СПОСОБ ПЕРЕБОТКИ ВОДНО-ОРГАНИЧЕСКОГО ОТХОДА МОЛИБДЕНОВОГО КАТАЛИЗАТОРА ОРГАНИЧЕСКОГО СИНТЕЗА
Вид РИД
Изобретение
Изобретение относится к технологии переработки отходов молибденового катализатора органического синтеза.
Известен способ извлечения молибдена из продуктов каталитического эпоксидирования олефинов (патент № RU 2268885), включающий обработку тяжелой фракции эпоксидата раствором щелочи, обработку образующегося отработанного щелочного потока экстрагентом с последующим осаждением трисульфида молибдена, в качестве осадителя трисульфида молибдена используют сернисто-щелочные стоки производств олефинов пиролизом углеводородного сырья. Продукты реакции эпоксидирования олефинов органическими гидропероксидами разделяют методом ректификации на легкие фракции и тяжелую фракцию, содержащую молибден, а также непрореагировавший гидропероксид и продукты побочных реакций. После промывки тяжелой фракции эпоксидата раствором каустической соды от побочных продуктов окисления и от отработанного молибденового катализатора образуется отработанный щелочной отход (ЩО). Образующийся ЩО - это раствор темно-коричневого цвета, с резким запахом, плотностью 1,11-1,18 г/см3, представляет собой сложную смесь натриевых солей органических кислот, фенолятов, смолистых и других веществ (до 35%). Молибден в количестве 0,1-0,3 мас.% представлен в виде органических комплексов неизвестного состава, рецикл молибденового катализатора невозможен ввиду непостоянства состава по ионам молибдена и натрия. Далее ЩО подвергают обработке экстрагентом, в качестве которого используют водный раствор минеральной кислоты (серной, соляной, азотной, фосфорной и др.) с концентрацией 2-2,5 N. Обработку проводят при температуре 50-100°С при объемном соотношении ЩО к экстрагенту от 1:1,5 до 1:2, после чего осуществляют осаждение молибдена из водной фазы. В водную фазу, помимо ионов молибдена, экстрагируются ионы натрия, которые не препятствуют процессу осаждения молибдена. Органическая фаза, очищенная от металлов, может быть подвергнута дальнейшей переработке с целью получения ряда полезных компонентов. В качестве осадителя трисульфида молибдена используют сернисто-щелочные стоки (СЩС) производств олефинов пиролизом углеводородного сырья, например, этиленового комплекса, являющиеся полноценным заменителем чистого сернистого натрия. Сернисто-щелочные стоки подают на осаждение при объемном соотношении к водной фазе, равном 0,19-1:1 в зависимости от содержания Na2S. Достигаемый технический результат - повышение степени извлечения молибдена и упрощение способа.
Недостатком данного способа является недостаточно эффективная переработка водно-органического отхода, включающая только выделение молибдена. При этом другие ценные продукты направляются на огневое обезвреживание.
Известен способ утилизации отхода процесса каталитического эпоксидирования олефинов (патент № RU 2393152), который включает выделение и обработку тяжелой фракции эпоксидата раствором щелочи и обработку образующегося отработанного щелочного потока экстрагентом. В соответствии с изобретением предложено в отработанный щелочной поток добавлять лиганд, образующий металлорганический комплекс молибдена, и выделять из отработанного щелочного потока фракцию, включающую пропиленгликоль, ацетофенон, этилбензол, фенол, метилфенилкарбинол и образовавшийся металлорганический комплекс молибдена, обработкой экстрагентом при Т≥Ткр и Р≥Ркр с последующим разделением экстракта на фракции путем ступенчатого снижения давления от Рэкстр до Р<Ркр с количеством ступеней снижения давления, равным количеству фракций компонентов, которые необходимо получить с учетом металлорганического комплекса молибдена, где Ткр, Ркр - критические значения температуры и давления экстрагента, Рэкстр - давление экстракции. Достигаемый технический результат - сохранение высокой степени извлечения молибдена из отработанного щелочного потока независимо от его состава и возможность извлечения фракций компонентов, содержащихся в отработанном щелочном потоке.
Недостатком известного способа также является недостаточно эффективная переработка водно-органического отхода, а выделение только двух ликвидных продуктов: молибдена и узкой фракции углеводородов.
Известен метод (патент № US 7854908) восстановления молибдена из сырья, содержащего молибден (в т.ч. в случае, когда таким сырьем является использованный катализатор), включающий нагрев и окисление сырья в вихревом реакторе с повышенным давлением при достаточно высокой температуре в окисляющей среде для того, чтобы получить окисление породы молибдена и сублимировать триоксид молибдена, выделение нелетучих веществ из газообразной фазы в выходящем потоке из вихревого реактора с повышенным давлением при помощи высокотемпературного циклонного сепаратора, подачей двух раздельных потоков, причем первый поток это нелетучие вещества, отделенные циклонным сепаратором из второго потока, который является газовым потоком, проходящим через циклонный сепаратор, при этом первый поток подает первый продукт, состоящий из нелетучих веществ, охлажденный второй поток подает второй продукт, т.е. конденсированный порошок триоксида молибдена и выделение второго продукта из газового потока.
Недостатком этого способа также является недостаточно эффективная переработка водно-органического отхода и сложность метода.
Задачей предлагаемого изобретения является разработка способа эффективной переработки водно-органического отхода молибденового катализатора с возможностью выделения большого количества ценных продуктов.
При этом способ переработки водно-органического отхода молибденового катализатора включает отгонку широкой фракции углеводородов с острым водяным паром, обработку кубового остатка серной кислотой, разделение продукта обработки на органическую и водную фазы, последовательную отгонку с острым водяным паром из органической фазы фенола и бензойной кислоты, выделение фенола из дистиллята экстракцией МТБЭ, очистку сырого фенола ректификацией, выделение бензойной кислоты из дистиллята фильтрацией, очистку выделенной бензойной кислоты возгонкой, экстракцию из водной фазы остатков фенола и бензойной кислоты МТБЭ, отделение МТБЭ от экстрагированных компонентов дистилляцией, обработку водной фазы сульфидом или гидросульфидом натрия для осаждения из трисульфида молибдена, фильтрационное отделение трисульфида молибдена от маточного раствора, его сушку при 100-120°С и окислительное прокаливание трисульфида молибдена до триоксида молибдена при 550-600°С, очистку маточного водного раствора сульфата натрия от примесей органической фазы сорбцией на активированном угле, выделение десятиводного сульфата натрия упариванием и последующей кристаллизацией.
Остатки осмоленной органической фазы направляют на огневое обезвреживание (сжигание).
Способ переработки водно-органического отхода молибденового катализатора органического синтеза включает отгонку углеводородов, обработку кубового остатка серной кислотой, разделение продукта обработки на водную и органическую фазы, выделение из водной фазы триоксида молибдена и десятиводного сульфата натрия, выделение из органической фазы фенола и бензойной кислоты. При этом с целью повышения комплексности переработки водно-органического отхода и выхода каждого выделяемого продукта отгонку широкой фракции углеводородов проводят острым водяным паром с температурой 110-130°С, обработку кубового остатка проводят концентрированной серной кислотой до рН 1-2 при температуре 25-40°С, расслаивание продукта обработки на водную и органическую фазы проводят при 30-40°С в течение 30-60 минут, после чего фазы разделяют, из охлажденной до 20-25°С водной фазы экстрагируют остатки фенола и бензойной кислоты метил-трет-бутиловым эфиром при соотношении объемов водной и органической фаз О:В=1:1, водную фазу отделяют от органической и обрабатывают водным раствором сульфида и/или гидросульфида натрия при мольном соотношении Mo:S=3:5 с выделением из раствора осадка трисульфида молибдена фильтрацией с последующей сушкой и окислительным обжигом трисульфида молибдена до триоксида молибдена при температуре 550-600°С, водный фильтрат после удаления трисульфида молибдена очищают от остатков органической фазы сорбцией на активированном угле с последующим упариванием водного раствора и кристаллизацией из упаренного раствора десятиводного сульфата натрия; из органической фазы, полученной после разделения кубового остатка последовательно выделяют фенол отгонкой с острым водяным паром с температурой 110-130°С, с последующей его экстракцией из дистиллята метил-трет-бутиловым эфиром при соотношении объемов водной и органической фаз О:В=1:1 и очищением от примесей ректификацией, и бензойную кислоту при температуре перегонки 105-110°С, выделение которой из дистиллята проводят фильтрацией, а очистку от примесей - возгонкой твердого высушенного продукта при температуре 105-110°С.
Отгонка широкой фракции углеводородов может проводиться острым водяным паром с исходным давлением от 4 до 12 ати, преимущественно 6 ати.
Дистиллят после охлаждения может расслаиваться на водную фазу, которую нагревают до необходимой температуры и возвращают в голову процесса отгонки углеводорода и органическую фазу, представляющую широкую фракцию углеводородов, которую возвращают в цикл органического синтеза на молибденовом катализаторе.
Кубовый остаток отгонки широкой фракции углеводородов с острым водяным паром может быть охлажден до 25-30°С и обработан концентрированной серной кислотой при температуре не превышающей 40°С до рН 1-2, после чего его расслаивают на водную и органическую фазы, которые разделяют для выделения из водной фазы триоксида молибдена и десятиводного сульфата натрия, а из органической фазы - фенола и бензойной кислоты.
Перед выделением триоксида молибдена из водной фазы можно экстрагировать остатки фенола и бензойной кислоты МТБЭ при температуре 20-25°С, О:В=1:1, времени контакта фаз 1-5 минут, времени расслаивания фаз перед разделением 10-20 минут.
Для выделения триоксида молибдена очищенный от фенола и бензойной кислоты водный раствор может быть нейтрализован до рН 6-8 10-20%-ным водным раствором NaOH, затем температура повышается до 80°С и раствор обрабатывается при перемешивании 10-20%-ным водным раствором сульфида и/или гидросульфида натрия при мольном соотношении Mo:S=1:5, раствор выдерживают при перемешивании 10-30 минут и нейтрализуют концентрированной серной кислотой до рН 2,5-3 и выдерживают при перемешивании еще 1-2 часа при 80°С до полного выделения в осадок трисульфида молибдена.
Осадок трисульфида молибдена может быть отделен от маточного раствора фильтрацией, промыт на фильтре водой до полного отсутствия в промывных водах сульфата натрия, высушен при 100-120°С и прокаливается в атмосфере воздуха при 550-600°С в течение 2-4-х часов до полного образования триоксида молибдена.
Фильтрат и промывные воды после отделения трисульфида молибдена объединяют, очищают от примесей органической фазы сорбцией на активированном угле, упаривают на 20-40% до образования десятиводного сульфата натрия и кристаллизуют Na2SO4·10H2O при охлаждении до 10-25°С, после чего отделяют от маточного раствора, который смешивают с исходным фильтратом и направляют на повторное упаривание, а выделившийся продукт - десятиводный сульфат натрия - высушивают при 100-120°С.
Из органической фазы, отделенной от молибденсодержащей водной фазы, выделяют фенол отгонкой с острым водяным паром при его температуре 110-130°С в дистиллят, который разделяют при отстаивании на водную фазу, представляющую собой раствор фенола в воде и органическую фазу - сырой фенол. Из водной фазы фенол извлекают экстракцией МТБЭ при температуре 15-25°С, O:В=1:1, за 2-3 контакта противоточной экстракции, времени контакта 1-5 минут и времени расслаивания 10-20 минут, экстракт фенола с МТБЭ отделяют от водного рафината и направляют на очистку фенола и выделение МТБЭ ректификацией. Сырой фенол направляют на очистку ректификацией.
Кубовый остаток после выделения фенола из органической фазы обрабатывают острым водяным паром при температуре 105-110°С и конденсируют пары воды и бензойной кислоты при 15-25°С, после чего отделяют кристаллы бензойной кислоты от водной фазы, высушивают и направляют на очистку возгонкой.
Кубовый остаток после выделения бензойной кислоты отгонкой с острым водяным паром обрабатывают 10-15%-ным водным раствором NaOH при соотношении объемов фаз О:В = 1:0,2-1,0 и образовавшийся водно-органический продукт направляют на огневое обезвреживание (сжигание).
Насыщенный органическими соединениями активированный уголь промывают 2-5 объемами горячей воды при температуре 60-80°С для удаления водного раствора сульфата натрия, после чего десорбируют органические соединения промывкой 2-5 объемами 10-15%-ного водного раствора NaOH при температуре 30-60°С и 2-5 объемами обессоленной воды, которые направляют на огневое обезвреживание, а подготовленный таким образом активированный уголь направляют на очистку новых потоков раствора сульфата натрия перед его выделением.
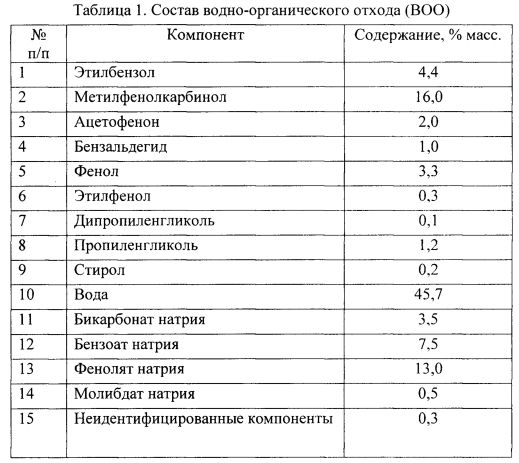
В лабораторных условиях для переработки использовался водно-органический отход следующего состава (см. Табл. 1).
Переработка BOO осуществлялась в шесть стадий.
Результатом первой стадии переработки исходного BOO массой 1500 г являлось выделение широкой фракции углеводородов (ШФУ).
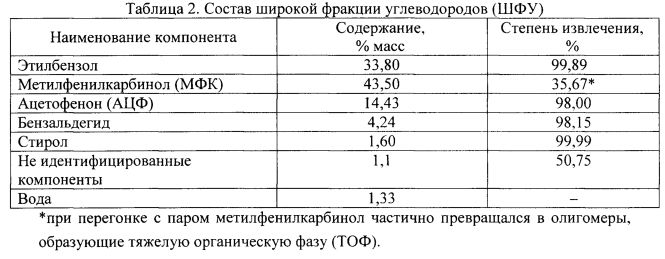
Кубовый остаток содержал BOO, из которого была удалена ШФУ (201 г), и небольшое количество сконденсированной воды. Степень извлечения всех компонентов в ШФУ представлена в табл. 2.
Второй стадией переработки BOO являлось выделение из кубового остатка перегонки с паром ШФУ суммарной органической фракции, содержащей фенол, бензойную кислоту и осмоленные органические продукты. С этой целью к кубовому остатку в количестве 1500 г порционно добавляли 300 г 92,5% серной кислоты при охлаждении до 25-30°С. Нейтрализацию проводили до рН 1-2 водной фазы, в результате чего из реакционной смеси выделялся углекислый газ, который сбрасывался в атмосферу. После прибавления всего количества серной кислоты смесь выдерживали 0,5 часа до полного расслоения водной и органической фаз. Верхний органический слой в количестве 383 г направляли на выделение фенола и бензойной кислоты. Нижний водный слой в количестве 1410 г направляли на выделение молибдена и сульфата натрия.
Третьей стадией переработки BOO являлось выделение молибдена из сернокислого раствора, полученного после нейтрализации кубового остатка отгонки с паром ШФУ. Водный сернокислый молибденсодержащий раствор, полученный после нейтрализации и отделения органической фазы, содержал остатки фенола, бензойной кислоты и тяжелой органической фазы (ТОФ), представляющей собой осмоленные органические соединения. Для отделения растворенных остатков перечисленных органических соединений проводили экстракционную очистку водного молибденсодержащего раствора. В качестве экстрагента использовался метил-трет-бутиловый эфир (МТБЭ). 1410 г сернокислого водного раствора молибдена обрабатывали в экстракторе 1828 г МТБЭ при комнатной температуре и интенсивном перемешивании в течение 15 минут, после чего водно-органическую эмульсию расслаивали в течение 0,5 часа. Водную фазу, содержащую молибден, отделяли от фазы МТБЭ, которую промывали 200 г воды, а также отделяли от фазы МТБЭ и объединяли с водным сернокислым раствором, содержащим молибден. В результате экстракционной очистки было получено 1492 г водного сернокислого раствора, который направлялся на выделение молибдена, и 1892 г экстракта МТБЭ, который направлялся на выделение фенола, бензойной кислоты и остатков ТОФ. Выделение этих продуктов из МТБЭ осуществлялось его отгонкой при температуре кипения 55,2°С. При отгонке МТБЭ сначала отгонялся его азеотроп с водой при температуре 52,6°С. В результате этой операции получалось 1828 г влажного МТБЭ, который возвращался на экстракционную очистку новых порций водного сернокислого молибденсодержащего раствора, и 118 г кубового остатка, содержащего фенол, бензойную кислоту и ТОФ, которые направляли на выделение фенола и бензойной кислоты.
Очищенный от органических примесей сернокислый раствор молибдена нейтрализовывался до рН 6 15%-ным водным раствором NaOH. В ходе нейтрализации серной кислоты щелочью температура реакционной смеси поднималась до 50-55°С, в результате чего происходила отгонка остатков растворенного в водном растворе МТБЭ, который улавливали охлаждением до 25°С и объединяли с МТБЭ, выделенным отгонкой на предыдущей стадии. Затем температуру этого раствора повышали до 80°С и подавали в него при перемешивании 220 г NaHS для осаждения сульфидных соединений молибдена. После подачи всего количества NaHS рН равновесного раствора повышалось до 10,5-11, что понижало выделение осадка трисульфида молибдена из раствора. Поэтому рН раствора дробно понижали до 2,5-3, добавляя концентрированную серную кислоту. После снижения рН до 2,5-3 из раствора выделялся осадок трисульфида молибдена. Для полной коагуляции и созревания осадка пульпу выдерживали при перемешивании 1 час при температуре 80°С, после чего горячую пульпу фильтровали, осадок на фильтре промывали трижды обессоленной водой при соотношении Т:Ж = 1:1, промывные воды и фильтрат объединяли. В результате получали 2022 г раствора сульфата натрия, который направлялся на стадию сорбционной очистки и выделения сульфата натрия. Твердый влажный осадок трисульфида молибдена в количестве 7,7 г отделяли от фильтра, высушивали и прокаливали в тигле в муфельной печи при температуре 600-650°С 2 часа. В результате окислительного прокаливания получалось 5,2 г триоксида молибдена, который является целевым продуктом переработки BOO. Выход молибдена в пересчете на его триоксид составил 99% от исходного содержания молибдена в BOO.
Четвертой стадией переработки BOO являлось выделение фенола из суммарной органической фазы, полученной после нейтрализации серной кислотой кубового остатка отгонки с паром ШФУ. Выделение фенола из суммарной органической фазы проводили также методом отгонки с паром.
Сырой фенол, выделенный из BOO в количестве 296 г, подавался в кубовую часть ректификационной колонны, где на первом этапе производили отгонку низкокипящей фракции МТБЭ, содержащегося в сыром феноле. Пары азеотропа МТБЭ конденсировались в теплообменнике и охлаждались до температуры 25°С, после чего направлялись на стадию экстракции фенола. Затем температуру в кубе поднимали до температуры кипения фенола 181-182°С и отгоняли фенол, пары которого конденсировались в теплообменнике, охлаждались до температуры 25°С и собирались в приемнике в количестве 210 г. Полученный продукт являлся еще одним целевым продуктом переработки BOO. С учетом экстракционного выделения фенола из дистиллята и его регенерации из растворов с МТБЭ, а также оборота МТБЭ в цикле сквозная степень извлечения фенола в товарный продукт достигал 98,5-99,0%.
Пятой стадией переработки BOO являлось выделение бензойной кислоты из суммарной органической фазы отгонкой с водяным паром, после отгонки из нее фенола. Окончание процесса отгонки фенола из суммарной органической фазы и начало отгонки бензойной кислоты определялось по образованию белых кристаллов бензойной кислоты в холодильнике конденсаторе паров воды и перегоняемого продукта. При этом температура перегоняемой смеси повышалась до 105-110°С. Для эффективной отгонки бензойной кислоты температуру в перегонном аппарате поднимали до 110-120°С. Конденсацию паров воды и бензойной кислоты проводили в том же холодильнике до температуры 40÷50°С, а образовавшуюся пульпу кристаллов бензойной кислоты в воде собирали в приемнике, снабженном мешалкой, и охлаждали до 25°С при перемешивании пульпы. Процесс отгонки проводился до тех пор, пока не прекратилось выпадение кристаллов бензойной кислоты на стенках холодильника конденсатора. Время отгонки составило от 6 до 10 часов. После полной отгонки бензойной кислоты и охлаждения пульпы до 25°С получили 6440 г водной пульпы бензойной кислоты, которую отфильтровали на воронке Бюхнера под вакуумом или на другом подходящем фильтре. Твердую фазу на фильтре промыли дважды 50 г обессоленной воды при Т:Ж = 2:1 и получили 82 г влажных кристаллов бензойной кислоты, которую после высушивания при 40-50°С под вакуумом в течение 1 часа направили на дополнительную очистку методом сублимации.
Фильтрат и промывные воды в количестве 6458 г объединяли и направляли на доизвлечение бензойной кислоты методом экстракции с МТБЭ по той же схеме, которая была описана при экстракции фенола. Кубовый остаток после отгонки МТБЭ в количестве 33 г, представляющий собой раствор бензойной кислоты и фенола в небольшом количестве МТБЭ, направлялся на разделение бензойной кислоты и фенола методом ректификации.
Рафинат экстракции, очищенный от МТБЭ, направлялся в парогенератор на образование пара для отгонки с паром фенола и бензойной кислоты.
Кубовый остаток ректификационной колонны после отгонки фенола представлял собой неочищенную бензойную кислоту, растворенную в примесях жидких органических соединений, таких как метилфенилкарбинол, ацетофенон, этилфенол, пропиленгликоль и др. Эту жидкотекучую массу в количестве 75 г направляли в подогреваемый реактор, снабженный мешалкой, где растворяли в 75 г обессоленной воды при перемешивании и температуре 90-95°С в течение 0,5-1 часа. В результате водно-органическая смесь расслаивалась на органическую фазу (верхний слой), которую направляли на огневое обезвреживание (сжигание), и водную фазу (нижний слой), которую направляли в кристаллизатор, где происходило выделение кристаллов бензойной кислоты из охлажденного водного раствора. Охлажденную пульпу кристаллов бензойной кислоты отфильтровывали на воронке Бюхнера, промывали на фильтре двумя порциями по 25 г обессоленной воды и получали 25 г влажных кристаллов бензойной кислоты, которые после сушки при 40-50°С под вакуумом в течение 1 часа направляли на очистку методом сублимации.
Объединенные фильтрат и промывные воды использовались повторно для перекристаллизации новых порций бензойной кислоты, после чего они выводились из процесса на огневое обезвреживание.
Высушенные кристаллы бензойной кислоты в количестве 96 г направлялись на очистку методом возгонки в сублиматоре.
Очищенные кристаллы бензойной кислоты после возгонки и охлаждения в сублиматоре в количестве 86 г представляли собой готовую продукцию, полученную из BOO. Остаток неочищенных кристаллов бензойной кислоты из сублиматора в количестве 10 г возвращали на повторную очистку на стадию перекристаллизации бензойной кислоты. С учетом оборотов бензойной кислоты на перекристаллизацию и экстракционное извлечение из водных растворов различного типа сквозной выход бензойной кислоты при извлечении из BOO достигал 98-99%.
На шестой стадией переработки BOO производилось выделение сульфата натрия из фильтрата и промывных вод процесса осаждения трисульфида молибдена. С этой целью объединенные фильтрат и промывные воды в количестве 2021 г направлялись на сорбционную очистку от остатков растворенной ТОФ на активированном угле марки БАУ. Очищенный от тяжелой органической фазы водный раствор, который содержал до 250 г/дм3 Na2SO4, собирался после сорбции в промежуточной емкости, из которой его подавали в выпарной аппарат. При упаривании такого раствора уже на 30% из него выкристаллизовывался десятиводный кристаллогидрат сульфата натрия Na2SO4×10H2O (мирабилит). Упаренный раствор сульфата натрия, представляющий собой пульпу десятиводного кристаллогидрата сульфата натрия в воде, в количестве 1090 г, направлялся в распылительную сушилку, в которую подавали нагретый в калорифере до температуры 100°С воздух. В распылительной сушилке происходило удаление остатков воды из упаренной пульпы кристаллогидрата сульфата натрия. Высушенный продукт Na2SO4×10H2O в количестве 845 г являлся еще одним целевым продуктом переработки BOO. Сквозной выход сульфата натрия с учетом отмывки сорбционных колонок на стадии регенерации от раствора сульфата натрия и возврата его в действующий цикл переработки BOO составил более 99%.
Техническим результатом изобретения является повышение эффективности выделения товарных ликвидных продуктов из водно-органического отхода: широкой фракции углеводородов (ШФУ), фенола, бензойной кислоты, триоксида молибдена и десятиводного сульфата натрия.

