ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ
Вид РИД
Изобретение
Настоящее изобретение касается 2-оксо-1-пирролидиновых производных, способов их получения, фармацевтических композиций, содержащих их, и их применения в качестве лекарственных средств.
ЕР 0162036 В1 раскрывает соединение (S)-α-этил-2-оксо-1-пирролидинацетамид, который имеет известное международное непатентованное название Леветирацетам.
Леветирацетам, левовращающее соединение, описано в качестве защитного агента для лечения и профилактики гипоксийного и ишемического типа заболеваний центральной нервной системы. Указанное соединение также эффективно при лечении эпилепсии, терапевтическое проявление которого, как было продемонстрировано, теряется полностью в случае его правовращающего энантиомера (R)-α-этил-2-оксо-1-пирролидинацетамида, также известного из ЕР 0165919 В1, (A.J.GOWER etal. Eur. J.Pharmacol., 222. (1992), 193-203).
Рацемический α-этил-2-оксо-1-пирролидинацетамид и его аналоги известны из Патента GB 1309692. US 3459738 раскрывает производные 2-оксо-1-пирролидинацетамида. ЕР 0645139 В1 раскрывает анксиолитическую активность Леветирацетама. РСТ/ЕРОО/1 1808 раскрывает применение Леветирацетама для методического и/или профилактического лечения биполярных заболеваний, мигрени, хронической и невропатической боли, также хорошо как и комбинация Леветирацетама с по крайней мере одним соединением, вызывающим невральное ингибирование, медицируемое GABAA рецепторами.
Неожиданно было найдено, что определенные аналоги Леветирацетама, особенно те, которые имеют дополнительное замещение в пирролидиновом кольце, демонстрируют значительно улучшенные терапевтические свойства.
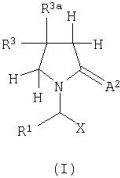
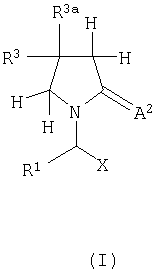
В первом аспекте, изобретение соответственно обеспечивает соединение, имеющее формулу I или его фармацевтически приемлемую соль,
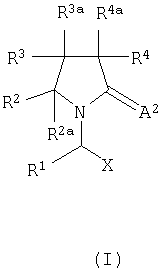
где X представляет собой -CA1NR5R6 или -CA1OR7 или -СА1-R8 или CN-;
А1 и А2 независимо являются кислородом, серой или -NR9;
R1 представляет собой водород, алкил, арил или -СН2-R1a, где R1a представляет собой арил, гетероцикл, галоген, гидрокси, амино, нитро или пиано;
R2, R3 и R4 являются одинаковыми или различными и каждый независимо представляет собой водород, галоген, гидрокси, тиол, амино, нитро, нитроокси, пиано, азидо, карбокси, амидо, сульфоновую кислоту, сульфонамид, алкил, алкенил, алкинил, сложный эфир, простой эфир, арил, гетероцикл или оксипроизводное, тиопроизводное, амин производное, ацилпроизводные, сульфонилпроизводное или сульфинилпроизводное;
R2a, R3a и R4a являются одинаковыми или различными и каждый независимо представляет собой водород, галоген, алкил, алкенил, алкинил или арил;
R5, R6, R7 и R9 являются одинаковыми или различными и каждый независимо представляет собой водород, гидрокси, алкил, арил, гетероцикл или оксипроизводное; и
R8 представляет собой водород, гидрокси, тиол, галоген, алкил, арил, гетероцикл или тиопроизводные;
при условии, что по крайней мере один из таких как R2, R3, R4, R2a, R3a и R4a отличен от водорода; и что когда соединение представляет собой смесь всех возможных изомеров, Х представляет собой CONR5R6, А2 является кислородом и R1 представляет собой водород, метил, этил или пропил, тогда замещение на пирролидиновом кольце отлично от моно-, ди- или триметила или моноэтила; и что когда R1, R2, R4, R2a, R3a и R4a каждый является водородом, А2 является кислородом и Х представляет собой CONR5R6, тогда R3 отличен от карбокси, сложного эфира, амидо, замещенного оксопирролидина, гидрокси, оксипроизводного, амино, аминопроизводных, метила, нафтила, фенила необязательно замещенного оксипроизводными или в пара положении атомом галогена.
В определениях, приведенных ниже, если другое не отмечено, R11 и R12 являются одинаковыми или различными и каждый независимо представляет собой амидо, алкил, алкенил, алкинил, ацил, сложный эфир, простой эфир, арил, аралкил, гетероцикл или оксипроизводное, тиопроизводное, ацилпроизводное, аминопроизводное, сульфонилпроизводное или сульфинилпроизводное, каждый необязательно замещен любой пригодной группой, включая, но не ограничивая, одним или более остатками, выбранными из низшего алкила или других групп, описанных ниже в качестве заместителей для алкила.
Термин «оксипроизводное» определен, как включающий -O-R11 группы, где R11 определен выше, за исключением "оксипроизводное". Неограничивающие примеры представляют собой алкокси, алкенилокси, алкинилокси, ацилокси, оксисложный эфир, оксиамидо, алкилсульфонилокси, алкилсульфинилокси, арилсульфонилокси, арилсульфинилокси, арилокси, аралкокси или гетероциклоокси, такие как пентилокси, аллилокси, метокси, этокси, фенокси, бензилокси, 2-нафтилокси, 2-пиридилокси, метилендиокси, карбонат.
Термин «тиопроизводное» определен, как включающий -S-R11 группы, где R11 определен выше, за исключением "тиопроизводное". Неограничивающие примеры представляют собой алкилтио, алкенилтио, алкинилтио и арилтио.
Термин «аминопроизводноое» определен, как включающий -NHR11 или - NHR11R12 группы, где R11 и R12 определены выше. Неограничивающие примеры представляют собой моно- или ди-алкил-, алкенил-, алкинил- и ариламино или смешанный амино.
Термин "ацилпроизводное» представляет собой радикал, полученный из карбоновой кислоты и, таким образом он определен, как включающий группы формулы R11-CO-, где R11 определен выше и он может также быть водородом. Неограничивающие примеры представляют собой формил, ацетил, пропинил, изобутирил, валерил, лауроил, гептандиоил, циклогексанкарбонил, кротонил, фумароил, акрилоил, бензоил, нафтоил, фуроил, никотиноил, 4-карбоксибутаноил, оксалил, этоксалил, цистеинил, оксамоил.
Термин "сульфонилпроизводное» определен, как включающий группу формулы -SO2-R11, где R11 определен выше, за исключением "сульфонилпроизводное". Неограничивающие примеры представляют собой алкилсульфонил, алкенилсульфонил, алкинилсульфонил и арилсульфонил.
Термин "сульфинилпроизводное» определен, как включающий группу формулы -SO-R11, где R11 определен выше, за исключением "сульфинилпроизводное". Неограничивающие примеры представляют собой алкилсульфинил, алкенилсульфинил, алкинилсульфинил и арилсульфинил.
Термин "алкил" определен, как включающий насыщенные, одновалентные углеводородные радикалы, имеющие линейные, разветвленные или циклические остатки или их комбинациии, содержащие 1-20 атомов углерода, предпочтительно 1-6 атомов углерода для нециклического алкила и 3-6 атомов углерода для циклоалкила (в этих двух предпочтительных случаях, если другое не определено, "низший алкил"). Алкильные остатки могут быть необязательно замещены от 1 до 5 заместителями независимо выбранными из группы, включающей галоген, гидрокси, тиол, амино, нитро, циано, тиоцианато, ацил, ацилокси, сульфонилпроизводное, сульфинилпроизводное, алкиламино, карбокси, сложный эфир, простой эфир, амидо, азидо, циклоалкил, сульфоновую кислоту, сульфонамид, тиопроизводное, оксисложный эфир, оксиамидо, гетероцикл, винил, С1-5-алкокси, С6-10-арилокси и С6-10-арил.
Предпочтительными алкильными группами являются метил, этил, пропил, изопропил, бутил, изо или трет-бутил, и 2,2,2-триметилэтил, каждый необязательно замещенный по крайней мере одним заместителем, выбранным из группы, включающей галоген, гидрокси, тиол, амино, нитро и циано, такой как трифторметил, трихлорметил, 2,2,2-трихлорэтил, 1,1-диметил-2,2-дибромэтил, 1,1-диметил-2,2,2-трихлорэтил.
Термин "алкенил" определен, как включающий оба разветвленные и неразветвленные, ненасыщенные углеводородные радикалы, имеющие по крайней мере одну двойную связь, такие как этенил (=винил), 1-метил-1-этенил, 2,2-диметил-1-этенил, "1-пропенил, 2-пропенил (=аллил), 1-бутенил, 2-бутенил, 3-бутенил, 4-пентенил, 1-метил-4-пентенил, 3-метил-1-пентенил, 1-гексенил, 2-гексенил, и тому подобное и которые могут быть необязательно замещены по крайней мере одним заместителем, выбранным из группы, включающей галоген, гидрокси, тиол, амино, нитро, пиано, арил и гетероцикл, такой как моно- и дигалогенвинил, где галоген представляет собой фтор, хлор или бром,
Термин "алкинил" определен, как включающий одновалентный разветвленный или неразветвленный углеводородный радикал, содержащий по крайней мере одну углерод-углеродную тройную связь, например этинил, 2-пропинил (=пропаргил), и тому подобное, и которые могут быть необязательно замещены по крайней мере одним заместителем, выбранным из группы, включающей галоген, гидрокси, тиол, амино, нитро, циано, арил и гетероцикл, такие как галогенэтинил.
Если присутствуют, в качестве мостиковых групп, алкил, алкенил и алкинил, то они представляют собой линейные- или разветвленные цепочки, С1-12, предпочтительно C1-4-алкилен или С2-12-, предпочтительно С2-4-алкенилен или - алкиниленовые остатки соответственно.
Группы, в которых разветвленные производные обычно определены префиксом, таким как «н», "втор.", "изо" и тому подобное (например, "н-пропил", "втор.-бутил") находятся в н-форме, если другое не отмечено.
Термин "арил" определен, как включающий органический радикал, полученный из ароматического углеводорода, содержащего 1-3 кольца и, содержащего 6-30 атомов углерода, путем удаления одного водорода, такой как фенил и нафтил, каждый необязательно замещенный от 1 до 5 заместителями независимо выбранными из галогена, гидрокси, тиола, амино, нитро, циано, ацила, ацилокси, сульфонила, сульфинила, алкиламино, карбокси, сложного эфира, простого эфира, амидо, азидо, сульфоновой кислоты, сульфонамида, алкилсульфонила, алкилсульфинила, алкилтио, оксисложного эфира, оксиамидо, арила, С1-6-алкокси, С6-10-арилокси, С1-6-алкила, С1-6-галогеналкила. Арильные радикалы предпочтительно являются моноциклическими, содержащими 6-10 атомов углерода. Предпочтительными арильными группами являются фенил и нафтил, каждый необязательно замещенный 1 до 5 заместителями независимо выбранными из галогена, нитро, амино, азидо, С1-6-алкокси, C1-6-алкилтио, C1-6-алкила, С1-6-галогеналкила и фенила. Термин "галоген" включает атом Cl, Br, F, I.
Термин "гидрокси" представляет собой группу формулы - ОН.
Термин "тиол" представляет собой группу формулы - SH.
Термин "пиано" представляет собой группу формулы - CN.
Термин "нитро" представляет собой группу формулы -NO2.
Термин "нитроокси" представляет собой группу формулы -ONO2-
Термин "амино" представляет собой группу формулы -NH2-.
Термин "азидо" представляет собой группу формулы -N3.
Термин "карбокси" представляет собой группу формулы -СООН.
Термин "сульфоновая кислота" представляет собой группу формулы -SO3Н.
Термин "сульфонамид" представляет собой группу формулы -SO2NH2-
Термин «сложный эфир» определен, как включающий группу формулы -COO-R11, где R11 определен выше, за исключением оксипроизводного, тиопроизводного или аминопроизводного.
Термин «простой эфир» определен, как включающий группу, выбранную из С1-50-линейного или разветвленного алкила, или С2-50-линейного или разветвленного алкенила или алкинильных групп или комбинации указанных групп, прерывающихся одним или более атомами кислорода.
Термин "амидо» определен, как включающий группу формулы -CONH2 или -CONHR11 или -CONR11R12, где R11 и R12 определены выше.
Термин "гетероцикл" определен, как включающий ароматический или неароматический циклический алкильный, алкенильный, или алкинильный остаток как определенный выше, имеющий по крайней мере один О, S и/или N атом, прерывающий карбоциклическую кольцевую структуру и необязательно, один атом углерода карбоциклической кольцевой структуры может быть замещен карбонилом. Неограничивающие примеры ароматических гетероциклов представляют собой пиридил, фурил, пирролил, тиенил, изотиазолил, имидазолил, бензимидазолил, тетразолил, хиназолинил, хинолизинил, нафтиридинил, пирадазинил, пиримидинил, пиразинил, хинолил, изохинолил, изобензофуранил, бензотиенил, пиразолил, индолил, индолизинил, пуринил, изоиндолил, карбазолил, тиазолил, 1,2,4-тиадиазолил, тиено(2,3-b)фуранил, фуропиранил, бензофуранил, бензоксепинил, изооксазолил, оксазолил, тиантренил, бензотиазолил или бензоксазолил, циннолинил, фталазинил, хиноксалинил, фенантридинил, акридинил, перимидинил, фенантролинил, фенотиазинил, фуразанил, изохроманил, индолинил, ксантенил, гипоксантинил, птеридинил, 5-азацитидинил, 5-азаурацилил, триазолопиридинил, имидазолопиридинил, пирролопиримидинил, и пиразолопиримидинил необязательно замещенные алкилом или как описано выше, для алкильных групп. Неограничивающие примеры неароматических гетероциклов представлюет собой тетрагидрофуранил, тетрагидропиранил, пиперидинил, пиперидинил, пиперазинил, имидазолидинил, морфолино, морфолинил, 1-оксаспиро(4.5)дек-2-ил, пирролидинил, 2-оксопирролидинил, остатки сахара (например глюкозы, пентозы, гексозы, рибозы, фруктозы, которые могут также быть замещены) или те же группы, которые могут необязательно быть замещены любой пригодной группой, включая, но не ограничивая одним или более остатков, выбранных из низшего алкила, или других групп как описано выше для алкильных групп. Термин "гетероцикл" также включает бициклические, трициклические и тетрациклические спирогруппы, в которых любое из приведенных выше гетероциклических колец сконденсировано с одним или более колец независимо выбранных из арильного кольца, циклогексанового кольца, циклогексенового кольца, циклопентанового кольца, циклопентенового кольца или другого моноциклического гетероциклического кольца или в том случае, если моноциклическая гетероциклическая группа связана мостиком, то такие как алкиленовая группа, такая как хинуклидинил, 7-азабицикло(2.2.1)гептанил, 7-оксабицикло(2.2.1)гептанил, 8-азабицикло(3.2.1)октанил.
Должно быть понятно, что в приведенных выше определениях, когда заместитель, такой как R2, R3, R4, R2a, R3a, R4a, R5, R6, R7, R8 присоединен к остальной части молекуле через гетероатом или карбонил, линейную- или разветвленную цепь, С 1-12-, предпочтительно C1-4-алкилен или С2-12, предпочтительно С2-4-алкенилен или - алкиниленовый мостик может необязательно быть вставлен между гетероатом или карбонилом и местом присоединения остальной части молекулы.
Предпочтительными примерами Х являются -COOR7 или-CONR5R6, где R5, R6 и R7 представляют собой предпочтительно водород, С1-4-алкил, фенил или алкилфенил.
Предпочтительно Х представляет собой карбокси или - CONR5R6, где R5 и R6 представляюет собой предпочтительно водород, С1-4-алкил, фенил или алкилфенил, особенно -CONH2.
Предпочтительно А1 и А2 каждый является кислородом.
Предпочтительно R1 представляют собой водород, алкил, особенно С1-12 алкил, в частности, низший алкил или арил, особенно фенил.
Примеры предпочтительных R1 групп являются метил, этил, пропил, изопропил, бутил, изо- или трет-бутил, 2,2,2-триметилэтил, каждый необязательно присоединенный через метиленовый мостик или те же группы, замещенные по крайней мере одним атомом галогена, такие как трифторметил, трихлорметил, 2,2,2-трихлорэтил, 1,1-диметил-2,2-дибромэтил, 1,1-диметил-2,2,2-три хлорэтил.
R1, как этил, особенно предпочтителен.
Предпочтительно R2 и R2a независимо представляют собой водород, галоген или алкил, особенно низший алкил.
Примеры предпочтительных R2 и R2a групп независимо представляют собой водород, галоген или метил, этил, пропил, изопропил, бутил, изо или трет-бутил, 2,2,2-триметилэтил или те же группы, замещенные по крайней мере одним атомом галогена, такие как трифторметил, трихлорметил, 2,2,2-трихлорэтил, 1,1-диметил-2,2-дибромэтил, 1,1-диметил-2,2,2-трихлорэтил.
Особенно по крайней мере один и наиболее предпочтительно оба R2 и R2a представляют собой водород.
Предпочтительно R3a, R4 и R4a независимо включает водород, алкил, особенно метил или этил или арил, особенно фенил, или аралкил, особенно бензил.
Примеры предпочтительных R3a, R4 и R4a групп независимо включают водород, галоген или метил, этил, пропил, изопропил, бутил, изо или трет-бутил, 2,2,2-триметилэтил или те же группы, замещенные по крайней мере одним атомом галогена, такие как трифторметил, трихлорметил, 2,2,2-трихлорэтил, 1,1-диметил-2,2-дибромэтил, 1,1-диметил-2,2,2-трихлорэтил.
Особенно по крайней мере один и наиболее предпочтительно оба R4 и R4a представляют собой водород.
R3a представляет собой, в частности, водород или алкил, особенно низший алкил и наиболее предпочтительно водород.
Предпочтительно R3 представляет собой водород, С1-12-алкил, особенно С1-6-алкил, каждый необязательно замещенный одним или более заместителями, выбранными из гидрокси, галогена, циано, тиопианато или алкокси и присоединенный к кольцу, либо непосредственно, либо через тио, сульфинильную, сульфонильную, карбонильную или оксикарбонильную группу и необязательно, С1-4-алкиленовый мостик, в частности, метиленовый; С2-6-алкенил или -алкинил, особенно С2-3-алкенил или -алкинил каждый необязательно замещенный одним или более галогенами; азидо; циано; амидо; карбокси; триазолилом, тетразолилом, пирролидинилом, пиридилом, 1-оксидопиридилом, тиоморфолинилом, бензодиоксолилом, фурилом, оксазолилом, пиримидинилом, пирролилом, тиадиазолилом, тиазолилом, тиенилом или пиперазинилом каждый необязательно замещенный одним или более заместителями, выбранными из галогена, C1-6-алкила и фенила и присоединенными к кольцу, либо непосредственно, либо через карбонильную группу или а С1-4-алкиленовый мостик, в частности, метиленовый; нафтил; или фенил, фенилалкил или фенилалкенил, каждый необязательно замещенный одним или более заместителями, выбранными из галогена, C1-6-алкила, С1-6 галогеналкила, С1-6-алкокси, C1-6-алкилтио, амино, азидо, фенила и нитро и каждый присоединенный к кольцу, либо непосредственно, либо через окси, сульфонильную, сульфонилокси, карбонильную или карбонилокси группу и необязательно дополнительно С1-4-алкиленовый мостик, в частности, метиленовый.
Также, предпочтительно, R3 представляет собой C1-6-алкил, необязательно замещенный одним или более заместителем, выбранным из галогена, тиоцианато, азидо, алкокси, алкилтио, фенилсульфонила; нитроокси; С2-3-алкенил или -алкинил, каждый необязательно замещенный одним или более галогенами или ацетилом; тетразолил, пиридил, фурил, пирролил, тиазолил или тиенил; или фенил или фенилалкил каждый необязательно замещенный одним или более заместителями, выбранными из галогена, C1-6-алкила, С1-6 галогеналкила, С1-6-алкокси, амино, азидо, фенила и нитро и каждый присоединенный к кольцу, либо непосредственно, либо через сульфонилокси и необязательно дополнительно С1-4-алкиленовый мостик, в частности метиленовый.
Другие примеры предпочтительных R3 группы включают водород, галоген или метил, этил, пропил, изопропил, бутил, изо или трет-бутил, 2,2,2-триметилэтил или те же группы, замещенные по крайней мере одним атомом галогена, такие как трифторметил, трихлорметил, 2,2,2-трихлорэтил, 1,1-диметил-2,2-дибромэтил, 1,1-диметил-2,2,2-трихлорэтил.
R3 представляет собой особенно C1-4-алкил, необязательно замещенный одним или более заместителями, выбранными из галогена, тиоцианато или азидо; С2-5-алкенил или -алкинила, каждый необязательно замещенный одним или более галогенами; тиенил; или фенил необязательно замещенный одним или более заместителями, выбранными из галогена, C1-6-алкила, С1-6 галогеналкила или азидо.
Дополнительные примеры предпочтительных R3 групп представляют собой С1-6 алкил и С2-6 галогеналкенил.
Предпочтительно R5 и R6 независимо представляют собой водород, метил, этил, пропил, изопропил, бутил, изо или трет-бутил, 2,2,2-триметилэтил, особенно водород или метил. Особенно по крайней мере один и наиболее предпочтительно оба R5 и R6 представляют собой водород.
Предпочтительно R7 представляет собой водород, метил, этил, пропил, изопропил, бутил, изо или трет-бутил, 2,2,2-триметилэтил, метокси, этокси, фенил, бензил или те же группы, замещенные по крайней мере одним атомом галогена, такие как трифторметил, хлорфенил.
Предпочтительно R7 представляет собой водород, метил или этил, особенно водород.
Предпочтительно R8 представляют собой водород, метил, этил, пропил, изопропил, бутил, изо или трет-бутил, 2,2,2-триметилэтил, фенил, бензил или те же группы, замещенные по крайней мере одним атомом галогена, такие как трифторметил, хлорбензил.
Предпочтительно R8 представляет собой водород или метил.
Комбинации одного или более указанных предпочтительных групп соединений особенно предпочтительны.
В особенности, группа соединений формулы I (Соединения 1 А) содержит те, в которых
А2 представляет собой кислород;
Х представляет собой -CONR5R6 или -COOR7 или -CO-R8 или CN;
R1 представляет собой водород или алкил, арил, галоген, гидрокси, амино, нитро, циано;
R2, R3, R4, являются одинаковыми или различными и каждый независимо представляет собой водород или галоген, гидрокси, амино, нитро, пиано, ацил, ацилокси, сульфонилпроизводное, сульфинилпроизводное, аминопроизводное, карбокси, сложный эфир, простой эфир, амидо, сульфоновую кислоту, сульфонамид, алкоксикарбонил, тиопроизводное, алкил, алкокси, оксисложный эфир, оксиамидо, арил, оксипроизводное, гетероцикл, винил и R3 может дополнительно представлять собой С2-5 алкенил, С2-5 алкинил или азидо, каждый необязательно замещенный одним или более галогеном, циано, тиоциано, азидо, циклопропилом, ацилом и/или фенилом; или фенилсульфонилокси, где любая фенильная часть может быть замещенной от одного или более галогеном, алкилом, галогеналкилом, алкокси, нитро, амино, и/или фенилом; наиболее предпочтительно метил, этил, пропил, изопропил, бутил или изобутил,
R2a, R3a и R4a представляют собой водород;
R5, R6, R7 являются одинаковыми или различными и каждый независимо представляет собой водород, гидрокси, алкил, арил, гетероцикл или оксипроизводное; и
R8 представляет собой водород, гидрокси, тиол, галоген, алкил, арил, гетероцикл, алкилтио или тиопроизводное.
Среди этих соединений 1А, R1 предпочтительно подставляет собой метил, этил, пропил, изопропил, бутил, или изобутил; наиболее предпочтительно метил, этил или н-пропил.
R2 и R4 предпочтительно представляют собой независимо водород или галоген или метил, этил, пропил, изопропил, бутил, изобутил; и, наиболее предпочтительно, каждый является водородом.
R3 представляет собой предпочтительно С1-5 алкил, С2-5 алкенил, С2-С5 алкинил, циклопропил, азидо, каждый необязательно замещенный одним или более галогеном, циано, тиоциано, азидо, алкилтио, циклопропилом, ацилом и/или фенилом; фенил; фенилсульфонил; фенилсульфонилокси, тетразол, тиазол, тиенил, фурил, пиррол, пиридин, где любая фенильная часть может быть замещенной от одного или более галогеном, алкилом, галогеналкилом, алкокси, нитро, амино; и/или фенилом; наиболее предпочтительно метил, этил, пропил, изопропил, бутил, или изобутил.
Х представляет собой предпочтительно -СООН или -СООМе или -COOEt или -CONH2; наиболее предпочтительно -CONH2.
Кроме того, в особенности группа соединений формулы I (Соединения IB) включает те, в которых:
Х представляет собой -CA1NH2, -СА1NHCH3 или -СА1N(СН3)2;
R1 представляет собой алкил или фенил;
R3 представляет собой алкил, алкенил, алкинил, циано, изотиоцианато, сложный эфир, карбоксил, амидо, арил, гетероцикл; или
R3 представляет собой СН2R10, где R10 представляют собой водород, циклоалкил, оксисложный эфир, оксиалкилсульфонил, оксиарилсульфонил, аминоалкилсульфонил, аминоарилсульфонил, нитроокси, циано, изотиоцианато, азидо, алкилтио, арилтио, алкилсульфинил, алкилсульфонил, гетероцикл, арилокси, алкокси или трифторэтил;
R3a представляет собой водород, алкил или арил (особенно при условии, что когда R3a представляет собой водород, R3 отличен от метила);
или R3R3a образуют циклоалкил;
и R2, R2a, R4 и R4a каждый является водородом.
Среди соединений формулы I,
R1 предпочтительно представляет собой алкил, особенно С1-12-, более предпочтительно, С1-6-алкил и наиболее предпочтительно этил;
R2, R2a, R3а и R4a представляют собой предпочтительно водород;
R3 предпочтительно выбрают из водорода; C1-12-алкила, особенно С1-6-алкила, каждый необязательно замещенный одним или более заместителями, выбранными из гидрокси, галогена, циано, тиоцианато или алкокси и присоединенными к кольцу, либо непосредственно, либо через тио, сульфинильную, сульфонильную, карбонильную или оксикарбонильную группу и необязательно дополнительно С1-4-алкиленовый мостик, в частности метиленовый; С2-6-алкенил или -алкинил, особенно С2-3-алкенил или - алкинил, каждый необязательно замещенный одним или более галогеномами;
азидо; циано; амидо; карбокси; триазолилом, тетразолилом, пирролидинилом, пиридилом, 1-оксидопиридилом, тиоморфолинилом, бензодиоксолилом, фурилом, оксазолилом, пиримидинилом, пирролилом, тиадиазолилом, тиазолилом, тиенилом или пиперазинилом, каждый необязательно замещенным одним или более заместителями, выбранными из галогена, C1-6-алкила и фенила, и присоединенных к кольцу, либо непосредственно, либо через карбонильную группу или С1-4-алкиленовый мостик, в частности метиленовый; нафтил; или фенил, фенилалкил или фенилалкенил, каждый необязательно замещенный одним или более заместителями, выбранными из галогена, C1-6-алкила, С1-6 галогеналкила, С1-6-алкокси, С1-6-алкилтио, амино, азидо, фенила и нитро и каждый присоединенный к кольцу, либо непосредственно, либо через окси, сульфонил, сульфонилокси, карбонил или карбонилокси группу и необязательно дополнительно С1-4-алкиленовый мостик, в частности метиленовый;
R3a предпочтительно представляет собой водород или С1-4-алкил;
R4 и R4a предпочтительно, независимо представляют собой водород, С1-4-алкил, фенил или бензил.
Другая группа соединений формулы I (соединения 1C) включают те из них в рацемической форме, в которых когда Х представляет собой -CONR5R6 и R1 представляет собой водород, метил, этил или пропил, тогда замещение на пирролидиновом кольце отлично от моно-, ди- или триметила или моноэтила.
Еще одна группа соединений формулы I (соединения ID) включают те из них в рацемической форме, в которых когда Х представляет собой -CONR5R6 и R1 представляет собой водород или С1-6-алкил, С2-6-алкенил или - алкинил или циклоалкил, каждый незамещенный, тогда замещение в кольце отлично от алкила, алкенила или алкинила, каждого незамещенного.
Следующая характерная группа соединений формулы I (соединения IE) содержит те, в которых
Х представляет собой -CA1NH2;
R1 представляет собой Н;
R3 представляет собой азидометил, иодометил, этил необязательно замещенные 1 до 5 атомами галогена, н-пропил, необязательно замещенный 1 до 5 атомами галогена, винил, необязательно замещенный одним или двумя метилами, и/или от 1 до 3 атомов галогена, ацетилен, необязательно замещенный С1-4-алкилом, фенилом или галогеном;
R3a представляет собой водород или галоген, предпочтительно фтор;
и R2, - R2a, R4 и R4a каждый является водородом;
и их рацематы или их энантиомерно обогащенную форму, предпочтительно чистые энантиомеры.
Еще одна характерная группа соединений формулы I (соединения IF) содержит те, в которых
Х представляет собой -СА1NH2;
R1 представляет собой Н;
R3 представляет собой C1-6-алкил, С2-6-алкенил или С2-6-алкинил, необязательно замещенный азидо, оксинитро, от 1 до 6 атомами галогена;
R3a представляет собой водород или галоген, предпочтительно фтор;
и R2, R2a, R4 и R4a каждый является водородом;
и их рацематы или их энантиомерно обогащенную форму образуют, предпочтительно чистые энантиомеры.
В перечисленных выше границах изобретения, когда атом углерода, к которому R1 присоедин является ассиметричным, предпочтительно соединение находится в "S" - конфигурации.
"фармацевтически приемлемые соли" согласно изобретению включают терапевтически активные, нетоксичные формы с основанием и кислотой соединений формулы I, способных образовывать такие формы.
Форма кислотно-аддитивной соли соединения формулы I, которое существует в свободной форме, в виде основания, может быть получена обработкой свободного основания, подходящей кислотой, такой как неорганическая кислота, например, гидрогалогеновой, такой как гидрохлорная или гидробромная, серной, азотной, фосфорной и тому подобное; или органической кислотой, такой как, например, уксусная, гидроксиуксусная, пропановая, молочная, пировиноградная, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, пальмоиновая и им подобные.
Соединения формулы I, содержащие кислотные протоны, могут быть превращены в их терапевтически активные, нетоксичные основные аддитивные солевые формы, например, соли с металлом или амином, обработкой подходящим органическим и неорганическим основаниями. Подходящие основные солевые формы включают, например, аммониевые соли, соли щелочного и щелочноземельного металла, например, соли лития, натрия, калия, магния, кальция и им подобные, соли с органическими основаниями, например, соли N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими, как, например, аргинин, лизин и им подобные.
Наоборот, указанные солевые формы могут быть превращены в свободные формы обработкой подходящим основанием или кислотой.
Соединения формулы I и их соли могут находиться в виде сольвата, что включено в объем настоящего изобретения. Такие сольваты включают, например, гидраты, алкоголяты и им подобные.
Многие соединения формулы I и некоторые их промежуточные соединения имеют по крайней мере один стереогенный центр в их структуре. Этот стереогенный центр может находиться в R- или S-конфигурации, указанное R- и S-обозначение используется в соответствии с правилами, описанными в Pure Appl. Chem., 45 (1976) 11-30.
Изобретение также относится ко всем стереоизомерным формам, таким, как энантиомерные и диастереоизомерные формы соединений формулы I или их смесям (включая все возможные смеси стереоизомеров).
Более того, определенные соединения формулы I, которые содержат алкенильные группы, могут существовать в виде Z (zusammen) или Е (entgegen) изомеров. В каждом случае изобретение включает как смесь, так и отдельные индивидуальные изомеры.
Составные заместители пирролидонового кольца могут также находиться в cis или trans положениях друг к другу относительно плоскости пирролидонового кольца.
Некоторые соединения формулы I могут также существовать в таутомерных формах. Такие формы, хотя и не подробно описаны в указанной выше формуле, подразумевается, включены в объем настоящего изобретения.
С точки зрения настоящего изобретения относительно соединения или соединений следует включать это соединение в любых его возможных изомерных формах и их смесях, если отдельная изомерная форма не указана особо.
Изобретение также включает в свой объем пролекарственные формы соединений формулы I и их различные подразделы и подгруппы.
Термин "пролекарство" как здесь используется включает формы соединения, которые легко трансформируются in vivo в родственное соединение по изобретению, например, при гидролизе в крови. Пролекарствами являются соединения, переносимые группы которых удаляются биотрансформацией до проявления их фармакологического действия. Такие группы включают группы, которые легко отщепляются in vivo от соединения их несущего, и соединение после отщепления остается или становится фармакологически активным. Метаболически отщепляемые группы составляют класс групп, хорошо известных специалисту данного уровня техники. Они включают, но не ограничиваются ими, такие группы, как алканоил (т.е. ацетил, пропионил, бутирил и им подобные), незамещенный и замещенный карбоциклический ароил (такой, как бензоил, замещенный бензоил и 1- и 2-нафтоил), алкоксикарбонил (такой, как этоксикарбонил), триалкилсилил (такой, как триметил- и триэтилсилил), моноэфиры, образованные с дикарбоновыми кислотами (такие, как сукцинил), фосфат, сульфат, сульфонат, сульфонил, сульфинил и им подобные. Соединения, несущие метаболически отщепляемые группы, имеют то преимущество, что они могут проявлять улучшенную биодоступностъ в результате повышенной растворимости и/или скорости абсорбции, придаваемыми родственному соединению благодаря наличию метаболически отщепляемой группы. Т.Higuchi и V.Stella, "Pro-drugs as Novel Delivery System", Vol.14 of the A.C.S. Symposium Series; "Bioreversible Carriers in Drug Design", ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
Соединения формулы I no изобретению могут быть получены аналогично стандартным способам, известным специалисту данного уровня техники в синтетической органической химии.
Следующее описание способа описывает далее некоторые пути синтеза пояснительным способом. Другие альтернативные и/или аналогичные способы могут быть легко понятны специалисту данного уровня техники. Как здесь используется относительно обозначений заместителя, "=" обозначает "представляет собой" и "≠" обозначает "иначе, чем".
А. ЦИКЛИЗАЦИЯ АМИНОЭФИРА.
Когда в формуле I А2=О, циклизуют аминоэфир формулы AA-II, в котором Q1 вместе с кислородом, к которому он присоединен, образует уходящую группу, особенно Q1 является алкильной группой, особенно линейной или разветвленной алкильной группой, имеющей от 1 до 4 атомов углерода.
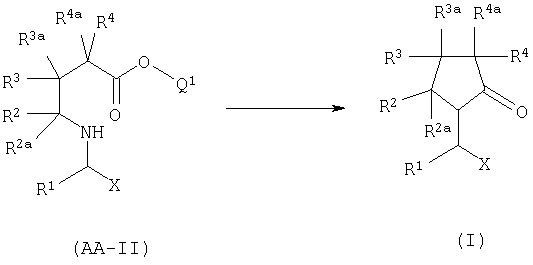
Q1 = метил или этил. Реакция известна и обычно проводится между комнатной температурой и 150°С, в присутствии или отсутствии катализатора, такого, как уксусная кислота, гидроксибензотриазол или 2-гидроксипиридин.
Q1 ≠ метил или этил. Эфир формулы AA-II гидролизуют в кислотных или основных условиях, затем циклизуют в обычных условиях пептидного синтеза, используя связующие агенты, например дициклогексилкарбодиимид (Bodanszky, М., Bodanszky, А., в "The Practice of Peptide Synthesis", Springer Verlag, 1984).
A.I Синтез AA-II присоединением по производному итаконата.
Соединения формулы АА-П, в которых R2a=R3a=Н и R3=COOQ2, в которых Q2 представляет собой линейную или разветвленную алкильную группу, необязательно оптически активную, получают реакцией соединения формулы AA-III с производным итаконата формулы AA-IV в соответствии с уравнением:
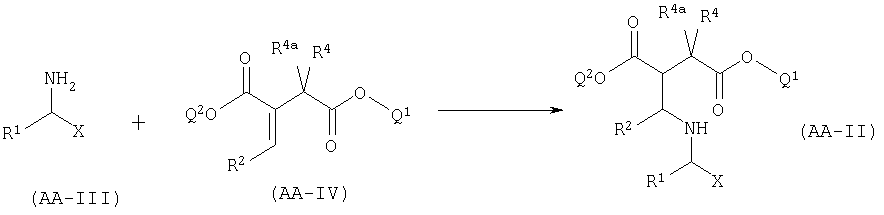
Эта реакция может быть осуществлена в соответствии с методикой, описанной в: Street, L.J., Baker, R., Book, Т., Kneen, CO., ManLeod, A.M., Merchant, K.J., Showell, G.A., Saunders, J., Herbert, R.H., Freedman, S.B., Harley, E.A., J. Med. Chem. (1990), 33, 2690-2697.
A.2 Синтез АА-II восстановительным аминированием.
Соединение формулы AA-II может быть получено восстановительным аминированием соединения формулы AA-V с соединением формулы AA-III в соответствии с уравнением:
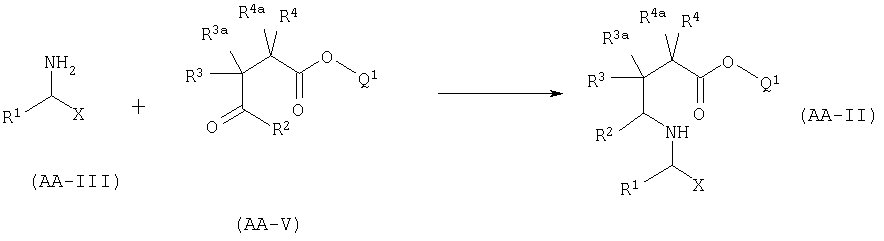
Эта реакция может быть осуществлена используя условия, описанные в Abdel-Magid, A.F., Harris, B.D., Maryanoff, C.A, Synlett (1994), 81-83. Альтернативно, когда X представляет собой CONR5R6, амин AA-III может быть присоединен через амидную группу к твердой подложке (например Rink-смоле). Соединения формулы AA-V могут быть получены одним из следующих способов:
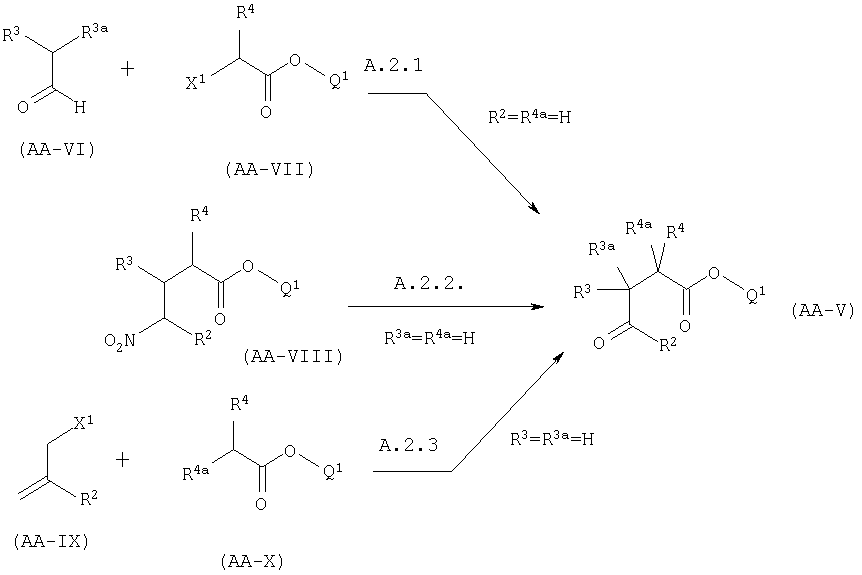
А.2.1. Альдегид формулы AA-VI алкилируют алкилгалогеноацетатом формулы AA-VII, в котором X1 представляет собой атом галогена, используя промежуточные енамины, как описано в Whitessell, J.K., Whitessell, M.A., Synthesis, (1983), 517-536 или используя гидразоны, как описано в Согеу, E.J., Enders, D., Tetrahedron Lett. (1976), 11 - 14 с последующим озонолизом.
А2.2. Нитроэфир формулы AA-VIII может быть превращен в соединение AA-V обработкой его основного конъюгата серной кислотой в метаноле и гидролизом промежуточного диметилацеталя (Nef реакция как в Urpi, F., Vilarrasa, J., Tetrahedron Lett. (1990), 31, 7499-7500). Нитроэфир формулы АА-VIII может быть получен как описано в Homi, A., Hubacek, I., Hesse, M., Helv. Chim. Acta (1994), 77, 579.
А.2.3. Эфир АА-Х алкилируют аллилгалогенидом AA-IX (X1=атом галогена) в присутствии сильного основания (например, диизопропиламида лития), с последующим восстановительным озонолизом ненасыщенного эфира как описано в Amruta Reggy Р., Hsiang B.C.H., Latffi T.N., Hill M.W., Woodward K.E., Rothman S.M., Ferrendelli J.A, Covey D.F., J. Med. Chem. (1996), 39, 1898-1906.
А.3. Синтез AA-II алкилированием у-галогеноэфира.
Соединение формулы AA-II, в котором Х=CONR^6, COOR7 или CN, может быть получено алкилированием γ-галогеноэфира AA-XI, в котором X2 представляет собой атом галогена, с амином AA-III.
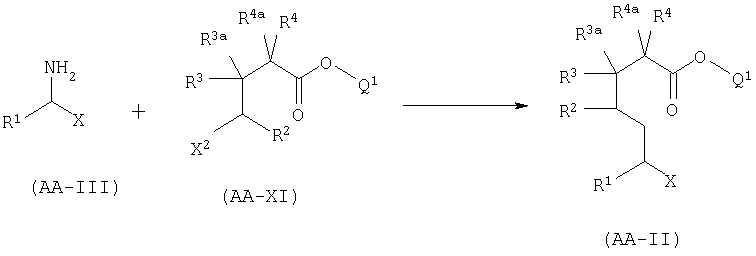
Эта реакция может быть проведена используя условия, описанные в GB 2225322 А. Синтез эфира AA-XI описан в части В.
А.4. Синтез AA-II восстановительным аминированием производных 5- гидроксилактона.
Соединение формулы AA-II, в котором Х=CONR5R6, COOR7 или CN, Q1=Н и R2a=Н может быть получено восстановительным аминированием 5-гидроксилактона формулы АА-XII с амином формулы AA-III в соответствии с уравнением:
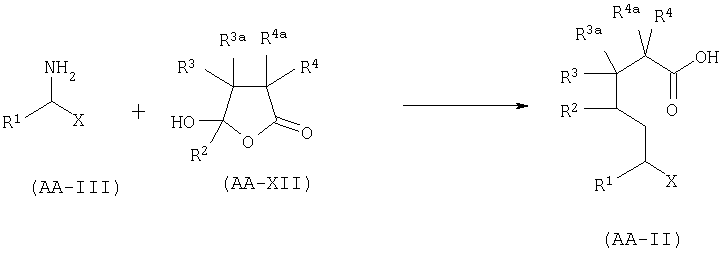
5-гидроксилактон формулы АА-ХП может быть синтезирован как описано в B.1.
В. КОНДЕНСАЦИЯ АМИНА С ПРОИЗВОДНЫМ γ-ГАЛОГЕНОКИСЛОТЫ.
Когда в формуле I A2=Q, Х=CONR7R8, COOR7 или CN и R2a=Н, соединение формулы AA-XIII реагирует с амином формулы AA-III в соответствии с уравнением:
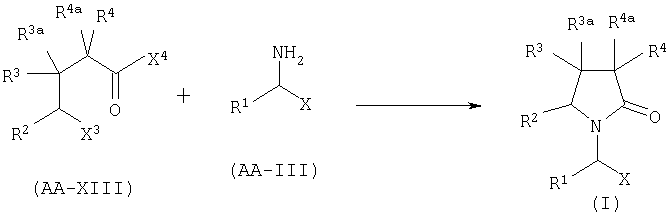
где X3 представляет собой атом галогена, предпочтительно атом йода или хлора, X4 представляет собой атом галогена, предпочтительно атом хлора. Эта реакция может быть проведена как описано в GB 2225322 А. Соединения формулы AA-XIII могут быть получены раскрытием лактона формулы AA-XIV в присутствии галогенирующего агента, например TMSI, SOCl2/ZnCl2 (при необходимости с последующим галогенированием полученной галогенокислоты (X4=ОН)) в соответствии с уравнением:
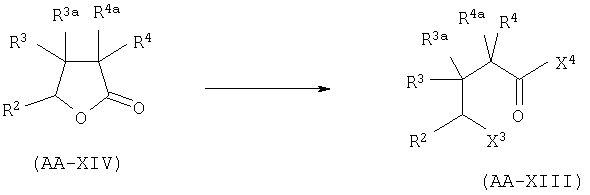
Раскрытие лактона AA-XIV может быть осуществлено в соответствии с методикой, описанной в: Mazzini, С., Lebreton, I, Alphand, V., Furstoss, R., Tetrahedron Lett. (1998), 38,1195-1196 и в Olah, G.A., Narang, S.C., Gupta, B.G.B., Malhotra, R., J. Org. Chem. (1979), 44,1247-1250. Галогенирование (X4=галоген) или этерификация (X4=OQ1) полученной галогенокислоты (X=ОН) может быть осуществлена в условиях, известных специалисту данного уровня техники. Лактоны формулы AA-XIV могут быть получены по одной из следующих методик:
B.I. Гидрирование или конъюгатное добавление металлоорганики.
Соединение AA-XIV, в котором R2a=R4a=Н, может быть получено гидрированием α,β-ненасыщенного лактона формулы AA-XV, или конъюгатным добавлением металлоорганического производного формулы R3M, в котором М представляет собой Li, Na, Mg или Zn, к соединению AA-XV, в конце катализируемого солями меди (I).
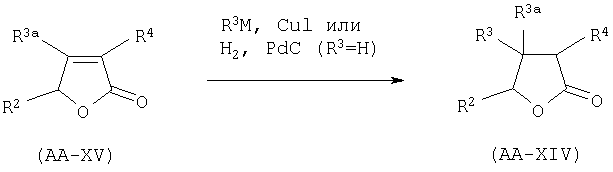
Эта реакция может быть проведена в соответствии с методиками, описанными в: Alexakis, A., Berlan, J., Besace, Y., Tetrahedron Lett. (1986), 27, 1047-1050; Upshutz, B.H., Ellsworth, E.L., Siahaan, Т., J. Amer. Chem. Soc. (1989), 111, 1351-1358, или при любом условии, известном среднему специалисту данного уровня техники.
В.2 Восстановление сукцинатного производного.
Когда в формуле AA-XIV R2=R2a=Н: восстановление карбоновой кислоты AA-XVI проводят в присутствии боргидридного реагента, предпочтительно LiBH4 или Са(ВН4)2, в спиртовом растворителе, в соответствии с уравнением:
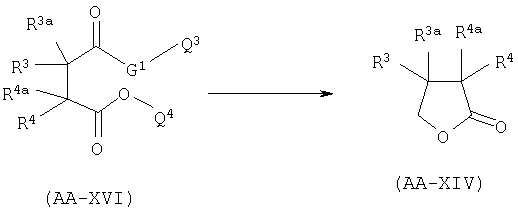
где Q3 представляет собой метильную или этильную группу, G1 представляет собой О или S и Q4 представляет собой атом водорода или линейный или разветвленный алкил, имеющий от 1 до 4 атомов углерода, при условии, что когда G1=S, Q4 = алкил и когда G1=О, Q4=Н.
С. АЛКИЛИРОВАНИЕ ПРОИЗВОДНОГО ЛАКТАМА.
Когда в формуле I А2=О и Х=COOR7, соединение формулы AA-XVII реагирует с соединением формулы АА-XVIII в соответствии с уравнением:
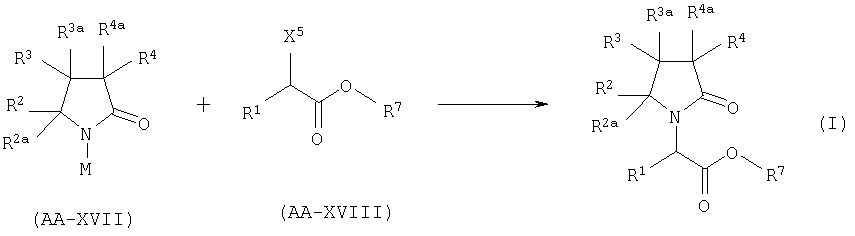
где X5 представляет собой атом галогена и М щелочной металл. Эта реакция может быть проведена при использовании методики, описанной в GB (case 15-09).
Соединения формулы AA-XVII могут быть получены в соответствии с методикой, описанной в Horni, A., Hubacek, I., Hesse, M., Helv. Chim. Acta (1994), 77,579.
D. ПРЕВРАЩЕНИЕ ПРОИЗВОДНОГО ЭФИРА.
Когда в формуле I А2=О и Х=CONR5R6, ни одна из групп R2, R2a, R3, R3a, R4 и R4a не является замещенной карбоксилом, сложным эфиром или сульфоновой кислотой, соответствующий эфир формулы
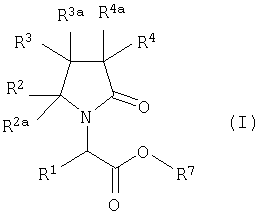
где R7 представляет собой атом водорода или линейную или разветвленную алкильную группу, имеющую от 1 до 4 атомов углерода, превращают в амин прямым аммонолизом или в условиях обычного пептидного синтеза, используя амин и связующие агенты, например алкилхлорформиат или дициклогексилкарбодиимид.
Е. ВОССТАНОВЛЕНИЕ α,β-НЕНАСЫЩЕННОГО ЛАКТАМА.
Когда в формуле I А2=О и R2a=R3a=R4a=Н, соединения формулы I могут быть получены восстановлением ненасыщенного лактама АА-XIX:
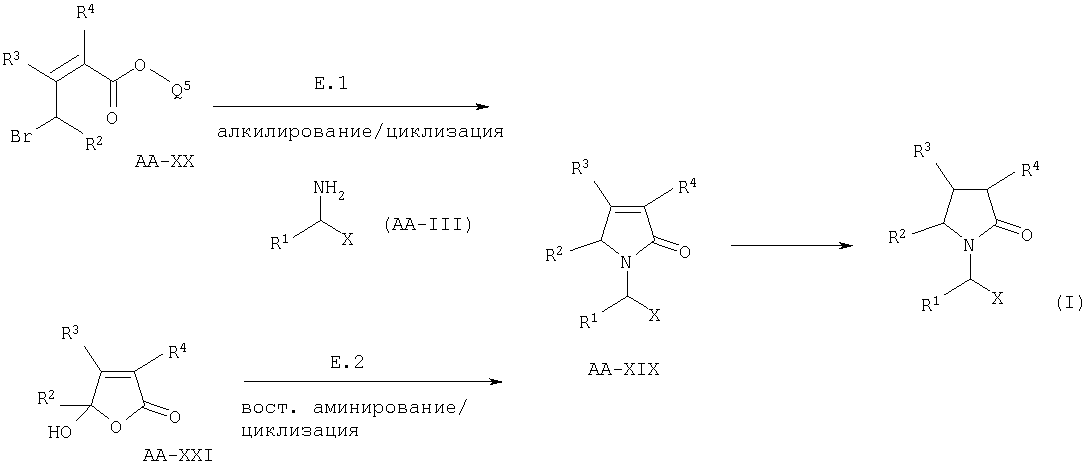
Стадия восстановления может быть осуществлена в классических условиях, известных среднему специалисту данного уровня техники, например водородом в присутствии Pd/C или необязательно в присутствии оптически активного катализатора. Когда R2, R3 или R4 чувствительны к гидрированию в условиях низкого давления, например, используя Pd/C в качестве катализатора, двойная связь смеси олефина может быть селективно восстановлена NaBH4 в присутствии CoCl2.
Соединения AA-XIX могут быть получены по одной из следующих методик:
E.1 Алкилированием
Соединение формулы АА-III алкилируют соединением формулы АА-ХХ, в которых Q5 представляет собой линейную или разветвленную алкильную группу, имеющую от 1 до 4 атомов углерода, и циклизуют. Стадия алкилирования может быть проведена в инертном растворителе, например, тетрагидрофуране, диметилформамиде или дихлорметане, между 0 и 50°С, в присутствии или отсутствии третичного амина. Реакция циклизации может протекать самопроизвольно или может быть проведена в соответствии с методикой, описанной в части А.
Е.2 Восстановительным аминированнем
Соединение формулы AA-XXI реагирует с соединением формулы AA-III в условиях восстановительного аминирования. Первая стадия этой реакции может быть проведена в инертном растворителе, например толуоле между 0 и 50°С, в присутствии восстановительных агентов, таких, как NaBH3CN, и в присутствии кислоты, например уксусной кислоты. Синтез соединения AA-XXI описан в Bourguignon, J.J. и др., J. Med. Chem. (1988), 31, 893-897.
F. ПРЕВРАЩЕНИЕ ФУНКЦИОНАЛЬНОЙ ГРУППЫ БОКОВОЙ ЦЕПИ.
F.1 Восстановление сложных эфиров в спирты.
Соединения формулы I, в которых А2=О, Х=CONR5R6 или COOR7, R7 является третичной алкильной группой, и одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-COOQ6, G2 является связью или алкиленовой группой и Q6 является линейной или разветвленной алкильной группой, имеющей от 1 до 4 атомов углерода, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2- СН2ОН. Эти превращения могут быть осуществлены в любых условиях, известных среднему специалисту данного уровня техники.
F.2 Активация и окисление спиртов.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a, R4 иR4a представляет собой -G2-CH2OH, G2 являются связью или алкиленовой группой, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-СН2Х6 или -G2-CHO, где X6 представляет собой атом хлора, брома или йода или группу формулы -О-SO2-Q7 или -О-Q8, Q7 является алкильной или арильной группой и Q8 является алкильной группой. Эти превращения могут быть осуществлены при любых условиях, известных среднему специалисту данного уровня техники.
F.3 Нуклеофильное замещение активированных спиртов.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-CH2X6, G2 является связью или алкиленовой группой и X6 является атомом хлора, брома или йода или группой формулы -O-SO2-O7 является таким, как описано в F.2, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-CH2X7, где X7 представляет собой азидо, галоген, нитро, амино, аминопроизводные, тиопроизводные и гетероциклы. Эти превращения могут быть осуществлены при любых условиях, известных среднему специалисту данного уровня техники.
F.4 Олефинированием альдегида.
Соединения формулы I, в которых А2=О, Х=CONR5R6, COOR7 или CN, и одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-CHO, G2 является связью или алкиленовой группой, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-Q9, где Q9 представляет собой винильную группу, не замещенную, моно- или ди- замещенную атомом галогена или алкильной группой. Эти превращения могут быть осуществлены при любых условиях, известных среднему специалисту данного уровня техники.
Альтернативно, соединения -G2-CN могут быть получены из соответствующего альдегида реакцией его оксима с SeO2 (как описано в Earl, R.A., Vollhardt, K.P.C., J. Org. Chem. (1984), 49, 4786).
F.5 Превращение производного кислоты в гетероциклы.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a, R4 иR4a представляет собой -G2-CN или -G2-COQ10, G2 является связью или алкиленовой группой и Q10 является алкокси, арилокси или аминогруппой, атомом галогена или аминопроизводным, при условии, что -COQ10 отличен от X, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-Q11, где Q11 представляет собой либо (i) -CO- арил/гетероцикл, палладий-катализируемым сочетанием хлорангидрида кислоты -G2-COCl и арил/гетероциклической металлоорганикой, например триметилпиридилоловом, или (ii) гетероцикл, например, тиазол (Friedman, B.S., Sparks, M., Adams, R., J. Amer. Chem. Soc. (1933), 55, 2262 или in Iroka, N., Hamada, Y., Shiori, Т., Tetrahedron (1992), 48, 7251), оксазол (in Street, LJ., Baker, R., Castro, J.L, Clamber, R.S., Guiblin, A.R., Hobbs, S.C., Metassa, V.G., Reeve, A.J., Beer, M.S., Migglemis, D.N., Noble, A.J., Stanton, J.A., Scholey, K., Hargreaves, R.J., J. Med. Chem. (1993), 36, 1529), оксадиазол (Ainsworth, С., J. Amer. Chem. Soc. (1955), 77, 1148), тетразол, начиная от нитрила (Goerlitzer, К., Kogt, R., Arch. Pharm. (1990), 323, 847) или тиадиазол (Lamattina, J. L., Mularski, C. J., J. Org. Chem. (1984). 49,4800).
F.6 Синтез производных кетона.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a R4 и R4a представляет собой -G2-CH=CQ12Q13 или -G2-CQ13=CHQ12, G2 является связью или алкиленовой группой, Q12 и Q13 являются атомом водорода или алкильной группой, при условии, что ни одна из других R1, X, R2, R2a, R3, R3a, R4 и R4a не является несущей функциональной группой, чувствительной к условиям окисления, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой соответственно -G2-CO-CHQ12Q13 или -G2-CHQ13-CO-Q12. Это превращение может быть осуществлено в любых подходящих условиях, известных среднему специалисту данного уровня техники, например, в присутствии О2 и PdCl2, в инертном растворителе, например, диметилформамиде или N-метилпирролидине, между 0 и 50°С (Bird, Transition Metals Intermediate in Organic Synthesis, Academic Press, NY, (1967), 88-111).
F.7 Дериватизация кетонов.
Соединения формулы I, в которых А2=О, Х=CONR5R6 или COOR7 и одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-CO-Q14, где G2 является связью или алкиленовой группой и Q14 представляет собой алкильную группу, являются ключевыми синтезируемыми промежуточными соединениями для синтеза (i) спиртов -G2-CHOH-Q14 восстановлением гидридным реагентом ((March, J., Advanced Organic Chemistry, Third Edition, John Wiley & Sons, (1985), 809), (ii) фторированной боковой цепи -G2-CF2-Q14, используя условия, описанные в Lal, G.S., Fez, G.P., Pesaresi, R.J., Prozonic, P.M., Chem. Commun. (1999), 215-216.
F.8 Синтез алкинильных производных.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a, R4 иR4a представляет собой -G2-C=C(X8)2, G2 является связью или алкиленовой группой и X8 является атомом галогена, при условии, что ни одна из других X, R1, R2, R2a, R3, R3a, R4 и R4a не является несущей функциональной группой, чувствительной к сильным основаниям, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-C=C-Q15, где Q15 представляет собой водород, галоген, алкил или арил. Эти превращения могут быть осуществлены:
- β-элиминированием, вызванным основанием (например, 1 эквивалент трет- ВиОК при низкой температуре, как описано в Michel, P., Rassat, A., Tetrahedron Lett. (1999), 40, 8579-8581) в галогенацетиленовое производное (Q15 = галоген) с последующим замещением, катализируемым металлом, галогена металлоорганическим соединением (например, MeZnCl в присутствии CuCN.LiCl, как описано в Micouin, L., Knochel, P., Synlett (1997), 327),
- прямым превращением в ацетиленид металла (например, с 2 экв. n-бутиллития) и алкилированием алкилгалогенидом или карбонильным производным (как описано Corey, E.J., Fuchs, P.L., Tetrahedron Lett. (1972), 36, 3769-3772).
F.9 Синтез алканов.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-C=C-Q16Q17, G2 является связью или алкиленовой группой, Q16 и Q17 являются алкилом или фтором, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-СН-СН-Q16Q17.
Стадия восстановления может быть осуществлена в стандартных условиях, известных среднему специалисту данного уровня техники, например водородом в присутствии Pd/C (March, J., "Advanced Organic Chemistry, Third Edition", John Wiley & Sons, (1985), HOI-1102).
F.10 Синтез (галоген)азидоарильных производных.
Соединения формулы I, в которых А2=О, Х=CONR5R6 или COOR7 или CN и одна из групп R2, R3 или R4 представляет собой G2-Q18, где Q18 представляет собой нитроарил или триазеноарил,
G2 является связью или алкиленовой группой, являются ключевыми промежуточными соединениями для синтеза соответствующих соединений, в которых одна из групп R2, R3 или R4 представляет собой G2-Q19, Q19 является азидоарилом, необязательно замещенным одним или несколькими атомами галогена, предпочтительно Br или F атомами. Превращение протекает через восстановление нитро или триазеновой группы в анилин любыми способами, известными среднему специалисту данного уровня техники, необязательно введение одного или нескольких атомов галогена (как у Xing-teng, D., Guo-bin, L., Synth. Commun. (1989), 19, 1261) и превращение амина в азид хорошо известными способами.
F.11 Синтез гетероциклов из аминов.
Соединения формулы I, в которых А1=О, Х=CONR5R6, COOR7 или CN, и одна из групп R2, R3 или R4 представляет собой G2-Q20, где G2 является связью или алкиленовой группой и Q20 представляет собой СООН, CONH2 или CN, являются ключевыми промежуточными соединениями для синтеза соответствующих соединений, в которых одна из групп R2, R3 или R4 представляет собой G2-NH2 или G2-CH2-NH2, что приводит к получению соответствующих соединений, в которых одна из групп R2, R3 или R4 представляет собой G2-Het или G2-CH2-Het, где Het представляет собой гетероцикл, связанный атомом азота, необязательно замещенный одним или несколькими атомами галогена.
- В случае, когда Х=CONR5R6, CN или COOR7 с R7, отличным от Н, и где R2, R3 или R4 представляют собой G2-COOH, превращение протекает через перегруппировку Курциуса (например, воздействием дифенилфосфоразидатом и триэтиламином и гашением in situ бензольным спиртом, как описано в: Kim, D., Weinreb, S.M., J. Org. Chem. (1978), 43, 125), снятие защиты с аминогруппы гидрогенолизом или любыми условиями, известными среднему специалисту данного уровня техники с получением R2, R3 или R4=G2-NH2, с последующим синтезом кольца с получением гетероцикла, такого, как пиррол (как в Jefford, C.W., Tang, Q., Zaslona, A., J. Amer. Chem. Soc. (1991), 113, 3513-3518), и необязательно введением одного или нескольких атомов галогена в кольцо (как в Gilow, H.M., Burton, D.E., J. Org. Chem. (1981), 46, 2221-2225).
- В случае, когда X=CONR5R6, COOR7 или CN и одна из групп R2, R3 или R4 представляет собой G2-CONH2, с Х отличным от CONR5R6, или G2-CN, с Х отличным от CN, превращение протекает через селективное восстановление амида или нитрила в аминометильную группу в любых условиях, известных среднему специалисту данного уровня техники, и синтез кольца с получением гетероцикла, такого, как триазол (как в Miles, R.W., Samano, V., Robins, M.J., J. Amer. Chem. Soc. (1995), 117, 5951-5957).
F.12 Синтез триазолов.
Соединения формулы I, в которых А2=О и одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2-СН2N3, G2 является связью или алкиленовой группой, являются ключевыми синтезируемыми промежуточными соединениями для соответствующих соединений, в которых одна из групп R2, R2a, R3, R3a, R4 и R4a представляет собой -G2- СН2-триазол. Эти превращения могут быть осуществлены длительным нагреванием в присутствии производного 1-(трифенилфосфоранилиден)-кетона (как описано в Hammerschmidt, R, Polsterer, J.P., Zbiral, E., Synthesis (1995), 415).
F.13 Заключение.
Когда соединения формулы I содержат один или несколько стереогенных центров и используются такие нестереоселективные способы синтеза, разделение на изомеры смеси стереоизомеров может наилучшим образом осуществляться в одну или несколько стадий, включая общее последовательное разделение смесей диастереоизомеров на их составляющие рацематы, используя предпочтительно хроматографические разделения на нехиральную или хиральную фазу обратимым или предпочтительно прямым способом, с последующей по крайней мере одной конечной стадией расщепления каждого рацемата на его энантиомеры, используя наиболее предпочтительно хроматографическое разделение на хиральную фазу обратимым или предпочтительно прямым способом. Альтернативно, когда используются частично стереоселективные способы синтеза, конечной стадией может быть разделение диастереомеров, используя предпочтительно хроматографическое разделение на нехиральную или хиральную фазу обратимым или предпочтительно прямым способом.
Некоторые промежуточные соединения, описанные выше, особенно соединения формулы AA-II, в которых различные заместители имеют значения, указанные выше, являются новыми и также составляют часть изобретения. Эти новые промежуточные соединения, в которых уходящая группа является фармацевтически приемлемой, обладают такой же полезностью, как описанные соединения формулы I здесь и далее.
Было обнаружено, что соединения формулы I и их фармацевтически приемлемые соли являются полезными в различных фармацевтических показаниях.
Например, соединения по изобретению полезны для лечения эпилепсии, эпилептогенеза, апоплексического удара и конвульсий.
Эти соединения могут также использоваться для лечения других неврологических заболеваний, включая биполярные заболевания, манию, депрессию, беспокойство, мигрень, тригеминальную и другую невралгию, хроническую боль, невропатическую боль, церебральную ишемию, сердечную аритмию, миотонию, кокаиновую зависимость, паралич, миоклонию, эссециальный тремор и другие расстройства движения, неонатальный церебральный паралич, амиотрофный латеральный склероз, спастичность, болезнь Паркинсона и другие дегенеративные заболевания.
К тому же соединения по изобретению могут быть полезны для лечения бронхиальной астмы, астматического состояния и аллергического бронхита, астматического синдрома, бронхиальной гиперреактивности и бронхоспастических синдромов так же, как и аллергического и вазомоторного ринита и риноконъюктивита.
Следовательно, настоящее изобретение, в следующем аспекте, относится к применению соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения неврологических и других заболеваний, таких как упомянутые выше.
В частности, настоящее изобретение относится к применению соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения эпилепсии, биполярных заболеваний, хронической боли или невропатической боли, мигрени, бронхиальных, астматических или аллергических состояний.
Активность и свойства активных соединений, оральная биодоступность и устойчивость in vitro или in vivo может в значительной степени меняться среди оптических изомеров описываемых соединений.
В предпочтительном воплощении активное соединение вводят в энантиомерно обогащенной форме, т.е. по большей части в форме одного изомера.
Например, в случае соединения формулы I, в котором R1 представляет собой этил, Х представляет собой -CONH2, А2 представляет собой кислород, когда R3 представляет собой пропил и все оставшиеся заместители представляют собой водород, предпочтительным является S (бутанамид), R (кольцо) энантиомер, и когда R3 представляет собой 2,2-дифторвинил и все оставшиеся заместители представляют собой водород, предпочтительным является S (бутанамид), S (кольцо) энантиомер.
Настоящее изобретение также относится к способу лечения эпилепсии, мигрени, биполярных заболеваний, хронической боли или невропатической боли или бронхиальных, астматических или аллергических состояний, у млекопитающего, нуждающегося в таком лечении, включающий введение терапевтической дозы по крайней мере одного соединения формулы I или его фармацевтически приемлемой соли пациенту.
Способы по изобретению включают введение млекопитающему (предпочтительно человеку), страдающему от вышеупомянутых состояний или заболеваний, соединения по изобретению в количестве, достаточном для облегчения или предотвращения заболевания или состояния.
Соединение обычно вводят в любой подходящей разовой дозированной форме, включая, но не ограничиваясь ею, содержание от 5 до 1000 мг, предпочтительно от 25 до 500 мг активного ингредиента на единицу дозированной формы.
Термин "лечение", как здесь используется, включает методичное лечение и профилактическое лечение.
Под "методичным" понимают эффективность для лечения текущего симптоматического случая заболевания или состояния.
Под "профилактическим" понимают предотвращение появления или повторения заболевания или состояния.
Термин "эпилепсия", как здесь используется, относится к заболеванию мозговой функции, характеризующегося периодическим и непредсказуемым появлением припадков. Припадок может быть "неэпилепсическим", когда вызван в нормальном мозге лечением, таким, как электрошок или химические конвульсанты или "эпилепсическим", когда вызван без доказательной причины.
Термин "припадок", как здесь используется, относится к скоротечному изменению поведения, обусловленному заболеванием, синхронным и ритмическим сжиганием популяций мозговых нейронов.
Термин "мигрень", как здесь используется, обозначает заболевание, характеризующееся повторяющимися приступами головной боли, которые очень широки по интенсивности, частоте и продолжительности. Приступы являются обычно односторонними и обычно связаны с анорексией, тошнотой, рвотой, фонофобией и/или фотофобией. В некоторых случаях они предшествуют или сопровождаются неврологическими и психологическими нарушениями. Мигреневая головная боль может длиться от 4 часов до примерно 72 часов. International Headache Society (IHS, 1988) классифицирует мигрень на имеющую ауру (классическая мигрень) и мигрень без ауры (общая мигрень) как главные типы мигрени. Мигрени с аурой состоят из фазы головной боли с предшествующими характерными визуальными, сенсорными, речевыми или моторными симптомами. При отсутствии таких симптомов головная боль называется мигренью без ауры.
Термин "биполярные заболевания", как здесь используется, обозначает заболевания, классифицированные как Mood заболевания по Diagnostic and Statistical Manual of Mental Disorders, 4th edition (Diagnostic and Statistical Manual of Mental Disorders (DSM-IV TM), American Psychiatry Association, Washington, DC, 1994). Биполярные заболевания обычно характеризуются спонтанно возбудимыми повторяющимися (т.е. по крайней мере двумя) случаями, в которых гипервозбудимость, активность и настроение пациента значительно нарушаются, это нарушение заключается в некоторых случаях в поднятии настроения и повышении энергии и активности (мания или гипомания) и в других случаях в ухудшении настроения и уменьшения энергии и активности (депрессия). Биполярные заболевания разделяют на четыре основных категории в DSM-IV (биполярные заболевания I, биполярные заболевания II, циклотимия, и биполярные заболевания, не определенные иначе).
Термин "случай мании", как здесь используется, относится к особому периоду, во время которого повышаются ненормальность и настойчивость, открытое или раздражительное настроение с проявлениями подавления речи и психомоторным возбуждением.
Термин "гипомания", как здесь используется, относится к менее выраженному случаю мании с пониженной степенью сложности.
Термин "главный депрессивный случай", как здесь используется, относится к периоду по крайней мере 2 недель, во время которых случается либо депрессивное настроение или снижение интереса или удовольствия в почти всех действиях с проявлениями рассеянной концентрации и психомоторным замедлением.
Термин "смешанный случай", как здесь используется, относится к периоду времени (длящемуся по крайней мере 1 неделю), в котором встречаются признаки как для случая мании, так и для главного депрессивного случая почти каждый день.
Термин "хроническая боль", как здесь используется, обозначает состояние, постепенно распознаваемое как протекающее заболевание в отличие от острой боли. Обычно определяемая как боль, которая продолжается после нормального времени для заживления, боль также может считаться хронической в том случае, когда станет ясно, что боль становится неотъемлемой частью жизни в предвидящемся будущем. Желательно, чтобы большинство синдромов хронической боли включало нейропатический компонент, который обычно тяжелее лечится, чем острая соматическая боль.
Термин "невропатическая боль", как здесь используется, обозначает боль, вызванную патологическими изменениями нерва, который показывает наличие вредных раздражителей, когда не существует такого распознаваемого раздражителя, обусловливая неправильную чувствительность боли. Другими словами, оказывается, система боли включается и не может сама отключиться.
Активность соединений формулы I или их фармацевтически приемлемых солей в качестве антиконвульсивных веществ может определяться на аудиогенной модели припадка. Целью этого теста является измерение антиконвульсивного потенциала соединения посредством аудиогенных припадков, вызванных у звукочувствительных мышей, генетической животной модели с рефлексом припадков. В этой модели первично вырабатываемой эпилепсии, припадки вызываются без электрической или химической стимуляции и типами припадка являются, по крайней мере частично, похожие по их клинической феноменологии на припадки, встречающиеся у людей (Loscher W. & Schmidt D., Epilepsia Res. (1998), 2, p.145-181; Buchhalter J.R., Epilepsia (1993), 34, S31-S41). Результаты, полученные с соединениями формулы I, показывают сильный фармакологический эффект.
Другие испытания, показывающие потенциальную антиконвульсивную активность, связаны с левитирацетам - связывающим сайтом (LBS), как описано далее.
Активность соединений формулы I или их фармацевтически приемлемых солей относительно хронической невропатической боли может определяться на моделях животных. Например, хроническая невропатическая боль может моделироваться на фармакологически вызванных диабетах у крыс. В этих моделях животные проявляют прогрессивную гипералгезию к болевым стимулам, симптом обычно наблюдается у пациентов с болезненной периферальной невропатией (Courteix С, Eschalier, А. и Lavarenne J., Pain, 53, (1993) 81-88). Было показано, что эта модель обладает высокой фармакологической предсказуемостью (Courteix С, Bardin M., Chantelauze С., Lavarenne J и Eschalier, A., Pain, 57 (1994) 153-160).
Активность соединений формулы I или их фармацевтически приемлемых солей при биполярных заболеваниях может быть испытана на моделях животных. Например, биполярные заболевания и особенно мания могут моделироваться на фармакологически вызванной гиперактивности у крыс и оценки их поведения в Y тупике. В такой ситуации терапевтические агенты, эффективные для человека, такие, как валпроат лития и натрия, повышают гиперактивность, следовательно, придавая предсказуемость модели (Сао В.J. и Peng N; A;, Eur. J; Pharmacol. 237 (1993) 177-181. Vale A.L. и Ratcliffe F. Psychopharmacology, 91 (1987) 352-355).
Потенциальные антиастматические свойства соединений формулы I или их фармацевтически приемлемых солей могут тестироваться на животной модели аллергической астмы, в которой гвинейским свиньям, чувствительным к овалбумину, вводят антиген и исследуют изменения легочной функции и клеточного содержания верхних дыхательных путей (Yamada и др. (1992) Development of an animal model of late asthmatic response in guinea pigs and effects anti-asthmatic drugs. Prostaglandins, 43: 507-521).
Активность любых упомянутых выше показателей может, конечно, определяться проведением подходящих клинических испытаний способами, известными среднему специалисту данного уровня техники, для определенного показателя и/или при разработке клинических испытаний вообще.
Для лечения заболеваний соединения формулы I или их фармацевтически приемлемые соли могут применяться в эффективной ежедневной дозе и вводиться в виде фармацевтической композиции.
Следовательно, другое воплощение настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения формулы I или его фармацевтически приемлемой соли в сочетании с фармацевтически приемлемым разбавителем или носителем.
Для приготовления фармацевтической композиции по изобретению одно или более соединений формулы I или его фармацевтически приемлемую соль тщательно перемешивают с фармацевтическим разбавителем или носителем в соответствии с обычными способами фармацевтических соединений, известными среднему специалисту.
Подходящие разбавители и носители могут иметь широкое разнообразие форм в зависимости от необходимого способа введения, например, орально, ректально или парентерально.
Фармацевтические композиции, включающие соединения по изобретению, могут, например, вводиться орально или парентерально, т.е. внутривенно, внутримышечно или подкожно, интратекально.
Фармацевтические композиции, пригодные для орального введения, могут быть твердыми или жидкими и могут, например, находиться в виде таблеток, пилюль, драже, желатиновых капсул, растворов, сиропов и им подобных.
Наконец, активный ингредиент можно смешать с инертным разбавителем или нетоксичным фармацевтически приемлемым носителем, таким как крахмал или лактоза. Необязательно эти фармацевтические композиции могут также включать связующее вещество, такое как микрокристаллическая целлюлоза, вязкое вещество или желатин, разделитель, такой как альгиновая кислота, лубрикант, такой как стеарат магния, пластификатор, такой как коллоидный диоксид кремния, подсластитель, такой как сахароза или сахарин, или красители, или отдушки, такие как мята перечная или метилсалицилат.
Изобретение также относится к композициям, которые могут высвобождать активный компонент контролируемым способом. фармацевтические композиции, которые могут использоваться для парентерального введения, находятся в стандартных видах, таких как водные или масляные растворы или суспензии, обычно содержащиеся в ампулах, одноразовых шприцах, стеклянных или пластиковых пузырьках или емкостях для жидкости.
В добавление к активному ингредиенту, эти растворы или суспензии могут необязательно также включать стерильный разбавитель, такой как вода для инъекции, физиологический соляной раствор, масла, полиэтиленгликоль, глицерин, пропиленгликоль или другие синтетические растворители, антибактериальные агенты, такие как бензиловый спирт, антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия, хелатирующие агенты, такие как этилендиаминтетрауксусная кислота, буферы, такие как ацетаты, цитраты или фосфаты, и агенты для поддержания осмолярности, такие как хлорид натрия или декстроза.
Эти фармацевтические формы получают, используя методики, которые обычно используются фармацевтами.
Количество активного ингредиента в фармацевтических композициях может варьироваться в широком диапазоне концентраций и зависит от различных факторов, таких как пол, возраст, вес пациента и медицинское состояние, так же как от способа введения. Это количество соединения формулы I в композициях для орального введения составляет, по крайней мере, 0.5% по весу и может достигать 80% по весу по отношению к общему весу композиции.
В соответствии с изобретением, также было обнаружено, что соединения формулы I или их фармацевтически приемлемые соли могут вводиться отдельно или в сочетании с другими фармацевтически активными компонентами. Неограничивающими примерами таких дополнительных соединений, которые могут быть названы для использования в сочетании с соединениями по изобретению, являются антивирусные средства, антиспатические средства (например, баклофен), антимиметики, антиманиакальные средства, стабилизирующие настроение, анальгетики (например, аспирин, ибупрофен, парацетамол), наркотические анальгетики, местные анестетика, опиоидные анальгетики, соли лития, антидепрессанты (например, миансерин, флуоксетин, тразодон), трициклические антидепрессанты (например, имипрамин, десипрамин), антиконвульсанты (например, валпроиновая кислота, карбамазепин, фенитоин), антипсихотические средства (например, рисперидон, галоперидол), невролептики, бензодиазепины (например, диазепам, клоназепам), фенотиазины (например, хлорпромазин), блокаторы кальциевого канала, амфетамин, клонидин, лидокаин, мексилетин, капсаицин, кофеин, куетиапин, антагонисты серотонина, β-блокаторы, антиаритмические средства, триптаны, производные спорыньи.
Особенный интерес в соответствии с настоящим изобретением вызывают комбинации по крайней мере одного соединения формулы I или его фармацевтически приемлемой соли или по крайней мере одного соединения, вызывающего нервное ингибирование, опосредованное GABAA рецепторами. Соединения формулы I проявляют потенциальную активность по отношению к соединениям, вызывающим нервное ингибирование, опосредованное GABAA рецепторами, облегчая, во многих случаях, эффективное лечение состояний и заболеваний при пониженном риске побочных эффектов.
Примеры соединений, нервное ингибирование, опосредованное GABAA рецепторами, включают следующие: бензодиазепины, барбитураты, стероиды и антиконвульсанты, такие, как валпроат, виагабатрин, тиагабин или их фармацевтически приемлемые соли.
Бензодиазепины включают 1, 4 бензодиазепины, такие, как диазепам и клоназепам, и 1, 5 бензодиазепины, такие, как клобазам. Предпочтительным соединением является клоназепам.
Барбитураты включают фенобарбитал и пентобарбитал. Предпочтительным соединением является фенобарбитал.
Стероиды включают адренокортикотропические гормоны, такие, как ацетат тетракосактида, и т.д. Антиконвульсанты включают гидантоины (фенитоин, этотоин и т.д.), оксазолидины (триметадион и т.д.), сукцинимиды (этосукцимид и т.д.), фенацемиды (фенацемид, ацетилфенетурид и т.д.), сульфонамиды (сультиам, ацетоазоламид и т.д.), аминобутановые кислоты (например, гаммааминобетагидроксибутановая кислота и т.д.), валпроат натрия и производные, карбамазепин и другие.
Предпочтительные соединения включают валпроиновую кислоту, валпромид, валпроатпивоксил, валпроат натрия, бивалпроат натрия, дивалпроекс, клоназепам, фенобарбитал, вигабатрин, тиагабин.
В предпочтительных оральных композициях ежедневная доза колеблется от 5 до 1000 миллиграмм (мг) соединения формулы I.
В композициях для парентерального введения количество настоящего соединения формулы I составляет, по крайней мере, 0.5% по весу и может достигать 33% по весу относительно общего веса композиции. В предпочтительных парентеральных композициях доза находится в пределах от 5 мг до 1000 мг соединения формулы I.
Ежедневная доза колеблется в широкой области разовых доз соединения формулы I и обычно лежит в пределах от 5 до 1000 мг. Однако ясно, что в особых случаях могут применяться специфические дозы в зависимости от индивидуальных требований, благоразумности врача.
Количество активных ингредиентов (соединение I и соединение, вызывающее нервное ингибирование, опосредованное GABAA рецепторами) в фармацевтической композиции по изобретению сильно зависит от млекопитающего, которому вводят композиции, излечиваемой болезни, других входящих активных ингредиентов и т.д. Обычно количество соединения, вызывающего нервное ингибирование, опосредованное GABAA рецепторами, и количество соединения I для данной композиции и дозу можно легко определить, используя стандартные методики.
Следующие примеры приведены только в целях иллюстрации и ни в коей мере не предназначены и не должны толковаться, как ограничивающие изобретение любым способом. Среднему специалисту данного уровня техники понятно, что могут быть выполнены стандартные варианты и модификации следующих примеров без выхода за рамки области или объема изобретения.
Если особо не указано в примерах, характеристику соединений проводили следующими методами:
ЯМР спектры записывали на BRUKER АС 250 Fourier Transform NMR Spectrometer, связанный с Aspect 3000 computer и 5 mm 1H/13С двойным зондом или BRUKER DRX 400 FT NMR, связанный с SG Indigo2 computer и 5 mm 1H/13C/15N тройным зондом обращенной геометрии. Соединение изучали в ДМСО-d6 (или CDCl3) растворе при температуре пробы 313 К и при концентрации 20 мг/мл. Инструмент настраивали на сигнал дейтерия ДМСО-d6 (или CDCl3). Химические сдвиги, указанные в ррм, отстающие от TMS, взятого в качестве внутреннего стандарта.
Масс-спектрометрические измерения LC/MS (жидкостная хроматография/масс спектр) способом осуществляют следующим способом: (Высокоэффективная жидкостная хроматография) HPLC условия
Анализы проводят, используя WATERS Alliance HPLC system, снабженную INERTSEL ODS 3, DP 5 μм, 250 X 4.6 мм колонкой.
Градиент изменяют от 100% растворителя А (ацетонитрил, вода, TFA (10/90/0.1, об/об/об)) до 100% растворителя В (ацетонитрил, вода, TFA (90/10/0.1, об/об/об)) в течение 7 мин при наличии 100% В в течение 4 мин. Скорость потока 2.5 мл/мин и расщепление 1/10 используют до API источника. Хромотографию проводят при 30°С. MS(масс-спектр) условия
Образцы растворяют в смеси ацетонитрил/вода, 70/30, об/об при концентрации около 250 (μгр/мл. API спектры (+ или -) измеряют используя FINNIGAN (San Jose, CA, USA) LCQ ионопроходный масс-спектрометр. APCI источник действует при 450°С и капиллярный нагреватель при 160°С. ESI источник действует при 3.5 кВ и капиллярный нагреватель при 210°С.
Масс-спектрометрические измерения DIP/EI методом - проводят следующим способом: образцы испаряют нагреванием пробы от 50°С до 250°С в течение 5 мин. El (Electron Impact) спектры записывают с помощью FINNIGAN (San Jose, CA, USA) TSQ 700 тандемного квадрупольного масс-спектрометра. Температура источника - 150°С.
Специфическую ротацию записывают на Perkin-Elmer MC241 или 341 поляриметре. Угол ротации записывают при 25°С в 1% растворе в МеОН. Для некоторых молекул растворителем является СН2Cl2 или ДМСО, в зависимости от растворимости.
Содержание воды определяют с помощью Metrohm microcoulometric Karl Fischer титратора.
Препаративную хроматографию проводят на силикагеле 60 Merck, размер частиц 15-40 μм, reference 1.15111.9025, с помощью на месте модифицированных Jobin Yvon-типа аксиальных компрессионных колонок (80 мм i.d.), скорости потока - между 70 и 150 мл/мин. Количество силикагеля и смесей растворителя описано в конкретных примерах.
Препаративную хиральную хроматографию проводят на DAICEL Chiralpak AD 20 μм, 100·500 мм колонке с помощью на месте собранного инструмента с различными смесями низших спиртов и от С5 до С8 линейных, разветвленных или циклических алканов при ±350 мл/мин. Смеси растворителей описаны в конкретных примерах.
Точки плавления определяют на Büchi 535 Totoli-типа плавильном устройстве и не корректируют или при начальной температуре на Perkin Elmer DSC7.
Измерения диффракции X-лучей порошка измеряют при подходящей температуре и атмосфере на контролируемом компьютером Philips PW 1710 оборудовании с PW3710 mpd контрольной единицей, используя монохроматор, Cu Kα радиацию (трубка, действующая при 40 k1N, 35 мА) и сцинтилляционный счетчик. Данные собирают в угловой области от 4° до 50° 2θ длительным сканирующим способом со скоростью сканирования 0.02 2θ/с.
В примерах используются следующие сокращения:
|
Если специально не оговорено в примерах, соединения получают в свободной (не солевой) форме.
ПРИМЕР 1. Синтез 4-замещенных 2-оксопирролидинбутанамидов путем восстановительного аминирования альдегидоэфира.
1.1. Синтез эфиров 3-замещенной-4-оксобутановой кислоты.
1.1.1. Путь А: Алкилированием енаминов
Синтез метилового эфира 5,5-диметил-3-формилгексановой кислоты 361 представлен следующим образом:
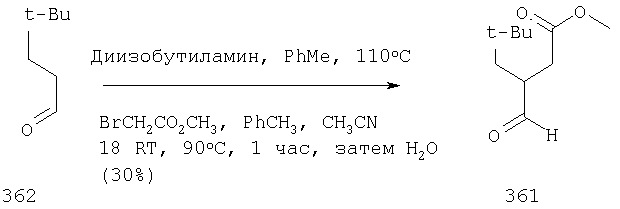
В трехгорлой колбе, оснащенной ловушкой Дина-Старка в атмосфере аргона нагревают раствор диизобутиламина (4.62 мл от Асгос), 4,4- диметилпенталя 362 (2.5 г, 0.021 моль) в толуоле (20 мл) при 130°С в течение 2 часов и воду экстрагируют. Желтый раствор охлаждают до комнатной температуры и добавляют метилбромацетат (3.7 г, 0.024 моль) за один раз. Розовый раствор перемешивают при комнатной температуре в течение ночи и 1 час при 90°С. Добавляют воду (10 мл) при указанной температуре и через 1 час, раствор охлаждают до комнатной температуры. Органический слой промывают HCl IN, насыщенным водным раствором бикарбоната натрия, сушат над сульфатом магния, фильтруют и упаривают, что дает масло, которое перегоняют при пониженном давлении (1 мм Hg), чтобы получить метиловый эфир 5,5-диметил-3-формилгексановой кислоты 361 в виде жидкости (1.1 г, 0.05 моль, Teb (1 мм Нд): 69-71°С). Альдегидоэфиры затем используют на стадии восстановительного аминирования. Альтернативно, алкилирование этилбромацетатом может быть осуществлено в присутствии толуола-ацетонитрила 1/1 (об/об) как растворителя. Конечный альдегид может также быть перегнан при пониженном давлении.
1.1.2. Другие синтетические пути.
Альдегидоэфиры могут также быть получены другими методами, включая:
(i) Алкилирование гидразона с помощью производного бромацетата. Например, 2,2-диметилэтиловый эфир 5-(фенил)-3-формилпентановой кислоты получают при взаимодействии N-(4-фенил)-пропилиден-N,N-диметилгидразона с трет-бутилбромацетатом и LDA с последующим озонолизом алкилированного гидразона.
(ii) Введение нитрометана к α,β-ненасыщенным эфирам. Этиловый эфир 3-(3-бром-фенил)-4-оксобутановой кислоты получают добавлением нитрометана к этиловому эфиру 3-(3-бромфенил)-акриловой кислоты в присутствии 1.8-диазабицикло[5.4.0]ундц-7-ена, окисления нитропроизводного в Nef условиях и контролируемого гидролиза метилапеталя с помощью HCl.
(iii) Озонолиз производных 4-пентеновой кислоты. Этиловый эфир 2-бензил-4-оксобутановой кислоты получают алкилированием с помощью амида диизопропиллития этилового эфира 3-фенилбутановой кислоты и аллилбромида с последующим озонолизом и восстановлением озонида с помощью PPh3.
1.2. Восстановительное аминирование эфиров 3-замещенной-4-оксобутановой кислоты и циклизация до пирролидин-2-она.
1.2.1. Восстановительное аминирование
Синтез метил 4-{[((1S)-1-аминокарбонил)пропил]амино}бутаноата 363 представлен.

В трехгорлую колбу, объемом 1 л, оснащенную парциальным конденсатором горячего орошения, в атмосфере аргона, нагревают суспензию альдегида 361 (1.7 г, 0.09 моль), (S)-2-аминобутанамид (1.58 г, 0.15 моль) и молекулярных сит (3 Å) в СН3ОН при 60°С в течение 0.5 часа. Суспензию охлаждают до 0°С и добавляют порциями боргидрид натрия (0.55 г). Через час при комнатной температуре реакционную смесь разбавляют этиловым эфиром, промывают водой, сушат над сульфатом магния, фильтруют и упаривают, что дает желтое масло. Метил 4-{[((1S)-1-аминокарбонил)пропил]амино}бутаноат 363 используют непосредственно на следующей стадии без дальнейшей очистки.
Альтернативно, восстановительное аминирование может быть осуществлено в тех же условиях с другим восстанавливающим агентом, таким как NaBH3CN или NaBH(OAc)3 (используя 1.4 моль. эквивалент в расчете на альдегидоэфир).
1.2.2. Циклизация бутановой кислоты (метилового или этилового) эфиров.
Синтез двух стереоизомеров (2S)-2-(4-неопентил-2-оксо-1-пирролидинил)бутанамида 149 и 148 представлен следующим образом:
reflux = нагревание при кипении
В трехгорлой колбе, оснащенной парциальным конденсатором горячего орошения, в атмосфере аргона, растворяют маслянистый 363 в смеси 1/1 толуола и 1,2-дихлорэтана (25 мл каждый) в присутствии гидроксибензотриазола (2.05 г, доступный от Aldrich) и раствор нагревают при 90°С в течение 2 часов, а затем охлаждают до комнатной температуры. Органическую фазу промывают последовательно насыщенным водным раствором бикарбоната натрия, водой, сушат над сульфатом магния, фильтруют и упаривают, что дает коричневое твердое вещество (1.8 г), которое очищают с помощью хроматографической колонки на силикагеле (Элюент: СН2Cl2/СН3ОН 95/05 (об/об)), что дает (2S)-2-(4-неопентил-2-оксо-1-пирролидинил)бутанамид (0.89 г, 0.0036 моль) в виде 1/1 смеси диастериоизомеров. Разделение 2-х изомеров осуществляют с помощью хроматографии на хиральной стационарной фазе (EtOH-гексан 1/1 (об/об)), что дает, после перекристаллизации в толуоле, два стереоизомера (соответственно 0.35 г и 0.37 г), физико-химические свойства описаны в таблице. Альтернативно, циклизация аминоэфира, может быть осуществлено с другими реагентами, отличными от гидрокси-бензотриазола, подобными уксусной кислоте (как растворитель) или 2-гидроксипиридину (1 эквивалент). Когда уксусную кислоту используют в качестве растворителя для циклизации, реакционную смесь упаривают под вакуумом досуха, разбавляют дихлорметаном и обрабатывают как описано выше.
1.2.3. Другие циклизации.
Альтернативно, циклизация может быть осуществлена в две стадии с помощью (i) кислотного или основного гидролиза эфира и (ii) циклизации активированного эфира в обычных условиях, описанных в пептидном синтезе.
1.3. Твердофазный синтез пирролидонов
1.3.1. Присоединение Fmoc защищенной аминокислоты на Rink амидную смолу.
DMF = ДМФА
4 г Rink амидной смолы (0.51 мек/г, 100-200 меш) помещают в стеклянный сосуд и перемешивают в 20% об/об пиперидина/ДМФА (40 мл) в течение 30 минут. Смолу осушают и полное снятие защиты повторяют. Смолу фильтруют, промывают (6 × ДМФА) и сушат. Смолу суспендируют в ДМФА (40 мл) и обрабатывают N-Fmoc-2-аминомасляной кислотой (3.02 г, 9.28 ммоль), а затем раствором 1,3-дипиклогексилкарбодиимида (1.4 г, 11.13 ммоль) в ДМФА (20 мл). Реакционную массу перемешивают в течение 1 часа при комнатной температуре, затем фильтруют, промывают (ДМФА) и процесс конденсации повторяют. Смолу фильтруют, промывают (6 × ДМФА, 6 × CH2Cl2), сушат и используют в том же состоянии на следующих стадиях.
1.3.2. Восстановительное аминирование с добавленным 5-гидрокси-4-пропилфуран-2-оном и циклизация.
100 мг смолы с N-Fmoc-2-аминомасляным амидом (0.051 ммоль) находится внутри фриттированного полипропиленового шприца. Удаление Fmoc группы достигается при использовании 20% пиперидина в ДМФА. К аминосмоле добавляют 5-гидрокси-4-пропилфуран-2-он (из 36.72 мг, 0.25 ммоль) в DCE (2 мл). Смолу затем обрабатывают уксусной кислотой (15 мкл) и триацетоксиборгидридом натрия (54 мг, 0.25 ммоль). Реакционную массу перемешивают в течение 18 часов при комнатной температуре, затем фильтруют и промывают в следующей последовательности растворителей: Н2О/ДМФА (1:1), ДМФА, CH2Cl2, СН3ОН и сушат. Смолу суспендируют в смеси трифторуксусной кислоты / СН2Cl2 (1/1) в течение 4 часов с энергичным перемешиванием, затем фильтруют, промывают (СН2Cl2×2). Фильтрат концентрируют, остаток растворяют в СН2Cl2 (2 мл) и концентрируют еще раз. Желаемые соединения очищают с помощью LC-MS (жидкостная хроматография/ масс-спектр) (Micromass - Gilson, LCZ - PlatforM, RP-18 колонка, градиент элюирования, СН3CN/Н2O/ТFА1%).
1.3.3. Восстановительное аминирование добавленным альдегидоэфирами и циклизация.
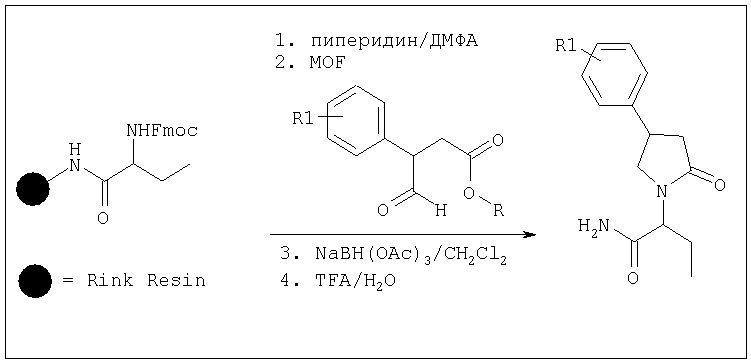
150 мг смолы с N-Fmoc-2-аминомасляным амидом (0.087 ммоль) содержится внутри фриттированного полипропиленового шприца. Удаление Fmoc группы достигается при использовании 20% пиперидина в ДМФА. К аминосмоле добавляют альдегид (0.5 ммоль) в TMOF (2 мл). Реакционную массу перемешивают в течение 18 часов при комнатной температуре, затем фильтруют и промывают (СН2Cl2). Смолу насыщают СН2Cl2 и затем обрабатывают триацетоксиборгидридом натрия (22 мг, 0.104 ммоль). Реакционную массу перемешивают еще 18 часов при комнатной температуре. Смолу затем промывают в следующей последовательности растворителей: Н2О × 6, СН3ОН × 6, СН2Cl2 × 6 и сушат. Смолу суспендируют в смеси трифторуксусной кислоты / воды (95/5) в течение 1 часа с орбитальным перемешиванием, затем фильтруют, промывают (СН2Cl2 × 2). Фильтрат концентрируют, остаток растворяют в СН2Cl2 (2 мл) и концентрируют еще раз. Желаемые соединения очищают с помощью LC-MS (Micromass-Gilson, LCZ - PlatforM, RP-18 колонку, градиент элюирования, СН3CN/Н2O/ТFА1%).
ПРИМЕР 2. Синтез 4-замещенных 2-оксопирролидинбутанамидов с помощью раскрытия кольца замещенных γ-Лактонов.
2.1. Синтез Лактонов
2.1.1. Путь А: Алкилирование 2,3-фуранона
Синтез 4-н-бутилбутиролактона 365 представлен следующим образом:
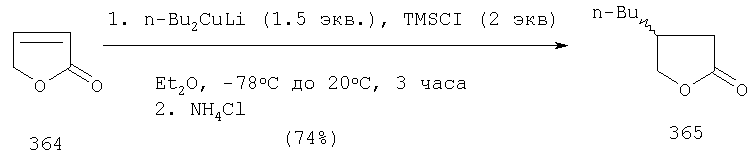
В трехгорлую колбу, в атмосфере аргона, добавляют н-бутиллитий (1.6 М в гексанах, 75 мл, 0.12 моль.) к суспензии CuI (11.42 г, 0.06 моль) в сухом ТГФ (80 мл), охлажденном при -30°С. После 0.5 час., раствор охлаждают до -78°С, по каплям вводят TMSC1 (4.75 г, 0.04 моль.), а затем 2,3-фуранон 364 (от Aldrich, 3.36 г, 0.04 моль.), растворенном в сухом ТГФ. Суспензии дают нагреться до комнатной температуры и гидролизуют насыщенным раствором хлорида аммония. Водный слой экстрагируют AcOEt (3х), промывают водой, сушат над сульфатом магния и упаривают досуха. Сырой лактон очищают диситиляцией (1 мм Hg; 73-80°С), что дает 2.7 г 4-н-бутилбутиролактона 365.
Альтернативно, реагент купрат может быть получен при замещении органолития с помощью органомагния, который может быть получен путем взаимодействия между алкилгалоидом и магниевыми стружками в обычных условиях для данного процесса. ТГФ может быть замещен диэтилэтиловым эфиром (Для общей информации смотри: Lipshutz, B.H.; Sengupta, S. Org. Reactions 1991, 41, 135).
2.1.2. Другие пути
Альтернативно, лактоны могут быть также получены:
(i) Восстановлением сукцинатных эфиров. 4-(циклопропил)метилбутиролактон получают алкилированием монометилсукцината с помощью циклопропилметилбромида с диизопропиламидом лития, с последующем восстановлением 2-(циклопропил)метил-сукциновой (янтарной) кислоты 1-метилового эфира с помощью NaBH4 и CaCl2.
(ii) Восстановлением сукциновой кислоты 1-алкилэфира 4-алкилтиоэфира. 4-аллилбутиролактон получают из этил 4-пентенового тиоэфира (синтезированного из 4-пентеновой кислоты и этантиола в присутствии дициклогексилкарбодиимида). Алкилирование этил 4-пентеновой кислоты тиоэфира этилбромацетатом с диизопропиламидом лития, позволяет получить 2-аллилсукциновой кислоты 1-метиловый эфир 4-этилтиоэфир, который затем переводят в 4-аллилбутиролактон путем взаимодействия последовательно с LiBH4 и серной кислотой.
2.2. Синтез пирролидонов
2.2.1. Ацилированием/алкилированием бутирамида.
Синтез двух стереоизомеров (2S)-2-(4-аллил-2-оксо-1-пирролидинил)бутанамида 228 и 224 представлен следующим образом:
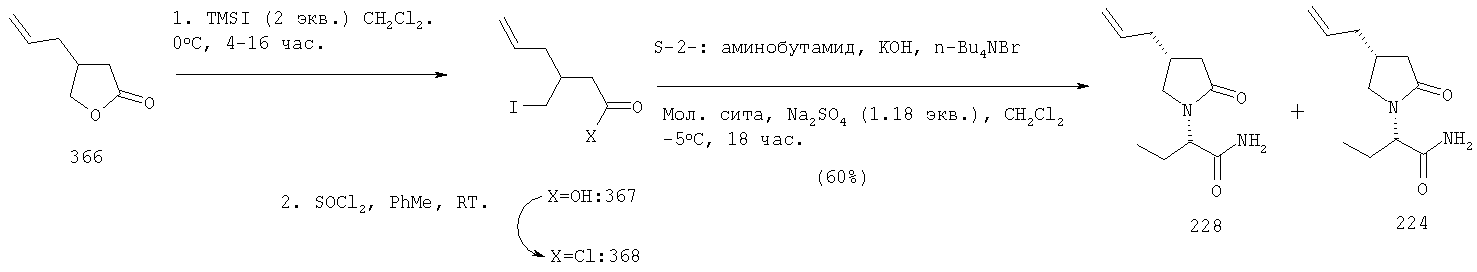
- Стадия 1: Раскрытие лактона
В трехгорлую колбу, в атмосфере аргона, добавляют TMSI (51 мл, Aldrich) к раствору сырого 4-аллилбутиролактона 366 (смотри прцедуру § 2.1.3., 22.9 г, 0.181 моль), охлажденному до 0°С. Раствор перемешивают в течение 2 часов при комнатной температуре и гидролизуют IN HCl (300 мл). Водный слой экстрагируют CH2Cl2 и объединенные органические фазы промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме, что дает сырой продукт 3-(иод)метил-5-гексеновую кислоту 367 (44.5 г). 1H ЯМР (250 МГц, CDCl3): 1.80-2.05 (м, 2Н), 2.20 (т, 2Н), 2.40-2.60 (т, 2Н), 5.10-5.20 (м, 2Н), 5.15-5.80 (м, 1Н).
- Стадия 2: Хлорирование иодкислоты.
В трехгорлой колбе, оснащенной парциальным конденсатором горячего орошения, в атмосфере аргона раствор тионилхлорида (25.5 мл) и сырой иодкислоты 367 (44.5 г, 0.175 моль) в бензоле (90 мл) перемешивают в течение 24 часов при комнатной температуре. Растворители упаривают под вакуумом, что дает сырой хлорангидрид 3-(иод)метил-5-гексеновой кислоты 368 (47 г), который используют непосредственно на следующей стадии без дальнейшей очистки. 1H ЯМР (250 МГц, CDCl3): 1.90-2.05 (м, 2Н), 2.15 (т, 2Н), 2.90-3.10 (м, 2Н), 3.25 (дд, 1Н), 3.35 (да, 1Н), 5.10-5.20 (м, 2Н), 5.15-5.80 (м, 1Н).
- Стадия 3: Ацилирование-алкилирование с S-2-аминобутирамидом.
В трехгорлой колбе, в атмосфере аргона, сырой хлорангидрид кислоты 368 (47 г, 0.172 моль) в СН2Cl2 (300 мл) добавляют по каплям к механически перемешиваемой суспензии молекулярных сит (29 г), порошка КОН (22.3 г), безводного Na2SO4 (28.8 г), тетра-н-бутиламмония бромида (2,8 г, 0.0086 моль) и S-2-аминобутирамида ([α]25 D=+19.35°; 26.3 г, 0.26 моль.) в CH2Cl2 (470 мл), охлажденного при 0°С. Раствор перемешивают в течение 5 час при -5°С, добавляют порошок КОН (6.2 г) и перемешивание продолжают 3 часа при -5°С. Реакционную смесь фильтруют на гифлоселе и растворитель упаривают в вакууме. Сырую реакционную смесь очищают последовательно хроматографией на силикагеле (AcOEt/i - PrOH: 97/03 (об/об)) и препаративной хроматографией на хиральной стационарной фазе (гексан/EtOH), что дает два изомера (2S)-2-(4-аллил-2-оксо-1-пирролидинил)бутанамида (соответственно 6.0 (228) и 5.48 г (224); 16 и 15%). Две небольшие примеси также выделяют, следуя хиральной хроматографии, определенные как два стереоизомера (2S)-2-[4-(2-иодпропил)-2-оксо-1-пирролидинил]бутанамида 225 (0.22 г) и 226 (0.27 г) в виде белых твердых веществ после перекристаллизации.
2.2.2. Алкилирование/ацилированием бутирамида.
Синтез двух стереоизомеров (2S)-2-(5-нонил-2-оксо-1-пирролидинил)бутанамида представлен следующим образом:
- Стадия 1: Раскрытие лактона
К раствору γ-ноналактона (0.32 мл, 2 ммоль) в тионилхлориде (164 мкл. 1,2.25 ммоль), добавляют хлорид цинка (12 мг, 0.088 ммоль) при комнатной температуре и смесь перемешивают в течение 24 часов. Добавляют избыток метанола и реакционную смесь перемешивают в течение 10 мин., после чего концентрируют при пониженном давлении, что дает 4-хлорнонановой кислоты метиловый эфир, который используют как таковой.
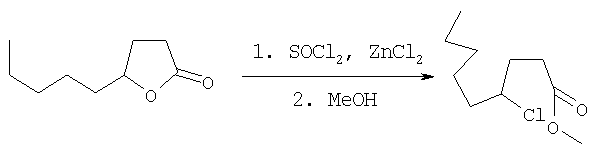
- Стадия 2: Алкилирование
К раствору метилового эфира 4-хлорнонановой кислоты (2 ммоль) в ДМ ФА (2 мл) последовательно добавляют 2-аминобутирамид (1 г, 10 ммоль), 300 мг иодида натрия (2 ммоль) и 276 мг карбоната калия (2 ммоль). Смесь перемешивают в течение ночи при 60°С. Твердые вещества фильтруют и промывают с помощью CH2Cl2 (2×2 мл). Фильтрат концентрируют при пониженном давлении, что дает производное эфира, которое используют как таковое для циклизации.
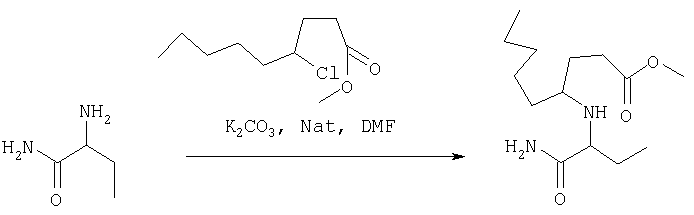
Стадия 3: циклизация: смотри условия § 1.2.2. и § 1.2.3.
2.3. Синтез кетопирролидин-2-онов
Синтез (2S)-2-[2-оксо-4-(2-оксопропил)-1-пирролидинил]бутанамида 230 представлен следующим образом:
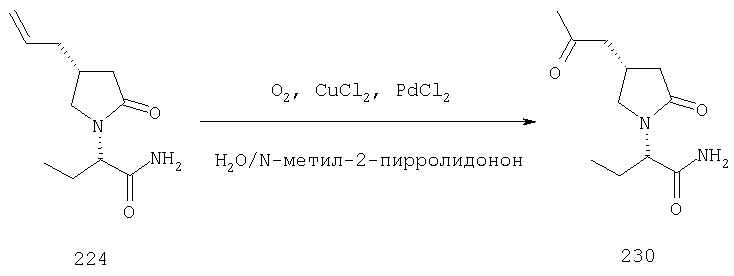
В трехгорлой колбе, пробулькивают кислород через раствор PdCl2 (0.68 г, 0.0039 моль.), CuCl2 (1.68 г, 0.0098 моль.) в N-метил-2-пирролидинона (NMP, 40 мл) и добавляют по каплям раствор (2S)-2-[2-оксо-4-(2-оксопропил)-1-пирролидинил]бутанамида 224 (4.13 г, 0.020 моль) в NMP (40 мл) (дополнительное время: 1.2 часа). Раствор перемешивают при пробулькивании газа в течение 0.75 часа, фильтруют 3 через целит и упаривают под вакуумом (1 мм Нд). Сырой кетон очищают хроматографией на силикагеле (СН2Cl2/метил-т-бутиловый эфир/i-PrOH 9/0.9/0.1 (об/об)), что дает (2S)-2-[2-оксо-4-(2-оксопропил)-1-пирролидинил]бутанамид 230 в виде белого твердого вещества после перекристаллизации из AcOEt.
2.4. Получение производных кетона 230
2.4.1. Синтез спиртов
Синтез (2S)-2-[(4S)-4-(2-гидроксипропил)-2-оксопирролидинил]бутанамида 233 представлен следующим образом:
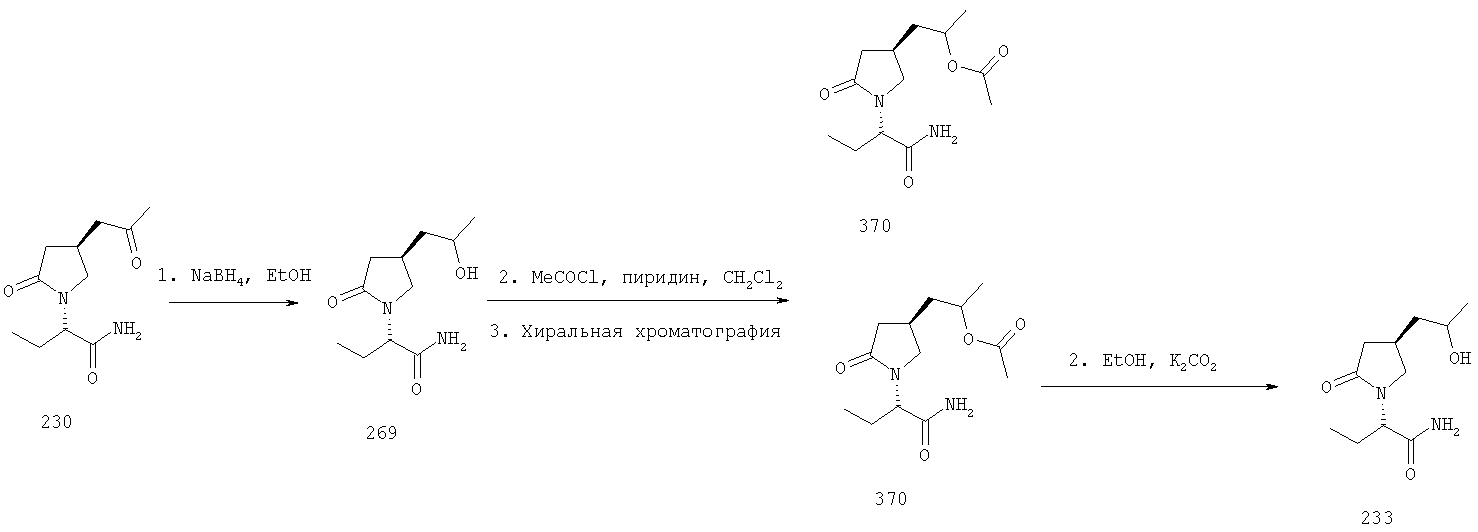
- Стадия 1: Восстановление.
В трехгорлой колбе, в атмосфере аргона, добавляют порциями NaBH4 к раствору 230 (9 г, 0.012 моль) в EtOH (140 мл), охлажденном при -5°С. Раствор перемешивают в течение 4 часов при указанной температуре, захолаживают насыщенным раствором хлорида аммония и упаривают досуха. Твердое вещество растворяют в СН3ОН/СН2Cl2, фильтруют и концентрируют в вакууме, Остаток очищают хроматографией на силикагеле (СН3ОН/СН2Cl2: 90/10 (об/об)), что дает эпимерную смесь спиртов 369 (2.2 г, 79%) в виде масла. Сырую смесь непосредственно ацетилируют на следующей стадии. 1Н ЯМР (400 МГц, (CD3)2SO): 0.70 (т, 3Н), 1.05 (д, 3Н), 1.30-1.45 (м, 1Н), 1.70-1.80 (м, 1Н), 1.80-2.05 (м, 1Н), 2.20-2.40 (м, 2Н, частично перекрыт с растворителем), 3.00-3.20 (м, 1Н), 3.30-3.35 (м, 2Н, частично перекрыт с растворителем), 3.50-3.65 (м, 1Н), 4.30 (м, 1Н), 4.45 (м, 1Н), 7.10 (синглет (широкий), 1Н), 7.20 (синглет(широкий), 1Н).
- Стадия 2: Ацетилирование.
В трехгорлой колбе, в атмосфере аргона, добавляют ацетил хлорид (0.91 г, 0.011 моль.) к раствору 4-N,N-диметиламинопиридина (0.11 г, 0.001 моль), пиридина (0.86 мл) и спирта в CH2Cl2 (90 мл) при комнатной температуре. Раствор перемешивают в течение 5 часов, захолаживают насыщенным раствором хлорида аммония и водный слой экстрагируют СН2Cl2 (3х), сушат над сульфатом магния и концентрируют в вакууме, что дает сырой ацетат, который очищают с помощью колоночной хроматографии на хиральный фазе (гексан/EtOH), что дает два эпимерных ацетата 370 и 371 (соответственно 1.143 и 1.17 г). Для смеси 1/1 370 и 371 перед хиральной хроматографией: 1Н ЯМР (400 МГц, CD3SOCD3): 0.90 (т, 3Н), 1.21-1.28 (м, 4Н), 1.51-1.82 (м, 4Н), 1.89-1.98 (м, 1Н 1.80-2.05 (м, 1Н),), 2.04 (с, 3Н), 2.16 (дд, 1Н), 2.38 (м, 1Н), 2.62 (дд, 1Н), 3.11 (дд, 1Н); 3.49 (дд, 1Н), 4.39-4.49 (м, 1Н), 4.89-4.99 (м, 1Н), 5.43 (с (широкий), 1Н), 6.24 (с (широкий), 1Н).
- Стадия 3: Деацетилирование.
В трехгорлой колбе, в атмосфере аргона, суспензию одного энантиомера ацетата 371 (1.11 г, 0.0042 моль.) и K2СО3 в EtOH перемешивают в течение 20 часов при 0°С, упаривают досуха и сырой спирт очищают хроматографией на силикагеле (СН3ОН/СН2Cl2: 85/15 (об/об)), что дает (2S)-2-[(4S)-4-(2-гидроксипропил)-2-оксопирролидинил]бутанамид 233 (0.67 г, 72%) в виде белого твердого вещества после перекристаллизации из ацетонитрила.
2.4.2. Фторирование 230
Фторирование кетона 230 используют для синтеза 2-[(4S)-4-(2,2-дифторпропил)-2-оксопирролидинил]бутанамида 265.
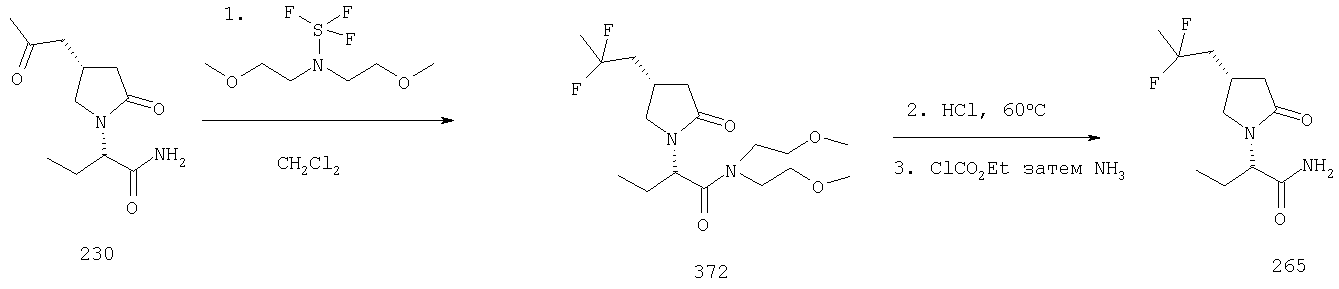
- Стадия 1: Фторирование
В тефлоновой колбе, в атмосфере аргона, добавляют порциями (MeOCH2CH2)2NSF3 (1.86 г, 0.009 моль) к раствору 230 (0.389 г, 0.0017 моль) в СН2Cl2 и нагревают 4 часа при 80°С. Раствор перемешивают в течение 4 часов при указанной температуре, захолаживают карбонатом натрия, экстрагируют СН2Cl2, промывают HCI 1N, сушат над MgSO4, фильтруют и концентрируют в вакууме, что дает третичный амид 372 (1.2 г). LC/MS: 365 (МН+). Сырую смесь непосредственно используют на следующей стадии.
- Стадия 2: Гидролиз и аммонолиз.
В трехгорлой колбе, в атмосфере аргона нагревают раствор сырого продукта 372 (0.28 г) в HCI 6N в течение 22 час при 60°С, охлаждают до комнатной температуры и водный раствор упаривают досуха.
Твердое вещество растирают в MeCN, фильтруют и сушат под вакуумом, что дает кислоту (1.2 г) в виде белого твердого вещества.
Сырую смесь амидируют в стандартных условиях, описанных в § 6.3.1. (стадия 2), что дает смесь (2S) и (2R)-2-[(4S)-4-(2,2-дифторпропил)-2-оксопирролидинил]бутанамида (соответственно 87 и 13%).
2.5. Синтез (2S)-2-(2-оксо-4-пропил-1-пирролидинил)бутанамида 158 и 159
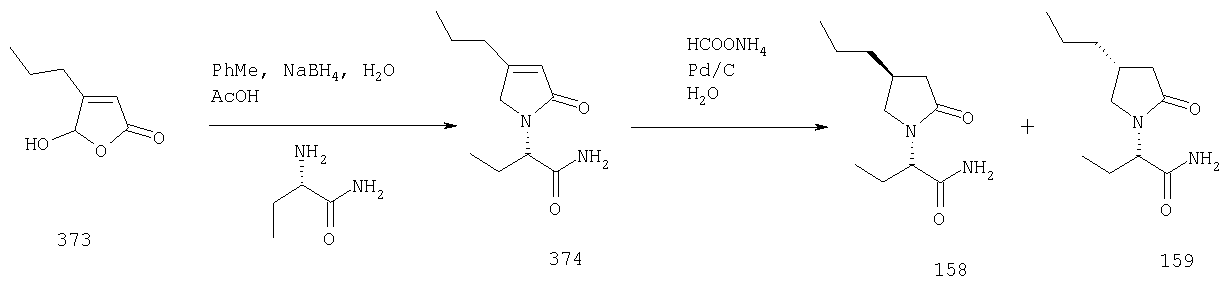
2.5.1. Стадия 1: Восстановительное аминирование
В трехгорлой колбе, в атмосфере аргона, 4-н-пропилгидроксифуранон 373 (35.5 г, 0.25 моль, синтезированный по Bourguignon JJ et al; J.Med. Chem, 1988, 31, 893-897), добавляют к раствору S-2-аминобутирамида (28.1 г, 0.275 моль.) в PhMe (355 мл) при 18°С. Раствор перемешивают в течение 0.5 час при указанной температуре и при этом появляется осадок. Реакционную смесь перемешивают в течение 2 час и добавляют по каплям NaOH 4N (37.5 мл) к суспензии, а затем водный раствор NaBH4 (6.2 г, 0.16 моль) в воде (62 мл). Через 1 час реакционную смесь тщательно захолаживают АсОН (30 мл), нагревают до 50°С в течение 3 час и охлаждают до комнатной температуры в течение ночи. Добавляют (20 мл) NaOH 50% об/об и водную фазу экстрагируют PhMe (2х). Органические фазы объединяют, промывают рассолом и концентрируют в вакууме, что дает сырой ненасыщенный пирролидон 374 (43.4 г) в виде оранжевого масла, которое используют на следующей стадии без дальнейшей очистки. Он может быть перекристаллизован в белое твердое вещество (DSC, начало; т.пл. = 72.9°С).
2.5.2. Стадия 2: Гидрогенолиз
В трехгорлой колбе, в атмосфере аргона, добавляют порциями водный раствор NH4COOH (8 г, 0.126 моль) к суспензии сырого продукта 374 (22 г, 0.105 моль) и 10% Pd/C (1.1 г) в воде (220 мл) и нагревают до 50°С. Суспензию перемешивают в течение 3 час при 50°С, охлаждают до комнатной температуры и перемешивают в течение ночи. Через 18 час, суспензию нагревают до 50°С и добавляют порциями водный раствор NH4COOH (8 г, 0.126 моль). Через 1.5 час добавляют третью порцию водного раствора NH4COOH (8 г, 0.126 моль.). Суспензию перемешивают в течение 0.5 час при 50°С и добавляют 10% Pd/C (1.1 г). Суспензию перемешивают в течение 5 час при указанной температуре и оставляют на ночь без перемешивания при комнатной температуре. Реакционную смесь фильтруют через целит, промывают водой (30 мл) и водный слой экстрагируют AcOEt (3х). Объединенные органический фазы промывают рассолом и концентрируют в вакууме, что дает сырой пирролидон в виде белых кристаллов (18.1 г). Два диастериозомера разделяют с помощью препаративной HPLC (высокоэффективной жидкостной хроматографии) на хиральной фазе (EtOH/гептан: 1/1), что дает после перекристаллизации из iPr2О, два пирролидона 158 (9.5 г) и 159 (7.2 г) в виде белых твердых веществ.
Две формы твердого вещества 159 наблюдают, называя их формой А и формой В. Форма А обычно характеризуеся пиками диафрагм 8.8, 9.8, 14.9, 15.0, 17.0, 17.1, 21.2, 21.4, 24.8 (26°). Форма В обычно характеризуеся пиками диафрагм 6.50, 11.25, 19.22, 23.44, 28.47, 29.94 (29°).
2.5.3. Синтез 5-гидрокси-4-пропилфуран-2-она
5-Гидрокси-4-пропил-5Н-фуран-2-он 373 (15 г, 0.1 моль), этилацетат (260 мл) и Pd/C 5% помещают в аппарат Парра. Смесь дегазируют и водород вводят при давление 35 пси. Указанную смесь затем энергично перемешивают при 25°С в течение 2 час. После фильтрования на целите, растворитель удаляют при пониженном давлении при 50°С, что дает 5-гидрокси-4-пропилфуран-2-он в виде сырого продукта (100% выходом). LC/MS: 145 (МН+).
ПРИМЕР 3. Синтез 4-замещенных 2-оксопирролидинбутанамидов алкилированием 2-оксопирролидин этил 2-бромбутаноатом.
3.1. Синтез 4-замещенных 2-оксопирролидинов
3.1.1.а.1. Получение этил 3-(3-хлорфенил)-2-пропеноата 375:
В 2-литровой трехгорлой колбе, оснащенной механической мешалкой и делительной воронкой, в инертной атмосфере, растворяют 106,6 г (755 молей, 1 экв.) 3-хлорбензальдегида в 1 л ТГФ и охлаждают до 0°С. Затем добавляют 341.9 г (980 ммолей, 1.3 экв) этил(трифенилфосфоранилиден) ацетата при энергичном перемешивании, температуру поднимают до 10°С. Смесь продолжают перемешивать один час при 0°С, а затем в течение ночи при комнатной температуре. Смесь концентрируют досуха, остаток суспендируют в диэтиловом эфире, оксид трифенилфосфина отфильтровывают и фильтрат концентрируют досуха. Остаток очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, петролейный эфир/EtOAc, 75:35), что дает 191.8 г чистого 375, 92% выходом. 1H ЯМР (250 МГц, (CD3)2SO): 1.30 (т, 3Н), 4.25 (кв, 2Н), 6.70 (д, 1Н), 7.40 (м, 2Н), 7.50-7.70 (м, 2Н), 7.85 (с(широкий), 1Н).
3.1.1.а.2. Другие способы:
Альтернативно, производные циннамата также синтезируют с помощью катализируемого палладием карбометаллирования акриловых производных.
Например, этил (2E)-3-(5-пиримидинил)-2-пропеноат 376 получают путем взаимодействия между этилакрилатом и 5-бромпиримидином в присутствии ацетата палладия.
3.1.1-b. Получение этил 3-(3-хлорфенил)-4-нитробутаноата 377:
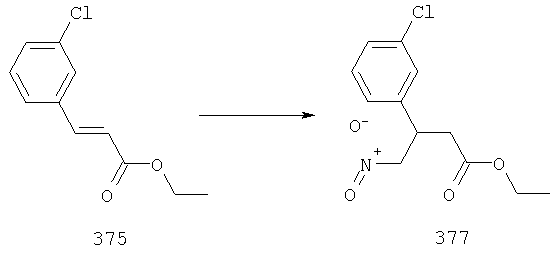
В трехгорлой колбе, объемом 500 мл, оснащенной парциальным конденсатором горячего орошения, магнитной мешалкой и капельной воронкой в инертной атмосфере, растворяют 100 г (447 ммолей, 1 экв) этил 3-(3-хлорфенил)-2-пропеноата 375 в 127 мл (2.37 молей, 5 экв) нитрометана. Затем добавляют по каплям 70.9 мл (447 ммолей, 1 экв) диазабициклоундецена при энергичном перемешивании, поддерживая температуру ниже 25°С (баня лед/вода). Темно-красную смесь перемешивают в течение ночи при комнатной температуре. Смесь разбавляют диэтиловым эфиром, промывают 1N HCl, водную фазу вновь экстрагируют дважды этиловым эфиром. Объединенные органические фазы сушат над сульфатом магния, фильтруют и концентрируют досуха, что дает 128.5 г сырого продукта 377, 99% выход, который используют как таковой на следующей стадии. 1H ЯМР (250 мгц, (CD3)2SO): 1.10 (т, 3Н), 2.70 (дд, 1Н), 2.75 (дд, 1Н). 3.95 (кв, 2Н), 4.95 (м, 2Н), 7.20-7.45 (м, 4Н).
3.1.1.c. Получение этил 4-амино-3-(3-хлорфенил)бутаноата 378:
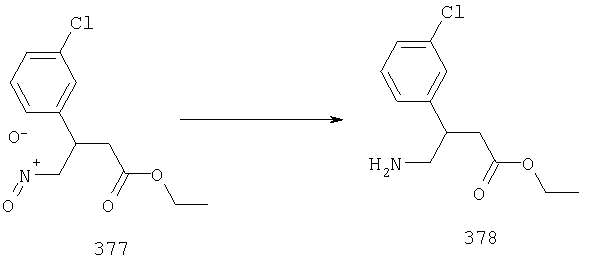
В 2-литровом автоклаве, в инертной атмосфере, растворяют 196 г (733 ммолей) этил 3-(3-хлорфенил)-4-нитробутаноата 377 в 200 мл этанола.
Суспензию 200 г предварительно высушенного (3 х, этанол) никеля Ренея добавляют в 700 мл этанола и смесь гидрируют в аппарате Парра при максимальном давлении Н2 20 пси (СИЛЬНО ЭКЗОТЕРМИЧНАЯ РЕАКЦИЯ, требуется охлаждение лед/вода). Смесь дегазируют, фильтруют на Целит/Норитовой подушке и фильтрат концентрируют в вакууме, что дает 136.7 г сырого продукта 378, 78% выход, который используют как таковой на следующей стадии.
3.1.1.d. Получение 4-(3-хлорфенил)-2-пирролидинона 379:
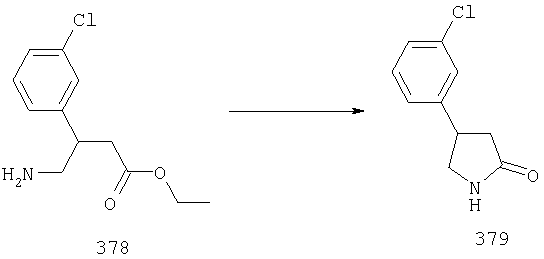
В колбе, объемом 500 мл, оснащенной парциальным конденсатором горячего орошения и магнитной мешалкой, растворяют 135.7 г (561 ммолей) этил 4-амино-3-(3-хлорфенил)бутаноат 378 в 200 мл толуола и смесь кипятят с обратным холодильником в течение 30 мин. Раствор концентрируют досуха и остаток очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, CH2Cl2/EtOH, 98:2 -> 95:5), что дает 54.4 г чистого 379 (49.2%). GC/MS: 197/197 M+
3.1.1.f. Получение этил 2-[4-(3-хлорфенил)-2-оксо-1-пирролидинил]бутаноата 380
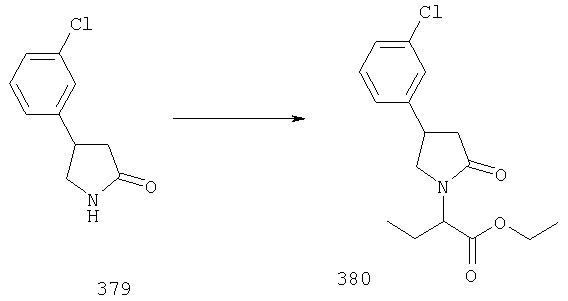
В 2-литровой трехгорлой колбе, оснащенной парциальным конденсатором горячего орошения, магнитной мешалкой и капельной воронкой в инертной атмосфере, растворяют 54.4 г (278 ммолей, 1 экв) 4-(3-хлорфенил)-2-пирролидинона 379 в 1.4 л ацетонитрила. Добавляют 64 мл (100.7 г, 556 ммолей, 2 экв) метил 2-бромбутаноата и температуру поднимают до 50°С. Затем добавляют порциями 22.24 г (556 ммолей, 2 экв) гидрида натрия, температуру поднимают до 65°С. Смесь перемешивают более одного часа при 50°С. Смесь концентрируют досуха, остаток суспендируют в этилацетат, промывают водой, водную фазу вновь экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, петролейный эфир/EtOAc, 70:30), что дает 56.7 г чистого 380, 69%. 1H ЯМР (250 МГц, (CD)3)2SO): 0.80-1.00 (м, 3Н), 1.60-1.90 (2Н, м), 2.35-2.55 (м, 1Н: частично перекрывается с растворителем), 2.60-2.90 (м, 1Н: частично перекрывается с растворителем), 3.70 (с, 3Н), 3.50-3.80 (м, 3Н), 4.50 (м, 1Н), 7.20-7.50 (м, 4Н).
3.1.1.g. Получение 2-[4-(3-хлорфенил)-2-оксо-1-пирролидинил]бутанамида 381:
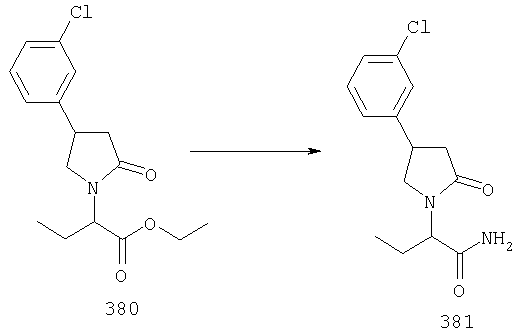
В трехгорлой колбе, объемом 1 л, оснащенной парциальным конденсатором горячего орошения, магнитной мешалкой, растворяют 56.7 г (192 ммолей) этил 2-[4-(3-хлорфенил)-2-оксо-1-пирролидинил] бутаноата 380 в 600 мл метанола. Газообразный аммиак пробулькивают через раствор и насыщенный раствор выдерживают при комнатной температуре в течение 5 дней, во время которых со временем улетучивается аммиак. После завершения реакции, раствор концентрируют досуха. Остаток очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, СН2Cl2/EtOH, 97:3), что дает 50 г чистого 381, 97.8%. 82.2 г Смеси диастереоизомеров разделяют с помощью хирально препаративной жидкостной хроматографии (Chiralpak AD, бензол/EtOH, 50:50) и каждую пару энантиомеров еще раз разделяют с помощью хиральной препаративной жидкостной хроматографии (Chiralpak AD, бензин/EtOH, 50:50).
Четыре соединения кристаллизуют из толуола, что дает 16.79 г, 13.9 г, 15.84 г, и 14.84 г 202, 203, 204 и 205 соответственно, 72% общий выход.
ПРИМЕР 4. Синтез 4-замещенных 2-оксопирролидинбутанамидов алкилированием/циклизацией эфиров 4-бром-3-замещенных-бут-2-еновой кислоты с 2-аминобутанамидами.
4.1. Синтез эфира 4-бром-3-замещенных-бут-2-еновой кислоты, алкилирование и восстановление
4.1.1 Бромирование этиловых эфиров 3-замещенной кротоновой кислоты
Синтез этилового эфира 4-бром-3-(2-тиофенил)-бут-2-еновой кислоты 382 представлен следующим образом:

В 2-литровой трехгорлой колбе, оборудованной механической мешалкой, кипятят с обратным холодильником, в атмосфере аргона, в течение 6 час дегазированный раствор этилового эфира 2-тиофен-3-ил-бут-2-еновой-кислоты 383 (32.88 г, 0.211 моль), N-бромсукцинимид (37.56 г, 0.211 моль) и 2,2'-аза-бис-изобутиронитрил (3.46 г, 0.021 моль) в CCl4 (600 мл), затем охлаждают до комнатной температуры и перемешивают в течение 20 час. Суспензию фильтруют и концентрируют в вакууме, что дает сырой бромид, который очищают хроматографией на силикагеле (Гексан/СН2Cl2: 65/35 (об/об)), что дает этиловый эфир 4-бром-3-(2-тиофенил)-бут-2-еновой кислоты 382 (36.72 г, 78%). 1Н ЯМР (250 МГц, (CDCl3): 3.80 (с, 3Н), 4.95 (с, 2Н), 6.25 (с, 1Н), 7.10 (дд, 1Н), 7.35 (д, 1Н), 7.45 (д, 1Н).
4.1.2. Алкилирование 2-аминобутанамида.
Синтез 2-[2-оксо-4-(2-тиенил)-1-пирролидинил]бутанамида 71 представлен следующим образом:
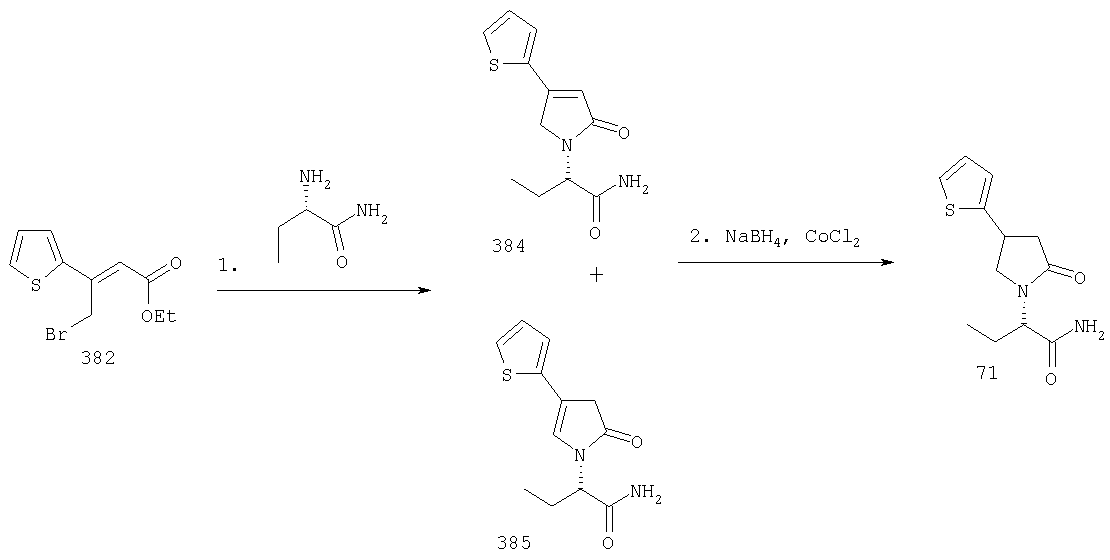
4.1.2.1. Стадия 1: Алкилирование-циклизация
В трехгорлой колбе, объемом 1 л в атмосфере аргона раствор метилового эфира 4-бром-2-тиофен-3-ил-бут-2-еновой-кислоты 382 (36.72 г, 0.134 моль), (S)-2-аминобутирамид ([α]25D: 19.09°; 31.6 г, 0.270 моль) в ТГФ (350 мл) перемешивают в течение 20 час при комнатной температуре. Суспензию фильтруют и концентрируют в вакууме, что дает сырые ненасыщенные пирролидоны 384 и 385 (43.47 г), которые используют на следующей стадии без дальнейшей очистки. Сырой пирролидон может быть выделен и обычно, представляет собой смесь изомеров двойной связи (олефин в 3,4 и 4,5, первый из которых является основным). 1H ЯМР (250 МГц, (CD3)2SO): 0.80 (т, 3Н), 1.30-1.90 (м, 2Н), 4,40 (д, 1Н), 4.45 (м. 1Н), 4.70 (д, 1Н), 6.30 (с, 2Н), 7.0 (с (широкий), 1Н), 7.15 (дд, 1Н), 7.40 (с (широкий), 1Н), 7.50 (д, 1Н), 7.85 (д, 1Н).
4.1.2.2. Стадия 2: Восстановление
В трехгорлой колбе, объемом 0.5 л в атмосфере аргона, добавляют порциями NaBH4 (1.75 г, 0.044 моль.) к раствору сырого ненасыщенного пирролидона 384/385 (14 г, 0.044 моль.), CoCCl2 (0.062 г, 0.0005 моль) в EtOH (100 мл) - диэтиленгликоль диметиловом эфире (65 мл), охлажденном при 0°С. Через 0.75 час, реакционную смесь нагревают при кипении с обратным холодильником в течение 48 час и в течение этого времени последовательно тремя порциями добавляют NaBH4 (1.75 г, 0.045 моль) и CoCl2 (0.062 г, 0.0005 моль) каждые 10 час, пока не исчезнет исходное вещество. Реакционную смесь охлаждают до комнатной температуры, гидролизуют насыщенным раствором хлорида аммония, экстрагируют AcOEt, сушат над сульфатом магния и концентрируют в вакууме, что дает сырой пирролидон, который очищают с помощью колоночной хроматографии на силикагеле (СН2Cl2/СН3ОН: 97/03 (об/об)), что дает 4.15 г 2-[2-оксо-4-(2-тиенил)-1-пирролидинил] бутанамида (38%). Смесь стереоизомеров очищают с помощью колоночной хроматографии на хиральной фазе (гексан/EtOH), что дает два диастериозомера (2S)-2-[2-оксо-4-(2-тиенил)-1-пирролидинил] бутанамид 71 (перекристал-лизовывают из AcOEt) и 72 (перекристаллизовывают из AcOEt). В указанном конкретном случае, две небольшие примеси, являющимися двумя диастериозомерами (2R)-2-[2-оксо-4-(2-тиенил)-1-пирролидинил]бутанамид 84 (0.25 г, перекристаллизовывают из AcOEt) и 85 (0.44 г, перекристаллизовывают из AcOEt) e также получают при очистке.
4.2. Синтез азидофенилпирролидонов
Синтез единственного энантиомера (2S)-2-[4-(3-азидофенил)-2-оксо-1-пирролидинил]бутанамида 86 представлен следующим образом:
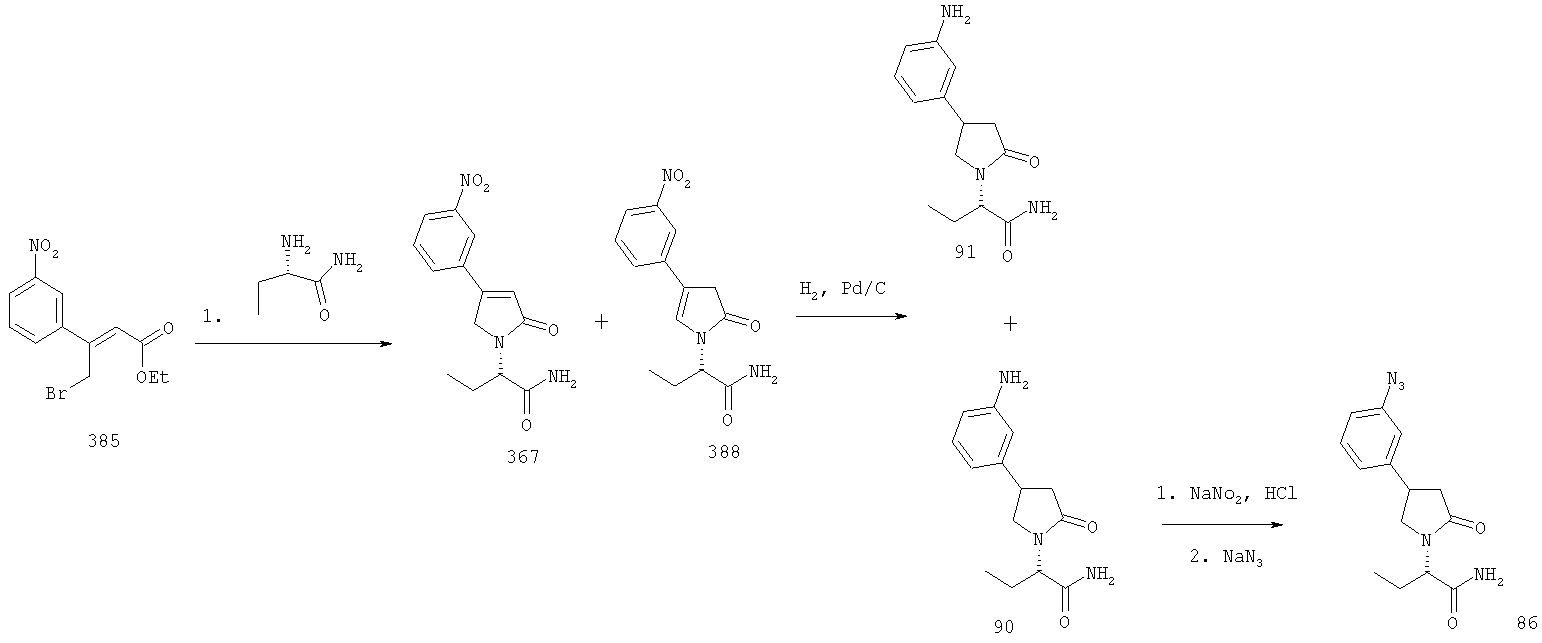
4.2.1. Синтез анилинов.
4.2.1.1. Стадия 1: Алкилирование (S)-2-аминобутирамида метиловым эфиром 4-бром-3-(3-нитрофенил)-бут-2-еновой-кислоты 386
Синтез 386 осуществляют as описано в § 4.1.1. 1H ЯМР (250 МГц, (CD3)2SO): 1.30 (т, 3Н), 4.20 (кв, 2Н), 5.15 (с, 2Н), 6.45 (с, 1Н), 7.75 (дд, 1Н), 8.10 (дд, 1Н), 8.25 (дд, 1Н), 8.45 (д, 1Н).
Алкилирование осуществляют, следуя экперементальной методике, описанной в § 4.1.2.1. (59%).LC/MS: 290 (МН+).
4.2.1.2. Стадия 2: Восстановление
В автоклаве, объемом 2,5 л, в инертной атмосфере, растворяют 7.22 г (0.025 моль) 387 и Pd на угле (10% об/об, 0.2 г) в EtOH (11) и смесь гидрируют в аппарате Парра при максимальном давлении Н2 в 20 пси. Через час смесь дегазируют, фильтруют на Целит/Норитовой подушке и фильтрат концентрируют в вакууме, что дает сырой пирролидон, который очищают с помощью хроматографической колонки на силикагеле (СН2Cl2/СН3ОН: 93/07 (об/об)), что дает смесь диастериозомеров, которые очищают с помощью колоночной хроматографии на хиральной фазе (гексан/EtOH), что дает, после реакции с HCl в EtOH (для синтеза гидрохлорида) два диастериозомера (2S)-2-[4-(3-аминофенил)-2-оксо-1-пирролидинил] бутанамида 90 (0.800 г, перекристаллизовывают из EtOH) и 91 (1.21 г, перекристаллизовывают из EtOH) в виде их гидрохлорной соли.
4.2.2. Синтез фенилазидо 86.
В трехгорлой колбе, в атмосфере аргона, добавляют по каплям NaNO2 (0.232 г, 0.0037 моль) в воде (1.5 мл) к раствору свободного основания (2S)-2-[4-(3-аминофенил)-2-оксо-1-пирролидинил] бутанамида 90 (0.8 г, 0.0031 моль) в HCl 10 M (6.5 мл), охлажденного до 0°С. Через 0.5 часов при комнатной температуре, добавляют NaN3 (0.220 г, 0.0037 моль) в воде (2 мл) и полученный раствор перемешивают в течение 0.5 час при 0°С. Реакционную смесь захолаживают NaOH (33% об/об) и разбавляют с помощью EtOAc. Водную фазу подкисляют до рН 5-6 и экстрагируют EtOAc. Объединенные органические фазы сушат над сульфатом магния и концентрируют в вакууме, что дает сырой пирролидон, который очищают с помощью хроматографической колонки на силикагеле (СН2Cl2/СН3ОН: 97/03 (об/об)), что дает, после перекристаллизации MeCN, 0.42 г единственный энантиомер (2S)-2-[2-оксо-4-(3-азидофенил)-1-пирролидинил]бутанамида 86 (48%).
4.3. Синтез (2S)-2-[4-(3-амино-2,4,6-трибромфенил)-2-оксо-1-пирролидинил]бутанамида 107
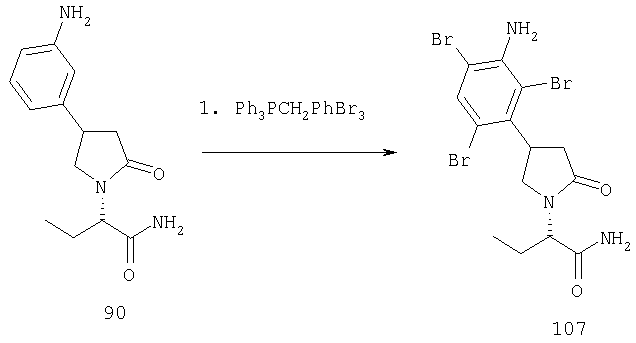
В трехгорлой колбе, в атмосфере аргона перемешивают раствор Ph3РСН2PhBr3 (2.870 г, 0.048 моль) и 90 (0.420 г, 0.0016 моль) в СН2Cl2 (10 мл) и СНзОН (5 мл) с NaHCO3 (0.407 г, 0.048 моль) в течение 4 час при комнатной температуре (оранжевый раствор). Реакционную смесь фильтруют и концентрируют в вакууме, что дает сырой анилин, который очищают с помощью хроматографической колонки на силикагеле (AcOEt/этанол 98/02 (об/об)), что дает 0.38 г ожидаемого анилина 107 (47%, перекристаллизация из Et2O).
4.4. Синтез (2S)-2-[4-метил-2-оксо-1-пирролидинил]бутанамида 35 и 36
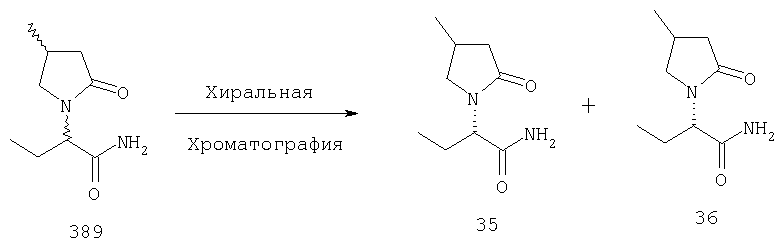
35 и 36 были получены путем хиральный очистки рацемата 389 на хиральной стационарной фазе, используя EtOH и гексан как растворители, 35 получают в виде белых кристаллов после перекристаллизации из i-Pr2OEt, 36 получают в виде белых кристаллов после перекристаллизации в Et2O,
ПРИМЕР 5. Синтез 4-замещенных 2-оксопирролидинбутанамидов путем дериватизации метил 1-[1-(аминокарбонил)пропил]-5-оксо-3-пирролидинкарбоксилата 11.
5.1. Синтез метил 1-[1-(аминокарбонил) пропил]-5-оксо-3-пирролидинкарбоксилата 11/12
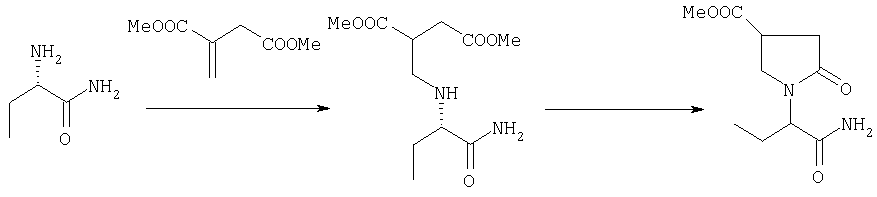
Указанное превращение описано также в § 7.0.1 для получения двух эфиров 11 и 12.
5.2. Синтез 1-[2S-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинкарбоновой кислоты 48
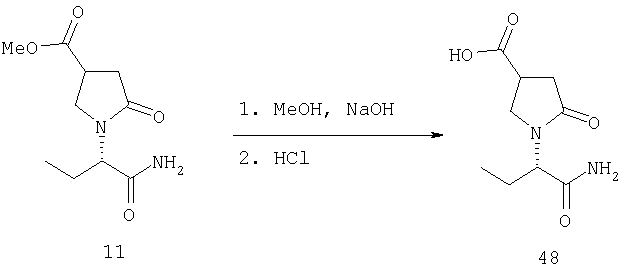
В трехгорлой колбе, в атмосфере аргона добавляют раствор 1N NaOH (126 мл) к раствору энантиомерно чистого эфира 11 (22.62 г, 0.1 моль) в СН3ОН, охлажденного при 0°С. Через 1.5 час при указанной температуре, реакционную массу подкисляют с помощью HCl (В (109 мл), растворители упаривают в вакууме. Остаток экстрагируют i-PrOH, фильтруют и фильтрат концентрируют в вакууме, чтобы получить сырую кислоту (17.82 г), которую перекристаллизовывают из MeCN, чтобы получить энантимерно чистую 1-[2S-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинкарбоновую кислоту 48.
5.3. Синтез (2S)-2-{4-(1,3,4-оксадиазол-2-ил)-2-оксо-1-пирролидинил]бутанамида 50
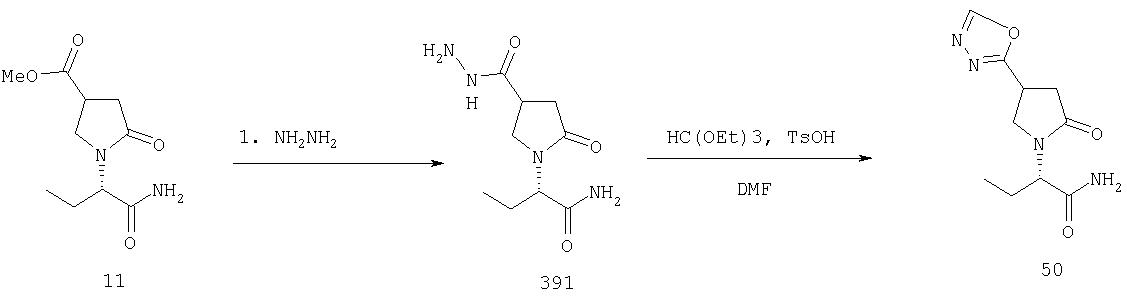
Стадия 1; Реакция с гидразином
В трехгорлой колбе в атмосфере аргона перемешивают раствор эфира 11 (3 г, 0.013 моль) и гидразингидрата (0.7 мл) в EtOH (3 мл) в течение 24 час. Затем желтый раствор концентрируют, что дает сырой гидразид 391, который кристаллизуется при стоянии (2.37 г, 79%). GC/MS: 228 (М+).
Стадия 2: Синтез оксадиазола
В трехгорлой колбе, в атмосфере аргона раствор сырого гидразида 391 (данный патент, 3 г, 0.013 моль), триэтил ортоформиата (2 мл) и п-толуолсульфоновой кислоты (0.010 г) нагревают при 110°С в течение 24 час. Реакционную смесь охлаждают до комнатной температуры, концентрируют под вакуумом, что дает сырой оксадиазол, который очищают хроматографией на силикагеле (СН2Cl2/СН3ОН: 95/05 (об/об)), что дает (2S)-2-[4-(1,3,4-оксадиазол-2-ил)-2-оксо-1-пирролидинил]бутанамид 50 (0.312 г) в виде масла.
5.4. Синтез производных 1,3,4-оксадиазола
Альтернативно, производные 1,3,4-оксадиазола могут быть получены из гидразина 391. Например, 2-[2-оксо-4-(5-сульфанил-1,3,4-оксадиазол-2-ил)-1-пирролидинил]бутанамид 51 получают при взаимодействии гидразина 391 с CS2 и КОН в EtOH.
5.5. Синтез 4-аминопирролидин-2-она 392
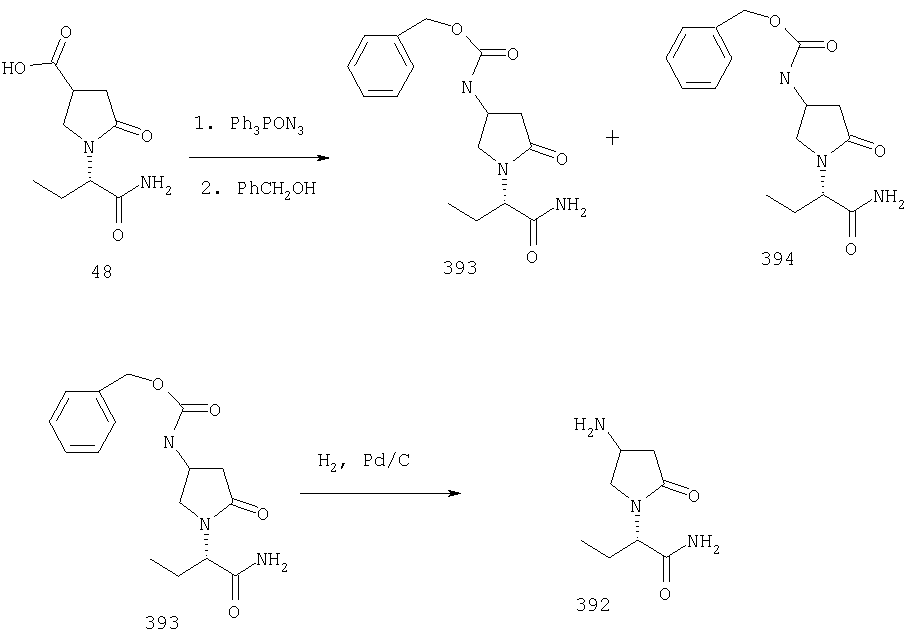
5.5.1. Стадия 1: Синтез карбамата 393
В трехгорлой колбе, в атмосфере аргона раствор энантиомерно чистой 1-[2S-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинкарбоновой кислоты 48 (19.06 г, 0.089 моль.), дифенилфосфорилазида (26.9 г, 0.097 моль.) и Et3N (13.5 мл) в MeCN (225 мл) нагревают при 55°С с образованием N2. Температуру выдерживают на уровне 55°С в течение 0.5 час, при 70°С в течение 2 часов и охлаждают до комнатной температуры. Добавляют бензиловый спирт (9.25 мл) и раствор кипятят с обратным холодильником в течение 4 час, охлаждают до комнатной температуры и концентрируют в вакууме. Сырой карбамат очищают хроматографией на силикагеле (AcOEt/СН3ОН/NH4OH: 95/04/01 (об/об)), что дает два диастереоизомерных карбамата 394 (2.64 г, 9.3%) и 393 (11.9 г, 42%). Для 393: 1H ЯМР (250 МГц, CDCl3): 0.90 (т, 3Н), 1.30-1.90 (м, 2Н), 2.35 (дд, 1Н), 2.75 (м, 1Н), 3.30 (м, 1Н), 3.75 (м, 1Н), 4.30-4.50 (м, 2Н), 5.10 (с, 2Н), 5.35 (с (широкий), 1Н), 5.55 (с (широкий), 1Н), 6.40 (с (широкий), 1Н), 7.30-7.45 (м, 5Н).
5.5.2. Стадия 2: Синтез 4-аминопирролидин-2-она 392
В автоклаве объемом 0.25 л, в инертной атмосфере, растворяют 11.9 г (0.037 ммоль) 393 и Pd на угле (10% об/об, 0.2 г) в EtOH (300 мл) и смесь гидрируют в аппарате Парра при максимальном давлении Н2 20 пси. Через 20 часов, смесь дегазируют, фильтруют на Целит/Норитовой подушке и фильтрат концентрируют в вакууме, что дает сырой амин, который перекристаллизовывают из PhMe, что дает 2-[4-амино-2-оксо-1-пирролидинил]бутанамид 392 (6.99 г, количественный).
5.6. Синтез 4-пиролпирролидин-2-она 223
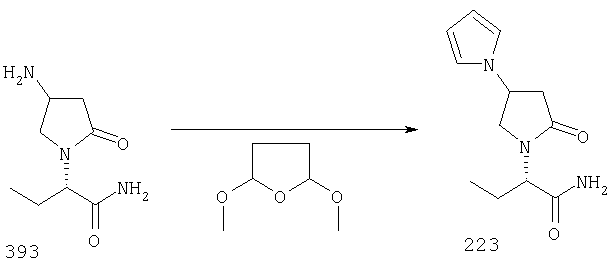
В трехгорлой колбе, в атмосфере аргона, суспензию 2-[4-амино-2-оксо-1-пирролидинил]бутанамида 393 (6.99 г, 0.037 моль), диметокситетрагидрофурана (5.53 г, 0.041 моль), пиридина (50.6 мл) и АсОН (36 мл) нагревают до 70°С и при этом происходит растворение. Через 2 час, при указанной температуре реакционную массу охлаждают до комнатной температуры, концентрируют в вакууме и сырой продукт очищают хроматографией на силикагеле [CH2Cl2/СН3ОН: 95/05 (об/об)), что дает 223 в виде масла (2.67 г, 30.1%).
5.7. Бромирование 4-пирролилдирролидин-2-она 223

В трехгорлой колбе, объемом 0.25 л с магнитной мешалкой в атмосфере аргона, дегазированный раствор 2S-4-пиролпирролидин-2-она 223 в виде единственного энантиомера (1.18 г, 0.0049 моль) в ТГФ (35 мл) охлаждают до -78°С и порциями добавляют N-бромсукцинимид (NBS) (0.877 г, 0.005 моль).
Реакционную смесь перемешивают в течение 0.5 час и добавляют Na2S2O3 (0.9 г) для гашения NBS. Реакционную смесь нагревают до комнатной температуры, концентрируют в вакууме и очищают хроматографией на силикагеле (EtOH/СН2Cl2: 05/95 (об/об)), что дает, после перекристаллизации в MeCN, (2S)-2-[4-(2-бром-1М-пиррол-1-ил)-2-оксо-1-пирролидинил]бутанамид 234 (1.05 г, 67%) в виде белого твердого вещества.
Альтернативно, используя ту же экперементальную методику и 2 экв. N-Бром-сукцинимида, может быть получен дибромпиррол 237.
5.8. Синтез производных тетразолила
Альтернативно к § 5.6, реакцией 2-[4-амино-2-оксо-1-пирролидинил]бутанамида с триэтилортоформиатом, NaN3 и АсОН получают 2-[2-оксо-4-(1N-тетразол-1-ил)-1-пирролидинил]бутанамид 67.
5.9. Синтез (4Н-1,2,4-триазол-4-ил) производных
Альтернативно к § 5.6, реакцией 2-[4-амино-2-оксо-1-пирролидинил]бутанамидов с пиридином и 1,2-бис((диметиламино)метилене)гидразином получают 2-[2-оксо-4-(4Н-1,2,4-триазол-4-ил)-1-пирролидинил]бутанамиды 65 и 66.
ПРИМЕР 6. Синтез 4-замещенных 2-оксо-пирролидинбутанамидов олефинацией 1-[1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396.
6.1. Синтез 1-[1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396
Стадия 1: Конденсирование 2-аминобутирата с метилитаконатом
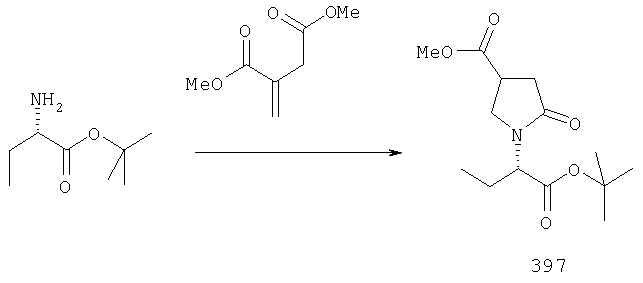
В трехгорлой колбе, объемом 1 л, в атмосфере аргона кипятят с обратным холодильником раствор 2,2-диметилэтил (S)-2-аминобутаноата (коммерчески доступный, 46.6 г, 0.268 моль) и диметилитаконата (83 мл, 0.59 моль) в СН3ОН (400 мл) в течение 20 час. Смесь перемешивают при комнатной температуре в течение 20 час, концентрируют в вакууме и остаток очищают хроматографией на силикагеле (СН2Cl2/СН3ОН: 97/3 (об/об)), что дает метил 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксилата 397 (81.6 г, количественный). Анализ смеси 1/1 метил 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксилата 397: 1H ЯМР (250 МГц, (CD3)2SO): 1.05 (т, 3Н), 1.44 (с,9Н), 1.60-1.65 (м, 1Н), 1.65-1.90 (м, 1Н), 2.40-2.65 (м, 2Н частично перекрывается с сигналами растворителя), 3.30-3.65 (м, 3Н), 3.70 (с, 3Н), 4.40 (дд, 1Н). Альтернативно, реакция может также быть осуществлена с рацемическим 2,2-диметилэтил-2-аминобутаноатом, что дает рацемическими бутанамид с аналогичным выходом.
Стадия 2: Синтез альдегида 396.
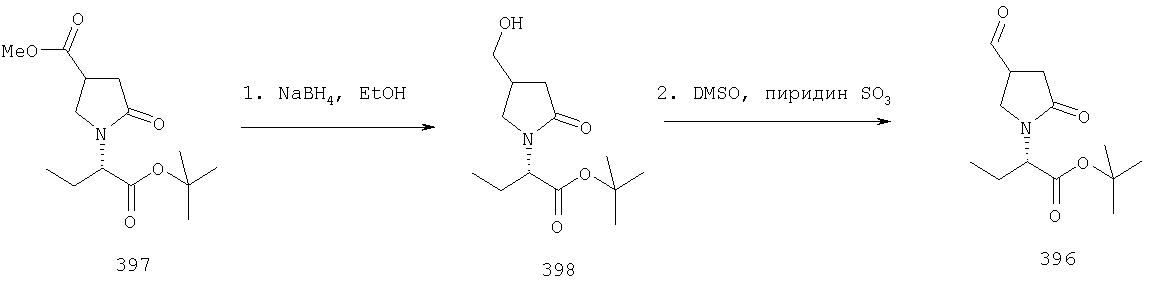
Восстановление эфира 397 до спирта 398
Это осуществляют, используя методику, описанную в § 7.0.2., используя 397 либо в виде единственного энантиомера, смесь двух диастериозомеров или смеси 1/1/1/1 4 стереоизомеров. Для диастереоизомерной смеси 1:1 трет-бутил (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутаноата 398: GC/MS:257 M+'
Окисление альдегида 396
В трехгорлой колбе, в атмосфере аргона добавляют раствор трет- бутил (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутаноата 398 (4.0 г, 0.016 моль.) в СН2Cl2 (8 мл) к суспензии CrO3 (6.2 г, 0.062 моль) в пиридине (11.3 мл/СН2Cl2 (80 мл)), перемешиваемой при комнатной температуре. Температуру поднимают до 30°С и суспензию перемешивают в течение 0.2 час. Суспензию фильтруют через целит и фильтрат промывают последовательно HCl 1N, рассолом, сушат над сульфатом магния и концентрируют в вакууме, что дает сырой альдегид, который очищают с помощью хроматографической колонки на силикагеле (гексан/ацетон 70/30 (об/об)), что дает 2.03 г 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегид 396 (41%). Альтернативно, реакцию можно также провести с рацемическим эфиром, что дает рацемический альдегид с аналогичным выход. Анализ смеси 1/1 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396: 1H ЯМР (250 МГц, (CDCl3): 0.91 (т, 3Н), 1.44 (с, 9Н), 1.55-1.77 (м, 1Н), 1.90-2.15 (м, 1Н), 2.63-2.82 (м, 2Н), 3.47-3.61 (м, 1Н), 3.65-3.79 (м, 1Н), 3.83-3.94 (м, 1Н одного из диастериозомеров), 4.48-4.62 (м, 1Н), 9.74 (с (широкий), 1Н).
6.2. Олефинация 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396
6.2.1. Синтез этиленовых производных.
Альтернативно к § 6.2.3., этиленовые производные могут быть получены олефинацией по Виттигу 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396 и соли фосфония в присутствии сильного основания. Например, (2S)-2-(2-оксо-4-винил-1-пирролидинил)бутановой кислоты 2,2-(диметил)этиловый эфир, получают путем взаимодействия альдегида 396 с Ph3РСН3Br и н-BuLi в ТГФ.
6.2.2. Олефинация с Ph3Р/CBr4
Альтернативно к § 6.2.3., производные галогенвинила могут быть получены олефинацией по Виттигу 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396 в присутствии фосфина и галогенометана. Например, (2S)-2-(2-оксо-4-(2,2-дибромвинил)-1-пирролидинил)бутановой кислоты 2,2-(диметил)этиловый эфир получают из альдегида 396 и CBr4 в присутствии трифенилфосфина.
6.2.3. Олефинация с (Me2N)3P/CF2Br2
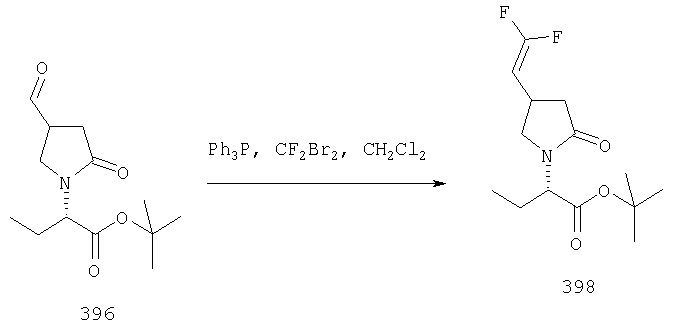
Представлен синтез двух диастериозомеров (2S)-2-(2-оксо-4-(2,2-дифторвинил)-1-пирролидинил)бутановой кислоты 2,2-(диметил)этилового эфира 399. В трехгорлой колбе, в атмосфере аргона, добавляют (Ме2N)3Р (89.8 г, 0.55 моль.) к раствору CF2Br2 (58 г, 0.25 моль.) в ТГФ (280 мл) при -78°С (появляется белый осадок) и нагревают до комнатной температуры. К образовавшейся соли фосфония добавляют по каплям раствор альдегида 396 в виде смеси 1/1 диастериозомеров (35.2 г, 0.138 моль) в ТГФ. Через 1 час реакционную смесь фильтруют через целит и концентрируют в вакууме, Реакционную смесь разбавляют гексаном, промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме, что дает сырой олефин, который очищают с помощью хроматографической колонки на силикагеле (СН2Cl2/СН3ОН 99/01 (об/об)), что дает 34.6 г. 1/1 смесь диастереоизомеров (2S)-2-(2-оксо-4-(2,2-дифторвинил)-1-пирролидинил)бутановой кислоты 2.2-(диметил)этилового эфира 399 (87%).: 1Н ЯМР (250 МГц, (CD3)2SO): 0.81-0.91 (м, 3Н), 1.44 (с, 9Н), 1.50-1.75 (м, 1Н), 1.80-1.95 (м, 1Н), 2.30-2.40 (м, 2Н частично перекрывается с растворителем), 3.00-3.35 (м, 2Н), 3.45-3.55 (м, 1Н), 4.20-4.40 (м, 1Н), 4.60 (ддд, 1Н для одного диастереоизомера), 4.75 (ддд, 1Н для другого диастереоизомера).
6.2.4. Олифенацией с (nBu)3Р/CCl3F
Альтернативно к § 6.2.3., производные галогенвинила могут быть получены олефинацией по Виттигу 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксальдегида 396 в присутствии фосфина и галогенметана. Например, 2-(2-оксо-4-(2-(Z)-фторвинил)-1-пирролидинил) бутановой кислоты 2,2-(диметил) этиловый эфир получают из альдегида 396 последовательной реакцией с CFCl3 и н-Bu3Р, а затем дефосфорилированием промежуточного винилового фосфония с помощью NaOH.
6.2.5. Синтез 4-цианопирролидона
Альтернативно, 4-цианопирролидоновые производные, получают при взаимодействии 1-[(1S)-1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкар-боксальдегида 396 с гидроксилом, а затем SeO2.
6.3. Аминирование 2.2-диметилэтилового эфира
6.3.1. Снятие защиты с помощью трифторуксусной кислоты и аминолиз
Синтез двух диастериозомеров (2S)-2-(2-оксо-4-(2,2-дифторвинил)-1-пирролидинил)бутанамидав 213 и 222 представлен следующим образом:
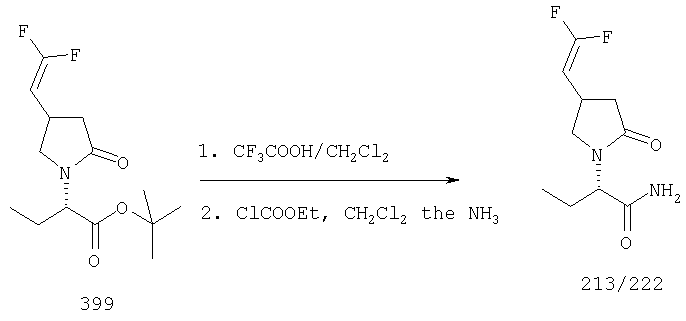
Стадия 1: Снятие защиты с 2.2-(диметил)этилового эфира
В трехгорлой колбе, в атмосфере аргона, раствор 1/1 смеси диастереоизомеров (2S)-2-(2-оксо-4-(2,2-дифторвинил)-1-пирролидинил)бутановой кислоты 2.2-(диметил) этилового эфира 399 (31.8 г, 0.110 моль.) в трифторуксусной кислоте (170 мл) и СН2Cl2 (500 мл) перемешивают в течение 20 час при комнатной температуре. Реакционную смесь упаривают досуха. Остаток растворяют в толуоле, вновь упаривают досуха, чтобы исключить присутствие трифторуксусной кислоты, что дает 32 г сырых кислот, которые используют на следующей стадии без дальнейшей очистки. LC/MS: 234 (МН+)
Стадия 2: Активация и аммонолиз
В трехгорлой колбе, в атмосфере аргона, оборудованной механической мешалкой, CICOOEt (23 мл, 0.24 моль) добавляют к раствору смеси кислот (25.6 г, 0.11 моль) в СН2Cl2 (250 мл) и триэтиламина (33.7 мл), охлажденного при -15°С. Реакционную смесь перемешивают в течение 1.5 час при -10°С, затем через раствор пропускают газообразный NH3, в то время как температуру поддерживают ниже 0°С. Суспензию перемешивают в течение 1 час при 0°С, нагревают до комнатной температуры, фильтруют и фильтрат упаривают под вакуумом, Сырые амиды очищают с помощью хроматографической колонки на силикагеле (СН2Cl2/EtOH 99/01 (об/об)), что дает 23 г 1/1 смеси диастереоизомеров (2S)-2-(2-оксо-4-(2,2-дифторвинил)-1-пирролидинил)бутановой кислоты 2.2-(диметил) этилового эфира, который очищают с помощью колоночной хроматографии на хиральной фазе (гексан/EtOH), что дает два диастериозомеров 213 (10.1 г, перекристаллизовывают из i-Pr2O) и 222 (11.2 г, перекристаллизовывают из i-Pr2O).
6.3.2. Альтернативно, снятие защиты может быть осуществлено с помощью бромкатехолборана.
4 диастериозомера 2-(2-оксо-4-(2,2-диметилвинил)-1-пирролидинил)бутанамида 163, получают при взаимодействии 1/1/ 1/1 смеси диастереоизомеров 2-(2-оксо-4-(2,2-диметилвинил)-1-пирролидинил)бутановой кислоты 2,2-(диметил) этилового эфира с бромокатехолбораном, что дает кислоту с последующим ее аминированием в условиях описанных в § 6.3.1 (стадия 2).
6.4. Синтез ацетиленовых производных
6.4.1. Синтез 2-(4-этинил-2-оксо-1-пирролидинил)бутанамида 206/207

В трехгорлой колбе, в атмосфере аргона, добавляют н-бутиллития (1.6 М в гексанах, 116 мл) к раствору 1/1 смеси двух диастериозомеров 2-[4-(2,2-дибромвинил)-2-оксо-1-пирролидинил]бутанамида (неопределенная стереохимия, 10.95 г, 0.031 моль) в ТГФ, охлажденного при -78°С. Белую суспензию перемешивают в течение 1.5 час при указанной температуре, захолаживают СН3ОН (120 мл), нагревают до комнатной температуры и концентрируют в вакууме. Сырой алкин растворяют в EtOH/СН2Cl2 (10/90 об/об)), фильтруют через целит, концентрируют в вакууме и полученное твердое вещество очищают последовательно с помощью хроматографии на силикагеле (EtOH/СН2Cl2: 10/90 (об/об)) и с помощью хроматографии на хиральной фазе (EtOH/гексан), что дает два диастериозомера 2-(4-этинил-2-оксо-1-пирролидинил) бутанамида 206 (0.84 г, перекристаллизовывают из PhMe) и 207 (0.44 г, перекристаллизовывают из PhMe).
Альтернативно, 2-(4-бром-этинил-2-оксо-1-пирролидинил)бутанамид 267 получают при взаимодействии 2-[4-(2,2- дибромвинил)-2-оксо-1-пирролидинил] бутанамида 47 с двумя эквивалентами трет.-бутилата калия в ТГФ при низкой температуре (от -50°С до 0°С).
6.4.2. Синтез 2-(4-пропин-1-ил-2-оксо-1-пирролидинил)бутанамида 280
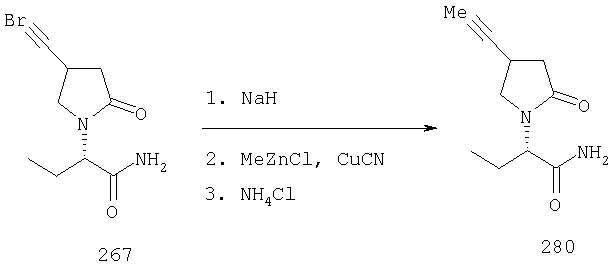
В трехгорлой колбе, в атмосфере аргона раствор метилхлорида цинка (полученного из метиллития (1.5 М в этиловым эфире, 6.14 мл) и ZnCl2 (1.25 г) в ТГФ (15 мл)) добавляют к раствору CuCN (0.82 г) и LiCl (0.78 г) в ТГФ (10 мл) при -10°С. В другой трехгорлой колбе, в атмосфере аргона, добавляют NaH (80% в масле, 0.097 г) к раствору 2-(4-бром-этинил-2-оксо-1-пирролидинил)бутанамида (1 г, 0.0036 моль.) в ТГФ (20 мл) при -10°С, а затем ZnCl2 (0.50 г). Указанный раствор амида затем добавляют по каплям в органокупрат, охлажденный при -78°С. Реакционную смесь перемешивают в течение 3 час при указанной температуре и дают нагреться до комнатной температуры в течение ночи. После гидролиза насыщенным водным раствором NH4Cl, водную слой экстрагируют СН2Cl2, сушат над MgSO4, фильтруют и концентрируют в вакууме, что дает сырой алкин, который очищают с помощью хроматографии на хиральной фазе (EtOH/гексан), что дает 2-(4-пропин-1-ил-2-оксо-1-пирролидинил)бутанамид 280.
6.5. Гидрирование олефиновых пирролидонов
Синтез 1/1/ 1/1 смеси 4 диастериозомеров 2-[4-(2,2-дифторэтил)-2-оксо-1-пирролидинил]бутанамида 157 представлен следующим образом:
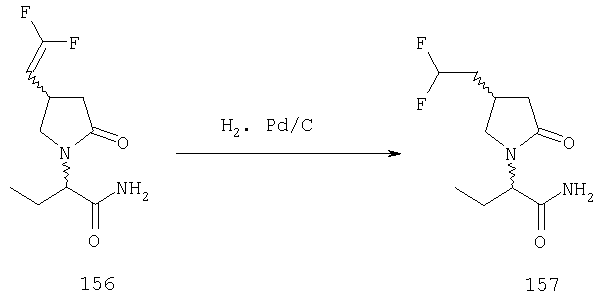
В автоклаве объемом 0.25 л, в инертной атмосфере, растворяют 1 г (0.0043 ммоль) 156 и Pd на угле (10% об/об, 0.2 г) в EtOH (50 мл) и смесь гидрируют в аппарате Парра. После 20 час, смесь дегазируют, фильтруют на Целит/Норитовой подушке и фильтрат концентрируют в вакууме, что дает сырой фторалкан, который перекристаллизовывают из PhMe, что дает 1/1/ 1/1 смесь 4 диастериозомеров 2-[4-(2,2-дифторэтил)-2-оксо-1-пирролидинил]бутанамидов в виде белого твердого вещества (0,75 г).
6.6. Синтез 2-[4-(5-метил-1,3-оксазол-2-ил)-2-оксо-1-пирролидинил]бутанамида 62 и 63
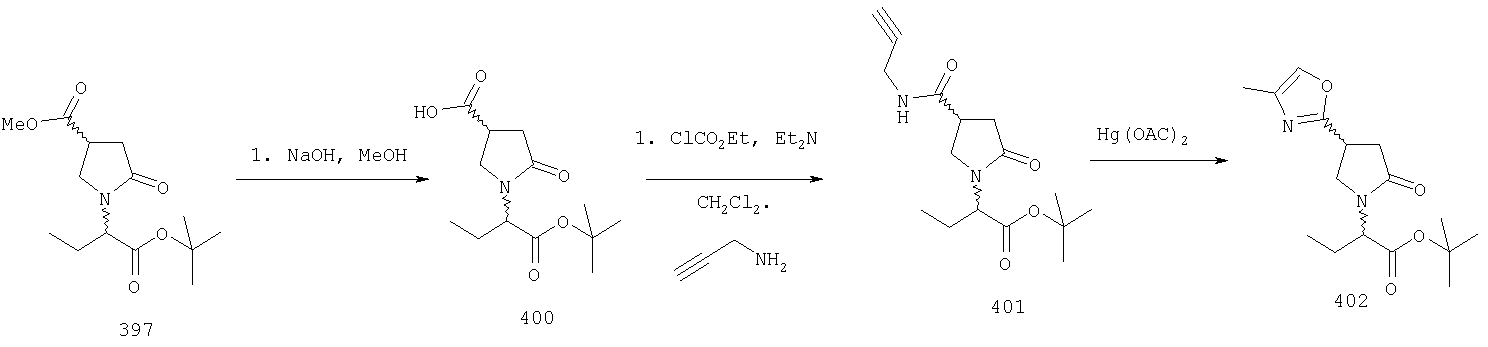
Стадия 1: Гидролиз эфира
В трехгорлой колбе, в атмосфере аргона, добавляют NaOH 1N (39 мл) к раствору метил 1-[1-(трет-бутоксикарбонил)пропил]-5-оксо-3-пирролидинкарбоксилата 397 в виде 1/1/ 1/1 смеси 4 стереоизомеров (10 г, 0.035 моль) в СН3ОН (100 мл) при 20°С. Раствор перемешивают в течение 0.5 час, упаривают досуха и подкисляют до рН 1 с помощью HCl 1N. Водный слой экстрагируют AcOEt, сушат над MgSO4, фильтруют и концентрируют в вакууме, что дает сырую кислоту 400 (8.45 г) в виде белого твердого вещества, которое используют без дальнейшей очистки на следующей стадии. 1Н ЯМР (250 МГц, (CD3)2SO): 0.80 (т, 3Н), 1.44 (с, 9Н), 1.55-1.60 (м, 1Н), 1.70-1.95 (м, 1Н), 2.40-2.55 (м, 2Н частично перекрывается с растворителем), 3.10-3.55 (м, 1Н частично перекрывается с растворителем), 4.45 (дд, 1Н).
Стадия 2: Синтез амида 401
В трехгорлой колбе, в атмосфере аргона, добавляют CICOOEt (0.50 мл, 0.005 моль) к раствору кислоты 400 (0.678 г, 0.0025 моль) в CH2Cl2 (10 мл) и триэтиламину (0.77 мл), охлажденному при -20°С. Реакционную смесь перемешивают в течение 1.5 час при -10°С, затем добавляют пропаргиламин (0.36 мл) к раствору, во то же время поддерживая температуру ниже 0°С. Суспензию перемешивают в течение 1 час при 0°С, нагревают до комнатной температуры, фильтруют и фильтрат упаривают под вакуумом. Сырой амид очищают с помощью хроматографической колонки на силикагеле (СН2Cl2/СН3ОН 98/02 (об/об)), что дает 0.8 г пропаргиламида 401 в виде 1/1/ 1/1 смеси четырех диастериозомеров. 1Н ЯМР (250 МГц, (CD3)2SO): 0.80 (т, 3Н), 1.44 (с, 9Н), 1.55-1.65 (м, 1Н), 1.70-1.95 (м, 1Н), 2.40-2.55 (м, 4Н частично перекрывается с растворителем), 3.0-3.70 (м, 3Н частично перекрывается с растворителем), 3.70-3.90 (м, 2Н), 4.45 (м, 1Н), 8.45 (м, 1Н).
Стадия 3: Синтез оксазола 402
В трехгорлой колбе, в атмосфере аргона раствор амида 402 (0.77 г, 0.0025 моль) в АсОН (40 мл) и Hg(ОАс)2 (0.048 г, 0.00015 моль) нагревают с обратным холодильником в течение 1 часа, реакционную массу охлаждают до комнатной температуры, концентрируют под вакуумом и гидролизуют насыщенным раствором Na2СО3. Водный слой экстрагируют СН2Cl2 и органическую фазу промывают рассолом, сушат над MgSO4, фильтруют и концентрируют в вакууме, что дает сырое соединение, которое очищают хроматографией на силикагеле (Гексан/AcOEt: 50/50 (об/об)), что дает чистый оксазол 402 (0.15 г, 20%). GC/MS: 308 (М-K), который может быть превращен в 62 и 63 аммонолизом аналогично к 6.3.1.
6.7. Синтез тетразолов.
6.7.1. Синтез незамещенных тетразолов
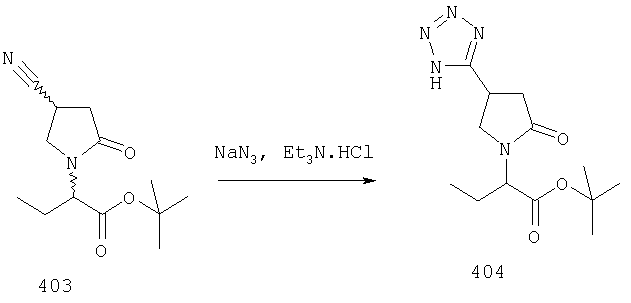
В трехгорлой колбе, в атмосфере аргона раствор рацемического нитрила 403 (2.66 г, 0.011 моль), NaN3 (4.8 г, 0.073 моль) и Et3N - гидрохлорида (10.12 г) нагревают при 110°С в ДМФА (60 мл) в течение 2 час, затем охлаждают до комнатной температуры и упаривают под вакуумом. Сырой продукт очищают хроматографией на силикагеле (CH2Cl2 / СН3ОН / АсОН: 90/08/02 (об/об)), что дает рацемический эфир тетразола 404 (3.42 г, 0.010 моль) в виде 1/1/ 1/1 смеси диастериозомеров. LC/MS: 295 (МН+).
6.7.2. Алкилирование тетразолов
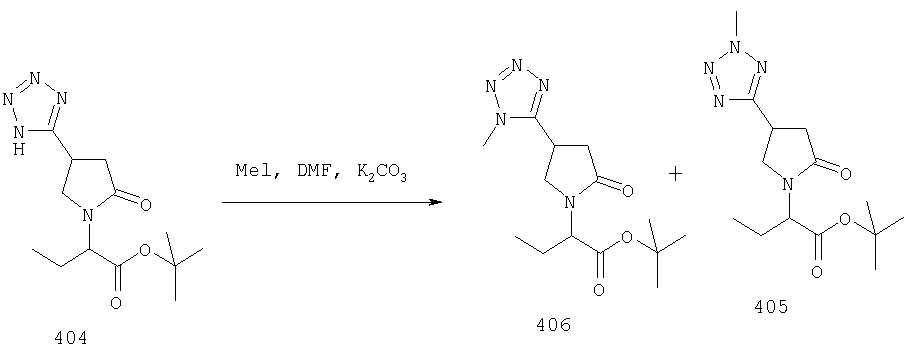
В трехгорлой колбе, в атмосфере аргона, перемешивают суспензию рацемического тетразола 404 (5.6 г, 0.019 моль), K2СО3 (2.88 г.) и Mel (1.3 мл) в ДМФА (60 мл) при комнатной температуре в течение 29 час и упаривают под вакуумом. Сырую смесь очищают хроматографией на силикагеле (МТВЕ/Гексан: 50/50 (об/об)), что дает два региоизомера тетразола 405 (1.98 г, 34%) и 406 (1.03 г, 17%) в виде масла. LC/MS: 309 (МН+).
6.8. Синтез тиазолов.
6.8.1. Синтез тиоамидов.
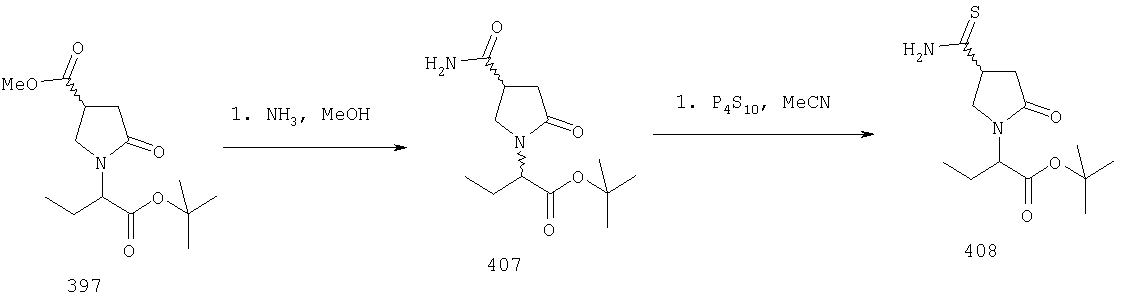
6.8.1.1. Аммонолиз 397
В трехгорлой колбе, объемом 0,5 л, оснащенной парциальным конденсатором горячего орошения, магнитной мешалкой и дополнительно трубкой для подвода газа в раствор, растворяют 10 г (0.035 ммолей) 397 в 100 мл метанола. Затем пропускают газообразный аммиак через раствор и насыщенный раствор выдерживают при комнатной температуре в течение 1 дня, в то время как аммиак постепенно улетучивается. После завершения реакции, раствор концентрируют в вакууме, что дает сырой амид 407 (9.6 г, 100%). 1H ЯМР (250 МГц, (CD3)2SO): 0.85 (т, 3Н), 1.44 (с, 9Н), 1.55-1.60 (м, 1Н), 1.70-1.95 (м, 1Н), 2.40-2.60 (м, 2Н частично перекрывается с растворителем), 3.00-3.70 (м, 1Н частично перекрывается с растворителем), 4.35-4.45 (м, 1Н), 6.95 (с (широкий), 1Н), 7.40 (с (широкий), 1Н).
6.8.1.2. Синтез тиоамида 408
В трехгорлой колбе, в атмосфере аргона, раствор сырого амида 407 (6 г, 0.022 моль), (4.93 г, 0.011 моль) и NaHCO3 (3.73 г) в MeCN (100 мл) перемешивают при 5°С в течение 6 час. Реакционную смесь фильтруют, концентрируют в вакууме и сырой тиоамид очищают хроматографией на силикагеле (AcOEt / гексан: 50/50 (об/об)), что дает после перекристаллизации из AcOEt тиоамид 408 (3.7 г, 60%). GC/MS: 286 (M+).
6.8.2. Синтез замещенных тиазолов
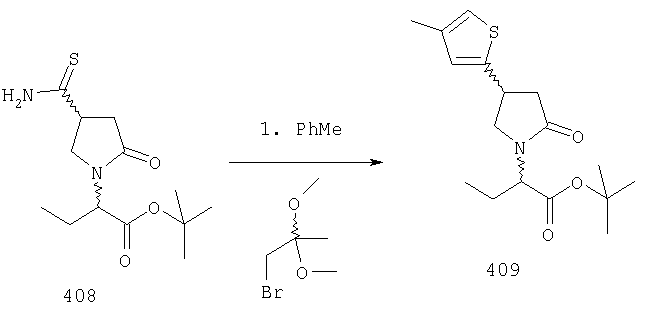
В трехгорлой колбе, в атмосфере аргона раствор тиоамида 408 в виде 1/1/ 1/1 смеси 4 диастериозомеров (в данном патенте, 1.5 г, 0.005 моль.), Al2О3 (12 г) и 1-бром-2-диметоксипроп-2-ена (0.85 мл) в PhMe (100 мл) кипятят с обратным холодильником в течение 3 час. Реакционную смесь охлаждают до комнатной температуры, фильтруют и концентрируют в вакууме, что дает сырой продукт тиазол 409 (0.5 г, 30%) который используют на следующих стадиях без дальнейшей очистки. GC/MS: 324 (М+')-
6.8.3. Синтез незамещенных тиазолов
Альтернативно незамещенные тиазолы могут быть получены при взаимодействии тиоамидов 408 с Al2O3 и бромацетальдегидом (полученным in situ из бром-2,2-диметоксиэтана в кислых условиях).
6.8.4. Синтез 1,2,4-гиадиазол-5-ил-производных
Альтернативно, 1,2,4-тиадиазол-5-ил-производные могут быть получены при взаимодействии тиоамида 408 последовательно с N,N-диметилацетамиддиметилацеталем, а затем с последующей циклизацией в присутствии пиридина.
6.9. Синтез 2-[2-оксо-4-(3-пирридинилкарбонил)-1-пирролидинил]бутановой кислоты 2.2-диметилэтилового эфира 410
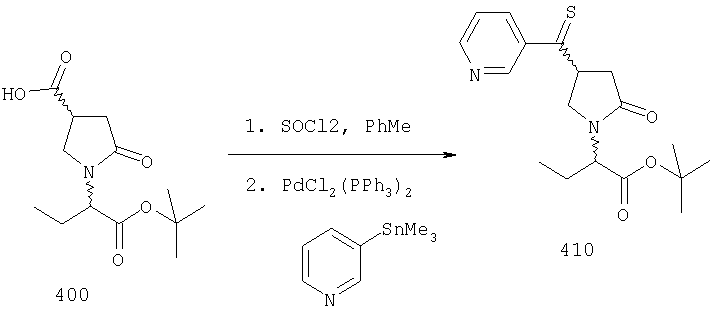
В трехгорлой колбе, в атмосфере аргона, добавляют SOCl2 (0.56 мл) к раствору кислоты 400 (1.90 г, 0.007 моль) в PhMe (20 мл) при комнатной температуре. Реакционную смесь кипятят с обратным холодильником 1.5 час, после чего она становится желтого цвета. После охлаждения до комнатной температуры, добавляют одну порцию PdCl2(PPh3)2 (0.25 г, 0.00035 моль) и 3-триметилстаннилпиридин (1.7 г, 0.007 моль), реакционную смесь кипятят с обратным холодильником в течение 0.5 час, охлаждают до комнатной температуры, захолаживают водой. Водный слой экстрагируют дихлорметаном и объединенные органический фазы промывают рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме (3.2 г). Сырой кетон очищают с помощью хроматографической колонки на силикагеле (СН2Cl2/СН3ОН 97/03 (об/об)), что дает 1.3 г кетона 410 в виде 1/1/ 1/1 смеси четырех диастериозомеров. LC/MS: 333 (МН+).
ПРИМЕР 7. Синтез 2-(4-замещенных-2-оксопирролидинил)-бутанамидов замещением активированного 2-(4-гидроксиметил-2-оксопирролидинил)-бутанамида
7.0. Синтез исходных стертов
7.0.1. Синтез эфироамида
7.0.1.а. Синтез метил 1-[(1S)-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинкарбоксилата 11/12.
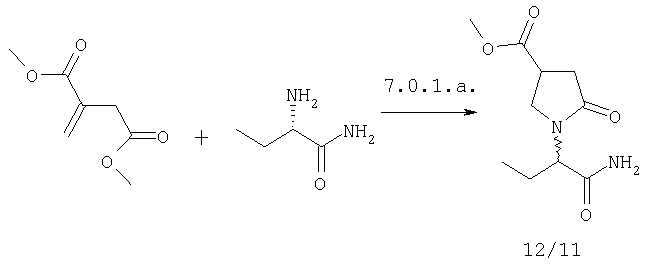
В трехгорлой колбе, объемом 10 л, оснащенной механической мешалкой и парциальным конденсатором горячего орошения, в инертной атмосфере, 1226 г (12 молей, 1 экв) (2S)-2-аминобутанамида и 1912 мл (2150 г, 13.2 молей, 1.1 экв) диметил итаконата растворяют в 6.13 л СН3ОН. Смесь выдерживают при кипении с обратным холодильником в течение 10 часов и медленно охлаждают до 20°С в течение более 4 часов. Затем ее фильтруют, осадок промывают СН3ОН и объединенные органические фазы концентрируют досуха, что дает 3.283 г сырого промежуточного продукта, 74%.
В трехгорлой колбе, объемом 20 л, оснащенной механической мешалкой, колонкой Рашинга и дистиллятором, в инертной атмосфере, сырой промежуточный продукт и 84.7 г (891 ммолей, 0.1 экв.) 2-гидроксипиридина растворяют в 11.6 л толуола. Смесь держат при кипении с обратным холодильником и образующийся метанол отгоняют в течение 8 часов, пока не соберут 480 мл. Температура в котле достигает 112°С. Смесь охлаждают и концентрируют досуха, что дает 2,187 г сырого амидоэфира в виде смеси диастереоизомеров в соотношении 57.5/42.5. 2 диастереоизомера разделяют с помощью Препаративной Жидкостной Хроматографии на хиральной фазе (Chiralpak AD 100х500 мм, EtOH/Н2О 99.9:0.1), элюаты концентрируют досуха, что дает 968 г сырого продукта 12 (первый элютируется) и 1,052 г сырого продукта 11 (второй элютируется). Сырой продукт 12 не является кристаллическим, его растворяют в 1.5 л EtOH и хранят как таковой, для дальнейшего применения.
Сырой продукт 11 перекристаллизовывают из 2 л EtOAc, что дает 676 г чистого 11.
Альтернативно, метил 1-[(1S)-2-амино-1-метил-2-оксоэтил]-5-оксо-3-пирролидинкарбоксилат, метил 1-[(15)-1-(аминокарбонил)бутил]-5-оксо-3-пирролидинкарбоксилат, метил 1-{(1S)-1-[(метиламино)карбонил]пропил}-5-оксо-3-пирролидинкарбоксилат, получают аналогичными путями.
7.0.2. Синтез спиртоамида
7.0.2.а. Синтез (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутанамида 6.
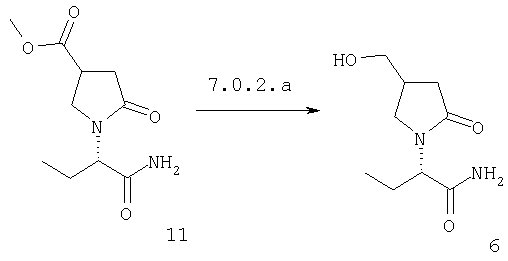
В трехгорлой колбе, объемом 2 л, оснащенной механической мешалкой и парциальным конденсатором горячего орошения, в инертной атмосфере раствор 133 г (583 ммолей, 1 экв) (2S)-2-(4-метоксикарбонил-2-оксо-1-пирролидинил)бутанамида 11 в 200 мл EtOH добавляют к 300 мл EtOH и смесь охлаждают до 0°С. Затем добавляют порциями 66.2 г (1.74 молей, 12 экв) твердого NaBH4, в течение более 1.5 час, все время поддерживая температуру между 2 и 4°С. После 2 часов, температуру поднимают до 12°С в течение 1 часа, и снова опускают до 2-4°С. Добавляют по каплям 240 мл насыщенного раствора NH4Cl в течение более 1 часа, а затем 120 мл ацетона, и смесь оставляют в течение ночи при комнатной температуре. Смесь фильтруют, осадок промывают 3×70 мл EtOH и объединенные органические фракции концентрируют досуха, что дает 148 г сырого продукта 6. Его суспендируют в 300 мл CH2Cl2 и перемешивают в течение 30 мин, фильтруют, промывают 2×100 мл СН2Cl2 и сушат, что дает 114 г чистого 6, 98%.
Альтернативно, (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]пропанамид, (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]пентанамид, (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]-N-метилбутанамид, получают по аналогичным путям.
7.1. Синтез непосредственным превращением спирта, используя PPh3
7.1.1. Синтез (2S)-2-[4-(иодометил)-2-оксо-1-пирролидинил]бутанамида 10
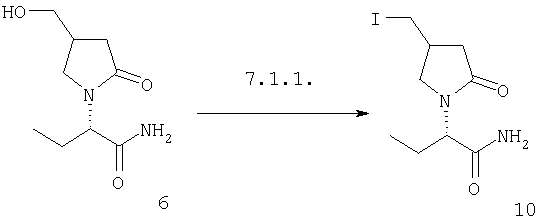
В трехгорлом сосуде, объемом 10 л, оснащенном механической мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, растворяют 400 г (2 молей, 1 экв) (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутанамид 6 в 3 л ацетонитрила. Добавляют 629 г (2.4 молей, 1.2 экв) трифенилфосфина, а затем 608 г (2.4 молей, 1.2 экв) иода в три порции в течение более 5 мин. Смесь нагревают до 60°С 30 мин и перемешивают при этой температуре в течение 5 часов. После охлаждения смесь концентрируют досуха, остаток суспендируют в растворе 750 г Na2S2О3 в 10 л воды и перемешивают при 50°С в течение 4 часов. Осадок отфильтровывают и промывают 3×1 л воды. Объединенные водные фазы обрабатывают 1 кг NaCl и экстрагируют 6×1 л СН2Cl2. Объединенные органические фазы сушат над MgSO4, фильтруют и концентрируют досуха, что дает 482 г сырого продукта 10. Его кристаллизуют из толуола. Несколько загрузок перекристаллизовывают вместе с этиловым эфиром из этилацетата, что дает 425 г чистого 10,68%.
Альтернативно, (2S)-2-[4-(иодометил)-2-оксо-1-пирролидинил]-N-метилбутанамид 146, (2S)-2-[4-(иодометил)-2-оксо-1-пирролидинил]пропанамид 110, (2S)-2-[4-(иодометил)-2-оксопирролидин-1-ил]пентанамид 105, (2S)-2-[4-(бромметил)-2-оксо-1-пирролидинил]бутанамид 8, (2S)-2-[4-(хлорметил)-2-оксо-1-пирролидинил]бутанамид 30 получают по аналогичным путям.
7.1.2. Синтез (2S)-2-[2-оксо-4-(феноксиметил)-1-пирролидинил]бутанамида 18
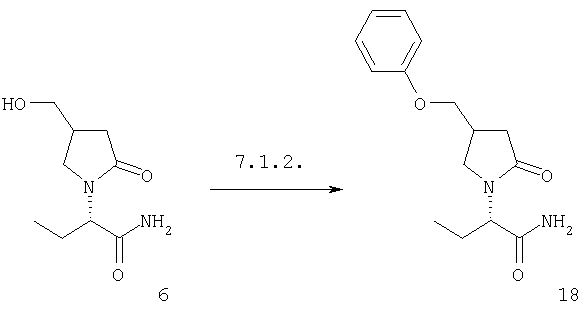
В трехгорлой колбе, объемом 50 мл, оснащенной магнитной мешалкой и капельной воронкой в инертной атмосфере, растворяют 1 г (5 ммолей, 1 экв) (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутанамид 6 в 20 мл ТГФ и охлаждают до 0°С. Последовательно добавляют 517 мг фенола, 0.87 мл (960 мг) диэтилазодикарбоксилата и 1.44 г трифенилфосфина (5.5 ммолей, 1.1 экв каждый) и смесь перемешивают в течение 2 часов. Смесь концентрируют досуха и очищают с помощью препаративной ЖХ (500 кг SiO2, CH2Cl2/EtOH, 97.5:2.5), что дает 1,1 г чистого 18, 80%, кристаллизуют из этилацетата.
7.2. Синтез замещением мезилата
7.2.1. Синтез {1-[(18)-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинил} метил метансульфоната 37
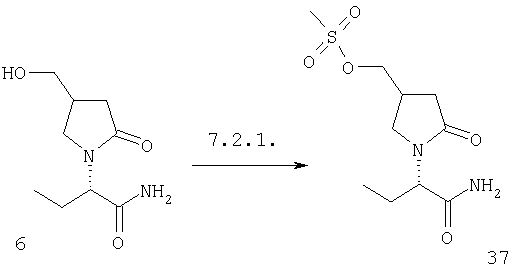
В трехгорлой колбе, объемом 4 л, оснащенной механической мешалкой, капельной воронкой и парциальным конденсатором горячего орошения в инертной атмосфере, растворяют 114 г (569 ммолей, 1 экв) (2S)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутанамид 6 в 2 л СН2Cl2 и охлаждают до 0°С. Добавляют одной порцией 158.5 мл (115 г, 2 экв) сухого триэтиламина, а затем добавляют по каплям раствор 66.3 мл (96.2 г, 1.5 экв) метан сульфонилхлорида в 190 мл СН2Cl2 в течение более 1 часа, все это время поддерживая температуру ниже 4°С. После 4 часов, добавляют 7.5 мл метансульфонил хлорида и 15 мл триэтиламина и смесь выдерживают в течение ночи в холодильнике. Смесь фильтруют, остаток промывают CH2Cl2 и объединенные органические фазы концентрируют досуха, что дает 216 г сырого продукта 37. Его очищают с помощью препаративной жидкостной хроматографии в нескольких объемах (1 кг SiO2, СН2Cl2/EtOH, 100:0 -> 96:4), что дает 109 г чистого 37, 69%. Альтернативно, {1-[(1S)-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинил}метил 4-метилбензолсульфонат 31, получают аналогичным путем.
7.2.2. Синтез (2S)-2-[4-(азидометил)-2-оксо-1-пирролидинил]бутанамида 32
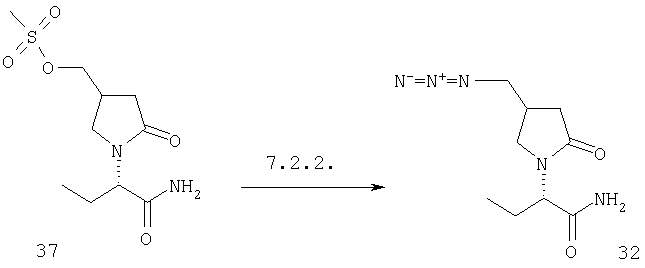
В трехгорлой колбе, объемом 3 л, оснащенной механической мешалкой и парциальным конденсатором горячего орошения, в инертной атмосфере, растворяют 89.7 г (322 ммолей, 1 экв) {1-[(1S)-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинил}метил метансульфоната 37 в 300 мл ацетонитрила. Добавляют одной порцией 27.3 г (419 ммолей, 1.3 экв) азида натрия в 150 мл ацетонитрила. Смесь поддерживают при кипении в течение 20 мин и перемешивают в течение ночи. Добавляют 3.1 г (48 ммолей, 0.2 экв) азида натрия и кипение продолжают еще 44 часа. После охлаждения до 10°С, смесь фильтруют, осадок промывают 3×50 мл ацетонитрила и объединенные органические фракции концентрируют досуха, что дает 77.3 г сырого продукта 32. Его кристаллизуют из 150 мл этилацетата при 10°С, что дает 60 г чистого 32, 82%.
Альтернативно, (2S)-2-[4-(фторметил)-2-оксо-1-пирролидинил]бутанамид 44, (2S)-2-[2-оксо-4-(1N-тетразол-1-илметил)-1-пирролидинил]бутанамид 39, (2S)-2-[2-оксо-4-(1N-тетразол-1-илметил)-1-пирролидинил]бутанамид 40, (2S)-2-[2-оксо-4-(1N-1,2,4-триазол-1-илметил)-1-пирролидинил]бутанамид 55, 2-[2-оксо-4-(1N-1,2,3-триазол-1-илметил)-1-пирролидинил]бутанамид 56, (2S)-2-{4-[(изопропилсульфанил)метил]-2-оксо-1-пирролидинил}бутанамид 24, (2S)-2-[2-оксо-4-(1-пирролидинилметил)-1-пирролидинил]бутанамид 15, (2S)-2-[2-оксо-4-(4-тиоморфолинилметил)-1-пирролидинил]бутанамид 17, получают аналогичными путями, из активированных производных спирта, таких как мезилаты, тозилаты или галоиды.
7.3. Другие синтезы
7.3.1. Синтез {1-[(1S)-1-(аминокарбонил)пропил]-5-оксо-3-пирролидинил}метил нитрата 38
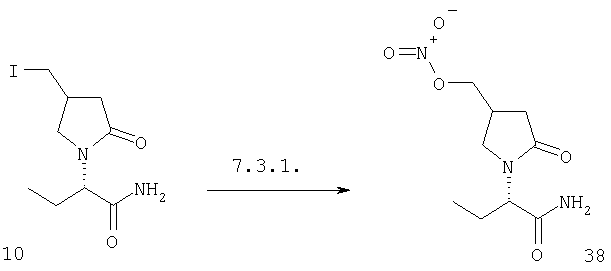
В трехгорлой колбе, объемом 500 мл, оснащенной механической мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, растворяют 8.10 г (26 ммолей, 1 экв) (2S)-2-[4-(иодометил)-2-оксо-1-пирролидинил]бутанамида 10 в 250 мл ацетонитрила. Добавляют 4.86 г (28.6 ммолей, 1.1 экв) нитрата серебра и смесь доводят до кипения. Через два часа, добавляют 440 мг (2.8 ммолей, 0.1 экв) нитрата серебра и кипение продолжают еще полных 4 часа. После охлаждения смесь концентрируют досуха и очищают с помощью препаративной жидкостной хроматографии (200 г SiO2, СН2Cl2/СН3ОН/NH4OH, 96:5.4:0.6), что дает 5.7 г сырого продукта 38. Его кристаллизуют из 50 мл этилацетата, что дает 4.13 г чистого 38, 65%.
7.3.2. Синтез 2-{4-[(бензилокси)метил]-2-оксо-1-пирролидинил}бутанамида 153/154
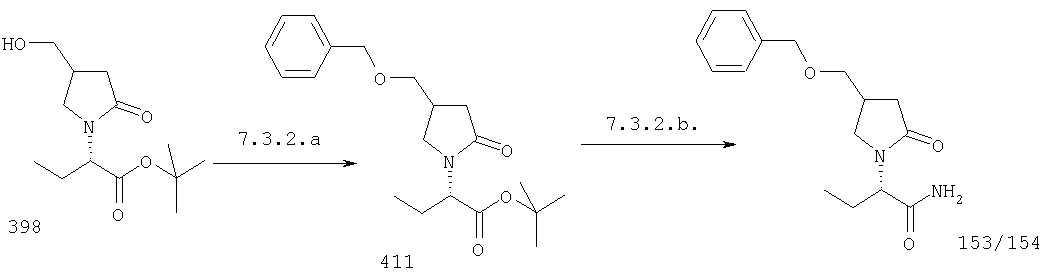
7.3.2.а. Синтез трет-бутил (2S)-2-{4-[(бензилокси)метил]-2-оксо-1-11ирролидинил]бутаноата
В трехгорлой колбе, объемом 100 мл, оснащенной магнитной мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, суспендируют 1.1 г (60%, 27.5 ммолей, 1.1 экв) гидрида натрия в 60 мл ДМФА и смесь охлаждают до 0°С. Осторожно добавляют 6.37 г (24.8 ммолей, 1 экв) трет-бутил (25)-2-[4-(гидроксиметил)-2-оксо-1-пирролидинил]бутаноата 398 в 10 мл ДМФА. Через 10 мин, добавляют 3.3 мл (4.75 г, 27.8 ммолей, 1 экв) бензилбромида в 10 мл ДМФА и перемешивание продолжают в течение 30 мин. при 0°С, а затем еще 3 часа при комнатной температуре. Смесь концентрируют досуха, остаток суспендируют в рассоле/СН2Cl2, декандируют и экстрагируют СН2Cl2. Объединенные органические фазы сушат над MgSO4, концентрируют досуха и остаток очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, гексан/МТВЕ, 40:60 -> 0:100), что дает 3.2 г смеси трет-Bu и бензилового эфира в две фракции, 37% общий выход. Его используют в качестве таковой для следующей стадии 7.3.1.b. 1H ЯМР (250 МГц, (CDCl3): 0.85 (т, 3Н), 1.44 (с, 9Н), 1.55-1.95 (м, 2Н), 2.10 (дд, 1Н), 2.45 (да., 1Н), 2.55-2.70 (м, 1Н), 3.45-3.55 (м, 1Н), 4.40 (дд., 1Н), 4.55 (с, 2Н), 7.20-7.40 (м, 5Н).
7.3.2.b. Синтез 2-{4-{(бензилокси)метил]-2-оксо-1-пирролидинил}бутанамид 153
В трехгорлой колбе, объемом 50 мл, оснащенной магнитной мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, растворяют 1.75 г бензилового эфира обогащенной фракции в 20 мл СН3ОН.
Затем пропускают через раствор газообразный аммиак и насыщенный раствор, выдерживают при комнатной температуре в течение 24 часов, во время которых со временем улетучивается аммиак. После завершения реакции, раствор концентрируют досуха и очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, СН2Cl2/СН3ОН, 98:2 -> 90:10), что дает два диастереоизомера.
В трехгорлой колбе, объемом 25 мл, оснащенной магнитной мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, растворяют 1.24 г трет-Bu эфира обогащенной фракции в 16 мл смеси 1:1 СН2Cl2/ТРА и выдерживают при 0-5°С в течение 24 часов. Раствор концентрируют досуха и остаток растворяют в 10 мл СН2Cl2. Добавляют 1.2 мл (2.2 экв по теории) триэтиламина и смесь охлаждают до -20°С. Добавляют по каплям 780 мкл этилхлорформиата и смесь оставляют медленно нагреваться до -10°С в течение более 1.5 час. Затем пропускают газообразный аммиак через раствор в течение 0.5 час и смесь выдерживают в течение ночи при комнатной температуре. Ее фильтруют, осадок промывают CH2Cl2, объединенные органические фракции концентрируют досуха и очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, СН2Cl2/СН3ОН, 98:2 -> 90:10), что дает два диастереомера. Первый и второй элюируемые диастереоизомеры из двух потоков объединяют и кристаллизуют из толуола, что дает соответственно 305 мг чистого 153 и 480 мг чистого 154, 11% общий выход.
7.3.3. Синтез (2S)-2-{4.[(5-метил-1N-1,2,3-триазол, 1-ил)метил]-2-оксо-1-пирролидинил]бутанамид 52
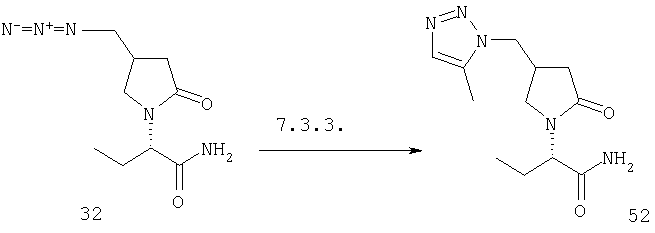
В трехгорлой колбе, объемом 50 мл, оснащенной магнитной мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, суспендируют 1 г (4.44 ммолей, 1 экв) (2S)-2-(4-(азидометил)-2-оксо-1-пирролидинил]бутанамида 32 в 20 мл толуола. Добавляют 1.55 г (4.88 ммолей, 1.1 экв) 1-(трифенилфосфоранилиден) ацетона и смесь нагревают до 80°С в течение 24 часов. После охлаждения, смесь концентрируют досуха и очищают с помощью препаративной жидкостной хроматографии (1 кг SiO2, СН2Cl2/СН3ОН/NH4OH, 94.5:5:0.5). Ее суспендируют в 15 мл воды и лиофилизуют, что дает 240 мг чистого 52 в виде прозрачного масла, 42%.
7.3.4. Синтез (2S)-2-[4-(изотиоцианатометил)-2-оксо-1-пирролидинил]бутанамида 49
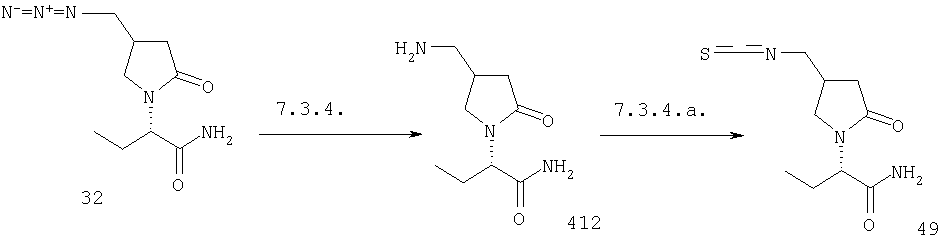
В автоклаве, объемом 500 мл, в инертной атмосфере, суспендируют 900 мг 10% Pd, адсорбированного на угле в 100 мл этанола. Добавляют раствор 8.7 г (38 ммолей) (2S)-2-[4-(азидометил)-2-оксо-1-пирролидинил]бутанамида 32 в 150 мл этанола и смесь гидрируют в аппарате Парра при максимальном давление Н2 в 30 пси в течение 2 часов. Смесь дегазируют, фильтруют на Целит/Норитовой подушке, остаток промывают 2×100 мл EtOH и объединенные фильтраты концентрируют досуха, что дает 7.93 г сырого продукта 412, 100% выходом, который используют как таковой на следующей стадии. GC/MS: 199 (М+).
7.3.4.а. Синтез (2S)-2-{4-(изотиоцианатометил)-2-оксо-1-пирролидинил]бутанамида 49
В трехгорлой колбе, объемом 100 мл, оснащенной магнитной мешалкой и парциальным конденсатором горячего орошения в инертной атмосфере, растворяют 4.5 г (22.7 ммолей, 1 экв) тиокарбонилимидазола в 25 мл ДМФА и смесь охлаждают до 0°С. Добавляют по каплям 4.53 г (22.7 ммолей, 1 экв) (2S)-2-[4-(аминометил)-2-оксо-1-пирролидинил]бутанамида 412 в 25 мл ДМФА в течение более 30 мин, смесь перемешивают 3 часа при комнатной температуре и оставляют в течение ночи. Смесь концентрируют досуха, остаток растворяют в 20 мл толуола, концентрируют снова досуха и остаток очищают с помощью препаративной жидкостной хроматографии (350 г SiO2 СН2Cl2/СН3ОН/NH4OH, 93.4:6:0.6), что дает 3.1 г сырого продукта 49. Его растирают в 20 мл этилового эфира, фильтруют и остаток (1.9 г) кристаллизуют из 15 мл ацетонитрила, что дает 1.2 г чистого 49 (22%).
Соединения формулы I представленные в следующей Таблице, могут быть получены аналогично или также как здесь описано.
В таблице, информация относительно стереохимии содержится в двух колонках под названием «данные по конфигурации». Вторая колонка показывает, является ли соединение, обладающим стереогенным центром (ACHIRAL), чистым энантиомером (PURE), рацематом (RAC) или представляет собой смесь двух или более стереоизомеров, возможно не в равных пропорциях (MIXT). Первая колонка, содержит стереохимический асигмент для каждого распознаваемого центра, по IUPAC рекомендациям, используемому в следующей колонке. Одно число показывет существование обоих конфигураций у этого центра. Число, а затем значки «R» или «S» указывают на известную абсолютную конфигурацию у этого центра. Число, за которым следует «§» указывает на существование одной, но неизвестной абсолютной конфигурации у этого центра. Буквы (А, В, С, Д) впереди являются указателем различных энантиомеров или рацематов одной структуры.
В таблице точки плавления в большинстве случаев определены с помощью DSC кривой. Когда визуальная (физионометрическая) точка плавления приведена, ее значение дано в круглых скобках.
В таблице, числа в колонке «синтез» относятся к синтезу в соответствии с которым получают большинство важных соединений. Небольшие варианты смогут потребоваться для получения аналогичных соединений. Такие модификации известны специалисту в этой области органического синтеза.
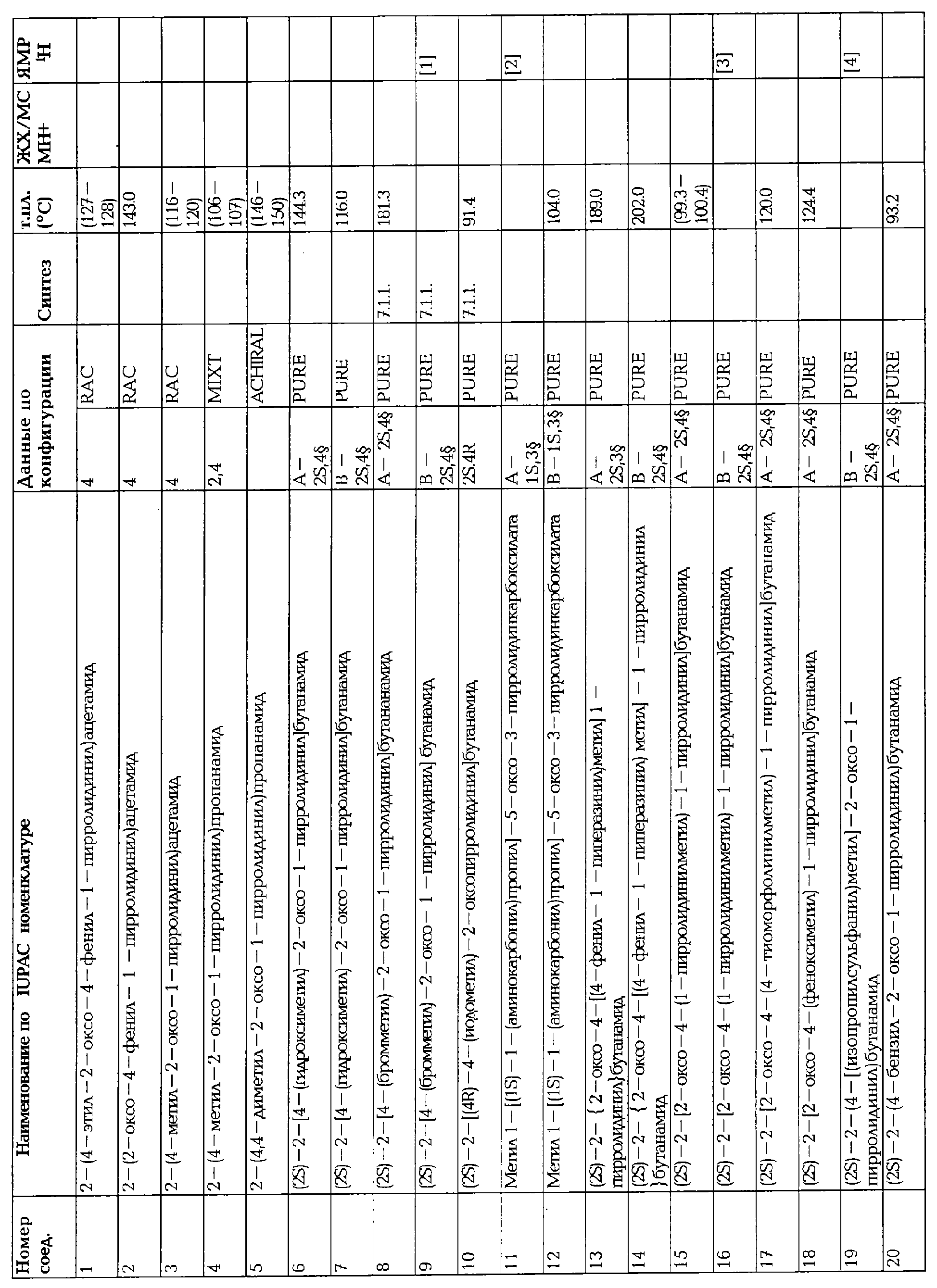
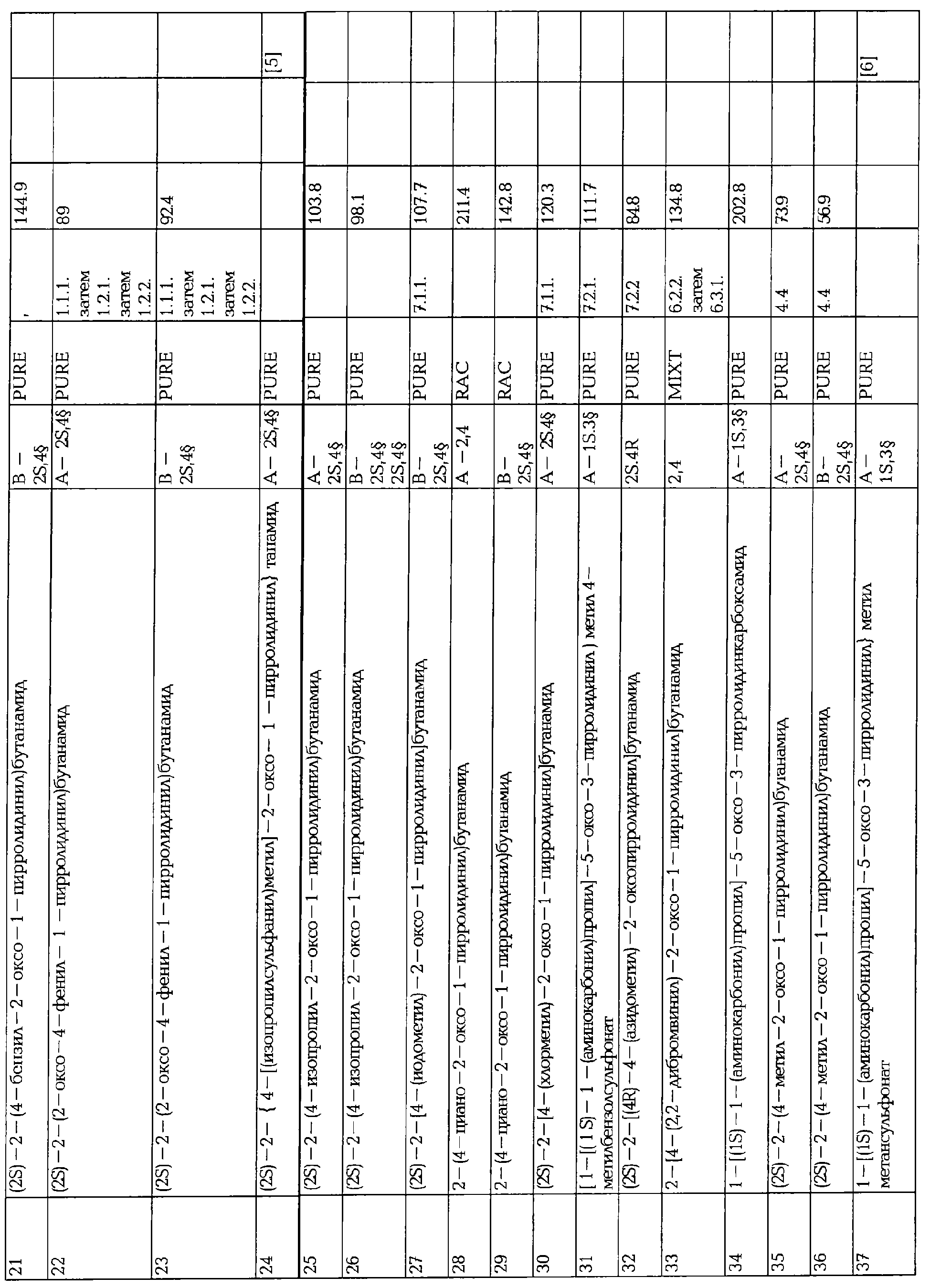
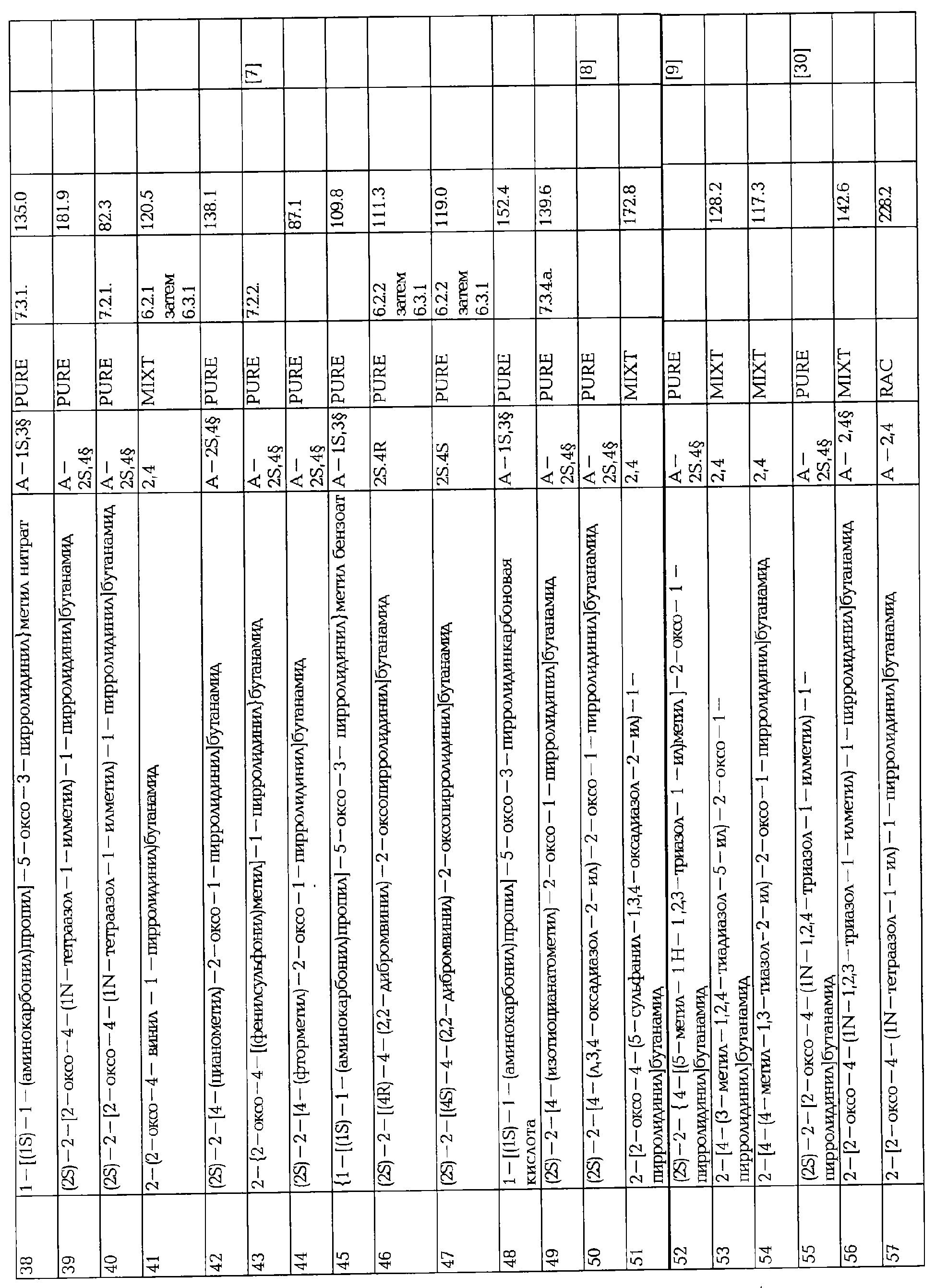
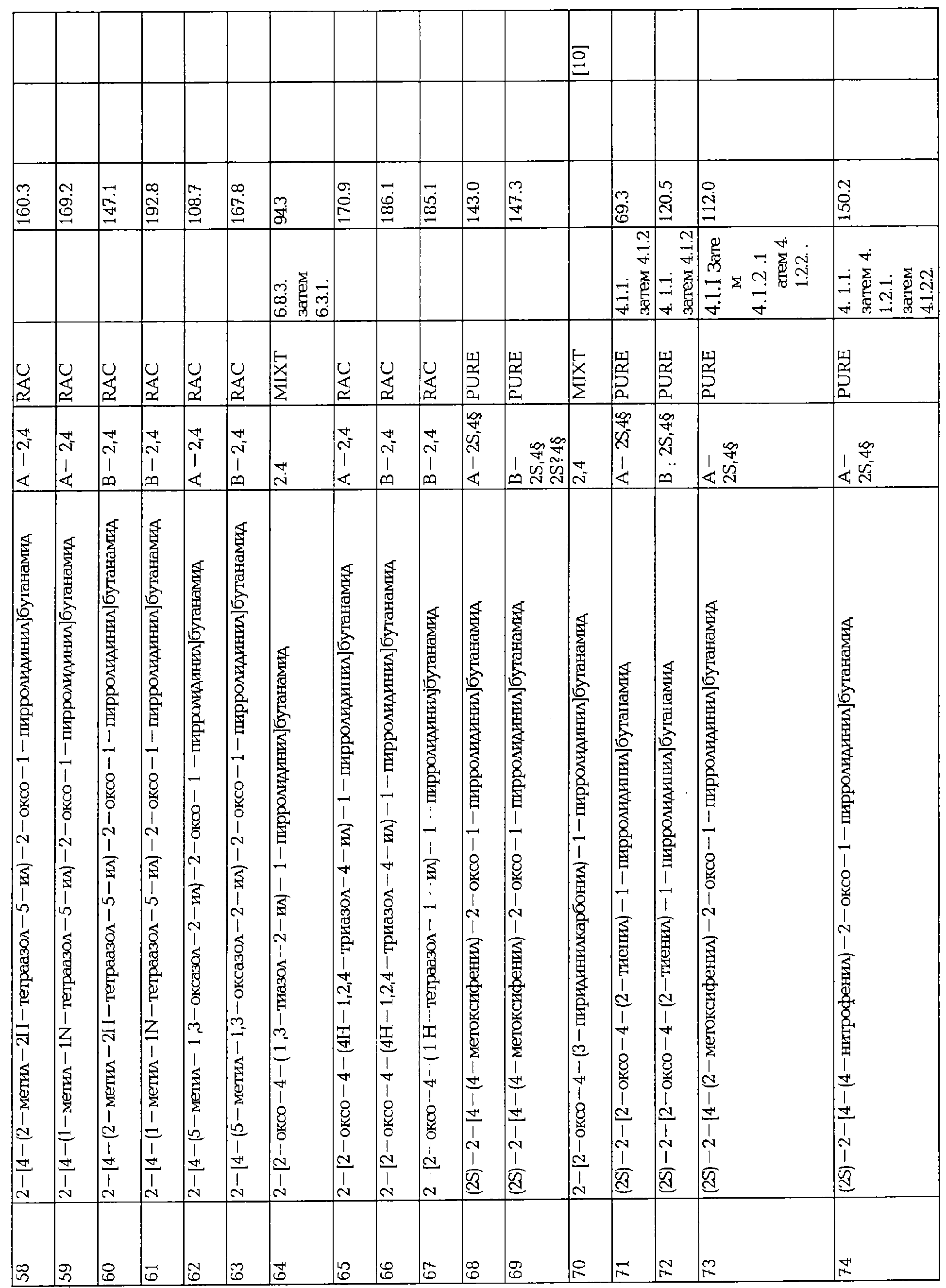
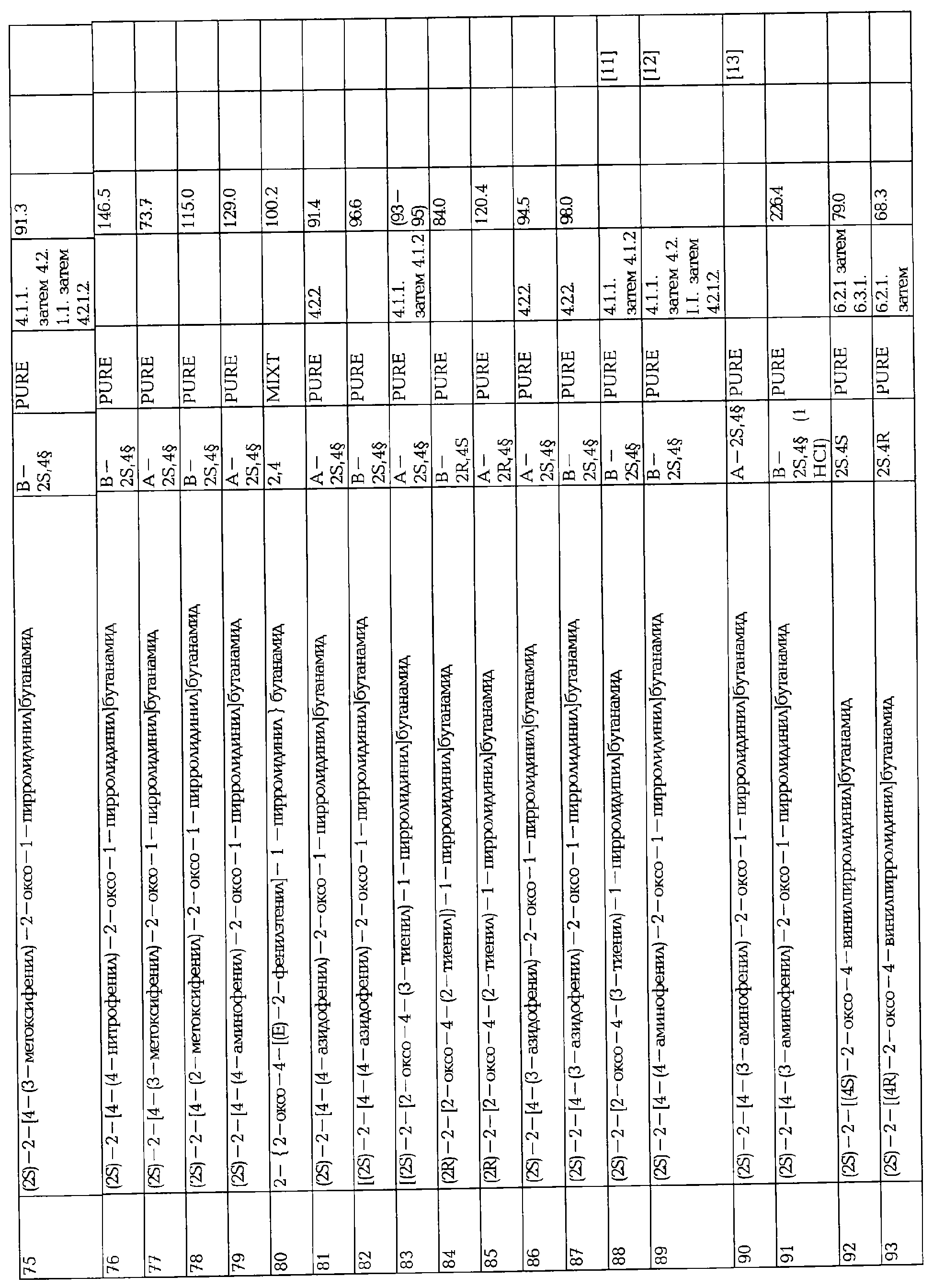
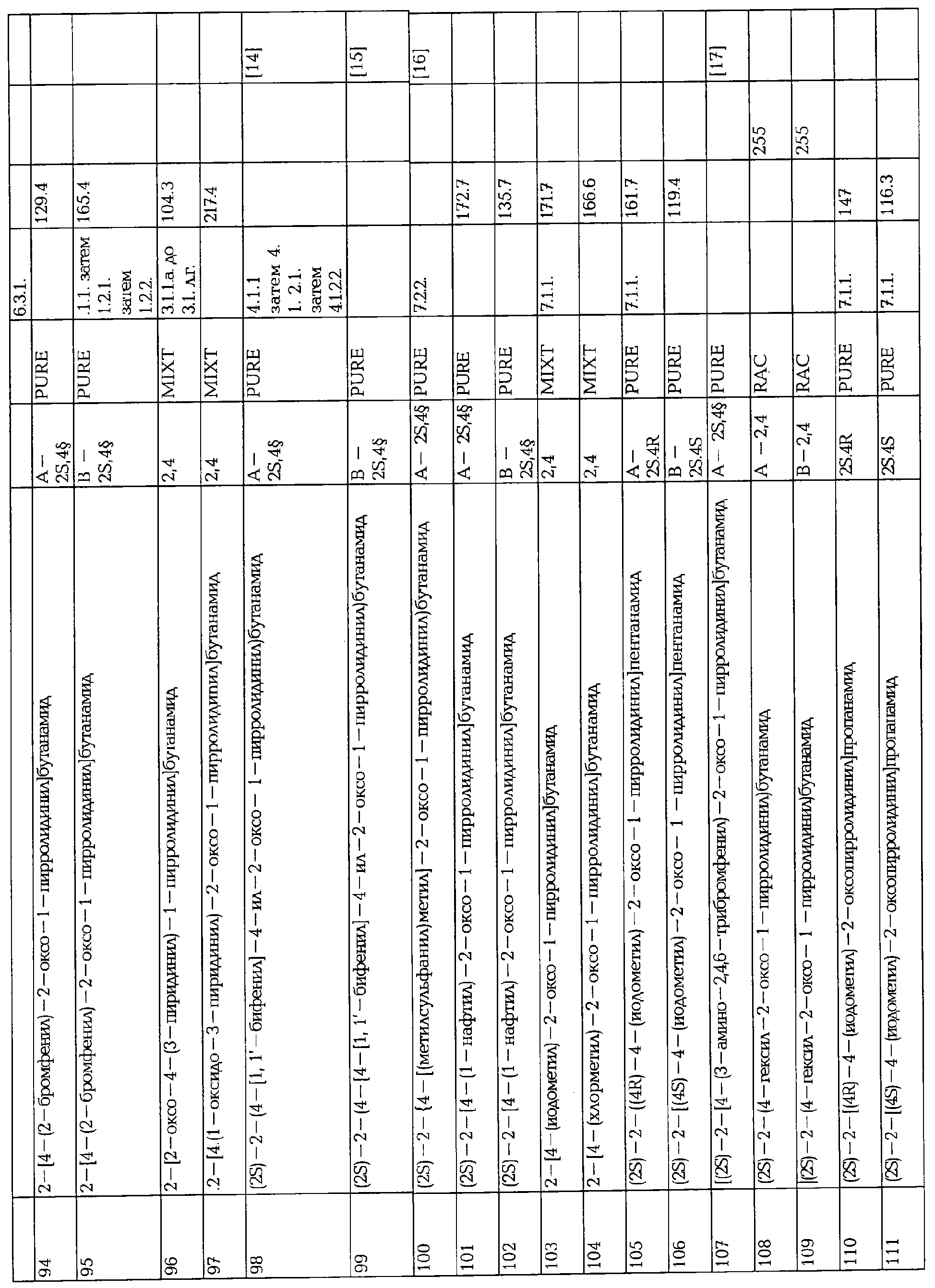
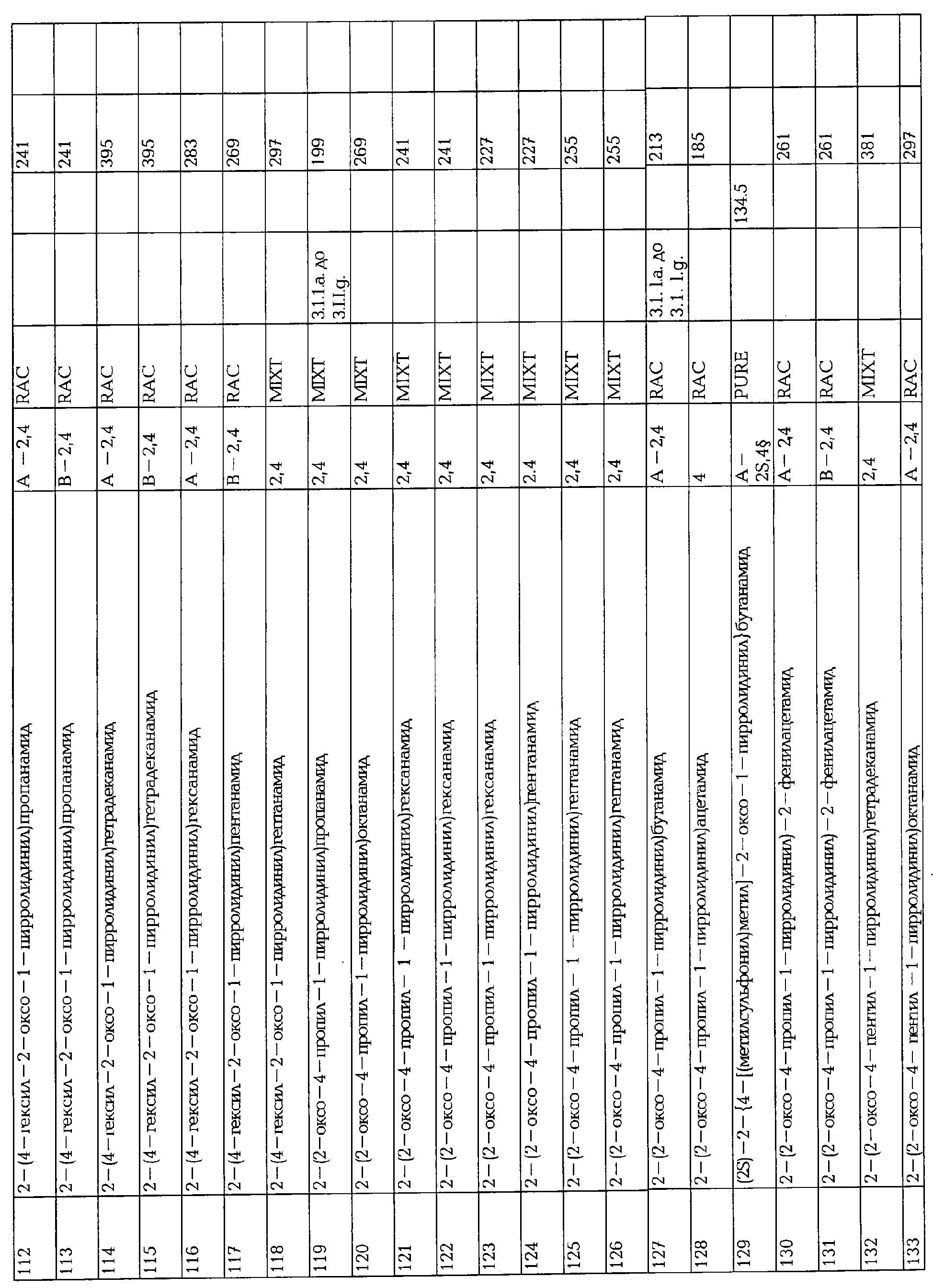
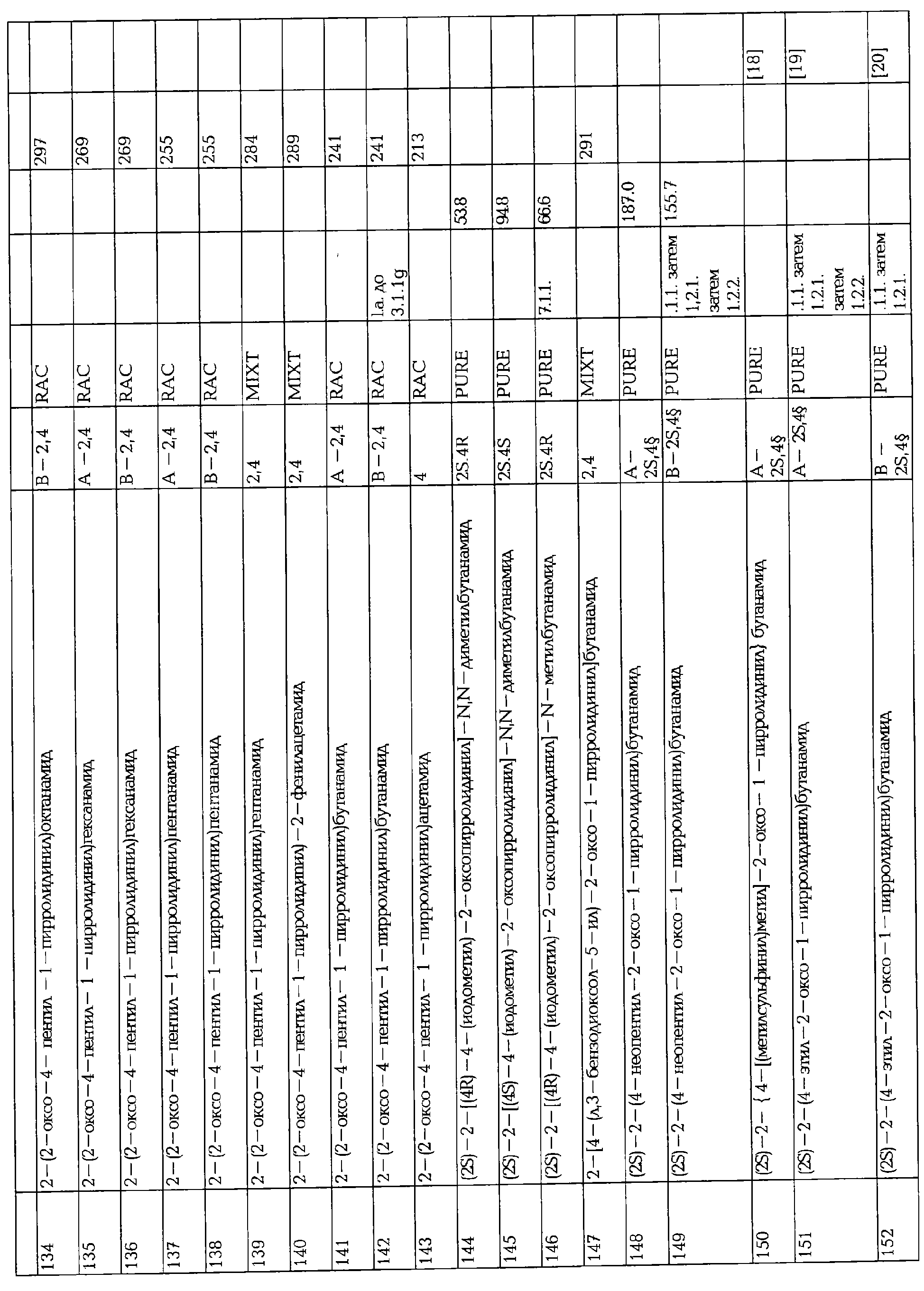
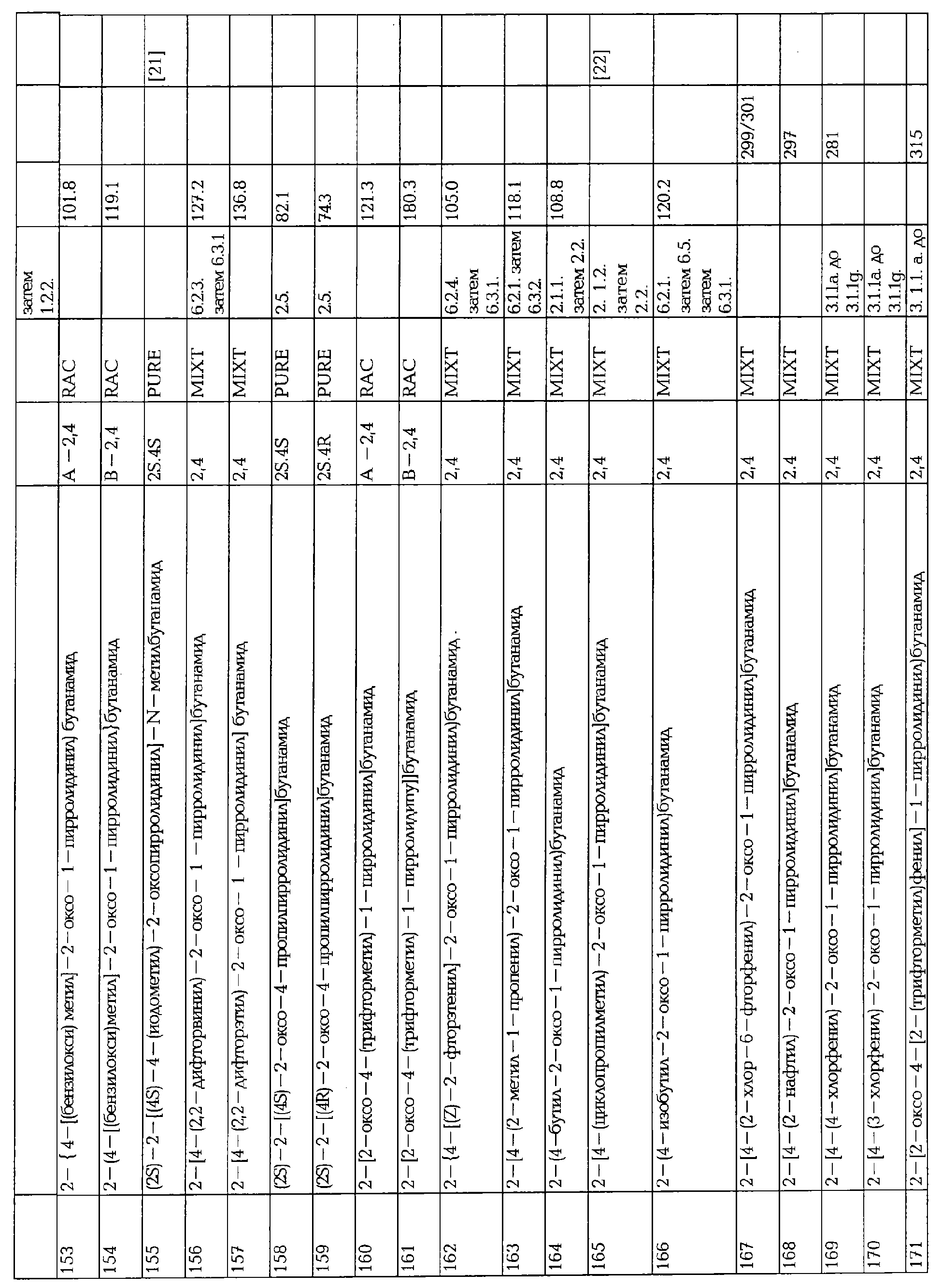
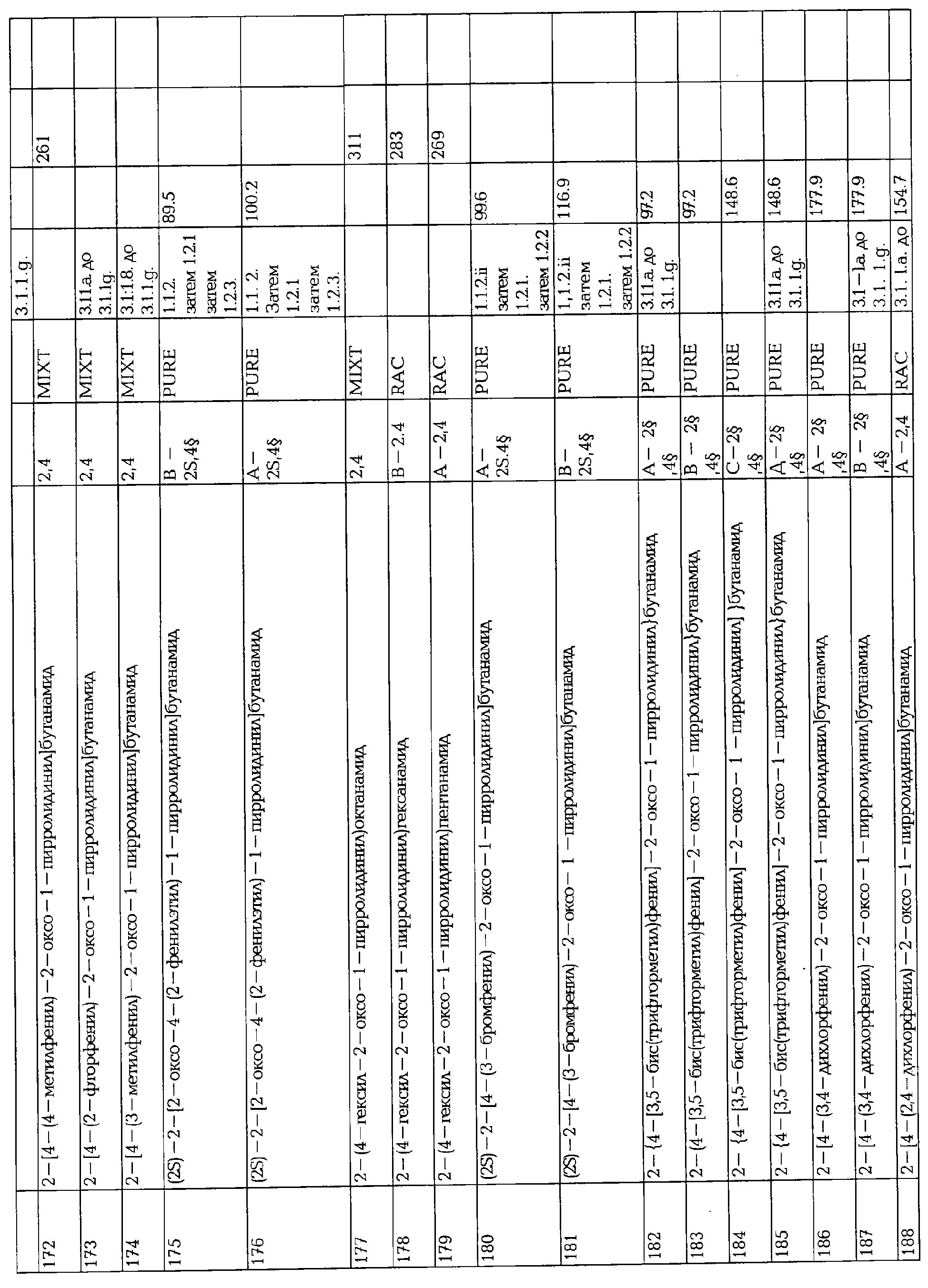
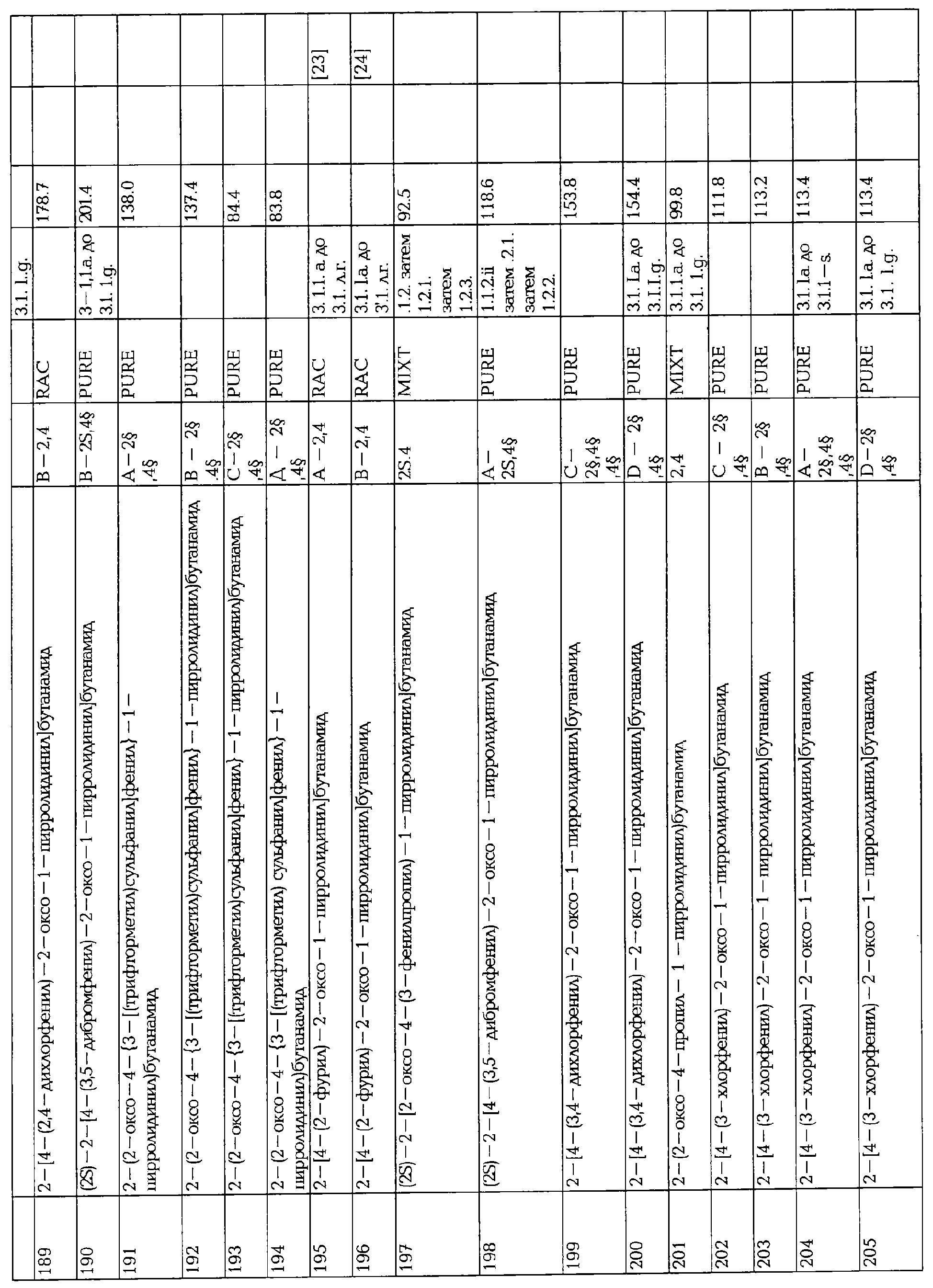
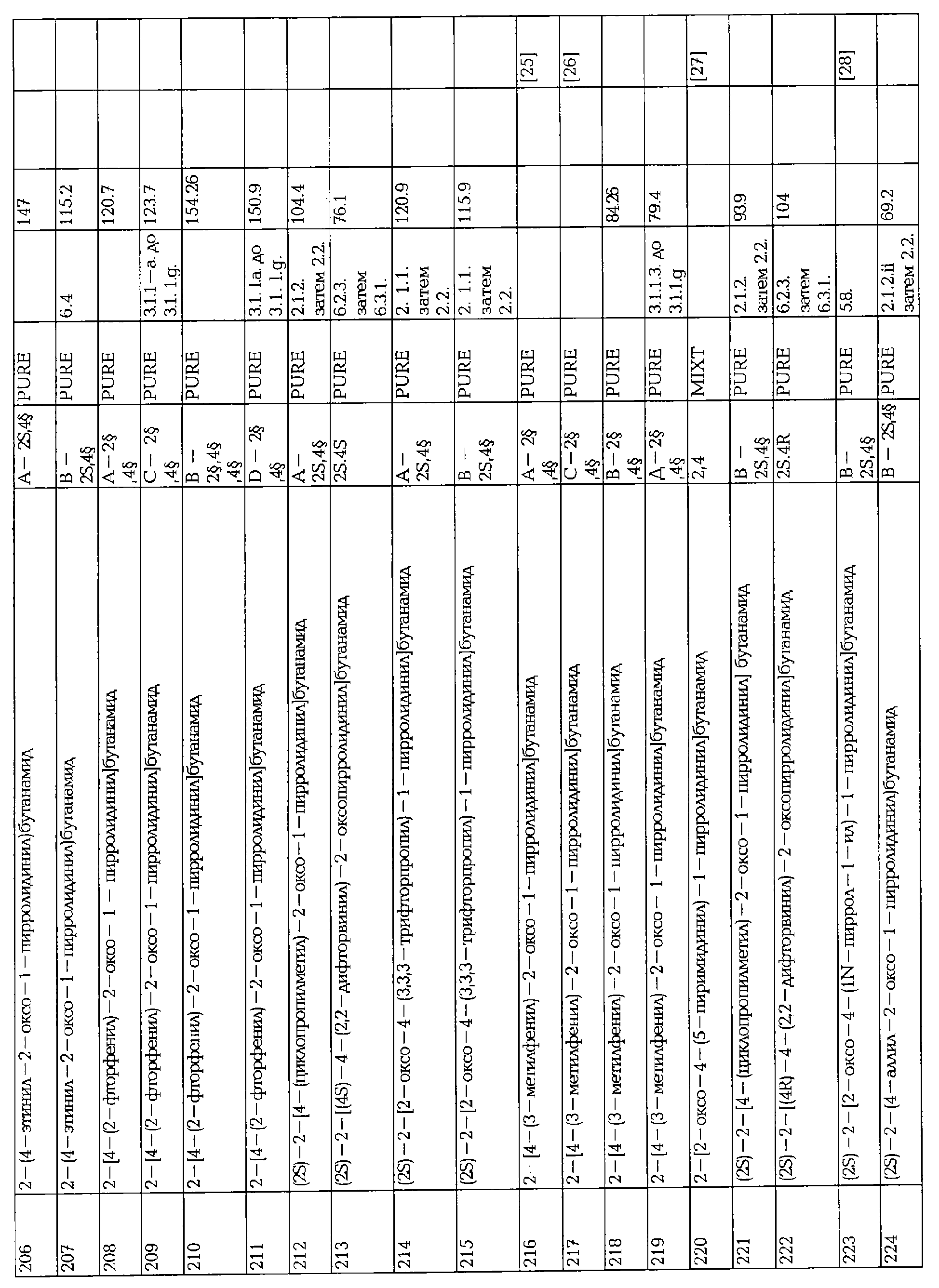
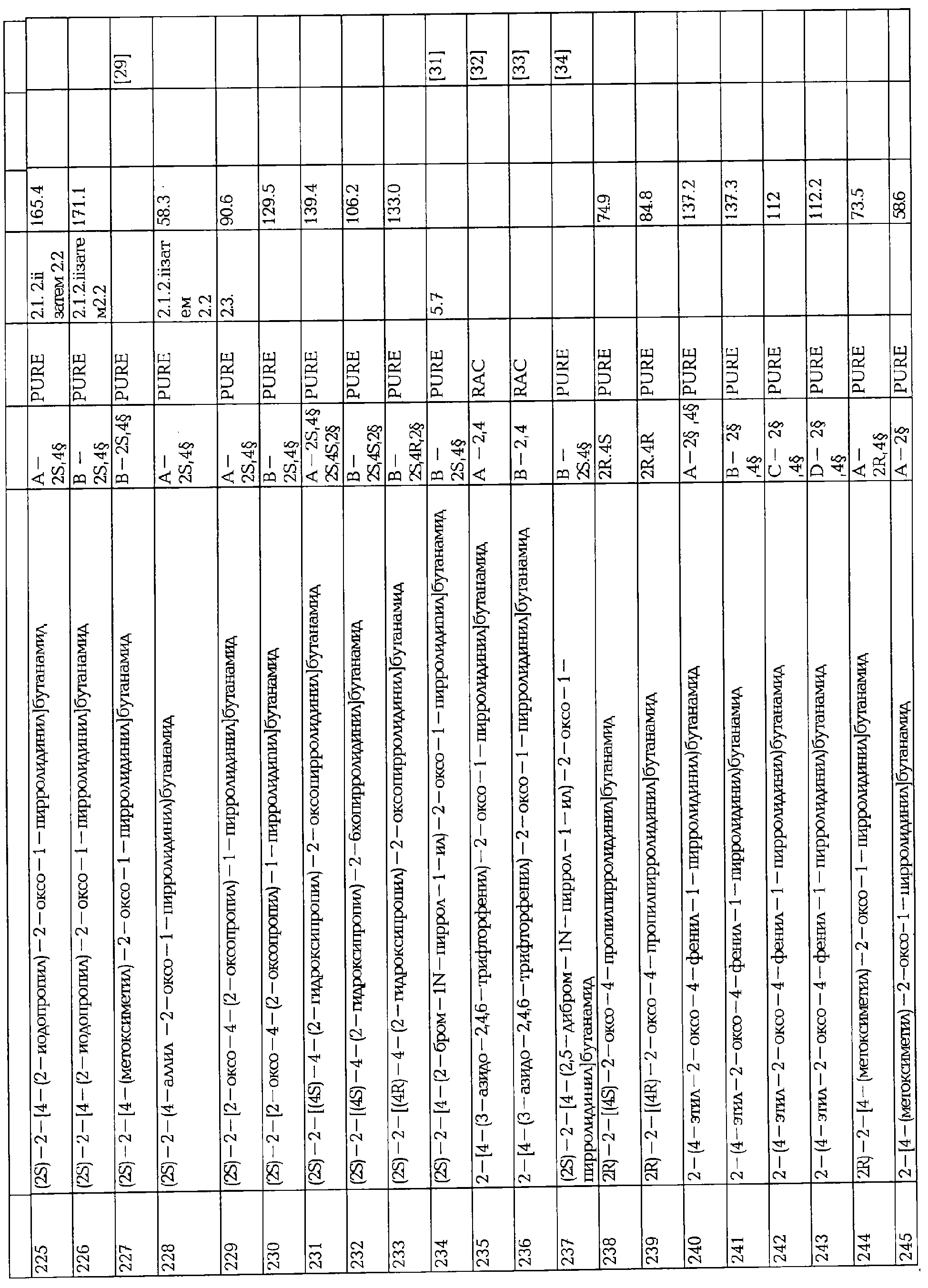
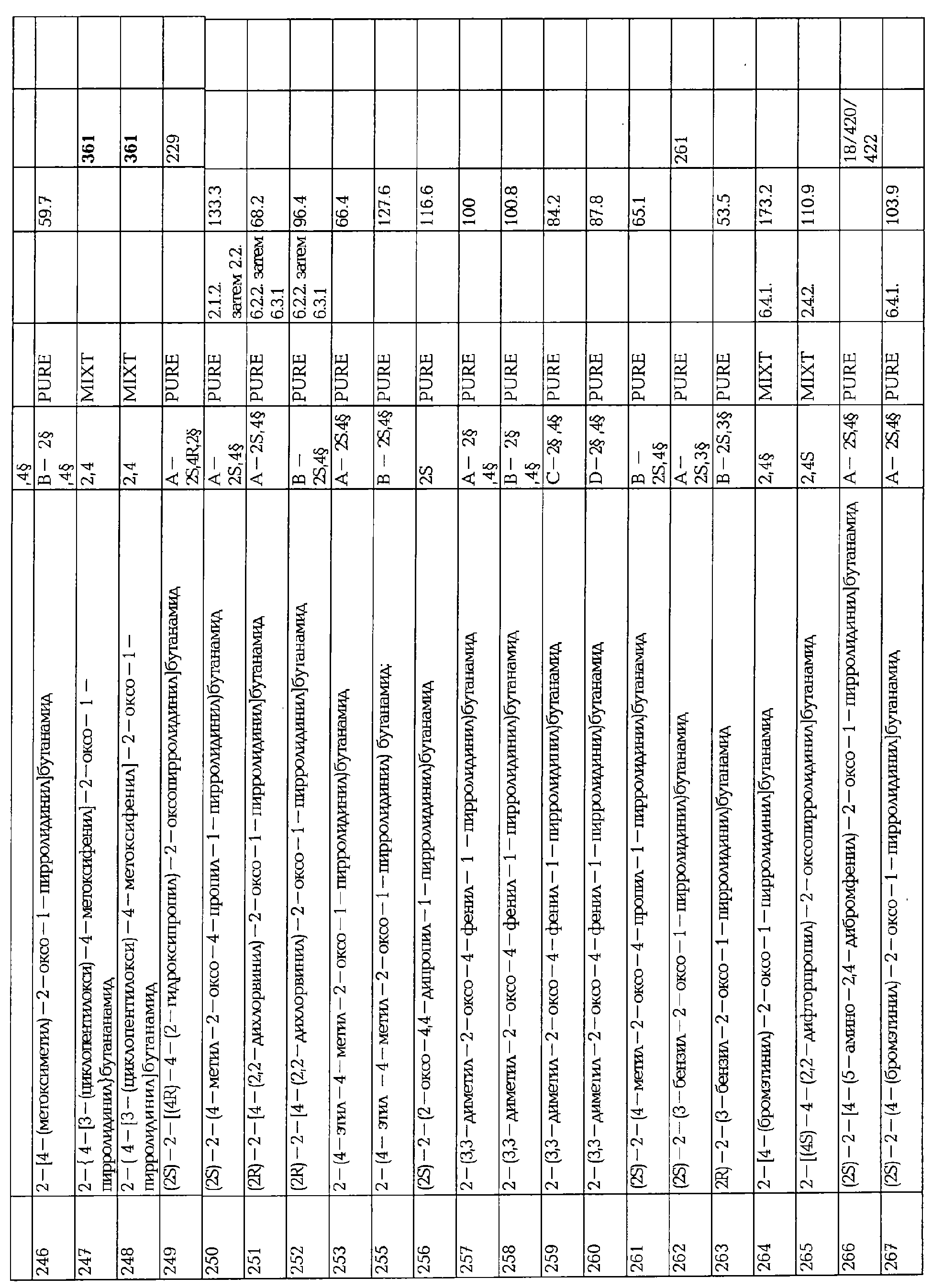

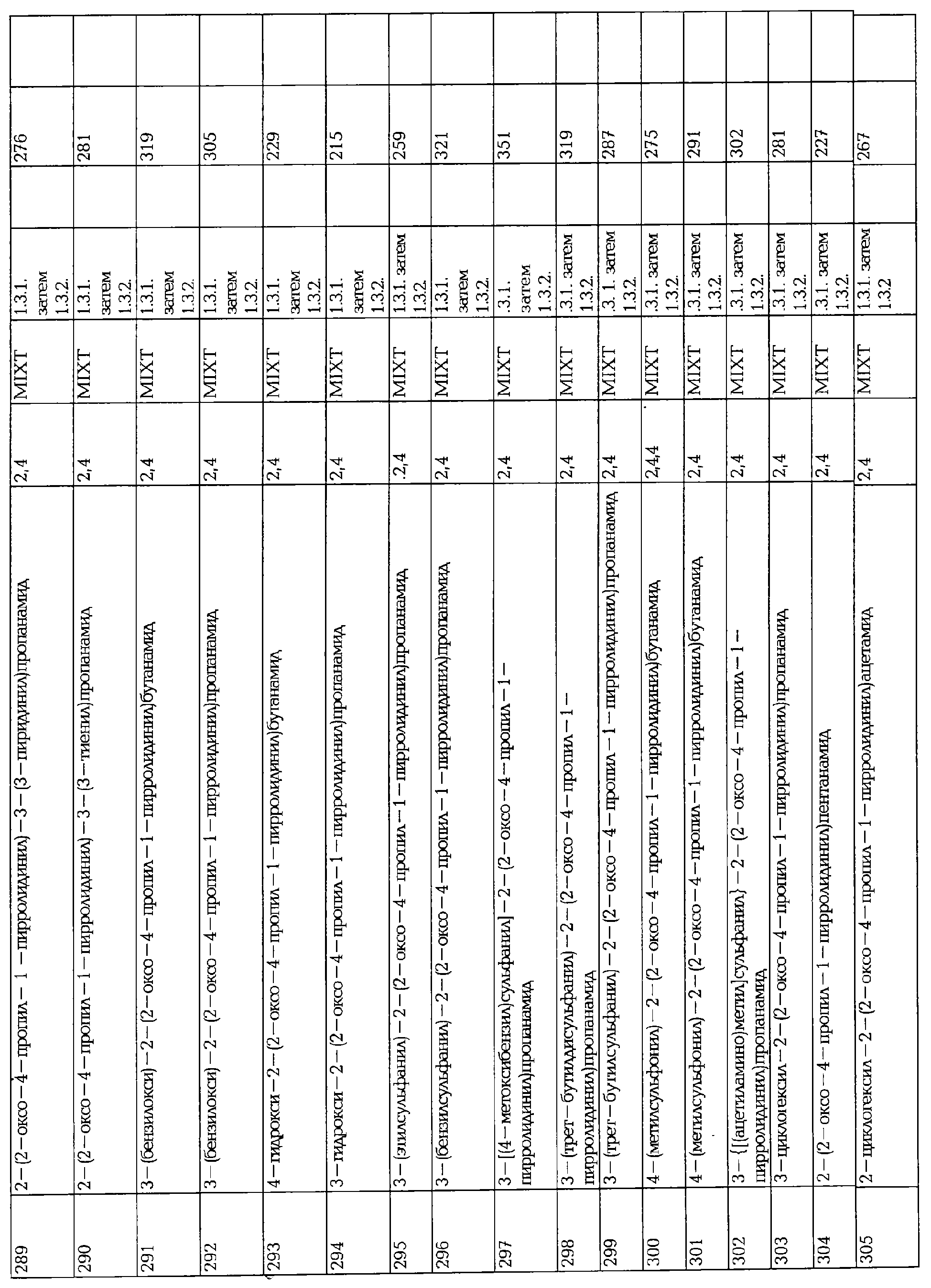
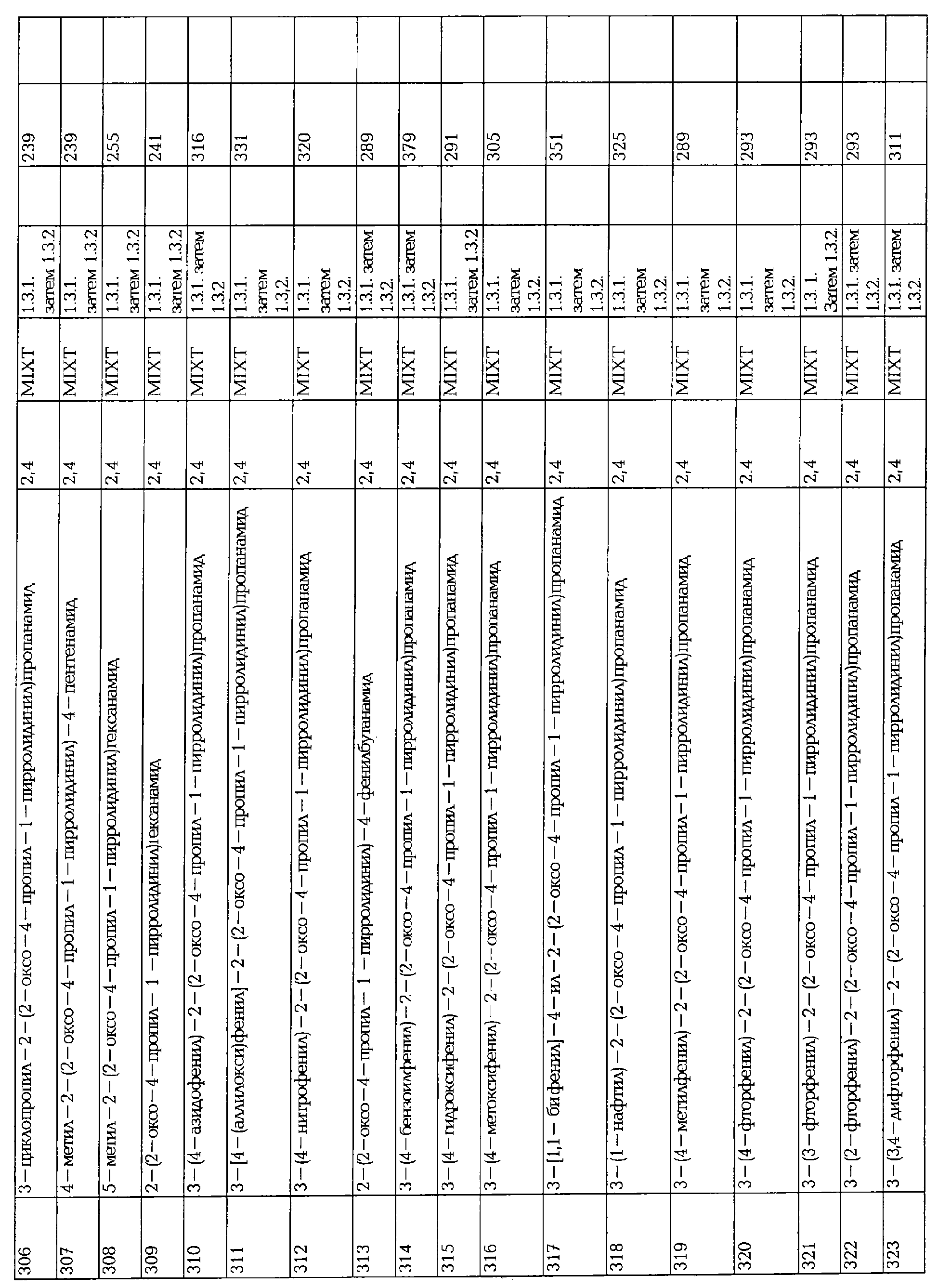
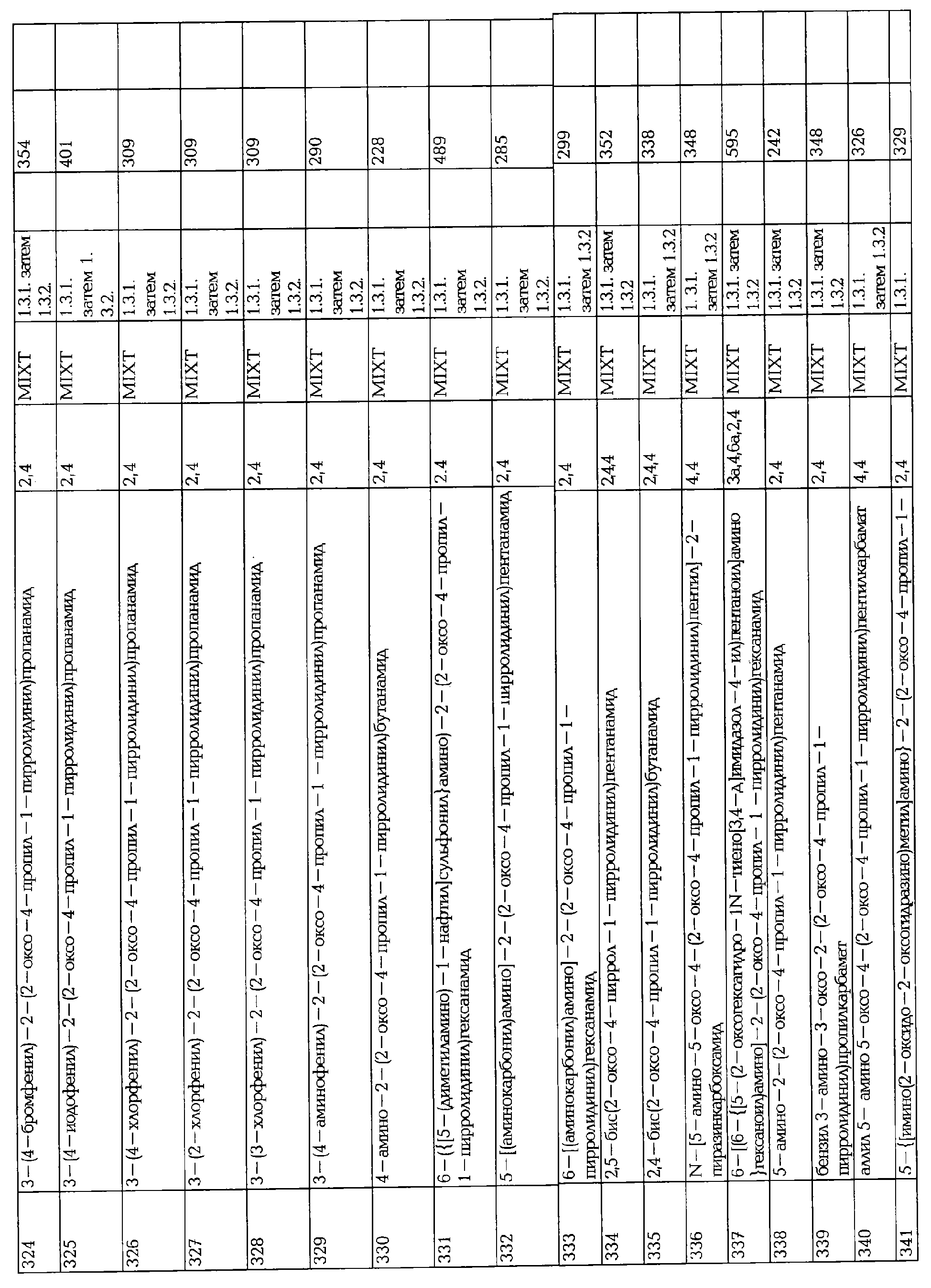
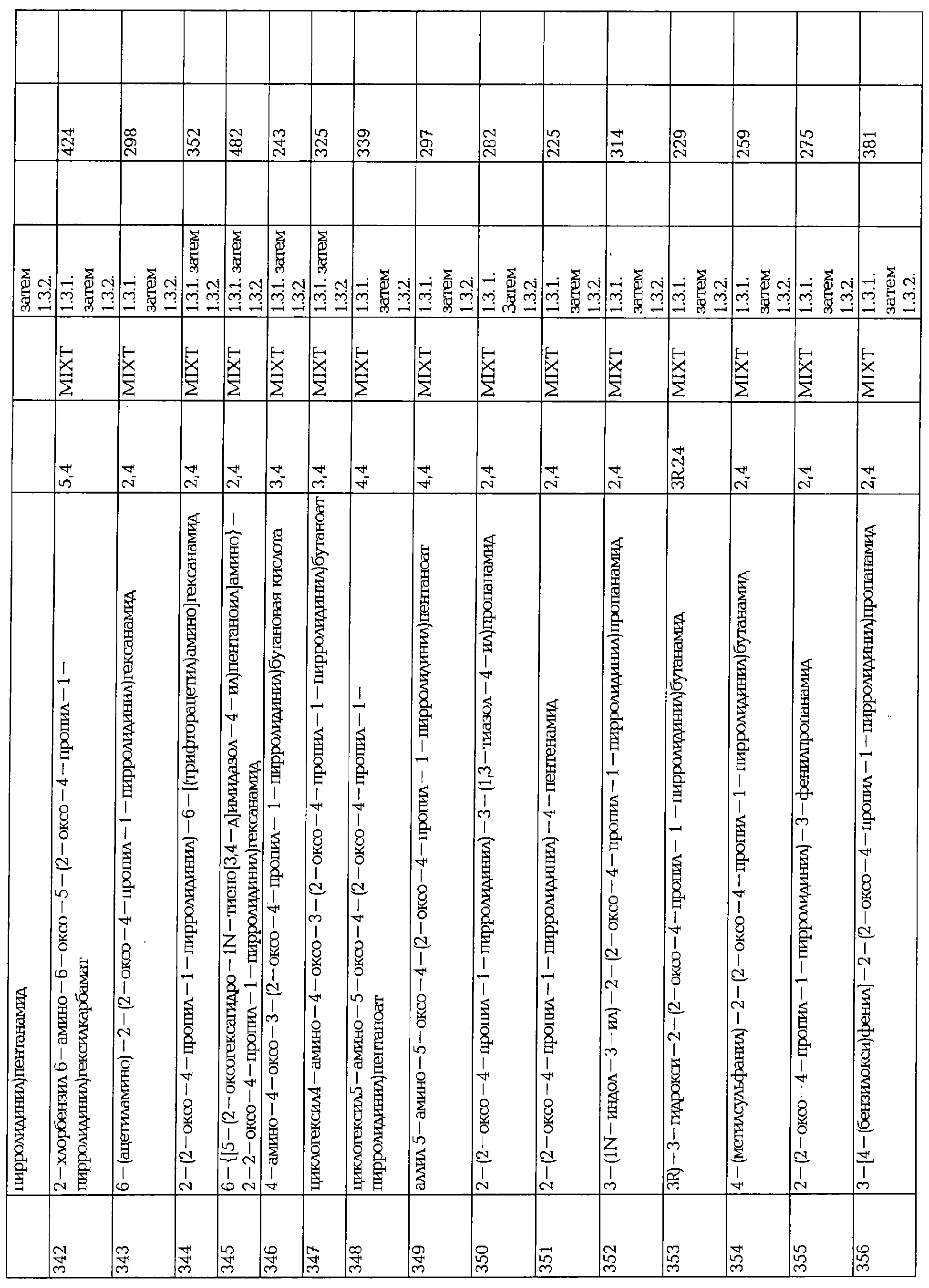
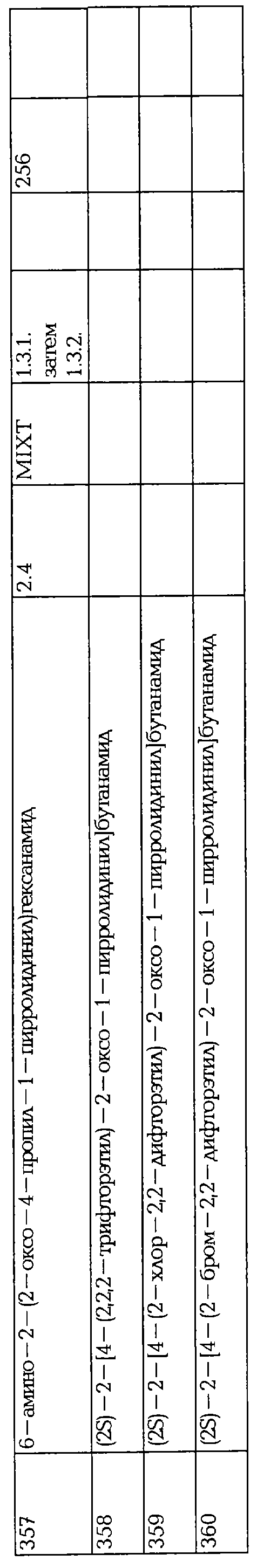




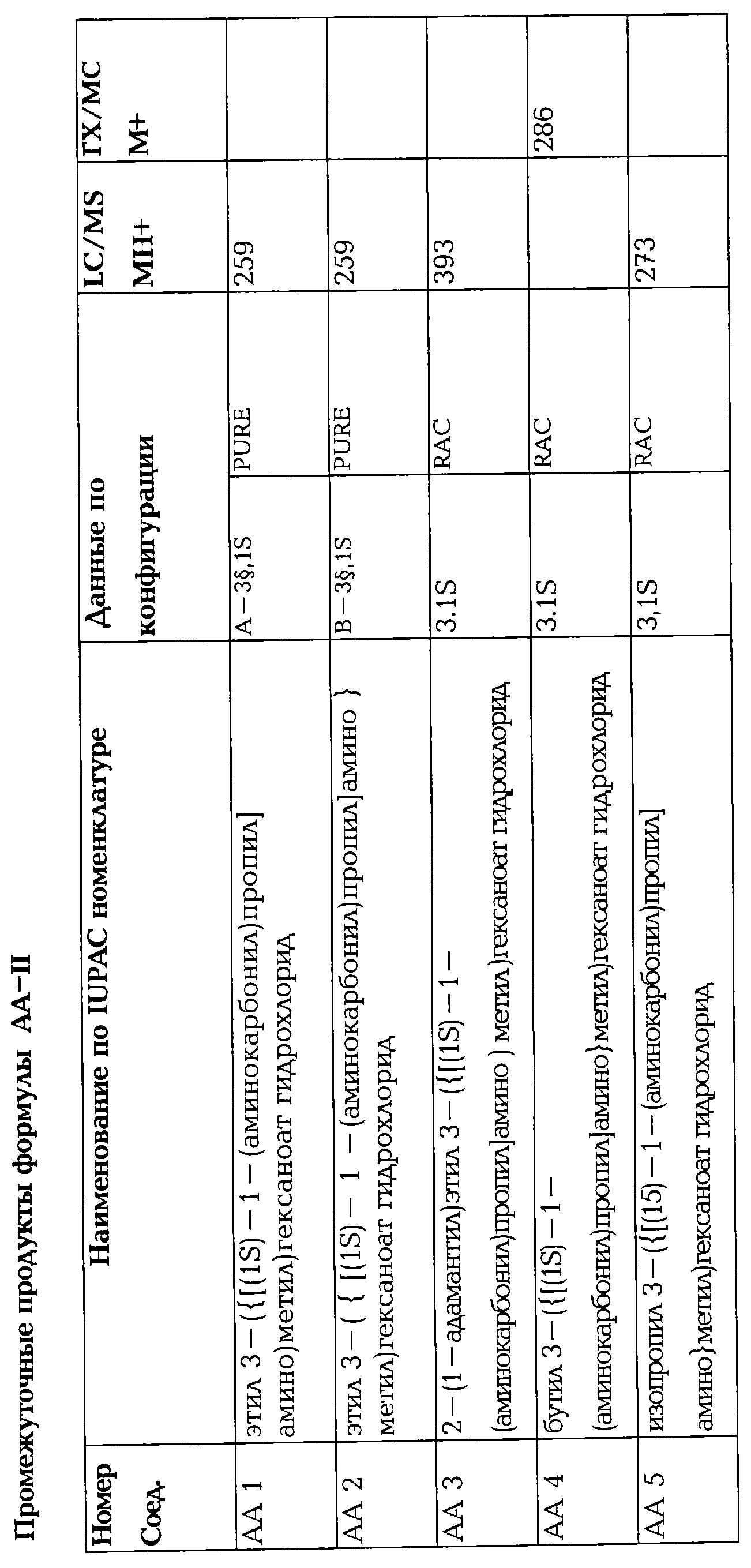
ПРИМЕР 8: Испытание по связыванию с LBS
[LBS обозначение для Леветирацетам-связывающего сайга, М. Noyer и др., Eur. J. Pharmacol., 286 (1995) 137-146.]
Константу ингибирования (Кi) соединения определяли в конкурентных экспериментах по связыванию путем измерения связывания единичной концентрации радиоактивного лиганда при равновесии с различными концентрациями немеченого тестируемого вещества. Концентрация тестируемого вещества, ингибирующая 50% специфического связывания радиолиганда обозначена IC50. Равновесная константа диссоциации Ki пропорциональна IC50 и рассчитывается по уравнению Cheng и Prusoff (Cheng Y. И др., Biochem, Pharmacol. 1972, 22, 3099-3108).
Область концентраций обычно включала 6 log единиц с различными шагами (от 0.3 до 0.5 log). Испытания проводили однократно или дважды, каждое определение KI проводилось на двух различных образцах тестируемого вещества.
Церебральную кору головного мозга от 200 до 250 г мужских Sprague - Dawley крыс гомогенизируют с помощью Potter S гомогенизатора (10 ударов при 1,000 rpm; Braun, Germany) в 20 ммоль/л Tris-HCl (pH 7.4), 250 ммоль/л сахарозы (буффер А); все операции проводят при 4°С. Гомогенат центрифугируют при 30,000 г в течение 15 мин. Полученные гранулы сырой мембраны повторно суспендируют в 50 ммоль/л Tris-HCl (pH 7.4), (буффер В) и инкубируют 15 мин при 37°С, центрифугируют при 30,000 × g в течение 15 мин и промывают дважды таким же буфером. Конечные гранулы повторно суспендируют в буфере А при концентрации белка в пределах от 15 до 25 мг/мл и хранят в жидком азоте.
Мембраны (150-200 мкг белка на испытание) инкубируют при 4°С в течение 120 мин в 0.5 мл 50 ммоль/л Tris-HCl буффере (pH 7.4), содержащем 2 ммоль/л MgCl2, от 1 до 2 10-9 моль/л [3H]-2-[4-(3-азидофенил)-2-оксо-л-пирролидинил]бутанамида и увеличивающиеся концентрации тестируемого вещества. Неспецифическое связывание (NSB) определяли как остаточное связывание, наблюдаемое в присутствии концентрации упомянутого вещества (например, 10-3 моль/л леветирацетама), которое связывается практически со всеми рецепторами. Мембраносвязанные и свободные радиолиганды отделяют быстрой фильтрацией через стекловолокнистые фильтры (эквивалентные Whatman GF/C или GF/B; VEL, Бельгия), предварительно смоченные 0.1% полиэтиленимином и 10-3 моль/л леветирацетамом для уменьшения неспецифического связывания. Образцы и фильтры промывают по крайней мере 6 мл 50 ммоль/л Tris - HCl (рН 7.4) буффером. Вся процедура фильтрации не превышает 10 секунд на образец. Радиоактивность, оставшуюся на фильтрах, вычисляют жидкостной сцинтилляцией в β-измерителе (Tri-Carb 1900 или TopCount 9206, Camberra Packard, Бельгия, или любом другом эквивалентной измерителе). Анализ данных проводят компьютерным нелинейным методом подходящего изгиба используя ряд уравнений, описывающих некоторые модели связывания, учитывая популяции независимых невзаимодействующих рецепторов, которые подчиняются закону масс.
Соединения по изобретению показали pKi величины от 6.0 и более. Особое сродство показано соединениями 8, 9, 10, 22, 23, 27, 30, 31, 32, 33, 38, 40, 41, 43, 46, 47, 49, 64, 71, 72, 73, 75, 81, 83, 86, 87, 88, 92, 93, 95, 96, 98, 100, 103, 105, 110, 119, 127, 142, 146, 149, 151, 152, 156, 157, 158, 159, 162, 163, 164, 165, 166, 169, 170, 171, 173, 174, 175, 176, 180, 181, 185, 187, 188, 195, 196, 197, 198, 200, 201, 204, 205, 207, 209, 211, 212, 213, 214, 215, 219, 221, 222, 223, 224, 225, 226, 228, 229, 234, 250, 251, 252, 264, 265, 267, 304, 306, 350 и 351.
ПРИМЕР 9: Животная модель мышей, чувствительных к звуку
Целью этого теста является измерение антиконвульсивной способности соединения на мышах, чувствительных к звуку, генетической животной модели с рефлекторными припадками. На этой модели первичной вырабатываемой эпилепсии, припадки вызывают без электрической или химической стимуляции и типы припадка являются, по крайней мере частично, подобными по их клинической феноменологии припадкам, встречающимся у человека (Loscher W. & Schmidt D., Epilepsy Rec, (1998), 2, 145-181; Buchhalter J.R., Epilepsia (1993), 34, S31-S41).
Используют генетически звукочувствительных мышей мужского или женского пола (14-28 г; N=10), произведенных от DBA породы, селекционированной Dr. Lehmann в Laboratory of Acoustic Physiology (Париж) и выращенных в UCB Pharma Sector husbandry unit с 1978. Экспериментальный проект состоял из нескольких групп, одной группе вводили контрольное вещество, а другим группам - различные дозы тестируемых соединений. Соединения вводят интраперитонеально за 60 минут до вызова аудиогенных припадков. Область вводимых доз имела логарифмическую прогрессию, обычно между 1.0×10-5 моль/кг и 1.0×10-3 моль/кг, но при необходимости тестировались более низкие или более высокие дозы.
Для тестирования животных помещают в маленькие клетки, по одной мыши в клетку, в звукопроницаемую камеру. После периода ориентации в течение 30 секунд передают акустический стимул (90 дБ, 10-20 кГц) в течение 30 секунд через репродукторы, расположенные над каждой клеткой. В течение этого времени мышей наблюдают и выявляют наличие 3 фаз активности припадка, а именно дикие метания, мышечные сокращения и тонизирующие конвульсии. Рассчитывают пропорцию мышей, защищенных от диких метаний, мышечных сокращений и тонизирующих конвульсий соответственно.
Для активных соединений рассчитывают величину ED50, т.е. дозу, производящую 50% защиту относительно контрольной группы, вместе с 95% доверительными интервалами, используя Probit Анализ (SAS/STAT® Software, version 6.09, PROBIT procedure) соотношений защищенных мышей для каждой из 3 фаз активности припадка.
Соединения по изобретению показали ED50 значения 10-4 (моль/кг) или менее. Особенно обещающую активность показали соединения 8, 9, 10, 22, 23, 27, 30, 31, 32, 33, 38, 40, 41, 46, 47, 64, 71, 72, 81, 86, 87, 88, 92, 93, 95, 96, 100, 105, 110, 146, 151, 152, 156, 158, 159, 162, 163, 164, 165, 166, 180, 181, 187, 188, 195, 196, 197, 198, 200, 201, 204, 205, 207, 209, 211, 212, 213, 214, 215, 219, 221, 222, 223, 224, 226, 228, 229, 234, 250, 251, 252, 264, 265, 267 АА 1, АА 2, АА 3, АА 4 и АА 5.
Далее представлены результаты испытаний ряда заявленных соединений.
Под испытуемыми соединениями ucb 34714 (бриварацетам) и ucb 44212 (сенетрацетам) значатся соединения формулы 1, где R3 и R3a означают соответственно С1-20 алкил и С2-6 алкенил.
Испытание 1.1. Противоположное действие ucb 34714 и лекарственных препаратов против эссенциального тремора на тремор, вызванный хармалином, по сравнению со спонтанным тремором и седативным эффектом у крыс.
Цель: Сравнить ucb 34714, новое производное пирролидона с высокой аффинностью к сайту связывания леветирацетама, с лекарственными веществами, предписанными для эссенциального тремора, антагонистичными вызванному хармалином (HARM) тремору у крыс.
Основание: HARM - индуцированный тремор используется в качестве модели эссенциального тремора. Однако HARM вызывает как седативный эффект, так и тремор. Таким образом, на этой модели лекарственные вещества могут снижать тремор только путем повышения седативного эффекта. Бихевиоральная (поведенческая) методология позволяет попытаться измерить независимый от седативности антитреморный эффект, чтобы найти лекарственные средства, которые более эффективно противостоят эссенциальному тремору, не вызывая побочных эффектов.
Методы: Тестируемые соединения вводят и.п. (интраперитонеально) за 30 минут перед и.п. введением 20 мг/кг HARM, которое вызывает тремор в течение 3 час. Тесты проводили через 15, 30 и 60 минут после HARM и поведенческие эффекты оценивали как: (1) индекс установленного (выявленного) тремора (ЕТ) (тесты движения при подъеме, в заданной позе, при наклоне); (2) индекс спонтанного тремора (ST) (тремор при спокойном положении головы/тела); и (3) седативный индекс (SED) (неподвижная опора головы/конечностей, локомоция, птоз).
Результаты: ucb 34714 в зависимости от дозы снижает как ЕТ, так и ST (максимальный эффект - 53 и - 43%, соответственно) в дозах (21-70 мг/кг), которые незначительно меняют SED. При дозе 120 мг/кг, которая уменьшает спонтанную локомоцию, снижение ЕТ (-56%) и ST (-68%) было ассоциировано с повышенным SED (57%). Пропранолол (2.5-20 мг/кг) снижал ЕТ (макс. - 49%), но сильнее снижал ST (макс. - 63%), а также повышал SED (28-37%), а при дозе 20 мг/кг вызывал постуральную гипотонию. Примидон (160 - 320 мг/кг) не оказывал влияния на ЕТ, но также снижал ST (-30 ÷ -37%) и повышал SED (макс. 46%). Клоназепам (0.3-3 мг/кг), который вызывал седативный эффект и атаксию, снижал ЕТ (макс. - 49%), но был также более эффективен в отношении ST (макс. - 77%) и заметно повышал SED (63-132%).
Выводы: В целом, когда лекарственное вещество снижает ST более, чем ЕТ, это коррелируется с повышением SED, что делает индекс установленного тремора необязательным для измерения независимого от седативности антитреморного эффекта. Из всех испытуемых лекарственных веществ ucb 34714 проявил наиболее значительную эффективность против ЕТ в дозах, которые не повышали индуцированный HARM SED и которые не давали побочных эффектов. Это наводит на мысль о перспективных терапевтических возможностях ucb 34714 при эссенциальном треморе.
Испытание 1.2
Сравнивалась способность ucb 34714 противостоять тремору, вызванному хармалином у крыс, с некоторыми лекарственными препаратами против тремора и против эпилепсии. Тремор, спровоцированный хармалином, оценивали с помощью 3 поведенческих (бихевиоральных) тестов (подъем, заданная поза, наклон), которые дают индекс установленного (выявленного) тремора (ЕТ). Мы применяли дозу 20 мг/кг хармалина и.п., которая вызывала тремор в течение 3 час. Тестируемые соединения вводили и.п. за 30 минут до введения хармалина. Тесты на тремор проводят через 15, 30 и 60 минут после введения хармалина. Леветирацетам (54-540 мг/кг) вызывал небольшие изменения ЕТ (3-25%) по сравнению с контрольной группой, принимавшей носитель. Напротив, ucb 34714, в дозах, вызывавших лишь пониженную эксплорацию, снижал ЕТ на 25, 53 и 66% после введения 38, 70 и 120 мг/кг и.п. соответственно. Пропранолол (2.5-20 мг/кг) и клоназепам (0.3- 3 мг/кг) были менее активны, чем ucb 34714, и вызывали заметную постуральную гипотонию. Примидон (160- 320 мг/кг) очень слабо снижал ЕТ (10%). Карбамазепин (5-40 мг/кг) в зависимости от дозы снижал ЕТ на 17-55%;
тогда как фенитоин (50-200 мг/кг) и вальпроат (150-450 мг/кг) были активны только в узком интервале доз. Габапентин (30-120 мг/кг) давал умеренный эффект (29-42%). Эти противоэпилептические лекарственные вещества были эффективны только в дозах, вызывающих заметные поведенческие побочные эффекты.
Выводы: по сравнению с другими лекарственными веществами ucb 34714 проявил более высокую эффективность и вызывал меньше побочных эффектов, наводя на мысль о перспективных терапевтических возможностях ucb 34714 при эссенциальном треморе
Испытание 1.3.
Испытывались ucb 38714 на крысах с индуцированным хармалином тремором, сравнивая активность соединения и побочные эффекты с активностью и побочными эффектами признанных лекарственных веществ против тремора и AED. Для того чтобы избежать или свести к минимуму влияние седативности, мы оценивали обработанных хармалином крыс во время намеренно вызванной поведенческой активности, получая 'индекс установленного тремора' (ЕТ).
Результаты:
Леветирацетам (50, 170, 340 и 540 мг/кг и.п.) вызывал небольшое, статистически незначимое уменьшение ЕТ (3-25%).
Напротив, ucb 38714 (10, 21, 38, 70 и 120 мг/кг, и.п.) значительно снижал ЕТ в интервале доз 38-120 мг/кг. При дозе 10 мг/кг ucb 38714 был подобен носителю. При дозе 38 мг/кг среднее значение ЕТ снижалось относительно собственного контрольного (носитель) на 33, 26 и 15% через 15, 30 и 60 мин после введения хармалина, соответственно, тогда как при дозе 70 мг/кг в среднем снижение достигало 64, 58 и 38%, соответственно. При дозе 120 мг/кг снижение составляло 86, 73 и 40%, соответственно. В течение общего времени наблюдения один час (AUC) дозы 38, 70 и 120 мг/кг ucb 38714 в зависимости от дозы (эффект дозы) снижали ЕТ на 25, 53 и 66%, соответственно. Пропранолол (2.5, 5, 10 и 20 мг/кг и.п.) статистически значимо снижал ЕТ от 5 мг/кг и выше. Снижение ЕТ при дозе 20 мг/кг составляло 64, 51 и 32% через 15, 30 и 60 мин после введения хармалина, соответственно. В течение общего времени наблюдения один час (AUC) пропранолол вызывал зависящее от дозы снижение ЕТ 12, 29 и 49% при дозах 2.5, 10 и 20 мг/кг, соответственно.
В случае примидона (160 и 320 мг/кг, и.п.) не удалось явственно снизить установленный тремор, несмотря на применение высоких доз. Максимальное среднее снижение ЕТ составляло 10% при дозе 320 мг/кг.
Клоназепам (0.3, 1 и 3 мг/кг, и.п.) вызывал зависящее от дозы снижение ЕТ. Среднее зависящее от дозы снижение ЕТ (AUC) для доз 0.3, 1 и 3 мг/кг составляло 1, 24 и 48%, соответственно.
Карбамазепин (5, 10, 20 и 40 мг/кг) в дозах 10, 20 и 40 мг/кг, статистически значимо снижали ЕТ на 17, 33 и 55%, соответственно.
Фенитоин (50, 100, 150 и 200 мг/кг, и.п.) в дозах 100, 150 и 200 мг/кг статистически значимо снижал ЕТ на 29, 42 и 49%, соответственно.
Вальпроат (150, 300 и 450 мг/кг, и.п.) снижал ЕТ в очень узком интервале доз. Значимое снижение на 22 и 49% получают при дозах 300 и 450 мг/кг, соответственно.
Габапентин (30, 60 и 120 мг/кг, и.п.) вызывал только умеренное, независимое от дозы снижение установленного тремора (30-40%).
ПОБОЧНЫЕ ЭФФЕКТЫ: ucb 38714 более эффективно снижал у крыс вызванный хармалином установленный тремор, чем современные стандартные лекарственные средства для лечения эссенциального тремора и AED, тогда как эффективность последних лекарственных веществ проявляется, по-видимому, только в дозах, вызывающих серьезные побочные эффекты, такие как постуральная гипотония (вялая поза, мышечная слабость) и нарушение походки (шатающаяся походка, атаксия). Этого не наблюдается в случае ucb 34714 или леветирацетама.
ВЫВОДЫ: Резистентность индуцированного хармалином тремора у крыс к фармакологическому лечению, по-видимому, является отражением частичной эффективности существующих фармакологических средств для лечения эссенциального тремора.
Испытание 2. Селетрацетам (ucb 44212) ингибирует вызывающие эпилепсию реакции в срезах гиппокампа in vitro.
Цель: Селетрацетам (ucb 44212) представляет собой новое производное пирролидона, родственное по структуре леветирацетаму (LEV, Keppra), которое проявляет более высокую аффинность, чем LEV, к сайту связывания LEV (синапс - везикулярный белок 2А - Lynch et al., PNAS, 101, 9861, 2004). Данное исследование посвящено оценке активности селетрацетама в двух in vitro моделях эпилепсии.
Методы. Действие на эпилептическую реакцию, индуцированную перфузией жидкостью с высоким К+- низким Са2+ (HKLCF) или добавлением к нормальной перфузии (ACSF) 5 мкМ бикукуллина метилиодида (BMI), оценивали с помощью диаграммы потенциала поля в области СА3 срезов гиппокампа крыс. Потенциалы поля вызывались стимуляцией фимбриального отдела постоянными импульсами, выявляющими единичный пик популяции (PS) при ACSF.
Результаты. HKLCF и BMI индуцируют вызывающие эпилепсию потенциалы поля в области СА3 при повышении амплитуды PS и продуцировании многократных PS в ответ на единичные раздражители. HKLCF, но не BMI, регулярно индуцировала спонтанные импульсы. Селетрацетам, 1-10 мкМ, заметно снижал индуцированное с помощью HKLCF повышение амплитуды PS (?PS) и амплитуд и числа многократных PS с максимальным эффектом при 3.2 мкМ. LEV проявлял показанную ранее аналогичную, но меньшую активность, максимальную при 32 мкМ (Margineanu & Klitgaard, Pharmacol. Res. 42, 281, 2000). Селетрацетам не снижал скорость спонтанных импульсов, аналогично LEV (Margineanu et al., Epilepsia 44, Suppl. 9, 261, 2003). Селетрацетам ингибировал индуцированный BMI PS со значимой величиной при 10 мкМ. Селетрацетам, 1-10 мкМ снижал число индуцированных многократных PS. Сообщалось, что LEV ингибирует эти маркеры появления эпилепсии с максимальным эффектом при 32 мкМ.
Заключение. Селетрацетам ингибировал вызывающие эпилепсию реакции в срезах биокампа у крыс с более высокой активностью и эффективностью, чем LEV, в двух in vitro моделях эпилепсии.
Испытание 3. Селетрацетам (ucb 44212) предупреждает приступы на животной модели хронической эпилепсии in vivo.
Данное исследование посвящено изучению возможной способности селетрацетама предупреждать приступы на in vivo моделях эпилепсии.
Методы. Активность по предупреждению приступов оценивают на самцах мышей линии NMRI с корнеальным возбуждением (25-35 г; n=10), самцах крыс линии Sprague Dawley с возбужденным гиппокампом (350-45 г; n=8), предрасположенных к аудиогенным приступам самцах мышей (20-25 г; n=10) и крысах с генетической эпилепсией абсансного типа из Страсбурга (GAERS), а также при максимальном электрошоке и приступах, индуцированных пентилентетразолом у самцов мышей линии NMRI (22-28 г; n=10). Для количественного определения побочных эффектов CNS использовали ротарод.
Результаты. Селетрацетам предупреждал моторные припадки вторично генерализованной эпилепсии у мышей с корнеальным возбуждением (ED50=0.31 мг/кг, и.п.) и крыс с возбуждением гиппокампа (минимальная активная доза = 0.23 мг/кг, п.о.). Аналогично, селетрацетам подавлял клонические конвульсии у мышей, склонных к аудиогенным припадкам (ED50=0.17 мг/кг, и.п.) и пик-волновые разряды у крыс линии GAERS (ED50=0.15 мг/кг и.п.). Это противоречит отсутствию защиты в тестах с максимальным электрошоком и с припадками, вызванными пентилентетразолом (ED50>232 мг/кг и.п.). Значения TD50 (ротарод) селетрацетама у мышей с возбужденной роговицей (корнеально возбужденных) и крыс GAERS составляли 325 и 449 мг/кг и.п., соответственно. По сравнению с протективными значениями ED50, полученными на тех же животных, это дает в результате высокую границу безопасности (безвредности) 1048 и 3075, соответственно.
Вывод: Данное исследование показало надежную защиту от приступов и высокую CNS переносимость селетрацетама на различных in vivo моделях эпилепсии.
Испытание 4. Антиконвульсивное действие селетрацетама (ucb 44212) на животных моделях эпилептического статуса
В данном исследовании изучаются возможные протективные свойства селетрацетама в отношении приступов у in vivo моделей самоподдерживающегося эпилептического статуса (SSSE).
Методы. SSSE вызывают с помощью 30-минутной периодической стимуляции перфорантного пути (PPS) с помощью постоянно имплантированных электродов у свободно двигающихся взрослых самцов крыс Wistar. Селетрацетам (100-300 мг/кг), носитель или леветирацетам (500 мг/кг) инъецировали внутривенно через 10 минут после индукции SSSE (раннее лечение установленного SSSE). Электрографические и поведенческие (бихевиоральные) проявления SSSE анализировали в режиме офлайн.
Результаты. Селетрацетам, инъецированный в.в. в ходе ранних стадий установленного SSSE (через 10 мин после окончания PPS), сокращал длительность припадков с эффектом дозы (в зависимости от дозы). Общее время пребывания в состоянии припадка после леветирацетама составляло 32+5 мин (500 мг/кг), общее время пребывания в состоянии припадка после введения селетрацетама составляло 3.5+0.7 мин (300 мг/кг), 11+1.7 мин (200 мг/кг) и 25±3.4 мин (100 мг/кг), тогда как у контрольных животных время пребывания в состоянии припадка (приступа) составляло 32.5+5.1 мин. Защита от припадков с помощью в.в. ведения селетрацетама (300 мг/кг) была значительно эффективнее, чем описанная ранее защита с помощью в.в. введения диазепама (10 мг/кг) или в.в. введения леветирацетама (300 мг/кг), и сравнима с защитой при в.в. введении фенитоина (50 мг/кг). Однако этот эффект получен при дозах селетрацетама, заметно более высоких (100-300 мг/кг в.в.), чем дозы, вызывающие защиту у животных моделей от частичной или генерализованной эпилепсии (0.1-0.3 мг/кг и.п.).
Выводы. Селетрацетам проявлял значительный протективный эффект в отношении припадков на этих животных моделях SSSE.
Испытание 5. Ucb 34714 эффективно на моделях нейропатической боли у крыс: сравнение с габапентином.
Целью данного исследования было сравнение действия ucb 34714 с действием габапентина на двух моделях нейропатической боли у крыс.
Влияние ucb 34714 на реакцию острой боли изучали с помощью теста "горячей пластинки" у крыс. Ucb 34714 (21-210 мг/кг) и габапентин (50-200 мг/кг) инъецируют интраперитонеально (и.п.) за 30 мин до тестирования (=исследование 3).
Действие ucb 34714 на гипералгезию изучали на крысах с мононейропатией (модель Беннетта, перевязка седалищного нерва = исследование 1) и с диабетом (стрептозоцин, 75 мг/кг, и.п. = исследование 2). Индуцированную гипералгезию определяют, надавливая на лапу. Через 2 недели после индукции мононейропатии или через 3 недели после индукции диабета крысам и.п.вводят ucb 34714 (2.1-68 мг/кг) или габапенин (30,60 мг/кг). Через интервалы после инъекции определяют порог вокализации.
Результаты показали, что ucb 34714 не изменяет время реакции в тесте "горячей пластинки". Напротив, габапентин значительно увеличивает время реакции при 200 мг/кг на 66%. Как у крыс с диабетом, так и у крыс с мононейропатией ucb 34714 значительно повышает порог вокализации и полностью отменяет (реверсирует) гипералгезию в дозе 21 мг/кг. Габапентин вызывает значительный антигипералгезивный эффект в дозе 30 мг/кг у крыс с диабетом, но только в дозе 60 мг/кг у крыс с мононейропатией.
Ucb 34714 не оказывал аналгезирующего действия при острых болевых рефлексах. Однако как у крыс с диабетом, так и у крыс с мононейропатией, ucb 34714 вызывал антигипералгезивный эффект, который вполне сравним с действием габапенина. Эти данные говорят о терапевтическом потенциале ucb 34714 у пациентов, страдающих от нейропатической боли.
Испытание 6. Ucb 34714 - сравнение с леветирацетамом на животных моделях хронической эпилепсии in vivo.
В данном исследовании сравниваются противоприпадочная и антиэпилептогенная активности ucb 34714 и леветирацетама (Lev) на многих in vivo моделях эпилепсии.
Исследование 1: Возбуждение коры (корнеальное) у мышей: Полностью возбужденным самцам NMRI мышей (20-25 г; N=10 в группе) предварительно утром дают физиологический раствор, затем стимулируют и наблюдают конвульсии. Аналогично утром после предварительной обработки им дают физиологический раствор, ucb 34714 (0.6-2.4 мг/кг и.п.) или Lev (1.8-10.0 мг/кг и.п.).
Величина ED50: 1.2 мг/кг и.п.для ucb 34714 и 7.3 мг/кг и.п. для Lev.
Исследование 2: Возбуждение гиппокампа у крыс: Полностью возбужденных самцов крыс Sprague-Dawley (250- 350 г; N=8 в группе) однократно стимулируют после перорального введения воды. Через два дня этот протокол повторяют с теми же животными после перорального введения либо воды, либо ucb 34714, либо Lev.
Минимальная активная доза, т.е. первая доза, вызывающая заметное уменьшение показателя (в баллах) тяжести припадка по сравнению с величиной перед введением препарата: 0.21 мг/кг п.о. для ucb 34714 и 54 мг/кг п.о. для Lev.
Исследование 3: Возбуждение миндалины у крыс: Полностью возбужденных самцов крыс Sprague-Daw-ley (250- 350 г; N=8 в группе) однократно стимулируют после введения физиологического раствора.
Через 2 дня и обрабатывают аналогичным образом после предварительного введения физиологического раствора, ucb 34714 (6.8, 21.2, 67.9 и 212.3 мг/кг и.п.) или Lev (17, 54, 170, 540 и 1700 мг/кг и.п.).
Ucb 34714 вызывал заметное подавления тяжести двигательно-припадочного состояния при дозе 21.2 мг/кг, тогда как Lev вызывал сходный эффект в дозе 170 мг/кг. Ucb 34714 также значительно снижал ADD в дозе 212.3 мг/кг, тогда как Lev был неактивен для этого показателя вплоть до дозы 1700 мг/кг.
Исследование 4: Аудиогенные припадки у мышей: Клонические судороги вызывали у мышей, генетически восприимчивых к звуку (165-28 г; N=10 в группе). Мышам предварительно вводили либо физиологический раствор, либо, ucb 34714 (1.4-4.0 мг/кг и.п.), либо Lev (24-48 мг/кг и.п.).
От появления кли(о?)нических судорог мышей защищали ucb 34714 с величиной ED50 2.4 мг/кг и Lev при 30 мг/кг.
Исследование 5: Спонтанные импульсно-волновые разряды (SWD) у крыс с генетической эпилепсией абсансного типа из Страсбурга (GAERS):
Каждому самцу крыс линии GAERS имплантировали 4 платиновых электрода в левую и правую кортикальные области. Через 20 минут, период привыкания, крысам инъецируют и.п.либо физиологический раствор, либо ucb 34714 (2,1, 6.8 и 67.9 мг/кг), либо Lev (5.4, 17.0 и 170 мг/кг), и непрерывно записывают EEG последовательно через 20-минутные интервалы вплоть до 120 минут.
Ucb 34714 заметно подавлял спонтанные SWD у крыс GAERS при дозе 2.1 мг/кг с полным ингибированием, наблюдаемым при 67.9 мг/кг. С другой стороны, Lev вызывал заметную супрессию SWD в дозе 5.4 мг/кг, но никогда не вызывал полного ингибирования - даже при дозе 170 мг/кг.
Исследования 6 и 7: Ротарод (вращательный) тест на возбужденных мышах и крысах: Полностью корнеально возбужденных мышей (N=10 в группе) или крыс с возбуждением миндалины (N=8 в группе) предварительно обучают и оставляют только тех животных, которые способны оставаться на вращающейся поверхности, по меньшей мере, 60 секунд в 3 последовательных испытаниях. На следующий день вводят либо физиологический раствор, либо ucb 34174, либо Lev и регистрируют число животных, не способных оставаться на вращающейся поверхности (стержне), по меньшей мере, 60 секунд.
Предыдущие исследования дали большую величину отношения TD50/ED50 для Lev (148) по сравнению с такими отношениями для других классических или более новых AED (2-21), что показывает необычайно высокий интервал между дозами Lev, которые вызывают значительное ухудшение при ротарод - тесте, и дозами, которые вызывают предупреждение двигательных припадков у корнеально возбужденных мышей. Отношение TD50/ED50 для ucb 34714 составляло 46. При аналогичной оценке крыс с возбуждением миндалины отношения для ucb 34714 и Lev составляли 4 и 2, соответственно.
Исследование 8: Развитие корнеального возбуждения у мышей: Возбуждение вызывали осуществляемой дважды в день стимуляцией в группах из 20 самцов мышей NMRI (20-25 г), которым предварительно давали либо физиологический раствор, либо ucb 34714 (0.21, 0.68, 2.1 и 6.8 мг/кг и.п.), либо Lev (1.7, 5.4, 17 и 54 мг/кг и.п.) перед каждой стимуляцией. Через 19 дней, в течение которых дважды в день проводили стимуляцию, введение лекарственного вещества прекращали, и в течение 2 дней проводили промывание без стимуляции. После промывания животных рестимулировали дважды в день еще в течение 5 дней без введения лекарственного вещества. Появление генерализованных припадков использовалось в качестве конечной точки для оценки развития корнеального возбуждения.
Предварительное введение ucb 34714 в процессе корнеального возбуждения мышей привела к значительному снижению частоты генерализованных двигательных припадков. Аналогичное снижение частоты генерализованных двигательных припадков в случае Lev наблюдается при более высоком интервале доз. Непрерывная корнеальная стимуляция с последующим прекращением лечения показала устойчивое снижение частоты генерализованных двигательных припадков в группе, ранее принимавшей наивысшую дозу ucb 34704. Подобный эффект не наблюдался ни в одной из групп, ранее принимавших Lev.
Выводы: Противоприпадочная активность, обнаруженная у ucb 34714 на животных моделях, которые, как полагают, имитируют неполную (возбужденные животные) и генерализованную (мыши, восприимчивые к аудиогенным припадкам, и мыши GAERS) эпилепсию у людей, более сильна и эффективна по сравнению с противоприпадочной активностью Lev.
Оценка на возбужденных животных наводит на мысль, что граница (предел) безопасности ucb 34714 аналогична границе (пределу) безопасности Lev.
Предварительное введение ucb 34714 в процессе корнеального возбуждения у мышей показывает сильную и устойчивую способность ингибировать развитие возбуждения, более высокую чем эта способность у Lev.
Настоящее исследование показывает, что ucb 34714 обладает более высокой активностью и эффективностью, чем Lev, в качестве противоприпадочного и антиэпилептогенного агента на различных in vivo моделях эпилепсии.