Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ БЛОК-СОПОЛИМЕРА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к получению мицелл, содержащих блок-сополимер и лекарственное средство, использующее указанный сополимер, и к способу получения блок-сополимера для противоракового лекарственного средства, содержащего указанную мицеллярную композицию в качестве активного ингредиента.
Предпосылки создания изобретения
Многие лекарственные средства (в частности, противораковые лекарственные средства) являются гидрофобными соединениями, большинство из которых не растворяется в воде. Чтобы получить желаемый терапевтический эффект при применении такого лекарственного средства, его обычно солюбилизируют и вводят пациенту. Поэтому солюбилизация лекарственных средств, слаборастворимых в воде (в частности, слаборастворимых в воде противораковых лекарственных средств), является важной для пероральных или парентеральных препаратов (в частности, для препаратов, предназначенных для внутривенного введения).
В качестве одного из способов солюбилизации слаборастворимых в воде противораковых лекарственных средств можно применять способ с добавлением поверхностно-активного вещества. Например, известно, что для солюбилизации паклитаксела применяют полиоксиэтиленовое производное касторового масла (кремофор). Кроме того, в качестве других способов, в патентном документе 1, патентном документе 2, патентном документе 3 и т.п. описано применение мицеллообразующего блок-сополимера в качестве носителя для лекарственного средства. В патентном документе 4, патентном документе 5 и патентном документе 6 описаны мицеллы с инкапсулированным паклитакселом, в которых в качестве носителей лекарственного средства применяют блок-сополимеры, имеющие полиэтиленгликолевые (PEG) структурные фрагменты и полиаминокислотные структурные фрагменты,
В патентном документе 5 описано, что мицеллы с инкапсулированным паклитакселом, обладающие исключительно высоким противораковым эффектом, получают, изменяя структуру полиаминокислотного структурного фрагмента блок-сополимера, который образует мицеллы, применяемые в патентном документе 4.
В патентном документе 6 описано, что когда применяют способ получения, отличный от способа, применяемого в патентном документе 5, уменьшается количество остаточных структур карбоновых кислот в структуре полиаминокислотного структурного фрагмента блок-сополимера, образующего мицеллы, и уменьшается токсичность, по сравнению с мицеллами, инкапсулирующими паклитаксел, описанными в патентном документе 5.
Список цитируемых патентных документов
Патентный документ 1: JP 6-107565 A
Патентный документ 2: JP 6-206815 A
Патентный документ 3: JP 11-335267 A
Патентный документ 4: JP 2001-226294 A
Патентный документ 5: WO 2004/082718 A
Патентный документ 6: WO 2006/033296 A
Сущность изобретения
Техническая задача
Способ получения блок-сополимера, описанный в патентном документе 6, включает первоначально введение арил(С1-С8)алкилового спирта, который может иметь заместитель, в конъюгат PEG с ацетилированной pAsp (полиаспарагиновая кислота), полученной способом, описанным в патентном документе 2, и выделение продукта. После этого, для уменьшения количества остаточных карбоксильных групп pAsp, вводят переносимый остаток мочевины pAsp и осуществляют реакцию циклизации. Однако арил(С1-С8)алкиловый спирт, который может иметь заместитель, частично отсоединяется этой, требующей нагревания реакцией второй стадии. Соответственно, было необходимо регулировать соотношения арил(С1-С8)алкилового спирта, который может иметь заместитель, вводимого как на первой, так и на второй стадии. Поэтому регулирование количества остаточных карбоксильных групп pAsp и соотношения вводимого арил(С1-С8)алкилового спирта, который может иметь заместитель, до сих пор оставалось трудной задачей.
Решение задачи
Для решения задач, описанных выше, авторы настоящего изобретения провели всестороннее исследование, в результате которого неожиданно нашли способ получения блок-сополимера, описанного в патентном документе 6, посредством способа, проводимого в одном реакторе, путем применения специфически ограниченных реакционных условий к способу получения блок-сополимера, описанному в патентном документе 5. Кроме того, авторы настоящего изобретения преодолели описанные выше производственные затруднения и, таким образом, завершили создание настоящего изобретения.
Это означает, что настоящее изобретение относится к следующему:
1) К способу получения блок-сополимера, представленного следующей формулой (1):
 ,
,
где R1 представляет собой атом водорода или (С1-С5)алкильную группу; R2 представляет собой (С1-С5)алкиленовую группу; R3 представляет собой метиленовую группу или этиленовую группу; R4 представляет собой атом водорода или (C1-C4)ацильную группу; R5 представляет собой гидроксильную группу, арил(C1-C8)алкоксигруппу, которая может иметь заместитель, или -N(R6)-CO-NHR7 (где R6 и R7, которые могут быть одинаковыми или отличаться один от другого, каждый представляет собой циклическую (С3-С6)алкильную группу или (C1-C5)алкильную группу, которая может быть замещена третичной аминогруппой); n составляет 20-500; m составляет 2-200; а составляет 0-100; и b составляет 0-100, при условии, что сумма а и b составляет больше чем или равна 1 и не больше чем m; доля группы R5, представляющей собой гидроксильную группу, составляет 0-5% от m; доля группы R5, представляющей собой арил(C1-C8)алкоксигруппу, у которой может быть заместитель, составляет 10-80% от m; и доля группы R5, представляющей собой -N(R6)-CO-NHR7, составляет 11-30% от m,
включающему взаимодействие соединения, представленного следующей формулой (2):
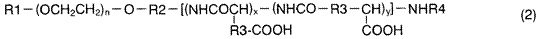 ,
,
где R1 представляет собой атом водорода или (С1-С5)алкильную группу; R2 представляет собой (С1-С5)алкиленовую группу; R3 представляет собой метиленовую группу или этиленовую группу; R4 представляет собой атом водорода или (C1-C4)ацильную группу; n составляет 20-500; x составляет 0-100; и y составляет 0-100, при условии, что сумма х и y составляет 2-200,
с арил(C1-C8)алкиловым спиртом, который может иметь заместитель, и с соединением на основе карбодиимида в количестве 2⋅(x+y) эквивалентов или более относительно количества карбоксильных групп в соединении формулы (2) (сумма х и y) в растворителе при 15-30°C в течение 2-48 часов.
2) Способ получения блок-сополимера, как описано выше в пункте 1), где R1 представляет собой метильную группу; R2 представляет собой триметиленовую группу; R3 представляет собой метиленовую группу; R4 представляет собой ацетильную группу; n составляет 80-400; m составляет 15-60; а составляет 5-60; и b составляет 5-60.
3) Способ получения блок-сополимера, как описано выше в пункте 1) или 2), где указанное соединение на основе карбодиимида представляет собой диэтилкарбодиимид, диизопропилкарбодиимид, дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид, или его соль с неорганической кислотой.
4) Способ получения блок-сополимера, как описано выше в любом из пунктов 1)-3), где указанное соединение на основе карбодиимида представляет собой диизопропилкарбодиимид.
Полезные эффекты изобретения
Способ получения блок-сополимера согласно настоящему изобретению является таким, что когда температуру реакции строго регулируют в способе получения, описанном в патентном документе 5, и реакцию осуществляют с использованием соединения на основе карбодиимида в количестве 2⋅(х+у) эквивалентов или более относительно количества карбоксильных групп в соединении формулы (2) (сумма х и у), тогда, вопреки ожиданиям, получают не блоксополимер, описанный в патентном документе 5, а блок-сополимер, описанный в патентном документе 6.
В способе получения блок-сополимера согласно настоящему изобретению можно регулировать скорость введения арил(С1-С8)алкилового спирта, который может иметь заместитель, в соединение, представленное формулой (2). Причиной этого является то, что указанный арил(С1-С8)алкиловый спирт, который уже был однажды введен, не отделяется при циклизации, как в случае способа получения, описанного в патентном документе 6, и количество свободного арил(С1-С8)алкилового спирта в реакционном растворе не увеличивается. В результате этого можно существенно уменьшить число непрореагировавших карбоксильных групп pAsp в соединении, представленном формулой (2). Поэтому в общепромышленном смысле и в сравнении со способом получения, описанном в патентном документе 6, способ получения блок-сополимера согласно настоящему изобретению представляет собой превосходный производственный способ, в котором получение продукта можно легко регулировать посредством одностадийной реакции.
В результате этого, по сравнению со способом получения блок-сополимера, описанным в патентном документе 6, каждую реакцию и каждый процесс выделения проводят только однократно, что дает возможность сократить производственный период и уменьшить количество используемого растворителя приблизительно до половины того количества, которое используют согласно патентному документу 6.
Описание вариантов осуществления изобретения
Настоящее изобретение относится к способу получения блок-сополимера, представленного следующей формулой (1) [где R1 представляет собой атом водорода или (С1-С5)алкильную группу; R2 представляет собой (С1-С5)алкиленовую группу; R3 представляет собой метиленовую группу или этиленовую группу; R4 представляет собой атом водорода или (С1-С4)ацильную группу; R5 представляет собой гидроксильную группу, арил(С1-С8)алкоксигруппу, которая может иметь заместитель, или -N(R6)-CO-NHR7 (где R6 и R7, которые могут быть одинаковыми или отличаться один от другого, каждый представляет собой циклическую (С3-С6)алкильную группу или (С1-С5)алкильную группу, которая может быть замещена третичной аминогруппой); n составляет 20-500; m составляет 2-200; а составляет 0-100; b составляет 0-100, при условии, что сумма а и b составляет больше чем или равна 1 и не больше чем m; доля группы R5, представляющей собой гидроксильную группу, составляет 0%-5% от m, доля группы R5, представляющей собой арил(С1-С8)алкоксигруппу, которая может иметь заместитель, составляет 10%-80% от m, и доля группы R5, представляющей собой -N(R6)-CO-NHR7, составляет 11%-30% от m], включающему взаимодействие соединения, представленного следующей формулой (2) [где R1 представляет собой атом водорода или (С1-С5)алкильную группу; R2 представляет собой (С1-С5)алкиленовую группу; R3 представляет собой метиленовую группу или этиленовую группу; R4 представляет собой атом водорода или (C1-C4)ацильную группу; n составляет 20-500; x составляет 0-100; и y составляет 0-100, при условии, что сумма х и y составляет 2-200; где численные значения являются средними значениями], которое имеет структурный фрагмент полиэтиленгликоля (PEG) и полиаминокислотный структурный фрагмент, с арил(C1-C8)алкиловым спиртом, который может иметь заместитель, и с соединением на основе карбодиимида в количестве 2⋅(x+y) эквивалентов или более относительно количества карбоксильных групп в соединении формулы (2) (сумма х и y), в растворителе при 15-30°C (предпочтительно, при 20-30°C) в течение 2-48 часов.
,
В соединениях, представленных формулами (1) и (2) и применяемых согласно настоящему изобретению, R1 может представлять собой атом водорода или (C1-C5)алкильную группу, но предпочтительной является (C1-C5)алкильная группа. Конкретные примеры (C1-C5)алкильной группы могут включать, но, но, не ограничиваясь ими, метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, втор-бутильную группу, трет-бутильную группу и н-пентильную группу, но особо предпочтительной является метильная группа.
Конкретные примеры (C1-C5)алкиленовой группы R2 могут включать, но, не ограничиваясь ими, метиленовую группу, этиленовую группу, триметиленовую группу и тетраметиленовую группу, причем предпочтительными являются этиленовая группа и триметиленовая группа.
R3 может представлять собой метиленовую группу или этиленовую группу, но предпочтительной является метиленовая группа.
R4 может представлять собой атом водорода или (C1-C4)ацильную группу, причем предпочтительной является (C1-C4)ацильная группа. Конкретные примеры могут включать, но, не ограничиваясь ими, формильную группу, ацетильную группу, пропионильную группу и бутироильную группу, причем особо предпочтительной является ацетильная группа.
В соединении, представленном формулой (2), n составляет 20-500 (предпочтительно, 80-400). x составляет 0-100 (предпочтительно, 5-60). y составляет 0-100 (предпочтительно, 5-60). Сумма х и у составляет 2-200 (предпочтительно, 10-100 и, особо предпочтительно, 5-60).
В соединении, представленном формулой (1), арил(C1-C8)алкоксигруппа для R5 может представлять собой линейную или разветвленную (C1-C8)алкоксигруппу, с которой связана ароматическая углеводородная группа, такая как фенильная группа или нафтильная группа. Конкретные примеры могут включать, но, не ограничиваясь ими, бензилоксигруппу, фенетилоксигруппу, фенилпропоксигруппу, фенилбутоксигруппу, фенилпентилоксигруппу, фенилгексилоксигруппу, фенилгептилоксигруппу, фенилоктилоксигруппу, нафтилэтоксигруппу, нафтилпропоксигруппу, нафтилбутоксигруппу и нафтилпентилоксигруппу.
Примеры заместителя для арил(C1-C8)алкоксигруппы, которая может иметь заместитель, могут включать, но, не ограничиваясь ими, низшие алкоксигруппы, такие как метоксигруппа, этоксигруппа, изопропоксигруппа, н-бутоксигруппа и трет-бутоксигруппа; атомы галогена, такие как атом фтора, атом хлора и атом брома; нитрогруппу; и цианогруппу. В настоящее изобретение включены замещенные формы, имеющие число замещений, составляющее от одного до максимально допустимого числа при каждом возможном положении замещения, но предпочтительной является незамещенная форма.
Указанная арил(C1-C8)алкоксигруппа, которая может иметь заместитель, может представлять собой незамещенную фенил(C1-C6)алкоксигруппу. Примеры могут включать, но, не ограничиваясь ими, незамещенную бензилоксигруппу, незамещенную фенетилоксигруппу, незамещенную фенилпропоксигруппу, незамещенную фенилбутоксигруппу, незамещенную фенилпентилоксигруппу и незамещенную фенилгексилоксигруппу. Предпочтительные примеры могут включать, но, не ограничиваясь ими, незамещенную бензилоксигруппу и незамещенную фенилбутоксигруппу.
Конкретные примеры циклической (C3-C6)алкильной группы или (C1-C5)алкильной группы, которая может быть замещена третичной аминогруппой, для R6 и R7 могут включать, но, не ограничиваясь ими, циклопропильную группу, циклопентильную группу, циклогексильную группу, метильную группу, этильную группу, изопропильную группу, н-бутильную группу, 3-диметиламинопропильную группу и 5-диметиламинопентильную группу. Предпочтительными среди них являются этильная группа, изопропильная группа, циклогексильная группа и 3-диметиламинопропильная группа, и особо предпочтительной является изопропильная группа.
Для соединения, представленного формулой (1), n, предпочтительно, находится в том же диапазоне, что и для соединения формулы (2), и m составляет 2-100 (предпочтительно, 10-100 и, особо предпочтительно, 15-60). Сумма а и b составляет больше, чем или равна 1 и не больше, чем m.
В формуле (1) m означает степень полимеризации аминокислотной структурной единицы полиаминокислотного структурного фрагмента. Полиаминокислотный структурный фрагмент включает разнообразные структурные единицы, где R5 в формуле (1) представляет собой гидроксильную группу, арил(C1-C8)алкоксигруппу, которая может иметь заместитель, или -N(R6)-CO-NHR7 и структурную единицу, имеющую структуру циклического имида.
Доля R5 в формуле (1), представляющего собой гидроксильную группу, составляет 0%-5% от m (предпочтительно, 0%-3%). Доля группы R5, представляющей собой арил(C1-C8)алкоксигруппу, которая может иметь заместитель, составляет 10%-80% от m (предпочтительно, 20%-80%). Доля группы R5, представляющей собой -N(R6)-CO-NHR7, составляет 11%-30% от m.
Особенно предпочтительно, когда доля R5, представляющего собой гидроксильную группу в соединении, представленном формулой (1), составляет 0% от m. Доля группы R5, представляющей собой гидроксильную группу, составляющая 0% от m, означает, что все карбоксильные группы полиаминокислотного структурного фрагмента соединения, представленного формулой (2), были замещены арил(C1-C8)алкоксигруппой, которая может иметь заместитель, и/или -N(R6)-CO-NHR7. При этом часть значения m, соответствующую гидроксильным группам, можно анализировать высокоэффективной жидкостной хроматографией с использованием анионообменной колонки, и ситуация, при которой рассматриваемое соединение не задерживается в колонке, означает, что эта часть m составляет 0%. Кроме того, согласно настоящему изобретению, часть значения m, соответствующую гидроксильным группам, анализируют потенциометрическим титрованием с основанием, и когда m составляет 0%, тогда часть значения m, соответствующую гидроксильным группам, находят равной 0,1 ммоль/г или менее.
В полиаминокислотном структурном фрагменте соединений, представленных формулой (1) и формулой (2), применяемых согласно настоящему изобретению, соответствующие фрагменты с аминокислотными структурными единицами могут быть связанными случайным образом или они могут быть связанными в блок-форме.
Указанный арил(C1-C8)алкиловый спирт, который может иметь заместитель, который применяют согласно настоящему изобретению, представляет собой спирт, соответствующий указанной выше арил(C1-C8)алкоксигруппе, которая может иметь заместитель,
Для арил(C1-C8)алкилового спирта, который может иметь заместитель, можно использовать соединения, которые являются коммерчески доступными. Кроме того, можно использовать соединения, получаемые известными способами органического синтеза, и соединения, получаемые с применением известных органических реакций.
Далее будет дано объяснение реакции между соединением, представленным формулой (2), и соединением на основе карбодиимида.
Данную реакцию осуществляют в определенных растворителях, и примеры используемого растворителя могут включать, но, не ограничиваясь ими, полярные растворители (такие как диметилформамид (ДМФ), диметилсульфоксид (ДМСО), ацетонитрил, тетрагидрофуран и диоксан) и неполярные растворители (такие как бензол, н-гексан и диэтиловый эфир). Дополнительные примеры могут включать воду и ее смешанные растворители, но, не ограничиваясь ими. Количество используемого растворителя обычно составляет приблизительно 1-100-кратное количество по массе в расчете на соединение исходного вещества.
Указанное соединение на основе карбодиимида, используемое в данной реакции, может представлять собой соединение на основе карбодиимида, имеющее циклическую (C3-C6)алкильную группу или (C1-C5)алкильную группу, которая может быть замещена третичной аминогруппой. Конкретные примеры могут включать, но, не ограничиваясь ими, диэтилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC⋅HCl), дициклогексилкарбодиимид (DCC) и диизопропилкарбодиимид (DIPCI). Среди этих соединений, предпочтительным соединением на основе карбодиимида является DCC или DIPCI, и особо предпочтительным является DIPCI.
Количество соединения на основе карбодиимида, используемого в данной реакции, составляет 2⋅(x+y) эквивалентов или больше и, более предпочтительно, от 2⋅(x+y) эквивалентов до 5⋅(x+y) эквивалентов относительно количества карбоксильных групп в соединении формулы (2) (сумма х и y). Когда избыточное количество соединения на основе карбодиимида используют при температуре реакции, составляющей 15-30°C, введение переносимого остатка мочевины в полиаминокислотный структурный фрагмент соединения, представленного формулой (2), и реакцию циклизации можно осуществлять, не вызывая отщепления арил(C1-C8)алкилового спирта, который может иметь заместитель. Соединение на основе карбодиимида можно использовать так, чтобы его общее количество можно было добавлять в начале реакции или это количество можно было подходящим образом добавлять раздельными порциями, вносимыми в ходе протекания реакции. Предпочтительно, реакцию введения арил(С1-С8)алкилового спирта, который может иметь заместитель, реакцию введения переносимого остатка мочевины и реакцию циклизации осуществляют, используя 2⋅(x+y) эквивалентов или более соединения на основе карбодиимида, затем добавляют 0,5⋅(x+y) эквивалентов или более соединения на основе карбодиимида, так чтобы все карбоксильные группы полиаминокислотного структурного фрагмента, представленного формулой (1), могли провзаимодействовать и, таким образом, реакция введения переносимого остатка мочевины и реакция циклизации будут завершены.
Во время реакции между соединением, представленным формулой (2), и соединением на основе карбодиимида могут быть включены вспомогательные вещества, такие как N-гидроксисукцинимид, 1-гидроксибензотриазол (HOBt), имид N-гидрокси-5-норборнен-2,3-дикарбоновой кислоты (HOBN), 4-диметиламинопиридин (DMAP), N,N-диизопропилэтиламин или триэтиламин, предпочтительным среди которых является DMAP. Когда используют вспомогательные вещества, их используемое количество составляет приблизительно от 0,1⋅(x+y) эквивалентов до 5⋅(x+y) эквивалентов (предпочтительно, приблизительно от 0,2⋅(x+y) эквивалентов до 2⋅(x+y) эквивалентов) относительно количества карбоксильных групп в соединении формулы (2) (сумма х и y).
Количество арил(C1-C8)алкилового спирта, используемого в данной реакции, составляет 0,4-1,0 молярных эквивалентов относительно 1 моля карбоксильных групп соединения, представленного формулой (2). Когда количество используемого арил(C1-C8)алкилового спирта устанавливают соответственно средней степени полимеризации соединения, представленного формулой (2), можно контролировать количество вводимого арил(C1-C8)алкилового спирта.
Температура реакции обычно составляет 15-30°C, но предпочтительно взаимодействие осуществляют при 20-30°C (особо предпочтительно, при 22-27°C). Время реакции составляет 2-48 часов (предпочтительно, 6-36 часов).
Примеры
Далее настоящее изобретение будет описано на конкретных примерах, но настоящее изобретение не должно ограничиваться следующими примерами.
Расчет реакционного отношения 4-фенил-1-бутанола (PhBuOH) в приведенных примерах выполняли следующим образом.
Расчет реакционного отношения 4-фенил-1-бутанола
Если массу всего реакционного раствора перед добавлением DIPCI обозначить как Q1,
общую массу реакционного раствора после добавления DIPCI обозначить как Q2,
значение площади пика, полученного для раствора образца перед добавлением DIPCI (количество отобранного образца: Р1) и определенного ВЭЖХ с обращенной фазой, как описано ниже, обозначить как AS, и
значение площади пика, полученного для раствора образца после добавления DIPCI (количество отобранного образца: Р2) и определенного ВЭЖХ с обращенной фазой, как описано ниже, обозначить как AT, то реакционное отношение выражается следующей формулой (при использовании такого же измерительного сосуда, как и сосуды, используемые для двух растворов образцов):
Реакционное отношение PhBuOH (%)={(1-AT×P1×Q2)/(AS×P2×Q1)}×100
Условия измерений, проводимых анионообменной ВЭЖХ в примерах, были следующими. Следует отметить, что при анионообменной ВЭЖХ, если реактив имеет карбоксильные группы, он удерживается в колонке.
Условия измерений при анионообменной ВЭЖХ
Колонка: TSKgel DEAE-5PW (производства Tosoh Corp.).
Концентрация образца: 5 мг/мл.
Инъецируемый объем: 20 мкл.
Температура колонки: 40°C.
Подвижная фаза:
(A) 20 мМ буферный раствор трис-хлористоводородная кислота (pH 8,0):ацетонитрил=80:20,
(B) 20 мМ буферный раствор трис-хлористоводородная кислота+1M водный раствор хлорида натрия (pH 8,0):ацетонитрил=80:20.
Скорость потока: 1 мл/мин.
Градиентные условия: B % (минуты): 10(0), 10(5), 100(40), 10(40,1), стоп (50,1).
Детектор: спектрофотометрический детектор ультрафиолетового и видимого света (детектируемая длина волны: 260 нм).
Условия измерений, осуществляемых при использовании ВЭЖХ с обращенной фазой в примерах, были следующими. Кроме того, такими же были условия измерений, используемые для определения реакционного отношения 4-фенил-1-бутанола.
Условия измерений при ВЭЖХ с обращенной фазой
[ВЭЖХ] - метод абсолютной калибровочной кривой -
Колонка: InertsilL ODS-3, 5 мкм (внутренний диаметр 4,6 мм, длина 150 мм).
Инъецируемый объем: 20 мкл.
Температура колонки: 40°C.
Подвижная фаза: 0,1% H3PO4/(H2O:CH3CN=60:40).
Скорость потока: 1,0 мл/мин.
Детектор: спектрофотометрический детектор ультрафиолетового и видимого света (детектируемая длина волны: 260 нм).
Пример 1
Получение блок-сополимера 1
ДМФ (1132 мл) добавляли к 65,0 г конъюгата PEG (средняя молекулярная масса: 12000) с ацетилированной pAsp (полиаспарагиновая кислота; средняя степень полимеризации: 41,6) (в формуле (2) R1 представляет собой метильную группу, R2 представляет собой триметиленовую группу, R3 представляет собой метиленовую группу, R4 представляет собой ацетильную группу, n составляет приблизительно 272, x составляет приблизительно 10,4, и y составляет приблизительно 31,2; далее в настоящем описании используется аббревиатура PEG-pAsp-Ac-1), полученного способом, описанным в патентном документе 2; соединение растворяли при 35°C и туда же добавляли DMAP (19,2 г) и 4-фенил-1-бутанол (15,9 г: 0,106 моль; 0,67 молярных эквивалентов относительно 1 моля карбоксильных групп в PEG-pAsp-Ac-1). Часть реагента, прилипшую во время добавления, смывали туда же диметилформамидом (66 мл). После подтверждения того, что соединения растворились, реакционный раствор доводили до 25°С и добавляли DIPCI (39,7 г: 2⋅(x+y) эквивалентов относительно карбоксильных групп в PEG-pAsp-Ac-1=83,2 эквивалента), диметилформамидом (60 мл) смывали туда же часть реагента, прилипшую при добавлении, и реакционному раствору давали возможность взаимодействовать в течение 22 часов при 25°C. В этом случае, через 20 часов после начала реакции, реакционное отношение сложноэфирных связей 4-фенил-1-бутанола становилось постоянным. С другой стороны, согласно анализу, проведенному анионообменной ВЭЖХ, реагенты все еще удерживались в колонке. Через 22 часа после начала реакции добавляли DIPCI (9,92 г: 0,5⋅(x+y) эквивалентов в расчете на карбоксильные группы в PEG-pAsp-Ac-1=20,8 эквивалентов), продолжая проведение реакции. Анионообменной ВЭЖХ проверяли прекращение удерживания реагентов в колонке, и через 26 часов после начала реакции ее останавливали. Реакционный раствор по каплям добавляли к смешанному растворителю из гептана и этилацетата, и смесь перемешивали. Смесь оставляли стоять в течение ночи, полученный при этом осадок собирали фильтрованием и сушили при пониженном давлении. Получали 76,2 г неочищенных кристаллов.
Полученные неочищенные кристаллы (75,0 г) растворяли в ДМФ (1050 мл) и затем туда же добавляли катионообменную смолу DOWEX 50w8 (248 мл). Кроме того, часть вещества, прилипшую во время добавления, смывали туда же ДМФ (75 мл), и полученную смесь перемешивали в течение 3 часов. Катионообменную смолу DOWEX 50w8 удаляли фильтрованием, промывая этилацетатом, и затем реакционный раствор, полученный таким образом, добавляли по каплям к смешанному растворителю из гептана и этилацетата. Смесь перемешивали. Смесь оставляли стоять в течение ночи и осадок, полученный таким образом, собирали фильтрованием и сушили при пониженном давлении. Получали 73,5 г блок-сополимера 1.
Блок-сополимер 1 (17,60 мг) растворяли в 1 мл ацетонитрила и туда же добавляли 1 мл воды и 2 мл 0,5н. водного раствора гидроксида натрия. Смесь перемешивали в течение 60 минут при комнатной температуре для гидролиза сложноэфирных связей, и затем смесь нейтрализовали добавлением 1 мл 4%-ного водного раствора фосфорной кислоты. Количество жидкости доводили до 25 мл добавлением 50%-ного гидратированного ацетонитрила. ВЭЖХ с обращенной фазой анализировали количество 4-фенил-1-бутанола, выделенного из полученной жидкости. По данным этого анализа, количество сложноэфирно связанного 4-фенил-1-бутанола составило 16,3% (масс./масс.) PEG-pAsp-Ac-1. Реакционное отношение сложноэфирных связей 4-фенил-1-бутанола составило 82,4%, и отношение введения 4-фенил-1-бутанола составило 55,2% от количества карбоксильных групп PEG-pAsp-Ac-1.
Полученный блок-сополимер 1 анализировали анионообменной ВЭЖХ в условиях измерений, описанных ниже, не обнаружив каких-либо пиков, которые указывали на удерживание в колонке.
Точно отвешивали навеску блок-сополимера 1 (501,4 мг), к которой добавляли 25 мл этанола для суспендирования сополимера. Затем, чтобы растворить полученную суспензию, к ней добавляли 35 мл воды. Полученный раствор блок-сополимера 1 титровали (по методике потенциометрического титрования) жидким гидроксидом калия (0,1 моль/л) и по следующей формуле рассчитывали число карбоксильных групп, приходящихся на 1 грамм блок-сополимера 1. В результате было обнаружено, что это число карбоксильных групп составило 0,05 ммоль/г. Это означает, что блок-сополимер 1 не имел каких-либо остаточных карбоксильных групп, поскольку, как описано выше, в том случае, когда доля карбоксильных групп составляет 0%, число карбоксильных групп не превышает 0,1 ммоль/г.
Число карбоксильных групп на грамм блок-сополимера=[(Титр образца, мл)-(Титр бланка, мл)]×0,1×f/образец (г) (ммоль/г)
Примечание: f - фактор 0,1 моль/л жидкого гидроксида калия
Чтобы проверить количество связывающихся переносимых остатков мочевины в блок-сополимере, в нем измеряли количество диизопропилмочевины. Точно отвешивали навеску блок-сополимера 1 (25,18 мг), к которой добавляли раствор внутреннего стандарта, доводя объем точно до 1 мл; полученную смесь использовали в качестве раствора образца. Отдельно в сосуде, в который предварительно вносили 5 мл раствора внутреннего стандарта, точно отвешивали изопропилизоцианат и туда же добавляли раствор внутреннего стандарта, доводя объем точно до 20 мл. Точно отвешивали 2,5 мл полученной жидкости и к ней добавляли раствор внутреннего стандарта, доводя объем точно до 50 мл. Полученный раствор использовали в качестве стандартного. Газовую хроматографию проводили с 1 мкл раствора образца и 1 мкл стандартного раствора в условиях, описанных ниже, определяя отношения QT и QS площади пика изопропилизоцианата относительно площади пика материала внутреннего стандарта, соответственно. Количество диизопропилмочевины (% (масс./масс.)) в блок-сополимере 1 рассчитывали по формуле, представленной ниже. В результате было установлено, что это количество составило 3,5% (масс./масс.).
Количество диизопропилмочевины (%)=(QT/QS)×[(масса изопропилизоцианата (г))/(масса образца (г)]×[(100-содержание воды в образце (%))/100]×1/4×1,695
Раствор внутреннего стандарта: Ацетонитрильный раствор этилацетата (1→2000)
Условия теста:
Детектор: Пламенно-ионизационный детектор.
Колонка: Внутренняя поверхность трубки из плавленого кварца с внутренним диаметром 0,53 мм и длиной 30 м покрыта слоем полиэтиленгликоля для газовой хроматографии толщиной 1,0 мкм.
Температура колонки: 50°C в течение 8 минут и последующее повышение температуры до 200°С со скоростью 25°С в минуту.
Температура в устройстве ввода: Постоянная температура около 270°C.
Температура детектора: Постоянная температура около 250°С.
Газ-носитель: Гелий.
Скорость потока: 3,5 мл/мин.
Отношение деления потока: 1:50.
Диапазон измерения площадей: 10 минут.
Пример 2
Получение блок-сополимера 2
ДМФ (536 мл) добавляли к 30,0 г конъюгата PEG (средняя молекулярная масса: 12000) с ацетилированной pAsp (полиаспарагиновая кислота; средняя степень полимеризации: 36,4) (в формуле (2) R1 представляет собой метильную группу, R2 представляет собой триметиленовую группу, R3 представляет собой метиленовую группу, R4 представляет собой ацетильную группу, n составляет приблизительно 272, x составляет приблизительно 9,1 и y составляет приблизительно 27,3; далее в настоящем описании используется аббревиатура PEG-pAsp-Ac-2), полученного способом, описанным в патентном документе 2; соединение растворяли при 35°C и туда же добавляли DMAP (8,18 г) и 4-фенил-1-бутанол (7,35 г: 0,049 моль; 0,73 молярных эквивалентов относительно 1 моля карбоксильных групп в PEG-pAsp-Ac-2). Часть реагента, прилипшую во время добавления, смывали туда же диметилформамидом (27 мл). После подтверждения того, что соединения растворились, реакционный раствор доводили до 25°С и добавляли DIPCI (16,9 г: 2⋅(x+y) эквивалентов относительно карбоксильных групп в PEG-pAsp-Ac-2=72,8 эквивалента). Диметилформамидом (27 мл) смывали туда же часть реагента, прилипшую при добавлении, и реакционному раствору давали возможность взаимодействовать в течение 22 часов при 25°C. В этом случае, через 18 часов после начала реакции, реакционное отношение сложноэфирных связей 4-фенил-1-бутанола становилось постоянным. С другой стороны, согласно анализу, проведенному анионообменной ВЭЖХ, реагенты все еще удерживались в колонке. Через 22 часа после начала реакции добавляли DIPCI (4,23 г: 0,5⋅(x+y) эквивалентов в расчете на карбоксильные группы в PEG-pAsp-Ac-2=18,2 эквивалентов), продолжая проведение реакции. ВЭЖХ на анионообменной колонке проверяли прекращение удерживания реагентов в колонке, и через 26 часов после начала реакции ее останавливали. Реакционный раствор по каплям добавляли к смешанному растворителю из гептана и этилацетата, и смесь перемешивали. Смесь оставляли стоять в течение ночи, полученный при этом осадок собирали фильтрованием и сушили при пониженном давлении. Получали 34,3 г неочищенных кристаллов.
Полученные неочищенные кристаллы (33,5 г) растворяли в ДМФ (469 мл) и затем туда же добавляли катионообменную смолу DOWEX 50w8 (111 мл). Кроме того, часть вещества, прилипшую во время добавления, смывали туда же ДМФ (34 мл), и полученную смесь перемешивали в течение 3 часов. Катионообменную смолу DOWEX 50w8 удаляли фильтрованием, промывая этилацетатом, и затем реакционный раствор, полученный таким образом, добавляли по каплям к смешанному растворителю из гептана и этилацетата. Смесь перемешивали. Смесь оставляли стоять в течение ночи и осадок, полученный таким образом, собирали фильтрованием и сушили при пониженном давлении. Получали 32,2 г блок-сополимера 2.
Блок-сополимер 2 гидролизовали способом, аналогично использованному в примере 1, и анализировали ВЭЖХ с обращенной фазой. Количество сложноэфирно связанного 4-фенил-1-бутанола составило 15,5% (масс./масс.) PEG-pAsp-Ac-2. Реакционное отношение сложноэфирных связей 4-фенил-1-бутанола составило 77,3%, и отношение введения 4-фенил-1-бутанола составило 56,4% количества карбоксильных групп PEG-pAsp-Ac-2.
Блок-сополимер 2 анализировали анионообменной ВЭЖХ в таких же условиях, как в примере 1, не обнаружив каких-либо пиков, которые указывали на удерживание в колонке.
Раствор блок-сополимера 2 титровали (по методике потенциометрического титрования) жидким гидроксидом калия (0,1 моль/л) способом, аналогично использованному в примере 1, и число карбоксильных групп составило 0,05 ммоль/г. Как описано выше, поскольку число карбоксильных групп, в случае, когда доля карбоксильных групп составляет 0%, составляло 0,1 ммоль/г или менее, блок-сополимер 2 не имел каких-либо остаточных карбоксильных групп.
Для блок-сополимера 2, количество диизопропилмочевины в блок-сополимере 2 рассчитывали по аналогичной методике, как в примере 1, которое составило 3,0% (масс./масс.).
Пример 3
Получение блок-сополимера 3
ДМФ (570 мл) добавляли к 30,0 г конъюгата PEG (средняя молекулярная масса: 12000) с ацетилированной pAsp (полиаспарагиновая кислота; средняя степень полимеризации: 46,8) (в формуле (1) R1 представляет собой метильную группу, R2 представляет собой триметиленовую группу, R3 представляет собой метиленовую группу, R4 представляет собой ацетильную группу, n составляет приблизительно 272, x составляет приблизительно 11,7, и y составляет приблизительно 35,1; далее в настоящем описании используется аббревиатура PEG-pAsp-Ac-3), полученного способом, описанным в патентном документе 2; соединение растворяли при 35°C и туда же добавляли DMAP (9,68 г) и 4-фенил-1-бутанол (7,29 г: 0,0486 моль; 0,61 молярных эквивалентов относительно 1 моля карбоксильных групп PEG-pAsp-Ac-3) и туда же диметилформамидом (32 мл) смывали часть реагента, прилипшего во время добавления. После подтверждения того, что соединения растворились, реакционный раствор доводили до 25°С и добавляли DIPCI (20,00 г: 2⋅(x+y) эквивалентов относительно карбоксильных групп в PEG-pAsp-Ac-3=93,6 эквивалента). Диметилформамидом (32 мл) туда же смывали часть реагента, прилипшего при добавлении, и реакционному раствору давали возможность взаимодействовать в течение 22 часов при 25°C. В этом случае, через 18 часов после начала реакции, реакционное отношение сложноэфирных связей 4-фенил-1-бутанола становилось постоянным. С другой стороны, согласно анализу, проведенному анионообменной ВЭЖХ, реагенты все еще удерживались в колонке. Через 22 часа после начала реакции добавляли DIPCI (5,0 г: 0,5⋅(x+y) эквивалентов в расчете на карбоксильные группы PEG-pAsp-Ac=23,4 эквивалентов), продолжая проведение реакции. ВЭЖХ на анионообменной колонке проверяли прекращение удерживания реагентов в колонке, и через 29 часов после начала реакции ее останавливали. Реакционный раствор по каплям добавляли к смешанному растворителю из гептана и этилацетата, и смесь перемешивали. Смесь оставляли стоять в течение ночи, полученный при этом осадок собирали фильтрованием и сушили при пониженном давлении. Получали 35,4 г неочищенных кристаллов.
Полученные неочищенные кристаллы (34,5 г) растворяли в ДМФ (483 мл) и затем туда же добавляли катионообменную смолу DOWEX 50w8 (114 мл). Кроме того, часть вещества, прилипшего во время добавления, смывали туда же ДМФ (34 мл), и полученную смесь перемешивали в течение 3 часов. Катионообменную смолу DOWEX 50w8 удаляли фильтрованием, промывая этилацетатом, и затем реакционный раствор, полученный таким образом, добавляли по каплям к смешанному растворителю из гептана и этилацетата. Смесь перемешивали. Смесь оставляли стоять в течение ночи, осадок, полученный таким образом, собирали фильтрованием и сушили при пониженном давлении. Получали 33,1 г блок-сополимера 3.
Блок-сополимер 3 гидролизовали способом, аналогично использованному в примере 1, и анализировали ВЭЖХ с обращенной фазой. Количество сложноэфирно связанного 4-фенил-1-бутанола составило 17,2% (масс./масс.) PEG-pAsp-Ac-3. Реакционное отношение сложноэфирных связей 4-фенил-1-бутанола составило 86,8%, и отношение введения 4-фенил-1-бутанола составило 53,0% количества карбоксильных групп PEG-pAsp-Ac-3.
Блок-сополимер 3 анализировали анионообменной ВЭЖХ в таких же условиях, как в примере 1, не обнаружив каких-либо пиков, которые указывали на удерживание в колонке.
Раствор блок-сополимера 3 титровали (по методике потенциометрического титрования) жидким гидроксидом калия (0,1 моль/л) способом, аналогично использованному в примере 1, и число карбоксильных групп составило 0,05 ммоль/г. Как описано выше, поскольку число карбоксильных групп, в случае, когда доля карбоксильных групп составляет 0%, составляло 0,1 ммоль/г или менее, блок-сополимер 3 не имел каких-либо остаточных карбоксильных групп.
Для блок-сополимера 3, количество диизопропилмочевины в блок-сополимере 3 рассчитывали по аналогичной методике, как в примере 1, и количество диизопропилмочевины в блок-сополимере 3 составило 3,8% (масс./масс.).
Пример 4
Получение блок-сополимера 4
ДМФ (1102 мл) добавляли к 62,0 г конъюгата PEG (средняя молекулярная масса: 12000) с ацетилированной pAsp (полиаспарагиновая кислота; средняя степень полимеризации: 41,6) (в формуле (1) R1 представляет собой метильную группу, R2 представляет собой триметиленовую группу, R3 представляет собой метиленовую группу, R4 представляет собой ацетильную группу, n составляет приблизительно 272, x составляет приблизительно 10,4, и y составляет приблизительно 31,2; далее в настоящем описании используется аббревиатура PEG-pAsp-AC-4), полученного способом, описанным в патентном документе 2; соединение растворяли при 35°C и туда же добавляли DMAP (18,69 г) и 4-фенил-1-бутанол (15,50 г: 0,103 моль; 0,67 молярных эквивалентов относительно 1 моля карбоксильных групп of PEG-pAsp-Ac-4), и туда же смывали диметилформамидом (61 мл) часть реагента, прилипшего во время добавления. После подтверждения того, что соединения растворились, реакционный раствор доводили до 25°С и добавляли DIPCI (38,62 г: 2⋅(x+y) эквивалентов относительно карбоксильных групп в PEG-pAsp-Ac-4=83,2 эквивалента). Диметилформамидом (61 мл) туда же смывали часть реагента, прилипшую при добавлении, и реакционному раствору давали возможность взаимодействовать в течение 22 часов при 25°C. В этом случае, через 20 часов после начала реакции, реакционное отношение сложноэфирных связей 4-фенил-1-бутанола становилось постоянным. С другой стороны, согласно анализу, проведенному анионообменной ВЭЖХ, реагенты все еще удерживались в колонке. Через 22 часа после начала реакции добавляли DIPCI (9,66 г: 0,5⋅(x+y) эквивалентов в расчете на карбоксильные группы в PEG-pAsp-Ac=20,4 эквивалентов), продолжая проведение реакции. ВЭЖХ на анионообменной колонке проверяли прекращение удерживания реагентов в колонке, и через 25 часов после начала реакции ее останавливали. Реакционный раствор по каплям добавляли к смешанному растворителю из гептана и этилацетата, и смесь перемешивали. Смесь оставляли стоять в течение ночи, полученный при этом осадок собирали фильтрованием и сушили при пониженном давлении. Получали 72,9 г неочищенных кристаллов.
Полученные неочищенные кристаллы (71,5 г) растворяли в ДМФ (1001 мл) и затем туда же добавляли катионообменную смолу DOWEX 50w8 (236 мл). Кроме того, часть вещества, прилипшую во время добавления, смывали туда же диметилформамидом (72 мл), и полученную смесь перемешивали в течение 3 часов. Катионообменную смолу DOWEX 50w8 удаляли фильтрованием, промывая этилацетатом, и затем реакционный раствор, полученный таким образом, добавляли по каплям к смешанному растворителю из гептана и этилацетата. Смесь перемешивали. Смесь оставляли стоять в течение ночи, осадок, полученный таким образом, собирали фильтрованием и сушили при пониженном давлении. Получали 69,7 г блок-сополимера 4.
Блок-сополимер 4 гидролизовали способом, аналогично использованному в примере 1, и анализировали ВЭЖХ с обращенной фазой. Количество сложноэфирно связанного 4-фенил-1-бутанола составило 16,6% (масс./масс.) PEG-pAsp-Ac-4. Поэтому реакционное отношение сложноэфирных связей 4-фенил-1-бутанола составило 81,4%, и отношение введения 4-фенил-1-бутанола составило 54,5% количества карбоксильных групп PEG-pAsp-Ac-4.
Блок-сополимер 4 анализировали анионообменной ВЭЖХ в таких же условиях, как в примере 1, не обнаружив каких-либо пиков, которые указывали на удерживание в колонке.
Раствор блок-сополимера 4 титровали (по методике потенциометрического титрования) жидким гидроксидом калия (0,1 моль/л) способом, аналогично использованному в примере 1, и число карбоксильных групп составило 0,05 ммоль/г. Как описано выше, поскольку число карбоксильных групп, в случае, когда доля карбоксильных групп составляет 0%, составляло 0,1 ммоль/г или менее, блок-сополимер 3 не имел каких-либо остаточных карбоксильных групп.
Для блок-сополимера 4, количество диизопропилмочевины в блок-сополимере 4 рассчитывали по аналогичной методике, как в примере 1, и количество диизопропилмочевины в блок-сополимере 4 составило 3,3% (масс./масс.).
Сравнительный пример 1 (Получение согласно способу получения, описанному в примерах патентного документа 5)
Получение блок-сополимера 5
ДМФ (70 мл) добавляли к 3,50 г конъюгата PEG (средняя молекулярная масса: 12000) с ацетилированной pAsp (полиаспарагиновая кислота; средняя степень полимеризации: 43,2) (в формуле (2) R1 представляет собой метильную группу, R2 представляет собой триметиленовую группу, R3 представляет собой метиленовую группу, R4 представляет собой ацетильную группу, n составляет приблизительно 272, x составляет приблизительно 10,8, и y составляет приблизительно 32,4; далее в настоящем описании используется аббревиатура PEG-pAsp-Ac-5), полученного способом, описанным в патентном документе 2; соединение растворяли при 35°C и туда же добавляли DMAP (0,87 г), 4-фенил-1-бутанол (1,34 г: 0,0089 моль; 1,00 молярных эквивалентов относительно 1 моля карбоксильных групп PEG-pAsp-AC-5) и DIPCI (1,12 г: 1 эквивалент относительно 1 моля карбоксильных групп PEG-pAsp-Ac-5), и смеси давали возможность взаимодействовать в течение 26 часов при 35°C. Реакционный раствор добавляли по каплям к смешанному растворителю из диизопропилового простого эфира и этанола, осадок собирали фильтрованием и сушили при пониженном давлении. Получали 3,70 г неочищенных кристаллов. Полученные неочищенные кристаллы растворяли в 50%-ном водном растворе ацетонитрила и затем раствор пропускали через катионообменную смолу DOWEX 50wS (40 мл), промывая 50%-ным ацетонитрилом. Элюат концентрировали при пониженном давлении и затем сушили вымораживанием и таким образом получали 3,72 г блок-сополимера 5.
Блок-сополимер гидролизовали способом, аналогично использованному в примере 1, и анализировали ВЭЖХ с обращенной фазой. Количество сложноэфирно связанного 4-фенил-1-бутанола составило 15,5% (масс./масс.) соединения формулы (2). Реакционное отношение сложноэфирных связей 4-фенил-1-бутанола составило 49,0%, и отношение введения 4-фенил-1-бутанола составило 49,0% количества карбоксильных групп PEG-pAsp-Ac-5.
Блок-сополимер 5 анализировали анионообменной ВЭЖХ в таких же условиях, как в примере 1, и детектировали пик со временем удерживания 14,3 минуты.
Блок-сополимер 5 титровали (по методике потенциометрического титрования) жидким гидроксидом калия (0,1 моль/л) способом, аналогично использованному в примере 1, и в результате расчета число карбоксильных групп составило 0,23 ммоль/г. Как описано выше, хотя число карбоксильных групп, в случае, когда доля карбоксильных групп составляет 0%, составляло 0,1 ммоль/г или менее, следует понимать, что блок-сополимер 5 имеет остаточные карбоксильные группы.
Для блок-сополимера 5, количество диизопропилмочевины в блок-сополимере 5 рассчитывали по аналогичной методике, как в примере 1, и количество диизопропилмочевины составило 2,3% (масс./масс.).
Сравнительный пример 2 (Получение согласно способу получения, описанному в примерах патентного документа 6)
Получение блок-сополимера 6
ДМФ (13,0 л) добавляли к 1,73 кг конъюгата PEG (средняя молекулярная масса: 12000) с ацетилированной pAsp (полиаспарагиновая кислота; средняя степень полимеризации: 41,0) (в формуле (2) R1 представляет собой метильную группу, R2 представляет собой триметиленовую группу, R3 представляет собой метиленовую группу, R4 представляет собой ацетильную группу, n составляет приблизительно 272, x составляет приблизительно 10,3, и y составляет приблизительно 30,8; далее в настоящем описании используется аббревиатура PEG-pAsp-Ac-6), полученного способом, описанным в патентном документе 2; соединение растворяли при 35°C и туда же добавляли DMAP (412 г, включая количество, смытое при ополаскивании 8,7 л ДМФ) и 4-фенил-1-бутанол (443 г: 2,95 моль; 0,70 молярных эквивалентов относительно 1 моля карбоксильных групп PEG-pAsp-Ac-6, включая количество, смытое при ополаскивании 2,2 л ДМФ) и реакционный раствор охлаждали до 22,5°C. Туда же добавляли DIPCI (532 г: 1 эквивалент относительно 1 моля карбоксильных групп PEG-pAsp-Ac-6, включая количество, смытое при ополаскивании 2,2 л ДМФ), и смеси давали возможность взаимодействовать в течение 22 часов при 22,5°C. В реакционный раствор добавляли этилацетат и гептан, перемешивали, осадок, полученный таким образом, собирали фильтрованием и сушили при пониженном давлении. Получали 2,08 кг неочищенных кристаллов.
Полученные неочищенные кристаллы гидролизовали способом, аналогично использованному в примере 1, и анализировали ВЭЖХ с обращенной фазой. Количество сложноэфирно связанного 4-фенил-1-бутанола составило 17,0% (масс./масс.) соединения формулы (2). Реакционное отношение сложноэфирных связей 4-фенил-1-бутанола составило 77,0%, и отношение введения 4-фенил-1-бутанола составило 53,9% количества карбоксильных групп PEG-pAsp-Ac-6.
Неочищенные кристаллы анализировали анионообменной ВЭЖХ в таких же условиях, как в примере 1, и детектировали пик со временем удерживания 16,9 минуты.
К неочищенным кристаллам (1,94 кг), полученным как описано выше, добавляли 14,6 л ДМФ, для растворения кристаллов при 31°С, и туда же добавляли DMAP (307 г, включая количество, смытое при ополаскивании 9,7 л ДМФ) и DIPCI (491 г: 1 эквивалент в расчете на карбоксильные группы в PEG-pAsp-Ac, включая количество, смытое при ополаскивании 4,9 л ДМФ), и смеси давали возможность взаимодействовать в течение 20 часов при 31°С. В реакционный раствор добавляли этилацетат и гептан, перемешивали, осадок, полученный таким образом, собирали фильтрованием и сушили при пониженном давлении. Получали 1,85 кг неочищенных кристаллов. 1,83 кг этих неочищенных кристаллов растворяли в ДМФ (22,0 л) и затем в раствор добавляли катионообменную смолу DOWEX 50w8 (6,0 л). Часть реагента, прилипшую во время добавления, смывали ДМФ (5,5 мл) и смесь перемешивали в течение одного часа. Катионообменную смолу DOWEX 50w8 удаляли фильтрованием, промывая этилацетатом (69 л), и затем в реакционный раствор, полученный таким образом, добавляли этилацетат и гептан. Смесь перемешивали. Полученный таким образом осадок собирали фильтрованием и сушили при пониженном давлении. Получали 1,79 кг блок-сополимера 6.
Блок-сополимер 6 гидролизовали способом, аналогично использованному в примере 1, и анализировали ВЭЖХ с обращенной фазой. Количество сложноэфирно связанного 4-фенил-1-бутанола составило 15,8% (масс./масс.) соединения формулы (2). После реакции второй стадии отношение введения 4-фенил-1-бутанола составило 50%.
Блок-сополимер 6 анализировали анионообменной ВЭЖХ в таких же условиях, как в примере 1, не обнаружив каких-либо пиков, которые указывали на удерживание в колонке.
Блок-сополимер 6 титровали (по методике потенциометрического титрования) жидким гидроксидом калия (0,1 моль/л) способом, аналогично использованному в примере 1, и число карбоксильных групп составило 0,04 ммоль/г. Как описано выше, поскольку число карбоксильных групп, в случае, когда доля карбоксильных групп составляет 0%, составляло 0,1 ммоль/г или менее, следует понимать, что блок-сополимер 6 не имеет остаточных карбоксильных групп.
Для блок-сополимера 6, количество диизопропилмочевины рассчитывали по аналогичной методике, как в примере 1, и количество диизопропилмочевины составило 3,6% (масс./масс.).
Результаты для блок-сополимеров, полученных в примерах 1-4 и в сравнительных примерах 1 и 2, приведены в таблице 1.
|
Как показано в таблице 1, все блок-сополимеры продемонстрировали сходное содержание 4-фенил-1-бутанола. С другой стороны, блок-сополимеры 1, 2, 3, 4 и 6 не удерживались в колонке при анализе анионообменной ВЭЖХ и демонстрировали почти одинаковое число остаточных карбоксильных групп. Эти результаты означают, что указанные блок-сополимеры по существу не имеют карбоксильных групп. В отношении количества диизопропилмочевины, только блок-сополимер 5 имеет более низкое ее содержание. На основании приведенных выше результатов было обнаружено, что блок-сополимеры согласно настоящему изобретению отличались от блок-сополимера, описанного в патентном документе 5, и были такими же, как блок-сополимер 6, описанный в патентном документе 6.
В способе получения блок-сополимера, описанном в сравнительном примере 2, во-первых, поскольку отношение введения регулировали, вводя 4-фенил-1-бутанол в количестве, превышающем желаемое содержание 4-фенил-1-бутанола, и уменьшая количество 4-фенил-1-бутанола посредством реакции второй стадии, можно осуществлять контроль за двумя разными реакциями. Кроме того, в реакции второй стадии, поскольку необходимо регулирование остаточных карбоксильных групп в pAsp, осложняется контроль над продукцией и практически затруднен. С другой стороны, в настоящем изобретении содержание 4-фенил-1-бутанола можно контролировать, получая блок-сополимер в одну реакцию. Поэтому, как можно видеть, способ получения согласно настоящему изобретению является превосходным промышленным способом получения с облегченным техническим контролем продукта по сравнению со способом получения, описанным в патентном документе 6.


Слоистый материал для многослойного стекла
Порфиразиновое красящее вещество, содержащая его композиция чернил и окрашенный продукт
Слоистый материал для многослойного стекла и межслойная пленка для многослойного стекла
Способ получения регуляторных дендритных клеток
Способ получения ненасыщенного альдегида и/или ненасыщенной карбоновой кислоты
Способ получения композита лекарственного средства и блок-сополимера и фармацевтический препарат, содержащий его
Экранизирующий инфракрасное излучение лист, способ его изготовления и его применение
Способ получения ненасыщенного альдегида и/или ненасыщенной карбоновой кислоты
Мицеллярный препарат, содержащий малорастворимый в воде противораковый агент, содержащее его противораковое средство и новый блок-сополимер
Блок-сополимер с пониженным содержанием примесей, полимерный носитель, фармацевтические препараты в полимерной форме и способы их получения
Катализатор и способ получения ненасыщенного альдегида и ненасыщенной карбоновой кислоты
Слоистый материал для многослойного стекла
Порфиразиновое красящее вещество, содержащая его композиция чернил и окрашенный продукт
Слоистый материал для многослойного стекла и межслойная пленка для многослойного стекла
Способ получения регуляторных дендритных клеток
Способ получения ненасыщенного альдегида и/или ненасыщенной карбоновой кислоты
Полимерное производное антиметаболита цитидина

