Результат интеллектуальной деятельности: СОЕДИНЕНИЯ
Вид РИД
Изобретение
Настоящее изобретение относится к новым производным 5-аминолевулиновой кислоты (5-АЛК) и их применению в качестве фотосенсибилизирующих агентов. В частности оно относится к соединениям общей формулы I и их фармацевтически приемлемым солям, к способам получения таких соединений и их применению в медицинских и косметических целях, например, в способах фотодинамической терапии и диагностики.
Фотодинамическая терапия (ФДТ) представляет собой способ лечения предраковых поражений, рака и нераковых заболеваний. ФДТ включает введение фотосенсибилизатора или его предшественника (т.е. "фотосенсибилизирующего агента") в целевой участок. Фотосенсибилизатор или его предшественник поглощается клетками, где предшественник фотосенсибилизатора превращается в фотосенсибилизатор. При подвергании целевого участка действию света фотосенсибилизатор возбуждается, как правило, из основного синглетного состояния в возбужденное синглетное состояние. Затем он претерпевает интеркомбинационную конверсию в долгоживущее возбужденное триплетное состояние. Одним из немногих химических соединений, присутствующих в тканях в основном триплетном состоянии, является молекулярный кислород. Когда фотосенсибилизатор и молекула кислорода находятся вблизи друг от друга, возможен перенос энергии, который обеспечивает релаксацию фотосенсибилизатора до его основного синглетного состояния и образование молекул кислорода в возбужденном синглетном состоянии. Синглетный кислород является очень агрессивным химическим соединением и очень быстро вступает в реакции с любыми близлежащими биомолекулами. В конечном счете эти разрушительные реакции убивают клетки за счет апоптоза или некроза, в результате чего, например, селективно уничтожаются раковые клетки. Эти механизмы еще не полностью выяснены, но исследования показывают, что клинический результат (т.е. селективность в отношении раковых клеток) не обусловлена селективным поглощением раковыми клетками. Скорее, уровни поглощения всеми типами клеток одинаковы, но процессы превращения и выведения различны в злокачественных клетках и в целом в метаболически активных клетках, таких как воспаленные или инфицированные клетки, что приводит к градиенту концентрации между опухолевой и нормальной тканями.
Некоторые фотосенсибилизирующие агенты известны и описаны в литературе, включая 5-аминолевулиновую кислоту (5-АЛК) и некоторые ее производные, например, сложные эфиры 5-АЛК, при этом и кислота, и ее эфиры представляют собой предшественники фотосенсибилизаторов. Они подвергаются внутриклеточному превращению в протопорфирины, такие как протопорфирин IX (ПпIХ), которые являются фотосенсибилизаторами. В настоящее время ряд фармацевтических продуктов, содержащих 5-АЛК или ее сложный эфир, применяют в клинической практике для ФДТ и фотодинамической диагностики (ФДЦ). Одним из них является Метвикс (Metvix®), продукт для кожи в форме крема, содержащий метиловый эфир 5-АЛК (разработанный PhotoCure ASA, Norway и продаваемый в настоящее время компанией "Галдерма" (Galderma, Switzerland)), для фотодинамической терапии актинического кератоза и базальноклеточной карциномы, Еще одним препаратом является Левулан Керастик (Levulan Kerastick®) (DUSA Pharmaceuticals, Canada), продукт для фотодинамической терапии актинического кератоза, содержащий 5-АЛК. Гексвикс (Hexvix®) (разработанный PhotoCure ASA) представляет собой водный раствор, содержащий гексиловый эфир 5-АЛК, для инсталляции в мочевой пузырь с целью диагностики рака мочевого пузыря.
Однако все еще существует потребность в альтернативных фотосенсибилизаторах или их предшественниках. Настоящее изобретение направлено на решение этой проблемы путем обеспечения предшественников фотосенсибилизаторов в соответствии с приведенной ниже общей формулой I.
Таким образом, согласно первому аспекту настоящего изобретения предложено соединение общей формулы I или его фармацевтически приемлемая соль:
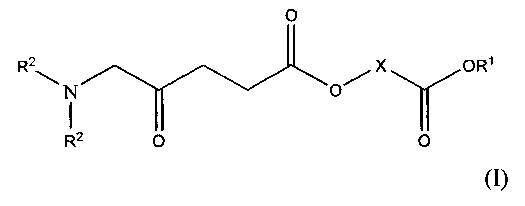
где
R1 представляет собой атом водорода или возможно замещенную алкильную или циклоалкильную группу;
каждый из R2, которые могут быть одинаковыми или различными, представляет собой атом водорода или возможно замещенную алкильную группу; и
X представляет собой связывающую группу.
Предпочтительно, что в соединениях формулы I фрагмент -X-CO2R1 является гидрофильным по своему характеру. Термин "гидрофильный" означает, что фрагмент молекулы -X-CO2R1 имеет склонность к взаимодействию с или растворению в воде или других полярных растворителях и/или веществах. Гидрофильные свойства этого фрагмента молекулы могут быть обусловлены свойствами группы X и/или свойствами группы -CO2R1. Соответственно, либо X может быть гидрофильным, либо группа -CO2R1 может быть гидрофильной, либо как X, так и группа -CO2R1 могут быть гидрофильными.
В первом варианте реализации гидрофильной является только связывающая группа X фрагмента -X-CO2R1 соединений общей формулы I.
Типовые примеры гидрофильных групп X представляют собой группы, содержащие один или более заместителей (т.е. боковых групп), которые обеспечивают гидрофильность группы, т.е. гидрофильных заместителей. Термин "гидрофильный заместитель" обозначает заместитель, способный к образованию водородных связей. Типовые и предпочтительные гидрофильные заместители представляют собой гидроксил, тиол, карбоксил, карбамоил, сложный эфир и амин, более предпочтительно гидроксил, амин и тиол. Альтернативно, гидрофильные группы X могут содержать один или более гетероатомов, обеспечивающих гидрофильность группы, т.е. гетероатомов, способных к образованию водородных связей. Предпочтительные гетероатомы представляют собой кислород или серу.
Примеры гидрофильных групп X включают, в частности, алкиленовые группы, разделенные одним или более гетероатомами, предпочтительно одним или более атомами кислорода. Такие группы включают полиэтиленгликолевые группы, предпочтительно полиэтиленгликолевые группы, содержащие 1-4 этиленоксидных звеньев. Другие примеры гидрофильных групп X представляют собой алкиленовые группы, предпочтительно С1-4 алкиленовые группы, содержащие в качестве заместителей один или более гидроксилов, тиолов или аминов.
Если гидрофильной является только группа X, группа R1 может представлять собой возможно замещенную алкильную группу, т.е. неразветвленную или разветвленную алкильную группу или циклоалкильную группу. В случае, когда R1 является замещенным, он замещен одним или более негидрофильными заместителями. Термин "негидрофильный заместитель" обозначает заместитель, который по существу не способен к образованию водородных связей. Негидрофильные заместители не включают никакую из групп, упомянутых в настоящем тексте в качестве примеров гидрофильных групп, таких как гидроксил, тиол, карбоксил, карбамоил, сложный эфир и амин. Предпочтительные негидрофильные заместители представляют собой галоген, предпочтительно F или Cl, нитро и арил. Если негидрофильный заместитель представляет собой арильную группу, указанная арильная группа может быть замещена одним или более галогеном, алкилом, галогеналкилом, алкокси (например, С1-3 алкокси) или нитрогруппами.
В одном из вариантов реализации R1 представляет собой незамещенную неразветвленную или разветвленную алкильную группу или незамещенную циклоалкильную группу, предпочтительно незамещенную неразветвленную алкильную группу, содержащую от 4 до 20 атомов углерода (предпочтительно от 4 до 10 атомов углерода, например, от 4 до 8 атомов углерода), незамещенную разветвленную алкильную группу, содержащую от 4 до 20 атомов углерода (предпочтительно от 4 до 10 атомов углерода, например, от 4 до 8 атомов углерода), или незамещенную циклоалкильную группу, содержащую от 3 до 7 атомов углерода (предпочтительно от 3 до 6 атомов углерода).
Если R1 представляет собой незамещенную алкильную группу, предпочтительными являются группы R1, представляющие собой неразветвленные алкильные группы. Более предпочтительными являются С4-10 неразветвленные алкильные группы, и наиболее предпочтительными являются С4-8 неразветвленные алкильные группы. Типовые примеры таких групп представляют собой н-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил и н-децил. В частности, предпочтительными являются н-бутил, н-пентил, н-гексил, н-гептил и н-октил.
Если R1 представляет собой незамещенную разветвленную алкильную группу, такие разветвленные алкильные группы предпочтительно содержат от 4 до 20 атомов углерода, более предпочтительно от 4 до 10 атомов углерода и наиболее предпочтительно от 4 до 8 атомов углерода. Типовые примеры таких разветвленных алкильных групп включают втор-бутил, трет-бутил, 2-метилбутил, 3,3-диметил-1-бутил и 1-этилбутил. Предпочтительные группы включают втор-бутил и трет-бутил.
Если R1 представляет собой незамещенную циклоалкильную группу, такие циклоалкильные группы предпочтительно состоят из 3-6 атомов углерода, например, циклопропил, циклобутил, циклопентил или циклогексил.
Если R1 представляет собой замещенную алкильную группу, эта группа может быть неразветвленной или разветвленной алкильной или циклоалкильной группой, содержащей один или более негидрофильных заместителей, предпочтительно один или два негидрофильных заместителя. В случае, когда присутствует более чем один негидрофильный заместитель, эти заместители могут быть одинаковыми или различными. Предпочтительные негидрофильные заместители представляют собой галоген, предпочтительно F или Cl, нитро и арил. Если негидрофильный заместитель представляет собой арильную группу, уазанная арильная группа может быть замещена одним или более галогеном, алкилом, галогеналкилом, алкокси (например, С1-3 алкокси) или нитрогруппами. Такие предпочтительные группы R1 представляют собой С1-2 алкил, замещенный одной или более арильными группами, предпочтительно одной или двумя арильными группами, которые сами по себе возможно замещены алкилом (например, С1-4 алкилом, галогеном, нитро, галогеналкилом или алкокси (например, С1-3 алкокси). Примеры таких групп R1 включают бензил, 4-изопропилбензил, 4-метилбензил, 2-метилбензил, 3-метилбензил, 4-[трет-бутил]бензил, 4-[трифторметил]бензил, 4-метоксибензил, 3,4-[дихлор]бензил, 4-хлорбензил, 4-фторбензил, 2-фторбензил, 3-фторбензил, 2,3,4,5,6-пентафторбензил, 3-нитробензил, 4-нитробензил, 2-фенилэтил, 4-фенилбутил и 4-дифенилметил. Такие более предпочтительные группы R1 представляют собой бензил, 4-изопропилбензил, 4-метилбензил, 4-нитробензил и 4-хлорбензил. Наиболее предпочтительным является бензил.
Во втором варианте реализации только группа -CO2R1, присутствующая во фрагменте -X-CO2R1 соединений общей формулы I, является гидрофильной группой. Типовой и предпочтительный пример такой группы представляет собой -СО2Н, т.е. случай, когда R1 представляет собой атом водорода. Альтернативными предпочтительными примерами являются группы -CO2R1, где R1 представляет собой короткоцепочечную неразветвленную или разветвленную алкильную группу, предпочтительно алкильную группу, содержащую 1-3 атома углерода, такую как метил, этил, н-пропил и изопропил.
Во втором варианте реализации настоящего изобретения связывающая группа X предпочтительно представляет собой неразветвленную или разветвленную алкиленовую группу, циклоалкиленовую группу, ариленовую группу или аралкиленовую группу, которая возможно замещена одним или более негидрофильными заместителями.
Если X представляет собой неразветвленную алкиленовую группу, она предпочтительно состоит из 1-16 атомов углерода, т.е. представляет собой метилен, этилен, пропилен, бутилен, пентилен, гексилен, гептилен, октилен, нонилен, децилен, ундецилен, додецилен, тридецилен, тетрадецилен, пентадецилен или гексадецилен, более предпочтительно из 1-6 атомов углерода, т.е. представляет собой метилен, этилен, пропилен, бутилен, пентилен или гексилен, еще более предпочтительно из 1-4 атомов углерода, т.е. представляет собой метилен, этилен, пропилен или бутилен.
Если X представляет собой разветвленную алкиленовую группу, она предпочтительно состоит из 2-10 атомов углерода, более предпочтительно из 2-6 атомов углерода. Предпочтительными примерами являются метилметилен, диметилметилен, 1-метилэтилен, 2-метилэтилен, 1,2-диметилэтилен, этилметилен, изопропилметилен, 1-этилэтилен и 2-этилэтилен. Наиболее предпочтительными примерами являются метилметилен, этилметилен и изопропилметилен.
Если X представляет собой циклоалкиленовую группу, она предпочтительно состоит из 3-8 атомов углерода, более предпочтительно из 5 или 6 атомов углерода. Предпочтительными примерами являются циклопентилен и циклогексилен.
Если X представляет собой ариленовую группу, она предпочтительно состоит из 6-12 атомов углерода. Предпочтительной группой является фенилен, т.е. -С6H4-, предпочтительно со свободными валентностями у атомов углерода 1 и 4.
Если X представляет собой аралкиленовую группу, она предпочтительно состоит из 7-15 атомов углерода. Предпочтительной группой является бензилен, т.е. -СН2-С6H4-, предпочтительно со свободной валентностью у атома углерода 4 ароматического кольца.
Любые из вышеописанных групп X, т.е. неразветвленные или разветвленные алкиленовые группы, циклоалкиленовые группы, ариленовые группы или аралкиленовые группы могут быть возможно замещены одним или более негидрофильными заместителями, например, любой из таких групп, описанных в настоящем тексте.
В третьем варианте реализации настоящего изобретения как X, так и группа -CO2R1 фрагмента -X-CO2R1 соединений общей формулы I являются гидрофильными. Типовые и предпочтительные примеры таких гидрофильных групп X и -CO2R1 предложены в предыдущих абзацах выше.
Каждый из R2, которые могут быть одинаковыми или различными, представляет собой атом водорода или возможно замещенную алкильную группу (предпочтительно С1-6 алкил, например, С1-3 алкильную группу). Если R2 представляет собой возможно замещенную алкильную группу, заместители могут быть гидрофильными заместителями или негидрофильными заместителями, как определено в настоящем тексте.
Предпочтительными соединениями в соответствии с настоящим изобретением являются те, в которых по меньшей мере один из R2 представляет собой атом водорода. В предпочтительном варианте реализации каждый из R2 представляет собой атом водорода.
В предпочтительном варианте реализации согласно настоящему изобретению предложено соединение общей формулы I или его фармацевтически приемлемая соль, где
R1 представляет собой атом водорода или короткоцепочечную неразветвленную или разветвленную алкильную группу, предпочтительно неразветвленную или разветвленную алкильную группу, содержащую 1-3 атома углерода, такую как метил, этил, н-пропил или изопропил;
каждый из R2, которые могут быть одинаковыми или различными, представляет собой атом водорода или возможно замещенную алкильную группу, предпочтительно водород;
и
связывающая группа X представляет собой
(a) возможно замещенную C1-6 алкиленовую группу, или
(b) возможно замещенную циклоалкиленовую, ариленовую или аралкиленовую группу.
В более предпочтительном варианте реализации согласно настоящему изобретению предложено соединение общей формулы I или его фармацевтически приемлемая соль, где
каждый из R1 и R2 представляет собой атом водорода; и
связывающая группа X представляет собой
(a) возможно замещенную C1-6 алкиленовую группу, или
(b) возможно замещенную циклоалкиленовую, ариленовую или аралкиленовую группу.
В более предпочтительном варианте реализации согласно настоящему изобретению предложено соединение общей формулы I или его фармацевтически приемлемая соль, где
R1 представляет собой атом водорода или короткоцепочечную неразветвленную или разветвленную алкильную группу, предпочтительно неразветвленную или разветвленную алкильную группу, содержащую 1-3 атома углерода, такую как метил, этил, н-пропил или изопропил;
каждый из R2, которые могут быть одинаковыми или различными, представляет собой атом водорода или возможно замещенную алкильную группу, предпочтительно водород;
и
связывающая группа X представляет собой
(a) возможно замещенную неразветвленную С1-4 алкиленовую группу или возможно замещенную разветвленную С2-6 алкиленовую группу, или
(b) возможно замещенную С5-6 циклоалкиленовую группу, возможно замещенную С6-12 ариленовую группу или возможно замещенную С7-15 аралкиленовую группу.
В более предпочтительном варианте реализации согласно настоящему изобретению предложено соединение общей формулы I или его фармацевтически приемлемая соль, где
каждый из R1 и R2 представляет собой атом водорода; и
связывающая группа X представляет собой
(a) возможно замещенную неразветвленную С1-4 алкиленовую группу или возможно замещенную разветвленную С2-6 алкиленовую группу, или
(b) возможно замещенную С5-6 циклоалкиленовую группу, возможно замещенную С6-12 ариленовую группу или возможно замещенную С7-15 аралкиленовую группу.
В одном из вариантов реализации такие группы X могут быть незамещенными. Альтернативно, такие группы могут быть замещены одним или более негидрофильными заместителями, описанными выше в настоящем тексте. Если связывающая группа X является замещенной, она может быть замещена одним или более негидрофильными заместителями. В случае, когда присутствует более чем один заместитель, они могут быть одинаковыми или различными и могут быть присоединены к одному и тому же или разным атомам углерода в алкиленовой цепи, циклоалкиленовом кольце, ариленовом кольце или цепи или кольце аралкиленовой группы.
В одном из вариантов реализации связывающая группа X представляет собой незамещенную неразветвленную С1-6 алкиленовую группу. Примерами таких групп являются незамещенная неразветвленная С1 алкиленовая группа, т.е. метиленовая группа, незамещенная неразветвленная С2 алкиленовая группа, т.е. этиленовая группа, незамещенная неразветвленная С3 алкиленовая группа, т.е. пропиленовая группа, незамещенная неразветвленная С4 алкиленовая группа, т.е. бутиленовая группа, незамещенная неразветвленная С5 алкиленовая группа, т.е. пентиленовая группа и незамещенная неразветвленная С6 алкиленовая группа, т.е. гексиленовая группа. Предпочтительные связывающие группы X представляют собой незамещенные неразветвленные С1-4 алкиленовые группы, т.е. метилен, этилен, пропилен и бутилен.
В другом варианте реализации связывающая группа X представляет собой замещенную неразветвленную С1-6 алкиленовую группу, предпочтительно замещенную неразветвленную С1-4 алкиленовую группу, более предпочтительно замещенную С1-2 алкиленовую группу. Предпочтительными заместителями являются галоген, предпочтительно F и Сl, и арил. В любом из случаев, когда заместитель представляет собой арильную группу, арил может быть незамещенным или замещенным одним или более галогеном, алкилом, галогеналкилом, алкокси (например, С1-3 алкокси) или нитрогруппами. В предпочтительном варианте реализации указанная арильная группа является незамещенной. Один или более таких заместителей (например, один или два) могут быть присоединены к алкиленовой цепи. Если присутствует более чем один такой заместитель, они могут быть связаны с одним и тем же атомом углерода или с разными атомами углерода, присутствующими в связывающей группе X. В одном из вариантов реализации два галогеновых заместителя могут быть связаны с одним и тем же атомом углерода. Неразветвленные С1-6 алкиленовые группы, более предпочтительно неразветвленные С1-4 алкиленовые группы и еще более предпочтительно неразветвленные С1-2 алкиленовые группы, моно- или дифторированные, составляют предпочтительный аспект настоящего изобретения. Неразветвленные C1-6 алкиленовые группы, более предпочтительно неразветвленные С1-4 алкиленовые группы и еще более предпочтительно неразветвленные С1-2 алкиленовые группы, замещенные арильным заместителем, предпочтительно фенилом, составляют другой предпочтительный аспект настоящнго изобретения.
В еще одном варианте реализации связывающая группа X представляет собой незамещенную разветвленную С2-6 алкиленовую группу. Предпочтительными примерами таких групп X являются метилметилен, т.е. -СН(СН3)-, этилметилен, т.е. -СН(СН2СН3)- и изопропилметилен, т.е. -СН(СН-(СН3)2)-.
В еще одном варианте реализации связывающая группа X представляет собой замещенную разветвленную С2-6 алкиленовую группу. Предпочтительными заместителями являются галоген, предпочтительно F и Сl, и арил. В случае, когда любой заместитель представляет собой любую арильную группу, указанный арил может быть незамещенным или замещенным одним или более галогеном, алкилом, галогеналкилом, алкокси (например, С1-3 алкокси) или нитрогруппами. В предпочтительном варианте реализации указанный арил является незамещенным. Галоген представляет собой предпочтительный заместитель, и один или более таких заместителей могут быть присоединены к разветвленной алкиленовой цепи. Если присутствует более чем один такой заместитель, они могут быть связаны с одним и тем же атомом углерода или с разными атомами углерода, присутствующими в разветвленной алкиленовой цепи. Предпочтительным примером такой группы X является трифторметилметилен.
В еще одном варианте реализации связывающая группа X представляет собой незамещенную циклоалкиленовую группу, такую как циклопропилен, циклобутилен, циклопентилен, циклогексилен, циклогептилен или циклооктилен. Предпочтительно, связывающая группа X представляет собой незамещенную циклоалкиленовую группу, состоящую из 5 или 6 атомов углерода, т.е. циклопентилен или циклогексилен.
В еще одном варианте реализации связывающая группа X представляет собой незамещенную ариленовую группу, состоящую из 6 или 12 атомов углерода. Предпочтительная связывающая группа X согласно этому варианту реализации представляет собой фенилен, т.е. -С6Н4-, предпочтительно со свободными валентностями у атомов углерода 1 и 4.
В еще одном варианте реализации связывающая группа X представляет собой замещенную ариленовую группу, при этом указанная ариленовая группа состоит из 6 или 12 атомов углерода, предпочтительно 6 атомов углерода. Предпочтительные заместители в кольце представляют собой алкил, галоген и нитро. Один или более таких заместителей (например, один или два) могут быть присоединены к ариленовому кольцу.
В еще одном варианте реализации связывающая группа X представляет собой незамещенную аралкиленовую группу, состоящую из 7-11 атомов углерода. Предпочтительная связывающая группа X согласно этому варианту реализации представляет собой бензилен, т.е. -СН2-С6Н4-, предпочтительно со свободной валентностью у атома углерода 4 ароматического кольца.
Предпочтительные примеры группы X включают -СН2-, -СН2СН2-, -СН2СН2СН2, -СН(СН3)-, -CF2-, циклогексилен, -СН2-С6H4-, -фенилен- и -CH(Ph)- (где Ph=фенил).
Предпочтительные соединения согласно настоящему изобретению включают те, в которых X представляет собой описанную выше группу и R1 представляет собой атом водорода или короткоцепочечную неразветвленную или разветвленную алкильную группу, предпочтительно алкильную группу, содержащую 1-3 атома углерода, такую как метил, этил, н-пропил и изопропил, и каждый из R2 одинаков и представляет собой водород.
В частности, можно привести следующие соединения, которые представляют собой предпочтительные соединения в соответствии с настоящим изобретением:
карбоксиметил-5-амино-4-оксопентаноат
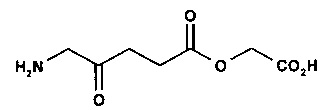
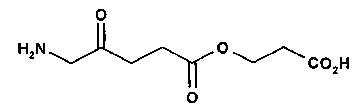
2-карбоксиэтил-5-амино-4-оксопентаноат
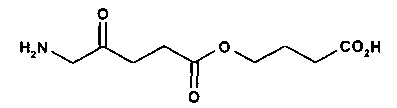
3-карбоксипропил-5-амино-4-оксопентаноат
карбоксидифторметил-5-амино-4-оксопентаноат
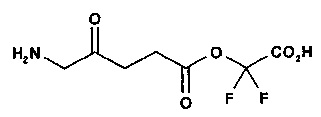
1-карбоксиэтил-5-амино-4-оксопентаноат
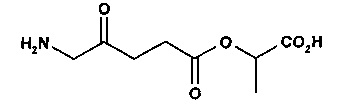
1-(изопропилкарбокси)этил-5-амино-4-оксопентаноат

(4-карбоксифенил)метил-5-амино-4-оксопентаноат
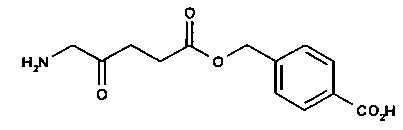
карбоксифенилметил-5-амино-4-оксопентаноат
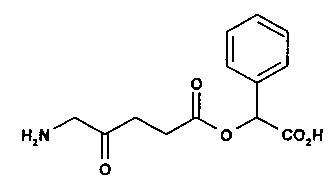
4-карбоксибутил-5-амино-4-оксопентаноат
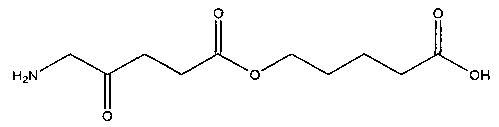
5-карбоксипентил-5-амино-4-оксопентаноат
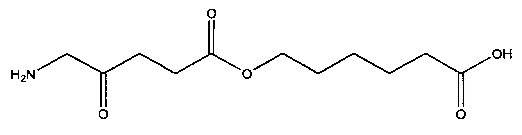
7-карбоксигептил-5-амино-4-оксопентаноат
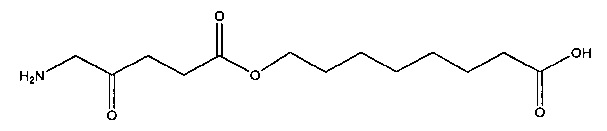
8-карбоксиоктил-5-амино-4-оксопентаноат
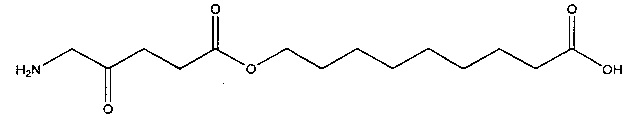
9-карбоксинонил-5-амино-4-оксопентаноат
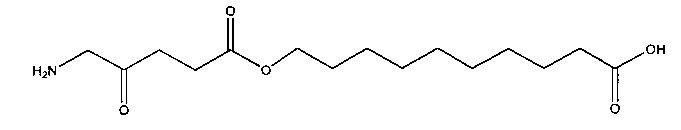
10-карбоксидецил-5-амино-4-оксопентаноат 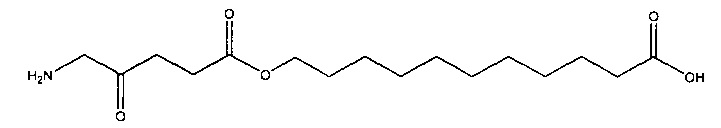
11-карбоксиундецил-5-амино-4-оксопентаноат
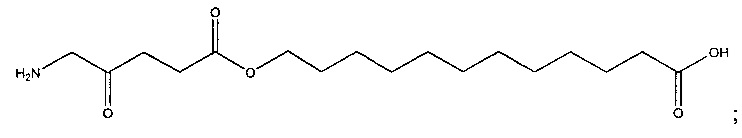 и
и
15-карбоксипентадецил-5-амино-4-оксопентаноат 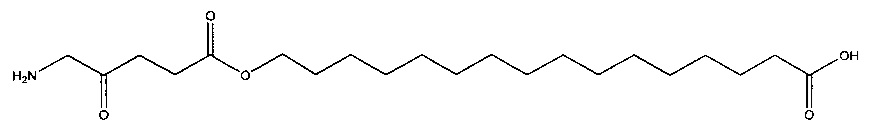
и их фармацевтитчески приемлемые соли.
В настоящем тексте термин "алкил", если не указано иное, относится к насыщенной углеводородной группе и включает любую длинноцепочечную или короткоцепочечную неразветвленную и разветвленную алкильную группу.
В настоящем тексте термин "алкилен" относится к двухвалентному радикалу, полученному из алкана, в котором свободные валентности образуют простые связи с остальной частью молекулы. Этот термин включает неразветвленные и разветвленные алкиленовые группы.
В настоящем тексте термин "циклоалкилен" относится к двухвалентному радикалу, полученному из циклоалкана, в котором свободные валентности образуют простые связи с остальной частью молекулы.
В настоящем тексте термин "арил" включает системы, содержащие ароматическое кольцо. Такие циклические системы могут быть моноциклическими или полициклическими (например, бициклическими) и содержат по меньшей мере одно ненасыщенное ароматическое кольцо. В случае, когда эти системы содержат полициклические структуры, последние могут быть конденсированными.
Термин "арилен" в настоящем тексте относится к двухвалентному радикалу, полученному из ароматического углеводорода, в котором свободные валентности образуют простые связи с остальной частью молекулы.
Термин "аралкилен" в настоящем тексте относится к двухвалентному радикалу, полученному из арилзамещенного алкила или алкилзамещенного арила, имеющего одну свободную валентность как в арильной, так и в алкильной части молекулы, в котором свободные валентности образуют простые связи с остальной частью молекулы.
Если не указано иное, термин "галоген" или "атом галогена" включает фтор, хлор, бром и йод.
Соединения согласно настоящему изобретению могут быть обеспечены в форме свободного амина, например, -NH2, -NHR2 или -NR2R2 или предпочтительно в форме фармацевтически приемлемой соли. Такие соли предпочтительно представляют собой соли присоединения кислоты с фармацевтически приемлемыми органическими или неорганическими кислотами.
Подходящие кислоты включают, например, соляную кислоту, азотную кислоту, бромоводородную кислоту, фосфорную кислоту, серную кислоту, сульфокислоту и производные сульфокислоты, уксусную кислоту, молочную кислоту, лимонную кислоту, винную кислоту, янтарную кислоту, малеиновую кислоту, фумаровую кислоту, аскорбиновую кислоту, олеиновую кислоту и стеариновую кислоту. Соответственно, подходящие соли включают, например, гидрохлорид, гидробромид, нитрат, фосфат, сульфат, сульфонат, мезилат, тозилат, напсилат, ацетат, лактат, цитрат, тартрат, сукцинат, малеат, фумарат, аскорбат, олеат и стеарат. Предпочтительные кислоты представляют собой соляную кислоту (НCl) и бромоводородную кислоту (НВr). Другими предпочтительными кислотами являются азотная кислота, сульфокислота и производные сульфокислоты (например, метансульфокислота, нафталинсульфокислота или толуолсульфокислота), как описано в публикации WO 2005/092838, PhotoCure ASA, полное содержание которой включено в настоящую заявку посредством ссылки.
Термин "фармацевтически приемлемая соль" обозначает соль, которая подходит для применения в составе фармацевтического продукта и которая отвечает требованиям, связанным, например, с безопасностью, биологической доступностью и переносимостью (см., например, P.Н. Stahl et al. (eds.) Handbook of Pharmaceutical Salts, Publisher Helvetica Chimica Acta, Zurich, 2002).
Соединения согласно изобретению могут быть получены с применением стандартных способов и процедур, хорошо известных в данной области техники для дериватизации соединений с несколькими функциональными группами, например, для дериватизации карбоновых кислот. Как известно в данной области техники, такие реакции могут включать защиту и снятие защиты для соответствующих групп таким образом, что только требуемые группы остаются активными и участвуют в реакции при выбранных условиях реакции. Так, например, можно защищать заместители, присутствующие в любом из реагентов, применяемых для получения соединений в соответствии с настоящим изобретением. Аналогично, группа -NR22 может быть защищена во время реакции и подвергнута снятию защиты в дальнейшем. Такие способы защиты/снятия защиты хорошо известны в данной области техники, см., например, McOmie в "Protective Groups in Organic Chemistry", Plenum, 1973 и T.W. Greene в "Protective Groups in Organic Chemistry", Wiley-Interscience, 1981.
Таким образом, в соответствии с еще одним аспектом настоящего изобретения предложен способ получения соединений согласно изобретению, включающий стадию дериватизации карбоксильной группы 5-аминолевулиновой кислоты или ее защищенного производного.
Таким образом, согласно настоящему изобретению предложен способ получения соединений в соответствии с изобретением, причем указанный способ включает по меньшей мере одну из следующих стадий:
(a) взаимодействие соединения формулы II:
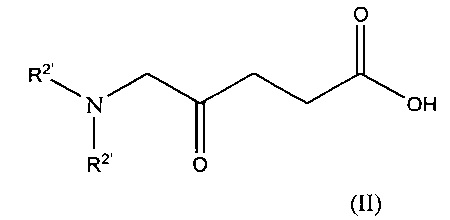
где каждый из R2’, которые могут быть одинаковыми или различными, представляет собой группу R2, определенную в настоящем тексте, или ее защищенное производное
с соединением формулы III:
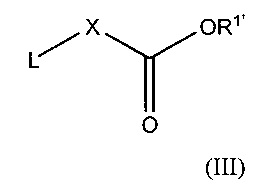
где X имеет значения, определенные в настоящем тексте;
L представляет собой уходящую группу, например, атом галогена; и
R1’ представляет собой группу R1, определенную в настоящем тексте, или ее защищенное производное;
(b) удаление защитной группы из защищенного производного соединения формулы I; и
(c) превращение соединения формулы I в его фармацевтически приемлемую соль.
Реакция согласно стадии (а) может подходящим образом быть проведена в растворителе или смеси растворителей, таких как вода, ацетон, этилацетат, диэтиловый эфир, метилформамид, тетрагидрофуран и т.д., при температурах не более температуры кипения смеси, предпочтительно при комнатных температурах. Точные условия реакции будет зависеть от применяемых реагентов, и условия могут быть выбраны таким образом, чтобы обеспечить достижение максимального выхода конечного продукта.
Реакцию согласно стадии (а) удобно проводить в присутствии катализатора, например, неорганической или органической кислоты или связывающего кислоту агента, такого как основание.
Соединения, применяемые в качестве исходных веществ, известны из литературы и во многих случаях коммерчески доступны, либо они могут быть получены с применением широко известных способов. Например, 5-АЛК доступна в Sigma-Aldrich или Biosynth AG, Switzerland.
Соединения согласно настоящему изобретению предпочтительно находятся в форме фармацевтически приемлемых и/или совместимых с кожей солей. Такие соли предпочтительно представляют собой соли присоединения кислоты с физиологически приемлемыми органическими или неорганическими кислотами, упомянутыми выше в настоящем тексте.
Соединения согласно настоящему изобретению являются предшественниками фотосенсибилизаторов, т.е. они могут поглощаться клетками и превращаться в протопорфирины, представляющие собой фотосенсибилизаторы. Следовательно, соединения согласно изобретению обладают ценными фармакологическими свойствами, а именно выступают в качестве предшественников фотосенсибилизаторов, что обеспечивает их пригодность для способов фотодинамической терапии и фотодинамической диагностики и для фотодинамических косметических методов.
Соответственно, согласно еще одному аспекту настоящего изобретения предложена композиция, содержащая описанное в настоящем тексте соединение формулы I или его фармацевтически приемлемую соль вместе с по меньшей мере одним фармацевтически приемлемым или косметически приемлемым носителем или вспомогательным веществом.
В предпочтительном варианте реализации изобретения предложена фармацевтическая композиция, содержащая описанное в настоящем тексте соединение формулы I или его фармацевтически приемлемую соль вместе с по меньшей мере одним фармацевтически приемлемым носителем или вспомогательным веществом.
В другом предпочтительном варианте реализации изобретения предложена косметическая композиция, содержащая описанное в настоящем тексте соединение формулы I или его фармацевтически приемлемую соль вместе с по меньшей мере одним косметически приемлемым носителем или вспомогательным веществом.
Согласно еще одному аспекту настоящего изобретения предложено описанное в настоящем тексте соединение или фармацевтическая композиция для применения в качестве лекарственного средства, например, в способе фотодинамической терапии или фотодинамической диагностики и, в частности, для лечения или диагностики расстройств или патологий наружных или внутренних поверхностей тела, которые восприимчивы к фотодинамической терапии или диагностике.
Согласно еще одному аспекту настоящего изобретения предложено применение описанного в настоящем тексте соединения формулы I или его фармацевтически приемлемой соли для получения фармацевтической композиции для применения в способе фотодинамической терапии или для получения фармацевтической композиции для применения в способе фотодинамической диагностики и, в частности, для лечения или диагностики расстройств или патологий наружных или внутренних поверхностей тела, которые восприимчивы к фотодинамической терапии или диагностике.
Применение соединений и фармацевтических композиций, описанных в настоящем тексте, для фотодинамической терапии или диагностики рака, инфекции, ассоциированной с раковым заболеванием, такой как вирусная инфекция (например, вирус папилломы человека, вирус гепатита В или вирус Эпштейна-Барр) или бактериальные инфекции (например, инфекция Helicobacter pylori), или для лечения или диагностики нераковых состояний представляет собой предпочтительные аспекты настоящего изобретения.
В настоящем тексте термины "рак" и "раковый" применяют по отношению к состояниям, при которых присутствуют злокачественные клетки. Таким образом, предзлокачественные состояния не охвачены этими терминами.
Термин "нераковый" включает доброкачественные и предзлокачественные состояния.
В настоящем тексте термин "лечение" или "терапия" включает как терапевтическое, так и профилактическое лечение или терапию.
Патологии и расстройства, которые можно лечить или диагностировать в соответствии с настоящим изобретением, включают любые злокачественные, предзлокачественные и доброкачественные патологии или расстройства, восприимчивые к фотодинамической терапии или диагностике.
В общем, клетки, которые являются метаболически активными, восприимчивы к фотодинамической терапии или диагностике с применением соединений согласно настоящему изобретению. Примеры метаболически активных клеток представляют собой клетки, которые претерпевают патологический тип роста. Такие патологические типы роста включают повышенное количество клеток/повышенную пролиферацию клеток (гиперплазию), аномальное созревание и дифференцировку клеток (дисплазию) и аномальную пролиферацию клеток (неоплазию). Клетки с гиперпластическим типом роста остаются подчиненными нормальным регуляторным механизмам контроля. Клетки с неопластическим типом роста являются генетически аномальными клетками, пролиферирующими нефизиологическим образом, при котором отсутствует реакция на нормальные стимулы. Другие примеры метаболически активных клеток представляют собой воспаленные клетки.
Соединения и фармацевтические композиции в соответствии с настоящим изобретением подходят, в частности, для применения в фотодинамической терапии и диагностике новообразований и опухолей (доброкачественных, предзлокачественных и злокачественных) внутренних поверхностей тела и наружных поверхностей тела (например, кожи). Примерами таких новообразований и опухолей наружных поверхностей тела являются актинический кератоз и болезнь Боуэна. Примерами таких новообразований и опухолей внутренних поверхностей тела являются рак мочевого пузыря и рак толстой кишки.
Также, соединения и фармацевтические композиции в соответствии с настоящим изобретением подходят, в частности, для применения в фотодинамической терапии и диагностике заболеваний и расстройств, ассоциированных с вирусными, бактериальными и грибковыми инфекциями, таких как угревая сыпь (ассоциированная с бактерией Propionibacterium acnes), вагинальная или цервикальная интраэпителиальная неоплазия (ассоциированная с вирусом папилломы человека), рак желудка (ассоциированный с бактерией Helicobacter pylori) и псевдомембранозный колит (ассоциированный с бактерией Clostridium difficile).
Также, соединения и фармацевтические композиции в соответствии с настоящим изобретением подходят, в частности, для применения в фотодинамической терапии инфекций кожи или ран с присутствием грамположительных бактерий. Такие инфекции часто вызваны Staphylococcus aureus (S. aureus) и обычно подлежат лечению антибиотиками, например, пенициллином. Существует неудовлетворенная медицинская потребность в поиске альтернативных способов лечения, поскольку многие штаммы S. aureus приобрели устойчивость к антибиотикам. Другие грамположительные бактерии, участвующие в развитии бактериальных раневых инфекций, представляют собой, например, Staphylococcus epidermis, Bacillus subtilis, Enterococcus faecalis и Micrococcus luteus.
Кроме того, соединения и фармацевтические композиции в соответствии с настоящим изобретением подходят, в частности, для применения в фотодинамической терапии и диагностике воспаленных клеток. Воспаление клеток обычно представляет собой защитную попытку организма удалить вредоносные раздражители и инициировать процесс заживления и, таким образом, зачастую ассоциировано с инфекцией. Примеры представляют собой воспалительную угревую сыпь, колит (например, воспалительное заболевание кишечника, язвенный колит и болезнь Крона) и инфекционный дерматит, т.е. воспаление кожи, вызванное бактериальной, вирусной или грибковой инфекцией.
Внутренние и наружные поверхности тела, которые можно лечить в соответствии с настоящим изобретением, включают кожу и все другие эпителиальные и серозные поверхности, включая, например, слизистую оболочку, выстилки органов, например, дыхательных путей, желудочно-кишечного и мочеполового тракта и железы с протоками, которые опорожняются на такие поверхности (например, печень, волосяные фолликулы с сальными железами, молочные железы, слюнные железы и семенные пузырьки). Помимо кожи, такие поверхности включают, например, выстилку влагалища, эндометрий и уротелий. Такие поверхности могут также включать полости, образующиеся в участках тела после иссечения пораженной или опухолевой ткани, например, полости мозга, образующиеся после иссечения опухолей, таких как глиомы.
Типовые поверхности, таким образом, включают: (i) кожу и конъюнктиву; (ii) выстилку ротовой полости, глотки, пищевода, желудка, отделов кишечника и придатков кишечника, прямой кишки и анального канала; (iii) выстилку носовых проходов, носовых пазух, носоглотки, трахеи, бронхов и бронхиол; (iv) выстилку мочеточников, мочевого пузыря и уретры; (v) выстилку вульвы, влагалища, шейки матки и матки; (vi) париетальную и висцеральную плевру; (vii) выстилку брюшной и тазовой полости и поверхности органов, содержащихся внутри этих полостей и (viii) твердую мозговую оболочку и мозговые оболочки.
Для получения фармацевтических композиций согласно настоящему изобретению соединения в соответствии с формулой I могут быть приготовлены любым обычным способом вместе с одним или более физиологически приемлемыми носителями или вспомогательными веществами в соответствии со способами, хорошо известными в данной области техники. При необходимости соединения или композиции согласно изобретению стерилизуют, например, путем облучения γ-лучами, автоклавирования или тепловой стерилизации, до или после добавления носителя или вспомогательного вещества, в случае их присутствия, с обеспечением стерильных составов.
Фармацевтические композиции могут быть введены системно (например, перорально или парентерально) или локально (например, посредством инъекции или местного введения) на пораженный участок или вблизи него. Фармацевтические композиции для местного введения являются предпочтительными и включают гели, кремы, мази, спреи, лосьоны, бальзамы, палочки, порошки, пессарии, суппозитории, аэрозоли, капли, растворы и любые другие обычные фармацевтические формы, применяемые в данной области техники. Местное введение в недоступные участки может быть обеспечено способами, известными в данной области техники, например, путем применения катетеров или других подходящих систем доставки лекарственных средств.
Мази, гели и кремы могут, например, быть приготовлены на водной или масляной основе с добавлением подходящих загустителей и/или гелеобразующих агентов. Любые применяемые загустители и гелеобразующие агенты должны быть нетоксичными, нераздражающими и не содержать выщелачиваемых примесей. Они должны быть инертными по отношению к активным ингредиентам, т.е. не должны способствовать их разложению. Составы для лечения ран, например, лечения бактериально инфицированных ран могут быть получены на основе гелевых составов, например, гидрогелей. Соединения согласно настоящему изобретению могут быть включены в такие гидрогелевые составы. Альтернативно, указанные соединения могут быть заключены в липосомы, которые включены в гидрогели (см., например, J. Hurler et al., J. Pharm. Sci. 101, No. 10, 2012, 3906-3915). Лосьоны могут быть приготовлены на водной или масляной основе и обычно также содержат один или более эмульгирующих, диспергирующих, суспендирующих агентов, загустителей или красителей. Порошки могут быть получены с применением любой подходящей порошковой основы. Капли, спреи и растворы могут быть приготовлены на водной или неводной основе, также содержащей один или более диспергирующих, солюбилизирующих или суспендирующих агентов. Аэрозоли обычно наносят из аэрозольных упаковок с применением подходящего пропеллента.
Альтернативно, фармацевтические композиции могут быть обеспечены в форме, приспособленной для перорального или парентерального введения, например, посредством внутрикожной, подкожной, интраперитонеальной или внутривенной инъекции. Альтернативные фармацевтические формы, таким образом, включают плоские или покрытые оболочкой таблетки, капсулы, суспензии и растворы, содержащие в качестве носителей или вспомогательных веществ кукурузный крахмал, лактозу, сахарозу, микрокристаллическую целлюлозу, стеарат магния, поливинилпирролидон, лимонную кислоту, винную кислоту, воду, воду/этанол, воду/глицерин, воду/сорбит, воду/полиэтиленгликоль, пропиленгликоль, стеариловый спирт, карбоксиметилцеллюлозу или жировые вещества, такие как твердый жир, или их подходящие смеси. Если фармацевтическая композиция в соответствии с изобретением необязательно содержит один или более фармацевтически приемлемых растворителей, такие растворители могут представлять собой свободную жирную кислоту, свободный жирный спирт, водный раствор, например, буфер, или воду.
Фармацевтические композиции могут дополнительно содержать общеизвестные фармацевтические вспомогательные вещества, такие как смазывающие агенты, загустители, увлажняющие агенты, эмульгирующие агенты, суспендирующие агенты, консервирующие средства, наполнители, связующие, консерванты, усилители адсорбции, например, агенты, способствующие проникновению через поверхность, упомянутые ниже, и т.п. Также могут применяться стабилизирующие и/или солюбилизирующие агенты, например, циклодекстрины (ЦД), α-, β-, γ- и 2-гидроксипропил-β-циклодекстрин (ГП-β-циклодекстрин). Специалисты в данной области техники имеют возможность выбора подходящих вспомогательных веществ в зависимости от своей цели. Общеизвестные вспомогательные вещества, которые могут применяться в описанных в настоящем тексте фармацевтических продуктах, приведены в различных руководствах (например, D.E. Bugay and W.P. Findlay (Eds) Pharmaceutical excipients (Marcel Dekker, New York, 1999), E-M Hoepfher, A. Reng and P.C. Schmidt (Eds) Fiedler Encyclopedia of Excipients for Pharmaceuticals, Cosmetics and Related Areas (Edition Cantor, Munich, 2002) и H.P. Fielder (Ed) Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik und angrenzende Gebiete (Edition Cantor Aulendorf, 1989)).
Все вышеупомянутые фармацевтически приемлемые вспомогательные вещества хорошо известны в данной области техники и коммерчески доступны у различных производителей.
Фармацевтические композиции согласно настоящему изобретению могут быть приготовлены таким образом, чтобы обеспечить быстрое, замедленное или отсроченное высвобождение активного ингредиента после введения пациенту, с применением способов, хорошо известных в данной области техники.
Концентрация описанных в настоящем тексте соединений в фармацевтических композициях зависит от природы соединения, композиции, способа введения, состояния, подлежащего лечению или диагностике, и субъекта, которому их вводят, и может быть изменена или скорректирована по выбору. В общем, однако, подходящими являются диапазоны концентрации, составляющие от 0,01 до 50% по массе, такие как от 0,05 до 20% по массе или от 1 до 10% по массе, например, от 1 до 5% по массе. Следует принять во внимание, что ФДТ может требовать применения более высоких концентраций соединений согласно изобретению, чем те, которые используют в способах диагностики.
В другом варианте реализации фармацевтические композиции могут дополнительно содержать один или более биоадгезивных агентов, например мукоадгезивных агентов, таких как описанные в публикации WO 02/09690, PhotoCure ASA, полное содержание которой включено в настоящую заявку посредством ссылки.
Соединения согласно изобретению могут быть приготовлены и/или введены вместе с другими активными компонентами, которые служат для усиления фотодинамического эффекта, например, агентами, способствующими проникновению через поверхность (или усилителями всасывания), и/или хелатирующими агентами. Подходящие агенты, способствующие проникновению через поверхность, и хелатирующие агенты и подходящие концентрации таких агентов описаны в публикации WO 2009/074811, PhotoCure ASA, полное содержание которой включено в настоящую заявку посредством ссылки. В зависимости от характера композиции, соединения могут быть введены совместно с другими такими необязательными агентами, например, в составе единой композиции или, альтернативно, они могут быть введены по отдельности (например, последовательно). В некоторых случаях может быть эффективной предварительная обработка наружной или внутренней поверхности тела, которую лечат, агентом, способствующим проникновению через поверхность, на отдельной стадии перед введением активного компонента. В случае, когда на стадии предварительной обработки используют агент, способствующий проникновению через поверхность, его можно применять в более высоких концентрациях, например, вплоть до 100% по массе. Если применяют такую стадию предварительной обработки, фармацевтическая композиция в соответствии с изобретением далее может быть введена не более чем через несколько часов после предварительной обработки, например, с интервалом от 5 до 60 минут после предварительной обработки.
Продукты и наборы, которые содержат описанную в настоящем тексте фармацевтическую композицию и, необязательно, агент, способствующий проникновению через поверхность, и/или хелатирующий агент в виде комбинированного препарата для одновременного, раздельного или последовательного применения в способе фотодинамической терапии или диагностики патологии наружной или внутренней поверхности тела, составляют еще один аспект настоящего изобретения.
В альтернативном рассмотрении, этот аспект настоящего изобретения относится к набору для применения в способе фотодинамической терапии или диагностики заболевания, расстройства или патологии наружной или внутренней поверхности тела, при этом указанный набор включает:
(a) первую емкость, содержащую описанное в настоящем тексте соединение или его фармацевтически приемлемую соль или описанную в настоящем тексте фармацевтическую композицию;
(b) вторую емкость, содержащую по меньшей мере один агент, способствующий проникновению через поверхность; и необязательно
(c) один или более хелатирующих агентов, содержащийся либо внутри указанной первой емкости, либо в необязательной третьей емкости.
Согласно еще одному аспекту настоящего изобретения предложен способ фотодинамической терапии или диагностики заболевания, расстройства или патологии наружной или внутренней поверхности тела, при этом указанный способ включает нанесение на пораженную поверхность описанной в настоящем тексте фармацевтической композиции и подвергание указанной поверхности действию света.
После нанесения фармацевтической композиции на наружную или внутреннюю поверхность тела целевой участок для лечения или для обследования (в случае диагностики) подвергают действию света (облучению) для достижения необходимой фотоактивации. Продолжительность периода времени между введением и действием света, т.е. время инкубации, будет зависеть от природы соединения, характера композиции, состояния, подлежащего лечению или диагностике, и способа введения. В общем, необходимо, чтобы соединение согласно изобретению в составе фармацевтической композиции высвобождалось в количестве, достаточном для поглощения клетками или тканью, которые лечат или диагностируют, превращения в фотосенсибилизатор и достижения эффективной концентрации в ткани в предполагаемом участке лечения/диагностики до фотоактивации. Как правило, время инкубации составляет не более 10 часов, например, от примерно 10 минут до 10 часов, например, от 30 минут до 7 часов или от 1 часа до 5 часов. Прямое местное введение может приводить к сокращению времени инкубации, например, полному отсутствию времени инкубации (т.е. незамедлительному подверганию действию света после введения), времени инкубации от примерно 1 минуты до 3 часов, например, от 5 минут до примерно 2 часов, или от 15 минут до 1,5 часов или от 30 минут до 1 часа. В случае, когда необходимо системное поглощение после перорального введения, время инкубации может быть большим, например, может составлять от примерно 30 минут до 6 часов, например, от 2 до 5 часов или от 3 до 4 часов. Оптимальное время инкубации, обеспечивающее максимальную концентрацию фотосенсибилизатора в участке-мишени, легко может быть определено, например, путем измерения флуоресценции в динамике.
Обычно применяют облучение в течение короткого интервала времени с высокой интенсивностью света, т.е. высокой плотностью потока, или в течение более длительного интервала времени с низкой интенсивностью света, т.е. низкой плотностью потока. Последнее может быть предпочтительным для процедуры ФДТ, в частности, когда пациента не подвергают анестезии. Процедуры ФДТ с низкой интенсивностью света приводят к уменьшению дискомфорта для пациента без какого-либо уменьшения эффективности лечения. Тем не менее, если процедура ФДТ или ФДД требует анестезии и/или иммобилизации пациента и/или должна быть проведена в операционной, может быть предпочтительным облучение, применяемое в течение короткого интервала времени с высокой интенсивностью света, т.е. высокой плотностью потока. Плотность потока зависит от вида устройства для облучения, т.е. от конкретной используемой лампы или лазера. Плотность потока может являться постоянным параметром, который не может быть выбран пользователем лампы/лазера. Однако, если плотность потока можно выбирать, для ламп, содержащих светоизлучающие диоды, могут быть выбраны обычные значения плотности потока, составляющие от 0,5 до 100 мВт/см2. Для процедуры с низкой плотностью потока предпочтительными являются значения плотности потока ниже 50 мВт/см2. Более предпочтительны значения плотности потока в диапазоне 1-30 мВт/см2, наиболее предпочтительны значения в диапазоне от 2 до 10 мВт/см2. Для процедуры с высокой плотностью потока предпочтительными являются значения плотности потока выше 50 мВт/см2, более предпочтительны значения в диапазоне от 50 до 70 мВт/см2.
Длина волны света, применяемого для облучения, может быть выбрана для достижения желаемого фотодинамического эффекта. Было обнаружено, что эффективным, в частности, является облучение длинами волн света, находящимися в диапазоне 300-800 нм, например, 400-700 нм и 500-700 нм. В случае ФДТ может быть, в частности, важным включение длин волн 630 и 690 нм. Красный свет (600-670 нм) является, в частности, предпочтительным, поскольку свет с этой длиной волны, как известно, хорошо проникает в ткань. В случае ФДД целевой участок может сначала быть обследован с применением белого света. Для достоверной фотодинамической диагностики обычно применяют синий свет, имеющий длину волны, находящуюся, как правило, в диапазоне от 360 до 450 нм, что вызывает флуоресценцию в красной области (например, от 550 до 750 нм), которая может быть обнаружена визуально или измерена с помощью подходящих спектрометров.
Облучение в случае ФДТ обычно применяют при плотности энергии от 10 до 200 Дж/см2, предпочтительно от 20 до 100 Дж/см2 и наиболее предпочтительно от 25 до 60 Дж/см2. В случае ФДД облучение предпочтительно проводят в течение всей процедуры диагностики или в течение ее части, например, в сочетании с диагностикой с применением белого света. Как для ФДТ, так и для ФДД может применяться однократное облучение или, альтернативно, может применяться дробная доза света, причем доставка этой дозы света осуществляется по частям, например, с интервалом от нескольких минут до нескольких часов между облучениями. Также может применяться многократное облучение.
Продолжительность облучения для ФДТ (т.е. время облучения) зависит от плотности потока из излучающего устройства и требуемой плотности энергии. По существу, плотность энергии является произведением времени облучения и плотности потока.
В данной области техники известны различные устройства для облучения, например, лазер и лампы. Последние могут представлять собой лампы, содержащие электрическую лампу, например, короткодуговую ксеноновую лампу или люминесцентную лампу. Альтернативно, лампа может содержать с вето излучающие диоды или органические светоизлучающие диоды (СИД и ОСИД). Для облучения внутренних поверхностей тела, например, выстилки мочевого пузыря или толстой кишки, могут применяться эндоскопы. Обычно эндоскопы содержат внешний источник света (например, лазер или лампу), соединенный с оптическим волокном, которое функционирует в качестве волновода для передачи света от внешнего источника света к целевому участку.
Таким образом, согласно еще одному аспекту настоящего изобретения предложен способ визуализации in vivo участка-мишени на внутренней или наружной поверхности тела, причем указанный способ включает следующие стадии:
(a) введение субъекту, например человеку или отличному от человека животному соединения, описанного в настоящем тексте, его фармацевтически приемлемой соли или фармацевтической композиции, содержащей указанное соединение или указанную соль;
(b) при необходимости, ожидание в течение периода времени, требующегося для поглощения указанного соединения или его фармацевтически приемлемой соли клетками указанного участка-мишени, превращения в фотосенсибилизатор и достижения эффективной концентрации в участке-мишени;
(c) подвергание участка-мишени действию света, за счет которого осуществляется фотоактивация фотосенсибилизатора;
(d) детекция флуоресценции, свидетельствующей о наличии патологии в участке-мишени; и
(e) необязательное преобразование зарегестрированной флуоресценции в изображение целевого участка.
В предпочтительном варианте реализации этого аспекта настоящего изобретения указанный способ визуализации может применяться к субъекту, которому предварительно было введено указанное соединение, его фармацевтически приемлемая соль или фармацевтическая композиция, содержащая указанное соединение или указанную соль.
Описанные в настоящем тексте процедуры ФДД также могут быть проведены во время хирургической операции, при которой соединение согласно изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят пациенту и затем выполняют хирургическую операцию под синим светом. То обстоятельство, что аномальные участки флуоресцируют в синем свете, помогает хирургу определить "границу хирургического вмешательства" и, таким образом, обеспечивает более избирательное иссечение пораженного участка (например, опухоли). Применение описанных в настоящем тексте соединений и фармацевтических композиций в способах хирургического лечения составляет еще один аспект настоящего изобретения.
Описанные в настоящем тексте способы терапии и диагностики также могут применяться в форме комбинированной терапии. Например, после курса ФДТ, проведенного в связи с патологией, расстройством или заболеванием внутренней или наружной поверхности тела с применением описанного в настоящем тексте соединения или композиции, может быть применен способ ФДД (например, для определения степени эффективности ФДТ и/или для обнаружения любого повторного возникновения состояния).
Таким образом, согласно еще одному аспекту настоящего изобретения предложено описанное в настоящем тексте соединение или композиция для применения в способе, который включает стадии: (i) проведения фотодинамической терапии патологии, расстройства или заболевания внутренней или наружной поверхности тела пациента и (ii) проведение фотодинамической диагностики у указанного пациента. По меньшей мере одну из стадий (i) и (ii) осуществляют после введения указанному пациенту соединения или фармацевтической композиции в соответствии с изобретением. Предпочтительно, обе стадии (i) и (ii) осуществляют после введения такого соединения или композиции.
После идентификации патологии, расстройства или заболевания внутренней или наружной поверхности тела с применением любого из описанных в настоящем тексте способов далее можно лечить его с помощью альтернативных терапевтических методов, например, хирургического лечения или химиотерапии. Примеры современных способов лечения включают хирургическое лечение, эндоскопическую абляционную терапию, химическую абляцию, термическую абляцию или механическую абляцию. В одном из вариантов реализации изобретения дополнительное применение соединения или фармацевтической композиции в целевом участке может быть осуществлено с целью обеспечения эффекта ФДТ.
Соединения согласно изобретению также могут применяться в способах диагностики in vitro, например, для исследования клеток, содержащихся в физиологических жидкостях. Большая величина флуоресценции, ассоциированная с присутствием метаболически активных клеток, может подходящим образом свидетельствовать о патологии или расстройстве. Этот способ обладает высокой чувствительностью и может применяться для ранней диагностики патологий или расстройств, например, рака мочевого пузыря или легкого путем исследования эпителиальных клеток в образцах мочи или мокроты, соответственно. Другие подходящие физиологические жидкости, которые помимо мочи и мокроты могут применяться для диагностики, включают кровь, семенную жидкость, слезы, спинномозговую жидкость и т.д. Также можно анализировать образцы или препараты тканей, например, биоптаты тканей или образцы костного мозга. Таким образом, настоящее изобретение распространяется на применение соединений согласно изобретению или их фармацевтически приемлемых солей для фотодинамической диагностики и на продукты и наборы для проведения указанной диагностики.
Еще один аспект настоящего изобретения относится к способу диагностики in vitro патологий, расстройств или заболеваний внутренней или наружной поверхности тела пациента путем анализа образца физиологической жидкости или ткани указанного пациента, при этом указанный способ включает по меньшей мере следующие стадии:
i) смешивание указанного образца физиологической жидкости или ткани с описанным выше в настоящем тексте соединением, его фармацевтически приемлемой солью или фармацевтической композицией, содержащей указанное соединение или указанную соль,
ii)подвергание указанной смеси действию света,
iii) определение уровня флуоресценции, и
iv) необязательное сравнения уровня флуоресценции с контрольными уровнями.
Соединения в соответствии с изобретением также могут применяться в фотодинамической терапии патологий, расстройств или заболеваний внутренней или наружной поверхности тела, вызванных или иным образом ассоциированных с бактериями, включая патогенные бактерии или условно-патогенные бактерии, т.е. бактерии, которые являются частью нормальной флоры человека, но могут быть патогенными при некоторых условиях.
Множественная лекарственная устойчивость представляет собой растущую проблему в области инфекционных заболеваний. Среди многих видов патогенных бактерий, которые приобрели устойчивость к традиционно применяемым антибиотикам, находится Staphylococcus aureus (S. aureus), который вызывает кожные инфекции, а также инфицирование ран и ожогов. Токсичные штаммы S. aureus могут попадать в кровоток в результате таких инфекций, а это в свою очередь может приводить к серьезным осложнениям и даже угрожающим жизни состояниям, таким как токсемия (синдром токсического шока), эндокардит и пневмония. Устойчивые штаммы S. aureus включают метициллинрезистентный S. aureus (MRSA).
Было обнаружено, что описанные в настоящем тексте соединения при применении в ФДТ эффективны для уничтожения или по меньшей мере уменьшения пролиферативного потенциала бактериальных клеток, в частности грамположительных бактерий, в частности Staphylococcus aureus.
Таким образом, согласно еще одному аспекту настоящего изобретения предложены соединение или фармацевтическая композиция, описанные в настоящем тексте, для применения в способе лечения и/или предотвращения бактериальной инфекции, например, для применения в способе лечения или предотвращения состояния, вызванного или ассоциированного с лекарственно-устойчивой бактерией. Способы медикаментозного лечения таких состояний, в соответствии с которыми эффективное количество соединения или композиции вводят пациенту, нуждающемуся в этом, составляют еще один аспект изобретения.
У младенцев инфекция S. aureus может вызывать тяжелое заболевание - стафилококковый синдром обожженной кожи (ССОК). Заболевание проявляется повсеместным образованием заполненных жидкостью волдырей, которые имеют тонкие стенки и легко разрушаются. Кроме того, S. aureus чрезвычайно преобладает у пациентов, страдающих атопическим дерматитом, которые менее устойчивы к нему, чем другие люди. Он зачастую вызывает осложнения, и это заболевание встречается преимущественно в зонах активного роста волос, включая подмышечные впадины, волосы и волосистую часть кожи головы.
Как было описано ранее, соединения в соответствии с настоящим изобретением, в частности, также находят применение в фотодинамических косметических методах. Соответственно, согласно еще одному аспекту изобретения предложена косметическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль, вместе с по меньшей мере одним совместимым с кожей носителем или вспомогательным веществом.
В настоящем тексте термин "косметический", применяемый в отношении любой композиции продукта, набора, способа или применения, обозначает продукт или способ лечения, который применяется или предназначен для применения в косметических целях, т.е. для укрепления, улучшения или поддержания общего внешнего вида кожи индивидуума, которому его вводят.
Термин "совместимый с кожей" обозначает вещество, которое подходит для применения в косметической композиции для кожи, например, в композиции для применения на коже млекопитающих, в частности, людей. "Совместимый с кожей" носитель или вспомогательное вещество обычно не вызывает раздражения и является хорошо переносимым.
Для получения косметических композиций в соответствии с настоящим изобретением соединения формулы I могут быть приготовлены любым обычным способом вместе с одним или более совместимыми с кожей носителями или вспомогательными веществами в соответствии со способами, хорошо известными в данной области техники. При необходимости соединения или композиции можно стерилизовать с использованием способов, описанных выше в настоящем тексте применительно к фармацевтическим композициям.
Косметические композиции наносят местно на кожу. Подходящие косметические композиции включают гели, кремы, мази, спреи, лосьоны, бальзамы, палочки, порошки, растворы и любые другие обычные косметические составы, применяемые в данной области техники.
Косметические мази, гели, лосьоны, бальзамы и кремы могут, например, быть приготовлены на водной или масляной основе (предпочтительно для мазей и бальзамов) с добавлением подходящих загустителей и/или гелеобразующих агентов. Любые применяемые загустители или гелеобразующие агенты должны быть нетоксичными, нераздражающими и не содержать выщелачиваемых примесей. Они должны быть инертными по отношению к соединениям согласно изобретению, т.е. не должны способствовать их разложению. Лосьоны и кремы могут быть приготовлены на водной или масляной основе и обычно также содержат один или более эмульгирующих, диспергирующих, суспендирующих агентов, загустителей или красителей. Порошки могут быть получены с применением любой подходящей порошковой основы. Спреи и растворы могут быть приготовлены на водной или неводной основе, также содержащей один или более диспергирующих, солюбилизирующих или суспендирующих агентов.
Косметические композиции могут дополнительно содержать общеизвестные косметические вспомогательные вещества, такие как смазывающие агенты, загустители, увлажняющие агенты, эмульгирующие агенты, суспендирующие агенты, консервирующие средства, отдушки, наполнители, связующие и консерванты. Они также могут содержать агенты, способствующие проникновению через поверхность, упомянутые ниже, и т.п. Специалисты в данной области техники имеют возможность выбора подходящих вспомогательных веществ в зависимости от своей цели. Общеизвестные вспомогательные вещества, которые могут применяться в описанных в настоящем тексте косметических продуктах, приведены в различных руководствах (например, Е-М Hoepfher, A. Reng and Р.С. Schmidt (Eds) Fiedler Encyclopedia of Excipients for Pharmaceuticals, Cosmetics and Related Areas (Edition Cantor, Munich, 2002) и H.P. Fielder (Ed) Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik und angrenzende Gebiete (Edition Cantor Aulendorf, 1989)). Кроме того, подходящие совместимые с кожей вспомогательные вещества и носители и их количества, подходящие для применения в косметических композициях в соответствии с настоящим изобретением, описаны в публикации WO 2011/107478, PhotoCure ASA, полное содержание которой включено в настоящую заявку посредством ссылки.
Все из вышеупомянутых косметических вспомогательных веществ хорошо известны в данной области техники и коммерчески доступны у различных производителей.
Необходимое количество соединений согласно настоящему изобретению в косметических композициях будет зависеть от ряда факторов, в том числе конкретного характера состава, того обстоятельства, применяется ли или нет источник света в косметической процедуре и, если применяется, типа источника света и выбранной длины волны, длительности косметической процедуры и общего количества процедур. С учетом этих различных факторов, количество легко может быть определено специалистами в данной области техники. Косметическая композиция в соответствии с настоящим изобретением содержит, как правило, 5% по массе или менее соединений согласно изобретению, предпочтительно от 0,02 до 3% по массе, более предпочтительно от 0,05 до 1,5% по массе, например, от 0,5 до 1,25% по массе, и наиболее предпочтительно от 0,1 до 1,0% по массе, при этом наиболее предпочтительным является диапазон от 0,25 до 0,75% по массе. При определении необходимого количества в пределах этих диапазонов также могут быть рассмотрены следующие критерии в рамках знаний и опыта специалистов в данной области техники:
- косметические композиции, которые способны к более глубокому проникновению в кожу, например, за счет характера композиции, природы выбранного соединения согласно изобретению или благодаря присутствию агентов, способствующих более глубокому проникновению, например, агентов, усиливающих проникновение через кожу, как правило, содержат более низкую концентрацию соединений согласно настоящему изобретению, чем композиции, демонстрирующие склонность оставаться преимущественно на поверхности кожи;
- косметические композиции, предназначенные для длительной обработки кожи (т.е. длительного выдерживания композиции на коже и/или длительного облучения светом) обычно содержат меньшие количества соединений согласно настоящему изобретению, чем композиции, предназначенные для кратковременной обработки;
- косметические композиции, предназначенные для более чем одного курса обработки кожи, например, для нескольких или многих процедур, таких как несколько процедур в течение периода времени или повторные процедуры, обычно содержат меньшие количества соединений согласно настоящему изобретению, чем композиции, предназначенные для однократного применения или предназначенные для применения ограниченное количество раз, зачастую с перерывом между каждой процедурой;
- косметические композиции, предназначенные для лечения кожи с лишь незначительными признаками (фото)старения могут содержать меньшие количества соединений согласно настоящему изобретению, чем композиции, предназначенные для лечения кожи, страдающей от тяжелой формы (фото)старения.
Косметические композиции в соответствии с настоящим изобретением могут применяться в способе косметической обработки. Такая косметическая обработка может быть проведена для укрепления и/или улучшения внешнего вида кожи субъекта-млекопитающего, предпочтительно человека. В частности, может быть обеспечено уменьшение признаков (фото)старения, например, уменьшение появления "гусиных лапок", темных кругов, мелких морщинок, морщин, уменьшение размера пор и улучшение плотности и эластичности кожи.
Подходящие источники света для таких косметических обработок, а также подходящие значения плотности потока, времени облучения, плотности энергии и длин волн подробно описаны в публикации WO 2011/107478, PhotoCure ASA, полное содержание которой включено в настоящую заявку посредством ссылки.
Настоящее изобретение проиллюстрировано следующими неограничивающими примерами со ссылками на прилагаемые чертежи, на которых:
на фигуре 1 показана флуоресценция кожи карликовых свиней после нанесения состава гидробромида карбоксиметил-5-амино-4-оксопентаноата в форме крема.
на фигуре 2 показана задержка роста бактерий S. aureus, инкубированных с гидробромидом карбоксиметил-5-амино-4-оксопентаноата и гидробромидом 2-карбоксиэтил-5-амино-4-оксопентаноата, за счет фотодинамических эффектов.
В примерах использованы следующие сокращения:
Воc - трет-бутоксикарбонил
Cbz - карбоксибензил
Разл. - разлагается
Пример 1 - Получение гидрогалоген идов карбоксиметил-5-амино-4-оксопентаноата (гидрохлорида и гидробромида)
1а - Получение трет-бутоксикарбонилметил-5-(Вос-амино)-4-оксопентаноата
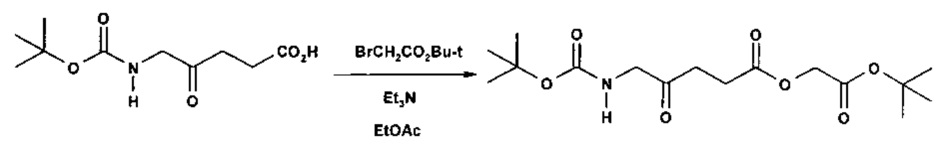
Триэтиламин (2,22 г; 22,0 ммоль) добавляли по каплям к перемешиваемому раствору 5-(Вос-амино)-4-оксопентановой кислоты (4,62 г; 20,0 ммоль) и трет-бутилбромацетата (4,30 г; 22,0 ммоль) в этилацетате (60 мл) в атмосфере аргона при комнатной температуре. Смесь (суспензию) кипятили с обратным холодильником в течение 17 ч, затем охлаждали до комнатной температуры, выливали в 0,5 М НСl (100 мл) и тщательно встряхивали. Водную часть экстрагировали этилацетатом (2×20 мл). Объединенные органические растворы промывали насыщенным раствором NaHCO3 (1×20 мл) и насыщенным раствором NaCl (1×20 мл), затем сушили (MgSO4), фильтровали и выпаривали. Выпаривание приводило к получению масла янтарного цвета, которое очищали посредством флэш-хроматографии на колонке 40×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (600 мл), собирая 5 фракций ×100 мл. Выпаривание фракций 1 и 2 приводило к получению 6,15 г (89%) продукта (желтое масло).
1Н ЯМР: (200 МГц; CDCl3): δ 1,44 (9Н, s), 1,47 (9Н, s), 2,77 (4Н, s), 4,06 (2Н, d, J=4,8 Гц), 4,49 (2Н, s), 5,25 (1H, уш s).
13С ЯМР: (50 МГц; CDCl3): δ 27,5, 28,0, 28,3, 34,2, 50,3, 61,3, 79,8, 82,5, 155,5, 166,5, 171,7, 203,9.
1b - Получение гидрохлорида карбоксиметил-5-амино-4-оксопентаноата

4 М раствор хлороводорода в диоксане (35 мл; 0,14 моль) добавляли к продукту 1а (3,3 г; 9,5 ммоль) и перемешивали до растворения масла с выделением газа. После выдерживания в течение одного часа растворитель выпаривали и остаток растирали с диэтиловым эфиром (4×20 мл); масло затвердевало до рыжевато-коричневого порошка. Остаток фильтровали и сушили в течение ночи в сушильном пистолете при 50°С и 13 мм рт.ст. Получали 2,05 г (96%) продукта (рыжевато-коричневый порошок).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 2,65 (2Н, t, J=6,2 Гц), 2,82 (2Н, t, J=5,9 Гц), 3,96 (2Н, уш s), 4,57 (2Н, s), 8,40 (3Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 26,7, 34,1, 46,5, 60,7, 168,8, 171,5, 202,3.
1с - Получение гидробромида карбоксиметил-5-амино-4-оксопентаноата
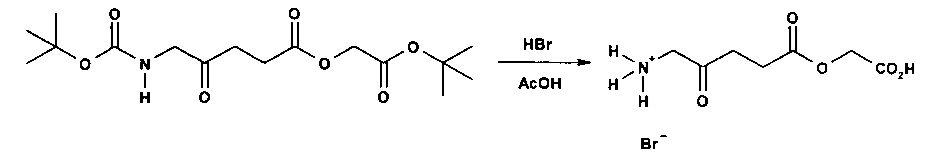
30% раствор бромоводорода в уксусной кислоте (12 мл) добавляли к продукту 1а (3,3 г; 9,5 ммоль), охлажденному с помощью водно-ледяной бани, и перемешивали до растворения масла с выделением газа. После перемешивания в течение 15 мин смесь растирали с гексаном (4×20 мл) и диэтиловым эфиром (4×20 мл); масло затвердевало до рыжевато-коричневого порошка. Остаток фильтровали и сушили в течение ночи в сушильном пистолете при ~50°С и 12 мм рт.ст. Получали 4,44 г (92%) продукта (рыжевато-коричневый порошок, т.пл. 110-114°С (разл.)).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 2,66 (2Н, t, J=6,6 Гц), 2,85 (2Н, t, J=6,6 Гц), 4,02 (2Н, d, J=4,8 Гц), 4,57 (2Н, s), 8,14 (3Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 26,7, 34,1, 46,6, 60,6, 168,8, 171,5, 202,3.
Пример 2 - Получение гидрохлорида 1-(изопропилкарбокси)этил-5-амино-4-оксопентаноата
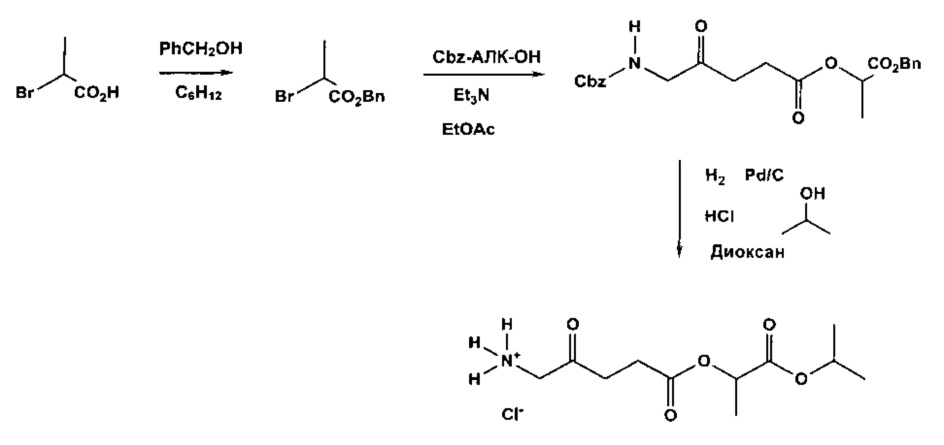
2а - Получение бензил-2-бромпропаноата
Смесь 2-бромпропановой кислоты (15,3 г; 100 ммоль), бензилового спирта (13,0 г; 120 ммоль) и моногидрата п-толуолсульфокислоты (50 мг) в циклогексане (200 мл) кипятили с обратным холодильником в аппарате Дина-Старка в течение 19 ч. Реакционную смесь охлаждали до комнатной температуры, промывали насыщенным раствором NaHCO3 (1×30 мл), водой (1×30 мл) и насыщенным раствором NaCl (1×30 мл). После высушивания (MgSO4), фильтрования и выпаривания остаток подвергали вакуумной перегонке. Получали 25,4 г (87%) продукта (бесцветная жидкость, т.кип. 111-113°С/1,1 ммрт.ст.).
1Н ЯМР: (200 МГц; CDCl3): δ 1,83 (3Н, d, J=7,0 Гц), 4,40 (1Н, q, J=7,0 Гц), 5,20 (2Н, s), 7,36 (5Н, s).
13С ЯМР: (50 МГц; CDCl3): δ 21,6, 39,9, 67,5, 128,0, 128,4, 128,5, 135,0, 169,9.
2b - Получение 1-(бензилоксикарбонил)этил-5-(Сbz-амино)-4-оксопентаноата
Триэтиламин добавляли по каплям к перемешиваемому раствору 5-(Сbz-амино)-4-оксопентановой кислоты (4,00 г; 15,0 ммоль) и продукта 2а (3,65 г; 15,0 ммоль) в этилацетате (50 мл). Перемешиваемую смесь кипятили с обратным холодильником в течение 2 дней в атмосфере аргона. После охлаждения до комнатной температуры добавляли 0,5 М раствор НСl (50 мл), и смесь тщательно встряхивали. Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные этилацетатные растворы промывали насыщенным раствором NaHCO3 (2×15 мл) и насыщенным раствором NaCl (1×15 мл), затем сушили (MgSO4). После фильтрования и выпаривания получали масло янтарного цвета, которое очищали посредством флэш-хроматографии на колонке 60×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 8 фракций ×100 мл. Выпаривание фракций 3-5 приводило к получению 4,92 г (77%) продукта (масло).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 1,41 (3Н, d, J=7,0 Гц), 2,56 (2Н, t, J=6,3 Гц), 2,68 (2Н, t, J=6,3 Гц), 4,03 (2Н, d, J=7,1 Гц), 5,04 (2Н, s), 5,06 (1Н, q, J=6,2 Гц), 5,16 (2Н, s), 7,36 (10Н, m), 7,54 (1Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 16,6, 26,9, 33,5, 49,6, 65,4, 66,1, 68,4, 127,6, 127,8, 128,3, 128,4, 135,6, 137,0, 156,3, 170,1, 171,6, 205,2.
2с - Получение гидрохлорида 1-(изопропилкарбокси)этил-5-амино-4-оксопентаноата
Перемешиваемую смесь продукта 2b (4,92 г; 11,5 ммоль), 10% палладия на активированном угле (100 мг), газообразного водорода и 2,0 М хлороводорода в диэтиловом эфире (10 мл; 20 ммоль) в 2-пропаноле (15 мл) и диоксане (15 мл) гидрировали при приблизительно 6 бар в течение 2 дней при комнатной температуре. Смесь фильтровали через фильтр с носителем Целит 545 (Celite® 545), и остаток промывали 2-пропанолом (2×5 мл). Объединенные фильтраты выпаривали и полученное масло растирали с диэтиловым эфиром (4×10 мл), далее хранили в морозильной камере. Масло не затвердевало. После вакуумной сушки (при 0,005 мм рт.ст.) в течение ночи получали 1,97 г (71%) продукта (рыжевато-коричневое вязкое масло).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 1,17 (6Н, dd, J=5,7 Гц), 1,37 (3Н, d, J=7,0 Гц), 2,60 (2Н, t, J=6,2 Гц), 2,80 (2Н, t, J=6,4 Гц), 3,93 (2Н, уш s), 4,88 (2Н, m), 8,38 (3Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 16,6, 21,3, 26,8, 34,1, 46,5, 68,5, 68,7, 169,7, 171,5, 202,3.
Пример 3 - Получение гидрохлорида карбоксидифторметил-5-амино-4-оксопентаноата
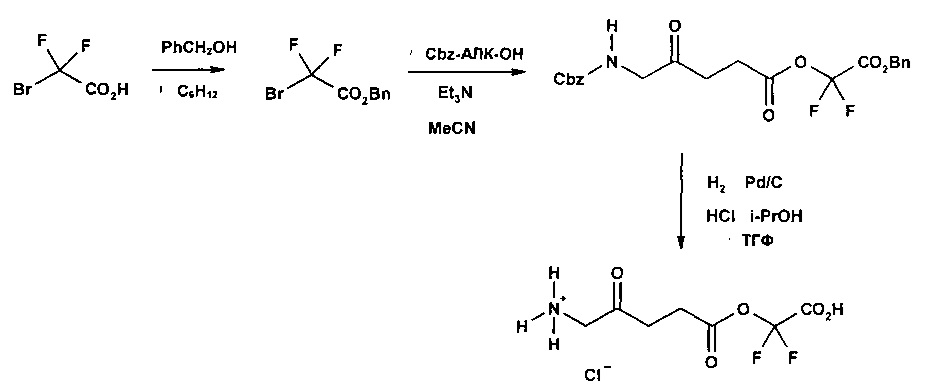
3а - Получение бензилбромдифторацетата
Это соединение получали в соответствии с примером 2а из бромдифторуксусной кислоты (9,82 г; 68,7 ммоль), бензилового спирта (7,0 г; 65 ммоль) и моногидрата п-толуолсульфокислоты (50 мг) в циклогексане (120 мл). Неочищенный продукт, полученный после выпаривания, использовали как есть на стадии 3b без какой-либо дополнительной обработки.
1Н ЯМР: (300 МГц; CDCl3): δ 5,35 (2Н, s), 7,39 (5Н, s).
13С ЯМР: (75 МГц; CDCl3): δ 69,8, 108,8 (t, JC-F=312 Гц), 128,6, 128,9, 129,2, 140,8, 159,5 (t, JC-F=31 Гц).
3b - Получение (бензилоксикарбонил)дифторметил-5-(Сbz-амино)-4-оксопентаноата
Триэтиламин (3,5 мл; 2,5 г; 25 ммоль) добавляли с помощью шприца к перемешиваемой смеси неочищенного продукта 3а (8,6 г; 20 ммоль) и 5-(Сbz-амино)-4-оксопентановой кислоты (6,63 г; 25 ммоль) в сухом ацетонитриле (100 мл). Смесь кипятили с обратным холодильником в течение 18 ч, затем растворитель выпаривали и темно-красный остаток растворяли в диэтиловом эфире (100 мл) и 0,5 М НСl (100 мл). После тщательного перемешивания слой эфира промывали водой (2×15 мл), насыщенным NaHCO3 (2×15 мл) и насыщенным раствором NaCl (1×15 мл), затем сушили (MgSO4). После фильтрования и выпаривания остаток очищали посредством флэш-хроматографии на колонке 60×55 мм с силикагелем 60, элюируемой смесью дихлорметан-диэтиловый эфир (9:1) (1000 мл), собирая 10 фракций ×75 Мл. После выпаривания фракций 2-5 и вакуумной сушки (0,02 мм рт.ст.) получали 6,7 г (74%) продукта (красно-оранжевое твердое вещество, т.пл. 48-50°С).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 2,56 (2Н, t, J=6,4 Гц), 2,73 (2Н, t, J=6,2 Гц), 3,90 (2Н, d, J=5,9 Гц), 5,05 (2H, s), 5,08 (2Н, s), 7,36 (10Н, s), 7,54 (1H, t, J=5,7 Гц).
13C ЯМР: (75 МГц; ДМСО-d6): δ 27,2, 33,7, 49,6, 65,42, 65,44, 101,1, 127,6, 127,7, 127,8, 127,9, 128,27, 128,33, 136,1, 137,0, 156,3, 172,0, 174,8, 205,5.
3с - Получение гидрохлорида карбоксидифторметил-5-амино-4-оксопентаноата
Это соединение получали из продукта 3b (4,0 г; 8,9 ммоль), 10% палладия на угле (0,20 г), газообразного водорода и 2 М НСl в диэтиловом эфире (10 мл; 20 ммоль) в 2-пропаноле (75 мл) и тетрагидрофуране (25 мл), как описано в примере 2с. После фильтрования через Целит раствор повторно фильтровали через складчатый бумажный фильтр. После выпаривания получали масло бледно-янтарного цвета, которое растирали с диэтиловым эфиром (4×10 мл) с получением 1,6 г (80%) продукта (бледного рыжевато-коричневого клейкого твердого вещества). Полученный продукт представлял собой смесь, которая содержала главным образом гидрохлорид карбоксидифторметил-5-амино-4-оксопентаноата, некоторое количество гидрохлорида изопропил-5-амино-4-оксопентаноата, а также некоторое количество гидрохлорида 1-(изопропилкарбокси)дифторметил-5-амино-4-оксопентаноата. Гидрохлорид карбоксидифторметил-5-амино-4-оксопентаноата можно было выделить из смеси стандартными способами, например, посредством препаративной ВЭЖХ.
1Н ЯМР: (300 МГц; ДМСО-d6): δ 2,63 (2Н, J, J=6,3 Гц), 2,83 (2Н, t, J=6,5 Гц), 3,96 (2Н, уш s), 8,37 (3Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 27,3, 34,2, 46,5, 171,4, 171,8, 202,5.
Пример 4 - Получение гидробромида 2-карбоксиэтил-5-амино-4-оксопентаноата
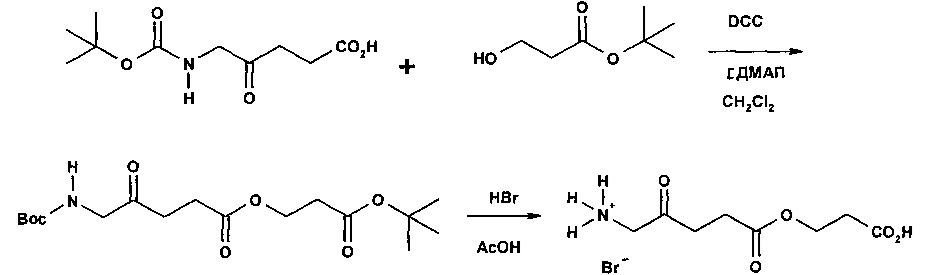
4а - Получение 2-(трет-бутоксикарбонил)этил-5-(Вос-амино)-4-оксопентаноата
Раствор N,N’-дициклогексилкарбодиимида (DCC, 3,34 г; 16,2 ммоль) в сухом дихлорметане (20 мл) добавляли с помощью шприца к перемешиваемой смеси 5-(Вос-амино)-4-оксопентановой кислоты (3,70 г; 16,0 ммоль), трет-бутил-3-гидроксипропаноата (2,31 г; 15,8 ммоль) и 4-диметиламинопиридина (ДМАП, 50 мг) в дихлорметане (40 мл) при комнатной температуре в атмосфере аргона. Умеренный экзотермический эффект реакции смягчали с помощью водно-ледяной бани. После перемешивания в течение 2 дней при комнатной температуре смесь фильтровали при пониженном давлении через фильтр из пористого стекла. Остаток промывали дихлорметаном (3×20 мл). Объединенные фильтраты выпаривали и остаток очищали посредством флэш-хроматографии на колонке 45×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (500 мл), собирая 7 фракций ×50 мл. После выпаривания фракций 2-4 получали 5,45 г (96%) продукта (бледно-желтое масло).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 1,38 (9Н, s), 1,41 (9Н, s), 2,49 (2Н, t, J=6,6 Гц), 2,53 (2Н, t, J=6,2 Гц), 2,66 (2Н, t, J=6,4 Гц), 3,76 (2Н, d, J=5,9 Гц), 4,16 (2Н, t, J=6,2 Гц), 7,06 (1H, t, J=5,8 Гц).
13С ЯМР: (75 МГц; ДМСО-d6): δ 27,1, 27,6, 28,1, 33,5, 34,3, 49,4, 59,9, 78,0, 80,1, 155,7, 169,6, 171,9, 205,8.
4b - Получение гидробромида 2-карбоксиэтил-5-амино-4-оксопентаноата
Это соединение получали из продукта 4а (5,43 г; 15,1 ммоль) и 30% НВr в уксусной кислоте (15 мл) в соответствии с примером 1с. Получали 3,38 г (79%) продукта (смолистое вещество яетарного цвета, которое не затвердевало).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 2,55 (4Н, t, J=6,4 Гц), 2,80 (2Н, t, J=6,3 Гц), 4,00 (2Н, d, J=3,5 Гц), 4,19 (2Н, t, J=6,3 Гц), 8,10 (3Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 26,9, 33,2, 34,1, 46,6, 60,1, 171,8 (2С), 202,6.
Пример 5 - Получение гидрохлорида 3-карбоксипропил-5-амино-4-оксопентаноата
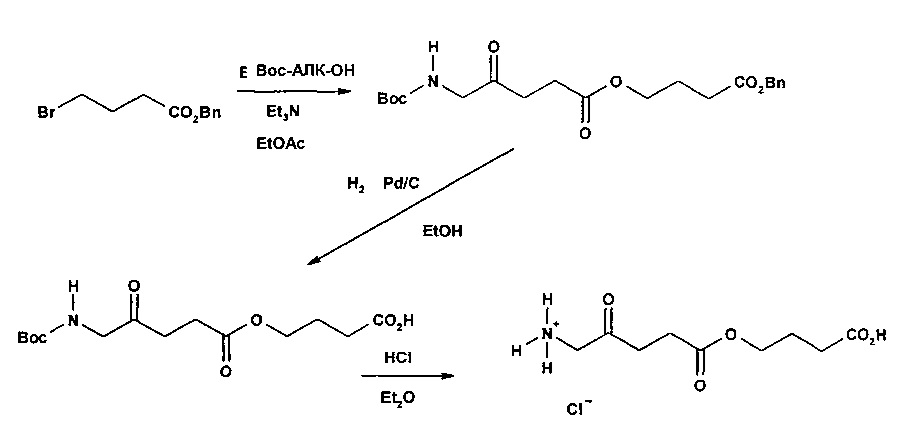
5а - Получение 3-(бензилоксикарбонил)пропил-5-(Вос-амино)-4-оксопентаноата
Это соединение получали из бензил-4-бромбутаноата (7,2 г; 28,0 ммоль), 5-(Вос-амино)-4-оксопентаноата (6,7 г; 29,0 ммоль), триэтиламина (2,95 ммоль) и этилацетата (100 мл), как описано в примере 1а. Неочищенный продукт очищали посредством флэш-хроматографии на колонке 60×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:2) (750 мл), собирая 6 фракций ×100 мл. После выпаривания фракций 3 и 4 получали 5,43 г (46%) продукта (желтое масло).
1Н ЯМР: (300 МГц; ДМСО-d6): δ 1,38 (9Н, s), 1,85 (2Н, m), 2,41-2,50 (4Н, m), 2,67 (2Н, t, J=6,7 Гц), 3,77 (2Н, d, J=5,8 Гц), 4,02 (2Н, t, J=6,4 Гц), 5,10 (2Н, s), 7,07 (1H, t, J=5,7 Гц), 7,37 (5Н, s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 23,6, 27,2, 28,1, 30,0, 33,6, 49,4, 63,0, 65,4, 78,0, 127,87, 127,93, 128,4, 136,1, 155,7, 172,1, 172,2, 206,0.
5b - Получение 3-карбоксипропил-5-(Вос-амино)-4-оксопентаноата
Это соединение получали из продукта 5а (5,40 г; 13,2 ммоль), 10% палладия на активированном угле (250 мг), газообразного водорода и 96% этанола (100 мл), как описано в примере 2с. Получали 4,30 г продукта (желтоватое масло), который использовали на стадии 5с без какой-либо дополнительной обработки.
5с - Получение гидрохлорида 3-карбоксипропил-5-амино-4-оксопентаноата
Это соединение получали из продукта 5b (4,20 г; 13,2 ммоль), 2 М НСl в диэтиловом эфире (7,5 мл; 15 ммоль) и диэтилового эфира (50 мл), как описано в примере 1b. После выпаривания растворителя маслянистый остаток растирали с диэтиловым эфиром (4×10 мл) и выдерживали в течение ночи в морозильной камере до его затвердевания в виде бледно-коричневого твердого вещества. Получали 1,90 г (57%) продукт после высушивания при 12 мм рт.ст. и комнатной температуре.
1Н ЯМР: (300 МГц; ДМСО-d6): δ 1,80 (2Н, т), 2,30 (2Н, t, J=7,4 Гц), 2,55 (2Н, t, J=6,7 Гц), 2,82 (2Н, t, J=6,4 Гц), 3,95 (2Н, d, J=5,1 Гц), 4,03 (2Н, t, J=6,5 Гц), 8,42 (3Н, уш s), 12,1 (1Н, уш s).
13С ЯМР: (75 МГц; ДМСО-d6): δ 23,6, 27,0, 30,1, 34,2, 46,4, 63,3, 171,9, 173,8, 202,6.
Пример 6 - Получение гидробромида карбоксифенилметил-5-амино-4-оксопентаноата
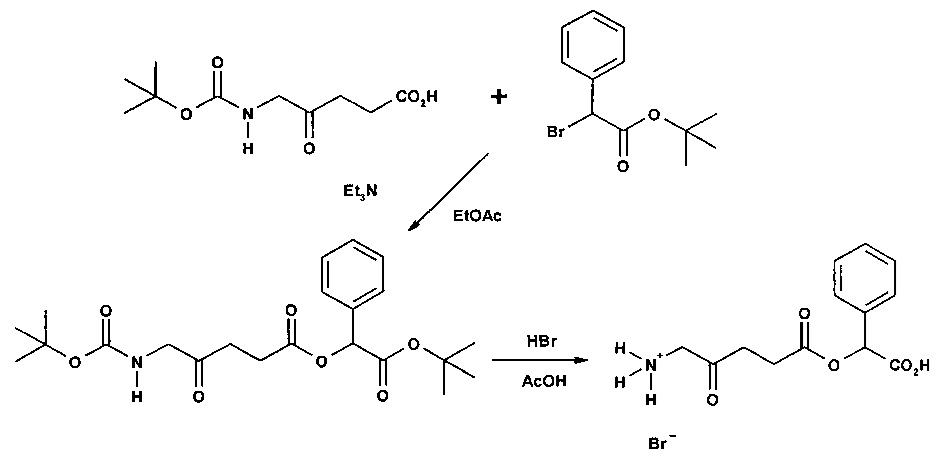
6а - Получение трет-бутил-α-бромфенилацетата
Концентрированную серную кислоту (0,55 мл; 10 ммоль) добавляли к перемешиваемой суспензии безводного MgSO4 (4,8 г; 40 ммоль) в сухом дихлорметане (40 мл) при комнатной температуре. После перемешивания в течение 15 мин добавляли α-бромфенилуксусную кислоту (2,15 г; 10,0 ммоль) с последующим добавлением трет-бутилового спирта (4,8 мл; 50 ммоль). Реакционную колбу закрывали пробкой, и смесь перемешивали в течение 7 дней при комнатной температуре. В реакционную смесь добавляли насыщенный раствор NaHCO3 (75 мл) и воду (75 мл). После разделения водный слой экстрагировали дихлорметаном (2×15 мл). Объединенные органические слои промывали водой и сушили (MgSO4). После фильтрования и выпаривания получали 2,39 г (88%) продукта (бесцветное масло).
1Н ЯМР: (200 МГц; CDCl3): δ 1,46 (9Н, s), 5,25 (1H, s), 7,30-7,38 (3Н, m), 7,48-7,56 (2Н, m).
13С ЯМР: (50 МГц; CDCl3): δ 27,7, 48,3, 83,0, 128,5, 128,6, 128,9, 136,2, 167,0.
6b - Получение трет-бутил-α-[5-(Вос-амино)-4-оксрпентаноилокси]-фенилацетата
Это соединение получали из 5-(Вос-амино)-4-оксопентановой кислоты (1,97 г; 8,5 ммоль), продукта 6а (2,35 г; 8,7 ммоль) и триэтиламина (0,88 г; 8,7 ммоль) в этилацетате (30 мл), как описано в примере 1а. Неочищенный продукт очищали посредством флэш-хроматографии на колонке 65×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:2) (1100 мл), собирая 18 фракций ×50 мл. После выпаривания фракций 3-8 получали 2,79 г (78%) продукта (масло янтарного цвета).
1Н ЯМР: (200 МГц; CDCl3): δ 1,39 (9Н, s), 1,44 (9Н, s), 2,75-2,85 (4Н, m), 4,05 (2Н, d, J=4,8 Гц), 5,22 (1H, уш s), 5,77 (1Н, s), 7,35-7,46 (5Н, m).
13С ЯМР: (50 МГц; CDCl3): δ 27,7, 27,8, 28,3, 34,2, 50,3, 75,2, 79,8, 82,5, 127,4, 128,8, 128,9, 134,0, 155,7, 167,5, 171,6, 203,7.
6с - Получение гидробромида карбоксифенилметил-5-амино-4-оксопентаноата
Это соединение получали из продукта 6b (2,75 г; 6,5 ммоль) и 33% НВr в уксусной кислоте (5 мл), как описано в примере 1с. Неочищенный продукт очищали посредством флэш-хроматографии на колонке 40×55 мм с силикагелем 60, элюируемой 10% метанолом в ацетонитриле (400 мл), собирая 20 фракций ×10 мл. После выпаривания фракций 4-15 и сушки в вакууме в течение 48 ч при 40°С и 0,01 мм рт.ст. получали 1,60 г (71%) продукта (оранжевая пена, т.пл. 77-80°С).
1Н ЯМР: (200 МГц; ДМСО-d6): δ 2,73 (2Н, m), 2,85 (2Н, m), 4,02 (2Н, s), 5,82 (1Н, s), 7,3-7,5 (5Н, m), 8,13 (3Н, уш s).
13С ЯМР: (50 МГц; ДМСО-d6): δ 26,8, 34,1, 46,5, 74,1, 127,4, 127,9, 128,5, 134,0, 169,4, 171,2, 202,1.
Пример 7 - Получение гидробромида (4-карбоксифенил)метил-5-амино-4-оксопентаноата
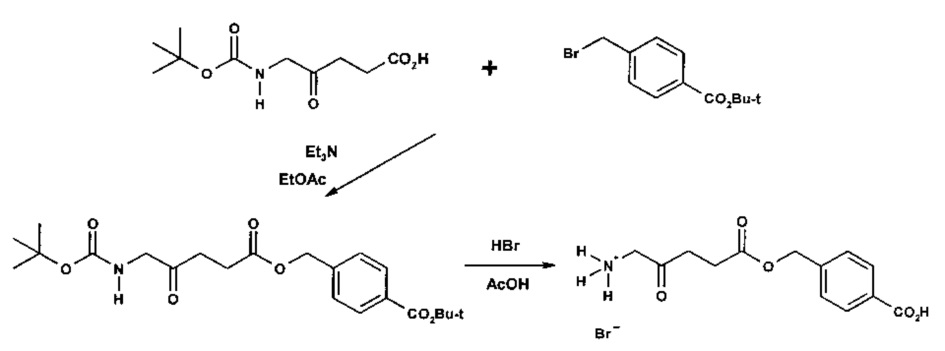
7а - Получение трет-бутил-4-(бромметил)бензоата
Это соединение получали из 4-(бромметил)бензойной кислоты (2,15 г; 10,0 ммоль), трет-бутилового спирта (4,8 мл; 50 ммоль), безводного MgSO4 (4,8 г; 40 ммоль) и концентрированной серной кислоты в дихлорметане (40 мл), как описано в примере 6а. Через 14 дней смесь обрабатывали, как описано в примере 6а. Получали 1,07 г (39%) продукта (белое твердое вещество). Продукт использовали на стадии 7b без дополнительной очистки.
1Н ЯМР: (200 МГц; CDCl3): δ 1,59 (9Н, s), 4,49 (2Н, s), 7,42 (2Н, d, J=8,4 Гц), 7,96 (2Н, d, J=8,4 Гц).
13С ЯМР: (50 МГц; CDCl3): δ 28,1, 32,3, 81,2, 128,7, 129,8, 130,6, 141,9, 165,0.
7b - Получение трет-бутил[4-[5-(Вос-амино)-4-оксопентаноилокси]метил]бензоата
Это соединение получали из 5-(Вос-амино)-4-оксопентановой кислоты (0,81 г; 3,50 ммоль), неочищенного продукта 7а (1,00 г; 3,7 ммоль) и триэтиламина (0,40 г; 4,0 ммоль) в этилацетате (15 мл), как описано в примере 1а. Неочищенный продукт очищали посредством флэш-хроматографии на колонке 65×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:2) (1200 мл), собирая 16 фракций ×50 мл. После выпаривания фракций 6-10 получали 0,59 г (40%) продукта (бесцветное масло).
1Н ЯМР: (200 МГц; CDCl3): δ 1,44 (9Н, s), 1,59 (9Н, s), 2,73 (4Н, s), 4,05 (2Н, d, J=5,0 Гц), 4,1 (1H, уш s), 5,15 (2Н, s), 7,37 (2Н, d, J=8,4 Гц), 7,98 (2Н, d, J=8,2 Гц).
13С ЯМР: (50 МГц; CDCl3): δ 27,7, 28,2, 28,3, 34,3, 50,3, 81,1 (2С), 127,4 (2С), 129,6 (2С), 131,8, 140,0, 165,2, 172,0, 203,9.
7с - Получение гидробромида (4-карбоксифенил)метил-5-амино-4-оксопентаноата
Это соединение получали из продукта 7b (0,50 г; 1,2 ммоль) и 33% НВr в уксусной кислоте (2 мл), как описано в примере 1 с. Добавление НВr приводило к немедленному осаждению белых твердых веществ. Осадок растирали с гексаном и диэтиловым эфиром, как описано в примере 1с. Остаток сушили в вакууме в течение 48 ч при 40°С и 0,01 мм рт.ст. Получали 0,37 г (88%) продукта (белый порошок, т.пл. 198-200°С (разл.)).
1Н ЯМР: (200 МГц; ДМСО-d6): δ 2,68 (2Н, t, J=6,2 Гц), 2,88 (2Н, t, J=6,2 Гц), 4,03 (2Н, s), 5,19 (2Н, s), 7,49 (2Н, d, J=8,0 Гц), 7,96 (2Н, d, J=8,4 Гц), 8,13 (3Н, уш s).
13С ЯМР: (50 МГц; ДМСО-d6): δ 27,0, 34,2, 46,6, 64,8, 127,3, 129,2, 130,1, 140,8, 166,8, 171,6, 202,4.
Примеры 8-15 - Получение неразветвленных гидрохлоридов карбоксиалкил-5-амино-4-оксопентаноата
Общая структура неразветвленных гидрохлоридов карбоксиалкил-5-амино-4-оксопентаноата, описанных в примерах 8-15:
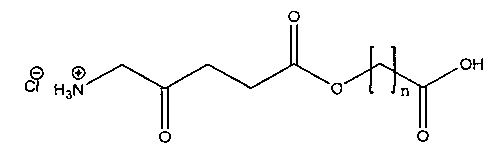
n=4, 5, 7, 8, 9, 10, 11, 15
Общие способы синтеза, применяемые для получения соединений в соответствии с примерами 8-15:
Способ синтеза А - Получение бензиловых сложных эфиров с применением карбодиимидов
Раствор N,N’-дициклогексилкарбодиимида (DCC) или N,N'-диизопропилкарбодиимида в сухом дихлорметане (10 мл) добавляли по каплям с помощью шприца к перемешиваемому раствору карбоновой кислоты, бензилового спирта или 2,2,2-трихлорэтанола и 4-пирролидинилпиридина в сухом дихлорметане (40 мл) в атмосфере аргона. После перемешивания в течение одного-двух дней при комнатной температуре смесь фильтровали под вакуумом, и остаток промывали небольшим количеством дихлорметана. Растворитель выпаривали и остаток сушили в вакууме при 50°С (температура бани) и 0,014 мм рт.ст. с применением устройства Kugelrohr для удаления избытка бензилового спирта.
Способ синтеза В - Получение бензиловых сложных эфиров с применением азеотропной перегонки
Смесь карбоновой кислоты, бензилового спирта и п-толуолсульфокислоты в толуоле кипятили с обратным холодильником в течение ночи с ловушкой Дина-Старка. После охлаждения до комнатной температуры смесь промывали насыщенным раствором NaHCO3 (1×15 мл) и насыщенным NaCl (1×15 Мл). После сушки (MgSO4), фильтрования и выпаривания сушили в вакууме при 50°С (температура бани) и 0,014 мм рт.ст. с применением устройства Kugelrohr.
Способ синтеза С - Сочетание сложных эфиров с N-защищенными производными 5-АЛК: ω-бромалканоаты и 5-(Сbz-амино)-4-оксопентаноат цезия
Бензил-ω-бромалканоат, 5-(Сbz-амино)-4-оксопентаноат цезия и Nal (10 мг) в сухом ДМСО (25 мл) нагревали до 100°С (температура бани) в атмосфере аргона в течение ночи. Смесь охлаждали до комнатной температуры, разбавляли водой (150 мл) и экстрагировали диэтиловым эфиром (5×25 мл). Объединенные эфирные растворы промывали водой (2×10 мл), насыщенным раствором NaCl (1×10 мл) и сушили (MgSO4). После фильтрования и выпаривания остаток очищали посредством флэш-хроматографии на колонке с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1). Фракции, содержащие продукт, выпаривали с выделением защищенного сложного эфира 5-АЛК.
Способ синтеза D - Сочетание сложных эфиров N-защищенных производных 5-АЛК: ω-гидроксиалканоаты и 5-(Сbz-амино)-4-оксопентановая кислота
Раствор N,N’-дициклогексилкарбодиимида (DCC) в сухом дихлорметане (10 мл) добавляли к перемешиваемому раствору ω-гидроксиалканоата, 5-(Сbz-амино)-4-оксопентановой кислоты и 4-пирролидинилпиридина (50 мг) в сухом дихлорметане (25 мл) в атмосфере аргона. После перемешивания при комнатной температуре в течение 1-3 дней смесь фильтровали под вакуумом. Фильтрат выпаривали и остаток очищали посредством флэш-хроматографии на колонке с силикагелм 60, элюируемой смесью этилацетат-гексан (1:1). Фракции, содержащие продукт, выпаривали с выделением защищенного сложного эфира 5-АЛК.
Способ синтеза Е - Сочетание сложных эфиров с N-защищенными производными 5-АЛК: трихлорэтил-ω-гидроксиалканоаты и 5-(Вос-амино)-4-оксопентановая кислота
Раствор N,N’-дициклогексилкарбодиимида (DCC, 2,41 г; 14,1 ммоль) в сухом дихлорметане (15 мл) добавляли по каплям в атмосфере аргона к раствору 2,2,2-трихлорэтил-ω-гидроксиалканоата, 5-(Вос-амино)-4-оксопентановой кислоты и пиридина в сухом дихлорметане, охлажденному до 0°С (температура бани). После перемешивания в течение одного часа при 0°С смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали под вакуумом, и к фильтрату добавляли уксусную кислоту (3 мл). После отстаивания в течение 30 мин смесь повторно фильтровали, и фильтрат разбавляли диэтиловым эфиром (150 мл). Раствор промывали 1 М НСl (3×25 мл), водой (3×25 мл), насыщенным раствором NaHCO3 (2×25 мл) и насыщенным раствором NaCl (1×25 мл). После сушки (MgSO4), фильтрования и выпаривания остаток очищали посредством флэш-хроматографии на колонке с силикагелем 60, элюируемой смесью этилацетат-гексан. Фракции, содержащие продукт, выпаривали с получением продукта.
Пример 8 - Получение гидрохлорида 4-карбоксибутил-5-амино-4-оксопентаноата
8а - Получение бензил-5-бромпентаноата
Это соединение получали из 5-бромпентановой кислоты (1,81 г; 10,0 ммоль), бензилового спирта (1,62 г; 15,0 ммоль) и п-толуолсульфокислоты (50 мг) в толуоле (100 мл) с применением способа синтеза В. Получали 2,60 г (96%) продукта (прозрачное масло). Продукт использовали на стадии 8с без дополнительной очистки.
1Н ЯМР (200 МГц; CDCl3): δ 1,8-1,9 (4Н, m, J=7,2 Гц), 2,39 (2Н, t, J=7,2 Гц); 3,39 (2Н, t, J=6,4 Гц); 5,12 (2Н, s), 7,35 (5Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 23,5, 31,9, 32,9, 33,2, 66,2, 128,1, 128,2, 128,4, 135,8, 172,7 м.д.
8b - Получение 5-(Сbz-амино)-4-оксопентаноата цезия
5-(Сbz-амино)-4-оксопентановую кислоту (2,66 г; 10,0 ммоль) добавляли к перемешиваемому раствору карбоната цезия (1,64 г; 5,0 ммоль) в деионизированной воде (40 мл). После прекращения выделения СО2 смесь замораживали в жидком азоте и лиофилизировали в течение ночи. Получали 4,2 г (~100%) продукта (бледно-коричневого твердого вещества). Продукт использовали на стадии 8с без дополнительной очистки.
8с - Получение 4-(бензилоксикарбонил)бутил-5-(Сbz-амино)-4-оксопентаноата
Это соединение получали из продуктов 8а (0,73 г; 2,7 ммоль) и 8b (1,0 г; 2,5 ммоль) в соответствии со способом синтеза С. Неочищенный продукт очищали на колонке 75×45 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 13 фракций ×50 мл. Собирали фракции, содержащие продукт (5-8), и после выпаривания получали 0,82 г (72%) продукта (желтоватое твердое вещество, т.пл. 53-56°С).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,58 (4Н, уш s), 2,3-2,5 (4Н, перекрывающиеся t), 2,69 (2Н, t, J=6 Гц), 3,89 (2Н, d, J=5,8 Гц), 3,99 (2Н, уш s); 5,04 (2Н, s), 5,09 (2Н, s), 7,36 (10Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 20,9, 23,6, 27,2, 32,8, 33,6, 49,6, 63,4, 65,2, 65,3, 127,5, 127,6, 127,7, 127,8, 128,1, 128,2, 136,0, 136,8, 156,2, 171,9, 172,3, 205,3 м.д.
8d - Получение гидрохлорида 4-карбоксибутил-5-амино-4-оксопентаноата
Перемешиваемую смесь продукта, полученного на стадии 8с (0,70 г; 1,54 ммоль), 12 М НСl (0,13 мл; 1,54 ммоль), 10% Pd/C (тип Degussa E101 NE/W), (100 мг) и 2-пропанола (25 мл) гидрировали при комнатной температуре и давлении 4 бар в течение ночи. Смесь фильтровали через фильтр с носителем Целит 545; фильтрат выпаривали и остаток растирали с сухим диэтиловым эфиром (3×5 мл). После высушивания остатка при комнатной температуре и 0,01 мм рт.ст. 0,38 г получали (97%) продукта (белое твердое вещество).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,4-1,7 (4Н, m), 2,25 (2Н, t, J=6,4 Гц), 2,55 (2Н, t, J=6,4 Гц), 2,82 (2Н, t, J=6,4 Гц), 3,9-4,1 (4Н, m), 8,43 (3Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 20,9, 23,5, 27,0, 30,0, 34,2, 46,4, 63,6, 171,8, 173,6, 202,3 м.д.
Пример 9 - Получение гидрохлорида 5-карбоксипентил-5-амино-4-оксопентаноата
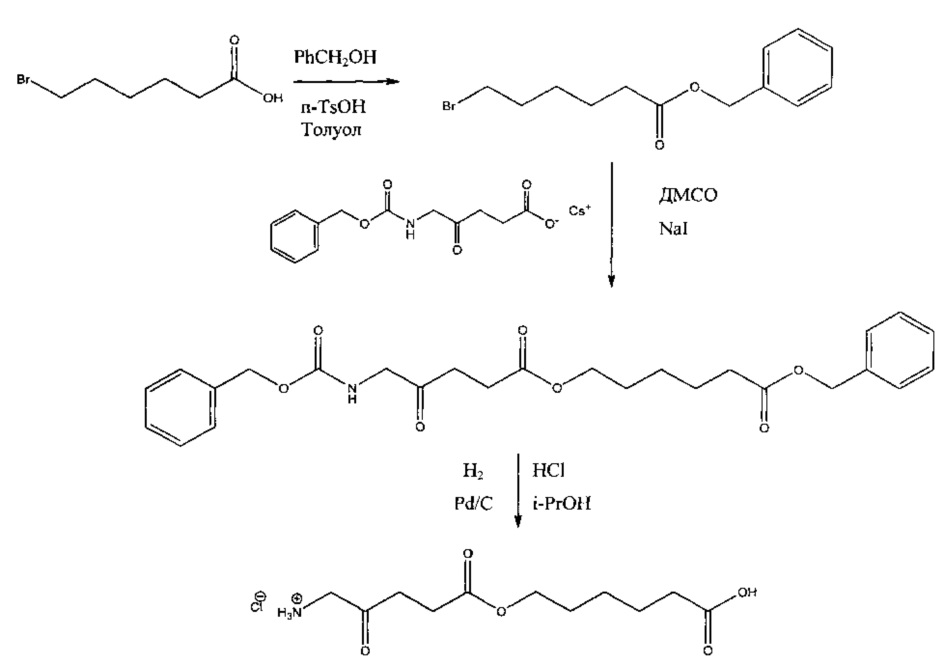
9а - Получение бензил-6-бромгексаноата
Это соединение получали из 6-бромгексановой кислоты (1,95 г; 10,0 ммоль), бензилового спирта (1,62 г; 15,0 ммоль) и п-толуолсульфокислоты (50 мг) в толуоле (100 мл) с применением способа синтеза В. Получали 2,80 г (98%) продукта (прозрачное масло). Продукт использовали на стадии 9b без дополнительной очистки.
1Н ЯМР (200 МГц; CDCl3): δ 1,4-1,5 (2Н, m, J=6,8 Гц), 1,6-1,7 (2Н, m, J=7,2 Гц), 1,8-1,9 (2Н, m, J=7,4 Гц), 2,37 (2Н, t, J=7,4 Гц); 3,38 (2Н, t, J=6,6 Гц); 5,12 (2Н, s), 7,35 (5Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,0, 27,7, 32,3, 33,4, 34,0, 66,1, 128,1, 128,4, 135,9, 173,0 м.д.
9b - Получение 5-(бензилоксикарбонил)пентил-5-(Сbz-амино)-4-оксопентаноата
Это соединение получали из продуктов 9а (0,77 г; 2,7 ммоль) и 8b (1,0 г; 2,5 ммоль) в соответствии со способом синтеза С. Время реакции составляло 2 дня. Неочищенный продукт очищали на колонке 75×45 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 14 фракций ×50 мл. Собирали фракции, содержащие продукт (5-7), и после выпаривания получали 0,52 г (44%) продукта (масло янтарного цвета, которое затвердевало до воскообразного твердого вещества при стоянии в морозильной камере).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,25-1,38 (2Н, m), 1,45-1,6 (4Н, m), 2,36 (2Н, t, J=7,2 Гц), 2,47 (2Н, t, J=6,4 Гц), 2,69 (2Н, t, J=6,2 Гц), 3,85-4,1 (4Н, перекрывающиеся d и t, J=6 и 6,4 Гц), 5,04 (2Н, s), 5,09 (2Н, s), 7,36 (10Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,0, 24,7, 27,2, 27,6, 33,2, 33,6, 49,6, 63,6, 65,2, 65,3, 127,5, 127,6, 127,7, 127,8, 128,1, 128,2, 136,1, 136,8, 156,2, 171,9, 172,4, 205,3 м.д.
9с - Получение гидрохлорида 5-карбоксипентил-5-амино-4-оксопентаноата
Это соединение получали из продукта 9b (0,45 г; 0,96 ммоль), 12 М НСl (0,08 мл; 0,96 ммоль), 10% Pd/C (100 мг), газообразного водорода и 2-пропанола (25 мл) с применением способа синтеза согласно примеру 8d. Получали 0,20 г (77%) продукта (белое твердое вещество).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,2-1,4 (2Н, m), 1,4-1,7 (4Н, m), 2,22 (2Н, t, J=7,2 Гц), 2,55 (2Н, t, J=6,4 Гц), 2,82 (2Н, t, J=6,2 Гц), 3,9-4,1 (4Н, m), 8,41 (3Н, s) м.д.
13С ЯМР (50 МГц; ДМСO-d6): δ 24,0, 24,8, 27,0, 27,7, 33,4, 34,2, 46,4, 63,8, 171,8, 174,1, 202,3 м.д.
Пример 10 - Получение гидрохлорида 7-карбоксигептил-5-амино-4-оксопентаноата
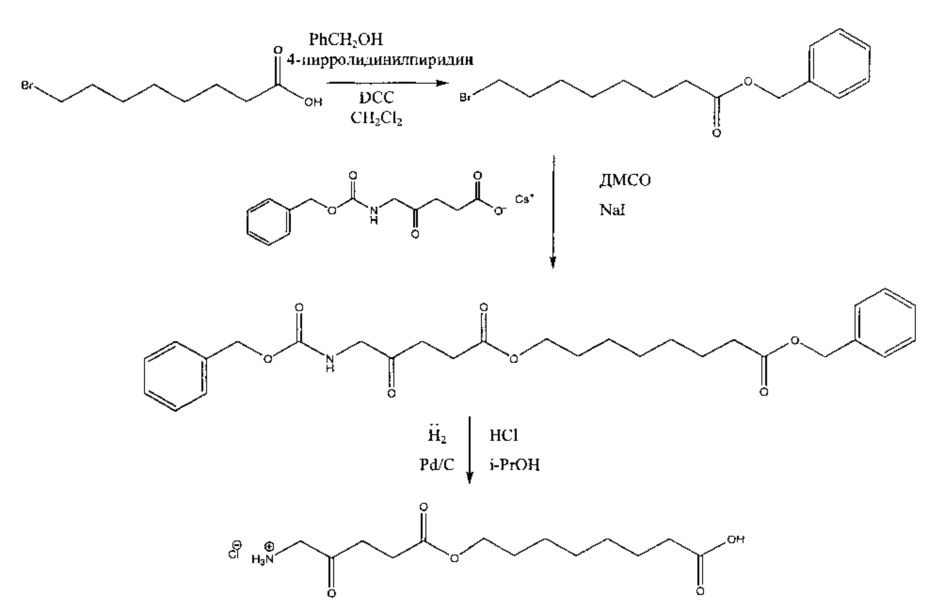
10а - Получение бензил-8-бромоктаноата
Это соединение получали из 8-бромоктановой кислоты (2,23 г; 10,0 ммоль), бензилового спирта (1,19 г; 11,0 ммоль), N,N’-дициклогексилкарбодиимида (DCC, 2,27 г; 11,0 ммоль) и 4-пирролидинилпиридина (50 мг) в сухом дихлорметане (50 мл) с применением способа синтеза А. Получали 3,14 г (100%) продукта (прозрачное масло, которое в конечном итоге затвердевало при стоянии).
1Н ЯМР (200 МГц; CDCl3): δ 1,33 (6Н, уш s), 1,6-1,7 (2Н, m), 1,75-1,9 (2Н, m), 2,35 (2Н, t, J=7,8 Гц); 3,38 (2Н, t, J=6,8 Гц); 5,11 (s, 2Н), 7,35 (s, 5Н) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,8, 27,9, 28,3, 28,9, 32,6, 33,8, 34,2, 66,0, 128,1, 128,4, 136,0, 173,3 м.д.
10b - Получение 7-(бензилоксикарбонил)гептил-5-(Сbz-амино)-4-оксопентаноата
Это соединение получали из продуктов 10а (0,79 г; 2,5 ммоль) и 8b (1,0 г; 2,5 ммоль) в соответствии со способом синтеза С. Время реакции составляло 2 дня. Неочищенный продукт очищали на колонке 80×45 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 13 фракций ×50 мл. Собирали фракции, содержащие продукт (3-6), и после выпаривания получали 0,83 г (67%) продукта (масло янтарного цвета, которое затвердевало до воскообразного твердого вещества при стоянии в морозильной камере, т.пл. 52-54°С).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,26 (6Н, уш s), 1,53 (4Н, уш s), 2,34 (2Н, t, J=7,2 Гц), 2,48 (2Н, t, J=5,8 Гц), 2,69 (2Н, t, J=6,4 Гц), 3,38 (1H, t, J=6,6 Гц), 3,89 (2Н, d, J=6 Гц), 3,98 (2Н, t, J=6,6 Гц), 5,04 (2Н, s), 5,08 (2Н, s), 7,35 (10Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,3, 25,1, 26,3, 27,2, 27,9, 33,4, 33,7, 49,6, 63,8, 65,1, 65,3, 127,4, 127,7, 128,2, 136,1, 136,8, 156,1, 171,8, 172,5, 205,2 м.д.
10с - Получение гидрохлорида 7-карбоксигептил-5-амино-4-оксопентаноата
Это соединение получали из продукта 10b (0,70 г; 1,4 ммоль), 12 М НСl (0,12 мл; 1,4 ммоль), 10% Pd/C (100 мг), газообразного водорода и 2-пропанола (25 мл) с применением способа синтеза согласно примеру 8d. Получали 0,30 г (70%) продукта (белое твердое вещество).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,28 (6Н, s), 1,4-1,7 (4Н, m), 2,20 (2Н, t, J=7 Гц), 2,55 (2Н, t, J=6,2 Гц), 2,81 (2Н, t, J=6,2 Гц), 3,9-4,1 (4Н, m), 8,42 (3Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,3, 25,1, 27,0, 27,9, 28,2, 28,3, 33,6, 34,2, 46,4, 63,9, 171,8, 174,2, 202,3 м.д.
Пример 11 - Получение гидрохлорида 8-карбоксиоктил-5-амино-4-оксопентаноата
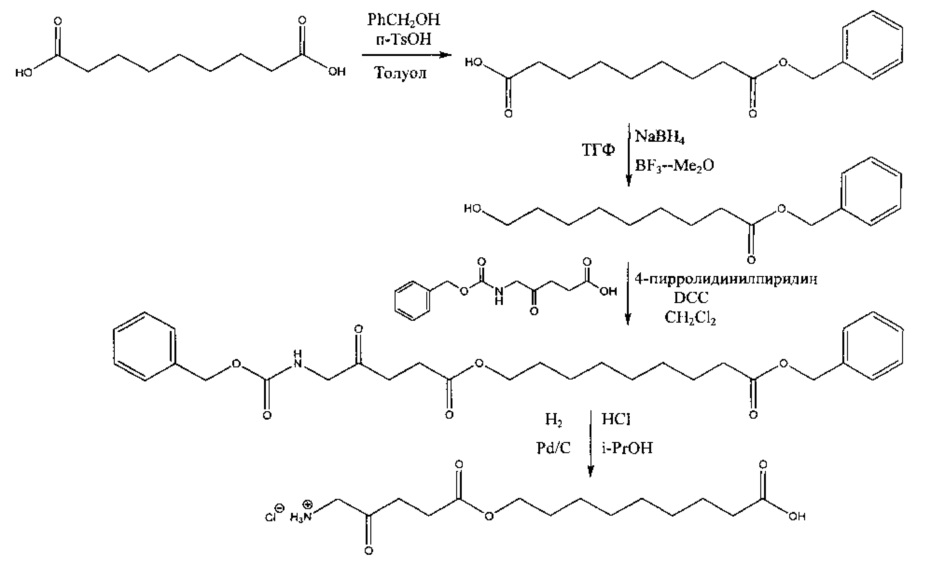
11а - Получение монобензилнонандиоата
Перемешиваемую смесь нонандикарбоновой кислоты (18,8 г; 0,10 моль), бензилового спирта (10,8 г; 0,10 моль) и п-толуолсульфокислоты (0,2 г) в толуоле (200 мл) кипятили с обратным холодильником с ловушкой Дина-Старка в течение ночи. Смесь охлаждали до комнатной температуры и экстрагировали 10% водным раствором N-метил-D-глюкамина (4×25 мл). Последний экстракт представлял собой мутную эмульсию. Первые три экстракта объединяли, подкисляли 1 М НСl (50 мл) и фильтровали с возвращением нонандикарбоновой кислоты (3,8 г). Эмульсию подкисляли 1 М НСl и оставляли расслаиваться на ночь. Органический слой выпаривали и остаток растворяли в гексане (200 мл) и диэтиловом эфире (50 мл). Раствор экстрагировали 10% N-метил-D-глюкамином (2×50 мл) (эмульсию разрушали путем добавления небольшого количества этанола). Органический раствор промывали водой (1×25 мл), и объединенный водный раствор подкисляли 1 М НСl (150 мл). После экстракции смесью дихлорметан-тетрагидрофуран (4:1) (5×10 мл) объединенные экстракты сушили (Nа2SO4 фильтровали и выпаривали. Получали 13,0 г (47%) продукта (масло, которое затвердевало при стоянии).
1Н ЯМР (200 МГц; CDCl3): δ 1,31 (6Н, уш s), 1,5-1,8 (4Н, m), 2,25-2,4 (4Н, m); 5,11 (s, 2Н), 7,34 (5Н, s), 10,4 (1H, уш s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,6, 28,8, 34,0, 34,2, 66,1, 128,1, 128,4, 136,0, 173,5, 179,9 м.д.
11b - Получение бензил-9-гидроксинонаноата
Продукт, полученный на стадии 11а (12,9 г; 46,3 ммоль) добавляли порциями к перемешиваемой смеси боргидрида натрия (1,70 г; 45,0 ммоль) в сухом тетрагидрофуране (25 мл). После прекращения выделения водорода добавляли по каплям раствор комплекса трифторид бора-диметиловый эфир (4,6 мл; 5,7 г; 50 ммоль) в сухом тетрагидрофуране (15 мл). Экзотермический эффект реакции смягчали с помощью бани с холодной водой. После перемешивания в течение 4 ч при комнатной температуре реакционную смесь гидролизовали холодной водой (20 мл). Смесь разделяли, и органическую фазу выпаривали; остаток смешивали с водой (20 мл) и дихлорметаном (70 мл) с получением эмульсии, которая разделялась после стояния в течение ночи. Органический слой промывали водой (4×15 мл) до тех пор, пока промывные воды не были нейтральными по показаниям индикаторной бумаги для определения рН. После высушивания (Na2SO4), фильтрования и выпаривания получали 11,95 г (98%) продукта (масло).
1Н ЯМР (200 МГц; CDCl3): δ 1,30 (8Н, уш s), 1,4-1,75 (4Н, m), 2,34 (2Н, t, J=7,6); 3,62 (2Н, t, J=6,6 Гц), 5,10 (2Н, s), 7,34 (5Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,9, 25,6, 29,0, 29,1, 32,6, 34,3, 63,0, 66,0, 128,0, 128,4, 136,0, 173,6 м.д.
11с - Получение 8-(бензилоксикарбонил)октил-5-(Сbz-амино)-4-оксопентаноата
Это соединение получали из продукта, полученного на стадии 11b (1,1 г; 4,2 ммоль), 5-(Сbz-амино)-4-оксопентановой кислоты (1,0 г; 3,8 ммоль), 4-пирролидинилпиридина (60 мг) и N,N'-дициклогексилкарбодиимида (DCC, 0,87 г; 4,2 ммоль) в дихлорметане (35 мл) в соответствии со способом синтеза D. Время реакции составляло 3 дня. Неочищенный продукт очищали на колонке 85×45 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 14 фракций ×50 мл. Фракции, содержащие продукт (4-7), выпаривали и получали 1,5 г (77%) продукта.
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,24 (8Н, уш s), 1,54 (4Н, перекрывающиеся t, J=6,2 Гц), 2,34 (2Н, t, J=7,2 Гц), 2,48 (2Н, t, J=6,2 Гц), 2,69 (2Н, t, J=6,2 Гц), 3,89 (2Н, d, J=6 Гц), 3,97 (2Н, t, J=6,6 Гц), 5,04 (2Н, s), 5,08 (2Н, s), 7,35 (10Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 23,6, 24,3, 25,0, 27,2, 27,9, 33,3, 33,6, 49,6, 63,8, 65,1, 65,3, 127,5, 127,6, 127,7, 128,1, 128,2, 136,1, 136,8, 156,2, 171,9, 172,5, 205,3 м.д.
11d - Получение гидрохлорида 8-карбоксиоктил-5-амино-4-оксопентаноата
Это соединение получали из продукта 11 с (1,4 г; 2,7 ммоль), 12 М НСl (0,25 мл; 3,0 ммоль), 10% Pd/C (100 мг), газообразного водорода и 2-пропанола (25 мл) с применением способа синтеза согласно примеру 8d. Получали 0,54 г (62%) продукта (белое твердое вещество).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,1-1,7 (12Н, m), 2,20 (2Н, t, J=7,4 Гц), 2,54 (2Н, t, J=6,4 Гц), 2,81 (2Н, t, J=6,4 Гц), 3,9-4,1 (4Н, m), 8,40 (3Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,4, 25,1, 27,0, 28,0, 28,4, 28,5, 33,5, 34,2, 46,4, 63,9, 171,8, 174,2, 202,3 м.д.
Пример 12 - Получение гидрохлорида 9-карбоксинонил-5-амино-4-оксопентаноата
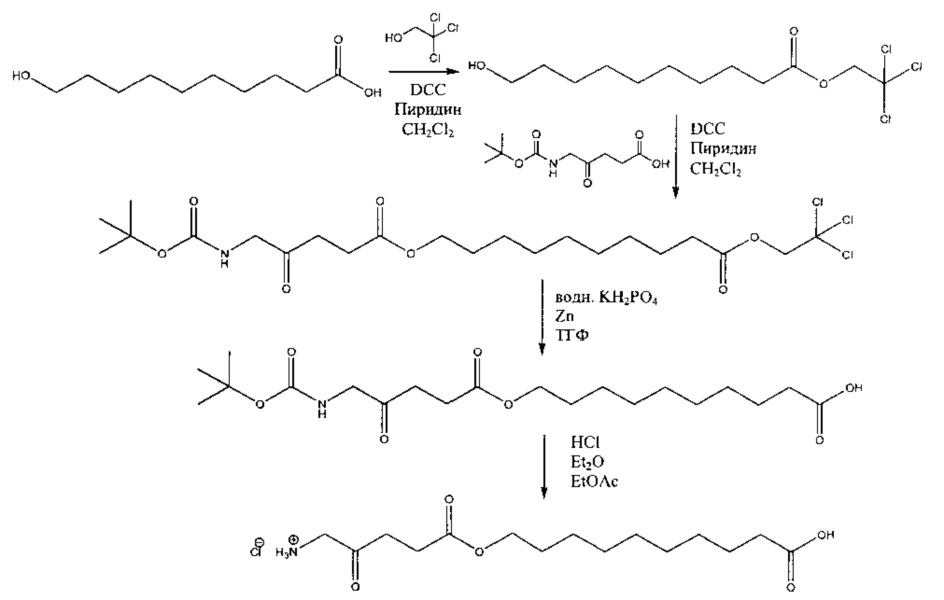
12а - Получение 2,2,2-трихлороэтил-10-гидроксидеканоата
Раствор N,N’-дициклогексилкарбодиимида (DCC, 2,41 г; 14,1 ммоль) в сухом дихлорметане (15 мл) добавляли по каплям в атмосфере аргона к перемешиваемому раствору 10-гидроксидекановой кислоты (1,88 г; 10,0 ммоль), 2,2,2-трихлорэтанола (6,13 г; 41,0 ммоль) и пиридина (4,1 мл) в сухом дихлорметане (35 мл), охлажденному до 0°С (температура бани). После перемешивания в течение одного часа при 0°С смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали под вакуумом, и к фильтрату добавляли уксусную кислоту (3 мл). После отстаивания в течение 30 мин смесь повторно фильтровали, и фильтрат разбавляли диэтиловым эфиром (150 мл). Раствор промывали 1 М НСl (3×25 мл), водой (3×25 мл), насыщенным раствором NаНСО3 (2×25 мл) и насыщенным раствором NaCl (1×25 мл). После высушивания (MgSO4), фильтрования и выпаривания остаток очищали посредством флэш-хроматографии на колонке 170×25 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1), собирая 12 фракций ×25 мл. Фракции 3-5 выпаривали и остаток сушили в вакууме при 50°С и 0,05 мм рт.ст. в устройстве Kugelrohr для удаления избытка 2,2,2-трихлорэтанола. Получали 2,1 г (66%) продукта (бесцветное масло).
1Н ЯМР (200 МГц; CDCl3): δ 1,32 (10Н, уш s), 1,4-1,8 (5Н, m), 2,47 (2Н, t, J=7,2 Гц); 3,63 (2Н, t, J=6,4 Гц), 4,74 (2Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,7, 25,6, 29,0, 29,1, 29,28, 29,32, 32,6, 33,9, 63,0, 73.8, 95,0, 172,1 м.д.
12b - Получение 2,2,2-трихлорэтил-10-[5-(Вос-амино)-4-оксопентаноилокси] деканоата
Это соединение получали из 5-(Вос-амино)-4-оксопентановой кислоты (1,00 г; 4,3 ммоль), продукта, полученного на стадии 12а (1,37 г; 4,3 ммоль), N,N'-дициклогексилкарбодиимида (DCC, 1,03 г; 5,0 ммоль) и пиридина (2,0 мл) в дихлорметане (40 мл) с применением способа синтеза Е. Неочищенный продукт очищали наколонке 100×50 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:2), собирая 16 фракций ×50 мл. Путем выпаривания фракций 4-9 получали 1,6 г (71%) продукта (желтоватое масло).
1Н ЯМР (200 МГц; CDCl3): δ 1,31 (10Н, уш s), 1,44 (9Н, s), 1,5-1,8 (4Н, m), 2,46 (2Н, t, J=7,2 Гц), 2,6-2,8 (4Н, m), 4,0-4,1 (4Н, m), 4,74 (2Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,7, 25,7, 25,8, 27,8, 28,3, 28,5, 28,9, 29,2, 29,3, 32,7, 33,9, 50,3, 64,9, 73,8, 95,0, 171,9, 172,3, 204 м.д.
12с - Получение 9-карбоксинонил-5-(Вос-амино)-4-оксопентаноата
Одномолярный раствор дигидрофосфата калия (КН2РO4) (4,0 мл; 4,0 ммоль) добавляли к перемешиваемому раствору продукта, полученного на стадии 12b (1,50 г; 2,8 ммоль), в тетрагидрофуране (25 мл) с последующим добавлением цинковой пыли (2,0 г; 30 ммоль). После перемешивания в течение 18 ч при комнатной температуре добавляли новую порцию 1 М раствора КН2РО4 (5 мл) и цинка (2,0 г; 30 ммоль). Через 5 ч добавляли дополнительное количество 1 М раствора КН2РО4 (25 мл), и смесь перемешивали в течение ночи при комнатной температуре. Смесь фильтровали под вакуумом, и остаток промывали этилацетатом. Этилацетат в фильтрате выпаривали и водный раствор экстрагировали дихлорметаном (4×5 мл). Объединенные экстракты сушили (Na2SO4), фильтровали и выпаривали. Получали 1,04 г (93%) продукта (бледно-желтое воскообразное твердое вещество).
1Н ЯМР (200 МГц; CDCl3): δ 1,30 (10Н, уш s), 1,44 (9Н, s), 1,5-1,7 (4Н, m), 2,34 (2Н, t, J=7,4 Гц), 2,6-2,8 (4Н, m), 4,0-4,1 (4Н, m) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,7, 25,7, 25,8, 27,8, 28,3, 28,5, 29,0, 29,1, 29,2, 29,3, 32,6, 34,0, 50,3, 65,0; 172,4, 179,1 м.д.
12d - Получение гидрохлорида 9-карбоксинонил-5-амино-4-оксопентаноата
1,0 М раствор НСl в диэтиловом эфире (7,0 мл; 7,0 ммоль) добавляли к перемешиваемому раствору продукта, полученного на стадии 12с (1,0 г; 2,5 ммоль), в этилацетате (10 мл) при комнатной температуре в атмосфере аргона. Через 7 ч избыток растворителя выпаривали и остаток растирали с диэтиловым эфиром (4×5 мл). Остаток сушили в течение ночи при 40°С и 15 мм рт.ст. с получением 0,11 г белого твердого вещества. Объединенные эфирные экстракты выпаривали и растворяли в 96% этаноле (25 мл) и 1 М НСl в диэтиловом эфире (5 мл). После перемешивания в течение 10 дней при комнатной температуре смесь фильтровали, и остаток растирали с диэтиловым эфиром, как описано выше, чтобы дать 0,22 г второй порции продукта. Анализ методом ЖХ-МС показал, что продукт содержал приблизительно 30% 5-амино-4-оксопентановой кислоты; объединенные порции продукта очищали посредством флэш-хроматографии на колонке 165×25 мм с силикагелем 60, элюируемой последовательно ацетонитрилом (100 мл), 2,5% метанолом в ацетонитриле (500 мл), 5% метанолом в ацетонитриле (500 мл), затем 7% метанолом в ацетонитриле (1000 мл), собирая 47 фракций ×50 мл. Фракции, содержащие продукт, выпаривали и остаток растворяли в воде (10 мл). Раствор лиофилизировали в течение ночи и получали 0,17 г (20%) продукта (белое твердое вещество, т.пл. 98-102°С, плавится с размягчением, не имеет точного значения т.пл.).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,26 (10Н, уш s), 1,4-1,6 (4Н, m), 2,19 (2Н, t, J=7,4 Гц), 2,54 (2Н, t, J=6,4), 2,81 (2Н, t, J=6,6 Гц), 3,9-4,1 (4Н, m), 8,5 (3Н, уш s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,4, 25,2, 27,0, 28,0, 28,4, 28,48, 28,54, 28,6, 33,6, 34,2, 46,4, 63,9, 171,8, 174,1, 202,3 м.д.
Пример 13 - Получение гидрохлорида 10-карбоксидецил-5-амино-4-оксопентаноата
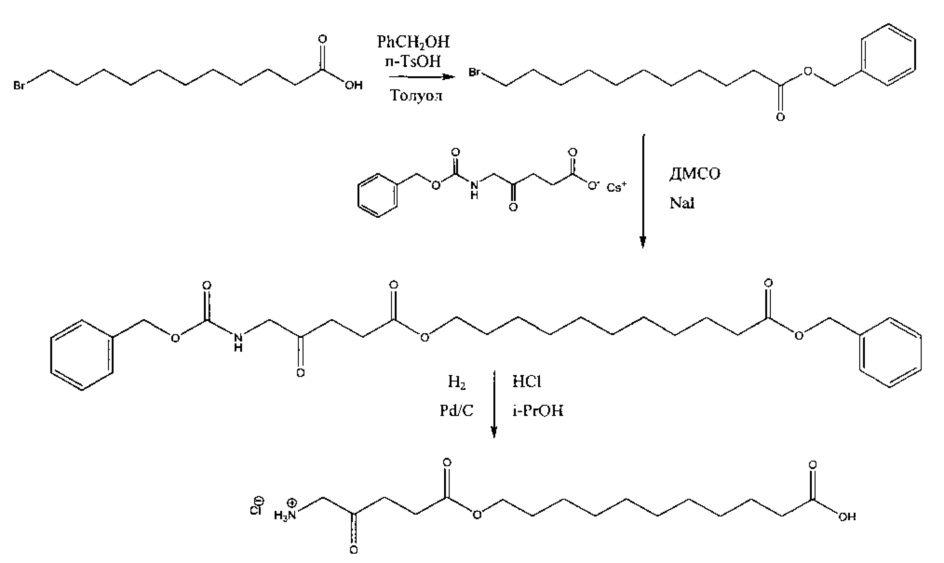
13а-Получение бензил-11-бромундеканоата
Это соединение получали из 11-бромундекановой кислоты (2,65 г; 10,0 ммоль), бензилового спирта (1,62 г; 15,0 ммоль) и п-толуолсульфокислоты (50 мг) в толуоле (100 мл) с применением способа синтеза В. Получали 3,62 г продукта, который использовали на стадии 13b без дополнительной очистки.
1Н ЯМР (200 МГц; CDCl3): δ 1,27 (12Н, уш s), 1,6-1,7 (2Н, m), 1,8-1,9 (2Н, m), 2,35 (2Н, t, J=7,4 Гц); 3,39 (2Н, t, J=6,8 Гц); 5,11 (2Н, s), 7,34 (5Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,9, 28,1, 28,7, 28,8, 29,0, 29,1, 29,3, 32,8, 33,9, 34,3, 66,0, 128,0, 128,4, 136,0, 173,4 м.д.
13b - Получение 10-(бензилоксикарбонил)децил-5-(Сbz-амино)-4-оксопентаноата
Это соединение получали из продуктов 13а (0,96 г; 2,7 ммоль) и 8b (1,0 г; 2,5 ммоль) в соответствии со способом синтеза С. Неочищенный продукт очищали на колонке 70×45 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 13 фракций ×50 мл. Фракцию, содержащую продукт (3), выпаривали и получали 0,69 г (47%) продукта (розоватое твердое вещество, т.пл. 70-72°С).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,23 (12Н, уш s), 1,54 (4Н, уш s), 2,34 (2Н, t, J=7,2 Гц), 2,48 (2Н, m), 2,69 (2Н, t, J=6,2 Гц), 3,89 (2Н, d, J=6,6 Гц), 3,98 (2Н, t, J=6,2 Гц), 5,04 (2Н, s), 5,08 (2Н, s), 7,35 (10Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 23,6, 24,3, 25,2, 27,2, 28,0, 28,3, 28,5, 28,7, 33,4, 33,6, 49,6, 63,8, 65,1, 65,3, 127,5, 127,6, 127,7, 128,1, 128,2, 136,1, 136,8, 156,2, 171,9, 172,5, 205,3 м.д.
13с - Получение гидрохлорида 10-карбоксидецил-5-амино-4-оксопентаноата
Это соединение получали из продукта 13b (0,60 г, 1,1 ммоль), 12 M НСl (0,09 мл; 1,1 ммоль), 10% Pd/C (100 мг), газообразного водорода и 2-пропанола (25 мл) с применением способа синтеза согласно примеру 8d. Получали 0,20 г (51%) продукта (белое твердое вещество).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,1-1,7 (16Н, m), 2,19 (2Н, t, J=7,4 Гц), 2,54 (2Н, t, J=6,4 Гц), 2,81 (2Н, t, J=6,4 Гц), 3,9-4,1 (4Н, m), 8,41 (3Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,4, 25,2, 27,0, 28,0, 28,4, 28,55, 28,62, 28,74, 33,6, 34,2, 46,4, 64,0, 170,6, 171,8, 202,3 м.д.
Пример 14 - Получение гидрохлорида 11-карбоксиундецил-5-амино-4-оксопентаноата
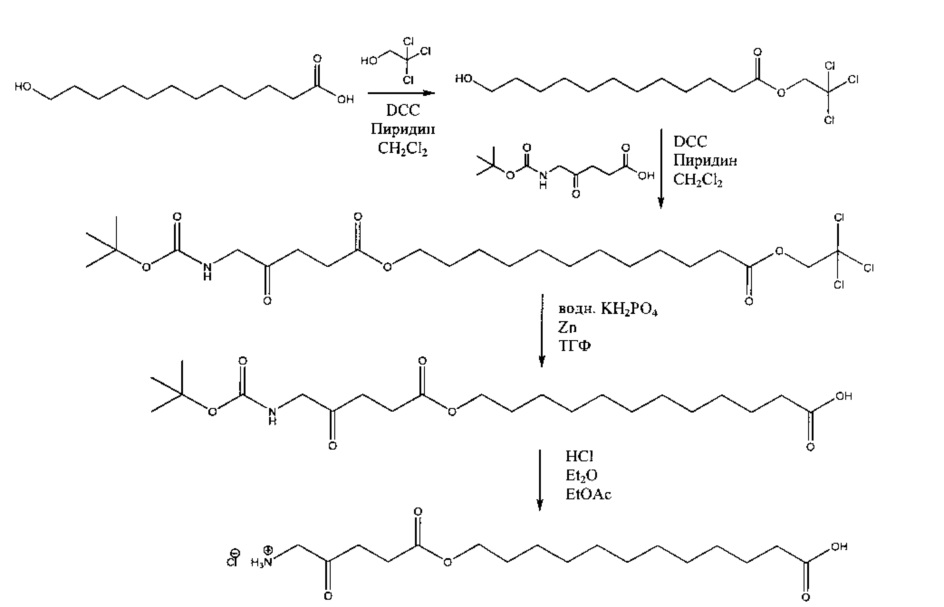
14а - Получение 2,2,2-трихлороэтил-12-гидроксидодеканоата
Это соединение получали из 12-гидроксидодекановой кислоты (2,0 г; 9,2 ммоль), 2,2,2-трихлорэтанола (6,0 г; 40 ммоль), N,N'-дициклогексилкарбодиимида (DCC, 4,1 г; 20 ммоль) и пиридина (5,0 мл) в сухом дихлорметане (50 мл) с применением способа синтеза согласно примеру 12а. Получали 3,63 г (66%) продукта (бесцветное масло).
1Н ЯМР (200 МГц; CDCl3): δ 1,28 (14Н, уш s), 1,5-1,75 (4Н, m), 2,46 (2Н, t, J=7,6 Гц); 3,63 (2Н, t, J=6,4 Гц), 4,74 (2Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,7, 25,7, 28,6, 29,0, 29,1, 29,2, 29,4, 32,7, 33,9, 62,9, 73,8, 95,0, 172,0 м.д.
14b - Получение 2,2,2-трихлорэтил-12-[5-(Вос-амино)-4-оксопентаноилокси] додеканоата
Это соединение получали из 5-(Вос-амино)-4-оксопентановой кислоты (2,0 г; 8,6 ммоль), продукта 14а (3,0 г; 8,6 ммоль), N,N'-дициклогексилкарбодиимида (DCC, 2,1 г; 10,0 ммоль) и пиридина (4,0 мл) в дихлорметане (75 мл) с применением способа синтеза Е. Неочищенный продукт очищали на колонке 90×50 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:3), собирая 13 фракций ×50 мл. После выпаривания фракций 6-11 получали 1,44 г (30%) продукта (желтоватое масло).
1Н ЯМР (200 МГц; CDCl3): δ 1,28 (14Н, уш s), 1,45 (9Н, s), 1,5-1,8 (4Н, m), 2,46 (2Н, t, J=1,4 Гц), 2,6-2,8 (4Н, m), 4,0-4,1 (4Н, m), 4,74 (2Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,7, 25,8, 28,3, 28,5, 29,1, 29,2, 29,3, 29,4, 29,5, 33,9, 34,3, 50,3, 64,9, 73,8, 74,1, 95,0, 172,0, 172,3, 204,1 м.д.
14с - Получение 11-карбоксиундецил-5-(Вос-амино)-4-оксопентаноата
Это соединение получали из продукта 14b (1,38 г; 2,5 ммоль) в тетрагидрофуране (25 мл) в соответствии со способом синтеза согласно примеру 12с и с применением тех же количеств 1 М раствора КН2РО4 и цинковой пыли. После обработки получали 1,0 г (93%) продукта (бледно-желтое масло).
1Н ЯМР (200 МГц; CDCl3): δ 1,28 (14Н, уш s), 1,44 (9Н, s), 1,5-1,7 (4Н, m), 2,33 (2Н, t, J=7,4 Гц), 2,6-2,8 (4Н, m), 4,0-4,1 (4Н, m) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,7, 25,8, 28,3, 28,5, 29,0, 29,2, 29,3, 29,4, 32,6, 34,0, 50,3, 65,0, 172,4, 178,9, 204,3 м.д.
14d - Получение гидрохлорида 11-карбоксиундецил-5-амино-4-оксопентаноата
1,0 М раствор НСl в диэтиловом эфире (5,0 мл; 5,0 ммоль) добавляли к перемешиваемому раствору продукта 14с (0,95 г; 2,2 ммоль) в этилацетате (10 мл) при комнатной температуре в атмосфере аргона. Через 5 дней смесь фильтровали и остаток растирали с диэтиловым эфиром (2×5 мл). Остаток сушили в течение ночи при 40°С и 15 мм рт.ст. с получением 0,11 г белого твердого вещества. Объединенные эфирные экстракты выпаривали. Анализ методом ЖХ-МС показал, что продукт содержал приблизительно 25% 5-амино-4-оксопентановой кислоты; объединенные порции продукта очищали посредством флэш-хроматографии на колонке 150×25 мм с силикагелем 60, элюируемой последовательно 3% метанолом в ацетонитриле (1000 мл), 6% метанолом в ацетонитриле (1000 мл), затем 9% метанолом в ацетонитриле (2000 мл), собирая 56 фракций ×50 мл. Фракции, содержащие продукт, выпаривали и остаток растворяли в воде (10 мл). Раствор лиофилизировали в течение ночи и получали 0,06 г (7,5%) продукта (белое твердое вещество, т.пл. 110-115°С (плавится с размягчением, не имеет точного значения т.пл.)).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,25 (14Н, уш s), 1,4-1,6 (4Н, m), 2,19 (2Н, t, J=7,4 Гц), 2,55 (2Н, t, J=6,2 Гц), 2,80 (2Н, t, J=6,4 Гц), 3,9-4,1 (4Н, m), 9,1 (3Н, уш s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,4, 25,2, 27,0, 28,0, 28,4, 28,5, 28,6, 28,8, 33,6, 34,1, 46,4, 64,0, 171,8, 174,2, 202,4 м.д.
Пример 15 - Получение гидрохлорида 15-карбоксипентадецил-5-амино-4-оксопентаноата 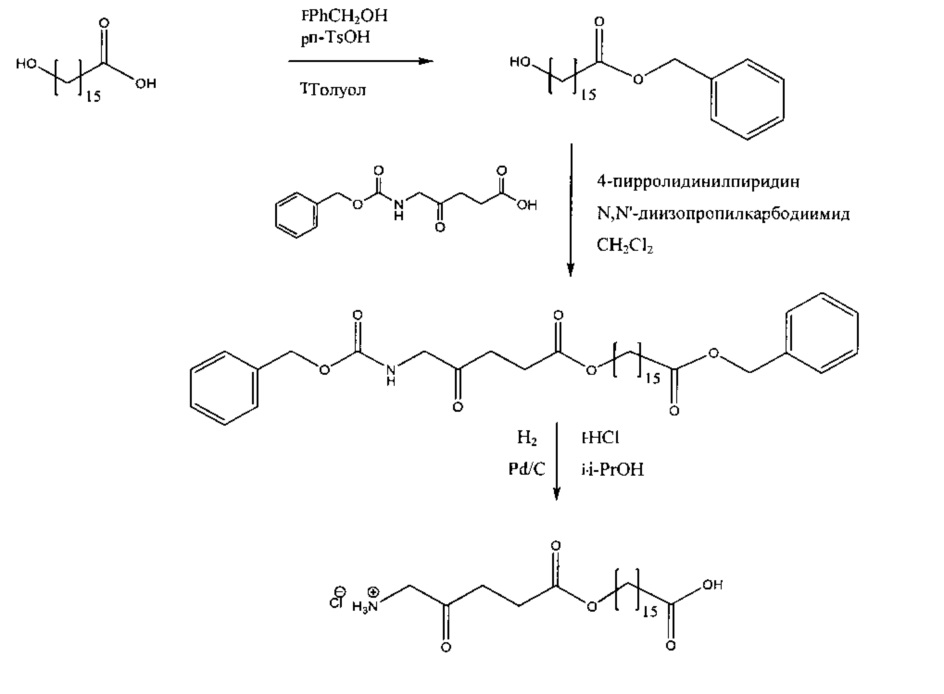
15а - Получение бензил-16-гидроксигексадеканоата
Это соединение получали из 16-гидроксигексадекановой кислоты (2,45 г; 9,0 ммоль), бензилового спирта (4,9 г; 45 ммоль) и п-толуолсульфокислоты (100 мг) в толуоле (100 мл) с применением способа синтеза В. Получали 2,82 г неочищенного продукта, который представлял собой смесь 1:1 ожидаемого продукта и сложного эфира двух молекул 16-гидроксигексадекановой кислоты. К неочищенному продукту добавляли раствор NaH (приблизительно 100 мг) (предварительно промытый три раза сухим гексаном) в бензиловом спирте (10 г), и смесь перемешивали при 100°С (температура бани) в атмосфере аргона в течение 3 ч. После охлаждения до комнатной температуры смесь разбавляли дихлорметаном (75 мл) и промывали 10% водным раствором лимонной кислоты (1×10 мл) и сушили (Na2SO4). После фильтрования и выпаривания остаток сушили в вакууме при 50°С (температура бани) и 0,014 мм рт.ст. с применением устройства Kugelrohr. Получали 2,56 г (76%) продукта (белое твердое вещество). Анализ методом ПМР-спектроскопии показал, что в продукте оставалось приблизительно 10% димера в форме сложного эфира.
1Н ЯМР (200 МГц; CDCl3): δ 1,25 (22Н, уш s), 1,55-1,65 (4Н, m, J=7,6 Гц), 1,9 (1Н, уш s), 2,35 (2Н, t, J=7,4 Гц); 3,63 (2Н, t, J=6,6 Гц); 5,11 (2Н, s), 7,34 (5Н, s) м.д.
13С ЯМР (50 МГц; CDCl3): δ 24,9, 25,7, 28,9, 29,1, 29,2, 29,4, 29,6, 32,8, 34,3, 63,0, 66,0, 128,0, 128,2, 128,4, 136,0, 173,6 м.д.
15b - Получение 15-(бензилоксикарбонил)пентадецил-5-(Сbz-амино)-4-оксопентаноата
Это соединение получали из продукта 15а (1,2 г; 3,2 ммоль), 5-(Сbz-амино)-4-оксопентановой кислоты (0,84 г; 3,2 ммоль), 4-пирролидинилпиридина (50 мг) и N,N'-диизопропилкарбодиимида (0,50 г; 4,0 ммоль) в дихлорметане (35 мл) в соответствии со способом синтеза D. Время реакции составляло 3 дня. Неочищенный продукт очищали на колонке 85×55 мм с силикагелем 60, элюируемой смесью этилацетат-гексан (1:1) (1000 мл), собирая 13 фракций ×50 мл. Фракции, содержащие продукт (3-6), выпаривали и получали 0,57 г (29%) продукта (белое твердое вещество, т.пл. 78-81°С).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,24 (22Н, уш s), 1,54 (4Н, уш s), 2,33 (2Н, t, J=7,4 Гц), 2,48 (2Н, уш s), 2,68 (2Н, уш s), 3,88 (2Н, уш s), 3,98 (2Н, уш s), 5,04 (2Н, s), 5,08 (2Н, s), 7,34 (10Н, s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,4, 25,3, 27,4, 28,1, 28,3, 28,6, 28,9, 33,5, 33,8, 49,8, 63,9, 65,2, 65,5, 127,4, 127,6, 127,7, 128,1, 128,2, 136,9, 171,8, 205,2 м.д.
15с - Получение гидрохлорида 15-карбоксипентадецил-5-амино-4-оксопентаноата
Это соединение получали из продукта 15b (0,45 г; 0,74 ммоль), 12 М НСl (0,062 мл; 0,74 ммоль), 10% Pd/C (100 мг), газообразного водорода и 2-пропанола (25 мл) с применением способа синтеза согласно примеру 8d. Получали 0,06 г (19%) продукта (белое твердое вещество).
1Н ЯМР (200 МГц; ДМСО-d6): δ 1,1-1,7 (28Н, m), 2,25 (2Н, m), 2,52 (2Н, m), 2,82 (2Н, m), 3,9-4,1 (4Н, m), 8,43 (3Н, s), 12,1 (1H, уш s) м.д.
13С ЯМР (50 МГц; ДМСО-d6): δ 24,4, 25,3, 27,0, 27,2, 27,6, 28,0, 28,4, 28,6, 28,9, 33,6, 34,2, 46,4, 63,9, 171,8, 174,1, 202,3 м.д.
Пример 16 - Исследование флуоресценции кожи in vivo на здоровых бестимусных мышах
Исследование проводили с целью оценки эффективности соединений согласно настоящему изобретению в качестве предшественников фотосенсибилизаторов для применения на коже. Соответственно, соединения согласно изобретению должны были обладать способностью проникать в кожу, поглощаться клетками кожи и превращаться в фотосенсибилизаторы. Результат этого процесса можно визуализировать путем подвергания кожи действию света, который возбуждает молекулы фотосенсибилизатора, и определения количества энергии, высвобождаемой в форме флуоресценции, во время которой возбужденные молекулы фотосенсибилизатора релаксируют в более низкое энергетическое состояние.
Следующие соединения согласно настоящему изобретению исследовали на коже здоровых бестимусных мышей:
Соединение 2: гидрохлорид 1-(изопропилкарбокси)этил-5-амино-4-оксопентаноата (полученный в соответствии с примером 2)
Соединение 4: гидробромид 2-карбоксиэтил-5-амино-4-оксопентаноата (полученный в соответствии с примером 4)
Соединение 7: гидробромид (4-карбоксифенил)метил-5-амино-4-оксопентаноата (полученный в соответствии с примером 7)
Вышеперечисленные соединения 2, 4 и 7 были приготовлены в виде кремов на основе препарата Unguentum Merck. Соединения исследовали в эквимолярных количествах:
16% (160 мг/г) соединения 2,
16% (160 мг/г) соединения 4 и
19% (190 мг/г) соединения 7.
В эксперимент были включены 3 группы мышей (5 мышей/группа, каждая из мышей одной группы получала один и тот же состав в форме крема). Примерно 100 мкл состава в форме крема наносили на спину каждой мыши. Мышей держали в темноте в течение 6 часов до измерения флуоресценции кожи во избежание фотообесцвечивания. Для оценки флуоресценции кожи применяли камеру для детекции флуоресценции (Medeikonos PDD/PDT, Medeikonos АВ, Gothenburg, Sweden) и получали снимки спины каждой мыши. Возбуждение осуществляли при длинах волн 365 и 405 нм и продолжительности облучения 2с. Каждое изображение калибровали относительно флуоресцентного стандарта (Uranyl Standard, J&M, Analytische Mess- und Regeltechnik GmbH, Hamburg, Germany) и корректировали с учетом фоновой флуоресценции. Среднюю величину флуоресценции кожи для каждого изображения рассчитывали с помощью анализатора изображений с программным обеспечением (MatLab 7.2.0.232, Math-Works, Natick, MA, USA) и рассчитывали среднюю величину флуоресценции кожи для каждой группы мышей (5 мышей/соединение).
Результаты:

Все 3 исследованных соединения вызывали флуоресценцию кожи, при этом соединение 2, гидрохлорид 1-(изопропилкарбокси)этил-5-амино-4-оксопентаноата демонстрировало наивысший уровень флуоресценции.
Пример 17 - Исследование флуоресценции кожи in vivo на карликовых свиньях
Соединение 1с (гидробромид карбоксиметил-5-амино-4-оксопентаноата, полученный в соответствии с примером 1с), был приготовлен в форме крема на основе препарата Unguentum Merck (15%, 150 мг/г; 0,56 ммоль).
Крем наносили на кожу спины карликовых свиней. Перед нанесением крема кожу вокруг при необходимости очищали с помощью стерильной воды и бинта. 0,5 г крема наносили на каждую исследуемую область (50 мм в диаметре) с получением однородного слоя крема и затем покрывали повязкой Тегадерм (Tegaderm®). Для удерживания повязки на месте применяли бинт Ветфлекс (Vet-Flex®).
Флуоресценцию кожи при 630 нм оценивали до нанесения крема и через 1,5, 5 и 12 часов после нанесения крема с применением устройства FluoDerm (DiaMedico ApS, Denmark) в соответствии с инструкциями производителя. Устройство FluoDerm представляет собой портативное устройство для объективного измерения in vivo в режиме реального времени средней величины флуоресценции в пределах круговой области диаметром 40 мм. Выполняли электронную корректировку измерений с учетом фактической интенсивности окружающего освещения. Результаты показаны на фигуре 1.
Результаты:
Как можно видеть на фигуре 1, флуоресценция кожи возрастала с течением времени.
Биопсия кожи:
Крем наносили, как описано выше. В момент времени t=0 (т.е. до нанесения крема), 1,5, 5 и 12 часов забирали образцы для биопсии с применением дерматома с шириной среза 10 мм (AcuPunch 10 mm, Acuderm, USA). Образцы забирали и обрабатывали в минимально освещенном помещении. Каждый образец зачищали от какой-либо подкожной жировой ткани и затем разрезали пополам. Каждый образец далее мгновенно замораживали в жидком азоте, в мешке из алюминиевой фольги или аналогичной упаковке, и переносили в морозильную камеру с температурой -80°С.
Из образцов для биопсии получали срезы толщиной 10 мкм, при этом также принимали меры для минимизации интенсивности окружающего освещения. В случаях, когда была возможна идентификация сальных желез и эпидермиса, получали серии из 3 последовательных срезов каждого образца. Срезы исследовали в течение 15 минут на микроскопе Leica DMRXE, приспособленного для обычной оптической микроскопии и флуоресцентной микроскопии. Местоположение эпидермиса и сальных желез определяли с применением стандартной оптической микроскопии. Процедуру проводили в течение 5 секунд, и затем источник света заменяли на ртутную лампу/режим флуоресценции с набором фильтров; фильтр возбуждения 390-447 нм, светоделитель 455 нм и эмиссионный фильтр >600 нм. Срезы незамедлительно фотографировали. Не перемещая образец, источник света заменяли обратно на источник для стандартной оптической микроскопии, и срезы фотографировали в точно таком же положении, как и для флуоресцентной микроскопии. Наряду с эпидермисом и сальными железами исследовали дерму и также фотографировали при обнаружении флуоресценции. Для дополнительного исследования локализации всех анализируемых структур срезы окрашивали гематоксилином/эозином.
Анализ изображений проводили с применением программного обеспечения Image J (версия Fiji-win32.exe). С помощью этой программы измеряли среднюю интенсивность флуоресценции на пиксель для сохраненных изображений.
Результаты:
Через 1,5 часа флуоресценция уже могла быть видна в сальных железах, а через 5 часов флуоресценция также была видна в эпидермисе. Таким образом, соединение 1 с демонстрировало селективность в отношении сальных желез и, следовательно, оно может подходить для фотодинамической терапии заболеваний, поражающих сальные железы, таких как угревая сыпь, фолликулярный кератоз, гиперплазия сальных желез, рак сальных желез или аденома сальных желез.
Пример 18 - Исследование флуоресценции кожи in vivo на здоровых бестимусных мышах и бестимусных мышах с УФ-индуцированным повреждением кожи
Эксперимент проводили с целью оценки способности соединения 6 к селективному накоплению в УФ-поврежденной коже. Для оценки измеряли флуоресценцию кожи in vivo после нанесения соединения на здоровую кожу мышей и на УФ-поврежденную кожу мышей. Модель бестимусных мышей с УФ-индуцированным повреждением кожи (актиническим повреждением) был разработана, как описано в К. Togsverd-Bo et al., Exp. Dermatol. 2012, 21, 260-264.
Эквимолярные количества соединения 2 (гидрохлорида 1-(изопропилкарбокси)этил-5-амино-4-оксопентаноата, полученного в соответствии с примером 2) и соединения 5 (гидрохлорида 3-карбоксипропил-5 амино-4-оксопентаноата, полученного в соответствии с примером 5) были приготовлены в форме крема на основе препарата Unguentum Merck: соединение 2: 16%, 160 мг/г и соединение 5: 14%, 140 мг/г, соответственно.
В эксперимент были включены одна группа здоровых бестимусных мышей (10 мышей, контрольная группа) и одна группа бестимусных мышей с УФ-индуцированным повреждением кожи (10 мышей). Примерно 125 мкл состава в форме крема наносили на спину каждой мыши. Перед нанесением крема кожу на спине каждой мыши пять раз обрабатывали липкими полосками для повышения проникновения соединения: клейкую пленку (скотч 3М) прижимали к коже в области нанесения, липкую полоску удаляли одним быстрым движением и использовали новую липкую полоску. Мышей содержали в темноте в течение 3 часов до измерения флуоресценции кожи во избежание фотообесцвечивания. Флуоресценцию кожи оценивали, как описано в примере 17, и рассчитывали среднюю величину флуоресценции кожи для каждой группы мышей. Результаты:
Определяли отношение величины флуоресценции кожи [отн. ед.] для УФ-поврежденной кожи мышей к величине флуоресценции кожи [отн. ед.] для здоровой кожи мышей.
Отношение для соединения 2 составляло 1,14, в то время как отношение для соединения 5 составляло 2,72. Следовательно, соединение 2 демонстрировало некоторую селективность в отношении УФ-поврежденной кожи, а соединение 5 демонстрировало выраженную селективность в отношении УФ-поврежденной кожи.
Пример 19 - Эффективность ФДТ в отношении бактерий
Эффективность различных соединений в соответствии с настоящим изобретением с точки зрения способности убивать бактерии за счет фотодинамической реакции исследовали на культуре грамположительной бактерии Staphylococcus aureus.
Был проведен анализ для следующих соединений согласно настоящему изобретению:
Соединение 1с: гидробромид карбоксиметил-5-амино-4-оксопентаноата (полученный в соответствии с примером 1с)
Соединение 4: гидробромид 2-карбоксиэтил-5-амино-4-оксопентаноата (полученный в соответствии с примером 4)
Бактериальный штамм: Штамм S. aureus DSM 20231 (АТСС 12600). Указанный бактериальный штамм выращивали на агаре с сердечным экстрактом (Difco) при 35-37°С в течение 24 часов до проведения эксперимента и ресуспендировали в 20 мМ буфере PIPES, рН 7,2-7,4 до концентрации, соответствующей стандарту мутности МакФарланда 0,5 (примерно 1-5×108 КОЕ/мл). Для обеспечения жизнеспособных культур клеток и проверки размера и чистоты инокулята исходные культуры бактерий готовили непосредственно перед проведением экспериментов и определяли величину КОЕ/мл посредством стандартного анализа колониеобразования.
Токсичность в темноте: "Токсичность в темноте", т.е. токсический эффект соединений в отсутствие света (и, следовательно, не связанный с эффектами ФДТ), может включать гибель бактериальных клеток и, таким образом, может препятствовать точному измерению эффекта ФДТ, который также приводит к гибели бактериальных клеток. Токсичность в темноте может быть измерена путем определения токсичности каждого соединения в тех же экспериментальных условиях, но в отсутствие света, что позволяет избежать какого-либо эффекта ФДТ. Применение соединений согласно настоящему изобретению в условиях, которые обеспечивают низкую токсичность в темноте, является предпочтительным, так как эффекты токсичности в темноте также могут включать повреждение или уничтожение клеток, не являющихся мишенями, и противодействие лечению методом ФДТ, как таковому. С учетом этих факторов, оптимальный агент для обеспечения ФДТ-опосредованной гибели бактериальных клеток должен демонстрировать как низкую токсичность в темноте, так и высокую эффективность индукции фотодинамического действия, с тем чтобы вызывать гибель бактериальных клеток-мишеней.
Токсичность в темноте определяли, как описано ниже в разделе "ФДТ-обработка", за исключением того, что планшет не облучали светом. Затем проводили анализ с применением микропланшетов, как описано ниже.
Токсичность в темноте соединений 1с и 4 исследовали для концентраций 0,001, 0,1, 1 и 10 мМ. Токсичность в темноте не была обнаружена ни для одной из этих концентраций.
ФДТ-обработка: Готовили базовые растворы соединений 1с и 4 в ДМСО с концентрацией 100 мМ. Перед началом эксперимента 0,02 мл ДМСО (контроль) или базового раствора соединений 2 и 3 вносили с помощью пипетки в лунки 24-луночного планшета VisiPlate-24 с получением концентраций 0,01 мМ, 0,1 мМ и 1 мМ. В каждую лунку добавляли 2 мл исходной культуры бактерий с последующим перемешиванием с помощью автоматической пипетки. Все лунки содержали ДМСО в конечной концентрации 1%. После инкубации в течение 4 часов планшет облучали красным светом (лампа Aktilite® CL128) в течение 32 минут (плотность энергии 148 Дж/см2). Для достижения равномерного освещения всех лунок планшет облучали снизу (лунки с плоским дном) с медленными круговыми движениями в пределах освещаемой лампой области.
Анализ микропланшетов: После освещения все содержимое лунок переносили в пробирки типа эппендорф, и 0,1 мл содержимого повторно вносили в лунки микропланшета. К повторно внесенному инокуляту добавляли 1,5 мл бульона с сердечным экстрактом (Difco) для получения кривых роста. Все переносы осуществляли с тщательным перемешиванием содержимого путем пипетирования.
Планшеты инкубировали при 37±1°С, используя функцию поддержания температуры в многозначном счетчике Victor 1420 (Perkin Elmer, Turku, Finland). Кривые роста получали путем измерения оптической плотности при 595 нм через регулярные промежутки времени до тех пор, пока бактериальные клетки не вступали в логарифмическую фазу роста. Измерения были выполнены автоматически с помощью многозначного счетчика.
Необработанные данные откладывали на осях в полулогарифмических координатах (log10 поглощения в зависимости от времени (1 единица = 15 мин)) для определения момента времени после посева, в который бактериальные клетки вступали в логарифмическую фазу роста (т.е. продолжительности лаг-фазы). Продолжительность лаг-фазы зависела от количества выживших клеток и возрастала пропорционально эффективности фотодинамической терапии.
Результаты:
Результаты показаны на фигуре 2, на которой можно видеть, что оба соединения эффективно убивали бактерии S. aureus и были способны эффективно задерживать повторный рост бактериальных клеток.
Пример 20 - Тест Эймса
Тест Эймса (см. B.N. Ames et al, Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutation Research 31, 347-364, 1975) представляет собой стандартный способ анализа, утвержденный регулирующими органами для выявления способности исследуемого химического соединения вызывать мутации в некоторых бактериальных штаммах. Если исследуемое химическое соединение вызывает такие мутации, оно не может применяться в фармацевтическом составе для использования у людей.
Цель данного исследования заключалась в оценке мутагенного потенциала и фотомутагенного потенциала соединения 1с, гидробромида карбоксиметил-5-амино-4-оксопентаноата (полученного в соответствии с примером 1с), путем проверки его способности вызывать реверсивную мутацию в двух гистидинзависимых штаммах Salmonella typhimurium, ТА98 и ТА100, в отсутствие и в присутствии метаболизирующей системы печени крысы (S-9) и также в отсутствие и в присутствии видимого света. Последнее оценивали, поскольку соединение 1с поглощалось бактериальными клетками и превращалось в фотосенсибилизатор, который, при облучении указанных клеток светом, мог вызывать образование мутагенных продуктов.
Результаты показывают, что соединение 1с не вызывало мутаций или фотомутаций в этих двух штаммах Salmonella typhimurium при проведении теста в условиях, применяемых для этого исследования.
Пример 21 - Исследование флуоресценции ПпIХ in vitro в раковых клетках
Исследование проводили с целью оценки эффективности соединений согласно настоящему изобретению в качестве предшественников фотосенсибилизаторов. Соответственно, соединения согласно изобретению должны были поглощаться клетками и должны были превращаться в фотосенсибилизаторы, т.е. ПпIХ. Результат этого процесса можно визуализировать путем подвергания клеток действию света, который возбуждает молекулы ПпIХ, и определения количества энергии, высвобождаемой в форме флуоресценции, во время которой возбужденные молекулы ПпIХ релаксируют в более низкое энергетическое состояние.
Были исследованы следующие соединения согласно настоящему изобретению:
Соединение 5 (гидрохлорид 3-карбоксипропил-5-амино-4-оксопентаноата, полученный в соответствии с примером 5)
Соединение 9 (гидрохлорид 5-карбоксипентил-5-амино-4-оксопентаноата, полученный в соответствии с примером 9)
Соединение 10 (гидрохлорид 7-карбоксигептил-5-амино-4-оксопентаноата, полученный в соответствии с примером 10)
Соединение 12 (гидрохлорид 9-карбоксинонил-5-амино-4-оксопентаноата, полученный в соответствии с примером 12)
Соединение 14 (гидрохлорид 11-карбоксиундецил-5-амино-4-оксопентаноата, полученный в соответствии с примером 14)
Соединение 15 (гидрохлорид 15-карбоксипентадецил-5-амино-4-оксопентаноата, полученный в соответствии с примером 15)
Следующие соединения применяли в качестве эталонных соединений:
Гидрохлорид 5-АЛК (АЛК)
Гидрохлорид гексилового эфира 5-АЛК (ГАЛ)
Базовые растворы вышеуказанных соединений готовили путем растворения их в ДМСО в концентрации 100 мМ. Концентрации, применяемые в экспериментах, получали путем разбавления базовых растворов фосфатно-солевым буфером (ФСБ) или средой для культивирования клеток.
Культивирование клеток и обработка клеток опытными и эталонными соединениями:
Клетки линии WiDr, полученные из первичной аденокарциномы ректосигмоидного отдела толстой кишки, пересевали в среду RPMI 1640 (Gibco), содержащую 10% фетальной телячьей сыворотки (ФТС), 100 ед/мл пенициллина, 100 мкг/мл стрептомицина и 1% глутамина. Клетки рассевали 1:100 по объему среды дважды в неделю и поддерживали при 37°С и 5% СO2 во влажной среде.
Для проведения экспериментов 5×105 клеток линии WiDr в 2 мл вышеописанной среды добавляли в каждую лунку 6-луночных пластиковых планшетов для культур тканей (Nunc) и оставляли на 48 ч при 37°С и 5% СO2 во влажной среде для надлежащего прикрепления к поверхности планшета. Затем клетки дважды промывали средой RPMI 1640 без сыворотки с последующим добавлением в лунки в дубликатах соответствующих разведений вышеуказанных опытных и эталонных соединений в 2 мл свежей среды для культивирования до конечных концентраций, составляющих 0,001, 0,003, 0,01, 0,03, 0,1, 0,3 и 1 мМ. В две лунки добавляли только 2 мл свежей среды для культивирования. Эти необработанные клетки служили в качестве контроля. Клетки инкубировали при 37°С в течение четырех часов в темноте.
Токсичность в темноте:
Токсичность в темноте, т.е. цитотоксичность в отсутствие света, измеряли посредством MTS-теста незамедлительно после вышеописанной инкубации в течение 4 часов с опытными и эталонными соединениями в различных концентрациях. Для гидрохлорида АЛК и соединений 5, 9, 10, 12 и 14 не наблюдали цитотоксичности ни для какой из исследуемых концентраций. Для гидрохлорида гексилового эфира наблюдали слабый цитотоксический эффект в диапазоне концентраций от 0,3 до 1 мМ при минимальной доле выживших клеток, составлявшей 85%. Соединение 15 демонстрировало токсичность в концентрациях выше 0,1 мМ.
Образование ПпIХ:
После обработки соединений согласно изобретению и эталонных соединений, как описано выше, клетки дважды промывали ФСБ и соскабливали с поверхности планшета с помощью скребка для клеток Costar в раствор 1М НСlO4 в 50% метаноле. Остатки от разрушенных клеток удаляли центрифугированием. Посредством этой процедуры ПпIХ был количественно экстрагирован из клеток. Содержание ПпIХ в каждом образце определяли флуорометрически с применением спектрофлуориметра Perkin Elmer LS50B. Возбуждение молекул ПпIХ осуществляли на длине волны 407 нм, а испускаемую флуоресценцию измеряли на 606 нм с применением длинноволнового отсекающего фильтра (530 нм) со стороны испускания. Стандарт с известной концентрацией ПпIХ добавляли к образцам в концентрациях, увеличивавших общую флуоресценцию на 50-100%. Концентрацию ПпIХ в каждом образце рассчитывали относительно содержания белка в контрольных клетках, измеренного методом количественного определения белка с применением бицинхониновой кислоты.
Результаты образования ПпIХ:
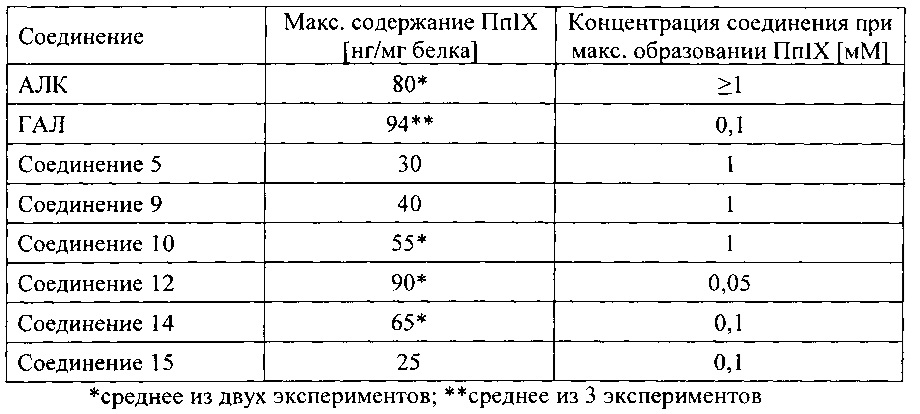
Результаты показывают, что все исследованные соединения являются подходящими фотосенсибилизирующими агентами в раковых клетках, т.е. имеют потенциал с точки зрения применения для фотодинамической терапии рака: все исследованные соединения превращались в ПпIХ в раковых клетках, и концентрации, соответствовавшие максимальному образованию ПпIХ, не являлись токсичными для клеток, т.е. токсичность в темноте не была обнаружена при таких концентрациях. Наблюдалась тенденция к тому, что максимальная концентрация ПпIХ соответствовала, как правило, более низким концентрациям для соединений с большей длиной цепи.
Пример 22 - Исследование флуоресценции ПпIХ in vitro в клетках мочевого пузыря крысы
Гидрохлорид гексилового эфира 5-АЛК (ГАЛ) представляет собой активный ингредиент препарата Гексвикс, коммерчески доступного лекарственного средства для фотодинамической диагностики рака мочевого пузыря. Экспериментальные исследования также показывают пригодность Гексвикса для фотодинамической терапии рака мочевого пузыря. ГАЛ превращается в ПпIХ в раковых клетках мочевого пузыря и может быть обнаружен по его характеристической флуоресценции. Первые исследования в процессе разработки Гексвикса были проведены на клетках и ткани мочевого пузыря крысы как модели клеток и ткани мочевого пузыря человека. Это исследование было выполнено в качестве первой стадии для оценки эффективности соединений согласно настоящему изобретению в качестве предшественников фотосенсибилизаторов, которые могут быть применены на мочевом пузыре. На этой первой стадии были определены подходящие концентрации соединений in vitro путем сравнения флуоресценции ПпIХ из соединений с флуоресценцией ПпIХ из ГАЛ. На основании результатов определяли концентрацию для последующего исследования in vivo.
Были исследованы следующие соединения:
Соединение 4: гидробромид 2-карбоксиэтил-5-амино-4-оксопентаноата (полученный в соответствии с примером 4)
Соединение 5: гидрохлорид 3-карбоксипропил-5-амино-4-оксопентаноата (полученный в соответствии с примером 5)
Соединение 8: гидрохлорид 2-карбоксибутил-5-амино-4-оксопентаноата (полученный в соответствии с примером 8)
Гидрохлорид гексилового эфира 5-АЛК (ГАЛ) применяли в качестве эталонного соединения.
Измерения уровня ПпIХ в клетках мочевого пузыря крысы:
Клетки мочевого пузыря крысы (AY-27) выращивали в среде для культивирования (RPMI 1640 с добавлением 9% ФТС, 1% L-глутамина (200 мМ) и 1% пенициллина (10000 ME), стрептомицина (10000 мкг/мл). Экспоненциально растущие клетки (AY-27) инкубировали в течение 2 часов со свежеприготовленными растворами 0,8 мМ ГАЛ (что представляет собой десятую часть концентрации, применяемой для обнаружения рака мочевого пузыря у пациентов-людей) и свежеприготовленными растворами соединений 4, 5 и 8 с концентрациями 3-15 мМ (см. таблицу ниже). Контрольные клетки получали не содержащую сыворотку среду без каких-либо опытных или эталонных соединений. После инкубации растворы или среду удаляли, и клетки промывали два раза ледяным ФСБ. ПпIХ экстрагировали из клеток с помощью ледяной лизирующей смеси для экстракции, состоящей из этанола/ДМСО/уксусной кислоты (80/20/1, об/об/об). Клеточный лизат центрифугировали (400 g, 10 мин, 2°С), осадок измельчали и суспензию обрабатывали ультразвуком в течение 15 мин перед центрифугированием. Сигнал флуоресценции ПпIХ измеряли с помощью спектрометра Perkin-Elmer LS 55 (Perkin-Elmer, Beaconsfield, UK) и выражали в виде зависимости от содержания белка. Интенсивность флуоресценции при 635 нм в каждом образце отмечали на калибровочной кривой, построенной по ПпIХ (Sigma-Aldrich, France), разведенному в описанной выше смеси для экстракции. Концентрацию ПпIХ выражали относительно содержания белка. Вкратце, смесь лизирующего буфера (1 мМ ЭДТА, 1 % Тритон Х-100 и 10 мМ Трис-HCl, рН 7,4) добавляли в чашки с замороженными клетками вместе с 500 мМ раствором ФМСФ (фенилметилсульфонилфторида, ингибитора протеаз) в ДМСО. Лизаты выдерживали на льду в течение 30 минут и центрифугировали при 4°С в течение 20 минут при 15000 g. Затем измеряли содержание белка с помощью построенной по БСА калибровочной кривой с применением набора для анализа белков Biorad DC (Bio-Rad, France) на основе метода, разработанного Лоури.
Все эксперименты проводили в 4-5 повторностях при приглушенном свете. Для концентрации соединений ≥1 мМ оценивали их цитотоксичность посредством МТТ-теста. Экспоненциально растущие клетки инкубировали со свежеприготовленными растворами или средой, как описано выше, в течение 2 часов в 96-луночных планшетах. После инкубации растворы или среду удаляли, и клетки дважды промывали ФСБ. Затем в каждую лунку добавляли 50 мкл МТТ (2,5 мг/мл) к 150 мкл не содержащей сыворотки среды RPMI и инкубировали в течение 3 часов. Оптическую плотность измеряли путем растворения кристаллов формазана в 50 мкл ДМСО. Поглощение нормализовали к таковому для контрольных клеток.
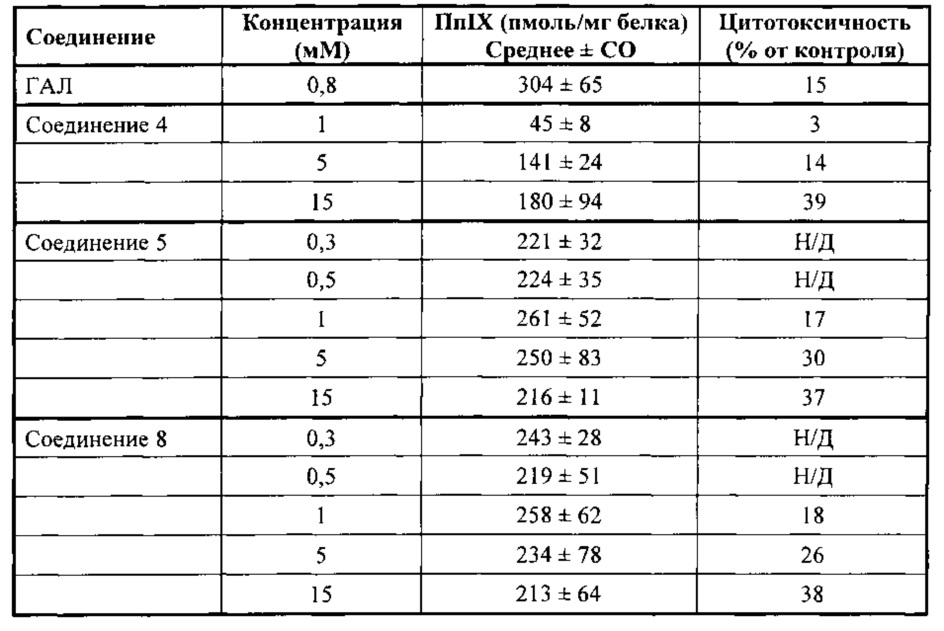
Как можно видеть из данных таблицы, ГАЛ, применяемый в качестве эталонного соединения, обеспечивал образование примерно 304 пмоль ПпIХ/мг белка с приемлемым уровнем цитотоксичности, составляющим 15%.
Соединение 4 вызывало образование ПпIХ в концентрациях, которые были значительно ниже по сравнению с таковыми для ГАЛ, примерно в той же концентрации (1 мМ). Примерно половина концентрации ПпIХ, соответствующей ГАЛ, была достигнута при концентрации, примерно в 6 раз превышающей концентрацию ГАЛ (т.е. 5 мМ). При более высокой концентрации соединение обладало цитотоксическим эффектом.
Соединения 5 и 8 вызывали образование Пп1Х в концентрациях, которые были сходны с таковыми для ГАЛ, в соизмеримых концентрациях (т.е. 1 мМ) и в концентрации 5 мМ. Уровни цитотоксичности были также сопоставимы и приемлемы при концентрации 1 мМ. Следовательно, соединения 5 и 8 являются перспективными кандидатами для применения в качестве предшественников фотосенсибилизаторов для ФДД и ФДТ в мочевом пузыре.
Эксперименты по определению рН:
Мочевой пузырь чувствителен к низким рН, тогда как соединения согласно настоящему изобретению (и, как правило, сложные эфиры АЛК) образуют пиразины при более высоких рН. Следовательно, раствор соединений согласно изобретению не должен приводить к рН, не допустимому для мочевого пузыря. Влияние исследуемых соединений на рН в среде проверяли путем добавления соединений в концентрациях 1, 5 и 25 мМ к среде RPMI 1640 с последующим измерением рН. Было обнаружено, что соединения в концентрации 1 мМ не изменяли рН в среде, в то время как соединения в концентрациях 5 и 25 мМ изменяли рН от 7,5 до 7,1 и 5,5 соответственно. После нанесения данных на график посредством интерполяции было установлено, что соединения в концентрации 15 мМ должны приводить к рН 6,0. При рН ниже 6 в мочевом пузыре могут возникать неблагоприятные эффекты (сокращения и т.д.). Концентрация 15 мМ намного превышает 10 мМ, предполагаемую концентрацию для исследований in vivo (основанную на экспериментах с ГАЛ и на том допущении, что для экспериментов in vivo будет применена 10-кратная концентрация из экспериментов in vitro).
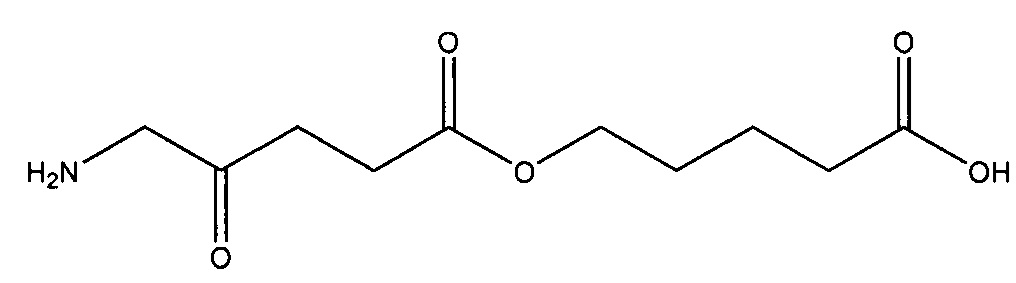
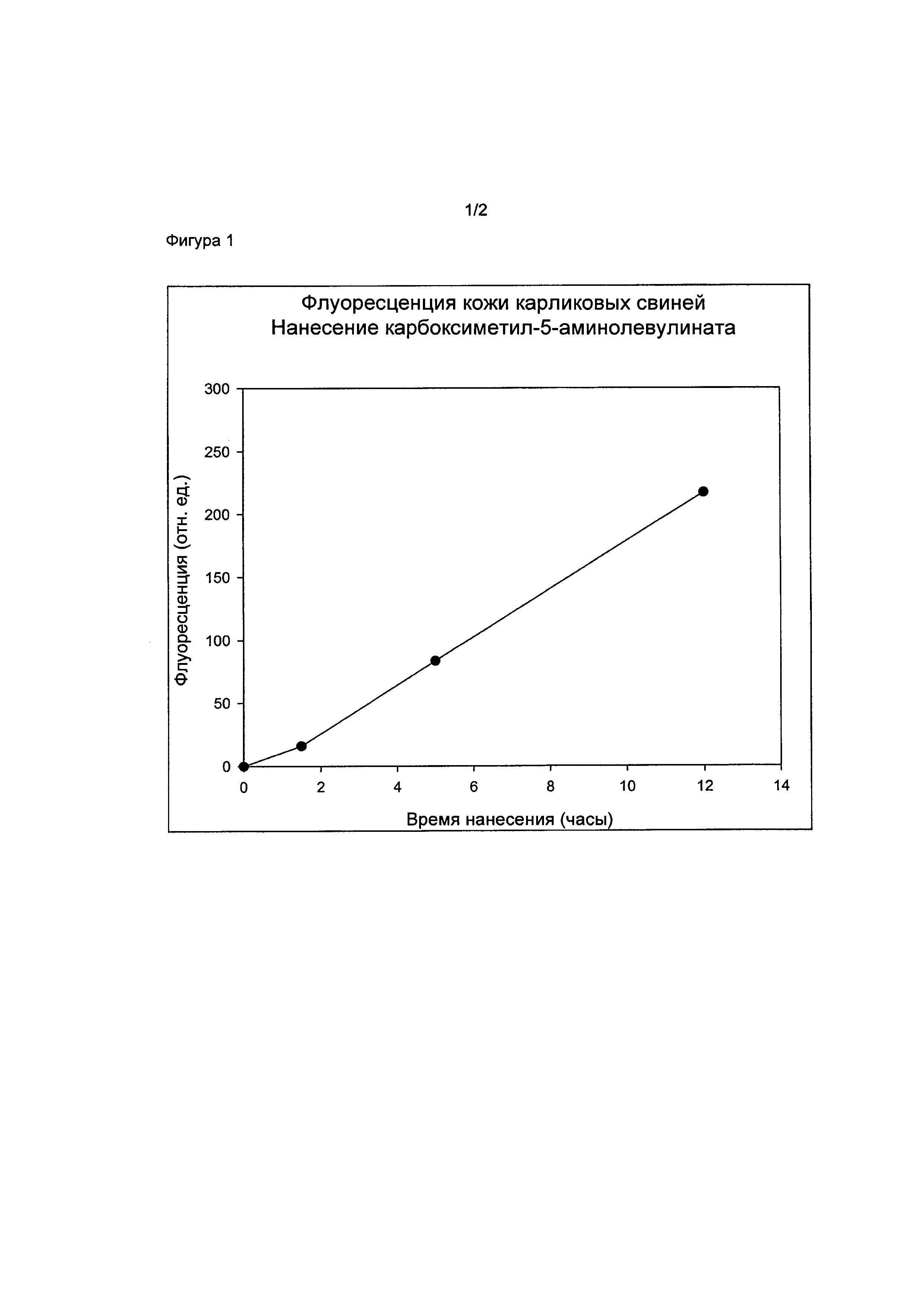
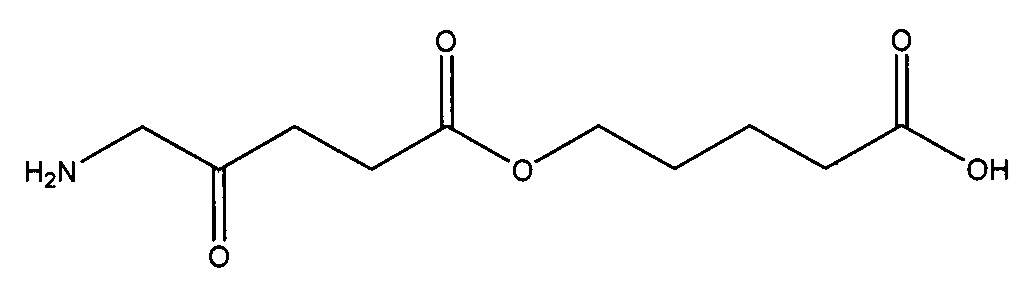
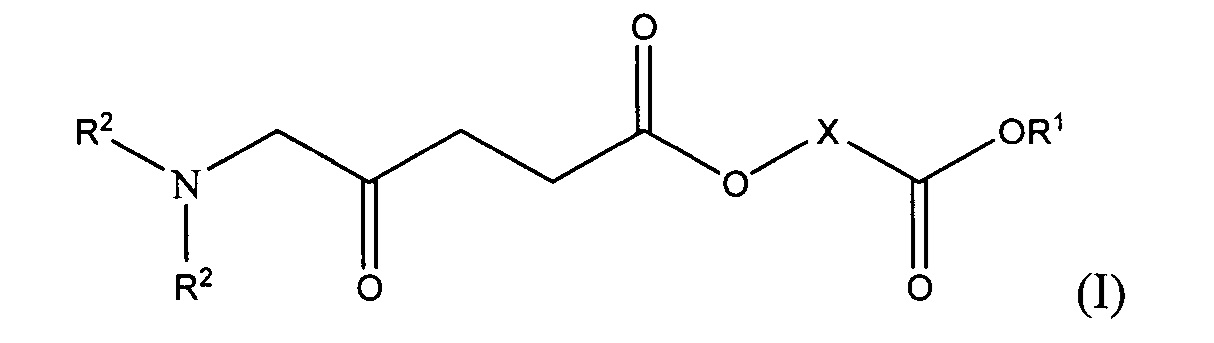
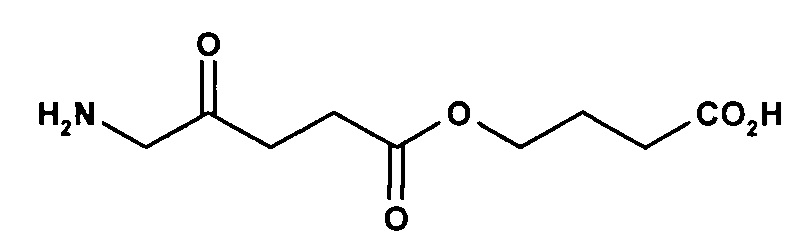
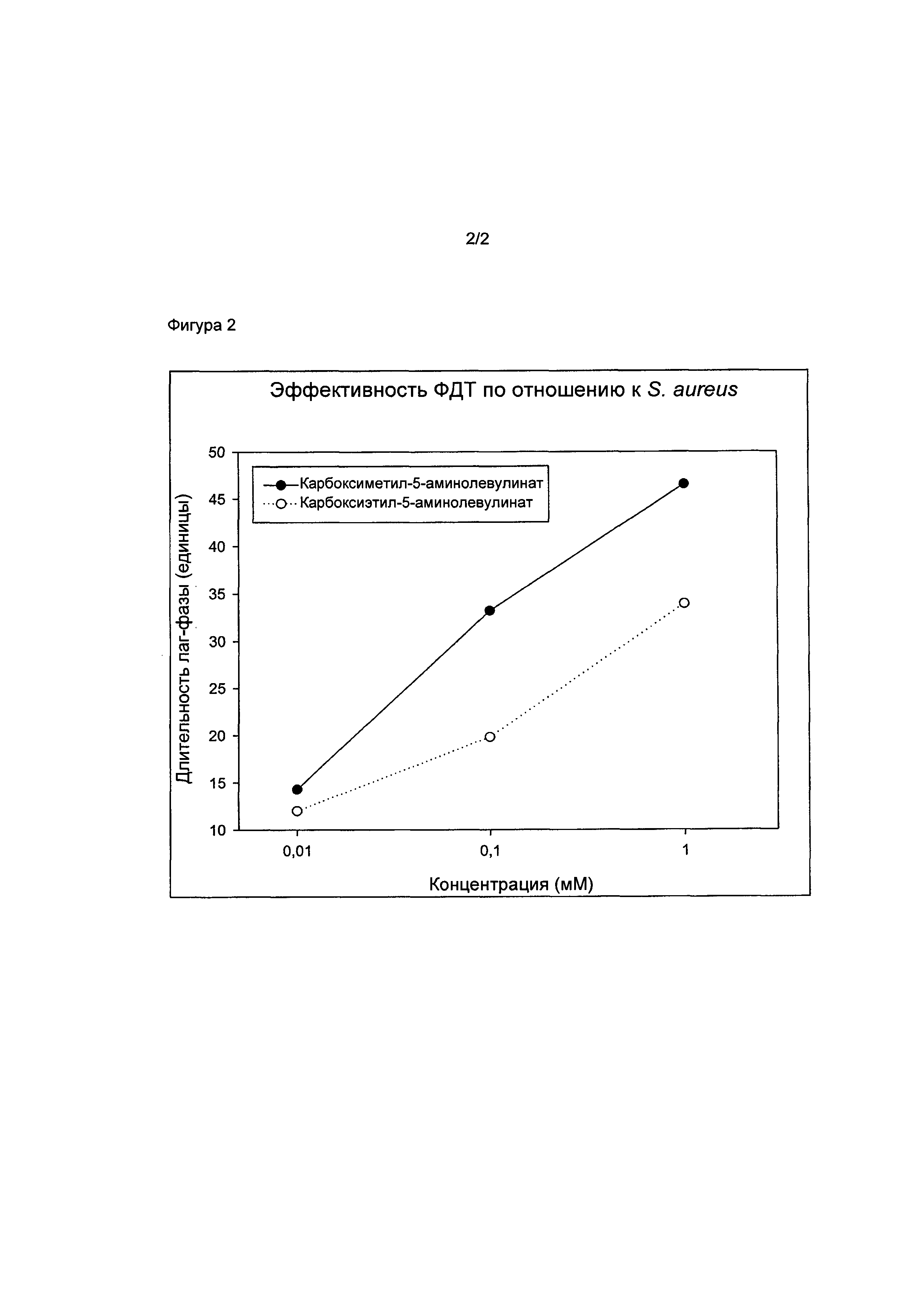
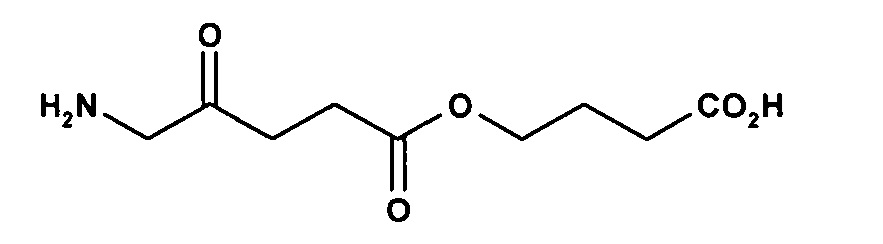
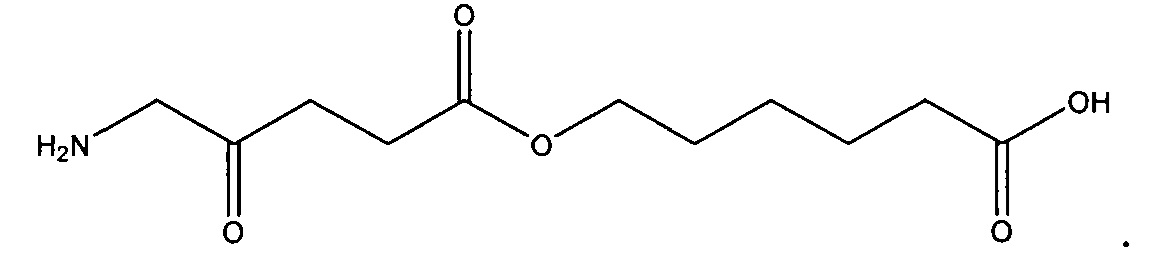
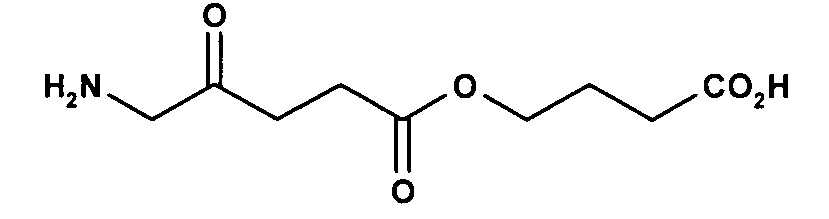
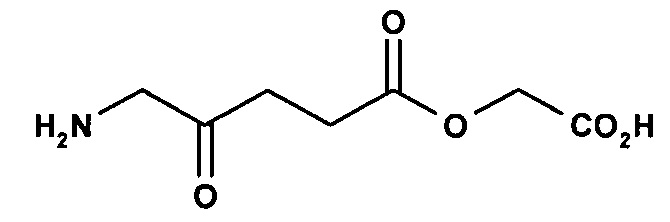
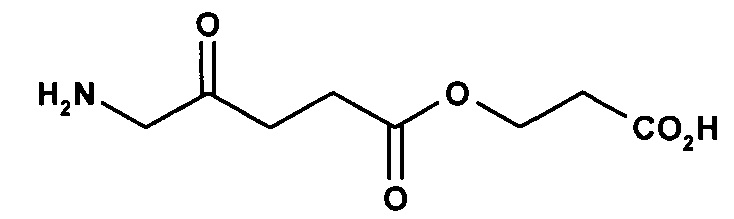
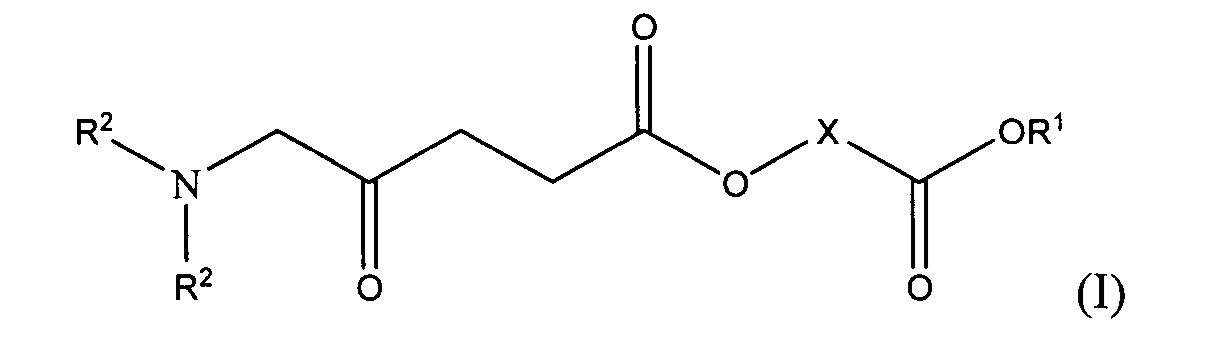
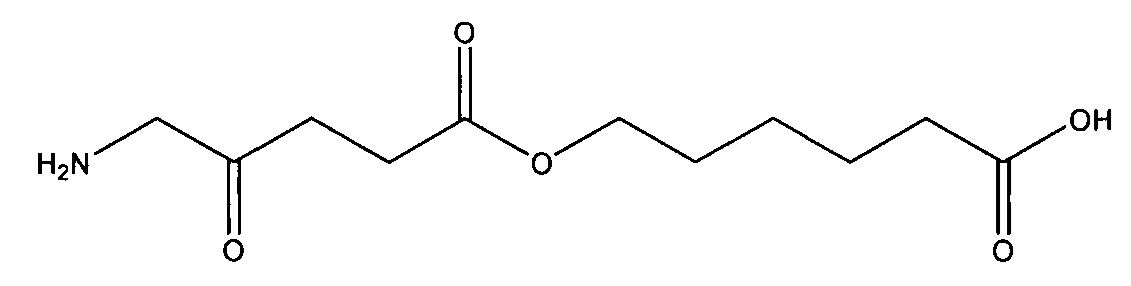
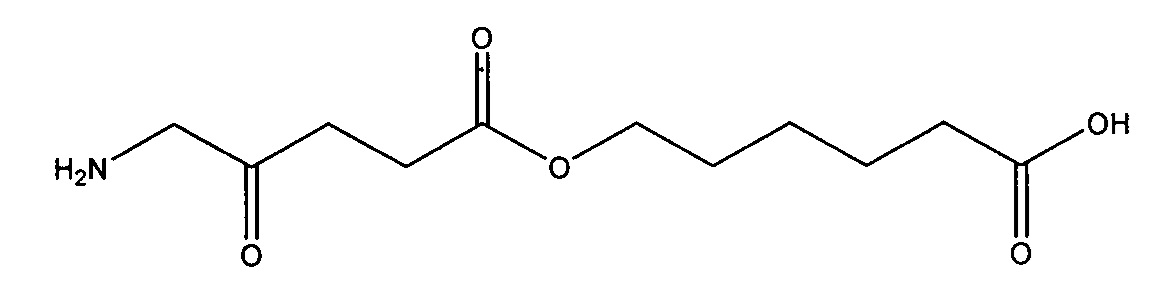
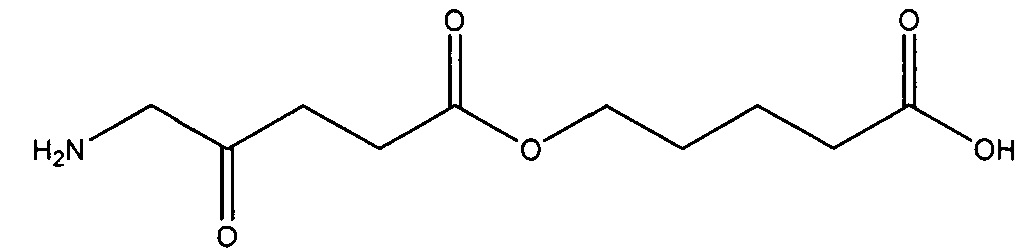
Устройство облучения
Применение 5-аминолевулиновой кислоты и ее производных в твердой форме для фотодинамического лечения и диагностики
Полутвердые композиции и фармацевтические продукты
Твердые композиции, содержащие 5-аминолевулиновую кислоту
Фотосенсибилизирующие композиции
Устройство облучения
Устройство облучения
Устройство облучения
Применение 5-аминолевулиновой кислоты и ее производных в твердой форме для фотодинамического лечения и диагностики
Полутвердые композиции и фармацевтические продукты
Твердые композиции, содержащие 5-аминолевулиновую кислоту
Фотосенсибилизирующие композиции
Устройство облучения
Устройство облучения
Лечение угрей с использованием производных 5-аминолевулиновой кислоты

