Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 2-ГИДРОКСИБУТИРОЛАКТОНА
Вид РИД
Изобретение
Настоящее изобретение относится к способу получения 2-гидроксибутиролактона (2HBL) из 2-гидрокси-4-метилтиомасляной кислоты (в равной степени сокращенно НМТВА, HMBA, АТ88 или Rhodimet АТ88), из ее оксо-аналога, 2-оксо-4-метилтиомасляной кислоты (сокращенно КМВ), 2-гидрокси-4-метилтиобутиронитрила, также как из их производных.
2HBL является важным промежуточным соединением синтеза. Его можно получать промышленным способом известным путем из яблочной кислоты в три стадии (DE 19735575A1, AU 2004200948A) или из γ-бутиролактона в две стадии (WO 2008/022953A1, Bull.Soc.Chim.Fr., 1971, 1, 294-301).
Проблема, возникающая в связи с данными двумя путями, заключается, с одной стороны, в сложности выделения 2HBL и, с другой стороны, в очень значительном образовании солей, которые затем нужно удалять. Первый путь синтеза дополнительно имеет недостатки вовлечения дорогих реагентов, т.е. ВН3 and TFAA; применение гидрида бора, в частности, требует особых условий безопасности. Во втором пути доступа используются токсичные реагенты, т.е. Br2 и PBr3, и эффективность, описанная согласно данной методике, не является очень высокой (RR=23-52%). В целом, оба из этих подходов остаются очень дорогими в промышленном масштабе и не очень производительными.
Согласно настоящему изобретению была предпринята попытка разработать способ синтеза 2HBL, который не имеет этих недостатков, в то же время оставаясь простым, недорогим и эффективным способом.
НМТВА представляет собой аналог метионина, необходимой аминокислоты, и находит весьма широкое применение, в особенности среди людей, в качестве пищевой добавки или лекарственного средства, так же как в кормлении животных, в качестве биологически доступного источника метионина. Производные этого аналога, особенно его сложные эфиры и его соли, также применяют по тем же показаниям, причем некоторые из них, такие как изопропиловый сложный эфир НМВА, имеют свойства, превосходящие свойства НМВА. НМВА производят в промышленном масштабе, согласно в полной мере общепринятым способам, в количестве нескольких сотен тысяч метрических тонн/год. Ее применение в качестве субстрата синтеза, таким образом, открывает дополнительную перспективу для этого.
Именно в этом контексте, согласно настоящему изобретению детально разработали способ синтеза 2HBL из НМТВА и ее производных, который по сравнению с вышеупомянутыми способами синтеза можно использовать в промышленных количествах. Разработанный способ проводят не больше, чем в три стадии, условия реакции которых являются гибкими и которые отличаются высокими скоростями превращений.
Он дополнительно имеет преимущества получения 2HBL, который можно легко выделить и очистить и который не образует солей в избытке.
Поэтому этот способ представляет собой действительное решение для промышленного синтеза 2HBL путем устранения всех трудностей, с которыми сталкиваются вышеупомянутые известные способы.
Таким образом, первой целью изобретения является способ получения 2HBL из соединения, или из его соли, или из его олигомеров, причем указанное соединение соответствует формуле (I)
CH3-S-CH2CH2CR1R2R3,
где
R1 представляет собой H,
R2 представляет собой группу, выбранную из ОН; OR4 и OCOR4, где R4 представляет собой группу, выбранную из линейных, циклических, эпициклических или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, и арильных групп, имеющих от 6 до 10 атомов углерода, возможно замещенных заместителем(лями), выбранным(и) из линейных или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, галогенов и гидроксильных, амино, нитро и алкокси групп, имеющие от 1 до 10 атомов углерода; и OSiRR'R”, где R, R' и R” выбраны независимо друг от друга из линейных, циклических, алициклических или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, арильных групп, имеющих от 6 до 10 атомов углерода, возможно замещенных заместителем(лями), выбранным(и) из линейных или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода или R1 и R2 вместе представляют собой =O,
R3 представляет собой COOH или COOR5 группу, где R5 представляет собой группу, выбранную из линейных, циклических, алициклических или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, бензильных групп, замещенных одним или двумя заместителями, выбранными из линейных или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, галогенов и гидроксильных, амино, нитро, алкокси групп, имеющих от 1 до 10 атомов углерода, или R3 представляет собой цианогруппу,
способ, согласно которому получают сульфоний указанного соединения, причем указанный сульфоний соответствует формуле (II)
[CH3][CH2CH2CR1R2CR3][CR6R7R8]S+ X-,
где R1, R2 и R3 имеют вышеприведенное определение, a R6 и R7 выбраны независимо друг от друга из Н, линейных, циклических, алициклических или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, и арильных групп, имеющих от 6 до 10 атомов углерода, необязательно замещенных заместителем(лями), выбранным(и) из линейных или разветвленных алкильных групп, имеющих от 1 до 6 атомов углерода, галогенидов и гидроксильных, амино, нитро и алкокси групп, имеющих от 1 до 10 атомов углерода; R8 выбран из H, линейных, циклических, алициклических или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, арильных групп, имеющих от 6 до 10 атомов углерода, необязательно замещенных заместителем(лями), выбранным(и) из линейных или разветвленных алкильных групп, имеющих от 1 до 10 атомов углерода, и притягивающих групп, в особенности групп, содержащих функциональную группу, выбранную из кислотных, сложноэфирных, циано- функциональных групп, и X представляет собой противоион, и
полученный таким образом сульфоний гидролизуют, и
2,4-гидроксимасляную кислоту или ее соль циклизируют в 2-гидроксибутиролактон.
Перед более подробным описанием изобретения ниже даны определения терминов, используемых в этом описании, и формула изобретения.
Определения
Под солью соединения формулы I подразумевается любое соединение формулы I, где водород карбоксильной группы замещен металлом, особенно щелочным металлом, щелочноземельным металлом или переходным металлом. Этот металл предпочтительно выбран из Na, Ca, Mn, Mg, Cr. Она может быть простой или сложной. Таким образом, кальциевая соль 2-гидрокси-4-метилтиомасляной кислоты может быть выбрана из солей формулы (НМТВА)n Ca, где n меняется от 2 до 10. Это понятие соли, безусловно, охватывает все смеси солей, входящих в вышеизложенное определение.
Под олигомером соединения формулы I подразумевается любой олигомер и в особенности димер, в том виде, в котором он может сосуществовать, включая следовые количества, с указанным соединением, когда последнее не используется в полностью очищенном виде.
Под получением 2-гидроксибутиролактона (2HBL) подразумевается охват всех форм 2HBL, или поодиночке, или в виде смесей, в особенности его стереоизомеров и его таутомеров. В зависимости от искомых форм, специалист в данной области выберет соответствующую форму(мы) исходной кислоты.
В объеме настоящего изобретения:
- Алкильная группа обозначает линейный, циклический, алициклический или разветвленный насыщенный углеводородный одновалентный радикал. Как показано, она имеет от 1 до 10 атомов углерода, предпочтительно от 1 до 6 атомов углерода. В качестве примеров, метильные, этильные, н-пропильные, изопропильные, н-бутильные, изобутильные, трет-бутильные, втор-бутильные, пентильные, неопентильные, н-гексильные, циклогексильные группы и т.д. входят в данное определение;
- Арильная группа обозначает ароматический углеводородный одновалентный радикал. В качестве примеров, фенильные, бензильные, толильные, нафтильные, бифенильные группы входят в данное определение.
- Алкокси группа обозначает О-алкильный радикал, где термин алкил соответствует вышеприведенному определению;
- Противоион X представляет собой частицу, которая обеспечит электрическую нейтральность сульфония формулы (II).
Этот способ включает стадии получения сульфония, гидролиза сульфония в 24 DHBA и циклизации 24 DHBA в 2HBL. Как будет показано ниже, эти стадии последовательные или сопутствующие, в зависимости от используемых реагентов.
Первая стадия состоит из получения активированной формы указанного соединения или его соли, неожиданно обнаружили, что форма сульфония может приводить к образованию 2HBL, как показано выше, в особенности в условиях, которые будут описаны позже.
В документах FR 2150605A1 и DE 2161991A1 описано получение сульфония из 2-гидрокси-4-алкилтиомасляной кислоты и в особенности из НМТВА воздействием галоидного алкила, предпочтительно в избытке, на указанную кислоту в присутствии воды, при температуре, составляющей между 10 и 100°C, и затем выделением сульфония экстракцией спиртом после удаления воды. Согласно изобретению, сульфоний можно получить этим способом или любой другой подходящей так называемой реакцией алкилирования на соединении (I) или его соли.
Для данной первой стадии реагент предпочтительно представляет собой агент формулы [CR6R7R8]X, где R6, R7 и R8 представляют собой такие, как определено выше, а X выбран из галогенов и OH, сульфатных, сульфонатных и фосфатных групп. Когда X обозначает галоген, предпочтительный агент для применения способа по изобретению в промышленном масштабе выбран из метилйодида, бромуксусной кислоты и бензилбромида. Когда X обозначает OH, агент выбран из спиртов, имеющих линейные или разветвленные цепи, имеющие от 2 до 6 атомов углерода, и предпочтительно применяется в кислой среде, например, в присутствии серной кислоты; выгодный агент, в особенности в отношении процесса изготовления, представляет собой трет-бутанол. В случае, когда агент представляет собой спирт и в особенности трет-бутанол, благоприятной реакционной средой является кислая водно-спиртовая среда.
Согласно другому альтернативному варианту, сульфоний получают воздействием на соединение агентом формулы [CR6R7R8]+X-, где [CR6R7R8]+ представляет собой карбокатион, образованный из соответствующего линейного или разветвленного алкена, имеющего от 2 до 10 атомов углерода, в присутствии кислоты. Эта кислота будет выбрана специалистом в данной области, исходя из его/ее общего знания относительно образования карбокатиона. Это предпочтительно неорганическая кислота, например серная кислота или соляная кислота.
Предпочтительно агент формулы [С(CH3)2H]+Х-, где X представляет собой HSO4 или Cl и образован из пропена в присутствии серной кислоты или соляной кислоты, соответственно. Согласно другому полезному альтернативному варианту, агент формулы [C(CH3)3]+Х-, где X представляет собой HSO4 или Cl и образован из изобутена в присутствии серной или соляной кислоты соответственно.
Конечно, можно использовать все другие агенты, приводящие к образованию сульфония. Более того, алкилирующий агент может поддерживаться.
Стадия гидролиза сульфония согласно способу по изобретению может рассматриваться во всех подходящих условиях. В качестве примера, ее проводят простым нагреванием реакционной среды непосредственно из предыдущей стадии алкилирования. Предпочтительно, чтобы pH был не слишком высоким, преимущественно он поддерживается на уровне порядка 6.
Последняя стадия способа по изобретению представляет собой циклизацию 24 DHB в 2HBL, которая может быть достигнута специалистом в данной области в условиях, например, описанных в вышеупомянутых документах AU 2004200948A и WO 2008/022953A1.
По меньшей мере две или даже все стадии образования сульфония, гидролиза сульфония в 24DHBA и циклизации 24 DHBA могут являться одновременными. Условия нагревания до температуры, меняющейся от 30 до 150°C, предпочтительно от 60 до 100°C, являются достаточными. Дополнительно наблюдают, что добавлением галогенидных солей, таких как NaBr, возможно увеличивать реакционную способность и избирательность этих реакций.
Как показано ранее, образованный таким образом 2HBL можно легко очистить и выделить из реакционной среды. Примеры, которые следуют, проиллюстрируют это, но специалист в данной области, вместе с тем, обратится к своему общему знанию в данной области о порядке осуществления действия. Таким образом, все хорошо известные методики декантации, дистилляции и т.д. могут применяться.
Настоящее изобретение и его преимущества проиллюстрированы в следующих примерах.
Ниже в экспериментальной части ТТ обозначает скорость превращения, RR - выход по реагенту, SAAT88 обозначает уксусный сульфоний АТ88, SBAT88, бензилсульфоний АТ88, МНАСа, кальциевая соль АТ88, SBMHACa, бензилсульфоний МНАСа, TDE и TMR - температуры рубашки и реакционной среды соответственно, DCM, дихлорметан.
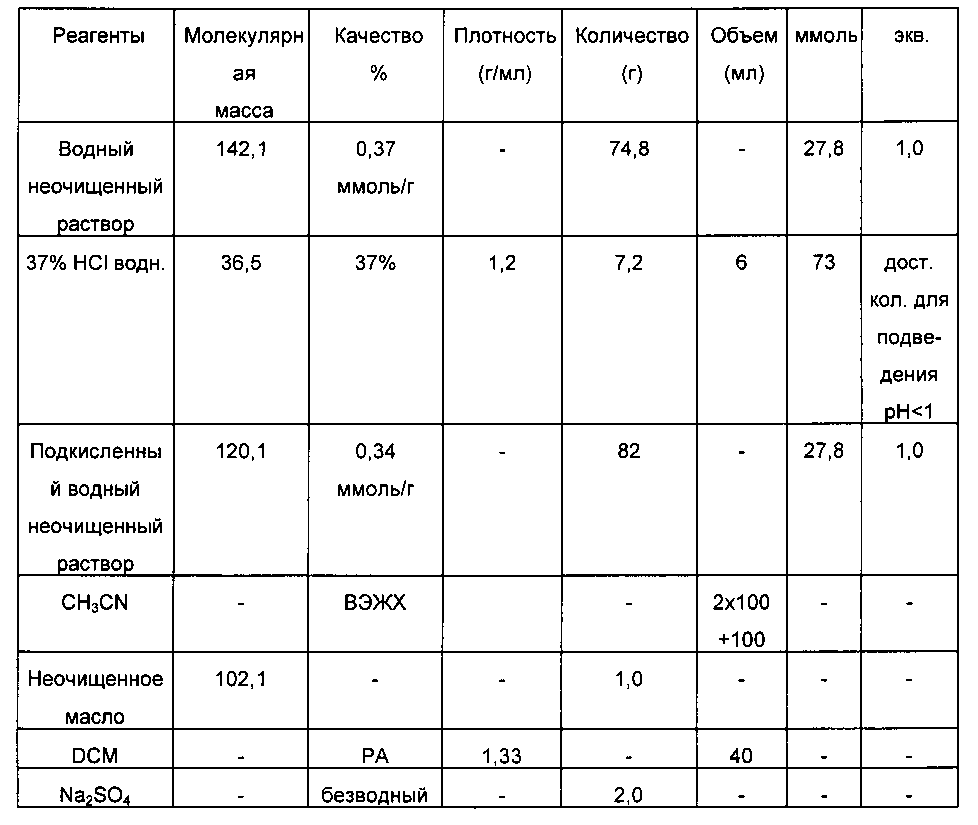
Пример 1: Получение 2HBL из НМТВА (или АТ88) через сульфоний, полученный воздействием бромуксусной кислоты
1.1. Получение 24 DHBA
Схема реакции
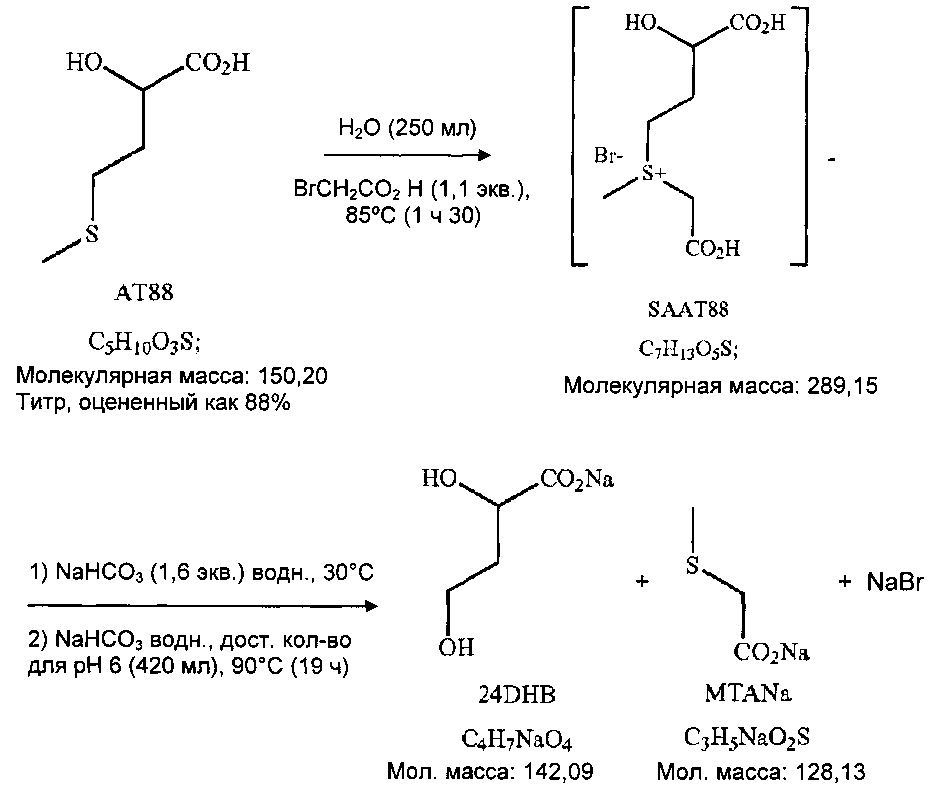
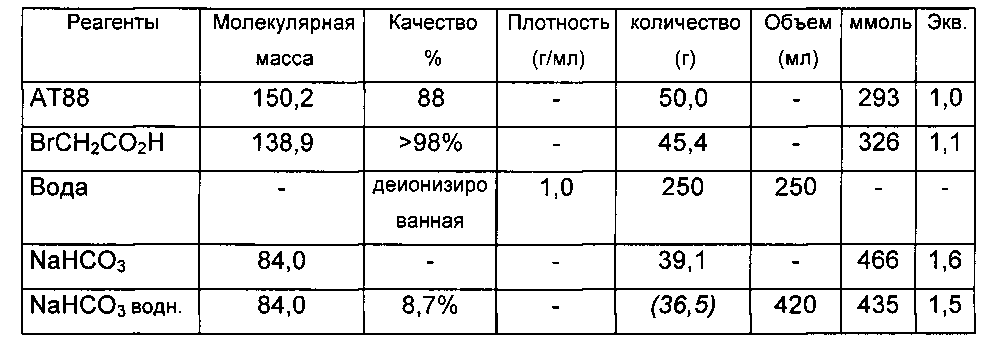
Реактивы и таблица загрузок
Действующие условия и результаты:
1.1.1. Алкилирование
В 500-мл реактор с рубашкой, оснащенный холодильником, термометром и механическим устройством для перемешивания с четырьмя наклоненными лопастями, при 20°C успешно вводят: 50 г НМТВА и 200 мл деионизированной воды (TDE=20°C). Среду перемешивают при 400 об/мин (непрозрачный раствор), и затем в течение 5 минут добавляют бромуксусную кислоту (45,4 г, 1,1 экв., не экзотермично), и промывание выполняют 50 мл деионизированной воды. Спустя 8 минут после добавления BrCH2CO2H получают светло-оранжевую прозрачную среду (перемешивание при 400 об/мин), не полностью экзотермично (TMR=20°C, TDE=20°C 10 мин после добавления бромуксусной кислоты).
Нагревание среды при перемешивании (400 об/мин) вплоть до 80-85°C (заданное значение TMR=80°C, достигаемое в течение 30 минут, TDE=95°C).
Поддержание нагревания и перемешивания в течение 1 ч 30 мин при 80-85°C (TDE=80°C (30 мин) и затем TDE=85°C (1 ч)). Получают светло-оранжевую прозрачную жидкость.
Отбор образцов для 1Н ЯМР анализа (100 мкл неочищенного раствора + 500 мкл D2O).
Критерий остановки процесса: остаточный АТ88<1 мол. % (триплет δ=2,4 млн-1, 2Н, D2O)  соответствующий результат.
соответствующий результат.
1.1.2. Гидролиз при pH 6
Предыдущую неочищенную реакционную смесь охлаждают до 30°C при перемешивании (300 об/мин, установленное значение TMR=30°C, достигаемое за 20 минут, TDE=20°C). pH пробу вводят в тот же самый реактор. Как только среда находится при 30°C, твердый NaHCO3 (39 г, 1,6 экв.) добавляют частями в течение 30 минут; сильно "замедленное" бурное вспенивание. В конце добавления измеренный pH 3,1 при 25°C. Светло-оранжевый прозрачный раствор.
Нагревание среды до 90°C (установленная величина TMR=90°C, достигаемая за 30 минут, TDE=95°C). Перемешивание при 400 об/мин.
При TMR=90°C (30 минут после начала нагревания; измеренный pH 3,0), регулирование pH начинают с установленного значения pH 6 добавлением 8,7%-ного водного раствора NaHCO3 (посредством шприцевого насоса, управляемого компьютером).
После 3 ч 30 мин регулирования количество добавленного NaHCO3 составляет 410 мл (pH 6,0). Перемешивание ослабляют до 100 об/мин, и нагревание поддерживают всю ночь (TDE=95°C, TMR=90°C).
После 19 ч регулирования pH среды составляет 6,1. Отбор образцов водной фазы (100 мкл + 500 мкл D2O) для 1Н ЯМР анализа.
Критерий остановки процесса: исчезновение характерного сигнала SAAT88 (синглет δ=2,81 млн-1 и мультиплет при δ=3,33 млн-1, D2O) соответствующий результат (SAAT88 не обнаружен).
Возвращение к 25°C в течение 1 ч 30 мин с легким помешиванием.
Получают 748 г водного неочищенного раствора
Результаты:
- TTAT88>99% (стадия 1, оценено 1Н ЯМР)
- RR24DHB=95% (проанализировано 1Н ЯМР)
- RRMTANa=95% (проанализировано 1Н ЯМР)
- Остаточный АТ88: <2 мол. % (оценено 1Н ЯМР)
1.2 Синтез и выделение 2HBL из 24 DHB
Схема реакции
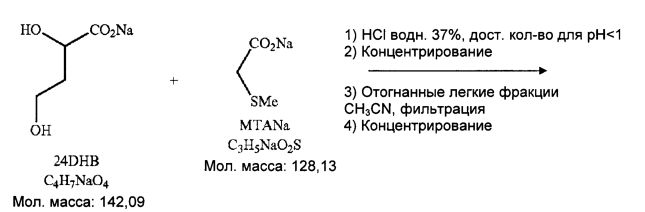
Неочищенный водный раствор
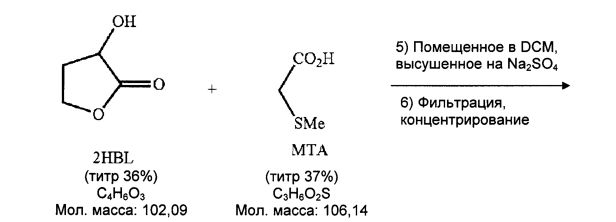
Неочищенное масло
Реагенты и таблица загрузок
Действующие условия и результаты:
а. Подкисление
В 250-мл 3-горлую колбу, обеспеченную устройством для магнитного перемешивания и оборудованную pH электродом, вводят 74,8 г водного раствора 24 DHB, и затем по каплям добавляют 37% HCl водн. до pH 0,5 (добавление 6 мл). Прозрачный светло-оранжевый раствор.
б. Концентрирование и отогнанные легкие фракции CH3CN
Предварительно полученный подкисленный раствор вводят в 250-мл колбу и концентрируют при пониженном давлении (20 мбар, 60°C). Неочищенный концентрат (масло + твердое вещество) объединяют с 100 мл ацетонитрила, и полученную суспензию затем концентрируют (60°C, 20 мбар).
Эту операцию повторяют один раз, и затем добавляют 100 мл ацетонитрила, и полученную суспензию фильтруют на фритте с пористостью No.3; соли и нерастворимые вещества промывают 2×10 мл ацетонитрила, и затем фильтрат концентрируют (17 мбар, 60°C).
Получают 6,6 г бледно-желто-оранжевого масла.
в. Высушивание на Na2SO4
В 10-мл пробирке Шотта, обеспеченной устройством для магнитного перемешивания, 1,0 г полученного ранее неочищенного масла растворяют 40 мл дихлорметана (мутный, непрозрачный раствор и присутствии незначительного количества смолистого остатка), и затем добавляют 2 г Na2SO4 при перемешивании. Перемешивание поддерживают в течение 30 минут с последующей фильтрацией на фритте с пористостью No.3 (прозрачный фильтрат); промывка солей 40 мл DCM. Фильтрат концентрируют при пониженном давлении (19 мбар, 35°C).
Получают 0,75 г бледно-желтого масла.
1Н ЯМР анализ (CDCl3)
Результаты (проанализировано 1Н ЯМР):
- RR2HBL=82%; титр 2HBL=47% (из 24 DHB)
- RRMTA=81%; титр МТА=48% (из MTANa)
Пример 2: Получение 2HBL из НМТВА (или АТ88) через сульфоний, полученный воздействием бензилбромида
2.1 Получение 24 DHBA
Схема реакции
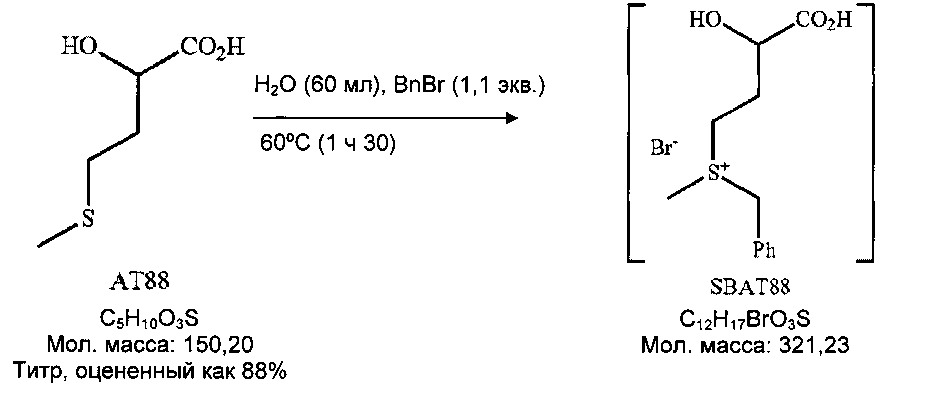
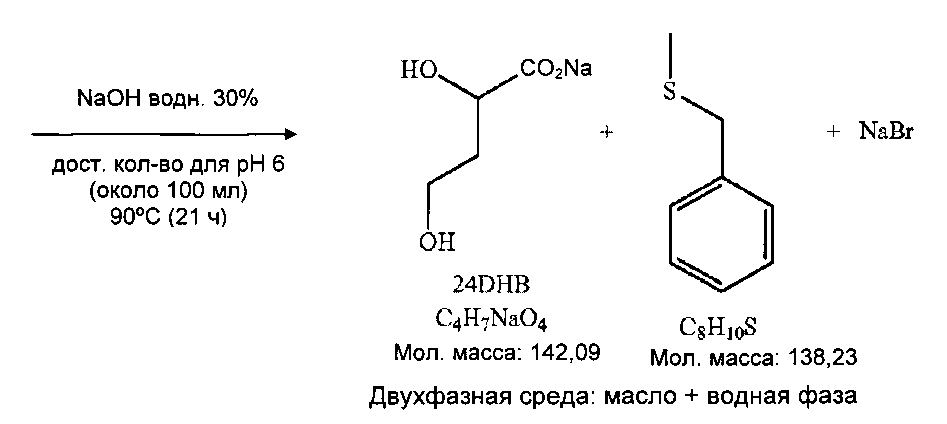
Реагенты и таблица загрузок
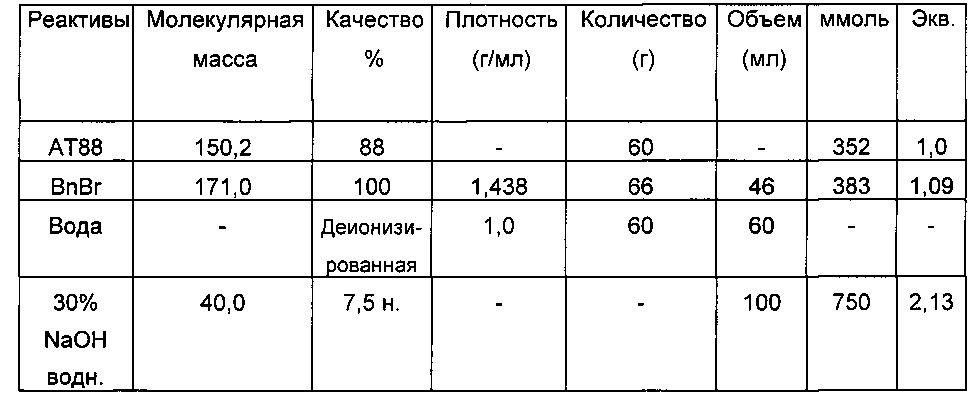
Действующие условия и результаты:
1. Алкилирование
В 250-мл реактор с рубашкой, оснащенный холодильником, термометром и 4-лопастным устройством для механического перемешивания, при 25°C последовательно вводят: 60 г АТ88 и 60 мл деионизированной воды (TDE=25°C). Среду перемешивают при 500 об/мин (бежевая эмульсия), и затем в течение 3 минут добавляют бензилбромид (46 мл, 1,1 экв.); двухфазная среда, перемешиваемая при 1000 об/мин для получения хорошо эмульгированной среды (бледно-коричневая непрозрачная среда). Добавление BnBr является экзотермическим (спустя 6 минут после добавления BnBr: TMR=36°C, TDE=25°C).
Нагревание среды при перемешивании (1000 об/мин) вплоть до 62°C (установленная величина TMR=62°C, достигаемая в течение 30 минут, TDE=65°C).
Поддержание нагревания и перемешивания в течение 1 ч 30 мин и при 62°C (TDE=65°C). Получают светло-оранжевый прозрачный раствор.
Отбор проб для 1Н ЯМР анализа (50 мкл неочищенного раствора + 500 мкл D2O).
Критерий остановки процесса: исчезновение характерного сигнала АТ88 (триплет δ=2,4 млн-1, D2O) соответствующий результат (АТ88 не обнаружен).
2. Гидролиз при pH 6
pH пробу вводят в тот же самый реактор. Нагревание среды до 90°C (установленная величина TMR=91°C, достигаемая за 30 минут, TDE=100°C и затем 93°C). Перемешивание при 700 об/мин.
При TMR=85°C (24 минуты после начала нагревания; измеренный pH -0,6), отбор образцов среды выполняют для 1Н ЯМР анализа (50 мкл + 500 мкл D2O) для проверки образования АТ88 перед началом регулирования подтвержденное наличие АТ88 (характерные сигналы: триплет δ=2,4 млн-1 и синглет δ=1,87 млн-1).
Регулирование pH начинают после отбора образцов с установленного значения pH 6 добавлением 30% соды (посредством шприцевого насоса, контролируемого компьютером). При pH 3,5 реакционная среда становится мутной и непрозрачной (3 минуты регулирования). После 7 минут регулирования (pH 6,0, достигнутая установленная величина) образование плавающего светло-оранжевого масла. После 1 ч регулирования значительное присутствие маслянистого супернатанта. Перемешивание при 400 об/мин.
После 5 ч регулирования добавленное количество соды почти больше не меняется (pH 6,13). Перемешивание ослабляют до 100 об/мин, и нагревание поддерживают всю ночь (TDE=93°C, TMR=90°C).
После 19 ч регулирования pH среды составляет 6,04. Отбор образцов водной фазы (50 мкл + 500 мкл D2O) и плавающего масла (30 мг + 600 мкл CDCl3) для 1Н ЯМР анализа.
Критерий остановки процесса: исчезновение характерного сигнала SBAT88 (синглет δ=2,58 млн-1, D2O) соответствующий результат (SBAT88 не определен).
Возвращение к 25°C в течение 3 ч при слабом перемешивании. Обе фазы разделяют простой декантацией и отводят.
Получают 142 г коричневого масла и 245 г водной неочищенной среды.
Результаты:
- TTAT88=100% (стадия 1, оценено 1Н ЯМР)
- RR24DHB=83% (проанализировано 1Н ЯМР)
- RRAT88=8% (проанализировано 1Н ЯМР)
2.2 Синтез и выделение 2HBL из 24 DHB
Схема реакции

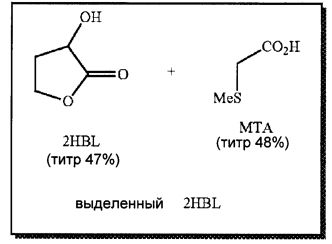
Неочищенный водный раствор
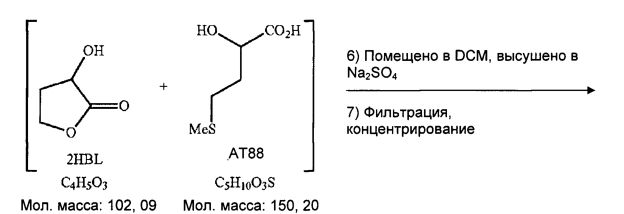
Неочищенное масло
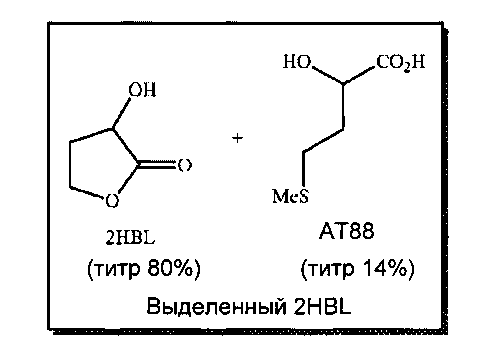
Реагенты и таблица загрузок
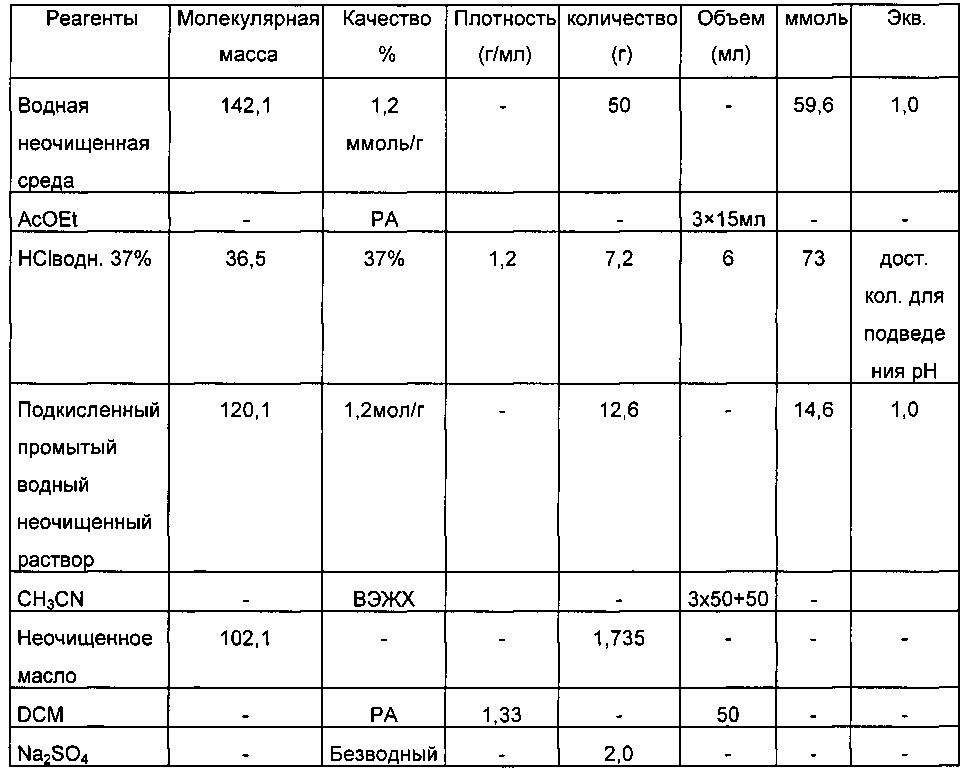
Действующие условия и результаты:
а. Промывки АсОЕТ
В 100-мл пробирку Шотта последовательно вводят 50 г неочищенной водной фазы, выделенной простой декантацией реакционной неочищенной среды от гидролиза, и затем 15 мл этилацетата.
Интенсивное перемешивание и декантация. Органическую фазу удаляют. Промывку водной фазы AcOEt повторяют дважды.
Получают 49,1 г водной неочищенной среды, прозрачный светло-оранжевый раствор.
1Н ЯМР анализ (100 мкл + 500 мкл D2O)
Критерий остановки процесса: бензильные остатки <1 мол. %, оцененные сравнением характерного бензильного сигнала (основная часть, δ=7,3 млн-1, D2O) с сигналом 24 DHB (характерный сигнал δ=3,62 млн-1) соответствующий результат (бензильные остатки <1 мол. %).
б. Подкисление
В 100-мл 3-горлую колбу, обеспеченную устройством для магнитного перемешивания и оснащенную pH электродом, вводят 49 г предшествующего водного раствора, и затем по капле добавляют 37% HCl водн. до pH -0.5 (добавление 6 мл). Прозрачный светло-оранжевый раствор.
в. Концентрирование и CH3CN отогнанные легкие фракции
12,3 г подкисленного раствора вводят в 100-мл колбу и концентрируют при пониженном давлении (20 мбар, 65°C). Концентрированный неочищенный продукт (масло + твердое вещество) объединяют с 50 мл ацетонитрила, и полученную суспензию затем концентрируют (65°C, 20 мбар). Эту операцию повторяют три раза, и затем добавляют 50 мл ацетонитрила, и полученную суспензию фильтруют на фритте с пористостью No.3, соли и нерастворимые вещества промывают 2×5 мл ацитонитрила, и фильтрат затем концентрируют (20 мбар, 65°C). Получают 1,73 г бледно-желто-оранжевого масла.
г. Высушивание на Na2SO4
В 100-мл 3-горлой колбе, обеспеченной устройством для магнитного перемешивания, 1,73 г неочищенного масла, полученного предварительно, растворяют 50 мл дихлорметана (мутный раствор, легкое образование белесоватых хлопьев, которые декантируют), и затем добавляют 2 г Na2SO4 при перемешивании.
Перемешивание поддерживают в течение 30 минут с последующей фильтрацией на фритте с пористостью No.3 (слегка мутный фильтрат); промывание солей 2×25 мл DCM. Фильтрат концентрируют при пониженном давлении (18 мбар, 35°C).
Получают 1,52 г бледно-желтое масло.
1Н ЯМР анализ (CDCl3)
Результаты (анализировано 1Н ЯМР):
- RR2HBL=81%; титр 2HBL=80%
- RRAT88=8%; титр АТ88=14%
Пример 3: Получение 2HBL из кальциевой соли НМТВА через сульфоний
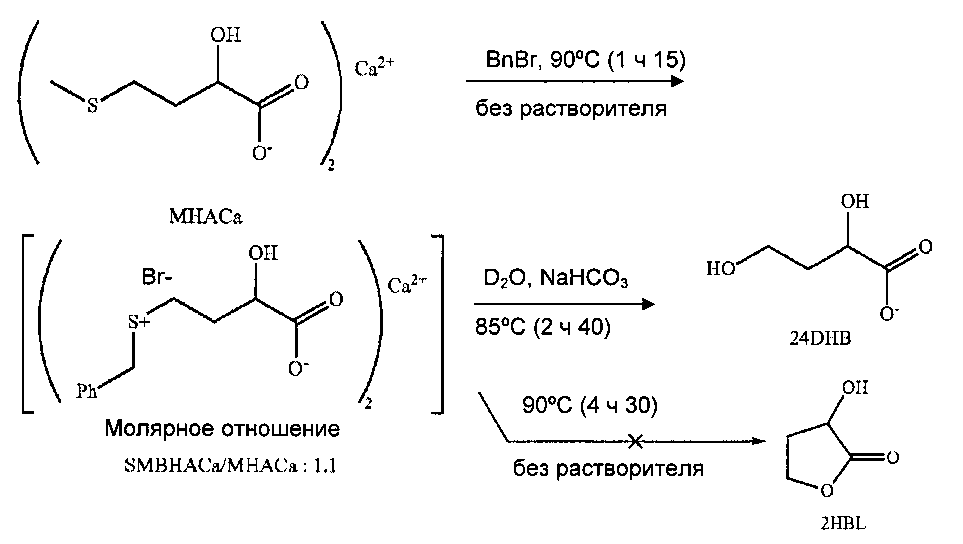
Образования 2HBL (или производных открытых форм) не наблюдается, даже после длительного нагревания до 90°C (4 ч 30 мин) неочищенного продукта алкилирования. Наоборот, воздействие добавления воды и кислого углекислого натрия на неочищенный продукт алкилирования (достаточное количество для подведения pH 8) с последующим нагреванием выражается значительным образованием 24 DHB. В проверяемых условиях, поэтому, требуется присутствие воды для замещения сульфония.
Пример 4: однореакторное получение 2HBL и влияние добавления соли (NaBr)
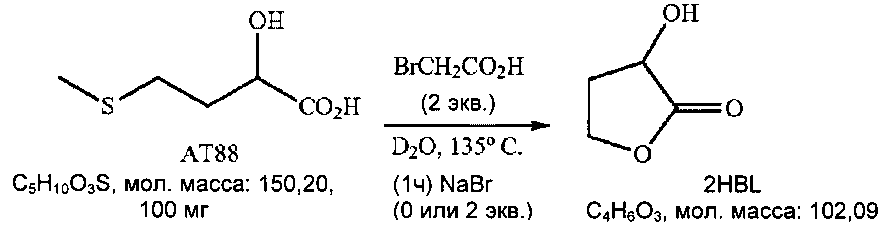
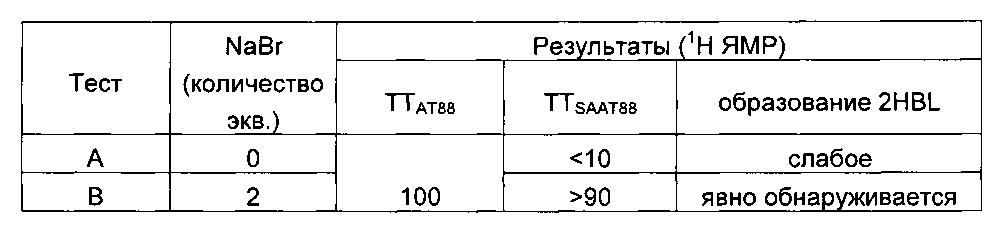
Используемые для теста В условия повторяли со следующей показателями эффективности (1Н ЯМР анализ, после 3 часов при 135°C): полная TTSAAT88, RR2HBL=33-39%.
Пример 5: Получение метилсульфония НМВА
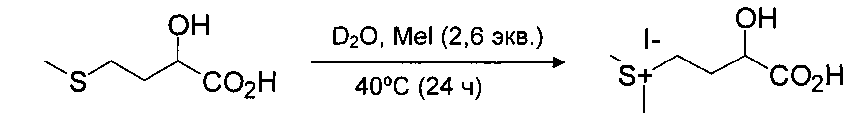
Действующие условия: 5-мл мини-реактор; при 25°C, введение АТ88 (644 мг), D2O (3 мл), Mel (478 мкл) и затем нагревание до 40°C в течение 24 часов. Концентрирование всего количества реакционной среды (18 мбар, 65°C).
Результаты (анализировано 1Н ЯМР): ТТ АТ88>95%,
RR выделенный(сульфоний)=89%, титр (сульфония)=70%
Пример 6: Получение НМВА трет-бутилсульфония
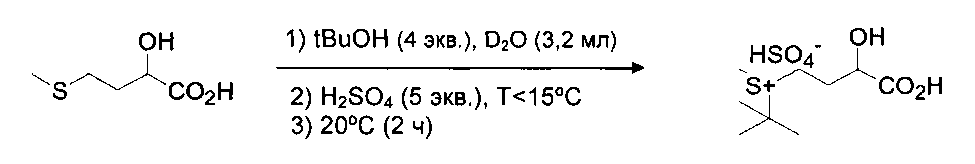
Действующие условия: 30-мл пробирка Шотта; при 25°C, введение АТ88 (3,4 г), D2O (3,2 мл), tBuOH (7,64 мл) и затем охлаждение до 10°C и добавление H2SO4 (2,5 мл, 5 экв.) в течение 20 минут поддержанием температуры Т<15°C; возвращение к 20°C после окончания добавления кислоты и 20°C поддерживают в течение 2 ч.
Результаты (оценено 1Н ЯМР): полная ТТ АТ88, RR анализированный(сульфоний)>90%.
Термостабильная композиция для животных, содержащая смесь ферментов
Способ получения акролеина из глицерола или глицерина
Способ получения аминокислоты из 2-аминобутиролактона
Способ получения комплекса кислоты и металла
Способ получения 2,4-дигидроксимасляной кислоты
Способ получения акролеина из глицерина
Способ получения 2,4-дигидроксибутирата
Способ ферментативного получения l-метионина из о-фосфо-l-гомосерина и метантиола
Способ получения олефина посредством каталитической конверсии по меньшей мере одного спирта
Способ получения метионина
Термостабильная композиция для животных, содержащая смесь ферментов
Способ получения акролеина из глицерола или глицерина
Способ получения аминокислоты из 2-аминобутиролактона
Способ получения комплекса кислоты и металла
Способ получения 2,4-дигидроксимасляной кислоты
Способ получения акролеина из глицерина
Способ получения 2,4-дигидроксибутирата

