СПОСОБ ПОЛУЧЕНИЯ СЕЛЕКТИВНОГО ИНГИБИТОРА ЦИКЛООКСИГЕНАЗЫ-2
Вид РИД
Изобретение
Область техники, к которой относится изобретение
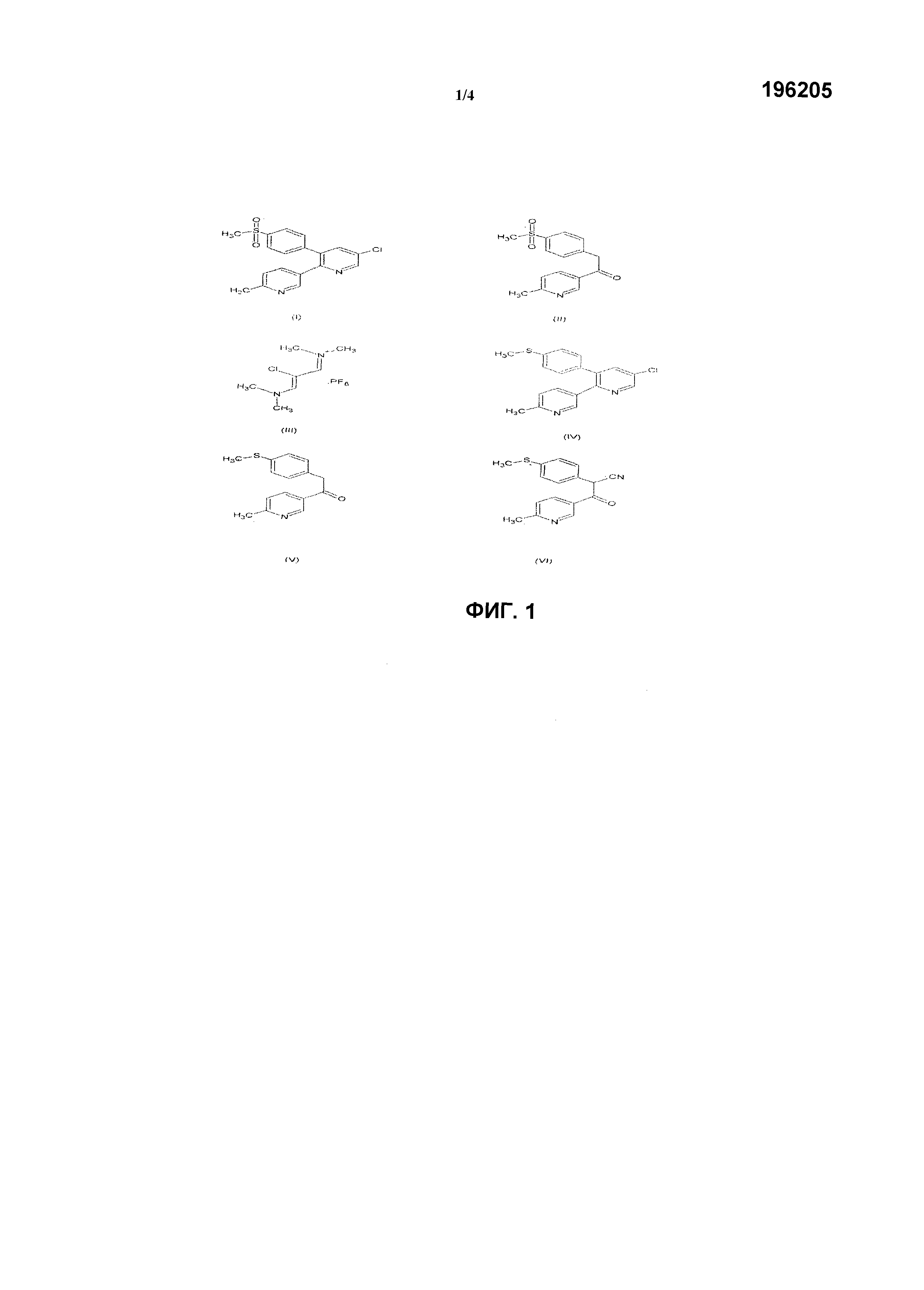
Настоящее изобретение относится к способу синтеза селективного ингибитора циклооксигеназы-2. В частности, изобретение относится к способу синтеза 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина по нижеследующей формуле (I) из нового соединения формулы (IV).
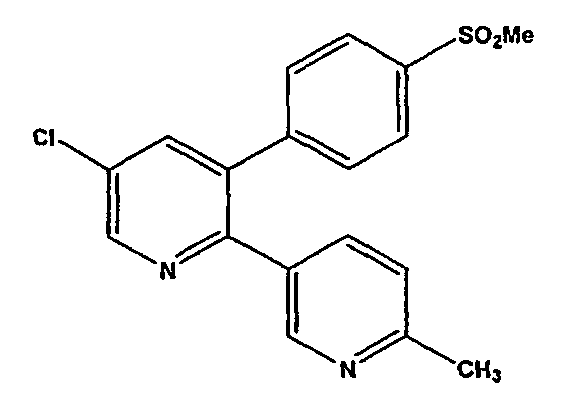
Эторикоксиб [формула (I)]
Настоящее изобретение далее относится к новому соединению (IV) и к способу его получения. Настоящее изобретение также относится к получению соединения формулы (II), используемого в синтезе указанного ингибитора циклооксигеназы-2.
Уровень техники
Селективные ингибиторы циклооксигеназы-2 образуют важный класс нестероидных противовоспалительных лекарственных средств (NSAID), отличающихся, в особенности, благодаря их улучшенному профилю безопасности. Известно, что широко применяемые NSAID, например, производные арилпропионовой кислоты или арилуксусной кислоты, вызывают раздражение желудка и появление язв после продолжительного применения данных лекарственных средств. Показано, что ингибиторы циклооксигеназы-2, которые действуют по механизму экспрессии изофермента в воспаленных тканях, в этом отношении более безопасны.
Эторикоксиб селективно ингибирует изоформу 2 фермента циклооксигеназы (COX-2) и в настоящее время одобрен более чем в 70 странах для назначения в терапии, такой как лечение ревматоидного артрита, псориатического аритрита, анкилозирующего спондилита, хронической поясничной боли, острой боли, остеоартрита и подагры.
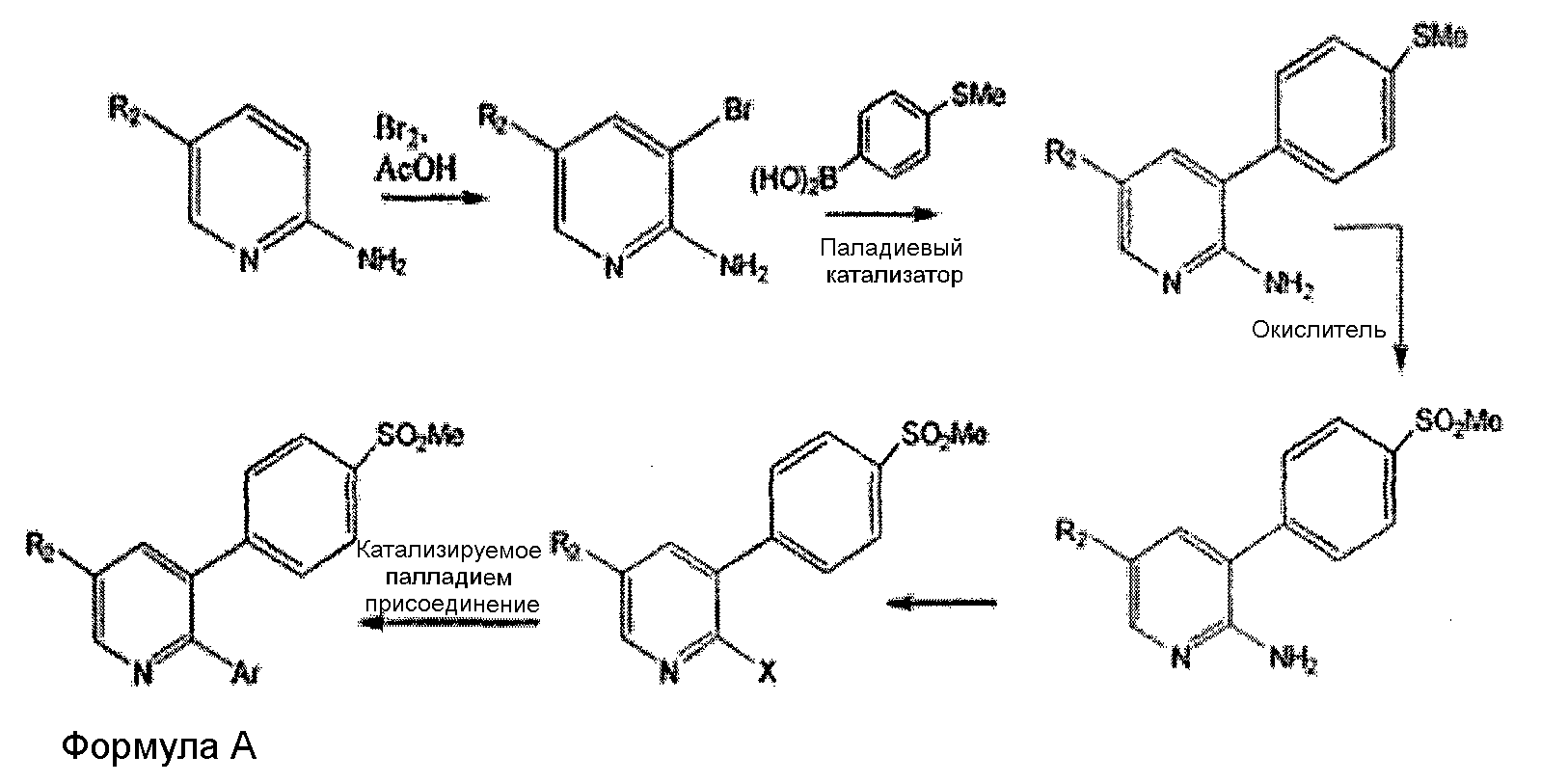
Соединения общей формулы (А), включая эторикоксиб, описаны в патенте США US5861419 в соответствии с нижеследующей схемой.
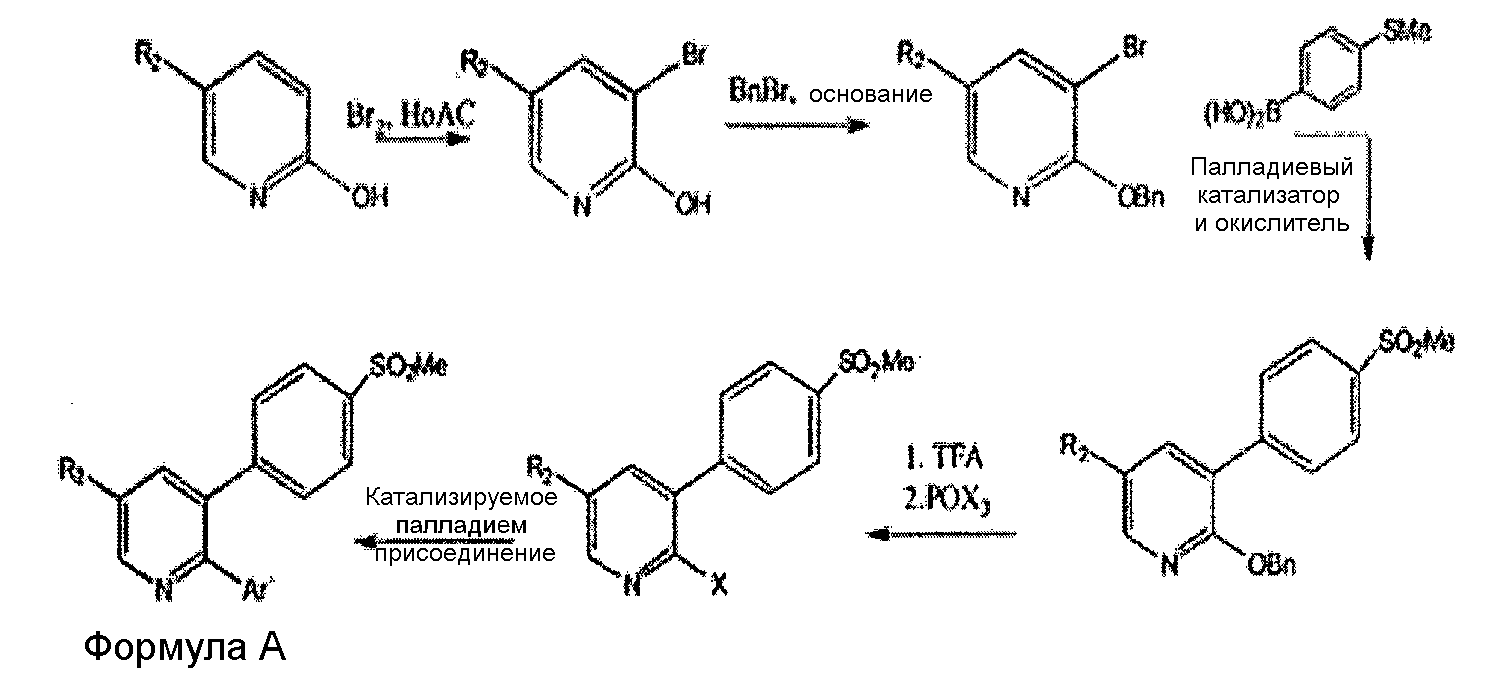
Другой способ также описан в патенте США US5861419 в соответствии с нижеследующей схемой.
Способ, описанный в патенте США US5861419, включает в себя многостадийный синтез с невысоким выходом конечного продукта.
Патент США US6040319 описывает другой способ получения эторикоксиба согласно нижеследующей схеме. Он относится к реакции конденсации между соединением формулы (II) и соединением формулы (IIIa), где Х может быть выбран из фосфатов, сульфатов, ацетатов, перхлоратов, боратов, бензоатов, напсилатов, в частности, гексафторфосфата, сульфата, мезилата, тозилата, трифлата, ацетата, трифторацетата, тетрафторбората, тетрафенилбората, гексафторантимоната, хлорида, бромида, фторида, иодида, бензоата и напсилата. Подходящими основаниями являются гидроксиды натрия или калия, карбонат цезия, алкоксиды, амиды и гидриды лития, натрия и калия.
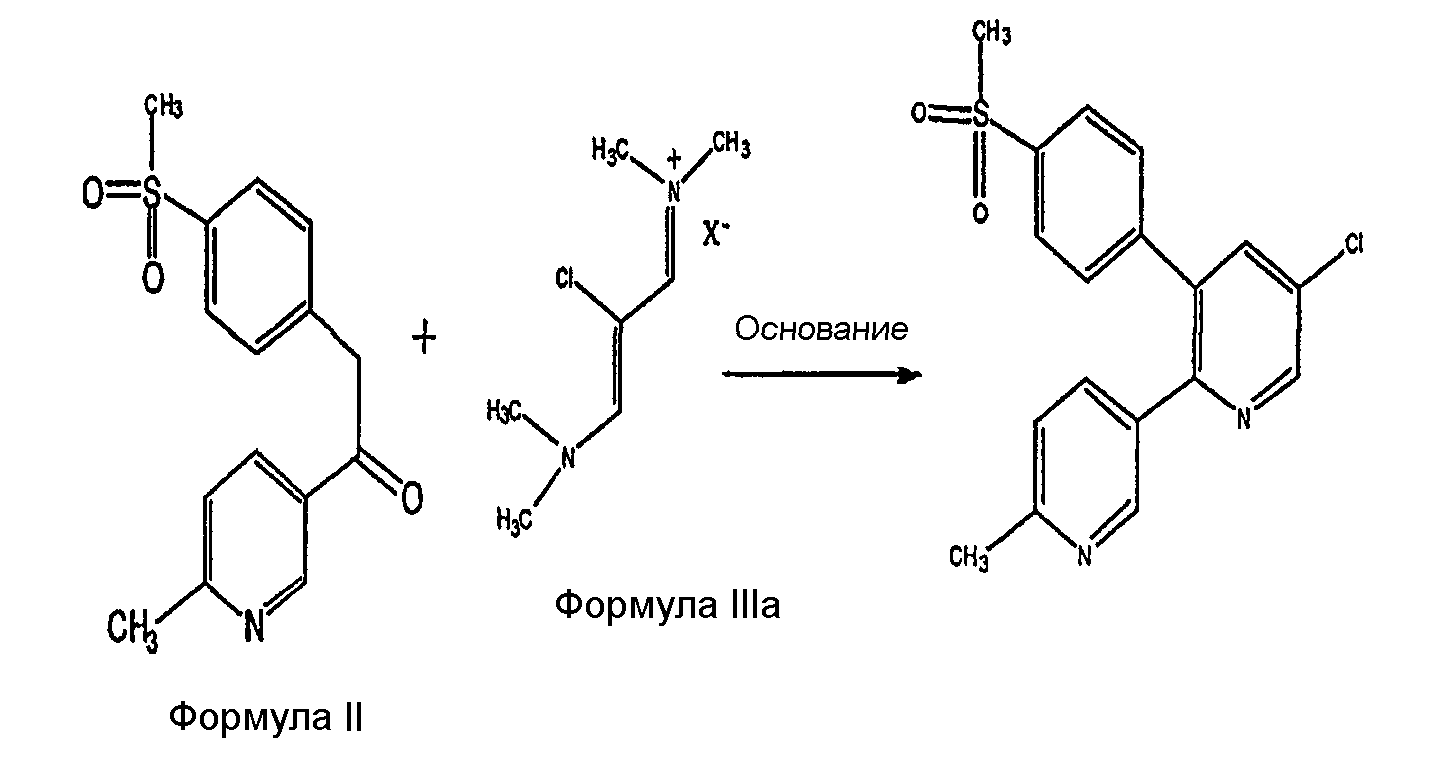
Патент WO 99/55830 описывает способ получения ингибиторов COX-2, включая эторикоксиб. Он содержит реакцию конденсации между солью винамидиния и производного замещенного бензилпиридилкетона, который в свою очередь получен в ходе реакции Гриньяра между производным пиридиламида и тиометилбензилгалида с последующим окислением.
Патент США US6071936 включает в себя производные замещенных пиридинов, их фармацевтические композиции и способ лечения СОХ-2-зависимых заболеваний. Различные соединения, включенные в указанный патент, имеют структуру производных 2,3-диарилпиридина с сульфоновым заместителем. Схема синтеза содержит катализируемое палладием диарильное присоединение между галоген-замещенным производным 2-аминопиридина и 4-(метилтио)-фенилбороновой кислотой с последующим окислением и дальнейшим присоединением другого арильного агента.
Патенты США US6001843, US6596736 B2 и US6812346 B2 также описывают структурно схожие замещенные производные пиридина и применяемую методику синтеза. Их использование в получении фармацевтических составов в качестве ингибиторов СОХ-2 также представлено в указанных изобретениях.
Патент WO2010/097802 А2 описывает способ получения лекарственного средства эторикоксиба. Хотя ранее было описано использование некоторых замещенных солей β-хлорвинамидиния, настоящее изобретение относится к получению других солей β-хлорвинамидиния с циклическими или гетероциклическими группами в качестве заместителя. Оно также описывает способ получения указанных других солей винамидиния, а также способ их очистки. Также в указанном патенте описаны разнообразные свойства одной из таких модифицированных солей β-хлорвинамидиния, а именно 2-хлор-1,3-(биспиперидил)триметинийгексафторфосфата, которая существует в форме I и форме II. Описан также и способ получения эторикоксиба с использованием данных солей винамидиния с циклическими заместителями.
Тем не менее, всегда существует необходимость разрабатывать более новые подходы к синтезу, которые могут привести к улучшению и повышению эффективности способа получения указанного лекарственного средства.
Описание изобретения
Таким образом, целью настоящего изобретения является предоставление простого и эффективного способа получения действенного лекарственного средства, а именно, 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина формулы (I), известного ингибитора циклооксигеназы-2 с установленной эффективностью.
Другой целью настоящего изобретения является предоставление простого и эффективного способа для ключевого промежуточного соединения формулы (II), а именно, 1-(6-метил-3-пиридинил)-2-[4-(метилсульфонил)фенил]этанона, используемого при получении 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, лекарственного средства, структура которого представлена формулой (I).
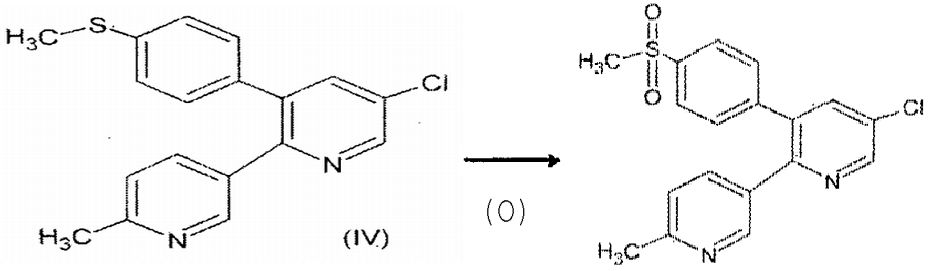
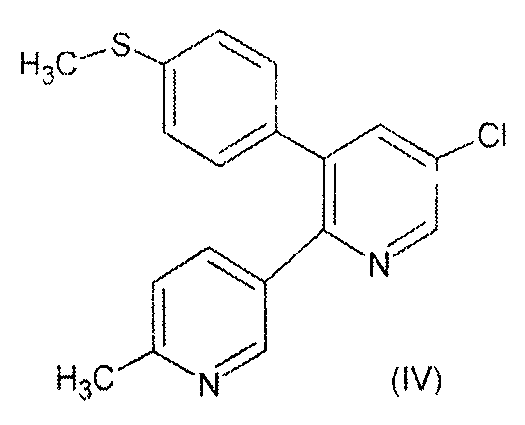
Другой целью настоящего изобретения является получение нового промежуточного соединения 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина, структура которого представлена формулой (IV), другого ключевого соединения для получения 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, структура которого представлена формулой (I).
Другой целью настоящего изобретения является предоставление способа получения соединения формулы (IV), которое представляет собой новое промежуточное соединение в получении лекарственного средства формулы (I).
Краткое описание изобретения
Согласно настоящему изобретению представлен способ синтеза ингибитора циклооксигеназы-2 эторикоксиба формулы (I) из соединения формулы (IV), включающий окисление соединения формулы (IV) в присутствии катализатора окисления и катализатора межфазного переноса.
В одном из аспектов настоящее изобретение относится к новому соединению формулы (IV).
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (IV), включающему реакцию между производным бензилпропилкетона формулы (V) и производным соли винамидиния формулы (III) в присутствии основания. Масса продукта далее проходит серии химических и манипуляционных стадий до получения целевого промежуточного соединения пиридина, представленного соединением формулы (IV).
Другой аспект настоящего изобретения относится к улучшенному способу получения эторикоксиба из 4-метилтиобензилцианида в качестве исходного соединения. Указанное соединение конденсируют с метил-6-метилникотинатом в присутствии основания до получения соединения формулы (VI), которое гидролизуется и декарбоксилируется до формы (V). Соединение (V) конденсируют с производным соли винамидиния формулы (III) в присутствии основания. Полученная масса продукта далее проходит серии химических и манипуляционных стадий до получения целевого промежуточного соединения пиридина, представленного соединением формулы (IV), которое далее проходит окисление с получением целевого соединения, эторикоксиба.
Краткое описание фигур
Настоящее изобретение детально описано со ссылками на сопутствующие фигуры. На фигурах:
Фиг.1 иллюстрирует описание структур.
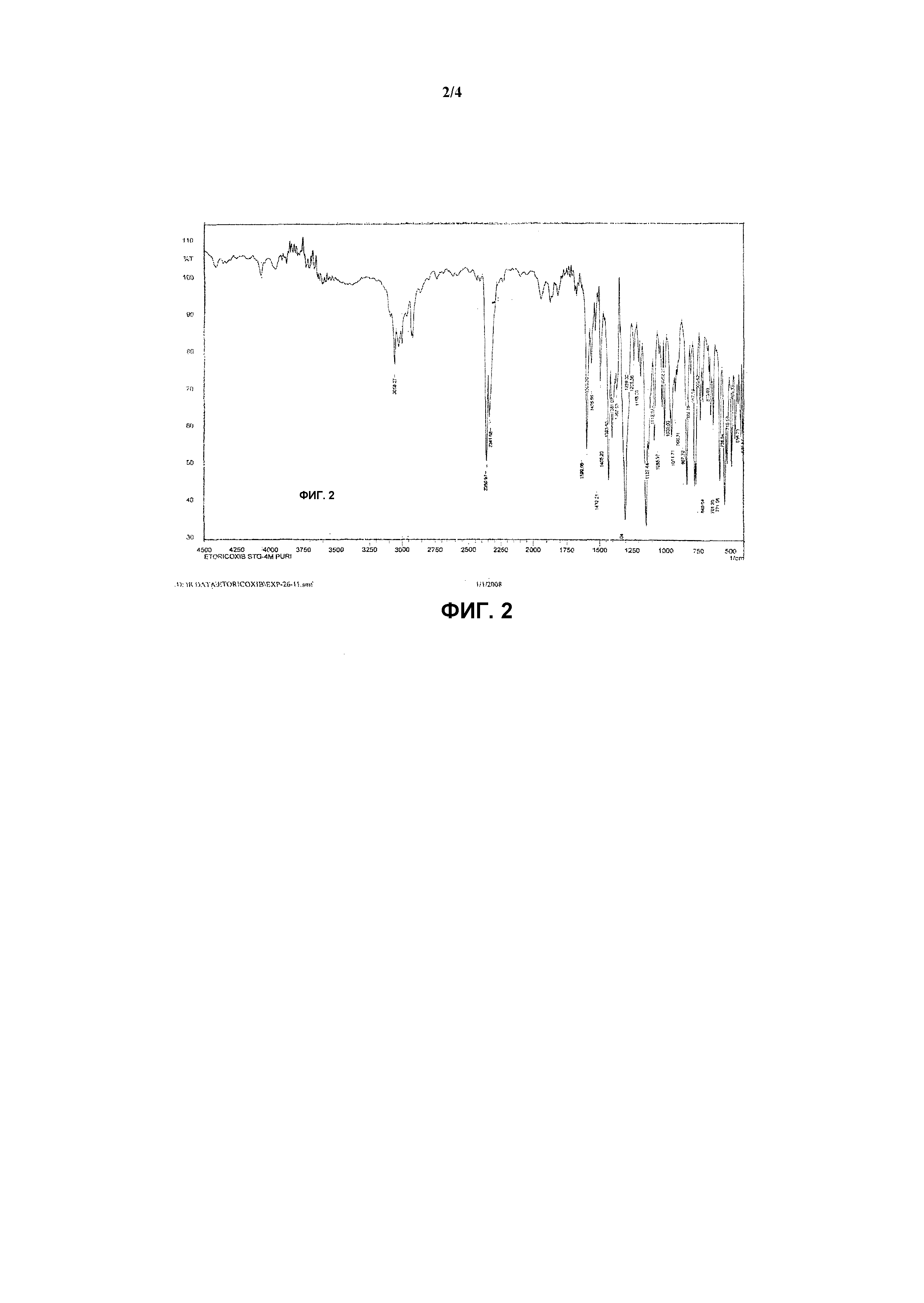
Фиг.2 иллюстрирует инфракрасный спектр соединения формулы (I) 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, т.е. эторикоксиба.
Фиг.3 иллюстрирует инфракрасный спектр промежуточного соединения формулы (IV) 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина.
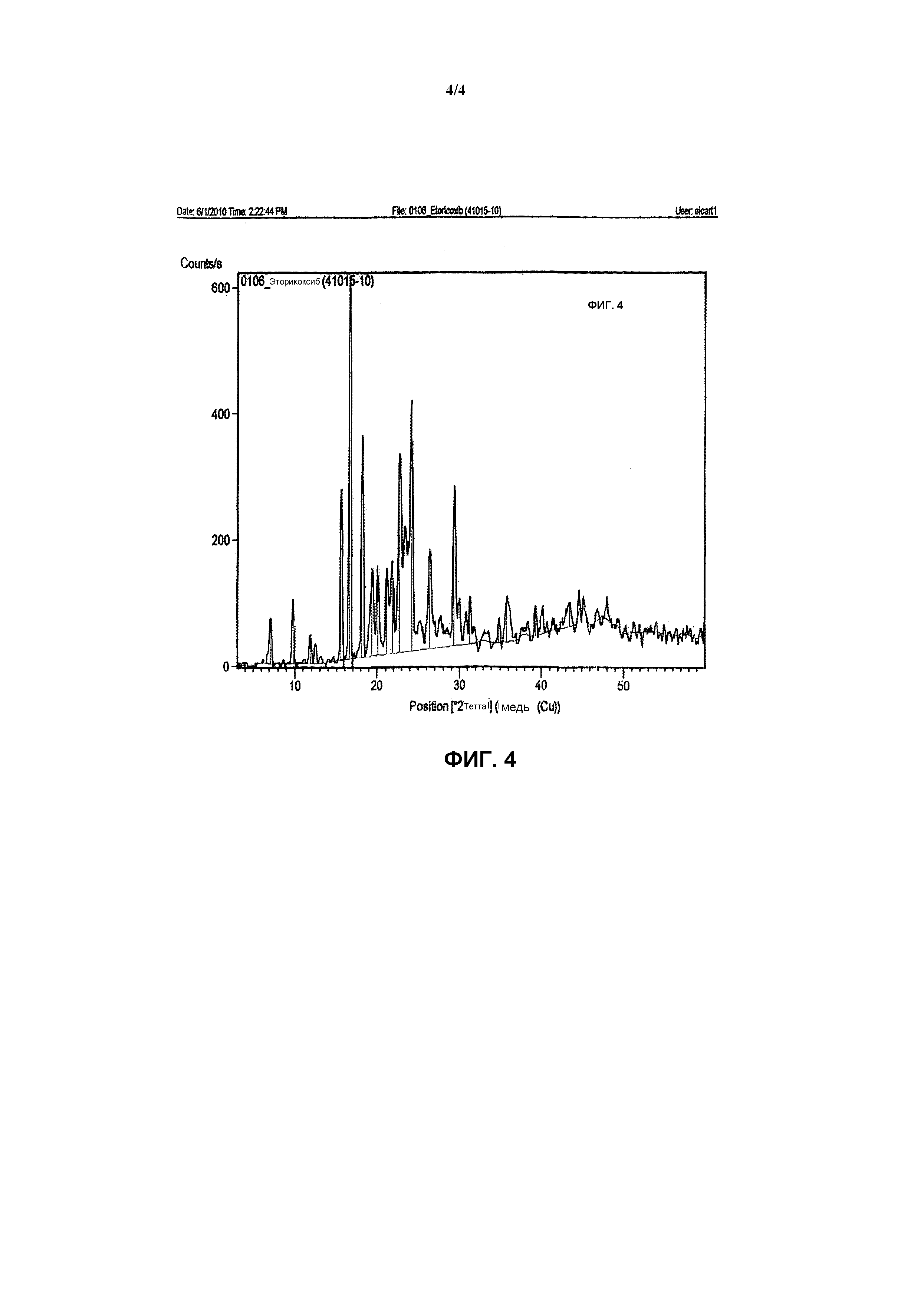
Фиг.4 иллюстрирует спектр порошковой рентгеновской дифракции 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, т.е. эторикоксиба.
Детальное описание изобретения
Далее настоящее изобретение будет детально описано со ссылкой на предпочтительные и дополнительные варианты осуществления для того, чтобы их различные аспекты могли быть полностью поняты и оценены.
Настоящее изобретение в соответствии с целью изобретения, указанной ранее, детально описывается в нижеследующих вариантах осуществления.
В предпочтительном варианте осуществления настоящее изобретение относится к способу получения эторикоксиба из соединения 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина (IV), включающему в себя окисление указанного соединения формулы (IV) в присутствии катализатора окисления и катализатора межфазного переноса с получением эторикоксиба формулы (I).
В соответствии с вышесказанным, соединение формулы (IV) 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридин окисляют в присутствии окисляющего агента, выбранного из пероксида водорода, пероксида натрия и т.д. с катализатором и в условиях двух фаз, включающих органическую и водную фазы.
Двухфазная масса обычно содержит воду, хлорированный углеводород, окисляющий агент, катализатор окисления и катализатор межфазного переноса. Обычно хлорированный углеводородный растворитель представлен не менее одним растворителем, выбранным из группы хлорированных углеводородных растворителей, содержащей такие растворители, как хлороформ, дихлорэтан, дихлорметан и тетрахлорметан. Обычно катализатор окисления представлен не менее одним, выбранным из группы солей, содержащей молибдат натрия, ванадат натрия и фольфрамат натрия. Катализатор межфазного переноса, как правило, представлен не менее одним, выбранным из группы таких катализаторов, содержащей метил-три-н-октилхлорид аммония, метил-три-н-бутилхлорид аммония, метил-три-н-бутилхлорид аммония, хлорид бензетония и хлорид метилбензетония. Реакцию окисления проводят водным раствором пероксида водорода при температуре в пределах 0-20°С, предпочтительно, при 10-14°С, с последующим повышением температуры до комнатной и контролем реакции с помощью ТСХ до ее завершения. Происходит разделение фаз, водная фаза проходит выделение тем же органическим растворителем, который использовался в реакции, все органические растворы смешивают, промывают водным раствором карбоната натрия и водой со значением рН, близким к нейтральному, высушивают над безводным сульфатом натрия, необязательно, очищают активированным углем, фильтруют и концентрируют для удаления растворителя под вакуумом. Остаточную массу обрабатывают спиртовым растворителем, как правило, представленным не менее одним из группы, содержащей метанол, ректифицированный спирт, изопропанол, н-пропанол и н-бутанол с объемным содержанием воды 0-4%. Массу продукта охлаждают до (-)2-2°С, фильтруют, промывают тем же холодным растворителем и высушивают до получения сырого продукта эторикоксиба формулы (I).
Очистка сырого эторикоксиба, полученного описанным способом, включает стадию кристаллизации. Сырой эторикоксиб формулы (I) очищают с помощью кристаллизации, которая включает растворение продукта в не менее одном спиртовом растворителе, как правило, выбранном из группы, содержащей н-пропанол, изопропанол, метанол, ректифицированный спирт, ацетон, с объемным содержанием воды 0-6%. Сырой продукт растворяют в растворителе при повышенной температуре, обрабатывают активированным углем в течение 15-30 минут, фильтруют и охлаждают до комнатной температуры и далее до 12-15°С в течение приблизительно часа. Твердый продукт выделяют фильтрацией, промывают охлажденным растворителем и высушивают.
В альтернативном случае сырой эторикоксиб формулы (I) может очищаться переводом его в соль. В соответствии с этим, способ включает в себя взаимодействие паратолуолсульфокислоты с эторикоксибом в присутствии органического растворителя до получения соли эторикоксиба и выделение полученной соли.
Выделенную соль эторикоксиба и паратолуолсульфокислоты далее растворяют в воде при комнатных условиях и обрабатывают водным раствором карбоната натрия до получения слабощелочной среды. Массу продукта далее обрабатывают толуолом при повышенной температуре для выделения массы продукта в толуоле. Водный слой выделяют в толуоле и смешивают с первым раствором толуола. Объединенный раствор толуола далее промывают водой, очищают активированным углем, высушивают над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения остатка. Остаток снова растворяют в не менее одном спиртовом растворителе, как правило, выбранном из группы, содержащей метанол, этанол, ректифицированный спирт, изопропанол и н-пропанол, с объемным содержанием воды 0-6%. Раствор охлаждают до 5-10°С в течение 1-2 часов, фильтруют, промывают охлажденным растворителем и высушивают до получения очищенного эторикоксиба.
Настоящее изобретение относится к новому промежуточному соединению формулы (IV)
В другом варианте осуществления настоящее изобретение относится к способу получения нового промежуточного соединения (IV), содержащему взаимодействие 1-(6-метил-3-пиридинил)-2-[4-(метилтио)фенил]этанона (V) с 2-хлор-N,N-диметиламинотриметиний гексафторфосфатом (III), в присутствии основания с последующим добавлением смеси спирта и кислоты при той же температуре, и добавлением водного раствора аммиака и далее безводной соли аммония до получения соединения формулы (IV).
В другом варианте осуществления настоящее изобретение относится к простому и эффективному способу получения соединения формулы (I), а именно 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина (эторикоксиба), с высокой чистотой. Описанный способ включает следующие стадии:
a. Реакцию конденсации между 4-метилтиобензилцианидом и метил-6-метилникотинатом в присутствии подходящего основания и подходящего растворителя при температуре кипения с обратным холодильником с получением 1-(6-метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанона (VI);
b. Гидролиз соединения (VI) со стадии (а) в присутствии кислоты при 40-50°С с последующим декарбоксилированием in situ при температуре кипения с обратным холодильником с получением 1-(6-метил-3-пиридинил)-2-[4-(метилтио)фенил]этанона формулы (V);
с. Взаимодействие соединения формулы (V) с солью 2-хлор-N,N-диметиламинотриметиний гексафторфосфатом (III) в присутствии основания при температуре в пределах от 0° до 10°С, с последующим добавлением смеси спирта и кислоты при той же температуре, и добавлением водного раствора аммиака и соли аммония, нагреванием, до получения промежуточного соединения 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина (IV); и
d. Проведение окисления соединения формулы (IV), полученного на стадии (с) в присутствии катализатора окисления и катализатора межфазного переноса в двухфазных условиях до получения эторикоксиба формулы (I).
Описанный процесс представлен ниже на схеме 1.
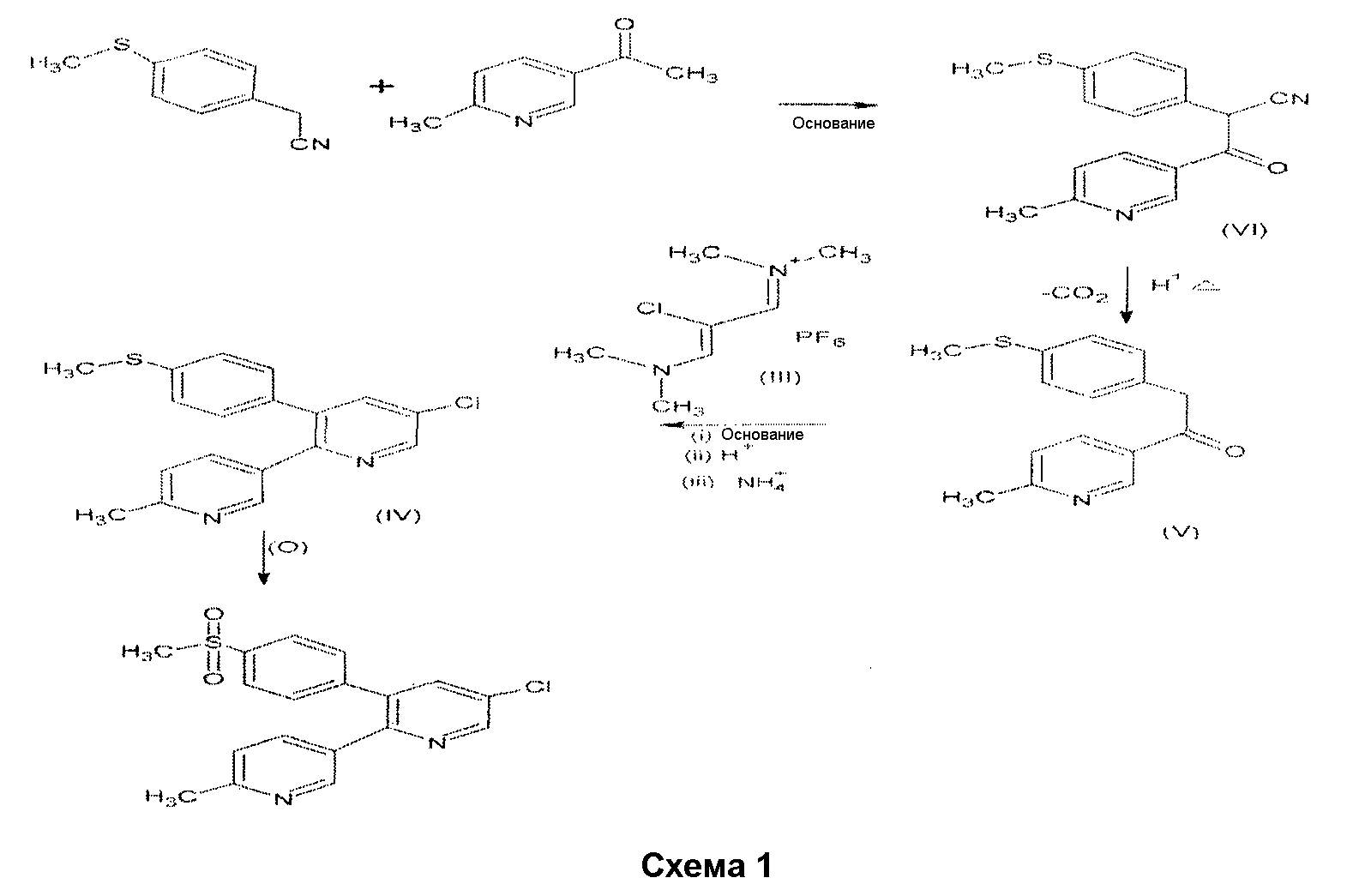
В соответствии со схемой 1, стадия (а) настоящего изобретения включает катализируемую основанием реакцию конденсации между 4-метилтиобензилцианидом и метил-6-метилникотинатом в углеводородном растворителе. Основание, как правило, выбирают из группы оснований, таких как метоксид натрия, амид натрия, гидрид натрия, трет-бутоксид калия и метоксид калия. Предпочтительно, используемыми основаниями являются метоксид натрия или трет-бутоксид калия. Используемые углеводороды, как правило, выбирают из гептана, толуола, ксилена или их смесей. Завершение реакции отслеживают с помощью анализа ТСХ, и реакцию останавливают перенесением реакционной массы в ледяную воду. Значение рН реакционной массы выравнивают разбавленной кислотой, предпочтительно, разбавленной соляной кислотой, до значения рН 5,0-6,5, предпочтительно 5,2-6,3, и продукт 1-(6-метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанон (VI) выделяют фильтрацией.
Соединение формулы (VI) со стадии (b) гидролизуют в присутствии кислого катализатора до получения соединения карбоновой кислоты, которое при нагревании декарбоксилируется in situ с получением 1-(6-метил-3-пиридинил)-2[4-(метилтио)фенил]этанона формулы (V). Реакционная среда состоит из смеси органической карбоновой кислоты, выбранной из группы кислот, таких как муравьиная кислота, ледяная уксусная кислота, пропионовая кислота, масляная кислота, пентановая кислота, и неорганической кислоты, такой, как концентрированная соляная кислота или концентрированная серная кислота. Обычно в описанном способе применяют сочетание ледяной уксусной кислоты и концентрированной соляной кислоты.
Соединение формулы (V) со стадии (b) выделяют с помощью углеводородного растворителя, выбранного из группы растворителей, таких как гексан, гептан, циклогексан и толуол, и далее значение рН выравнивают до 6,8-7,3 водным раствором аммиака. Предпочтительно, использование гексана в качестве растворителя для выделения, и предпочтительное значение рН находится в промежутке от 6,9 до 7,2. Массу продукта выделяют фильтрацией и высушивают при 45-60°С.
Способ выделения соединения формулы (V) (стадия b) может необязательно содержать выделение реакционной массы стадии (b) после завершения реакции и выравнивания значения рН до 6,8-7,3 растворителями, выбранными из галогенированных углеводородов, таких как хлороформ, дихлорметан, эфиров, таких как этилацетат или пропилацетат. Обычно в качестве предпочтительного растворителя используют дихлорметан. Промежуточное соединение (V) выделяют концентрированием для удаления растворителя, и далее добавляют другой растворитель, выбранный из низших спиртов, предпочтительно, изопропанол, с охлаждением массы до 10-12°С, фильтрацией продукта и последующим высушиванием при 45-50°С.
Промежуточное соединение (V), полученное вышеописанным способом, необязательно очищают перекристаллизацией из метанола.
К соединению 1-(6-метил-3-пиридинил)-2-[4-(метилсульфонил)фенил]этанону формулы (V) в соответствии со стадией (с) добавляют основной катализатор в органическом растворителе при температуре в промежутке от 0 до 20°С, предпочтительно, от 0 до 10°С; более предпочтительно, от 5 до 8°С. Основной катализатор выбирают из группы, содержащей метоксид натрия, метоксид калия, трет-бутоксид калия, амид натрия и гидрид натрия. Обычно в качестве основного катализатора используют трет-бутоксид калия. Органический растворитель выбирают из группы трет-бутанола, изопропанола, тетрагидрофурана и метил-трет-бутилового эфира. В качестве растворителя обычно используют изопропанол. Затем реакционная масса взаимодействует с промежуточной солью 2-хлор-N,N-диметиламинотриметиний гексафторфосфатом, соединением формулы (III), при температуре в пределах от 0 до 10°С в течение 2-4 часов, при этом протекание реакции контролируют с помощью ТСХ до тех пор, пока одно из реагирующих промежуточных соединений, а именно соединение формулы (V), не будет использовано практически полностью. Реакционную массу далее обрабатывают смесью спиртового растворителя, выбранного из группы, содержащей метанол, этанол, изопропанол, трет-бутанол и н-пропанол; и карбоновой кислоты, выбранной из группы кислот, состоящей из муравьиной кислоты, уксусной кислоты, н-пропионовой кислоты и н-масляной кислоты. В качестве спиртового растворителя, как правило, используют изопропанол, и в качестве карбоновой кислоты используют уксусную кислоту. Реакционную массу выдерживают при температуре в пределах от 0 до 10°С, предпочтительно, от 5 до 10°С, в течение 2,5-3 часов. Далее добавляют водный раствор аммиака с последующим добавлением соли аммония, выбранной из группы солей, включающей ацетат аммония, карбонат аммония и т.д. Обычно в реакции используют ацетат аммония. Массу нагревают с обратным холодильником до завершения реакции, охлаждают до комнатной температуры, и далее смешивают с раствором аммиака и раствором формальдегида. Реакционный растворитель удаляют перегонкой при пониженном давлении с добавлением другого растворителя толуола при нагревании реакционной массы вновь до 60-65°С. Разделяются органический и водный слой. Водный слой промывают толуолом, объединенные слои толуола промывают водным раствором карбоната натрия, потом водой, очищают активированным углем, фильтруют, высушивают над безводным сульфатом натрия и концентрируют под вакуумом для удаления растворителя. К остаточной массе добавляют спиртовой растворитель, выбранный из группы метанола, изопропанола, ректифицированного спирта, н-пропанола или их смеси. Добавляют, как правило, изопропанол, и далее охлаждением достигают переход продукта в твердое состояние. Массу продукта фильтруют, промывают изопропанолом и высушивают с получением соединения формулы (IV).
Соединение формулы (IV) кристаллизуют из органического растворителя, выбранного из группы спиртовых растворителей, включающей этиловый спирт, ректифицированный спирт, н-пропанол, изопропанол или их смеси, с объемным содержанием воды 0-6%. Соединение формулы (IV) растворяют в достаточном количестве растворителя, нагревают с обратным холодильником до растворения, фильтруют, охлаждают до получения твердого продукта и фильтруют для выделения продукта. В качестве растворителя для очистки, как правило, используют изопропанол. Продукт выделяют и высушивают при 45-50°С.
Соединение формулы (IV) 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридин окисляют в присутствии окисляющего агента, выбранного из пероксида водорода, пероксида натрия и т.д. с катализатором и в условиях двух фаз, включающих органическую и водную фазы.
Двухфазная масса обычно содержит воду, хлорированный углеводород, окисляющий агент, катализатор окисления и катализатор межфазного переноса. Обычно хлорированный углеводородный растворитель представлен не менее одним растворителем, выбранным из группы хлорированных углеводородных растворителей, содержащей такие растворители, как хлороформ, дихлорэтан, дихлорметан и тетрахлорметан. Обычно катализатор окисления представлен не менее одним, выбранным из группы солей, содержащей молибдат натрия, ванадат натрия и фольфрамат натрия. Катализатор межфазного переноса, как правило, представлен не менее одним, выбранным из группы таких катализаторов, содержащей метил-три-н-октилхлорид аммония, метил-три-н-бутилхлорид аммония, метил-три-н-бутилхлорид аммония, хлорид бензетония и хлорид метилбензетония. Реакция окисления проводится водным раствором пероксида водорода при температуре в пределах 0-20°С, предпочтительно при 10-14°С, с последующим повышением температуры до комнатной и контролем реакции с помощью ТСХ до ее завершения. Происходит разделение фаз, водную фазу выделяют тем же органическим растворителем, который использовался в реакции, все органические растворы смешивают, промывают водным раствором карбоната натрия и водой со значением рН, близким к нейтральному, высушивают над безводным сульфатом натрия, необязательно, очищают активированным углем, фильтруют и концентрируют для удаления растворителя под вакуумом. Остаточную массу обрабатывают спиртовым растворителем, как правило, представленным не менее одним из группы, содержащей метанол, ректифицированный спирт, изопропанол, н-пропанол и н-бутанол с объемным содержанием воды 0-4%. Массу продукта охлаждают до (-)2-2°С, фильтруют, промывают тем же холодным растворителем и высушивают до получения сырого продукта эторикоксиба формулы (I).
Очистка сырого эторикоксиба, полученного описанным способом, включает стадию кристаллизации. Сырой эторикоксиб формулы (I) очищают с помощью кристаллизации, которая включает растворение продукта в не менее одном спиртовом растворителе, как правило, выбранном из группы, содержащей н-пропанол, изопропанол, метанол, ректифицированный спирт, ацетон, с объемным содержанием воды 0-6%. Сырой продукт растворяют в растворителе при повышенной температуре, обрабатывают активированным углем в течение 15-30 минут, фильтруют и охлаждают до комнатной температуры и далее до 12-15°С в течение приблизительно часа. Отвердевший продукт выделяют фильтрацией, промывают охлажденным растворителем и высушивают.
Альтернативно, сырой эторикоксиб формулы (I) может быть очищен путем перевода в его соль. В соответствии с этим, способ включает в себя взаимодействие паратолуолсульфокислоты с эторикоксибом в присутствии органического растворителя до получения соли эторикоксиба и выделение полученной соли.
Выделенную соль эторикоксиба и паратолуолсульфокислоты далее растворяют в воде при комнатных условиях и обрабатывают водным раствором карбоната натрия до получения слабощелочной среды. Массу продукта далее обрабатывают толуолом при повышенной температуре для выделения массы продукта в толуоле. Водную фазу выделяют в толуоле и смешивают с первым раствором толуола. Объединенный раствор толуола далее промывают водой, очищают активированным углем, высушивают над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения остатка. Остаток снова растворяют в не менее одном спиртовом растворителе, как правило, выбранном из группы, содержащей метанол, этанол, ректифицированный спирт, изопропанол и н-пропанол, с содержанием воды 0-6%. Раствор охлаждают до 5-10°С в течение 1-2 часов, фильтруют, промывают охлажденным растворителем и высушивают до получения очищенного эторикоксиба.
Настоящее изобретение относится к получению соли винамидиния 2-хлор-N,N-диметиламинотриметиний гексафторфосфата формулы (III). Обычно N,N-диметилформамид связывается с хлорацетилхлоридом при температуре приблизительно 50-55°С, и далее добавляют хлорокись фосфора при повышенной температуре 65-70°С в течение 5-6 часов. Далее проводят охлаждение до комнатной температуры и добавление ледяной водной смеси, содержащей гексафторфосфорную кислоту с добавлением водного раствора гидрохлорида натрия для выравнивания рН до значения приблизительно от 2,0 до 2,8, и выдерживают при перемешивании в течение приблизительно 30 минут. Промежуточное соединение формулы (V) выделяют фильтрацией, промывают и затем очищают водным изопропанолом и высушивают. Оно также может быть необязательно заново очищено растворением в метаноле при повышении температуры до кипения, очисткой активированным углем, фильтрацией раствора, частичной концентрацией с удалением раствора перегонкой, охлаждением массы до достижения кристаллизации, фильтрацией и высушиванием.
В другом варианте осуществления настоящее изобретение относится к способу получения 1-(6-метил-3-пиридинил)-2-[4-(метилсульфонил)фенил]этанона формулы (II), включающему:
a. Реакцию конденсации между 4-метилтиобензилцианидом и метил-6-метилникотинатом в присутствии подходящего основания и подходящего растворителя при температуре кипения с обратным холодильником до получения влажной массы 1-(6-метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанона (VI);
b. Гидролиз влажной массы соединения формулы (VI) со стадии (а) в присутствии кислоты при 40-50°С с последующим декарбоксилированием in situ при температуре кипения с обратным холодильником до получения 1-(6-метил-3-пиридинил)-2-[4-(метилтио)фенил]этанона формулы (V); и
с. Проведение окисления соединения формулы (V), полученного на стадии (b), в присутствии катализатора окисления и катализатора межфазного переноса с последующим добавлением смеси спирта и кислоты, добавлением водного раствора аммиака и далее соли аммония, нагревания, до получения соединения формулы (II).
Согласно описанному способу, между 4-метилтиобензилцианидом и метил-6-метилникотинатом проводится катализируемая основанием реакция конденсации, как описано выше. Масса продукта в виде влажного осадка проходит катализируемый кислотой гидролиз и далее термическое декарбоксилирование с получением 1-(6-метил-3-пиридинил)-2-[4-(метилсульфонил)фенил]этанона, соединения формулы (V), детали которого описаны выше. Указанное промежуточное соединение (V), выделенное из реакционной массы как масса влажного продукта, растворяют в органическом растворителе для проведения следующей стадии окисления. Растворитель выбирают из хлорированных углеводородов, таких как хлороформ, дихлорэтан, дихлорметан и тетрахлорметан. Катализатор окисления выбирают из группы солей, содержащей молибдат натрия, ванадат натрия или фольфрамат натрия. Катализатор межфазного переноса представлен не менее одним, выбранным из группы таких катализаторов, содержащей метил-три-н-октилхлорид аммония, метил-три-н-бутилхлорид аммония, метил-три-н-бутилхлорид аммония, хлорид бензетония и хлорид метилбензетония. Реакция окисления проводится в присутствии пероксида, такого как пероксид водорода, пероксид натрия и т.д. в водном растворе при температуре в пределах 0-20°С, предпочтительно, при 10-14°С, с последующим повышением температуры до комнатной и контролем реакции с помощью ТСХ до ее завершения. Происходит разделение фаз, водную фазу выделяют тем же органическим растворителем, который использовался в реакции, все органические растворы смешивают, промывают водным раствором карбоната натрия и водой со значением рН, близким к нейтральному, высушивают над безводным сульфатом натрия, необязательно очищают активированным углем, фильтруют и концентрируют для удаления растворителя под вакуумом. Остаточную массу обрабатывают спиртовым растворителем, представленным не менее одним из группы, содержащей метанол, ректифицированный спирт, изопропанол, н-пропанол и н-бутанол, с объемным содержанием воды 0-4%. Массу продукта охлаждают до (-)2-2°С, фильтруют, промывают тем же холодным растворителем и высушивают до получения сырого продукта 1-(6-метил-3-пиридинил)-2-[4-(метилсульфонил)фенил]этанона формулы (II).
В другом случае промежуточное соединение формулы (II) получают способом, включающим в себя реакцию конденсации между 4-метилсульфонилфенилуксусной кислотой и метил-6-метилникотинатом в присутствии металлорганического соединения, такого как трет-бутилхлорид магния, как описано в данной области.
Далее настоящее изобретение будет проиллюстрировано нижеследующими примерами, которые не ограничивают эффективный объем заявленного в формуле изобретения. Следовательно, любые варианты осуществления настоящего изобретения, описанные выше, не должны расцениваться как удаление от сущности и объема настоящего изобретения, как оно заявлено. Настоящее изобретение было описано в рамках его конкретных вариантов осуществления, и для специалистов в данной области различные модификации, параллели и эквиваленты будут очевидными, и подразумеваются включенными в объем настоящего изобретения.
Пример 1
Получение 1-(6-метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанона (VI)
4-Метилтиобензилцианид (10,0 г) растворяют в (100 мл) толуоле, и реакционную массу нагревают с обратным холодильником. Метил-6-метилникотинат (10,87 г) медленно добавляют при температуре кипения с обратным холодильником в течение приблизительно 30 минут. Реакционную массу перемешивают в течение 10 минут. Раствор метоксида натрия (30% масс/масс) 19,96 г добавляют в течение 30 минут. Температуру реакции поддерживают на уровне температуры кипения до подтверждения анализом ТСХ практически полного исчезновения пятна, соответствующего метил-6-метилникотинату. Реакционную массу охлаждают до 25-30°C при перемешивании. Реакцию в реакционной массе останавливают в смеси измельченного льда (75 г) и воды (10 мл). Значение рН реакционной массы доводят до 5,2-6,2 с помощью разбавленной соляной кислоты. Реакционную массу перемешивают в течение часа, фильтруют, промывают водой и высушивают при температуре 60-70°С (выход 16 г, чистота 96,3% по данным ВЭЖХ).
Пример 2
Получение 1-(6-метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанона (VI)
4-Метилтиобензилцианид (10,0 г) растворяют в (80 мл) толуоле с 20 мл н-гептана, и реакционная масса нагревают с обратным холодильником. Метил-6-метилникотинат (11,30 г) медленно добавляют при температуре кипения в течение приблизительно 30 минут. Реакционную массу перемешивают в течение 10 минут. Раствор метоксида натрия (30% масс/масс) 19,96 г добавляют в течение 30 минут. Температуру реакции поддерживают на уровне температуры кипения до подтверждения анализом ТСХ практически полного исчезновения пятна, соответствующего метил-6-метилникотинату. Реакционную массу охлаждают до 25-30°C при перемешивании. Реакцию в реакционной массе останавливают в смеси измельченного льда (75 г) и воды (15 мл). Значение рН реакционной массы доводят до 5,2-6,2 с помощью разбавленной соляной кислоты. Реакционную массу перемешивают в течение часа, фильтруют, промывают водой и высушивают при температуре 60-70°С (выход 15,7 г, чистота 95,7% по данным ВЭЖХ).
Пример 3
Получение 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V)
1-(6-Метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанон (VI) (10 г) добавляют к смеси концентрированной соляной кислоты (70 мл) и ледяной уксусной кислоты (25 мл) при температуре 40-50°С, с последующим декарбоксилированием при температуре кипения. Протекание реакции контролируют с помощью ТСХ до завершения реакции. Реакционную массу охлаждают до комнатной температуры и промывают выделением гексаном.
Указанную реакционную массу медленно выливают в смесь (31,75 мл) концентрированного раствора аммиака и (10,00 мл) воды. Реакционную массу перемешивают в течение 10 минут, и значение рН доводят до 6,80-7,20 разбавленным раствором аммиака при температуре 0-5°С. Погашенную реакционную массу перемешивают в течение 30-60 мин и фильтруют. Полученный продукт промывают водой, высушивают в лотковой сушилке при 45-50°С до содержания влаги менее 2% до получения (V) в виде кремово-желтого порошка (выход 8,8 г, чистота по ВЭЖХ=93,89%).
Пример 4
Получение 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V)
1-(6-Метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанон (VI) (10 г) добавляют к смеси концентрированной соляной кислоты (80 мл) и ледяной уксусной кислоты (30 мл) при температуре 40-50°С, с последующим декарбоксилированием при температуре кипения. Протекание реакции контролируют с помощью ТСХ до завершения реакции. Реакционную массу охлаждают до комнатной температуры и промывают экстракцией гексаном. Указанную выше реакционную массу медленно выливают в смесь (31,75 мл) концентрированного раствора аммиака и (10,00 мл) воды. Реакционную массу перемешивают в течение 10 минут, и значение рН доводят до 7,00-7,20 разбавленным раствором аммиака при температуре 0-5°С. Погашенную реакционную массу перемешивают в течение 30-60 минут и фильтруют. Полученный продукт промывают водой, высушивают в лотковой сушилке при 45-50°С до содержания влаги менее 2% до получения (V) в виде кремово-желтого порошка (выход 8,70 г, чистота по ВЭЖХ=95,00%).
Пример 5
Получение 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V)
К соединению (VI) (10 г) (полученному способом, описанным выше в примере 4) добавляют смесь концентрированной соляной кислоты (75 мл) и ледяной уксусной кислоты (25 мл) при комнатной температуре, и нагревают до 70-80°C. После завершения реакции, ее охлаждают до комнатной температуры и выделяют (12 мл) толуолом.
Указанную реакционную массу далее добавляют к смеси (31,75 мл) раствора аммиака и (10,00 мл) воды и перемешивают. Значение рН реакционной массы доводят до 6,80-7,20 с помощью разбавленного раствора карбоната натрия (5% масса/объем) при температуре 0-5°С. Реакционную массу перемешивают в течение 60 минут и фильтруют до получения влажного осадка. Влажный продукт промывают дважды (10 мл) водой. Влажный осадок растворяют в (70 мл) дихлорметане с последующим добавлением (20 мл) воды, перемешивают в течение 10 минут и разделением фаз. Водную фазу дважды выделяют дихлорметаном. Объединенную органическую фазу, содержащую продукт, промывают (10 мл) водой (10,00 мл), высушивают над безводным сульфатом натрия и очищают активированным углем.
Слой с продуктом концентрируют путем перегонки дихлорметана при атмосферном давлении до получения полутвердого остатка. Остаточную массу дегазируют под вакуумом в течение 30 минут. К массе добавляют изопропиловый спирт (5 мл), и ее охлаждают до 10-12°С. После перемешивания в течение 1 часа при температуре 10-12°С, массу фильтруют и промывают охлажденным изопропиловым спиртом (2,00 мл). Продукт высушивают в вакуумном сушильном шкафу при температуре 45-50°С (7 г, чистота по ВЭЖХ 93,5%)
Полученный высушенный продукт (7 г) очищают с помощью обработки метилизобутилкетоном (17,5 мл). Суспензию продукта перемешивают в течение 30 минут, фильтруют и промывают метилизобутилкетоном (2 мл) и высушивают в вакуумном сушильном шкафу при температуре 40-45°С. Очищенный продукт (5 г) показал чистоту по ВЭЖХ 95,00%.
Пример 6
Получение 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V)
К соединению (VI) (10 г) (полученному способом, описанным выше в примере 4) добавляют смесь концентрированной соляной кислоты (75 мл) и ледяной уксусной кислоты (25 мл). После завершения реакции, ее охлаждают до комнатной температуры и выделяют (12 мл) толуолом.
Указанную реакционную массу далее добавляют к смеси (31,75 мл) раствора аммиака и (10,00 мл) воды и перемешивают. Значение рН реакционной массы доводят до 6,9-7,1 с помощью разбавленного раствора карбоната натрия (5% масса/объем) при температуре 0-5°С. Реакционную массу перемешивают в течение 60 минут и фильтруют до получения влажного осадка. Влажный продукт промывают дважды (10 мл) водой. Влажный осадок продукта растворяют в (70 мл) дихлорметане с последующим добавлением (20 мл) воды, перемешиванием в течение 10 минут и разделением слоев. Водный слой дважды выделяют дихлорметаном. Объединенный органический слой, содержащий продукт, промывают водой (10,00 мл), высушивают над безводным сульфатом натрия и очищают активированным углем.
Слой с продуктом концентрируют путем перегонки дихлорметана при атмосферном давлении с получением полутвердого остатка. Остаточную массу дегазируют под вакуумом в течение 30 минут. К массе добавляют изопропиловый спирт (5 мл), и ее охлаждают до 8-10°С. После перемешивания в течение 1 часа при температуре 8-10°С, массу фильтруют и промывают охлажденным изопропиловым спиртом (2,00 мл). Продукт высушивают в вакуумном сушильном шкафу при температуре 45-50°С с получением высушенного продукта (7 г, чистота по ВЭЖХ 93,24%)
Полученный высушенный продукт (7 г) очищают с помощью обработки метилизобутилкетоном (17,5 мл). Суспензию продукта перемешивают в течение 30 минут, фильтруют и промывают метилизобутилкетоном (2 мл), и высушивают в вакуумном сушильном шкафу при температуре 40-45°С. Очищенный продукт (5,2 г) показал чистоту по ВЭЖХ 95,40%.
Пример 7
Очистка 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V)
Высушенный продукт 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V) (10 г) и метанол (150 мл) нагревают до растворения. Добавляют активированный уголь (0,5 г), реакционную массу выдерживают при перемешивании в течение 30 минут, а затем фильтруют. Фильтрат охлаждают при перемешивании при 0-3°С в течение часа, массу продукта отфильтровывают и промывают охлажденным метанолом (2,5 мл). Очищенный продукт, полученный таким образом, высушивают под вакуумом при 45-50°С (выход 4,5 г, чистота по ВЭЖХ 98,65%).
Пример 8
Очистка 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V)
Высушенный продукт 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанона (V) (10 г) и метанол (98 мл) с (2 мл) воды нагревают до растворения. Добавляют активированный уголь (0,5 г), реакционную массу выдерживают при перемешивании в течение 30 минут, а затем фильтруют. Фильтрат охлаждают при перемешивании при 0-3°С в течение часа, массу продукта отфильтровывают и промывают охлажденным метанолом (2,5 мл). Очищенный продукт, полученный таким образом, высушивают под вакуумом при 45-50°С (выход 5,5 г, чистота по ВЭЖХ 98,22%).
Пример 9
Получение соли винамидиния 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III)
N,N-диметилформамид (440 мл) нагревают до температуры 50-55°C, и к нему медленно добавляют хлорацетилхлорид (99 г) в течение 3-4 часов. Реакционную массу далее нагревают до 65-70°С, и оксихлорид фосфора (140 г), постепенно добавляют в течение приблизительно 5-6 часов. Массу выдерживают при перемешивании в течение 5 часов при температуре 65-70°С, охлаждают до 25-30°С с последующим внесением в измельченный лед и воду, содержащую гексафторфосфорную кислоту (232 г) и гидроксид натрия, при поддержании рН около 2,0-2,2. Погашенную массу перемешивают в течение 30 минут, фильтруют для выделения твердого продукта и промывают холодной водой (выход влажного продукта 260-300 г) Соль хлорида винамидиния (III), полученную как описано выше, очищают с помощью обработки водным раствором изопропилового спирта при температуре 75-80°С, с последующим охлаждением до 20-23°С. Суспензию массы продукта фильтруют, промывают изопропиловым спиртом и высушивают при температуре 55-65°С до содержания влаги менее 0,5% (выход 174 г), т.пл. 124-127°С.
Пример 10
Получение соли винамидиния 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III)
N,N-диметилформамид (400 мл) нагревают до температуры 50-55°C и к нему медленно добавляют хлорацетилхлорид (99 г) в течение 3-4 часов. Реакционную массу далее нагревают до 65-70°С, и оксихлорид фосфора (132 г), постепенно добавляют в течение приблизительно 5-6 часов. Массу выдерживают при перемешивании в течение 5 часов при температуре 65-70°С, охлаждают до 25-30°С с последующим внесением в измельченный лед и воду, содержащую гексафторфосфорную кислоту (232 г) и гидроксид натрия, при поддержании рН около 2,1-2,5. Погашенную массу перемешивают в течение 30 минут, фильтруют для выделения твердого продукта и промывают холодной водой (выход влажного продукта 260-300 г).
Соль хлорида винамидиния (III), полученную, как описано выше, очищают с помощью обработки водным раствором изопропилового спирта при температуре 75-80°С, с последующим охлаждением до 20-23°С. Суспензию массы продукта фильтруют, промывают изопропиловым спиртом и высушивают при температуре 55-65°С до содержания влаги менее 0,5% (выход 165 г), т.пл. 124-127°С.
Пример 11
Очистка 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III)
Соединение (III), полученное как описано выше в примере 10, очищают кристаллизацией из метанола. К материалу (10 г) добавляют метанол (150 мл) и нагревают до температуры кипения с обратным холодильником. Массу обрабатывают активированным углем (0,5 г) в течение 30 минут, фильтруют в горячем состоянии и затем концентрируют с помощью перегонки 60-70 мл метанола. Концентрированный материал далее постепенно охлаждают до комнатной температуры и затем до 2-5°С. Очищенный продукт фильтруют и промывают холодным метанолом (5 мл). Его высушивают при 50-60°С до неизменного веса (выход 8,2 г, чистота по ВЭЖХ 98,80%).
Пример 12
Очистка 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III)
Соединение (III), полученное как описано выше в примере 10, очищают кристаллизацией из метанола. К материалу (10 г) добавляют метанол (125 мл) и нагревают до температуры кипения. Массу обрабатывают активированным углем (0,5 г) в течение 30 минут, фильтруют в горячем состоянии и затем концентрируют с помощью перегонки 60-70 мл метанола. Концентрированный материал далее постепенно охлаждают до комнатной температуры и затем до 2-5°С. Очищенный продукт фильтруют и промывают холодным метанолом (5 мл). Его высушивают при 50-60°С до неизменного веса (выход 8,5 г, чистота по ВЭЖХ 98,95%).
Пример 13
Очистка 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III)
Соединение (III), полученное как описано выше в примере 10, очищают кристаллизацией из метанола. К материалу (10 г) добавляют метанол (200 мл) и нагревают до температуры кипения. Массу обрабатывают активированным углем (0,5 г) в течение 30 минут, фильтруют в горячем состоянии и затем концентрируют с помощью перегонки 60-70 мл метанола. Концентрированный материал далее постепенно охлаждают до комнатной температуры и затем до 2-5°С. Очищенный продукт фильтруют и промывают холодным метанолом (5 мл). Его высушивают в лотковой сушилке при 50-60°С до неизменного веса (выход 7,2 г, чистота по ВЭЖХ 99,14%).
Пример 14
Получение 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина (IV)
1-(6-Метил-3-пиридинил)-2-[4-(метилтио)фенил]этанон, соединение формулы (V) (10 г), и изопропанол (150 мл) смешивают в суспензию и выдерживают при 5-8°С в течение 20-30 минут. Порошок трет-бутоксида калия (5,01 г) постепенно добавляют к реакционной смеси при поддержании температуры реакционной массы 5-8°С в течение получаса. Реакционную массу выдерживают при той же температуре в течение 3 часов при перемешивании. Соль 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III, 13 г) добавляют к реакционной смеси при поддержании температуры реакционной массы 5-8°С. Реакция проходит в течение от 2,5 до 4,0 часов до завершения в соответствии с контролем ТСХ в отношении отсутствия исходного соединения формулы (V).
Реакционную массу, полученную описанным выше способом, добавляют в течение приблизительно 15-30 минут к смеси изопропанола (13,0 мл) и ледяной уксусной кислоты (15 мл), выдержанной при 5-10°C. Далее реакционную массу перемешивают в течение 2,5-3,0 часов. Затем к ней добавляют раствор аммиака (27 мл), и массу перемешивают в течение 10 минут. После этого добавляют безводный ацетат аммония (2,4 г), и реакционную массу медленно нагревают до температуры кипения в течение 5-6 часов. Образование целевого продукта и завершение реакции контролируют с помощью ТСХ. Реакционную массу охлаждают до комнатной температуры, и к ней заново добавляют дополнительный объем раствора аммиака (27 мл), с последующим добавлением раствора формальдегида (0,85 мл). Изопропанол удаляют из реакционной массы при 45-55°С под вакуумом. Добавляют толуол (80 мл), реакционную массу снова нагревают до 60-65°С в течение 30 минут, отстаивают, и затем проводят разделение органического и водного слоев. Водный слой повторно выделяют толуолом (2 раза по 40 мл), экстракты толуола смешивали, промывали 10%-ным раствором карбоната натрия (60 мл) и водой (60 мл). Далее их обрабатывают активированным углем (1,0 г), высушивают над безводным сульфатом натрия, фильтруют и концентрируют для удаления растворителя при температуре 55-60°С под вакуумом. К остаточной массе добавляют изопропанол (35 мл), и ее постепенно охлаждают до 0-3°С в течение 1-2 часов. Массу продукта отфильтровывают, промывают 3-5 мл холодного изопропанола и высушивают при 60-70° до получения (I) (выход 7,3 г, т.пл. 98-102°С, чистота по ВЭЖХ 97,60%).
Пример 15
Получение 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина (IV)
1-(6-Метил-3-пиридинил)-2-[4-(метилтио)фенил]этанон, соединение формулы (V) (10 г) и изопропанол (150 мл) смешивают в суспензию и выдерживают при 5-8°С в течение 20-30 минут. Порошок метоксида калия (2,91 г) постепенно добавляют к реакционной смеси при поддержании температуры реакционной массы 5-8°С в течение получаса. Реакционную массу выдерживают при той же температуре в течение 3 часов при перемешивании. Соль 2-хлор-N,N-диметиламинотриметиний гексафторфосфата (III, 12,6 г) добавляют к реакционной смеси при поддержании температуры реакционной массы 5-8°С. Реакция проходит в течение от 2,5 до 4,0 часов до завершения в соответствии с контролем ТСХ в отношении отсутствия исходного соединения формулы (V).
Реакционную массу, полученную описанным выше способом, добавляют в течение приблизительно 15-30 минут к смеси изопропанола (13,0 мл) и ледяной уксусной кислоты (14,4 мл), выдержанной при 5-10°C. Далее реакционную массу перемешивают в течение от 2,5 до 3,0 часов. Затем к ней добавляют раствор аммиака (27 мл), и массу перемешивают в течение 10 минут. После этого добавляют безводный ацетат аммония (2,4 г), и реакционную массу медленно нагревают до температуры кипения в течение 5-6 часов. Образование целевого продукта и завершение реакции контролируют с помощью ТСХ. Реакционную массу охлаждают до комнатной температуры, и к ней добавляют дополнительный объем раствора аммиака (27 мл), с последующим добавлением раствора формальдегида (0,9 мл). Изопропанол удаляют из реакционной массы при 45-55°С под вакуумом. Добавляют толуол (80 мл), реакционную массу снова нагревают до 60-65°С в течение 30 минут, отстаивают, и затем проводят разделение органического и водного слоев. Водный слой повторно выделяют толуолом (2 раза по 40 мл), экстракты толуола смешивают, промывают 10%-ным раствором карбоната натрия (60 мл) и водой (60 мл). Далее их обрабатывают активированным углем (1,0 г), высушивают над безводным сульфатом натрия, фильтруют концентрируют для удаления растворителя при температуре 55-60°С под вакуумом. К остаточной массе добавляют изопропанол (35 мл), и ее постепенно охлаждают до 0-3°С в течение 1-2 часов. Массу продукта отфильтровывают, промывают 3-5 мл холодного изопропанола и высушивают при 60-70°С до получения (I) (выход 7,0 г, т.пл. 98-102°С, чистота по ВЭЖХ 97,34%).
Пример 16
Очистка 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина (IV)
Соединение (IV), полученное описанным выше способом (18 г), растворяют в изопропаноле (54 мл) при 60-65°C при перемешивании. Горячий раствор фильтруют, и фильтрат постепенно охлаждают до комнатной температуры, а затем до 0-5°С. Суспензию продукта выдерживают в холоде в течение 2,0 часов, отфильтровывают, промывают охлажденным (5-10 мл) изопропанолом, высушивают при 45-50°С (выход 15,5 г, т.пл. 103-106°С, чистота по ВЭЖХ 98,6%).
Пример 17
Очистка 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина (IV)
Соединение (IV), полученное описанным выше способом (18 г), растворяют в смеси изопропанола (45 мл) и воды (0,90 мл) при 60-65°C при перемешивании. Горячий раствор фильтруют, и фильтрат постепенно охлаждают до комнатной температуры, а затем до 0-5°С. Суспензию продукта выдерживают в холоде в течение 2,0 часов, фильтруют, промывают охлажденным (5-10 мл) изопропанолом, высушивают при 45-50° (выход 16 г, т.пл. 103-107°С, чистота по ВЭЖХ 98,8%).
Пример 18
Очистка 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридинил)пиридина (IV)
Соединение (IV), полученное описанным выше способом (18 г), растворяют в изопропаноле (90 мл) и воде (3,60 мл) при 60-65°C при перемешивании. Горячий раствор фильтруют, и фильтрат постепенно охлаждают до комнатной температуры, а затем до 0-5°С. Суспензию продукта выдерживают в холоде в течение 2,0 часов, фильтруют, промывают охлажденным (5-10 мл) изопропанолом, высушивают при 60-70° (выход 14 г, т.пл. 102-105°С, чистота по ВЭЖХ 98,2%).
Пример 19
Получение 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридил)пиридина (I)
Соединение формулы (IV), а именно 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридил)пиридин (10 г), растворяют в дихлорметане (100 мл) и перемешивают при 25-28°C. Растворенная в воде (0,5 мл) серная кислота (0,94 г) медленно добавляют к вышеуказанному раствору при температуре 10-15°С. Затем к нему постепенно добавляют раствор вольфрамата натрия (0,17 г) в воде (1,5 мл), с последующим добавлением метил-три-н-октилхлорида аммония (0,25 г) и дихлорметана (2 мл). Реакционную массу затем окисляют путем постепенного добавления 50% раствора перекиси водорода (6,87 г) в воде (2 мл) при поддержании температуры реакционной смеси 8-10°С в течение приблизительно от 1,5 до 2,0 часов. Температуру реакционной смеси постепенно повышают до 28-30°С и поддерживают в течение нескольких часов с контролем ТСХ до завершения реакции. К реакционной массе добавляют воду (50 мл) и 10% раствор бикарбоната натрия для поддержания рН на уровне 6,8-7,0. Отделяют слой дихлорметана, содержащий продукт, и водная масса проходит выделение дихлорметаном два раза (25 мл). Слои дихлорметана смешивают, дважды промывают водой (20 мл), высушивают над безводным сульфатом натрия и обрабатывают активированным углем (0,8 г). Отфильтрованный раствор продукта концентрируют с помощью перегонки до получения остатка. Остатки растворителя удаляют с применением вакуума, и добавляют изопропанол (30 мл). Массу охлаждают до (-)2-2°C в течение 2 часов. Продукт выделяют фильтрованием, промывают охлажденным ИПС (3 мл) с последующей сушкой при температуре 60-70°С (выход 7,2 г, т.пл. 129-131°С, чистота по ВЭЖХ 97,14%, порошок светло-кремового цвета).
Пример 20
Получение 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридил)пиридина (I)
Соединение формулы (IV), а именно 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридил)пиридин (10 г), растворяют в дихлорметане (100 мл) и перемешивают при 25-28°C. Растворенная в воде (0,5 мл) серная кислота (0,94 г) медленно добавляют к вышеуказанному раствору при температуре 10-15°С. Затем к нему постепенно добавляют раствор вольфрамата натрия (0,17 г) в воде (1,5 мл) с последующим добавлением метил-три-н-октилхлорида аммония (0,25 г) и дихлорметана (2 мл). Реакционную массу затем окисляют путем постепенного добавления 50% раствора перекиси водорода (5,62 г) в воде (2 мл) при поддержании температуры реакционной смеси 10-12°С в течение приблизительно от 1,5 до 2,0 часов. Температуру реакционной смеси постепенно повышают до 28-30°С и поддерживают в течение нескольких часов с контролем ТСХ до завершения реакции. К реакционной массе добавляют воду (50 мл) и 10% раствор бикарбоната натрия для поддержания рН на уровне 6,6-6,8. Отделяют слой дихлорметана, содержащий продукт, и водную массу выделяют дихлорметаном два раза (25 мл). Слои дихлорметана смешивают, дважды промывают водой (20 мл), высушивают над безводным сульфатом натрия и обрабатывают активированным углем (0,8 г). Отфильтрованный раствор продукта концентрируют с помощью перегонки до получения остатка. Остатки растворителя удаляют с применением вакуума, и добавляют изопропанол (30 мл). Массу охлаждают до (-)2-2°C в течение 2 часов. Продукт выделяют фильтрованием, промывают охлажденным ИПС (3 мл) с последующей сушкой при температуре 60-70°С (выход 7,1 г, т.пл. 129-132°С, чистота по ВЭЖХ 97,0%, порошок светло-кремового цвета).
Пример 21
Получение 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридил)пиридина (I)
Соединение формулы (IV), а именно 5-хлор-3-(4-метилтио)фенил-2-(2-метил-5-пиридил)пиридин (10 г), растворяют в дихлорметане (100 мл) и перемешивают при 25-28°C. Растворенную в воде (0,5 мл) серную кислоту (0,94 г) медленно добавляют к вышеуказанному раствору при температуре 10-15°С. Затем к нему постепенно добавляют раствор вольфрамата натрия (0,17 г) в воде (1,5 мл) с последующим добавлением метил-три-н-октилхлорида аммония (0,25 г) и дихлорметана (2 мл). Реакционную массу затем окисляют путем постепенного добавления 50% раствора перекиси водорода (8,12 г) в воде (2 мл) при поддержании температуры реакционной смеси 12-14°С в течение приблизительно от 1,5 до 2,0 часов. Температуру реакционной смеси постепенно повышают до 28-30°С и поддерживают в течение нескольких часов с контролем ТСХ до завершения реакции. К реакционной массе добавляют воду (50 мл) и 10% раствор бикарбоната натрия для поддержания рН на уровне 6,95-7,15. Отделяют слой дихлорметана, содержащий продукт, и водную массу выделяют дихлорметаном два раза (25 мл). Слои дихлорметана смешивают, дважды промывают водой (20 мл), высушивают над безводным сульфатом натрия и обрабатывают активированным углем (0,8 г). Отфильтрованный раствор продукта концентрируют с помощью перегонки до получения остатка. Остатки растворителя удаляют с использованием вакуума, и добавляют изопропанол (30 мл). Массу охлаждают до (-)2-2°C в течение 2 часов. Продукт выделяют фильтрованием, промывают охлажденным ИПС (3 мл) с последующей сушкой при температуре 60-70°С (выход 7,5 г, т.пл. 131-134°С, чистота по ВЭЖХ 96,89%, порошок светло-кремового цвета).
Пример 22
Очистка 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 19, очищают с помощью кристаллизации из водного изопропанола. Соединение (20 г) растворяют в изопропаноле (70 мл) с содержанием воды 4% масс/масс при температуре кипения с обратным холодильником и при перемешивании. Добавляют активированный уголь (1,9 г), и раствор продукта фильтруют после 15-20 минут нагревания с обратным холодильником. Фильтрат медленно охлаждают до комнатной температуры и затем до 12-15°С в течение приблизительно 1 часа. Кристаллический продукт собирают путем фильтрации с последующей промывкой охлажденным изопропанолом (5-10 мл). Массу высушивают при температуре 60-70°С до получения светло-кремового продукта (выход 14,4 г, т.пл. 133-137°С, чистота по ВЭЖХ 99,10%).
Пример 23
Очистка 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 19, очищают с помощью кристаллизации из водного изопропанола. Соединение (20 г) растворяют в изопропаноле (90 мл) с содержанием воды 4% масс/масс при температуре кипения и при перемешивании. Добавляют активированный уголь (1,9 г), и раствор продукта фильтруют после 15-20 минут нагревания с обратным холодильником. Фильтрат медленно охлаждают до комнатной температуры и затем до 12-15°С в течение приблизительно 1 часа. Кристаллический продукт собирают путем фильтрации с последующей промывкой охлажденным изопропанолом (5-10 мл). Массу высушивают при температуре 60-70°С до получения светло-кремового продукта (выход 14,0 г, т.пл. 134-137°, чистота по ВЭЖХ 99,40%).
Пример 24
Очистка 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 19, очищают с помощью кристаллизации из водного изопропанола. Соединение (20 г) растворяют в изопропаноле (70 мл) с содержанием воды 3% масс/масс при температуре кипения и при перемешивании. Добавляют активированный уголь (1,9 г), и раствор продукта фильтруют после 15-20 минут нагревания с обратным холодильником. Фильтрат медленно охлаждают до комнатной температуры и затем до 12-15°С в течение приблизительно 1 часа. Кристаллический продукт собирают путем фильтрации с последующей промывкой охлажденным изопропанолом (5-10 мл). Массу высушивают при температуре 60-70°С до получения светло-кремового продукта (выход 15,3 г, т.пл. 135-137°С, чистота по ВЭЖХ 99,18%).
Пример 25
Очистка 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 19, очищают с помощью кристаллизации из водного изопропанола. Соединение (20 г) растворяют в смеси изопропанола (60 мл) и ацетона (15 мл) с содержанием воды 6% масс/масс при температуре кипения с обратным холодильником и при перемешивании. Добавляют активированный уголь (1,9 г), и раствор продукта фильтруют после 15-20 минут нагревания с обратным холодильником. Фильтрат медленно охлаждают до комнатной температуры и затем до 12-15°С в течение приблизительно 1 часа. Кристаллический продукт собирают путем фильтрации с последующей промывкой охлажденным изопропанолом (5-10 мл). Массу высушивают при температуре 60-70°С до получения светло-кремового продукта (выход 15 г, т.пл. 135-137°С, чистота по ВЭЖХ 99,35%).
Пример 26
Очистка 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 19 (20 г), растворяют в этилацетате (110 мл) при комнатной температуре и при перемешивании. К нему медленно, небольшими порциями добавляют пара-толуолсульфокислоту (16 г). Массу нагревают до 55-60°С до получения прозрачного раствора, а затем обрабатывают активированным углем (0,5 г) в течение 15-20 минут. Горячий раствор фильтруют и охлаждают при перемешивании до 10-12°С в течение примерно одного часа. Соль эторикоксиба и пара-толуолсульфокислоты выделяют фильтрованием, промывают охлажденным этилацетатом (10-15 мл) и высушивают при 50-60°С до неизменного веса (выход 28 г).
К соли эторикоксиб-ПТСК, полученной как описанию выше (28 г), добавляют воду (250 мл) и перемешивают при комнатной температуре в течение 15-20 минут. Постепенно добавляют 10% раствор бикарбоната натрия до значения рН приблизительно 7,90-8,10, а затем толуол (50 мл). Реакционную смесь нагревают до 50-55°С, перемешивают в течение 10 минут и отстаивают. Слой с толуолом, содержащий продукт, разделяют, и водную слой дважды выделяют толуолом (80 мл), таким же образом объединенный слой с толуолом промывают водой (100 мл), обрабатывают активированным углем (2,5 г), высушивают над безводным сульфатом натрия, фильтруют и концентрируют при 60-65°С при пониженном давлении с получением остатка. Изопропанол (94 мл), содержащий 4% воды, добавляют к остатку и нагревают до 55-65°С для растворения. Раствор медленно охлаждают до комнатной температуры и затем до 5-10°С в течение 1,5 часов. Массу продукта отфильтровывают, промывают холодным изопропанолом (10 мл) и высушивают при 60-65°C до неизменного веса (выход 11 г, т.пл. 135-137°С, чистота по ВЭЖХ 99,23%).
Пример 27
Получение 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 19 (20 г) растворяют в изопропиловом спирте (70 мл) и воде (4,20 мл) при температуре кипения с обратным холодильником и при перемешивании. Добавляют активированный уголь (1,9 г), и раствор продукта фильтруют через 15-20 минут. Фильтрат медленно охлаждают до комнатной температуры и затем до 12-15°С в течение приблизительно 1 часа. Кристаллический продукт собирают путем фильтрации с последующей промывкой охлажденным изопропанолом (5-10 мл). Массу высушивают при температуре 60-70°С до получения светло-кремового продукта (выход 15,3 г, т.пл. 135-137°С, чистота по ВЭЖХ 99,10%).
Пример 28
Получение 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина, Эторикоксиба (I)
Эторикоксиб (I), полученный в примере 21 (20 г) растворяют в чистом этаноле (80 мл) при температуре кипения с обратным холодильником и при перемешивании. Добавляют активированный уголь (1,9 г), и раствор продукта фильтруют через 15-20 минут. Фильтрат медленно охлаждают до комнатной температуры и затем до 12-15°С в течение приблизительно 1 часа. Кристаллический продукт собирают путем фильтрации с последующей промывкой охлажденным изопропанолом (5-10 мл). Массу высушивают при температуре 60-70°С до получения светло-кремового продукта (выход 15,1 г, т.пл. 136-138°С, чистота по ВЭЖХ 99,28%).
Пример 29
Получение 1-(6-метил-3-пиридинил)-2-[4-(метилсульфонил)фенил]этанон (II)
4-Метилтиобензилцианид (20,0 г) растворяют в (100 мл) толуоле, и реакционную массу нагревают с обратным холодильником. Метил-6-метилникотинат (21,74 г) медленно добавляют в течение 45 минут. В течение 30 минут добавляют порошок метоксида натрия 12,0 г. Температуру реакционной смеси поддерживают на уровне кипения в течение приблизительно 4-6 часов, до практически полного исчезновения пятна, соответствующего метил-6-метилникотинату в ТСХ. Реакционную массу охлаждают до 25-30°С при перемешивании с последующим медленным добавлением в смесь измельченного льда (190 г) и воды (45 мл). Значение рН реакционной массы доводят до 5,2-6,2 с помощью разбавленной соляной кислоты, реакционную массу перемешивают в течение часа, фильтруют, промывают водой и высушивают с помощью отсоса на воронке Бюхнера. Полученную влажную массу продукта 1-(6-метил-3-пиридинил)-2-циано-2-[(4-метилтио)фенил]этанона (VI) добавляют к смеси концентрированной соляной кислоты (140 мл) и ледяной уксусной кислоты (60 мл) при температуре 40-50°С, а затем декарбоксилируют при температуре кипения с обратным холодильником. Реакционную массу охлаждают до комнатной температуры после завершения реакции по результатам ТСХ, и промывают путем выделения гексаном. Реакционную массу, полученную выше, медленно выливают в смесь (70 мл) концентрированного раствора аммиака и (25,00 мл) воды. Реакционную массу перемешивают в течение 10 минут, и значение рН доводят до 6,80-7,20 разбавленным раствором аммиака при 0-5°С. Массу продукта выдерживают в течение 30 минут и фильтруют. Полученный продукт промывают водой и высушивают центрифугированием.
Массу (влажного) продукта 1-(6-метил-3-пиридинил)-2-[4-(метилтио)фенил]этанона (V) растворяют в дихлорметане (185 мл), перемешивают и отстаивают до разделения водного слоя. Раствор продукта в дихлорметане направляют непосредственно в следующую стадию синтеза для окисления смесью концентрата серной кислотой (4,10 г) и воды (4,50 мл) и перемешивают в течение 15-20 минут. Также добавляют раствор вольфрамата натрия (0,74 г) в воде (14,0 мл), и реакционную массу перемешивают в течение 10 минут с последующим добавлением смеси метил-три-н-октилхлорида (0,80 г) в дихлорметане (8,0 мл). Реакционную смесь перемешивают в течение 10 минут и охлаждают до 18-20°С. Смесь 50% перекиси водорода (19,50 г) и воды (12,50 мл) постепенно добавляют в течение 45 минут при температуре 18-20°C. Реакционную массу выдерживают в течение нескольких часов с контролем ТСХ до практически полного исчезновения пятна, соответствующего 1-(6-метил-3-пиридинил)-2-[(4-метилтио)фенил]этанону (V) в ТСХ. Значение рН реакционной массы доводят до 6,95-7,10 с использованием смеси разбавленного раствора аммиака при перемешивании в течение 30 минут при 25-30°С. Смесь перемешивают в течение 10 минут, водный слой разделяют и выделяют дихлорметаном (130 мл). Объединенные слои дихлорметана, содержащие продукт, промывают водой (110 мл), высушивают над безводным сульфатом натрия и фильтруют. Дихлорметан получают при атмосферном давлении. В конце при помощи вакуума массу дегазируют. Затем добавляют изопропиловый спирт (96 мл), массу перемешивают и охлаждают до 25-30°С с последующим охлаждением до 0-5°С, выдерживают в течение часа, фильтруют, промывают охлажденным изопропанолом и высушивают при температуре 50-60°С (выход 21 г, т.пл. 176-180°С, чистота по ВЭЖХ 92,50%).