Результат интеллектуальной деятельности: КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ МИЕЛОФИБРОЗА
Вид РИД
Изобретение
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Эта заявка испрашивает приоритет предварительной заявки США №61/410924, поданной 7 ноября 2010, которая включена в этот документ путем ссылки в полном ее объеме.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
В этом документе предоставлены композиции и способы для лечения миелофиброза. Композиции и способы, представленные в этом документе, относятся к лечению миелофиброза соединениями, которые ингибируют JAK2-киназы, или их фармацевтически приемлемыми солями, или их гидратами.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Миелофиброз («MF») представляет собой редкое заболевание, в основном поражающее людей старческого возраста. MF представляет собой BCR-ABL1-негативную миелопролиферативную неоплазию («MPN»), которая является вновь возникшим заболеванием (первичным заболеванием) или может предваряться истинной полицитемией («PV») или эссенциальной тромбоцитемией («ET»). Клинические признаки включают прогрессирующую анемию, выраженную спленомегалию, конституциональные симптомы (например, утомляемость, ночные приливы, боль в костях, зуд и кашель) и потерю веса (Tefferi A, N. Engl. J. Med. 342:1255-1265, 2000). Медиана выживаемости колеблется в диапазоне от менее 2 лет до более 15 лет, что основано на выявленных в последнее время прогностических факторах (Cervantes F. et al., Blood 113:2895-2901, 2009; Hussein K. et al. Blood 115:496-499, 2010; Patnaik MM et al., Eur. J. Haematol 84:105-108, 2010). Мутации, вовлекающие JAK2-киназу (James C. et al., Nature 434:1144-1148, 2005; Scott L.M. et al., N. Engl. J. Med. 356:459-468, 2007), ген MPL (монофосфорил липид А) (Pikman Y. et al., PLoS. Med. 3:e270, 2006), ген TET2 (Delhommeau F. et al., N. Engl. J. Med. 360:2289-2301, 2009), ген ASXL1 (Carbuccia N. et al., Leukemia 23:2183-2186, 2009), ген IDH1/IDH2 (изоцитратдегидрогеназы) (Green A. et al., N. Engl. J. Med. 362:369-370, 2010; Tefferi A. et al., Leukemia 24:1302-1309, 2010), ген CBL (Grand FH et al., Blood 113:6182-6192, 2009), ген IKZF1 (Jager R. et al., Leukemia 24:1290-1298, 2010), ген LNK (Oh. ST. et al., Blood 116:988-992, 2010) или ген EZH2 (Ernst T. et al., Nat. Genet. 42:722-726) были описаны у пациентов с MNP, в том числе у пациентов с MF. Некоторые мутации возникают с высокой частотой при MF (например, мутации JAK2-киназ у ~50% пациентов) и либо непосредственно (например, мутации JAK2-киназы или MPL), либо косвенно (например, мутации LNK или CBL) индуцируют гиперактивацию JAK-STAT-киназ ((сигнальный трансдуктор и активатор транскрипции)-киназа).
Доступные в настоящее время методы лечения являются неэффективными в реверсировании течения процесса MF, будь то первичное или повторное/вторичное заболевание. Единственной возможностью вылечить заболевание на сегодняшний день является трансплантация костного мозга. Однако большинство пациентов не являются подходящими кандидатами для трансплантации костного мозга ввиду их старческого среднего возраста на момент постановки диагноза, где клинические проявления заболевания и смертность, связанные с трансплантацией, возрастают. Таким образом, возможности управления течением миелофиброза на настоящий момент являются неадекватными в том, чтобы удовлетворять потребностям всех пациентов. Основные возможности для активного вмешательства включают циторедуктивную терапию, например, с помощью гидроксимочевины, лечение анемии с помощью андрогенов, эритропоэтина и спленэктомию. Эти возможности, как было показано, не улучшают выживаемость и большей частью рассматриваются как паллиативные (временно облегчающие или ослабляющие проявление заболевания) (Cervantes F., Myelofibrosis: Biology and treatment options, European Journal of Haematology, 2007, 79 (suppl.68) 13-17). Таким образом, существует потребность в обеспечении дополнительных методов терапии для пациентов, страдающих миелофиброзом.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В этом документе предоставлены капсулы, подходящие для перорального введения. В некоторых вариантах осуществления капсулы содержат смесь, включающую (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия, где смесь содержится в капсуле.
В некоторых вариантах осуществления капсула содержит приблизительно 10 мг – приблизительно 680 мг соединения, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула содержит приблизительно 10 мг – приблизительно 500 мг соединения. В некоторых вариантах осуществления капсула содержит любое количество, выбираемое из 10 мг, 40 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг или 600 мг соединения. В некоторых вариантах осуществления массовое соотношение соединения к микрокристаллической целлюлозе в капсуле находится от приблизительно 1:1,5 до 1:15, где масса для соединения в массовом соотношении равна массе фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления массовое соотношение соединения к стеарилфумарату натрия в капсуле составляет от приблизительно 5:1 до приблизительно 50:1, где масса для соединения в массовом соотношении равна массе фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу. В некоторых вариантах осуществления силикатированная микрокристаллическая целлюлоза представляет собой комбинацию 98% микрокристаллической целлюлозы и 2% коллоидного диоксида кремния.
Также предоставлены в этом документе стандартные лекарственные формы, содержащие смесь, включающую (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия. В некоторых вариантах осуществления, стандартные лекарственные формы предназначены для лечения миелофиброза, а именно для лечения миелофиброза в соответствии со способом, описанным в этом документе.
В некоторых вариантах осуществления стандартная лекарственная форма содержит смесь, включающую (i) от приблизительно 10 мг до приблизительно 680 мг (или от приблизительно 10 мг до приблизительно 500 мг) соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где указанная масса соответствует массе фрагмента соединения, составляющего свободное основание, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия. В некоторых вариантах осуществления стандартная лекарственная форма имеет форму капсулы, и смесь содержится в капсуле. В некоторых вариантах осуществления соединение в смеси составляет от приблизительно 10 мг до приблизительно 500 мг, где указанная масса соответствует массе фрагмента соединения, составляющего свободное основание. В некоторых вариантах осуществления добавка содержит (i) приблизительно 10 мг (или приблизительно любое количество, выбираемое из 40 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг) соединения, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления массовое соотношение соединения к микрокристаллической целлюлозе в капсуле находится в диапазоне от приблизительно 1:1,5 до приблизительно 1:15, где масса соединения в массовом соотношении дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления массовое соотношение соединения к стеарилфумарату натрия в капсуле находится в диапазоне между приблизительно 5:1 и приблизительно 50:1, где масса соединения в массовом соотношении дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу. В некоторых вариантах осуществления силикатированная микрокристаллическая целлюлоза представляет собой комбинацию 98% микрокристаллической целлюлозы и 2% коллоидного диоксида кремния.
В некоторых вариантах осуществления стеарилфумарат натрия составляет приблизительно 1% (масс./масс.) от массы содержимого капсулы. В некоторых вариантах осуществления массовое соотношение соединения к микрокристаллической целлюлозе, например, к силикатированной микрокристаллической целлюлозе, составляет от приблизительно 40:60 до приблизительно 10:90 (например, приблизительно 40:60 или приблизительно 1:1,5 или приблизительно 10:90 или приблизительно 1:9).
В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления стандартная лекарственная форма или капсула содержит смесь, включающую приблизительно 12 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида, приблизительно 122 мг силикатированной микрокристаллической целлюлозы и приблизительно 1 мг стеарилфумарата натрия. В некоторых вариантах осуществления стандартная лекарственная форма или капсула содержит смесь, включающую приблизительно 47 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида, приблизительно 448 мг силикатированной микрокристаллической целлюлозы и приблизительно 5 мг стеарилфумарата натрия. В некоторых вариантах осуществления стандартная лекарственная форма или капсула содержит смесь, включающую приблизительно 117 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления стандартная лекарственная форма или капсула содержит смесь, включающую приблизительно 235 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида, приблизительно 357 мг силикатированной микрокристаллической целлюлозы и приблизительно 6,00 мг стеарилфумарата натрия. В некоторых вариантах осуществления капсула представляет собой твердую желатиновую капсулу.
Также предоставлены в этом документе способы получения капсульного лекарственного продукта, включающие в себя а) смешение смазывающего вещества с соединением, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, для получения гранул и b) смешение гранул, полученных на стадии а), с эксципиентом. В некоторых вариантах осуществления смазывающее вещество представляет собой стеарилфумарат натрия. В некоторых вариантах осуществления эксципиент представляет собой микрокристаллическую целлюлозу, например, силикатированную микрокристаллическую целлюлозу. В некоторых вариантах осуществления стеарилфумарат натрия составляет приблизительно 1% (масс./масс.) от массы содержимого капсулы. В некоторых вариантах осуществления массовое соотношение соединения к силикатированной микрокристаллической целлюлозе составляет от приблизительно 1:1,5 до приблизительно 1:9. В некоторых вариантах осуществления массовое соотношение соединения к силикатированной микрокристаллической целлюлозе составляет приблизительно 1:1,5. В некоторых вариантах осуществления массовое соотношение соединения к силикатированной микрокристаллической целлюлозе составляет приблизительно 1:9. В некоторых вариантах осуществления капсула представляет собой твердую желатиновую капсулу.
Также предоставлены в этом документе способы лечения миелофиброза у субъекта, включающие в себя пероральное введение соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где соединение содержится в смеси, включающей в себя (i) соединение, (ii) эксципиент (например, микрокристаллическую целлюлозу) и (iii) смазывающее вещество (например, стеарилфумарат натрия). Могут быть использованы любые из стандартных лекарственных форм или капсул, описанных в этом документе. В некоторых вариантах осуществления предоставляют способ лечения миелофиброза у субъекта, включающий в себя пероральное введение соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где соединение находится в капсуле, содержащей смесь, включающую в себя (i) соединение, (ii) микрокристаллическую целлюлозу (например, силикатированную микрокристаллическую целлюлозу) и (iii) стеарилфумарат натрия.
Также предоставлены в этом документе способы лечения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект дает отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в гене Янус-Киназы 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 гена JAK2-киназы человека.
Также предоставлены в этом документе способы лечения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект ранее получил другое лечение миелофиброза. В некоторых вариантах осуществления предыдущее лечение представляет собой метод лечения с помощью ингибитора JAK2-киназы, который представляет собой не N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления предыдущее лечение включает в себя введение INCB018424 (руксолитиниб). В некоторых вариантах осуществления субъект не восприимчив к предыдущему лечению. В некоторых вариантах осуществления предыдущее лечение представляет собой метод лечения с помощью N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида или его фармацевтически приемлемой соли или гидрата соли. В некоторых вариантах осуществления предыдущее лечение было прервано при показании, включающем повышенные уровни амилазы, липазы, аспартат-аминотрансферазы («AST»), аланин-аминотрансферазы («ALT») и/или креатинина. В некоторых вариантах осуществления предыдущее лечение было прервано при показании гематологического состояния, выбранного из группы, состоящей из анемии, тромбоцитопении и нейтропении.
Также предоставлены в этом документе способы уменьшения интенсивности симптомов состояния насыщенности клетками ткани костного мозга или фиброза костного мозга, сопутствующего миелофиброзу, у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат.
Также предоставлены в этом документе способы уменьшения зуда, сопутствующего миелофиброзу, у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат.
Также предоставлены в этом документе способы контроля за проведением лечения миелофиброза у субъекта, включающие в себя (а) введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат; (b) контроль негематологического параметра, выбранного из группы, состоящей из уровня амилазы, уровня липазы, уровня аспартат-аминотрансферазы (AST) и уровня креатинина, у субъекта; и (с) определение того, что следует ли субъекту продолжать лечение или следует прервать его. Также предоставлены в этом документе способы контроля за проведением лечения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат; и прерывание лечения при показании повышенных уровней одного или более ферментов или молекул, выбранных из группы, состоящей из амилазы, липазы, аспартат-аминотрансферазы (AST), аланин-аминотрансферазы (ALT) и креатинина в сыворотке субъекта, без предварительного снижения дозы. В некоторых вариантах осуществления один или более повышенных уровней составляют осложнения 4 степени.
Также предоставлены в этом документе способы контроля за проведением лечения миелофиброза у субъекта, включающие в себя (а) введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат; (b) контроль гематологического параметра, выбранного из группы, состоящей из анемии, тромбоцитопении и нейтропении, в сыворотке субъекта; и (с) определение того факта, следует ли субъекту продолжать лечение или следует прервать его. Также предоставлены в этом документе способы контроля за проведением лечения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид или его фармацевтически приемлемую соль или его гидрат; и прерывание лечения при показании одного или более гематологических состояний, выбранных из группы, состоящей из анемии, тромбоцитопении и нейтропении, без предварительного снижения дозы. В некоторых вариантах осуществления одно или более гематологических состояний составляют осложнения 4 степени.
В некоторых вариантах осуществления способов контроля за проведением лечения, предоставляемых в этом документе, способы дополнительно включают в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, после прерывания субъекту лечения, по меньшей мере, на 2 недели. В некоторых вариантах осуществления субъекту было прервано лечение, по меньшей мере, на 3 недели. В некоторых вариантах осуществления субъекту было прервано лечение, по меньшей мере, на 4 недели. В некоторых вариантах осуществления лечение было прервано без предварительного снижения дозы.
В некоторых вариантах осуществления соединение вводят субъекту-человеку в дозе от приблизительно 240 мг в день до приблизительно 680 мг в день, где указанная масса соответствует массе фрагмента соединения, составляющего свободное основание. В некоторых вариантах осуществления соединение вводят в дозе от приблизительно 300 мг в день до приблизительно 500 мг в день (например, от приблизительно 300 мг в день до приблизительно 400 мг в день или от приблизительно 400 мг в день до приблизительно 500 мг в день), где указанная масса соответствует массе фрагмента соединения, составляющего свободное основание. В некоторых вариантах осуществления соединение вводят в дозе, равной приблизительно любой дозе из приблизительно 240 мг в день, 250 мг в день, 300 мг в день, 350 мг в день, 400 мг в день, 450 мг в день, 500 мг в день, 550 мг в день, 600 мг в день, 650 мг в день или 680 мг в день, где указанная масса соответствует массе фрагмента соединения, составляющего свободное основание. В некоторых вариантах осуществления соединение вводят ежедневно и/или перорально. В некоторых вариантах осуществления соединение вводят в течение периода, по меньшей мере, 1 цикла, по меньшей мере, 2 циклов, по меньшей мере, 3 циклов, по меньшей мере, 4 циклов, по меньшей мере, 5 циклов или, по меньшей мере, 6 циклов (например, по меньшей мере, 7 циклов, по меньшей мере, 8 циклов, по меньшей мере, 9 циклов, по меньшей мере, 10 циклов, по меньшей мере, 11 циклов, по меньшей мере, 12 циклов, по меньшей мере, 15 циклов, по меньшей мере, 18 циклов, по меньшей мере, 24 циклов), где 1 цикл представляет собой 28-дневный цикл лечения. В некоторых вариантах осуществления соединение находится в капсуле и вводится перорально. В некоторых вариантах осуществления соединение находится в стандартной лекарственной форме. Могут быть введены любые капсулы или стандартные лекарственные формы, описанные в этом документе. В некоторых вариантах осуществления способов, предоставляемых в этом документе, соединение находится в смеси, включающей (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия. В некоторых вариантах осуществления массовое соотношение соединения к микрокристаллической целлюлозе в смеси находится в диапазоне от приблизительно 1:1,5 до 1:15, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления массовое соотношение соединения к стеарилфумарату натрия в смеси имеет значение в диапазоне от приблизительно 5:1 до приблизительно 50:1, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу. В некоторых вариантах осуществления субъектом является человек.
В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект имеет первичный миелофиброз. В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект имеет миелофиброз, развившийся на фоне предшествующей истинной полицитемии. В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект имеет миелофиброз, развившийся на фоне предшествующей эссециальной тромбоцитемии. В некоторых вариантах осуществления субъект имеет высокий риск возникновения миелофиброза. В некоторых вариантах осуществления субъект имеет средний риск возникновения миелофиброза (например, как промежуточный 2 уровень риска возникновения заболевания). В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект дает положительный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в Янус-киназе 2 (JAK2) человека или дает положительный результат в отношении мутации, соответствующей мутации с заменой валина на фенил аланин в позиции 617 JAK2-киназы человека. В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект дает отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в Янус-киназе 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 JAK2-киназы человека. В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект имеет пальпируемую спленомегалию. В некоторых вариантах осуществления субъект с миелофиброзом имеет селезенку, расположенную, по меньшей мере, на 5 см ниже реберного края, что определено пальпацией. В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект является трансфузионно-зависимым (зависим от поддерживающей трансфузионной терапии). В некоторых вариантах осуществления композиций и способов, предоставляемых в этом документе, субъект не является трансфузионно-зависимым.
В некоторых вариантах осуществления способов, предоставляемых в этом документе, приведении соединения субъекту-человеку, Сmax соединения достигается в пределах от приблизительно 2 до приблизительно 4 часов после введения дозы. В некоторых вариантах осуществления после введения соединения субъекту-человеку, период полувыведения соединения составляет от приблизительно 16 до приблизительно 34 часа. В некоторых вариантах осуществления среднее значение AUC (площади под кривой), полученной для соединения, увеличивается более, чем пропорционально с повышением доз, находящихся в диапазоне от приблизительно 30 мг до приблизительно 800 мг в день. В некоторых вариантах осуществления накопление соединения находится в диапазоне от приблизительно 1,25-кратного до приблизительно 4,0-кратного в стационарном состоянии, когда дозу соединения вводят один раз в день. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия. В некоторых вариантах осуществления массовое соотношение соединения к микрокристаллической целлюлозе в смеси находится в диапазоне между приблизительно 1:1,5 и 1:15, где масса соединения соответствует массе фрагмента соединения, составляющего свободное основание. В некоторых вариантах осуществления массовое соотношение соединения к стеарилфумарату натрия в смеси имеет значение в диапазоне от приблизительно 5:1 до приблизительно 50:1, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу.
Также предоставлены в этом документе изделия производства или наборы, содержащие (а) любое изделие из капсул, обеспечиваемых в данной заявке, и (b) листок-вкладыш в упаковке или этикетку, где указано, что капсула полезна для лечения миелофиброза у субъекта. Также предоставлены в этом документе изделия производства или наборы, содержащие (а) любое изделие из стандартных лекарственных форм, обеспечиваемых в этой заявке, и (b) листок-вкладыш в упаковке или этикетку, указывающий(ую), что капсула полезна в лечении миелофиброза у субъекта. В некоторых вариантах осуществления предоставляется изделие производства или набор, содержащий (а) смесь, включающую в себя (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия, и (b) листок-вкладыш в упаковке или этикетку, указывающий(ую), что добавка полезна в лечении миелофиброза у субъекта.
Также предоставлены в этом документе изделия производства или наборы, содержащие (а) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и (b) листок-вкладыш в упаковке или этикетку, указывающий(ую), что соединение может быть применено для лечения миелофиброза у субъекта, где субъект дает отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 Янус-киназы 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 JAK2-киназы человека.
Также предоставлены в этом документе изделия производства или наборы, содержащие (а) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и (b) листок-вкладыш в упаковке или этикетку, указывающий(ую), что соединение может быть применено для лечения миелофиброза у субъекта, где субъект ранее получил другое лечение миелофиброза. В некоторых вариантах осуществления предыдущее лечение представляет собой метод лечения с применением ингибитора JAK2-киназы, который не представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат.
Также предоставлены в этом документе изделия производства или наборы, содержащие (а) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и (b) листок-вкладыш в упаковке или этикетку, указывающий(ую), что соединение может быть применено для уменьшения интенсивности симптомов состояния насыщенности клетками ткани костного мозга и/или фиброза костного мозга.
Также предоставлены в этом документе изделия производства или наборы, содержащие (а) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и (b) листок-вкладыш в упаковке или этикетку, указывающий(ую), что соединение может быть применено для уменьшения зуда, сопутствующего миелофиброзу.
Также предоставлены в этом документе изделия производства или наборы, содержащие соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и листок вкладыш-инструкцию в упаковке или этикетку, указывающую на упаковке, что соединение может быть применено для лечения миелофиброза у субъекта и что субъект должен прервать лечение при показании повышенных уровней одного или более ферментов или молекул, выбранных из группы, состоящей из: амилазы, липазы, аспартат-аминотрансферазы (AST), аланин-аминотрансфкразы (ALT) и креатинина, в сыворотке субъекта и/или при показании одного или более гематологических состояний, выбранных из группы, состоящей из анемии, тромбоцитопении и нейтропении. В некоторых вариантах осуществления листок-вкладыш в упаковке или этикетка на упаковке дополнительно указывает, что введение соединения может быть прекращено без предварительного снижения дозы. В некоторых вариантах осуществления один или несколько показателей повышенных уровней ферментов или молекул представляют собой осложнения 4 степени. В некоторых вариантах осуществления одно или несколько гематологических состояний представляют собой осложнения 4 степени.
В некоторых вариантах осуществления листок-вкладыш в упаковке или этикетка на упаковке размещен(а) так, чтобы бы его/ее могли увидеть предполагаемые покупатели. В некоторых вариантах осуществления соединение находится в стандартной лекарственной форме или капсульной форме.
В некоторых вариантах осуществления листок-вкладыш или этикетка показывает, что при введении субъекту-человеку Сmax соединения достигается в пределах от приблизительно 2 до приблизительно 4 часов после введения дозы. В некоторых вариантах осуществления листок-вкладыш в упаковке или этикетка на упаковке показывает, что при введении соединения субъекту-человеку, период полувыведения соединения составляет от приблизительно 16 до приблизительно 34 часа. В некоторых вариантах осуществления листок-вкладыш показывает, что AUC (средняя площадь под кривой), полученной для соединения, увеличивается более, чем пропорционально с увеличением доз, находящихся в диапазоне от приблизительно 30 мг до приблизительно 800 мг в день. В некоторых вариантах осуществления листок-вкладыш в упаковке или этикетка на упаковке указывает, что накопление соединения находится в диапазоне от приблизительно 1,25-кратного до приблизительно 4,0-кратного в стационарном состоянии, когда дозу соединения вводят один раз в день. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия. В некоторых вариантах осуществления массовое соотношение соединения к микрокристалличекой целлюлозе в смеси составляет от приблизительно 1:1,5 до 1:15, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления массовое соотношение соединения к стеарилфумарату натрия в смеси имеет значение в диапазоне от приблизительно 5:1 до приблизительно 50:1, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу.
В некоторых вариантах осуществления предоставляется применение соединения в изготовлении лекарственного препарата для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, (ii) эксципиент (например, микрокристаллическую целлюлозу, такую как силикатированную микрокристаллическую целлюлозу) и (iii) смазывающее вещество (например, стеарилфумарат натрия). В некоторых вариантах осуществления соединение вводят перорально. В некоторых вариантах осуществления применение осуществляют в соответствии со способом, описанным в этом документе.
В некоторых вариантах осуществления предоставляется применение соединения в изготовлении лекарственного препарата для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект дает отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в Янус-киназе 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 в JAK2-киназе человека. В некоторых вариантах осуществления предоставляется применение соединения в изготовлении лекарственного препарата для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект ранее получил другое лечение миелофиброза. В некоторых вариантах осуществления предыдущее лечение включает в себя введение ингибитора JAK2-киназы, который не представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления применение осуществляют в соответствии со способом, описанным в этом документе.
В некоторых вариантах осуществления предоставляют соединение для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, (ii) эксципиент (например, микрокристаллическую целлюлозу, такую как силикатированную микрокристаллическую целлюлозу) и (iii) смазывающее вещество (например, стеарилфумарат натрия). В некоторых вариантах осуществления соединение вводят перорально. В некоторых вариантах осуществления применение осуществляют в соответствии со способом, описанным в этом документе.
В некоторых вариантах осуществления предоставляют соединение для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект дает отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в Янус-киназе 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 в JAK2-киназе человека. В некоторых вариантах осуществления предоставляется соединение для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект ранее получил другое лечение миелофиброза. В некоторых вариантах осуществления предыдущее лечение включает в себя введение ингибитора JAK2-киназы, который не представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления лечение осуществляют в соответствии со способом, описанным в этом документе.
Следует понимать, что одно, несколько или все свойства различных вариантов осуществления, описанных в этом документе, могут быть объединены с получением других вариантов осуществления композиций и способов, предоставляемых в этой заявке. Эти и другие аспекты композиций и способов, предоставляемых в этом документе, станут понятны специалисту в данной области.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 показывает снижение пальпируемого размера селезенки за цикл лечения для пациентов, которых лечат с помощью TG101348 дозой 680 мг/день (исходная доза) (N=37). Дозы для цикла 1 были 520–800 мг/день, и дозы для циклов 2–6 были 360–680 мг/день. Для ≥50% субъектов цикла 6, было 22-47%-ное увеличение у 3 субъектов, которые принимали лекарственное средство в течение ~2–3 недель непосредственно перед измерением.
Фигура 2 показывает уровень лейкоцитов в крови (WBC = белые кровяные тельца) у субъектов, которые получали лечение с помощью TG101348. Исходный уровень лейкоцитов в крови составлял >11×109/л. Дозы в фазе контрольного наблюдения пациента находились в диапазоне от 360 до 680 мг/день. Последний визит контрольного наблюдения попадал в диапазон от 8 до 24 недель (медианное значение 24 недели). «ULN» означает верхнюю границу нормы.
Фигура 3 показывает уровень тромбоцитов у субъектов, которые получали лечение с помощью TG101348. Исходный уровень тромбоцитов в крови составлял >450×109/л. Дозы в фазе контрольного наблюдения пациента находились в диапазоне от 360 до 680 мг/день. Последний визит контрольного наблюдения попадал в диапазон от 12 до 24 недель (медианное значение 24 недели). «ULN» означает верхнюю границу нормы.
Фигура 4 показывает выраженное в процентах количество субъектов с ухудшенными, неизмененными, улучшенными или устраненными конституциональными симптомами (утомляемость, чувство быстрого насыщения, кашель, ночные приливы и зуд) у субъектов, которые получали лечение с помощью TG101348. Последний визит попадал в диапазон от 4 до 24 недель (медианное значение 20 недель). Данные здесь отражали изменения относительно симптомов, присутствующих на исходном уровне. 18 субъектов сообщили о впервые выявленном симптоме (≥1) во время исследования; из них, симптомы у 12 субъектов были устранены к моменту последнего визита контрольного наблюдения. Тяжесть оценивалась субъектами по шкале 1–10:0 = отсутствие симптома; 1–3 = слабо выраженный симптом; 4–7 = умеренно выраженный симптом; 8–10 = сильно выраженный симптом. Улучшение приравнивается снижению степени выраженности симптома до отсутствия такового или до слабой или умеренной степени выраженности относительно оценки степени выраженности на исходном уровне.
Фигура 5 показывает уровни цитокинов (IL-6, IL-8, IL-2 (интерлейкины) и TNF-α (фактор некроза опухоли альфа)) у субъектов, которые получали лечение посредством TG101348.
Фигура 6 показывает изменение аллельной нагрузки при мутации с заменой валина на фенилаланин в позиции 617 (V617F) относительно исходного уровня в виде доли исходного уровня аллельной нагрузки у субъектов с исходным уровнем >20% (N=22), которых лечили с применением TG101348. Фигура показывает подгруппу субъектов с положительным результатом в отношении мутации JAK2V617F в общей группе пациентов (N=48). Дозы в фазе контрольного наблюдения пациента составляли 360-680 мг/день. Последний визит контрольного наблюдения попадал в диапазон от 20 до 72 недель (медианное значение 24 недели).
Фигура 7 показывает насыщенность клетками ткани костного мозга на исходном уровне (насыщенность 60%) и после 18 циклов лечения с применением TG101348 (насыщенность 5-10%) у 76-летнего субъекта мужского пола, имеющего первичный миелофиброз (PMF) c отрицательным результатом в отношении мутации с заменой V617F. Исходная доза была 30 мг/день, и доза в фазе контрольного наблюдения пациента составила 520 мг/день.
Фигура 8 показывает фиброз ткани костного мозга на исходном уровне (3+) и после 18 циклов лечения с применением TG101348 (0) у 56-летнего субъекта мужского рода, имеющего первичный миелофиброз (PMF) c отрицательным результатом в отношении мутации с заменой V617F. Исходная доза была 240 мг/день, и доза в фазе контрольного наблюдения пациента составила 440 мг/день.
Фигура 9 показывает различные показатели для субъекта, имеющего первичный миелофиброз (PMF) c положительным результатом в отношении мутации с заменой V617F JAK2-киназы, которого лечат с применением TG101348 (исходная доза на уровне 680 мг/день).
Фигуры 10А-10G показывают распределение доз TG101348 в конце каждого цикла лечения для субъектов, у которых первоначальная доза составляла 30 мг/день, 60 мг/день, 120 мг/день, 240 мг/день, 360 мг/день, 520 мг/день и 800 мг/день, соответственно, (n=25).
Фигура 11 показывает распределение доз TG101348 в конце каждого цикла лечения субъектов, у которых первоначальная доза составляла 680 мг/день (n=34).
Фигура 12А показывает график средних значений концентраций TG101348 в плазме крови в зависимости от времени в полулогарифмическом масштабе (Цикл 1, День 1). Фигура 12В показывает график средних значений концентраций TG101348 в плазме крови в зависимости от времени в полулогарифмическом масштабе (Цикл 1, День 28).
Фигура 13 показывает касающуюся спленомегалии реакцию на лечение с применением TG101348. Эта фигура показывает снижение пальпируемого размера селезенки относительно исходного уровня после цикла лечения для субъектов в когорте с максимальной переносимой дозой (n=37). Показана доля субъектов с ≥50%-ным и 100%-ным снижением пальпируемой спленомегалии. Что касается субъектов, которые завершили 6 циклов лечения, 90% субъектов имели ≥25%-ное снижение пальпируемого размера селезенки, 66% субъектов имели ≥50%-ное снижение пальпируемого размера селезенки, и у 31% субъектов селезенка стала непальпируемой.
Фигуры 14А-14С показывают действие TG101348 в отношении симптомов миелофиброза. (А): Доля субъектов в когорте, где получали максимальную переносимую дозу, с полной регрессией чувства быстрого насыщения за цикл лечения относительно исходной оценки по шкале симптомов: «слабо выраженный симптом» (оценка = 1-3), «умеренно выраженный симптом» (оценка = 4-7) или «сильно выраженный симптом» (оценка = 8-10). Двадцать семь (79%) и 19 (56%) пациентов подлежали оценке в отношении уменьшения чувства быстрого насыщения в конце 1 и 6 циклов лечения, соответственно. После 2 циклов лечения, 56% сообщили о полной регрессии этого симптома с долговременной пользой. (В): Доля субъектов в когорте, где получали максимальную переносимую дозу, с полной регрессией утомляемости за цикл лечения относительно исходной оценки по шкале симптомов: «слабо выраженный симптом» (оценка = 1-3) или с уменьшением или с полной регрессией утомляемости относительно исходной оценки по шкале симптомов: «умеренно выраженный симптом» (оценка = 4-7) или «сильно выраженный симптом» (оценка = 8-10). Двадцать четыре (71%) и 16 (47%) пациентов подлежали оценке в отношении уменьшения утомляемости в конце 1 и 6 циклов лечения, соответственно. После 6 циклов лечения, 63% пациентов сообщали об уменьшении и 25% имели полную регрессию этого симптома. (С): Доля субъектов в когорте, где получали максимальную переносимую дозу, с полной регрессией ночных приливов за цикл лечения относительно исходной оценки по шкале симптомов: «слабо выраженный симптом» (оценка = 1-3), «умеренно выраженный симптом» (оценка = 4-7) или «сильно выраженный симптом» (оценка = 8-10). Четырнадцать (40%) и 9 (26%) пациентов подлежали оценке в отношении уменьшения ночных приливов в конце 1 и 6 циклов лечения, соответственно. После 1 цикла лечения, 64% субъектов имели полную регрессию этого симптома; после 6 циклов, эта доля увеличилась до 89%.
Фигура 15 показывает реакцию, касающуюся лейкоцитоза, на лечение с применением TG101348. Изменения уровня лейкоцитов в крови (WBC = белые кровяные тельца) после 6 циклов у субъектов, которые поступили на исследование с лейкоцитозом (уровень WBC >11×109/л). После 6 циклов лечения 16 субъектов, получавшие дозы во всем диапазоне доз (57%), и 13 субъектов в когорте, где получали максимальную переносимую дозу (MTD), (72%) достигали нормального уровня WBC, с долговременной пользой.
Фигуры 16А-16D показывают эффект лечения с применением TG101348 в отношении аллельной нагрузки мутации с заменой валина на фенилаланин в позиции 617 (V617F) JAK2-киназы. Представление данных по аллельной нагрузке мутации с заменой V617F JAK2-киназы в виде коробчатой диаграммы для всех субъектов с положительной реакцией на мутацию (n=51; фигуры А и В) и для подгруппы с исходной аллельной нагрузкой >20% (n=23; фигуры 16С и 16D). y-Ось отражает аллельную нагрузку мутации с заменой V617F JAK2-киназы от 1,0 (100%) до 0,0 (0%). Изменение аллельной нагрузки мутации с заменой V617F JAK2-киназы за цикл лечения (вплоть до конца 12 цикла; то есть С13D1) в сравнении с полученным в ходе предварительного исследования исходным уровнем показано для 2 групп (фигуры 16А и 16С); изменение в конце 6 цикла (то есть С7D1) и 12 цикла показано на фигурах 16В и 16D. Значительное снижение аллельной нагрузки мутации с заменой V617F JAK2-киназы в сравнении с полученным в ходе предварительного исследования исходным уровнем наблюдали в конце 6 цикла для группы, показывающей положительный результат в отношении мутации (фигура 16В; р=0,04), и для подгруппы с исходной аллельной нагрузкой >20% (фигура 16D; р=0,002); похожее значительное снижение наблюдали в конце 12 цикла для предыдущей (фигура 16В; р=0,01) и последующей (фигура 16D; р=0,002) групп. Для выполнения сравнения медианной аллельной нагрузки мутации с заменой V617А JAK2-киназы используют ранговый критерий согласованных пар Вилкоксона.
Фигура 17 показывает абсолютные изменения уровней провоспалительных цитокинов относительно исходного уровня на стадии цикла 6: IL-6 (Интерлейкин-6) (A), TNF-α (фактор некроза опухоли альфа) (B), IL-8 (Интерлейкин-8) (С) и IL-2 (Интерлейкин-2) (D). Абсолютные разницы IL-6 (-4719 пг/мл) и IL-2 (-1827 пг/мл) опущены на фигурах 17А и 17D, соответственно, для 1 субъекта (101-039), так как они искажают представление данных для других субъектов.
Фигура 18 показывает зависимость средних концентраций TG101348 в плазме крови от времени на линейной диаграмме после перорального введения доз один раз в день (Цикл 1; День 28).
ПОДРОБНОЕ ОПИСАНИЕ
I. Определения
«Лечение» или «проведение лечения» означает подход для получения благотворных или желательных результатов, в том числе клинических результатов. Благотворные или желательные клинические результаты могут включать один или более результатов, выбранных из следующего: снижение симптомов, возникших вследствие заболевания, повышение качества жизни субъектов, страдающих от заболевания, снижение дозы других лекарственных препаратов, необходимых для лечения заболевания, замедление прогрессирования заболевания и/или продление выживаемости индивидуумов, но не ограничиваются этим. В некоторых вариантах осуществления для лечения миелофиброза, благотворные клинические результаты включают один или более результатов, выбранных из снижения спленомегалии, уменьшения конституциональных симптомов (таких как чувство быстрого насыщения, утомляемость, ночные приливы, кашель и зуд), снижения лейкоцитоза, снижения тромбоцитоза, снижения аллельной нагрузки мутации с заменой V617F JAK2-киназы, снижения фиброза ткани костного мозга и/или снижения насыщенности клетками ткани костного мозга.
«Замедление развития заболевания» означает отсрочивание, задерживание, замедление, торможение, стабилизирование и/или отдаление развития заболевания (такого как миелофиброза) или симптомов заболевания и может включать «выживаемость без прогрессирования заболевания». Такое замедление может иметь различную продолжительность во времени, в зависимости от истории заболевания и/или от индивидуума, подлежащего вылечиванию. Специалисту в данной области будет ясно, что достаточное или значительное замедление может, по существу, охватывать предупреждение (профилактику), в том смысле, что индивидуум не развивает заболевание.
«Эффективная дозировка» или «эффективное количество» лекарственного средства, соединения или фармацевтической композиции составляет количество, достаточное для получения благотворных или желаемых результатов. Для профилактического применения, благотворные или желательные результаты могут включать, например, один или более результатов, таких как устранение или снижение риска, понижения тяжести или отсрочивание возникновение заболевания, включая биохимические, гистологические и/или поведенческие симптомы заболевания, его осложнения и промежуточные патологические фенотипы, обнаруживаемые во время развития заболевания. Для терапевтического применения, благотворные или желаемые результаты могут включать, например, один или более клинических результатов, таких как снижение одного или более симптомов и патологических состояний, обусловленных или связанных с заболеванием, повышение качества жизни индивидуумов, страдающих от заболевания, снижение дозы других лекарственных препаратов, необходимых для лечения заболевания, усиление действия другого лекарственного препарата, например, с помощью таргетинга (направленной доставки лекарственного средства к органу-мишени), отсрочивания прогрессирования заболевания и/или продления выживаемости. В случае миелофиброза эффективное количество лекарственного средства может иметь эффект снижения одного или более симптомов спленомегалии, ослабления конституциональных симптомов (таких как чувство быстрого насыщения, утомляемость, ночные приливы, кашель и зуд), снижения лейкоцитоза, снижения тромбоцитоза, снижения аллельной нагрузки мутации с заменой V617F JAK2-киназы, снижения фиброза ткани костного мозга и/или снижения насыщенности клетками ткани костного мозга. Эффективная дозировка может быть введена одним или несколькими введениями. Эффективная дозировка лекарственного средства, соединения или фармацевтической композиции может представлять собой, например, количество, достаточное для выполнения профилактического или терапевтического лечения, либо прямого, либо опосредованного. Как ясно из клинического контекста, эффективная дозировка лекарственного средства, соединения или фармацевтической композиции может быть достигнута или может быть не достигнута в сочетании с другим(ой) лекарственным средством, соединением или фармацевтической композицией. Таким образом, «эффективная дозировка» может быть рассмотрена в контексте введения одного или более терапевтических средств, и может быть предусмотрено, что отдельно взятое средство должно быть введено в эффективном количестве, если в сочетании с одним или несколькими другими средствами, может быть достигнут или достигается желаемый результат.
«Уменьшение интенсивности симптомов» насыщенности клетками ткани костного мозга или фиброза ткани костного мозга относится к снижению уровня насыщенности клетками ткани костного мозга или фиброза ткани костного мозга у субъекта в сравнении с уровнем насыщенности клетками ткани костного мозга или фиброза ткани костного мозга до начала лечения соединением, предоставляемым в этой заявке. Снижение насыщенности клетками ткани костного мозга или фиброза ткани костного мозга может составлять, по меньшей мере, 5, 10, 20, 30, 40, 50, 60, 70, 80 или 90%.
«В сочетании с» относится к введению одного метода лечения в дополнение к другому методу лечения. Само по себе, выражение «в сочетании с» может относиться к введению одного метода лечения до, во время или после введения другого метода лечения индивидууму.
Как используется в этом документе, «пациент» или «субъект» относится к млекопитающему, включающему в себя человека, собаку, лошадь, корову или кошку и так далее.
Термин «фармацевтически приемлемый» относится к тому факту, что носитель, разбавитель или эксципиент должен быть совместим с другими ингредиентами состава и может быть введен субъекту.
«Фармацевтически приемлемые соли» относятся к производным раскрываемых соединений, где исходное родоначальное соединение является модифицированным в результате изготовления его солей присоединения кислоты или основания.
Формы единственного числа включают ссылку на формы множественного числа, если контекст не указывает ясно иное.
Упоминание значения или параметра вместе с «приблизительно» включает (и описывает) варианты осуществления, которые направлены на то значение или тот параметр сам по себе. Например, описание, относящееся к «приблизительно Х», включает описание «Х».
Понятно, что аспекты и вариации композиций и способов, предоставляемых в этом документе, могут включать «состоящий(ая) из» и/или «в основном состоящий(ая) из» аспектов и вариаций.
II. Соединения и Фармацевтические композиции
В этой заявке предоставляется соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. Также в этой заявке предоставляются фармацевтические композиции, содержащие N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и фармацевтически приемлемый эксципиент или носитель. Соединение и фармацевтические композиции, описанные в этом документе, могут быть применены для лечения или замедления развития миелофиброза у субъекта. N-трет-Бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид имеет следующую химическую структуру:
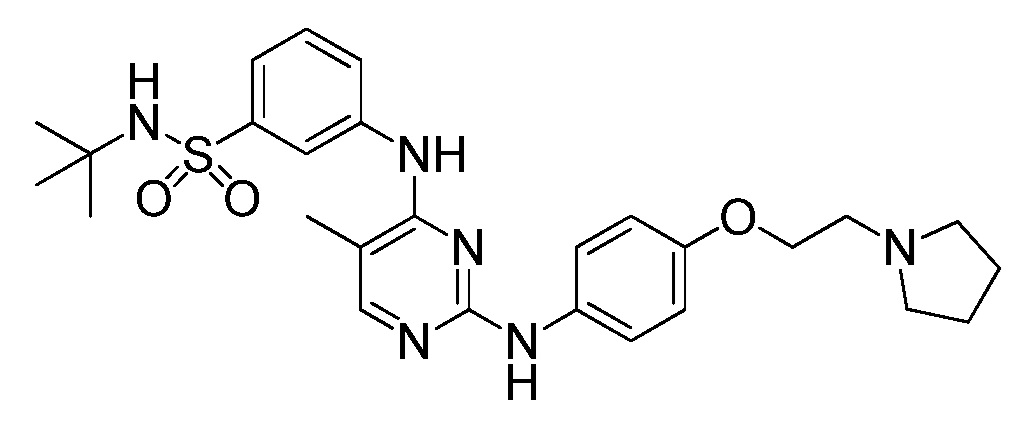
Соединение, предоставляемое в этой заявке, может быть введено в состав терапевтических композиций в виде природных или солевых форм. Фармацевтически приемлемые нетоксичные соли включают соли присоединения основания (образованные свободными карбоксильными или другими анионными группами), которые могут быть получены из неорганических оснований, таких как, например, гидроксид натрия, гидроксид калия, гидроксид аммония, гидроксид кальция или гидроксид трехвалентного железа, и таких органических оснований, как изопропиламин, триметиламин, 2-этиламино-этанол, гистидин, прокаин и тому подобное. Такие соли также могут быть образованы как соли присоединения кислоты с любыми свободными катионными группами и, как правило, будут образованы неорганическими кислотами, такими как, например, хлористоводородная кислота, серная кислота или фосфорная кислота, или органическими кислотами, такими как уксусная кислота, лимонная кислота, пара-толуолсульфокислота, метансульфокислота, щавелевая кислота, винная кислота, миндальная кислота и тому подобное.
Соли соединений, предоставляемых в этой заявке, могут включать аминные соли, образованные протонированием аминогруппы с помощью неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, фосфорная кислота и тому подобное. Соли соединений, предоставляемых в этой заявке, также могут включать аминные соли, образованные протонированием аминогруппы с помощью подходящих органических кислот, таких как пара-толуолсульфокислота, уксусная кислота, метансульфокислота и тому подобное. Дополнительные эксципиенты, которые предусмотрены для использования при практическом применении композиций и способов, предоставляемых в этой заявке, представляют собой эксципиенты, доступные среднему специалисту в данной области, например, эксципиенты, обнаруживаемые в Фармакопее Соединенных Штатов, том XXII, и в Национальном Формуляре Соединенных Штатов, том XVII, в Фармакопейной конвенции США, Inc., Rockville, Md. (1989), релевантное содержание которых включено в этот документ посредством ссылок.
В дополнение к тому, соединения, предоставляемые в этой заявке, могут включать полиморфные модификации. Соединение, описанное в этом документе, может находиться в альтернативных формах. Например, соединение, описанное в этом документе, может включать гидратную форму. «Гидрат» относится к соединению, предоставляемому в этой заявке, которое связано с водой в молекулярной форме, то есть в которой связь Н-ОН не расщеплена и может быть представлена, например, формулой R.H2O, где R представляет собой соединение, предоставляемое в этом документе. Данное соединение может образовывать более одного гидрата, включающего, например, моногидраты (R.H2O) или полигидраты (R.nH2O, где n является целым числом, которое превышает 1), в том числе, например, дигидраты (R.2H2O), тригидраты (R.3H2O) и тому подобное, или частичные гидраты, такие как, например, R.n/2H2O, R.n/3H2O, R.n/4H2O и тому подобное, где n представляет собой целое число.
Соединения, описанные в этом документе, также могут включать гидратные формы соли присоединения кислоты. Как используют в этом документе, «гидрат соли присоединения кислоты» относится к комплексу, который может быть образован через связывание соединения, имеющего один или более основных фрагментов, по меньшей мере, с одним соединением, имеющим один или более кислотных фрагментов, или через связывание соединения, имеющего один или более кислотных фрагментов, по меньшей мере, с одним соединением, имеющим один или более основных фрагментов, где упомянутый комплекс дополнительно связан с молекулами воды с образованием гидрата, где упомянутый гидрат является таким же, как он определен ранее, и R означает комплекс, описанный в этом документе выше.
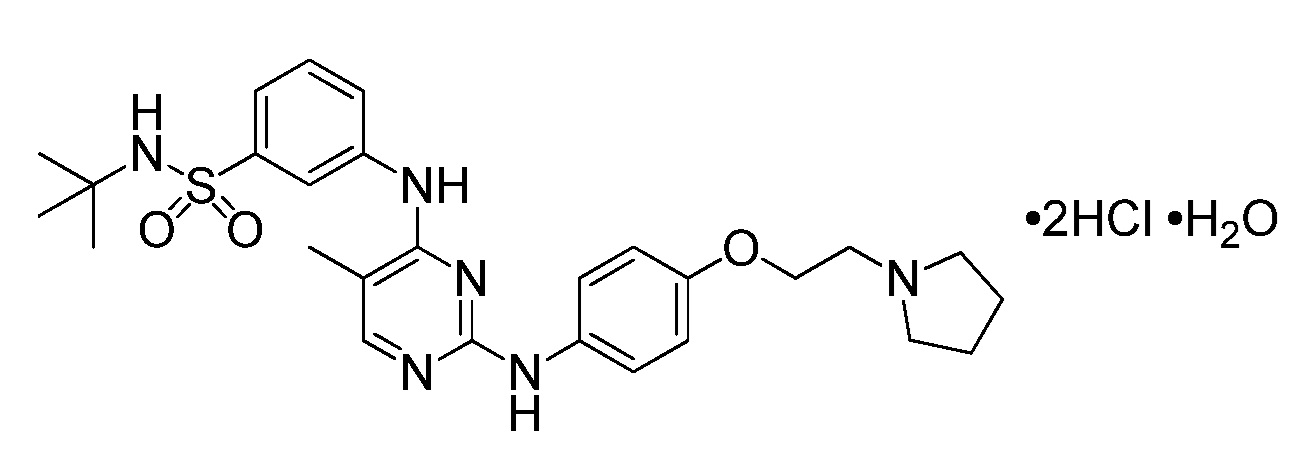
В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида и имеет следующую химическую структуру:
Фармацевтические композиции для введения соединения, описанного в этом документе, либо как такового, либо в комбинации с другими терапевтическими средствами, могут быть удобно представлены в дозированной единичной форме и могут быть приготовлены любым способом из способов, хорошо известных в области фармацевтики, и способами, описанными в Примерах 4, 5 и 6. Такие способы могут включать приведение активного ингредиента к соединению с носителем, который состоит из одного или более вспомогательных ингредиентов. В общем, фармацевтические композиции приготавливают путем приведения активного ингредиента к однородному и тщательному соединению с жидким носителем или с тонкоизмельченным твердым носителем или и с тем, и с другим и затем, при необходимости, путем формования продукта с получением желаемого состава. В фармацевтической композиции активное целевое соединение включено в количестве, достаточном для того, чтобы производить желательное действие на патологический процесс или на патологическое состояние. Фармацевтические композиции, содержащие активный ингредиент, могут иметь форму, подходящую для перорального применения, например, как твердые или мягкие капсулы. Подходящая оболочка капсулы может представлять собой твердый желатин или гидроксипропилметил-целлюлозу («HPMC»).
В этой заявке предоставляют составы, содержащие (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) один или более эксципиентов и (iii) одно или более смазывающих веществ. Составы могут находиться в капсульной форме и вводятся перорально. Составы могут находиться в стандартной лекарственной форме. В некоторых вариантах осуществления, эксципиент представляет собой лактозу (такую как Fast-Flo), маннит (такой как Parteck M200), микрокристаллическую целлюлозу («MCC») (такую как Avicel PH102), MCC (такую как ProSolv 90 HD). В некоторых вариантах осуществления смазывающее вещество представляет собой стеарат магния, стеарилфумарат натрия (такой как Pruv) или лаурилфумарат натрия. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу. В некоторых вариантах осуществления капсула представляет собой твердую желатиновую капсулу.
В некоторых вариантах осуществления предоставляют капсулу, подходящую для перорального введения, содержащую смесь, включающую (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) эксципиент (например, микрокристаллическую целлюлозу, такую как силикатированная микрокристаллическая целлюлоза) и (iii) смазывающее вещество (например, стеарилфумарат натрия), где смесь содержится в капсуле. Для изготовления капсул могут быть использованы способы, известные в данной области и описанные в этом документе. См., например, Пример 3. Микрокристаллическая целлюлоза может быть использована в качестве наполнителя и/или разбавителя в капсулах, предоставляемых в этой заявке. Стеарилфумарат натрия может быть использован в качестве смазывающего вещества в капсулах, обеспечиваемых в этом документе. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу. Например, силикатированная микрокристаллическая целлюлоза может состоять из микрокристаллической целлюлозы и частиц коллоидного диоксида кремния. В некоторых вариантах осуществления силикатированная микрокристаллическая целлюлоза представляет собой комбинацию 98% микрокристаллической целлюлозы и 2% коллоидного диоксида кремния.
В некоторых вариантах осуществления капсула содержит от приблизительно 10 мг до приблизительно 680 мг соединения, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула содержит от приблизительно 10 мг до приблизительно 650 мг (или от приблизительно 10 мг до приблизительно 550 мг или от приблизительно 10 мг до приблизительно 500 мг), где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула содержит от приблизительно 100 мг до приблизительно 600 мг (или от приблизительно 200 мг до приблизительно 550 мг или от приблизительно 300 мг до приблизительно 500 мг), где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула содержит приблизительно 10 мг, приблизительно 20 мг, приблизительно 40 мг, приблизительно 100 мг, приблизительно 150 мг, приблизительно 200 мг, приблизительно 250 мг, приблизительно 300 мг, приблизительно 350 мг, приблизительно 400 мг, приблизительно 450 мг, приблизительно 500 мг, приблизительно 550 мг, приблизительно 600 мг и приблизительно 650 мг соединения, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула представляет собой твердую желатиновую капсулу. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида.
В некоторых вариантах осуществления массовое соотношение соединения к эксципиенту (например, к микрокристаллической целлюлозе, такой как силикатированная микрокристаллическая целлюлоза) в капсуле составляет от приблизительно 1:1,5 до приблизительно 1:15 (например, от приблизительно 1:5 до приблизительно 1:10, от приблизительно 1:5 до приблизительно 1:12 или от приблизительно 1:10 до приблизительно 1:15), где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления массовое соотношение соединения к смазывающему веществу (например, к стеарилфумарату натрия) в капсуле находится в диапазоне от приблизительно 5:1 до приблизительно 50:1 (например, между приблизительно 5:1 и приблизительно 10:1, между приблизительно 5:1 и приблизительно 25:1, между приблизительно 5:1 и приблизительно 40:1, между приблизительно 7:1 и приблизительно 34:1 или между приблизительно 8:1 и приблизительно 34:1), где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию.
В некоторых вариантах осуществления капсула содержит от приблизительно 5% до приблизительно 50% (например, от приблизительно 5% до приблизительно 10% или от приблизительно 5% до приблизительно 35%) соединения от общей массы содержимого капсулы, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула содержит от приблизительно 40% до приблизительно 95% (например, от приблизительно 50% до приблизительно 90% или от приблизительно 60% до приблизительно 90%) эксципиента (например, микрокристаллической целлюлозы, такой как силикатированная микрокристаллическая целлюлоза) от общей массы содержимого капсулы. В некоторых вариантах осуществления капсула содержит от приблизительно 0,2% до приблизительно 5% (например, от приблизительно 0,2% до приблизительно 2% ,или от приблизительно 0,5% до приблизительно 1,5%, или приблизительно 0,5%, или приблизительно 1%, или приблизительно 1,5%) смазывающего вещества (например, стеарилфумарата натрия) от общей массы содержимого капсулы.
Также в этой заявке предоставляют стандартные лекарственные формы, содержащие смесь, включающую (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) эксципиент (такой как микрокристаллическая целлюлоза) и (iii) смазывающее вещество (такое как стеарилфумарат натрия). Любая из капсул, описанных в этом документе, может быть использована в стандартной лекарственной форме. В некоторых вариантах осуществления стандартная лекарственная форма предназначена для лечения миелофиброза. В некоторых вариантах осуществления лечение поводится в соответствии со способом, описанным в этом документе.
В некоторых вариантах осуществления стандартная лекарственная форма содержит смесь, включающую (i) от приблизительно 10 мг от приблизительно 680 мг (или от приблизительно 10 мг от приблизительно 500 мг) соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где указанная масса соответствует массе фрагмента соединения, составляющего свободное основание, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия. В некоторых вариантах осуществления соединение в смеси составляет от приблизительно 10 мг от приблизительно 500 мг, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию.
В некоторых вариантах осуществления стандартная лекарственная форма имеет форму капсулы, и смесь содержится в капсуле. В некоторых вариантах осуществления стандартная лекарственная форма содержит приблизительно 10 мг, приблизительно 20 мг, приблизительно 40 мг, приблизительно 100 мг, приблизительно 150 мг, приблизительно 200 мг, приблизительно 250 мг, приблизительно 300 мг, приблизительно 350 мг, приблизительно 400 мг, приблизительно 450 мг, приблизительно 500 мг, приблизительно 550 мг, приблизительно 600 мг или приблизительно 650 мг соединения, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления добавка содержит (i) приблизительно 10 мг (или приблизительно любое количество из 40 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг) соединения, (ii) микрокристаллическую целлюлозу и (iii) стеарилфумарат натрия, где указанная масса дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида.
В некоторых вариантах осуществления массовое соотношение соединения к эксципиенту (например, к микрокристаллической целлюлозе, такой как силикатированная микрокристаллическая целлюлоза) в стандартной лекарственной форме находится в диапазоне от приблизительно 1:1,5 до приблизительно 1:15 (например, между приблизительно 1:5 и приблизительно 1:10, между приблизительно 1:5 и приблизительно 1:12 или между приблизительно 1:10 и приблизительно 1:15), где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления массовое соотношение соединения к смазывающему веществу (например, к стеарилфумарату натрия) в стандартной лекарственной форме составляет от приблизительно 5:1 до приблизительно 50:1 (например, в диапазоне от приблизительно 5:1 до приблизительно 10:1, от приблизительно 5:1 до приблизительно 25:1, от приблизительно 5:1 до приблизительно 40:1, от приблизительно 7:1 до приблизительно 34:1 или от приблизительно 8:1 до приблизительно 34:1), где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления микрокристаллическая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу. В некоторых вариантах осуществления силикатированная микрокристаллическая целлюлоза представляет собой комбинацию 98% микрокристаллической целлюлозы и 2% коллоидного диоксида кремния.
В некоторых вариантах осуществления смазывающее вещество (например, стеарилфумарат натрия) составляет от приблизительно 0,1% до приблизительно 10%, от приблизительно 0,5% до приблизительно 5%, от приблизительно 0,5% до приблизительно 3%, от приблизительно 0,5% до приблизительно 2%, от приблизительно 0,75% до приблизительно 1,5% от массы содержимого капсулы. В некоторых вариантах осуществления смазывающее вещество (например, стеарилфумарат натрия) составляет, по меньшей мере, приблизительно любое количество из 0,1%, 0,25%, 0,5%, 0,75%, 1%, 1,25%, 1,5%, 1,75%, 2%, 2,5%, 3%, 3,5%, 4%, 4,5% или 5% массы содержимого капсулы. В некоторых вариантах осуществления смазывающее вещество (например, стеарилфумарат натрия) составляет приблизительно любое количество из 0,1%, 0,25%, 0,5%, 0,75%, 1%, 1,25%, 1,5%, 1,75%, 2%, 2,5%, 3%, 3,5%, 4%, 4,5% или 5% от массы содержимого капсулы.
В некоторых вариантах осуществления массовое соотношение соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, к эксципиенту (например, к микрокристаллической целлюлозе, такой как силикатированная микрокристаллическая целлюлоза) в капсуле или в стандартной лекарственной форме составляет от приблизительно 40:60 до приблизительно 10:90. В некоторых вариантах осуществления массовое соотношение соединения к эксципиенту (например, к микрокристаллической целлюлозе, такой как силикатированная микрокристаллическая целлюлоза) в капсуле или в стандартной лекарственной форме составляет приблизительно любое соотношение, выбранное из 95:5, 90:10, 85:15, 80:20, 75:25, 70:30, 65:35, 60:40, 55:45, 50:50, 45:55, 40:60, 35:65, 30:70, 25:75, 20:80, 15:85, 10:90 или 5:95. В некоторых вариантах осуществления массовое соотношение соединения к эксципиенту (например, к микрокристаллической целлюлозе, такой как силикатированная микрокристаллическая целлюлоза) составляет от приблизительно 1:1,5 до приблизительно 1:9,5, например приблизительно любое соотношение, выбранное из 1:1,5, 1:2, 1:2,5, 1:3, 1:3,5, 1:4, 1:4,5, 1:5, 1:5,5, 1:6, 1:6,5, 1:7, 1:7,5, 1:8, 1:8,5, 1:9 или 1:9,5. В некоторых вариантах осуществления соединение представляет собой моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида.
В некоторых вариантах осуществления капсула содержит от приблизительно 5% до приблизительно 50% (например, от приблизительно 5% до приблизительно 10% или от приблизительно 5% до приблизительно 35%) соединения от общей массы добавки, где масса соединения дана в расчете на массу фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления капсула содержит от приблизительно 40% до приблизительно 95% (например, от приблизительно 50% до приблизительно 90% или от приблизительно 60% до приблизительно 90%) микрокристаллической целлюлозы (такой как силикатированная микрокристаллическая целлюлоза) от общей массы добавки. В некоторых вариантах осуществления капсула содержит от приблизительно 0,2% до приблизительно 5% (например, от приблизительно 0,2% до приблизительно 2% или от приблизительно 0,5% до приблизительно 1,5% или приблизительно 0,5%, приблизительно 1% или приблизительно 1,5%) стеарилфумарата натрия от общей массы добавки.
В некоторых вариантах осуществления капсула или стандартная лекарственная форма содержит смесь, включающую приблизительно 12 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида, приблизительно 122 мг силикатированной микрокристаллической целлюлозы и приблизительно 1 мг стеарилфумарата натрия. В некоторых вариантах осуществления капсула или стандартная лекарственная форма содержит смесь, включающую приблизительно 47 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида, приблизительно 448 мг силикатированной микрокристаллической целлюлозы и приблизительно 5 мг стеарилфумарата натрия. В некоторых вариантах осуществления капсула или стандартная лекарственная форма содержит смесь, включающую приблизительно 117 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. В некоторых вариантах осуществления капсула или стандартная лекарственная форма содержит смесь, включающую приблизительно 235 мг моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида, приблизительно 357 мг силикатированной микрокристаллической целлюлозы и приблизительно 6,00 мг стеарилфумарата натрия.
Также в этом документе предоставляют пероральные составы в форме раствора, содержащие соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления пероральный состав в форме раствора дополнительно содержит метилцеллюлозу. В некоторых вариантах осуществления пероральный состав в форме раствора дополнительно содержит метилцеллюлозу и Твин 80. В некоторых вариантах осуществления, пероральный состав в форме раствора содержит соединение в концентрации от приблизительно 1 мг/мл до приблизительно 25 мг/мл, от приблизительно 2 мг/мл до приблизительно 20 мг/мл, от приблизительно 3 мг/мл до приблизительно 15 мг/мл, от приблизительно 5 мг/мл до приблизительно 10 мг/мл. В некоторых вариантах осуществления пероральный состав в форме раствора содержит соединение в приблизительно любой концентрации из 2 мг/мл, 3 мг/мл, 4 мг/мл, 5 мг/мл, 6 мг/мл, 6,25 мг/мл, 6,5 мг/мл, 7 мг/мл, 8 мг/мл, 9 мг/мл, 10 мг/мл, 12,5 мг/мл или 15 мг/мл. В некоторых вариантах осуществления пероральный состав в форме раствора содержит от приблизительно 0,1% до приблизительно 5%, от приблизительно 0,2% до приблизительно 3%, от приблизительно 0,25% до приблизительно 2%, от приблизительно 0,25% до приблизительно 1% или приблизительно 0,5% по массе метилцеллюлозы. В некоторых вариантах осуществления пероральный состав в форме раствора содержит от приблизительно 0,01% до приблизительно 0,5%, от приблизительно 0,02% до приблизительно 0,3%, от приблизительно 0,025% до приблизительно 0,2%, от приблизительно 0,025% до приблизительно 0,1% или приблизительно 0,05% по массе Твин 80.
В некоторых вариантах осуществления капсула не содержит способствующее всасыванию вещество. В некоторых вариантах осуществления капсула содержит способствующее всасыванию вещество (например, витамин E TPGS, Gelucire 44/14, Pluronic F127 или глицерилмоностеарат).
Капсула или стандартная лекарственная форма, предоставляемая в этой заявке, может обладать одним или более из следующих свойств: (1) при введении субъекту, такому как человеческий субъект, Сmax соединения достигается в пределах от приблизительно 2 до приблизительно 4 часов после введения дозы; (2) при введении человеческому субъекту, период полувыведения соединения составляет от приблизительно 16 до приблизительно 34 часа; (3) среднее значение AUC (площади под кривой), полученной для соединения, увеличивается более чем пропорционально с повышением доз, находящихся в диапазоне от приблизительно 30 мг до приблизительно 800 мг в день; (4) накопление соединения находится в диапазоне от приблизительно 1,25-кратного до приблизительно 4,0-кратного в стационарном состоянии в том случае, когда дозу соединения вводят один раз в день.
Также предоставляют способы получения капсульного лекарственного продукта, включающие а) смешение смазывающего вещества с соединением, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, для получения гранул и b) смешение гранул, полученных на стадии а), с эксципиентом. В некоторых вариантах осуществления смазывающее вещество представляет собой стеарилфумарат натрия. В некоторых вариантах осуществления эксципиент представляет собой микрокристаллическую целлюлозу, такую как силикатированная микрокристаллическая целлюлоза. Такой способ может быть использован для изготовления капсулы или стандартной лекарственной формы, описанной в этом документе. Масса (например, как массовое соотношение или массовое процентное содержание) и компоненты, что касается соединения, эксципиента и/или смазывающего вещества, могут соответствовать любым массам и компонентам, описанным в этом документе.
III. Способы лечения и предупреждения миелофиброза
В этом документе предоставляют способы лечения, замедления развития и/или предупреждения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат (например, моногидрат дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида). В некоторых вариантах осуществления субъект имеет миелофиброз. В некоторых вариантах осуществления субъект находится в группе риска развития миелофиброза. В некоторых вариантах осуществления субъект является человеческим субъектом. Любой из составов, описанных в этом документе, такой как капсулы или стандартные лекарственные формы, описанные в этом документе, могут быть использованы для лечения субъекта с миелофиброзом. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, которое представляет собой (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) эксципиент (такой как микрокристаллическая целлюлоза) и (iii) смазывающее вещество (такое как стеарилфумарат натрия).
Миелофиброз, который может быть вылечен соединениями, описанными в этом документе, включает первичный миелофиброз (MF) и вторичный миелофиброз (например, миелофиброз, развившийся на фоне предшествующей истинной полицитемии (post-PV MF) или эссенциальной тромбоцитемии (post-ET MF)). Миелофиброз, который может быть вылечен соединениями, описанными в этом документе, также включает в себя миелофиброз высокого риска развития, промежуточного риска развития, такого как промежуточного 2 уровня риска развития заболевания. Способы диагностирования различных типов миелофиброза известны в данной области. См., например, Cervantes et al., Blood 2009. В некоторых вариантах осуществления субъект с миелофиброзом имеет селезенку, расположенную, по меньшей мере, на 5 см ниже реберного края, что определено пальпацией.
В некоторых вариантах осуществления субъект имеет точечную мутацию с заменой валина на фенилаланин в позиции 617 в Янус-киназе 2 (JAK2-киназа) (JAK2V617F), если субъект является человеком, или точечную мутацию, соответствующую замене валина на фенилаланин в позиции 617 в Янус-киназе 2 (JAK2-киназа) (JAK2V617F), если субъект не является человеком. В некоторых вариантах осуществления субъект имеет отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 JAK2-киназы в том случае, если субъект является человеком, или отрицательный результат в отношении мутации, соответствующей замене валина на фенилаланин в позиции 617 Янус-киназы 2 (JAK2-киназы) в том случае, если субъект не является человеком. Имеет ли субъект положительный результат или отрицательный результат в отношении мутации JAK2V617F, можно определить с помощью анализа методом полимеразной цепной реакции («PCR») с использованием геномной ДНК из клеток ткани костного мозга или клеток крови (например, лейкоцитов цельной крови). Анализ методом ПЦР может представлять собой аллель-специфичную ПЦР (например, аллель-специфичную количественную ПЦР) или ПЦР секвенировние. См. публикации: Kittur J. et al., Cancer 2007, 109(11):2279-84, и McLornan D. et al., Ulster Med J. 2006, 75(2):112-9, каждая из которых явным образом включена в этот документ посредством ссылок.
В некоторых вариантах осуществления субъект, которого лечат способами, описанными в данном документе, ранее получил другую терапию или лечение миелофиброза. В некоторых вариантах осуществления субъект не дает клинического ответа на другую терапию миелофиброза или имеет возврат болезни (рецидив) после получения другой терапии миелофиброза. Предыдущая терапия может включать ингибитор JAK2-киназы (например, INCB018424 (также известный как руксолитиниб, доступный для приобретения в Incyte), CEP-701 (лестауртиниб, доступный для приобретения в Сephalon) или XL019 (доступный для приобретения в Exelixis)) (См. публикацию Verstovsek S., Hematology Am. Soc. Hematol. Educ. Program. 2009:636-42) или ингибитор не-JAK2-киназы (такой как гидроксимочевина). В некоторых вариантах осуществления субъект получил лечение руксолитинибом первичного миелофиброза, миелофиброза, развившегося на фоне истинной полицитемии (Post-PV MF), миелофиброза, развившегося на фоне эссенциальной тромбоцитемии (Post-ET MF), истинной полицитемии или эссенциальной тромбоцитемии, в течение, по меньшей мере, 14 дней и имел перерыв в лечении в течение, по меньшей мере, 30 дней. В некоторых вариантах осуществления предыдущая терапия представляет собой лечение соединением, описанным в данном документе, и предыдущая терапия была прервана при показании, включающем один или более повышенных уровней амилазы, липазы, аспартат-аминотрансферазы (AST), аланин-аминотрансферазы (ALT) и/или креатинина в сыворотке субъекта, и/или при показании гематологического состояния, выбранного из группы, состоящей из анемии, тромбоцитопении и нейтропении. В некоторых вариантах осуществления доза соединения во втором лечении является аналогичной или ниже, чем доза в предыдущей терапии.
Субъект может получать пероральное и/или ежедневное лечение. Субъект (такой как человек) может получить лечение путем введения соединения в дозе от приблизительно 240 мг в день до приблизительно 680 мг в день (или от приблизительно 300 мг в день до приблизительно 500 мг в день), где указанная масса является массой фрагмента соединения, соответствующего свободному основанию. В некоторых вариантах осуществления соединение вводят в дозе, составляющей приблизительно любое количество, выбранное из 240 мг/день, 250 мг/день, 300 мг/день, 350 мг/день, 400 мг/день, 450 мг/день, 500 мг/день, 550 мг/день, 600 мг/день, 650 мг/день или 680 мг/день. Соединение может находиться в капсуле и/или в стандартной лекарственной форме, описанной в данном документе. В некоторых вариантах осуществления, вводимое соединение находится в смеси вместе с микрокристаллической целлюлозой и стеарилфумаратом натрия, а смесь содержится в капсуле. В некоторых вариантах осуществления соединение вводят перорально.
Также в данной заявке предоставляют способы уменьшения интенсивности одного или более симптомов, сопутствующих миелофиброз. Например, лечение с применением соединения, описанного в данном документе, является эффективным в уменьшении размера селезенки, уменьшении интенсивности конституциональных симптомов (таких как чувство быстрого насыщения, утомляемость, ночные приливы, кашель и зуд), снижении лейкоцитоза, снижении тромбоцитоза, снижении аллельной нагрузки мутации JAK2V617F, уменьшении фиброза костного мозга, уменьшении зуда, уменьшении кахексии и/или снижении насыщенности клетками ткани костного мозга. Снижение, понижение, уменьшение интенсивности или улучшение состояния может быть, по меньшей мере, на 5, 10, 20, 30, 40, 50, 60, 70, 80 или 90%, по сравнению с уровнем до начала лечения соединением, предоставляемым в данном документе. В некоторых вариантах осуществления селезенка становится непальпируемой у субъекта после лечения. В некоторых вариантах осуществления субъект имеет полную регрессию лейкоцитоза и/или тромбоцитоза после лечения. В некоторых вариантах осуществления субъект имеет полную регрессию зуда после лечения.
В некоторых вариантах осуществления соединение вводят субъекту ежедневно в течение, по меньшей мере, 1 цикла, по меньшей мере, 2 циклов, по меньшей мере, 3 циклов, по меньшей мере, 4 циклов, по меньшей мере, 5 циклов или в течение, по меньшей мере, 6 циклов, где цикл составляет 28 дней. В некоторых вариантах осуществления соединение вводят субъекту ежедневно в течение, по меньшей мере, 6 циклов, где цикл составляет 28 дней, в течение, по меньшей мере, 8 циклов, где цикл составляет 28 дней, в течение, по меньшей мере, 10 циклов, где цикл составляет 28 дней, в течение, по меньшей мере, 12 циклов, где цикл составляет 28 дней, в течение, по меньшей мере, 15 циклов, где цикл составляет 28 дней, в течение, по меньшей мере, 18 циклов, где цикл составляет 28 дней или в течение, по меньшей мере, 24 циклов, где цикл составляет 28 дней. В некоторых вариантах осуществления соединение вводят субъекту ежедневно в течение, по меньшей мере, одного месяца, в течение, по меньшей мере, двух месяцев, в течение, по меньшей мере, трех месяцев, в течение, по меньшей мере, четырех месяцев, в течение, по меньшей мере, пяти месяцев, в течение, по меньшей мере, шести месяцев, в течение, по меньшей мере, восьми месяцев или в течение, по меньшей мере, одного года. В некоторых вариантах осуществления соединение вводят один раз в день.
В некоторых вариантах осуществления при введении соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, субъекту, такому как субъект-человек, Сmax соединения достигается в пределах от приблизительно 1 до приблизительно 5 часов, от приблизительно 1,5 до приблизительно 4,5 часов, от приблизительно 2 до приблизительно 4 часов или от приблизительно 2,5 до приблизительно 3,5 часов после введения дозы. В некоторых вариантах осуществления при введении соединения субъекту-человеку, период полувыведения соединения составляет от приблизительно 12 до приблизительно 40 часов, от приблизительно 16 до приблизительно 34 часа или от приблизительно 20 до приблизительно 30 часов. В некоторых вариантах осуществления среднее значение AUC (площади под кривой), полученной для соединения, увеличивается более чем пропорционально с повышением доз, находящихся в диапазоне от приблизительно 30 мг до приблизительно 800 мг в день. В некоторых вариантах осуществления накопление соединения находится в диапазоне от приблизительно 1,1-кратного до приблизительно 5-кратного, от приблизительно 1,25-кратного до приблизительно 4,0-кратного, от приблизительно 1,5-кратного до приблизительно 3,5-кратного, от приблизительно 2-кратного до приблизительно 3-кратного в стационарном состоянии в том случае, когда дозу соединения вводят один раз в день.
В некоторых вариантах осуществления способ включает выдачу предписания субъекту принимать внутрь эффективное количество соединения на пустой желудок. В некоторых вариантах осуществления способы дополнительно включают выдачу предписания субъекту избегать приема внутрь средств, которые являются, по меньшей мере, умеренно действующими индукторами или ингибиторами фермента CYP3A4 (цитохром Р450 3А4). В некоторых вариантах осуществления субъект не получает сопутствующую терапию с применением лекарственных средств – растительных средств, известных как, по меньшей мере, умеренно действующие ингибиторы или индукторы фермента CYP3A4. С учетом оценок in vitro, N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид метаболизируется ферментом CYP3A4 человека. Средства, которые могут увеличивать концентрации N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида в плазме (то есть ингибиторы CYP3А4) или уменьшать концентрации N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида в плазме (то есть индукторы CYP3А4), включая растительные средства и пищевые продукты (например, грейпфрут/грейпфрутовый сок), следует избегать субъектам, которые проходят лечение таким образом, как описано в данном документе. В дополнение к тому, данные, полученные in vitro, показали, что N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид ингибирует фермент CYP3A4 время-зависимым образом. Средства, которые являются чувствительными субстратами в отношении метаболизма с участием фермента CYP3A4, следует использовать с осторожностью, поскольку совместное введение с N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамидом может приводить к более высоким концентрациям совместно вводимого средства в плазме. Перечень клинически релевантных субстратов фермента CYP3A4 включает алфентанил, Циклоспорин, Диерготамин, этинил-эстрадиол, эрготамин, фентанил, пимозид, квинидин, сиролимус, такролимус, кларитромицин, эритромицин, телитромицин, алпразолам, диазепам, мидазолам, триазолам, индинавир, ритонавир, саквинавир, прокинетик, цизаприд, астемизол, хлорфенирамин, амлодипин, дилтиазем, фелодипин, нифедипин, верапамил, аторвастатин, церивастатин, ловастатин, симвастатин, арипипразол, гливек, галоперидол, силденафил, тамоксифен, таксанес, тразодон и Винкристин. Перечень клинически релевантных индукторов фермента CYP3A4 включает карбамазепин, фенобарбитал, фенитоин, пиоглитазон, рифабутин, рифампин, зверобой обыкновенный (продырявленный) и троглитазон. Перечень клинически релевантных ингибиторов фермента CYP3А4 включает индинавир, нелфинавир, ритонавир, кларитромицин, итраконазол, кетоконазол, нефазодон, эритромицин, грейпфрутовый сок, верапамил, дилтиазем, циметидин, амиодарон, флувоксамин, мибефрадил и тролеандомицин. См., ссылку на публикацию Flockhart et al., http://medicine.iupui.edu/clinpharm/ddis/clinicaltable.aspx., 2009.
Также предоставляют в этой заявке способы мониторинга (контроля за проведением) лечения миелофиброза у субъекта, включающие в себя (а) введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат; (b) мониторинг (контроль) гематологического параметра и/или негематологического параметра у субъекта; и (с) определение того, следует ли субъекту продолжать лечение или следует прервать его. В некоторых вариантах осуществления гематологический параметр выбирают из группы, состоящей из анемии, тромбоцитопении и нейтропении. В некоторых вариантах осуществления негематологический параметр представляет собой фермент или молекулу в крови или в сыворотке, где повышенный уровень фермента или молекулы служит признаком повреждения ткани или органа. В некоторых вариантах осуществления сывороточный(ая) фермент или молекула может представлять собой, например, амилазу, липазу, аспартат-аминотрансферазу (AST), аланин-аминотрансферазу (ALT), креатинин, щелочную фосфатазу и кальций. Способы мониторинга этих параметров известны в данной области и описаны в данном документе. См., Примеры 1–3. В некоторых вариантах осуществления способ дополнительно включает в себя введение субъекту эффективного количества соединения, описанного в данном документе, после того, как субъекту прервали лечение, по меньшей мере, на 2 недели, по меньшей мере, на 3 недели или, по меньшей мере, на 4 недели. В некоторых вариантах осуществления предыдущее лечение было прервано без предварительного снижения дозы.
Также в данной заявке предоставлены способы мониторинга лечения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и прерывание лечения при показании повышенных уровней одного или более ферментов или молекул, выбранных из группы, состоящей из амилазы, липазы, аспартат-аминотрансферазы (AST), аланин-аминотрансферазы (ALT) и креатинина, и/или сниженного уровня кальция в крови или в сыворотке субъекта без предварительного снижения дозы. Также предоставлены в данном документе способы мониторинга лечения миелофиброза у субъекта, включающие в себя введение субъекту эффективного количества соединения, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и прерывание лечения при показании одного или более гематологических состояний, выбранных из группы, состоящей из анемии, тромбоцитопении и нейтропении без предварительного снижения дозы. В некоторых вариантах осуществления лечение прерывают в том случае, когда один или более параметров (в том числе гематологические и негематологческие параметры) составляют осложнения 3 или 4 степени.
Нежелательные явления 3 или 4 степени в отношении гематологических и негематологических параметров известны в данной области и показаны в Таблице ниже. См., например, публикацию: Common Terminology Criteria for Adverse Events (CTCAE), Вариант 4.0, Опубликовано: 28 мая 2009 г. (в4.03: 14 июня 2010 г.).
|
IV. Изделия производства и наборы
Также предоставлены в данном документе изделия производства или наборы, содержащие соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления изделие производства или набор дополнительно включает инструкции по применению соединений, описанных в данном документе, способами, предоставляемыми в данном документе. В некоторых вариантах осуществления изделие производства или набор дополнительно включает этикетку или листок вкладыш в упаковке, обеспечивающую(ий) инструкции. В некоторых вариантах осуществления соединение находится в капсуле и/или в стандартной лекарственной форме, описанной в данном документе.
В некоторых вариантах осуществления изделие производства или набор может дополнительно включать контейнер. Подходящие контейнеры включают, например, бутылки, флаконы (например, двухкамерные флаконы), шприцы (такие как шприц или двухкамерные шприцы) и тест-тубы. Контейнер может быть изготовлен из разнообразных материалов, таких как стекло или пластик, и контейнер может вмещать соединение, например, в составе, который должен быть введен. Изделие производства или набор может дополнительно включать этикетку или листок вкладыш, которая(ый) находится на контейнере или прилагается к контейнеру, где могут быть приведены указания по перерастворению и/или по применению соединения. В некоторых вариантах осуществления листок-вкладыш или этикетка находится в таком месте, которое является видимым для потенциального покупателя.
Этикетка или листок-вкладыш может дополнительно указывать, что соединение является полезным или предназначено для лечения или предупреждения миелофиброза у субъекта. В некоторых вариантах осуществления листок вкладыш или этикетка указывает, что соединение может быть использовано для уменьшения интенсивности насыщенности клетками ткани костного мозга и/или фиброза ткани костного мозга. В некоторых вариантах осуществления листок-вкладыш или этикетка указывает, что соединение может быть использовано для лечения миелофиброза у субъекта, где субъект имеет отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в гене JAK2-Киназы (JAK2V617F) человека или отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 гена JAK2-киназы человека. В некоторых вариантах осуществления листок вкладыш или этикетка указывает, что соединение может быть использовано для лечения миелофиброза у субъекта и что субъект должен прервать лечение при показании повышенных уровней одного или более веществ, выбранных из амилазы, липазы, аспартат-аминотрансферазы (AST), аланин-аминотрансферазы (ALT), креатинина и/или щелочной фосфатазы, и/или сниженного уровня кальция в сыворотке субъекта, и/или при показании одного или более состояний выбранных из анемии, тромбоцитопении и/или нейтропении. В некоторых вариантах осуществления листок вкладыш или этикетка дополнительно указывает, что введение соединения может быть прекращено без предварительного снижения дозы.
В некоторых вариантах осуществления предоставляют набор или изделие производства, содержащий(ее) (а) смесь, включающую (i) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, (ii) эксципиент (например, микрокристаллическую целлюлозу, такую как силикатированную микрокристаллическую целлюлозу) и (iii) смазывающее вещество (например, стеарилфумарат натрия), и (b) листок вкладыш или этикетку, указывающий(ую), что добавка является полезной для лечения миелофиброза у субъекта. В некоторых вариантах осуществления предоставляют набор или изделие производства, содержащий(ее) (а) соединение, которое представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, и (b) листок-вкладыш или этикетку, указывающую, что соединение может быть использовано для лечения миелофиброза у субъекта, где субъект ранее получил другую терапию миелофиброза с применением ингибитора JAK2-киназы, который не представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат.
В некоторых вариантах осуществления листок вкладыш или этикетка указывает, что при введении соединения субъекту-человеку, Сmax соединения достигается в пределах от приблизительно 1 до приблизительно 5 часов, приблизительно 1,5–4,5 часов, приблизительно 2–4 часов или приблизительно 2,5–3,5 часов после введения дозы. В некоторых вариантах осуществления листок-вкладыш или этикетка указывает, что при введении соединения субъекту-человеку, период полувыведения соединения составляет от приблизительно 12 до приблизительно 40 часов, от приблизительно 16 до приблизительно 34 часа или от приблизительно 20 до приблизительно 30 часов. В некоторых вариантах осуществления среднее значение AUC (площади под кривой), полученной для соединения, увеличивается более чем пропорционально с повышением доз, находящихся в диапазоне от приблизительно 30 мг до приблизительно 800 мг в день. В некоторых вариантах осуществления накопление соединения находится в диапазоне от приблизительно 1,1-кратного до приблизительно 5-кратного, от приблизительно 1,25-кратного до приблизительно 4,0-кратного, от приблизительно 1,5-кратного до приблизительно 3,5-кратного, от приблизительно 2-кратного до приблизительно 3-кратного в стационарном состоянии в том случае, когда дозу соединения вводят один раз в день.
В некоторых вариантах осуществления листок вкладыш или этикетка предоставляет предписания субъекту принимать внутрь эффективное количество соединения на пустой желудок. В некоторых вариантах осуществления листок вкладыш или этикетка дает предписания субъекту избегать приема внутрь средств, которые являются, по меньшей мере, умеренно действующими индукторами или ингибиторами фермента CYP3A4. В некоторых вариантах осуществления индуктор или ингибитор фермента CYP3A4 представляет собой любой из индукторов или ингибиторов фермента CYP3A4, описанных в данном документе.
Также обеспечены применения соединения в изготовлении лекарственного препарата для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления применение проводят в соответствии со способом, описанным в данном документе. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, (ii) эксципиент (например, микрокристаллическую целлюлозу, такую как силикатированную микрокристаллическую целлюлозу) и (iii) смазывающее вещество (например, стеарилфумарат натрия). В некоторых вариантах осуществления соединение вводят перорально. В некоторых вариантах осуществления применение осуществляют в соответствии со способом, описанным в данном документе. В некоторых вариантах осуществления предоставляют применение соединения в изготовлении лекарственного препарата для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект имеет отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в гене Янус-Киназы 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 гена JAK2-киназы человека. В некоторых вариантах осуществления обеспечивают применение соединения в изготовлении лекарственного препарата для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект ранее получил другую терапию миелофиброза. В некоторых вариантах осуществления предыдущая терапия включает в себя применение ингибитора JAK2-киназы, который не представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат.
Также предоставлено соединение для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат. В некоторых вариантах осуществления лечение проводят в соответствии со способом, описанным в данном документе. В некоторых вариантах осуществления соединение находится в смеси, включающей в себя (i) соединение, (ii) эксципиент (например, микрокристаллическую целлюлозу, такую как силикатированную микрокристаллическую целлюлозу) и (iii) смазывающее вещество (например, стеарилфумарат натрия). В некоторых вариантах осуществления соединение вводят перорально. В некоторых вариантах осуществления лечение проводят в соответствии со способом, описанным в данном документе. В некоторых вариантах осуществления предоставляют соединение для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект имеет отрицательный результат в отношении мутации с заменой валина на фенилаланин в позиции 617 в гене Янус-Киназы 2 (JAK2) человека или дает отрицательный результат в отношении мутации, соответствующей мутации с заменой валина на фенилаланин в позиции 617 гена JAK2-киназы человека. В некоторых вариантах осуществления обеспечивают соединение для лечения миелофиброза у субъекта, где соединение представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат, где субъект ранее получил другую терапию миелофиброза. В некоторых вариантах осуществления предыдущая терапия включает в себя применение ингибитора JAK2-киназы, который не представляет собой N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид, или его фармацевтически приемлемую соль, или его гидрат.
Следующее представляет собой примеры способов и композиций, предоставляемых в данном документе. Ясно, что на практике могут быть применены различные другие варианты осуществления, с учетом общего описания, предоставленного выше.
ПРИМЕРЫ
Пример 1. Оценивание действия TG101348 при миелофиброзе
Как используют в данном документе, «TG101348» относится к моногидрату дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида. Субъектам в этом исследовании вводят капсульную форму TG101348, которая описана в Примере 5. TG101348 оценивают в исследовании I фазы в отношении лечения миелофиброза. Это исследование происходит в тот момент времени, когда данные собраны.
Предпосылки: TG101348 является сильно действующим, перорально биодоступным, JAK2-селективным ингибитором с небольшой молекулой, который оценивают в исследовании I фазы в отношении лечения миелофиброза. Ограничивающая дозу токсичность проявляется как асимптоматическая амилаземия/липаземия 3 или 4 степени, которая является обратимой, и максимальная переносимая доза («MTD») составляет 680 мг. Наиболее частые негематологические токсичности составляют слабо выраженную тошноту, рвоту и/или диарею, что легко контролируется или разрешается спонтанно. Нейтропению и тромбоцитопению 3/4 степени наблюдают у 14% и 25% пациентов, соответственно. TG101348 имеет активность в уменьшении размера селезенки, количества лейкоцитов и аллельной нагрузки мутации JAK2V617F («VF»). Этот пример описывает результаты с акцентом внимания на данных, полученных для когорты в фазе подтверждения выбранной дозы, лечение пациентов которой начали с дозы 680 мг/день.
Результаты: Пятьдесят девять пациентов (медианный возраст = 66 лет; диапазон 43-86) получают лечение – 28 в фазе повышения дозы и 31 в фазе подтверждения дозы. В целом, 44 пациента имеют PMF (первичный миелофиброз), 12 - миелофиброз, развившийся на фоне предшествующей истинной полицитемии (post-PV MF), и 3 – миелофиброз, развившийся на фоне эссенциальной тромбоцитемии (post-ET MF); 86% имеют положительный результат в отношении мутации с заменой валина на фенилаланин (VF). Средний пальпируемый размер селезенки равен 18 см, и для 22 пациентов требуется переливание красных кровяных клеток («RBC»=эритроциты) в фазе включения пациентов в исследование. После медианного контрольного наблюдения, проходившего через 12 недель (диапазон <1-76), 18 (31%) пациентов прекращают лечение по причине токсичности (n=7; тромбоцитопения = 3, нейтропения = 1), сочетанных заболеваний (n=5), отзыва информированного согласия (n=4) или нарушения/отсутствия ответной реакции (по 1). В том случае, когда собраны данные результатов в этом примере, оставшийся 41 пациент получает следующие уровни доз: 680 мг (n=14), 520–600 мг (n=16), 360-440 мг (n=10) и 240 мг (n=1). Кумулятивное время воздействия лекарственного средства к тому моменту времени, когда собраны данные в этом примере, составляет 362 пациент-месяца; время воздействия максимальной переносимой дозы (MTD) или дозы, превышающей MTD (≥680 мг) составляет 154 пациент-месяца. Сорок пациентов (68%) начинают лечение с дозы ≥680 мг.
Токсичность: TG101348 хорошо переносится. Из числа пациентов, которые начинают лечение с дозы ≥680 мг, наблюдают нейтропению ¾ степени в 15/0% случаев и тромбоцитопению ¾степени в 20/10% случаев. Двадцати четырем (60%) пациентам не требуются переливания RBC в исходный момент (медианное значение гемоглобина («Hgb»)=9,6 г/дл); диапазон 7,4–13,1; у 42% и 8% пациентов развивается анемия 3 Степени («Ст3») и 4 Степени («Ст4»), соответственно. Большинство пациентов, которые начинают лечение с дозы ≥680 мг, развивают слабо выраженную тошноту (1 Ст3), рвоту (1 Ст3) и/или диарею (3 Ст3), которые являются самокупируемыми или легко контролируются. Другие негематологические токсичности включают повышение активности трансаминаз 1/2 Степени («Ст1/2») (38%), повышение уровня сывороточного креатинина Ст1/2 (38%) и асимптоматическую гиперлипаземию (33%).
Эффективность: Тридцать три пациента, которые начинают лечение с дозы ≥680 мг, проходят, по меньшей мере, 3 цикла лечения; через 3 месяца, снижение пальпируемого размера селезенки (исходное медианное значение = 18 см; диапазон 6-32) составляет, по меньшей мере, 50% у 22 (67%) пациентов; селезенка становится непальпируемой у 9 (27%) пациентов. Все пациенты в количестве 21 с лейкоцитозом на исходном уровне (диапазон WBC (содержание лейкоцитов в крови) 11–203×109/л), которые начинают лечение с дозы ≥680 мг, показывают заметное снижение своего уровня WBC (диапазон 4-90); 70% имеют нормальный уровень WBC на стадии своего последнего визита контрольного наблюдения. В общем, 48 из 51 пациента с положительным результатом в отношении мутации с заменой валина на фенилаланин (VF) проходят, по меньшей мере, 1 цикл и подлежат оцениванию в отношении ответной реакции аллельной нагрузки мутации с заменой VF; в фазе последнего предусмотренного контрольного наблюдения, медианное снижение нагрузки мутантного аллеля на гранулоциты составляет 48%; 21 (44%) пациент имеет ≥50%-ное снижение, и в группе, которая начала лечение с дозы ≥680 мг, 48% имеют ≥50%-ное снижение. Для пациентов, подлежащих оценке, имеет место клинически значимая польза или регрессия конституциональных симптомов, включая чувство быстрого насыщения, утомляемость, кашель, зуд и ночные приливы.
Заключения: TG101348 хорошо переносится пациентами с миелофиброзом. Ответные реакции размера селезенки и уровня лейкоцитов проявляются часто/постоянно, наблюдаются быстро и дают существенную клиническую пользу для пациентов. Такие ответные реакции связаны со значительным снижением аллельной нагрузки мутации с заменой VF и указывают на активность TG101348 в отношении клона злокачественных опухолевых клеток при миелофиброзе.
Пример 2. Оценивание действия TG101348 при миелофиброзе
Субъектам в этом исследовании вводят капсульную форму TG101348. TG101348 оценивают в исследовании I фазы в отношении лечения миелофиброза. Это исследование также описано в Примере 1. В этом примере описывают данные, доступные на момент времени сбора данных.
Это исследование представляет собой открытое, многоцентровое исследование с увеличением дозы, которое проводят с расширенной когортой в фазе подтверждения дозы при MTD. Основной целью этого исследования является определение безопасности/переносимости, DLT (Ограничивающей дозу токсичности), MTD (максимальной переносимой дозы) и фармакокинетики TG101348 у субъектов с MF. Второстепенной целью этого исследования является оценивание предварительных данных клинической и фармакодинамической активности.
Ключевые критерии отбора субъектов-участников исследования включают: Миелофиброз (PMF (первичный миелофиброз или post-PV/ET MF (миелофиброз, развившийся на фоне предшествующей истинной полицитемии или эссенциальной тромбоцитемии)); Высокий риск или промежуточный риск с симптоматической спленомегалией/ отсутствием отвечаемости на предоставляемую терапию; показатель общего состояния больного по шкале оценки ECOG ≤2; ANC (абсолютное число нейтрофилов) ≥1×109/л; Количество тромбоцитов ≥50×109/л; Сывороточный креатинин ≤2 мг/дл; Общий билирубин ≤2 мг/дл; AST/ALT (аспартат-аминотрансфераза/аланин-аминотрансфераза) ≤3X верхней границы нормы.
Распределение субъектов-участников для этого исследования включено в Таблицу 1.
|
В Таблицу 2 включены демографические показатели и исходные характеристики для субъектов.
|
Это исследование представляет собой исследование с увеличением дозы для подтверждения выбора дозы для расширенной когорты при MTD. Ниже описываются данные с акцентированием внимания на когорте в фазе подтверждения выбора дозы, которая начинает лечение с дозы 680 мг/день.
Снижение размера пальпируемой селезенки за цикл лечения для субъектов, которые получают лечение соединением TG101348 в дозе 680 мг/день (исходная доза) (N=37) показано на Фигуре 1. Исходный размер селезенки составляет: медианное значение = 18 см; диапазон = 6–32 см. 49% субъектов достигают клинического улучшения, что следует из результатов снижения пальпируемой спленомегалии (ответная реакция IWG) (56% субъектов через 12 недель; 100% субъектов через 20 недель). К моменту времени сбора данных отсутствует возврат (рецидив) или прогрессирование заболевания.
На Фигуре 2 показано действие TG101348 на лейкоцитоз. Исходный уровень WBC составляет >11×109/л. 73% субъектов имеют нормальные уровни WBC на момент свего визита контрольного наблюдения. На Фигуре 3 показано действие TG101348 на тромбоцитоз (исходный уровень тромбоцитов >450×109/л). TG101348 может снижать уровни тромбоцитов. На Фигуре 4 показаны воздействия TG101348 на конституциональные симптомы (исходный уровень относительно уровня в последнем визите). TG101348 может снижать конституциональные симптомы, сопутствующие MF. TG101348 не дает значительных изменений в отношении уровней цитокинов (см. Фигуру 5, все показанные значения являются медианами). Фигура 6 показывает действие TG101348 в отношении аллельной нагрузки мутации с заменой V617F у субъектов с исходным уровнем >20% (N=22). Фигура 6 показывает, что TG101348 может снижать аллельную нагрузку мутации JAK2V617F у 59% субъектов с исходным уровнем >20%.
Фигура 7 показывает воздействия TG101348 на состояние насыщенности клетками ткани костного мозга у 76-летнего субъекта мужского пола с PMF при отрицательном результате в отношении мутации с заменой V617F. Исходная доза составляет 30 мг/день, и доза на момент времени контрольного наблюдения пациента составляет 520 мг/день. Фигура 7 показывает, что TG101348 может снижать насыщенность клетками ткани костного мозга у этого субъекта от 60%-ной насыщенности клетками ткани костного мозга в исходный момент до 5–10%-ной насыщенности клетками ткани костного мозга после 18 циклов лечения. Фигура 8 показывает действие TG101348 в отношении фиброза ткани костного мозга у 56-летнего субъекта мужского пола с PMF при отрицательном результате в отношении мутации с заменой V617F. Исходная доза составляет 240 мг/день, и доза на момент времени контрольного наблюдения составляет 440 мг/день. Фигура 8 показывает, что TG101348 может снижать фиброз ткани костного мозга у субъекта от 3+ в исходный момент до 0 после 18 циклов лечения.
Появившиеся за время лечения гематологические токсичности 3&4 степени у Субъектов, принимающих MTD, (N=40) показаны в Таблице 3. Появившиеся за время лечения негематологические нежелательные явления (по сообщению, по меньшей мере, 5 субъектов) у Субъектов, принимающих MTD, показаны в Таблице 4.
|
|
Появившиеся за время лечения негематологические лабораторные показатели степени ≥2 у субъектов, принимающих MTD, (N=40) показаны в Таблице 5.
|
Лабораторные показатели имеют преходящие и обратимые изменения и имеют возврат к нормальным величинам самопроизвольно или после прекращения введения дозы и/или снижения дозы.
Фигура 9 показывает различные измерения у субъекта с PMF при положительном результате в отношении мутации JAK2V617F, который начинает лечение соединением TG101348 в дозе 680 мг/день. TG101348 может снизить размер пальпируемой селезенки с 9 см до 0 см и приводит к полной регрессии зуда у этого субъекта.
Заключения: TG101348, как правило, хорошо переносится, при наличии управляемых желудочно-кишечных эффектов 1 степени, в особенности при более высоких дозах. Данные указывают на непродолжительно действующие токсичности. Оказывается, что ожидаемое целевое миелосуппрессивное действие в наибольшей степени ограничено эритропоэзом, который может быть ослаблен при более низких, но по-прежнему эффективных, дозах. TG101348 имеет заметную активность в MF-сопутствующей спленомегалии: ~две трети достигают ≥50%-ного снижения пальпируемой спленомегалии; ~30% имеют полную регрессию. TG101348 имеет значительную анти-миелопролиферативную активность, где фактически все прошедшие лечение субъекты демонстрируют полную регрессию лейкоцитоза и тромбоцитоза. TG101348 имеет заметную активность в отношении MF-сопутствующих конституциоальных симптомов, зуда и кахексии. TG101348 вызывает значительное снижение аллельной нагрузки мутации JAK2V617F у изрядной доли прошедших лечение субъектов. TG101348 имеет минимальное действие в отношении сывороточных уровней провоспалительных цитокинов; это согласуется с отсутствием мгновенного нежелательного феномена цитокиновой отдачи при прекращении приема исследуемого лекарственного средства. Не желая быть связанными какой бы то ни было теорией, полагают, что активность TG101348, по-видимому, является прямым следствием его ингибиторной активности в отношении JAK2-киназы, а не непрямого эффекта от неспецифической антицитокиновой активности. Более того, предварительные наблюдения показывают снижение насыщенности клетками ткани костного мозга (BM) и ретикулярного фиброза при длительном лечении.
Пример 3. Оценивание действия TG101348 при миелофиброзе
Субъектам в этом исследовании вводят капсульную форму TG101348.
Дизайн клинического исследования: Исследование включает Фазу 1, испытание с увеличением дозы (MF-TG101348-001). Это исследование также описано в Примерах 1 и 2. Исследуемые подходящие для участия в исследовании пациенты имеют возраст ≥18 лет и входят в группу высокого или промежуточного риска возникновения первичного миелофиброза (PMF), миелофиброза, развившегося на фоне предшествующей истинной полицитемии (post-PV MF) или эссенциальной тромбоцитемии (post-ET MF) (Tefferi A et al., Leukemia 22:14-22, 2008). Дополнительные критерии отбора и центры, принимающие участие в исследовании, перечислены в Таблице 6. Все пациенты предоставляют информированное согласие в письменном виде. Первичными конечными точками исследования являются определение безопасности и переносимости, определение ограничивающей дозу токсичности («DLT»), максимальной переносимой дозы («MTD») и фармакокинетические («PK») свойства TG101348. Вторичной конечной точкой исследования является оценка терапевтической активности.
|
Пациентов приписывают одной из 8 дозовых когорт, отличающихся дозами, которые находятся в диапазоне от 30 до 800 мг в день, с использованием когортного дизайна 3+3. TG101348 вводят перорально один раз в день, где план лечения предусматривает непрерывную ежедневную терапию в течение 24 недель (6×28-дневные циклы). Увеличение дозы внутри субъекта разрешается после завершения, по меньшей мере, 3 циклов лечения лекарственным средством в исходной дозе. Как только определена DLT (ограничивающая дозу токсичность), когорта в фазе подтверждения выбора дозы начинает лечение лекарственным средством в MTD (в максимальной переносимой дозе). Лечение более 6 циклов разрешается в расширенном клиническом исследовании (MF-TG101348-002; NCT00724334), если оно признано благотворным для пациента и, если хорошо переносится.
Оценивание токсичности и ответной реакции: Оценивания безопасности проводятся еженедельно во время 1 цикла, через неделю во время 2 и циклов и после этого один раз в четыре недели. Токсичность распределяют по степеням токсичности в соответствии с общей терминологией критериев нежелательных явлений Национального института рака США (NCI-CTCAE) (версия 3.0).
Международная Рабочая Группа измеряет каждые 4 недели ответные реакции при использовании Исследования MNP (наиболее вероятного количества) и Критериев Лечения (IWG-MRT) (Tefferi A et al., Blood 108:1497-1503, 2006). Оценивание гистологии костного мозга проводят в исходный момент и каждые 24 недели терапии. Изменения аллельной нагрузки мутации JAK2V617F во фракции гранулоцитов периферической крови измеряют так же, как описано ранее (Kittur J. et al., Cancer 109:2279-2284, 2007); оценивания производят в исходный момент и каждые 4 недели во время первых 6 циклов и в каждом 6-м цикле терапии в расширенном клиническом исследовании.
Фармакокинетические характеристики: Кривые концентрация-время для TG101348 в плазме крови оценивают с помощью анализа без учета компартментов (с использованием программного обеспечения WinNonlin®, версия 5.2).
Оценивание уровней цитокинов: Образцы для измерения содержания цитокинов собирают в исходный момент исследования и после этого каждые 4 недели. Уровни цитокинов измеряют с использованием мультиплексного анализа с применением «сэндвичевого» метода ELISAs (Millipore, St. Charles, MO).
Результаты
Включение пациентов в исследование: Всего включено в исследование 59 субъектов; 28 – в фазе увеличения дозы и 31 – в фазе подтверждения выбора дозы (Таблица 7). Сорок четыре субъекта имеют PMF (первичный миелофиброз), 12 - post-PV MF (миелофиброз, развившийся на фоне предшествующей истинной полицитемии) и 3 post-ET MF (миелофиброз, развившийся на фоне предшествующей эссенциальной тромбоцитемии); 86% имеют положительный результат в отношении мутации JAK2V617F. Медианная длительность заболевания составляет 3,4 года (диапазон 0,06–25,8). В фазе включения в исследование, медианный пальпируемый размер селезенки равен 18 см, где селезенка расположена ниже левого реберного края (83% имеют пальпируемый размер селезенки >10 см), медианный уровень гемоглобина равен 9,2 г/дл (диапазон 6,6–15,2) и 21 (36%) субъект имеет зависимость от трансфузии эритроцитов по критериям IWG-MRT.
|
В фазе увеличения дозы, исходная доза TG101348 составляет 30 мг/день, и последующие уровни дозы составляют 60, 120, 240, 360, 520, 680 и 800 мг/день (Таблица 7). При дозе 800 мг/день, 2 из 6 пациентов испытывают ограничивающую дозу токсичность; как следствие, MTD (максимальная переносимая доза) заявлена при 680 мг/день. В фазе подтверждения выбора дозы, все пациенты начинают лечение при MTD. «Когорта c MTD» (n=40; Таблица 7) включает пациентов, которые получают 680 мг/день в качестве своей исходной дозы (когорта в фазе увеличения дозы, n=3; когорта в фазе подтверждения выбора дозы, n=31), и пациентов, чья доза лекарственного средства снижена от 800 мг/день (n=6) до 680 мг/день после объявления MTD.
Медианное значение (диапазон) воздействия TG101348 для всех когорт (n=59) и для когорт с MTD (n=40) составляет 155 (2-172) и 147 (8-171) дней, соответственно. Дозы TG101348 в конце каждого цикла для дозовой когорты проиллюстрированы на Фигурах 10 и 11. В когорте с MTD, для 28 субъектов (70%) требуется снижение дозы в течение первых 6 циклов; основными причинами являются: цитопения(и) (20%), желудочно-кишечные нежелательные явления (12,5%), повышение уровня амилазы/липазы (10%), повышение уровня ALT (аланин-аминотрансферазы) (7,5%), усмотрение исследователя (7,5%) или другие нежелательные явления (12,5%). Медианным циклом в фазе снижения дозы для когорты с MTD является 3 цикл (диапазон 1-7); медианное значение (диапазон) дозы в конце 3 цикла составляет 680 мг/день (360–680 мг/день); и 520 мг/день (360–680 мг/день) в конце 6 цикла.
Сорок три (73%) субъекта, включая 28 (70%) из когорты с MTD, продолжают лечение в расширенном исследовании; в фазе вхождения в расширенное исследование, 31 (72%) субъект получает <680 мг/день лекарственного средства (медианное значение 520 мг/день; диапазон 120–680 мг/день). На момент окончания сбора данных, медианное значение (диапазон) кумулятивного воздействия TG101348 для 43 субъектов составляет 380 дней (170-767). Число завершенных циклов лечения находится в диапазоне от 7–29; 39 субъектов (66%), включая 27 (68%) из когорты с MTD, завершают 12 циклов лечения. На момент окончания сбора данных, 28% и 14% субъектов, которые вошли в расширенное исследование, завершили 18 и 24 цикла лечения, соответственно. Медианное значение (диапазон) дозы лечения во время расширенной фазы составляет 440 мг/день (120–680 мг/день).
Фармакокинетические свойства: Максимальная (пиковая) концентрация TG101348 в плазме достигается через 1–4 часа после введения дозы. TG101348 показывает более, чем пропорциональные дозы, увеличения фармакокинетических параметров плазмы (Сmax и AUC0-t) (Таблица 8 и Фигура 12). Средние стационарные значения Cmax и AUC0-t увеличиваются приблизительно 54-кратно и 88-кратно, соответственно, по сравнению с 27-кратным увеличением дозы. Период полувыведения в терминальной фазе в стационарном состоянии остается схожим для всех доз (16–34 часа), согласующимся с линейным выведением лекарственного средства. Фигура 18 показывает график зависимости средних концентраций TG101348 в плазме относительно времени на линейном графике, полученном после перорального введения доз один раз в день (цикл 1; День 28). Фигура показывает значения IC50, IC90 и 3-кратные значения IC90 (3×IC90) для TG101348 в отношении концентрации TG101348 в плазме в зависимости от времени. Доза 520 мг/день позволяет получить концентрацию TG101348 в плазме, которая превышает значения 3×IC90 в течение, по меньшей мере, 24 часов после введения дозы. Доза 360 мг/день позволяет получить Сmax, превышающую значение 3×IC90, и концентрацию TG101348 в плазме, которая превышает IC90 в течение, по меньшей мере, 24 часов после введения дозы.
|
Профиль безопасности: Ограничивающая дозу токсичность (DLT) у 2 из 6 пациентов, получивших лечение в дозе 800 мг/день, представляет собой асимптоматическую гиперамилаземию 3 или 4 степени (наряду с гиперлипаземией или без таковой), которая является обратимой. Наиболее распространенные негематологические нежелательные явления, по меньшей мере, возможно связанные с TG101348, преимущественно включают тошноту 1 степени, диарею и рвоту; осложнения 3 степени подтверждаются в общей когорте/когорте с MTD для 3%/5%, 10%/13% и 3%/3% субъектов, соответственно, и отсутствуют осложнения 4 степени (Таблица 9). Эти нежелательные явления являются дозо-зависимыми, где появление нежелательного явления 3 степени наблюдается почти исключительно при исходной дозе TG101348 ≥680 мг/день. Желудочно-кишечные симптомы в основном являются самокупируемыми или контролируются симптоматическим лечением и/или с помощью снижения дозы. Другие нежелательные явления (Степени 3/4; общая когорта/когорта с MTD) включает асимптоматические увеличения уровней сывороточной липазы (10%/15%), AST (аспартат-аминотрансферазы) (2%/3%), ALT (аланин-аминотрансферазы) (7%/8%), креатинина (0%/0%) и щелочной фосфатазы (0%/0%) (Таблица 9).
|
Гематологические нежелательные явления 3/4 степени, рассматриваемые касательно TG101348, включают анемию (35% из 37 субъектов, которые не являются трасфузионно-зависимыми на исходном уровне), тромбоцитопению (24%) и/или нейтропению (10%) (Таблица 10). Большая часть появившихся во время лечения цитопений отмечена в первых трех циклах лечения. Из 13 субъектов, которые заболели анемией 3/4 степени (все в когорте с MTD), 67% участвуют в исследовании с анемией 2 степени. Возникновение необходимости трансфузии значительно ниже для субъектов, которые начинали лечение в дозе 240–520 мг/день (33%) в противоположность субъектам, начавшим лечение в дозе 680 мг/день (72%). Из 14 субъектов с тромбоцитопенией 3/4 степени, 4 и 5 субъектов участвуют в исследовании с тромбоцитопенией 1 и 2 степени, соответственно.
|
На момент окончания сбора данных, никакие специфические с точки зрения безопасности данные не проявляются в случае непрерывного введения дозы TG101348 более 6 циклов терапии.
Серьезные нежелательные явления, рассматриваемые, по меньшей мере, как возможно связанные с TG101348, возникают у 8 субъектов и включают асимптоматическую гиперлипаземию, тромбоцитопению/нейтропению, депрессию, синдром лизиса опухоли, инсульт (острое нарушение мозгового кровообращения) и обезвоживание (Таблица 11). Один субъект прекращает лечение ввиду тромбоцитопении 4 степени; все другие явления являются обратимыми, и субъекты могут продолжать/возобновлять лечение при более низкой дозе после регрессии нежелательного явления.
|
Один субъект имел тяжелую легочную гипертензию и сердечную недостаточность в области правых отделов сердца во время 4 цикла (при 240 мг/день); явление рассматривалось исследователем как несвязанное с TG101348.
Пятнадцать (25%) субъектов прекращают лечение во время первых 6 циклов терапии (Таблица 12). Причины прекращения приема лекарственного средства включают связанные с проводимым лечением нежелательные явления (n=6); принятие исследователем решения/ интеркуррентную болезнь (n=3) или отзыв информационного согласия (n=6). Восемь из 43 субъектов (19%) прерывают лечение во время расширенного исследования, в том числе 3 субъекта – из-за нежелательных явлений, возникающих после проведения терапии в течение в целом 24–46 недель (Таблица 12).
|
|
|
Три субъекта имеют прогрессирование заболевания (указаны дозы в фазе начала и прекращения исследования): каждый из них имеет прогрессирующую гепатоспленомегалию и асцит на фоне эндокардита (2 цикл; 680 и 520 мг/день), острый миелофиброз (13 цикл; 520 и 200 мг/день) и лейкемическая трансформация (2 цикл; 520 и 520 мг/день).
Ответные реакции показаны ниже.
Спленомегалия: Начало ответной реакции селезенки возникает быстро и, как правило, наблюдается в пределах первых двух циклов. К 6 циклу, 36 субъектов (61%) показывают минимум 25%-ное уменьшение пальпируемого размера селезенки, в том числе 65% в когорте с MTD (анализ выборки «намеренных лечиться»). К этому моменту времени, ≥50%-ное уменьшение пальпируемого размера селезенки, сохраняющегося в течение, по меньшей мере, 8 недель, (то есть Клиническое Улучшение («CI») согласно критериям IWG-MRT) наблюдается у 39% и 45% субъектов общей когорты и в когорте с MTD, соответственно. Ответные реакции селезенки на цикл лечения для когорты с MТD показаны на Фигуре 13. Трое из 4 субъектов (75%) с MF при наличии отрицательного результата в отношении мутации JAK2V617F, которые завершили 6 циклов лечения, достигают клинического улучшения (CI). Самая низкая исходная доза, при которой наблюдается CI, составляет 240 мг/день. Медианное время (диапазон) достижения CI по всем дозам составляет 141 день (41-171) и 113 дней (41–170) для когорты с MTD. К 12 циклу, ответные реакции селезенки (CI) наблюдаются у 48% и 50% субъектов, для общей когорты и для когорты с MTD, соответственно. Среднее значение (± стандартное отклонение (SD)) продолжительности ответной реакции размера селезенки согласно критериям IWG-MRT составляет 315 (±129) дней и 288 (±76) дней для общей когорты и когорты с MTD, соответственно.
Конституциональные симптомы: Тридцать пять субъектов в когорте с MTD подтверждают наличие и тяжесть чувства быстрого насыщения, утомляемости, ночных приливов, кашля и зуда по 11-балльной шкале (0 = отсутствие симптомов – 10 = наихудшие из возможных симптомы) на исходном уровне и в конце, по меньшей мере, одного цикла. Симптомы подразделяют по таким группам, как «отсутствие симптома» (оценка в баллах = 0), «слабо выраженный симптом» (оценка в баллах = 1-3), «умеренно выраженный симптом» (оценка в баллах = 4-7) или «сильно выраженный симптом» (оценка в баллах = 8-10).
Чувство быстрого насыщения подтверждают 29 (85%) субъектов на исходном уровне. После 2 циклов лечения (n=27), 56% сообщают о полной регрессии этого симптома (Фигура 14А). Утомляемость подтверждают на исходном уровне 26 (76%) субъектов. После 6 циклов (n=16), 63% сообщают об улучшении состояния, и 25% сообщают о полной регрессии этого симптома (Фигура 14В). Ночные приливы подтверждают на исходном уровне 14 (40%) субъектов. После 1 цикла, 64% субъектов имеют полную регрессию этого симптома; после 6 циклов, эта доля увеличивается до 89% (n=9) (Фигура 14С). Кашель подтверждают на исходном уровне 13 (37%) субъектов. После 1 цикла (n=12), 75% подтверждают улучшение состояния, и 67% подтверждают полную регрессию этого симптома. Зуд подтверждают 8 (23%) субъектов на исходном уровне. После 1 цикла лечения, 75% имеют улучшение состояния, где 50% сообщают о полной регрессии. Ответные реакции конституциональных симптомов являются длительными в большинстве случаев.
Масса тела: В конце 6 и 12 циклов, медианное значение массы тела является стабильным относительно исходного уровня для общей когорты и для когорты с MTD (Таблица 13).
|
Лейкоцитоз и Тромбоцитоз: Лейкоцитоз (уровень WBC>11×109/л) имеется на исходном уровне у 33 субъектов (56%), 28 из которых завершают 6 циклов лечения; из них, 18 находятся в когорте с MTD. После 6 циклов, 16 субъектов по всем дозам (57%) и 13 субъектов в когорте с MTD (72%) достигают нормального уровня WBC (лейкоциты = белые кровяные тельца) (Фигура 15); после 12 циклов, 14 субъектов из 25 (56%) по всем дозам и 10 субъектов из 17 (59%) в когорте с MTD имеют нормальные уровни WBC.
Тромбоцитоз (количество тромбоцитов >450×109/л) отмечают на исходном уровне для 10 (17%) субъектов по всем дозам и для 7 субъектов (19%) в когорте с MTD (n=37), все из которых завершили 6 циклов терапии. К этому моменту времени, 90% и 100% субъектов по всем дозам и в когорте с MTD, соответственно, достигают нормального уровня тромбоцитов; после 12 циклов, 7 из 8 субъектов (88%) по всем дозам и все 6 субъектов в когорте с MTD имеют нормальное количество тромбоцитов.
Аллельная нагрузка мутации JAK2V617F: Пятьдесят один субъект (86%) имеют положительный результат в отношении мутации JAK2V617F, с медианным значением (диапазоном) аллельной нагрузки 20% (3-100%); из них, 23 (45%) имеют «значительную» аллельную нагрузку (определенную на исходном уровне как >20%) с медианным значением (диапазоном) аллельной нагрузки 60% (23-100%). Для всех субъектов с положительным результатом в отношении мутации, имеет место значительное снижение аллельной нагрузки мутации JAK2V617F после 6 циклов (р=0,04) и после 12 циклов лечения (р=0,01) (Фигуры 16А и 16В). После 6 и 12 циклов лечения, медианное значение (диапазон) аллельной нагрузки составляет 17% (0-100%) и 19% (0-100%), соответственно. Подобно тому, для 23 субъектов с аллельной нагрузкой мутации JAK2V617F на исходном уровне >20%, имеет место значительное и даже более резко выраженное снижение аллельной нагрузки мутации JAK2V617F после 6 циклов (р=0,002) и 12 циклов лечения (р=0,002) (Фигуры 16С и 16D). После 6 и 12 циклов лечения, медианное значение (диапазон) аллельной нагрузки составляет 31% (4-100%) и 32% (7-100%), соответственно. После 6 циклов, 16 из 20 субъектов (80%) с аллельной нагрузкой на исходном уровне >20%, которые достигали этой временной точки, показывают медианное 61%-ное (диапазон 6-96%) снижение, и 9 субъектов (45%) имеют ≥50%-ное снижение аллельной нагрузки мутации JAK2V617F. В противоположность тому, 4 субъекта (20%) демонстрируют повышение (18%, 21%, 30% и 58%). Восемнадцать субъектов (78%) группы с аллельной нагрузкой >20% завершают 12 циклов лечения с медианным 50%-ным (диапазон 29-82%) снижением, и 7 (39%) субъектов имеют ≥50%-ное снижение мутации JAK2V617F. Три (17%) субъекта показывают повышение аллельной нагрузки (7%, 18% и 22%), и 2 других со 100%-ной аллельной нагрузкой на исходном уровне не демонстрируют никакого изменения.
Обсуждение: Значительная доля пациентов, получивших лечение в этом исследовании, испытывает быстрое, существенное и длительное регулирование симптоматической спленомегалии, лейкоцитоза, тромбоцитоза и конституциональных симптомов. В дополнение к тому, также имеется подтверждение значительного снижения бремени геномного заболевания, что указывает на потенциал в отношении активности, изменяющей течение заболевания. Ответные реакции имеются у пациентов с MF, которые давали отрицательный результат в отношении мутации JAK2V617F. Неизвестно, имеют ли субъекты, участвующие в этом исследовании, другие мутации в пути сигнальной трансдукции JAK-STAT (Янус-Киназа – Сигнальный трансдуктор и активатор пути транскрипции), такие как мутации MPL, LNK или еще неизвестных аллелей (Pardanani AD et al., Blood 108:3472-3476, 2006; Oh ST et al., Blood First Edition Paper, prepublished online April 19, 2010; DOI 101182/blood-2010-02-270108 2010; Pardanani A et al., Leukemia In press: 2010).
Результаты клинического исследования показывают, что терапия с применением TG101348 может быть прекращена без предварительного снижения дозы или постепенного снижения дозы. Субъекты, которые прекращают лечение (с возобновлением лечения на более поздней стадии или без того), не испытывают «цитокиновую отдачу». Это указывает на то, что лечение может быть прервано без предварительного снижения дозы.
Цитокиновая отдача в контексте миелофиброза представляет собой феномен, который возникает у пациентов, которые получают терапию, отличающуюся от терапии посредством TG101348, и которым по какой-либо причине прерывают лечение. В некоторых случаях, пациенты, прекратившие лечение, испытывают тяжелые симптомы, включающие сильное увеличение размера селезенки и возврат конституциональных симптомов. В некоторых случаях, пациенты, прекратившие лечение, испытывают жизнеугрожающие гемодинамические нарушения (Wadleigh and Tefferi, Clinical Advances in Hematology & Oncology, 8:557-563, 2010).
Следует отметить, что из числа имеющих небольшую молекулу ингибиторов пути JAK-STAT-киназ при MF, TG101348, по-видимому, является уникальным по своей способности индуцировать значительное и устойчивое снижение нагрузки мутантного аллеля JAK2V617F. Не желая быть связанными какой бы то ни было теорией, полагают, что, по-видимому, эффект ингибирования JAK2-киназы в отношении бремени заболевания, является основой подтверждения клинической эффективности лечения миелофиброза посредством TG101348, в отличие от непрямого антицитокинового эффекта, который может играть главную роль в ответных реакциях на действие антагонистов семейства JAK-киназ, которые имеют нецелевую активность в отношении JAK1-киназы, а также в отношении JAK2-киназы. В поддержку этого, отсутствуют согласующиеся между собой изменения уровней провоспалительных цитокинов (интерлейкин («IL»)-6, IL-2, IL-8 и TNF-α) относительно исходного уровня во время лечения с применением соединения TG101348 (Фигура 17). Напротив, и в соответствии с целевой активностью TG101348 в отношении JAK2-киназы, после начала лечения можно увидеть повышения сывороточных уровней EPO (эритропоэтина) и в меньшей степени TPO (тромбопоэтина) относительно исходного уровня (данные не показаны).
DLT (асимптоматическую гиперамилаземию, иногда в сочетании с гиперлипаземией) для TG101348 наблюдают относительно других имеющих небольшую молекулу ингибиторов, включающих нилотиниб (Kantarjian H.M. et al., Blood 110:3540-3546, 2007). Желудочно-кишечные нежелательные явления в этом исследовании проявляются часто, но являются причиной прекращения лечения только у одного субъекта. Эти симптомы возникают уже после первой введенной дозы и показывают четкую зависимость от дозы. Миелосупрессивные эффекты TG101348 также являются дозозависимыми.
Хотя MTD (680 мг/день) соединения TG101348 является наиболее эффективной дозой, она также имеет связь с самым высоким числом новых случаев нежелательных явлений. Следовательно, более низкая исходная доза (например, 400-500 мг/день) может обеспечить оптимальный баланс риск/польза. Кроме того, так как миелофиброз является гетерогенным заболеванием, динамический режим введения дозы может максимально повысить возможность для определения индивидуальной для пациента оптимальной дозы.
Эти наблюдения позволяют предположить, что в дополнение к MF, TG101348 также может иметь потенциальную роль в лечении PV (истинной полицитемии) и ET (эссенциальной тромбоцитемии).
Пример 4. Синтез TG101348
Пример 4.1. N-трет-Бутил-3-(2-хлор-5-метил-пиримидин-4-иламино)бензолсульфонамид (Промежуточное соединение)
Пример 4.1(а)
Смесь 2-хлор-5-метил-пиримидин-4-иламина (1) (0,4 г, 2,8 ммоль), 3-бром-N-трет-бутил-бензолсульфонамида (2) (1,0 г, 3,4 ммоль), Pd2(dba)3 (0,17 г, 0,19 ммоль), Xantphos (0,2 г, 3,5 ммоль) и карбоната цезия (2,0 г, 6,1 ммоль) суспендируют в диоксане (25 мл) и греют при кипячении с обратным холодильником в атмосфере аргона в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и разбавляют дихлорметаном (DCM) (30 мл). Смесь фильтруют и фильтрат концентрируют в вакууме. Остаток растворяют в этилацетате (EtOAc) и добавляют гексаны до тех пор, пока не выпадет в осадок твердое вещество. После фильтрации, указанное в заглавии соединение (1,2 г, 98%) получают в виде твердого вещества светло-коричневого цвета. Его используют на следующей стадии без очистки. Масс-спектрометрия с ионизацией электрораспылением (MS (ES+)): m/z 355 (M+H)+.
Пример 4.1(b)
Промежуточное соединение синтезируют из 2,4-дихлор-5-метилпиримидина (SM1) и N-t-бутил-3-аминобензолсульфонамида (SM2) на следующих стадиях: (1) Смешивают MeOH (6,7 UOa) и SM1 (Combi Blocks) (UOa); (2) Добавляют SM2 (1,15 UOa, 0,82 экв.) и H2O (8,5 UOa); (3) Нагревают при 45°C, 20 часов, под N2, IPC CPL SM2<2%; (4) Охлаждают до 20°C; (5) Подвергают центрифугированию, под N2; (6) Промывают Н2О (2,1 UOa) + MeOH (1,7 UOa); (7) Смешивают твердое вещество в H2O (4,3 UOa) + MeOH (3,4 UOa); (8) Подвергают центрифугированию, под N2; (9) Промывают H2O (2,1 UOa) + MeOH (1,7 UOa); и (10) Сушат при 45°C, в вакууме, 15 часов. Получают Промежуточное соединение, масса 49,6 кг (UOb); Выход 79%; Оптическая чистота (OP): 99,6%.
Пример 4.2. N-трет-Бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамид
Пример 4.2(а)
Смесь N-трет-Бутил-3-(2-хлор-5-метил-пиримидин-4-иламино)бензолсульфонамида (Промежуточное соединение) (0,10 г, 0,28 ммоль) и 4-(2-пирролидин-1-ил-этокси)-фениламина (3) (0,10 г, 0,49 ммоль) в уксусной кислоте (3 мл) герметизируют в пробирке для проведения реакции в условиях микроволнового излучения и облучают микроволновым излучением при 150°C в течение 20 минут. После охлаждения до комнатной температуры, крышку удаляют и смесь концентрируют. Остаток очищают с помощью высокоэффективной жидкостной хроматографии (HPLC) и собранные фракции объединяют и выливают в насыщенный раствор NaHCO3 (30 мл). Объединенные водные слои экстрагируют этилацетатом (2×30 мл) и объединенные органические слои промывают рассолом, сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют и получающееся в результате твердое вещество растворяют в минимальном количестве EtOAc и гексаны добавляют до тех пор, пока твердое вещество не выпадет в осадок. После фильтрации, указанное в заглавии соединение получают в виде твердого вещества белого цвета (40 мг, 27%). 1Н ЯМР (500 МГц, DMSO-d6): δ 1,12 (с, 9Н), 1,65-1,70 (м, 4Н), 2,12 (с, 3Н), 2,45-2,55 (м, 4Н), 2,76 (т, J=5,8 Гц, 2Н), 3,99 (т, J=6,0 Гц, 2Н), 6,79 (д, J=9,0 Гц, 2Н), 7,46-7,53 (м, 4Н). 7,56 (с, 1Н), 7,90 (с, 1Н), 8,10-8,15 (м, 2Н), 8,53 (с, 1Н), 8,77 (с, 1Н). Масс-спектрометрия с ионизацией электрораспылением (MS (ES+)): m/z 525 (M+H)+.
Пример 4.2(b)
Моногидрат дигидрохлорида N-трет-Бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида получают из 4-[2-(1-пирролидинил)этокси]анилин-дигидрохлорида (SM3) и Промежуточного соединения после стадий (А) и (В).
Стадия (А), получение свободного основания SM3 (3) из SM3, включает стадии (1)–(9): (1) Солюбилизируют NaOH (0,42 UOb) в Н2О (9 UOb); (2) Охлаждают до <20°C, под N2; (3) Добавляют TBME (6 UOb), затем SM3 (Malladi Drugs) (1,06 UOb); (4) Смешивают >20 минут, затем прекращают смешение; (5) Дренируют Водную фазу, затем экстрагируют посредством TBME (3 UOb); (6) Объединяют органическую фазу; (7) Концентрируют, в вакууме, Т<40°C, с получением Масла; (8) Солюбилизируют в IPA (2,5 UOb); и (9) Вычисляют сухой экстракт 23%.
Стадия (В) включает стадии (1)–(6): (1) Смешивают IPA (10,5 UOb) и Промежуточное Соединение (UOb); (2) Добавляют свободное основание SM3 (0,75 UOb, 1,33 экв./пром.соед.); (3) добавляют концентрированную HCl (0,413 UOb); (4) Нагревают при 70°C, 20 часов, под N2, IPC CPL Промежуточное соединение <2%; (5) Охлаждают до <20°C; (2) Подвергают центрифугированию, N2; (3) Промывают IPA (3 UOb); (4) Сушат при 50°C, в вакууме, 26 часов; (5) Распределяют массу в измельчителе Fitzmill; и (6) Помещают в полиэтиленовый пакет (х2)/ барабан с полиэтиленовым вкладышем. Получают моногидрат дигидрохлорида TG101348, масса 83,8 кг; Выход 98%; Оптическая чистота (OP): 99,5%.
Пример 5. Капсульная форма TG101348 и Способ изготовления TG101348
Лекарственные продукты на основе TG101348 предоставляют в виде капсул с 10-мг-, 40-мг- и 200-мг-дозировкой (содержанием действующего вещества в лекарственной форме), где массы заданы по количеству активного (то есть свободного основания) фрагмента TG101348. Количественный состав для каждой дозировки TG101348 в случае капсульного лекарственного продукта, показан в Таблице 14.
|
Компоненты, которые используют в процессе изготовления капсулы с каждой дозировкой, на основе одной партии, показаны в Таблице 15.
|
Способ изготовления капсул с TG101348 описывают ниже.
А. Сухое гранулирование интрагранулярных компонентов (осуществляют для всех трех дозировок лекарственного продукта): 1. TG101348 и интрагранулярный стеарилфумарат натрия смешивают в V-образном смесителе в течение 5 минут. 2. Смесь пропускают через измельчитель конической формы, оснащенный круглым сетчатым фильтром 18-меш и круглой лопастной мешалкой. Смесь повторно загружают в V-образный смеситель. 3. Интрагранулярную силикатированную микрокристаллическую целлюлозу просеивают через сетчатый фильтр 20-меш и добавляют в смеситель. Смесь смешивают в течение 15 минут. 4. Смесь пропускают через роликовый пресс. 5. Полученные с помощью роликового пресса полоски пропускают через конический измельчитель, оснащенный круглым сетчатым фильтром 16-меш и круглой лопастной мешалкой. 6. Измельченный материал смешивают в V-образном смесителе в течение 5 минут. 7. Образцы, прошедшие междуоперационную проверку (IPC), извлекают из V-образного смесителя с помощью пробоотборника. Образцы подвергают анализу на их действенность.
В. Добавление экстрагранулярных компонентов (осуществляют для капсул с дозировкой 10 мг и 40 мг): 1. В том случае, где действенность гранул (полученных на Стадии 7 Стадии А) имеет величину вне диапазона номинальных доз 98-102% (масс./масс.), экстрагранулярную силикатированную микрокристаллическую целлюлозу регулируют соответственно. 2. В V-образный смеситель загружают моногидрат дигидрохлорида TG101348/силикатированную микрокристаллическую целлюлозу/гранулы стеарилфумарата натрия (полученные на стадии А). 3. Экстрагранулярную силикатированную микрокристаллическую целлюлозу просеивают через сетчатый фильтр 20-меш и добавляют в V-образный смеситель. 4. В V-образный смеситель добавляют экстрагранулированный стеарилфумарат натрия. 5. Интрагранулярные и экстрагранулярные компоненты смешивают в течение 15 минут. 6. Образцы, прошедшие междуоперационную проверку, извлекают из V-образного смесителя с помощью пробоотборника и анализируют на их действенность.
С. Заполнение капсул (осуществляют для всех трех дозировок лекарственного продукта): 1. Если действенность (для капсул с дозировкой 200 мг, получаемых на стадии 7 стадии А, или для капсул с дозировкой 10 мг и 40 мг, получаемых на стадии 6 стадии В) имеет величину вне диапазона номинальных доз 98-102% (масс./масс.), массу содержимого в капсуле корректируют соответственно. 2. Полученный материал инкапсулируют с использованием автоматической машины для заполнения капсул. Полученные капсулы раскладывают по бутылочкам и хранят при 20-28°F (68-82°C) и при влажности окружающей среды.
Исследуют однородность состава (дозирования по содержанию) и растворение. Валидацию метода HPLC (высокоэффективная жидкостная хроматография) выполняют с помощью одного химика-аналитика, с применением дизайна один процесс – один химик-аналитик, и он отвечает всем необходимым критериям по специфичности, чувствительности, воспроизводимости (прецизионности), точности, линейности и стабильности образца. Специфичность оценивают и подтверждают путем сравнения разрешения пиков между TG101348 и всеми его родственными соединениями, промежуточными соединениями и деградантами (устанавливают в ходе исследований форсированной деградации). Предел количественного определения и предел обнаружения устанавливают на уровне 0,10 мкг/мл и 0,03 мкг/мл TG101348, соответственно. Воспроизводимость результатов (прецизионность) по однородности состава оценивают с помощью шести инъекционных препаратов из капсул с дозировкой 10 мг и 200 мг, приготовленных в целевой аналитической концентрации. Результаты RSD соответствуют 3,7% и 5,8% для капсул с дозировками 10 мг и 200 мг, соответственно. Воспроизводимость результатов по растворению оценивают по шести инъекционным препаратам в каждой временной точке растворения капсул с дозировками 10 мг и 200 мг. Данные результатов по относительному стандартному отклонению («RSD») для всех дозировок и соответствующих временных точек хорошо попадают в диапазон критериев приемлемости (±10%), задаваемый в валидационном протоколе. Точность (определяют в результате извлечения анализируемого вещества, добавленного в известном количестве в раствор-плацебо, в случае капсул с дозировками 10 мг и 200 мг) оценивают при 70%, 100% и 130% относительно целевой аналитической стандартной концентрации. Значения по извлечению для всех измерений находятся в пределах диапазона критериев приемлемости (93-105%), задаваемого в валидационном протоколе. Линейность показывают в диапазоне от 50% до 120% относительно целевой аналитической стандартной концентрации и дают показатель r2, равный 1,00. Во время валидации метода также продемонстрированы стабильность образца и устойчивость метода.
Пример 6. Исследование состава для TG101348
Проводят исследование состава для моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида.
Совместимость моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида с капсульными оболочками
Было неизвестно, могла ли быть несовместима щелочная/кислотная природа моногидрата дигидрохлорида N-трет-бутил-3-[(5-метил-2-{[4-(2-пирролидин-1-илэтокси)фенил]амино}пиримидин-4-ил)амино]бензолсульфонамида (моногидрат ди-HCl TG101348) с капсулами вследствие упомянутой возможной щелочной/кислотной природы дигидрохлорида.
Твердые желатиновые и гидроксипропилметил-целлюлозные (HPMC) капсульные оболочки (размер №00250) наполняют 250 миллиграммами моногидрата дигидрохлорида TG101348. Заполненные капсулы подвергают ускоренному испытанию на стабильность (40°C/75%-ная относительная влажность (RH) и 25°C/60%-ная относительная влажность). Капсулы упаковывают в 30-миллилитровые (1 oz) бутылочки из полиэтилена высокой плотности (HDPE) темно-янтарного цвета. Краткое описание составов и протокола ускоренного испытания на стабильность касательно исследования стабильности показано в Таблице 16.
|
Было обнаружено, что моногидрат дигидрохлорида TG101348 совместим с твердыми желатиновыми капсулами. Никаких заметных изменений в характеристиках (внешний вид, количественный состав пробы, примеси) не было отмечено во временных точках исследования (t=1, 2 и 3 недели).
Совместимость смеси лекарственных веществ с наполнителями и смазывающими веществами
Составляют матрицу приготовления смесей для исследования совместимости моногидрата дигидрохлорида TG101348 с четырьмя наполнителями и двумя смазывающими веществами (Таблица 17). Смеси приготавливают в количестве 2,5 г каждую путем предварительного просеивания через сетчатые фильтры (500 мкм), смешения всех компонентов за исключением смазывающего вещества с помощью смесителя Turbula T2B в течение 10 минут при скорости вращения мешалки 22 оборота в минуту (rpm), просеивания смеси через сетчатый фильтр (500 мкм), смешения в течение 10 минут, добавления смазывающего вещества (масса регулируется) и смешения в течение 5 минут. Смеси изготавливают и после этого хранят в банках из HDPE (ПЭВП) темного цвета емкостью 30 мл в основных (60°C/ влажность окружающей среды) и резервных (40°C/75% RH, 25°C/60% RH и 5°C) условиях. Краткое изложение протокола ускоренного испытания на стабильность показано в Таблице 18. Никаких заметных изменений характеристик (внешний вид, количественный состав пробы, примеси) не было отмечено во время исследования.
|
|
|
Разработка лекарственной формы Порошок-в-Капсуле
Выбор эксципиента
Испытание на совместимость с эксципиентами, проводимое для сухих смесей моногидрата дигидрохлорида TG101348 c четырьмя наполнителями (лактозой, маннитом, микрокристаллической целлюлозой (MCC) Avicel PH102 и SMCC ProSolv 90 HD) и с двумя смазывающими веществами (стеаратом магния и стеарилфумаратом натрия (Pruv)) (Таблица 17) не выявило несовместимости. ProSolv SMCC 90HD (то есть силикатированную микрокристаллическую целлюлозу) и лактозу Fast-Flo (то есть высушенный распылением моногидрат лактозы) выбирают в качестве наполнителей для дополнительного испытания, основываясь на свойствах, подходящих для технологий прямого сухого смешения. Стеарат магния (растительного происхождения) и стеарилфумарат натрия (Pruv) выбирают в качестве смазывающих веществ для дополнительного испытания. Все эксципиенты одобрены органами нормативно-правового регулирования для применения в твердых пероральных лекарственных формах (США, Евросоюз, Япония).
Пригодность для разработки технологии с применением Сухого Порошка
Истинные плотности содержащих моногидрат дигидрохлорида TG101348 частиц округлого/гранулярного внешнего вида, силикатированной микрокристаллической целлюлозы (ProSolv SMCC 90HD) и стеарилфумарата натрия (Pruv) измеряют с помощью гелиевого пикнометра (Micromeritics Accupyc 1340). Истинная плотность лекарственного вещества и эксципиентов (наполнителей/разбавителей) оказываются хорошо совпадающими.
Капсульные составы
Составляют «матрицу» капсульных составов прототипа для оценивания стабильности и данные сводят вместе в Таблице 19. Выбирают две величины дозировок, 10 и 125 мг.
|
Протокол Ускоренного Испытания на Стабильность
Таблица 20 кратко излагает протокол ускоренного испытания на стабильность, применяемый для капсульных прототипов. Никаких заметных изменений характеристик (внешний вид, количественный состав пробы, примеси, растворение in vitro) не было отмечено во временных точках исследования (t=1, 2, 4 и 8 недель при 40°C/75% RH и при 25°C/60% RH). Основываясь на этих результатах, Прототипы Р2 и Р6 выбирают для дополнительного оценивания.
|
Рассмотрение характеристик растворения in vitro
В испытании растворения in vitro, составы прототипов, введенные в желатиновые капсульные оболочки (Р1, Р2, Р5, Р6) демонстрируют > 85%-ное высвобождение лекарственного средства в пределах 15 минут. Составы прототипов, введенных в гидроксипропилметил-целлюлозные (HPMC) капсульные оболочки (например, Р3, Р4, Р7, Р8) обычно показывают <60%-ное высвобождение лекарственного средства по истечении 60 минут. Прототипы в HPMC-капсулах, следовательно, не проходят из капсул в ходе испытания после t=0.
Разработка капсул, содержащих вещество, способствующее всасыванию
TG101348 имеет промежуточную проницаемость между «низкой» и «высокой» проницаемостью, на основе данных по проницаемости в клетках СаСо-2. В дополнение к тому, биодоступность в нескольких образцах обычно составляет 20-25%. Следовательно, не известно, будет ли необходимо «способствующее всасыванию вещество» в составе для достижения адекватной биодоступности.
Выбор эксципиента
На основе совместимости с эксципиентами, описанной выше, силикатированную микрокристаллическую целлюлозу (ProSolv SMCC 90 HD) используют в качестве основного наполнителя/ эксципиента-носителя для состава, который должен обладать свойством облегчения всасывания. Четыре кандидата на роль эксципиента, обладающего свойством облегчения всасывания, выбирают для дополнительного испытания (Таблица 21).
|
Составы и способы изготовления составов
Таблица 22 сводит вместе испытанные составы, обладающие свойством способствовать всасыванию. Для изготовления состава выбирают гранулирование в расплаве для сравнения со способом изготовления прямым смешением.
|
Перекрестное Исследование фармакокинетических свойств (PK), проведенное на собаке породы Бигль
Перекрестное исследование фармакокинетических свойств (PK) на собаке породы Бигль проводят с испытанием пяти составов: перорального раствора, который описан ниже, двух капсульных составов без вещества, способствующего всасыванию, и двух капсульных составов с веществом, способствующим всасыванию.
Пяти собакам породы Бигль вводят каждый состав с дозой TG101348 125 мг или приблизительно 11 мг/кг в расчете на средние массы тела, с периодом вымывания в одну неделю между дозами. Введенные составы сведены вместе в Таблице 23.
|
Все четыре капсульных состава имеют характеристики мгновенного высвобождения наряду с демонстрацией биоэквивалентности дозе эталонного раствора. Таким образом, несмотря на промежуточную (между высокой и низкой) проницаемость в клетках CaCo-2 человека и 20-25%-ную биодоступность у различных видов животных, капсульные составы без состава веществ, способствующих всасыванию, демонстрируют характеристики мгновенного высвобождения.
Разработка технологии
Морфология частиц лекарственного вещества
Различную морфологию частиц, от округленных, гранулярных частиц (средний размер частиц ≈ 25 мкм), до частиц в форме небольших иголок (средний размер частиц ≈ 7-10 мкм) обнаружен среди различных лотов лекарственного вещества. Игольчатая форма, как обнаружено, имеет высокий статический заряд, что может негативно влиять на изготовление лекарственного продукта и также негативно влиять на однородность состава лекарственного продукта.
Технология сухого гранулирования
Первоначальный состав, разработанный с лекарственным веществом, имеющим округлые, гранулярные частицы со средним размером частиц 25 мкм, имеет соотношение 50:50 по массе лекарственного вещества TG101348 и наполнителя, с 0,5% (масс./масс.) смазывающего вещества. Перед вальцеванием, приготавливают смесь лекарственного вещества, наполнителя и смазывающего вещества. Как описано в этом документе, лекарственное вещество пропускают через установку для совместного измельчения для деагломерации перед смешением с эксципиентами состава. Лекарственное вещество, имеющее частицы в форме небольших иголок, демонстрирует высокую агломерируемость при хранении. После деагломерирования лекарственного вещества, имеющего частицы в форме небольших иголок, значительная повторная агломерация или 'комкование' может происходить почти мгновенно. Такую повторную агломерацию значительно снижают с помощью смешения лекарственного средства со смазывающим веществом перед измельчением.
Первоначальный состав с моногидратом дигидрохлорида TG101348 имеет соотношение по массе лекарственного вещества TG101348 и наполнителя 50:50, с 0,5% (масс./масс.) смазывающего вещества. Этот состав показывает плохую сыпучесть и значительное прилипание к металлическим вальцам внутри роликового пресса.
Количество смазывающего вещества стеарата магния может быть увеличено в составе, однако повышение концентрации в составе может оказать отрицательное влияние на кинетику высвобождения лекарственного средства. Смазывающее вещество лаурилфумарат натрия, как также показано, совместимо с моногидратом дигидрохлорида TG101348 и менее гигроскопично, чем стеарат магния, и его добавляют (в массовом соотношении 2,0% масс./масс.) вместо стеарата магния, что минимизирует прилипание состава к металлическим вальцам роликового пресса. Однако, сыпучесть порошка остается плохой.
Соотношение лекарственного вещества TG101348 к наполнителю снижают с приблизительно 50:50 до приблизительно 40:60. Содержание смазывающего вещества (Pruv) также снижают до 1% масс./масс., что обеспечивает приемлемую сыпучесть и минимальное прилипание внутри роликового пресса.
Разработка многодозовых составов
При испытаниях заполнения капсул вручную методом энергичного набивания желатиновых капсульных оболочек размером №00 выявлено, что приблизительно 600 мг гранул, по-видимому, составляют максимально достигаемую массу содержимого капсул. При вводе в состав 40% (масс./масс.) лекарственного вещества и при использовании порции моногидрата дигидрохлорида TG101348, имеющей содержание свободного основания 83,78%, оказалось, что верхний уровень дозировки 200 мг для капсулы является практически достижимым.
Описанная в этом документе технология сухой грануляции, разработанная для получения частиц, содержащих интрагранулярные моногидрат дигидрохлорида TG101348 и лаурилфумарат натрия, позволяет получить ряд дозировок капсул с применением технологий сухого смешения.
Средний размер гранул составляет приблизительно 300 мкм, и средний размер частиц силикатированной микрокристаллической целлюлозы составляет приблизительно 100 мкм. Следовательно, составы для капсул с дозировками 40 мг и 10 мг получают в результате разбавления гранул экстрагранулярной микрокристаллической целлюлозой. В общем, размеры частиц с учетом Размера гранул и частиц экстрагранулярной силикатированной микрокристаллической целлюлозы являются достаточно сходными, что позволяет обеспечить гомогенное смешение.
Капсулы с дозировкой 40 мг получают с использованием объема состав-включающего содержимого, сравнимого с 200 мг состава, в аналогичной капсульной оболочке (твердая желатиновая капсула размером №00). Для получения капсул с дозировкой 10 мг, используют обычную смесь с составом для получения дозировки капсулы 40 мг, для заполнения капсулы меньшего размера.
Состав в форме раствора для перорального введения
Разрабатывают состав в форме раствора для перорального введения, который содержит лекарственное вещество, 0,5% метилцеллюлозы (МС) и 0,05% Твин 80. Исследование рН-стабильности проводят при 60°C на составах, прошедших через фильтр из полиэфирсульфона (PES) с размером ячеек 0,22 мкм. Никаких заметных изменений характеристик (внешний вид, количественный состав, примеси) не было отмечено во время исследования (14 дней). Разрабатывают второй состав в форме раствора для перорального введения, который содержит лекарственное вещество и 0,5% МС. Второй состав в форме раствора для перорального введения используют в исследовании фармакокинетических свойств на собаке.
Хотя вышеприведенные примеры описаны довольно подробно путем иллюстрации примеров с целью прояснения понимания, описания и примеры не следует истолковывать как ограничивающие объем изобретения.
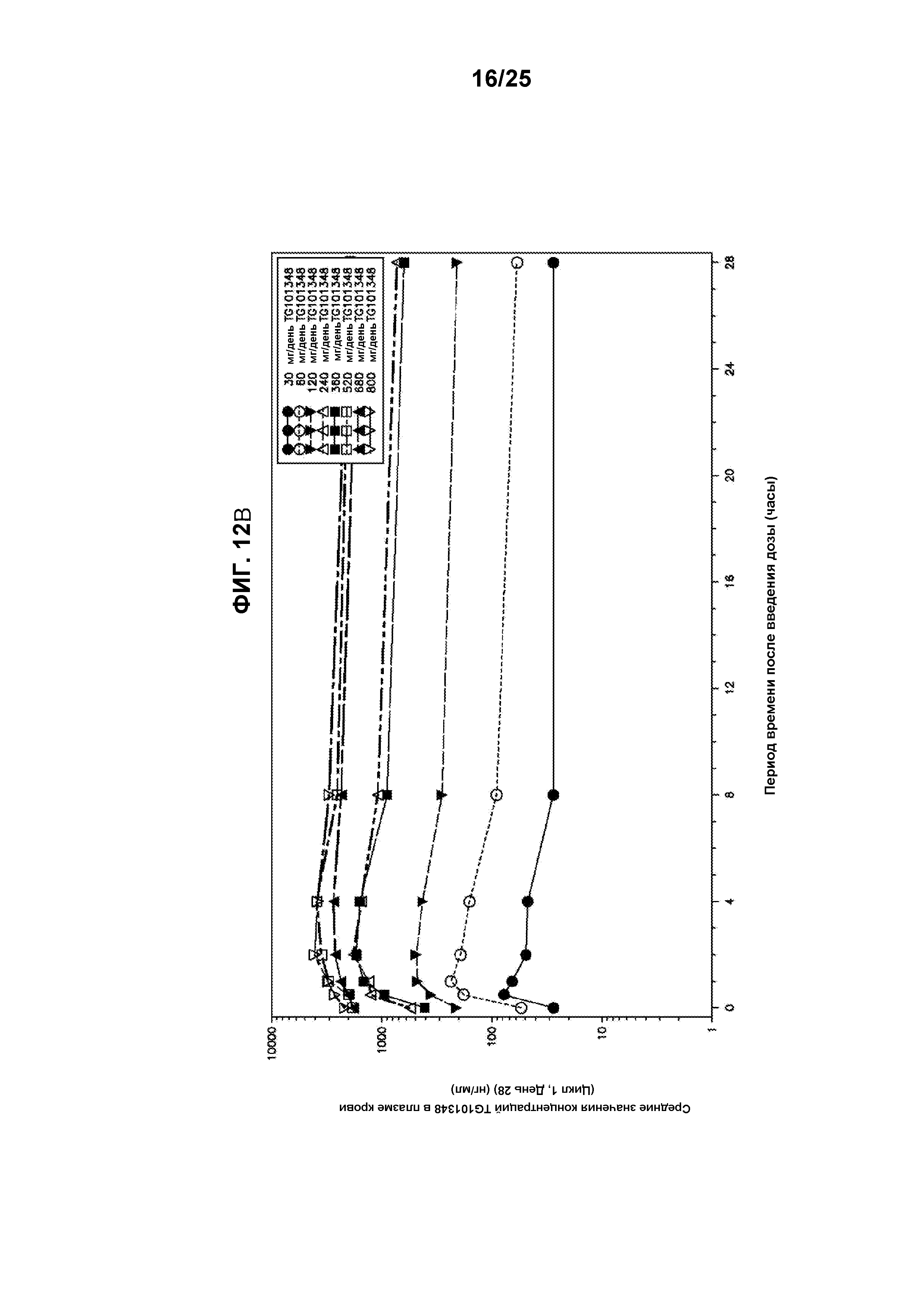
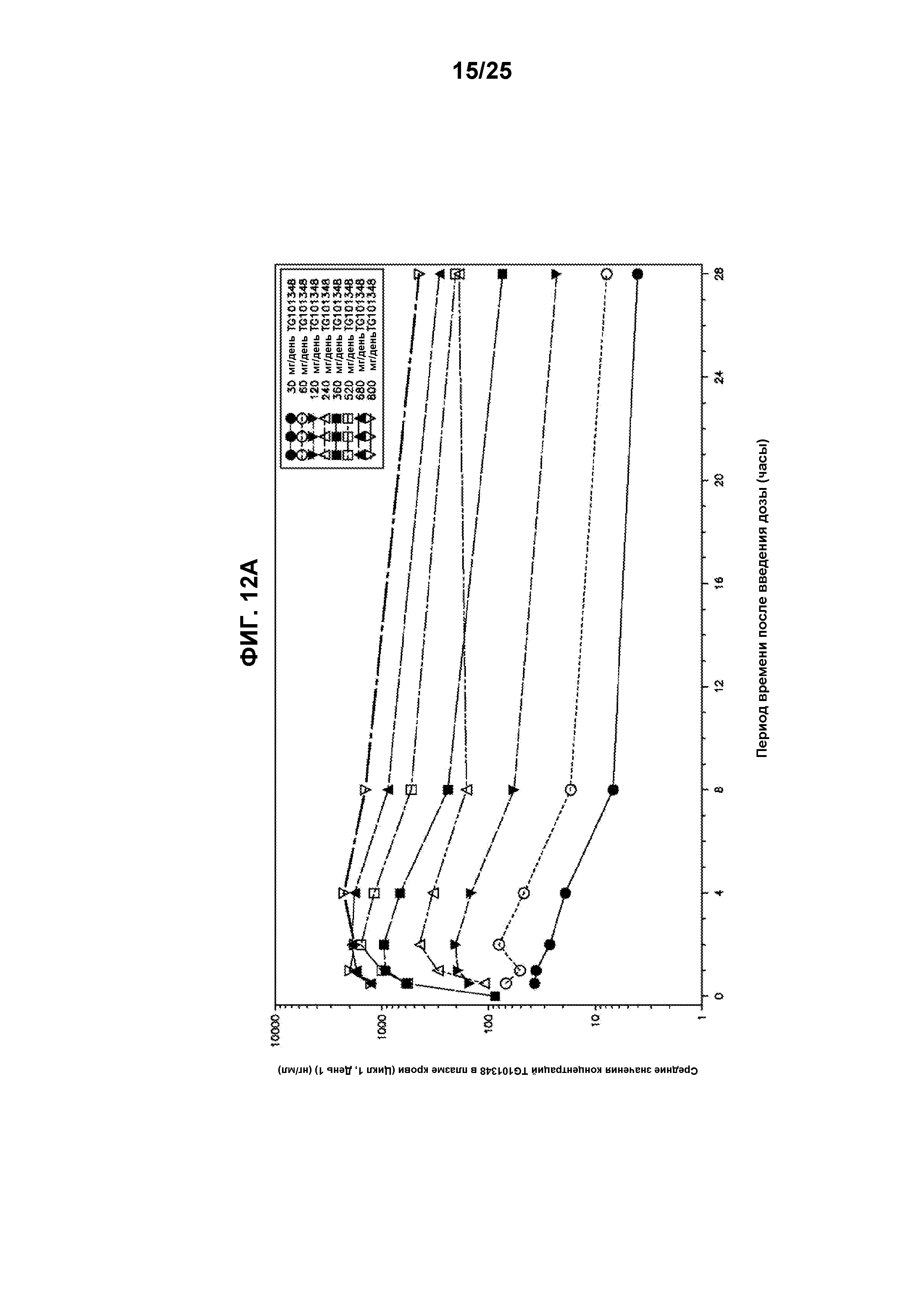

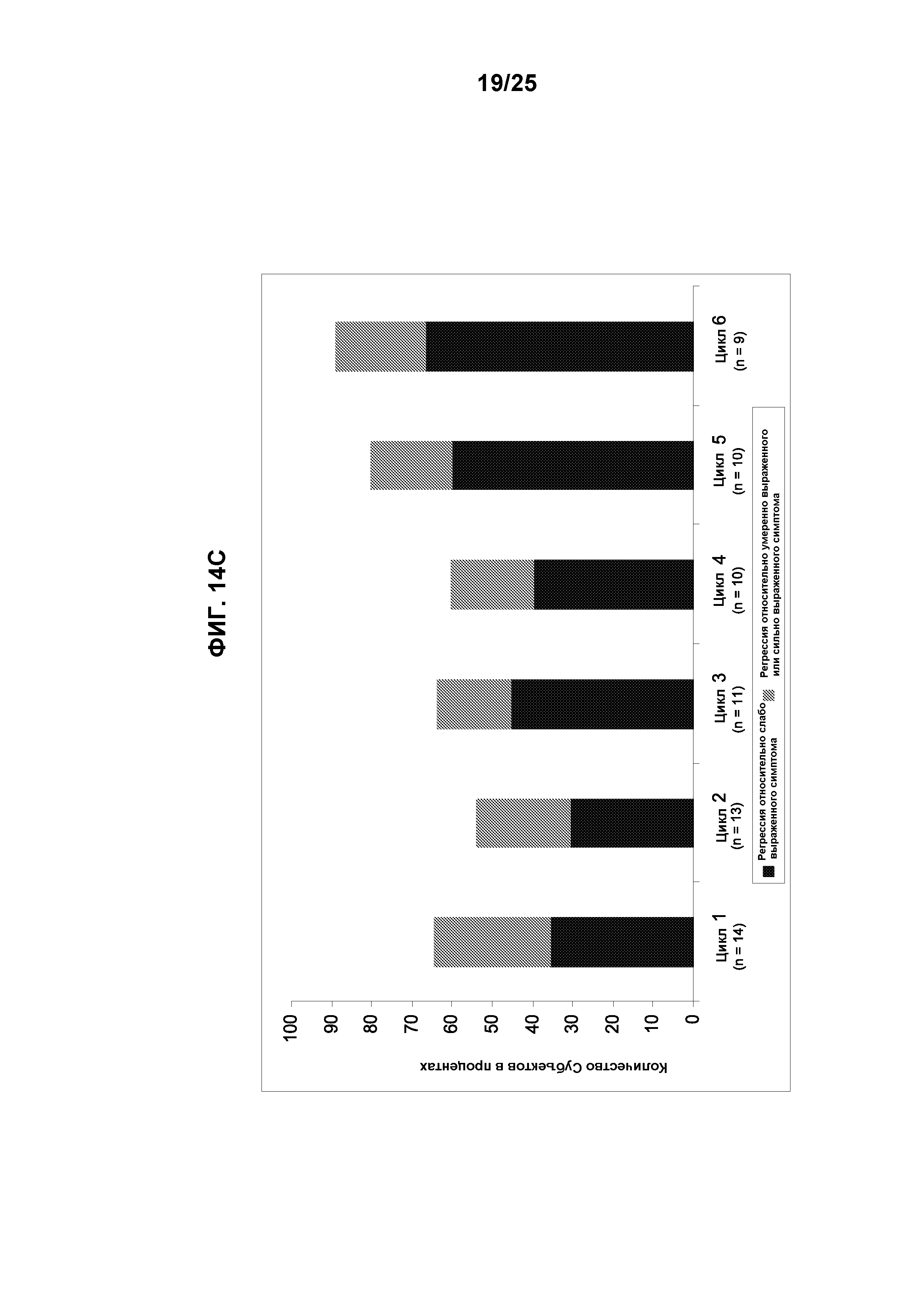
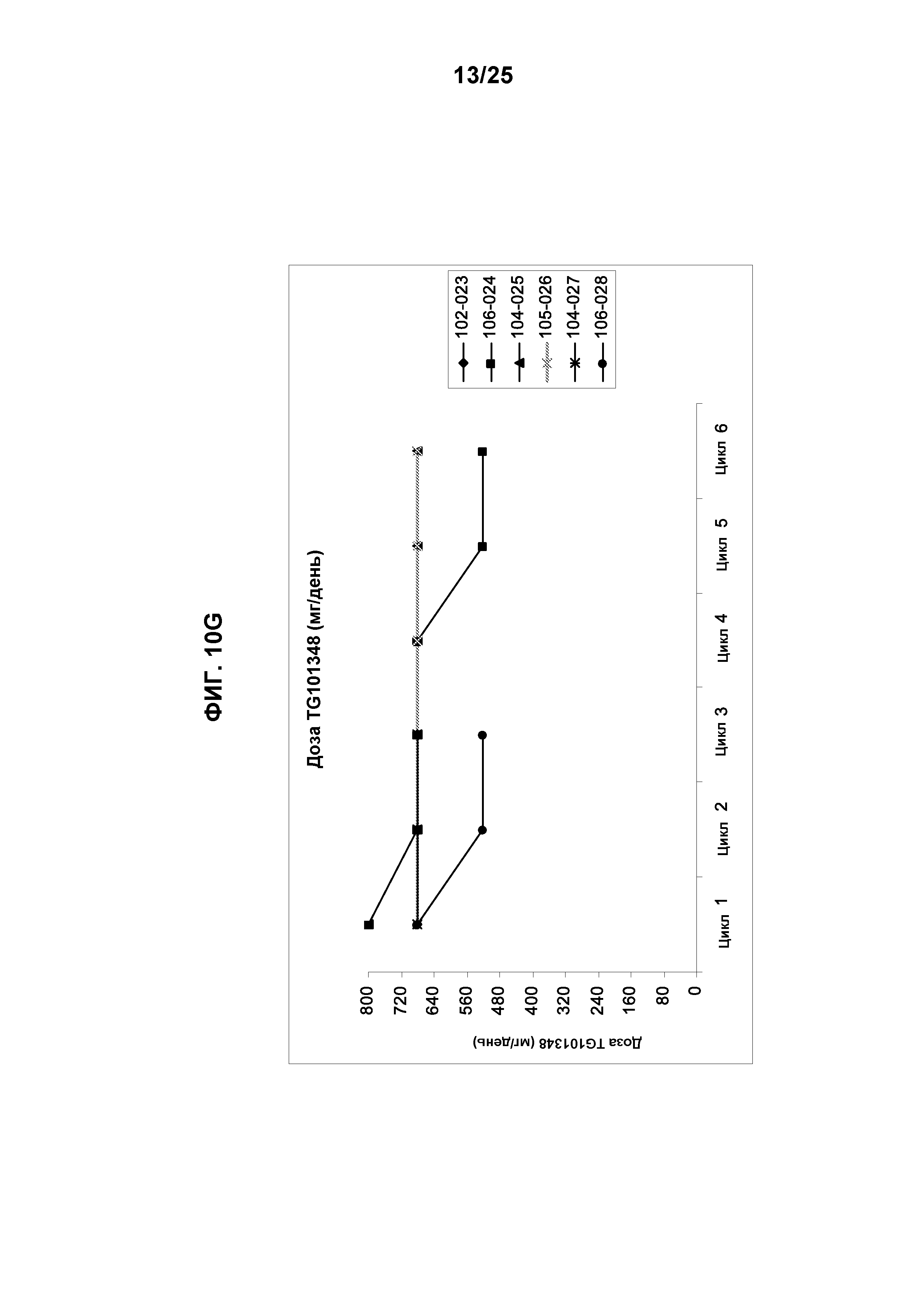
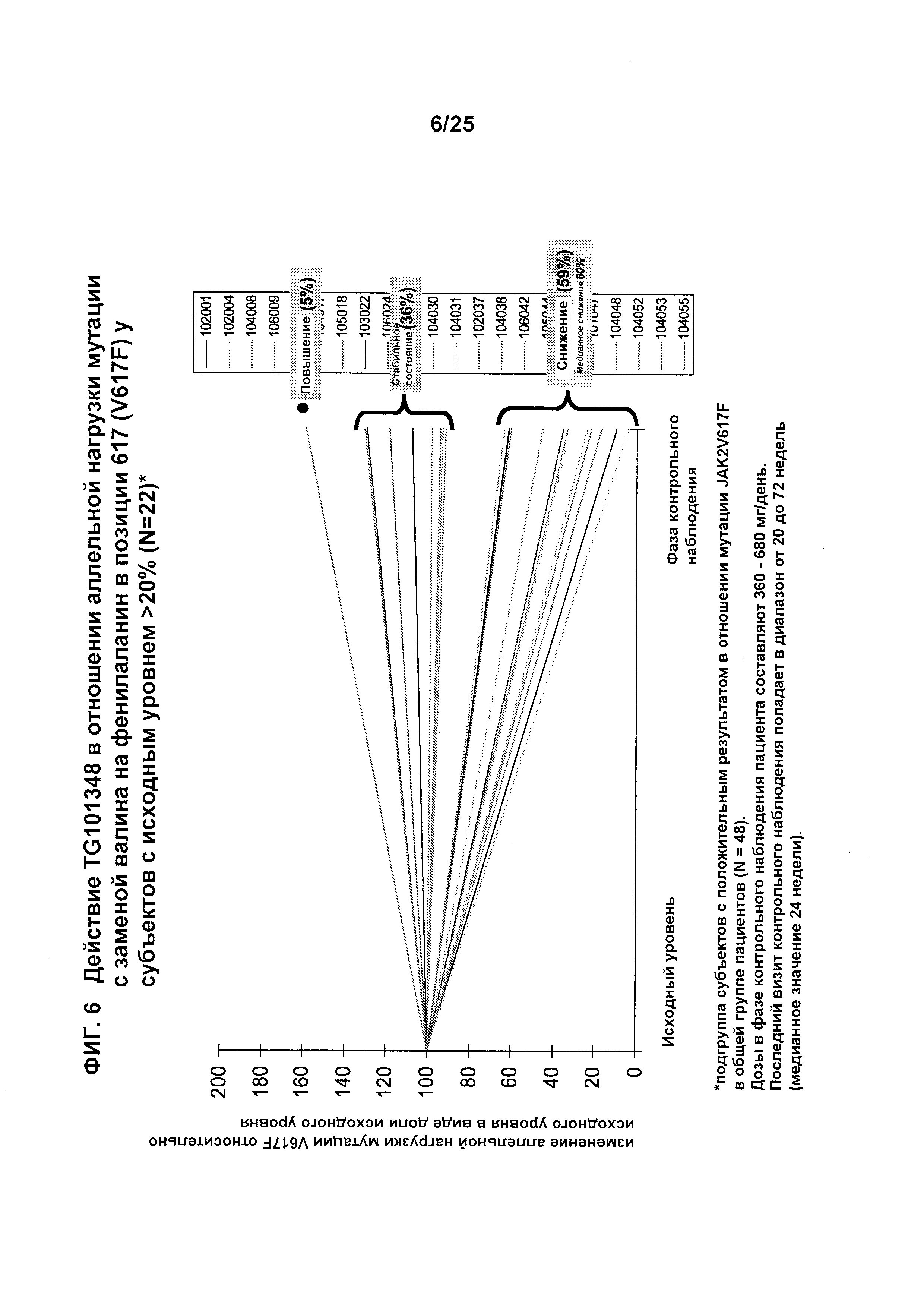
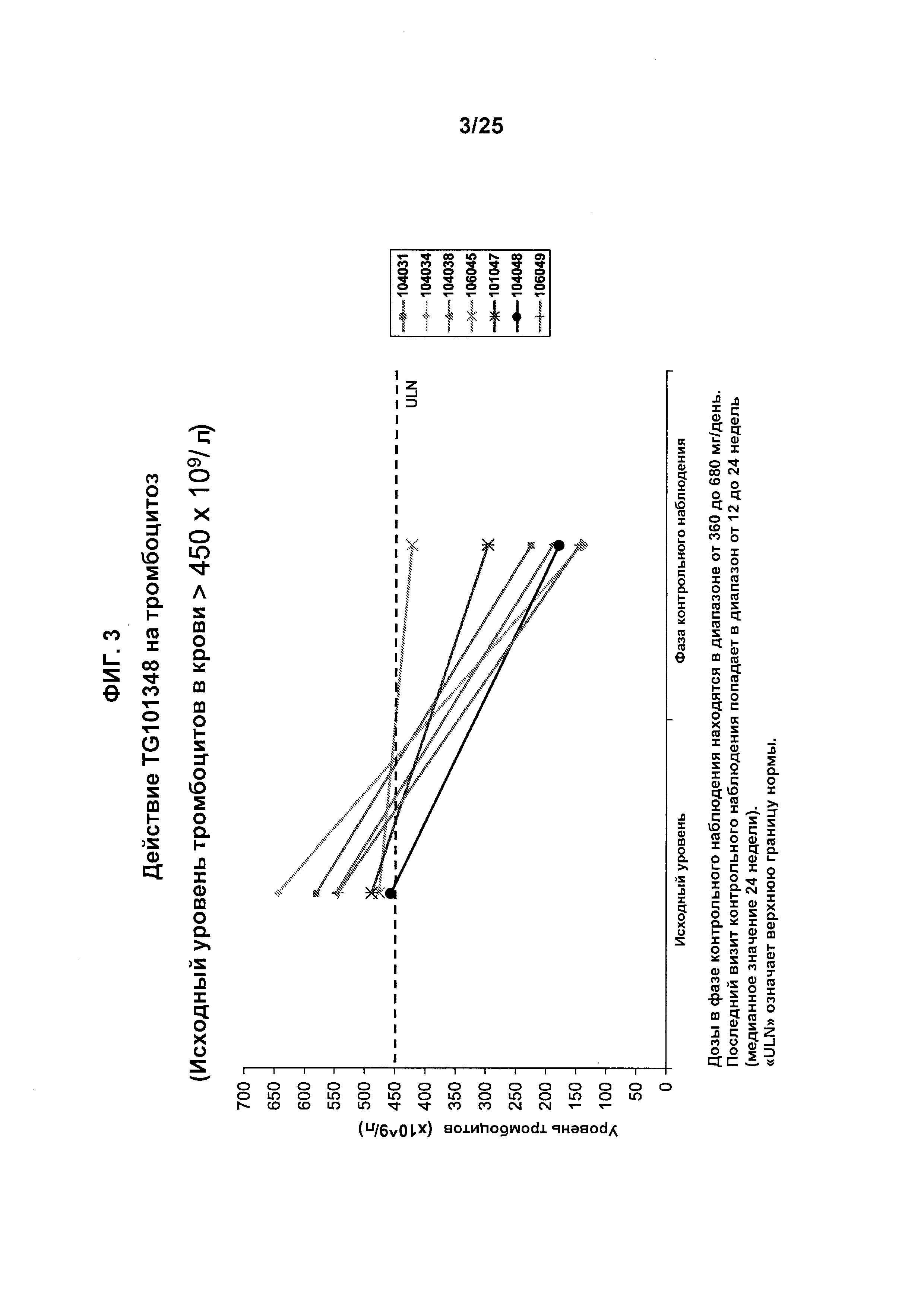

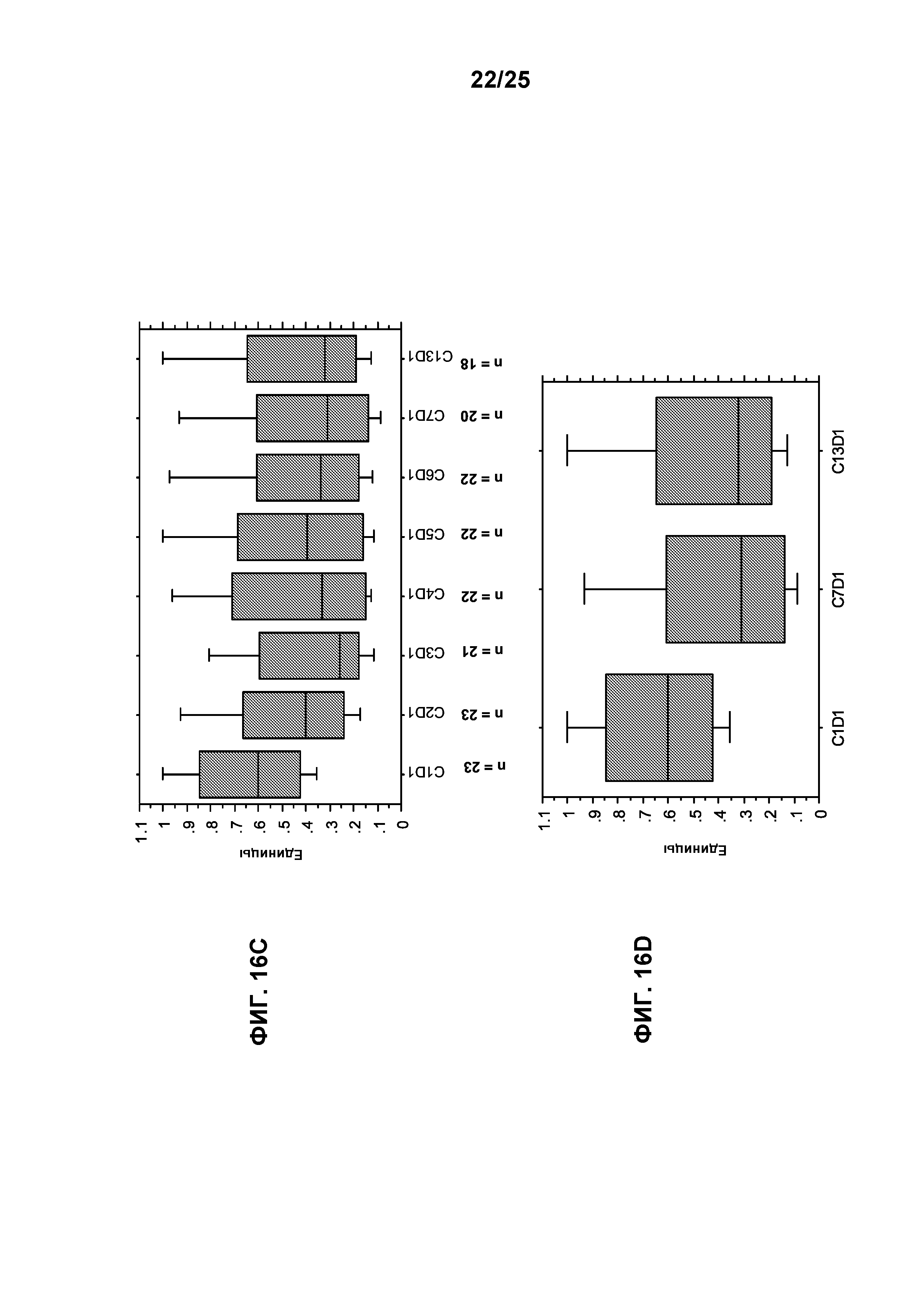

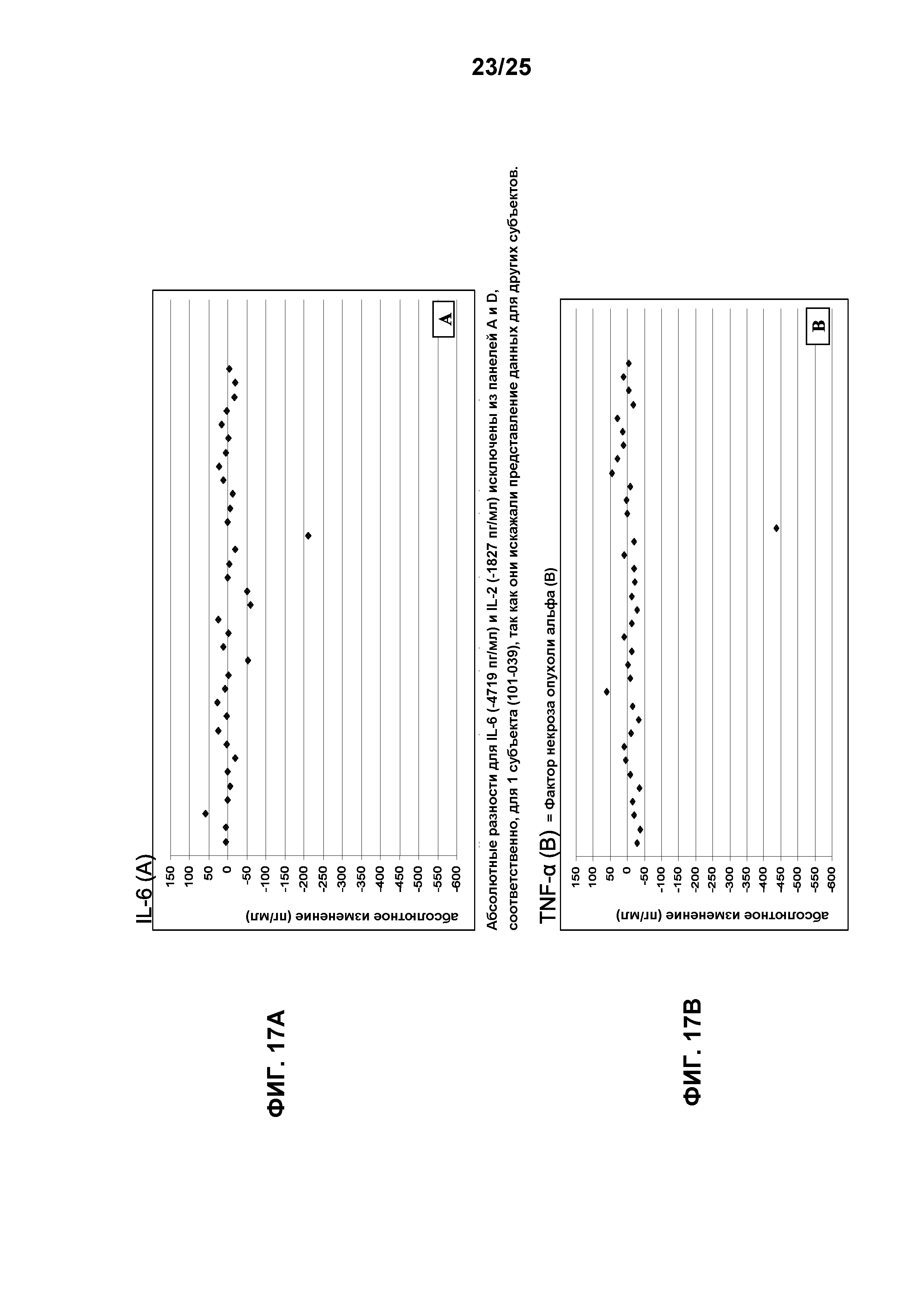

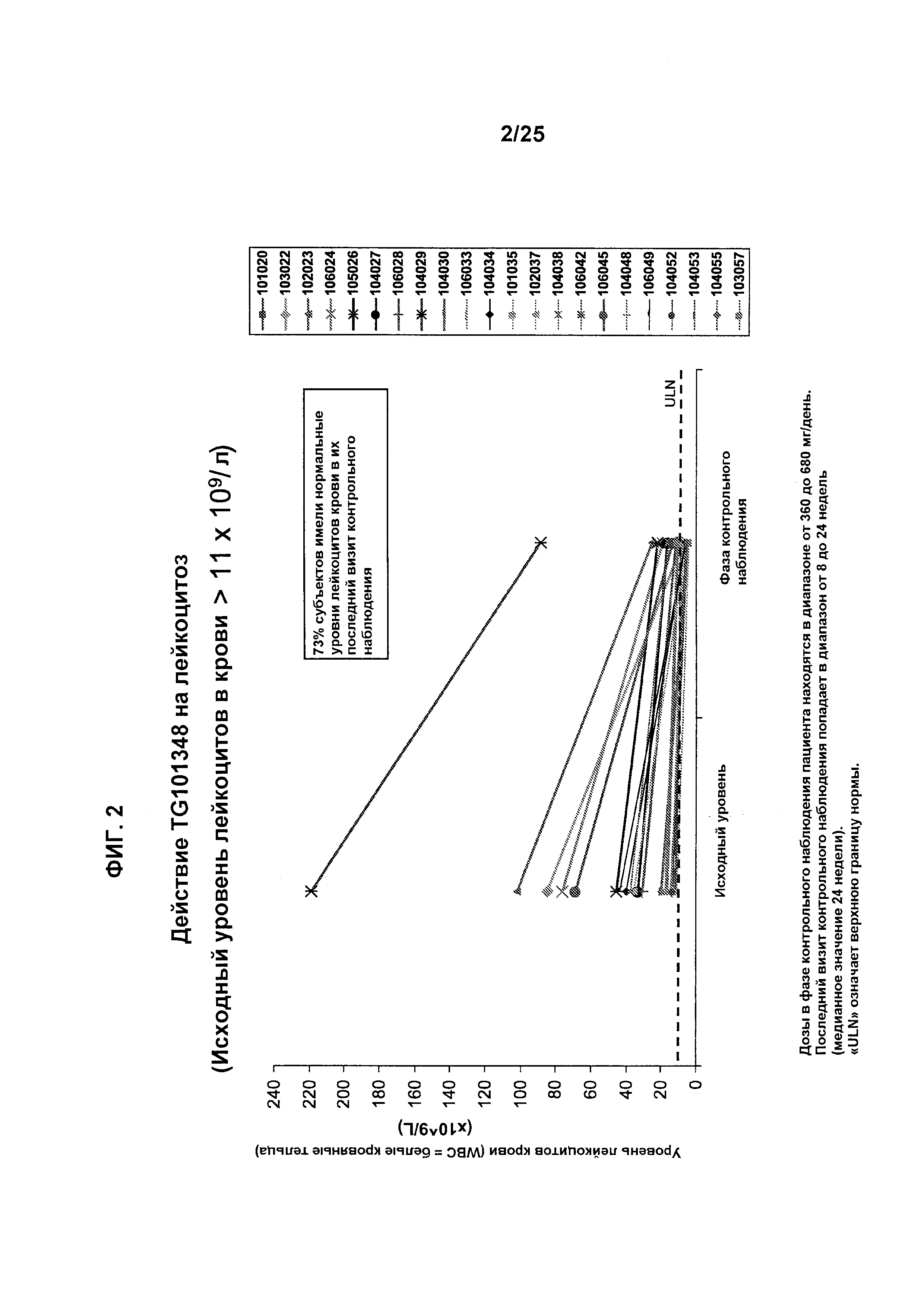
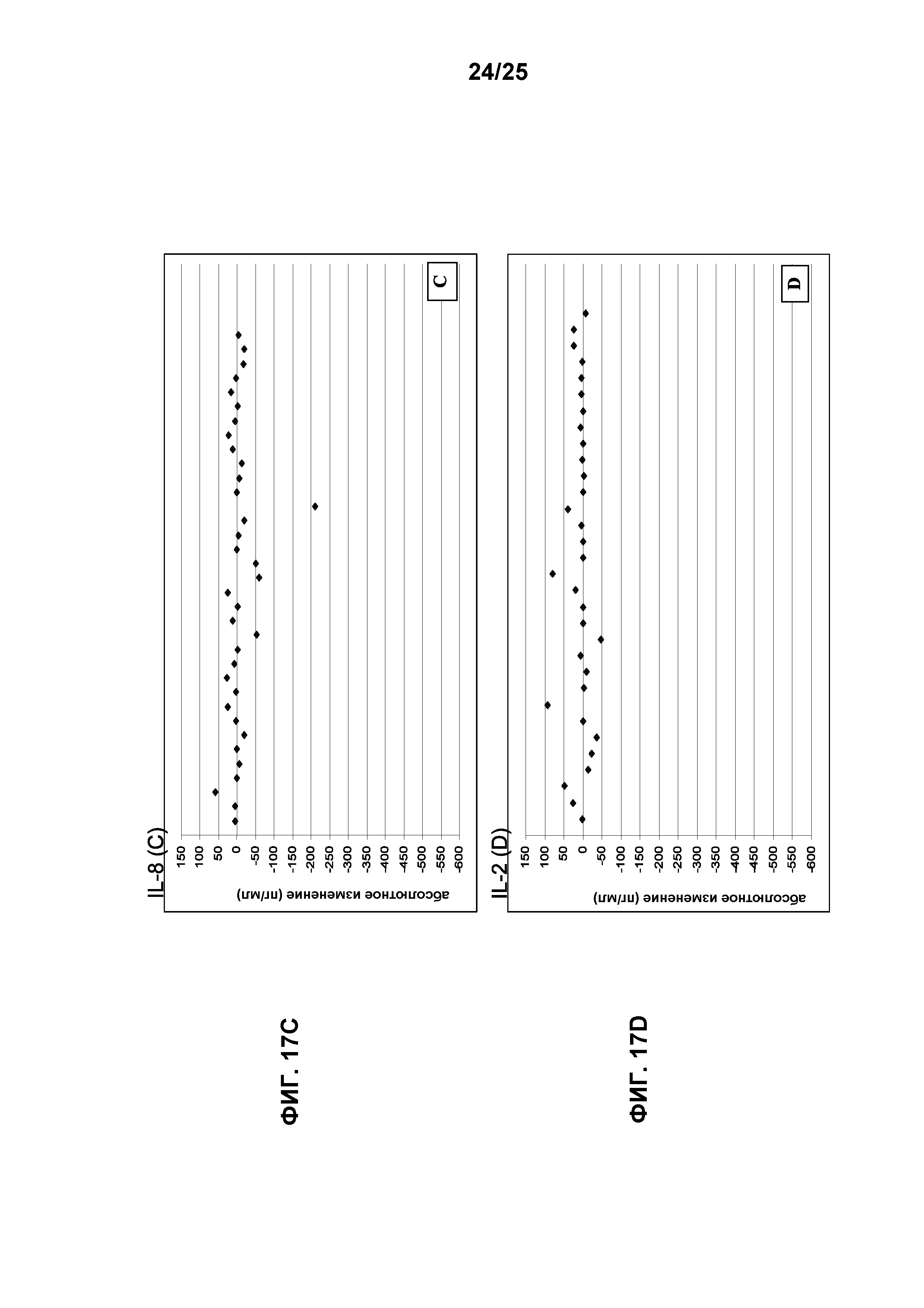
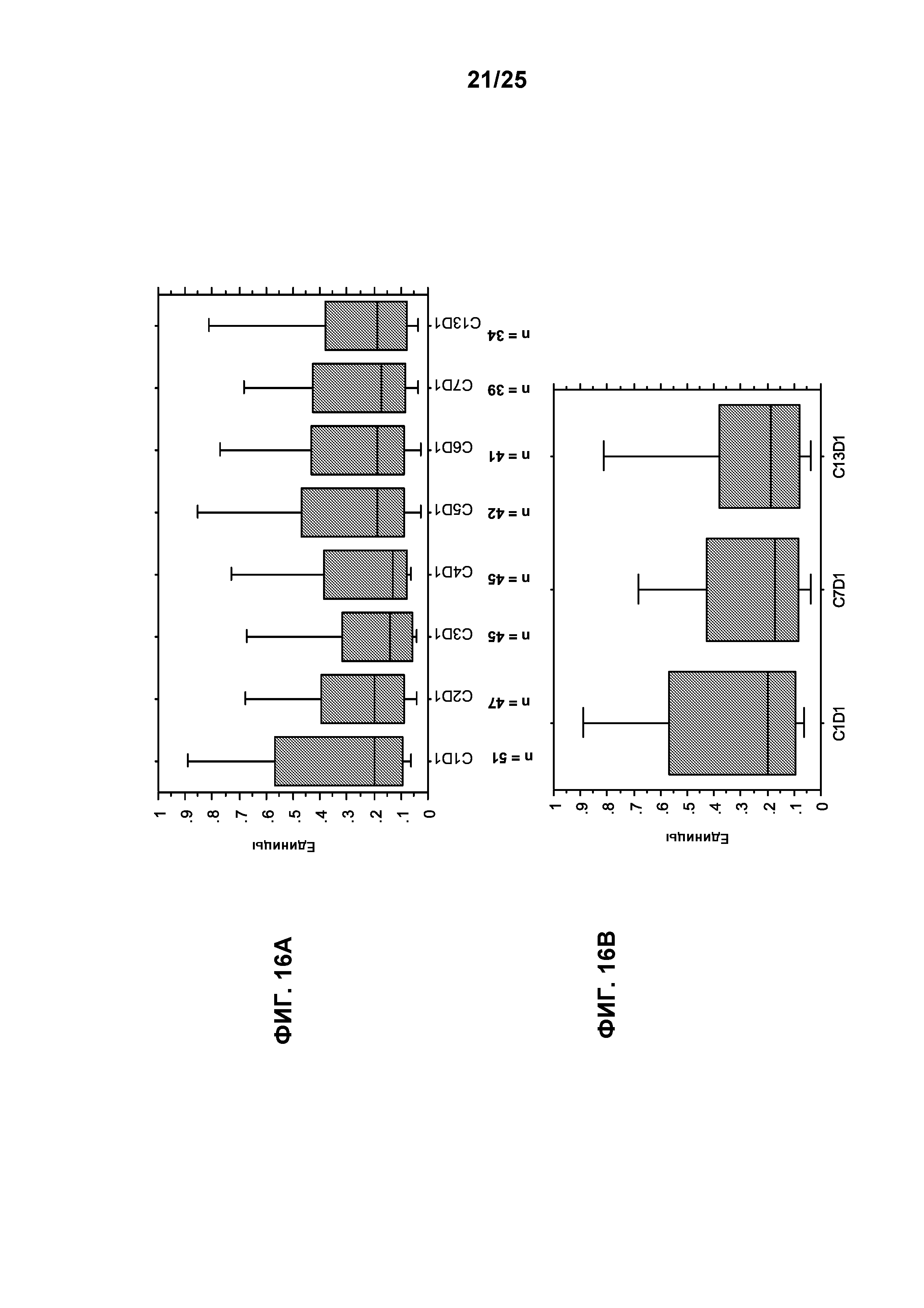
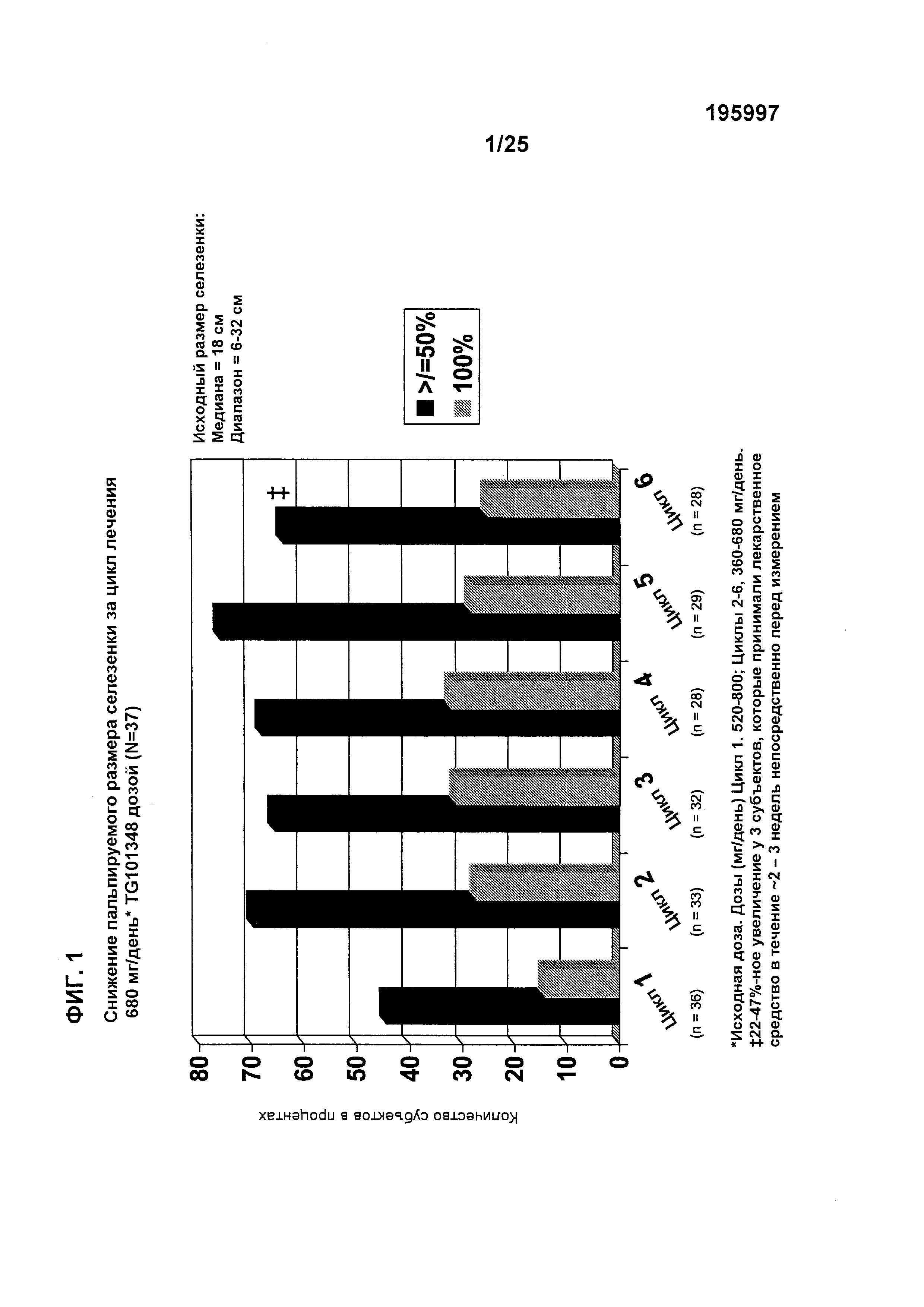
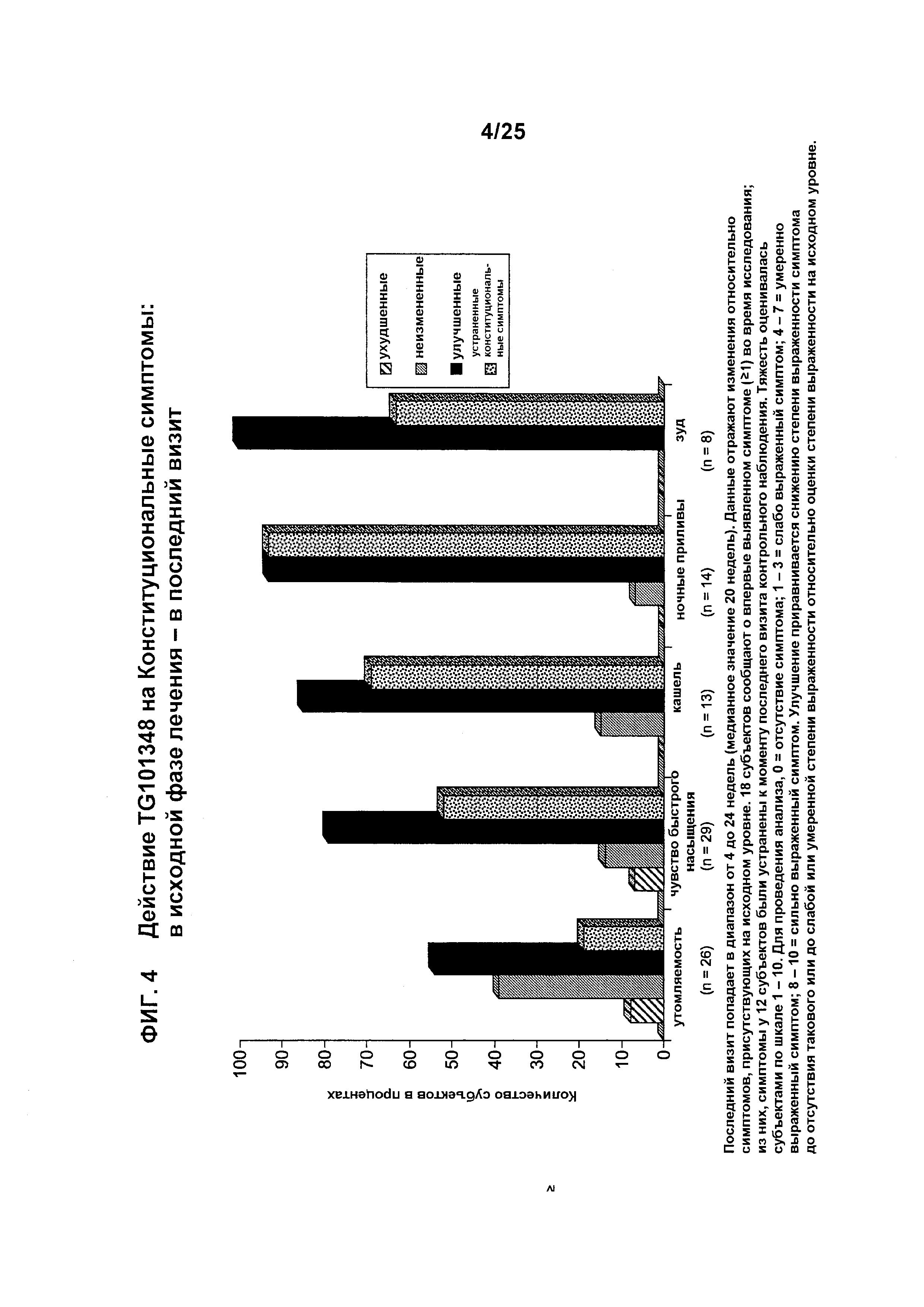

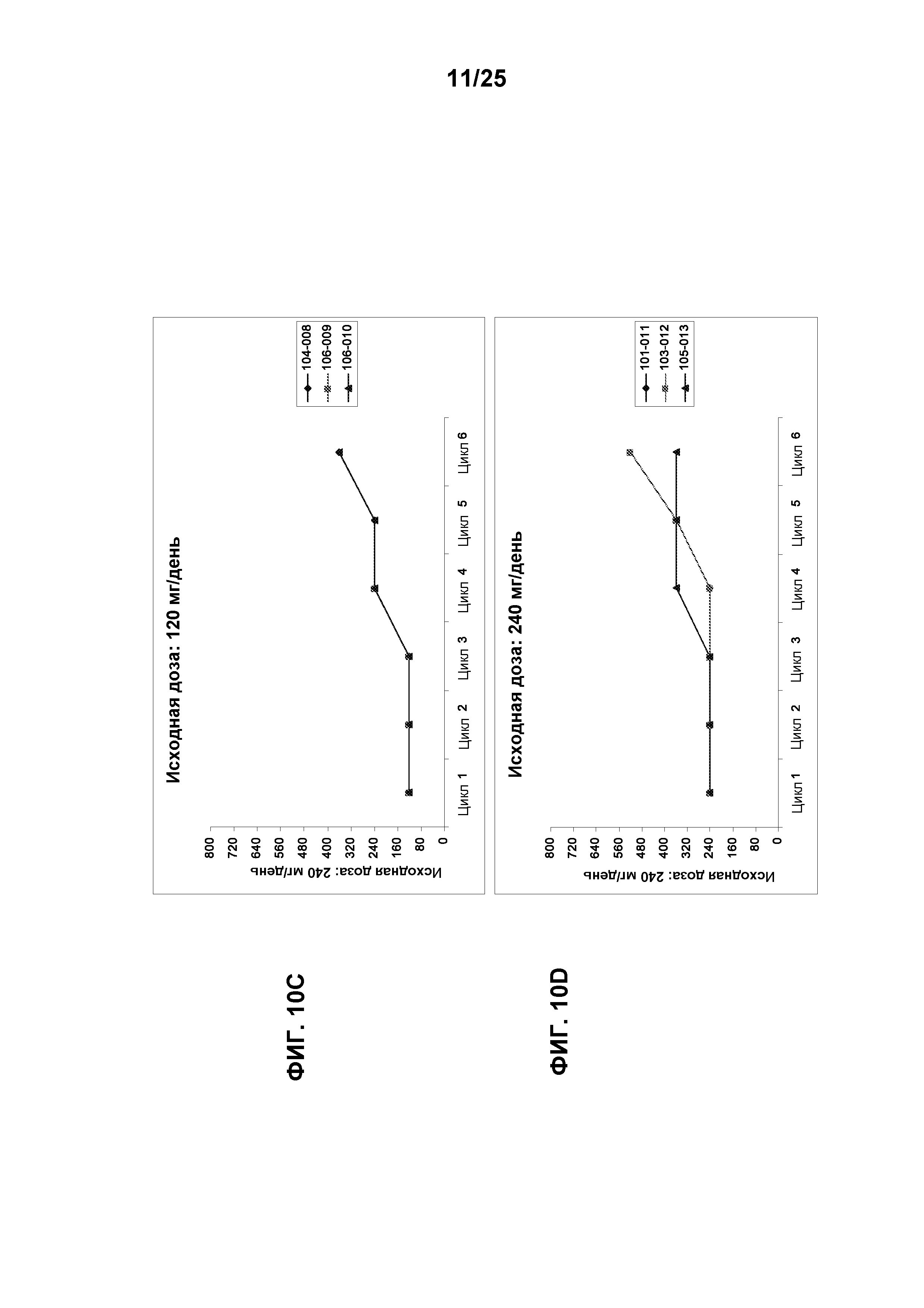
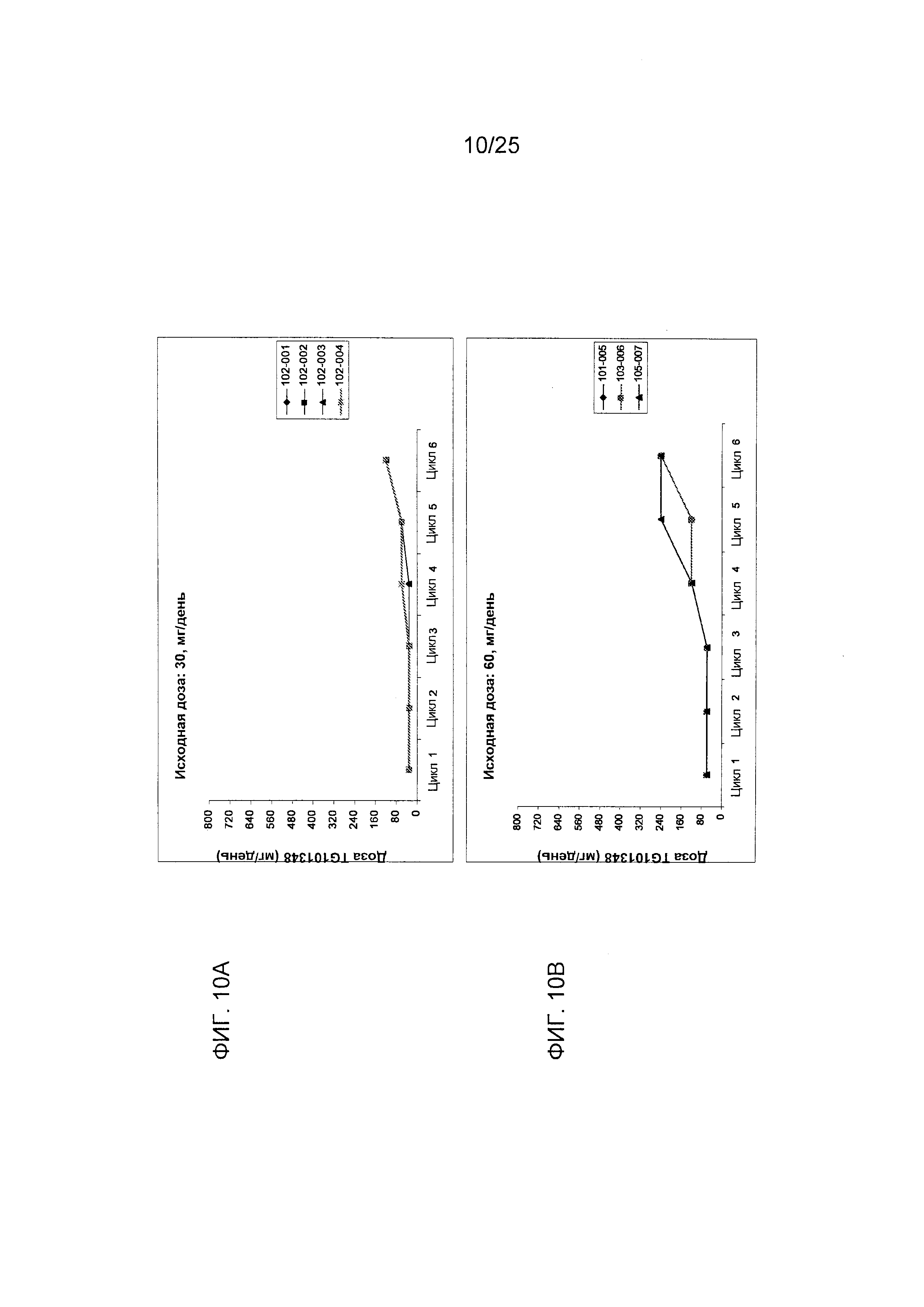
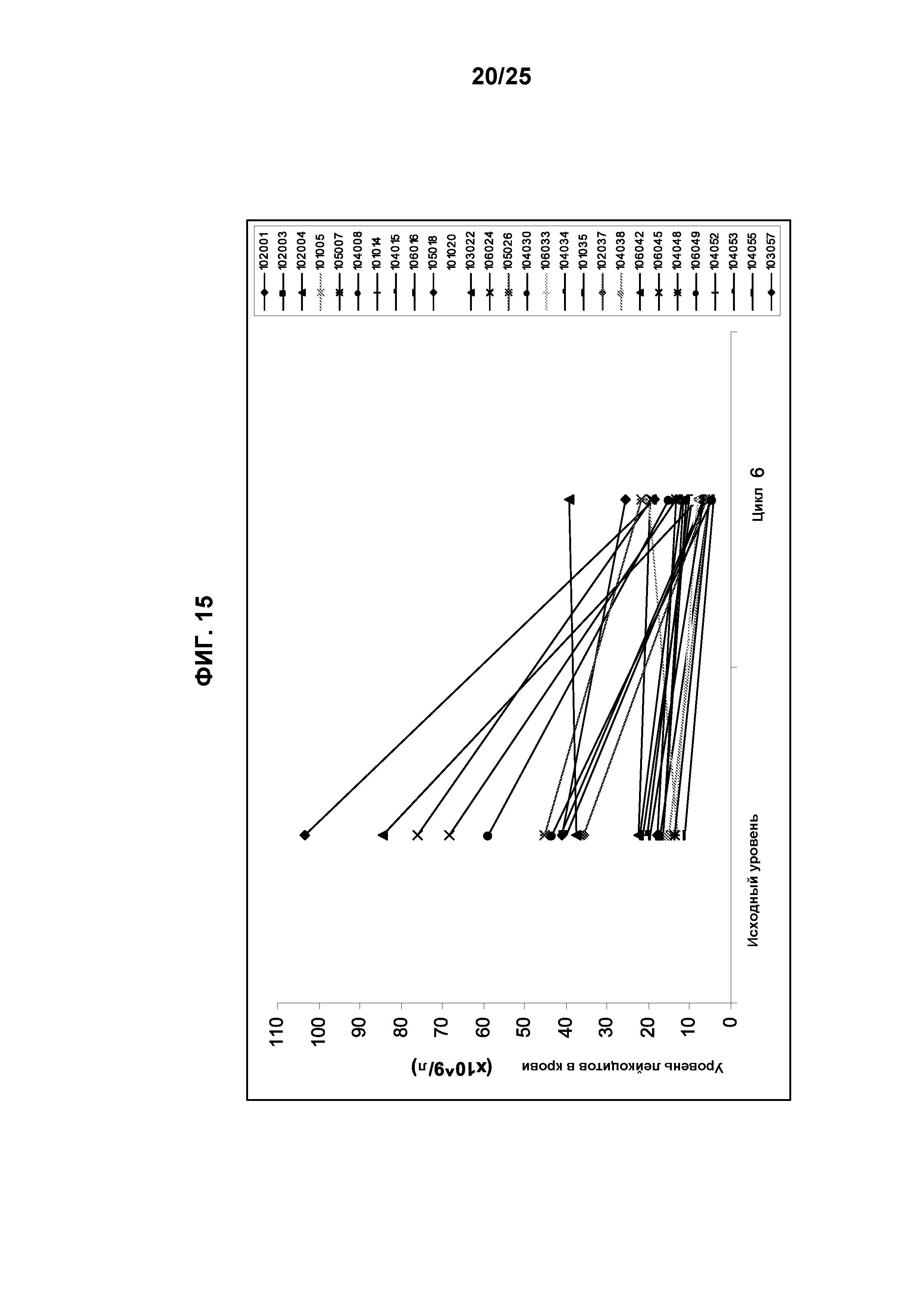


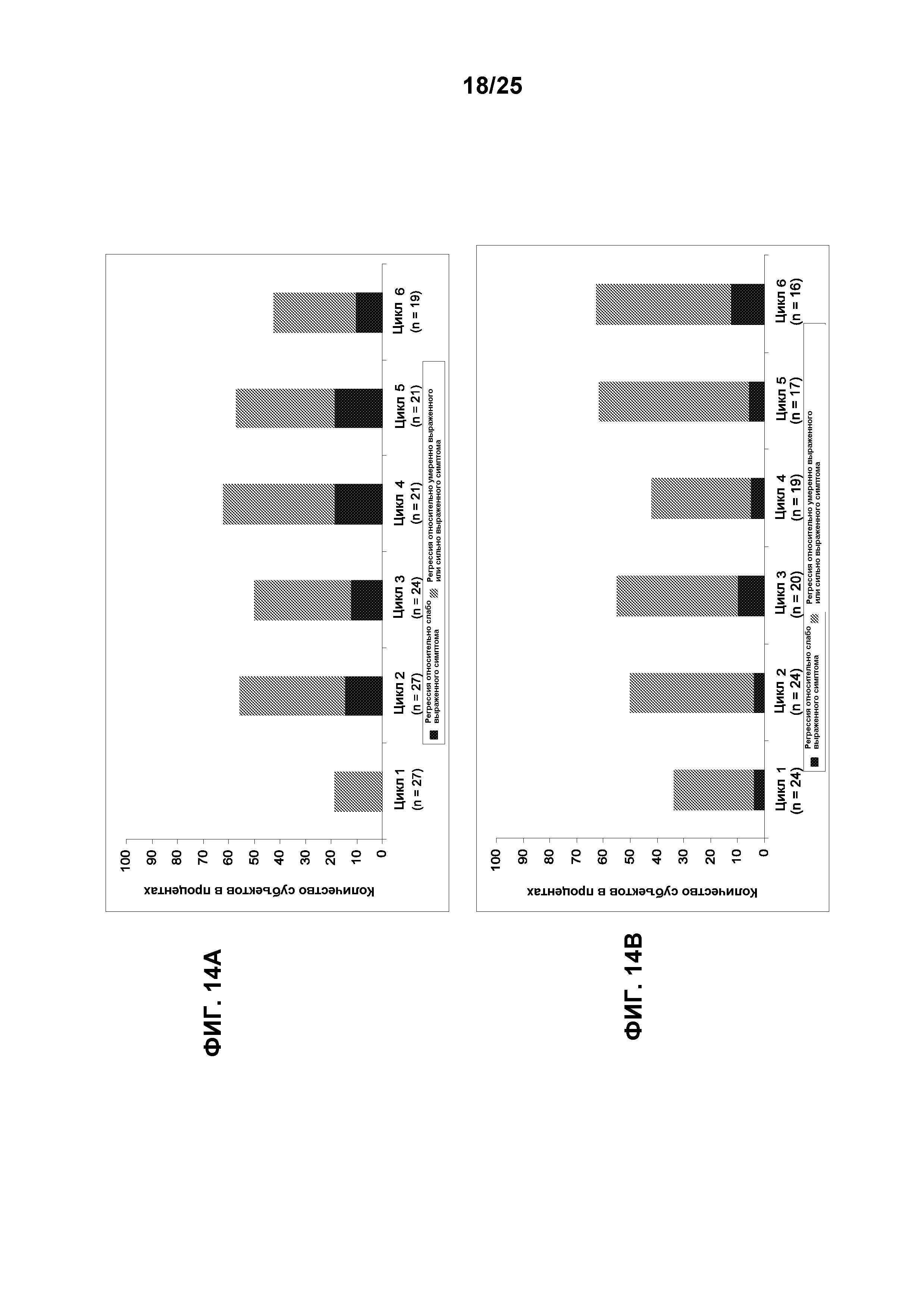

Би-арил-мета-пиримидиновые ингибиторы киназ
Би-арил-мета-пиримидиновые ингибиторы киназ
Би-арил-мета-пиримидиновые ингибиторы киназ
Би-арил-мета-пиримидиновые ингибиторы киназ

