Результат интеллектуальной деятельности: СПОСОБЫ СЕКВЕНИРОВАНИЯ ТРЕХМЕРНОЙ СТРУКТУРЫ ИССЛЕДУЕМОЙ ОБЛАСТИ ГЕНОМА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к области молекулярной биологии, в частности к методам рекомбинантных ДНК. В более детальном изложении настоящее изобретение относится к секвенированию ДНК. Настоящее изобретение относится к способам определения (части) последовательности ДНК исследуемой области генома. В частности, настоящее изобретение относится к определению последовательности частей генома, совместно образующих пространственную конфигурацию. Настоящее изобретение далее относится к применению способов по настоящему изобретению для разработки методов индивидуальной диагностики и лечения путем исследования тканей на наличие злокачественных новообразований и других состояний.
Уровень техники
Значительные усилия были направлены на разработку методов “обогащения мишени” с целью секвенирования, в соответствии с которыми области генома из образца ДНК избирательно улавливают и/или избирательно амплифицируют и затем секвенируют (см. в публикации Mamanova et al., Nature Methods, 2010, (2):111-118). Методы обогащения генома имеют важное значение, так как позволяют сфокусировать внимание на конкретной области генома, анализ которой является более эффективным с точки зрения затрат времени и экономичности, а также гораздо менее сложным по сравнению с анализом всего генома. Существуют разные методы обогащения генома. Например, область генома можно амплифицировать при помощи реакции ПЦР с использованием одной пары праймеров и, таким образом, обогатить данную область генома. Однако размер продукта, получаемого с помощью ПЦР, является весьма ограниченным. В настоящее время могут быть амплифицированы продукты ПЦР длиной не более 10-40 т.п.о. (Cheng et al., Proc. Natl. Acad. Sci. USA, 1994; 91(2): 5695-5699), но такие методы не являются достаточно надежными, и каждая реакция ПЦР требует оптимизации и проверки при ограниченном размере продукта. Для увеличения размера областей, которые могут быть амплифицированы, а также для повышения надежности данного анализа были разработаны мозаичные методы с использованием нескольких пар праймеров для ПЦР, специально предназначенных для исследуемой области генома. Указанные праймеры использованы, например, в методе мультиплексной ПЦР или ПЦР RainDance. С методами целенаправленной амплификации совместимы разные ферментативные методы, такие как образование кольцевой молекулы мишени. В других методах использованы улавливающие зонды, локализованные на матрице или в растворе, которые, имея в длину 60-120 оснований, улавливают исследуемую область генома путем гибридизации.
Из приведенных выше примеров следует, что для обогащения исследуемой области генома сначала необходимо получить информацию о последовательности исследуемой области генома, которая нужна для создания зондов и/или праймеров для улавливания и/или амплификации исследуемой области генома. Например, для обогащения последовательности длиной 30 Мб обычно необходимо выполнить 6000 отдельных ПЦР. При использовании улавливающих зондов необходимо получить еще больше информации о последовательности, при этом потребуется по меньшей мере 250000 зондов длиной 120 п.о. для улавливания последовательности длиной 30 Мб. При выполнении указанных анализов возникают ошибки вследствие использования недостаточно точных данных о последовательности для зондов и/или праймеров, в значительной степени охватывающих исследуемую область генома. При выполнении указанных анализов невозможно выделить последовательности, которые существенно отличаются от созданных матричных последовательностей, и поэтому нельзя обнаружить инсерции. Кроме того, указанные методы требуют фрагментации ДНК на последовательности, состоящие из нескольких сотен пар оснований, до выполнения анализа. Из вышеизложенного следует, что исследуемая область генома разрывается на множество частей, в результате чего утрачивается информация, относящаяся, например, к реаранжировке в исследуемой области. Следовательно, существует потребность в усовершенствованных методах обогащения генома, которые являются гораздо более точными, не требуют использования тысяч коротких последовательностей и позволяют выполнять полное секвенирование исследуемой области.
Для исследования структуры ядра млекопитающих были разработаны анализы определения конформации хромосомы (3С/4С), с помощью которых можно анализировать структурную организацию области генома (WO 2007/004057, WO 2008/08845). Указанные методы включают перекрестное сшивание клеток in vivo, например, с помощью формальдегида, в результате чего пространственное расположение хроматина, включая ДНК, фиксируется в виде трехмерной укладки цепей. Затем хроматин фрагментируют, например, с помощью рестрикционного фермента и лигируют перекрестно-сшитые фрагменты ДНК. В результате происходит лигирование фрагментов ДНК, расположенных в непосредственной близости друг от друга. Лигированные продукты затем могут быть амплифицированы при помощи ПЦР и подвергнуты анализу с целью определения частоты взаимодействия лигированных фрагментов ДНК, что является показателем близкого расположения фрагментов. Амплификация при помощи ПЦР может быть выполнена в отношении последовательности-мишени в исследуемой области генома. Высокая частота взаимодействия с исследуемой областью генома свидетельствует о непосредственной близости расположения, низкая частота взаимодействия указывает на удаленное расположение. Для идентификации фрагментов ДНК необходимо получить информацию о последовательности. Такая информация о последовательности может быть получена в результате обнаружения амплифицированных фрагментов при помощи микроматрицы, включающей зонды, или путем секвенирования небольшой части амплифицированных фрагментов (обычно 20-30 пар оснований бывает достаточно для идентификации соответствующего положения в геноме). В любом случае число идентифицированных фрагментов ДНК, то есть частота взаимодействия, указывает на близость фрагмента к исследуемой точке, и полученная информация может быть использована для определения внутрихромосомных и межхромосомных взаимодействий.
Сущность изобретения
В настоящее время установлено, что перекрестное сшивание и фрагментация ДНК в клетке и последующее лигирование перекрестно-сшитых фрагментов ДНК может быть идеальным началом анализа исследуемой области генома, включающей последовательность нуклеотида-мишени, то есть линейной матрицы хромосомы, окружающей последовательность нуклеотида-мишени. Настоящее изобретение создано на основе концепции, предполагающей, что в результате перекрестного сшивания ДНК будут предпочтительно перекрестно сшиты последовательности, расположенные на линейной матрице хромосомы в непосредственной близости от последовательности нуклеотида-мишени. В качестве перекрестно-сшивающего агента может быть использован, например, формальдегид. После сшивания ДНК может быть подвергнута (ферментативной) обработке, то есть фрагментации, и лигированию, при этом ДНК остается в перекрестно-сшитом состоянии. Могут быть лигированы только перекрестно-сшитые фрагменты, расположенные в непосредственной близости друг от друга. Фрагменты ДНК, лигируемые с фрагментом ДНК, включающим последовательность нуклеотида-мишени, фактически представляют собой исследуемую область генома, включающую последовательность нуклеотида-мишени. При этом вероятность внутрихромосомного перекрестного сшивания в среднем всегда выше частоты межхромосомного перекрестного сшивания. Как правило, вероятность перекрестного сшивания разных фрагментов обратно пропорциональна линейному расстоянию между указанными фрагментами. В зависимости от конкретных условий перекрестного сшивания 20-30% фрагментов, лигированных с исследуемым нуклеотидом-мишенью, расположены в пределах 0,5 Мб от последовательности нуклеотида-мишени, при этом 50-80% фрагментов, лигированных с исследуемым нуклеотидом-мишенью, находятся в хромосоме, включающей указанную последовательность нуклеотида-мишени. Лигированные фрагменты ДНК, включающие последовательность нуклеотида-мишени и, следовательно, исследуемую область генома, могут быть амплифицированы, то есть обогащены, в результате использования одного или нескольких олигонуклеотидных праймеров, узнающих последовательность нуклеотида-мишени. Последовательность исследуемой области генома затем может быть определена при помощи методов (высокопроизводительного) секвенирования, хорошо известных в данной области. Данный метод характеризуется незначительными ошибками, так как не требует обширной информации о последовательности для анализа исследуемой области генома. Например, исследуемая область генома может включать представляющий интерес аллель. Последовательность нуклеотида-мишени может быть выбрана без включения в последовательность представляющего интерес аллеля. Исследуемая область генома затем может быть амплифицирована при использовании последовательности нуклеотида-мишени без необходимости получения информации о последовательности представляющего интерес аллеля. Таким образом, представляющий интерес аллель может быть обогащен без необходимости использования любой последовательности данного аллеля. При выполнении указанного метода обогащения не возникают ошибки из-за использования олигонуклеотидов и/или зондов, охватывающих последовательность представляющего интерес аллеля. Кроме того, поскольку на стадии лигирования происходит лигирование фрагментов, расположенных в непосредственной близости друг от друга, указанный метод позволяет производить анализ последовательностей отдельных аллелей. Например, когда образец перекрестно-сшитой ДНК включает несколько аллелей (например, при получении образца ДНК из популяции гетерогенных клеток или при значении плоидности больше единицы), каждый аллель может иметь разное окружение в геноме. Фрагмент ДНК, включающий последовательность нуклеотида-мишени, будет взаимодействовать только с фрагментами ДНК, находящимися в том же пространстве. Таким образом, лигированные фрагменты ДНК представляют собой геномную среду, из которой происходят указанные фрагменты. В результате определения по меньшей мере части последовательности всех лигированных фрагментов ДНК можно связать последовательности фрагментов ДНК, используя информацию о последовательностях разных лигированных фрагментов ДНК, и создать последовательность для отдельных исследуемых областей генома.
Определения терминов
В приведенном ниже описании изобретения и примерах использован ряд терминов. Для достижения четкого и единообразного понимания описания изобретения и формулы изобретения в объеме, охватываемом такими терминами, далее даны определения использованных терминов. За исключением особо оговоренных случаев все использованные технические и научные термины имеют значения, известные специалисту в области, к которой относится настоящее изобретение. Все публикации, заявки на патенты, патенты и другие научные материалы полностью включены в настоящее описание изобретения в качестве ссылки.
Квалифицированному специалисту должны быть очевидны способы выполнения стандартных методов, использованных в способах по настоящему изобретению. Способы выполнения стандартных методов в молекулярной биологии, биохимии, вычислительной химии, культивировании клеток, методах рекомбинантных ДНК, биоинформатике, геномике, секвенировании и родственных областях хорошо известны специалистам в данной области и описаны, например, в следующих публикациях: Sambrook et al., Molecular Cloning. A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989; Ausubel et al., Current Protocols in Molecular Biology, John Wiley & Sons, New York, 1987 и периодически публикуемые дополненные издания, а также серия Methods in Enzymology, Academic Press, San Diego.
В использованном здесь значении термины в единственном числе включают значения терминов во множественном числе за исключением тех случаев, когда из контекста следует обратное. Например, способ выделения молекулы ДНК включает выделение множества молекул (например, десятков, сотен, тысяч, десятков тысяч, сотен тысяч, миллионов или более молекул).
Термин ”исследуемая область генома” по настоящему изобретению означает последовательность ДНК организма, в которой желательно определить по меньшей мере часть указанной последовательности ДНК. Например, исследуемая область генома может быть областью генома, предположительно включающей аллель, ассоциированный с заболеванием. В использованном здесь значении термин ”аллель (аллели)” означает любую одну или более альтернативных форм гена в определенном локусе. В диплоидной клетке организма аллели данного гена локализованы в определенном месте или локусе на хромосоме. На каждой хромосоме пары гомологичных хромосом присутствует один аллель. Таким образом, в диплоидной клетки могут существовать два аллеля и, следовательно, две отдельные (разные) исследуемые области генома.
Термин ”нуклеиновая кислота” по настоящему изобретению может означать любой полимер или олигомер пиримидиновых и пуриновых оснований, предпочтительно цитозина, тимина, урацила, аденина и гуанина (см. публикацию Albert L. Lehninger, Principles of Biochemistry, at 793-800 (Worth Pub. 1982), которая полностью включена в настоящее описание изобретения в качестве ссылки). В объем настоящего изобретения входит любой дезоксирибонуклеотидный, рибонуклеотидный или пептидный компонент нуклеиновой кислоты и их любые химические варианты, в частности метилированные, гидроксиметилированные или гликозилированные формы указанных оснований и тому подобные. Полимеры или олигомеры могут быть гетерогенными или гомогенными и могут быть выделены из природных источников или могут быть получены искусственным или синтетическим путем. Кроме того, нуклеиновые кислоты могут представлять собой ДНК или РНК либо их смесь и могут существовать постоянно или временно в одноцепочечной или двухцепочечной форме, включая гомодуплесные, гетеродуплексные и гибридные состояния.
Термин ”образец ДНК” означает образец, полученный из организма или из ткани организма либо из культуры ткани и/или клеток, включающей ДНК. Образец ДНК может быть получен из организма любого типа, например микроорганизмов, вирусов, растений, грибов, животных, человека и бактерий или их комбинаций. Например, образец ткани человека с подозрением на наличие бактериальной и/или вирусной инфекции может включать клетки человека, а также вирусы и/или бактерии. Образец может включать клетки и/или ядра клеток. Образец ДНК может быть получен у больного или субъекта, подвергающегося риску заражения или с подозрением на наличие конкретного заболевания, например рака или любого другого состояния, требующего исследования ДНК организма.
Термин ”перекрестное сшивание” по настоящему изобретению означает взаимодействие ДНК в двух разных положениях с достижением соединения в двух указанных разных положениях. Соединение ДНК в двух разных положениях может быть произведено путем образования прямой ковалентной связи между цепями ДНК. Две цепи ДНК могут быть перекрестно сшиты при помощи УФ-излучения, образующего ковалентные связи непосредственно между цепями ДНК. Соединение ДНК в двух разных положениях может быть непрямым, образуемым при помощи агента, такого как, например, молекула перекрестно-сшивающего агента. Первая часть ДНК может быть соединена с первой реакционно-способной группой молекулы перекрестно-сшивающего агента, включающей две реакционно-способные группы, при этом вторая реакционно-способная группа молекулы перекрестно-сшивающего агента может быть присоединена ко второй части ДНК, в результате чего достигается непрямое перекрестное сшивание первой и второй частей ДНК при помощи молекулы перекрестно-сшивающего агента. Непрямая перекрестная связь между двумя цепями ДНК может быть образована при помощи нескольких молекул. Например, типичной молекулой перекрестно-сшивающего агента, которая может быть использована в настоящем изобретении, является формальдегид. Формальдегид вызывает образование белок-белковых и ДНК-белковых перекрестных связей. Таким образом, формальдегид может перекрестно сшивать друг с другом разные цепи ДНК при помощи ассоциированных с ними белков. Например, формальдегид может взаимодействовать с белком и ДНК, соединяя белок и ДНК при помощи молекулы перекрестно-сшивающего агента. Следовательно, две части ДНК могут быть перекрестно сшиты с использованием формальдегида, образующего соединение между первой частью ДНК и белком, при этом белок может образовывать второе соединение с другой молекулой формальдегида, соединяющейся со второй частью ДНК, в результате чего образуется перекрестная связь, которая может быть представлена как ДНК1 - перекрестно-сшивающий агент - белок - перекрестно-сшивающий агент - ДНК2. В любом случае должно быть понятно, что перекрестное сшивание по настоящему изобретению включает образование (прямых или непрямых) соединений между цепями ДНК, находящимися в физической близости друг от друга. Поскольку ДНК является высокоорганизованным элементом, цепи ДНК могут находиться в физической близости друг от друга в клетке, даже будучи отделенными от исследуемой точки последовательности, например, 100 т.п.о. Метод перекрестного сшивания входит в объем настоящего изобретения, если указанный метод совместим с последующими стадиями фрагментации и лигирования.
Термин ”образец перекрестно-сшитой ДНК” означает образец ДНК, который был подвергнут перекрестному сшиванию. Результатом перекрестного сшивания образца ДНК является сохранение в неповрежденном виде трехмерной структуры ДНК в образце. Цепи ДНК, находящиеся в физической близости друг от друга, сохраняют занимаемые ими положения.
Термин ”реверсия перекрестного сшивания” по настоящему изобретению означает разрыв перекрестных связей, в результате чего ранее перекрестно-сшитая ДНК больше не является перекрестно-сшитой и пригодна для выполнения последующих стадий амплификации и/или секвенирования. Например, обработка образца ДНК, перекрестно-сшитого формальдегидом, протеазой К вызывает расщепление белка, присутствующего в образце. Так как перекрестно-сшитая ДНК имеет непрямое соединение, образованное белком, обработка протеазой может устранить перекрестное сшивание ДНК. Однако фрагменты белка, оставшиеся присоединенными к ДНК, могут препятствовать последующему секвенированию и/или амплификации. Поэтому реверсия соединений между ДНК и белком может также вызвать ”реверсию перекрестного сшивания”. Соединение ДНК - перекрестно-сшивающий агент - белок может быть реверсировано в результате выполнения стадии нагревания, например, путем инкубации при 70°С. Так как в образце ДНК присутствуют большие количества белка, часто бывает желательно дополнительно расщепить белок протеазой. Поэтому в объем настоящего изобретения входит любой метод ”реверсии перекрестного сшивания”, позволяющий сделать цепи ДНК, соединенные в перекрестно-сшитом образце, пригодными для секвенирования и/или амплификации.
Термин ”фрагментация ДНК” означает любой метод, который при применении к ДНК, являющейся или не являющейся перекрестно-сшитой ДНК, или к любой другой ДНК позволяет получить фрагменты ДНК. Такими методами, хорошо известными в данной области, являются обработка ультразвуком, гидродинамическое фрагментирование и/или ферментативная рестрикция, но в настоящем изобретении также могут быть использованы другие методы.
Термин ”рестрикционная эндонуклеаза” или “рестрикционный фермент” означает фермент, который узнает специфическую нуклеотидную последовательность (сайт узнавания) в молекуле двухцепочечной ДНК и расщепляет обе цепи молекулы ДНК на каждом и почти на каждом сайте узнавания, оставляя тупой конец или липкий 3'- или 5'-конец. Узнаваемая специфическая нуклеотидная последовательность может определять частоту расщепления, например нуклеотидная последовательность из 6 нуклеотидов встречается в среднем через каждые 4096 нуклеотидов, при этом нуклеотидная последовательность из 4 нуклеотидов встречается гораздо чаще, в среднем через каждые 256 нуклеотидов.
Термин ”лигирование” по настоящему изобретению означает соединение отдельных фрагментов ДНК. Фрагменты ДНК могут иметь тупые концы или совместимые липкие концы, которые могут гибридизировать друг с другом. Фрагменты ДНК могут быть соединены ферментом, например лигазой, ДНК-лигазой. Однако также может быть использовано неферментативное лигирование, если фрагменты ДНК соединены с образованием ковалентной связи. Обычно между гидроксильной и фосфатной группой отдельных цепей образуется сложная фосфодиэфирная связь.
Термин ”олигонуклеотидные праймеры” обычно означает цепи нуклеотидов, способных стимулировать синтез ДНК. ДНК-полимераза не может синтезировать ДНК de novo без праймеров. Праймер гибридизирует с ДНК, образуя пары оснований. Нуклеотиды, способные образовывать пары оснований, комплементарные друг другу, представляют собой, например, цитозин и гуанин, тимин и аденин, аденин и урацил, гуанин и урацил. Комплементарность между праймером и существующей цепью ДНК может быть не равна 100%, то есть не все основания праймера должны образовывать пару оснований с существующей цепью ДНК. Нуклеотиды из 3'-конца праймера гибридизируют с существующей цепью ДНК, используя данную цепь в качестве матрицы (матричный синтез ДНК). В качестве праймеров могут быть использованы молекулы синтетических олигонуклеотидов, применяемые при выполнении реакции амплификации.
Термин ”амплификация” означает реакцию амплификации полинуклеотидов, а именно популяции полинуклеотидов, которые реплицируются из одной или нескольких исходных последовательностей. Амплификация может означать разные реакции амплификации, которые включают, не ограничиваясь ими, полимеразную цепную реакцию (ПЦР), линейные полимеразные реакции, амплификацию на основе последовательности нуклеиновой кислоты, кольцевую амплификацию и подобные реакции.
Термин ”секвенирование” означает определение порядка следования нуклеотидов (последовательностей оснований) в образце нуклеиновой кислоты, например ДНК или РНК. Существует много методов секвенирования, таких как, например, секвенирование Сангера и методы высокопроизводительного секвенирования, разработанные компаниями Roche, Illumina и Applied Biosystems.
Термин ”совокупность смежных фрагментов” использован в связи с анализом последовательностей ДНК и означает перегруппированные смежные фрагменты ДНК, выделенные из двух или более фрагментов ДНК, имеющих смежные нуклеотидные последовательности. Таким образом, совокупность смежных фрагментов может представлять собой набор перекрывающихся фрагментов ДНК, образующих (неполную) смежную последовательность исследуемой области генома. Совокупность смежных фрагментов также может представлять собой набор фрагментов ДНК, которые при выполнении сравнительного анализа с эталонной последовательностью могут образовывать последовательность смежных нуклеотидов. Например, термин ”совокупность смежных фрагментов” означает серию (лигированных) фрагментов ДНК, расположенных с образованием последовательности, в которой каждый (лигированный) фрагмент ДНК перекрывает по меньшей мере один из соседних фрагментов. Связанные (лигированные) фрагменты ДНК могут быть упорядочены вручную или, предпочтительно, при помощи соответствующих компьютерных программ, таких как FPC, PHRAP, CAP3 и т.д., а также могут быть сгруппированы в отдельные совокупности смежных фрагментов.
Термин ”адаптор” означает молекулу короткого двухцепочечного олигонуклеотида с ограниченным числом пар оснований, например, имеющий от около 10 до около 30 пар оснований, которые созданы с возможностью лигирования с концами фрагментов. Адапторы обычно состоят из двух синтетических олигонуклеотидов, нуклеотидные последовательности которых частично комплементарны друг другу. При смешивании двух синтетических олигонуклеотидов в растворе в соответствующих условиях указанные олигонуклеотиды гибридизируют друг с другом, образуя двухцепочечную структуру. После гибридизации один конец молекулы адаптора может быть совместим с концом рестрикционного фрагмента и лигирован с указанным концом, другой конец адаптора может не обладать способностью лигирования, но данное условие не является обязательным, например, в том случае, когда адаптор должен быть лигирован между фрагментами ДНК.
Термин ”идентификатор” означает короткую последовательность, которая может быть добавлена к адаптору или праймеру, включена в последовательность или каким-либо другим образом использована в качестве метки, представляя собой уникальный идентификатор. Такой идентификатор (или метка) последовательности может представлять собой уникальную последовательность оснований, имеющую разную, но определенную длину, обычно включающую 4-16 п.о., предназначенную для идентификации конкретного образца нуклеиновой кислоты. Например, метки, состоящие из 4 п.о., позволяют получить 4(exp4)=256 разных меток. Типичными примерами являются последовательности ZIP, известные в данной области в качестве меток, обычно используемых для обнаружения путем гибридизации (Lannone et al., Cytometry, 39:131-140, 2000). Идентификаторы входят в объем настоящего изобретения, так как благодаря использованию такого идентификатора можно определить происхождение образца (ПЦР) при последующей обработке. В случае объединения обработанных продуктов, полученных из разных образцов нуклеиновой кислоты, при помощи разных идентификаторов можно определить разные образцы нуклеиновой кислоты. Например, при выполнении секвенирования по настоящему изобретению методом высокопроизводительного секвенирования можно объединить несколько образцов. Идентификаторы позволяют идентифицировать последовательности, соответствующие разным образцам. Идентификаторы также могут быть включены в адапторы для лигирования с фрагментами ДНК для идентификации последовательностей фрагментов ДНК. Идентификаторы предпочтительно отличаются друг от друга по меньшей мере двумя парами оснований и предпочтительно не содержат двух идентичных смежных оснований во избежание ошибочного считывания. Функция идентификатора иногда может быть объединена с другими функциями, такими как адапторы или праймеры.
Термин ”выбор размера молекул” по настоящему изобретению означает методы, с помощью которых могут быть выбраны размеры молекул, например, (лигированных) фрагментов ДНК или амплифицированных (лигированных) фрагментов ДНК. Методы, которые могут быть использованы для указанной цели, включают, не ограничиваясь ими, например, гель-электрофорез, гель-хроматографию, экстракционную хроматографию, и при осуществлении настоящего изобретения может быть использован любой такой метод, позволяющий выбрать молекулы определенного размера.
Термин ”сравнительный анализ” означает сравнение двух или более нуклеотидных последовательностей на основании наличия коротких или длинных участков идентичных или подобных нуклеотидов. В данной области хорошо известны методы и компьютерные программы, предназначенные для выполнения сравнительного анализа. Одной компьютерной программой, которая может быть использована или адаптирована для выполнения сравнительного анализа, является программа ”Align 2”, созданная в компании Genentech, Inc., которая была зарегистрирована с документацией пользователя в Ведомстве по охране авторского права США, Вашингтон, D.C. 20559, 10 декабря 1991 г.
Краткое описание чертежей
На фиг.1 схематически показан способ определения последовательности исследуемой области генома по настоящему изобретению. Указанный способ включает:
(а) перекрестное сшивание, в соответствии с которым фиксация формальдегидом вызывает перекрестное сшивание близко расположенных последовательностей ДНК в ядре (N) (такими последовательностями часто являются последовательности, близко расположенные на хромосоме (Ch), например последовательности одного гена) при помощи ассоциированных с ними белком (например, гистонов). Показаны 5 гипотетических фрагментов исследуемой области генома А, В, С, D и Е;
(b) фрагментацию перекрестно-сшитого образца ДНК, например, путем расщепления рестрикционным ферментом (например, мелкощепящей рестриктазой) (такой как NlaIII);
(с) лигирование перекрестно-сшитых рестрикционных фрагментов с образованием кольцевой ДНК;
(d) реверсию перекрестного сшивания и последующее выполнение амплификации, например, при помощи ПЦР с использованием набора праймеров для (обратной) ПЦР в исследуемой точке, расположенной рядом или внутри исследуемой области генома. Фрагменты (A, B, C, D и Е), перекрестно-сшитые в указанной исследуемой точке, амплифицируют и обогащают по сравнению с остальной частью генома.
Амплифицированные фрагменты секвенируют, например, путем секвенирования всех кольцевых молекул (длинные транскрипты), при этом продукт, амплифицированный при помощи ПЦР, также может быть сначала фрагментирован для создания библиотеки секвенирования, совместимой с методами секвенирования компаний Illumina или SOLiD;
(е) создание совокупности смежных фрагментов из полученных транскриптов, при этом может быть произведено сравнение последовательностей с эталонным геномом для выявления генетичесих изменений.
На фиг.2 схематически показан ген BRCA1 с 5 разными исследуемыми точками (А, В, С, D и Е). Черная стрелка показывает направление считывания. Числа в кружках со стрелками показывают положение в последовательности гена. Точка Е находится в начале гена и точка А расположена в конце гена. Указанные исследуемые точки разделены примерно 15-25 т.п.о.
На фиг.3 показан гель-электрофорез образцов ДНК, выполняемый во время получения перекрестно-сшитого образца ДНК для секвенирования гена BRCA1, описанного в примерах.
(А) В полосе М показан маркер Pstl лямбда-ДНК, в полосе 1 показан нерасщепленный контрольный образец, в полосе 2 показан контрольный образец, расщепленный NlaIII, в полосе 3 показан лигированный контрольный образец, полученный после лигирования образца, предварительно расщепленного NlaII, в полосе 4 показано второе расщепление Nspl.
(В) В полосе М показан маркер Pstl лямбда-ДНК. В полосах А, В, С, D и Е показаны продукты амплификации ДНК разными методами, соответствующие образцам, полученным на стадии 67, описанные в разделе ”Примеры”, и соответствующие исследуемые точки, показанные на фиг.2.
Подробное описание изобретения
Одним объектом настоящего изобретения является способ определения последовательности исследуемой области генома, содержащей последовательность нуклеотида-мишени, который включает фрагментацию перекрестно-сшитой ДНК, лигирование фрагментированной перекрестно-сшитой ДНК, реверсию перекрестного сшивания и определение по меньшей мере части последовательностей лигированных фрагментов ДНК, включающих последовательность нуклеотида-мишени, и использование определенных последовательностей для создания последовательности исследуемой области генома.
Образец перекрестно-сшитой ДНК включает образец ДНК, который был подвергнут перекрестному сшиванию. Перекрестное сшивание ДНК, присутствующей в образце, в значительной степени сохраняет трехмерную структуру ДНК. В качестве стандартного перекрестно-сшивающего агента может быть использован, например, формальдегид. Образцы могут быть получены у субъекта и/или из больной ткани, а также могут быть выделены из других организмов или из разных участков одного организма, например все образцы могут быть получены у одного субъекта, один образец может быть получен из здоровой ткани и один образец может быть получен из больной ткани. Образцы могут быть подвергнуты анализу по настоящему изобретению и сравнены с эталонным образцом либо разные образцы могут быть подвергнуты анализу и сравнены друг с другом. Например, у субъекта с подозрением на рак молочной железы один биопсийный образец может быть получен из предполагаемой опухоли. Другой биопсийный образец может быть получен из здоровой ткани. Оба биопсийных образца могут быть подвергнуты анализу по настоящему изобретению. Исследуемые области генома могут представлять собой гены BRCA1 и BRCA2, которые состоят из 83 и 86 т.п.о. (см. публикацию Mazoyer, 2005, Human Mutation 25:415-422). Определив последовательность исследуемой области генома по настоящему изобретению и сравнив последовательности области генома разных биопсийных образцов друг с другом и/или с эталонной последовательностью гена BRCA, можно обнаружить генетические мутации, которые помогут поставить диагноз субъекту, назначить лечение и/или прогнозировать развитие болезни.
В результате фрагментации образца перекрестно-сшитой ДНК фрагменты ДНК, находящиеся в исследуемой области генома, остаются в непосредственной близости друг от друга, поскольку являются перекрестно-сшитыми. При последующем лигировании перекрестно-сшитых фрагментов ДНК происходит лигирование фрагментов ДНК исследуемой области генома, расположенных в непосредственной близости друг от друга благодаря наличию перекрестных сшивок. Лигирование подобного типа можно также определить как проксимальное лигирование. Фрагменты ДНК, включающие последовательность нуклеотида-мишени, могут быть лигированы с фрагментами ДНК в пределах большого линейного расстояния на уровне последовательности. Определив (по меньшей мере часть) последовательности лигированных фрагментов, включающих фрагмент, содержащий последовательность нуклеотида-мишени, можно получить последовательности фрагментов ДНК в пространственном окружении исследуемой области генома. Каждая отдельная последовательность нуклеотида-мишени, по-видимому, может быть перекрестно сшита с несколькими другими фрагментами ДНК. Благодаря этому с фрагментом, включающим последовательность нуклеотида-мишени, часто можно лигировать несколько фрагментов ДНК. Объединяя (части) последовательностей (амплифицированных) лигированных фрагментов ДНК, которые были лигированы с фрагментом, включающим последовательность нуклеотида-мишени, можно создать последовательность исследуемой области генома. Фрагмент ДНК, лигированный с фрагментом, включающим последовательность нуклеотида-мишени, содержит любой фрагмент, который может присутствовать в лигированных фрагментах ДНК.
В данной области известны методы, включающие перекрестное сшивание ДНК, а также фрагментацию и лигирование фрагментов ДНК (например, WO 2007/004057 или WO 2008/08845). Целью таких методов является выявление частоты взаимодействия между разными фрагментами ДНК, а не идентификация нуклеотидной последовательности фрагментов, расположенных рядом с последовательностью нуклеотида-мишени. Первоначальная идея использования 4С для обнаружения частоты взаимодействия требовала только короткого транскрипта. Данные о частоте взаимодействия коротких транскриптов могут быть представлены в виде графика зависимости от положений транскриптов на хромосоме. С помощью такого графика можно определить взаимодействие конкретной исследуемой области генома с другой областью в любом месте генома или наличие транслокаций между хромосомами. Например, обнаружение большой частоты взаимодействия в хромосоме, отличной от хромосомы, содержащей последовательность нуклеотида-мишени, указывает на транслокацию. Настоящее изобретение не относится к определению частоты взаимодействия. В соответствии с настоящим изобретением установлено, что в результате фрагментации перекрестно-сшитой ДНК и последующего лигирования фрагментов ДНК можно определить область генома, окружающую последовательность нуклеотида-мишени, и после секвенирования реконструировать совокупность смежных фрагментов данной области генома. В методах, известных в данной области, основное внимание уделяется определению частоты взаимодействия коротких транскриптов с последовательностью нуклеотида-мишени, а в настоящем изобретении внимание сфокусировано на определении всей или по меньшей мере большой части последовательности лигированных фрагментов ДНК (включающих фрагмент ДНК с нуклеотидом-мишенью), благодаря чему при использовании последовательностей фрагментов ДНК и связывании лигированных фрагментов ДНК могут быть созданы совокупности смежных фрагментов для исследуемой области генома.
Линеаризованные лигированные фрагменты
Один вариант осуществления изобретения относится к способу определения последовательности исследуемой области генома, содержащей последовательность нуклеотида-мишени, который включает стадии:
а) получение образца перекрестно-сшитой ДНК;
b) фрагментацию перекрестно-сшитой ДНК;
с) лигирование фрагментированной перекрестно-сшитой ДНК;
d) реверсию перекрестного сшивания;
е) необязательную фрагментацию ДНК, полученной на стадии d), предпочтительно при помощи рестрикционного фермента;
f) необязательное лигирование фрагментированной ДНК, полученной на стадии d) или е), по меньшей мере с одним адаптором;
g) необязательную амплификацию ДНК, полученной на стадии d) или е), содержащей последовательность нуклеотида-мишени, с использованием по меньшей мере одного олигонуклеотидного праймера, гибридизирующего с последовательностью нуклеотида-мишени, или амплификацию ДНК, полученной на стадии f), с использованием по меньшей мере одного дополнительного праймера, гибридизирующего по меньшей мере с одним адаптором;
h) определение по меньшей мере части последовательности (амплифицированных) лигированных фрагментов ДНК, полученных на стадии d), e) или g), содержащих последовательность нуклеотида-мишени, предпочтительно в результате выполнения высокопроизводительного секвенирования;
i) создание совокупности смежных фрагментов исследуемой области генома из определенных последовательностей.
Образец перекрестно-сшитой ДНК, используемый на стадии а), получают в соответствии с настоящим описанием изобретения. Образец перекрестно-сшитой ДНК фрагментируют на стадии b). В результате фрагментации перекрестно-сшитой ДНК образуются фрагменты ДНК, удерживаемые вместе благодаря наличию перекрестных сшивок. Стадия фрагментации b) может включать обработку ультразвуком и последующую ферментативную репарацию концов ДНК. Обработка ультразвуком вызывает фрагментацию ДНК на произвольных сайтах, при этом фрагменты могут иметь тупые концы или липкие 3'- или 5'-концы; поскольку точечные разрывы ДНК являются произвольными, ДНК может быть репарирована (ферментативно) путем заполнения возможных липких 3'- или 5'-концов, в результате чего образуются фрагменты ДНК с тупыми концами, позволяющими лигировать указанные фрагменты с адапторами и/или друг с другом на следующей стадии с). Альтернативно липкие концы также можно сделать тупыми, удалив нуклеотиды липких концов, например, при помощи экзонуклеаз. Стадия фрагментации b) также может включать фрагментацию одним или несколькими рестрикционными ферментами или их комбинациями. Фрагментация рестрикционным ферментом является предпочтительной, так как позволяет контролировать средний размер фрагментов. Образовавшиеся фрагменты могут иметь совместимые липкие или тупые концы, позволяющие лигировать указанные фрагменты на следующей стадии с). Кроме того, для деления образца перекрестно-сшитой ДНК на множество частей могут быть использованы рестрикционные ферменты с разными сайтами узнавания. Такой метод является предпочтительным, так как при использовании разных рестрикционных ферментов, имеющих разные сайты узнавания, из каждой части образца могут быть получены разные фрагменты ДНК.
На следующей стадии с) фрагменты лигируют. Так как фрагмент, включающий последовательность нуклеотида-мишени, может быть перекрестно сшит с несколькими другими фрагментами ДНК, с фрагментом, содержащим последовательность нуклеотида-мишени, могут быть лигированы несколько фрагментов ДНК. В результате этого могут образоваться комбинации фрагментов ДНК, расположенных в непосредственной близости друг от друга, поскольку указанные фрагменты удерживаются вместе благодаря перекрестным сшивкам. В лигированных фрагментах ДНК могут иметь место разные комбинации и/или порядок следования фрагментов ДНК. При получении фрагментов ДНК с помощью рестрикционных ферментов сайт узнавания рестрикционного фермента известен, что позволяет идентифицировать фрагменты на основании остатков сайтов или восстановленных сайтов узнавания рестрикционных ферментов, указывающих на разделение между разными фрагментами ДНК. При получении фрагментов ДНК путем произвольной фрагментации под воздействием ультразвука и последующей ферментативной репарации концов ДНК труднее отличить один фрагмент от другого. Независимо от использованного метода фрагментации стадия лигирования с) может быть выполнена в присутствии адаптора путем лигирования адапторных последовательностей между фрагментами. Альтернативно адаптор может быть лигирован при выполнении отдельной стадии. Преимуществом лигирования адаптора на отдельной стадии является простота идентификации разных фрагментов путем обнаружения адапторных последовательностей, расположенных между фрагментами. Например, в случае тупых концов фрагментов ДНК адапторные последовательности связываются с обоими концами фрагментов ДНК, определяя границу между отдельными фрагментами ДНК. Затем перекрестное сшивание реверсируется на стадии d), в результате чего образуется группа лигированных фрагментов ДНК, состоящая из двух или более фрагментов. Субпопуляция группы лигированных фрагментов ДНК включает фрагмент ДНК, содержащий последовательность нуклеотида-мишени. В результате реверсии перекрестного сшивания разрушается структурная/пространственная фиксация ДНК, и последовательность ДНК может быть использована на последующих стадиях, например, амплификации и/или секвенирования, так как перекрестно-сшитая ДНК не может быть приемлемым субстратом для таких стадий. Последующие стадии е) и/или f) могут быть выполнены после реверсии перекрестного сшивания, однако стадии е) и/или f) также могут быть выполнены при нахождении лигированных фрагментов ДНК в перекрестно-сшитом состоянии.
Лигированные фрагменты ДНК могут быть необязательно фрагментированы на стадии е) предпочтительно при помощи рестрикционного фермента. Целью выполнения первой стадии фрагментации и необязательной второй стадии фрагментации является получение лигированных фрагментов ДНК, размер которых пригоден для выполнения следующей стадии амплификации и/или стадии определения последовательности. Кроме того, в результате выполнения второй стадии фрагментации, предпочтительно с использованием фермента, могут быть получены концы лигированных фрагментов, пригодные для необязательного лигирования адаптора на стадии f). Вторая стадия фрагментации может быть выполнена после реверсии перекрестного сшивания, однако вторая стадия фрагментации е) и/или стадия лигирования f) также могут быть выполнены при нахождении фрагментов ДНК в перекрестно-сшитом состоянии.
При использовании на стадиях фрагментации b) и е) рестрикционных ферментов желательно, чтобы сайт узнавания рестрикционного фермента на стадии е) был длиннее сайта узнавания на стадии b). Таким образом, фермент, используемый на стадии е), разрезает последовательность с меньшей частотой, чем на стадии b). Из вышеизложенного следует, что средний размер фрагмента ДНК, полученного на стадии b), меньше среднего размера фрагмента, полученного на стадии е), после рестрикции ДНК. Поэтому на первой стадии фрагментации образуются относительно мелкие фрагменты, которые затем лигируются. Так как второй рестрикционный фермент, используемый на стадии е), разрезает последовательность менее часто, чем на стадии b), в большинстве фрагментов ДНК может отсутствовать сайт узнавания рестрикционного фермента, использованного на стадии е). Таким образом, при последующей фрагментации лигированных фрагментов ДНК на второй стадии многие фрагменты ДНК, полученные на стадии b), могут оставаться интактными. Такое состояние фрагментов может быть полезно, так как объединенные последовательности фрагментов ДНК, полученных на стадии b), могут быть использованы для создания совокупности смежных фрагментов для исследуемой области генома. Если фрагментация на стадии b) происходит менее часто, чем на стадии с), фрагменты, полученные на стадии b), будут снова фрагментированы, в результате чего могут быть потеряны относительно большие последовательности ДНК, пригодные для создания совокупности смежных фрагментов. Таким образом, независимо от метода фрагментации, использованного на стадии b) и е), желательно, чтобы фрагментация на стадии b) происходила чаще, чем на стадии е), благодаря чему фрагменты ДНК, полученные на стадии b), в основном остаются интактными, то есть не фрагментируются на стадии е).
С лигированными фрагментами ДНК, полученными на стадии d) или е), может быть необязательно лигирован по меньшей мере один адаптор. Концы лигированных фрагментов ДНК должны быть совместимы с лигированием такого адаптора. Так как лигированные фрагменты ДНК, полученные на стадии d) или е), могут представлять собой линейную ДНК, в результате лигирования адаптора может быть получена последовательность, гибридизирующая с праймером. Адапторная последовательность, лигированная с лигированными фрагментами ДНК, включающими последовательность нуклеотида-мишени, образует молекулу ДНК, которая может быть амплифицирована при помощи ПЦР.
На следующей стадии g) ДНК, полученная на стадии f), включающая последовательность нуклеотида-мишени, может быть амплифицирована с использованием по меньшей мере одного олигонуклеотидного праймера, гибридизирующего с последовательностью нуклеотида-мишени, и по меньшей мере одного дополнительного праймера, гибридизирующего по меньшей мере с одним адаптором. Так как стадия f) лигирования адаптора является необязательной, ДНК, полученная на стадии d) или е), включающая нуклеотид-мишень, также может быть амплифицирована на стадии g) с использованием по меньшей мере одного олигонуклеотидного праймера, гибридизирующего с последовательностью нуклеотида-мишени.
Затем определяют последовательность (амплифицированных) лигированных фрагментов ДНК, полученных на стадии d), e), f) или g), включающих последовательность нуклеотида-мишени. Указанную последовательность предпочтительно определяют при помощи метода высокопроизводительного секвенирования, так как данная технология является более удобной и позволяет определить большое число последовательностей во всей исследуемой области генома. Из определенных последовательностей может быть создана совокупность смежных фрагментов исследуемой области генома. После определения последовательностей фрагментов ДНК могут быть получены перекрывающиеся транскрипты, из которых может быть создана исследуемая область генома. При получении фрагментов ДНК методом произвольной фрагментации уже на стадии фрагментации могут образоваться фрагменты ДНК, которые при секвенировании позволяют получить перекрывающиеся транскрипты. При увеличении размера образца, например при увеличении числа анализируемых клеток, может быть повышена достоверность созданной исследуемой области генома. Альтернативно, при анализе на стадии b) множества частей образцов с использованием разных рестрикционных ферментов также могут быть получены перекрывающиеся транскрипты. При увеличении числа частей образцов увеличивается количество перекрывающихся фрагментов, что может повысить достоверность совокупности смежных фрагментов созданной исследуемой области генома. Из определенных перекрывающихся последовательностей может быть создана совокупность смежных фрагментов. Альтернативно, если последовательности не перекрывают друг друга, например, при использовании одного рестрикционного фермента на стадии b), созданию совокупности смежных фрагментов исследуемой области генома может способствовать сравнение (лигированных) фрагментов ДНК с эталонной последовательностью.
Кольцевые лигированные фрагменты
Альтернативный вариант осуществления изобретения относится к способу определения последовательности исследуемой области генома, содержащей последовательность нуклеотида-мишени, который включает стадии:
а) получение образца перекрестно-сшитой ДНК;
b) фрагментацию перекрестно-сшитой ДНК;
с) лигирование фрагментированной перекрестно-сшитой ДНК;
d) реверсию перекрестного сшивания;
е) необязательную фрагментацию ДНК, полученной на стадии d), предпочтительно при помощи рестрикционного фермента;
f) образование кольцевой ДНК, полученной на стадии d) или е);
g) необязательную и предпочтительную амплификацию кольцевой ДНК, включающей последовательность нуклеотида-мишени, при использовании по меньшей мере одного праймера, гибридизирующего с последовательностью нуклеотида-мишени;
h) определение по меньшей мере части последовательности (амплифицированных) лигированных фрагментов ДНК, включающих нуклеотид-мишень, методом высокопроизводительного секвенирования;
i) создание из определенных последовательностей совокупности смежных фрагментов исследуемой области генома.
Образец перекрестно-сшитой ДНК, используемый на стадии а), получают в соответствии с настоящим описанием изобретения. Образец перекрестно-сшитой ДНК фрагментируют на стадии b). В результате фрагментации перекрестно-сшитой ДНК образуются фрагменты ДНК, удерживаемые вместе благодаря наличию перекрестных сшивок. Стадия фрагментации b) может включать обработку ультразвуком и последующую ферментативную репарацию концов ДНК. Обработка ультразвуком вызывает фрагментацию ДНК на произвольных сайтах, при этом фрагменты могут иметь тупые концы или липкие 3'- или 5'-концы; поскольку точечные разрывы ДНК являются произвольными, ДНК может быть репарирована (ферментативно) путем заполнения возможных липких 3'- или 5'-концов, в результате чего образуются фрагменты ДНК с тупыми концами, позволяющими лигировать указанные фрагменты с адапторами и/или друг с другом на следующей стадии с). Альтернативно липкие концы также можно сделать тупыми, удалив нуклеотиды липких концов, например, при помощи экзонуклеаз. Стадия фрагментации b) также может включать фрагментацию одним рестрикционным ферментом или комбинациями рестрикционных ферментов. Фрагментация рестрикционным ферментом является предпочтительной, так как позволяет контролировать средний размер фрагментов. Образовавшиеся фрагменты могут иметь совместимые липкие или тупые концы, позволяющие лигировать указанные фрагменты на следующей стадии с) без необходимости дальнейшей модификации. Кроме того, для деления образца перекрестно-сшитой ДНК на множество частей могут быть использованы рестрикционные ферменты с разными сайтами узнавания. Такой метод является предпочтительным, так как при использовании разных рестрикционных ферментов, имеющих разные сайты узнавания, из каждой части образца могут быть получены разные фрагменты ДНК.
На следующей стадии с) фрагменты лигируют. При получении фрагментов ДНК с помощью рестрикционных ферментов сайт узнавания рестрикционного фермента известен, что позволяет идентифицировать фрагменты на основании остатков сайтов или восстановленных сайтов узнавания рестрикционных ферментов, указывающих на разделение между разными фрагментами ДНК. При получении фрагментов ДНК путем произвольной фрагментации под воздействием ультразвука и последующей ферментативной репарации концов ДНК труднее отличить один фрагмент от другого. Независимо от использованного метода фрагментации стадия лигирования с) может быть выполнена в присутствии адаптора путем лигирования адапторных последовательностей между фрагментами. Альтернативно адаптор может быть лигирован при выполнении отдельной стадии. Преимуществом лигирования адаптора на отдельной стадии является простота идентификации разных фрагментов путем обнаружения адапторных последовательностей, расположенных между фрагментами. Например, в случае тупых концов фрагментов ДНК адапторные последовательности связываются с концами фрагментов ДНК, определяя границу между отдельными фрагментами ДНК.
Затем перекрестное сшивание реверсируется на стадии d), в результате чего образуется группа лигированных фрагментов ДНК, состоящая из двух или более фрагментов. Субпопуляция группы лигированных фрагментов ДНК включает фрагмент ДНК, содержащий последовательность нуклеотида-мишени. В результате реверсии перекрестного сшивания разрушается структурная/пространственная фиксация ДНК, и последовательность ДНК может быть использована на следующих стадиях, например, амплификации и/или секвенирования, так как перекрестно-сшитая ДНК не может быть приемлемым субстратом для таких стадий. Последующие стадии е) и/или f) могут быть выполнены после реверсии перекрестного сшивания, однако стадии е) и/или f) также могут быть выполнены при нахождении лигированных фрагментов ДНК в перекрестно-сшитом состоянии.
Лигированные фрагменты ДНК могут быть необязательно фрагментированы на стадии е) предпочтительно при помощи рестрикционного фермента. Указанная фрагментация может быть выполнена после реверсии перекрестного сшивания, но при этом предполагается, что до реверсии перекрестного сшивания может быть выполнена вторая фрагментация. Фрагментацию желательно выполнять при помощи рестрикционного фермента, так как рестрикционный фермент позволяет контролировать стадию фрагментации и получить при выборе соответствующего рестрикционного фермента совместимые концы лигированных фрагментов ДНК, пригодные для образования кольцевых лигированных фрагментов ДНК, полученных на стадии f). Однако фрагментация может быть выполнена другими методами, такими как, например, гидродинамическое фрагментирование и/или обработка ультразвуком, с последующим выполнением ферментативной репарации концов ДНК, благодаря чему образуется двухцепочечная ДНК с тупыми концами, которая также может быть лигирована с образованием кольцевой ДНК.
Целью выполнения первой стадии фрагментации и необязательной второй стадии фрагментации является получение лигированных фрагментов ДНК, пригодных для последующего выполнения стадии образования кольцевой молекулы, стадии амплификации и/или стадии определения последовательности. При выполнении стадий фрагментации b) и е) при помощи рестрикционных ферментов желательно, чтобы в результате выполнения стадии фрагментации е) были получены в среднем более длинные фрагменты по сравнению с фрагментами, полученными на стадии фрагментации b). При использовании на стадиях фрагментации b) и е) рестрикционных ферментов желательно, чтобы сайт узнавания рестрикционного фермента на стадии е) был длиннее сайта узнавания на стадии b). Таким образом, фермент, используемый на стадии е), разрезает последовательность с меньшей частотой, чем на стадии b). Из вышеизложенного следует, что средний размер фрагмента ДНК, полученного на стадии е), меньше среднего размера фрагмента, полученного на стадии е), после рестрикции ДНК. Поэтому на первой стадии фрагментации образуются относительно мелкие фрагменты, которые затем лигируются. Так как второй рестрикционный фермент, используемый на стадии е), разрезает последовательность менее часто, чем на стадии b), в большинстве фрагментов ДНК может отсутствовать сайт узнавания рестрикционного фермента, используемого на стадии е). Таким образом, при последующей фрагментации лигированных фрагментов ДНК на второй стадии многие фрагменты ДНК, полученные на стадии b), могут оставаться интактными. Такое состояние фрагментов может быть полезно, так как объединенные последовательности фрагментов ДНК, полученных на стадии b), могут быть использованы для создания совокупности смежных фрагментов для исследуемой области генома. Если фрагментация на стадии b) происходит менее часто, чем на стадии с), фрагменты, полученные на стадии b), будут снова фрагментированы, в результате чего могут быть потеряны относительно большие последовательности ДНК, пригодные для создания совокупности смежных фрагментов. Таким образом, независимо от метода фрагментации, использованного на стадии b) и е), желательно, чтобы фрагментация на стадии b) происходила чаще, чем на стадии е), благодаря чему фрагменты ДНК, полученные на стадии b), в основном остаются интактными, то есть не фрагментируются на стадии е).
Лигированные фрагменты ДНК, полученные на стадии d) или е), у которых было реверсировано перекрестное сшивание, используются для образования кольцевых молекул на стадии f). Перекрестное сшивание желательно реверсировать до образования кольцевых молекул, так как образование кольцевых молекул из перекрестно-сшитых ДНК может дать неблагоприятные результаты. Однако кольцевые молекулы могут быть также образованы из перекрестно-сшитых лигированных фрагментов ДНК. Вполне возможно, что не потребуется дополнительная стадия образования кольцевых молекул, так как кольцевые молекулы лигированных фрагментов ДНК могут быть образованы уже на стадии лигирования, поэтому стадия образования кольцевых молекул f) может быть выполнена одновременно со стадией с). Однако желательно выполнить дополнительную стадию образования кольцевых молекул. Образование кольцевых молекул предполагает лигирование концов лигированных фрагментов ДНК с образованием замкнутого кольца. Кольцевые ДНК, включающие лигированные фрагменты ДНК, содержащие последовательность нуклеотида-мишени, затем могут быть амплифицированы с использованием по меньшей мере одного праймера, гибридизирующего с последовательностью нуклеотида-мишени. Для выполнения стадии амплификации необходимо реверсировать перекрестное сшивание, так как перекрестно-сшитая ДНК может затруднять или препятствовать амплификации. Предпочтительно используют два праймера, гибридизирующих с последовательностью нуклеотида-мишени при осуществлении обратной реакции ПЦР. Подобным образом могут быть амплифицированы фрагменты кольцевой ДНК, лигированные с фрагментом ДНК, включающим последовательность нуклеотида-мишени.
Затем определяют последовательность (амплифицированных) лигированных фрагментов ДНК, полученных на стадии d), e), f) или g), включающих последовательность нуклеотида-мишени. Указанную последовательность предпочтительно определяют при помощи метода высокопроизводительного секвенирования, так как данная технология является более удобной и позволяет определить большое число последовательностей во всей исследуемой области генома. Из определенных последовательностей может быть создана совокупность смежных фрагментов исследуемой области генома. После определения последовательностей фрагментов ДНК могут быть получены перекрывающиеся транскрипты, из которых может быть создана исследуемая область генома. При получении фрагментов ДНК методом произвольной фрагментмации уже на стадии фрагментации могут образоваться фрагменты ДНК, которые при секвенировании позволяют получить перекрывающиеся транскрипты. При увеличении размера образца, например при увеличении числа анализируемых клеток, может быть повышена достоверность созданной исследуемой области генома. Альтернативно, когда на стадии b) подвергают анализу множество частей образцов при использовании разных рестрикционных ферментов, также могут быть получены перекрывающиеся транскрипты. При увеличении числа частей образцов увеличивается количество перекрывающихся фрагментов, что может повысить достоверность совокупности смежных фрагментов созданной исследуемой области генома. Из указанных определенных перекрывающихся последовательностей может быть создана совокупность смежных фрагментов. Альтернативно, если последовательности не перекрывают друг друга, например, при использовании одного рестрикционного фермента на стадии b), созданию совокупности смежных фрагментов исследуемой области генома может способствовать сравнение (лигированных) фрагментов ДНК с эталонной последовательностью.
Несколько последовательностей-мишеней
Один вариант осуществления изобретения относится к способу определения последовательности исследуемой области генома, включающей две последовательности нуклеотидов-мишеней. Указанный способ может включать выполнение вышеуказанных стадий до стадии амплификации. В данном случае при выполнении стадии амплификации используют не одну последовательность нуклеотида-мишени, а две. Для двух последовательностей нуклеотидов-мишеней при выполнении реакции ПЦР используют два разных праймера, по одному праймеру для каждой последовательности нуклеотида-мишени. При наличии в лигированном фрагменте ДНК двух сайтов связывания праймеров из двух последовательностей нуклеотидов-мишеней два праймера амплифицируют последовательность между двумя сайтами связывания праймеров при условии, что указанные сайты связывания праймеров правильно ориентированы. Использование кольцевого лигированного фрагмента ДНК может быть предпочтительным, так как вероятность правильной ориентации двух сайтов связывания праймеров выше по сравнению с линейным лигированным фрагментом ДНК (в кольцевой молекуле амплификация будет произведена в двух из четырех ориентаций по сравнению с одной из четырех ориентаций в линейном лигированном фрагменте ДНК). В другом варианте осуществления изобретения помимо двух последовательностей нуклеотидов-мишеней исследуемая область генома включает дополнительные нуклеотиды-мишени, и при выполнении амплификации при помощи реакции ПЦР используют отдельный праймер для каждого нуклеотида-мишени. Объединение нескольких нуклеотидов-мишеней и соответствующих праймеров на одной стадии амплификации увеличивает вероятность образования ампликона комбинациями праймеров.
Например, как описано в разделе ”примеры”, для гена BRCA1 были использованы 5 разных нуклеотидов-мишеней (см., например, фиг.2). ПЦР может быть выполнена в результате выбора праймера для одной последовательности нуклеотида-мишени (также определяемой как исследуемая точка), например исследуемая точка А с другой исследуемой точкой В. Кроме того, ПЦР может быть выполнена при использовании праймера для каждой последовательности нуклеотида-мишени, А, В, С, D и Е. Так как указанные нуклеотиды-мишени находятся в физической близости друг от друга, выполнение такой амплификации обогащает исследуемую область генома при условии, что сайты связывания праймеров заканчиваются в лигированных фрагментах ДНК, благодаря чему может быть образован ампликон.
Таким образом, настоящее изобретение относится к способам определения последовательности исследуемой области генома, включающей одну или более последовательностей нуклеотидов-мишеней, в которых на стадии амплификации может быть использован один праймер, гибридизирующий с последовательностью нуклеотида-мишени, и один или более праймеров, гибридизирующих с одним или несколькими дополнительными нуклеотидами-мишенями, при амплификации с использованием праймеров лигированных фрагментов ДНК или кольцевой ДНК.
Определение последовательности лигированных фрагментов ДНК
Стадию определения последовательности лигированных фрагментов ДНК предпочтительно выполняют методами высокопроизводительного секвенирования. Методы высокопроизводительного секвенирования хорошо известны в данной области, и, в принципе, при осуществлении настоящего изобретения может быть использован любой метод. Методы высокопроизводительного секвенирования могут быть выполнены в соответствии с инструкциями производителя (предоставляемыми, например, компаниями Roche, Illumina или Applied Biosystems). Как правило, с (амплифицированными) лигированными фрагментами ДНК могут быть лигированы адапторы секвенирования. В случае амплификации линейного или кольцевого фрагмента методом ПЦР, рассмотренным в настоящем описании изобретения, амплифицированный продукт является линейным, позволяя лигировать адапторы. Для лигирования адапторных последовательностей могут быть получены соответствующие концы (например, тупые, комплементарно неустойчивые концы). Альтернативно праймеры, используемые к для ПЦР или другого метода амплификации, могут включать адапторные последовательности с целью образования на стадии амплификации g) амплифицированных продуктов с адапторными последовательностями. Кольцевой фрагмент, не подлежащий амплификации, может быть фрагментирован, предпочтительно при помощи рестрикционного фермента, между сайтами связывания праймеров при выполнении обратной реакции ПЦР, в результате чего фрагменты ДНК, лигированные с фрагментом ДНК, включающим последовательность нуклеотида-мишени, остаются интактными. При выполнении стадий с) и f) способов по настоящему изобретению также могут быть использованы адапторы секвенирования. Указанные адапторы секвенирования могут быть частью адапторных последовательностей, необязательно используемых при выполнении указанных стадий, и/или на указанных стадиях могут быть использованы отдельные адапторы секвенирования.
При использовании метода высокопроизводительного секвенирования могут быть предпочтительно получены длинные транскрипты. Длинные транскрипты позволяют считывать несколько лигированных фрагментов ДНК. Подобным образом могут быть идентифицированы фрагменты ДНК, полученные на стадии b). Последовательности фрагментов ДНК можно сравнить с эталонной последовательностью и/или друг с другом. Например, как показано ниже, такие последовательности фрагментов ДНК могут быть использованы для определения соотношения фрагментов клеток, содержащих генетическую мутацию. В результате секвенирования последовательностей фрагментов ДНК, расположенных рядом с такими последовательностями, можно идентифицировать уникальные лигированные фрагменты ДНК. Это особенно важно при получении фрагментов ДНК на стадии b) путем произвольной фрагментации. Вероятность того, что две клетки содержат одинаковые фрагменты ДНК, является очень низкой, не говоря уже о том, что будут одинаковыми концы фрагмента ДНК, с которым лигирован такой фрагмент. Следовательно, идентифицируя фрагменты ДНК подобным образом, можно определить соотношение клеток и/или исследуемых областей генома, включающих определенную мутацию.
Таким образом, нет необходимости производить полное секвенирование лигированных фрагментов ДНК. Желательно секвенировать по меньшей мере (несколько) фрагментов ДНК, поскольку последовательности фрагментов ДНК уже определены.
Можно также считывать даже более короткие последовательности, например короткие транскрипты, состоящие из 50-100 нуклеотидов. В таком случае желательно фрагментировать (амплифицированную) лигированную ДНК с образованием фрагментов меньшей длины, которые затем могут быть лигированы с соответствующим адаптором, пригодным для выполнения высокопроизводительного секвенирования. При использовании стандартного метода секвенирования может быть потеряна информация о лигированных фрагментах ДНК. При наличии коротких транскриптов невозможно идентифицировать полную последовательность фрагментов ДНК. При использовании таких коротких транскриптов могут быть предусмотрены дополнительные стадии обработки, в соответствии с которыми отдельные лигированные фрагменты ДНК после фрагментации лигируют или снабжают идентификаторами, благодаря чему из коротких транскриптов могут быть созданы совокупности смежных фрагментов для лигированных фрагментов ДНК. Такие методы высокопроизводительного секвенирования, включающие короткие транскрипты, могут быть использованы для секвенирования спаренных концов. При секвенировании спаренных концов и использовании коротких транскриптов можно связать лигированные фрагменты ДНК, используя короткие транскрипты с обоих концов секвенируемой молекулы ДНК, которая может включать разные фрагменты ДНК. Подобная операция возможна благодаря тому, что два транскрипта могут быть связаны путем заполнения относительно большой последовательности ДНК на основании последовательности, определенной с обоих концов. Таким образом могут быть созданы совокупности смежных фрагментов для (амплифицированных) лигированных фрагментов ДНК.
Однако короткие транскрипты могут быть использованы без идентификации фрагментов ДНК, так как из коротких транскриптов может быть создана исследуемая область генома, в частности, если указанная исследуемая область генома была амплифицирована. Информация, относящаяся к фрагментам ДНК и/или отдельной исследуемой области генома (например, диплоидной клетки), может быть потеряна, но мутации ДНК все же могут быть идентифицированы.
Таким образом, стадия определения по меньшей мере части (амплифицированной) лигированной последовательности ДНК может быть выполнена с использованием коротких транскриптов, но желательно определять более длинные транскрипты, чтобы можно было идентифицировать последовательности фрагментов ДНК. Кроме того, могут быть использованы другие методы высокопроизводительного секвенирования (амплифицированных) лигированных фрагментов ДНК, например объединение коротких транскриптов, полученных в результате секвенирования спаренных концов, с концами, относительно удаленных более длинных транскриптов, благодаря чему могут быть созданы совокупности смежных фрагментов для (амплифицированных) лигированных фрагментов ДНК.
Один вариант осуществления изобретения относится к контролю качества созданной последовательности. При анализе последовательностей методом высокопроизводительного секвенирования могут возникнуть ошибки секвенирования. Ошибка секвенирования может возникнуть, например, во время удлинения цепи ДНК при включении неправильного (то есть некомплементарного матрице) основания в цепь ДНК. Ошибка секвенирования отличается от мутации, так как исходная ДНК, подвергаемая амплификации и/или секвенированию, не должна содержать такую мутацию. В соответствии с настоящим изобретением последовательности фрагментов ДНК могут быть определены вместе с (по меньшей мере с частью) последовательностями фрагментов ДНК, лигированными с указанной последовательностью, которые могут быть уникальными. Уникальность лигированных фрагментов ДНК, полученных на стадии с), позволяет произвести контроль качества последовательности, определенной на стадии h). При амплификации и секвенировании лигированных фрагментов ДНК на достаточную глубину будет секвенировано множество копий одного и того же уникального (лигированного) фрагмента ДНК. Можно сравнить последовательности копий, полученных из одного исходного лигированного фрагмента ДНК, и выявить ошибки амплификации и/или секвенирования.
Другие варианты осуществления изобретения
Используя образец перекрестно-сшитой ДНК, способами по настоящему изобретению можно определить последовательности нескольких исследуемых областей генома. Каждая исследуемая область генома содержит последовательность нуклеотида-мишени, для которой могут быть созданы один или более соответствующих праймеров. Несколько исследуемых областей генома могут представлять собой перекрывающиеся исследуемые области генома, в результате чего увеличивается длина последовательности, на основании которой может быть определена требуемая последовательность. Например, если последовательность исследуемой области генома, включающая последовательность нуклеотида-мишени, обычно имеет длину, равную 1 Мб, то при объединении 5 частично перекрывающихся исследуемых областей генома, каждая из которых содержит соответствующую последовательность нуклеотида-мишени, при перекрывании, равном, например, 0,1 Мб, будет получена последовательность длиной 4,6 Мб (0,9+3*(0,1+0,8)+0,1+0,9=4,6 Мб), благодаря чему значительно увеличивается размер исследуемой области генома, в которой может быть определена или подвергнута какому-либо другому анализу требуемая последовательность. Для увеличения среднего охвата и/или однородности охвата области генома можно использовать несколько последовательностей нуклеотидов-мишеней, расположенных на определенных расстояниях в исследуемой области генома.
Кроме того, по меньшей мере в один из олигонуклеотидных праймеров на стадии g) может быть введен идентификатор. Идентификаторы также могут быть введены в адапторные последовательности, используемые для лигирования между фрагментами на стадии лигирования с). В результате введения идентификатора в олигонуклеотидный праймер при одновременном анализе множества образцов или множества частей образцов перекрестно-сшитой ДНК можно легко определить происхождение каждого образца. Части образцов перекрестно-сшитой ДНК могут быть обработаны по-разному, несмотря на наличие одного исходного образца перекрестно-сшитой ДНК, и/или образцы ДНК могут быть получены из разных организмов или у разных субъектов. Идентификаторы позволяют объединять по-разному обработанные образцы для последующей одинаковой обработки образцов, например, при выполнении одинаковых стадий. Такая совместная обработка образцов может быть особенно предпочтительной на стадии секвенирования h), выполняемой методом высокопроизводительного секвенирования.
До или после стадии амплификации g) в соответствии со способами по настоящему изобретению может быть выполнена стадия выбора размера молекул. Стадия выбора размера молекул может быть выполнена при помощи экстракционной хроматографии, гель-электрофореза или центрифугирования в градиенте плотности, которые являются методами, хорошо известными в данной области. Обычно выбирают ДНК, размер которой равен 20-20000 пар оснований, предпочтительно 50-10000 пар оснований, наиболее предпочтительно 100-3000 пар оснований. На стадии выбора размера молекул можно выбрать (амплифицированные) лигированные фрагменты ДНК в диапазоне размеров, оптимальных для амплификации методом ПЦР и/или секвенирования длинных транскриптов на следующей стадии секвенирования. В настоящее время коммерчески доступными методами можно секвенировать транскрипты длиной 500 нуклеотидов; новые технологии, такие как метод секвенирования одной молекулы ДНК в реальном времени (SMRT™), разработанный компанией Pacific Biosciences (http://www.pacificbiosciences.com/), позволяют секвенировать транскрипты, включающие от 1000 до 10000 нуклеотидов.
Если плоидность клетки исследуемой области генома больше 1, способами по настоящему изобретению на стадии h) создается совокупность смежных фрагментов для каждой плоидности. Так как среда любого данного сайта-мишени в геноме в основном состоит из последовательностей ДНК, расположенных на расстоянии физической близости от последовательности-мишени на линейной матрице хромосомы, можно реконструировать любую конкретную матрицу хромосомы. Если плоидность исследуемой области генома больше 1, в клетке имеется несколько исследуемых областей генома (или их эквивалентов). Исследуемые области генома обычно не находятся в одном пространстве, то есть указанные области пространственно отделены друг от друга. При фрагментации образца перекрестно-сшитой ДНК такой клетки из каждой исследуемой области генома в клетке будет образован соответствующий фрагмент ДНК, включающий последовательность нуклеотида-мишени. Указанные фрагменты ДНК будут лигированы с фрагментами ДНК, расположенными в непосредственной близости от них. Таким образом, лигированные фрагменты ДНК будут представлять разные исследуемые области генома. Например, если плоидность равна двум и два фрагмента, содержащие уникальные мутации и отделенные друг от друга расстоянием, равным 1 Мб, будут обнаружены вместе в лигированных фрагментах ДНК, можно сделать вывод о том, что указанные два фрагмента находятся в одной исследуемой области генома. Таким образом были идентифицированы два фрагмента, относящиеся к одной области генома. При создании совокупности смежных фрагментов из последовательностей идентифицированных фрагментов два указанных фрагмента, содержащих мутацию, будут использованы для создания совокупности смежных последовательностей для одной конкретной области генома, в то время как совокупность смежных последовательностей, созданная для другой области генома, не будет содержать таких мутаций.
Таким образом, в соответствии со способами по настоящему изобретению стадия h) создания совокупности смежных фрагментов включает следующие стадии:
1) идентификацию фрагментов, полученных на стадии b);
2) соотнесение указанных фрагментов с областью генома;
3) создание совокупности смежных фрагментов для области генома из последовательностей указанных фрагментов.
Возможна ситуация, когда три фрагмента содержат уникальную мутацию (А*, В* и С*) и плоидность исследуемой области генома равна двум. В таком случае будут идентифицированы лигированные продукты, включающие два мутированных фрагмента, один из которых включает А*В* и другой включает А*С*. Кроме того, будут обнаружены лигированные продукты, включающие немутированные фрагменты, ВС и АС. В данном случае лигированные фрагменты ДНК А*В и А*С* связаны фрагментом А* и лигированные фрагменты ДНК ВС и АС связаны фрагментом С. Фрагменты ДНК А*, В* и С* относятся к одной области генома, в то время как А, В и С относятся к другой области генома. Таким образом, соотнесение фрагментов с областью генома на стадии 2) включает идентификацию разных лигированных продуктов и связывание разных лигированных продуктов, включающих фрагменты ДНК.
Аналогичным образом вышеизложенное относится к популяциям гетерогенных клеток. Например, при наличии образца перекрестно-сшитой ДНК, включающего популяцию гетерогенных клеток (например, клеток разного происхождения или клеток из организма, включающего нормальные клетки и генетически мутированные клетки (например, раковые клетки)), могут быть созданы совокупности смежных фрагментов для каждой исследуемой области генома, соответствующей разным средам генома (которые могут быть разными средами генома в клетке или разными средами генома из разных клеток).
Идентификация мутаций
Альтернативные варианты осуществления изобретения относятся к способам идентификации наличия или отсутствия генетической мутации.
Первый вариант осуществления изобретения относится к способу идентификации наличия или отсутствия генетической мутации, который включает стадии а)-h) любого из вышеописанных способов по настоящему изобретению, относящихся к созданию совокупности смежных фрагментов для множества образцов, и дополнительные стадии:
i) сравнение совокупности смежных фрагментов множества образцов;
j) идентификацию наличия или отсутствия генетической мутации в исследуемых областях генома из множества образцов.
Альтернативно настоящее изобретение относится к способу идентификации наличия или отсутствия генетической мутации, который включает стадии а)-h) любого из вышеописанных способов по настоящему изобретению и дополнительные стадии:
i) сравнение совокупности смежных фрагментов с эталонной последовательностью;
j) идентификацию наличия или отсутствия генетической мутации в исследуемой области генома.
Генетические мутации могут быть идентифицированы, например, путем сравнения совокупностей смежных фрагментов нескольких образцов, при этом, если один (или более) образцов включают генетическую мутацию, можно сделать вывод о том, что данная последовательность совокупности смежных фрагментов отличается от последовательности других образцов, то есть будет идентифицирована генетическая мутация. При отсутствии различий в последовательностях совокупностей смежных фрагментов будет идентифицировано отсутствие генетической мутации. Альтернативно может быть использована эталонная последовательность, с которой может быть произведено сравнение последовательности совокупности смежных фрагментов. Отличие последовательности совокупности смежных фрагментов образца от эталонной последовательности указывает на наличие генетической мутации, то есть в данном случае идентифицирована генетическая мутация. Отсутствие различий между совокупностью смежных фрагментов образца или образцов и эталонной последовательностью свидетельствует об отсутствии генетической мутации.
Для идентификации наличия или отсутствия генетической мутации не нужно создавать совокупность смежных фрагментов. Наличие или отсутствие генетической мутации можно идентифицировать, сравнивая последовательности фрагментов ДНК друг с другом или с эталонной последовательностью. Таким образом, альтернативные варианты осуществления изобретения относятся к способу идентификации наличия или отсутствия генетической мутации в соответствии с любым из вышеописанных способов без выполнения стадии h) создания совокупности смежных фрагментов.
Такой способ включает стадии а)-g) любого из вышеописанных способов и дополнительные стадии:
h) сравнение определенных последовательностей (амплифицированных) лигированных фрагментов ДНК с эталонной последовательностью;
i) идентификацию наличия или отсутствия генетической мутации в определенных последовательностях.
Альтернативно настоящее изобретение относится к способу идентификации наличия или отсутствия генетической мутации путем определения последовательностей (амплифицированных) лигированных фрагментов ДНК множества образцов, который включает стадии а)-g) любого из вышеописанных способов и дополнительные стадии:
h) сравнение определенных последовательностей (амплифицированных) лигированных фрагментов ДНК множества образцов;
i) идентификацию наличия или отсутствия генетической мутации в определенных последовательностях.
Соотношение аллелей или клеток,
содержащих генетическую мутацию
Как было указано выше, при получении образца перекрестно-сшитой ДНК из популяций гетерогенных клеток (например, клеток разного происхождения или клеток из организма, включающего нормальные клетки и генетически мутированные клетки (например, раковые клетки)), могут быть созданы совокупности смежных фрагментов для каждой исследуемой области генома, соответствующей разным средам генома (которые могут быть разными средами генома из разных аллелей в клетке или разными средами генома из разных клеток). Кроме того, может быть определено соотношение фрагментов или лигированных фрагментов ДНК, содержащих генетическую мутацию, которое может соответствовать соотношению аллелей или клеток, содержащих генетическую мутацию. Лигирование фрагментов ДНК является произвольным процессом, сборка и порядок следования фрагментов ДНК, являющихся частью лигированных фрагментов ДНК, могут быть уникальными и могут представлять одну клетку и/или одну исследуемую область генома клетки. Кроме того, при выполнении фрагментации на стадии b) методом произвольной фрагментации, таким как, например, обработка ультразвуком, точечные разрывы ДНК могут представлять собой дополнительный уникальный признак, особенно в контексте других фрагментов ДНК, с которыми лигирован данный фрагмент (которые также могут иметь уникальные концы).
Таким образом, идентификация лигированных фрагментов ДНК, включающих фрагмент с генетической мутацией, может также включать идентификацию лигированных фрагментов ДНК с уникальным порядком следования и сборкой фрагментов ДНК. Соотношение аллелей или клеток, содержащих генетическую мутацию, может иметь важное значение для оценки методов лечения, например, в случае субъектов, проходящих курс лечения от рака. Раковые клетки могут содержать определенную генетическую мутацию. Процентное значение клеток, содержащих такую мутацию, может быть мерой успешного или безуспешного лечения. Альтернативные варианты осуществления изобретения относятся к способу определения соотношения фрагментов, содержащих генетическую мутацию, и/или соотношения лигированных фрагментов ДНК, содержащих генетическую мутацию. В указанном варианте осуществления изобретения генетическая мутация может быть определена в виде конкретной генетической мутации или путем отбора конкретных генетических мутаций.
Первый вариант осуществления изобретения относится к способу определения соотношения фрагментов, содержащих генетическую мутацию, предположительно в популяции гетерологичных клеток, который включает стадии а)-h) любого из вышеописанных способов и дополнительные стадии:
i) идентификации фрагментов, полученных на стадии b);
j) идентификацию наличия или отсутствия генетической мутации в полученных фрагментах;
k) определение числа фрагментов, содержащих генетическую мутацию;
l) определение числа фрагментов, не содержащих генетическую мутацию;
m) вычисление соотношение фрагментов, содержащих генетическую мутацию.
Альтернативный вариант осуществления изобретения относится к способу определения соотношения лигированных продуктов, содержащих фрагмент с генетической мутацией, предположительно в популяции гетерологичных клеток, который включает стадии а)-h) любого из вышеописанных способов и дополнительные стадии:
i) идентификацию фрагментов, полученных на стадии b);
j) идентификацию наличия или отсутствия генетической мутации в полученных фрагментах;
k) идентификацию лигированных продуктов, полученных на стадии f), включающих фрагменты, содержащие или не содержащие генетическую мутацию;
l) определение числа лигированных продуктов, включающих фрагменты с генетической мутацией;
m) определение числа лигированных продуктов, включающих фрагменты без генетической мутации;
n) вычисление соотношения лигированных продуктов, содержащих генетическую мутацию.
В способах, определяемых указанными вариантами осуществления изобретения, наличие или отсутствие генетической мутации может быть выявлено на стадии j) в результате сравнения с эталонной последовательностью и/или сравнения последовательностей фрагментов ДНК из множества образцов.
В способах по настоящему изобретению идентифицированная генетическая мутация может представлять собой полиморфизм одного нуклеотида (SNP), инсерцию, инверсию и/или транслокацию. При обнаружении делеции и/или инсерции число фрагментов и/или лигированных продуктов в образце, содержащих указанную делецию и/или инсерцию, можно сравнить с эталонным образцом для идентификации обнаруженной делеции и/или инсерции. Делецию, инсерцию, инверсию и/или транслокацию можно также идентифицировать на основании наличия точечных разрывов хромосомы в анализируемых фрагментах.
В другом варианте осуществления изобретения вышеописанными способами определяют наличие или отсутствие метилированных нуклеотидов во фрагментах ДНК, лигированных фрагментах ДНК и/или исследуемых областях генома. Например, ДНК, полученная на стадиях а)-f), может быть обработана бисульфитом. В результате обработки ДНК бисульфитом остатки цитозина превращаются в урацил, при этом остатки 5-метилцитозина не подвергаются воздействию. Таким образом, обработка бисульфитом вызывает определенные изменения в последовательности ДНК, зависящие от состояния метилирования отдельных остатков цитозина, что позволяет получить информацию о метилировании отдельных нуклеотидов сегмента ДНК. В результате деления образцов на части, из которых один из образцов подвергают обработке, а другой не подвергают обработке, можно идентифицировать метилированные нуклеотиды. Альтернативно можно сравнить последовательности из множества образцов, обработанных бисульфитом, или последовательность из одного образца, обработанного бисульфитом, можно сравнить с эталонной последовательностью.
При анализе (коротких) транскриптов можно избежать секвенирования использованных праймеров. В альтернативном способе по настоящему изобретению последовательность праймера может быть удалена до выполнения стадии высокопроизводительного секвенирования. Таким образом, альтернативный вариант осуществления изобретения относится к способу определения последовательности исследуемой области генома, содержащей последовательность нуклеотида-мишени, который включает следующие стадии:
а) получение образца перекрестно-сшитой ДНК;
b) фрагментацию перекрестно-сшитой ДНК;
с) лигирование фрагментированной перекрестно-сшитой ДНК;
d) реверсию перекрестного сшивания;
е) необязательную фрагментацию ДНК, полученной на стадии d), предпочтительно при помощи рестрикционного фермента;
f) необязательное лигирование фрагментированной ДНК, полученной на стадии d) или е), по меньшей мере с одним адаптором;
g) амплификацию лигированных фрагментов ДНК, полученных на стадии d) или е), включающих последовательность нуклеотида-мишени, с использованием по меньшей мере одного праймера, который (1) имеет липкий 5'-конец, содержащий сайт узнавания рестрикционного фермента типа III, и (2) гибридизирует с последовательностью нуклеотида-мишени, или амплификацию лигированных фрагментов ДНК, полученных на стадии f), с использованием по меньшей мере одного праймера, который (1) предпочтительно имеет липкий 5'-конец, содержащий сайт узнавания рестрикционного фермента типа III, и (2) гибридизирует с последовательностью нуклеотида-мишени, и по меньшей мере одного праймера, который гибридизирует по меньшей мере с одним адаптором;
h) расщепление исследуемых амплифицированных нуклеотидных последовательностей рестрикционным ферментом типа III с последующим выполнением стадии выбора размера молекул для удаления высвобожденных двухцепочечных последовательностей праймеров;
i) фрагментацию ДНК предпочтительно путем обработки ультразвуком;
j) необязательное лигирование двухцепочечных последовательностей адаптора, необходимых для следующего секвенирования;
k) определение по меньшей мере части последовательности (амплифицированных) лигированных фрагментов ДНК, полученных на стадии d), e), f) или g), включающих последовательность нуклеотида-мишени, предпочтительно при помощи высокопроизводительного секвенирования;
l) идентификацию генетических изменений и создание совокупности смежных фрагментов исследуемой области генома из определенных последовательностей.
В альтернативном варианте осуществления изобретения в любых вышеописанных способах на стадии g) используют праймеры, содержащие часть, например биотин, для необязательной очистки (амплифицированных) лигированных фрагментов ДНК путем связывания с твердым носителем.
В одном варианте осуществления изобретения лигированные фрагменты ДНК, включающие последовательность нуклеотида-мишени, могут быть уловлены гибридизирующим зондом (или улавливающим зондом), который гибридизирует с последовательностью нуклеотида-мишени. Гибридизирующий зонд может быть присоединен непосредственно к твердому носителю или может включать часть, например биотин, обеспечивающую связывание с твердым носителем, пригодным для улавливания биотина (например, гранулы, покрытые стрептавидином). В любом случае происходит улавливание лигированных фрагментов ДНК, включающих последовательность нуклеотида-мишени, что позволяет отделить лигированные фрагменты ДНК, содержащие последовательность нуклеотида-мишени, от лигированных фрагментов ДНК, не содержащих последовательность нуклеотида-мишени. Такие стадии улавливания позволяют обогатить лигированные фрагменты ДНК, включающие последовательность нуклеотида-мишени. Следовательно, при осуществлении настоящего изобретения может быть выполнена стадия амплификации, которая также является стадией обогащения, альтернативно стадией улавливания при помощи зонда, специфичного к последовательности нуклеотида-мишени. При создании исследуемой области генома может быть использован по меньшей мере один улавливающий зонд для улавливания последовательности нуклеотида-мишени. При создании исследуемой области генома могут быть использованы несколько зондов для улавливания нескольких последовательностей нуклеотидов-мишеней. Например, аналогично описанию, приведенному для гена BRCA1, в качестве улавливающего зонда можно использовать по одному праймеру для каждой из 5 последовательностей нуклеотидов-мишеней (А, В, С, D или Е). Альтернативно можно объединить 5 праймеров (А, В, С, D и Е) для улавливания исследуемой области генома.
В одном варианте осуществления изобретения стадия амплификации и стадия улавливания могут быть объединены, при этом сначала выполняют стадию улавливания и затем стадию амплификации или наоборот.
В одном варианте осуществления изобретения может быть использован улавливающий зонд, который гибридизирует с адапторной последовательностью, введенной в (амплифицированные) лигированные фрагменты ДНК.
ПРИМЕР
Ниже приведен пример способа секвенирования гена по настоящему изобретению, который был использован для определения полной последовательности гена Brca1. Были использованы клетки SUM149PT, представляющие собой линию клеток рака молочной железы, с делецией Т в положении 2288 локуса Brca1 (Elstrodt et al., Cancer Res., 2006). Указанный способ схематически изображен на фиг.1.
Культивирование клеток
Клетки SUM149PT культивировали на чашках площадью 150 см2 до получения полных клеточных пластинок в среде RPMI, содержащей 10% FCS и пенициллин-стрептомицин. Предварительный подсчет клеток на чашке показал, что полная чашка площадью 150 см2 содержит ~20×106 клеток SUM149PT.
Фиксация и лизис клеток
Культивируемые клетки промывали PBS и фиксировали PBS/10% FCS/2% формальдегида в течение 10 минут при комнатной температуре. Затем клетки промывали, собирали, вводили в лизирующий буфер (50 мМ трис-буфера с HCl, рН 7,5, 150 мМ NaCl, 5 мМ EDTA, 0,5% NP-40, 1% TX-100 и однократные ингибиторы протеазы (Roche № 11245200)) и инкубировали в течение 10 минут на льду. Затем клетки промывали и вводили в Milli-Q.
Фрагментация 1: расщепление
Фиксированные лизированные клетки расщепляли ферментом NlaIII (New England Biolab № R0125).
Лигирование 1
Фермент NlaIII инактивировали нагреванием и стадию лигирования выполняли, используя ДНК-лигазу Т4 (Roche, № 799009).
Реверсия перекрестного сшивания
К образцу добавляли Prot K (10 мг/мл) и инкубировали при 65°С. Затем добавляли РНКазу А (10 мг/мл, Roche № 10109169001) и образец инкубировали при 37°С. Затем производили экстракцию фенолхлороформом, супернатант, содержащий ДНК, осаждали и гранулировали. Гранулу растворяли в 10 мМ трис-буфера с HCl, рН 7,5.
Фрагментация 2: второе расщепление
Расщепленный и лигированный образец расщепляли ферментом Nspl (New England Biolabs № R0602S).
Лигирование 2: второе лигирование и очистка
К образцу добавляли Prot K (10 мг/мл) и инкубировали при 65°С. Затем добавляли РНКазу А (10 мг/мл, Roche № 10109169001) и образец инкубировали при 37°С. Затем производили экстракцию фенолхлороформом, супернатант, содержащих ДНК, осаждали и гранулировали. Гранулу растворяли в 10 мМ трис-буфера с HCl, рН 7,5. Была получена матрица обогащения, которую можно хранить или сразу же использовать.
Амплификация лигированных фрагментов ДНК: ПЦР
Праймеры, используемые для обогащения методом ПЦР локуса Brca1, были созданы в виде инвертированных уникальных праймеров, расположенных рядом (<50 п.о.) с сайтами рестрикции рестриционного фрагмента NlaIII, при этом расстояние между наборами праймеров, то есть ”исследуемыми точками”, равно примерно 20 т.п.о. (см. фиг.2 и таблицу 1).
|
Таблица 1. Обзор использованных последовательностей праймеров. Названия праймеров относятся к гену BRCA1 и включают указание положения на карте последовательности (например, 50,1 (т.п.о.)) и определение: верхний праймер (fw) или нижний праймер (rev), vp означает исследуемые точки, ID означает SEQ ID NO:, то есть SEQ ID NO:1-10. Также указана последовательность гена BRCA1, к которой соответствует данный праймер (начальная (5') последовательность и концевая (3') последовательность), следует отметить, что праймеры имеют наружную ориентацию, то есть обратную ориентацию, указанные праймеры не могут образовать ампликон при использовании нормальной ДНК в качестве матрицы.
При выполнении типичной реакции обогащения методом ПЦР используют 25 мкл смеси:
- 2,5 мкл 10-кратного буфера 3 для ПЦР (поставляется вместе с удлиненной матричной полимеразой);
- 0,5 мкл dNTP (10 мМ);
- 0,5 мкл верхнего праймера (разведенного в отношении 1/7 из 1 мкг/мкл исходного праймера);
- 0,5 мкл нижнего праймера (разведенного в отношении 1/7 из 1 мкг/мл исходного праймера);
- 0,375 мкл удлиненной матричной полимеразы (Roche № 11759060001);
- 100 нг матрицы обогащения;
- Х мкл Milli-Q до общего объема, равного 25 мкл.
Секвенирование амплифицированных лигированных фрагментов ДНК
Была создана библиотека для секвекнирования методом SOLiD в соответствии со стандартными методами компании SOLiD.
Результаты
Распределение транскриптов от разных исследуемых точек было наибольшим вокруг сайта исследуемой точки. Статистические данные представлены в таблице 2. В последовательностях транскриптов из библиотек исследуемых точек С, D и Е была обнаружена мутация 2288delT. Также было установлено, какие последовательности гена BRCA1 не были охвачены: 15807 пар оснований от исследуемой точки А, 50124 п.о. от точки В. От исследуемых точек С, D и Е были охвачены все последовательности BRCA1.
|
Таблица 2. Статистические данные для последовательностей транскриптов каждой исследуемой точки. Представлены статистические данные последовательностей транскриптов для библиотеки исследуемых точек (vp А-Е). М (транскрипты, совместимые с геном BRCA1), TR (общее число транскриптов), % MtT (% от общего числа транскриптов, совместимых с мишенью), среднее значение (средний охват), срединное значение (срединный охват), % nt 20x (% нуклеотидов из гена BRCA1, охваченных более 20 раз).
Таким образом, из отдельных исследуемых точек С, D и Е был охвачен весь ген BRCA1 длиной 100 т.п.о., из исследуемой точки А было охвачено 85 т.п.о. гена BRCA1, из исследуемой точки В было охвачено 50 т.п.о. гена BRCA1 и из исследуемых точек С, D и Е была подтверждена мутация 2288delT.


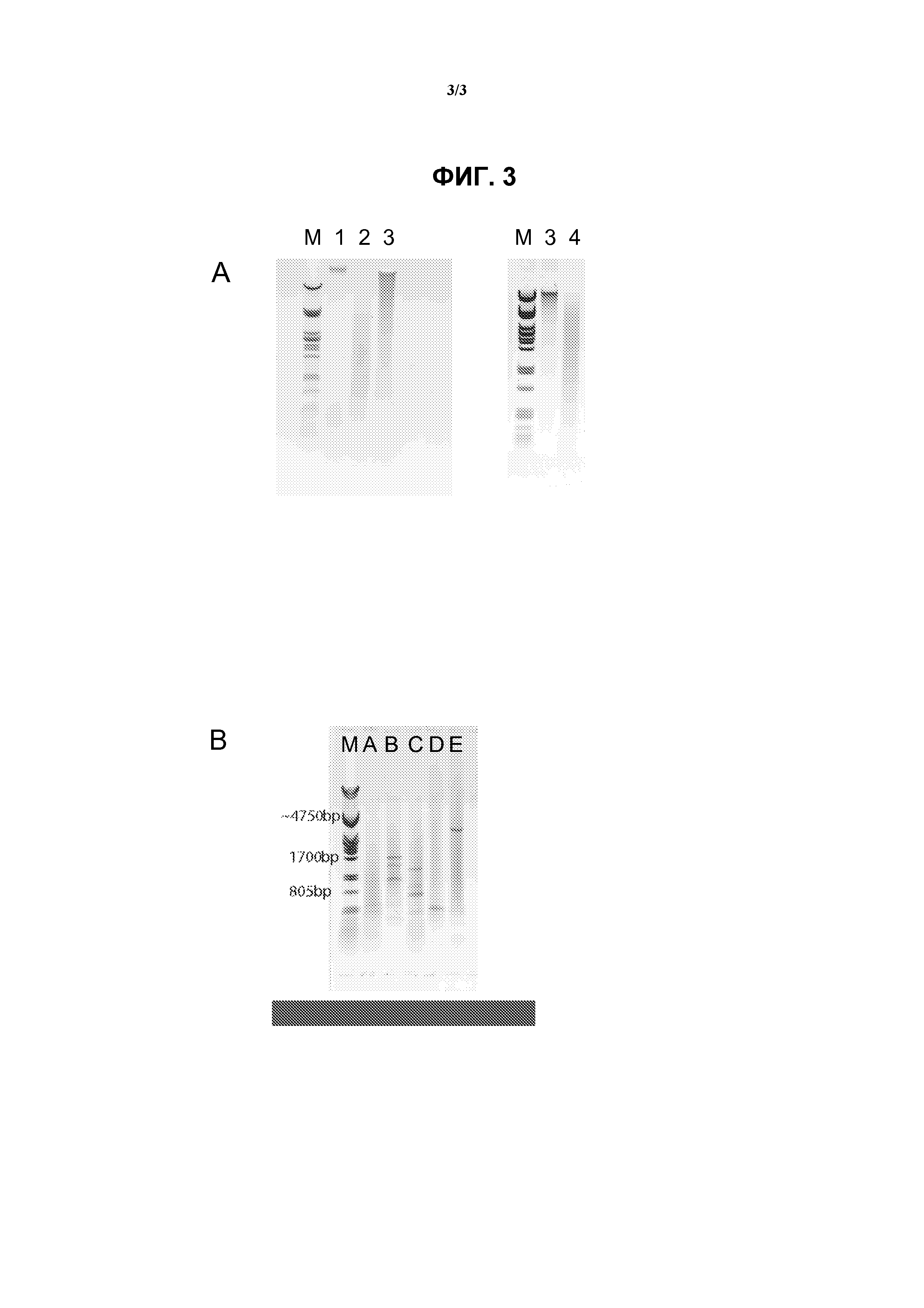

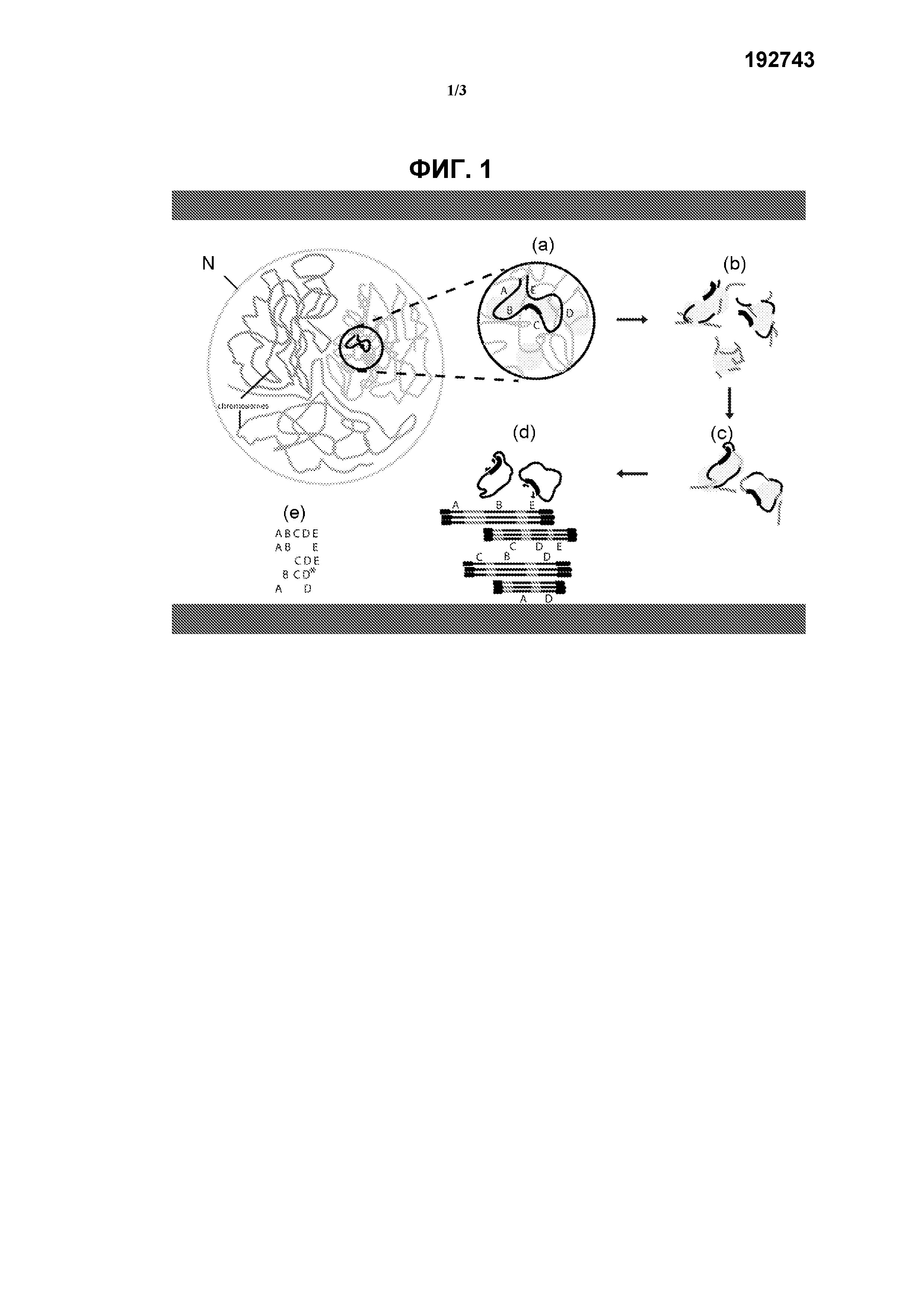
Культуральная среда для эпителиальных стволовых клеток и органоидов, содержащих указанные стволовые клетки
Органоид печени, варианты его применения и способ культивирования для его получения
Улучшенный способ дифференцировки
Культуральная среда для эпителиальных стволовых клеток и органоидов, содержащих указанные стволовые клетки
Органоид печени, варианты его применения и способ культивирования для его получения

