Результат интеллектуальной деятельности: ПОСЛЕДУЮЩАЯ ОБРАБОТКА ДЕБОРИРОВАННОГО ЦЕОЛИТА БЕТА
Вид РИД
Изобретение
Настоящее изобретение относится к способу последующей обработки цеолитного материала, обладающего каркасной структурой ВЕА, включающей Х2О3 и YO2, с помощью которого цеолитный материал, обладающий каркасной структурой ВЕА получают и затем обрабатывают жидкой системой растворителей и затем обрабатывают жидкой водной системой. Кроме того, настоящее изобретение относится к цеолитному материалу, который получают или можно получить этим способом. Кроме того, настоящее изобретение относится к применению указанного цеолитного материала, в частности, в качестве катализатора или компонента катадидадора.
Цеолиты широко применяют в химической промышленности, например, в качестве гетерогенных катализаторов в различных химических и нефтехимических технологиях. Обычно цеолиты представляют собой кристаллические алюмосиликаты, обладающие микропористой структурой. Особые свойства цеолитов приписывают, в частности, их пористой структуре, обычно представляющей собой регулярную систему пор молекулярных размеров, и их особому химическому составу. Существует много известных структур цеолитов, натуральных цеолитов или синтетических цеолитов, которые можно использовать в качестве гетерогенных катализаторов в различных случаях.
Для изменения характеристик цеолитных материалов, таких как их структура или их состав, можно использовать способы последующей обработки. Самыми распространенными способами последующей обработки, описанными в литературе, являются обработки паром, обработки кислотой или обработки основанием.
Обработку паром часто используют для повышения активности и стабильности цеолита при различных селективных реакциях. Например, в ЕР 0013433 А1 описано применение обработки паром для повышения активности цеолита за счет увеличения отношения Si/Al. Эта обработка паром влияет не только на отношение Si/Al, но и на кислотно/основные характеристики и гидрофильность/гидрофобность цеолита.
Обработка кислотой может оказывать аналогичное влияние и также может привести к изменению состава каркаса. Например, в WO 02/057181 А2 описано деборирование силиката, при котором для проведения стадии деборирования используют кислоту. В конкретных примерах, приведенных в этом документе, используют ледяную уксусную кислоту, а в возможных вариантах осуществления, которые дополнительно не конкретизированы, описаны хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота и винная кислота.
WO 2009/016153 А2 описана комбинация обработки паром и обработки кислотой. По данным этого документа модифицированные фосфором молекулярные сита обрабатывают паром при высоких температурах и затем проводят стадию выщелачивания кислым раствором для удаления Al из цеолитного материала.
И обработка паром, и обработка кислотой могут оказать значительное влияние на характеристики цеолитного материала. При проведении для цеолитного материала, включающего и трехвалентные, и четырехвалентные структурные компоненты X и Y в виде Х2О3 и YO2 соответственно, обработки паром и/или кислотой, молярное отношение X2O3:YO2 уменьшается. Однако установлено, что, например, степень кристалличности цеолитных материалов можно уменьшить путем обработки паром и/или обработки кислотой. Кроме того, установлено, что гидрофобность цеолитных материалов, характеризующаяся поглощением воды цеолитным материалом, уменьшается. Поэтому предполагают, что и обработка паром, и обработка кислотой приводят к частичному превращению цеолитного материала в аморфный материал и к изменению гидрофобности.
Таким образом, объектом настоящего изобретения является способ последующей обработки цеолитного материала, который не обладает указанным недостатком.
Кроме того, объектом настоящего изобретения является последующая обработка цеолитного материала, обладающего низким молярным отношением X2O3:YO2 и одновременно высокой гидрофобностью. Объектом настоящего изобретения также является последующая обработка цеолитного материала, обладающего низким молярным отношением Х2О3:YO2, высокой гидрофобностью и одновременно уменьшенной концентрацией внутренних дефектов, таких как силанольные скопления. В частности, было установлено, что обработка цеолитного материала с целью уменьшения молярного отношения X2O3:YO2, такая как обработка паром и/или кислотой, приводит к усилению образования силанольных скоплений, которые, например, характеризуются полосой поглощения в ИК спектре цеолитного материала, находящейся в диапазоне от 3500 до 3550 см-1. При использовании в настоящем изобретении, термин "силанольные скопления" предпочтительно означает связанные водородной связью группы Si-ОН, характерное поглощение которых в ИК спектрах, например, силикалитов находится в диапазоне от 3200 до 3650 см-1, как это описано в публикации Zecchina et al. in J. Phys. Chem. 1992, 96, pp. 4991-4997.
Неожиданно было установлено, что способ последующей обработки, который включает обработку цеолитного материала жидкой системой растворителей для уменьшения молярного отношения Х2О3:YO2 с последующей обработкой жидкой водной системой, обладающей значением рН, находящимся в диапазоне от 5,5 до 8, при повышенных температурах, равных не ниже 75°С, лишен указанных недостатков. Также неожиданно было установлено, что указанная обработка жидкой водной системой приводит к уменьшению концентрации внутренних дефектов в полученном цеолитном материале.
Поэтому настоящее изобретение относится к способу последующей обработки цеолитного материала, обладающего каркасной структурой ВЕА, способ включает
(i) получение цеолитного материала, обладающего каркасной структурой ВЕА, где каркасная структура цеолитного материала включает Х2О3 и YO2, в котором молярное отношение X2O3:YO2 превышает 0,02:1.
(ii) обработку цеолитного материала, полученного на стадии (i), жидкой системой растворителей и тем самым получение цеолитного материала, обладающего молярным отношением X2O3:YO2, составляющим не более 0,02:1, и по меньшей мере частичное выделение цеолитного материала из жидкой системы растворителей;
(iii) обработку цеолитного материала, полученного на стадии (ii), жидкой водной системой, обладающей значением рН, находящимся в диапазоне от 5,5 до 8, и при температуре не ниже 75°С;
где значение рН водной системы, использующейся на стадии (iii), определяют с помощью стеклянного электрода для измерения рН.
Стадия (i)
В настоящем изобретении Х2О3 и YO2, содержащиеся в каркасной структуре ВЕА цеолитного материала, полученного на стадии (i), содержатся в ней в качестве структурообразующих элементов в отличие от некаркасных элементов, которые могут находиться в порах и полостях, образованных каркасной структурой.
Обычно не налагаются особые ограничения на то, как на стадии (i) получают цеолитный материал, обладающий каркасной структурой ВЕА. Например, может быть возможным приобретение подходящего имеющегося в продаже цеолитного материала, обладающего каркасной структурой ВЕА. Кроме того, например, для получения цеолитного материала можно использовать любую возможную методику синтеза такого цеолита. Предпочтительно, если цеолитный материал получают способом с использованием в качестве исходных веществ подходящих источников Х2О3 и YO2 в присутствии подходящего соединения-шаблона, также называющегося направляющим реагентом для образования структуры.
Обычно каркасная структура цеолитного материала, полученного на стадии (i), включает X2O3 и YO2. Предпочтительно, если подходящие источники X2O3 и YO2 используют в таком количестве, что не менее 75 мас.%, более предпочтительно не менее 90 мас.%, более предпочтительно не менее 95 мас.%, более предпочтительно не менее 98 мас.%, более предпочтительно не менее 99 мас.% каркасной структуры цеолитного материала, полученного на стадии (i), составляют Х2О3 и YO2.
Обычно X2O3 и YO2 могут содержаться в цеолитном материале, обладающем каркасной структурой ВЕА и молярным отношением X2O3:YO2, превышающем 0,02:1, предпочтительно составляющим не менее 0,03:1, более предпочтительно находящимся в диапазоне от 0,03:1 до 0,07:1, более предпочтительно от 0,03:1 до 0,06:1, более предпочтительно от 0,03:1 до 0,05:1.
Хотя не налагаются особые ограничения на химическую природу четырехвалентных элементов Y, предпочтительные четырехвалентные элементы Y, соответствующие настоящему изобретению, включают, но не ограничиваются только ими, Si, Sn, Ti, Zr, Ge и комбинации двух или большего количества из них. Более предпочтительно, если Y выбран из группы, включающей Si, Sn, Ti, Zr, Ge и комбинации двух или большего количества из них. Более предпочтительно, если Y выбран из группы, включающей Si, Sn, Ti, Zr, и комбинации двух или большего количества из них. Более предпочтительно, если Y означает Si, Sn или комбинацию Si и Sn. Более предпочтительно, если Y означает Si.
Хотя не налагаются особые ограничения на химическую природу трехвалентных элементов X, предпочтительные трехвалентные элементы, соответствующие настоящему изобретению, включают, но не ограничиваются только ими, Al, В, In, Ga, Fe и их комбинации. Более предпочтительно, если X выбран из группы, включающей Al, В, In, Ga, Fe и комбинации двух или большего количества из них. Более предпочтительно, если X выбран из группы, включающей Al, В, In и комбинации двух или большего количества из них. Более предпочтительно, если X означает Al, В или их комбинацию. Более предпочтительно, если X означает В.
Поэтому настоящее изобретение относится к описанному выше способу, в котором Y выбран из группы, включающей Si, Sn, Ti, Zr, Ge и комбинации двух или большего количества из них, Y предпочтительно означает Si, и в котором X выбран из группы, включающей Al, В, In, Ga, Fe и комбинации двух или большего количества из них, X предпочтительно означает В.
Поэтому в предпочтительном варианте осуществления на стадии (i) получают цеолитный материал, обладающий каркасной структурой ВЕА, в котором не менее 90 мас.%, более предпочтительно не менее 95 мас.%, более предпочтительно не менее 98 мас.%, более предпочтительно не менее 99 мас.% каркасной структуры составляют B2O3 и SiO2, и в котором молярное отношение B2O3:SiO2 превышает 0,02:1, более предпочтительно составляет не менее 0,03:1, более предпочтительно находится в диапазоне от 0,03:1 до 0,07:1, более предпочтительно от 0,03:1 до 0,06:1, более предпочтительно от 0,03:1 до 0,05:1. Это вещество также обозначают, как В-ВЕА.
Таким образом, в предпочтительном варианте осуществления настоящего изобретения на стадии (i) получают цеолитный материал, обладающий каркасной структурой ВЕА, в котором не менее 95 мас.%, предпочтительно не менее 98 мас.% каркасной структуры составляют В2О3 и SiO2, и в котором молярное отношение В2О3 и SiO2 находится в диапазоне от 0,03:1 до 0,07:1, предпочтительно от 0,03:1 до 0,06:1.
В предпочтительном варианте осуществления настоящего изобретения цеолитный материал, полученный на стадии (i), получают способом синтеза, включающим
(1) приготовление смеси, содержащей по меньшей мере одно соединение-мишень, по меньшей мере один источник YO2 и по меньшей мере один источник X2O3, и
(2) кристаллизацию цеолитного материала из смеси, приготовленной на стадии (1).
В настоящем изобретении по меньшей мере одно соединение-шаблон, использующееся на стадии (1) может представлять собой любое подходящее соединение-шаблон (направляющий реагент для образования структуры). Подходящие соединения-шаблоны включают пиперидин, гексаме-тиленимин, гидроксид N,N,N-триметил-1-адамантанаммония, пиперидин, гексаметиленимин, дибензил-1,4-диазабицикло[2.2.2]октан, гидроксид ди-бензилметиламмония, гидроксид тетраэтиламмония и их смесь. В предпочтительном варианте осуществления настоящего изобретения используют гидроксид тетраэтиламмония.
Кроме того, YO2 можно получить на стадии (1) в любой возможной форме при условии, что цеолитный материал, обладающий каркасной структурой ВЕА, включающей YO2, можно закристаллизовать на стадии (2). Предпочтительно, если YO2 получают в чистом виде и/или в виде соединения, которое включает YO2 в виде химического фрагмента и/или в виде соединения, которое частично или полностью химически превращается в YO2 во время проведения стадии (1). В предпочтительных вариантах осуществления настоящего изобретения, в которых Y означает Si или комбинацию Si с одним или несколькими другими четырехвалентными элементами, источником SiO2, использующимся на стадии (1), может быть любой возможный источник. Поэтому можно использовать, например, все типы диоксида кремния и силикаты, предпочтительно тонкодисперсный диоксид кремния, гидрозоли диоксида кремния, реакционноспособный аморфный твердый диоксид кремния, силикагель, кремниевую кислоту, растворимое стекло, гидрат метасиликата, сесквисиликат или дисиликат натрия, коллоидный диоксид кремния, пирогенный диоксид кремния, эфиры кремниевой кислоты или тетраалкоксисиланы, или смеси по меньшей мере двух из этих соединений.
В предпочтительных вариантах осуществления настоящего изобретения, в которых смесь, соответствующая стадии (1), включает по меньшей мере один источник SiO2, указанный источник предпочтительно включает по меньшей мере одно соединение, выбранное из группы, включающей диоксид кремния и силикаты, предпочтительно силикаты, более предпочтительно силикаты щелочных металлов. Из числа предпочтительных силикатов щелочных металлов по меньшей мере один источник предпочтительно включают растворимое стекло, более предпочтительно силикат натрия и/или калия и более предпочтительно силикат натрия. В особенно предпочтительных вариантах осуществления настоящего изобретения источником SiO2 является силикат натрия. Кроме того, в вариантах осуществления, включающих диоксид кремния, тонкодисперсный диоксид кремния является предпочтительным.
В предпочтительном варианте осуществления настоящего изобретения, в которых цеолитный материал, обладающий каркасной структурой ВЕА, включает Х2О3, по меньшей мере один источник Х2О3 получают на стадии (1). Обычно Х2О3 можно получить в любой возможной форме при условии, что цеолитный материал, обладающий каркасной структурой ВЕА, включающей Х2О3, можно закристаллизовать на стадии (2). Предпочтительно, если Х2О3 получают в чистом виде и/или в виде соединения, которое содержит Х2О3 в виде химического фрагмента и/или в виде соединения, которое частично или полностью химически превращается в X2O3 во время проведения способа, соответствующего настоящему изобретению.
В более предпочтительных вариантах осуществления настоящего изобретения, в которых X означает В или комбинацию В с одним или несколькими другими трехвалентными элементами, например, свободную борную кислоту и/или бораты, и/или эфиры борной кислоты, такие как, например, триэтилборат или триметилборат, можно использовать в качестве исходных веществ и в качестве по меньшей мере одного источника Х2О3, соответственно.
В настоящем изобретении не налагаются особые ограничения на комбинации Х2О3 и YO2, которые содержатся в цеолитном материале, обладающем каркасной структурой ВЕА. Таким образом, в принципе, любая возможная комбинация одного или нескольких четырехвалентных элементов Y в YO2 может содержаться в цеолитном материале, в комбинации с одним или несколькими трехвалентными элементами X в Х2О3, где указанные выше элементы X и Y соответственно означают составляющие элементы каркасной структуры ВЕА.
Обычно процедуру кристаллизации в соответствии со стадией (2) можно провести любым возможным образом при условии, что цеолитный материал, обладающий каркасной структурой ВЕА, кристаллизуется из смеси, соответствующей стадии (1). Смесь можно закристаллизовать в сосуде любого типа, в котором предпочтительно используют средства смешивания, предпочтительно путем вращения сосуда и/или перемешивания и более предпочтительно путем перемешивания смеси.
Предпочтительно, если смесь нагревают в течение по меньшей мере части процедуры кристаллизации на стадии (2). Обычно смесь можно нагревать до любой возможной температуры кристаллизации при условии, что цеолитный материал, обладающий каркасной структурой ВЕА кристаллизуется из смеси. Предпочтительно, если смесь нагревают до температуры кристаллизации, находящейся в диапазоне от 80 до 200°С, более предпочтительно от 90 до 190°С, более предпочтительно от 100 до 185°С, более предпочтительно от 120 до 180°С, более предпочтительно от 140 до 175°С, еще более предпочтительно от 150 до 165°С.
Предпочтительное нагревание на стадии (2) в настоящем изобретении можно провести любым возможным образом, подходящим для кристаллизации цеолитного материала, обладающего каркасной структурой ВЕА. Обычно нагревание можно провести при одной температуре кристаллизации или при разных температурах. Предпочтительно, если для установления температуры кристаллизации используют линейную скорость нагревания, где скорость нагревания предпочтительно находится в диапазоне от 5 до 100°С/ч, более предпочтительно от 10 до 70°С/ч, более предпочтительно от 15 до 50°С/ч и еще более предпочтительно от 20 до 30°С/ч.
Обычно не налагаются особые ограничения на продолжительность процедуры кристаллизации на стадии (2) способа, соответствующего настоящему изобретению. В предпочтительных вариантах осуществления, включающих нагревание смеси в соответствии со стадией (1), указанную процедуру кристаллизации проводят в течение времени, находящегося в диапазоне от 10 до 200 ч, более предпочтительно от 20 до 190 ч, и еще более предпочтительно от 40 до 170 ч. В способе, соответствующем настоящему изобретению, также предпочтительно, если кристаллизацию проводят в течение времени, находящегося в диапазоне от 60 до 160 ч, более предпочтительно от 80 до 150 ч, и еще более предпочтительно от 110 до 130 ч.
В предпочтительном варианте осуществления настоящего изобретения, в котором смесь нагревают на стадии (2), указанное нагревание можно провести во время всей процедуры кристаллизации или только во время одной или нескольких ее частей при условии, что цеолитный материал, обладающий каркасной структурой ВЕА, кристаллизуется. Предпочтительно, если нагревание проводят в течение всего времени проведения кристаллизации.
Обычно способ, соответствующий настоящему изобретению, необязательно может включать дополнительные стадии обработки и/или дополнительное физическое и/или химическое превращение цеолитного материала, обладающего каркасной структурой ВЕА, кристаллизующегося на стадии (2) из смеси, полученной на стадии (1). Закристаллизованный материал можно, например, обработать с помощью любой последовательности процедур выделения и/или промывки, где цеолитный материал, полученный кристаллизацией на стадии (2), обрабатывают с помощью по меньшей мере одной процедуры выделения и по меньшей мере одной процедуры промывки.
Выделение закристаллизованного цеолитного материала можно провести по любой возможной методике. Эти методики включают, например, методики фильтрования, ультрафильтрования, диафильтрования и центрифугирования и/или декантации или, например, методики распылительной сушки и методики распылительного гранулирования, где методики фильтрования могут включать стадии отсасывания и/или фильтрования под давлением. Можно использовать комбинацию двух или большего количества этих методик.
В одной или нескольких необязательных процедурах промывки можно использовать любой подходящий растворитель. Средствами для промывки, которые можно использовать, являются, например, вода, спирты, такие как метанол, этанол или пропанол или смеси двух или большего количества из них. Примерами смесей являются смеси двух или более спиртов, таких как метанол и этанол или метанол и пропанол, или этанол и пропанол, или метанол и этанол и пропанол, или смеси воды и по меньшей мере одного спирта, такие как вода и метанол или вода и этанол, или вода и пропанол, или вода и метанол и этанол, или вода и метанол и пропанол, или вода и этанол и пропанол, или вода и метанол и этанол и пропанол. Вода или смесь воды и по меньшей мере одного спирта, предпочтительно вода и этанол, является предпочтительной, дистиллированная вода является особенно предпочтительной в качестве единственного средства для промывки.
В настоящем изобретении закристаллизованный цеолитный материал предпочтительно выделяют из суспензии фильтрованием и получают осадок на фильтре, который предпочтительно промывают, предпочтительно водой. Если проводят промывку, то процедуру промывки предпочтительно продолжать, пока электропроводность воды для промывки, не достигнет значения не выше 1000 мкСм/см, более предпочтительно не выше 850 мкСм/см, более предпочтительно не выше 700 мкСм/см.
После выделения цеолитного материала из суспензии, предпочтительно фильтрованием и предпочтительно после промывки, промытый цеолитный материал необязательно предварительно сушат, например, путем обработки потоком подходящего газа, такого как воздух, обедненный воздух или технический азот, в течение времени, предпочтительно находящегося в диапазоне от 4 до 10 ч, более предпочтительно от 5 до 8 ч.
Затем необязательно предварительно высушенный осадок на фильтре предпочтительно сушат. Сушку предпочтительно проводят при температуре, находящейся в диапазоне от 100 до 300°С, более предпочтительно от 150 до 275°С, более предпочтительно от 200 до 250°С, в подходящей атмосфере, такой как технический азот, воздух или обедненный воздух. Такую сушку можно провести, например, в подходящей печи для сушки или с помощью распылительной сушки. Если сушку проводят с помощью распылительной сушки, то температура осушающего газа на входе предпочтительно находится в диапазоне от 200 до 250°С, более предпочтительно от 220 до 250°С, и температура осушающего газа на выходе предпочтительно находится в диапазоне от 100 до 175°С, более предпочтительно от 120 до 150°С. Если проводят распылительную сушку, то можно направить маточный раствор, содержащий цеолитный материал, необязательно после концентрирования, прямо на распылительную сушку. Кроме того, можно направить выделенный и промытый цеолитный материал на распылительную сушку, необязательно после подходящего повторного суспендирования промытого и необязательно предварительно высушенного цеолитного материала.
Поэтому настоящее изобретение также относится к описанному выше способу, в котором до стадии (ii) цеолитный материал, полученный на стадии (i), подвергают распылительной сушке, в котором во время распылительной сушки температура осушающего газа на входе предпочтительно находится в диапазоне от 200 до 250°С и температура осушающего газа на выходе предпочтительно находится в диапазоне от 100 до 175°С.
Предпочтительный способ синтеза предпочтительно включает прокаливание цеолитного материала, полученного на стадии (i), в котором цеолитный материал необязательно можно предварительно подвергнуть распылительной сушке. В предпочтительном варианте осуществления настоящего изобретения до стадии (ii) цеолитный материал, полученный на стадии (i), необязательно после распылительной сушки, прокаливают. Во время прокаливания по меньшей мере одно соединение-шаблон предпочтительно по меньшей мере частично, более предпочтительно в основном удаляется из каркасной структуры.
Прокаливание обычно включает нагревание цеолитного материала, полученного на стадии (i), при температуре выше 350°С. Процедура прокаливания, соответствующая настоящему изобретению, обычно включает нагревание цеолитного материала, полученного на стадии (i), при температуре, находящейся в диапазоне от 400 до 700°С, предпочтительно от 450 до 550°С, в подходящей атмосфере, такой как технический азот, воздух или обедненный воздух. Кроме того, процедуру прокаливания цеолитного материала, полученного на стадии (i), проводят в течение времени, находящегося в диапазоне от 1 до 10 ч, предпочтительно от 3 до 6 ч. Таким образом, в предпочтительном варианте осуществления настоящего изобретения прокаливание проводят при температуре, находящейся в диапазоне от 400 до 700°С, предпочтительно от 450 до 550°С, в течение времени, находящегося в диапазоне от 1 до 10 ч, предпочтительно от 3 до 6 ч.
В настоящем изобретении не налагаются особые ограничения на степень кристалличности цеолитного материала, полученного на стадии (i). Для степени кристалличности значения, приведенные в контексте настоящего изобретения, следует понимать, как определенные по методике, описанной в эталонном примере 2. Например, степень кристалличности цеолитного материала, полученного на стадии (i), может находиться в диапазоне от 40 до 100%, в частности от 50 до 90% или от 55 до 80%.
Для поглощения воды цеолитным материалом, полученным на стадии (i), значения, приведенные в контексте настоящего изобретения, следует понимать, как определенные по методике, описанной в эталонном примере 1. Например, поглощение воды цеолитным материалом, полученным на стадии (i), может находиться в диапазоне от 10 до 50 мас.%, в частности от 10 до 40 мас.% или от 10 до 30 мас.%.
Стадия (ii)
В настоящем изобретении цеолитный материал, обладающий каркасной структурой ВЕА, особенно предпочтительно выделенный, высушенный с помощью распылительной сушки и прокаленный цеолитный материал, полученный на стадии (i), обрабатывают на стадии (ii) жидкой системой растворителей и в результате получают цеолитный материал, обладающий молярным отношением Х2О3:YO2, составляющим не более 0,02:1, и цеолитный материал по меньшей мере частично выделяют из жидкой системы растворителей.
Обычно не налагаются особые ограничения на химическую природу жидкой системы растворителей, использующейся на стадии (ii). Таким образом, можно использовать кислую водную систему для уменьшения молярного отношения X2O3:YO2 для цеолитного материала, полученного на стадии (i), до значения, составляющего не более 0,02:1. В качестве кислот жидкая система растворителей может включать, например, хлористоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, щавелевую кислоту или винную кислоту. Предпочтительно, если жидкая система растворителей, использующаяся на стадии (ii) выбрана из группы, включающей воду, одноатомные спирты, многоатомные спирты и смеси двух или большего количества из них. Касательно одноатомных спиртов и многоатомных спиртов не налагаются особые ограничения. Предпочтительно, если эти спирты содержат от 1 до 6 атомов углерода, более предпочтительно от 1 до 5 атомов углерода, более предпочтительно от 1 до 4 атомов углерода и более предпочтительно от 1 до 3 атома углерода. Многоатомные спирты предпочтительно содержат от 2 до 5 гидроксигрупп, более предпочтительно от 2 до 4 гидроксигрупп, предпочтительно 2 или 3 гидроксигруппы. Особенно предпочтительными одноатомными спиртами являются метанол, этанол и пропанол, такой как 1-пропанол и 2-пропанол. Особенно предпочтительными многоатомными спиртами являются этан-1,2-диол, пропан-1,2-диол, пропан-1,3-диол, пропан-1,2,3-триол. Если используют смеси двух или более описанных выше соединений, то предпочтительно, чтобы эти смеси содержали воду и по меньшей мере один одноатомный и/или по меньшей мере один многоатомный спирт. Наиболее предпочтительно, если жидкая система растворителей состоит из воды. Поэтому настоящее изобретение относится к определенному выше способу и цеолитному материалу, который можно получить или получают этим способом, в котором жидкая система растворителей выбрана из группы, включающей воду, метанол, этанол, пропанол, этан-1,2-диол, пропан-1,2-диол, пропан-1,3-диол, пропан-1,2,3-триол и смеси двух или более из них, предпочтительно воду.
Кроме того, особенно предпочтительно, если жидкая система растворителей не содержит неорганическую кислоту или органическую кислоту или ее соль, кислота выбрана из группы, включающей хлористоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, щавелевую кислоту и винную кислоту. Поэтому настоящее изобретение также относится к описанному выше способу, в котором жидкая система растворителей выбрана из группы, включающей воду, метанол, этанол, пропанол, этан-1,2-диол, пропан-1,2-диол, пропан-1,3-диол, пропан-1,2,3-триол и смеси двух или более из них, предпочтительно воду, и в котором жидкая система растворителей не содержит неорганическую или органическую кислоту или ее соль, кислота выбрана из группы, включающей хлористоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, щавелевую кислоту и винную кислоту. Еще более предпочтительно настоящее изобретение также относится к описанному выше способу, в котором жидкая система растворителей выбрана из группы, включающей воду, метанол, этанол, пропанол, этан-1,2-диол, пропан-1,2-диол, пропан-1,3-диол, пропан-1,2,3-триол и смеси двух или более из них, предпочтительно воду, и в котором жидкая система растворителей не содержит неорганическую или органическую кислоту или ее соль.
Не налагаются особые ограничения на условия проведения реакции, использующиеся на стадии (ii), при условии, что система растворителей, описанная выше, находится в своем жидком состоянии и что молярное отношение X2O3:YO2 уменьшается до значения, составляющего не более 0,02:1. В частности, для описанных ниже предпочтительных температур специалист в данной области техники должен выбрать соответствующее давление, при котором проводят обработку для поддержания системы растворителей в ее жидком состоянии.
На продолжительность обработки в соответствии со стадией (ii) не налагаются особые ограничения. Указанная выше продолжительность означает время, в течение которого выдерживают жидкую систему растворителей при указанной ниже температуре обработки. Предпочтительно, если на стадии (ii) обработку проводят в течение времени, равного от 6 до 20 ч, предпочтительно от 7 до 17 ч, более предпочтительно от 8 до 12 ч. Предпочтительные температуры, при которых проводят обработку, находятся в диапазоне от 50 до 125°С, предпочтительно от 90 до 115°С, более предпочтительно от 95 до 105°С. Наиболее предпочтительно, если обработку в соответствии со стадией (ii) проводят при температуре кипения системы растворителей. Если система растворителей состоит из двух или большего количества компонентов, то обработку в соответствии со стадией (ii) предпочтительно проводят при температуре кипения компонента, обладающего самой низкой температурой кипения.
В предпочтительном варианте осуществления настоящего изобретения обработку в соответствии со стадией (ii) проводят при кипячении с возвратом флегмы. Таким образом, предпочтительный сосуд, представляющий собой открытую систему, использующийся для обработки в соответствии со стадией (ii), предпочтительно снабжают обратным холодильником. Во время проведения стадии (ii) температуру жидкой системы растворителей поддерживают в основном постоянной или меняют, таким образом, обработку жидкой системой растворителей проводят при двух или большем количестве разных температур. Наиболее предпочтительно, если температуру поддерживают в основном постоянной в пределах определенных выше диапазонов.
Поэтому настоящее изобретение относится к описанному выше способу, включающему
(ii) обработку цеолитного материала, полученного на стадии (i), жидкой системой растворителей, предпочтительно водой, и тем самым получение цеолитного материала, обладающего молярным отношением X2O3:YO2, предпочтительно B2O3:SiO2, составляющим не более 0,02:1, в открытой системе при кипячении с возвратом флегмы при температуре, находящейся в диапазоне от 95 до 105°С, и по меньшей мере частичное выделение цеолитного материала из жидкой системы растворителей.
На количество цеолитного материала, которое используется в пересчете на количество жидкой системы растворителей, не налагаются особые ограничения. Предпочтительно, если отношение массы использующегося цеолитного материала к массе жидкой системы растворителей находится в диапазоне от 1:5 до 1:50, более предпочтительно от 1:10 до 1:35, более предпочтительно от 1:10 до 1:20, еще более предпочтительно от 1:12 до 1:18.
Во время обработки в соответствии со стадией (ii) также предпочтительно надлежащим образом перемешивать жидкую систему растворителей. Во время проведения стадии (ii) скорость перемешивания поддерживают в основном постоянной или меняют, таким образом, обработку проводят при двух или большем количестве разных скоростей перемешивания. Наиболее предпочтительно, если цеолитный материал суспендируют в жидкой системе растворителей при первой скорости перемешивания и во время проведения стадии (ii) при указанных выше температурах скорость перемешивания меняют, предпочтительно увеличивают. Сами скорости перемешивания можно выбирать соответствующим образом в зависимости, например, от объема жидкой системы растворителей, количества использующегося цеолитного материала, желательной температуры и т.п. Предпочтительно, если скорость перемешивания, при которой цеолитный материал суспендируют в жидкой системе растворителей, находится в диапазоне от 5 до 200 об/мин (оборотов в минуту) более предпочтительно от 10 до 200 об/мин, более предпочтительно от 20 до 55 об/мин, более предпочтительно от 30 до 50 об/мин. Скорость перемешивания, при которой проводят обработку при указанных выше температурах, предпочтительно находится в диапазоне от 50 до 100 об/мин, более предпочтительно от 55 до 90 об/мин, более предпочтительно от 60 до 80 об/мин.
После обработки в соответствии со стадией (ii) полученный цеолитный материал выделяют из суспензии. Можно использовать все методики выделения цеолитного материала из суспензии. Эти методики включают, например, методики фильтрования, ультрафильтрования, диафильтрования и центрифугирования или, например, методики распылительной сушки и методики распылительного гранулирования, где методики фильтрования могут включать стадии отсасывания и/или фильтрования под давлением. Можно использовать комбинацию двух или большего количества этих методик.
В одной или нескольких необязательных процедурах промывки можно использовать любой подходящий растворитель. Средствами для промывки, которые можно использовать, являются, например, вода, спирты, такие как метанол, этанол или пропанол или смеси двух или более из них. Примерами смесей являются смеси двух или более спиртов, таких как метанол и этанол или метанол и пропанол, или этанол и пропанол, или метанол и этанол и пропанол, или смеси воды и по меньшей мере одного спирта, такие как вода и метанол или вода и этанол, или вода и пропанол, или вода и метанол и этанол, или вода и метанол и пропанол, или вода и этанол и пропанол, или вода и метанол и этанол и пропанол. Вода или смесь воды и по меньшей мере одного спирта, предпочтительно вода и этанол, является предпочтительной, дистиллированная вода является особенно предпочтительной в качестве единственного средства для промывки. Если проводят промывку, то может быть предпочтительно продолжать процедуру промывки, пока электропроводность воды для промывки не достигнет значения не выше 1000 мкСм/см, более предпочтительно не выше 850 мкСм/см, более предпочтительно не выше 700 мкСм/см.
В настоящем изобретении цеолитный материал предпочтительно выделяют из суспензии фильтрованием и получают осадок на фильтре, который предпочтительно промывают, предпочтительно водой.
Обычно способ, соответствующий настоящему изобретению, необязательно может включать дополнительные стадии обработки и/или дополнительное физическое и/или химическое превращение цеолитного материала, обладающего каркасной структурой ВЕА, полученного на стадии (ii). Полученный цеолитный материал, например, можно обработать с помощью любой последовательности процедур выделения и/или промывки, на которой цеолитный материал обрабатывают с помощью по меньшей мере одной процедуры выделения и по меньшей мере одной процедуры промывки.
После выделения цеолитного материала, обладающего каркасной структурой ВЕА из суспензий, предпочтительно проводимого фильтрованием, и после промывки, цеолитный материал, полученный на стадии (ii), необязательно сушат. Процедура сушки необязательно может включать одну или более стадий сушки. Обычно можно использовать любые возможные средства сушки. Процедуры сушки предпочтительно включают нагревание и/или обработку в вакууме цеолитного материала, обладающего каркасной структурой ВЕА.
Предпочтительно, если выделенный и промытый цеолитный материал сушат, например, путем обработки осадка на фильтре потоком подходящего газа, такого как воздух, обедненный воздух или азот, в течение времени, предпочтительно находящегося в диапазоне от 4 до 10 ч, более предпочтительно от 5 до 8 ч.
Также предпочтительно, если до стадии (iii) цеолитный материал, полученный после обработки в соответствии со стадией (ii), подвергают распылительной сушке. Если сушку проводят с помощью распылительной сушки, то температура осушающего газа на входе предпочтительно находится в диапазоне от 200 до 250°С, более предпочтительно от 220 до 250°С, и температура осушающего газа на выходе предпочтительно находится в диапазоне от 100 до 175°С, более предпочтительно от 120 до 150°С. Если проводят распылительную сушку, то можно направить маточный раствор, содержащий цеолитный материал, необязательно после концентрирования, прямо на распылительную сушку. Кроме того, можно направить выделенный и промытый цеолитный материал на распылительную сушку, необязательно после подходящего повторного суспендирования промытого и необязательно предварительно высушенного цеолитного материала.
В особенно предпочтительном варианте осуществления настоящего изобретения цеолитный материал, полученный на стадии (ii), находится в форме порошка, предпочтительно в форме порошка, полученного распылением, где полученный распылением порошок можно получить с помощью распылительной сушки на стадии (i) и/или распылительной сушки на стадии (ii).
В соответствии со стадией (ii) предпочтительно, если высушенный цеолитный материал необязательно прокаливают. Прокаливание предпочтительно проводят в подходящей атмосфере, такой как воздух, обедненный воздух или азот, при температуре, находящейся в диапазоне от 400 до 700°С, предпочтительно от 500 до 600°С, в течение времени, находящегося в диапазоне от 1 до 10 ч, предпочтительно от 2 до 6 ч.
В предпочтительном варианте осуществления настоящего изобретения цеолитный материал, полученный на стадии (ii), не прокаливают до стадии (iii).
В настоящем изобретении обработка в соответствии со стадией (ii) жидкой системой растворителей уменьшает молярное отношение X2O3:YO2 для цеолитного материала; таким образом, она является процедурой удаления по меньшей мере части X из каркасной структуры ВЕА. Поэтому молярное отношение X2O3:YO2 для цеолитного материала, обладающего каркасной структурой ВЕА, полученного на стадии (ii), больше, чем молярное отношение X2O3:YO2 для цеолитного материала, обладающего каркасной структурой ВЕА, полученного на стадии (i). В предпочтительном варианте осуществления настоящего изобретения молярное отношение X2O3:YO2, полученное на стадии (ii), составляет не более 0,02:1, предпочтительно не более 0,01:1, более предпочтительно находится в диапазоне от 0,0005:1 до 0,01:1, более предпочтительно от 0,0009:1 до 0,003:1.
Стадия (iii)
После стадии (ii) цеолитный материал, обладающий каркасной структурой ВЕА, предпочтительно выделенный и высушенный цеолитный материал, обладающий каркасной структурой ВЕА, обрабатывают жидкой водной системой, обладающей значением рН, находящимся в диапазоне от 5,5 до 8, и при температуре не ниже 75°С.
В настоящем изобретении в жидкой водной системе может содержаться любое возможное количество воды при условии, что содержание воды в жидкой системе растворителей превышает 50 мас.% и значение рН жидкой водной системы находится в указанном выше диапазоне. Обычно содержание воды в жидкой системе растворителей составляет не менее 85 мас.%, предпочтительно не менее 90 мас.%, более предпочтительно не менее 95 мас.%, более предпочтительно не менее 99 мас.%, еще более предпочтительно не менее 99,9 мас.%. Еще более предпочтительно, если жидкая водная система, использующаяся на стадии (iii), в основном состоит из воды и может содержать лишь некоторые примеси, которые содержатся в воде, предпочтительно содержатся в предпочтительно использующейся деионизированной воде. Таким образом, в предпочтительном варианте осуществления настоящего изобретения на стадии (iii) жидкая водная система содержит не менее 90 мас.%, предпочтительно не менее 99 мас.%, более предпочтительно не менее 99,9 мас.% воды.
Для значения рН водной системы, использующейся на стадии (iii), значение рН в диапазоне от 5,5 до 8 является предпочтительным. Значение рН жидкой водной системы измеряют с помощью стеклянного электрода для определения рН. В другом предпочтительном варианте осуществления настоящего изобретения жидкая водная система, использующаяся на стадии (iii), обладает значением рН, находящимся в диапазоне от 6 до 7,5, предпочтительно от 6,5 до 7,5.
Не налагаются особые ограничения на продолжительность обработки жидкой водной системой, соответствующей стадии (iii). Предпочтительно, если на стадии (iii) цеолитный материал обрабатывают жидкой водной системой в течение времени, находящегося в диапазоне от 0,5 до 24 ч, предпочтительно от 1 до 18 ч, более предпочтительно от 8 до 14 ч.
Кроме того, не налагаются особые ограничения на температуру жидкой водной системы при условии, что водная система находится в своем жидком состоянии. Поэтому для описанных ниже предпочтительных температур специалист в данной области техники должен выбрать соответствующее давление, при котором проводят обработку в соответствии со стадией (ii) для поддержания системы растворителей в ее жидком состоянии.
Предпочтительно, если на стадии (iii) цеолитный материал обрабатывают жидкой водной системой при температуре, находящейся в диапазоне от 75 до 200°С, предпочтительно от 90 до 180°С, более предпочтительно от 100 до 160°С, более предпочтительно от 110 до 160°С, более предпочтительно от 110 до 150°С.
Хотя не налагаются особые ограничения на тип сосуда, в котором проводят нагревание стадии (iii), сосуд предпочтительно выбрать так, чтобы обеспечить обработку цеолитного материала при указанных выше температурах, при которых водная система находится в своем жидком состоянии. Поэтому при более высоких температурах обработку в соответствии со стадией (iii) проводят в закрытой системе при автогенном давлении.
В возможном варианте осуществления настоящего изобретения нагревание на стадии (iii) проводят в открытой системе. В этом случае при проведении реакции налагается ограничение на температуру, поскольку обработку проводят при температуре ниже температуры кипения использующейся водной системы для сохранения водной системы в ее жидком состоянии.
В предпочтительном варианте осуществления нагревание на стадии (iii) проводят в закрытой системе. Таким образом, нагревание на стадии (iii) предпочтительно проводят при сольвотермических условиях и это означает, что цеолитный материал обрабатывают жидкой водной системой при автогенном давлении жидкой водной системы, например, в автоклаве или других сосудах, пригодных для образования сольвотермических условий.
Поэтому, в особенно предпочтительном варианте осуществления настоящего изобретения на стадии (iii) цеолитный материал обрабатывают жидкой водной системой в закрытой системе, предпочтительно в автоклаве, при автогенном давлении.
Следовательно, в предпочтительном варианте осуществления настоящего изобретения обработку в соответствии со стадией (ii) проводят в открытой системе, предпочтительно при кипячении с возвратом флегмы, и обработку в соответствии со стадией (iii) проводят в закрытой системе, предпочтительно при автогенном давлении, где на стадии (ii) система растворителей находится в своем жидком состоянии и на стадии (iii) водная система находится в своем жидком состоянии. Поэтому настоящее изобретение также относится к способу, определенному выше, включающему
(ii) обработку цеолитного материала, полученного на стадии (i), жидкой системой растворителей, предпочтительно водой, и тем самым получение цеолитного материала, обладающего молярным отношением Х2О3:YO2, предпочтительно B2O3:SiO2, составляющим не более 0,02:1, в открытой системе при кипячении с возвратом флегмы при температуре, находящейся в диапазоне от 95 до 105°С, и по меньшей мере частичное выделение цеолитного материала из жидкой системы растворителей;
(iii) обработку цеолитного материала, полученного на стадии (ii) жидкой водной системой, предпочтительно водой, обладающей значением рН, находящимся в диапазоне от 6,5 до 7,5, и при температуре, находящейся в диапазоне от 110 до 160°С, в закрытой системе при автогенном давлении.
Во время обработки в соответствии со стадией (iii) также предпочтительно надлежащим образом перемешивать жидкую водную систему. Во время проведения стадии (iii) скорость перемешивания поддерживают в основном постоянной или меняют, таким образом, обработку проводят при двух или большем количестве разных скоростей перемешивания. Сами скорости перемешивания можно выбирать соответствующим образом в зависимости, например, от объема жидкой водной системы, количества использующегося цеолитного материала, желательной температуры и т.п. Предпочтительно, если скорость перемешивания, использующаяся на стадии (iii), находится в диапазоне от 10 до 200 об/мин (оборотов в минуту), более предпочтительно от 50 до 180 об/мин, более предпочтительно от 80 до 160 об/мин, более предпочтительно от 110 до 130 об/мин.
На количество жидкой водной системы, использующейся на стадии (iii), в пересчете на количество использующегося цеолитного материала не налагаются особые ограничения. Предпочтительно, если на стадии (iii) отношение массы жидкой водной системы к массе цеолитного материала находится в диапазоне от 35:1 до 5:1, предпочтительно от 30:1 до 10:1, более предпочтительно от 25:1 до 15:1.
После обработки в соответствии со стадией (iii) цеолитный материал, обладающий каркасной структурой ВЕА, предпочтительно выделяют из суспензии. Выделение цеолитного материала можно провести по любой возможной методике, включая, например, методики фильтрования, ультрафильтрования, диафильтрования и центрифугирования и/или декантации, где методики фильтрования могут включать стадии отсасывания и/или фильтрования под давлением. Затем выделенный цеолитный материал обрабатывают с помощью одной или большего количества необязательных процедур промывки. Средствами для промывки, которые можно использовать, являются, например, воду, спирты, такие как метанол, этанол или пропанол или смеси двух или большего количества из них. Примерами смесей являются смеси двух или большего количества спиртов, таких как метанол и этанол или метанол и пропанол, или этанол и пропанол, или метанол и этанол и пропанол, или смеси воды и по меньшей мере одного спирта, такие как вода и метанол или вода и этанол, или вода и пропанол, или вода и метанол и этанол, или вода и метанол и пропанол, или вода и этанол и пропанол, или вода и метанол и этанол и пропанол. Вода или смесь воды и по меньшей мере одного спирта, предпочтительно вода и этанол, является предпочтительной, дистиллированная вода является особенно предпочтительной в качестве единственного средства для промывки. Если проводят промывку, то может быть предпочтительно продолжать процедуру промывки, пока электропроводность воды для промывки не достигнет значения не выше 1000 мкСм/см, более предпочтительно не выше 850 мкСм/см, более предпочтительно не выше 700 мкСм/см.
В настоящем изобретении цеолитный материал предпочтительно выделяют из суспензии фильтрованием и получают осадок на фильтре, который предпочтительно промывают, предпочтительно дистиллированной водой.
Обычно способ, соответствующий настоящему изобретению, необязательно может включать дополнительные стадии обработки и/или дополнительное физическое и/или химическое превращение цеолитного материала, обладающего каркасной структурой ВЕА, полученного на стадии (iii). Полученный цеолитный материал можно, например, обработать с помощью любой последовательности процедур выделения и/или промывки, где цеолитный материал, полученный на стадии (iii), обрабатывают с помощью по меньшей мере одной процедуры выделения и по меньшей мере одной процедуры промывки.
После выделения цеолитного материала, обладающего каркасной структурой ВЕА, из суспензий, предпочтительно проводимого фильтрованием, и после промывки, цеолитный материал, полученный на стадии (iii), направляют по меньшей мере на одну стадию сушки. Процедура сушки необязательно может включать одну или более стадий сушки. Обычно можно использовать любые возможные средства сушки. Процедуры сушки предпочтительно включают нагревание и/или обработку в вакууме цеолитного материала, обладающего каркасной структурой ВЕА.
После выделения цеолитного материала, обладающего каркасной структурой ВЕА, из суспензий, предпочтительно проводимого фильтрованием, и после промывки промытый осадок на фильтре, содержащий цеолитный материал, обладающий каркасной структурой ВЕА, необязательно сушат, например, путем обработки осадка на фильтре потоком подходящего газа, предпочтительно потоком азота. Таким образом, в особенно предпочтительном варианте осуществления настоящего изобретения на стадии (iii) цеолитный материал сушат. Не налагаются особые ограничения на продолжительность и температуру сушки. Сушку предпочтительно проводят в подходящей атмосфере, такой как воздух, обедненный воздух или азот, при температуре, находящейся в диапазоне от 100 до 180°С, предпочтительно от 120 до 150°С, в течение времени, находящегося в диапазоне от 10 до 70 ч, предпочтительно от 15 до 25 ч.
После выделения цеолитного материала из суспензии, предпочтительно фильтрованием и предпочтительно после промывки и до сушки, промытый цеолитный материал можно предварительно высушить, например, путем обработки потоком подходящего газа, такого как воздух, обедненный воздух или азот, в течение времени, предпочтительно находящегося в диапазоне от 4 до 10 ч, более предпочтительно от 5 до 8 ч.
Кроме того, в предпочтительном варианте осуществления настоящего изобретения цеолитный материал, необязательно после сушки, прокаливают на стадии (iii). Прокаливание предпочтительно проводят при температуре, находящейся в диапазоне от 350 до 600°С, предпочтительно от 400 до 500°С, в течение времени, находящегося в диапазоне от 1 до 10 ч, предпочтительно от 2 до 7 ч. Предпочтительная атмосферы для прокаливания включают азот, воздух или обедненный воздух.
В особенно предпочтительном варианте осуществления настоящего изобретения, после стадии (i) и/или после стадии (ii), и/или после стадии (iii) цеолитный материал до прокаливания сушат, где сушку предпочтительно проводят при температуре, находящейся в диапазоне от 100 до 180°С, предпочтительно от 120 до 150°С, в течение времени, находящегося в диапазоне от 10 до 70 ч, предпочтительно от 15 до 25 ч.
Неожиданно было установлено, что способом, соответствующим настоящему изобретению, можно получить цеолитный материал с молярным отношением X2O3:YO2, уменьшенным по сравнению со значением для цеолитного материала, полученного на стадии (i), где одновременно гидрофобность цеолитного материала, характеризующуюся результатами измерения поглощения воды, описанными в настоящем изобретении, можно сохранить постоянной или даже увеличить. Такое увеличение гидрофобности желательно для многих возможных случаев применения цеолитного материала, например, в качестве каталитически активных материалов. Поэтому установлено, что комбинация стадии деборирования в соответствии со стадией (ii) и последующей обработки жидкой водной системой позволяет преодолеть недостатки способов предшествующего уровня техники. Таким образом, в настоящем изобретении поглощение воды цеолитным материалом, полученным на стадии (iii), меньше, чем поглощение воды цеолитным материалом, полученным на стадии (ii). Предпочтительно, если поглощение воды цеолитным материалом, полученным на стадии (iii), не менее, чем на 40%, предпочтительно не менее, чем на 50%, более предпочтительно не менее, чем на 55%, еще более предпочтительно не менее, чем на 60% меньше, чем поглощение воды цеолитным материалом, полученным на стадии (i).
Кроме того, установлено, что обработка жидкой водной системой, соответствующей стадии (iii), влияет на удельную площадь поверхности цеолитного материала. Предпочтительно, если удельная площадь поверхности цеолитного материала, полученного на стадии (iii), не менее, чем на 100 м2/г, предпочтительно не менее, чем на 200 м2/г, меньше, чем удельная площадь поверхности цеолитного материала, полученного на стадии (ii). Термин "удельная площадь поверхности" при использовании в контексте настоящего изобретения означает удельную площадь поверхности БЭТ во многих точках (методика Брунауэра - Эметта - Теллера), которую определяют путем адсорбции азота в соответствии со стандартом DIN 66131.
Кроме того, неожиданно было установлено, что дополнительная обработка жидкой водной системой, соответствующей стадии (iii) способа, соответствующего настоящему изобретению, оказывает благоприятное влияние на характеристики силанольных групп цеолитного материала. В частности, в инфракрасном спектре цеолитного материала, соответствующего настоящему изобретению, силанольные группы первого типа характеризуются первой полосой поглощения с максимумом в области от 3701 до 3741 см-1, где указанную первую полосу поглощения можно приписать поверхностным силанольным группам, и силанольные группы второго типа характеризуются второй полосой поглощения с максимумом в области от 3500 до 3550 см-1, где указанную вторую полосу поглощения можно приписать силанольным скоплениям. И в этом случае при использовании в настоящем изобретении, термин "поверхностный силанол" или "поверхностные силанольные группы" предпочтительно означает Si-OH, которые не связаны водородными связями и для которых характеристическое поглощение в ИК спектрах, например, силикалитов находится в диапазоне от 3650 до 3800 см-1, как это описано в публикации Zecchina et al. in J. Phys. Chem. 1992, 96, pp. 4991-4997. Точнее, можно видеть, что обработка цеолитного материала для уменьшения молярного отношения Х2О3:YO2, такая как обработка паром и/или кислотой, приводит к усилению образования силанольных скоплений, которые характеризуются увеличением интенсивности второй полосы поглощения, тогда как концентрация поверхностных силанольных групп, которая характеризуются интенсивностью первой полосы поглощения, остается относительно постоянной после обработки для уменьшения молярного отношения X2O3:YO2. В результате отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения, определенное для данного ИК спектра цеолитного материала, является надежным показателем относительной концентрации силанольных скоплений в данном цеолитном материале и, в частности, изменений концентрации силанольных скоплений после обработки цеолитного материала, например, в результате его обработки паром и/или кислотой. Точнее, уменьшение отношения интенсивности первой полосы поглощения к интенсивности второй полосы поглощения указывает на увеличение относительной концентрации силанольных скоплений в цеолитном материале, тогда как его увеличение соответственно указывает на уменьшение относительной концентрации силанольных скоплений.
Таким образом, в настоящем изобретении, установлено, что отношение интенсивности полосы поглощения в ИК спектре, характеризующей силанольные группы первого типа, к интенсивности полосы поглощения в ИК спектре, характеризующей силанольные группы второго типа цеолитного материала, полученного на стадии (iii), не менее, чем на 5%, предпочтительно не менее, чем на 10%, более предпочтительно не менее, чем на 20%, более предпочтительно не менее, чем на 30%, более предпочтительно не менее, чем на 50%, более предпочтительно, чем на 60% и более предпочтительно, чем на 70% больше, чем соответствующее отношение для цеолитного материала, полученного на стадии (ii). Как отмечено выше, для отношения интенсивности первой полосы поглощения в ИК спектре к интенсивности второй полосы поглощения в ИК спектре, увеличение указанного отношения указывает на уменьшение относительной концентрации внутренних дефектов (т.е. силанольных скоплений) в цеолитном материале, что обусловлено обработкой жидкой водной системой на стадии (iii). Таким образом, неожиданный технический эффект способа, соответствующего настоящему изобретению, можно видеть по изменению отношения интенсивностей указанных выше полос в ИК спектре цеолитного материала до и после обработки на стадии (iii), где увеличение указанного отношения, которое можно видеть на указанной стадии, указывает на неожиданные регенерирующие эффекты, обеспечиваемые способом, соответствующим настоящему изобретению, после разложения цеолитной структуры в результате обработки на стадии (ii) для удаления Х2О3 из каркаса цеолита, например, путем обработки паром и/или кислотой.
Кроме того, установлено, что с помощью обработки жидкой водной системой, соответствующей стадии (iii) способа, соответствующего настоящему изобретению, степень кристалличности цеолитного материала, полученного на стадии (ii) можно сохранить постоянной или даже немного уменьшить. В частности, гидротермальная обработка оказывает слабое влияние на степень кристалличности цеолитного материала, так что в значительной степени можно сохранить начальную степень кристалличности цеолитного материала, полученного на стадии (ii). Например, степень кристалличности цеолитного материала, полученного на стадии (iii), определенная путем анализа с помощью РГГ (рентгенография), может быть менее, чем на 30%, предпочтительно менее, чем на 20%, более предпочтительно менее, чем на 15%, более предпочтительно менее, чем на 10%, более предпочтительно менее, чем на 5%, более предпочтительно менее, чем на 2% и более предпочтительно менее, чем на 1% меньше, чем степень кристалличности цеолитного материала, полученного на стадии (ii).
Особенно предпочтительно, если способ, соответствующий настоящему изобретению, не включает обработку паром, в особенности обработку водяным паром, ни до, ни во время, ни после стадии (iii). Таким образом, предпочтительно, если цеолитный материал, полученный способом, соответствующим настоящему изобретению, не обрабатывают паром в течение всего времени проведения способа.
Также предпочтительно, если способ, соответствующий настоящему изобретению, не включает обработку цеолитного материала водным раствором, обладающим значением рН, равным меньше 5,5 или больше 8, ни до, ни во время, ни после стадии (iii). Таким образом, предпочтительно, если цеолитный материал, полученный способом, соответствующим настоящему изобретению, не обрабатывают кислотой или основанием в течение всего времени проведения способа.
Настоящее изобретение также относится к цеолитному материалу, который можно получить или получают способом, соответствующим настоящему изобретению.
Предпочтительные цеолитные материалы
Как отмечено выше, способ, соответствующий настоящему изобретению, характеризуется значительными преимуществами, в частности, в отношении степени кристалличности и также гидрофобности цеолитных материалов, которые предпочтительно представляют собой порошок или полученный распылением порошок, обладающий молярным отношением X2O3:YO2, меньшим, чем в исходном цеолитном материале. Как определено выше, Y предпочтительно выбран из группы, включающей Si, Sn, Ti, Zr, Ge и комбинации двух или большего количества из них, Y предпочтительно означает Si, и X предпочтительно выбран из группы, включающей Al, В, In, Ga, Fe и комбинации двух или большего количества из них, X предпочтительно означает В.
Поэтому настоящее изобретение также относится к цеолитному материалу, обладающего каркасной структурой ВЕА, в котором каркасная структура включает YO2 и X2O3, где Y означает четырехвалентный элемент и X означает трехвалентный элемент, указанный цеолитный материал обладает молярным отношением Х2О2:YO2, составляющим не более 0,02:1, предпочтительно не более 0,002:1, и поглощением воды, составляющим не более 7 мас.%, предпочтительно не более 6 мас.%, где предпочтительно, если не менее 95 мас.%, более предпочтительно не менее 98 мас.%, более предпочтительно не менее 99 мас.% каркасной структуры цеолитного материала составляют X2O3 и YO2. В частности, настоящее изобретение относится к цеолитному материалу, обладающему каркасной структурой ВЕА, в котором каркасная структура включает SiO2 и B2O3, указанный цеолитный материал обладает молярным отношением B2O3:SiO2, составляющим не более 0,02:1, предпочтительно не более 0,002:1, и поглощением воды, составляющим не более 7 мас.%, предпочтительно не более 6 мас.%, и где предпочтительно, если не менее 95 мас.%, более предпочтительно не менее 98 мас.%, более предпочтительно не менее 99 мас.% каркасной структуры цеолитного материала составляют B2O3 и SiO2.
Предпочтительно, если молярное отношение Х2О3:YO2, предпочтительно молярное отношение В2О3:SiO2, составляет не более 0,002:1, более предпочтительно находится в диапазоне от 0,0001:1 до 0,002:1, более предпочтительно от 0,0005:1 до 0,002:1, и поглощение воды цеолитным материалом находится в диапазоне от 3 до 7 мас.%, предпочтительно от 4 до 6 мас.%.
Кроме того, установлено, что цеолитные материалы, соответствующие настоящему изобретению, предпочтительно обладают инфракрасным спектром для первого типа силанольных групп, которые характеризуются первой полосой поглощения с максимумом в области от 3701 до 3741 см-1, и второго типа силанольных групп, которые характеризуются второй полосой поглощения с максимумом в области от 3500 до 3550 см-1. Предпочтительно, если отношение интенсивности указанной первой полосы поглощения к интенсивности указанной второй полосы поглощения равно не менее 1,0, предпочтительно находится в диапазоне от 1,0 до 3,0, более предпочтительно в диапазоне от 1,3 до 2,2, более предпочтительно в диапазоне от 1,2 до 2,8, более предпочтительно в диапазоне от 1,4 до 2,6, более предпочтительно в диапазоне от 1,5 до 2,5, более предпочтительно в диапазоне от 1,6 до 2,4, более предпочтительно в диапазоне от 1,7 до 2,3 и более предпочтительно в диапазоне от 1,8 до 2,2.
Кроме того, установлено, что цеолитные материалы, соответствующие настоящему изобретению, предпочтительно характеризуются степенью кристалличности, определенной путем анализа с помощью РГГ, составляющей не менее 60%, предпочтительно находящейся в диапазоне от 60 до 90%, более предпочтительно в диапазоне от 65 до 80%.
Кроме того, установлено, что цеолитные материалы, соответствующие настоящему изобретению, предпочтительно обладают удельной площадью поверхностью (площадь поверхности БЭТ), равной не более 600 м2/г, предпочтительно находящейся в диапазоне от 60 до 600 м2/г, более предпочтительно от 100 до 600 м2/г, более предпочтительно от 200 до 600 м2/г, определенной в соответствии со стандартом DIN 66131.
Предпочтительные случаи применения
Цеолитный материал, обладающий каркасной структурой ВЕА, соответствующий настоящему изобретению, который предпочтительно можно получить или получают способом, соответствующим настоящему изобретению, предпочтительно используют в качестве катализатора, в качестве подложки для катализатора или в качестве компонента катализатора. Например, можно отметить применение в качестве подложки для катализатора на основе благородного металла в катализаторах окисления дизельного топлива (DOC) или применение в качестве компонента покрытия в DOC. Кроме того, цеолитный материал, соответствующий настоящему изобретению, предпочтительно полученный способом, соответствующим настоящему изобретению, можно использовать в качестве катализатора в реакции гидрирования или дегидрирования, таких как дегидрирование изопропанола. Кроме того, его можно использовать в качестве молекулярного сита, адсорбента или наполнителя.
Поэтому настоящее изобретение также относится к способу катализа, в котором цеолитный материал, соответствующий настоящему изобретению или который можно получить или получают способом, соответствующим настоящему изобретению, используют в качестве катализатора, в качестве подложки для катализатора или в качестве компонента катализатора.
Обычно и в частности, в случае, когда цеолитный материал, соответствующий настоящему изобретению, используют в качестве катализатора, можно изготовить формованное изделие, содержащее цеолитный материал, например, путем надлежащего смешивания цеолитного материала по меньшей мере с одним связующим и/или по меньшей мере с одним предшественником связующего и необязательно по меньшей мере с одним порообразующим агентом и/или по меньшей мере с одним пластифицирующим агентом. Формованному изделию можно придать любую возможную форму, такую как нити, например, обладающие прямоугольным, треугольным шестиугольным, квадратным, овальным или круговым сечением, звездообразные структуры, таблетки, сферы, полые цилиндры и т.п. Примерами таких связующих являются оксиды металлов, такие как, например, SiO2, Al2O3, TiO2, ZrO2 или MgO или глины, или смеси двух или большего количества этих оксидов, или смешанные оксиды по меньшей мере двух из элементов Si, Al, Ti, Zr и Mg. Порообразующие агенты, такие как образующие мезопоры агенты, включают полимерные виниловые соединения, такие как полиалкиленоксиды, такие как полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и сложные полиэфиры. Клеящие агенты включают органические, в частности, гидрофильные полимеры, такие как углеводы, такие как целлюлоза, производные целлюлозы, такие как метилцеллюлоза, и крахмал, такой как картофельный крахмал, клей для обоев, полиакрилаты, полиметакрилаты, поливиниловый спирт, поливинилпирролидон, полиизобутен или политетрагидрофуран. Можно отметить применение в качестве клеящих агентов воды, спиртов или гликолей или их смесей, таких как смеси воды и спирта или воды и гликоля, таких как, например, вода и метанол или вода и этанол, или вода и пропанол, или вода и пропиленгликоль.
Настоящее изобретение иллюстрируется следующими примерами.
Примеры
Эталонный пример 1. Определение поглощения воды
Изотермы адсорбции/десорбции воды получали с помощью прибора VTI SA, выпускающегося фирмой ТА Instruments, по ступенчатой программе для получения изотерм. Эксперимент состоял из опыта или группы опытов, проводимых для образца материала, который был помещен на чашку микровесов внутри прибора. Перед проведением измерения остаточную влагу из образца удаляли путем нагревания образца до 100°С (линейная скорость нагревания равна 5°С/мин) и его выдерживания в течение 6 ч в потоке азота. После проведения программы сушки температуру в камере снижали до 25°С и при проведении измерении поддерживали постоянной. Микровесы калибровали и массу высушенного образца уравновешивали (максимальное отклонение массы составляло 0,01 мас.%). Поглощение воды образцом измеряли, как увеличение массы по сравнению с массой сухого образца. Сначала строили кривую адсорбции путем увеличения относительной влажности (ОВ) (выраженной, как содержание в мас.% воды в атмосфере внутри камеры) атмосферы, действующей на образец, и измерения поглощения воды образцом при равновесии. Значение ОВ увеличивали с шагом, равным 10 мас.%, от 5% до 85% и на каждой ступени определяли ОВ в системе и массу образца до установления равновесия, и регистрировали увеличение массы. Полное количество воды, поглощенной образцом, определяли после воздействия на образец атмосферы с ОВ, равной 85 мас.%. При исследовании десорбции ОВ снижали с 85 мас.% до 5 мас.% с шагом, равным 10%, и определяли, и регистрировали массу образца (поглощение воды).
Эталонный пример 2. Определение степени кристалличности
Степень кристалличности цеолитных материалов, соответствующих настоящему изобретению, определяли путем анализа с помощью РГГ, где степень кристалличности данного материала выражали по сравнению с эталонным цеолитным материалом, при котором сопоставляли поверхности отражения этих двух цеолитных материалов. Эталонным цеолитным материалом, обладающим кристалличностью 100% являлась аммониевая форма цеолита бета в порошкообразном виде, продающаяся под номером CAS 1318-O2-1. Определение степени кристалличности проводили с помощью дифрактометра D8 Advance series 2, выпускающегося фирмой Bruker AXS. Дифрактометр обладал щелью расхождения в 0,1° и использовался детектор Lynxeye. Образцы, а также эталонный цеолитный материал исследовали в диапазоне от 19° до 25° (углов 2 тета). После введения поправки на базовую линию поверхности отражения исследовали с помощью программного обеспечения EVA (выпускающегося фирмой Bruker AXS). Отношения для поверхностей отражения выражали в процентах.
Эталонный пример 3. Исследования ИК спектров
Исследования И К спектров проводили с помощью спектрометра Nicolet 6700. Цеолитные материалы прессовали в прочные пеллеты без использования добавок. Пеллету помещали в высоковакуумную ячейку, находящуюся в ИК спектрометре. Перед исследованием образец предварительно обрабатывали в высоком вакууме (105 мбар) в течение 3 ч при 300°С. Спектры снимали после охлаждения ячейки до 50°С. Спектры регистрировали в диапазоне от 4000 см-1 до 800 см-1 с разрешением 2 см-1. Полученные спектры представляли в виде графиков, на оси х которых отложено волновое число (см1) и на оси у отложена оптическая плотность (произвольные единицы). Для количественного определения высот пиков и отношения высот пиков вводили поправку на базовую линию. Изменения в области от 3000 см-1 до 3900 см-1 анализировали и сопоставляли для множества образцов, полосу при 1800±5 см-1 использовали для сравнения.
Пример 1:
(i) Получение исходного материала (цеолитного материала, обладающего каркасной структурой ВЕА)
209 кг Деионизованной воды помещали в сосуд. При перемешивании при 120 об/мин (оборотов в минуту) добавляли 355 кг гидроксида тетраэтиламмония и суспензию перемешивали в течение 10 мин при комнатной температуре. Затем 61 кг борной кислоты суспендировали в воде и суспензию перемешивали в течение еще 30 мин при комнатной температуре. Затем добавляли 555 кг Ludox® AS-40 и полученную смесь перемешивали при 70 об/мин в течение еще 1 ч при комнатной температуре. Жидкий гель по данным измерения с помощью стеклянного электрода для определения рН, обладал значением рН, равным 11,8. Конечную полученную смесь переносили в сосуд для кристаллизации и нагревали при 160°С в течение 6 ч под давлением, равным 7,2 бар, и при перемешивании (140 об/мин). Затем смесь охлаждали до комнатной температуры. Смесь повторно нагревали при 160°С в течение 6 ч и перемешивали при 140 об/мин в течение еще 55 ч. Смесь охлаждали до комнатной температуры и затем смесь нагревали в течение еще 45 ч при температуре, равной 160°С при перемешивании при 140 об/мин. К 380 кг этой суспензии добавляли 7800 кг деионизированной воды. Суспензию перемешивали при 70 об/мин и добавляли 100 кг 10 мас.% водного раствора HNO3. Из этой суспензии содержащий бор цеолитный материал, обладающий каркасной структурой ВЕА, выделяли фильтрованием. Затем осадок на фильтре промывали деионизированной водой при комнатной температуре, пока электропроводность воды для промывки, не достигала значения менее 150 мкСм/см. Полученный таким образом осадок на фильтре сушили в потоке азота.
Полученный таким образом осадок на фильтре смешивали с водой и получали суспензию, обладающую содержанием твердых веществ, равным 40 мас.%. Эту суспензию подвергали распылительной сушке в башне с распылительным орошением при следующих условиях распылительной сушки:
осушающий газ, газ для сопла: технический азот
температура осушающего газа:
- температура в башне с распылительным орошением (на входе): 235°С
- температура в башне с распылительным орошением (на выходе): 140°С сопло:
- верхний компонент сопла:поставщик Gerig; размер 0
- температура газа для сопла:комнатная температура
- давление газа для сопла: 1 бар
режим работы: прямая подача азота
использующийся аппарат: башня с распылительным орошением с одним соплом
конфигурация: башня с распылительным орошением - фильтр - скруббер
скорость потока газа: 1500 кг/ч
материал фильтра: Nomex® иглопробивной материал 20 м2
подача с помощью насоса с гибкой трубой: SP VF 15 (поставщик: Verder).
Башня с распылительным орошением представляла собой вертикально расположенный цилиндр, обладающий длиной, равной 2650 мм, диаметром, равным 1200 мм, и этот цилиндр на дне обладал коническим сужением. Длина конуса равнялась 600 мм. В верхней части цилиндра находились средства распыления (двухкомпонентное сопло). Высушенный с помощью распылительной сушки материал отделяли от осушающего газа на фильтре, расположенном после башни с распылительным орошением, и после этого осушающий газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла и газ сопла пропускали через кольцеобразную щель, окружающую отверстие.
Затем высушенный с помощью распылительной сушки материал прокаливали при 500°С в течение 5 ч. Прокаленный материал обладал молярным отношением B2O3:SiO2, равным 0,045, степенью кристалличности, составляющей 100%, и поглощением воды, составляющим 20,9 мас.%. ИК спектр полученного продукта содержал первую полосу поглощения с максимумом в диапазоне от 3701 см-1 до 3741 см-1 и вторую полосу поглощения с максимумом в диапазоне от 3500 см-1 до 3550 см-1, где отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения равно 2,97.
(ii) Обработка жидкой системой растворителей - деборирование
840 кг Деионизованной воды помещали в сосуд, снабженный обратным холодильником. При перемешивании при 40 об/мин использовали 28 кг высушенного с помощью распылительной сушки материала, полученного в соответствии со стадией (i). Затем сосуд закрывали и кипятили с обратным холодильником. Скорости перемешивания увеличивали до 70 об/мин. При перемешивании при 70 об/мин содержимое сосуда нагревали при 100°С в течение 1 ч и выдерживали при этой температуре в течение 20 ч. Затем содержимое сосуда охлаждали до температуры, равной менее 50°С.
Полученный деборированный цеолитный материал, обладающий структурой типа ВЕА, выделяли из суспензии фильтрованием под давлением азота, равным 2,5 бар, и четырежды промывали деионизированной водой при комнатной температуре. После фильтрования осадок на фильтре сушили в потоке азота в течение 6 ч.
Полученный деборированный цеолитный материал подвергали распылительной сушке при условиях, описанных на стадии (i). Полученный деборированный цеолитный материал обладал молярным отношением В2О3:SiO2, равным менее 0,002, степенью кристалличности, составляющей 77%, и поглощением воды, составляющим 15 мас.%. ИК спектр деборированного продукта содержит первую полосу поглощения с максимумом в диапазоне от 3701 см-1 до 3741 см-1 и вторую полосу поглощения с максимумом в диапазоне от 3500 см-1 до 3550 см-1, где отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения равно 1,27.
По сравнению с исходным цеолитным материалом, полученным на стадии (i), деборированный цеолитный материал характеризуется уменьшенным молярным отношением B2O3:SiO2 (т.е. от начального, равного 0,045, до равного менее 0,002), уменьшенной степенью кристалличности (т.е. от начальной, равной 100%, до равной 77%) и уменьшенным поглощением воды (т.е. от начального, составляющего 20,9 мас.%, до составляющего 15 мас.%). Кроме того, отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения в ИК спектре уменьшается от 2,97 до 1,27 и это показывает, что в результате процедуры деборирования в цеолитном материале образовалось значительное количество внутренних дефектов (т.е. силанольных скоплений).
(iii) Обработка жидкой водной системой
1600 г Деионизованной воды помещали в сосуд и при перемешивании добавляли 80 г деборированного цеолитного материала, полученного (ii). Суспензию перемешивали в течение 10 мин при комнатной температуре. Затем суспензию нагревали при 140°С при автогенном давлении в течение 12 ч.
Полученный цеолитный материал выделяли из суспензии фильтрованием и промывали деионизированной водой при комнатной температуре. После фильтрования осадок на фильтре сушили при 120°С в течение 16 ч.
Затем высушенный цеолитный материал прокаливали. Цеолитный материал нагревали до 450°С в течение 5,5 ч и грели при этой температуре в течение 2 ч.
Обработку водой проводили во второй раз, как описано выше. Полученный материал обладал молярным отношением B2O3:SiO2, равным 0,0009, степенью кристалличности, составляющей 68%, и поглощением воды, составляющим 5,4 мас.%. ИК спектр полученного продукта содержал первую полосу поглощения с максимумом в диапазоне от 3701 см-1 до 3741 см-1 и вторую полосу поглощения с максимумом в диапазоне от 3500 см-1 до 3550 см-1, где отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения равно 2,17. Удельная площадь поверхности БЭТ во многих точках, которую определяли путем адсорбции азота при 77 K в соответствии со стандартом DIN 66131, равнялась 229 м2/г.
Результаты примера 1
В настоящем изобретении использовали комбинацию процедуры деборирования с использованием жидкой системы растворителей (воды) с обработкой жидкой водной системой (водой). Эта комбинация, с одной стороны, давала цеолитный материал, обладающий каркасной структурой ВЕА с уменьшенным молярным отношением B2O3:SiO2 (т.е. от начального, составляющего 0,045:1, до составляющего 0,0009:1), уменьшенной степенью кристалличности (т.е. от начальной, равной 100%, до равной 68%), уменьшенным поглощением воды (т.е. от начального, составляющего 20,9 мас.%, до составляющего 5,4 мас.%) и, следовательно, обладающий увеличенной гидрофобностью. Что касается отношения интенсивности первой полосы поглощения к интенсивности второй полосы поглощения в ИК спектре, то указанное отношение для конечного продукта примера 1 намного больше, чем для деборированного продукта в примере 1-(ii), и это показывает, что относительная концентрация силанольных скоплений в цеолитном материале значительно уменьшается при обработке жидкой водной системой. Такое уменьшение относительной концентрации внутренних дефектов (т.е. силанольных скоплений) является весьма неожиданным, в особенности с учетом того факта, что степень кристалличности цеолитного материала уменьшается от 77% (для деборированного продукта в примере 1-(ii)) до 68% (для конечного продукта примера 1) после обработки жидкой водной системой.
Пример 2.
(iii) Обработка жидкой водной системой
1600 г Деионизованной воды помещали в сосуд и при перемешивании добавляли 80 г деборированного цеолитного материала, полученного в соответствии с примером 1, стадия (ii), обладающего молярным отношением В2О3:SiO2, равным 0,002, и поглощением воды, составляющим 15 мас.%. Суспензию перемешивали в течение 10 мин при комнатной температуре. Затем суспензию нагревали при 160°С при автогенном давлении в течение 13 ч.
Полученный цеолитный материал выделяли из суспензии фильтрованием и промывали деионизированной водой при комнатной температуре. После фильтрования осадок на фильтре сушили при 120°С в течение 5 ч.
Затем высушенный цеолитный материал прокаливали при 450°С в течение 2 ч.
Обработку водой проводили во второй раз, как описано выше. Полученный материал обладал молярным отношением В2О3:SiO2, равным 0,0008, степенью кристалличности, составляющей 64%, и поглощением воды, составляющим 4,3 мас.%. ИК спектр полученного продукта содержал первую полосу поглощения с максимумом в диапазоне от 3701 см-1 до 3741 см-1 и вторую полосу поглощения с максимумом в диапазоне от 3500 см-1 до 3550 см-1, где отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения равно 1,8. Удельная площадь поверхности БЭТ во многих точках, которую определяли путем адсорбции азота при 77 К в соответствии со стандартом DIN 66131, равнялась 61 м2/г.
Результаты примера 2
В настоящем изобретении использовали комбинацию процедуры деборирования с использованием жидкой системы растворителей (воды) с обработкой жидкой водной системой (водой). Эта комбинация, с одной стороны, давала цеолитный материал, обладающий каркасной структурой ВЕА с уменьшенным молярным отношением В2О3:SiO2 (т.е. от начального, составляющего 0,045:1, до составляющего 0,0008:1), уменьшенной степенью кристалличности (т.е. от начальной, равной 100%, до равной 64%), уменьшенным поглощением воды (т.е. от начального, составляющего 20,9 мас.%, до составляющего 4,3 мас.%) и, следовательно, обладающий увеличенной гидрофобностью. Что касается отношения интенсивности первой полосы поглощения к интенсивности второй полосы поглощения в ИК спектре, то указанное отношение для конечного продукта примера 2, близкое к наблюдающемуся для конечного продукта примера 1, также намного больше, чем для деборированного продукта в примере 1-(ii), и это показывает, что относительная концентрация силанольных скоплений в цеолитном материале значительно уменьшается при обработке жидкой водной системой. Такое уменьшение относительной концентрации внутренних дефектов (т.е. силанольных скоплений) также является неожиданным, в особенности с учетом того факта, что степень кристалличности цеолитного материала уменьшается от 77% (для деборированного продукта в примере 1-(ii)) до 64% (для конечного продукта примера 2) после обработки жидкой водной системой.
Сравнительный пример 1. Обработка паром
Тонкий слой образца из 100 г цеолитного материала, полученного в соответствии с примером 1, стадия (ii), обладающего молярным отношением В2О3:SiO2, равным менее 0,002, и поглощением воды, составляющим 15 мас.%, помещали в муфельную печь и нагревали до 650°С (линейная скорость повышения температуры равна 5 K/мин). Для обработки паром использовали поток газа со скоростью, равной 6 л/мин (10% пара в воздухе), причем введение воды начинали при температуре, равной 200°С. Обработку паром проводили в течение 1 ч при 650°С.
Полученный материал обладал молярным отношением В2О3:SiO2, равным 0,0014, степенью кристалличности, составляющей 70%, и поглощением воды, составляющим 13,0 мас.%. Удельная площадь поверхности БЭТ во многих точках, которую определяли путем адсорбции азота при 77 K в соответствии со стандартом DIN 66131, равнялась 518 м2/г. ИК спектр продукта, полученного после обработки паром, содержит первую полосу поглощения с максимумом в диапазоне от 3701 см-1 до 3741 см-1 и вторую полосу поглощения с максимумом в диапазоне от 3500 см-1 до 3550 см-1, где отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения равно 1,42.
Таким образом, по сравнению с цеолитным материалом, полученным способом, соответствующим настоящему изобретению, хотя в обработанных паром материалах в более или менее сохранялась степень кристалличности, обработка паром не оказывала восстанавливающего воздействия на внутренние дефекты в форме силанольных скоплений, отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения в ИК спектре вследствие обработки паром увеличивалось лишь незначительно. Кроме того, обработка паром мало влияла на поглощение воды, которое также сохранялось более или менее постоянным и лишь немного уменьшалось, что явно отличается от данным для цеолитных материалов, полученных способом, соответствующим настоящему изобретению, для которого в результате обработки жидкой водной системой на стадии (iii) поглощение воды уменьшалось до части исходного значения.
Сравнительный пример 2. Обработка паром
Тонкий слой образца из 100 г цеолитного материала, полученного в соответствии с примером 1, стадия (ii), обладающего молярным отношением В2О3:SiO2, равным менее 0,002, и поглощением воды, составляющим 15 мас.%, помещали в муфельную печь и нагревали до 850°С (линейная скорость повышения температуры равна 5 К/мин). Для обработки паром использовали поток газа со скоростью, равной 6 л/мин (10% пара в воздухе), причем введение воды начинали при температуре, равной 200°С. Обработку паром проводили в течение 1 ч при 850°С.
Полученный материал обладал молярным отношением В2О3:SiO2, равным 0,0016, и поглощением воды, составляющим 8,1 мас.%. Удельная площадь поверхности БЭТ во многих точках, которую определяли путем адсорбции азота при 77 K в соответствии со стандартом DIN 66131, равнялась 405 м2/г. ИК спектр продукта, полученного после обработки паром, содержит первую полосу поглощения с максимумом в диапазоне от 3701 см-1 до 3741 см-1 и вторую полосу поглощения с максимумом в диапазоне от 3500 см-1 до 3550 см-1, где отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения равно 1,4.
Однако рентгенограмма РГГ полученного цеолитного материала, отличалась от полученной для цеолита бета и это показывает, что обработка паром в жестких условиях сильно изменяет каркас цеолита. Таким образом, условия, использованные в представленном примере, явно привели к изменению каркаса цеолита, так что его нельзя сравнивать с цеолитным материалом, полученным способом, соответствующим настоящему изобретению. Несмотря на указанное изменение структуры, в полученном цеолитном материале также не наблюдались восстанавливающие изменения, которые можно было наблюдать при использовании способа, соответствующего настоящему изобретению. В частности, как в сравнительном примере 1, отношение интенсивности первой полосы поглощения к интенсивности второй полосы поглощения в ИК спектре оставалось практически постоянным. С другой стороны, поглощение воды уменьшалось немного больше, однако не наблюдались результаты, полученные для цеолитных материалов, обработанных жидкой водной системой в соответствии со стадией (iii) способа, соответствующего настоящему изобретению.
Пример 3. Исследования дегидратации изопропанола
Общая методика приготовления порошкообразных катализаторов
Общая методика приготовления катализатора
До исследования катализатор получали в виде экструдата. 18 г Воды добавляли к 20 г высушенного порошка в смесителе Stephan-Werke GmbH (Model No.: 0ZDe042/4s) при скорости перемешивания, равной 80 об/мин. Эту смесь перемешивали до получения однородной смеси, для чего требовалось примерно 10 мин. Затем добавляли 4 г Ludox AS40 и 1 г Walocel перемешивали до получения однородной смеси, для чего требовалось примерно 2 мин. К смеси добавляли Ludox® AS40 (20 мас.%) в качестве связующего, к смеси добавляли Walocel™ (5 мас.%) в качестве смазывающего вещества. Затем медленно добавляли 2 г воды и пасту перемешивали в течение примерно 5 мин до однородности. Затем эту пасту прессовали с помощью ручного пресса с отверстием для экструзии диаметром 2 мм длиной 10 см. Полученный экструдат сушили при 120°С в течение 5 ч и прокаливали при 500°С в течение 5 ч. Затем экструдат измельчали и просеивали для отделения частиц размером от 0,5 до 0,6 мм. Эту фракцию использовали для исследования в реакторе. Использовали сита, выпускающиеся фирмой Retsch (сито 500 мкм 60.122.000500 и сито 600 мкм 60.122.000600), оба обладающие диаметром, равным 200 мм, и высотой, равной 25 мм).
Общая методика дегидратации изопропанола
Активности катализаторов определяли по разложению изопропанола, проводимому при атмосферном давлении. 4,0 г Катализатора, приготовленного в соответствии с примером 3 - Общая методика приготовления катализатора (размер частиц от 500 до 800 мкм) помещали в U-образный реактор (49,5 см длина, 0,6 см диаметр). Реактор закрепляли в регулируемой печи. В поток азота (6 л/ч) подавали пары изопропанола (200°С) с помощью насоса для ВЭЖХ (высокоэффективная жидкостная хроматография) при скорости, равной 8,021 г изопропанола в час. Для исключения конденсации изопропанола или любого продукта реакции все трубы от сатуратора до криостата нагревали до температуры выше 120°С. Газообразную реакционную смесь загружали в реактор при среднечасовой скорости подачи, равной 2,0 г/(г*ч). Температуру в печи поддерживали в течение 1 ч при 200°С, в течение 1 ч при 250°С, в течение 1 ч при 300°С и в течение 1 ч при 350°С. При каждой температуре реагенты и продукты собирали путем ожижения при температуре, равной 5°С, в криостате после прохождения через реактор. Концентрации реагентов и продуктов определяли с помощью газовой хроматографии (Perkin Elmer Autosystem GC с метилсиликоновой капиллярной колонкой НР-1 длиной 50 м и пламенным ионизационным детектором).
Цеолитные материалы, изученные при исследованиях дегидратации изопропанола:
- цеолитный материал, полученный в соответствии с примером 1, стадия (iii), т.е. материал, который обрабатывали с помощью соответствующей настоящему изобретению комбинации деборирования жидкой системой растворителей (водой) и обработки жидкой водной системой (водой);
- цеолитный материал, полученный в соответствии с примером 1, стадия (ii), т.е. материал, который только деборировали жидкой системой растворителей (водой) без проведения последующей стадии обработки жидкой водной системой; после деборирования, это вещество прокаливали путем нагревания до 550°С (линейная скорость повышения температуры равна 2 К/мин) и нагревания при этой температуре в течение 4 ч;
- цеолитный материал, полученный в соответствии с примером 1, стадия (ii), т.е. материал, который только деборировали жидкой системой растворителей (водой), без проведения последующей стадии обработки жидкой водной системой; после деборирования, это вещество прокаливали путем нагревания до 650°С (линейная скорость повышения температуры равна 2 К/мин) и нагревания при этой температуре в течение 3 ч.
Результаты примера 3
Результаты, рассмотренные ниже, представлены на фиг. 1.
При использовании цеолитного материала, соответствующего настоящему изобретению, полученного в соответствии с примером (iii), при температуре, равной 250°С, наблюдалась конверсия изопропанола, составляющая примерно 33%. Кроме того, при температуре, равной 300°С, использование этого цеолитного материала приводило к конверсии изопропанола, составляющей примерно 99%. Однако при использовании цеолитных материалов, полученных в соответствии с примером 1, стадия (ii), при температуре, равной 250°С, превращение изопропанола не протекало. При температуре, равной 300°С, при использовании цеолитного материала, полученного в соответствии с примером 1, стадия (ii), который прокаливали при 550°С, наблюдалась конверсия изопропанола, составляющая лишь примерно 90%. Использование цеолитного материала, полученного в соответствии с примером 1, стадия (ii), который прокаливали при температуре, равной 650°С, приводило к конверсии изопропанола, составляющей лишь примерно 65% при температуре 300°С.
Сопоставление рабочих характеристик катализатора при 300°С показывает, что катализатор, полученный в настоящем изобретении, приводит к конверсии изопропанола, которая на 9% или 34% больше, чем при использовании катализаторов, которые не подвергают обработке, соответствующей настоящему изобретению. Кроме того, катализатор, полученный в настоящем изобретении, является единственным из трех исследованных катализаторов, который способен катализировать реакцию уже при температуре, равной 250°С.Значения конверсии изопропанола показывают, что катализатор, полученный способом, соответствующим настоящему изобретению, т.е. с использованием комбинации (ii) процедуры деборирования с использованием жидкой системы растворителей и (iii) обработкой жидкой водной системой, обладает наилучшими рабочими характеристиками.
Краткое описание чертежа
На фиг. 1 представлены результаты исследований дегидратации изопропанола, в которых использовали
- цеолитный материал, полученный способом, соответствующим настоящему изобретению (пример 1 (iii)) (ромбы,  )
)
- цеолитный материал, полученный в соответствии с примером 1, стадия (ii), который дополнительно прокаливают путем нагревания до 550°С (линейная скорость повышения температуры равна 2 К/мин) и нагревают при этой температуре в течение 4 ч (квадраты,  );
);
- цеолитный материал, полученный в соответствии с примером 1, стадия (ii), который дополнительно прокаливают путем нагревания до 650°С (линейная скорость повышения температуры равна 2 К/мин) и нагревают при этой температуре в течение 3 ч (треугольники,  ).
).
По оси х отложена температура в °С, по оси у отложена конверсия изопропанола в %.
Цитированная литература
- ЕР 0013433 А1
- WO 02/057181 А2
- WO 2009/016153 А2
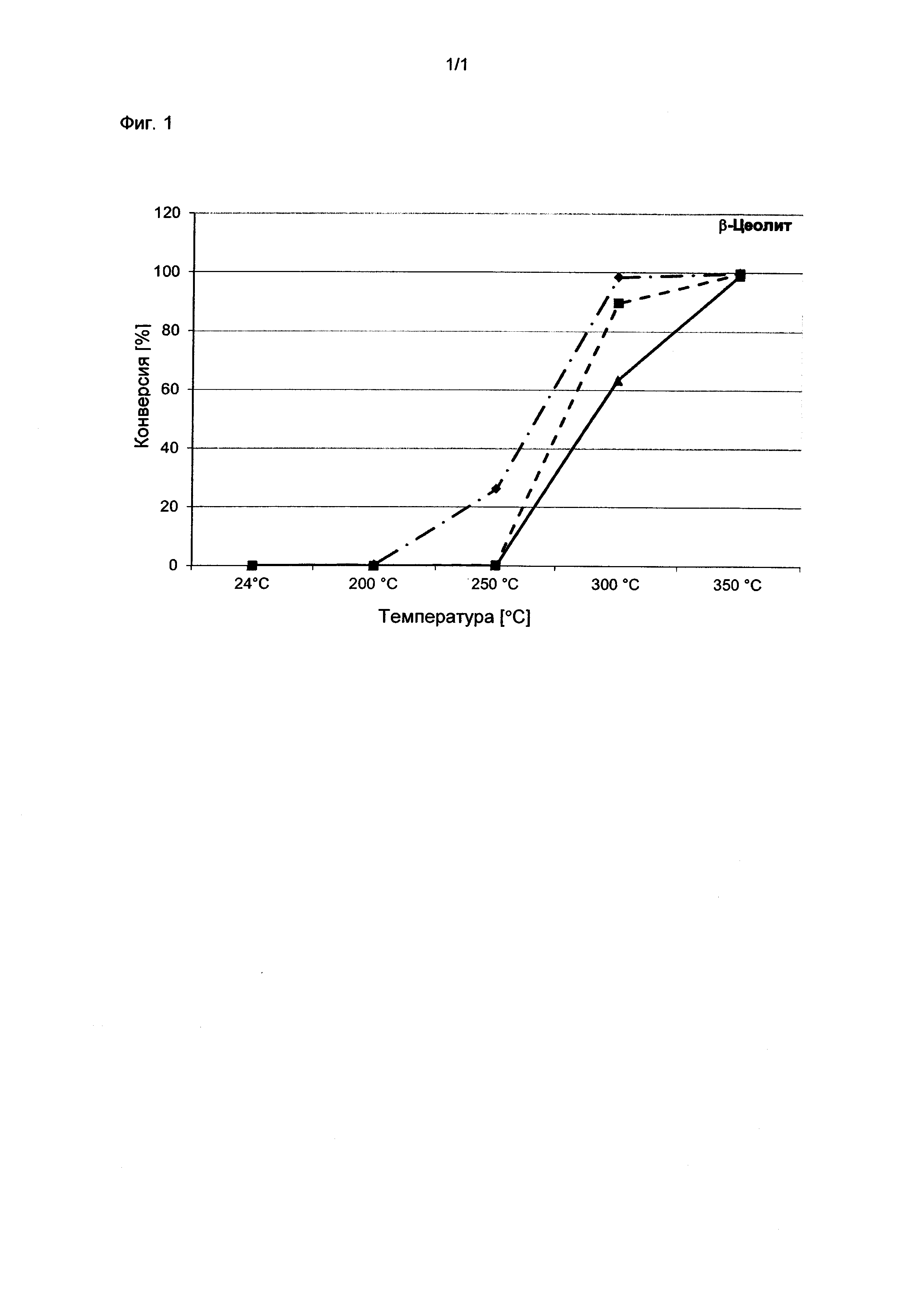
Способ хранения жидкой в условиях хранения мономерной фазы
Способ транспортировки жидкой мономерной фазы, извлеченной из резервуара для хранения, в резервуаре автозаправщика или танкера
Способ получения механически стабильных водопоглощающих полимерных частиц
Способ получения акриловой кислоты
Способ получения триэтилентетрамина (тэта) через этилендиаминдиацетонитрил (эддн)
Способ получения смеси этиленаминов
Средство для нанесения покрытий на вспенивающиеся частицы стирольного полимеризата
Способ для обнаружения и подсчета жизнеспособных микроорганизмов вида legionella pneumophila и набор для его осуществления
Способ получения смесей этиленаминов
Эластичный пеноматериал из частиц на основе смесей полиолефина/полимера стирола
Способ хранения жидкой в условиях хранения мономерной фазы
Способ транспортировки жидкой мономерной фазы, извлеченной из резервуара для хранения, в резервуаре автозаправщика или танкера
Способ получения механически стабильных водопоглощающих полимерных частиц
Способ получения акриловой кислоты
Способ получения триэтилентетрамина (тэта) через этилендиаминдиацетонитрил (эддн)
Способ получения смеси этиленаминов
Средство для нанесения покрытий на вспенивающиеся частицы стирольного полимеризата
Способ для обнаружения и подсчета жизнеспособных микроорганизмов вида legionella pneumophila и набор для его осуществления
Способ получения смесей этиленаминов
Эластичный пеноматериал из частиц на основе смесей полиолефина/полимера стирола

