Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА
Вид РИД
Изобретение
Настоящее изобретение относится к непрерывному способу получения пропиленоксида. В соответствии со способом изобретения пропен взаимодействует с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 для получения пропиленоксида. Сырье для реакции, которое вводят в по меньшей мере один реактор, в котором осуществляют способ непрерывного эпоксидирования по изобретению, содержит пропен, метанол и пероксид водорода. Дополнительно данное сырье для реакции содержит конкретное количество катионов калия и дополнительно фосфор в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты.
На конверсию и селективность реакций эпоксидирования можно влиять, например, через температуру реакции эпоксидирования, pH реакционной смеси эпоксидирования и/или добавление одного или нескольких соединений к реакционной смеси, отличных от таких реагентов, как пропен и пероксид водорода.
Clerici et al. (J. Catal. 140 (1993) pp.71-83) описывают эффект от добавления некоторых соединений при эпоксидировании пропена и олефинов с помощью пероксида водорода в метаноле в качестве растворителя и в присутствии силикалита титана-1 в качестве катализатора. В этой научной статье описаны исследования эффектов разнообразных основных, нейтральных или кислотных добавок. Однако никакой явной и недвусмысленной связи между химической природой добавки и наблюдаемым эффектом на конверсию и селективность не было обнаружено. Утверждалось, что малые количества гидроксидов щелочных металлов и ацетатов щелочных металлов обладают положительным влиянием на селективность, несмотря на то, что добавление ацетата лития полностью ингибирует окисление 1-бутена.
EP 0230949 A2 описывает способ получения пропиленоксида посредством эпоксидирования пропена с пероксидом водорода в присутствии силикалита титана-1 в качестве катализатора, в котором перед реакцией или во время реакции кислотность используемого катализатора подходящим образом нейтрализуют нейтрализующим агентом. Нейтрализации перед реакцией достигают посредством использования подходящих силилирующих агентов. Однако, этот способ имеет недостаток, состоящий в том, что катализатор должен быть подвергнут реакции силилирования после каждого цикла регенерации. В соответствии с EP 0230949 A2 нейтрализация во время реакции может быть достигнута посредством добавления сильноосновных или слабоосновных химических соединений. Например, гидроксид натрия или гидроксид калия указаны в качестве сильных оснований. В качестве слабых оснований указаны гидроксид аммония, карбонат натрия, гидрокарбонат натрия, гидрофосфат натрия и соответствующие калиевые и литиевые соли, а также соли щелочных металлов соли щелочноземельных металлов органических кислот с одним-десятью атомами углерода и/или алкоголят щелочного металла или алкоголяты щелочноземельных металлов с одним-десятью атомами углерода. В соответствии с этим документом эти добавки могут применяться в очень широком интервале концентраций. Например, раствор пероксида водорода, используемый для реакции эпоксидирования, может быть смешан с основанием в количестве 0,0001 и 0,1 масс.%. В соответствии с этим документом никаких особенных эффектов не было показано для конкретных добавок. Основным аспектом этого документа является нейтрализация катализатора, которая может быть достигнута с каждой подходящей основной добавкой.
EP 0712852 A1 также описывает способ эпоксидирования пропена с пероксидом водорода в присутствии силикалита титана-1 в качестве катализатора. В соответствии с этим документом добавление неосновных солей может приводить к улучшенным селективностям, где эти неосновные соли добавляют к используемому раствору пероксида водорода. В соответствии с этим документом, неосновные соли представляют собой соли, которые при концентрации, равной 0,1 моль/л в воде при 25°C имеют pH меньше чем 8, но больше чем 4. Однозначно указаны соли аммония, щелочных металлов и щелочноземельных металлов, в которых анионы представляют собой, например, галогениды, нитраты, сульфаты, формиаты, ацетаты, гидрокарбонаты или анионы кислородсодержащих кислот, которые содержат фосфор, мышьяк, сурьму или олово. Также в этом контексте добавки могут быть добавлены в очень широких интервалах концентраций, в которых предлагаются концентрации между 0,00001 и 0,02 моль/л. Дополнительно этот документ умалчивает о каких-либо особенных эффектах, которые могут быть достигнуты посредством использования конкретных солей.
EP 0757043 A1 также описывает такой способ эпоксидирования для получения пропиленоксида с использованием пероксида водорода в присутствии силикалита титана-1 в качестве катализатора. В соответствии с этим документом перед реакцией или во время реакции используют нейтральные или кислые соли. Однозначно указанные соли содержат катионы из группы, состоящей из лития, натрия, калия, аммония,  или
или 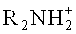 с анионами из группы, состоящей из сульфата, нитрата, хлората, хлорида или дигидрофосфата. Концентрация такой соли может находиться в интервале между 0,0001 и 1 моль/л. В противоположность документам, цитируемым выше, раскрывается, что использование основных солей может приводить к более высоким каталитическим активностям. Однако использование этих основных солей может, в то же самое время, приводить к сниженной селективности.
с анионами из группы, состоящей из сульфата, нитрата, хлората, хлорида или дигидрофосфата. Концентрация такой соли может находиться в интервале между 0,0001 и 1 моль/л. В противоположность документам, цитируемым выше, раскрывается, что использование основных солей может приводить к более высоким каталитическим активностям. Однако использование этих основных солей может, в то же самое время, приводить к сниженной селективности.
WO 99/48882 A1 также описывает способ эпоксидирования пропена с пероксидом водорода в присутствии силикалита титана-1 в качестве катализатора, в котором pH реакционной смеси поддерживают между 4,8 и 6,5. Чтобы регулировать pH в этом интервале, раскрывается, что должны быть добавлены основные соединения. В качестве особенно предпочтительного основания указан ацетат натрия. В соответствии с этим документом, не является важным то, какие конкретные основные добавки используются. Совсем наоборот, описано, что независимо от природы добавленного основания, более лучших результатов достигают, если pH реакционной смеси поддерживают в указанном конкретном интервале.
WO 2004/029032 A1 относится к непрерывному способу эпоксидирования олефинов с использованием гетерогенного катализатора для инициации реакции эпоксидирования, конкретно силикалита титана-1. Чтобы предотвратить дезактивацию катализатора, поясняется, что водная реакционная смесь должна содержать олефин, пероксид водорода, менее чем 100 массовых частей на миллион (массовых частей на миллион) щелочных металлов, щелочноземельные металлы, независимо от того, находятся ли они в ионной или комплексной форме, основания или катионы оснований, имеющих pKB менее чем 4,5, или их комбинации; и по меньшей мере 100 массовых частей на миллион оснований или катионов оснований, имеющих pKB равную по меньшей мере 4,5 или их комбинации, где массовые частей на миллион приведены в расчете от общей массы пероксида водорода в реакционной смеси. В соответствии с предпочтительным вариантом выполнения, реакционная смесь должна дополнительно содержать по меньшей мере 100 массовых частей на миллион анионов или соединений, которые могут диссоциировать с образованием анионов, в целом в расчете от массы пероксида водорода в котором такие анионы, предпочтительно присутствующие в обычных стабилизирующих количествах, предпочтительно представляют собой любой вид оксофосфорных анионов, таких как ортофосфат, гидрофосфат, гидрофосфат, пирофосфат, нитрат. Что касается этих стабилизирующих анионов или соединений, которые могут диссоциировать в растворе пероксида водорода для продуцирования этих стабилизирующих анионов, WO 2004/029032 A1 раскрывает, что они должны присутствовать в количестве не более чем 1000 массовых частей на миллион, предпочтительно 100-1000 массовых частей на миллион, более предпочтительно 200-800 массовых частей на миллион, наиболее предпочтительно 200-600 массовых частей на миллион, в расчете от массы пероксида водорода. Катионы, присутствующие в соединениях, используемых в соответствии с WO 2004/029032 A1, представляют собой натрий и литий, раскрытые в изобретательских примерах.
EP 1085017 A1 также описывает способ эпоксидирования пропена с пероксидом водорода в присутствии силикалита титана-1 в качестве катализатора. В соответствии с этим документом, является важным поддерживать pH раствора пероксида водорода, используемое в интервале от 4 до 6,5, или pH реакционной среды в интервале от 5 до 9,5. Чтобы достичь желательного значения pH, указаны разнообразные основания. Например, раскрыты гидроксиды щелочных металлов, карбонаты щелочных металлов, гидрокарбонаты щелочных металлов, фосфаты щелочных металлов, карбоксилаты щелочных металлов и аммиак. Из этого документа, специалист в данной области техники может узнать, что специфичным является не химическая природа используемых оснований, но исключительно значение pH, которое приводит к желательному эффекту.
Целью настоящего изобретения являлось обеспечить улучшенный способ эпоксидирования пропена с пероксидом водорода в присутствии силикалита титана-1 в качестве катализатора, обеспечивающий низкую селективность по отношению к побочным продуктам и побочным продуктам реакции эпоксидирования, в то же самое время, обеспечивающий очень высокие степени конверсии по отношению к материалу пероксида водорода.
Неожиданно, было обнаружено, что данная проблема может быть решена, если при непрерывном эпоксидировании пропена в метанольном растворе в присутствии силикалита титана-1 в качестве катализатора, катион конкретного щелочного металла присутствует в реакционной смеси в конкретном и узком концентрационном интервале по отношению к пероксиду водорода, где реакционная смесь дополнительно содержит фосфор в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты.
По этой причине настоящее изобретение относится к непрерывному способу получения пропиленоксида, включающему в себя взаимодействие пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 для получения пропиленоксида, в котором сырье для реакции, содержащее пропен, метанол и пероксид водорода, вводят в реактор, причем указанное сырье для реакции содержит катионы калия (K+) в количестве от 110 до 190 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции, и дополнительно содержит фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты.
Термин "анионы фосфорной кислородсодержащей кислоты", как используют в контексте настоящего изобретения, относится к аниону или смеси двух или нескольких анионов, которые могут одновременно присутствовать в равновесии, для каждой возможной фосфорной кислородсодержащей кислоты. Посредством примера могут быть указаны пероксофосфорная кислота (H3PO5), фосфорная кислота (Н3РО4), фосфоновая кислота (Н3РО3), фосфиновая кислота (H3PO2), гиподифосфоновая кислота (H4P2O4), дифосфоновая кислота (H4P2O5), гиподифосфорная кислота (H4P2O6), дифосфорная кислота (H4P2O7), пероксодифосфорная кислота (H4P2O8), трифосфорная кислота (H5P3O10). Предпочтительные анионы представляют собой анионы, производные от фосфорной кислоты (H3PO4) и дифосфорной кислоты (H4P2O7). Дополнительно, термин "фосфорная кислородсодержащая кислота" включает в себя органические фосфоновые кислоты формулы (R-P(=O)(OH)2 такие как этидроновая кислота CH3C(ОН)(P(=O)(ОН)2)2.
Сырье для реакции
В соответствии с настоящим изобретением указанные катионы калия и указанные анионы по меньшей мере одной фосфорной кислородсодержащей кислоты содержатся в сырье для реакции. Термин "сырье для реакции", как используется в этом контексте настоящего изобретения, относится к сырью, содержащему полное количество пропена, полное количество пероксида водорода и полное количество метанола, вводимых в реактор в качестве исходных материалов. Это сырье для реакции может вводиться в реактор в качестве одного единственного потока сырья или в качестве индивидуальных сырьевых потоков, таких как, например, поток сырья, содержащий метанол и пероксид водорода, и поток сырья, содержащий пропен, или поток сырья, содержащий метанол и пропен и поток сырья, содержащий пероксид водорода, или поток сырья, содержащий метанол, и поток сырья, содержащий пропен, и поток сырья, содержащий пероксид водорода. Если используется более одного потока сырья, индивидуальные сырьевые потоки либо смешивают перед тем, как их вводят в реактор или подходящим образом смешивают после того, как их вводят в реактор.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения используют по меньшей мере три индивидуальных сырьевых потока, по меньшей мере один из которых представляет собой сырье, вместе с которым метанол вводят в реактор, по меньшей мере один из которых представляет собой сырье, вместе с которым пропен вводят в реактор, и по меньшей мере один из которых представляет собой сырье, вместе с которым пероксид водорода вводят в реактор. По этой причине суммарное метанольное сырье может быть составлено из одного или нескольких индивидуальных потоков метанольного сырья, суммарное пропеновое сырье может быть составлено из одного или нескольких индивидуальных потоков пропенового сырья, и суммарный сырьевой пероксид водорода может быть составлен из одного или нескольких индивидуальных сырьевых потоков пероксида водорода. Эти потоки подходящим образом смешивают, как описано выше.
По этой причине настоящее изобретение направлено на описанный выше способ, в котором сырье для реакции получают из сырья пероксида водорода, метанольного сырья и пропенового сырья.
Что касается рассматриваемого метанольного сырья, оно может быть составлено из потока сырья свежего метанола и по меньшей мере одного метанольного потока, полученного на по меньшей мере одной стадии нисходящей переработки суммарного способа получения пропиленоксида. Такой последний метанольный поток именуется в настоящем изобретении ниже как "рециркулированный метанольный поток" или "поток рециркулированного метанольного сырья". Дополнительно, метанольное сырье может быть составлено только из свежего метанольного потока или составлено только из потока рециркулированного метанола. Предпочтительно, оно составлено из смеси свежего метанольного потока и потока рециркулированного метанола.
Что касается рассматриваемого пропенового сырья, оно может быть составлено из потока сырья свежего пропена и по меньшей мере одного пропенового потока, полученного на по меньшей мере одной стадии нисходящей переработки суммарного способа получения пропиленоксида. Такой последний пропеновый поток именуется в настоящем изобретении ниже как "рециркулированный пропеновый поток" или "поток рециркулированного пропенового сырья". Дополнительно, пропеновое сырье может быть составлено только из свежего пропенового потока. Предпочтительно, оно составлено из смеси свежего пропенового потока и рециркулированного пропенового потока.
Предпочтительно, индивидуальные потоки подходящим образом смешивались перед тем, как их вводят в реактор. Никакого особенного порядка смешивания не требуется. Предпочтительно, чтобы индивидуальные потоки, более предпочтительно свежий метанольный поток, поток рециркулированного метанола, свежий пропеновый поток, поток рециркулированного пропена и поток пероксида водорода, смешивались бы таким образом, чтобы не образовывались твердые вещества. Термин "отсутствие твердых веществ", как используется в этом конкретном контексте настоящего изобретения, относится к содержанию твердых веществ, равному в лучшем случае, предпочтительно менее чем 0,2 масс. частей на миллион в расчете от массы полного сырья для реакции. В соответствии с настоящим изобретением дополнительно является предпочтительным смешивать подходящим образом индивидуальные потоки для получения исходного сырья для реакции, которое содержит жидкую фазу. Предпочтительно, чтобы индивидуальные потоки подходящим образом смешивались бы для получения исходного сырья для реакции, которое состоит из по меньшей мере одной жидкой фазы. Даже более предпочтительно, чтобы индивидуальные потоки подходящим образом смешивались бы для получения исходного сырья для реакции, которое состоит из одной жидкой фазы. По этой причине настоящее изобретение направлено на описанный выше способ, в котором сырье для реакции, когда его вводят в реактор, состоит из одной жидкой фазы.
Пропеновое сырье
В общем случае пропен может использоваться в способе настоящего изобретения, в особенности в виде потока свежего пропена, в виде потока чистого пропена, содержащего только минорные количества примесей, таких как пропан или т.п. Предпочтительно, пропеновое сырье, как описано выше, содержит по меньшей мере 90 об.%, более предпочтительно по меньшей мере 95 об.% пропена. Таким образом, для обозначения категорий качества пропена могут использоваться термины "химически чистый пропен" или "пропен полимерной степени чистоты". Обычно, пропан является основным компонентом, содержащимся в пропене с такими категориями качества, помимо пропена. Объемное отношение пропена к пропану в потоке пропенового исходного сырья, предпочтительно используемое в соответствии с настоящим изобретением, находится в интервале от 99,99:0,01 до 95:5. По этой причине настоящее изобретение направлено на вышеописанный способ, в котором пропеновое сырье, в особенности, поток свежего пропенового исходного сырья, дополнительно содержит пропан, где объемное отношение пропена к пропану находится предпочтительно в интервале от 99,99:0,01 до 95:5. Более предпочтительно, пропеновое сырье, в особенности, поток свежего пропенового исходного сырья, дополнительно содержит пропан, где объемное отношение пропена к пропану находится в интервале от 99:1 до 95:5, более предпочтительно от 97:3 до 95:5.
Как описано выше, в соответствии с предпочтительным вариантом выполнения настоящего изобретения является возможным отделять пропен, который не участвовал во взаимодействии во время реакции эпоксидирования по настоящему изобретению, на по меньшей мере одной подходящей стадии нисходящей переработки и подходящим образом рециркулировать такой отделенный пропен в виде части пропенового исходного сырья, а именно, как поток рециркулированного пропена, направляемый на изобретательскую реакцию эпоксидирования. Такая подходящая и предпочтительная стадия нисходящей переработки и предпочтительные композиции такого потока рециркулированного пропена описаны подробно в настоящем изобретении ниже.
В соответствии с предпочтительным вариантом выполнения пропеновое сырье, используемое в способе в соответствии с настоящим изобретением, предпочтительно содержащее поток свежего пропенового исходного сырья и поток исходного сырья из рециркулированного пропена, не содержит катионы калия (K+) и не содержит фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты. Термин "не содержит катионы калия (K+)", как используется в этом контексте настоящего изобретения, относится к пропеновому сырью, содержащему катионы калия (K+) в количестве менее чем 1 масс. частей на миллион, предпочтительно менее чем 0,1 масс. частей на миллион. Термин "не содержит фосфор (Р) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты" как используется в этом контексте настоящего изобретения, относится к пропеновому сырью, содержащему фосфор (Р) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты в количестве менее чем 1 масс. частей на миллион, предпочтительно менее чем 0,1 масс. частей на миллион, в расчете от общей массы потока пропенового исходного сырья.
Что касается рассматриваемого объемного отношения потока свежего пропена по отношению к потоку рециркулированного пропена, то не существует никаких конкретных ограничений. Подходящие объемные отношения находятся в интервале от 0,5 до 20, предпочтительно от 1 до 10, более предпочтительно от 2 до 5.
Метанольное сырье
Обычно свежий метанольный поток может использоваться в виде потока чистого метанола, содержащего только минорные количества примесей. Предпочтительно, чтобы метанольное сырье, как описано выше, представляло бы собой технически чистый метанол, который кроме метанола, обычно содержит только следы воды в виде примеси в количествах, равных не более 0,5 масс.%, предпочтительно не более 0,2 масс.%. Поток рециркулированного метанола, как описано выше, обычно содержит по меньшей мере 95 масс.% метанола, причем основной примесью является вода. Этот поток рециркулированного метанола может также содержать небольшие количества органических примесей, таких как ацетальдегид, пропиональдегид, диметоксиметан, 1,1-диметоксиэтан, 1,1-диметоксипропан, ацетон, 4-метил-1,3-диоксолан, 2,4-диметил-1,3-диоксолан и метилформиат. Общее количество этих компонентов в потоке рециркулированного метанола составляет обычно не более 1 масс.%, предпочтительно не более 0,5 масс.%.
В соответствии с предпочтительным вариантом выполнения метанольное сырье, используемое в способе настоящего изобретения, предпочтительно состоящее из потока свежего метанольного исходного сырья и потока рециркулированного метанольного сырья, не содержит катионы калия (K+) и не содержит фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты. Термин "не содержит катионы калия (K+)" как используется в этом контексте настоящего изобретения, относится к метанольному сырью, содержащему катионы калия (K+) в количестве менее чем 1 частей на миллион, предпочтительно менее чем 0,1 масс. частей на миллион. Термин "не содержит фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты", как используется в этом контексте настоящего изобретения, относится к метанольному сырью, содержащему фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты в количестве менее чем 1 масс. частей на миллион, предпочтительно менее чем 0,1 масс. частей на миллион, в расчете от общей массы потока метанольного исходного сырья.
Что касается рассматриваемого объемного отношения потока рециркулированного метанола по отношению к свежему метанольному потоку, то не существует никаких конкретных ограничений. Подходящие объемные отношения находятся в интервале от 10 до 2000, предпочтительно от 50 до 1000, более предпочтительно от 100 до 500.
Сырье пероксид водорода
Что касается рассматриваемого исходного сырья пероксида водорода, то не существует никаких конкретных ограничений при условии, что сырье для реакции, содержащее пропен, метанол и пероксид водорода, которое вводят в реактор, содержит катионы калия (K+) в количестве в соответствии с настоящим изобретением, и дополнительно содержит фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты.
Конкретно, сырье пероксид водорода может быть получено в соответствии с каждым возможным способом. Возможно получать пероксид водорода посредством превращения серной кислоты в пероксодисерную кислоту посредством анодного окисления с одновременным выделением водорода на катоде. Гидролиз пероксодисерной кислоты затем приводит через пероксомоносерную кислоту к пероксиду водорода и серной кислоте, которую таким образом получают обратно. Получение пероксида водорода из элементов также является возможным. В зависимости от конкретного способа получения сырье пероксид водорода может представлять собой, например, водное или водное/метанольное сырье пероксид водорода. В случае применения водного сырья пероксида водорода, содержание сырья по отношению к пероксиду водорода обычно находится в интервале от 3 до 85 масс.%, предпочтительно от 25 до 75 масс.%, более предпочтительно от 30 до 50 масс.%, такой как от 30 до 40 масс.% или от 35 до 45 масс.%, от 40 до 50 масс.%. В случае применения водного/метанольного сырья пероксида водорода, содержание сырья по отношению к пероксиду водорода обычно находится в интервале от 3 до 85 масс.%, предпочтительно от 4 до 25 масс.%, более предпочтительно от 5 до 15 масс.%, и массовое отношение пероксида водорода по отношению к воде составляет обычно по меньшей мере 0,4, предпочтительно в интервале от 0,4 до 17, более предпочтительно в интервале от 0,6 до 6. В соответствии с предпочтительным вариантом выполнения настоящего изобретения, используют сырье в виде водного пероксида водорода.
По этой причине настоящее изобретение относится к описанному выше способу, в котором сырье пероксид водорода представляет собой водный или метанольный или водный/метанольный, предпочтительно водный раствор пероксида водорода, содержащий пероксид водорода предпочтительно в количестве от 25 до 75 масс.%, более предпочтительно от 30 до 50 масс.%.
В соответствии с настоящим изобретением предпочтительным является использовать сырье пероксид водорода, которое получают в виде раствора неочищенного пероксида водорода посредством экстракции смеси, которая является результатом процесса, известного как антрахиноновый процесс, посредством которого практически получают все мировое производство пероксида водорода (см., например, Ullmann′s Encyclopedia of Industrial Chemistry, 5th edition, volume A 13 (1989) pages 443-466), в котором применяют раствор антрахинона, содержащий алкильную группу, предпочтительно имеющую от 2 до 10 атомов углерода, более предпочтительно по меньшей мере 5 атомов углерода, как, например, 5 атомов углерода или 6 атомов углерода, и где используемый растворитель обычно состоит из смеси двух различных растворителей. Этот раствор антрахинона обычно называют рабочим раствором. В данном процессе, пероксид водорода, образующийся в ходе антрахинонового процесса, обычно отделяют посредством экстракции из соответствующего рабочего раствора после цикла гидрирования/реокисления. Указанную экстракцию можно осуществить предпочтительно с использованием по существу чистой воды, и получают неочищенный водный раствор пероксида водорода. В то же время в целом является возможным дополнительно очистить таким образом полученный неочищенный водный раствор пероксида водорода посредством перегонки, предпочтительным является, в соответствии с настоящим изобретением, использовать такой неочищенный водный раствор пероксида водорода, который не был подвергнут очистке посредством перегонки. Дополнительно, в целом является возможным подвергнуть неочищенный водный раствор пероксида водорода дополнительной стадии экстракции, в которой применяют подходящий экстракционный реагент, предпочтительно органический растворитель. Более предпочтительно, органический растворитель, используемый для этой дополнительной стадии экстракции, является таким же растворителем, который применяется в антрахиноновом процессе. Предпочтительно, экстракцию осуществляют, используя один из растворителей в рабочем растворе и наиболее предпочтительно с использованием наиболее неполярного растворителя рабочего раствора. В случае, когда неочищенный водный раствор пероксида водорода подвергают такой дополнительной стадии экстракции, получают так называемый неочищенный промытый раствор пероксида водорода. В соответствии с предпочтительным вариантом выполнения настоящего изобретения, неочищенный промытый раствор пероксида водорода применяют в качестве сырья пероксида водорода. Получение неочищенного раствора описано, например, в Европейской патентной заявке EP 1122249 A1. Что касается термина "по существу чистая вода", то делается ссылка на параграф 10, страница 3 EP 1122249 A1, которая включена посредством ссылки.
По этой причине настоящее изобретение также относится к описанному выше способу, в котором в качестве сырья пероксида водорода используют неочищенный водный раствор пероксида водорода, полученный посредством экстрагирования реакционной смеси антрахинонового процесса с использованием воды, предпочтительно по существу чистой воды, причем указанный неочищенный водный раствор пероксида водорода содержит пероксид водорода предпочтительно в количестве от 25 до 75 масс.%, более предпочтительно от 30 до 50 масс.%.
Дополнительно, настоящее изобретение также относится к описанному выше способу, в котором в качестве сырья пероксида водорода, используют неочищенный водный раствор пероксида водорода, полученный посредством экстрагирования реакционной смеси антрахинонового процесса водой, предпочтительно по существу чистой водой, указанный неочищенный водный раствор пероксида водорода содержит пероксид водорода предпочтительно в количестве от 25 до 75 масс.%, более предпочтительно от 30 до 50 масс.%, и в котором после экстракции водой, предпочтительно по существу чистой водой, неочищенный водный раствор пероксида водорода не подвергают стадии перегонки, а подвергают дополнительной стадии экстракции.
Чтобы обеспечить достаточную стабильность пероксида водорода во время экстракции водой, предпочтительно по существу чистой водой, обычно добавляют подходящие стабилизирующие агенты к воде, предпочтительно используемой по существу чистой воде. Конкретно, следует упомянуть сильные неорганические кислоты и/или хелатирующие агенты. В соответствии с предпочтительными способами экстракции, небольшие количества нитратов и/или фосфатов и пирофосфатов, соответственно, добавляют в качестве стабилизирующих агентов, либо в виде кислот или в виде натриевых солей. Эти стабилизирующие агенты обычно добавляют в таких количествах, что неочищенный водный раствор пероксида водорода содержит от 50 до 400 масс. частей на миллион катионов натрия, от 100 до 700 масс. частей на миллион фосфора, рассчитанного как фосфат 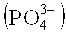 , и от 50 до 400 масс. частей на миллион нитратных анионов, в каждом случае рассчитанных по отношению к пероксиду водорода, содержащемуся в неочищенном водном растворе пероксида водорода. Предпочтительные интервалы составляют, например, от 50 до 200 масс. частей на миллион или от 50 до 100 масс. частей на миллион катионов натрия, от 100 до 500 масс. частей на миллион или от 100 до 300 масс. частей на миллион фосфора, и от 50 до 200 масс. частей на миллион или от 50 до 100 масс. частей на миллион нитрата. Как правило, молярное отношение натрия по отношению к фосфору, рассчитанному как фосфат, составляет от 0,75 до 1,25 и молярное отношение фосфора, рассчитанного как фосфат, по отношению к нитрату составляет от 0,5 до 2,5. Дополнительно, возможно использование других стабилизирующих агентов, таких как станниты, как, например, станнит натрия (Na2SnO2) и/или органических фосфоновых кислот, конкретно органических дифосфоновых кислот таких, как этидроновая кислота.
, и от 50 до 400 масс. частей на миллион нитратных анионов, в каждом случае рассчитанных по отношению к пероксиду водорода, содержащемуся в неочищенном водном растворе пероксида водорода. Предпочтительные интервалы составляют, например, от 50 до 200 масс. частей на миллион или от 50 до 100 масс. частей на миллион катионов натрия, от 100 до 500 масс. частей на миллион или от 100 до 300 масс. частей на миллион фосфора, и от 50 до 200 масс. частей на миллион или от 50 до 100 масс. частей на миллион нитрата. Как правило, молярное отношение натрия по отношению к фосфору, рассчитанному как фосфат, составляет от 0,75 до 1,25 и молярное отношение фосфора, рассчитанного как фосфат, по отношению к нитрату составляет от 0,5 до 2,5. Дополнительно, возможно использование других стабилизирующих агентов, таких как станниты, как, например, станнит натрия (Na2SnO2) и/или органических фосфоновых кислот, конкретно органических дифосфоновых кислот таких, как этидроновая кислота.
Как описано выше, было неожиданно обнаружено, что эти виды сырья для реакции показывают неожиданные и преимущественные эффекты, когда они содержат катионы калия (K+) в количестве от 110 до 190 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции, и дополнительно содержат фосфор (Р) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты. Дополнительно еще предпочтительными являются виды сырья для реакции, которые содержат K+ в количестве от 120 до 175 микромолей, предпочтительно от 130 до 160 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции.
Что касается рассматриваемого молярного отношения K+ по отношению к P в сырье для реакции, то не существует никаких конкретных ограничений. Однако, было дополнительно обнаружено, что предпочтительным является, чтобы это молярное отношение находилось в определенном интервале. Предпочтительно, в сырье для реакции, молярное отношение K+ по отношению к P находится в интервале от 0,75 до 1,0, предпочтительно от 0,8 до 0,95.
Конкретно вследствие того факта, что в соответствии с предпочтительным вариантом выполнения, неочищенный водный раствор пероксида водорода, более предпочтительно неочищенный промытый водный раствор пероксида водорода используют в качестве сырья пероксида водорода, который смешивают/смешивали с метанольным сырьем и пропеновым сырьем для получения сырья для реакции, настоящее изобретение также относится к описанному выше способу, в котором сырье для реакции дополнительно содержит ионы натрия (Na+).
Предпочтительно, чтобы в сырье для реакции, молярное отношение K+ по отношению к Na+ являлось бы большим или равным 0,1, более предпочтительно большим, чем или равным 0,2, более предпочтительно большим, чем или равным 0,5, более предпочтительно большим, чем или равным 1,0. Предпочтительно, чтобы в сырье для реакции, молярное отношение K+ по отношению к Na+ составляло бы менее чем или равно 3,0, более предпочтительно менее чем или равно 2,5, более предпочтительно больше чем или равно 2,2, более предпочтительно больше чем или равно 2,0.
В соответствии с еще более предпочтительными вариантами выполнения настоящего изобретения, в сырье для реакции молярное отношение K+ по отношению к Na+ находится в интервале от 1,0 до 3,0, предпочтительно от 1,2 до 1,75, более предпочтительно от 1,3 до 1,6.
Соответственно, особенно предпочтительно, чтобы в сырье для реакции молярное отношение K+ плюс Na+ по отношению к P находилось бы в интервале от 1,43 до 1,53, предпочтительно от 1,44 до 1,52, более предпочтительно от 1,45 до 1,51.
Как обсуждалось выше, предпочтительным вариантом выполнения настоящего изобретения является, чтобы описанный выше неочищенный водный раствор пероксида водорода, более предпочтительно неочищенный промытый водный раствор пероксида водорода использовали в качестве сырья пероксида водорода. Такие неочищенные водные растворы пероксида водорода, более предпочтительно неочищенные промытые водные растворы пероксида водорода обычно имеют содержание K+ менее чем 110 микромолей, более предпочтительно менее чем 70 микромолей, более предпочтительно менее чем 30 микромолей, конкретно менее чем 5 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырьевом пероксиде водорода.
По этой причине настоящее изобретение относится к описанному выше способу, в котором сырье пероксид водорода содержит K+ в количестве менее чем 110 микромолей, предпочтительно менее чем 70 микромолей, более предпочтительно менее чем 30 микромолей, конкретно менее чем 5 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье пероксиде водорода.
По этой причине настоящее изобретение относится к описанному выше способу, в котором сырье пероксид водорода содержит K+ в количестве менее чем 110 микромолей, предпочтительно менее чем 70 микромолей, более предпочтительно менее чем 30 микромолей, конкретно менее чем 5 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье пероксиде водорода, и дополнительно содержит от 50 до 400 масс. частей на миллион катионов натрия, от 100 до 700 масс. частей на миллион фосфора, рассчитанного как фосфат 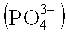 , и от 50 до 400 масс. частей на миллион нитратных анионов, в каждом случае рассчитанных по отношению к пероксиду водорода, содержащемуся в неочищенном водном растворе пероксида водорода.
, и от 50 до 400 масс. частей на миллион нитратных анионов, в каждом случае рассчитанных по отношению к пероксиду водорода, содержащемуся в неочищенном водном растворе пероксида водорода.
Принимая во внимание предпочтительный вариант выполнения настоящего изобретения, в соответствии с которым описанный выше неочищенный раствор пероксида водорода, предпочтительно описанный выше неочищенный промытый водный раствор пероксида водорода, полученный в антрахиноновом процессе с последующей экстракцией водой, предпочтительно по существу чистой водой, используется в качестве сырья пероксида водорода, было обнаружено, что конкретно с учетом достижения содержания K+ в сырье для реакции, необходимо подходящим образом увеличить содержание K+, вследствие того факта, что такие неочищенные растворы пероксида водорода, предпочтительно такие неочищенные промытые водные растворы пероксида водорода, как правило, имеют слишком низкое содержание K+, а именно содержание, которое обычно составляет менее чем 110 микромолей, более предпочтительно менее чем 70 микромолей, более предпочтительно менее чем 30 микромолей, конкретно менее чем 5 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье пероксиде водорода. Увеличение содержания K+ является возможным посредством увеличения содержания K+ в сырье пероксида водорода или посредством увеличения содержания K+ в метанольном сырье или посредством увеличения содержания K+ в пропеновом сырье или посредством увеличения содержания K+ в сырье пероксида водорода и метанольном сырье или посредством увеличения содержания K+ в метанольном сырье и пропеновом сырье или посредством увеличения содержания K+ в сырье пероксиде водорода и пропеновом сырье или посредством увеличения содержания K+ в сырьевом пероксиде водорода и метанольном сырье и пропеновом сырье. В соответствии с предпочтительным вариантом выполнения, увеличивают по меньшей мере содержание K+ в метанольном сырье. В соответствии с особенно предпочтительным вариантом выполнения, увеличивают содержание K+только в метанольном сырье. Даже более предпочтительно, увеличивают содержание K+ только в метанольном сырье посредством увеличения содержания K+ в потоке рециркулированного метанола, как описано выше.
K+-содержащий поток
Увеличение содержания K+ может осуществляться в соответствии с каждым возможным способом. В целом, предпочтительным является добавлять по меньшей мере один раствор, содержащий по меньшей мере одну, по меньшей мере частично растворимую соль калия к по меньшей мере одному из указанных видов сырья, предпочтительно к по меньшей мере метанольному сырью, конкретно только к потоку сырья рециркулированного метанола. Вне зависимости от сырья, в котором увеличивают содержание K+, предпочтительным является использовать по меньшей мере один водный раствор, содержащий K+ и дополнительно содержащий по меньшей мере один подходящий анион. В качестве подходящих анионов, анионы по меньшей мере одной фосфорной кислородсодержащей кислоты являются предпочтительными.
По этой причине настоящее изобретение относится к описанному выше способу, в котором по меньшей мере один раствор, содержащий K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты добавляют к сырью пероксида водорода или к пропеновому сырью или к метанольному сырью или смешанному сырью из двух или трех видов, в таком количестве, что сырье для реакции содержит K+ и Р в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, конкретно K+, Na+, и Р в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, в количествах, как описано в настоящем изобретении выше.
В соответствии с настоящим изобретением, особенно предпочтительным является использовать такие растворы, содержащие K+ и Р в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, в которых молярное отношение K+ по отношению к Р находится в интервале от 1,8 до 2,2, предпочтительно в интервале от 1,9 до 2,1. Наиболее предпочтительно, по меньшей мере один раствор представляет собой раствор гидрофосфата дикалия (K2HPO4), более предпочтительно водный раствор гидрофосфата дикалия или метанольный раствор гидрофосфата дикалия или водный/метанольный раствор гидрофосфата дикалия, более предпочтительно водный раствор гидрофосфата дикалия.
Что касается рассматриваемых концентраций растворов, содержащих K+, предпочтительно растворов, содержащих K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, более предпочтительно растворов, содержащих растворы гидрофосфата дикалия, более предпочтительно водных растворов гидрофосфата дикалия, то не существует никаких конкретных ограничений. Например, концентрация может быть выбрана в соответствии с масштабом процесса эпоксидирования, в котором, например, в процессе промышленного масштаба, концентрация может быть сравнительно высокой или в котором, в процессе лабораторного масштаба, концентрация может быть сравнительно низкой. Использование высоких концентраций может быть преимуществом, в случае, например, когда сравнительно низкие количества растворителя будут дополнительно вводиться в процесс эпоксидирования. Такие высокие концентрации, например, гидрофосфата дикалия находятся в интервале по меньшей мере 40 масс.%, более предпочтительно по меньшей мере 45 масс.%, более предпочтительно по меньшей мере 50 масс.%, рассчитанных от общей массы раствора. Однако, разбавленные растворы с концентрациями вплоть до 1 масс.% также являются в равной степени подходящими и особенно в случае, когда раствор добавляют к метанольному сырью, концентрации в интервале от 1 до 15 масс.% являются предпочтительными, причем интервал от 1 до 10 масс.% является более предпочтительным и интервал от 1 до 5 масс.% является даже более предпочтительным.
В то время как предпочтительным является добавлять эти растворы, содержащие K+, предпочтительно K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, к сырью пероксида водорода для увеличения его содержания K+, также возможным является добавлять подходящий раствор в ходе процесса, который приводит к сырью пероксида водорода, например в антрахиноновый процесс и/или последующую стадию экстракции. Если, например, водный неочищенный раствор пероксида водорода подвергают дополнительной стадии экстракции, из которой получают водный неочищенный промытый раствор пероксида водорода, также является возможным добавлять подходящий раствор, содержащий K+, предпочтительно K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, в ходе этой дополнительной стадии экстракции.
По этой причине настоящее изобретение относится к непрерывному способу получения пропиленоксида, включающему в себя взаимодействие пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 для получения пропиленоксида, в котором сырье для реакции, содержащее пропен, метанол и пероксид водорода вводят в реактор, причем указанное сырье для реакции содержит катионы калия (K+) в количестве от 110 до 190 микромолей по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции, и дополнительно содержит фосфор (Р) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты,
- в котором сырье для реакции получают из сырья пероксида водорода, метанольного сырья и пропенового сырья;
- в котором сырье пероксида водорода содержит K+ в количестве менее чем 110 микромолей, предпочтительно менее чем 70 микромолей, более предпочтительно менее чем 30 микромолей, конкретно менее чем 5 микромолей по отношению к 1 молю пероксида водорода, содержащегося в сырье пероксида водорода, указанное сырье пероксида водорода предпочтительно представляет собой неочищенный раствор пероксида водорода, полученный в антрахиноновом процессе с последующей экстракцией водой, предпочтительно по существу чистой водой или неочищенный промытый раствор пероксида водорода, полученный при подвергании неочищенного раствора пероксида водорода дополнительной стадии экстракции;
- в котором по меньшей мере один раствор, содержащий K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты добавляют к сырье пероксиду водорода или к пропеновому сырью или к метанольному сырью или смешанному сырью из двух или трех из них, предпочтительно к метанольному сырью, в таком количестве, чтобы сырье для реакции содержало бы K+ и Р в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты в количествах, определенных в любом из пунктов 1-7, указанный по меньшей мере один раствор, содержащий K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты предпочтительно является водным раствором гидрофосфата дикалия.
Дополнительно, настоящее изобретение также относится к следующим вариантам выполнения, включая комбинации вариантов выполнения, являющиеся результатом соответствующих обратных ссылок:
1. Способ получения пероксида водорода, включающий в себя
- получение реакционной смеси посредством антрахинонового процесса, в котором антрахиноновое соединение предпочтительно содержит алкильный остаток имеющий от 2 до 10, более предпочтительно по меньшей мере 5 атомов углерода, более предпочтительно 5 или 6 атомов углерода;
- получение неочищенного раствора пероксида водорода посредством подвергания реакционной смеси стадии экстракции с использованием воды, предпочтительно по существу чистой воды в качестве экстракционного реагента, указанный неочищенный раствор пероксида водорода имеет содержание K+ менее чем 110 микромолей, предпочтительно менее чем 70 микромолей, более предпочтительно менее чем 30 микромолей, более предпочтительно менее чем 5 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в растворе пероксида водорода;
- необязательно подвергание неочищенного раствора пероксида водорода дополнительной стадии экстракции для получения неочищенного промытого раствора пероксида водорода;
- добавление раствора, содержащего K+, предпочтительно содержащего K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, в котором молярное отношение K+ по отношению к Р находится предпочтительно в интервале от 1,8 до 2,2, более предпочтительно в интервале от 1,9 до 2,1, к неочищенному раствору пероксида водорода или к неочищенному промытому раствору пероксида водорода.
2. Способ варианта выполнения 1, в котором раствор, содержащий K+, предпочтительно содержащий K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, добавляют в таком количестве, что содержание K+ полученного в результате раствора пероксида водорода находится в интервале от 110 до 190 микромолей, предпочтительно от 120 до 175 микромолей, более предпочтительно от 130 до 160 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в растворе пероксида водорода.
3. Способ варианта выполнения 1 или 2, в котором раствор, содержащий K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты представляет собой водный раствор гидрофосфата дикалия (K2HPO4).
Что касается рассматриваемого содержания сырья для реакции по отношению к ионам других металлов, ионам щелочных металлов, отличных от натрия и калия, и/или ионам щелочноземельных металлов, предпочтительным является в контексте настоящего изобретения, чтобы общее количество (a) ионов щелочных металлов, отличных от K+ и Na+, (b) ионов щелочноземельных металлов, и (c) ионов других металлов, содержащихся в сырье для реакции, составляло бы не более чем 5 микромолей, предпочтительно не более чем 3 микромоля, более предпочтительно не менее чем 1 микромоль по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции.
В качестве ионов щелочных металлов, отличных от K+ и Na+, могут быть указаны Li+, Rb+ или Cs+ посредством примера. В качестве ионов щелочноземельных металлов могут быть указаны Mg2+, Са2+, Sr2+, Ba2+ посредством примера. В качестве ионов других металлов, могут быть посредством примера упомянуты железо, алюминий, олово, палладий, хром, никель, марганец, молибден, ванадий и кобальт.
Как обсуждалось выше, предпочтительным является добавлять обсуждаемые выше растворы, содержащие K+, предпочтительно K+ и P в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты, для получения сырья для реакции содержащего K+ в концентрации от 110 до 190 микромолей. Количество K+ может легко регулироваться специалистом в данной области техники посредством подходящим образом определенного количества калия. В контексте настоящего изобретения следует понимать, что содержание металлов определяют посредством ИСП-ОЭС (оптическая эмиссионная спектрометрия с индуктивно-связанной плазмой), оптической эмиссионной спектроскопией после возбуждения в индуктивно связанной аргоновой плазме. Следует понимать, что количество Р определяют посредством ИСП-ОЭС.
Реакция эпоксидирования
Как описано в настоящем изобретении выше, предпочтительным является смешивать подходящим образом индивидуальные потоки, а именно поток метанольного сырья, поток сырья пероксида водорода и поток пропенового сырья, для получения сырья для реакции, которое содержит жидкую фазу. Предпочтительно, чтобы индивидуальные потоки подходящим образом смешивались бы для получения сырья для реакции, которое состоит из жидких фаз. Даже более предпочтительно, чтобы индивидуальные потоки подходящим образом смешивались бы для получения сырья для реакции, которое состоит из одной жидкой фазы. По этой причине настоящее изобретение направлено на описанный выше способ, в котором сырье для реакции, когда его вводят в реактор, состоит из одной жидкой фазы.
В сырье для реакции молярное отношение пропена по отношению к пероксиду водорода обычно находится в интервале от 0,9 до 2,5, предпочтительно от 1,0 до 2,0, более предпочтительно от 1,05 до 1,5.
В соответствии с настоящим изобретением это сырье для реакции предпочтительно вводят в реактор, где реакцию эпоксидирования осуществляют в присутствии катализатора силикалита титана-1. В то время как обычно возможно применять катализатор в качестве суспензионного катализатора, предпочтительным является использовать катализатор в качестве неподвижного катализатора. Более предпочтительно, чтобы катализатор силикалита титана-1 содержал бы силикалит титана-1 в виде каталитически активного материала, погруженного в пористый матрикс, предпочтительно в мезопористый матрикс, более предпочтительно матрикс из мезопористого диоксида кремния. Катализатор, предпочтительно используемый в соответствии с настоящим изобретением, подробно описан в настоящем изобретении ниже. Что касается дополнительных условий реакции, при которых осуществляют реакцию эпоксидирования, то не существует никаких конкретных ограничений.
Предпочтительно, чтобы давление, при котором реакцию пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 осуществляют в реакторе, подходящим образом выбиралась бы так, чтобы в реакторе не присутствовало никакой газообразной фазы. Более предпочтительно, чтобы давление, при котором реакцию пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 осуществляют в реакторе, составляло бы по меньшей мере 10 бар, предпочтительно по меньшей мере 15 бар, более предпочтительно по меньшей мере 20 бар и конкретно в интервале от 20 до 40 бар. Обычно реакционную смесь в реакторе подходящим образом охлаждают. Предпочтительно, реакционную смесь в реакторе снаружи и/или изнутри охлаждают таким образом, чтобы максимальная температура реакционной смеси в реакторе находилась бы в интервале от 30 до 70°C.
Как правило, реакцию эпоксидирования в соответствии с настоящим изобретением можно осуществлять в по меньшей мере одном реакторе. Если применяют более одного реактора, два или несколько реакторов могут эксплуатироваться, последовательно и/или два или несколько реакторов могут эксплуатироваться параллельно.
Если два или несколько реакторов эксплуатируют последовательно, реакционная смесь, взятая из данного реактора, может быть подвергнута по меньшей мере одной промежуточной обработке, перед подачей в следующий реактор. При такой промежуточной обработке, физические и/или химические свойства потока могут изменяться. Например, могут изменяться температура и/или давление потока и/или химический состав потока, например, посредством реакции, перегонки и т.п.
Предпочтительно, реакцию эпоксидирования в соответствии с настоящим изобретением осуществляют посредством способа, включающего в себя
(i) взаимодействие пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 в по меньшей мере одном реакторе R1, где сырье для реакции, содержащее пропен, метанол и пероксид водорода вводят в R1, причем указанное сырье для реакции содержит катионы калия (K+) в количестве от 110 до 190 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции, и дополнительно содержит фосфор (Р) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты;
(ii) отделение потока, содержащего непрореагировавший пероксид водорода, от реакционной смеси, полученной на (i) и удаленной из R1;
(iii) смешивание потока, содержащего непрореагировавший пероксид водорода, с потоком пропена, направление смешанного потока в по меньшей мере 1 реактор R2, содержащий катализатор силикалит титана-1, и взаимодействие пропена с пероксидом водорода в R2.
В соответствии с предпочтительным вариантом выполнения, способ эпоксидирования настоящего изобретения состоит из этих трех стадий. В соответствии с даже более предпочтительным вариантом выполнения обе стадии реакции (i) и (iii) осуществляют в непрерывном режиме. Реакции эпоксидирования на стадиях (i) и (iii) предпочтительно осуществляют в режиме с неподвижным слоем.
В способе изобретения возможно использовать такие же или различные типы реакторов на стадиях (i) и (iii). Таким образом, возможным является осуществлять одну из стадий реакции в изотермическом или адиабатическом реакторе, а другую стадию реакции, независимо от других, в изотермическом или адиабатическом реакторе. Термин "реактор", как используют в этой связи, включает в себя, как описано выше, одиночный реактор, каскад из по меньшей мере двух соединенных последовательно реакторов, по меньшей мере двух реакторов, которые эксплуатируют параллельно, или множество реакторов, в котором по меньшей мере два реактора соединены последовательно и в котором по меньшей мере два реактора эксплуатируют параллельно. В соответствии с предпочтительным вариантом выполнения стадию (i) настоящего изобретения осуществляют в по меньшей мере двух реакторах R1, которые эксплуатируют параллельно, и стадию (iii) настоящего изобретения осуществляют в одиночном реакторе R2.
Каждый из реакторов, описанных выше, особенно реакторов в соответствии с предпочтительным вариантом выполнения, может эксплуатироваться в режиме эксплуатации с нисходящим потоком или восходящим потоком.
В том случае, когда реакторы эксплуатируют в режиме с нисходящим потоком, предпочтительным является использовать реакторы с неподвижным слоем, которые являются трубчатыми, мультитрубчатыми или мультитарельчатыми реакторами, предпочтительно оборудованными по меньшей мере одной охлаждающей рубашкой. В этом случае, реакцию эпоксидирования осуществляют при температуре от 30 до 80°C, и температурный профиль в реакторах поддерживают на таком уровне, что температура охлаждающей среды в охлаждающих рубашках составляет по меньшей мере 40°C, и максимальная температура в слое катализатора составляет 60°C. В случае эксплуатации реакторов с нисходящим потоком, возможно выбрать условия реакции, такие как температура, давление, скорость подачи сырья и относительные количества исходных материалов, таким образом, что реакцию осуществляют в одиночной фазе, более предпочтительно в одиночной жидкой фазе или в многофазной системе содержащей, например, 2 или 3 фазы. Что касается режима эксплуатации с нисходящим потоком, является особенно предпочтительным проводить реакцию эпоксидирования в мультифазной реакционной смеси, содержащей жидкий водный пероксид водорода в обогащенной фазе, содержащей метанол, и жидкую органическую обогащенную олефинами фазу, предпочтительно фазу, обогащенную пропеном.
В случае, когда реакторы эксплуатируют в режиме восходящего потока, предпочтительным является использовать по меньшей мере два реактора с неподвижным слоем R1 на стадии (i) и по меньшей мере один реактор R2, предпочтительно в точности один реактор R2 на стадии (iii). В соответствии с еще дополнительным вариантом выполнения по меньшей мере два реактора R1, используемые на стадии (i), соединены последовательно или их эксплуатируют параллельно, более предпочтительно эксплуатируют параллельно. Обычно, необходимо оборудовать по меньшей мере один из реакторов, используемых на стадии (i) и/или (iii) средством охлаждения, таким как охлаждающая рубашка. Особенно предпочтительно, когда по меньшей мере два реактора R1, которые используют на стадии (i), соединяются параллельно и могут эксплуатироваться попеременно. В случае, когда реакторы эксплуатируют в режиме с восходящим потоком, два или несколько реакторов R1, соединенных параллельно на стадии (i), являются особенно предпочтительно трубчатыми реакторами, мультитрубчатыми реакторами или мультитарельчатыми реакторами, более предпочтительно мультитрубчатыми реакторами и особенно предпочтительно кожухотрубными реакторами, содержащими множество трубок, такое как от 1 до 20000, предпочтительно от 10 до 10000, более предпочтительно от 100 до 8000, более предпочтительно от 1000 до 7000 и особенно предпочтительно от 3000 до 6000 трубок. Для регенерации катализатора силикалита титана-1, используемого в реакции эпоксидирования, является возможным для по меньшей мере одного из реакторов, соединенных параллельно на стадии (i), быть выведенным из эксплуатации для соответствующей стадии реакции с регенерацией катализатора, присутствующего в этом реакторе, причем по меньшей мере один реактор R1 всегда является доступным для реакции исходного материала или исходных материалов на каждой стадии во время протекания непрерывного способа.
Что касается охлаждающей среды, применяемой для охлаждения реакционной смеси в указанных выше реакторах, оборудованных охлаждающими рубашками, то в настоящем изобретении отсутствуют конкретные ограничения. Особенно предпочтительными являются масла, спирты, жидкие соли или вода, такая как речная вода, солоноватая вода и/или морская вода, которая может в каждом случае, например, предпочтительно забираться из реки и/или озера и/или моря, близкого к химическому заводу, на котором реактор изобретения и способ изобретения применяют, и после любого необходимого подходящего удаления суспендированного материала посредством фильтрации и/или осаждения, использоваться непосредственно без дополнительной обработки для охлаждения реакторов. Вторичная охлаждающая вода, которая предпочтительно переносится вокруг замкнутого контура, является особенно применимой для целей охлаждения. Эта вторичная охлаждающая вода является как правило по существу деионизированной или деминерализованной водой, к которой предпочтительно по меньшей мере добавляют одно средство против биологического обрастания. Более предпочтительно, эта вторичная охлаждающая вода циркулирует между реактором и, например, башней охлаждения. Предпочтение аналогично отдается вторичной охлаждающей воде, являющейся, например, охлаждаемой в противотоке в по меньшей мере одном противоточном теплообменнике посредством, например, речной воды, солоноватой воды и/или морской воды.
На стадии (iii) особенное предпочтение отдается использованию непрерывно эксплуатируемому реактору шахтного типа, и особенно предпочтительно непрерывно эксплуатируемому адиабатическому реактору шахтного типа.
По этой причине настоящее изобретение также относится к способу, как описано выше, в котором на стадии (i), используют по меньшей мере два кожухотрубных реактора R1, каждый из которых имеет от 1 до 20000 внутренних трубок и которые непрерывно эксплуатируют в режиме с восходящим потоком, причем указанные реакторы R1 эксплуатируют параллельно, и в котором на стадии (iii) используют адиабатический реактор шахтного типа R2, который непрерывно эксплуатируют в режиме с восходящим потоком. Еще более предпочтительно, реакцию в по меньшей мере одном из этих реакторов, более предпочтительно в по меньшей мере двух реакторах R1 стадии (i) и еще более предпочтительно во всех реакторах R1 и R2, используемых на стадиях (i) и (iii) проводят таким образом, что в соответствующем реакторе отсутствует газообразная фаза и предпочтительно присутствует одна одиночная жидкая фаза.
Применяемое давление в реакторах R1 и R2 обычно составляет по меньшей мере 10 бар, предпочтительно по меньшей мере 15 бар, более предпочтительно по меньшей мере 20 бар и конкретно в интервале от 20 до 40 бар. Применяемая температура охлаждающей среды, предпочтительно охлаждающей воды для охлаждения реакционной смеси в реакторе или реакторах R1, находится в интервале предпочтительно от 20 до 70°C, более предпочтительно от 25 до 65°C и особенно предпочтительно от 30 до 60°C. Максимальное различие между температурой охлаждающей среды, перед охлаждением, и максимальной температурой в неподвижном слое катализатора составляет предпочтительно не более чем 25 K, более предпочтительно менее чем 25 K, более предпочтительно не более чем 12 K, более предпочтительно менее чем 12 K.
В соответствии с предпочтительным вариантом выполнения изобретения, в соответствии с которым реактор или реакторы R1 на стадии (i) представляют собой реакторы с неподвижным слоем, смесь продуктов, получаемая из них, по существу состоит из пропиленоксида, непрореагировавшего пропилена, метанола, воды и пероксида водорода.
В соответствии с дополнительным предпочтительным вариантом выполнения настоящего изобретения, конверсия пероксида водорода в R1 находится предпочтительно в интервале от 85 до 95%, более предпочтительно в интервале от 87 до 93%, в таком как в интервале от 87 до 90% или от 88 до 91% или от 89 до 92% или от 90 до 93%.
В соответствии с предпочтительным вариантом выполнения, смесь продуктов, полученная со стадии (i), имеет содержание метанола в интервале от 55 до 75 масс.%, особенно предпочтительно от 60 до 70 масс.%, в расчете от общей массы смеси продуктов, содержание воды в интервале от 5 до 25 масс.%, особенно предпочтительно от 10 до 20 масс.%, в расчете от общей массы смеси продуктов, содержание пропиленоксида в интервале от 5 до 20 масс.%, особенно предпочтительно от 8 до 15 масс.%, в расчете от общей массы смеси продуктов, и содержание пропилена в интервале от 1 до 10 масс.%, особенно предпочтительно от 1 до 5 масс.%, в расчете от общей массы смеси продуктов.
Температура смеси продуктов, полученной со стадии (i), находится предпочтительно в интервале от 30 до 70°C. Перед подачей в ректификационную колонну (b) температуру смеси продуктов предпочтительно регулируют в по меньшей мере одном теплообменнике до температуры в интервале от 50 до 80°C, более предпочтительно от 60 до 70°C.
В соответствии со стадией (ii) непрореагировавший пероксид водорода, содержащийся в реакционной смеси, полученной со стадии (i) и удаленной из R1, отделяют из реакционной смеси, полученной со стадии (i). Это отделение предпочтительно осуществляют посредством перегонки с использованием по меньшей мере одной, предпочтительно в точности одной ректификационной колонны K1. Реакционную смесь, полученную из по меньшей мере одного реактора, предпочтительно из по меньшей мере двух реакторов R1, применяемых на стадии (i), содержащую пропиленоксид, метанол, воду и непрореагировавший пероксид водорода, и дополнительно содержащую непрореагировавший пропен, вводят в ректификационную колонну K1.
Ректификационную колонну K1, сконфигурированную как традиционная ректификационная колонна, предпочтительно эксплуатируют при верхнем давлении от 1 до 10 бар, более предпочтительно от 1 до 5 бар, более предпочтительно от 1 до 3 бар и еще более предпочтительно от 1 до 2 бар, как 1, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9 или 2 бар. В соответствии с особенно предпочтительным вариантом выполнения ректификационная колонна K1 имеет от 5 до 60, предпочтительно от 10 до 50 и особенно предпочтительно от 15 до 40 теоретических тарелок. В верхней части ректификационной колонны К1 стадии (ii), предпочтительно получают поток, по существу состоящий из пропиленоксида, метанола и пропена, имеющий содержание воды, равное не более чем 0,5 масс.%, предпочтительно не более чем 0,4 масс.% и еще более предпочтительно не более чем 0,3 масс.%, и имеющий содержание пероксида водорода не более чем 100 частей на миллион, предпочтительно не более чем 20 частей на миллион и еще более предпочтительно не более чем 10 частей на миллион, в каждом случае в расчете от общей массы смеси, полученной в верхней части колонны. В нижней части ректификационной колонны K1 стадии (ii), предпочтительно получают поток, по существу состоящий из метанола, воды и пероксида водорода, имеющий содержание пропена, равное не более чем 50 частей на миллион, предпочтительно не более чем 10 частей на миллион и еще более предпочтительно не более чем 5 частей на миллион, и имеющий содержание пропиленоксида, равное не более чем 50 частей на миллион, предпочтительно не более чем 20 частей на миллион и еще более предпочтительно не более чем 10 частей на миллион, в каждом случае в расчете от общей массы смеси, полученной в нижней части колонны.
В соответствии с предпочтительным вариантом выполнения ректификационную колонну K1, используемую на стадии (ii), конфигурируют как колонну с разделительной стенкой, имеющую по меньшей мере один боковой отвод, предпочтительно один боковой отвод. Предпочтительно, чтобы колонна с разделительной стенкой предпочтительно имела бы от 20 до 60, более предпочтительно от 30 до 50 теоретических тарелок. Верхняя комбинированная область ввода потока и отводной части колонны с разделительной стенкой предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, от общего числа теоретических тарелок в колонне, секция обогащения части ввода потока предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, отпарная секция части ввода потока предпочтительно имеет от 15 до 70%, более предпочтительно от 20 до 60%, отпарная секция отводной части предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, секция обогащения отводной части предпочтительно имеет от 15 до 70%, более предпочтительно от 20 до 60%, и нижняя комбинированная область вводной и отводной части колонны предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, в каждом случае от общего числа теоретических тарелок в колонне. Аналогично является преимущественным для ввода, через который смесь продуктов, полученная с (a), подается в колонну, и бокового отвода, через который часть метанола, предпочтительно от 0 до 50%, более предпочтительно от 1 до 40%, еще более предпочтительно от 5 до 30% и особенно предпочтительно от 10 до 25% метанола, отбирают в качестве промежуточной кипящей фракции и, еще более предпочтительно, непосредственно подают назад на стадию (i), регулироваться на различных высотах в колонне по отношению к положению теоретических тарелок. Ввод предпочтительно расположен в положении, которое находится от 1 до 25, более предпочтительно от 5 до 15 теоретических тарелок выше или ниже бокового отвода.
Колонну с разделительной стенкой, предпочтительно применяемую в способе настоящего изобретения, предпочтительно конфигурируют либо как насадочную колонну, содержащую статистическую насадку или упорядоченную насадку или как тарельчатую колонну. Например, возможно использовать листовую металлическую или сетчатую насадку, имеющую удельную поверхность от 100 до 1000 м2/м3, предпочтительно от около 250 до 750 м2/м3, в качестве упорядоченной насадки. Такая насадка обеспечивает высокую эффективность разделения в сочетании с низким перепадом давления на теоретическую тарелку. При указанной выше конфигурации колонны, область колонны, разделенная разделительной стенкой, которая состоит из секции обогащения части ввода потока, отпарная секция отводной части, отпарная секция части ввода потока и секция обогащения отводной части, или их части обеспечена/обеспечены упорядоченной насадкой или статистической насадкой. Разделительная стенка может быть термически изолирована в этих областях. Дифференциальное давление над разделительной стенкой колонны может использоваться в качестве регулирующего параметра для мощности нагрева.
Перегонку в колонне с разделительной стенкой K1 преимущественно осуществляют при давлении в верхней части от 1 до 10 бар, предпочтительно от 1 до 5 бар, более предпочтительно от 1 до 3 бар и еще более предпочтительно от 1 до 2 бар, как 1, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9 или 2 бар. Перегонку предпочтительно осуществляют в температурном интервале от 65 до 100°C, более предпочтительно от 70 до 85°C. Температуру перегонки измеряют в нижней части колонны. В верхней части ректификационной колонны K1 стадии (ii), конфигурированной как колонна с разделительной стенкой, получают поток, по существу состоящий из пропиленоксида, метанола и пропена, имеющий содержание воды равное не более чем 500 частей на миллион, предпочтительно не более чем 400 частей на миллион, и еще более предпочтительно не более чем 300 частей на миллион, и имеющий содержание пероксида водорода не более чем 50 частей на миллион, предпочтительно не более чем 20 частей на миллион и еще более предпочтительно не более чем 10 частей на миллион, в каждом случае в расчете от общей массы смеси, полученной в верхней части колонны. Кроме того, полученный верхний поток имеет содержание пропена от 15 до 35 масс.%, предпочтительно от 20 до 30 масс.% и еще более предпочтительно от 20 до 25 масс.%, содержание пропиленоксида от 50 до 80 масс.%, предпочтительно от 55 до 75 масс.% и особенно предпочтительно от 60 до 70 масс.%, и содержание метанола от 5 до 20 масс.%, более предпочтительно от 7,5 до 17,5 масс.% и особенно предпочтительно от 10 до 15 масс.%, в каждом случае в расчете от общей массы верхнего потока. Из бокового отвода ректификационной колонны с разделительной стенкой К1, получают поток, по существу состоящий из метанола и воды, имеющий содержание метанола, равное по меньшей мере 95 масс.%, предпочтительно по меньшей мере 96 масс.% и еще более предпочтительно по меньшей мере 97 масс.%, и имеющий содержание воды, равное не более чем 5 масс.%, предпочтительно не более чем 3,5 масс.% и еще более предпочтительно не более чем 2 масс.%, в каждом случае в расчете от общей массы смеси, полученной из бокового отвода колонны. В нижней части ректификационной колонны с разделительной стенкой K1, получают поток, по существу состоящий из метанола, воды и непрореагировавшего пероксида водорода, имеющий содержание пропена, равное не более чем 50 частей на миллион, предпочтительно не более чем 10 частей на миллион и еще более предпочтительно не более чем 5 частей на миллион, и имеющий содержание пропиленоксида, равное не более чем 50 частей на миллион, предпочтительно не более чем 20 частей на миллион и еще более предпочтительно не более чем 10 частей на миллион, в каждом случае в расчете от общей массы смеси, полученной в нижней части колонны.
По меньшей мере часть потока, отбираемая со стороны колонны с разделительной стенкой K1, может быть рециркулирована в качестве растворителя на стадию (i) способа изобретения. Предпочтительно, по меньшей мере 90%, более предпочтительно по меньшей мере 95% потока, отбираемого из бокового отвода, рециркулирует на стадию (i). В соответствии с настоящим изобретением нагревание потока продукта, полученного со стадии (i), осуществляют, используя по меньшей мере частично поток кубового остатка ректификационной колонны K1 стадии (ii). Предпочтительно, от 50 до 100%, более предпочтительно от 80 до 100% и особенно предпочтительно от 90 до 100% потока кубового остатка, полученного с ректификационной колонны K1, применяемой на (ii) используют для нагревания потока продукта, полученного с (a) от температуры в интервале от 45 до 55°C до температуры в интервале от 65 до 70°C.
Поток кубового остатка, отбираемый из ректификационной колонны K1, предпочтительно ректификационной колонны с разделительной стенкой К1, по существу состоящий из метанола, воды и непрореагировавшего пероксида водорода, затем подается в реактор R2 стадии (iii). Предпочтительно, поток кубового остатка охлаждают перед введением в реактор посредством, например, одностадийного охлаждения или двухстадийного охлаждения, более предпочтительно до температуры от 20 до 40°C, еще более предпочтительно до температуры от 30 до 40°C. Еще более предпочтительно, свежий пропен дополнительно добавляют непосредственно в реактор R2 стадии (iii) или добавляют к потоку кубового остатка, полученному с K1 стадии (ii) перед введением его в реактор R2 стадии (iii).
Селективность суммарного процесса со стадиями от (i) до (iii) по отношению к пероксиду водорода находится предпочтительно в интервале от 78 до 99%, более предпочтительно в интервале от 88 до 97% и особенно предпочтительно в интервале от 90 до 96%. Общая конверсия пероксида водорода составляет предпочтительно по меньшей мере 99,5%, более предпочтительно по меньшей мере 99,6%, более предпочтительно по меньшей мере 99,7% и особенно предпочтительно по меньшей мере 99,8%.
Реакционная смесь, полученная со стадии (iii), предпочтительно имеет содержание метанола от 50 до 90 масс.%, более предпочтительно от 60 до 85 масс.% и особенно предпочтительно от 70 до 80 масс.%, в расчете от общей массы реакционной смеси. Содержание воды находится предпочтительно в интервале от 5 до 45 масс.%, более предпочтительно от 10 до 35 масс.% и особенно предпочтительно от 15 до 25 масс.%, в расчете от общей массы реакционной смеси. Содержание пропиленоксида находится предпочтительно в интервале от 1 до 5 масс.%, более предпочтительно от 1 до 4 масс.% и особенно предпочтительно от 1 до 3 масс.%, в расчете от общей массы реакционной смеси. Содержание пропилена находится предпочтительно в интервале от 0 до 5 масс.%, более предпочтительно от 0 до 3 масс.% и особенно предпочтительно от 0 до 1 масс.%, в расчете от общей массы реакционной смеси.
Как уже упоминалось выше, было обнаружено, что селективности к побочным продуктам являются крайне низкими в случае, когда сырье для реакции имеет состав в соответствии с изобретением. Конкретно, общая селективность по отношению к сумме 1-метокси-2-пропанола, 2-метокси-1-пропанола, пропиленгликоля и кислорода составляет предпочтительно не более чем 8,5%, более предпочтительно не более чем 8,0%, в расчете от пероксида водорода.
По этой причине настоящее изобретение также относится к описанному выше способу, в котором реакцию пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 осуществляют посредством способа, включающего в себя
(i) взаимодействие пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 в по меньшей мере одном реакторе R1, который предпочтительно эксплуатируют в изотермическом режиме, где сырье для реакции, содержащее пропен, метанол и пероксид водорода вводят в R1, указанное сырье для реакции содержит катионы калия (K+) в количестве от 110 до 190 микромолей по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции, и дополнительно содержит фосфор (Р) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты;
(ii) разделение потока, содержащего непрореагировавший пероксид водорода из реакционной смеси, полученной с (i) и удаленной из R1, указанное разделение предпочтительно осуществляют посредством перегонки в по меньшей мере 1, предпочтительно 1 ректификационной колонне K1, более предпочтительно 1 ректификационной колонне К1 с разделительной стенкой;
(iii) смешивание потока, содержащего непрореагировавший пероксид водорода с потоком пропена, пропускание смешанного потока в по меньшей мере 1, предпочтительно 1 реактор R2, более предпочтительно 1 реактор шахтного типа R2, содержащий катализатор силикалит титана-1 и предпочтительно эксплуатируемый в адиабатическом режиме, и взаимодействие пропена с пероксидом водорода в R2,
в котором конверсия пероксида водорода в R1 находится предпочтительно в интервале от 85 до 95%, более предпочтительно в интервале от 87 до 93%, и
в котором полная конверсия пероксида водорода после R2 составляет предпочтительно по меньшей мере 99,5%, более предпочтительно по меньшей мере 99,6%, более предпочтительно по меньшей мере 99,7% и особенно предпочтительно по меньшей мере 99,8%;
и в котором, в соответствии с еще дополнительным предпочтительным вариантом выполнения, полная селективность по отношению к сумме 1-метокси-2-пропанола, 2-метокси-1-пропанола, пропиленгликоля и кислорода составляет предпочтительно не более чем 8,5%, более предпочтительно не более чем 8,0%, в расчете на пероксид водорода.
Катализатор силикалита титана-1
В соответствии с настоящим изобретением катализатор силикалит титана-1, предпочтительно катализатор неподвижного слоя силикалит титана-1, используют в качестве катализатора для эпоксидирования пропена с пероксидом водорода в метаноле в качестве растворителя. Силикалит титана-1 представляет собой микропористый цеолит структуры типа MFI, который не содержит алюминия и в котором Si(IV) в кристаллической решетке силиката частично заменен на титан в виде Ti(IV). Термин "микропоры", как используют в контексте настоящего изобретения, относится к порам, имеющим размер пор меньше чем 2 нм, определенный в соответствии с DIN 66134.
Цеолит катализатора силикалита титана-1, используемый на стадиях (i) и (iii), может в принципе быть получен любым возможным способом. В типовом случае синтез по меньшей мере одного титанового цеолита в соответствии с настоящим изобретением осуществляют в гидротермических системах, включающих комбинацию активного источника оксида кремния и источника титана, такого как оксида титана, с по меньшей мере одним матричным соединением, способным к образованию желательного титанового цеолита в водной суспензии, например в основной суспензии. Обычно используют органические матрицы. Предпочтительно, синтез осуществляют при повышенных температурах, например температурах в интервале от до 150 до 200°C, предпочтительно от 160 до 180°C.
В принципе, любое подходящее соединение может применяться в качестве источника оксида кремния. Типичные источники оксида кремния (SiO2) включают силикаты, гидрогель кремнезема, кремниевую кислоту, коллоидный кремнезем, коллоидальная оксид кремния, тетраалкоксисиланы, гидроксиды кремния, осажденный кремнезем и глины. Могут использоваться как так называемый диоксид кремния "влажного процесса", так и так называемый диоксид кремния "сухого процесса". В этих случаях, диоксид кремния является особенно предпочтительно аморфным, в котором размер частиц диоксида кремния находится, например, в интервале от 5 до 100 нм и удельная поверхность частиц диоксида кремния находится, например, в интервале от 50 до 500 м2/г. Коллоидный диоксид кремния является, среди прочих, доступным для приобретения как Ludox®, Syton®, Nalco® или Snowtex®. Диоксид кремния "влажного процесса" является, среди прочих, доступным для приобретения как Hi-Sil®, Ultrasil®, Vulcasil®, Santocel®, Valron-Estersil®, Tokusil® или Nipsil®. Диоксид кремния "сухого процесса" является доступным для приобретения, среди прочих, как Aerosil®, Reolosil®, Cab-O-Sil®, Fransil® или ArcSilica®. В пределах объема настоящего изобретения благоприятно использовать соединение-предшественник диоксид кремния в качестве источника оксида кремния.
Например, тетраалкоксиланы, такие как, например, тетраэтоксисилан или тетрапропоксисилан, могут быть упомянуты в качестве соединения-предшественника.
В качестве матрицы может применяться любая матрица, подходящая для обеспечения желательной цеолитной структуры MFI. Конкретно, используют гидроксид тетрапропиламмония, более предпочтительно гидроксид тетра-н-пропиламмония. В предпочтительном варианте выполнения способа в соответствии с настоящим изобретением по меньшей мере один порообразующий агент удаляют на последней стадии посредством кальцинирования, как описано ниже.
Обычно, синтез силикалита титана-1 осуществляют периодически в автоклаве таким образом, что реакционную суспензию подвергают аутогенному давлению в течение ряда часов или ряда дней пока не получат цеолит силикалит титана-1. В соответствии с предпочтительным вариантом выполнения настоящего изобретения, синтез обычно протекает при повышенных температурах, где температура во время стадии гидротермической кристаллизации обычно находятся в интервале от 150 до 200°C, предпочтительно в интервале от 160 до 180°C. Обычно, реакцию осуществляют в течение времени в интервале от нескольких часов до нескольких дней, предпочтительно в течение времени в интервале от 12 ч до 48 ч, более предпочтительно от 20 ч до 30 ч. Дополнительно является возможным добавлять затравочные кристаллы для синтеза загрузок.
В соответствии с вариантом выполнения настоящего изобретения, полученный кристаллический силикалит титана-1 отделяют из реакционной суспензии, т.е. из маточного раствора, необязательно промывают и сушат.
Могут применяться все методы, известные для отделения кристаллического силикалита титана-1 из суспензии. Среди прочих, необходимо упомянуть методы фильтрации, ультрафильтрации, диафильтрации и центрифугирования.
В случае, когда полученный кристаллический силикалит титана-1 промывают, указанная стадия промывки может осуществляться с использованием любого подходящего промывочного вещества, такого как, например, вода, спирты, такие как, например, метанол, этанол или метанол и пропанол или этанол и пропанол или метанол и этанол и пропанол, или смесей воды и по меньшей мере одного спирта, таких как, например, вода и этанол или вода и метанол или вода и этанол, или вода и пропанол или вода и метанол и этанол или вода и метанол и пропанол или вода и этанол и пропанол или вода и этанол и метанол и пропанол. Воду или смесь воды и по меньшей мере одного спирта, предпочтительно воды и этанола применяют в качестве промывочного вещества.
Сушку кристаллического силикалита титана-1 проводят при температурах, обычно, в интервале от 80 до 160 C, предпочтительно от 90 до 145°C, особенно предпочтительно от 100 до 130 C.
Вместо указанных выше методов разделения, таких как, среди прочих, методы фильтрации, ультрафильтрации, диафильтрации и центифугирования, суспензия может, в соответствии с альтернативным вариантом выполнения, также подвергаться методам распыления, как, например распылительная грануляция и распылительная сушка.
Если отделение кристаллического силикалита титана-1 осуществляют посредством метода распыления, стадии отделения и сушки могут объединяться в единственную стадию. В таком случае может использоваться либо реакционная суспензия как таковая или концентрированная реакционная суспензия. Дополнительно, возможно добавлять подходящую добавку, как, например, по меньшей мере одно подходящее связующее вещество и/или по меньшей мере один порообразующий агент к суспензии - либо к реакционной суспензии как таковой, либо к концентрированной суспензии - перед распылительной сушкой или распылительной грануляцией. Подходящие связующие вещества описаны подробно ниже. В качестве порообразующего агента могут применяться все порообразующие агенты, описанные выше. В случае, когда суспензию сушат с распылением, порообразующий агент - если он добавлен - может добавляться двумя путями. Во-первых, порообразующий агент может быть добавлен к реакционной смеси перед распылительной сушкой. Однако, также возможно добавлять часть порообразующего агента к реакционной смеси перед распылительной сушкой, причем оставшуюся часть порообразующего агента добавляют к материалу, высушенному распылительной сушкой.
В случае, когда суспензию сначала концентрируют для увеличения содержания силикалита титана-1 в суспензии, концентрирование может быть достигнуто, например, посредством упаривания, как например, упаривания при пониженном давлении или посредством фильтрации в тангенциальном потоке. Аналогично, суспензия может быть концентрирована посредством разделения указанной суспензии на две фракции, где твердое вещество, содержащееся в одной из обеих фракций, отделяют методами фильтрации, диафильтрации, ультрафильтрации или центрифугирования, и суспендируют после необязательной стадии промывки и/или стадии сушки другой фракции суспензии. Полученная таким образом концентрированная суспензия может затем подвергаться распылительным методам, таким как, например, распылительная грануляция и распылительная сушка.
В соответствии с альтернативным вариантом выполнения, концентрирования достигают посредством отделения по меньшей мере одного титанового цеолита из суспензии, и ресуспендирования титанового цеолита, необязательно вместе с по меньшей мере одной подходящей добавкой, как уже описано выше, в котором титановый цеолит может быть подвергнут по меньшей мере одной стадии промывки и/или по меньшей мере одной стадии сушки перед ресуспендированием. Ресуспендированный титановый цеолит может затем использоваться для распылительных методов, предпочтительно распылительной сушки.
Распылительная сушка является прямым методом сушки взвесей, суспензий или растворов посредством подачи хорошо диспергированных взвесей жидкость-твердое вещество, суспензии или раствора, часто дополнительно содержащих связующее вещество, в распылитель и последующей быстрой сушки в потоке горячего воздуха. Распылитель может принадлежать к нескольким различным типам. Наиболее обычной является колесное распыление, при которой используют высокоскоростное вращение колеса или диска для раздробления взвеси на капельки, которые выносятся из колеса в камеру и подвергаются быстрой сушке перед столкновением со стенками камеры. Распыление может также осуществляться посредством одиночных жидкостных сопел, действие которых основано на гидростатическом давлении для принудительного продавливания взвеси через малое сопло. Также применяют мультижидкостные сопла, где применяют давление газа для принудительного продавливания взвеси через сопло. Распыленный материал, полученный с использованием методов распылительной сушки и распылительной грануляции, как, например, сушки в псевдоожиженном слое, может содержать твердые и/или полые сферы и может в значительной степени состоять из таких сфер, которые имеют, например, диаметр в интервале от 5 до 500 мкм или 5 до 300 мкм. Могут применяться сопла для одного компонента или множества компонентов. Применение вращающегося распылителя также является возможным. Возможные температуры ввода для применяемого газа-носителя находятся, например, в интервале от 200 до 600°C, предпочтительно в интервале от 300 до 500°C. Температура вывода газа носителя находится, например, в интервале от 50 до 200°C. Воздух, обедненный воздух или кислород-азотные смеси с содержанием кислорода до 10 об.%, предпочтительно до 5 об.%, более предпочтительно менее чем 5 об.%, как, например, до 2 об.%, могут быть указаны в качестве газов носителей. Распылительные методы могут осуществляться в противотоке или в параллельном потоке.
Предпочтительно, силикалит титана-1 отделяют из реакционной суспензии посредством традиционной фильтрации или центрифугирования, необязательно сушат и/или кальцинируют и ресуспендируют, предпочтительно в смеси, предпочтительно водной смеси из материала по меньшей мере одного связующего вещества и/или одного порообразующего агента. Полученную в результате суспензию затем предпочтительно подвергают распылительной сушке или распылительной грануляции. Полученный распыленный материал может быть подвергнут дополнительной стадии промывки, указанную стадию промывки осуществляют, как описано выше. Необязательно промытый распыленный материал затем сушат и кальцинируют, где сушку и кальцинирование предпочтительно осуществляют, как описано выше.
В соответствии с альтернативным вариантом выполнения, кристаллизацию силикалита титана-1 осуществляют не перед тем, как описанная выше суспензия была высушена с распылением. По этой причине сначала образуют суспензию, содержащую источник оксида кремния, предпочтительно диоксида кремния, источник оксида титана, и соединение матрицы, способное образовывать силикалит титана-1. Затем, суспензию сушат с распылением, где в последующем необязательно дополнительный порообразующий агент добавляют к высушенному распылением силикалиту титана-1.
Высушенный распылением силикалит титана-1, полученный в соответствии с указанными выше способами, может необязательно быть подвергнут по меньшей мере одному процессу промывки, если далее осуществляют по меньшей мере один процесс промывки, предпочтительно по меньшей мере одной стадии сушки и/или по меньшей мере одной стадии кальцинирования.
Силикалит титана-1, необязательно полученный посредством распылительных методов, может дополнительно подвергаться по меньшей мере одной стадии кальцинирования, которую осуществляют в соответствии с предпочтительным вариантом выполнения изобретения последовательно за стадией сушки или вместо стадии сушки. По меньшей мере одну стадию кальцинирования осуществляют при температурах, как правило, в интервале от 350-750°C, предпочтительно от 400-700°C, особенно предпочтительно от 450-650°C.
Кальцинирование силикалита титана-1 может осуществляться в атмосфере любого подходящего газа, где воздух и/или обедненный воздух является предпочтительным. Кроме того, кальцинирование предпочтительно осуществляют в муфельной печи, вращаемой конической и/или ленточной печи для кальцинирования, в которых кальцинирование обычно осуществляют в течение одного часа или более, например, в течение времени в интервале от 1 до 24 или от 4 до 12 часов. Возможным в способе в соответствии с настоящим изобретением является, например, кальцинировать силикалит титана-1 однократно, дважды или более часто в течение в каждом случае по меньшей мере одного часа, например в каждом случае от 4 ч до 12 ч, предпочтительно от 4 ч до 8 ч, где является возможным поддерживать температуры во время стадии кальцинирования постоянными или изменять температуры непрерывно или прерывисто. Если кальцинирование проводят дважды или более часто, температуры кальцинирования на индивидуальных стадиях могут быть различными или идентичными.
Таким образом, предпочтительный вариант выполнения настоящего изобретения относится к способу, как описано выше, в котором силикалит титана-1 отделенный от суспензии, например, посредством фильтрации или распылительной сушки, промывают подходящим промывочным веществом, и в последующем подвергают по меньшей мере одной стадии сушки. Сушку осуществляют при температурах, как правило, в интервале от 80 до 160°C, предпочтительно от 90 до 145°C, особенно предпочтительно от 100 до 130°C. Наиболее предпочтительно, после сушки осуществляют стадию кальцинирования. Стадию осуществляют при температурах, как правило в интервале от 350-750°C, предпочтительно от 400-700°C, особенно предпочтительно от 450-650°C.
Силикалит титана-1, полученный, как описано выше, обычно может непосредственно использоваться в качестве катализатора на стадиях (i) и (iii). Однако, особенно предпочтительным является применять неподвижный катализатор на обеих стадиях (i) и (iii), т.е. не использовать кристаллический цеолитный материал per se в качестве катализатора, но кристаллический материал, обработанный с получением литого изделия, содержащего силикалит титана-1. Таким образом, в соответствии с предпочтительным вариантом выполнения, литое изделие, содержащее силикалит титана-1, как описано выше, используют в качестве катализатора.
Обычно, в случае, когда литое изделие используют в качестве катализатора, указанный катализатор может содержать все возможные дополнительные соединения в дополнение к силикалиту титана-1 в соответствии с изобретением, например, среди прочих, по меньшей мере одно связующее вещество и/или по меньшей мере один порообразующий агент. Кроме того, катализатор может содержать по меньшей мере один склеивающий агент вместо по меньшей мере одного связующего вещества и/или по меньшей мере одного порообразующего агента или в дополнение к по меньшей мере одному связующему веществу и/или по меньшей мере одному порообразующему агенту.
В качестве связующего вещества являются подходящими все соединения, которые обеспечивают адгезию и/или сцепление между силикалитом титана-1, подлежащим формованию, которые выходят за пределы физической сорбции, которая может присутствовать без связующего вещества. Примерами таких связующих веществ являются оксиды металлов, такие как, например, SiO2, Al2O3, TiO2, ZrO2 или MgO или глины или смеси двух или нескольких из этих соединений. Глинистые минералы и природные или синтетически полученные глиноземы, такие как, например, альфа-, бета-, гамма-, дельта-, эта-, каппа-, хи- или тета-глинозем и их неорганические или металлоорганические соединения-предшественники, такие как, например, гиббсит, байерит, бемит или псевдобемит или триалкоксиалюминаты, такие как, например, триизопропилат алюминия, являются особенно предпочтительными в качестве веществ, связующих Al2O3. Дополнительные предпочтительные связующие вещества представляют собой амфифильные соединения, имеющие полярный и неполярный фрагмент и графит. Дополнительные связующие вещества представляют собой, например, глины, такие как, например, монтмориллониты, каолины, метакаолин, гекторит, бентониты, галлоиситы, дикиты, накриты или анакситы.
Эти связующие вещества могут применяться как таковые. Также в пределах объема настоящего изобретения возможно применять соединения, из которых связующее вещество образуется на по меньшей мере одной дополнительной стадии при получении литых изделий. Примеры таких предшественников связующих веществ представляют собой тетраалкоксиланы, тетраалкоксититанаты, тетраалкоксицирконаты или смесь двух или нескольких различных тетраалкоксиланов или смесь двух или нескольких различных тетраалкоксититанатов или смесь двух или нескольких различных тетраалкоксицирконатов или смесь по меньшей мере одного тетраалкоксилана и по меньшей мере одного тетраалкоксититаната или по меньшей мере одного тетраалкоксилана и по меньшей мере одного тетраалкоксицирконата или по меньшей мере одного тетраалкоксититаната и по меньшей мере одного тетраалкоксицирконата или смесь по меньшей мере одного тетраалкоксилана и по меньшей мере одного тетраалкоксититаната и по меньшей мере одного тетраалкоксицирконата.
В контексте настоящего изобретения связующие вещества, которые либо полностью, либо частично содержат SiO2, или которые представляют собой предшественник SiO2, из которого образуется SiO2 на по меньшей мере одной дополнительной стадии, являются особенно предпочтительными. В этом контексте могут применяться как коллоидный кремнезем, так и кремнезем так называемого "влажного процесса" и кремнезем так называемого "сухого процесса". Особенно предпочтительно, когда этот кремнезем представляет собой аморфный кремнезем, размер частиц которого находится, например, в интервале от 5 до 100 нм, и удельная поверхность частиц которого находится в интервале от 50 до 500 м2/г.
Коллоидный кремнезем, предпочтительно в виде щелочного и/или аммиачного раствора, более предпочтительно как аммиачный раствор, является доступным для приобретения, среди прочих, например, как Ludox®, Syton®, Nalco® или Snowtex®. Кремнезем "влажного процесса" является доступным для приобретения, среди прочих, например как Hi-Sil®, Ultrasil®, Vulcasil®, Santocel®, Valron-Estersil®, Tokusil® или Nipsil®. Кремнезем "сухого процесса" является доступным для приобретения, среди прочих, например как Aerosil®, Reolosil®, Cab-O-Sil®, Fransil® или ArcSilica®. Среди прочих аммиачный раствор коллоидного кремнезема является предпочтительным в настоящем изобретении. Соответственно, настоящее изобретение также описывает катализатор, содержащий литое изделие, как описано выше, указанное литое изделие содержит силикалит титана-1, как описано выше, и дополнительно SiO2 как материал связующего вещества, в котором связующее вещество применяют в соответствии с (I) и представляет собой связующее вещество, содержащее или образующее SiO2. Как правило, титановый цеолит может также формоваться без использования связующего вещества.
Таким образом, настоящее изобретение также относится к способу, в котором на стадиях (i) и (iii) катализатор силикалита титана-1 получают посредством формования силикалита титана-1 с получением литого изделия, содержащего силикалит титана-1 и предпочтительно по меньшей мере одно связующее вещество, конкретно, кремнеземное связующее вещество.
Если желательно, по меньшей мере один порообразующий агент может быть добавлен к смеси силикалита титана-1 и по меньшей мере одного связующего вещества или по меньшей мере предшественника связующего вещества для дополнительной обработки и для образования формованной основы катализатора для использования в качестве неподвижного катализатора. Порообразующие агенты, которые могут применяться, все являются соединениями, которые по отношению к полученному литому изделию обеспечивают удельный размер пор и/или распределение удельного размера пор и/или определенных объемов пор. Конкретно, порообразующие агенты, которые обеспечивают по отношению к полученному литому изделию, микропоры и/или микропоры, конкретно мезопоры и микропоры.
Таким образом, настоящее изобретение также относится к способу, в котором на стадиях (i) и (iii), катализатор силикалита титана-1 получают посредством формования силикалита титана-1 с получением литого изделия, содержащего силикалит титана-1 и предпочтительно по меньшей мере одно связующее вещество, конкретно кремнеземное связующее вещество, литого изделия, конкретно имеющего микропоры и мезопоры.
Что касается примеров для порообразующих агентов, которые могут применяться, делается ссылка на порообразующие агенты, уже упомянутые выше. Предпочтительно, порообразующие агенты, применяемые в способе формования изобретения, представляют собой полимеры, которые являются диспергируемыми, суспендируемыми или эмульгируемыми в воде или в водных смесях растворителей. Особенно предпочтительные полимеры представляют собой полимерные винильные соединения, такие как, например, полиалкиленоксиды, такие как полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и сложные полиэфиры, углеводы, такие как, например, целлюлоза или производные целлюлозы, такие как, например, метилцеллюлоза или сахара или природные волокна. Дополнительными подходящими порообразующими агентами являются, например, пульпа или графит.
Если желательно достичь распределения размера пор, может применяться смесь двух или нескольких порообразующих агентов. В особенно предпочтительном варианте выполнения способа в соответствии с изобретением, как описано ниже, порообразующие агенты удаляют посредством кальцинирования с получением пористой формованной основы катализатора. Предпочтительно, порообразующие агенты, которые обеспечивают мезопоры и/или микропоры, особенно предпочтительно мезопоры, добавляют к смеси по меньшей мере одного связующего вещества и силикалита титана-1 для формования силикалита титана-1. Обычно, силикалит титана-1 может также формоваться для получения формованной основы катализатора без использования порообразующего агента.
Помимо связующего вещества и необязательно порообразующего агента, также возможно добавлять дополнительные компоненты, например, по меньшей мере одно склеивающее средство к смеси, которую формуют для получения формованной основы катализатора.
Если по меньшей мере одно склеивающее средство применяют в способе изобретения, указанное склеивающее средство применяют либо в дополнение к по меньшей мере одному порообразующему агенту или вместо него. Конкретно, соединения, которые действуют также как порообразующие агенты, могут применяться в качестве склеивающего средства. Склеивающие средства, которые могут применяться, все являются соединениями, известными как подходящие для данной цели. Они представляют собой предпочтительно органические, конкретно гидрофильные полимеры, такие как, например, целлюлоза, производные целлюлозы, такие как, например, метилцеллюлоза, и крахмал, такие как, например, картофельный крахмал, обойная штукатурка, полиакрилаты, полиметакрилаты, поливиниловый спирт, поливинилпирролидон, полиизобутелен или политетрагидрофуран. Можно упомянуть применение воды, спиртов или гликолей или их смесей, таких как смеси воды и спирта, или воды и гликоля, такие как, например, вода и метанол или вода и этанол или вода и пропанол или вода и пропиленгликоль в качестве склеивающих средств. Предпочтительно, целлюлоза, производные целлюлозы, вода и смеси двух или нескольких из этих соединений, таких как вода и целлюлоза или вода и производные целлюлозы, применяют в качестве склеивающих средств. В особенно предпочтительном варианте выполнения способа в соответствии с изобретением, по меньшей мере одно склеивающее средство удаляют посредством кальцинирования, как дополнительно описано ниже, с получением литого изделия.
В соответствии с дополнительным вариантом выполнения настоящего изобретения, по меньшей мере одна кислотная добавка может быть добавлена к смеси, которую формуют с получением литого изделия. Если применяют кислотную добавку, органические кислотные соединения, которые могут удаляться посредством кальцинирования, являются предпочтительными. В этом контексте можно упомянуть карбоновые кислоты, такие как, например, муравьиная кислота, щавелевая кислота и/или лимонная кислота. Также возможно применять два или несколько из этих кислотных соединений.
Порядок добавления компонентов к смеси, которую формуют с получением литого изделия, не является критическим. Если, например, используют комбинацию связующего вещества, порообразующего агента, склеивающего средства и необязательно по меньшей мере одного кислотного соединения, возможно сначала добавить по меньшей мере одно связующее вещество, затем по меньшей мере один порообразующий агент, по меньшей мере одно кислотное соединение и окончательно по меньшей мере одно склеивающее средство и заменить последовательность по отношению к по меньшей мере одному связующему веществу, по меньшей мере одному порообразующему агенту, по меньшей мере одному кислотному соединению и по меньшей мере одному склеивающему средству.
После добавления по меньшей мере одного связующего вещества и/или по меньшей мере одного склеивающего средства и/или по меньшей мере одного порообразующего агента и/или по меньшей мере одной кислотной добавки к смеси, содержащей силикалит титана-1, смесь обычно гомогенизируют в течение от 10 до 180 минут. Среди прочих, месильные машины, смешивающие бегуны или экструдеры особенно предпочтительно применяют для гомогенизации. Смесь предпочтительно замешивают. В промышленном масштабе, измельчение в смешивающем бегуне является предпочтительным для гомогенизации. Гомогенизацию, как правило, осуществляют при температурах в интервале от около 10°C до температуры кипения склеивающего средства и атмосферном давлении или слегка выше атмосферного давления. При необходимости может затем добавляться по меньшей мере одно из соединений, описанных выше. Смесь, полученную таким образом, гомогенизируют, предпочтительно замешивают, пока не образуется экструдируемый пластический материал.
Гомогенизированную смесь затем формуют с получением литого изделия. Могут использоваться все известные подходящие методы формования, такие как экструзия, распылительная сушка, распылительная грануляция, брикетирование, т.е. механическое прессование с добавлением дополнительного связующего вещества или таблетирование или без него, т.е. компактирование посредством круговых и/или вращательных движений.
Предпочтительными методами формования являются методы, в которых используются общепринятые экструдеры для формования смеси, содержащей силикалит титана-1. Таким образом, например, получают экструдаты, имеющие диаметр от 1 до 10 мм и предпочтительно от 2 до 5 мм. В дополнение к экструдеру, может также использоваться экструзионный пресс для получения литых изделий. Форма литых изделий, полученных в соответствии с изобретением, может быть выбрана по желанию. Конкретно, среди прочих, возможными являются сферы, овальные формы, цилиндры или таблетки.
Аналогично, можно упомянуть полые структуры, как, например полые цилиндры или структуры, образованные в виде сот или также звездообразных геометрических фигур.
Формование может происходить при давлении окружающей среды или при давлении выше давления окружающей среды, например в интервале давления от 1 бар нескольких сотен бар. Кроме того, компактирование может происходить при температуре окружающей среды или при температуре выше температуры окружающей среды, например, в интервале температур от 20 до 300°C. Если сушка и/или кальцинирование являются частью стадии формования, возможными являются температуры вплоть до 600°C.Окончательно, компактирование может происходить в окружающей атмосфере или в регулируемой атмосфере. Регулируемыми атмосферами являются, например, атмосферы инертного газа, восстанавливающие атмосферы и/или окисляющие атмосферы.
Стадия формования предпочтительно сопровождается по меньшей мере одной стадией сушки. Эту по меньшей мере одну стадию сушки осуществляют при температурах в интервале как правило от 80 до 160°C, предпочтительно от 90 до 145°C и особенно предпочтительно от 100 до 130°C, обычно в течение 6 ч или более, например в интервале от 6 до 24 ч. Однако, в зависимости от содержания влаги в материале, подлежащем сушке, более короткие значения для времени сушки, такие как, например, около 1, 2, 3, 4 или 5 ч также являются возможными.
Прежде и/или после стадии сушки, предпочтительно полученный экструдат может, например, тонко измельчаться. Предпочтительно, посредством этого получают гранулы или кусочки, имеющие диаметр частиц от 0,1 до 5 мм, конкретно от 0,5 до 2 мм.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения, сушка литых изделий, соответственно, предпочтительно сопровождается по меньшей мере одной стадией кальцинирования. Кальцинирование осуществляют при температурах обычно в интервале от 350-750 С, предпочтительно от 400-700 С, особенно предпочтительно от 450-650°C.
Кальцинирование может осуществляться в атмосфере любого подходящего газа, где воздух и/или обедненный воздух являются предпочтительными. Кроме того, кальцинирование предпочтительно осуществляют в муфельной печи, вращающейся печи и/или ленточной кальцинирующей печи, где продолжительность кальцинирования составляет в целом 1 ч или более, например, в интервале от 1 до 24 ч или в интервале от 3 до 12 ч. В способе в соответствии с настоящим изобретением, соответственно является возможным, например, кальцинировать формованную основу катализатора однократно, дважды или более часто в течение в каждом случае по меньшей мере 1 ч, как, например, в каждом случае в интервале от 3 до 12 ч, в котором является возможным для температур во время стадии кальцинирования оставаться постоянными или изменяться непрерывно или прерывисто. Если кальцинирование осуществляют дважды или более часто, температуры кальцинирования на индивидуальных стадиях могут быть различными или идентичными.
В соответствии с особенно предпочтительным вариантом выполнения, формованную основу катализатора подвергают гидротермической обработке. Гидротермическая обработка может осуществляться с использованием любого подходящего метода, известного специалистам в данной области техники. Таким образом, катализатор или формованный катализатор обычно контактирует с водой или водяным паром. Типично, указанную гидротермическую обработку осуществляют посредством зарядки катализатора или в соответствии с изобретением вместе с водой в автоклав, нагревания взвеси до температуры в интервале от 100 до 200°C, предпочтительно в интервале от 120 до 150°C при давлении в интервале от 1,5 до 5 бар, предпочтительно в интервале от 2 до 3 бар, в течение периода в интервале от 1 до 48 часов, предпочтительно в интервале от 24 до 48 часов. Типично следует по меньшей мере одна стадия промывки, предпочтительно с водой в качестве промывочного вещества. После обработки водой катализатор предпочтительно сушат и/или кальцинируют, где сушку и кальцинирование осуществляют, как уже описано выше. В соответствии с предпочтительным вариантом выполнения, гидротермическую обработку осуществляют посредством перемешивания формованной основы катализатора в автоклаве, в котором скорость перемешивания регулируют до скорости перемешивания таким образом, чтобы избежать возможного истирания. Если катализатор применяют в форме цилиндрических экструдатов, однако, некоторое истирание является желательным для достижения цилиндрических экструдатов, имеющих закругленные края. С такими экструдатами, имеющими закругленные края, может достигаться более высокая насыпная плотность, например для использования экструдатов в виде неподвижного катализатора в трубчатом реакторе R1 и/или в реакторе шахтного типа R2. Кроме того, снижается образование пыли указанных катализаторов в способе эпоксидирования на стадиях (i) и (iii).
Дополнительно, в способе эпоксидирования настоящего изобретения используют катализатор силикалит титана-1, как описано выше, имеющий микропоры и мезопоры, содержащий от 49,5 до 80%, предпочтительно от 69,5 до 80% масс. силикалита титана-1, в расчете от общей массы катализатора, и от 19,5 до 50%, предпочтительно от 19,5 до 30% масс. по меньшей мере одного связующего вещества, предпочтительно кремнеземного связующего вещества, в расчете от общей массы формованной основы катализатора.
Стадии нисходящей переработки
Смесь продуктов, взятая из реактора R2 стадии (iii), может подаваться на дополнительные стадии нисходящей переработки, пропиленоксид высокой степени чистоты подходящим образом отделяют от указанной смеси продуктов. Дополнительно поток, взятый из верхней части ректификационной колонны K1 стадии (ii), может комбинироваться со смесью продуктов, взятой из реактора R2 стадии (iii), которую затем подают на указанные стадии нисходящей очистки. Альтернативно, возможно раздельно подавать смесь продуктов, взятую из реактора R2 стадии (iii), и верхний поток ректификационной колонны K1 стадии (ii) на указанные стадии нисходящей очистки. Предпочтительно, объединенный поток (М), состоящий из реакционной смеси, полученной с (iii) и удаленной из R2, и потока верхней фракции, полученного из K1, подают на указанные стадии нисходящей очистки. В последующем, указанный поток (М) также называют смесью (М).
Стадия (iv)
В соответствии со стадией (iv), непрореагировавший пропен предпочтительно отделяют от смеси (М) посредством перегонки с получением смеси (M-iv1), содержащей по меньшей мере 80 масс.% пропена, и смеси (M-iv2), содержащей метанол, воду и по меньшей мере 7 масс.% пропиленоксида.
Разделение в соответствии со стадией (iv) предпочтительно осуществляют в по меньшей мере одной ректификационной колонне K2, более предпочтительно в одной ректификационной колонне K2. Предпочтительно, эта колонна K2 имеет от 5 до 40, более предпочтительно от 10 до 35 и особенно предпочтительно от 15 до 30 теоретических тарелок. Ректификационную колонну K2 предпочтительно эксплуатируют при верхнем давлении от 1 до 5 бар, более предпочтительно от 1 до 4 бар, более предпочтительно от 1 до 3 бар и еще более предпочтительно от 1 до 2 бар, как 1, 1,1,1,2,1,3, 1,4,1,5, 1,6, 1,7, 1,8, 1,9 или 2 бар.
Предпочтительно, смесь (M-iv1), получаемая в верхней части ректификационной колонны K2, содержит по меньшей мере 85 масс.% пропена, еще более предпочтительно от 85 до 90 масс.% пропена. Как обсуждалось выше, в контексте настоящего изобретения возможно вводить свежий пропен в виде пропена химической степени чистоты в изобретательскую реакцию эпоксидирования. В случае, когда применяют такой химически чистый пропен, смесь (M-iv1) может дополнительно содержать до 15 масс.%, предпочтительно от 5 до 10 масс.% пропана, в расчете от общей массы смеси (M-iv1). Поток верхней фракции, полученный из ректификационной колонны K2, предпочтительно имеет содержание пропиленоксида, равное не более чем 200 об. частей на миллион, предпочтительно не более чем 150 об. частей на миллион, более предпочтительно не более чем 100 об. частей на миллион.
Как обсуждалось выше, пропен, который не прореагировал в ходе реакции эпоксидирования, предпочтительно отделяют на по меньшей мере одной стадии нисходящей переработки и рециркулируют в виде потока рециркулированного пропена. Более предпочтительно, он является смесью (M-iv1), которую рециркулируют в виде потока рециркулированного пропена, необязательно после подвергания по меньшей мере одной дополнительной стадии очистки. Такая дополнительная стадия очистки может включать, например, способ, включающий в себя
(aa) сжатие и охлаждение газообразной смеси (M-iv1);
(bb) отделение пропена от полученной в результате смеси посредством абсорбирования пропена в абсорбент;
(cc) отделение пропена от абсорбента посредством десорбции;
в котором сжатие или охлаждение или сжатие и охлаждение на (aa) осуществляют по меньшей мере дважды, сжатие и охлаждение на (aa) более предпочтительно осуществляют три раза.
Предпочтительно, данный способ включает в себя
(aa) сжатие газообразной смеси (M-iv1) при давлении от 13 до 18 бар и охлаждение сжатой смеси при температуре от 30 до 45°C и повтор сжатия и охлаждения однократно или дважды, где от 50 до 90 масс процентов пропена, от 60 до 99 масс процентов метанола и/или от 70 до 99,5 масс процентов воды, содержащейся в смеси (M-iv1), конденсируют и предпочтительно рециркулируют в реакцию эпоксидирования;
(bb) отделение пропена от сжатой и охлажденной смеси посредством абсорбции пропена при давлении от 13 до 18 бар на абсорбент, указанный абсорбент имеет температуру кипения от 200 до 300°C при стандартном давлении и является смесью углеводородов CnH2n+2, где n равно от 13 до 15, указанная смесь содержит углеводород C14H30 в количестве 30 масс. процентов или более от смеси;
(cc) отделение пропена от абсорбента посредством десорбции в разделительной колонне при давлении от 16 до 25 бар и температуре от 50 до 200°C, и
рециркуляцию пропена, полученного на (cc), в указанную реакцию эпоксидирования и рециркуляцию абсорбента в (bb).
Предпочтительно, после стадии десорбции (cc) полученную смесь предпочтительно подают на по меньшей мере одну дополнительную стадию очистки. На этой дополнительной стадии очистки смесь предпочтительно фракционируют на компоненты, пропен и пропан. Указанное фракционирование предпочтительно осуществляют в C3 сплиттере, как описано, например, в Ullmann′s Encyclopedia of Industrial Chemistry, 5е Издание, Том А22, стр.214. Разделение может осуществляться в колонне при давлении от около 15 до 25 бар. Разделение может также осуществляться с использованием термически связанных колонн, и они, например, эксплуатируются при давлении около 15 или 25 бар. Пропен отводят из верхней части C3 сплиттера, сконфигурированного как колонна, а пропан отводят из нижней части. Этот пропен, отведенный из верхней части, предпочтительно рециркулируют в виде потока рециркулированного пропена в реакцию эпоксидирования.
Указанный способ, включающий в себя от (aa) до (cc), описан подробно в WO 2005/103024 A1, которая включена в настоящем изобретении посредством ссылки. Конкретно, пример WO 2005/103024 A1, на стр.27, строка 8 до стр.28, строки 22 включен в настоящем изобретении посредством ссылки, а также варианты выполнения 1-20 и их конкретные комбинации, раскрытые на стр.24, строке 28 до страницы 26, строка 41.
В соответствии с еще одним предпочтительным вариантом выполнения, вместо стадий (bb) и (cc), осуществляют стадии (bb′) и (cc′), включающие в себя
(bb′) добавление водорода к сжатой и охлаждаемой смеси и снижение кислорода, содержащегося в смеси (GII) по меньшей мере частично посредством реакции с водородом в присутствии катализатора, содержащего медь в элементарной и/или оксидной форме на носителе, в котором медь присутствует на носителе в количестве от 30 до 80 масс.%, предпочтительно от 40 до 50 масс.%, в расчете от целого катализатора и рассчитанная как CuO;
(cc′) отделение пропена от смеси, являющейся результатом (aa′), и повторное введение отделенного пропена в реакцию эпоксидирования.
Указанный способ, включающий в себя стадии (aa), (bb′) и (cc′), подробно описан в WO 2010/130610 A1, которая включена в настоящее изобретение посредством ссылки, касающейся общего и конкретного раскрытия стадий (bb′) и (cc′), в WO 2010/130610 A1, называемых как стадии (III) и (IV). Конкретно, примеры из WO 2010/130610 A1 на стр.48, II. 19-22 (получение медного катализатора), на стр.48, строка 24 до стр.49, строка 24 (1ый пример гидрогенизации), и на стр.49, строка 26 до стр.50, строка 16 (2ой пример гидрогенизации) включены в настоящее изобретение посредством ссылки.
Предпочтительно, смесь (M-iv2), полученная как поток кубового остатка, содержит от 55 до 80 масс.%, более предпочтительно от 60 до 75 масс.% и особенно предпочтительно от 65 до 70 масс.% метанола, от 13 до 25 масс.%, более предпочтительно от 15 до 20 масс.% воды, и по меньшей мере 7 масс.%, более предпочтительно по меньшей мере 8 масс.%, более предпочтительно по меньшей мере 9 масс.% и особенно предпочтительно по меньшей мере 10 масс.%, например от 10 до 15 масс.%, как около 10, около 11, около 12, около 13, около 14 или около 15 масс.% пропиленоксида. Поток кубового остатка, полученный с ректификационной колонны К2, предпочтительно имеет содержание пропена, равное не более чем 200 масс. частей на миллион, предпочтительно не более чем 150 масс. частей на миллион, более предпочтительно не более чем 100 масс. частей на миллион.
В соответствии с предпочтительным вариантом выполнения, ректификационная колонна K2 имеет наружное орошение, где по меньшей мере один подходящий растворитель добавляют к колонне. Наиболее предпочтительно, метанол применяют в качестве дополнительного орошения. Еще более предпочтительно, по меньшей мере часть смеси (M-v2), как подробно описано в следующем разделе "Стадия (v)", используют в качестве дополнительного орошения.
Стадия (v)
Предпочтительно, в соответствии со стадией (v), смесь (M-iv2), полученную со стадии (iv) как поток кубового остатка, подвергают дополнительному процессу дистилляционного разделения, в котором получают смесь (M-v1), содержащую по меньшей мере 98 масс.% пропиленоксида, и смесь (M-v2), содержащую воду, по меньшей мере 55 масс.% метанола и не более чем 100 масс. частей на миллион пропиленоксида.
Разделение в соответствии с стадией (v) предпочтительно осуществляют в по меньшей мере одной ректификационной колонне K3, более предпочтительно в одной ректификационной колонне K3. Предпочтительно, эта колонна K3 имеет от 30 до 110, более предпочтительно от 40 до 100 и особенно предпочтительно от 50 до 90 теоретических тарелок.
Ректификационную колонну K3 предпочтительно эксплуатируют при верхнем давлении от 1 бар или менее. Особенно предпочтительно, ректификационную колонну K3 эксплуатируют в виде вакуумной колонны при верхнем давлении менее чем 1 бар, более предпочтительно не более чем 0,9 бар, более предпочтительно при не более чем 0,8 бар, более предпочтительно при не более чем 0,7 бар, и еще более предпочтительно при не более чем 0,6 бар. Предпочтительные интервалы верхнего давления составляют, например, от 0,3 до 0,9 бар, более предпочтительно от 0,4 бар до 0,8 бар. Предпочтительные верхние давления составляют, например, около 0,4 бар или около 0,5 бар или около 0,6 бар или около 0,7 бар или около 0,8 бар.
В соответствии с предпочтительным вариантом выполнения, смесь (M-v1), полученная как верхний поток из K3, содержит по меньшей мере 99 масс.%, предпочтительно по меньшей мере 99,1 масс.%, более предпочтительно по меньшей мере 99,2 масс.%, более предпочтительно по меньшей мере 99,3 масс.%, более предпочтительно по меньшей мере 99,4 масс.%, и еще более предпочтительно по меньшей мере 99,5 масс.% пропиленоксида. Предпочтительные содержания (M-v1) по отношению к пропиленоксиду составляют, например, в интервале от 99,1 до 99,9, более предпочтительно от 99,2 до 99,9, более предпочтительно от 99,3 до 99,9, более предпочтительно от 99,4 до 99,9 и еще более предпочтительно от 99,5 до 99,9 масс.%, в расчете от общей массы смеси (M-v1).
В соответствии с предпочтительным вариантом выполнения, смесь (M-v2), полученная как поток кубового остатка из K3, содержит от 55 до 85 масс.%, более предпочтительно от 65 до 80 масс.% и особенно предпочтительно от 75 до 80 масс.% метанола и от 15 до 45 масс.%, более предпочтительно от 20 до 35 масс.% и особенно предпочтительно от 20 до 25 масс.% воды, где содержание смеси (M-v2), касающееся метанола, а также воды составляет больше, чем соответствующее содержание смеси (M-iv2). Дополнительно поток кубового остатка, полученный с K3, предпочтительно имеет содержание пропиленоксида, равное не более чем 200 масс. частей на миллион, более предпочтительно не более чем 150 масс. частей на миллион, более предпочтительно не более чем 100 масс. частей на миллион.
Предпочтительно, указанный поток (M-v2) подают в качестве дополнительного орошения в ректификационную колонну К2, используемую на стадии (iv) как описано выше.
В соответствии с дополнительным вариантом выполнения настоящего изобретения, отделение пропиленоксида на стадии (v) осуществляют в по меньшей мере двух, более предпочтительно в двух ректификационных колоннах K3, а именно, К31 и К32.
По этой причине настоящее изобретение также относится к способу, как описано выше, в котором на (v) пропиленоксид отделяют в двух ректификационных колоннах К31 и К32, в котором из первой ректификационной колонны К31, получают смесь, содержащую по меньшей мере 98 масс.% пропиленоксида, указанную смесь вводят во вторую ректификационную колонну К32, из которой получают поток пропиленоксида, содержащий по меньшей мере 99,8 масс.% пропиленоксида. Еще более предпочтительно, поток пропиленоксида, полученный из второй ректификационной колонны К32, содержит по меньшей мере 99,9 масс.% пропиленоксида, еще более предпочтительно по меньшей мере 99,99 масс.% пропилена.
Предпочтительно, первая колонна К31 имеет от 30 до 110, более предпочтительно от 40 до 100 и особенно предпочтительно от 50 до 90 теоретических тарелок. Первую колонну K3 предпочтительно эксплуатируют при верхнем давлении от 1 бар или менее. Особенно предпочтительно, ректификационную колонну K3 эксплуатируют как вакуумную колонну при верхнем давлении менее, чем 1 бар, более предпочтительно при не более чем 0,9 бар, более предпочтительно при не более чем 0,8 бар, более предпочтительно при не более чем 0,7 бар, и еще более предпочтительно при не более чем 0,6 бар. Предпочтительные интервалы верхнего давления составляют, например, от 0,3 до 0,9 бар, более предпочтительно от 0,4 бар до 0,8 бар. Предпочтительно, верхние давления составляют, например, около 0,4 бар или около 0,5 бар или около 0,6 бар или около 0,7 бар или около 0,8 бар.
Предпочтительно, вторая колонна К32 имеет от 25 до 60, более предпочтительно от 30 до 55 и особенно предпочтительно от 35 до 50 теоретических тарелок. Вторую колонну К32 предпочтительно эксплуатируют при верхнем давлении от 1 до 7 бар, более предпочтительно от 2 до 6 бар и особенно предпочтительно от 3 до 5 бар.
Смесь, полученная с верхней части первой колонны К31, которая подается в качестве потока сырья на вторую колонну К32, может дополнительно содержать некоторые побочные продукты, полученные в результате с одной или более стадий суммарного способа эпоксидирования. Примерами таких побочных продуктов являются альдегиды, такие как, например, ацетальдегид и/или формальдегид. Эти побочные продукты могут содержаться в верхнем потоке первой колонны К31 в количестве до 0,3 масс.%, предпочтительно до 0,20 масс.% и особенно предпочтительно до 0,15 масс.%, в расчете от общей массы (M-v2) и в расчете от суммы соответствующих масс этих низкокипящих соединений.
По этой причине настоящее изобретение относится к описанному выше способу, дополнительно включающему в себя
(iv) перегонку комбинированного потока, состоящего из реакционной смеси, полученной с (iii) и удаленной из R2, и потока верхней фракции, полученного из К1, в ректификационной колонне К2 для получения потока верхней фракции, имеющего содержание пропиленоксида, равное не более чем 100 об. частей на миллион, и потока кубового остатка, имеющего содержание пропена, равное не более чем 100 масс. частей на миллион, указанную перегонку предпочтительно осуществляют с использованием метанольного потока в качестве дополнительного орошения;
(v) перегонку потока кубового остатка, полученного с (iv) в ректификационной колонне K3 для получения смесевого потока верхней фракции, содержащего по меньшей мере 98 масс.% пропиленоксида и потока кубового остатка, имеющего содержание пропиленоксида, равное не более чем 100 масс. частей на миллион.
Дополнительно, настоящее изобретение относится к смеси, содержащей по меньшей мере 98 масс.% пропиленоксида, получаемой или полученной посредством описанного выше способа, дополнительно включающего в себя
(iv) перегонку комбинированного потока, состоящего из реакционной смеси, полученной с (iii) и удаленной из R2, и потока верхней фракции, полученного из K1, в ректификационной колонне К2 для получения потока верхней фракции, имеющего содержание пропиленоксида, равное не более чем 100 об. частей на миллион и потока кубового остатка, имеющего содержание пропена, равное не более чем 100 масс. частей на миллион, указанную перегонку предпочтительно осуществляют с использованием метанольного потока в качестве дополнительного орошения;
(v) перегонку потока кубового остатка, полученного с (iv), в ректификационной колонне K3 для получения смесевого потока верхней фракции, содержащего по меньшей мере 98 масс.% пропиленоксида и потока кубового остатка, имеющего содержание пропиленоксида, равное не более чем 100 масс. частей на миллион.
Предпочтительно, указанная смесь, содержащая по меньшей мере 98 масс.% пропиленоксида, которая является получаемой или полученной посредством вышеуказанного способа на стадии (v) в качестве смесевого верхнего потока, имеет очень низкое содержание побочных продуктов, таких как ацетальдегид, метилформиат и вода. Обычно, указанная смесь содержит не более чем 50 частей на миллион, предпочтительно не более чем 30 частей на миллион, более предпочтительно не более чем 25 частей на миллион ацетальдегида, не более чем 100 частей на миллион, предпочтительно не более чем 80 частей на миллион, более предпочтительно не более чем 75 частей на миллион метилформиата и не более чем 50 частей на миллион, предпочтительно не более чем 40 частей на миллион, более предпочтительно не более чем 30 частей на миллион воды.
По этой причине настоящее изобретение также относится к смеси, содержащей по меньшей мере 98 масс.%, предпочтительно по меньшей мере 99 масс.%, более предпочтительно по меньшей мере 99,9 масс.%, более предпочтительно по меньшей мере 99,99 масс.% пропиленоксида, получаемой или полученной посредством способа включающего в себя и необязательно состоящего из
(i) взаимодействия пропена с пероксидом водорода в метанольном растворе в присутствии катализатора силикалита титана-1 в по меньшей мере одном реакторе R1, который предпочтительно эксплуатируют в изотермическом режиме, в котором сырье для реакции, содержащее пропен, метанол и пероксид водорода, вводят в R1, указанное сырье для реакции содержит катионы калия (K+) в количестве от 110 до 190 микромолей, по отношению к 1 молю пероксида водорода, содержащегося в сырье для реакции, и дополнительно содержит фосфор (P) в форме анионов по меньшей мере одной фосфорной кислородсодержащей кислоты;
(ii) разделения потока, содержащего непрореагировавший пероксид водорода из реакционной смеси полученной с (i) и удаленной из R1, указанное разделение предпочтительно осуществляют посредством перегонки в по меньшей мере 1, предпочтительно 1 ректификационной колонне K1, более предпочтительно 1 ректификационной колонне с разделительной стенкой K1;
(iii) смешивания потока, содержащего непрореагировавший пероксид водорода, с потоком пропена, пропускание смешанного потока в по меньшей мере 1, предпочтительно 1 реактор R2, более предпочтительно 1 реактор шахтного типа R2, содержащий катализатор силикалита титана-1 и предпочтительно эксплуатируемый в адиабатическом режиме, и взаимодействие пропена с пероксидом водорода в R2;
(iv) перегонки комбинированного потока, состоящего из реакционной смеси полученной с (iii) и удаленной из R2, и потока верхней фракции, полученного из К1, в ректификационной колонне К2 для получения потока верхней фракции, имеющего содержание пропиленоксида, равное не более чем 100 об. частей на миллион, и потока кубового остатка, имеющего содержание пропена, равное не более чем 100 масс. частей на миллион, указанную перегонку предпочтительно осуществляют с использованием метанольного потока в качестве дополнительного орошения;
(v) перегонки потока кубового остатка, полученного с (iv) в ректификационной колонне K3 для получения смесевого верхнего потока, содержащего по меньшей мере 98 масс.% пропиленоксида, и потока кубового остатка, имеющего содержание пропиленоксида, равное не более чем 100 масс. частей на миллион, указанную смесь получают в качестве смеси верхнего потока (v).
Предпочтительно, эта смесь верхнего потока содержит не более чем 10 масс. частей на миллион, предпочтительно не более чем 5 масс. частей на миллион, более предпочтительно не более чем 2 масс. частей на миллион формальдегида; не более чем 10 масс. частей на миллион, предпочтительно не более чем 7 масс. частей на миллион, более предпочтительно не более чем 4 масс. частей на миллион метилформиата; не более чем 10 масс. частей на миллион, предпочтительно не более чем 8 масс. частей на миллион, более предпочтительно не более чем 6 масс. частей на миллион метанола; не более чем 75 масс. частей на миллион, предпочтительно не более чем 70 масс. частей на миллион, более предпочтительно не более чем 65 масс. частей на миллион воды.
По этой причине настоящее изобретение также относится к смеси, содержащей по меньшей мере 99,99 масс.% пропиленоксида, предпочтительно получаемой или полученной посредством описанного выше способа, включающего в себя стадии (i)-(v), указанная смесь содержит не более чем 2 масс. частей на миллион формальдегида, не более чем 4 масс. частей на миллион, метилформиата, не более чем 6 масс. частей на миллион метанола и не более чем 65 масс. частей на миллион воды.
В соответствии с дополнительным предпочтительным вариантом выполнения настоящего изобретения указанная смесь содержит менее чем 5 масс. частей на миллион, предпочтительно менее чем 4 масс. частей на миллион, более предпочтительно менее чем 3 масс. частей на миллион ацетальдегида. Конкретно, содержание ацетальдегида смесей настоящего изобретения находятся ниже предела обнаружения и, таким образом, считают, что они не содержит ацетальдегид.
По этой причине настоящее изобретение также относится к смеси, содержащей по меньшей мере 99,99 масс.% пропиленоксида, предпочтительно получаемой или полученной посредством описанного выше способа, включающего в себя стадии (i)-(v), указанная смесь содержит менее чем 3 масс. частей на миллион ацетальдегида, предпочтительно не содержит ацетальдегид и, более предпочтительно, дополнительно содержит не более чем 2 масс. частей на миллион формальдегида, не более чем 4 масс. частей на миллион метилформиата, не более чем 6 масс. частей на миллион метанола и не более чем 65 масс. частей на миллион воды.
В соответствии с дополнительным предпочтительным вариантом выполнения настоящего изобретения, указанная смесь содержит менее чем 5 масс. частей на миллион, предпочтительно менее чем 4 масс. частей на миллион, более предпочтительно менее чем 3 масс. частей на миллион пропена; менее чем 5 масс. частей на миллион, предпочтительно менее чем 4 масс. частей на миллион, более предпочтительно менее чем 3 масс. частей на миллион пропана; менее чем 20 масс. частей на миллион, предпочтительно менее чем 15 масс. частей на миллион, более предпочтительно менее чем 10 масс. частей на миллион 1,1-диметоксиэтана; менее чем 10 масс. частей на миллион, предпочтительно менее чем 7 масс. частей на миллион, более предпочтительно менее чем 5 масс. частей на миллион диметоксиметана; менее чем 20 масс. частей на миллион, предпочтительно менее чем 15 масс. частей на миллион, более предпочтительно менее чем 10 масс. частей на миллион 1,1-диметоксипропана; менее чем 20 масс. частей на миллион, предпочтительно менее чем 15 масс. частей на миллион, более предпочтительно менее чем 10 масс. частей на миллион 4-метил-1,3-диоксолана; и менее чем 10 масс. частей на миллион, предпочтительно менее чем 7 масс. частей на миллион, более предпочтительно менее чем 5 масс. частей на миллион пропионаля. Конкретно, каждое из этих соединений находится ниже предела обнаружения и, таким образом, считают, что смесь настоящего изобретения не содержит каждого из этих соединений.
В соответствии с дополнительной стадией (vi), смесь (M-v2), полученную со стадии (v) как поток кубового остатка, предпочтительно подвергают дополнительному процессу дистилляционного разделения. Необязательно, после стадии (v) и перед стадией (vi), может располагаться стадия каталитического гидрирования. На такой стадии каталитического гидрирования, смесь (M-v2), полученную со стадии (v), предпочтительно подвергают гидрированию в присутствии катализатора гидрирования, который содержит каталитически активный металл, выбранный из группы, состоящей из Pd, Pt, Rh, Ir, Os и смеси двух или нескольких этих металлов. Предпочтительно, такую реакцию гидрирования можно осуществлять при температуре от 65 до 85°C и при парциальном давлении водорода от 3 до 20 бар, предпочтительно от 3 до 13 бар. Подробности, касающиеся предпочтительных условий реакции и предпочтительных катализаторов, и также получения, активации и регенерации предпочтительных катализаторов можно найти в WO 2007/074101 A1, которая по отношению к каталитическому гидрированию и используемым для него катализаторам, включена в настоящем изобретении посредством ссылки. Если осуществляют такую стадию гидрирования, смесь, полученную с этой стадии, называемую в последующем смесью (M-v2′), подвергают стадии (vi) настоящего изобретения.
Стадия (vi)
В соответствии со стадией (vi) смесь (M-v2), полученная со стадии (v) как поток кубового остатка, или необязательно (M-v2′), полученная со стадии гидрирования, предпочтительно подвергают дополнительному способу дистилляционного разделения, в котором получают смесь (M-vi1), содержащую по меньшей мере 85 масс.% метанола и до 10 масс.% воды, и смесь (M-vi2), содержащую по меньшей мере 90 масс.% воды.
Перегонка на стадии (vi) может осуществляться в одной, двух, трех или более ректификационных колоннах K4.
В соответствии с одним вариантом выполнения, перегонку на стадии (vi) осуществляют в одной ректификационной колонне K4. Предпочтительно, эта ректификационная колонна K4 имеет от 10 до 100, более предпочтительно от 20 до 90 и особенно предпочтительно от 30 до 70 теоретических тарелок. Ректификационную колонну К4 эксплуатируют при давлении предпочтительно от 1 до 12 бар, более предпочтительно от 2 до 11 бар и особенно предпочтительно от 3 до 10 бар. Смесь (M-vi1), полученная из верхней части колонны К4, содержит по меньшей мере 85 масс.% метанола и до 10 масс.% воды, более предпочтительно по меньшей мере 90 масс.% метанола и до 10 масс.% воды, более предпочтительно по меньшей мере 95 масс.% метанола и до 5 масс.% воды, более предпочтительно по меньшей мере 96 масс.% метанола и до 4 масс.% воды, более предпочтительно по меньшей мере 97 масс.% метанола и до 3 масс.% воды, более предпочтительно по меньшей мере 98 масс.% метанола и до 2 масс.% воды, более предпочтительно по меньшей мере 98,5 масс.% метанола и до 1,5 масс.% воды, более предпочтительно по меньшей мере 98,5 масс.% метанола и до 1 масс.% воды. Флегмовое число этой колонны находится предпочтительно в интервале от 1 до 10, более предпочтительно в интервале от 2 до 8.
В соответствии с предпочтительным вариантом выполнения перегонку на стадии (vi) осуществляют в способе перегонки с двумя давлениями, где в первой ректификационной колонне К41 перегонку осуществляют при верхнем давлении, которое отличается от верхнего давления второй ректификационной колонны К42. В соответствии с еще дополнительным предпочтительным вариантом выполнения, колонны К41 и k42 являются термически связанными. В соответствии с одним вариантом выполнения холодильник, используемый для конденсации верхнего потока первой или второй ректификационной колонны, используют одновременно в качестве испарителя второй или первой ректификационной колонны. Предпочтительно, холодильник, применяемый для конденсации потока верхней фракции, полученного из второй ректификационной колонны, используют одновременно в качестве испарителя первой ректификационной колонны. В соответствии с еще дополнительным вариантом выполнения поток кубового остатка, полученный с колонны К41, который подают в качестве входного потока в колонну К42, нагревают, перед введением в К42, с потоком кубового остатка, полученного с колонны К42. В соответствии с предпочтительным вариантом выполнения, эти возможности термического сочетания объединяют.
Перегонку в первой колонне К41 предпочтительно осуществляют при верхнем давлении в интервале от 2 до 8 бар, более предпочтительно от 2 до 6 бар и особенно предпочтительно в интервале от 2,5 до 6 бар. Перегонку во второй колонне (K2) предпочтительно осуществляют при верхнем давлении в интервале от 8 до 15 бар, более предпочтительно от 8,5 до 14 бар, и особенно предпочтительно в интервале от 9 до 13 бар. Ректификационная колонна К41 имеет предпочтительно от 5 до 30, более предпочтительно от 7 до 25 и особенно предпочтительно от 10 до 20 теоретических тарелок. Предпочтительно, поток кубового остатка, полученный с К41, нагревают при температуре от 110 до 180°C, более предпочтительно от 120 до 180°C, более предпочтительно от 130 до 175°C и еще более предпочтительно от 140 до 170°C.
Флегмовое число колонны К42 находится предпочтительно в интервале от 1 до 5, более предпочтительно от 2 до 4. Флегмовое число определяют как массовый расход потока верхней фракции, полученного из колонны К42, разделенный на массовый расход фракции этого потока, подаваемого назад в верхнюю часть К42. Ректификационная колонна К42 имеет предпочтительно от 5 до 60, более предпочтительно от 10 до 55 и особенно предпочтительно от 15 до 50 теоретических тарелок.
Предпочтительно, ректификационную колонну К42 конфигурируют как колонну с разделительной стенкой, имеющую по меньшей мере один боковой отвод, предпочтительно один боковой отвод. Предпочтительно, колонна с разделительной стенкой К42 имеет от 10 до 60, более предпочтительно от 15 до 50 теоретических тарелок. Верхняя комбинированная область входной и отводной части колонны с разделительной стенкой предпочтительно имеет от 10 до 70%, более предпочтительно от 15 до 55%, секция обогащения входной части предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, отпарная секция входной части предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, отпарная секция отводной части предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, секция обогащения отводной части предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, и нижняя комбинированная область входной и отводной части колонны предпочтительно имеет от 5 до 50%, более предпочтительно от 15 до 30%, в каждом случае от общего числа теоретических тарелок в колонне. Колонну с разделительной стенкой предпочтительно конфигурируют как насадочную колонну, содержащую статистически расположенную насадку или упорядоченную насадку или как тарельчатую колонну. Например, возможно применять листовую металлическую или сетчатую насадку, имеющую удельную поверхность от 100 до 1000 м2/м3, предпочтительно от около 250 до 750 м2/м3, в качестве упорядоченной насадки. Такая насадка обеспечивает высокую эффективность разделения в сочетании с низким перепадом давления на теоретическую тарелку. При указанной выше конфигурации колонны, обеспечены область колонны, отделенная разделительной стенкой, которая состоит из секции обогащения входной части, отпарной секции отводной части, отпарной секции входной части и секции обогащения отводной части или ее части с упорядоченной насадкой или статистической насадкой. Разделительная стенка может быть термически изолирована в этих областях.
По сравнению с традиционной ректификационной колонной, колонна с разделительной стенкой, используемая на стадии (d), имеет то преимущество, что некоторые побочные продукты, являющиеся результатом одной или нескольких стадий суммарного способа эпоксидирования, могут быть легко отделены от метанола, что является преимуществом, поскольку смесь (M-vi1) наиболее предпочтительно подается обратно как поток рециркулированного метанольного сырья на стадию (i) способа эпоксидирования, как описано выше.
В случае, когда стадию (vi) осуществляют как перегонку при двух давлениях, используя традиционную ректификационную колонну К41 и колонну с разделительной стенкой К42, поток верхней фракции, полученный из верхней части колонны К41, содержит по меньшей мере 85 масс.% метанола и до 10 масс.% воды, более предпочтительно по меньшей мере 90 масс.% метанола и до 10 масс.% воды, более предпочтительно по меньшей мере 95 масс.% метанола и до 5 масс.% воды, более предпочтительно по меньшей мере 96 масс.% метанола и до 4 масс.% воды и особенно предпочтительно по меньшей мере 97 масс.% метанола и до 3 масс.% воды. В соответствии с особенно предпочтительным вариантом выполнения, верхний поток содержит менее чем 3 масс.% воды, как, например, от 1 до 2 масс.% воды. Температура верхнего потока находится предпочтительно в интервале от 90 до 130°C, более предпочтительно от 95 до 120°C и особенно предпочтительно от 100 до 110°C.
Поток кубового остатка, полученный с К41, имеет предпочтительную температуру от 100 до 140°C, более предпочтительно в интервале от 110 до 130°C.Этот поток кубового остатка предпочтительно подают в К42 и перед подачей нагревают с потоком кубового остатка К42 до температуры от 110 до 180°C, более предпочтительно от 120 до 180°C, более предпочтительно от 130 до 175°C и еще более предпочтительно от 140 до 170°C.Поток кубового остатка, полученный с К41, имеет предпочтительное содержание метанола от 40 до 70 масс.% и предпочтительное содержание воды от 30 до 60 масс.%.
Верхний поток (M-vi1), полученный с колонны К42, содержит по меньшей мере 85 масс.% метанола и до 10 масс.% воды, более предпочтительно по меньшей мере 90 масс.% метанола и до 10 масс.% воды, более предпочтительно по меньшей мере 95 масс.% метанола и до 5 масс.% воды, более предпочтительно по меньшей мере 96 масс.% метанола и до 4 масс.% воды, более предпочтительно по меньшей мере 97 масс.% метанола и до 3 масс.% воды, более предпочтительно по меньшей мере 98 масс.% метанола и до 2 масс.% воды, более предпочтительно по меньшей мере 98,5 масс.% метанола и до 1,5 масс.% воды, более предпочтительно по меньшей мере 98,5 масс.% метанола и до 1 масс.% воды.
Смесь (M-vi2), полученная с нижней части колонны К42, содержит по меньшей мере 90 масс.% воды, более предпочтительно по меньшей мере 95 масс.% воды и особенно предпочтительно по меньшей мере 97 масс.% воды. Предпочтительно, (M-dii) по существу не содержит метанол, т.е. она имеет содержание метанола, равное менее чем 0,5 масс.%, предпочтительно менее чем 5 частей на миллион, более предпочтительно менее чем 1 частей на миллион. В дополнение к воде (М-vi2) может содержать некоторые побочные продукты, являющиеся результатом одной или нескольких стадий суммарного способа эпоксидирования. Примерами таких побочных продуктов являются гликолевые соединения, такие как пропиленгликоли. Эти побочные продукты могут содержаться в (M-vi2) в количестве до 4 масс.%, предпочтительно до 3 масс.%.
В соответствии со способом настоящего изобретения возможно, что смесь (M-v2), вводимая на стадию (vi), содержит побочные продукты, полученные на по меньшей мере одной стадии суммарного способа эпоксидирования, такие как простые гликолевые эфиры, как метоксипропанолы. Что касается этих смесей, было неожиданно обнаружено, что описанная выше перегонка при двух давлениях дополнительно содержит колонну с разделительной стенкой, обеспечивает одновременное отделение этих побочных продуктов от метанольного потока, который подают обратно в качестве растворителя на стадию (а) и получают смесь (M-vi2), как описано выше, имеющую очень низкое содержание, касающееся побочных продуктов, не более чем 4 масс.%, предпочтительно не более чем 3 масс.%.
Смесь (M-vi3), отбираемая из бокового отвода колонны с разделительной стенкой К42, содержит по меньшей мере 10 масс.% простых эфиров гликоля, более предпочтительно по меньшей мере 15 масс.% простых эфиров гликоля и особенно предпочтительно по меньшей мере 20 масс.% простых эфиров гликоля. Еще более предпочтительно, (M-diii) имеет а содержание метанола, равное не более чем 5 масс.%, более предпочтительно менее чем 2 масс.%, более предпочтительно не более чем 2 масс.% и особенно предпочтительно менее чем 2 масс.%.
Как упоминалось выше, верхний поток, полученный либо из традиционной ректификационной колонны K4, или из колонны с разделительной стенкой К42, получаемый как поток (M-vi2), предпочтительно рециркулируют в качестве потока метанольного сырья на стадию (i) способа эпоксидирования. В настоящем изобретении выше приводится ссылка на соответствующий раздел "Метанольное сырье".
Дополнительно, возможно подавать указанный поток (M-vi2) в качестве наружного метанольного орошения ректификационной колонны K2, используемой на стадии (iv), как описано выше.
По этой причине настоящее изобретение также относится к описанному выше способу, дополнительно включающему в себя
(vi) перегонку потока кубового остатка, полученного с (v) в по меньшей мере одной ректификационной колонне К4 для получения потока кубового остатка, имеющего содержание метанола, равное не более чем 0,5 масс.%, и потока верхней фракции, имеющего содержание метанола, равное по меньшей мере 98,5 масс.%, и содержание воды, равное не более чем 1 масс.%;
(vii) рециркуляцию верхнего потока, полученного с (vi) в качестве исходного материала, для непрерывного способа эпоксидирования.
Настоящее изобретение дополнительно иллюстрируется следующей Фиг.1 и Примерами и Сравнительными Примерами.
Краткое описание чертежей
Фиг.1 показывает значения селективности по отношению к побочным продуктам (в %), полученные в примерах и Сравнительных Примерах, нанесенные на график зависимости от средней температуры T (средней) (в °C) охлаждающей среды, пропускаемой через охлаждающую рубашку реактора R1, как описано в Ссылочном Примере.
Примеры
Ссылочный Пример: Получение катализатора и испытание способа
Пропиленоксид получали в основном реакторе R1 и реакторе R2 нисходящей переработки, разделенными ректификационной колонной K1. В качестве катализатора в реакторах R1 и R2, использовали катализатор силикалит титана-1.
Получение катализатора TS-1
Порошковый синтез
|
TEOS (300 кг) загружали в перемешиваемый корпусной реактор при комнатной температуре, и начинали перемешивание (100 об/ мин.). Во втором аппарате, сначала смешивали 60 кг TEOS и 13,5 кг TEOT и затем добавляли TEOS в первый аппарат. Далее, еще 360 кг TEOS добавляли к смеси в первом аппарате. Затем, содержимое первого аппарата перемешивали в течение 10 мин перед добавлением 950 г TPAOH. Перемешивание продолжали в течение 60 мин. Этанол, высвобождаемый при гидролизе, отделяли посредством перегонки при температуре кубового остатка, равной 95°C. 300 кг воды затем добавляли к содержимому первого аппарата, и дополнительно добавляли воду в количестве, эквивалентном количеству дистиллята. Полученную смесь перемешивали в течение 1 ч. Кристаллизацию осуществляли при 175°C в течение 12 ч при аутогенном давлении. Полученные кристаллы силикалита титана-1 отделяли, сушили и кальцинировали при температуре 500°C на воздухе в течение 6 ч.
Синтез кремнеземного золя
|
В аппарат наливали 1096 г воды и добавляли 2,5 л водного раствора аммиака. Полученную смесь перемешивали в течение 15 мин. Далее содержимое аппарата нагревали до температуры 40°C. Затем, добавляли 360 кг TEOS, и содержимое аппарата нагревали до температуры 80°C. Эту температуру поддерживали в течение 2 ч (при кипячении с обратным холодильником). Окончательно, спирт, полученный при гидролизе, отгоняли посредством нагревания содержимого аппарата до температуры 95°C. После перегонки, содержимое аппарата охлаждали до температуры, равной 40°C.
Эту методику повторяли 4 раза.
Формование
|
|
Порошок TS-1, Aerosil и Walocel смешивали в течение 20 мин в бегуне. Затем добавляли кремнеземный золь. Через 35 мин после первого добавления TS-1, добавляли 70 л дистиллированной воды. Через еще 35 мин добавляли 2 кг PEO. Через еще 20 мин, добавляли 10 л воды. Еще после 10 мин, добавляли 2,9 кг PEO. Формуемую массу экструдировали через матрицу, имеющую круговые поры с диаметром 1,5 мм. Полученные нити кальцинировали в ленточном кальцинаторе при температуре 550°C.
Эту методику повторяли 4 раза.
В целом, получали 1740 кг нитей с насыпной плотностью от 470 до 480 г/л. Содержание титана в нитях составляло 0,71 масс.%, содержание Si было равно 44 масс.%. Объем пор нитей, определенный посредством Hg порометрии, был равен 73 мл/г.
Реактор R1
Основной реактор представлял собой вертикально расположенный трубчатый реактор, изготовленный из нержавеющей стали с длиной, равной 2000 мм и внутренним диаметром трубы, равным 28,5 мм. Через трубу сырье для реакции направляли от нижней части к верхней части, т.е. в режиме восходящего потока. Дополнительно, реактор R1 был оборудован центрированным термокарманом с наружным диаметром 8 мм, проходящим от низа реакционной трубы к верхней части реакционной трубы. 10-Кратную термопару с равномерно расположенными термоэлементами помещали в термокарман, что обеспечивало измерение температурного профиля вдоль оси реакционной трубы. Давление в реакторе R1 поддерживали постоянным при 20 бар.
Реактор R1 был дополнительно оборудован охлаждающей рубашкой. В качестве охлаждающей среды смесь этиленгликоль/вода пропускали через охлаждающую рубашку в режиме восходящего потока. Скорость потока охлаждающей среды регулировали таким образом, что температурное различие между температурой ввода и температурой вывода охлаждающей среды составляло не более чем 2°C. Обычно это температурное различие составляло только около 0,5°C.
Реактор R1 дополнительно содержал 620 г нитей гетерогенного катализатора силикалита титана-1, полученного как описано выше. Длина нитей находилась в интервале от 3 до 5 мм. Пустое пространство в верхней части R1 было заполнено стеклянными шариками, имеющими диаметр 4 мм.
Сырье для реакции получали посредством смешивания пяти индивидуальных потоков:
- Первого потока (F1), который содержал около 99 масс.% метанола и около 1 масс.% воды. Этот поток обеспечивали скоростью потока 1700 г/ч. Этот поток не содержал ионы калия, не содержал ионы натрия и не содержал фосфор.
- Второго потока (F2), потока водного пероксида водорода. Этот поток содержал 40 масс.% пероксида водорода. В качестве стабилизирующих агентов, этот поток пероксида водорода содержал 98,6 микромолей ионов натрия на 1 моль пероксида водорода, 91,8 микромолей фосфатов (выраженных как фосфор, Р) на 1 моль пероксида водорода, и 45 мг нитрата на кг пероксида водорода. Отдельно от натрия, поток пероксида водорода содержал только следы (менее чем 10 масс. частей на миллиард) других металлов (железа, алюминия, олова, палладия). Такой водный раствор пероксида водорода является доступным для приобретения, например, от Solvay как неочищенный промытый пероксид водорода, полученный в соответствии с антрахиноновым процессом с использованием системы растворителя, по существу не содержащей органических соединений азота и фосфора. Этот поток обеспечивали скоростью потока, равной 278 г/ч.
- Третьего потока (F3), пропиленовый поток (пропилена полимерной степени чистоты), содержал 99,9 масс.% пропена, оставшаяся часть по существу является пропиленом. Этот поток обеспечивали скоростью потока, равной 142 г/ч.
- Четвертого потока (F4), представляющего собой водный поток, содержащий различные гидрофосфатные соли в соответствии с изобретательскими примерами и Сравнительными Примерами, приведенными в настоящем изобретении ниже. Этот четвертый поток добавляли в таком количестве, чтобы индивидуальные молярные отношения в сырье для реакции получали в соответствии с изобретательскими примерами и Примерами, приведенными в настоящем изобретении ниже.
- Пятого потока (F5, составленный MeOH), который являлся результатом петли рециркуляции метанола, как описано в настоящем изобретении ниже, обеспечивали скоростью потока таким образом, чтобы запас метанола на заводе оставался постоянным.
Все потоки обеспечивали постоянными скоростями потока в жидкой форме, смешивали, и смешанный поток, а именно сырье для реакции, состоящее из одной одиночной жидкой фазы, направляли в реактор R1 при комнатной температуре. Жидкая реакционная смесь в реакторе R1 также состояла из одной единственной фазы.
Каждые 20 минут содержание пероксида водорода в жидкой фракции исходящего из реактора потока, полученного после разгерметизации реакционной смеси, автоматически определяли посредством колориметрии, используя титратор Metrohm ADI 2015, соединенный с диодной матрицей Zeiss MCS521 УФ/видимого спектрометра, в соответствии с титанилсульфатным методом. На основании этого содержания пероксида водорода и массовых расходов потоков через R1, рассчитывали конверсию пероксида водорода. Температуру охлаждающей среды, пропускаемой через охлаждающую рубашку R1, как обсуждалось выше, регулировали так образом, что значение конверсии пероксида водорода, определенное таким образом, поддерживалось постоянным при значении около 90%, т.е. в интервале от около 87 до около 93%.
Ректификационная колонна K1
Исходящий поток из реактора R1, его жидкую а также газообразную часть, направляли в ректификационную колонну и перегоняли при давлении окружающей среды. Ректификационная колонна (DN25×2600 мм длиной, PN 10) была изготовлена из нержавеющей стали и оборудована насадкой Sulzer CY. Перегонку проводили при давлении окружающей среды и другие условия эксплуатации (нагрев кубовых остатков, флегмовое число) регулировали таким образом, чтобы по существу весь пропиленоксид содержался в верхнем потоке. Поток кубового остатка содержал около 85 масс.% метанола и по существу весь непрореагировавший пероксид водорода.
Этот поток кубового остатка пропускали через теплообменник и доводили до температуры 35°C. Перед введением этого потока в реактор R2 его смешивали с потоком пропилена (пропилен полимерной степени чистоты, содержащий 99,9 масс.% пропена, оставшаяся часть по существу является пропаном; скорость потока: 22,5 г/ч).
Реактор R2
Реактор R2 был идентичен R1, но эксплуатировался как по существу адиабатический реактор. R2 содержал 292 г такого же катализатора силикалита титана-1, как применяли в R1. Для достижения адиабатических условий, рубашку R2 вакуумировали и R2 дополнительно изолировали. В противоположность термопаре, используемой в R1, R2 содержал 5-кратную термопару. Как и R1, R2 эксплуатировали в режиме восходящего потока, при котором давление регулировали до значения 15 бар, так, чтобы никакой газообразной фазы не образовывалось в R2. Конверсию пероксида водорода в R2 определяли, как описано для R1. На выходе из R2, конверсия пероксида водорода составляла по меньшей мере 99,8%.
Дополнительная переработка потока, выходящего из R2
Поток реакционной смеси, удаленный из R2, и поток верхней фракции, полученный из K1, объединяли и направляли как поток сырья в ректификационную колонну K2 нисходящей переработки. К2 была изготовлена из нержавеющей стали (DN25×3600 мм длиной, PN 10), оборудована насадкой Sulzer CY и эксплуатировалась при давлении окружающей среды, условия перегонки (нагрев кубовых остатков, внешнее флегмовое число) регулировали таким образом, что содержание пропена потока кубового остатка составляло ниже 100 масс. частей на миллион. Метанол, имеющий температуру 2°C, применяли в качестве дополнительного орошения и направляли в верхнюю часть K2. Содержание пропиленоксида в потоке верхней фракции, полученном из К2, составляло ниже 100 об. частей на миллион. Поток кубового остатка, полученный с K2, затем направляли в ректификационную колонну K3 нисходящей переработки.
К3 была изготовлена из стекла (DN50, 3300 мм длиной), ее оборудовали насадкой Sulzer EX и эксплуатировали при давлении окружающей среды. Условия перегонки (нагрев кубовых остатков, флегмовое число) регулировали таким образом, чтобы содержание пропиленоксида в потоке кубового остатка составляло ниже 100 масс. частей на миллион. Содержание пропиленоксида потока верхней фракции, полученного из K2, было равно по меньшей мере 98 масс.%. Этот верхний поток, неочищенная смесь пропиленоксида, может быть дополнительно очищен или использован как таковой, в зависимости от спецификаций по чистоте. Поток кубового остатка, полученный с K3, затем направляли в ректификационную колонну K4 нисходящей переработки.
K4 изготавливали из стекла (DN50, 2200 мм длиной), оборудовали насадкой Sulzer CY и эксплуатировали при давлении окружающей среды. Условия перегонки (нагрев кубовых остатков, флегмовое число) регулировали таким образом, чтобы поток кубового остатка имел содержание метанола ниже 0,5 масс.%, и верхний поток имел содержание метанола, равное по меньшей мере 98,5 масс.%, и содержание воды, равное не более чем 1 масс.%. Основную часть этого верхнего потока рециркулировали в качестве исходного материала в R1 (первого потока, содержащегося в сырье для реакции, как описано в настоящем изобретении выше); минорную часть этого верхнего потока использовали в качестве дополнительного орошения для ректификационной колонны K2.
Для определения выходов желательного продукта пропиленоксида и побочных продуктов 1-метокси-2-пропанола, 2-метокси-1-пропанола, пропиленгликоля, ацетальдегида и кислорода, определяли количество и состав всех сырьевых потоков и выходящих потоков. Для минимизации всех ошибок эксперимента, все жидкие потоки, поступающие на завод и покидающие его, собирали, и их количество определяли посредством взвешивания. Количество газообразного потока, выходящего из верхней части ректификационной колонны K2, определяли, используя прецизионные барабанные измерители газового потока с влажным тестом (Ritter, Series TG).
Состав жидких потоков определяли посредством количественной газовой хроматографии с методом внутреннего стандарта. Содержание воды определяли титрованием по Карлу-Фишеру. Содержание пероксида водорода в водном растворе, используемом в качестве исходного материала, определяли по марганцеметрическому содержанию гидропероксипропанолов, которое опосредованно определяли по содержанию пропиленгликоля перед и после восстановления указанных гидропероксипропанолов трифенилфосфином. Поскольку этот побочный продукт содержался только во внутренних потоках, его определение не имело значения для определения суммарных выходов.
Состав газообразного потока также определяли посредством газовой хроматографии.
Все выходы и селективности относятся к пероксиду водорода. Поскольку конверсия пероксида водорода является полной (пероксид водорода, непрореагировавший в R2 будет разлагаться во время обработки), выход и селективность для продукта являются равными по отношению друг к другу. Поскольку эксперименты проводят в течение длительного времени, и используемое устройство находится в состоянии устойчивого режима эксплуатации, накоплением реагентов или продуктов внутри устройства можно пренебречь. Массовые балансы в течение периода экспериментов согласуются в интервале, большем чем 2%.
Выход данного продукта A в расчете от пероксида водорода (YA) обычно определяют по общему выражению:
YA=nA×(моль продукта A в выходящих потоках) / (моль H2O2 в сырьевом потоке F2)
где
- nA представляет собой стехиометрический фактор, определяемый как моли H2O2, необходимые для получения одного моля продукта А. Этот фактор равен 1 для пропиленоксида, 1-метокси-2-пропанола, 2-метокси-1-пропанола и пропиленгликоля и 2 для кислорода и ацетальдегида, который образуется под действием катализируемого кислотой разложения гидропероксипропанолов.
- "моль продукта A в выходящих потоках", как используют в уравнении выше, определяют как общее количество продукта A, содержащегося во всех потоках, выходящих из завода в течение времени эксперимента, выражаемое в молях.
- "моль of H2O2 в F2", как используют в уравнении выше, определяют как общее количество H2O2, подаваемое на завод с потоком F2 в течение времени эксперимента, выражаемое в молях.
Как указано ниже, для осуществления следующих экспериментов, реакторы R1 и R2 были заполнены свежим катализатором. Затем начинали подачу метанольного сырья (F1), и установка эксплуатировалась настолько долго, насколько было необходимым для установления устойчивой метанольной петли через реактор R1→ кубовые остатки колонны K2→ реактор R2→ кубовые остатки колонны K2→ кубовые остатки колонны K3 → верхняя часть колонны K4 и обратно в реактор R1. Температуру охлаждающей среды, пропускаемой через охлаждающую рубашку R1, устанавливали до 29°C и температуру сырьевого потока в R2 устанавливали до 35°C и поддерживали постоянной на протяжении экспериментов. Затем начинали подачу потока пероксида водорода (F2), потока пропена (F3), добавление потока (F4), и составление метанола (F5). Если во время эксперимента, катализатор дезактивировался, температуру охлаждающей среды в R1 постепенно увеличивали, чтобы компенсировать эту дезактивацию. Решение о необходимости повышать или нет температуру охлаждающей среды принимали на основании конверсии пероксида водорода, определяемой, как описано выше. Установление баланса начиналось, как только конверсия пероксида водорода в R1 достигала значения, равного около 90%, и была устойчивой в интервале от 87 до 93%, и завод в целом находился в устойчивом состоянии. После проведения данного первого эксперимента с использованием конкретной добавки, содержащейся в добавленном потоке (F4), данный эксперимент с использованием отличного добавленного потока проводили в большинстве случаев без смены катализатора в R1 и R2. Чтобы минимизировать различие в температурах между экспериментами, эксперименты с более крупным количеством добавки проводили сначала, а после проводили эксперименты с меньшими количествами добавок. После смены добавки или ее количества температуру охлаждающей среды регулировали для достижения желательной конверсии пероксида водорода в R1. Обычно, менее 48 часов было необходимо для достижения нового устойчивого состояния после изменения количества и/или природы добавки.
Пример 1: Получение пропиленоксида с содержанием калия в сырье для реакции, равным 133 микромолей на 1 моль пероксида водорода
Эксперимент осуществляли со свежеприготовленным катализатором. Гидрофосфат дикалия (1,25 масс.%, водный раствор) добавляли как поток F4. Количество добавленного раствора гидрофосфата дикалия выбирали таким образом, что в сырье для реакции в R1 молярное отношение калия к пероксиду водорода составляло 133 микромолей K на моль пероксида водорода, молярное отношение (калий + натрий) к фосфору составляло 1,46, молярное отношение калия к натрию составляло 1,35, и молярное отношение калия к фосфору составляло 0,84. После достижения постоянной конверсии пероксида водорода, равной 90% в R1 (через 90 ч, температура охлаждающей среды в R1 31,8°C), начинался баланс и продолжался в течение 250 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,35°C в день. В течение 250 ч конверсия пероксида водорода после R2 составляла 99,9%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 8,0%. При этом балансе некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Поэтому вышеуказанные значения выходов не достигают 100%. Эти побочные продукты представляют собой, например, монометиловый эфир дипропиленгликоля, монометиловый эфир трипропиленгликоля, дипропиленгликоль, трипропиленгликоль, формальдегид, гидроксиацетон, пропиональдегид, ацетон, метилформиат, 1,1-диметоксиметан, 1,1-диметоксиэтан, 1,1-диметоксипропан, 4-метил-1,3-диоксолан, 2,4-диметил-1,3-диоксолан, 2-этил-4-метил-1,3-диоксолан, 2,2,4-триметил-1,3-диоксолан. Если учитывать эти побочные продукты, ошибка в балансе составляет <1%.
Сравнительный Пример 1: Получение пропиленоксида с содержанием калия в сырье для реакции, равным 100 микромолей на 1 моль пероксид водорода
Эксперимент в соответствии с Примером 1 продолжали. Гидрофосфат дикалия (1,25 масс.%, водный раствор) добавляли как поток F4. Количество добавленного раствора гидрофосфата дикалия выбирали таким образом, что в сырье для реакции в R1, молярное отношение калия к пероксиду водорода составляло 100 микромолей K на моль пероксида водорода. Молярное отношение (калий + натрий) к фосфору составляло затем 1,40, молярное отношение калия к натрию составляло 1,01, и молярное отношение калия к фосфору составляло 0,70. Через 40 ч, достигали постоянной конверсии пероксида водорода, равной 90% в R1, при температуре охлаждающей среды, равной 33,9°C. При этом значении баланс устанавливался и продолжался в течение 340 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,46°C в день. В течение 340 ч, конверсия пероксида водорода после R2 составляла 99,9%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 9,3%. При этом балансе некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Сравнительный Пример 2: Получение пропиленоксида с содержанием цезия в сырье для реакции, равным 129 микромолей на 1 моль пероксид водорода
Эксперимент в соответствии со Сравнительным Примером 1 был продолжен. Добавку, содержащуюся в потоке F4, заменили. Вместо гидрофосфата дикалия добавляли гидрофосфат дицезия (1,25 масс.%, водный раствор) как поток F4. Количество добавленного раствора гидрофосфата дицезия выбирали таким образом, что в сырье для реакции в R1, молярное отношение цезия к пероксиду водорода составляло 129 микромолей Cs на моль пероксида водорода. Молярное отношение натрия к фосфору составляло тогда 0,63, молярное отношение калия к натрию составляло 0, и молярное отношение калия к фосфору было равно 0. Через 38 ч, достигали постоянную конверсию пероксида водорода, равную 90% в R1, при температуре охлаждающей среды, равной 41,1°C. При этом значении баланс устанавливался и продолжался в течение 140 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,25°C в день. В течение 140 ч, конверсия пероксида водорода после R2 составляла 99,9%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 10,1%. При этом балансе, некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Сравнительный Пример 3: Получение пропиленоксида с содержанием натрия в сырье для реакции, равным 270,6 микромолей на 1 моль пероксид водорода
Эксперимент осуществляли со свежеприготовленным катализатором, как в Примере 1. Гидрофосфат динатрия (1,25 масс.%, водный раствор) добавляли как поток F4. Количество добавленного раствора гидрофосфата динатрия выбирали таким образом, что в сырье для реакции в R1, молярное отношение натрия к пероксиду водорода составляло 270,6 микромолей Na на моль пероксида водорода (т.е. в добавление к натрию, содержащемуся в потоке F2, 98,6 микромолей Na на моль пероксида водорода, еще другие 172 микромолей натрия на 1 моль пероксида водорода добавляли через поток F4). Молярное отношение натрия к фосфору составляло затем 1,52, молярное отношение калия к натрию составляло 0, и молярное отношение калия к фосфору было равно 0. После достижения постоянной конверсии пероксида водорода, равной 90% в R1 (через 120 ч, температура охлаждающей среды 31,0°C), баланс устанавливался и продолжался в течение 260 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,31°C в день. В течение 260 ч, конверсия пероксида водорода после R2 составляла 99,85%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 9,1%. При этом балансе, некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Сравнительный Пример 4: Получение пропиленоксида с содержанием калия в сырье для реакции, равным 195 микромолей на 1 моль пероксид водорода
Эксперимент в соответствии со Сравнительным Примером 3 был продолжен. Гидрофосфат дикалия (1,25 масс.%, водный раствор) добавляли как поток F4.
Количество добавленного раствора гидрофосфата дикалия выбирали таким образом, что в сырье для реакции в R1, молярное отношение калия к пероксиду водорода составляло 195 микромолей К на моль пероксида водорода. Молярное отношение (калий + натрий) к фосфору составляло затем 1,55, молярное отношение калия к натрию составляло 1,98, и молярное отношение калия к фосфору было равно 1,03. Через 10 ч, достигали постоянную конверсию пероксида водорода, равную 90%, при температуре охлаждающей среды, равной 38,0°C. При этом значении баланс устанавливался и продолжался в течение 180 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,31°C в день. В течение 180 ч конверсия пероксида водорода после R2 составляла 99,85%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 8,8%. При этом балансе некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Пример 2: Получение пропиленоксида с содержанием калия в сырье для реакции, равным 155 микромолей на 1 моль пероксида водорода
Эксперимент продолжали в соответствии со Сравнительным Примером 4. Гидрофосфат дикалия (1,25 масс.%, водный раствор) добавляли как поток F4. Количество добавленного раствора гидрофосфата дикалия выбирали таким образом, что в сырье для реакции в R1, молярное отношение калия к пероксиду водорода составляло 155 микромолей K на моль пероксида водорода. Молярное отношение (калий + натрий) к фосфору составляло затем 1,50, молярное отношение калия к натрию составляло 1,57, и молярное отношение калия к фосфору составляло 0,91. Через 110 ч, достигали постоянной конверсии пероксида водорода, равной 90%, при температуре охлаждающей среды, равной 38,0°C. При этом значении баланс устанавливался и продолжался в течение 170 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,31°C в день. В течение 170 ч конверсия пероксида водорода после R2 составляла 99,85%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 8,0%. При этом балансе некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Сравнительный Пример 5: Получение пропиленоксида с содержанием натрия в сырье для реакции, равным 198,6 микромолей на 1 моль пероксида водорода
Эксперимент осуществляли со свежеприготовленным катализатором, как в Примере 1. Гидрофосфат динатрия (1,25 масс.%, водный раствор) добавляли как поток F4 Количество добавленного раствора гидрофосфата динатрия выбирали таким образом, что в сырье для реакции в R1, молярное отношение натрия к пероксиду водорода составляло 198,6 микромолей Na на моль пероксида водорода (т.е. в добавление к натрию, содержащемуся в потоке F2 98,6 микромолей Na на моль пероксида водорода, еще 100 микромолей натрий на 1 моль пероксида водорода добавляли через поток F4). Молярное отношение натрия к фосфору составляло затем 1,40, молярное отношение калия к натрию было равно 0, и молярное отношение калия к фосфору было равно 0. После достижения постоянной конверсии пероксида водорода, равной 90% в R1 (через 120 ч, температура охлаждающей среды 29,5°C), баланс устанавливался и продолжался в течение 230 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 0,64°C в день. В течение 230 ч, конверсия пероксида водорода после R2 была равна 99,8%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составлял 9,9%. При этом балансе, некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Сравнительный Пример 6: Получение пропиленоксида с содержанием аммония в сырье для реакции, равным 171 микромолей на 1 моль пероксид водорода
Эксперимент осуществляли со свежеприготовленным катализатором, как в Примере 1. Гидрофосфат диаммония (1,25 масс.%, водный раствор) добавляли как поток F4. Количество добавленного раствора гидрофосфата диаммония выбирали таким образом, что в сырье для реакции в R1, молярное отношение аммония к пероксиду водорода составляло 171 микромолей аммония на моль пероксида водорода (поскольку аммоний не содержался в потоке F2, весь аммоний добавляют через поток F4). Молярное отношение натрия к фосфору составляло затем 0,56, молярное отношение калия к натрию было равно 0, и молярное отношение калия к фосфору было равно 0. После достижения постоянной конверсии пероксида водорода, равной 90% в R1 (через 100 ч, температура охлаждающей среды 29,3°C), баланс устанавливался и продолжался в течение 216 ч. Чтобы поддерживать конверсию пероксида водорода в R1 постоянной, охлаждающая среда в R1 должна увеличиваться со средней скоростью, равной 1,1°C в день. В течение 216 ч, конверсия пероксида водорода после R2, составляла 99,9%. Были получены следующие выходы:
|
Общий выход по 1-метокси-2-пропанолу, 2-метокси-1-пропанолу, пропиленгликолю, ацетальдегиду и кислороду составляло 9,7%. При этом балансе некоторые побочные продукты, присутствующие только в минорных количествах, не были однозначно указаны. Делается ссылка на соответствующее обсуждение в Примере 1. Если учитывать побочные продукты, ошибка в балансе составляет <1%.
Обобщение примеров и Сравнительных Примеров
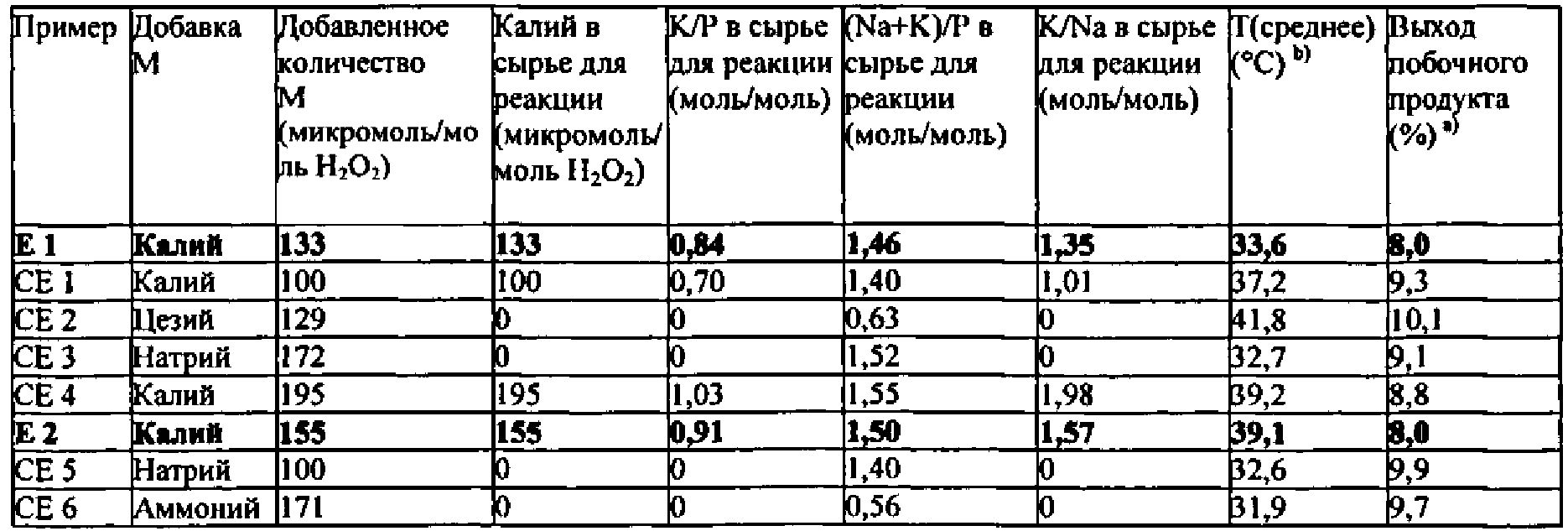
a) Сумма выходов 1-метокси-2-пропанола, 2-метокси-1-пропанола, пропиленгликоля, ацетальдегида и кислорода. Выход рассчитывали относительно пероксида водорода.
b) T (средняя) определяют как (температура охлаждающей среды в R1 в начале баланса + температура охлаждающей среды в R1 в конце баланса)/2
Сравнение примеров, в которых добавляют калий
(расположенное в соответствии с увеличивающимся количеством добавленного калия)
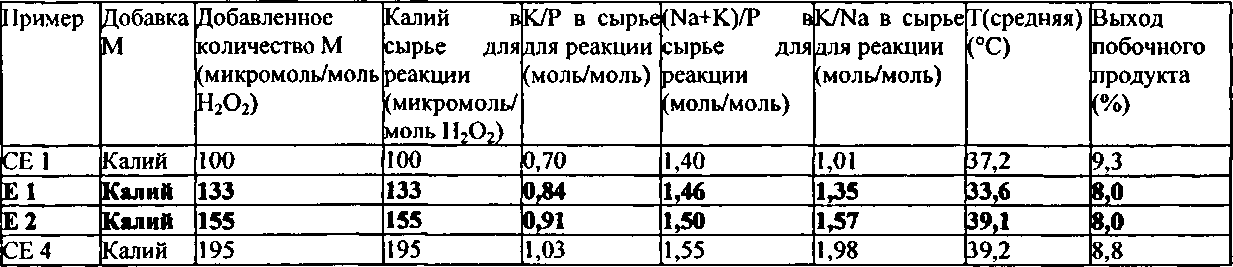
Это сравнение показывает, что только при свыше 100 микромолей и ниже 195 микромолей добавленного количество калия, получали предпочтительную низкую селективность к побочному продукту. По этой причине было показано, что существует предпочтительный интервал для калия, содержащегося в сырье для реакции, который приводит к низкой селективности к побочному продукту.
Сравнение примеров, касающихся замены калия на цезий

Сравнение Примера 1 и Сравнительного Примера 2 показывает, что в то время как количество добавленного М по существу является постоянным, замена калия на по существу такое же количество цезия - приводит к отношениям K/H2O2, K/P, (Na+K)/P и K/Na в сырье для реакции, которые выходят за пределы изобретательских, предпочтительных интервалов - приводит к явно увеличенной селективности по отношению к побочному продукту.
Сравнение примеров, касающихся замены калия на натрий
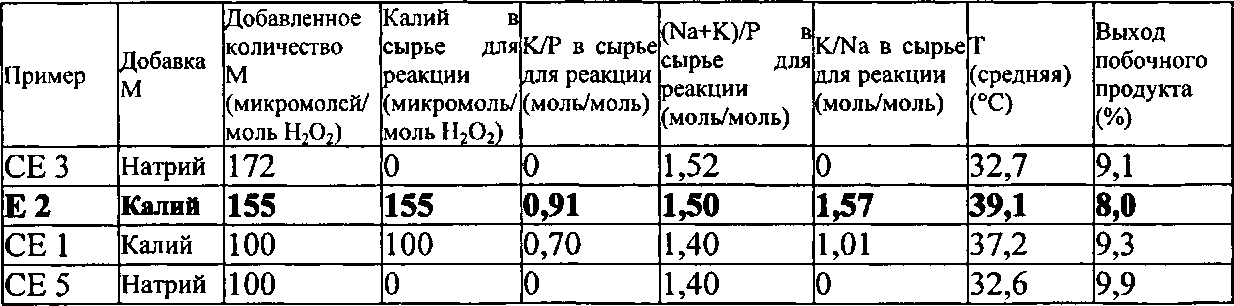
Сравнение Сравнительного Примера 3 с Примером 2, оба из которых имеют аналогичное количество добавленного M, показывает, что замена калия на натрий приводит к увеличенной селективности к побочному продукту. Более того, это сравнение показывает, что не только отношение (Na+K)/P приводит к низким селективностям по отношению к побочным продуктам (оба значения находятся в предпочтительном интервале); необходимо, чтобы добавка М была калием для достижения предпочтительных результатов.
Несмотря на то, что Сравнительный Пример 1 показывает количество добавленного калия, которое выходит за пределы изобретательского интервала, сравнение CE 1 с CE 5 тем не менее показывает, что при рассмотрении калия и натрия, калий является предпочтительной добавкой.
Сравнение примеров, касающихся замены калия на аммоний
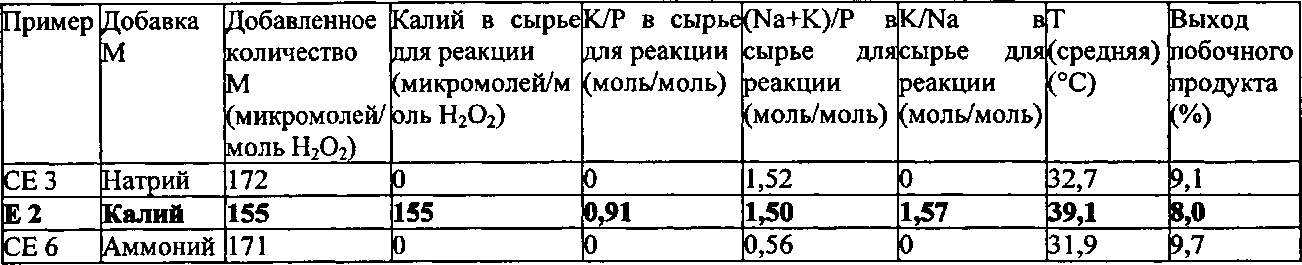
Сравнение Сравнительных Примеров 3 и 6 (и Примера 2) показывает, что замена непредпочтительного натрия на по существу такое же количество аммония приводит к даже более увеличенной селективности к побочному продукту. Также делается ссылка на сравнение натрия с калием, приведенное выше. Как обсуждалось выше, все эксперименты по балансу осуществляли при постоянном значении конверсии пероксида водорода, равной 90% в реакторе R1. Чтобы показать, что селективности по отношению к побочному продукту, полученные из примеров и Сравнительных Примеров, не коррелируют с T (средняя) - средними температурами, находящимися внутри сравнительно узкого интервала, равного около 10°C - дается ссылка на Фиг.1. Там, селективности по отношению к побочному продукту (в %) нанесены на график зависимости от T (средней) (в °C). Данный факт доказывает, что выбор постоянного значения конверсии пероксида водорода в качестве исходного параметра является наилучшим выбором, тем более, что известно, что данная конверсия является той конверсией, которая имеет значительное влияние на селективности по отношению к побочным продуктам, поскольку реакции, приводящие к побочным продуктам являются по существу последовательными реакциями.
Цитируемые ссылки
Clerici et al., J. Catal. 140 (1993) pages 71-83
EP 0230949 A2
EP 0712852 A1
EP 0757043 A1
WO 99/48882 A1
WO 2004/029032 A1
EP 1085017 A1
EP 1122249 A1
Ullmann′s Encyclopedia of Industrial Chemistry, 5th edition, volume A13 (1989) pages 443-466
Ullmann′s Encyclopedia of Industrial Chemistry, 5th Edition, Volume A22, page 214
WO 2005/103024 A1
WO 2010/130610 A1
WO 2007/074101 A1
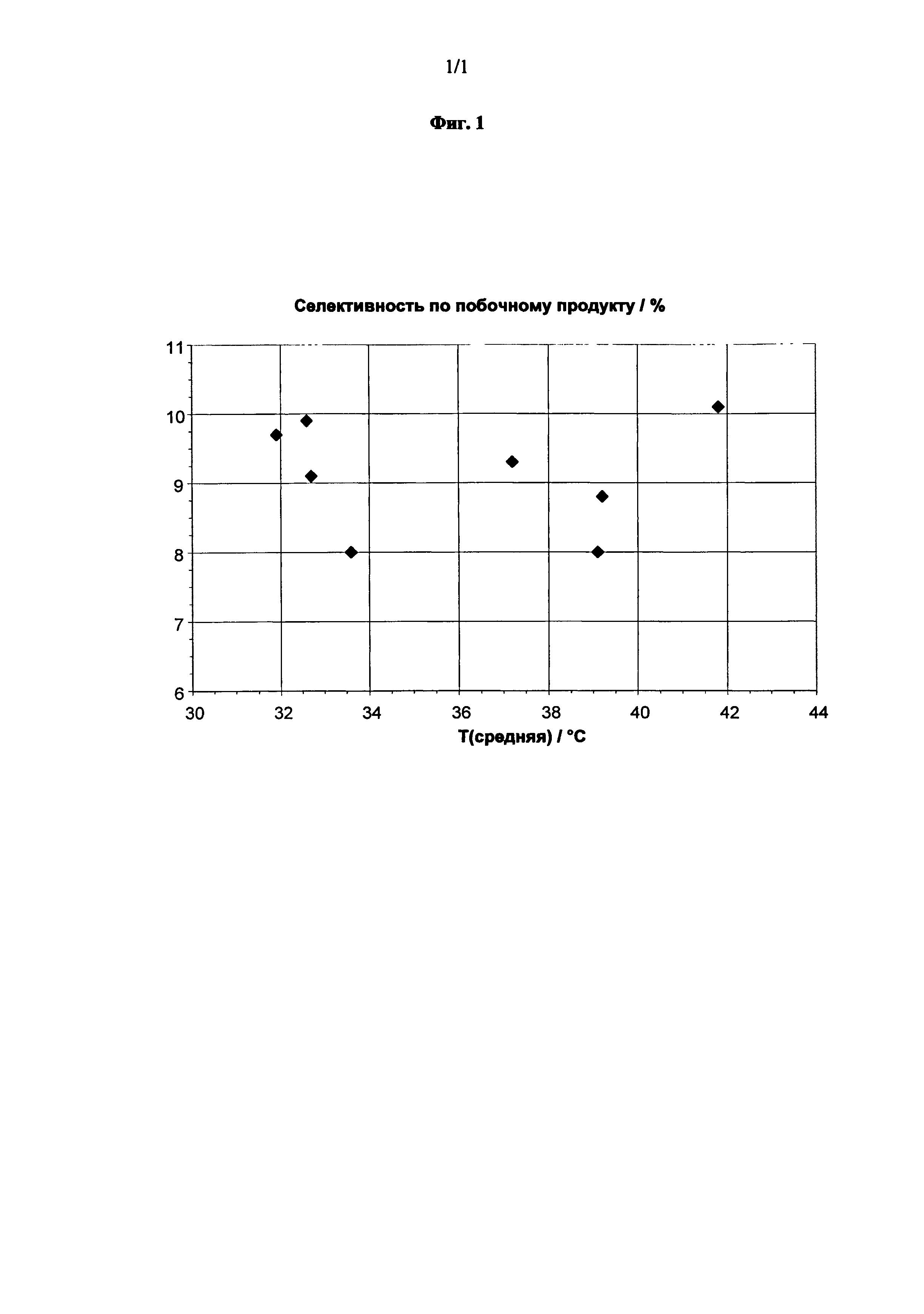
Комбинированный способ получения порошка карбонила железа и углеводородов
Способ изготовления каталитически активных геометрических формованных изделий
Способ изготовления каталитически активных геометрических формованных изделий
Амфифильные водорастворимые алкоксилированные полиалкиленимины, имеющие внутренний полиэтиленоксидный блок и наружный полипропиленоксидный блок
Способ получения синильной кислоты
Привитой сополимер как ингибитор газовых гидратов
Способ надежного предотвращения обратного потока при перекачивании жидкости
Применение алкоксилированных полиалканоламинов для деэмульгирования эмульсий типа "масло в воде"
Улучшенный способ получения синильной кислоты посредством каталитической дегидратации газообразного формамида
Косметические препараты на основе молекулярно впечатанных полимеров
Неэлектролитическое осаждение барьерных слоев
Способ изготовления композитных элементов на базе пенопластов на изоцианатной основе
Выделяемые и передиспергируемые наночастицы переходных металлов, их получение и применение в качестве ик-излучателей
Стеклянные фритты
Способ электрохимического расщепления лигнина на алмазном электроде
Комбинированный способ получения порошка карбонила железа и углеводородов
Способ изготовления каталитически активных геометрических формованных изделий
Способ изготовления каталитически активных геометрических формованных изделий
Амфифильные водорастворимые алкоксилированные полиалкиленимины, имеющие внутренний полиэтиленоксидный блок и наружный полипропиленоксидный блок
Способ получения синильной кислоты

