Результат интеллектуальной деятельности: ПОЛУЧЕНИЕ АМФИФИЛЬНЫХ БЛОК-СОПОЛИМЕРОВ ПУТЕМ КОНТРОЛИРУЕМОЙ РАДИКАЛЬНОЙ МИЦЕЛЛЯРНОЙ ПОЛИМЕРИЗАЦИИ
Вид РИД
Изобретение
Настоящее изобретение относится к конкретному способу полимеризации, с помощью которого обеспечивают доступ к амфифильным блок-сополимерам, которые имеют конкретную контролируемую структуру, то есть, в общих чертах, основанную на главной цепи, образованной из гидрофильных единиц (водорастворимых или диспергируемых в воде единиц), прерываемых в различных местах небольшими гидрофобными блоками, причем все эти гидрофобные блоки практически имеют одинаковый размер и присутствуют практически в том же количестве и соотношении во всех полимерных цепях. Настоящее изобретение также относится к амфифильным полимерам контролируемой структуры, полученным в соответствии с данным способом, средняя молярная масса которых может точно контролироваться посредством способа получения согласно настоящему изобретению, и которые могут применяться, в частности, в качестве ассоциативных загустителей, поверхностно-активных веществ или модификаторов свойств поверхности.
Для получения водорастворимых или диспергируемых в воде полимеров, в том числе гидрофобных блоков, применяют один известный на данный момент способ, который известен как "мицеллярная радикальная полимеризация". Примеры мицеллярной полимеризации были описаны, в частности, в US 4432881 или в Polymer, vol. 36, No. 16, pp. 3197-3211 (1996), на которые для дополнительных сведений может быть сделана ссылка.
В соответствии с конкретной методикой "мицеллярной радикальной полимеризации", которая в остальной части описания в целях краткости будет обозначаться как "мицеллярная полимеризация", блок-сополимеры мультиблочного типа синтезируются путем сополимеризации гидрофильных мономеров и гидрофобных мономеров в водной диспергирующей среде (как правило, воде или смеси вода/спирт), которая содержит:
- гидрофильные мономеры в растворенной форме или диспергированные в указанной среде
и
- гидрофобные мономеры в мицеллах поверхностно-активных веществ, образованных в указанной среде путем введения в нее данного поверхностно-активного вещества при концентрации выше критической концентрации мицеллообразования (cmc).
В соответствии с определенным вариантом осуществления гидрофобные мономеры, присутствующие в мицеллах поверхностно-активного вещества и применяемые при мицеллярной полимеризации, могут быть мономерами, которые сами по себе обладают свойством образования мицелл без необходимости добавления дополнительных поверхностно-активных веществ (мономеры, называемые в остальной части описания "способными к самостоятельному мицеллообразованию"). В соответствии с этим определенным вариантом осуществления применяемое поверхностно-активное вещество само по себе может представлять собой гидрофобный мономер, способный к самостоятельному мицеллообразованию и применяемый без каких-либо других поверхностно-активных веществ, однако присутствие какого-то дополнительного поверхностно-активного вещества данного типа не исключено. Таким образом, для целей данного описания, когда упоминаются гидрофобные мономеры в мицеллах поверхностно-активного вещества, такое понятие включает как (i) гидрофобные мономеры, присутствующие в мицеллах поверхностно-активного вещества, отличного от таких мономеров, так и (ii) мономеры, содержащие по меньшей мере одну гидрофобную часть или блок и образующие сами по себе мицеллы в водной среде. Эти две вышеуказанные формы (i) и (ii) являются совместимыми и могут применяться совместно (гидрофобные мономеры в мицеллах, образованные другим мономером, способным к самостоятельному мицеллообразованию, например, или, альтернативно, мицеллы, содержащие комбинацию поверхностно-активных веществ и мономеров, способных к самостоятельному мицеллообразованию).
Считается, что при мицеллярной полимеризации гидрофобные мономеры, содержащиеся в мицеллах, находятся в "мицеллярном растворе".
Мицеллярный раствор, на который ссылаются, представляет собой микрогетерогенную систему, которая обычно является изотропной, оптически прозрачной и термодинамически стабильной.
Следует также отметить, что мицеллярный раствор типа, применяемого при мицеллярной полимеризации, следует отличать от микроэмульсии. В частности, в противоположность микроэмульсии, мицеллярный раствор образуется при любой концентрации, превышающей критическую концентрацию мицеллообразования применяемого поверхностно-активного вещества, при единственном условии, что гидрофобный мономер является растворимым по меньшей мере до определенной степени во внутреннем пространстве мицелл. Кроме того, мицеллярный раствор отличается от эмульсии отсутствием внутренней гомогенной фазы: мицеллы содержат очень малое количество молекул (обычно менее 1000, как правило, менее 500 и обычно от 1 до 100, чаще всего от 1 до 50 мономеров и не более нескольких сотен молекул поверхностно-активного вещества, если поверхностно-активное вещество присутствует), а мицеллярный раствор, как правило, обладает физическими свойствами, схожими со свойствами мицелл поверхностно-активного вещества без мономеров. Кроме того, как правило, мицеллярный раствор является прозрачным относительно видимого света, учитывая небольшой размер мицелл, который не приводит к преломлению света, в отличие от капель эмульсии, которые отражают свет и придают эмульсии характерный мутный или белый внешний вид.
Методика мицеллярной полимеризации приводит к образованию характерных блок-сополимеров, каждый из которых содержит несколько гидрофобных блоков практически одинакового размера, при этом данный размер можно контролировать. В частности, с учетом того, что гидрофобные мономеры заключены в мицеллах, каждый из образованных гидрофобных блоков контролируемого размера по сути содержит определенное количество nH гидрофобных мономеров, причем количество nH можно вычислить следующим образом (Macromolecular Chem. Physics, 202, 8, 1384-1397, 2001):
nH=Nagg⋅[MH]/([поверхностно-активное вещество]-cmc),
где
Nagg представляет собой число агрегации поверхностно-активного вещества, которое отражает количество поверхностно-активных веществ, присутствующих в каждой мицелле,
[MH] представляет собой молярную концентрацию гидрофобного мономера в среде, и
[поверхностно-активное вещество] представляет собой молярную концентрацию поверхностно-активного вещества в среде,
cmc представляет собой критическую (молярную) концентрацию мицеллообразования.
Таким образом, методика мицеллярной полимеризации позволяет осуществлять эффективный контроль гидрофобных единиц, введенных в образованные полимеры, то есть:
- полностью контролировать молярную долю гидрофобных единиц в полимере (путем модуляции соотношения концентраций двух мономеров) и
- более точно контролировать количество гидрофобных единиц, присутствующих в каждом из гидрофобных блоков (путем модификации параметров, которые влияют на nH, определенный выше).
Мультиблочные сополимеры, полученные путем мицеллярной полимеризации, также имеют ассоциативную природу, что в абсолютном выражении делает их хорошими кандидатами для применения в качестве средств для повышения вязкости.
Однако, как правило, мицеллярная полимеризация приводит к образованию полимерных цепей с высокой степенью неоднородности по размеру. Действительно, хотя они позволяют осуществлять тонкий контроль размера гидрофобных блоков, современные методики мицеллярной полимеризации приводят к более неравномерной полимеризации единиц гидрофильного мономера. Таким образом, известные на данный момент способы мицеллярной полимеризации приводят к образованию полимерных цепей, часто обладающие высокой неоднородностью состава, с, как правило, в значительной степени полидисперсным распределением молекулярной массы без возможного предопределения полученной средней молекулярной массы Mn. Эта неоднородность особенно раскрывается в вышеуказанной статье Polymer, vol. 36, No. 16, pp. 3197-3211 (1996).
Более того, полученная микроструктура полимеров (то есть распределение гидрофобных блоков в различных цепях) не является контролируемой, что обусловлено, в частности, очень коротким временем жизни растущих цепей относительно общего времени полимеризации в сочетании с отличиями в реакционной способности растущих активных центров по отношению к гидрофильным мономерам и гидрофобным мономерам (а также отличиями в их концентрации).
Другими словами, мицеллярная полимеризация в действительности делает возможной, в большинстве случаев, интеграцию гидрофобных блоков контролируемого размера в гидрофильные цепи, что позволяет синтезировать самоассоциативные полимеры, но без контроля общего размера синтезированных полимеров или микроструктуры этих полимеров, что не позволяет осуществлять тонкий контроль свойств этих самоассоциативных полимеров.
Кроме того, отсутствие контроля микроструктуры не позволяет в полной мере осуществлять тонкую модуляцию и контроль свойств синтезированных полимеров путем мицеллярной полимеризации. Более того, это препятствует доступу к coполимерам контролируемого строения.
В дополнение, способы мицеллярной полимеризации обычно ограничены крайне разбавленными системами для обеспечения добавления и смешивания реагентов. Молекулярные массы, получаемые при мицеллярной радикальной полимеризации, как правило, составляют примерно от 500000 до 5000000 г/моль, например, от 500000 до 3000000.
Для уменьшения отклонения состава в полимерах, полученных в результате мицеллярной полимеризации, в US 6207771 предложен способ полунепрерывного добавления гидрофобных мономеров. Тем не менее, интересен тот факт, что этот способ, однако, не позволяет осуществлять эффективный контроль микроструктуры и совершенно не позволяет осуществлять контроль молекулярных масс.
Одной из целей настоящего изобретения является создание блок-сополимеров, содержащих гидрофобные блоки контролируемого размера, типа, полученного при стандартной мицеллярной полимеризации, но за счет улучшения контроля средней молекулярной массы синтезированных цепей, а также обеспечения контроля микроструктуры полимеров, то есть однородности, от одной полимерной цепи к другой, распределения гидрофобных блоков в гидрофильной главной цепи.
С этой целью, согласно первому аспекту настоящего изобретения, одним из объектов настоящего изобретения является способ получения блок-coполимера, который включает этап (E) мицеллярной радикальной полимеризации, при котором в водной среде (M) в контакт приводят следующее:
- гидрофильные мономеры, растворенные или диспергированные в указанной водной среде (M);
- гидрофобные мономеры в виде мицеллярного раствора, то есть содержащего в диспергированной форме в среде (M) мицеллы, содержащие эти гидрофобные мономеры (данное диспергированное состояние, в частности, может быть достигнуто с помощью по меньшей мере одного поверхностно-активного вещества);
- по меньшей мере один инициатор радикальной полимеризации, при этом данный инициатор, как правило, является водорастворимым или диспергируемым в воде; и
- по меньшей мере одно средство контроля радикальной полимеризации.
Водная среда (M), применяемая на этапе (E) представляет собой среду, содержащую воду, относительное содержание которой предпочтительно составляет по меньшей мере 50% по массе, или даже по меньшей мере 80%, например, по меньшей мере 90%, или даже по меньшей мере 95%. Данная водная среда может необязательно содержать растворители, отличные от воды, например, спирт, смешиваемый с водой. Таким образом, средой (M) может быть, например, водно-спиртовая смесь. В соответствии с возможным вариантом среда (M) может содержать другие растворители, предпочтительно в концентрации, в которой указанный растворитель является смешиваемым с водой, что, в частности, может позволить уменьшение количества применяемых стабилизирующих поверхностно-активных веществ. Таким образом, например, среда (M) может содержать пентанол или любую другую добавку для модуляции числа агрегации поверхностно-активных веществ. В целом, предпочтительно, чтобы среда (M) представляла собой непрерывную фазу на основе воды, состоящую из одного или нескольких растворителей и/или добавок, которые являются смешиваемыми друг с другом, а также с водой в концентрациях, при которых они применяются.
Для целей описания настоящего изобретения, выражение "средство контроля радикальной полимеризации" означает соединение, которое способно увеличивать время жизни растущих полимерных цепей в реакции полимеризации и придавать полимеризации живой или контролируемый характер. Данное средство контроля, как правило, представляет собой средство обратимого переноса, применяемое в контролируемой радикальной полимеризации и известное под терминами RAFT или MADIX, которое обычно применяют в способе обратимого переноса по механизму присоединения-фрагментации, такое как описанное, например, в WO 96/30421, WO 98/01478, WO 99/35178, WO 98/58974, WO 00/75207, WO 01/42312, WO 99/35177, WO 99/31144, FR 2794464 или WO 02/26836.
В соответствии с предпочтительным вариантом осуществления средство контроля радикальной полимеризации, применяемое на этапе (E), представляет собой соединение, содержащее тиокарбонилтиогруппу -S(C=S)-. Таким образом, например, оно может представлять собой соединение, содержащее ксантогенатную группу (несущую функциональные группы -SC=S-O-), например, ксантогенат. Могут быть предусмотрены другие типы средства контроля (например, типа, применяемого в CRP или ATRP).
В соответствии с определенным вариантом осуществления средство контроля, применяемое на этапе (E), может представлять собой полимерную цепь, полученную в результате контролируемой радикальной полимеризации и несущую группу, которая способна контролировать радикальную полимеризацию (полимерная цепь "живого" типа, который представляет собой тип, хорошо известный сам по себе). Таким образом, например, средство контроля может представлять собой полимерную цепь (предпочтительно гидрофильную или диспергируемую в воде), функционализированную на конце цепи с помощью ксантогенатной группы или, в частности, содержащую группу -SC=S-, например, полученную в соответствии с методикой MADIX.
В качестве альтернативы, средство контроля, применяемое на этапе (E), представляет собой соединение, не относящееся к полимерам и несущее группу, обеспечивающую контроль радикальной полимеризации, в частности, тиокарбонилтиогруппу -S(C=S)-.
Исследования, которые проводили авторы настоящего изобретения в контексте настоящего изобретения, позволили на данный момент продемонстрировать, что мицеллярная радикальная полимеризация, осуществляемая в присутствии средства контроля радикальной полимеризации вышеуказанного типа, в дополнение к преимуществам, обычно наблюдаемым при мицеллярной полимеризации (то есть (i) контролю молярной доли гидрофобных единиц в полимерах и (ii) контролю количества гидрофобных единиц в каждом гидрофобном блоке) приводит к:
- контролю средней молекулярной массы и
- контролю распределения гидрофобных блоков в различных цепях;
- получению "живых" по природе полимерных цепей, обеспечивая возможность получения сложных полимеров контролируемого строения.
Такой эффект наиболее особенно заметен, когда применяемое средство контроля представляет собой соединение, которое является растворимым или диспергируемым в водной среде (M), применяемой на этапе (E), и/или когда данное средство контроля не способно проникать в мицеллы мицеллярного раствора. Такой эффект также можно наблюдать в случае, когда средство контроля не является растворимым/диспергируемым в среде (M) или когда средство контроля способно проникать в мицеллы.
В соответствии с конкретным вариантом осуществления средство контроля радикальной полимеризации, применяемое на этапе (E), представляет собой полимер, предпочтительно олигомер, водорастворимый или диспергируемый в воде по природе и несущий тиокарбонилтиогруппу -S(C=S)-, например, ксантогенатную группу -SC=S-O-. Такой полимер, способный действовать как в качестве средства контроля полимеризации, так и в качестве мономера на этапе (E), в остальной части описания также называют "форполимером". Как правило, такой форполимер получают в результате радикальной полимеризации гидрофильных мономеров в присутствии средства контроля несущего тиокарбонилтиогруппу -S(C=S)-, например, ксантогенат. Таким образом, например, в соответствии с предпочтительным вариантом осуществления, который проиллюстрирован в конце данного описания, средство контроля, применяемое на этапе (E), предпочтительно может представлять собой форполимер, несущий тиокарбонилтиогруппу -S(C=S)-, например, ксантогенатную группу -SC=S-O-, полученный после этапа (E0) контролируемой радикальной полимеризации перед этапом (E). На данном этапе (E0), как правило, могут приводить в контакт гидрофильные мономеры, предпочтительно идентичные применяемым на этапе (E), инициатор радикальной полимеризации и средство контроля, несущее тиокарбонилтиогруппу -S(C=S)-, например, ксантогенат.
Осуществление вышеуказанного этапа (E0) перед этапом (E) позволяет, в общих чертах, гидрофилизировать огромное число средств контроля, несущих функциональные тиокарбонилтиогруппы (например, ксантогенатов, которые по своей природе являются скорее гидрофобными), путем преобразования их из форполимеров, которые являются растворимыми или диспергируемыми в среде (M) из этапа (E). Предпочтительно, форполимер, синтезированный на этапе (E0), имеет короткую полимерную цепь, например, содержащую последовательность из менее 50 или даже менее 25 мономерных единиц, например, от 2 до 15.
Неожиданно было обнаружено, что условия этапа (E) позволяют объединить преимущества как контролируемой радикальной полимеризации, так и мицеллярной полимеризации. В данном контексте авторы настоящего изобретения, в частности, теперь продемонстрировали, что присутствие мицелл в среде для полимеризации не влияет на действие средств контроля, что делает возможным осуществление контролируемой полимеризации мономеров, присутствующих в водной среде, аналогично контролируемой радикальной полимеризации, осуществляемой в однородной среде, позволяя, таким образом, очень легко предсказывать и контролировать среднюю молекулярную массу синтезированного полимера (данная масса находится в пропорциональной зависимости от начальной концентрации средства контроля в среде, и она тем выше, чем меньше последняя, при этом данная концентрация задает количество растущих полимерных цепей). В то же время присутствие средства контроля не оказывает отрицательное влияние на какой-либо полезный эффект, наблюдаемый при полимеризации, то есть точный контроль размера гидрофобных блоков.
Дополнительно к данному контролю полимеризации мономеров, который не достигается при большинстве традиционных способов мицеллярной полимеризации, выполнение этапа (E) способа согласно настоящему изобретению также позволяет, снова совершенно неожиданно, получить доступ к полимерам как большого, так и контролируемого размера, что оказывается очень неожиданным ввиду максимальных размеров, которые могут быть достигнуты в настоящее время при помощи способов контролируемой радикальной полимеризации или мицеллярной радикальной полимеризации в отсутствие средств контроля.
В условиях этапа (E) оказывается возможным контроль среднечисловой молярной массы полимеров до очень высоких значений; таким образом, в соответствии с определенным вариантом осуществления полимеры, синтезированные согласно способу по настоящему изобретению, могут иметь молекулярную массу более 300000 г/моль. В частности, за счет корректировки начальной концентрации средства контроля в среде (M) этап (E) может, как правило, привести к синтезу блок-сополимера с молекулярной массой Mn более 400000 г/моль. В соответствии с предпочтительным вариантом осуществления способа по настоящему изобретению на этапе (E) начальную концентрацию средства контроля в среде выбирают таким образом, чтобы средняя молекулярная масса синтезированного гидрофильного блок-сополимера представляла собой среднечисловую молекулярную массу Mn, превышающую или равную 500000 г/моль, например, от 500000 до 1000000 г/моль, при этом возможно достижение размеров до 2000000.
Способ по настоящему изобретению также позволяет получить полимеры меньшей массы. В соответствии с предпочтительным вариантом осуществления синтезированный полимер представляет собой полимер с массой от 1000 до 100000 г/моль и предпочтительно от 2000 до 25000 г/моль. Как правило, такие полимеры с меньшей массой могут быть получены при концентрации ниже их критической концентрации перекрывания. С учетом их небольших размеров такие полимеры могут диффундировать на границах раздела фаз и принимать участие в модификации свойств этих границ раздела фаз или поверхностей.
Независимо от размера полимеров, синтезированных на этапе (E), эти полимеры также имеют в высокой степени контролируемую микроструктуру с цепями, практически все из которых являются одинаковыми, содержащую гидрофобные блоки, распределенные, по сути, одинаково от одной полимерной цепи к другой. Данная однородность распределения гидрофобных блоков от одной цепи к другой позволяет получить множество полимеров, все из которых обладают одинаковыми свойствами, что позволяет получить композиции, обладающие превосходными целевыми и воспроизводимыми свойствами, что является преимуществом для определенных путей применения полимеров, например, когда их пытаются использовать для достижения эффекта точно измеренного повышения вязкости. Полимеры, полученные в соответствии с настоящим изобретением, отличаются этим от полимеров, полученных при мицеллярной полимеризации, которые чаще всего имеют очень широкое и очень неоднородное распределение гидрофобных блоков в различных цепях.
Таким образом, осуществление этапа (E) обеспечивает доступ, в частности, к новым и предпочтительным полимерам. В соответствии со вторым аспектом настоящего изобретения эти полимеры представляют собой другой объект настоящего изобретения. С учетом выполнения условий этапа (E) такие полимеры обычно имеют линейную структуру с гидрофобными блоками, расположенными в неизменном градиенте, то есть при постоянно снижающейся или постоянно возрастающей концентрации от начала до конца образуемой полимерной цепи, что, в частности, объясняется тем фактом, что гидрофобные мономеры, присутствующие в мицеллярном растворе, через некоторое время становятся истощенными.
Полимеры, полученные в соответствии с настоящим изобретением, могут применяться во многих областях. Наиболее особенно они могут применяться в качестве поверхностно-активных веществ и/или модификаторов реологических свойств, в частности, в качестве средств повышения вязкости или загустителей, а именно, в водных средах.
В соответствии с третьим аспектом объектом настоящего изобретения также является такое определенное применение конкретных полимеров, полученных в соответствии с настоящим изобретением. Объектом настоящего изобретения также являются способы модификации водных сред с применением таких полимеров в качестве модификаторов реологических свойств. Настоящее изобретение также относится к водным композициям, содержащим полимеры согласно настоящему изобретению, которые можно применять, в частности, при разработке месторождений нефти и/или газа. Объектом настоящего изобретения также являются способы применения водных композиций данного типа для разработки месторождений нефти и/или газа, в частности, способы с применением циркуляции или размещения такой композиции в скважине.
Конкретные полимеры, полученные в соответствии с настоящим изобретением, могут также применяться для осуществления функционализации поверхности (гидрофилизации или гидрофобизации в зависимости от природы поверхности с учетом амфифильной природы полимеров), стабилизации границ раздела фаз масло/вода или вода/газ.
Различные признаки и варианты осуществления настоящего изобретения далее будут описаны более подробно.
Средство контроля радикальной полимеризации
Средство контроля, применяемое на этапе (E) или, при необходимости, на этапе (E0) способа по настоящему изобретению, предпочтительно представляет собой соединение, несущее тиокарбонилтиогруппу -S(C=S)-. В соответствии с конкретным вариантом осуществления настоящего изобретения средство контроля может нести несколько тиокарбонилтиогрупп. Оно необязательно может представлять собой полимерную цепь, несущую такую группу.
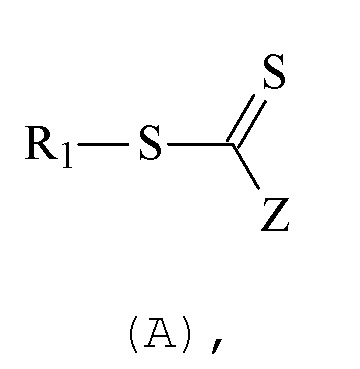
Таким образом, данное средство контроля может, например, соответствовать нижеприведенной формуле (A):
в которой
- Z представляет:
. атом водорода,
. атом хлора,
. необязательно замещенный алкильный или необязательно замещенный арильный радикал,
. необязательно замещенный гетероцикл,
. необязательно замещенный алкилтиорадикал,
. необязательно замещенный арилтиорадикал,
. необязательно замещенный алкоксирадикал,
. необязательно замещенный арилоксирадикал,
. необязательно замещенный аминорадикал,
. необязательно замещенный гидразиновый радикал,
. необязательно замещенный алкоксикарбонильный радикал,
. необязательно замещенный арилоксикарбонильный радикал,
. необязательно замещенный карбоксильный или ацилоксильный радикал,
. необязательно замещенный ароилоксирадикал,
. необязательно замещенный карбамоильный радикал,
. цианорадикал,
. диалкильный радикал или диарилфосфонаторадикал,
. диалкилфосфинато- или диарилфосфинаторадикал, или
. полимерную цепь,
и
- R1 представляет:
. необязательно замещенную алкильную, ацильную, арильную, аралкильную, алкеновую или алкиновую группу,
. насыщенный или ненасыщенный, ароматический, необязательно замещенный карбоцикл или гетероцикл, или
. полимерную цепь, которая предпочтительно является гидрофильной или диспергируемой в воде, когда средство применяют на этапе (E).
Группы R1 или Z, когда они являются замещенными, могут быть замещенными необязательно замещенными фенильными группами, необязательно замещенными ароматическими группами, насыщенными или ненасыщенными карбоциклами, насыщенными или ненасыщенными гетероциклами или группами, выбранными из следующих: алкоксикарбонил или арилоксикарбонил (-COOR), карбоксил (-COOH), ацилокси (-O2CR), карбамоил (-CONR2), циано (-CN), алкилкарбонил, алкиларилкарбонил, арилкарбонил, арилалкилкарбонил, фталимидо, малеимидо, сукцинимидо, амидино, гуанидино, гидроксил (-OH), амино (-NR2), галоген, перфторалкил CnF2n+1, аллил, эпокси, алкокси (-OR), S-алкил, S-арил, группами гидрофильной или ионной природы, такими как соли щелочных металлов карбоновых кислот, соли щелочных металлов сульфокислоты, цепями полиалкиленоксида (PEO, PPO), катионными заместителями (солями четвертичного аммония), при этом R представляет собой алкильную или арильную группу или полимерную цепь.
Для средств контроля с формулой (A), применяемых на этапе (E), как правило, предпочтительно, чтобы группа R1 имела гидрофильную природу. Предпочтительно она представляет собой водорастворимую или диспергируемую в воде полимерную цепь.
Группа R1, в качестве альтернативы, может быть амфифильной, то есть иметь как гидрофильную, так и липофильную природу. Предпочтительно R1 не является гидрофобной.
В отношении средств контроля с формулой (A), применяемых на этапе (E0), R1 может, как правило, представлять собой замещенную или незамещенную, предпочтительно замещенную, алкильную группу. Средство контроля с формулой (A), применяемое на этапе (E0), может, однако, содержать другие типы группы R1, в частности, кольцо или полимерную цепь.
Необязательно замещенные алкильные, ацильные, арильные, аралкильные или алкиновые группы обычно содержат от 1 до 20 атомов углерода, предпочтительно от 1 до 12 и более предпочтительно от 1 до 9 атомов углерода. Они могут быть линейными или разветвленными. Они могут также быть замещены атомами кислорода, в частности, в виде сложных эфиров, или атомами серы или азота.
Среди алкильных радикалов можно упомянуть, в частности, метильный, этильный, пропильный, бутильный, пентильный, изопропильный, трет-бутильный, пентильный, гексильный, октильный, децильный или додецильный радикалы.
Алкиновые группы представляют собой радикалы, обычно содержащие от 2 до 10 атомов углерода, при этом они содержат по меньшей мере одну ацетиленовую ненасыщенную группу, такую как ацетиленильный радикал.
Ацильная группа представляет собой радикал, обычно содержащий от 1 до 20 атомов углерода, с карбонильной группой.
Среди арильных радикалов можно упомянуть, в частности, фенильный радикал, необязательно замещенный, в частности, нитро- или гидроксильной функциональной группой.
Среди аралкильных радикалов можно упомянуть, в частности, бензильный или фенэтильный радикал, необязательно замещенный, в частности, нитро- или гидроксильной функциональной группой.
Когда R1 или Z представляют собой полимерную цепь, данная полимерная цепь может быть получена в результате радикальной или ионной полимеризации или получена в результате поликонденсации.
Предпочтительно соединения, несущие ксантогенатную -S(C=S)O-, тритиокарбонатную, дитиокарбаматную или дитиокарбазатную функциональную группу, например, несущие O-этилксантогенатную функциональную группу формулы -S(C=S)OCH2CH3, применяются в качестве средства контроля на этапе (E), а также, при необходимости, на этапе (E0).
При осуществлении этапа (E0) особенно предпочтительно применять в качестве средств контроля на данном этапе соединение, выбранное из ксантогенатов, тритиокарбонатов, дитиокарбаматов и дитиокарбазатов. Ксантогенаты, в частности, несущие O-этилксантогенатную функциональную группу -S(C=S)OCH2CH3, такие как O-этил-S-(1-метоксикарбонилэтил)ксантогенат CH3CH(CO2CH3)S(C=S)OEt, оказались наиболее особенно предпочтительными. Другим возможным средством контроля на этапе (E0) является дибензилтритиокарбонат формулы PhCH2S(C=S)SCH2Ph (в которой Ph = фенил).
"Живые" форполимеры, полученные на этапе (E0) с применением вышеуказанных средств контроля, оказались особенно предпочтительными при осуществлении этапа (E).
Гидрофильные мономеры
Способ согласно настоящему изобретению можно использовать с очень большим количеством гидрофильных мономеров.
Как правило, мономеры могут включать мономеры, выбранные из
- этиленненасыщенных карбоновых кислот, сульфокислот и фосфоновых кислот и/или их производных, таких как акриловая кислота (AA), метакриловая кислота, этакриловая кислота, α-хлоракриловая кислота, кротоновая кислота, малеиновая кислота, малеиновый ангидрид, итаконовая кислота, цитраконовая кислота, мезаконовая кислота, глутаконовая кислота, аконитовая кислота, фумаровая кислота, сложные моноэфиры моноэтиленненасыщенной дикарбоновой кислоты, содержащие от 1 до 3 и предпочтительно от 1 до 2 атомов углерода, например, монометилмалеат, винилсульфокислота, (мет)аллилсульфокислота, сульфоэтилакрилат, сульфоэтилметакрилат, сульфопропилакрилат, сульфопропилметакрилат, 2-гидрокси-3-акрилоилоксипропилсульфокислота, 2-гидрокси-3-метакрилоилоксипропилсульфокислота, стиролсульфокислоты, 2-акриламидо-2-метилпропансульфокислота, винилфосфоновая кислота, α-метилвинилфосфоновая кислота и аллилфосфоновая кислота;
- сложных эфиров α,β-этиленненасыщенных одноосновных и двухосновных карбоновых кислот и C2-C3-алкандиолов, например, 2-гидроксиэтилакрилата, 2-гидроксиэтилметакрилата, 2-гидроксиэтилэтакрилата, 2-гидроксипропилакрилата, 2-гидроксипропилметакрилата, 3-гидроксипропилакрилата, 3-гидроксипропилметакрилата и (мет)акрилатов полиалкиленгликоля;
- амидов α,β-этиленненасыщенных одноосновных кислот, а также их N-алкильных и N,N-диалкильных производных, таких как акриламид, метакриламид, N-метил(мет)акриламид, N-этил(мет)акриламид, N-пропил(мет)акриламид, N,N-диметил(мет)акриламид, N,N-диэтил(мет)акриламид, морфолинил(мет)акриламид и метилолакриламид (акриламид и N,N-диметил(мет)акриламид оказались особенно предпочтительными);
- N-виниллактамов и их производных, например, N-винилпирролидона и N-винилпиперидона;
- N-виниламидных соединений с открытой цепью, например, N-винилформамида, N-винил-N-метилформамида, N-винилацетамида, N-винил-N-метилацетамида, N-винил-N-этилацетамида, N-винилпропионамида, N-винил-N-метилпропионамида и N-винилбутирамида;
- сложных эфиров α,β-этиленненасыщенных одноосновных и двухосновных карбоновых кислот и аминоспиртов, например, N,N-диметиламинометил(мет)акрилата, N,N-диметиламиноэтил(мет)акрилата, N,N-диэтиламиноэтилакрилата и N,N-диметиламинопропил(мет)акрилата;
- амидов α,β-этиленненасыщенных одноосновных и двухосновных карбоновых кислот и диаминов, содержащих по меньшей мере одну первичную или вторичную аминогруппу, таких как N-[2-(диметиламино)этил]акриламид, N[2-(диметиламино)этил]метакриламид, N-[3-(диметиламино)пропил]акриламид, N-[3-(диметиламино)пропил]метакриламид, N-[4-(диметиламино)бутил]акриламид и N-[4-(диметиламино)бутил]метакриламид;
- N-диаллиламинов, N,N-диаллил-N-алкиламинов, их солей присоединения кислоты и их продуктов кватернизации, причем алкил, применяемый в данной случае, преимущественно представляет собой C1-C3-алкил;
- соединений N,N-диаллил-N-метиламина и N,N-диаллил-N,N-диметиламмония, например, хлоридов и бромидов;
- азотсодержащих гетероциклов, замещенных винилом и аллилом, например, N-винилимидазола, N-винил-2-метилимидазола, гетероароматических соединений, замещенных винилом и аллилом, например 2- и 4-винилпиридина, 2- и 4-аллилпиридина, а также их солей;
- сульфобетаинов, а также
- смесей и комбинаций двух или нескольких вышеуказанных мономеров.
В соответствии с определенным вариантом осуществления такие мономеры могут, в частности, включать акриловую кислоту (AA). В соответствии с возможным вариантом осуществления все мономеры представляют собой акриловую кислоту, но также может предусматриваться применение смеси, содержащей, помимо прочего, акриловую кислоту в виде смеси с другими гидрофильными мономерами, в качестве мономеров.
В соответствии с предпочтительным вариантом осуществления гидрофильные мономеры из этапа (E) включают (мет)акриловую кислоту и/или мономеры (мет)акриламидо.
Для целей описания настоящего изобретения выражение "(мет)акриловая кислота" включает метакриловую кислоту и акриловую кислоту, а также их смеси.
Подобным образом, для целей описания настоящего изобретения, выражение "(мет)акрилат" включает метакрилат и акрилат, а также их смеси.
Подобным образом, для целей описания настоящего изобретения, выражение "(мет)акриламид/(мет)акриламидо" включает метакриламид/метакриламидо и акриламид/акриламидо, а также их смеси.
Мономеры, содержащие кислотные группы, могут применяться для полимеризации в форме свободной кислоты или в частично или полностью нейтрализованной форме. Для нейтрализации могут применяться, например, KOH, NaOH, аммиак или другие основания.
В соответствии с другим определенным вариантом осуществления мономеры, применяемые в способе по настоящему изобретению, представляют собой, в частности, акриловую кислоту, метакриловую кислоту и/или их соли и/или их смеси.
В соответствии с другим вариантом осуществления мономеры, применяемые на этапе (E), включают (мет)акриламидные мономеры или, в общем, мономеры (мет)акриламидо (и, как правило, состоят из них), включая
- мономеры акриламидо, то есть акриламид, его сульфонатное производное (AMPS), соединение четвертичного аммония (APTAC) и сульфопропилдиметиламмонийпропилакриламид;
- мономеры метакриламидо, такие как сульфопропилдиметиламмонийпропилметакриламид (SPP) и сульфогидроксипропилдиметиламмонийпропилметакриламидо.
В соответствии с определенным вариантом осуществления мономеры из этапа (E) представляют собой акриламиды. Акриламид, применяемый на этапе (E), предпочтительно представляет собой акриламид, который не стабилизирован с помощью меди. В случае присутствия меди предпочтительно вводить средство, образующее комплекс с медью, такое как EDTA, при необходимости, предпочтительно в относительном содержании от 20 до 2000 частей на миллион. Когда на этапе (E) применяют акриламиды, то они могут, как правило, применяться в форме порошка или водного раствора (возможно, но не обязательно, стабилизированного монометиловым эфиром гидрохинона HQME или, альтернативно, солями меди (предпочтительно с добавлением EDTA, при необходимости)).
Независимо от их точной природы, мономеры из этапа (E) могут применяться при относительно высоких концентрациях, как правило, при концентрациях, которые могут быть достаточными для обеспечения образования геля, если этап (E) осуществляли в отсутствие средства контроля. Авторы настоящего изобретения теперь неожиданно продемонстрировали, что полимеризация на этапе (E) в случае необходимости может осуществляться при условиях, которые соответствуют таковым для полимеризации в геле, и что это необязательно приводит к гелеобразованию в реакционной среде в ходе полимеризации благодаря наличию средства контроля. Независимо от того, наблюдается ли гелеобразование в среде или нет, этап (E) способствует полимеризации контролируемого типа, в отличие от полимеризации, осуществляемой без дополнительного средства контроля.
Как правило, начальная концентрация мономера в реакционной среде из этапа (E) может доходить до 40% по массе или вплоть до 50% по массе, при этом данная концентрация, как правило, остается при значении менее 30% по массе относительно общей массы реакционной среды. Например, начальная концентрация мономера в реакционной среде из этапа (E) составляет 0,5%-35% и, в частности, 1%-20% по массе относительно общей массы реакционной среды.
В соответствии с конкретным вариантом осуществления гидрофильные мономеры, применяемые на этапе (E), представляют собой чувствительные к нагреванию макромономеры, которые являются нерастворимыми в воде при температуре выше определенной (точки помутнения), но являются растворимыми при более низкой температуре, при этом этап (E) осуществляют при температуре выше точки помутнения. Макромономеры данного типа, как правило, содержат полимеризуемую функциональную группу типа акриламидо и боковую цепь, включающую последовательности этиленоксида или пропиленоксида (со случайным чередованием или в блоках), или, альтернативно, таковую на основе N-изопропилакриламида или N-винилкапролактама. В данном варианте осуществления, в частности, обеспечивается доступ к получению полимеров, обладающих свойствами загущения при нагревании, которые могут применяться, например, в нефтеперерабатывающей промышленности.
Предпочтительно на этапе (E) все гидрофильные мономеры растворены и/или диспергированы в водной среде (M).
Гидрофобные мономеры
Такие мономеры применяются на этапе (E) в виде мицеллярного раствора, то есть содержащего в диспергированной форме в среде (M) мицеллы, содержащие такие гидрофобные мономеры. На этапе (E) может быть предусмотрен любой мономер гидрофобной природы при условии, что он может включаться в мицеллы данного типа.
В качестве неограничивающих примеров гидрофобных мономеров, которые могут применяться в соответствии с настоящим изобретением, в частности, могут быть указаны:
- винилароматические мономеры, такие как стирол, α-метилстирол, пара-хлорметилстирол, винилтолуол, 2-метилстирол, 4-метилстирол, 2-(н-бутил)стирол или 4-(н-децил)стирол (стирол, в частности, оказался особенно предпочтительным);
- галогенированные виниловые соединения, такие как галогениды винила или винилидена, например, хлориды или фториды винила или винилидена, соответствующие формуле RbRcC=CX1X2,
где X1=F или Cl,
X2=H, F или Cl,
каждый из Rb и Rc независимо представляет
- H, Cl, F; или
- алкильную группу, предпочтительно хлорированную и/или фторированную, более предпочтительно перхлорированную или перфторированную;
- сложные эфиры α,β-этиленненасыщенных одноосновных или дикарбоновых кислот и C2-C30-алканолов, например, метилэтакрилат, этил(мет)акрилат, этилэтакрилат, н-пропил(мет)акрилат, изопропил(мет)акрилат, н-бутил(мет)акрилат, втор-бутил(мет)акрилат, трет-бутил(мет)акрилат, трет-бутилэтакрилат, н-гексил(мет)акрилат, н-гептил(мет)акрилат, н-октил(мет)акрилат, 1,1,3,3-тетраметилбутил(мет)акрилат, этилгексил(мет)акрилат, н-нонил(мет)акрилат, н-децил(мет)акрилат, н-ундецил(мет)акрилат, тридецил(мет)акрилат, миристил(мет)акрилат, пентадецил(мет)акрилат, пальмитил(мет)акрилат, гептадецил(мет)акрилат, нонадецил(мет)акрилат, арахидил(мет)акрилат, бегенил(мет)акрилат, лигноцерил(мет)акрилат, церотинил(мет)акрилат, мелиссинил(мет)акрилат, пальмитолеил(мет)акрилат, олеил(мет)акрилат, линолеил(мет)акрилат, линоленоил(мет)акрилат, стеарил(мет)акрилат и лаурил(мет)акрилат и их смеси;
- сложные эфиры винилового или аллилового спирта и одноосновных карбоновых кислот C1-C30, например, винилформиат, винилацетaт, винилпропионат, винилбутирaт, виниллаурат, винилстеарат, винилпропионат или винилверсатат и их смеси;
- этиленненасыщенные нитрилы, такие как акрилoнитрил или метакрилoнитрил, и их смеси;
- сложные эфиры α,β-этиленненасыщенных одноосновных или двухосновных карбоновых кислот и C3-C30-алкандиолов, например, 2-гидроксипропилакрилат, 2-гидроксипропилметакрилат, 3-гидроксипропилакрилат, 3-гидроксипропилметакрилат, 3-гидроксибутилакрилат, 3-гидроксибутилметакрилат, 4-гидроксибутилакрилат, 4-гидроксибутилметакрилат, 6-гидроксигексилакрилат, 6-гидроксигексилметакрилат, 3-гидрокси-2-этилгексилакрилат и 3-гидрокси-2-этилгексилметакрилат;
- первичные амиды α,β-этиленненасыщенных одноосновных и двухосновных карбоновых кислот и N-алкильные и N,N-диалкильные производные, такие как N-пропил(мет)акриламид, N-(н-бутил)(мет)акриламид, N-(трет-бутил)(мет)акриламид, N-(н-октил)(мет)акриламид, N-(1,1,3,3-тетраметилбутил)(мет)акриламид, N-этилгексил(мет)акриламид, N-(н-нонил)(мет)акриламид, N-(н-децил)(мет)акриламид, N-(н-ундецил)(мет)акриламид, N-тридецил(мет)акриламид, N-миристил(мет)акриламид, N-пентадецил(мет)акриламид, N-пальмитил(мет)акриламид, N-гептадецил(мет)акриламид, N-нонадецил(мет)акриламид, N-арахидил(мет)акриламид, N-бегенил(мет)акриламид, N-лигноцерил(мет)акриламид, N-церотинил(мет)акриламид, N-мелиссинил(мет)акриламид, N-пальмитолеил(мет)акриламид, N-олеил(мет)акриламид, N-линолеил(мет)акриламид, N-линоленоил(мет)акриламид, N-стеарил(мет)акриламид и N-лаурил(мет)акриламид;
- N-виниллактамы и их производные, такие как N-винил-5-этил-2-пирролидон, N-винил-6-метил-2-пиперидон, N-винил-6-этил-2-пиперидон, N-винил-7-метил-2-капролактам и N-винил-7-этил-2-капролактам;
- сложные эфиры α,β-этиленненасыщенных одноосновных и двухосновных карбоновых кислот и аминоспиртов, например, N,N-диметиламиноциклогексил(мет)акрилат;
- амиды α,β-этиленненасыщенных одноосновных и двухосновных карбоновых кислот и диаминов, содержащих по меньшей мере одну первичную или вторичную аминогруппу, такие как N-[4-(диметиламино)бутил]акриламид, N-[4-(диметиламино)бутил]метакриламид, N-[2-(диэтиламино)этил]акриламид, N-[4-(диметиламино)циклогексил]акриламид и N-[4-(диметиламино)циклогексил]метакриламид; и
- моноолефины и неароматические углеводороды C2-C8, содержащие по меньшей мере две сопряженные двойные связи, например, этилен, пропилен, изобутилен, изопрен или бутадиен.
В соответствии с предпочтительным вариантом осуществления применяемые гидрофобные мономеры в соответствии с настоящим изобретением могут быть выбраны из:
- α,β-ненасыщенных сложных эфиров C1-C30-алкила и предпочтительно C4-C22-алкила, в частности, алкилакрилатов и метакрилатов, например, метил-, этил-, бутил-, 2-этилгексил-, изооктил-, лаурил-, изодецил- или стеарилакрилатов или метакрилатов (лаурилметакрилат, в частности, оказался особенно предпочтительным);
- α,β-ненасыщенных амидов C1-C30-алкила и предпочтительно C4-C22-алкила, в частности, алкилакриламидов и метакриламидов, например, метил-, этил-, бутил-, 2-этилгексил-, изооктил-, лаурил-, изодецил- или стеарилакриламида или метакриламида (лаурилметакриламид, в частности, оказался особенно предпочтительным);
- сложных эфиров винилового или аллилового спирта и насыщенных карбоновых кислот, таких как винил- или аллилацетaт, пропионат, версатат или стеарат;
- α,β-ненасыщенных нитрилов, содержащих 3-12 атомов углерода, таких как акрилoнитрил, или
- акрилoнитрила,
- α-олефинов и диенов с сопряженными двойными связями;
- смесей и комбинаций двух или нескольких вышеуказанных мономеров.
Предпочтительно мицеллы мицеллярного раствора из этапа (E) не содержат каких-либо мономеров, гидрофильных или диспергируемых в воде по природе. Кроме того, предпочтительно все гидрофобные мономеры, применяемые на этапе (E), содержатся в мицеллах мицеллярного раствора.
Инициирование и осуществление радикальной полимеризации на этапах (E) и (E0)
При применении на этапе (E) инициатор радикальной полимеризации предпочтительно является водорастворимым или диспергируемым в воде. Помимо данного предпочтительного условия, любой инициатор радикальной полимеризации (источник свободных радикалов), который сам по себе известен и приспособлен к условиям, выбранным для таких этапов, может применяться на этапе (E) и этапе (E0) способа по настоящему изобретению.
Таким образом, применяемый инициатор радикальной полимеризации в соответствии с настоящим изобретением может быть выбран, например, из инициаторов, традиционно применяемых при радикальной полимеризации. Он может представлять собой, например, один из следующих инициаторов:
- пероксиды водорода, такие как трет-бутилгидропероксид, гидропероксид кумола, трет-бутилпероксиацетaт, трет-бутилпероксибензоат, трет-бутилпероксиоктоат, трет-бутилпероксинеодеканоат, трет-бутилпероксиизобутират, лауроилпероксид, трет-амилпероксипивалат, трет-бутилпероксипивалат, дикумилпероксид, бензоилпероксид, персульфат калия, персульфат аммония,
- азосоединения, такие как 2-2ʹ-азобис(изобутирoнитрил), 2,2ʹ-азобис(2-бутаннитрил), 4,4ʹ-азобис(4-пентановая кислота), 1,1ʹ-азобис(циклогексанкарбонитрил), 2-(трет-бутилазо)-2-цианопропан, 2,2ʹ-азобис[2-метил-N-(1,1)-бис(гидроксиметил)-2-гидроксиэтил]пропионамид, 2,2ʹ-азобис(2-метил-N-гидроксиэтил]пропионамид, 2,2ʹ-азобис(N,Nʹ-диметиленизобутирaмидин)дихлорид, 2,2ʹ-азобис(2-амидинопропан)дихлорид, 2,2ʹ-азобис(N,Nʹ-диметиленизобутирамид), 2,2ʹ-азобис(2-метил-N-[1,1-бис(гидроксиметил)-2-гидроксиэтил]пропионамид), 2,2ʹ-азобис(2-метил-N-[1,1-бис(гидроксиметил)этил]пропионамид), 2,2ʹ-азобис[2-метил-N-(2-гидроксиэтил)пропионамид], дигидрат 2,2ʹ-азобис(изобутирамида),
- окислительно-восстановительные системы, включающие комбинации, такие как
- смеси пероксида водорода, алкилпероксида, перэфиров, перкарбонатов и т.п. и любых солей железа, солей титана, формальдегид-сульфоксилата цинка или формальдегид-сульфоксилата натрия, а также восстанавливающих сахаров,
- персульфаты, пербораты или перхлораты щелочных металлов или аммония в комбинации с бисульфитом щелочного металла, таким как метабисульфит натрия, и восстанавливающими сахарами и
- персульфаты щелочных металлов в комбинации с арилфосфиновой кислотой, такой как бензолфосфоновая кислота и т.п., и восстанавливающими сахарами.
Как правило, количество применяемого инициатора предпочтительно определяют таким образом, чтобы количество образованных радикалов составляло не более 50 мол. % и предпочтительно не более 20 мол. % относительно количества средства контроля или средства переноса.
Наиболее особенно, на этапе (E), как правило, предпочтительным оказывается применение радикального инициатора окислительно-восстановительного типа, который обладает, помимо прочего, преимуществом, заключающимся в отсутствии необходимости нагревания реакционной среды (без термического инициирования), и в отношении которого в данный момент авторы настоящего изобретения также обнаружили, что он оказался подходящим для мицеллярной полимеризации на этапе (E).
Таким образом, инициатор радикальной полимеризации, применяемый на этапе (E), может, как правило, представлять собой окислительно-восстановительный инициатор, как правило, не нуждающийся в нагревании для термического инициирования. Как правило, он представляет собой смесь по меньшей мере одного окислителя с по меньшей мере одним восстановителем.
Окислитель, присутствующий в данной окислительно-восстановительной системе, предпочтительно представляет собой водорастворимое средство. Данный окислитель может быть выбран, например, из пероксидов, таких как пероксид водорода, трет-бутилгидропероксид, гидропероксид кумола, трет-бутилпероксиацетaт, трет-бутилпероксибензоат, трет-бутилпероксиоктоат, трет-бутилпероксинеодеканоат, трет-бутилпероксиизобутират, лауроилпероксид, трет-амилпероксипивалат, трет-бутилпероксипивалат, дикумилпероксид, бензоилпероксид, персульфат натрия, персульфат калия, персульфат аммония или бромат калия.
Восстановитель, присутствующий в данной окислительно-восстановительной системе, также предпочтительно представляет собой водорастворимое средство. Данный восстановитель может, как правило, быть выбран из формальдегид-сульфоксилата натрия (в частности, в форме его дигидрата, известного под названием Rongalit, или в форме ангидрида), аскорбиновой кислоты, эриторбиновой кислоты, сульфитов, бисульфитов или метасульфитов (в частности, сульфитов, бисульфитов или метасульфитов щелочных металлов), нитрило-трис-пропионамидов и третичных аминов и этаноламинов (которые предпочтительно являются водорастворимыми).
Возможные окислительно-восстановительные системы включают комбинации, такие как:
- смеси водорастворимых персульфатов и водорастворимых третичных аминов,
- смеси водорастворимых броматов (например, броматов щелочных металлов) и водорастворимых сульфитов (например, сульфитов щелочных металлов),
- смеси пероксида водорода, алкилпероксида, перэфиров, перкарбонатов и т.п. и любых солей железа, солей титана, формальдегид-сульфоксилата цинка или формальдегид-сульфоксилата натрия, а также восстанавливающих сахаров,
- персульфаты, пербораты или перхлораты щелочных металлов или аммония в комбинации с бисульфитом щелочного металла, таким как метабисульфит натрия, и восстанавливающими сахарами и
- персульфаты щелочных металлов в комбинации с арилфосфиновой кислотой, такой как бензолфосфоновая кислота и т.п., и восстанавливающими сахарами.
Предпочтительная окислительно-восстановительная система включает комбинацию персульфата аммония и формальдегид-сульфоксилата натрия (и предпочтительно состоит из нее).
В общем, и в частности, в случае применения окислительно-восстановительной системы типа персульфат аммония/формальдегид-сульфоксилат натрия, оказалось предпочтительным, чтобы реакционная среда из этапа (E) не содержала медь. В случае присутствия меди, как правило, желательно добавлять средство, образующее комплекс с медью, такое как EDTA, в количестве, достаточном для того, чтобы скрыть ее присутствие.
Независимо от природы применяемого инициатора радикальная полимеризация на этапе (E0) может осуществляться в любой подходящей физической форме, например, в растворе в воде или в растворителе, например, в спирте или THF, или в эмульсии в воде (способ с применением латекса), в массе, при необходимости, путем контроля температуры и/или pH так, чтобы получить растворимые и/или нерастворимые формы жидкостей.
После осуществления этапа (E), учитывая конкретный способ применения средства контроля, получают полимеры, функционализированные с помощью переносимых групп ("живые" полимеры). Такая "живая" природа позволяет при желании применять такие полимеры в последующей реакции полимеризации посредством методики, которая сама по себе хорошо известна. Альтернативно, если необходимо, возможно осуществлять деактивацию или разрушение групп переноса, например, путем гидролиза, озонолиза или реакции с аминами, посредством способов, которые известны сами по себе. Таким образом, в соответствии с определенным вариантом осуществления способ по настоящему изобретению после этапа (E) может включать этап (E1) гидролиза, озонолиза или реакции с аминами, который способен обеспечить деактивацию и/или разрушение всех или некоторых переносимых групп, присутствующих в полимере, полученном на этапе (E).
Поверхностно-активные вещества
Для получения мицеллярного раствора гидрофобных мономеров, применяемого на этапе (E), может применяться любое подходящее поверхностно-активное вещество, при этом, без ограничения, возможно применение, например, поверхностно-активных веществ, выбранных из следующего списка:
- анионные поверхностно-активные вещества, которые могут быть выбраны из:
сульфонатов сложных алкиловых эфиров, например, формулы R-CH(SO3M)-CH2COORʹ, или сульфатов сложных алкиловых эфиров, например, формулы R-CH(OSO3M)-CH2COORʹ, в которой R представляет собой C8-C20- и предпочтительно C10-C16-алкильный радикал, Rʹ представляет собой C1-C6- и предпочтительно C1-C3-алкильный радикал, и M представляет собой катион щелочного металла, например, натрия, или катион аммония; при этом наиболее особенно можно упомянуть сульфонаты сложных метиловых эфиров, в которых радикал R представляет собой C14-C16;
алкилбензолсульфонатов, в частности, C9-C20, первичных или вторичных алкилсульфонатов, в частности, C8-C22, сульфонатов алкилглицерина;
алкилсульфатов, например, формулы ROSO3M, в которой R представляет C10-C24- и предпочтительно C12-C20-алкильный или гидроксиалкильный радикал; M представляет собой катион, определенный так же, как указано выше;
сульфатов алкиловых эфиров, например, формулы RO(OA)nSO3M, в которой R представляет C10-C24- и предпочтительно C12-C20-алкильный или гидроксиалкильный радикал; OA представляет этоксилированную и/или пропоксилированную группу; M представляет катион, определенный так же, как указано выше, n, как правило, находится в диапазоне от 1 до 4, например, сульфата лаурилового эфира с n=2;
сульфатов алкиламидов, например формулы RCONHRʹOSO3M, в которой R представляет C2-C22- и предпочтительно C6-C20-алкильный радикал, Rʹ представляет C2-C3-алкильный радикал, M представляет катион, определенный так же, как указано выше, а также их полиалкоксилированных (этоксилированных и/или пропоксилированных) производных (сульфатов алкиламидоэфиров);
солей насыщенных или ненасыщенных жирных кислот, например C8-C24 и предпочтительно C14-C20, и катиона щелочноземельного металла, N-ацил-N-алкилтауратов, алкилизетионатов, алкилсукцинаматов и алкилсульфосукцинатов, алкилглутаматов, сложных моноэфиров или сложных диэфиров сульфосукцината, N-ацилсаркозинатов и полиэтоксикарбоксилатов;
фосфатов сложных моноэфиров и сложных диэфиров, например, имеющих следующую формулу: (RO)x-P(=O)(OM)x, в которой R представляет необязательно полиалкоксилированный алкильный, алкиларильный, арилалкильный или арильный радикал, x и xʹ равны 1 или 2, при условии, что сумма x и xʹ равна 3, M представляет катион щелочноземельного металла;
- неионогенные поверхностно-активные вещества, которые могут быть выбраны из:
алкоксилированных жирных спиртов, например, лаурета-2, лаурета-4, лаурета-7, олета-20, алкоксилированных триглицеридов, алкоксилированных жирных кислот, алкоксилированных сложных эфиров сорбитана, алкоксилированных жирных аминов, алкоксилированных бис(1-фенилэтил)фенолов, алкоксилированных трис(1-фенилэтил)фенолов, алкоксилированных алкилфенолов, продуктов, полученных в результате конденсации этиленоксида с гидрофобным соединением, полученным в результате конденсации пропиленоксида с пропиленгликолем, таких как продукты Pluronic, реализуемые BASF; продуктов, полученных в результате конденсации этиленоксида с соединением, полученным в результате конденсации пропиленоксида с этилендиамином, таких как продукты Tetronic, реализуемые BASF; алкилполигликозидов, например, описанных в US 4565647, или алкилглюкозидов; амидов жирных кислот, например C8-C20, в частности, моноалканоламидов жирных кислот, например, кокамида MEA или кокамида MIPA;
- амфотерные поверхностно-активные вещества (истинные амфотерные формы, содержащие ионную группу и возможную ионную группу с противоположным зарядом, или цвиттер-ионные формы, одновременно содержащие два противоположных заряда), которые могут представлять собой:
бетаины в общем, в частности, карбоксибетаины, например, лаурилбетаин (Mirataine BB от компании Rhodia), или октилбетаин, или кокоилбетаин (Mirataine BB-FLA от Rhodia); амидоалкилбетаины, например, кокамидопропилбетаин (CAPB) (Mirataine BDJ от компании Rhodia или Mirataine BET C-30 от Rhodia);
сульфобетаины или сультаины, например, кокамидопропилгидроксисультаин (Mirataine CBS от компании Rhodia);
алкиламфоацетaты и алкиламфодиацетaты, например, содержащие цепь кокоила или лаурила (Miranol C2M Conc. NP, C32, L32 в частности, от компании Rhodia);
алкиламфопропионаты или алкиламфодипропионаты (Miranol C2M SF);
алкиламфогидроксипропилсультаины (Miranol CS),
алкиламиноксиды, например, лаураминоксид (INCI);
- катионные поверхностно-активные вещества, которые могут представлять собой необязательно полиэтоксилированные соли первичных, вторичных или третичных жирных аминов, соли четвертичного аммония, такие как хлориды или бромиды тетраалкиламмония, алкиламидоалкиламмония, триалкилбензиламмония, триалкилгидроксиалкиламмония или алкилпиридиния, производные имидазолинов и аминоксиды катионной природы; при этом примером катионного поверхностно-активного вещества является хлорид или бромид цетримония (INCI);
- поверхностно-активные вещества, применяемые в соответствии с настоящим изобретением, которые могут быть блок-coполимерами, содержащими по меньшей мере один гидрофильный блок и по меньшей мере один гидрофобный блок, отличный от гидрофильного блока, предпочтительно полученными согласно способу полимеризации, при котором:
(a0) в водной фазе приводят в контакт по меньшей мере один гидрофильный мономер (или, соответственно, гидрофобный мономер), по меньшей мере один источник свободных радикалов и по меньшей мере одно средство контроля радикальной полимеризации типа -S(C=S)-;
(a1) полимер, полученный после этапа (a0), приводят в контакт с по меньшей мере одним гидрофобным мономером (или, соответственно, гидрофильным мономером), отличным от мономера, применяемого на этапе (a0), и по меньшей мере одним источником свободных радикалов;
посредством чего получают диблок-coполимер.
Полимеры трехблочного типа или таковые, содержащие больше блоков, могут необязательно быть получены путем осуществления после этапа (a1) этапа (a2), на котором полимер, полученный после этапа (a1), приводят в контакт с по меньшей мере одним мономером, отличным от мономера, применяемого на этапе (a1), и по меньшей мере одним источником свободных радикалов; а также, в более общем смысле, путем осуществления (n+1) этапов такого типа, как вышеуказанные этапы (a1) и (a2), причем n представляет собой целое число, как правило, находящееся в диапазоне от 1 до 3, при этом на каждом этапе (an), где n≥1, полимер, полученный после этапа (an-1), приводят в контакт с по меньшей мере одним мономером, отличным от мономера, применяемого на этапе (an-1), и по меньшей мере одним источником свободных радикалов. Возможно, например, применение в соответствии с настоящим изобретением сополимеров типа, описанного в WO 03/068827, WO 03/068848 и WO 2005/021612.
Применение полимеров по настоящему изобретению
Полимеры, полученные после этапа (E) и после необязательного этапа (E1), описанные в предыдущем абзаце, являются, помимо прочего, применимыми для регуляции реологических свойств жидкой среды, в частности, водной среды. Они могут также применяться в качестве ассоциативных загустителей, в качестве средств, повышающих вязкость, гелеобразователей или модификаторов свойств поверхности или для изготовления наногибридных материалов. Они могут также применяться в качестве средств векторизации.
В данном контексте полимер в соответствии с настоящим изобретением может, в частности, применяться для загущения или адаптации реологических свойств очень большого числа композиций, например, композиций, свойства которых выражают принципы косметологии, фармацевтики, ветеринарии или защиты растений, или, альтернативно, например, в качестве моющих средств. Таким образом, полимер в соответствии с настоящим изобретением может применяться, например, для модификации реологических свойств косметической композиции, товаров бытовой химии, композиции моющего средства или состава, предназначенного для области сельского хозяйства.
Более конкретно, полимеры, полученные в соответствии с настоящим изобретением, оказались предпочтительными в качестве регуляторов реологических свойств в области добычи нефти и природного газа. Они могут применяться, в частности, для изготовления буровых жидкостей, для разрыва пласта, для интенсификации добычи и для повышения коэффициента извлечения нефти.
В области повышения коэффициента извлечения нефти (EOR) полимеры, полученные согласно способу по настоящему изобретению, как правило, обладают способностью к быстрой гидратации, а также свойствами хорошей впрыскиваемости и стабильности при сдвиге, в частности, с учетом контролируемого характера полимеризации, которая приводит к образованию партий полимера однородного состава и структуры с показателями полидисперсности, которые ниже таковых для "неконтролируемых" систем.
Кроме того, природа полимеров, которые могут быть синтезированы в соответствии с настоящим изобретением, является чрезвычайно изменчивой, что приводит к очень широкому выбору в отношении как главной цепи, так и наличия заместителей, которые могут быть осмысленно выбраны в зависимости от предполагаемых путей применения полимера.
Например, в случае применения в EOR для мономеров, являющихся составляющими полимера, преимущественным является придание ему устойчивости к действию высокой температуры. С этой целью полимеры, предназначенные для применения в EOR, могут быть, например, типа, полученного из мономеров, выбранных из акриламидо, метакриламидо, виниловых или аллиловых мономеров. Как правило, не является преимущественным применение акрилатов или метакрилатов из-за их чувствительности к гидролизу. Например, для улучшения термической устойчивости главной цепи могут применяться мономеры, такие как N-метилолакриламид, диметилакриламид, N-морфолинакриламидо, винилпирролидон, виниламид, производные акриламидо, такие как AMPS или APTAC, стиролсульфонат натрия и их производные или винилсульфонат натрия.
В соответствии с конкретным вариантом осуществления, который лучше всего подходит для применения в области EOR, полимеры несут функциональные группы, которые обеспечивают устойчивость к солям и которые противодействуют эффектам потери вязкости, которые часто встречаются в EOR при отсутствии таких функциональных групп в полимере. Полимеры в соответствии с настоящим изобретением, которые являются стабильными в отношении солей, могут быть, в частности, синтезированы путем осуществления одного или нескольких из следующих способов:
- применение дополнительных мономеров типа 3-акриламидо-3-метилбутаноата натрия (например, в соответствии с методикой, описанной в US 4584358);
- применение дополнительных мономеров типа сульфокислот или сульфонатов, например, AMPS (акриламидометилпропансульфокислота), и их солей (в частности, солей натрия) или стиролсульфоната и его солей;
- полученные полимеры могут быть полиамфолитического типа с гидрофильной главной цепью, содержащей смесь (i) мономерных единиц, несущих по меньшей мере один отрицательный заряд (например, сульфонаты вышеуказанного типа); и (ii) мономерных единиц, несущих по меньшей мере один положительный заряд (например, APTAC, MAPTAC, дикват, (дихлорид метакриламидопропилпентаметил-1,3-пропилен-2-оламмония), DADMAC (хлорид диаллилдиметиламмония), N-винилформамид (предшественник аминов, катионизируемый после гидролиза) или винилпиридин или его кватернизированное производное);
- применение дополнительных мономеров типа сульфобетаинов, например, пропилакриламида сульфопропилдиметиламмония, пропилметакриламида сульфопропилдиметиламмония (SPP), сульфогидроксипропилдиметиламмонийпропилметакриламидо (SHPP), 2-винил(3-сульфопропил)пиридинийбетаина, 4-винил(3-сульфопропил)пиридинийбетаина, 1-винил-3-(3-сульфопропил)имидазолийбетаина или сульфопропилметилдиаллиламмонийбетаина.
Различные аспекты и преимущества настоящего изобретения будут дополнительно проиллюстрированы с помощью нижеприведенных примеров, в которых полимеры получали согласно способу по настоящему изобретению.
ПРИМЕРЫ
В следующих примерах синтез полимеров осуществляли с применением:
- раствора A, содержащего "живой" форполимер P1 на основе поли(акриламида), полученный в условиях этапа (E0);
- раствора B, который представляет собой мицеллярный раствор лаурилметакрилата (LMA) и додецилсульфата натрия (SDS);
- раствора C, который представляет собой мицеллярный раствор лаурилакрилата (LA) и додецилсульфата натрия (SDS);
- раствора D, который представляет собой мицеллярный раствор лаурилметакриламида (LMAM) и додецилсульфата натрия (SDS); или
- раствора E, который представляет собой мицеллярный раствор LMAM и додецилсульфата натрия (SDS);
при этом их получают при следующих условиях.
Раствор A, содержащий форполимер P1
40 г водного 50% раствора акриламида (не содержащего медь), 29,9 г дистиллированной воды, 8,25 г O-этил-S-(1-метоксикарбонилэтил)ксантогената (CH3CH(CO2CH3))S(C=S)OEt, 26,6 г этанола и 0,537 мг инициатора V-50 (2,2ʹ-азобис(2-метилпропионамидин)дигидрохлорида) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C).
Смесь дегазировали путем барботирования азотом в течение 30 минут. Колбу затем помещали в масляную баню, термостатически поддерживаемую при 60°C, и реакцию полимеризации оставляли протекать при перемешивании в течение 3 часов при 60°C.
Достигали 100% превращения (определяемого с помощью 1H-ЯМР). Среднечисловая молярная масса форполимера P1, определенная с помощью 1H-ЯМР, составляла 750 г/моль.
Растворитель выпаривали в вакууме (роторный испаритель; 15 мбар, 50°C) и сушили в течение 120 минут при 50°C (содержание твердых веществ, измеренное после данного высушивания: 37%, 115°C, 60 мин.).
Затем добавляли дистиллированную воду таким образом, чтобы получить приблизительно 40% раствор форполимера, называемый в данном документе ниже раствором A.
Раствор B: мицеллярный раствор LMA/SDS
146,2 г 30% раствора SDS (nSDS=0,152 моль), 168,9 г дистиллированной воды, 3,9 г LMA (nLMA=0,0153 моль) и 5,4 г сульфата натрия помещали в круглодонную колбу на 500 мл при комнатной температуре (20°C).
Смесь перемешивали при помощи стержневого магнита в течение 6 часов, пока не получили чистый мицеллярный раствор.
В данном мицеллярном растворе nH=(0,0153×62)/0,145=6,5
(nSDS-CMC=0,152-0,007=0,145; NAgg=62).
Раствор C: мицеллярный раствор LA/SDS
146,2 г 30% раствора SDS (nSDS=0,152 моль), 169,0 г дистиллированной воды, 3,55 г LA (nLA=0,0153 моль) и 5,4 г сульфата натрия помещали в круглодонную колбу на 500 мл при комнатной температуре (20°C).
Смесь перемешивали при помощи стержневого магнита в течение 6 часов, пока не получили чистый мицеллярный раствор.
В данном мицеллярном растворе nH=(0,0153×62)/0,145=6,5
(nSDS-CMC=0,152-0,007=0,145; NAgg=62).
Раствор D: мицеллярный раствор LMAM/SDS
146,2 г 30% раствора SDS (nSDS=0,152 моль), 169,1 г дистиллированной воды, 3,9 г LMAM (nLMAM=0,0153 моль) и 5,4 г сульфата натрия помещали в круглодонную колбу на 500 мл при комнатной температуре (20°C).
Смесь нагревали до 60°C и перемешивали при помощи стержневого магнита в течение 2 часов, пока не получили чистый мицеллярный раствор.
В данном мицеллярном растворе nH=(0,0153×62)/0,145=6,5
(nSDS-CMC=0,152-0,007=0,145; NAgg=62).
Раствор E: мицеллярный раствор LMAM/SDS
146,2 г 30% раствора SDS (nSDS=0,152 моль), 169,1 г дистиллированной воды, 2,5 г LMAM (nLMAM=0,0098 моль) и 5,4 г сульфата натрия помещали в круглодонную колбу на 500 мл при комнатной температуре (20°C).
Смесь нагревали до 60°C и перемешивали при помощи стержневого магнита в течение 2 часов, пока не получили чистый мицеллярный раствор.
В данном мицеллярном растворе nH=(0,0098×62)/0,145=4,2
(nSDS-CMC=0,152-0,007=0,145; NAgg=62).
В нижеприведенных примерах среднечисловую молярную массу синтезированных полимеров сравнивали для наглядности с теоретической среднечисловой молярной массой (Mnth), получаемой в контексте полностью контролируемой полимеризации, которая позволяет подтвердить, в частности, хорошо контролируемый характер полимеризации, осуществляемой в соответствии с настоящим изобретением, при этом абсолютно противоположные результаты обычно получают, когда применяют условия полимеризации в геле.
Теоретическую среднечисловую молярную массу, на которую ссылаются в данном документе, рассчитывают согласно следующей формуле:
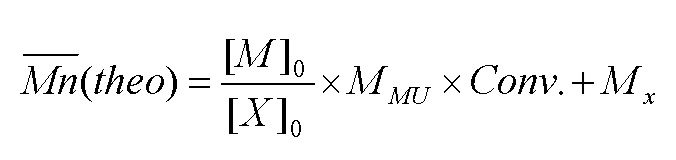 ,
,
где
Mn(theo) = теоретическая среднечисловая масса,
[M]0 = начальная концентрация мономера,
[X]0 = начальная концентрация средства контроля,
MMU = молярная масса мономера,
Conv. = превращение мономера (выход реакции),
MX = молярная масса средства контроля.
СРАВНИТЕЛЬНЫЙ ПРИМЕР: неконтролируемая мицеллярная полимеризация
Сополимер акриламида и LMA: 97,5/2,5-nH 6,3
25 г раствора B, 6,54 г водного раствора акриламида при 50% по массе и 3,14 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 3,138 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Данный водный раствор формальдегид-сульфоксилата натрия предварительно дегазировали путем барботирования азотом.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 24 часов при комнатной температуре (20°C).
Через 24 часа протекания реакции достигали 98% превращения (определяемого с помощью 1H-ЯМР). С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором углов Malls 3 получали следующее значение среднемассовой молярной массы (MW):
Mw=>1500000 г/моль.
Образец было очень трудно отфильтровать даже при концентрации 10 мг в 10 мл. Значение представляет собой среднее для двух впрыскиваний и, таким образом, представляет собой только один порядок величины элюированной фракции.
Пример 1
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 148000 г/моль
50 г раствора B, 13,08 г акриламида (водный раствор при 50% по массе), 0,088 г раствора A и 3,14 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 3,138 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Данный водный раствор формальдегид-сульфоксилата натрия предварительно дегазировали путем барботирования азотом.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 24 часов при комнатной температуре (20°C).
Через 24 часа протекания реакции достигали 98% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 2 г геля и 8 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. pH раствора доводили до 10 путем добавления раствора NaOH при 50% по массе. Колбу затем нагревали при 65°C в течение 5 дней. Раствор полимера, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 3 угла получали следующее значение среднемассовой молярной массы (MW): 230000 г/моль.
Пример 2
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 740000 г/моль
50 г раствора B, 13,08 г акриламида (водный раствор при 50% по массе), 0,018 г раствора A и 3,14 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 3,138 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Данный водный раствор формальдегид-сульфоксилата натрия предварительно дегазировали путем барботирования азотом.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 24 часов при комнатной температуре (20°C).
Через 24 часа протекания реакции достигали 98% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 2 г геля и 8 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. pH раствора доводили до 10 путем добавления раствора NaOH при 50% по массе. Колбу затем нагревали при 65°C в течение 5 дней. Раствор полимера, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 3 угла получали следующее значение среднемассовой молярной массы (MW): 602000 г/моль.
Пример 3
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 15000 г/моль
50 г раствора B, 13,08 г акриламида (водный раствор при 50% по массе), 0,88 г раствора A и 6,26 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 6,26 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Данный водный раствор формальдегид-сульфоксилата натрия предварительно дегазировали путем барботирования азотом.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 24 часов при комнатной температуре (20°C).
Через 24 часа протекания реакции достигали 98% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 2 г геля и 8 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. pH раствора доводили до 10 путем добавления раствора NaOH при 50% по массе. Колбу затем нагревали при 65°C в течение 5 дней. Раствор полимера, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 3 угла получали следующее значение среднемассовой молярной массы (MW): 29800 г/моль.
Пример 4
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 74000 г/моль
50 г раствора B, 13,08 г акриламида (водный раствор при 50% по массе), 0,176 г раствора A и 6,26 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 6,26 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Данный водный раствор формальдегид-сульфоксилата натрия предварительно дегазировали путем барботирования азотом.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 24 часов при комнатной температуре (20°C).
Через 24 часа протекания реакции достигали 98% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 2 г геля и 8 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. pH раствора доводили до 10 путем добавления раствора NaOH при 50% по массе. Колбу затем нагревали при 65°C в течение 5 дней. Раствор полимера, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 3 угла получали следующее значение среднемассовой молярной массы (MW): 65000 г/моль.
Пример 5
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 370000 г/моль
50 г раствора B, 13,08 г акриламида (водный раствор при 50% по массе), 0,035 г раствора A и 6,26 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 6,26 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Данный водный раствор формальдегид-сульфоксилата натрия предварительно дегазировали путем барботирования азотом.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 24 часов при комнатной температуре (20°C).
Через 24 часа протекания реакции достигали 98% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 2 г геля и 8 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. pH раствора доводили до 10 путем добавления раствора NaOH при 50% по массе. Колбу затем нагревали при 65°C в течение 5 дней. Раствор полимера, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 3 угла получали следующее значение среднемассовой молярной массы (MW): 352000 г/моль.
Пример 6
Сополимер акриламида и LA: 97,5/2,5 мол. %, nH 6,3, Mnth 10000 г/моль
50 г раствора C, 13,17 г акриламида (водный раствор при 50% по массе), 0,137 г Rhodixan A1 и 6,32 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 6,32 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 92% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 25,0 г геля и 27,4 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. Затем добавляли 9,6 г раствора NaOH при 25% по массе. Колбу затем нагревали при 60°C в течение 24 часов. Раствор полимера, полученный таким образом, осаждали из 250 мл MeOH, повторно растворяли в 10 мл воды и повторно осаждали из 100 мл MeOH. Полимер, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 18 углов получали следующее значение среднемассовой молярной массы (MW): 81400 г/моль.
Пример 7
Сополимер акриламида и LA: 97,5/2,5 мол. %, nH 6,3, Mnth 250000 г/моль
25 г раствора C, 6,522 г акриламида (водный раствор при 50% по массе), 0,283 г Rhodixan A1 (раствор в этаноле при 1,0% по массе) и 1,6 г персульфата аммония (водный раствор при 1,0% по массе) помещали в стеклянную колбу на 50 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 20 минут. 1,6 г формальдегид-сульфоксилата натрия в форме водного раствора при 1,0% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 95% превращения (определяемого с помощью ВЭЖХ-анализа остаточного акриламида).
Гель, полученный таким образом, подвергали щелочному гидролизу. 15,0 г геля и 25,0 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. Затем добавляли 9,6 г раствора NaOH при 25% по массе. Колбу затем нагревали при 60°C в течение 24 часов. Раствор полимера, полученный таким образом, осаждали из 250 мл EtOH, повторно растворяли в 10 мл воды и повторно осаждали из 200 мл EtOH. Полимер, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 18 углов получали следующее значение среднемассовой молярной массы (MW): 310000 г/моль.
Пример 8
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 10000 г/моль
50 г раствора B, 13,07 г акриламида (водный раствор при 50% по массе), 0,135 г Rhodixan A1 и 6,28 г персульфата аммония (водный раствор при 0,5% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 6,28 г формальдегид-сульфоксилата натрия в форме водного раствора при 0,5% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 46% превращения (определяемого с помощью 1H-ЯМР).
Гель, полученный таким образом, подвергали щелочному гидролизу. 25,0 г геля и 27,4 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. Затем добавляли 9,6 г раствора NaOH при 25% по массе. Колбу затем нагревали при 60°C в течение 24 часов. Раствор полимера, полученный таким образом, осаждали из 250 мл MeOH, повторно растворяли в 10 мл воды и повторно осаждали из 100 мл MeOH. Полимер, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 3 угла получали следующее значение среднемассовой молярной массы (MW): 109900 г/моль.
Пример 9
Сополимер акриламида и LMA: 97,5/2,5 мол. %, nH 6,3, Mnth 250000 г/моль
25 г раствора B, 6,530 г акриламида (водный раствор при 50% по массе), 0,277 г Rhodixan A1 (раствор в этаноле при 1,0% по массе) и 1,6 г персульфата аммония (водный раствор при 1,0% по массе) помещали в стеклянную колбу на 50 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 20 минут. 1,6 г формальдегид-сульфоксилата натрия в форме водного раствора при 1,0% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 93% превращения (определяемого с помощью ВЭЖХ-анализа остаточного акриламида).
Гель, полученный таким образом, подвергали щелочному гидролизу. 15,0 г геля и 25,0 г дистиллированной воды помещали в круглодонную колбу на 100 мл при комнатной температуре. Затем добавляли 9,6 г раствора NaOH при 25% по массе. Колбу затем нагревали при 60°C в течение 24 часов. Раствор полимера, полученный таким образом, осаждали из 250 мл EtOH, повторно растворяли в 10 мл воды и повторно осаждали из 200 мл EtOH. Полимер, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 18 углов получали следующее значение среднемассовой молярной массы (MW): 357000 г/моль.
Пример 10
Сополимер акриламида и LMAM: 97,5/2,5 мол. %, nH 6,3, Mnth 10000 г/моль
50,0 г раствора D, 13,0 г акриламида (водный раствор при 50% по массе) и 0,146 г Rhodixan A1 смешивали в пластмассовой бутылке на 150 мл. 5 мл данного раствора помещали в стеклянную колбу на 5 мл. Добавляли 0,3 мл раствора персульфата аммония (водный раствор при 5,0% по массе) и 0,3 мл формальдегид-сульфоксилата натрия в форме водного раствора при 0,25% по массе. Смесь дегазировали путем барботирования азотом в течение 5 минут.
Реакцию полимеризации затем оставляли протекать без перемешивания в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 97% превращения (определяемого с помощью ВЭЖХ-анализа остаточного акриламида).
Полимер осаждали из 15 мл этанола и затем повторно растворяли в 15 мл воды, к которой добавляли 1 г NaOH. Данный раствор затем оставляли для протекания реакции в течение 72 часов, и затем осаждали полимер из 50 мл этанола. Полимер, полученный таким образом, анализировали с помощью пространственно-эксклюзионной хроматографии.
С помощью анализа с применением пространственно-эксклюзионной хроматографии в воде, дополненной буферным раствором с pH 7, содержащим 100 мМ NaCl, 25 мМ NaH2PO4, 25 мМ Na2HPO4, с детектором Malls на 18 углов получали следующее значение среднемассовой молярной массы (MW): 25600 г/моль.
Пример 11
Сополимер акриламида и LMAM: 97,5/2,5 мол. %, nH 6,3, Mnth 500000 г/моль
100 г раствора D, 26,0 г акриламида (водный раствор при 50% по массе), 0,541 г Rhodixan A1 (раствор в этаноле при 1,0% по массе) и 6,24 г персульфата аммония (водный раствор при 1,0% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 30 минут. 6,26 г формальдегид-сульфоксилата натрия в форме водного раствора при 1,0% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 90,3% превращения (определяемого с помощью ВЭЖХ-анализа остаточного акриламида).
Гель, полученный таким образом, не может быть гидролизован вследствие устойчивости метакриламидной группы к гидролизу.
Пример 12
Сополимер акриламида, AMPS и LMAM: 79,6/19,9/0,5 мол. %, nH 4,0, Mnth 500000 г/моль
30,3 г раствора E, 69,9 г воды, 20,8 г акриламида (водный раствор при 50% по массе), 16,3 г AMPS (водный раствор при 50% по массе), 0,540 г Rhodixan A1 (раствор в этаноле при 1,0% по массе) и 6,00 г персульфата аммония (водный раствор при 5,0% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 20 минут. 1,5 г формальдегид-сульфоксилата натрия в форме водного раствора при 1,0% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 64,6% превращения (определяемого с помощью ВЭЖХ-анализа остаточного акриламида и AMPS).
Гель, полученный таким образом, не может быть гидролизован вследствие устойчивости метакриламидной группы к гидролизу.
Пример 13
Сополимер акриламида, AMPS и LMAM: 79,6/19,9/0,5 мол. %, nH 4,0, Mnth 500000 г/моль
30,3 г раствора E, 69,7 г воды, 20,8 г акриламида (водный раствор при 50% по массе), 16,3 г AMPS (водный раствор при 50% по массе), 1,111 г меркаптоэтанола (водный раствор при 0,5% по массе) и 6,00 г персульфата аммония (водный раствор при 5,0% по массе) помещали в круглодонную колбу на 250 мл при комнатной температуре (20°C). Смесь дегазировали путем барботирования азотом в течение 20 минут. 1,5 г формальдегид-сульфоксилата натрия в форме водного раствора при 1,0% по массе добавляли в среду одной порцией. Смесь дегазировали путем барботирования азотом в течение 15 минут.
Реакцию полимеризации затем оставляли протекать при перемешивании в течение 16 часов при комнатной температуре (20°C).
Через 16 часов протекания реакции достигали 80,7% превращения (определяемого с помощью ВЭЖХ-анализа остаточного акриламида и AMPS).
Гель, полученный таким образом, не может быть гидролизован вследствие устойчивости метакриламидной группы к гидролизу.
Пример 14: фильтруемость раствора полимера
Полимеры, полученные в примере 12 (синтез, контролируемый с помощью средства обратимого переноса) и примере 13 (с помощью средства необратимого переноса), растворяли при 0,2% активного полимера в водном растворе хлорида калия при 1% по массе (раствор получали из 10 г KCl, доведенного до 1 кг деионизированной водой). Растворы перемешивали с помощью магнитной мешалки в течение 24 часов и затем кондиционировали в течение 6 часов при 60°C для обеспечения максимально возможного растворения или диспергирования.
Проводили тестирование фильтруемости, которое заключалось в пропускании раствора, применяемого для промывки пористой среды, через керамический фильтр с известной пористостью. Керамические диски, применяемые в данном испытании, имели толщину 6,2 мм и проницаемость 775 мД (измеренную по проницаемости/впрыскиваемости для ртути), то есть размеры пор в среднем составляли 10 мкм (такие диски доступны от OFITE под регистрационным номером 170-55).
В фильтровальный патрон из нержавеющей стали (доступный от OFITE под регистрационным номером 170-46: двухконечный HTHP-блок для керамических дисков), способный вмещать 175 мл раствора, помещали 100 мл 0,2% раствора полимера. Температуру стабилизировали при 60°C на период 15 минут перед началом тестирования фильтруемости.
Применяли давление азота 0,35 бар, и массу, собранную на выходе из фильтра, регистрировали в зависимости от времени.
Изменение значений собранной массы (называемой отфильтрованной массой) в зависимости от времени для растворов, содержащих 0,2% полимеров из примеров 12 и 13, в ходе фильтрации при 60°C приведено на прилагаемых фигурах 1 и 2.
В ходе данного тестирования было очень четко видно, что полимер из примера 12 может впрыскиваться в пористую среду. Значения собранной массы возрастает линейно. Накопление полимера или забивание им фильтра отсутствует, и весь раствор может быть отфильтрован за период менее 2 минут. Напротив, полимер из примера 13 демонстрирует типичную тенденцию к забиванию фильтра и накоплению осадка на фильтре при скорости прохождения жидкости, которая снижается в зависимости от времени. Кроме того, невозможно отфильтровать весь раствор за разумно необходимый период времени, и тестирование останавливали через один час при менее 10% отфильтрованного раствора. Наконец, в ходе разборки патрона на фильтре наблюдали слой гелеобразного осадка толщиной 2 мм.
В свете данного примера преимущество контроля полимеризации с помощью средства обратимого контроля очевидно в контексте применения растворов полимеров для промывки пористой среды.
Полиамидная огнестойкая композиция
Способ получения соединений, содержащих нитрильные группы
Композиция на основе оксида циркония, оксида титана или смешанного оксида циркония и титана, нанесенная на носитель из оксида алюминия или оксигидроксида алюминия, способы ее получения и ее применение в качестве катализатора
Способ получения соединений, содержащих функциональные нитрильные группы
Вязкоупругая композиция с улучшенной вязкостью
Композиция, содержащая лантансодержащий перовскит на подложке из алюминия или из оксигидроксида алюминия, способ получения и применение в катализе
Способ получения соединений, содержащих нитрильные группы
Способ получения алкилгидропероксидных соединений
Новый способ получения осажденных кремнеземов, осажденные кремнеземы с особой морфологией, гранулометрическим составом и пористостью и их применение, в частности, для усиления полимеров
Способ получения дифторуксусной кислоты
Способ получения оксисульфированных и фторсодержащих органических производных
Способ получения трифторметансульфиновой кислоты
Полиамидная огнестойкая композиция
Способ получения соединений, содержащих нитрильные группы
Композиция на основе оксида циркония, оксида титана или смешанного оксида циркония и титана, нанесенная на носитель из оксида алюминия или оксигидроксида алюминия, способы ее получения и ее применение в качестве катализатора
Способ получения соединений, содержащих функциональные нитрильные группы
Вязкоупругая композиция с улучшенной вязкостью
Установка для кристаллизации адипиновой кислоты
Способ переработки углеводородных соединений, содержащих нитрильные или аминные функциональные группы
Способ получения аминов

