Результат интеллектуальной деятельности: СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ ИНГИБИТОРОВ ПРОТЕАЗЫ HCV
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к синтезу и промежуточным соединениям для синтеза макроциклических ингибиторов протеазы вируса гепатита С (HCV).
Уровень техники
Вирус гепатита С (HCV) является главной причиной хронического гепатита, который может развиться в фиброз печени, приводящий к циррозу, конечной стадии заболевания печени, и к НСС (гепатоцеллюлярная карцинома), что делает его главной причиной пересадки печени. Современная анти-HCV терапия, основанная на пегилированном интерфероне-альфа (IFN-α) в комбинации с рибавирином, имеет ограниченную эффективность, значительные побочные эффекты и плохо переносится многими пациентами. Это побуждает к поиску более эффективного, более удобного и лучше переносимого лечения.
Репликация генома HCV опосредуется рядом ферментов, среди которых находится сериновая протеаза NS3 HCV и ее ассоциированный кофактор, NS4A. Описаны различные средства, которые ингибируют этот фермент. Публикация WO 05/073195 раскрывает линейные и макроциклические ингибиторы сериновой протеазы NS3 с центральным замещенным пролиновым остатком, а публикация WO 05/073216 - с центральным циклопентановым остатком. Среди них макроциклические производные являются привлекательными из-за их заметной активности против HCV и привлекательного фармакокинетического профиля.
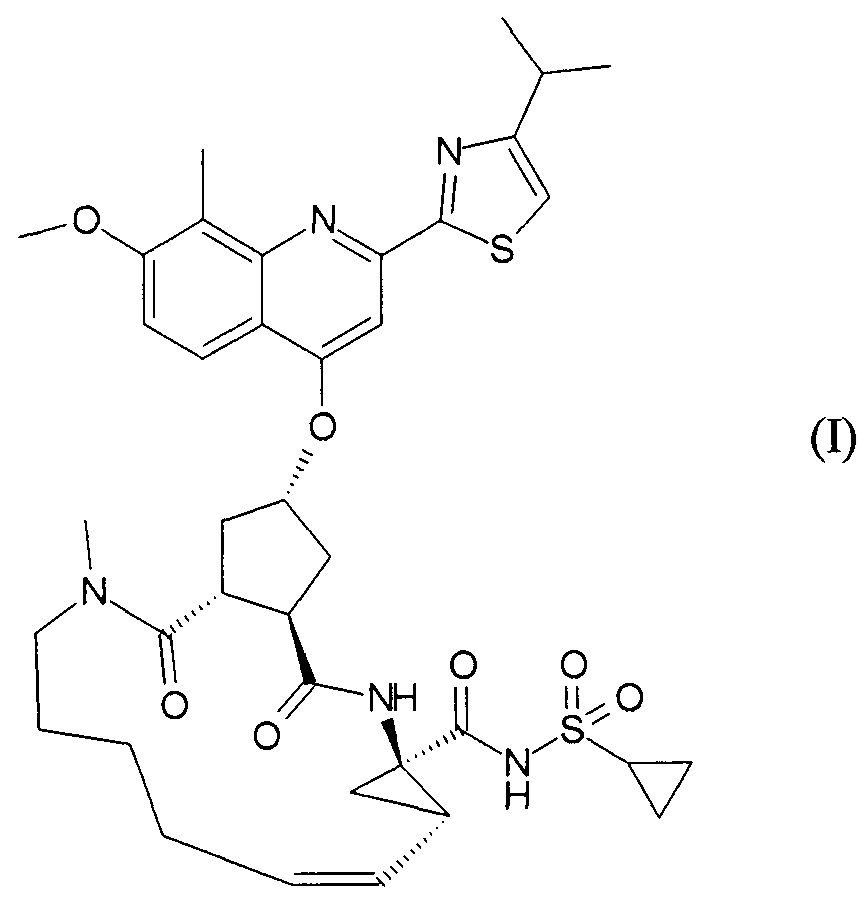
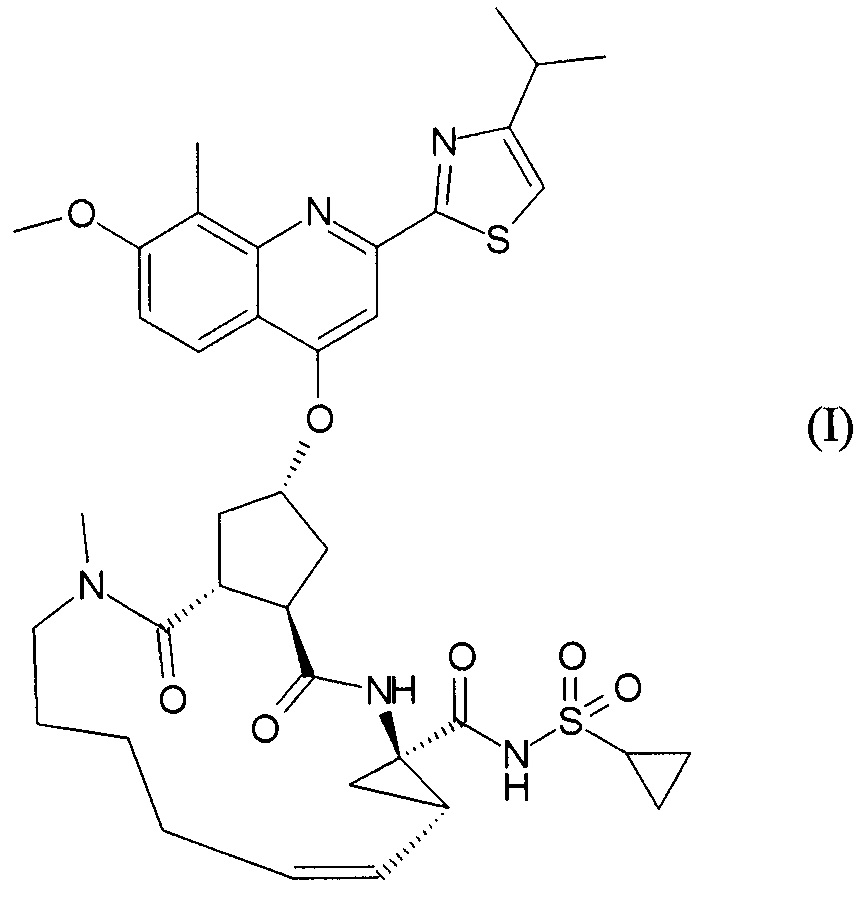
Публикация WO 2007/014926 описывает макроциклические циклопентановые и пролиновые производные, в том числе соединение формулы I, со структурой, представленной далее. Соединение формулы I является очень эффективным ингибитором сериновой протеазы HCV, и особенно привлекательно с точки зрения фармакокинетики. Благодаря своим благоприятным свойствам оно было выбрано в качестве потенциального кандидата для разработки лекарственного средства против HCV. Соответственно существует необходимость производства больших количеств такого активного ингредиента на основе способов, которые обеспечивают продукт с высоким выходом и с высокой степенью чистоты. В публикации WO 2008/092955 описаны способы и промежуточные соединения для получения соединения формулы I.

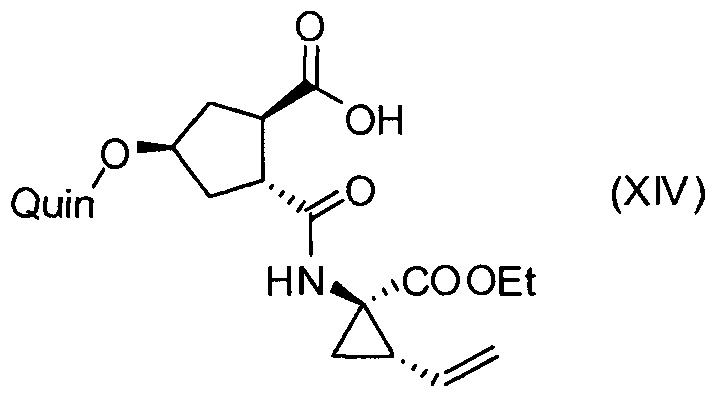
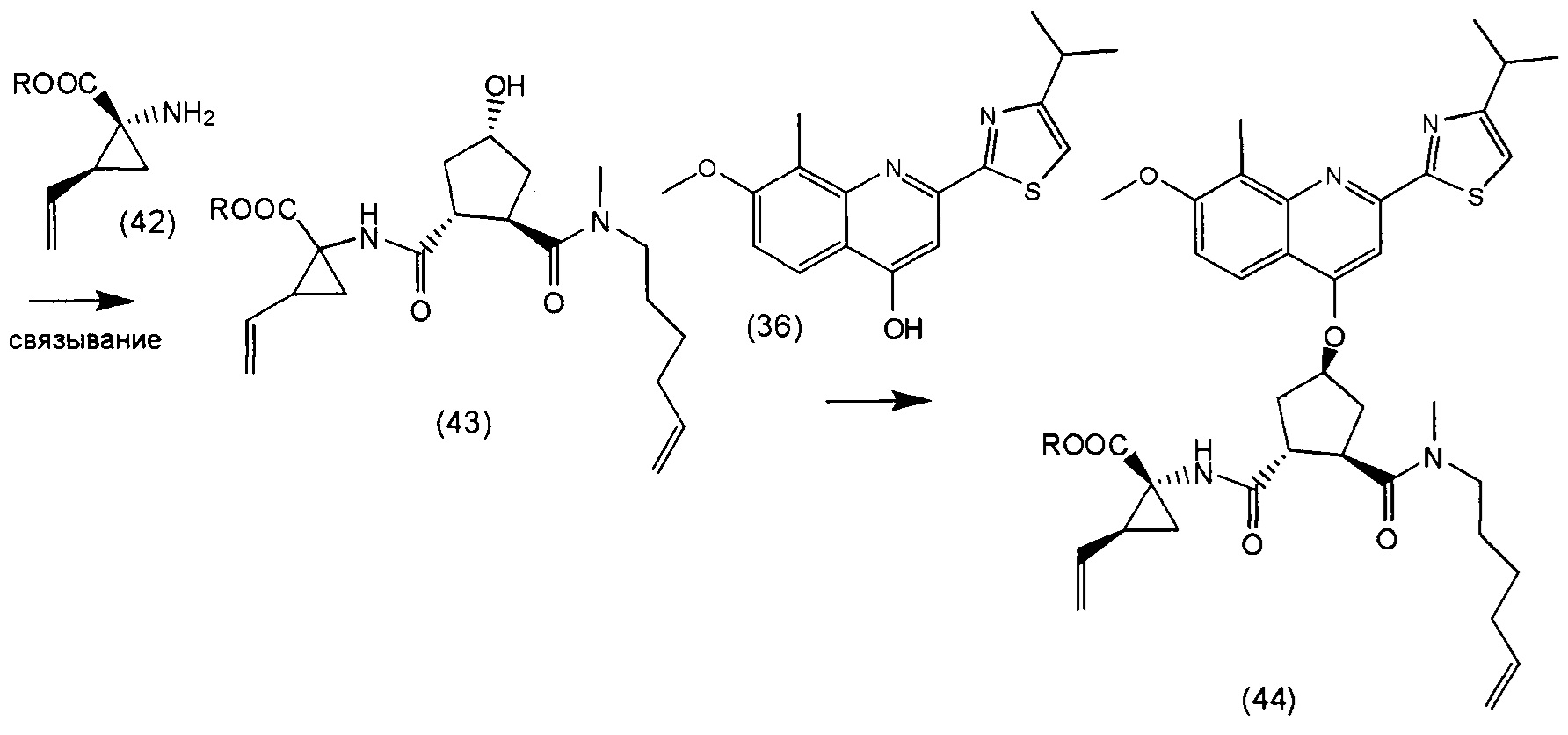
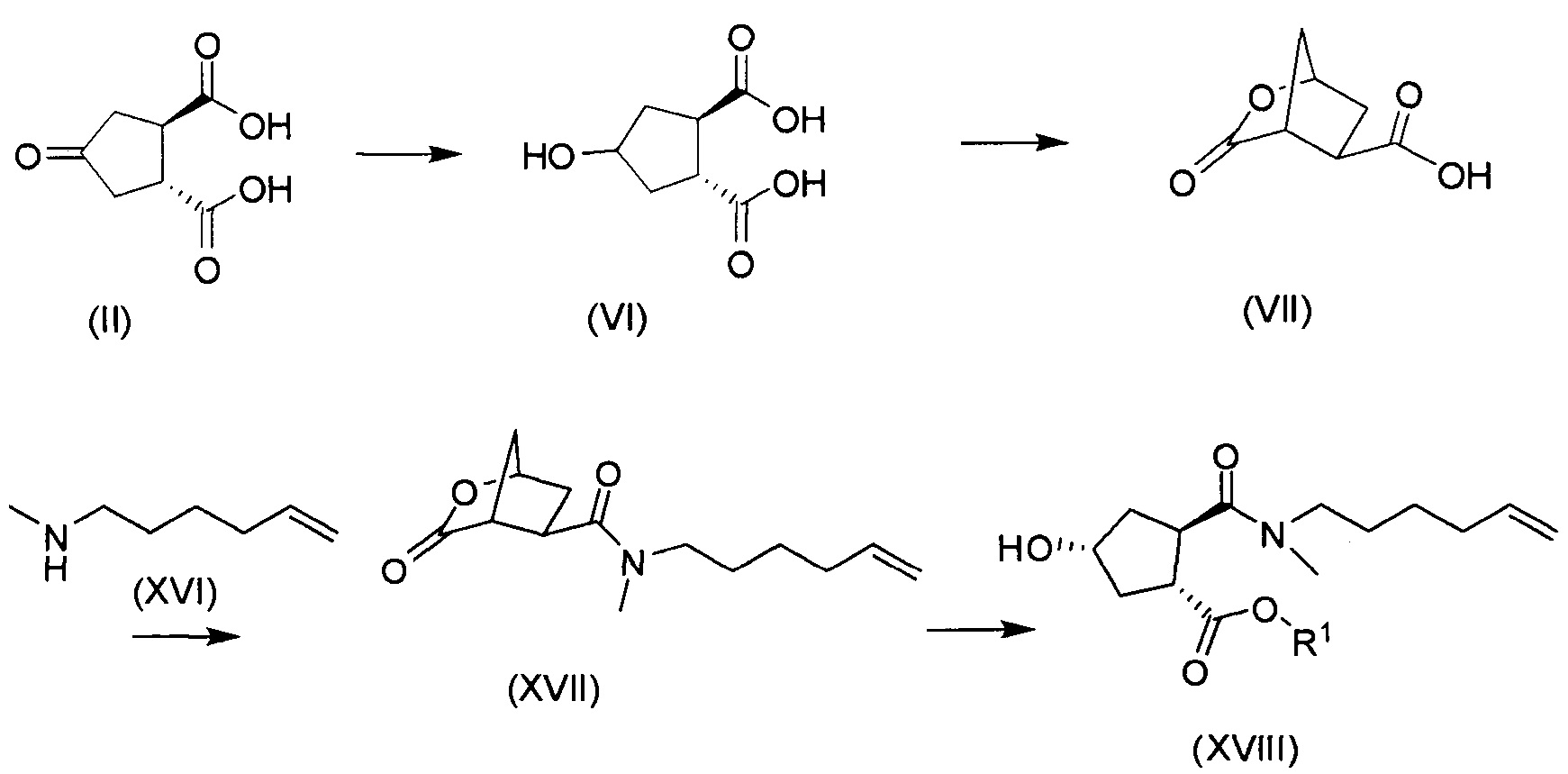
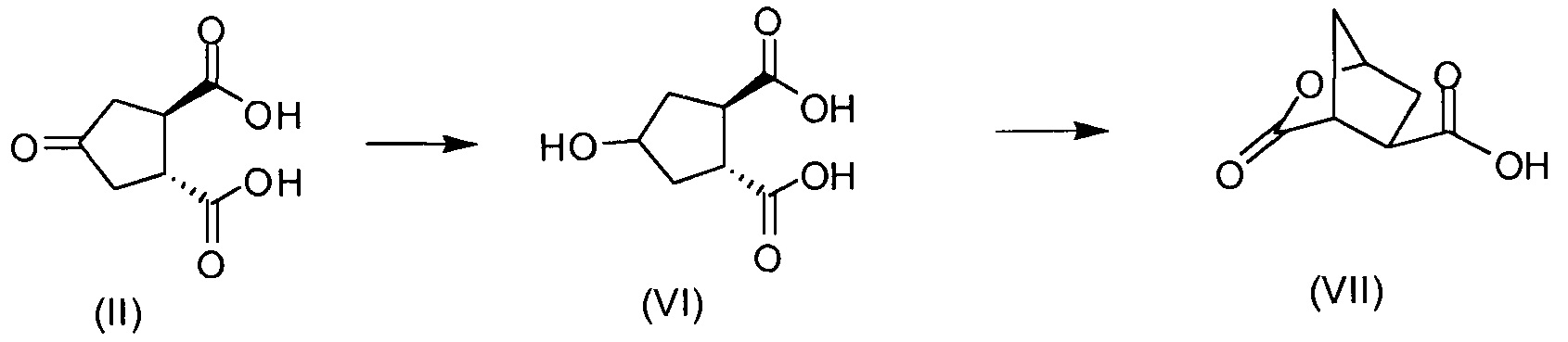
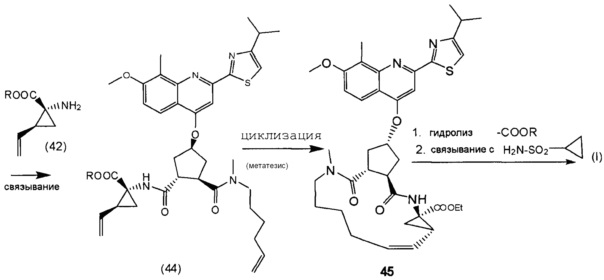
В соответствии с публикацией WO 2007/014926 соединение формулы I может быть получено исходя из бициклического лактона карбоновой кислоты, обозначенного как соединение 39 в примере 4, или в общем описании этой ссылки как соединение 17b, или как соединение VII в настоящем описании и в формуле изобретения. Карбоновую кислоту в бициклическом лактоне карбоновой кислоты сочетают с N-метилгекс-5-ениламином 38, после чего следует раскрытие лактона до 4-гидроксициклопентанового производного 41. Последнее производное 41 затем сочетают с аминоциклопропилкарбоксильным эфиром до диамида циклопентандикарбоновой кислоты 43, который сочетают с хинолином 36 по реакции образования простого эфира Мицуноби (Mitsunobu), которая включает инверсию у несущего гидрокси-группу атома углерода. Полученное промежуточное соединение 44 подвергают циклизации по реакции метатезиса до макроциклического производного, в котором сложноэфирную группу гидролизуют и сочетают с циклопропилсульфониламидом, получая целевой конечный продукт формулы I. Такие реакции проиллюстрированы на приведенной ниже схеме, в которой R представляет собой С1-4-алкил, а в примере 4 заместитель R представляет собой этил.


Энантиомерно чистый бициклический лактон 39 получают исходя из энантиомера 3,4-бис(метоксикарбонил)циклопентанона, обозначенного как (17а) в публикации WO 2007/014926. Последний получают, как описано в публикации Rosenquist el al., Acta Chemica Scandinavica 46 (1992), 1127-1129. Рацемический метиловый эфир циклогександикарбоновой кислоты синтезируют по реакции Дильса-Альдера 3-сульфолена и диметилфумарата, после чего следует окислительное расщепление двойной связи, циклизация и декарбоксилирование, приводящие к диметиловому эфиру (±)-4-кетоциклопентандикарбоновой кислоты. Разрешение последнего путем гидролиза с использованием эстеразы свиной печени приводит к соответствующей (+)-монокислоте и (-)-диэфиру, который представляет собой промежуточное соединение (17а) публикации WO 2007/014926.

После удаления (+)-монокислоты диэфир транс-(3R,4R)-3,4-бис(метоксикарбонил)циклопентанона (17а) превращают в бициклический лактон 17b (также обозначаемый как соединение VII, см. выше), вначале путем восстановления кето-группы в спирт, после чего следует гидролиз сложных эфиров и образование лактона.
Методика синтеза для получения соединения I, описанная в публикации WO 2008/092955, начинается с промежуточного соединения D, где сложноэфирную группу гидролизуют и сочетают со сложным эфиром циклопропиламинокислоты С. Полученное промежуточное соединение В циклизуют по реакции олефинового метатезиса до макроциклического сложного эфира А, который гидролизуют и сочетают с циклопропилсульфониламидом до конечного продукта I. Такие реакции представлены в общих чертах на приведенной ниже реакционной схеме. В этой и в следующих реакционных схемах R представляет собой С1-4-алкил, в частности, R представляет собой этил. R1 представляет собой С1-4-алкил, в частности, R1 представляет собой метил или этил.


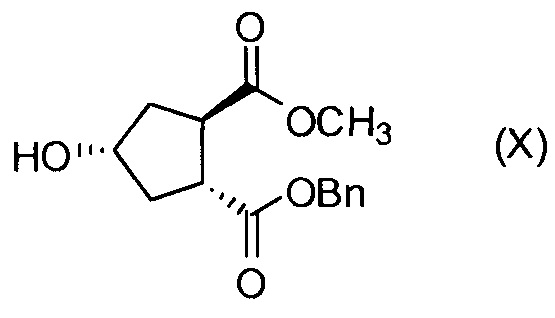
Промежуточное соединение D, в свою очередь, получают, начиная с гидроксициклопентилового бис-эфира формулы Н1, или:
(а) взаимодействием Н1 с тиазолил-замещенным хинолинолом Е с получением хинолинилоксициклопентилового бис-эфира формулы К, после чего следует расщепление группы бензилового эфира до монокарбоновой кислоты J, которую, в свою очередь, сочетают с N-метилгексенамином с образованием промежуточного соединения D; или
(b) расщеплением бензилового эфира в соединении Н1 до монокарбоновой кислоты G, сочетанием последней с N-метилгексенамином с получением гидроксициклопентиламида F, который, в свою очередь, вводят в реакцию с соединением E, получая в результате соединение D; что показано в общих чертах на приведенной ниже реакционной схеме:

Каждый R1 на схеме принимает определенные выше значения, и Bn означает бензил.
Публикация WO 2008/092955 также описывает методики получения промежуточного соединения Н1, начиная с 4-оксо-1,2-циклопентандикарбоновой кислоты О, путем восстановления кето-группы в спиртовую группу, в результате получают 4-гидрокси-1,2-циклопентандикарбоновую кислоту N, которую, в свою очередь, циклизуют до бициклического лактона М. Этерификация карбоксильной группы в последнем дает бензиловый эфир лактона L, где лактон раскрывают реакцией переэтерификации в присутствии С1-4-алканола, получая в результате промежуточное соединение Н, которое разрешают по его энантиомерам Н1 и Н2; что показано в общих чертах на приведенной ниже реакционной схеме:

Недостаток описанного выше способа состоит в том, что он включает разрешение энантиомеров соединения Н с помощью хиральной колоночной хроматографии, обременительной процедуры, которую трудно проводить при крупномасштабном производстве. Другой недостаток состоит в том, что разрешение имеет место на последней стадии синтеза, в результате чего половина структурного блока соединения Н выбрасывается. Присутствие различных хиральных центров в соединении формулы I и его предшественниках представляет собой особую проблему в том, что энантиомерная чистота является существенной для того, чтобы иметь продукт, который приемлем для терапевтического применения. Следовательно, способы получения соединения D должны приводить к продуктам приемлемой энантиомерной чистоты без применения обременительных методик с потерей значительных количеств нецелевых стереоизомерных форм.
В публикации Honda et al., Tetrahedron Letters, Vol. 22, № 28, pp. 2679-2682, 1981, описан синтез (±)-брефелдина А, с использованием следующих исходных материалов:

Синтез Honda с соавторами начинается от dl-транс-4-оксо-циклопентан-1,2-дикарбоновой кислоты 2, которую этерифицируют до соответствующего метилового эфира 3 и восстанавливают с помощью никеля Ренея до спирта 4. Частичный гидролиз соединения 4 до монокарбоновой кислоты и бензилирование с помощью бензилбромида дает преимущественно диастереоизомер 5, а именно диастереоизомер, где гидрокси-группа и бензильная сложноэфирная группа находятся в цис-положении. Последний сложный эфир 5 в публикации Honda с соавторами и соединение Н, оба, являются рацематами, но являются диастереоизомерами друг друга, более точно эпимерами по атому углерода № 4, несущему гидрокси-группу. Соединение Н1 является одним из двух энантиомеров, полученных путем отделения от рацемического соединения Н. Другим энантиомером является соединение Н2.
Бициклический лактон (17b) представляет собой значимый структурный блок в синтезе соединения формулы I. Обнаружение пути синтеза для получения этого лактона с хорошим выходом и высокой энантиомерной чистотой является желаемой для достижения целью. Настоящее изобретение предлагает способ получения (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты, которая может быть легко превращена в бициклический лактон (17b).
Способы настоящего изобретения имеют преимущество в том, что они приемлемы для крупномасштабного производства. Обременительные стадии очистки, в особенности с помощью хроматографии, исключены.
Описание изобретения
Обзор структур, описанных в данном описании и в формуле изобретения.
|



|


 идентично с
идентично с 

 идентично с
идентично с 



|





 идентично с
идентично с 

|




|




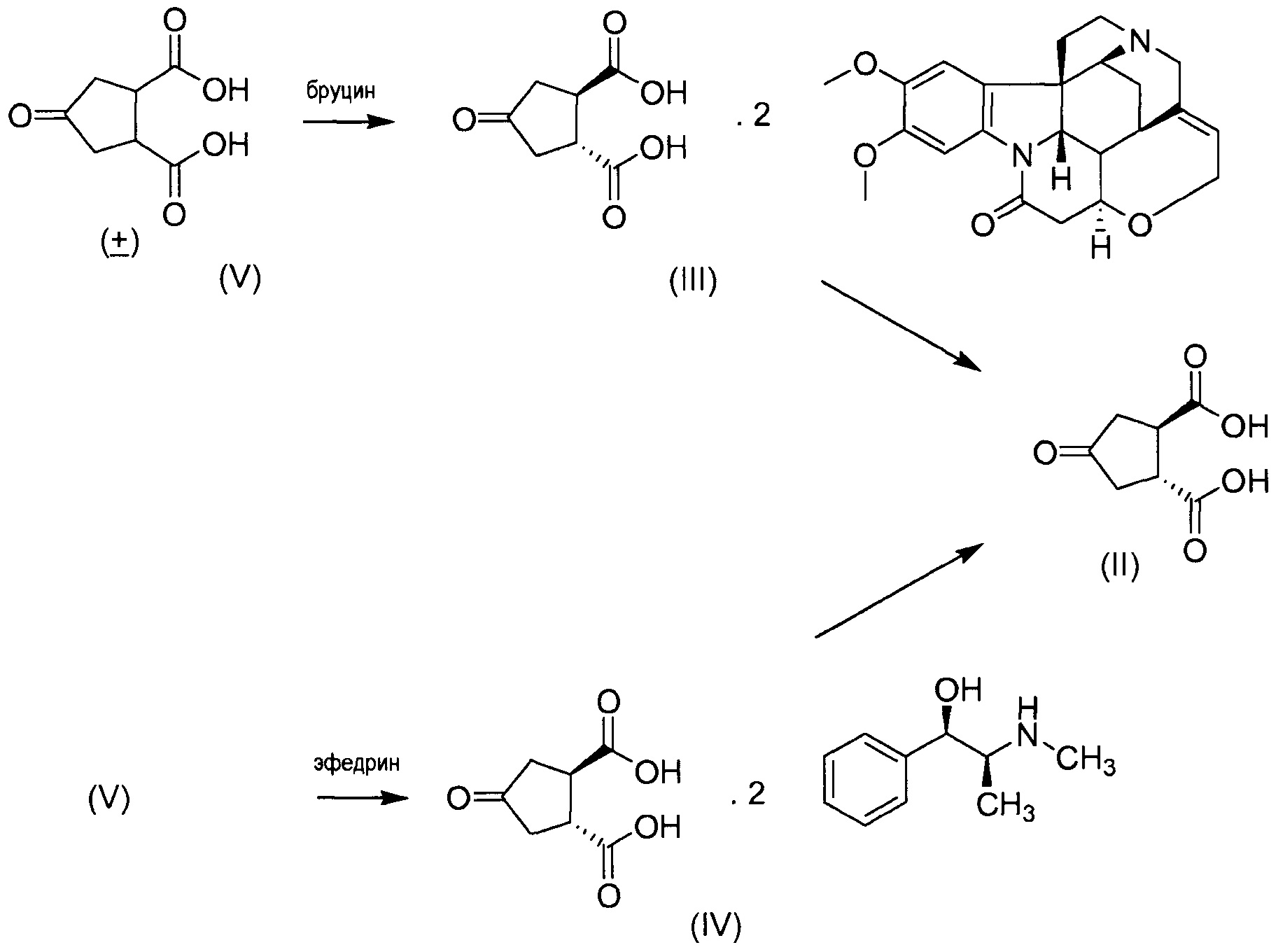
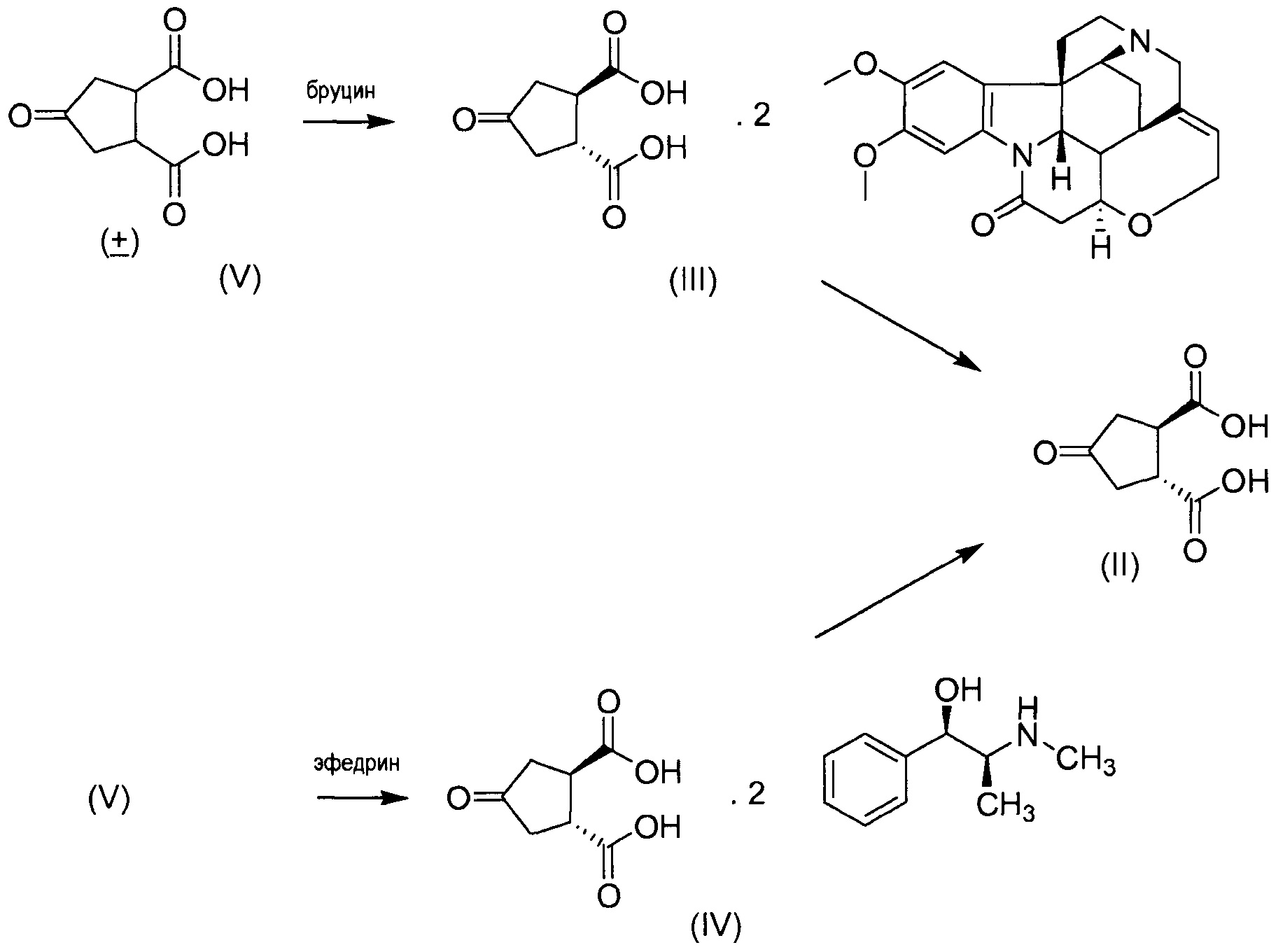
В одном аспекте настоящее изобретение относится к способу получения (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты II путем разрешения рацемической 4-оксо-1,2-циклопентандикарбоновой кислоты (V), причем указанный способ включает:
(а) взаимодействие 4-оксо-1,2-циклопентандикарбоновой кислоты (V) с бруцином или (1R,2S)-(-)-эфедрином, с получением в результате бис-бруциновой или бис-(1R,2S)-(-)-эфедриновой соли (V), и
(b) селективное осаждение бис-бруциновой или бис-(1R,2S)-(-)-эфедриновой соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты II, тогда как бис-бруциновая или бис-(1R,2S)-(-)-эфедриновая соль (1S,2S)-4-оксо-1,2-циклопентандикарбоновой кислоты остается в растворе;
(с) высвобождение кислоты II путем удаления бруцина или (1R,2S)-(-)-эфедрина из осажденной соли, полученной на стадии (b).
Этот способ в общих чертах представлен на приведенной ниже реакционной схеме:

Изобретение также относится к промежуточному соединению соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с бруцином (1:2), имеющей структуру III, и соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с (1R,2S)-(-)-эфедрином (1:2), имеющей структуру IV.
Бруциновая соль III может быть получена путем смешения бруцина и транс-4-оксо-1,2-циклопентандикарбоновой кислоты в присутствии растворителя, нагревания смеси до тех пор, пока все твердые вещества не растворяться, и обеспечения возможности смеси охладиться, в результате чего бруциновая соль кристаллизуется.
Растворителями являются спирты, такие как метанол или этанол, и водные спирты, такие как водный метанол или этанол. Интерес представляют смеси спирт/вода с небольшим количеством воды, например, с содержанием воды в интервале от приблизительно 2 до приблизительно 20%, или от приблизительно 5 до приблизительно 10% (масс./масс.). В частности, можно использовать смесь вода/метанол с содержанием воды в интервале от приблизительно 5 до приблизительно 10%, например, приблизительно 5% (масс./масс.). В одном варианте осуществления готовят смесь водного спирта и бруцина и слегка нагревают до температуры в интервале от приблизительно 30 до приблизительно 50°С, например, до приблизительно 40°С, после чего добавляют раствор транс-4-оксо-1,2-циклопентандикарбоновой кислоты. Полученную смесь нагревают до тех пор, пока все твердые вещества не растворяться, в частности, путем нагревания при температуре кипения с обратным холодильником. Затем смеси дают охладиться, предпочтительно медленно, до комнатной температуры. Образовавшиеся твердые вещества отфильтровывают. Они могут быть перекристаллизованы, например, из воды.
Аналогично может быть получена (1R,2S)-(-)-эфедриновая соль IV, но вместо водных спиртов могут быть использованы водные кетоны, такие как водный ацетон. Интерес представляют смеси ацетон/вода с небольшим количеством воды, например, с содержанием воды в интервале от приблизительно 2 до приблизительно 25%, или от приблизительно 10 до приблизительно 20% (масс./масс.), например, приблизительно 16% (масс./масс.).
Разрешение может быть осуществлено с помощью (1S,2R)-(+)-эфедрина с получением (1S,2R)-(+)-эфедриновой соли (1S,2S)-4-оксо-1,2-циклопентандикарбоновой кислоты (2:1) в виде белого твердого вещества.
Разрешение транс-4-оксо-1,2-циклопентандикарбоновой кислоты также предпринимали с помощью цинхонидина с получением цинхонидиновой соли (1S,2S)-4-оксо-1,2-циклопентандикарбоновой кислоты (1:2), выделяемой в виде твердого вещества.
Разрешение также предпринимали с помощью цинхонина, однако соль, в ее рацемической форме, может быть выделена только в виде стекловидного твердого вещества.
Разрешение также предпринимали с помощью никотина, и снова неуспешно, так как соль транс-4-оксо-1,2-циклопентандикарбоновой кислоты была выделена в виде масла.
Бруциновая и (1R,2S)-(-)-эфедриновая соль (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты может быть превращена в свободную кислоту формулы II. На первой стадии бруцин и (1R,1S)-(-)-эфедрин удаляют обработкой водных суспензий соли основанием, например, гидроксидом аммония. Это предпочтительно проводят при повышенной температуре, например, при температуре, которая находится в интервале от 60 до 100°С, например, при 80°С. Охлаждение до комнатной температуры приводит к кристаллизации твердого бруцина, который может быть выделен фильтрованием. Обработка оставшегося раствора может быть проведена упариванием досуха и повторным растворением остатка в воде. К этому раствору добавляют кислоту, например, HCl, получая при осаждении (1R,2R)-4-оксо-1,2-циклопентандикарбоновую кислоту. Полученное твердое вещество может быть отфильтровано и промыто холодной водой.
Также может быть получена и выделена бис-соль щелочного металла.
Выделенный бруцин может быть высушен и перекристаллизован из смеси вода-этанол (50:50), чтобы очистить бруцин, который может быть использован повторно при разрешении.
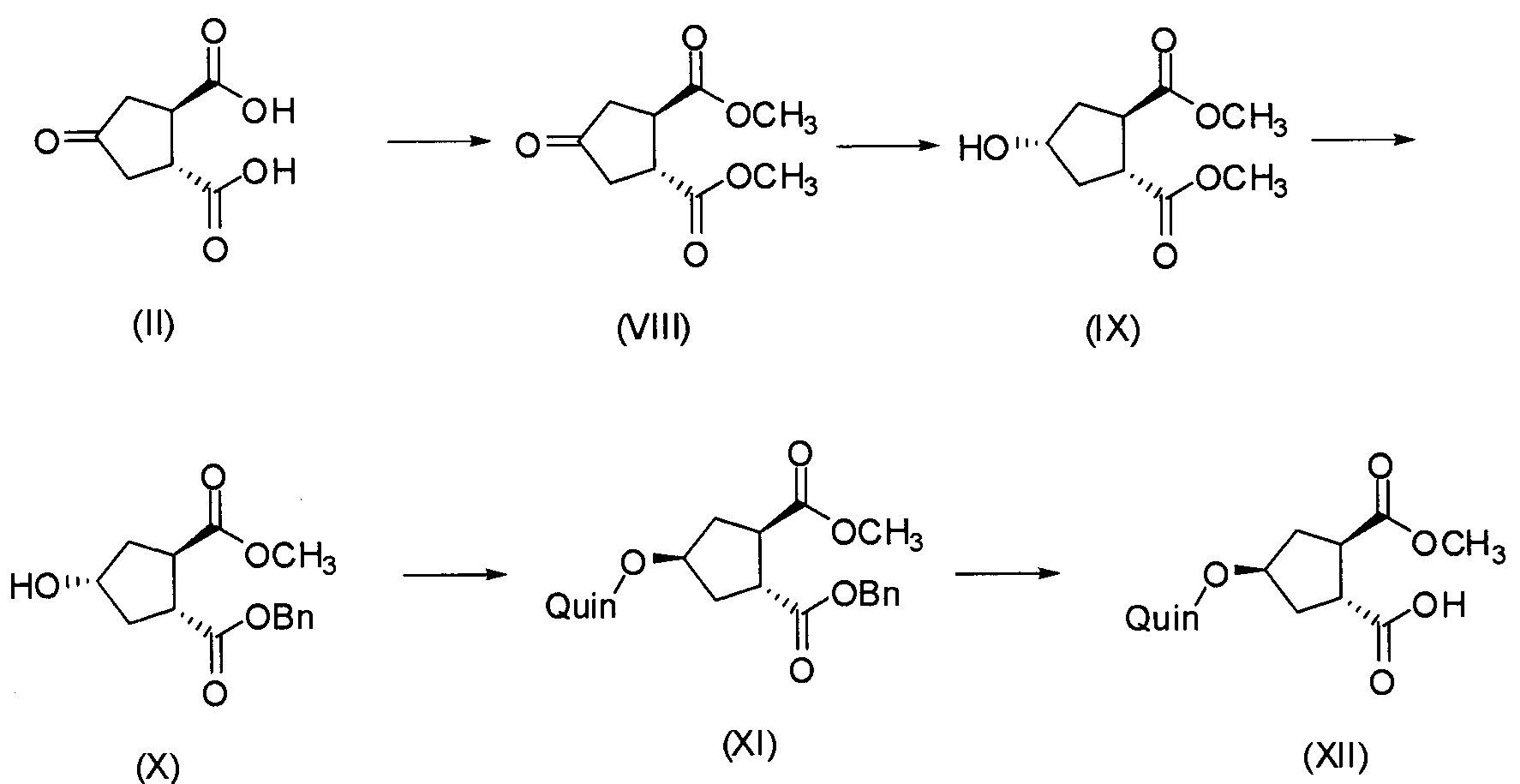
В другом аспекте (R,R)-4-оксо-1,2-циклопентандикарбоновую кислоту II, полученную описанным выше способом, или бруциновую соль III, или (1R,2S)-(-)-эфедриновую соль (IV) используют в качестве исходного материала в способе получения бициклического лактона (VII) путем восстановления кето-группы до спиртовой группы, получая 4-гидрокси-1,2-циклопентандикарбоновую кислоту (VI), которую циклизуют до лактона (VII).

Рацемическая 4-оксо-1,2-циклопентандикарбоновая кислота V в качестве исходного материала может быть получена, как описано выше в разделе изобретения «Уровень техники». Восстановление кето-группы в гидрокси-группу, соединения II до соединения VI, может быть осуществлено с использованием подходящего восстановителя, в частности, с помощью водорода в присутствии металлического катализатора, например, родия на угле или на оксиде алюминия или никеля Ренея, в инертном растворителе, например, в водной среде, такой как вода, в присутствии основания, например, NaOH, KOH, или органического основания, такого как триэтиламин, N-метилморфолин или основание Ханига (диизопропилэтиламин).
Полученная 4-гидроксициклопентан-1,2-дикарбоновая кислота VI может быть превращена в соль, например, бис-соль третичного амина, такую как бис(триэтиламинная) соль, или бис-соль щелочного металла, такую как бис-натриевая или бис-калиевая соли.
Промежуточное соединение VI может быть подвергнуто циклизации с образованием лактона VII по реакции с хлорформиатом, например, этил- или метилхлорформиатом. Эту реакцию можно провести в инертном для реакции растворителе, таком как кетон, в особенности ацетон, или простой эфир, такой как ТГФ, или MeТГФ, или ацетонитрил. Может быть добавлено основание, например, третичный амин, такой как триэтиламин или N-метилморфолин (NMM). В альтернативном варианте осуществления лактон-образующим агентом является 2,4,6-трихлор-1,3,5-триазин (TCT) или его производное.
В конкретном варианте осуществления промежуточное соединение II превращают в 4-гидрокси-1,2-циклопентандикарбоновую кислоту VI по реакции восстановления, описанной выше, которая циклизует лактон VII с использованием триазинового производного, по методике «одного горшка» без выделения промежуточных продуктов. Соединение VI получают в воде после стадии восстановления, к которой может быть добавлен органический со-растворитель на второй стадии, например, ацетон, метилэтилкетон (МЕК), тетрагидрофуран (ТГФ) или 2-метилтетрагидрофуран (МеТГФ). Триазиновые производные для этой реакции включают такие агенты, как 2,4,6-трихлор-1,3,5-триазин (TCT), хлордиметокситриазин (CDMT), N-(3,5-диметокситриазинил)-N-метилморфолинийхлорид (DMTMM) или дихлорметокситриазин (DCMT). Такая реакционная последовательность дает простую, короткую и экономичную методику получения лактона VII с высоким выходом и высокой чистотой. Воду, используемую в качестве растворителя на стадии восстановления, нет необходимости удалять и никакого отделения промежуточного соединения 4-гидрокси-1,2-циклопентандикарбоновой кислоты VI не требуется.
Бруциновая и (1R,2S)-(-)-эфедриновая соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты III или IV могут быть превращены непосредственно в диметиловый эфир (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты. На первой стадии удаляют бруцин и (1R,2S)-(-)-эфедрин, как описано выше, и выделяют твердый бруцин и (1R,2S)-(-)-эфедрин. Маточные жидкости после выделения бруцина и (-)-эфедрина упаривают с получением энантиомерно чистой (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты. Она может быть использована для получения соединения I, как описано в публикации WO 2007/014926. Таким образом, остаток, который получен после упаривания, забирают в смесь метанол/толуол и добавляют сильную кислоту, например, серную кислоту. Реакционную смесь нагревают, предпочтительно, до кипения с обратным холодильником, после чего растворитель отгоняют из реакционной смеси, пока не будет достигнута внутренняя температура >70°С. Затем смесь охлаждают приблизительно до 30°С и добавляют воду. Полученную смесь перемешивают при комнатной температуре и дополнительно обрабатывают, выделяя образовавшийся органический слой. Упаривание дает целевой диметиловый эфир.
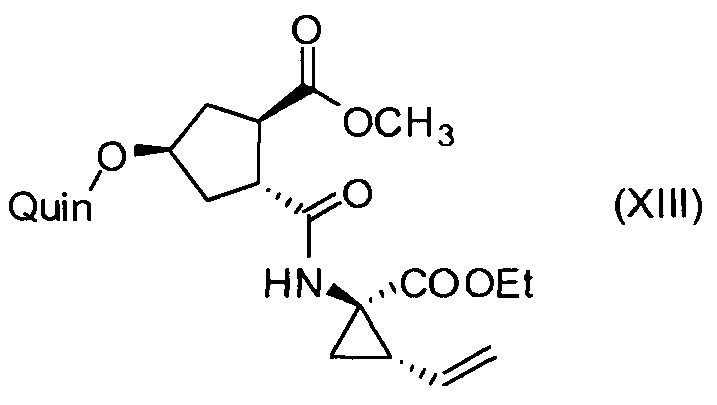
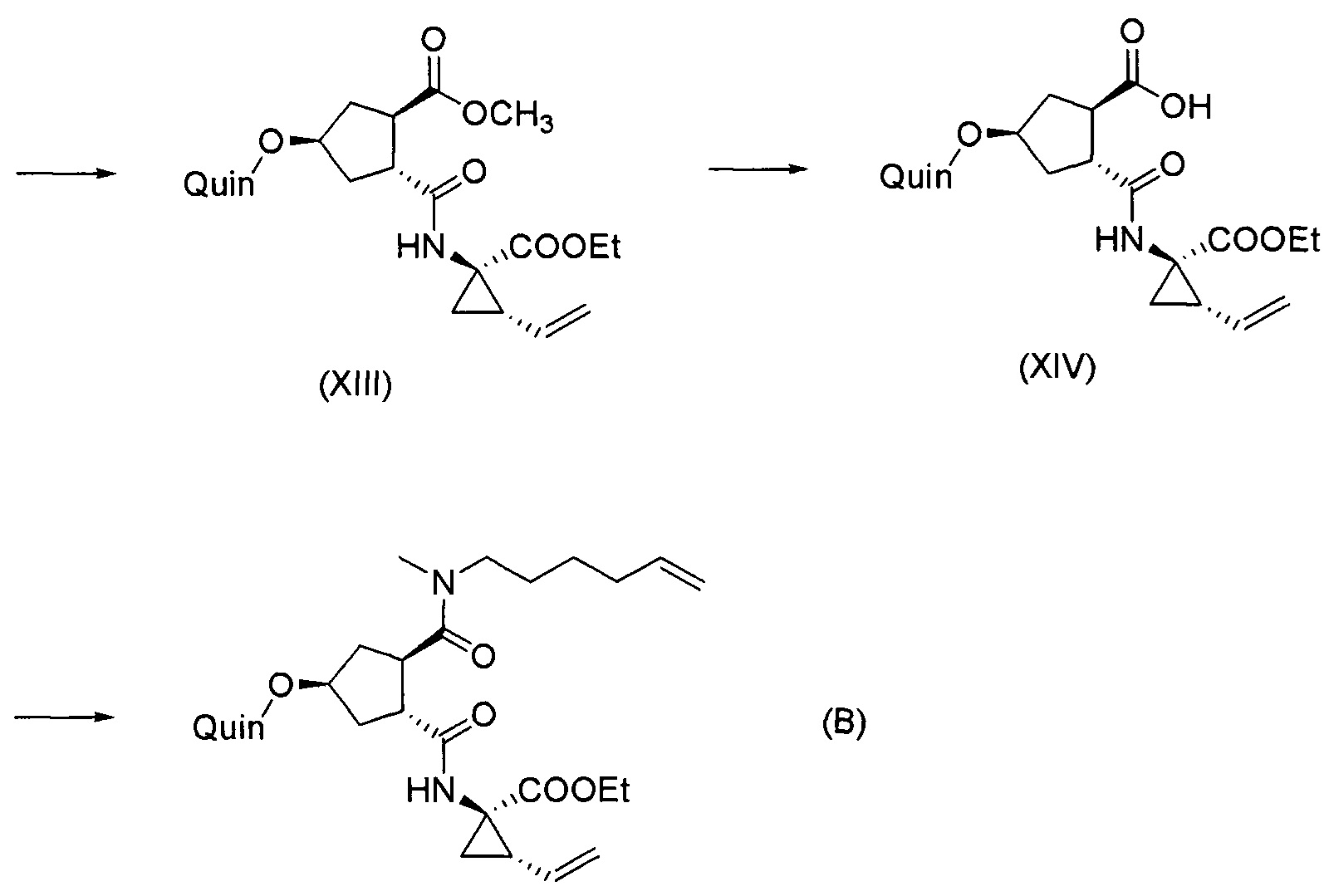
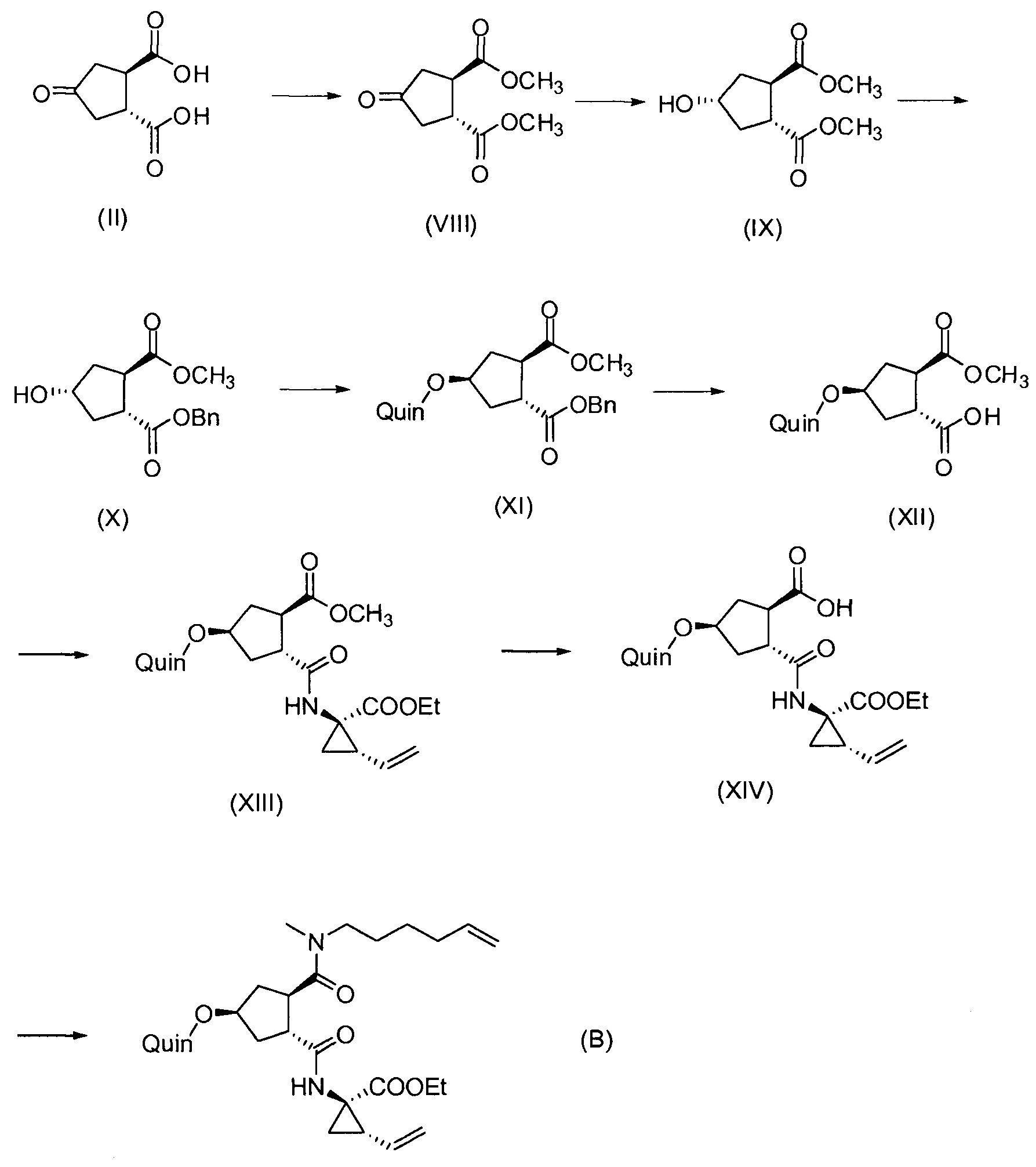
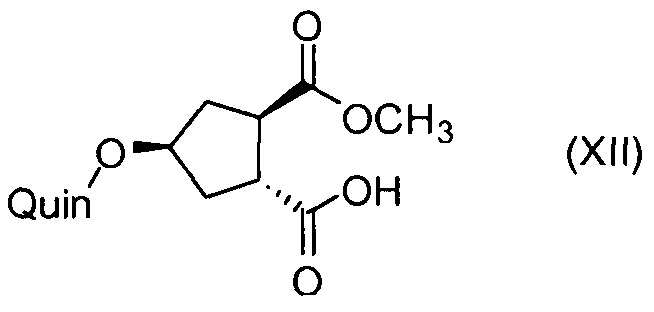
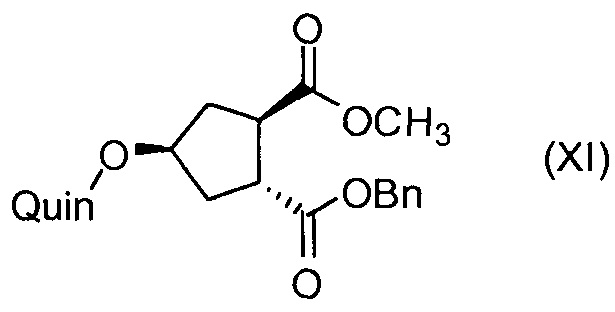
Последнее соединение может быть превращено в диметиловый эфир (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты VIII, как описано выше, и затем превращено в другие промежуточные соединения в синтезе соединения формулы I. Кето-группу в соединении VIII восстанавливают, получая промежуточное соединение IX, которое превращают в соединение Х по реакции переэтерификации по ОН-группе. Гидроксильную группу в соединении Х превращают в простую хинолинилэфирную группу в соединении XI по реакции Мицуноби, которая включает инверсию гидрокси-несущего атома углерода. Бензильную группу в соединении XI снимают, получая затем промежуточное соединение XII, и последнее сочетают со сложным эфиром циклопропиламинокислоты XXII, получая соединение XIII, в котором метиловый эфир гидролизуют с получением соединения XIV. Последнее сочетают с N-метил-5-гексен-1-амином XVI, получая промежуточное соединение XV, которое представляет собой соединение, описанное выше, и которое может быть подвергнуто циклизации до макроциклического промежуточного соединения А, которое превращают в конечный продукт I.
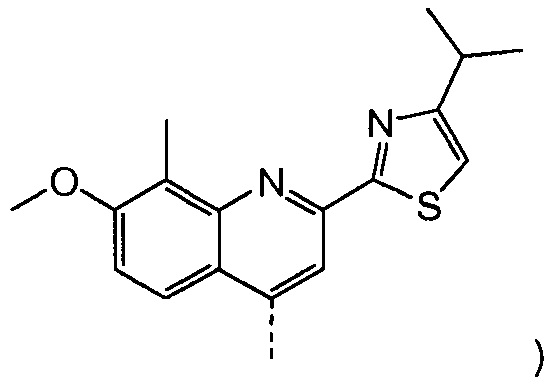
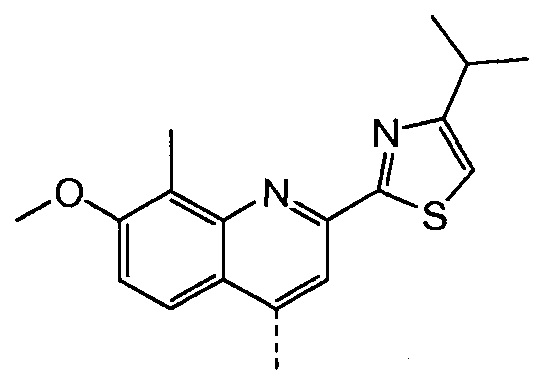
Такая реакционная последовательность проиллюстрирована на приведенной ниже схеме, где Quin представляет собой хинолиновую группу формулы:


Такая методика синтеза обеспечивает преимущество в том, что некоторые промежуточные соединения могут быть кристаллизованы, что дает возможность исключить примеси. Конечный продукт получают с высоким выходом и высокой чистотой, в частности, с высокой стереохимической чистотой. Промежуточные соединения, которые могут быть выделены в виде твердого вещества, представляют собой промежуточные соединения XI, XII, XIII и XIV.
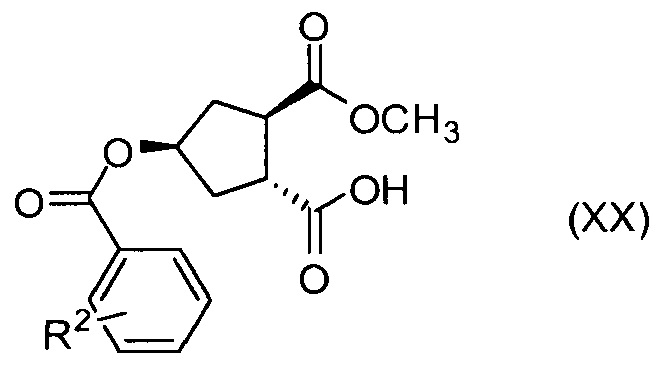
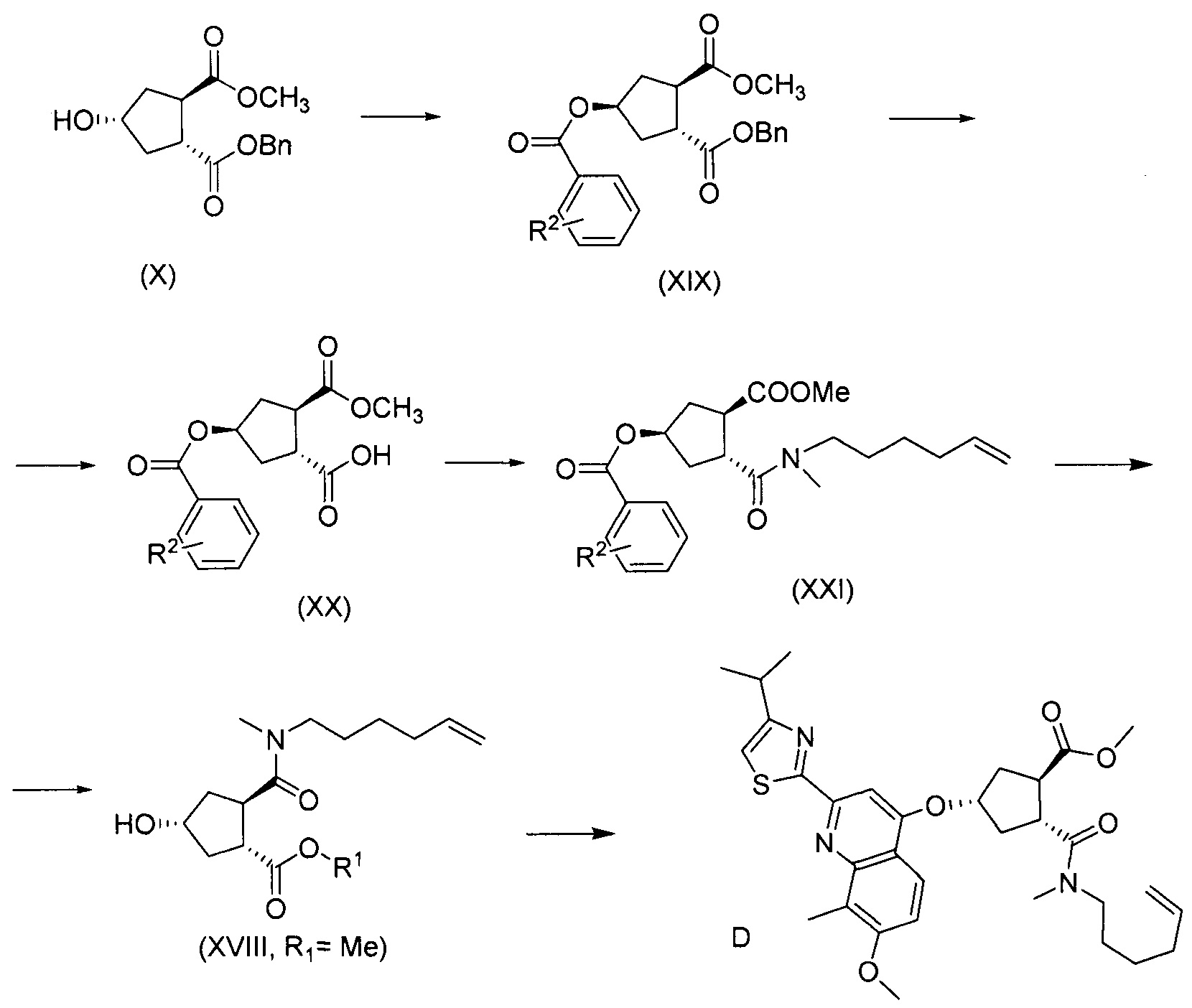
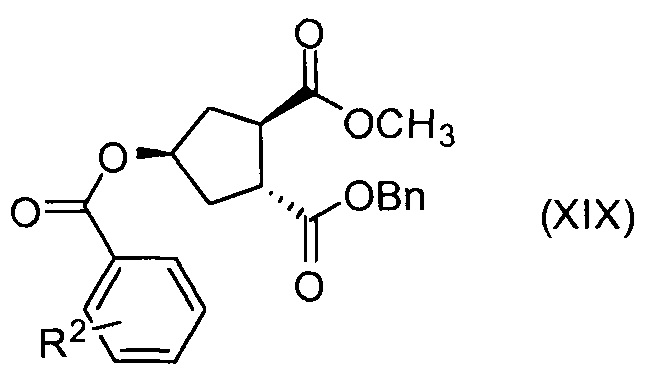
В другом аспекте, соединение XVIII, полезное промежуточное соединение для получения соединения I, готовят из промежуточного соединения Х по схеме, описанной ниже:
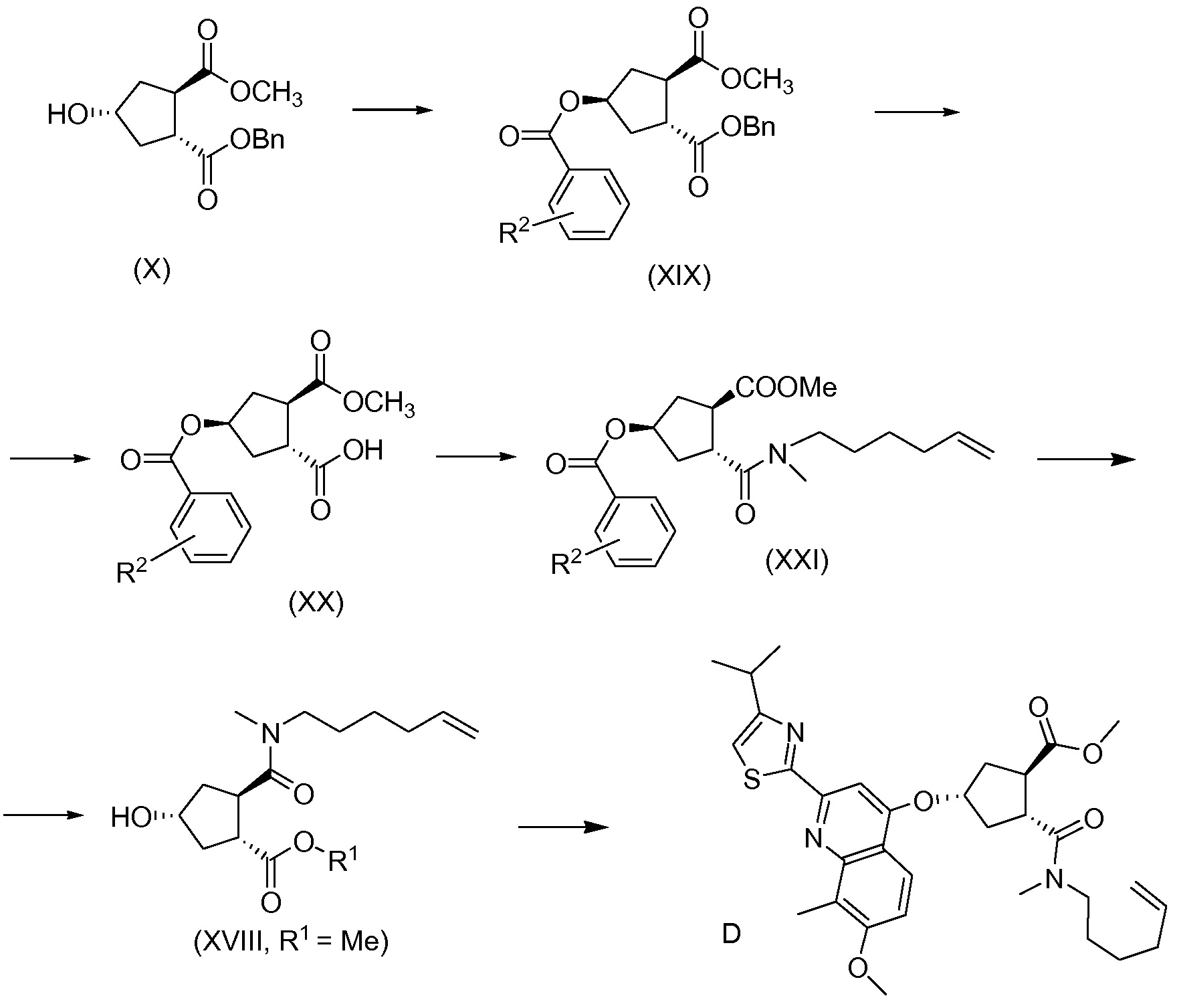
В приведенной выше схеме и далее заместитель R2 представляет собой атом водорода, С1-4-алкил, арил, атом галогена, -SO2-C1-4-алкил, CN или NO2. R2 может быть замещен в о-, м- или, в особенности, в п-положении. Интерес представляет группа NO2, в особенности п-NO2 (4-NO2). R1 имеет значения, определенные выше, и в частности, представляет собой метил.
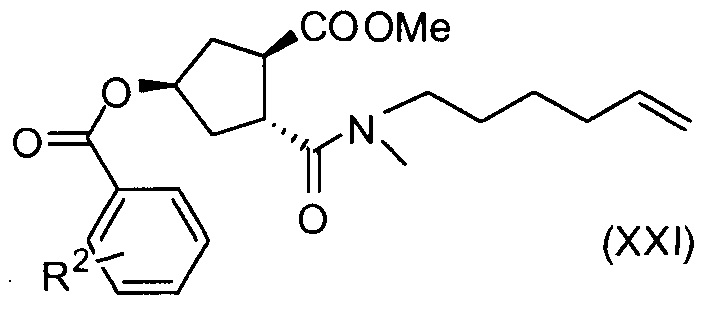
Соединение Х вводят в реакцию с ароматической кислотой, азодикарбоксилатом и фосфином в реакции Мицуноби с получением соединения XIX, которое выделяют из реакционной смеси в виде кристаллического твердого вещества, обеспечивая в результате эффективную очистку от обеих примесей в соединении Х, а также примесей от реакции Мицуноби. Соединение XIX обрабатывают ацетатом палладия и формиатом натрия, получают соединение ХХ, которое сочетают с N-метил-5-гексен-1-амином XVI, получая соединение XXI, которое обрабатывают основанием в метаноле, получая соединение XVIII (R1 = Ме).
Методики синтеза настоящего изобретения обеспечивают преимущество в том, что получают правильную стереохимию при циклопентановом остатке без использования хиральной хроматографии. Бруциновая соль III и эфедриновая соль IV, как установлено, селективно кристаллизуются с высокой диастереомерной чистотой (содержат кислоту II с высокой энантиомерной чистотой).
Открытие, что соли III и IV могут быть выделены путем кристаллизации, дает элегантный путь получения (R,R)-4-оксо-1,2-циклопентандикарбоновой кислоты II и затем бициклического лактона VII с высокой энантиомерной чистотой. Перекристаллизация или повторное суспендирование обеспечивают дополнительную очистку таких солей. Соли III и IV могут быть использованы в качестве исходного материала в дальнейшем синтезе кислоты VI, как описано выше. Последняя, в свою очередь, может быть превращена в лактон VII, важный структурный блок при получении соединения формулы I.
В другом варианте осуществления настоящее изобретение относится к соединениям per se формулы III или IV.
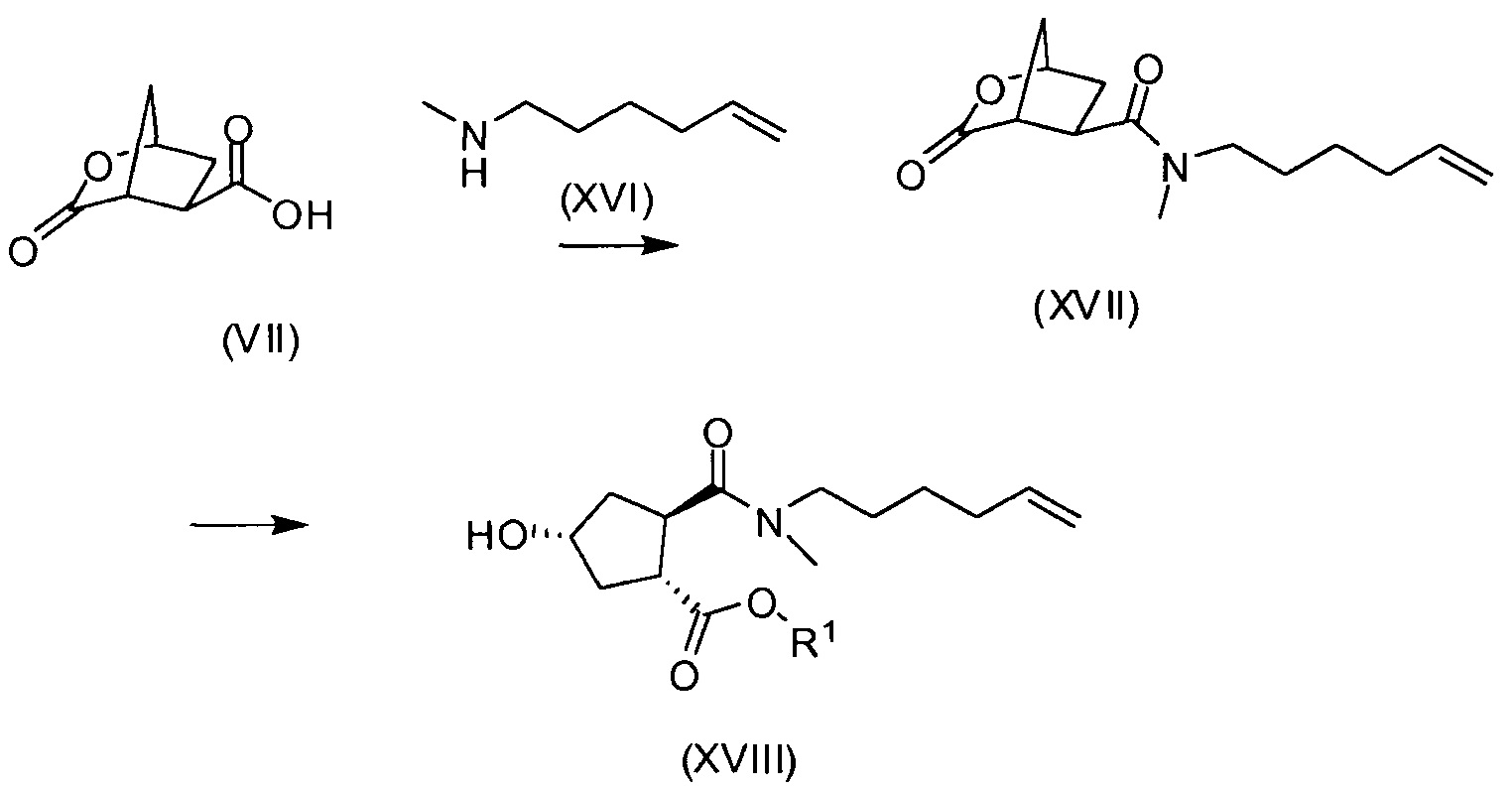
Циклический лактон кислоты VII может быть выделен либо в виде кислоты, либо в виде ее соли, но получаемый водно-органический раствор соединения VII, полученного в реакционной последовательности, описанной выше, может быть использован непосредственно в реакции сочетания с амином XVI с получением амида XVII.
В еще одном аспекте бициклический лактон кислоты VII или его соль, либо выделенные, либо невыделенные, используют в качестве исходного материала в способе получения циклопентанового производного XVIII реакцией бициклического лактона VII с N-метил-5-гексен-1-амином (NMHA) по реакции амидообразования с получением амида бициклического лактона XVII, в котором лактоновую группу открывают, получая целевой продукт XVIII. Такие реакции проиллюстрированы ниже на схеме, где R1 имеет значения, определенные выше.

Дальнейшую обработку соединения формулы XVIII до конечных продуктов формулы I проводят, как описано в приведенных выше реакционных схемах, и в особенности, как описано в публикации WO 2008/092955. Взаимодействие бициклического лактона VII с N-метил-5-гексен-1-амином XVI представляет собой реакцию амидообразования, которая включает взаимодействие исходных веществ с амид-сочетающим реагентом в растворителе необязательно в присутствии основания, как описано в публикациях WO 05/073195 и WO 2007/014926. Такая реакция может быть проведена, например, с использованием N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (EEDQ) в качестве сочетающего агента в дихлорметане (DCM), тетрагидрофуране (ТГФ) или 2-метилтетрагидрофуране (МеТГФ) в качестве растворителя; или может быть проведена с использованием TCT или его производных (CDMT, DCMT, DMTMM) в воде или в смеси воды и органического растворителя. Органическими растворителями для такой реакции являются ацетон, метилэтилкетон (MEK), тетрагидрофуран (ТГФ), МеТГФ, CPME (циклопентилметиловый эфир), С1-4-алкилацетат, С1-4-алкилпропионат, С1-4-алкилбутират и толуол.
Выше и далее используют следующие обозначения, если не указано другое. Определение «С1-4-алкил» означает линейные или разветвленные насыщенные углеводородные радикалы, содержащие от 1 до 4 атомов углерода, такие как, например, метил и этил; а также 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил.
Общепринятое обозначение для представления стереохимических соединений, которого также придерживаются в данном описании, состоит в следующем:
- Соединение, представленное без стереосвязей, является рацемическим, или конфигурация стереогенного(ых) центра(ов) не определена.
- Соединение, представленное со стереосвязями и одним из дескрипторов «(±)», «rel» или «рац», является рацемическим, и стереохимия является относительной.
- Соединение, представленное со стереосвязями, но без одного из дескрипторов «(±)», «rel» или «рац», относится к нерацемическому соединению (скалемическое вещество), то есть, к энантиообогащенному соединению.
Например, в публикации Honda с соавторами ссылку на обозначение «(±)» используют в названии статьи, показывая, что описан рацемический синтез с рацемическими промежуточными соединениями. Однако приведенному выше правилу не обязательно следовать во всех публикациях.
Энантиомерную чистоту указывают в виде энантиомерного отношения (э.о.). Для солей значение «э.о.» относится к отношению двух энантиомеров кислоты в смеси диастереомерных солей.
Примеры
Приведенные ниже примеры предназначены для иллюстрации настоящего изобретения и их не следует рассматривать в качестве ограничения объема настоящего изобретения.
Пример 1: Разрешение 4-оксо-1,2-циклопентандикарбоновой кислоты с помощью бруцина
Получение бис-бруциновой соли 4-оксо-1,2-циклопентандикарбоновой кислоты, то есть, соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с бруцином (1:2)

Бруцин (288,7 г, 0,73 моль) добавляют к раствору воды (87 мл) в метаноле (1653 мл). Смесь нагревают до 40°С и по каплям в течение 15 минут добавляют раствор рац-транс-4-оксо-1,2-циклопентандикарбоновой кислоты (60 г, 0,35 моль) в метаноле (665 мл) и воде (35 мл). Полученную суспензию кипятят с обратным холодильником до тех пор, пока все твердые вещества не растворятся. Смеси дают медленно охладиться до 22°С. Твердые вещества отфильтровывают и промывают небольшим количеством воды. Твердый материал сушат в вакууме при 50°С в течение 16 часов, получают 265 г бруциновой соли (смесь диастереомеров, приблизительно 1:1). Затем эту соль перекристаллизовывают из воды (1026 мл), после сушки получают 120,6 г (36%) соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с бруцином (1:2) в виде не совсем белого кристаллического вещества.
[α]D: -91,4.
Пример 2: Разрешение 4-оксо-1,2-циклопентандикарбоновой кислоты с помощью (-)-эфедрина
Получение бис-эфедриновой соли 4-оксо-1,2-циклопентандикарбоновой кислоты, то есть, с (1R,2S)-(-)-эфедрином [соединение (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с (1R,2S)-(-)-эфедрином (1:2)]
(1R,2S)-(-)-Эфедрин (20,16 г, 0,12 моль) добавляют к суспензии транс-4-оксо-1,2-циклопентандикарбоновой кислоты (10 г, 58 ммоль) в ацетоне (200 мл) и воды (26 мл). Смесь кипятят с обратным холодильником до тех пор, пока не образуется гомогенный раствор. Смеси дают медленно охладиться до 22°С. Твердые вещества отфильтровывают и промывают ацетоном. Твердое вещество сушат в вакууме при 50°С, получают 10,3 г (35%) соединения (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с (1R,2S)-(-)-эфедрином (1:2) в виде белого твердого вещества.
[α]D: -69,1.
1H-ЯМР (400 МГц, ДМСО-d6) δ м.д. 0,86 (д, J=6,8 Гц, 6H), 2,29-2,39 (м, 1H), 2,39-2,47 (м, 1H), 2,49 (с, 6H), 2,96-3,12 (м, 4H), 4,89 (д, J=3,3 Гц, 2H), 7,19-7,32 (м, 2H), 7,33-7,54 (м, 8H).
13C-ЯМР (150 МГц, ДМСО-d6) 11,17, 31,81, 42,12, 44,06, 59,55, 71,08, 125,97, 126,92, 127,93, 142,17, 175,74, 176,47, 215,40.
Разрешение также проводят с помощью (1S,2R)-(+)-эфедрина, получают с выходом 32% соединение (1S,2S)-4-оксо-1,2-циклопентандикарбоновой кислоты с (1S,2R)-(+)-эфедрином (1:2) в вид белого твердого вещества.
[α]D: +66,3.
Пример 3: (1R,2R)-4-оксо-1,2-циклопентандикарбоновая кислота и выделение эфедрина
К раствору соли (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты с (1R,2S)-(-)-эфедрином (1:2) (251 г, 0,5 моль) добавляют 8 н. водный раствор КОН (199,21 г, 1,10 моль) и смесь перемешивают 5 минут. Добавляют 2-метилтетрагидрофуран (688 мл) и смесь интенсивно перемешивают 20 минут. Два слоя разделяют и водную фазу подкисляют с помощью HCl. Органическая фаза 2-метилтетрагидрофурана содержит эфедрин, который может быть рециркулирован из этой фазы. Водный слой затем упаривают досуха на роторном испарителе и остаток перекристаллизовывают из воды (50 мл), получают 55,15 г (выход 64%) (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты в виде не совсем белого твердого вещества.
2-Метилтетрагидрофурановый раствор эфедрина упаривают досуха на роторном испарителе, получают сырой эфедрин в виде желтого масла, которое затвердевает при стоянии через три дня. Сырой эфедрин растворяют в 2-метилтетрагидрофуране (400 мл) и смесь подкисляют с помощью HCl в изопропаноле. Твердое вещество отфильтровывают и промывают 2-метилтетрагидрофураном (50 мл). Твердый гидрохлорид эфедрина сушат в вакууме при 50°С перед его растворением в воде (300 мл) при 40°С. К теплому водному раствору порциями добавляют достаточное количество карбоната калия, пока не образуется двухфазная система. Добавляют 2-метилтетрагидрофуран (200 мл) и смесь интенсивно перемешивают 5 минут. Два слоя разделяют и водную фазу экстрагируют 2-метилтетрагидрофураном (200 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и упаривают досуха, получают 124,7 г (выход 75%) выделенного эфедрина в виде белого твердого вещества.
Пример 4: Получение бициклического лактона карбоновой кислоты VII

К суспензии 32,7 г (0,19 моль) (1R,2R)-4-оксо-1,2-циклопентандикарбоновой кислоты (промежуточное соединение II) в 237,5 мл воды в атмосфере азота добавляют 1,0 мл (0,019 моль) 50%-ного (масс./масс.) водного раствора NaOH. Смесь нагревают до 60°С и добавляют 2,5 г Ph/C (5% масс./масс.). Затем реакционную колбу продувают водородом и выдерживают в атмосфере водорода при перемешивании до тех пор, пока не будет достигнута полная конверсия. Теплую реакционную смесь фильтруют через целлит (Celite) и осадок на фильтре дважды промывают 10 мл воды. Добавляют триэтиламин (55,61 мл, 0,49 моль) и 80% объема растворителя отгоняют при давлении 30 мбар. Реакционную колбу оборудуют насадкой Дина-Старка, заполненной 2-метилтетрагидрофураном, и к реакционной смеси добавляют 2-метилтетрагидрофуран (100 мл). Смесь кипятят с обратным холодильником 4 часа, чтобы удалить оставшуюся воду. Затем 80% объема растворителя отгоняют при обычном давлении. Смесь охлаждают до 50°С и добавляют ацетон. Смесь еще охлаждают до 22°С и добавляют еще ацетон (760 мл). Полученную суспензию охлаждают в атмосфере азота до -5°С и добавляют триэтиламин (27,8 мл, 20,24 г, 0,2 моль). Затем по каплям добавляют этилхлорформиат (22,68 г, 0,21 моль) и смесь перемешивают при 0°С в течение 3 часов, затем при 22°С еще 12 часов. Реакционную смесь фильтруют через дикалит (Dicalite) и твердые вещества промывают ацетоном (100 мл). Полученный раствор соединения VII в ацетоне может быть использован в других методиках для получения промежуточного соединения XVII.
Пример 5: Синтез соединения VIII, диметил-(1R,2R)-4-оксо-1,2-циклопентандикарбоксилата

Суспензию бис-бруциновой соли 4-оксо-1,2-циклопентандикарбоновой кислоты (144 г, 0,15 моль) в воде (750 мл) нагревают до 80°С. Добавляют по каплям гидроксид аммония (50%-ный раствор в воде, 11,8 мл, 0,32 моль) и полученную смесь перемешивают при 80°С в течение 30 минут. Суспензию охлаждают до 22°С и фильтруют, затем твердый материал промывают водой (37 мл). Объединенный фильтрат и промывные жидкости упаривают досуха на роторном испарителе. К остатку добавляют метанол (300 мл) и толуол (750 мл). Добавляют серную кислоту (4,3 мл) и смесь кипятят с обратным холодильником 2 часа. Растворитель отгоняют из реакционной смеси до тех пор, пока не будет достигнута внутренняя температура >70°С. Смесь охлаждают до 30°С и добавляют воду (150 мл). Полученную смесь перемешивают при 22°С в течение 60 минут. Два слоя разделяют и органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха, получают 27 г диметил-(1R,2R)-4-оксо-1,2-циклопентандикарбоксилата в виде светло-желтого масла, которое затвердевает при стоянии.
ГХ-МС: m/z=200 (M+).
1H-ЯМР (600 МГц, CDCl3) δ м.д. 2,35-2,40 (м, 2H), 2,5-2,56 (м, 2H), 3,25-3,31 (м, 2H), 3,75 (с, 6H).
13C-ЯМР (150 МГц, CDCl3) 40,56, 43,23, 52,09, 172,87, 212,03.
[α]D:-192.2
Пример 6: Синтез соединения IX, диметил-(1R,2R)-4-гидрокси-1,2-циклопентандикарбоксилата
Добавляют родий на оксиде алюминия (5%, влажный, 25 г) к раствору диметил-(1R,2R)-4-оксо-1,2-циклопентандикарбоксилата (100 г, 0,5 моль) в тетрагидрофуране (1000 мл). Реакционный сосуд продувают водородом и затем перемешивают в атмосфере водорода до полной конверсии. Реакционную смесь фильтруют через дикалит и осадок на фильтре промывают тетрагидрофураном (10 мл). Объединенные фильтраты и промывные жидкости упаривают досуха, получают 95,9 г диметил-(1R,2R)-4-гидрокси-1,2-циклопентандикарбоксилата в виде бесцветного масла.
Восстановление также может быть проведено с использованием Ni-Ренея в качестве катализатора в атмосфере водорода при давлении 6 бар.
Пример 7: Синтез промежуточного соединения Х, бензил-метил-(1R,2R,4R)-4-гидрокси-1,2-циклопентандикарбоксилат

Толуол (435 мл), карбонат натрия (23,6 г, 0,22 моль) и бензиловый спирт (48,13 г, 0,45 моль) добавляют в колбу, оборудованную насадкой Дина-Старка. Смесь кипятят с обратным холодильником 90 минут для удаления любых следов воды. Смесь охлаждают до 80°С и реакционную колбу продувают азотом. Добавляют диметил-(1R,2R)-4-гидрокси-1,2-циклопентандикарбоновую кислоту (30 г, 0,15 моль) и смесь кипятят с обратным холодильником при слабом токе азота над реакционной смесью в течение 6 часов. По каплям добавляют раствор HCl (42 мл, 0,46 моль) в воде (0,56 мл) и смесь перемешивают при 22°С в течение 15 минут. Два слоя разделяют и водную фазу экстрагируют толуолом (30 мл) и дихлорметаном (30 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и упаривают на роторном испарителе, получают сырое промежуточное соединение Х.
Остаток очищают колоночной хроматографией на силикагеле, элюируя смесью CH2Cl2/этилацетат (85:15), получают 23,9 г (выделенный выход 58%) бензил-метил-(1R,2R,4R)-4-гидрокси-1,2-циклопентандикарбоксилата в виде бесцветного масла.
ГХ-МС: m/z=278 (M+).
1H-ЯМР (600 МГц, CDCl3) δ м.д. 1,90-2,02 (м, 2H), 2,10-2,16 (м, 1H), 2,26 (ддд, 1H), 2,37 (д, J=4,91 Гц, 1H), 3,25-3,29 (м, 1H), 3,45 (кв, J=4,9 Гц, 1H), 3,66 (с, 3H), 4,37-4,40 (м, 1H), 5,15 (с, 2H), 7,30-7,37 (м, 5H).
13C-ЯМР (150 МГц, CDCl3) 38,59, 39,82, 45,21, 45,49, 52,12, 66,91, 72,86, 128,05, 128,28, 128,58, 135,73, 175,21.
Пример 8: Синтез соединения XI, бензил-метил-(1R,2R,4S)-4-([2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси)-1,2-циклопентандикарбоксилат
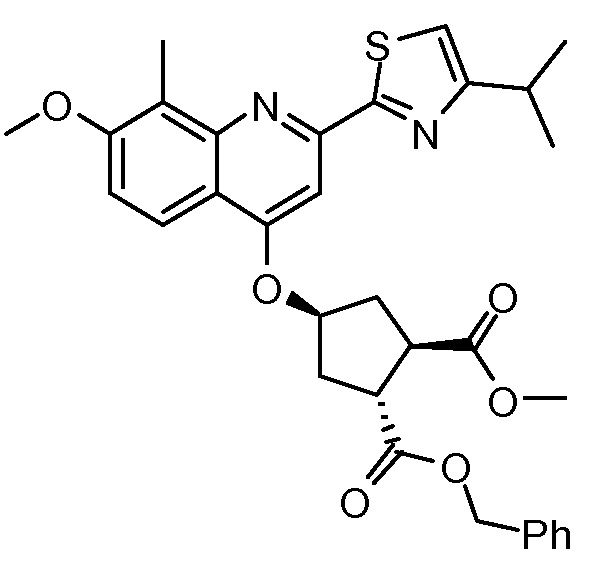
Раствор бензил-метил-(1R,2R,4R)-4-гидрокси-1,2-циклопентандикарбоксилата (промежуточное соединение Х, 4,1 г, 14,7 ммоль), 2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4(1Н)-хинолинон (Quin-ОН, 4,40 г, 14,0 ммоль) и трифенилфосфин (5,80 г, 22,1 ммоль) в толуоле (74 мл) кипятят с обратным холодильником 90 минут при наличии насадки Дина-Старка. Смесь охлаждают до -5°С и по каплям добавляют диизопропилазодикарбоксилат (4,47 г, 22,1 ммоль) при такой скорости, чтобы температура оставалась ниже 5°С. Реакционную смесь перемешивают при температуре от 0°С до 5°С в течение 3 часов, перед тем как дать ей медленно нагреться до 22°С, и перемешивают 16 часов. Добавляют воду (14,7 мл) и смесь перемешивают 10 минут. Реакционную смесь фильтруют, и твердые вещества промывают небольшим количеством толуола. Две фазы фильтрата разделяют и органическую фазу упаривают досуха. К остатку добавляют 1-бутанол (74 мл) и полученный раствор перемешивают при 22°С в течение 2 часов. Твердые вещества отфильтровывают и промывают 1-бутанолом (5 мл), получают после сушки 6,8 г бензил-метил-(1R,2R,4S)-4-([2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси)-1,2-циклопентандикарбоксилата в виде белого твердого вещества.
1H-ЯМР (600 МГц, CDCl3) δ м.д. 1,38 (д, J=7,1 Гц, 6H), 1,68 2,26-2,31 (м, 1H), 2,47-2,49 (м, 1H), 2,56-2,64 (м, 2H), 2,69 (с, 3H), 3,19 (гептет, J=6,8 Гц, 1H), 3,35-3,39 (м, 1H), 3,57 (с, 3H), 3,68-3,72 (м, 1H), 3,97 (с, 3H), 5,18 (AB J=12,5 Гц, 2H), 5,27-5,34 (м, 1H), 7,01 (с, 1H), 7,21 (д, J=9,1 Гц, 1H), 7,30-7,39 (м, 5H), 7,44-7,49 (м, 1H), 7,94 (д, J=9,1 Гц, 1H).
13C-ЯМР (150 МГц, CDCl3) 9,89, 22,50, 31,11, 35,92, 36,79, 45,32, 45,73, 52,21, 56,25, 66,86, 78,26, 95,70, 112,34, 114,20, 116,81, 120,28, 121,92, 128,16, 128,34, 128,48, 132,08, 135,71, 148,71, 151,80, 158,13, 160,57, 164,97, 169,93, 174,06.
Т.пл.: 112,4°С.
[α]D: -19,6.
Масс-спектрометрия высокого разрешения (МСВР): 575,22095 -масса соответствует формуле C32H35N2O6S.
Рассчитанная масса: 575,22158.
Пример 9: Синтез соединения XII, (1R,2R,4R)-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]-2-(метоксикарбонил)циклопентанкарбоновая кислота

Добавляют ацетат палладия(II) (152 мг, 0,68 ммоль) к раствору бензил-метил-(1R,2R,4S)-4-([2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси)-1,2-циклопентандикарбоксилата (XI, 7,8 г, 13,6 ммоль) в 2-метилтетрагидрофуране (27 мл) в атмосфере азота. Смесь нагревают до 45°С и добавляют триэтилсилан (6,52 мл, 40,7 ммоль). Полученную смесь нагревают до 60°С и перемешивают 16 часов. Добавляют соляную кислоту (115 мг, 1,1 ммоль), активированный уголь (0,4 г) и Celite® (0,4 г) и смесь перемешивают еще 60 минут при 60°С. Реакционную смесь фильтруют теплой, и твердые вещества промывают 2-метилтетрагидрофураном (6,8 мл). Объединенный фильтрат и промывные жидкости упаривают досуха. Остаток растворяют в метаноле и кипятят с обратным холодильником. Добавляют воду (13,6 г) и смесь снова кипятят с обратным холодильником. Смеси дают медленно охладиться до 22°С и твердый материал отфильтровывают и промывают холодным метанолом (5,5 мл). Твердые вещества сушат в вакууме при 50°С, получают 5,6 г (1R,2R,4R)-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]-2-(метоксикарбонил)циклопентанкарбоновой кислоты в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,39 (д, J=6,8 Гц, 6H), 2,30-2,39 (м, 1H), 2,48-2,69 (м, 2H), 2,70 (с, 3H), 3,16-3,29 (м, 1H), 3,36-3,44 (м, 1H), 3,64 (с, 3H), 3,69-3,77 (м, 1H), 3,99 (с, 3H), 5,32 (т, J=4,9 Гц, 1H), 7,03 (д, J=0,76 Гц, 1H), 7,24 (д, J=9,1 Гц, 1H), 7,51 (с, 1H), 7,95 (д, J=9,6 Гц, 1H), 10,03 (ушир.с, 1H).
13C-ЯМР (100 МГц, CDCl3) 9,85, 22,42, 22,53, 30,90, 35,92, 36,57, 45,01, 45,55, 52,31, 56,22, 78,25, 95,69, 112,46, 114,27, 116,77, 120,24, 121,95, 148,70, 151,49, 158,16, 160,63, 164,89, 170,36, 174,10, 178,76.
Т.пл.: 134,8°С.
[α]D: -13,8.
МСВР: 485,17776 - масса соответствует формуле C25H29N2O6S.
Рассчитанная масса: 485,17463.
Пример 10: Синтез соединения XIII, метил-(1R,2R,4R)-2-[[(1R,2S)-1-(этоксикарбонил)-2-винилциклопропил]карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоксилат

К раствору (1R,2R,4R)-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]-2-(метоксикарбонил)циклопентанкарбоновой кислоты (XII, 3 г, 6,19 ммоль) в ТГФ (31 мл) добавляют 1 М водный раствор NaHCO3 (6,0 мл) и полученный раствор перемешивают при 22°С в течение 15 минут. Добавляют 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (1,61 г, 6,50 ммоль) и этил-(1R,2S)-1-амино-2-винилциклопропанкарбоксилат·4-метилбензолсульфонат (XXII·TsOH, 2,03 г, 6,19 ммоль) и смесь перемешивают при 22°С в течение 16 часов. К реакционной смеси добавляют 1 М водный раствор HCl (12,4 мл) и смесь перемешивают несколько минут. К реакционной смеси добавляют 2-метилтетрагидрофуран (31 мл) и две фазы разделяют. Органический слой промывают 1 М водным раствором NaOH (12,4 мл) и водой (9,3 мл) перед сушкой над Na2SO4, фильтруют и фильтрат упаривают досуха. Остаток перекристаллизовывают из изопропанола (18,6 мл), после сушки получают 2,2 г метил-(1R,2R,4R)-2-[[(1R,2S)-1-(этоксикарбонил)-2-винилциклопропил]карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоксилата (XIII) в виде не совсем белого твердого вещества.
1H-ЯМР (600 МГц, CD2Cl2) δ м.д. 1,11 (т, J=7,8 Гц, 3H), 1,29 (д, J=6,8 Гц, 6H), 1,36 (дд, J=9,44, 5,29 Гц, 1H), 1,72 (дд, J=7,93, 5,29 Гц, 1H), 2,01-2,06 (м, 1H), 2,25-2,34 (м, 2H), 2,34-2,39 (м, 1H), 2,50-2,55 (м, 1H), 2,56 (с, 3H), 3,04-3,12 (м, 1H), 3,14-3,19 (м, 1H), 3,29-3,34 (м, 1H), 3,53 (с, 3H), 3,88 (с, 3H), 3,96-4,07 (м, 2H), 5,02 (дд, J=10,39, 1,70 Гц, 1H), 5,17-5,23 (м, 2H), (м, 2H), 5,61-5,69 (м, 1H), 6,68 (с, 1H), 6,96 (д, J=0,76 Гц, 1H), 7,15 (д, J=9,06 Гц, 1H), 7,42 (с, 1H), 7,83 (д, J=9,06 Гц, 1H).
13C-ЯМР (150 МГц, CD2Cl2) 10,17, 14,52, 22,75, 23,50, 31,69, 33,99, 36,22, 36,32, 40,77, 46,25, 46,73, 52,79, 56,68, 61,91, 79,45, 96,27, 112,76, 114,80, 117,34, 117,93, 120,70, 122,12, 134,41, 149,15, 152,41, 158,76, 161,25, 165,59, 170,18, 170,53, 174,58, 175,65.
[α]D: -6,5.
Т.пл.: 155,4°С.
Пример 11: Синтез соединения XIV, (1R,2R,4R)-2-[[(1R,2S)-1-(этоксикарбонил)-2-винилциклопропил]карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоновая кислота

К раствору метил-(1R,2R,4R)-2-[[(1R,2S)-1-(этоксикарбонил)-2-винилциклопропил]карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоксилата (XIII, 0,97 г, 1,5 ммоль) в ТГФ (6 мл) добавляют раствор LiOH (66 мг, 1,57 ммоль) в воде (1,5 мл). Полученную смесь перемешивают при 22°С в течение 16 часов. Добавляют воду (6 мл) и 2-метилтетрагидрофуран (10 мл) и два слоя разделяют. Водную фазу экстрагируют 2-метилтетрагидрофураном (5 мл). Объединенные органические слои промывают 1 н. водным раствором HCl (5 мл), сушат над Na2SO4, фильтруют и упаривают досуха, получают 0,74 г (1R,2R,4R)-2-[[(1R,2S)-1-(этоксикарбонил)-2-винилциклопропил]карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоновой кислоты (XIV) в виде желтого твердого вещества. Анализ показывает, что продукт имеет чистоту >90%, и его используют на следующей стадии без дополнительной очистки.
МСВР: 608,24262.
Рассчитанная масса: 608,24304.
Пример 12: Синтез соединения XV, этил-(1R,2S)-1-([[(1R,2R,4R)-2-[гекс-5-ен-1-ил(метил)карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метилхинолинил-4-ил]окси]циклопентил]карбонил]амино)-2-винилциклопропанкарбоксилат
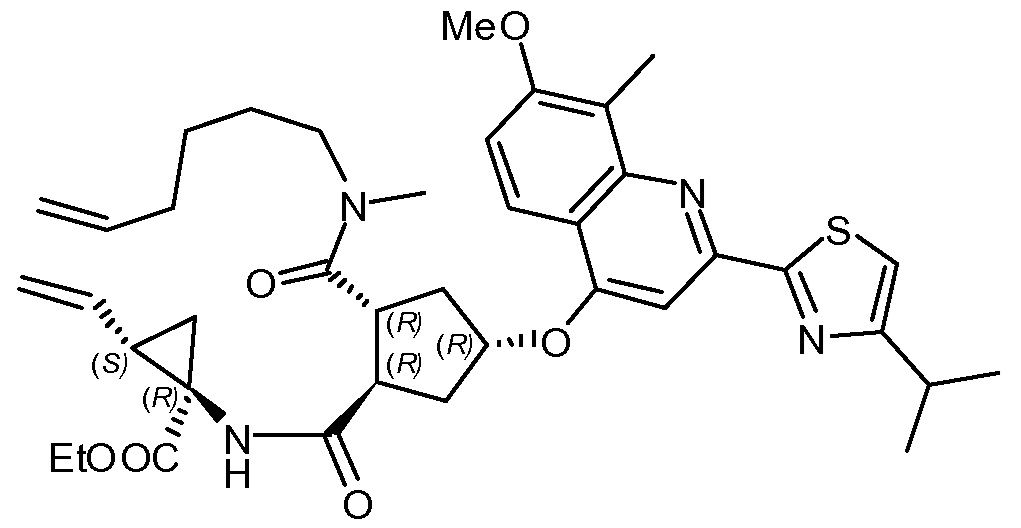
К раствору (1R,2R,4R)-2-[[(1R,2S)-1-(этоксикарбонил)-2-винилциклопропил]карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоновой кислоты (XIV, 0,64 г, 1 ммоль) и N-метил-5-гексен-1-амина (131 мг, 1,2 ммоль) в ТГФ (10 мл) добавляют 1-этоксикарбонил-2-этокси-1,2-дигидрохинолин (300 мг, 1,2 ммоль). Полученную смесь кипятят с обратным холодильником 4 часа. Раствору дают охладиться до 22°C и добавляют 2-метилтетрагидрофуран (10 мл). Органический раствор перед сушкой над Na2SO4 промывают 1 н. водным раствором HCl (4,2 мл, 4,2 ммоль) и водой (2,1 мл), фильтруют и упаривают досуха, получают 480 мг этил-(1R,2S)-1-([[(1R,2R,4R)-2-[гекс-5-ен-1-ил(метил)карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метилхинолинил-4-ил]окси]циклопентил]карбонил]амино)-2-винилциклопропанкарбоксилата в виде стеклоподобного масса. Данные ЖХ и ЯМР показывают чистоту >90%.
ЖХ-МС: m/z=703 ([М+Н]+).
1H-ЯМР (400 МГц, ДМСО-d6 - смесь ротамеров) δ м.д. 0,87 (т, J=7,30 Гц, 1H), 1,06-1,19 (м, 3H), 1,19-1,31 (м, 2H), 1,33 (д, J=6,80 Гц, 6H), 1,35-1,45 (м, 2H), 1,46-1,66 (м, 2H), 1,84-2,00 (м, 2H), 2,00-2,18 (м, 3H), 2,25-2,36 (м, 1H), 2,58 (с, 3H), 2,64-2,77 (м, 1H), 2,80 (с, 3H - один ротамер), 3,00 (с, 3H - один ротамер), 3,08-3,30 (м, 2H), 3,34-3,52 (м, 3H), 3,97 (с, 3H), 3,98-4,12 (м, 2H), 4,82-5,13 (м, 3H), 5,18-5,37 (м, 2H), 5,55-5,86 (м, 2H), 7,39-7,50 (м, 3H), 8,06 (т, J=8,94 Гц, 1H), 8,59 (с, 1H - один ротамер), 8,73 (с, 1H - один ротамер).
13C-ЯМР (100 МГц, ДМСО-d6 - смесь ротамеров) δ м.д. 9,79, 13,80, 13,96, 13,99, 14,12, 14,53, 18,59, 22,21, 22,30, 25,25, 25,32, 26,02, 27,64, 30,40, 32,07, 32,14, 32,81, 33,19, 34,64, 34,68, 36,31, 36,76, 36,95, 36,98, 42,28, 46,01, 46,43, 46,74, 48,74, 56,10, 60,33, 60,51, 60,59, 78,73, 78,80, 95,34, 95,38, 112,67, 112,76, 114,67, 114,88, 115,47, 116,20, 116,23, 117,34, 120,03, 120,05, 120,59, 120,62, 134,13, 134,18, 138,41, 138,49, 147,85, 151,22, 157,98, 158,0, 160,75, 164,25, 168,66, 168,69, 169,82, 169,85, 172,48, 172,51, 173,66, 173,83.
Пример 13: Синтез соединения XIX (R2 = п-NO2), бензил-метил-(1R,2R,4S)-4-[(4-нитробензоил)окси]-1,2-циклопентандикарбоксилат

Суспензию бензил-метил-(1R,2R,4R)-4-гидрокси-1,2-циклопентандикарбоксилата (Х, 96 г, 0,34 моль), 4-нитробензойной кислоты (69,2 г, 0,41 моль) и трифенилфосфина (120,3 г, 0,46 моль) в толуоле (1380 мл) кипятят с обратным холодильником в сосуде, оборудованном насадкой Дина-Старка в течение 30 минут, чтобы удалить все следы воды. Смесь охлаждают до -5°С и по каплям добавляют диизопропилазодикарбоксилат (92,8 г, 0,46 ммоль). Полученную смесь перемешивают при 0°С еще 60 минут перед тем как дать смеси медленно нагреться до 22°С и перемешивают 12 часов. Добавляют воду (345 мл) и полученную смесь перемешивают 10 минут. Твердые вещества отфильтровывают и промывают толуолом (86 мл). Из фильтрата отгоняют 1200 мл растворителя. Смесь охлаждают до 70°С и добавляют изопропанол (1380 мл). Отгоняют 1460 мл растворителя и смесь охлаждают до 70°С. Добавляют изопропанол (690 мл) и смесь кипятят с обратным холодильником. Смеси дают медленно охладиться до 22°С и перемешивают еще 2 часа. Отфильтровывают твердые вещества и промывают изопропанолом (69 мл). Твердое вещество сушат в вакууме, получают 80,2 г бензил-метил-(1R,2R,4S)-4-[(4-нитробензоил)окси]-1,2-циклопентандикарбоксилата в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,15-2,36 (м, 2H), 2,36-2,46 (м, 1H), 2,46-2,59 (м, 1H), 3,28-3,38 (м, 1H), 3,57-3,64 (м, 1H), 3,65 (с, 3H), 5,17 (д, J=2,5 Гц, 2H), 5,49-5,53 (м, 1H), 7,33-7,39 (м, 5H), 8,15 (д, J=9,1 Гц, 2H), 8,28 (д, J=8,8 Гц, 2H).
[α]D: -16,3.
Т.пл.: 79,5°С.
МСВР: 428,13632.
Рассчитанная масса: 428,13454.
Пример 14: Удаление бензилового спирта из сырого промежуточного соединения X путем окисления
Сырое промежуточное соединение Х, бензил-метил-(1R,2R,4R)-4-гидрокси-1,2-циклопентандикарбоксилат, полученное в примере 7 (3,1 г, 111 ммоль) в бензиловом спирте (12,5 мл, 0,12 моль), растворяют в толуоле (246 мл). К полученному раствору добавляют соль 2,2’-[1,2-этандиилбис(иминометандиил)]дифенол-медь2+ (см. Velusamy S., Punniyamurthy T., Eur. J. Org. Chem., 2003, 3913) (2,05 г, 6,15 ммоль) и 2,2,6,6-тетраметилпиперидин-N-оксид (0,96 г, 6,15 ммоль). Смесь нагревают до 80°С и через раствор 3 часа барботируют атмосферный воздух. Смеси дают остыть до 22°С и фильтруют через дикалит. Осадок на фильтре промывают толуолом. К фильтрату добавляют воду (246 мл) и смесь перемешивают 5 минут. Слои разделяют и к органической фазе добавляют воду (123 мл) и метабисульфит натрия (46,76 г, 0,24 ммоль). Полученную смесь перемешивают при 22°С в течение 10 минут и затем фильтруют через дикалит. Два слоя фильтрата разделяют и органическую фазу промывают водой (123 мл) и затем 2М водным раствором HCl (240 мл). Органический раствор сушат над Na2SO4, фильтруют и упаривают досуха на роторном испарителе, получают 2,7 г бензил-метил-(1R,2R,4R)-4-гидрокси-1,2-циклопентандикарбоксилата в виде светло-коричневого масла, которое используют в следующей реакции напрямую.
Анализ продукта показывает, что удалено 95% бензилового спирта.
Примечание: Рассмотренная реакция может быть проведена путем барботирования вместо атмосферного воздуха смеси 5% кислорода в 95% азота. Несмотря на то, что такая смесь делает реакцию опасной, она также увеличивает время, требуемое для достижения в реакции полного превращения.
Пример 15: Синтез соединения XX (R2 = п-NO2), (1R,2R,4R)-2-(метоксикарбонил)-4-[(4-нитробензоил)окси]циклопентанкарбоновая кислота

К суспензии бензил-метил-(1R,2R,4S)-4-[(4-нитробензоил)окси]-1,2-циклопентандикарбоксилата (XIX, 29 г, 67,8 ммоль) и формиата натрия (6,92 г, 102 ммоль) в ДМФА (204 мл) в атмосфере азота добавляют ацетат палладия(II) (762 мг, 3,4 ммоль). Смесь нагревают до 100°С в течение 3 часов. Реакционную смесь охлаждают до 70°С и фильтруют через Celite®. Осадок на фильтре промывают ДМФА (10 мл). Объединенные фильтрат и промывные жидкости подкисляют до рН 1 с помощью HCl и полученный раствор выливают в воду (610 мл). Полученную смесь перемешивают при 22°C в течение 10-15 минут. Твердые вещества отфильтровывают и промывают водой (14 мл), после сушки получают 18,4 г (1R,2R,4R)-2-(метоксикарбонил)-4-[(4-нитробензоил)окси]циклопентанкарбоновой кислоты в виде красно-коричневого твердого вещества.
1H-ЯМР (600 МГц, CDCl3) δ м.д. 2,24-2,36 (м, 2H), 2,40-2,45 (м, 1H), 2,50-2,55 (м, 1H), 3,34-3,38 (м, 1H), 3,59-3,64 (м, 1H), 3,70 (с, 3H), 5,51-5,54 (м, 1H), 8,16 (д, J=9,0 Гц, 2H), 8,29 (д, J=8,9 Гц, 2H).
13C-ЯМР (150 МГц, CDCl3) 36,06, 36,65, 45,11, 45,11, 52,41, 77,02, 123,58, 130,77, 135,47, 150,62, 164,02, 174,40, 177,64.
[α]D: -22,6.
Т.пл.: 111°С.
МСВР: 338,08726.
Рассчитанная масса: 338,08759.
Пример 16: Синтез соединения XXI (R2 = п-NO2), (1R,3R,4R)-3-[5-гексен-1-ил(метил)карбамоил]-4-(метоксикарбонил)циклопентил-4-нитробензоат

К раствору (1R,2R,4R)-2-(метоксикарбонил)-4-[(4-нитробензоил)окси]циклопентанкарбоновой кислоты (ХХ, 15,3 г, 39,0 ммоль) и N-метил-5-гексен-1-амина (5,30 г, 46,8 ммоль) в ТГФ (78 мл) добавляют 1-этоксикарбонил-2-этокси-1,2-дигидрохинолин (12,06 г, 48,76 ммоль) и смесь нагревают до 50°С в течение 8 часов. Смесь охлаждают до 22°С, добавляют 5 н. водный раствор HCl (23,4 мл, 117,0 ммоль) и смесь интенсивно перемешивают 2 минуты. Добавляют 2-метилтетрагидрофуран (78 мл) и два слоя разделяют. Органический слой перед сушкой над Na2SO4 промывают 1 М водным раствором бикарбоната натрия (57 мл) и водой (29 мл), фильтруют и упаривают досуха. Сырой материал очищают колоночной хроматографией на силикагеле, элюируя смесью гептан/этилацетат (1:1), получают 11,5 г (1R,3R,4R)-3-[5-гексен-1-ил(метил)карбамоил]-4-(метоксикарбонил)циклопентил-4-нитробензоата в виде светло-желтого масла.
1H-ЯМР (600 МГц, CDCl3 - смесь ротамеров) δ м.д. 1,24 (т, J=7,2 Гц, 2H), 1,36-1,44 (м, 2H), 1,52-1,57 (м, 1H), 1,6-1,67 (м, 1H), 2,08 (кв, J=7,2 Гц, 2H), 2,17-2,24 (м, 1H), 2,27-2,32 (м, 1H), 2,54-2,61 (м, 1H), 2,96 (с, 3H - один ротамер), 3,10 (с, 3H - один ротамер), 3,41 (т, J=7,5 Гц, 1H), 3,44-3,50 (м, 1H), 3,66 (с, 3H), 3,68-3,72 (м, 1H), 3,76-3,80 (м, 1H), 4,94-5,02 (м, 2H), 5,53-5,59 (м, 1H), 5,72-5,81 (м, 1H), 8,16 (д, J=9,1 Гц, 2H), 8,29 (д, J=8,3 Гц, 2H).
13C-ЯМР (150 МГц, CDCl3 - смесь ротамеров) 25,86, 26,53, 28,13, 33,41, 34,08, 35,40, 36,22, 37,16, 37,83, 41,64, 42,17, 46,01, 48,04, 49,86, 52,18, 77,88, 114,74, 115,14, 123,59, 130,71, 135,61, 137,96, 138,48, 150,57, 164,02, 173,12, 173,38, 175,07.
МСВР: 433,19708.
Рассчитанная масса: 433,19748.
Пример 17: Синтез соединения XVIII (R1 = Ме), метил-(1R,2R,4R)-2-[5-гексен-1-ил(метил)карбамоил]-4-гидроксициклопентанкарбоксилат

Добавляют карбонат натрия (980 мг, 9,25 ммоль) к раствору (1R,3R,4R)-3-[5-гексен-1-ил(метил)карбамоил]-4-(метоксикарбонил)циклопентил-4-нитробензоата (XXI, 4,0 г, 9,25 ммоль) в метаноле (46 мл) и гетерогенную смесь перемешивают при 22°С в течение 90 минут. Реакционную смесь фильтруют и твердые вещества промывают метанолом (20 мл). Объединенные метанольные растворы упаривают досуха. Остаток растирают с метанолом (9 мл) и твердые вещества отфильтровывают. Фильтрат упаривают досуха с получением 2,7 г желтого масла, которое затвердевает при стоянии. Данные ЖХ показывают, что выделенным продуктом является метил-(1R,2R,4R)-2-[5-гексен-1-ил(метил)карбамоил]-4-гидроксициклопентанкарбоксилат (52% масс./масс.) и метил-4-нитробензоат. Этот продукт используют на следующей стадии без дополнительной очистки.
Пример 18: Синтез соединения D, метил-(1R,2R,4S)-2-[5-гексен-1-ил(метил)карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоксилат

Добавляют 2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4(1Н)-хинолинон (Quin-ОН, 1,27 г, 4,0 ммоль) к сырому раствору метил-(1R,2R,4R)-2-[5-гексен-1-ил(метил)карбамоил]-4-гидроксициклопентанкарбоксилата (XVIII, 2,2 г, 4,0 ммоль) в толуоле (28 мл). Добавляют трифенилфосфин (1,11 г, 4,25 ммоль) и смесь охлаждают до -5°С. По каплям добавляют диизопропилазодикарбоксилат (860 мг, 4,25 ммоль) при такой скорости, чтобы температура оставалась ниже 5°С. Смесь перемешивают при 0°С еще 60 минут, перед тем как дать ей нагреться до 22°С, и перемешивают 16 часов. Реакционную смесь фильтруют и твердые вещества промывают толуолом. Объединенный толуольный раствор упаривают досуха, и остаток очищают колоночной хроматографией на силикагеле, элюируя смесью гептан/этилацетат (7:3), с получением 1,6 г метил-(1R,2R,4S)-2-[5-гексен-1-ил(метил)карбамоил]-4-[[2-(4-изопропил-1,3-тиазол-2-ил)-7-метокси-8-метил-4-хинолинил]окси]циклопентанкарбоксилата в виде бежевого твердого вещества.
Данные спектров ЖХ, МС и ЯМР идентичны уже представленным спектрам.
Как используется в данном случае % масс. (% масс./масс.) соли в растворе всегда относится к % масс./масс. исходной кислоты в растворе. Например, 25%-ный масс./масс. раствор VI.2NMM (VI.2NMM, относится к (2:1) N-морфолиновой соли соединения VI) в растворителе относится к раствору VI.2NMM, в котором 25 г исходной кислоты VI присутствует в 100 г раствора).
Пример 19: В 4 мл воды разбавляют 735 мг 23,7%-ного масс./масс. водного раствора соединения VI, бис-калиевая соль (1 ммоль), и смешивают с 364 мкл NMM (3,3 ммоль). Добавляют 203 мг (1,1 ммоль) TCT и реакционную смесь перемешивают в течение ночи при комнатной температуре, затем разбавляют до конечного объема 10 мл, получают 63 мМ водный раствор соединения VII (выход 63%).
Пример 20: В 4 мл воды разбавляют 735 мг 23,7%-ного масс./масс. водного раствора соединения VI, бис-калиевая соль (1 ммоль), и смешивают с 728 мкл NMM (6,6 ммоль). Добавляют 406 мг (2,2 ммоль) TCT и реакционную смесь перемешивают в течение ночи при комнатной температуре, затем разбавляют до конечного объема 10 мл, получают 78 мМ водный раствор соединения VII (выход 78%).
Пример 21: Смешивают 728 мкл NMM (6,6 ммоль) с 4 мл воды и добавляют 406 мг (2,2 ммоль) TCT. Перед добавлением 735 мг 23,7%-ного масс./масс. водного раствора соединения VI, бис-калиевая соль (1 ммоль), смесь перемешивают несколько минут. Полученную реакционную смесь перемешивают еще в течение ночи при комнатной температуре, затем разбавляют до конечного объема 10 мл, получают 57 мМ водный раствор соединения VII (выход 57%).
Пример 22: В 4 мл воды разбавляют 735 мг 23,7%-ного масс./масс. водного раствора соединения VI, бис-калиевая соль (1 ммоль), и смешивают с 221 мкл NMM (2 ммоль). Добавляют 648 мг (2,2 ммоль) DMTMM·H2O и реакционную смесь перемешивают в течение ночи при комнатной температуре, затем разбавляют до конечного объема 10 мл, получают 54 мМ водный раствор соединения VII (выход 54%).
Пример 23: В 4 мл ацетона растворяют 386 мг (2,2 ммоль) CDMT и добавляют 463 мкл (4,2 ммоль) NMMR. Смесь перемешивают несколько минут, затем добавляют 735 мг 23,7%-ного масс./масс. водного раствора соединения VI, бис-калиевая соль. Полученную смесь дополнительно перемешивают в течение ночи при комнатной температуре, затем разбавляют до конечного объема 10 мл, получают 69 мМ раствор соединения VII (выход 69%).
Пример 24: В 4 мл МеТГФ растворяют 368 мг (2,2 ммоль) CDMT и добавляют 463 мкл (4,2 ммоль) NMM. Смесь перемешивают несколько минут, затем добавляют 735 мг 23,7%-ного масс./масс. водного раствора соединения VI, бис-калиевая соль. Полученную смесь дополнительно перемешивают в течение ночи при комнатной температуре, затем разбавляют до конечного объема 10 мл, получают 54 мМ раствор соединения VII (выход 54%).
Пример 25: В 59 мл МеТГФ растворяют 5,66 г (32,2 ммоль) CDMT. Добавляют 3,7 мл (33,7 ммоль) NMM и смесь перемешивают 1 час при 25°С. Добавляют 10 г 25,5%-ного масс./масс. водного раствора соединения VI.2NMM (14,6 ммоль) и полученную смесь перемешивают еще несколько часов при 25°С. Добавляют 15 мл воды и 3 мл концентрированной HCl. Смесь перемешивают несколько минут, нерастворимые вещества отфильтровывают, фильтрат декантируют и водный слой экстрагируют 15 мл МеТГФ. Органические слои объединяют и промывают 7 мл рассола, получают 53,1 г 2,59%-ного масс./масс. раствора соединения VII в МеТГФ, который также содержит 0,23% масс./масс. соединения VI (выход 60%).
Пример 26: В 59 мл изопропилацетата растворяют 5,66 г (32,2 ммоль) CDMT. Добавляют 3,7 мл (33,7 ммоль) NMM и смесь перемешивают 1 час при 25°С. Добавляют 10 г 25,5%-ного масс./масс. водного раствора бис-N-метилморфолиновой соли соединения VI (14,6 ммоль) и полученную смесь дополнительно перемешивают несколько часов при 25°С. Добавляют 15 мл воды и 3 мл концентрированной HCl. Смесь перемешивают несколько минут, выделяемые материалы отфильтровывают, фильтрат декантируют и водный слой экстрагируют 15 мл изопропилацетата. Органические слои объединяют и промывают 7 мл рассола, получают 56,6 г 1,3%-ного масс./масс. раствора соединения VII в изопропилацетате, который также содержит с 0,18% масс./масс. соединения VI (выход 32%).
Пример 27: В 59 мл ацетона растворяют 5,66 г (32,2 ммоль) CDMT. Добавляют 3,7 мл (33,7 ммоль) NMM и смесь перемешивают 1 час при 25°С. Добавляют 10 г 25,5%-ного масс./масс. водного раствора бис-N-метилморфолиновой соли соединения VI (14,6 ммоль) и полученную смесь перемешивают еще несколько часов при 25°С. Нерастворимые вещества отфильтровывают, к фильтрату добавляют 1 мл концентрированной HCl и фильтрат декантируют. Органический слой промывают 7 мл рассола, получают 44,4 г 1,44%-ного масс./масс. раствора соединения VII в МеТГФ, который также содержит 0,04% масс./масс. соединения VI (выход 28%).
Пример 28: В 205 мл МеТГФ растворяют 19,80 г (113 ммоль) CDMT. Добавляют 13 мл (118 ммоль) NMM и смесь перемешивают при 25°С в течение 2 часов. Добавляют 35 г 25,5%-ного масс./масс. водного раствора бис-N-метилморфолиновой соли соединения VI (51,3 ммоль) и реакционную смесь перемешивают в течение ночи при 25°С. Добавляют 51 мл воды и 10,6 мл концентрированной HCl и смесь перемешивают несколько минут при 25°С. Полученное твердое вещество отфильтровывают и фильтрат декантируют. Органический слой промывают 51 мл воды и 26 мл рассола, получают 181,7 г 2,13%-ного масс./масс. раствора соединения VII в МеТГФ (выход 48%).
Пример 29: В 205 мл МеТГФ растворяют 19,80 г (113 ммоль) CDMT. Добавляют 13 мл (118 ммоль) NMM и смесь перемешивают при 25°С в течение 2 часов. Смешивают 35 г 25,5%-ного масс./масс. водного раствора бис-N-метилморфолиновой соли соединения VI (51,3 ммоль) с 14,3 мл (102,5 ммоль) триэтиламина, затем добавляют к смеси CDMT и бис-N-метил-морфолиновую соль соединения VI (NMM), и реакционную смесь перемешивают в течение ночи при 25°С. Добавляют 51 мл воды и 19,9 мл концентрированной HCl и смесь перемешивают несколько минут при 25°С. Полученное твердое вещество отфильтровывают и фильтрат декантируют. Органический слой промывают 51 мл воды и 26 мл рассола, получают 163,6 г 2,56%-ного масс./масс. раствора соединения VII в МеТГФ (выход 52%).
Пример 30: В 29 мл МеТГФ растворяют 2,83 г (16 ммоль) CDMT. Добавляют 1,9 мл (18 ммоль) NMM и смесь перемешивают при 20°С в течение 1 часа. Смешивают 52,01 г 2,59%-ного масс./масс. раствора соединения VII в МеТГФ (8,7 ммоль) (из примера 25) с 1,82 г NMHA и 232 мкл (2,9 ммоль) NMM и полученный раствор добавляют к смеси CDMT-NMM в МеТГФ. Реакционную смесь перемешивают в течение ночи при 20°С. Добавляют 14,6 мл воды и 1 мл концентрированной HCl. Смесь декантируют и органический слой последовательно промывают 14,6 мл воды, 14,6 мл воды, содержащей 150 мг NaOH, и 7,3 мл воды, затем сушат над сульфатом магния. Нерастворимые вещества отфильтровывают, получают 83,1 г 2,51%-ного масс./масс. раствора соединения XVII в МеТГФ (выход 96%; 57% от N-метил-морфолиновой соли VI).
Пример 31: В 29 мл изопропилацетата растворяют 2,83 г (16 ммоль) CDMT. Добавляют 1,9 мл (18 ммоль) NMM и смесь перемешивают при 20°С в течение 1 часа. Смешивают 55,5 г 1,3%-ного масс./масс. раствора соединения VII в изопропилацетате (4,7 ммоль) (из примера 26) с 1,82 г N-метил-5-гексен-1-амина (NMHA) и 232 мкл (2,9 ммоль) NMM и полученный раствор добавляют к смеси CDMT-NMM в изопропилацетате. Реакционную смесь перемешивают в течение ночи при 20°С. Добавляют 14,6 мл воды и 1 мл концентрированной HCl. Смесь декантируют, и органический слой последовательно промывают 14,6 мл воды, 14,6 мл воды, содержащей 150 мг NaOH, и 7,3 мл воды, затем сушат над сульфатом магния. Нерастворимые вещества отфильтровывают, получают 82,51 г 1,43%-ного масс./масс. раствора соединения XVII в изопропилацетате (выход количественный; 33% от бис-N-метилморфолиновой соли VI).
Пример 32: В 29 мл ацетона растворяют 2,83 г (16 ммоль) CDMT. Добавляют 1,9 мл (18 ммоль) NMM и смесь перемешивают при 20°С в течение 1 часа. Смешивают 43,3 г 1,44%-ного масс./масс. раствора соединения VII в ацетоне (4,7 ммоль) (из примера 27) с 1,82 г NMHA и 232 мкл (2,9 ммоль) NMM и полученный раствор добавляют к смеси CDMT-NMM в ацетоне. Реакционную смесь перемешивают в течение ночи при 20°С. Ацетон удаляют в вакууме и остаток распределяют в смеси [толуол (59 мл) - вода (14,6 мл) - концентрированная HCl (1 мл)]. Слои разделяют и органический слой последовательно промывают 14,6 мл воды, 14,6 мл воды, содержащей 150 мг NaOH, и 7,3 мл воды, затем сушат над сульфатом магния. Нерастворимые вещества отфильтровывают, получают 74,27 г 1,41%-ного масс./масс. раствора соединения XVII в толуоле (выход количественный, 29% от бис-N-метилморфолиновой соли VI).
Пример 33: В 103 мл МеТГФ растворяют 9,90 г (56,4 ммоль) CDMT. Добавляют 6,8 мл (61,5 ммоль) NMM и смесь перемешивают при 25°С в течение 2 часов. Смешивают 162,2 г 2,13%-ного масс./масс. раствора соединения VII в МеТГФ (из примера 28) с 0,71 мл (5,1 ммоль) триэтиламина и затем к смеси CDMT-NMM добавляют 6,38 г (56,4 ммоль) NMHA. Реакционную смесь перемешивают 3 часа при 25°С. Добавляют 51 мл воды и 3 мл концентрированной HCl и смесь перемешивают несколько минут. Полученное твердое вещество отфильтровывают и фильтрат декантируют. Органический слой последовательно промывают 26 мл воды, 51 мл воды, содержащей 0,6 г NaOH, и 26 мл воды, затем сушат над сульфатом магния. Нерастворимые вещества отфильтровывают, получают 265,4 г 2,51%-ного масс./масс. раствора соединения XVII в МеТГФ (выход количественный; 54% от бис-N-метилморфолиновой соли VI).
Пример 34: В 103 мл МеТГФ растворяют 9,90 г (56,4 ммоль) CDMT. Добавляют 6,8 мл (61,5 ммоль) NMM и смесь перемешивают при 25°С в течение 2 часов. Смешивают 180,2 г 2,56%-ного масс./масс. раствора соединения VII в МеТГФ (из примера 29) с 0,71 мл (5,1 ммоль) триэтиламина и затем к смеси CDMT-NMM добавляют 6,38 г (56,4 ммоль) NMHA. Реакционную смесь перемешивают 3 часа при 25°С. Добавляют 51 мл воды и 3 мл концентрированной HCl, и смесь перемешивают несколько минут. Полученное твердое вещество отфильтровывают и фильтрат декантируют. Органический слой последовательно промывают 26 мл воды, 51 мл воды, содержащей 0,6 г NaOH, и 26 мл воды, затем сушат над сульфатом магния. Нерастворимые вещества отфильтровывают, получают 255,4 г 2,91%-ного масс./масс. раствора соединения XVII в МеТГФ (выход количественный; 58% от бис-N-метилморфолиновой соли VI).
Пример 35: К 82,1 г 2,51%-ного масс./масс. раствора соединения XVII в МеТГФ (из примера 30) добавляют 14,6 мл метанола и 76 мкл метансульфоновой кислоты. Раствор кипятят с обратным холодильником в течение ночи. Добавляют 125 мг карбоната натрия и смесь кипятят с обратным холодильником еще один час. Отгоняют 51 мл растворителя (удаление метанола) и концентрат последовательно промывают 7,3 мл воды и 3,5 мл рассола, сушат над сульфатом магния и фильтруют. Получают 31,1 г 5,3%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в МеТГФ (выход 71%; 41% от бис-N-метилморфолиновой соли VI).
Пример 36: Концентрируют в вакууме 81,4 г 1,43%-ного масс./масс. раствора соединения XVII в изопропилацетате (из примера 31) и остаток снова растворяют в 59 мл толуола. Добавляют 14,6 мл метанола и 76 мкл метансульфоновой кислоты и раствор кипятят с обратным холодильником в течение ночи. Добавляют 125 мг карбоната натрия и смесь кипятят с обратным холодильником еще 1 час. Отгоняют 38 мл растворителя (удаление метанола) и концентрат последовательно промывают 7,3 мл воды и 3,5 мл рассола, сушат над сульфатом магния и фильтруют, получают 28,9 г 3,9%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в толуоле (выход 86%; 28% от бис-N-метил-морфолиновой соли VI).
Пример 37: Концентрируют в вакууме 85,4 г 1,45%-ного масс./масс. раствора соединения XVII в метилизопропилкетоне (MIK) (из примера 31) и остаток снова растворяют в 59 мл толуола. Добавляют 14,6 мл метанола и 76 мкл метансульфоновой кислоты и раствор кипятят с обратным холодильником в течение ночи. Добавляют 125 мг карбоната натрия и смесь кипятят с обратным холодильником еще 1 час. Отгоняют 28 мл растворителя (удаление метанола) и концентрат последовательно промывают 7,3 мл воды и 3,5 мл рассола, сушат над сульфатом магния и фильтруют, получают 38,0 г 3,2%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в толуоле (выход 86%; 30% от бис-N-метилморфолиновой соли VI).
Пример 38: К 73 г 1,41%-ного масс./масс. раствора соединения XVII в толуоле (из примера 32) добавляют 14,6 мл метанола и 76 мкл метансульфоновой кислоты. Раствор кипятят с обратным холодильником в течение ночи. Добавляют 125 мг карбоната натрия и смесь кипятят с обратным холодильником еще 1 час. Отгоняют 31 мл растворителя (удаление метанола) и концентрат последовательно промывают 7,3 мл воды и 3,5 мл рассола, сушат над сульфатом магния и фильтруют, получают 63,0 г 1,75%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в толуоле (выход 95%; 28% от бис-N-метилморфолиновой соли VI).
Пример 39: К 135 г 2,51%-ного масс./масс. раствора соединения XVII в МеТГФ (из примера 33) добавляют 26 мл метанола и 84 мкл метансульфоновой кислоты. Раствор кипятят с обратным холодильником в течение ночи. Добавляют 136 мг карбоната натрия и смесь кипятят с обратным холодильником еще 1 час. Отгоняют приблизительно 52 мл растворителя (удаление метанола) и концентрат промывают 26 мл воды, сушат над сульфатом магния и фильтруют, получают 55,16 г 5,30%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в МеТГФ (выход 76%; 41% от бис-N-метилморфолиновой соли VI).
Пример 40: К 135 г 2,51%-ного масс./масс. раствора соединения XVII в МеТГФ (из примера 33) добавляют 154 мл толуола. Отгоняют 32 мл растворителя и концентрат охлаждают до 60°С. Добавляют 26 мл метанола и 84 мкл метансульоновой кислоты и раствор кипятят с обратным холодильником в течение ночи. Добавляют 136 мг карбоната натрия и смесь кипятят с обратным холодильником еще 1 час. Отгоняют приблизительно 52 мл растворителя (удаление метанола) и концентрат промывают 26 мл воды, сушат над сульфатом магния и фильтруют, получают 88,74 г 5,21%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в МеТГФ (выход количественный; 65% от бис-N-метилморфолиновой соли VI).
Пример 41: К 126 г 2,91%-ного масс./масс. раствора соединения XVII в МеТГФ (из примера 34) добавляют 154 мл толуола. Отгоняют 190 мл растворителя и концентрат охлаждают до 60°С. Добавляют 26 мл метанола и 84 мкл метансульфоновой кислоты и раствор кипятят с обратным холодильником в течение ночи. Добавляют 136 мг карбоната натрия и смесь кипятят с обратным холодильником еще 1 час. Отгоняют 20 мл растворителя (удаление метанола) и концентрат промывают 26 мл воды, сушат над сульфатом магния и фильтруют, получают 92,6 г 2,20%-ного масс./масс. раствора соединения XVIII (R1 = Ме) в МеТГФ (выход 49%; 29% от бис-N-метилморфолиновой соли VI).
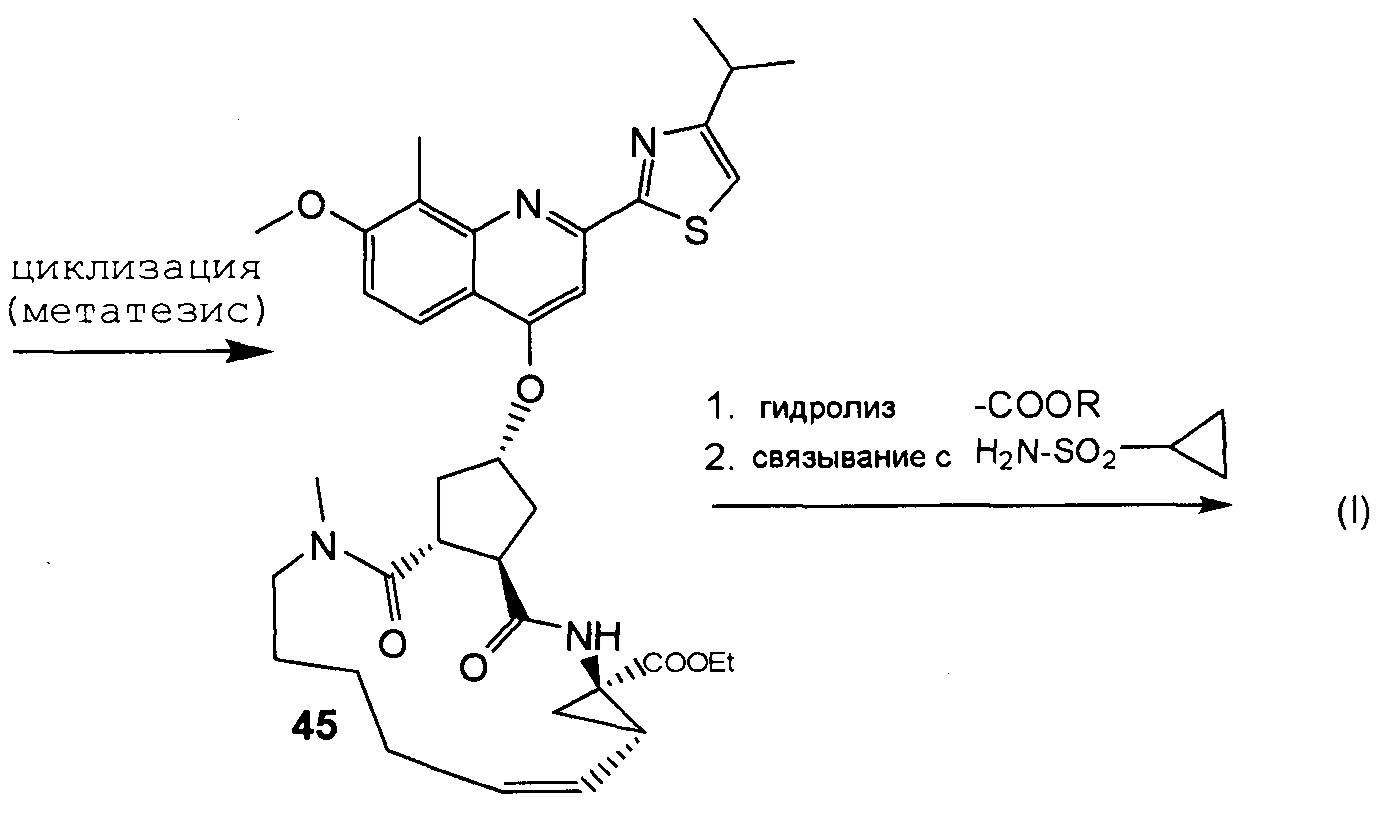
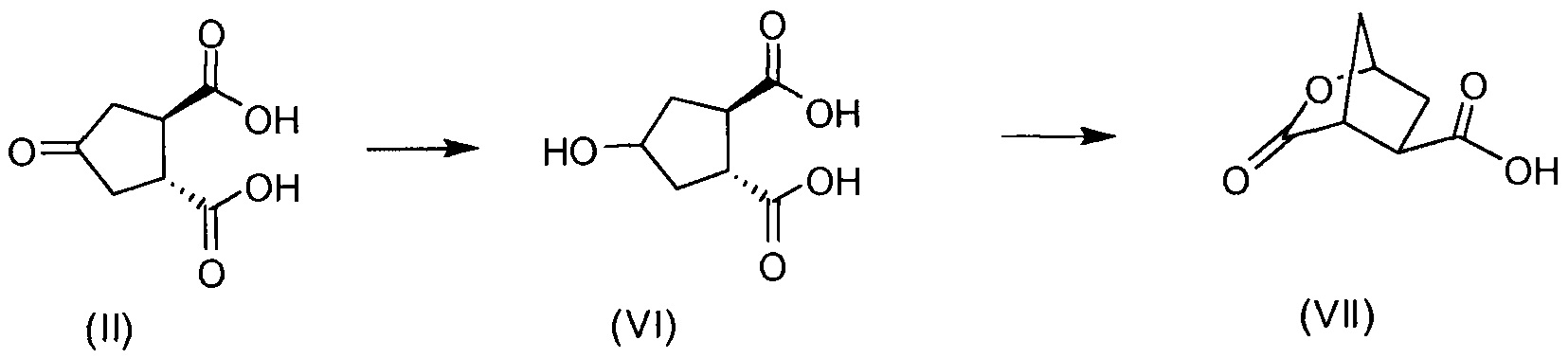
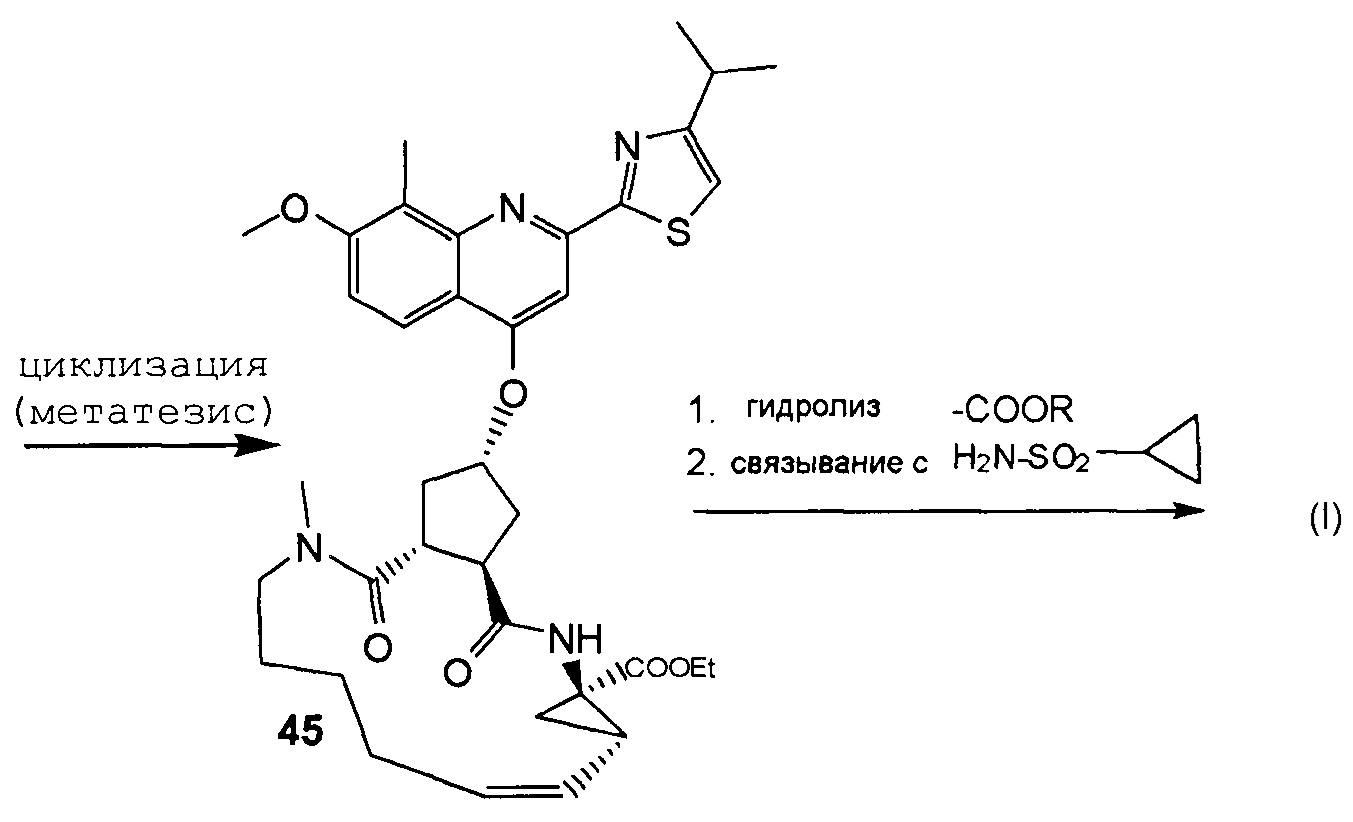
Способы и промежуточные продукты для получения макроциклического ингибитора протеазы вируса гепатита с
Полиморфные формы макроциклического ингибитора hcv
Способы и промежуточные соединения для получения макроциклического ингибитора протеазы вируса гепатита с
Тромбин-связывающие молекулы антител и их применение
Колпачок для инъекционного устройства
Захватное приспособление для ручного инъекционного устройства
Захватное приспособление для ручного инъекционного устройства
Захватное приспособление для ручного инъекционного устройства
Захватное приспособление для ручного инъекционного устройства
Способы и промежуточные продукты для получения макроциклического ингибитора протеазы вируса гепатита с
Полиморфные формы макроциклического ингибитора hcv
Способы и промежуточные соединения для получения макроциклического ингибитора протеазы вируса гепатита с
Тромбин-связывающие молекулы антител и их применение

