Результат интеллектуальной деятельности: ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к производному пиразино[2,3-d]изоксазола, которое является пригодным в качестве промежуточного соединения и т.п. для получения 6-фтор-3-гидрокси-2-пиразинкарбоксамида (далее обозначаемого как “T-705"), пригодного для лечения, такого как профилактика и терапия инфекции вирусом гриппа, и к способу его получения. Кроме того, настоящее изобретение относится к способу получения производного пиразинкарбонитрила и производного пиразинкарбоксамида с использованием производного пиразино[2,3-d]изоксазола.
Уровень техники
T-705 представляет собой соединение, пригодное для профилактики, лечения и т.п. вирусной инфекции и, в частности, инфекции вирусом гриппа. Известно, что T-705 получают, например, из 6-фтор-3-гидрокси-2-пиразинкарбонитрила (далее называемого T-705A) (патентные документы 1 и 2). В патентном документе 2 описано, что T-705A можно эффективно выделять в форме солей с различными аминами.
Примеры известного способа получения T-705A включают: (1) способ, включающий обеспечение реакции 3,6-дифтор-2-пиразинкарбонитрила с бензиловым спиртом, а затем дебензилирование продукта реакции; (2) способ, включающий обеспечение реакции 3,6-дифтор-2-пиразинкарбонитрила с водой; и (3) способ, включающий обеспечение реакции 3,6-дифтор-2-пиразинкарбонитрила с карбоксилатом, а затем получение T-705A гидролизом (патентные документы 1 и 2).
Однако поскольку 3,6-дифтор-2-пиразинкарбонитрил в высокой степени раздражает кожу и легко испаряется вследствие того, что он представляет собой низкомолекулярную жидкость, с его производством связаны проблемы, которые требуют специализированного оборудования и осторожного обращения.
Более того, что касается синтеза пиразино[2,3-d]изоксазола, имеющего карбонильную группу в положении 3, известны примеры, описанные в непатентных документах 1 и 2. Однако пиразино[2,3-d]изоксазол по настоящему изобретению нельзя синтезировать такими способами синтеза.
Документы уровня техники
Патентные документы
Патентный документ 1: международная публикация WO 01/60834
Патентный документ 2: международная публикация WO 09/41473
Непатентные документы
Непатентный документ 1: Journal of Organic Chemistry. 1972, Vol. 37, #15, pp. 2498-2502
Непатентный документ 2: Journal of Organic Chemistry. 1988. Vol. 53. #9, pp. 2052-2055
Сущность изобретения
Задача, решаемая с помощью изобретения
Задачей настоящего изобретения является предоставление промежуточного соединения для получения T-705 и способа его получения, который обеспечивает высокую безопасность и простоту обращения, и, кроме того, предоставление безопасного и легкого способа получения T-705 и т.п.
Средства для решения задачи
Таким образом, настоящее изобретение относится к следующим [1]-[15].
[1] Производное пиразино[2,3-d]изоксазола, соответствующее следующей формуле (I):
[Химическая формула 1]
формула (I)
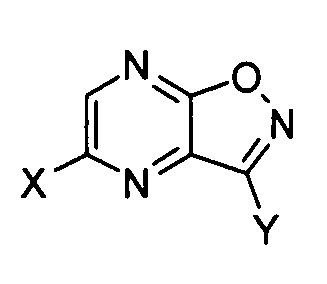
где X обозначает атом галогена, гидроксильную группу или сульфамоилоксигруппу, и Y обозначает -C(=O)R или -CN; где R обозначает атом водорода, алкоксигруппу, арилоксигруппу, алкильную группу, арильную группу или аминогруппу; где сульфамоилоксигруппа, алкоксигруппа, арилоксигруппа, алкильная группа, арильная группа и аминогруппа могут быть необязательно замещенными.
[2] Производное пиразино[2,3-d]изоксазола согласно [1], где Y обозначает -C(=O)R, где R обозначает алкоксигруппу или аминогруппу, и алкоксигруппа и аминогруппа могут быть необязательно замещенными.
[3] Производное пиразино[2,3-d]изоксазола согласно [1] или [2], где X обозначает гидроксильную группу, атом хлора или атом фтора.
[4] Производное пиразино[2,3-d]изоксазола согласно [1], где X обозначает атом фтора или атом хлора, и Y обозначает -C(=O)R, где R обозначает необязательно замещенную алкоксигруппу.
[5] Производное пиразино[2,3-d]изоксазола согласно [1], где X обозначает атом фтора или атом хлора, и Y обозначает -C(=O)R, где R обозначает метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу или н-бутоксигруппу.
[6] Способ получения производного пиразино[2,3-d]изоксазола, соответствующего следующей формуле (I-1):
[Химическая формула 3]
формула (I-1)
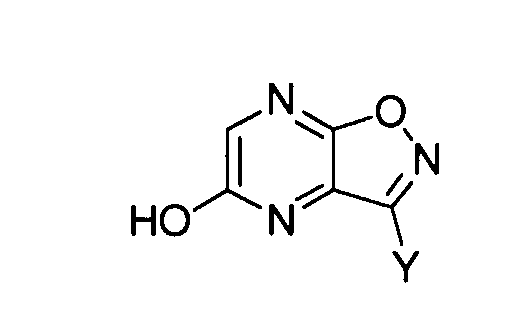
где Y имеет такое же значение, как описано ниже,
который включает обработку кислотой производного изоксазола, соответствующего следующей формуле (II):
[Химическая формула 2]
формула (II)
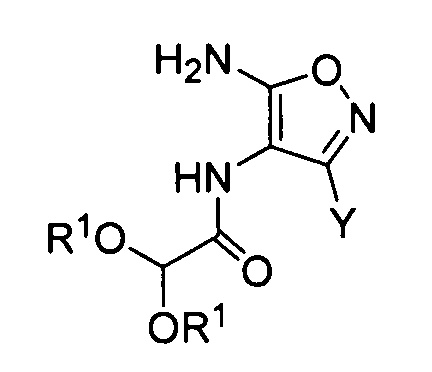
где Y обозначает -C(=O)R или -CN; где R обозначает атом водорода, алкоксигруппу, арилоксигруппу, алкильную группу, арильную группу или аминогруппу; и R1 обозначает атом водорода или алкильную группу; где алкоксигруппа, арилоксигруппа, алкильная группа, арильная группа и аминогруппа могут быть необязательно замещенными.
[7] Способ получения производного пиразинкарбонитрила, соответствующего следующей формуле (III):
[Химическая формула 5]
формула (III)
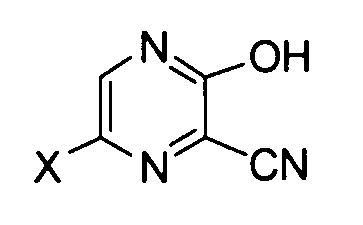
где X имеет те же значения, как описано ниже,
который включает обработку основанием производного пиразино[2,3-d]изоксазола, соответствующего следующей формуле (I):
[Химическая формула 4]
формула (I)
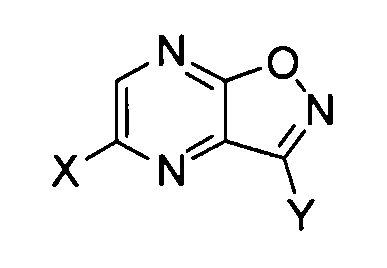
где X обозначает атом галогена, гидроксильную группу или сульфамоилоксигруппу, и Y обозначает -C(=O)R или -CN; где R обозначает атом водорода, алкоксигруппу, арилоксигруппу, алкильную группу, арильную группу или аминогруппу; где сульфамоилоксигруппа, алкоксигруппа, арилоксигруппа, алкильная группа, арильная группа и аминогруппа могут быть необязательно замещенными.
[8] Способ получения соединения, соответствующего следующей формуле (IV):
[Химическая формула 8]
формула (IV)
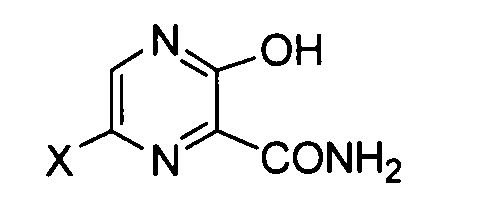
где X имеет те же значения, как описано ниже,
который включает стадию обработки основанием производного пиразино[2,3-d]изоксазола, соответствующего следующей формуле (I):
[Химическая формула 6]
формула (I)
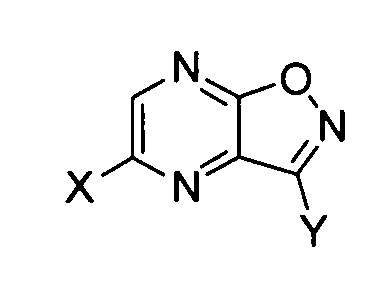
где X представляет собой атом галогена, гидроксильную группу или сульфамоилоксигруппу, и
Y обозначает -C(=O)R или -CN; где R обозначает атом водорода, алкоксигруппу, арилоксигруппу, алкильную группу, арильную группу или аминогруппу; где сульфамоилоксигруппа, алкоксигруппа, арилоксигруппа, алкильная группа, арильная группа и аминогруппа могут быть необязательно замещенными.
так чтобы получить соединение, соответствующее следующей формуле (III):
[Химическая формула 7]
формула (III)
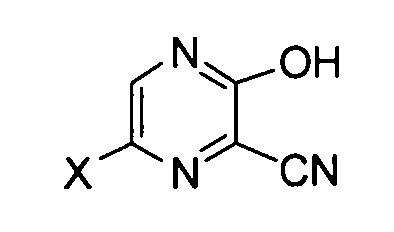
где X имеет те же значения, как описано выше,
и стадию добавления воды к соединению, соответствующему формуле (III).
[9] Способ получения согласно [7] или [8], где X обозначает атом фтора, и Y обозначает -C(=O)R, где R обозначает необязательно замещенную алкоксигруппу.
[10] Способ получения по п.[7] или [8], где X обозначает атом фтора, и Y обозначает -C(=O)R, где R обозначает метоксигруппу, этоксигруппу н-пропоксигруппу, изопропоксигруппу или н-бутоксигруппу.
[11] Соединение, соответствующее следующей формуле (C-2):
[Химическая формула 9]
формула (C-2)
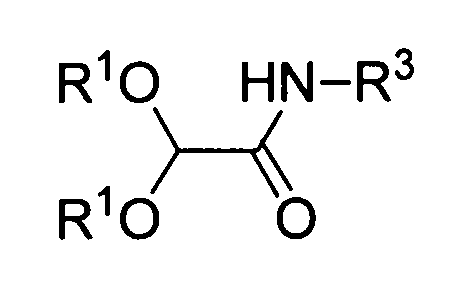
где R1 обозначает алкильную группу, R3 обозначает -CH2CN, соответствующее следующей формуле (C-2a):
[Химическая формула 10]
формула (C-2a)
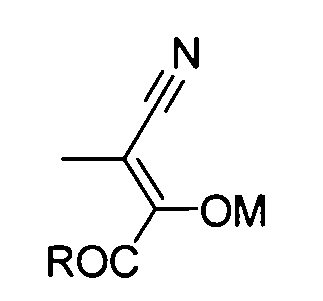
или следующей формуле (C-2b)
[Химическая формула 11]
формула (C-2b)
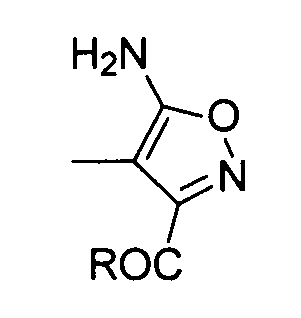
где R обозначает алкоксигруппу, M обозначает H, Li, K или Na; где алкокси и алкильная группа могут быть необязательно замещенными.
[12] Способ получения производного пиразино[2,3-d]изоксазола, соответствующего следующей формуле (J-4):
[Химическая формула 13]
формула (J-4)
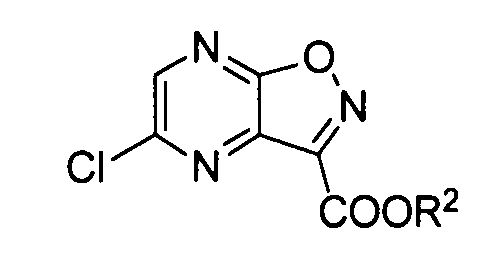
где R2 имеет те же значения, как описано ниже,
который включает обеспечение реакции соединения, соответствующего следующей формуле (J-3):
[Химическая формула 12]
формула (J-3)
где R2 обозначает алкильную группу или арильную группу; где алкильная группа и арильная группа могут быть необязательно замещенными,
с хлорирующим агентом.
[13] Способ получения производного пиразино[2,3-d]изоксазола, соответствующего следующей формуле (J-5):
[Химическая формула 15]
формула (J-5)
где R2 обозначает алкильную группу или арильную группу; где алкильная группа и арильная группа могут быть необязательно замещенными,
который включает обеспечение реакции производного пиразино[2,3-d]изоксазола, соответствующего следующей формуле (J-4):
[Химическая формула 14]
формула (J-4)
где R2 обозначает алкильную группу или арильную группу; где алкильная группа и арильная группа могут быть необязательно замещенными,
с фторирующим агентом в присутствии 2,4-динитрохлорбензола или 2,4-динитрофторбензола.
[14] Соединение, соответствующее следующей формуле (J-1):
[Химическая формула 16]
формула (J-1)
где R2 обозначает алкильную группу или арильную группу; где алкильная группа и арильная группа могут быть необязательно замещенными.
[15] Соединение, соответствующее следующей формуле (J-2a):
[Химическая формула 17]
формула (J-2a)
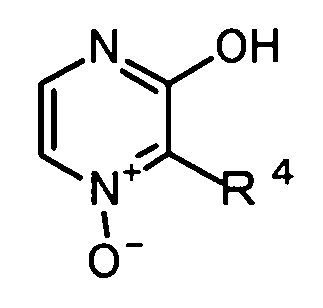
где R4 обозначает -CH2COOR2, или следующей формуле (J-2b):
[Химическая формула 18]
формула (J-2b)
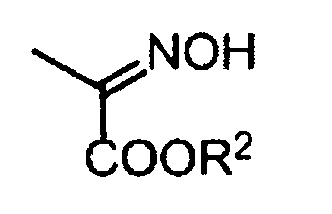
где R2 обозначает алкильную группу или арильную группу; где алкильная группа и арильная группа могут быть необязательно замещенными.
Соединение формулы (I-1), соединение формулы (III), соединение формулы (IV), соединение формулы (J-2) и соединение формулы (J-3) могут существовать в качестве таутомера. Настоящее изобретение включает эти таутомеры. Кроме того, в рамках настоящего изобретения можно использовать гидраты, сольваты и все кристаллические формы.
Также соединения, описанные в настоящем описании, могут образовывать соль.
Соли в таком случае могут включать, например, широко известные соли, образующиеся на основной группе, такой как аминогруппа, или образующиеся на кислотной группе, такой как гидроксильная группа или карбоксильная группа.
Соли, образующиеся на основной группе, могут включать, например, соль, образующуюся с минеральной кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, азотная кислота и серная кислота; соль с органической карбоновой кислотой, такой как муравьиная кислота, уксусная кислота, лимонная кислота, щавелевая кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, яблочная кислота, виннокаменная кислота, аспарагиновая кислота, трихлоруксусная кислота и трифторуксусная кислота; и соли с сульфоновой кислотой, такой как метансульфоновая кислота, бензолсульфоновый кислота, п-толуолсульфоновая кислота, меситиленсульфоновая кислота и нафталинсульфоновая кислота.
Соли, образующиеся на кислотной группе, могут включать, например, соли, образующиеся со щелочными металлами, такими как натрий и калий; соли со щелочноземельными металлами, такими как кальций и магний; соли аммония; и соли, образующиеся с азотсодержащими органическими основаниями, такие как триметиламин, триэтиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, диэтиламин, дициклогексиламин, прокаин, дибензиламин, N-бензил-β-фенэтиламин, 1-эфенамин и N,N'-дибензилэтилендиамин.
Среди вышеупомянутых солей предпочтительные соли включают фармакологически приемлемые соли.
Эффект изобретения
В соответствии с настоящим изобретением T-705 и т.п. можно получать безопасно и без труда.
Способ осуществления изобретения
Описано соединение, соответствующее формуле (I).
В соединении, соответствующем формуле (I), X обозначает атом галогена, гидроксильную группу или сульфамоилоксигруппу. Когда X обозначает атом галогена, примеры атома галогена включают атом фтора, атом хлора, атом брома и атом йода. Когда X обозначает сульфамоилоксигруппу, атом азота сульфамоилоксигруппы может быть замещен гидроксильной группой, аминогруппой, алкильной группой, арильной группой, гетероциклической группой или алкиленовой группой с гетероатомом в качестве посредника или без него. Заместитель на атоме азота содержит предпочтительно 0-10, более предпочтительно 2-8 и наиболее предпочтительно 2-6 атомов углерода. Такая группа, кроме того, может иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, приведенные в группе заместителей A, как описано ниже. Примеры сульфамоилоксигруппы, которая может быть необязательно замещенной, включают сульфамоилоксигруппу, N,N-диметилсульфамоилоксигруппу, N,N-диэтилсульфамоилоксигруппу и морфолиносульфонилоксигруппу.
X обозначает предпочтительно атом фтора, атом хлора, атом брома или гидроксильную группу, более предпочтительно атом фтора, атом хлора или гидроксильную группу и наиболее предпочтительно атом фтора.
Y обозначает -C(=O)R или -CN. В рамках настоящего изобретения R обозначает атом водорода, алкоксигруппу, арилоксигруппу, алкильную группу, арильную группу или аминогруппу. Когда R обозначает алкоксигруппу, он предпочтительно представляет собой линейную, разветвленную или циклическую алкоксигруппу, содержащую от 1 до 10 атомов углерода. Алкоксигруппа более предпочтительно содержит 1-8 и наиболее предпочтительно 1-6 атомов углерода. Алкоксигруппа может дополнительно иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры алкоксигруппы, которая может быть необязательно замещенной, включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, 2-метоксиэтоксигруппу, н-бутоксигруппу, изобутоксигруппу, трет-бутоксигруппу, изоамилоксигруппу, н-амилоксигруппу, неопентилоксигруппу, н-гексилоксигруппу, циклогексилоксигруппу, бензилоксигруппу и 2-этилгексилоксигруппу.
Когда R обозначает арилоксигруппу, предпочтительной является арилоксигруппа, содержащая 6-12 атомов углерода, более предпочтительной является арилоксигруппа, содержащая 6-10 атомов углерода, и наиболее предпочтительной является арилоксигруппа, содержащая 6-8 атомов углерода. Кроме того, арилоксигруппа может иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры арилоксигруппы, которая может быть необязательно замещенной, включают феноксигруппу, 4-метоксифеноксигруппу, 4-диметиламинофеноксигруппу, 3-метилфеноксигруппу, 2,6-диметилфеноксигруппу и 4-трет-амилфеноксигруппу.
Когда R обозначает алкильную группу, он предпочтительно представляет собой линейную, разветвленную или циклическую алкильную группу, содержащую 1-10 атомов углерода. Более предпочтительно алкильная группа содержит 1-8 и наиболее предпочтительно 1-6 атомов углерода. Кроме того, алкильная группа может иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры алкильной группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, трет-бутильную группу, изобутильную группу, н-бутильную группу, н-пентильную группу, циклопентильную группу, циклогексильную группу и 1-этилпропильную группу.
Когда R обозначает арильную группу, он предпочтительно представляет собой арильную группу, содержащую предпочтительно 6-12, более предпочтительно 6-10 и наиболее предпочтительно 6-8 атомов углерода. Кроме того, арильная группа может иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры арильной группы, которая может быть необязательно замещенной, включают фенильную группу, 4-хлорфенильную группу, метоксифенильную группу, 3,4-диметилфенильную группу и 4-фторфенильную группу.
Когда R обозначает аминогруппу, аминогруппа может быть замещена гидроксильной группой, аминогруппой, алкильной группой, арильной группой, гетероциклической группой или алкиленовой группой с гетероатомом в качестве посредника или без него. Заместитель на аминогруппе содержит предпочтительно 0-10, более предпочтительно 2-8, и наиболее предпочтительно 2-6 атомов углерода. Кроме того, заместитель может иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры аминогруппы, которая может быть необязательно замещенной, включают аминогруппу, N,N-диметиламиногруппу, N,N-диэтиламиногруппу, N,N-диизопропиламиногруппу, N,N-дипропиламиногруппу, морфолиногруппу, пиперидиногруппу, 4-метилпиперазиногруппу, пирролидиногруппу и N-метил-N-фениламиногруппу.
Y предпочтительно обозначает -C(=O)R, где R представляет собой алкоксигруппу.
Группа заместителей A: алкильная группа, содержащая 1-10 атомов углерода, алкенильная группа, содержащая 2-10 атомов углерода, алкинильная группа, содержащая 2-10 атомов углерода, алкоксигруппа, содержащая 1-10 атомов углерода, арилоксигруппа, содержащая 6-10 атомов углерода, атом галогена, арильная группа, содержащая 6-10 атомов углерода, гидроксильная группа, аминогруппа, ациламиногруппа, содержащая 1-10 атомов углерода, алкилсульфониламиногруппа, содержащая 1-10 атомов углерода, карбамоильная группа, содержащая 1-10 атомов углерода, сульфамоильная группа, содержащая 0-10 атомов углерода, карбоксильная группа, алкоксикарбонильная группа, содержащая 2-10 атомов углерода, ацилоксигруппа, содержащая 2-12 атомов углерода, гетероциклическая группа, цианогруппа и нитрогруппа.
Примеры алкенильной группы, содержащей 2-10 атомов углерода, включают винильную группу, аллильную группу, пропенильную группу, изопропенильную группу, бутенильную группу, изобутенильную группу, 1,3-бутадиенильную группу, пентильную группу, гексенильную группу, гептенильную группу и октенильную группу.
Примеры алкинильной группы, содержащей 2-10 атомов углерода, включают этинильную группу, пропинильную группу, бутинильную группу, пентинильную группу, гексинильную группу, гептинильную группу и октинильную группу.
Примеры ациламиногруппы, содержащей 1-12 атомов углерода, включают ацетиламиногруппу, пропиониламиногруппу, бензоиламиногруппу и нафтоиламиногруппу.
Примеры алкилсульфониламиногруппы, содержащей 1-10 атомов углерода, включают метансульфониламиногруппу, бензолсульфониламиногруппу и толуолсульфониламиногруппу.
Примеры карбамоильной группы, содержащей 1-10 атомов углерода, включают карбамоильную группу, N,N-диметилкарбамоильную группу, N,N-диэтилкарбамоильную группу и морфолинокарбонильную группу.
Примеры сульфамоильной группы, содержащей 0-10 атомов углерода, включают сульфамоильную группу, N,N-диметилсульфамоильную группу, N,N-диэтилсульфамоильную группу и морфолиносульфонильную группу.
Примеры алкоксикарбонильной группы, содержащей 2-10 атомов углерода, включают метоксикарбонильную группу, этоксикарбонильную группу, н-пропоксикарбонильную группу, изопропоксикарбонильную группу, 2-метоксиэтоксикарбонильную группу, н-бутоксикарбонильную группу, изобутоксикарбонильную группу и трет-бутоксикарбонильную группу.
Примеры ацилоксигруппы, содержащей 2-12 атомов углерода, включают ацетилоксигруппу, пропионилоксигруппу, бензоилоксигруппу и нафтоилоксигруппу.
Примеры гетероциклической группы включают пирролильную группу, пирролинильную группу, пирролидинильную группу, пиперидинильную группу, пиперанизинильную группу, имидазолильную группу, пиразолильную группу, пиридильную группу, тетрагидропиридильную группу, пиридазинильную группу, пиразинильную группу, пиримидинильную группу, тетразолильную группу, имидазолинильную группу, имидазолидинильную группу, пиразолинильную группу, пиразолидинильную группу, фурильную группу, пиранильную группу, тиенильную группу, оксазолильную группу, оксадиазолильную группу, изоксазолильную группу, морфолинильную группу, тиазолильную группу, изотиазолильную группу, тиадиазолильную группу, тиоморфолинильную группу, тиоксанильную группу, пиррол-1-ильную группу, пирролин-1-ильную группу, пирролидин-1-ильную группу, пиперидин-1-ильную группу, пиперазин-1-ильную группу, имидазол-1-ильную группу, пиразол-1-ильную группу, тетразол-1-ильную группу, имидозолин-1-ильную группу, имидазолидин-1-ильную группу, пиразолин-1-ильную группу, пиразолидин-1-ильную группу, морфолин-4-ильную группу, тиоморфолин-4-ильную группу, индолильную группу, индолинильную группу, 2-оксоиндолинильную группу, изоиндолильную группу, индолизинильную группу, бензимидазолильную группу, бензотриазолильную группу, индазолильную группу, хинолильную группу, тетрагидроквинелильную группу, тетрагидроизохинолинильную группу, хинолизинильную группу, изохинолильную группу, фталазинильную группу, нафтиридинильную группу, хиноксалинильную группу, дигидрохиноксалинильную группу, хиназолинильную группу, циннолинильную группу, хинуклидинильную группу, пирролопиридильную группу, 2,3-дигидробензопирролильную группу, бензофуранильную группу, изобензофуранильную группу, хроменильную группу, хроманильную группу, изохроманильную группу, бензо-1,3-диоксолильную группу, бензо-1,4-диоксанильную группу, 2,3-дигидробензофуранильную группу, бензотиенильную группу, 2,3-дигидробензотиенильную группу, бензоморфолинильную группу, бензоморфолинильную группу, бензотиазолильную группу, бензотиадиазолильную группу, индол-1-ильную группу, индолин-1-ильную группу, изоиндол-2-ильную группу, бензимидазол-1-ильную группу, бензотриазол-1-ильную группу, бензотриазол-2-ильную группу, индазол-1-ильную группу, бензоморфолин-4-ильную группу, тиантренильную группу, ксантенильную группу, феноксатиинильную группу, карбозолильную группу, β-карболинильную группу, фенантридинильную группу, акридинильную группу, перимидинильную группу, фенантролинильную группу, феназинильную группу, фенотиозинильную группу и феноксазинильную группу.
Примеры алкильной группы, содержащей 1-10 атомов углерода, алкоксигруппы, содержащей 1-10 атомов углерода, арилоксигруппы, содержащей 6-10 атомов углерода, атома галогена, арильной группы, содержащей 6-10 атомов углерода, и аминогруппы включают группы, описанные в отношении заместителя в описаниях формулы (I).
Заместители, включенные в группу заместителей A, могут быть далее замещены одним или несколькими заместителями, выбранными из группы заместителей A.
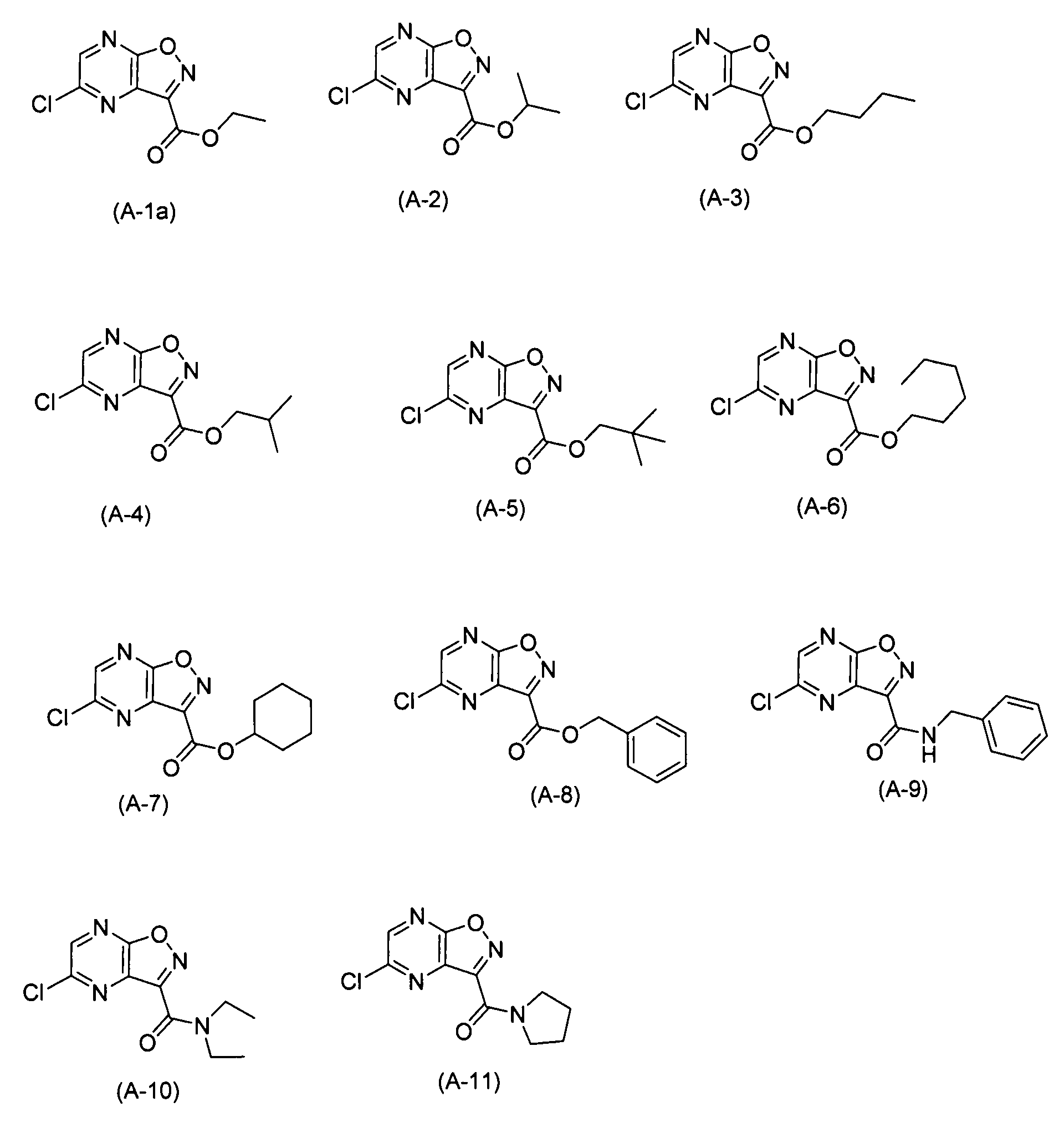
С точки зрения практической ценности соединения по настоящему изобретению в качестве промежуточного соединения для получения T-705A и T-705 предпочтительно, чтобы в формуле (I) X представлял собой атом фтора, атом хлора или гидроксильную группу, и Y представлял собой -C(=O)R, где R обозначает алкоксигруппу или аминогруппу, где алкоксигруппа и аминогруппа могут быть замещенными; более предпочтительно, чтобы X представлял собой атом фтора, атом хлора или гидроксильную группу, и Y представлял собой -C(=O)R, где R обозначает необязательно замещенную алкоксигруппу; кроме того, предпочтительно, чтобы X представлял собой атом фтора и Y представлял собой -C(=O)R, где R обозначает необязательно замещенную алкоксигруппу; и наиболее предпочтительно, чтобы X представлял собой атом фтора и Y представлял собой -C(=O)R, где R обозначает метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу или н-бутоксигруппу.
Далее описаны соединения, соответствующие формуле (II), формуле (I-1), формуле (III) и формуле (IV).
Определения и предпочтительные диапазоны X и Y в формуле (II), формуле (I-1), формуле (III) и формуле (IV) являются такими же, как и диапазоны, описанные для формулы (I).
В формуле (II) R1 обозначает атом водорода или алкильную группу, где алкильная группа может быть необязательно замещенной. Когда R1 обозначает алкильную группу, она предпочтительно представляет собой линейную, разветвленную или циклическую алкильную группу, содержащую 1-10 атомов углерода. Более предпочтительно алкильная группа содержит 1-8 и наиболее предпочтительно 1-6 атомов углерода. Кроме того, алкильная группа может иметь один или несколько заместителей. В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры алкильной группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, трет-бутильную группу, изобутильную группу, н-бутильную группу, н-пентильную группу, циклопентильную группу, циклогексильную группу и 1-этилпропильную группу. Предпочтительно R1 представляет собой метильную группу или этильную группу.
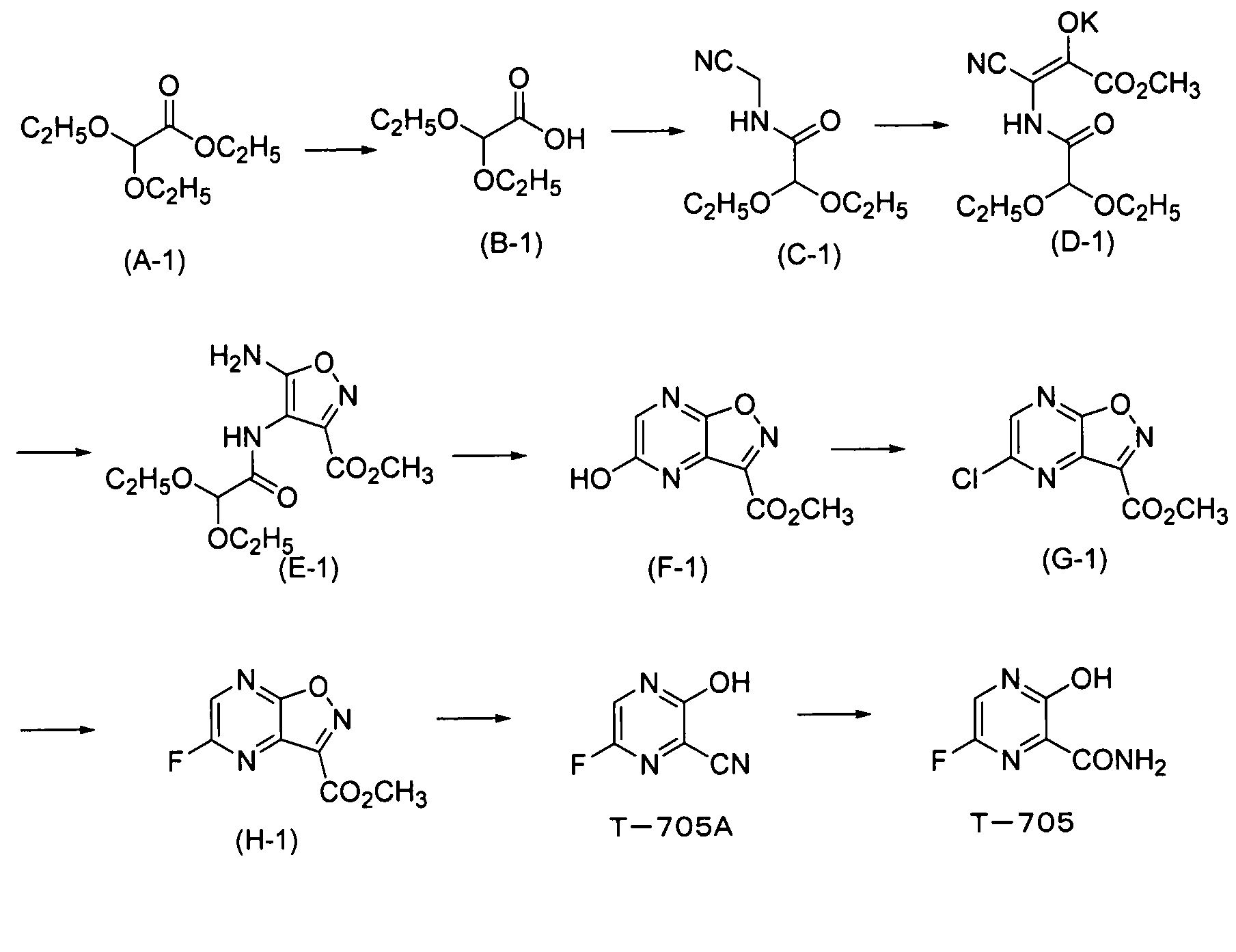
Соединение, соответствующее формуле (I), и соединение, соответствующее формуле (II), можно синтезировать по схеме, описанной ниже. В формуле, как показано ниже, определения и предпочтительные диапазоны для R и R1 являются такими же, как описано для формулы (I) или формулы (II), и M обозначает H, Li, K или Na.
[Химическая формула 19]
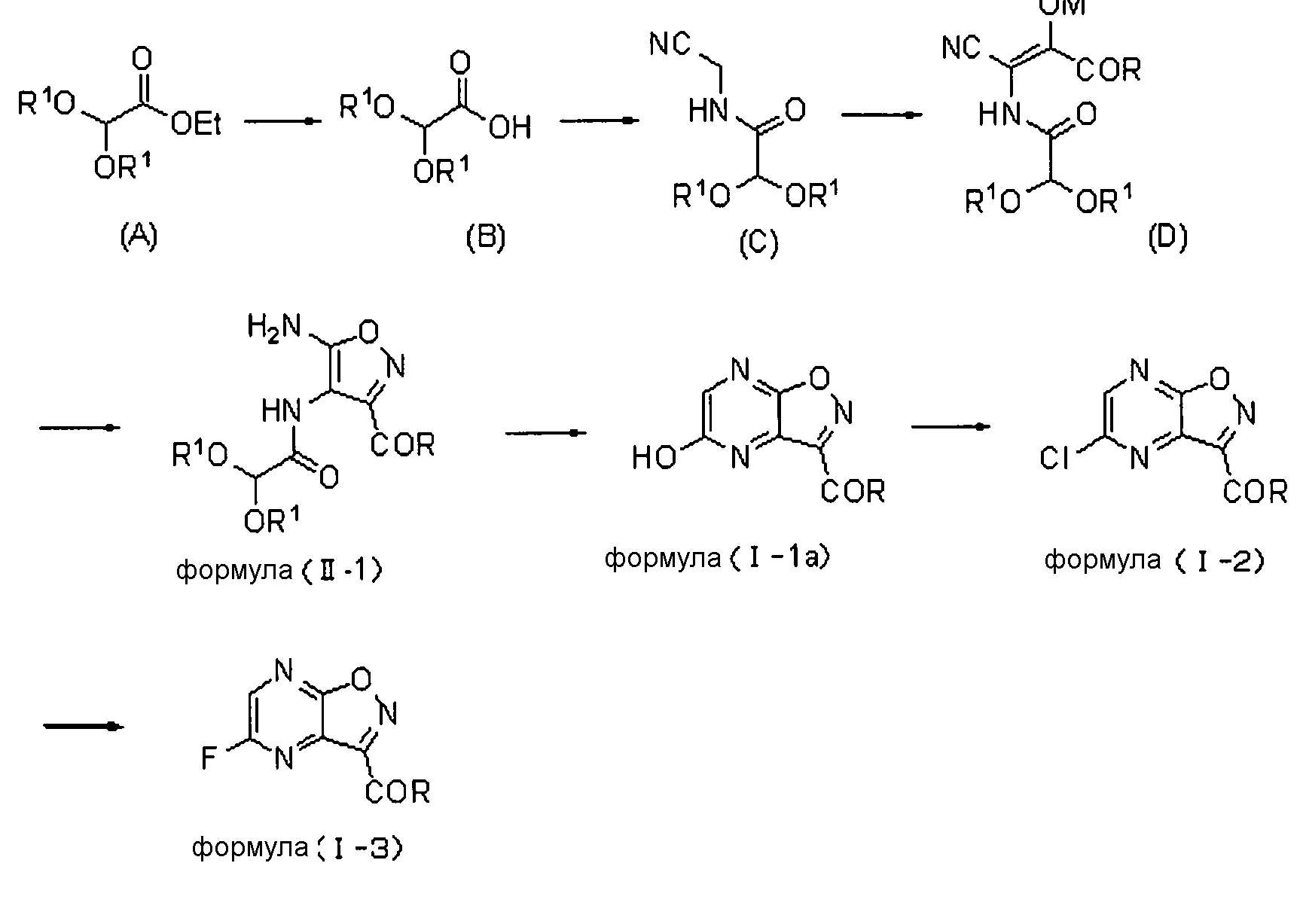
Сложный эфир уксусной кислоты (A) подвергают гидролизу с получением карбоновой кислоты (B). В этой реакции в качестве растворителей можно использовать различные типы растворителей. Главным образом, можно использовать воду или смешанный растворитель из воды и органического растворителя, смешивающегося с водой. В качестве оснований можно использовать различные типы неорганических оснований или органических оснований. Предпочтительными являются гидроксиды металлов, такие как гидроксид натрия, гидроксид лития или гидроксид калия. Предпочтительно используют температуру реакции от -20 до 100°C. Более предпочтительно температура реакции составляет от 0 до 80°C.
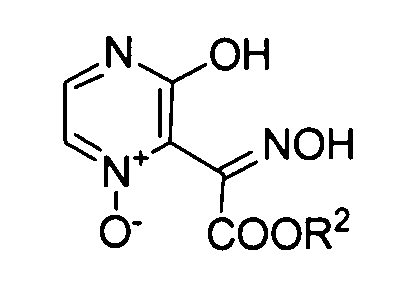
Полученной карбоновой кислоте (B) позволяют реагировать с аминоацетонитрилом в диапазоне от основного до нейтрального, так чтобы преобразовать его в амид (C). Примеры конденсирующего агента, используемого в ходе этой реакции, включают: карбодиимиды, такие как дициклогексилкарбодиимид или гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида; активаторы, такие как карбонилдиимидазол или N,N'-дисукцинимидилкарбонат; и катионные агенты дегидратации-конденсации, такие как йодид 2-хлор-1-метилпиридиния, хлорид 2-хлор-1,3-диметилимидазолиния или гексафторфосфонат хлор-N,N,N',N'-тетраметилформамидиния. Также можно использовать способ, включающий обеспечение реакции полученного соединения с галогенангидридами, такими как сложный эфир хлоркарбоновой кислоты или метансульфонилхлорид, с получением смешанного ангидрида кислоты, а затем обеспечение его реакции с ацетонитрилом. Температура реакции варьирует в зависимости от используемого конденсирующего агента, как правило, она предпочтительно составляет от -20°C до комнатной температуры. Растворитель, который можно использовать в этой реакции, конкретно не ограничивается, при условии, что он не влияет на реакцию, и его примеры включают: нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как хлороформ, метиленхлорид или 1,2-дихлорэтан; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и N-этилпирролидон; сложные эфиры, такие как этилацетат, изопропилацетат или бутилацетат; сульфоксиды, такие как диметилсульфоксид; сульфолан и тетрагидрофуран. Эти растворители можно использовать в комбинации. Также реакцию предпочтительно проводят в двухфазной системе из органического растворителя и воды.
Реакцию конденсации амида (C) в амид (D) можно проводить путем реакции амида (C) с диэфиром щавелевой кислоты или сходными с ним с использованием алкоксида металла в качестве основания в растворителе, таком как тетрагидрофуран или толуол. Температура реакции предпочтительно составляет от 0 до 60°С и более предпочтительно от 10 до 40°С. Продукт реакции, как правило, осаждают из реакционной системы в форме соли. Эту соль можно собирать фильтрацией, а затем можно использовать в последующей реакции или ее можно прямо использовать в последующей реакции без проведения специальных действий. В ином случае отфильтрованный кристалл можно нейтрализовывать и полученный продукт затем можно использовать в последующей реакции.
Преобразование амида (D) в изоксазол (формула (II-1)) можно осуществлять сначала путем реакции амида (D) с гидроксиламином с образованием оксима и проведением реакции замыкания его кольца с катализатором, таким как кислота или основание. В качестве гидроксиламина можно использовать любой из водного раствора 50% гидроксиламина, гидроксиламина гидрохлорида и гидроксиламина сульфата. В качестве растворителя предпочтительно используют диметилсульфоксид, метанол, этанол, воду или сходные с ними. Температура реакции предпочтительно составляет от 0 до 100°C и более предпочтительно от комнатной температуры до 80°C.
Соединение, соответствующее формуле (II-1), обрабатывают кислотой, так чтобы получить 5-гидроксипиразино[2,3-d]изоксазол (формула (I-1a)). Примеры кислоты, используемой в этом способе, включают протонные кислоты, такие как хлористоводородная кислота, серная кислота, п-толуолсульфоновая кислота, камфорсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота; и кислоты Льюиса, такие как хлорид алюминия, хлорид цинка или хлорид железа. Кислотами, которые предпочтительно используют в этом способе, являются протонные кислоты. Из них более предпочтительными являются хлористоводородная кислота, серная кислота и п-толуолсульфоновая кислота, и особенно предпочтительной является п-толуолсульфоновая кислота. Количество кислоты, используемой в качестве катализатора, предпочтительно в 0,0001-1000 раз, более предпочтительно в 0,001-100 раз и наиболее предпочтительно в 0,01-10 раз превышает молярное количество соединения, соответствующего формуле (II-1).
Тип растворителя, используемого в рамках настоящего изобретения, конкретно не ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают: нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол, простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля, кетоны, такие как ацетон или 2-бутанон; спирты, такие как метанол, этанол или 2-пропанол; амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; карбоновые кислоты, такие как уксусная кислота, пропионовая кислота или трифторуксусная кислота; сложные эфиры, такие как этилацетат или изопропилацетат; и сульфоксиды, такие как диметилсульфоксид. Эти растворители можно использовать в комбинации. Примеры предпочтительного растворителя включают ароматические углеводороды, простые эфиры, спирты, карбоновые кислоты, сложные эфиры и сульфоксиды. Из них более предпочтительными являются карбоновые кислоты, спирты и сложные эфиры и еще более предпочтительной является уксусная кислота. Такой растворитель может также действовать в качестве кислотного катализатора.
Используемое количество растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно в 1-50 раз (об./масс.) и более предпочтительно в 1-15 раз (об./масс.) превышающем количество соединения, соответствующего формуле (II-1).
Температура реакции, как правило, варьирует в зависимости от используемого кислотного катализатора и растворителя. Предпочтительно она составляет 200°C или ниже и более предпочтительно от 0 до 150°C. Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 50 часов, более предпочтительно от 5 минут до 24 часов и особенно предпочтительно от 5 минут до 5 часов.
В этой реакции R в соединении формулы (II-1) особенно предпочтительно представляет собой алкоксигруппу.
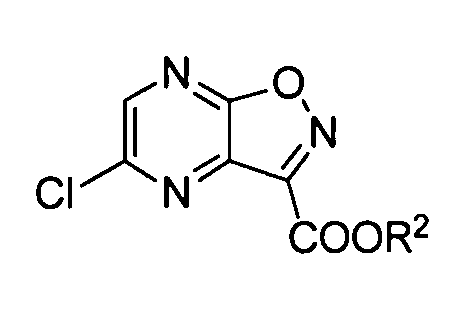
Преобразование соединения формулы (I-1a) в 5-хлорпиразин[2,3-d]изоксазол (формула (I-2)) можно осуществлять с использованием различных типов хлорирующих агентов, с растворителем или без него. Хлорирующий агент может быть выбран из фосфорилхлорида, пентахлорида фосфора, трихлорида фосфора и т.п. Когда используют растворитель, предпочтительные примеры растворителя включают N,N-диметилформамид, N,N-диметилацетамид, ацетонитрил, сульфолан, N-метилпирролидон, этилацетат и их смешанный растворитель. При необходимости также можно добавлять триэтиламин, пиридин, гидрохлорид триэтиламина и т.п. Температура реакции предпочтительно составляет от комнатной температуры до 130°C и, как правило, более предпочтительно она составляет от 50 до 110°C.
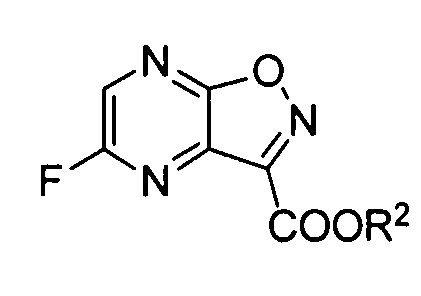
В реакции преобразования соединения формулы (I-2) в 5-фторпиразино[2,3-d]изоксазол (формула (I-3)) в качестве фторирующих агентов можно использовать различные типы фторирующих реагентов. Предпочтительные примеры фторирующего реагента включают фторид калия, фторид цезия, фторид тетра-н-бутиламмония, фторид тетраметиламмония и фторид тетрафенилфосфония. Среди них предпочтительными являются фторид калия и фторид цезия. Что касается фторида калия, особенно предпочтительным является высушенный распылительной сушкой фторид калия. Фторирующий агент добавляют в количестве, предпочтительно в 1-10 раз, более предпочтительно в 1,1-5 раз и наиболее предпочтительно в 1,1-3 раз превышающем молярное количество субстрата реакции. Также можно добавлять дегидратирующий фторирующий агент, такой как 2,2-дифтор-1,3-диметилимидазолин (DFI) или 1,1,2,3,3,3-гексафтор-1-диэтиламинопропан (реагент Ишикава). Предпочтительные примеры растворителя включают апротонные растворители, такие как ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид, сульфолан, диметилсульфоксид, N-метилпирролидон, N-этилпирролидон или тетрагидрофуран. Среди них более предпочтительными являются ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид, сульфолан и диметилсульфоксид и еще более предпочтительными являются ацетонитрил, N,N-диметилформамид и диметилсульфоксид. Используемое количество растворителя конкретно не ограничено и предпочтительно оно в 0,5-20 раз (об./масс.), более предпочтительно 1-10 раз (об./масс.) и наиболее предпочтительно 1-3 раз (об./масс.) превышает объем соединения формулы (I-2). Верхний предел температуры реакции варьирует в зависимости от температуры кипения растворителя. Как правило, предпочтительно он составляет 0-130°C, более предпочтительно от комнатной температуры до 110°C и наиболее предпочтительно от 50 до 100°C. Предпочтительно, чтобы концентрация содержащейся воды в реакционной системе была низкой. Более предпочтительно концентрация содержащейся воды составляет от 0,01 до 1000 м.д., более предпочтительно от 0,01 до 500 м.д. и наиболее предпочтительно от 0,01 до 300 м.д. Для уменьшения содержания воды в реакционной системе можно осуществлять различные типы дегидратирующих воздействий перед реакцией. Например, предпочтительно, чтобы фторирующий реагент, подлежащий применению, сушили нагреванием (80-500°C) и вакуумным отсосом (0,001-100 торр). Более того, когда используют растворитель с высокой температурой кипения (диметилсульфоксид, сульфолан, N-метилпирролидон, N,N-диметилформамид и т.д.), предпочтительно проводят азеотропную дегидратацию с использованием толуола или ксилола. Более того, предпочтительно отгонять растворитель с высокой температурой кипения при пониженном давлении, так чтобы снизить содержание воды в системе. Более того, для снижения содержания воды в системе можно добавлять молекулярные сита или сходные с ними. В этом случае предпочтительными являются молекулярные сита, которые были дегидратированы и высушены при высокой температуре. Для цели ускорения реакции предпочтительно можно использовать катионные катализаторы фазового переноса, такие как хлорид тетра-н-бутиламмония, бромид тетра-н-бутиламмония, хлорид тетрафенилфосфония, хлорид тетраметиламмония или бромид триметилбензиламмония, и неионные катализаторы фазового переноса, такие как 18-краун-6, полиэтиленгликоль 400, полиэтиленгликоль 1000 или трис(2-(2-метоксиэтокси)этил)амин. Время реакции предпочтительно составляет от 5 минут до 50 часов, более предпочтительно от 10 до 10 часов и наиболее предпочтительно от 15 минут 5 часов.
Когда соединение формулы (J-5) синтезируют из соединения формулы (J-4), как описано ниже, в реакционную смесь предпочтительно добавляют 2,4-динитрохлорбензол или 2,4-динитрофторбензол. С использованием этих добавок количество черного смолистого компонента, образующегося в результате реакции фторирования, может быть снижено, и таким образом качество соединения формулы (J-5) или, более того, соединения, полученного в последующем процессе, может быть улучшено.
[Химическая формула 20]
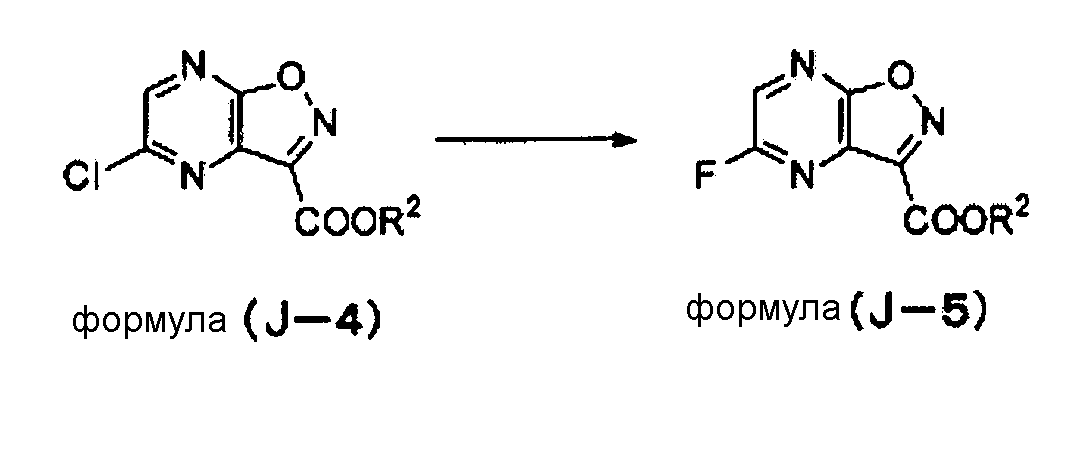
2,4-динитрохлобензол или 2,4-динитрофторбензол добавляют в количестве, предпочтительно в 0,001-10 раз, более предпочтительно в 0,01-1 раз и наиболее предпочтительно в 0,01-0,2 раз превышающем молярное количество соединения формулы (J-4).
Кроме того, можно преобразовывать соединение формулы (I-1a) в соединение формулы (I-3) без посредничества соединения формулы (I-2) в соответствии со способом обеспечения воздействия 2,2-дифтор-1,3-диметилимидазолина (DFI) на соединение формулы (I-1a) в ацетонитриле.
Кроме того, также можно преобразовывать соединение формулы (I-1a) в соединение формулы (I-3) путем преобразования группы в положении 5 соединения формулы (I-1a) в уходящую группу, такую как сульфамоилоксигруппа, в соответствии со способом обеспечения реакции соединения формулы (I-1a) с сульфамоилхлоридом в присутствии основания, а затем замещения группы в положении 5 с использованием фторида калия, фторида тетрабутиламмония или сходных с ними в качестве источника аниона фтора.
Иными словами, производное пиразино[2,3-d]изоксазола, имеющее в его 5 положении группу, поддающуюся замещению атомом фтора или группой, которую можно легко индуцировать в такую группу, также является важным в качестве промежуточного соединения для получения T-705A.
Когда Y представляет собой -C(=O)R и R представляет собой аминогруппу в формуле (I), соединение можно синтезировать способом обеспечения реакции соединения, соответствующего формуле (I), где R представляет собой алкоксигруппу, с амином. В этой реакции предпочтительно добавлять подходящее основание (например, триэтиламин, диизопропилэтиламин, пиридин, карбонат калия или бикарбонат натрия). Тип используемого растворителя в этом способе конкретно не ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля, кетоны, такие как ацетон или 2-бутанон; спирты, такие как метанол, этенол или 2-пропанол: амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; сложные эфиры, такие как этилацетат или изопропилацетат; и сульфоксиды, такие как диметилсульфоксид. Эти растворители можно использовать в комбинации. Примеры предпочтительного растворителя включают ароматические углеводороды, простые эфиры, спирты, сложные эфиры и сульфоксиды. Используемое количество растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно в 1-50 раз (об./масс.) и более предпочтительно в 1-15 раз (об./масс.) превышающем количество соединения формулы (I). Температура реакции предпочтительно составляет 200°C или ниже и более предпочтительно от 0 до 150°C. Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 50 часов, более предпочтительно от 5 минут до 24 часов и особенно предпочтительно от 5 минут до 5 часов.
Среди соединений, соответствующих формуле (I), соединение, соответствующее формуле (J-4), как показано ниже, можно синтезировать по следующей схеме, например.
[Химическая формула 21]
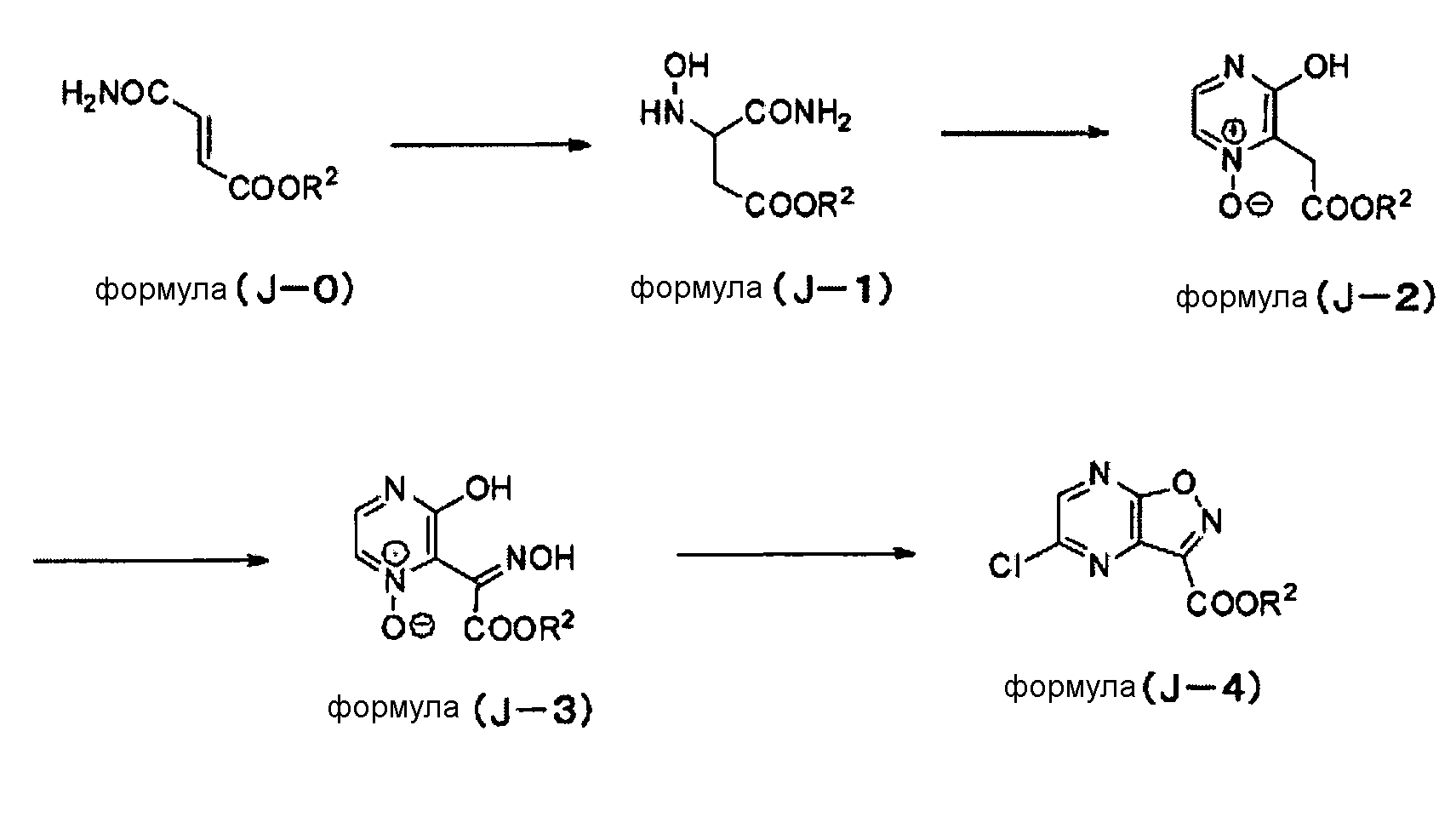
В соединениях формул (J-0)-(J-4) R2 обозначает алкильную группу или арильную группу. Алкильная группа и арильная группа могут быть необязательно замещенными. Следует отметить, что то же самое применимо к R2 в соединении описанной выше формулы (J-5).
Когда R2 обозначает алкильную группу, она предпочтительно представляет собой линейную, разветвленную или циклическую алкильную группу, содержащую 1-10 атомов углерода. Алкильная группа более предпочтительно содержит 1-8 и наиболее предпочтительно 1-6 атомов углерода. Кроме того, алкильная группа может иметь заместитель(и). В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры алкильной группы, которая может быть необязательно замещенной, включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, 2-метоксиэтильную группу, трет-бутильную группу, изобутильную группу, н-бутильную группу, изоамильную группу, н-амильную группу, неопентильную группу, н-гексильную группу, циклогексильную группу, бензильную группу и 2-этилгексильную группу.
Когда R2 обозначает арильную группу, она предпочтительно представляет собой арильную группу, содержащую предпочтительно 6-12, более предпочтительно 6-10и наиболее предпочтительно 6-8 атомов углерода. Кроме того, арильная группа может иметь заместитель(и). В качестве таких заместителей предпочтительными являются заместители, перечисленные в группе заместителей A. Примеры арильной группы, которая может быть необязательно замещенной, включают фенильную группу, 4-метоксифенильную группу, 4-диметиламинофенильную группу, 3-метилфенильную группу, 2,6-диметилфенильную группу и 4-трет-аминофенильную группу.
В рамках настоящего изобретения R2 в соединениях формул (J-0)-(J-4) предпочтительно представляет собой алкильную группу, содержащую 1-6 атомов углерода. Особенно предпочтительными являются метильная группа, этильная группа, н-пропильная группа, изопропильная группа, н-бутильная группа, изобутильная группа, втор-бутильная группа и трет-бутильная группа.
В соединении формулы (J-3) гидроксильная группа оксима может иметь структуры как анти-, так и син-изомеров. В рамках настоящего изобретения оно может представлять собой либо один изомер, либо их смесь.
Соединение формулы (J-0) можно синтезировать известным способом. Например, ангидриду малеиновой кислоты позволяют реагировать со спиртом для синтеза сложного моноэфира малеиновой кислоты. Затем индуцируют его преобразование в хлорангидрид кислоты с использованием хлорирующего агента, такого как тионилхлорид, а затем хлорангидриду кислоты позволяют реагировать с аммиаком, так чтобы преобразовать его в амидную структуру. Альтернативно описанному выше сложному моноэфиру малеиновой кислоты позволяют реагировать, например, с метансульфонилхлоридом или эфиром хлормуравьиной кислоты, так чтобы индуцировать его преобразование в смешанный ангидрид кислоты, а затем смешанному ангидриду кислоты позволяют реагировать с аммиаком, так чтобы преобразовать его в амидную структуру. В ходе этих реакций в дополнение к аммиаку можно использовать соли, такие как карбонат аммония или ацетат аммония. Более того, в качестве альтернативного способа ангидриду малеиновой кислоты позволяют реагировать с аммиаком для синтеза моноамида малеиновой кислоты, а затем ему позволяют реагировать со спиртом в присутствии кислотного катализатора, такого как концентрированная серная кислота, для этерификации его, так чтобы получить соединение формулы (J-0).
В рамках настоящего изобретения соединение формулы (J-0) может представлять собой либо цис-, либо транс-изомер.
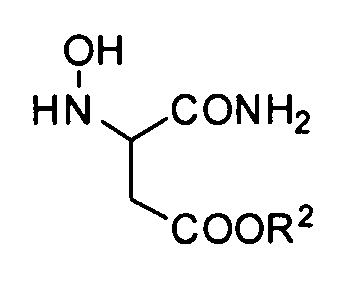
Преобразование соединения формулы (J-0) в соединение формулы (J-1) можно осуществлять путем сопряженного присоединения гидроксиламина.
В качестве гидроксиламина можно использовать 50% водный раствор гидроксиламина, гидрохлорид гидроксиламина, сульфат гидроксиламина или сходные с ними.
Когда используют гидрохлорид гидроксиламина или сульфат гидроксиламина, предпочтительно добавляют различные типы органических оснований или неорганических оснований. Примеры основания, которое можно использовать в этом способе, включают триэтиламин, пиридин, гидроксид натрия, гидроксид калия, карбонат калия, бикарбонат натрия и фосфат натрия. Основание используют в количестве, предпочтительно в 0,1-10 раз, более предпочтительно в 0,5-2 раз и наиболее предпочтительно в 1-1,2 раз превышающем количество гидроксиламина.
Гидроксиламин используют в количестве, предпочтительно в 1-10 раз, более предпочтительно в 1-2 раза и наиболее предпочтительно в 1-1,2 раз превышающем молярное количество соединения формулы (J-0).
Тип растворителя, используемого в этом способе, конкретно не ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают воду, нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля, кетоны, такие как ацетон или 2-бутанон; спирты, такие как метанол, этанол или 2-пропанол; амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; сложные эфиры, такие как этилацетат или изопропилацетат; и сульфоксиды, такие как диметилсульфоксид. Эти растворители можно использовать в комбинации. Примеры предпочтительного растворителя включают ароматические углеводороды, простые эфиры, спирты, сложные эфиры и сульфоксиды. Среди них более предпочтительными являются ароматические углеводороды, спирты и сложные эфиры.
Используемое количество растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно в 1-50 раз (об./масс.), более предпочтительно в 1-10 раз (об./масс.) и наиболее предпочтительно в 1-3 раза (об./масс.) превышающем количество соединения формулы (J-0).
Температура реакции варьирует в зависимости от используемого растворителя. Предпочтительно она составляет от 0 до 130°C, более предпочтительно от комнатной температуры до 100°C и особенно предпочтительно от комнатной температуры до 50°C.
Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 10 часов, более предпочтительно, от 10 минут до 5 часов и особенно предпочтительно от 10 минут до 1 часов.
Соединение формулы (J-1) можно выделять, а затем можно подвергать последующему процессу. В ином случае его можно подвергать последующему процессу без выделения.
Преобразование соединения формулы (J-1) в соединение формулы (J-2) можно осуществлять, позволяя соединению формулы (J-1) реагировать с глиоксалем в присутствии кислоты или основания. В качестве глиоксаля предпочтительно используют недорогостоящий 40% водный раствор глиоксаля. Также в качестве продукта, эквивалентного глиоксалю, можно использовать, например, ацетальную структуру или аддукт сульфитных ионов.
Глиоксаль используют в количестве, предпочтительно в 1-10 раз, более предпочтительно, в 1-5 раз и наиболее предпочтительно в 1-3 раза превышающем молярное количество соединения формулы (J-1).
Тип растворителя, используемого в этом способе, конкретно не ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают: воду, нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля; кетоны, такие как ацетон или 2-бутанон; спирты, такие как метанол, этанол или 2-пропанол; амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; сложные эфиры, такие как этилацетат или изопропилацетат; и сульфоксиды, такие как диметилсульфоксид. Эти растворители можно использовать в комбинации. Примеры предпочтительного растворителя включают воду, нитрилы, простые эфиры, кетоны, спирты и амиды. Из них более предпочтительными являются вода, простые эфиры и спирты и наиболее предпочтительной является вода.
Используемое количество растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно в 1-50 раз (об./масс.), более предпочтительно в 1-20 раз (об./масс.) и наиболее предпочтительно в 1-10 раз (об./масс.) превышающем количество соединения формулы (J-1).
Для повышения выхода предпочтительно добавлять кислоту или основание в данную реакцию. Примеры кислоты, используемой в этой реакции, включают протонные кислоты, такие как хлористоводородная кислота, серная кислота, п-толуолсульфоновая кислота, камфорсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота; и кислоты Льюиса, такие как хлорид алюминия, хлорид цинка или хлорид железа. Из них предпочтительными являются протонные кислоты и более предпочтительными являются хлористоводородная кислота, серная кислота и уксусная кислота.
В качестве оснований можно использовать различные типы неорганических оснований или органических оснований. Примеры предпочтительного неорганического основания включают бикарбонат натрия, карбонат калия, карбонат натрия, гидроксид лития, гидроксид натрия, гидроксид калия, фосфат калия и моногидрофосфат натрия. Примеры предпочтительного органического основания включают триэтиламин, N,N-диизопропилэтиламин, пиридин и пиколин.
Кислоту или основание используют в количестве, предпочтительно в 0,01-100 раз, более предпочтительно в 0,1-10 раз и наиболее предпочтительно в 1-5 раз превышающем молярное количество соединения формулы (J-1).
Температура реакции варьирует в зависимости от используемого растворителя. Предпочтительно она составляет от 0 до 130°C, более предпочтительно от комнатной температуры до 100°C и особенно предпочтительно от 40 до 80°C.
Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 10 часов, более предпочтительно от 10 минут до 5 часов и особенно предпочтительно от 30 минут до 2 часов.
Преобразование соединение формулы (J-2) в соединение формулы (J-3) можно осуществлять, позволяя соединению формулы (J-2) реагировать с нитритным сложным эфиром в присутствии кислоты. В качестве нитритного сложного эфира можно использовать этилнитрит, н-пропилнитрит, изопропилнитрит, н-бутилнитрит, изобутилнитрит, трет-бутилнитрит, изоамилнитрит или сходные с ними. Из них особенно предпочтительным является изоамилнитрит ввиду его доступности.
Более того, соединение формулы (J-3) также можно получать путем добавления водного раствора нитрита натрия к смеси соединения формулы (J-2) и кислоты.
Нитритный сложный эфир или нитрит натрия используют в количестве, предпочтительно в 1-10 раз, более предпочтительно в 1-5 раз и наиболее предпочтительно в 1-3 раз превышающем молярное количество соединения формулы (J-2).
Примеры кислоты, используемой в этом способе, включают протонные кислоты, такие как хлористоводородная кислота, серная кислота, уксусная кислота, п-толуолсульфоновая кислота, камфорсульфоновая кислота, трифторукусусная кислота или трифторметансульфоновая кислота; и кислоты Льюиса, такие как хлорид алюминия, хлорид цинка или хлорид железа. Кислоты, которые предпочтительно используют в этом способе, представляют собой протонные кислоты. Среди них более предпочтительными являются хлористоводородная кислота, серная кислота и уксусная кислота и наиболее предпочтительной является хлористоводородная кислота. Когда используют хлористоводородную кислоту, хлорангидрид кислоты, такой как ацетилхлорид, можно добавлять к спиртам, таким как этанол, так чтобы в системе образовалась хлористоводородная кислота. Кислоту используют в количестве, предпочтительно в 0,01-100 раз, более предпочтительно в 0,1-10 раз и наиболее предпочтительно в 1-5 раз превышающем количество соединения формулы (J-2).
Тип растворителя, используемого в этом способе, конкретно не ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают воду, нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол, простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля, кетоны, такие как ацетон или 2-бутанон; спирты, такие как метанол, этанол или 2-пропанол; амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; карбоновые кислоты, такие как уксусная кислота, пропионовая кислота или трифторуксусная кислота; сложные эфиры, такие как этилацетат или изопропилацетат; и сульфоксиды, такие как диметилсульфоксид. Эти растворители можно использовать в комбинации. Примеры предпочтительного растворителя включают воду, простые эфиры, спирты, амиды и карбоновые кислоты. Из них более предпочтительными являются карбоновые кислоты и спирты.
Используемое количество растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно 1-50 (об./масс.), более предпочтительно в 1-10 раз (об./масс.) и более предпочтительно в 1-5 раз (об./масс.) превышающем количество соединения формулы (J-2).
Температура реакции варьирует в зависимости от используемого растворителя. Предпочтительно она составляет от 0 до 130°C, более предпочтительно от комнатной температуры до 100°C и особенно предпочтительно от комнатной температуры до 70°C.
Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 10 часов, более предпочтительно от 10 минут до 5 часов и особенно предпочтительно от 30 минут до 3 часов.
Для преобразования соединения формулы (J-3) в соединение формулы (J-4) можно одновременно проводить хлорирование кольца пиразина и образование кольца изоксазола или эти реакции можно проводить последовательно.
В этой реакции в качестве реагента(ов) используют оксихлорид фосфора, тионилхлорид, пентахлорид фосфора, трихлорид фосфора, пирокатехилфосфотрихлорид, дихлортрифенилфосфоран и оксалилхлорид по отдельности или в комбинации из двух или более типов. Из них более предпочтительными являются оксихлорид фосфора и тионилхлорид с точки зрения выхода и затрат и особенно предпочтительным является оксихлорид фосфора. Реагент используют в количестве, предпочтительно в 1-20 раз, более предпочтительно в 2-10 раз и наиболее предпочтительно в 2-5 раз превышающем количество соединения формулы (J-3).
Тип растворителя, используемого в этом способе, конкретно не ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают нитрилы, такие как ацетонитрил; ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля; кетоны, такие как ацетон или 2-бутанон; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон; мочевины, такие как 1,3-диметил-2-имидазолидинон; и сложные эфиры, такие как этилацетат или изопропилацетат. Эти растворители можно использовать в комбинации. Примеры предпочтительного растворителя включают нитрилы, ароматические углеводороды, простые эфиры, амиды, мочевины и сложные эфиры. Из них более предпочтительными являются ароматические углеводороды и амиды.
Для повышения скорости реакции предпочтительно добавляют диметилформамид.
Используемое количество растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно в 1-50 раз (об./масс.), более предпочтительно 1-10 раз (об./масс.) и наиболее предпочтительно 1-5 раз (об./масс.) превышающем соединение формулы (J-3).
Температура реакции варьирует в зависимости от используемого растворителя. Предпочтительно она составляет от 0 до 130°C, более предпочтительно от комнатной температуры до 100°C и особенно предпочтительно от 50 до 80°C. Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 20 часов, более предпочтительно от 30 минут до 10 часов и особенно предпочтительно от 1 до 5 часов.
Далее описана реакция получения соединения формулы (III) с использованием соединения формулы (I). В этой реакции в качестве оснований можно использовать различные типы неорганических оснований или органических оснований. Примеры предпочтительного неорганического основания включают фторид калия, фторид цезия, бикарбонат натрия, карбонат калия, карбонат натрия, гидроксид лития, гидроксид натрия, гидроксид калия, фосфат натрия, фосфат калия, гидрофосфат натрия и борат натрия. Примеры или предпочтительного органического основания включают триэтиламин, этил(диизопропил)амин, пиридин и пиколин. Более предпочтительные основания включают бикарбонат натрия, карбонат калия, карбонат натрия, гидроксид натрия, гидроксид калия, фосфат натрия, фосфат калия и моногидрофосфат натрия.
Основание используют в количестве, предпочтительно в 0,1-100 раз, более предпочтительно в 0,5-30 раз и наиболее предпочтительно в 1-10 раз превышающем молярное количество соединения формулы (I).
Тип растворителя, используемого в этом способе, конкретно на ограничен, при условии, что он не влияет на реакцию. Примеры растворителя включают воду, нитрилы, такие как ацетонитрил, ароматические углеводороды, такие как бензол, толуол или ксилол, простые эфиры, такие как диоксан, тетрагидрофуран или диметиловый эфир этиленгликоля, кетоны, такие как ацетон или 2-бутанон; спирты, такие как метанол, этанол или 2-пропанол; амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; сложные эфиры, такие как этилацетат или изопропилацетат; и сульфоксиды, такие как диметилсульфоксид. Эти растворители можно использовать в комбинации. В качестве такого растворителя предпочтительным является использование исключительно воды или использование смешанного растворителя из воды и органических растворителей (спирты, нитрилы, простые эфиры или сульфоксиды), смешивающихся с водой. Более того, также предпочтительно использовать растворители, которые являются несмешивающимися с водой, такие как ароматические углеводороды, сложные эфиры или простые эфиры, и чтобы реакцию проводили в двухфазной системе из такого не смешивающегося растворителя и воды. Более того, растворитель, смешивающийся с водой, можно смешивать с растворителем, не смешивающимся с водой, а затем смешанный таким образом растворитель можно использовать. Примеры более предпочтительного растворителя включают ароматические углеводороды, простые эфиры, спирты, сложные эфиры и воду. Более предпочтительной является двухфазная система из ароматического углеводорода и воды. Количество используемого растворителя конкретно не ограничено. Растворитель используют в количестве, предпочтительно в 1-50 раз (об./масс.) и более предпочтительно в 1-15 раз (об./масс.) превышающем количество соединения формулы (I).
Температура реакции предпочтительно составляет 200°C или ниже и более предпочтительно от 0 до 150°C. Время реакции конкретно не ограничено. Предпочтительно оно составляет от 5 минут до 50 часов, более предпочтительно от 5 минут до 24 часов и особенно предпочтительно от 5 минут до 5 часов.
В такой реакции также можно использовать описанные выше катионные катализаторы фазового переноса или неионные катализаторы фазового переноса.
В этой реакции особенно предпочтительно, чтобы X в соединении формулы (I) представлял собой атом фтора, и Y представлял собой -C(=O)R, где R обозначает необязательно замещенную алкоксигруппу.
В соответствии со способом, описанным в Shin Jikken Kagaku Koza (New Experimental Chemistry Course). Vol. 14, pp. 1151-1154 (под редакцией Chemical Society of Japan. 1977), воду добавляют к соединению формулы (III) (1) в кислых условиях, (2) в основных условиях в присутствии или в отсутствие гиперацидных условий, или (3) в нейтральных условиях, тем самым, получая соединение формулы (IV).
В этой реакции особенно предпочтительно, чтобы X представлял собой атом фтора.
ПРИМЕРЫ
Далее способ безопасного и легкого получения T-705A и T-705 с использованием соединения формулы (I) по настоящему изобретению в качестве промежуточного соединения описан с помощью следующих конкретных примеров. Следует отметить, что в данных спектров ЯМР в представленных ниже примерах “с” обозначает синглет, “д” обозначает дублет, “т” обозначает триплет, “кв" обозначает квартет, "квинт" обозначает квинтет, “септ” обозначает септет, “н” обозначает нонуплет, “дд” обозначает квартет с неравными расстояниями, “м” обозначает мультиплет, и “ушир.с.” обозначает уширенную линию.
[Химическая формула 22]
Пример синтеза 1: Синтез (B-1)
11,3 л воды и 1090 г гидроксида натрия добавляли в 30-л реакционную емкость, изготовленную из стекла, и растворяли в ней. К полученному раствору добавляли 4000 г (A-1) (реагент Tokyo Chemical Industry Co. Ltd.), а затем полученную смесь перемешивали при внутренней температуре 70°C в течение 30 минут. После этого к реакционному раствору добавляли 2270 г хлорида натрия и растворяли в нем, а затем полученную реакционную смесь охлаждали до 0°C или ниже. К реакционной смеси медленно добавляли 2270 мл концентрированной хлористоводородной кислоты и 11,3 л этилацетата. После завершения разделения водный слой удаляли. К полученному органическому слою добавляли 11,3 л насыщенного солевого раствора и после завершения разделения жидкости водный слой отделяли. Полученный органический слой концентрировали при пониженном давлении. К полученному таким образом осадку добавляли 5,00 л толуола, а затем раствор толуола концентрировали пониженном давлении. К полученному веществу вновь добавляли 5,00 л толуола с последующим вакуумным концентрированием. В результате получали 3200 г масла светло-желтого цвета (B-1). Выход: 95,1%.
1H-ЯМР (400 МГц, CDCl3) δ: 9,09 (уш., 1H), 4,97 (с, 1H), 3,64-3,77 (м, 4H), 1,28 (т, J=7,1 Гц, 6H).
Пример синтеза 2: Синтез (C-1)
1,48 кг (10,0 моль) (B-1) растворяли в 7,40 л ацетонитрила, а затем к полученному раствору добавляли 1,10 кг (5,25 моль) сульфата аминоацетонитрила. Поддерживая внутреннюю температуру на уровне 5°C или ниже, к смеси добавляли 1,91 кг (10,0 моль) 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида. Поддерживая внутреннюю температуру от 0°C до 6°C, к смеси добавляли 2,07 кг (20,0 моль) триэтиламина в течение 90 минут. Полученную реакционную оставляли при комнатной температуре в течение ночи. К реакционной смеси добавляли 3,00 л воды, а затем растворитель отгоняли при пониженном давлении. К осадку добавляли 7,40 л этилацетата, а затем полученную смесь перемешивали в течение 10 минут. После этого реакционный раствор оставляли стоять, а затем водный слой удаляли. Органический слой охлаждали. Затем при поддержании внутренней температуры 6°C или ниже к нему добавляли приблизительно 2,00 л хлористоводородной кислоты в концентрации 1,00 моль/л, так чтобы pH водного слоя довести до pH 5. Далее, к нему добавляли 1,00 л воды, а затем смесь перемешивали и оставляли стоять, так чтобы удалить водный слой. К органическому слою добавляли 3,00 л насыщенного солевого раствора, а затем полученную смесь перемешивали и оставляли стоять, так чтобы удалить водный слой. Из полученного органического слоя отгоняли растворитель при пониженном давлении, а затем к осадку добавляли 2,00 л толуола с последующей вакуумной дистилляцией. Далее к полученному веществу добавляли 1,00 л толуола с последующей вакуумной дистилляцией с получением 1,11 кг светло-коричневого масла (C-1). Выход: 59,7%.
1H ЯМР (CDCl3) δ: 1,27 (6H, т, J=7,2 Гц), 3,65 (2H, кв, J=7,2 Гц), 3,69 (2H, кв, J=7,2 Гц), 4,22 (1H, с), 4,23 (1H, с), 4,86 (1H, с), 6,80-7,10 (1H, уш.).
Пример синтеза 3: Синтез (E-1)
В атмосфере азота 1,44 кг (12,8 моль) или трет-бутоксида калия растворяли в 10,4 л тетрагидрофурана. После этого, поддерживая внутреннюю температуру 10°C или ниже, раствор 2,08 кг (11,2 моль) (C-1), растворенного в 2,08 л тетрагидрофурана, капельно добавляли к раствору в течение 1 часа. Затем к раствору добавляли 1,58 кг (13,4 моль) диметилоксалата, а затем полученную смесь перемешивали при 40°C в течение 2 часов. После этого в реакционный раствор дополнительно добавляли 16,0 л метанола с последующим концентрированием с получением раствора (D-1) в метаноле. Полученный таким образом раствор использовали непосредственно в следующей реакции.
1H ЯМР (ДМСО-d6) δ: 1,15 (6H, т, J=7,2 Гц), 3,52-3,58 (4H, м), 3,60 (3H, с), 4,75 (1H, с), 7,88 (1H, уш.).
В атмосфере азота, поддерживая внутреннюю температуру 10°C или ниже, 0,979 л (13,2 моль) трифторуксусной кислоты капельно добавляли к полученному раствору (D-1) в метаноле, а затем к смеси добавляли 0,815 кг (11,7 моль) гидроксиламина гидрохлорида. При перемешивании полученную смесь нагревали до температуры кипячения с обратным холодильником в течение 5 часов. После этого реакционный раствор охлаждали до комнатной температуры, а затем метанол отгоняли при пониженном давлении. После этого к осадку добавляли 10,4 л этилацетата и 8,30 л 20,0% водного раствора хлорида натрия. После перемешивания проводили разделение жидкостей, а затем водный слой удаляли. К оставшемуся органическому слою добавляли 8,30 л или 20,0% водного раствора хлорида натрия и 0,260 кг бикарбоната натрия. После перемешивания проводили разделение жидкостей, а затем водный слой удаляли. К органическому слою вновь добавляли 8,30 л 20,0% водного раствора хлорида натрия добавляли. После перемешивания проводили разделение жидкостей, а затем водный слой удаляли. Органический слой концентрировали с получением 3,14 кг (чистота: 49,0%) коричневого масла (E-1). Выход из (C-1): 47,9%.
1H ЯМР (CDCl3) δ: 1,32 (6H, т, J=6,8 Гц), 3,50-3,80 (м, 4H), 3,98 (3H, с), 4,93 (1H, с), 5,82 (2H, уш.), 9,29 (1H, уш.).
Пример синтеза 4: синтез (F-1)
1,55 кг (5,40 моль) (E-1) растворяли в 3,90 л уксусной кислоты, а затем к раствору добавляли 213 г (1,12 моль) п-толуолсульфоновой кислоты моногидрата. Полученную смесь подвергали реакции при внутренней температуре от 77°C до 80°C в течение 2 часов. После этого полученную реакционную смесь охлаждали до комнатной температуры, а затем к смеси добавляли 8,00 л воды с последующим перемешиванием в течение 20 минут. После этого осадок собирали фильтрацией, а затем его промывали водой до тех пор, пока значение pH фильтрата не составляло pH 5 или более. После этого полученное вещество сушили при 40°C в течение ночи с получением 615 г твердого вещества светло-желтого цвета (F-1). Выход: 58,4%.
1H ЯМР (ДМСО-d6) δ: 3,99 (3H, с), 8,26 (1H, с), 12,75-13,00 (1H, уш.).
Следует отметить, что, поскольку (F-1) представляло собой твердое вещество и имело низкую летучесть и низкую способность вызывать раздражение кожи, его можно было безопасно и без труда использовать в последующей реакции.
Пример синтеза 5: Синтез (G-1)
156 г (0,800 моль) (F-1) смешивали с 373 мл (4,00 моль) оксихлорида фосфора, а затем к смеси добавляли 110 г (0,800 моль) гидрохлорида триэтиламина. Полученную смесь подвергали реакции при внутренней температуре 85°C в течение 4 часов. После этого реакционный раствор охлаждали до комнатной температуры. Смешанный раствор 800 мл толуола и 1600 мл воды охлаждали на льду, а затем полученную, как описано выше, реакционную смесь добавляли к смешанному раствору в течение 1 часа при поддержании внутренней температуры от 25°C до 30°C. Реакционную смесь далее перемешивали при внутренней температуре от 22°C до 23°C в течение 1 часа, а затем ее оставляли стоять. Водный слой удаляли и к органическому слою добавляли 800 мл насыщенного солевого рассола. После этого реакционную смесь перемешивали, а затем оставляли стоять и водный слой удаляли. Это действие повторяли четыре раза. На 4-ый раз значение pH водного слоя составляло pH 6. К полученному органическому слою добавляли безводный сульфат натрия с последующим перемешиванием. После удаления сульфата натрия растворитель отгоняли при пониженном давлении с получением 152 г твердого вещества светло-коричневого цвета (G-1). Выход: 88,9%.
1H ЯМР (CDCl3) δ: 4,14(3H, с), 8,65(1H, с).
Следует отметить, что, поскольку (G-1) представляло собой твердое вещество и имело низкую летучесть и низкую способность вызывать раздражение кожи, его можно было безопасно и без труда использовать в последующей реакции.
Пример синтеза 6: Синтез (H-1)
В атмосфере азота перемешивали смешанный раствор 2,00 г (10,3 ммоль) (F-1) и 40,0 мл ацетонитрила, а затем к нему капельно добавляли 1,88 мл (15,4 ммоль) 2,2-дифтор-1,3-диметилимидазолидина. После завершения капельного добавления полученную смесь перемешивали при температуре от 80 до 90°C в течение 3 часов. После этого реакционный раствор концентрировали при пониженном давлении, а затем полученный осадок отделяли и очищали хроматографией на силикагеле (элюент: гексан/этилацетат=2/1 (объемное соотношение)). В результате было получено 0,900 г белого твердого вещества (H-1). Выход: 44,4%.
1H ЯМР (CDCl3) δ: 8,53(1H, д, J=6,6 Гц), 4,14 (3H, с)
19F-ЯМР (CDCl3) δ: -78,74 (1F, д, J=6,6 Гц).
Следует отметить, что, поскольку (H-1) представляло собой твердое вещество и имело низкую летучесть и низкую способность вызывать раздражение кожи, его можно было безопасно и без труда использовать в последующей реакции.
Пример синтеза 7: Синтез (H-1)
1,80 г (31,0 ммоль) фторида калия смешивали с 22,0 мл диметилсульфоксида, а затем к смеси добавляли 15,0 мл толуола с последующим перемешиванием. После этого толуол отгоняли при пониженном давлении при внешней температуре 80°C при 70 мм рт.ст. и к осадку добавляли 1,07 г (5,00 ммоль) (G-1) с последующей реакцией при внутренней температуре 80°C в течение 3 часов. После этого продукт реакции охлаждали до комнатной температуры и к нему добавляли 300 мл этилацетата и 200 мл воды. Реакционную смесь перемешивали, а затем оставляли стоять и водный слой удаляли. Это действие повторяли два раза. Затем к органическому слою добавляли 50,0 мл насыщенного солевого раствора, а затем полученную смесь перемешивали и оставляли стоять, так чтобы удалить водный слой. Полученное вещество сушили над сульфатом магния, а затем фильтровали. Отфильтрованное вещество концентрировали с получением смеси 0,830 г твердого вещества коричневого цвета (H-1) и 0,03 г (G-1). Выход: 84,0%.
(Соотношение ингредиентов в смеси было вычислено исходя из целочисленных значений спектров ЯМР).
Пример синтеза 8: Синтез T-705A
3,00 мл тетрагидрофурана, 3,00 мл воды и 55,0 мг (1,38 ммоль) гидроксида натрия добавляли к 200 мг (1,01 ммоль) (H-1) и при перемешивании полученную смесь нагревали при 80°C в течение 3 часов. После этого реакционный раствор охлаждали до комнатной температуры и к нему добавляли ионобменную смолу DOWEX (зарегистрированный торговый знак) 50W×2 - 200 (H). После этого полученное вещество фильтровали и концентрировали с получением 126 мг T-705A в форме твердого вещества желтого цвета. Выход: 89,7%.
1H ЯМР (ДМСО-d6) δ: 8,22 (1H, д, J=8,1 Гц), 13,85 (1H, уш.).
19F-ЯМР (ДМСО-d6) δ: -94,13(1H, уш.).
Понятно, что в соответствии со способом обработки T-705A основным водным раствором, описанным в примере получения 4 патентного документа 1 или в примере получения 2 патентного документа 2 или сходных с ними, T-705 можно получать с использованием T-705A, синтезированного способом по настоящему изобретению.
Кроме того, ниже описаны примеры синтеза производного пиразино[2,3-d]изоксазола по настоящему изобретению и т.п.
[Химическая формула 23]
Пример синтеза 9: Синтез (A-1a)
10,7 г (50,0 ммоль) (G-1) смешивали с 50,0 мл этилового спирта, а затем к смеси добавляли и 17,4 мл (100 ммоль) диизопропилэтиламина и 0,610 г (5,00 ммоль) 4-диметиламинопиридина. Полученную смесь подвергали реакции при внутренней температуре 80°C в течение 2,5 часов, а затем ее охлаждали до комнатной температуры. Реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1), с получением 7,39 г белого твердого вещества (A-1a). Выход: 64,8%.
1H ЯМР (CDCl3) δ: 8,62 (1H, с), 4,60 (2H, кв, J=7,0 Гц), 1,51 (3H, т, J=7,0 Гц).
Пример синтеза 10: Синтез (A-2)
42,7 г (0,200 моль) (G-1) смешивали с 500 мл изопропилового спирта, а затем к смеси добавляли 42,0 мл (0,300 моль) триэтиламина. Полученную смесь подвергали реакции при внутренней температуре 80°C в течение 2 часов, а затем ее охлаждали до комнатной температуры. Реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 41,6 г белого твердого вещества (A-2). Выход: 86,0%.
1H ЯМР (CDCl3) δ: 8,63 (1H, с), 5,45 (1H, кв, J=6,0 Гц), 1,49 (6H, с).
Пример синтеза 11: Синтез (A-3)
32,0 г (150 ммоль) (G-1) смешивали со 150 мл 1-бутилового спирта, а затем к смеси добавляли 52,3 мл (300 ммоль) диизопропилэтиламина и 1,83 г (15,0 ммоль) 4-диметиламинопиридина. Полученную смесь подвергали реакции при внутренней температуре 90°C в течение 2 часов, а затем ее охлаждали до комнатной температуры. Реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 25,1 г белого твердого вещества (A-3). Выход: 65,4%.
1H ЯМР (CDCl3) δ: 8,63 (1H, с), 4,55 (2H, т, J=6,8 Гц), 1,81-1,89 (2H, м), 1,47-1,57 (2H, м), 1,00 (3H, т, J=7,2 Гц).
Пример синтеза 12: Синтез (A-4)
1,00 г (4,39 ммоль) (A-1a) растворяли в 10,0 мл изобутилового спирта, а затем к раствору добавляли 107 мг (0,878 ммоль) 4-диметиламинопиридина. Полученную смесь перемешивали при нагревании при 100°C в течение 5 часов. После этого реакционный раствор охлаждали до комнатной температуры, а затем концентрировали. Осадок очищали хроматографией на силикагеле (гексан:этилацетат - 4:1) с получением 0,780 г (A-4) в форме светло-желтого масла. Выход: 69,4%.
1H ЯМР (CDCl3) δ: 1,07 (6H, д, J=6,8 Гц), 2,19 (1H, h, J=6,7 Гц), 4,32 (2H, д, J=6,6 Гц), 8,63 (1H, с).
Пример синтеза 13: Синтез (A-5)
1,07 г (5,00 ммоль) (G-1) смешивали с 5,00 г неопентилового спирта, а затем к смеси добавляли 1,70 мл (10,0 ммоль) диизопропилэтиламина. Полученную смесь подвергали реакции при внутренней температуре 100°C в течение 5 часов. После этого реакционный раствор охлаждали до комнатной температуры и к нему добавляли 30,0 мл или этилацетата и 20,0 мл воды. Реакционную смесь перемешивали, а затем оставляли стоять, и водный слой удаляли. Это действие повторяли два раза. Органический слой концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат - 4:1) с получением 0,750 г или белого твердого вещества (A-5). Выход: 55,6%.
1H ЯМР (CDCl3) δ: 8,63 (1H, д, J=6,6 Гц), 4,23 (2H, с), 1,09 (9H, с).
Пример синтеза 14: Синтез (A-6)
2,28 г (10,0 ммоль) или (A-1a) смешивали с 10,0 г 1-гексилового спирта, а затем к смеси добавляли 3,48 мл (20,0 ммоль) диизопропилэтиламина и 0,120 г 4-диметиламинопиридина. Полученную смесь подвергали реакции при внутренней температуре 80°C в течение 3,5 часов. После этого реакционный раствор охлаждали до комнатной температуры. Реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан: этилацетат=4:1) с получением 2,30 г белого твердого вещества (A-6). Выход: 81,0%.
1H ЯМР (CDCl3) δ: 8,63 (1H, с), 4,54 (2H, т, J=6,8 Гц), 1,81-1,90 (2H, м), 1,31-1,53(6H, м), 0,91 (3H, т, J=7,2 Гц).
Пример синтеза 15: Синтез (A-7)
2,14 г (10,0 ммоль) (G-1) смешивали с 10,0 г циклогексилового спирта и 2,00 мл (10,0 ммоль) диизопропилэтиламина, а затем к смеси добавляли 0,210 г 4-диметиламинопиридина. Полученную смесь подвергали реакции при внутренней температуре 100°C в течение 1 часа. После этого реакционный раствор охлаждали до комнатной температуры и к нему добавляли 100 мл этилацетата и 50,0 мл хлористоводородной кислоты (1 моль/л). Реакционную смесь перемешивали, а затем оставляли стоять и водный слой удаляли. Это действие повторяли два раза. Затем к органическому слою добавляли 20,0 мл насыщенного солевого раствора, а затем полученную смесь стерилизовали и оставляли стоять, чтобы удалить водный слой. Полученное вещество сушили над сульфатом магния, а затем фильтровали. Фильтрат концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат - 4:1) с получением 1,30 г белого твердого вещества (A-7). Выход: 46,1%.
1H ЯМР (CDCl3) δ: 8,62 (1H, с), 5,19-5,28 (1H, м), 1,31-2,08 (10H, м).
Пример синтеза 16: Синтез (A-8)
2,28 г (10,0 ммоль) (A-1a) смешивали с 2,08 мл (20,0 ммоль) бензилового спирта, а затем к смеси добавляли 20,0 мл диизопропилэтиламина. Полученную смесь подвергали реакции при внутренней температуре 80°C в течение 1 часа. После этого реакционный раствор охлаждали до комнатной температуры. Реакционный раствор концентрировали, а затем осадок перекристаллизовывали (гексан/этилацетат) с получением 0,780 г белого твердого вещества (A-8). Выход: 26,9%.
1H ЯМР (CDCl3) δ: 8,62 (1H, с), 7,32-7,57 (5H, м), 5,57 (2H, с).
Пример синтеза 17: Синтез (A-9)
0,430 г (2,00 ммоль) (G-1) смешивали с 4,00 мл этилового спирта, а затем к смеси добавляли 0,220 мл (2,00 ммоль) бензилового спирта. Полученную смесь подвергали реакции при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали и осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 0,490 г твердого вещества желтого цвета (A-9). Выход: 84,8%.
1H ЯМР (CDCl3) δ: 8,64 (1H, с), 7,31-7,42 (5H, м), 4,77(2H, д, J=6,0 Гц).
Пример синтеза 18: Синтез (A-10)
6,41 г (30,0 ммоль) (G-1) смешивали с 16,0 мл (150 ммоль) диэтиламина, а затем полученную смесь подвергали реакции при 50°C в течение 45 минут. Реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 6,33 г желтого твердого вещества (A-10). Выход: 82,7%.
1H ЯМР (CDCl3) δ: 8,60 (1H, с), 3,67 (2H, т, J=7,2 Гц), 3,47 (2H, т, J=7,2 Гц), 1,34 (3H, т, J=7,2 Гц), 1,26 (3H, т, J=7,2 Гц).
Пример синтеза 19: Синтез (A-11)
2,14 г (10,0 ммоль) (G-1) смешивали с 15,0 мл метилового спирта, а затем к смеси добавляли 0,860 мл (10,5 ммоль) пирролидина. Полученную смесь подвергали реакции при комнатной температуре в течение 40 минут. После этого раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат - 4:1) с получением 2,27 г твердого вещества желтого цвета (A-11). Выход: 89,7%.
1H ЯМР (CDCl3) δ: 8,61 (1H, с), 3,72-3,81 (4H, м), 1,98-2,05 (4H, м).
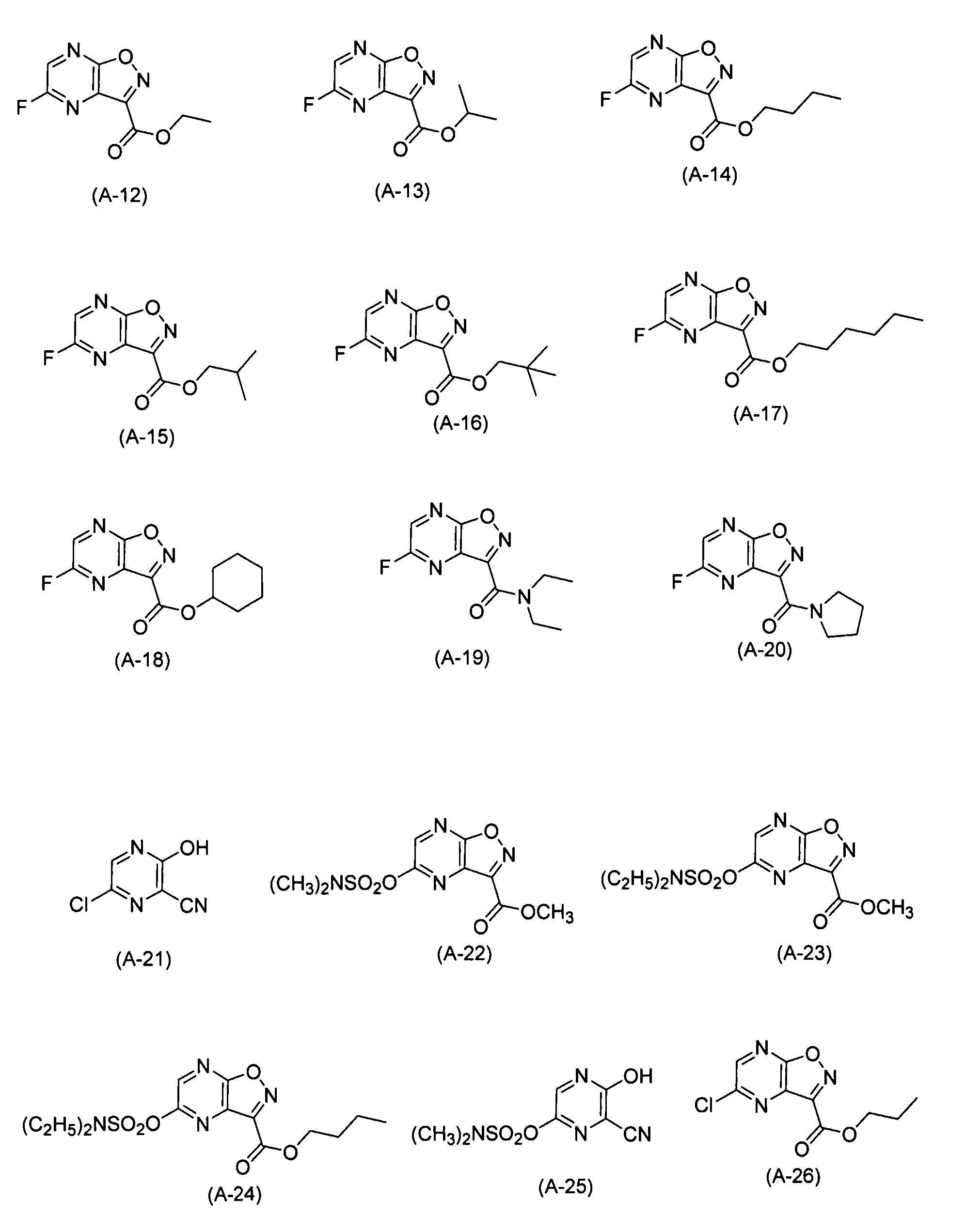
Пример синтеза 20: Синтез (A-12)
0,630 г (10,8 ммоль) или фторида калия смешивали с 14,4 мл диметилсульфоксида, а затем растворитель отгоняли при пониженном давлении при внешней температуре 80°C при 3-5 гПа. После этого к осадку добавляли 15,0 мл сухого диметилсульфоксида и 0,820 г (3,60 ммоль) (A-1a), а затем полученную смесь подвергали реакции при внутренней температуре 90°C в течение 4 часов. В соответствии с анализом с использованием высокоэффективной жидкостной хроматографии, было выявлено, что уровень продукции составляет 97,0%. (В качестве внутреннего стандарта использовали дифениловый эфир.)
Пример синтеза 21: Синтез (A-13)
3,50 г (60,0 ммоль) фторида калия смешивали с 250 мл диметилсульфоксида, а затем растворитель отгоняли при пониженном давлении при внешней температуре 130°C при 21 мм рт.ст. После этого к осадку добавляли 80,0 мл сухого диметилсульфоксида и 4,83 г (20,0 ммоль) (A-2), а затем полученную смесь подвергали реакции при внутренней температуре 90°C в течение 4 часов. Реакционный раствор охлаждали до комнатной температуры и к нему добавляли 100 мл толуола и 100 мл воды. Затем реакционную смесь перемешивали и оставляли стоять и водный слой удаляли. Это действие повторяли два раза. Затем к полученному органическому слою добавляли 100 мл насыщенного солевого раствора, а затем полученную смесь перемешивали и оставляли стоять, так чтобы удалить водный слой. Полученное вещество сушили над сульфатом магния, а затем фильтровали. Фильтрат концентрировали с получением 3,97 г твердого вещества (A-13). Выход: 88,2%.
1H ЯМР (CDCl3) δ: 8,50 (1H, д, J=6,4 Гц), 5,46 (1H, кв, J=6,4 Гц), 1,49 (6H, д, J=6,4 Гц).
19F-ЯМР (CDCl3) δ: -79,16 (IF, д, J=6,4 Гц).
Пример синтеза 22: Синтез (A-14)
0,360 г (6,20 ммоль) фторида калия смешивали с 8,00 мл диметилсульфоксида и к смеси добавляли 14,0 мл толуола с последующим перемешиванием. После этого толуол отгоняли при пониженном давлении при внешней температуре 80°C при 70 мм рт.ст. После этого к осадку добавляли 0,510 г (2,00 ммоль) (A-3), а затем полученную смесь подвергали реакции при внутренней температуре 80°C в течение 2 часов и при внутренней температуре 90°C в течение 1,5 часов. После этого реакционный раствор охлаждали до комнатной температуры и к нему добавляли 30,0 мл толуола и 20,0 мл воды. Реакционную смесь перемешивали, а затем оставляли стоять и водный слой удаляли. Это действие повторяли три раза. Затем к органическому слою добавляли 20,0 мл насыщенного солевого раствора, а затем полученную смесь оставляли стоять, так чтобы удалить водный слой. Полученное вещество сушили над сульфатом магния, а затем фильтровали. Фильтрат концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан: этилацетат - 4:1) с получением 0,400 г белого твердого вещества (A-14). Выход: 83,7%.
1H ЯМР (CDCl3) δ: 8,51 (1H, д, J=6,4 Гц), 4,54 (2H, т, J=6,8 Гц), 1,81-1,89 (2H, м), 1,47-1,57 (2H, м), 1,00 (3H, т, J=7,2 Гц).
19F-ЯМР (CDCl3) δ: -79,02(1F, д, J=6,4 Гц).
Пример синтеза 23: Синтез (A-15)
В атмосфере азота 4,68 мл диметилсульфоксида и 10,0 мл толуола добавляли к 203 мг (3,51 ммоль) фторида калия, а затем полученную смесь нагревали до 70°C. После этого толуол отгоняли при пониженном давлении. Далее к осадку добавляли 0,300 г (1,17 ммоль) (A-4), и при перемешивании полученную смесь подвергали реакции при 80°C в течение 2 часов. В результате анализа ВЭЖХ реакционного раствора было выявлено, что уровень продукции составляет 84,0%. (В качестве внутреннего стандарта использовали дифениловый эфир.)
Пример синтеза 24: Синтез (A-16)
0,190 г белого твердого вещества (A-16) получали из 0,230 г (4,00 ммоль) фторида калия, 6,00 мл диметилсульфоксида и 0,400 г (1,50 ммоль) (A-5) с помощью тех же действий, как и действия, использованные в примере синтеза 22. Выход: 50,1%.
1H ЯМР (CDCl3) δ: 8,51 (1H, д, J=6,6 Гц), 4,22 (2H, с), 1,09 (9H, с).
19F-ЯМР (CDCl3) δ: -79,11(1F, д, J=6,6 Гц).
Пример синтеза 25: Синтез (A-17)
0,620 г (10,7 ммоль) фторида калия смешивали с 14,4 мл диметилсульфоксида, а затем к смеси добавляли 14,0 мл толуола с последующим перемешиванием. После этого толуол отгоняли при пониженном давлении при внешней температуре 80°C при 70 мм.рт.ст. К осадку добавляли 0,510 г (A-6), а затем полученную смесь подвергали реакции при внутренней температуре 80°C в течение 2 часов и при внутренней температуре 90°C в течение 2 часов. В соответствии с анализом с использованием высокоэффективной жидкостной хроматографии, уровень продукции составлял 89,0%. (В качестве внутреннего стандарта использовали дифениловый эфир.)
Пример синтеза 26: Синтез (A-18)
0,310 г белого твердого вещества (A-18) получали из 0,470 г (8,10 ммоль) фторида калия, 11,0 мл диметилсульфоксида и 0,760 г (2,70 ммоль) (A-7) с помощью тех же действий, как и действия, использованные в примере синтеза 22. Выход: 43,4%.
1H ЯМР (CDCl3) δ: 88,49 (1H, д, J=6,6 Гц), 5,20-5,27 (1H, м), 1,99-2,07 (2H, м), 1,33-1,90 (8H, м).
Пример синтеза 27: Синтез (A-19)
0,590 г (10,0 ммоль) фторида калия, 12,0 мл диметилсульфоксида и 0,760 г (3,00 ммоль) (A-10) подвергали реакции при 80°C в течение 4 часов с помощью тех же действий, как и действия, использованные в примере синтеза 25. В результате было выявлено, что уровень продукции составляет 65,0%.
Пример синтеза 28: Синтез (A-20)
0,520 г (9,00 ммоль) фторида калия, 12,0 мл диметилсульфоксида и 0,76 г (3,00 ммоль) (A-11) подвергали реакции при 80°C в течение 4 часов с помощью тех же действий, как и действия, использованные в примере синтеза 25. В результате было выявлено, что уровень продукции составляет 81,0%.
Пример синтеза 29: Синтез (A-21)
1,00 мл тетрагидрофурана, 1,00 мл воды и 20,0 мг (0,491 ммоль) или гидроксида натрия добавляли к 100 мг (0,468 ммоль) (G-1) и при перемешивании полученную смесь нагревали при 80°C в течение 1 часа. После этого реакционный раствор охлаждали до комнатной температуры и к нему добавляли ионообменную смолу DOWEX (зарегистрированный торговый знак) 50W×2 - 200 (H). После этого полученное вещество фильтровали и концентрировали с получением 70,0 мг (A-21) в форме твердого вещества желтого цвета. Выход: 95,9%.
1H ЯМР (ДМСО-d6) δ: 8,44 (1Н, с), 13,85 (1Н, уш.).
Пример синтеза 30: Синтез T-705A
2,00 мл толуола, 1,00 мл воды и 0,224 г (2,66 ммоль) бикарбоната натрия добавляли к 0,500 г (2,22 ммоль) (A-15) и при перемешивании полученную смесь подвергали реакции при 80°C в течение 3 часов, и при 100°C в течение 5 часов. В результате анализа с использованием высокоэффективной жидкостной хроматографии было выявлено, что уровень продукции составил 92,0%.
Пример синтеза 31: Синтез (A-22)
Смешанный раствор 1,36 г (7,00 ммоль) (F-1), 20,0 мл ацетонитрила и 1,49 мл (9,00 ммоль) диизопропилэтиламина охлаждали на льду, а затем к раствору добавляли 0,860 мл (8,00 ммоль) диметилкарбаминовой кислоты. Полученную смесь подвергали реакции при комнатной температуре в течение 1 часа. После этого к реакционному раствору добавляли 100 мл или этилацетата и 100 мл воды. Затем реакционную смесь оставляли стоять, и водный слой удаляли. Это действие повторяли два раза. Затем органический слой сушили над сульфатом магния, а затем концентрировали. Затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 0,740 г белого твердого вещества (A-22). Выход: 35,1%.
1H ЯМР (CDCl3) δ: 8,57 (1H, с), 4,10 (3H, с), 3,17 (6H, с).
Пример синтеза 32: Синтез (A-23)
Смешанный раствор 6,66 г (34,0 ммоль) (F-1), 50,0 мл ацетонитрила и 6,80 мл (41,0 ммоль) диизопропилэтиламина охлаждали на льду, а затем к раствору добавляли 5,10 мл (41,0 ммоль) хлорангидрида диметилкарбаминовой кислоты и 0,370 г (3,00 ммоль) 4-диметиламинопиридина. Полученную смесь подвергали реакции при комнатной температуре в течение ночи. После этого реакционный раствор концентрировали, а затем к концентрату добавляли 100 мл этилацетата и 100 мл разбавленной хлористоводородной кислоты (1 моль/л). Реакционную смесь перемешивали, а затем оставляли стоять и водный слой удаляли. Это действие повторяли два раза. Затем органический слой сушили над сульфатом магния, а затем концентрировали. Затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 9,97 г жидкости желтого цвета (A-23). Выход: 88,4%.
1H ЯМР (CDCl3) δ: 8,56 (1H, с), 4,11 (3H, с), 3,59 (4H, кв, J=7,2 Гц), 1,31 (6H, т, J=7,2 Гц).
Пример синтеза 33: Синтез (A-24)
1,65 г (5,00 ммоль) (A-23) смешивали с 5,00 мл 1-бутилового спирта, а затем к смеси добавляли 1,70 мл (10,0 ммоль) диизопропилэтиламина. Полученную смесь подвергали реакции при внутренней температуре 80°C в течение 2 часов. После этого реакционный раствор охлаждали до комнатной температуры. Реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле (гексан:этилацетат=4:1) с получением 1,47 г жидкости желтого цвета (A-24). Выход: 79,0%.
1H ЯМР (CDCl3) δ: 8,55 (1H, с), 4,52 (2H, т, J=6,8 Гц), 3,57 (4H, кв, J=7,2 Гц), 1,80-1,88 (2H, м), 1,48-1,57 (2H, м), 1,30 (6H, т, J=7,2 Гц), 1,00 (3H, т, J=7,2 Гц).
Пример синтеза 34: Синтез (A-25)
10,0 мл тетрагидрофурана, 10,0 мл воды и 0,270 г (6,75 ммоль) гидроксида натрия добавляли к 1,51 г (5,00 ммоль) (A-22) и при перемешивании полученную смесь нагревали при 80°C в течение 40 минут. После этого реакционный раствор охлаждали до комнатной температуры и к нему добавляли ионообменную смолу DOWEX (зарегистрированный торговый знак) 50W×2 - 200 (H). Полученное вещество фильтровали и концентрировали с получением 0,610 г (A-25) в форме желтого твердого вещества. Выход: 50,0%.
1H ЯМР (ДМСО-d6) δ: 8,04 (1H, с), 2,88 (3H, с).
Пример синтеза 35: Синтез (A-21)
2,00 мл толуола, 1,00 мл воды и 0,340 г (4,00 ммоль) бикарбоната натрия добавляли к 0,510 г (2,00 ммоль) (A-10) и при перемешивании смесь подвергали реакции при 100°C в течение 2 часов. В результате анализа с использованием высокоэффективной жидкостной хроматографии реакционного раствора было выявлено, что скорость продукции составляет 13,0%.
Пример синтеза 36: Синтез T-705A
0,750 мл N,N-диметилформамида, 0,120 мл воды и 114 мг (1,16 ммоль) или ацетата калия добавляли к 185 мг (0,773 ммоль) (A-14). При перемешивании полученную смесь нагревали при 80°C в течение 3 часов, а затем ее охлаждали до комнатной температуры. В результате анализа HPLC реакционной смеси было выявлено, что скорость продукции составляет 57,0%.
Пример синтеза 37: Синтез T-705A
2,00 мл тетрагидрофурана, 1,00 мл воды и 37,0 мг (0,930 ммоль) гидроксида натрия добавляли к 185 мг (0,773 ммоль) (A-14). При перемешивании полученную смесь нагревали при 80°C в течение 1 часа, а затем ее охлаждали до комнатной температуры. В результате анализа ВЭЖХ реакционной смеси было выявлено, что скорость продукции составляет 94,9%.
Пример синтеза 38: Синтез T-705A
2,00 мл изопропилового спирта, 1,00 мл воды и 37,0 мг (0,930 ммоль) гидроксида натрия добавляли к 185 мг (0,773 ммоль) (A-14). При перемешивании полученную смесь нагревали при 80°C в течение 1 часа, а затем ее охлаждали до комнатной температуры. В результате анализа ВЭЖХ реакционной смеси было выявлено, что скорость продукции составляет 85,6%.
Пример синтеза 39: Синтез (T-705A)
В атмосфере азота 10,0 мл диметилсульфоксида и 15,0 мл N,N-диметилформамида добавляли к 460 мг (7,91 ммоль) фторида калия и N,N-диметилформамид отгоняли. Кроме того, к нему добавляли 0,460 г (2,61 ммоль) (A-22) и при перемешивании полученную смесь подвергали реакции при 80°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и к нему добавляли 50,0 мл этилацетата и 30,0 мл воды. Реакционную смесь перемешивали, а затем оставляли стоять. После разделения жидкостей полученный органический слой промывали 30 мл воды, а затем 30 мл насыщенного солевого раствора и растворитель отгоняли с помощью испарителя. К осадку добавляли 2,0 мл диметилсульфоксида, 1,0 мл воды и 0,120 г (3,00 ммоль) гидроксида натрия и при перемешивании полученную смесь подвергали реакции при 80°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и к нему добавляли 0,48 мл (2,41 ммоль) дициклогексиламина. После доведения значения pH раствора до pH=9 с помощью концентрированной хлористоводородной кислоты, к нему добавляли 2,0 мл ацетона и 3,0 мл воды. Выпавшие в осадок кристаллы отфильтровывали с получением 0,25 г соли T-705A с дициклогексиламином в виде твердого вещества светло-коричневого цвета.
Пример синтеза 40: Синтез (A-26)
5,00 г (23,4 ммоль) (G-1) смешивали с 23,0 мл 1-пропанола и после этого к смеси добавляли 8,00 мл (46,8 ммоль) диизопропилэтиламина и 0,250 г (2,00 ммоль) 4-диметиламинопиридина. Полученную смесь подвергали реакции при 80°C в течение 70 минут и при 90°C в течение 110 минут. После этого реакционный раствор концентрировали, а затем осадок подвергали хроматографии на силикагеле с получением 3,50 г твердого вещества светло-желтого цвета (A-26). Выход: 61,8%.
1H ЯМР (CDCl3) δ: 8,64 (1H, с), 4,51 (2H, кв, J=6,8 Гц), 1,85-1,94 (2H, м) , 1.08 (3H, т, J=7,2 Гц).
Поскольку соединения (A-1a)-(A-20), (A-22)-(A-24) и (A-26) имели низкую летучесть и низкую способность вызывать раздражение кожи, с ними можно обращаться безопасно и без труда.
[Химическая формула 26]
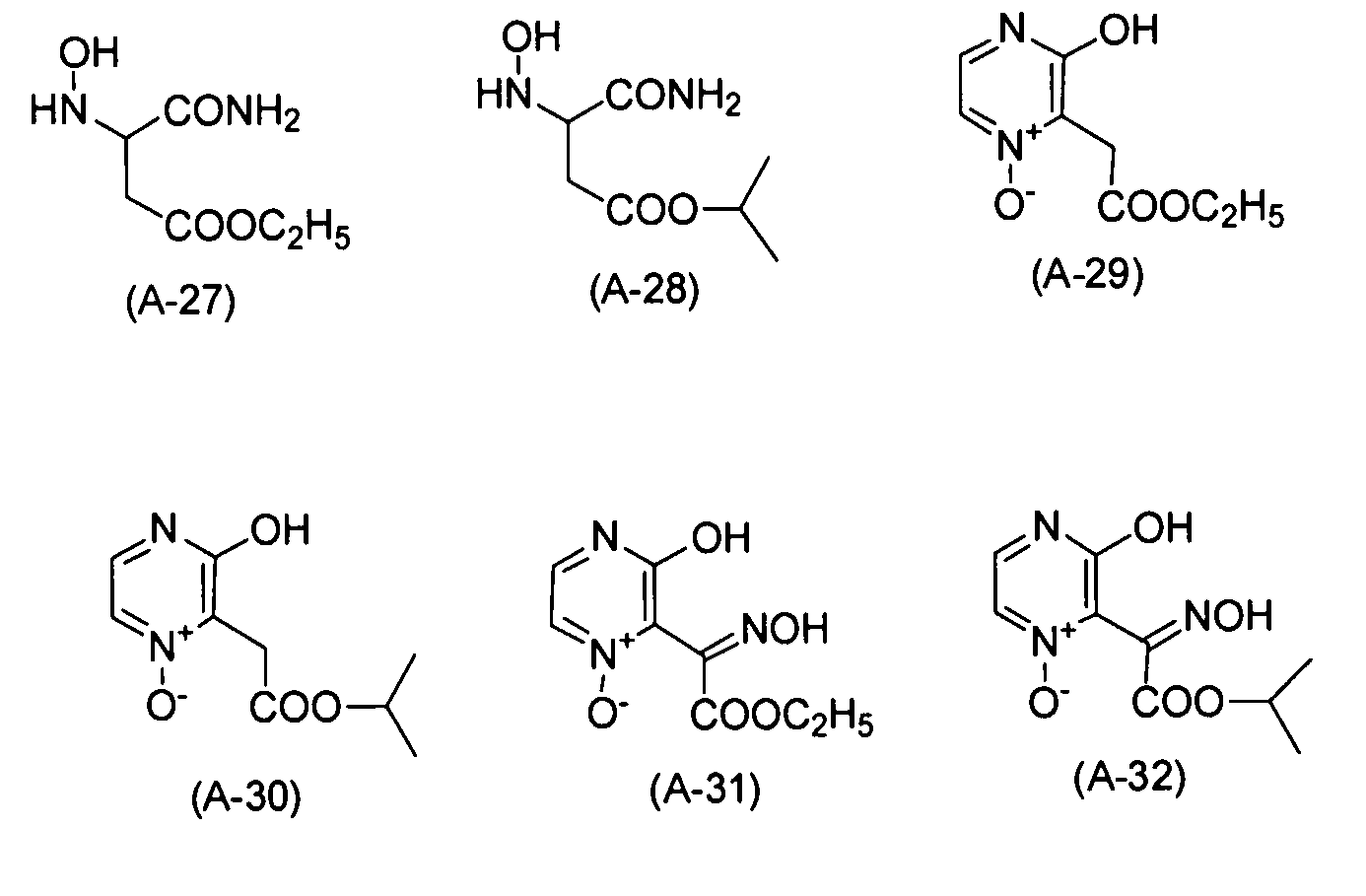
Пример синтеза 41: Синтез (A-27)
В атмосфере азота 54 г (0,377 моль) этил-(Z)-4-амино-4-оксо-2-бутеноата растворяли в 300 мл этанола и при поддержании внутренней температуры при 15-25°C к раствору капельно добавляли 26,2 г (0,396 моль) 50% водного раствора гидроксиламина. Полученную смесь перемешивали при 20°C в течение 4,5 часов, а затем реакционный раствор охлаждали до -20°C. Выпавшее в осадок твердое вещество отфильтровывали. Полученное таким образом твердое вещество промывали 50,0 мл охлажденного этилацетата с получением 42,4 г белого твердого вещества (A-27). Выход: 63,8%.
1H ЯМР (ДМСО-d6) δ: 1,18 (3H, т, J=7,2 Гц), 2,40 (1H, дд, J=7,6, 15,6 Гц), 2,59 (1H, дд, J=6,0, 16,0 Гц), 3,63 (1H, дд, J=6,0, 7,6 Гц), 4,05 (2H, кв, J=6,8 Гц), 5,80 (1H, уш.), 7,12 (1H, уш.), 7,31 (1H, уш.), 7,51 (1H, уш.).
Пример синтеза 42: Синтез (A-29)
56,0 г (0,386 моль) 40% водного раствора глиоксаля, 125 мл тетрагидрофурана, 125 мл воды и 13,3 г (0,0955 моль) или карбоната калия смешивали, а затем полученную смесь охлаждали до 12°C. Затем к реакционному раствору добавляли 33,7 г (0,191 моль) (A-27), а затем полученную смесь перемешивали при 20°C в течение 3 часов. После этого к реакционному раствору добавляли 11,6 г уксусной кислоты, а затем полученную смесь концентрировали до 80,0 г. К концентрату добавляли 30,0 мл насыщенного солевого раствора, а затем полученную смесь перемешивали. Выпавшее в осадок твердое вещество фильтровали и фильтрат промывали 30 мл насыщенного солевого раствора, а затем сушили с получением 14,0 г твердого вещества светло-розового цвета (A-29). Выход: 36,7%.
1H ЯМР (ДМСО-d6) δ: 1,16 (3H, т, J=6,8 Гц), 3,69 (2H, с), 4,06 (2H, кв, J=7,2 Гц), 7,24 (1H, д, J=6,0 Гц), 7,54 (1H, д, J=5,6 Гц), 12,3 (1H, уш.).
Пример синтеза 43: Синтез (A-31)
Раствор, полученный добавлением 1,60 мл (22,5 ммоль) ацетилхлорида к 30,0 мл этанола, добавляли к смеси 5,00 г (0,0252 моль) (A-29) и 65,0 мл или этанола, а затем полученную таким образом смесь перемешивали. После этого к реакционному раствору добавляли 3,70 мл (27,5 ммоль) изоамилнитрита, а затем полученную смесь перемешивали при комнатной температуре в течение 4 часов. После этого к реакционному раствору добавляли 0,500 мл (3,72 ммоль) изоамилнитрита и полученную смесь далее перемешивали при комнатной температуре в течение 3,3 часов. К этому реакционному раствору добавляли раствор, полученный добавлением 0,500 мл (7,04 ммоль) ацетилхлорида к 5,00 мл или этанола, и 0,500 мл (3,72 ммоль) или изоамилнитрита, и полученную таким образом смесь оставляли на ночь. Затем к реакционному раствору добавляли раствор, полученный добавлением 1,60 мл (22,5 ммоль) или ацетилхлорида к 20,0 мл этанола, и 1,50 мл (11,1 ммоль) изоамилнитрита, а затем полученную таким образом смесь перемешивали при 35°C. После этого растворитель концентрировали при пониженном давлении. К полученному веществу добавляли ацетонитрил, а затем полученную смесь охлаждали на льду. Выпавшее в осадок твердое вещество фильтровали с получением 4,10 г белого твердого вещества (A-31). Выход: 71,5%.
1H ЯМР (ДМСО-d6) δ: 13,0 (1H, уш.), 12,4 (1H, уш.), 7,66 (1H, д, J=6,0 Гц), 7,29 (1H, д, J=6,0 Гц), 4,20 (2H, кв, J=7,0 Гц), 1,21 (3H, т, J=7,0 Гц).
Пример синтеза 44: Синтез (A-1a)
12,0 мл толуола и 12,0 мл диметилформамида охлаждали на льду, а затем добавляли 4,60 мл (49,3 ммоль) оксихлорида фосфора. После этого к смеси добавляли 2,27 г (10,0 ммоль) (A-31) и полученную таким образом смесь перемешивали при 70°C в течение 4,5 часов. После этого реакционный раствор охлаждали до комнатной температуры, а затем к нему добавляли этилацетат и воду. Полученную смесь перемешивали, а затем оставляли стоять. После этого водный слой удаляли и органический слой концентрировали при пониженном давлении. Полученный осадок разделяли хроматографией на силикагеле (элюент: гексан/этилацетат - 9/1). В результате было получено 1,60 г белого твердого вещества (A-1a). Выход: 70,3%.
Пример синтеза 45: Синтез изопропил-(Z)-4-амино-4-оксо-2-бутеноата
196 г (2,00 моль) ангидрида малеиновой кислоты растворяли в 123 г (2,05 моль) 2-пропанола и 800 мл этилацетата. После этого к раствору капельно добавляли 300 мл (2,15 моль) триэтиламина при внутренней температуре 10°C или ниже в течение 1,5 часов, а затем полученную смесь перемешивали в течение 1 часа. После этого к реакционной смеси капельно добавляли 193 мл (2,03 моль) этилхлорформиата при внутренней температуре -5°C или ниже в течение 2 часов. После перемешивания смеси в течение 30 минут полученную реакционную смесь капельно добавляли к водному раствору, содержащему 300 мл (2,16 моль) 28% аммиачной воды и 250 г льда. Полученную реакционную смесь оставляли при комнатной температуре в течение ночи. После этого к продукту реакции добавляли 400 мл этилацетата, а затем перемешивали. После этого проводили разделение жидкостей в реакционном растворе, так чтобы удалить водный слой. Это действие повторяли три раза. Полученные органические слои собирали, а затем концентрировали. Проводили перекристаллизацию из смеси гексан/этилацетат с получением 50,5 г изопропил-(Z)-4-амино-4-оксо-2-бутеноата в форме белого твердого вещества. Выход: 16,1%.
1H ЯМР (ДМСО-d6) δ: 1,20 (6H, д, J=6,0 Гц), 4,94 (1H, с, J=6,4 Гц), 6,15 (1H, д, J=11,6 Гц), 6,26 (1H, д, J=12,0 Гц), 7,18 (1H, уш.), 7,57 (1H, уш.).
Пример синтеза 46: Синтез (A-28)
13,9 г (0,210 моль) 50% водного раствора гидроксиламина растворяли в 200 мл 2-пропанола. При поддержании внутренней температуры при 3,5-6°C на ледяной бане к раствору добавляли 31,4 г (0,200 моль) изопропил-(Z)-4-амино-4-оксо-2-бутеноата в течение 15 минут, а затем к нему добавляли 20,0 мл 2-пропанола. Полученный реакционный раствор хранили при комнатной температуре в течение 3 часов, а затем его оставляли стоять в холодильнике. Выпавшее в осадок твердое вещество собирали фильтрацией, а затем промывали холодным 2-пропанолом. Полученное вещество сушили при пониженном давлении при комнатной температуре с получением 22,3 г белого твердого вещества (A-28). Выход: 58,6%.
1H ЯМР (ДМСО-d6) δ: 1,18 (6H, д, J=6,4 Гц), 2,36 (1H, дд, J=8,0, 16,0 Гц), 2,55 (1H, дд, J=6,0, 16,0 Гц), 3,61 (1H, т, J=6,8 Гц), 4,87 (1H, с, J=6,4, 6,4 Гц), 5,70-5,90 (1H, уш.), 7,00-7,18 (1H, уш.), 7,20-7,35 (1H, уш.), 7,46 (1H, с).
Пример синтеза 47: Синтез (A-28)
9,43 г (60,0 ммоль) изопропил-(E)-4-амино-4-оксо-2-бутеноата растворяли в 28,3 мл тетрагидрофурана, а затем полученный раствор нагревали на водяной бане, которая была установлена на 42°C. К раствору капельно добавляли 4,16 г (63,3 ммоль) водного раствора 50% гидроксиламина в течение 20 минут, а затем полученный раствор перемешивали при 42°C в течение 1 часа. После этого к реакционному раствору добавляли 9,40 мл воды, а затем тетрагидрофуран отгоняли при пониженном давлении. С помощью 1H-ЯМР было подтверждено, что исходный материал исчезал из полученного раствора, и в нем содержалось (A-28).
1H ЯМР (D2O) δ: 1,26 (6H, д, J=6,4 Гц), 2,68 (1H, дд, J=6,8, 16,4 Гц), 2,77 (1H, дд, J=7,2, 16,0 Гц), 3,96 (1H, т, J=6,8 Гц), 5,01 (1H, с, J=6,4, 6,4 Гц).
Пример синтеза 48: Синтез (A-30)
3,72 г (25,0 ммоль) 39% водного раствора глиоксаля растворяли в 30,0 мл 2-пропанола. Внутренняя температура была установлена на 41°C на горячей водяной бане. После этого 2,38 г (12,5 ммоль) (A-28) растворяли в 2,00 мл воды и 4,00 мл 2-пропанола, а затем в полученный раствор капельно добавляли к раствору, полученному как описано выше. В ходе этого действия вместе с описанным выше раствором капельно добавляли 1 моль/л водного раствора карбоната натрия, так чтобы pH реакционного раствора можно было поддерживать при pH 8,9-9,1. Полученную реакционную смесь подвергали реакции при внутренней температуре 41°C в течение 2 часов. Внутреннюю температуру снижали до 20°C и к реакционному продукту добавляли уксусную кислоту, так чтобы довести pH до pH 6,0. Растворитель отгоняли при пониженном давлении, а затем к осадку добавляли насыщенный солевой раствор. Полученное твердое вещество собирали фильтрацией, а затем его промывали холодным насыщенным солевым раствором, а затем сушили с получением 2,89 г светло-коричневого твердого вещества (A-30). Выход: 62,3% (чистота: 57,2%).
1H ЯМР (ДМСО-d6) δ: 1,17 (6H, д, J=6,0 Гц), 3,66 (2H, с), 4,87 (1H, с, J=6,4, 6,4 Гц), 7,24 (1H, д, J=5,6 Гц), 7,53 (1H, д, J=5,6 Гц), 12,00-12,50 (1H, уш.).
Пример синтеза 49: Синтез (A-30)
12,22 г (77,8 ммоль) изопропил-(E)-4-амино-4-оксо-2-бутеноата растворяли в 19,8 мл THF и полученный раствор охлаждали до 15-20°C на водяной бане. К раствору капельно добавляли 5,14 г (77,8 ммоль) 50% водного раствора гидроксиламина в течение 1 минуты, а затем полученный реакционный раствор перемешивали при 27-30°C в течение 3 часов. С помощью 1H-ЯМР было подтверждено, что исходный материал исчез из полученного раствора, и в нем содержалось (A-28).
0,118 г бикарбоната натрия растворяли в 18,3 мл воды. К нему капельно добавляли 20,31 г (140,0 ммоль) 40% водного раствора глиоксаля и упомянутого выше раствора (A-28) в THF в течение 60 минут. В ходе этого действия вместе с описанным выше раствором также капельно добавляли 50% водный раствор гидроксида натрия, чтобы pH реакционного раствора можно было поддерживать на уровне pH 8,2-8,4 (три раствора одновременно капельно добавляли). Полученную реакционную смесь подвергали реакции при внутренней температуре 50°C в течение 1 часа. В ходе этого действия к нему капельно добавляли 50% водный раствор гидроксида натрия, так чтобы pH реакционного раствора можно было поддерживать при pH 8,4. THF отгоняли при пониженном давлении, а затем к осадку добавляли 5,0 г солевого раствора. Добавляли концентрированную хлористоводородную кислоту при внутренней температуре 40-50°C так чтобы довести pH до pH 3,0. Раствор охлаждали до 5°C в течение 1 ч и фильтровали. Твердое вещество на сите промывали два раза 10 мл воды при 5°C или ниже с последующей сушкой с получением 10,80 г твердого вещества светло-коричневого цвета (A-30) (чистота: 90%). Выход из A-28: 58,9%.
Пример синтеза 50: Синтез (A-32)
В атмосфере азота 20,0 мл изопропилового спирта добавляли к 4,60 г (21,7 ммоль) (A-30) и при перемешивании полученную смесь охлаждали до 5°C. Далее к реакционному раствору добавляли 2,86 мл (40,3 ммоль) ацетилхлорида при поддержании внутренней температуры 10°C или ниже. Температуру реакционной смеси увеличивали до 40°C и к ней капельно добавляли 5,41 мл (40,3 ммоль) изоамилнитрита. После завершения капельного добавления полученную смесь перемешивали при 25°C в течение 1,5 часов, а затем ее охлаждали до -10°C. Выпавшее в осадок твердое вещество фильтровали, а затем его два раза промывали 5,00 мл толуола. Полученное вещество сушили с получением 4,59 г твердого вещества светло-желтого цвета (A-32). Выход: 87,9%.
1H ЯМР (ДМСО-d6) δ: 1,22 (6H, д, J=6,0 Гц), 5,01 (1H, с, J=6,4 Гц), 7,28 (1H, д, J=5,6 Гц), 7,65 (1H, д, J=5,6 Гц), 12,4 (1H, уш.), 13,0 (1H, уш.).
Пример синтеза 51: Синтез (A-2)
В атмосфере азота при перемешивании смешанного раствора 25,0 г (0,104 моль) (A-32), 62,5 мл N,N-диметилформамида и 62,5 мл толуола, внутреннюю температуру поддерживали при 15°C или ниже и к смешанному раствору капельно добавляли 47,3 мл (0,510 моль) оксихлорида фосфора. После завершения капельного добавления температуру реакционного раствора увеличивали до 70°C, а затем реакционный раствор перемешивали в течение 7 часов. После этого реакционный раствор охлаждали до комнатной температуры, а затем полученную таким образом реакционную смесь медленно капельно добавляли к смешанному раствору 62,5 мл толуола и 300 мл 10% солевого раствора при внутренней температуре 10°C или ниже. После завершения разделения жидкостей органический слой два раза промывали 100 мл 10% солевого раствора, а затем 100 мл 10% раствора бикарбоната натрия и 100 мл 10% солевого раствора. Этот органический слой концентрировали, а затем осадку добавляли 7,50 мл изопропилового спирта и 150 мл гексана. Выпавшее в осадок твердое вещество фильтровали, а затем его промывали два раза 15,0 мл смешанного растворителя изопропиловый спирт/гексан=5/95 (объемное соотношение) с получением 12,6 г светло-розового твердого вещества (A-2) (чистота: 98,3%). Выход: 49,3%.
Пример синтеза 52: Синтез (A-13)
0,219 г (2,00 ммоль) хлорида тетраметиламмония, 2,32 г (40,0 ммоль) фторида калия, 9,70 мл сухого диметилсульфоксида и 38,6 мл сухого толуола смешивали. После этого толуол отгоняли при пониженном давлении при внешней температуре 120°C. После охлаждения смеси до комнатной температуры к реакционному раствору добавляли 0,203 г (1,00 ммоль) 2,4-динитрохлорбензола и 4,83 г (20,0 ммоль) (A-2), а затем полученную смесь подвергали реакции при внутренней температуре 90°C в течение 2 часов. После охлаждения смеси до комнатной температуры к реакционному раствору добавляли 0,180 мл воды, а затем полученную смесь перемешивали в течение 2,5 часов. После этого к реакционному раствору далее добавляли 0,180 мл воды, а затем полученную смесь перемешивали в течение 1 часа. После этого к реакционному раствору добавляли 14,5 мл толуола и 14,2 мл воды и полученную смесь перемешивали, а затем оставляли стоять, так чтобы удалить водный слой. Затем к органическому слою добавляли 14,5 мл насыщенного раствора бикарбоната натрия и полученную смесь перемешивали, а затем оставляли стоять, так чтобы удалить водный слой. В результате получали светло-желтый раствор (A-13), и не было выявлено черного смолянистого компонента. Этот раствор прямо использовали в дальнейшем процессе.
Пример синтеза 53: Синтез соли T-705 с дициклогексиламином
14,5 мл воды и 3,36 г (40,0 ммоль) бикарбоната натрия добавляли к раствору (A-13), полученному в описанном выше примере синтеза 52, а затем полученную смесь подвергали реакции при внешней температуре 100°C в течение 4 часов. После этого органический слой удаляли, а затем к водному слою добавляли 3,43 мл (60,0 ммоль) уксусной кислоты. Полученную смесь кипятили с обратным холодильником при пониженном давлении при внешней температуре 70°C при 100 мм рт.ст. в течение 1,5 часа. Смесь охлаждали до комнатной температуры, а затем к реакционному раствору добавляли 5,00 мл воды, 9,60 л ацетона и 3,30 мл 28% аммиачной воды. Затем к смешанному раствору капельно добавляли 3,78 мл (19,0 ммоль) дициклогексиламина в течение 10 минут, а затем полученную смесь перемешивали при комнатной температуре в течение 1 часа. После этого к реакционному раствору добавляли 9,60 мл воды, а затем полученную смесь перемешивали при внутренней температуре 5°C в течение 1 часа, а затем твердое вещество отфильтровывали. Твердое вещество на Nutsche последовательно промывали 10,0 мл воды, смешанным раствором из 5,00 мл ацетона и 5,00 мл воды, и 10,0 мл ацетона при 10°C или ниже. Полученное вещество сушили с получением 5,37 г соли T-705A с дициклогексиламином в форме твердого вещества светло-коричневого цвета. Выход: 83,0%; и чистота ВЭЖХ: 99,0%.
Пример синтеза 54: Синтез T-705
10,0 мл толуола и водный раствор гидроксида натрия (полученный растворением 0,656 г гидроксида натрия в 20,0 мл воды) добавляли к 5,00 г (15,6 ммоль) соли T-705A с дициклогексиламином, а затем полученную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор оставляли стоять в течение 10 минут, а затем верхний слой удаляли. К нижнему слою добавляли 10,0 мл толуола, а затем его перемешивали и оставляли стоять в течение 10 минут. После этого верхний слой удаляли. К нижнему слою добавляли водный раствор гидроксида натрия (полученный растворением 0,593 г гидроксида натрия в 5,00 мл воды). Затем при поддержании внутренней температуры при 15-20°C к смеси капельно добавляли 2,68 мл (31,5 ммоль) 40,0% об./масс. пероксида водорода. Полученную смесь перемешивали при 25°C в течение 30 минут и pH раствора доводили до pH 6,5-8,0 хлористоводородной кислотой. После этого смесь нагревали до 40°C, так чтобы твердое вещество полностью растворялось в растворе. После этого к реакционному раствору добавляли 0,250 г активированного угля (SHIRASAGI A), а затем полученную смесь перемешивали при 40°C в течение 30 минут с последующей фильтрацией. Твердое вещество на Nutsche промывали 5,00 мл воды, а затем к смешанному раствору фильтрата и промывочного раствора добавляли хлористоводородную кислоту при внутренней температуре 35-45°C, так чтобы его pH довести до pH 3-4. Смешанный раствор охлаждали до 0-5°C, а затем его перемешивали в течение 1 часа. После этого выпавшее в осадок твердое вещество фильтровали, а затем его промывали 5,00 мл воды и 5,00 мл изопропилового спирта с получением 2,06 г белого твердого вещества (T-705). Выход: 84,0%.
Промышленная применимость
Настоящее изобретение пригодно для получения T-705, который пригоден для лечения, такого как профилактика и терапия, инфекции вирусом гриппа, и т.п.
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/8bacb13550faa95992860361a2c5d0c6.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/044d77ee6887305473355695fbaaad95.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/efe8b114509786aa6cc088846128902a.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/ede84d5fc7ff78fc7887a19f7512fabf.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/0a314b47e7c93bba1c4785429b498f31.jpg)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/4516e947601be9f057cfdcf65af43fd2.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/400faab28e48592a5667c2a83810cce3.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/e6212fc9c352669b3e71931706b7bad3.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/df8207b267f1b0cc2f4205bb18bd6ed4.jpg)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/4a1897e8bbb0d99a1a79daf5a7ec7a60.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/0792e4a67e1f5e340c01d38d21cf310c.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/e5cd4d68c86d12d3be7fb631f29ae71c.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/75259111c8868a47612b245077500421.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/4eb17dbd102687a3bd7d25c896b9c8ea.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/c019f563ea115de3ee03437e7e67600c.jpg)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/34b876c91f1fbfb0a1ea537bce8b5071.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/5a7c54ca4a5a026a539a1916f56a2163.jpg)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/bbafe9da9d06d8b163b5e379ef5a1b4a.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/cdd0eb7493232f6bb3a70401466ca2be.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/d9c9ca033a1823ee02ded22f7696983e.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/75cb57b501d2c74b4dceabbd789a746b.jpg)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/1d9ef436a0d6f18c07e31a38bf8f9318.jpg)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/ce69a66e3086d6cd29c271101f22f730.png)
![ПРОИЗВОДНОЕ ПИРАЗИНО[2,3-d]ИЗОКСАЗОЛА](https://fips.edrid.ru/images/rid/80/a6/02/94689ff1609ada077ff470b292727226.png)
Новый моногидрат производного нафтиридина и способ его получения
Способ применения комбинации производного бензофенона или его соли и иммуносупрессора и фармацевтическая композиция, содержащая данные компоненты
Применение бензофенонового производного или его соли и ингибитора tnf-α в комбинации, и фармацевтическая композиция, содержащая данное производное или его соль и ингибитор
Таблетки и гранулированные порошки, содержащие 6-фтор-3-гидрокси-2-пиразинкарбоксамид
Производное n-ацилантраниловой кислоты или его соль
Соединение, представляющее собой простой полиэфир, отверждающее средство, использующее соединение, представляющее собой простой полиэфир, и способ получения соединения, представляющего собой простой полиэфир
Устройство формирования изображений и способ управления устройством формирования изображений
Устройство обработки изображений, способ и устройство формирования изображения
Элемент формирования цветного изображения
Элемент формирования цветных изображений
Новый моногидрат производного нафтиридина и способ его получения
Способ применения комбинации производного бензофенона или его соли и иммуносупрессора и фармацевтическая композиция, содержащая данные компоненты
Применение бензофенонового производного или его соли и ингибитора tnf-α в комбинации, и фармацевтическая композиция, содержащая данное производное или его соль и ингибитор
Таблетки и гранулированные порошки, содержащие 6-фтор-3-гидрокси-2-пиразинкарбоксамид
Производное n-ацилантраниловой кислоты или его соль
Соединение, представляющее собой простой полиэфир, отверждающее средство, использующее соединение, представляющее собой простой полиэфир, и способ получения соединения, представляющего собой простой полиэфир
Устройство формирования изображений и способ управления устройством формирования изображений
Устройство обработки изображений, способ и устройство формирования изображения
Элемент формирования цветного изображения
Элемент формирования цветных изображений

