Результат интеллектуальной деятельности: МНОГОКОМПОНЕНТНЫЙ ОКСИДНЫЙ КАТАЛИЗАТОР, СПОСОБ ЕГО ИЗГОТОВЛЕНИЯ И СПОСОБ ИЗГОТОВЛЕНИЯ НЕНАСЫЩЕННОГО НИТРИЛА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение предлагает многокомпонентный оксидный катализатор, способ изготовления многокомпонентного оксидного катализатора и способ изготовления ненасыщенного нитрила с использованием многокомпонентного оксидного катализатора.
Уровень техники
В настоящее время ненасыщенный нитрил, который представляет собой общедоступный товарный продукт, изготавливают, главным образом, осуществляя каталитическое окисление в среде аммиака (аммоксидирование) и используя в этой реакции олефин, аммиак и кислород. С другой стороны, в последние годы всеобщее внимание привлек способ введения алкана, такого как пропан или изобутан, в качестве исходного материала вместо олефина в реакцию парофазного каталитического аммоксидирования, в которой образуется соответствующий ненасыщенный нитрил. Кроме того, были предложены разнообразные катализаторы для использования в такой реакции.
Патентный документ 1 описывает катализатор, в котором многокомпонентный оксид металла, содержащий Mo, V, Nb и B, нанесен на диоксид кремния, составляющий от 20 до 60 мас. % в пересчете на SiO2 по отношению к суммарной массе оксид металла и диоксида кремния, в качестве катализатора для парофазного каталитического окисления или парофазного каталитического аммоксидирования пропана или изобутана.
Патентный документ 2 описывает нанесенный на диоксид кремния катализатор, используемый, когда пропан или изобутан вводится в реакцию парофазного каталитического аммоксидирования, в которой образуется ненасыщенный нитрил, или пропан или изобутан вводится в реакцию парофазного каталитического окисления, в которой образуется ненасыщенная карбоновая кислота, причем нанесенный на диоксид кремния катализатор имеет определенный состав металлического компонента, относительное содержание диоксида кремния и объем пор.
Как правило, если рассматривать относительное содержание V и Sb в катализаторе аммоксидирования типа MoVSbOx, содержание V составляет более чем содержание Sb. Их соотношение хорошо известно для катализатора аммоксидирования, полученного путем гидротермального синтеза или аналогичным способом (см., например, непатентный документ 1). Таким образом, в многокомпонентном оксидном катализаторе аммоксидирования типа MoV, изготовленном способом, который не представляет собой гидротермальный синтез, соотношение содержания V и Sb также регулируется таким образом, чтобы оно не уменьшалось, насколько это возможно, или чтобы оно увеличивалось.
Патентный документ 3 описывает катализатор, обеспечивающий повышенный выход в реакции и срок службы катализатора. Однако используемое содержание V все же остается высоким. С другой стороны, для фактической реакции известно, что Mo в активной структуре образует комплекс с водой, образующейся в каталитической реакции или побочной реакции, что вызывает удаление Mo. В случае многокомпонентного кристалла MoV удаление Mo приводит к повышению относительного содержания V в многокомпонентном кристалле, что чрезмерно увеличивает активность по отношению к исходному материалу.
Список цитируемой литературы
Патентная литература
Патентный документ 1: публикация японской выложенной патентной заявки №2001-276618
Патентный документ 2: публикация японской выложенной патентной заявки №2002-219362
Патентный документ 3: публикация международной патентной заявки №WO 2012-090979
Непатентная литература
Непатентный документ 1: Ind. Eng. Chem. Res., 2006 г., т. 45, №2, с. 607-614
Сущность изобретения
Техническая проблема
По вышеупомянутым причинам, для промышленного производства требуется дополнительное повышение выхода. Даже если используются катализаторы, описанные в вышеупомянутых патентных документах 1-3, образуются многочисленные побочные продукты, такие как диоксид углерода (CO2) и монооксид углерода (CO), которые приводят к недостаточному выходу ненасыщенного нитрила. CO2 и CO представляют собой соединения, которые не находят применения. Подавление их образования также приводит к эффективному использованию исходного материала.
Настоящее изобретение было выполнено для решения вышеупомянутых проблем. Задача настоящего изобретения заключается в том, чтобы предложить многокомпонентный оксидный катализатор, который может подавлять образование CO2 и CO и повышать выход ненасыщенного нитрила в процессе введения пропана или изобутана в реакцию парофазного каталитического аммоксидирования, в которой образуется соответствующий ненасыщенный нитрил, а также способ изготовления многокомпонентного оксидного катализатора и способ изготовления ненасыщенного нитрила с использованием многокомпонентного оксидного катализатора.
Решение проблемы
Авторы настоящего изобретения выполнили исследования по вышеупомянутым проблемам. Авторы настоящего изобретения обнаружили, что ванадий (V) принимает участие в образовании CO2 и CO. Стало очевидным, что CO2 и CO образуются в больших количествах при использовании традиционного катализатора, потому что присутствует в определенной степени избыток V, с которым не образуют комплексы другие металлы, такие как Mo или Nb. Кроме того, было обнаружено, что Mo удаляется в процессе реакции, повышая относительное содержание V в многокомпонентном оксидном катализаторе и чрезмерно увеличивая активность по отношению к исходному материалу, и в результате этого ускоряются реакции образования побочных продуктов. С другой стороны, уменьшение содержания V в многокомпонентном оксидном катализаторе может вызывать отклонение соотношения металлов в составе активных частиц, которые рассматривались ранее, и чрезмерно уменьшать активность по отношению к исходному материалу. Таким образом, уменьшение содержания V в многокомпонентном оксидном катализаторе не рассматривалось.
Кроме того, чтобы решить вышеупомянутые проблемы, авторы настоящего изобретения выполнили всесторонние исследования для достижения низкого содержания V по отношению к Mo и повышения выхода. Авторы настоящего изобретения обнаружили, что когда соотношение Mo и V и соотношение V и Sb находятся в определенном интервале, эксплуатационные характеристики улучшаются, в то время как уменьшается избыток V. В результате этого было выполнено настоящее изобретение.
Таким образом, настоящее изобретение заключается в следующем.
1. Многокомпонентный оксидный катализатор, используемый для реакции парофазного каталитического окисления или реакции парофазного каталитического аммоксидирования пропана или изобутана, причем данный многокомпонентный оксидный катализатор содержит многокомпонентный оксид, состав которого представляет следующая формула (1):
Mo1VaSbbNbcWdZeOn (1),
в которой компонент Z представляет собой один или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют собой атомные соотношения элементов; и 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, 0≤e≤0,2 и 0,60<a/b<1,00.
2. Многокомпонентный оксидный катализатор по пункту 1, дополнительно содержащий от 20 до 70 масc. % диоксида кремния в пересчете на SiO2.
3. Способ изготовления многокомпонентного оксидного катализатора, содержащего многокомпонентный оксид, состав которого представляет следующая формула (1):
Mo1VaSbbNbcWdZeOn (1),
в которой компонент Z представляет собой один или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют собой атомные соотношения элементов; и 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, 0≤e≤0,2 и 0,60<a/b<1,00,
причем данный способ включает следующие стадии:
(I) стадия смешивания исходных материалов и изготовления жидкой смеси исходных материалов, которая содержит Mo, V, Sb, Nb, W и Z, и в которой атомные соотношения представляют собой 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, 0≤e≤0,2 и 0,60<a/b<1,00;
(II) стадия высушивания, на которой высушивается жидкая смесь исходных материалов, и получается высушенный порошок;
(III) стадия прокаливания, на которой прокаливается высушенный порошок, и получается прокаленный материал; и
(IV) стадия удаления покровного вещества, на которой удаляется покровное вещество, присутствующее на поверхности частиц прокаленного материала.
4. Способ изготовления многокомпонентного оксидного катализатора по пункту 3, в котором стадия смешивания исходных материалов (I) включает следующие стадии:
(a) изготовление водной жидкой смеси, содержащей Mo, V, Sb и компонент Z;
(b) добавление золя диоксида кремния и водного раствора пероксида водорода в водную жидкую смесь, полученную на стадии (a);
(c) смешивание раствора, полученного на стадии (b), с водным раствором, содержащим Nb, дикарбоновую кислоту, пероксид водорода и содержащее W соединение; и
(d) добавление содержащей порошок диоксида кремния жидкой суспензии в раствор, полученный на стадии (c), для старения раствора.
5. Способ изготовления соответствующего ненасыщенного нитрила посредством введения пропана или изобутана в реакцию парофазного каталитического окисления или реакцию парофазного каталитического аммоксидирования, в которой соответствующий ненасыщенный нитрил производится из пропана и изобутана, и используется многокомпонентный оксидный катализатор по пункту 1 или 2.
Полезные эффекты изобретения
Настоящее изобретение может предложить многокомпонентный оксидный катализатор, который может подавлять образование CO2 и CO и повышать выход ненасыщенного нитрила. Настоящее изобретение может также предложить способ изготовления многокомпонентного оксидного катализатора, в котором многокомпонентный оксидный катализатор можно легко изготавливать при низкой себестоимости. Кроме того, можно осуществлять способ изготовления ненасыщенного нитрила, в котором можно подавлять образование CO2 и CO и повышать выход ненасыщенного нитрила посредством использования данного многокомпонентного оксидного катализатора.
Описание вариантов осуществления
Далее будет подробно описан способ осуществления настоящее изобретение (далее называется просто "вариант осуществления настоящего изобретения"). Следующий вариант осуществления настоящего изобретения представлен, чтобы проиллюстрировать настоящее изобретение. Настоящее изобретение не следует истолковывать как ограниченное следующим описанием. Настоящее изобретение можно осуществлять при внесении соответствующих изменений в пределах объема настоящего изобретения.
Многокомпонентный оксидный катализатор
Многокомпонентный оксидный катализатор согласно варианту осуществления настоящего изобретения используется для реакции парофазного каталитического окисления или реакция парофазного каталитического аммоксидирования пропана или изобутана и содержит многокомпонентный оксид, состав которого представляет следующая формула (1):
Mo1VaSbbNbcWdZeOn (1),
в которой компонент Z представляет собой один или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют собой атомные соотношения элементов; и 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, 0≤e≤0,2 и 0,60<a/b<1,00.
Для многокомпонентного оксидного катализатора, с точки зрения предотвращения реакции разложения образующегося ненасыщенного нитрила, оказывается предпочтительным, что компонент Z однородно распределяется в частицах катализатора. Термин "однородно" означает, что распределение компонента Z не приводит к диспропорционированию в частицах катализатора. Предпочтительно термин "однородно" означает, что 80 мас. % или более оксидных частиц, содержащих компонент Z, присутствуют в частицах катализатора как мелкие частицы, все из которых имеют диаметр, составляющий 1 мкм или менее. Когда многокомпонентный оксидный катализатор содержит диоксид кремния, с точки зрения однородности, значение дисперсии (значение, полученное делением среднеквадратического отклонения на среднее значение) соотношения уровней сигнала компонентов Z и Si, во время анализа состава поперечного сечения частицы катализатора, находится предпочтительно в интервале от 0 до 0,5, предпочтительнее в интервале от 0 до 0,4 и еще предпочтительнее в интервале от 0 до 0,3. В настоящем документе значение дисперсии соотношения уровней сигнала обозначено как "Dx", как будет описано ниже.
Для многокомпонентного оксидного катализатора согласно варианту осуществления настоящего изобретения, с точки зрения повышения выхода ненасыщенного нитрила, важно установить атомное соотношение (а) содержания V и Мо и соотношение (a/b) содержания V и Sb в интервале 0,1≤а<0,2 и интервале 0,60<a/b<1,00, соответственно. По результатам исследования авторов настоящего изобретения становится очевидным, что в традиционном катализаторе присутствует чрезмерный избыток V, который не соединяется в достаточной степени в комплекс с другими металлическими компонентами, и присутствие чрезмерного избытка V превращается в фактор образования CO2 и СО и уменьшения выхода ненасыщенного нитрила. После этого в результате рассмотрения соотношения содержания Мо и V и соотношения содержания V и Sb, а также продолжения всестороннего исследования, стало очевидным, что чрезмерный избыток V можно подавлять, устанавливая атомное соотношение содержания V и Мо и соотношение содержания V и Sb в интервале 0,1≤а<0,2 и интервале 0,60<a/b<1, 00, соответственно. Таким образом, чрезмерный избыток V подавляется, и в результате
этого образование CO2 и CO может подавляться без уменьшения активности по отношению к исходному материалу, и выход ненасыщенного нитрила может повышаться.
Авторы настоящего изобретения выполнили исследования и обнаружили, что выход целевого продукта повышается, когда атомное соотношение содержания V и Mo и соотношение содержания V и Sb находится в пределах вышеупомянутых определенных интервалов. Когда атомное соотношение содержания V и Mo находится в интервале от 0,1≤a<0,2, положение, в котором должен, главным образом, присутствовать Mo, замещает V, и увеличивается содержание V в кристаллической структуре, что требуется для активности по отношению к пропану, и при этом повышается активность. Когда соотношение содержания V и Sb находится в интервале от 0,60<a/b<1,00, уменьшается вероятность того, что V и Mo окисляются в процессе прокаливания катализатора, и при этом подавляется осаждение оксидов V и Mo. Таким образом, предполагается, что увеличивается относительное количество активных частиц в многокомпонентном оксидном катализаторе, что повышает селективность в отношении целевого продукта. С вышеупомянутой точки зрения, значение a находится предпочтительно в интервале 0,12≤a<0,2 и предпочтительнее в интервале 0,13≤a<0,2. Значение a/b находится предпочтительно в интервале 0,60<a/b<0,95, и предпочтительнее в интервале 0,60<a/b<0,85. Когда значения a и a/b находятся в вышеупомянутых интервалах, может дополнительно подавляться образование CO2 и CO, и выход ненасыщенного нитрила может дополнительно улучшаться.
Значение b находится в интервале 0,15≤b≤0,5, предпочтительно в интервале 0,15≤b≤0,4 и предпочтительнее в интервале 0,15≤b≤0,3. Значение c находится в интервале 0,01≤c≤0,5, предпочтительно в интервале 0,01≤c≤0,4 и предпочтительнее в интервале от 0,01≤c≤0,35. Кроме того, значение d находится в интервале 0≤d≤0,4, предпочтительно в интервале 0≤d≤0,3 и предпочтительнее в интервале 0≤d≤0,25. Кроме того, значение e находится в интервале 0≤e≤0,2, предпочтительно в интервале 0≤e≤0,15 и предпочтительнее в интервале 0≤e≤0,1. Когда значения b, c, d и e находятся в вышеупомянутых интервалах, может дополнительно подавляться образование CO2 и CO, и выход ненасыщенного нитрила может дополнительно улучшаться.
Число n представляет собой относительное количество атомов кислорода, которое определяется значениями a, b, c, d и e. Z представляет собой один или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba. Среди них Yb, Y, La, и Ce являются предпочтительными, и Ce является более предпочтительным.
Многокомпонентный оксидный катализатор согласно варианту осуществления настоящего изобретения дополнительно содержит предпочтительно от 20 до 70 мас. % диоксида кремния, предпочтительнее 40 до 65 мас. % диоксида кремния и еще предпочтительнее 40 до 60 мас. % диоксида кремния в пересчете на SiO2 по отношению к суммарной массе катализатора, содержащего многокомпонентный оксид и диоксид кремния. Содержание диоксида кремния составляет 20 мас. % или более, и в результате этого дополнительно улучшается активность катализатора. Когда содержание диоксида кремния составляет 70 мас. % или менее, катализатор, как правило, проявляет повышенную активность.
Способ измерения концентрации составляющего катализатор элемента не ограничивается определенным образом. Можно использовать обычный способ измерения концентрации металла. Например, можно использовать рентгенофлуоресцентный анализ (XRF). Когда измеряется концентрация металла в твердых частицах катализатор, можно надлежащим образом использовать рентгенофлуоресцентный анализ с точки зрения простоты измерения и точности количественного определения и т. д. Когда анализируется небольшое количество металла, катализатор переводится в соответствующий раствор, и его количество можно определять методом индуктивно-связанной плазмы (ICP) или атомной абсорбции с использованием жидкого раствора. Когда является желательным определение количества углерода, водорода и азота, можно надлежащим образом использовать количественный анализ соответствующих элементов (CHN). Упомянутое выше значение Dx можно определять методом электронно-зондового микроанализа (EPMA) и т. д.
Способ изготовления многокомпонентного оксидного катализатора
Способ изготовления многокомпонентного оксидного катализатора согласно варианту осуществления настоящего изобретения представляет собой способ изготовления многокомпонентного оксидного катализатора, содержащего многокомпонентный оксид, состав которого представляет следующая формула (1):
Mo1VaSbbNbcWdZeOn (1),
в которой компонент Z представляет собой один или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют собой атомные соотношения элементов; и 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, 0≤e≤0,2 и 0,60<a/b<1,00,
причем данный способ включает следующие стадии:
(I) стадия смешивания исходных материалов и изготовление жидкой смеси исходных материалов, которая содержит Mo, V, Sb, Nb, W и Z, и в которой атомные соотношения представляют собой 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, 0≤e≤0,2 и 0,60<a/b<1,00;
(II) стадия высушивания, на которой высушивается жидкая смесь исходных материалов, и получается высушенный порошок;
(III) стадия прокаливания, на которой прокаливается высушенный порошок, и получается прокаленный материал; и
(IV) стадия удаления покровного вещества, на которой удаляется покровное вещество, присутствующее на поверхности частиц прокаленного материала.
(I) Стадия смешивания исходных материалов
На стадии смешивания исходных материалов согласно варианту осуществления настоящего изобретения, жидкую смесь исходных материалов изготавливают таким образом, что например, атомное соотношение V (a), атомное соотношение Sb (b), атомное соотношение Nb (c), атомное соотношение W (d) и атомное соотношение Z (e) в расчете на один атом Mo находятся в интервалах 0,1≤a<0,2, 0,15≤b≤0,5, 0,01≤c≤0,5, 0≤d≤0,4, и 0≤e≤0,2, соответственно, и 0,60<a/b<1,00. Соотношение компонентов устанавливают таким образом, чтобы его значение отличалось от соотношения компонентов получаемого, в конечном счете, многокомпонентного оксидного катализатора. Это объясняется тем, что покровное вещество катализатора, которое будет описано далее, имеет состав, отличающийся от состава основного материала катализатора; соотношение компонентов катализатора в целом также изменяется посредством отделения покровного вещества от основного материала катализатора; и соотношение компонентов на стадии смешивания исходных материалов устанавливается с учетом этого изменения. В настоящем описании термин "покровное вещество" означает вещество, которое попадает и/или прикрепляется на поверхность прокаленного материала, полученного посредством заключительного прокаливания, которое будет описано далее. "Покровное вещество" означает вещество, которое выступает с поверхности прокаленного материала или прикрепляется на поверхность.
Изготовление катализатора
Исходный материал-источник Mo не ограничивается определенным образом. В частности, могут быть использованы гептамолибдат аммония (NH4)6Mo7O24·4H2O, триоксид молибдена MoO3, фосфорномолибденовая кислота H3PMo12O40, кремнемолибденовая кислота H4SiMo12O40, и пентахлорид молибдена MoCl5 и т. д. Среди них особенно предпочтительным является гептамолибдат аммония (NH4)6Mo7O24·4H2O.
Исходный материал-источник V не ограничивается определенным образом. В частности, могут быть использованы метаванадат аммония NH4VO3, оксид ванадия(V) V2O5, хлориды ванадия VCl4 и VCl3 и т. д. Среди них особенно предпочтительным является метаванадат аммония NH4VO3.
Исходный материал-источник Sb не ограничивается определенным образом. В частности, могут быть использованы оксиды сурьмы Sb2O3 и Sb2O5, сурьмянистая кислота HSbO2, сурьмяная кислота HSbO3, антимонат аммония NH4SbO3, хлорид сурьмы SbCl3, соль органической кислоты, такая как тартрат сурьмы, металлическая сурьма и т. д. Среди них особенно предпочтительным является оксид сурьмы(III) Sb2O3.
Исходный материал-источник Nb не ограничивается определенным образом. В частности, могут быть использованы ниобиевая кислота, неорганический ниобат и органический ниобат. Среди них особенно предпочтительной является ниобиевая кислота. Ниобиевая кислота имеет формулу Nb2O5·nH2O и называется также терминами "гидроксид ниобия" или "гидрат оксида ниобия". Кроме того, в качестве исходного материала-источника Nb предпочтительно используется раствор содержащего Nb вещества, в котором молярное соотношение дикарбоновой кислоты и ниобия составляет от 1 до 4. В качестве дикарбоновой кислоты предпочтительной является щавелевая кислота.
Исходный материал-источник W не ограничивается определенным образом. В частности, могут быть использованы содержащая вольфрам соль, такая как соль аммония, нитрат, карбоксилат, карбоксилат аммония, пероксокарбоксилат, пероксокарбоксилат аммония, галогенированная соль аммония, галогенид, ацетилацетонат, алкоксид, трифенильное соединение, полиоксометаллат, аммония полиоксометаллат; триоксид вольфрама, диоксид вольфрама, вольфрамовая кислота, водный раствор метавольфрамата аммония, паравольфрамат аммония, кремневольфрамовая кислота, кремневольфрамомолибденовая кислота и т.д. Среди них особенно предпочтительным является водный раствор метавольфрамата аммония.
Исходный материал-источник Z (одного или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba) не ограничивается определенным образом, при том условии, что данный исходный материал содержит какие-либо из этих элементов. Могут быть использованы соединения, содержащие какие-либо их этих элементов, а также материалы, в которых какие-либо из этих элементов-металлов солюбилизированы соответствующим реагентом. В качестве соединения, содержащего какие-либо из этих элементов, обычно могут быть использованы соль аммония, нитрат, карбоксилат, карбоксилат аммония, пероксокарбоксилат, пероксокарбоксилат аммония, галогенированная соль аммония, галогенид, ацетилацетонат, алкоксид и т. д. Среди них предпочтительно используется водный раствор исходного материала, такого как нитрат или карбоксилат.
При смешивании исходных материалов процедура растворения, процедура смешивания или процедура диспергирования исходных материалов-источников составляющих катализатор элементов не ограничивается определенным образом. Исходные материалы можно растворять, смешивать или диспергировать в одной и той же водной среде. В качестве альтернативы, исходные материалы можно индивидуально растворять, смешивать или диспергировать в водных средах, а затем можно смешивать эти водные среды. В случае необходимости можно осуществлять нагревание и/или перемешивание.
Когда многокомпонентные оксидные катализаторы согласно варианту осуществления настоящего изобретения представляет собой катализатор, содержащий диоксид кремния, и предпочтительно нанесенный на диоксид кремния катализатор, носителем которого является диоксид кремния, жидкую смесь исходных материалов предпочтительно изготавливают таким образом, чтобы в ней содержался исходный материал-источник диоксида кремния. Исходный материал-источник диоксид кремния не ограничивается определенным образом. В частности, можно использовать золь диоксида кремния. Порошок диоксида кремния можно использовать в качестве части или всего исходного материала-источника диоксида кремния.
Стадия смешивания исходных материалов согласно варианту осуществления настоящего изобретения предпочтительно включает следующие стадии:
(a) изготовление водной жидкой смеси, содержащей Mo, V, Sb и компонент Z;
(b) добавление золя диоксида кремния и водного раствора пероксида водорода в водную жидкую смесь, полученную на стадии (a);
(c) смешивание раствора, полученного на стадии (b), с водным раствором, содержащим Nb, дикарбоновую кислоту, пероксид водорода и содержащее W соединение; и
(d) добавление содержащей порошок диоксида кремния жидкой суспензии в раствор, полученный на стадии (c), для старения раствора. В результате этого степени окисления металлов становятся более подходящими на стадии перед прокаливанием.
Далее вышеупомянутая стадия смешивания исходных материалов будет описана с использованием примера, в котором жидкую смесь исходных материалов-источников нанесенного на диоксид кремния катализатора, содержащего Mo соединения, содержащего V соединения, содержащего Sb соединения, содержащего Nb соединения, содержащего W соединения и содержащего Z соединения изготавливают, используя воду в качестве растворителя и/или дисперсионной среды. Следует отметить, что стадия смешивания исходных материалов не ограничивается этим.
Содержащее Mo соединение, содержащее V соединение, содержащее Sb соединение и содержащее Z соединение добавляют в воду и нагревают, получая водную жидкую смесь (A). Температура нагревания и продолжительность нагревания для изготовления водной жидкой смеси (A) предпочтительно устанавливаются таким образом, что исходные соединения могут растворяться в достаточной степени. Температура нагревания предпочтительно составляет от 70°C до 100°C. Продолжительность нагревания предпочтительно составляет от 30 минут до 5 часов. В процессе растворения водную жидкую смесь (A) предпочтительно перемешивают таким образом, что повышается вероятность растворения исходных материалов. При этом внутреннее пространство резервуара может быть заполнено воздухом. Однако, с точки зрения регулирования степени окисления элементов в получаемом многокомпонентном оксидном катализаторе, внутреннее пространство резервуара может быть заполнено азотом. Состояние водной жидкой смеси (A) после окончания нагревания определяется как водная жидкая смесь (A′). Температура водной жидкой смеси (A′) составляет предпочтительно 20°C или более и 80°C или менее и предпочтительнее 40°C или более и 80°C или менее. Температура водной жидкой смеси (A′) составляет 20°C или более, и в результате этого уменьшается вероятность того, что происходит осаждение соединений металлов, растворенных в водной жидкой смеси (A′).
После этого золь диоксида кремния добавляется в водную жидкую смесь (A) или водную жидкую смесь (A′), когда завершается нагревание. Золь диоксида кремния выполняет функцию носителя. Температура при добавлении золя диоксида кремния предпочтительно составляет 80°C или менее. Когда золь диоксида кремния добавляется при температуре, составляющей 80°C или менее, устойчивость золя диоксида кремния является относительно высокой, что, как правило, подавляет гелеобразование в жидкой смеси исходных материалов. Золь диоксида кремния можно добавлять, когда начинается старение, которое будет описано далее, в процессе старения или непосредственно перед тем, как жидкая смесь исходных материалов высушивается. Золь диоксида кремния предпочтительно добавляется в водную жидкую смесь (A′). Кроме того, с точки зрения регулирования степени окисления элементов в получаемом многокомпонентном оксиде, соответствующее количество водного раствора пероксида водорода предпочтительно добавляется в водную жидкую смесь (A′) по мере необходимости. Водный раствор пероксида водорода можно добавлять в готовую водную жидкую смесь (A′). Водный раствор пероксида водорода можно добавлять в процессе изготовления водной жидкой смеси (A′) или после или до того, как добавляется золь диоксида кремния. При этом, с точки зрения регулирования степени окисления элементов в получаемом многокомпонентном оксиде в надлежащем интервале, водный раствор пероксида водорода добавляется в таком количестве, что молярное соотношение H2O2/Sb составляет предпочтительно от 0,01 до 5, предпочтительнее от 0,5 до 3 и еще предпочтительнее от 1 до 2,5.
Температура нагревания и продолжительность нагревания после добавления водного раствора пероксида водорода в водную жидкую смесь (A′) предпочтительно устанавливаются таким образом, чтобы в достаточной степени происходила жидкофазная реакция окисления, в которой участвует водный раствор пероксида водорода. Температура нагревания предпочтительно составляет от 30°C до 70°C. Продолжительность нагревания предпочтительно составляет от 5 минут до 4 часов. Аналогичным образом, скорость вращения при перемешивании в процессе нагревания можно устанавливать в зависимости от скорости вращения, при которой становится вероятным жидкофазная реакция окисления, в которой участвует водный раствор пероксида водорода. С точки зрения осуществления в достаточной степени жидкофазной реакции окисления, в которой участвует водный раствор пероксида водорода, в процессе нагревания предпочтительно сохраняется состояние перемешивания. Водная жидкая смесь, изготовленная таким образом, определяется как (A′′).
После этого содержащее Nb соединение и дикарбоновая кислота нагреваются в процессе перемешивания в воде, и получается жидкая смесь (B0). Пример дикарбоновой кислоты представляет собой щавелевая кислота (COOH)2. После этого водный раствор пероксида водорода предпочтительно добавляется в жидкую смесь (B0), и получается водная жидкая смесь (C). При этом, с точки зрения образования комплекса с содержащим Nb соединением и стабилизации содержащего Nb соединение в растворенном состоянии, надлежащего регулирования окислительно-восстановительного состояния составляющего катализатор элемента и изготовления катализатора, имеющего надлежащие эксплуатационные характеристики, молярное соотношение H2O2/Sb составляет предпочтительно от 0,5 до 20 и предпочтительнее от 1 до 10.
После этого, в зависимости от целевого состава, водная жидкая смесь (A′′), водная жидкая смесь (C), содержащее W соединение и порошок диоксида кремния надлежащим образом смешиваются, и получается водная жидкая смесь (D). Затем полученная водная жидкая смесь (D) подвергается обработке путем старения, и получается жидкая смесь исходных материалов. С точки зрения изготовления катализатора, имеющего надлежащие эксплуатационные характеристики, используемый здесь порошок диоксида кремния предпочтительно добавляется в раствор, полученный путем смешивания водной жидкой смеси (A′′), водной жидкой смеси (C) и содержащего W соединения. Порошок диоксида кремния можно добавлять в неизменном виде. Предпочтительнее порошок диоксида кремния добавляется в виде жидкости, в которой порошок диоксида кремния диспергирован в воде, т.е. в виде содержащей порошок диоксида кремния жидкой суспензии. При этом концентрация порошка диоксида кремния в содержащей порошок диоксида кремния жидкой суспензии составляет предпочтительно от 1 до 30 мас. % и предпочтительнее от 3 до 20 мас. %. Когда концентрация порошка диоксида кремния составляет 1 мас. % или более, в результате этого, как правило, может подавляться искажение формы частиц катализатора, вызываемое низкой вязкостью суспензии. При этом, как правило, можно подавлять также снижение активности частиц катализатора и аналогичные явления. Концентрация порошка диоксида кремния составляет 30 мас. % или менее, и в результате этого возникает возможность предотвращения гелеобразования в жидкой смеси исходных материалов и закупоривания трубопровода, вызываемого высокой вязкостью жидкой смеси исходных материалов, что делает возможным легкое получение высушенного порошка. Кроме того, эксплуатационные характеристики катализатора также проявляют тенденцию к дальнейшему улучшению.
Старение водной жидкой смеси (D) означает, что водная жидкая смесь (D) выдерживается в неподвижном состоянии или перемешивается в течение заданного времени. Продолжительность старения составляет предпочтительно 90 минут или более и 50 часов или менее, и предпочтительнее 90 минут или более и 6 часов или менее. Когда продолжительность старения находится в пределах вышеупомянутого интервала, в результате этого становится вероятным образование водной жидкой смеси (D), имеющей подходящее окислительно-восстановительное состояние (электрический потенциал), что, как правило, дополнительно улучшает эксплуатационные характеристики полученного многокомпонентного оксида в качестве катализатора. При этом, когда многокомпонентный оксидный катализатор производится в промышленном масштабе после высушивания с помощью распылительная сушилка, скорость обработки методом распылительной сушки обычно определяет производительность. Требуется продолжительное время для осуществления распылительной сушки всей жидкой смеси после распылительной сушки части водной жидкой смеси (D). При этом старение продолжается старение водной жидкой смеси, которая не подвергается обработке посредством распылительной сушки. Таким образом, продолжительность старения включает не только продолжительность старения перед высушиванием на стадии (II), которое будет описано далее, но также период времени от начала до окончания высушивания. С точки зрения предотвращения конденсации содержащего Mo компонента и осаждения оксидов металлов, включая соединения V и другого металла или множества металлов, температура старения предпочтительно составляет 25°C или более. С точки зрения предотвращения чрезмерной скорости гидролиза комплекса, содержащего Nb и пероксид водорода, чтобы получалась суспензия в предпочтительной форме, температура старения составляет предпочтительно 65°C или менее, предпочтительнее 25°C или более и 65°C или менее и еще предпочтительнее 45°C или более и 60°C или менее. Катализатор может дополнительно восстанавливаться в процессе прокаливания посредством увеличения продолжительности старения, повышения температуры старения и т.д., или посредством осуществления увеличения продолжительности и температуры старения в сочетании. Авторы настоящего изобретения провели всесторонние исследования и в результате этого обнаружили, что степень восстановления катализатора после прокаливания и окислительно-восстановительный потенциал суспензии имеют некоторую корреляцию. Если окислительно-восстановительный потенциал суспензии увеличивается, катализатор после прокаливания становится окислительным. Если окислительно-восстановительный потенциал суспензии уменьшается, катализатор после прокаливания становится восстановительным. Таким образом, окислительно-восстановительный потенциал суспензии составляет предпочтительно от 400 до 600 мВ, предпочтительнее от 450 до 550 мВ и еще предпочтительнее от 470 до 510 мВ.
(II) Стадия высушивания
Стадия высушивания согласно варианту осуществления настоящего изобретения представляет собой стадию, на которой высушивается жидкая смесь исходных материалов, и получается высушенный порошок. Высушивание можно осуществлять, используя известный способ. Например, высушивание можно осуществлять посредством распылительной сушки или выпаривания досуха. Когда осуществляется реакционный процесс в псевдоожиженном слое для реакции парофазного каталитического окисления или реакции парофазного каталитического аммоксидирования, высушенный порошок предпочтительно получается в состоянии микросфер, которое является предпочтительным с точки зрения обеспечения текучести внутри реактора, или в подобном состоянии, и, таким образом, предпочтительно осуществляется распылительная сушка. В качестве способа распыления в процессе распылительной сушки можно использовать способ центрифугирования, способ двухкомпонентного распыления или способ распыления при высоком давлении. В качестве источника тепла для высушивания можно использовать воздух, нагреваемый паром, или электрический нагреватель.
Скорость распыления, скорость поступающей жидкой смеси исходных материалов, скорость вращения распылителя в случае способа центрифугирования и другие параметры предпочтительно устанавливаются таким образом, чтобы обеспечивать подходящий размер частиц получаемого высушенного порошка. Средний диаметр частиц высушенного порошка составляет предпочтительно от 35 до 75 мкм, предпочтительнее от 40 до 70 мкм и еще предпочтительнее от 45 до 65 мкм. Средний диаметр частиц не изменятся в значительной степени даже после прокаливания.
(III) Стадия прокаливания
Стадия прокаливания согласно варианту осуществления настоящего изобретения представляет собой стадию, на которой прокаливается высушенный порошок, и получается прокаленный материал. Например, вращающаяся печь (вращающаяся обжиговая печь) может использоваться в качестве прокаливающего устройства для прокаливания высушенного порошка. Форма прокаливающего устройства для прокаливания высушенного порошка согласно настоящему изобретению не ограничивается определенным образом. В качестве данной формы предпочтительно используется трубчатая форма (трубчатая печь) с точки зрения возможности осуществления непрерывного прокаливания, и особенно предпочтительной является цилиндрическая форма. С точки зрения возможности регулирования температуры прокаливания предпочтительным способом повышения температуры или другим способом, способ нагревание предпочтительно представляет собой способ внешнего нагревания. Может быть надлежащим образом использована электрическая печь. Размер, качество и другие параметры трубчатой печи для прокаливания можно выбирать соответствующим образом в зависимости от условий прокаливания или объема производства.
Прокаливание желательно осуществлять в два этапа на стадии прокаливания. Когда первое прокаливание определяется как предварительное прокаливание, и последующее прокаливание определяется как заключительное прокаливание, оказывается предпочтительным, что предварительное прокаливание осуществляется в температурном интервале от 250 до 400°C, и заключительное прокаливание осуществляется в температурном интервале от 450 до 700°C. Предварительное прокаливание и заключительное прокаливание можно осуществлять в непрерывном режиме. Заключительное прокаливание можно осуществлять немедленно после того, как завершается предварительное прокаливание. Предварительное прокаливание и заключительное прокаливание можно соответствующим образом разделять на несколько этапов.
Прокаливание можно осуществлять в условиях атмосферы или циркуляции воздуха. Однако, с точки зрения получения многокомпонентного оксидного катализатора в предпочтительном окислительно-восстановительном состоянии, по меньшей мере, частично прокаливание предпочтительно осуществляется в условиях циркуляции инертного газа, в котором практически отсутствует кислород, например, в условиях циркуляции азота. Когда прокаливание осуществляется в периодическом режиме, количество поступающего инертного газа составляет предпочтительно 50 Нл/час или более на 1 кг высушенного порошка, предпочтительнее от 50 до 5000 Нл/час и еще предпочтительнее от 50 до 3000 Нл/час с точки зрения получения многокомпонентного оксидного катализатора в предпочтительном окислительно-восстановительном состоянии. При этом "Нл" означает объем газа, измеренный в литрах в стандартных условиях температуры и давления, которые составляют, соответственно, 0°C и 1 атм. (0,1 МПа).
В способе изготовления многокомпонентного оксидного катализатора согласно варианту осуществления настоящего изобретения степень восстановления предварительно прокаленного материала составляет предпочтительно от 7 до 15%, предпочтительнее от 8 до 12% и еще предпочтительнее 9 до 12%. Когда степень восстановления в способе изготовления катализатора находится в пределах данного интервала, например, дополнительно повышается выход. Конкретные примеры способа регулирования степени восстановления в желательном интервале включают способ изменения температуры предварительного прокаливания, способ введения окисляющего компонента, такого как кислород, в атмосферу в процессе прокаливания или способ введения восстанавливающего компонента в атмосферу в процессе прокаливание. Данные способы могут сочетаться друг с другом.
(IV) Стадия удаления покровного вещества
Стадия удаления покровного вещества согласно варианту осуществления настоящего изобретения представляет собой стадию, на которой удаляется покровное вещество, который присутствует на поверхности частиц прокаленного материала. Многие покровные вещества представляют собой выступающие на поверхности кристаллы оксидов и другие примеси. В частности, в случае прокаленного материала, содержащий множество металлов, оксид, имеющий состав, который отличается от состава кристалла, образующего основную часть прокаленного материала, может образовываться в такой форме, что данный оксид выходит из основной части прокаленного материала. Поскольку такое покровное вещество превращается в фактор, снижающий текучесть, покровное вещество необходимо удалять с поверхность катализатора. Когда покровное вещество удаляется в граммовых количествах, можно использовать следующее устройство. Таким образом, может быть использована перпендикулярная трубка, в которой перфорированная пластина, имеющая одно или несколько отверстий, установлена в нижней часть, и бумажный фильтр установлен в верхней части. Прокаленные материалы помещаются в перпендикулярную трубку, и воздух циркулирует из нижней части. Таким образом, воздушный поток выходит из каждого отверстия, вступая в контакт с прокаленными материалами, и в результате этого происходит удаление покровных веществ.
Способ изготовления ненасыщенного нитрила
Способ изготовления ненасыщенного нитрила согласно варианту осуществления настоящего изобретения представляет собой способ изготовления соответствующего ненасыщенного нитрила посредством введения пропана или изобутана в реакцию парофазного каталитического окисления или реакцию парофазного каталитического аммоксидирования, в которой соответствующий ненасыщенный нитрил производится из пропана и изобутана, и в которой используется многокомпонентный оксидный катализатор согласно варианту осуществления настоящего изобретения. Далее будет описан способ введения пропана в реакцию парофазного каталитического аммоксидирования в состоянии, в котором пропан, аммиак и содержащий кислород газ приводятся в контакт с многокомпонентным оксидным катализатором согласно варианту осуществления настоящего изобретения, который заполняет реактор, и в результате этого производится акрилонитрил.
Исходные материалы
Используемые в качестве исходных материалов пропан и аммиак не обязательно должны иметь высокую чистоту, но должны быть технически чистыми, например, можно использовать пропан, содержащий не более чем приблизительно 3 об. % примесей, таких как, например, этан, этилен, n-бутан и изобутан, и аммиак должен содержать не более чем приблизительно 3 об. % примесей, таких как, например, вода. Примеры содержащего кислород газа представляют собой, но не ограничиваются этим, воздух, обогащенный кислородом воздух, чистый кислород или газ, такой как любой из перечисленных выше газов, разбавленный инертным газом, таким как гелий, аргон, диоксид углерода или азот, или водяной пар. Когда газы используются в промышленном масштабе, среди них этих газов предпочтительно используется воздух по соображениям простоты.
Условия реакции
Реакция парофазного каталитического окисления пропана или изобутана не ограничивается определенным образом. В частности, реакция парофазного каталитического окисления может осуществляться в следующих условиях. Молярное соотношение кислорода, который поступает для реакции, и пропана или изобутана составляет предпочтительно от 0,1 до 6 и предпочтительнее от 0,5 до 4. Температура реакции составляет предпочтительно от 300 до 500°C и предпочтительнее от 350 до 500°C. Давление реакции составляет предпочтительно от 5·104 до 5·105 Па, и предпочтительнее от 1·105 до 3·105 Па. Продолжительность контакта составляет предпочтительно от 0,1 до 10 с·г/см3 и предпочтительнее от 0,5 до 5 с·г/см3. Когда условия реакции находятся в вышеупомянутых интервалах, в результате этого, как правило, появляется дополнительная возможность подавления образование CO2 и CO, и, соответственно, может, как правило, дополнительно повышаться выход ненасыщенного нитрила.
Согласно варианту осуществления настоящего изобретения, продолжительность контакта определяется следующей формулой.
Продолжительность контакта (с·г/см3)=(W/F)·273/(273+T)
При этом W, F и T определяются следующим образом:
W = заполняющее количество (г) катализатора;
F = скорость потока (Н·см3/с) исходного смешанного газа в стандартных условиях (0°C, 1,013·105 Па); и
T = температура реакции (°C).
Реакция парофазного каталитического аммоксидирования пропана или изобутана с использованием многокомпонентный оксидный катализатор согласно варианту осуществления настоящего изобретения не ограничивается определенным образом. В частности, реакция парофазного каталитического аммоксидирования может осуществляться в следующих условиях. Молярное соотношение кислорода, который поступает для реакции, и пропана или изобутана составляет предпочтительно от 0,1 до 6 и предпочтительнее от 0,5 до 4. Молярное соотношение аммиака, который поступает для реакции, и пропана или изобутана составляет предпочтительно от 0,3 до 1,5 и предпочтительнее от 0,7 до 1,2. Температура реакции составляет предпочтительно от 350 до 500°C и предпочтительнее от 380 до 470°C. Давление реакции составляет предпочтительно от 5·104 до 5·105 Па, и предпочтительнее от 1·105 до 3·105 Па. Продолжительность контакта составляет предпочтительно от 0,1 до 10 с·г/см3 и предпочтительнее от 0,5 до 5 с·г/см3. Когда условия реакции находятся в вышеупомянутых интервалах, в результате этого, как правило, появляется дополнительная возможность подавления образование CO2 и CO, и, соответственно, может, как правило, дополнительно повышаться выход ненасыщенного нитрила.
Традиционные способы, такие как способ неподвижного слоя, способ псевдоожиженного слоя и способ подвижного слоя, можно использовать в качестве способа осуществления реакции парофазного каталитического окисления и реакции парофазного каталитического аммоксидирования. Среди них, вследствие простоты отвода выделяющегося в реакции тепла, предпочтительным оказывается реактор с псевдоожиженным слоем. Реакция парофазного каталитического аммоксидирования может осуществляться в прямоточной системе или рециркуляционной системе.
Примеры
Далее вариант осуществления настоящего изобретения будет описан более подробно со ссылкой на примеры и сравнительные примеры, но вариант осуществления настоящего изобретения не ограничивается данными примерами.
Выход акрилонитрила (ненасыщенного нитрила)
В данных примерах и сравнительных примерах выход акрилонитрила определяли следующим образом. Число моль полученного акрилонитрила измеряли, осуществляя предварительный анализ газа, содержащего акрилонитрил в известной концентрации, методом газовой хроматографии, используя газовый хроматограф модели GC2014 (производитель Shimadzu Corporation), получая калибровочную кривую, а затем вводя в газовый хроматограф определенное количество газа, образующегося в реакции аммоксидирования.
Выход акрилонитрила (%) = (число моль полученного акрилонитрила)/(число моль введенного пропана)·100
Выход CO2 и CO
В данных примерах и сравнительных примерах выход CO2 и CO определяли следующим образом. Число моль полученных CO2 и CO измеряли, осуществляя предварительный анализ газа, содержащего CO2 и CO в известных концентрациях, методом газовой хроматографии, используя газовый хроматограф модели GC2014 (производитель Shimadzu Corporation), получая калибровочную кривую, а затем вводя в газовый хроматограф определенное количество газа, образующегося в реакции аммоксидирования.
Выход CO2 или CO (%) = (число моль полученного CO2 или CO)/{(число моль введенного пропана или изобутана) · (число атомов углерода пропана или изобутана)} · 100
Пример 1
Изготовление высушенного порошка
Высушенный порошок (D1) изготавливали следующим образом.
К 1,771 г воды добавляли 340,5 г гептамолибдата аммония (NH4)6Mo7O24·4H2O, 46,9 г метаванадата аммония NH4VO3, 66,2 г оксида сурьмы(III) Sb2O3 и 4,2 г нитрата церия Ce(NO3)3·6H2O, и смесь нагревали при 95°C в течение одного часа в процессе перемешивания, получая водный исходный жидкий материал (A1).
К 257,6 г содержащей ниобий жидкой смеси (B0), в которой молярное соотношение щавелевой кислоты и ниобия составляло 2,5, добавляли 40 г водного раствора пероксида водорода, содержащего 30 мас. % H2O2, и осуществляли перемешивание при комнатной температуре в течение 10 минут, получая водный исходный жидкий материал (B1).
После того, как полученный водный исходный жидкий материал (A1) охлаждали до 70°C, к нему добавляли 564,7 г золя диоксида кремния, содержащего 34,0 мас. % SiO2, а затем к смеси дополнительно добавляли 80 г водного раствора пероксида водорода, содержащего 30 мас. % H2O2. После этого полученную в результате смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем водный исходный жидкий материал (B1), 26,5 г водного раствора метавольфрамата аммония (имеющего концентрацию 50%) и жидкую дисперсию, полученную посредством диспергирования 192 г порошка диоксида кремния в 1728 г воды, последовательно добавляли в водный исходный жидкий материал (A1), и полученную в результате смесь затем перемешанный и подвергали старению при 50°C в течение 2,5 часов, получая жидкую смесь (C1) в форме водной суспензии в качестве жидкой смеси исходных материалов.
Полученную водную жидкую смесь (C1) направляли в центрифужную распылительную сушилку (здесь и далее источник тепла для высушивания представлял собой воздух), и в результате высушивания получался высушенный порошок (D1) в форме микросфер. Температуры на впуске и выпуске сушилки составляли, соответственно, 210°C и 120°C.
Операция просеивания
Полученный высушенный порошок (D1) просеивали, используя сито, имеющее отверстия размером 25 мкм, и получался высушенный порошок (E1) как просеянный материал. В полученном высушенном порошке (E1) относительное содержание частиц, диаметр которых составлял 25 мкм или менее, составляло 0,2 мас. %, и средний диаметр частицы составлял 54 мкм. Здесь и далее относительное содержание частиц и средний диаметр частиц измеряли, используя лазерный дифракционный прибор под торговым наименованием LS230 (производитель Beckman Coulter).
Прокаливание высушенного порошка (E1)
Полученный высушенный порошок (E1) вводили в исходном количестве 80 г/час в непрерывном режиме в изготовленную из нержавеющей стали цилиндрическую вращающуюся трубчатую печь для прокаливания, у которой диаметр (здесь и далее внутренний диаметр) составлял 3 дюйма (7,62 см), и длина составляла 89 см. Газообразный азот поступал со скоростью 1,5 Нл/мин в направлении, противоположном направлению движения высушенного порошка (то есть в противоточном режиме, что применяется и далее), и с такой же скоростью в том же направлении (то есть в прямоточном режиме, что применяется и далее), соответственно, в трубчатой печи для прокаливания, и суммарная скорость потока составляла 3,0 Нл/мин. Температуру трубчатой печи для прокаливания устанавливали таким образом, что данная температура повышалась в течение 4 часов до максимальной температуры прокаливания, составляющей 360°C, и при этом трубчатая печь для прокаливания вращалась со скоростью 4 об/мин, и температура поддерживалась на уровне 360°C в течение одного часа для осуществления предварительного прокаливания. Небольшое количество предварительно прокаленного материала отбирали в качестве образца на выпуске трубчатой печи для прокаливания и нагревали до 400°C в атмосфере азота. После этого измеряли степень восстановления. Степень восстановления составляла 10,2%. Собранный предварительно прокаленный материал вводили в исходном количестве 60 г/час в непрерывном режиме в изготовленную из нержавеющей стали цилиндрическую вращающуюся трубчатую печь для прокаливания, у которой диаметр (здесь и далее внутренний диаметр) составлял 3 дюйма (7,62 см), и длина составляла 89 см. Газообразный азот поступал со скоростью 1,1 Нл/мин в направлении, противоположном направлению движения высушенного порошка и с такой же скоростью в том же направлении, соответственно, в трубчатой печи для прокаливания, и суммарная скорость потока составляла 2,2 Нл/мин. Температуру трубчатой печи для прокаливания устанавливали таким образом, что данная температура повышалась до 680°C в течение 2 часов, выдерживалась на уровне 680°C в течение 2 часов и затем снижалась до 600°C в течение 8 часов, чтобы осуществлялось заключительное прокаливание.
Удаление покровного вещества
Помещали 50 г прокаленного материала (F1) в перпендикулярную трубку, имеющую внутренний диаметр 41,6 мм и длину 70 см, в нижней части которой был установлен перфорированный диск, имеющий три отверстия, каждое диаметром 1/64 дюйма (0,39685 мм), и в верхней части был установлен бумажный фильтр. После этого воздух циркулировал при комнатной температуре через каждое отверстие в направлении верхней части из нижней части перпендикулярной трубки и вступал в контакт с прокаленными материалами. При этом длина пути воздушного потока в направлении движения воздушного потока составляла 56 мм, и средняя линейная скорость воздушного потока составляла 332 м/с. Покровное вещество отсутствовало в многокомпонентном оксидном катализаторе (G1), полученном после 24 часов.
Соотношение a/b компонентов многокомпонентного оксидного катализатора (G1) измеряли, осуществляя рентгенофлуоресцентный анализ и используя устройство под торговым наименованием RINT1000 (производитель Rigaku Corporation, здесь и далее напряжение рентгеновской трубки 50 кВ, ток трубки 50 мА). Полученные результаты представлены в таблице 1. При этом состав полученного многокомпонентного оксидного катализатора (G1) представлял собой Mo1V0,190Sb0,200Nb0,100W0,03Ce0,005On/53,2 мас. % SiO2.
Реакция аммоксидирования пропана
Пропан вводили в реакцию парофазного каталитического аммоксидирования следующим способом, используя многокомпонентный оксидный катализатор (G1), полученный, как описано выше. В используемый для реакции в псевдоожиженном слое трубчатый реактор из высококремнеземистого стекла викор (Vycor), имеющий внутренний диаметр 2 5 мм, помещали 35 г многокомпонентного оксидного катализатора. В этот трубчатый реактор вводили смешанный газ, содержащий пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, при продолжительности контакта 3,0 (с·г/см3) в условиях температуры реакции 440°C и атмосферного давления (0,1 МПа). Выходы акрилонитрила (AN) при осуществлении реакции в непрерывном режиме в течение 30 суток с использованием катализатора представлены в таблице 1.
Примеры 2-9 и сравнительные примеры 1-8
Были изготовлены катализаторы, имеющие составы, представленные в таблице 1, и реакцию аммоксидирования пропана осуществляли таким же способом, как в примере 1, используя катализаторы. Выходы реакций с использованием катализаторов
представлены в таблице 1.
|
На основании результатов примеров было показано, что катализаторы согласно варианту осуществления настоящего изобретения обеспечивают высокий выход целевого продукта и низкий выход CO2 и CO выход. В частности, выходы CO2 и CO значительно уменьшаются в сопоставлении со сравнительными примерами, и, таким образом, можно предположить, что подавляется не только разложение акрилонитрила, но также образование полезных побочных продуктов, таких как ацетонитрил или цианистоводородная кислота.
Основанием для составления настоящей заявки служит японская патентная заявка (японская патентная заявка №2012-214867, поданная в патентное ведомство Японии 27 сентября 2012 г.), содержание которой включается в настоящий документ посредством ссылки.
Промышленная применимость
Многокомпонентный оксидный катализатор согласно настоящему изобретению можно надлежащим образом использовать в качестве катализатора, который может подавлять образование CO2 и CO, а также повышать выход ненасыщенного нитрила в процессе введения пропана или изобутана в реакцию парофазного каталитического аммоксидирования, в которой образуется соответствующий ненасыщенный нитрил.
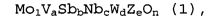
Половолоконный мембранный модуль с покрытой внешней периферией мембраны
Способ получения изоцианата
Метакриловая смола, литое изделие из нее и способ получения метакриловой смолы
Способ улавливания электроосаждаемой краски
Катионообменная мембрана, электролизер с ее использованием и способ изготовления катионообменной мембраны
Способ получения изоцианатов с использованием диарилкарбоната
Способ получения сложного эфира n-замещенной карбаминовой кислоты и способ получения изоцианата с использованием сложного эфира n-замещенной карбаминовой кислоты
Устройство для удаления поверхностных веществ катализатора
Устройство для получения смешанного раствора и способ приготовления смешанного раствора
Способ получения ненасыщенного нитрила
Половолоконный мембранный модуль с покрытой внешней периферией мембраны
Способ получения изоцианата
Метакриловая смола, литое изделие из нее и способ получения метакриловой смолы
Способ улавливания электроосаждаемой краски
Катионообменная мембрана, электролизер с ее использованием и способ изготовления катионообменной мембраны
Способ получения изоцианатов с использованием диарилкарбоната
Способ получения сложного эфира n-замещенной карбаминовой кислоты и способ получения изоцианата с использованием сложного эфира n-замещенной карбаминовой кислоты
Устройство для удаления поверхностных веществ катализатора
Устройство для получения смешанного раствора и способ приготовления смешанного раствора
Способ получения ненасыщенного нитрила

