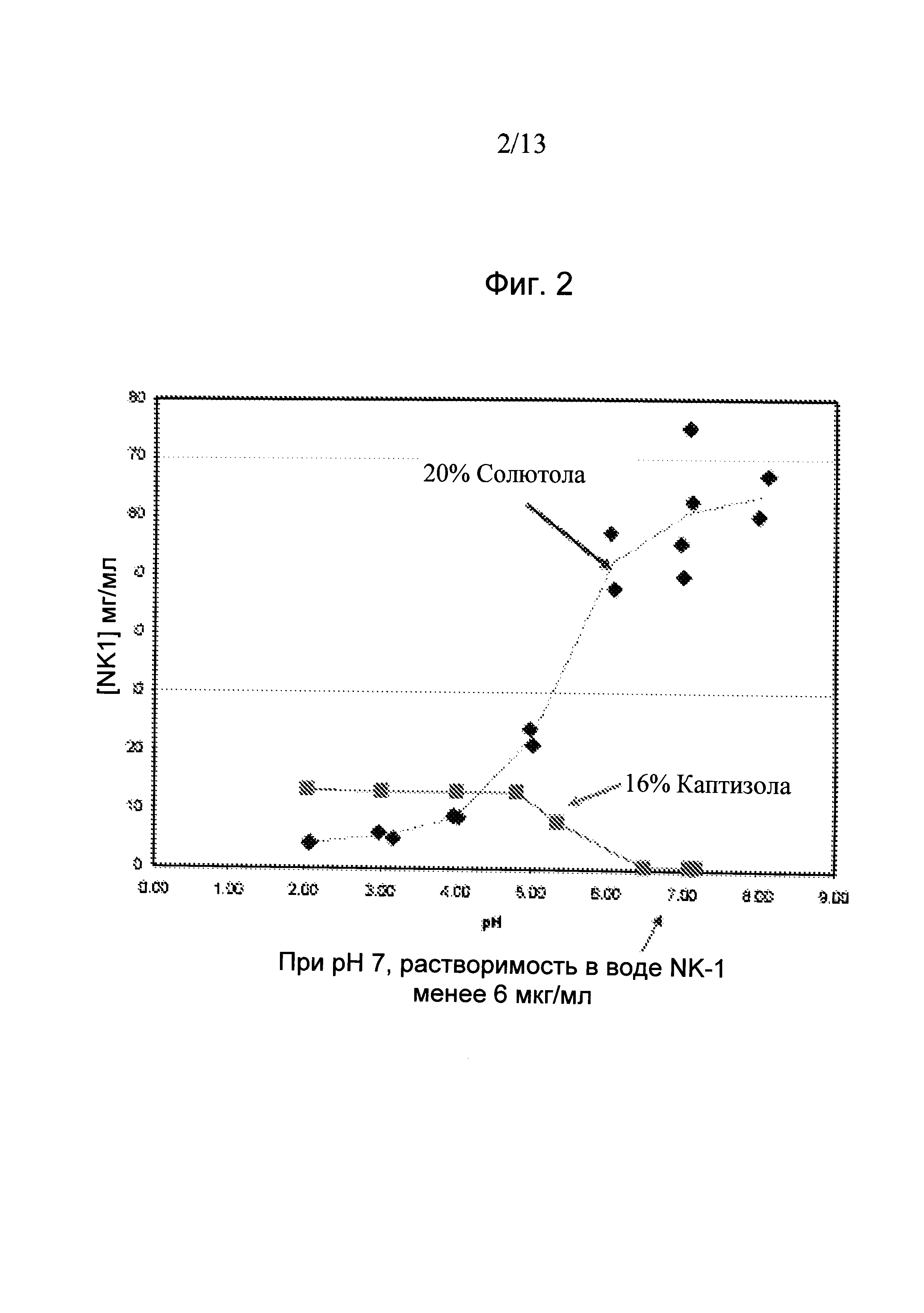
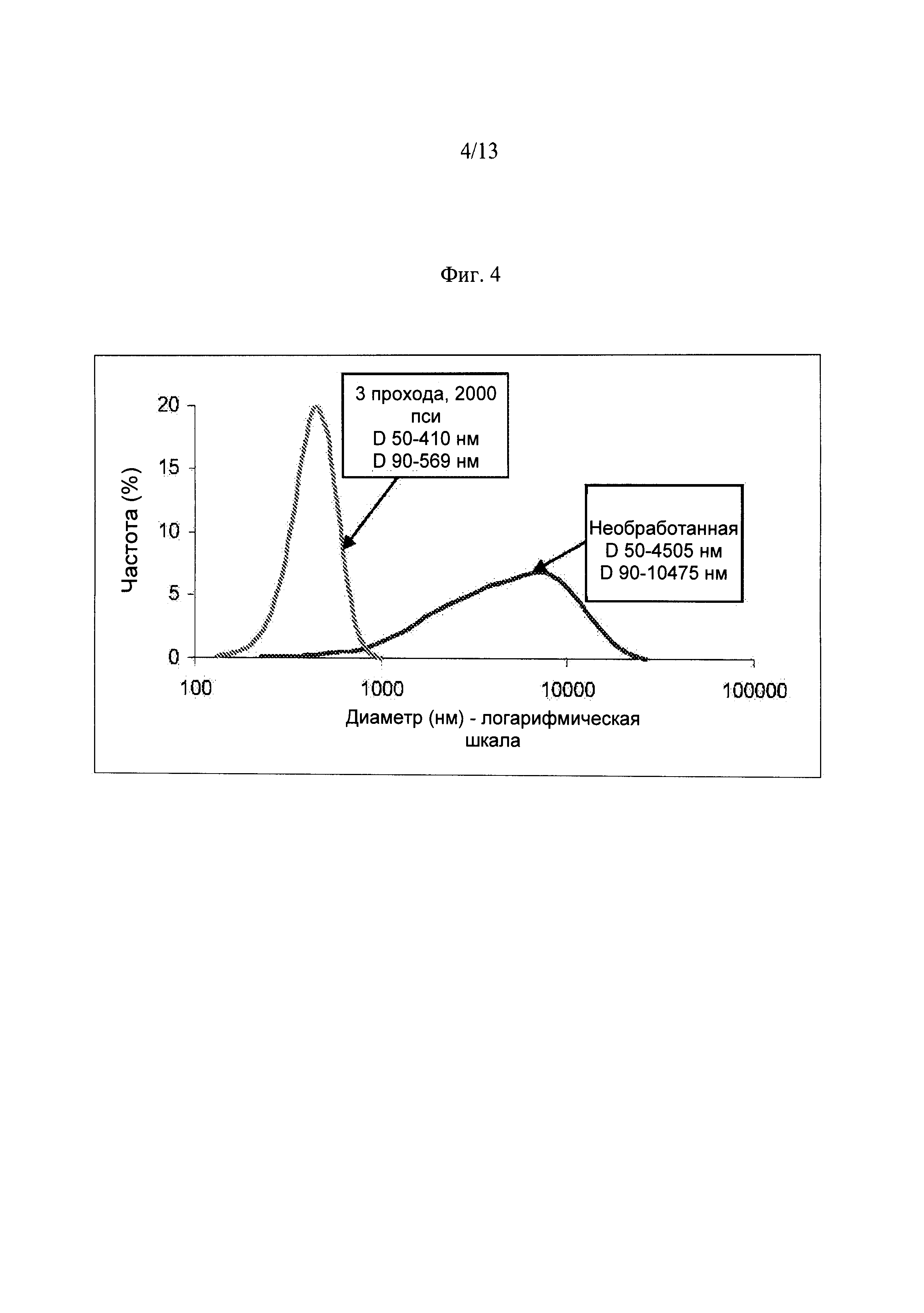


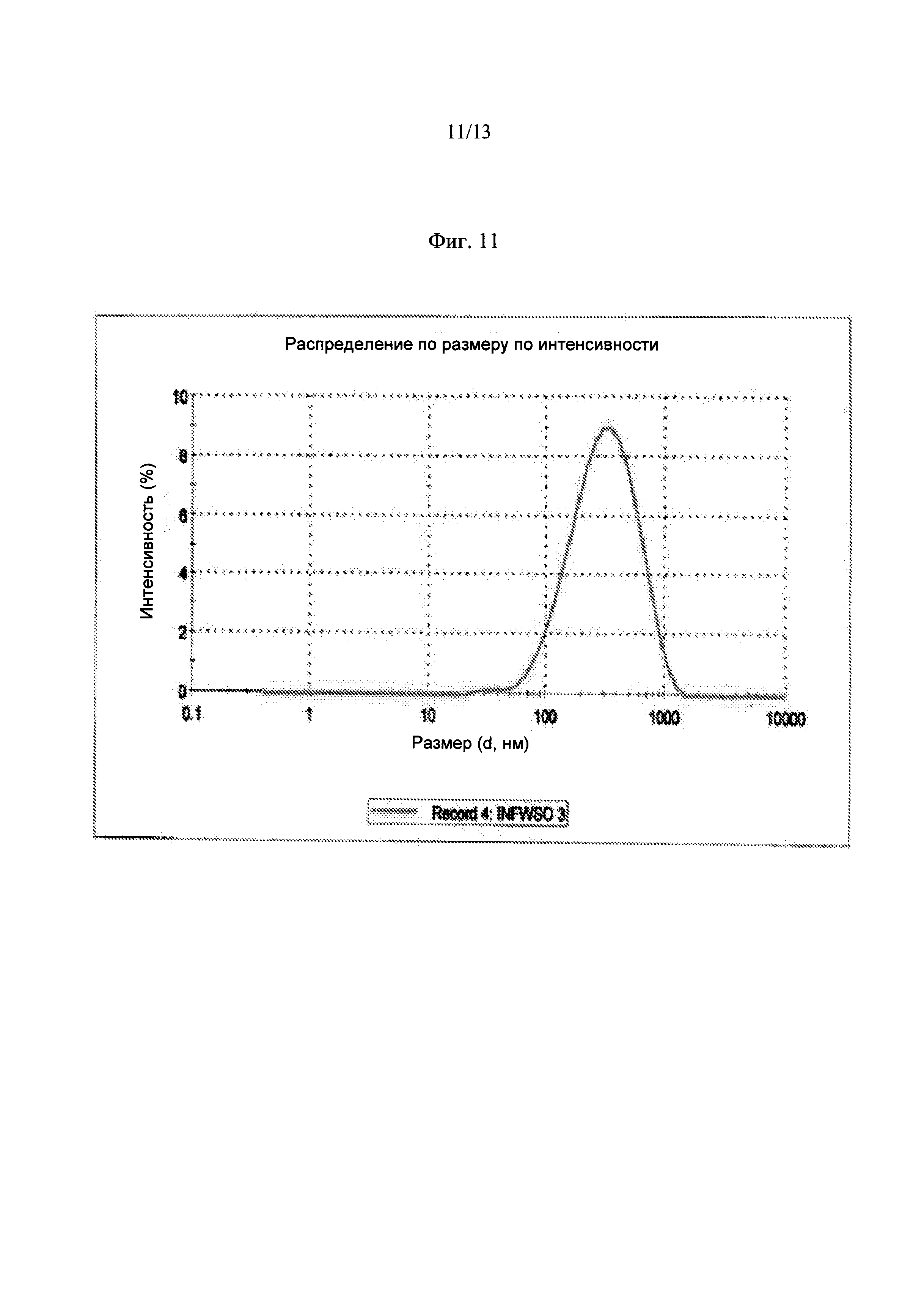
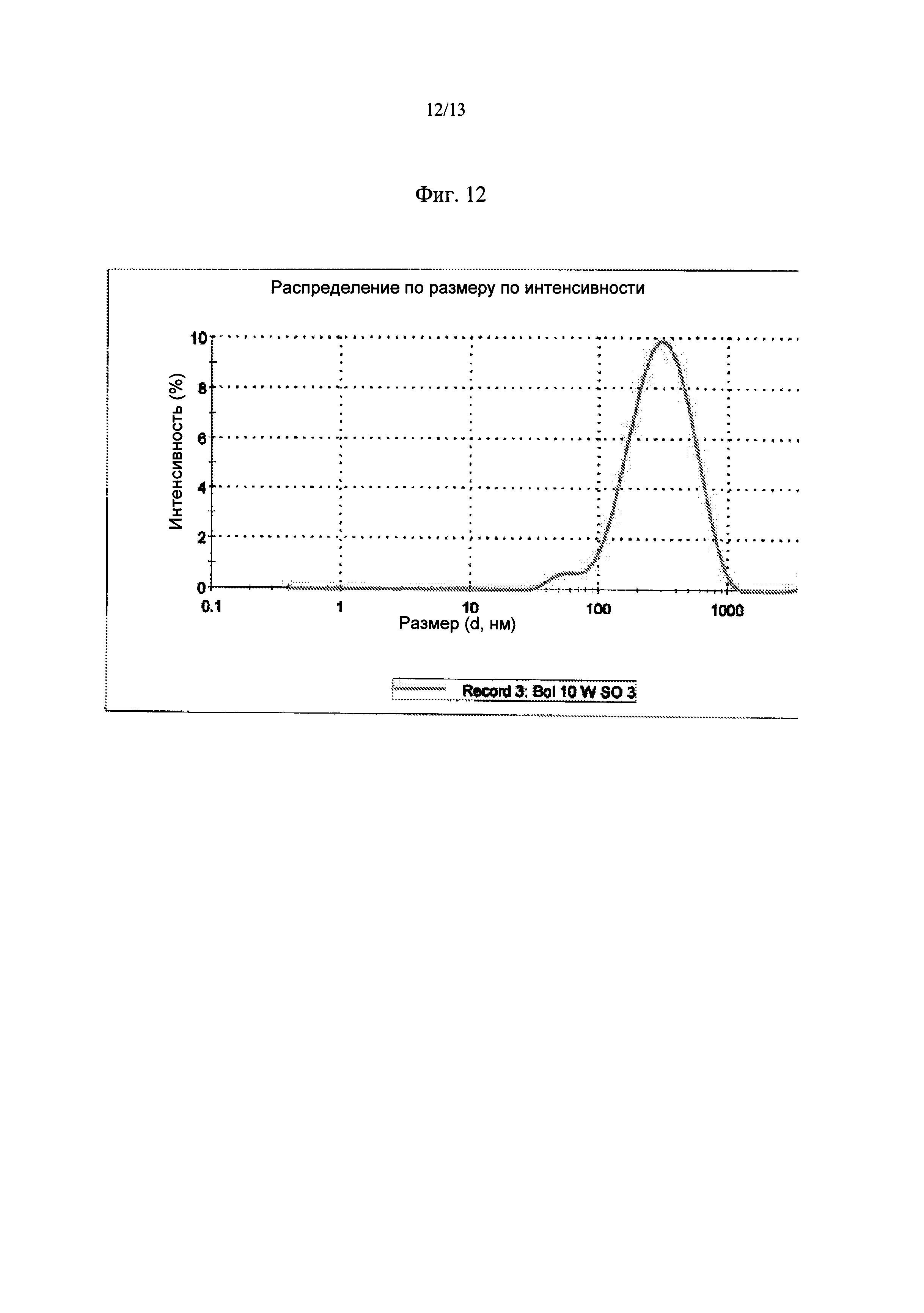
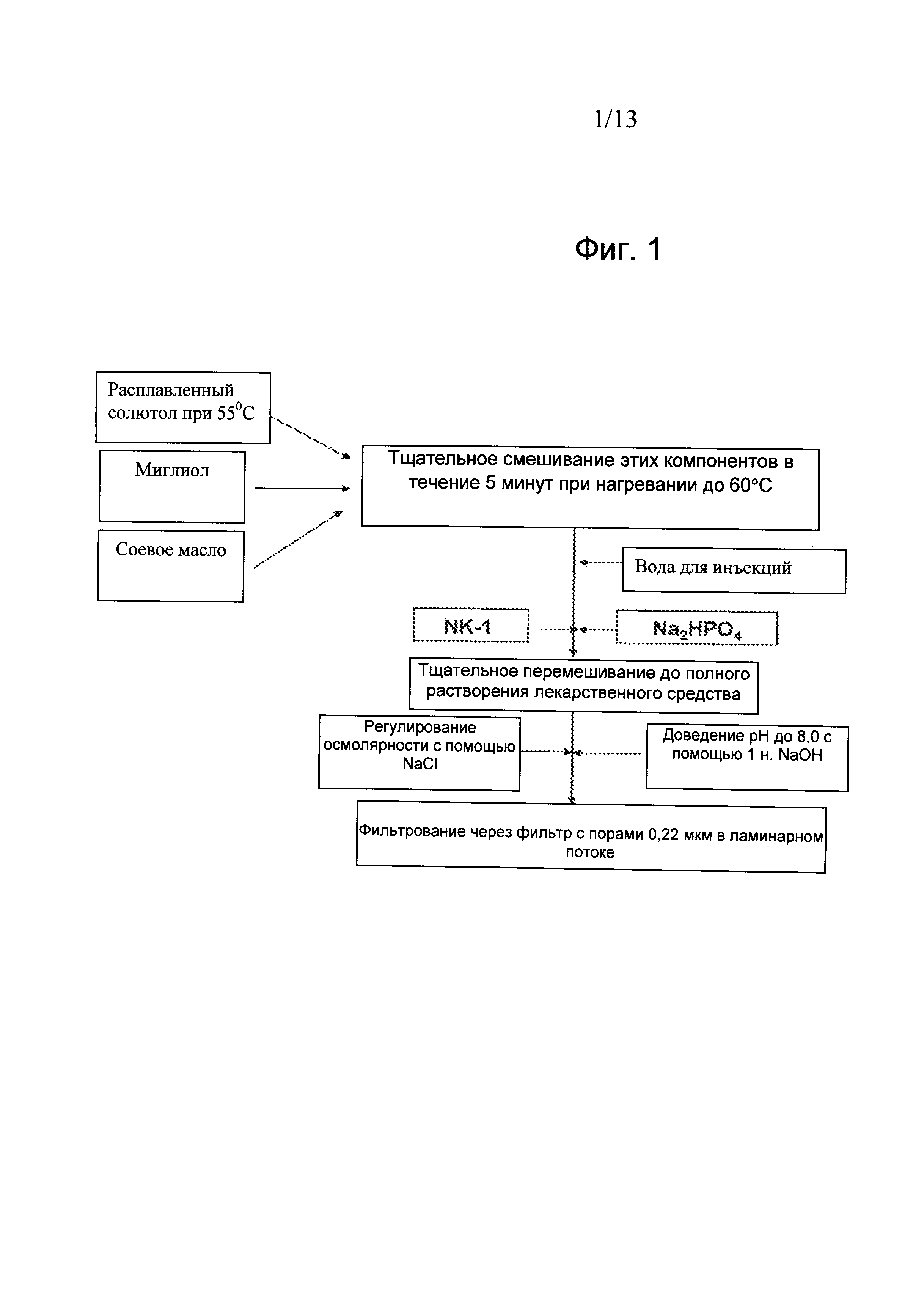
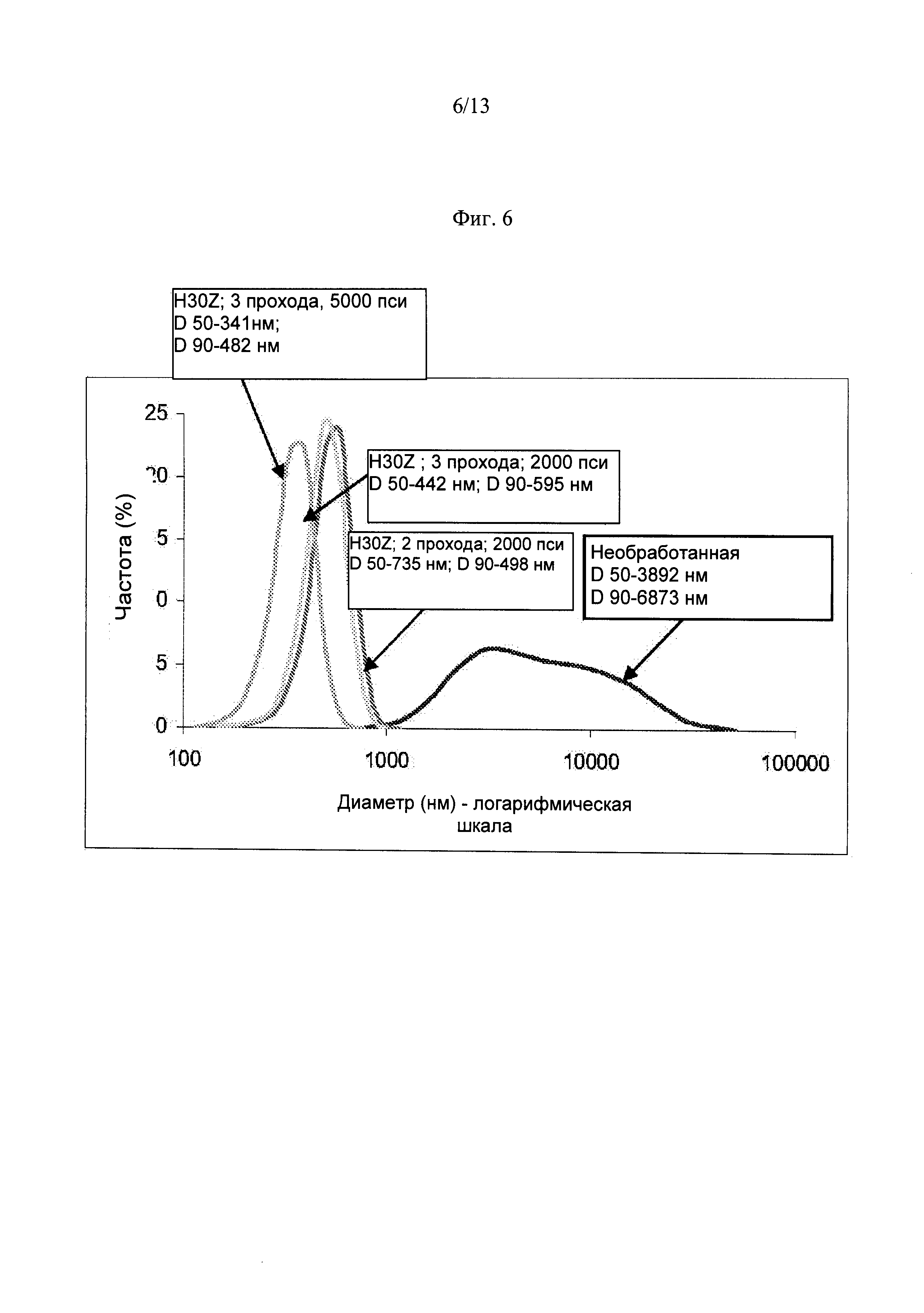
Фармацевтические лекарственные формы и композиции фенилэфрина для абсорбции в ободочной кишке

