Результат интеллектуальной деятельности: НОВЫЙ ЭФФЕКТИВНЫЙ ИНГИБИТОР КИНАЗЫ 4, АССОЦИИРОВАННОЙ С ИНТЕРЛЕЙКИНОМ-1 (IRAK4)
Вид РИД
Изобретение
Область техники
Изобретение относится к производному N-(пиридинил)тиазол-2-амина селективному ингибитору киназы ИРАК4, который может применяться в терапии и профилактике иммунных воспалительных заболеваний, в частности, ревматоидного артрита, бронхиальной астмы, демиелинизирующих заболеваний центральной нервной системы, включающих аутоиммунный миелосклероз, а также для лечения ряда гематологических онкологических заболеваний, сопровождающихся MyD88 мутациями.
Уровень техники
ИРАК4 киназа является ключевым звеном в передаче сигнала от рецептора интерлейкина-1 (тип I) и некоторых толл-подобных рецепторов, что приводит к выработке цитокинов и активации других генов, находящихся под контролем фактора транскрипции NF-kB, в ответ на проникновение в организм инфекции. Недостаточность этой киназы приводит к нарушению иммунного ответа на бактериальную, грибковую, паразитарную или вирусную инфекции из-за блокады сигнального пути рецептора TLR4.
Эксперименты с нокаутированными по гену ИРАК4 мышами или мутантными мышами, у которых отсутствует киназная активность ИРАК4 (IRAK4 KDKI), а также эксперименты на клетках человеческих субъектов, для которых было обнаружено нарушение экспрессии киназы ИРАК4, продемонстрировали блокирование всех сигнальных путей через рецепторы интерлейкина-1, интерлейкина-18 и толл-подобных рецепторов, за исключением толл-подобного рецептора-3 (Ku, С. et al., 2007, J. Exp. Med. 204, р. 2407-2422; Suzuki, N. et. al. 2002, Nature, 416, p. 750-754, Koziczak-Holbro, M. et al., 2007, J. Biol. Chem. 282, p. 13552-13560; Kawagoe Τ et. al. 2007, J. Exp. Med. 204, p. 1013-1024).
Вместе с этим было обнаружено, что у животных со сниженной активностью ИРАК4 киназы (IRAK4 KDKI) в меньшей степени развивается множественный склероз (Stascke, К.А. et. al., 2009, J. Immunol. 183, p. 568-577), ревматоидный артрит (Koziczak-Holbro, M. et al., 2009, Arthritis Rheum. 60, p. 1661-1671), атеросклероз (Kim, T.W. et. al, 2011, J. Immunol. 186, p. 2871-2880; Rekhter, M. et. al, 2008, Bioch. Bioph. Res. Comm. 376, p. 642-648) и инфаркт миокарда (Maekawa, Y. et. al., 2009, Circulation 120, p. 1401-1414). Эти данные предполагают возможность использования ингибиторов ИРАК4 киназы для лечения указанных заболеваний.
Кроме того, были получены обнадеживающие данные о перспективности использования ингибиторов ИРАК4 киназы для лечения гематологических видов рака с онкогенными мутациями адаптерного белка MyD88, например, диффузной В-крупноклеточной лимфомы (ABC-DLBCL) и макроглобулинемии Вальденстрема (WM) (Yang, G. et. al, 2013, Blood, 122, p. 1222-1232; Ngo, V.N. et. al, 2011, Nature, 470, p. 115-119; Srivastava, R. et. al., 2012, Cancer Res., 72, p. 6209).
Работы по разработке низкомолекулярных ингибиторов ИРАК4 ведутся в настоящее время в таких компаниях, как Nimbus Discovery (ND-2158, ND-2110), Bristol-Meyer Squibb, Astellas (AS2444697), Aurigene, TGTherapeutics (LG 0224912) и Merck (PF-05387252), однако, все эти работы находятся на стадии доклинических исследований, и ни одна из них не доведена до клинических испытаний. Имеющиеся на сегодняшний день низкомолекулярные ингибиторы ИРАК4 описаны в обзорах последнего времени (Hynes, J. Jr. and Nair, S.K., 2014 Annual Reports in Medicinal Chemistry, 49, p. 117-133; Chaudhary, D. et. al, 2015, J. Med. Chem., p. 96-110). К настоящему времени также известны модуляторы ИРАК4 киназы, включающие производные пиразол[1,5А]пиримидина и тиено[3,2В]пиримидина (RU 2013103539), макроциклические соединения, применяющиеся для лечения воспалительных заболеваний (WO 2014143672) и другие соединения. Данные, полученные на моделях животных, свидетельствуют о потенциальном применении низкомолекулярных ингибиторов ИРАК4 киназы для лечения ревматоидного артрита, хронического заболевания почек, псориаза и некоторых видов рака.
Раскрытие изобретения
Задачей (техническим результатом) настоящего изобретения является разработка нового эффективного селективного ингибитора ИРАК4 киназы, обладающего высокой биодоступностью и низкой токсичностью и являющегося перспективным для лечения заболеваний, связанных с аберрантной активностью ИРАК4 киназы, а именно, иммунных воспалительных заболеваний, в частности, ревматоидного артрита, бронхиальной астмы и демиелинизирующих заболеваний центральной нервной системы, в том числе аутоиммунного миелосклероза, а также онкологических заболеваний, сопровождающихся MyD88 мутациями.
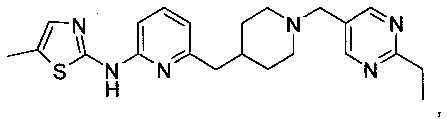
Указанный технический результат достигается путем получения нового соединения N-(6-((1-((2-этилпиримидин-5-ил)метил)пиперидин-4-ил)метил)пиридин-2-ил)-5-метилтиазол-2-амина (ИР-1) следующей структуры:

или его фармацевтически приемлемых солей, гидратов или сольватов, обладающих способностью эффективно селективно ингибировать ИРАК4 киназу.
Достижение указанного технического результата обеспечивается также в результате применения соединений по изобретению для получения фармацевтической композиции для лечения и/или предотвращения патологического состояния, связанного с аберрантной активностью ИРАК4 киназы.
В некоторых вариантах изобретения соединения по изобретению применяют при патологических состояниях, представляющих собой иммунное воспалительное заболевание, в частности, при таких заболеваниях, как ревматоидный артрит, бронхиальная астма или демиелинизирующее заболевание центральной нервной системы, в том числе при аутоиммунном миелосклерозе.
В некоторых других вариантах изобретения соединения по изобретению применяют при патологических состояниях, представляющих собой онкологическое заболевание, в частности, при таких заболеваниях, как В-крупноклеточная лимфома или макроглобулинемия Вальденстрема.
Достижение указанного технического результата обеспечивается также за счет создания фармацевтической композиции для лечения и/или предотвращения иммунных воспалительных заболеваний, онкологических заболеваний, связанных с аберрантной активностью ИРАК4 киназы, характеризующейся тем, что она содержит терапевтически эффективное количество соединения по изобретению и фармацевтически приемлемый носитель, растворитель и/или наполнитель.
В результате проведенных исследований, было синтезировано соединение N-(6-((1-((2-этилпиримидин-5-ил)метил)пиперидин-4-ил)метил)пиридин-2-ил)-5-метилтиазол-2-амин:

Проведенные эксперименты показали, что полученное соединение представляет собой селективный ингибитор ИРАК4 киназы, который проявляет высокую активность (IC50 7-12 нМ) и селективность к ИРАК4 киназе по сравнению с другими киназами (ИРАК-1, р38α, МК-2, SYK), а также с панелью других киназ.
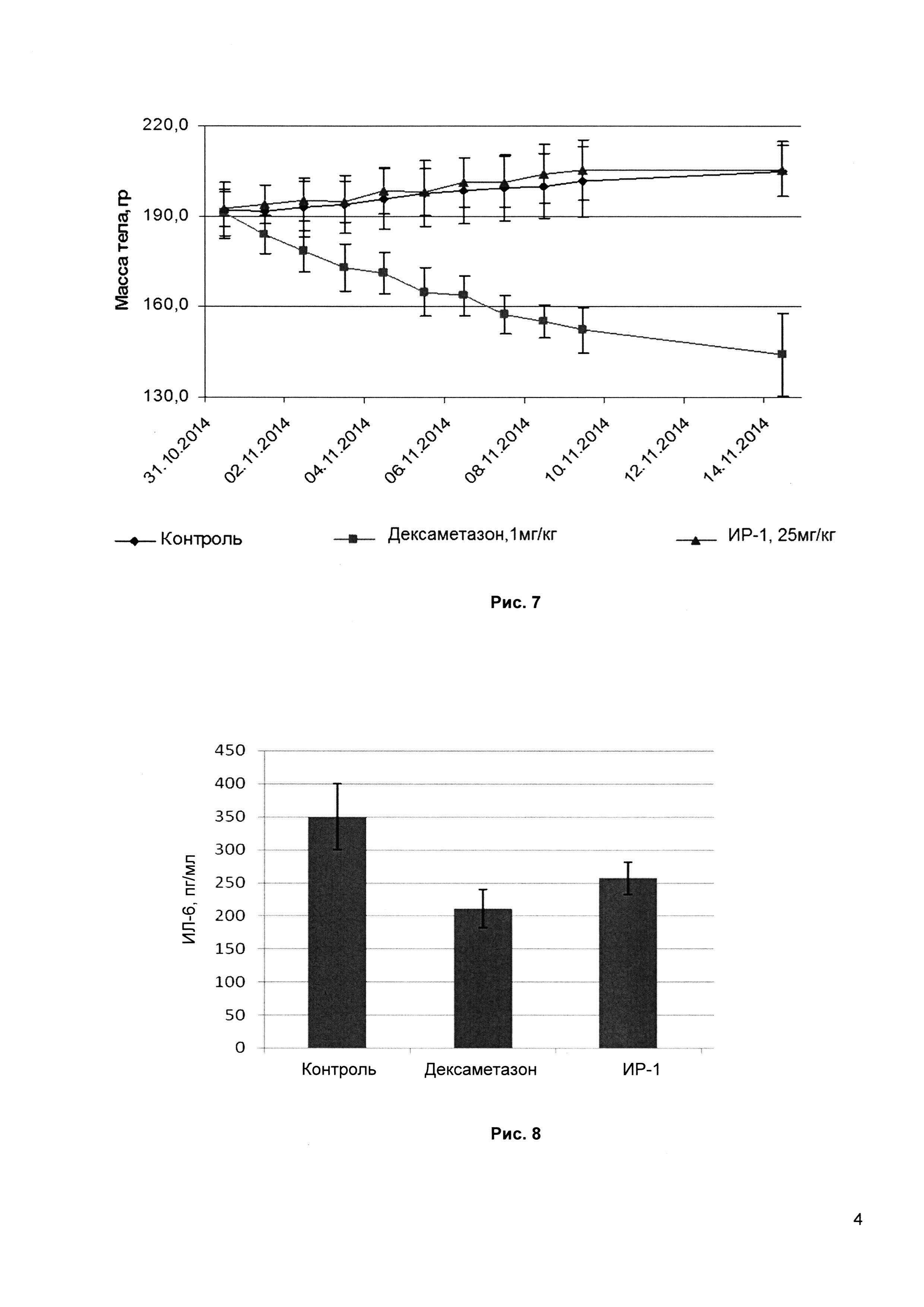
Кроме того, ИР-1 имеет благоприятный ADME профиль (всасывание, распределение, метаболизм и выведение): высокую растворимость в воде, низкое ингибирование цитохромов Сур450, отсутствие блокирования hERG канала, высокую (>90%) биодоступность на грызунах (мыши, крысы) и низкую токсичность (LD50>500 мг/кг на мышах). Субхроническое пероральное введение ИР-1 в максимально переносимой дозе также не выявило токсического действия этого соединения. Было проведено изучение субхронической токсичности на мышах в дозе 100 мг/кг и крысах в дозе 25 мг/кг в течение 14 дней, при этом не было отмечено никаких токсических проявлений соединения ИР-1, состояние животных не отличалось от контрольной группы, а потери массы тела не отмечены. При вскрытии патологий внутренних органов обнаружено не было.
В результате экспериментов также установлено, что ИР-1 ингибирует активность сигнального каскада NF-kB и экспрессию ФНО-альфа на клеточных моделях острой моноцитарной лейкемии ТНР-1. IC50 для ИР-1 составляют 0,244 мкМ по отношению к активности сигнального каскада NF-kB и 0,283 мкМ по отношению к экспрессии ФНО-альфа.
Были исследованы противовоспалительные, анальгетические и жаропонижающие свойства соединения ИР-1 на крысах. В модели «формалинового отека» при однократном введении флогогена ИР-1 проявил более выраженный противовоспалительный эффект, чем Диклофенак (уменьшение отека через 2 часа на 42% по сравнению с 12% для Диклофенака). ИР-1 обладает способностью повышать порог болевой чувствительности (ПБЧ) при гиперальгезии, вызванной введением 4% раствора формалина. На пике действия препарата (через 1 час после введения) ПБЧ снижался на 68,6% по отношению к исходному уровню. В тесте «горячая пластинка», отражающем эффективность в подавлении острой соматической поверхностной боли, ИР-1 практически в 3 раза (189,7%) превосходил показания контрольной группы и более чем в два раза - эффективность стандартного препарата Диклофенак (90,12% по отношению к контролю). Кроме того, ИР-1 проявляет выраженный гипотермический эффект на фоне гипертермии у крыс, вызванной внутрибрюшинным введением раствора пирогенала 10 мгк/мл, значительно превосходящий таковой у Диклофенака. Через два часа после введения исследуемого вещества ИР-1 и Диклофенака отмечено достоверное снижение уровня температуры тела по отношению к собственной исходной температуре на 1,33°С у группы, получавшей соединение ИР-1, на 0,6°С - у группы, получавшей Диклофенак, и на 0,36°С - у контрольной группы.
В проведенных экспериментах ИР-1 в дозах 5, 10 и 30 мг/кг проявил противовоспалительные свойства в модели каррагинанового воспаления у мышей и достоверное подавление отека.
В модели адъювантного артрита у крыс, вызванного введением адъюванта Фрейнда, соединение ИР-1 в дозе 25 мг/кг обладает противовоспалительным действием.
Было обнаружено, что соединение ИР-1 достоверно снижает концентрацию интерлейкина-6 в суставах с индуцированным артритом у крыс (Дексаметазон - на 40%, ИР-1 - на 27%). Однако не обнаружено аналогичного достоверного снижения концентрации фактора некроза опухоли и интерлейкина-1b в модели с индуцированным артритом у крыс, что свидетельствует о том что только ингибирования IRAK4 недостаточно для ингибирования этих факторов, что еще раз подтверждает механизм действия ИР-1 как ингибитора ИРАК4 киназы.
Эксперименты на нокаутированных мышах продемонстрировали, что ингибитор ИРАК4 киназы ИР-1 эффективен в моделях ревматоидного артрита и аутоиммунных заболеваний, а также для лечения гематологических видов рака с онкогенными мутациями адаптерного белка MyD88, например, диффузной В-крупноклеточной лимфомы (ABC-DLBCL) и макроглобулинемии Вальденстрема (WM).
Описание чертежей
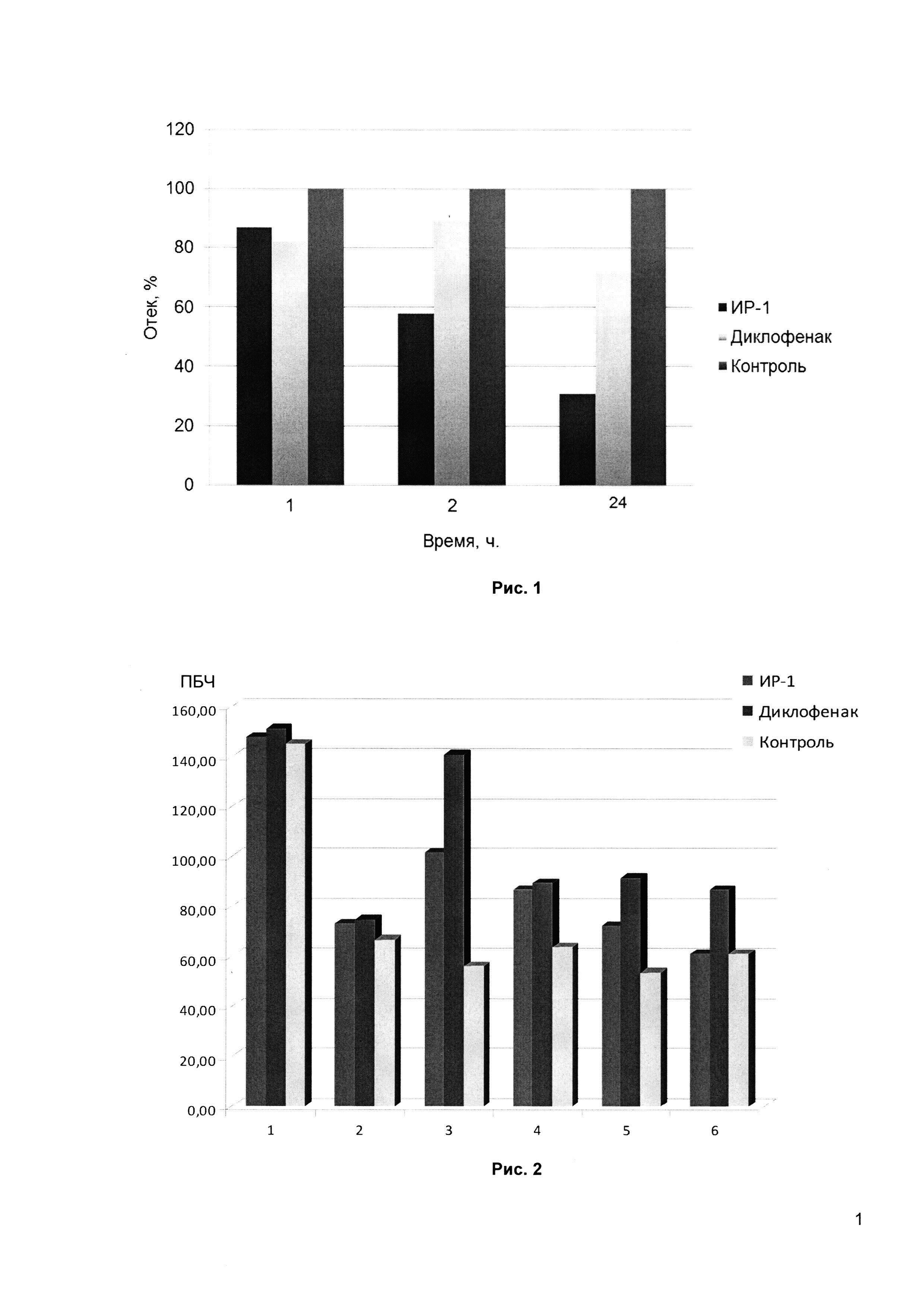
Рис. 1 - динамика противоотечного действия соединения ИР-1 в сравнении со стандартным препаратом Диклофенак и контрольной группой.
Рис. 2 - влияние ИР-1 на динамику изменения порога болевой чувствительности (ПБЧ, в абсолютных показателях приложенного усилия (грамм)). Обозначения: 1 - до введения формалина; 2 - через 2 часа после введения формалина; 3 - через 1 час после введения исследуемых веществ; 4 - через 2 часа после введения исследуемых веществ; 5 - через 3 часа после введения исследуемых веществ; 6 - через 4 часа после введения исследуемых веществ.
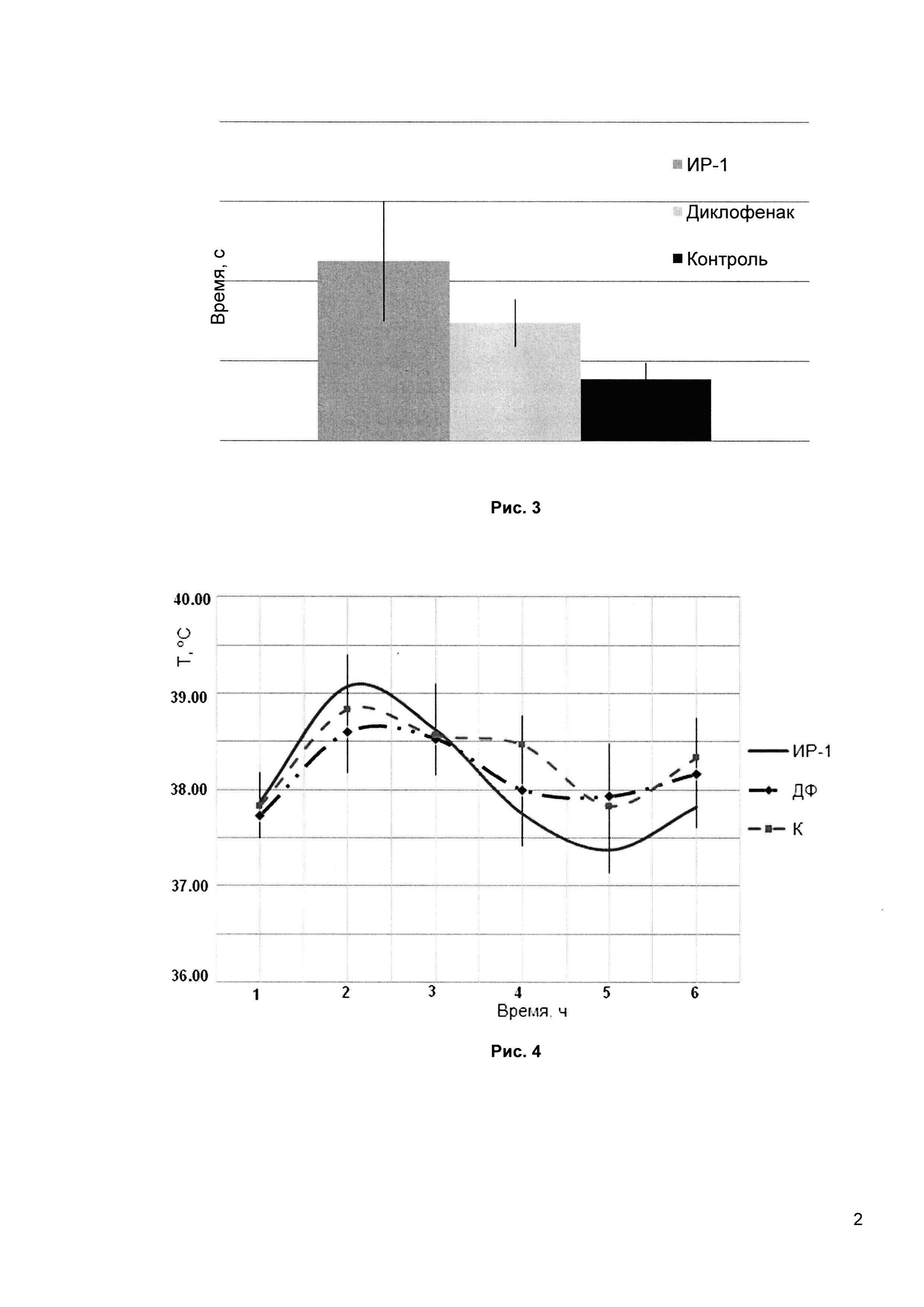
Рис. 3 - анальгетическое действие ИР-1 в модели «горячая пластинка» (сравнение латентного времени первого облизывания задней лапы).
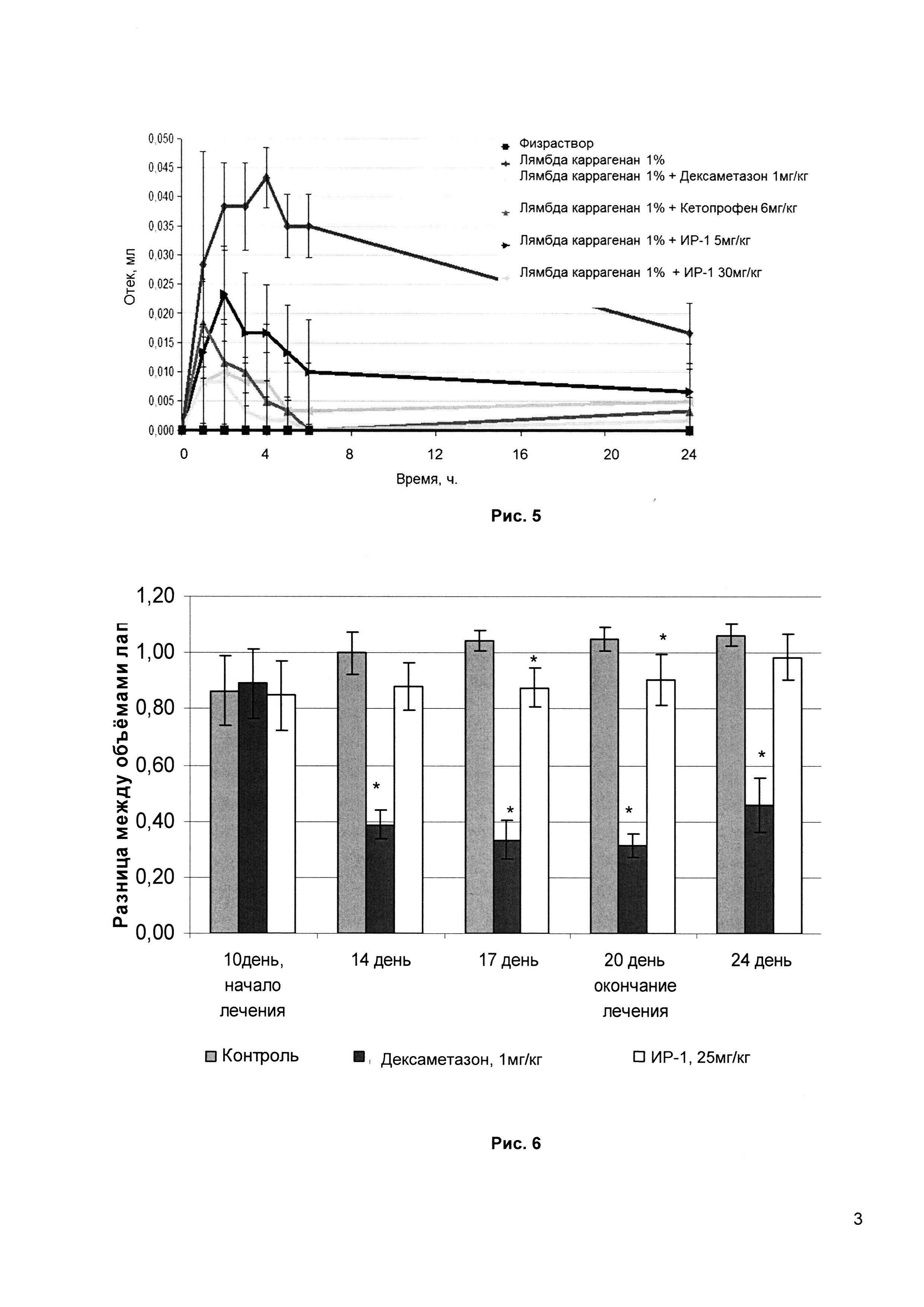
Рис. 4 - жаропонижающее действие ИР-1 в модели пирогеналовой лихорадки у крыс (сравнение динамики температуры тела крыс после введения пирогенала при действии препарата ИР-1 в сравнении со стандартным препаратом Диклофенак и контрольной группой, в °С).
Рис. 5 - противовоспалительное действие ИР-1 в модели каррагинанового воспаления на мышах линии SHK.
Рис. 6 - влияние Дексаметазона и ИР-1 при пероральном введении на адъювант-индуцированный отек лап у крыс (* - при p<0, 05).
Рис. 7 - изменение массы тела крыс при введении ИР-1 в сравнении со стандартным препаратом Дексаметазон и контрольной группой в тесте изучения токсичности.
Рис. 8 - изучение влияния ИР-1 на течение адъювантного артрита у крыс (сравнение концентрации ИЛ-6 в суставах с индуцированным артритом при введении ИР-1, препарата сравнения (Дексаметазона) и контрольной группой).
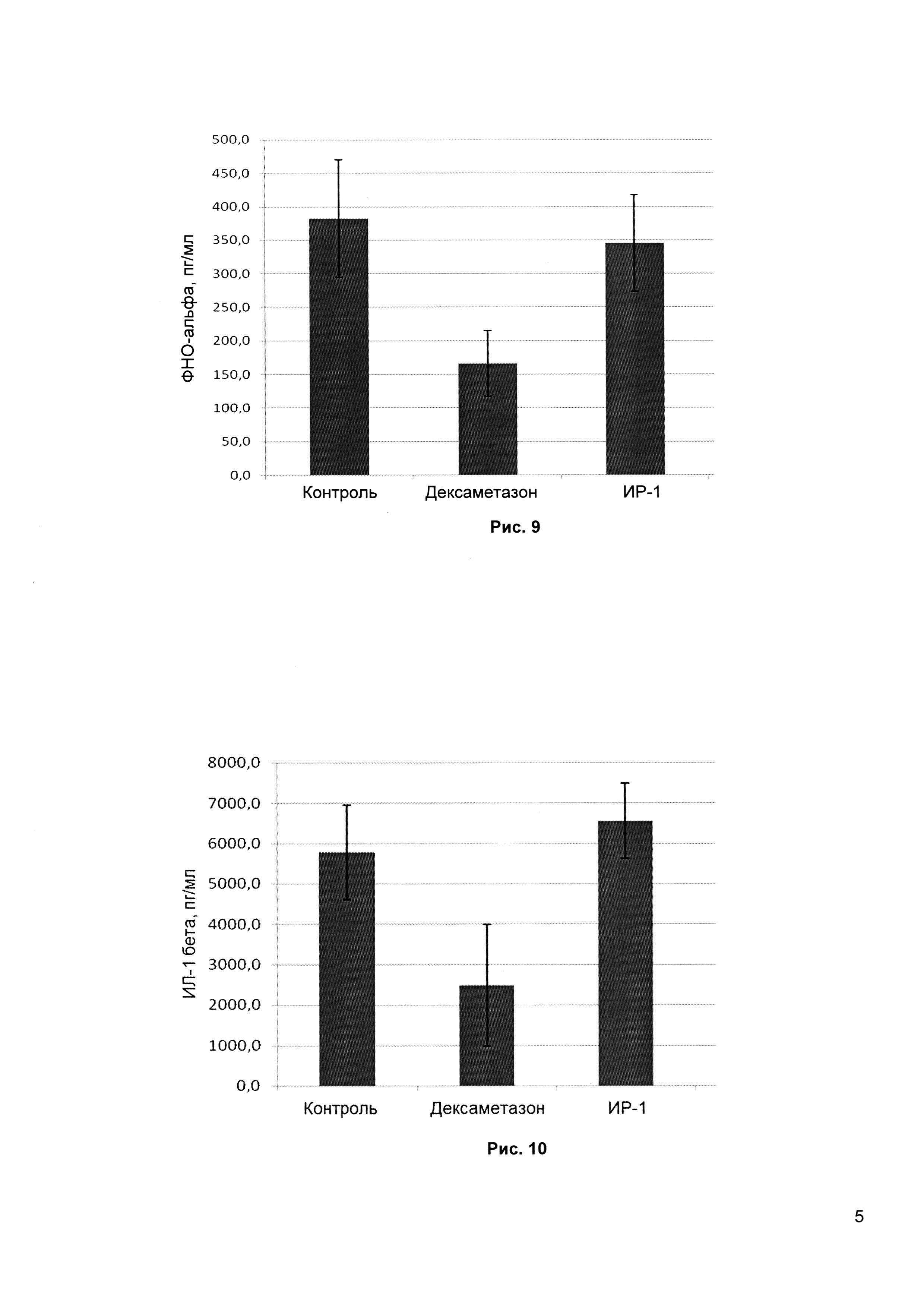
Рис. 9 - изучение влияния ИР-1 на течение адъювантного артрита у крыс (сравнение концентрации ФНО-альфа в суставах с индуцированным артритом при введении ИР-1, препарата сравнения (Дексаметазона) и контрольной группой).
Рис.10 - изучение влияния ИР-1 на течение адъювантного артрита у крыс (сравнение концентрации ИЛ-1b в суставах с индуцированным артритом при введении ИР-1, препарата сравнения (Дексаметазона) и контрольной группой).
Подробное раскрытие изобретения
Термины и определения
Термин «сольват» относится к ассоциации или комплексу из одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин «гидрат» относится к комплексу, где молекулами растворителя является вода.
Соединения настоящего изобретения могут существовать в свободной форме или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, валериат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкил сульфонаты и арил сульфонаты.
Способ терапевтического применения соединений
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения по изобретению. Под терапевтически эффективным количеством подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение (профилактику). Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Соединение по изобретению, или фармацевтическая композиция, содержащая соединение, может быть введено в организм пациента в любом количестве и любым путем введения, эффективным для лечения или профилактики заболевания.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, парентерально, местно и т.п.
Эффективная системная дозировка соединения, вводимая разово или в виде нескольких отдельных доз, как правило, лежит в диапазоне от 0,01 до 500 мг/кг, предпочтительно от 0,1 до 125 мг/кг, еще более предпочтительно, от 1 до 100 мг/кг. Обычно соединение вводится пациенту, нуждающемуся в таком лечении, в дневной дозировке ориентировочно от 50 до 2000 мг на пациента. Введение может осуществляться как разово, так и несколько раз в день, неделю (или любой другой временной интервал), или время от времени. Кроме того, соединение может вводиться в организм пациента ежедневно в течение определенного периода дней (например, 2-10 дней), а затем следует период без приема вещества (например, 1-30 дней).
В том случае, когда соединение по изобретению используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов.
Фармацевтические композиции
Изобретение также относится к фармацевтическим композициям, которые содержат ИР-1 (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, таких, которые могут быть введены в организм пациента совместно с соединением, составляющем суть данного изобретения, и которые не разрушают фармакологической активности этого соединения, и являются нетоксичными при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахариды, а также их производные; желатин; тальк; эксципиенты, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе, например, для введения в организм орально, местно, внутриглазным способом, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, подкожно, внутримышечно, а также инфузионным способом, в рекомендованных дозировках.
Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом, методами микрокапсулирования, методами приготовления наноформ препарата, или другими методами, известными в фармацевтике.
При получении композиции, например в форме таблетки, активное начало смешивают с одним или несколькими фармацевтическими эксципиентами, такими как желатин, крахмал, лактоза, стеарат магния, тальк, кремнезем, аравийская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки можно покрыть сахарозой, целлюлозным производным или другими веществами, подходящими для нанесения оболочки. Таблетки могут быть получены различными способами, такими как непосредственное сжатие, сухое или влажное гранулирование или горячее сплавление в горячем состоянии.
Фармацевтическую композицию в форме желатиновой капсулы можно получить, смешивая активное начало с растворителем и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным путем используются водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат фармакологически совместимые агенты, например пропиленгликоль или бутиленгликоль.
Для примера, стандартная лекарственная форма, содержащая соединение по изобретению в форме таблетки, может содержать следующие ингредиенты:

Синтез соединений по изобретению
Общая схема синтеза ИР-1:

Схема синтеза интермедиата (1)

Схема синтеза интермедиата (2)

Материалы и методы
Для синтеза соединения ИР-1 использовались коммерчески доступные реагенты (с чистотой не ниже 98%) и растворители (российское представительство Sigma Aldrich, г. Москва) без дополнительной очистки, если не указано иное. Растворители для хроматографии имели чистоту «для ВЭЖХ» и применялись без дополнительной очистки. Протекание реакций контролировали при помощи тонкослойной хроматографии (ТСХ) с использованием пластин Merck Silica Gel 60 F-254. Колоночную флэш-хроматографию выполняли на силикагеле Merck silica gel 60 (0.015-0.040 мм).
ЖХ/МС (LC/MS) анализ соединения выполняли на приборе Surveyor MSQ (Thermo Fisher Scientific) с использованием химической ионизации при атмосферном давлении при следующих условиях: колонка ZORBAX Eclipse XDB-C18 2.1×15 мм, 1.8 мкм; 0.1% раствор муравьиной кислоты в воде/ацетонитрил.
Спектр 1Н-ЯМР был снят на «MERCURY plus 400 MHz» спектрометре (Varian). Значения химических сдвигов в массовых долях (м.д.) даны относительно тетраметилсилана (ТМС).
Препаративная ВЭЖХ очистка выполнялась на Agilent 1100 Series, с использованием колонки Luna С18(2) (Phenomenex, USA), 250×21.2 мм, мкм.
Синтез интермедиата (1)
Синтез соединения (3), синтетическая стадия а:
К суспензии метилтрифенилфосфоний иодида (50 ммоль) в безводном тетрагидрофуране (ТГФ) (100 мл) небольшими порциями добавляют гидрид натрия (75 ммоль, 40% суспензия в масле) в инертной атмосфере аргона при 5°С. Реакционную смесь перемешивают 1 час при 10°С. После этого к реакционной смеси при 5°С добавляют раствор трет-бутил-4-оксопиперидин-1-карбоксилата (45 ммоль) в безводном ТГФ (20 мл). Получившуюся реакционную смесь перемешивают при комнатной температуре 12 часов. После окончания реакции реакционную смесь вылиют в воду (150 мл) и экстрагируют дихлорметаном (2×70 мл). Органический слой отделяют, высушивают над безводным сульфатом натрия и упаривают в вакууме на роторном испарителе (LABOROTA 4000, Heidolph, Германия). Маслообразный остаток очищают колоночной хроматографией на силикагеле (элюент: смесь гексана и этилацетата 10:1). Выход: 80.2%.
Синтез соединения (4), синтетическая стадия b:
К раствору алкена (3) (0.1 моль) в 80 мл безводного ТГФ под атмосферой аргона через капельную воронку в течение 5 минут добавляют 9-борабицикло[3.3.1]нонан (0.1 моль, 200 мл, 0.5М раствор в ТГФ). Полученный бесцветный раствор перемешивают при комнатной температуре 24 часа. После этого добавляют 2,6-дибромпиридин (0.12 моль), карбонат калия (110.4 г, 0.8 моль), воду (80 мл) и Pd(PPh3)4 (5.5 г, 0.0067 моль). Полученную реакционную смесь кипятят 3 часа, затем охлаждают до комнатной температуры и выливают в воду со льдом (500 г). Полученную смесь несколько раз экстрагируют этилацетатом (3×100 мл). Объединенные органические экстракты высушивают над безводным сульфатом натрия и упаривают в вакууме роторного испарителя. Маслообразный остаток очищают колоночной хроматографией на силикагеле (элюент: этилацетат-гексан 1:10). Выход 81%.
Синтез соединения (5), синтетическая стадия с:
Смесь 5-метил-тиазол-2-иламина (10 ммоль), соединения (4) (10 ммоль), 9,9 диметил-4,5-бис(дифенилфосфино)ксантена (4% моль), комплексного катализатора трис(дибензилиденацетон)дипалладий (0) - хлороформ (2% моль), карбоната натрия (15 ммоль) в смеси толуол/вода (8:1, 80 мл) нагревают при температуре 100°С 16 часов при интенсивном перемешивании (ТСХ контроль) в атмосфере аргона. После охлаждения реакционной смеси до комнатной температуры к ней добавляют воду (100 мл) и экстрагируют этилацетатом (1×40 мл). Объединенные органические экстракты высушивают над безводным сульфатом натрия и отфильтровывают через тонкий слой силикагеля. Фильтрат упаривают в вакууме роторного испарителя (LABOROTA 4000, Heidolph, Германия). Маслообразный остаток очищают колоночной хроматографией на силикагеле (элюент: н-гексан - этилацетат 3:2). Выход: 35%.
Синтез соединения (1), синтетическая стадия d:
В плоскодонную колбу на 100 мл, снабженную магнитной мешалкой, помещают раствор соединения (5) (3.5 ммоль) в безводном метаноле (20 мл), добавляют 18% раствор (по массе) хлороводорода в 1,4-диоксане (20 мл). Смесь перемешивают 24 часа при комнатной температуре. Затем растворитель отгоняют в вакууме роторного испарителя (LABOROTA 4000, Heidolph). Остаток затирают с холодным диэтиловым эфиром (50 мл). Образовавшийся белый осадок дигидрохлорида (1) отфильтровывают, промывают небольшим количеством диэтилового эфира, высушивают в вакуумном шкафу до постоянной массы при комнатной температуре. Выход: 95%.
Синтез интермедиата (2) 2-этилпиримидин-5-карбоксальдегида
Интермедиат (2) был синтезирован в соответствии с методом, описанным в литературе (J. Med. Chem. 2004, 47, 4829-4837):
В трехгорлую круглодонную колбу емкостью 2 л, снабженную магнитной мешалкой, иммерсионным термометром и дополнительной капельной воронкой, помещают ДМФА (209 мл, 2700 ммоль) и охлаждают до 0°С. К реакционной смеси через капельную воронку осторожно добавляют оксихлорид фосфора (54,8 мл, 590 ммоль), поддерживая температуру реакционной смеси в интервале 5-10°С. Через 2 часа получившийся красно-оранжевый раствор обрабатывают бромуксусной кислотой (25,0 г, 184 ммоль) и нагревают до 90°С в течение 6 часов. По мере нагревания смеси реакция становится экзотермической и выделяется углекислый газ. Смесь охлаждают и к колбе присоединяют короткий холодильник. Под высоким вакуумом при 120°С отгоняют ДМФА, получая красно-оранжевое масло. Смолистый остаток охлаждают до комнатной температуры и выливают в лед (5 г). Добавляют водный NaBF4 (40 г в 80 мл H2O). При растворении твердого остатка происходит бурная экзотермическая реакция. Экзотермический процесс контролируют периодическим погружением смеси в ледяную баню. Смесь охлаждают до 0°С, и образовавшийся желто-оранжевый осадок отфильтровывают, а затем, для того, чтобы удалить избыток NaBF4, растворяют в горячем ацетонитриле (1 л) и снова отфильтровывают. Фильтрат охлаждают до -30°С, и выпавший кристаллический осадок (28 г) отфильтровывают. Выпавшая вторая порция осадка составляет 7 г винамидиниевой соли (всего 35 г, выход 53%).
Суспензию винамидиниевой соли (20,0 г, 78,5 ммоль) и пропанимидамида гидрохлорида (86.2 ммоль) в этаноле (2000 мл) обрабатывают раствором этилата натрия (246 ммоль) при комнатной температуре. Смесь кипятят 3 часа, охлаждают до комнатной температуры и упаривают на роторном испарителе (температура бани 30°С; давление 40 мм рт.ст.). Остаток растворяют в воде (100 мл), экстрагируют дихлорметаном (3×200 мл). Объединенные органические экстракты высушивают над безводным сульфатом натрия, отфильтровывают и упаривают.
Остаток очищают при помощи флэш-хроматографии на силикагеле (элюент: этилацетат - гексан 2:5). Было получено 5.87 г (55%) продукта (масло, Rf 0.6, этилацетат - гексан 2:5).
Синтез соединения ИР-1
В плоскодонную колбу объемом 100 мл, снабженную магнитной мешалкой, помещают интермедиат (1) (3 ммоль), интермедиат (2) (3,3 ммоль), триацетоксиборогидрид натрия (4 ммоль) в смеси с 50 мл безводного дихлорметана и 50 мл ТГФ. Реакционную смесь перемешивают 24 часа при комнатной температуре (контроль протекания реакции осуществляют по ТСХ), затем добавляют концентрированный раствор карбоната калия (100 мл) и отделяют нижний органический слой. Водную фаза экстрагируют дихлорметаном (2×40 мл). Объединенные органические экстракты высушивают над безводным сульфатом натрия и отфильтровывают через тонкий слой силикагеля. Фильтрат упаривают в вакууме роторного испарителя (LABOROTA 4000, Heidolph, Германия). Маслообразный остаток очищают колоночной хроматографией на силикагеле (элюент: дихлорметан - метанол 20:1).
В результате проведенного синтеза было получено белое кристаллическое вещество с Тпл174-176°С. 73%. 1Н-ЯМР, м.д., J (Гц), d6-ДМСО: 10,77 (уш.с, 1Н), 8.56 (с, 2Н), 7,51 (дд, J=8,2; 7,3 Гц, 1Н), 6,98 (кв, J=1,3 Гц, 1Н), 6.67 (д, J=7.2 Гц, 1Н), 3.43 (с, 2Н), 2,85 (кв, J=7,6 Гц, 2Н), 2,79-2,70 (м, 2Н), 2.61 (д, J=7,1 Гц, 2Н), 2,30 (д, J=1,3 Гц, 3Н), 2,00-1,88 (м, 2Н), 1,68-1,47 (м, 2Н), 1,25 (т, J=7,6 Гц, 3Н). 1,29-1,19 (м, 2Н); 13С-ЯМР, м.д., d6-ДМСО: 169,65; 158,05; 157,46; 157,01; 151,10; 137,49; 134,10; 128,21; 123,01; 114,62; 107,34; 56,55; 52,81; 43,60; 35,06; 31,59; 31,30; 12,17; 10,85; Элементный анализ, рассчитано для C22H28N6S: Найдено %: С 63,96, H 6,95, N 20,51. Вычислено %: С 64,18, H 6,91, N 20,57.
Характеристика биологической активности соединения ИР-1
1. Определение ингибирующего действия ИР-1 на киназную активность ИРАК4
Взаимодействие соединения ИР-1 с киназой ИРАК4 определяли методом HTRF анализа (homogeneous time resolved fluorescence - гомогенная флуоресценция с временным разрешением).
Реакцию по определению киназной активности проводили в 96-луночных планшетах (Costar, 3694) в реакционном буфере (20 мМ HEPES, рН 7.0, 15 мМ MgCl2, 2 мМ DTT, 0.2 мМ Na3VO4, 0.005% Triton Х-100) в течение 60 минут при 30°С и интенсивном перемешивании. Конечная концентрация компонентов реакции (в одной лунке): ИРАК4 киназы - 5 нг, биотинилированный субстрат S1-STK (CisBio, 62ST2PEC) - 200 нМ, АТР (аденозинтрифосфат) (Sigma, А6419) - 10 мкМ, ИР-1 - 10 мкМ, 5% ДМСО - 10 мкМ.
Для детекции фосфорилированного субстрата к реакционной смеси добавляли буферную смесь (CisBio, 62SDBRDF), содержащую антитела, меченные европием (CisBio, 62ST2PEC), и флюорофор Sa-XL665 (CisBio, 610SAXLG). Конечные концентрации: 0.5Х буферной смеси, 0.05Х антитела, 50 нМ Sa-XL665. Инкубацию с антителами проводили в течение 2 часов при комнатной температуре и интенсивном перемешивании. Флюоресценцию определяли при λвозбуждения = 340 нм и λэмиссии = 615 нм и λэмиссии = 665 нм с использованием планшетного спектрофотометра (Tecan Infinite F500).
Полученные результаты приведены в таблице 1:

Также в этом эксперименте определяли ингибирующую активность в отношении активности ИРАК4 киназы ближайших структурных аналогов соединения ИР-1 (см. таблицу 2). В результате проведенного эксперимента неожиданно было установлено, что соединение ИР-1 обладает активностью в десятки раз большей, чем его аналоги.

2. Изучение влияния ИР-1 на острое экссудативное воспаление в модели формалинового отека лапы у крыс
Острую воспалительную реакцию (отек) воспроизводили субплантарным (подподошвенный или плантарный апоневроз) введением 0,05 мл 4% раствора формалина. Выраженность воспалительной реакции оценивали через 1, 3 и 24 часа после индукции воспаления по изменению объема лапы онкометрически (определяли объем воды, вытесненной из полностью заполненного сосуда лапой животного). Исследуемое вещество (ИР-1) и вещество сравнения (Диклофенак) вводили в помощью зонда в желудок животных за 1 час до введения формалина. ИР-1 вводили в дозе 10% от ЛД50. Контрольную группу составляли животные, которым ни исследуемое вещество, ни препарат сравнения не вводились.
Противовоспалительный эффект оценивали по уменьшению отека, выраженного в процентах к контролю. Расчет производили по следующей формуле:
100-(ΔVИР-1/ΔVконтроль)*100%;
где ΔVИP-1 - средний прирост объема лапы для данной временной точки по группе ИР-1,
ΔVконтроль - средний прирост объема лапы для данной временной точки по группе контроля.
Соответствующий расчет был произведен и в группе, получавшей Диклофенак.
Критерием эффективности по данному тесту может служить достоверное уменьшение отека лапы не менее чем на 30% по сравнению с контролем. Исследуемое вещество ИР-1 проявляет достоверную противовоспалительную активность по результатам данного теста, и в сравнении с контрольной группой, и по отношению к Диклофенаку.
По результатам теста «формалинового отека» ИР-1 при однократном введении флогогена обладает более выраженным противовоспалительным действием, чем Диклофенак. Уменьшение отека в группе животных, получавшей препарат ИР-1 по сравнению с контрольной группой, через 1 час составляет 12,2%, через 2 часа - 42% и через 24 часа - 68,37%, а в группе животных, получавших Диклофенак, - 17,1%, 12% и 28,74% соответственно (Рис. 1).
3. Изучение анальгетического действия ИР-1 в модели воспалительной гиперальгезии у крыс
Воспалительную гиперальгезию (повышение болевой чувствительности воспаленных тканей) вызывали субплантарным введением 0,05 мл 4% раствора формалина (контрольной группе вводили эквивалентное количество изотонического раствора NaCl) и оценивали по снижению порога болевой чувствительности (ПБЧ) на механическое раздражение тканей лапы животного до введения формалина и после него. Измерение проводили на воспаленной лапе с использованием прибора «Давление на лапу Рандалд-Селито» производства фирмы IIТС Incorporated Life Science, USA, позволяющего обеспечить плавное увеличение нагрузки на воспаленную лапу до появления болевой реакции (оценивается по писку животного). Приложенная нагрузка в граммах выводится на дисплей прибора. Препараты вводили внутрижелудочно через 2 часа после введения формалина. Исследуемое вещество вводили в средней эффективной дозе ЕД50=25 мг/кг. Стандартный препарат Диклофенак вводили в дозе 8 мг/кг крысы.
Оценку ПБЧ проводили до введения формалина, через 2 часа после введения формалина (до введения препаратов), через 1, 2, 3 и 4 часа после введения препаратов. Анальгетический эффект оценивали по снижению гиперальгезии через 1, 2, 3 и 4 часа после введения препарата по отношению к исходному уровню ПБЧ. Критерием эффективности данного теста считается снижение гиперальгезии не менее чем на 50% от исходного уровня.
Исследуемое вещество ИР-1 обладает способностью повышать порог болевой чувствительности при гиперальгезии, вызванной введением 4% раствора формалина. На пике действия препарата (через 1 час после введения) ПБЧ снижался на 68,6% по отношению к исходному уровню, а у диклофенака - на 93,17% (Рис. 2), что свидетельствует о большем анальгетическом эффекте ИР-1.
4. Изучение анальгетического действия ИР-1 в модели «горячая пластинка» у крыс
Тест является базисным для исследования анальгетической активности и выявляет активность соединений в подавлении острой соматической поверхностной боли. После помещения на горячую поверхность, при достижении порога болевой чувствительности, животное проявляет беспокойство в виде двигательных реакций: отдергивания лап, облизывания подушечек лап и подпрыгивания. Возможные критерии эффективности: латентное время облизывания передних лап, латентное время облизывания задних лап, латентное время первого подпрыгивания (любая попытка оторвать от поверхности одновременно все четыре конечности).
Температуру устанавливали на уровне 55°С, что соответствует стандартной методике проведения теста. Регистрировали латентное время первого облизывания задней конечности.
Препараты вводили внутрижелудочно за 1 час до проведения теста в средней терапевтической дозе: соединение ИР-1 - 25 мг/кг; препарат Диклофенак - 8 мг/кг, группе контроля вводили эквивалентное количество очищенной воды.
По результатам данного теста эффективность препарата ИР-1 практически в 3 раза (189,7%) превосходит показания контрольной группы и более чем в два раза превышает эффективность стандартного препарата Диклофенак (90,12% по отношению к контролю) (Рис. 3). Препарат ИР-1 по результатам теста «Горячая пластинка» проявил достоверную и высокую эффективность в подавлении острой соматической поверхностной боли.
5. Изучение жаропонижающего действия ИР-1 в модели пирогеналовой лихорадки у крыс
Лихорадочную реакцию вызывали внутрибрюшинным введением раствора пирогенала в дозе 0,2 мл раствора на 100 г массы тела животного. Ректальную температуру измеряли электротермометром до введения пирогена (исходная температура) и через 2 часа после его введения (предполагаемый пик лихорадочной реакции). Разница этих измерений представляет собой оцениваемую гипертермическую реакцию. Исследуемое вещество и препарат сравнения вводили сразу после получения достоверной гипертермической реакции. Исследуемое вещество вводили внутрижелудочно в средней эффективной дозе ЕД50=25 мг/кг. Препарат сравнения Диклофенак вводили в дозе 8 мг/кг крысы. Контрольную группу составляли животные, которым ни исследуемое вещество, ни препарат сравнения не вводились.
Жаропонижающее действие оценивали по уменьшению гипертермии через 1, 2, 3 и 4 часа после введения исследуемого вещества (препарата сравнения).
Через два часа отмечено достоверное снижение уровня температуры тела на 1,33°С у группы, получавшей препарат ИР-1, на 0,6°С - у группы, получившей Диклофенак, и на 0,36°С - у контрольной группы по отношению к собственной исходной температуре (Рис. 4). Это позволяет сделать однозначный вывод о наличии жаропонижающего эффекта исследуемого препарата ИР-1, который превосходит аналогичный у Диклофенака.
6. Оценка противовоспалительного действия препарата ИР-1 в сравнении с Кетопрофеном и Дексаметазоном в модели каррагинанового воспаления на мышах линии SHK.
Дозы вводимых веществ:
Лямбда Каррагенан - 1%, 0,1% или 3% р-р, подкожно в подушечку левой задней лапки;
исследуемые вещества:
Дексаметазон - 1 мг/кг (внутрибрюшинно);
Кетопрофен - 6 мг/кг (перорально);
ИР-1 - 5 мг/кг и 30 мг/кг (перорально);
Объем введения: 10 мл/кг.
Описание исследования: мышей пометили и распределили по группам - 6-8 самцов на исследуемое вещество. Перед началом исследования у животных измерили объем обеих лап (мл) с помощью плетизмометра (Vgo Basile North America Inc. PA 19473, Model №7140). Дексаметазон и Кетопрофен вводили за 30 мин до введения Лямбда Каррагенана (по 0,025 мл/лапа), ИР-1 вводили одновременно с Лямбда Каррагенаном.
Контроль исследования осуществляли по лапам животных, в которые исследуемые вещества не вводились; отрицательный контроль - группа, которой вместо Лямбда Каррагенана вводился физраствор и исследуемые вещества не вводились.
Первое измерение делали спустя один час после введения Лямбда Каррагенана, далее один раз в час в течение 6 часов, последнее измерение производили через 24 часа.
Данные вносились в протокол и далее обрабатывались в программе Libreofice 3.3.
Брали разницу между показаниями левой (в которую вводили Лямбда Каррагенан) и правой лапы (контроль), высчитывали средние и стандартные отклонения по группам, строили кривые зависимости отека лапы от времени (Рис. 5).
Также проводили сравнения между группами, которым вводили исследуемые вещества, и контролем (Лямбда Каррагенан, 1% раствор), с использованием t - теста для независимых групп (значимость различий при р<0,05).
Полученные результаты демонстрируют достоверное подавление отека у мышей при введении ИР-1 (р<0,05, кроме 1 часа), сравнимое с показаниями для препаратов сравнения - Дексаметазона и Кетопрофена.
7. Изучение влияния соединений ИР-1 и Дексаметазона на течение адъювантного артрита у крыс.
Материалы и методы
Экспериментальное исследование провели на 21 крысах-самцах стока Wistar. Длительность акклиматизационного периода для всех животных составила 7 дней. В течение периода акклиматизации проводили ежедневный осмотр каждого животного. Перед началом исследования животные без признаков отклонений внешнего вида были распределены на 3 экспериментальные группы. Маркировка клетки кодировала пол животных, дату начала эксперимента, дату начала ввода препаратов, название группы. Каждому отобранному в исследование животному был присвоен индивидуальный номер. Животные содержались в стандартных условиях.
В период акклиматизации и в ходе эксперимента животных кормили брикетированными кормами для содержания крыс фирмы «Мэст» со свободным доступом к воде и корму. В качестве подстила использовались опилки той же фирмы.
Световой режим составлял 12 часов света и 12 часов темноты. Температура воздуха поддерживалась в пределах 23°С, относительная влажность - 70%. Никаких существенных отклонений этих параметров в период акклиматизации и в ходе эксперимента зафиксировано не было.
Воспалительную реакцию моделировали путем субплантарного введения в подушечку левой задней лапы крыс 0,2 мл полного адъюванта Фрейнда (Sigma-Aldrich, США), содержащего 1 мг Mycobacterium tuberculosis в 1 мл парафинового масла.
Исследуемые вещества вводили внутрижелудочно с помощью зонда в виде суспензии (0,5% раствора метилцеллюлозы и 1% Твин 80).
На 10-й день после введения адъюванта начали введение веществ экспериментальным группам (в каждой по 7 животных): группа 1 - контроль, получавший 0,5% раствор метилцеллюлозы + 1% Твин 80; группа 2 - Дексаметазон в дозе 1 мг/кг; группа 3 - ИР-1 в дозе 25 мг/кг. Вещества вводили один раз в день в течение 10 дней. Выраженность воспалительной реакции оценивали на 10, 14, 17 и 24-й дни эксперимента.
В качестве критериев оценки эффективности исследуемых веществ использовали онкометрический метод - определение объема лап по количеству вытесненной из сосуда жидкости с помощью плетизмометра.
Токсическое действие исследуемых веществ оценивали по внешнему виду, потери массы тела и гибели животных.
На 25-й день эксперимента все животные были подвержены эвтаназии 70%-ной углекислотой. После чего они были вскрыты для макроскопической оценки внутренних органов, и у каждого животного были отрезаны голеностопные суставы обеих задних лап. Суставы взвесили, полученное значение массы записали в ручной протокол, после чего измельчили в ступке и гомогенизировали в 1 мл фосфатно-солевого буфера / 200 мг ткани, содержащего ингибитор протеазы cOmplete™ (Roche, Швейцария) с целью их подготовки для определения концентрации IL-1b, IL-6, ФНО-альфа. Полученный гомогенат центрифугировали, супернатант слили и заморозили при -80°С. Все работы с суставами проводились на сухом льду.
Анализ данных
После измерений объема лап рассчитывали разницу между левой и правой лапой на основании данных плетизмометрии. Считали средние значения, стандартные отклонения по группам в программе Excel 2003. Также проводили сравнения между группами, которым вводили исследуемые вещества, и контролем, с использованием t-теста для независимых групп (значимость различий при р<0,05).
Результаты
1. Оценка противоотечного эффекта
При моделировании адъювантного артрита крыс у животных отек левой лапы сформировался уже на третий день после введения адъюванта и к 10 дню (день начала лечения) превышал объем правой лапы на 86%.
В начале лечения разницы между контрольной группой и группой, получавшей ИР-1 в дозе 25 мг/кг, отмечено не было. Дексаметазон в дозе 1 мг/кг снижал воспалительную реакцию и этот эффект держался на протяжении всего эксперимента примерно одинаково. Эффективность ИР-1 была отмечена в середине, а также к концу лечения (Рис. 6).
2. Оценка токсичности исследуемых соединений
В ходе эксперимента у животных в группе 2 (Дексаметазон, 1 мг/кг), были отмечены токсические эффекты: шерсть вздыблена, двигательная активность снижена, изогнутость, потери массы тела животных составили от 13 до 33%. После окончания введений одно животное погибло.
В контрольной группе и в группе 3 (ИР-1, 25 мг/кг) состояние животных было в норме на протяжении всего эксперимента (Рис. 7).
При вскрытии животных, получавших Дексаметазон, отмечено повреждение печени, селезенки, поджелудочной железы, кишечника.
При макроскопии остальных животных других групп повреждений внутренних органов не обнаружено.
3. Определение содержания IL-6, Il-1b, ФНО-альфа в гомогенате суставов крыс.
Приготовление растворов (все концентраты входят в состав комплекта реагентов) 50 мл двадцатикратного промывочного буфера разводили до однократного добавлением до 1000 мл деионизованной воды, рН доводили до 7.4. 5 мл двадцатикратного тестового буфера разводили до 100 мл дистиллированной водой. Концентрированный конъюгат биотина разводился непосредственно перед использованием в тестовом буфере в концентрации 1:100, стрептавидин-пероксидаза хрена разводилась непосредственно перед использованием в тестовом буфере в концентрации 1:200. Стандарты IL-6, Il-1b, ФНО-альфа разводились до концентрации 1нг/мл за 30 минут до использования в дистиллированной воде, последующие серийные разведения проводились непосредственно в плате в соотношении 1:2.
Проведение эксперимента
Лунки промывали 400 мкл промывочного буфера, оставляя его на 10-15 секунд, в первые два ряда на плате добавляли стандартные разведения стандартов IL-6, Il-1b и ФНО-альфа (концентрации 500 пг/мл, 250 пг/мл, 125 пг/мл, 62,5 пг/мл, 31,3 пг/мл, 15,6 пг/мл, 7,8 пг/мл и пустой раствор). В каждую лунку добавляли по 50 мкл растворителя для проб и в лунки для проб - по 50 мкл проб и 50 мкл биотинового конъюгата. Платы накрывали адгезионной пленкой и инкубировали при комнатной температуре в течение 2 часов, при покачивании на качалке 100 об/мин. После окончания инкубации раствор удаляли из платы, и платы промывали 4 раза 400 мкл промывочного буфера. В каждую лунку затем добавляли 100 мкл ТМБ - (тетраметилбензидиновый субстрат) раствора субстрата и инкубировали при комнатной температуре дополнительно 10 минут. Реакцию останавливали добавлением 100 мкл стоп-раствора, входящего в набор реагентов для проведения анализа. Адсорбцию замеряли на спектрофотометре при длине волны 450 нм и референсной длине волны 620 нм.
Обработка результатов
Уровни IL-6, IL-1b и ФНО-альфа вычисляли по стандартной кривой, построенной с использованием стандартов с использованием программы Excel 2003. Считали средние значения, стандартные отклонения по группам в программе Excel 2003. Также проводили сравнения между группами, которым вводили исследуемые вещества, и контролем, с использованием t-теста для независимых групп (значимость различий при р<0,05) (Рисунки 8-10).
Выводы
1. В ходе эксперимента было установлено наличие противовоспалительного эффекта Дексаметазона и ИР-1 в процессе лечения животных на модели адъювантного артрита крыс. Через 4 дня после последней инъекции разницу между контрольной и группой, получавшей ИР-1 в дозе 25 мг/кг, не наблюдали. Эффект Дексаметазона сохранился.
2. Дексаметазон оказался токсичен. О чем свидетельствует потеря массы тела и гибель одного из семи (14%) животных, после 10-ти пероральных введений. ИР-1 в дозе 25 мг/кг не токсичен.
3. Дексаметазон и ИР-1 достоверно снижают концентрацию интерлейкина-6 в суставах с индуцированным артритом у крыс (Дексаметазон - на 40%, ИР-1 - на 27%).
4. Только дексаметазон достоверно снижает концентрацию фактора некроза опухоли и интерлейкина-1b в модели с индуцированным артритом у крыс, что подтверждает различия в механизмах действия Дексаметазона и ИР-1, а также свидетельствует о том, что только ингибирования ИРАК4 киназы недостаточно для ингибирования вышеуказанных факторов.
Таким образом, проведенные эксперименты показали, что ИР-1 является эффективным селективным ингибитором ИРАК4 киназы, обладающим высокой биодоступностью и низкой токсичностью, проявляющим эффективность в моделях иммунных воспалительных и онкологических заболеваний, что делает ИР-1 перспективным лекарственным средством для лечения и/или предотвращения заболеваний, связанных с ИРАК4 опосредованной передачей сигнала.
Хотя настоящее изобретение описано выше со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области будет очевидным, что конкретные подробно описанные эксперименты должны рассматриваться как иллюстративные, а не как ограничительные. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
Селективные ингибиторы haspin киназы
Новый класс селективных агонистов соматостатиновых рецепторов
Новые ингибиторы серин-треониновых киназ, в том числе для лечения онкологических заболеваний и туберкулеза
Селективные ингибиторы haspin киназы
Новый класс селективных агонистов соматостатиновых рецепторов
Новые ингибиторы серин-треониновых киназ, в том числе для лечения онкологических заболеваний и туберкулеза
Замещенные 2-метилиден-5-(фениламино)-2,3-дигидротиофен-3-оны для лечения лейкозов с транслокациями mll-гена и других онкологических заболеваний

