КОМБИНИРОВАННАЯ СИСТЕМА ДОСТАВКИ С НЕМЕДЛЕННЫМ/ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ С КОРОТКИМ ПЕРИОДОМ ПОЛУВЫВЕДЕНИЯ, В ТОМ ЧИСЛЕ ДЛЯ РЕМОГЛИФЛОЗИНА
Вид РИД
Изобретение
Уровень техники
Широкая распространенность сахарного диабета вызывает рост общественной обеспокоенности во всем мире. По данным 2007 года, примерно 246 миллионов человек страдают этим заболеванием, и ежегодно выявляют дополнительно 7 миллионов человек, у которых возникло это заболевание. Предполагается, что к 2025 году число больных диабетом достигнет 380 миллионов человек.
Сахарный диабет является метаболическим синдромом, для которого характерна гипергликемия, возникающая в результате абсолютного дефицита секреции инсулина (диабет 1 типа) или в результате резистентности к действию инсулина, с сопутствующим неадекватным компенсаторным повышением секреции инсулина (диабет 2 типа). Хроническая гипергликемия является основным фактором риска развития микроваскулярных осложнений, таких как ретинопатия, нефропатия и нейропатия. Если усилия по соблюдению здорового образа жизни не могут привести к достижению и поддержанию целевого гликемического уровня, то необходимо дополнительное лечение.
Сущность изобретения
Настоящее изобретение относится к новой лекарственной форме препаратов с коротким периодом полувыведения, таких как антидиабетический препарат ремоглифлозина этабонат, которая обеспечивает немедленное высвобождение лекарственного средства, а также замедленное высвобождение второго болюса лекарственного средства, что, таким образом, позволяет достичь режима введения дозы, по меньшей мере, 250 мг ремоглифлозина этабоната один раз в сутки и может обеспечить эффективный контроль уровня глюкозы в плазме крови; также изобретение относится к способу лечения диабета с применением такой лекарственной формы.
Краткое описание фигур
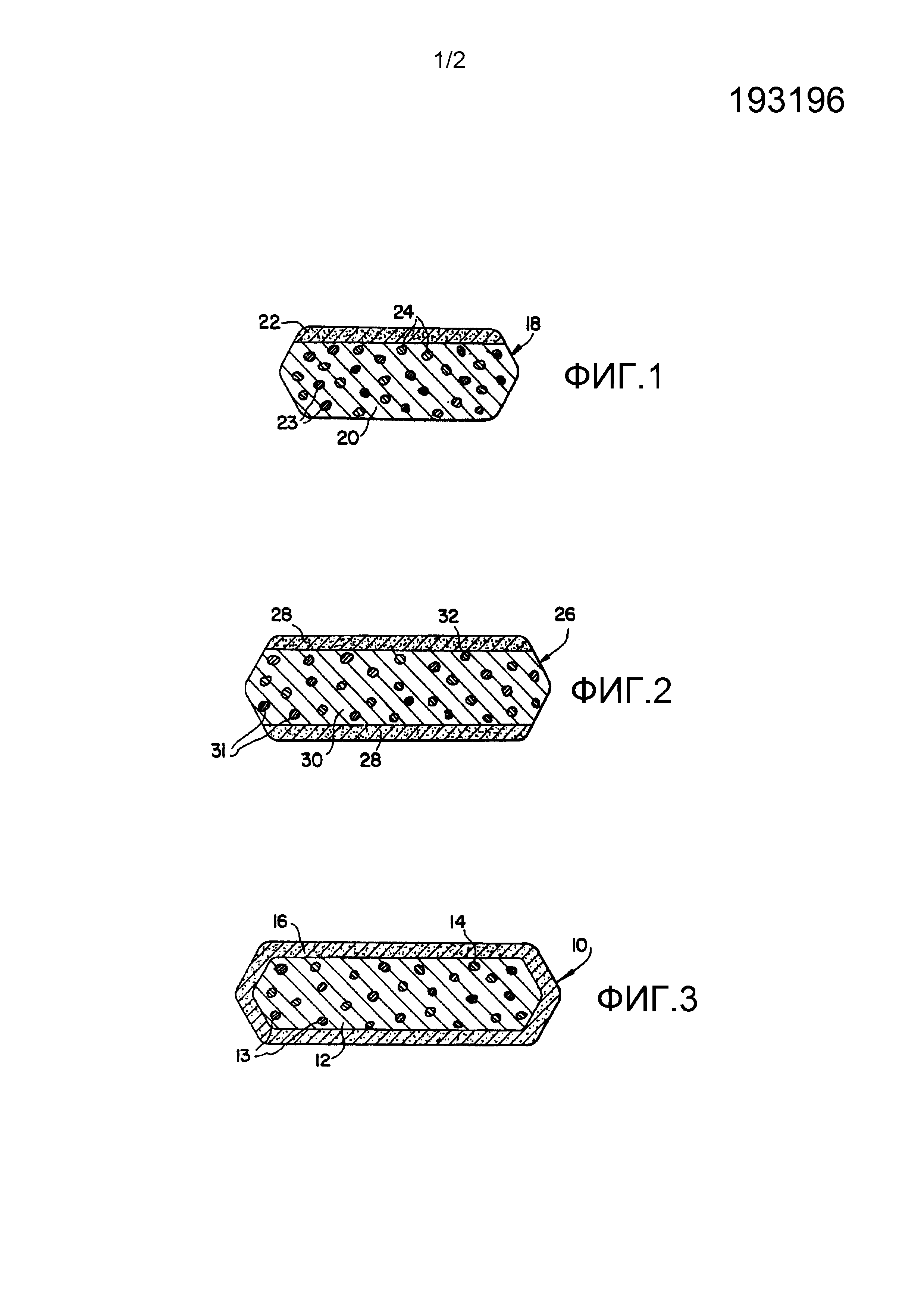
На фиг.1 представлено изображение в разрезе таблетки с фармацевтической композицией, описанной в данном описании.
На фиг.2 представлено изображение в разрезе таблетки с фармацевтической композицией, описанной в данном описании, согласно второму варианту осуществления.
На фиг.3 представлено изображение в разрезе таблетки с фармацевтической композицией, описанной в данном описании, согласно третьему варианту осуществления.
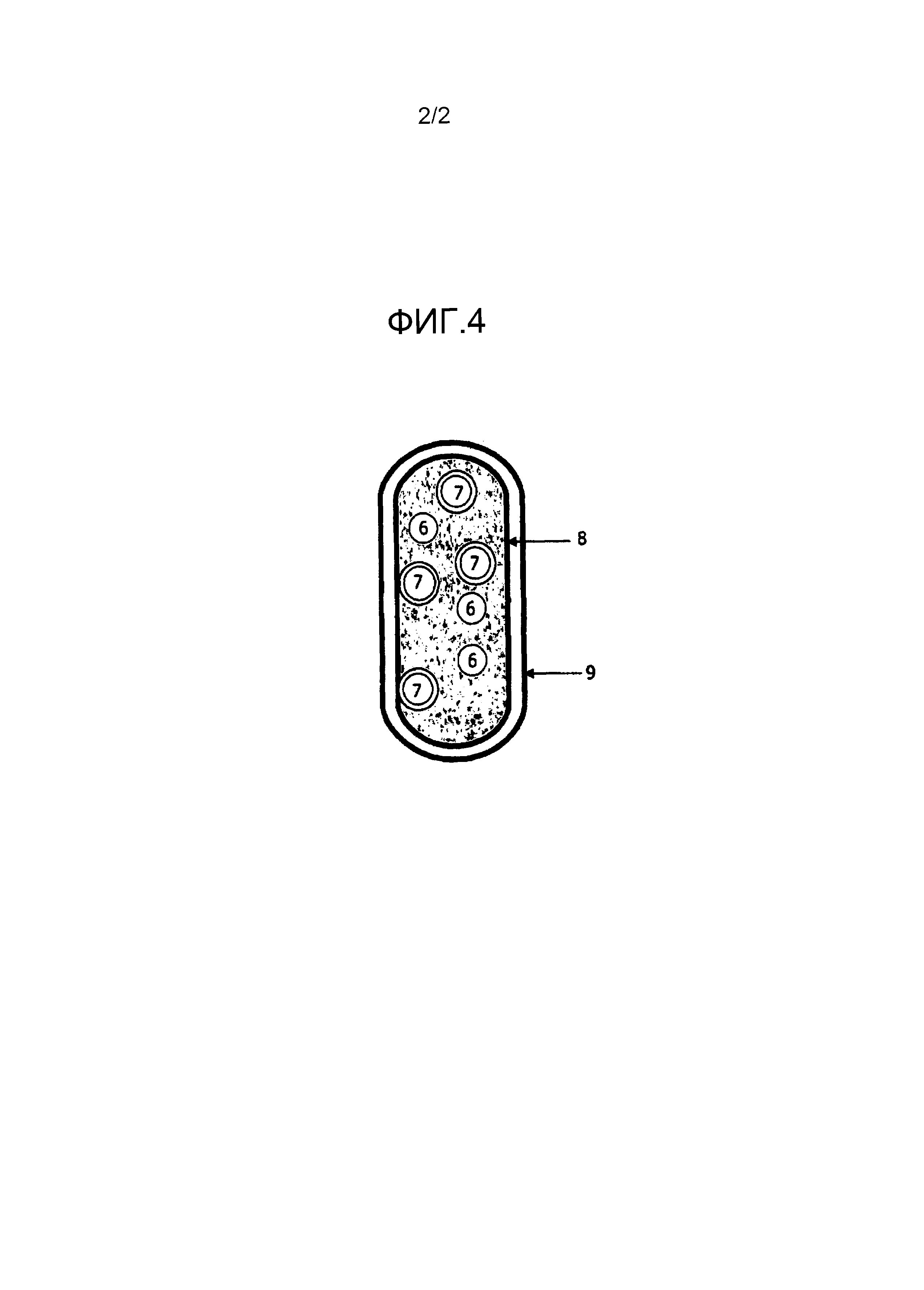
На фиг.4 представлено изображение в разрезе капсулы с фармацевтической композицией, описанной в данном описании.
Сокращения
SGLT (натрий-глюкозный котранспортер)
A1C (гемоглобин A1C)
UKPDS (Британское проспективное исследование диабета)
ЛПВП (липопротеины высокой плотности)
ADA (Американская диабетическая ассоциация)
ОВС (один раз в сутки)
ДВС (два раза в сутки)
ЖК (желудочно-кишечный)
СД 2 типа (сахарный диабет 2 типа)
ГПП-1 (глюкагон-подобный пептид-1)
DPP-IV (дипептидилпротеаза IV)
HMG-CoA (3-гидрокси-3-метилглутарил-коэнзим А)
АСАТ (ацил-CoA:холестерин O-ацилтрансфераза)
LHRH (лютеинизирующий-рилизинг-гормон)
AUC (площадь под кривой)
IR (немедленное высвобождение)
MR (модифицированное/замедленное высвобождение)
MTP (микросомальный белок транспорта триглицеридов)
FPG (уровень глюкозы в плазме натощак)
ДИАБЕТ
В последнее время большое внимание сосредоточено на возможности применения натрий-глюкозного котранспортера-2 (SGLT2) в качестве новых лекарственных средств для лечения сахарного диабета (1). В семейство SGLT входит несколько изоформ, которые активно транспортируют глюкозу и галактозу через кишечные и почечные мембраны, причем этот процесс связан с транспортом ионов натрия (2). SGLT2 представляет собой низкоаффинный, высокопроизводительный натрий-глюкозный котранспортер, который находится в основном в S1-сегменте проксимальных канальцев почек (3). У здорового человека более 99% глюкозы из плазмы крови, которая фильтруется в почках, подвергается реабсорбции. Такая реабсорбция примерно на 90% происходит посредством SGLT2. Остальные 10% реабсорбции, вероятно, опосредованы высокоаффинным котранспортером SGLT1, который находится в кишечнике и почечных проксимальных канальцах. У людей с мутантным инактивированным SGLT2 выявляют стойкую почечную глюкозурию, но в остальном они остаются здоровыми (4, 5). Таким образом, ингибирование SGLT2 может считаться перспективным способом улучшения гомеостаза глюкозы. Предполагается, что при ингибировании SGLT2 будет происходить выведение глюкозы из кровотока за счет повышения экскреции глюкозы с мочой, механизм которого не требует секреции инсулина бета-клетками поджелудочной железы с ограниченной функцией.
В идеале, ингибирование такого транспортера глюкозы должно происходить в течение фазы колебания постпрандиального уровня глюкозы, когда глюкоза поступает в кровь после приема пищи. У здорового, не страдающего диабетом субъекта постпрандиальная концентрация глюкозы в крови через 2 часа обычно составляет менее 120 мг/дл и редко превышает 140 мг/дл. Уровень глюкозы достигает пика примерно через 1 час после начала приема пищи, и затем возвращается к допрандиальному уровню в течение 2-3 часов (6, 7). Это повышение и снижение постпрандиального уровня глюкозы опосредовано первой фазой инсулиновой реакции, при которой в ответ на поступление питательных веществ в течение 10 минут обычно высвобождается большое количество эндогенного инсулина. У лиц с диабетом 2 типа значительно уменьшена или отсутствует первая фаза инсулиновой реакции, в результате чего постпрандиальный уровень глюкозы постоянно повышен на протяжении большей части суток (8).
Чтобы ответить на вопрос, улучшает ли воздействие на постпрандиальную гипергликемию общий контроль гликемии, Feinglos et al. (9) в исследовании пациентов с сахарным диабетом 2 типа с вторичной недостаточностью при терапии сульфонилмочевиной показали, что при улучшении постпрандиальной гипергликемии с помощью инсулина лизпро (Humalog) во время приема пищи в сочетании с препаратами сульфонилмочевины не только снижается колебание постпрандиального уровня глюкозы через 2 часа, но также снижается и уровень глюкозы натощак и уровень гемоглобина A1C с 9,0% до 7,1% (P<0,0001). В группе субъектов, получавших лизпро, также выявлен положительный эффект значительного снижения уровня общего холестерина и повышения концентрации холестерина ЛПВП.
Улучшения уровня A1C были также отмечены в исследованиях Bastyr et al. (10), показавших, что терапия, направленная на снижение постпрандиального уровня глюкозы по сравнению с уровнем глюкозы натощак, может вызывать улучшение в отношении снижения уровня гликированного гемоглобина. Дополнительно, в исследовании пациентов с гестационным диабетом De Veciana et al. (11) показали, что лечение, направленное не на уровень глюкозы натощак, а на постпрандиальный уровень глюкозы через 1 час, снижает уровень гликированного гемоглобина и улучшает неонатальный исход.
Не прекращаются споры о том, в какой степени постпрандиальный уровень глюкозы способствует развитию микроваскулярных и макроваскулярных осложнений. Доклад консенсусной конференции Американской диабетической ассоциации (ADA) по постпрандиальному уровню глюкозы подтвердил выводы исследования по контролю диабета и его осложнений, исследования Кумамото и Британского проспективного исследования диабета UKPDS, которые показали, что терапия, направленная на достижение нормального уровня гликемии, уменьшает возникновение и задерживает прогрессирование отдаленных микроваскулярных осложнений (12). Дополнительно, как указано ранее, эпидемиологический анализ данных UKPDS показал, что благодаря снижению уровня гликемии также наблюдалось улучшение макроваскулярных исходов (13). Таким образом, если постпрандиальный уровень глюкозы участвует в общей гликемии, то контроль постпрандиального уровня глюкозы должен вносить вклад в развитие осложнений диабета.
Многочисленные эпидемиологические исследования показали, что повышенные постпрандиальные уровни/уровни после нагрузки глюкозой являются независимыми и значительными факторами риска макроваскулярных осложнений и повышают риск смертности. Исследование сердца Гонолулу (14) выявило выраженную корреляцию между уровнями глюкозы после нагрузки глюкозой и частотой сердечно-сосудистой смертности. В интервенционном исследовании диабета (15), в котором наблюдали вновь диагностированных пациентов с сахарным диабетом 2 типа, выявлено, что умеренная постпрандиальная гипергликемия является более показательным признаком атеросклероза, чем уровень гликемии натощак, и выявлено, что не уровень глюкозы натощак, а постпрандиальный уровень представляет собой независимый фактор риска сердечно-сосудистой смертности. В исследовании DECODE (16), в котором наблюдали более 25000 субъектов в течение в среднем 7,3 лет, показано, что повышенный риск смертности гораздо более тесно связан с уровнем в плазме через 2 часа после нагрузки глюкозой, чем с уровнем глюкозы натощак. Аналогично этим выводам, de Vegt et al. (17) обнаружили, что степень риска, обусловленная постпрандиальной концентрацией глюкозы через 2 часа, была почти в два раза выше, чем степень риска, обусловленная уровнем A1C.
Безопасность контроля постпрандиального уровня глюкозы зависит как от применяемой терапии, так и от конкретных способностей каждого пациента распознавать и лечить гипогликемию при ее возникновении. Несмотря на то, что у пациентов с сахарным диабетом 2 типа редко наблюдается тяжелая гипогликемия, страх гипогликемии (у пациентов и поставщиков) остается одним из основных препятствий на пути достижения контроля постпрандиального уровня глюкозы и, вероятно, более строгого общего контроля гликемии. Предлагаемая композиция ремоглифлозина обеспечивает безопасное, эффективное дозирование ингибитора SGLT2, который можно принимать один раз в сутки, таким образом, это способствует обеспечению соблюдения режима и схемы лечения со стороны пациента.
Достижение строгого контроля постпрандиального уровня глюкозы у пациентов с сахарным диабетом 2 типа может вызывать затруднения, но, тем не менее, современные новые препараты инсулина и пероральные лекарственные средства могут отчасти являться решением этой проблемы. Аналоги инсулина быстрого действия, такие как инсулин аспарт (Novolog) и инсулин лизпро, за более короткий период времени создают высокий уровень инсулина в сыворотке и обладают более короткой продолжительностью действия, чем обычный человеческий инсулин, что приводит к снижению колебаний постпрандиального уровня глюкозы с уменьшением продолжительности постпрандиального уровня гипергликемии, а также к снижению частоты тяжелой гипогликемии у пациентов с диабетом 2 типа (18, 19). В этой связи, комбинация композиции ремоглифлозина этабоната и приведенных выше препаратов инсулина может обеспечить превосходный механизм контроля постпрандиального уровня глюкозы.
Можно утверждать, что частота и тяжесть гипогликемии, по данным UKPDS, могли быть ниже, если пациенты применяли новые аналоги инсулина и пероральные препараты в комбинации с технологией мониторинга уровня глюкозы в домашних условиях (которая не была широко доступной на момент начала исследования), но вместе с тем, нельзя не учитывать риск развития гипогликемии при диабете 2 типа. Все гипогликемические препараты (средства, усиливающие секрецию и инсулин) могут вызывать тяжелую гипогликемию. Таким образом, важно, чтобы работники сферы здравоохранения понимали степень риска, связанного с каждым применяемым лечением, и чтобы каждый метод лечения должным образом соответствовал способности каждого пациента распознавать гипогликемию и реагировать на ее возникновение. При введении ремоглифлозина выявлены очень редкие доказанные случаи возникновения гипогликемии, вероятно, связанные с механизмом действия.
Масштабные рандомизированные интервенционные исследования дали убедительные доказательства того, что достижение и поддержание жесткого контроля уровня гликемии (<6,5% A1C) значительно снижает риск диабетических микроваскулярных и макроваскулярных осложнений. К сожалению, масштабные эпидемиологические исследования показали, что диабет 2 типа не только часто плохо контролируется, но и наличие в Соединенных Штатах в настоящее время эпидемии диабета. Поскольку максимальный рост распространенности диабета 2 типа наблюдается у взрослых людей в возрасте от 30 до 39 лет, будет расти число людей, живущих с диабетом 2 типа более продолжительное время. Поэтому крайне важно, чтобы работники системы здравоохранения нашли способы повышения эффективности лечения сахарного диабета в целях предотвращения многолетних истощающих осложнений и огромной финансовой нагрузки на систему здравоохранения.
Утверждается, что новые цели в области контроля гликемии являются неадекватными по той причине, что они небезопасны или слишком трудны для достижения, что противоречит результатам тщательной клинической оценки. Основной задачей должно стать достижение наилучшего контроля уровня гликемии у каждого пациента, поскольку любое снижение уровня A1C значительно снижает риск развития диабетических осложнений. Чтобы помочь пациентам в достижении наилучших показателей контроля их уровня гликемии, потребуется применение адекватной терапии, адекватного мониторинга и комплексного обучения пациентов самоконтролю диабета.
Список литературы
1. Marsenic, O. Glucose control by the kidney: an emerging target in diabetes. Am. J. Kidney Dis. 2009, 53, 875-883.
2. Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab. Pharmacokinet. 2005, 20, 452-477.
3. Kanai, Y.; Lee, W.S.; You, G.; Brown, D.; Hediger, M.A. The human kidney low affinity Na+/glucose cotransporter SGLT2: delineation of the major renal reabsorptive mechanism for D-glucose. J. Clin. Invest. 1994, 93, 397-404.
4. Van den Heuvel, L.P.; Assink, K.; Willemsen, M.; Monnens, L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum. Genet. 2002, 111, 544-547.
5. Calado, J.; Soto, K.; Clemente, C; Correia, P.; Rueff, J. Novel compound heterozygous mutations in SLC5A2 are responsible for autosomal recessive renal glucosuria. Hum. Genet. 2004, 114, 314-316.
6. American Diabetes Association: Postprandial blood glucose (Consensus Statement). Diabetes Care 24:775-778, 2001.
7. Polonsky KS, Given BD, Hirsch LJ, Tillil H, Shapiro ET, Beebe C, Frank BH, Galloway JA, Van Cauter E: Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med 318:1231-1239, 1988.
8. Pfeifer MA, Halter JB, Porte D Jr: Insulin secretion in diabetes mellitus. Am J Med 70:579-88, 1981.
9. Feinglos MN, Thacker CH, English J, Bethel MA, Lane JD: Modification of postprandial hyperglycemia with insulin lispro improves glucose control in patients with type 2 diabetes. Diabetes Care 20:1539-1542, 1997.
10. Bastyr EJ, Stuart CA, Brodows RG, Schwartz S, Graf CJ, Zagar A, Robertson KE (IOEZ Study Group): Therapy focused on lowering postprandial glucose, not fasting glucose, may be superior for lowering HbA1c. Diabetes Care 23:1236-1241, 2000.
11. De Veciana M, Major CA, Morgan MA, Asrat T, Toohey JS, Lien JM, Evans AT: Postprandial versus preprandial blood glucose monitoring in women with gestational diabetes mellitus requiring insulin therapy. N Engl J Med 333:1239-1241, 1995.
12. American Diabetes Association: Postprandial blood glucose (Consensus Statement). Diabetes Care 24:775-778, 2001.
13. (Stratton IM, Adler Al, Neil HA, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR: Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321:405-412, 2000).
14. (Donahue RP, Abbott RD, Reed DM, Yano K: Postchallenge glucose concentration and coronary heart disease in men of Japanese ancestry (Honolulu Heart Program). Diabetes 36:689-692, 1987).
15. (Ziegelasch HJ, Lindner J (The DIS Group): Risk factors for myocardial infarction and death in newly detected NIDDM: the Diabetes Intervention Study, 11-year follow-up. Diabetologia 39:1577-1583, 1996).
16. (DECODE Study Group: Glucose tolerance and mortality: comparison of WHO and American Diabetic Association diagnostic criteria. Lancet 354:617-621, 1999).
17. (de Vegt F, Dekker JM, Ruhe HG, Stehouwer CD, Nijpels G, Bouter LM, Heine RJ: Hyperglycaemia is associated with all-cause and cardiovascular mortality in the Hoorn population: the Hoorn Study. Diabetologia 42:926-931, 1999).
18. Rosenfalck AM, Thorsby P, Kjems L, Birkeland K, Dejgaard A, Hanssen KF, Madsbad S: Improved postprandial glycaemic control with insulin aspart in type 2 diabetic patients treated with insulin. Acta Diabetol 37:41-46, 2000.
19. Anderson JH Jr, Brunelle RL, Keohane P, Koivisto VA, Trautmann ME, Vignati L, DiMarchi R: Mealtime treatment with insulin analog improves postprandial hyperglycemia and hypoglycemia in patients with non-insulin-dependent diabetes mellitus. Multicenter Insulin Lispro Study Group. Arch Intern Med 157:1249-1255, 1997).
20. Moller, DE, New drug targets for type 2 diabetes and the metabolic syndrome, Nature 414, 821-827, 2001.
21. Leth, A, Changes in Plasma and Extracellular Fluid Volumes in Patients with Essential Hypertension During Long-Term Treatment with Hydrochlorothiazide, Circulation 42,479-485, 1970.
22. Rohifing JJ, Brunzell JD: The effects of diuretics and adrenergic-blocking agents on plasma lipids. West J Med. 145:210-218, 1986.
23. Fisher, M.C, Morella, A.M. (1994), European Patent 609961.
24. Hansraj, B.R., Bashir, R.H. (1992), European Patent 502642.
25. Rollet, M. (1987), French Patent 2594693.
26. Howard, S.A., Kotwal, P.M. (1997) U.S. Pat. No. 5645858.
27. Macrae, R.J., Smith J.S. (1997), World Patent WO 9718814.
28. Belenduik, G.W., McCarty, J.A., Rudnic, E.M. (1996), U.S. Pat. No. 5484608.
29. Bhatti, G.K., Edgren, D.E., Hatamkhani, Z., Wong, P.S., Wong, P.S.L. (1994), World Patent WO 9427589.
30. Palepu, N.R., Venkatesh, G.M., (1997) European Patent 701436.
РЕМОГЛИФЛОЗИНА ЭТАБОНАТ
Ремоглифлозина этабонат, также называемый 5-метил-4-[4-(1-метилэтокси)бензил]-1-(1-метилэтил)-1Н-пиразол-3-ил-6-O-(этоксикарбонил)-β-D-глюкопиранозидом, имеет следующую формулу (I):
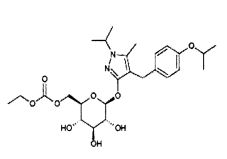
Согласно другому номенклатурному соглашению, указанная молекула представляет собой 6-О-(этоксикарбонил)-бета-D-глюкопиранозилокси)-4-[(4-изопропоксифенил)метил]-1-изопропил-5-метилпиразол. Ремоглифлозина этабонат также известен как GSK189075 или KGT-1681. Соли соединений формулы (I) являются полезными в качестве активного ингредиента в фармацевтической композиции по изобретению. Такими солями могут быть соли, описанные в патенте США 7084123, зарегистрированном 1 августа 2006 года, который включен в данное описание посредством ссылки. Примеры таких солей включают кислотно-аддитивные соли неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., кислотно-аддитивные соли органических кислот, таких как муравьиная кислота, уксусная кислота, метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, пропионовая кислота, лимонная кислота, янтарная кислота, винная кислота, фумаровая кислота, масляная кислота, щавелевая кислота, малоновая кислота, малеиновая кислота, молочная кислота, яблочная кислота, угольная кислота, глютаминовая кислота, аспарагиновая кислота, адипиновая кислота, олеиновая кислота, стеариновая кислота и т.п., и соли неорганических оснований, таких как натриевая соль, калиевая соль, кальциевая соль, магниевая соль и тому подобное.
Соединения, представленные приведенной выше формулой (I), включают сольваты этих соединений с фармацевтически приемлемыми растворителями, такими как этанол и вода.
Ремоглифлозина этабонат можно получать способами, описанными в патентах США 7084123 и 7375087, в частности, в примере 1 патента США 7084123, и каждый из приведенных патентов включен в данное описание посредством ссылки.
Ремоглифлозина этабонат является пролекарством ремоглифлозина (также называемого GSK189074 или KGT-1650).
Ремоглифлозина этабонат обладает потенциалом для применения в качестве монотерапии при лечении сахарного диабета (СД) 2 типа. На сегодняшний день были проведены исследования по оценке эффективности, безопасности и переносимости в течение до 12 недель, с разной эффективностью, таким образом, существует необходимость охарактеризовать профиль ряда выбранных доз лекарственного средства в течение 12-недельного периода.
План исследования предусматривал группу пациентов, получавших плацебо, чтобы дополнительно охарактеризовать профиль лекарственного средства, и для достижения максимального гликемического эффекта. Вместе с тем, чтобы минимизировать время, в течение которого субъекты могли иметь субоптимальный контроль уровня гликемии, продолжительность двойного слепого исследования лекарственного средства была ограничена до 12 недель. Дополнительно, были включены критерии для возможности введения через 6 недель резервного лечения для тех субъектов, у которых выявлен высокий уровень глюкозы натощак (FP).
В 12-недельном исследовании было достигнуто улучшение гликемического контроля как при введении один раз в сутки (250, 500 и 1000 мг ОВС), так и при введении два раза в сутки (от 50 мг до 1000 мг ДВС). Кроме того, доказано, что при указанных дозах может наблюдаться клинически значимая потеря массы, что является ключевым требованием для новых способов лечения СД 2 типа (20). Дозы выше 250 мг ДВС могут быть связаны с относительно небольшими дополнительными преимуществами в отношении гликемии и потери массы, и более низкие дозы показывали тенденцию к лучшей переносимости, в дальнейших исследованиях могли применяться общие суточные дозы менее 500 мг в сутки. Анализ показывает, что воздействие ремоглифлозина, которое значительно ингибирует перенос SGLT2 в период сна, коррелирует с небольшим подъемом холестерина липопротеинов низкой плотности (ЛПНП). В частности, при дозах 250 мг и 500 мг два раза в сутки выявляют значительное повышение уровня холестерина ЛПНП. Считается, что основной механизм такого изолированного повышения уровня холестерина ЛПНП обусловлен сочетанием 1) гемоконцентрации в результате диуретического действия, подобно эффекту, который вызывается мочегонными средствами (21, 22), и 2) краткосрочным ингибированием SGLT. Это также подтверждается более высокими дозами ОВС, при которых наблюдается повышение гематокрита (суррогатного маркера гемоконцентрации), но отсутствует соответствующее повышение уровня холестерина ЛПНП. Дополнительно, доза комбинированного немедленного/замедленного высвобождения (IR/SR), которая вводится один раз в сутки утром, также может показывать значимые преимущества в отношении уровня гликемии и потери массы. С точки зрения безопасности, кроме небольшого эффекта, наблюдаемого в отношении липидного профиля и гематокрита, наблюдались слабые отличия в профиле безопасности на основе частоты введения.
Лекарственные средства, абсорбция которых ограничена верхними отделами желудочно-кишечного тракта, и сочетается с плохой абсорбцией в дистальных отделах тонкой кишки, толстой кишки и ободочной кишки, обычно считаются неподходящими кандидатами для получения композиций пероральных систем с регулируемой доставкой. Эти границы абсорбции (например, в верхних отделах желудочно-кишечного тракта), называются "окном абсорбции".
Функцией желудочно-кишечного тракта является продвижение проглоченного материала из желудка (где происходит пищеварение) в тонкую кишку (где в основном происходит абсорбция) и в толстую кишку (где происходит абсорбция/выделение воды, как часть процессов регуляции жидкости в организме). Время нахождения в желудке не перевариваемых материалов зависит от того, находится ли субъект в состоянии сытости или натощак. Обычные периоды времени опорожнения желудка для твердого содержимого (с диаметром частиц более нескольких миллиметров) варьируют от нескольких десятков минут в состоянии натощак до нескольких часов в состоянии насыщения. Периоды времени транзита по тонкой кишке составляют порядка от 3 до 4 часов.
Пероральные системы доставки с регулируемым высвобождением действуют путем высвобождения их действующего вещества из лекарственного средства в течение длительного периода времени после введения. Таким образом, лекарственные формы с регулируемым высвобождением могут находиться только относительно короткий период в отделах желудочно-кишечного тракта, где может происходить хорошее всасывание некоторых лекарственных средств. Лекарственная форма будет продвигаться дальше к отделам кишечника, в которых абсорбция некоторых лекарственных средств является плохой или отсутствует, и при этом продолжать высвобождение содержащегося в нем лекарственного средства, хотя значительная часть его действующего вещества не будет доставлена. При высвобождении лекарственного средства из лекарственной формы при описанных обстоятельствах не будет происходить его абсорбция. Таким образом, введение лекарственного средства с соблюдением окна абсорбции в общепринятой системе доставки с регулируемым высвобождением может приводить к достижению субтерапевтического уровня в крови и к отсутствию эффективности при патологических состояниях, для лечения которых было предназначено данное лекарственное средство.
В лекарственной форме с регулируемым высвобождением авторы композиции пытались снизить скорость растворения, например, путем включения лекарственного средства в полимерную матрицу или путем окружения лекарственного средства полимерной барьерной мембраной, через которую должна происходить диффузия лекарственного средства при высвобождении для абсорбции. Для уменьшения скорости высвобождения лекарственного средства из лекарственной формы до подходящего уровня, соответствующего желаемому профилю его концентрации в крови, имеющего очень высокую растворимость в воде, для матрицы или барьерной мембраны потребуются очень большое количество полимера. Это можно осуществить, если общая суточная доза лекарственного средства, предназначенная для доставки, составляет всего нескольких миллиграммов, но для многих лекарственных средств, обладающих описанными свойствами растворимости, требуется общая суточная доза порядка многих сотен миллиграммов. Для таких продуктов можно создать пероральные лекарственные формы с регулируемым высвобождением путем использования большого количества полимера, но вместе с тем, в результате можно получить недопустимо большую лекарственную форму.
Терапевтические схемы применения ремоглифлозина этабоната можно улучшить с помощью лекарственной формы, которая позволит уменьшить частоту введения, а именно, один раз в сутки, по сравнению с введением два раза в сутки, что обеспечит удобство для пациентов, и, вероятно, улучшит соблюдение пациентами режима и схемы лечения. В общепринятых композициях с модифицированным высвобождением выявлено отсутствие компенсации характерного для этой молекулы короткого периода полувыведения, и это означает, что единственным способом доставки ремоглифлозина этабоната является введение два раза в сутки. Для снижения частоты введения необходимо создать такой тип высвобождения из лекарственной формы, чтобы повысился эффективный уровень в плазме, но возможность эффективной доставки на этом уровне уменьшится благодаря сочетанному влиянию значительного уменьшения проницаемости для лекарственного средства при транзите от проксимального отдела тонкой кишки к ободочной кишке и ограниченному времени пребывания в отделах желудочно-кишечного тракта с хорошей абсорбцией лекарственного средства. Это время транзита вниз к "полезному" отделу желудочно-кишечного тракта составляет, скорее всего, только нескольких часов. Сохранение или даже улучшение биодоступности из лекарственной формы, которая высвобождает ремоглифлозина этабонат комбинированным способом, обеспечивает желаемую концентрацию лекарственного средства в плазме крови в течение желаемого периода времени, которым обычно являются часы бодрствования пациента.
КОМПОЗИЦИИ
Общепринятые на предшествующем уровне технологии получения пероральной лекарственной формы с регулируемым высвобождением охватывают или матричные системы, или системы в форме отдельных частиц. Матричные системы можно получать путем гомогенного смешивания лекарственного средства с гидрофильными полимерами, такими как гидроксипропиметилцеллюлоза, гидроксипропилцеллюлоза, полиэтиленоксид, карбомер, с некоторыми полимерами, полученными из метакриловой кислоты, альгината натрия, или смеси компонентов, выбранных из перечисленного выше, и прессование полученной смеси в таблетки (с использованием при необходимости некоторых других эксципиентов). При соблюдении однородности с вышеуказанными материалами можно включать в композицию гидрофобные полимеры, такие как этилцеллюлоза, некоторые полимерные эфиры метакриловой кислоты, ацетатбутират целлюлозы, поли(этилен-ко-винилацетат), чтобы получить дополнительный контроль высвобождения. Дополнительная альтернатива предполагает включение лекарственного средства внутрь таблетки на восковой основе, путем гранулирования или простого смешивания лекарственного средства с воском, таким как карнаубский воск, микрокристаллический воск или коммерчески доступные очищенные эфиры жирных кислот. Как указано выше, невозможно применять такие подходы к лекарственным средствам с очень высокой растворимостью в воде.
Системы с отдельными частицами состоят из лекарственных форм на основе множества сфер, несущих лекарственное средство, которые получены путем наслаивания лекарственного средства на ядро, обычно на сферу из сахаро-крахмальной смеси диаметром примерно 0,8 мм, до достаточного уровня, и создания барьера для высвобождения лекарственного средства вокруг сферы, несущей лекарственное средство. Также сферы, несущие лекарственное средство, могут быть изготовлены путем грануляции влажной массы смеси лекарственного средства и эксципиентов, пропуская влажную массу через перфорированную сетку для образования коротких нитей, которые закругляют в аппарате для сферонизации перед сушкой и нанесением барьера для высвобождения лекарственного средства. Барьером для высвобождения лекарственного средства может быть воск, такой как карнаубский воск или глицериновые эфиры жирных кислот, или полимерный барьер, например, смесь этилцеллюлозы и гидроксипропилметилцеллюлозы. Такая технология подходит для умеренно растворимых лекарственных средств в дозах, в единицах измерения от миллиграммов до менее чем несколько сотен миллиграммов в сутки.
В некоторых примерах системы предполагают получение композиции с регулируемым высвобождением из лекарственного средства с высокой растворимостью в воде путем оптимизации подхода к системе с отдельными частицами. В публикации Fisher раскрыта система с отдельными частицами для лекарственных средств с высокой растворимостью, в частности, агонистов опиатов (23) на основе содержащего лекарственное средство ядра, окруженного барьером, регулирующим высвобождение лекарственного средства, которое обладает свойством частичной растворимости при очень кислом рН.
Hansraj (24) описывает покрытие ядер, несущих лекарственное средство, полимерами, полученными из метакриловой или акриловой кислоты, свойства которых модифицировали путем включения, по меньшей мере, одного анионного поверхностно-активного вещества. В такой системе высвобождение лекарственных средств с высокой растворимостью в воде регулируют, не прибегая к использованию толстых оболочек в слое, регулирующем скорость высвобождения.
Rollet (25) описано получение замедленного высвобождения лекарственного средства из композиции с отдельными частицами на основе тонких частиц гидрофильных и гидрофобных кремнеземов или силикатов. Предположительно, эта система будет функционировать для лекарственных средств с высокой растворимостью в воде.
Мультисистемы с отдельными частицами обычно помещают в капсулы, для обеспечения единичных лекарственных форм, поскольку происходит разрушение таких частиц при попытке спрессовать их в таблетки. Общая доза, содержащаяся в одной лекарственной форме, ограничена возможным заполнением в твердой желатиновой капсуле легко проглатываемого размера, и обычно составляет не более нескольких сотен миллиграммов.
Одна лекарственная форма системы регулируемого высвобождения используемая для высоко растворимых в воде лекарственных средств, включают нанесение многослойного покрытия вокруг лекарственной формы, как описано Howard (26). Если оболочка не используется, применяют специальные смеси полимеров или образование комплекса с лекарственным средством. Macrae (27) описано использование смесей полиэтиленоксида, гидроксипропилметилцеллюлозы необязательно с энтеросолюбильными полимерами, для достижения постоянной скорости высвобождения лекарственных средств с высокой растворимостью в воде. Belenduik (28) объединил высоко растворимое в воде лекарственное средство и гидрофильный полимер на основе акриловой кислоты, и диспергировал полученную смесь в гидрофобную матрицу. Описаны варианты осмотических систем ALZA, подходящие для лекарственных средств с высокой растворимостью в воде, таких как венлафаксина гидрохлорид (29). Для этих систем нужно два слоя, слой лекарственного средства и осмотически регулируемый смещаемый слой, все окруженные проницаемой для воды/непроницаемой для лекарственного средства мембраной с выпускным каналом для лекарственного средства в этой мембране.
Были получены гранулы высоко растворимого в воде клавуланата (30), с необходимостью использовать барьерный слой из гидрофобного воскового материала, чтобы обеспечить регулируемое высвобождение из этого материала при формулировании совместной композиции гранул амоксициллина тригидрата с регулируемым высвобождением в капсуле или прессованной таблетке.
Подробное описание изобретения
Настоящее изобретение относится к составлению композиции лекарственного средства с относительно коротким периодом полувыведения и ограниченным окном абсорбции, которое представляет собой, например, ремоглифлозина этабонат или его соль, имеющие окно абсорбции в верхнем отделе желудочно-кишечного тракта, для получения лекарственной формы, которая может обеспечить эффективное воздействие в течение периода бодрствования пациента. Композиция по изобретению (а) обеспечивает немедленное высвобождение лекарственного средства, обычно, до завтрака или во время завтрака, и (b) адекватное замедленное высвобождение для обеспечения достаточного покрытия при последующих колебаниях уровня глюкозы в течение суток. Композиции по изобретению относятся к композиции с немедленным/замедленным высвобождением лекарственного средства.
В случае ремоглифлозина этабоната, композиция по изобретению позволяет пациенту применять схему введения, по меньшей мере, от 250 мг ремоглифлозина этабоната один раз в сутки до 500 мг ремоглифлозина этабоната один раз в сутки, в форме одной единичной лекарственной формы, такой как одна таблетка или одна капсула, обеспечивая, при этом, эффективный контроль уровня глюкозы в плазме крови. Композиции ремоглифлозина этабоната по изобретению можно вводить один раз в сутки в указанных выше дозах для эффективного лечения диабета, избегая, при этом, проблем, связанных с высокой концентрацией в плазме ремоглифлозина этабоната в ночное время, которая может возникать при схеме приема ремоглифлозина этабоната два раза в сутки, и одновременно обеспечивая оптимальный терапевтический контроль и оптимальный профиль безопасности.
Настоящее изобретение применимо ко всем лекарственным средствам, обладающим коротким периодом полувыведения и ограниченным окном абсорбции, для лечения сахарного диабета и, в частности, в случаях, когда механизм действия охватывает ослабление или сглаживание колебаний прандиальной глюкозы.
Комбинированная система доставки с немедленным высвобождением по изобретению представляет собой гетерогенную двухфазную систему, которая включает (1) твердую дисперсную фазу, состоящую из отдельных гранул или частиц, содержащих (а) лекарственное средство, которое имеет короткий период полувыведения, предпочтительно, ремоглифлозина этабонат или его соль, и ограниченное окно абсорбции (например, как окно абсорбции в верхнем отделе желудочно-кишечного тракта), и (b) материал замедленного высвобождения, полученный из одного или более гидрофильных полимеров, и/или одного или более гидрофобных полимеров, и/или одного или более гидрофобных веществ другого типа (например, из одного или более из воска, жирного спирта и/или эфира жирной кислоты), и (2) твердую фазу, в которой диспергированы и в которую заключены гранулы или частицы из твердой фазы композиции с замедленным высвобождением, причем указанная твердая фаза в основном состоит из материала немедленного высвобождения, полученного из одного или более гидрофильных полимеров и/или из одного или более гидрофобных полимеров, и/или из одного или более гидрофобных веществ другого типа (например, из одного или более из воска, жирного спирта и/или эфира жирных кислот).
Изобретение особенно подходит для доставки лекарственных средств с коротким периодом полувыведения, таких как ремоглифлозина этабонат и его фармацевтически приемлемые соли, в регулируемом режиме со значительным начальным выбросом лекарственного средства, и последующим замедленным высвобождением лекарственного средства (которое освобождается из отдельных частиц, образующих твердую дисперсную фазу) в течение некоторого времени, соответствующего окну абсорбции в верхних отделах ЖКТ. Лекарственное средство при высвобождении из частиц композиции с замедленным высвобождением, по сути, становится доступным для абсорбции в верхнем отделе желудочно-кишечного тракта.
Твердая фаза композиции с немедленным высвобождением представляет собой, в частности, фазу или матрицу, имеющую частицы или гранулы, включающие лекарственное средство (образующие твердую фазу замедленного высвобождения), диспергированные или заключенные в твердую фазу композиции с немедленным высвобождением.
Кроме того, согласно настоящему изобретению предоставлен способ снижения резистентности к инсулину или способ лечения сахарного диабета, в котором пациенту, нуждающемуся в лечении, вводят комбинированную композицию немедленного/замедленного высвобождения по изобретению, содержащую антидиабетический лекарственный препарат.
Термин «сахарный диабет», используемый в данном описании, относится к диабету 2 типа и диабету 1 типа, обычно к диабету 2 типа.
Применяемый антидиабетический фармацевтический препарат предпочтительно представляет собой SGLT2 ингибитор, например, ремоглифлозина этабонат или его фармацевтически приемлемую соль, такую как гидрохлорид, которые имеют общее название ремоглифлозина этабонат. Хлористоводородная соль ремоглифлозина этабоната является особенно активным ингредиентом настоящего изобретения.
В другом аспекте настоящего изобретения предоставлен способ снижения резистентности к инсулину или способ лечения диабета, в котором комбинированная композиция по изобретению с немедленным/замедленным высвобождением содержит ремоглифлозина этабонат и вводится пациенту, нуждающемуся в таком лечении, по схеме в дозе, по меньшей мере, примерно 250 мг ремоглифлозина этабоната один раз в сутки, в частности, примерно от 250 мг до 500 мг один раз в сутки.
Термин "высвобождаемый материал", относящийся к материалу, который присутствует в твердой дисперсной фазе композиции с замедленным высвобождением и в твердой фазе композиции с немедленным высвобождением, относится к одному или более гидрофильным полимерам, и/или к одному или более гидрофобным полимерам, и/или к одному или более гидрофобным материалам другого типа, таким как, например, один или более восков, жирных спиртов и/или эфиров жирной кислоты. "Высвобождаемый материал", присутствующий в твердой дисперсной фазе замедленного высвобождения, может быть таким, как "высвобождаемый материал", присутствующий в твердой фазе немедленного высвобождения, или отличаться от этого материала. Вместе с тем, обычно "высвобождаемый материал", присутствующий в дисперсной фазе замедленного высвобождения, может отличаться от "высвобождаемого материала" из композиции немедленного высвобождения.
Термин "ограниченное окно абсорбции" или аналогичный термин, относящийся к свойствам лекарственного средства, лекарственного препарата или фармацевтического средства, предназначенного для использования в композиции по изобретению, относится к биодоступности при пероральном введении, которая составляет менее 75%, обычно менее 60%, и обычно снижается при увеличении дозы, и почти неизменно имеет абсорбцию, ограниченную проницаемостью/временем транзита.
Комбинированная система немедленного/замедленного высвобождения по изобретению может включать твердую дисперсную фазу замедленного высвобождения и твердую фазу немедленного высвобождения в массовом соотношении в пределах приблизительно от 0,5:1 приблизительно до 4:1, например, приблизительно от 0,8:1 приблизительно до 2:1.
Твердая дисперсная фаза замедленного высвобождения будет содержать лекарственное средство в количестве в диапазоне приблизительно от 10 приблизительно до 98% масс., например, приблизительно от 15 приблизительно до 95% масс., и материал замедленного высвобождения в виде гидрофильных полимеров и/или гидрофобных полимеров, и/или других гидрофобных материалов, в количестве в диапазоне приблизительно от 5 приблизительно до 95% масс., предпочтительно приблизительно от 7 приблизительно до 85% масс., при этом вышеуказанные % приведены исходя из массы твердой дисперсной фазы замедленного высвобождения. При использовании смесей гидрофильный полимер должен использоваться в массовом соотношении к гидрофобному полимеру и/или к другому гидрофобному материалу в диапазоне примерно от 0,05:1 примерно до 19:1, в частности, примерно от 0,1:1 примерно до 10:1.
Средний размер частиц или гранул из твердой дисперсной фазы замедленного высвобождения должен составлять диапазон примерно от 30 мкм до 0,8 мм, и в частности, примерно от 50 мкм до 0,5 мм.
Твердая фаза немедленного высвобождения должна содержать материал немедленного высвобождения (который обычно отличается от материала, используемого в твердой дисперсной фазе замедленного высвобождения) в виде одного или более гидрофильных полимеров, и/или гидрофобных полимеров и/или других гидрофобных материалов в количестве в диапазоне приблизительно от 40 приблизительно до 100%, в частности, приблизительно от 60 приблизительно до 100% (исходя из массы твердой фазы немедленного высвобождения).
Общее содержание полимерного материала замедленного высвобождения в фармацевтической композиции настоящего изобретения (включающего гидрофильные полимеры, и/или гидрофобные полимеры, и/или другой гидрофобный материал, присутствующий в твердой дисперсной фазе замедленного высвобождения, и гидрофильный полимер, и/или гидрофобные полимеры, и/или другой гидрофобный материал, присутствующий в твердой фазе немедленного высвобождения) должно быть в диапазоне приблизительно от 25 приблизительно до 75% масс., предпочтительно приблизительно от 30 приблизительно до 65%, более предпочтительно приблизительно от 35 приблизительно до 60% масс., исходя из общей массы фармацевтической композиции.
Гидрофильные полимеры, которые можно использовать в твердой дисперсной фазе замедленного высвобождения и/или твердой фазе немедленного высвобождения, включают, но без ограничения, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, натрийкарбоксиметилцеллюлозу, кальцийкарбоксиметилцеллюлозу, альгинат аммония, альгинат натрия, альгинат калия, альгинат кальция, альгинат пропиленгликоля, альгиновую кислоту, поливиниловый спирт, повидон, карбомер, пектат калия, пектинат калия и тому подобное.
Гидрофобные полимеры, которые можно использовать в твердой дисперсной фазе замедленного высвобождения и/или в твердой фазе немедленного высвобождения, включают, но без ограничения, этилцеллюлозу, гидроксиэтилцеллюлозу, сополимер аммония метакрилата (Eudragit RL.TM. или Eudragit RS.TM.), сополимеры метакриловой кислоты (Eudragit L.TM. или Eudragit S.TM.), сополимер этилового эфира метакриловой кислоты и акриловой кислоты (Eudragit L 100-5.TM.), нейтральный сополимер сложных эфиров метакриловой кислоты (Eudragit NE 30D.TM.), сополимер сложных эфиров диметиламиноэтилметакрилата-метакриловой кислоты (Eudragit E 100.TM.), сополимеры винилметилового эфира/малеинового ангидрида, их соли и сложные эфиры (Gantrez ТМ.).
Другие гидрофобные материалы, которые можно использовать в твердой дисперсной фазе замедленного высвобождения и/или в твердой фазе немедленного высвобождения, включают, но без ограничения, воски, такие как пчелиный воск, карнаубский воск, микрокристаллический воск и озокерит; жирные спирты, такие как цетостеариловый спирт, стеариловый спирт; цетиловый спирт и миристиловый спирт; и сложные эфиры жирных кислот, такие как глицерилмоностеарат, глицеролмоноолеат, ацетилированные моноглицериды, тристеарин, трипальмитин, воск цетиловых эфиров, глицерилпальмитостеарат, глицерилбегенат и гидрогенизированное касторовое масло.
Гидрофильные полимеры и/или гидрофобные полимеры, используемые в твердой дисперсной фазе замедленного высвобождения и/или в твердой фазе немедленного высвобождения, могут представлять собой ионные или неионные полимеры, в частности, ионные полимеры используют в твердой дисперсной фазе замедленного высвобождения, и неионные полимеры используют в твердой фазе немедленного высвобождения.
Предпочтительные ионные полимеры, предназначенные для использования в твердой дисперсной фазе замедленного высвобождения, включают альгинат натрия, карбомер (Carbopol.TM.), кальцийкарбоксиметилцеллюлозу или натрийкарбоксиметилцеллюлозу, ксантановую камедь, сополимер этилового эфира метакриловой кислоты и акриловой кислоты, сополимер сложных эфиров диметиламиноэтилметакрилата-метакриловой кислоты, ацетатфталат целлюлозы, фталат гидроксипропилметилцеллюлозы, тримеллитат гидроксипропилметилцеллюлозы и малеат гидроксипропилметилцеллюлозы, и особенно предпочтительной является натрийкарбоксиметилцеллюлоза. Предпочтительные двухфазные системы доставки с немедленным/замедленным высвобождением согласно настоящему изобретению указаны в разделе примеров.
Особенно предпочтительным активным ингредиентом является гидрохлоридная соль ремоглифлозина этабоната.
КОМБИНАЦИИ
Если это желательно, ремоглифлозина этабонат или его соль можно использовать в комбинации с другим антигипергликемическим средством, и/или гиполипидемическим средством, и/или средством против ожирения, которые можно вводить перорально в одной лекарственной форме согласно изобретению, в отдельных пероральных лекарственных формах или в форме инъекции. Ремоглифлозина этабонат или его соль должны применяться в массовом соотношении в диапазоне приблизительно от 0,01:1 приблизительно до 300:1, предпочтительно приблизительно от 0,05:1 приблизительно до 250:1 от массы другого антигипергликемического средства, и/или гиполипидемического средства, и/или средства против ожирения.
Применение ремоглифлозина этабоната или его соли в комбинации с другим антигипергликемическим средством может быть особенно полезным для достижения результатов против гипергликемии, по сравнению с применением каждого из этих лекарственных средств по отдельности, и эффект такого применения может превышать совокупные аддитивные антигипергликемические эффекты, которые оказывают эти лекарственные средства.
Дополнительно, согласно настоящему изобретению, предоставлен способ снижения резистентности к инсулину или лечения гипергликемии, включая диабет 2 типа (NIDDM - инсулиннезависимый сахарный диабет) и/или диабета 1 типа (IDDM - инсулинзависимый сахарный диабет), в котором пациенту, нуждающемуся в лечении, вводят терапевтически эффективное количество комбинированной композиции по изобретению, содержащей ремоглифлозина этабонат или его соль, необязательно в комбинации с другим антигипергликемическим средством, и/или гиполипидемическим средством, и/или средством против ожирения.
Другое антигипергликемическое средство может представлять собой пероральное антигипергликемическое средство, в частности, бигуанид, такой как метформин (под общепринятым торговым названием глюкофаг) или другие известные бигуаниды, которые улучшают лечение гипергликемии в первую очередь посредством подавление продукции глюкозы в печени.
Другое антигипергликемическое средство может представлять собой пероральное антигипергликемическое средство, в частности, сульфонилмочевину, например, глибурид (также известный как глибенкламид), глимепирид (описанный в патенте США 4379785), глипизид, гликлазид или хлорпропамид, другие известные препараты сульфонилмочевины или другие антигипергликемические средства, которые действуют на АТФ-зависимые каналы β-клеток.
Ремоглифлозина этабонат или его соль применяют в массовом соотношении к сульфонилмочевине в диапазоне приблизительно от 300:1 приблизительно до 50:1, предпочтительно приблизительно от 250:1 приблизительно до 75:1.
Пероральным антигипергликемическим средством может быть ингибитор глюкозидазы, например, акарбоза (описанная в патенте США 4904769) или миглитол (описанный в патенте США 4639436), которые можно вводить в отдельной пероральной лекарственной форме.
Ремоглифлозина этабонат или его соль применяют в массовом соотношении к ингибитору глюкозидазы в диапазоне примерно от 300:1 примерно до 2:1, например, примерно от 200:1 примерно до 25:1.
Ремоглифлозина этабонат или его соль можно применять в комбинации с тиазолидиндионовым пероральным антидиабетическим средством (который действует на чувствительность к инсулину у больных NIDDM), таким как троглитазон (Rezulin RTM, Warner-Lambert, описанный в патенте США 4572912), розиглитазон (SKB), пиоглитазон (Takeda), MCC-555 (Mitsubishi, описанный в патенте США 5594016), GL-262570 (Glaxo-Welcome), энглитазон (CP-68722, Pfizer) или дарглитазон (CP-86325, Pfizer).
Ремоглифлозина этабонат или его соль применяют в массовом соотношении к тиазолидиндиону в количестве в диапазоне примерно от 75:1 примерно до 0,1:1, например, в соотношении примерно от 5:1 примерно до 0,5:1.
Можно включать сульфонилмочевину и тиазолидиндион в количестве перорального антидиабетического средства менее 150 мг в одну таблетку с композицией по изобретению в качестве отдельного быстрорастворимого слоя.
Ремоглифлозина этабонат или его соль также можно применять в комбинации с непероральным антигипергликемическим средством, таким как инсулин или глюкагон-подобный пептид-1 (ГПП-1), например, ГПП-1 (1-36) амид, ГПП-1 (7-36) амид, ГПП-1 (7-37) (как описано в патенте США 5614492, Habener, описание которого включено в данное описание посредством ссылки), которые можно вводить путем инъекций или с помощью трансдермального или трансбуккального устройства.
Пероральным антигипергликемическим средством также может быть ингибитор дипептидилпротеазы IV (DPP-IV), например, ситиглиптин, вилдаглиптин, саксаглиптин, линаглиптин (разрабатываемый компанией Boehringer Ingelheim) или алоглиптин.
В случае присутствия в описанных выше композициях препаратов сульфонилмочевины, таких как глибенкламид, глимепирид, глипирид, глипизид, глипизид, хлорпропамид и гликлазид, можно использовать ингибиторы глюкозидазы акарбозу или миглитол, применяя их в количестве и по схеме введения согласно справочнику лекарственных средств (Physician's Desk Reference).
В случае присутствия тиазолидиндионовых антидиабетических средств, их можно применять в количестве в диапазоне примерно от 0,01 примерно до 2000 мг/в сутки, которое можно вводить одной или раздельными дозами от одного до четырех раз в сутки.
В случае присутствия инсулина его можно вводить, применяя композиции, количество и схему введения, указанные в справочнике лекарственных средств.
В случае присутствия ГПП-1 можно вводить пептиды в пероральной трансбуккальной композиции, назальным путем или парентеральным путем, как описано в патенте США 5346701 (TheraTech), патентах США 5614492 и 5631224, которые включены в данное описание посредством ссылки.
Применение ремоглифлозина этабоната или его соли в комбинации с другим конкретным антигипергликемическим средством может привести к более выраженным антигипергликемическим результатам, чем возможный эффект каждого из этих лекарственных средств по отдельности, и превышающему комбинированному аддитивному антигипергликемическому эффекту, вызываемому этими лекарственными средствами.
Кроме того, согласно настоящему изобретению, предоставлен способ снижения резистентности к инсулину или способ лечения гипергликемии, включая диабет 2 типа (NIDDM) и/или диабет 1 типа (IDDM), в котором пациенту, нуждающемуся в лечении, вводят терапевтически эффективное количество комбинированной композиции по изобретению, содержащей ремоглифлозина этабонат или его соль, необязательно в комбинации со средством против ожирения.
Дополнительно, согласно настоящему изобретению предлагается способ снижения массы, в котором нуждающемуся в лечении пациенту вводят терапевтически эффективное количество композиции по изобретению, содержащей ремоглифлозина этабонат или его соль, необязательно в комбинации со средством против ожирения.
Средство против ожирения может представлять собой пероральное средство против ожирения, в частности, ингибитор панкреатической липазы, анорексическое средство, антагонист каннабиноидных рецепторов (CB-1), агонист 5HTC или антагонист дофаминовых рецепторов, примерами которых являются ксеникал, сибутрамин, фентермин, фенфлюрамин, римонабант, лоркасерин или бупропион.
Средство против ожирения может представлять собой пероральное средство против ожирения, в частности, ингибитор панкреатической липазы, например, ксеникал (орлистат/Alli).
Ремоглифлозина этабонат или соль применяют в массовом соотношении к средству против ожирения в диапазоне примерно от 300:1 примерно до 50:1, например, в соотношении примерно от 250:1 примерно до 75:1.
Пероральное средство против ожирения может также представлять собой анорексическое средство, например, фентермин, фенфлюрамин или лоркасерин.
Ремоглифлозина этабонат или его соль применяют в массовом соотношении к анорексическому средству в диапазоне примерно от 300:1 примерно до 2:1, например, в соотношении примерно от 200:1 примерно до 25:1.
Ремоглифлозина этабонат или его соль можно применять в комбинации с пероральным средством против ожирения, например, с антагонистом каннабиноидного рецептора-1.
Ремоглифлозина этабонат или его соль применяют в массовом соотношении к антагонисту каннабиноидного рецептора-1 в количестве в диапазоне примерно от 75:1 примерно до 0,1:1, например, в соотношении примерно от 5:1 примерно до 0,5:1.
Ремоглифлозина этабонат или его соль также можно применять в комбинации с непероральным средством против ожирения, таким как лептин, который можно вводить путем инъекции или с помощью трансдермального или трансбуккального устройства.
Гиполипидемическое средство, которое можно необязательно применять в комбинации с ремоглифлозина этабонатом или его солью, может включать MTP-ингибиторы, ингибиторы HMG-CoA-редуктазы, ингибиторы сквален-синтетазы, производные фиброевой кислоты, ингибиторы АСАТ, ингибиторы абсорбции холестерина, ингибиторы котранспортера Na.sup.+/желчных кислот в подвздошной кишке, секвестранты желчных кислот и/или никотиновую кислоту и ее производные.
MTP-ингибиторы, применяемые в изобретении, включают MTP-ингибиторы, описанные в следующих патентах: патенте США 5595872, патенте США 5739135, патенте США 5712279, патенте США 5760246, патенте США 5827875, патенте США 5885983 и в заявке США № 09/175180, поданной 20 октября 1998 года, в настоящее время патент США 5563440.
Предпочтительные ингибиторы MTP, применяемые согласно настоящему изобретению, включают ингибиторы MTP, описанные в патентах США 5739135 и 5712279 и в патенте США 5760246.
Гиполипидемическое средство может представлять собой ингибитор HMG-CoA-редуктазы, который включает, но без ограничения, мевастатин и родственные соединения, описанные в патенте США 3983140, ловастатин (мевинолин) и родственные соединения, описанные в патенте США 4231938, правастатин и родственные соединения, например, описанные в патенте США 4346227, симвастатин и родственные соединения, описанные в патентах США№ 4448784 и 4450171. Другие ингибиторы HMG-CoA-редуктазы, которые можно применять согласно изобретению, включают, но без ограничения, флувастатин, описанный в патенте США 5354772, церивастатин, описанный в патентах США 5006530 и 5177080, аторвастатин, описанный в патентах США 4681893, 5273995, 5385929 и 5686104, пиразольные аналоги производных мевалонолактона, описанные в патенте США 4613610, инденовые аналоги производных мевалонолактона, описанные в заявке PCT WO 86/03488, 6-[2-(замещенный пиррол-1-ил)алкил)пиран-2-оны и их производные, описанные в патенте США 4647576, SC-45355, Searle дихлорацетат (3-замещенного производного пентандионовой кислоты), имидазольные аналоги мевалонолактона, описанные в заявке PCT WO 86/07054, производные 3-карбокси-2-гидроксипропанфосфоновой кислоты, описанные во французском патенте 2596393, производные 2,3-дизамещенного пиррола, фурана и тиофена, описанные в Европейской патентной заявке N 0221025, нафтильные аналоги мевалонолактона, описанные в патенте США 4686237, октагидронафталены, описанные в патенте США 4499289, кето-аналоги мевинолина (ловастатин), описанные в Европейской патентной заявке 142146 A2, а также другие известные ингибиторы HMG CoA-редуктазы.
Дополнительно, в патенте GB 2205837 раскрыты соединения фосфиновой кислоты, полезные для ингибирования HMG-CoA редуктазы, которые подходят для применения в настоящем изобретении.
Ингибиторы сквален-синтетазы, подходящие для применения в настоящем изобретении, включают, но без ограничения, альфа-фосфоносульфонаты, описанные в патенте США 5712396, соединения, раскрытые Biller et al, J. Med. Chem., 1988, 1988, Vol. 31, № 10, стр. 1869-1871, включая изопреноид(фосфинилметил)фосфонаты, а также другие ингибиторы сквален-синтетазы, описанные в патентах США 4871721 и 4924024 и авторами Biller, S.A., Neuenschwander, K., Ponpipom, M.M., и Poulter, C.D., Current Pharmaceutical Design, 2, 1-40 (1996).
Кроме того, другие ингибиторы сквален-синтетазы, подходящие для использования в изобретении, включают терпеноидные пирофосфаты, раскрытые P. Ortiz de Montellano et al, J. Med. Chem., 1977, 20, 243-249, аналог А фарнезилдифосфата и аналоги прескваленпирофосфата (PSQ-РР), описанные Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293, фосфинилфосфонаты, описанные McClard, RW et al., JACS, 1987, 109, 5544, и циклопропаны, описанные Capson, Т.L, PhD диссертация, июнь 1987 года, кафедра медицинской химии Университета Юты, реферат, содержание, стр. 16, 17, 40-43, 48-51, обзор.
Другие гиполипидемические средства, подходящие для применения в настоящем изобретении, включают, но без ограничения, производные фиброевой кислоты, такие как фенофибрат, гемфиброзил, клофибрат, безафибрат, ципрофибрат, клинофибрат и т.п., пробукол и родственные соединения, описанные в патенте США 3674836, примерами которых являются пробукол и гемфиброзил, секвестранты желчных кислот, такие как холестирамин, колестипол и DEAE-сефадекс (Secholex.RTM, Policexide.RTM), а также липостабил (Rhone-Poulenc), Eisai E-5050 (N-замещенные производные этаноламина), иманиксил (HOE-402), тетрагидролипстатин (THL), истигмастанилфосфорилхолин (SPC, Roche), аминоциклодекстрин (Tanabe Seiyoku), Ajinomoto AJ-814 (производное азулена), мелинамид (Sumitomo), Sandoz 58-035, американский цианамид CL-277,082 и CL-283,546 (дизамещенные производные мочевины), никотиновая кислота, аципимокс, ацифран, неомицин, п-аминосалициловая кислота, аспирин, производные поли(диаллилметиламина), например, описанные в патенте США 4759923, четвертичный амин поли(диаллилдиметиламмонийхлорида) и иононы, такие, как описано в патенте США 4027009, а также другие известные средства, снижающие уровень холестерина в сыворотке крови.
Гиполипидемическое средство может представлять собой ингибитор АСАТ, описанный в следующих публикациях: "The ACAT inhibitor, CI-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB 100-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; "RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor", Smith, C, et al, Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals", Krause et al, Editor(s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation: Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, Fla.; "ACAT inhibitors: potential anti-atherosclerotic agents", Sliskovic et al, Curr. Med. Chem. (1994), 1(3), 204-25; "Inhibitors of acyl-CoA:cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(1-phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity", Stout et al, Chemtracts: Org. Chem. (1995), 8(6), 359-62.
Ингибитор абсорбции холестерина может представлять собой SCH 48461 Schering-Plough или соединение, описанное в журнале Atherosclerosis 115, 45-63 (1995) или J. Med. Chem. 41, 973 (1998).
Ингибитор котранспортера Na.sup.+/желчных кислот подвздошной кишки может представлять собой соединение, описанное в Drugs of the Future, 24, 425-430 (1999). Предпочтительными гиполипидемическими средствами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин и церивастатин.
Приведенные выше патенты США включены в данное описание посредством ссылки. Применяемое количество и дозы соответствуют указаниям справочника лекарственных средств и/или патентов, приведенных выше.
Соединения формулы (I) настоящего изобретения используют в массовом соотношении к гиполипидемическому средству (когда присутствует) в диапазоне примерно от 500:1 до 1:500, в частности, примерно от 100:1 примерно до 1:100.
Вводимую дозу необходимо тщательно подбирать в зависимости от возраста, массы и состояния пациента, а также от пути введения, лекарственной формы, схемы введения и желаемого результата.
Дозировки и формы гиполипидемического средства соответствуют описанию в различных патентах, документах и заявках, приведенных выше.
Дозировки и формы другого гиполипидемического средства, при необходимости его применения, соответствуют указаниям последнего издания справочника лекарственных средств.
При пероральном введении достичь удовлетворительного результата, применяя ингибитор МТР от одного раза в сутки до четырех раз в сутки в количестве в диапазоне примерно от 0,01 мг/кг примерно до 100 мг/кг, например, в диапазоне примерно от 0,1 мг/кг примерно до 75 мг/кг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор MTP в количестве примерно от 1 примерно до 500 мг, в частности, примерно от 2 примерно до 400 мг, например, примерно от 5 примерно до 250 мг и применяется от одного до четырех раз в сутки.
При парентеральном введении ингибитор MTP применяют от одного до четырех раз в сутки в количестве в диапазоне примерно от 0,005 мг/кг массы тела примерно до 10 мг/кг, и в частности, примерно от 0,005 мг/кг примерно до 8 мг/кг.
При пероральном введении можно достичь удовлетворительного результата, применяя ингибитор HMG CoA-редуктазы, например, правастатин, ловастатин, симвастатин, аторвастатин, флувастатин или церивастатин, при этом применяемые дозы соответствуют указаниям справочника лекарственных средств, например, дозы находятся в диапазоне примерно от 1 до 2000 мг, и в частности, примерно от 4 примерно до 200 мг.
Ингибитор сквален-синтетазы можно применять в дозах в диапазоне примерно от 10 мг примерно до 2000 мг, в частности примерно от 25 мг примерно до 200 мг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор HMG CoA-редуктазы в количестве примерно от 0,1 примерно до 100 мг, например, примерно от 5 примерно до 80 мг, и более предпочтительно примерно от 10 примерно до 40 мг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор сквален-синтетазы в количестве примерно от 10 примерно от 500 мг, предпочтительно примерно от 25 примерно до 200 мг.
Ремоглифлозина этабонат или его соль и гиполипидемическое средство можно применять совместно в одной пероральной лекарственной форме или в отдельных пероральных лекарственных формах, принимаемых одновременно.
Описанные выше композиции можно вводить в лекарственных формах, как описано выше, в виде одной или в виде раздельных доз, принимаемых от одного раза в сутки до четырех раз в сутки. Целесообразным может быть начальное лечение пациента низкой комбинированной дозой и постепенный переход к высокой комбинированной дозе.
Предпочтительными гиполипидемическими средствами являются правастатин, симвастатин, ловастатин, аторвастатин, флувастатин или церивастатин.
В системе доставки по изобретению можно использовать следующие дополнительные типы высоко растворимых в воде лекарственных средств: правастатин; антигипертензивные препараты и антидепрессанты, относящиеся к гуанетидину (как описано в патенте США 2928829); и относящиеся к гуаноксифену (как описано в BE612362); антибиотики и противовирусные препараты, например, относящиеся к амидиномицину (как описано в патенте JP 21418); к сталлимицину (как описано в DE 1039198); к арфаменину B (как описано в опубликованной европейской патентной заявке 85/133550A2); к хитиноворину-A (как описано в опубликованной европейской патентной заявке 85/150378A2 и в патенте США 4723004); к стрептомицину (как описано в патенте США 2868779); SB-59 (как описано в Justus Liebigs, Ann. Chem. (1973) 7, 1112-1140); TAN-1057-A (как описано в патенте США 4971965); к стрептониазиду (как описано в J. Am. Chem. Soc. (1953) 75, 2261); иммуностимуляторы, относящиеся к ST-789 (как описано в опубликованной заявке на европейски патент 88/260588); ингибиторы пептид гидролазы, относящиеся к нафамастату (как описано в патенте США 4454338); габексату (как описано в патенте США 3751447); сепимостату (как описано в патентах США 4777182 и 4820730); ингибиторы фактора Ха, относящиеся к DX-9065a (как описано в опубликованной европейской патентной заявке 92/0540051); противовоспалительные средства, относящиеся к паранилину, как описано в патенте США 2877269; пептидилальдегиды (как описано в WO 94/13693); противоанафилактические средства, относящиеся к GMCHA-ТВР (Батебуласт/Batebulast) (как описано в патенте США 4465851); противоязвенные средства, относящиеся к бенексату (как описано в патенте США 4348410); дезоксиспергуалин (как описано в патентах США 4518532, 4658058 и 4983328); и аргинин.
Другие водорастворимые препараты, подходящие для использования в изобретении, включают пептиды, имеющие молекулярную массу от 100 до 10000, более предпочтительно примерно от 100 примерно до 6000, и имеющие от 2 до 35 аминокислотных остатков. В композиции настоящего изобретения также могут входить пептиды с высокой молекулярной массой, даже с молекулярной массой свыше 10000 примерно до 50000.
Подходящие небольшие пептиды имеют примерно от 2 примерно до 10, более предпочтительно примерно от 2 примерно до 6 аминокислотных остатков. Небольшие пептиды включают антагонисты фибриногеновых рецепторов (RGD-содержащие пептиды), которые представляют собой тетрапептиды со средней молекулярной массой примерно 600. Эти пептидные антагонисты являются мощными ингибиторами агрегации тромбоцитов при концентрации в плазме крови ниже 1 пмоль/мл. Предпочтительные антагонисты фибриногена включают пептид цикло(S,S)(Na-ацетил-Cys-(N.sup.a-метил)Arg-Gly-Asp-Pen-NH.sub.2 (Ali et al., EP 0341915, описание которого включено в данное описание посредством ссылки) и пептид цикло(S,S)-(2-меркапто)бензоил-(N.sup.a-метил)Arg-Gly-Asp-(2-меркапто)фениламид (EP 0423212, описание которого включено в данное описание посредством ссылки). Другие антагонисты фибриногена, используемые в настоящем изобретении, представляют собой пептиды, раскрытые в патенте Pierschbacher et al., WO 89/05150 (патент США 8804403); Marguerie, EP 0275748; Adams et al, патент США 4857508; Zimmerman et al, патент США 4683291; Nutt et al, EP 0410537, EP 0410539, EP 0410540, EP 0410541, EP 0410767, EP 0410833, EP 0422937 and EP 0422938; Ali et al, EP 0372486; Ohba et al, WO 90/02751 (PCT/JP89/00926); Klein et al, патент США 4,952,562; Scarborough et al, WO 90/15620 (PCT/US90/03417), Ali et al. PCT/US90/06514 и PCT/US92/00999; пептидо-подобные соединения, раскрытые Ali et al., ЕР 0381033 и ЕР 0384362; и RGD пептид цикло-N.sup.a-ацетил-Cys-Asn-Gly-Dtc-Amf-Asp-Cys-OH (в котором Dtc обозначает 4,4'-диметилтиазолидин-5-карбоновую кислоту, и Amf обозначает 4-аминометилфенилаланин).
RGD-пептид может давать преимущество при включении его в композицию по изобретению в количестве примерно до 600 мг/г массы гидрофильной фазы или от 0,1 до 60 мг/г массы композиции.
Другие пептиды, используемые в настоящем изобретении, включают, но без ограничения, другие RGD-содержащие пептиды, например, пептиды, раскрытые Momany, патент США 4411890 и патент США 4410513; Bowers et al., патент США 4880778, патент США 4880777, патент США 4839344 и WO 89/10933 (PCT/US89/01829); пептид Ala-His-D-Nal-Ala-Trp-D-Phe-Lys-NH.sub.2 (в котором Nal обозначает B-нафтилаланин), и пептиды, раскрытые Momany, патент США 4228158, патент США 4228157, патент США 4228156, патент США 4228155, патент США 4226857, патент США 4224316, патент США 4223021, патент США 4223020, патент США 4223019 и патент США 4410512.
Другие подходящие пептиды включают гексапептиды, такие как рилизинг-пептид гормона роста (GHRP) His-D-Trp-Ala-Trp-D-Phe-Lys-NH.sub.2 (Momany, патент США 4411890, описание которого включено в данное описание во всей полноте посредством ссылки). Указанный пептид может быть полезным при включении его в композицию в количестве примерно до 250 мг/г массы гидрофильной фазы или от 0,1 до 25 мг/кг массы композиции.
Большие полипептиды и белки, подходящие для использования в композиции с регулируемым высвобождением согласно настоящему изобретению, включают инсулин, кальцитонин, элкатонин, пептид, относящийся к кальцитонин-гену, и свиной соматостатин, а также их аналоги и гомологи. Другие подходящие большие полипептиды включают пептиды, раскрытые Pierschbacher et al., патент США 4589881 (>30 остатков); Bittle et al., патент США 4544500 (20-30 остатков) и Dimarchi et al., EP 0204480 (>34 остатков).
Другие типы соединений, используемых в настоящем изобретении, включают аналоги или гомологи лютеинизирующего-рилизинг-гормона (LHRH), которые проявляют высокую активность высвобождения ЛГ или ингибируют действие LHRH; аналоги или гомологи НР5, которые обладают гемопоэтической активностью; аналоги или гомологи эндотелина, которые обладают гипотензивной активностью; аналоги или гомологи энкефалина, которые обладают антиноцицептивной активностью; аналоги или гомологи хлорцистокинина; аналоги или гомологи циклоспорина А, которые обладают иммунодепрессивной активностью; аналоги или гомологи атриального натрийуретического фактора; пептидергические противоопухолевые агенты; аналоги или гомологи пептида, высвобождающего гастрин; аналоги или гомологи соматостатина; антагонисты гастрина, антагонисты брадикининов; антагонисты нейротензина; антагонисты бомбезина; агонисты и антагонисты окситоцина; агонисты и антагонисты вазопрессина; аналоги и гомологи гирудина; аналоги и гомологи цитозащитного пептид-циклолинопептида; аналоги альфа-меланоцитстимулирующего гормона (MSH); аналоги и гомологи MSH-рилизинг-фактора (Pro-Leu-Gly-NH.sub.2); пептиды, ингибирующие коллагеназу; пептиды, ингибирующие эластазу; пептиды, ингибирующие ренин; пептиды, ингибирующие протеазу ВИЧ; пептиды, ингибирующие ангиотензин-превращающий фермент; пептиды, ингибирующие химазы и триптазы, и пептиды, ингибирующие ферменты свертывания крови.
Другие подходящие лекарственные средства включают непептидные терапевтические агенты, такие как антибиотики, противомикробные средства, противоопухолевые средства, сердечно-сосудистые и почечные средства, противовоспалительные, иммуносупрессивные и иммуностимулирующие средства и средства, влияющие на центральную нервную систему (ЦНС).
КОМПОЗИЦИИ
Комбинированную композицию с немедленным/замедленным высвобождением согласно настоящему изобретению можно вводить различным видам млекопитающих, таким как собаки, кошки, люди и т.д., которые нуждаются в таком лечении.
Систему по изобретению можно включать в общепринятую системную лекарственную форму, например, в таблетки или капсулы. Приведенные выше лекарственные формы могут также включать необходимое физиологически приемлемое вещество-носитель, эксципиент, лубрикант, буфер, антибактериальный агент, объемообразующий агент (например, маннит), антиоксиданты (аскорбиновая кислота или бисульфит натрия) и тому подобное.
Вводимую дозу можно тщательно подбирать в зависимости от возраста, массы и состояния пациента, а также от пути введения, лекарственной формы, схемы введения и желаемого результата. В общем, описанные выше лекарственные формы композиции, содержащей ремоглифлозина этабонат или его соль (или по отдельности, или вместе с другим антигипергликемическим средством, и/или гиполипидемическим средством, и/или средством против ожирения), можно вводить в количествах, указанных выше для ремоглифлозина этабоната гидрохлорида.
Комбинация ремоглифлозина этабоната или его соли и другого антигипергликемического средства, и/или гиполипидемического средства, и/или средства против ожирения может быть сформулирована в форме раздельных или, если это возможно, в единой композиции используя общепринятые методики создания композиций.
Различные композиции по изобретению могут содержать один или более наполнителей или эксципиентов в количестве в диапазоне примерно до 90% масс., и предпочтительно примерно от 1 примерно до 80% масс., например, лактозу, сахар, кукурузный крахмал, модифицированный кукурузный крахмал, маннит, сорбит, неорганические соли, такие как карбонат кальция, и/или производные целлюлозы, такие как древесная целлюлоза и микрокристаллическая целлюлоза (также известная как добавка для прессования).
В композиции может присутствовать один или более связующих агентов в дополнение к наполнителям или вместо эксципиентов в количестве в диапазоне примерно от 0 примерно до 35%, и предпочтительно примерно от 0,5 примерно до 30% масс. композиции. Примеры таких связующих агентов, которые подходят для использования в изобретении, включают поливинилпирролидон (с молекулярной массой в диапазоне примерно от 5000 примерно до 80000, и предпочтительно примерно 40000), лактозу, крахмалы, такие как кукурузный крахмал, модифицированный кукурузный крахмал, сахар, камедь и т.п., а также восковое связующее вещество в тонкодисперсном виде (менее 500 мкм), например, карнаубский воск, парафин, спермацет, полиэтилены или микрокристаллический воск.
Если композиция имеет лекарственную форму таблетки, она может содержать один или более лубрикантов для таблеток в количестве в диапазоне примерно от 0,2 примерно до 8%, например, примерно от 0,5 примерно до 2% от массы композиции, например, стеарат магния, стеариновую кислоту, пальмитиновую кислоту, стеарат кальция, тальк, карнаубский воск и тому подобное. Другие общепринятые ингредиенты, которые необязательно могут присутствовать, включают консерванты, стабилизаторы, противоадгезивные агенты или кондиционеры текучести на основе диоксида кремния, или глиданты, такие как диоксид кремния марки Syloid, а также красители FD&C.
Таблетки по изобретению могут также необязательно включать слой дополнительного покрытия, который может составлять примерно до 15% от массы композиции таблетки. Слой покрытия (который может фактически содержать действующее вещество с немедленным высвобождением), который можно наносить поверх твердой фазы немедленного высвобождения, содержащей частицы твердой фазы замедленного высвобождения, заключенные внутри, может содержать любые общепринятые композиции для покрытий и включает один или более пленкообразователей или связующих веществ, таких как гидрофильный полимер, например, гидроксипропилметилцеллюлоза, и/или гидрофобный полимер, например, нейтральный полимер сложных эфиров метакриловой кислоты, этилцеллюлозу, ацетат целлюлозы, поливиниловый спирт и малеиновый ангидрид, полимеры бета-пинена, сложные эфиры глицерина из древесных смол и т.п., и один или более пластификаторов, например, триэтилцитрат, диэтилфталат, пропиленгликоль, глицерин, бутилфталат, касторовое масло и тому подобное. Как ядро таблеток, так и композиции для покрытия могут содержать алюминиевый лак для придания окраски.
Пленкообразователи наносят из системы растворителей, содержащих один или более растворителей, включающих воду, спирты, такие как метиловый спирт, этиловый спирт или изопропиловый спирт, кетоны, такие как ацетон или этилметилкетон, хлорированные углеводороды, такие как метиленхлорид, дихлорэтан и 1,1,1-трихлорэтан.
При использовании красителя его можно наносить вместе с композициями пленкообразователя, пластификатора и растворителя композиции или это может быть совершенно отдельный верхний слой.
Капсулы по изобретению, такие, как показано в фиг.4, могут включать эксципиент или фармацевтически приемлемый носитель, находящийся внутри, в котором во взвешенном состоянии находятся частицы немедленного и замедленного высвобождения. Выражение «фармацевтически приемлемый носитель», используемое в изобретении, означает фармацевтически приемлемый материал, композицию или носитель, например, жидкий или твердый фильтр, разбавитель, эксципиент, растворитель или инкапсулирующий материал. Каждый носитель должен быть "приемлемым" в плане совместимости с другими ингредиентами композиции и не оказывать вреда пациенту. Некоторые примеры веществ, которые можно использовать в качестве фармацевтически приемлемых носителей, включают (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как какао-масло и воски для суппозиториев; (9) масла, такие как арахисовое масло, масло семян хлопка, саффлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатно-буферный раствор и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Композиции капсул могут содержать обычные эксципиенты для добавления в капсулу, включающие, но без ограничения: наполнители, такие как микрокристаллическая целлюлоза, полисахариды сои, дигидрат фосфата кальция, сульфат кальция, лактоза, сахароза, сорбит, или любой другой инертный наполнитель. Дополнительно, могут присутствовать агенты текучести, такие как коллоидный диоксид кремния, силикагель, стеарат магния, стеарат кальция или любые другие вещества, которые придают свойства хорошей текучести. При желании также можно добавлять лубриканты, такие как полиэтиленгликоль, лейцин, глицерилбегенат, стеарат магния или стеарат кальция.
Композиции могут быть общепринято представлены в единичной лекарственной форме и могут быть изготовлены любым из способов, хорошо известных в области фармацевтики. Все способы включают связывание лекарственного средства с носителем или разбавителем, которые состоят из одного или более дополнительных ингредиентов. Обычно композиции готовят путем равномерного и тщательного перемешивания вещества с носителями и затем, если это необходимо, разделяют продукт на единичные дозы. Специалистам в данной области техники будет очевидно, что для получения и введения фармацевтических композиций согласно настоящему изобретению можно использовать любую основу лекарственной формы или носитель, которые обычно используют, и которые являются инертными по отношению к действующему веществу, и предпочтительно не препятствуют биоадгезии в вариантах осуществления с использованием биоадгезивного покрытия. Примеры таких основ лекарственной формы и носители описаны, например, в Remington's Pharmaceutical Sciences, 18-е изд. (1990), описание которого включено в данное описание посредством ссылки.
Конкретные примеры носителей и разбавителей включают фармацевтически приемлемые гидрогели, такие как альгинат, хитозан, метилметакрилаты, целлюлоза и ее производные (микрокристаллическая целлюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, этилцеллюлоза), агарозы и повидон TM., каолин, магния стеарат, крахмал, лактоза, сахароза, агенты регуляции плотности, например, сульфат бария и масла, усилители растворения, такие как аспарагиновая кислота, лимонная кислота, глютаминовая кислота, винная кислота, бикарбонат натрия, карбонат натрия, фосфат натрия, глицин, трицин, трометамин и TRIS.
Специалистам в данной области будет очевидно, что количество лекарственного средства, необходимое для терапевтического эффекта при его введении, безусловно, варьирует в зависимости от выбранного средства, характера и тяжести состояния и вида животного, проходящего курс лечения, и, в конечном счете, зависит от решения врача. Кроме того, оптимальное количество и интервалы между приемом отдельных доз лекарственного средства зависят от характера и степени состояния, которое подвергают лечению, от лекарственной формы, пути и места введения, от конкретного пациента, которого лечат, и такой оптимальный вариант можно определять с помощью общепринятых технологий. Следует понимать, что оптимальный курс лечения, а именно, количество принимаемых доз, может быть установлено специалистами в данной области техники с использованием общепринятых тестов для определения курса лечения.
Ремоглифлозина этабонат можно вводить один раз в сутки в количестве, по меньшей мере, примерно 200 мг, предпочтительно примерно от 250 мг до 500 мг в одной твердой лекарственной форме, такой как одна таблетка или одна капсула.
Дополнительно, согласно настоящему изобретению, композиция ремоглифлозина этабоната с немедленным/замедленным высвобождением по изобретению достигает концентрации ремоглифлозина этабоната в плазме (Cmax), по меньшей мере, в течение одного часа, и увеличивает время достижения максимальной концентрации в плазме ремоглифлозина этабоната (Tmax), по меньшей мере, в течение примерно 3 часов (но в диапазоне от 2 до 4 часов), и при этом имеет умеренный эффект на площадь под кривой (AUC) концентрации в плазме ремоглифлозина этабоната. Таким образом, композиции ремоглифлозина этабоната немедленного/замедленного высвобождения по изобретению можно применять ремоглифлозина этабонат для приема один раз в сутки при лечении сахарного диабета.
ФИГУРЫ
На фиг.1 представлена таблетка 18 по изобретению, которая может отличаться 3 отдельными конкретными вариантами осуществления. Таблетка 18 может иметь гомогенный слой немедленного высвобождения первого фармацевтически активного ингредиента 22, отдельные частицы с замедленным высвобождением первого фармацевтически активного ингредиента 23, связующее вещество 20 с замедленным высвобождением, и отдельные частицы второго фармацевтически активного ингредиента 24, которые, в данном случае, имеют замедленное высвобождение в связи с их включением в связующее вещество 20.
Альтернативно и во втором варианте, таблетка 18 может иметь слой 22 немедленного высвобождения второго активного ингредиента, отдельные частицы замедленного высвобождения для замедленного высвобождения первого фармацевтически активного ингредиента 23, связующее вещество 20 немедленного высвобождения и отдельные частицы первого активного ингредиента 24.
Третий вариант таблетки 18 может иметь цветной слой 22, отдельные частицы замедленного высвобождения для замедленного высвобождения первого активного ингредиента 23, связующее вещество 20 немедленного высвобождения и частицы 24 немедленного высвобождения первого активного ингредиента.
Фиг.2 может отличаться двумя вариантами. В первом варианте таблетка 26 может иметь гомогенные слои 28 для немедленного высвобождения первого активного ингредиента, отдельные частицы замедленного высвобождения первого активного ингредиента 31, связующее вещество 30 немедленного высвобождения и отдельные частицы второго активного ингредиента 32.
Альтернативно и во втором варианте, таблетка 26 может иметь гомогенный слой немедленного высвобождения первого активного ингредиента 28, а также частицы немедленного высвобождения первого активного ингредиента 32. Замедленное высвобождение первого активного ингредиента осуществляется как замедленное высвобождение из отдельных частиц 31 в связующем веществе немедленного высвобождения 30.
Фиг.3 может отличаться двумя вариантами. В первом варианте таблетка 10 может содержать слой оболочки 16, полностью покрывающий твердое ядро 12, при этом слой 16 представляет собой гомогенную композицию немедленного высвобождения первого активного ингредиента. В ядре 12 расположены частицы 13 с замедленным высвобождением первого активного ингредиента и частицы 14 с замедленным высвобождением второго активного ингредиента.
Альтернативно и во втором варианте, таблетка 10 может содержать слой оболочки 16, который быстро растворяется при приеме внутрь. Внутри слоя 16 находится связующее вещество 12 немедленного высвобождения, отдельные частицы 13 с немедленным высвобождением первого активного ингредиента и дискретные частицы 14 с замедленным высвобождением первого активного ингредиента.
Фиг.4 может отличаться двумя вариантами. В первом варианте желатиновая капсула или каплета 9 может содержать внутреннюю часть 8, содержащую сферы/пеллеты/частицы 6 с немедленным высвобождением и сферы/пеллеты/частицы 7 с контролируемым/замедленным/постоянным высвобождением. Каждая из таких сфер 6 и 7 представляет собой отдельную частицу с первым фармацевтическим ингредиентом или с первым и вторым фармацевтическими ингредиентами. В остальном внутреннем пространстве 8 капсулы 9 содержится эксципиент или носитель, который может быть твердым или текучим твердым веществом, несущим сферы 6 и 7.
Альтернативно и во втором варианте, капсула или каплета 9 может содержать внутреннюю часть 8, содержащую гомогенную смесь первого фармацевтически активного ингредиента для немедленного высвобождения при проглатывании. Также в матрице 8 содержатся две отдельные сферы/пеллеты/частицы 6 и 7 с активным ингредиентом, которые высвобождают его в разное время. Например, высвобождение из сферы 6 может происходить с Tmax примерно через 2-3 часа после проглатывания, тогда как высвобождение из гранул 7 может происходить примерно через 4-5 часов после проглатывания. Таким образом, капсула 9 может иметь три отдельные и отличающиеся композиции ремоглифлозина этабоната, чтобы таким образом достигать концентрации в крови ремоглифлозина, по меньшей мере, примерно 50 нг/мл в течение периода, по меньшей мере, примерно 5 часов.
Во всех приведенных выше описаниях фиг.1, 2, 3 и 4, первым фармацевтически активным ингредиентом является ремоглифлозина этабонат или его соль. Второй активный ингредиент, когда он присутствует, выбирают согласно описанию изобретения. Дополнительно, в описание второго активного ингредиента можно легко вносить изменения путем полного устранения второго активного ингредиента.
Ссылочный пример 1
Ремоглифлозина этабонат является пролекарством ремоглифлозина (также известного, как GSK189074), активное вещество, которое ингибирует натрий-зависимый глюкозный транспортер 2 (SGLT2). Тем не менее, короткий период полувыведения ремоглифлозина (примерно от 1,5 до 2 ч), скорее всего, потребует введения два раза в сутки в целях эффективности. Клиническое испытание проводили для определения возможности пролонгации фармакодинамического (PD) эффекта одной из нескольких композиций с модифицированным/постоянным (MR) высвобождением в течение интервала введения и, следовательно, возможности снижения общей суточной дозы при введении два раза в сутки, по сравнению с дозой, необходимой для композиции IR. Было некоторое предположение, что любая из композиций MR покажет возможность введения один раз в сутки (таблица 1).
|
Сравнительный пример 2
После проведения клинических испытаний было предположение, что ремоглифлозин этабонат только абсорбируется в верхних отделах ЖКТ (таблица 2). После перорального приема ремоглифлозина этабонат в композиции немедленного высвобождения (IR), исходный препарат быстро всасывался и превращался в активное вещество ремоглифлозин. Аналогично, ремоглифлозина этабонат подвергался быстрой абсорбции и превращению в ремоглифлозин, когда биоусиленный раствор или суспензию композиции ремоглифлозина этабоната вводили непосредственно в середину тонкой кишки. Тем не менее, выявлена ограниченная абсорбция ремоглифлозина этабоната и/или его превращение в ремоглифлозин, если биоусиленный раствор или суспензию композиции вводили непосредственно в слепую кишку/ободочную кишку.
На основании значений Tmax сравнивали скорость появления ремоглифлозина этабоната и ремоглифлозина в циркуляторном русле во всех примерах лечения, вместе с тем, у двух субъектов наблюдали небольшую задержку Tmax ремоглифлозина при введении суспензии в слепую кишку/ободочную кишку. Применение IR пероральной таблетки ремоглифлозина этабоната в качестве эталонного лечения показало степень биодоступности активного соединения, ремоглифлозина, которую определяли по AUC (0-бесконечность) или AUC (0-конец), была примерно на 4% или на 12% ниже, если биоусиленный раствор или суспензию композиции вводили непосредственно в середину тонкой кишки, соответственно; и была примерно на 83% или на 96% ниже, когда раствор или суспензию вводили непосредственно в слепую кишку/ободочную кишку, соответственно. Степень биодоступности ремоглифлозина была несколько ниже (8%), когда суспензию вводили в середину тонкой кишки, по сравнению с биоусиленным раствором; но значительно ниже (80%), когда суспензию вводили в слепую кишку/ободочную кишку, по сравнению с биоусиленным раствором. Из всех способов лечения IR таблетки при пероральном введении показывали самую высокую степень биодоступности ремоглифлозина, при этом суспензия при введении в слепую кишку/ободочную кишку, показывала самую низкую степень биодоступности ремоглифлозина.
Из полученных данных следует, что при пероральном введении ремоглифлозина этабоната экстенсивно и в первую очередь всасывается в середине тонкой кишки и/или там же превращается в ремоглифлозин, при этом в слепой кишке/ободочной кишке выявлена небольшая абсорбция и/или метаболическое превращение в ремоглифлозин. Соотношение AUC метаболитов и исходного вещества было намного меньше при непосредственной доставке композиций ремоглифлозина этабоната в слепую кишку/ободочную кишку, по сравнению с пероральными IR таблетками или доставкой лекарственного средства в середину тонкой кишки. С использованием IR таблетки в качестве эталона определяли Cmax ремоглифлозина этабоната, которая была примерно на 22% или на 40% ниже, если биоусиленный раствор или суспензию композиции вводили непосредственно в середину тонкой кишки, соответственно; и была примерно на 82% или 96% ниже, если раствор или суспензию вводили непосредственно в слепую кишку/ободочную кишку, соответственно.
Ремоглифлозина этабонат имеет свойственную ему плохую проницаемость в нижнем отделе желудочно-кишечного тракта, что приводит к его абсорбции почти исключительно в верхнем отделе желудочно-кишечного тракта. Он также имеет короткий период полувыведения (t½=1,5 часа). Это может вызвать проблемы в обеспечении высвобождения необходимого количества соединения во время трех основных приемов пищи и последующие колебания уровня глюкозы в плазме крови.
|
Пример 1
Капсулы ремоглифлозина этабоната (с немедленным высвобождением/замедленным высвобождением)
Получали композиции с различными концентрациями ремоглифлозина этабоната согласно следующей таблице 3, и в результате получали капсулы, содержащие от 250 до 500 мг активного вещества в одной капсуле:
|
Поскольку ремоглифлозин имеет плохую проницаемость в нижнем отделе желудочно-кишечного тракта, таким образом, абсорбцию можно наблюдать почти исключительно в верхних отделах желудочно-кишечного тракта. Это окно абсорбции находится в диапазоне от 3 до 6 часов в зависимости от приема пищи, индивидуальных различий и т.д. Ремоглифлозин также имеет короткий период полувыведения (t=1,5 часа). Для решения этих аспектов получали таблетки в соответствии с примерами 1а-1j, которые высвобождали низкую дозу лекарственного средства с немедленным высвобождением (IR) и высокую дозу лекарственного средства с замедленным высвобождением (DR) для достижения профиля воздействия, который будет максимально ингибировать SGLT2 в течение трех основных колебаний уровня глюкозы (завтрак, обед и ужин), при этом сводя к минимуму воздействие соединения во время сна. Таким образом, в примере 1c представлена прессованная таблетка примерно 350 мг, которая содержит композицию IR, позволяющую достигнуть максимальной концентрации в плазме ремоглифлозина 160 нг/мл через 1 ч после приема внутрь. Проявление Cmax из плазмы до показателя 40 нг/мл происходит через 3 часа. В этот период времени композиция DR, которая увеличивает Tmax от приема пищи до 4-5 часов, высвобождает 250 мг активного вещества для достижения Cmax 450 нг/мл, и затем выводится из плазмы крови примерно до концентрации 10 нг/мл примерно 10 PM.
Ремоглифлозина этабонат, микрокристаллическую целлюлозу и натрийкроскармеллозу гранулируют с раствором вода/повидон. Гранулы подвергают сушке и измельчению, затем смешивают их с маннитом, микрокристаллической целлюлозой и кроскармеллозой. Смесь смазывают стеаратом магния и прессуют.
Гранулы с наслоенным лекарством и энтеросолюбильным покрытием изготавливают путем покрытия сфер микрокристаллической целлюлозы водной суспензией, содержащей микронизированный ремоглифлозина этабонат, повидон и очищенную воду, затем водной суспензией Opadry, и затем водной энтеросолюбильной суспензией из сополимера метакриловой кислоты, моностеарата глицерина, полисорбата 80, триэтилцитрата и очищенной воды. Получение композиции осуществляют по стандартным методикам и с использованием общепринятых эксципиентов.
Каждые из гранул 50 и 100 мг с IR композицией смешивают с массой гранул (DR композиция) 200, 250, 300 и 400 мг с энтеросолюбильным покрытием и заполняют ими оболочки капсулы из гипромеллозы (гидроксипропилметилцеллюлозы, HPMC). Альтернативно, гранулы с IR- и DR-композициями можно прессовать в таблетки, как указано в примере 2.
Пример 2
Таблетки ремоглифлозина этабоната (с немедленным высвобождением/замедленным высвобождением)
Получали композиции с различными концентрациями ремоглифлозина этабоната и прессовали в таблетки, согласно следующей таблице 4, и в результате получали таблетки с немедленным высвобождением/замедленным высвобождением, каждая из которых содержала от 350 до 700 мг ингредиентов, включающих активное вещество:
|
Таким образом, новые композиции по изобретению представляют значительное преимущество в отношении введения людям ремоглифлозина этабоната один в раз в сутки при лечении сахарного диабета.
Композиции ремоглифлозина этабоната, описанные в приведенных выше примерах, можно вводить один раз в сутки, как описано выше, в одной, двух или более таблетках и/или капсулах, чтобы обеспечить оптимальный терапевтический контроль.
Настоящее изобретение включает комбинацию аспектов и вариантов осуществления, а также конкретные варианты осуществления, описанные во всем тексте настоящего описания.
Если не указано иное, тот факт, что конкретный термин или выражение не определены конкретно, не должен ассоциироваться с неопределенностью или отсутствием ясности, но другие термины в данном описании используются в их обычном значении. При использовании в описании торговых названий авторы имеют ввиду независимое включение торговой марки продуктов и активного фармацевтического ингредиента(ов) данного продукта под торговой маркой.
Выявленные конкретные фармакологические ответы могут варьировать в соответствии и в зависимости от конкретного выбранного активного соединения, или от присутствия фармацевтических носителей, а также от типа композиции и применяемого способа введения, и такие предполагаемые варианты или различия в результатах предусмотрены в соответствии с практическим осуществлением настоящего изобретения.
Хотя в данном описании проиллюстрированы и подробно описаны конкретные варианты осуществления настоящего изобретения, настоящее изобретение не ограничивается ими. Подробное описание, приведенное выше, предоставлено в качестве примера настоящего изобретения и не должно рассматриваться, каким-либо образом, как ограничивающее изобретение. Специалистам в данной области техники будут очевидны модификации, и все модификации, которые не отступают от существа изобретения, предназначены для включения в объем прилагаемой формулы изобретения.