Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСНЫХ КИСЛЫХ СОЛЕЙ ДВУХВАЛЕНТНЫХ МЕТАЛЛОВ ДИКАРБОНОВЫХ КИСЛОТ
Вид РИД
Изобретение
Изобретение относится к химической промышленности, а более конкретно - к способам промышленного производства солей, в частности высокочистых комплексных солей двухвалентных металлов дикарбоновых кислот.
Комплексные соли двухвалентных металлов дикарбоновых кислот общей формулы: Ме(АсН)2⋅nH2O, (I), где Me - двухвалентный катион, Ас - анион дикарбоновой кислоты, Н - водород, n≥0, применяются в медицине, косметологии, ветеринарии и для пищевых целей. В частности, в медицине и ветеринарии такие соединения используют для обеспечения эффективной трансмембранной доставки катионов двухвалентных металлов (см. патент RU 2228183 [1]), что необходимо при профилактике и лечении широкого спектра заболеваний.
В настоящее время известно достаточно большое количество способов получения таких солей, однако все эти способы имеют характер лабораторных методик. Дело в том, что относительно сложно и дорого получить такие соединения высокой чистоты, так как обычно при синтезе формируется целый спектр соединений - от средних солей (биологически неэффективных) до сложносочлененых солей переменного состава, биологическое действие которых не изучено и разрешения на применение в пищевой, фармацевтической и ветеринарной областях не имеющих.
Также оказалось, что технология получения таких солей осложняется процессом их сушки. Большая часть известных из уровня техники решений не позволяет получить соли такой структуры, которая позволяла бы после сушки перевести их в безводное состояние. Это может быть связано с тем, что в процессе таких синтезов одна из молекул воды встает в «координатное» положение в структуре соли, а далее определяет степень гидратированности.
Комплексные соединения двухвалентных (Cd, Са, Mg, Ni, Zn, Cu, Fe и др.) катионов с янтарной, аспарагиновой и фумаровой кислотами получают путем взаимодействия соответствующей кислоты (в виде расплава, водного раствора или суспензии) или ее натриевой (калиевой) соли с солями (карбонатами, хлоридами, нитратами и т.д.), оксидами, гидроксидами и комплексными соединениями (в том числе аквахелатами) соответствующих катионов (смотри статью Stephenson Т.A. et al. "J. Chem.Soc", 1964, Aug. 2538-2541 [2], статью Tomita Takeshi et al. "Bull. Chem. Soc. Japan", 1968, 41, №5, 1130-1132 [3], патент JP 31606 [4], патент FR 27M, 28.11.60 (РЖХ, 1962, 20Л280) [5] и патент FR 3CAM, 6.11.61 (РЖХ, 1963, 23Н238П) [6]).
В одном из известных решений [7] предлагается получать соли ЯК гидрированием соответствующих солей малеиновой, фумаровой кислот, однако таким способом нельзя получить соли непредельных кислот, кроме того не обеспечивается возможность синтеза комплексных кислых солей. Продуктом такого синтеза являются средние соли структуры АсМе, где Ас - анион янтарной кислоты, Me - двухвалентный металл.
Известные решения, описанные в патенте GB 1129544 [8] и в патенте JP 13012 [9], касающихся способов синтеза солей дикарбоновых кислот нагреванием смеси спирта (например, бутандиола), гидроокиси металла и воды при температуре от 250 до 400°С и давлении от 50 до 500 атм [8] или обработкой смеси ацетата и карбоната металла окисью углерода при температуре от 300 до 450°С [9] являются достаточно экзотическими и осуществляются в жестких условиях.
При всех описанных выше вариантах синтеза получают смесь комплексных соединений, отличающихся одно от другого не только различным соотношением М и L, но и обладающих различной структурой. Этот факт подтвержден и проведенными авторами исследованиями: в зависимости от условий синтеза и химических свойств катиона и аниона их соотношение Ас:Ме варьируется в широких пределах (1:1, 1:2, 1:3, 1:4, 1:5, 2:1, 2:3). Константы устойчивости комплексов с различным соотношением Ас:Ме и их растворимость в воде различаются иногда значительно, что затрудняет возможность получения в водной среде однородного по составу соединения. Например, растворимость в воде сукцинатов двухвалентных катионов (средних солей MeSuc) значительно ниже растворимости комплексных соединений, в которых соотношение Me:Suc меньше 1, при этом, например, для комплексов CaSucn (где n принимает значения от 2 до 5) зависимость растворимости в воде от n проходит через максимум, соответствующий n порядка 3. В то же время для фумаратов имеет место обратная закономерность: растворимость MeFum выше растворимости комплексов с соотношением Me:Fum меньше 1.
Если для получения комплексной соли состава Ме(АсН)2 в водную среду исходные компоненты ("щелочная" и "кислая") вводятся в соотношении 1:2, как, например, в патент JP 24954 [7], то в продукте реакции кроме Ме(АсН)2 присутствуют также соединения состава МеАс и Ме(АсН)3 вследствие, например, следующих реакций:
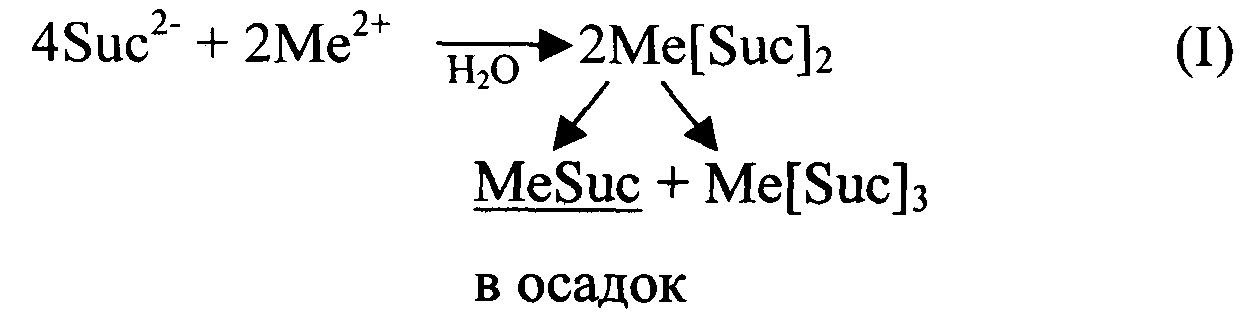
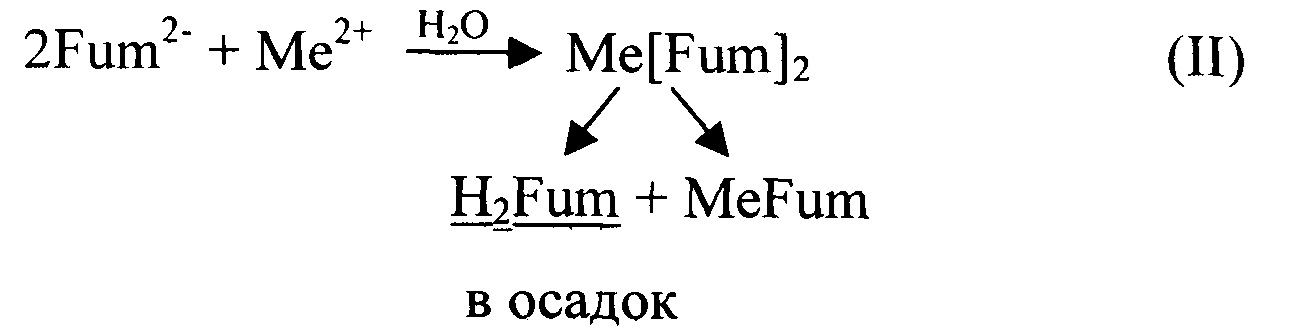
В результате в реакции (I) при кристаллизации вместе с целевым соединением Me [Suc] 2 соосаждается плохо растворимая средняя соль MeSuc (практически не усваиваемая организмом животных), тогда как эквивалентное количество более растворимого Me[Suc]3 в основном уходит в раствор. Рекомбинация фумаратов Me[Fum]2 в реакции (II) приводит к тому, что осадок кроме целевого соединения содержит примесь фумаровой кислоты, в раствор частично уходит эквивалентное ей количество более растворимого, чем H2Fum и Me[Fum]2, соединения MeFum. При этом достоверная дифференциация отхода и целевого продукта либо крайне трудоемка и дорогостояща, либо невозможна.
Обычно, если желательно получить соединение с соотношением Ме:Ас меньше 1, в раствор "кислой" компоненты дозируется "щелочная" компонента или ее раствор (если порядок обратный - однозначно преимущественно образуются труднорастворимые комплексы состава МеАс, которые с большим трудом взаимодействуют даже с избытком "кислой" компоненты). Но даже если синтез ведется "обычным" способом, предлагаемым в большинстве перечисленных информационных источников, в том числе ближайшем аналоге (патент JP 49-8849) [10], - в реакционной зоне имеют место постоянно изменяющиеся концентрации и соотношение реагентов, что также не способствует получению индивидуального комплекса с определенным соотношением Ме:Ас, в результате продукт реакции представляет собой смесь комплексов, причем соотношение комплексов достоверно не воспроизводится. Это делает невозможным применение данных способов для промышленного получения целевого продукта - комплексных кислых солей двухвалентных металлов дикарбоновых кислот.
Кроме того, известно, что в водных растворах многозарядные катионы всегда образуют аквакомплексы [Ме(H2O)n]2+, содержащие в качестве лиганда одну или несколько молекул воды (в зависимости от координационного числа данного катиона). Эти аквакомплексы образуются в результате присоединения молекул воды или гидратации катионов - если "щелочная" компонента предварительно растворяется (суспендируется) в воде. Они также могут образовываться из других координационных соединений в результате внутрисферного замещения при другом способе введения в реакцию исходных компонентов.
Поэтому комплексные соли MenAcm⋅xH2O содержат не только кристаллизационную, связанную с анионом, но и прочносвязанную, координированную воду, определенным образом расположенную вокруг катиона. Характер связи определяется в том числе и условиями синтеза (температурой, скоростью и порядком дозировки компонентов, условиями кристаллизации, продолжительностью выдержки, условиями сушки и т.д.).
Для того чтобы свести к минимуму присутствие в конечном целевом комплексе примесей солей другого состава, появляющихся в результате гидролиза, рекомбинации, внутрисферного обмена и т.д., предложены в патенте FR 27М, 28.11.60 (РЖХ, 1962, 20Л280) [5], в статье, опубликованной в "J. Indian Chem. Soc", 1961, 38, №3, 153-154 [6] и в патент FR 3САМ, 6.11.61 (РЖХ, 1963, 23Н238П) [11] способы синтеза в неводной среде (например, в абсолютированных спиртах или хлороформе). Такие методы синтеза приемлемы как препаративные, однако при масштабных наработках использование органического растворителя усложняет технологию (регенерация растворителя), снижает производительность (низкая растворимость исходных компонентов в органических растворителях) и удорожает, таким образом, конечный продукт. Более того, присутствие остаточных спиртов (кроме этилового) и хлороформа в конечном продукте не допускается по требованиям пищевой безопасности, а полное их выделение из конечного продукта весьма затруднительно. Все это делает применение таких известных технологий для промышленных целей невозможным.
Близким по существу к предлагаемому изобретению является патент RU 2115657 [12], автор которого предлагает синтез и применение аквахелатов, которые "включают биогенный металл и, как минимум, один органический лиганд … Металл выбирается из группы … металлов "f", "d", "р", более предпочтительно Cs, Mg, Са, In, Se, Те, Fe(II), Fe(III), Co(II), Co(III), Mn(II), Cu, Ni, Zn. Лиганд выбирается из аминокислот …, карбоновых кислот …".
При этом известный из [12] способ получения "аквахелатов" сводится к взаимодействию водных растворов соли металла и лиганда, "количество которого тщательно проверяется для регулирования стехиометрии между лигандом и металлом ,… соотношение металла к лиганду устанавливаются равным 1:0,5; 1:1; 1:1,5; 1:2, … после образования "аквахелатов" часть воды или вся вода может быть удалена из раствора … до тех пор, пока аквахелат не примет форму сухого порошка, … можно обезвоживать аквахелат для удаления всей лабильной воды, при этом лабильная вода восстанавливается".
Главный недостаток, присущий способу [12], - невозможность получить индивидуальное комплексное соединение с определенным соотношением металла и лиганда (что подробно обсуждалось выше) - по предлагаемому способу можно получить только смесь комплексов различного состава.
Наиболее близкими по решаемой задаче к заявляемому способу являются способы по патенту [10], в соответствии с которыми сукцинатные комплексы кальция Ca(Suc)n, где n=2÷5, получают следующим образом: карбонат кальция и янтарную кислоту в мольном соотношении 1:2÷5 (в зависимости от соотношения Ca:Suc в целевом комплексе) растворяют в 5-ти кратном (по отношению к ЯК) количестве воды, выдерживают в течение 24 часов при комнатной температуре, фильтруют, полученный раствор постепенно нагревают до 100°С при перемешивании и медленно выпаривают всю загруженную воду. Через 3 часа после начала выпаривания начинается образование кристаллов, через 6 часов выпаривание воды завершается.
Недостатки описанного выше способа [10]:
1. Длительность процедуры: выдержка в течение 24 часов, затем продолжительная медленная выпарка воды. Для получения 129 г продукта реакционную массу объемом <1 л выпаривают 6 часов, при этом состав и качественные характеристики полученного осадка не обсуждаются.
2. Неравномерность по составу осадка, образующегося в процессе выпарки: в осадке, выпадающем в начале выпарки (по данным ЯМР и ИК-спектроскопии), присутствуют менее растворимые комплексы. Например, при синтезе комплекса Ca(Suc)3 вместе с ним сначала кристаллизуются и соосаждаются менее растворимые CaSuc, Ca(Suc)2, Ca(Suc)4, Ca(Suc)5, а при синтезе Ca(Suc)2 - CaSuc, Ca(Suc)4 и Ca(Suc)5.
3. В процессе предварительной фильтрации из реакционной смеси вместе с механическими примесями может быть частично удален один из реагентов. Вследствие даже небольшого изменения исходного соотношения реагентов неравномерность состава полученного продукта, описанная в вышеприведенном пункте 2, усугубляется.
Технической задачей, на решение которой направлено настоящее изобретение, является разработка более эффективного и промышленно применимого способа синтеза высокочистых комплексных кислых солей общей формулы: Ме(АсН)2⋅nH2O, (I), где Me - двухвалентный катион, Ас - анион дикарбоновой кислоты, Н - водород, n≥0. Необходимость синтеза комплексов конкретного состава обусловлена тем, что комплексы одинакового качественного состава, но различные по соотношению Ме:Ас, имеют отличающиеся физико-химические и биологические характеристики, а комплексные кислые соли, имеющие одинаковый состав, но разную степень гидратации, имеют различную фармакокинетику и разные способности к взаимодействию с другими компонентами лекарств или пищи.
Поставленная задача решается за счет того, что синтез ведется таким образом, чтобы в реакционном объеме сохранялось постоянное, равное исходному соотношение между всеми компонентами реакции.
Для этого в качестве «щелочного» агента реакции используют оксид двухвалентного металла, который разводят в воде непосредственно перед проведением реакции. Реакцию проводят при перемешивании таким образом, чтобы перемешивание реакционной смеси в реакторе происходило во всем его объеме наполнения, при этом режим скорости потока обеспечивает переходное или турбулентное движение жидкости.
На практике в соответствии с заявляемым техническим решением для получения соли с заданным соотношением Ме:Ас=1:2 в реактор вводят компоненты не в стехиометрическом соотношении 1:2, а при молярном соотношении МеО:AcH2=1:2,005-2,10. Снижение этого соотношения ниже 2,01 приводит к образованию средних солей, увеличение этого соотношения выше 2,1 приводит к появлению солей, у которых количество анионов дикарбоновой кислоты в структуре получаемых солей будет более двух. Таким образом, наиболее эффективным решением является обеспечение молярной концентрации дикарбоновой кислоты и оксида двухвалентного металла в водном растворе в соотношении 2,005-2,1:1.
В качестве оксидов металлов чаще всего применяют оксиды цинка, магния и кальция, в качестве дикарбоновых кислот - янтарную, фумаровую, L - аспарагиновую, L,D - аспарагиновую кислоты.
Заявляемый способ позволяет получать комплексные кислые соли двухвалентных металлов дикарбоновых кислот заданного состава с содержанием основного вещества более 95 масс. %. При этом полученный продукт может быть либо полностью обезвожен при сушке, либо иметь заранее заданное содержание воды.
Получаемые по заявляемому изобретению соли сами по себе обладают биологической активностью, а их комбинация делает чрезвычайно перспективными их применение для активации или регулирования целого ряда основных обменных процессов животных организмов. В сочетании с другими компонентами биологическая активность комплексных кислых солей по заявляемому изобретению становится необычно высокой и несопоставимой с другими веществами с известной целевой биологической активностью. При этом наблюдается выраженный синергетический эффект, который невозможно получить другими средствами.
Ниже дана расшифровка символов, приведенных в примерах:
SucH2 НООС-СН2-СН2-СООН - янтарная кислота
SucH НООС-СН2-СН2-СОО- - анион янтарной кислоты FumH2
НООС-СН=СН-СООН - фумаровая кислота FumH
НООС-СН=СН-СОО- - анион фумаровой кислоты
AspH2 HOOC-CH(NH2)-СН2-СООН - аспарагиновая кислота
AspH HOOC-CH(NH2)-CH2-COO- - анион аспарагиновой кислоты
Пример 1. Сравнительный
118 г янтарной кислоты и 25 г карбоната кальция растворяют в 600 мл воды при комнатной (24-25°С) температуре (концентрация соли 17,6% масс.). Раствор пропускают через бумажный фильтр и выдерживают 24 часа при комнатной температуре, затем медленно (в течение 15-20 минут) подогревают до 100°С и выпаривают при этой температуре в течение 6 часов.
Полученный осадок высушивают до постоянной массы. Выход (127,5 г) - 100% от теоретического.
Содержание Са (комплексонометрия) соответствует соотношению Са:Suc=1:4, что вполне объяснимо, так как исходные компоненты были загружены именно в таком соотношении, а вся вода из реакционной массы удалена. Однако по данным ЯМР (метка по углероду и водороду) получена смесь комплексных сукцинатных солей кальция, в которой около 84% - тетрасукцинат кальция, а остальные 16% - смесь, где присутствуют менее кислые сукцинаты кальция (CaSuc, CaSuc2, CaSuc3) и комплексные соли кальция с соотношением Са:Suc=1: 5 и выше.
Пример 2. Синтез кислого сукцината магния.
2SucH2+MgO+3H2O→Mg(SucH)2⋅4H2O
В реактор загружают 3,7 л воды, нагревают до 80-85°С и при перемешивании суспендируют 5,4 кг янтарной кислоты (5%-ный избыток от стехиометрии). При этой температуре получают суспензию 3,2-3,4 кг янтарной кислоты в насыщенном водном растворе ЯК (концентрация 41,2-44,5% масс.).
После выдержки для стабилизации системы в течение 30-40 мин порциями при постоянном перемешивании дозируют 925 г окиси магния (содержание основного вещества 95%). Перемешивание при 80-85°С продолжают до получения раствора комплексной соли - кислого сукцината магния (концентрация не менее 71% масс.), затем медленно, со скоростью менее 0,5°С в минуту, реакционную массу охлаждают до температуры (70-80°С), при которой начинается кристаллизация соли из насыщенного раствора и дозировка исходных компонентов.
Сушка при температуре 60-80°С на полочной сушилке обеспечивает получению безводной соли, а при сушке в потоке теплого (30-40°С) воздуха готовый продукт является тетрагидратом.
При сушке током теплого (менее 50°С) воздуха получают соль в виде четырехводного кристаллогидрата с содержанием основного вещества до 98,8% масс.(комплексонометрия). Содержание магния в данном эксперименте составило 7,27% масс.(атомно-абсорбционная спектрометрия).
Пример 3. Синтез кислого сукцината кальция.
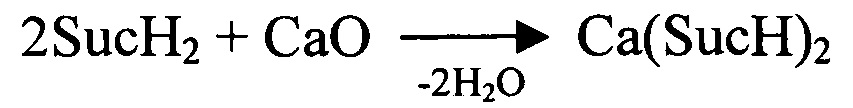
7,7 л воды подогревают до 90-95°С, при перемешивании растворяют 2,04 кг янтарной кислоты (3%-ный избыток от стехиометрии) и постепенно вводят в реакционную зону 577 г оксида кальция (содержание основного вещества 96-97%), добавляя каждую следующую порцию после растворения предыдущей. Полученный в результате раствор соли (концентрация не менее 22% масс.) охлаждают до температуры 79-80°С, при которой начинается кристаллизация осадка. Далее процесс ведут по Примеру 2.
Можно получить как безводную соль (сушка при 105-110°С), так и моногидрат (сушка током теплого воздуха или вакуумная сушка при 30-40°С и остаточном давлении 15-20 мм рт.ст). Содержание основного вещества - кислого сукцината кальция - не менее 97% (в описанном примере от 99,7% до 100%), содержание средней соли (CaSuc⋅H2O) - не более 3% масс. (в описанном примере средней соли не обнаружено).
Пример 4. Синтез кислого фумарата цинка.
2FumH2+ZnO→Zn(FumH)2⋅0,5H2O+0,5H2O
В 9,62 л воды, подогретой до 94-95°С при перемешивании, растворяют 299,8 г фумаровой кислоты (1%-ный избыток от стехиометрии). В раствор (концентрация не менее 3% масс.) фумаровой кислоты медленно дозируют 108 г окиси цинка, перемешивая постоянно до получения раствора соли, который затем медленно (со скоростью 0,8-1,0°С) охлаждают до начала кристаллизации (84-86°С). Далее синтез соли ведут при температуре 80°С, отделяя выпадающий осадок на фильтре, контролируя в реакционном объеме концентрацию фумарат-аниона (или катиона цинка) и поддерживая их постоянные величины (аналогично Примеру 2).
После вакуумной сушки (при 45-50°С, 15-20 мм рт.ст) получают соль состава Zn(FumH)2⋅0,5H2O с содержанием основного вещества до 98,5% масс. Содержание Zn в данном эксперименте составило 21,16% (рентгенофлуоресцентный анализ).
Пример 5. Синтез кислого аспарагината магния.
2AspH2+MgO+3H2O→Mg(AspH)2⋅4H2O
8,6 л воды подогревают до 90-95°С, загружают 1,3 кг (0,4-0,6% избыток от стехиометрического) аспарагиновой кислоты, перемешивают и получают суспензию (740-750 г) аспарагиновой кислоты в ее насыщенном водном растворе (концентрация 5-7%), куда при постоянном перемешивании дозируют 198,5 г окиси магния (содержание основного вещества не менее 97-99% масс.). После перемешивания получают раствор аспарагината магния (концентрация 16-18% масс.), который охлаждают до начала кристаллизации (65-70°С). Далее согласно Примеру 2.
После сушки в полочной сушилке при 80°С получают аспарагинат магния в виде четырехводного кристаллогидрата с содержанием основного вещества не менее 99,5% масс.
Пример 6. Синтез сукцината кальция кислого дигидрата
2SucH2+CaO+H2O→Ca(SucH)2⋅2H2O
В реактор загружают 640 л воды и нагревают ее до 70-75°С, после чего загружают порциями 480 кг 100%-ной янтарной кислоты (исходя из мольного соотношения ЯК : СаО=2,1:1) и продолжают нагрев до 80-85°С до ее полного растворения.
Медленно и равномерно тонкой струйкой дозируют при перемешивании водную суспензию гидроксида кальция, полученного гашением 108,5 кг оксида кальция в 385 л воды в рабочей, предпочтительно эмалированной, емкости.
Реакционную массу выдерживают в реакторе при перемешивании и температуре от 85 до 90°С в течение 1 часа для завершения реакции.
По окончании выдержки нагрев отключают и при непрерывном перемешивании охлаждают реакционную массу в два этапа: сначала до 65-75°С (самоохлаждением), затем до 16-18°С путем подачи в рубашку реактора холодной воды. При этом кристаллизуется сукцинат кальция кислый в виде двухводного кристаллогидрата.
По завершении кристаллизации охлажденную суспензию фильтруют в несколько приемов порциями по 130-160 л. Весь выгруженный влажный осадок дополнительно отжимают от маточного раствора на центрифуге до остаточной влажности 5-7% масс, после чего направляют на сушку.
Высушивание влажного осадка соли осуществляют в сушильном шкафу при температуре от 40 до 50°С с получением сукцината кальция кислого в виде двухводного кристаллогидрата.
Общее количество сухого продукта - сукцината кальция кислого двухводного, - получаемого с одной операции синтеза составляет от 485 до 506 кг.
Пример 7. Моногидрат фумарата цинка кислого.
2FumH2+ZnO→Zn(FumH)2⋅H2O
В реактор загружают 13 л воды и при постоянном нагреве и перемешивании суспендируют 11,9 кг фумаровой кислоты. Суспензию нагревают до температуры в 60-65°С, после чего дозируют приготовленную непосредственно перед дозировкой в отдельной емкости суспензию 4,17 кг окиси цинка в 10 л воды. После этого реакционную массу выдерживают в течение 2 часов при перемешивании (с одновременной частичной упаркой), продолжая ее нагрев до температуры в 80-90°С.
По завершении выдержки нагрев отключают и ведут охлаждение реакционной массы до температуры в 60-65°С при перемешивании, а затем ее выгружают в кристаллизатор и охлаждают до температуры в 16-18°С.
Образовавшуюся суспензию фильтруют порциями, осадок отжимают и получают от 18 до 20 кг осадка с влажностью не более 30-35% масс. Влажный осадок сушат в потоке теплого (с температурой от 40 до 50°С) воздуха. Получают от 14 до 15 кг сухого продукта с содержанием основного вещества 99,2% (содержание цинка 20,86%)
Пример 8. Тестирование включения анионов дикарбоновых кислот, входящих в полученные соединения, в обменные процессы.
Для проведения исследования по примерам 2-7 были синтезированы соединения, в которых вместо анионов обычных янтарной, аспарагиновой и фумаровой кислот были использованы меченные по одному атому 13С кислоты. Достоверным сигналом включения их в обменные процессы служит выход меченого атома 13С в дыхании в виде 13CO2. Эксперимент проводили следующим образом: группам крыс (в каждой по 8 особей весом от 225 до 250 г) с помощью пипетки вводили водные растворы в количестве, эквивалентном 1 мг содержащихся в них анионов кислот, следующих соединений: янтарная кислота, фумарат натрия (фумаровая кислота практически нерастворима в воде), аспарагиновая кислота, Ca(SucH)2, Mg(SucH)2⋅4H2O, Mg(AspH)2⋅4H2O, Zn(FumH)2⋅0,5Н2О, Zn(FumH)2⋅H2O. Затем крысу быстро помещали в герметичную камеру, воздух из которой отбирается кратно подаче воздуха дозирующим насосом в камеру. Отобранный в течение 12 часов после введения пробы воздух анализировали на содержание 13С в выделенном CO2, при этом фоновой концентрацией пренебрегали, так как она не превышает 0,1%. Далее определяли массовый процент 13С, выделенного в дыхании, к введенному. Получены следующие результаты: янтарная кислота - 99,4%, фумарат натрия - 97,8%, аспарагиновая кислота - 59,4%, Ca(SucH)2 - 49,7%, Mg(SucH)2⋅4H2O - 47,7%, Mg(AspH)2⋅4H2O - 47,4%, Zn(FumH)2⋅0,5Н2О - 43,5%, Zn(FumH)2⋅H2O - 41,4%. Как видно из полученных результатов, анионы всех тестированных соединений активно включаются во внутриклеточные обменные процессы.
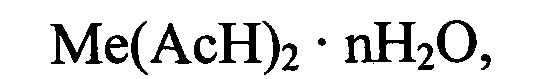
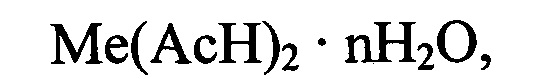
Способ получения аммонийных солей фумаровой или янтарной кислоты
Средство и способы культивирования, хранения и криоконсервации стволовых и дифференцированных клеток человека и животных
Антиостеопорозное средство
Способ получения аммонийных солей фумаровой или янтарной кислоты
Средство и способы культивирования, хранения и криоконсервации стволовых и дифференцированных клеток человека и животных
Антиостеопорозное средство
Средство для мезотерапии
1,20-дибром-3,6,9,12,15,18-гексаоксаперфтор-4,7,10,11,14,17-гексаметилэйкозан в качестве исходного соединения для эмульсий медико-биологического назначения и способ его получения
2-бромтетрафторэтилперфторалкиловые эфиры для получения эмульсий медико-биологического назначения
Α-бром-ω-галогенперфторполиэфиры в качестве основы газотранспортных композиций медико-биологического назначения
Α,ω-дибромполиоксаперфторалканы, способ их получения и эмульсии медико-биологического назначения на их основе

