Результат интеллектуальной деятельности: [1,2,4]ТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым производным [1,2,4]триазолопиридина с ингибирующей фосфодиэстеразу активностью, а также их применению в качестве терапевтических агентов при лечении воспалительных заболеваний и состояний.
Уровень техники изобретения
Фосфодиэстеразы представляют собой ферменты, которые катализируют гидролиз циклического АМФ и/или циклического ГМФ в клетках в 5-АМФ и 5-ГМФ, соответственно, и как таковые они являются определяющими для клеточной регуляции уровней цАМФ или цГМФ. Из 11 фосфодиэстераз, идентифицированных до сих пор, фосфодиэстеразы (PDE) 4, PDE7 и PDE8 являются селективными для цАМФ. PDE4 является наиболее важным модулятором цАМФ, экспрессируемым в иммунных и воспалительных клетках, таких как нейтрофилы, макрофаги и Т-лимфоциты (Z. Huang and J.A. Mancini, Current Med. Chem. 13, 2006, pp 3253-3262). Поскольку цАМФ является ключевым вторичным мессенджером при модуляции воспалительных реакций, было обнаружено, что PDE4 регулирует воспалительные реакции воспалительных клеток путем модуляции провоспалительных цитокинов, таких как TNF-α, IL-2, IFN-y, GM-CSF и LTB4. Ингибирование PDE4, таким образом, становится привлекательной мишенью для лечения воспалительных заболеваний, таких как астма, хроническая обструктивная болезнь легких (COPD), ревматоидный артрит, атопический дерматит, воспалительное заболевание кишечника, такое как болезнь Крона и т.д. (M.D. Houslay et al., Drug Discovery Today 10 (22), 2005, рр. 1503-1519). Когда пациенты с атопическим дерматитом (AD) имеют увеличенную PDE-активность, оказывается, что ингибирование PDE4 может быть также эффективным способом лечения AD (Journal of Investigative Dermatology (1986), 87 (3), 372-6).
Семейство генов PDE4 состоит по меньшей мере из четырех генов, A, B, C и D, которые имеют высокую степень гомологии (V. Boswell Smith and D. Spina, Curr. Opinon Investig. Drugs 6(11), 2006, pp. 1136-1141). Четыре изоформы PDE4 дифференциально экспрессируются в различных тканях и типах клеток. Так, PDE4B преимущественно экспрессируется в моноцитах и нейтрофилах, но не экспрессируется в коре головного мозга и эпителиальных клетках, тогда как PDE4D экспрессируется в легких, коре головного мозга, мозжечке и Т-клетках (C. Kroegel and М. Foerster, Exp. Opinion Investig. Drugs 16(1), 2007, рр. 109-124). Было предположено, что ингибирование PDE4D в головном мозге связано с отрицательными действиями, обнаруженными при введении ингибиторов PDE4 клинически, главным образом, тошноты и рвоты, в то время как ингибирование PDE4B связано с противовоспалительными действиями (В. Lipworth, Lancet 365, 2005, рр. 167-175). Однако не считается, что ингибиторы PDE, разработанные до сих пор, являются специфическими для любой из четырех изоформ PDE4.
Изучены многочисленные ингибиторы PDE4 относительно их терапевтического действия на воспалительные заболевания, в основном астмы и COPD.
Первый из них, теофиллин, является слабым неселективным ингибитором фосфодиэстеразы, применяемым при лечении респираторных заболеваний, таких как астма и COPD. Лечение теофиллином может, однако, вызывать как слабые, так и тяжелые побочные действия, например аритмию и судороги, ограничивая тем самым клиническую применимость теофиллина (Kroegel and Foerster, см. выше). Поскольку фосфодиэстераза оставалась привлекательной мишенью для противовоспалительной терапии, были разработаны и исследованы в клинических условиях несколько других, более селективных ингибиторов PDE4. Клиническую разработку многих из первого поколения ингибиторов PDE4, таких как ролипрам, прекратили вследствие ограничивающих дозу побочных действий, в основном тошноты и рвоты. Тем не менее рофлумиласт в 2010 г. был одобрен для тяжелого COPD, связанного с хроническим бронхитом, после того как ограничивающие дозу побочные действия, тошнота, диарея и головная боль, были сведены к минимуму. Ингибиторы PDE4 второго поколения с явно менее выраженными побочными действиями в настоящее время находятся в стадии клинических испытаний (Houslay, см. выше). Ингибиторы PDE4 описаны, например, в ЕР 0771794 и ЕР 0943613.
В WO 2008/125111, LEO Pharma A/S, описываются производные триазолопиридина с сильной ингибирующей PDE4 активностью. Эти соединения включают в себя линкер, включающий в себя карбонильную группу между бициклической гетероциклической системой колец и моноциклической системой колец. Для родственного соединения, пикламиласта, было показано, что линкер является чрезвычайно важным для положения моноциклического кольца, так чтобы он мог взаимодействовать с ферментом PDE4 (Card G.L., et al., "Структурная основа для активности лекарственных средств, которые ингибируют фосфодиэстеразы", Structure 2004 Dec; 12(12); 2233-47), тем самым обеспечивая желаемое ингибирующее действие.
В WO 2010/069322, LEO Pharma A/S, описываются производные триазолопиридина без карбонильного линкера между бициклической и моноциклической системами колец. Обнаружено, что данные соединения проявляют ингибирующую PDE4 активность.
Существует постоянная потребность в разработке новых ингибиторов PDE4, которые имеют более благоприятное терапевтическое окно, то есть обладают более слабыми побочными действиями, сохраняя при этом свое терапевтическое противовоспалительное действие.
Сущность изобретения
Задачей настоящего изобретения является создание новых соединений, которые являются сильнодействующими ингибиторами PDE4, имеющими профиль стабильности в биологической ткани, которая означает, что только очень низкое системное воздействие соединений будет наблюдаться, например, при местном введении. Более точно, соединения настоящего изобретения обладают высоким клиренсом в микросомах печени человека. Они быстро гидролизуются в цельной крови человека, но в то же самое время проявляют стабильность по отношению к ферментативному гидролизу в кератиноцитах человека
В одном аспекте изобретения предложено соединение формулы (I)
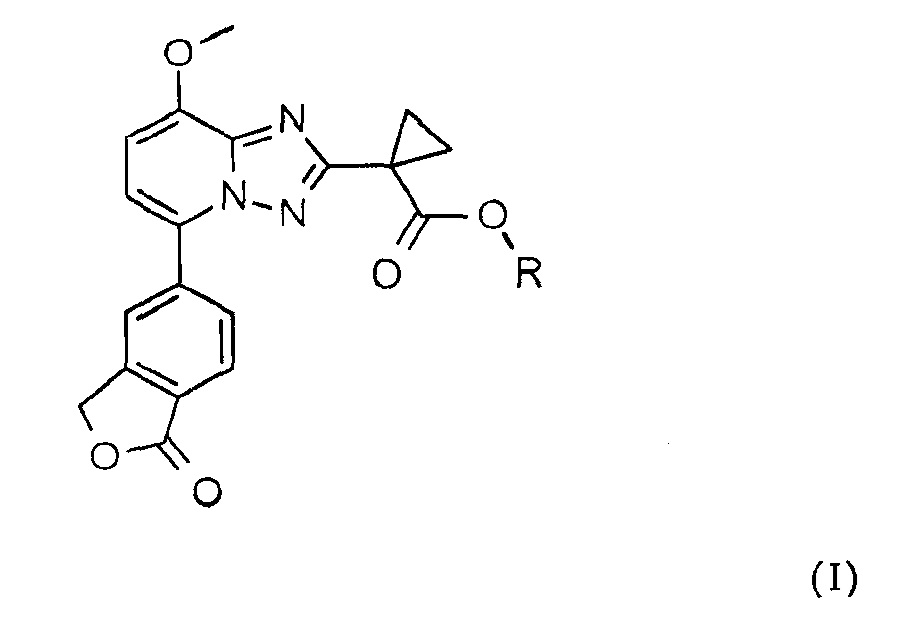 ,
,
в которой R имеет значения, указанные ниже.
В другом аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединение общей формулы (I), определяемой выше, вместе с фармацевтически приемлемым носителем, или эксципиентом, или фармацевтически приемлемым носителем(ями), необязательно вместе с одним или несколькими другим терапевтически активным соединением(ями).
В другом аспекте изобретение относится к применению соединения изобретения для изготовления фармацевтических композиций для профилактики, лечения, предотвращения или уменьшения интенсивности симптомов заболевания, нарушения или состояния, восприимчивого к ингибирующей активности PDE4.
В еще одном аспекте изобретение относится к способу профилактики, лечения, предотвращения или уменьшения интенсивности симптомов заболеваний, нарушений или состояний, восприимчивых к ингибирующей активности PDE4, причем способ содержит стадию введения в организм живого животного терапевтически эффективного количества соединения формула (I) настоящего изобретения.
Другие задачи настоящего изобретения будут очевидны специалисту в данной области техники из следующего подробного описания и примеров.
Подробное описание изобретения
В одном аспекте данное изобретение относится к соединению формулы (I)
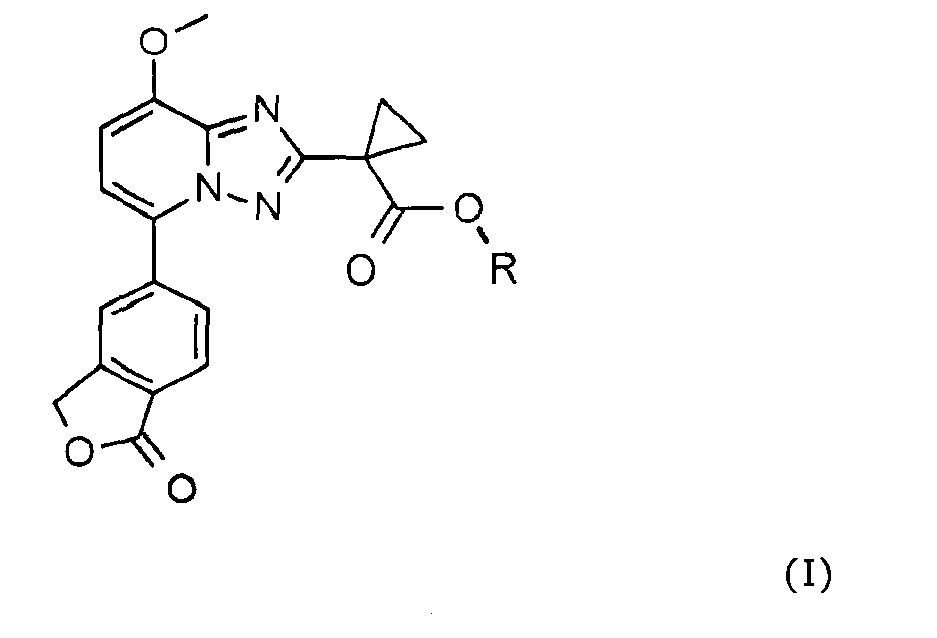
любому из его стереоизомеров или любой смеси его стереоизомеров или его фармацевтически приемлемой соли, у которых R представляет собой разветвленный бутил.
В одном варианте осуществления настоящего изобретения R представляет собой 1-метилпропил, 2-метилпропил или трет-бутил.
В другом варианте осуществления R представляет собой 1-метилпропил.
В другом варианте осуществления R представляет собой 2-метилпропил.
В другом варианте осуществления R представляет собой трет-бутил.
Конкретные примеры соединений формулы (I) можно выбрать из группы, состоящей из
[(1S)-1-метилпропил]-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксилата;
[(1R)-1-метилпропил]-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксилата;
[2-метилпропил]-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксилата;
трет-бутил-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксилата или
их фармацевтически приемлемых солей.
Определения
Применяемые на всем протяжении настоящего описания и прилагаемой формулы изобретения следующие термины имеют указанные значения.
Термин "лечение", применяемый в контексте, означает способ лечения пациента и уход за пациентом с целью борьбы с заболеванием, нарушением или болезненным состоянием. Предполагается, что термин включает в себя задержку развитие заболевания, нарушения или состояния, облегчение или ослабление симптомов и осложнений и/или лечения или ликвидации заболевания, нарушения или состояния. Пациент, подвергаемый лечению, предпочтительно является млекопитающим, в частности человеком.
Термины "заболевание", "состояние" и "нарушение", используемые в контексте, применяют взаимозаменяемым образом для описания состояния пациента, которое является ненормальным физиологическим состоянием человека.
Термин "лекарственное средство", применяемый в контексте, означает фармацевтическую композицию, пригодную для введения фармацевтически активного соединения пациенту.
Термин "фармацевтически приемлемый", применяемый в контексте, означает, что средство является подходящим для обычных фармацевтических применений, то есть не вызывает никаких побочных действий у пациентов и т.д.
Предполагается, что термин "фармацевтически приемлемая соль" означает соли, полученные взаимодействием соединения формулы I с подходящей неорганической или органической кислоты, такой как хлористоводородная, бромистоводородная, иодистоводородная, серная, азотная, фосфорная, муравьиная, уксусная, 2,2-дихлоруксусная, адипиновая, аскорбиновая, L-аспарагиновая, L-глутаминовая, галактаровая, молочная, малеиновая, L-яблочная, фталевая, лимонная, пропионовая, бензойная, глутаровая, глюконовая, D-глюкуроновая, метансульфоновая, салициловая, янтарная, малоновая, винная, бензолсульфоновая, этан-1,2-дисульфоновая, 2-гидроксиэтансульфоновая, толуолсульфоновая, сульфаминовая или фумаровая кислота.
Соединения изобретения можно получить в кристаллической форме либо непосредственно концентрированием из органического растворителя или кристаллизацией или перекристаллизацией из органического растворителя или смеси указанного растворителя и сорастворителя, который может быть органическим или неорганическим, таким как вода. Кристаллы можно выделить в форме по существу без растворителя или в виде сольвата, такого как гидрат. Изобретение охватывает все кристаллические модификации и формы, а также их смеси.
Соединения формулы (I) могут содержать или могут не содержать асимметрично замещенные (хиральные) атомы углерода, которые обуславливают существование изомерных форм, например энантиомеров. Настоящее изобретение относится ко всем таким изомерам, либо в чистом виде, либо в виде их смесей (например, рацематов). Чистые стереоизомерные формы соединений и промежуточных продуктов данного изобретения можно получить с применением процедур, известных в данной области. Различные изомерные формы можно разделить физическими методами разделения, такими как селективная кристаллизация и хроматографические методы, например жидкостная хроматография с применением хиральных стационарных фаз. Указанные чистые стереоизомерные формы можно также получить из соответствующих чистых стереоизомерных форм подходящих исходных веществ, при условии, что реакция протекает стереоселективным или стереоспецифическим образом. Если желаемым является конкретный стереоизомер, указанное соединение предпочтительно будет синтезировано стереоселективными или стереоспецифическими способами получения. В таких способах преимущественно будут применять хиральные чистые исходные вещества.
Терапевтическое применение
Когда соединения изобретению обладают ингибирующей PDE4 активностью, соединения могут быть применимыми в качестве терапевтических агентов для воспалительных аллергических заболеваний, таких как бронхиальная астма, COPD, аллергический ринит и нефрит; аутоиммунных заболеваний, таких как ревматоидный артрит, рассеянный склероз, болезнь Крона и системная красная волчанка; острых или хронических кожных болезней; заболеваний центральной нервной системы, таких как депрессия, амнезия и деменция; органопатии, связанной с ишемическим рефлюксом, вызванным сердечной недостаточностью, шоком и цереброваскулярными заболеваниями и т.д.; инсулин-резистентного сахарного диабета; ран; СПИДа и тому подобного.
В одном варианте осуществления соединения настоящего изобретения считаются применимыми для лечения, предотвращения или ослабления симптомов кожных заболеваний или состояний.
В другом варианте осуществления соединения настоящего изобретения считаются применимыми для лечения, предотвращения или ослабления симптомов кожных заболеваний или состояний, выбранных из группы, состоящей из пролиферативных и воспалительных заболеваний кожи, дерматита, атопического дерматита, себорейного дерматита, контактного дерматита, псориаза, рака, эпидермального воспаления, алопеции, кожной атрофии, индуцированной стероидами кожной атрофии, старения кожи, фотостарения кожи, угревой сыпи, крапивницы, зуда и экземы.
В другом варианте осуществления соединения настоящего изобретения считаются применимыми для лечения или ослабления симптомов атопического дерматита.
В другом варианте осуществления соединения настоящего изобретения считаются применимыми для лечения или ослабления псориаза.
Соединения изобретения, необязательно в комбинации с другими активными соединениями, могут быть применимыми для лечения кожных заболеваний или состояний, в частности для лечения пролиферативных и воспалительных заболеваний кожи, дерматита, атопического дерматита, себорейного дерматита, контактного дерматита, псориаза, рака, эпидермального воспаления, алопеции, атрофии кожи, индуцированной стероидами атрофии кожи, старения кожи, фотостарения кожи, угревой сыпи, крапивницы, зуда и экземы.
Помимо того что соединения настоящего изобретения являются применимыми для лечения человека, они могут быть также применимыми в ветеринарии для лечения животных, включая млекопитающих, таких как лошади, крупный рогатый скот, овцы, свиньи, собаки и кошки.
Для применения в терапии соединения настоящего изобретения обычно находятся в форме фармацевтической композиции. Поэтому изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), необязательно вместе с одним или несколькими другими терапевтически активного соединениями, вместе с фармацевтически приемлемым эксципиентом или носителем. Наполнитель должен быть "приемлемым" в смысле совместимости с другими ингредиентами композиции и не быть вредным для его реципиента.
В форме унифицированной дозы соединение можно вводить один или несколько раз в день с соответствующими интервалами времени в зависимости всегда, однако, от состояния пациента и в соответствии с рецептом, выписанным практикующим врачом. Унифицированная доза препарата для местного применения преимущественно содержит от 0,1 мг до 1000 мг, предпочтительно от 1 мг до 100 мг, например 5-50 мг соединения формулы (I).
Подходящая доза соединения настоящего изобретения будет зависеть, помимо прочего, от возраста и состояния пациента, тяжести заболевания, подвергаемого лечению, и других факторов, хорошо известных практикующему врачу. Соединение можно вводить либо перорально, парентерально, либо местно согласно различным схемам применения, например, ежедневно или с недельными интервалами. В общем разовая доза будет в диапазоне от 0,001 до 10 мг/кг массы тела, например в диапазоне от 0,01 до 1 мг/кг массы тела. Соединение можно вводить в виде болюса (т.е. всю суточную дозу вводить сразу) или в виде дробных доз два или более раза в день.
В контексте местного лечения дозу можно более подходящим образом называть "унифицированной дозой", которая означает унитарную, т.е. разовую дозу, которую можно вводить пациенту и которую можно легко изготовить и упаковать, причем она остается как физически, так и химически стабильной унифицированной дозой, содержащей либо активное вещество как таковое, либо его смесь с твердыми или жидкими фармацевтическими разбавителями или носителями. "Унифицированную дозу" можно вводить местно пациенту при нанесении на квадратный сантиметр кожи в количестве от 0,1 мг до 50 мг, предпочтительно от 0,2 мг до 5 мг конечной рассматриваемой композиции.
Предполагается также, что при некоторых схемах лечения может быть благоприятным введение с более длительными интервалами, например через день, каждую неделю или даже с более длительными интервалами.
Если лечение включает в себя введение другого терапевтически активного соединения, рекомендуется принимать во внимание публикацию Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 9th Ed., J.G. Hardman and L.E. Limbird (Eds.), McGraw-Hill 1995, для определения пригодных дозировок указанных соединений.
Введение соединения настоящего изобретения с одним или несколькими другими активными соединениями можно проводить либо одновременно, либо последовательно.
Препараты включают в себя, например, препараты в форме, подходящей для перорального (в том числе препараты со стойко поддерживаемым или регулируемым во времени высвобождением), ректального, парентерального (в том числе подкожного, внутрибрюшинного, внутримышечного, внутрисуставного и внутривенного), трансдермального, офтальмического, местного, дермального, назального или трансбуккального введения. Местное введение заявленного препарата является особенно подходящим.
Препараты можно преимущественно представлять в дозированной лекарственной форме и его можно получать любым из способов, хорошо известных в области фармации, например, как описано в публикации Remington, The Science and Practice of Pharmacy, 20th ed., 2000. Все способы включают в себя стадию смешивания активного ингредиента с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. Обычно препараты получают однородным и тесным смешиванием активного ингредиента с жидким носителем или тонкоизмельченным твердым носителем или обоими такими носителями и затем, если необходимо, формованием продукта в нужный препарат.
Препараты настоящего изобретения, подходящие для перорального введения, могут быть в форме дискретных единиц, таких как капсулы, саше, таблетки или лепешки, причем каждая из них содержит заранее определенное количество активного ингредиента; в форме порошка или гранул; в форме раствора или суспензии в водной жидкости или неводной жидкости, такой как этанол или глицерин; или в форме эмульсии масло в воде или эмульсии вода в масле. Такими маслами могут быть пищевые масла, такие как, например, хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают в себя синтетические или природные смолы, такие как трагакант, альгинат, аравийская камедь, декстран, натриевая соль карбоксиметилцеллюлозы, желатин, метилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, карбомеры и поливинилпирролидон. Активные ингредиенты можно также вводить в виде болюса, лекарственной кашки или пасты.
Таблетку можно изготовить прессованием или формованием активного ингредиента, необязательно с одним или несколькими дополнительными ингредиентами. Прессованные таблетки можно получать прессованием в подходящей машине активного ингредиента(ов) в сыпучей форме, такой как порошок или гранулы, необязательно смешанного со связывающим веществом, таким как, например, лактоза, глюкоза, крахмал, желатин, аравийская камедь, трагакантовая камедь, альгинат натрия, карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, полиэтиленгликоль, воски или тому подобное; смазывающим веществом, таким как, например, олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия или тому подобное; дезинтегрирующим агентом, таким как, например, крахмал, метилцеллюлоза, агар, бентонит, натриевая соль кроскармеллозы, натриевая соль гликолята крахмала, кросповидон или тому подобное, или диспергирующим агентом, таким как полисорбат 80. Формованные таблетки можно получить формованием в подходящей машине смеси порошкообразного активного ингредиента и подходящего носителя, увлажненного инертным жидким разбавителем.
Препараты для ректального введения могут быть в форме суппозиториев, в которых соединение настоящего изобретения смешано с низкоплавящимися, растворимыми или нерастворимыми в воде твердыми веществами, такими как масло-какао, гидрогенизированные растительные масла, полиэтиленгликоль или эфиры жирных кислот и полиэтиленгликолей, тогда как эликсиры можно получить с применением миристилпальмитата.
Препараты, подходящие для парентерального введения, преимущественно содержат стерильный масляный или водный препарат активных ингредиентов, который предпочтительно является изотоническим с кровью реципиента, например изотонический солевой раствор, изотонический раствор глюкозы или буферный раствор. Препарат можно преимущественно стерилизовать, например, фильтрованием через задерживающий бактерии фильтр, добавлением стерилизующего агента к препарату, облучением препарата или нагреванием препарата. Липосомные препараты, описываемые, например, в Encyclopedia of Pharmaceutical Technology, vol. 9, 1994, также являются пригодными для парентерального введения.
Альтернативно, соединения формулы (I) можно представить в виде стерильного твердого препарата, например лиофилизированного порошка, который легко растворяют в стерильном растворителе непосредственно перед применением.
Трансдермальные препараты могут быть в форме пластыря или наклейки.
Препараты, пригодные для офтальмического введения, могут быть в форме стерильного водного препарата активных ингредиентов, которые могут быть в микрокристаллической форме, например в форме водной микрокристаллической суспензии. Липосомные составы или биоразлагаемые полимерные системы, например, описываемые в Encyclopedia of Pharmaceutical Technology, vol. 2, 1989, можно также применять для предоставления активного ингредиента для офтальмического введения.
Препараты, пригодные для местного или офтальмологического введения, включают в себя жидкие или полужидкие препараты, такие как линименты, лосьоны, гели, аппликации, эмульсии масло в воде или вода в масле, такие как кремы, мази или пасты; или растворы или суспензии, такие как капли. Композиции для офтальмологического лечения могут предпочтительно дополнительно содержать циклодекстрин.
Для местного введения соединение формулы (I) обычно может присутствовать в количестве от 0,01 до 5% (в расчете на массу композиции), например от 0,01% до 1% (расчете на массу композиции).
Препараты, подходящие для назального или трансбуккального введения, включают в себя порошок, самодвижущиеся и распыляющиеся препараты, такие как аэрозоли и распылители. Такие препараты описаны более подробно, например, в Modern Pharmaceutics, 2nd ed., G.S. Banker and C.T. Rhodes (Eds.), page 427-432, Marcel Dekker, New York; Modern Pharmaceutics, 3th ed., G.S. Banker and C.Т. Rhodes (eds.), page 618-619 and 718-721, Marcel Dekker, New York, and Encyclopedia of Pharmaceutical Technology, vol. 10, J. Swarbrick and J.C. Boylan (Eds.), page 191-221, Marcel Dekker, New York.
Помимо вышеуказанных ингредиентов, препараты соединения формулы (I) могут включать в себя один или несколько дополнительных ингредиентов, таких как разбавители, буферы, корригенты, красящее вещество, поверхностно-активные вещества, загустители, консерванты, например метилгидроксибензоат (в том числе антиоксиданты), эмульгирующие агенты и тому подобное.
Фармацевтическая композиция может дополнительно содержать один или несколько других активных компонентов, обычно применяемых при лечении кожных заболеваний или состояний, например, выбранных из группы, состоящей из глюкокортикоидов, витамина D и аналогов витамина D, антигистаминов, антагонистов фактора активации тромбоцитов (PAF), антихолинергических агентов, метилксантинов, β-адренергических агентов, ингибиторов СОХ-2, салицилатов, индометацина, флуфенамата, напроксена, тимегадина, солей золота, пеницилламина, агентов, снижающих содержание холестерина сыворотки крови, ретиноидов, солей цинка, салицилазосульфапиридина и ингибиторов кальциневрина.
Способы получения
Соединения настоящего изобретения можно получить несколькими путями, хорошо известными специалистам в области синтеза. Соединения формулы (I) можно, например, получить с применением реакций и методов, описанных ниже, вместе с методами, известными в области синтетической органической химии, или их вариантами, известными специалистам в данной области. Предпочтительные способы включают в себя, но не ограничиваются способами, описанными ниже. Реакции проводят в растворителях, подходящих для реагентов и веществ, используемых и подходящих для осуществляемых превращений. Кроме того, в синтетических способах, описанных ниже, должно быть понятно, что все предлагаемые условия реакций, в том числе выбор растворителя, атмосферы реакции, температуры реакции, продолжительности эксперимента и процедуры обработки, выбирают так, чтобы они были условиями, стандартными для данной реакции, которые должны быть легко признаны специалистом в данной области органического синтеза. Не все соединения, относящиеся к данному классу, могут быть совместимы с некоторыми из условий реакции, необходимых в некоторых из описанных методов. Такие ограничения для заместителей, которые совместимы с условиями реакции, будут легко очевидны для специалистов в данной области, можно применять альтернативные методы.
Исходные вещества являются либо известными, либо коммерчески доступными соединениями или их можно получить с помощью обычных синтетических способов, хорошо известных специалистам в данной области.
Способ ЖХ-МС "XE Metode 7 CM"
Контроль свойств проводили на приборе Waters LCT Premier MS и Waters Aquity UPLC.
Колонка: Waters Aquity UPLC HSS T3 1,8 мкм, 2,1×50 мм, при 40°С.
Растворители: A=10 мМ ацетата аммония+0,1% HCOOH, B=MeCN+0,1% НСООН.
Расход потока: 0,7 мл/мин. Объем впрыска 2 мкл. Диапазон УФ-детектирования 240-400 нм.
|
Подтверждение молекулярной массы и чистоту получали и проверяли OpenLynx.
Спектры 1H ядерного магнитного резонанса (ЯМР) регистрировали при 400 или 600 МГц. Величины химических сдвигов (δ, в миллионных долях) получали при измерении в указанном растворителе относительно внутренних стандартов тетраметилсилана (δ=0,00) или хлороформа (δ=7,25). Величина мультиплета, либо определенного (дублет (д), триплет (т), квартет (к)), либо неопределенного (м), указана приблизительно для средней точки, если не указан диапазон. (bs) означает уширенный синглет. Применяемые органические растворители обычно были безводными. Хроматографию проводили на силикагеле Мерк 60 (0,040-0,063 мм). Указанные отношения растворителей относятся к отношениям об.:об., если не указано иное.
На всем протяжении контекста применяли следующие аббревиатуры:
DBU 1,8-диазабицикло[5.4.0]ундец-7-ен
DCE 1,2-дихлорэтан
DCM дихлорметан
DIAD диизопропилазодикарбоксилат
DMAP N,N-диметилпиридин-4-амин
ДМФА N,N-диметилформамид
ДМСО диметилсульфоксид
EDCI (3-диметиламинопропил)этилкарбодиимид
EtOH этанол
MeOH метанол
EtOAc этилацетат
л литр
Ме метил
ЯМР ядерный магнитный резонанс
КТ комнатная температура
ТГФ тетрагидрофуран
Пет. петролейный эфир
Общие методы
Соединения изобретения можно получить, например, согласно следующим неограничивающим общим методам и примерам. R имеет значения, указанные ранее для соединений формулы (I).
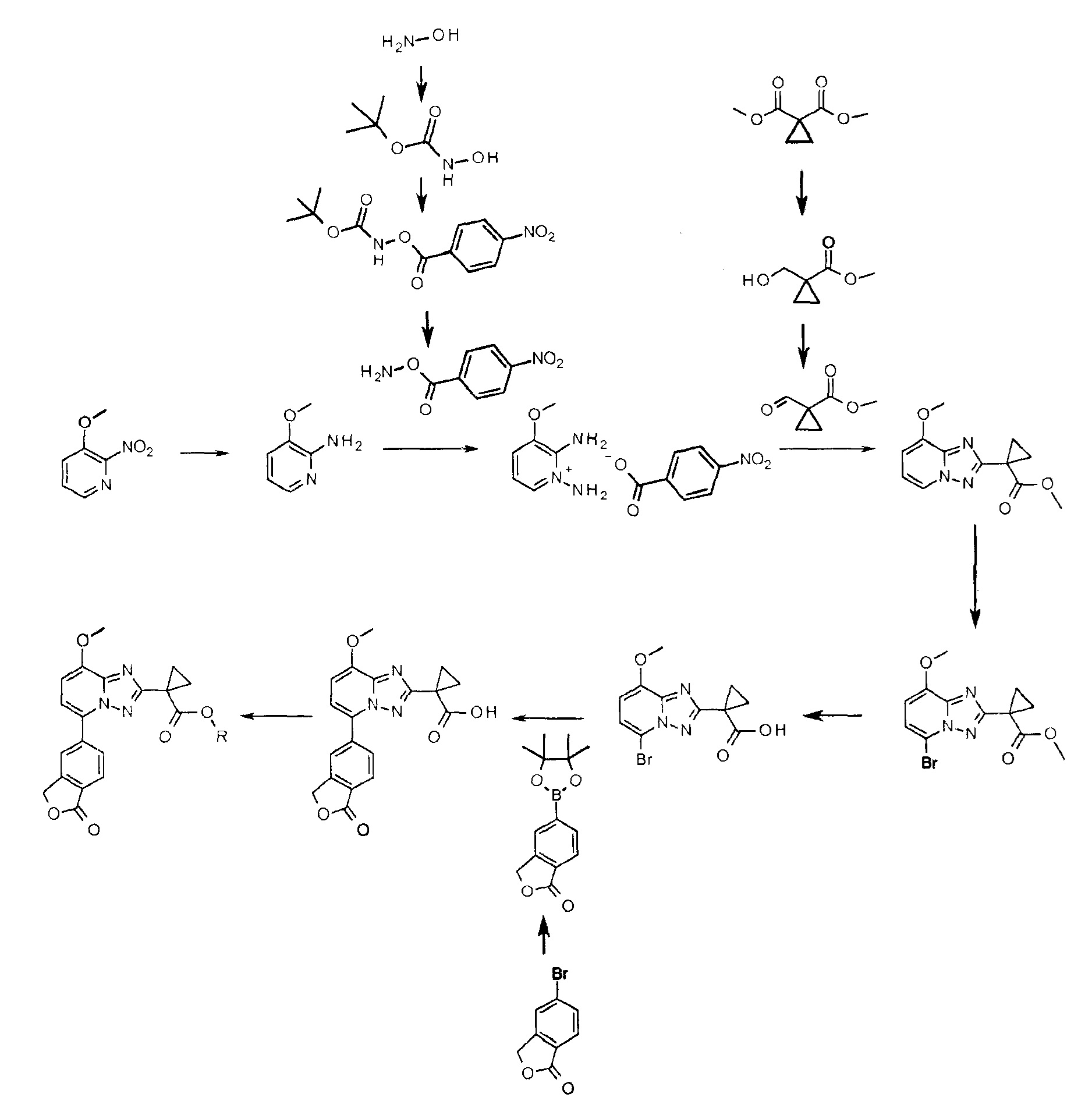
Получение 1
трет-Бутилгидроксикарбамат
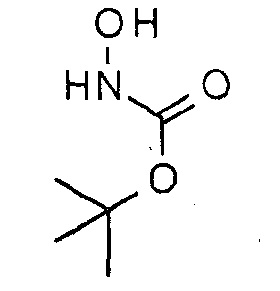
К перемешиваемой суспензии соли гидроксиламин⋅HCl (150 г, 2,17 моль) и K2C03 (150 г, 1,09 моль) в диэтиловом эфире (940 мл) и воде (30 мл) при 0°С медленно добавляли раствор ди-трет-бутилдикарбонат (308 г, 1,41 ммоль) в диэтиловом эфире (600 мл) в течение 15 мин. После добавления реакционную смесь перемешивали при КТ в течение 2 часов. Реакционную смесь фильтровали и фильтрат сушили над безводным Na2S04 и концентрировали. Полученный сырой продукт промывали циклогексаном (50 мл×3) и сушили, получая при этом указанное в заголовке соединение (150 г, 52%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3): δ=7,18 (ушир., 2H), 1,47 (с, 9Н) м.д.
Получение 2
трет-Бутил-4-нитробензоилоксикарбамат
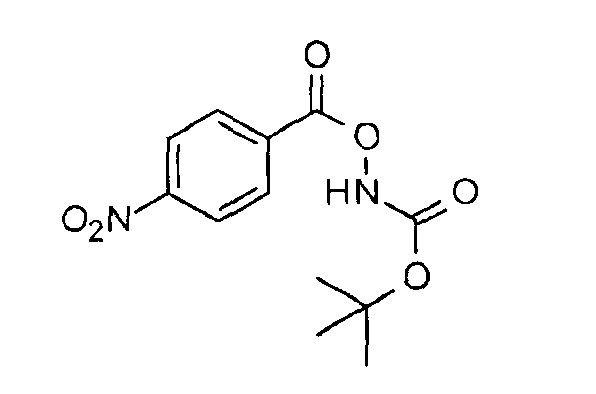
К перемешиваемому раствору трет-бутилгидроксикарбамата (150 г, 1,128 моль) в дихлорметане (2 л) при 0°С добавляли триэтиламин (174 мл, 1,24 моль) с последующим добавлением 4-нитробензоилхлорида (205 г, 1,105 моль) в равных отношениях. После того как добавление было завершено, реакционную смесь перемешивали при КТ течение 1 часа. Реакционную смесь гасили водой (500 мл) и экстрагировали. Отделенный слой дихлорметана промывали насыщенным раствором соли (200 мл), сушили над безводным Na2S04 и концентрировали. Полученный неочищенный продукт промывали гексаном (100 мл×2) и сушили, получая при этом указанное в заголовке соединение (300 г, 94%, желтое твердое вещество). 1H ЯМР (400 МГц, CDCl3): δ=8,34-8,27 (м, 4H), 2,97-2,92 (м, 1Н), 1,53 (с, 9Н) м.д.
Получение 3
О-(4-Нитробензоил)гидроксиламин
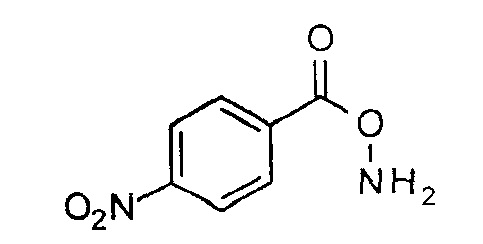
К перемешиваемому раствору трет-бутил-4-нитробензоилоксикарбамата (300 г, 1,06 моль) в дихлорметане (2 л) при 0°С медленно добавляли метансульфоновую кислоту (69 мл, 1,06 моль). После того как добавление было закончено, реакционную смесь оставляли для перемешивания при КТ в течение 16 часов. Реакционную смесь разбавляли дихлорметаном (1 л), промывали 10% водным раствором NaHCO3 (300 мл), водой (200 мл), насыщенным раствором соли (200 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт промывали гексаном (100 мл×2) и сушили, получая при этом указанное в заголовке соединение (150 г, 77%, бледно-желтое твердое вещество). 1H ЯМР (400 МГц, CDCl3): δ=8,33-8,30 (м, 2H), 8,22-8,19 (м, 2H), 6,73 (ушир.с, 2H) м.д.
Получение 4
Этиловый эфир 1-гидроксиметилциклопропанкарбоновой кислоты
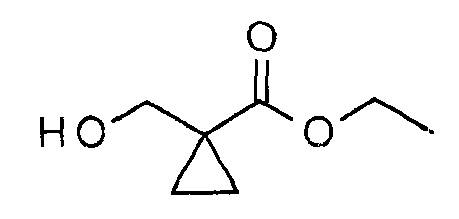
К перемешиваемому раствору диэтилциклопропан-1,1-дикарбоксилата (2,13 г, 11,4 ммоль) в ТГФ (80 мл) при КТ медленно добавляли литийалюминийтри-трет-бутоксигидрид (38,76 мл, 38,76 ммоль, 1,0М раствор в ТГФ). После того как добавление было завершено, реакционную смесь перемешивали при КТ в течение 18 часов. Реакционную смесь разбавляли этилацетатом (100 мл), промывали 1 н водным HCl (20 мл), водой (20 мл), 5% водным NaHCO3 (25 мл), насыщенным раствором соли (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали, получая при этом указанное в заголовке соединение (1,3 г, 79%, желтое масло). 1H ЯМР (400 МГц, CDCl3): δ=4,20-4,13 (м, 2Н), 3,62 (м, 2Н), 2,61 (м, 1H), 1,29-1,24 (м, 5H), 0,88-0,85 (м, 2Н) м.д.
Получение 5
Этиловый эфир 1-формилциклопропанкарбоновой кислоты
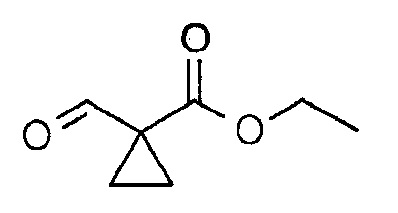
К перемешиваемому раствору этилового эфира 1-гидроксиметилциклопропанкарбоновой кислоты (1,1 г, 7,63 ммоль) в дихлорметане (45 мл) добавляли NaHCO3 (2,5 г, 29,76 ммоль) и периодинан Десс-Мартина (6,46 г, 15,23 ммоль). Затем суспензию перемешивали при КТ в течение 30 мин. Реакционную смесь гасили смесью 1:1 10% водного раствора Na2S2O3 и 10% водного раствора NaHCO3 (20 мл), поддерживая температуру ниже 20°С, перемешивали в течение 30 мин. Затем реакционную смесь разбавляли дихлорметаном (100 мл) и экстрагировали. Органический слой промывали насыщенным раствором соли (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (0-10% EtOAc в петролейном эфире в качестве элюента), получая при этом указанное в заголовке соединение (800 мг, 76%, желтое масло). 1Н ЯМР (400 МГц, CDCl3): δ=10,40 (с, 1H), 4,25 (м, 2H), 1,68-1,65 (м, 2H), 1,62-1,59 (м, 2H), 1,33-1,26 (м, 3H) м.д.
Получение 6
3-Метоксипиридин-2-иламин
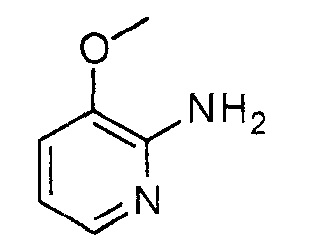
Суспензию 3-метокси-2-нитропиридина (30 г, 194,8 ммоль) и 10% Pd/C (10 г) в этаноле (1 л) гидрировали в обычном гидрогенизаторе (Н2, давление 275790 Па (40 фунтов на квадратный дюйм))) при КТ в течение 4 часов. Реакционную смесь фильтровали через целит и фильтрат концентрировали, получая при этом указанное в заголовке соединение (22 г, 91% твердого коричневого вещества). 1H ЯМР (400 МГц, CDCl3): δ=7,66 (д, J=5,2 Гц; 1Н), 6,91 (д, J=7,6 Гц, 1Н), 6,63-6,60 (м, 1Н), 4,65 (ушир., 2H), 3,84 (с, 3H) м.д.
Получение 7
1,2-Диамино-3-метоксипиридиниевая соль 4-нитробензойной кислоты
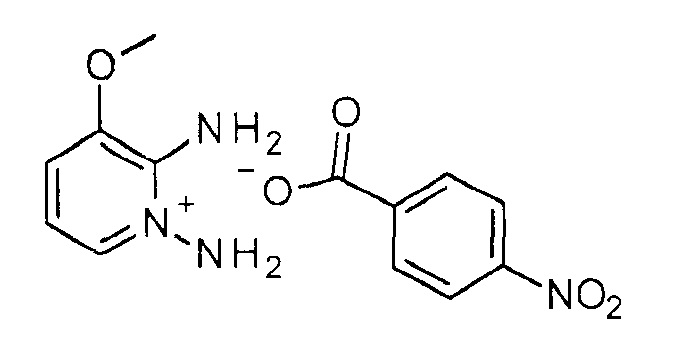
К перемешиваемому раствору 3-метоксипиридин-2-иламина (30 г, 164,8 ммоль) в дихлорметане (400 мл) при 10°С добавляли О-(4-нитробензоил)гидроксиламин (13,2 г, 214,2 ммоль). После добавления реакционную смесь перемешивали при КТ в течение 16 часов. Полученный осадок отфильтровывали, промывали дихлорметаном (25 мл×2) и сушили, получая при этом указанное в заголовке соединение (40 г, 91%, коричневое твердое вещество). (Предупреждение: соль является термически нестабильной).
1H ЯМР (400 МГц, ДМСО): δ=8,53 (шир. 2Н), 8,12 (д, J=8 Гц, 2Н), 8,01 (д, J=8,8 Гц, 2Н), 7,73 (д, J=6,4 Гц, 1Н), 7,33 (д, J=7,2 Гц, 1Н), 7,17 (шир., 2Н), 6,77 (т, J=6,8 Гц, 1Н), 3,93 (с, 3H) м.д.
Получение 8
Этиловый эфир 1-(8-метокси-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоновой кислоты
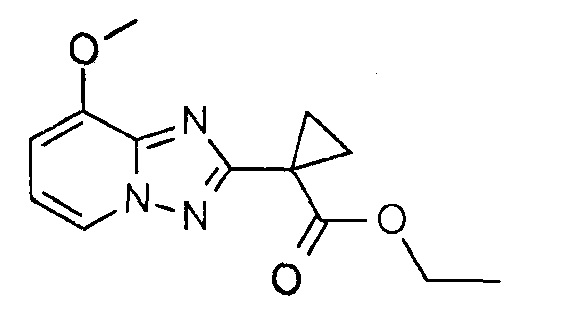
К перемешиваемому раствору диамино-3-метоксипиридиниевой соли 4-нитробензойной кислоты (1 г, 7,14 ммоль) в этаноле (10 мл) при 0°С добавляли DBU (2,1 мл) с последующим добавлением этилового эфира 1-формилциклопропанкарбоновой кислоты (1,5 г, 10,71 ммоль). После добавления реакционную смесь перемешивали при КТ в течение 2 часов. Реакционную смесь концентрировали, полученный остаток разбавляли EtOAc (100 мл), промывали водой (20 мл×2), насыщенным раствором соли (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (с применением 0-15% раствора EtOAc в CH2Cl2 в качестве элюента), получая при этом указанное в заголовке соединение (800 мг, 53%, белое твердое вещество).
1H ЯМР (400 МГц, CDCl3): δ=8,18-8,16 (д, J=6,4 Гц; 1Н), 6,89 (д, J=7,2 Гц, 1Н), 6,76 (д, J=8 Гц, 1Н), 4,20-4,14 (м, 2Н), 4,03 (с, 3H), 1,73-1,70 (м, 2H), 1,59-1,56 (м, 2H), 1,20 (т, J=6,8 Гц; 3Н) м.д.
Получение 9
Этиловый эфир 1-(5-бром-8-метокси-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоновой кислоты
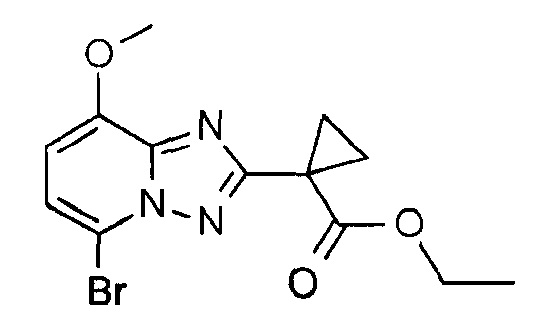
К перемешиваемому раствору этилового эфира 1-(8-метокси-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоновой кислоты (25 г, 95,7 ммоль) в ацетонитриле (300 мл) при КТ порциями добавляли N-бромсукцинимид (34 г, 191,5 ммоль). После добавления реакционную смесь перемешивали при КТ в течение 6 часов. Реакционную смесь разбавляют EtOAc (600 мл), промывали водой (100 мл×2), насыщенным раствором соли (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (0-10% EtOAc в дихлорметане в качестве элюента), получая при этом указанное в заголовке соединение (25 г, 77%, бесцветное твердое вещество).
1H ЯМР (400 МГц, ДМСО): δ=7,44 (д, J=8,4 Гц; 1Н), 7,07 (д, J=8 Гц; 1Н), 4,43-4,08 (м, 2Н), 3,97 (с, 3H),,60-1,57 3,93 (м, 2H), 1,48-1,45 (м, 2H), 1,13 (т, J=7,4 Гц; 3Н) м.д.
Получение 10
1-(5-Бром-8-метокси-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоновая кислота
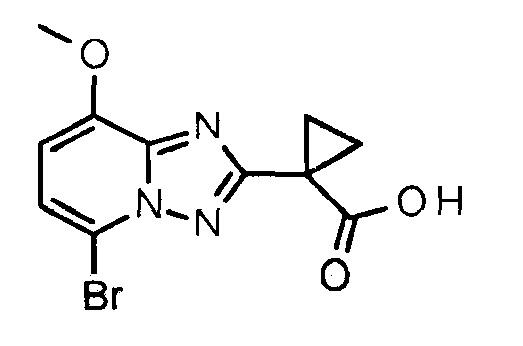
К раствору этил-1-(5-бром-8-метокси-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоксилата (3,00 г, 8,82 ммоль) в ТГФ (25 мл) добавляли водный 1 М раствор LiOH (25 мл). Смесь перемешивали при 80°С в течение 30 минут, охлаждали до комнатной температуры и разбавляли EtOAc (50 мл) и водой (50 мл). Органическую фазу экстрагировали водным 0,1М раствором NaOH (25 мл) и объединенные водные фазы подкисляли конц. HCI до рН 0-1 и экстрагировали четыре раза DCM (30 мл). Выпаривание досуха объединенных органических фаз давало указанное в заголовке соединение (2,54 г, 94%). 1H ЯМР (ДМСО, 400 МГц): δ=12,61 (с, 1H), 7,42 (д, 1H, J=8,3 Гц), 7,06 (д, 1H, J=8,3 Гц), 3,97 (с, 3H), 1,53 (д, 2Н, J=3,9 Гц), 1,40 (д, 2Н, J=3,9 Гц) м.д.
Получение 11
1-[8-Метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоновая кислота
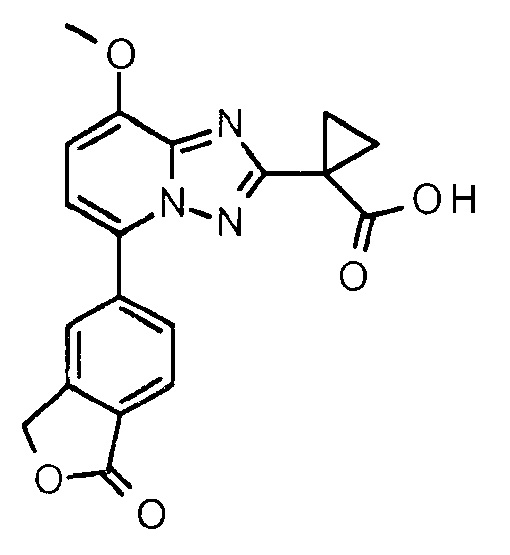
1-(5-Бром-8-метокси-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоновую кислоту (1,00 г, 3,20 ммоль) и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3Н-изобензофуран-1-1-он (получение бороната описано в WO2011/134468) (1,67 г, 6,40 ммоль) растворяли в дегазированном диоксане (16 мл). Pd2(dba)3 (29 мг, 32 мкмоль)), PCy3 (18 мг, 64 мкмоль) и K3PO4 (2,38 г, 11,2 ммоль) смешивали в дегазированной воде (10 мл). Два раствора смешивали и затем нагревали в микроволновой печи до 110°С в течение 10 минут, охлаждали до комнатной температуры и разбавляли EtOAc (40 мл). Органическую фазу экстрагировали
водой (25 мл) и водным 0,1 М раствором NaOH (25 мл) и объединенные водные фазы подкисляли конц. HCl до рН 0-1 и экстрагировали DCM (30 мл×4). После упаривания объединенных органических фаз указанное в заголовке соединение кристаллизовалось (746 мг, 64%). Время удерживания при ВЭЖХ (XE Metode 7 СМ): 1,97 минут. Определенная "М+1"-масса: 366,11. Вычисленная "М+1"-масса: 366,11.
1H ЯМР (ДМСО, 300 МГц): δ=8,25-8,20 (м, 1H), 8,13 (дд, 1H, J=8,2 Гц, 1,4 Гц), 8,00 (д, 1H, J=8,0 Гц), 7,40 (д, 1H, J=8,2 Гц), 7,24 (д, 1H, J=8,3 Гц), 5,51 (с, 2H), 4,04 (с, 3H), 1,58-1,48 (м, 2H), 1,48-1,39 (м, 2H) м.д.
Пример 1
[(1S)-1-Метилпропил]-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксилат (соединение 1)
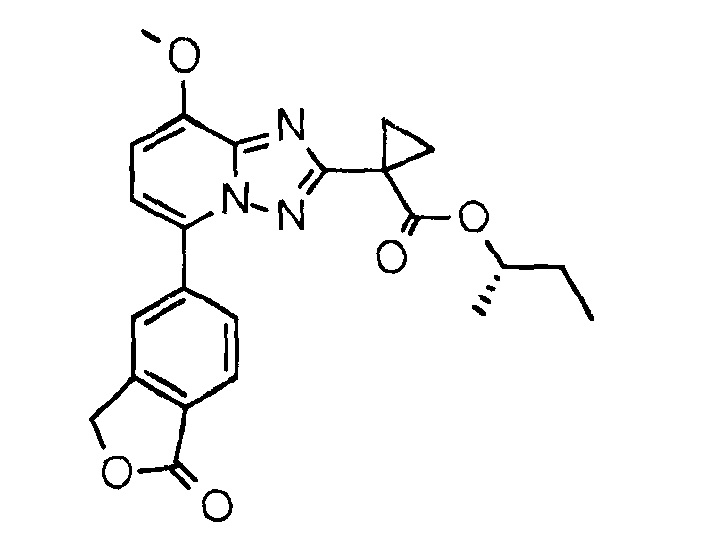
Смесь 1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоновой кислоты (10 мг, 27 мкмоль), (1S)-1-метилпропанола (10 мкл, 108 мкмоль), DMAP (6,7 мг, 54 мкмоль) и EDC⋅HCl (10,5 мг, 54 мкмоль) в DCM (0,5 мл) перемешивали в герметизированной ампуле при КТ на протяжении ночи, затем добавляли еще (1S)-1-метилпропанол (10 мкл, 108 мкмоль) и DMAP (6,7 мг, 54 мкмоль) и смесь нагревали до 50°С в течение 3 часов. Выпаривание досуха и очистка кислотной препаративной ВЭЖХ давали указанное в заголовке соединение. Время удерживания по ВЭЖХ (XE Metode 7 СМ): 2,35 минуты. Определенная "М+1"-масса: 422,16. Вычисленная "M+1"-масса: 422,17.
1H ЯМР (ДМСО, 600 МГц): δ=8,25-8,21 (м, 1H), 8,14 (дд, 1H, J=8,0 Гц, 1,2 Гц), 8,01-7,97 (м, 1H), 7,41 (д, 1H, J=8,2 Гц), 7,23 (д, 1H, J=8,2 Гц), 5,51 (с, 2H), 4,78 (h, 1H, J=6,3 Гц), 4,04 (с, 3H), 1,61-1,40 (м, 6H), 1,13 (д, 3H, J=6,2 Гц), 0,78 (т, 3H, J=7,4 Гц) м.д.
Пример 2
[(1R)-1-Метилпропил]-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксилат (соединение 2)
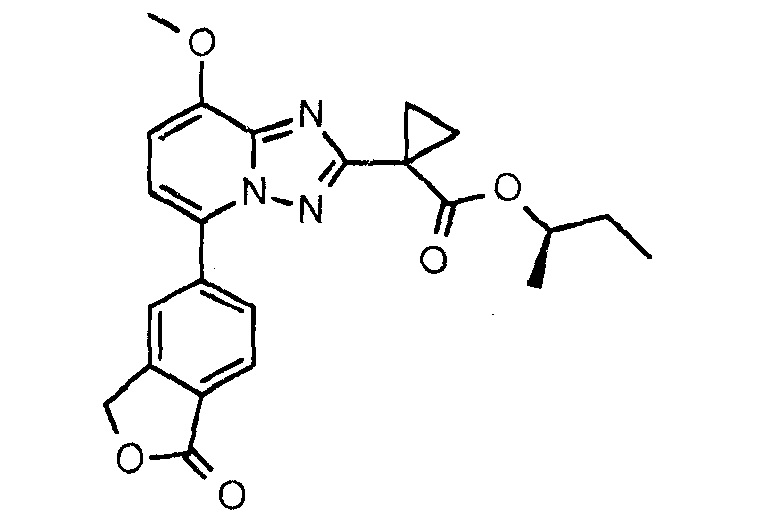
Смесь 1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоновой кислоты (400 мг, 1,095 ммоль), (2R)-бутан-2-ола (122 мг, 1,64 ммоль), DMAP (147 мг, 1,20 ммоль) и EDCl⋅HCl (231 мг, 1,20 ммоль) в DCM (10 мл) перемешивали при КТ в течение ночи перед тем, как его выпаривали досуха. Колоночная хроматография (градиент 0-5% МеОН в DCM) с последующей перекристаллизацией в EtOH и лиофилизацией давали указанное в заголовке соединение в виде бесцветного порошка (138 мг, 30%). Время удерживания при ВЭЖХ (XE Metode 7 СМ): 2,33 минуты. Определенная "М+1"-масса: 422,15, вычисленная "M+1"-масса: 422,17.
1H ЯМР (ДМСО, 600 МГц): δ=8,23 (шир.с, 1H), 8,14 (дд, 1H, J=8,0 Гц, 1,6 Гц), 7,99 (д, 1H, J=8,0 Гц), 7,41 (д, 1H, J=8,2 Гц), 7,24 (д, 1H, J=8,2 Гц), 5,51 (с, 2H), 4,78 (h, 1H, J=6,3 Гц), 4,04 (с, 3H), 1,62-1,38 (м, 6H), 1,13 (д, 3H, J=6,3 Гц), 0,78 (т, 3H, J=7,4 Гц) м.д.
Пример 3
[2-Метилпропил]-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[,5-а]пиридин-2-ил]циклопропанкарбоксилат (соединение 3)
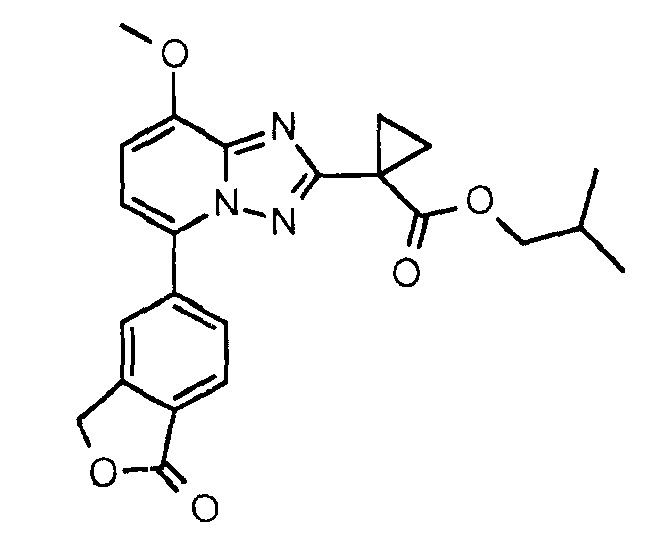
Смесь 1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоновой кислоты (250 мг, 685 мкмоль), изобутанола (100 мкл, 1,37 ммоль), DMAP (250 мг, 2,05 ммоль) и EDCI⋅HCl (262 мг, 1,37 ммоль) в DCM (14 мл) перемешивали при 50°С в течение 2 часов в герметизированной ампуле, затем ее разбавляли DCM (80 мл), промывали водным 1 М раствором HCl (40 мл) и упаривали досуха. Неочищенную смесь снова растворяли в MeCN (~2 мл) и неочищенный продукт кристаллизовали при добавлении воды (~2 мл). Колоночная хроматография (градиент 20-100% раствор EtOAc в петр. эфире) и последующая перекристаллизация в MeCN и воде давали указанное в заголовке соединение в виде бесцветных кристаллов (178 мг, 62%). Время удерживания при ВЭЖХ (XE Metode 7 СМ): 2,34 минуты. Определенная "М+1"-масса: 422,16. Вычисленная "M+1"-масса: 422,17.
1H ЯМР (ДМСО, 600 МГц): δ=8,22 (шир.с, 1H), 8,13 (дд, 1H, J=8,1 Гц, 1,5 Гц), 8,00 (д, 1H, J=8,0 Гц), 7,40 (д, 1H, J=8,2 Гц), 7,24 (д, 1H, J=8,2 Гц), 5,51 (с, 2H), 4,04 (с, 3H), 3,84 (д, 2H, J=6,5 Гц), 1,79 (м, 1H), 1,61-1,54 (м, 2H), 1,54-1,46 (м, 2H), 0,78 (д, 6H, J=6,7 Гц) м.д.
Пример 4
трет-Бутил-1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[,5-а]пиридин-2-ил]циклопропанкарбоксилат (соединение 4)
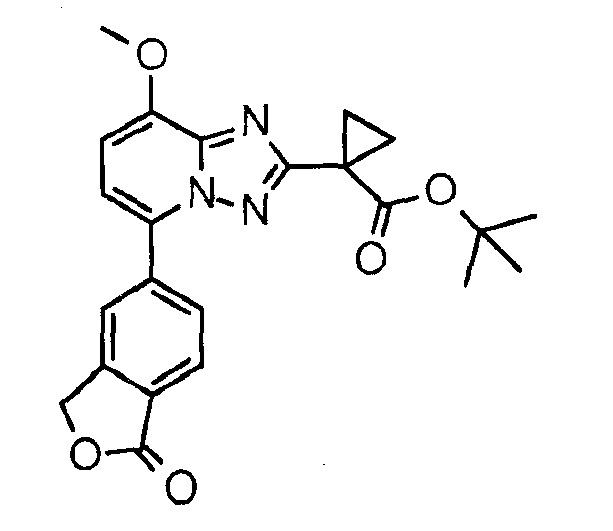
Суспензию 1-[8-метокси-5-(1-оксо-3Н-изобензофуран-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоновой кислоты (30 мг, 83 мкмоль) и хлорида бензилтриэтиламмония (19 мг, 83 мкмоль) в ДМФА (1,0 мл) осторожно нагревали до тех пор, пока она не становилась раствором. Добавляли трет-бутилбромид (297 мкл, 2,64 ммоль) и K2C03 (171 мг, 1,24 ммоль) и смесь перемешивали при 55°С в течение трех дней. Добавляли дополнительный трет-бутилбромид (139 мкл, 1,24 ммоль) и K2C03 (171 мг, 1,24 ммоль) и смесь перемешивали при 55°С в течение еще одного дня. Очистка препаративной ВЭЖХ давала указанное в заголовке соединение. Время удерживания по ВЭЖХ (XE Metode 7 СМ): 2,29 минуты. Определенная "М+1"-масса: 422,18. Вычисленная "M+1"-масса: 422,17.
1H ЯМР (ДМСО, 300 МГц): δ=8,27-8,22 (м, 1H), 8,16 (дд, 1H, J=8,0 Гц, 1,5 Гц), 8,00 (д, 1H, J=8,2 Гц), 7,40 (д, 1H, J=8,2 Гц), 7,22 (д, 1H, J=8,3 Гц), 5,51 (м, 2H), 4,04 (с, 3H), 1,54-1,40 (м, 4H), 1,38 (с, 9H) м.д.
АНАЛИЗЫ
Анализ PDE4
Рекомбинантную PDE4 человека (GenBank acession no NM_006203) инкубировали в течение 1 часа с тестируемым соединением при концентрации вплоть до 10 мкΜ, с цАМФ (1×10-5М) и с небольшим количеством (0,021 МВк) радиоактивно меченного цАМФ. В конце инкубации расщепление субстрата оценивали связыванием продукта АМФ со SPA-гранулами, которые генерируют хемилюминесценцию при связывании с радиоактивной меткой. Продукт АМФ ингибировал связывание радиоактивной метки с гранулами, и обнаруживали конкуренцию с люминесцентным сигналом.
Результаты вычисляли как молярные концентрации, приводящие к 50% ингибированию расщепления субстрата, по сравнению с контрольными образцами, и выражали в диапазоне величин IC50 (нМ).
Соединения настоящего изобретения испытывали в анализе PDE4, IC50 (нМ): соединение 1, 10,6 нМ; соединение 2, 13,0 нМ; соединение 3, 12,3 нМ; соединение 4, 20,7 нМ (на основе средней величины 2-5 испытаний для каждого соединения).
Высвобождение TNF-α
Мононуклеарные клетки периферической крови (РВМС) человека выделяли из лейкоцитарных пленок. Кровь смешивали с физиологическим раствором в отношении 1:1, и РВМС выделяли с использованием пробирок Lymphoprep TM (Nycomed, Норвегия). РВМС суспендировали в RPMI1640 с 0,5% сывороточным альбумином человека, пенициллин/стрептомицин (pen/strep) и 2 мМ L-глутамином при концентрации 5×105 c/мл. Клетки предварительно инкубировали в течение 30 минут с испытуемыми соединениями в 96-луночных планшетах для культуры тканей и стимулировали в течение 18 часов с липополисахаридом в количестве 1 мг/мл (Sigma). Концентрацию TNF-α в супернатантах измеряли с использованием гомогенного флуоресцентного резонанса с временным разрешением (TR-FRET). Количественные данные анализа получали измерением флуоресценции при 665 нм (пропорционально концентрации TNF-α) и 620 нм (контроль).
Результаты представлены в виде величин IC50 (нМ), вычисленных из кривых ингибирования с применением в качестве положительных контролей секрецию в стимулированных LPS лунках и в качестве отрицательных контролей секрецию в нестимулированных клетках.
Соединения настоящего изобретения испытывали в анализе высвобождения TNF-α, IC50(нМ): соединение 1 12,8 нМ; соединение 2, 15,7 нМ; соединение 3, 14,6 нМ; соединение 4, 15,3 нМ (на основе средней величины 2-5 испытаний для каждого соединения).
Анализ HLM (микросом печени человека)
Инкубации испытуемых соединений в ДМСО, разбавленном фосфатным буфером, рН 7,4, при концентрации 0,5 мкΜ проводили с микросомами печени человека (0,5 мг/мл). Процент органическом растворителе при инкубации составлял 1%. Суспензию микросом печени человека в фосфатном буфере смешивали с NADPH (1 мМ) и предварительно нагревали до 37°С перед добавлением испытуемого соединения. Аликвоты брали через 0, 5, 10, 20 и 30 минут и реакции терминировали добавлением метанола, содержащего аналитический внутренний стандарт (IS).
Результаты выражали в виде аппарентного клиренса (CIapp) (мл/мин/кг) и индекса экстракции печени (Eh) (%), вычисленного из константы скорости (к) (мин-1) истощения тестируемого соединения.
Соединения настоящего изобретения испытывали в анализе HLM, Eh (%): соединение 1, >91%; соединение 2, >91%; соединение 3, >91%; соединение 4, >91% (на основе среднего значения 2-3 испытаний для каждого соединения).
Анализ цельной крови (WB) человека
Инкубацию испытуемых соединений в ДМСО, разбавленном фосфатным буфером, рН 7,4, при концентрации 1 мкΜ проводили с цельной кровью человека. Процент органическом растворителе при инкубациях был 1%. Инкубацию проводили при 37°C с аликвотами, отобранными через 0, 15, 30, 60 и 120 минут, и реакции останавливали добавлением метанола, содержащего аналитический внутренний стандарт (IS).
Результаты выражали как период полураспада (T1/2) в минутах, вычисленный из константы скорости (к) (мин-1) истощения испытуемого соединения.
Образцы примеров соединений настоящего изобретения испытывали в анализе WB, Т1/2 (минуты): соединение 1, 10,7 минуты; соединение 2, 12,6 минуты; соединение 3, 16,6 минуты; соединение 4, <11,2 минуты (на основе средней величины 2-4 испытаний для каждого соединения).
Анализ стабильности кератиноцитов (КС)
Инкубацию испытуемых соединений в ДМСО, разбавленном средой для роста, рН ~7,4, при инкубации 1 мкΜ проводили с посеянными кератиноцитами человека. Процент органического растворителе при инкубации был 0,5%. Инкубации проводили при 37°C с аликвотами, отобранными через 0, 60, 120, 240 и 1440 минут, и реакции останавливали добавлением метанола, содержащего аналитический внутренний стандарт (IS).
Результаты выражали как период полураспада (T1/2) в минутах, вычисленный из константы скорости (к) (мин-1) истощения испытуемого соединения.
Образцы соединений примеров настоящего изобретения испытывали в анализе KC, T1/2 (минуты): соединение 1, >720 минут; соединение 2, >720 минут; соединение 3, >720 минут; соединение 4, >720 минут (на основе средней величины 2-4 испытаний для каждого соединения).
![[1,2,4]ТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ](https://fips.edrid.ru/images/rid/67/ee/05/4b9c84f136a3bbfd6775a40293ca5f6f.jpg)
![[1,2,4]ТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ](https://fips.edrid.ru/images/rid/67/ee/05/5c0c3fccec7c2ed22cb8f1fce467e597.jpg)
Замещенные метилфенилкетоны, пригодные для использования в качестве ингибиторов pde4
Новые ингибиторы фосфодиэстераз
Фармацевтическая композиция, содержащая аналог витамина d и смесь сорастворитель - поверхностно-активное вещество
Новые циклические углеводородные соединения для лечения заболеваний
Триазолопиридины в качестве ингибиторов фосфодиэстеразы для лечения кожных заболеваний
Нанокристаллы кальципотриола моногидрита
Фармацевтическая композиция, включающая растворяющую смесь и производное или аналог витамина d
Кожная композиция, включающая аналог витамина d и смесь растворителя и поверхностно-активных веществ
Кристаллический ингенол мебутат
Композиция фармацевтического аэрозоля, включающая аналог витамина d и кортикостероид
Замещенные метилфенилкетоны, пригодные для использования в качестве ингибиторов pde4
Новые ингибиторы фосфодиэстераз
Фармацевтическая композиция, содержащая аналог витамина d и смесь сорастворитель - поверхностно-активное вещество
Новые циклические углеводородные соединения для лечения заболеваний
Триазолопиридины в качестве ингибиторов фосфодиэстеразы для лечения кожных заболеваний
Нанокристаллы кальципотриола моногидрита
Фармацевтическая композиция, включающая растворяющую смесь и производное или аналог витамина d
Кожная композиция, включающая аналог витамина d и смесь растворителя и поверхностно-активных веществ
Кристаллический ингенол мебутат
Композиция фармацевтического аэрозоля, включающая аналог витамина d и кортикостероид

